Attached files
| file | filename |
|---|---|
| EX-23.3 - EXHIBIT 23.3 - Cellectar Biosciences, Inc. | v234598_ex23-3.htm |
| EX-23.2 - EXHIBIT 23.2 - Cellectar Biosciences, Inc. | v234598_ex23-2.htm |
Registration No. 333-175284
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Amendment No. 2 to
FORM S-1
REGISTRATION STATEMENT UNDER THE SECURITIES ACT OF 1933
NOVELOS THERAPEUTICS, INC.
(Exact name of registrant as specified in its charter)
|
Delaware
|
2834
|
04-3321804
|
|
(State or other jurisdiction
of incorporation or organization)
|
(Primary Standard Industrial
Classification Code Number)
|
(I.R.S. Employer
Identification Number)
|
One Gateway Center
Suite 504
Newton, Massachusetts 02458
(617) 244-1616
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive offices)
Harry S. Palmin
President and Chief Executive Officer
Novelos Therapeutics, Inc.
One Gateway Center, Suite 504
Newton, Massachusetts 02458
(617) 244-1616
(Name, address, including zip code, and telephone number, including area code, of agent for service)
Copies to:
|
Paul Bork, Esq.
Foley Hoag LLP
155 Seaport Boulevard
Boston, Massachusetts 02210
(617) 832-1000
|
Harvey Kesner, Esq.
Sichenzia Ross Friedman Ference LLP
61 Broadway
New York, New York 10006
(212) 930-9700
|
Approximate date of commencement of proposed sale to the public: As soon as practicable after this Registration Statement is declared effective.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, check the following box. ¨
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer,” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
(Check one):
|
Large accelerated filer ¨
|
Accelerated filer ¨
|
|
Non-accelerated filer ¨
|
Smaller reporting company x
|
(Do not check if a smaller reporting company)
The registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933 or until the Registration Statement shall become effective on such date as the Commission, acting pursuant to said Section 8(a), may determine.
The information in this preliminary prospectus is not complete and may be changed. We may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities and it is not soliciting offers to buy these securities in any jurisdiction where the offer or sale is not permitted.
|
PRELIMINARY PROSPECTUS
|
SUBJECT TO COMPLETION, DATED SEPTEMBER 14, 2011
|
Novelos Therapeutics, Inc.
shares of common stock
This is a firm commitment public offering of shares of our common stock.
Our common stock is quoted on the OTC Bulletin Board under the symbol “NVLT.OB”. We intend to apply for listing of our common stock on The NASDAQ Capital Market under the symbol “NVLT”. No assurance can be given that our application will be approved. Prior to the effectiveness of the registration statement of which this prospectus is a part, we anticipate that we will effect a reverse stock split within a range of 1:2 to 1:10 (the “Offering Reverse Split”). The proposal for the Offering Reverse Split has been approved by our stockholders and our board of directors is authorized to determine the time and ratio at which the reverse split will be effected by filing the appropriate amendment to our certificate of incorporation. On September 13, 2011, the last reported sale price for our common stock was $1.38 per share.
Investing in the offered securities involves a high degree of risk. See “Risk Factors” beginning on page 7 of this prospectus for a discussion of information that you should consider before investing in our securities.
NEITHER THE SECURITIES AND EXCHANGE COMMISSION NOR ANY STATE SECURITIES COMMISSION HAS APPROVED OR DISAPPROVED OF THESE SECURITIES OR DETERMINED IF THIS PROSPECTUS IS TRUTHFUL OR COMPLETE. ANY REPRESENTATION TO THE CONTRARY IS A CRIMINAL OFFENSE.
|
Per Share
|
Total
|
|||||||
|
|
|
|
|
|
|
|
||
|
Public offering price
|
|
$
|
|
|
|
$
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Underwriting discounts and commissions (1)
|
|
$
|
|
|
|
$
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Proceeds, before expenses, to us
|
|
$
|
|
|
|
$
|
|
|
(1) See “Underwriting” for a description of compensation payable to the underwriter.
We have granted a 45-day option to Rodman & Renshaw, LLC, the underwriter, to purchase up to an additional shares from us solely to cover over-allotments, if any. The shares issuable upon exercise of the underwriter option are identical to those offered by this prospectus and have been registered under the registration statement of which this prospectus forms a part.
In addition, we have also agreed to issue to the underwriter a warrant to purchase a number of shares equal to 5% of the shares of common stock sold (excluding the over-allotment) exercisable at $ per share.
The underwriter expects to deliver the shares to purchasers in the offering on or about , 2011.
Rodman & Renshaw, LLC
The date of this prospectus is , 2011.

Time-Lapse Photography Illustrating Tumor Regression in Mouse After a Single Dose of HOT

LIGHT Compared to FDG as a Cancer PET Imaging Agent in Mouse Xenograft Model
(Same mouse: first imaged with FDG, then with LIGHT 24 hours later)

T = Cancerous Tumor, B = Bladder, I = Inflammation site, H = Heart
The images provided above are for illustrative purposes only and may not be indicative of all results.
The above illustrations do not refer to products approved by the FDA.
Novelos has not received any revenue from the sale of its products.
NOVELOS THERAPEUTICS, INC.
|
|
Page
|
|
|
|
|
PROSPECTUS SUMMARY
|
1
|
|
|
|
|
RISK FACTORS
|
7
|
|
|
|
|
FORWARD-LOOKING STATEMENTS
|
19
|
|
|
|
|
USE OF PROCEEDS
|
19
|
|
|
|
|
CAPITALIZATION
|
20
|
|
|
|
|
MARKET FOR COMMON EQUITY AND RELATED STOCKHOLDER MATTERS
|
20
|
|
|
|
|
DILUTION
|
22
|
|
|
|
|
MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
|
22
|
|
|
|
|
BUSINESS
|
27
|
|
|
|
|
LITIGATION
|
38
|
|
|
|
|
PROPERTIES
|
39
|
|
|
|
|
MANAGEMENT
|
39
|
|
|
|
|
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT
|
45
|
|
|
|
|
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS
|
47
|
|
|
|
|
UNDERWRITING
|
48
|
|
|
|
|
DESCRIPTION OF SECURITIES
|
52
|
|
|
|
|
DISCLOSURE OF COMMISSION POSITION ON INDEMNIFICATION FOR SECURITIES ACT LIABILITIES
|
54
|
|
|
|
|
WHERE YOU CAN FIND MORE INFORMATION
|
54
|
|
|
|
|
LEGAL MATTERS
|
54
|
|
|
|
|
EXPERTS
|
54
|
|
|
|
|
GLOSSARY OF SCIENTIFIC TERMS
|
55
|
|
FINANCIAL STATEMENTS (POST-ACQUISITION)
|
F-1
|
|
|
|
|
FINANCIAL STATEMENTS (PRE-ACQUISITION)
|
F-26
|
|
UNAUDITED PRO FORMA CONDENSED COMBINED FINANCIAL STATEMENTS
|
F-60
|
No dealer, salesperson or other person has been authorized to give any information or to make any representations other than those contained in this prospectus in connection with the offer contained in this prospectus and, if given or made, such information or representations must not be relied upon as having been authorized by us.
Neither the delivery of this prospectus nor any sale made hereunder shall under any circumstances create an implication that there has been no change in our affairs since the date hereof. This prospectus does not constitute an offer to sell or a solicitation of an offer to buy securities other than those specifically offered hereby or of any securities offered hereby in any jurisdiction where, or to any person to whom, it is unlawful to make such offer or solicitation. The information contained in this prospectus speaks only as of the date of this prospectus unless the information specifically indicates that another date applies.
This prospectus has been prepared based on information provided by us and by other sources that we believe are reliable. This prospectus summarizes certain documents and other information in a manner we believe to be accurate, but we refer you to the actual documents, if any, for a more complete understanding of what we discuss in this prospectus. In making a decision to invest in the common stock, you must rely on your own examination of us and the terms of the offering and securities offered in this prospectus, including the merits and risks involved.
We are not making any representation to you regarding the legality of an investment in the securities offered in this prospectus under any legal investment or similar laws or regulations. You should not consider any information in this prospectus to be legal, business, tax or other advice. You should consult your own attorney, business advisor and tax advisor for legal, business and tax advice regarding an investment in our common stock.
You may only rely on the information contained in this prospectus or that we have referred you to. We have not authorized anyone to provide you with different information. This prospectus does not constitute an offer to sell or a solicitation of an offer to buy any securities other than the securities offered by this prospectus. This prospectus does not constitute an offer to sell or a solicitation of an offer to buy any securities in any circumstances in which such offer or solicitation is unlawful or in any state or to any person within any state to whom such offer would be unlawful under the laws or securities regulations of such state. Neither the delivery of this prospectus nor any sale made in connection with this prospectus shall, under any circumstances, create any implication that there has been no change in our affairs since the date of this prospectus or that the information contained by reference to this prospectus is correct as of any time after its date.
PROSPECTUS SUMMARY
This summary highlights information contained elsewhere in this prospectus and does not contain all of the information that you should consider in making your investment decision. Before investing in our securities, you should carefully read this entire prospectus, including our financial statements and the related notes and the information set forth under the headings “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” in each case included elsewhere in this prospectus.
References in this prospectus to “Cellectar” relate to the activities and financial information of Cellectar, Inc. prior to the Acquisition, references to “Novelos” relate to the activities and financial information of Novelos Therapeutics, Inc. prior to the Acquisition and references to “we” “us” or “the Company” relate to the activities and obligations of the combined company following the Acquisition.
Please refer to the Glossary of Scientific Terms on page 55 of this prospectus for definitions of certain technical and scientific terms used throughout this prospectus.
Overview
Our Business
On April 8, 2011, Novelos Therapeutics, Inc. (“we, the “Company”, “Novelos”) completed a business combination with Cellectar, Inc. (“Cellectar”), a privately held Wisconsin corporation that designed and developed products to detect, treat and monitor a wide variety of human cancers (the “Acquisition”). Following the Acquisition, we are developing novel drugs for the treatment and diagnosis of cancer based on our cancer-targeting technology platform: CLR1401(“COLD”), 131 I-CLR1404 (“HOT”, iodine-131 radiolabeled compound) and 124 I-CLR1404 (“LIGHT”, labeled with a shorter-lived radioisotope, iodine-124). A radiolabeled compound is a radioactive compound containing radioisotopes as a part of its structure. A radioisotope (also referred to as radioactive isotopes or radionuclides) is a variant of a particular chemical element (e.g. iodine) that has an unstable nucleus and can undergo radioactive decay, during which ionizing radiation (e.g. gamma rays or subatomic particles) is emitted. We believe our compounds are selectively taken up and retained in a wide variety of cancer cells (including cancer stem cells) versus normal cells. We believe our therapeutic compounds directly kill cancer cells while minimizing harm to normal cells, offering the potential for a paradigm shift in cancer therapy by providing efficacy against all three major drivers of mortality in cancer: primary tumors, metastases and cancer stem cell-based relapse. More specifically, we believe our technology enables targeted delivery to cancer cells of apoptosis-inducing Akt inhibition or, when a radioactive molecule is attached, of ionizing radiation sufficient to kill cancer cells. Apoptosis is a tightly regulated form of cell death by which organisms can eliminate damaged or aberrant cells. Apoptosis is often absent in cancer cells, contributing to their uncontrolled growth. Akt is a molecule that can regulate cell growth and survival and is present at high levels in many cancer types. When radiolabeled with iodine-124 for PET imaging, we believe our agent can provide an accurate and quantitative diagnosis of cancer, including metastases, and we also believe our agent can objectively predict and measure therapeutic success. Together, we believe this platform is capable of yielding multiple, distinct oncology product opportunities in a broad spectrum of cancers that would enable us to “find, treat and follow” cancer anywhere in the body in a novel, effective and highly selective way.
COLD is a cancer-targeted chemotherapy that in pre-clinical experiments inhibits the phosphatidylinosotol 3-kinase (PI3K)/Akt survival pathway, which is overexpressed in many types of cancer. As a result, COLD selectively inhibits Akt activity, induces apoptosis through caspase activation and inhibits cell proliferation in cancer cells versus normal cells. Caspases are molecules that can stimulate apoptosis. COLD also exhibits significant in vivo efficacy in mouse xenograft tumor models, including non-small cell lung cancer and triple-negative breast cancers, producing long-lasting tumor growth suppression and significantly increased survival. We believe COLD has the potential to be best-in-class versus other Akt inhibitors in development due to a) cancer cell/cancer stem cell targeting, resulting in cancer-selective inhibition of Akt and cell proliferation or b) suitability for intravenous administration that we believe offers the prospect of greater systemic exposure and hence Akt inhibition in cancer cells, which we believe would result in superior efficacy. We expect to submit an Investigational New Drug (“IND”) application to the United States Food and Drug Administration (“FDA”) in late 2012.
HOT (iodine-131 radiolabeled compound) is a small-molecule, broad-spectrum, cancer-targeted molecular radiotherapeutic that we believe has first-in-class potential. HOT is comprised of a small quantity of COLD (too little for significant Akt inhibition), acting as a cancer-targeted delivery and retention vehicle, and incorporating a cytotoxic (cell-killing) dose of radiotherapy (in the form of iodine-131, a radioisotope that is already in common use to treat thyroid and other cancer types). It is this “intracellular radiation” mechanism of cancer cell killing, coupled with selective delivery to a wide range of malignant tumor types, that imbues HOT with broad-spectrum anti-cancer activity. In 2009, we filed an IND with the FDA to study HOT in humans. In early 2010, we successfully completed a Phase 1a dosimetry trial in humans demonstrating initial safety and establishing dosing parameters for a Phase 1b dose-escalation trial. Radiation dosimetry measures how much radiation is absorbed by tumors and body organs in order to optimize delivery of radiation therapy. The Phase 1b dose-escalation trial is aimed at determining the Maximum Tolerated Dose, and we expect it to begin in the fourth quarter of 2011. In parallel, we expect to initiate Phase 2 efficacy trials in solid tumors in 2012 as soon as a minimal efficacious dose is established. We may determine such an effective dose upon seeing a tumor response in the Phase 1b trial or calculating it from parallel imaging trials in patients (see LIGHT below). Preclinical experiments in vitro (in cell culture) and in vivo (in animals) have demonstrated selective killing of cancer cells along with a benign safety profile. HOT’s anti-tumor/survival-prolonging activities have been demonstrated in over a dozen different xenograft models (human tumor cells implanted into animals) including breast, prostate, lung, glioma (brain), pancreatic, melanoma, ovarian, uterine, renal and colorectal cancers. In all but two models, a single administration of HOT was sufficient for efficacy. In view of HOT’s selective uptake and retention in a wide range of solid tumors, its single-agent efficacy in xenograft models and its non-specific mechanism of cancer-killing (radiation), we expect to first develop HOT as a monotherapy, initially for solid tumors.
1
LIGHT (labeled with a shorter-lived radioisotope, iodine-124) is a small-molecule imaging agent that we believe has first-in-class potential in detecting and quantifying cancerous tumors and metastases. LIGHT is comprised of a small quantity of COLD (too little for Akt inhibition), acting as a cancer-targeted delivery and retention vehicle, and incorporating iodine-124, a recently discovered positron emission tomography (PET) imaging isotope. PET imaging used in conjunction with CT scanning has now become the imaging method of choice in oncology. In studies to date, LIGHT selectively illuminated malignant tumors in 52 of 54 animal models of cancer, reflecting broad-spectrum, cancer-selective uptake and retention. We expect investigator-sponsored Phase 1/2 trials of LIGHT as a PET imaging agent to begin in the fourth quarter of 2011. The trials will initially include brain metastases, lung and breast cancers. These human trials, if successful, will serve two important purposes:
|
|
·
|
To provide proof-of-concept for LIGHT itself as a PET imaging agent with the potential to supplant the current “gold standard” agent, 18-fluoro-deoxyglucose (FDG), due to what we believe to be LIGHT’s superior cancer-specificity and more favorable logistics of clinical use; and
|
|
|
·
|
To accelerate clinical development of HOT by predicting efficacy and enabling estimation of efficacious doses of HOT for Phase 2 trials.
|
Our core technology platform is based upon the research conducted by Cellectar’s founder and our Chief Scientific Officer, Dr. Jamey Weichert, commencing in 1994 at the University of Michigan (“U. Mich.”) where alkyphospholipid analogs were initially designed, synthesized, radiolabeled, and evaluated. Since 1998, Dr. Weichert has continued his research at the University of Wisconsin (“U. Wisc.”) and founded Cellectar in 2002 to further develop and commercialize the technology. Cellectar obtained exclusive rights to the related technology patents in North America owned by U. Mich. in 2003, and continued development of the platform while obtaining ownership of numerous additional patents and patent applications in North America, Europe and Japan (lasting until 2025, 2028 and 2030, without extensions) prior to the Acquisition. The license granted by U. Mich. to Cellectar (“the U.Mich license”) gives us the exclusive right to develop, manufacture, market and sublicense our HOT and LIGHT compounds, and provides, among other things, that we pay a royalty equal to 3% of net sales of any licensed products sold by us or our sublicensees, with a reduction in such royalties in the case of certain incoming or outgoing sublicense arrangements. The license also requires us to make payments to U. Mich., ranging from $50,000 to $200,000, upon the achievement of certain key regulatory and commercial milestones, for an aggregate of up to $400,000 in milestone payments. The license expires upon the expiration of the last covered Michigan patent (currently 2016 without extensions). U. Mich. may terminate the agreement if we cease operations, if we fail to make any required payments under the agreement, or if we otherwise materially breach the agreement, subject to the applicable notice and cure periods. If the U. Mich. license expires or terminates for any reason, it will not impact the ownership of the additional patents that we have independently obtained, however the early termination of the U. Mich. license would result in the loss of our rights to use the covered patents.
Key Risks and Uncertainties
We are subject to numerous risks and uncertainties, including the following:
|
|
·
|
We will require additional capital in order to continue our operations, and may have difficulty raising additional capital.
|
|
|
·
|
We are a development stage company with a history of losses and can provide no assurance of our future operating results;
|
|
|
·
|
We and our Chief Executive Officer are defendants in a securities fraud class action lawsuit. We are also defending counterclaims in another lawsuit that we initiated, and if we are not successful in defending claims against us, the resulting liability could be substantial;
|
|
|
·
|
At present, our success depends solely on the successful commercialization of Cellectar compounds;
|
|
|
·
|
The integration of Novelos and Cellectar may be costly and difficult;
|
|
|
·
|
We have a history of recurring losses and an accumulated deficit which, among other factors, raise substantial doubt about our ability to continue as a going concern, which may hinder our ability to obtain future financing;
|
|
|
·
|
The failure to complete development of our therapeutic technology, to obtain government approvals, including required FDA approvals, or to comply with ongoing governmental regulations could prevent, delay or limit introduction or sale of proposed products and result in failure to achieve revenues or maintain our ongoing business;
|
|
|
·
|
Clinical trials involve a lengthy and expensive process with an uncertain outcome, and results of earlier studies and trials may not be predictive of future trial results;
|
|
|
·
|
We may be required to suspend or discontinue clinical trials due to unexpected side effects or other safety risks that could preclude approval of our product candidates;
|
2
|
|
·
|
We have limited in-house research and manufacturing capacity and will rely, to some extent, on research and manufacturing facilities at various universities, hospitals, contract research organizations and contract manufacturers for a portion of our research, development, and manufacturing. In the event we exceed our in-house capacity or lose access to those facilities, our ability to gain FDA approval and commercialization of our drug delivery technology and products could be delayed or impaired; and
|
|
|
·
|
We are exposed to product, clinical and preclinical liability risks that could create a substantial financial burden should we be sued.
|
For a more detailed description of the material risks and uncertainties we face, please see “Risk Factors” beginning on page 7 of this prospectus.
Reverse Stock Split
At a special meeting of our stockholders held on June 30, 2011, our stockholders approved a proposal to amend our Amended and Restated Certificate of Incorporation, or Charter, to effect a reverse split of our common stock within a range of 1:2 to 1:10 (“the Offering Reverse Split”) in order to satisfy requirements for the listing of our common stock on The NASDAQ Capital Market. The exact ratio will be set within such range in the discretion of our board of directors without further approval or authorization of our stockholders, with our board of directors having the ultimate discretion as to whether or not to proceed with the Offering Reverse Split. Our board of directors has determined that, immediately prior to the effective date of this registration statement of which this prospectus is a part, the shares of our common stock then outstanding will be subject to a reverse split on a one-for- basis.
Recent Changes in Capital
Immediately prior to the Acquisition, we completed a 1-for-153 reverse split of our common stock (the “April Reverse Split”). We then issued to the former shareholders of Cellectar 17,001,596 shares of our common stock as consideration for the Acquisition. The shares issued to Cellectar shareholders in the Acquisition constituted approximately 85% of our outstanding common stock after giving effect to the Acquisition. Upon the closing of the Acquisition, we completed the private placement of 6,846,537 shares of our common stock and warrants to purchase an additional 6,846,537 shares of our common stock for gross proceeds of approximately $5,135,000. As a result of the Acquisition, we are implementing a revised business plan focused on the development of the Cellectar compounds. Development of our other compounds (NOV-002 and NOV-205) has been suspended.
Company Information
Our headquarters and manufacturing operation, which is compliant with current Good Manufacturing Practices (cGMP), is located at 3301 Agriculture Drive, Madison, WI 53716. Our principal executive offices are located at One Gateway Center, Suite 504, Newton, MA 02458. Our telephone number is (617) 244-1616 and our web address is www.novelos.com. The information included or referred to on, or accessible through, our website does not constitute part of, and is not incorporated by reference into, this prospectus.
3
|
Proposed Reverse Split
|
|
Prior to the effectiveness of the registration statement of which this prospectus is a part, we anticipate that we will effect the Offering Reverse Split within a range of 1:2 to 1:10 in order to satisfy the minimum price requirement for our common stock to be listed on The NASDAQ Capital Market. The proposal for the Offering Reverse Split has been approved by our stockholders and our board of directors is authorized to determine the time and ratio at which the reverse split will be effected by filing the appropriate amendment to our certificate of incorporation.
|
|
Securities offered by us:
|
|
Up to shares of common stock (up to shares if the underwriter exercises its over-allotment option).
|
|
Common Stock to be outstanding after this offering:
|
|
shares.
|
|
|
||
|
Use of Proceeds:
|
We expect to use the net proceeds received from this offering to fund our research and development activities, including furthering development of LIGHT, HOT and COLD and for general corporate purposes, including capital expenditures, working capital, and, potentially, acquisition activities. For a more complete description of our anticipated use of proceeds from this offering, see “Use of Proceeds.”
|
|
|
Risk Factors:
|
|
See “Risk Factors” beginning on page 7 and the other information included in this prospectus for a discussion of factors you should carefully consider before deciding whether to purchase our securities.
|
|
OTC Bulletin Board symbol for our Common Stock:
|
|
NVLT.OB
|
|
Proposed NASDAQ Capital Market listing symbol for our common stock:
|
We are applying for listing of our common stock on The NASDAQ Capital Market under the symbol “NVLT”. No assurance can be given that our application will be approved.
|
|
4
The number of shares of our common stock to be outstanding after this offering is based on 26,826,157 shares of common stock outstanding as of September 9, 2011 and excludes, as of that date:
|
|
·
|
an aggregate of 3,632,638 shares of common stock issuable upon the exercise of outstanding stock options issued to employees, directors and consultants, including under our 2006 Stock Incentive Plan;
|
|
|
·
|
an aggregate of 3,476,112 additional shares of common stock reserved for future issuance under our 2006 Stock Incentive Plan;
|
|
|
·
|
an aggregate of 7,039,468 additional shares of common stock reserved for issuance under outstanding warrant agreements entered into in connection with the private placement of our securities completed on April 8, 2011 expiring on March 31, 2016 at an exercise price of $0.75 per share; and
|
|
|
·
|
an aggregate of 287,854 additional shares of common stock reserved for issuance under various outstanding warrant agreements, with expiration dates between May 7, 2012 and December 31, 2016, at exercise prices ranging from $0.75 to $191.25.
|
Unless we specifically state otherwise, the share information in this prospectus is as of September 9, 2011 and reflects or assumes no exercise of outstanding options or warrants to purchase shares of our common stock.
Since the former stockholders of Cellectar retained the majority voting interest in the combined business following the Acquisition, the Acquisition has been accounted for as a reverse acquisition whereby Cellectar, Inc. is treated as the acquirer for accounting and financial reporting purposes. As such, the historical financial information presented in this prospectus represents the historical financial information of Cellectar, except for the capital structure which represents the historical amounts of Cellectar retroactively adjusted to reflect the legal capital structure of Novelos by applying the exchange ratio established in the Acquisition.
5
Summary Historical Financial Information
The following table summarizes our financial data. We have derived the following summary of our statements of operations data for the six months ended June 30, 2011 and 2010 and the summary of our balance sheet data as of June 30, 2011 from our unaudited consolidated financial statements appearing elsewhere in this prospectus. We have derived the following summary of our statements of operations data for the fiscal years ended December 31, 2010 and 2009 and the summary of our balance sheet data as of December 31, 2010 and 2009 from our audited financial statements appearing elsewhere in this prospectus. The following summary of our financial data set forth below should be read together with our financial statements and the related notes to those statements, as well as the section titled “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” appearing elsewhere in this prospectus.
|
Six Months Ended
June 30,
|
Year Ended
December 31,
|
|||||||||||||||
|
2011
|
2010
|
2010
|
2009
|
|||||||||||||
|
Statement of Operations Data:
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
Research and development costs
|
|
$
|
1,433,356
|
|
|
$
|
2,100,116
|
|
|
$
|
2,984,207
|
|
|
$
|
4,351,983
|
|
|
General and administrative costs
|
|
|
928,440
|
|
|
|
923,666
|
|
|
|
1,156,549
|
|
|
|
1,824,302
|
|
|
Merger costs
|
746,207
|
-
|
52,925
|
-
|
||||||||||||
|
Total costs and expenses
|
|
|
3,108,003
|
|
|
|
3,023,782
|
|
|
|
4,193,681
|
|
|
|
6,176,285
|
|
|
Other expense
|
|
|
(452,460
|
)
|
|
|
(374,425
|
)
|
|
|
(366,582
|
)
|
|
|
(43,588
|
)
|
|
Net loss attributable to common stockholders
|
|
|
(3,560,463
|
)
|
|
|
(3,398,207
|
)
|
|
|
(4,560,263
|
)
|
|
|
(6,219,873
|
)
|
| June 30, |
December 31,
|
|||||||||||
| 2011 | 2010 | 2009 | ||||||||||
|
Balance Sheet Data:
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|||
|
Current assets
|
$
|
3,479,213
|
|
$
|
1,279,781
|
|
|
$
|
1,704,212
|
|
||
|
Working capital (deficit)
|
|
3,029,721
|
|
|
374,964
|
|
|
|
795,891
|
|
||
|
Total assets
|
|
8,431,231
|
|
|
4,802,142
|
|
|
|
5,824,706
|
|
||
|
Long term debt, including current portion
|
|
450,000
|
|
|
3,846,728
|
|
|
|
866,532
|
|
||
|
Total stockholders’ equity
|
|
7,408,068
|
|
|
133,762
|
|
|
|
4,126,893
|
|
||
The following table summarizes certain pro forma statement of operations and balance sheet data to give effect for the Acquisition and the private placement of our securities that occurred immediately following the Acquisition as though it occurred at the beginning of the periods presented for the purpose of statement of operations data. We have derived the following summary pro forma financial information from the unaudited pro forma financial statement data appearing elsewhere in this prospectus and the below information should be read together with the unaudited pro forma information.
|
Six Months
Ended
June 30,
|
Year Ended
December 31,
|
|||||||
|
2011
|
2010
|
|||||||
|
Pro Forma Statement of Operations Data:
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
||
|
Research and development costs
|
|
$
|
1,970,811
|
|
|
$
|
5,982,191
|
|
|
General and administrative costs
|
|
|
1,507,549
|
|
|
|
3,618,501
|
|
|
Total costs and expenses
|
|
|
3,478,360
|
|
|
|
9,600,692
|
|
|
Other income
|
|
|
96,857
|
|
|
|
7,895,726
|
|
|
Net loss
|
(3,381,503
|
)
|
(1,704,966
|
)
|
||||
|
Net loss attributable to common stockholders
|
|
|
(3,381,503
|
)
|
|
|
(1,704,966
|
)
|
6
RISK FACTORS
Investing in our securities involves a high degree of risk. Before you invest in our securities, you should be aware that our business faces numerous financial and market risks, including those described below, as well as general economic and business risks. The following discussion provides information concerning the material risks and uncertainties that we have identified and believe may adversely affect our business, financial condition and results of operations. Before you decide whether to invest in our securities, you should carefully consider these risks and uncertainties, together with all of the other information included in this prospectus.
Risks Related to Our Business and Industry
We will require additional capital in order to continue our operations, and may have difficulty raising additional capital.
We expect that we will continue to generate significant operating losses for the foreseeable future. At June 30, 2011, our consolidated cash balance was approximately $3,313,000. We believe our cash on hand, together with the net proceeds from this offering (without exercise of the underwriters over-allotment option), would be adequate to fund operations through the end of 2012. During that time, we anticipate that we will incur approximately $16,000,000 in costs, subject to the availability of funds. Of this amount, approximately $11,400,000 is estimated for research and development activities and $4,600,000 is estimated for general and administrative costs. The research and development costs consist of approximately $2,500,000 for the development of HOT (including approximately $1,600,000 in Phase 1b trial costs), $1,100,000 for the development of LIGHT, $3,200,000 for the development of COLD (principally activities preceding an IND) and $4,600,000 for our general, unallocated research and development costs including salaries, overhead, patent and other costs that are not specific to a certain development project. In addition to these estimated costs, we may incur up to $800,000 in clinical development costs during 2013, if necessary, to complete the Phase 1b dose-escalation trial in HOT. We have expended and expect to continue to expend substantial funds on the research, development and clinical and pre-clinical testing of our drug compounds. We will require additional funds to conduct research and development, establish and conduct clinical and pre-clinical trials, establish commercial-scale manufacturing arrangements and provide for the marketing and distribution of our products. Our ability to execute our operating plan beyond that time depends on our ability to obtain additional funding via the sale of equity and/or debt securities, a strategic transaction or otherwise. Although we have not entered into negotiations for a financing or strategic transaction (other than the offering contemplated in this prospectus), we plan to actively pursue financing alternatives. However, there can be no assurance that we will obtain the necessary funding or that it will be available on a timely basis or upon terms acceptable to us. If we obtain capital by issuing debt or preferred stock, the holders of such securities would likely obtain rights that are superior to those of holders of our common stock.
Our capital requirements and our ability to meet them depend on many factors, including:
|
|
·
|
the number of potential products and technologies in development;
|
|
|
·
|
continued progress and cost of our research and development programs;
|
|
|
·
|
progress with pre-clinical studies and clinical trials;
|
|
|
·
|
the time and costs involved in obtaining regulatory clearance;
|
|
|
·
|
costs involved in preparing, filing, prosecuting, maintaining and enforcing patent claims;
|
|
|
·
|
costs of developing sales, marketing and distribution channels and our ability to sell our drugs;
|
|
|
·
|
costs involved in establishing manufacturing capabilities for clinical trial and commercial quantities of our drugs;
|
|
|
·
|
competing technological and market developments;
|
|
|
·
|
market acceptance of our products;
|
|
|
·
|
costs for recruiting and retaining management, employees and consultants;
|
|
|
·
|
costs for educating physicians regarding the application and use of our products;
|
|
|
·
|
our status as a Bulletin Board-listed company and the prospects for our stock being listed on a national exchange;
|
|
|
·
|
uncertainty and economic instability resulting from terrorist acts and other acts of violence or war; and
|
|
|
·
|
the condition of capital markets and the economy generally, both in the U.S. and globally.
|
We may consume available resources more rapidly than currently anticipated, resulting in the need for additional funding sooner than expected. We may seek to raise any necessary additional funds through the issuance of warrants, equity or debt financings or executing collaborative arrangements with corporate partners or other sources, which may be dilutive to existing stockholders or have a material effect on our current or future business prospects. In addition, in the event that additional funds are obtained through arrangements with collaborative partners or other sources, we may have to relinquish economic and/or proprietary rights to some of our technologies or products under development that we would otherwise seek to develop or commercialize by ourselves. If we cannot secure adequate financing when needed, we may be required to delay, scale back or eliminate one or more of our research and development programs or to enter into license or other arrangements with third parties to commercialize products or technologies that we would otherwise seek to develop ourselves and commercialize ourselves. In such event, our business, prospects, financial condition, and results of operations may be adversely affected.
7
We are a development stage company with a history of losses and can provide no assurance of our future operating results.
We are a development stage company and have incurred net losses and negative cash flows since inception. We currently have no product revenues, and may not succeed in developing or commercializing any products which will generate product or licensing revenues. We do not expect to have any marketable products on the market for several years. Our primary activity to date has been research and development. In addition, development of our product candidates requires a process of pre-clinical and clinical testing, during which our product candidates could fail. We may not be able to enter into agreements with one or more companies experienced in the manufacturing and marketing of therapeutic drugs and, to the extent that we are unable to do so, we will not be able to market our product candidates. Eventual profitability will depend on our success in developing, manufacturing, and marketing our product candidates. Cellectar has experienced net losses and negative cash flows from operating activities since inception and we expect such losses and negative cash flows to continue in the foreseeable future. As of December 31, 2009 and 2010, Cellectar had working capital of $795,891 and $374,964, respectively, and stockholders’ equity of $4,126,893 and $133,762, respectively. As of June 30, 2011, we had working capital of $3,029,721 and stockholders’ equity of $7,408,068. For the period from Cellectar’s inception in November 2002 through June 30, 2011, the years ended December 31, 2009 and 2010, and for the six months ended June 30, 2011, we incurred net losses of $(27,605,467), $(6,219,873), $(4,560,263), and $(3,560,463), respectively. We may never achieve profitability.
We and our Chief Executive Officer are defendants in a securities fraud class action lawsuit. We are also defending counterclaims in another lawsuit that we initiated. If we are not successful in defending claims against us, the resulting liability could be substantial.
A putative class action complaint was filed on March 5, 2010 in the U.S. District Court for the District of Massachusetts by an alleged shareholder on behalf of himself and all others who purchased or otherwise acquired our common stock in the period between December 14, 2009 and February 24, 2010, against us and our President and Chief Executive Officer, Harry S. Palmin. The complaint claimed, among other things, that the defendants violated Section 10(b) of the Securities Exchange Act of 1934, as amended, and Rule 10b-5 promulgated thereunder in connection with alleged misleading disclosures related to the progress of the Phase 3 trial of NOV-002 in advanced non-small cell lung cancer. On June 23, 2011, the case was dismissed without prejudice. On August 5, 2011, the plaintiffs filed a second amended complaint realleging that the defendants violated Section 10(b) of the Exchange Act and Rule 10b-5 in connection with alleged misleading disclosures related to the Phase 3 clinical trial for NOV-002 in non-small cell lung cancer. On September 9, 2011, the defendants filed a motion to dismiss the second amended complaint. The plaintiff’s opposition to the motion is due by October 7, 2011.
In addition, on June 28, 2010, we received a letter from counsel to ZAO BAM and ZAO BAM Research Laboratories (Russian companies, collectively referred to as “BAM”) alleging that we modified the chemical composition of NOV-002 without prior notice to or approval from BAM, constituting a material breach of a technology and assignment agreement we had entered into with BAM on June 20, 2000 (the “June 2000 Agreement”). On September 24, 2010, we filed a complaint in Suffolk Superior Court seeking a declaratory judgment by the court that the June 2000 Agreement has been replaced by a subsequent agreement between the parties dated April 1, 2005 (the “April 2005 Agreement”), that Novelos’ obligations to BAM are governed solely by the April 2005 Agreement and that the obligations of the June 2000 Agreement have been performed and fully satisfied. BAM answered the complaint, denying the material allegations, and stating its affirmative defenses and certain counterclaims. In June 2011, BAM filed an amended counterclaim alleging additional claims related to Novelos’ acquisition of Cellectar. On June 15, 2011 we filed our response to their amended counterclaim. On August 5, 2011, we filed a motion for judgment on the pleadings as to our declaratory judgment count and all counts of BAM’s amended counterclaim. The motion has been opposed by BAM and a hearing on the motion is scheduled on September 27, 2011.
While we intend to vigorously defend ourselves in these actions, the uncertainties of litigation and the uncertainties related to insurance coverage and collection as well as the actual value of claims make it difficult to accurately predict the financial effect these claims may ultimately have on us. We may not be successful in defending such claims, and the resulting liability could be substantial and may not be covered by insurance. At the time the class action complaint was filed, we carried a total of $10 million in directors and officers liability insurance coverage, consisting of $5 million in primary coverage (including costs to defend) and $5 million in excess liability coverage. The BAM dispute is not covered by insurance. In addition, the lawsuits divert management’s attention and resources, whether or not the claims are ultimately successful, and this could adversely affect our business. As a result, there can be no assurance as to the long-term effect litigation will have on our business, prospects, financial condition or results of operations.
At present, our success depends solely on the successful commercialization of Cellectar compounds.
Prior to the Acquisition, Novelos had for over ten years been developing oxidized glutathione-based compounds for the treatment of cancer, including NOV-002, an injectable small-molecule compound based on a proprietary formulation of oxidized glutathione that Novelos had been developing for use in combination with standard-of-care chemotherapies for the treatment of solid tumors, and NOV-205, a hepatoprotective agent with immunomodulating and anti-inflammatory properties.
Following the Acquisition, development of NOV-002 and NOV-205 has been suspended and we are now focused on the development of novel drugs for the treatment and diagnosis of cancer based on the cancer-targeting technologies of Cellectar: CLR1401 (“COLD”), 131 I-CLR1404 (“HOT”, a radiolabeled compound) and 124 I-CLR1404 (“LIGHT”, labeled with a shorter-lived radioisotope, iodine-124). As a result the successful commercialization of HOT, COLD and LIGHT is crucial for our success. Our proposed products and their potential applications are in an early stage of clinical and manufacturing/process development and face a variety of risks and uncertainties. Principally, these risks include the following:
8
|
|
·
|
future clinical trial results may show that the cancer-targeting technologies of Cellectar are not well tolerated by recipients at its effective doses or are not efficacious;
|
|
|
·
|
future clinical trial results may be inconsistent with Cellectar’s previous preliminary testing results and data from Cellectar’s earlier studies may be inconsistent with clinical data;
|
|
|
·
|
even if the cancer-targeting technologies of Cellectar are shown to be safe and effective for their intended purposes, we may face significant or unforeseen difficulties in obtaining or manufacturing sufficient quantities at reasonable prices or at all;
|
|
|
·
|
our ability to complete the development and commercialization of the cancer-targeting technologies of Cellectar for our intended use is substantially dependent upon our ability to obtain and maintain experienced and committed partners to assist us with obtaining clinical and regulatory approvals for, and the manufacturing, marketing and distribution of, our products;
|
|
|
·
|
even if the cancer-targeting technologies of Cellectar are successfully developed, commercially produced and receive all necessary regulatory approvals, there is no guarantee that there will be market acceptance of our products; and
|
|
|
·
|
our competitors may develop therapeutics or other treatments which are superior or less costly than our own with the result that our product candidates, even if they are successfully developed, manufactured and approved, may not generate sufficient revenues to offset the development and manufacturing costs of our product candidates.
|
If we are unsuccessful in dealing with any of these risks, or if we are unable to successfully commercialize the cancer-targeting technologies of Cellectar for some other reason, our business, prospects, financial condition, and results of operations may be adversely affected.
The integration of Novelos and Cellectar may be costly and difficult.
The successful integration of independent businesses or assets can be a complex, costly and time-consuming process. The difficulties of integrating Novelos and Cellectar include, among others:
|
|
·
|
consolidating research and development operations;
|
|
|
·
|
preserving important research and development, manufacturing and supply, and other relationships;
|
|
|
·
|
minimizing the diversion of management’s attention from ongoing business concerns;
|
|
|
·
|
coordinating geographically separate organizations; and
|
|
|
·
|
optimizing the functioning of a newly constituted Board of Directors.
|
We may not accomplish the integration of Novelos and Cellectar smoothly or successfully. The diversion of the attention of our management from current operations to integration efforts and any difficulties encountered in combining operations could prevent the combined company from realizing the full benefits anticipated to result from the Acquisition and may adversely affect the combined business. Additionally, the costs associated with the integration of Novelos and Cellectar may be significant. To the extent that we incur integration costs that are not anticipated, these unexpected costs could adversely impact our liquidity and force us to seek additional funding sooner than would otherwise be necessary. To date, there have been no notable difficulties encountered during integration.
We have a history of recurring losses and an accumulated deficit which, among other factors, raise substantial doubt about our ability to continue as a going concern, which may hinder our ability to obtain future financing.
Our financial statements as of December 31, 2010 included elsewhere in this prospectus were prepared under the assumption that we will continue as a going concern. The independent registered public accounting firm that audited our 2010 financial statements, in their report, included an explanatory paragraph referring to our recurring losses since inception and expressing substantial doubt in our ability to continue as a going concern. Our financial statements do not include any adjustments that might result from the outcome of this uncertainty. Our ability to continue as a going concern depends on our ability to obtain additional equity or debt financing, attain further operating efficiencies, reduce expenditures, and, ultimately, to generate revenue.
9
The failure to complete development of our therapeutic technology, to obtain government approvals, including required FDA approvals, or to comply with ongoing governmental regulations could prevent, delay or limit introduction or sale of proposed products and result in failure to achieve revenues or maintain our ongoing business.
Our research and development activities and the manufacture and marketing of our intended products are subject to extensive regulation for safety, efficacy and quality by numerous government authorities in the U.S. and abroad. Before receiving clearance to market our proposed products by the FDA, we will have to demonstrate that our products are safe and effective for the patient population for the diseases that are to be treated. Clinical trials, manufacturing and marketing of drugs are subject to the rigorous testing and approval process of the FDA and equivalent foreign regulatory authorities. The Federal Food, Drug and Cosmetic Act and other federal, state and foreign statutes and regulations govern and influence the testing, manufacturing, labeling, advertising, distribution and promotion of drugs and medical devices. As a result, clinical trials and regulatory approval can take many years to accomplish and require the expenditure of substantial financial, managerial and other resources.
In order to be commercially viable, we must successfully research, develop, obtain regulatory approval for, manufacture, introduce, market and distribute our technologies. This includes meeting a number of critical developmental milestones including:
|
|
·
|
demonstrating benefit from delivery of each specific drug for specific medical indications;
|
|
|
·
|
demonstrating through pre-clinical and clinical trials that each drug is safe and effective; and
|
|
|
·
|
demonstrating that we have established viable Good Manufacturing Practices capable of potential scale-up.
|
The timeframe necessary to achieve these developmental milestones may be long and uncertain, and we may not successfully complete these milestones for any of our intended products in development.
In addition to the risks previously discussed, our technology is subject to developmental risks that include the following:
|
|
·
|
uncertainties arising from the rapidly growing scientific aspects of drug therapies and potential treatments;
|
|
|
·
|
uncertainties arising as a result of the broad array of alternative potential treatments related to cancer and other diseases; and
|
|
|
·
|
anticipated expense and time believed to be associated with the development and regulatory approval of treatments for cancer and other diseases.
|
In order to conduct the clinical trials that are necessary to obtain approval by the FDA to market a product, it is necessary to receive clearance from the FDA to conduct such clinical trials. The FDA can halt clinical trials at any time for safety reasons or because we or our clinical investigators do not follow the FDA’s requirements for conducting clinical trials. If we are unable to receive clearance to conduct clinical trials for a product, or the trials are halted by the FDA, we will not be able to achieve any revenue from such product in the U.S. as it is illegal to sell any drug for use in humans in the U.S. without FDA approval.
Even if we do ultimately receive FDA approval for any of our products, these products will be subject to extensive ongoing regulation, including regulations governing manufacturing, labeling, packaging, testing, dispensing, prescription and procurement quotas, record keeping, reporting, handling, shipment and disposal of any such drug. Failure to obtain and maintain required registrations or to comply with any applicable regulations could further delay or preclude development and commercialization of our drugs and subject us to enforcement action.
Clinical trials involve a lengthy and expensive process with an uncertain outcome, and results of earlier studies and trials may not be predictive of future trial results.
In order to receive regulatory approval for the commercialization of our product candidates, we must conduct, at our own expense, extensive clinical trials to demonstrate safety and efficacy of these product candidates. Clinical testing is expensive, it can take many years to complete and its outcome is uncertain. Failure can occur at any time during the clinical trial process. For example, we incurred costs of over $35 million in clinical trial expenses over a period of 4 years in connection with the Phase 3 trial of NOV-002 for non-small cell lung cancer, and NOV-002 did not ultimately demonstrate efficacy for that indication.
10
We may experience delays in clinical testing of our product candidates. We do not know whether planned clinical trials will begin on time, will need to be redesigned or will be completed on schedule, if at all. Clinical trials can be delayed for a variety of reasons, including delays in obtaining regulatory approval to commence a trial, in reaching agreement on acceptable clinical trial terms with prospective sites, in obtaining institutional review board approval to conduct a trial at a prospective site, in recruiting patients to participate in a trial or in obtaining sufficient supplies of clinical trial materials. Many factors affect patient enrollment, including the size of the patient population, the proximity of patients to clinical sites, the eligibility criteria for the trial, competing clinical trials and new drugs approved for the conditions we are investigating. Prescribing physicians will also have to decide to use our product candidates over existing drugs that have established safety and efficacy profiles. Any delays in completing our clinical trials will increase our costs, slow down our product development and approval process and delay our ability to generate revenue.
In addition, the results of preclinical studies and early clinical trials of our product candidates do not necessarily predict the results of later-stage clinical trials. Product candidates in later stages of clinical trials may fail to show the desired safety and efficacy traits despite having progressed through initial clinical testing. The data collected from clinical trials of our product candidates may not be sufficient to support the submission of a new drug application or to obtain regulatory approval in the United States or elsewhere. Because of the uncertainties associated with drug development and regulatory approval, we cannot determine if or when we will have an approved product for commercialization or achieve sales or profits.
Our clinical trials may not demonstrate sufficient levels of efficacy necessary to obtain the requisite regulatory approvals for our drugs, and our proposed drugs may not be approved for marketing. As discussed elsewhere in this prospectus, we suffered significant setbacks in the development of NOV-002 and NOV-205, as some of the promising results of earlier trials were not demonstrated in later stage trials. As a result, following the Acquisition, development of these compounds has been suspended.
We may be required to suspend or discontinue clinical trials due to unexpected side effects or other safety risks that could preclude approval of our product candidates.
Our clinical trials may be suspended at any time for a number of reasons. For example, we may voluntarily suspend or terminate our clinical trials if at any time we believe that they present an unacceptable risk to the clinical trial patients. In addition, regulatory agencies may order the temporary or permanent discontinuation of our clinical trials at any time if they believe that the clinical trials are not being conducted in accordance with applicable regulatory requirements or that they present an unacceptable safety risk to the clinical trial patients.
Administering any product candidates to humans may produce undesirable side effects. These side effects could interrupt, delay or halt clinical trials of our product candidates and could result in the FDA or other regulatory authorities denying further development or approval of our product candidates for any or all targeted indications. Ultimately, some or all of our product candidates may prove to be unsafe for human use. Moreover, we could be subject to significant liability if any volunteer or patient suffers, or appears to suffer, adverse health effects as a result of participating in our clinical trials.
We have limited in-house research and manufacturing capacity and will rely, to some extent, on research and manufacturing facilities at various universities, hospitals, contract research organizations and contract manufacturers for a portion of our research, development, and manufacturing. In the event we exceed our in-house capacity or lose access to those facilities, our ability to gain FDA approval and commercialization of our drug delivery technology and products could be delayed or impaired.
We remain in the research and development and clinical and pre-clinical trial phase of product commercialization and have limited experience in establishing, supervising and conducting commercial manufacturing. Accordingly, if our products are approved for commercial sale, we will need to establish the capability, or engage a contract manufacturer that has the capability, to commercially manufacture our products in accordance with FDA and other regulatory requirements. There can be no assurance that we would be able to successfully establish any such capability, or indentify a suitable manufacturing partner on acceptable terms.
At the present time, we have limited research, development or manufacturing capabilities within our facilities. Our manufacturing facility in Madison, WI has adequate capacity to supply drug product for Phase 2 studies of HOT, but we will need to expand for larger Phase 3 studies. We are exploring scaling up production capacity of COLD, via contract manufacturers or at our facility, to support an IND filing and clinical trials. LIGHT is currently manufactured by our collaborator, the University of Wisconsin at Madison in small quantities, at no cost to us, for use in investigator-sponsored clinical trials pursuant to a materials transfer agreement expiring in June 2013, but which may be terminated at any time by either party. We rely and expect to continue to rely, to some extent, on contracting with third parties to use their facilities to conduct research, development and manufacturing. The limited facilities of our own in which to conduct research, development and manufacturing may delay or impair our ability to gain FDA approval and commercialization of our drug delivery technology and products.
11
We may rely on third-party contract research organizations, service providers and suppliers to support development and clinical testing of our products. This may expose us to the risks of not being able to directly oversee the production and quality of the manufacturing process. Furthermore, these contractors, whether foreign or domestic, may experience regulatory compliance difficulties, mechanical shutdowns, employee strikes or other unforeseeable acts that may delay production. Failure of any of these contractors to provide the required services in a timely manner or on commercially reasonable terms could materially delay the development and approval of our products, increase our expenses and materially harm our business, prospects, financial condition and results of operations.
We believe that we have a good working relationship with our contractors. However, should the situation change, we may be required to relocate these activities on short notice, and we do not currently have access to alternate facilities to which we could relocate our research, development and/or manufacturing activities. The cost and time to establish or locate an alternate research, development and/or manufacturing facility to develop our technology would be substantial and would delay obtaining FDA approval and commercializing our products.
We are exposed to product, clinical and preclinical liability risks that could create a substantial financial burden should we be sued.
Our business exposes us to potential product liability and other liability risks that are inherent in the testing, manufacturing and marketing of pharmaceutical products. In addition, the use, in our clinical trials, of pharmaceutical products that we or our current or potential collaborators may develop and then subsequently sell may cause us to bear a portion of or all product liability risks. While we carry an insurance policy covering up to $5,000,000 per occurrence and $5,000,000 in the aggregate of liability incurred in connection with such claims should they arise, there can be no assurance that our insurance will be adequate to cover all situations. Moreover, there can be no assurance that such insurance, or additional insurance, if required, will be available in the future or, if available, will be available on commercially reasonable terms. Furthermore, our current and potential partners with whom we have collaborative agreements or our future licensees may not be willing to indemnify us against these types of liabilities and may not themselves be sufficiently insured or have a net worth sufficient to satisfy any product liability claims. A successful product liability claim or series of claims brought against us could have a material adverse effect on our business, prospects, financial condition and results of operations.
Acceptance of our products in the marketplace is uncertain and failure to achieve market acceptance will prevent or delay our ability to generate revenues.
Our future financial performance will depend, at least in part, on the introduction and customer acceptance of our proposed products. Even if approved for marketing by the necessary regulatory authorities, our products may not achieve market acceptance. The degree of market acceptance will depend on a number of factors including:
|
|
·
|
receiving regulatory clearance of marketing claims for the uses that we are developing;
|
|
|
·
|
establishing and demonstrating the advantages, safety and efficacy of our technologies;
|
|
|
·
|
pricing and reimbursement policies of government and third-party payers such as insurance companies, health maintenance organizations and other health plan administrators;
|
|
|
·
|
our ability to attract corporate partners, including pharmaceutical companies, to assist in commercializing our intended products; and
|
|
|
·
|
our ability to market our products.
|
Physicians, patients, payers or the medical community in general may be unwilling to accept, use or recommend any of our products. If we are unable to obtain regulatory approval or commercialize and market our proposed products as planned, we may not achieve any market acceptance or generate revenue.
We may face litigation from third parties who claim that our products infringe on their intellectual property rights, particularly because there is often substantial uncertainty about the validity and breadth of medical patents.
We may be exposed to future litigation by third parties based on claims that our technologies, products or activities infringe on the intellectual property rights of others or that we have misappropriated the trade secrets of others. This risk is exacerbated by the fact that the validity and breadth of claims covered in medical technology patents and the breadth and scope of trade-secret protection involve complex legal and factual questions for which important legal principles are unresolved. Any litigation or claims against us, whether or not valid, could result in substantial costs, could place a significant strain on our financial and managerial resources and could harm our reputation. The U. Mich license does require and license agreements that we may enter into in the future would likely require that we pay the costs associated with defending this type of litigation. In addition, intellectual property litigation or claims could force us to do one or more of the following:
|
|
·
|
cease selling, incorporating or using any of our technologies and/or products that incorporate the challenged intellectual property, which would adversely affect our ability to generate revenue;
|
|
|
·
|
obtain a license from the holder of the infringed intellectual property right, which license may be costly or may not be available on reasonable terms, if at all; or
|
12
|
|
·
|
redesign our products, which would be costly and time-consuming.
|
If we are unable to protect or enforce our rights to intellectual property adequately or to secure rights to third-party patents, we may lose valuable rights, experience reduced market share, assuming any, or incur costly litigation to protect our intellectual property rights.
Our ability to obtain licenses to patents, maintain trade secret protection and operate without infringing the proprietary rights of others will be important to commercializing any products under development. Therefore, any disruption in access to the technology could substantially delay the development of our technology.
The patent positions of biotechnology and pharmaceutical companies, such as ours, that involve licensing agreements, are frequently uncertain and involve complex legal and factual questions. In addition, the coverage claimed in a patent application can be significantly reduced before the patent is issued or in subsequent legal proceedings. Consequently, our patent applications and any issued and licensed patents may not provide protection against competitive technologies or may be held invalid if challenged or circumvented. To the extent we license patents from third parties, as in the case of the U. Mich. license, the early termination of any such license agreement would result in the loss of our rights to use the covered patents which could severely delay, inhibit or eliminate our ability to develop and commercialize compounds based on the licensed patents. Our competitors may also independently develop products similar to ours or design around or otherwise circumvent patents issued or licensed to us. In addition, the laws of some foreign countries may not protect our proprietary rights to the same extent as U.S. law.
We also rely on trade secrets, technical know-how and continuing technological innovation to develop and maintain our competitive position. Although we generally require our employees, consultants, advisors and collaborators to execute appropriate confidentiality and assignment-of-inventions agreements, our competitors may independently develop substantially equivalent proprietary information and techniques, reverse engineer our information and techniques, or otherwise gain access to our proprietary technology. We may be unable to meaningfully protect our rights in trade secrets, technical know-how and other non-patented technology.
We may have to resort to litigation to protect our rights for certain intellectual property, or to determine their scope, validity or enforceability. Enforcing or defending our rights is expensive, could cause diversion of our resources and may not prove successful. Any failure to enforce or protect our rights could cause us to lose the ability to exclude others from using our technology to develop or sell competing products.
Confidentiality agreements with employees and others may not adequately prevent disclosure of our trade secrets and other proprietary information and may not adequately protect our intellectual property, which could limit our ability to compete.
We operate in the highly technical field of research and development of small molecule drugs, and rely in part on trade secret protection in order to protect our proprietary trade secrets and unpatented know-how. However, trade secrets are difficult to protect, and we cannot be certain that our competitors will not develop the same or similar technologies on their own. We have taken steps, including entering into confidentiality agreements with our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors, to protect our trade secrets and unpatented know-how. These agreements generally require that the other party keep confidential and not disclose to third parties all confidential information developed by the party or made known to the party by us during the course of the party’s relationship with us. We also typically obtain agreements from these parties which provide that inventions conceived by the party in the course of rendering services to us will be our exclusive property. However, these agreements may not be honored and may not effectively assign intellectual property rights to us. Enforcing a claim that a party has illegally obtained and is using our trade secrets or know-how is difficult, expensive and time consuming, and the outcome is unpredictable. In addition, courts outside the United States may be less willing to protect trade secrets or know-how. The failure to obtain or maintain trade secret protection could adversely affect our competitive position.
We may be subject to claims that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
As is common in the biotechnology and pharmaceutical industry, we employ individuals who were previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that we or these employees have used or disclosed trade secrets or other proprietary information of their former employers, either inadvertently or otherwise. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
13
The use of hazardous materials, including radioactive materials, in our research and development imposes certain compliance costs on us and may subject us to liability for claims arising from the use or misuse of these materials.
Our research and development, manufacturing and administration of our drugs involves the controlled use of hazardous materials, including chemicals and radioactive materials, such as radioactive isotopes. We are subject to federal, state and local laws and regulations governing the storage, use and disposal of these materials and some waste products and are required to maintain both a manufacturer’s license and a radioactive materials license with State of Wisconsin agencies. We believe that our safety procedures for the storage, use and disposal of these materials comply with the standards prescribed by federal, state and local regulations. However, we cannot completely eliminate the risk of accidental contamination or injury from these materials. If there were to be an accident, we could be held liable for any damages that result, which could exceed our financial resources. We currently maintain insurance coverage, with limits of up to $2,500,000 depending on the nature of the claim, for damages resulting from the hazardous materials we use; however, future claims may exceed the amount of our coverage. Also, we do not have insurance coverage for pollution cleanup and removal. Currently the costs of complying with federal, state and local regulations are not significant, and consist primarily of waste disposal expenses and permitting fees. However, they could become expensive, and current or future environmental regulations may impair our research, development, production and commercialization efforts. If we are unable to maintain the required licenses and permits for any reason, it will negatively impact our research and development activities.
Due to our limited marketing, sales and distribution experience, we may be unsuccessful in our efforts to sell our products, enter into relationships with third parties or develop a direct sales organization.
We have not established marketing, sales or distribution capabilities for our proposed products. Until such time as our products are further along in the development process, we will not devote any meaningful time and resources to this effort. At the appropriate time, we intend to develop our own sales and marketing capabilities or enter into agreements with third parties to sell our products.
We have limited experience in developing, training or managing a sales force. If we choose to establish a direct sales force, we may incur substantial additional expenses in developing, training and managing such an organization. We may be unable to build a sales force on a cost-effective basis or at all. Any such direct marketing and sales efforts may prove to be unsuccessful. In addition, we will compete with many other companies that currently have extensive marketing and sales operations. Our marketing and sales efforts may be unable to compete against these other companies. We may be unable to establish a sufficient sales and marketing organization on a timely basis, if at all.
If we choose to enter into agreements with third parties to sell our products, we may be unable to establish or maintain third-party relationships on a commercially reasonable basis, if at all. In addition, these third parties may have similar or more established relationships with our competitors.
We may be unable to engage qualified distributors. Even if engaged, these distributors may:
|
|
·
|
fail to adequately market our products;
|
|
|
·
|
fail to satisfy financial or contractual obligations to us;
|
|
|
·
|
offer, design, manufacture or promote competing products; or
|
|
|
·
|
cease operations with little or no notice.
|
If we fail to develop sales, marketing and distribution channels, we would experience delays in product sales and incur increased costs, which would have a material adverse effect on our business, prospects, financial condition, and results of operation.
If we are unable to convince physicians of the benefits of our intended products, we may incur delays or additional expense in our attempt to establish market acceptance.
Achieving use of our products in the target market of cancer diagnosis and treatment may require physicians to be informed regarding these products and their intended benefits. The time and cost of such an educational process may be substantial. Inability to successfully carry out this physician education process may adversely affect market acceptance of our products. We may be unable to timely educate physicians regarding our intended products in sufficient numbers to achieve our marketing plans or to achieve product acceptance. Any delay in physician education may materially delay or reduce demand for our products. In addition, we may expend significant funds towards physician education before any acceptance or demand for our products is created, if at all.
The market for our products is rapidly changing and competitive, and new therapeutics, new drugs and new treatments that may be developed by others could impair our ability to maintain and grow our business and remain competitive.
The pharmaceutical and biotechnology industries are subject to rapid and substantial technological change. Developments by others may render our technologies and intended products noncompetitive or obsolete, or we may be unable to keep pace with technological developments or other market factors. Technological competition from pharmaceutical and biotechnology companies, universities, governmental entities and others diversifying into the field is intense and is expected to increase. Most of these entities have significantly greater research and development capabilities and budgets than we do, as well as substantially more marketing, manufacturing, financial and managerial resources. These entities represent significant competition for us. Acquisitions of, or investments in, competing pharmaceutical or biotechnology companies by large corporations could increase our competitors’ financial, marketing, manufacturing and other resources.
14
We operate with limited day-to-day business management, serve as a vehicle to hold certain technology for possible future exploration, and have been and will continue to be engaged in the development of new drugs and therapeutic technologies. As a result, our resources are limited and we may experience management, operational or technical challenges inherent in such activities and novel technologies. Competitors have developed or are in the process of developing technologies that are, or in the future may be, the basis for competition. Some of these technologies may accomplish therapeutic effects similar to those of our technology, but through different means. Our competitors may develop drugs and drug delivery technologies that are more effective than our intended products and, therefore, present a serious competitive threat to us.
For example, Perifosine, an alkylphospholipid that is being developed by Keryx Biopharmaceuticals, which has licensed it in North America from Æterna Zentaris Inc., is a possible future competitor to COLD. We do not know of any current or potential direct competitors for HOT and LIGHT. Marketed drugs Zevalin (Spectrum Pharmaceuticals) and Bexxar (Glaxo Smith Kline) provide examples of targeted radiotherapeutics specifically for lymphoma indication. FDG is the current standard for PET imagining for cancer and may be an alternative diagnostic imaging agent to LIGHT.
The potential widespread acceptance of therapies that are alternatives to ours may limit market acceptance of our products even if they are commercialized. Many of our targeted diseases and conditions can also be treated by other medication or drug delivery technologies. These treatments may be widely accepted in medical communities and have a longer history of use. The established use of these competitive drugs may limit the potential for our technologies and products to receive widespread acceptance if commercialized.
If users of our products are unable to obtain adequate reimbursement from third-party payers, or if additional healthcare reform measures are adopted, it could hinder or prevent our product candidates’ commercial success.
The continuing efforts of government and insurance companies, health maintenance organizations and other payers of healthcare costs to contain or reduce costs of healthcare may adversely affect our ability to generate future revenues and achieve profitability, including by limiting the future revenues and profitability of our potential customers, suppliers and collaborative partners. For example, in certain foreign markets, pricing or profitability of prescription pharmaceuticals is subject to government control. The U.S. government and other governments have shown significant interest in pursuing healthcare reform. Any government-adopted reform measures could adversely affect the pricing of healthcare products and services in the U.S. or internationally and the amount of reimbursement available from governmental agencies or other third-party payers. The continuing efforts of the U.S. and foreign governments, insurance companies, managed care organizations and other payers of healthcare services to contain or reduce healthcare costs may adversely affect our ability to set prices for our products, should we be successful in commercializing them, and this would negatively affect our ability to generate revenues and achieve and maintain profitability.
New laws, regulations and judicial decisions, or new interpretations of existing laws, regulations and decisions, that relate to healthcare availability, methods of delivery or payment for healthcare products and services, or sales, marketing or pricing of healthcare products and services, also may limit our potential revenue and may require us to revise our research and development programs. The pricing and reimbursement environment may change in the future and become more challenging for several reasons, including policies advanced by the current or future executive administrations in the U.S., new healthcare legislation or fiscal challenges faced by government health administration authorities. Specifically, in both the U.S. and some foreign jurisdictions, there have been a number of legislative and regulatory proposals to change the healthcare system in ways that could affect our ability to sell our products profitably. In the U.S., changes in federal healthcare policy were passed into law in 2010 and are being considered by Congress again this year. Some of these proposed reforms could result in reduced reimbursement rates for our product candidates, which would adversely affect our business strategy, operations and financial results.
Our ability to commercialize our products will depend in part on the extent to which appropriate reimbursement levels for the cost of our products and related treatment are obtained by governmental authorities, private health insurers and other organizations, such as health maintenance organizations (HMOs). Third-party payers are increasingly challenging the prices charged for medical drugs and services. Also, the trend toward managed healthcare in the U.S. and the concurrent growth of organizations such as HMOs that could control or significantly influence the purchase of healthcare services and drugs, as well as legislative proposals to reform healthcare or change government insurance programs, may all result in lower prices for or rejection of our drugs. The cost containment measures that healthcare payers and providers are instituting and the effect of any healthcare reform could materially harm our ability to operate profitably.
We depend on key personnel who may terminate their employment with us at any time, and our success will depend on our ability to hire additional qualified personnel.
Our success will depend to a significant degree on the continued services of our chief executive officer, Harry Palmin, Cellectar founder and our chief scientific officer, Jamey Weichert and our senior vice president of research and development, Christopher Pazoles. There can be no assurance that these individuals will continue to provide services to us. In addition, our success may depend on our ability to attract and retain other highly skilled personnel. We may be unable to recruit such personnel on a timely basis, if at all. Our management and other employees may voluntarily terminate their employment with us at any time. The loss of services of key personnel, or the inability to attract and retain additional qualified personnel, could result in delays in development or approval of our products, loss of sales and diversion of management resources. To date, we have not experienced difficulties attracting and retaining highly qualified personnel, but there can be no assurance we will be successful in doing so in the future.
15
Risks Related to our Common Stock
Our stock price has experienced price fluctuations.
There can be no assurance that the market price for our common stock will remain at its current level and a decrease in the market price could result in substantial losses for investors. The market price of our common stock may be significantly affected by one or more of the following factors:
|
|
·
|
announcements or press releases relating to the biopharmaceutical sector or to our own business or prospects;
|
|
|
·
|
regulatory, legislative, or other developments affecting us or the healthcare industry generally;
|
|
|
·
|
sales by holders of restricted securities pursuant to effective registration statements or exemptions from registration; and
|
|
|
·
|
market conditions specific to biopharmaceutical companies, the healthcare industry and the stock market generally.
|
Our five largest stockholders beneficially own approximately 64% of our outstanding common stock, which limits the influence of other shareholders.
As of September 9, 2011, 64% of our outstanding common stock is beneficially owned by our five largest shareholders, all of whom are former shareholders of Cellectar. The interests of these stockholders may differ from those of other stockholders. These stockholders will likely continue to have the ability to significantly affect the outcome of all corporate actions requiring stockholder approval, including the following actions:
|
|
·
|
the election of directors;
|
|
|
·
|
the amendment of charter documents; and
|
|
|
·
|
the approval of certain mergers and other significant corporate transactions, including a sale of substantially all of our assets.
|
There may be a limited public market for our securities; we presently fail to qualify for listing on any national securities exchanges.
Our common stock currently does not meet all of the requirements for initial listing on a registered stock exchange. Specifically, the bid price of our common stock is less than the minimum bid price required to obtain a listing, which is why we have undertaken to effect a Reverse Split prior to the effectiveness of the registration statement of which this prospectus forms a part. Trading in our common stock continues to be conducted on the electronic bulletin board in the over-the-counter market. As a result, an investor may find it difficult to dispose of or to obtain accurate quotations as to the market value of our common stock, and our common stock may be less attractive for margin loans, for investment by financial institutions, as consideration in future capital raising transactions or other purposes. In connection with this offering, we have applied for listing of our common stock on The NASDAQ Capital Market under the symbol “NVLT”. No assurance can be given that our application will be approved.
Our common stock has historically been a “penny stock” under SEC rules. It may be more difficult to resell shares of common stock classified as “penny stock”.
Our common stock has historically been a “penny stock” under applicable SEC rules (generally defined as non-exchange traded stock with a per share price below $5.00). These rules impose additional sales practice requirements on broker-dealers that recommend the purchase or sale of penny stocks to persons other than those who qualify as “established customers” or “accredited investors.” For example, broker-dealers must determine the appropriateness for non-qualifying persons of investments in penny stocks. Broker-dealers must also provide, prior to a transaction in a penny stock not otherwise exempt from the rules, a standardized risk disclosure document that provides information about penny stocks and the risks in the penny stock market. The broker-dealer also must provide the customer with current bid and offer quotations for the penny stock, disclose the compensation of the broker-dealer and its salesperson in the transaction, furnish monthly account statements showing the market value of each penny stock held in the customer’s account, provide a special written determination that the penny stock is a suitable investment for the purchaser, and receive the purchaser’s written agreement to the transaction.
Legal remedies available to an investor in "penny stocks" may include the following:
|
·
|
if a "penny stock" is sold to the investor in violation of the requirements listed above, or other federal or states securities laws, the investor may be able to cancel the purchase and receive a refund of the investment.
|
|
·
|
if a “penny stock” is sold to the investor in a fraudulent manner, the investor may be able to sue the persons and firms that committed the fraud for damages.
|
However, investors who have signed arbitration agreements may have to pursue their claims through arbitration.
These requirements may have the effect of reducing the level of trading activity, if any, in the secondary market for a security that becomes subject to the penny stock rules. The additional burdens imposed upon broker-dealers by such requirements may discourage broker-dealers from effecting transactions in our securities, which could severely limit the market price and liquidity of our securities. These requirements may restrict the ability of broker-dealers to sell our common stock and may affect your ability to resell our common stock.
Many brokerage firms will discourage or refrain from recommending investments in penny stocks. Most institutional investors will not invest in penny stocks. In addition, many individual investors will not invest in penny stocks due, among other reasons, to the increased financial risk generally associated with these investments.
16
For these reasons, penny stocks may have a limited the market and, consequently, limited liquidity. We have applied for a listing of our common stock on the NASDAQ Capital Market in connection with this offering. Upon obtaining such listing, our common stock would cease to be a penny stock. However, we can give no assurance at what time, if ever, our common stock will not be classified as a “penny stock” In the future.
If we fail to maintain effective internal controls over financial reporting, the price of our common stock may be adversely affected.
Our internal control over financial reporting may have weaknesses and conditions that could require correction or remediation, the disclosure of which may have an adverse impact on the price of our common stock. We are required to establish and maintain appropriate internal controls over financial reporting. Failure to establish those controls, or any failure of those controls once established, could adversely affect our public disclosures regarding our business, prospects, financial condition or results of operations. In addition, management’s assessment of internal controls over financial reporting may identify weaknesses and conditions that need to be addressed in our internal controls over financial reporting or other matters that may raise concerns for investors. Any actual or perceived weaknesses and conditions that need to be addressed in our internal control over financial reporting or disclosure of management’s assessment of our internal controls over financial reporting may have an adverse impact on the price of our common stock. During the year ended December 31, 2005, a material weakness existed in Novelos’ internal controls over financial reporting with respect to the identification of and accounting for the beneficial conversion feature of a preferred stock financing transaction. This material weakness was remediated during 2006. As a result of this material weakness, we were required to restate our financial statements as of and for the quarter ended September 30, 2005 and as of and for the year ended December 31, 2005.
We are required to comply with certain provisions of Section 404 of the Sarbanes-Oxley Act of 2002 and if we fail to comply in a timely manner, our business could be harmed and our stock price could decline.
Rules adopted by the SEC pursuant to Section 404 of the Sarbanes-Oxley Act of 2002 require an annual assessment of internal controls over financial reporting, and for certain issuers an attestation of this assessment by the issuer’s independent registered public accounting firm. The standards that must be met for management to assess the internal controls over financial reporting as effective are evolving and complex, and require significant documentation, testing, and possible remediation to meet the detailed standards. We expect to incur significant expenses and to devote resources to Section 404 compliance on an ongoing basis. It is difficult for us to predict how long it will take or costly it will be to complete the assessment of the effectiveness of our internal control over financial reporting for each year and to remediate any deficiencies in our internal control over financial reporting. As a result, we may not be able to complete the assessment and remediation process on a timely basis. In addition, although attestation requirements by our independent registered public accounting firm are not presently applicable to us we could become subject to these requirements in the future and we may encounter problems or delays in completing the implementation of any resulting changes to internal controls over financial reporting. In the event that our Chief Executive Officer or Chief Financial Officer determine that our internal control over financial reporting is not effective as defined under Section 404, we cannot predict how regulators will react or how the market prices of our shares will be affected; however, we believe that there is a risk that investor confidence and share value may be negatively affected.
Our common stock could be further diluted as the result of the issuance of additional shares of common stock, convertible securities, warrants or options.
In the past, we have issued common stock, convertible securities (such as convertible preferred stock and notes) and warrants in order to raise money. We have also issued options and warrants as compensation for services and incentive compensation for our employees and directors. We have shares of common stock reserved for issuance upon the exercise of certain of these securities and may increase the shares reserved for these purposes in the future. Our issuance of additional common stock, convertible securities, options and warrants could affect the rights of our stockholders, could reduce the market price of our common stock or could result in adjustments to exercise prices of outstanding warrants (resulting in these securities becoming exercisable for, as the case may be, a greater number of shares of our common stock), or could obligate us to issue additional shares of common stock to certain of our stockholders.
Shares eligible for future sale may adversely affect the market.
From time to time, certain of our stockholders may be eligible to sell all or some of their shares of common stock by means of ordinary brokerage transactions in the open market pursuant to Rule 144 promulgated under the Securities Act, subject to certain limitations. In general, pursuant to amended Rule 144, non-affiliate stockholders may sell freely after six months subject only to the current public information requirement. Affiliates may sell after six months subject to the Rule 144 volume, manner of sale (for equity securities), current public information and notice requirements. Of the approximately 26,826,000 shares of our common stock outstanding as of September 9, 2011, approximately 1.1 million shares are freely tradable without restriction as of that date. Any substantial sales of our common stock pursuant to Rule 144 may have a material adverse effect on the market price of our common stock.
17
Provisions of our charter, bylaws, and Delaware law may make an acquisition of us or a change in our management more difficult.
Certain provisions of our amended restated certificate of incorporation and bylaws could discourage, delay or prevent a merger, acquisition or other change in control that stockholders may consider favorable, including transactions in which you might otherwise receive a premium for your shares. These provisions also could limit the price that investors might be willing to pay in the future for shares of our common stock or warrants, thereby depressing the market price of our common stock. Stockholders who wish to participate in these transactions may not have the opportunity to do so.
Furthermore, these provisions could prevent or frustrate attempts by our stockholders to replace or remove our management. These provisions:
|
|
provide for the division of our board into three classes as nearly equal in size as possible with staggered three-year terms and further limit the removal of directors and the filling of vacancies;
|
|
|
authorize our board of directors to issue without stockholder approval blank check preferred stock that, if issued, could operate as a “poison pill” to dilute the stock ownership of a potential hostile acquirer to prevent an acquisition that is not approved by our board of directors;
|
|
|
require that stockholder actions must be effected at a duly called stockholder meeting and prohibit stockholder action by written consent;
|
|
|
establish advance notice requirements for stockholder nominations to our board of directors or for stockholder proposals that can be acted on at stockholder meetings;
|
|
|
require the approval of the holders of 75% of the outstanding shares of our capital stock entitled to vote in order to amend certain provisions of our restated certificate of incorporation and restated bylaws.
|
In addition, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which may, unless certain criteria are met, prohibit large stockholders, in particular those owning 15% or more of our outstanding voting stock, from merging or combining with us for a prescribed period of time.
We have not paid dividends in the past and do not expect to pay dividends for the foreseeable future. Any return on investment may be limited to the value of our common stock.
No cash dividends have been paid on Novelos common stock. We do not expect to pay cash dividends in the near future. Payment of dividends would depend upon our profitability at the time, cash available for those dividends, and other factors as our board of directors may consider relevant. If we do not pay dividends, our common stock may be less valuable because a return on an investor’s investment will only occur if our stock price appreciates.
Risks Related to this Offering
Our management team will have immediate and broad discretion over the use of the net proceeds from this offering and we may use the net proceeds in ways with which you disagree.
There is no minimum offering amount required as a condition to closing this offering and therefore net proceeds from this offering will be immediately available to our management to use at their discretion. We currently intend to use the net proceeds from this offering to fund our research and development activities, for general corporate purposes, and possibly for acquisitions of other companies, products or technologies, though no such acquisitions are currently contemplated. See “Use of Proceeds.” We have not allocated specific amounts of the net proceeds from this offering for any of the foregoing purposes. Accordingly, our management will have significant discretion and flexibility in applying the net proceeds of this offering. You will be relying on the judgment of our management with regard to the use of these net proceeds, and you will not have the opportunity, as part of your investment decision, to assess whether the proceeds are being used appropriately. It is possible that the net proceeds will be invested in a way that does not yield a favorable, or any, return for us or our stockholders. The failure of our management to use such funds effectively could have a material adverse effect on our business, prospects, financial condition, and results of operation.
You will experience immediate and substantial dilution as a result of this offering and may experience additional dilution in the future.
You will incur immediate and substantial dilution as a result of this offering. After giving effect to the sale by us of up to shares offered in this offering at an assumed public offering price of $ per share, and after deducting the underwriter ’ s discounts and commissions and estimated offering expenses payable by us, investors in this offering can expect an immediate dilution of $ per share, or %, at the assumed public offering price. In addition, in the past, we issued options and warrants to acquire shares of common stock. To the extent these options are ultimately exercised, you will sustain future dilution. We may also acquire or license other technologies or finance strategic alliances by issuing equity, which may result in additional dilution to our stockholders.
18
This prospectus, including the documents that we incorporate by reference, contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended (the “Securities Act”), and Section 21E of the Exchange Act. Such forward-looking statements include those that express plans, anticipation, intent, contingency, goals, targets or future development and/or otherwise are not statements of historical fact. These forward-looking statements are based on our current expectations and projections about future events and they are subject to risks and uncertainties known and unknown that could cause actual results and developments to differ materially from those expressed or implied in such statements.
In some cases, you can identify forward-looking statements by terminology, such as “expects,” “anticipates,” “intends,” “estimates,” “plans,” “believes,” “seeks,” “may,” “should”, “could” or the negative of such terms or other similar expressions. Accordingly, these statements involve estimates, assumptions and uncertainties that could cause actual results to differ materially from those expressed in them. Any forward-looking statements are qualified in their entirety by reference to the factors discussed throughout this prospectus.
You should read this prospectus and the documents that we reference herein and therein and have filed as exhibits to the registration statement, of which this prospectus is part, completely and with the understanding that our actual future results may be materially different from what we expect. You should assume that the information appearing in this prospectus is accurate as of the date on the front cover of this prospectus or such prospectus supplement only. Because the risk factors referred to above could cause actual results or outcomes to differ materially from those expressed in any forward-looking statements made by us or on our behalf, you should not place undue reliance on any forward-looking statements. Further, any forward-looking statement speaks only as of the date on which it is made, and we undertake no obligation to update any forward-looking statement to reflect events or circumstances after the date on which the statement is made or to reflect the occurrence of unanticipated events. New factors emerge from time to time, and it is not possible for us to predict which factors will arise. In addition, we cannot assess the impact of each factor on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements. We qualify all of the information presented in this prospectus and any accompanying prospectus supplement, and particularly our forward-looking statements, by these cautionary statements.
Based on an assumed public offering price of $ per share, we estimate that the net proceeds to us from the sale of the shares that we are offering, assuming gross proceeds of $ million, will be approximately $ million, after deducting underwriting discounts and commissions and estimated offering expenses, or approximately $ if the underwriter’s over-allotment is exercised in full.
We expect to use any proceeds received from this offering as follows:
|
|
·
|
to fund our research and development activities, including the further development of our LIGHT, HOT and COLD compounds in a wide range of cancers; and
|
|
|
·
|
for general corporate purposes, such as general and administrative expenses, capital expenditures, working capital, prosecution and maintenance of our intellectual property, and the potential investment in technologies or products that complement our business.
|
We believe that our cash on hand, together with the net proceeds from this offering (without exercise of the underwriters over-allotment option) would be adequate to fund operations through the end of 2012. We estimate that our costs through the end of 2012 will be approximately $16 million. This amount consists of approximately $2.5 million for HOT development, approximately $1.1 million for LIGHT development, approximately $3.2 million for COLD development, approximately $4.6 million for general, fixed and overhead research and development expenses that are not allocated to specific projects, and approximately $4.6 million in general and administrative costs.
The above estimates contemplate the conduct of the following clinical trials: the initiation of HOT Phase 1b dose-escalation trial in the fourth quarter of 2011, the initiation of two HOT Phase 2a proof-of-concept trials in second half of 2012, the initiation of investigator sponsored LIGHT Phase 1/2 imaging trials in multiple solid tumors (such as brain metastasis, lung and breast cancers) starting in the fourth quarter of 2011, and the initiation of COLD Phase 1 trial in late 2012. We anticipate that the planned trials will generate initial proof-of-concept data for LIGHT in the first quarter of 2012 and initial proof-of concept data for HOT in late 2012. The HOT Phase 1b dose-escalation trial may not be completed until late-2013.
We have no current understandings, commitments or agreements with respect to any acquisition of or investment in any technologies or products.
Pending the application of the net proceeds as described above or otherwise, we may invest the proceeds in short-term, investment-grade, interest-bearing securities or guaranteed obligations of the U.S. government or other securities.
19
The following table sets forth our cash and cash equivalents and capitalization, each as of June 30, 2011:
|
|
·
|
on an actual basis; and
|
|
|
·
|
on a pro forma as adjusted basis to give effect to the issuance of the shares offered hereby.
|
You should consider this table in conjunction with our financial statements and the notes to those financial statements and the pro forma financial information included elsewhere in this prospectus.
|
As of June 30, 2011
|
|||||
|
Actual
|
Pro Forma As
Adjusted (1)
|
||||
|
Cash and cash equivalents
|
|
$
|
3,313,398
|
|
$
|
|
|
|
|
|
|
|
|
Wisconsin Department of Commerce Loan
|
|
|
450,000
|
|
|
|
Capital lease obligations
|
|
|
7,387
|
|
|
|
Total debt obligations
|
|
|
457,387
|
|
|
|
|
|
|
|
|
|
|
Stockholders’ equity (deficit):
|
|
|
|
|
|
|
Common stock, par value $0.0001 per share: 150,000,000 shares authorized; 26,826,157 issued as of June 30, 2011
|
|
|
268
|
|
|
|
Additional paid in capital
|
|
|
35,013,267
|
|
|
|
Deficit accumulated during the development stage
|
|
|
(27,605,467
|
)
|
|
|
Total stockholders’ equity
|
|
|
7,408,068
|
|
|
|
|
|
|
|
|
|
|
Total capitalization
|
|
$
|
7,865,455
|
|
$
|
|
(1)
|
Assumes that $ million of our shares is sold in this offering at an assumed offering price of $ per share and that the net proceeds thereof are approximately $ million after deducting underwriting discounts and commissions and our estimated offering expenses.
|
Market Information
Our common stock has been quoted on the OTC Bulletin Board under the symbol “NVLT” since June 14, 2005. The following table provides, for the periods indicated, the range of high and low bid prices for our common stock. These over-the-counter market quotations reflect inter-dealer prices, without retail mark-up, mark-down or commission, and may not represent actual transactions.
|
Fiscal Year 2009
|
|
High
|
|
|
Low
|
|
||
|
First Quarter
|
|
$
|
91.74
|
|
|
$
|
45.87
|
|
|
Second Quarter
|
|
|
143.73
|
|
|
|
50.45
|
|
|
Third Quarter
|
|
|
149.84
|
|
|
|
87.15
|
|
|
Fourth Quarter
|
|
|
443.42
|
|
|
|
99.38
|
|
|
Fiscal Year 2010
|
|
High
|
|
|
Low
|
|
||
|
First Quarter
|
|
$
|
466.36
|
|
|
$
|
26.16
|
|
|
Second Quarter
|
|
|
42.66
|
|
|
|
14.67
|
|
|
Third Quarter
|
|
|
23.08
|
|
|
|
6.17
|
|
|
Fourth Quarter
|
|
|
9.17
|
|
|
|
2.82
|
|
20
|
Fiscal Year 2011
|
|
High
|
|
|
Low
|
|
||
|
First Quarter
|
|
$
|
8.10
|
|
|
$
|
1.52
|
|
|
Second Quarter
|
|
3.82
|
|
|
0.95
|
|
||
|
Third Quarter (through September 9, 2011)
|
1.55
|
1.25
|
||||||
The above share prices have been adjusted to reflect the April Reverse Split, but have not been adjusted to reflect the Offering Reverse Split anticipated to be effected prior to the effectiveness of the registration statement of which this prospectus is a part.
On September 9, 2011 there were 232 holders of record of our common stock. This number does not include stockholders for whom shares were held in a “nominee” or “street” name.
We have not declared or paid any cash dividends on our common stock and do not anticipate declaring or paying any cash dividends in the foreseeable future. We currently expect to retain future earnings, if any, for the development of our business.
Our transfer agent and registrar is American Stock Transfer and Trust Company, 59 Maiden Lane, New York, NY 10038.
Equity compensation plans
The following table provides information as of December 31, 2010, giving effect to the April Reverse Split regarding shares authorized for issuance under our equity compensation plans, including individual compensation arrangements.
We have two equity compensation plans approved by our stockholders: the 2000 Stock Option and Incentive Plan and the 2006 Stock Incentive Plan. During 2004 and 2005, we also issued options to our directors and consultants that were not approved by our stockholders. These options are exercisable within a ten-year period from the date of the grant and vest at various intervals with all options being fully vested within three years of the date of grant. The option price per share is not less than the fair market value of our common stock on the date of grant.
Equity compensation plan information
|
Plan category
|
Number of shares to
be issued upon
exercise of outstanding
options, warrants and
rights (#)
|
Weighted-average
exercise price of
outstanding options,
warrants and rights
($)
|
Number of shares
remaining available for
future issuance under
equity compensation plans
(excluding shares reflected
in column (a)) (#)
|
|||||||||
|
|
|
(a)
|
|
|
(b)
|
|
|
(c)
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|||
|
Equity compensation plans approved by stockholders
|
|
|
38,657
|
|
|
$
|
100.98
|
|
|
|
25,555
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Equity compensation plans not approved by stockholders
|
|
|
10,569
|
|
|
$
|
100.98
|
|
|
|
0
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
|
49,226
|
|
|
$
|
100.98
|
|
|
|
25,555
|
|
21
Our pro forma net tangible book value as of June 30, 2011 was $5,732,606 or $0.21 per share of common stock, based upon 26,826,157 shares outstanding as of that date. Net tangible book value per share is determined by dividing such number of outstanding shares of common stock into our net tangible book value, which is our total tangible assets less total liabilities. After giving effect to the sale of the shares in this offering at the assumed public offering price of $ per share, at June 30, 2011, after deducting underwriting discounts and commissions and other estimated offering expenses payable by us, our pro forma as adjusted net tangible book value at June 30, 2011 would have been approximately , or $ per share. This represents an immediate increase in net tangible book value of approximately $ per share to our existing stockholders, and an immediate dilution of $ per share to investors purchasing shares in the offering.
The following table illustrates the per share dilution to investors purchasing shares in the offering:
|
Assumed public offering price per share
|
|
$
|
|
|
|
Pro forma net tangible book value per share as of June 30, 2011
|
|
$
|
|
|
|
Increase per share attributable to sale of shares to investors
|
|
$
|
|
|
|
Pro forma as adjusted net tangible book value per share after the offering
|
|
$
|
|
|
|
Dilution per share to investors
|
|
$
|
|
|
The foregoing illustration does not reflect potential dilution from the exercise of outstanding options or warrants to purchase shares of our common stock. The foregoing illustration also does not reflect the dilution that would result from the underwriter warrants.
OPERATIONS
The following discussion and analysis should be read together with our financial statements and the related notes appearing elsewhere in this prospectus. This discussion contains forward-looking statements reflecting our current expectations that involve risks and uncertainties. See “Forward-Looking Statements” for a discussion of the uncertainties, risks and assumptions associated with these statements. Actual results and the timing of events could differ materially from those discussed in our forward-looking statements as a result of many factors, including those set forth under “Risk Factors” and elsewhere in this prospectus.
Acquisition
On April 8, 2011, we entered into a business combination with Cellectar (the “Acquisition”). Immediately prior to the Acquisition, we completed a 1-for-153 reverse split of our common stock (the “April Reverse Split”). We then issued 17,001,596 shares of our common stock to the former shareholders of Cellectar as consideration for the Acquisition, constituting approximately 85% of our outstanding common stock after giving effect to the Acquisition. Upon the closing of the Acquisition, we completed the private placement of 6,846,537 shares of our common stock and warrants to purchase an additional 6,846,537 shares of our common stock (in each case after giving effect to the April Reverse Split). As a result of the Acquisition, we are implementing a revised business plan focused on the development of the Cellectar compounds. We conduct our operations from Cellectar’s headquarters in Madison, WI and our executive offices are in Newton, MA. Further development of our other compounds (NOV-002 and NOV-205) has been suspended. The following discussion for activity prior to April 8, 2011 corresponds to the results of operations of Cellectar prior to Acquisition.
On April 8, 2011, immediately prior to the Acquisition, Cellectar paid approximately $627,000 in full settlement of a note payable to a bank. The payment was made in order to avoid an event of default that would have occurred as a result of the change of control that occurred at the time of the Acquisition. On April 8, 2011, the holders of Cellectar convertible notes converted outstanding principal of $2,720,985 and unpaid interest thereon into a total of 4,181,535 shares of common stock.
Overview
We are a pharmaceutical company developing novel drugs for the treatment and diagnosis of cancer. We currently have three cancer-targeted compounds, which we believe are selectively taken up and retained in cancer cells (including cancer stem cells) versus normal cells. Thus, we believe our therapeutic compounds directly kill cancer cells while minimizing harm to normal cells offering the potential for a paradigm shift in cancer therapy – efficacy versus all three major drivers of mortality in cancer: primary tumors, metastases and stem cell-based relapse. LIGHT is a small-molecule cancer imaging agent. We believe LIGHT has first-in-class potential and we expect it to enter Phase 1/2 clinical trials in the fourth quarter of this year. HOT is a small-molecule, broad-spectrum, cancer-targeted molecular radiotherapeutic that delivers radiation directly and selectively to cancer cells and cancer stem cells. We believe HOT also has first-in-class potential, and we expect it to enter a Phase 1b dose escalation trial in the fourth quarter of this year and Phase 2 trials in 2012 as a monotherapy for solid tumors with significant unmet medical need. COLD, a cancer-targeted chemotherapy that we expect to submit an Investigational New Drug (“IND”) application to the FDA late in 2012, works primarily through Akt inhibition. Together, we believe our compounds are able to “find, treat and follow” cancer anywhere in the body in a novel, effective and highly selective way.
22
Prior to the Acquisition, for more than ten years, Novelos had been developing oxidized glutathione-based compounds for the treatment of cancer, including NOV-002, an injectable small-molecule compound based on a proprietary formulation of oxidized glutathione that we had been developing for use in combination with standard of care chemotherapies for the treatment of solid tumors. From 2005 through 2010 Novelos raised approximately $67 million in capital for the development of our compounds. From November 2006 through January 2010, Novelos conducted a Phase 3 trial of NOV-002 plus first-line chemotherapy in advanced non-small cell lung cancer which, when completed in February 2010, did not meet its primary and secondary efficacy endpoints. Following the completion of the Phase 3 trial during 2010, Novelos continued clinical development of NOV-002 in breast cancer and NOV-205 in hepatitis C, although further development of those compounds has now been suspended. We also explored strategic alternatives which resulted in the completion of the Acquisition in April 2011.
Results of Operations
Executive summary. In March 2010, Cellectar completed a Phase 1a dosimetry trial of HOT in humans (the “Phase 1a Trial”), demonstrating initial safety and establishing dosing parameters for a Phase 1b dose-escalation trial. Following the completion of the Phase 1a Trial and as a result of limited funding, Cellectar suspended research and manufacturing activities, terminated certain non-key personnel and implemented salary reductions in an effort to contain costs while Cellectar concentrated on its fund raising efforts. The decreases in research and development costs for the six months ended June 30, 2011 compared to the six months ended June 30, 2010 and the decrease in operating costs in the year ended December 31, 2010 compared to the year ended December 31, 2009 are primarily attributable to the cost reduction efforts implemented in 2010. The decreases in general and administrative expense for the six -month period ended June 30, 2011 were offset by increases in professional fees associated with Cellectar becoming a public company. Following the Acquisition, we are resuming development activities in preparation for planned clinical trials in HOT and LIGHT scheduled to begin in the fourth quarter of 2011.
Research and development expense. Research and development expense consists of costs incurred in identifying, developing and testing and manufacturing product candidates, which primarily include salaries and related expenses for personnel, costs of our research and manufacturing facility, cost of manufacturing materials, fees paid to professional service providers for independent monitoring and analysis of our clinical trials, and costs to secure intellectual property. The Company analyzes its research and development expenses based on four categories as follows: clinical projects, preclinical projects, chemistry and manufacturing costs, and general fixed and overhead costs that are not allocated to the functional project costs, including personnel costs, manufacturing facility costs, related overhead costs and patent costs.
General and administrative expense. General and administrative expense consists primarily of salaries and other related costs for personnel in executive, finance and administrative functions. Other costs include insurance, costs for public and investor relations, directors’ fees and professional fees for legal and accounting services.
Six Months Ended June 30, 2011 and 2010
Research and Development. Research and development expense for the six months ended June 30, 2011 was approximately $1,433,000 (comprised of $1,000 in clinical project costs, $68,000 of preclinical project costs, $50,000 of manufacturing and related costs and $1,314,000 in general unallocated research and development costs) compared to approximately $2,100,000 (comprised of $189,000 in clinical project costs, $445,000 of preclinical project costs, $65,000 of manufacturing and related costs and $1,401,000 in general unallocated research and development costs) for the same period in 2010. The approximately $667,000, or 32%, decrease in research and development expense occurred in several categories. The approximately $188,000 decrease in clinical projects in the six months ended June 30, 2011 versus the comparable period in 2010 was related to the completion of the Phase 1a trial in March 2010 and the approximately $377,000 decrease in preclinical projects, for the six months ended June 30, 2011 versus the same period in 2010 was primarily related to an approximately $335,000 decrease in subcontracted preclinical research as those activities had increased in the first half of 2010 in preparation for future clinical trials. Manufacturing costs decreased related to manufacturing and laboratory supplies as a result of the cost reduction efforts described above. General unallocated research and development costs decreased primarily as a result of an approximately $159,000 decrease in salary and overhead costs partially offset by increases in consulting, maintenance and permitting costs as we prepared to resume manufacturing and research activities following the Acquisition. Stock-based compensation increased by approximately $30,000 as a result of stock options granted in May 2011 following the Acquisition.
General and Administrative. General and administrative expense for the six months ended June 30, 2011 was approximately $928,000 compared to approximately $924,000 in the same period of 2010. While total costs remained relatively consistent, there were fluctuations in three categories of costs. The cost of subcontracted services increased by approximately $54,000 as a result of increased investor relations activities and costs associated with public company reporting. Insurance costs increased approximately $20,000. Salaries and related costs decreased by approximately $65,000 as a result of the cost cutting initiatives described above that remained in effect for the first three months of 2011.
Merger Costs. Merger costs during the six months ended June 30, 2011 consisted of approximately $450,000 in investment banking fees, approximately $286,000 in legal fees and approximately $10,000 in insurance costs.
23
Grant income. Qualifying therapeutic discovery projects, among others, include those designed to treat or prevent diseases or conditions by conducting pre-clinical or clinical activities for the purpose of securing FDA approval of a product. We received payments of approximately $44,000 in the first six months of 2011 under a cash grant from the U.S. Internal Revenue Service as a qualifying therapeutic discovery project credit pursuant to Patient Protection and Affordable Care Act. The payments have been recorded as a component of other income.
Loss on Derivative Warrants. We recorded a loss on derivative warrants of approximately $70,000 in the six months ended June 30, 2011. This amount represents the change in fair value, during the respective period, of outstanding warrants which contain “down-round” anti-dilution provisions whereby the number of shares for which the warrants are exercisable and/or the exercise price of the warrants is subject to change in the event of certain issuances of stock at prices below the then-effective exercise prices of the warrants.
Interest expense, net. Interest expense, net for the six months ended June 30, 2011 and 2010 consists approximately of the following:
|
Six Months Ended June 30,
|
||||||||
|
2011
|
2010
|
|||||||
|
|
|
|
|
|
|
|
||
|
Interest expense, convertible notes
|
|
$
|
(159,000
|
)
|
$
|
(140,000
|
)
|
|
|
Beneficial conversion feature, convertible notes
|
(258,000
|
)
|
(214,000
|
)
|
||||
|
Interest expense, bank note
|
(6,000
|
)
|
(29,000
|
)
|
||||
|
Interest expense, other
|
(7,000
|
)
|
(1,000
|
)
|
||||
|
Interest income
|
|
3,000
|
10,000
|
|||||
|
|
$
|
(427,000
|
)
|
$
|
(374,000
|
)
|
||
Since the convertible notes were converted based on revised conversion terms that resulted in the issuance of an additional 343,963 shares of common stock than would have been issued if the convertible notes had been converted in accordance with their original terms, the value of these additional shares of approximately $258,000 was recorded as a component of interest expense in the quarter ended June 30, 2011. Since the convertible notes were convertible into common stock at the date of issuance at a price per share which is less than the estimated fair value of our common stock at that date, the estimated intrinsic value of the beneficial conversion feature of approximately $214,000 was recorded as a component of interest expense on the date of issuance in the first three months of 2010. The increase in interest expense on the convertible notes was a result of the increased effective interest rate in effect during the first three months of 2011 until the conversion that occurred on April 8, 2011. The decrease in interest expense on the bank note was a result of the settlement of that obligation in connection with the Acquisition. The increase in other interest expense is principally a result of the issuance of notes payable to the Wisconsin Department of Commerce in September 2010. The reduction of interest income was a result of a decrease in average cash balances and interest rates.
Years Ended December 31, 2010 and 2009
Research and Development. Research and development expense for the year ended December 31, 2010 was approximately $2,984,000, (comprised of $200,000 in clinical project costs, $458,000 of preclinical project costs, $76,000 of chemistry and manufacturing and related costs and $2,250,000 in general unallocated research and development costs) compared to approximately $4,352,000 (comprised of $468,000 in clinical project costs, $267,000 of preclinical project costs, $437,000 of chemistry and manufacturing and related costs and $3,180,000 in general unallocated research and development costs) for the year ended December 31, 2009. The approximately $1,368,000, or 31%, decrease in research and development expense occurred in several categories. The approximately $268,000 decrease in clinical project costs is primarily related to an approximately $237,000 decrease in clinical trial costs associated with the Phase 1a Trial in 2010 compared to 2009 as a result of the completion of the trial in March 2010. Preclinical project costs increased by approximately $191,000 related to an approximately $340,000 increase in subcontracted preclinical research activities in preparation for future clinical trials offset by an approximately $150,000 decrease in lab supplies as a result of the cost reduction efforts described above. General unallocated research and development costs decreased by approximately $930,000 primarily related to an approximately $715,000 decrease in salary and overhead costs and an approximately $117,000 decrease in patent costs in 2010 compared to 2009 as a result of the cost reduction efforts described above. Chemistry and manufacturing costs decreased by approximately $361,000 due to the completion of the Phase 1a trial in early 2010 and as a result of the cost reduction efforts described above.
24
General and Administrative. General and administrative expense for the year ended December 31, 2010 was approximately $1,157,000 compared to approximately $1,824,000 in the year ended December 31, 2009. The approximately $667,000, or 37%, decrease is primarily due to a decrease of approximately $668,000 in salary and overhead costs and a decrease in consulting of approximately $83,000 as a result of cost reduction efforts. This decrease was offset by an increase in legal and other professional fees of approximately $277,000 associated with fund raising efforts and the Acquisition.
Merger Costs. Merger costs during the twelve months ended December 31, 2010 consisted of approximately $53,000 in legal fees.
Grant income. Qualifying therapeutic discovery projects, among others, include those designed to treat or prevent diseases or conditions by conducting pre-clinical or clinical activities for the purpose of securing FDA approval of a product. We received a cash grant during 2010 of approximately $200,000 from the U.S. Internal Revenue Service as a qualifying therapeutic discovery project credit pursuant to Patient Protection and Affordable Care Act. This grant has been recorded as a component of other income.
Interest expense, net. Interest expense, net for the years ended December 31, 2010 and 2009 consists approximately of the following:
|
Year ended December 31,
|
||||||||
|
2010
|
2009
|
|||||||
|
Interest expense, convertible notes
|
$ | (305,000 | ) | $ | — | |||
|
Beneficial conversion feature, convertible notes
|
(214,000 | ) | — | |||||
|
Interest expense, bank note
|
(55,000 | ) | (68,000 | ) | ||||
|
Interest expense, other
|
(7,000 | ) | — | |||||
|
Interest income
|
15,000 | 24,000 | ||||||
| $ | (566,000 | ) | $ | (44,000 | ) | |||
Since the convertible notes were convertible into common stock at the date of issuance at a price per share which is less than the estimated fair value of our common stock at that date, the estimated intrinsic value of the beneficial conversion feature of approximately $214,000 was recorded as a component of interest expense on the date of issuance. In addition, we recorded approximately $305,000 in interest expense on the convertible notes based on the stated interest rate. The increase in other interest expense is principally a result of the issuance of notes payable to the Wisconsin Department of Commerce in September 2010. The approximately $9,000, or 37% decrease in interest income is attributable to the lower average cash and cash equivalents balance during the year end December 31, 2010.
Liquidity and Capital Resources
We have financed our operations since inception primarily through the sale of equity securities and securities convertible into equity securities. To date, Cellectar and Novelos have raised capital aggregating approximately $105 million. Novelos has raised capital aggregating approximately $78 million, including proceeds from the April 2011 private placement. Since its inception, Cellectar raised capital aggregating approximately $27 million. As of June 30, 2011, we had approximately $3,313,000 in cash and cash equivalents.
During the six months ended June 30, 2011, approximately $2,903,000 in cash was used in operations. During this period we reported a net loss of approximately $3,560,000. However, this loss included the following non-cash items: an approximately $70,000 loss on derivative warrants, approximately $258,000 in stock-based compensation, approximately $291,000 in depreciation and amortization expense and approximately $258,000 of interest expense associated with convertible notes. After adjustment for these non-cash items, we used approximately $407,000 in cash for the payment of accounts payable and accrued liabilities resulting from the payment of vendor liabilities that had accumulated leading up to the Acquisition and private placement. Other changes in working capital provided cash of approximately $29,000. We incurred approximately $158,000 of accrued interest associated with notes payable that were converted to common stock on April 8, 2011. During the six months ended June 30, 2010, we used only $116,000 in cash to pay vendor liabilities, since Cellectar had taken action to reduce costs and conserve cash during that time.
During the six months ended June 30, 2011, we purchased approximately $23,000 in fixed assets. As described above, on April 8, 2011, we completed the Acquisition. In connection with the Acquisition, we acquired cash of approximately $905,000.
During the six months ended June 30, 2011, we repaid approximately $676,000 in long-term obligations, including the payment, immediately prior to the Acquisition, of approximately $627,000 in full settlement of a Cellectar note payable to a bank. In connection with that repayment, restrictions were released on $500,000 of cash equivalents. On April 8, 2011, the holders of Cellectar convertible notes converted outstanding principal of $2,720,985 and unpaid interest thereon into a total of 4,181,535 shares of common stock.
Upon the closing of the Acquisition, we completed the private placement of our common stock and warrants for net proceeds of approximately $4,866,000.
Deferred issuance costs increased by approximately $29,000 due to payments made in connection with a proposed underwritten offering.
25
The financial statements included elsewhere in this prospectus have been prepared on a basis that assumes that we will continue as a going concern and that contemplates the continuity of operations, realization of assets and the satisfaction of liabilities and commitments in the normal course of business. We have incurred losses since inception in devoting substantially all of our efforts toward research and development and have an accumulated deficit of $27,605,467 at June 30, 2011. During the six months ended June 30, 2011, we generated a net loss of $3,560,463 and we expect that we will continue to generate operating losses for the foreseeable future. At June 30, 2011, our cash balance was approximately $3,313,000. We believe our cash on hand is adequate to fund operations into the fourth quarter of 2011, including the commencement of a Phase 1b clinical trial for HOT. Our ability to execute our operating plan beyond that time depends on our ability to obtain additional funding via the sale of equity and/or debt securities, a strategic transaction or otherwise. We estimate that our costs through the end of 2012 will be approximately $16 million. This amount consists of approximately $2.5 million for HOT development, approximately $1.1 million for LIGHT development, approximately $3.2 million for COLD development, approximately $4.6 million for general, fixed and overhead research and development expenses that are not allocated to specific projects, and approximately $4.6 million in general and administrative costs. On July 1, 2011, we filed a Registration Statement on Form S-1 for an underwritten public offering of our common stock with proceeds of up to $15,000,000, excluding the underwriter’s over-allotment. There can be no assurance that the registration statement will become effective or that any securities will be sold pursuant to it. We plan to actively pursue this and other financing alternatives, however we have not entered into negotiations for any such alternative transaction. There can be no assurance that we will obtain the necessary funding. Other than the uncertainties regarding our ability to obtain additional funding, there are currently no known trends, demands, commitments, events or uncertainties that are likely to materially affect our liquidity.
The preparation of financial statements and related disclosures in conformity with accounting principles generally accepted in the United States, or GAAP, requires management to make certain estimates, judgments and assumptions that affect the reported amounts of assets and liabilities as of the date of the financial statements, as well as the reported amounts of revenues and expenses during the periods presented. Actual results could differ from those estimates. We review these estimates and assumptions periodically and reflect the effects of revisions in the period that they are determined to be necessary.
We believe that the following accounting policies reflect our more significant judgments and estimates used in the preparation of our financial statements.
Accrued Liabilities. As part of the process of preparing financial statements, we are required to estimate accrued liabilities. This process involves identifying services that have been performed on our behalf, and estimating the level of service performed and the associated cost incurred for such service as of each balance sheet date in our financial statements. Examples of estimated expenses for which we accrue include: contract service fees such as amounts paid to clinical research organizations and investigators in conjunction with clinical trials; fees paid to vendors in conjunction with the manufacturing of clinical materials; and professional service fees, such as for lawyers and accountants. In connection with such service fees, our estimates are most affected by our understanding of the status and timing of services provided relative to the actual levels of services incurred by such service providers. The majority of our service providers invoice us monthly in arrears for services performed. In the event that we do not identify certain costs that have begun to be incurred, or we over- or underestimate the level of services performed or the costs of such services, our reported expenses for such period would be too high or too low. The date on which certain services commence, the level of services performed on or before a given date and the cost of such services are often determined based on subjective judgments. We make these judgments based on the facts and circumstances known to us in accordance with GAAP.
Stock-based Compensation . We account for stock-based compensation by measuring the cost of employee services received in exchange for an award of equity instruments based on the grant-date fair value of the award. That cost is recognized over the period during which an employee is required to provide service in exchange for the award, the requisite service period (usually the vesting period). We account for transactions in which services are received from non-employees in exchange for equity instruments based on the fair value of such services received or of the equity instruments issued, whichever is more reliably measured.
Accounting for equity instruments granted or sold by us under accounting guidance requires fair-value estimates of the equity instrument granted or sold. If our estimates of the fair value of these equity instruments are too high or too low, our expenses may be over- or understated. For equity instruments granted or sold in exchange for the receipt of goods or services, we estimate the fair value of the equity instruments based on consideration of factors that we deem to be relevant at that time.
The following table sets forth information regarding all options to purchase common stock granted since the Acquisition:
|
Date of Grant
|
Number of Options
Granted
|
Exercise
Price
|
Fair value of
Underlying Stock
on Grant Date
|
Fair Value of
Instrument on
Grant Date
|
Compensation
Expense
Recognized
through June 30,
2011
|
|||||||||||||||
|
April 25, 2011
|
100,000 | $ | 3.00 | $ | 3.00 | $ | 2.79 | $ | 5,000 | |||||||||||
|
May 18, 2011
|
1,581,100 | $ | 1.40 | $ | 1.40 | $ | 1.17 | 57,324 | ||||||||||||
|
May 18, 2011
|
670,200 | $ | 1.40 | $ | 1.40 | $ | 1.18 | 23,365 | ||||||||||||
|
May 18, 2011
|
750,000 | $ | 1.40 | $ | 1.40 | $ | 1.15 | 51,186 | ||||||||||||
|
May 18, 2011
|
405,100 | $ | 1.40 | $ | 1.40 | $ | 1.14 | 18,126 | ||||||||||||
|
May 18, 2011
|
150,000 | $ | 1.40 | $ | 1.40 | $ | 1.30 | 39,181 | ||||||||||||
|
May 23, 2011
|
20,000 | $ | 1.88 | $ | 1.88 | $ | 1.57 | 1,095 | ||||||||||||
|
May 25, 2011
|
20,000 | $ | 1.65 | $ | 1.65 | $ | 1.34 | 4,340 | ||||||||||||
| 3,696,400 | $ | 199,617 | ||||||||||||||||||
There were no option grants during 2010 or in 2011 prior to the Acquisition.
Off-Balance Sheet Arrangements
As of December 31, 2010 and June 30, 2011, we had no off-balance sheet arrangements.
26
Business of Novelos
Novelos Therapeutics, Inc. (“Novelos” or the “Company”) is a pharmaceutical company developing compounds for the treatment of cancer. On April 8, 2011, Novelos entered into a business combination with Cellectar, Inc. (“Cellectar”), a privately held Wisconsin corporation that designed and developed products to detect, treat and monitor a wide variety of human cancers, and Cell Acquisition Corp. (the “Merger Subsidiary”), a wholly owned subsidiary of Novelos, pursuant to which Cellectar was merged into the Merger Subsidiary (the “Acquisition”).
We are developing novel drugs for the treatment and diagnosis of cancer based on our cancer-targeting technology: COLD, HOT (iodine-131 radiolabeled compound) and LIGHT (labeled with a shorter-lived radioisotope, iodine-124). We believe our compounds are selectively taken up and retained in a wide variety of cancer cells (including cancer stem cells) versus normal cells. We believe our therapeutic compounds directly kill cancer cells while minimizing harm to normal cells, offering the potential for a paradigm shift in cancer therapy by providing efficacy against all three major drivers of mortality in cancer: primary tumors, metastases and cancer stem cell-based relapse. More specifically, we believe our technology enables targeted delivery to cancer cells of apoptosis-inducing Akt inhibition or, when a radioactive molecule is attached, of ionizing radiation sufficient to kill cancer cells. When radiolabeled with iodine-124 for PET imaging, we believe our agent can provide an accurate and quantitative diagnosis of cancer, including metastases, and can also objectively measure therapeutic success. Together, we believe this platform is capable of yielding multiple, distinct oncology product opportunities in a broad spectrum of cancers which enable us to “find, treat and follow” cancer anywhere in the body in a novel, effective and highly selective way.
Market Overview
Our target market is broad and represents the market for the treatment and diagnosis of cancer. According to Drug Discovery News (April 2009), Datamonitor (July 3, 2006) and PharmaLive (October 8, 2009), the global market for cancer pharmaceuticals reached an estimated $66 billion in 2007, nearly doubling from $35 billion in 2005 and is expected to grow to $80 billion by 2012. Furthermore, the US National Cancer Institute (January 12, 2011) estimates that the overall cost of treating cancer in the US will increase to $158 billion by 2020 from $125 billion in 2010.
Technology Overview
Our compounds are alkylphospholipids (“APLs”) that interact with lipid rafts, which are specialized microdomains within cell membranes. Importantly, the core chemical structure shared across all three products provides selective targeting of cancer cells in preference to normal cells (due to enrichment of lipid rafts in the former). COLD was deliberately designed to contain iodine (in the form of the stable, non-radioactive isotope, 127 I), thus enabling additional, distinct products differing only with respect to the form of iodine they contain – HOT contains short-lived radioactive 131 I and LIGHT contains the even more short-lived radioactive 124 I. As a result, three cancer-targeted product profiles have been generated from a single chemical structure that is the foundation of our technology platform – a chemotherapeutic (COLD), a molecular radiotherapeutic (HOT) and a diagnostic/imaging agent (LIGHT).
Our core technology platform is based upon the research conducted by Cellectar’s founder and our Chief Scientific Officer, Dr. Jamey Weichert beginning in 1994 at U. Mich. where alkyphospholipid analogs were initially designed, synthesized, radiolabeled, and evaluated. Since 1998, Dr. Weichert has continued his research at U. Wisc. and subsequently founded Cellectar in 2002 to further develop and commercialize the technology. Cellectar obtained exclusive rights to the related technology patents owned by U. Mich. in 2003 and continued development of the platform while obtaining ownership of numerous additional patents and patent applications (lasting until 2025, 2028 and 2030 without extensions) prior to the Acquisition.
COLD is a cancer-targeted chemotherapy that, in pre-clinical experiments, inhibits the phosphatidylinosotol 3-kinase (PI3K)/Akt survival pathway, which is overexpressed in many types of cancer. As a result, COLD selectively inhibits Akt activity, induces apoptosis through caspase activation and inhibits cell proliferation in cancer cells versus normal cells. COLD also exhibits significant in vivo efficacy in mouse xenograft tumor models, including non-small cell lung cancer and triple-negative breast cancers, producing long-lasting tumor growth suppression and significantly increased survival. We believe COLD has the potential to be best-in-class versus other Akt inhibitors in development due to a) cancer cell/cancer stem cell targeting, resulting in cancer-selective inhibition of Akt and cell proliferation or b) suitability for intravenous administration which offers the prospect of greater systemic exposure and hence Akt inhibition in cancer cells that we believe results in superior efficacy. We expect to submit an Investigational New Drug (“IND”) application to the FDA in late 2012.
27
HOT (iodine-131 radiolabeled compound) is a small-molecule, broad-spectrum, cancer-targeted molecular radiotherapeutic that we believe has first-in-class potential. HOT is comprised of a small quantity of COLD (too little for significant Akt inhibition), acting as a cancer-targeted delivery and retention vehicle, and incorporating a cytotoxic dose of radiotherapy (in the form of iodine-131, a radioisotope that is already in common use to treat thyroid and other cancer types). It is this “intracellular radiation” mechanism of cancer cell killing, coupled with selective delivery to a wide range of malignant tumor types, that imbues HOT with broad-spectrum anti-cancer activity. In 2009, we opened an IND with the FDA to study HOT in humans. In early 2010, we successfully completed a Phase 1a dosimetry trial in humans demonstrating initial safety and establishing dosing parameters for a Phase 1b dose-escalation trial. The Phase 1b dose-escalation trial is aimed at determining the Maximum Tolerated Dose, and we expect it to begin in the fourth quarter of 2011. In parallel, we expect to initiate Phase 2 efficacy trials in solid tumors in 2012 as soon as a minimal efficacious dose is established. We may determine such an effective dose upon seeing a response in the Phase 1b trial or calculating it from imaging trials in patients (see LIGHT below). Preclinical experiments in vitro (in cell culture) and in vivo (in animals) have demonstrated selective killing of cancer cells along with a benign safety profile. HOT’s anti-tumor/survival-prolonging activities have been demonstrated in over a dozen different xenograft models (human tumor cells implanted into animals) including breast, prostate, lung, glioma (brain), pancreatic, melanoma, ovarian, uterine, renal and colorectal cancers. In all but two models, a single administration of HOT was sufficient for efficacy. In view of HOT’s selective uptake and retention in a wide range of solid tumors, its single-agent efficacy in xenograft models and its non-specific mechanism of cancer-killing (radiation), we expect to first develop HOT as a monotherapy, initially for solid tumors.
LIGHT (labeled with a shorter-lived radioisotope, iodine-124) is a small-molecule imaging agent that we believe has first-in-class potential in detecting and quantifying cancerous tumors and metastases. LIGHT is comprised of a small quantity of COLD (too little for Akt inhibition), acting as a cancer-targeted delivery and retention vehicle, and incorporating 124 I, a new positron emission tomography (PET) imaging isotope. PET imaging used in conjunction with CT scanning has now become the imaging method of choice in oncology. In studies to date, LIGHT selectively illuminated malignant tumors in 52 of 54 animal models of cancer, demonstrating evidence of broad-spectrum, cancer-selective uptake and retention. PET imaging studies with LIGHT in cancer patients are being conducted at the University of Wisconsin under an investigator-sponsored IND. The investigator-sponsored IND for LIGHT was submitted on March 28, 2003 and was approved by the FDA on April 25, 2003. The IND is held by Dr. Anne Traynor at the University of Wisconsin, who both initiates and conducts the investigation and under whose immediate direction the investigational drug is administered. Novelos provides funding for the studies and the data is shared with Novelos while the study progresses and at the conclusion of the study. We expect additional investigator-sponsored Phase 1/2 trials of LIGHT as a PET imaging agent to begin in the fourth quarter of 2011, and that the trials will initially include brain metastases, lung and breast cancers. These human trials, if successful, will serve two important purposes. First, to provide proof-of-concept for LIGHT itself as a PET imaging agent with the potential to supplant the current “gold standard” agent, 18-fluoro-deoxyglucose (FDG), due to what we believe to be LIGHT’s superior cancer-specificity and more favorable logistics of clinical use. Second, to accelerate clinical development of HOT by predicting efficacy and enabling estimation of efficacious doses of HOT for Phase 2 trials.
Products in Development
COLD
COLD is a cancer-targeted chemotherapy that, in pre-clinical experiments, inhibits the phosphatidylinosotol 3-kinase (PI3K)/Akt survival pathway, which is overexpressed in many types of cancer. We believe COLD has the potential to be best-in-class versus other Akt inhibitors in development. We believe that COLD has important advantages over competitor agents including:
|
•
|
Selective uptake and retention by cancer cells/cancer stem cells compared to normal cells/stem cells. This results in significantly greater potency of COLD as an inhibitor of cell proliferation in cancer cells vs. normal cells (greater than a 10-fold difference), or
|
|
•
|
Suitability for intravenous administration, avoiding dose-limiting gastrointestinal toxicity seen with orally administered Akt-inhibiting APCs and potentially enabling greater systemic drug exposure and, hence, Akt-inhibition in cancer cells, resulting in superior efficacy.
|
We expect to submit an IND application to the FDA in late 2012.
Chemically, COLD is 18-(p-[127 I] iodophenyl) octadecyl phosphocholine, an alkyl phosphocholine (APC) subtype within the alkyl-phospholipid (APL) class of anti-tumor agents that includes perifosine, miltefosine and eldefosine. The iodine atom in its structure is the stable, non-radioactive (“cold”) isotope, 127 I.
COLD exhibits significant in vivo efficacy in mouse xenograft tumor models, including non-small cell lung cancer and triple-negative breast cancers. In these models, human cancer cells are transplanted to and grow/metastasize in immunosuppressed animals. Tumor-bearing mice treated therapeutically (i.e., after primary tumors were established) with COLD i.v. (100-times the mass dose used as a carrier in the radiotherapy agent, HOT) once a week for 5 weeks, showed almost complete suppression of tumor growth compared to saline-treated control animals. Tumor growth suppression by COLD was maintained long after the end of the treatment period. Importantly, survival in COLD-treated groups at experiment termination (100-200 days post tumor-cell injection) was 90% or more compared to 20% or less in control groups. Additionally, in a side-by-side comparison, COLD was much more effective in suppressing tumor growth and increasing survival in the lung cancer model than a standard dosing regimen of erlotinib (Tarceva, a marketed epidermal growth factor receptor kinase inhibitor).
28
The in vivo efficacy of COLD is believed to be at least in part the result of selective inhibition of the apoptosis-suppressing PI3K/Akt signaling pathway in cancer cells. This pathway, which is activated by growth factors such as PDGF (platelet-derived growth factor), EGF (epidermal growth factor), and insulin, is overactive in many human cancers and contributes to cell growth, proliferation, survival and resistance to radiation and chemotherapeutics. COLD selectively inhibits Akt activation in human cancer cells compared to normal proliferating cells (e.g., human fibroblasts). At the same concentrations, COLD induces apoptosis through caspase activation and suppresses proliferation in a wide range of human cancer cell lines including prostate carcinoma, ovarian carcinoma, triple-negative breast carcinomas, pancreatic adenocarcinoma and non-small cell lung cancer. At these concentrations, COLD does not inhibit proliferation of normal cells.
Other cancer targeting APCs have also been reported to be active in xenograft models and to selectively inhibit tumor cell proliferation via a mechanism that involves induction of apoptosis through caspase activation subsequent to inhibition of Akt activation and signaling. However, APCs are generally dose-limited in vivo (including in man) by side effects stemming from the necessity for their oral administration (due to their hemolytic properties), thus limiting Akt inhibition and anti-tumor efficacy. In contrast, data to date support the contention that COLD can be safely administered intravenously at doses that we believe will result in greater drug exposure compared to other APCs and, thus, in greater Akt inhibition and improved efficacy.
Non-APC Akt inhibitors in development are not cancer-targeting and thus have the potential for an unfavorable therapeutic index (due to non-selective inhibition of Akt, and hence proliferation) in normal vs. cancer cells. In contrast, selective uptake and retention of COLD results in more than 10-fold more potent inhibition of Akt activity and cell proliferation in cancer cells vs. normal cells.
The development path for COLD includes evaluation in a standard battery of IND-enabling pre-clinical tests and scaled-up manufacture. In parallel, we intend to test COLD in mouse xenograft tumor models in combination with standard chemotherapeutic agents to demonstrate synergies as have been reported for perifosine. These additional pre-clinical data will enable estimation of COLD plasma levels associated with in vivo efficacy, establish a starting dose for the initial Phase 1 clinical trial and facilitate selection of target indications. We expect to file an IND with the FDA for COLD in late 2012.
HOT ( iodine-131 radiolabeled compound )
HOT is a small-molecule, broad-spectrum, cancer-targeted molecular radiotherapeutic that we believe has first-in-class potential. HOT is comprised of a small quantity of COLD (too little for Akt inhibition), acting as a cancer-targeted delivery and retention vehicle and incorporating a cytotoxic dose of radiotherapy (in the form of iodine-131, a radioisotope that is already in common use to treat thyroid and other cancer types). It is this “intracellular radiation” mechanism of cancer cell killing, coupled with delivery to a wide range of malignant tumor types, that imbues HOT with broad-spectrum anti-cancer activity. In 2009, we opened an IND with the FDA to study HOT in humans. In early 2010, we successfully completed a Phase 1a dosimetry trial in humans demonstrating initial safety and establishing dosing parameters for a Phase 1b dose-escalation trial. The Phase 1b dose-escalation trial is aimed at determining the Maximum Tolerated Dose, and we expect it to begin in the fourth quarter of 2011. In parallel, we expect to initiate Phase 2 efficacy trials in solid tumors in 2012 as soon as a minimal efficacious dose is established. We may determine such an effective dose upon seeing a tumor response in the Phase 1b trial or calculating it from imaging trials in patients (see LIGHT below). HOT’s anti-tumor/survival-prolonging activity has been demonstrated in multiple animal xenograft models (human tumor cells implanted into mice). In all but two models, a single administration of HOT was sufficient for efficacy. In view of HOT’s selective uptake and retention in a wide range of solid tumors, its single-agent efficacy in xenograft models and its non-specific mechanism of cancer-killing (radiation), we expect to first develop HOT as a monotherapy, initially for solid tumors.
Chemically, HOT is 18-(p-[ 131 I]iodophenyl) octadecyl phosphocholine, identical to COLD except that the iodine in its structure is the radioactive (“hot”) isotope, 131 I, which has a radiation half-life of eight days.
Single intravenous doses of HOT administered therapeutically (i.e., after primary tumors were established) have resulted in significant anti-tumor and/or survival benefit compared to control animals in mouse xenograft tumor models including ovarian, pancreatic, non-small cell lung, triple-negative breast, prostate, glioma, colorectal and kidney cancers. Survival benefit generally reflected the degree of tumor growth suppression. Efficacy was also seen in a xenograft model employing human uterine sarcoma cells which over-express efflux pumps known to underlie resistance to many standard chemotherapeutic drugs. The broad in vivo efficacy profile of HOT across many tumor types is reflected in the fact that selective tumor localization of LIGHT (which uses the same cancer-targeting drug delivery and retention vehicle as HOT) has been demonstrated in over 50 xenograft, spontaneous and transgenic cancer models. HOT was also tested in combination with a standard efficacious dose of gemcitabine in a pancreatic cancer xenograft model. Single doses of HOT or gemcitabine given alone were equally efficacious while the combination therapy was significantly more efficacious than either treatment alone (additive). In each xenograft study, the dose of HOT was ~100 µCi, which is at least 50% less than the maximum tolerated dose in mice.
Extensive, IND-enabling, Good Laboratory Practices (GLP) in vivo and in vitro pre-clinical pharmacokinetic/distribution, toxicology and drug safety studies were successfully completed using non-pharmacological concentrations/doses of COLD consistent with its role as a delivery/retention vehicle in HOT. Tissue distribution studies supported prediction of acceptable human organ exposures and body clearance for HOT. Importantly, and in sharp distinction from biological products labeled with 131 I, the small molecule HOT showed very minimal variation in excretion kinetics and tissue distribution between individuals within species or across a 500-fold variation in dose. Single- and repeated-dose animal toxicology studies indicated very high margins of safety (80-200x) over the anticipated maximum human therapy dose of HOT.
29
In February 2010 we completed a Phase 1a dosimetry trial with a single intravenous doses of 10 mCi HOT in eight patients with relapsed or refractory advanced solid tumors. Single doses of HOT were well tolerated. The reported adverse events were all considered minimal, manageable and either not dose limiting or not related to HOT. There were no serious adverse events reported. Analysis of total body imaging and blood and urine samples collected over 42 days following injection indicated that doses of HOT expected to be therapeutically effective can be administered without harming vital organs. Two subjects (one with colorectal cancer metastasized to lung and another with prostate cancer) had tumors that were imaged with 3D nuclear scanning (SPECT/CT) on day 6 after administration of HOT. Uptake of HOT into tumor tissue (but not adjacent normal tissue or bone marrow) was clearly demonstrated in both subjects. Echoing animal studies, pharmacokinetic analyses demonstrated a prolonged halflife of radioactivity in the plasma after HOT administration (approximately 200 hours) and that there was no significant variation in excretion or radiation dosimetry between subjects. The trial established an initial dose of 12.5 mCi / m 2 (for example, 20 mCi dose for a patient with 1.6m 2 body surface area) for the Phase 1b escalating dose trial that is expected to begin in the fourth quarter of 2011.
The primary objective of this Phase 1b dose-escalation trial in patients with a range of advanced solid tumors is to define the Maximum Tolerated Dose (MTD) of HOT. In addition to determining the MTD, the Phase 1b trial will evaluate overall tumor response (using standard RESIST I criteria) and safety. Concurrently, separate studies will generate quantitative imaging data in cancer patients using LIGHT (see below). These imaging trials with LIGHT will predict efficacy and enable calculation of a minimal efficacious dose of HOT for Phase 2 trials, expected to begin in 2012, with an initial focus on solid tumors with significant unmet medical need. Based on its broad-spectrum mechanism of action and wide-ranging single agent activity in animal cancer models, HOT is anticipated to be used as monotherapy through proof-of-concept clinical trials, with subsequent exploration of combination with chemotherapeutic agents (a number of which are known to be radiosensitizers and thus with potential to enhance the efficacy of HOT).
Tumor treatment with radioactive isotopes has been used as a fundamental cancer therapeutic for decades. The goals of targeted cancer therapy — selective delivery of effective doses of isotopes that destroy tumor tissue, sparing of surrounding normal tissue, non-accumulation in vital organs such as the liver and kidneys — remain goals of novel therapies as well. We believe our isotope delivery technology is poised to achieve these goals. Because, to date, HOT has been shown to reliably and near-universally accumulate in cancer cells and because the therapeutic properties of the iodine-131 are well known, we believe the risk of non-efficacy in human clinical trials is less than that of other cancer therapies at this stage of development, although no assurance can be given.
Other targeted radiotherapies include the marketed drugs Zevalin® (90 Y, Spectrum Pharmaceuticals) and Bexxar® (131 I, GSK). In both cases, tumor-targeting is monoclonal antibody-based and limited to non-Hodgkins lymphoma, which is a type of cancer involving cells of the immune system. Thus, these agents are not appropriate comparators for HOT because of their limited therapeutic utility (only one type of tumor) and because their target indication is often well-managed by other drugs (unlike HOT which has potential to treat tumor types for which the current standard of care is associated with very poor outcomes). Notably, both Zevalin® and Bexxar® were approved on the basis of objective response rates (shrinking of tumors) without data to support improvement in survival, suggesting that regulatory approval of radiopharmaceuticals can be based on relatively shorter and smaller pivotal clinical trials than is often the case in oncology.
In conclusion, we believe that HOT is not subject to the full extent of development risk typically associated with early-stage cancer therapeutics for the following reasons:
|
|
·
|
HOT is selectively taken up by and retained in cancer vs. normal cells and its delivery vehicle (COLD) is intended to be given to patients in sub-pharmacological doses, resulting in an improved safety profile compared to standard chemotherapy.
|
|
|
·
|
HOT does not rely on inhibition or enhancement of a specific pathway; it works by exposing cancer cells to sustained lethal radiation from within.
|
|
|
·
|
To date, HOT (as demonstrated with LIGHT studies) has shown near-universal cancer-specific retention in more than 50 in vivo tumor models, making the molecule potentially effective in numerous cancer types (broad-spectrum) as compared to type-specific therapies.
|
|
|
·
|
We believe we have completed all preclinical safety, pharmacology and toxicology studies required for an NDA including both single-dose and multi-dose studies.
|
|
|
·
|
HOT is a small molecule that is easily characterized and synthesized and is therefore not subject to scale-up and manufacturing risks typically associated with large molecules such as monoclonal antibodies.
|
|
|
·
|
HOT exploits a new cancer-selective delivery and retention mechanism, but is paired with a proven and effective radioisotope ( 131 I) for therapy.
|
LIGHT ( labeled with a shorter-lived radioisotope, iodine-124 )
LIGHT is a small-molecule imaging agent that we believe has first-in-class potential in detecting and quantifying cancerous tumors and metastases. LIGHT is comprised of a small quantity of COLD (too little for Akt inhibition), acting as a cancer-targeted delivery and retention vehicle, and incorporating 124 I, a new positron emission tomography (PET) imaging isotope. PET imaging used in conjunction with CT scanning has now become the imaging method of choice in oncology. In studies to date, LIGHT selectively illuminated malignant tumors in 52 of 54 animal models of cancer, demonstrating evidence of broad-spectrum, cancer-selective uptake and retention. We expect investigator-sponsored Phase 1/2 trials of LIGHT as a PET imaging agent to begin in the fourth quarter of 2011, and that the trials will initially include brain metastases, lung and breast cancers. These human trials, if successful, will serve two important purposes. First, they will provide proof-of-concept for LIGHT itself as a PET imaging agent with the potential to supplant the current “gold standard” agent, 18-fluoro-deoxyglucose (FDG), due to what we believe to be LIGHT’s superior cancer-specificity and more favorable logistics of clinical use. Second, they will accelerate clinical development of HOT by predicting efficacy and enabling estimation of efficacious doses of HOT for Phase 2 trials.
30
Chemically, LIGHT is 18-(p-[ 124 I]iodophenyl) octadecyl phosphocholine, identical to COLD except that the iodine is the radioactive isotope, 124 I, which has a radiation half-life of 4 days.
Studies demonstrating the utility of LIGHT for imaging primary tumors and metastases as well as cancer stem cells are described above (Technology Overview).
FDG is the current gold standard for PET imaging. According to Bio-Tech Systems (November 2010), sales of FDG in the US in 2009 were approximately $300 million and projected to grow to approximately $900 million in 2017. FDG accumulates in any tissue having increased glucose metabolism compared to surrounding tissue. As a result and in contrast to LIGHT, FDG is not selective for malignant tumors. FDG localizes in certain normal tissue such as heart, kidney and brain tissues that also have high glucose metabolism. FDG is also known to localize in inflammatory sites. Other major limitations to the use of FDG are found in pelvic imaging due to the high renal (kidney) clearance of the compound. These characteristics of FDG, therefore, decrease its diagnostic specificity for certain malignancies. FDG is no longer covered by patent and is typically manufactured onsite at PET imaging medical facilities because of its limited (110 minute) half life.
We compared LIGHT and FDG side by side (24 hours apart) in the same tumor-bearing mouse that was also treated with carageenan to induce inflammation. As expected, FDG demonstrated significant uptake into the inflammatory lesion and organs such as heart and bladder compared to the malignant tumors which were poorly imaged. LIGHT, on the other hand, showed no uptake into the inflammatory lesion and organs, yet clear and demonstrable uptake into the tumors.
Additionally, the radioisotopic half-life of only 110 minutes for fluorine-18 labeled agents, such as FDG, severely limits their delivery range relative to the point of manufacture. 124 I has a four-day half-life that permits worldwide distribution of LIGHT from one manufacturing location. Additionally, the longer half-life affords a longer imaging window of up to seven days following injection.
We expect to begin investigator sponsored Phase 1/2 trials of LIGHT aimed at demonstrating and quantifying selective uptake and retention in human solid tumors in the fourth quarter of 2011. We expect initial indications to include brain metastases, lung and breast cancers, with extension to other cancer types to follow.
Technology
COLD, HOT and LIGHT are alkylphospholipids (“APLs”) that interact with specialized microdomains within cell membranes (called “lipid rafts”) and, as a result, whose molecular targets are located at cellular membranes. Importantly, the core chemical structure shared across all three products provides selective targeting of cancer cells in preference to normal cells (due to enrichment of lipid rafts in the former). COLD was deliberately designed to contain iodine (in the form of the stable, non-radioactive isotope, 127 I), thus enabling additional, distinct products differing only with respect to the form of iodine they contain – HOT contains short-lived radioactive 131 I and LIGHT contains even more short-lived 124 I. As a result, three cancer-targeted product profiles have been generated from a single chemical structure — a chemo-therapeutic agent (COLD), a molecular radiotherapeutic agent (HOT) and a diagnostic/imaging agent (LIGHT).
31
Using a fluorescent-labeled analog of COLD (CLR1501 or “GLOW1”), selective uptake and retention has been demonstrated in cancer cells in vitro . Twenty-four hours after treatment, a variety of human tumor cell types (melanoma, colorectal, uterine, pancreatic, ovarian, glioblastoma) show six to ten-fold more staining with GLOW1 relative to normal cells (e.g., skin fibroblasts) do not. Significantly, uptake/retention was also seen in cancer stem cells which are known to be relatively resistant to both chemotherapy and radiation and may therefore contribute to eventual relapse of disease following conventional chemotherapy.
Malignant tumor targeting, including targeting of cancer stem cells, has also been demonstrated in vivo . For example, mice without intact immune systems, and inoculated with Panc-1 (pancreatic carcinoma), were injected with CLR1502 (“GLOW2”, a fluorescent-labeled analog of COLD that is active in the near-infrared range) 24 and 96 hours prior to imaging. In vivo optical imaging showed pronounced accumulation of GLOW2 in tumors versus non-target organs and tissues. Similarly, PET imaging of tumor-bearing animals (colon, glioma, triple negative breast and pancreatic tumor xenograft models) administered the imaging agent LIGHT clearly shows selective uptake and retention by both primary tumors and metastases, including cancer stem cells. Furthermore, PET/CT analysis following co-injection of HOT (for therapy) and LIGHT (for imaging) revealed time-dependent tumor shrinkage and disappearance (over 9 days) in a cancer xenograft model. Finally, we believe that the capability of our technology in targeting cancer stem cells in vivo was demonstrated by treating tumor-bearing mice with GLOW1 and then removing the tumor and isolating cancer stem cells, which continued to display GLOW1 labeling even after three weeks in cell culture.
The basis for selective tumor targeting of our compounds lies in differences between the plasma membranes of cancer cells as compared to those of most normal cells. Specifically, cancer cell membranes are highly enriched in “lipid rafts”. Lipid rafts are specialized regions of the membrane phospholipid bilayer that contain high concentrations of cholesterol and sphingolipids and serve to organize cell surface and intracellular signaling molecules (e.g. growth factor and cytokine receptors, the phophatidylinosotol 3-kinase (P13K)/Akt survival pathway. Lipid rafts are central to the activity of our compounds in two ways:
|
|
1.
|
Lipid rafts are portals of entry for APLs such as COLD, HOT and LIGHT. The marked selectivity of our compounds for cancer cells versus non-cancer cells is due to the fact that cancer cells have far more lipid rafts. In addition to accumulating in lipid rafts, COLD, HOT and LIGHT are transported into the cytoplasm, where they distribute to organelle membranes (mitochondria, ER, lysosomes) but not the nucleus.
|
|
|
2.
|
Lipid rafts also regulate signaling-based cell functions including apoptosis and cell proliferation, and COLD disrupts this regulation. For example, one key signaling pathway that is regulated by interactions with lipid rafts and phospholipids is the phosphatidylinosotol 3-kinase (PI3K)/Akt pathway. Akt (a serine/threonine protein kinase) is activated in lipid raft regions via phosphorylation by PI-dependent kinases and goes on to phosphorylate anti-apoptotic proteins (e.g., Bcl-xL and FLIP) resulting in their inactivation and, thus, promotion of tumor cell survival. COLD pharmacologically inhibits the activation of Akt. In cancer cells, Akt inhibition is associated with induction of apoptosis and decreased cell proliferation/survival.
|
The pivotal role played by lipid rafts is underscored by the fact that disruption of lipid raft architecture suppresses uptake of GLOW1 and radiolabeled COLD into cancer cells.
Legacy Products
NOV-002, our legacy compound, is a small-molecule immunomodulating and anti-cancer compound based on a proprietary formulation of oxidized glutathione. NOV-002 has been administered to approximately 1,000 cancer patients in clinical trials and was in Phase 2 development for solid tumors in combination with chemotherapy.
From November 2006 through January 2010, we conducted a Phase 3 trial of NOV-002 plus first-line chemotherapy in advanced non-small cell lung cancer (“NSCLC”) following three Phase 2 trials (two conducted in Russia and one conducted by us in the U.S.) that had demonstrated clinical activity and safety. The Phase 3 trial enrolled 903 patients, 452 of whom received NOV-002. In February 2010, we announced that the primary endpoint of improvement in overall survival compared to first-line chemotherapy alone was not met in this pivotal Phase 3 trial. Following evaluation of the detailed trial data, we announced in March 2010 that the secondary endpoints also were not met in the trial and that adding NOV-002 to paclitaxel and carboplatin chemotherapy was not statistically or meaningfully different in terms of efficacy-related endpoints or recovery from chemotherapy toxicity versus chemotherapy alone. However, NOV-002 was safe and did not add to the overall toxicity of chemotherapy. Based on the results from the Phase 3 trial, we have discontinued development of NOV-002 for NSCLC in combination with first-line paclitaxel and carboplatin chemotherapy. The aggregate costs incurred in connection with our development of NOV-002, including administrative overhead, were approximately $70 million.
32
Novelos had also been developing NOV-205, a second oxidized glutathione-based compound. NOV-205 had been administered to approximately 200 hepatitis patients in clinical trials and was in Phase 2 development for chronic hepatitis C non-responders. An IND for NOV-205 as a monotherapy for chronic hepatitis C was accepted by the FDA in 2006. A U.S. Phase 1b clinical trial with NOV-205 in patients who previously failed treatment with pegylated interferon plus ribavirin was completed in December 2007. Based on favorable safety results of that trial, in March 2010 Novelos initiated a multi-center U.S. Phase 2 trial evaluating NOV-205 as monotherapy in up to 40 chronic hepatitis C genotype 1 patients who previously failed treatment with pegylated interferon plus ribavirin. Safety was established in twenty patients receiving either 30mg or 60mg of NOV-205 daily for 49 days; however, no viral load reduction was observed.
Further development of NOV-002 and NOV-205 has been suspended. At this time, we expect to devote our resources to the development and commercialization of the Cellectar compounds, and we do not expect to conduct any further development of the oxidized glutathione compounds. The IND for NOV-205 was withdrawn on July 5, 2011. We anticipate that the IND for NOV-002 will be placed on inactive status.
Manufacturing
We manufacture HOT and COLD at our current Good Manufacturing Practices (cGMP)-compliant radiopharmaceutical manufacturing facility in Madison, WI. This facility, consisting of approximately 19,500 square feet, contains offices, laboratories, a radiopharmaceutical research lab, a cGMP radiopharmaceutical manufacturing suite and a cGMP analytical laboratory for product release. Our manufacturing facility holds a State of Wisconsin Department of Health Services Radioactive Materials License which authorizes the use and possession of radioactive material for both manufacturing and distribution activities. This license establishes a possession limit of 9 Curies of iodine-131. The facility also holds a State of Wisconsin DHS Radioactive Materials License which authorizes the use and possession of radioactive materials by Cellectar for research and development. The research and development license permits the use and possession of iodine-125, iodine-131 and iodine-124 in quantities sufficient to support in-house HOT manufacturing and other research needs. To date, small quantities of LIGHT have been manufactured by our collaborator, the University of Wisconsin in Madison, at no cost to the Company, in connection with investigator-sponsored clinical trials, pursuant to a materials transfer agreement expiring in June 2013. The materials transfer agreement contains standard provisions for the protection of data and intellectual property and may be terminated by either party at any time before expiration. We are in the process of negotiating a fee-based arrangement with the University of Wisconsin covering the manufacture of LIGHT in the future. If the University was unable to manufacture LIGHT for any reason, we could manufacture it at our Madison facility, without material additional investment. We are exploring scaling up production capacity of COLD, via contract manufacturers or at our facility, to support an IND filing and clinical trials, but we have not yet entered into any agreements for the manufacture of COLD. The drug substance is identical for all three products with the exception of the different iodine isotope used in each. The base molecule is a dry powder produced via a six-step synthetic scheme. The release specifications for drug substance have been established and validated. The impurity levels at small scale are very low suggesting that larger scale production should be feasible. We have also demonstrated 24-month stability for the drug substance in desiccated and refrigerated form. We believe our laboratories are well equipped with the appropriate equipment for manufacturing pilot and small-scale batches to cGMP. We believe we have adequate capacity for any Phase 2 trials of HOT and the potential for larger scale build-out for larger Phase 3 trials. All investigational drug substance and product intended for human use during clinical studies will be manufactured according to ICH guidelines, FDA requirements (CFR part 211) and cGMP.
Sales and Marketing
COLD, HOT and LIGHT have not been partnered to date. We plan to pursue and evaluate all available options to develop, launch and commercialize our compounds. These options presently include, but are not limited to, entering into a partnering arrangement with a pharmaceutical company or various pharmaceutical companies with strong development and commercial expertise and infrastructure in the U.S, Europe and/or Japan. While we currently do not plan to build our own sales force or utilize a contract sales organization for launch and commercialization of our compounds, we may reconsider.
Competition
COLD
We believe COLD has the potential to be best-in-class versus other Akt inhibitors in development. Important advantages of COLD over competitor agents are believed to include:
|
•
|
Selective uptake and retention by cancer cells/cancer stem cells compared to normal cells/stem cells. This results in significantly greater potency of COLD as an inhibitor of cell proliferation in cancer cells vs. normal cells (>10-fold difference), or
|
|
•
|
Suitability for intravenous administration, avoiding dose-limiting gastrointestinal toxicity seen with orally administered Akt-inhibiting APCs and potentially enabling greater systemic drug exposure and, hence, Akt-inhibition in cancer cells, resulting in superior efficacy.
|
Perifosine, an alkylphospholipid that is being being developed by Keryx Biopharmaceuticals, which has licensed it in North America from Æterna Zentaris Inc., is a possible future competitor to COLD.
33
HOT
HOT’s “intracellular radiation” mechanism of cancer cell killing, coupled with delivery to a wide range of malignant tumor types, that imbues HOT with broad-spectrum anti-cancer activity. Other targeted radiotherapies include the marketed drugs Zevalin® (manufactured by Spectrum Pharmaceuticals) and Bexxar® (manufactured by GlaxoSmithKline). In both cases, tumor-targeting is monoclonal antibody-based and limited to non-Hodgkins lymphoma, which is a type of cancer involving cells of the immune system. Thus, these agents are not appropriate comparators for HOT because of their limited therapeutic utility (only one type of tumor) and because their target indication is often well-managed by other drugs (unlike HOT which has potential to treat tumor types for which the current standard of care is associated with very poor outcomes). Notably, both Zevalin® and Bexxar® were approved on the basis of objective response rates (shrinking of tumors) without data to support improvement in survival, suggesting that regulatory approval of radiopharmaceuticals can be based on relatively shorter and smaller pivotal clinical trials than is often the case in oncology. We do not believe Zevalin or Bexxar would be competing products of HOT in any material respect.
LIGHT
FDG is the current gold standard for cancer PET imaging. According to Bio-Tech Systems (November 2010), sales of FDG in the US in 2009 were approximately $300 million and projected to grow to approximately $880 million in 2017. FDG accumulates in any tissue having increased glucose metabolism compared to surrounding tissue. As a result and in contrast to LIGHT, FDG is not selective for malignant tumors. FDG localizes in certain normal tissue such as heart, kidney and brain tissues that also have high glucose metabolism. FDG is also known to localize in inflammatory sites. Other major limitations to the use of FDG are found in pelvic imaging due to the high renal (kidney) clearance of the compound. We believe these characteristics of FDG, therefore, decrease its diagnostic specificity for certain malignancies. FDG is no longer covered by patent and is typically manufactured onsite at PET imaging medical facilities because of its limited (110 minute) half-life.
We compared LIGHT and FDG side by side (24 hours apart) in the same tumor-bearing mouse that was also treated with carageenan to induce inflammation. As expected, FDG demonstrated significant uptake into the inflammatory lesion and organs such as heart and bladder compared to the malignant tumors which were poorly imaged. LIGHT, on the other hand, showed no uptake into the inflammatory lesion and organs, yet clear and demonstrable uptake into the tumors.
Additionally, the radioisotopic half-life of only 110 minutes for fluorine-18 labeled agents, such as FDG, severely limits their delivery range relative to the point of manufacture. 124 I has a four-day half-life that permits worldwide distribution of LIGHT from one manufacturing location. Additionally, the longer half-life affords a longer imaging window of up to seven days following injection.
Intellectual Property
We have established a broad U.S. and international intellectual property rights portfolio around our cancer-targeting alkylphospholipid technology platform including COLD, HOT and LIGHT.
Our proprietary rights include patents and patent applications that are either owned by us or exclusively licensed to us by the U. Mich. (the “U. Mich. patents”) HOT and LIGHT are covered by the U. Mich. patents that provide compound (composition of matter) coverage in the US and Canada and expire in 2016. Our patents and applications cover methods of use, composition and method of manufacture related to COLD, HOT and LIGHT. Many of these patents and applications are filed in key commercial markets worldwide. These will generally expire between 2025 and 2030 unless extended.
In particular, HOT is covered by three additional series of our patents and applications aside from the U. Mich. patents. The first is directed to a method of use for cancer therapy and has also been filed in Europe and Japan, in addition to the U.S. These will be expected to expire in 2025. The second application is directed to modified forms of HOT and is pending in the U.S., and will be filed/nationalized in foreign countries in 2012. These patents, once issued, would expire in most jurisdictions in 2030. Lastly, an application directed to cancer stem cell therapy is pending in the U.S. and will be filed/nationalized in foreign countries in 2011 and is expected to expire in 2030. Some of these resulting patents may be extendable on a country-by-country basis.
COLD is covered by a series of pending applications directed to methods of using COLD for cancer therapy and will be filed/nationalized in foreign countries in 2012. These patents, once issued, would expire in 2030. Some of these resultant patents may be extendable on a country-by-country basis.
Separate from any patent protection and following product approval by regulatory authorities, data exclusivity may be available for HOT and COLD for up to 10 years on a country-by-country basis (e.g., up to 5 years in the U.S.).
The early termination of the U. Mich. license would result in the loss of our rights to use the covered patents which could severely delay, inhibit or eliminate our ability to develop and commercialize compounds based on the licensed patents.
34
LIGHT is covered by the U. Mich. patents as well as two of our U.S. patents, one of which is directed to its use for virtual colonoscopy (expiring 2025) and one of which is directed to its use for in vitro diagnostics (expiring 2025). LIGHT is also covered by pending U.S. and European patent applications directed to its use for in vivo diagnostics and once issued should expire in 2025. Lastly, the use of LIGHT for diagnostics purposes with cancer stem cells is pending in the U.S. and will be filed/nationalized in foreign countries in 2011. These patents are expected to expire in 2030.
In addition to the above noted patents/applications directed to HOT, COLD and LIGHT, we own other patents/applications directed to different forms of alkylphospholipids and methods of manufacturing of alkylphospholipids.
We also own all intellectual property rights worldwide (excluding Russia and the other states of the former Soviet Union, the “Russian Territory”) related to our clinical-stage pipeline compound, NOV-002, and other pre-clinical compounds based on oxidized glutathione. Issued composition of matter patents cover proprietary formulations of oxidized glutathione that do not expire until 2019, and these patents include methods of manufacture for oxidized glutathione formulated with various metals. In our dispute with BAM, one of the remedies BAM is seeking is the revocation of our rights in these compounds.
Licenses / Collaborations
In September 2003, Cellectar entered into the U. Mich license which granted Cellectar exclusive rights to the development, manufacture and marketing of products under several composition of matter patents in North America which expire at varying dates in 2016. The U. Mich. license expires upon the expiration of the last covered patent. We are responsible for an annual license fee of $10,000 and are required to pay costs associated with the maintenance of the patents covered by the U. Mich. license. Additionally, we are required to make milestone payments of $50,000 upon the filing of a New Drug Application (NDA) for a licensed product intended for use in a therapeutic or diagnostic application (such milestone fees may be deferred and paid within twelve months of the first commercial sale of such product) and make certain milestone payments within a year following the first commercial sale of any licensed products. The sales milestones range from $100,000 to $200,000, dependent upon whether the drug is for use in a therapeutic or diagnostic application, provided that if sales in the first 12 months are less than the amount of the milestone, then we are required to pay 50% of all sales until the milestone is satisfied. The milestone payments may total up to $400,000. The U. Mich. license provides that we pay a royalty equal to 3% of net sales of any licensed products sold by us or our sublicensees for such licensed products, provided however if the sublicense fee payable to us is between 4%-5% of net sales then the royalties payable to U. Mich. shall be equal to 50% of the sublicense fee. Furthermore, the U. Mich. license provides for a reduction in the royalties owed by up to 50% if we are required to pay royalties to any third parties related to the sale of the licensed products. If we receive any revenue in consideration for rights to the licensed technology that is not based on net sales, excluding any funded research and development, we are required to pay U.Mich. 10% of amounts received. During 2003, pursuant to the U. Mich. license, Cellectar paid approximately $54,000 of back patent costs and issued 203,483 shares of common stock to U. Mich. as partial consideration for the rights described above. U. Mich. may terminate the agreement if we cease operations, if we fail to make any required payment under the agreement, or if we otherwise materially breach the agreement, subject to applicable notice and cure periods. To date, we have made all payments as they have become due, there have been no defaults under the U. Mich. license, nor have we ever been notified of a default by U. Mich. We may terminate the agreement with six months notice to U. Mich. and the return of licensed product and related data. The U. Mich. license contained milestones that required certain development activities to be completed by specified dates. All such development milestones have been either completed or removed by subsequent amendment to the agreement. U. Mich. has provided no warranties as to validity or otherwise with respect to the licensed technology.
Employees
As of September 9, 2011 we had 16 full time employees. We believe our relationships with our employees are good.
Regulation
The production, distribution, and marketing of products employing our technology, and our development activities, are subject to extensive governmental regulation in the United States and in other countries. In the United States, we are subject to the Federal Food, Drug, and Cosmetic Act, as amended, and the regulations of the FDA, as well as to other federal, state, and local statutes and regulations. These laws, and similar laws outside the United States, govern the clinical and preclinical testing, manufacture, safety, effectiveness, approval, labeling, distribution, sale, import, export, storage, record-keeping, reporting, advertising, and promotion of drugs. Product development and approval within this regulatory framework, if successful, will take many years and involve the expenditure of substantial resources. Violations of regulatory requirements at any stage may result in various adverse consequences, including the FDA’s and other health authorities’ delay in approving or refusal to approve a product. Violations of regulatory requirements also may result in enforcement actions.
The following paragraphs provide further information on certain legal and regulatory issues with a particular potential to affect our operations or future marketing of products employing its technology.
Research, Development, and Product Approval Process
The research, development, and approval process in the United States and elsewhere is intensive and rigorous and generally takes many years to complete. The typical process required by the FDA before a therapeutic drug may be marketed in the United States includes:
35
|
·
|
preclinical laboratory and animal tests performed under the FDA’s Good Laboratory Practices regulations, referred to herein as GLP;
|
|
·
|
submission to the FDA of an IND, which must become effective before human clinical trials may commence;
|
|
·
|
human clinical studies performed under the FDA’s Good Clinical Practices regulations, to evaluate the drug’s safety and effectiveness for its intended uses;
|
|
FDA review of whether the facility in which the drug is manufactured, processed, packed, or held meets standards designed to assure the product’s continued quality; and
|
|
·
|
submission of a marketing application to the FDA, and approval of the application by the FDA.
|
Preclinical Testing
During preclinical testing, studies are performed with respect to the chemical and physical properties of candidate formulations. These studies are subject to GLP requirements. Biological testing is typically done in animal models to demonstrate the activity of the compound against the targeted disease or condition and to assess the apparent effects of the new product candidate on various organ systems, as well as its relative therapeutic effectiveness and safety. An IND must be submitted to the FDA and become effective before studies in humans may commence.
Clinical Trials
Clinical trial programs in humans generally follow a three-phase process. Typically, Phase 1 studies are conducted in small numbers of healthy volunteers or, on occasion, in patients afflicted with the target disease. Phase 1 studies are conducted to determine the metabolic and pharmacological action of the product candidate in humans and the side effects associated with increasing doses, and, if possible, to gain early evidence of effectiveness. In Phase 2, studies are generally conducted in larger groups of patients having the target disease or condition in order to validate clinical endpoints, and to obtain preliminary data on the effectiveness of the product candidate and optimal dosing. This phase also helps determine further the safety profile of the product candidate. In Phase 3, large-scale clinical trials are generally conducted in patients having the target disease or condition to provide sufficient data for the statistical proof of effectiveness and safety of the product candidate as required by United States regulatory agencies.
In the case of products for certain serious or life-threatening diseases, the initial human testing may be done in patients with the disease rather than in healthy volunteers. Because these patients are already afflicted with the target disease or condition, it is possible that such studies will also provide results traditionally obtained in Phase 2 studies. These studies are often referred to as “Phase 1/2” studies. However, even if patients participate in initial human testing and a Phase 1/2 study carried out, the sponsor is still responsible for obtaining all the data usually obtained in both Phase 1 and Phase 2 studies.
Before proceeding with a study, sponsors may seek a written agreement from the FDA regarding the design, size, and conduct of a clinical trial. This is known as a Special Protocol Assessment (“SPA”). Among other things, SPAs can cover clinical studies for pivotal trials whose data will form the primary basis to establish a product’s efficacy. SPAs help establish upfront agreement with the FDA about the adequacy of a clinical trial design to support a regulatory approval, but the agreement is not binding if new circumstances arise. There is no guarantee that a study will ultimately be adequate to support an approval even if the study is subject to an SPA.
United States law requires that studies conducted to support approval for product marketing be “adequate and well controlled.” In general, this means that either a placebo or a product already approved for the treatment of the disease or condition under study must be used as a reference control. Studies must also be conducted in compliance with good clinical practice requirements, and informed consent must be obtained from all study subjects.
The clinical trial process for a new compound can take ten years or more to complete. The FDA may prevent clinical trials from beginning or may place clinical trials on hold at any point in this process if, among other reasons, it concludes that study subjects are being exposed to an unacceptable health risk. Trials may also be prevented from beginning or may be terminated by institutional review boards, who must review and approve all research involving human subjects. Side effects or adverse events that are reported during clinical trials can delay, impede, or prevent marketing authorization. Similarly, adverse events that are reported after marketing authorization can result in additional limitations being placed on a product’s use and, potentially, withdrawal of the product from the market.
36
Following the completion of clinical trials, the data are analyzed to determine whether the trials successfully demonstrated safety and effectiveness and whether a product approval application may be submitted. In the United States, if the product is regulated as a drug, an NDA must be submitted and approved before commercial marketing may begin. The NDA must include a substantial amount of data and other information concerning the safety and effectiveness of the compound from laboratory, animal, and human clinical testing, as well as data and information on manufacturing, product quality and stability, and proposed product labeling.
Each domestic and foreign manufacturing establishment, including any contract manufacturers we may decide to use, must be listed in the NDA and must be registered with the FDA. The application generally will not be approved until the FDA conducts a manufacturing inspection, approves the applicable manufacturing process and determines that the facility is in compliance with cGMP requirements.
Under the Prescription Drug User Fee Act, as amended, the FDA receives fees for reviewing an NDA and supplements thereto, as well as annual fees for commercial manufacturing establishments and for approved products. These fees can be significant. For fiscal year 2011, the NDA review fee alone is $1,542,000, although certain limited deferral, waivers, and reductions may be available.
Each NDA submitted for FDA approval is usually reviewed for administrative completeness and reviewability within 45 to 60 days following submission of the application. If deemed complete, the FDA will “file” the NDA, thereby triggering substantive review of the application. The FDA can refuse to file any NDA that it deems incomplete or not properly reviewable. The FDA has established performance goals for the review of NDAs— six months for priority applications and 10 months for standard applications. However, the FDA is not legally required to complete its review within these periods and these performance goals may change over time.
Moreover, the outcome of the review, even if generally favorable, typically is not an actual approval but an “action letter” that describes additional work that must be done before the application can be approved. The FDA’s review of an application may involve review and recommendations by an independent FDA advisory committee. Even if the FDA approves a product, it may limit the approved therapeutic uses for the product as described in the product labeling, require that warning statements be included in the product labeling, require that additional studies be conducted following approval as a condition of the approval, impose restrictions and conditions on product distribution, prescribing, or dispensing in the form of a risk management plan, or otherwise limit the scope of any approval.
Post NDA Regulation
Significant legal and regulatory requirements also apply after FDA approval to market under an NDA. These include, among other things, requirements related to adverse event and other reporting, product advertising and promotion and ongoing adherence to cGMPs, as well as the need to submit appropriate new or supplemental applications and obtain FDA approval for certain changes to the approved product labeling, or manufacturing process. The FDA also enforces the requirements of the Prescription Drug Marketing Act which, among other things, imposes various requirements in connection with the distribution of product samples to physicians.
The regulatory framework applicable to the production, distribution, marketing, and/or sale, of our product pipleline may change significantly from the current descriptions provided herein in the time that it may take for any of our products to reach a point at which an NDA is approved.
Overall research, development, and approval times depend on a number of factors, including the period of review at FDA, the number of questions posed by the FDA during review, how long it takes to respond to the FDA’s questions, the severity or life-threatening nature of the disease in question, the availability of alternative treatments, the availability of clinical investigators and eligible patients, the rate of enrollment of patients in clinical trials, and the risks and benefits demonstrated in the clinical trials.
Other United States Regulatory Requirements
In the United States, the research, manufacturing, distribution, sale, and promotion of drug and biological products are potentially subject to regulation by various federal, state, and local authorities in addition to the FDA, including the Centers for Medicare and Medicaid Services (formerly the Heath Care Financing Administration), other divisions of the United States Department of Health and Human Services (e.g., the Office of Inspector General), the United States Department of Justice and individual United States Attorney offices within the Department of Justice, and state and local governments. For example, sales, marketing, and scientific/educational grant programs must comply with the anti-fraud and abuse provisions of the Social Security Act, the False Claims Act, the privacy provision of the Health Insurance Portability and Accountability Act, and similar state laws, each as amended. Pricing and rebate programs must comply with the Medicaid rebate requirements of the Omnibus Budget Reconciliation Act of 1990 and the Veterans Health Care Act of 1992, each as amended. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements apply. All of these activities are also potentially subject to federal and state consumer protection, unfair competition, and other laws.
37
Moreover, we are now, and may become subject to, additional federal, state, and local laws, regulations, and policies relating to safe working conditions, laboratory practices, the experimental use of animals, and/or the use, storage, handling, transportation, and disposal of human tissue, waste, and hazardous substances, including radioactive and toxic materials and infectious disease agents used in conjunction with our research work.
Foreign Regulatory Requirements
We and any future collaborative partners may be subject to widely varying foreign regulations, which may be quite different from those of the FDA, governing clinical trials, manufacture, product registration and approval, and pharmaceutical sales. Whether or not FDA approval has been obtained, we or any future collaboration partners must obtain a separate approval for a product by the comparable regulatory authorities of foreign countries prior to the commencement of product marketing in these countries. In certain countries, regulatory authorities also establish pricing and reimbursement criteria. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. In addition, under current United States law, there are restrictions on the export of products not approved by the FDA, depending on the country involved and the status of the product in that country.
Reimbursement and Pricing Controls
In many of the markets where we or any future collaborative partners would commercialize a product following regulatory approval, the prices of pharmaceutical products are subject to direct price controls (by law) and to drug reimbursement programs with varying price control mechanisms. Public and private health care payors control costs and influence drug pricing through a variety of mechanisms, including through negotiating discounts with the manufacturers and through the use of tiered formularies and other mechanisms that provide preferential access to certain drugs over others within a therapeutic class. Payors also set other criteria to govern the uses of a drug that will be deemed medically appropriate and therefore reimbursed or otherwise covered. In particular, many public and private health care payors limit reimbursement and coverage to the uses of a drug that are either approved by the FDA or that are supported by other appropriate evidence (for example, published medical literature) and appear in a recognized drug compendium. Drug compendia are publications that summarize the available medical evidence for particular drug products and identify which uses of a drug are supported or not supported by the available evidence, whether or not such uses have been approved by the FDA. For example, in the case of Medicare coverage for physician-administered oncology drugs, the Omnibus Budget Reconciliation Act of 1993, with certain exceptions, prohibits Medicare carriers from refusing to cover unapproved uses of an FDA-approved drug if the unapproved use is supported by one or more citations in the American Hospital Formulary Service Drug Information the American Medical Association Drug Evaluations, or the United States Pharmacopoeia Drug Information. Another commonly cited compendium, for example under Medicaid, is the DRUGDEX Information System.
During 2009 and 2010, there have been positive developments regarding medical reimbursement for therapeutic radiopharmaceuticals in the United States. The Centers for Medicare and Medicaid Services have proposed that the reimbursement for radiopharmaceuticals shift from a cost-to-charge ratio (“CCR”) to an average selling price (“ASP”) plus 4% model. There has been support expressed for this proposal by government agencies, key industry members and the industry consortium, The Council on Radionuclides and Radiopharmaceuticals. The historical CCR model resulted in a reimbursement rate that was lower than the cost to purchase the drugs, thus creating a disincentive for hospitals to prescribe radiopharmaceuticals. The current ASP proposal is a solution to this reimbursement problem. Furthermore, there are proposals pending, which, if adopted, would decrease the physician reimbursement for chemotherapy and conventional radiation therapy. The proposed reduction in physician reimbursement for chemotherapy could likely result in the movement of a large volume of cancer care from the physician’s office to the hospital environment. The proposed reduction in physician reimbursement for radiation therapy would result in a gap in revenue for radiation oncologists. Both proposed reductions favor an increase in opportunities to prescribe therapeutic radiopharmaceuticals. In fact, Zevalin® had an 84% increase in 2010 revenues versus 2009, according to Spectrum’s Form 10-K for the year ended December 31, 2010.
LITIGATION
A putative federal securities class action complaint was filed on March 5, 2010 in the United States District Court for the District of Massachusetts by an alleged shareholder of Novelos, on behalf of himself and all others who purchased or otherwise acquired our common stock in the period between December 14, 2009 and February 24, 2010, against Novelos and our President and Chief Executive Officer, Harry S. Palmin. On October 1, 2010, the court appointed lead plaintiffs (Boris Urman and Ramona McDonald) and appointed lead plaintiffs’ counsel. On October 22, 2010, an amended complaint was filed. The amended complaint claims, among other things, that the defendants violated Section 10(b) of the Securities Exchange Act of 1934, as amended, and Rule 10b-5 promulgated thereunder in connection with alleged misleading disclosures related to the progress of the Phase 3 clinical trial of NOV-002 for non-small cell lung cancer. On December 6, 2010, we filed a motion to dismiss the complaint with prejudice. On January 20, 2011, the plaintiffs filed their opposition to our motion and on March 3, 2011, we filed our response to their opposition. On June 23, 2011, the motion to dismiss was granted and the case was dismissed without prejudice. On August 5, 2011, the plaintiffs filed a second amended complaint realleging that the defendants violated Section 10(b) of the Exchange Act and Rule 10b-5 in connection with alleged misleading disclosures related to the Phase 3 clinical trial for NOV-002 in non-small cell lung cancer. On September 9, 2011, the defendants filed a motion to dismiss the second amended complaint. The plaintiff’s opposition to the motion is due by October 7, 2011. We believe the allegations are without merit and intend to vigorously defend against them.
38
On June 28, 2010, we received a letter from counsel to ZAO BAM and ZAO BAM Research Laboratories (Russian companies, collectively referred to as “BAM”) alleging that we modified the chemical composition of NOV-002 without prior notice to or approval from BAM, constituting a material breach of a technology and assignment agreement we had entered into with BAM on June 20, 2000 (the “June 2000 Agreement”). The letter references our amendment, submitted to the FDA on August 30, 2005, to our investigational new drug application dated August 1999 as the basis for BAM’s claims and demands the transfer of all intellectual property rights concerning NOV-002 to BAM. Mark Balazovsky, a director of Novelos from June 1996 until November 2006 and a shareholder of Novelos through at least June 25, 2010, is, to our knowledge, still the general director and principal shareholder of ZAO BAM. On September 24, 2010, we filed a complaint in Suffolk Superior Court seeking a declaratory judgment by the court that the June 2000 Agreement has been replaced by a subsequent agreement between the parties dated April 1, 2005 (the “April 2005 Agreement”), that Novelos’ obligations to BAM are governed solely by the April 2005 Agreement and that the obligations of the June 2000 agreement have been performed and fully satisfied. On November 29, 2010, BAM answered the complaint, denying the material allegations, and stating its affirmative defenses and certain counterclaims. On January 14, 2011, we responded to the counterclaims, denying BAM’s material allegations and stating our affirmative defenses. On June 9, 2011, BAM filed an amended counterclaim alleging additional claims related to Novelos’ acquisition of Cellectar. In that amended counterclaim, BAM alleges that the acquisition evidences Novelos’ abandonment of the technology assigned to it by BAM constituting a breach of the June 2000 Agreement or, if that agreement is determined to no longer be in effect, a breach of the April 2005 Agreement and/or a breach of the implied duty of good faith and fair dealing with respect to the April 2005 Agreement. On June 15, 2011 we filed our response to their amended counterclaim. On August 5, 2011, we filed a motion for judgment on the pleadings as to our declaratory judgment count and all counts of BAM’s amended counterclaim. The motion has been opposed by BAM and a hearing on the motion is scheduled on September 27, 2011. We believe BAM’s allegations and counterclaims are without merit and intend to defend vigorously against them.
We lease our executive office in Newton, Massachusetts. Our office consists of approximately 2,000 square feet and is rented for approximately $5,300 per month. This lease may be terminated by either party with one month notice.
We lease office, laboratory and manufacturing space in Madison, WI. The space consists of approximately 19,500 square feet and is rented for approximately $12,500 per month and expires on September 14, 2012. The lease may be renewed for two-year periods through 2024 with an increase of 3% in annual rent.
We believe that our present facilities are adequate to meet our current needs. If new or additional space is required, we believe that adequate facilities are available at competitive prices.
MANAGEMENT
As of September 9, 2011, our directors and executive officers are:
|
Name
|
Age
|
Position
|
||
|
Stephen A. Hill, B.M. B.Ch., M.A., F.R.C.S. (3)
|
53
|
Chairman of the Board (term expiring at 2012 annual meeting or upon his successor being duly elected and qualified)
|
||
|
Harry S. Palmin
|
41
|
President, Chief Executive Officer and Director (term as Director expiring at 2013 annual meeting or upon his successor being duly elected and qualified)
|
||
|
Kimberly A. Hawkins
|
39
|
Vice President of Clinical Development
|
||
|
Christopher J. Pazoles, Ph.D.
|
61
|
Senior Vice President of Research and Development
|
||
|
Joanne M. Protano
|
42
|
Vice President, Chief Financial Officer and Treasurer
|
||
|
Jamey P. Weichert, Ph.D.
|
54
|
Chief Scientific Officer and Director (term as Director expiring at 2013 annual meeting or upon his successor being duly elected and qualified)
|
||
|
Thomas Rockwell Mackie, Ph.D. (2)
|
56
|
Director (term expiring at 2011 annual meeting or upon his successor being duly elected and qualified)
|
||
|
James S. Manuso, Ph.D. (2)(3)
|
62
|
Director (term expiring at 2011 annual meeting or upon his successor being duly elected and qualified)
|
||
|
John Neis (1)(2)
|
56
|
Director (term expiring at 2012 annual meeting or upon his successor being duly elected and qualified)
|
||
|
John E. Niederhuber, M.D. (1)(3)
|
73
|
Director (term expiring at 2011 annual meeting or upon his successor being duly elected and qualified)
|
||
|
Howard M. Schneider (1)
|
67
|
Director (term expiring at 2013 annual meeting or upon his successor being duly elected and qualified)
|
||
|
Michael F. Tweedle, Ph.D. (2)
|
60
|
Director (term expiring at 2012 annual meeting or upon his successor being duly elected and qualified)
|
|
|
(1)
|
Member of the audit committee.
|
|
|
(2)
|
Member of the compensation committee
|
|
|
(3)
|
Member of the nominating and corporate governance committee
|
39
Our executive officers are appointed by, and serve at the discretion of, our board of directors.
Stephen A. Hill. Dr. Hill was elected the chairman of the board of directors of Novelos in September 2007. Dr. Hill has been the President and CEO of 21CB since March 2011. 21CB is a nonprofit initiative of UPMC designed to provide the United States government with a domestic solution for its biodefense and infectious disease biologics portfolio. Dr. Hill served as the President and Chief Executive Officer of Solvay Pharmaceuticals, Inc. since April 2008 until its acquisition by Abbott Laboratories in 2010. Prior to joining Solvay, Dr. Hill had served as ArQule's President and Chief Executive Officer since April 1999. Prior to his tenure at ArQule, Dr. Hill was the Head of Global Drug Development at F. Hoffmann-La Roche Ltd. from 1997 to 1999. Dr. Hill joined Roche in 1989 as Medical Adviser to Roche Products in the United Kingdom. He held several senior positions at Roche, including Medical Director where he was responsible for clinical trials of compounds across a broad range of therapeutic areas, including CNS, HIV, cardiovascular, metabolic and oncology products. Subsequently, he served as Head of International Drug Regulatory Affairs at Roche headquarters in Basel, Switzerland, where he led the regulatory submissions for seven major new chemical entities. Dr. Hill also was a member of Roche's Portfolio Management, Research, Development and Pharmaceutical Division Executive Boards. Prior to Roche, Dr. Hill served seven years with the National Health Service in the United Kingdom in General and Orthopedic Surgery. Dr. Hill is a Fellow of the Royal College of Surgeons of England and holds his scientific and medical degrees from St. Catherine's College at Oxford University. Dr. Hill’s extensive experience in a broad range of senior management positions with companies in the life sciences sector make him a highly qualified member of our board of directors.
Harry S. Palmin. Mr. Palmin has served as our president and a director since 1998 and our chief executive officer since January 2005. From 1998 to September 2005, he served as our acting chief financial officer. From 1996 to 1998, he was a vice president at Lehman Brothers and from 1993 to 1996, he was an associate at Morgan Stanley & Co. Mr. Palmin earned a B.A. in economics and business and a M.A. in international economics and finance from the International Business School at Brandeis University. He has also studied at the London School of Economics and the Copenhagen Business School. Mr. Palmin’s experience managing the funding and development of our product candidates for 13 years and his knowledge of capital markets are strong qualifications to serve on our board of directors.
Kimberly A. Hawkins. Ms. Hawkins has served as our vice president of clinical development since November 2010 and served as our director of clinical development since May 2006. She has worked for 17 years in the biopharmaceutical industry managing and overseeing clinical operations for multiple global Phase 1, 2 and 3 clinical studies. From 2001 to 2006, Ms. Hawkins was a senior manager in clinical development at Antigenics, Inc., a cancer biotechnology company where she managed multiple Phase 1 and 2 studies. From 1994 to 2001 she was employed by Genzyme Corporation, Center for Clinical Research Practice where she held the positions of clinical research associate, trainer of good clinical practice and study coordinator. From 1993 to 1994 she held the position of clinical research coordinator at Boston Medical Center. Ms. Hawkins has a B.S. degree in Human Physiology from Boston University and a Masters Degree in Public Health from Boston University School of Public Health.
Christopher J. Pazoles. Dr. Pazoles has served as our vice president of research and development since July 2005. He has 30 years of biopharmaceutical research and development and senior management experience. From May 2004 to June 2005, he held a senior research and development position at the Abbott Bioresearch Center, a division of Abbott Laboratories. From October 2002 to January 2004, he served as chief operating officer and head of research and development at ALS Therapy Development Foundation. From 1994 to October 2002, Dr. Pazoles served as vice president of research for Phytera, Inc. From 1981 to 1994, he served as a researcher and senior manager with Pfizer. Dr. Pazoles holds a Ph.D. in microbiology from the University of Notre Dame.
Joanne M. Protano. Ms. Protano was appointed our vice president, chief financial and accounting officer, and treasurer in December 2007. She has 20 years of finance and senior management experience. She previously held the position of Senior Director of Finance and Controller of the Company from June 2006 to December 2007. From 1996 to 2006, she held various management and senior management positions with Ascential Software, Inc. and predecessor companies including Assistant Controller, Reporting for Ascential Software, Vice President and Chief Financial Officer for the Ascential Software Division of Informix Software, Inc. and Corporate Controller of Ardent Software, Inc. Prior to her tenure in the technology industry, from 1990 to 1996 she was employed by Deloitte and Touche LLP as an audit manager, serving technology and healthcare clients. Ms. Protano received a B.S. in business administration from Bryant College.
Jamey P. Weichert. Dr. Weichert was the primary founder of Cellectar serving as Cellectar’s Chairman and Chief Scientific Officer since 2002. He was appointed as the Chief Scientific Officer and a director of Novelos at the time of the Acquisition. Dr. Weichert is an Associate Professor of the Departments of Radiology, Medical Physics, Pharmaceutics and member of the Comprehensive Cancer Center at the University of Wisconsin, Madison. He has a bachelors degree in chemistry from the University of Minnesota and a doctorate in medicinal chemistry from U. Mich.. His research interests include the design, synthesis and evaluation of biomimetic CT and MRI imaging agents and diapeutic radiopharmaceuticals. He has been involved in molecularly targeted imaging agent development his entire professional career and has developed or co-developed several imaging agents nearing clinical trial status. Dr. Weichert serves or has served on the editorial boards of numerous scientific journals and has authored more than 40 peer reviewed publications and 150 abstracts. He also has 20 issued or pending patents related to drug delivery, imaging and contrast agent development. Dr. Weichert’s experience founding and managing the development of Cellectar’s product candidates and his knowledge of radiation technology are strong qualifications to serve on our board of directors.
40
Thomas Rockwell Mackie. Dr. Mackie became a director of the Company at the time of the Acquisition. He served as a director of Cellectar since December 2006. In 1997, he co-founded TomoTherapy Incorporated, a maker of advanced radiation therapy solutions for the treatment of cancer and other diseases and served as Chairman of its board of directors from 1999 until its Acquisition by Accuray Incorporated in June 2011. Dr. Mackie also served as President of TomoTherapy Inc. from 1997 until 1999 and as Treasurer from 1997 until 2000. Since 1987, Dr. Mackie has been a professor in the departments of Medical Physics and Human Oncology at the University of Wisconsin, where he established the TomoTherapy research program. Dr. Mackie also co-founded Geometrics Corporation (now merged with ADAC Corp.), which developed a radiotherapy treatment planning system. Dr. Mackie currently serves as a director of Shine Medical Technologies and Bioionix Inc. and served on the management committee of Wisconsin Investment Partners from 2006 to 2009. Dr. Mackie has a B.Sc. in Physics from the University of Saskatchewan and a Ph.D. in Physics from the University of Alberta in Edmonton. Dr. Mackie’s qualifications to serve on our board of directors include his extensive senior management experience with radiation technology companies.
James S. Manuso. Dr. Manuso has served as one of our directors since August 2007. Since January 2005, Dr. Manuso has served as Chairman and Chief Executive Officer of SuperGen, Inc. (now renamed Astex Pharmaceuticals, Inc.), served as President of SuperGen from January 2005 through July 2011 and has served as a director of SuperGen since February 2001. Dr. Manuso is co-founder and former president and chief executive officer of Galenica Pharmaceuticals, Inc. Dr. Manuso co-founded and was general partner of PrimeTech Partners, a biotechnology venture management partnership, from 1998 to 2002, and Managing General Partner of The Channel Group LLC, an international life sciences corporate advisory firm. He was also president of Manuso, Alexander & Associates, Inc., management consultants and financial advisors to pharmaceutical and biotechnology companies. Dr. Manuso was a vice president and Director of Health Care Planning and Development for The Equitable Companies (now Group Axa), where he also served as Acting Medical Director. He currently serves on the board of directors of the Biotechnology Industry Organization (BIO) and its Health Section Governing Board and serves on the board of privately-held KineMed, Inc. He previously served on the boards of Merrion Pharmaceuticals Ltd. (Dublin, Ireland) Inflazyme Pharmaceuticals, Inc. (Vancouver, Canada), Symbiontics, Inc., (ZyStor, Inc., sold to BioMarin), Quark Biotech, Inc., Galenica Pharmaceuticals, Inc., Supratek Pharma, Inc., and EuroGen, Ltd. (London, UK). Dr. Manuso earned a B.A. in economics and chemistry from New York University, a Ph.D. in experimental psychophysiology from the Graduate Faculty of The New School University, a certificate in health systems management from Harvard Business School, and an executive M.B.A. from Columbia Business School. Dr. Manuso’s experience founding, leading and serving as a director for pharmaceutical companies makes him a highly qualified member of our board of directors.
John Neis. Mr. Neis became a director of our Company at the time of the Acquisition. He served as director of Cellectar since February 2008. Mr. Neis has been Managing Director of Venture Investors LLC since 1986 and heads the firm’s Healthcare practice. He has over 23 years in the venture capital industry and serves on the Board of Directors of companies from formation through initial public offering or sale. Mr. Neis also currently serves on the boards of directors of Virent Energy Systems, Deltanoid Pharmaceuticals, Inviragen, Inc. and Mithridion, Inc. He is a former member of the Boards of Directors of several firms including TomoTherapy, Third Wave Technologies (acquired by Hologic) and NimbleGen Systems (acquired by Roche). Mr. Neis was appointed to the Board of the Wisconsin Technology Council and he also serves on the advisory boards for the Weinert Applied Ventures Program, the University of Wisconsin, Madison Business School and Tandem Press. Mr. Neis has a B.S. in Finance from the University of Utah, and a M.S. in Marketing and Finance from the University of Wisconsin, Madison. He is a Chartered Financial Analyst. Mr. Neis’ extensive experience leading emerging companies make him a highly qualified member of our board of directors.
John E. Niederhuber. Dr. Niederhuber became a director of our Company at the time of the Acquisition. Dr. Niederhuber served as Director of the National Cancer Institute (NCI) from 2005 to 2010. He has also served as NCI's Chief Operating Officer and Deputy Director for Translational and Clinical Sciences. Dr. Niederhuber served as Chair of the National Cancer Advisory Board (NCAB) from 2002 to 2004. In addition to his management and advisory roles, Dr. Niederhuber has remained involved in research, through his laboratory on the National Institutes of Health (NIH) campus. Under his leadership, the Tumor and Stem Cell Biology Section, which is a part of the Cell and Cancer Biology Branch of NCI's Center for Cancer Research, is studying tissue stem cells as the cell-of-origin for cancer. Dr. Niederhuber also holds a clinical appointment on the NIH Clinical Center Medical Staff. As a surgeon, Dr. Niederhuber's clinical emphasis is on gastrointestinal cancer, hepatobiliary (liver, bile duct, and gall bladder) cancer, and breast cancer. He is recognized for his pioneering work in hepatic artery infusion chemotherapy and was the first to demonstrate the feasibility of totally implantable vascular access devices. Dr. Niederhuber is a graduate of Bethany College in West Virginia and the Ohio State University School of Medicine. He was an NIH Academic Trainee in Surgery at the University of Michigan from 1969 to 1970 and was a Visiting Fellow in the Division of Immunology at The Karolinska Institute in Stockholm, Sweden from 1970 to 1971. He completed his training in surgery at the University of Michigan in 1973 and was a member of the faculty of the University of Michigan from 1973 to 1987, being promoted to Professor of Microbiology/Immunology and Professor of Surgery in 1980. During 1986 and 1987, he was Visiting Professor in the Department of Molecular Biology and Genetics at The Johns Hopkins University School of Medicine in Baltimore, MD. Dr. Niederhuber’s qualifications to serve on our board of directors include his extensive experience with cancer research.
41
Howard M. Schneider. Mr. Schneider has served as one of our directors since February 2005. Mr. Schneider is currently retired. From January to December 2003, he served as chief executive officer of Metrosoft, Inc., and had been an advisor to such company from July to December 2002. From May 2000 to May 2001, he served as president of Wofex Brokerage, Inc. and from 1965 to 1999, he served as an executive at Bankers Trust Company holding a variety of positions in the commercial banking and investment banking businesses. Mr. Schneider received a B.A. in economics from Harvard College and a M.B.A. from New York University. Mr. Schneider’s extensive senior management experience in the financial sector makes him a highly qualified member of our board of directors.
Michael F. Tweedle. Dr. Tweedle became a director of Novelos at the time of the Acquisition. He is currently Professor and Stefanie Spielman Chair in Cancer Imaging in Radiology and the James Comprehensive Cancer Center of Ohio State University, Director of the Wright Center Molecular Imaging (MI) Agents Laboratory of Ohio State University, and has an adjunct appointment in the Chemistry Department of Ohio State University. Prior to joining Ohio State University, his academic appointments included Adjunct Associate Professor at University of Pennsylvania and the Science Advisory Board of New York University. Dr. Tweedle was the President of Bracco Research USA Inc. from 1995 to 2009 where he was the lead scientist and chief executive for creation of new molecular imaging pharmaceuticals. His industrial experience in drug discovery research also includes appointments at the Diagnostics Drug Discovery Division at Bristol-Myers Squibb, New England Nuclear, NEN/DuPont Pharmaceuticals, and The Squibb Institute for Medical Research. He has invented and led translational development of diagnostic imaging pharmaceuticals for nuclear medicine, one of the first Gd-based MRI agents (ProHance TM ), X ray, Optical and US agents, and a radiotheranostic. In 2005 he won the Harry Fisher Medal. Dr. Tweedle holds a B.A from Knox College, B.A. (1973), a Ph.D. from Rice University (1978) and was a Stanford University NRS Fellow. Dr. Tweedle’s qualifications to serve on our board of directors include his extensive experience with radiation and cancer research and drug discovery.
Code of Ethics
The board of directors has adopted a Code of Ethics applicable to all of our directors, officers and employees, including our principal executive officer, principal financial officer and principal accounting officer. A copy of the Code of Ethics is available at our website www.novelos.com.
Compensation of Directors and Executive Officers
Summary Compensation: The following table sets forth certain information about the compensation we paid or accrued with respect to our principal executive officer and our two most highly compensated executive officers (other than our chief executive officer) who served as executive officers during the year ended December 31, 2010 and whose annual compensation exceeded $100,000 for that year.
Other annual compensation in the form of perquisites and other personal benefits has been omitted as the aggregate amount of those perquisites and other personal benefits was less than $10,000 for each person listed.
|
Name and Principal Position
|
Year
|
Salary
($)
|
Bonus
($)
(4)
|
Option
Awards ($)
(5)
|
Total ($)
|
|||||||||||||
|
Harry S. Palmin (1)
|
2010
|
$ | 270,000 | $ | 0 | $ | 0 | $ | 270,000 | |||||||||
|
President, Chief Executive Officer
|
2009
|
$ | 270,000 | $ | 40,500 | $ | 131,650 | $ | 442,150 | |||||||||
|
Christopher J. Pazoles (2)
|
2010
|
$ | 235,000 | $ | 39,167 | $ | 0 | $ | 274,167 | |||||||||
|
Vice President of Research and Development
|
2009
|
$ | 235,000 | $ | 35,250 | $ | 105,320 | $ | 375,570 | |||||||||
|
Elias B. Nyberg (3)
|
2010
|
$ | 225,000 | $ | 37,500 | $ | 0 | $ | 262,500 | |||||||||
|
Vice President of Regulatory, Quality and Compliance
|
2009
|
$ | 225,000 | $ | 33,750 | $ | 78,990 | $ | 337,740 | |||||||||
|
|
(1)
|
There has been no increase to Mr. Palmin’s annual salary for 2011. On May 18, 2011, Mr. Palmin was granted an option to purchase 1,340,400 shares of common stock at an exercise price of $1.40 per share, which option will vest with respect to: 670,200 such shares in equal quarterly installments over a four-year period; 167,550 such shares upon the closing of one or more financings with total gross proceeds of at least $10 million before December 31, 2011; 167,550 such shares upon the closing of one or more financings with total gross proceeds of at least $20 million before December 31, 2012; 167,550 such shares upon the availability of proof of concept data in man for LIGHT by December 31, 2011; and 167,550 such shares upon the initiation of a Phase 2a clinical trial for HOT by August 31, 2012.
|
|
|
(2)
|
On May 18, 2011, Dr. Pazoles’ annual salary was increased to $250,000 and he was granted an option to purchase 200,000 shares of common stock at an exercise price of $1.40 per share, which option will vest in equal quarterly installments over a three-year period.
|
|
|
(3)
|
On March 10, 2011, the Company terminated Dr. Nyberg’s employment. In connection with that termination, which was without cause, Dr. Nyberg received a payment of approximately $83,000 pursuant to the terms of the executive retention agreement between him and the Company dated May 14, 2010.
|
42
|
|
(4)
|
Bonus amounts for 2009 were paid in 2010. Bonus amounts for Dr. Pazoles and Dr. Nyberg in 2010 represent retention bonuses paid as of October 1, 2010 pursuant to their respective retention agreements dated May 14, 2010.
|
|
|
(5)
|
The fair value of each stock award was estimated on the grant date using the Black-Scholes option-pricing model. See Note 9 to the financial statements for a description of the assumptions used in estimating the fair value of stock options. There were no option grants during 2010.
|
Employment Agreements
On January 31, 2006, we entered into an employment agreement with Harry Palmin effective January 1, 2006, whereby he agreed to serve as our president and chief executive officer for an initial term of two years at an annual salary of $225,000. The agreement is automatically renewed for successive one-year terms unless notice of termination is provided by either party at least 90 days prior to the end of such term. The agreement was renewed for an additional one-year term on January 1, 2011 in accordance with its terms. On December 17, 2007, the Board of Directors approved an increase in Mr. Palmin’s annual salary to $270,000 effective January 1, 2008. He is eligible to receive an annual cash bonus at the discretion of the compensation committee and he is entitled to participate in our employee fringe benefit plans or programs generally available to our senior executives. The agreement provides that in the event that we terminate Mr. Palmin without cause or he resigns for good reason (as defined below), we will (i) pay Mr. Palmin his pro rata share of the average of his annual bonus paid during the two fiscal years preceding his termination; (ii) pay Mr. Palmin his base salary for 11 months after the date of termination; (iii) continue to provide him benefits for 11 months after the date of termination; and (iv) fifty percent of his unvested stock options will vest. The agreement provides for the vesting of unvested options upon a Change of Control, defined as the sale of all or substantially all of the assets or issued and outstanding capital stock of the Company, (ii) merger or consolidation involving the Company in which stockholders of the Company immediately before such merger or consolidation do not own immediately after such merger or consolidation capital stock or other equity interests of the surviving corporation or entity representing more than fifty percent (50%) in voting power of capital stock or other equity interests of such surviving corporation or entity outstanding immediately after such merger or consolidation, or (iii) a change, without the approval of the board of directors, in the composition of the board of directors such that directors who were serving as of the date of the agreement cease to constitute a majority of the board of directors. The agreement also contains a non-compete provision, which prohibits Mr. Palmin from competing with us for one year after termination of his employment with us.
“Cause” means (i) gross neglect of duties for which employed; (ii) committing fraud, misappropriation or embezzlement in the performance of duties as our employee; (iii) conviction or guilty or nolo plea of a felony or misdemeanor involving moral turpitude; or (iv) willfully engaging in conduct materially injurious to us or violating a covenant contained in the employment agreement.
“Good Reason” means (i) the failure of our board of directors to elect Mr. Palmin to the offices of president and chief executive officer; (ii) the failure by our stockholders to continue to elect Mr. Palmin to our board of directors; (iii) our failure to pay Mr. Palmin the compensation provided for in the employment agreement, except for across-the-board cuts applicable to all of our officers on an equal percentage basis, provided that such reduction is approved by our board of directors; (iv) relocation of Mr. Palmin’s principal place of employment to a location beyond 50 miles of Newton, Massachusetts; (v) a reduction of base salary or material reduction in other benefits or any material change by us to Mr. Palmin’s function, duties, authority, or responsibilities, which change would cause Mr. Palmin’s position with us to become one of lesser responsibility, importance, or scope; and (vi) our material breach of any of the other provisions of the employment agreement.
On June 1, 2011, the employment agreement between the Company and Harry Palmin dated January 31, 2006 was amended to remove the obligation of the Company to continue to pay Mr. Palmin’s salary and benefits for a period of 11 months following termination by the Company without Cause or termination by Mr. Palmin with Good Reason. The Company may elect that the obligation of Mr. Palmin not to compete with the Company survive for a period of one year from his termination, provided however that Mr. Palmin would continue to receive his base salary during that one-year noncompetition period.
We have entered into retention agreements with each of our four vice presidents. The agreements provide for the lump-sum payment of six months’ base salary and benefits to each such officer following a termination without cause or a resignation with good reason occurring on or before November 14, 2011. Certain of the agreements provide that if the executives were employed by us as of October 1, 2010, they would receive a payment of two months’ base salary as a retention bonus on that date. The retention bonus was paid in October 2010 and will be deducted from the severance amounts that may become payable upon a subsequent involuntary termination. The total remaining amount that may become payable to our Named Executive Officers pursuant to the retention agreements is approximately $86,000 to Christopher Pazoles.
43
Outstanding Equity Awards at Fiscal Year-End
The following table sets forth certain information regarding stock options held as of December 31, 2010 by the executive officers named in the summary compensation table and gives effect to the April Reverse Split. There were no option grants during 2010.
|
Individual Grants
|
||||||||||||||||
|
Name
|
Year
of Grant
|
Number of
securities
underlying
unexercised
options
(#
exercisable)
|
Number of
securities
underlying
unexercised
options
(#
unexercisable)
|
Exercise or
base price
($/share)
|
Expiration
date
|
|||||||||||
|
Harry S. Palmin
|
2009
|
(1) | 544 | 1,089 | $ | 114.75 |
12/8/2019
|
|||||||||
|
2008
|
(2) | 1,743 | 871 | 65.79 |
12/15/2018
|
|||||||||||
|
2007
|
(2) | 1,307 | — | 68.85 |
12/17/2017
|
|||||||||||
|
2006
|
(2) | 980 | — | 139.23 |
12/11/2016
|
|||||||||||
|
2005
|
(3) | 1,633 | — | 1.53 |
1/31/2015
|
|||||||||||
|
2005
|
(3) | 980 | — | 1.53 |
3/31/2015
|
|||||||||||
|
2004
|
(4) | 2,156 | — | 1.53 |
4/1/2014
|
|||||||||||
|
2003
|
(5) | 46 | — | 107.10 |
8/1/2013
|
|||||||||||
|
Christopher J. Pazoles
|
2009
|
(1) | 435 | 872 | $ | 114.75 |
12/8/2019
|
|||||||||
|
2008
|
(2) | 872 | 435 | 65.79 |
12/15/2018
|
|||||||||||
|
2007
|
(2) | 816 | — | 68.85 |
12/17/2017
|
|||||||||||
|
2006
|
(2) | 653 | — | 139.23 |
12/11/2016
|
|||||||||||
|
2005
|
(6) | 653 | — | 1.53 |
4/8/2015
|
|||||||||||
|
Elias B. Nyberg
|
2009
|
(1) | 326 | 654 | $ | 114.75 |
12/8/2019
|
|||||||||
|
2008
|
(2) | 436 | 217 | 65.79 |
12/15/2018
|
|||||||||||
|
2008
|
(7) | 653 | — | 88.74 |
4/1/2018
|
|||||||||||
|
(1)
|
These shares vest quarterly in increments of one-twelfth over three years from the date of grant. The exercise price equals the closing price on the date of grant.
|
|
(2)
|
These shares vest annually in increments of one-third over three years from the date of grant. The exercise price equals the closing price on the date of grant.
|
|
(3)
|
These shares initially vested over a two-year period. Pursuant to their terms, the shares fully vested upon the completion of a non-bridge loan financing, which occurred in the second quarter of 2005. The exercise price equals the fair market value of our common stock on the date of grant as determined by our board of directors.
|
|
(4)
|
These shares initially vested one-third upon grant and one-third annually over the following two years. Pursuant to their terms, one additional year of vesting occurred upon the completion of a non-bridge loan financing, which occurred in the second quarter of 2005. The exercise price equals the fair market value of our common stock on the date of grant as determined by our board of directors.
|
|
(5)
|
These shares vest annually in increments of one-third over three years from the date of grant. The exercise price equals the fair market value of our common stock on the date of grant as determined by our board of directors.
|
|
(6)
|
These shares vested in increments of one-fourth every six months over two years from the date of grant. The exercise price equals the fair market value of our common stock on the date of grant as determined by our board of directors.
|
|
(7)
|
These shares were fully vested upon grant. The exercise price equals the closing price on the date of grant.
|
Options granted pursuant to the 2006 Stock Incentive Plan will become fully vested upon a termination event within one year following a change in control, as defined. A termination event is defined as either termination of employment other than for cause or constructive termination resulting from a significant reduction in either the nature or scope of duties and responsibilities, a reduction in compensation or a required relocation.
Director Compensation
Summary Compensation: The following table sets forth certain information about the compensation we paid or accrued with respect to our directors who served during the year ended December 31, 2010.
44
|
Name and Principal Position
|
Year
|
Director
Fees
($) (3)
|
Total ($)
|
|||||||
|
Stephen A. Hill, Chairman (1)
|
2010
|
$ | 39,500 | $ | 39,500 | |||||
|
Michael J. Doyle, Director (1)(2)
|
2010
|
33,250 | 33,250 | |||||||
|
Sim Fass, Director (1)(2)
|
2010
|
32,500 | 32,500 | |||||||
|
James S. Manuso, Director (1)
|
2010
|
23,000 | 23,000 | |||||||
|
David B. McWilliams, Director (1)(2)
|
2010
|
24,500 | 24,500 | |||||||
|
Howard M. Schneider, Director (1)
|
2010
|
39,000 | 39,000 | |||||||
|
(1)
|
As of December 31, 2010, outstanding options to purchase common stock held by directors were as follows: Dr. Hill 2,287; Mr. Doyle 2,287; Dr. Fass 2,287; Dr. Manuso 1,960; Mr. McWilliams 2,643; Mr. Schneider 1,633.
|
|
(2)
|
In connection with the Acquisition, Mr. Doyle, Dr. Fass and Mr. McWilliams resigned from the board of directors.
|
|
(3)
|
Director fees include all fees earned for director services including quarterly fees, meeting fees and committee chairman fees.
|
|
(4)
|
There were no option grants during 2010.
|
During 2010, we paid our non-employee directors a cash fee of $5,000 per quarter. The non-employee directors also received a fee of $1,500 for any board or committee meeting attended and $750 for each telephonic board or committee meeting in which the director participated. We also paid our chairman an additional annual fee in the amount of $15,000, our non-employee director who serves as the chair of the audit committee an additional annual fee of $10,000 and our non-employee directors who served as the chairman of the compensation and the nominating and corporate governance committees an additional annual fee of $5,000. We reimbursed directors for reasonable out-of-pocket expenses incurred in attending board and committee meetings and undertaking certain matters on our behalf. Directors who are our employees do not receive separate fees for their services as directors. There has been no change to cash fees payable to non-employee directors for 2011. On May 18, 2011 options to purchase 150,000 shares of common stock at $1.40 per share, vesting quarterly over two years were granted to our chairman Stephen Hill. On that same date, options purchase 100,000 shares of common stock at $1.40 per year, vesting quarterly over two years were granted to each of the six non-employee directors other than the chairman.
SECURITY OWNERSHIP OF CERTAIN
BENEFICIAL OWNERS AND MANAGEMENT
At the close of business on September 9, 2011, there were 26,826,157 shares of our common stock outstanding. The following table provides information regarding beneficial ownership of our common stock as of September 9, 2011:
|
|
·
|
Each person known by us to be the beneficial owner of more than five percent of our common stock;
|
|
|
·
|
Each of our directors;
|
|
|
·
|
Each executive officer named in the summary compensation table; and
|
|
|
·
|
All of our current directors and executive officers as a group.
|
The address of each executive officer and director is c/o Novelos Therapeutics, Inc., One Gateway Center, Suite 504, Newton, Massachusetts 02458. The persons named in this table have sole voting and investment power with respect to the shares listed, except as otherwise indicated. The inclusion of shares listed as beneficially owned does not constitute an admission of beneficial ownership. Shares included in the “Right to Acquire” column consist of shares that may be purchased through the exercise of options or warrants that are exercisable within 60 days of September 9, 2011.
45
|
Shares Beneficially Owned
|
||||||||||||||||
|
Name and Address of Beneficial Owner
|
Outstanding
|
Right to Acquire
|
Total
|
Percentage
|
||||||||||||
|
Venture Investors LLC (1) (2)
University Technology Park
505 S. Rosa Road; Suite 201
Madison, WI 53719
|
4,534,308 | 2,000,000 | 6,534,308 | 22.7 | ||||||||||||
|
Jamey P. Weichert (3)
c/o Cellectar Inc.
3301 Agriculture Drive
Madison, WI 53716
|
4,706,730 | 11,333 | 4,718,063 | 17.6 | ||||||||||||
|
MEG-Cellectar II, LLC (2) (4)
3001 West Beltline Highway, Suite 202
Madison, WI 53713
|
2,150,401 | 160,000 | 2,310,401 | 8.6 | ||||||||||||
|
Greenway Properties Inc. (2)(5)
725 Heartland Trail, Suite 102
Madison, WI 53707
|
1,337,400 | 1,000,000 | 2,337,400 | 8.4 | ||||||||||||
|
Continuum Investment Limited Partnership (6)
P.O. Box 620557
Middleton, WI 53562
|
1,808,524 | 0 | 1,808,524 | 6.7 | ||||||||||||
|
Harry S. Palmin (7)
|
4,190 | 51,683 | 55,873 | * | ||||||||||||
|
Christopher J. Pazoles
|
0 | 20,422 | 20,422 | * | ||||||||||||
|
Stephen A. Hill
|
0 | 20,969 | 20,969 | * | ||||||||||||
|
Thomas Rockwell Mackie
|
116,122 | 12,500 | 128,622 | * | ||||||||||||
|
James S. Manuso
|
0 | 14,392 | 14,392 | * | ||||||||||||
|
John Neis (1) (8)
|
4,534,308 | 2,012,500 | 6,546,808 | 22.7 | ||||||||||||
|
John E. Neiderhuber
|
0 | 12,500 | 12,500 | * | ||||||||||||
|
Howard M. Schneider
|
654 | 14,063 | 14,717 | * | ||||||||||||
|
Michael F. Tweedle
|
0 | 12,500 | 12,500 | * | ||||||||||||
|
All directors and officers as a group (12 persons)
|
9,362,004 | 2,212,307 | 11,574,311 | 39.9 | ||||||||||||
|
(1)
|
Ownership consists of shares of common stock held by Venture Investors Early Stage Fund IV Limited Partnership and Advantage Capital Wisconsin Partners I, Limited Partnership. VIESF IV GP LLC is the general partner of Venture Investors Early Stage Fund IV Limited Partnership and Venture Investors LLC is the submanager and special limited partner of Advantage Capital Wisconsin Partners I, Limited Partnership. The investment decisions of VIESF IV GP LLC and Venture Investors LLC are made collectively by six managers, including Mr. Neis. Each such manager and Mr. Neis disclaim such beneficial ownership except to the extent of his pecuniary interest therein. The address of Mr. Neis is c/o Venture Investors LLC, 505 South Rosa Road, #201, Madison, Wisconsin 53719.
|
|
(2)
|
Shares in the “Right to Acquire” column consist of warrants to purchase common stock at a price of $0.75, expiring on March 31, 2016.
|
|
(3)
|
Dr. Weichert serves as a director and our Chief Scientific Officer following the Acquisition. The shares beneficially owned by him have been included in the total of directors and officers as a group.
|
|
(4)
|
Ownership consists of shares of common stock held by MEG-Cellectar II, LLC and approximately 184,000 shares owned by Bradley L. Hutter. Mr. Hutter is the managing director of Mortenson Equity Management LLC, which is the manager of MEG-Cellectar II, LLC and has dispositive and voting power over shares held by MEG-Cellectar II, LLC.
|
|
(5)
|
Jeffrey Straubel is the President and principal owner of Greenway Properties, Inc. and has sole dispositive and voting power over shares held by Greenway Properties, Inc.
|
|
(6)
|
Ownership includes shares of common stock held by Cellectar Investor I, LLC. Continuum Investment Limited Partnership is the manager of Cellectar Investor I, LLC. Bruce Neviaser is the president of Continuum Investment Limited Partnership and has sole dispositive and voting power over shares held by Continuum Investment Limited Partnership and Cellectar Investor I, LLC.
|
|
(7)
|
Ownership of H. Palmin includes shares owned by his wife, Deanna Palmin.
|
|
(8)
|
Shares in the “Right to Acquire” column consist of warrants to purchase 2,000,000 shares of common stock at a price of $0.75, expiring on March 31, 2016, held by Venture Investors Early Stage Fund IV Limited Partnership and options to purchase 12,500 shares of common stock at $1.40 per share, held by Mr. Neis.
|
46
We do not have a written policy for the review, approval or ratification of transactions with related parties or conflicted transactions. When such transactions arise they are referred to the Audit Committee for consideration or for referral to the Board of Directors for its consideration.
One of our directors, John Neis, is a managing director of Venture Investors LLC which beneficially owns approximately 23% of our common stock.
Jamey Weichert, our Chief Scientific Officer, director, shareholder and principal founder of Cellectar is a faculty member at the University of Wisconsin-Madison (U. Wisc.). Cellectar paid $16,082 to the U. Wisc. during 2009 for research related activities. No payments were made to UW during the year ended December 31 2010. During the six months ended June 30, 2011, we made contributions totaling $62,500 to the UW Foundation for use towards research activities associated with the development of our compounds.
We are obligated to ZAO BAM, a Russian company engaged in the pharmaceutical business, under a royalty and technology transfer agreement. Mark Balazovsky, a director until November 2006, is the majority shareholder of ZAO BAM. Pursuant to the royalty and technology transfer agreement between Novelos and ZAO BAM, we are required to make royalty payments of 1.2% of net sales of oxidized glutathione-based products. We are also required to pay ZAO BAM $2 million for each new oxidized glutathione-based drug within eighteen months following FDA approval of such drug.
If a royalty is not being paid to ZAO BAM on net sales of oxidized glutathione products, then we are required to pay ZAO BAM 3% of all license revenues. If license revenues exceed our cumulative expenditures including, but not limited to, preclinical and clinical studies, testing, FDA and other regulatory agency submission and approval costs, general and administrative costs, and patent expenses, then we would be required to pay ZAO BAM an additional 9% of the amount by which license revenues exceed our cumulative expenditures. During 2008, we paid ZAO BAM $15,000, which was 3% of the upfront license payment received under the collaboration agreement with Lee’s Pharm, described in Note 5 to the financial statements.
On June 28, 2010, we received a letter from counsel to ZAO BAM and ZAO BAM Research Laboratories (collectively, “BAM”) alleging that we modified the chemical composition of NOV-002 without prior notice to or approval from BAM, constituting a material breach of a technology and assignment agreement we had entered into with BAM on June 20, 2000 (the “June 2000 Agreement”). The letter references our amendment, submitted to the FDA on August 30, 2005, to our investigational new drug application dated August 1999 as the basis for BAM’s claims and demands the transfer of all intellectual property rights concerning NOV-002 to BAM. Mark Balazovsky, a director of Novelos from June 1996 until November 2006 and a shareholder of Novelos through at least June 25, 2010, is, to our knowledge, still the general director and principal shareholder of ZAO BAM. On September 24, 2010, we filed a complaint in Suffolk Superior Court seeking a declaratory judgment by the court that the June 2000 Agreement has been replaced by a subsequent agreement between the parties dated April 1, 2005 (the “April 2005 Agreement”), that the Company’s obligations to BAM are governed solely by the April 2005 Agreement and that the obligations of the June 2000 agreement have been performed and fully satisfied. On November 29, 2010, BAM answered our complaint, denying the material allegations and stating its affirmative defenses and certain counterclaims. On January 14, 2011, we responded to the counterclaims, denying BAM’s material allegations and stating our affirmative defenses. On June 9, 2011, BAM filed an amended counterclaim alleging additional claims related to Novelos’ acquisition of Cellectar. In that amended counterclaim, BAM alleges that the acquisition evidences Novelos’ abandonment of the technology assigned to it by BAM constituting a breach of the June 2000 Agreement or, if that agreement is determined to no longer be in effect, a breach of the April 2005 Agreement and/or a breach of the implied duty of good faith and fair dealing with respect to the April 2005 Agreement. On June 15, 2011 we filed our response to their amended counterclaim. On August 5, 2011, we filed a motion for judgment on the pleadings as to our declaratory judgment count and all counts of BAM’s amended counterclaim. The motion has been opposed by BAM and a hearing on the motion is scheduled on September 27, 2011. We believe BAM’s allegations and counterclaims are without merit and intend to defend vigorously against them.
As a result of the assignment to Novelos of the exclusive worldwide intellectual property and marketing rights of oxidized glutathione (excluding the Russian Territory), Novelos is obligated to the Oxford Group, Ltd., or its assignees, for future royalties. Simyon Palmin, a founder of Novelos, a director until August 15, 2008 and the father of our president and chief executive officer, is president of Oxford Group, Ltd. Mr. Palmin was also an employee of Novelos until September 2008 and performed consulting services to the Company through December 2009. Pursuant to the agreement, as revised May 26, 2005, Novelos is required to pay Oxford Group, Ltd., or its assignees, a royalty in the amount of 0.8% of our net sales of oxidized glutathione-based products.
Director Independence
Each member of the Audit Committee, the Compensation Committee and the Nominating and Corporate Governance Committee and seven of our nine directors meet the independence requirements of the Nasdaq Stock Market for membership on the committees on which he serves. The board of directors considered the information included in transactions with related parties as outlined above along with other information the board considered relevant, when considering the independence of each director. Harry S. Palmin and Jamey P. Weichert are not independent directors.
47
Rodman & Renshaw, LLC is acting as the sole managing underwriter of this offering. Under the terms and subject to the conditions contained in an underwriting agreement dated the date of this prospectus, Rodman & Renshaw, LLC, or the underwriter, has agreed to purchase, and we have agreed to sell to them, all shares offered by this prospectus.
Nature of Underwriting Commitment
The underwriting agreement provides that the underwriter is committed to purchase all shares offered in this offering, other than those covered by the over-allotment option described below, if the underwriter purchases any of these securities. The underwriting agreement provides that the obligations of the underwriter to purchase the shares offered hereby is conditional and may be terminated at its discretion based on its assessment of the state of the financial markets. The obligations of the underwriter may also be terminated upon the occurrence of other events specified in the underwriting agreement. Furthermore, pursuant to the underwriting agreement, the underwriters obligations are subject to various other customary conditions, representations and warranties contained in the underwriting agreement, such as receipt by the underwriter of officers’ certificates and legal opinions of our counsel.
Pricing of Securities
The underwriter has advised us that it proposes to offer the shares directly to the public at the public offering price set forth on the cover page of this prospectus, and to certain dealers that are members of the Financial Industry Regulatory Authority (FINRA), at such price less a concession not in excess of $ per share. The underwriter may allow, and the selected dealers may reallow, a concession not in excess of $ per share to certain brokers and dealers. After this offering, the offering price and concessions and discounts to brokers and dealers and other selling terms may from time to time be changed by the underwriter. These prices should not be considered an indication of the actual value of our shares of common stock and are subject to change as a result of market conditions and other factors. No variation in those terms will change the amount of proceeds to be received by us as set forth on the cover page of this prospectus.
Our common stock is quoted on the OTC Bulletin Board under the symbol “NVLT.OB.” On , 2011, the closing market price of our common stock as quoted on OTC Bulletin Board was . The public offering price for the shares was determined by negotiation between us and the underwriter. The principal factors considered in determining the public offering price of the shares included:
|
|
·
|
the information in this prospectus and otherwise available to the underwriters;
|
|
|
·
|
the history and the prospects for the industry in which we will compete;
|
|
|
·
|
our current financial condition and the prospects for our future cash flows and earnings;
|
|
|
·
|
the general condition of the economy and the securities markets at the time of this offering;
|
|
|
·
|
the recent market prices of, and the demand for, publicly-traded securities of generally comparable companies; and
|
|
|
·
|
the public demand for our securities in this offering
|
We cannot be sure that the public offering price will correspond to the price at which our shares of common stock will trade in the public market following this offering or that an active trading market for our shares of common stock will develop and continue after this offering.
Commissions and Discounts
The following table summarizes the compensation to be paid to the underwriter by us and the proceeds, before expenses, payable to us, assuming a $ offering price. The information assumes either no exercise or full exercise by the underwriter of the over-allotment option.
48
|
Total
|
||||||||||||
|
Per Share
|
Without
Over-Allotment
|
With
Over-Allotment
|
||||||||||
|
Public offering price
|
$ | $ | $ | |||||||||
|
Underwriting discount (7%) (1)
|
$ | $ | $ | |||||||||
|
Non-accountable expense allowance (1%) (2)
|
$ | $ | $ | |||||||||
|
Proceeds, before expenses, to us (3)
|
$ | $ | $ | |||||||||
|
|
(1)
|
Underwriting discount is $ per share (7% of the price of the shares sold in the offering).
|
|
|
(2)
|
The expense allowance of 1% is not payable with respect to the shares sold upon exercise of the underwriter’s over-allotment option. Includes $50,000 which was previously paid to the underwriter as an advance.
|
|
|
(3)
|
We estimate that the total expenses of this offering, excluding the underwriter’s discount and the non-accountable expense allowance are approximately $ .
|
We have granted the underwriter an option, exercisable for 45 days after the closing date of this offering, to purchase up to 15% of the shares sold in the offering ( additional shares) solely to cover over-allotments, if any, at the same price as the initial shares offered. If the underwriter fully exercises the over-allotment option, the total public offering price, underwriting discount and expenses and net proceeds (before expenses) to us will be $ , $ , and $ respectively.
Lock-ups
All of our directors and executive officers and our significant stockholders will enter into lock-up agreements that prevent them from selling any shares of our common stock or any securities convertible into or exercisable or exchangeable for shares of our common stock, subject to certain exceptions, for a period of not less than six months from the date of this prospectus without the prior written consent of the underwriter. The underwriter may in its sole discretion and at any time without notice release some or all of the shares subject to lock-up agreements prior to the expiration of the lock-up period. When determining whether or not to release shares from the lock-up agreements, the underwriter will consider, among other factors, the stockholder’s reasons for requesting the release, the number of shares for which the release is being requested and market conditions at the time.
Underwriter’s Warrant
We have also agreed to issue to the underwriter, a warrant to purchase a number of shares equal to 5% of the shares of common stock sold (excluding the over-allotment). The shares issuable upon exercise of this warrant are identical to those offered by this prospectus. This warrant is exercisable at $ per share (125% of the price of the shares sold in this offering), at any time after the one-year anniversary of the effective date of the offering and no more than five years from the effective date of the offering in compliance with Rule 5110(f)(2)(H)(i) of FINRA. The warrant may also be exercised on a cashless basis. The warrant and the shares of common stock underlying the warrant have been deemed compensation by the FINRA and are therefore subject to a 180-day lock-up pursuant to Rule 5110(g)(1) of FINRA. The underwriter (or permitted assignees under the Rule) will not sell, transfer, assign, pledge, or hypothecate this warrant or the securities underlying this warrant, except to any underwriter or selected dealer participating in the offering or their bona fide officers or partners, nor will the underwriter engage in any hedging, short sale, derivative, put, or call transaction that would result in the effective economic disposition of this warrant or the underlying securities for a period of 180 days from the effective date of the offering, except as provided in Rule 5110(g)(2) of FINRA. The warrant provides for unlimited “piggy back” registration rights at any time after the one-year anniversary of the effective date of the offering and no more than five years from the effective date of the offering in compliance with Rule 5110(f)(2)(H)(i) of FINRA. These rights apply to all of the securities directly and indirectly issuable upon exercise of the warrant. We will bear all fees and expenses attendant to registering the securities issuable on exercise of the warrant, other than underwriting commissions incurred and payable by the holders. The exercise price and number of shares issuable upon exercise of the warrant may be adjusted in certain circumstances including in the event of a stock dividend, extraordinary cash dividend or our recapitalization, reorganization, merger or consolidation. However, the warrant exercise price or underlying shares will not be adjusted for issuances of common stock at a price below the warrant exercise price.
Right of First Refusal
We have granted to the underwriter a right of first refusal to purchase for its account or to sell for our account, or any subsidiary or successor, any securities of the Company or any such subsidiary or successor which we or any subsidiary or successor may seek to sell in public or private equity and public debt offerings whether with or without or through an underwriter, placement agent or broker-dealer.
We may, however, in lieu of granting a right of first refusal, designate the underwriter as lead underwriter or co-manager of any underwriting group or co-placement agent of any proposed financing, and the underwriter shall be entitled to receive as its compensation 50% of the compensation payable to the underwriting or placement agent group when serving as co-manager or co-placement agent and 33% of the compensation payable to the underwriting or placement agent group when serving as co-manager or co-placement agent with respect to a proposed financing in which there are three co-managing or lead underwriters or co-placement agents.
49
In connection with this offering, the underwriter or certain of the securities dealers may distribute prospectuses electronically. No forms of prospectus other than printed prospectuses and electronically distributed prospectuses that are printable in Adobe PDF format will be used in connection with this offering.
The underwriter previously acted as placement agent for the private placement which closed simultaneously with the Acquisition. For such services, the underwriter was paid a cash fee of $200,000 and we issued to the underwriter warrants to purchase 192,931 shares of our common stock. The cash fee was used to acquire 266,667 shares of our common stock and a warrant to purchase 266,667 shares of our common stock upon the same terms as other purchasers in the private placement. These shares and warrants are subject to a 180-day lock-up pursuant to Rule 5110(g)(1) of FINRA.
In connection with the offering we are applying to have our common stock approved for listing on the NASDAQ Capital Market under the symbol “NVLT”.
The underwriter has informed us that it does not expect to confirm sales of shares offered by this prospectus to accounts over which they exercise discretionary authority without obtaining the specific approval of the account holder.
Stabilization
Until the distribution of the shares offered by this prospectus is completed, rules of the SEC may limit the ability of the underwriter to bid for and to purchase our securities. As an exception to these rules, the underwriter may engage in transactions effected in accordance with Regulation M under the Securities Exchange Act of 1934 that are intended to stabilize, maintain or otherwise affect the price of our common stock. The underwriter may engage in over-allotment sales, syndicate covering transactions, stabilizing transactions and penalty bids in accordance with Regulation M.
|
|
·
|
Stabilizing transactions permit bids or purchases for the purpose of pegging, fixing or maintaining the price of the common stock, so long as stabilizing bids do not exceed a specified maximum.
|
|
|
·
|
Over-allotment involves sales by the underwriter of shares in excess of the number of shares the underwriter is obligated to purchase, which creates a short position. The short position may be either a covered short position or a naked short position. In a covered short position, the number of shares over-allotted by the underwriter is not greater than the number of shares that it may purchase in the over-allotment option. In a naked short position, the number of shares involved is greater than the number of shares in the over-allotment option. The underwriter may close out any covered short position by either exercising its over-allotment option or purchasing shares in the open market.
|
|
|
·
|
Covering transactions involve the purchase of securities in the open market after the distribution has been completed in order to cover short positions. In determining the source of securities to close out the short position, the underwriter will consider, among other things, the price of securities available for purchase in the open market as compared to the price at which it may purchase securities through the over-allotment option. If the underwriter sells more shares of common stock than could be covered by the over-allotment option, creating a naked short position, the position can only be closed out by buying securities in the open market. A naked short position is more likely to be created if the underwriter is concerned that there could be downward pressure on the price of the securities in the open market after pricing that could adversely affect investors who purchase in this offering.
|
|
|
·
|
Penalty bids permit the underwriter to reclaim a selling concession from a selected dealer when the shares of common stock originally sold by the selected dealer are purchased in a stabilizing or syndicate covering transaction.
|
These stabilizing transactions, covering transactions and penalty bids may have the effect of raising or maintaining the market price of our securities or preventing or retarding a decline in the market price of our securities. As a result, the price of our securities may be higher than the price that might otherwise exist in the open market.
Neither we nor the underwriters make any representation or prediction as to the effect that the transactions described above may have on the prices of our securities. These transactions may occur on the over the counter market or on any other trading market. If any of these transactions are commenced, they may be discontinued without notice at any time.
Foreign Regulatory Restrictions on Purchase of the Shares
We have not taken any action to permit a public offering of shares of our shares outside the United States or to permit the possession or distribution of this prospectus outside the United States. Persons outside the United States who come into possession of this prospectus must inform themselves about and observe any restrictions relating to this offering of shares and the distribution of the prospectus outside the United States.
50
In addition to the public offering of the shares in the United States, the underwriters may, subject to the applicable foreign laws, also offer the shares to certain institutions or accredited persons in the following countries:
Australia . If this document is issued or distributed in Australia it is issued or distributed to “wholesale clients” only, not to “retail clients”. For the purposes of this paragraph, the terms “wholesale client” and “retail client” have the meanings given in section 761 of the Australian Corporations Act 2001 (Cth). This document is not a disclosure document under the Australian Corporations Act, has not been lodged with the Australian Securities & Investments Commission and does not purport to include the information required of a disclosure document under the Australian Corporations Act. Accordingly, (i) the offer of securities under this document is only made to persons to whom it is lawful to offer such securities under one or more exemptions set out in the Australian Corporations Act, (ii) this document is only made available in Australia to those persons referred to in clause (i) above, and (iii) the offeree must be sent a notice stating in substance that, by accepting this offer, the offeree represents that the offeree is such a person as referred to in clause (i) above, and, unless permitted under the Australian Corporations Act, agrees not to sell or offer for sale within Australia any of the securities sold to the offeree within 12 months after its transfer to the offeree under this document.
China . THIS PROSPECTUS HAS NOT BEEN AND WILL NOT BE CIRCULATED OR DISTRIBUTED IN THE PRC, AND ADSs MAY NOT BE OFFERED OR SOLD, AND WILL NOT BE OFFERED OR SOLD TO ANY PERSON FOR RE-OFFERING OR RESALE, DIRECTLY OR INDIRECTLY, TO ANY RESIDENT OF THE PRC EXCEPT PURSUANT TO APPLICABLE LAWS AND REGULATIONS OF THE PRC
DIFC . DIFC and UAE have different requirements and, as a result, a generic legend for each is provided below
UAE . The offering has not been approved or licensed by the Central Bank of the United Arab Emirates (the “UAE”), Securities and Commodities Authority of the UAE and/or any other relevant licensing authority in the UAE including any licensing authority incorporated under the laws and regulations of any of the free zones established and operating in the territory of the UAE, in particular the Dubai Financial Services Authority (the “DFSA”), a regulatory authority of the Dubai International Financial Centre (the “DIFC”).
The offering does not constitute a public offer of securities in the UAE, DIFC and/or any other free zone in accordance with the Commercial Companies Law, Federal Law No.8 of 1984 (as amended), DFSA Offered Securities Rules and NASDAQ Dubai Listing Rules, accordingly, or otherwise. The securities offered hereby may not be offered to the public in the UAE and/or any of the free zones, including, in particular, the DIFC.
The securities offered hereby may be offered and issued only to a limited number of investors in the UAE or any of its free zones (including, in particular, the DIFC) who qualify as sophisticated investors under the relevant laws and regulations of the UAE or the free zone concerned, including, in particular, the DIFC.
The Company represents and warrants that the securities offered hereby will not be offered, sold, transferred or delivered to the public in the UAE or any of its free zones, including, in particular, the DIFC.”
Dubai . The issuer is not licensed by the Dubai Financial Services Authority (“DFSA”) to provide financial services in the Dubai International Financial Centre (“DIFC”). The offering has not been approved or licensed by the Central Bank of the United Arab Emirates (the “UAE”), Securities and Commodities Authority of the UAE and/or any other relevant licensing authority in the UAE including any licensing authority incorporated under the laws and regulations of any of the free zones established and operating in the territory of the UAE, in particular the DFSA, a regulatory of the DIFC.
The offering does not constitute a public offer of securities in the UAE, DIFC and/or any other free zone in accordance with the Commercial Companies Law, Federal Law No.8 of 1984 (as amended), DFSA Offered Securities Rules and NASDAQ Dubai Listing Rules, accordingly, or otherwise. The securities offered hereby may not be offered to the public in the UAE and/or any of the free zones, including, in particular, the DIFC.
The securities offered hereby may be offered and issued only to a limited number of investors in the UAE or any of its free zones (including, in particular, the DIFC) who qualify as sophisticated investors under the relevant laws and regulations of the UAE or the free zone concerned, including, in particular, the DIFC.
The Company represents and warrants that the securities offered hereby will not be offered, sold, transferred or delivered to the public in the UAE or any of its free zones, including, in particular, the DIFC.
Israel . The shares offered by this prospectus have not been approved or disapproved by the Israeli Securities Authority (the ISA), nor have such shares been registered for sale in Israel. The shares may not be offered or sold, directly or indirectly, to the public in Israel, absent the publication of a prospectus. The ISA has not issued permits, approvals or licenses in connection with the offering or publishing the prospectus; nor has it authenticated the details included herein, confirmed their reliability or completeness, or rendered an opinion as to the quality of the common stock being offered. Any resale, directly or indirectly, to the public of the shares offered by this prospectus is subject to restrictions on transferability and must be effected only in compliance with the Israeli securities laws and regulations.
51
Pakistan . The investors / subscribers in Pakistan will be responsible for ensuring their eligibility to invest under the applicable laws of Pakistan and to obtain any regulatory consents if required for such purpose.
Saudi Arabia . NO OFFERING OF SHARES IS BEING MADE IN THE KINGDOM OF SAUDI ARABIA, AND NO AGREEMENT RELATING TO THE SALE OF THE SHARES WILL BE CONCLUDED IN SAUDI ARABIA. THIS DOCUMENT IS PROVIDED AT THE REQUEST OF THE RECIPIENT AND IS BEING FORWARDED TO THE ADDRESS SPECIFIED BY THE RECIPIENT. NEITHER THE AGENT NOR THE OFFERING HAVE BEEN LICENSED BY THE SAUDI’S SECURITIES AND EXCHANGE COMMISSION OR ARE OTHERWISE REGULATED BY THE LAWS OF THE KINGDOM OF SAUDI ARABIA.
THEREFORE, NO SERVICES RELATING TO THE OFFERING, INCLUDING THE RECEIPT OF APPLICATIONS AND/OR THE ALLOTMENT OF THE SHARES, MAY BE RENDERED WITHIN THE KINGDOM BY THE AGENT OR PERSONS REPRESENTING THE OFFERING.
UK . The content of this prospectus has not been issued or approved by an authorised person within the meaning of the United Kingdom Financial Services and Markets Act 2000 (“FSMA”). Reliance on this prospectus for the purpose of engaging in any investment activity may expose an Investor to a significant risk of losing all of the property or other assets invested. This prospectus does not constitute a Prospectus within the meaning of the FSMA and is issued in reliance upon one or more of the exemptions from the need to issue such a prospectus contained in section 86 of the FSMA.
Indemnification
The underwriting agreement provides for indemnification between us and the underwriter against specified liabilities, including liabilities under the Securities Act, and for contribution by us and the underwriter to payments that may be required to be made with respect to those liabilities. We have been advised that, in the opinion of the SEC, indemnification for liabilities under the Securities Act is against public policy as expressed in the Securities Act, and is therefore, unenforceable.
Under our amended and restated certificate of incorporation, our authorized capital stock consists of 150,000,000 shares of common stock, $0.00001 par value per share, and 7,000 shares of preferred stock, $0.00001 par value per share.
Our amended and restated certificate of incorporation authorizes us to issue shares of our preferred stock from time to time in one or more series without stockholder approval. No shares of preferred stock are outstanding,
All outstanding shares of our common stock are duly authorized, validly issued, fully-paid and non-assessable.
Common Stock
Voting. Holders of our common stock are entitled to one vote per share held of record on all matters to be voted upon by our stockholders. Our common stock does not have cumulative voting rights. Persons who hold a majority of the outstanding common stock entitled to vote on the election of directors can elect all of the directors who are eligible for election.
Dividends. Subject to preferences that may be applicable to the holders of any outstanding shares of our preferred stock, the holders of our common stock are entitled to receive such lawful dividends as may be declared by our board of directors.
Liquidation and Dissolution. In the event of our liquidation, dissolution or winding up, and subject to the rights of the holders of any outstanding shares of our preferred stock, the holders of shares of our common stock will be entitled to receive pro rata all of our remaining assets available for distribution to our stockholders.
Other Rights and Restrictions. Our amended and restated certificate of incorporation prohibits us from granting preemptive rights to any of our stockholders. All outstanding shares are fully paid and nonassessable.
Provisions of our charter and our by-laws could make it more difficult to acquire us by means of a merger, tender offer, proxy contest, open market purchases, removal of incumbent directors and otherwise. These provisions, which are summarized below, are expected to discourage types of coercive takeover practices and inadequate takeover bids and to encourage persons seeking to acquire control of us to first negotiate with us. We believe that the benefits of increased protection of our potential ability to negotiate with the proponent of an unfriendly or unsolicited proposal to acquire or restructure us outweigh the disadvantages of discouraging takeover or acquisition proposals because negotiation of these proposals could result in an improvement of their terms.
52
Authorized but Unissued Stock . We have shares of common stock and preferred stock available for future issuance, in some cases, without stockholder approval. We may issue these additional shares for a variety of corporate purposes, including public offerings to raise additional capital, corporate acquisitions, stock dividends on our capital stock or equity compensation plans. The existence of unissued and unreserved common stock and preferred stock may enable our board of directors to issue shares to persons friendly to current management or to issue preferred stock with terms that could render more difficult or discourage a third-party attempt to obtain control of us, thereby protecting the continuity of our management. In addition, if we issue preferred stock, the issuance could adversely affect the voting power of holders of common stock and the likelihood that such holders will receive dividend payments and payments upon liquidation.
Amendments to by-laws . Our certificate of incorporation and by-laws authorize the Board to amend, repeal, alter or rescind the by-laws at any time without stockholder approval. Allowing the Board to amend our by-laws without stockholder approval enhances Board control over our by-laws.
Classification of Board; removal of directors; vacancies. Our certificate of incorporation provide for the division of the Board into three classes as nearly equal in size as possible with staggered three-year terms; that directors may be removed only for cause by the affirmative vote of the holders of two-thirds of our shares of capital stock entitled to vote; and that any vacancy on the Board, however occurring, including a vacancy resulting from an enlargement of the board, may be filled only by the vote of a majority of the directors then in office. The limitations on the removal of directors and the filling of vacancies could have the effect of making it more difficult for a third party to acquire, or of discouraging a third party from acquiring, control of us. Our certificate of incorporation requires the affirmative vote of the holders of at least 75% of our shares of capital stock issued and outstanding and entitled to vote to amend or repeal any of these provisions.
Notice Periods for Stockholder Meetings . Our by-laws provide that for business to be brought by a stockholder before an annual meeting of stockholders, the stockholder must give written notice to the corporation not less than 90 nor more than 120 days prior to the one year anniversary of the date of the annual meeting of stockholders of the previous year; provided, however, that in the event that the annual meeting of stockholders is called for a date that is not within 30 days before or after such anniversary date, notice by the stockholder must be received not later than the close of business on the tenth day following the day on which the corporation's notice of the date of the meeting is first given or made to the stockholders or disclosed to the general public, whichever occurs first.
Stockholder action; special meetings . Our certificate of incorporation provides that stockholder action may not be taken by written action in lieu of a meeting and provides special meetings of the stockholders may only be called by our president or by our Board. These provisions could have the effect of delaying until the next stockholders' meeting stockholder actions that are favored by the holders of a majority of our outstanding voting securities. These provisions may also discourage another person or entity from making a tender offer for our common stock, because that person or entity, even if it acquired a majority of our outstanding voting securities, would be able to take action as a stockholder only at a duly called stockholders' meeting, and not by written consent. Our certificate of incorporation requires the affirmative vote of the holders of at least 75% of our shares of capital stock issued and outstanding and entitled to vote to amend or repeal the provisions relating to prohibition on action by written consent and the calling of a special meeting of stockholders.
Nominations . Our by-laws provide that nominations for election of directors may be made only by (i) the Board or a committee appointed by the Board; or (ii) a stockholder entitled to vote on director election, if the stockholder provides notice to the Secretary of the Corporation presented not less than 90 days nor more than 120 days prior to the anniversary of the last annual meeting (subject to the limited exceptions set forth in the bylaws). These provisions may deter takeovers by requiring that any stockholder wishing to conduct a proxy contest have its position solidified well in advance of the meeting at which directors are to be elected and by providing the incumbent Board with sufficient notice to allow them to put an election strategy in place.
53
DISCLOSURE OF COMMISSION POSITION ON
INDEMNIFICATION FOR SECURITIES ACT LIABILITIES
Our charter contains provisions to indemnify our directors and officers to the maximum extent permitted by Delaware law. We believe that indemnification under our charter covers at least negligence on the part of an indemnified person. Our charter permits us to advance expenses incurred by an indemnified person in connection with the defense of any action or proceeding arising out of the person’s status or service as our director, officer, employee or other agent upon an undertaking by the person to repay those advances if it is ultimately determined that the person is not entitled to indemnification.
Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act of 1933 and is, therefore, unenforceable.
WHERE YOU CAN FIND MORE INFORMATION
We are a reporting company and file annual, quarterly and special reports, and other information with the Securities and Exchange Commission. Copies of the reports and other information may be read and copied at the SEC’s Public Reference Room at 100 F Street NE, Washington, D.C. 20549. You can request copies of such documents by writing to the SEC and paying a fee for the copying cost. You may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC maintains a web site at http://www.sec.gov that contains reports, proxy and information statements and other information regarding registrants that file electronically with the SEC.
This prospectus is part of a registration statement on Form S-1 that we filed with the SEC. Certain information in the registration statement has been omitted from this prospectus in accordance with the rules and regulations of the SEC. We have also filed exhibits and schedules with the registration statement that are excluded from this prospectus. For further information you may:
|
|
·
|
read a copy of the registration statement, including the exhibits and schedules, without charge at the SEC’s Public Reference Room; or
|
|
|
·
|
obtain a copy from the SEC upon payment of the fees prescribed by the SEC.
|
The validity of the securities being offered by this prospectus is being passed upon for us by Foley Hoag LLP, Boston, Massachusetts. Sichenzia Ross Friedman Ference LLP, New York, New York, is acting as counsel to the underwriter in this offering.
EXPERTS
The audited financial statements of Novelos Therapeutics, Inc. included in this prospectus and elsewhere in the registration statement have been so included in reliance upon the report of Grant Thornton LLP, independent registered public accountants, upon the authority of said firm as experts in accounting and auditing in giving said report.
The audited financial statements of Novelos Therapeutics, Inc. (Pre-Acquisition) included in this prospectus and elsewhere in the registration statement have been so included in reliance upon the report of Stowe & Degon LLC, independent registered public accountants, upon the authority of said firm as experts in accounting and auditing in giving said report.
54
GLOSSARY OF SCIENTIFIC TERMS
Akt - Akt (also known as Akt/PKB) is an important cell signaling enzyme (a serine/threonine protein kinase) that plays a key role in multiple cellular processes such as glucose metabolism, cell proliferation, apoptosis, transcription and cell migration.
Apoptosis - A highly regulated, normal cell process leading to programmed cell death by which organisms can eliminate damaged or aberrant cells. Apoptosis is often abnormally suppressed in cancer cells, contributing to their uncontrolled proliferation.
Caspases - Caspases are a family of enzymes (cysteine-aspartic proteases or cysteine-dependent aspartate-directed proteases) that play essential roles in apoptosis (programmed cell death), necrosis, and inflammation.
Cytotoxic - Cytotoxicity is the quality of being toxic to cells (i.e. cell-killing). Many cancer chemotherapeutic drugs are cytotoxic to cancer cells (and, to some extent, normal cells) thus resulting in unwanted side-effects e.g. nausea/vomiting, hair loss, suppression of the immune system.
Dosimetry - Radiation dosimetry is the calculation of absorbed dose and optimization of dose delivery in radiation therapy.
Lipid Rafts - Specialized regions of the membrane phospholipid bilayer that contain high concentrations of cholesterol and sphingolipids and serve to organize cell surface and intracellular signaling molecules (e.g. growth factor and cytokine receptors, the phophatidylinosotol 3-kinase (P13K)/Akt survival pathway).
Radiolabeled - Refers to a molecule containing a radioisotope as a part of its structure.
Radioisotope - Also referred to as radioactive isotopes or radionuclides. These are variants of atoms of particular chemical elements (e.g. iodine) with an unstable nucleus that can undergo radioactive decay during which ionizing radiation (e.g. gamma rays, subatomic particles) is emitted.
Uptake – An act of taking in or absorbing, especially into a living organism, tissue or cell.
Xenograft - Tissue, organs or cells from an individual of one species transplanted into or grafted onto an individual of another species.
55
FINANCIAL STATEMENTS
INDEX TO FINANCIAL STATEMENTS FOR NOVELOS THERAPEUTICS, INC.
(A Development Stage Company)
|
Page
|
||||
|
Report of Independent Registered Public Accounting Firm
|
F-2
|
|||
|
Balance Sheets at June 30, 2011, December 31, 2010 and 2009
|
F-3
|
|||
|
Statements of Operations for the Six Months Ended June 30, 2011 and 2010, the Years Ended December 31, 2010 and 2009, and the Cumulative Development-Stage Period from November 7, 2002 (date of inception) to December 31, 2010
|
F-4
|
|||
|
Statements of Stockholders’ Equity for the Six Months Ended June 30, 2011 and Cumulative Development-Stage Period from November 7, 2002 (date of inception) to December 31, 2010
|
F-5
|
|||
|
Statements of Cash Flows for the Six Months Ended June 30, 2011 and 2010, the Years Ended December 31, 2010 and 2009, and the Cumulative Development-Stage Period from November 7, 2002 (date of inception) to December 31, 2010
|
F-6
|
|||
|
Notes to Financial Statements
|
F-7
|
|||
F-1
Board of Directors and Stockholders
Novelos Therapeutics, Inc.
We have audited the accompanying balance sheets of Novelos Therapeutics, Inc. (a Development Stage Company) (a Delaware corporation) (the “Company”) as of December 31, 2010 and 2009, and the related statements of operations, stockholders’ equity (deficiency), and cash flows for each of the two years in the period ended December 31, 2010 and the period from November 7, 2002 (date of inception) through December 31, 2010. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Novelos Therapeutics, Inc. as of December 31, 2010 and 2009, and the results of its operations and its cash flows for each of the two years in the period ended December 31, 2010 and the period from November 7, 2002 (date of inception) through December 31, 2010, in conformity with accounting principles generally accepted in the United States of America.
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the financial statements, the Company has incurred losses since its inception and, as of December 31, 2010, had an accumulated deficit of $24,045,004. These conditions, along with other matters as set forth in Note 1, raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ GRANT THORNTON LLP
Chicago, Illinois
June 14, 2011
F-2
NOVELOS THERAPEUTICS, INC.
(a Development Stage Company)
BALANCE SHEETS
|
Consolidated
June 30,
2011
|
December 31,
2010
|
December 31,
2009
|
||||||||||
|
(unaudited)
|
(audited)
|
(audited)
|
||||||||||
|
ASSETS
|
||||||||||||
|
CURRENT ASSETS:
|
||||||||||||
|
Cash and cash equivalents
|
$
|
3,313,398
|
$
|
673,739
|
$
|
980,125
|
||||||
|
Restricted cash
|
55,000
|
555,000
|
555,000
|
|||||||||
|
Prepaid expenses and other current assets
|
53,115
|
51,042
|
69,626
|
|||||||||
|
Deferred issuance costs
|
57,700
|
—
|
99,461
|
|||||||||
|
Total current assets
|
3,479,213
|
1,279,781
|
1,704,212
|
|||||||||
|
FIXED ASSETS, NET
|
3,249,334
|
3,510,489
|
4,088,951
|
|||||||||
|
INTANGIBLE ASSETS
|
—
|
—
|
19,671
|
|||||||||
|
EXCESS PURCHASE PRICE OVER NET ASSETS ACQUIRED
|
1,675,462
|
—
|
—
|
|||||||||
|
OTHER ASSETS
|
27,222
|
11,872
|
11,872
|
|||||||||
|
TOTAL ASSETS
|
$
|
8,431,231
|
$
|
4,802,142
|
$
|
5,824,706
|
||||||
|
CURRENT LIABILITIES:
|
||||||||||||
|
Accounts payable and accrued liabilities
|
$
|
365,793
|
$
|
392,881
|
$
|
715,588
|
||||||
|
Accrued interest
|
—
|
305,049
|
—
|
|||||||||
|
Derivative liability
|
81,540
|
—
|
—
|
|||||||||
|
Notes payable, current portion
|
—
|
204,802
|
190,789
|
|||||||||
|
Capital lease obligations, current portion
|
2,159
|
2,085
|
1,944
|
|||||||||
|
Total current liabilities
|
449,492
|
904,817
|
908,321
|
|||||||||
|
LONG-TERM LIABILITIES:
|
||||||||||||
|
Convertible debt (Note 7)
|
—
|
2,720,985
|
—
|
|||||||||
|
Notes payable, net of current portion
|
450,000
|
920,941
|
675,743
|
|||||||||
|
Deferred rent
|
118,443
|
115,311
|
105,338
|
|||||||||
|
Capital lease obligations, net of current portion
|
5,228
|
6,326
|
8,411
|
|||||||||
|
Total long-term liabilities
|
573,671
|
3,763,563
|
789,492
|
|||||||||
|
COMMITMENTS AND CONTINGENCIES
|
||||||||||||
|
STOCKHOLDERS’ EQUITY:
|
||||||||||||
|
Common stock, $0.00001 par value; 150,000,000 shares authorized; 26,826,157 shares issued and outstanding at June 30, 2011, 12,820,102 shares issued and outstanding at December 31, 2010 and December 31, 2009
|
268
|
128
|
128
|
|||||||||
|
Additional paid-in capital
|
35,013,267
|
24,178,638
|
23,611,506
|
|||||||||
|
Deficit accumulated during the development stage
|
(27,605,467
|
)
|
(24,045,004
|
)
|
(19,484,741
|
)
|
||||||
|
Total stockholders’ equity
|
7,408,068
|
133,762
|
4,126,893
|
|||||||||
|
TOTAL LIABILITIES AND STOCKHOLDERS’ EQUITY
|
$
|
8,431,231
|
$
|
4,802,142
|
$
|
5,824,706
|
||||||
The accompanying notes are an integral part of these financial statements.
F-3
NOVELOS THERAPEUTICS, INC.
(a Development Stage Company)
STATEMENTS OF OPERATIONS
|
Six Months Ended June 30,
|
Year Ended
December 31,
|
Cumulative
Development-
Stage Period
from
November 7,
2002 (date of
inception)
through
December 31,
2010
|
||||||||||||||||||
|
Consolidated
2011
(unaudited)
|
2010
(unaudited)
|
2010
(audited)
|
2009
(audited)
|
(audited)
|
||||||||||||||||
|
COSTS AND EXPENSES:
|
||||||||||||||||||||
|
Research and development
|
$
|
1,433,356
|
$
|
2,100,116
|
$
|
2,984,207
|
$
|
4,351,983
|
$
|
17,205,959
|
||||||||||
|
General and administrative
|
928,440
|
923,666
|
1,156,549
|
1,824,302
|
6,970,179
|
|||||||||||||||
|
Merger costs
|
746,207
|
—
|
52,925
|
—
|
52,925
|
|||||||||||||||
|
Total costs and expenses
|
3,108,003
|
3,023,782
|
4,193,681
|
6,176,285
|
24,229,063
|
|||||||||||||||
|
LOSS FROM OPERATIONS
|
(3,108,003
|
)
|
(3,023,782
|
)
|
(4,193,681
|
)
|
(6,176,285
|
)
|
(24,229,063
|
)
|
||||||||||
|
OTHER INCOME (EXPENSE):
|
||||||||||||||||||||
|
Grant income
|
44,479
|
—
|
200,000
|
—
|
200,000
|
|||||||||||||||
|
Loss on derivative liability
|
(70,393
|
)
|
—
|
—
|
—
|
—
|
||||||||||||||
|
Interest expense, net
|
(426,546
|
)
|
(373,852
|
)
|
(566,156
|
)
|
(43,588
|
)
|
(17,102
|
)
|
||||||||||
|
Other income
|
—
|
(573
|
)
|
(426
|
)
|
—
|
1,161
|
|||||||||||||
|
Total other income (expense)
|
(452,460
|
)
|
(374,425
|
)
|
(366,582
|
)
|
(43,588
|
)
|
184,059
|
|||||||||||
|
NET LOSS ATTRIBUTABLE TO COMMON STOCKHOLDERS
|
$
|
(3,560,463
|
)
|
$
|
(3,398,207
|
)
|
$
|
(4,560,263
|
)
|
$
|
(6,219,873
|
)
|
$
|
(24,045,004
|
)
|
|||||
|
BASIC AND DILUTED NET LOSS ATTRIBUTABLE TO COMMON STOCKHOLDERS PER COMMON SHARE
|
$
|
(0.18
|
)
|
$
|
(0.27
|
)
|
$
|
(0.36
|
)
|
$
|
(0.49
|
)
|
$
|
(2.53
|
)
|
|||||
|
SHARES USED IN COMPUTING BASIC AND DILUTED NET LOSS ATTRIBUTABLE TO COMMON STOCKHOLDERS PER COMMON SHARE
|
19,317,642
|
12,820,102
|
12,820,102
|
12,820,102
|
9,513,115
|
|||||||||||||||
The accompanying notes are an integral part of these financial statements.
F-4
NOVELOS THERAPEUTICS, INC.
(a Development Stage Company)
STATEMENTS OF STOCKHOLDERS’ EQUITY
|
Common Stock
|
Additional
Paid-in
Capital
|
Deficit
Accumulated
During the
Development
Stage
|
Total
Stockholders’
Equity
|
|||||||||||||||||
|
Shares
|
Par
Amount
|
|||||||||||||||||||
|
BALANCE AT NOVEMBER 7, 2002
|
—
|
$
|
—
|
$
|
—
|
$
|
—
|
$
|
—
|
|||||||||||
|
Issuance of common stock for cash
|
6,440,123
|
64
|
590,205
|
—
|
590,269
|
|||||||||||||||
|
Issuance of common stock in exchange for professional services
|
101,220
|
1
|
9,107
|
—
|
9,108
|
|||||||||||||||
|
Net loss
|
—
|
—
|
—
|
—
|
—
|
|||||||||||||||
|
BALANCE AT DECEMBER 31, 2002
|
6,541,343
|
65
|
599,312
|
—
|
599,377
|
|||||||||||||||
|
Issuance of common stock for cash, net of issuance costs
|
37,958
|
—
|
4,937
|
—
|
4,937
|
|||||||||||||||
|
Issuance of common stock in exchange for licensed technology
|
203,483
|
2
|
80,410
|
—
|
80,412
|
|||||||||||||||
|
Net loss
|
—
|
—
|
—
|
(295,790)
|
(295,790
|
)
|
||||||||||||||
|
BALANCE AT DECEMBER 31, 2003
|
6,782,784
|
67
|
684,659
|
(295,790
|
)
|
388,936
|
||||||||||||||
|
Net loss
|
—
|
—
|
—
|
(342,761
|
)
|
(342,761
|
)
|
|||||||||||||
|
BALANCE AT DECEMBER 31, 2004
|
6,782,784
|
67
|
684,659
|
(638,551
|
)
|
46,175
|
||||||||||||||
|
Issuance of common stock for cash, net of issuance costs
|
610,664
|
6
|
835,862
|
—
|
835,868
|
|||||||||||||||
|
Net loss
|
—
|
—
|
—
|
(481,837
|
)
|
(481,837
|
)
|
|||||||||||||
|
BALANCE AT DECEMBER 31, 2005
|
7,393,448
|
73
|
1,520,521
|
(1,120,388
|
)
|
400,206
|
||||||||||||||
|
Issuance of common stock for cash, net of issuance costs
|
2,202,179
|
22
|
7,097,050
|
—
|
7,097,072
|
|||||||||||||||
|
Common stock repurchased
|
(43,819
|
)
|
—
|
(31,667
|
)
|
—
|
(31,667
|
)
|
||||||||||||
|
Stock-based compensation
|
—
|
—
|
43,994
|
—
|
43,994
|
|||||||||||||||
|
Net loss
|
—
|
—
|
—
|
(963,440
|
)
|
(963,440
|
)
|
|||||||||||||
|
BALANCE AT DECEMBER 31, 2006
|
9,551,808
|
95
|
8,629,898
|
(2,083,828
|
)
|
6,546,165
|
||||||||||||||
|
Issuance of common stock for cash, net of issuance costs
|
60,250
|
1
|
249,999
|
—
|
250,000
|
|||||||||||||||
|
Exercise of warrant to purchase common stock
|
75,045
|
1
|
249,999
|
—
|
250,000
|
|||||||||||||||
|
Stock-based compensation
|
—
|
—
|
570,392
|
—
|
570,392
|
|||||||||||||||
|
Net loss
|
—
|
—
|
—
|
(5,090,325
|
)
|
(5,090,325
|
)
|
|||||||||||||
|
BALANCE AT DECEMBER 31, 2007
|
9,687,103
|
97
|
9,700,288
|
(7,174,153
|
)
|
2,526,232
|
||||||||||||||
|
Issuance of common stock for cash, net of issuance costs
|
3,132,999
|
31
|
12,931,531
|
—
|
12,931,562
|
|||||||||||||||
|
Stock-based compensation
|
—
|
—
|
477,488
|
—
|
477,488
|
|||||||||||||||
|
Net loss
|
—
|
—
|
—
|
(6,090,715
|
)
|
(6,090,715
|
)
|
|||||||||||||
|
BALANCE AT DECEMBER 31, 2008
|
12,820,102
|
128
|
23,109,307
|
(13,264,868
|
)
|
9,844,567
|
||||||||||||||
|
Stock-based compensation
|
—
|
—
|
502,199
|
—
|
502,199
|
|||||||||||||||
|
Net loss
|
—
|
—
|
—
|
(6,219,873
|
)
|
(6,219,873
|
)
|
|||||||||||||
|
BALANCE AT DECEMBER 31, 2009
|
12,820,102
|
128
|
23,611,506
|
(19,484,741
|
)
|
4,126,893
|
||||||||||||||
|
Stock-based compensation
|
—
|
—
|
353,340
|
—
|
353,340
|
|||||||||||||||
|
Intrinsic value of beneficial conversion feature associated with convertible debt issued in exchange for cash
|
—
|
—
|
213,792
|
—
|
213,792
|
|||||||||||||||
|
Net loss
|
—
|
—
|
—
|
(4,560,263
|
)
|
(4,560,263
|
)
|
|||||||||||||
|
BALANCE AT DECEMBER 31, 2010
|
12,820,102
|
128
|
24,178,638
|
(24,045,004
|
)
|
133,762
|
||||||||||||||
|
Issuance of common stock upon conversion of convertible notes
|
4,181,535
|
42
|
3,184,665
|
—
|
3,184,707
|
|||||||||||||||
|
Issuance of common stock in a business combination
|
2,959,871
|
30
|
2,219,873
|
—
|
2,219,903
|
|||||||||||||||
|
Cash paid in lieu of fractional shares in a business combination
|
(41
|
)
|
—
|
(145
|
)
|
—
|
(145
|
)
|
||||||||||||
|
Issuance of common stock and warrants, net of issuance costs
|
6,846,537
|
68
|
4,866,338
|
—
|
4,866,406
|
|||||||||||||||
|
Intrinsic value of beneficial conversion feature associated with the conversion of convertible debt
|
—
|
—
|
257,973
|
—
|
257,973
|
|||||||||||||||
|
Issuance of common stock upon the cashless exercise of warrants
|
18,153
|
—
|
48,339
|
—
|
48,339
|
|||||||||||||||
|
Stock-based compensation
|
—
|
—
|
257,586
|
—
|
257,586
|
|||||||||||||||
|
Net loss
|
—
|
—
|
—
|
(3,560,463
|
)
|
(3,560,463
|
)
|
|||||||||||||
|
BALANCE AT JUNE 30, 2011 (CONSOLIDATED) (unaudited)
|
26,826,157
|
$
|
268
|
$
|
35,013,267
|
$
|
(27,605,467
|
)
|
$
|
7,408,068
|
||||||||||
The accompanying notes are an integral part of these financial statements.
F-5
NOVELOS THERAPEUTICS, INC.
(a Development Stage Company)
STATEMENTS OF CASH FLOWS
|
Six Months Ended
June 30,
|
Year Ended
December 31,
|
Cumulative Development-Stage
Period from November 7, 2002
through December 31, 2010
|
||||||||||||||||||
|
Consolidated
2011
(unaudited)
|
2010
(unaudited)
|
2010
(audited)
|
2009
(audited)
|
(audited)
|
||||||||||||||||
|
Net loss
|
$ | (3,560,463 | ) | $ | (3,398,207 | ) | $ | (4,560,263 | ) | $ | (6,219,873 | ) | $ | (24,045,004 | ) | |||||
|
Adjustments to reconcile net loss to cash used in operating activities:
|
||||||||||||||||||||
|
Depreciation and amortization
|
290,739 | 291,677 | 580,114 | 576,745 | 1,831,197 | |||||||||||||||
|
Stock-based compensation
|
257,586 | 241,353 | 353,340 | 502,199 | 1,947,413 | |||||||||||||||
|
Intrinsic value of beneficial conversion feature associated with convertible debt
|
257,973 | 213,792 | 213,792 | — | 213,792 | |||||||||||||||
|
Issuance of stock for technology and services
|
— | — | — | — | 89,520 | |||||||||||||||
|
Impairment of intangible assets
|
— | — | 19,671 | — | 19,671 | |||||||||||||||
|
Loss on disposal of fixed assets
|
— | — | — | 1,607 | 30,468 | |||||||||||||||
|
Loss on derivative warrants
|
70,393 | — | — | — | — | |||||||||||||||
|
Changes in:
|
||||||||||||||||||||
|
Prepaid expenses and other current assets
|
25,969 | (19,189 | ) | 18,584 | 23,152 | (62,914 | ) | |||||||||||||
|
Accounts payable and accrued liabilities
|
(407,216 | ) | (115,989 | ) | (322,707 | ) | 278,131 | 392,881 | ||||||||||||
|
Accrued interest
|
158,672 | 140,448 | 305,049 | — | 305,049 | |||||||||||||||
|
Deferred rent
|
3,132 | 289 | 9,973 | 62,789 | 115,311 | |||||||||||||||
|
Cash used in operating activities
|
(2,903,215 | ) | (2,645,826 | ) | (3,382,447 | ) | (4,775,250 | ) | (19,162,616 | ) | ||||||||||
|
CASH FLOWS FROM INVESTING ACTIVITIES:
|
||||||||||||||||||||
|
Cash acquired in a business combination
|
905,649 | — | — | — | — | |||||||||||||||
|
Purchases of fixed assets
|
(23,069 | ) | — | (1,652 | ) | (143,347 | ) | (5,368,181 | ) | |||||||||||
|
Proceeds from sale of fixed assets
|
— | — | — | — | 7,000 | |||||||||||||||
|
Purchases of short-term certificates of deposit
|
— | — | — | — | (5,500,730 | ) | ||||||||||||||
|
Proceeds from short-term certificates of deposit
|
— | — | — | — | 5,500,730 | |||||||||||||||
|
Change in restricted cash
|
500,000 | — | — | — | (555,000 | ) | ||||||||||||||
|
Payment for intangible assets
|
— | — | — | — | (19,671 | ) | ||||||||||||||
|
Cash provided by (used in) investing activities
|
1,382,580 | — | (1,652 | ) | (143,347 | ) | (5,935,852 | ) | ||||||||||||
|
CASH FLOWS FROM FINANCING ACTIVITIES:
|
||||||||||||||||||||
|
Proceeds from issuance of convertible notes
|
— | 2,720,985 | 2,720,985 | — | 2,720,985 | |||||||||||||||
|
Proceeds from long-term obligations
|
— | — | 450,000 | — | 1,677,945 | |||||||||||||||
|
Payments on long-term obligations
|
(675,743 | ) | (93,818 | ) | (190,789 | ) | (177,807 | ) | (552,201 | ) | ||||||||||
|
Payments on capital lease obligations
|
(1,024 | ) | (956 | ) | (1,944 | ) | (619 | ) | (2,563 | ) | ||||||||||
|
Proceeds from issuance of common stock, net of issuance costs
|
4,866,406 | — | — | — | 21,709,708 | |||||||||||||||
|
Proceeds from exercise of warrant
|
— | — | — | — | 250,000 | |||||||||||||||
|
Repurchase of common stock
|
— | — | — | — | (31,667 | ) | ||||||||||||||
|
Cash paid in lieu of fractional shares in a business combination
|
(145 | ) | — | — | — | — | ||||||||||||||
|
Change in deferred issuance costs
|
(29,200 | ) | 89,098 | 99,461 | (99,461 | ) | — | |||||||||||||
|
Cash provided by (used in) financing activities
|
4,160,294 | 2,715,309 | 3,077,713 | (277,887 | ) | 25,772,207 | ||||||||||||||
|
INCREASE (DECREASE) IN CASH AND EQUIVALENTS
|
2,639,659 | 69,483 | (306,386 | ) | (5,196,484 | ) | 673,739 | |||||||||||||
|
CASH AND EQUIVALENTS AT BEGINNING OF PERIOD
|
673,739 | 980,125 | 980,125 | 6,176,609 | — | |||||||||||||||
|
CASH AND EQUIVALENTS AT END OF PERIOD
|
$ | 3,313,398 | $ | 1,049,608 | $ | 673,739 | $ | 980,125 | $ | 673,739 | ||||||||||
|
SUPPLEMENTAL DISCLOSURE OF CASH FLOW INFORMATION
|
||||||||||||||||||||
|
Interest paid
|
$ | 13,700 | $ | 29,300 | $ | 55,454 | $ | 68,436 | $ | 194,973 | ||||||||||
|
Fair value of derivative warrants reclassified to additional paid-in capital upon cashless exercise
|
$ | 48,339 | $ | — | $ | — | $ | — | $ | — | ||||||||||
|
Issuance of common stock upon conversion of notes payable and accrued interest
|
$ | 3,184,706 | $ | — | $ | — | $ | — | $ | — | ||||||||||
|
Fair value of assets acquired in exchange for securities in a business combination
|
$ | 984,057 | $ | — | $ | — | $ | — | $ | — | ||||||||||
|
Fair value of liabilities assumed in exchange for securities in a business combination
|
$ | (439,616 | ) | $ | — | $ | — | $ | — | $ | — | |||||||||
|
Excess of purchase price over net assets acquired in a business combination
|
$ | 1,675,462 | $ | — | $ | — | $ | — | $ | — | ||||||||||
The accompanying notes are an integral part of these financial statements.
F-6
NOTES TO FINANCIAL STATEMENTS
(ALL INFORMATION AS OF AND FOR THE SIX MONTHS ENDED
JUNE 30, 2011 AND 2010 IS UNAUDITED)
1. NATURE OF BUSINESS, ORGANIZATION AND GOING CONCERN
Novelos Therapeutics, Inc. (“Novelos” or the “Company”) is a pharmaceutical company developing compounds for the treatment of cancer. On April 8, 2011, Novelos completed a business combination with Cellectar, Inc. (“Cellectar”), a privately held Wisconsin corporation that designed and developed products to detect, treat and monitor a wide variety of human cancers, and Cell Acquisition Corp. (the “Merger Subsidiary”), a Wisconsin corporation and a wholly owned subsidiary of Novelos. Pursuant to the transaction Cellectar was merged into the Merger Subsidiary (the “Acquisition”, see Note 4). References in these financial statements and notes to “Cellectar” relate to the activities and financial information of Cellectar prior to the Acquisition, references to “Novelos” relate to the activities and financial information of Novelos prior to the Acquisition and references to “the Company” or “we” or “us” or “our” relate to the activities and obligations of the combined Company following the Acquisition.
Immediately prior to the Acquisition, Novelos completed a 1-for-153 reverse split of its common stock (the “Reverse Split”). Novelos then issued to the shareholders of Cellectar at that date 17,001,596 shares of its common stock as consideration for the Acquisition, representing a ratio of 0.8435 shares of Novelos common stock in exchange for one share of Cellectar common stock (the “Exchange Ratio”) as set forth in the Agreement and Plan of Merger (the “Merger Agreement”) dated April 8, 2011. The shares issued to Cellectar shareholders in the Acquisition constituted approximately 85% of Novelos’ outstanding common stock after giving effect to the Acquisition. Upon the closing of the Acquisition, the Company completed the private placement of 6,846,537 shares of its common stock and warrants to purchase an additional 6,846,537 shares of its common stock for gross proceeds of approximately $5,135,000.
Accounting principles generally accepted in the United States require that a company whose security holders retain the majority voting interest in the combined business be treated as the acquirer for financial reporting purposes. Accordingly, the Acquisition will be accounted for as a reverse acquisition whereby Cellectar, Inc. will be treated as the acquirer for accounting and financial reporting purposes. The financial statements presented herein as of and for the twelve months ended December 31, 2010 and 2009 represent the historical financial information of Cellectar, except for the capital structure which represents the historical amounts of Cellectar, retroactively adjusted to reflect the legal capital structure of Novelos by applying the Exchange Ratio. On April, 8, 2011, Cellectar was merged into the Merger Subsidiary a wholly owned subsidiary of Novelos; as such, the financial statements presented herein as of and for the six months ended June 30, 2011 include the historical results of Cellectar from January 1, 2011 through April 8, 2011, except for the capital structure which represents the historical amounts of Cellectar, retroactively adjusted to reflect the legal capital structure of Novelos by applying the Exchange Ratio, and include the consolidated results of the combined company from April 9, 2011 through June 30, 2011. All per-share amounts and outstanding shares, including all common stock equivalents, and stock options, have been retroactively restated in these financial statements and notes for all periods presented to reflect the capital structure of Novelos by applying the Exchange Ratio. The cumulative capital activity from the date of inception (November 7, 2002) up to the closing of the Acquisition, as presented in the accompanying statement of stockholders’ equity, equals 17,001,596 shares of common stock, which represents the equity interests the legal parent (Novelos) issued to effect the Acquisition. The number of authorized shares of common stock disclosed on the balance sheet (150,000,000) represents the number of authorized shares of Novelos common stock following the Acquisition. Additionally, on the accompanying balance sheets as of December 31, 2010 and 2009 and statements of stockholders’ equity for the period from inception (November 7, 2002) to December 31, 2010 the aggregate par value of the issued common stock was reduced to reflect the $0.00001 par value of Novelos common stock associated with the shares of Cellectar common stock adjusted for the Exchange Ratio and the difference was reclassified to additional paid-in capital.
As a result of the Acquisition, the Company has implemented a revised business plan focused on the development of the Cellectar compounds. Development of Novelos’ other compounds (NOV-002 and NOV-205) has been suspended. The Company is conducting its operations from Cellectar’s headquarters in Madison, WI and the Company’s executive offices are in Newton, MA.
The Company is subject to a number of risks similar to those of other small pharmaceutical companies. Principal among these risks are dependence on key individuals, competition from substitute products and larger companies, the successful development and marketing of its products in a highly regulated environment and the need to obtain additional financing necessary to fund future operations.
The accompanying financial statements have been prepared on a basis that assumes that the Company will continue as a going concern and that contemplates the continuity of operations, realization of assets and the satisfaction of liabilities and commitments in the normal course of business. Cellectar has incurred losses since inception in devoting substantially all of its efforts toward research and development and has an accumulated deficit of $24,045,004 at December 31, 2010. During the year ended December 31, 2010, Cellectar generated a net loss of $4,560,263 and the Company expects that it will continue to generate operating losses for the foreseeable future. The Company believes that its cash on hand following the Acquisition, plus the proceeds from the private placement completed in connection with the Acquisition, is adequate to fund operations into the fourth quarter of 2011. The Company’s ability to execute its operating plan beyond that time depends on its ability to obtain additional funding via the sale of equity and/or debt securities, a strategic transaction or otherwise. The Company plans to continue to actively pursue financing alternatives, but there can be no assurance that it will obtain the necessary funding. The accompanying financial statements do not include any adjustments that might result from the outcome of this uncertainty.
F-7
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
The accompanying financial statements reflect the application of certain accounting policies, as described in this note and elsewhere in the accompanying notes to the financial statements. The unaudited financial statements as of and for the year ended June 30, 2011 are presented on a consolidated basis to reflect the Acquisition described in Note 4.
The accompanying consolidated unaudited June 30, 2011 balance sheet, the statements of operations and cash flows for the six months ended June 30, 2011 and 2010, and the statements of stockholders’ equity for the six months ended June 30, 2011 and the related interim information contained within the notes to the financial statements have been prepared in accordance with the rules and regulations of the Securities and Exchange Commission (“SEC”) for interim financial information. Accordingly, they do not include all of the information and the notes required by U.S. generally accepted accounting principles for complete financial statements. In the opinion of management, the unaudited interim financial statements reflect all adjustments, consisting of normal and recurring adjustments, necessary for the fair presentation of the Company’s financial position at June 30, 2011 and results of its operations and its cash flows for the six months ended June 30, 2011 and 2010 and the period from November 7, 2002 (inception) to June 30, 2011. The results for the six months ended June 30, 2011 are not necessarily indicative of future results.
Development Stage Company — Cellectar has been in the development stage since its inception. The primary activities since inception have been organizational activities, research and development and raising capital. No significant revenues have been generated from planned principal operations. As of June 30, 2011 and December 31, 2010 and 2009 the Company continues to be in the development stage.
The summary unaudited condensed statement of operations for the cumulative development-stage period from November 7, 2002 (date of inception) through June 30, 2011 is as follows:
|
COSTS AND EXPENSES:
|
||||
|
Research and development
|
$
|
18,639,315
|
||
|
General and administrative
|
7,898,618
|
|||
|
Merger costs
|
799,133
|
|||
|
Total costs and expenses
|
27,337,066
|
|||
|
LOSS FROM OPERATIONS
|
(27,337,066
|
)
|
||
|
OTHER EXPENSE, NET
|
(268,401
|
)
|
||
|
NET LOSS ATTRIBUTABLE TO COMMON STOCKHOLDERS
|
$
|
(27,605,467
|
)
|
|
|
BASIC AND DILUTED NET LOSS ATTRIBUTABLE TO COMMON STOCKHOLDERS PER COMMON SHARE
|
$
|
(2.74
|
)
|
|
|
SHARES USED IN COMPUTING BASIC AND DILUTED NET LOSS ATTRIBUTABLE TO COMMON STOCKHOLDERS PER COMMON SHARE
|
10,075,059
|
The summary unaudited condensed statement of cash flow for the cumulative development-stage period from November 7, 2002 (date of inception) through June 30, 2011 is as follows:
|
Net loss
|
$
|
(27,605,467
|
)
|
|
|
Adjustments to reconcile net loss to cash used in operating activities
|
5,008,752
|
|||
|
Changes in working capital
|
530,884
|
|||
|
Cash used in operating activities
|
(22,065,831
|
)
|
||
|
CASH FLOWS FROM INVESTING ACTIVITIES:
|
||||
|
Cash acquired in a business combination
|
905,649
|
|||
|
Purchases of fixed assets, net of $7,000 proceeds from sale of fixed assets
|
(5,384,250
|
)
|
||
|
Change in restricted cash
|
(55,000
|
)
|
||
|
Payment for intangible assets
|
(19,671
|
)
|
||
|
Cash used in investing activities
|
(4,553,272
|
)
|
||
|
CASH FLOWS FROM FINANCING ACTIVITIES:
|
||||
|
Proceeds from issuance of common stock and warrants, net of issuance costs, repurchase of common stock, and cash paid in lieu of fractional shares in a business combination
|
26,794,302
|
|||
|
Change in deferred issuance costs
|
(29,200
|
)
|
||
|
Proceeds from issuance of long-term obligations
|
4,398,930
|
|||
|
Payments on long-term obligations
|
(1,231,531
|
)
|
||
|
Cash provided by financing activities
|
29,932,501
|
|||
|
INCREASE IN CASH AND EQUIVALENTS
|
3,313,398
|
|||
|
CASH AND EQUIVALENTS AT BEGINNING OF PERIOD
|
—
|
|||
|
CASH AND EQUIVALENTS AT END OF PERIOD
|
$
|
3,313,398
|
||
|
SUPPLEMENTAL DISCLOSURE OF CASH FLOW INFORMATION
|
||||
|
Interest paid
|
$
|
208,689
|
||
|
Fair value of derivative warrants reclassified to additional paid-in capital upon cashless exercise
|
$
|
48,339
|
||
|
Issuance of common stock upon conversion of notes payable and accrued interest
|
$
|
3,184,706
|
||
|
Fair value of assets acquired in exchange for securities a business combination
|
$
|
984,057
|
||
|
Fair value of liabilities assumed in exchange for securities in a business combination
|
$
|
(439,616
|
)
|
|
|
Excess of purchase price over net assets acquired in a business combination
|
$
|
1,675,462
|
F-8
Use of Estimates — The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and judgments that may affect the reported amounts of assets, liabilities, revenue and expenses and disclosure of contingent assets and liabilities. On an on-going basis, management evaluates its estimates including those related to unbilled vendor amounts and share-based compensation. Management bases its estimates on historical experience and on various other assumptions that are believed to be reasonable, the results of which form the basis for making judgments about the carrying values of assets and liabilities. Actual results may differ from those estimates under different assumptions or conditions. Changes in estimates are reflected in reported results in the period in which they become known.
Cash and Cash Equivalents — All short-term investments purchased with original maturities of three months or less are considered to be cash equivalents.
Restricted Cash — Restricted cash at June 30, 2011 consists of a certificate of deposit required under the Company’s lease agreement (see Note 14). Restricted cash at December 31, 2010 and December 31, 2009 consists of a certificate of deposit required for collateral for a promissory note with a bank (see Note 8) and a certificate of deposit required under the Company’s lease agreement (see Note 14).
Fixed Assets — Property and equipment are stated at cost. Depreciation on property and equipment is provided using the straight-line method over the estimated useful lives of the assets (5 years). Due to the significant value of leasehold improvements purchased during the initial 3-year lease term and the economic penalty for not extending the building lease, leasehold improvements are depreciated over 17 years (their estimated useful life) which represents the full term of the lease, including all extensions (Note 14).
Intangible Assets — Intangible assets at June 30, 2011 consist of the excess of purchase price over net assets acquired in connection with the Acquisition and will be allocated to intangibles, which could potentially include the fair value of the compounds developed prior to the Acquisition by Novelos, with the remainder allocated to goodwill once the Company completes the final allocation of purchase price (Note 4). Intangible assets at December 31, 2009 consisted of costs incurred to obtain trademarks. These costs were capitalized when the expense was incurred and at which time the assets were deemed to have an indefinite life. During 2010, following a reduction in staff and suspension of research and manufacturing activities in order to reduce operating costs, it was determined that the trademarks had been impaired and the carrying value was reduced to zero.
Impairment of Long - Lived Assets — Whenever events or circumstances change, an assessment is made as to whether there has been an impairment in the value of long-lived assets by determining whether projected undiscounted cash flows generated by the applicable asset exceed its net book value as of the assessment date.
Stock-Based Compensation — Employee stock-based compensation is accounted for in accordance with the guidance of Financial Accounting Standards Board Accounting Standards Codification (“FASB ASC”) Topic 718, Compensation – Stock Compensation which requires all share-based payments to employees, including grants of employee stock options, to be recognized in the financial statements based on their fair values. Non-employee stock-based compensation is accounted for in accordance with the guidance of FASB ASC Topic 505, Equity. As such, the Company recognizes expense based on the estimated fair value of options granted to non-employees over their vesting period, which is generally the period during which services are rendered and deemed completed by such non-employees.
F-9
Research and Development — Research and development costs are expensed as incurred.
Income Taxes — Income taxes are accounted for using the liability method of accounting. Under this method, deferred tax assets and liabilities are determined based on temporary differences between the financial statement and tax basis of assets and liabilities and net operating loss and credit carryforwards using enacted tax rates in effect for the year in which the differences are expected to reverse. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. Valuation allowances are established when it is more likely than not that some portion of the deferred tax assets will not be realized. Management has provided a full valuation allowance against the Company’s gross deferred tax asset. Tax positions taken or expected to be taken in the course of preparing tax returns are required to be evaluated to determine whether the tax positions are “more likely than not” of being sustained by the applicable tax authority. Tax positions deemed not to meet a more-likely-than-not threshold would be recorded as tax expense in the current year. There were no uncertain tax positions that require accrual or disclosure to the financial statements as of December 31, 2010 and 2009.
Comprehensive Loss — There were no components of comprehensive loss other than net loss in all of the periods presented.
Grant Income — Cellectar received a cash grant of $44,000 and $200,000 for the six-month period ended June 30, 2011 and the year ended December 31, 2010, respectively from the U.S. Internal Revenue Service as a qualifying therapeutic discovery project credit pursuant to the Patient Protection and Affordable Care Act. This grant has been recorded as a component of other income.
Fair Value of Financial Instruments — The guidance under FASB ASC Topic 825, Financial Instruments , requires disclosure of the fair value of certain financial instruments. Financial instruments in the accompanying financial statements consist of cash equivalents, accounts payable, convertible debt and long-term obligations. The carrying amount of cash equivalents, investments and accounts payable, approximates fair value due to their short-term nature. The estimated fair value of the convertible debt, determined on an as-converted basis including conversion of accumulated unpaid interest, was approximately $0 and $3,264,000 at June 30, 2011 and December 31, 2010, respectively. The carrying value of long-term obligations, including the current portion, approximates fair value because the fixed interest rate approximates current market rate of interest available in the market.
Derivative Instruments – The Company generally does not use derivative instruments to hedge exposures to cash flow or market risks. However, certain warrants to purchase common stock that do not meet the requirements for classification as equity, in accordance with the Derivatives and Hedging Topic of the Financial Accounting Standards Board Accounting Standards Codification (“FASB ASC”), are classified as liabilities. In such instances, net-cash settlement is assumed for financial reporting purposes, even when the terms of the underlying contracts do not provide for a net-cash settlement. These warrants are considered derivative instruments because the agreements contain “down-round” provisions whereby the number of shares for which the warrants are exercisable and/or the exercise price of the warrants are subject to change in the event of certain issuances of stock at prices below the then-effective exercise price of the warrants. The number of shares issuable under such warrants was 77,729 at June 30, 2011. The primary underlying risk exposure pertaining to the warrants is the change in fair value of the underlying common stock. Such financial instruments are initially recorded at fair value with subsequent changes in fair value recorded as a component of gain or loss on derivatives in each reporting period. If these instruments subsequently meet the requirements for equity classification, the Company reclassifies the fair value to equity. At June 30, 2011, these warrants represented the only outstanding derivative instruments issued or held by the Company. There were no outstanding derivative instruments at December 31, 2010 or 2009.
Concentration of Credit Risk — Financial instruments that subject the Company to credit risk consist of cash and equivalents on deposit with financial institutions, which may exceed federally insured limits. Excess cash is invested on an overnight basis in an investment account that is fully collateralized principally by government-backed obligations. Cash and equivalent balances are maintained with a stable and well-capitalized financial institution.
New Accounting Pronouncements — In January 2010, the FASB issued ASU No. 2010-06, Improving Disclosures about Fair Value Measurements , which requires additional disclosures about the amounts of and reasons for significant transfers in and out of Level 1 and Level 2 fair value measurements. This standard also clarifies existing disclosure requirements related to the level of disaggregation of fair value measurements for each class of assets and liabilities and disclosures about inputs and valuation techniques used to measure fair value for both recurring and non-recurring Level 2 and Level 3 measurements. Since this new accounting standard only required additional disclosure, the adoption of the standard in the first quarter of 2010 did not impact the accompanying financial statements. Additionally, effective for interim and annual periods beginning after December 15, 2010, this standard will require additional disclosure and require an entity to present disaggregated information about activity in Level 3 fair value measurements on a gross basis, rather than one net amount. The adoption of this accounting standard did not impact the accompanying financial statements.
In December 2010, the FASB issued ASU No. 2010-29, Disclosures of Supplementary Pro Forma Information for Business Combinations, which, if comparative financial statements are presented, requires the supplemental pro forma disclosure of revenue and earnings to be presented as if the business combination had occurred at the beginning of the comparable prior annual reporting period only. This standard also expands the supplemental pro forma disclosures required under FASB ASC Topic 850, Business Combinations , to include a description of the nature and amount of material nonrecurring pro forma adjustments directly attributable to the business combination in the reported pro forma revenue and earnings. This standard is effective for the Company for any business combinations completed after January 1, 2011. The Company adopted the provisions of this standard during the first quarter of 2011.
F-10
In May 2011, the FASB issued ASU No. 2011-04, Amendments to Achieve Common Fair Value Measurement and Disclosure Requirements in U.S. Generally Accepted Accounting Principles (“GAAP”) and International Financial Reporting Standards (“IFRSs”). This standard updates accounting guidance to clarify the measurement of fair value to align the guidance and improve the comparability surrounding fair value measurement within GAAP and IFRSs. The standard also updates requirements for measuring fair value and expands the required disclosures. The standard does not require additional fair value measurements and was not intended to establish valuation standards or affect valuation practices outside of financial reporting. This standard will become effective for the Company on January 1, 2012. The Company does not expect that the adoption of this standard will have a material impact when applied prospectively on the Company’s financial statements or required disclosures.
In June 2011, the FASB issued ASU No. 2011-05, Presentation of Comprehensive Income. This standard eliminates the current option to report other comprehensive income and its components in the statement of changes in equity. The standard is intended to enhance comparability between entities that report under US GAAP and those that report under IFRS, and to provide a more consistent method of presenting non-owner transactions that affect an entity's equity. Under the ASU, an entity can elect to present items of net income and other comprehensive income in one continuous statement, referred to as the statement of comprehensive income, or in two separate, but consecutive, statements. Each component of net income and each component of other comprehensive income, together with totals for comprehensive income and its two parts, net income and other comprehensive income, would need to be displayed under either alternative. The statement(s) would need to be presented with equal prominence as the other primary financial statements. The ASU does not change items that constitute net income and other comprehensive income, when an item of other comprehensive income must be reclassified to net income or the earnings-per-share computation (which will continue to be based on net income). The new US GAAP requirements are effective for public entities as of the beginning of a fiscal year that begins after December 15, 2011 and interim and annual periods thereafter. Early adoption is permitted, but full retrospective application is required under the accounting standard. The Company does not expect that the adoption of this standard will have a material impact on our results of operations, cash flows, and financial position.
Reclassifications — Certain prior-period amounts have been reclassified to conform to the current-period presentation.
In accordance with Fair Value Measurements and Disclosures Topic of the FASB ASC, the Company groups its financial assets and financial liabilities generally measured at fair value in three levels, based on the markets in which the assets and liabilities are traded and the reliability of the assumptions used to determine fair value.
|
|
·
|
Level 1: Input prices quoted in an active market for identical financial assets or liabilities.
|
|
|
·
|
Level 2: Inputs other than prices quoted in Level 1, such as prices quoted for similar financial assets and liabilities in active markets, prices for identical assets and liabilities in markets that are not active or other inputs that are observable or can be corroborated by observable market data.
|
|
|
·
|
Level 3: Input prices quoted that are significant to the fair value of the financial assets or liabilities which are not observable or supported by an active market.
|
Assets and liabilities measured at fair value on a recurring basis are summarized below:
|
June 30, 2011
|
||||||||||||||||
|
Level 1
|
Level 2
|
Level 3
|
Fair Value
|
|||||||||||||
|
Liabilities:
|
||||||||||||||||
|
Warrants
|
$ | - | $ | 81,540 | $ | - | $ | 81,540 | ||||||||
|
December 31, 2010
|
||||||||||||||||
|
Level 1
|
Level 2
|
Level 3
|
Fair Value
|
|||||||||||||
|
Liabilities:
|
||||||||||||||||
|
Warrants
|
$ | - | $ | - | $ | - | $ | - | ||||||||
The Company uses the Black-Scholes option pricing model and assumptions that consider, among other variables, the fair value of the underlying stock, risk-free interest rate, volatility, expected life and dividend rates in estimating fair value for the warrants considered to be derivative instruments. Assumptions used are generally consistent with those disclosed for stock-based compensation (see Note 11).
F-11
|
4.
|
ACQUISITION
|
Merger Agreement
On April 8, 2011, Novelos acquired Cellectar through a merger with and into the Merger Subsidiary, pursuant to the merger Agreement entered into on that date. As a result of the Acquisition, the Merger Subsidiary, which has been renamed Cellectar, Inc., owns all assets of and operates the business previously owned and operated by Cellectar.
In the Acquisition, the former stockholders of Cellectar received an aggregate number of shares of Novelos common stock constituting approximately 85% of the outstanding shares of Novelos common stock, after giving effect to the Acquisition but before giving effect to the concurrent private placement of Novelos securities described below. Prior to the Acquisition, Novelos amended and restated its certificate of incorporation and in connection therewith, among other things, effected a 1-for-153 reverse split of its common stock (the “Reverse Split”) resulting in 2,959,871 shares of Novelos common stock outstanding. Novelos then issued 17,001,596 shares of Novelos common stock to the stockholders of Cellectar upon the effective date of the Acquisition. Warrants and options to purchase Novelos common stock that were outstanding prior to the Acquisition remained outstanding following the Acquisition. These consist of warrants to purchase a total of 315,164 shares of Novelos common stock with prices ranging from $16.07 to $191.25 and options to purchase a total of 49,159 shares of Novelos common stock with prices ranging from $1.53 to $1,072.53.
XMS Capital Partners, the financial advisor to Cellectar in connection with the Acquisition, received a cash fee of $200,000 upon the completion of the Acquisition in consideration of their services. Rodman & Renshaw, LLC (“Rodman”), financial advisor to Novelos in connection with the Acquisition, received a cash fee of $250,000 upon the completion of the Acquisition in consideration of their services. These amounts were recorded as merger costs and expensed as incurred on the date of the Acquisition. In addition to the investment banking fees, the Company also expensed an additional $296,207 of merger-related legal and other costs during the six months ended June 30, 2011.
The Acquisition was completed principally to leverage synergies between Novelos’ strategic focus and experience in developing and funding the development of cancer drugs and Cellectar’s portfolio of cancer-targeted compounds.
Purchase Accounting
The Acquisition was accounted for using the purchase method of accounting as a reverse acquisition. In a reverse acquisition, the post-acquisition net assets of the surviving combined company includes the historical cost basis of the net assets of the accounting acquirer (Cellectar) plus the fair value of the net assets of the accounting acquiree (Novelos). Further, under the purchase method, the purchase price is allocated to the assets acquired and liabilities assumed based on their estimated fair values and the excess of the purchase price over the estimated fair value of the identifiable net assets is presented as excess purchase price over net assets acquired. The cost of acquisition and related purchase-price allocation is based on preliminary evaluation of the fair value of assets and liabilities assumed from Novelos and may change when the final valuation of certain intangible assets is determined. The evaluation is preliminary principally due to the pending evaluation of the Company’s intangible assets. The excess of purchase price over net assets acquired will be allocated to intangibles and goodwill once the Company completes the final allocation of purchase price.
The fair value of the consideration transferred in the Acquisition was $2,219,903 and was calculated as the number of shares of common stock that Cellectar would have had to issue (adjusted for the Exchange Ratio) in order for Novelos shareholders to obtain a 15% equity interest in the combined Company post-acquisition, multiplied by the estimated fair value of the Company’s common stock on the acquisition date. The estimated fair value of the Company’s common stock was based on the offering price of the common stock sold in the private placement which was both completed concurrently with and conditioned upon the closing of the Acquisition. This price was determined to be the best indication of fair value on that date since the price was based on an arm’s length negotiation with a group consisting of both new and existing investors that had been advised of the pending Acquisition and assumed similar liquidity risk as those investors holding the majority of shares being valued as purchase consideration.
The following table summarizes the Company’s preliminary estimated fair values of the assets acquired and the liabilities assumed at the date of acquisition.
|
Consideration - issuance of securities
|
$ | 2,219,903 | ||
|
Prepaid expenses and other assets
|
$ | 71,892 | ||
|
Fixed assets
|
6,515 | |||
|
Accrued liabilities
|
(380,130 | ) | ||
|
Derivative liability
|
(59,485 | ) | ||
|
Excess of purchase price over net assets acquired
|
1,675,462 | |||
|
Total purchase price – net of cash acquired of $905,649
|
$ | 1,314,254 |
F-12
The excess of purchase price over net assets acquired will be allocated to intangibles, which could potentially include the fair value of the compounds developed prior to the Acquisition by Novelos, with the remainder allocated to goodwill once the Company completes the final allocation of purchase price. The estimated fair values of assets acquired and liabilities assumed are provisional and are based on the information that was available as of the acquisition date to estimate the fair value of assets acquired and liabilities assumed. The Company believes that the information provides a reasonable basis for estimating the fair values of assets acquired and liabilities assumed, but the Company is waiting for additional information necessary to finalize those fair values. Therefore, the provisional measurements of fair value reflected are subject to change and such changes may be significant. The Company expects to finalize the valuation and complete the purchase price allocation as soon as practicable but no later than one year from the acquisition date.
|
5.
|
FIXED ASSETS
|
Fixed assets consisted of the following at December 31:
| 2010 | 2009 | |||||||
|
Office and laboratory equipment
|
$
|
2,984,375
|
$
|
2,982,723
|
||||
|
Leasehold improvements
|
2,317,597
|
2,317,597
|
||||||
|
Total fixed assets
|
5,301,972
|
5,300,320
|
||||||
|
Less accumulated depreciation and amortization
|
(1,791,483
|
)
|
(1,211,369
|
)
|
||||
|
Fixed assets, net
|
$
|
3,510,489
|
$
|
4,088,951
|
During the six months ended June 30, 2011, the change to net fixed assets consisted of an increase of $291,000 related to accumulated depreciation and amortization, $23,000 related to the purchase of equipment, and $6,000 related to the fair value of fixed assets acquired as a result of the Acquisition (see Note 4).
|
6.
|
LICENSE AGREEMENTS
|
2003 License Agreement with the University of Michigan
In September 2003, Cellectar entered into an exclusive license agreement (the “U. Mich. Agreement”) with the Regents of the University of Michigan, (“U. Mich.”) for the development, manufacture and marketing of products under several composition of matter patents in North America which expire at varying dates in 2016. The U. Mich. Agreement expires upon the expiration of the last covered patent. The Company is responsible for an annual license fee of $10,000 and is required to pay costs associated with the maintenance of the patents covered by the U. Mich. Agreement. Additionally, the Company is required to make milestone payments of $50,000 upon the filing of a New Drug Application (“NDA”) for a licensed product intended for use in a therapeutic or diagnostic application (such milestone fees may be deferred and paid within 12 months of the first commercial sale of such products) and make certain milestone payments within a year following the first commercial sale of any licensed products. The sales milestones range from $100,000 to $200,000, dependent upon whether the drug is for use in a therapeutic or diagnostic application, provided that if sales in the first 12 months are less than the amount of the milestone, then we are required to pay 50% of all sales until the milestone is satisfied. The milestone payments may total up to $400,000. The U. Mich. Agreement provides that the Company pay a royalty equal to 3% of net sales of any licensed products sold by the Company or its sublicensees for such licensed products, provided however if the sublicense fee payable to the Company is between 4%-5% of net sales, then the royalties payable to U. Mich. shall be equal to 50% of the sublicense fee. Furthermore, the U. Mich. Agreement provides for a reduction in the royalties owed by up to 50% if the Company is required to pay royalties to any third parties related to the sale of the licensed products. If we receive any revenue in consideration for rights to the licensed technology that is not based on net sales, excluding any funded research and development, we are required to pay U.Mich. 10% of amounts received. U. Mich. may terminate the agreement if the Company ceases operations, if the Company fails to make any required payment under the agreement, or if the Company otherwise materially breaches the agreement, subject to the applicable notice and cure periods. To date, the Company has made all payments as they have become due, there have been no defaults under the U. Mich. Agreement, nor has the Company been notified of a default by U. Mich. The Company may terminate the agreement with six months notice to U. Mich. and the return of licensed product and related data. The U. Mich. Agreement contained milestones that required certain development activities to be completed by specified dates. All such development milestones have either been completed or have been removed by subsequent amendment to the agreement. U. Mich. has provided no warranties as to validity or otherwise with respect to the licensed technology.
Cellectar paid $300 and $5,000 to U. Michigan for the reimbursement of patent maintenance fees during the years ended December 31, 2010 and 2009, respectively. There were no payments made in the six-month period ended June 30, 2011. As of December 31, 2010, all annual license fees have been paid in a timely manner.
In connection with the Michigan Agreement, during 2003 Cellectar paid approximately $54,000 of back patent costs and issued 203,483 shares of common stock to U. Michigan as partial consideration for the rights described above. The estimated fair-market value of the issuance was $80,412 and was recorded as a license cost and is included as a component of stockholders equity in the accompanying balance sheets.
F-13
2005 License Agreement with Wisconsin Alumni Research Foundation
In January 2005, Cellectar entered into a license agreement (the “WARF Agreement”) with the Wisconsin Alumni Research Foundation (“WARF”) under which Cellectar received a license, with a right to grant sublicenses, to develop, manufacture, and market products with respect to certain patents. The WARF Agreement required an initial license fee of $8,800 and provided that Cellectar pay royalties equal to 0.3% of sales of any licensed products. Cellectar was also required to reimburse WARF for patent filing fees and related costs. During the years ended December 31, 2010 and 2009, there were no costs related to the patents under the WARF Agreement. In June 2010, the WARF Agreement was terminated.
Novelos License Agreements
During 2007, Novelos entered into a Collaboration Agreement with Lee’s Pharmaceutical (HK) Ltd. (“Lee’s Pharm”) whereby Lee’s Pharm obtained an exclusive license to develop, manufacture and commercialize NOV-002 and NOV-205 in China, Hong Kong, Taiwan and Macau (the “Chinese Territory”). Under the terms of the agreement the Company is entitled to receive up to $1,700,000 in future milestone payments upon the completion of development and marketing milestones by Lee’s Pharm and to receive royalty payments of between 12-25% of net sales of NOV-205 and NOV-002, as applicable, in the Chinese Territory and receive royalty payments of 12-15% of net sales of NOV-205 in the Chinese Territory. The agreement expires upon the expiration of the last patent covering any of the licensed products, or twelve years from the date of the first commercial sale in China, whichever occurs later.
During 2009, Novelos entered into a collaboration agreement (the “Collaboration Agreement”) with Mundipharma International Corporation Limited (“Mundipharma”) to develop, manufacture and commercialize, on an exclusive basis, Licensed Products (as defined in the Collaboration Agreement), including NOV-002, in Europe (other than the Russian Territory), Asia (other than the Chinese Territory) and Australia (collectively referred to as the “Mundipharma Territory”). Mundipharma is an independent associated company of Purdue Pharma, L.P. (“Purdue”). The Collaboration Agreement provides for Mundipharma to pay the Company royalties and fixed milestone payments based on sales and commercial launches in the licensed territories. For countries in which patents are held, the Collaboration Agreement expires on a country-by-country basis within the Mundipharma Territory on the earlier of (1) expiration of the last applicable Novelos patent within the country or (2) the determination that any patents within the country are invalid, obvious or otherwise unenforceable. For countries in which no patents are held, the Collaboration Agreement expires the earlier of 15 years from its effective date or upon generic product competition in the country resulting in a 20% drop in Mundipharma’s market share. The Company may terminate the Collaboration Agreement upon breach or default by Mundipharma. Mundipharma may terminate the Collaboration Agreement upon breach or default, filing of voluntary or involuntary bankruptcy by Novelos, the termination of certain agreements with companies associated with the originators of the licensed technology, or 30-day notice for no reason.
The Company has suspended development of the products covered by the collaboration agreements described above. The Company does not anticipate that the collaboration agreements will have a material effect on its results of operations, cash flows, and financial position following the Acquisition.
|
7.
|
CONVERTIBLE DEBT
|
On January 25, 2010, Cellectar issued nine convertible promissory notes (“Convertible Notes”) in an aggregate principal amount of $2,720,985. The Convertible Notes provided for interest of 12% compounded annually with a maturity date of the earlier of (i) the date on which Cellectar’s cash reserves fall below $250,000 or (ii) January 20, 2011. Upon an event of default, as defined, the interest rate increased by 10% to 22%. The outstanding principal balance, together with any unpaid interest, was convertible immediately, by the lender, into common stock of the Company at $0.82987 per share (giving effect to the Exchange Ratio). Furthermore, the Convertible Notes were subject to an automatic conversion feature, equal to 70% of the per share price of the qualified financing, should the Company complete a qualified financing transaction which raises at least $20,000,000 in proceeds to the Company. Since the Convertible Notes were convertible into common stock at date of issuance at a per share price which was less than the estimated fair value of the Company’s common stock at that date, the Convertible Notes contained a beneficial conversion feature (“BCF”). The estimated intrinsic value of the BCF of $213,792 was determined as the difference between the conversion price and the estimated fair value of Cellectar common stock on the date of issuance, multiplied by the 3,278,786 shares of common stock into which the Convertible Notes were convertible at issuance. This amount was recorded as a component of interest expense on the date of issuance. The estimated per-share fair value of Cellectar common stock was determined by management based on a number of factors including an independent valuation, which was determined to be the best indication of the fair value as of the issuance date of the Convertible Notes. Since the conversion price was subject to adjustment in the event of a qualified transaction, as defined, the Convertible Notes also contain a contingent beneficial conversion feature (“CBCF”). This contingency was not resolved; therefore no intrinsic value was allocated to the CBCF. As of December 31, 2010 and 2009, principal of $2,720,985 and $0 was outstanding, respectively, on the Convertible Notes.
As of December 31, 2010, the Convertible Notes are classified as a long-term obligation on the accompanying balance sheets as a result of the conversion of the short-term obligation through the issuance of equity securities in connection with the Acquisition.
F-14
On January 20, 2011, the Convertible Notes matured but remained unpaid. Following the maturity and default of the Convertible Notes, the holders of the Convertible Notes agreed that all of the outstanding notes would be automatically converted simultaneous with the completion of an acquisition and financing (the “Conversion Time”), if completed. The amount of shares issued upon such conversion would be dependent on the amount of investment made by the note holders at the Conversion Time and were negotiated based on outstanding principal and projected accrued interest based on an assumed closing date for the acquisition and financing. Since the number of shares to be issued upon conversion could not be determined until the Conversion Time the Convertible Notes contained a CBCF. On April 1, 2011, Cellectar’s Board of Directors voted to accept the note holders consent to convert the Convertible Notes into 4,181,535 shares of common stock immediately prior to the Acquisition. On April 8, 2011, immediately prior to the Acquisition, the principal and unpaid interest on the Convertible Notes was converted into the agreed total of 4,181,535 shares of common stock. Upon conversion of the Convertible Notes, the Company reclassified the aggregate outstanding principal and interest totaling $3,184,706 to a component of additional paid-in capital. The revised conversion terms resulted in the issuance of an additional 343,963 shares of common stock over the 3,837,572 shares of common stock that would have been issued if the unpaid principal and accrued interest on the Convertible Notes had been converted on that date in accordance with their original terms at the stated conversion price. On the date of conversion, the Company determined that the value of these additional shares was $257,973, based on the $0.75 per share offering price of the common stock sold in the private placement completed concurrently with the Acquisition, which is the best indication of fair value on the date of conversion. Since the conversion was not completed until April 8, 2011, the value of the additional shares of $257,973 was recorded as a component of interest expense in the three-months ended June 30, 2011.
8. LONG-TERM NOTES PAYABLE
On January 11, 2008, Cellectar entered into a loan agreement with a bank to borrow up to $1,200,000. The borrowing, evidenced by a note (the “Bank Note”), bore interest at a rate of 7.01% per annum, could be prepaid without penalty and was payable in 48 monthly principal and interest payments of $20,520 with a balloon payment of any remaining unpaid principal and interest on March 28, 2012. In the event of default of payment, Cellectar would be required to pay a late charge equal to 5% of the delinquent payment and the interest rate on the unpaid principal would be increased by 3%. The Bank Note was collateralized by substantially all assets of Cellectar and a deposit account in the amount of $500,000. The cash collateral is classified as restricted cash in the accompanying balance sheet. As of December 31, 2010 and 2009, $470,941 and $675,743 are classified as a long-term note payable in the accompanying balance sheet, respectively. On April 8, 2011, immediately prior to the Acquisition, Cellectar paid $627,000 in full settlement of the Bank Note. The payment was made in order to avoid an event of default that would have occurred as a result of the change of control that occurred at the time of the Acquisition.
On September 15, 2010, Cellectar entered into certain loan agreements with the Wisconsin Department of Commerce (“WDOC Notes”) to borrow a total of $450,000. The WDOC Notes bear interest at 2% per annum beginning on the date of disbursement and allow for the deferral of interest and principal payments until April 30, 2015. In the event of default of payment, interest on the delinquent payment is payable at a rate equal to 12% per annum. Monthly payments of $20,665 for principal and interest shall commence on May 1, 2015 and continue for 23 equal installments with the final installment of any remaining unpaid principal and interest due on April 1, 2017. As of December 31, 2010, $450,000 is classified as a long-term note payable in the accompanying balance sheet.
Long-term notes payable consists of the following as of December 31:
|
2010
|
2009
|
|||||||
|
Bank Note, 7.01% interest
|
$
|
675,743
|
$
|
866,532
|
||||
|
Wisconsin Department of Commerce, 2% interest
|
450,000
|
—
|
||||||
|
1,125,743
|
866,532
|
|||||||
|
Less current portion
|
(204,802
|
)
|
(190,789
|
)
|
||||
|
Long-term note payable, net of current portion
|
$
|
920,941
|
$
|
675,743
|
||||
As of December 31, 2010, long-term notes payable matures as follows:
|
Years ended December 31,
|
||||
|
$
|
204,802
|
|||
|
2012
|
470,941
|
|||
|
2013
|
—
|
|||
|
2014
|
—
|
|||
|
2015
|
119,957
|
|||
|
Thereafter
|
330,043
|
|||
|
$
|
1,125,743
|
|||
F-15
During the six-months ended June 30, 2011, payments of $675,743 were made in connection with the Bank Note including a payment of approximately $627,000 in full settlement of the Bank Note made immediately prior to the Acquisition in order to avoid an event of default that would have occurred as a result of the change in control that occurred at the time of the Acquisition.
As of June 30, 2011, the outstanding principal on the WDOC Notes of $450,000 is classified as long-term debt outstanding in the accompanying balance sheet.
|
9.
|
LINE OF CREDIT
|
In 2009, Cellectar had a $100,000 line of credit with a bank. Borrowings under the line of credit bore interest at LIBOR plus 3.25% with a 4.5% minimum rate. The line of credit expired on January 10, 2010 and was not renewed. There were no amounts outstanding under the line as of December 31, 2009.
10. STOCKHOLDERS’ EQUITY
On January 1, 2008, Cellectar converted from a Wisconsin limited liability company to a Wisconsin corporation (the “Conversion”). Each issued and outstanding unit of equity in the limited liability company immediately prior to the Conversion was converted into one issued and outstanding share of Cellectar common stock and each unexercised unit option outstanding immediately prior to the Conversion was converted into an option to acquire the same number of shares of the corporation’s common stock. For purposes of presentation in these financial statements, all amounts and disclosures related to equity issuances prior to the Acquisition have been retroactively restated by applying the Exchange Ratio in order to reflect the capital structure of Novelos and therefore the issuance of Novelos common stock, rather than member units in the limited liability company or common stock of Cellectar, as applicable.
From inception until December 31, 2010, Cellectar issued 12,559,218 shares of common stock for net proceeds of approximately $21,710,000.
April 2011 Private Placement
Concurrently with and conditioned upon the execution of the Merger Agreement, the Company entered into a Securities Purchase Agreement with certain accredited investors under which the Company sold an aggregate of 6,846,537 units, each unit consisting of one share of its common stock and a warrant to purchase one share of its common stock, at a price of $0.75 per unit, for gross proceeds of approximately $5,135,000 (the “April Private Placement”). The warrants have an exercise price of $0.75 and expire on March 31, 2016. The warrant exercise price and/or the common stock issuable pursuant to such warrant will be subject to adjustment only for stock dividends, stock splits or similar capital reorganizations so that the rights of the warrant holders after such event will be equivalent to the rights of warrant holders prior to such event. The relative fair value of the warrants issued to the investors was $2,124,286 at issuance and has been included as a component of stockholders’ equity. The Company uses the Black-Scholes option pricing model to value warrants and applies assumptions that consider, among other variables, the fair value of the underlying stock, risk-free interest rate, volatility, expected life and dividend rates in estimating fair value for the warrants. Assumptions used are generally consistent with those disclosed for stock-based compensation (see Note 11).
The Securities Purchase Agreement includes a requirement that the Company file with the SEC no later than October 5, 2011, a registration statement covering the resale of the shares of common stock, and the shares of common stock underlying the warrants, issued pursuant to the Securities Purchase Agreement that are not otherwise saleable under an available exemption from registration requirements. The Company is also required to use our commercially reasonable efforts to have the registration statement declared effective by December 4, 2011, and to keep the registration statement continuously effective under the Securities Act of 1933, as amended (the “Securities Act”), until the earlier of the date when all the registrable securities covered by the registration statement have been sold or the second anniversary of the closing.
In the event the Company fails to file the registration statement within the timeframe specified by the Securities Purchase Agreement, or if it fails to obtain effectiveness of this registration on or prior to the December 4, 2011 (if there is no review by the SEC) or by January 3, 2012 (if there is review by the SEC) with respect to the maximum number of shares permitted to be registered by the SEC, the Company will be required to pay to the purchasers liquidated damages equal to 1.5% per month (pro-rated on a daily basis for any period of less than a full month) of the aggregate purchase price of the units purchased until the registration statement is filed or declared effective, as applicable, not to exceed 5% of the aggregate purchase price. The Company will be allowed to suspend the use of the registration statement for not more than 30 consecutive days, on not more than two occasions, in any 12-month period. The Company has also granted piggy-back registration rights with respect to any shares of common stock that it is required to exclude from the registration statement as a condition of its effectiveness, and has also agreed to file further registration statements with respect to any such shares six months after the effective date of the initial registration statement. As of June 30, 2011, and through the date of this filing, the Company has not concluded that it is probable that damages will become due; therefore, no accrual for damages has been recorded.
F-16
The Company paid to Rodman, the placement agent for the financing, a cash fee equal to $200,000 and issued warrants to purchase 192,931 shares of its common stock (having an exercise price of $0.75 and which expire March 31, 2016) in consideration for their advisory services with respect to the financing pursuant to the placement agency agreement between Rodman and the Company. Rodman is entitled to registration rights with respect to the shares of common stock issuable upon exercise of these warrants. The cash fee was recorded as a reduction of gross proceeds received. The estimated fair value of the warrants issued to the placement agent was $112,096 and was recorded as a component of stockholders’ equity.
Common Stock Warrants — The following table summarizes information with regard to outstanding warrants to purchase common stock as of June 30, 2011. There were no outstanding common stock purchase warrants as of December 31, 2010 and 2009. The Company issued warrants to purchase 7,039,468 shares of common stock in connection with the April Private Placement. In addition, outstanding warrants to purchase 315,164 shares of common stock, originally issued in connection with Novelos equity and debt financings from 2007 through 2010, remained outstanding subsequent to the Acquisition (Note 4).
|
Offering
|
Number of Shares
Issuable Upon
Exercise of
Outstanding
Warrants
|
Exercise
Price
|
Expiration Date
|
||||||
|
April 8, 2011 Private Placement
|
7,039,468
|
$
|
0.75
|
March 31, 2016
|
|||||
|
Series B Preferred Stock – placement agents
|
5,392
|
$
|
191.25
|
May 2, 2012
|
|||||
|
Series C Exchange
|
8,169
|
$
|
191.25
|
May 2, 2012
|
|||||
|
Series E Preferred Stock
|
60,330
|
$
|
99.45
|
December 31, 2015
|
|||||
|
August 2009 Private Placement
|
31,194
|
$
|
100.98
|
December 31, 2015
|
|||||
|
July 2010 Direct Offering (1)
|
77,729
|
$
|
0.75
|
July 27, 2015
|
|||||
|
Preferred Incentive Warrants
|
105,040
|
$
|
16.065
|
July 27, 2015
|
|||||
|
Total
|
7,327,322
|
||||||||
(1) The exercise price of these warrants was adjusted to $0.75 per share in connection with the private placement completed on April 8, 2011. This warrant has been accounted for as a derivative instrument as described in Note 2.
On May 4, 2011, 18,153 shares of common stock were issued in connection with the cashless exercise of warrants to purchase 27,310 shares of common stock at $0.75 per share. The Company reclassified $48,339 from the derivative liability to additional paid-in capital upon the exercise of the warrants.
The following shares were reserved for future issuance upon exercise of stock options and warrants or conversion of debt:
|
June 30,
|
December 31,
|
|||||||||||
|
2011
(unaudited)
|
2010
|
2009
|
||||||||||
|
Warrants
|
7,327,322 | — | — | |||||||||
|
Stock options
|
3,632,638 | 769,189 | 991,736 | |||||||||
|
Convertible notes
|
— | 3,646,370 | — | |||||||||
|
Total number of shares reserved for future issuance
|
10,959,960 | 4,415,559 | 991,736 | |||||||||
11. STOCK-BASED COMPENSATION
Cellectar’s stock-based compensation plans prior to the Acquisition are summarized below:
2006 Unit Option Plan. The 2006 Unit Option Plan (the “2006 Cellectar Plan”), as amended and restated, provided Cellectar the ability to grant to employees, directors, consultants, and other non-employees units of interest in Cellectar. The maximum aggregate number of shares that were subject to grant under the 2006 Cellectar Plan was 1,012,200.
F-17
Cellectar granted 631,360 unit options under the 2006 Cellectar Plan and no additional grants will be made thereunder. In connection with the Conversion described in Note 10, each issued and outstanding unexercised unit option outstanding immediately prior to the Conversion was converted into an option to acquire the same number of shares of Cellectar’s common stock. A total of 606,889 and 691,248 options to purchase shares of Cellectar’s common stock were outstanding under the 2006 Cellectar Plan as of December 31, 2010 and 2009, respectively. These options generally vested annually over four years and expire on the eighth anniversary of the grant date. No options were granted under the 2006 Cellectar Plan during 2010 or 2009. There have been no exercises of options issued under the 2006 Cellectar Plan. On March 17, 2011, in contemplation of the Acquisition, Cellectar terminated the remaining options outstanding under the 2006 Cellectar Plan as of that date.
2008 Stock Incentive Plan. The 2008 Stock Incentive Plan (the “2008 Plan”) provided Cellectar the ability to grant to employees, directors, consultants and other non-employees of Cellectar options to purchase common stock. The maximum aggregate number of shares that were subject to grant under the 2008 Plan was 823,930. Cellectar granted a total of 382,223 options under the 2008 Plan. A total of 162,300 and 300,488 options to purchase shares of Cellectar’s common stock were outstanding as of December 31, 2010 and 2009, respectively. These options generally vested annually over four years and expire on the tenth anniversary of the grant date. During 2009, 90,929 options were granted under the 2008 Plan. During 2010, no options were granted under the 2008 Plan. No options have been exercised. On March 17, 2011, in contemplation of the Acquisition, Cellectar terminated the remaining options outstanding under the 2008 Plan as of that date.
As of December 31, 2010, an aggregate of 1,066,941 shares were available for grant under the 2006 Cellectar Plan and 2008 Plan.
Cellectar’s Board of Directors determined exercise prices and vesting periods on the date of grant, subject to the provisions of the 2006 Cellectar Plan and 2008 Plan. Options have been granted at or above the estimated fair-market value of the common stock at the grant date. Options granted pursuant to the 2006 Cellectar Plan and 2008 Plan generally would have become fully vested in the event of a business combination whereby the options are not assumed or replaced by the surviving company, as defined.
In connection with the Acquisition, the Company assumed options to purchase 49,159 shares of common stock at exercise prices ranging from $1.53 to $1,072.53.
2006 Novelos Stock Option Plan. Following the Acquisition, option grants to directors and employees will be made under the Novelos Therapeutics 2006 Stock Incentive Plan (the “Plan”). On May 18, 2011, the Board of Directors of the Company approved certain amendments to the Plan to, among other things, increase the aggregate number of shares of the Company’s common stock reserved for issuance under the Plan (including any shares that have already been issued thereunder), to 7,000,000 and remove the 750,000 share annual individual limitation on grants under the Plan. On June 30, 2011, the Company’s stockholders ratified those amendments.
A total of 7,000,000 shares of common stock are reserved for issuance under the Plan for grants of incentive or nonqualified stock options, rights to purchase restricted and unrestricted shares of common stock, stock appreciation rights and performance share grants. A committee of the board of directors determines exercise prices, vesting periods and any performance requirements on the date of grant, subject to the provisions of the Plan. Options are granted at or above the fair market value of the common stock at the grant date and expire on the tenth anniversary of the grant date. Vesting periods are generally between one and four years. Options granted pursuant to the Plan generally will become fully vested upon a termination event occurring within one year following a change in control, as defined. A termination event is defined as either termination of employment or services other than for cause or constructive termination of employees or consultants resulting from a significant reduction in either the nature or scope of duties and responsibilities, a reduction in compensation or a required relocation. There are an aggregate of 3,476,112 shares available for grant under the Plan.
Accounting for Stock-Based Compensation
Employee stock-based compensation is accounted for in accordance with the guidance of FASB ASC Topic 718, Compensation – Stock Compensation which requires all share-based payments to employees, including grants of employee stock options, to be recognized in the financial statements based on their fair values. Non-employee stock-based compensation is accounted for in accordance with the guidance of FASB ASC Topic 505, Equity. As such, the Company recognizes expense based on the estimated fair value of options granted to non-employees over their vesting period, which is generally the period during which services are rendered and deemed completed by such non-employees.
F-18
The following table summarizes amounts charged to expense for stock-based compensation related to employee and director stock option grants and stock-based compensation recorded in connection with stock options granted to non-employee consultants:
|
Six Months Ended
June 30,
(unaudited)
|
Year Ended
December 31,
|
Cumulative
Development-
Stage Period
from
November 7,
2002 through
December 31,
|
||||||||||||||||||
|
Consolidated
2011
|
2010
|
2010
|
2009
|
2010
|
||||||||||||||||
|
Employee and director stock option grants:
|
||||||||||||||||||||
|
Research and development
|
$ | 54,991 | $ | 42,048 | $ | 61,791 | $ | 87,598 | $ | 298,386 | ||||||||||
|
General and administrative
|
136,455 | 199,305 | 291,549 | 414,401 | 1,577,303 | |||||||||||||||
| 191,446 | 241,353 | 353,340 | 501,999 | 1,875,689 | ||||||||||||||||
|
Non-employee consultant stock option grants:
|
||||||||||||||||||||
|
Research and development
|
17,287 | — | — | 200 | 71,724 | |||||||||||||||
|
General and administrative
|
48,853 | — | — | — | — | |||||||||||||||
| 66,140 | — | — | 200 | 71,724 | ||||||||||||||||
|
Total stock-based compensation
|
$ | 257,586 | $ | 241,353 | $ | 353,340 | $ | 502,199 | $ | 1,947,413 | ||||||||||
On July 14, 2010, the expiration date of vested options held by a former employee was extended until July 8, 2015. The extension constituted a modification to the terms of the award and additional stock-based compensation was measured as the excess of the fair value of the modified award over the fair value of the original award immediately before the modification. Accordingly, incremental stock-based compensation expense of $20,000 was recorded in connection with the modification.
The Company granted 3,496,400 stock options to employees and non-employees during the six months ended June 30, 2011 under the Plan. The Company issued options to purchase a total of 200,000 shares of common stock to non-employees outside of any formalized plan, but 100,000 were forfeited as a result of the cancelation and replacement as described below. Exercise prices for all grants made in the during the six months ended June 30, 2011 were equal to the market value of the Company’s common stock on the date of grant.
On May 18, 2011, the Company canceled 100,000 options originally granted on April 25, 2011 with an exercise price of $3.00 per share and issued 100,000 replacement stock option awards with an exercise price of $1.40. The cancelation and replacement constituted a modification to the terms of the award and additional stock-based compensation was measured as the excess of the fair value of the modified award over the fair value of the original award immediately before the modification. Accordingly, incremental stock-based compensation expense of $4,494 was recorded in connection with the modification.
Valuation and amortization method. The fair value of each stock award is estimated on the grant date using the Black-Scholes option-pricing model. The estimated fair value of employee stock options is amortized to expense using the straight-line method over the vesting period.
Volatility. Cellectar estimated volatility based on a review of volatility estimates of publicly held drug development companies in a similar stage of development. Subsequent to the Acquisition, the Company estimates volatility based on an average of (1) the Company’s historical volatility since its common stock has been publicly traded and (2) review of volatility estimates of publicly held drug development companies with similar market capitalizations.
Risk-free interest rate. The risk-free interest rate is based on the U.S. Treasury yield curve in effect at the time of grant commensurate with the expected term assumption.
Expected term . The expected term of stock options granted is based on an estimate of when options will be exercised in the future. The Company applied the simplified method of estimating the expected term of the options, as described in the SEC’s Staff Accounting Bulletins 107 and 110, as the Company has had a significant change in its business operations as result of the Acquisition and the historical experience is not indicative of the expected behavior in the future. The expected term, calculated under the simplified method, is applied to groups of stock options that have similar contractual terms. Using this method, the expected term is determined using the average of the vesting period and the contractual life of the stock options granted. The Company applied the simplified method to non-employees who have a truncation of term based on termination of service and utilizes the contractual life of the stock options granted for those non-employee grants which do not have a truncation of service.
F-19
Forfeitures. Stock-based compensation expense is recorded only for those awards that are expected to vest. FASB ASC Topic 718 requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. The term “forfeitures” is distinct from “cancellations” or “expirations” and represents only the unvested portion of the surrendered option. An annual forfeiture rate of 0% was applied to all unvested options as of December 31, 2010 as Cellectar had experienced very few forfeitures through 2009 and there was insufficient history to develop an accurate estimate of future forfeitures. An annual forfeiture rate of 0% was applied to all unvested options as of June 30, 2011 as the historical experience of forfeitures is not representative of expected future forfeiture rates as a result of the significant changes in the business operations as a result of the Acquisition. This analysis will be re-evaluated semi-annually and the forfeiture rate will be adjusted as necessary. Ultimately, the actual expense recognized over the vesting period will be for only those shares that vest.
The following table summarizes weighted-average values and assumptions used for options granted to employees, directors and consultants in the periods indicated:
|
Six Months
Ended June
30, 2011
(unaudited)
|
Year Ended
December 31,
2009
|
|||||||
|
Volatility
|
110 | % | 85 | % | ||||
|
Risk-free interest rate
|
1.84% – 3.17 | % | 1.72%-1.91 | % | ||||
|
Expected life (years)
|
5.5 – 6.25 | 6.25 | ||||||
|
Dividend
|
0 | % | 0 | % | ||||
|
Weighted-average exercise price
|
$ | 1.45 | $ | 0.76 | ||||
|
Weighted-average grant-date fair value
|
$ | 1.22 | $ | 0.55 | ||||
There were no stock options granted in the six months ended June 30, 2010 or the year ended December 31, 2010.
Stock Option Activity
A summary of stock option activity under stock option plans is as follows:
|
Number of
Shares
Issuable Upon
Exercise of
Outstanding
Options
|
Weighted
Average
Exercise Price
|
Weighted
Average
Remaining
Contracted
Term in
Years
|
Aggregate
Intrinsic
Value
|
|||||||||||||
|
Outstanding at November 7, 2002
|
—
|
|||||||||||||||
|
Granted
|
922,654
|
$
|
2.52
|
|||||||||||||
|
Forfeited
|
(12,653
|
)
|
$
|
3.04
|
||||||||||||
|
Outstanding at January 1, 2009
|
910,001
|
$
|
2.86
|
|||||||||||||
|
Granted
|
90,929
|
$
|
0.76
|
|||||||||||||
|
Forfeited
|
(9,194
|
)
|
$
|
2.72
|
||||||||||||
|
Outstanding at December 31, 2009
|
991,736
|
$
|
2.68
|
|||||||||||||
|
Canceled
|
(222,547
|
)
|
$
|
2.63
|
||||||||||||
|
Outstanding at December 31, 2010
|
769,189
|
$
|
2.69
|
|||||||||||||
|
Canceled
|
(769,189
|
)
|
$
|
2.69
|
||||||||||||
|
Options acquired in connection with a business combination
|
49,159
|
$
|
100.52
|
|||||||||||||
|
Granted
|
3,696,400
|
$
|
1.45
|
|||||||||||||
|
Canceled
|
(12,921
|
)
|
$
|
112.21
|
||||||||||||
|
Forfeited
|
(100,000
|
)
|
$
|
3.00
|
||||||||||||
|
Outstanding at June 30, 2011 (unaudited)
|
3,632,638
|
$
|
2.35
|
|||||||||||||
|
Vested, December 31, 2010
|
743,450
|
$
|
2.69
|
4.07
|
$
|
—
|
||||||||||
|
Unvested, December 31, 2010
|
25,739
|
$
|
2.62
|
5.08
|
$
|
—
|
||||||||||
|
Exercisable at December 31, 2010
|
743,450
|
$
|
2.69
|
4.07
|
$
|
—
|
||||||||||
|
Vested, June 30, 2011 (unaudited)
|
30,896
|
$
|
96.59
|
5.78
|
$
|
—
|
||||||||||
|
Unvested, June 30, 2011 (unaudited)
|
3,601,742
|
$
|
1.54
|
9.88
|
$
|
—
|
||||||||||
|
Exercisable at June 30, 2011 (unaudited)
|
30,896
|
$
|
96.59
|
5.78
|
$
|
—
|
||||||||||
F-20
The aggregate intrinsic value of options outstanding is calculated based on the positive difference between the estimated per share fair value of Cellectar’s common stock at the end of the respective period and the exercise price of the underlying options. As of December 31, 2010, the estimated fair-market value of Cellectar’s common stock at the end of the periods shown was less than the exercise price of the underlying options, as such, the aggregate intrinsic value is $0. As of June 30, 2011, the fair value of the Company’s common stock was less than the exercise price of the underlying options, as such, the aggregate intrinsic value was $0. There have been no option exercises to date. Shares of common stock issued upon the exercise of options are from authorized but unissued shares.
The weighted-average grant-date fair value of options granted during the year ended December 31, 2009 and for the period November 2, 2002 to December 31, 2009 was $0.55 and $1.90, respectively. There were no options granted during the year ended December 31, 2010. The total fair value of shares vested during December 31, 2010 and 2009 and for the period November 2, 2007 (date of inception) to December 31, 2010 was $199,600, $185,600 and $2,050,000, respectively. The weighted-average grant-date fair value of vested and unvested options outstanding at December 31, 2010 and 2009 was $2.01 and $1.91 and $1.85 and $1.88, respectively.
As of December 31, 2010, there was approximately $58,000 and $0 of total unrecognized compensation cost related to unvested stock-based compensation arrangements related to employees and non-employees, respectively. Of the total unrecognized amount as of December 31, 2010, all was recognized in the six months ended June 30, 2011.
On March 4, 2011, in contemplation of the Acquisition and in accordance with terms of the applicable option agreements, Cellectar accelerated the vesting on all outstanding and unvested options at that date and notified all option holders that any unexercised options as of March 17, 2011 would then be terminated. On March 17, 2011, Cellectar terminated all outstanding options. The remaining unamortized compensation expense of $58,000 was recorded related to the acceleration of outstanding options in the quarter ended March 31, 2011. No additional compensation expense was recorded related to the acceleration of unvested shares as the acceleration did not represent a modification to the original terms of the options.
As of June 30, 2011, there was $3,008,021 of total unrecognized compensation cost related to unvested stock-based compensation arrangements. Of this total amount, the Company expects to recognize $561,084, $1,125,400, $858,452, $387,397 and $75,688 during 2011, 2012, 2013, 2014 and 2015, respectively. The Company expects 3,601,742 in unvested options to vest in the future. The weighted-average grant-date fair value of vested and unvested options outstanding at June 30, 2011 was $3.98 and $1.17, respectively.
12. INCOME TAXES
Deferred tax assets consisted of the following at December 31:
|
2010
|
2009
|
|||||||
|
Deferred tax assets
|
||||||||
|
Federal net operating loss
|
$
|
6,116,804
|
$
|
4,754,001
|
||||
|
Federal research and development tax credit carryforwards
|
390,600
|
273,788
|
||||||
|
Wisconsin net operating loss credit carryforwards
|
814,492
|
589,548
|
||||||
|
Wisconsin research and development tax credit carryforwards
|
220,738
|
171,552
|
||||||
|
Stock-based compensation expense
|
552,859
|
415,060
|
||||||
|
Charitable contribution carryforwards
|
49,725
|
49,725
|
||||||
|
Accrued liabilities
|
25,327
|
75,711
|
||||||
|
Total deferred tax assets
|
8,170,545
|
6,329,385
|
||||||
|
Deferred tax liabilities
|
||||||||
|
Depreciable and intangible assets
|
(434,056
|
)
|
(475,524
|
)
|
||||
|
Total deferred tax liabilities
|
(434,056
|
)
|
(475,524
|
)
|
||||
|
Net deferred tax assets
|
7,736,489
|
5,853,861
|
||||||
|
Less valuation allowance
|
(7,736,489
|
)
|
(5,853,861
|
)
|
||||
|
Total deferred tax assets
|
$
|
—
|
$
|
—
|
||||
F-21
As of December 31, 2010, Cellectar had federal and state net operating loss carryforwards (“NOLs”) of approximately $15,684,000 and $15,633,000 respectively, which expire beginning in 2030 and 2025, respectively. In addition, Cellectar has federal and state research and development and investment tax credits of approximately $391,000 and $335,000, respectively. The amount of NOLs which may be utilized annually in future periods will be limited pursuant to Section 382 of the Internal Revenue Code as a result of substantial changes in the Company’s ownership that have occurred or that may occur in the future. The Company has not quantified the amount of such limitations.
Because of the limited operating history, continuing losses and uncertainty associated with the utilization of the NOLs in the future, management has provided a full allowance against the gross deferred tax asset.
Cellectar did not have unrecognized tax benefits or accrued interest and penalties at any time during the years ended December 31, 2010 and 2009, and does not anticipate having unrecognized tax benefits over the next twelve months. The Company is subject to audit by the IRS for tax periods commencing January 1, 2007.
For the six-month period ended June 30, 2011, the federal and state NOLs increased by approximately $3,560,000 as a result of the loss recorded.
13. NET LOSS PER SHARE
Basic net loss per share is computed by dividing net loss attributable to common stockholders by the weighted average number of shares of common stock outstanding during the period. Diluted net loss per share is computed by dividing net loss attributable to common stockholders, as adjusted, by the sum of the weighted average number of shares of common stock and the dilutive potential common stock equivalents then outstanding. Potential common stock equivalents consist of stock options and convertible debt. Since there is a net loss attributable to common stockholders for the six months ended June 30, 2011 and 2010 and the years ended December 31, 2010 and 2009, the inclusion of common stock equivalents in the computation for those periods would be antidilutive. Accordingly, basic and diluted net loss per share is the same for all periods presented.
The following potentially dilutive securities have been excluded from the computation of diluted net loss per share since their inclusion would be antidilutive:
|
Six Months Ended June 30,
(unaudited)
|
Twelve Months Ended
December 31,
|
Cumulative
Development-
Stage Period
from November
7, 2002
(inception)
through
December 31,
|
||||||||||||||||||
|
Consolidated
2011
|
2010
|
2010
|
2009
|
2010
|
||||||||||||||||
|
Convertible debt
|
—
|
3,448,026
|
3,646,370
|
—
|
3,646,370
|
|||||||||||||||
|
Warrants
|
7,327,322
|
—
|
—
|
—
|
—
|
|||||||||||||||
|
Stock options
|
3,632,638
|
991,061
|
769,189
|
991,736
|
769,189
|
|||||||||||||||
14. COMMITMENTS
Property Lease
On September 5, 2007, Cellectar entered into a 36-month lease for office and manufacturing space, commencing September 15, 2007. The lease provides for the option to extend the lease under its current terms for seven additional two-year terms. Rent is $8,050 per month for the first year and then escalates by 3% per year for the duration of the term including any lease extension terms. The lease also requires the payment of monthly rent of $1,140 for approximately 3,400 square feet of expansion space. The monthly rent for the expansion space is fixed until such time as the expansion space is occupied at which time the rent would increase to the current per square foot rate in effect under the original lease terms. The Company is responsible for certain building-related costs such as property taxes, insurance, and repairs and maintenance. Rent expense is recognized on a straight-line basis and accordingly the difference between the recorded rent expense and the actual cash payments has been recorded as deferred rent as of each balance sheet dates. Due to the significant value of leasehold improvements purchased during the initial 3-year lease term and the economic penalty for not extending the building lease, straight-line rent expense and the associated deferred rent has been calculated over 17 years, which represents the full term of the lease, including all extensions.
F-22
The Company is required to remove certain alterations, additions and improvements upon termination of the lease that altered a portion of the rentable space. In no event shall the cost of such removal, at commercially reasonable rates, paid by the Company exceed $55,000 (“Capped Amount”) and any amount in excess of the Capped Amount shall be the obligation of the landlord. The Company is required to maintain a certificate of deposit equal to the Capped Amount during the term of the lease, which amount is shown as restricted cash in the accompanying balance sheets.
Effective June 1, 2010, Cellectar entered into a seven-month extension of its office space and effective December 13, 2010 amended the extension to increase the lease extension an additional five-months, expiring May 31, 2011. In connection with the extension, the monthly rent was adjusted to fifty percent of the rent due immediately prior to the extension and the Company could terminate the lease at the end of a month with 10-day written notice. The option was retained, prior to May 31, 2011, to further extend the lease through September 14, 2012, in accordance with the original lease terms provided that it pay the unpaid rent for June 1, 2010 through March 31, 2011, based on the original terms of the lease, plus interest at 10% per annum. As of December 31, 2010, $45,000 was accrued in the accompanying balance sheet for the unpaid rent and accrued interest.
On April 15, 2011, the Company extended its building lease for the Madison, WI headquarters through September 14, 2012 according to the terms in the original lease and paid all unpaid rent and related accrued interest.
Future minimum lease payments under this non-cancelable lease are approximately $196,000 and $124,000 during 2011 and 2012.
As a result of the Acquisition, the Company assumed the lease agreement for Novelos’ office space in Newton, MA, which has a term that is month-to-month and requires monthly rental payments of $5,300.
Rent expense was $98,000 and $75,000 for the six months ended June 30, 2011 and 2010, respectively, $159,000 and $180,000 for the years ended December 31, 2010 and 2009, respectively and $945,000 from inception to December 31, 2010.
Capital Lease
Certain equipment is leased under a capital lease. The lease agreement requires monthly principal and interest payments of $217 and expires on September 3, 2014. The outstanding obligation is being amortized using a 7% interest rate based on comparable borrowing rates.
The following table provides the estimated future minimum rental payments under all capital leases together with the present value of the net minimum lease payments as of December 31, 2010:
|
Minimum
lease
payments
|
Less interest
|
Present value
of net
minimum
lease
payments
|
||||||||||
|
2011
|
$
|
2,608
|
$
|
523
|
$
|
2,085
|
||||||
|
2012
|
2,608
|
373
|
2,235
|
|||||||||
|
2013
|
2,608
|
211
|
2,397
|
|||||||||
|
2014
|
1,739
|
45
|
1,694
|
|||||||||
|
$
|
9,563
|
$
|
1,152
|
$
|
8,411
|
|||||||
The equipment recorded under capitalized leases is included in fixed assets as of December 31:
|
2010
|
2009
|
|||||||
|
Office equipment
|
$
|
10,973
|
$
|
10,973
|
||||
|
Less accumulated amortization
|
(2,928
|
)
|
(732
|
)
|
||||
|
$
|
8,045
|
$
|
10,241
|
|||||
Retention Agreements
Following the Acquisition, the Company has retention agreements with each of its four vice presidents, three of such agreements were executed by Novelos during 2010. The agreements provide for the lump-sum payment of six months’ base salary and benefits to each such officer following a termination without cause or a resignation with good reason occurring on or before November 14, 2011. Certain of the agreements provide that if the executives were employed with Novelos as of October 1, 2010, they would receive a payment of two months’ base salary as a retention bonus on that date. The retention bonus of $68,000 was paid in October 2010 and will be deducted from the severance amounts that may become payable upon a subsequent involuntary termination. The total remaining amount that may become payable to the Company’s executive officers pursuant to the retention agreements is approximately $350,000.
F-23
On May 18, 2011, the Company’s board of directors approved the entry into a retention agreement with each of 5 individuals that were employees of Cellectar at the time of the Acquisition. The agreements provide for the lump-sum payment of six months’ base salary and benefits following a termination without cause or a resignation with good reason occurring on or before November 18, 2011. The agreements expire November 18, 2011.
|
15.
|
LITIGATION
|
Following the Acquisition, the Company is party to certain legal matters that existed with Novelos prior to the Acquisition. The following summarizes the status of those matters.
Class Action
A putative federal securities class action complaint was filed on March 5, 2010 in the United States District Court for the District of Massachusetts by an alleged shareholder of Novelos, on behalf of himself and all others who purchased or otherwise acquired Novelos common stock in the period between December 14, 2009 and February 24, 2010, against Novelos and its President and Chief Executive Officer, Harry S. Palmin. On October 1, 2010, the court appointed lead plaintiffs (Boris Urman and Ramona McDonald) and appointed lead plaintiffs’ counsel. On October 22, 2010, an amended complaint was filed. The amended complaint claims, among other things, that Novelos violated Section 10(b) of the Securities Exchange Act of 1934, as amended, and Rule 10b-5 promulgated thereunder in connection with alleged misleading disclosures related to the progress of the Phase 3 clinical trial of NOV-002 for non-small cell lung cancer. On December 6, 2010, the defendants filed a motion to dismiss the complaint with prejudice. On January 20, 2011, the plaintiffs filed their opposition to our motion and on March 3, 2011, the defendants filed their response to the opposition. On June 23, 2011, the motion to dismiss was granted and the case was dismissed without prejudice. Because the dismissal was without prejudice, the plaintiffs could reinstitute the proceeding by filing an amended complaint. On August 5, 2011, the plaintiffs filed a second amended complaint realleging that the defendants violated Section 10(b) of the Exchange Act and Rule 10b-5 in connection with alleged misleading disclosures related to the Phase 3 clinical trial for NOV-002 in non-small cell lung cancer. On September 9, 2011, the defendants filed a motion to dismiss the second amended complaint. The plaintiff’s opposition to the motion is due by October 7, 2011. The Company believes the allegations are without merit and intends to vigorously defend against them.
BAM Dispute
On June 28, 2010, Novelos received a letter from counsel to ZAO BAM and ZAO BAM Research Laboratories (Russian companies, collectively referred to as “BAM”) alleging that it modified the chemical composition of NOV-002 without prior notice to or approval from BAM, constituting a material breach of a technology and assignment agreement Novelos had entered into with BAM on June 20, 2000 (the “June 2000 Agreement”). The letter references the amendment, submitted to the FDA on August 30, 2005, to Novelos’ investigational new drug application dated August 1999 as the basis for BAM’s claims and demands the transfer of all intellectual property rights concerning NOV-002 to BAM. Mark Balazovsky, a director of Novelos from June 1996 until November 2006 and a shareholder of Novelos through at least June 25, 2010, is, to our knowledge, still the general director and principal shareholder of ZAO BAM. On September 24, 2010, Novelos filed a complaint in Suffolk Superior Court seeking a declaratory judgment by the court that the June 2000 Agreement has been replaced by a subsequent agreement between the parties dated April 1, 2005 (the “April 2005 Agreement”), that Novelos’ obligations to BAM are governed solely by the April 2005 Agreement and that the obligations of the June 2000 agreement have been performed and fully satisfied. On November 29, 2010, BAM answered the complaint, denying the material allegations, and stating its affirmative defenses and certain counterclaims. On January 14, 2011, Novelos responded to the counterclaims, denying BAM’s material allegations and stating its affirmative defenses. On June 9, 2011, BAM filed an amended counterclaim alleging additional claims related to Novelos’ acquisition of Cellectar. In that amended counterclaim, BAM alleges that the acquisition evidences Novelos’ abandonment of the technology assigned to it by BAM constituting a breach of the June 2000 Agreement or, if that agreement is determined to no longer be in effect, a breach of the April 2005 Agreement and/or a breach of the implied duty of good faith and fair dealing with respect to the April 2005 Agreement. On June 15, 2011 the Company filed its response to their amended counterclaim. On August 5, 2011, the Company filed a motion for judgment on the pleadings as to its declaratory judgment count and all counts of BAM’s amended counterclaim. The motion has been opposed by BAM and a hearing on the motion is scheduled on September 27, 2011. The Company believes BAM’s allegations and counterclaims are without merit and intend to defend vigorously against them.
We do not anticipate that these litigation contingencies will have a material impact on the Company’s future financial position, results of operations or cash flows.
16. EMPLOYEE RETIREMENT PLAN
On January 1, 2009, Cellectar adopted a Safe Harbor defined contribution plan under Section 401(k) of the Internal Revenue Code which covered eligible employees who meet minimum age requirements and allowed participants to contribute a portion of their annual compensation on a pre-tax basis. Cellectar contributed 3% of each participant’s compensation. Contributions made for the years ended December 31, 2010 and 2009 were $23,000 and $56,000, respectively. Cellectar paid administrative expenses for the plan of $3,300 and $2,800 for the years ended December 31, 2010 and 2009, respectively. The plan was canceled effective August 30, 2010.
F-24
Following the Acquisition, the Company has a defined contribution plan under Section 401(k) of the Internal Revenue Code which allows eligible employees who meet minimum age requirements to contribute a portion of their annual compensation on a pre-tax basis. The Company has not made any matching contributions under this plan.
17. RELATED PARTY TRANSACTIONS
Jamey Weichert, the Company’s Chief Scientific Officer, director, shareholder and principal founder is a faculty member at the University of Wisconsin-Madison (“UW”). Cellectar paid $16,082 to the UW during 2009 for research related activities.
Cellectar made contributions of $25,000 to the UW Foundation, a not-for-profit, tax-exempt Wisconsin corporation, which serves as the official fundraising and gift-receiving organization for the UW and other donor-designated units of the UW System during the year ended December 31, 2009. During the six months ended June 30, 2011, the Company made contributions totaling $62,500 to the UW Foundation for use towards research activities associated with the development of the Company’s compounds. No payments were made to UW during the six months ended June 30, 2010 or the year ended December 31, 2010.
18. (UNAUDITED) SUPPLEMENTAL PRO FORMA INFORMATION
The table below summarizes net loss for the periods shown as though the Acquisition occurred as of January 1, 2010:
|
For the Six Months Ended June 30,
|
For the Twelve
Months Ended
December 31,
|
|||||||||||
|
2011
|
2010
|
2010
|
||||||||||
|
Net loss
|
$
|
(3,381,503
|
)
|
$
|
(730,792
|
)
|
$
|
(1,704,966
|
)
|
|||
The pro forma net loss has been adjusted for the following:
|
1)
|
Elimination of $165,000, $169,000, and $361,000 of interest expense for the six months ended June 30, 2011 and 2010 and the twelve months ended December 31, 2010, respectively; such amounts relate to interest accrued on the Convertible Notes which were converted immediately prior to the Acquisition (see Note 7) and the Bank Note which was paid in full settlement of the note immediately prior to the Acquisition (see Note 8).
|
|
2)
|
Recognition of a additional BCF of $463,000 in the six months ended June 30, 2010 and the year ended December 31, 2010 and the elimination of BCF of $258,000 in the six months ended June 30, 2011 in connection with the conversion of the Convertible Notes, which is assumed to have occurred on January 1, 2010 for the purpose of pro forma presentation (see Note 7).
|
|
3)
|
Elimination of Acquisition costs incurred during the six months ended June 30, 2011 and the twelve months ended December 31, 2010, which are assumed to have been incurred prior to January 1, 2010 for the purpose of presentation in the pro forma statements of operations.
|
|
4)
|
Elimination of $450,000 of investment banking fees incurred upon the consummation of the Acquisition on April 8, 2011 from the six months ended June 30, 2011.
|
|
5)
|
Elimination of dividends and deemed dividends on Novelos’ preferred convertible stock, which is assumed to have been exchanged for common stock at January 1, 2010 in order to reflect the post-acquisition capital structure for the purpose of pro forma presentation.
|
|
6)
|
Elimination of Novelos historical revenue related to the amortization of deferred revenue that was determined to have no fair value in purchase accounting.
|
|
7)
|
Elimination of liquidated damages accrued in 2010 related to Novelos’ convertible preferred stock. The liquidated damages are assumed not to have accrued as the preferred stock is assumed to have been exchanged for common stock at January 1, 2010 in order to reflect the post-acquisition capital structure for the purpose of pro forma presentation.
|
|
19.
|
PROPOSED REVERSE STOCK SPLIT
|
On June 30, 2011, the Company held a special meeting of stockholders. At the meeting, the stockholders approved, among other things, separate amendments to the certificate of incorporation that would effect a reverse split of the Company’s common stock within a range of 1:2 to 1:10, and authorized the Company’s board of directors to determine the ratio at which the reverse split will be effected by filing the appropriate amendment to the certificate of incorporation. The purpose of the proposed reverse split is to increase the price per share of the Company’s common stock in order to exceed the minimum price per share required to secure a listing on a national securities exchange. On July 1, 2011, the Company filed a Registration Statement on Form S-1 for an underwritten public offering of its common stock with proceeds of up to $15,000,000, excluding the underwriters over-allotment. In connection with that proposed offering, the Company has applied for a listing of its common stock on the NASDAQ Capital Market.
F-25
FINANCIAL STATEMENTS
FINANCIAL STATEMENTS FOR NOVELOS THERAPEUTICS, INC. (PRE-ACQUISITION)
Table of Contents
|
Page
|
||
|
Report of Independent Registered Public Accounting Firm
|
F-27 | |
|
Balance Sheets at December 31, 2010 and 2009
|
F-28 | |
|
Statements of Operations for the Years Ended December 31, 2010 and 2009
|
F-29 | |
|
Statements of Redeemable Preferred Stock and Stockholders’ Equity (Deficiency) for the Years Ended December 31, 2010 and 2009
|
F-30 | |
|
Statements of Cash Flows for the Years Ended December 31, 2010 and 2009
|
F-31 | |
|
Notes to Financial Statements
|
F-32 | |
|
Balance Sheet at March 31, 2011
|
F-48 | |
|
Statements of Operations for the Three Months Ended March 31, 2011 and 2010
|
F-49 | |
|
Statements of Cash Flows for the Three Months Ended March 31, 2011 and 2010
|
F-50 | |
|
Notes to Interim Financial Statements
|
F-51 |
F-26
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and Board of Directors
Novelos Therapeutics, Inc.
Newton, Massachusetts
We have audited the accompanying balance sheets of Novelos Therapeutics, Inc. as of December 31, 2010 and 2009 and the related statements of operations, redeemable preferred stock and stockholders’ equity (deficiency), and cash flows for the years then ended. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Novelos Therapeutics, Inc. as of December 31, 2010 and 2009 and the results of its operations, changes in redeemable preferred stock and stockholders’ equity (deficiency), and cash flows for the years then ended in conformity with accounting principles generally accepted in the United States.
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the financial statements, the Company has generated insignificant revenues and has incurred continuing losses in the development of its products. These factors raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in this regard are described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ Stowe & Degon LLC
Westborough, Massachusetts
April 11, 2011
F-27
NOVELOS THERAPEUTICS, INC. (PRE-ACQUISITION)
BALANCE SHEETS
|
December 31,
|
December 31,
|
|||||||
|
2010
|
2009
|
|||||||
|
ASSETS
|
||||||||
|
CURRENT ASSETS:
|
||||||||
|
Cash and equivalents
|
$
|
2,372,951
|
$
|
8,769,529
|
||||
|
Prepaid expenses and other current assets
|
63,526
|
102,923
|
||||||
|
Total current assets
|
2,436,477
|
8,872,452
|
||||||
|
FIXED ASSETS, NET
|
8,755
|
44,097
|
||||||
|
DEPOSITS
|
15,350
|
15,350
|
||||||
|
TOTAL ASSETS
|
$
|
2,460,582
|
$
|
8,931,899
|
||||
|
LIABILITIES, REDEEMABLE PREFERRED STOCK AND STOCKHOLDERS’ DEFICIENCY
|
||||||||
|
CURRENT LIABILITIES:
|
||||||||
|
Accounts payable and accrued liabilities
|
$
|
565,723
|
$
|
3,299,217
|
||||
|
Accrued compensation
|
—
|
245,711
|
||||||
|
Accrued dividends
|
—
|
2,902,963
|
||||||
|
Derivative liability (see Note 2)
|
288,250
|
10,486,594
|
||||||
|
Deferred revenue – current
|
33,333
|
33,333
|
||||||
|
Total current liabilities
|
887,306
|
16,967,818
|
||||||
|
DEFERRED REVENUE – NONCURRENT
|
366,667
|
400,000
|
||||||
|
COMMITMENTS AND CONTINGENCIES
|
||||||||
|
REDEEMABLE PREFERRED STOCK:
|
||||||||
|
Series E convertible preferred stock, $0.00001 par value; 735 shares designated, 0 and 548.26078125 shares issued and outstanding at December 31, 2010 and 2009, respectively (see Note 6)
|
—
|
18,459,619
|
||||||
|
STOCKHOLDERS’ EQUITY (DEFICIENCY):
|
||||||||
|
Preferred Stock, $0.00001 par value; 7,000 shares authorized: Series C 8% cumulative convertible preferred stock; 272 shares designated; 0 and 204 issued and outstanding at December 31, 2010 and 2009, respectively
|
—
|
—
|
||||||
|
Common stock, $0.00001 par value; 150,000,000 shares authorized; 2,959,914 and 455,281 shares issued and outstanding at December 31, 2010 and 2009, respectively
|
30
|
5
|
||||||
|
Additional paid-in capital
|
75,183,275
|
49,176,545
|
||||||
|
Accumulated deficit
|
(73,976,696
|
)
|
(76,072,088
|
)
|
||||
|
Total stockholders’ equity (deficiency)
|
1,206,609
|
(26,895,538
|
)
|
|||||
|
TOTAL LIABILITIES, REDEEMABLE PREFERRED STOCK AND STOCKHOLDERS’ EQUITY (DEFICIENCY)
|
$
|
2,460,582
|
$
|
8,931,899
|
||||
See report of independent registered public accounting firm and notes to financial statements.
F-28
NOVELOS THERAPEUTICS, INC. (PRE-ACQUISITION)
STATEMENTS OF OPERATIONS
|
Year Ended December 31,
|
||||||||
|
2010
|
2009
|
|||||||
|
REVENUES
|
$
|
33,334
|
$
|
33,334
|
||||
|
COSTS AND EXPENSES:
|
||||||||
|
Research and development
|
2,997,984
|
8,080,242
|
||||||
|
General and administrative
|
2,486,032
|
2,182,253
|
||||||
|
Total costs and expenses
|
5,484,016
|
10,262,495
|
||||||
|
LOSS FROM OPERATIONS
|
(5,450,682
|
)
|
(10,229,161
|
)
|
||||
|
OTHER INCOME (EXPENSE):
|
||||||||
|
Interest income
|
2,421
|
1,013
|
||||||
|
Grant income
|
244,479
|
62,980
|
||||||
|
Gain (loss) on derivative warrants (see Note 2)
|
8,118,174
|
(12,114,371
|
)
|
|||||
|
Liquidated damages (see Note 6)
|
(819,000
|
)
|
—
|
|||||
|
Miscellaneous
|
—
|
6,233
|
||||||
|
Total other income (expense)
|
7,546,074
|
(12,044,145
|
)
|
|||||
|
NET INCOME (LOSS)
|
2,095,392
|
(22,273,306
|
)
|
|||||
|
PREFERRED STOCK DIVIDENDS
|
(2,207,827
|
)
|
(3,296,289
|
)
|
||||
|
PREFERRED STOCK DEEMED DIVIDENDS
|
(12,541,201
|
)
|
(714,031
|
)
|
||||
|
NET LOSS ATTRIBUTABLE TO COMMON STOCKHOLDERS
|
$
|
(12,653,636
|
)
|
$
|
(26,283,626
|
)
|
||
|
BASIC AND DILUTED NET LOSS ATTRIBUTABLE TO COMMON STOCKHOLDERS PER COMMON SHARE
|
$
|
(15.36
|
)
|
$
|
(80.57
|
)
|
||
|
SHARES USED IN COMPUTING BASIC AND DILUTED NET LOSS ATTRIBUTABLE TO COMMON STOCKHOLDERS PER COMMON SHARE
|
823,933
|
326,209
|
||||||
See report of independent registered public accounting firm and notes to financial statements.
F-29
NOVELOS THERAPEUTICS, INC. (PRE-ACQUISITION)
STATEMENTS OF REDEEMABLE PREFERRED STOCK AND STOCKHOLDERS’ EQUITY (DEFICIENCY)
|
REDEEMABLE
PREFERRED STOCK
Series D and E
Convertible
Preferred Stock
|
Common Stock
|
Series C Cumulative
Convertible
Preferred Stock
|
Additional
Paid-in
Capital
|
Accumulated
Deficit
|
Total
Stockholders’
Equity
(Deficiency)
|
|||||||||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Par
Amount
|
Shares
|
Par
Amount
|
|||||||||||||||||||||||||||||||
|
BALANCE AT JANUARY 1, 2009
|
413.5 | $ | 13,904,100 | 287,423 | $ | 3 | 272 | $ | — | $ | 40,204,549 | $ | (59,693,153 | ) | $ | (19,488,601 | ) | |||||||||||||||||||
|
Reclassification of warrants to derivative liability (see Note 2)
|
— | — | — | — | — | — | (6,893,316 | ) | 5,894,371 | (998,945 | ) | |||||||||||||||||||||||||
|
Conversion of Series C convertible preferred stock and accumulated dividends into common stock
|
— | — | 10,058 | — | (68 | ) | — | 184,246 | — | 184,246 | ||||||||||||||||||||||||||
|
Conversion of Series E convertible preferred stock and accumulated dividends into common stock
|
(97.18209375 | ) | (3,213,056 | ) | 51,889 | 1 | — | — | 3,514,313 | — | 3,514,314 | |||||||||||||||||||||||||
|
Cashless exercise of warrants
|
— | — | 3,162 | — | — | — | 1,000,962 | — | 1,000,962 | |||||||||||||||||||||||||||
|
Issuance of common stock in exchange for warrants
|
— | — | 13,623 | — | — | — | 1,625,760 | — | 1,625,760 | |||||||||||||||||||||||||||
|
Issuance of common stock and warrants in a private placement, net of issuance costs of $61,116
|
— | — | 89,126 | 1 | — | — | 8,938,883 | — | 8,938,884 | |||||||||||||||||||||||||||
|
Compensation expense associated with options issued to employees
|
— | — | — | — | — | — | 437,066 | — | 437,066 | |||||||||||||||||||||||||||
|
Compensation expense associated with options issued to non-employees
|
— | — | — | — | — | — | 427,271 | — | 427,271 | |||||||||||||||||||||||||||
|
Issuance of Series E redeemable convertible preferred stock and warrants, net of issuance costs of $795,469
|
200 | 6,297,323 | — | — | — | — | 2,907,208 | — | 2,907,208 | |||||||||||||||||||||||||||
|
Issuance of Series E redeemable convertible preferred stock in payment of accumulated dividends
|
31.942875 | 1,597,144 | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
|
Adjustment to record the carrying value of Series E redeemable convertible preferred stock at fair value on the date of sale
|
— | (125,892 | ) | — | — | — | — | 125,892 | — | 125,892 | ||||||||||||||||||||||||||
|
Fair value of the extension of expiration date of warrants
|
— | — | — | — | — | — | 839,923 | — | 839,923 | |||||||||||||||||||||||||||
|
Accretion of deemed dividend associated with the extension of expiration date of warrants
|
— | — | — | — | — | — | (839,923 | ) | — | (839,923 | ) | |||||||||||||||||||||||||
|
Dividends accrued on preferred stock
|
— | — | — | — | — | — | (3,296,289 | ) | — | (3,296,289 | ) | |||||||||||||||||||||||||
|
Net loss
|
— | — | — | — | — | — | — | (22,273,306 | ) | (22,273,306 | ) | |||||||||||||||||||||||||
|
BALANCE AT DECEMBER 31, 2009
|
548.26078125 | 18,459,619 | 455,281 | 5 | 204 | — | 49,176,545 | (76,072,088 | ) | (26,895,538 | ) | |||||||||||||||||||||||||
|
Exercise of stock options
|
— | — | 5,991 | — | — | — | 158,567 | — | 158,567 | |||||||||||||||||||||||||||
|
Conversion of Series E convertible preferred stock and accumulated dividends into common stock
|
(139.99673625 | ) | (4,689,593 | ) | 76,770 | 1 | — | — | 5,324,517 | — | 5,324,518 | |||||||||||||||||||||||||
|
Cashless exercise of warrants
|
— | — | 53,478 | 1 | — | — | 2,584,396 | — | 2,584,397 | |||||||||||||||||||||||||||
|
Compensation expense associated with options issued to employees
|
— | — | — | — | — | — | 586,321 | — | 586,321 | |||||||||||||||||||||||||||
|
Compensation expense (benefit) associated with options issued to non-employees
|
— | — | — | — | — | — | (249,023 | ) | — | (249,023 | ) | |||||||||||||||||||||||||
|
Issuance of common stock and warrants in a public offering (net of issuance costs of $250,862)
|
— | — | 140,056 | 1 | — | — | 744,910 | — | 744,911 | |||||||||||||||||||||||||||
|
Fair value of warrants issued to preferred shareholders
|
— | — | — | — | — | — | 586,050 | — | 586,050 | |||||||||||||||||||||||||||
|
Accretion of deemed dividend associated with the issuance of warrants to preferred stockholders
|
— | — | — | — | — | — | (586,050 | ) | — | (586,050 | ) | |||||||||||||||||||||||||
|
Exchange of preferred stock and dividends for common stock
|
(408.264045 | ) | (13,770,026 | ) | 2,228,338 | 22 | (204 | ) | — | 19,064,869 | — | 19,064,891 | ||||||||||||||||||||||||
|
Fair value of incremental shares issued to preferred stockholders in connection with exchange of preferred stock for common stock
|
— | — | — | — | — | — | 11,955,151 | — | 11,955,151 | |||||||||||||||||||||||||||
|
Accretion of deemed dividend associated with the incremental shares of common stock issued in connection with the exchange of preferred stock
|
— | — | — | — | — | — | (11,955,151 | ) | — | (11.955,151 | ) | |||||||||||||||||||||||||
|
Dividends accrued on preferred stock
|
— | — | — | — | — | — | (2,207,827 | ) | — | (2,207,827 | ) | |||||||||||||||||||||||||
|
Net income
|
— | — | — | — | — | — | — | 2,095,392 | 2,095,392 | |||||||||||||||||||||||||||
|
BALANCE AT DECEMBER 31, 2010
|
— | $ | $— | 2,959,914 | $ | 30 | — | $ | — | $ | 75,183,275 | $ | (73,976,696 | ) | $ | 1,206,609 | ||||||||||||||||||||
See report of independent registered public accounting firm and notes to financial statements.
F-30
NOVELOS THERAPEUTICS, INC. (PRE-ACQUISITION)
STATEMENTS OF CASH FLOWS
|
Year Ended December 31,
|
||||||||
|
2010
|
2009
|
|||||||
|
CASH FLOWS FROM OPERATING ACTIVITIES:
|
||||||||
|
Net income (loss)
|
$
|
2,095,392
|
$
|
(22,273,306
|
)
|
|||
|
Adjustments to reconcile net income (loss) to cash used in operating activities:
|
||||||||
|
Depreciation and amortization
|
35,342
|
32,354
|
||||||
|
Stock-based compensation
|
337,298
|
864,337
|
||||||
|
(Gain) loss on derivative warrants
|
(8,118,174
|
)
|
12,114,371
|
|||||
|
Non-cash settlement of liquidated damages
|
819,000
|
—
|
||||||
|
Changes in:
|
||||||||
|
Prepaid expenses and other current assets
|
39,397
|
26,862
|
||||||
|
Accounts payable and accrued liabilities
|
(2,733,494
|
)
|
(1,354,695
|
)
|
||||
|
Accrued compensation
|
(245,711
|
)
|
5,072
|
|||||
|
Deferred revenue
|
(33,333
|
)
|
(33,333
|
)
|
||||
|
Cash used in operating activities
|
(7,804,283
|
)
|
(10,618,338
|
)
|
||||
|
CASH FLOWS FROM INVESTING ACTIVITIES:
|
||||||||
|
Purchases of fixed assets
|
—
|
(18,000
|
)
|
|||||
|
Cash used in investing activities
|
—
|
(18,000
|
)
|
|||||
|
CASH FLOWS FROM FINANCING ACTIVITIES:
|
||||||||
|
Proceeds from issuance of common stock, net
|
1,249,138
|
8,938,884
|
||||||
|
Proceeds from issuance of Series E convertible preferred stock and warrants, net
|
—
|
9,204,531
|
||||||
|
Proceeds from exercise of stock options
|
158,567
|
—
|
||||||
|
Cash provided by financing activities
|
1,407,705
|
18,143,415
|
||||||
|
INCREASE (DECREASE) IN CASH AND EQUIVALENTS
|
(6,396,578
|
)
|
7,507,077
|
|||||
|
CASH AND EQUIVALENTS AT BEGINNING OF YEAR
|
8,769,529
|
1,262,452
|
||||||
|
CASH AND EQUIVALENTS AT END OF YEAR
|
$
|
2,372,951
|
$
|
8,769,529
|
||||
|
SUPPLEMENTAL DISCLOSURES OF NON-CASH FINANCING ACTIVITIES
|
||||||||
|
Dividends accumulated on shares of Series E preferred stock exchanged for or converted into shares of common stock
|
$
|
5,110,790
|
$
|
1,898,402
|
||||
|
Dividends accumulated on shares of Series C preferred stock converted into shares of common stock
|
$
|
—
|
$
|
184,246
|
||||
|
Fair value of derivative warrants upon adoption of new accounting principle
|
$
|
—
|
$
|
998,945
|
||||
|
Fair value of common stock issued in exchange for tender of derivative warrants
|
$
|
—
|
$
|
1,625,760
|
||||
|
Fair value of derivative warrants upon cashless exercise
|
$
|
2,584,397
|
$
|
1,000,962
|
||||
|
Exchange of Series D for Series E preferred stock
|
$
|
—
|
$
|
13,904,100
|
||||
|
Relative fair value of warrants issued to stockholders
|
$
|
504,227
|
$
|
4,835,727
|
||||
See report of independent registered public accounting firm and notes to financial statements.
F-31
NOVELOS THERAPEUTICS, INC. (PRE-ACQUISITION)
NOTES TO FINANCIAL STATEMENTS
1. NATURE OF BUSINESS, ORGANIZATION AND GOING CONCERN
Novelos Therapeutics, Inc. (“Novelos” or the “Company”) is a biopharmaceutical company developing compounds for the treatment of cancer.
On April 8, 2011, the Company entered into a business combination with Cellectar, Inc. (“Cellectar”), a privately held Wisconsin corporation that designed and developed products to detect, treat and monitor a wide variety of human cancers (the “Acquisition”, see Note 12). Immediately prior to the Acquisition, the Company completed a 1-for-153 reverse split of its common stock (the “Reverse Split”). The Company then issued 17,001,596 shares of its common stock to the former shareholders of Cellectar as consideration for the Acquisition, constituting approximately 85% of Novelos’ outstanding common stock after giving effect to the Acquisition. Upon the closing of the Acquisition, the Company completed the private placement of 6,846,537 shares of its common stock and warrants to purchase an additional 6,846,537 shares of its common stock (in each case after giving effect to the Reverse Split) for gross proceeds of approximately $5,135,000. As a result of the Acquisition, the Company is implementing a revised business plan focused on the development of the Cellectar compounds. Development of Novelos’ other compounds (NOV-002 and NOV-205) has been suspended. The Company will conduct its operations from Cellectar’s headquarters in Madison, WI and the Company’s executive offices will remain in Newton, MA.
The Reverse Split reduced the number of outstanding shares of Common Stock from 452,866,983 shares to 2,959,914 (2,959,871 after the cash settlement of fractional shares). On the accompanying balance sheet, the aggregate par value of the issued common stock was reduced by reclassifying the par value amount of the eliminated shares of common stock to additional paid-in capital. All per share amounts and outstanding shares, including all common stock equivalents, stock options and warrants, have been retroactively restated in these financial statements and notes for all periods presented to reflect the Reverse Split. Additionally, the number of authorized shares of common stock disclosed on the balance sheet has been reduced to 150,000,000 from 750,000,000 to reflect the reduction in authorized shares of common stock that became effective concurrent with the Reverse Split.
Accounting principles generally accepted in the United States require that a company whose security holders retain the majority voting interest in the combined business be treated as the acquirer for financial reporting purposes. Accordingly, the Acquisition will be accounted for as a reverse acquisition whereby Cellectar, Inc. will be treated as the acquirer for accounting and financial reporting purposes. The financial statements presented herein represent the historical financial information of Novelos, prior to the acquisition.
The Company is subject to a number of risks similar to those of other small biopharmaceutical companies. Principal among these risks are dependence on key individuals, competition from substitute products and larger companies, the successful development and marketing of its products in a highly regulated environment and the need to obtain additional financing necessary to fund future operations.
On February 24, 2010, the Company announced that its Phase 3 clinical trial for NOV-002 in non-small cell lung cancer (the “Phase 3 Trial”) did not meet its primary endpoint of a statistically significant increase in median overall survival. Following evaluation of the detailed trial data, on March 18, 2010, the Company announced that the secondary endpoints had also not been met in the Phase 3 Trial and that it had discontinued development of NOV-002 for NSCLC in combination with first-line paclitaxel and carboplatin chemotherapy.
These financial statements have been prepared on the basis that the Company will continue as a going concern. The Company has generated insignificant revenues and has incurred operating losses since inception in devoting substantially all of its efforts toward research and development. The Company expects that it will continue to generate operating losses for the foreseeable future. The Company believes that its cash on hand at December 31, 2010, plus the proceeds from the private placement completed in connection with the Acquisition, is adequate to fund operations into the fourth quarter of 2011. The Company’s ability to execute its operating plan beyond the fourth quarter of 2011 depends on its ability to obtain additional funding via the sale of equity and/or debt securities, a strategic transaction or otherwise. The Company plans to continue to actively pursue financing alternatives, but there can be no assurance that it will obtain the necessary funding.
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
The accompanying financial statements reflect the application of certain accounting policies, as described in this note and elsewhere in the accompanying notes to the financial statements.
Use of Estimates — The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires the Company’s management to make estimates and judgments that may affect the reported amounts of assets, liabilities, revenue and expenses and disclosure of contingent assets and liabilities. On an on-going basis, the Company’s management evaluates its estimates including those related to unbilled research and development costs, valuation of derivatives and share-based compensation. The Company bases its estimates on historical experience and on various other assumptions that are believed to be reasonable, the results of which form the basis for making judgments about the carrying values of assets and liabilities. Actual results may differ from those estimates under different assumptions or conditions. Changes in estimates are reflected in reported results in the period in which they become known.
F-32
Cash Equivalents — The Company considers all short-term investments purchased with original maturities of three months or less to be cash equivalents.
Fixed Assets — Property and equipment are stated at cost. Depreciation on property and equipment is provided using the straight-line method over the estimated useful lives of the assets, which range from three to five years. Leasehold improvements are depreciated over the lesser of the estimated useful lives of the assets or the remaining lease term.
Impairment of Long-Lived Assets — Whenever events or circumstances change, the Company assesses whether there has been an impairment in the value of long-lived assets by determining whether projected undiscounted cash flows generated by the applicable asset exceed its net book value as of the assessment date. There were no impairments of the Company’s assets at the end of each period presented.
Stock-Based Compensation — The Company accounts for employee stock-based compensation in accordance with the guidance of FASB ASC Topic 718, Compensation – Stock Compensation which requires all share-based payments to employees, including grants of employee stock options, to be recognized in the financial statements based on their fair values. The Company accounts for non-employee stock-based compensation in accordance with the guidance of FASB ASC Topic 505, Equity which requires that companies recognize compensation expense based on the estimated fair value of options granted to non-employees over their vesting period, which is generally the period during which services are rendered by such non-employees.
Revenue Recognition — Revenue is recognized when persuasive evidence of an arrangement exists, the price is fixed and determinable, delivery has occurred, and there is reasonable assurance of collection. Upfront payments received in connection with technology license or collaboration agreements are recognized over the estimated term of the related agreement. The Company has not yet received milestone or royalty payments in connection with license or collaboration agreements.
Research and Development — Research and development costs are expensed as incurred.
Income Taxes — The Company uses the liability method of accounting for income taxes. Under this method, deferred tax assets and liabilities are determined based on temporary differences between the financial statement and tax basis of assets and liabilities and net operating loss and credit carryforwards using enacted tax rates in effect for the year in which the differences are expected to reverse. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. Valuation allowances are established when it is more likely than not that some portion of the deferred tax assets will not be realized. Tax positions taken or expected to be taken in the course of preparing the Company’s tax returns are required to be evaluated to determine whether the tax positions are “more likely than not” of being sustained by the applicable tax authority. Tax positions deemed not to meet a more-likely-than-not threshold would be recorded as tax expense in the current year. There were no uncertain tax positions that require accrual or disclosure to the financial statements as of December 31, 2010 and 2009.
Comprehensive Income (Loss) — The Company had no components of comprehensive income other than net income (loss) in all of the periods presented.
Fair Value of Financial Instruments — The guidance under FASB ASC Topic 825, Financial Instruments , requires disclosure of the fair value of certain financial instruments. The Company’s financial instruments consist of cash equivalents, accounts payable, accrued expenses and redeemable preferred stock. The estimated fair value of the redeemable preferred stock, determined on an as-converted basis including conversion of accumulated unpaid dividends, was $114,780,000 at December 31, 2009. The estimated fair value of the remaining financial instruments approximates their carrying value due to their short-term nature.
Concentration of Credit Risk — Financial instruments that subject the Company to credit risk consist of cash and equivalents on deposit with financial institutions. The Company’s excess cash is on deposit in an overnight investment account that is fully collateralized by government-backed obligations.
Derivative Instruments — The Company generally does not use derivative instruments to hedge exposures to cash flow or market risks; however, starting January 1, 2009, certain warrants to purchase common stock that do not meet the requirements for classification as equity, in accordance with the Derivatives and Hedging Topic of the FASB ASC, are classified as liabilities. In such instances, net-cash settlement is assumed for financial reporting purposes, even when the terms of the underlying contracts do not provide for a net-cash settlement. These warrants are considered derivative instruments as the agreements contain “down-round” provisions whereby the number of shares for which the warrants are exercisable and/or the exercise price of the warrants is subject to change in the event of certain issuances of stock at prices below the then-effective exercise price of the warrants. The number of such warrants was 91,524 at January 1, 2009, 48,489 at December 31, 2009 and 138,611 at December 31, 2010. The primary underlying risk exposure pertaining to the warrants is the change in fair value of the underlying common stock. Such financial instruments are initially recorded at fair value, or relative fair value when issued with other instruments, with subsequent changes in fair value recorded as a component of gain or loss on derivatives in each reporting period. If these instruments subsequently meet the requirements for equity classification, the Company reclassifies the fair value to equity. At December 31, 2010 and 2009, these warrants represent the only outstanding derivative instruments issued or held by the Company. As a result of the significant decline in the Company’s stock price following the announcement of the results of the Phase 3 Trial, the Company recorded a gain of approximately $8,118,000 during the year ended December 31, 2010 in connection with the revaluation of the derivative liability balance at December 31, 2010.
F-33
New Accounting Pronouncements — In September 2009, the Financial Accounting Standards Board (“FASB”) amended the accounting standards related to revenue recognition for arrangements with multiple deliverables and arrangements that include software elements (“new accounting principles”). The new accounting principles permit prospective or retrospective adoption, and the Company elected prospective adoption at the beginning of the first quarter of 2010. The adoption of the standard in 2010 had no impact on the Company’s financial statements.
In January 2010, the FASB issued ASU No. 2010-06, Improving Disclosures about Fair Value Measurements, which requires additional disclosures about the amounts of and reasons for significant transfers in and out of Level 1 and Level 2 fair value measurements. This standard also clarifies existing disclosure requirements related to the level of disaggregation of fair value measurements for each class of assets and liabilities and disclosures about inputs and valuation techniques used to measure fair value for both recurring and non-recurring Level 2 and Level 3 measurements. Since this new accounting standard only required additional disclosure, the adoption of the standard in the first quarter of 2010 did not impact the Company’s financial statements. Additionally, effective for interim and annual periods beginning after December 15, 2010, this standard will require additional disclosure and require an entity to present disaggregated information about activity in Level 3 fair value measurements on a gross basis, rather than one net amount.
Adoption of New Accounting Principle — Effective January 1, 2009, the Company adopted the guidance of FASB ASC 815-40-15, Derivatives and Hedging , which establishes a framework for determining whether certain freestanding and embedded instruments are indexed to a company’s own stock for purposes of evaluation of the accounting for such instruments under existing accounting literature. As a result of this adoption, certain warrants that were previously determined to be indexed to the Company’s common stock upon issuance were determined not to be indexed to the Company’s common stock because they include “down-round” anti-dilution provisions whereby the number of shares for which the warrants are exercisable and/or the exercise price of the warrants is subject to change in the event of certain issuances of stock at prices below the then-effective exercise price of the warrants. The fair value of the warrants at the dates of issuance totaling $6,893,000 was initially recorded as a component of additional paid-in capital. Upon adoption of this guidance on January 1, 2009, the Company recorded a derivative liability of $999,000, a decrease to the opening balance of additional paid-in capital of approximately $6,893,000 and recorded a decrease to accumulated deficit totaling approximately $5,894,000, representing the decrease in the fair value of the warrants from the date of issuance to December 31, 2008. The increase in fair value of the warrants of approximately $12,114,000 during the year ended December 31, 2009 and the decrease in fair value of the warrants of $8,118,000 during the year ended December 31, 2010 have been included as a component of other income (expense) in the accompanying statement of operations. Certain of the warrants that had been recorded as a derivative liability were exchanged or exercised for shares of the Company’s common stock during the years ended December 31, 2010 and 2009. See Note 6 for a description of those transactions. The fair value of the warrants of $288,000 and $10,487,000 at December 31, 2010 and 2009 is included as a current liability in the accompanying balance sheet as of that date.
Reclassifications — Certain prior year amounts have been reclassified to conform to the current year presentation.
3. FIXED ASSETS
Fixed assets consisted of the following at December 31:
|
2010
|
2009
|
|||||||
|
Office and computer equipment
|
$
|
61,907
|
$
|
73,261
|
||||
|
Computer software
|
43,896
|
43,896
|
||||||
|
Leasehold improvements
|
4,095
|
4,095
|
||||||
|
Total fixed assets
|
109,898
|
121,252
|
||||||
|
Less accumulated depreciation and amortization
|
(101,143
|
)
|
(77,155
|
)
|
||||
|
Fixed assets, net
|
$
|
8,755
|
$
|
44,097
|
||||
4. FAIR VALUES OF ASSETS AND LIABILITIES
In accordance with Fair Value Measurements and Disclosures Topic of the FASB ASC, the Company groups its financial assets and financial liabilities generally measured at fair value in three levels, based on the markets in which the assets and liabilities are traded and the reliability of the assumptions used to determine fair value.
|
|
·
|
Level 1: Input prices quoted in an active market for identical financial assets or liabilities.
|
|
|
·
|
Level 2: Inputs other than prices quoted in Level 1, such as prices quoted for similar financial assets and liabilities in active markets, prices for identical assets and liabilities in markets that are not active or other inputs that are observable or can be corroborated by observable market data.
|
F-34
|
|
·
|
Level 3: Input prices that are significant to the fair value of the financial assets or liabilities which are not observable or supported by an active market.
|
Assets and liabilities measured at fair value on a recurring basis are summarized below:
|
December 31, 2010
|
||||||||||||||||
|
Level 1
|
Level 2
|
Level 3
|
Fair Value
|
|||||||||||||
|
Liabilities:
|
||||||||||||||||
|
Warrants
|
$
|
-
|
$
|
288,250
|
$
|
-
|
$
|
288,250
|
||||||||
|
December 31, 2009
|
||||||||||||||||
|
Level 1
|
Level 2
|
Level 3
|
Fair Value
|
|||||||||||||
|
Liabilities:
|
||||||||||||||||
|
Warrants
|
$
|
-
|
$
|
10,487,000
|
$
|
-
|
$
|
10,487,000
|
||||||||
The Company uses valuation methods and assumptions that consider among others the fair value of the underlying stock, risk-free interest rate, volatility, expected life and dividend rates in estimating fair value for the warrants considered to be derivative instruments. Assumptions used are generally consistent with those disclosed for stock-based compensation (see Note 7).
|
5.
|
COLLABORATION AGREEMENTS
|
2007 Collaboration Agreement with Lee’s Pharmaceutical (HK) Ltd.
In December 2007 the Company entered into a Collaboration Agreement with Lee’s Pharmaceutical (HK) Ltd. (“Lee’s Pharm”). Pursuant to this agreement, Lee’s Pharm obtained an exclusive license to develop, manufacture and commercialize NOV-002 and NOV-205 in China, Hong Kong, Taiwan and Macau (the “Chinese Territory”). The Company has suspended further development of NOV-205; however, this suspension may not impact the development strategy of Lee’s Pharm. Under the terms of the agreement the Company received a license fee of $500,000 in March 2008 and is entitled to receive up to $1,700,000 in future milestone payments upon the completion of development and marketing milestones by Lee’s Pharm. This initial $500,000 payment received is being amortized over the estimated term of this agreement, 15 years. Accordingly, $33,000 of license revenue was recognized in each of the years ended December 31, 2010 and 2009.
The Lee’s Pharm agreement provides that the Company receive royalty payments of 20-25% of net sales of NOV-002 in the Chinese Territory and receive royalty payments of 12-15% of net sales of NOV-205 in the Chinese Territory. Lee’s Pharm is obligated to reimburse the Company for the manufacturing cost of pharmaceutical products provided to Lee’s Pharm in connection with the agreement. Lee’s Pharm has committed to spend a minimum amount on development in the first four years of the agreement. The agreement expires upon the expiration of the last patent covering any of the licensed products, or twelve years from the date of the first commercial sale in China, whichever occurs later.
2009 Collaboration Agreement with Mundipharma
On February 11, 2009, Novelos entered into a collaboration agreement (the “Collaboration Agreement”) with Mundipharma International Corporation Limited (“Mundipharma”) to develop, manufacture and commercialize, on an exclusive basis, Licensed Products (as defined in the Collaboration Agreement), which includes the Company’s lead compound, NOV-002, in Europe (other than the Russian Territory), Asia (other than the Chinese Territory) and Australia (collectively referred to as the “Mundipharma Territory”). Mundipharma is an independent associated company of Purdue Pharma, L.P. (“Purdue”). The Collaboration Agreement provides for Mundipharma to pay the Company royalties and fixed milestone payments based on sales and commercial launches in the licensed territories.
For countries in which patents are held, the Collaboration Agreement expires on a country-by-country basis within the Mundipharma Territory on the earlier of (1) expiration of the last applicable Novelos patent within the country or (2) the determination that any patents within the country are invalid, obvious or otherwise unenforceable. For countries in which no patents are held, the Collaboration Agreement expires the earlier of 15 years from its effective date or upon generic product competition in the country resulting in a 20% drop in Mundipharma’s market share. Novelos may terminate the Collaboration Agreement upon breach or default by Mundipharma. Mundipharma may terminate the Collaboration Agreement upon breach or default, filing of voluntary or involuntary bankruptcy by Novelos, the termination of certain agreements with companies associated with the originators of the licensed technology, or 30-day notice for no reason. If any regulatory approval within the Mundipharma Territory is suspended as a result of issues related to the safety of the Licensed Products, then Mundipharma’s obligations under the Collaboration Agreement will be suspended until the regulatory approval is reinstated. If that reinstatement does not occur within 12 months of the suspension, then Mundipharma may terminate the Collaboration Agreement.
F-35
Concurrently with the execution of the Collaboration Agreement, Novelos completed a private placement of Series E preferred stock and common stock purchase warrants to Purdue.
The Company expects that the negative results of its Phase 3 Trial will adversely affect development and commercialization of NOV-002 under the collaboration agreements with Lee’s Pharm and Mundipharma.
6. STOCKHOLDERS’ DEFICIENCY
Exchange of Outstanding Preferred Stock for Common Stock
On November 30, 2010, the Company entered into an Exchange Agreement with each of the holders of its Series E convertible preferred stock (the “Series E Preferred Stock”) and Series C convertible preferred stock (the “Series C Preferred Stock”) pursuant to which each such holder exchanged all of the holder’s shares of Series E Preferred Stock or Series C Preferred Stock, as applicable, and all rights, preferences and privileges associated therewith (including but not limited to any accrued but unpaid dividends thereon) and any rights of the holder to liquidated damages under agreements to register the Company’s capital stock, for an aggregate of 2,228,338 shares of common stock, representing 75.3% of the Company’s common stock outstanding effective immediately following the exchange. As a result of the exchange, all of the liquidation preference applicable to the preferred stock, approximately $27,337,000 as of November 30, 2010, was eliminated. Furthermore, future dividends totaling $2,327,000 annually were eliminated, special voting rights applicable to the preferred stock are no longer applicable, and the former holders of Series E Preferred Stock have released any rights to require the registration of shares of the Company’s common stock for resale under the Securities Act. The effective price per share at which the common stock was issued in connection with the exchange (based on the aggregate liquidation preference of all of the preferred stock divided by the total number of shares of common stock issued in exchange for such preferred stock) was approximately $12.27. The market price of the Company’s common stock as of the last trading day immediately preceding the exchange was $6.12.
The exchange was accounted for as a recapitalization and the carrying value of the Series E Preferred Stock of $13,770,000, accumulated dividends totaling $4,476,000 and estimated liquidated damages of $819,000 for failure to timely file a resale registration statement (see “Registration Rights” below) were reclassified to additional paid-in-capital as of the date of the exchange. If the preferred stock had been converted according to its terms, the holders would have received a total of 274,882 shares of common stock. At the date of issuance the fair market value totaling $11,955,151 of the additional 1,953,456 shares issued in the exchange has been recorded as a deemed dividend to preferred stockholders in the year ended December 31, 2010.
Issuance of Series C Preferred Stock
During 2007, the Company issued 272 shares of Series C Preferred Stock in exchange for all 3,264 shares of Series A preferred stock, originally issued in 2005. The Series C Preferred Stock was initially convertible at $153.00 per share into 21,333 shares of common stock. In connection with the sale of Series D preferred stock in 2008, the conversion price of the Series C Preferred Stock was reduced to $99.45 per share, according to its terms.
Terms of the Series C Preferred Stock
The Series C Preferred Stock had an annual dividend rate of 8% until October 1, 2008 and 20% thereafter. The dividends were payable quarterly. Such dividends were to be paid only after all outstanding dividends on the Series D Preferred Stock (with respect to the current fiscal year and all prior fiscal years) had been paid to the holders of the Series D Preferred Stock. No dividends were paid on Series C Preferred Stock during 2009 and 2010. The conversion price was subject to adjustment for stock dividends, stock splits or similar capital reorganizations and upon the occurrence of certain dilutive issuances of securities. The Series C Preferred Stock did not have voting rights and was redeemable only at the option of the Company upon 30 days’ notice at a 20% premium plus any accrued but unpaid dividends. In the event of any voluntary or involuntary liquidation, dissolution or winding up of the Company’s affairs, the Series C Preferred Stock would have been treated as senior to Novelos common stock. After all required payments were made to holders of Series E Preferred Stock, the holders of Series C Preferred Stock would have been entitled to receive first, $12,000 per share and all accrued and unpaid dividends. If, upon any winding up of the Company’s affairs, the Company’s remaining assets available to pay the holders of Series C Preferred Stock were not sufficient to permit the payment in full, then all of the Company’s assets would have been distributed to the holders of Series C Preferred Stock (and any remaining holders of Series E Preferred Stock as may be required) on a pro rata basis.
Conversions of Series C Preferred Stock
During the year ended December 31, 2009, 68 shares of the Company’s Series C Preferred Stock, having an aggregate stated value of $816,000, and accumulated dividends thereon of $184,000 were converted into shares of the Company’s common stock, leaving 204 shares of Series C Preferred Stock outstanding which were convertible into 24,615 shares of common stock as of December 31, 2009. During 2010, all shares of Series C Preferred Stock were exchanged for shares of common stock (see “Exchange of Outstanding Preferred Stock for Common Stock” above).
F-36
Series E Preferred Stock Private Placement
Sale of Series E Preferred Stock to Purdue
Concurrently with the execution of the Collaboration Agreement on February 11, 2009, Novelos sold to Purdue 200 shares of a newly created series of the Company’s preferred stock, designated “Series E Convertible Preferred Stock,” par value $0.00001 per share (the “Series E Preferred Stock”), and a warrant (the “Series E Warrant”) to purchase 60,330 shares of Novelos common stock for an aggregate purchase price of $10,000,000 (the “Series E Financing”).
The Series E Warrant is exercisable for an aggregate of 60,330 shares of Novelos common stock at an exercise price of $99.45 per share. The warrant expires on December 31, 2015. The warrant exercise price and/or the common stock issuable pursuant to such warrant are subject to adjustment for stock dividends, stock splits or similar capital reorganizations so that the rights of the warrant holder after such event will be equivalent to the rights of the warrant holder prior to such event.
Exchange of Series D Preferred Stock for Series E Preferred Stock
The Company also entered into an exchange agreement with the holders (the “Series D Investors”) of the Company’s Series D preferred stock, issued during 2008, under which all 413.5 outstanding shares of Series D preferred stock and accumulated but unpaid dividends thereon totaling $1,597,144 were exchanged for 445.442875 shares of Series E Preferred Stock. The rights and preferences of the Series E Preferred Stock were substantially the same as the Series D preferred stock. In addition, the Series D Investors waived liquidated damages through the date of the exchange as a result of the Company’s failure to file a registration statement covering the shares of common stock underlying the Series D preferred stock and warrants not otherwise registered. In connection with the execution of this exchange agreement, warrants held by the Series D Investors to purchase a total of 77,551 shares of the Company’s common stock were amended to extend the expiration of the warrants to December 31, 2015 (from April 11, 2013) and to remove a forced exercise provision.
Terms of Series E Preferred Stock
The shares of Series E Preferred Stock had a stated value of $50,000 per share and were convertible into shares of common stock at any time after issuance at the option of the holder at $99.45 per share of common. If there was an effective registration statement covering the shares of common stock underlying the Series E Preferred Stock and the VWAP, as defined in the Series E Certificate of Designations, of Novelos common stock exceeded $306.00 for 20 consecutive trading days, then the outstanding shares of Series E Preferred Stock would automatically convert into common stock at the conversion price then in effect. The conversion price was subject to adjustment for stock dividends, stock splits or similar capital reorganizations.
The Series E Preferred Stock has an annual dividend rate of 9%, payable semi-annually on June 30 and December 31. Such dividends were payable in cash, in shares of Series E Preferred Stock or in registered shares of Novelos common stock at the Company’s option, subject to certain conditions. The Company has not paid any cash dividends on Series E Preferred Stock.
The terms of the Series E Preferred Stock provided that for as long as any shares of Series E Preferred Stock remained outstanding, Novelos was prohibited without the prior consent of holders of a majority of the outstanding shares of Series E preferred stock (including in such majority the Xmark Funds and Purdue) from (i) paying dividends to its common stockholders, (ii) amending its certificate of incorporation or by-laws, (iii) issuing any equity security or any security convertible into or exercisable for any equity security at a price of $99.45 or less or with rights senior to the Series E Preferred Stock (except for certain exempted issuances), (iv) increasing the number of shares of Series E Preferred Stock or issuing any additional shares of Series E Preferred Stock, (v) selling or otherwise granting rights with respect to all or substantially all of its assets (or in the case of licensing, any material intellectual property) or the Company's business and/or entering into a merger or consolidation with another company unless Novelos would be the surviving corporation, the Series E Preferred Stock would remain outstanding, there would be no changes to the rights and preferences of the Series E Preferred Stock and there would not be created any new class of capital stock senior to the Series E Preferred Stock, (vi) redeeming or repurchasing any capital stock other than the Series E Preferred Stock, (vii) incurring any new debt for borrowed money in excess of $500,000 and (viii) changing the number of the Company’s directors. As of December 31, 2010, no shares of Series E preferred stock remained outstanding, and accordingly, none of these restrictions applied. See “Exchange of Outstanding Preferred Stock for Common Stock” above.
Advisor Fees
Ferghana Partners, Inc. (“Ferghana”), a New York consulting firm, received a cash fee for their services in connection with the negotiation and execution of the Collaboration Agreement equal to $700,000 (or seven percent (7%) of the gross proceeds to the Company resulting from the sale of Series E Preferred Stock and common stock purchase warrants to Purdue in connection with the Collaboration Agreement). Novelos is also obligated to pay Ferghana six percent (6%) of all payments to Novelos by Mundipharma under the Collaboration Agreement other than royalties on net sales.
F-37
Accounting Treatment of Series E Financing
The terms of the Series E Preferred Stock contained provisions that required redemption in circumstances that were beyond the Company’s control, such as the acquisition of more than 50% of our outstanding stock by any person or entity. Therefore, the shares were recorded as redeemable preferred stock outside of permanent equity in the balance sheet as of December 31, 2009. The gross proceeds of $10,000,000 received in conjunction with the Series E Financing were allocated on a relative fair value basis between the Series E Preferred Stock and the warrants. The relative fair value of the warrants issued to investors of $2,907,000 (determined using the Black-Scholes option pricing model, estimated volatility of 80%, a risk-free interest rate of 2.17% and a term equal to the term of the warrant) was recorded as additional paid-in capital while the relative fair value of the Series E Preferred Stock of $7,093,000 was recorded as temporary equity. The carrying value of the Series E Preferred Stock was immediately adjusted to its fair value of $7,385,000 based on the fair value of the as-converted common stock. The difference of $292,000 represents a beneficial conversion feature and was recorded as a deemed dividend to preferred stockholders. Issuance costs related to the Series E Financing of $795,000 were netted against temporary equity. The Series E Preferred Stock that was issued in payment of dividends was initially recorded in temporary equity at the value of the dividends that had accrued totaling $1,597,000. This amount was then adjusted to the fair value of $1,179,000 based on the fair value of the as-converted common stock. The difference of $418,000 was recorded as an offset to the deemed dividends recorded. The Series E Preferred Stock that was issued in exchange for outstanding shares of Series D preferred stock was recorded at $13,904,000, the carrying value of the shares of Series D preferred stock as of the date of the exchange.
As a result of the modification to the warrants to extend their expiration by approximately 32 months that occurred in connection with the exchange of all outstanding shares of Series D preferred stock for shares of Series E Preferred Stock, in the year ended December 31, 2009, a deemed dividend of $840,000 was recorded. This amount represented the incremental fair value of the warrants immediately before and after modification using the Black-Scholes option pricing model, volatility of 80%, discount rates of 1.54% and 2.17% and the remaining warrant term.
Conversions of Series E Preferred Stock
During the year ended December 31, 2009, 97.18209375 shares of the Company’s Series E Preferred Stock, having an aggregate stated value of $4,859,000 and accumulated dividends thereon of $301,000, were converted into 51,889 shares of common stock. The associated carrying value of the converted shares totaling approximately $3,213,000 was reclassified to permanent equity from temporary equity. During the year ended December 31, 2010, 140 shares of the Company’s Series E Preferred Stock, having an aggregate stated value of $7,000,000, and accumulated dividends thereon totaling $635,000, were converted into 76,770 shares of common stock. The associated carrying value of the converted shares totaling approximately $4,690,000 was reclassified to permanent equity from temporary equity. In November 2010, all outstanding shares of Series E Preferred Stock were exchanged for shares of common stock. See “Exchange of Preferred Stock” below.
August 2009 Common Stock Private Placement
Securities Purchase Agreement
On August 25, 2009, the Company entered into the August 2009 Purchase Agreement with Purdue to sell 89,126 shares of its common stock, $0.00001 par value and warrants to purchase 31,194 shares of its common stock at an exercise price of $100.98 per share, expiring December 31, 2015, for an aggregate purchase price of $9,000,000 (the “August 2009 Private Placement”). Concurrent with the execution and delivery of the August 2009 Purchase Agreement, the Company sold Purdue 34,660 shares of its common stock and a warrant to purchase 12,131 shares of its common stock at $100.98 per share for approximately $3,500,000 (the “Initial Closing”). On November 10, 2009, the Company completed the final closing under the August 2009 Purchase Agreement and sold Purdue 54,466 shares of Novelos common stock and warrants to purchase 19,063 shares of Novelos common stock for gross proceeds of $5,500,000. Issuance costs associated with the transactions totaled $61,000 and such amount was recorded as a reduction of additional paid-in capital.
Pursuant to the August 2009 Purchase Agreement, Purdue acquired a right of first refusal (the “Right of First Refusal”) with respect to bona fide offers for the license or other acquisition of NOV-002 Rights (as defined in the August 2009 Purchase Agreement) in the U.S. (the “U.S. License”) received from third parties and approved by the Company’s board of directors. Under the Right of First Refusal, Novelos will be required to communicate to Purdue the terms of any such third-party offers received and Purdue will have 30 days to enter into a definitive agreement with Novelos on substantially similar terms that provide no lesser economic benefit to Novelos as provided in the third-party offer. The Right of First Refusal terminates upon business combinations, as defined in the August 2009 Purchase Agreement. Novelos has separately entered into letter agreements with Mundipharma and its independent associated company providing for a conditional exclusive right to negotiate for, and a conditional right of first refusal with respect to, NOV-002 Rights for Latin America, Mexico and Canada.
F-38
Pursuant to the August 2009 Purchase Agreement, Purdue has the right to either designate one member to Novelos’ Board or designate an observer to attend all meetings of the Board and committees thereof and to have access to all information made available to members of the Board. This right lasts until the later of such time as Purdue or its independent associated companies no longer hold at least one-half of the common stock purchased pursuant to the August 2009 Purchase Agreement and no longer hold at least one-half of the Series E Preferred Stock issued to them on February 11, 2009. The right to designate a Board observer had previously been granted in connection with the financing that occurred on February 11, 2009 and Purdue appointed such an observer in February 2009. Purdue also has the right to participate in future equity financings in proportion to their pro rata ownership of common stock.
Common Stock Purchase Warrant
The common stock purchase warrants have an exercise price of $100.98 per share and expire on December 31, 2015. The warrant exercise price and/or the number of shares of common stock issuable pursuant to such warrant will be subject to adjustment for stock dividends, stock splits or similar capital reorganizations so that the rights of the warrant holders after such event will be equivalent to the rights of warrant holders prior to such event. The relative fair value of the warrants issued to Purdue totaled $1,929,000 and was recorded as a component of additional paid-in capital. The fair value of the warrants was determined based on the market value of the Company’s common stock on the dates of issuance using the Black-Scholes method of valuation, estimated volatility of 90%, risk-free interest rates ranging from 2.02% to 2.7% and a term equal to the term of the warrant.
Registration Rights
As part of the August 2009 Private Placement, the Company entered into a registration rights agreement with Purdue (the “Purdue Registration Agreement”). The Purdue Registration Agreement required the Company to have filed with the SEC no later than May 17, 2010, a registration statement covering the resale of all the shares of common stock issued pursuant to the August 2009 purchase agreement and all shares of common stock issuable upon exercise of the warrants issued pursuant to the August 2009 purchase agreement. The registration rights agreement provided for liquidated damages equal to 1.5% per month (pro-rated on a daily basis for any period of less than a full month) of the aggregate purchase price of the common stock until the delinquent registration statement is filed. The Company did not file the registration statement and accrued $819,000 as a component of other income (expense) during the year ended December 31, 2010, representing management’s best estimate of the probable total liquidated damages that may be settled. In connection with the exchange of their shares of Series E Preferred Stock for common stock on November 30, 2010, Purdue released the Company from any requirement to register shares of its common stock for resale and forfeited any rights to receive liquidated damages pursuant to any registration agreements. The accrued liquidated damages were settled in connection with the exchange and, accordingly, the amount that had been accrued was reclassified to additional paid-in capital.
July 2010 Registered Offering
On July 27, 2010, pursuant to securities purchase agreements entered into with institutional investors on July 21, 2010, the Company completed the sale, in an offering registered under the Securities Act of 1933, as amended, of an aggregate of 140,056 shares of its common stock and five-year warrants to purchase up to an aggregate of 150,040 shares of its common stock at an exercise price of $10.71 per share, for gross proceeds of $1,500,000 and net proceeds of $1,249,000 after deducting transaction costs. The warrant exercise price is subject to adjustment in certain circumstances and therefore, the relative fair value of the warrants at the date of issuance, $504,000, has been bifurcated from the proceeds and recorded as a derivative liability. The Company uses valuation methods and assumptions that consider among others the fair value of the underlying stock, risk-free interest rate, volatility, expected life and dividend rates in estimating fair value for the warrants considered to be derivative instruments. The assumptions used to value these warrants at the time of issuance are generally consistent with those disclosed for stock-based compensation (see Note 7).
Since the securities in this financing were issued at a price less than $100.98 per share, the Company obtained the consent of its preferred stockholders pursuant to a consent and waiver dated July 6, 2010, as amended on July 21, 2010. In connection with obtaining this consent, the Company issued five-year warrants (the “Incentive Warrants”) to its preferred stockholders for the purchase of up to an aggregate of 150,039 shares of common stock at an exercise price of $16.065 per share. No adjustments to the conversion price of the preferred stock or warrants held by preferred stockholders were made in connection with the financing. The fair value of the Incentive Warrants at date of issuance, $586,000, is reflected as a deemed dividend to the preferred stockholders on the statement of operations. The Company used valuation methods and assumptions that consider among others the fair value of the underlying stock, risk-free interest rate, volatility, expected life and dividend rates in estimating fair value of the Incentive Warrants. The assumptions used to value these warrants are generally consistent with those disclosed for stock-based compensation (see Note 7).
The financing resulted in adjustments to certain warrants pursuant to their terms. Warrants issued in 2005 that were exercisable for 1,591 shares at an exercise price of $99.45 per share as of immediately prior to the transaction became exercisable for 14,776 shares at an exercise price of $10.71 per share, and warrants issued in 2006 that were exercisable for 29,787 shares at an exercise price of $263.16 per share as of immediately prior to the transaction became exercisable for 33,569 shares at an exercise price of $234.09 per share. The 2005 warrants expired unexercised on August 9, 2010, and the 2006 warrants will expire in March 2011.
F-39
Common Stock Warrants — The following table summarizes information with regard to outstanding warrants as of December 31, 2010, issued in connection with equity and debt financings since 2005.
|
Offering
|
Outstanding
(as adjusted)
|
Exercise
Price
(as adjusted)
|
Expiration Date
|
||||||
|
2006 Issuance of Common Stock
|
33,569
|
$
|
234.09
|
March 7, 2011
|
|||||
|
Series B Preferred Stock – placement agents
|
5,392
|
$
|
191.25
|
May 2, 2012
|
|||||
|
Series C Exchange
|
8,169
|
$
|
191.25
|
May 2, 2012
|
|||||
|
Series E Preferred Stock
|
60,330
|
$
|
99.45
|
December 31, 2015
|
|||||
|
August 2009 Private Placement
|
31,194
|
$
|
100.98
|
December 31, 2015
|
|||||
|
July 2010 Direct Offering (1)
|
105,039
|
$
|
10.71
|
July 27, 2015
|
|||||
|
Preferred Incentive Warrants
|
105,040
|
$
|
16.065
|
July 27, 2015
|
|||||
|
Total
|
348,733
|
||||||||
(1) The exercise price of these warrants was adjusted in connection with the private placement completed on April 8, 2011. See Note 12.
During the year ended December 31, 2010, warrants to purchase 14,776 shares of common stock at $10.71 and 5,941 shares of common stock at $99.45 expired unexercised. On March 7, 2011, warrants to purchase 33,569 shares of common stock at $234.09 expired unexercised.
During the year ended December 31, 2010, 53,478 shares of the Company’s common stock were issued upon the cashless exercise of warrants to purchase 89,752 shares of the Company’s common stock. The Company reclassified $2,584,000 from derivative liability to additional paid-in capital upon the exercise of warrants. The following is a summary of the exercises:
|
Original private placement
|
Shares of
Common Stock
Issued
|
Warrants
Exercised
|
Exercise
Price
|
|||||||||
|
2005 Bridge Financing
|
2,059 | 2,614 | $ | 95.625 | ||||||||
|
2005 Issuance of Common Stock – placement agents
|
1,481 | 2,074 | $ | 99.45 | ||||||||
|
2006 Issuance of Common Stock
|
2,395 | 6,480 | $ | 263.16 | ||||||||
|
Series B Preferred Stock – purchasers
|
29,709 | 49,019 | $ | 99.45 | ||||||||
|
Series B Preferred Stock – placement agents
|
229 | 490 | $ | 191.25 | ||||||||
|
Series D Preferred Stock
|
17,292 | 28,531 | $ | 99.45 | ||||||||
|
Series C Exchange
|
313 | 544 | $ | 191.25 | ||||||||
|
Total
|
53,478 | 89,752 | ||||||||||
During the year ended December 31, 2009, a total of 3,162 shares of the Company’s common stock were issued upon the cashless exercise of warrants to purchase 6,975 shares of common stock. The Company reclassified a total of $1,001,000 from derivative liability to additional paid-in capital upon the exercise of warrants. The following is a summary of the exercises:
|
Original private placement
|
Shares of
Common Stock
Issued
|
Warrants
Exercised
|
Exercise
Price
|
|||||||||
|
2005 Bridge Financing
|
1,429
|
2,091
|
$
|
95.625
|
||||||||
|
2005 Common Stock
|
1,310
|
3,172
|
$
|
99.45
|
||||||||
|
Series A Preferred Stock
|
250
|
396
|
$
|
99.45
|
||||||||
|
2006 Issuance of Common Stock
|
173
|
1,316
|
$
|
263.16
|
||||||||
|
Total
|
3,162
|
6,975
|
||||||||||
F-40
On August 21, 2009, the Company entered into exchange agreements with certain accredited investors who held warrants, issued in the 2006 private placement, to purchase 45,409 shares of its common stock. Pursuant to the exchange agreements, an aggregate of 13,623 shares of the Company’s common stock with a fair value of $1,626,000 were issued in exchange for these warrants. The holders agreed not to transfer or dispose of the shares of common stock before February 18, 2010. The warrants had been recorded as a derivative liability on the Company’s balance sheet at their estimated fair value of $1,109,000 at the date of exchange. The difference of $517,000 between the estimated fair value of the warrants at the date of exchange and the common stock issued to settle the derivative liability has been included as a component of the loss on derivative warrants for the year ended December 31, 2009. Following the exchange, warrants expiring on March 7, 2011 to purchase a total of 35,504 shares of common stock at $278.46 per share remained outstanding. Following the final closing of the August 2009 Private Placement, described above, the number of these outstanding warrants was increased to 37,584 and the exercise price was reduced to $263.16, as a result of anti-dilution provisions in the warrants.
Authorized and Reserved Shares — On October 18, 2010 the Company’s stockholders approved an amendment to the certificate of incorporation to increase the total number of authorized shares of the Company’s common stock from 225,000,000 to 750,000,000. On April 8, 2011, the Company completed the Reverse Split described in Note 12, and amended the certificate of incorporation to decrease the total number of authorized shares to 150,000,000 from 750,000,000.
The following shares were reserved for future issuance upon exercise of stock options or warrants or conversion of preferred stock as of the dates indicated:
|
December 31,
|
||||||||
|
2010
|
2009
|
|||||||
|
2000 Stock Option Plan
|
291
|
366
|
||||||
|
2006 Stock Incentive Plan
|
38,366
|
43,856
|
||||||
|
Options issued outside of formalized plans
|
10,569
|
16,037
|
||||||
|
Warrants
|
348,733
|
232,164
|
||||||
|
Preferred stock
|
—
|
329,451
|
||||||
|
Total shares reserved for future issuance
|
397,959
|
621,874
|
||||||
7. STOCK-BASED COMPENSATION
The Company’s stock-based compensation plans are summarized below:
2000 Stock Option Plan. As of December 31, 2010, there are options to purchase 291 shares of the Company’s common stock outstanding under a stock option plan established in August 2000 (the “2000 Plan”). There will be no further grants made under the 2000 Plan. Options generally vested annually over three years and expire on the tenth anniversary of the grant date. No options were granted or exercised under the 2000 Plan during 2009. During 2010, options to purchase 65 shares of common stock under the 2000 Plan were exercised and options to purchase 9 shares of common stock were canceled.
2006 Stock Incentive Plan. On May 1, 2006, the Company’s board of directors adopted, and on July 21, 2006 the Company’s stockholders approved, the 2006 Stock Incentive Plan (the “2006 Plan”). A total of 65,359 shares of common stock are reserved for issuance under the 2006 Plan for grants of incentive or nonqualified stock options, rights to purchase restricted and unrestricted shares of common stock, stock appreciation rights and performance share grants. A committee of the board of directors determines exercise prices, vesting periods and any performance requirements on the date of grant, subject to the provisions of the 2006 Plan. Options are granted at or above the fair market value of the common stock at the grant date and expire on the tenth anniversary of the grant date. Vesting periods are generally two to three years. In the year ended December 31, 2009, stock options for the purchase of 12,679 shares of common stock were granted under the 2006 Plan. During the year ended December 31, 2010, options to purchase 1,438 shares of common stock were exercised and options to purchase 3,921 shares of common stock were canceled. As of December 31, 2010 and December 31, 2009, 25,555 and 21,503 shares remain available for grant under the 2006 Plan. Options granted pursuant to the 2006 Plan generally will become fully vested upon a termination event occurring within one year following a change in control, as defined. A termination event is defined as either termination of employment or services other than for cause or constructive termination of employees or consultants resulting from a significant reduction in either the nature or scope of duties and responsibilities, a reduction in compensation or a required relocation.
Other Stock Option Activity. During 2005 and 2004, the Company issued options to purchase a total of 17,345 shares of common stock to employees, directors and consultants outside of any formalized plan. These options are exercisable within a ten-year period from the date of grant, and vest at various intervals with all options being fully vested within two to three years of the grant date. The options are not transferable except by will or domestic relations order. The option price per share is not less than the fair market value of the shares on the date of the grant. During the year ended December 31, 2010, options to purchase 4,488 shares of common stock were exercised and options to purchase 980 shares of common stock were canceled.
F-41
Accounting for Stock-Based Compensation
The Company accounts for employee stock-based compensation in accordance with the guidance of FASB ASC Topic 718, Compensation – Stock Compensation which requires all share-based payments to employees, including grants of employee stock options, to be recognized in the financial statements based on their fair values. The Company accounts for non-employee stock-based compensation in accordance with the guidance of FASB ASC Topic 505, Equity which requires that companies recognize compensation expense based on the estimated fair value of options granted to non-employees over their vesting period, which is generally the period during which services are rendered by such non-employees.
The following table summarizes amounts charged to expense for stock-based compensation related to employee and director stock option grants and stock-based compensation recorded in connection with stock options and restricted stock awards granted to non-employee consultants:
|
Year Ended
December 31,
|
||||||||
|
2010
|
2009
|
|||||||
|
Employee and director stock option grants:
|
||||||||
|
Research and development
|
$
|
230,101
|
$
|
148,030
|
||||
|
General and administrative
|
356,220
|
289,036
|
||||||
|
586,321
|
437,066
|
|||||||
|
Non-employee consultant stock option grants and restricted stock awards:
|
||||||||
|
Research and development
|
(220,969
|
)
|
328,614
|
|||||
|
General and administrative
|
(28,054
|
)
|
98,657
|
|||||
|
(249,023
|
)
|
427,271
|
||||||
|
Total stock-based compensation
|
$
|
337,298
|
$
|
864,337
|
||||
On December 31, 2009, the expiration of options held by a former employee was extended until January 31, 2010 and incremental stock-based compensation expense for non-employees of $15,000 was recorded in connection with the one-month extension.
In January 2009, the Company modified the terms of options to purchase 261 shares of common stock held by two employees to vest all unvested options and to extend the expiration dates of the options. The modification was made in connection with the termination of the two employees to reduce costs. During the year ended December 31, 2009, incremental stock-based compensation expense of $8,000 was recorded in connection with the modification of the option terms.
Determining Fair Value
Valuation and amortization method. The fair value of each stock award is estimated on the grant date using the Black-Scholes option-pricing model. The estimated fair value of employee stock options is amortized to expense using the straight-line method over the vesting period.
Volatility. The Company estimates volatility based on an average of (1) the Company’s historical volatility since its common stock has been publicly traded and (2) review of volatility estimates of publicly held drug development companies with similar market capitalizations.
Risk-free interest rate. The risk-free interest rate is based on the U.S. Treasury yield curve in effect at the time of grant commensurate with the expected term assumption.
Expected term. The expected term of stock options granted is based on the Company’s estimate of when options will be exercised in the future as there have been limited stock option exercises to date. The expected term is generally applied to one group as a whole as the Company does not expect substantially different exercise or post-vesting termination behavior within its population of option holders.
Forfeitures. The Company records stock-based compensation expense only for those awards that are expected to vest. FASB ASC Topic 718 requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. The term “forfeitures” is distinct from “cancellations” or “expirations” and represents only the unvested portion of the surrendered option. The Company has applied an annual forfeiture rate of 0% to all unvested options as of December 31, 2010 as the Company has experienced very few forfeitures to date and believes that there is insufficient history to develop an accurate estimate of future forfeitures. This analysis will be re-evaluated semi-annually and the forfeiture rate will be adjusted as necessary. Ultimately, the actual expense recognized over the vesting period will be for only those shares that vest.
F-42
The following table summarizes weighted average values and assumptions used for options granted to employees, directors and consultants in the periods indicated:
|
Year Ended
December 31,
2009
|
||||
|
Volatility
|
90
|
%
|
||
|
Weighted-average volatility
|
90
|
%
|
||
|
Risk-free interest rate
|
2.12
|
%
|
||
|
Expected life (years)
|
5
|
|||
|
Dividend
|
0
|
%
|
||
|
Weighted-average exercise price
|
$
|
0.75
|
||
|
Weighted-average grant-date fair value
|
$
|
0.53
|
||
There were no stock option grants during the year ended December 31, 2010.
Stock Option Activity
A summary of stock option activity under the 2000 Plan, the 2006 Plan and outside of any formalized plan is as follows:
|
Options
Outstanding
|
Weighted
Average
Exercise Price
|
Weighted
Average
Remaining
Contracted
Term in
Years
|
Aggregate
Intrinsic
Value
|
|||||||||||||
|
Outstanding at January 1, 2009
|
47,580
|
$
|
91.80
|
7.9
|
$
|
989,718
|
||||||||||
|
Options granted
|
12,679
|
$
|
114.75
|
|||||||||||||
|
Outstanding at December 31, 2009
|
60,259
|
$
|
96.39
|
7.5
|
$
|
17,650,255
|
||||||||||
|
Options exercised
|
(5,991
|
)
|
$
|
26.01
|
$
|
663,600
|
||||||||||
|
Options canceled
|
(5,042
|
)
|
$
|
139.23
|
$
|
0
|
||||||||||
|
Outstanding at December 31, 2010
|
49,226
|
$
|
100.98
|
6.9
|
$
|
24,842
|
||||||||||
|
Exercisable at December 31, 2010
|
39,270
|
$
|
100.98
|
6.4
|
$
|
24,842
|
||||||||||
The aggregate intrinsic value of options outstanding is calculated based on the positive difference between the closing market price of the Company’s common stock at the end of the respective period and the exercise price of the underlying options. During the year ended December 31, 2010, the total intrinsic value of options exercised was $663,000 and the total amount of cash received from exercise of these options was $158,600. Shares of common stock issued upon the exercise of options are from authorized but unissued shares.
As of December 31, 2010, there was approximately $647,000 of total unrecognized compensation cost related to unvested stock-based compensation arrangements. Of this total amount, 70% and 30% are expected to be recognized during 2011 and 2012, respectively. The Company expects 9,956 in unvested options to vest in the future. The weighted-average grant-date fair value of vested and unvested options outstanding at December 31, 2010 was $62.73 and $68.85, respectively.
8. INCOME TAXES
The Company’s deferred tax assets consisted of the following at December 31:
|
2010
|
2009
|
|||||||
|
Net operating loss carryforwards
|
$
|
12,872,000
|
$
|
9,543,000
|
||||
|
Research and development expenses
|
14,180,000
|
14,906,000
|
||||||
|
Tax credits
|
1,711,000
|
1,563,000
|
||||||
|
Capital loss carryforward
|
340,000
|
340,000
|
||||||
|
Stock-based compensation
|
365,000
|
650,000
|
||||||
|
Gross deferred tax asset
|
29,468,000
|
27,002,000
|
||||||
|
Valuation allowance
|
(29,468,000
|
)
|
(27,002,000
|
)
|
||||
|
Net deferred tax asset
|
$
|
—
|
$
|
—
|
||||
F-43
As of December 31, 2010, the Company had federal and state net operating loss carryforwards of approximately $33,180,000 and $25,812,000 respectively, which expire through 2030. In addition, the Company has federal and state research and development and investment tax credits of approximately $1,382,000 and $449,000, respectively, which expire through 2030. The amount of net operating loss carryforwards which may be utilized annually in future periods may be limited pursuant to Section 382 of the Internal Revenue Code as a result of substantial changes in the Company’s ownership that have occurred or that may occur in the future.
The capital loss carryforward relates to the loss recorded in prior years for Novelos’ investment in an unrelated company.
Because of the Company’s limited operating history, continuing losses and uncertainty associated with the utilization of the net operating loss carryforwards in the future, management has provided a 100% allowance against the Company’s gross deferred tax asset. In 2010, the difference between the Company’s total statutory tax rate of approximately 40% and its effective tax rate of 0% is due to the nontaxable gain of $8,118,000 on derivative warrants, an increase in the valuation allowance of $2,466,000 and nondeductible liquidated damages accrued of $819,000. In 2009, the difference between the Company’s total statutory tax rate of approximately 40% and its effective tax rate of 0% is due equally to the increase in valuation allowance and the reduction in tax loss resulting from the nondeductible loss on derivative warrants.
The Company did not have any unrecognized tax benefits or accrued interest and penalties at any time during the years ended December 31, 2010 and 2009, and does not anticipate having any unrecognized tax benefits over the next twelve months. The Company is subject to audit by the IRS for tax periods commencing January 1, 2007.
9. NET INCOME (LOSS) PER SHARE
Basic net loss per share is computed by dividing net loss attributable to common stockholders by the weighted average number of shares of common stock outstanding during the period. Diluted net income per share is computed by dividing net loss attributable to common stockholders, as adjusted, by the sum of the weighted average number of shares of common stock and the dilutive potential common stock equivalents then outstanding. Potential common stock equivalents consist of stock options, warrants and convertible preferred stock and accumulated dividends. Since the Company has a net loss attributable to common stockholders for the years ended December 31, 2010 and 2009, the inclusion of common stock equivalents in the computation for those periods would be antidilutive. Accordingly, basic and diluted net loss per share is the same for the periods presented.
The following potentially dilutive securities have been excluded from the computation of diluted net income (loss) per share since their inclusion would be antidilutive:
|
Year Ended
December 31,
|
||||||||
|
2010
|
2009
|
|||||||
|
Stock options
|
50,074
|
60,260
|
||||||
|
Warrants
|
356,925
|
232,164
|
||||||
|
Conversion of preferred stock
|
2,042,108
|
329,451
|
(1)
|
|||||
(1) Includes shares of common stock that may become issuable upon conversion of preferred stock dividends accumulated at the respective date.
10. COMMITMENTS
Property Lease
Effective September 1, 2010, the Company entered into a six-month extension to its lease for office space, at a rate of $5,275 per month, expiring February 28, 2011. Rent expense was $65,000 and $87,000 for the years ended December 31, 2010 and 2009, respectively. Future minimum lease payments under this non-cancelable lease are approximately $10,600 during 2011. After February 28, 2011, the lease may be canceled with 30-day notice by either party.
Royalty Arrangements
The Company is obligated to a Russian company, ZAO BAM, under a royalty and technology transfer agreement. Mark Balazovsky, a director of the Company until November 2006, is the majority shareholder of ZAO BAM. Pursuant to the royalty and technology transfer agreement between the Company and ZAO BAM, the Company is required to make royalty payments of 1.2% of net sales of oxidized glutathione-based products. The Company is also required to pay ZAO BAM $2 million for each new oxidized glutathione-based drug within eighteen months following FDA approval of such drug.
F-44
If a royalty is not being paid to ZAO BAM on net sales of oxidized glutathione products, then the Company is required to pay ZAO BAM 3% of all license revenues. If license revenues exceed the Company’s cumulative expenditures including, but not limited to, preclinical and clinical studies, testing, FDA and other regulatory agency submission and approval costs, general and administrative costs, and patent expenses, then the Company would be required to pay ZAO BAM an additional 9% of the amount by which license revenues exceed the Company’s cumulative expenditures.
As a result of the assignment to Novelos of the exclusive worldwide intellectual property and marketing rights of oxidized glutathione (excluding the Russian Territory), Novelos is obligated to the Oxford Group, Ltd., or its assignees, for future royalties. Simyon Palmin, a founder of Novelos, a director until August 12, 2008 and the father of the Company’s president and chief executive officer, is president of Oxford Group, Ltd. Mr. Palmin was also an employee of the Company until September 2008 and performed consulting services through December 2009. Pursuant to the agreement, as revised May 26, 2005, Novelos is required to pay Oxford Group, Ltd., or its assignees, a royalty in the amount of 0.8% of the Company’s net sales of oxidized glutathione-based products.
Employment Agreements
The Company entered into an employment agreement with Harry Palmin effective January 1, 2006, whereby he agreed to serve as the Company’s president and chief executive officer for an initial term of two years. The agreement is automatically renewed for successive one-year terms unless notice of termination is provided by either party at least 90 days prior to the end of such term. The agreement was renewed most recently for an additional one-year term on January 1, 2011 in accordance with its terms. The agreement provides for an initial salary of $225,000 in 2006, participation in standard benefit programs and an annual cash bonus at the discretion of the compensation committee. The agreement further provides that upon resignation for good reason or termination without cause, both as defined in the agreement, Mr. Palmin will receive his pro rata share of the average of his annual bonus paid during the two fiscal years preceding his termination, his base salary and benefits for 11 months after the date of termination and fifty percent of his unvested stock options will vest. The agreement also contains a non-compete provision, which prohibits Mr. Palmin from competing with the Company for one year after termination of his employment with the Company.
Retention Agreements
On May 14, 2010, the Company entered into retention agreements with each of its four vice-presidents. The agreements provide for the lump-sum payment of six months’ base salary and benefits to each such officer following a termination without cause or a resignation with good reason occurring on or before November 14, 2011. The agreements further provide that if the executives remain employed with the Company as of October 1, 2010, they will receive a payment of two months’ base salary as a retention bonus on that date. The retention bonus, an aggregate of $140,000, was paid in October 2010 and will be deducted from the severance amounts that may become payable upon a subsequent involuntary termination. Elias Nyberg’s employment was terminated on March 10, 2011, without cause, and he received a payment of approximately $83,000 pursuant to the executive retention agreement. The agreements expire November 14, 2011. The total remaining amount that may become payable to the Company’s Named Executive Officers pursuant to the retention agreements is approximately $86,000 to Christopher Pazoles. Concurrently with the execution of the retention agreements, the employment agreement between the Company and Christopher Pazoles dated July 15, 2005 was terminated.
On May 14, 2010, the Company also entered into retention agreements with each of its three non-executive employees. The agreements provide for the lump-sum payment of six months’ base salary and benefits to each employee following a termination without cause or a resignation with good reason occurring on or before November 14, 2011. The agreements expire November 14, 2011.
11. LITIGATION
A purported class action complaint was filed on March 5, 2010 in the United States District Court for the District of Massachusetts by an alleged shareholder of the Company, on behalf of himself and all others who purchased or otherwise acquired the Company’s common stock in the period between December 14, 2009 and February 24, 2010, against the Company and its President and Chief Executive Officer, Harry S. Palmin. On October 1, 2010, the court appointed lead plaintiffs (Boris Urman and Ramona McDonald) and appointed lead plaintiffs’ counsel. On October 22, an amended complaint was filed. The amended complaint claims that the Company violated Section 10(b) of the Securities Exchange Act of 1934, as amended, and Rule 10b-5 promulgated thereunder in connection with alleged disclosures related to the Phase 3 clinical trial of NOV-002 for non-small cell lung cancer. On December 6, 2010, the Company filed a motion to dismiss the complaint with prejudice. On January 20, 2011, the plaintiffs filed their opposition to our motion and on March 3, 2011, we filed our response to their opposition. Our motion to dismiss remains pending. The Company believes the allegations are without merit and intends to defend vigorously against the allegations.
F-45
On June 28, 2010, the Company received a letter from counsel to ZAO BAM and ZAO BAM Research Laboratories (collectively, “BAM”) alleging that the Company modified the chemical composition of NOV-002 without prior notice to or approval from BAM, constituting a material breach of a technology and assignment agreement the Company had entered into with BAM on June 20, 2000 (the “June 2000 Agreement”). The letter references the Company’s amendment, submitted to the FDA on August 30, 2005, to its investigational new drug application dated August 1999 as the basis for BAM’s claims and demands the transfer of all intellectual property rights concerning NOV-002 to BAM. Mark Balazovsky, a director of Novelos from June 1996 until November 2006 and a shareholder of Novelos through at least June 25, 2010, is, to the Company’s knowledge, still the general director and principal shareholder of ZAO BAM. The Company believes the allegations are without merit and intends to defend vigorously against any proceedings that BAM may initiate as to these allegations. On September 24, 2010, the Company filed a complaint in Suffolk Superior Court seeking a declaratory judgment by the court that the June 2000 Agreement has been replaced by a subsequent agreement between the parties dated April 1, 2005 (the “April 2005 Agreement”), that Novelos’ obligations to BAM are governed solely by the April 2005 Agreement and that the obligations of the June 2000 agreement have been performed and fully satisfied. On November 29, 2010, BAM answered the complaint, denying the material allegations, and stating its affirmative defenses and certain counterclaims. On January 14, 2011, the Company responded to the counterclaims, denying BAM’s material allegations and stating our affirmative defenses. The Company believes the counterclaims are without merit and intends to vigorously defend against them.
|
12.
|
SUBSEQUENT EVENTS
|
Merger Agreement
On April 8, 2011, the Company entered into an Agreement and Plan of Merger (the “Merger Agreement”) with Cellectar, Inc. (“Cellectar”) and Cell Acquisition Corp. (the “Merger Subsidiary”), a wholly owned subsidiary of Novelos, pursuant to which Cellectar was merged into the Merger Subsidiary (the “Acquisition”). As a result of the Acquisition, the Merger Subsidiary, which has been renamed Cellectar, Inc., owns all assets and operates the business previously owned and operated by Cellectar. Prior to the Acquisition, Cellectar was in the business of developing drugs for the treatment and diagnosis of cancer. The Company will continue to develop Cellectar’s compounds following the Acquisition.
As consideration for the Acquisition, the former stockholders of Cellectar received aggregate consideration consisting of a number of shares of Novelos common stock constituting, after giving effect to the Acquisition but before giving effect to the concurrent private placement of our securities described below, approximately 85% of the outstanding shares of Novelos common stock. Prior to the Acquisition, the Company amended and restated its certificate of incorporation and in connection therewith, among other things, effected a 1-for-153 reverse split of its common stock (the “Reverse Split”). Immediately following the effectiveness of the Reverse Split, there were approximately 2,959,871 shares of our common stock outstanding, and the Company issued 17,001,596 shares of our common stock to the former stockholders of Cellectar upon the effectiveness of the Acquisition.
Rodman & Renshaw, LLC (“Rodman”), financial advisor to Novelos in connection with the Acquisition, received a cash fee of $250,000 upon the completion of the Acquisition in consideration of their services. XMS Capital Partners, the financial advisor to Cellectar in connection with the Acquisition, received a cash fee of $200,000 upon the completion of the Acquisition in consideration of their services.
Securities Purchase Agreement
Concurrently with the execution of the Merger Agreement, the Company entered into a Securities Purchase Agreement with certain accredited investors under which the Company sold an aggregate of 6,846,537 units, each unit consisting of one share of its common stock and a warrant to purchase one share of its common stock, at a price of $0.75 per unit, for gross proceeds of approximately $5,135,000. The warrants have an exercise price of $0.75 and expire on March 31, 2016. The warrant exercise price and/or the common stock issuable pursuant to such warrant will be subject to adjustment for stock dividends, stock splits or similar capital reorganizations so that the rights of the warrant holders after such event will be equivalent to the rights of warrant holders prior to such event.
The Securities Purchase Agreement includes a requirement that the Company file with the Securities and Exchange Commission (“SEC”) no later than October 5, 2011, a registration statement covering the resale of the shares of common stock, and the shares of common stock underlying the warrants, issued pursuant to the Securities Purchase Agreement. The Company is also required to use our commercially reasonable efforts to have the registration statement declared effective by December 4, 2011, and to keep the registration statement continuously effective under the Securities Act of 1933, as amended (the “Securities Act”) until the earlier of the date when all the registrable securities covered by the registration statement have been sold or the second anniversary of the closing.
In the event the Company fails to file the registration statement within the timeframe specified by the Securities Purchase Agreement, or if it fails to obtain effectiveness of this registration on or prior to the December 4, 2011 (if there is no review by the SEC) or by January 3, 2012 (if there is review by the SEC) with respect to the maximum number of shares permitted to be registered by the SEC, the Company will be required to pay to the purchasers liquidated damages equal to 1.5% per month (pro-rated on a daily basis for any period of less than a full month) of the aggregate purchase price of the units purchased until the registration statement is filed or declared effective, as applicable. The Company will be allowed to suspend the use of the registration statement for not more than 30 consecutive days, on not more than two occasions, in any 12 month period. The Company has also granted piggy-back registration rights with respect to any shares of common stock that it is required to exclude from the registration statement as a condition of its effectiveness, and has also agreed to file further registration statements with respect to any such shares six months after the effective date of the initial registration statement.
F-46
The Company paid to Rodman, the placement agent for the financing, a cash fee equal to $200,000 and warrants to purchase 192,931 shares of its common stock in consideration for their advisory services with respect to the financing pursuant to the placement agency agreement between Rodman and the Company. Rodman is entitled to registration rights with respect to the shares of common stock issuable upon exercise of these warrants. The warrants have the same terms as those issued to the investors in the private placement.
Changes in Directors and Executive Officers
Effective April 8, 2011, prior to the completion of the Acquisition, Michael J. Doyle, Sim Fass and David B. McWilliams resigned from the Company’s board of directors and their respective committee appointments.
Effective April 8, 2011, as a condition to the completion of the Acquisition, Jamey P. Weichert, Thomas Rockwell Mackie, John Neis, John E. Niederhuber and Michael F. Tweedle were appointed to the Company’s board of directors. Committee assignments have not yet been determined. Jamey P. Weichert, Thomas Rockwell Mackie and John Neis, previously served on the board of directors of Cellectar.
Amendment of Certificate of Incorporation
Effective April 7, 2011 the Company’s certificate of incorporation was amended to eliminate the Certificate to Set Forth Designations, Voting Powers, Preferences, Restrictions and Relative Rights of Series C 8% Cumulative Convertible Preferred Stock. There had not been any shares of Series C preferred stock outstanding since December 2010.
Effective April 7, 2011 the Company’s certificate of incorporation was amended to eliminate the Certificate of Designations, Preferences and Rights of Series E Convertible Preferred Stock. There had not been any shares of Series E preferred stock outstanding since December 2010.
Prior to the closing of the Acquisition on April 8, 2011, the Company amended and restated its certificate of incorporation in order (a) to effect the reverse split; (b) to reduce the number of shares of our authorized common stock from 750,000,000 to 150,000,000; (c) to eliminate the right of the stockholders to act by written consent; and (d) to classify the board of directors into three classes. Class I directors will stand for re-election at the Company’s next annual meeting of stockholders, Class II directors will stand for re-election at the 2012 annual meeting of stockholders, and Class III directors will stand for re-election at the 2013 annual meeting of stockholders. Thomas Rockwell Mackie, James S. Manuso and John Niederhuber serve as Class I directors, Stephen A. Hill, Michael F. Tweedle and John Neis serve as Class II directors, and Harry S. Palmin, Jamey P. Weichert and Howard M. Schneider serve as Class III directors.
F-47
NOVELOS THERAPEUTICS, INC. (PRE-ACQUISITION)
BALANCE SHEET
|
March 31,
|
||||
|
2011
(unaudited)
|
||||
|
ASSETS
|
||||
|
CURRENT ASSETS:
|
||||
|
Cash and equivalents
|
$ | 1,030,942 | ||
|
Prepaid expenses and other current assets
|
28,042 | |||
|
Deferred transaction costs
|
28,500 | |||
|
Total current assets
|
1,087,484 | |||
|
FIXED ASSETS, NET
|
6,515 | |||
|
DEPOSITS
|
15,350 | |||
|
TOTAL ASSETS
|
$ | 1,109,349 | ||
|
LIABILITIES, REDEEMABLE PREFERRED STOCK AND STOCKHOLDERS’ EQUITY
|
||||
|
CURRENT LIABILITIES:
|
||||
|
Accounts payable and accrued liabilities
|
$ | 250,008 | ||
|
Derivative liability (see Note 2)
|
162,760 | |||
|
Deferred revenue – current
|
33,333 | |||
|
Total current liabilities
|
446,101 | |||
|
DEFERRED REVENUE – NONCURRENT
|
358,333 | |||
|
COMMITMENTS AND CONTINGENCIES
|
||||
|
STOCKHOLDERS’ EQUITY:
|
||||
|
Common stock, $0.00001 par value; 150,000,000 shares authorized; 2,959,871 shares issued and outstanding at March 31, 2011
|
30 | |||
|
Additional paid-in capital
|
75,291,653 | |||
|
Accumulated deficit
|
(74,986,768 | ) | ||
|
Total stockholders’ equity
|
304,915 | |||
|
TOTAL LIABILITIES, REDEEMABLE PREFERRED STOCK AND STOCKHOLDERS’ EQUITY
|
$ | 1,109,349 | ||
See notes to financial statements.
Share totals give retroactive effect to the 1-for-153 reverse split of our common stock completed
on April 8, 2011.
F-48
NOVELOS THERAPEUTICS, INC. (PRE-ACQUISITION)
STATEMENTS OF OPERATIONS
(Unaudited)
|
Three Months Ended March 31,
|
||||||||
|
2011
|
2010
|
|||||||
|
REVENUE
|
$ | 8,333 | $ | 8,333 | ||||
|
COSTS AND EXPENSES:
|
||||||||
|
Research and development
|
532,686 | 1,910,889 | ||||||
|
General and administrative
|
611,877 | 644,763 | ||||||
|
Total costs and expenses
|
1,144,563 | 2,555,652 | ||||||
|
LOSS FROM OPERATIONS
|
(1,136,230 | ) | (2,547,319 | ) | ||||
|
OTHER INCOME:
|
||||||||
|
Interest income
|
668 | — | ||||||
|
Gain on derivative warrants (see Note 2)
|
125,490 | 7,897,441 | ||||||
|
Total other income
|
126,158 | 7,897,441 | ||||||
|
NET INCOME (LOSS)
|
(1,010,072 | ) | 5,350,122 | |||||
|
PREFERRED STOCK DIVIDENDS
|
— | (656,635 | ) | |||||
|
NET INCOME (LOSS) ATTRIBUTABLE TO COMMON STOCKHOLDERS
|
$ | (1,010,072 | ) | $ | 4,693,487 | |||
|
BASIC NET INCOME (LOSS) ATTRIBUTABLE TO COMMON STOCKHOLDERS PER COMMON SHARE
|
$ | (0.34 | ) | $ | 8.99 | |||
|
SHARES USED IN COMPUTING BASIC NET INCOME (LOSS) ATTRIBUTABLE TO COMMON STOCKHOLDERS PER COMMON SHARE
|
2,959,871 | 522,350 | ||||||
|
DILUTED NET INCOME (LOSS) ATTRIBUTABLE TO COMMON STOCKHOLDERS PER COMMON SHARE
|
$ | (0.34 | ) | $ | 3.41 | |||
|
SHARES USED IN COMPUTING DILUTED NET INCOME (LOSS) ATTRIBUTABLE TO COMMON STOCKHOLDERS PER COMMON SHARE
|
2,959,871 | 881,861 | ||||||
See notes to financial statements.
Share totals and share-based calculations give retroactive effect to the 1-for-153 reverse split of our common stock completed on April 8, 2011.
F-49
NOVELOS THERAPEUTICS, INC. (PRE-ACQUISITION)
STATEMENTS OF CASH FLOWS
(Unaudited)
|
Three Months Ended March 31,
|
||||||||
|
2011
|
2010
|
|||||||
|
CASH FLOWS FROM OPERATING ACTIVITIES:
|
||||||||
|
Net income (loss)
|
$ | (1,010,072 | ) | $ | 5,350,122 | |||
|
Adjustments to reconcile net income (loss) to cash used in operating activities:
|
||||||||
|
Depreciation and amortization
|
2,240 | 27,290 | ||||||
|
Stock-based compensation
|
108,378 | (97,479 | ) | |||||
|
Gain on derivative warrants
|
(125,490 | ) | (7,897,441 | ) | ||||
|
Changes in:
|
||||||||
|
Prepaid expenses and other current assets
|
35,483 | (47,574 | ) | |||||
|
Accounts payable and accrued liabilities
|
(315,715 | ) | (403,760 | ) | ||||
|
Accrued compensation
|
— | (238,022 | ) | |||||
|
Deferred revenue
|
(8,333 | ) | (8,333 | ) | ||||
|
Cash used in operating activities
|
(1,313,509 | ) | (3,315,197 | ) | ||||
|
CASH FLOWS FROM FINANCING ACTIVITIES:
|
||||||||
|
Deferred financing costs
|
(28,500 | ) | — | |||||
|
Proceeds from exercise of stock options
|
— | 157,400 | ||||||
|
Cash provided by financing activities
|
(28,500 | ) | 157,400 | |||||
|
DECREASE IN CASH AND EQUIVALENTS
|
(1,342,009 | ) | (3,157,797 | ) | ||||
|
CASH AND EQUIVALENTS AT BEGINNING OF PERIOD
|
2,372,951 | 8,769,529 | ||||||
|
CASH AND EQUIVALENTS AT END OF PERIOD
|
$ | 1,030,942 | $ | 5,611,732 | ||||
|
SUPPLEMENTAL DISCLOSURES OF NON-CASH FINANCING ACTIVITIES
|
||||||||
|
Dividends accumulated on shares of Series E preferred stock exchanged or converted into shares of common stock
|
$ | — | $ | 634,925 | ||||
|
Fair value of derivative warrants reclassified to additional paid-in capital upon cashless exercise
|
$ | — | $ | 2,584,397 | ||||
|
Carrying value of redeemable preferred stock converted into common stock
|
$ | — | $ | 4,689,593 | ||||
See notes to financial statements.
F-50
NOVELOS THERAPEUTICS, INC. (PRE-ACQUISITION)
NOTES TO FINANCIAL STATEMENTS
(Unaudited)
|
1.
|
NATURE OF BUSINESS, BASIS OF PRESENTATION
|
Novelos Therapeutics, Inc. (“Novelos” or the “Company”) is a biopharmaceutical company developing compounds for the treatment of cancer.
On April 8, 2011, the Company entered into a business combination with Cellectar, Inc. (“Cellectar”), a privately held Wisconsin corporation that designed and developed products to detect, treat and monitor a wide variety of human cancers (the “Acquisition”, see Note 10). Immediately prior to the Acquisition, the Company completed a 1-for-153 reverse split of its common stock (the “Reverse Split”). The Company then issued to the former shareholders of Cellectar 17,001,596 shares of its common stock as consideration for the Acquisition, which shares constituted approximately 85% of Novelos’ outstanding common stock after giving effect to the Acquisition. Upon the closing of the Acquisition, the Company completed the private placement of 6,846,537 shares of its common stock and warrants to purchase an additional 6,846,537 shares of its common stock (in each case after giving effect to the Reverse Split) for gross proceeds of approximately $5,135,000. As a result of the Acquisition, the Company is implementing a revised business plan focused on the development of the Cellectar compounds. Development of Novelos’ other compounds (NOV-002 and NOV-205) has been suspended.
The Reverse Split reduced the number of outstanding shares of Common Stock from 452,866,983 shares to 2,959,871 shares. On the Company's balance sheet, the aggregate par value of the issued common stock was reduced by reclassifying the par value amount of the eliminated shares of common stock to additional paid-in capital. All per share amounts and outstanding shares, including all common stock equivalents, stock options and warrants, have been retroactively restated in these financial statements and notes for all periods presented to reflect the Reverse Split. Additionally, the number of authorized shares of common stock disclosed on the balance sheet has been reduced to 150,000,000 from 750,000,000 to reflect the reduction in authorized shares of common stock that became effective concurrent with the Reverse Split.
Accounting principles generally accepted in the United States require that a company whose security holders retain the majority voting interest in the combined business be treated as the acquirer for financial reporting purposes. Accordingly, the Acquisition will be accounted for as a reverse acquisition whereby Cellectar, Inc. will be treated as the acquirer for accounting and financial reporting purposes. The financial statements presented herein represent the historical financial information of Novelos, prior to the Acquisition.
The Company is subject to a number of risks similar to those of other small biopharmaceutical companies. Principal among these risks are dependence on key individuals, competition from substitute products and larger companies, the successful development and marketing of its products in a highly regulated environment and the need to obtain additional financing necessary to fund future operations.
On February 24, 2010, the Company announced that its Phase 3 clinical trial for NOV-002 in non-small cell lung cancer (“NSCLC”) (the “Phase 3 Trial”) did not meet its primary endpoint of a statistically significant increase in median overall survival. Following evaluation of the detailed trial data, on March 18, 2010, the Company announced that the secondary endpoints had also not been met in the Phase 3 Trial and that it had discontinued development of NOV-002 for NSCLC in combination with first-line paclitaxel and carboplatin chemotherapy.
These financial statements have been prepared on the basis that the Company will continue as a going concern. The Company has generated insignificant revenues and has incurred operating losses since inception in devoting substantially all of its efforts toward research and development. The Company expects that it will continue to generate operating losses for the foreseeable future. The Company believes that its cash on hand at March 31, 2011, plus the proceeds from the private placement completed in connection with the Acquisition, is adequate to fund operations into the fourth quarter of 2011. The Company’s ability to execute its operating plan beyond that time depends on its ability to obtain additional funding via the sale of equity and/or debt securities, a strategic transaction or otherwise. The Company plans to continue to actively pursue financing alternatives, but there can be no assurance that it will obtain the necessary funding.
F-51
The accompanying unaudited financial statements of the Company have been prepared in accordance with accounting principles generally accepted in the United States (“GAAP”) for interim financial information and with the instructions to Form 10-Q. Accordingly, they do not include all of the information and footnotes required by GAAP for complete financial statements. In the opinion of management, all adjustments (consisting of normal recurring accruals) considered necessary for the fair presentation of these financial statements have been included. The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Interim results are not necessarily indicative of results to be expected for other quarterly periods or for the entire year ending December 31, 2011. These unaudited financial statements should be read in conjunction with the audited financial statements and related notes thereto included in the Company’s latest annual report for the year ended December 31, 2010 on Form 10-K, which was filed with the Securities and Exchange Commission (“SEC”) on April 14, 2011. The report from the Company’s independent registered public accounting firm dated April 11, 2011 and included with its annual report on Form 10-K indicated that factors exist that raised substantial doubt about the Company’s ability to continue as a going concern.
Comprehensive Income (Loss) – The Company had no components of comprehensive income (loss) other than the net income (loss) in all periods presented.
Derivative Instruments – The Company generally does not use derivative instruments to hedge exposures to cash flow or market risks. However, certain warrants to purchase common stock that do not meet the requirements for classification as equity, in accordance with the Derivatives and Hedging Topic of the Financial Accounting Standards Board Accounting Standards Codification (“FASB ASC”), are classified as liabilities. In such instances, net-cash settlement is assumed for financial reporting purposes, even when the terms of the underlying contracts do not provide for a net-cash settlement. These warrants are considered derivative instruments because the agreements contain “down-round” provisions whereby the number of shares for which the warrants are exercisable and/or the exercise price of the warrants are subject to change in the event of certain issuances of stock at prices below the then-effective exercise price of the warrants. The number of shares issuable under such warrants was 105,042 at March 31, 2011. The primary underlying risk exposure pertaining to the warrants is the change in fair value of the underlying common stock. Such financial instruments are initially recorded at fair value with subsequent changes in fair value recorded as a component of gain or loss on derivatives in each reporting period. If these instruments subsequently meet the requirements for equity classification, the Company reclassifies the fair value to equity. At March 31, 2011, these warrants represented the only outstanding derivative instruments issued or held by the Company.
|
2.
|
FAIR VALUES OF ASSETS AND LIABILITIES
|
In accordance with Fair Value Measurements and Disclosures Topic of the FASB ASC, the Company groups its financial assets and financial liabilities generally measured at fair value in three levels, based on the markets in which the assets and liabilities are traded and the reliability of the assumptions used to determine fair value.
|
|
·
|
Level 1: Input prices quoted in an active market for identical financial assets or liabilities.
|
|
|
·
|
Level 2: Inputs other than prices quoted in Level 1, such as prices quoted for similar financial assets and liabilities in active markets, prices for identical assets and liabilities in markets that are not active or other inputs that are observable or can be corroborated by observable market data.
|
|
|
·
|
Level 3: Input prices quoted that are significant to the fair value of the financial assets or liabilities which are not observable or supported by an active market.
|
F-52
Assets and liabilities measured at fair value on a recurring basis are summarized below:
|
March 31, 2011
|
||||||||||||||||
|
Level 1
|
Level 2
|
Level 3
|
Fair Value
|
|||||||||||||
|
Liabilities:
|
||||||||||||||||
|
Warrants
|
$ | - | $ | 162,760 | $ | - | $ | 162,760 | ||||||||
The Company uses valuation methods and assumptions that consider among others the fair value of the underlying stock, risk-free interest rate, volatility, expected life and dividend rates in estimating fair value for the warrants considered to be derivative instruments. Assumptions used are generally consistent with those disclosed for stock-based compensation (see Note 5).
|
3.
|
COLLABORATION AGREEMENTS
|
In December 2007 the Company entered into a Collaboration Agreement with Lee’s Pharmaceutical (HK) Ltd. (“Lee’s Pharm”). Pursuant to this agreement, Lee’s Pharm obtained an exclusive license to develop, manufacture and commercialize NOV-002 and NOV-205 in China, Hong Kong, Taiwan and Macau (the “Chinese Territory”). The Company has suspended further development of NOV-205; however, this suspension may not impact the development strategy of Lee’s Pharm. Under the terms of the agreement the Company received a license fee of $500,000 in March 2008 and is entitled to receive up to $1,700,000 in future milestone payments upon the completion of development and marketing milestones by Lee’s Pharm. This initial $500,000 payment received is being amortized over the estimated term of this agreement, 15 years. Accordingly, $8,333 of license revenue was recognized in each of the three-month periods ended March 31, 2011 and 2010.
The Lee’s Pharm agreement provides that the Company receive royalty payments of 20-25% of net sales of NOV-002 in the Chinese Territory and receive royalty payments of 12-15% of net sales of NOV-205 in the Chinese Territory. Lee’s Pharm is obligated to reimburse the Company for the manufacturing cost of pharmaceutical products provided to Lee’s Pharm in connection with the agreement. Lee’s Pharm has committed to spend a minimum amount on development in the first four years of the agreement. The agreement expires upon the expiration of the last patent covering any of the licensed products, or twelve years from the date of the first commercial sale in China, whichever occurs later.
On February 11, 2009, Novelos entered into a collaboration agreement (the “Collaboration Agreement”) with Mundipharma International Corporation Limited (“Mundipharma”) to develop, manufacture and commercialize, on an exclusive basis, Licensed Products (as defined in the Collaboration Agreement), which includes the Company’s compound, NOV-002, in Europe (other than the Russian Territory), Asia (other than the Chinese Territory) and Australia (collectively referred to as the “Mundipharma Territory”). Mundipharma is an independent associated company of Purdue Pharma, L.P. (“Purdue”). The Collaboration Agreement provides for Mundipharma to pay the Company royalties and fixed milestone payments based on sales and commercial launches in the licensed territories.
For countries in which patents are held, the Collaboration Agreement expires on a country-by-country basis within the Mundipharma Territory on the earlier of (1) expiration of the last applicable Novelos patent within the country or (2) the determination that any patents within the country are invalid, obvious or otherwise unenforceable. For countries in which no patents are held, the Collaboration Agreement expires the earlier of 15 years from its effective date or upon generic product competition in the country resulting in a 20% drop in Mundipharma’s market share. Novelos may terminate the Collaboration Agreement upon breach or default by Mundipharma. Mundipharma may terminate the Collaboration Agreement upon breach or default, filing of voluntary or involuntary bankruptcy by Novelos, the termination of certain agreements with companies associated with the originators of the licensed technology, or 30-day notice for no reason. If any regulatory approval within the Mundipharma Territory is suspended as a result of issues related to the safety of the Licensed Products, then Mundipharma’s obligations under the Collaboration Agreement will be suspended until the regulatory approval is reinstated. If that reinstatement does not occur within 12 months of the suspension, then Mundipharma may terminate the Collaboration Agreement.
Concurrently with the execution of the Collaboration Agreement, Novelos completed a private placement of Series E preferred stock and common stock purchase warrants to Purdue.
F-53
The Company expects that the negative results of its Phase 3 Trial will adversely affect development and commercialization of NOV-002 under the collaboration agreements with Lee’s Pharm and Mundipharma.
|
4.
|
STOCKHOLDERS’ EQUITY
|
Common Stock Warrants — The following table summarizes information with regard to outstanding warrants as of March 31, 2011, issued in connection with equity and debt financings since 2005.
|
Offering
|
Number of
Number of Shares
Issuable Upon
Exercise of
Outstanding
Warrants
|
Exercise
Price
|
Expiration Date
|
||||||
|
Series B Preferred Stock – placement agents
|
5,392
|
$
|
191.25
|
May 2, 2012
|
|||||
|
Series C Exchange
|
8,169
|
$
|
191.25
|
May 2, 2012
|
|||||
|
Series E Preferred Stock
|
60,331
|
$
|
99.45
|
December 31, 2015
|
|||||
|
August 2009 Private Placement
|
31,194
|
$
|
100.98
|
December 31, 2015
|
|||||
|
July 2010 Direct Offering (1)
|
105,042
|
$
|
10.71
|
July 27, 2015
|
|||||
|
Preferred Incentive Warrants
|
105,042
|
$
|
16.065
|
July 27, 2015
|
|||||
|
Total
|
315,170
|
||||||||
(1) The exercise price of these warrants was adjusted to $0.75 per share in connection with the private placement completed on April 8, 2011. See Note 10.
On March 7, 2011, warrants to purchase 33,569 shares of common stock at $234.09 per share expired unexercised.
On May 3, 2011, 18,153 shares of common stock were issued in connection with the cashless exercise of warrants to purchase 27,311 shares of common stock at $0.75 per share.
|
5.
|
STOCK-BASED COMPENSATION
|
The following table summarizes amounts charged (credited) to expense for stock-based compensation related to employee and director stock option grants and stock-based compensation recorded in connection with stock options granted to non-employee consultants:
|
Three Months Ended
March 31,
|
||||||||
|
2011
|
2010
|
|||||||
|
Employee and director stock option grants:
|
||||||||
|
Research and development
|
$ | 49,298 | $ | 57,113 | ||||
|
General and administrative
|
59,682 | 82,928 | ||||||
| 108,980 | 140,041 | |||||||
|
Non-employee consultant stock option grants:
|
||||||||
|
Research and development
|
(545 | ) | (210,825 | ) | ||||
|
General and administrative
|
(57 | ) | (26,695 | ) | ||||
| (602 | ) | (237,520 | ) | |||||
|
Total stock-based compensation
|
$ | 108,378 | $ | (97,479 | ) | |||
F-54
There were no stock option grants during the three months ended March 31, 2011 or 2010.
|
Number of
Shares
Issuable Upon
Exercise of
Outstanding
Options
|
Weighted
Average
Exercise Price
|
Weighted
Average
Remaining
Contracted
Term in
Years
|
Aggregate
Intrinsic
Value
|
|||||||||||||
|
Outstanding at December 31, 2010
|
49,227 | $ | 100.61 | 6.9 | $ | 24,842 | ||||||||||
|
Outstanding at March 31, 2011
|
49,227 | $ | 100.61 | 6.6 | 12,421 | |||||||||||
|
Exercisable at March 31, 2011
|
40,348 | $ | 101.03 | 6.2 | $ | 12,421 | ||||||||||
The aggregate intrinsic value of options outstanding is calculated based on the positive difference between the closing market price of the Company’s common stock at the end of the respective period and the exercise price of the underlying options. There were no options exercised during the three months ended March 31, 2011. Shares of common stock issued upon the exercise of options are from authorized but unissued shares.
As of March 31, 2011, there was approximately $538,000 of total unrecognized compensation cost related to unvested stock-based compensation arrangements. Of this total amount, 64% and 36% are expected to be recognized during 2011 and 2012, respectively. The Company expects 8,879 in unvested options to vest in the future. The weighted-average grant-date fair value of vested and unvested options outstanding at March 31, 2011 was $62.75 and $68.07, respectively.
|
6.
|
NET INCOME (LOSS) PER SHARE
|
Basic net income (loss) per share is computed by dividing net income (loss) attributable to common stockholders by the weighted average number of shares of common stock outstanding during the period. Diluted net income per share is computed by dividing net income attributable to common stockholders by the sum of weighted average number of shares of common stock and the dilutive potential common stock equivalents then outstanding. Potential common stock equivalents consist of stock options, warrants and convertible preferred stock and accumulated dividends. Since the Company has a net loss for the three months ended March 31, 2011, the inclusion of common stock equivalents in the computation would be antidilutive. Accordingly, basic and diluted net loss per share are the same for the three months ended March 31, 20011.
The following table sets forth the shares and net income used in the diluted earnings per share computation for the three months ended March 31, 2010:
|
Numerator:
|
||||
|
Net income available to common stockholders used in basic earnings per share calculation
|
$ | 4,693,487 | ||
|
Derivative gain recorded on dilutive warrants
|
(2,340,515 | ) | ||
|
Dividends on convertible preferred stock
|
656,635 | |||
|
Net income available to common stockholders used in diluted earnings per share calculation
|
$ | 3,009,607 | ||
|
Denominator:
|
||||
|
Weighted average shares of common stock used in the computation of basic earnings per share
|
522,350 | |||
|
Dilutive effect of stock options
|
26,430 | |||
|
Dilutive effect of warrants to purchase common stock
|
79,646 | |||
|
Dilutive effect of convertible preferred stock
|
253,435 | |||
|
Shares used in computation of diluted earnings per share
|
881,861 | |||
F-55
The following potentially dilutive securities have been excluded from the computation of diluted net income (loss) per share since their inclusion would be antidilutive:
|
Three Months Ended
March 31,
|
||||||||
|
|
2011
|
2010
|
||||||
|
Stock options
|
49,227 | 3,970 | ||||||
|
Warrants
|
315,170 | 43,349 | ||||||
|
7.
|
INCOME TAXES
|
The Company accounts for income taxes in accordance with the Income Taxes Topic of the FASB ASC. Under this guidance, deferred tax assets or liabilities are computed based on the difference between the financial statement and income tax basis of assets and liabilities, and net operating loss carryforwards, using the enacted tax rates. Deferred income tax expense or benefit is based on changes in the asset or liability from period to period. The Company did not record a provision or benefit for federal, state or foreign income taxes for the three months ended March 31, 2011 or 2010 because the Company has experienced losses on a tax basis since inception. The net income reported for the three months ended March 31, 2010 was a result of the gain recorded on the revaluation of derivative warrant liability during that period, which is a nontaxable item. The Company has not recorded deferred tax assets as their realization is uncertain.
|
8.
|
LITIGATION
|
A purported class action complaint was filed on March 5, 2010 in the United States District Court for the District of Massachusetts by an alleged shareholder of the Company, on behalf of himself and all others who purchased or otherwise acquired the Company’s common stock in the period between December 14, 2009 and February 24, 2010, against the Company and its President and Chief Executive Officer, Harry S. Palmin. On October 1, 2010, the court appointed lead plaintiffs (Boris Urman and Ramona McDonald) and appointed lead plaintiffs’ counsel. On October 22, 2010, an amended complaint was filed. The amended complaint claims that the Company violated Section 10(b) of the Securities Exchange Act of 1934, as amended, and Rule 10b-5 promulgated thereunder in connection with alleged disclosures related to the Phase 3 clinical trial of NOV-002 for non-small cell lung cancer. On December 6, 2010, the Company filed a motion to dismiss the complaint with prejudice. On January 20, 2011, the plaintiffs filed their opposition to our motion and on March 3, 2011, the Company filed its response to their opposition. The motion to dismiss remains pending. The Company believes the allegations are without merit and intends to defend vigorously against the allegations.
F-56
On June 28, 2010, the Company received a letter from counsel to ZAO BAM and ZAO BAM Research Laboratories (Russian companies, collectively referred to as “BAM”) alleging that the Company modified the chemical composition of NOV-002 without prior notice to or approval from BAM, constituting a material breach of a technology and assignment agreement the Company had entered into with BAM on June 20, 2000 (the “June 2000 Agreement”). The letter references the Company’s amendment, submitted to the FDA on August 30, 2005, to its investigational new drug application dated August 1999 as the basis for BAM’s claims and demands the transfer of all intellectual property rights concerning NOV-002 to BAM. Mark Balazovsky, a director of Novelos from June 1996 until November 2006 and a shareholder of Novelos through at least June 25, 2010, is, to the Company’s knowledge, still the general director and principal shareholder of ZAO BAM. The Company believes the allegations are without merit and intends to defend vigorously against any proceedings that BAM may initiate as to these allegations. On September 24, 2010, the Company filed a complaint in Suffolk Superior Court seeking a declaratory judgment by the court that the June 2000 Agreement has been replaced by a subsequent agreement between the parties dated April 1, 2005 (the “April 2005 Agreement”), that Novelos’ obligations to BAM are governed solely by the April 2005 Agreement and that the obligations of the June 2000 agreement have been performed and fully satisfied. On November 29, 2010, BAM answered the complaint, denying the material allegations, and stating its affirmative defenses and certain counterclaims. On January 14, 2011, the Company responded to the counterclaims, denying BAM’s material allegations and stating its affirmative defenses. The Company believes the counterclaims are without merit and intends to vigorously defend against them.
|
9.
|
COMMITMENTS
|
Retention Agreements
The Company has entered into retention agreements with each of its three vice presidents. The agreements provide for the lump-sum payment of six months’ base salary and benefits to each such officer following a termination without cause or a resignation with good reason occurring on or before November 14, 2011. Certain of the agreements provide that if the executives were employed with the Company as of October 1, 2010, they would receive a payment of two months’ base salary as a retention bonus on that date. The retention bonus was paid in October 2010 and will be deducted from the severance amounts that may become payable upon a subsequent involuntary termination. The total remaining amount that may become payable to the Company’s Named Executive Officers pursuant to the retention agreements is approximately $86,000 to Christopher Pazoles.
During the three months ended March 31, 2011, pursuant to retention agreements, the Company paid a total of approximately $218,000 in severance payments to employees terminated during that period, including $83,000 to Elias Nyberg, the Company’s former vice president of regulatory, quality and compliance.
|
10.
|
SUBSEQUENT EVENTS
|
Merger Agreement
On April 8, 2011, the Company entered into an Agreement and Plan of Merger (the “Merger Agreement”) with Cellectar, Inc. (“Cellectar”) and Cell Acquisition Corp. (the “Merger Subsidiary”), a wholly-owned subsidiary of Novelos, pursuant to which Cellectar was merged into the Merger Subsidiary (the “Acquisition”). As a result of the Acquisition, the Merger Subsidiary, which has been renamed Cellectar, Inc., owns all assets of and operates the business previously owned and operated by Cellectar. Prior to the Acquisition, Cellectar was in the business of developing drugs for the treatment and diagnosis of cancer. The Company will continue to develop Cellectar’s compounds following the Acquisition.
As consideration for the Acquisition, the former stockholders of Cellectar received aggregate consideration consisting of a number of shares of Novelos common stock constituting, after giving effect to the Acquisition but before giving effect to the concurrent private placement of the Company’s securities described below, approximately 85% of the outstanding shares of Novelos common stock. Prior to the Acquisition, the Company amended and restated its certificate of incorporation and in connection therewith, among other things, effected a 1-for-153 reverse split of its common stock (the “Reverse Split”). Immediately following the effectiveness of the Reverse Split, there were 2,959,871 shares of common stock outstanding, and the Company issued 17,001,596 shares of its common stock to the former stockholders of Cellectar upon the effectiveness of the Acquisition.
Rodman & Renshaw, LLC (“Rodman”), financial advisor to Novelos in connection with the Acquisition, received a cash fee of $250,000 upon the completion of the Acquisition in consideration for their services. XMS Capital Partners, the financial advisor to Cellectar in connection with the Acquisition, received a cash fee of $200,000 upon the completion of the Acquisition in consideration for their services.
Accounting principles generally accepted in the United States require that a company whose security holders retain the majority voting interest in the combined business be treated as the acquirer for financial reporting purposes. Accordingly, the Acquisition will be accounted for as a reverse acquisition whereby Cellectar, Inc. will be treated as the accounting acquirer.
F-57
Securities Purchase Agreement
Concurrently with the execution of the Merger Agreement, the Company entered into a Securities Purchase Agreement with certain accredited investors under which the Company sold an aggregate of 6,846,537 units, each unit consisting of one share of its common stock and a warrant to purchase one share of its common stock, at a price of $0.75 per unit, for gross proceeds of approximately $5,135,000. The warrants have an exercise price of $0.75 and expire on March 31, 2016. The warrant exercise price and/or the common stock issuable pursuant to such warrant will be subject to adjustment for stock dividends, stock splits or similar capital reorganizations so that the rights of the warrant holders after such event will be equivalent to the rights of warrant holders prior to such event.
The Securities Purchase Agreement includes a requirement that the Company file with the SEC no later than October 5, 2011, a registration statement covering the resale of the shares of common stock, and the shares of common stock underlying the warrants, issued pursuant to the Securities Purchase Agreement. The Company is also required to use commercially reasonable efforts to have the registration statement declared effective by December 4, 2011, and to keep the registration statement continuously effective under the Securities Act of 1933, as amended (the “Securities Act”), until the earlier of the date when all the registrable securities covered by the registration statement have been sold or the second anniversary of the closing.
In the event the Company fails to file the registration statement within the timeframe specified by the Securities Purchase Agreement, or if it fails to obtain effectiveness of this registration on or prior to the December 4, 2011 (if there is no review by the SEC) or by January 3, 2012 (if there is review by the SEC) with respect to the maximum number of shares permitted to be registered by the SEC, the Company will be required to pay to the purchasers liquidated damages equal to 1.5% per month (pro-rated on a daily basis for any period of less than a full month) of the aggregate purchase price of the units purchased until the registration statement is filed or declared effective, as applicable. The Company will be allowed to suspend the use of the registration statement for not more than 30 consecutive days, on not more than two occasions, in any 12 month period. The Company has also granted piggy-back registration rights with respect to any shares of common stock that it is required to exclude from the registration statement as a condition of its effectiveness, and has also agreed to file further registration statements with respect to any such shares six months after the effective date of the initial registration statement.
The Company paid to Rodman, the placement agent for the financing, a cash fee equal to $200,000 and warrants to purchase 192,931 shares of its common stock in consideration for their advisory services with respect to the financing pursuant to the placement agency agreement between Rodman and the Company. Rodman is entitled to registration rights with respect to the shares of common stock issuable upon exercise of these warrants. The warrants have the same terms as those issued to the investors in the private placement.
As a result of the financing, warrants to purchase 105,042 shares of common stock at $10.71 per share, giving effect to the Reverse Split, became exercisable for $0.75 per share according to their terms. On May 3, 2011, 18,153 shares of common stock were issued in connection with the cashless exercise 27,311 of these warrants.
Changes in Directors and Executive Officers
Effective April 8, 2011, prior to the completion of the Acquisition, Michael J. Doyle, Sim Fass and David B. McWilliams resigned from the Company’s board of directors and their respective committee appointments.
Effective April 8, 2011, as a condition to the completion of the Acquisition, Jamey P. Weichert, Thomas Rockwell Mackie, John Neis, John E. Niederhuber and Michael F. Tweedle were appointed to the Company’s board of directors. Jamey P. Weichert, Thomas Rockwell Mackie and John Neis previously served on the board of directors of Cellectar.
On April 25, 2011, the Company’s board of directors appointed the following individuals to serve on the following committees of the board of directors.
F-58
Howard M. Schneider, John Neis and John E. Niederhuber were appointed to the audit committee of the board of directors. Mr. Schneider was appointed as the chairman of that committee, a position that he held prior to the Acquisition.
Thomas Rockwell Mackie, James S. Manuso, John Neis and Michael F. Tweedle were appointed to the compensation committee of the board of directors. Dr. Mackie was appointed as the chairman of that committee.
Stephen A. Hill, John E. Niederhuber and James S. Manuso were appointed to the nominating and corporate governance committee of the board of directors. Dr. Hill was appointed as the chairman of that committee.
Amendment of Certificate of Incorporation
Effective April 7, 2011 the Company’s certificate of incorporation was amended to eliminate the Certificate to Set Forth Designations, Voting Powers, Preferences, Restrictions and Relative Rights of Series C 8% Cumulative Convertible Preferred Stock. There had not been any shares of Series C preferred stock outstanding since December 2010.
Effective April 7, 2011 the Company’s certificate of incorporation was amended to eliminate the Certificate of Designations, Preferences and Rights of Series E Convertible Preferred Stock. There had not been any shares of Series E preferred stock outstanding since December 2010.
Prior to the closing of the Acquisition on April 8, 2011, the Company amended and restated its certificate of incorporation in order to (a) effect the reverse split; (b) reduce the number of shares of authorized common stock from 750,000,000 to 150,000,000; (c) eliminate the right of the stockholders to act by written consent; and (d) classify the board of directors into three classes. Class I directors will stand for re-election at the Company’s next annual meeting of stockholders, Class II directors will stand for re-election at the 2012 annual meeting of stockholders, and Class III directors will stand for re-election at the 2013 annual meeting of stockholders. Thomas Rockwell Mackie, James S. Manuso and John Niederhuber serve as Class I directors, Stephen A. Hill, Michael F. Tweedle and John Neis serve as Class II directors, and Harry S. Palmin, Jamey P. Weichert and Howard M. Schneider serve as Class III directors.
F-59
UNAUDITED PRO FORMA CONDENSED COMBINED FINANCIAL STATEMENTS
Table of Contents
|
Page
|
||
|
Unaudited Pro Forma Condensed Combined Statement of Operations for the Six Months Ended June 30, 2011
|
F-61 | |
|
Unaudited Pro Forma Condensed Combined Statement of Operations for the Twelve Months Ended December 31, 2010
|
F-62 | |
|
Notes to Unaudited Pro Forma Condensed Combined Financial Statements
|
F-63 |
On April 8, 2011, Novelos completed the acquisition of Cellectar, Inc. (“Cellectar”) pursuant to an Agreement and Plan of Merger (the “Merger Agreement”) between Novelos, Cellectar and Cell Acquisition Corp., a wholly owned subsidiary of Novelos (the “Merger Subsidiary”). As a result, Cellectar was merged into the Merger Subsidiary (the “Acquisition”) and the Merger Subsidiary, which has been renamed Cellectar, Inc., owns all assets and operates the business previously owned and operated by Cellectar. References in these unaudited pro forma combined financial statements and notes to “Cellectar” relate to the activities and financial information of Cellectar prior to the Acquisition, references to “Novelos” relate to the activities and financial information of Novelos prior to the Acquisition and references to “the Company” or “we” or “us” or “our” relate to the activities of the combined Company following the Acquisition.
The following unaudited pro forma condensed combined financial statements combine the historical financial statements of Novelos and Cellectar after giving effect to the Acquisition. The unaudited pro forma condensed combined financial statements are provided for informational purposes only and are subject to a number of uncertainties and assumptions and do not purport to represent what the combined companies’ actual performance or financial position would have been if Novelos and Cellectar had been operating as combined entities for the periods presented and does not purport to indicate the financial position or results of operations as of any future date or for any future period. These unaudited condensed combined financial statements should be read in conjunction with the historical financial statements, including the notes thereto, of Novelos included in our Form 10-K for the year ended December 31, 2010, our Form 10-Q for the six-month period ended June 30, 2011 and in the historical financial statements included elsewhere in this prospectus.
The unaudited pro forma condensed combined statement of operations for the six-month period ended June 30, 2011 and the twelve-month period ended December 31, 2010 give effect to the Acquisition as if it had occurred January 1, 2010.
The pro forma adjustments are based on available information, preliminary estimates and certain assumptions that the Company believes are reasonable and are described in the accompanying notes to the unaudited pro forma condensed combined financial statements. The unaudited pro forma condensed combined financial statements assume that the Acquisition will be accounted for using the purchase method of accounting in accordance with ASC Topic 805 – Business Combinations. The total purchase price has been preliminarily allocated based on available information and the preliminary estimates and assumptions that management believes are reasonable. The evaluation is prelimary principally as a result of the pending evaluation of the Company’s intangible assets. The allocation of the purchase price has not been finalized and the actual adjustments may change when the final valuation of certain intangible assets is determined. Accordingly, there can be no assurance that the final allocation of purchase price will not materially differ from the preliminary allocations reflected in the unaudited pro forma condensed combined financial statements.
F-60
NOVELOS THERAPEUTICS, INC.
(a Development Stage Company)
UNAUDITED PRO FORMA CONDENSED COMBINED STATEMENT OF OPERATIONS
for the Six Months Ended June 30, 2011
|
Historical
|
||||||||||||||||
|
Novelos, (Pre-
Acquisition) |
Combined
Company as Reported |
Pro forma
Adjustments
|
Pro forma
Combined |
|||||||||||||
|
REVENUE
|
$ | 8,333 | $ | — | $ | (8,333 | )(a) | $ | — | |||||||
|
COSTS AND EXPENSES:
|
||||||||||||||||
|
Research and development
|
537,455 | 1,433,356 | — | 1,970,811 | ||||||||||||
|
General and administrative
|
862,546 | 1,674,647 | (1,029,644 | )(b) | 1,507,549 | |||||||||||
|
Total costs and expenses
|
1,400,001 | 3,108,003 | (1,029,644 | ) | 3,478,360 | |||||||||||
|
LOSS FROM OPERATIONS
|
(1,391,668 | ) | (3,108,003 | ) | 1,021,311 | (3,478,360 | ) | |||||||||
|
OTHER INCOME (EXPENSE):
|
||||||||||||||||
|
Grant income
|
— | 44,479 | — | 44,479 | ||||||||||||
|
Gain (loss) on derivative instruments
|
125,490 | (70,393 | ) | — | 55,097 | |||||||||||
|
Interest income (expense), net
|
668 | (426,546 | ) | 423,159 | (c) | (2,719 | ) | |||||||||
|
Total other income (expense)
|
126,158 | (452,460 | ) | 423,159 | 96,857 | |||||||||||
|
Net (loss)
|
$ | (1,265,510 | ) | $ | (3,560,463 | ) | $ | 1,444,470 | $ | (3,381,503 | ) | |||||
|
NET LOSS ATTRIBUTABLE TO COMMON STOCKHOLDERS
|
$ | (1,265,510 | ) | $ | (3,560,463 | ) | $ | 1,444,470 | $ | (3,381,503 | ) | |||||
|
BASIC AND DILUTED NET LOSS ATTRIBUTABLE TO COMMON STOCKHOLDERS PER COMMON SHARE
|
$ | (0.43 | ) | $ | (0.18 | ) | $ | (0.13 | ) | |||||||
|
SHARES USED IN COMPUTING BASIC AND DILUTED NET LOSS ATTRIBUTABLE TO COMMON STOCKHOLDERS PER COMMON SHARE
|
2,959,871 | 19,317,642 | 26,813,921 | |||||||||||||
See accompanying notes to the unaudited pro forma condensed combined financial statements.
F-61
NOVELOS THERAPEUTICS, INC.
(a Development Stage Company)
UNAUDITED PRO FORMA CONDENSED COMBINED STATEMENT OF OPERATIONS
for the Twelve Months Ended December 31, 2010
|
Historical
|
|
|
||||||||||||||
|
Novelos
|
Cellectar
|
Pro forma
Adjustments
|
Pro forma
Combined |
|||||||||||||
|
REVENUE
|
$ | 33,334 | $ | — | $ | (33,334 | )(a) | $ | — | |||||||
|
COSTS AND EXPENSES:
|
||||||||||||||||
|
Research and development
|
2,997,984 | 2,984,207 | — | 5,982,191 | ||||||||||||
|
General and administrative
|
2,486,032 | 1,209,474 | (77,005 | )(b) | 3,618,501 | |||||||||||
|
Total costs and expenses
|
5,484,016 | 4,193,681 | (77,005 | ) | 9,600,692 | |||||||||||
|
LOSS FROM OPERATIONS
|
(5,450,682 | ) | (4,193,681 | ) | 43,671 | (9,600,692 | ) | |||||||||
|
OTHER INCOME (EXPENSE):
|
||||||||||||||||
|
Grant income
|
244,479 | 200,000 | — | 444,479 | ||||||||||||
|
Gain on derivative instruments
|
8,118,174 | — | — | 8,118,174 | ||||||||||||
|
Liquidated damages
|
(819,000 | ) | — | 819,000 | (d) | — | ||||||||||
|
Interest income (expense), net
|
2,421 | (566,156 | ) | (102,766 | )(e) | (666,501 | ) | |||||||||
|
Other income
|
— | (426 | ) | — | (426 | ) | ||||||||||
|
Total other income (expense)
|
7,546,074 | (366,582 | ) | 716,234 | 7,895,726 | |||||||||||
|
Net income (loss)
|
2,095,392 | (4,560,263 | ) | 759,905 | (1,704,966 | ) | ||||||||||
|
Preferred stock dividend
|
(2,207,827 | ) | — | 2,207,827 | (f) | — | ||||||||||
|
Preferred stock deemed dividend
|
(12,541,201 | ) | — | 12,541,201 | (f) | — | ||||||||||
|
NET LOSS ATTRIBUTABLE TO COMMON STOCKHOLDERS
|
$ | (12,653,636 | ) | $ | (4,560,263 | ) | $ | 15,508,933 | $ | (1,704,966 | ) | |||||
|
BASIC AND DILUTED NET LOSS ATTRIBUTABLE TO COMMON STOCKHOLDERS PER COMMON SHARE
|
$ | (15.36 | ) | $ | (0.36 | ) | $ | (0.06 | ) | |||||||
|
SHARES USED IN COMPUTING BASIC AND DILUTED NET LOSS ATTRIBUTABLE TO COMMON STOCKHOLDERS PER COMMON SHARE
|
823,933 | 12,820,102 | 26,808,004 | |||||||||||||
See accompanying notes to the unaudited pro forma condensed combined financial statements
F-62
NOVELOS THERAPEUTICS, INC.
(a Development Stage Company)
NOTES TO UNAUDITED PRO FORMA CONDENSED COMBINED FINANCIAL STATEMENTS
1. BASIS OF PRESENTATION
The unaudited pro forma condensed combined statement of operations for the six-month period ended June 30, 2011 has been prepared based on the unaudited consolidated statement of operations of the Company for the six-month period ended June 30, 2011 as reported on Form 10-Q as filed with the Securities and Exchange Commission (the “SEC”) on August 12, 2011 and the historical information of Novelos for the period from January 1, 2011 through April 8, 2011 giving effect to the Acquisition as if it had occurred January 1, 2010. The unaudited pro forma condensed combined statements of operations for the twelve-month period ended December 31, 2010 has been prepared based on the historical information of Novelos and Cellectar giving effect to the Acquisition as if it had occurred January 1, 2010.
In accordance with the rules and regulations of the SEC, unaudited statements may omit or condense information and disclosures normally required for a complete set of financial statements prepared in accordance with generally accepted accounting principles. Management believes that the notes to the unaudited pro forma financial statements as presented contain disclosures adequate to make the information presented useful and not misleading.
The pro forma adjustments are based on available information, preliminary estimates and certain assumptions that the Company believes are reasonable and are described in the accompanying notes to the unaudited pro forma condensed combined financial statements, which should be read in conjunction with these unaudited pro forma condensed combined financial statements.
As consideration for the Acquisition, the stockholders of Cellectar received aggregate consideration consisting of a number of shares of Novelos common stock constituting approximately 85% of the outstanding shares of Novelos common stock, after giving effect to the Acquisition but before giving effect to the concurrent private placement of our securities. Since the former shareholders of Cellectar retained the majority voting interest in the combined business, the Acquisition (see Note 2) will be accounted for as a reverse acquisition whereby Cellectar will be treated as the acquirer for accounting and financial reporting purposes.
The unaudited pro forma condensed combined financial statements assume that the Acquisition will be accounted for using the purchase method of accounting in accordance with ASC Topic 805 – Business Combinations. The total purchase price has been preliminarily allocated based on available information and the preliminary estimates and assumptions that management believes are reasonable. The allocation of the purchase price has not been finalized and the actual adjustments may change when the final valuation of certain intangible assets is determined. The evaluation is preliminary principally as a result of the pending evaluation of the Company’s intangible assets. Accordingly, there can be no assurance that the final allocation of purchase price will not materially differ from the preliminary allocations reflected in the unaudited pro forma combined financial statements.
2. ACQUISITION
Conversion of Convertible Notes and Payment of Long-Term Debt
On April 8, 2011, immediately prior to the Acquisition, Cellectar paid the note payable to a bank in full settlement of the note. The payment was made in order to avoid an event of default that would have occurred as a result of the change of control that occurred at the time of the Acquisition.
Following the maturity and default of the Convertible Notes, the holders of the Convertible Notes agreed that all of the outstanding notes would be automatically converted simultaneous with the completion of an acquisition and financing (the “Conversion Time”), if completed. The amount of shares issued upon such conversion would be dependent on the amount of investment made by the note holders at the Conversion Time and were negotiated based on outstanding principal and projected accrued interest based on an assumed closing date for the acquisition and financing. On April 1, 2011, Cellectar’s Board of Directors voted to accept the note holders consent to convert the Convertible Notes into 4,181,535 shares of common stock immediately prior to the Acquisition. On April 8, 2011, immediately prior to the Acquisition, the principal and unpaid interest on the Convertible Notes was converted into the agreed total of 4,181,535 shares of common stock. The revised conversion terms resulted in the issuance of an additional 343,963 shares of common stock over the 3,837,572 shares of common stock that would have been issued if the unpaid principal and accrued interest on the Convertible Notes had been converted on that date in accordance with their original terms at the stated conversion price.
Merger Agreement
On April 8, 2011, Novelos completed the acquisition of Cellectar, Inc. (“Cellectar”) pursuant to an Agreement and Plan of Merger (the “Merger Agreement”) between Novelos, Cellectar and Cell Acquisition Corp., a wholly owned subsidiary of Novelos (the “Merger Subsidiary”). As a result, Cellectar was merged into the Merger Subsidiary (the “Acquisition”) and the Merger Subsidiary, which has been renamed Cellectar, Inc., owns all assets and operates the business previously owned and operated by Cellectar.
F-63
As consideration for the Acquisition, the stockholders of Cellectar received aggregate consideration consisting of a number of shares of Novelos common stock constituting, after giving effect to the Acquisition but before giving effect to the concurrent private placement of Novelos securities described below, approximately 85% of the outstanding shares of Novelos common stock. Prior to the Acquisition, Novelos amended and restated its certificate of incorporation and in connection therewith, among other things, effected a 1-for-153 reverse split of its common stock (the “Reverse Split”), which has been retroactively reflected in the accompanying pro forma financial statements. Immediately prior to the Acquisition, there were 2,959,871 shares of Novelos common stock outstanding. Novelos then issued 17,001,596 shares of Novelos common stock to the former stockholders of Cellectar upon the effective date of the Acquisition. Warrants and options to purchase Novelos common stock that were outstanding prior to the Acquisition remained outstanding following the Acquisition. These consist of warrants to purchase a total of 315,164 shares of Novelos common stock with prices ranging from $16.07 to $191.25 and options to purchase a total of 49,159 shares of Novelos common stock with prices ranging from $1.53 to $1,072.53.
XMS Capital Partners, the financial advisor to Cellectar in connection with the Acquisition, received a cash fee of $200,000 upon the completion of the Acquisition in consideration of their services. Rodman & Renshaw, LLC (“Rodman”), financial advisor to Novelos in connection with the Acquisition, received a cash fee of $250,000 upon the completion of the Acquisition in consideration of their services. These amounts were recorded to general and administrative expense as of the date of the Acquisition.
Securities Purchase Agreement
Concurrently with the execution of the Merger Agreement, Novelos entered into a Securities Purchase Agreement with certain accredited investors under which it sold an aggregate of 6,846,537 units, each unit consisting of one share of its common stock and a warrant to purchase one share of its common stock, at a price of $0.75 per unit, for gross proceeds of approximately $5,135,000. The warrants have an exercise price of $0.75 and expire on March 31, 2016. The warrant exercise price and/or the common stock issuable pursuant to such warrant will be subject to adjustment for stock dividends, stock splits or similar capital reorganizations so that the rights of the warrant holders after such event will be equivalent to the rights of warrant holders prior to such event.
Rodman, the placement agent for the financing, was paid a cash fee equal to $200,000 and issued warrants to purchase 192,931 shares of the Company’s common stock (having an exercise price of $0.75 and which expire March 31, 2016) in consideration for their advisory services with respect to the financing pursuant to the placement agency agreement between Rodman and Novelos. Rodman is entitled to registration rights with respect to the shares of common stock issuable upon exercise of these warrants. These amounts were recorded as a reduction of gross proceeds received.
Purchase Accounting
The Acquisition will be accounted for using the purchase method of accounting as a reverse acquisition. In a reverse acquisition, the post-acquisition net assets of the surviving combined company includes the historical cost basis of the net assets of the accounting acquirer, Cellectar, plus the fair value of the net assets of the accounting acquiree, Novelos. Further, under the purchase method, the purchase price is allocated to the assets acquired and liabilities assumed based on their estimated fair values and the excess of the purchase price over the estimated fair value of the identifiable net assets is presented as excess purchase price over net assets acquired. The cost of acquisition and related purchase price allocation is based on preliminary evaluation of the fair value of assets and liabilities assumed of Novelos and may change when the final valuation of certain intangible assets is determined. The preliminary evaluation principally relates to the evaluation of the Company’s intangible assets.
The fair value of the consideration transferred in the Acquisition was $2,219,903 and was calculated as the number of shares of common stock that Cellectar would have had to issue (adjusted for the Exchange Ratio) in order for Novelos shareholders to obtain a 15% equity interest in the combined Company post-acquisition, multiplied by the estimated fair value of the Company’s common stock on the acquisition date. The estimated fair value of the Company’s common stock was based on the offering price of the common stock sold in the private placement which was both completed concurrently with and conditioned upon the closing of the Acquisition. This price was determined to be the best indication of fair value on that date since the price was based on an arm's length negotiation with a group consisting of both new and existing investors that were aware of the pending Acquisition and assumed similar liquidity risk as those investors holding the majority of shares being valued as purchase consideration.
The following table summarizes the Company’s preliminary estimated fair values of the assets acquired and the liabilities assumed at the date of acquisition.
|
Consideration - issuance of securities
|
$ | 2,219,903 | ||
|
Prepaid expenses and other assets
|
$ | 71,892 | ||
|
Fixed assets
|
6,515 | |||
|
Accrued liabilities
|
(380,130 | ) | ||
|
Derivative liability
|
(59,485 | ) | ||
|
Excess of purchase price over net assets acquired
|
1,675,462 | |||
|
Total purchase price – net of cash acquired of $905,649
|
$ | 1,314,254 |
F-64
The excess of purchase price over net assets acquired will be allocated to intangibles, which could potentially include the fair value of the compounds developed prior to the Acquisition by Novelos, with the remainder allocated to goodwill once the Company completes the final allocation of purchase price. The estimated fair values of assets acquired and liabilities assumed are provisional and are based on the information that was available as of the acquisition date to estimate the fair value of assets acquired and liabilities assumed. The Company believes that the information provides a reasonable basis for estimating the fair values of assets acquired and liabilities assumed, but the Company is waiting for additional information necessary to finalize those fair values. Therefore, the provisional measurements of fair value reflected are subject to change and such changes may be significant. The Company expects to finalize the valuation and complete the purchase price allocation as soon as practicable but no later than one year from the acquisition date.
3. PRO FORMA ADJUSTMENTS
Statement of operations adjustments
|
(a)
|
Represents the elimination of Novelos historical revenue related to the amortization of an advance payment made to Novelos in connection with a collaboration agreement. The payment was recorded as deferred revenue and recognized as revenue on a straight-line basis over the life of the collaboration agreement. As a result of the Acquisition, the Company determined as part of the purchase price allocation that the estimated fair value of the deferred revenue was $0. As such, for purposes of the pro forma presentation the historical revenue associated with deferred revenue has been eliminated.
|
|
(b)
|
Represents the adjustment to reflect transaction-related costs in connection with the Acquisition. For purposes of the pro forma unaudited statement of operations presentation for the six and twelve month period ended June 30, 2011 and December 31, 2010, the Company prepared the pro forma as if the Acquisition occurred on January 1, 2010. As such, the Company eliminated transaction-related costs that were incurred prior to the consummation of the Acquisition and therefore excluded for pro forma presentation. Additionally, the Company paid $450,000 related to investment banking fees as described in Note 2 on April 8, 2011. For purposes of the pro forma presentation, these investment banking fees were eliminated from the six month period ended June 30, 2011. See adjustments as follows:
|
|
For the Six
Months Ended June 30,
2011 |
For the
Twelve Months Ended December
31, 2010 |
|||||||
|
Adjustment to eliminate transaction related costs that were incurred prior to the consummation of the Acquisition.
|
$
|
(579,644
|
)
|
$
|
(77,005
|
)
|
||
|
Adjustment to eliminate the merger costs as a result of the consummation of the Acquisition (as described in Note 2)
|
(450,000
|
)
|
—
|
|||||
|
Total adjustment to merger costs
|
$
|
(1,029,644
|
)
|
$
|
(77,005
|
)
|
||
|
(c)
|
Represents the adjustment to eliminate interest expense associated with the convertible notes and the bank note for the six-month period ended June 30, 2011 and to eliminate interest expense associated with a beneficial conversion feature that was recorded as a result of the conversion of convertible notes immediately prior to the Acquisition. For purposes of the pro forma unaudited statement of operations for the six months ended June 30, 2011 presentation, the conversion of the notes would have occurred immediately before the Acquisition, or January 1, 2010 and therefore the associated interest expense has been excluded from the pro forma presentation. A detail of the components of the adjustment is presented below:
|
|
Adjustment to eliminate interest expense associated with the convertible notes
|
$
|
158,680
|
||
|
Adjustment to eliminate interest expense associated with the bank note
|
6,506
|
|||
|
Adjustment to reflect the impact of the beneficial shares that were issued on the date of the conversion of the convertible notes in connection with the acquisition
|
257,973
|
|||
|
Total adjustment to interest income (expense), net
|
$
|
423,159
|
F-65
|
(d)
|
Represents the adjustment to eliminate the estimated liquidated damages recorded for failure to file a registration statement for the resale of common stock issuable upon exercise of shares of Novelos’ convertible preferred stock. For pro forma presentation, the preferred stock is assumed to have been exchanged for common stock at January 1, 2010 in order to reflect the post-acquisition capital structure and liquidated damages are therefore assumed not to have accrued. |
|
(e)
|
Represents the adjustment to eliminate the interest expense for the year ended December 31, 2010 associated with the convertible notes and the bank note, net of an adjustment to reflect the additional interest expense associated with beneficial shares that were issued on the assumed date of conversion (January 1, 2010) in connection with the Acquisition associated with the outstanding principal balance of $2,720,985 on convertible notes. For the purpose of pro forma presentation, the fair value of the beneficial shares was calculated assuming the notes were converted on the date of issuance (January 2010); at which time no interest would have accrued or converted resulting in a higher fair value for the pro forma presentation in the statement of operations. The Company determined the interest expense for pro forma purposes of $677,062 as the difference between the conversion price and the estimated fair value of Novelos common stock as of the Acquisition date, or $0.75 per share, multiplied by the beneficial shares assumed converted of 902,749 less the amount of interest expense already included in the statement of operations of $213,793.
|
|
Adjustment to eliminate interest expense associated with the convertible notes
|
$
|
305,049
|
||
|
Adjustment to eliminate interest expense associated with the bank note
|
55,454
|
|||
|
Adjustment to eliminate interest expense associated with a beneficial conversion feature resulting from the conversion of convertible notes on April 8, 2011.
|
213,793
|
|||
|
Adjustment to reflect interest expense related to beneficial shares that were issued on the date of the conversion of the convertible notes, for pro forma purposes, in connection with the acquisition as if it occurred on January 1, 2010
|
(677,062
|
)
|
||
|
Total adjustment to interest income (expense), net
|
$
|
(102,766
|
)
|
|
(f)
|
Represents the elimination of the accruing dividends and deemed dividends on Novelos’ convertible preferred stock, which is assumed to have been exchanged for common stock at January 1, 2010 in order to reflect the post-acquisition capital structure for the purpose of presentation in the pro forma statements of operations.
|
F-66
In Humans, HOT Targets Cancerous Tumors - Not Normal Tissue or Bone Marrow
(SPECT/CT imaging of HOT in Phase 1a trial patients; Bright areas = HOT uptake)

Our Therapeutic Compounds (HOT and COLD) Selectively Target Cancer Stem Cells
(Fluorescence photomicrographs; Green = uptake of fluorescent-labeled COLD)
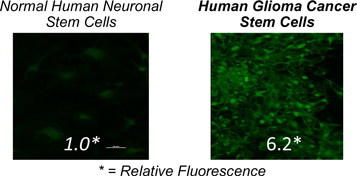
cGMP Radiopharmaceutical Manufacturing Facility at Our Headquarters in Madison, WI

The images provided above are for illustrative purposes only and may not be indicative of all results.
The above illustrations do not refer to products approved by the FDA.
Novelos has not received any revenue from the sale of its products.
Novelos Therapeutics, Inc.
shares of common stock
PROSPECTUS
Rodman & Renshaw, LLC
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 13. Other Expenses of Issuance and Distribution
The following table provides information regarding the various actual and anticipated expenses (other than placement agent fees) payable by us in connection with the issuance and distribution of the securities being registered hereby. All amounts shown are estimates except the Securities and Exchange Commission registration fee.
|
Nature of Expense
|
Amount
|
|||
|
SEC registration fee
|
$
|
2,003
|
||
|
Accounting fees and expenses
|
20,000
|
|||
|
Legal fees and expenses
|
150,000
|
|||
|
Transfer agent’s fees and expenses
|
3,000
|
|||
|
Printing and related fees
|
15,000
|
|||
|
Miscellaneous
|
10,000
|
|||
|
Total
|
$
|
200,003
|
||
Item 14. Indemnification of Directors and Officers.
Section 102(b)(7) of the Delaware General Corporation Law allows us to adopt a charter provision eliminating or limiting the personal liability of directors to us or our stockholders for breach of fiduciary duty as directors, but the provision may not eliminate or limit the liability of directors for (a) any breach of the director's duty of loyalty to us or our stockholders, (b) any acts or omissions not in good faith or which involve intentional misconduct or a knowing violation of law, (c) unlawful payments of dividends or unlawful stock repurchases or redemptions under Section 174 of the Delaware General Corporation Law or (d) any transaction from which the director derived an improper personal benefit. Article Seventh of our charter provides that none of our directors shall be personally liable to us or our stockholders for monetary damages for any breach of fiduciary duty as a director, subject to the limitations imposed by Section 102(b)(7). Article Seventh also provides that no amendment to or repeal of Article Seventh shall apply to or have any effect on the liability or the alleged liability of any director with respect to any acts or omissions of such director occurring prior to such amendment or repeal. A principal effect of Article Seventh is to eliminate or limit the potential liability of our directors for monetary damages arising from breaches of their duty of care, unless the breach involves one of the four exceptions described in (a) through (d) above.
Section 145 of the Delaware General Corporation Law provides, in general, that a corporation incorporated under the laws of the State of Delaware, such as us, may indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action, suit or proceeding (other than a derivative action by or in the right of the corporation) by reason of the fact that such person is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee or agent of another enterprise, against expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by such person in connection with such action, suit or proceeding if such person acted in good faith and in a manner such person reasonably believed to be in or not opposed to the best interests of the corporation, and, with respect to any criminal action or proceeding, had no reasonable cause to believe such person's conduct was unlawful. In the case of a derivative action, a Delaware corporation may indemnify any such person against expenses (including attorneys’ fees) actually and reasonably incurred by such person in connection with the defense or settlement of such action or suit if such person acted in good faith and in a manner such person reasonably believed to be in or not opposed to the best interests of the corporation, except that no indemnification will be made in respect of any claim, issue or matter as to which such person will have been adjudged to be liable to the corporation unless and only to the extent that the Court of Chancery of the State of Delaware or any other court in which such action was brought determines such person is fairly and reasonably entitled to indemnity for such expenses.
Article Eighth of our amended and restated certificate of incorporation and Section 5.1 of our bylaws provide that we will indemnify our directors, officers, employees and agents to the extent and in the manner permitted by the provisions of the Delaware General Corporation Law, as amended from time to time, subject to any permissible expansion or limitation of such indemnification, as may be set forth in any shareholders’ or directors’ resolution or by contract.
II-1
The effect of these provisions would be to permit indemnification by us for, among other liabilities, liabilities arising out of the Securities Act of 1933.
Item 15. Recent Sales of Unregistered Securities
In the last three years we have sold the following securities in reliance on, unless otherwise indicated, the exemption under Section 4(2) of the Securities Act of 1933, as amended, as transactions not involving any public offering. All share and per share amounts have been adjusted to give effect to the 1-for-153 reverse stock split that occurred immediately prior to the Acquisition.
2011
From April 1, 2011 through June 30, 2011:
On April 8, 2011, we issued an aggregate of 17,001,596 shares of our common stock as merger consideration to the former shareholders of Cellectar.
Concurrently with the Acquisition, on April 8, 2011, we entered into a Securities Purchase Agreement with certain accredited investors under which we sold an aggregate of 6,846,537 units, each unit consisting of one share of our common stock and a warrant to purchase one share of our common stock, at a price of $0.75 per unit for gross proceeds of $5,134,903. The warrants have an exercise price of $0.75 and expire on March 31, 2016. We also issued a warrant to purchase 192,931 shares of our common stock for $0.75, expiring March 31, 2016, to the placement agent in the financing.
On May 3, 2011, we issued 18,153 shares of our common stock in connection with the cashless exercise of warrants to purchase 27,310 shares of common stock at $0.75 per share.
From January 1, 2011 through March 31, 2011:
None.
2010
From October 1, 2010 through December 31, 2010:
On November 30, 2010, we issued 2,228,338 shares of common stock in exchange for all outstanding shares of Series E preferred stock and all outstanding shares of Series C preferred stock. The issuance was made pursuant to an exchange agreement between the Company and all holders of its preferred stock.
From July 1, 2010 through September 30, 2010:
On July 27, 2010, we issued five-year warrants (the “Incentive Warrants”) to our preferred stockholders for the purchase of up to an aggregate of 105,042 shares of common stock at an exercise price of $16.07 per share pursuant to a consent and waiver dated July 6, 2010, as amended on July 21, 2010. The Incentive Warrants were issued in connection with an offering registered under the Securities Act of 1933, as amended, of an aggregate of 140,056 shares of its common stock and five-year warrants to purchase up to an aggregate of 105,042 shares of its common stock, for gross proceeds of $1,500,000.
From April 1, 2010 through June 30, 2010:
None.
From January 1, 2010 through March 31, 2010:
|
|
·
|
We issued 76,769 shares of our common stock upon conversion of approximately 140 shares of our Series E preferred stock, having an aggregate stated value of approximately $7,000,000, and accumulated undeclared dividends thereon.
|
II-2
|
|
·
|
We issued 47,000 shares of our common stock upon the cashless exercise of warrants to purchase 77,551 shares of common stock. The warrants had an expiration date of December 31, 2015 and an exercise price of $99.45 per share.
|
|
|
·
|
We issued 1,480 shares of our common stock upon the cashless exercise of warrants to purchase 2,075 shares of common stock. The warrants had an expiration date of August 9, 2010 and an exercise price of $99.45 per share.
|
|
|
·
|
We issued 229 shares of our common stock upon the cashless exercise of warrants to purchase 490 shares of common stock. The warrants had an expiration date of May 2, 2012 and an exercise price of $191.25 per share.
|
|
|
·
|
We issued 2,395 shares of our common stock upon the cashless exercise of warrants to purchase 6,480 shares of common stock. The warrants had an expiration date of March 7, 2011 and an exercise price of $263.16 per share.
|
|
|
·
|
We issued 313 shares of our common stock upon the cashless exercise of warrants to purchase 544 shares of common stock. The warrants had an expiration date of May 2, 2012 and an exercise price of $191.25 per share.
|
|
|
·
|
We issued 2,058 shares of our common stock upon the cashless exercise of warrants to purchase 2,614 shares of common stock. The warrants had an expiration date of April 1, 2010 and an exercise price of $95.62 per share.
|
2009
From October 1, 2009 through December 31, 2009:
|
|
·
|
We issued 31,384 shares of our common stock upon conversion of approximately 58 shares of our Series E preferred stock, having an aggregate stated value of approximately $2,907,000, and accumulated undeclared dividends thereon.
|
|
|
·
|
We issued 4,330 shares of our common stock upon conversion of 28 shares of our Series C preferred stock having an aggregate stated value of $336,000, and accumulated undeclared dividends thereon.
|
|
|
·
|
We issued 172 shares of our common stock upon the cashless exercise of warrants to purchase an aggregate of 1,316 shares of common stock. The warrants had an expiration date of March 7, 2011 and an exercise price of $263.16 per share.
|
|
|
·
|
We issued 793 shares of our common stock upon the cashless exercise of warrants to purchase an aggregate of 1,320 shares of common stock. The warrants had an expiration date of August 9, 2010 and an exercise price of $0.65 per share.
|
|
|
·
|
We issued 218,648 shares of our common stock upon the cashless exercise of warrants to purchase an aggregate of 320,000 shares of common stock. The warrants had an expiration date of April 1, 2010 and an exercise price of $99.45 per share.
|
|
|
·
|
We issued 249 shares of our common stock upon the cashless exercise of warrants to purchase an aggregate of 396 shares of common stock. The warrants had an expiration date of October 3, 2010 and an exercise price of $99.45 per share.
|
|
|
·
|
We sold 54,466 shares of our common stock and warrants to purchase 19,063 shares of common stock at an exercise price of $100.98 per share for gross proceeds of approximately $5,500,000.
|
From July 1, 2009 through September 30, 2009:
|
|
·
|
We sold 34,660 shares of our common stock and warrants to purchase 12,131 shares of common stock at an exercise price of $100.98 per share for gross proceeds of approximately $3,500,000.
|
|
|
·
|
We issued 13,622 shares of our common stock in exchange for outstanding warrants to purchase 45,409 shares of common stock at an exercise price of $278.46 per share. These warrants had been issued in a March 2006 financing. The issuance was made pursuant to an exchange agreement with each warrant holder and was exempt from registration under Section 3(a)(9) of the Securities Act.
|
II-3
|
|
·
|
We issued 747 shares of our common stock upon conversion of approximately 39 shares of our Series E preferred stock, having an aggregate stated value of approximately $1,952,000, and accumulated undeclared dividends thereon.
|
|
|
·
|
We issued 747 shares of our common stock upon conversion of 5 shares of our Series C preferred stock, having an aggregate stated value of $60,000, and accumulated dividends thereon.
|
|
|
·
|
We issued 476 shares of our common stock upon the cashless exercise of warrants to purchase an aggregate of 1,715 shares of common stock. The warrants had an expiration date of August 9, 2010 and an exercise price of $99.45 per share.
|
From April 1, 2009 through June 30, 2009:
|
|
·
|
We issued 39 shares of our common stock upon the cashless exercise of warrants to purchase an aggregate of 136 shares of common stock. The warrants had an expiration date of August 9, 2010 and an exercise price of $99.45 per share.
|
|
|
·
|
We issued 4,979 shares of our common stock upon conversion of 35 shares of our Series C preferred stock, having an aggregate stated value of $420,000, and accumulated dividends thereon.
|
From January 1, 2009 through March 31, 2009:
|
|
·
|
We sold 200 shares of our Series E preferred stock and warrants to purchase 60,331 shares of our common stock at an exercise price of $99.45 per share for gross proceeds of approximately $10,000,000 and paying approximately $800,000 in fees and expenses. In addition, 413.5 shares of our Series D preferred stock and accumulated undeclared dividends thereon were exchanged for 445.442875 shares of our Series E preferred stock.
|
2008
From July 1, 2008 through September 30, 2008:
In August 2008, we sold 30,165 shares of our common stock to two related accredited investors at $99.45 per share, for gross proceeds of approximately $3,000,000.
II-4
Item 16. Exhibits and Financial Statement Schedules
|
Incorporated by Reference
|
||||||||||
|
Exhibit
No. |
Description
|
Filed with
this Form S-1 |
Form
|
Filing Date
|
Exhibit
No. |
|||||
|
1.1
|
Form of Underwriting Agreement*
|
|||||||||
|
2.1
|
Agreement and Plan of Merger by and among Novelos Therapeutics, Inc., Cell Acquisition Corp. and Cellectar, Inc. dated April 8, 2011
|
8-K
|
April 11, 2011
|
2.1
|
||||||
|
3.1
|
Second Amended and Restated Certificate of Incorporation
|
8-K
|
April 11, 2011
|
3.1
|
||||||
|
3.2
|
Amended and Restated By-laws
|
8-K
|
June 1, 2011
|
3.1
|
||||||
|
3.3
|
Form of Certificate of Amendment to the Second Amended and Restated Certificate of Incorporation of Novelos Therapeutics, Inc.
|
S-1
|
July 1, 2011
|
3.3
|
||||||
|
5.1
|
Legal Opinion of Foley Hoag LLP*
|
|||||||||
| 4.1 | Form of Underwriter’s Warrant* | |||||||||
|
10.1
|
Employment Agreement with Harry S. Palmin dated January 31, 2006
|
8-K
|
February 6, 2006
|
99.1
|
||||||
|
10.2
|
Second Amendment to Employment Agreement between the Company and Harry Palmin
|
8-K
|
June 1, 2011
|
10.1
|
||||||
|
10.3
|
2000 Stock Option and Incentive Plan
|
SB-2
|
November 16, 2005
|
10.2
|
||||||
|
10.4
|
Form of 2004 non-plan non-qualified stock option
|
SB-2
|
November 16, 2005
|
10.3
|
||||||
|
10.5
|
Form of non-plan non-qualified stock option used from February to May 2005
|
SB-2
|
November 16, 2005
|
10.4
|
||||||
|
10.6
|
Form of non-plan non-qualified stock option used after May 2005
|
SB-2
|
November 16, 2005
|
10.5
|
||||||
|
10.7
|
Consideration and new technology agreement dated April 1, 2005 with ZAO BAM
|
10-QSB
|
August 15, 2005
|
10.2
|
||||||
|
10.8
|
Form of common stock purchase warrant dated March 2006
|
8-K
|
March 3, 2006
|
99.3
|
||||||
|
10.9
|
2006 Stock Incentive Plan, as amended
|
8-K
|
May 23,2011
|
10.1
|
||||||
|
10.10
|
Form of Incentive Stock Option under Novelos Therapeutics, Inc.’s 2006 Stock Incentive Plan
|
8-K
|
December 15, 2006
|
10.1
|
||||||
|
10.11
|
Form of Non-Statutory Stock Option under Novelos Therapeutics, Inc.’s 2006 Stock Incentive Plan
|
8-K
|
December 15, 2006
|
10.2
|
||||||
|
10.12
|
Form of Non-Statutory Director Stock Option under Novelos Therapeutics, Inc.’s 2006 Stock Incentive Plan
|
8-K
|
December 15, 2006
|
10.3
|
||||||
II-5
|
10.13
|
Form of Common Stock Purchase Warrant dated May 2, 2007 issued pursuant to the Agreement to Exchange and Consent dated May 2, 2007
|
10-QSB
|
May 8, 2007
|
4.2
|
||||||
|
10.14
|
Collaboration Agreement dated February 11, 2009**
|
10-K
|
March 30, 2009
|
10.39
|
||||||
|
10.15
|
Common Stock Purchase Warrant dated February 11, 2009
|
8-K
|
February 18, 2009
|
4.2
|
||||||
|
10.16
|
Form of Warrant Exchange Agreement dated August 21, 2009
|
8-K
|
August 26, 2009
|
10.5
|
||||||
|
10.17
|
Securities Purchase Agreement dated August 25, 2009
|
S-1
|
September 15, 2009
|
10.41
|
||||||
|
10.18
|
Common Stock Purchase Warrant dated August 25, 2009
|
S-1
|
September 15, 2009
|
10.43
|
||||||
|
10.19
|
Letter Agreement with LP Clover Limited dated August 25, 2009
|
S-1
|
September 15, 2009
|
10.44
|
||||||
|
10.20
|
Letter Agreement with Mundipharma International Corporation Limited dated August 25, 2009
|
S-1
|
September 15, 2009
|
10.45
|
||||||
|
10.21
|
Consent and Amendment Agreement dated January 21, 2010
|
S-1/A
|
January 26, 2010
|
10.47
|
||||||
|
10.22
|
Form of Executive Retention Agreement dated May 14, 2010
|
10-Q
|
May 17, 2010
|
10.3
|
||||||
|
10.23
|
Form of Placement Agent Agreement Between the Company and Rodman and Renshaw LLC
|
S-1A
|
June 25, 2010
|
10.50
|
||||||
|
10.24
|
Written Consent and Waiver of Holders of Series C Convertible Preferred Stock and Series E Convertible Preferred Stock dated July 6, 2010
|
S-1A
|
July 7, 2010
|
10.52
|
||||||
|
10.25
|
Form of Common Stock Purchase Warrant to be issued pursuant to the Consent and Waiver of Holders of Series C Convertible Preferred Stock and Series E Convertible Preferred Stock dated July 6, 2010
|
S-1A
|
July 7, 2010
|
10.53
|
||||||
|
10.26
|
Form of Securities Purchase Agreement dated July 21, 2010
|
8-K
|
July 22, 2010
|
10.1
|
||||||
|
10.27
|
Amendment to Consent and Waiver of Holders of Series C Convertible Preferred Stock and Series E Convertible Preferred Stock dated July 21, 2010
|
8-K
|
July 22, 2010
|
10.2
|
||||||
|
10.28
|
Exchange Agreement dated November 30, 2010 between the Company and the holders of Series C Convertible Preferred Stock and Series E Convertible Preferred Stock
|
8-K
|
November 30, 2010
|
10.1
|
II-6
|
10.29
|
Form of Common Stock Purchase Warrant dated April 8, 2011
|
8-K
|
April 11, 2011
|
4.3
|
||||||
|
10.30
|
Securities Purchase Agreement dated April 8, 2011
|
8-K
|
April 11, 2011
|
10.1
|
||||||
|
10.31
|
Placement Agency Agreement dated April 1, 2011
|
8-K
|
April 11, 2011
|
99.1
|
||||||
|
10.32
|
License Agreement between Cellectar, LLC and the Regents of the University of Michigan dated September 14, 2003, as amended through June 2010
|
S-1
|
July 1, 2011
|
10.31
|
||||||
|
10.33
|
Lease Agreement between Cellectar, LLC and McAllen Properties LLC, as amended and extended to date
|
S-1
|
July 1, 2011
|
10.32
|
||||||
|
10.34
|
Loan Agreement between the Wisconsin Department of Commerce and Cellectar, Inc. dated September 15, 2010
|
S-1
|
July 1, 2011
|
10.33
|
||||||
|
10.35
|
General Business Security Agreement dated September 15, 2010
|
S-1
|
July 1, 2011
|
10.34
|
||||||
|
23.1
|
Consent of Foley Hoag LLP (included in Exhibit 5.1)*
|
|||||||||
|
23.2
|
Consent of Grant Thornton LLP
|
X
|
||||||||
|
23.3
|
Consent of Stowe & Degon LLC
|
X
|
||||||||
|
24.1
|
Powers of Attorney (included on signature page)
|
S-1
|
July 1, 2011
|
24.1
|
* To be filed by amendment
** Portions of this exhibit have been omitted pursuant to a confidential treatment order.
Item 17. Undertakings.
(a) The undersigned registrant hereby undertakes to:
(1) File, during any period in which it offers or sells securities, a post-effective amendment to this Registration Statement to:
(i) Include any prospectus required by Section 10(a)(3) of the Securities Act;
(ii) Reflect in the prospectus any facts or events which, individually or together, represent a fundamental change in the information in the Registration Statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than a 20 percent change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective Registration Statement.
(iii) Include any additional or changed material information on the plan of distribution.
(2) For determining liability under the Securities Act, treat each post-effective amendment as a new registration statement of the securities offered, and the offering of the securities at that time to be the initial bona fide offering.
(3) File a post-effective amendment to remove from registration any of the securities that remain unsold at the end of the offering.
II-7
(b) Insofar as indemnification for liabilities arising under the Securities Act of 1933 (the “Act”) may be permitted to directors, officers and controlling persons of the registrant pursuant to foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Act and is, therefore, unenforceable.
(c) Each prospectus filed pursuant to Rule 424(b)(Sec.230.424(b) of this chapter) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A (Sec.230.430A of this chapter), shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness. Provided that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use.
(d) For purposes of determining any liability under the Securities Act of 1933, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant pursuant to Rule 424(b) or under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective.
(e) For the purpose of determining any liability under the Securities Act of 1933, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
II-8
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, as amended, the registrant has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Newton, Commonwealth of Massachusetts, on September 14, 2011.
|
NOVELOS THERAPEUTICS, INC.
|
||
|
By:
|
/s/ Harry S. Palmin
|
|
|
Harry S. Palmin
|
||
|
President and Chief Executive Officer
|
||
SIGNATURES AND POWER OF ATTORNEY
Pursuant to the requirements of the Securities Act of 1933, as amended, this Registration Statement has been signed by the following persons in the capacities and on the dates indicated.
|
Signature
|
Title
|
Date
|
||
|
/s/ Harry S. Palmin
|
Chief Executive Officer and Director
|
September 14, 2011
|
||
|
Harry S. Palmin
|
( principal executive officer )
|
|||
|
/s/ Joanne M. Protano
|
Chief Financial Officer
|
September 14, 2011
|
||
|
Joanne M. Protano
|
( principal financial officer and principal accounting officer)
|
|||
|
/s/ *
|
Chairman of the Board of Directors
|
September 14, 2011
|
||
|
Stephen A. Hill
|
||||
|
/s/ *
|
Director
|
September 14, 2011
|
||
|
T. Rockwell Mackie
|
||||
|
/s/ *
|
Director
|
September 14, 2011
|
||
|
James S. Manuso
|
||||
|
/s/ *
|
Director
|
September 14, 2011
|
||
|
John Neis
|
||||
|
/s/ *
|
Director
|
September 14, 2011
|
||
|
John E. Niederhuber
|
||||
|
/s/ *
|
Director
|
September 14, 2011
|
||
|
Howard M. Schneider
|
||||
|
/s/ *
|
|
Director
|
|
September 14, 2011
|
|
Michael F. Tweedle
|
||||
|
/s/ *
|
Director
|
September 14, 2011
|
||
|
Jamey P. Weichert
|
*/s/ Harry S. Palmin as attorney-in-fact.
EXHIBIT INDEX
|
Incorporated by Reference
|
||||||||||
|
Exhibit
No. |
Description
|
Filed with
this Form S-1 |
Form
|
Filing Date
|
Exhibit
No. |
|||||
|
1.1
|
Form of Underwriting Agreement*
|
|||||||||
|
2.1
|
Agreement and Plan of Merger by and among Novelos Therapeutics, Inc., Cell Acquisition Corp. and Cellectar, Inc. dated April 8, 2011
|
8-K
|
April 11, 2011
|
2.1
|
||||||
|
3.1
|
Second Amended and Restated Certificate of Incorporation
|
8-K
|
April 11, 2011
|
3.1
|
||||||
|
3.2
|
Amended and Restated By-laws
|
8-K
|
June 1, 2011
|
3.1
|
||||||
|
3.3
|
Form of Certificate of Amendment to the Second Amended and Restated Certificate of Incorporation of Novelos Therapeutics, Inc.
|
S-1
|
July 1, 2011
|
3.3
|
||||||
|
5.1
|
Legal Opinion of Foley Hoag LLP*
|
|||||||||
| 4.1 | Form of Underwriter’s Warrant* | |||||||||
|
10.1
|
Employment Agreement with Harry S. Palmin dated January 31, 2006
|
8-K
|
February 6, 2006
|
99.1
|
||||||
|
10.2
|
Second Amendment to Employment Agreement between the Company and Harry Palmin
|
8-K
|
June 1, 2011
|
10.1
|
||||||
|
10.3
|
2000 Stock Option and Incentive Plan
|
SB-2
|
November 16, 2005
|
10.2
|
||||||
|
10.4
|
Form of 2004 non-plan non-qualified stock option
|
SB-2
|
November 16, 2005
|
10.3
|
||||||
|
10.5
|
Form of non-plan non-qualified stock option used from February to May 2005
|
SB-2
|
November 16, 2005
|
10.4
|
||||||
|
10.6
|
Form of non-plan non-qualified stock option used after May 2005
|
SB-2
|
November 16, 2005
|
10.5
|
||||||
|
10.7
|
Consideration and new technology agreement dated April 1, 2005 with ZAO BAM
|
10-QSB
|
August 15, 2005
|
10.2
|
||||||
|
10.8
|
Form of common stock purchase warrant dated March 2006
|
8-K
|
March 3, 2006
|
99.3
|
||||||
|
10.9
|
2006 Stock Incentive Plan, as amended
|
8-K
|
May 23,2011
|
10.1
|
||||||
|
10.10
|
Form of Incentive Stock Option under Novelos Therapeutics, Inc.’s 2006 Stock Incentive Plan
|
8-K
|
December 15, 2006
|
10.1
|
||||||
|
10.11
|
Form of Non-Statutory Stock Option under Novelos Therapeutics, Inc.’s 2006 Stock Incentive Plan
|
8-K
|
December 15, 2006
|
10.2
|
||||||
|
10.13
|
Form of Common Stock Purchase Warrant dated May 2, 2007 issued pursuant to the Agreement to Exchange and Consent dated May 2, 2007
|
10-QSB
|
May 8, 2007
|
4.2
|
||||||
|
10.14
|
Collaboration Agreement dated February 11, 2009**
|
10-K
|
March 30, 2009
|
10.39
|
||||||
|
10.15
|
Common Stock Purchase Warrant dated February 11, 2009
|
8-K
|
February 18, 2009
|
4.2
|
||||||
|
10.16
|
Form of Warrant Exchange Agreement dated August 21, 2009
|
8-K
|
August 26, 2009
|
10.5
|
||||||
|
10.17
|
Securities Purchase Agreement dated August 25, 2009
|
S-1
|
September 15, 2009
|
10.41
|
||||||
|
10.18
|
Common Stock Purchase Warrant dated August 25, 2009
|
S-1
|
September 15, 2009
|
10.43
|
||||||
|
10.19
|
Letter Agreement with LP Clover Limited dated August 25, 2009
|
S-1
|
September 15, 2009
|
10.44
|
||||||
|
10.20
|
Letter Agreement with Mundipharma International Corporation Limited dated August 25, 2009
|
S-1
|
September 15, 2009
|
10.45
|
||||||
|
10.21
|
Consent and Amendment Agreement dated January 21, 2010
|
S-1/A
|
January 26, 2010
|
10.47
|
||||||
|
10.22
|
Form of Executive Retention Agreement dated May 14, 2010
|
10-Q
|
May 17, 2010
|
10.3
|
||||||
|
10.23
|
Form of Placement Agent Agreement Between the Company and Rodman and Renshaw LLC
|
S-1A
|
June 25, 2010
|
10.50
|
||||||
|
10.24
|
Written Consent and Waiver of Holders of Series C Convertible Preferred Stock and Series E Convertible Preferred Stock dated July 6, 2010
|
S-1A
|
July 7, 2010
|
10.52
|
||||||
|
10.25
|
Form of Common Stock Purchase Warrant to be issued pursuant to the Consent and Waiver of Holders of Series C Convertible Preferred Stock and Series E Convertible Preferred Stock dated July 6, 2010
|
S-1A
|
July 7, 2010
|
10.53
|
||||||
|
10.26
|
Form of Securities Purchase Agreement dated July 21, 2010
|
8-K
|
July 22, 2010
|
10.1
|
||||||
|
10.27
|
Amendment to Consent and Waiver of Holders of Series C Convertible Preferred Stock and Series E Convertible Preferred Stock dated July 21, 2010
|
8-K
|
July 22, 2010
|
10.2
|
||||||
|
10.28
|
Exchange Agreement dated November 30, 2010 between the Company and the holders of Series C Convertible Preferred Stock and Series E Convertible Preferred Stock
|
8-K
|
November 30, 2010
|
10.1
|
|
10.29
|
Form of Common Stock Purchase Warrant dated April 8, 2011
|
8-K
|
April 11, 2011
|
4.3
|
||||||
|
10.30
|
Securities Purchase Agreement dated April 8, 2011
|
8-K
|
April 11, 2011
|
10.1
|
||||||
|
10.31
|
Placement Agency Agreement dated April 1, 2011
|
8-K
|
April 11, 2011
|
99.1
|
||||||
|
10.32
|
License Agreement between Cellectar, LLC and the Regents of the University of Michigan dated September 14, 2003, as amended through June 2010
|
S-1
|
July 1, 2011
|
10.31
|
||||||
|
10.33
|
Lease Agreement between Cellectar, LLC and McAllen Properties LLC, as amended and extended to date
|
S-1
|
July 1, 2011
|
10.32
|
||||||
|
10.34
|
Loan Agreement between the Wisconsin Department of Commerce and Cellectar, Inc. dated September 15, 2010
|
S-1
|
July 1, 2011
|
10.33
|
||||||
|
10.35
|
General Business Security Agreement dated September 15, 2010
|
S-1
|
July 1, 2011
|
10.34
|
||||||
|
23.1
|
Consent of Foley Hoag LLP (included in Exhibit 5.1)*
|
|||||||||
|
23.2
|
Consent of Grant Thornton LLP
|
X
|
||||||||
|
23.3
|
Consent of Stowe & Degon LLC
|
X
|
||||||||
|
24.1
|
Powers of Attorney (included on signature page)
|
S-1
|
July 1, 2011
|
24.1
|
* To be filed by amendment
** Portions of this exhibit have been omitted pursuant to a confidential treatment order.