Attached files
| file | filename |
|---|---|
| EXCEL - IDEA: XBRL DOCUMENT - HALOZYME THERAPEUTICS, INC. | Financial_Report.xls |
| EX-32 - CERTIFICATION OF CHIEF EXECUTIVE OFFICER AND CHIEF FINANCIAL OFFICER - HALOZYME THERAPEUTICS, INC. | d250665dex32.htm |
| EX-31.1 - CERTIFICATION OF CHIEF EXECUTIVE OFFICER - HALOZYME THERAPEUTICS, INC. | d250665dex311.htm |
| EX-31.2 - CERTIFICATION OF CHIEF FINANCIAL OFFICER - HALOZYME THERAPEUTICS, INC. | d250665dex312.htm |
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-Q
(Mark One)
[X] QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the quarterly period ended September 30, 2011
OR
[ ] TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from to .
Commission File Number 001-32335
HALOZYME THERAPEUTICS, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 88-0488686 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) | |
| 11388 Sorrento Valley Road, San Diego, CA | 92121 | |
| (Address of principal executive offices) | (Zip Code) | |
(858) 794-8889
(Registrant’s telephone number, including area code)
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes [X] No [ ]
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes [X] No [ ]
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer ¨ | Accelerated filer x | Non-accelerated filer ¨ (Do not check if a smaller reporting company) |
Smaller reporting company ¨ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes [ ] No [X]
The number of outstanding shares of the registrant’s common stock, par value $0.001 per share, was 103,691,643 as of November 2, 2011.
Table of Contents
INDEX
2
Table of Contents
PART I — FINANCIAL INFORMATION
CONDENSED CONSOLIDATED BALANCE SHEETS
| September 30, 2011 |
December 31, 2010 |
|||||||
| (Unaudited) | (Note) | |||||||
| ASSETS | ||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 66,329,358 | $ | 83,255,848 | ||||
| Accounts receivable |
6,237,904 | 2,328,268 | ||||||
| Inventory |
103,443 | 193,422 | ||||||
| Prepaid expenses and other assets |
4,297,814 | 3,720,896 | ||||||
|
|
|
|
|
|||||
| Total current assets |
76,968,519 | 89,498,434 | ||||||
| Property and equipment, net |
1,274,753 | 1,846,899 | ||||||
|
|
|
|
|
|||||
| Total Assets |
$ | 78,243,272 | $ | 91,345,333 | ||||
|
|
|
|
|
|||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 1,219,905 | $ | 3,820,368 | ||||
| Accrued expenses |
8,864,705 | 8,605,569 | ||||||
| Deferred revenue |
3,707,795 | 2,917,129 | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
13,792,405 | 15,343,066 | ||||||
| Deferred revenue, net of current portion |
36,667,445 | 55,176,422 | ||||||
| Deferred rent, net of current portion |
728,259 | 474,389 | ||||||
| Commitments and contingencies (Note 11) |
||||||||
| Stockholders’ equity: |
||||||||
| Preferred stock — $0.001 par value; 20,000,000 shares authorized; no shares issued and outstanding |
- | - | ||||||
| Common stock — $0.001 par value; 150,000,000 shares authorized; 103,647,930 and 100,580,849 shares issued and outstanding at September 30, 2011 and December 31, 2010, respectively |
103,648 | 100,581 | ||||||
| Additional paid-in capital |
253,557,547 | 245,502,670 | ||||||
| Accumulated deficit |
(226,606,032) | (225,251,795) | ||||||
|
|
|
|
|
|||||
| Total stockholders’ equity |
27,055,163 | 20,351,456 | ||||||
|
|
|
|
|
|||||
| Total Liabilities and Stockholders’ Equity |
$ | 78,243,272 | $ | 91,345,333 | ||||
|
|
|
|
|
|||||
| Note: | The condensed consolidated balance sheet at December 31, 2010 has been derived from audited financial statements at that date. It does not include, however, all of the information and notes required by U.S. generally accepted accounting principles for complete financial statements. |
See accompanying notes to condensed consolidated financial statements.
3
Table of Contents
CONDENSED CONSOLIDATED STATEMENTS OF OPERATIONS
(UNAUDITED)
| Three Months Ended September 30, |
Nine Months Ended September 30, |
|||||||||||||||
| 2011 | 2010 | 2011 | 2010 | |||||||||||||
| Revenues: |
||||||||||||||||
| Product sales |
$ | 1,156,903 | $ | 98,100 | $ | 1,487,822 | $ | 695,440 | ||||||||
| Revenues under collaborative agreements |
21,785,525 | 3,298,407 | 52,187,447 | 9,356,151 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total revenues |
22,942,428 | 3,396,507 | 53,675,269 | 10,051,591 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Operating expenses: |
||||||||||||||||
| Cost of product sales |
11,723 | 7,214 | 201,675 | 96,413 | ||||||||||||
| Research and development |
13,514,352 | 12,448,865 | 42,647,265 | 35,840,475 | ||||||||||||
| Selling, general and administrative |
4,263,520 | 3,374,069 | 12,237,152 | 10,488,568 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total operating expenses |
17,789,595 | 15,830,148 | 55,086,092 | 46,425,456 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Operating income (loss) |
5,152,833 | (12,433,641) | (1,410,823) | (36,373,865) | ||||||||||||
| Interest and other income, net |
12,360 | 24,065 | 56,586 | 25,889 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net income (loss) |
$ | 5,165,193 | $ | (12,409,576) | $ | (1,354,237) | $ | (36,347,976) | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net income (loss) per share: |
||||||||||||||||
| Basic |
$ | 0.05 | $ | (0.13) | $ | (0.01) | $ | (0.39) | ||||||||
| Diluted |
$ | 0.05 | $ | (0.13) | $ | (0.01) | $ | (0.39) | ||||||||
| Shares used in computing net income (loss) per share: |
||||||||||||||||
| Basic |
103,223,352 | 93,626,893 | 102,282,904 | 92,342,665 | ||||||||||||
| Diluted |
105,009,189 | 93,626,893 | 102,282,904 | 92,342,665 | ||||||||||||
See accompanying notes to condensed consolidated financial statements.
4
Table of Contents
CONDENSED CONSOLIDATED STATEMENTS OF CASH FLOWS
(UNAUDITED)
| Nine Months Ended September 30, |
||||||||
| 2011 | 2010 | |||||||
| Operating activities: |
||||||||
| Net loss |
$ | (1,354,237) | $ | (36,347,976) | ||||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||
| Share-based compensation |
3,916,329 | 3,745,976 | ||||||
| Depreciation and amortization |
851,613 | 1,164,162 | ||||||
| (Gain) loss on disposal of equipment |
(992) | 8,431 | ||||||
| Changes in operating assets and liabilities: |
||||||||
| Accounts receivable |
(3,909,636) | 1,621,833 | ||||||
| Inventory |
89,979 | 82,971 | ||||||
| Prepaid expenses and other assets |
(576,918) | (3,317,779) | ||||||
| Accounts payable and accrued expenses |
(2,183,864) | (2,184,007) | ||||||
| Deferred rent |
89,453 | (226,699) | ||||||
| Deferred revenue |
(17,718,311) | (2,557,349) | ||||||
|
|
|
|
|
|||||
| Net cash used in operating activities |
(20,796,584) | (38,010,437) | ||||||
|
|
|
|
|
|||||
| Investing activities: |
||||||||
| Purchases of property and equipment |
(271,521) | (315,807) | ||||||
|
|
|
|
|
|||||
| Net cash used in investing activities |
(271,521) | (315,807) | ||||||
|
|
|
|
|
|||||
| Financing activities: |
||||||||
| Proceeds from exercises of stock options |
4,141,615 | 737,267 | ||||||
| Proceeds from issuance of common stock, net |
- | 59,965,059 | ||||||
|
|
|
|
|
|||||
| Net cash provided by financing activities |
4,141,615 | 60,702,326 | ||||||
|
|
|
|
|
|||||
| Net (decrease) increase in cash and cash equivalents |
(16,926,490) | 22,376,082 | ||||||
| Cash and cash equivalents at beginning of period |
83,255,848 | 67,464,506 | ||||||
|
|
|
|
|
|||||
| Cash and cash equivalents at end of period |
$ | 66,329,358 | $ | 89,840,588 | ||||
|
|
|
|
|
|||||
| Supplemental disclosure of non-cash investing and financing activities: |
||||||||
| Accounts payable for purchases of property and equipment |
$ | 6,954 | $ | 109,705 | ||||
See accompanying notes to condensed consolidated financial statements.
5
Table of Contents
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(UNAUDITED)
| 1. | Organization and Business |
Halozyme Therapeutics, Inc. (“Halozyme” or the “Company”) is a biopharmaceutical company dedicated to the development and commercialization of recombinant human enzymes that either transiently modify tissue under the skin to facilitate injection of other therapies or correct diseased tissue structures for clinical benefit. The Company’s existing products and its products under development are based primarily on intellectual property covering the family of human enzymes known as hyaluronidases.
The Company’s operations to date have involved: (i) organizing and staffing its operating subsidiary, Halozyme, Inc.; (ii) acquiring, developing and securing its technology; (iii) undertaking product development for its existing products and a limited number of product candidates; and (iv) supporting the development of partnered product candidates. The Company currently has multiple proprietary programs in various stages of research and development. In addition, the Company has collaborative partnerships with F. Hoffmann-La Roche, Ltd. and Hoffmann-La Roche, Inc. (“Roche”), Baxter Healthcare Corporation (“Baxter”), ViroPharma Incorporated (“ViroPharma”) and Intrexon Corporation (“Intrexon”) to apply the Company’s proprietary Enhanze™ Technology to the partners’ biological therapeutic compounds. The Company also had a partnership with Baxter, under which Baxter had worldwide marketing rights for the Company’s marketed product, Hylenex® recombinant (hyaluronidase human injection), (the “Hylenex Partnership”). Hylenex recombinant is a human recombinant formulation of hyaluronidase that has received approval from the U.S. Food and Drug Administration (“FDA”) to facilitate subcutaneous fluid administration for achieving hydration; to increase the dispersion and absorption of other injected drugs; and in subcutaneous urography for improving resorption of radiopaque agents. In January 2011, the Company and Baxter mutually agreed to terminate the Hylenex Partnership. The Company’s technology is also being used in ICSI Cumulase®, a third party’s marketed product used for in vitro fertilization (“IVF”). Currently, the Company has received only limited revenue from the sales of active pharmaceutical ingredients (“API”) to the third party that produces ICSI Cumulase, in addition to other revenues from its collaborative partnerships.
| 2. | Summary of Significant Accounting Policies |
Basis of Presentation
The accompanying interim unaudited condensed consolidated financial statements have been prepared in accordance with United States generally accepted accounting principles (“U.S. GAAP”) and with the rules and regulations of the U.S. Securities and Exchange Commission (“SEC”) related to a quarterly report on Form 10-Q. Accordingly, they do not include all of the information and disclosures required by U.S. GAAP for a complete set of financial statements. These interim unaudited condensed consolidated financial statements and notes thereto should be read in conjunction with the audited consolidated financial statements and notes thereto included in the Company’s Annual Report on Form 10-K for the year ended December 31, 2010, filed with the SEC on March 11, 2011. The unaudited financial information for the interim periods presented herein reflects all adjustments which, in the opinion of management, are necessary for a fair presentation of the financial condition and results of operations for the periods presented, with such adjustments consisting only of normal recurring adjustments. Operating results for interim periods are not necessarily indicative of the operating results for an entire fiscal year.
The condensed consolidated financial statements include the accounts of Halozyme Therapeutics, Inc. and its wholly owned subsidiary, Halozyme, Inc. All intercompany accounts and transactions have been eliminated.
The preparation of consolidated financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the amounts reported in the Company’s consolidated financial statements and accompanying notes. On an ongoing basis, the Company evaluates its estimates and judgments, which are based on historical and anticipated results and trends and on various other assumptions that management
6
Table of Contents
believes to be reasonable under the circumstances. By their nature, estimates are subject to an inherent degree of uncertainty and, as such, actual results may differ from management’s estimates.
Adoption of Recent Accounting Pronouncements
Effective January 1, 2011, the Company adopted on a prospective basis Financial Accounting Standards Board’s (“FASB”) Accounting Standards Update (“ASU”) No. 2010-17, Revenue Recognition (Topic 605): Milestone Method of Revenue Recognition (“Milestone Method”). ASU No. 2010-17 states that the Milestone Method is a valid application of the proportional performance model when applied to research or development arrangements. Accordingly, an entity can make an accounting policy election to recognize a payment that is contingent upon the achievement of a substantive milestone in its entirety in the period in which the milestone is achieved. The Milestone Method is not required and is not the only acceptable method of revenue recognition for milestone payments. The adoption of ASU No. 2010-17 did not have a material impact on the Company’s consolidated financial position or results of operations.
Effective January 1, 2011, the Company adopted on a prospective basis FASB’s ASU No. 2009-13, Revenue Recognition (Topic 605): Multiple-Deliverable Revenue Arrangements. ASU No. 2009-13 requires an entity to allocate arrangement consideration at the inception of an arrangement to all of its deliverables based on their relative selling prices. ASU No. 2009-13 eliminates the use of the residual method of allocation and requires the relative-selling-price method in all circumstances in which an entity recognizes revenue for an arrangement with multiple deliverables subject to Accounting Standards Code 605-25. The Company accounted for the collaborative arrangements with ViroPharma and Intrexon under the provisions of ASU No. 2009-13, which resulted in revenue recognition patterns that are materially different from those recognized for the Company’s existing multiple-element arrangements.
Pending Adoption of Accounting Pronouncements
In June 2011, the FASB issued ASU No. 2011-05, Comprehensive Income (Topic 220): Presentation of Comprehensive Income. In ASU No. 2011-05, an entity has the option to present the total of comprehensive income, the components of net income, and the components of other comprehensive income either in a single continuous statement of comprehensive income or in two separate but consecutive statements. In both choices, an entity is required to present each component of net income along with total net income, each component of other comprehensive income along with a total for other comprehensive income, and a total amount for comprehensive income. ASU No. 2011-05 eliminates the option to present the components of other comprehensive income as part of the statement of changes in stockholders’ equity. The amendments in ASU No. 2011-05 do not change the items that must be reported in other comprehensive income or when an item of other comprehensive income must be reclassified to net income. The amendments in ASU No. 2011-05 are effective for fiscal years, and interim periods within those years, beginning after December 15, 2011. The Company does not expect the adoption of ASU No. 2011-05 to have a material impact on its consolidated financial position or results of operations.
Revenue Recognition
The Company generates revenues from product sales and collaborative agreements. The Company recognizes revenues in accordance with the authoritative guidance for revenue recognition. The Company recognizes revenue when all of the following criteria are met: (1) persuasive evidence of an arrangement exists; (2) delivery has occurred or services have been rendered; (3) the seller’s price to the buyer is fixed or determinable; and (4) collectibility is reasonably assured.
Product Sales — Revenue from the sales of API for ICSI Cumulase is recognized when the transfer of ownership occurs, which is upon shipment to the Company’s distributor. The Company is obligated to accept returns for product that does not meet product specifications. Historically, the Company has not had any product returns as a result of not meeting product specifications.
Prior to the termination of the Hylenex Partnership with Baxter in January 2011, the Company supplied Baxter with API for Hylenex recombinant at its fully burdened cost plus a margin. Baxter filled and finished Hylenex recombinant and held it for subsequent distribution, at which time the Company ensured it met product specifications and released it as available for sale. Because of the Company’s continued involvement in the
7
Table of Contents
development and production process of Hylenex recombinant, the earnings process was not considered to be complete. Accordingly, the Company deferred the revenue and related product costs on the API for Hylenex recombinant until the product was filled, finished, packaged and released. Baxter could only return the API for Hylenex recombinant to the Company if it did not conform to the specified criteria set forth in the Hylenex Partnership or upon termination of such agreement. In addition, the Company received product-based payments upon the sale of Hylenex recombinant by Baxter, in accordance with the terms of the Hylenex Partnership. Product-based revenues were recognized as the Company earned such revenues based on Baxter’s shipments of Hylenex recombinant to its distributors when such amounts could be reasonably estimated. Effective January 7, 2011, the Company and Baxter mutually agreed to terminate the Hylenex Partnership and the associated agreements. See Note 7, “Deferred Revenue,” for further discussion.
Revenues under Collaborative Agreements — The Company entered into license and collaboration agreements under which the collaborative partners obtained worldwide exclusive rights for the use of the Company’s proprietary recombinant human PH20 enzyme (“rHuPH20”) in the development and commercialization of the collaborators’ biologic compounds. The collaborative agreements contain multiple elements including nonrefundable payments at the inception of the arrangement, license fees, exclusivity fees, payments based on achievement of specific milestones designated in the collaborative agreements, reimbursements of research and development services, payments for supply of rHuPH20 API for the collaborator and/or royalties on sales of products resulting from collaborative agreements. The Company analyzes each element of its collaborative agreements and considers a variety of factors in determining the appropriate method of revenue recognition of each element.
Prior to the adoption of ASU No. 2009-13 on January 1, 2011, in order for a delivered item to be accounted for separately from other deliverables in a multiple-element arrangement, the following three criteria had to be met: (i) the delivered item had standalone value to the customer, (ii) there was objective and reliable evidence of fair value of the undelivered items and (iii) if the arrangement included a general right of return relative to the delivered item, delivery or performance of the undelivered items was considered probable and substantially in the control of the vendor. For the collaborative agreements entered into prior to January 1, 2011, there was no objective and reliable evidence of fair value of the undelivered items. Thus, the delivered licenses did not meet all of the required criteria to be accounted for separately from undelivered items. Therefore, the Company recognizes revenue on nonrefundable upfront payments and license fees from these collaborative agreements over the period of significant involvement under the related agreements.
For new collaborative agreements or material modifications of existing collaborative agreements entered into after December 31, 2010, the Company follows the provisions of ASU No. 2009-13. In order to account for the multiple-element arrangements, the Company identifies the deliverables included within the agreement and evaluates which deliverables represent units of accounting. Analyzing the arrangement to identify deliverables requires the use of judgment, and each deliverable may be an obligation to deliver services, a right or license to use an asset, or another performance obligation. The deliverables under the Company’s collaborative agreements include (i) the license to the Company’s rHuPH20 technology, (ii) at the collaborator’s request, research and development services which are reimbursed at contractually determined rates, and (iii) at the collaborator’s request, supply of rHuPH20 API which is reimbursed at the Company’s cost plus a margin. A delivered item is considered a separate unit of accounting when the delivered item has value to the collaborator on a standalone basis based on the consideration of the relevant facts and circumstances for each arrangement. Factors considered in this determination include the research capabilities of the collaborator and the availability of research expertise in this field in the general marketplace.
Arrangement consideration is allocated at the inception of the agreement to all identified units of accounting based on their relative selling price. The relative selling price for each deliverable is determined using vendor specific objective evidence (“VSOE”), of selling price or third-party evidence of selling price if VSOE does not exist. If neither VSOE nor third-party evidence of selling price exists, the Company uses its best estimate of the selling price for the deliverable. The amount of allocable arrangement consideration is limited to amounts that are fixed or determinable. The consideration received is allocated among the separate units of accounting, and the applicable revenue recognition criteria are applied to each of the separate units. Changes in the allocation of the sales price between delivered and undelivered elements can impact revenue recognition but do not change the total revenue recognized under any agreement.
8
Table of Contents
Upfront license fee payments are recognized upon delivery of the license if facts and circumstances dictate that the license has standalone value from the undelivered items, which generally include research and development services and the manufacture of rHuPH20 API, the relative selling price allocation of the license is equal to or exceeds the upfront license fee, persuasive evidence of an arrangement exists, the Company’s price to the collaborator is fixed or determinable, and collectability is reasonably assured. Upfront license fee payments are deferred if facts and circumstances dictate that the license does not have standalone value. The determination of the length of the period over which to defer revenue is subject to judgment and estimation and can have an impact on the amount of revenue recognized in a given period.
The terms of the Company’s collaborative agreements provide for milestone payments upon achievement of certain development and regulatory events and/or specified sales volumes of commercialized products by the collaborator. Prior to the Company’s adoption of the Milestone Method, the Company recognized milestone payments upon the achievement of specified milestones if: (1) the milestone was substantive in nature and the achievement of the milestone was not reasonably assured at the inception of the agreement, (2) the fees were nonrefundable and (3) the Company’s performance obligations after the milestone achievement would continue to be funded by the Company’s collaborator at a level comparable to the level before the milestone achievement.
Effective January 1, 2011, the Company adopted on a prospective basis the Milestone Method. Under the Milestone Method, the Company recognizes consideration that is contingent upon the achievement of a milestone in its entirety as revenue in the period in which the milestone is achieved only if the milestone is substantive in its entirety. A milestone is considered substantive when it meets all of the following criteria:
| 1. | The consideration is commensurate with either the entity’s performance to achieve the milestone or the enhancement of the value of the delivered item(s) as a result of a specific outcome resulting from the entity’s performance to achieve the milestone, |
| 2. | The consideration relates solely to past performance, and |
| 3. | The consideration is reasonable relative to all of the deliverables and payment terms within the arrangement. |
A milestone is defined as an event (i) that can only be achieved based in whole or in part on either the entity’s performance or on the occurrence of a specific outcome resulting from the entity’s performance, (ii) for which there is substantive uncertainty at the date the arrangement is entered into that the event will be achieved and (iii) that would result in additional payments being due to the Company.
Reimbursements of research and development services are recognized as revenue during the period in which the services are performed as long as there is persuasive evidence of an arrangement, the fee is fixed or determinable and collection of the related receivable is probable. Revenue from the manufacture of rHuPH20 API is recognized when the API has met all specifications required for the collaborator acceptance and title and risk of loss have transferred to the collaborator. The Company does not directly control when any collaborator will request research and development services or supply of rHuPH20 API; therefore, the Company cannot predict when it will recognize revenues in connection with research and development services and supply of rHuPH20 API. Royalties to be received based on sales of licensed products by the Company’s collaborators incorporating the Company’s rHuPH20 API will be recognized as earned.
The collaborative agreements typically provide the collaborators the right to terminate such agreement in whole or on a product-by-product or target-by-target basis at any time upon 90 days prior written notice to the Company. There are no performance, cancellation, termination or refund provisions in any of the Company’s collaborative agreements that contain material financial consequences to the Company.
See Note 3, “Collaborative Agreements,” and Note 7, “Deferred Revenue,” for further discussion.
Cost of Product Sales
Cost of product sales consists primarily of raw materials, third-party manufacturing costs, fill and finish costs and freight costs associated with the sales of API for ICSI Cumulase and API for Hylenex recombinant. Cost of product sales also consists of the write-down of obsolete inventory.
9
Table of Contents
Research and Development Expenses
Research and development expenses include salaries and benefits, facilities and other overhead expenses, external clinical trials, research-related manufacturing services, contract services and other outside expenses. Research and development expenses are charged to operations as incurred when these expenditures relate to the Company’s research and development efforts and have no alternative future uses. Advance payments, including nonrefundable amounts, for goods or services that will be used or rendered for future research and development activities are deferred and capitalized. Such amounts will be recognized as an expense as the related goods are delivered or the related services are performed or such time when the Company does not expect the goods to be delivered or services to be performed.
Milestone payments that the Company makes in connection with in-licensed technology or product candidates are expensed as incurred when there is uncertainty in receiving future economic benefits from the in-licensed technology or product candidates. The Company considers the future economic benefits from the in-licensed technology or product candidates to be uncertain until such in-licensed technology or product candidates are approved for marketing by the FDA or comparable regulatory agencies in foreign countries or when other significant risk factors are abated. Management has viewed future economic benefits for all of the Company’s in-licensed technology or product candidates to be uncertain and has expensed these amounts for accounting purposes.
Clinical Trial Expenses
Expenses related to clinical trials are accrued based on the Company’s estimates and/or representations from service providers regarding work performed, including actual level of patient enrollment, completion of patient studies and clinical trials progress. Other incidental costs related to patient enrollment or treatment are accrued when reasonably certain. If the contracted amounts are modified (for instance, as a result of changes in the clinical trial protocol or scope of work to be performed), the Company modifies its accruals accordingly on a prospective basis. Revisions in the scope of a contract are charged to expense in the period in which the facts that give rise to the revision become reasonably certain. Historically, the Company has had no material changes in its clinical trial expense accruals that would have had a material impact on its consolidated results of operations or financial position.
Share-Based Compensation
Share-based compensation expense is measured at the grant date, based on the estimated fair value of the award, and is recognized as expense, net of estimated forfeitures, over the employee’s requisite service period. Total share-based compensation expense related to all of the Company’s share-based awards was allocated as follows:
| Three Months Ended September 30, |
Nine Months Ended September 30, |
|||||||||||||||
| 2011 | 2010 | 2011 | 2010 | |||||||||||||
| Research and development |
$ | 890,276 | $ | 691,266 | $ | 1,944,552 | $ | 2,049,134 | ||||||||
| Selling, general and administrative |
867,083 | 634,424 | 1,971,777 | 1,696,842 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Share-based compensation expense |
$ | 1,757,359 | $ | 1,325,690 | $ | 3,916,329 | $ | 3,745,976 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Share-based compensation expense per basic and diluted share |
$ | 0.02 | $ | 0.01 | $ | 0.04 | $ | 0.04 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Share-based compensation expense from: |
||||||||||||||||
| Stock options |
$ | 806,078 | $ | 1,035,496 | $ | 2,318,728 | $ | 3,131,890 | ||||||||
| Restricted stock awards and restricted stock units |
951,281 | 290,194 | 1,597,601 | 614,086 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| $ | 1,757,359 | $ | 1,325,690 | $ | 3,916,329 | $ | 3,745,976 | |||||||||
|
|
|
|
|
|
|
|
|
|||||||||
10
Table of Contents
Since the Company has a net operating loss carryforward as of September 30, 2011, no excess tax benefits for the tax deductions related to share-based awards were recognized in the interim unaudited condensed consolidated statements of operations. For the three months ended September 30, 2011 and 2010, employees exercised stock options to purchase 38,511 and 73,112 shares of common stock, respectively, for aggregate proceeds of approximately $79,000 and $240,000, respectively. For the nine months ended September 30, 2011 and 2010, employees exercised stock options to purchase 2,713,573 and 281,654 shares of common stock, respectively, for aggregate proceeds of approximately $4.1 million and $737,000, respectively.
As of September 30, 2011, total unrecognized estimated compensation cost related to non-vested stock options and non-vested restricted stock awards and restricted stock units granted prior to that date was approximately $6.2 million, and $1.8 million, respectively, which is expected to be recognized over a weighted-average period of approximately 2.5 years and eight months, respectively.
In May 2011, the Company’s stockholders approved the Company’s 2011 Stock Plan, which provides for the granting of up to a total of 6,000,000 shares of common stock (subject to certain limitations as described in the 2011 Stock Plan) to selected employees, consultants and non-employee members of the Company’s Board of Directors (“Outside Directors”) as stock options, stock appreciation rights, restricted stock awards (“RSAs”), restricted stock unit awards (“RSUs”) and performance awards. The 2011 Stock Plan is being utilized for the initial equity awards for new hires of the Company as well as for annual and performance equity awards for existing employees. Options granted under the 2011 Stock Plan generally have a 10-year term and vest at the rate of one-fourth of the shares on the first anniversary of the date of grant and 1/48 of the shares monthly thereafter. As of September 30, 2011, 5.4 million shares were available for future grants of share-based awards under the 2011 Stock Plan.
The 2011 Stock Plan replaced the Company’s prior stock plans, consisting of the Company’s 2008 Stock Plan, 2006 Stock Plan and 2004 Stock Plan (“Prior Plans”). The Prior Plans were terminated such that no additional awards could be granted thereunder but the terms of the Prior Plans remain in effect with respect to outstanding awards until they are exercised, settled, forfeited or otherwise canceled in full.
Comprehensive Income (Loss)
Comprehensive income (loss) is defined as the change in equity during a period from transactions and other events and circumstances from non-owner sources. Comprehensive income (loss) was the same as the Company’s net income (loss) for the reporting periods.
Fair Value Measurements
The Company follows the authoritative guidance for fair value measurements and disclosures which, among other things, defines fair value, establishes a consistent framework for measuring fair value and expands disclosure for each major asset and liability category measured at fair value on either a recurring or nonrecurring basis. Fair value is defined as an exit price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants. As such, fair value is a market-based measurement that should be determined based on assumptions that market participants would use in pricing an asset or liability.
The framework for measuring fair value provides a hierarchy that prioritizes the inputs to valuation techniques used in measuring fair value as follows:
| Level 1 |
Quoted prices (unadjusted) in active markets for identical assets or liabilities; | |
| Level 2 |
Inputs other than quoted prices included within Level 1 that are either directly or indirectly observable; and | |
| Level 3 |
Unobservable inputs in which little or no market activity exists, therefore requiring an entity to develop its own assumptions about the assumptions that market participants would use in pricing. | |
11
Table of Contents
Cash equivalents of approximately $65.0 million and $79.8 million at September 30, 2011 and December 31, 2010, respectively, are carried at fair value and are classified within Level 1 of the fair value hierarchy because they are valued based on quoted market prices for identical securities. The Company has no instruments that are classified within Level 2 or Level 3.
| 3. | Collaborative Agreements |
Roche Partnership
In December 2006, the Company and Roche entered into the Roche Partnership, under which Roche obtained a worldwide, exclusive license to develop and commercialize product combinations of rHuPH20 and up to thirteen Roche target compounds resulting from the partnership. Under the terms of the Roche Partnership, Roche paid $20.0 million as an initial upfront license fee for the application of rHuPH20 to three pre-defined Roche biologic targets. Due to the Company’s continuing involvement obligations (for example, support activities associated with rHuPH20 enzyme), revenues from the upfront payment, exclusive designation fees and annual license maintenance fees were deferred and are being recognized over the term of the Roche Partnership. Roche may pay the Company further payments which could potentially reach a value of up to $111.0 million for the initial three exclusive targets dependent upon the achievement of specified clinical, regulatory and sales-based milestones.
Under the terms of the Roche Partnership, Roche will also pay the Company royalties on product sales for these first three targets. Through September 30, 2011, Roche has elected two additional exclusive targets. In 2010, Roche did not pay the annual license maintenance fee on five target slots. As a result, Roche has an option to select only three additional targets under the Roche Partnership, provided that Roche continues to pay annual exclusivity maintenance fees to the Company. For each of the additional five targets, Roche may pay the Company further upfront and milestone payments of up to $47.0 million per target, as well as royalties on product sales for each of these additional five targets. Additionally, Roche will obtain access to the Company’s expertise in developing and applying rHuPH20 to Roche targets. Under the terms of the Roche Partnership, the Company was obligated to scale up the production of rHuPH20 and to identify a second source manufacturer that would help meet anticipated production obligations arising from the Roche Partnership.
The Company has determined that the clinical and regulatory milestones are substantive; therefore, the Company expects to recognize such clinical and regulatory milestone payments as revenue upon achievement of the milestones. Given the challenges inherent in developing and obtaining approval for pharmaceutical and biologic products, there was substantive uncertainty whether any of the clinical and regulatory milestones would be achieved at the time the Roche Partnership was entered into. In addition, the Company has determined that the sales-based milestone payments are similar to royalty payments; therefore, the Company will recognize such sales-based milestone payments as revenue upon achievement of the milestones. In the three and nine months ended September 30, 2011, the Company recognized zero and $5.0 million, respectively, as revenue under collaborative agreements in accordance with the Milestone Method related to the achievement of certain clinical milestones pursuant to the terms of the Roche Partnership.
Gammagard Partnership
In September 2007, the Company entered into the Gammagard Partnership with Baxter, under which Baxter obtained a worldwide, exclusive license to develop and commercialize product combinations of rHuPH20, with a current Baxter product, GAMMAGARD LIQUID. Under the terms of the Gammagard Partnership, Baxter paid the Company a nonrefundable upfront payment of $10.0 million. Due to the Company’s continuing involvement obligations (for example, support activities associated with rHuPH20 enzyme), the $10.0 million upfront payment was deferred and is being recognized over the term of the Gammagard Partnership. Baxter may make further milestone payments totaling $37.0 million to the Company upon the achievement of regulatory approval for the licensed product candidate and specified sales volumes of commercialized product by Baxter. In addition, Baxter will pay royalties on the sales, if any, of the product that result from the collaboration. The Gammagard Partnership is applicable to both kit and formulation combinations. Baxter assumes all development, manufacturing, clinical, regulatory, sales and marketing costs under the Gammagard Partnership, while the Company is responsible for the supply of rHuPH20 enzyme. The Company performs research and development activities at the request of Baxter, which are reimbursed by Baxter under the terms of the Gammagard Partnership.
12
Table of Contents
In addition, Baxter has certain product development and commercialization obligations in major markets identified in the Gammagard Partnership.
The Company has determined that the regulatory milestones are substantive; therefore, the Company expects to recognize such regulatory milestone payments as revenue upon achievement of such milestones. Given the challenges inherent in developing and obtaining approval for pharmaceutical and biologic products, there was substantive uncertainty whether any of the regulatory events would be achieved at the time the Gammagard Partnership was entered into. In addition, the Company has determined that sales-based milestone payments are similar to royalty payments and, therefore, will be recognized as revenue upon achievement of the milestones. In the three and nine months ended September 30, 2011, the Company recognized zero and $3.0 million, respectively, as revenue under collaborative agreements in accordance with the Milestone Method related to the achievement of regulatory milestones pursuant to the terms of the Gammagard Partnership.
ViroPharma and Intrexon Partnerships
Effective May 10, 2011, the Company and ViroPharma entered into a collaboration and license agreement “ViroPharma Partnership”, under which ViroPharma obtained a worldwide exclusive license for the use of rHuPH20 enzyme in the development of a subcutaneous injectable formulation of ViroPharma’s commercialized product, Cinryze® (C1 esterase inhibitor [human]). In addition, the license provides ViroPharma with exclusivity to C1 esterase inhibition and to the Hereditary Angioedema, along with three additional orphan indications. Under the terms of the ViroPharma Partnership, ViroPharma paid a nonrefundable license fee of $9.0 million. In addition, the Company is entitled to receive an annual exclusivity fee of $1.0 million commencing on May 10, 2012 and on each anniversary of the effective date of the agreement thereafter until a certain development event occurs. ViroPharma is solely responsible for the development, manufacturing and marketing of any products resulting from this partnership. The Company is entitled to receive payments for research and development services and supply of rHuPH20 API if requested by ViroPharma. In addition, the Company is entitled to receive additional cash payments potentially totaling $44.0 million for a product for treatment of Hereditary Angioedema and $10.0 million for each product for treatment of each of three additional orphan indications upon achievement of development and regulatory milestones. The Company is also entitled to receive royalties on future product sales by ViroPharma. ViroPharma may terminate the agreement prior to expiration for any reason on a product-by-product basis upon 90 days’ prior written notice to the Company. Upon any such termination, the license granted to ViroPharma (in total or with respect to the terminated product, as applicable) will terminate and revert to the Company.
Effective June 6, 2011, the Company and Intrexon entered into a collaboration and license agreement “Intrexon Partnership”, under which Intrexon obtained a worldwide exclusive license for the use of rHuPH20 enzyme in the development of a subcutaneous injectable formulation of Intrexon’s recombinant human alpha 1-antitrypsin (rHuA1AT). Under the terms of the Intrexon Partnership, Intrexon paid a nonrefundable upfront license fee of $9.0 million. In addition, the Company is entitled to receive an annual exclusivity fee of $1.0 million commencing on June 6, 2012 and on each anniversary of the effective date of the agreement thereafter until a certain development event occurs. Intrexon is solely responsible for the development, manufacturing and marketing of any products resulting from this partnership. The Company is entitled to receive payments for research and development services and supply of rHuPH20 API if requested by Intrexon. In addition, the Company is entitled to receive additional cash payments potentially totaling $44.0 million for each product for use in the exclusive field and $10 million for each product for use in the non-exclusive field upon achievement of development and regulatory milestones. The Company is also entitled to receive escalating royalties on product sales and a cash payment of $10.0 million upon achievement of a specified sales volume of product sales by Intrexon. Intrexon may terminate the agreement prior to expiration for any reason on a product-by-product basis upon 90 days’ prior written notice to the Company. Upon any such termination, the license granted to Intrexon (in total or with respect to the terminated product, as applicable) will terminate and revert to the Company. Intrexon’s chief executive officer and chairman of its board of directors is also a member of the Company’s board of directors.
In accordance with ASU No. 2009-13, the Company identified the deliverables at the inception of the ViroPharma and Intrexon agreements which are the license, research and development services and API supply. The Company has determined that the license, research and development services and API supply individually represent separate units of accounting, because each deliverable has standalone value. The estimated selling prices for these units of accounting was determined based on market conditions, the terms of comparable collaborative arrangements for similar technology in the pharmaceutical and biotech industry and entity-specific factors such as the terms of the
13
Table of Contents
Company’s previous collaborative agreements, the Company’s pricing practices and pricing objectives and the nature of the research and development services to be performed for the partners. The arrangement consideration was allocated to the deliverables based on the relative selling price method. Based on the results of the Company’s analysis, the Company determined that the upfront payment was earned upon the granting of the worldwide, exclusive right to the Company’s technology to the collaborator in both the ViroPharma Partnership and Intrexon Partnership. However, the amount of allocable arrangement consideration is limited to amounts that are fixed or determinable; therefore, the amount allocated to the license was only to the extent of cash received. As a result, the Company recognized the $9.0 million upfront license fee received under the ViroPharma Partnership and the $9.0 million upfront license fee received under the Intrexon Partnership as revenues under collaborative agreements upon receipt of the upfront license fees in the quarter ended June 30, 2011.
The Company will recognize the exclusivity fees as revenues under collaborative agreements when they are earned. The Company will recognize reimbursements for research and development services as revenues under collaborative agreements as the related services are delivered. The Company will recognize revenue from sales of API as revenues under collaborative agreements when such API has met all required specifications by the partners and the related title and risk of loss and damages have passed to the partners. The Company cannot predict the timing of delivery of research and development services and API as they are at the partners’ requests.
The Company is eligible to receive additional cash payments upon the achievement by the partners of specified development, regulatory and sales-based milestones. The Company has determined that each of the development and regulatory milestones is substantive; therefore, the Company expects to recognize such development and regulatory milestone payments as revenues under collaborative agreements upon achievement in accordance with the Milestone Method. Given the challenges inherent in developing and obtaining approval for pharmaceutical and biologic products, there was substantive uncertainty whether any of the development and regulatory milestones would be met at the time these partnerships were entered into, and the milestones are based in part on the occurrence of a separate outcome resulting from the Company’s performance. In addition, the Company has determined that the sales-based milestone payment is similar to a royalty payment; therefore, the Company will recognize the sales-based milestone payment as revenue upon achievement of the milestone because the Company has no future performance obligations associated with the milestone. In September 2011, ViroPharma announced the initiation of a Phase 2 clinical study to evaluate the safety, pharmacokinetics and pharmacodynamics of subcutaneous administration of Cinryze in combination with rHuPH20 in subjects with hereditary angioedema. As a result, in the three months ended September 30, 2011 the Company recognized a $3.0 million milestone payment from ViroPharma as revenue under collaborative agreements in accordance with the Milestone Method related to the achievement of this development milestone pursuant to the terms of the ViroPharma Partnership.
| 4. | Inventory |
Inventory at September 30, 2011 consists of approximately $77,000 in raw materials used in the manufacture of ICSI Cumulase products and approximately $26,000 in work-in-process inventory for Hylenex recombinant. Inventory at December 31, 2010 consists of raw materials used in the manufacture of the Company’s Hylenex recombinant and ICSI Cumulase products. As of December 31, 2010, the reserve for Hylenex recombinant API inventory obsolescence was approximately $875,000. There was no such reserve as of September 30, 2011.
| 5. | Property and Equipment |
Property and equipment, net consists of the following:
| September 30, 2011 |
December 31, 2010 |
|||||||
| Research equipment |
$ | 4,534,207 | $ | 4,308,654 | ||||
| Computer and office equipment |
1,243,448 | 1,215,894 | ||||||
| Leasehold improvements |
1,002,912 | 998,368 | ||||||
|
|
|
|
|
|||||
| 6,780,567 | 6,522,916 | |||||||
| Accumulated depreciation and amortization |
(5,505,814) | (4,676,017) | ||||||
|
|
|
|
|
|||||
| $ | 1,274,753 | $ | 1,846,899 | |||||
|
|
|
|
|
|||||
14
Table of Contents
Depreciation and amortization expense totaled approximately $239,000 and $365,000 for the three months ended September 30, 2011 and 2010, respectively, and approximately $852,000 and $1.2 million for the nine months ended September 30, 2011 and 2010, respectively.
| 6. | Accrued Expenses |
Accrued expenses consist of the following:
| September 30, 2011 |
December 31, 2010 |
|||||||
| Accrued outsourced research and development expenses |
$ | 6,077,588 | $ | 3,647,762 | ||||
| Accrued compensation and payroll taxes |
2,182,913 | 3,045,950 | ||||||
| Accrued expenses |
604,204 | 1,911,857 | ||||||
|
|
|
|
|
|||||
| $ | 8,864,705 | $ | 8,605,569 | |||||
|
|
|
|
|
|||||
| 7. | Deferred Revenue |
Deferred revenue consists of the following:
| September 30, 2011 |
December 31, 2010 |
|||||||
| Collaborative agreements |
$ | 40,375,240 | $ | 48,761,361 | ||||
| Product sales |
- | 9,332,190 | ||||||
|
|
|
|
|
|||||
| Total deferred revenue |
40,375,240 | 58,093,551 | ||||||
| Less current portion |
3,707,795 | 2,917,129 | ||||||
|
|
|
|
|
|||||
| Deferred revenue, net of current portion |
$ | 36,667,445 | $ | 55,176,422 | ||||
|
|
|
|
|
|||||
Roche Partnership - In December 2006, the Company and Roche entered into the Roche Partnership under which Roche obtained a worldwide, exclusive license to develop and commercialize product combinations of rHuPH20and up to thirteen Roche target compounds. Under the terms of the Roche Partnership, Roche paid $20.0 million to the Company in December 2006 as an initial upfront payment for the application of rHuPH20 to three pre-defined Roche biologic targets. Through September 30, 2011, Roche has paid an aggregate of $19.25 million in connection with Roche’s election of two additional exclusive targets and annual license maintenance fees for the right to designate the remaining targets as exclusive targets. In 2010, Roche did not pay the annual license maintenance fees on five of the remaining eight target slots. As a result, Roche currently retains the option to exclusively develop and commercialize rHuPH20 with three additional targets through the payment of annual license maintenance fees.
Due to the Company’s continuing involvement obligations (for example, support activities associated with rHuPH20 enzyme), revenues from the upfront payment, exclusive designation fees and annual license maintenance fees were deferred and are being recognized over the term of the Roche Partnership. The Company recognized revenue from the upfront payment, exclusive designation fees and annual license maintenance fees under the Roche Partnership in the amounts of approximately $491,000 and $458,000 for the three months ended September 30, 2011 and 2010, respectively, and approximately $1.5 million for the nine months ended September 30, 2011 and 2010. Deferred revenue relating to the upfront payment, exclusive designation fees and annual license maintenance fees under the Roche Partnership was $31.4 million and $32.9 million as of September 30, 2011 and December 31, 2010, respectively.
Baxter Partnerships - In September 2007, the Company and Baxter entered into the Gammagard Partnership, under which Baxter obtained a worldwide, exclusive license to develop and commercialize product combinations of rHuPH20 with GAMMAGARD LIQUID. Under the terms of the Gammagard Partnership, Baxter paid the Company a nonrefundable upfront payment of $10.0 million. Due to the Company’s continuing involvement obligations (for example, support activities associated with rHuPh20 enzyme), the $10.0 million upfront payment was deferred and is being recognized over the term of the Gammagard Partnership. The Company recognized revenue from the upfront payment under the Gammagard Partnership in the amounts of approximately
15
Table of Contents
$121,000 for the three months ended September 30, 2011 and 2010 and approximately $362,000 and $400,000 for the nine months ended September 30, 2011 and 2010, respectively. Deferred revenue relating to the upfront payment under the Gammagard Partnership was $7.7 million and $8.1 million as of September 30, 2011 and December 31, 2010, respectively.
In February 2007, the Company and Baxter amended certain existing agreements for Hylenex recombinant and entered into the Hylenex Partnership for kits and formulations with rHuPH20. Under the terms of the Hylenex Partnership, Baxter paid the Company a nonrefundable upfront payment of $10.0 million. In addition, Baxter would make payments to the Company based on sales of the products covered under the Hylenex Partnership. Baxter had prepaid nonrefundable product-based payments totaling $10.0 million in connection with the execution of the Hylenex Partnership. Due to the Company’s continuing involvement obligations (for example, support activities associated with rHuPh20 enzyme), the $10.0 million upfront payment was initially deferred and was being recognized over the term of the Hylenex Partnership. The prepaid product-based payments were also deferred and were being recognized as product sales revenues as the Company earned such revenues from the sales of Hylenex recombinant by Baxter.
Effective January 7, 2011, the Company and Baxter mutually agreed to terminate the Hylenex Partnership and the associated agreements. The termination of these agreements does not affect the other relationships between the parties, including the application of the Company’s Enhanze Technology to Baxter’s GAMMAGARD LIQUID.
On July 18, 2011, the Company and Baxter entered into an agreement (the “Transition Agreement”) setting forth certain rights, data and assets to be transferred by Baxter to the Company during a transition period. Effective July 18, 2011, the Company has no future performance obligations to Baxter in connection with the Hylenex Partnership. Therefore, the Company recognized the unamortized deferred revenue of approximately $9.3 million relating to the prepaid product-based payments and the unamortized deferred revenue of approximately $7.6 million relating to deferred upfront payment from the Hylenex Partnership as revenues under collaborative agreements for the three months ended September 30, 2011. For the three months ended September 30, 2010, the Company recognized revenues under the Hylenex Partnership from the upfront payment in the amounts of approximately $117,000. No revenue relating to the prepaid product-based payments was recognized for the three months ended September 30, 2010. For the nine months ended September 30, 2011 and 2010, the Company recognized revenues under the Hylenex Partnership from the upfront payment in the amounts of approximately $7.8 million and $387,000, respectively, and from the product-based payments in the amounts of approximately $9.3 million and $332,000, respectively. Deferred revenues relating to the upfront payment and product-based payments under the Hylenex Partnership was $7.8 million and $9.3 million at December 31, 2010. There were no deferred revenues relating to the Hylenex Partnership at September 30, 2011.
In addition, pursuant to the terms of the Transition Agreement, Baxter no longer has the right to return the Hylenex recombinant API previously delivered to Baxter. Accordingly, the Company recognized approximately $991,000 of deferred revenue related to such Hylenex recombinant API as product sales revenue during the three months ended September 30, 2011.
| 8. | Net Income (Loss) Per Share |
Basic net income (loss) per common share (“EPS”) is computed by dividing net income (loss) for the period by the weighted-average number of common shares outstanding during the period, without consideration for common stock equivalents. Diluted net income (loss) per common share is computed by dividing net income (loss) for the period by the weighted-average number of common shares outstanding and potentially dilutive common shares outstanding. Dilutive potential common shares outstanding, determined using the treasury stock method, principally include: shares that may be issued under the Company’s stock options, RSAs and RSUs. For the three months ended September 30, 2011 and 2010, the Company has excluded approximately 1.9 million and 8.6 million shares, respectively, of stock options, unvested RSAs and unvested RSUs from the computation of diluted EPS because their impact would have been anti-dilutive. For the nine months ended September 30, 2011 and 2010, the Company has excluded approximately 5.9 million and 8.6 million shares of stock options, unvested RSAs and unvested RSUs from the computation of diluted EPS as their impact would have been anti-dilutive because of the Company’s net loss in these reporting periods.
16
Table of Contents
The following table sets forth the computation of basic and diluted EPS:
| Three Months Ended September 30, |
Nine Months Ended September 30, |
|||||||||||||||
| 2011 | 2010 | 2011 | 2010 | |||||||||||||
| Net Income (Loss) — Numerator: |
||||||||||||||||
| Net income (loss) for basic and diluted EPS |
$ | 5,165,193 | $ | (12,409,576) | $ | (1,354,237) | $ | (36,347,976) | ||||||||
| Shares — Denominator: |
||||||||||||||||
| Weighted-average shares for basic EPS |
103,223,352 | 93,626,893 | 102,282,904 | 92,342,665 | ||||||||||||
| Effect of dilutive options, RSAs and RSUs |
1,785,837 | - | - | - | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Weighted-average shares for diluted EPS |
105,009,189 | 93,626,893 | 102,282,904 | 92,342,665 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Basic and diluted net income (loss) per share |
$ | 0.05 | $ | (0.13) | $ | (0.01) | $ | (0.39) | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| 9. | Stockholders’ Equity |
During the nine months ended September 30, 2011 and 2010, the Company issued an aggregate of 2,713,573 and 281,654 shares of common stock, respectively, in connection with the exercises of stock options at a weighted average exercise price of $1.70 and $2.62 per share, respectively, for net proceeds of approximately $4.1 million and $737,000, respectively. During the nine months ended September 30, 2011 and 2010, the Company granted 353,508 and 120,000 RSAs, with a weighted average grant-date fair value of $6.51 and $7.67 per share, respectively. Options to purchase approximately 5.4 million and 8.0 million shares of the Company’s common stock were outstanding as of September 30, 2011 and December 31, 2010, respectively. RSUs to purchase 163,000 shares of the Company’s common stock were outstanding as of September 30, 2011. There were no RSUs outstanding as of December 31, 2010.
During the nine months ended September 30, 2010, the Company also issued 8.3 million shares of common stock in a public offering at a price of $7.25 per share, generating approximately $60.0 million in net proceeds. In connection with this financing, the Company granted to an underwriter an option to purchase 1,245,000 shares of common stock at a price of $7.25 per share. The option was exercisable in the event that the underwriter sold more than 8.3 million shares of common stock. The option expired unexercised in October 2010.
| 10. | Restructuring Liability |
In October 2010, the Company completed a corporate reorganization to focus its resources on advancing its core proprietary programs and supporting strategic alliances with Roche and Baxter. This reorganization resulted in a reduction in the workforce of approximately 25 percent primarily in the discovery research and preclinical areas. The restructuring liability was approximately $117,000 as of December 31, 2010 and was paid in full as of September 30, 2011.
| 11. | Commitments and Contingencies |
Operating Leases - The Company’s administrative offices and research facilities are located in San Diego, California. The Company leases an aggregate of approximately 58,000 square feet of office and research space.
17
Table of Contents
In July 2007, the Company entered into a lease agreement (the “Original Lease”) with BC Sorrento, LLC (“BC Sorrento”) for the facilities located at 11388 Sorrento Valley Road, San Diego, California (“11388 Property”) for 27,575 square feet of office and research space commencing in September 2008 through January 2013. Under the terms of the Original Lease, the initial monthly rent payment was approximately $37,000 net of costs and property taxes associated with the operation and maintenance of the leased facilities, commencing in September 2008 and increased to approximately $73,000 starting in March 2009. Thereafter, the annual base rent was subject to approximately 4% annual increases each year throughout the term of the Original Lease. In addition, the Company received a certain tenant improvement allowance and free rent under the terms of the Original Lease. Effective September 2010, BMR-11388 Sorrento Valley Road LP (“BMR-11388”) acquired the 11388 Property and became the new landlord of the 11388 Property.
In June 2011, the Company entered into an amended and restated lease (the “11388 Lease”) with BMR-11388 for the 11388 Property commencing from June 2011 through January 2018. The 11388 Lease superseded the Original Lease. Under the terms of the 11388 Lease, the initial monthly rent payment is approximately $38,000 net of costs and property taxes associated with the operation and maintenance of the leased facilities, commencing in December 2011 and increasing to approximately $65,000 starting in January 2013. Thereafter, the annual base rent is subject to approximately 2.5% annual increases each year throughout the term of the 11388 Lease. In addition, the Company received a cash incentive of approximately $98,000, a tenant improvement allowance of $300,000 and free and reduced rent totaling approximately $744,000. Combined with the unamortized deferred rent under the Original Lease, unamortized deferred rent associated with the 11388 Lease of $733,000 and $545,000 was included in deferred rent as of September 30, 2011 and December 31, 2010, respectively.
In July 2007, the Company entered into a sublease agreement (the “11404 Sublease”) with Avanir Pharmaceuticals, Inc. (“Avanir”) for Avanir’s excess leased facilities located at 11404 Sorrento Valley Road, San Diego, California for 21,184 square feet of office and research space (“11404 Property”) for a monthly rent payment of approximately $54,000, net of costs and property taxes associated with the operation and maintenance of the subleased facilities. The 11404 Sublease expires in January 2013. The annual base rent is subject to approximately 4% annual increases each year throughout the terms of the 11404 Sublease. In addition, the Company received free rent totaling approximately $492,000, of which approximately $182,000 and $266,000 was included in deferred rent as of September 30, 2011 and December 31, 2010, respectively.
In April 2009, the Company entered into a sublease agreement (the “11408 Sublease”) with Avanir for 9,187 square feet located at 11408 Sorrento Valley Road, San Diego, California for office and research space (“11408 Property”), which expires in January 2013. The monthly rent payments, which commenced in January 2010, were approximately $21,000 and are subject to an annual increase of approximately 3%. Under terms of the 11408 Sublease, the Company received a tenant improvement allowance of $75,000, of which approximately $34,000 and $49,000 was included in deferred rent at September 30, 2011 and December 31, 2010, respectively.
In June 2011, the Company entered into a lease agreement (the “11404/11408 Lease”) with BMR-Sorrento Plaza LLC (“BMR-Sorrento”) for the 11404 Property and 11408 Property commencing in January 2013 through January 2018. Pursuant to the terms of the 11404/11408 Lease, the initial monthly rent payment is approximately $71,000 net of costs and property taxes associated with the operation and maintenance of the leased facilities, commencing in January 2013 and is subject to approximately 2.5% annual increases each year throughout the term of the 11404/11408 Lease.
The Company pays a pro rata share of operating costs, insurance costs, utilities and real property taxes incurred by the landlords for the subleased facilities.
18
Table of Contents
Additionally, the Company leases certain office equipment under operating leases. Approximate annual future minimum operating lease payments as of September 30, 2011 are as follows:
| Operating Leases | ||||
| Three months ending December 31, 2011 |
$ | 287,000 | ||
| Twelve months ending December 31, 2012 |
1,470,000 | |||
| Twelve months ending December 31, 2013 |
1,654,000 | |||
| Twelve months ending December 31, 2014 |
1,677,000 | |||
| Twelve months ending December 31, 2015 |
1,715,000 | |||
| Twelve months ending December 31, 2016 |
1,758,000 | |||
| Thereafter |
1,870,000 | |||
|
|
|
|||
| Total minimum lease payments |
$ | 10,431,000 | ||
|
|
|
|||
Legal Contingencies - From time to time, the Company is involved in legal actions arising in the normal course of its business. The Company is not presently subject to any material litigation nor, to management’s knowledge, is any litigation threatened against the Company that collectively is expected to have a material adverse effect on the Company’s consolidated cash flows, financial condition or results of operations.
In May 2010, the Company delivered a notice of breach to Baxter due to Baxter’s failure to provide Hylenex recombinant in accordance with the terms of the Hylenex Partnership. Baxter had contested the claims made in the Company’s initial notice of breach and asserted their own breach claims against the Company. Pursuant to the terms of the Transition Agreement, signed on July 18, 2011, Baxter’s breach claims against the Company were discharged.
19
Table of Contents
| Item 2. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
As used in this report, unless the context suggests otherwise, the terms “we,” “our,” “ours,” and “us” refer to Halozyme Therapeutics, Inc., and its wholly owned subsidiary, Halozyme, Inc., which are sometimes collectively referred to herein as “the Company.”
The following information should be read in conjunction with the unaudited condensed consolidated financial statements and notes thereto included in Item 1 of this Quarterly Report on Form 10-Q. Past financial or operating performance is not necessarily a reliable indicator of future performance, and our historical performance should not be used to anticipate results or future period trends.
Except for the historical information contained herein, this report contains “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995. Such statements reflect management’s current forecast of certain aspects of our future. Words such as “expect,” “anticipate,” “intend,” “plan,” “believe,” “seek,” “estimate,” “think,” “may,” “could,” “will,” “would,” “should,” “continue,” “potential,” “likely,” “opportunity” and similar expressions or variations of such words are intended to identify forward-looking statements, but are not the exclusive means of indentifying forward-looking statements in this report. Additionally, statements concerning future matters such as the development or regulatory approval of new products, enhancements of existing products or technologies, third party performance under key collaboration agreements, revenue and expense levels and other statements regarding matters that are not historical are forward-looking statements. Such statements are based on currently available operating, financial and competitive information and are subject to various risks, uncertainties and assumptions that could cause actual results to differ materially from those anticipated or implied in our forward-looking statements due to a number of factors including, but not limited to, those set forth below under the section entitled “Risks Factors” and elsewhere in this Quarterly Report on Form 10-Q and our most recent Annual Report on Form 10-K.
Overview
We are a biopharmaceutical company dedicated to the development and commercialization of recombinant human enzymes that either transiently modify tissue under the skin to facilitate injection of other therapies or correct diseased tissue structures for clinical benefit. Our existing products and our products under development are based primarily on intellectual property covering the family of human enzymes known as hyaluronidases. Hyaluronidases are enzymes (proteins) that break down hyaluronan, or HA, which is a naturally occurring space-filling, gel-like substance that is a major component of both normal tissues throughout the body, such as skin and cartilage, and abnormal tissues such as tumors. Our primary technology, Enhanze™, is a proprietary delivery platform using our first approved recombinant human PH20 enzyme, or rHuPH20, a human synthetic version of hyaluronidase. rHuPH20 is a naturally occurring enzyme that temporarily degrades HA, thereby facilitating the penetration and diffusion of other drugs and fluids that are injected under the skin or in the muscle. Our proprietary rHuPH20 technology is applicable to multiple therapeutic areas and may be used to both expand existing markets and create new ones through the development of our own proprietary products. Through partnerships or other collaborations, the rHuPH20 technology may also be applied to existing and developmental products of biopharmaceutical companies that market drugs requiring or benefiting from injection via the subcutaneous route of administration.
Our operations to date have involved: (i) organizing and staffing our operating subsidiary, Halozyme, Inc.; (ii) acquiring, developing and securing our technology; (iii) undertaking product development for our existing products and a limited number of product candidates; and (iv) supporting the development of partnered product candidates. We continue to increase our focus on our proprietary product pipeline and have expanded investments in our proprietary product candidates. We currently have multiple proprietary programs in various stages of research and development. In addition, we currently have collaborative partnerships with F. Hoffmann-La Roche, Ltd and Hoffmann-La Roche, Inc., or Roche, Baxter Healthcare Corporation, or Baxter, ViroPharma Incorporated, or ViroPharma, and Intrexon Corporation, or Intrexon, to apply Enhanze™ Technology to the partners’ biological therapeutic compounds. We also had another partnership with Baxter, under which Baxter had worldwide marketing rights for our marketed product, Hylenex® recombinant (hyaluronidase human injection), or Hylenex Partnership. Hylenex recombinant is a recombinant formulation of hyaluronidase that has received the approval from the U.S. Food and Drug Administration, or FDA, to facilitate subcutaneous fluid administration for achieving hydration; to increase the dispersion and absorption of other injected drugs; and in subcutaneous urography for improving
20
Table of Contents
resorption of radiopaque agents. We and Baxter mutually agreed to terminate the Hylenex Partnership in January 2011. Our rHuPH20 technology is also being used in ICSI Cumulase®, a third party’s marketed product used for in vitro fertilization, or IVF. Currently, we have received only limited revenue from the sales of active pharmaceutical ingredients, or API, to the third party that produces ICSI Cumulase, in addition to other revenues from our partnerships.
In February 2007, we and Baxter amended certain existing agreements relating to Hylenex recombinant and entered into the Hylenex Partnership for kits and formulations with rHuPH20. In October 2009, Baxter commenced the commercial launch of Hylenex recombinant. Hylenex recombinant was voluntarily recalled in May 2010, because a portion of the Hylenex recombinant manufactured by Baxter was not in compliance with the requirements of the underlying Hylenex recombinant agreements. During the second quarter of 2011, we submitted the data that the FDA had requested to support the reintroduction of Hylenex recombinant. The FDA has approved the submitted data and has granted the reintroduction of Hylenex recombinant. We expect to reintroduce Hylenex recombinant by the end of 2011.
Effective January 7, 2011, we and Baxter mutually agreed to terminate the Hylenex Partnership and the associated agreements. In June 2011, we entered into a commercial manufacturing and supply agreement with Baxter, under which Baxter will fill and finish Hylenex recombinant for us. On July 18, 2011, we and Baxter entered into an agreement setting forth certain rights, data and assets to be transferred by Baxter to us during a transition period, or the Transition Agreement. The termination of these agreements does not affect the other relationships between the parties, including the application of our Enhanze Technology to Baxter’s GAMMAGARD LIQUID.
We and our partners have product candidates in the research, preclinical and clinical stages, but future revenues from the sales and/or royalties of these product candidates will depend on our partners’ abilities and ours to develop, manufacture, obtain regulatory approvals for and successfully commercialize product candidates. It may be years, if ever, before we and our partners are able to obtain regulatory approvals for these product candidates. We have incurred net operating losses each year since inception, with an accumulated deficit of approximately $226.6 million as of September 30, 2011.
We currently have a shelf registration statement on Form S-3 (Registration No. 333-164215) which allows us, from time to time, to offer and sell up to approximately $39.8 million of equity or debt securities. We may utilize this universal shelf in the future to raise capital to fund the continued development of our product candidates, the commercialization of our products or for other general corporate purposes.
21
Table of Contents
Product and Product Candidates
We have one marketed product and multiple product candidates targeting several indications in various stages of development. The following table summarizes our proprietary product and product candidates as well as our partnered product candidates:
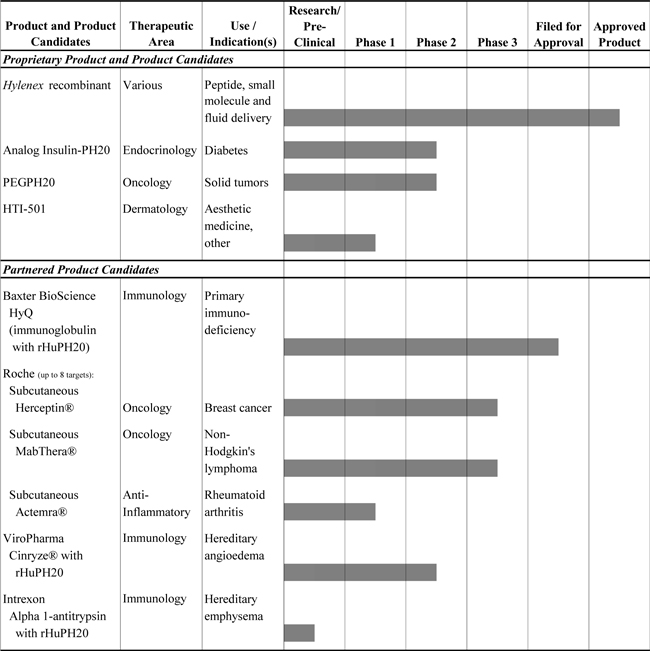
Ultrafast Insulin Program
Our lead proprietary program focuses on the formulation of rHuPH20 with prandial (mealtime) insulins for the treatment of diabetes mellitus. Diabetes mellitus is an increasingly prevalent, costly condition associated with substantial morbidity and mortality. Attaining and maintaining normal blood sugar levels to minimize the long-term clinical risks is a key treatment goal for diabetic patients. Combining rHuPH20 with a rapid acting analog insulin,
22
Table of Contents
i.e., insulin lispro (Humalog®), or Lispro-PH20, insulin aspart (Novolog®), or Aspart-PH20, and insulin glulisine (Apidra®), or collectively PH20 Analog, facilitates faster insulin dispersion in, and absorption from, the subcutaneous space into the vascular compartment leading to faster insulin response. By making mealtime insulin onset faster, i.e., providing earlier insulin to the blood and thus earlier glucose lowering activity, a combination of analog insulin with rHuPH20 may yield a better profile of insulin effect, more like that found in healthy, non-diabetic people.
The primary goal of our ultrafast insulin program is to develop a best-in-class insulin product, with demonstrated clinical benefits for type 1 and 2 diabetes mellitus patients, in comparison to the current standard of care analog products. With a more rapidly absorbed, faster acting insulin product, we seek to demonstrate one or more significant improvements relative to existing treatment, such as improved glycemic control, less hypoglycemia, and less weight gain. A number of Phase 1 and Phase 2 clinical pharmacology trials and registration trial-enabling treatment studies in connection with our ultrafast insulin program, that will investigate the various attributes of our insulin candidates, have been completed or are ongoing or planned. The status of some of these trials is summarized below:
| • | In June 2011, we reported results from the first stage of an insulin pump study comparing insulin aspart co-mixed with rHuPH20 versus aspart alone at the Scientific Sessions of the American Diabetes Association in San Diego, California. The results demonstrated that aspart mixed with rHuPH20 has pharmacokinetic and glucodynamic profiles that were more consistent over infusion set life as compared to analog alone, and the combination also provided a reduction of post-meal glycemic excursions relative to aspart alone. |
In October 2011, we announced positive results from the second stage of the insulin pump study in patients with type 1 diabetes at the Diabetes Technology Meeting in San Francisco, California on October 27 – 29, 2011. This Phase 1b study was conducted as a randomized, double-blind, crossover design, to determine insulin pharmacokinetics, glucodynamics, safety and tolerability of rHuPH20 as a single injection prior to the start of three days of commercially available mealtime insulin aspart pump infusion therapy. The data demonstrated that pre-administration of rHuPH20 led to consistent insulin exposure over the infusion set life and superior glucose control following meals. Compared to insulin aspart alone, pre-administration with rHuPH20 reduced the variability in insulin exposure and action profiles observed with continuous insulin infusion and provided a consistent ultrafast profile over three days of use. In the test meal setting, the consistent ultrafast profile with pre-administration of rHuPH20 led to consistently reduced postprandial excursions. Insulin aspart infusion with and without rHuPH20 pretreatment was similarly well tolerated.
| • | In October 2011, we announced the positive results from two Phase 2 clinical trials of our ultrafast PH20 insulin analog formulations in patients with type 1 and type 2 diabetes. Both trials met the primary endpoint of non-inferiority of HbA1C, which reflects average blood sugar level over a prolonged period of time, compared to the insulin analog comparator, with superior reductions in post-prandial glucose excursions in the PH20 Analog arms. Compared to insulin analog alone, PH20 Analog use resulted in a greater than 50% increase in the proportion of patients able to consistently achieve AACE (American Association of Clinical Endocrinologists) guidelines for post-prandial |
23
Table of Contents
| glucose targets in both type 1 and type 2 patients. Across all of the treatment groups, there was no meaningful difference in hypoglycemia incidence or event rates. Hypoglycemia events were generally mild, and adverse events with PH20 Analog formulations were similar to those observed during the insulin analog comparator phase. Results are from two Phase 2 ultrafast insulin treatment studies, one in type 1 diabetes patients and one in type 2 patients, that compared two ultrafast insulin analog products formulated with rHuPH20 (Lispro-PH20 or Aspart-PH20) to an active comparator, Humalog. More than 110 patients enrolled in each of the trials and received an insulin analog alone and one of the Analog-PH20 treatments for 12 weeks along with basal insulin glargine. The primary endpoint of each study was a comparison of glycemic control, the main measurement that diabetes patients use to assess treatment effectiveness, as assessed by the change in HbA1C from baseline. Data regarding post-prandial glucose levels, the proportion of patients that safely achieve HbA1C targets, rates of hypoglycemia, weight change and additional endpoints were collected as well. We currently expect to present the results of these studies at a major medical meeting in June 2012. |
We view insulin pens and pumps as distinct product opportunities that could be pursued separately. Based on the data we’ve seen thus far, we believe that a large biotech or pharmaceutical company with global access to the primary care markets would be best positioned to maximize the value of the pen market. We believe that the pre-administration of rHuPH20 would be the best product offering for the pump market. The next step will be for Halozyme to evaluate this opportunity using Hylenex recombinant in a clinical study. We would expect to have results from this study by the end of the first quarter 2012.
PEGPH20
We have developed an investigational PEGylated form of rHuPH20, or PEGPH20, a new molecular entity as a candidate for the systemic treatment of tumors which accumulate HA. PEGylation refers to the attachment of polyethylene glycol to our FDA-approved rHuPH20 enzyme, now known as PEGPH20, which converts rHuPH20 from transient and short lived enzyme to a more stable entity in blood which can be used to treat systemic disease.
Certain cancers, including pancreatic, lung, breast, colon and prostate, have been shown to accumulate high levels of HA. Aberrant accumulation of this component of the tumor’s infrastructure supports a protective network that surrounds certain tumors. This pathologic accumulation of HA along with other matrix components creates a unique microenvironment for the growth of tumor cells compared to normal cells. Depleting the HA component of the tumor architecture with PEGPH20 disrupts this unique tumor microenvironment and opens the previously constricted vessels to allow accumulation of cancer therapy. Increased blood flow also enhances cancer radiotherapy. Our scientists have also shown that disrupting the specialized environment around tumors will directly inhibit the growth. Because HA accumulates in about 25% of all solid tumors, PEGPH20 has the potential to help patients with many different kinds of cancer.
We initiated the first Phase 1 clinical trial in 2009 to assess PEGPH20 over a range of doses and frequencies to investigate the safety and tolerability of the treatment in patients with solid tumor malignancies refractory to prior therapies. The study was closed to further enrollment in March 2011. In July 2010, we initiated a second Phase 1 clinical trial with PEGPH20 in the treatment of solid tumors. This trial incorporates the use of oral dexamethasone as prophylactic treatment for all patients prior to receiving intravenous, or IV, administration of PEGPH20
24
Table of Contents
and subsequent post-dose oral dexamethasone. The second Phase 1 study is ongoing and actively enrolling. We presented the results from these Phase 1 studies at the 2011 EORTC-NCI-SCO Annual Meeting on October 27-29 in Brussels, Belgium. The pharmacodynamics of PEGPH20 were first evaluated by measuring the concentration of circulating HA catabolites. HA plasma levels increased, in a dose-dependent manner, after PEGPH20 administration. Freeing up of HA by PEGPH20 is consistent with the expected mechanism of PEGPH20. Tumor imaging from subsets of patients pre- and post-administration of PEGPH20 demonstrated that PEGPH20 treatment rapidly restored tumor perfusion and decreased metabolic activity in tumors. Dynamic Contrast Enhanced Magnetic Resonance Imaging (DCE-MRI) showed that the imaging agent was able to access a pancreatic tumor more easily post-administration of PEGPH20. [18F]-fluorodeoxyglucose positron emission tomography (FDG-PET) imaging showed a reduction in metabolic activity of lung metastases in a patient with metastatic rectal carcinoma after PEGPH20 treatment. The expected mechanism of action of PEGPH20 was further confirmed by histochemical analysis of pre- and post-treatment tumor biopsies. Biopsies of liver metastases from two patients with colorectal carcinoma were stained for HA before and after one cycle of treatment with PEGPH20. Reduced HA staining was observed in biopsies from both patients after treatment.
A Phase 2 clinical trial, with a Phase 1b run-in period, was initiated in October 2011 for patients with metastatic pancreatic cancer. In the Phase 1b portion, the patients will receive the standard of care, gemcitabine, with PEGPH20. The objective of the first phase is to identity a safe and well-tolerated dose that will be selected for the second phase. The Phase 2 portion of the trial will compare gemcitabine alone versus gemcitabine with PEGPH20 with regard to various clinical endpoints. The second phase will be a randomized, double-blind, placebo-controlled study to assess safety, tolerability, and efficacy of chemotherapy either with or without PEGPH20.
HTI-501
HTI-501, a recombinant human cathepsin L, is a lysosomal proteinase that acts by degrading collagen and is our first conditionally active biologic. Collagen is an abundant protein in the body, particularly in connective tissue, and is present in high amounts in the extracellular matrix in the form of collagen fibers. Collagens are a class of helical proteins that are assembled into macromolecular fibrils and fibers. The collagen fiber network provides a structural scaffolding framework in the extracellular matrix. In the skin, these collagen fibers connect the superficial epithelial tissues to the underlying connective tissues. Collagen abnormalities contribute to a number of medical conditions, including frozen shoulder, Dupuytren’s contracture, Peyronie’s disease and cellulite.
A conditionally active biologic is a molecule that is only active under certain physiological conditions. HTI-501 is active under mildly acidic conditions and inactive at the pH normally found in the tissue. The enzyme is combined with a low pH buffer and injected in its active state. The enzyme is only active locally and for a short period of time as once the mildly acidic conditions of the HTI-501 administration have been neutralized by the body, the enzyme becomes inactive. We are harnessing this conditional activity to exert control over the duration and location of the enzyme’s therapeutic activity, potentially improving the efficacy or safety of this product candidate for both medical and aesthetic conditions.
In September 2011, we initiated a proof-of-concept study with HTI-501 in the treatment of edematous fibrosclerotic panniculopathy, also known as cellulite. The condition affects 80 to 90 percent of post-adolescent women and is prevalent in all races. The collagen fibers, or fibrous septae, anchor the epidermis against the swelling of subcutaneous fat, which creates the dimpled appearance associated with the condition. HTI-501 is thought to act by releasing the tension in the collagenous fibrous septae and smooth the dimpled appearance of the skin.
HTI-501 has the potential to be studied as a treatment for other medical conditions involving collagen, such as frozen shoulder, Dupuytren’s contracture, Peyronie’s disease, keloids and hypertrophic scarring.
Enhanze Technology
Enhanze Technology is a proprietary delivery platform using our first approved enzyme – recombinant human hyaluronidase - rHuPH20. This enzyme temporarily degrades HA. This temporary degradation creates an opportunistic window for the improved subcutaneous delivery of injectable biologics such as monoclonal antibodies and other large therapeutic molecules, as well as small molecules and fluids. The HA reconstitutes its normal density within several days and, therefore, any effect of the rHuPH20 on the architecture of the subcutaneous space is temporary. By using our rHuPH20 enzyme, many therapeutics that could normally only be injected intravenously can now be administered subcutaneously. This change in the route of delivery to subcutaneous from IV can
25
Table of Contents
often improve patient convenience, enhance pharmacokinetics, boost efficacy, extend the product lifecycle and reduce cost. We currently have Enhanze Technology partnerships with Roche, Baxter, ViroPharma and Intrexon. We are currently pursuing additional partnerships with biopharmaceutical companies that market drugs requiring or benefiting from injection via the subcutaneous route of administration.
Roche Partnership
In December 2006, we and Roche entered into the Roche Partnership, under which Roche obtained a worldwide, exclusive license to develop and commercialize product combinations of rHuPH20 with up to thirteen Roche target compounds. Roche initially had the exclusive right to apply rHuPH20 to only three pre-defined Roche biologic targets with the option to exclusively develop and commercialize rHuPH20 with an additional ten targets. Roche elected to add a fourth exclusive target in December 2008 and a fifth exclusive target in June 2009. In 2010, Roche did not pay the annual license maintenance fee on five of the remaining eight additional target slots. As a result, Roche currently retains the option to exclusively develop and commercialize rHuPH20 with an additional three targets through the payment of annual license maintenance fees. Pending the successful completion of various clinical, regulatory and sales events, Roche will be obligated to make milestone payments to us, as well as royalty payments on the sales of products that result from the partnership.
Compounds directed at three of the five Roche exclusive targets have previously commenced clinical trials. One compound formulated with rHuPH20 (subcutaneous MabThera®) is in Phase 3 clinical trial, one compound formulated with rHuPH20 (subcutaneous Herceptin®) has completed a Phase 3 clinical trial and one compound formulated with rHuPH20 (subcutaneous Actemra®) has completed a Phase 1 clinical trial. Subcutaneous formulation offers patient convenience by avoiding insertion of an IV line.
In October 2011, Roche announced positive top line results from the Phase 3 clinical trial for a subcutaneously delivered version of Roche’s anticancer biologic, Herceptin (trastuzumab), in women with early HER2-positive breast cancer who received a new, investigational subcutaneous injection of Herceptin. The study experienced comparable results to Herceptin given as an IV infusion. The subcutaneous administration takes around 5 minutes to administer whereas the IV formulation (the current standard) takes around 30 minutes to infuse. Roche is also developing an auto-injector device that should further simplify the process and could enable patients to be dosed at home or in the doctor’s office rather than at an infusion clinic or hospital. The ready to use formulation may also significantly reduce pharmacy time as no medicine preparation time is required. This Phase 3 clinical trial was an open-label trial involving 596 women with HER2-positive early breast cancer. The trial was designed to compare trastuzumab concentration in the blood (pharmacokinetics), efficacy (pathologic complete response) and safety of Herceptin SC to that of Herceptin IV. The trial met its co-primary endpoints that were trastuzumab concentration in the blood (serum concentrations) and efficacy. Secondary endpoints included event-free survival and overall survival. No new safety signals were observed and adverse events were overall consistent with Herceptin IV. Herceptin is approved to treat HER2-positive breast cancer and currently is given intravenously. Breast cancer is the most common cancer among women worldwide. Each year, more than 1.4 million new cases of breast cancer are diagnosed worldwide, and nearly 450,000 people will die of the disease annually. In HER2-positive breast cancer, increased quantities of the HER2 protein are present on the surface of the tumor cells. This is known as ‘HER2 positivity’ and affects approximately 15-20% of people with breast cancer. Roche has stated that data from this trial will be submitted for presentation at an upcoming medical meeting and will support a marketing application to regulatory authorities in the European Union in 2012.
In February 2011, Roche began a Phase 3 clinical trial for a subcutaneous formulation of MabThera (rituximab). The study investigates pharmacokinetics, efficacy and safety of MabThera SC. IV administered MabThera is approved for the treatment of non-Hodgkin’s lymphoma (NHL) and Chronic Lymphocytic Leukemia (CLL), types of cancer that affects lymphocytes, or white blood cells. An estimated 66,000 new cases of NHL were diagnosed in the U.S. in 2009 with approximately 125,000 new cases reported worldwide.
In 2009, Roche completed a Phase 1 clinical trial for a subcutaneous formulation of Actemra. This trial investigated the safety and pharmacokinetics of subcutaneous Actemra in patients with rheumatoid arthritis. The results from this Phase 1 trial suggest that further exploration may be warranted. Actemra administered intravenously is approved for the treatment of rheumatoid arthritis. Roche is separately developing a subcutaneous form of Actemra that does not use rHuPH20 and is being investigated for weekly or biweekly administration.
Additional information about the Phase 3 subcutaneous Herceptin and Phase 3 subcutaneous MabThera clinical trials can be found at www.clinicaltrials.gov and www.roche-trials.com.
26
Table of Contents
Baxter Gammagard Partnership
GAMMAGARD LIQUID is a current Baxter product that is indicated for the treatment of primary immunodeficiency disorders associated with defects in the immune system. In September 2007, Halozyme and Baxter entered into an Enhanze Technology partnership, or the Gammagard Partnership. Under the terms of this partnership, Baxter obtained a worldwide, exclusive license to develop and commercialize product combinations of rHuPH20 with GAMMAGARD LIQUID, or HyQ. Pending the successful completion of various regulatory and sales milestones, Baxter will be obligated to make milestone payments to us, as well as royalty payments on the sales of products that result from the partnership. Baxter is responsible for all development, manufacturing, clinical, regulatory, sales and marketing costs under the Gammagard Partnership, while we will be responsible for the supply of the rHuPH20 enzyme. We perform research and development activities at the request of Baxter, which are reimbursed by Baxter under the terms of the Gammagard Partnership. In addition, Baxter has certain product development and commercialization obligations in major markets identified in the Gammagard License. Baxter filed for regulatory approval of HyQ in the US in the second quarter of 2011. In September 2011, Baxter announced that it had submitted an application to the European Medicines Agency’s Committee for Human Medicinal products seeking marketing approval for HyQ.
ViroPharma Partnership
Effective May 10, 2011, we and ViroPharma entered into a collaboration and license agreement, or ViroPharma Partnership, under which ViroPharma obtained a worldwide exclusive license for the use of rHuPH20 enzyme in the development of a subcutaneous injectable formulation of ViroPharma’s commercialized product, Cinryze® (C1 esterase inhibitor [human]). In addition, the license provides ViroPharma with exclusivity to C1 esterase inhibition and to the Hereditary Angioedema, along with three additional orphan indications. Under the terms of the ViroPharma Partnership, ViroPharma paid a nonrefundable upfront license fee of $9.0 million. In addition, we are entitled to receive an annual exclusivity fee of $1.0 million commencing on May 10, 2012 and on each anniversary of the effective date of the agreement thereafter until a certain development event occurs. ViroPharma is solely responsible for the development, manufacturing, regulatory approval and marketing of any products resulting from this partnership. We are entitled to receive payments for research and development services and supply of rHuPH20 API if requested by ViroPharma. In addition, we are entitled to receive additional cash payments potentially totaling $44.0 million for a product for treatment of Hereditary Angioedema and $10.0 million for each product for treatment of each of the three additional orphan indications upon achievement of development and regulatory milestones. We are also entitled to receive royalties on product sales by ViroPharma. ViroPharma may terminate the agreement prior to expiration for any reason on a product-by-product basis upon 90 days’ prior written notice to us. Upon any such termination, the license granted to ViroPharma (in total or with respect to the terminated product, as applicable) will terminate and revert to us.
In September 2011, ViroPharma announced that they had initiated a Phase 2 clinical study to evaluate the safety, pharmacokinetics and pharmacodynamics of subcutaneous administration of Cinryze in combination with rHuPH20 in subjects with hereditary angioedema.
Intrexon Partnership
Effective June 6, 2011, we and Intrexon entered into a collaboration and license agreement, or Intrexon Partnership, under which Intrexon obtained a worldwide exclusive license for the use of rHuPH20 enzyme in the development of a subcutaneous injectable formulation of Intrexon’s recombinant human alpha 1-antitrypsin (rHuA1AT). Under the terms of the Intrexon Partnership, Intrexon paid a nonrefundable upfront license fee of $9.0 million. In addition, we are entitled to receive an annual exclusivity fee of $1.0 million commencing on June 6, 2012 and on each anniversary of the effective date of the agreement thereafter until a certain development event occurs. Intrexon is solely responsible for the development, manufacturing, regulatory approval and marketing of any products resulting from this partnership. We are entitled to receive payments for research and development services and supply of rHuPH20 API if requested by Intrexon. In addition, we are entitled to receive additional cash payments potentially totaling $44.0 million for each product for use in the exclusive field and $10.0 million for each product for use in the non-exclusive field upon achievement of development and regulatory milestones. We are also entitled to receive escalating royalties on product sales and a cash payment of $10.0 million upon achievement of a specified sales volume of product sales by Intrexon. Intrexon may terminate the agreement prior to expiration for any reason on a product-by-product basis upon 90 days’ prior written notice to us. Upon any such termination, the
27
Table of Contents
license granted to Intrexon (in total or with respect to the terminated product, as applicable) will terminate and revert to us. Intrexon’s chief executive officer and chairman of its board of directors is also a member of the Company’s board of directors.
HYLENEX recombinant
Hylenex recombinant is a recombinant formulation of hyaluronidase that has received the FDA approval to facilitate subcutaneous fluid administration for achieving hydration; to increase the dispersion and absorption of other injected drugs; and in subcutaneous urography for improving resorption of radiopaque agents. In February 2007, we and Baxter amended certain existing agreements relating to Hylenex recombinant and entered into the Hylenex Partnership for kits and formulations with rHuPH20. Pending the successful completion of a series of regulatory and sales events, Baxter would have been obligated to make milestone payments to us, as well as royalty payments on the sales of products that result from the partnership. Baxter was responsible for development, manufacturing, clinical, regulatory, sales and marketing costs of the products covered by the Hylenex Partnership. We supplied Baxter with API for Hylenex recombinant, and Baxter prepared, filled, finished and packaged Hylenex recombinant and held it for subsequent distribution.
In October 2009, Baxter commenced the commercial launch of Hylenex recombinant for use in pediatric rehydration at the 2009 American College of Emergency Physicians (ACEP) scientific assembly. In addition, under the Hylenex Partnership, Baxter had a worldwide, exclusive license to develop and commercialize product combinations of rHuPH20 with Baxter hydration fluids and generic small molecule drugs, with the exception of combinations with (i) bisphosphonates, (ii) cytostatic and cytotoxic chemotherapeutic agents and (iii) proprietary small molecule drugs, the rights to which had been retained by us.
In May 2010, Hylenex recombinant was voluntarily recalled because a portion of the Hylenex recombinant manufactured by Baxter was not in compliance with the requirements of the underlying Hylenex recombinant agreements. During the second quarter of 2011, we submitted the data that the FDA had requested to support the reintroduction of Hylenex recombinant. The FDA has approved the submitted data and has granted the reintroduction of Hylenex recombinant. We expect to reintroduce Hylenex recombinant by the end of 2011.
Effective January 7, 2011, we and Baxter mutually agreed to terminate the Hylenex Partnership and the associated agreements. In June 2011, we entered into a commercial manufacturing and supply agreement with Baxter in June 2011, under which Baxter will fill and finish Hylenex recombinant for us. On July 18, 2011, we and Baxter entered into the Transition Agreement setting forth certain rights, data and assets to be transferred by Baxter to us during a transition period. The termination of these agreements does not affect the other relationships between the parties, including the application of our Enhanze Technology to Baxter’s GAMMAGARD LIQUID.
API for ICSI Cumulase
API for ICSI Cumulase is an ex vivo (used outside of the body) formulation of rHuPH20 to replace the bovine (bull) enzyme currently used for the preparation of oocytes (eggs) prior to IVF during the process of intracytoplasmic sperm injection (ICSI), in which the enzyme is an essential component. API for ICSI Cumulase strips away the HA that surrounds the oocyte, allowing the clinician to then perform the ICSI procedure.
Revenues
We generate revenues from product sales and collaborative agreements. Revenue from product sales depends on our ability to develop, manufacture, obtain regulatory approvals for and successfully commercialize our products and product candidates. Payments received under collaborative agreements may include nonrefundable payment at the inception of the arrangement, license fees, exclusivity fees, payments based on achievement of specified milestones designated in the collaborative agreements, reimbursements of research and development services, payments for the manufacture of rHuPH20 API for the partner and/or royalties, as applicable, on sales of products resulting from collaborative agreements. We analyze each element of our collaborative agreements and consider a variety of factors in determining the appropriate method of revenue recognition. See “Critical Accounting Policies and Estimates — Revenue Recognition — Revenue Under Collaborative Agreements” below for our revenue recognition policies for payments received under collaborative agreements.
28
Table of Contents
Costs and Expenses
Cost of Product Sales. Cost of product sales consists primarily of raw materials, third-party manufacturing costs, fill and finish costs, and freight costs associated with the sales of API for ICSI Cumulase and API for Hylenex recombinant. Cost of product sales also consists of the write-down of obsolete inventory.
Research and Development. Our research and development expenses include salaries and benefits, research-related manufacturing services, clinical trials, contract research services, supplies and materials, facilities and other overhead costs and other outside expenses. We charge all research and development expenses to operations as they are incurred. Our research and development activities are primarily focused on the development of our various product candidates.
Since our inception in 1998 through September 30, 2011, we have incurred research and development expenses of $244.1 million. From January 1, 2008 through September 30, 2011, approximately 28% and 16% of our research and development expenses were associated with the development of our ultrafast insulin and PEGPH20 product candidates, respectively. Research and development expenses incurred for the nine months ended September 30, 2011 and 2010 were as follows:
| Nine Months Ended September 30, | ||||||||
| Programs | 2011 | 2010 | ||||||
| Product Candidates: |
||||||||
| Ultrafast Insulin |
$ | 14,926,849 | $ | 9,654,004 | ||||
| PEGPH20 |
5,833,649 | 5,389,602 | ||||||
| HTI-501 |
3,368,582 | 3,534,908 | ||||||
| HYLENEX |
2,638,597 | 1,157,212 | ||||||
| Enhanze partnerships |
5,054,061 | 6,058,343 | ||||||
| rHuPH20 platform (1) |
8,917,103 | 5,888,249 | ||||||
| Other |
1,908,424 | 4,158,157 | ||||||
|
|
|
|
|
|||||
| Total research and development expenses |
$ | 42,647,265 | $ | 35,840,475 | ||||
|
|
|
|
|
|||||
| (1) | Includes research, development and manufacturing expenses related to rHuPH20 that were not allocated to a specific program at the time the expenses were incurred. |
Due to the uncertainty in obtaining the FDA and other regulatory approvals, our reliance on third parties and competitive pressures, we are unable to estimate with any certainty the additional costs we will incur in the continued development of our proprietary product candidates for commercialization. However, we expect our research and development expenses to increase as our product candidates advance into later stages of clinical development.
Clinical development timelines, likelihood of success and total costs vary widely. We anticipate that we will make ongoing determinations as to which research and development projects to pursue and how much funding to direct to each project on an ongoing basis in response to existing resource levels, the scientific and clinical progress of each product candidate, and other market and regulatory developments. We plan on focusing our resources on those proprietary and partnered product candidates that represent the most valuable economic and strategic opportunities.
Product candidate completion dates and costs vary significantly for each product candidate and are difficult to estimate. The lengthy process of seeking regulatory approvals and the subsequent compliance with applicable regulations require the expenditure of substantial resources. Any failure by us to obtain, or any delay in obtaining, regulatory approvals could cause our research and development expenditures to increase and, in turn, have a material adverse effect on our results of operations. We cannot be certain when, or if, our product candidates will
29
Table of Contents
receive regulatory approval or whether any net cash inflow from our other product candidates, or development projects, will commence.
Selling, General and Administrative. Selling, general and administrative, or SG&A, expenses consist primarily of compensation and other expenses related to our corporate operations and administrative employees, accounting and legal expenses, other professional services expenses, marketing expenses, as well as other expenses associated with operating as a publicly traded company.
Critical Accounting Policies and Estimates
Our discussion and analysis of our financial position and results of operations are based on our consolidated financial statements, which have been prepared in accordance with U.S. generally accepted accounting principles, or U.S. GAAP. The preparation of our consolidated financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses and related disclosure of contingent assets and liabilities. We review our estimates on an ongoing basis. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities. Actual results may differ from these estimates under different assumptions or conditions. We believe the following accounting policies to be critical to the judgments and estimates used in the preparation of our consolidated financial statements.
Revenue Recognition
We generate revenues from product sales and collaborative agreements. We recognize revenues in accordance with the authoritative guidance for revenue recognition. Revenue is recognized when all of the following criteria are met: (1) persuasive evidence of an arrangement exists; (2) delivery has occurred or services have been rendered; (3) the seller’s price to the buyer is fixed or determinable; and (4) collectibility is reasonably assured.
Product Sales
Revenue from the sales of API for ICSI Cumulase is recognized when the transfer of ownership occurs, which is upon shipment to our distributor. We are obligated to accept returns for product that does not meet product specifications. Historically, we have not had any product returns as a result of not meeting product specifications.
Prior to the termination of the Hylenex Partnership with Baxter in January 2011, we supplied Baxter with API for Hylenex recombinant at our fully burdened cost plus a margin. Baxter filled and finished Hylenex recombinant and held it for subsequent distribution, at which time we ensured it met product specifications and released it as available for sale. Because of our continued involvement in the development and production process of Hylenex recombinant, the earnings process was not considered to be complete. Accordingly, we deferred the revenue and related product costs on the API for Hylenex recombinant until the product was filled, finished, packaged and released. Baxter could only return the API for Hylenex recombinant to us if it did not conform to the specified criteria set forth in the Hylenex Partnership or upon termination of such agreement.
Effective January 7, 2011, we and Baxter mutually agreed to terminate the Hylenex Partnership and the associated agreements. On July 18, 2011, we and Baxter entered into the Transition Agreement setting forth the transfer of certain rights, data and assets by Baxter to us during a transition period. Pursuant to the terms of the Transition Agreement, Baxter no longer has the right to return Hylenex recombinant API previously delivered to Baxter. Accordingly, we recognized approximately $991,000 of deferred revenue related to such Hylenex recombinant API as product sales revenue during the three months ended September 30, 2011.
Revenues under Collaborative Agreements
We entered into license and collaboration agreements under which the collaborative partners obtained worldwide exclusive rights for the use of our proprietary rHuPH20 enzyme in the development and commercialization of the partners’ biologic compounds. The collaborative agreements contain multiple elements including nonrefundable payments at the inception of the arrangement, license fees, exclusivity fees, payments based on achievement of specified milestones designated in the collaborative agreements, reimbursements of research and development services, payments for supply of rHuPH20 API for the partner and/or royalties on sales of
30
Table of Contents
products resulting from collaborative agreements. We analyze each element of the collaborative agreements and consider a variety of factors in determining the appropriate method of revenue recognition of each element.
Prior to the adoption of ASU No. 2009-13, Multiple-Deliverable Revenue Arrangements, on January 1, 2011, in order for a delivered item to be accounted for separately from other deliverables in a multiple-element arrangement, the following three criteria had to be met: (i) the delivered item had standalone value to the customer, (ii) there was objective and reliable evidence of fair value of the undelivered items and (iii) if the arrangement included a general right of return relative to the delivered item, delivery or performance of the undelivered items was considered probable and substantially in the control of the vendor. For the collaborative agreements entered into prior to January 1, 2011, there was no objective and reliable evidence of fair value of the undelivered items. Thus, the delivered licenses did not meet all of the required criteria to be accounted for separately from undelivered items. Therefore, we recognize revenue on nonrefundable upfront payments and license fees from these collaborative agreements over the period of significant involvement under the related agreements.
For new collaborative agreements or material modifications of existing collaborative agreements entered into after December 31, 2010, we follow the provisions of ASU No. 2009-13. In order to account for the multiple-element arrangements, we identify the deliverables included within the agreement and evaluate which deliverables represent units of accounting. Analyzing the arrangement to identify deliverables requires the use of judgment, and each deliverable may be an obligation to deliver services, a right or license to use an asset, or another performance obligation. The deliverables under our collaborative agreements include (i) the license to rHuPH20 technology, (ii) at the collaborator’s request, research and development services which are reimbursed at contractually determined rates, and (iii) at the collaborator’s request, supply of rHuPH20 API which is reimbursed at our cost plus a margin. A delivered item is considered a separate unit of accounting when the delivered item has value to the collaborator on a standalone basis based on the consideration of the relevant facts and circumstances for each arrangement. Factors considered in this determination include the research capabilities of the partner and the availability of research expertise in this field in the general marketplace.
Arrangement consideration is allocated at the inception of the agreement to all identified units of accounting based on their relative selling price. The relative selling price for each deliverable is determined using vendor specific objective evidence, or VSOE, of selling price or third-party evidence of selling price if VSOE does not exist. If neither VSOE nor third-party evidence of selling price exists, we use our best estimate of the selling price for the deliverable. The amount of allocable arrangement consideration is limited to amounts that are fixed or determinable. The consideration received is allocated among the separate units of accounting, and the applicable revenue recognition criteria are applied to each of the separate units. Changes in the allocation of the sales price between delivered and undelivered elements can impact revenue recognition but do not change the total revenue recognized under any agreement.
Upfront license fee payments are recognized upon delivery of the license if facts and circumstances dictate that the license has standalone value from the undelivered items, which generally include research and development services and the manufacture of rHuPH20 API, the relative selling price allocation of the license is equal to or exceeds the upfront license fee, persuasive evidence of an arrangement exists, our price to the partner is fixed or determinable, and collectability is reasonably assured. Upfront license fee payments are deferred if facts and circumstances dictate that the license does not have standalone value. The determination of the length of the period over which to defer revenue is subject to judgment and estimation and can have an impact on the amount of revenue recognized in a given period.
The terms of our collaborative agreements provide for milestone payments upon achievement of certain development and regulatory events and/or specified sales volumes of commercialized products by the collaborator. Prior to our adoption of the milestone method of revenue recognition, or Milestone Method, we recognized milestone payments upon the achievement of specified milestones if: (1) the milestone was substantive in nature and the achievement of the milestone was not reasonably assured at the inception of the agreement, (2) the fees were nonrefundable and (3) our performance obligations after the milestone achievement would continue to be funded by our collaborator at a level comparable to the level before the milestone achievement.
Effective January 1, 2011, we adopted on a prospective basis the Milestone Method. Under the Milestone Method, we recognize consideration that is contingent upon the achievement of a milestone in its entirety as revenue in the period in which the milestone is achieved only if the milestone is substantive in its entirety. A milestone is
31
Table of Contents
considered substantive when it meets all of the following criteria:
| 1. | The consideration is commensurate with either the entity’s performance to achieve the milestone or the enhancement of the value of the delivered item(s) as a result of a specific outcome resulting from the entity’s performance to achieve the milestone, |
| 2. | The consideration relates solely to past performance, and |
| 3. | The consideration is reasonable relative to all of the deliverables and payment terms within the arrangement. |
A milestone is defined as an event (i) that can only be achieved based in whole or in part on either the entity’s performance or on the occurrence of a specific outcome resulting from the vendor’s performance, (ii) for which there is substantive uncertainty at the date the arrangement is entered into that the event will be achieved and (iii) that would result in additional payments being due to the vendor.
Reimbursements of research and development services are recognized as revenue during the period in which the services are performed as long as there is persuasive evidence of an arrangement, the fee is fixed or determinable and collection of the related receivable is probable. Revenue from the manufacture of rHuPH20 API is recognized when the API has met all specifications required for the collaborator acceptance and title and risk of loss have transferred to the collaborator. We do not directly control when any collaborator will request research and development services or supply of rHuPH20 API; therefore, we cannot predict when we will recognize revenues in connection with research and development services and supply of rHuPH20 API. Royalties to be received based on sales of licensed products by our collaborators incorporating rHuPH20 will be recognized as earned.
The collaborative agreements typically provide the partners the right to terminate such agreement in whole or on a product-by-product or target-by-target basis at any time upon 90 days prior written notice to us. There are no performance, cancellation, termination or refund provisions in any of our collaborative agreements that contain material financial consequences to us.
Roche Partnership
In December 2006, we and Roche entered into the Roche Partnership, under which Roche obtained a worldwide, exclusive license to develop and commercialize product combinations of our proprietary rHuPH20 enzyme and up to thirteen Roche target compounds resulting from the partnership. Under the terms of the Roche Partnership, Roche paid $20.0 million as an initial upfront license fee for the application of rHuPH20 to three pre-defined Roche biologic targets. Due to our continuing involvement obligations (for example, support activities associated with rHuPH20 enzyme), revenues from the upfront payment, exclusive designation fees and annual license maintenance fees were deferred and are being recognized over the term of the Roche Partnership. Roche may pay us further payments which could potentially reach a value of up to $111.0 million for the initial three exclusive targets upon achievement of specified clinical, regulatory and sales-based milestones
Under the terms of the Roche Partnership, Roche will also pay us royalties on product sales for these first three targets. Through September 30, 2011, Roche has elected two additional exclusive targets. In 2010, Roche did not pay the annual license maintenance fee on five target slots. As a result, Roche has an option to select only three additional targets under the Roche partnership agreement, provided that Roche continues to pay annual exclusivity maintenance fees to us. For each of the additional five targets, Roche may pay us further upfront and milestone payments of up to $47.0 million per target, as well as royalties on product sales for each of these additional five targets. Additionally, Roche will obtain access to our expertise in developing and applying rHuPH20 to Roche targets.
We have determined that the clinical and regulatory milestones are substantive; therefore, we expect to recognize such clinical and regulatory milestone payments as revenue upon achievement. Given the challenges inherent in developing and obtaining approval for pharmaceutical and biologic products, there was substantive uncertainty whether any of the clinical and regulatory milestones would be achieved at the time the Roche Partnership was entered into. In addition, we have determined that the sales-based milestone payments are similar to royalty payments; therefore, we will recognize such sales-based milestone payments as revenue upon achievement of the milestone. In the three and nine months ended September 30, 2011, we recognized zero and $5.0 million,
32
Table of Contents
respectively, as revenue under collaborative agreements in accordance with the Milestone Method related to the achievement of certain clinical milestones pursuant to the terms of the Roche Partnership.
Gammagard Partnership
In September 2007, we entered into the Gammagard Partnership with Baxter, under which Baxter obtained a worldwide, exclusive license to develop and commercialize product combinations of rHuPH20, with a current Baxter product, GAMMAGARD LIQUID. Under the terms of the Gammagard Partnership, Baxter paid us a nonrefundable upfront payment of $10.0 million. Due to our continuing involvement obligations (for example, support activities associated with rHuPH20 enzyme), the $10.0 million upfront payment was deferred and is being recognized over the term of the Gammagard Partnership. Baxter may make further milestone payments totaling $37.0 million to us upon the achievement of regulatory approval for the licensed product candidate and specified sales volumes of commercialized product by Baxter. In addition, Baxter will pay royalties on the sales, if any, of the product that result from the collaboration. The Gammagard Partnership is applicable to both kit and formulation combinations. Baxter assumes all development, manufacturing, clinical, regulatory, sales and marketing costs under the Gammagard Partnership, while we are responsible for the supply of the rHuPH20 enzyme. We perform research and development activities at the request of Baxter, which are reimbursed by Baxter under the terms of the Gammagard Partnership. In addition, Baxter has certain product development and commercialization obligations in major markets identified in the Gammagard Partnership.
We have determined that the regulatory milestones are substantive; therefore, we expect to recognize such regulatory milestone payments as revenue upon achievement. Given the challenges inherent in developing and obtaining approval for pharmaceutical and biologic products, there was substantive uncertainty whether any of the regulatory events would be achieved at the time the Gammagard Partnership was entered into. In addition, we have determined that sales-based milestone payments are similar to royalty payments and, therefore, will be recognized as revenue upon achievement of the milestone. In the three and nine months ended September 30, 2011, we recognized zero and $3.0 million payment as revenue under collaborative agreements in accordance with the Milestone Method related to the achievement of a regulatory milestone pursuant to the terms of the Gammagard Partnership.
ViroPharma and Intrexon Partnerships
Effective May 10, 2011, we and ViroPharma entered into the ViroPharma Partnership, under which ViroPharma obtained a worldwide exclusive license for the use of rHuPH20 enzyme in the development of a subcutaneous injectable formulation of ViroPharma’s commercialized product, Cinryze® (C1 esterase inhibitor [human]). In addition, the license provides ViroPharma with exclusivity to C1 esterase inhibition and to the Hereditary Angioedema, along with three additional orphan indications. Under the terms of the ViroPharma Partnership, ViroPharma paid a nonrefundable upfront license fee of $9.0 million. In addition, we are entitled to receive an annual exclusivity fee of $1.0 million commencing on May 10, 2012 and on each anniversary of the effective date of the agreement thereafter until a certain development event occurs. ViroPharma is solely responsible for the development, manufacturing and marketing of any products resulting from this partnership. We are entitled to receive payments for research and development services and supply of rHuPH20 API if requested by ViroPharma. In addition, we are entitled to receive additional cash payments potentially totaling $44.0 million for a product for treatment of Hereditary Angioedema and $10.0 million for each product for treatment of each of the three additional orphan indications upon achievement of development and regulatory milestones. We are also entitled to receive royalties on product sales by ViroPharma. ViroPharma may terminate the agreement prior to expiration for any reason on a product-by-product basis upon 90 days’ prior written notice to us. Upon any such termination, the license granted to ViroPharma (in total or with respect to the terminated product, as applicable) will terminate and revert to us.
Effective June 6, 2011, we and Intrexon entered into the Intrexon Partnership, under which Intrexon obtained a worldwide exclusive license for the use of rHuPH20 enzyme in the development of a subcutaneous injectable formulation of Intrexon’s recombinant human alpha 1-antitrypsin (rHuA1AT). Under the terms of the Intrexon Partnership, Intrexon paid a nonrefundable upfront license fee of $9.0 million. In addition, we are entitled to receive an annual exclusivity fee of $1.0 million commencing on June 6, 2012 and on each anniversary of the effective date of the agreement thereafter until a certain development event occurs. Intrexon is solely responsible for the development, manufacturing and marketing of any products resulting from this partnership. We are entitled to receive payments for research and development services and supply of rHuPH20 API if requested by Intrexon. In
33
Table of Contents
addition, we are entitled to receive additional cash payments potentially totaling $44.0 million for each product for use in the exclusive field and $10.0 million for each product for use in the non-exclusive field upon achievement of development and regulatory milestones. We are also entitled to receive escalating royalties on product sales and a cash payment of $10.0 million upon achievement of a specified sales volume of product sales by Intrexon. Intrexon may terminate the agreement prior to expiration for any reason on a product-by-product basis upon 90 days’ prior written notice to us. Upon any such termination, the license granted to Intrexon (in total or with respect to the terminated product, as applicable) will terminate and revert to us. Intrexon’s chief executive officer and chairman of its board of directors is also a member of the Company’s board of directors.
In accordance with ASU No. 2009-13, we identified the deliverables at the inception of the ViroPharma and Intrexon Partnerships which are the license, research and development services and API supply. We have determined that the license, research and development services and API supply individually represent separate units of accounting, because each deliverable has standalone value. The estimated selling prices for these units of accounting was determined based on market conditions, the terms of comparable collaborative arrangements for similar technology in the pharmaceutical and biotech industry and entity-specific factors such as the terms of our previous collaborative agreements, our pricing practices and pricing objectives and the nature of the research and development services to be performed for the partners. The arrangement consideration was allocated to the deliverables based on the relative selling price method. Based on the results of our analysis, we determined that the upfront payment was earned upon the granting of the worldwide exclusive right to our technology to the collaborator in both the ViroPharma Partnership and Intrexon Partnership. However, the amount of allocable arrangement consideration is limited to amounts that are fixed or determinable; therefore, the amount allocated to the license was only to the extent of cash received. As a result, we recognized the $9.0 million upfront license fee received under the ViroPharma Partnership and $9.0 million upfront license fee received under the Intrexon Partnership as revenues under collaborative agreements upon the receipt of such upfront license fees in the three months ended June 30, 2011.
We will recognize the exclusivity fees as revenue under collaborative agreements when they are earned. We will recognize reimbursements for research and development services as revenue under collaborative agreement as the related services are delivered. We will recognize payments from sales of API as revenue under collaborative agreements when such API has met all required specifications by the partners and the related title and risk of loss and damages have passed to the partners. We cannot predict the timing of delivery of research and development services and API as they are at the partners’ requests.
We are eligible to receive additional cash payments upon the achievement by the partners of specified development, regulatory and sales-based milestones. We have determined that each of the development and regulatory milestones is substantive; therefore, we expect to recognize such development and regulatory milestone payments as revenue under collaborative agreements upon achievement in accordance with the Milestone Method. Given the challenges inherent in developing and obtaining approval for pharmaceutical and biologic products, there was substantive uncertainty whether any of the development and regulatory milestones would be met at the time these partnerships were entered into, and the milestones are based in part on the occurrence of a separate outcome resulting from our performance. In addition, we have determined that the sales-based milestone payment is similar to a royalty payment; therefore, we will recognize the sales-based milestone payment as revenue upon achievement of the milestone because we have no future performance obligations associated with the milestone. In the three months ended September 30, 2011, we recognized the $3.0 million payment from ViroPharma as revenue under collaborative agreements in accordance with the Milestone Method related to the achievement of a regulatory milestone pursuant to the terms of the ViroPharma Partnership.
Hylenex Partnership
Under the terms of the Hylenex Partnership, Baxter paid us a nonrefundable upfront payment of $10.0 million in 2007. Due to our continuing involvement obligations (for example, support activities associated with rHuPh20 enzyme), the $10.0 million upfront payment was deferred and was being recognized over the term of the Hylenex Partnership. In addition, we received product-based payments upon the sale of Hylenex recombinant by Baxter, in accordance with the terms of the Hylenex Partnership. Baxter had prepaid $10.0 million of non-refundable product-based payments. Revenues from product-based payments were recognized based on Baxter’s shipments of Hylenex recombinant to its distributors when such amounts could be reasonably estimated. The prepaid
34
Table of Contents
product-based payments were initially deferred and were being recognized as product sales revenue as we earned such revenue from the sales of Hylenex recombinant by Baxter.
On January 7, 2011, we and Baxter mutually agreed to terminate the Hylenex Partnership and associated agreements. In June 2011, we entered into a commercial manufacturing and supply agreement with Baxter, under which Baxter will fill and finish Hylenex recombinant for us. On July 18, 2011, we and Baxter entered into the Transition Agreement setting forth the transfer of certain rights, data and assets by Baxter to us during a transition period. Effective July 18, 2011, we have no future performance obligations to Baxter in connection with the Hylenex Partnership. Therefore, we recognized as revenues under collaborative agreements approximately $7.6 million relating to the unamortized deferred upfront payment and approximately $9.3 million relating to the unamortized deferred product-based payments during the quarter ended September 30, 2011. The termination of these agreements does not affect the other relationships between the parties, including the application of our Enhanze Technology to Baxter’s GAMMAGARD LIQUID.
Share-Based Payments
We use the fair value method to account for share-based payments in accordance with the authoritative guidance for stock compensation. The fair value of each option award is estimated on the date of grant using a Black-Scholes-Merton option pricing model, or Black-Scholes model, that uses assumptions regarding a number of complex and subjective variables. These variables include, but are not limited to, our expected stock price volatility, actual and projected employee stock option exercise behaviors, risk-free interest rate and expected dividends. Expected volatilities are based on the historical volatility of our common stock. The expected term of options granted is based on analyses of historical employee termination rates and option exercises. The risk-free interest rates are based on the U.S. Treasury yield in effect at the time of the grant. Since we do not expect to pay dividends on our common stock in the foreseeable future, we estimated the dividend yield to be 0%. Forfeitures are estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. We estimate pre-vesting forfeitures based on our historical experience.
If factors change and we employ different assumptions for determination of fair value in future periods, the share-based compensation expense that we record may differ significantly from what we have recorded in the current period. There is a high degree of subjectivity involved when using option pricing models to estimate share-based compensation. Certain share-based payments, such as employee stock options, may expire worthless or otherwise result in zero intrinsic value as compared to the fair values originally estimated on the grant date and reported in our consolidated financial statements. Alternatively, values may be realized from these instruments that are significantly in excess of the fair values originally estimated on the grant date and reported in our consolidated financial statements. There is currently no market-based mechanism or other practical application to verify the reliability and accuracy of the estimates stemming from these valuation models, nor is there a means to compare and adjust the estimates to actual values. Although the fair value of employee share-based awards is determined in accordance with authoritative guidance on stock compensation using an option-pricing model, that value may not be indicative of the fair value observed in a willing buyer/willing seller market transaction.
Research and Development Expenses
Research and development expenses include salaries and benefits, facilities and other overhead expenses, clinical trials, research-related manufacturing services, contract services and other outside expenses. Research and development expenses are charged to operations as they are incurred. Advance payments, including nonrefundable amounts, for goods or services that will be used or rendered for future research and development activities are deferred and capitalized. Such amounts will be recognized as an expense as the related goods are delivered or the related services are performed or such time that we do not expect the goods to be delivered or services to be rendered.
Milestone payments that we make in connection with in-licensed technology or product candidates are expensed as incurred when there is uncertainty in receiving future economic benefits from the licensed technology or product candidates. We consider the future economic benefits from the licensed technology or product candidates to be uncertain until such licensed technology or product candidates are approved for marketing by regulatory bodies such as the FDA or when other significant risk factors are abated. Management has viewed future economic
35
Table of Contents
benefits for all of our licensed technology or product candidates to be uncertain and has expensed these amounts for accounting purposes.
Payments in connection with our clinical trials are often made under contracts with multiple contract research organizations that conduct and manage clinical trials on our behalf. The financial terms of these agreements are subject to negotiation and vary from contract to contract and may result in uneven payment flows. Generally, these agreements set forth the scope of work to be performed at a fixed fee, unit price or on a time-and-material basis. Payments under these contracts depend on factors such as the successful enrollment or treatment of patients or the completion of other clinical trial milestones. Expenses related to clinical trials are accrued based on our estimates and/or representations from service providers regarding work performed, including actual level of patient enrollment, completion of patient studies and clinical trials progress. Other incidental costs related to patient enrollment or treatment are accrued when reasonably certain. If the contracted amounts are modified (for instance, as a result of changes in the clinical trial protocol or scope of work to be performed), we modify our accruals accordingly on a prospective basis. Revisions in scope of contract are charged to expense in the period in which the facts that give rise to the revision become reasonably certain. Because of the uncertainty of possible future changes to the scope of work in clinical trials contracts, we are unable to quantify an estimate of the reasonably likely effect of any such changes on our consolidated results of operations or financial position. Historically, we have had no material changes in our clinical trial expense accruals that would have had a material impact on our consolidated results of operations or financial position.
Inventory
Inventory consists of raw materials used in production, work in process and finished goods inventory on hand related to our Hylenex recombinant and ICSI Cumulase products. Inventory is valued at lower of cost or market (net realizable value) using the first-in, first-out method. Inventory is reviewed periodically for slow-moving or obsolete status. To the extent that its net realizable value is lower than cost, an impairment would be recorded. As of December 31, 2010, the reserve for Hylenex recombinant API inventory obsolescence was approximately $875,000. There was no such reserve as of September 30, 2011.
The above listing is not intended to be a comprehensive list of all of our accounting policies. In many cases, the accounting treatment of a particular transaction is specifically dictated by U.S. GAAP. There are also areas in which our management’s judgment in selecting any available alternative would not produce a materially different result. Please see our audited consolidated financial statements and notes thereto included in Part II — Item 8 of our Annual Report on Form 10-K for the year ended December 31, 2010, which contain accounting policies and other disclosures required by U.S. GAAP.
Results of Operations
Three Months Ended September 30, 2011 Compared to Three Months Ended September 30, 2010
Product Sales – Product sales were $1.2 million for the three months ended September 30, 2011 compared to $98,000 for the three months ended September 30, 2010. The increase of $1.1 million was primarily due to the recognition of approximately $991,000 of deferred revenue in the three months ended September 30, 2011, related to API for Hylenex recombinant previously delivered to Baxter, because the earnings process related to these product sales was complete in the three months ended September 30, 2011. Based on the termination of the Hylenex Partnership in January 2011, we expect only product sales of API for ICSI Cumulase in future periods until Hylenex recombinant is reintroduced to the market. We expect to reintroduce Hylenex recombinant by the end of 2011.
36
Table of Contents
Revenues Under Collaborative Agreements – Revenues under collaborative agreements for the three months ended September 30, 2011 and 2010 were as follows:
| Three Months Ended September 30, |
Increase (Decrease) | |||||||||||||||
| 2011 | 2010 | Amount | Percent | |||||||||||||
| Amortization of deferred upfront, license fees and product-based payments: |
||||||||||||||||
| Baxter |
$ | 17,027,136 | $ | 237,333 | $ | 16,789,803 | 7074% | |||||||||
| Roche |
491,286 | 457,803 | 33,483 | 7% | ||||||||||||
|
|
|
|
|
|
|
|||||||||||
| 17,518,422 | 695,136 | 16,823,286 | 2420% | |||||||||||||
|
|
|
|
|
|
|
|||||||||||
| Milestone payment from ViroPharma |
3,000,000 | - | 3,000,000 | 100% | ||||||||||||
| Reimbursements for research and development services: |
||||||||||||||||
| Baxter |
399,297 | 850,892 | (451,595) | (53%) | ||||||||||||
| Roche |
559,756 | 1,692,379 | (1,132,623) | (67%) | ||||||||||||
| ViroPharma |
273,050 | - | 273,050 | 100% | ||||||||||||
| Other |
35,000 | 60,000 | (25,000) | (42%) | ||||||||||||
|
|
|
|
|
|
|
|||||||||||
| 1,267,103 | 2,603,271 | (1,336,168) | (51%) | |||||||||||||
|
|
|
|
|
|
|
|||||||||||
| Total revenues under collaborative agreements |
$ | 21,785,525 | $ | 3,298,407 | $ | 18,487,118 | 560% | |||||||||
|
|
|
|
|
|
|
|||||||||||
Pursuant to the terms of the Transition Agreement between us and Baxter, signed on July 18, 2011, we have no future performance obligations to Baxter in connection with the Hylenex Partnership. Accordingly, we recognized approximately $9.3 million related to the deferred prepaid product-based payments and approximately $7.6 million related to the deferred upfront payment under the Hylenex Partnership in the three months ended September 30, 2011. The decrease in reimbursements for research and development services was due to the decrease in services requested by the partners. Research and development services rendered by us on behalf of our partners are at the request of the partners; therefore, the amount of future revenues related to reimbursable research and development services is uncertain. We expect the non-reimbursement revenues under our collaborative agreements to continue to fluctuate in future periods based on our partners’ abilities to meet various clinical and regulatory milestones set forth in such agreements and our abilities to obtain new collaborative agreements.
Cost of Product Sales – Cost of product sales were $12,000 for the three months ended September 30, 2011 compared to $7,000 for the three months ended September 30, 2010. Based on the termination of the Hylenex Partnership in January 2011, we expect only cost of product sales of API for ICSI Cumulase in future periods until Hylenex recombinant is reintroduced to the market.
Research and Development – Research and development expenses were $13.5 million for the three months ended September 30, 2011 compared to $12.4 million for the three months ended September 30, 2010. The increase of $1.1 million, or 9%, was primarily due to a $1.4 million increase in manufacturing activities and a $551,000 increase in clinical trial activities mainly supporting our PEGPH20 program. These increases were partially offset by a $694,000 decrease in compensation cost mainly due to the reduction in force in October 2010. We expect research and development costs to increase slightly in future periods as we continue with our clinical trial programs and continue to develop and manufacture our product candidates.
Selling, General and Administrative – SG&A expenses were $4.3 million for the three months ended September 30, 2011 compared to $3.4 million for the three months ended September 30, 2010. The increase of $889,000, or 26%, was primarily due to increases of $216,000 in marketing expenses, $263,000 in legal expenses and
37
Table of Contents
$232,000 in stock-based compensation mainly due to the grants of restricted stock awards and restricted stock units to certain employees in May 2011. In connection with our plan to reintroduce Hylenex recombinant by the end of 2011, we expect SG&A expenses to increase in future periods as we plan to increase sales and marketing activities.
Interest and Other Income, net – Interest and other income, net consisted of interest income of $12,000 for the three months ended September 30, 2011 compared to $23,000 for the three months ended September 30, 2010. The decrease in interest income was primarily due to lower interest rates, offset in part by higher average cash and cash equivalent balances, in 2011 as compared to the same period in 2010.
Net Income (Loss) – Net income for the three months ended September 30, 2011 was $5.2 million, or $0.05 per basic and diluted common share, compared to a net loss of ($12.4) million, or ($0.13) per basic and diluted common share for the three months ended September 30, 2010. The increase in net income was primarily due to the recognition of deferred upfront license fees, deferred product-based payments and deferred product sales in connection with the termination of the Hylenex Partnership totaling $17.9 million in the three months ended September 30, 2011. In addition, we received a $3.0 million milestone payment under the ViroPharma Partnership that was earned in the three months ended September 30, 2011. The increase in net income was offset in part by an increase in operating expenses in 2011 as compared to the same period in 2010.
Nine Months Ended September 30, 2011 Compared to Nine Months Ended September 30, 2010
Product Sales – Product sales were $1.5 million for the nine months ended September 30, 2011 compared to $695,000 for the nine months ended September 30, 2010. The increase of $792,000 was primarily due to the recognition of $991,000 of deferred revenues related to API for Hylenex recombinant previously delivered to Baxter in the nine months ended September 30, 2011, because the earnings process related to these product sales was complete in July 2011 pursuant to the terms of the Transition Agreement between us and Baxter, signed on July 18, 2011. Based on the termination of the Hylenex Partnership in January 2011, we expect only product sales of API for ICSI Cumulase in future periods until Hylenex recombinant is reintroduced to the market. We expect to reintroduce Hylenex recombinant by the end of 2011.
38
Table of Contents
Revenues Under Collaborative Agreements – Revenues under collaborative agreements for the nine months ended September 30, 2011 and 2010 were as follows:
| Nine Months Ended September 30, |
Increase (Decrease) | |||||||||||||||
| 2011 | 2010 | Amount | Percent | |||||||||||||
| Upfront payments and amortization of deferred upfront, license fees and product-based payments: |
||||||||||||||||
| Baxter |
$ | 17,501,802 | $ | 786,597 | $ | 16,715,205 | 2125% | |||||||||
| ViroPharma |
9,000,000 | - | 9,000,000 | 100% | ||||||||||||
| Intrexon |
9,000,000 | - | 9,000,000 | 100% | ||||||||||||
| Roche |
1,473,858 | 1,517,304 | (43,446) | (3%) | ||||||||||||
|
|
|
|
|
|
|
|||||||||||
| 36,975,660 | 2,303,901 | 34,671,759 | 1505% | |||||||||||||
|
|
|
|
|
|
|
|||||||||||
| Milestone payments: |
||||||||||||||||
| Roche |
5,000,000 | - | 5,000,000 | 100% | ||||||||||||
| Baxter |
3,000,000 | - | 3,000,000 | 100% | ||||||||||||
| ViroPharma |
3,000,000 | - | 3,000,000 | 100% | ||||||||||||
|
|
|
|
|
|
|
|||||||||||
| 11,000,000 | - | 11,000,000 | 100% | |||||||||||||
|
|
|
|
|
|
|
|||||||||||
| Reimbursements for research and development services: |
||||||||||||||||
| Baxter |
949,360 | 3,268,340 | (2,318,980) | (71%) | ||||||||||||
| Roche |
2,929,772 | 3,473,660 | (543,888) | (16%) | ||||||||||||
| ViroPharma |
276,535 | - | 276,535 | 100% | ||||||||||||
| Others |
56,120 | 310,250 | (254,130) | (82%) | ||||||||||||
|
|
|
|
|
|
|
|||||||||||
| 4,211,787 | 7,052,250 | (2,840,463) | (40%) | |||||||||||||
|
|
|
|
|
|
|
|||||||||||
| Total revenues under collaborative agreements |
$ | 52,187,447 | $ | 9,356,151 | $ | 42,831,296 | 458% | |||||||||
|
|
|
|
|
|
|
|||||||||||
Pursuant to the terms of the Transition Agreement between us and Baxter, signed on July 18, 2011, we have no future performance obligations to Baxter in connection with the Hylenex Partnership. Accordingly, we recognized approximately $9.3 million related to the deferred prepaid product-based payments and approximately $7.6 million related to the deferred upfront payment under the Hylenex Partnership in the three months ended September 30, 2011. The decrease in reimbursements for research and development services was due to the decrease in services requested by the partners. Research and development services rendered by us on behalf of our partners are at the partners’ request; therefore, the amount of future revenues related to reimbursable research and development services is uncertain. We expect the non-reimbursement revenues under our collaborative agreements to continue to fluctuate in future periods based on our partners’ abilities to meet various clinical and regulatory milestones set forth in such agreements and our abilities to obtain new collaborative agreements.
Cost of Product Sales – Cost of product sales were $202,000 for the nine months ended September 30, 2011 compared to $96,000 for the nine months ended September 30, 2010. The increase of $106,000, or 110%, is mainly due to the write-off of obsolete API for Hylenex recombinant inventory. Based on the termination of the Hylenex Partnership in January 2011, we expect only cost of product sales of API for ICSI Cumulase in future periods until Hylenex recombinant is reintroduced to the market.
Research and Development – Research and development expenses were $42.6 million for the nine months ended September 30, 2011 compared to $35.8 million for the nine months ended September 30, 2010. The increase of
39
Table of Contents
$6.8 million, or 19%, was primarily due to a $9.2 million increase in clinical trial activities mainly supporting our ultrafast insulin and PEGPH20 programs and a $3.3 million increase in manufacturing activities. These increases were offset in part by a $3.9 million decrease in compensation costs mainly due to the reduction in force in October 2010 and an $862,000 decrease in research supplies. We expect research and development costs to increase slightly in future periods as we continue with our clinical trial programs and continue to develop and manufacture our product candidates.
Selling, General and Administrative – SG&A expenses were $12.2 million for the nine months ended September 30, 2011 compared to $10.5 million for the nine months ended September 30, 2010. The increase of $1.7 million, or 16%, was primarily due to increases of $797,000 in marketing expenses and $335,000 in legal expenses. In connection with our plan to reintroduce Hylenex recombinant by the end of 2011, we expect SG&A expenses to increase in future periods as we plan to increase sales and marketing activities.
Interest and Other Income, net – Interest and other income, net consisted primarily of interest income of $56,000 for the nine months ended September 30, 2011 compared to $32,000 for the nine months ended September 30, 2010. The increase in interest income was primarily due to higher average cash and cash equivalent balances and investing in higher interest rate investments in 2011 as compared to the same period in 2010.
Net Loss – Net loss for the nine months ended September 30, 2011 was $1.4 million, or $0.01 per common share, compared to net loss of $36.3 million, or $0.39 per common share for the nine months ended September 30, 2010. The decrease in net loss was primarily due to an increase in revenues under collaborative agreements resulting from $18.0 million of upfront license fees we received from the ViroPharma and Intrexon Partnerships and $11.0 million in milestone payments from Roche, Baxter and ViroPharma which were earned in the nine months ended September 30, 2011. The increase was also due to the recognition of deferred upfront license fees, deferred product-based payments and deferred product sales in connection with the termination of the Hylenex Partnership totaling $17.9 million in the three months ended September 30, 2011. The decrease in net loss was offset in part by an increase in operating expenses in 2011 as compared to the same period in 2010.
Liquidity and Capital Resources
Overview
Our principal sources of liquidity are our existing cash and cash equivalents. As of September 30, 2011, we had cash and cash equivalents of approximately $66.3 million. We will continue to have significant cash requirements to support product development activities. The amount and timing of cash requirements will depend on the success of our clinical development programs, regulatory and market acceptance, and the resources we devote to research and other commercialization activities.
We believe that our current cash and cash equivalents will be sufficient to fund our operations for at least the next twelve months. Currently, we anticipate total net cash burn of approximately $30.0 to $35.0 million for the year ending December 31, 2011, depending on the progress of various preclinical and clinical programs, the timing of our manufacturing scale up and the achievement of various milestones under our existing collaborative agreements. We do not expect our revenues to be sufficient to fund operations for several years. We expect to fund our operations going forward with existing cash resources, anticipated revenues from our existing collaborations and cash that we will raise through future transactions. We may finance future cash needs through any one of the following financing vehicles: (i) the public offering of securities; (ii) new collaborative agreements; (iii) expansions or revisions to existing collaborative relationships; (iv) private financings; and/or (v) other equity or debt financings.
We currently have the ability to offer and sell up to $39.8 million of additional equity or debt securities under an effective universal shelf registration statement. In January 2010, we filed the shelf registration statement on Form S-3 (Registration No. 333-164215) which allows us, from time to time, to offer and sell up to $100.0 million of equity or debt securities. In September 2010, we sold approximately $60.2 million of our common stock in an underwritten public offering at a net price of $7.25 per share. We may utilize this universal shelf in the future to raise capital to fund the continued development of our product candidates, the commercialization of our products or for other general corporate purposes.
Our existing cash and cash equivalents may not be adequate to fund our operations until we become cash flow positive, if ever. We cannot be certain that additional financing will be available when needed or, if available,
40
Table of Contents
financing will be obtained on favorable terms. If we are unable to raise sufficient funds, we will need to delay, scale back or eliminate some or all of our research and development programs, delay the launch of our product candidates, if approved, and/or restructure our operations. If we raise additional funds by issuing equity securities, substantial dilution to existing stockholders could result. If we raise additional funds by incurring debt financing, the terms of the debt may involve significant cash payment obligations, the issuance of warrants that may ultimately dilute existing stockholders when exercised, and covenants that may restrict our ability to operate our business.
Cash Flows
Net cash used in operations was $20.8 million during the nine months ended September 30, 2011 compared to $38.0 million of net cash used in operations during the nine months ended September 30, 2010. This change was primarily due to the decrease in net loss of $35.0 million adjusted for non-cash items including stock-based compensation and depreciation and amortization in addition to changes in working capital. The decrease in net loss after adjustments for non-cash items was mainly due to the receipts of $18.0 million in upfront license fees from the ViroPharma and Intrexon Partnerships and milestone payments totaling $8.0 million received from Roche and Baxter; offset in part by an increase in R&D and SG&A expenses.
Net cash used in investing activities was $272,000 during the nine months ended September 30, 2011 compared to $316,000 during the nine months ended September 30, 2010. This was primarily due to a decrease in purchases of property and equipment during 2011.
Net cash provided by financing activities was $4.1 million during the nine months ended September 30, 2011 compared to $60.7 million during the nine months ended September 30, 2010. Net cash provided by financing activities in 2011 consisted of net proceeds from stock option exercises. Net cash provided by financing activities in 2010 primarily consisted of $60.0 million in net proceeds from the sale of our common stock in September 2010 and $737,000 in net proceeds from stock option exercises.
Off-Balance Sheet Arrangements
As of September 30, 2011, we did not have any relationships with unconsolidated entities or financial partnerships, such as entities often referred to as structured finance or special purpose entities, which would have been established for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or limited purposes. In addition, we did not engage in trading activities involving non-exchange traded contracts. As such, we are not materially exposed to any financing, liquidity, market or credit risk that could arise if we had engaged in these relationships.
Contractual Obligations
In July 2007, we entered into a lease agreement, or the Original Lease, with BC Sorrento, LLC, or BC Sorrento for the facilities located at 11388 Sorrento Valley Road, San Diego, California, or the 11388 Property, for 27,575 square feet of office and research space commencing in September 2008 through January 2013. Under the terms of the Lease, the initial monthly rent payment was approximately $37,000 net of costs and property taxes associated with the operation and maintenance of the leased facilities, commencing in September 2008 and increased to approximately $73,000 starting in March 2009. Thereafter, the annual base rent was subject to approximately 4% annual increases each year throughout the term of the Lease. Effective September 2010, BMR-11388 Sorrento Valley Road LLC, or BMR-11388, acquired the 11388 Property and became the new landlord of the 11388 Property.
In June 2011, we entered into an amended and restated lease, or the 11388 Lease, with BMR-11388 for the 11388 Property commencing from June 2011 through January 2018. The 11388 Lease superseded the Original Lease. Under the terms of the 11388 Lease, the initial monthly rent payment is approximately $38,000 net of costs and property taxes associated with the operation and maintenance of the leased facilities, commencing in December 2011 and increasing to approximately $65,000 starting in January 2013. Thereafter, the annual base rent is subject to approximately 2.5% annual increases each year throughout the term of the 11388 Lease. In addition, we received a cash incentive of approximately $98,000, a tenant improvement allowance of $300,000 and free and reduced rent totaling approximately $744,000.
41
Table of Contents
In June 2011, we entered into a lease agreement or the 11404/11408 Lease, with BMR-Sorrento Plaza LLC, or BMR-Sorrento, for the 11404 Property and 11408 Property commencing in January 2013 through January 2018. Pursuant to the terms of the 11404/11408 Lease, the initial monthly rent payment is approximately $71,000 net of costs and property taxes associated with the operation and maintenance of the leased facilities, commencing in January 2013 and is subject to approximately 2.5% annual increases each year throughout the term of the 11404/11408 Lease.
Recent Accounting Pronouncements
See Note 2, Summary of Significant Accounting Policies — Adoption of Recent Accounting Pronouncements and Pending Adoption of Accounting Pronouncements, in the Notes to Condensed Consolidated Financial Statements for discussions of new accounting pronouncements and their effect, if any, on us.
Risk Factors
The following information sets forth factors that could cause our actual results to differ materially from those contained in forward-looking statements we have made in this Quarterly Report on Form 10-Q, our most recent Annual Report on Form 10-K and those we may make from time to time. In addition to the risk factors discussed below, we are also subject to additional risks and uncertainties not presently known to us or that we currently deem immaterial. If any of these known or unknown risks or uncertainties actually occurs, our business, financial position and results of operations could be materially and adversely affected and the value of our securities could decline significantly.
Risks Related To Our Business
We have generated only minimal revenue from product sales to date; we have a history of net losses and negative cash flow, and we may never achieve or maintain profitability.
Relative to expenses incurred in our operations, we have generated only minimal revenue from product sales, licensing fees and milestone payments to date and we may never generate sufficient revenues from future product sales, licensing fees and milestone payments to offset expenses. Even if we ultimately do achieve significant revenues from product sales, licensing fees and/or milestone payments, we expect to incur significant operating losses over the next few years. We have never been profitable, and we may never become profitable. Through September 30, 2011, we have incurred aggregate net losses of approximately $226.6 million.
If our proprietary and partnered product candidates do not receive and maintain regulatory approvals, or if approvals are not obtained in a timely manner, such failure or delay would substantially impair our ability to generate revenues.
Approval from the FDA is necessary to manufacture and market pharmaceutical products in the United States and the other countries in which we anticipate doing business have similar requirements. The process for obtaining FDA and other regulatory approvals is extensive, time-consuming and costly, and there is no guarantee that the FDA or other regulatory bodies will approve any applications that may be filed with respect to any of our proprietary or partnered product candidates, or that the timing of any such approval will be appropriate for the desired product launch schedule for a product candidate. We, and our partners, attempt to provide guidance as to the timing for the filing and acceptance of such regulatory approvals, but such filings and approvals may not occur on the originally anticipated timeline, or at all. Only one of our partnered product candidates is currently in the regulatory approval process and there are no proprietary product candidates currently in the regulatory approval process. We and our partners may not be successful in obtaining such approvals for any potential products in a timely manner, or at all (please also refer to the risk factor titled “Our proprietary and partnered product candidates may not receive regulatory approvals for a variety of reasons, including unsuccessful clinical trials.” for additional information relating to the approval of product candidates).
Additionally, in order to continue to manufacture and market pharmaceutical products, we must maintain our regulatory approvals. If we or any of our partners are unsuccessful in maintaining our regulatory approvals our ability to generate revenues would be adversely affected.
42
Table of Contents
If our contract manufacturers are unable to manufacture significant amounts of the API used in our products and product candidates, our product development and commercialization efforts could be delayed or stopped and our collaborative partnerships could be damaged.
We have existing supply agreements with contract manufacturing organizations Avid Bioservices, Inc., or Avid, and Cook Pharmica LLC, or Cook, to produce bulk API. These manufacturers each produce API under current Good Manufacturing Practices, or cGMP, for clinical uses. In addition, Avid currently produces API for commercialized products. Avid and Cook will also provide support for the chemistry, manufacturing and controls sections for FDA and other regulatory filings. We rely on their ability to successfully manufacture these batches according to product specifications and Cook has relatively limited experience manufacturing our API. In addition, as a result of our contractual obligations to Roche, we will be required to significantly scale up our commercial API production at Cook during the next two years. If Cook is unable to obtain status as a cGMP-approved manufacturing facility, or if either Avid or Cook: (i) are unable to retain status as cGMP-approved manufacturing facilities; (ii) are unable to otherwise successfully scale up our API production; or (iii) fail to manufacture the API required by our proprietary and partnered products and product candidates for any other reason, our business will be adversely affected. We have not established, and may not be able to establish, favorable arrangements with additional API manufacturers and suppliers of the ingredients necessary to manufacture the API should the existing manufacturers and suppliers become unavailable or in the event that our existing manufacturers and suppliers are unable to adequately perform their responsibilities. We have attempted to mitigate the impact of supply interruption through the establishment of excess API inventory, but there can be no assurances that this safety stock will be maintained or that it will be sufficient to address any delays, interruptions or other problems experienced by Avid and/or Cook. Any delays, interruptions or other problems regarding the ability of Avid and/or Cook to supply API on a timely basis could: (i) cause the delay of clinical trials or otherwise delay or prevent the regulatory approval of proprietary or partnered product candidates; (ii) delay or prevent the effective commercialization of proprietary or partnered products and/or (iii) cause us to breach contractual obligations to deliver API to our partners. Such delays would likely damage our relationship with our partners under our key collaboration agreements and they would have a material adverse effect on our business and financial condition.
If any party to a key collaboration agreement, including us, fails to perform material obligations under such agreement, or if a key collaboration agreement, or any other collaboration agreement, is terminated for any reason, our business could significantly suffer.
We have entered into multiple collaboration agreements under which we may receive significant future payments in the form of maintenance fees, milestone payments and royalties. In the event that a party fails to perform under a key collaboration agreement, or if a key collaboration agreement is terminated, the reduction in anticipated revenues could delay or suspend our product development activities for some of our product candidates, as well as our commercialization efforts for some or all of our products. In addition, the termination of a key collaboration agreement by one of our partners could materially impact our ability to enter into additional collaboration agreements with new partners on favorable terms, if at all. In certain circumstances, the termination of a key collaboration agreement would require us to revise our corporate strategy going forward and reevaluate the applications and value of our technology.
For example, Hylenex recombinant was voluntarily recalled in May 2010 because a portion of the Hylenex recombinant manufactured by Baxter was not in compliance with the requirements of the underlying Hylenex recombinant agreements. In January 2011, we and Baxter mutually agreed to terminate the Hylenex Partnership and we reacquired all rights to Hylenex recombinant. During the second quarter of 2011, we submitted the data that the FDA had requested to support the reintroduction of Hylenex recombinant. The FDA has approved the submitted data and has granted the reintroduction of Hylenex recombinant. We expect to reintroduce Hylenex recombinant by the end of 2011.
Most of our current proprietary and partnered products and product candidates rely on the rHuPH20 enzyme.
The rHuPH20 enzyme is a key technological component of Enhanze Technology, our ultrafast insulin program, our PEGPH20 program, Hylenex recombinant and other proprietary and partnered products and product candidates. An adverse development for rHuPH20 (e.g., an adverse regulatory determination relating to rHuPH20, we are unable to obtain sufficient quantities of rHuPH20, we are unable to obtain or maintain material proprietary
43
Table of Contents
rights to rHuPH20 or we discover negative characteristics of rHuPH20) would substantially impact multiple areas of our business, including current and potential partnerships, as well as proprietary programs.
Our proprietary and partnered product candidates may not receive regulatory approvals for a variety of reasons, including unsuccessful clinical trials.
Clinical testing of pharmaceutical products is a long, expensive and uncertain process and the failure or delay of a clinical trial can occur at any stage. Even if initial results of preclinical studies or clinical trial results are promising, we or our partners may obtain different results that fail to show the desired levels of safety and efficacy, or we may not, or our partners may not, obtain applicable regulatory approval for a variety of other reasons. Clinical trials for any of our proprietary or partnered product candidates could be unsuccessful, which would delay or prohibit regulatory approval and commercialization of the product candidates. In the United States and other jurisdictions, regulatory approval can be delayed, limited or not granted for many reasons, including, among others:
| • | regulatory review may not find a product candidate safe or effective enough to merit either continued testing or final approval; |
| • | regulatory review may not find that the data from preclinical testing and clinical trials justifies approval, or they may require additional studies that would significantly delay or make continued pursuit of approval commercially unattractive; |
| • | a regulatory agency may reject our trial data or disagree with our interpretations of either clinical trial data or applicable regulations; |
| • | the cost of a clinical trial may be greater than what we originally anticipate, and we may decide to not pursue regulatory approval for such a trial; |
| • | a regulatory agency may not approve our manufacturing processes or facilities, or the processes or facilities of our partners, our contract manufacturers or our raw material suppliers; |
| • | a regulatory agency may identify problems or other deficiencies in our existing manufacturing processes or facilities, or the existing processes or facilities of our partners, our contract manufacturers or our raw material suppliers; |
| • | a regulatory agency may change its formal or informal approval requirements and policies, act contrary to previous guidance, adopt new regulations or raise new issues or concerns late in the approval process; or |
| • | a product candidate may be approved only for indications that are narrow or under conditions that place the product at a competitive disadvantage, which may limit the sales and marketing activities for such product candidate or otherwise adversely impact the commercial potential of a product. |
If a proprietary or partnered product candidate is not approved in a timely fashion on commercially viable terms, or if development of any product candidate is terminated due to difficulties or delays encountered in the regulatory approval process, it could have a material adverse impact on our business and we will become more dependent on the development of other proprietary or partnered product candidates and/or our ability to successfully acquire other products and technologies. There can be no assurances that any proprietary or partnered product candidate will receive regulatory approval in a timely manner, or at all.
We anticipate that certain proprietary and partnered products will be marketed, and perhaps manufactured, in foreign countries. The process of obtaining regulatory approvals in foreign countries is subject to delay and failure for the reasons set forth above, as well as for reasons that vary from jurisdiction to jurisdiction. The approval process varies among countries and jurisdictions and can involve additional testing. The time required to obtain approval may differ from that required to obtain FDA approval. Foreign regulatory agencies may not provide approvals on a timely basis, if at all. Approval by the FDA does not ensure approval by regulatory authorities in other countries or jurisdictions, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other foreign countries or jurisdictions or by the FDA.
44
Table of Contents
Our key partners are responsible for providing certain proprietary materials that are essential components of our partnered product candidates, and any failure to supply these materials could delay the development and commercialization efforts for these partnered product candidates and/or damage our collaborative partnerships.
Our partners are responsible for providing certain proprietary materials that are essential components of our partnered product candidates. For example, Roche is responsible for producing the Herceptin and MabThera required for its subcutaneous product candidates and Baxter is responsible for producing the GAMMAGARD LIQUID for its product candidate. If a partner, or any applicable third party service provider of a partner, encounters difficulties in the manufacture, storage, delivery, fill, finish or packaging of either components of the partnered product candidate or the partnered product candidate itself, such difficulties could: (i) cause the delay of clinical trials or otherwise delay or prevent the regulatory approval of partnered product candidates; and/or (ii) delay or prevent the effective commercialization of partnered products. Such delays could have a material adverse effect on our business and financial condition. For example, Baxter received a Warning Letter from the FDA in January 2010 regarding Baxter’s GAMMAGARD LIQUID manufacturing facility in Lessines, Belgium. The FDA indicated in March 2010 that the issues raised in the Warning Letter had been addressed by Baxter and we do not expect these issues to impact the development of the GAMMAGARD LIQUID product candidate.
If we have problems with third parties that either distribute API on our behalf or prepare, fill, finish and package our products and product candidates for distribution, our commercialization and development efforts for our products and product candidates could be delayed or stopped.
We rely on third parties to store and ship API on our behalf and to also prepare, fill, finish and package our products and product candidates prior to their distribution. If we are unable to locate third parties to perform these functions on terms that are acceptable to us, or if the third parties we identify fail to perform their obligations, the progress of clinical trials could be delayed or even suspended and the commercialization of approved product candidates could be delayed or prevented. For example, Hylenex recombinant was voluntarily recalled in May 2010 because a portion of the Hylenex recombinant manufactured by Baxter was not in compliance with the requirements of the underlying Hylenex recombinant agreements. During the second quarter of 2011, we submitted the data that the FDA had requested to support the reintroduction of Hylenex recombinant. The FDA has approved the submitted data and has granted the reintroduction of Hylenex recombinant. We expect to reintroduce Hylenex recombinant by the end of 2011. In June 2011, we entered into a commercial manufacturing and supply agreement with Baxter, under which Baxter will fill, finish and package Hylenex recombinant product for us. Any delay in manufacturing Hylenex recombinant by Baxter would likely delay our ability to reintroduce the product by the end of 2011. Under our commercial manufacturing and supply agreement with Baxter, Baxter has agreed to fill and finish Hylenex recombinant product for us for a limited period of time. The initial term of the commercial manufacturing and supply agreement with Baxter expires on December 31, 2012 and is renewable for one additional year. In June 2011, we entered into a services agreement with a third party manufacturer for the technology transfer of Hylenex recombinant manufacture. While we expect to enter into a commercial manufacturing and supply agreement with a new manufacturer of Hylenex recombinant, if we are unable to find a suitable manufacturer of Hylenex recombinant prior to the expiration of the commercial manufacturing and supply agreement with Baxter or if a new manufacturer encounters difficulties in the manufacture, fill, finish or packaging of Hylenex recombinant, our business and financial condition could be adversely effected.
We may wish to raise additional capital in the next twelve months and there can be no assurance that we will be able to obtain such funds.
During the next twelve months, we may wish to raise additional capital to continue the development of our product candidates or for other current corporate purposes. Our current cash position and expected revenues during the next few years may not constitute the amount of capital necessary for us to continue the development of our proprietary product candidates and to fund general operations. In addition, if we engage in acquisitions of companies, products or technology in order to execute our business strategy, we may need to raise additional capital. We will need to raise additional capital in the future through one or more financing vehicles that may be available to us. Potential financing vehicles include: (i) the public or private issuance of securities; (ii) new collaborative agreements; and/or (iii) expansions or revisions to existing collaborative relationships.
45
Table of Contents
Considering our stage of development, the nature of our capital structure and general market conditions, if we are required to raise additional capital in the future, the additional financing may not be available on favorable terms, or at all. If additional capital is not available on favorable terms when needed, we will be required to significantly reduce operating expenses through the restructuring of our operations. If we are successful in raising additional capital, a substantial number of additional shares may be issued and these shares will dilute the ownership interest of our current investors.
If we are unable to sufficiently develop our sales, marketing and distribution capabilities or enter into successful agreements with third parties to perform these functions, we will not be able to fully commercialize our products.
We may not be successful in marketing and promoting our existing product, Hylenex recombinant, product candidates or any other products we develop or acquire in the future. Our sales, marketing and distribution capabilities are very limited. In order to commercialize any products successfully, we must internally develop substantial sales, marketing and distribution capabilities or establish collaborations or other arrangements with third parties to perform these services. We do not have extensive experience in these areas, and we may not be able to establish adequate in-house sales, marketing and distribution capabilities or engage and effectively manage relationships with third parties to perform any or all of such services. To the extent that we enter into co-promotion or other licensing arrangements, our product revenues are likely to be lower than if we directly marketed and sold our products, and any revenues we receive will depend upon the efforts of third parties, whose efforts may not meet our expectations or be successful. These third parties would be largely responsible for the speed and scope of sales and marketing efforts, and may not dedicate the resources necessary to maximize product opportunities. Our ability to cause these third parties to increase the speed and scope of their efforts may also be limited. In addition, sales and marketing efforts could be negatively impacted by the delay or failure to obtain additional supportive clinical trial data for our products. In some cases, third party partners are responsible for conducting these additional clinical trials and our ability to increase the efforts and resources allocated to these trials may be limited. For example, in January 2011 we and Baxter mutually agreed to terminate the Hylenex Partnership and the associated agreements.
If we or our partners fail to comply with regulatory requirements, regulatory agencies may take action against us or them, which could significantly harm our business.
Any approved products, along with the manufacturing processes, post-approval clinical data, labeling, advertising and promotional activities for these products, are subject to continual requirements and review by the FDA and other regulatory bodies. Regulatory authorities subject a marketed product, its manufacturer and the manufacturing facilities to continual review and periodic inspections. We, and our partners, will be subject to ongoing regulatory requirements, including required submissions of safety and other post-market information and reports, registration requirements, cGMP regulations, requirements regarding the distribution of samples to physicians and recordkeeping requirements. The cGMP regulations include requirements relating to quality control and quality assurance, as well as the corresponding maintenance of records and documentation. We rely on the compliance by our contract manufacturers with cGMP regulations and other regulatory requirements relating to the manufacture of our products. We and our partners are also subject to state laws and registration requirements covering the distribution of our products. Regulatory agencies may change existing requirements or adopt new requirements or policies. We or our partners may be slow to adapt or may not be able to adapt to these changes or new requirements.
Regulatory requirements applicable to pharmaceutical products make the substitution of suppliers and manufacturers costly and time consuming. We have minimal internal manufacturing capabilities and are, and expect to be in the future, entirely dependent on contract manufacturers and suppliers for the manufacture of our products and for their active and other ingredients. The disqualification of these manufacturers and suppliers through their failure to comply with regulatory requirements could negatively impact our business because the delays and costs in obtaining and qualifying alternate suppliers (if such alternative suppliers are available, which we cannot assure) could delay clinical trials or otherwise inhibit our ability to bring approved products to market, which would have a material adverse effect on our business and financial condition.
46
Table of Contents
Later discovery of previously unknown problems with our proprietary or partnered products, manufacturing processes or failure to comply with regulatory requirements, may result in any of the following:
| • | restrictions on our products or manufacturing processes; |
| • | warning letters; |
| • | withdrawal of the products from the market; |
| • | voluntary or mandatory recall; |
| • | fines; |
| • | suspension or withdrawal of regulatory approvals; |
| • | suspension or termination of any of our ongoing clinical trials; |
| • | refusal to permit the import or export of our products; |
| • | refusal to approve pending applications or supplements to approved applications that we submit; |
| • | product seizure; or |
| • | injunctions or the imposition of civil or criminal penalties. |
For example, Hylenex recombinant was voluntarily recalled in May 2010 because a portion of the HYLENEX manufactured by Baxter was not in compliance with the requirements of the underlying Hylenex recombinant agreements. During the second quarter of 2011, we submitted the data that the FDA had requested to support the reintroduction of Hylenex recombinant. The FDA has approved the submitted data and has granted the reintroduction of Hylenex recombinant. We expect to reintroduce Hylenex recombinant by the end of 2011.
If proprietary or partnered product candidates are approved by regulatory bodies such as the FDA but do not gain market acceptance, our business may suffer and we may not be able to fund future operations.
Assuming that our proprietary or partnered product candidates obtain the necessary regulatory approvals, a number of factors may affect the market acceptance of these existing product candidates or any other products which are developed or acquired in the future, including, among others:
| • | the price of products relative to other therapies for the same or similar treatments; |
| • | the perception by patients, physicians and other members of the health care community of the effectiveness and safety of these products for their prescribed treatments; |
| • | our ability to fund our sales and marketing efforts and the ability and willingness of our partners to fund sales and marketing efforts; |
| • | the degree to which the use of these products is restricted by the approved product label; |
| • | the effectiveness of our sales and marketing efforts and the effectiveness of the sales and marketing efforts of our partners; |
| • | the introduction of generic competitors; and |
| • | the extent to which reimbursement for our products and related treatments will be available from third party payors. |
47
Table of Contents
If these products do not gain market acceptance, we may not be able to fund future operations, including the development or acquisition of new product candidates and/or our sales and marketing efforts for our approved products, which would cause our business to suffer.
In addition, our proprietary and partnered product candidates will be restricted to the labels approved by applicable regulatory bodies such as the FDA, and these restrictions may limit the marketing and promotion of the ultimate products. If the approved labels are restrictive, the sales and marketing efforts for these products may be negatively affected.
Developing and marketing pharmaceutical products for human use involves product liability risks, for which we currently have limited insurance coverage.
The testing, marketing and sale of pharmaceutical products involves the risk of product liability claims by consumers and other third parties. Although we maintain product liability insurance coverage, product liability claims can be high in the pharmaceutical industry and our insurance may not sufficiently cover our actual liabilities. If product liability claims were to be made against us, it is possible that our insurance carriers may deny, or attempt to deny, coverage in certain instances. If a lawsuit against us is successful, then the lack or insufficiency of insurance coverage could materially and adversely affect our business and financial condition. Furthermore, various distributors of pharmaceutical products require minimum product liability insurance coverage before purchase or acceptance of products for distribution. Failure to satisfy these insurance requirements could impede our ability to achieve broad distribution of our proposed products and the imposition of higher insurance requirements could impose additional costs on us. In addition, since many of our partnered product candidates include the pharmaceutical products of a third party, we run the risk that problems with the third party pharmaceutical product will give rise to liability claims against us.
Our inability to attract, hire and retain key management and scientific personnel could negatively affect our business.
Our success depends on the performance of key management and scientific employees with biotechnology experience. Given our relatively small staff size relative to the number of programs currently under development, we depend substantially on our ability to hire, train, motivate and retain high quality personnel, especially our scientists and management team. If we are unable to retain existing personnel or identify or hire additional personnel, we may not be able to research, develop, commercialize or market our products and product candidates as expected or on a timely basis and we may not be able to adequately support current and future alliances with strategic partners.
Furthermore, if we were to lose key management personnel, such as Gregory Frost, Ph.D., our President and Chief Executive Officer, we would likely lose some portion of our institutional knowledge and technical know-how, potentially causing a substantial delay in one or more of our development programs until adequate replacement personnel could be hired and trained. For example, Dr. Frost has been with us from soon after our inception, and he possesses a substantial amount of knowledge about our development efforts. If we were to lose his services, we would experience delays in meeting our product development schedules. In 2008, we adopted a severance policy applicable to all employees and a change in control policy applicable to senior executives. We have not adopted any other policies or entered into any other agreements specifically designed to motivate officers or other employees to remain with us.
We do not have key man life insurance policies on the lives of any of our employees, including Dr. Frost.
Our operations might be interrupted by the occurrence of a natural disaster or other catastrophic event.
Our operations, including laboratories, offices and other research facilities, are located in a three building campus in San Diego, California. We depend on our facilities and on our partners, contractors and vendors for the continued operation of our business. Natural disasters or other catastrophic events, interruptions in the supply of natural resources, political and governmental changes, wildfires and other fires, floods, explosions, actions of animal rights activists, earthquakes and civil unrest could disrupt our operations or those of our partners, contractors and vendors. Even though we believe we carry commercially reasonable business interruption and liability insurance, and our contractors may carry liability insurance that protect us in certain events, we might suffer losses as a result of business interruptions that exceed the coverage available under our and our contractors’ insurance policies or for
48
Table of Contents
which we or our contractors do not have coverage. Any natural disaster or catastrophic event could have a significant negative impact on our operations and financial results. Moreover, any such event could delay our research and development programs.
If we or our partners do not achieve projected development goals in the timeframes we publicly announce or otherwise expect, the commercialization of our products and the development of our product candidates may be delayed and, as a result, our stock price may decline.
We publicly articulate the estimated timing for the accomplishment of certain scientific, clinical, regulatory and other product development goals. The accomplishment of any goal is typically based on numerous assumptions and the achievement of a particular goal may be delayed for any number of reasons both within and outside of our control. If scientific, regulatory, strategic or other factors cause us to not meet a goal, regardless of whether that goal has been publicly articulated or not, the commercialization of our products and the development of our proprietary and partnered product candidates may be delayed. In addition, the consistent failure to meet publicly announced milestones may erode the credibility of our management team with respect to future milestone estimates.
Future acquisitions could disrupt our business and harm our financial condition.
In order to augment our product pipeline or otherwise strengthen our business, we may decide to acquire additional businesses, products and technologies. As we have limited experience in evaluating and completing acquisitions, our ability as an organization to make such acquisitions is unproven. Acquisitions could require significant capital infusions and could involve many risks, including, but not limited to, the following:
| • | we may have to issue convertible debt or equity securities to complete an acquisition, which would dilute our stockholders and could adversely affect the market price of our common stock; |
| • | an acquisition may negatively impact our results of operations because it may require us to amortize or write down amounts related to goodwill and other intangible assets, or incur or assume substantial debt or liabilities, or it may cause adverse tax consequences, substantial depreciation or deferred compensation charges; |
| • | we may encounter difficulties in assimilating and integrating the business, products, technologies, personnel or operations of companies that we acquire; |
| • | certain acquisitions may impact our relationship with existing or potential partners who are competitive with the acquired business, products or technologies; |
| • | acquisitions may require significant capital infusions and the acquired businesses, products or technologies may not generate sufficient value to justify acquisition costs; |
| • | an acquisition may disrupt our ongoing business, divert resources, increase our expenses and distract our management; |
| • | acquisitions may involve the entry into a geographic or business market in which we have little or no prior experience; and |
| • | key personnel of an acquired company may decide not to work for us. |
If any of these risks occurred, it could adversely affect our business, financial condition and operating results. We cannot assure you that we will be able to identify or consummate any future acquisitions on acceptable terms, or at all. If we do pursue any acquisitions, it is possible that we may not realize the anticipated benefits from such acquisitions or that the market will not view such acquisitions positively.
49
Table of Contents
Risks Related To Ownership of Our Common Stock
Our stock price is subject to significant volatility.
We participate in a highly dynamic industry which often results in significant volatility in the market price of common stock irrespective of company performance. As a result, our high and low sales prices of our common stock during the twelve months ended September 30, 2011 were $8.31 and $5.54, respectively. We expect our stock price to continue to be subject to significant volatility and, in addition to the other risks and uncertainties described elsewhere in this Quarterly Report on Form 10-Q, in the annual report on Form 10-K for the year ended December 31, 2010 and all other risks and uncertainties that are either not known to us at this time or which we deem to be immaterial, any of the following factors may lead to a significant drop in our stock price:
| • | a dispute regarding our failure, or the failure of one of our third party partners, to comply with the terms of a collaboration agreement; |
| • | the termination, for any reason, of any of our collaboration agreements; |
| • | the sale of common stock by any significant stockholder, including, but not limited to, direct or indirect sales by members of management or our Board of Directors; |
| • | the resignation, or other departure, of members of management or our Board of Directors; |
| • | general negative conditions in the healthcare industry; |
| • | general negative conditions in the financial markets; |
| • | the failure, for any reason, to obtain regulatory approval for any of our proprietary or partnered product candidates; |
| • | the failure, for any reason, to secure or defend our intellectual property position; |
| • | for those products that are waiting to be approved by the FDA, the failure of the FDA to approve such products in a timely manner consistent with the FDA’s historical approval process; |
| • | the suspension of any clinical trial due to safety or patient tolerability issues; |
| • | the suspension of any clinical trial due to market and/or competitive conditions; |
| • | our failure, or the failure of our third party partners, to successfully commercialize products approved by applicable regulatory bodies such as the FDA; |
| • | our failure, or the failure of our third party partners, to generate product revenues anticipated by investors; |
| • | problems with an API contract manufacturer or a fill and finish manufacturer for any product or product candidate; |
| • | the sale of additional debt and/or equity securities by us; |
| • | our failure to obtain financing on acceptable terms; or |
| • | a restructuring of our operations. |
50
Table of Contents
Future sales of shares of our common stock pursuant to our universal shelf registration statement may negatively affect our stock price.
We currently have the ability to offer and sell up to $39.8 million of additional equity or debt securities under an effective universal shelf registration statement. Sales of substantial amounts of shares of our common stock or other securities under our universal shelf registration statement could lower the market price of our common stock and impair our ability to raise capital through the sale of equity securities. In the future, we may issue additional options, warrants or other derivative securities convertible into our common stock.
Trading in our stock has historically been limited, so investors may not be able to sell as much stock as they want to at prevailing market prices.
Our stock has historically traded at a low daily trading volume. If low trading volume continues, it may be difficult for stockholders to sell their shares in the public market at any given time at prevailing prices.
Risks Related To Our Industry
Compliance with the extensive government regulations to which we are subject is expensive and time consuming and may result in the delay or cancellation of product sales, introductions or modifications.
Extensive industry regulation has had, and will continue to have, a significant impact on our business. All pharmaceutical companies, including ours, are subject to extensive, complex, costly and evolving regulation by the federal government, principally the FDA and, to a lesser extent, the U.S. Drug Enforcement Administration, or DEA, and foreign and state government agencies. The Federal Food, Drug and Cosmetic Act, the Controlled Substances Act and other domestic and foreign statutes and regulations govern or influence the testing, manufacturing, packaging, labeling, storing, recordkeeping, safety, approval, advertising, promotion, sale and distribution of our products. Under certain of these regulations, we and our contract suppliers and manufacturers are subject to periodic inspection of our or their respective facilities, procedures and operations and/or the testing of products by the FDA, the DEA and other authorities, which conduct periodic inspections to confirm that we and our contract suppliers and manufacturers are in compliance with all applicable regulations. The FDA also conducts pre-approval and post-approval reviews and plant inspections to determine whether our systems, or our contract suppliers’ and manufacturers’ processes, are in compliance with cGMP and other FDA regulations. If we, or our contract supplier, fail these inspections, we may not be able to commercialize our product in a timely manner without incurring significant additional costs, or at all.
In addition, the FDA imposes a number of complex regulatory requirements on entities that advertise and promote pharmaceuticals including, but not limited to, standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities, and promotional activities involving the internet.
We are dependent on receiving FDA and other governmental approvals prior to manufacturing, marketing and shipping our products. Consequently, there is always a risk that the FDA or other applicable governmental authorities will not approve our products, or will take post-approval action limiting or revoking our ability to sell our products, or that the rate, timing and cost of such approvals will adversely affect our product introduction plans or results of operations.
We may be required to initiate or defend against legal proceedings related to intellectual property rights, which may result in substantial expense, delay and/or cessation of the development and commercialization of our products.
We primarily rely on patents to protect our intellectual property rights. The strength of this protection, however, is uncertain. For example, it is not certain that:
| • | our patents and pending patent applications cover products and/or technology that we invented first; |
| • | we were the first to file patent applications for these inventions; |
51
Table of Contents
| • | others will not independently develop similar or alternative technologies or duplicate our technologies; |
| • | any of our pending patent applications will result in issued patents; and |
| • | any of our issued patents, or patent pending applications that result in issued patents, will be held valid and infringed in the event the patents are asserted against others. |
We currently own or license several patents and also have pending patent applications applicable to rHuPh20 and other proprietary materials. There can be no assurance that our existing patents, or any patents issued to us as a result of our pending patent applications, will provide a basis for commercially viable products, will provide us with any competitive advantages, or will not face third party challenges or be the subject of further proceedings limiting their scope or enforceability. For example, a European patent, EP1603541, claiming rHuPH20 was granted to us on November 11, 2009. Claims to the human PH20 glycoprotein, PEGylated variants, the glycoprotein produced by recombinant methods, and pharmaceutical compositions with other agents, including antibodies, insulins, cytokines, anti-infectives and additional therapeutic classes were awarded in this patent and additional claims are in prosecution. On August 13, 2010, however, we learned that an opposition to this patent was filed with the European Patent Office. We have contested the opposition with written submissions to the European Patent Office and we expect to obtain European patent protection that is equal or superior to claims previously issued in a counterpart United States patent (U.S. Patent No. 7,767,429). Any limitations in our patent portfolio could have a material adverse effect on our business and financial condition. In addition, if any of our pending patent applications do not result in issued patents, or result in issued patents with narrow or limited claims, this could have a material adverse effect on our business and financial condition.
We may become involved in interference proceedings in the U.S. Patent and Trademark Office, or other proceedings in other jurisdictions, to determine the priority, validity or enforceability of our patents. In addition, costly litigation could be necessary to protect our patent position.
We also rely on trademarks to protect the names of our products. These trademarks may not be acceptable to regulatory agencies. In addition, these trademarks may be challenged by others. If we enforce our trademarks against third parties, such enforcement proceedings may be expensive. We also rely on trade secrets, unpatented proprietary know-how and continuing technological innovation that we seek to protect with confidentiality agreements with employees, consultants and others with whom we discuss our business. Disputes may arise concerning the ownership of intellectual property or the applicability or enforceability of these agreements, and we might not be able to resolve these disputes in our favor.
In addition to protecting our own intellectual property rights, third parties may assert patent, trademark or copyright infringement or other intellectual property claims against us based on what they believe are their own intellectual property rights. If we become involved in any intellectual property litigation, we may be required to pay substantial damages, including but not limited to treble damages, for past infringement if it is ultimately determined that our products infringe a third party’s intellectual property rights. Even if infringement claims against us are without merit, defending a lawsuit takes significant time, may be expensive and may divert management’s attention from other business concerns. Further, we may be stopped from developing, manufacturing or selling our products until we obtain a license from the owner of the relevant technology or other intellectual property rights. If such a license is available at all, it may require us to pay substantial royalties or other fees.
Patent protection for protein-based therapeutic products and other biotechnology inventions is subject to a great deal of uncertainty, and if patent laws or the interpretation of patent laws change, our competitors may be able to develop and commercialize products based on our discoveries.
Patent protection for protein-based therapeutic products is highly uncertain and involves complex legal and factual questions. In recent years, there have been significant changes in patent law, including the legal standards that govern the scope of protein and biotechnology patents. Standards for patentability of full-length and partial genes, and their corresponding proteins, are changing. Recent court decisions have made it more difficult to obtain patents, by making it more difficult to satisfy the requirement of non-obviousness, have decreased the availability of injunctions against infringers, and have increased the likelihood of challenging the validity of a patent through a declaratory judgment action. Taken together, these decisions could make it more difficult and costly for us to obtain,
52
Table of Contents
license and enforce our patents. In addition, the Leahy-Smith America Invents Act (HR 1249) was signed into law in September 2011, which among other changes to the U.S. patent laws, changes patent priority from “first to invent” to “first to file,” implements a post-grant opposition system for patents and provides for a prior user defense to infringement. These judicial and legislative changes have introduced significant uncertainty in the patent law landscape and may potentially negatively impact our ability to procure, maintain and enforce patents to provide exclusivity for our products.
There also have been, and continue to be, policy discussions concerning the scope of patent protection awarded to biotechnology inventions. Social and political opposition to biotechnology patents may lead to narrower patent protection within the biotechnology industry. Social and political opposition to patents on genes and proteins may lead to narrower patent protection, or narrower claim interpretation, for genes, their corresponding proteins and inventions related to their use, formulation and manufacture. Patent protection relating to biotechnology products is also subject to a great deal of uncertainty outside the United States, and patent laws are evolving and undergoing revision in many countries. Changes in, or different interpretations of, patent laws worldwide may result in our inability to obtain or enforce patents, and may allow others to use our discoveries to develop and commercialize competitive products, which would impair our business.
If third party reimbursement and customer contracts are not available, our products may not be accepted in the market.
Our ability to earn sufficient returns on our products will depend in part on the extent to which reimbursement for our products and related treatments will be available from government health administration authorities, private health insurers, managed care organizations and other healthcare providers.
Third-party payors are increasingly attempting to limit both the coverage and the level of reimbursement of new drug products to contain costs. Consequently, significant uncertainty exists as to the reimbursement status of newly approved healthcare products. Third party payors may not establish adequate levels of reimbursement for the products that we commercialize, which could limit their market acceptance and result in a material adverse effect on our financial condition.
Customer contracts, such as with group purchasing organizations and hospital formularies, will often not offer contract or formulary status without either the lowest price or substantial proven clinical differentiation. If our products are compared to animal-derived hyaluronidases by these entities, it is possible that neither of these conditions will be met, which could limit market acceptance and result in a material adverse effect on our financial condition.
The rising cost of healthcare and related pharmaceutical product pricing has led to cost containment pressures that could cause us to sell our products at lower prices, resulting in less revenue to us.
Any of the proprietary or partnered products that have been, or in the future are, approved by the FDA may be purchased or reimbursed by state and federal government authorities, private health insurers and other organizations, such as health maintenance organizations and managed care organizations. Such third party payors increasingly challenge pharmaceutical product pricing. The trend toward managed healthcare in the United States, the growth of such organizations, and various legislative proposals and enactments to reform healthcare and government insurance programs, including the Medicare Prescription Drug Modernization Act of 2003, could significantly influence the manner in which pharmaceutical products are prescribed and purchased, resulting in lower prices and/or a reduction in demand. Such cost containment measures and healthcare reforms could adversely affect our ability to sell our products.
In March 2010, the United States adopted the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or the Healthcare Reform Act. This law substantially changes the way health care is financed by both governmental and private insurers, and significantly impacts the pharmaceutical industry. The Healthcare Reform Act contains a number of provisions that are expected to impact our business and operations, in some cases in ways we cannot currently predict. Changes that may affect our business include those governing enrollment in federal healthcare programs, reimbursement changes, fraud and abuse and enforcement. These changes will impact existing government healthcare programs and will result in the development of new programs, including Medicare payment for performance initiatives and improvements to the physician quality reporting system and feedback program.
53
Table of Contents
Additional provisions of the Healthcare Reform Act, some of which become effective in 2011, may negatively affect our revenues in the future. For example, the Healthcare Reform Act imposes a non-deductible excise tax on pharmaceutical manufacturers or importers that sell branded prescription drugs to U.S. government programs that we believe will impact our revenues from our products. In addition, as part of the Healthcare Reform Act’s provisions closing a funding gap that currently exists in the Medicare Part D prescription drug program (commonly known as the “donut hole”), we will also be required to provide a 50% discount on branded prescription drugs dispensed to beneficiaries within this donut hole. We expect that the Healthcare Reform Act and other healthcare reform measures that may be adopted in the future could have a material adverse effect on our industry generally and on our ability to maintain or increase our product sales or successfully commercialize our product candidates or could limit or eliminate our future spending on development projects.
Furthermore, individual states have become increasingly aggressive in passing legislation and implementing regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access, importation from other countries and bulk purchasing. Legally mandated price controls on payment amounts by third party payors or other restrictions could negatively and materially impact our revenues and financial condition. We anticipate that we will encounter similar regulatory and legislative issues in most other countries outside the United States.
We face intense competition and rapid technological change that could result in the development of products by others that are superior to our proprietary and partnered products under development.
Our proprietary and partnered products have numerous competitors in the United States and abroad including, among others, major pharmaceutical and specialized biotechnology firms, universities and other research institutions that have developed competing products. Pending the reintroduction of Hylenex recombinant, the competitors for Hylenex recombinant will include, but are not limited to ISTA Pharmaceuticals, Inc. and Amphastar Pharmaceuticals, Inc. among others. For our analog insulin with rHuPH20 product candidates, such competitors may include Biodel Inc., Novo Nordisk Inc. and Mannkind Corporation. These competitors may develop technologies and products that are more effective, safer, or less costly than our current or future proprietary and partnered product candidates or that could render our technologies and product candidates obsolete or noncompetitive. Many of these competitors have substantially more resources and product development, manufacturing and marketing experience and capabilities than we do. In addition, many of our competitors have significantly greater experience than we do in undertaking preclinical testing and clinical trials of pharmaceutical product candidates and obtaining FDA and other regulatory approvals of products and therapies for use in healthcare.
Item 3. Quantitative and Qualitative Disclosures About Market Risk
There have been no material changes in our market risks during the three months ended September 30, 2011. For additional information on market risks, refer to “Quantitative and Qualitative Disclosures About Market Risk” included in Part II, Item 7A of our annual report on Form 10-K for the year ended December 31, 2010.
Item 4. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in our Exchange Act reports is recorded, processed, summarized and reported within the timelines specified in the Securities and Exchange Commission’s rules and forms, and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure. In designing and evaluating the disclosure controls and procedures, management recognized that any controls and procedures, no matter how well designed and operated, can only provide reasonable assurance of achieving the desired control objectives, and in reaching a reasonable level of assurance, management necessarily was required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
Under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, we conducted an evaluation of our disclosure controls and procedures, as such term is defined under Rule 13a-15(e) promulgated under the Securities Exchange Act of 1934, as amended. Based
54
Table of Contents
on this evaluation, our principal executive officer and our principal financial officer concluded that our disclosure controls and procedures were effective as of the end of the period covered by this Quarterly Report on Form 10-Q.
Changes in Internal Control Over Financial Reporting
There were no significant changes in our internal control over financial reporting that occurred during the three months ended September 30, 2011, that have materially affected, or are reasonably likely to materially affect our internal control over financial reporting.
From time to time, we may be involved in disputes, including litigation, relating to claims arising out of operations in the normal course of our business. Any of these claims could subject us to costly legal expenses and, while we generally believe that we have adequate insurance to cover many different types of liabilities, our insurance carriers may deny coverage or our policy limits may be inadequate to fully satisfy any damage awards or settlements. If this were to happen, the payment of any such awards could have a material adverse effect on our consolidated results of operations and financial position. Additionally, any such claims, whether or not successful, could damage our reputation and business. We currently are not a party to any legal proceedings, the adverse outcome of which, in management’s opinion, individually or in the aggregate, would have a material adverse effect on our consolidated results of operations or financial position.
In May 2010, the Company delivered a notice of breach to Baxter due to Baxter’s failure to provide Hylenex recombinant in accordance with the terms of the Hylenex Partnership. Baxter had contested the claims made in our initial notice of breach and asserted their own breach claims against us. Pursuant to the terms of the Transition Agreement, signed on July 18, 2011, Baxter’s breach claims against us were discharged.
We have provided updated Risk Factors in the section labeled “Risk Factors” in Part I, Item 2, “Management’s Discussion and Analysis of Financial Condition and Results of Operations”. The “Risk Factors” section provides updated information in certain areas, particularly with respect to uncertainties regarding the regulatory approval of proprietary and partnered product candidates. We do not believe the updates have materially changed the type or magnitude of the risks we face in comparison to the disclosure provided in our most recent Annual Report on Form 10-K.
Item 2. Unregistered Sales of Equity Securities and Use of Proceeds
Not applicable.
Item 3. Defaults Upon Senior Securities
Not applicable.
Item 4. (Removed and Reserved)
Not applicable.
| Exhibit | Title | |
| 31.1 | Certification of Chief Executive Officer pursuant to Rule 13a-14(a) and 15d-14(a) of the Securities Exchange Act of 1934, as amended | |
55
Table of Contents
| 31.2 | Certification of Chief Financial Officer pursuant to Rule 13a-14(a) and 15d-14(a) of the Securities Exchange Act of 1934, as amended | |
| 32 | Certification of Chief Executive Officer and Chief Financial Officer pursuant to 18 U.S.C. 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | |
| 101 | The following materials from the Halozyme Therapeutics, Inc. Quarterly Report on Form 10-Q for the quarter ended September 30, 2011 formatted in eXtensible Business Reporting Language (XBRL): (i) the Condensed Consolidated Balance Sheets, (ii) the Condensed Consolidated Statements of Operations, (iii) the Condensed Consolidated Statements of Cash Flows and (iv) related notes, tagged as block of text*. |
| * | Pursuant to Rule 406T of Regulation S-T, the Interactive Data Files on Exhibit 101 hereto are deemed not filed or part of a registration statement or prospectus for purposes of Sections 11 or 12 of the Securities Act of 1933, as amended, are deemed not filed for purposes of Section 18 of the Securities and Exchange Act of 1934, as amended, and otherwise are not subject to liability under those sections. |
56
Table of Contents
Pursuant to the requirements of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
| Halozyme Therapeutics, Inc., | ||||
| a Delaware corporation | ||||
| Dated: November 8, 2011 |
/s/ Gregory I. Frost, Ph.D. |
|||
| Gregory I. Frost, Ph.D. | ||||
| President and Chief Executive Officer | ||||
| (Principal Executive Officer) | ||||
| Dated: November 8, 2011 |
/s/ Kurt A. Gustafson |
|||
| Kurt A. Gustafson | ||||
| Vice President, Secretary and Chief Financial Officer | ||||
| (Principal Financial and Accounting Officer) | ||||
57