Attached files
| file | filename |
|---|---|
| EX-23.1 - 22nd Century Group, Inc. | v229053_ex23-1.htm |
| EX-10.20 - 22nd Century Group, Inc. | v229053_ex10-20.htm |
As filed with the Securities and Exchange Commission on July 20, 2011
Registration No. 333-173420
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Amendment No. 3
to
Form S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
22nd CENTURY GROUP, INC.
(Exact name of registrant as specified in its charter)
|
Nevada
(State or other jurisdiction of
Incorporation or organization)
|
5194
(Primary Standard Industrial
Classification Code Number)
|
98-0468420
(I.R.S. Employer
Identification No.)
|
8201 Main Street, Suite 6
Williamsville, New York 14221
(716) 270-1523
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive offices)
Joseph Pandolfino
Chief Executive Officer
22nd Century Group, Inc.
8201 Main Street, Suite 6
Williamsville, New York 14221
(716) 270-1523
(Address, including zip code, and telephone number, including area code, of agent for service)
Copies to:
Patrick G. Quick, Esq.
Thomas L. James, Esq.
Teri L. Champ, Esq.
Foley & Lardner LLP
777 East Wisconsin Avenue
Milwaukee, Wisconsin 53202-5306
(414) 271-2400
Approximate date of commencement of proposed sale to the public: As soon as practicable after the Registration Statement becomes effective.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, check the following box. x
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer ¨ Accelerated filer ¨ Non-accelerated filer ¨ Smaller reporting company x
CALCULATION OF REGISTRATION FEE
|
Title of each class of
securities to be registered
|
Amount to be
registered (1)
|
Proposed
maximum
offering price
per share (2)
|
Proposed
maximum
aggregate
offering
price(2)
|
Amount of
registration fee
(3)
|
||||||||||||
|
Common stock, par value $0.00001 per share
|
5,434,446 | $ | 1.25 | $ | 6,793,057.50 | $ | 788.68 | |||||||||
|
(1)
|
Pursuant to Rule 416 under the Securities Act of 1933, as amended, the number of shares of common stock registered hereby is subject to adjustment to prevent dilution resulting from stock splits, stock dividends or similar transactions.
|
|
(2)
|
Estimated solely for the purpose of determining the amount of the registration fee, based on the average of the high and low sale prices of the common stock as reported by the OTC Bulletin Board on April 6, 2011 in accordance with Rule 457(c) under the Securities Act of 1933.
|
|
(3)
|
The registration fee was previously paid on April 8, 2011.
|
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933, as amended, or until the Registration Statement shall become effective on such date as the Securities and Exchange Commission, acting pursuant to said Section 8(a), may determine.
|
The information in this prospectus is not complete and may be changed. The selling stockholders may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This prospectus is not an offer to sell these securities and the selling stockholders are not soliciting an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
|
SUBJECT TO COMPLETION, DATED JULY 20, 2011
PROSPECTUS
22nd CENTURY GROUP, INC.
5,434,446 Shares of Common Stock
This prospectus relates to the offering by the selling stockholders of 22nd Century Group, Inc. of up to 5,434,446 shares of common stock, par value $0.00001 per share. These shares were privately issued to the selling stockholders in connection with a private placement and merger transaction. We will not receive any proceeds from the sale of common stock by the selling stockholders.
The selling stockholders have advised us that they will sell the shares of common stock from time to time in broker’s transactions, in the open market, on the OTC Bulletin Board, in privately negotiated transactions or a combination of these methods, at market prices prevailing at the time of sale, at prices related to the prevailing market prices or at negotiated prices. We will pay the expenses incurred to register the shares for resale, but the selling stockholders will pay any underwriting discounts, commissions or agent’s commissions related to the sale of their shares of common stock.
Our common stock is traded on the OTC Bulletin Board under the symbol “XXII.OB”. On July 15, 2011, the closing sale price of our common stock was $1.25 per share.
Investing in our common stock involves risks. Before making any investment in our securities, you should read and carefully consider risks described in the “Risk Factors” section beginning on page 9 of this prospectus.
You should rely only on the information contained in this prospectus or any prospectus supplement or amendment thereto. We have not authorized anyone to provide you with different information. This prospectus may only be used where it is legal to sell these securities. The information in this prospectus is only accurate on the date of this prospectus, regardless of the time of any sale of securities.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
This date of this prospectus is July___, 2011
You should rely only on the information contained in this prospectus. We have not authorized any other person to provide you with information that is different from that contained in this prospectus. If anyone provides you with different or inconsistent information, you should not rely on it. The selling stockholders are offering to sell and seeking offers to buy these securities only in jurisdictions where offers and sales are permitted. You should assume that the information contained in this prospectus is accurate only as of the date of this prospectus, regardless of the time of delivery of this prospectus or of any sale of common stock. Our business, financial condition, results of operations and prospects may have changed since that date.
TABLE OF CONTENTS
|
Page
|
||
|
PROSPECTUS SUMMARY
|
1
|
|
|
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
|
8
|
|
|
RISK FACTORS
|
9
|
|
|
PRINCIPAL AND SELLING STOCKHOLDERS
|
28
|
|
|
USE OF PROCEEDS
|
32
|
|
|
DIVIDEND POLICY
|
32
|
|
|
DETERMINATION OF OFFERING PRICE
|
32
|
|
|
MARKET FOR COMMON EQUITY AND RELATED STOCKHOLDER MATTERS
|
32
|
|
|
SECURITIES AUTHORIZED FOR ISSUANCE UNDER EQUITY COMPENSATION PLANS
|
33
|
|
|
BUSINESS
|
35
|
|
|
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
|
57
|
|
|
DIRECTORS AND EXECUTIVE OFFICERS
|
64
|
|
|
EXECUTIVE COMPENSATION
|
67
|
|
|
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS
|
71
|
|
|
PLAN OF DISTRIBUTION
|
73
|
|
|
DESCRIPTION OF SECURITIES
|
75
|
|
|
LEGAL MATTERS
|
79
|
|
|
EXPERTS
|
79
|
|
|
WHERE YOU CAN FIND MORE INFORMATION
|
79
|
|
|
CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
|
80
|
|
|
INDEX TO FINANCIAL STATEMENTS
|
F-1
|
|
|
EXHIBIT INDEX
|
E-1
|
i
PROSPECTUS SUMMARY
This summary highlights information contained elsewhere in this prospectus. This summary is not complete and does not contain all the information that should be considered before investing in our common stock. Investors should read the entire prospectus carefully, including the more detailed information contained herein under the “Risk Factors” and “Cautionary Note Regarding Forward-Looking Statements” sections and our consolidated financial statements and the notes to those financial statements.
As used in this prospectus, unless the context otherwise requires, the “Company,” “we,” “us” and “our” refer to 22nd Century Group, Inc., a Nevada corporation, as well as its subsidiaries, 22nd Century Limited, LLC, a Delaware limited liability company, and Goodrich Tobacco Company, LLC, a Delaware limited liability company, taken as a whole, and also refer to the operations of 22nd Century Limited, LLC prior to the merger on January 25, 2011, as discussed below, which resulted in 22nd Century Limited, LLC becoming our wholly-owned subsidiary. Hereinafter, 22nd Century Limited, LLC is sometimes referred to as “22nd Century.”
Our Company
Overview
We are a plant biotechnology company and a global leader in modifying the content of nicotinic alkaloids in tobacco plants through genetic engineering and plant breeding. We control 90 issued patents, of which we own 8 issued patents and we have the exclusive license to an additional 90 issued patents in an aggregate of 79 countries where at least 75% of the world’s smokers reside. Additionally, we control 45 pending patent applications, of which we own 29 and have an exclusive license to 16. In the U.S., patents that currently cover our products expire in 2019 through 2022. Also in the U.S., patent applications that we expect will cover our products would, if patents are granted in connection with such applications, expire in 2024 through 2028. However, there is no guarantee that patents will be granted in response to such patent applications and therefore cover our products. Our two worldwide exclusive licenses, one from North Carolina State University (“NCSU”) and the other from National Research Council of Canada, Plant Biotechnology Institute in Saskatoon, Canada (“NRC”), each involve multiple patent families, patent applications and patents. The exclusive rights under each of these two license agreements expire on the date on which the last patent covered by the subject license expires in the country or countries where such patents are in effect, which is expected to be from 2022 through 2028, respectively. We believe our proprietary technology provides us with opportunities in the global market for approved smoking cessation aids and the emerging market for modified risk tobacco products, which is a term that we explain below; however, we have not yet identified any specific countries or developed any timelines or cost estimates for international expansion of our business.
Our January 25, 2011 Private Placement Offering (as defined under the “Recent Developments” section on page 5) of 5,434,446 Units (as defined under the “Recent Developments” section), which we collectively refer to in this prospectus as “PPO Securities,” resulted in approximately $3.4 million in net cash proceeds and a reduction of debt obligations to shareholders that were on the balance sheet at December 31, 2010 of approximately $614,000, which were exchanged for equity interests in the Private Placement Offering. This offering involves the resale of common stock, and we have registered that resale to satisfy registration rights related to the PPO Securities. We will not receive any proceeds from the sale of common stock under this prospectus.
We plan to use a material portion of the proceeds from the Private Placement Offering to complete a Phase II-B clinical trial which is necessary to seek approval from the U.S. Food and Drug Administration (“FDA”) for X-22, our prescription smoking cessation aid in development. We believe the cost of completing the Phase II-B trial is approximately $1.1 million, of which we have paid approximately one-third as of March 31, 2011. We have designated our upcoming phase II clinical trial as a “Phase II-B” clinical trial because (i) we are looking for continued indications of clinical efficacy and safety in larger groups of patients in our clinical trial, and (ii) very low nicotine (VLN) cigarettes made from the Company’s VLN tobacco have already demonstrated efficacy in previous Phase II trials, specifically the Phase II trial at the University of Minnesota which had a very similar protocol compared to our upcoming phase II clinical trial. However, “Phase II-B” is not an official FDA designation.
1
There is no guarantee that X-22 will obtain the necessary approvals from the FDA to be marketed as a smoking cessation aid. We estimate the cost of completing the Phase III trials is approximately $10 million. We currently plan to seek to raise approximately $15 million in the fourth quarter of 2011 through the sale of our common stock and/or securities convertible into common stock to fund Phase III trials, modified risk exposure studies and our general working capital requirements. At this time, we have not yet attempted to obtain any approval for X-22 outside of the U.S., and there is no guarantee that we will be able to obtain any approval for X-22 outside of the U.S. (for those countries that require such regulatory approvals). We are currently in default under the terms of our exclusive worldwide license agreement with North Carolina State University (“NCSU”) for failure to timely pay certain amounts owed under the license agreement. We have requested that NCSU defer payment of these amounts. Should NCSU choose instead to invoke its right to terminate this license agreement, we would have 60 days notice to cure the payment default, and if we did not cure the payment default within such 60-day period and the license terminated, our business would be materially and adversely affected. If NCSU does not agree to a deferment of the unpaid balance owing under the license agreement and we pay such balance, we may not have sufficient funds to pay in full the cost of our Phase II-B clinical trial for X-22 (as described in more detail throughout this prospectus).
Our net sales for the three month period ended March 31, 2011 were $117,456 mainly from our research cigarette program. For the year ended December 31, 2010, we had total sales of $49,784 from our research cigarette program. Our net loss for the three month period ended March 31, 2011 was approximately $652,000, or $0.03 per diluted common share. For the year ended December 31, 2010, our net loss was $1,423,647, or $0.11 per diluted common share. Total assets were $3,410,431 at March 31, 2011, compared to $2,800,520 at December 31, 2010. Total current liabilities were $1,458,997 at March 31, 2011, compared to $4,823,635 at December 31, 2010. Total liabilities, including long term liabilities, were $5,197,747 at March 31, 2011, including our warrant derivative liability of $3,061,750, compared to $4,889,192 at December 31, 2010, which did not include any warrant derivative liability. As of March 31, 2011, we had working capital of approximately $0.5 million compared to negative working capital of approximately $4.1 million at December 31, 2010. The improvement in our working capital position of $4.6 million was mainly a result of proceeds from the January 25, 2011 Private Placement Offering and the transfer from accounts payable of $587,000 to notes payable as part of payment arrangements with two vendors for past due amounts we owed, offset by our net loss for the three months ended March 31, 2011. As reported in our Annual Report on Form 8-K/A as filed with the SEC on March 23, 2011, our independent registered public accounting firm expressed substantial doubt as to whether we can continue as a going concern. Even in light of our receipt of the proceeds of the Private Placement Offering, we cannot guarantee our ability to continue as a going concern.
We have met with the FDA regarding the remaining clinical trials for X-22 and based on the FDA’s guidance, we plan to conduct a Phase II-B trial and two larger and concurrent Phase III trials with the same protocols. X-22 will be a prescription-only kit containing very low nicotine (“VLN”) cigarettes made from our proprietary tobacco, which has approximately 95% less nicotine compared to tobacco in existing “light” cigarettes. The therapy protocol allows the patient to smoke our VLN cigarettes without restriction over the six-week treatment period to facilitate the goal of the patient quitting smoking by the end of the treatment period. Although further research and clinical trials are needed to determine efficacy of X-22, we believe this therapy protocol has thus far been successful because VLN cigarettes made from our proprietary tobacco satisfy smokers’ cravings for cigarettes while (i) greatly reducing nicotine exposure and nicotine dependence and (ii) extinguishing the association between the act of smoking and the rapid delivery of nicotine. We believe X-22 will be more attractive to smokers than other therapies since it smokes and tastes like a typical cigarette, involves the same smoking behavior, and does not expose the smoker to any new drugs or new side effects (other than the exposure and side effects from smoking cigarettes in general) that may occur when a person is exposed to a new drug. There is no guarantee that X-22 will obtain the necessary approvals from the FDA to be marketed as a smoking cessation aid.
Independent studies, including two Phase II clinical trials, have demonstrated that VLN cigarettes made from our proprietary VLN tobacco are at least as effective as FDA-approved smoking cessation aids. As we describe further in the Technology Platform and Intellectual Property section, X-22 is a cigarette that contains our proprietary VLN tobacco. We believe X-22 will be well-positioned as a smoking cessation product by motivating and encouraging more smokers to attempt to quit smoking, although there is no assurance that these results will occur.
The 2009 Family Smoking Prevention and Tobacco Control Act (“Tobacco Control Act”) granted the FDA authority over the regulation of all tobacco products. While it prohibits the FDA from banning cigarettes outright, it allows the FDA to require the reduction of nicotine or any other compound in tobacco and cigarette smoke. The Tobacco Control Act also banned all sales in the U.S. of cigarettes with flavored tobacco (other than menthol). As of June 2010, all cigarette companies were required to cease the use of the terms “low tar,” “light” and “ultra light” in describing cigarettes sold in the U.S. Besides numerous other regulations, including certain marketing restrictions, for the first time in history, a U.S. regulatory agency will scientifically evaluate cigarettes that may pose lower health risks as compared to conventional cigarettes.
The Tobacco Control Act establishes procedures for the FDA to regulate the labeling and marketing of modified risk tobacco products, which includes cigarettes that (i) reduce exposure to tobacco smoke toxins and/or (ii) pose lower health risks, as compared to conventional cigarettes (“Modified Risk Cigarettes”). The Tobacco Control Act requires the FDA to issue specific regulations and guidance regarding applications that must be submitted to the FDA for the authorization to label and market Modified Risk Cigarettes. The FDA has yet to release its regulations regarding modified risk tobacco products. However, based in part on the timelines contained in the Tobacco Control Act, we expect the FDA to issue such regulations and guidance within the next year.
2
Although we believe that two of our cigarette products, which we refer to as BRAND A and BRAND B, will qualify as Modified Risk Cigarettes, further research and exposure studies are needed for the FDA to make this determination. Compared to commercial cigarettes, we believe the tobacco in BRAND A has approximately 95% less nicotine than tobacco in cigarettes previously marketed as “light” cigarettes, and BRAND B’s smoke contains the lowest amount of “tar” per milligram of nicotine. There is no guarantee that X-22 will obtain the necessary approvals from the FDA to be marketed as a smoking cessation aid, and there is no guarantee that Brand A or Brand B will obtain the necessary authorizations from the FDA to be classified as Modified Risk Cigarettes. We have not attempted to obtain approval of X-22 or authorization of our Modified Risk Cigarettes outside of the United States, and there is no guarantee that we will be able to gain such approval or such authorization for these products outside of the United States (for those countries that require such regulatory authorizations).
The net proceeds of the Private Placement Offering will not be sufficient to enable us to complete the FDA approval process for X-22 and/or the FDA authorization process for our Modified Risk Cigarettes. We will require additional capital beyond the net proceeds of the Private Placement Offering to complete these activities. We may not be able to obtain additional debt or equity financing on favorable terms, if at all. If we raise additional funds through the issuance of equity securities, our stockholders may experience substantial dilution, or the equity securities may have rights, preferences or privileges senior to those of existing stockholders. If we raise additional funds through debt financings, these financings may involve significant cash payment obligations and covenants that restrict our ability to operate our business and make distributions to our stockholders. We also could elect to seek funds through arrangements with collaborators. To the extent that we raise additional funds through collaboration and licensing arrangements, it may be necessary to relinquish some rights to our technologies or our potential products or grant licenses on terms that are not favorable to us. We currently plan to seek to raise approximately $15 million in the fourth quarter of 2011 through the sale of our common stock and/or securities convertible into common stock to fund Phase III trials, modified risk exposure studies and our general working capital requirements.
Within our two product categories, the Tobacco Control Act offers us the following specific advantages:
Smoking Cessation Aids
FDA approval must be obtained, as has been the case for decades, before a product can be marketed for quitting smoking. The Tobacco Control Act provides that products for quitting smoking or smoking cessation be considered for “Fast Track” designation by the FDA. The “Fast Track” programs of the FDA are intended to facilitate development and expedite review of drugs to treat serious and life-threatening conditions so that an approved product can reach the market expeditiously. We believe that X-22 will qualify for “Fast Track” designation by the FDA. However, there is no guarantee that X-22 will qualify for “Fast Track” designation at the FDA.
Modified Risk Cigarettes
We intend to seek FDA authorization to market BRAND A and BRAND B as Modified Risk Cigarettes. We believe that BRAND A and BRAND B will achieve significant market share in the global cigarette market among smokers who will not quit but are interested in reducing the harmful effects of smoking. We believe this new regulatory environment represents a paradigm shift for the tobacco industry. The Tobacco Control Act allows the FDA to mandate the use of reduced-risk technologies across all conventional tobacco products or cigarettes. We expect this to create opportunities for us to license our proprietary technology and/or tobaccos to larger competitors. There is no guarantee that Brand A or Brand B will obtain the necessary authorizations from the FDA to be classified as Modified Risk Cigarettes. Further research and exposure studies are needed for the FDA to make this determination
3
RED SUN and MAGIC Cigarettes
Our subsidiary, Goodrich Tobacco Company, LLC (f/k/a Xodus, LLC), has introduced two super-premium priced cigarette brands, RED SUN and MAGIC, into the U.S. market in the first quarter of 2011. Super-premium priced means brands that are priced higher than national premium brands such as Marlboro® Newport® and Camel®. Both brands are available in regular and menthol and all four brand styles are king size, packaged in hinge-lid hard packs. We intend to focus our marketing efforts on tobacconists, smoke shops and tobacco outlets. We believe that the ban in 2009 by the FDA of all flavored cigarettes (with the exception of menthol) has resulted in a product void in these tobacco channels. According to Tobacco Merchants Association, flavored cigarettes (besides menthol), mainly clove cigarettes, were approximately 1% of the U.S. cigarette market (3.25 billion cigarettes equating to $0.7 billion) before the ban on such cigarettes in June 2009, when the Tobacco Control Act became law. Since these were super-premium priced products (priced up to 60% higher than the best selling national brands), they were mainly distributed in specialty tobacco channels such as tobacconists, smoke shops and tobacco outlets, which collectively are about 10,000 stores in the U.S., a small fraction of the hundreds of thousands of stores that sell cigarettes in the U.S.
Certain wholesalers and retailers are now seeking other specialty cigarettes to replace the banned flavored cigarettes. We believe that certain U.S. cigarette wholesalers and retailers will, among other reasons, purchase RED SUN and MAGIC to replace their lost sales of flavored cigarettes as well as potential lost sales of “light” and “ultra light” cigarettes, since as of June 2010, all cigarette companies were required to cease the use of the terms “low tar,” “light” and “ultra light.”
Government Research Cigarettes
The National Institute on Drug Abuse (“NIDA”), a component of the National Institutes of Health (“NIH”), provides the scientific community with controlled and uncontrolled research chemicals and drug compounds in its Drug Supply Program. In 2009, NIDA included an option to develop and produce research cigarettes with ten different levels of nicotine, including a minimal (placebo) level, or Research Cigarette Option, in its request for proposals for a five-year contract for Preparation and Distribution of Research and Drug Products. We have agreed, as a subcontractor to RTI International (“RTI”) pursuant to RTI’s contract with NIDA for the Research Cigarette Option, to supply modified nicotine cigarettes to NIDA. In August 2010, we met with officials from NIDA, FDA, RTI, the National Cancer Institute and the Centers for Disease Control and Prevention to finalize certain aspects of the design of these research cigarettes. These research cigarettes are distributed under the mark SPECTRUM.
In 2010, we received our first purchase order of $152,660 which included a design phase fee of $40,604. In March 2011, the Company received a revised purchase order and authorization to bill $112,000 for manufacture planning and material procurement for 9 million cigarettes from RTI. Payment was received on May 2, 2011. The Company received an additional purchase order from RTI for approximately $680,000 in May 2011 to ship the 9 million research cigarettes. Approximately 4 million cigarettes out of the 9 million were shipped to and received by RTI on July 6, 2011. We believe that the revenue from the sale of SPECTRUM, plus direct orders of other cigarettes from researchers, will be approximately $825,000 in 2011 and $3 million over the next 5 years. As a subcontractor, we do not have a formal contractual arrangement at this time with RTI to purchase additional SPECTRUM cigarettes or with other researchers, and there is no guarantee that we will receive future orders from RTI or other researchers.
Technology Platform and Intellectual Property
Our proprietary technology enables us to decrease or increase the level of nicotine in tobacco plants by decreasing or increasing the expression of gene(s) responsible for nicotine production in the tobacco plant using genetic engineering. For example, we believe that one of our proprietary tobacco varieties contains the lowest nicotine content of any tobacco ever commercialized, with approximately 95% less nicotine than tobacco in leading “light” cigarette brands. We believe that, as opposed to extracting nicotine from tobacco, this proprietary tobacco grows with virtually no nicotine without adversely affecting the other leaf constituents important to a cigarette’s characteristics, including taste and aroma. Our genetic engineering processes suppress an enzyme involved in synthesis of a nicotine precursor in the roots of the tobacco plant. Roots of the tobacco plant produce nicotine which is transported to the rest of the plant. We believe levels of certain compounds in the tobacco leaf of our genetically engineered variety are not substantially different than that of conventional tobacco, except for the large reduction in nicotine and related compounds. We believe, based on analyses of a range of chemical constituents in smoke from cigarettes made from VLN tobacco, that the smoke is highly similar to smoke from conventional cigarettes, except for the large reduction in nicotine and related compounds. Furthermore, subjective ratings of our products’ smoking characteristics approximate those of conventional cigarettes. Therefore, we believe the taste and aroma characteristics are within the range of typical cigarettes.
4
Our proprietary technology is covered by 12 patent families consisting of 98 issued patents in 79 countries, 8 of which we own and 90 of which are exclusively licensed to us, and approximately 45 pending patent applications, of which we own 29 pending patent applications and have an exclusive license to 16 pending patent applications. A “patent family” is a set of patents granted in various countries to protect a single invention. Our patent coverage in the United States, which we believe is the most valuable smoking cessation market and cigarette market, consists of 14 issued patents and 7 pending applications. In the U.S., patents that currently cover our products expire in 2019 through 2022. Also in the U.S., patent applications that we expect will cover our products would, if patents are granted in connection with such applications, yield patents that expire in 2024 through 2028. However, there is no guarantee that patents will be granted in response to such patent applications and therefore cover our products. Our two worldwide exclusive licenses, one from North Carolina State University (“NCSU”) and the other from National Research Council of Canada, Plant Biotechnology Institute in Saskatoon, Canada (“NRC”), each involve multiple patent families, patent applications and patents. The exclusive rights under each of these two license agreements expire on the date on which the last patent covered by the subject license expires in the country or countries where such patents are in effect, which is expected to be from 2022 through 2028, respectively. In China, which we consider the world’s largest cigarette market, we own or have the exclusive license to use 5 issued patents and 3 pending patent applications. We also have the exclusive right to license and sublicense these patent rights. The patents owned by or exclusively licensed to us are issued in countries where we believe approximately 75% of the world’s smokers reside. As a result of these patents, patent applications and licenses, we believe we have exclusive worldwide rights to all uses of the following genes responsible for nicotine content in tobacco plants: QPT, A622, NBB1, MPO and genes for several transcription factors. Similarly, we believe we have exclusive rights to plants with altered nicotine content produced from modifying expression of these genes and tobacco products produced from these plants.
We own various registered trademarks in the United States. We also have exclusive rights to plant variety protection, or PVP, certificates in the United States (issued by the U.S. Department of Agriculture) and Canada. A PVP certificate prevents anyone other than the owner/licensee from planting a plant variety for 20 years in the U.S. or 18 years in Canada. The protections of PVP are independent of, and in addition to, patent protection.
While we have performed searches for third-party intellectual property rights that may affect our ability to commercialize our potential products, we have not performed searches for those third-party intellectual property rights that may raise freedom-to-operate issues, nor have we obtained legal opinions regarding the commercialization of our potential products. Therefore, there may be patents of which we are unaware that may affect our ability to commercialize our potential products.
Recent Developments
On January 25, 2011, we entered into an Agreement and Plan of Merger and Reorganization with 22nd Century Acquisition Subsidiary, our wholly-owned Delaware limited liability company subsidiary, or Acquisition Sub, and 22nd Century. On that date, Acquisition Sub merged with and into 22nd Century, and 22nd Century, as the surviving entity, became our wholly-owned subsidiary. In this prospectus, we refer to the merger and reorganization transactions consummated on January 25, 2011 as the “Merger.”
Prior to the consummation of the Merger, 22nd Century completed a private placement of units of its securities, or “Units”, with each Unit consisting of one limited liability company membership interest of 22nd Century and a five-year warrant to purchase one half of one (1/2) limited liability company membership interest of 22nd Century at an exercise price of $1.50 per whole limited liability company membership interest of 22nd Century. We refer to the offering in this prospectus as the “Private Placement Offering.” 22nd Century also issued warrants to purchase its limited liability company membership interests to a financial advisor for financial advisory services rendered in connection with the Private Placement Offering.
Prior to the closing of the Merger, we transferred all of our pre-Merger operating assets and liabilities pursuant to the terms of a split-off agreement (the “Split-Off Agreement”), to our wholly-owned subsidiary, Touchstone Split Corp., a Delaware corporation (the “Split-Off Subsidiary”). Thereafter, pursuant to the Split-Off Agreement, we transferred all of the outstanding capital stock of the Split-Off Subsidiary to our then-sole director in exchange for $1.00, such consideration being deemed to be adequate by our pre-Merger Board of Directors (the “Board”).
5
At the closing of the Merger, each limited liability company membership interest of 22nd Century issued and outstanding immediately prior to the closing of the Merger was exchanged for one share of our common stock, and each warrant to purchase limited liability company membership interests of 22nd Century was exchanged for one warrant of like tenor and term to purchase shares of our common stock. An aggregate of 21,434,446 shares of common stock and warrants to purchase an aggregate of 8,151,980 shares of common stock were issued to the holders of Units of 22nd Century, and immediately following the closing of the Merger an aggregate of 26,759,646 shares of common stock were issued and outstanding and an aggregate of 8,651,980 shares are common stock were reserved for issuance pursuant to the exercise of warrants to purchase shares of common stock.
In connection with the Merger, our Board was expanded to five members. The sole officer and sole member of the Board prior to the closing of the Merger, David Rector, resigned as an officer and a director after the closing of the Merger. The current members of our Board are Joseph Pandolfino, Henry Sicignano III, Joseph Alexander Dunn, Ph.D., James W. Cornell and Steven Katz. Messrs. Cornell and Katz and Dr. Dunn qualify as “independent” directors under the applicable definition of the NASDAQ Global Market (“NASDAQ”) listing standards, so that a majority of the Company’s Board members are “independent.” Although the Company’s securities are not currently traded on the NASDAQ or any other exchange, which would require that the Company’s Board include a majority of directors that are “independent,” the Company has elected to do so anyhow as part of its corporate governance policies. Our current executive officers are Joseph Pandolfino, Chief Executive Officer, Henry Sicignano III, President and Secretary, Michael R. Moynihan, Ph.D., Vice President of Research & Development, and C. Anthony Rider, Chief Financial Officer and Treasurer. Each of Messrs. Pandolfino, Sicignano and Rider was an executive officer of 22nd Century prior to the closing of the Merger.
Following the closing of the Merger, there were 26,759,646 shares of Common Stock issued and outstanding. Approximately 59.8% of such issued and outstanding shares were held by individuals and entities that were holders of limited liability company membership interests of 22nd Century prior to consummation of the Private Placement Offering, approximately 20.3% were held by the investors in the Private Placement Offering and approximately 19.9% were held by the pre-Merger stockholders of Parent.
On April 1, 2011, under our 2010 equity incentive plan (“EIP”), the Board granted an aggregate of 1,150,000 shares of our common stock to our officers and directors and options to purchase an aggregate of 35,000 shares of our common stock to our employees.
The Merger is being accounted for as a reverse acquisition and recapitalization of 22nd Century for financial accounting purposes whereby 22nd Century is deemed to be the acquirer for accounting and financial reporting purposes. Consequently, the assets and liabilities and the historical operations that will be reflected in the financial statements prior to the Merger will be those of 22nd Century and will be recorded at the historical cost basis of 22nd Century, and the consolidated financial statements after completion of the Merger will include the assets and liabilities of the Company and 22nd Century, historical operations of 22nd Century and operations of the Company beginning on the closing date of the Merger. As a result, all the historical financial information reported in this prospectus is the financial information of 22nd Century.
We have entered into an agreement with a federally licensed cigarette manufacture to produce RED SUN, MAGIC, SPECTRUM, BRAND A, BRAND B and the clinical trial cigarettes for X-22.
We were incorporated under the laws of the State of Nevada on September 12, 2005 under the name Touchstone Mining Limited. We changed our name to 22nd Century Group, Inc. on November 23, 2010 in anticipation of the Merger with 22nd Century. Our principal executive offices are located at 8201 Main Street, Suite 6, Williamsville, New York 14221. The telephone number at our principal executive offices is (716) 270-1523. Our website address is www.xxiicentury.com. Information contained on our website is not deemed part of this prospectus.
6
|
Common stock currently outstanding
|
27,909,646 shares (1) (2)
|
|
Common stock offered by us
|
None
|
|
Common stock offered by the selling stockholders
|
5,434,446 shares
|
|
Use of Proceeds
|
We will not receive any proceeds from the sale of common stock offered by this prospectus.
|
|
Risk Factors
|
See “Risk Factors” and other information included in this prospectus for a discussion of factors that you should consider before deciding to invest in shares of our common stock.
|
|
OTC Bulletin Board Symbol
|
XXII.OB
|
|
(1)
|
As of July 15, 2011
|
|
(2)
|
Assumes that all other outstanding warrants and options are not exercised
|
7
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus contains forward-looking statements. This prospectus includes statements regarding our plans, goals, strategies, intentions, beliefs or current expectations. These statements are expressed in good faith and based upon a reasonable basis when made, but there can be no assurance that these expectations will be achieved or accomplished. These forward looking statements can be identified by the use of terms and phrases such as “believe,” “plan,” “intend,” “anticipate,” “target,” “estimate,” “expect,” and the like, and/or future-tense or conditional constructions “may,” “could,” “should,” etc. Items contemplating or making assumptions about, actual or potential future sales, market size, collaborations, and trends or operating results also constitute forward-looking statements.
These forward-looking statements are only predictions, are uncertain and involve substantial known and unknown risks, uncertainties and other factors which may cause our (or our industry’s) actual results, levels of activity or performance to be materially different from any future results, levels of activity or performance expressed or implied by these forward-looking statements. The “Risk Factors” section of this prospectus sets forth detailed risks, uncertainties and cautionary statements regarding our business and these forward-looking statements.
Since our common stock is considered a “penny stock,” we are ineligible to rely on the safe harbor for forward-looking statements provided in Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act.
We cannot guarantee future results, levels of activity or performance. You should not place undue reliance on these forward-looking statements, which speak only as of the date that they were made. These cautionary statements should be considered with any written or oral forward-looking statements that we may issue in the future. Except as required by applicable law, including the securities laws of the United States, we do not intend to update any of the forward-looking statements to conform these statements to reflect actual results, later events or circumstances or to reflect the occurrence of unanticipated events. You should carefully review and consider the various disclosures made by us in our reports filed with the Securities and Exchange Commission which attempt to advise interested parties of the risks and factors that may affect our business, financial condition, results of operation and cash flows. If one or more of these risks or uncertainties materialize, or if the underlying assumptions prove incorrect, our actual results may vary materially from those expected or projected.
8
RISK FACTORS
An investment in shares of our common stock is highly speculative and involves a high degree of risk. We face a variety of risks that may affect our operations or financial results and many of those risks are driven by factors that we cannot control or predict. The following discussion addresses those risks that management believes could materially adversely affect our business, financial condition and results of operation. Only those investors who can bear the risk of loss of their entire investment should participate in this offering. Prospective investors should carefully consider the following risk factors in evaluating an investment in our common stock.
Risks Related to Our Business and Operations
We may not be able to continue as a going concern.
Recurring losses from operations, our limited working capital of approximately $503,000 as of March 31, 2011 (approximately $4.1 million at December 31, 2010), shareholders’ deficit of $1.8 million as of March 31, 2011 ($2.1 million at December 31, 2010) and the uncertainty of obtaining additional financing on a timely basis, raise doubt about our ability to continue as a going concern. The report of our independent registered public accounting firm on our financial statements for the year ended December 31, 2010, includes an emphasis of a matter paragraph expressing substantial doubt whether we can continue as a going concern. Even in light of our receipt of the proceeds of the Private Placement Offering, we cannot guarantee our ability to continue as a going concern.
We have had a history of losses, and we may be unable to achieve or sustain profitability.
We experienced net losses of approximately $652,000 for the first three months of 2011 and $1.4 million and $1.2 million during the years ended December 31, 2010 and 2009, respectively. We expect to continue to incur net losses and negative operating cash flows in the foreseeable future and cannot be certain that we will ever achieve profitability. Since 2007, we have received only limited licensing revenue from a former licensee and have achieved limited revenue of product sales. We will need to spend significant capital to fulfill planned operating goals and conduct clinical studies, achieve regulatory approvals and, subject to such approvals, successfully produce products for commercialization. In addition, as a public company, we will incur significant legal, accounting and other expenses that we did not incur as a private company.
We have a history of negative cash flow, and our ability to generate positive cash flow is uncertain.
We had negative cash flow before financing activities of approximately $2,177,000 during the first three months of 2011 and $1,018,000 and $172,000 during the years ended December 31, 2010 and 2009, respectively. We anticipate that we will continue to have negative cash flow for the foreseeable future as we will continue to incur increased expenses from seeking regulatory approvals, including clinical trials and exposure studies, sales and marketing, and general and administrative expenses, as well as to purchase inventory. Our business will also require significant amounts of working capital to support our growth. Therefore, we may need to raise additional investment capital to achieve growth, and we may not achieve sufficient revenue growth to generate positive future cash flow. An inability to generate positive cash flow for the foreseeable future or raise additional capital on reasonable terms may decrease our long-term viability.
Our limited operating history makes it difficult to evaluate our current business and future prospects.
We have been in existence since 1998, but our activities have been limited primarily to licensing and funding research and development activities. Our limited operating history may make it difficult to evaluate our current business and our future prospects. We have encountered and will continue to encounter risks and difficulties frequently experienced by growing companies in rapidly changing industries, including increasing expenses as we continue to grow our business. If we do not manage these risks successfully, our business will be harmed.
9
We have no experience in managing growth. If we fail to manage our growth effectively, we may be unable to execute our business plan or address competitive challenges adequately.
We currently have six employees. Any growth in our business will place a significant strain on our managerial, administrative, operational, financial, information technology and other resources. We intend to further expand our overall business, customer base, employees and operations, which will require substantial management effort and significant additional investment in our infrastructure. We will be required to continue to improve our operational, financial and management controls and our reporting procedures and we may not be able to do so effectively. As such, we may be unable to manage our growth effectively.
Our working capital requirements involve estimates based on demand expectations and may decrease or increase beyond those currently anticipated, which could harm our operating results and financial condition.
We have no experience in selling smoking cessation products or Modified Risk Cigarettes on a commercial basis. As a result, we intend to base our funding and inventory decisions on estimates of future demand. If demand for our products does not increase as quickly as we have estimated or drops off sharply, our inventory and expenses could rise, and our business and operating results could suffer. Alternatively, if we experience sales in excess of our estimates, our working capital needs may be higher than those currently anticipated. Our ability to meet any demand for our products may depend on our ability to arrange for additional financing for any ongoing working capital shortages, since it is likely that cash flow from sales will lag behind our investment requirements.
The net proceeds of the Private Placement Offering will not be sufficient to enable us to complete the FDA approval process for our X-22 smoking cessation product and the FDA authorization process for our Modified Risk Cigarettes.
We will require additional capital in the future beyond the net proceeds of the Private Placement Offering to complete the FDA approval process for our X-22 smoking cessation product and the FDA authorization process for our Modified Risk Cigarettes. In particular, we currently plan to seek to raise approximately $15 million in the fourth quarter of 2011 through the sale of our common stock and/or securities convertible into common stock to fund Phase III trials, modified risk exposure studies and our general working capital requirements. However, we may not be able to obtain additional debt or equity financing on favorable terms, if at all. We believe the cost of the Phase II-B trial is approximately $1.1 million, of which we have paid approximately 45 percent as of July 15, 2011. We estimate the cost of completing the Phase III trials to be approximately $10 million. We estimate that the cost of completing the FDA authorization process for each of our potential Modified Risk Cigarettes is estimated to be at least $1 million. However, this depends on the regulations expected to be released by FDA within the next year. If we raise additional funds through the issuance of equity securities as we currently plan to seek to do, our stockholders may experience substantial dilution, or the equity securities may have rights, preferences or privileges senior to those of existing stockholders. If we raise additional funds through debt financings, these financings may involve significant cash payment obligations and covenants that restrict our ability to operate our business and make distributions to our stockholders. We also could elect to seek funds through arrangements with collaborators. To the extent that we raise additional funds through collaboration and licensing arrangements, it may be necessary to relinquish some rights to our technologies or our potential products or grant licenses on terms that are not favorable to us.
Due to market conditions and the status of our product development activities, additional funding may not be available to us on acceptable terms, or at all. Having insufficient funds may require us to delay, scale back or eliminate some or all of our clinical programs or to relinquish greater rights to potential products at an earlier stage of development or on less favorable terms than we would otherwise choose. Our failure to raise additional financing would adversely affect our ability to maintain, develop, enhance or grow our business, take advantage of future opportunities or respond to competitive pressures. If we cannot raise additional capital on acceptable terms, we may not be able to, among other things:
|
|
·
|
continue or complete clinical trials of our X-22 smoking cessation aid;
|
|
|
·
|
continue or complete the steps necessary to seek FDA authorization of our Modified Risk Cigarettes;
|
|
|
·
|
develop or enhance our potential products or introduce new products;
|
|
|
·
|
expand our development, sales and marketing and general and administrative activities;
|
|
|
·
|
attract tobacco growers, customers or manufacturing and distribution partners;
|
|
|
·
|
acquire complementary technologies, products or businesses;
|
|
|
·
|
expand our operations in the United States or internationally;
|
|
|
·
|
hire, train and retain employees; or
|
10
|
|
·
|
respond to competitive pressures or unanticipated working capital requirements.
|
Continued instability in the credit and financial market conditions may negatively impact our business, results of operations, and financial condition.
Financial markets in the United States, Canada, Europe and Asia continue to experience disruption, including, among other things, significant volatility in security prices, declining valuations of certain investments, severely diminished liquidity and credit availability. Business activity across a wide range of industries and regions continues to be reduced and local governments and many businesses are still in serious difficulty due to the lack of consumer spending and the lack of liquidity in the credit markets. As a clinical-stage biotechnology company, we rely on third parties for several important aspects of our business, including the supply of tobacco, manufacturing and distribution of our products, development of our potential products, and conduct of our clinical trials. Such third parties may be unable to satisfy their commitments to us due to tightening of global credit from time to time, which would adversely affect our business. The continued instability in the credit and financial market conditions may also negatively impact our ability to access capital and credit markets and our ability to manage our cash balance. While we are unable to predict the continued duration and severity of the adverse conditions in the United States and other countries, any of the circumstances mentioned above could adversely affect our business, financial condition, operating results and cash flow or cash position.
We will depend on the success of our X-22 smoking cessation aid and our Modified Risk Cigarettes and we may not be able to successfully commercialize these potential products.
Our goal is to develop products whose potential for risk reduction can be substantiated and that meet adult smokers’ taste expectations. We may not succeed in these efforts. If we do not succeed, but one or more of our competitors do, we may be at a competitive disadvantage. The success of our business depends in part on our ability to obtain FDA approval for our X-22 smoking cessation aid and FDA authorization under the Tobacco Control Act to market our BRAND A and BRAND B cigarettes as Modified Risk Cigarettes. The FDA has yet to release its regulations regarding modified risk tobacco products. We have not obtained approval to market X-22 in any jurisdiction, nor have we obtained authorization to market our BRAND A or BRAND B cigarettes as Modified Risk Cigarettes, and we cannot predict whether we will be able to obtain such approval or authorizations, or if regulators will permit the marketing of tobacco products with claims of reduced risk to consumers. Any failure to obtain such approval or authorizations would significantly undermine the commercial viability of the applicable product. If we fail to successfully commercialize these products in the United States, we may be unable to generate sufficient revenue to sustain and grow our business, and our business, financial condition, results of operations and cash flows will be adversely affected.
We will depend on third parties to manufacture our products.
We currently do not intend to manufacture any of our products and depend on contract manufacturers to produce our products according to our specifications, in sufficient quantities, on time, in compliance with appropriate regulatory standards and at competitive prices. We currently do not have an arrangement with any contract manufacturer to produce our final version of X-22 smoking cessation aid once it is approved by the FDA.
Manufacturers supplying our potential products must comply with FDA regulations which require, among other things, compliance with the FDA’s evolving regulations on Current Good Manufacturing Practices (“cGMP(s)”), which are enforced by the FDA through its facilities inspection program. The manufacture of products at any facility will be subject to strict quality control, testing and record keeping requirements, and continuing obligations regarding the submission of safety reports and other post-market information. We cannot guarantee that our current contract manufacturer will pass FDA and/or similar inspections in foreign countries to produce the final version of our X-22 smoking cessation aid, or that future changes to cGMP manufacturing standards will not also affect the manufactures of our other products. Therefore, we may have to build our own manufacturing facility which would require additional capital.
11
We will mainly depend on third parties to market, sell and distribute our products, and we currently have no commercial arrangements for the marketing, sale or distribution of our X-22 smoking cessation aid.
We expect to depend on third parties to a great extent to market, sell and distribute our products and we currently have no arrangements with third parties in place to provide such services for our X-22 smoking cessation aid. We cannot be sure that we will be able to enter into such arrangements on acceptable terms, or at all.
If we are unable to enter into marketing, sales and distribution arrangements with third parties for our X-22 smoking cessation aid, we would need to incur significant sales, marketing and distribution expenses in connection with the commercialization of X-22 and any future potential products. We do not currently have a dedicated sales force, and we have no experience in the sales, marketing and distribution of pharmaceutical products. Developing a sales force is expensive and time-consuming, and we may not be able to develop this capacity. If we are unable to establish adequate sales, marketing and distribution capabilities, independently or with others, we may not be able to generate significant revenue and may not become profitable.
If our X-22 smoking cessation aid does not gain market acceptance among physicians, patients, third-party payers and the medical community, we may be unable to generate significant revenue.
Our X-22 smoking cessation aid may not achieve market acceptance among physicians, patients, third-party payers and others in the medical community. If we receive FDA approval for the marketing of X-22 as a smoking cessation aid in the U.S., the degree of market acceptance could depend upon a number of factors, including:
|
|
·
|
limitations on the indications for use for which X-22 may be marketed ;
|
|
|
·
|
the establishment and demonstration in the medical community of the clinical efficacy and safety of our potential products and their potential advantages over existing products;
|
|
|
·
|
the prevalence and severity of any side effects;
|
|
|
·
|
the strength of marketing and distribution support; and/or
|
|
|
·
|
sufficient third-party coverage or reimbursement.
|
The market may not accept our X-22 smoking cessation aid, based on any number of the above factors. Even if the FDA approves the marketing of X-22 as a smoking cessation aid, there are other FDA-approved products available and there will also be future competitive products which directly compete with X-22. The market may choose to continue utilizing such existing or future competitive products for any number of reasons, including familiarity with or pricing of such products. The failure of any of our potential products to gain market acceptance could impair our ability to generate revenue, which could have a material adverse effect on our future business, financial condition, results of operations and cash flows.
Our principal competitors in the smoking cessation market have, and any future competitors may have, greater financial and marketing resources than we do, and they may therefore develop products or other technologies similar or superior to ours or otherwise compete more successfully than we do.
We have no experience in selling smoking cessation products. Competition in the smoking cessation aid products industry is intense, and we may not be able to successfully compete in the market. In the market for FDA-approved smoking cessation aids, our principal competitors include Pfizer Inc., GlaxoSmithKline PLC, Perrigo Company, Novartis International AG, and Niconovum AB, a subsidiary of Reynolds American Inc. The industry consists of major domestic and international companies, most of which have existing relationships in the markets into which we plan to sell, as well as financial, technical, marketing, sales, manufacturing, scaling capacity, distribution and other resources and name recognition substantially greater than ours. In addition, we expect new competitors will enter the markets for our products in the future. Potential customers may choose to do business with our more established competitors, because of their perception that our competitors are more stable, are more likely to complete various projects, can scale operations more quickly, have greater manufacturing capacity, are more likely to continue as a going concern and lend greater credibility to any joint venture. If we are unable to compete successfully against manufacturers of other smoking cessation products, our business could suffer, and we could lose or be unable to obtain market share.
12
We face intense competition in the market for our RED SUN and MAGIC cigarettes and our BRAND A and BRAND B cigarettes, and our failure to compete effectively could have a material adverse effect on our profitability and results of operations.
Cigarette companies compete primarily on the basis of product quality, brand recognition, brand loyalty, taste, innovation, packaging, service, marketing, advertising, retail shelf space and price. We are subject to highly competitive conditions in all aspects of our business and we may not be able to effectively market and sell our RED SUN and MAGIC cigarettes or other cigarettes we may introduce to the market, even if we are able to market our BRAND A and BRAND B cigarettes as Modified Risk Cigarettes. The competitive environment and our competitive position can be significantly influenced by weak economic conditions, erosion of consumer confidence, competitors’ introduction of low-price products or innovative products, higher cigarette taxes, higher absolute prices and larger gaps between price categories, and product regulation that diminishes the ability to differentiate tobacco products. Domestic competitors include Philip Morris USA, Reynolds American Inc., Lorillard Inc., Commonwealth Brands, Inc., Liggett Group LLC, Vector Tobacco Inc. and Star Scientific Inc. International competitors include Philip Morris International, British American Tobacco, Japan Tobacco Inc. and regional and local tobacco companies; and, in some instances, government-owned tobacco enterprises, principally in China, Egypt, Thailand, Taiwan, Vietnam and Algeria.
Our MAGIC cigarettes and our BRAND A cigarettes may have relatively low sales volume in the future.
A third-party, former licensee marketed cigarettes containing our proprietary VLN tobacco in the past on a limited basis that yielded low sales volumes. Our marketing and sales of our MAGIC cigarettes and BRAND A cigarettes which contain our proprietary VLN tobacco may also result in low sales volume.
Our competitors may develop products that are less expensive, safer or more effective, which may diminish or eliminate the commercial success of any potential product that we may commercialize.
If our competitors market products that are less expensive, safer or more effective than our potential products, or that reach the market before our potential products, we may not achieve commercial success. The market may choose to continue utilizing existing products for any number of reasons, including familiarity with or pricing of these existing products. The failure of our X-22 smoking cessation aid or our cigarette brands to compete with products marketed by our competitors would impair our ability to generate revenue, which would have a material adverse effect on our future business, financial condition, results of operations and cash flows. Our competitors may:
|
|
·
|
develop and market products that are less expensive or more effective than our proposed products;
|
|
|
·
|
commercialize competing products before we or our partners can launch our proposed products;
|
|
|
·
|
operate larger research and development programs or have substantially greater financial resources than we do;
|
|
|
·
|
initiate or withstand substantial price competition more successfully than we can;
|
|
|
·
|
have greater success in recruiting skilled technical and scientific workers from the limited pool of available talent;
|
|
|
·
|
more effectively negotiate third-party licenses and strategic relationships; and
|
|
|
·
|
take advantage of acquisition or other opportunities more readily than we can.
|
In addition, if we fail to stay at the forefront of technological change, we may be unable to compete effectively. Our competitors may render our technologies obsolete by advances in existing technological approaches or the development of new or different approaches, potentially eliminating the advantages that we believe we derive from our research approach and proprietary technologies.
13
Government mandated prices, production control programs, shifts in crops driven by economic conditions and adverse weather patterns may increase the cost or reduce the quality of the tobacco and other agricultural products used to manufacture our products.
We depend upon independent tobacco producers to grow our specialty proprietary tobaccos with specific nicotine contents for our products. As with other agricultural commodities, the price of tobacco leaf can be influenced by imbalances in supply and demand, and crop quality can be influenced by variations in weather patterns, diseases and pests. We must also compete with other tobacco companies for contract production with independent tobacco growers. Tobacco production in certain countries is subject to a variety of controls, including government mandated prices and production control programs. Changes in the patterns of demand for agricultural products could cause farmers to plant less tobacco. Any significant change in tobacco leaf prices, quality and quantity could affect our profitability and our business.
Our future success depends on our ability to retain key personnel.
Our success will depend to a significant extent on the continued services of our senior management team, and in particular Joseph Pandolfino, our Chief Executive Officer, Henry Sicignano III, our President, and Michael Moynihan, Ph.D., our Vice President of R&D. The loss or unavailability of any of these individuals may significantly delay or prevent the development of our potential products and other business objectives by diverting management’s attention to transition matters. Identification of suitable management replacements, if any, could have a material adverse effect on our business, operating results, cash flows and financial condition. While each of these individuals is party to employment agreements with us, they could terminate their relationships with us at any time, and we may be unable to enforce any applicable employment or non-compete agreements.
We also rely on consultants and advisors to assist us in formulating our research and development, manufacturing, distribution, marketing and sales strategies. All of our consultants and advisors are either self-employed or employed by other organizations, and they may have conflicts of interest or other commitments, such as consulting or advisory contracts with other organizations, that may affect their ability to contribute to us.
Product liability claims, product recalls or other claims could cause us to incur losses or damage our reputation.
The risk of product liability claims or product recalls, and associated adverse publicity, is inherent in the development, manufacturing, marketing and sale of cigarettes and smoking cessation products. We do not currently have product liability insurance for our products or our potential products and do not expect to be able to obtain product liability insurance at reasonable commercial rates for these products. Any product recall or lawsuit seeking significant monetary damages may have a material adverse affect on our business and financial condition. A successful product liability claim against us could require us to pay a substantial monetary award. We cannot assure you that such claims will not be made in the future.
We may be unable to complete or integrate acquisitions effectively, which may adversely affect our growth, profitability and results of operations.
We may pursue acquisitions as part of our business strategy. However, we cannot be certain that we will be able to identify attractive acquisition targets, obtain financing for acquisitions on satisfactory terms or successfully acquire identified targets. Additionally, we may not be successful in integrating acquired businesses into our existing operations or achieving projected synergies. Competition for acquisition opportunities in the industries in which we operate may rise, thereby increasing our costs of making acquisitions or causing us to refrain from making further acquisitions. These and other acquisition-related factors could negatively and adversely impact our growth, profitability and results of operations.
Risks Related to Regulatory Approvals and Insurance Reimbursement
If we fail to obtain FDA and foreign regulatory approvals of X-22 as a smoking cessation aid and FDA authorization to market BRAND A and BRAND B as Modified Risk Cigarettes, we will be unable to commercialize these potential products in and outside the U.S., other than the sale of our BRAND A and BRAND B cigarettes as conventional cigarettes.
There can be no assurance that our X-22 smoking cessation aid will be approved by the FDA, European Medicines Agency (“EMA”), or any other governmental body. In addition, there can be no assurance that all necessary approvals will be granted for our potential products or that review or actions will not involve delays caused by requests for additional information or testing that could adversely affect the time to market for and sale of our potential products. Even if X-22 is approved by the FDA, the FDA may require the product to only be prescribed to patients who have already failed to quit smoking with another approved therapy. Further, failure to comply with applicable regulatory requirements can, among other things, result in the suspension of regulatory approval as well as possible civil and criminal sanctions.
14
The development, testing, manufacturing and marketing of our potential products are subject to extensive regulation by governmental authorities in the United States and throughout the world. In particular, the process of obtaining approvals by the FDA, EMA and other international FDA-equivalent agencies in targeted countries is costly and time consuming, and the time required for such approval is uncertain. Our X-22 smoking cessation aid must undergo rigorous clinical testing and an extensive regulatory approval process mandated by the FDA or EMEA. Such regulatory review includes the determination of manufacturing capability and product performance. Generally, only a small percentage of pharmaceutical products are ultimately approved for commercial sale.
The scope of review, including product testing and exposure studies, to be required by the FDA under the Tobacco Control Act in order for cigarettes such as BRAND A and BRAND B to be marketed as Modified Risk Cigarettes has not yet been fully established. We may be unsuccessful in establishing that BRAND A or BRAND B are Modified Risk Cigarettes, and we may fail to demonstrate that either BRAND A or BRAND B significantly reduces exposure to certain tobacco smoke toxins. Even if we are able to demonstrate reduced exposure to certain tobacco smoke toxins, the FDA may decide that allowing a reduced risk claim is not in the best interest of the public health, and the FDA may not allow us to market our BRAND A and/or BRAND B cigarettes as Modified Risk Cigarettes. The FDA may prevent us from selling BRAND A or BRAND B or both products in the U.S. market before the FDA makes a determination of whether to authorize us to market our BRAND A or BRAND B cigarettes as Modified Risk Cigarettes. Furthermore, the FDA could force us to remove from the U.S. market our other tobacco products such as RED SUN or MAGIC.
If we fail to comply with extensive regulations enforced by the FDA and other agencies, the commercialization of our potential products could be prevented, delayed or halted.
There is no guarantee that we will obtain the necessary approvals and authorizations from the FDA for X-22 to be marketed as a smoking cessation aid or for Brand A or Brand B to be classified as Modified Risk Cigarettes. Clinical trials and the manufacturing and marketing of X-22, BRAND A and BRAND B are subject to extensive regulation by various government authorities. We have not received marketing approval for our X-22 smoking cessation aid, nor have we applied for or received FDA authorization to market BRAND A or BRAND B cigarettes as Modified Risk Cigarettes. The process of obtaining FDA and other required regulatory approvals and authorizations is lengthy and expensive, and the time required for such approvals and authorizations is uncertain. The processes are affected by such factors as:
|
|
·
|
the severity of the disease involved;
|
|
|
·
|
the quality of submissions relating to the potential product;
|
|
|
·
|
the potential product’s clinical efficacy and safety;
|
|
|
·
|
the strength of the chemistry and manufacturing control of the process;
|
|
|
·
|
the manufacturing facility’s compliance;
|
|
|
·
|
the availability of alternative treatments;
|
|
|
·
|
the risks and benefits demonstrated in clinical trials; and
|
|
|
·
|
the patent status and marketing exclusivity rights of certain innovative products.
|
Any regulatory approval or authorization that we receive for our potential products may also be subject to limitations on the indicated uses for which the product may be marketed or contain requirements for potentially costly post-marketing follow-up studies. The subsequent discovery of previously unknown problems with the product, including adverse events of unanticipated severity or frequency, may result in restrictions on the marketing of the product and/or withdrawal of the product from the market.
15
Manufacturing, labeling, storage and distribution activities in the United States also are subject to strict regulation and licensing by the FDA. The manufacturing facilities for biopharmaceutical products and tobacco products are subject to periodic inspection by the FDA and other regulatory authorities and from time to time, these agencies may send notice of deficiencies as a result of such inspections. Our failure, or the failure of our contractors’ manufacturing facilities, to continue to meet regulatory standards or to remedy any deficiencies could result in corrective action by the FDA or these other authorities, including the interruption or prevention of marketing, closure of our contractors’ manufacturing facilities, and fines or penalties.
Regulatory authorities also could require post-marketing surveillance to monitor and report to the FDA potential adverse effects of our potential products. The U.S. Congress or the FDA in specific situations can modify the regulatory process. If approved, any of our potential products’ subsequent failure to comply with applicable regulatory requirements could, among other things, result in warning letters, fines, suspension or revocation of regulatory approvals, product recalls or seizures, operating restrictions, injunctions and criminal prosecutions.
The FDA’s policies may change and additional government regulations may be enacted that could prevent or delay regulatory approval of our potential products. We cannot predict the likelihood, nature or extent of adverse government regulation that may arise from future legislation or administrative action. If we are not able to maintain regulatory compliance, we might not be permitted to market our potential products and our business could suffer.
In the future, we intend to distribute and sell our potential products outside of the United States, which will subject us to further regulatory risk.
In addition to seeking approval from the FDA for our X-22 smoking cessation aid in the United States, we intend to seek governmental approvals required to market X-22 and our other potential products in other countries. Marketing of our X-22 smoking cessation aid is not permitted in certain countries until we have obtained required approvals or exemptions in the individual country. The regulatory review process varies from country to country, and approval by foreign governmental authorities is unpredictable, uncertain and generally expensive. Our ability to market our potential products could be substantially limited due to delays in receipt of, or failure to receive, the necessary approvals or clearances. We anticipate commencing the applications required in some or all of these countries following approval by the FDA; however, we may decide to file applications in advance of the FDA approval if we determine such filings to be both time and cost effective. If we export any of our potential products that have not yet been cleared for commercial distribution in the United States, such products may be subject to FDA export restrictions. Failure to obtain necessary regulatory approvals could impair our ability to generate revenue from international sources.
Market acceptance of our X-22 smoking cessation aid could be limited if users are unable to obtain adequate reimbursement from third-party payers.
Government health administration authorities, private health insurers and other organizations generally provide reimbursement for FDA-approved smoking cessation products, and our commercial success could depend in part on these third-party payers agreeing to reimburse patients for the costs of our X-22 smoking cessation aid. Even if we succeed in bringing our X-22 smoking cessation aid to market, there is no assurance that third-party payers will consider X-22 cost effective or provide reimbursement in whole or in part for its use.
Significant uncertainty exists as to the reimbursement status of newly approved health care products. Our X-22 smoking cessation aid is intended to replace or alter existing therapies or procedures. These third-party payers may conclude that our X-22 smoking cessation aid is less safe, effective or cost-effective than these existing therapies or procedures. Therefore, third-party payers may not approve X-22 for reimbursement.
If third-party payers do not approve our potential products for reimbursement or fail to reimburse for them adequately, sales could suffer as some physicians or their patients could opt for a competing product that is approved for reimbursement or is adequately reimbursed. Even if third-party payers make reimbursement available, these payers’ reimbursement policies may adversely affect our ability and the ability of our potential collaborators to sell our potential products on a profitable basis.
The trend toward managed healthcare in the United States, the growth of organizations such as health maintenance organizations and legislative proposals to reform healthcare and government insurance programs could significantly influence the purchase of healthcare services and products, resulting in lower prices and reduced demand for our potential products which could adversely affect our business, financial condition, results of operations and cash flows.
16
In addition, legislation and regulations affecting the pricing of our potential products may change in ways adverse to us before or after the FDA or other regulatory agencies approve any of our potential products for marketing. While we cannot predict the likelihood of any of these legislative or regulatory proposals, if any government or regulatory agency adopts these proposals, they could materially adversely affect our business, financial condition, results of operations and cash flows.
We could be negatively impacted by the application or enforcement of federal and state fraud and abuse laws, including anti-kickback laws and other federal and state anti-referral laws.
We will need to establish a program to ensure compliance with all potentially applicable laws in connection with the development, manufacturing, marketing and sales of our potential products. For example, all product marketing efforts must be strictly scrutinized to assure that they are not associated with improper remunerations to referral sources in violation of the federal Anti-Kickback Statute and similar state statutes. Remunerations may include potential future activities for our potential products, including discounts, rebates and bundled sales, which must be appropriately structured to take advantage of statutory and regulatory “safe harbors.” From time to time, we may engage physicians in consulting activities. In addition, we may decide to sponsor continuing medical education activities for physicians or other medical personnel. We also may award or sponsor study grants to physicians from time to time. All relationships with physicians, including consulting arrangements, continuing medical education and study grants, must be similarly reviewed for compliance with the Anti-Kickback Statute to assure that remuneration is not provided in return for referrals. Patient inducements may also be unlawful. Inaccurate reports of product pricing, or a failure to provide product at an appropriate price to various governmental entities, could also serve as a basis for an enforcement action under various theories.
Claims which are “tainted” by virtue of kickbacks or a violation of self-referral rules may be alleged as false claims if other elements of a violation are established. The federal False Claims Act, which includes a provision allowing whistleblowers to bring actions on behalf of the federal government and receive a portion of the recovery, applies to those who submit a false claim and those who cause a false claim to be submitted. Because our potential customers may seek payments from the federal healthcare programs for our potential products, even during the clinical trial stages, we must ensure that we take no actions which could result in the submission of false claims. For example, free product samples which are knowingly or with reckless disregard billed to the federal healthcare programs could constitute false claims. If the practice was facilitated or fostered by us, we could be liable. Similarly, inadequate accounting for or a misuse of any federal grant funds used for product research and development could be alleged as a violation of the False Claims Act or other relevant statutes.
The risk of us being found in violation of these laws is increased by the fact that many of them have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations, and additional legal or regulatory change.
Delays in clinical testing could result in increased costs to us and delay our ability to generate revenue.
Significant delays in clinical testing could materially increase our product development costs. Clinical trials can be delayed for a variety of reasons, including delays in obtaining regulatory approval to commence and continue a study, delays in reaching agreement on acceptable clinical study terms with prospective sites, delays in obtaining institutional review board approval to conduct a study at a prospective site and delays in recruiting patients to participate in a study.
In addition, we plan to rely on third-party clinical investigators to conduct our clinical trials and other third-party organizations to oversee the operations of these clinical trials and to perform data collection and analysis. As a result, we may face additional delays outside of our control if these parties do not perform their obligations in a timely fashion. Significant delays in testing or regulatory approvals or authorizations for any of our current or future potential products, including our X-22 smoking cessation aid or our BRAND A and BRAND B cigarettes as Modified Risk Cigarettes, could prevent or cause delays in the commercialization of such potential products, reduce potential revenues from the sale of such potential products and cause our costs to increase.
17
Our clinical trials for any of our potential products may produce negative or inconclusive results and we may decide, or regulators may require us, to conduct additional clinical and/or preclinical testing for these potential products or cease our trials.
We do not know whether clinical trials of our potential products will demonstrate safety and efficacy sufficiently to result in marketable products. Because our clinical trials for our X-22 smoking cessation aid and any other potential products may produce negative or inconclusive results, we may decide, or regulators may require us, to conduct additional clinical and/or preclinical testing for these potential products or cease our clinical trials. If this occurs, we may not be able to obtain approval for these potential products or our anticipated time of bringing these potential products to the market may be substantially delayed and we may also experience significant additional development costs. We may also be required to undertake additional clinical testing if we change or expand the indications for our potential products.
The use of hazardous materials in our operations may subject us to environmental claims or liabilities.
Our research and development activities involve the use of hazardous materials. Injury or contamination from these materials may occur and we could be held liable for any damages, which could exceed our available financial resources. This liability could materially adversely affect our business, financial condition, results of operations and cash flows.
We are subject to federal, state and local laws and regulations governing the use, manufacture, storage, handling and disposal of hazardous materials and waste products. We may be required to incur significant costs to comply with environmental laws and regulations in the future that could materially adversely affect our business, financial condition, results of operations and cash flows.
The degree of public acceptance or perceived public acceptance of our genetically modified tobacco may affect our sales and operations.
Some opponents of genetically modified crops have actively raised public concern about the potential adverse effects these crops, and the products made from them, may have on human and animal health, other plants, and the environment. Public concern may affect the timing of, and whether we are able to obtain, government approvals. Even after approvals are granted, public concern may lead to increased regulation or legislation, which could affect our sales and profitability, and may adversely affect sales of our products, due to concerns about products derived from biotechnology. In addition, opponents of agricultural biotechnology have attacked farmers’ fields and facilities used by agricultural biotechnology companies, and may launch future attacks against farmers’ fields and our research, production or other facilities, which could affect our sales and our costs.
Risks Related to the Tobacco Industry
Our business faces significant governmental action aimed at increasing regulatory requirements with the goal of preventing the use of tobacco products.
Cigarette companies face significant governmental action, especially in the United States pursuant to the Tobacco Control Act, including efforts aimed at reducing the incidence of tobacco use, restricting marketing and advertising, imposing regulations on packaging, warnings and disclosure of flavors or other ingredients, prohibiting the sale of tobacco products with certain characterizing flavors or other characteristics, limiting or prohibiting the sale of tobacco products by certain retail establishments and the sale of tobacco products in certain packaging sizes, and seeking to hold them responsible for the adverse health effects associated with both smoking and exposure to environmental tobacco smoke. Governmental actions, combined with the diminishing social acceptance of smoking and private actions to restrict smoking, have resulted in reduced industry volume in the United States and other countries, and we expect that these factors will continue to reduce consumption levels in these countries.
18
Certain of such actions may have a favorable impact on our X-22 smoking cessation aid, or on our BRAND A and BRAND B cigarettes if we are able to market them as Modified Risk Cigarettes. However, there is no assurance of such favorable impact and such actions may have a negative impact on our ability to market RED SUN and MAGIC.
Significant regulatory developments will take place over the next few years in many markets, driven principally by the World Health Organization’s Framework Convention on Tobacco Control (“FCTC”). The FCTC is the first international public health treaty on tobacco, and its objective is to establish a global agenda for tobacco regulation with the purpose of reducing initiation of tobacco use and encouraging cessation. In addition, the FCTC has led to increased efforts by tobacco control advocates and public health organizations to reduce the palatability and appeal of tobacco products. Partly because of some or a combination of these efforts, unit sales of tobacco products in certain markets, principally Western Europe and Japan, have been in general decline and we expect this trend to continue. Our operating results could be significantly affected by any significant decrease in demand for cigarettes, any significant increase in the cost of complying with new regulatory requirements and requirements that lead to a commoditization of tobacco products.
The FDA requirement regarding graphic health warnings on cigarette packaging and in cigarette advertising in September 2012 is likely to have a negative impact on sales of our products.
In November 2010, as required by the Tobacco Control Act, the FDA issued a proposed rule to modify the required warnings that appear on cigarette packages and in cigarette advertisements. These warning were finalized on June 21, 2011 and consist of nine new textual warning statements accompanied by color graphics depicting the negative health consequences of smoking. The FDA selected nine images from the originally proposed 36 images after reviewing the relevant scientific literature, analyzing the results from an 18,000 person study and considering more than 1,700 comments from a variety of groups. The graphic health warnings will be located beneath the cellophane wrapping on cigarette packages, and will comprise the top 50 percent of the front and rear panels of cigarette packages. The graphic health warnings will occupy 20 percent of a cigarette advertisement and will be located at the top of the advertisement. Each warning is accompanied by a smoking cessation phone number, 1-800-QUIT-NOW. When implemented in September 2012, all cigarettes manufactured for sale or distribution in the United States will need to include these new graphic health warnings on their packages. Any reduction in the number smokers will probably reduce the demand for MAGIC and RED SUN, as well as X-22, Brand A and B, if and when approved/authorized by the FDA.
We may become subject to litigation related to cigarette smoking and exposure to environmental tobacco smoke, or ETS, which could severely impair our results of operations and liquidity.
Although we are not currently subject to legal proceedings, we may become subject to litigation related to the sale of our RED SUN and MAGIC cigarettes and our BRAND A and BRAND B cigarettes. Legal proceedings covering a wide range of matters related to tobacco use are pending or threatened in various U.S. and foreign jurisdictions. Various types of claims are raised in these proceedings, including product liability, consumer protection, antitrust, tax, contraband shipments, patent infringement, employment matters, claims for contribution and claims of competitors and distributors.
Litigation is subject to uncertainty and it is possible that there could be adverse developments in pending cases. An unfavorable outcome or settlement of pending tobacco related litigation could encourage the commencement of additional litigation. The variability in pleadings, together with the actual experience of management in litigating claims, demonstrates that the monetary relief that may be specified in a lawsuit bears little relevance to the ultimate outcome.
Damages claimed in some tobacco-related litigation are significant and, in certain cases range into the billions of dollars. We anticipate that new cases will continue to be filed. The FCTC encourages litigation against tobacco product manufacturers. It is possible that our results of operations, cash flows or financial position could be materially affected by an unfavorable outcome or settlement of litigation, whether or not we are a party to such litigation.
Cigarettes are subject to substantial taxes. Significant increases in cigarette-related taxes have been proposed or enacted and are likely to continue to be proposed or enacted in numerous jurisdictions. These tax increases may affect our sales and profitability and make us less competitive versus certain of our competitors.
Tax regimes, including excise taxes, sales taxes and import duties, can disproportionately affect the retail price of manufactured cigarettes versus other tobacco products, or disproportionately affect the relative retail price of our RED SUN and MAGIC cigarettes and our BRAND A and BRAND B cigarettes versus lower-priced cigarette brands manufactured by our competitors. Increases in cigarette taxes are expected to continue to have an adverse impact on sales of cigarettes resulting in (i) lower consumption levels, (ii) a shift in sales from manufactured cigarettes to other tobacco products or to lower-price cigarette categories, (iii) a shift from local sales to legal cross-border purchases of lower price products, and (iv) illicit products such as contraband and counterfeit.
We may become subject to governmental investigations on a range of matters.
Cigarette companies are often subject to investigations, including allegations of contraband shipments of cigarettes, allegations of unlawful pricing activities within certain markets, allegations of underpayment of custom duties and/or excise taxes, and allegations of false and misleading usage of descriptors such as “lights” and “ultra lights.” We cannot predict the outcome of any to which we may become subject, and we may be materially affected by an unfavorable outcome of future investigations.
19
Risks Related to Intellectual Property
Our proprietary rights may not adequately protect our intellectual property, products and potential products, and if we cannot obtain adequate protection of our intellectual property, products and potential products, we may not be able to successfully market our products and potential products.
Our commercial success will depend in part on obtaining and maintaining intellectual property protection for our technologies, products and potential products. We will only be able to protect our technologies, products and potential products from unauthorized use by third parties to the extent that valid and enforceable patents cover them, or other market exclusionary rights apply.
The patent positions of life sciences companies, like ours, can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. No consistent policy regarding the breadth of claims allowed in such companies’ patents has emerged to date in the United States. The general patent environment outside the United States also involves significant uncertainty. Accordingly, we cannot predict the breadth of claims that may be allowed or that the scope of these patent rights could provide a sufficient degree of future protection that could permit us to gain or keep our competitive advantage with respect to these products and technology. Additionally, life science companies like ours are often dependent on creating a pipeline of products. We may not be able to develop additional potential products, or proprietary technologies that produce commercially viable products or that are themselves patentable.
Our issued patents may be subject to challenge and possibly invalidated by third parties. Changes in either the patent laws or in the interpretations of patent laws in the United States or other countries may diminish the value of our intellectual property.
In addition, others may independently develop similar or alternative products and technologies that may be outside the scope of our intellectual property. Should third parties obtain patent rights to similar products or technology, this may have an adverse effect on our business.
We also rely on trade secrets to protect our technology, products and potential products, especially where we do not believe patent protection is appropriate or obtainable. Trade secrets, however, are difficult to protect. While we believe that we use reasonable efforts to protect our trade secrets, our own or our strategic partners’ employees, consultants, contractors or advisors may unintentionally or willfully disclose our information to competitors. We seek to protect this information, in part, through the use of non-disclosure and confidentiality agreements with employees, consultants, advisors and others. These agreements may be breached, and we may not have adequate remedies for a breach. In addition, we cannot ensure that those agreements will provide adequate protection for our trade secrets, know-how or other proprietary information or prevent their unauthorized use or disclosure.
To the extent that consultants or key employees apply technological information independently developed by them or by others to our products and potential products, disputes may arise as to the proprietary rights of the information, which may not be resolved in our favor. Consultants and key employees that work with our confidential and proprietary technologies are required to assign all intellectual property rights in their discoveries to us. However, these consultants or key employees may terminate their relationship with us, and we cannot preclude them indefinitely from dealing with our competitors. If our trade secrets become known to competitors with greater experience and financial resources, the competitors may copy or use our trade secrets and other proprietary information in the advancement of their products, methods or technologies. If we were to prosecute a claim that a third party had illegally obtained and was using our trade secrets, it could be expensive and time consuming and the outcome could be unpredictable. In addition, courts outside the United States are sometimes less willing to protect trade secrets than courts in the United States. Moreover, if our competitors independently develop equivalent knowledge, we would lack any contractual claim to this information, and our business could be harmed.
20
Our ability to commercialize our potential products will depend on our ability to sell such products without infringing the patent or proprietary rights of third parties. If we are sued for infringing intellectual property rights of third parties, such litigation could be costly and time consuming and an unfavorable outcome could have a significant adverse effect on our business.
Our ability to commercialize our potential products will depend on our ability to sell such products without infringing the patents or other proprietary rights of third parties. Third-party intellectual property rights in our field are complicated, and third-party intellectual property rights in these fields are continuously evolving. While we have conducted searches for such third-party intellectual property rights, and purchased or licensed those we have discovered, we have not performed specific searches for third-party intellectual property rights that may raise freedom-to-operate issues, and we have not obtained legal opinions regarding commercialization of our potential products. As such, there may be existing patents that may affect our ability to commercialize our potential products.
In addition, because patent applications are published up to 18 months after their filing, and because patent applications can take several years to issue, there may be currently pending third-party patent applications that are unknown to us, which may later result in issued patents.
If a third-party claims that we infringe on its patents or other proprietary rights, we could face a number of issues that could seriously harm our competitive position, including:
|
|
·
|
infringement claims that, with or without merit, can be costly and time consuming to litigate, can delay the regulatory approval process and can divert management’s attention from our core business strategy;
|
|
|
·
|
substantial damages for past infringement which we may have to pay if a court determines that our products or technologies infringe upon a competitor’s patent or other proprietary rights;
|
|
|
·
|
a court order prohibiting us from commercializing our potential products or technologies unless the holder licenses the patent or other proprietary rights to us, which such holder is not required to do;
|
|
|
·
|
if a license is available from a holder, we may have to pay substantial royalties or grant cross licenses to our patents or other proprietary rights; and
|
|
|
·
|
redesigning our process so that it does not infringe the third-party intellectual property, which may not be possible, or which may require substantial time and expense including delays in bringing our potential products to market.
|
Such actions could harm our competitive position and our ability to generate revenue and could result in increased costs.
Our patent applications may not result in issued patents, which may have a material adverse effect on our ability to prevent others from commercially exploiting products similar to ours.
We own 8 issued patents and we have the exclusive license to an additional 90 issued patents in an aggregate of 79 countries. In addition, we own or exclusively license approximately 45 pending patent applications, of which we own 29 such patent applications and have an exclusive license with regard to 16 such patent applications. We cannot assure you these patent applications will issue, in whole or in part, as patents. Patent applications in the United States are maintained in secrecy until the patents are published or are issued. Since publication of discoveries in the scientific or patent literature tends to lag behind actual discoveries by several months, we cannot be certain that we are the first creator of inventions covered by pending patent applications or the first to file patent applications on these inventions. We also cannot be certain that our pending patent applications will result in issued patents or that any of our issued patents will afford protection against a competitor. In addition, patent applications filed in foreign countries are subject to laws, rules and procedures that differ from those of the United States, and thus we cannot be certain that foreign patent applications related to U.S. patents will be issued. Furthermore, if these patent applications issue, some foreign countries provide significantly less effective patent enforcement than in the United States.
21
The status of patents involves complex legal and factual questions and the breadth of claims allowed is uncertain. Accordingly, we cannot be certain that the patent applications that we file will result in patents being issued, or that our patents and any patents that may be issued to us in the near future will afford protection against competitors with similar technology. In addition, patents issued to us may be infringed upon or designed around by others and others may obtain patents that we need to license or design around, either of which would increase costs and may adversely affect our operations.
We license certain patent rights from third-party owners. If such owners do not properly maintain or enforce the patents underlying such licenses, our competitive position and business prospects could be harmed.
We license rights to third-party intellectual property that is necessary or useful for our business, and we may enter into additional licensing agreements in the future. Our success could depend in part on the ability of some of our licensors to obtain, maintain and enforce patent protection for their intellectual property, in particular, those patents to which we have secured exclusive rights. Our licensors may not successfully prosecute the patent applications to which we are licensed. Even if patents are issued with respect to these patent applications, our licensors may fail to maintain these patents, may determine not to pursue litigation against other companies that are infringing these patents, or may pursue such litigation less aggressively than we could. In addition, our licensors may terminate their agreements with us in the event we breach the applicable license agreement and fail to cure the breach within a specified period of time. Without protection for the intellectual property we license, other companies might be able to offer substantially identical products for sale, which could adversely affect our competitive business position and harm our business prospects.
We are currently in default pursuant to the terms of an intellectual property license to which we are a party.
We are currently in payment default for certain patent-related costs pursuant to the terms of our exclusive worldwide license agreement with North Carolina State University (“NCSU”), dated as of March 6, 2009 (the “License Agreement”). We are required to reimburse NCSU for such patent-related costs within 30 days of being invoiced, and a portion of such reimbursements has become past due mainly as a result of (i) an interference proceeding invoked by the U.S. Patent and Trademark Office between NCSU and a former licensee of 22nd Century and (ii) an arbitration proceeding brought by 22nd Century against the same former licensee. Both of these actions involved the License Agreement. 22nd Century at its sole option decided to defend NCSU in this patent interference and pay all related expenses including legal fees. This resulted in NCSU obtaining international rights to a patent family that was incorporated into the License Agreement, thereby mutually benefiting NCSU and 22nd Century. Separately, the arbitration decision was decisively in 22nd Century’s favor containing multiple awards to 22nd Century, some of which also benefited NCSU since its technology was involved in the proceeding. The arbitration award was not disputed and was fulfilled by the defendant in its entirety in 2009. We believe the results of both of these actions, the interference proceeding and arbitration proceeding, greatly benefited NCSU and 22nd Century beyond 22nd Century’s cumulative net cost of these actions which was approximately $800,000. Consequently, as of March 31, 2011, the balance we owed to NCSU for these patent-related costs was approximately $728,000, after our $400,000 payment in February 2011. We have entered into negotiations with NCSU regarding the timing of payment for the balance, and we have requested NCSU to defer payment of the balance until 2012. We have not received any termination notice from NCSU. However, NCSU may have the right to terminate the License Agreement upon 60 days prior written notice, which includes the opportunity for us to cure the payment default within this timeframe. The intellectual property licensed to us under the License Agreement is crucial to our business, and if NCSU chooses to invoke any right it may have to terminate the License Agreement and we are unable to cure the default, our business would be materially and adversely affected. Furthermore, if NCSU does not agree to continue to defer payment of the balance, we may not have sufficient funds to pay for the Phase II-B clinical trial for X-22 in full.
22
Risks Related to Ownership of Our Common Stock
The Securities issued in the Merger are “restricted securities” and, as such, may not be sold except in limited circumstances.
None of the shares of common stock or warrants issued in the Merger or the shares of common stock issuable upon exercise of such warrants, which we refer to collectively as the “Securities,” have been registered under the Securities Act, or registered or qualified under any state securities laws. The Securities were sold and/or issued pursuant to exemptions contained in and under those laws. Accordingly, the Securities are “restricted securities” as defined in Rule 144 under the Securities Act and must, therefore, be held indefinitely unless registered under applicable federal and state securities laws, or an exemption from the registration requirements of those laws is available. The securities purchase agreements, warrants and certificates representing the Securities contain legends reflecting their restricted status.
Although we are required to register the shares of common stock issued to the investors in the Private Placement Offering in exchange for the 22nd Century limited liability company membership interests included in the Units purchased by such investors in the Private Placement Offering, we cannot assure that the SEC will declare the registration statement effective, thereby enabling the shares of common stock to be freely tradable. Rule 144 under the Securities Act, which permits the resale, subject to various terms and conditions, of limited amounts of restricted securities after they have been held for six months will not immediately apply to our common stock because we were at one time designated as a “shell company” under SEC regulations. Pursuant to Rule 144(i), securities issued by a current or former shell company that otherwise meet the holding period and other requirements of Rule 144 nevertheless cannot be sold in reliance on Rule 144 until one year after the date on which the issuer filed current “Form 10 information” (as defined in Rule 144(i)) with the SEC reflecting that it ceased being a shell company, and provided that at the time of a proposed sale pursuant to Rule 144, the issuer has satisfied certain reporting requirements under the Exchange Act. Because, as a former shell company, the reporting requirements of Rule 144(i) will apply regardless of holding period, the restrictive legends on certificates for the shares of common stock issued to the investors in the Private Placement Offering in exchange for the 22nd Century limited liability company membership interests included in the Units sold in the Private Placement Offering or issued upon exercise of the warrants cannot be removed except in connection with an actual sale that is subject to an effective registration statement under, or an applicable exemption from the registration requirements of, the Securities Act.
Because the Merger was a reverse merger, we may not be able to attract the attention of major brokerage firms if we seek to raise additional capital in the future.
Additional risks may exist since the Merger was a “reverse merger.” Certain SEC rules are more restrictive when applied to reverse merger companies, such as the ability of stockholders to resell their shares of common stock pursuant to Rule 144. In addition, securities analysts of major brokerage firms may not provide coverage of our common stock following the Merger since there may be little incentive for brokerage firms to recommend the purchase of our common stock. We cannot assure you that brokerage firms will want to conduct any secondary offerings on our behalf if we seek to raise additional capital in the future.
We will incur increased costs and demands upon management as a result of complying with the laws and regulations affecting public companies, which could harm our operating results.
As a public company, we will continue to incur significant legal, accounting and other expenses, including costs associated with public company reporting requirements. We will also incur substantial expenses in connection with the preparation and filing of the registration statement and responding to SEC comments in connection with its review of the registration statement. We also incur costs associated with current corporate governance requirements, including requirements under Section 404 and other provisions of the Sarbanes-Oxley Act, as well as rules implemented by the SEC and the OTC Bulletin Board or any stock exchange on which our common stock may be listed in the future. The expenses incurred by public companies for reporting and corporate governance purposes have increased dramatically in recent years and are likely to continue to increase. We expect these rules to substantially increase our legal and financial compliance costs and to make some activities more time-consuming and costly. We estimate that the cost we will incur to comply with laws and regulations affecting public companies will be approximately $300,000 on an annual basis.
23
If we fail to maintain proper and effective internal controls, our ability to produce accurate and timely financial statements could be impaired, which could harm our operating results, our ability to operate our business and investors’ views of us.
Ensuring that we have adequate internal financial and accounting controls and procedures in place so that we can produce accurate financial statements on a timely basis is a costly and time-consuming effort that will need to be evaluated frequently. Section 404 of the Sarbanes-Oxley Act requires public companies to conduct an annual review and evaluation of their internal controls and attestations of the effectiveness of internal controls by independent auditors. Our failure to maintain the effectiveness of our internal controls in accordance with the requirements of the Sarbanes-Oxley Act could have a material adverse effect on our business. We could lose investor confidence in the accuracy and completeness of our financial reports, which could have an adverse effect on the price of our common stock. In addition, if our efforts to comply with new or changed laws, regulations, and standards differ from the activities intended by regulatory or governing bodies due to ambiguities related to practice, regulatory authorities may initiate legal proceedings against us and our business may be harmed.
An active trading market for our common stock may not develop or be sustained, and you may not be able to resell your shares at or above the price at which you purchased them.
An active trading market for our shares may never develop or be sustained. In the absence of an active trading market for our common stock, shares of common stock may not be able to be resold at or above the purchase price of such shares. Although there can be no assurances, we expect that our common stock will continue to be quoted on the OTC Bulletin Board, an over-the-counter quotation system, on which the shares of our common stock are currently quoted. However, even if our common stock continues to be quoted on the OTC Bulletin Board, it is unlikely that an active market for our common stock will develop in the foreseeable future. It may be more difficult to dispose of shares or obtain accurate quotations as to the market value of our common stock compared to securities of companies whose shares are traded on the NASDAQ or other stock exchanges.
Trading in our common stock is currently limited and our stock price may be highly volatile and could decline in value.
The number of shares of common stock and warrants issued as a result of the Merger bears no relationship to our assets, book value or historical results of operations or any other established criterion of value on a stand alone or pro forma combined basis with 22nd Century, or the trading price of the shares of our common stock prior to the Merger, and may not bear any continuing relationship to the trading price of our common stock following the Merger.
In addition, our common stock is currently traded on the OTC Bulletin Board, and, therefore, the trading volume is currently more limited and sporadic than if our common stock were traded on a national stock exchange such as the NASDAQ Stock Market or the NYSE. Further, the market prices for securities in general have been highly volatile and may continue to be highly volatile in the future. The following factors, in addition to other risk factors described in this section, may have a significant impact on the market price of our common stock:
|
|
·
|
results from and any delays in any clinical trials programs;
|
|
|
·
|
failure or delays in entering potential products into clinical trials;
|
|
|
·
|
failure or discontinuation of any of our research programs;
|
|
|
·
|
delays in establishing new strategic relationships;
|
|
|
·
|
delays in the development of our potential products and commercialization of our potential products;
|
|
·
|
market conditions in our sector and issuance of new or changed securities analysts’ reports or recommendations;
|
|
|
·
|
general economic conditions, including recent adverse changes in the global financial markets;
|
|
|
·
|
actual and anticipated fluctuations in our quarterly financial and operating results;
|
|
|
·
|
developments or disputes concerning our intellectual property or other proprietary rights;
|
|
|
·
|
introduction of technological innovations or new commercial products by us or our competitors;
|
|
|
·
|
issues in manufacturing or distributing our products or potential products;
|
|
|
·
|
market acceptance of our products or potential products;
|
|
|
·
|
third-party healthcare reimbursement policies;
|
|
|
·
|
FDA or other United States or foreign regulatory actions affecting us or our industry;
|
|
|
·
|
litigation or public concern about the safety of our products or potential products;
|
|
|
·
|
additions or departures of key personnel;
|
24
|
|
·
|
third-party sales of large blocks of our common stock;
|
|
|
·
|
sales of our common stock by our executive officers, directors or significant stockholders; and
|
|
·
|
equity sales by us of our common stock or securities convertible into common stock to fund our operations.
|
These and other external factors may cause the market price and demand for our common stock to fluctuate substantially, which may limit or prevent investors from readily selling their shares of common stock and may otherwise negatively affect the liquidity of our common stock. In addition, in the past, when the market price of a stock has been volatile, holders of that stock have instituted securities class action litigation against the company that issued the stock. If any of our stockholders brought a lawsuit against us, we could incur substantial costs defending the lawsuit. Such a lawsuit could also divert the time and attention of our management.
A significant portion of the total outstanding shares of common stock may be sold into the public market in the near future, which could cause the market price of our common stock to drop significantly, even if our business is doing well.
Sales of a substantial number of shares of our common stock in the public market could occur at any time. These sales, or the perception in the market that the holders of a large number of shares intend to sell shares, could reduce the market price of our common stock.
We currently have outstanding 27,909,646 shares of common stock, of which 5,434,446 are included in the registration statement of which this prospectus is a part and of which 5,317,920 shares can be freely sold in the public market after the effective date of the registration statement. Of the 27,909,646 shares of common stock outstanding, unless they are registered for resale pursuant to the registration statement, or included in a subsequent registration statement declared effective by the SEC, 22,584,446 shares are restricted shares owned by “affiliates” and by “non-affiliates” who have held such shares for less than one year (including investors in the Private Placement Offering), which could be sold after meeting certain requirements of Rule 144 of the Securities Act. Shares of common stock held by “non-affiliates” (including investors in the Private Placement Offering) may be resold after such shares have been held for longer than one year, without meeting such requirements. In addition, 16,074,903 of such restricted shares held by our directors, executive officers, and beneficial owners of 10% of more of our issued and outstanding common stock are subject to lock-up agreements preventing the re-sale of such shares for 18 months following the date of the closing of the Merger, except to another individual or entity that is subject to a similar lock-up agreement. These lock-up agreements do not apply to the 422,544 shares of common stock or warrants to purchase 211,272 shares of common stock issued to Clearwater Partners, LLC and Angelo Tomasello upon consummation of the Merger in exchange for the securities contained in the PPO Securities (5,434,446 equity securities of the Private Placement Offering) purchased by Clearwater Partners, LLC and Angelo Tomasello in the Private Placement Offering nor to any shares of common stock issued to Clearwater Partners, LLC or Angelo Tomasello upon the exercise of such warrants.
On March 31, 2011, we registered all of the shares of common stock that we may issue under the 2010 Equity Incentive Plan or EIP, including 4,250,000 shares reserved for future issuance under such plan. On April 1, 2011, the Board granted an aggregate of 1,150,000 shares of our common stock to our officers and directors and options to purchase an aggregate of 35,000 shares of our common stock to our employees under the EIP. These shares can be freely sold in the public market upon issuance, subject to the lock-up agreements of certain recipients.
Our common stock is a “penny stock,” which is likely to limit its liquidity.
The market price of our common stock is, and will likely remain for the foreseeable future, less than $5.00 per share, and therefore will be a “penny stock” according to SEC rules, unless our common stock is listed on a national securities exchange. The OTC Bulletin Board is not a national securities exchange. Designation as a “penny stock” requires any broker or dealer selling these securities to disclose certain information concerning the transaction, obtain a written agreement from the purchaser and determine that the purchaser is reasonably suitable to purchase the securities. These rules may restrict the ability of brokers or dealers to sell our common stock and may affect the ability of current holders of our common stock to sell their shares. Such rules may also deter broker-dealers from recommending or selling our common stock, which may further limit its liquidity. This may also make it more difficult for us to raise additional capital in the future. Accordingly, although we will undertake to register under the Securities Act the resale of the shares of common stock issued in the Merger, these shares will be highly illiquid. Because of such expected illiquidity, it will likely be difficult to re-sell shares of our common stock as desired.
25
If securities or industry analysts do not publish or cease publishing research or reports about us, our business or our market, or if they change their recommendations regarding our stock adversely, our stock price and trading volume could decline.
No securities or industry analysts currently publish research or reports about us. The trading market for our common stock will be influenced by the research and reports that industry or securities analysts may publish about us, our business, our market or our competitors. If any of the analysts who may cover us in the future change their recommendation regarding our stock adversely, or provide more favorable relative recommendations about our competitors, our stock price would likely decline. If any analyst who may cover us were to cease coverage of us or fail to regularly publish reports on us, we could lose visibility in the financial markets, which in turn could cause our stock price or trading volume to decline.
We are controlled by our current officers, directors and principal stockholders.
As of July 15, 2011, our directors and executive officers beneficially owned approximately 33.8% of the shares of our common stock actually issued and outstanding as of such date, not including securities exercisable or convertible within sixty days of such date. Accordingly, our directors and executive officers will have substantial influence over, and may have the ability to control, the election of our board of directors and the outcome of issues submitted to a vote of our stockholders.
We do not expect to declare any dividends in the foreseeable future.
We have not paid cash dividends to date. We currently intend to retain our future earnings, if any, to fund the development and growth of our business, and we do not anticipate paying any cash dividends on our capital stock for the foreseeable future. In addition, the terms of any future debt facilities may preclude us from paying dividends on the common stock. As a result, capital appreciation, if any, of our common stock could be the sole source of gain for the foreseeable future.
Anti-takeover provisions contained in our articles of incorporation and bylaws, as well as provisions of Nevada law, could impair a takeover attempt.
Our amended and restated articles of incorporation and bylaws currently contain provisions that, together with Nevada law, could have the effect of rendering more difficult or discouraging an acquisition deemed undesirable by our board of directors. Our corporate governance documents presently include the following provisions:
|
·
|
authorizing blank check preferred stock, which could be issued with voting, liquidation, dividend and other rights superior to our common stock; and
|
|
|
·
|
limiting the liability of, and providing indemnification to, our directors and officers
|
These provisions, alone or together, could delay hostile takeovers and changes in control of us or changes in our management.
As a Nevada corporation, we also may become subject to the provisions Nevada Revised Statutes Sections 78.378 through 78.3793, which prohibit an acquirer, under certain circumstances, from voting shares of a corporation’s stock after crossing specific threshold ownership percentages, unless the acquirer obtains the approval of the stockholders of the issuer corporation. The first such threshold is the acquisition of at least one-fifth, but less than one-third of the outstanding voting power of the issuer. We may become subject to the above referenced Statutes if we have 200 or more stockholders of record, at least 100 of whom are residents of the State of Nevada, and do business in the State of Nevada directly or through an affiliated corporation.
26
As a Nevada corporation, we are subject to the provisions of Nevada Revised Statutes Sections 78.411 through 78.444, which prohibit an “interested stockholder” from entering into a combination with the corporation, unless certain conditions are met. An “interested stockholder” is a person who, together with affiliates and associates, beneficially owns (or within the prior three years did own) 10 percent or more the corporation’s voting stock.
Any provision of our amended and restated articles of incorporation, our bylaws or Nevada law that has the effect of delaying or deterring a change in control of our Company could limit the opportunity for our stockholders to receive a premium for their shares of our common stock, and could also affect the price that some investors are willing to pay for our common stock.
27
The following table sets forth information regarding the beneficial ownership of our common stock as of July 15, 2011, except as noted below, by:
|
|
·
|
each of our directors;
|
|
|
·
|
each of our Named Executive Officers;
|
|
|
·
|
each person, or group of affiliated persons, who is known by us to beneficially own more than 5% of our common stock;
|
|
|
·
|
all of our directors and executive officers as a group; and
|
|
|
·
|
each selling stockholder.
|
Beneficial ownership is determined in accordance with rules of the SEC. These rules generally attribute beneficial ownership of securities to persons who possess sole or shared voting power or investment power with respect to those securities and include shares of common stock issuable upon exercise of warrants that are immediately exercisable or exercisable within 60 days of July 15, 2011. Except as otherwise indicated in the footnotes to the table below, all of the shares reflected in the table are shares of common stock and all persons listed below have sole voting and investment power with respect to the shares beneficially owned by them, subject to applicable community property laws. The information is not necessarily indicative of beneficial ownership for any other purpose.
Percentage ownership calculations for beneficial ownership prior to this offering are based on 27,909,646 shares of common stock outstanding as of July 15, 2011. Beneficial ownership calculations for after the offering assume that the selling stockholder disposes of all shares of common stock covered by this registration statement and does not acquire or dispose of any additional shares of common stock. The selling stockholder is not, representing, however, that any of the shares covered by this registration statement will be offered for sale, and the selling stockholder reserves the right to accept or reject, in whole or in part, any proposed sale of shares.
In computing the number of shares of common stock beneficially owned by a person and the percentage ownership of that person, we deemed outstanding shares of common stock subject to options held by that person that are immediately exercisable or exercisable within 60 days of July 15, 2011. We did not deem these shares outstanding, however, for the purpose of computing the percentage ownership of any other person. Beneficial ownership representing less than 1% is denoted with an asterisk (*). Except as otherwise indicated in the footnotes to the table below, the address of named beneficial owners that are officers, directors or owners of 5% or more of our common stock is: c/o 22nd Century Group, Inc., 8201 Main Street, Suite 6, Williamsville, New York 14221.
28
SELLING STOCKHOLDERS’ TABLE
|
Shares Beneficially Owned Prior to Offering
|
Shares Beneficially Owned After Offering
|
|||||||||||||||||||||||
|
Name of Beneficial Owner
|
Number of Shares Held
|
Shares Issued Under 2010 Equity Incentive Plan (“EIP”)
|
Percent
|
Shares
Offered
|
Number
|
Percent
|
||||||||||||||||||
|
Management & Directors
|
||||||||||||||||||||||||
|
Joseph Pandolfino (1)
|
6,010,396 | 90,000 | 20.78 | % | - | 6,100,396 | 20.78 | % | ||||||||||||||||
|
Henry Sicignano, III (2)
|
3,757,427 | 700,000 | 15.49 | % | - | 4,457,427 | 15.49 | % | ||||||||||||||||
|
Michael R. Moynihan, Ph.D. (3)
|
1,017,645 | 100,000 | 3.97 | % | - | 1,117,645 | 3.97 | % | ||||||||||||||||
|
C. Anthony Rider (4)
|
243,473 | 75,000 | 1.14 | % | - | 318,473 | 1.14 | % | ||||||||||||||||
|
Joseph A. Dunn, Ph.D. (5)
|
- | 25,000 | * | - | 25,000 | * | ||||||||||||||||||
|
James W. Cornell (5)
|
- | 25,000 | * | - | 25,000 | * | ||||||||||||||||||
|
Steven Katz (5)
|
- | 25,000 | * | - | 25,000 | * | ||||||||||||||||||
|
All directors and executive officers as a group (7 persons) (1)-(5)
|
11,028,941 | 1,040,000 | 39.54 | % | - | 12,068,941 | 39.54 | % | ||||||||||||||||
|
Other 5% Owners
|
||||||||||||||||||||||||
|
Clearwater Partners, LLC (6)
|
5,144,279 | - | 17.65 | % | 97,544 | 5,046,735 | 17.31 | % | ||||||||||||||||
|
Angelo J. Tomasello (7)
|
4,193,881 | - | 14.48 | % | 325,000 | 3,868,881 | 13.36 | % | ||||||||||||||||
|
Centrum Bank AG (8)
|
1,500,000 | - | 5.28 | % | 1,000,000 | 500,000 | 1.76 | % | ||||||||||||||||
|
Rodman & Renshaw LLC (9)
|
1,487,819 | - | 5.13 | % | 395,376 | 1,092,443 | 3.58 | % | ||||||||||||||||
|
Other Selling Stockholders
|
||||||||||||||||||||||||
|
Joseph Anderson (10)
|
955,417 | - | 3.39 | % | 150,000 | 805,417 | 2.86 | % | ||||||||||||||||
|
Mark Tompkins (11)
|
750,000 | - | 2.64 | % | 500,000 | 250,000 | * | |||||||||||||||||
|
Paul Tompkins (12)
|
750,000 | - | 2.64 | % | 500,000 | 250,000 | * | |||||||||||||||||
|
Fors Family Limited Partnership (13)
|
375,000 | - | 1.34 | % | 250,000 | 125,000 | * | |||||||||||||||||
|
Gibralt US, Inc. (14)
|
375,000 | - | 1.34 | % | 250,000 | 125,000 | * | |||||||||||||||||
|
Cranshire Capital, L.P. (15)
|
300,000 | - | 1.07 | % | 200,000 | 100,000 | * | |||||||||||||||||
|
Adrien Ellul (16)
|
300,000 | - | 1.07 | % | 200,000 | 100,000 | * | |||||||||||||||||
|
Rosalind Master Fund L.P. (17)
|
270,000 | - | * | 180,000 | 90,000 | * | ||||||||||||||||||
|
Rosalind Capital Partners L.P. (18)
|
270,000 | - | * | 180,000 | 90,000 | * | ||||||||||||||||||
|
Robert C. Torch (19)
|
225,000 | - | * | 150,000 | 75,000 | * | ||||||||||||||||||
|
Dennis Chugh (20)
|
150,000 | - | * | 100,000 | 50,000 | * | ||||||||||||||||||
|
Nicholas N. Branca (21)
|
150,000 | - | * | 100,000 | 50,000 | * | ||||||||||||||||||
|
Craig T. Schorr (22)
|
150,000 | - | * | 100,000 | 50,000 | * | ||||||||||||||||||
|
Nadine Smith (23)
|
309,756 | - | 1.11 | % | 100,000 | 209,756 | * | |||||||||||||||||
|
John D. Long, Jr. (24)
|
150,000 | - | * | 100,000 | 50,000 | * | ||||||||||||||||||
|
Kingsbrook Opportunities Master Fund LP (25)
|
150,000 | - | * | 100,000 | 50,000 | * | ||||||||||||||||||
|
Iroquois Master Fund, Ltd. (26)
|
150,000 | - | * | 100,000 | 50,000 | * | ||||||||||||||||||
|
Peter & Ruth G. Julian Living Trust (27)
|
75,000 | - | * | 50,000 | 25,000 | * | ||||||||||||||||||
|
Songin CPAs PLLC Profit Sharing Plan (28)
|
75,000 | - | * | 50,000 | 25,000 | * | ||||||||||||||||||
|
Kanski Holdings Limited Partnership, LLLP (29)
|
75,000 | - | * | 50,000 | 25,000 | * | ||||||||||||||||||
|
Cojack Investment Opportunities, LLC (30)
|
37,500 | - | * | 25,000 | 12,500 | * | ||||||||||||||||||
|
Nora B. Sullivan (31)
|
37,500 | - | * | 25,000 | 12,500 | * | ||||||||||||||||||
|
Michael K. Landi (32)
|
37,500 | - | * | 25,000 | 12,500 | * | ||||||||||||||||||
|
Ann L. Hautamaki and Raymond Dale Hautamaki (33)
|
22,500 | - | * | 15,000 | 7,500 | * | ||||||||||||||||||
(1) Includes 1,441,761 shares of common stock issuable upon exercise of warrants. All of the shares owned by Mr. Pandolfino are subject to restrictions on transfer or sale as contained in a lock-up agreement with the Company which expires on July 25, 2012.
(2) Consists of 332,603 shares of common stock held by Henry Sicignano III, 2,542,347 shares of common stock held by Henry Sicignano III Group, LLC, 69,564 shares of common stock issuable to Mr. Sicignano upon exercise of warrants, and 800,413 shares of common stock issuable to Henry Sicignano III Group, LLC upon exercise of warrants. Mr. Sicignano is Managing Member of Henry Sicignano III Group, LLC and, accordingly, exercises voting and investment power with respect to the shares held by Henry Sicignano III Group, LLC. The 31,626 shares of common stock being sold in this offering are held in the name of Henry Sicignano III Group, LLC. Mr. Sicignano’s 600,000 shares issued as equity incentive awards under the Company’s EIP are time-based awards subject to vesting over the next 4 years on April 1 of 2012 to 2015, such that 150,000 shares shall vest on April 1 of each such year. Mr. Sicignano also holds 100,000 performance based shares of restricted stock issued as equity incentive awards under the Company’s EIP, which are subject to forfeiture unless certain performance milestones are achieved. All of the shares owned and/or controlled by Mr. Sicignano are subject to restrictions on transfer or sale as contained in a lock-up agreement with the Company which expires on July 25, 2012.
29
(3) Includes 243,711 share of common stock issuable upon exercise of warrants. All of the shares owned by Dr. Moynihan are subject to restrictions on transfer or sale as contained in a lock-up agreement with the Company which expires on July 25, 2012.
(4) Includes 57,970 share of common stock issuable upon exercise of warrants. All of the shares owned by Mr. Rider are subject to restrictions on transfer or sale as contained in a lock-up agreement with the Company which expires on July 25, 2012.
(5) All of the shares owned by this holder are subject to restrictions on transfer or sale as contained in a lock-up agreement with the Company which expires on July 25, 2012.
(6) Includes 1,238,764 shares of common stock issuable upon exercise of warrants. Richard G. Saffire, Managing Member of Clearwater Partners, LLC exercises voting and investment power with respect to shares owned by Clearwater Partners, LLC. The address of Clearwater Partners, LLC is 34 Sunburst Circle, East Amherst, New York 14051. All of the shares owned by Clearwater Partners, LLC are subject to restrictions on transfer or sale as contained in a lock-up agreement with the Company which expires on July 25, 2012, except that 97,544 of such shares are not subject to restrictions on transfer or sale pursuant to such lock-up agreement.
(7) Includes 1,044,972 shares of common stock issuable upon exercise of warrants. The address of Angelo Tomasello is 4720 Spaulding Drive, Clarence, New York 14031. All of the shares owned by Mr. Tomasello are subject to restrictions on transfer or sale as contained in a lock-up agreement with the Company which expires on July 25, 2012, except that 325,000 of such shares are not subject to restrictions on transfer or sale pursuant to such lock-up agreement.
(8) Includes 500,000 shares of common stock issuable upon exercise of warrants. Juerg Muehlethaler and Alessandra Waibel, Executive Director and Authorized Officer, respectively, of Centrum Bank AG exercise voting and investment power with respect to shares owned by Centrum Bank AG. The address of Centrum Bank AG is Kirchstrasse 3, Postfach 1168, 9490 Vaduz Liechtenstein.
(9) Includes 1,003,623 shares of common stock issuable upon exercise of warrants. Rodman & Renshaw, LLC is a registered broker-dealer that acted as placement agent to the Company in connection with the Private Placement Offering and continues to provide financial advisory services to the Company. Thomas Pinou, Controller of Rodman & Renshaw, LLC, exercises voting and investment power with respect to shares owned by Rodman & Renshaw, LLC.
(10) Includes 248,909 shares of common stock issuable upon exercise of warrants.
(11) Includes 250,000 shares of common stock issuable upon exercise of warrants.
(12) Includes 250,000 shares of common stock issuable upon exercise of warrants.
(13) Includes 125,000 shares of common stock issuable upon exercise of warrants. Richard D. Fors, III and Patricia Fors, each a general partner of Fors Family Limited Partnership possess shared voting and investment power with respect to shares owned by Fors Family Limited Partnership.
(14) Includes 125,000 shares of common stock issuable upon exercise of warrants.
(15) Includes 100,000 shares of common stock issuable upon exercise of warrants. Downsview Capital, Inc. is the general partner of Cranshire Capital, L.P. Mitchell P. Kopin is President of Downsview Capital, Inc. Mr. Kopin possesses voting and investment power with respect to shares owned by Cranshire Capital, L.P.
(16) Includes 100,000 shares of common stock issuable upon exercise of warrants.
(17) Includes 90,000 shares of common stock issuable upon exercise of warrants. Rosalind Advisors, Inc. is adviser to Rosalind Master Fund L.P. and Steven Salamon is President of Rosalind Advisors, Inc. Mr. Salamon possesses voting and investment power with respect to shares owned by Rosalind Master Fund L.P.
(18) Includes 90,000 shares of common stock issuable upon exercise of warrants. Rosalind Advisors, Inc. is adviser to Rosalind Capital Partners L.P. and Steven Salamon is President of Rosalind Advisors, Inc. Mr. Salamon possesses voting and investment power with respect to shares owned by Rosalind Capital Partners L.P.
(19) Includes 75,000 shares of common stock issuable upon exercise of warrants.
(20) Includes 50,000 shares of common stock issuable upon exercise of warrants.
30
(21) Includes 50,000 shares of common stock issuable upon exercise of warrants.
(22) Includes 50,000 shares of common stock issuable upon exercise of warrants.
(23) Includes 50,000 shares of common stock issuable upon exercise of warrants.
(24) Includes 50,000 shares of common stock issuable upon exercise of warrants.
(25) Includes 50,000 shares of common stock issuable upon exercise of warrants. Kingsbrook Partners LP is the investment manager of Kingsbrook Opportunities Master Fund LP. Kingsbrook Opportunities GP LLC is the General Partner of Kingsbrook Opportunities Master Fund LP. KB GP LLC is the General Partner if Kingsbrook Partners LP. Ari J. Storch, Adam J. Chill, and Scott M. Wallace are the sole Managing Members of Kingsbrook Opportunities GP LLC and KB GP LLC. Messers. Storch, Chill, and Wallace possess voting and investment power with respect to shares owned by Kingsbrook Opportunities Master Fund LP. Each of Kingsbrook Partners LP, Kingsbrook Opportunities GP LLC, KB GP LLC and Messrs. Stroch, Chill, and Wallace disclaim beneficial ownership of the shares owned by Kingsbrook Opportunities Master Fund LP.
(26) Includes 50,000 shares of common stock issuable upon exercise of warrants. Iroquois Capital Management L.L.C. is the investment manager of Iroquois Master Fund, Ltd. Joshua Silverman and Richard Abbe are managing members of Iroquois Capital Management L.L.C. Messers. Silverman and Abbe possess voting and investment power with respect to shares owned by Iroquois Master Fund, Ltd. Each of Messers. Silverman and Abbe disclaim beneficial ownership of the shares owned by Iroquois Master Fund, Ltd.
(27) Includes 25,000 shares of common stock issuable upon exercise of warrants. Ruth G. Julian is trustee of the Peter & Ruth G. Julian Living Trust. Mrs. Julian possesses voting and investment power with respect to shares owned by the Peter & Ruth G. Julian Living Trust.
(28) Includes 25,000 shares of common stock issuable upon exercise of warrants. Songin & Company, CPAs, PLLC Retirement Trust holds, investments and administers funds contributed under the Songin CPAs PLLC Profit Sharing Plan. Michael J. Songin is Trustee of Songin & Company, CPAs, PLLC Retirement Trust. Mr. Songin possesses voting and investment power with respect to shares owned by Songin CPAs PLLC Profit Sharing Plan.
(29) Includes 25,000 shares of common stock issuable upon exercise of warrants. Jeffery W. Kanski and Dr. James R. Kanski, Trustee of the Dr. James R. Kanski Irrevocable Trust under Agreement dated June 6, 2005, each a General Partner of Kanski Holdings Limited Partnership, LLLP, possess voting and investment power with respect to shares owned by Kanski Holdings Limited Partnership, LLLP.
(30) Includes 12,500 shares of common stock issuable upon exercise of warrants. Raymond Dean Hautamaki, Managing Member of Cojack Investment Opportunities LLC possess voting and investment power with respect to shares owned by Cojack Investment Opportunities LLC.
(31) Includes 12,500 shares of common stock issuable upon exercise of warrants.
(32) Includes 12,500 shares of common stock issuable upon exercise of warrants.
(33) Includes 7,500 shares of common stock issuable upon exercise of warrants.
31
USE OF PROCEEDS
We will not receive proceeds from the sale of common stock under this prospectus. We have agreed to bear the expenses (other than any underwriting discounts or commissions or agent’s commissions) in connection with the registration of the common stock being offered hereby by the selling stockholders.
DIVIDEND POLICY
We have not previously and do not plan to declare or pay any dividends on our common stock. Our current policy is to retain all funds and any earnings for use in the operation and expansion of our business. Payment of future dividends, if any, will be at the discretion of our board of directors after taking into account various factors, including current financial condition, operating results and current and anticipated cash needs.
DETERMINATION OF OFFERING PRICE
All shares of our common stock being offered will be sold by the selling stockholders without our involvement. As a result, the selling stockholders will determine at what price they may sell the offered shares, and these sales may be made at prevailing market prices or at privately negotiated prices.
Our common stock is quoted on the OTC Bulletin Board under the symbol “XXII.OB.” On November 23, 2010, we changed the name of our Company to 22nd Century Group, Inc. and on December 3, 2010 our symbol changed from “THSM.OB” to “XXII.OB.” As of July 15, 2011, there were 27,909,646 shares of our common stock issued and outstanding and 8,686,980 shares issuable upon exercise of outstanding stock options and warrants. On that date, there were approximately 41 holders of record of shares of our common stock.
The trading market for our common stock has been extremely limited and sporadic. As of July 15, 2011, the last reported sale price of our shares on the OTC Bulletin Board was $1.25 per share. For the periods indicated, the following table sets forth the high and low bid prices per share of our common stock, as derived from quotations provided by the OTC Bulletin Board Information Center.
|
Quarter Ended
|
High Bid
|
Low Bid
|
||||||
|
June 30, 2011
|
$ |
1.30
|
$ |
1.10
|
||||
|
March 31, 2011
|
$ | 1.41 | $ | 0.005 | ||||
|
December 31, 2010
|
$ | 0.005 | $ | 0.005 | ||||
|
September 30, 2010
|
$ | 0.005 | $ | 0.005 | ||||
|
June 30, 2010
|
$ | 0.007 | $ | 0.005 | ||||
|
March 31, 2010
|
$ | 0.007 | $ | 0.005 | ||||
|
December 31, 2009
|
$ | 0.005 | $ | 0.005 | ||||
|
September 30, 2009
|
$ | 0.005 | $ | 0.005 | ||||
|
June 30, 2009
|
$ | 0.005 | $ | 0.005 | ||||
Trades in our common stock may be subject to Rule 15g-9 of the Exchange Act, which imposes requirements on broker/dealers who sell securities subject to the rule to persons other than established customers and accredited investors. For transactions covered by the rule, broker/dealers must make a special suitability determination for purchasers of the securities and receive the purchaser’s written agreement to the transaction before the sale.
The SEC also has rules that regulate broker/dealer practices in connection with transactions in “penny stocks.” Penny stocks generally are equity securities with a price of less than $5.00 (other than securities listed on some national exchanges, provided that the current price and volume information with respect to transactions in that security is provided by the applicable exchange or system). The penny stock rules require a broker/dealer, before effecting a transaction in a penny stock not otherwise exempt from the rules, to deliver a standardized risk disclosure document prepared by the SEC that provides information about penny stocks and the nature and level of risks in the penny stock market. The broker/dealer also must provide the customer with current bid and offer quotations for the penny stock, the compensation of the broker/dealer and its salesperson in the transaction, and monthly account statements showing the market value of each penny stock held in the customer’s account. The bid and offer quotations, and the broker/dealer and salesperson compensation information, must be given to the customer orally or in writing before effecting the transaction, and must be given to the customer in writing before or with the customer’s confirmation. These disclosure requirements may have the effect of reducing the level of trading activity in the secondary market for shares of common stock. As a result of these rules, investors may find it difficult to sell their shares.
32
SECURITIES AUTHORIZED FOR ISSUANCE UNDER OUR EQUITY INCENTIVE PLAN
On October 21, 2010, we established our 2010 Equity Incentive Plan (“EIP”) for officers, employees, directors, consultants and advisors to the Company and its affiliates, consisting of 4,250,000 shares of common stock. The EIP has a term of ten (10) years and is administered by our Board or a committee to be established by our Board (the “Administrator”), to determine the various types of incentive awards that may be granted to recipients under this plan and the number of shares of common stock to underlie each such award under the EIP. The EIP also contains a provision which restricts the plan to granting awards relating to no more than 1,600,000 shares of common stock during the first twelve (12) months following the effective date of the Merger or January 25, 2011. On April 1, 2011, under our EIP, the Board granted an aggregate of 1,150,000 shares of our common stock to our officers and directors and options to purchase an aggregate of 35,000 shares of our common stock to our employees.
The purpose of the EIP is to attract and retain the best available personnel for positions of substantial responsibility, to provide incentives to individuals who perform services for the Company, and to promote the success of the Company’s business.
If an incentive award granted under the EIP expires, terminates, is unexercised or is forfeited, or if any shares are surrendered to us in connection with an incentive award, the shares subject to such award and the surrendered shares will become available for further awards under the EIP.
The number of shares of our common stock subject to the EIP, any number of shares subject to any numerical limit in the EIP, and the number of shares and terms of any incentive award will be adjusted in the event of any change in our outstanding common stock by reason of any stock dividend, spin-off, split-up, stock split, reverse stock split, recapitalization, reclassification, merger, consolidation, liquidation, business combination or exchange of shares or similar transaction.
The EIP authorizes the grant of incentive stock options, nonqualified stock options, stock appreciation rights, restricted stock, restricted stock units, performance shares, restricted stock or restricted stock units.
Stock Options. Stock options entitle the participant, upon exercise, to purchase a specified number of shares of the Company’s common stock at a specified price and for a specified period of time. The exercise price for each stock option shall be determined by the Administrator but shall not be less than 100% of the fair market value of the Company’s common stock on the date of grant. The “fair market value” means the value of the common stock as the Administrator may determine in good faith, by reference to the closing price of such stock on any established stock exchange or national market system on which the Company’s common stock is listed on the day of determination, or if the Company’s common stock is not so listed, the value of such stock as may be determined by the Administrator in good faith.
Any stock options granted in the form of an incentive stock option will be intended to comply with the requirements of Section 422 of the Internal Revenue Code of 1986, as amended. Only options granted to employees qualify for incentive stock option treatment.
Each stock option shall expire at such time as the Administrator shall determine at the time of grant. No stock option shall be exercisable later than the tenth anniversary of its grant. A stock option may be exercised in whole or in installments. A stock option may not be exercisable for a fraction of a share. Shares of the Company’s common stock purchased upon the exercise of a stock option must be paid for in full at the time of exercise in cash or such other consideration determined by the Administrator.
Stock Appreciation Rights. A stock appreciation right (“SAR”) is the right to receive a payment equal to the excess of the fair market value of a specified number of shares of common stock on the date the SAR is exercised over the exercise price of the SAR. The exercise price for each SAR shall not be less than 100% of the fair market value of the Company’s common stock on the date of grant, and the term shall be no more than ten years from the date of grant. At the discretion of the Administrator, the payment upon an SAR exercise may be in cash, in shares equivalent thereof, or in some combination thereof.
33
Upon exercise of an SAR, the participant shall be entitled to receive payment from the Company in an amount determined by multiplying the excess of the fair market value of a share of the Company’s common stock on the date of exercise over the exercise price of the SAR by the number of shares with respect to which the SAR is exercised. The payment may be made in cash or stock, at the discretion of the Administrator.
Restricted Stock and Restricted Stock Units. Restricted stock and restricted stock units may be awarded or sold to participants under such terms and conditions as shall be established by the Administrator. Restricted stock and restricted stock units shall be subject to such restrictions as the Administrator determines, including a prohibition against sale, assignment, transfer, pledge or hypothecation, and a requirement that the participant forfeit such shares or units in the event of termination of employment. A restricted stock unit provides a participant the right to receive payment at a future date after the lapse of restrictions or achievement of performance criteria or other conditions determined by the Administrator.
Performance Stock. The Administrator shall designate the participants to whom long-term performance stock are to be awarded and determine the number of shares, the length of the performance period and the other vesting terms and conditions of each such award; provided the stated performance period will not be less than twelve (12) months. Each award of performance stock shall entitle the participant to a payment in the form of shares of common stock of the Company upon the attainment of performance goals and other vesting terms and conditions specified by the Administrator. The Administrator may, in its discretion, make a cash payment equal to the fair market value of shares of common stock otherwise required to be issued to a participant pursuant to a Performance Stock Award.
All awards made under the EIP may be subject to vesting and other contingencies as determined by the Administrator and will be evidenced by agreements approved by the Administrator which set forth the terms and conditions of each award.
34
BUSINESS
Unless the context otherwise requires, references in this Business section to the “Company,” “we,” “us” and “our” refer to 22nd Century Group, Inc., a Nevada corporation, as well as its subsidiaries 22nd Century Limited, LLC, a Delaware limited liability company, and Goodrich Tobacco Company, LLC, a Delaware limited liability company, taken as a whole, and also refer to the operations of 22nd Century Limited, LLC prior to the closing of the Merger on January 25, 2011.
Background
We were incorporated under the laws of the State of Nevada on September 12, 2005, to engage in the acquisition, exploration and development of mineral deposits and reserves. We had been in a development stage since our inception and had minimal business operations prior to the Merger. Prior to the closing of the Merger, we (i) obtained forgiveness of all our outstanding promissory notes in the aggregate principal amount of $162,327, (ii) cancelled the 386,389 shares of our common stock held by Milestone Enhanced Fund Ltd. and the 10,015,200 shares of our common stock held by Nanuk Warman, (iii) entered into contractual agreements with certain of our stockholders pursuant to which an aggregate of 139,800 shares of our common stock were cancelled following the closing of the Merger (such 139,800 shares of our common stock being deemed to be no longer issued and outstanding as of January 25, 2011) and (iv) effected a 2.782-for-one forward stock split by way of dividend and subsequent cancellation to ensure that the pre-Merger shareholders of the Company owned an aggregate of 5,325,200 shares of our common stock immediately prior to the closing of the Merger.
In addition, prior to the closing of the Merger, we transferred all of our pre-Merger operating assets and remaining liabilities to the Split-Off Subsidiary pursuant to the terms of the certain Split-Off Agreement, entered into by and among the Company, David Rector, and the Split-Off Subsidiary. Prior to the Merger and pursuant to the terms of the Split-Off Agreement, the Company transferred and sold all of the issued and outstanding shares of capital stock of the Split-Off Subsidiary to Mr. Rector in exchange for $1.00, such consideration being deemed to be adequate by our Board.
22nd Century was formed as a New York limited liability company on February 20, 1998 as 21st Century Limited, LLC, which merged with a newly-formed Delaware limited liability company, 22nd Century Limited, LLC on November 29, 1999, with 22nd Century being the surviving company. 22nd Century’s offices are located in Williamsville, New York. Since beginning operations, 22nd Century has worked to modify the content of nicotinic alkaloids in tobacco plants through genetic engineering and plant breeding.
Prior to the closing of the Merger, 22nd Century completed the Private Placement Offering of 5,434,446 PPO Securities at the purchase price of $1.00 per Unit, each such Unit consisting of one limited liability company membership interest and a five-year warrant to purchase one-half of one (1/2) limited liability company membership interest, at an exercise price of $1.50 per whole Unit.
After the Merger with the Company, we succeeded to the business of 22nd Century as our sole line of business, including the business conducted by Goodrich Tobacco Company, LLC, a 96% owned subsidiary of 22nd Century prior to the Merger.
Overview
We are a plant biotechnology company and a global leader in modifying the content of nicotinic alkaloids in tobacco plants through genetic engineering and plant breeding. We control 98 issued patents of which we own 8 issued patents and we have the exclusive license to an additional 90 issued patents in an aggregate of 79 countries where at least 75% of the world’s smokers reside. Additionally, we control 45 pending patent applications, of which we own 29 and have an exclusive license to 16. In the U.S., patents that currently cover our products expire in 2019 through 2022. Also in the U.S., patent applications that we expect will cover our products would, if patents are granted in connection with such applications, yield patents that expire in 2024 through 2028. However, there is no guarantee that patents will be granted in response to such patent applications and therefore cover our products. Our two worldwide exclusive licenses, one from North Carolina State University (“NCSU”) and the other from National Research Council of Canada, Plant Biotechnology Institute in Saskatoon, Canada (“NRC”), each involve multiple patent families, patent applications and patents. The exclusive rights under each of these two license agreements expire on the date on which the last patent covered by the subject license expires in the country or countries where such patents are in effect, which is expected to be from 2022 through 2028, respectively. We believe our proprietary technology provides us with opportunities in the global market for approved smoking cessation aids and the emerging market for modified risk tobacco products; however, we have not yet identified any specific countries or developed any timelines or cost estimates for international expansion of our business.
35
We plan to use a material portion of the proceeds of the Private Placement Offering to complete a Phase II-B clinical trial which is necessary to seek approval from the U.S. Food and Drug Administration (“FDA”) for X-22, our prescription smoking cessation aid in development. We have met with the FDA regarding the remaining clinical trials for X-22, and based on the FDA’s guidance, we plan to conduct a Phase II-B trial and two larger and concurrent Phase III trials with the same protocols. We have designated our upcoming phase II clinical trial as a “Phase II-B” clinical trial because (i) we are looking for continued indications of clinical efficacy and safety in larger groups of patients in our clinical trial, and (ii) very low nicotine (VLN) cigarettes made from the Company’s VLN tobacco have already demonstrated efficacy in previous Phase II trials, specifically the Phase II trial at the University of Minnesota which had a very similar protocol compared to our upcoming phase II clinical trial. However, “Phase II-B” is not an official FDA designation.
X-22 will be a prescription-only kit containing very low nicotine (“VLN”) cigarettes made from our proprietary tobacco, which has approximately 95% less nicotine compared to tobacco in existing “light” cigarettes. The therapy protocol allows the patient to smoke our VLN cigarettes without restriction over the six-week treatment period to facilitate the goal of the patient quitting smoking by the end of the treatment period. Although further research and clinical trials are needed to determine efficacy of X-22, we believe this therapy protocol has thus far been successful because VLN cigarettes made from our proprietary tobacco satisfy smokers’ cravings for cigarettes while (i) greatly reducing nicotine exposure and nicotine dependence and (ii) extinguishing the association between the act of smoking and the rapid delivery of nicotine. We believe X-22 will be more attractive to smokers than other therapies since it smokes and tastes like a typical cigarette, involves the same smoking behavior, and does not expose the smoker to any new drugs or new side effects (other than the exposure and side effects from smoking cigarettes in general) that may occur when a person is exposed to a new drug.
Independent studies, including two Phase II clinical trials, have demonstrated that VLN cigarettes made from our proprietary VLN tobacco are at least as effective as FDA-approved smoking cessation aids. As we describe further in the Technology Platform and Intellectual Property section of this prospectus, X-22 is a cigarette that contains our proprietary VLN tobacco. We believe X-22 will be well positioned as a smoking cessation product by motivating and encouraging more smokers to attempt to quit smoking, although there is no assurance that these results will occur.
The 2009 Family Smoking Prevention and Tobacco Control Act (“Tobacco Control Act”) granted the FDA authority over the regulation of all tobacco products. While it prohibits the FDA from banning cigarettes outright, it allows the FDA to require the reduction of nicotine or any other compound in tobacco and cigarette smoke. The Tobacco Control Act also banned all sales in the U.S. of cigarettes with flavored tobacco (other than menthol). As of June 2010, all cigarette companies were required to cease the use of the terms “low tar,” “light” and “ultra light” in describing cigarettes sold in the U.S. Besides numerous other regulations, including certain marketing restrictions, for the first time in history, a U.S. regulatory agency will scientifically evaluate cigarettes that may pose lower health risks as compared to conventional cigarettes.
The Tobacco Control Act establishes procedures for the FDA to regulate the labeling and marketing of modified risk tobacco products, which includes cigarettes that (i) reduce exposure to tobacco smoke toxins and/or (ii) pose lower health risks, as compared to conventional cigarettes (“Modified Risk Cigarettes”). The Tobacco Control Act requires the FDA to issue specific regulations and guidance regarding applications that must be submitted to the FDA for the authorization to label and market Modified Risk Cigarettes. The FDA has yet to release its regulations regarding modified risk tobacco products. Based in part on the timelines contained in the Tobacco Control Act, we expect the FDA to issue such regulations and guidance within the next year.
We believe that two of our cigarette products, which we refer to as BRAND A and BRAND B, will qualify as Modified Risk Cigarettes. Compared to commercial cigarettes evaluated by Michael J. Morton and Susan W. Laffoon in the research paper, titled, Cigarette smoke chemistry market maps under Massachusetts Department of Public Health smoking conditions (Regul Toxicol Pharmacol, 2008 Jun;51(1):1-30.), we believe that the tobacco in BRAND A has approximately 95% less nicotine than tobacco in cigarettes previously marketed as “light” cigarettes, and BRAND B’s smoke contains the lowest amount of “tar” per milligram of nicotine.
Within our two product categories, the Tobacco Control Act offers us the following specific advantages:
Smoking Cessation Aids
FDA approval must be obtained, as has been the case for decades, before a product can be marketed for quitting smoking. The Tobacco Control Act provides that products for quitting smoking or smoking cessation be considered for “Fast Track” designation by the FDA. The “Fast Track” programs of the FDA are intended to facilitate development and expedite review of drugs to treat serious and life-threatening conditions so that an approved product can reach the market expeditiously. We believe that X-22 will qualify for “Fast Track” designation by the FDA. However, there is no guarantee that X-22 will qualify for “Fast Track” designation at the FDA.
36
Modified Risk Cigarettes
We intend to seek FDA authorization to market BRAND A and BRAND B as Modified Risk Cigarettes. We believe that BRAND A and BRAND B will achieve significant market share in the global cigarette market among smokers who will not quit but are interested in reducing the harmful effects of smoking. We believe this new regulatory environment represents a paradigm shift for the tobacco industry. The Tobacco Control Act allows the FDA to mandate the use of reduced-risk technologies across all conventional tobacco products or cigarettes. We expect this to create opportunities for us to license our proprietary technology and/or tobaccos to larger competitors. There is no guarantee that Brand A or Brand B will obtain the necessary authorizations from the FDA to be classified as Modified Risk Cigarettes.
RED SUN and MAGIC Cigarettes
Our subsidiary, Goodrich Tobacco Company, LLC (f/k/a Xodus, LLC), has introduced two super-premium priced cigarette brands, RED SUN and MAGIC, into the U.S. market in the first quarter of 2011. Both brands are available in regular and menthol and all four brand styles are king size, packaged in hinge-lid hard packs. We intend to focus our marketing efforts on tobacconists, smoke shops and tobacco outlets. We believe the ban in 2009 by the FDA of all flavored cigarettes (with the exception of menthol) has resulted in a product void in these tobacco channels. Certain wholesalers and retailers are now seeking other specialty cigarettes to replace the banned flavored cigarettes. We believe that certain U.S. cigarette wholesalers and retailers will, among other reasons, purchase RED SUN and MAGIC to replace their lost sales of flavored cigarettes as well as potential lost sales of “light” and “ultra light” cigarettes, since as of June 2010, all cigarette companies were required to cease the use of the terms “low tar,” “light” and “ultra light.”
Government Research Cigarettes
The National Institute on Drug Abuse (“NIDA”), a component of the National Institutes of Health (“NIH”), provides the scientific community with controlled and uncontrolled research chemicals and drug compounds in its Drug Supply Program. In 2009, NIDA included an option to develop and produce research cigarettes with ten different levels of nicotine, including a minimal (placebo) level, or Research Cigarette Option, in its request for proposals for a five-year contract for Preparation and Distribution of Research and Drug Products. We have agreed, as a subcontractor to RTI International (“RTI”) in RTI’s contract with NIDA for the Research Cigarette Option, to supply modified nicotine cigarettes to NIDA. In August 2010, we met with officials from NIDA, FDA, RTI, the National Cancer Institute and the Centers for Disease Control and Prevention to finalize certain aspects of the design of these research cigarettes. These research cigarettes are distributed under the mark SPECTRUM.
In 2010, we received our first purchase order of $152,660 which included a design phase fee of $40,604. In March 2011, the Company received a revised purchase order and authorization to bill $112,000 for manufacture planning and material procurement for 9 million cigarettes from RTI International (“RTI”). Payment was received on May 2, 2011. The Company received an additional purchase order from RTI for approximately $680,000 in May 2011 to ship the 9 million research cigarettes. These government research cigarettes are distributed under the mark SPECTRUM. Approximately 4 million cigarettes out of the 9 million were shipped to and received by RTI on July 6, 2011. We believe that the revenue from the sale of SPECTRUM, plus direct orders of other cigarettes from researchers, will be approximately $825,000 in 2011 and $3 million over the next 5 years. As a subcontractor, we do not have a formal contractual arrangement at this time with RTI to purchase additional SPECTRUM cigarettes or with other researchers, and there is no guarantee that we will receive future orders from RTI or other researchers.
“Tar” Nicotine and Smoking Behavior
The dependence of many smokers on tobacco is largely due to the properties of nicotine, but the adverse effects of smoking on health are mainly due to other components present in tobacco smoke, including “tar” and carbon monoxide. “Tar” is the common name for the (resinous) total particulate matter minus nicotine and water produced by the burning of tobacco (or other plant material) during the act of smoking. “Tar” and nicotine are commonly measured in milligrams per cigarette trapped on a Cambridge filter pad under standardized conditions using smoking machines. These results are referred to as “yields” or, more specifically, “tar” yield and nicotine yield.
37
Individual smokers generally seek a certain amount of nicotine per cigarette and can easily adjust how intensely each cigarette is smoked to obtain a satisfactory amount of nicotine. Smoking of low yield (e.g., “light” or “ultra light”) cigarettes compared to high yield (“full flavor”) cigarettes often results in taking more puffs per cigarette, larger puffs and/or smoking more cigarettes per day to obtain a satisfactory amount of nicotine, a phenomenon known as “compensation” or “compensatory smoking.” “Full flavor” cigarettes have relatively higher “tar” and nicotine yields (from smoking machines) and taste more robust than “light” and “ultra lights.” A report by the National Cancer Institute in 2001 stated that due to compensatory smoking, low yield cigarettes are not safer than high yield cigarettes, which is the reason that the Tobacco Control Act has banned the use of the terms “low tar,” “light” and “ultra light” in the U.S. market. Studies have shown that smokers do not compensate when smoking cigarettes made with our VLN tobacco, and that smoking VLN cigarettes, such as BRAND A, actually assist smokers to smoke fewer cigarettes per day and reduce their exposure to certain tobacco smoke toxins, including nicotine. Other studies have shown that non-commercial cigarettes with low tar-to-nicotine ratios (“tar” yield divided by nicotine yield from smoking machines), such as BRAND B, result in smokers inhaling less of certain tobacco smoke toxins, including carbon monoxide (CO).
Market
Cigarettes and Smoking Cessation Aids
The U.S. cigarette market consists of approximately 46 million adult smokers who spent approximately $80 billion in 2010 on approximately 310 billion cigarettes. The World Health Organization (“WHO”) predicts that the current 1.3 billion smokers worldwide will increase to 1.7 billion smokers by the year 2025. Worldwide manufacturer sales in 2010 were over 5.0 trillion cigarettes, resulting in annual retail sales of approximately $600 billion. Our products address unmet needs of smokers; for those who want to quit, an innovative smoking cessation aid, and for those who do not quit, cigarettes that can reduce the level of exposure to tobacco toxins.
In 2009, annual sales of smoking cessation aids in the U.S., all of which must be approved by the FDA, were approximately $1.0 billion. Outside the United States, the smoking cessation market is in its infancy. Visiongain estimates the 2008 global smoking cessation market at approximately $3.0 billion.
According to The Institute of Medicine (IOM), the health arm of the National Academy of Sciences, in a 2007 report, (Ending the Tobacco Problem) approximately 43% of U.S. smokers attempt to quit smoking each year and: “There is an enormous opportunity to increase population prevalence of smoking cessation by reaching and motivating the 57 percent of smokers who currently make no quit attempt per year.” The report also concludes that approximately 95% of “self-quitters” (i.e., those who attempt to quit smoking without any treatment) relapse and resume smoking. According to the 2010 report of the U.S. Surgeon General on smoking, each year 5% or less of U.S. smokers achieve long-lasting abstinence. According to the U.S. Centers for Disease Control and Prevention, it takes smokers an average of 8 to 11 “quit attempts” before achieving long-term success, and according to a 2006 research paper entitled, “Varenicline, an ά4β2 Nicotinic Acetylcholine Receptor Partial Agonist, vs Sustained-Release Bupropion and Placebo for Smoking Cessation, A Randomized Controlled Trial,” by David Gonzales, PhD. et al., in the Journal of the American Medical Association, over 40% of smokers in the United States have already failed to quit smoking at least once with NRT. Thus, we believe that upon FDA approval, our X-22 smoking cessation aid will be attractive to smokers, especially with those who have previously attempted to quit and failed.
Use of existing smoking cessation aids results in relapse rates that can be as high as 90% in the first year after a smoker initially “quits,” according to the American Lung Association in a 2007 report. Smokers currently have the following limited choices of FDA-approved products to help them quit smoking:
|
|
•
|
varenicline (Chantix®/Champix® outside the U.S.), manufactured by Pfizer,
|
|
|
•
|
bupropion (Zyban®), manufactured by GlaxoSmithKline, and
|
|
|
•
|
nicotine replacement therapy, or “NRT,” which is available in the U.S. in several forms: gums, patches, nasal sprays, inhalers and lozenges.
|
Chantix® and Zyban® are pills and are nicotine free. Chantix®, Zyban®, the nicotine nasal spray and the nicotine inhaler are available by prescription only. Nicotine gums, nicotine patches, and lozenges are available over-the-counter.
Chantix® was introduced in the U.S. market in the fourth quarter 2006. Since 2007, Chantix® has been the best-selling smoking cessation aid in the United States, with sales of $701 million in 2007, $489 million in 2008 and $386 million in 2009 and $330 million in 2010. In July 2009, the FDA required a “Boxed Warning,” the most serious type of warning in prescription drug labeling, for both Chantix® and Zyban® based on the potential side effects of these drugs. Despite this Boxed Warning, worldwide sales of Chantix® in 2009 and 2010 were approximately $700 million and $755 million, respectively. There is no guarantee that, even if approved by the FDA, X-22 will have comparable sales to Chantix®, or that the FDA will not require X-22 to have a Boxed Warning.
Other than Chantix® and Zyban®, the only FDA-approved smoking cessation therapy in the United States is NRT. These products consist of gums, patches, nasal sprays, inhalers and lozenges. Nicotine gums and nicotine patches have been sold in the U.S. for 26 years and 18 years, respectively, and millions of smokers have already tried NRT products and failed to stop smoking. According to Perrigo Company, a pharmaceutical company that sells NRT products, sales of NRT products in the United States have averaged approximately $500 million annually from 2007 to 2009.
38
Modified Risk Tobacco Products
A substantial number of adult smokers are unable or unwilling to quit smoking. For example, each year one-half of the adult smokers in the United States do not attempt to quit. Nevertheless, we believe the majority of these smokers are interested in reducing the harmful effects of smoking.
In a 2005 analyst report, The Third Innovation, Potentially Reduced Exposure Cigarettes, JP Morgan examined the effects of FDA regulation of tobacco, including the market for safer cigarettes. JP Morgan’s proprietary survey of over 600 smokers found that 90% of smokers are willing to try a safer cigarette. Among JP Morgan’s other conclusions, it stated: “FDA oversight would imbue PREPS [‘potential reduced exposure products’ which essentially equate to potential modified risk tobacco products] with a regulatory ‘stamp of approval’ and allow for more explicit comparative health claims with conventional cigarettes. Consumers should trust the FDA more than industry health claims.” Prior to the Tobacco Control Act becoming law in 2009, no regulatory agency or body had the authority to assess potential modified risk tobacco products.
Some major cigarette manufacturers have developed and marketed alternative cigarette products. For example, Philip Morris USA developed an alternative cigarette, called Accord®, in which the tobacco is heated rather than burned. R.J. Reynolds Tobacco Company has developed and is marketing an alternative cigarette, called Eclipse®, in which the tobacco is primarily heated, with only a small amount of tobacco burned. Philip Morris and RJ Reynolds have indicated that their products may deliver fewer smoke components compared to conventional cigarettes. Vector Tobacco Inc. (“Vector Tobacco”), which is our former licensee, has marketed a cigarette offered in three brand styles with reduced levels of nicotine, called Quest®. Both Accord® and Eclipse®, which are not conventional cigarettes (e.g., they do not burn down) and have only achieved limited sales. With the exception of Eclipse®, the above products are no longer being manufactured.
Complete cessation from all tobacco and medicinal nicotine products is the ultimate goal of the public health community. However, some public health officials desire to migrate cigarette smokers en masse to medicinal nicotine (also known as NRT) or smokeless tobacco products to replace cigarettes. We believe this is unattainable in the foreseeable future for many reasons, including because the smoking experience is much more complex than simply seeking nicotine. In a 2009 WHO report, statistics demonstrate that approximately 90% of global tobacco users smoke cigarettes. Worldwide cigarette sales (in U.S. dollars) are approximately 12 times greater than sales of smokeless tobacco products and approximately 200 times greater than sales of NRT products. A study published in 2011 in Tobacco Control shows that approximately 6% of the smoking population uses smokeless tobacco products in conjunction with cigarettes. Such smokers are known as “dual users.”
There are newer forms of smokeless tobacco products that have been introduced in the market that are less messy to use than chewing tobacco or dry snuff (since spitting is not involved). These products include Swedish-style snus and dissolvable tobacco products such as Ariva® and Stonewall® tablets made by Star Scientific Inc., and Camel® Orbs, Camel® Strips and Camel® Sticks recently introduced by R.J. Reynolds Tobacco Company. Although use of such products may be more discreet and convenient than traditional forms of smokeless tobacco, they have the same route of delivery of nicotine as nicotine gum and nicotine lozenges, which have been available over-the-counter in the United States for 16 years and 8 years, respectively, and have not significantly replaced cigarettes.
Products
X-22 Smoking Cessation Aid
X-22 is a tobacco-based botanical medical product for use as a smoking cessation therapy. X-22 will be a prescription-only kit containing VLN cigarettes made from our proprietary tobacco, which has approximately 95% less nicotine compared to tobacco in existing “light” cigarettes. The therapy protocol allows the patient to smoke our VLN cigarettes without restriction over the six-week treatment period to facilitate the goal of the patient quitting smoking by the end of the treatment period. Although further research and clinical trials are needed to determine efficacy of X-22, we believe this therapy protocol has been successful thus far because VLN cigarettes made from our proprietary tobacco satisfy smokers’ cravings for cigarettes while also: (i) greatly reducing nicotine exposure and nicotine dependence and (ii) extinguishing the association between the act of smoking and the rapid delivery of nicotine.
39
We further believe that X-22 offers the following advantages over existing smoking cessation products:
|
|
•
|
X-22 separates the act of smoking from the rapid delivery of nicotine;
|
|
|
•
|
X-22 is more attractive than other therapies since it smokes, tastes and smells like a typical cigarette and involves the same smoking behavior;
|
|
|
•
|
X-22 does not expose smokers to any new drugs or new side effects (other than the exposure and side effects from smoking cigarettes in general) that may occur when a person is exposed to a new drug; and
|
|
|
•
|
X-22 is more effective than other smoking cessation aids because:
|
|
|
•
|
X-22 provides greater relief from withdrawal symptoms than the FDA-approved nicotine lozenge;
|
|
|
•
|
X-22 reduces cravings more than the FDA-approved prescription nicotine inhaler; and
|
|
|
•
|
X-22 decreases the likelihood of relapse (in the case of Chantix®, approximately half of those who quit relapse within 8 weeks after the end of treatment).
|
We have met with the FDA regarding the remaining X-22 clinical trials and, based on the FDA’s guidance, we plan to conduct a Phase II-B trial and two larger and concurrent Phase III trials with the same protocols, all of which entail measuring the quitting efficacy of the X-22 cigarette against a typical cigarette with conventional nicotine content that is visually indistinguishable from X-22. We plan to complete the FDA-approval process for our X-22 smoking cessation aid in the fourth quarter of 2012 at the earliest (as a prescription), and upon such approval launch X-22 in the U.S. market and in other top smoking cessation markets thereafter. However, the process of obtaining FDA approval is lengthy and uncertain and we can offer no assurances that this timeline will be met. To date, we have not taken any steps to obtain any necessary approval for our X-22 smoking cessation aid outside of the United States.
Our Modified Risk Cigarettes
Although we believe that two of our cigarette products, which we refer to as BRAND A and BRAND B, will qualify as Modified Risk Cigarettes, further research and exposure studies are needed for the FDA to make this determination. We believe that our BRAND A and BRAND B cigarettes will benefit smokers who are unable or unwilling to quit smoking and who may be interested in cigarettes which reduce exposure to certain tobacco smoke toxins and/or pose a lower health risk than conventional cigarettes. This group of smokers may include the approximate one-half of the 46 million adult smokers in the United States, according to the IOM, who do not attempt to quit in a given year. Compared to commercial cigarettes evaluated by Michael J. Morton and Susan W. Laffoon in the research paper, titled, Cigarette smoke chemistry market maps under Massachusetts Department of Public Health smoking conditions (Regul Toxicol Pharmacol, 2008 Jun;51(1):1-30.), we believe the tobacco in BRAND A has approximately 95% less nicotine than tobacco in cigarettes previously marketed as “light” cigarettes and BRAND B’s smoke contains the lowest amount of “tar” per milligram of nicotine. We believe that BRAND A and BRAND B will qualify as Modified Risk Cigarettes and we intend to seek FDA authorization to market BRAND A and BRAND B as Modified Risk Cigarettes. However, the FDA has not yet issued comprehensive guidance regarding applications that must be submitted to the FDA for Modified Risk Cigarettes, including the criteria for such authorizations. We believe the FDA will issue such guidance in 2011. The FDA has yet to release its regulations regarding modified risk tobacco products, and there is no guarantee that Brand A or Brand B will obtain the necessary authorizations from the FDA to be classified as Modified Risk Cigarettes.
40
BRAND A Cigarettes
Compared to other commercial tobacco cigarettes, we believe that BRAND A has the lowest nicotine content. The tobacco in BRAND A contains approximately 95% less nicotine than tobacco in leading “light” cigarette brands. Although studies have demonstrated that smokers who smoke VLN cigarettes containing our proprietary tobacco smoke fewer cigarettes per day resulting in significant reductions in smoke exposure, including “tar,” nicotine and carbon monoxide, further research and exposure studies are needed to validate these previous studies. The Company believes that there is a possibility that this means smokers might not be interested in smoking the product in the long term. Due to the very low nicotine levels, compensatory smoking does not occur with VLN cigarettes containing our proprietary tobacco.
In a June 16, 2010 press release, Dr. David Kessler, the former FDA Commissioner, recommended that “[t]he FDA should quickly move to reduce nicotine levels in cigarettes to non-addictive levels. If we reduce the level of the stimulus, we reduce the craving. It is the ultimate harm reduction strategy.” Shortly thereafter in a Washington Post article, Dr. Kessler said that the amount of nicotine in a cigarette should drop from about 10 milligrams to less than 1 milligram. BRAND A contains approximately 0.4 milligram of nicotine.
A Phase II smoking cessation clinical trial at the University of Minnesota Masonic Comprehensive Cancer Center, which is further described below, also measured exposure of various smoke compounds in smokers from smoking a VLN cigarette containing our proprietary tobacco over a six-week period. Smokers significantly reduced their smoking as compared to their usual brand of cigarettes. As depicted below, the number of VLN cigarettes smoked per day on average decreased from 19 (the baseline number of cigarettes of smokers’ usual brand) to 12 by the end of the six-week period, even though participants were instructed to smoke ad libitum (as many cigarettes as desired) during treatment. Furthermore, besides significant reductions in other biomarkers, carbon monoxide (CO) levels, an indicator of smoke exposure, significantly decreased from 20 parts per million (baseline) to 15 parts per million. Cotinine, a metabolite and biomarker of nicotine, significantly decreased from 4.2 micrograms/mL (baseline) to 0.2 micrograms/mL. All differences were statistically significant (P<0.05). Further research and exposure studies are needed, however, to validate the findings of this study.

We believe these and other results and future exposure studies the FDA may require will result in a Modified Risk Cigarette claim for BRAND A. We further believe smokers who desire to smoke fewer cigarettes per day while also satisfying cravings and reducing exposure to nicotine will find BRAND A beneficial. We intend that BRAND A will be available in regular and menthol; with both styles being king size (85 mm) cigarettes.
41
BRAND B Cigarettes
Compared to other commercial tobacco cigarettes, we believe that BRAND B’s smoke contains the lowest amount of “tar” per milligram of nicotine. Using a proprietary high nicotine tobacco blend in conjunction with specialty cigarette components, BRAND B allows the smoker to achieve a satisfactory amount of nicotine per cigarette while inhaling less “tar” and carbon monoxide. (Specialty cigarette components means filters and cigarette papers that are not usually found on typical brands and generally cost more than conventional filters and cigarette papers.) At the same time, we do not expect exposure to nicotine from BRAND B to be significantly higher than some full flavor cigarette brands (cigarettes that have relatively higher “tar” and nicotine yields (from smoking machines) and taste more robust than “light” and “ultra lights.”). Although studies have demonstrated less “tar” inhalation from low tar-to-nicotine ratio cigarettes other than Brand B, human exposure studies have not been completed on Brand B. Further research and exposure studies are needed to be validate these previous studies. We believe smokers who desire to reduce smoke exposure (meaning inhaling less smoke either per cigarette or over a fixed time period such as per day), but are less concerned about nicotine will find BRAND B beneficial. We intend that BRAND B will be available in regular and menthol; with both styles being king size (85 mm) cigarettes.
BRAND B has a “tar” yield between typical “light” and “ultra-light” cigarettes, but a nicotine yield of typical full flavor cigarettes. The graph below compares the tar-to-nicotine ratios (yields from smoking machines) of BRAND B and BRAND B menthol to those of the leading cigarette brands. As shown, for example, BRAND B has approximately 47% less “tar” per milligram of nicotine compared to the smoke from Marlboro Light®.
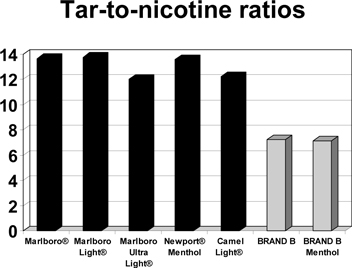
In a 2001 report, entitled Clearing the Smoke, Assessing the Science Base for Tobacco Harm Reduction, the Institute of Medicine notes that a low “tar”/moderate nicotine cigarette is a viable strategy for reducing the harm caused by smoking. The report states: “Retaining nicotine at pleasurable or addictive levels while reducing the more toxic components of tobacco is another general strategy for harm reduction.” Although short-term human exposure studies have not been completed on Brand B and further research and exposure studies are needed, we believe based on previous studies of low tar-to-nicotine ratio cigarettes, that evaluation of BRAND B in short-term human exposure studies will confirm that exposure to smoke from BRAND B, including certain tobacco smoke toxins and carbon monoxide, will be significantly reduced as compared to exposure to smoke from the leading brands of cigarettes. We believe results from these exposure studies will warrant a Modified Risk Cigarette claim for BRAND B.
RED SUN and MAGIC Cigarettes
Our subsidiary, Goodrich Tobacco Company, LLC (f/k/a Xodus, LLC), has introduced two super-premium priced cigarette brands, RED SUN and MAGIC, into the U.S. market in the first quarter of 2011. Both brands are available in regular and menthol and all four brand styles are king size, packaged in hinge-lid hard packs. We intend to focus our marketing efforts on tobacconists, smoke shops and tobacco outlets. We believe the ban in 2009 by the FDA of all flavored cigarettes (with the exception of menthol) has resulted in a product void in these tobacco channels. Certain wholesalers and retailers are now seeking other specialty cigarettes to replace the banned flavored cigarettes. We believe that certain U.S. cigarette wholesalers and retailers will, among other reasons, purchase these cigarettes to replace their lost sales of flavored cigarettes as well as lost sales of “light” cigarettes, since as of June 2010, all cigarette companies were required to cease the use of the terms “low tar,” “light” and “ultra light.”
42
Clinical Trials with Cigarettes Containing our VLN Tobacco
VLN cigarettes containing our proprietary tobacco have been the subject of various independent studies, including two Phase II clinical trials for smoking cessation which were not funded by us. Both of these Phase II clinical trials were “intent to treat” trials, meaning that any patients who dropped out of the trials for any reason at any time during treatment or during the follow-up periods were considered failures (still smoking and not abstinent). Dropout rates during smoking cessation trials are generally high since patients either quit smoking or resume smoking their usual brand. In either case, they may believe there is no reason to continue.
One of these two Phase II clinical trials compared the quitting efficacy of a VLN cigarette containing our proprietary tobacco versus a low nicotine cigarette and an FDA-approved nicotine lozenge (4 mg) in a total of 165 patients treated for six weeks (Hatsukami et al. 2010). This clinical trial was led by Dr. Dorothy Hatsukami, Director of the National Transdisciplinary Tobacco Use Research Center, at the University of Minnesota Masonic Comprehensive Cancer Center. Dr. Hatsukami was selected in 2010 as one of the nine voting members of the 12-person Tobacco Products Scientific Advisory Committee (“TPSAC”), within the FDA’s Center for Tobacco Products created under the Tobacco Control Act. (TPSAC will make recommendations and issue reports to the FDA Commissioner on tobacco regulatory matters, including but not limited to, the impact of the use of menthol in cigarettes, altering levels of nicotine in tobacco products, and applications submitted to the FDA for modified risk tobacco products.)
Results from this Phase II trial conclude that patients exclusively using the VLN cigarette containing our proprietary tobacco achieved a 43% quit rate (confirmed four-week continuous abstinence) as compared to a quit rate of 35% for the group exclusively using the FDA-approved nicotine lozenge and a 21% quit rate for the group exclusively using the low nicotine cigarette. Smoking abstinence at the 6-week follow-up after the end of treatment was 47% for the VLN cigarette group, 37% for the nicotine lozenge group and 23% for the low nicotine cigarette group. Furthermore, the VLN cigarette was also associated with greater relief from withdrawal symptoms and cravings of usual brand cigarettes than the nicotine lozenge. Carbon monoxide (CO) levels in patients were tested at each treatment clinic visit to verify smoking abstinence. Although these results of this study are positive, further research and clinical trials are needed to determine efficacy of VLN cigarettes for quitting smoking, including X-22.
|
Treatments
|
||||||||||||||||||||||||||||
|
Low
|
FDA-Approved
|
Very Low
|
||||||||||||||||||||||||||
|
Nicotine Cigarette
|
Nicotine Lozenge
|
Nicotine Cigarette
|
||||||||||||||||||||||||||
|
0.3 mg nicotine/cig.
|
4 mg nicotine
|
0.05 mg nicotine/cig.
|
||||||||||||||||||||||||||
|
n = 52
|
n = 60
|
n = 53
|
||||||||||||||||||||||||||
|
Number
|
Number
|
Number
|
||||||||||||||||||||||||||
|
Abstinent
|
Percent
|
Abstinent
|
Percent
|
Abstinent
|
Percent
|
P-value
|
||||||||||||||||||||||
|
4-Week Continuous Abstinence
|
||||||||||||||||||||||||||||
|
- Standard Threshold in Cessation
|
11 | 21.2 | % | 21 | 35.0 | % | 23 | 43.4 | % | 0.0508 | ||||||||||||||||||
|
- Carbon Monoxide Verified
|
||||||||||||||||||||||||||||
|
- Post Treatment
|
||||||||||||||||||||||||||||
|
6-Week Point Prevalence Abstinence
|
||||||||||||||||||||||||||||
|
- Six Weeks from the End of Treatment
|
12 | 23.1 | % | 22 | 36.7 | % | 25 | 47.2 | % | 0.0357 | ||||||||||||||||||
|
- Carbon Monoxide Verified
|
||||||||||||||||||||||||||||
Unlike Phase III clinical trials for other FDA-approved smoking cessation aids, four week continuous abstinence in the University of Minnesota Phase II trial was measured after the treatment period, when patients were “off” medication as shown in the chart below, rather than during the last four weeks of the treatment period. For example, according to the prescription Chantix® label, four-week continuous abstinence in the Chantix® Phase III clinical trials (the 44 percent quit rate advertised by Pfizer) was measured during the last four weeks of the 12-week treatment period, while patients were still taking Chantix®. In one of these Chantix® Phase III clinical trials, approximately one-third of those who had been abstinent during the last week of treatment returned to smoking within four weeks after they stopped taking Chantix®, and approximately 45% returned to smoking within eight weeks after they stopped taking Chantix®.
43
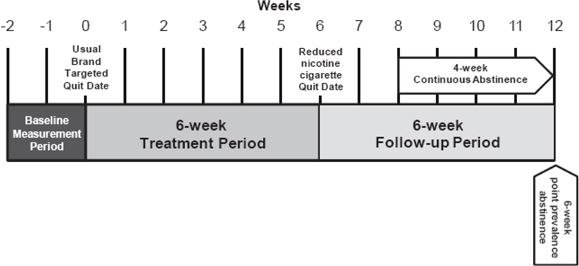
Patients who used the VLN cigarette containing our proprietary tobacco over the six-week treatment period significantly reduced their smoking as compared to their usual brand of cigarettes. The number of VLN cigarettes smoked per day on average decreased from 19 (the baseline number of cigarettes of the smoker’s usual brand) to 12 by the end of the six-week treatment period, even though participants in this clinical trial were instructed to smoke ad libitum (as many cigarettes as desired) during treatment. Carbon monoxide (CO) levels, an indicator of smoke exposure, significantly decreased from 20 parts per million (baseline) to 15 parts per million. Cotinine, a metabolite and biomarker of nicotine, significantly decreased from 4.2 micrograms/mL (baseline) to 0.2 micrograms/mL. All differences in the above three measurements were statistically significant (P<0.05). Although these results are positive, further research and clinical trials are needed to determine the effectiveness of VLN cigarettes.
Additional biomarkers of smoke exposure were significantly reduced on average from baseline measurements (taken before the six-week treatment period) in patients who used the VLN cigarette containing our proprietary tobacco:
44
|
BIOMARKER
|
DESCRIPTION
|
LEVEL IN PATIENTS
|
REDUCTION
|
|||||||||||
|
(pmol/mg creatinine)
|
||||||||||||||
|
Baseline
|
VLN Cigarette
|
|||||||||||||
|
NNAL
|
Metabolites of the tobacco-specific carcinogen NNK
|
0.92 | 0.2 | 78 | % | |||||||||
|
NNN
|
Metabolites of the tobacco-specific carcinogen NNN
|
0.09 | 0.03 | 67 | % | |||||||||
|
1-HOP
|
Metabolite of pyrene, a marker for uptake of carcinogenic polycyclic aromatic hydrocarbons
|
0.89 | 0.57 | 36 | % | |||||||||
|
3-HPMA
|
Metabolite of the smoke toxicant acrolein
|
3320 | 1453 | 56 | % | |||||||||
|
S-PMA
|
Metabolite of the carcinogen benzene
|
2.46 | 0.76 | 69 | % | |||||||||
|
All differences were statistically significant (P<0.05).
|
||||||||||||||
In a separate Phase II clinical trial funded by Vector Tobacco, our former licensee, under Investigational New Drug (“IND”) Application 69,185, a randomized double-blind, active controlled, parallel group, multi-center Phase II smoking cessation clinical trial was conducted to evaluate the quitting efficacy of Quest® reduced-nicotine cigarettes as a smoking cessation treatment in 346 patients (Becker et al. 2008). Treatment consisted of smoking three reduced-nicotine cigarette styles (Quest 1®, Quest 2® and Quest 3®) for two weeks each, with nicotine yields per cigarette of 0.6 mg (a low nicotine cigarette made with a blend of regular tobacco and our proprietary VLN tobacco), 0.3 mg (an extra low nicotine cigarette made with a blend of regular tobacco and our proprietary VLN tobacco) and 0.05 mg (a VLN cigarette made with tobacco only from our proprietary VLN tobacco variety) either in combination with nicotine patch therapy (a nicotine replacement therapy or NRT product) or placebo patches.
In this three-arm clinical trial in which patients were treated over a period of sixteen (16) weeks, use of reduced-nicotine cigarettes in combination with nicotine patches was more effective (the difference was statistically significant) in achieving four-week continuous abstinence than use of nicotine patches alone (32.8% vs. 21.9%), and use of reduced-nicotine cigarettes without nicotine patches yielded an abstinence rate similar (the difference was not statistically significant) to that of nicotine patches (16.4% vs. 21.9%). No serious adverse events were attributable to the investigational product.
The major differences between the Vector Tobacco Phase II clinical trial and the University of Minnesota Phase II clinical trial is the duration of time during treatment that VLN cigarettes are smoked and the use of nicotine replacement therapy (“NRT’) in combination with VLN cigarettes. In the Vector Tobacco trial, VLN cigarettes were smoked by patients (in two arms of the study) for only two weeks, either in combination with using a nicotine patch or placebo patch, followed by continued use of nicotine patches for the subsequent ten (10) weeks. In the University of Minnesota Phase II clinical trial, VLN cigarettes (in one arm of the study) were smoked for six weeks without any use of NRT before, during or after this 6-week treatment period. We believe that the effectiveness of VLN cigarettes for use in smoking cessation is higher when they are used alone (without NRT or another therapy) and for a longer time period, as in the University of Minnesota trial. We have therefore decided to have patients use VLN cigarettes alone and for 6 weeks in our upcoming clinical trials.
A 2008 binding arbitration award, which was completely fulfilled in 2009 by our former licensee, Vector Tobacco, provided us with copies of all of Vector Tobacco’s FDA submissions relating to Vector Tobacco’s IND for Quest® and awarded to us a right of reference to Vector Tobacco’s IND for Quest®, including all results of Vector’s Phase II clinical trial. This arbitration award allows us to use all such information in our IND with the FDA for our VLN cigarette that contains our same proprietary tobacco that Vector Tobacco used in its IND submissions to the FDA. This arbitration award has been helpful to us with the FDA, since analytical reports produced by Vector Tobacco pertaining to our proprietary tobacco and cigarettes made from our proprietary tobacco are being utilized by us with the FDA.
45
Another smoking cessation clinical trial using VLN cigarettes containing our proprietary tobacco was a randomized controlled trial conducted at Roswell Park Cancer Institute, Buffalo, New York, to investigate the effect of smoking a very low nicotine cigarette (“VLN”) in combination with a nicotine patch for 2 weeks prior to the quit date (Rezaishiraz et al. 2007). Ninety-eight adult smokers were randomized to two treatments: (i) two weeks of a VLN (Quest 3®) and 21-mg nicotine patch before the quit date and (ii) a reduced nicotine cigarette (Quest 1®) during the two weeks before the quit date. After the quit date, all subjects received counseling for smoking cessation and nicotine patch therapy for up to eight weeks (four weeks of 21-mg patches, two weeks of 14-mg patches, and two weeks of 7-mg patches). Group 1, which used the VLN cigarette and a nicotine patch before quitting, had lower combined craving scores during the two weeks before and after the quit date. Self-reported point prevalence of smoking abstinence at the three- and six-month follow-up points was higher in Group 1 (43% vs. 34% and 28% vs. 21%).
A study at Dalhousie University, Halifax, Nova Scotia (Barrett 2010), compared the effects of low nicotine cigarettes and an FDA-approved nicotine inhaler on cravings and smoking behavior of smokers who did not intend to quit. In separate laboratory sessions, each of 22 participants used a VLN cigarette (Quest 3®), a reduced nicotine cigarette (Quest 1®, which contains approximately two-thirds conventional tobacco and one-third VLN tobacco), a nicotine inhaler (10 mg; 4 mg deliverable, Pharmacia), or a placebo inhaler (identical in appearance to the nicotine inhaler, but containing no nicotine). Cravings, withdrawal and mood descriptors were rated before and after a 20-minute treatment session during which subjects were instructed to smoke two cigarettes or to use an inhaler every 10 seconds. The reduction in the rating of intent to smoke (usual cigarette brand) after using the VLN cigarette (-10.0) was significantly greater than the reduction with the nicotine inhaler (-1.9). Use of the VLN cigarette was also associated with significantly increased satisfaction and relaxation compared to the nicotine inhaler.
Although the results of these studies indicate effectiveness of VLN cigarettes for quitting smoking, further research and clinical trials are needed to validate the efficacy of VLN cigarettes, including X-22, for quitting smoking.
Technology Platform
Our proprietary technology enables us to decrease or increase the level of nicotine (and other nicotinic alkaloids) in tobacco plants by decreasing or increasing the expression of gene(s) responsible for nicotine production in the tobacco plant using genetic engineering. The basic techniques are the same as those used in the production of genetically modified varieties of other crops, which in 2009 were planted on 330 million acres in 25 countries according to the International Service for the Acquisition of Agri-Biotech Applications. This includes approximately 85% of the corn and soybeans grown in the United States. The only components of the technology that are distinct from those in commercialized genetically modified varieties of major crops are segments of tobacco genes (DNA sequences) that are also present in all conventional tobacco plants. Genetically modified or transgenic tobacco that we use in our products has been deregulated by the USDA. Thus, plants may be grown and used in products in the United States without legal restrictions or labeling requirements related to the genetic modification. Nevertheless, our proprietary tobacco is grown only by farmers under contracts that require segregation and prohibit transfer of material to other parties.
During the development of genetically-engineered plant varieties, many candidate plant lines are evaluated in the field in multiple locations over several years, as in any other variety development program. This is carried out in order to identify lines that have not only the specific desired trait, e.g., very low nicotine, but have overall characteristics that are suitable for commercial production of the desired product. This process allows us to see if there are undesirable effects of the genetic modification approach or the specific genetic modification event, regardless of whether the effects are anticipated or unanticipated. For example, since nicotine is known to be an insecticide that is effective against a wide range of insects, reduction of nicotine content in the plants may be expected to affect susceptibility to insect pests. While there are differences in the susceptibility of VLN tobacco to some insects, all tobacco is attacked by a number of insects. The measures taken to control insect pests of conventional tobacco are adequate to control insect pests in VLN tobacco.
Once a genetically-engineered tobacco plant with the desired characteristics is obtained, each plant can produce hundreds of thousands of seeds. When each seed is germinated, the resulting tobacco plant has identical characteristics as the parent and sibling plants and the nicotine content of these plants generally fall within a narrow range. Tobacco products with either low or high nicotine content are easily produced through this method. For example, we believe that one of our proprietary tobacco varieties contains the lowest nicotine content of any tobacco ever commercialized, with approximately 95% less nicotine than tobacco in leading “light” cigarette brands. We believe this proprietary tobacco grows with virtually no nicotine without adversely affecting the other leaf constituents important to a cigarette’s characteristics, including taste and aroma.
46
Sources of Raw Materials
We obtain a large portion of our tobacco leaf requirements from farmers in multiple U.S. states that are under direct contracts with us. The contracts prohibit the transfer of our proprietary seeds and plant materials to other parties. The total delivered tobacco leaf from these farmers was approximately 50 percent greater in 2010 than 2009 and we plan to increase leaf production with farmers in 2011. We purchase the balance of our tobacco requirements through third parties. As we expand our sales and distribution of our current commercial brands, RED SUN and MAGIC, and proceed to market with our X-22 smoking cessation aid and BRAND A and BRAND B cigarettes, we plan to continue to scale up the amount of tobacco leaf we obtain directly from farmers under contract.
Intellectual Property
Our proprietary technology is covered by 12 patent families consisting of 98 issued patents in 79 countries, of which we own 8 issued patents and exclusively license 90 issued patents, and approximately 45 pending patent applications, of which we own 29 pending patent applications and exclusively license 16 pending patent applications. A “patent family” is a set of patents granted in various countries to protect a single invention. Our patent coverage in the United States, which we believe is the most valuable smoking cessation market and cigarette market, consists of 14 issued patents and 7 pending applications. In the U.S., patents that currently cover our products expire in 2019 through 2022. Also in the U.S., patent applications that we expect will cover our products would, if patents are granted in connection with such applications, yield patents that expire in 2024 through 2028. However, there is no guarantee that patents will be granted in response to such patent applications and therefore cover our products. Our two worldwide exclusive licenses, one from North Carolina State University (“NCSU”) and the other from National Research Council of Canada, Plant Biotechnology Institute in Saskatoon, Canada (“NRC”), each involve multiple patent families, patent applications and patents. The exclusive rights under each of these two license agreements expire on the date on which the last patent covered by the subject license expires in the country or countries where such patents are in effect, which is expected to be from 2022 through 2028, respectively. In China, which we consider the world’s largest cigarette market, we own or exclusively license 5 issued patents and 3 pending patent applications. We also have the exclusive right to license and sublicense these patent rights. The patents we own or exclusively license are issued in countries where we believe at least 75% of the world’s smokers reside. As a result, we believe we have exclusive worldwide rights to all uses of the following genes responsible for nicotine content in tobacco plants: QPT, A622, NBB1, MPO and genes for several transcription factors. We also believe we have exclusive rights to plants with altered nicotine content produced from modifying expression of these genes and tobacco products produced from these plants.
We own various registered trademarks in the United States. We also have exclusive rights to plant variety protection, or PVP, certificates in the United States (issued by the U.S. Department of Agriculture) and Canada. A PVP certificate prevents anyone other than the owner/licensee from planting a plant variety for twenty (20) years in the U.S. or (18) years in Canada. The protections of PVP are independent of, and in addition to, patent protection.
Sales and Marketing
X-22 Smoking Cessation Aid
Before completion of the Phase III clinical trials, we intend to enter into arrangements in both the U.S. and international markets with pharmaceutical companies to market and sell X-22. We plan to seek marketing partners in the U.S. with existing pharmaceutical sales forces that already call on medical and dental offices in their geographic markets. The level of marketing support for X-22 in the U.S. will depend on many factors such as the type of partner we can attract and the timeliness of obtaining FDA approval. In the event we market the product internally, we expect to expend no more than $500,000 on marketing in the first year in the U.S. Internationally, we expect to license parties that have the financial strength and regulatory experience to market X-22. We have not yet identified specific countries where we intend to market X-22 and, thus, we have not developed timelines or costs for such international expansion.
There are approximately 700,000 physicians in the U.S., including approximately 80,000 general practitioners, many of whom are aware of new medications, even before they achieve FDA approval. There are also approximately 170,000 dentists in the U.S. who can write prescriptions for smoking cessation aids. We plan to develop awareness for X-22 by educating physicians and dentists about our X-22 smoking cessation aid. We intend to advertise in professional journals, use direct mail campaigns to medical professionals, and attend trade shows and professional conferences. We also intend to use internet advertising and pharmacy circulars to reach consumers and to encourage them to ask their physicians and dentists about our X-22 smoking cessation aid. We expect to use public relations to increase public awareness about X-22. We will seek to use federal and state-funded smoking cessation programs and clinics to inform clinicians and patients about, and encourage the use of, X-22 as a smoking cessation aid. We will also seek to participate in various government-funded programs which purchase approved smoking cessation aids and then distribute these to smokers at no charge or at greatly reduced prices.
47
BRAND A and BRAND B
The Tobacco Control Act establishes procedures for the FDA to regulate the labeling and marketing of modified risk tobacco products, which includes cigarettes that (i) reduce exposure to tobacco smoke toxins and/or (ii) pose lower health risks, as compared to conventional cigarettes (“Modified Risk Cigarettes”). The Tobacco Control Act requires the FDA to issue specific regulations and guidance regarding applications that must be submitted to the FDA for the authorization to label and market Modified Risk Cigarettes. The FDA has yet to release its regulations regarding modified risk tobacco products. However, based in part on the timelines contained in the Tobacco Control Act, we expect the FDA to issue such regulations and guidance within the next year. Although we believe that two of our cigarette products, which we refer to as BRAND A and BRAND B, will qualify as Modified Risk Cigarettes, further research and exposure studies are needed for the FDA to make this determination. Compared to commercial cigarettes, we believe that the tobacco in BRAND A has approximately 95% less nicotine than tobacco in cigarettes previously marketed as “light” cigarettes, and BRAND B’s smoke contains the lowest amount of “tar” per milligram of nicotine. Upon FDA authorization to classify these products as modified risk cigarettes, the earliest timing of which we estimate to be in approximately 2 years, we expect to expend $250,000 in the first year following such authorization on marketing each brand in the U.S. We expect to license parties internationally that have the financial strength and regulatory experience to market our brands. We have not yet identified specific countries where we intend to market our brands and, thus, we do not know at this time the regulatory approval processes that will be applicable to our products in specific countries.
RED SUN and MAGIC Cigarettes
Our subsidiary, Goodrich Tobacco Company, intends to focus its marketing efforts for RED SUN and MAGIC on tobacconists, smoke shops and tobacco outlets. Until we are profitable, we do not expect to spend more than the total annual gross margin of RED SUN and MAGIC on marketing efforts in the U.S. We believe the ban in 2009 by the FDA of all flavored cigarettes (with the exception of menthol) has resulted in a product void in these tobacco channels. Certain wholesalers and retailers are now seeking other specialty cigarettes to replace the banned flavored cigarettes. We believe that certain U.S. cigarette wholesalers and retailers will, among other reasons, purchase these cigarettes to replace their lost sales of flavored cigarettes as well as potential lost sales of “light” and “ultra light” cigarettes, since as of June 2010, all cigarette companies were required to cease the use of the terms “low tar,” “light” and “ultra light”.
Government Research Cigarettes
The National Institute on Drug Abuse (“NIDA”), a component of the National Institutes of Health (“NIH”), provides the scientific community with controlled and uncontrolled research chemicals and drug compounds in its Drug Supply Program. In 2009, NIDA included an option to develop and produce research cigarettes with ten different levels of nicotine, including a minimal (placebo) level, or Research Cigarette Option, in its request for proposals for a five-year contract for Preparation and Distribution of Research and Drug Products. We have agreed, as a subcontractor to RTI International (“RTI”) in RTI’s contract with NIDA for the Research Cigarette Option, to supply modified nicotine cigarettes to NIDA. In August 2010, we met with officials from NIDA, FDA, RTI, the National Cancer Institute and the Centers for Disease Control and Prevention to finalize certain aspects of the design of these research cigarettes. These research cigarettes will be distributed under the mark SPECTRUM and will not be marketed.
In 2010, we received our first purchase order of $152,660 which included a design phase fee of $40,604. In March 2011, the Company received a revised purchase order and authorization to bill $112,000 for manufacture planning and material procurement for 9 million cigarettes from RTI. Payment was received on May 2, 2011. The Company received an additional purchase order from RTI for approximately $680,000 in May 2011 to ship the 9 million research cigarettes. Approximately 4 million cigarettes out of the 9 million were shipped to and received by RTI on July 6, 2011. We believe that the revenue from the sale of SPECTRUM, plus direct orders of other cigarettes from researchers, will be approximately $825,000 in 2011 and $3 million over the next 5 years. As a subcontractor, we do not have a formal contractual arrangement at this time with RTI to purchase additional SPECTRUM cigarettes or with other researchers, and there is no guarantee that we will receive future orders from RTI or other researchers.
48
Healthcare Reimbursement
Government and private sector initiatives to limit the growth of healthcare costs, including price regulation, competitive pricing, coverage and payment policies, and managed-care arrangements, are continuing in many countries where we intend to sell our X-22 smoking cessation aid, including the United States. These changes are causing the marketplace to put increased emphasis on the delivery of more cost-effective medical products.
Government healthcare programs in the United States, including Medicare and Medicaid, private healthcare insurance and managed-care plans have attempted to control costs by limiting the amount of reimbursement for which they will pay for particular procedures or treatments. This may create price sensitivity among potential customers for our X-22 smoking cessation aid, even if we obtain FDA approval for it. Some third-party payers must also approve coverage for new or innovative devices or therapies before they will reimburse healthcare providers who use the medical devices or therapies. Even though a new medical product may have been cleared for commercial distribution, we may find limited demand for X-22 until reimbursement approval has been obtained from governmental and private third-party payers.
Approximately 160 million Americans have private health insurance with prescription coverage and the majority, and an increasing number of these plans, cover pharmacologic treatments for smoking cessation. Healthcare payers, including governmental bodies, are increasingly willing to fund smoking cessation treatments due to the expected savings from reducing the incidence of smoking-related illnesses. Approximately 46 million Americans were covered by Medicare in 2009. Medicare provides insurance coverage for up to two smoking cessation attempts per year and each attempt may include four counseling sessions.
Approximately 47 million Americans were covered by state Medicaid programs in 2009. Approximately 30% of Medicaid recipients are smokers. Medicaid programs in 42 states and the District of Columbia cover at least one form of pharmacologic treatment for smoking cessation (Chantix®, Zyban® or NRT). The new healthcare legislation is expanding Medicaid coverage to all 50 states. The current retail price of the 12-week prescription of Chantix® is over $450.We expect X-22 to be price competitive with any FDA-approved smoking cessation aid, especially Chantix®. which we believe will not only encourage governmental and private third-party payers to cover X-22, but will encourage smokers to attempt to quit with X-22 since they will not have to purchase their usual brand of cigarettes over the 6-week treatment period.
Manufacturing
We have entered into an agreement with a federally licensed cigarette manufacture to produce RED SUN, MAGIC, SPECTRUM, BRAND A, BRAND B and the clinical trial cigarettes for X-22.
Competition
In the market for FDA-approved smoking cessation aids, our principal competitors include Pfizer Inc., GlaxoSmithKline PLC, Novartis International AG, and Niconovum AB, a subsidiary of Reynolds American Inc. The industry consists of major domestic and international companies, most of which have existing relationships in the markets into which we plan to sell, as well as financial, technical, marketing, sales, manufacturing, scaling capacity, distribution and other resources, and name recognition substantially greater than ours.
Cigarette companies compete primarily on the basis of product quality, brand recognition, brand loyalty, taste, innovation, packaging, service, marketing, advertising, retail shelf space and price. Cigarette sales can be significantly influenced by weak economic conditions, erosion of consumer confidence, competitors’ introduction of low-price products or innovative products, higher cigarette taxes, higher absolute prices and larger gaps between price categories, and product regulation that diminishes the ability to differentiate tobacco products. Domestic competitors include Philip Morris USA, Reynolds American Inc., Lorillard Inc., Commonwealth Brands, Inc., Liggett Group LCC, Vector Tobacco Inc., and Star Scientific Inc. International competitors include Philip Morris International, British American Tobacco, Japan Tobacco Inc., Imperial Tobacco Group and regional and local tobacco companies; and, in some instances, government-owned tobacco enterprises, principally in China, Egypt, Thailand, Taiwan, Vietnam and Algeria.
49
Potential Smoking Cessation Aids
Nicotine Vaccines
Nicotine vaccines are under development in clinical trials. However, they have not yet achieved the efficacy of other FDA-approved smoking cessation therapies. Nicotine itself is not recognized by the body as a foreign compound since the molecule is too small. In order to stimulate the production of antibodies, nicotine must be attached to a carrier to make the vaccine work. Different vaccine development programs use different carriers. Four companies, Cytos Biotechnology AG, Celtic Pharmaceuticals Holdings, Nabi Biopharmaceuticals, L.P. and Independent Pharmaceutica AB have or have had vaccine candidates in clinical trials. Cytos exclusively licensed its nicotine vaccine candidate to Novartis in 2007 for 35 million Swiss Francs ($30 million) and up to 565 million Swiss Francs ($492 million) in milestone payments and royalties. In October 2009, it was announced that Cytos’ nicotine vaccine candidate failed to show efficacy in a Phase II trial.
GlaxoSmithKline Biologicals SA exclusively licensed Nabi’s nicotine vaccine candidate, NicVAX®, in an agreement which was approved by Nabi’s shareholders in March 2010. Together with an upfront non-refundable fee of $40 million paid by GlaxoSmithKline, Nabi is eligible to receive over $500 million in option fees and milestones, not including potential royalties on global sales. Phase III NicVAX® clinical trials commenced in 2010.
According to Nabi Pharmaceuticals (www.nabi.com), these vaccine treatments entail seven consecutive monthly injections. Increases in abstinence rates have been reported but only among a minority of trial subjects with the highest levels of anti-nicotine antibodies. To date, not all subjects of such vaccine treatments develop sufficient antibody levels despite receiving multiple injections. Even in those who do develop sufficient antibody levels, cravings for cigarettes are not addressed by this treatment, although the pharmacological reward of nicotine is suppressed. Expectations are that the treatment, if approved, would need to be repeated every 12 to 18 months to assist in preventing relapse.
Electronic or E-cigarettes
Although the FDA has not evaluated electronic cigarettes, or e-cigarettes, for quitting smoking, and we are not aware of any published result of a controlled clinical trial of e-cigarettes as a smoking cessation aid, e-cigarettes are included here since there have been unconfirmed claims that these products facilitate cessation. E-cigarettes have been the subject of much controversy for this and various other reasons, including the fact that these products are actually not cigarettes or tobacco products at all but are battery-operated devices filled with nicotine, flavor and other chemicals. They turn nicotine and other chemicals into a vapor that is inhaled. E-cigarettes have very similar nicotine kinetics and delivery as nicotine inhalers, a prescription NRT product already approved by the FDA, which is the reason we believe that using e-cigarettes to quit smoking is not likely to be any more effective than other nicotine replacement products.
In a September 9, 2010 press release, the FDA issued warning letters to five e-cigarette distributors for various violations of the Federal Food, Drug, and Cosmetic Act, including unsubstantiated claims and poor manufacturing practices. The FDA said these e-cigarette companies are illegally marketing their products as tools to help people quit using cigarettes. The FDA believes e-cigarettes “[m]eet the definition of a combination drug-device product under the Federal Food, Drug and Cosmetic Act.” In a letter to the Electronic Cigarette Association of the same date, the FDA said the agency intends to regulate electronic cigarettes and related products in a manner consistent with its mission of protecting the public health.
The FDA has also been confiscating imports of e-cigarettes and has been in litigation with importers of these products. A federal appeals court ruled on December 7, 2010 that the FDA can only regulate electronic cigarettes as tobacco products rather than as a drug-delivery device. The FDA is appealing this decision; however, the U.S. Court of Appeals for the District of Columbia Circuit on January 2011 rejected the FDA’s request to have the entire court review the December 7, 2010 decision that went against the Agency. The FDA, which has always contended that e-cigarettes should be regulated as drug-delivery devices not tobacco products, now has the option of asking the U.S. Supreme Court to take up the case. An FDA spokesman said that the Agency is evaluating the latest court ruling “and considering its legal and regulatory options.” Many countries have already banned e-cigarettes as has the state of Oregon and other states are in the process of banning them.
50
Government Regulation
Smoking Cessation Aids
Government authorities in the U.S. and foreign countries extensively regulate the research, development, testing, manufacture, labeling, promotion, advertising, distribution, sampling, marketing and import and export of pharmaceutical products. FDA approval must be obtained, as has been the case for decades, before a product can be marketed for quitting smoking or reducing withdrawal symptoms. In addition, as with all FDA-approved prescription drugs, the FDA must approve the brand name of our X-22 smoking cessation aid. The FDA approval process for smoking cessation aids is similar to that required by the FDA for new drug approvals, although the cost to complete clinical trials for a smoking cessation aid such as X-22 are generally far less than clinical trials for drugs. The primary endpoint of the clinical trial for smoking cessation aids is smoking abstinence, which is generally confirmed by inexpensive, noninvasive biomarker tests. Since potential quitters are already smokers, X-22 will not expose participants in the clinical trials to any new compounds, unlike a new chemical entity, such as Chantix®.
The process of obtaining governmental approvals and complying with ongoing regulatory requirements requires the expenditure of substantial time and financial resources. In addition, statutes, rules, regulations and policies may change and new legislation or regulations may be issued that could delay such approvals. If we fail to comply with applicable regulatory requirements at any time during the product development process, approval process, or after approval, we may become subject to administrative or judicial sanctions. These sanctions could include the FDA’s refusal to approve pending applications, withdrawals of approvals, clinical holds, warning letters, product recalls, product seizures, total or partial suspension of our operations, injunctions, fines, civil penalties or criminal prosecution. Any agency enforcement action could have a material adverse effect on us.
We believe that, in many foreign countries, a product for which a smoking cessation claim is made (e.g., X-22) generally can be sold legally without having to be approved by a regulatory agency, as the presence of a smoking cessation claim in many cases does not trigger regulatory approval requirements. In contrast, the U.S. FDA considers tobacco/nicotine addiction a disease, with the result that smoking cessation products are regulated because their purpose it to cure or mitigate the disease of smoking. Therefore, given the regulatory landscape of many foreign countries, we believe it is unlikely that the Company would need to seek regulatory approval or authorization for our products in such countries. We believe the same is generally true in many foreign countries regarding claims for reduced exposure/risk tobacco products (modified risk tobacco products).
The U.S. regulatory scheme for the development and commercialization of new drugs can be divided into three distinct phases: an investigational phase including both preclinical and clinical investigations leading up to the submission of a New Drug Application (“NDA”); a period of FDA review culminating in the approval or refusal to approve the NDA; and the post-marketing period.
Preclinical Phase
The preclinical phase involves the characterization, product formulation and animal testing necessary to prepare an IND Application for submission to the FDA. The IND must be reviewed and authorized by the FDA before the drug can be tested in humans. Once a new drug agent has been identified and selected for further development, preclinical testing is conducted to confirm pharmacological activity, to generate safety data, to evaluate prototype dosage forms for appropriate release and activity characteristics, and to confirm the integrity and quality of the material to be used in clinical trials. A bulk supply of the active ingredient to support the necessary dosing in initial clinical trials must be secured. Data from the preclinical investigations and detailed information on proposed clinical investigations are compiled in an IND submission and submitted to the FDA before human clinical trials may begin. If the FDA does not formally communicate an objection to the IND within 30 days, the specific clinical trials outlined in the IND may go forward.
Clinical Phase
The clinical phase of drug development follows an IND submission and involves the activities necessary to demonstrate the safety, tolerability, efficacy, and dosage of the substance in humans, as well as the ability to produce the substance in accordance with the FDA’s cGMP requirements. Data from these activities are compiled in an NDA requesting approval to market the drug for a given use, or indication. Clinical trials must be conducted under the supervision of qualified investigators in accordance with good clinical practice, and according to IND-approved protocols detailing, among other things, the study objectives and the parameters, or endpoints, to be used in assessing safety and efficacy. Each trial must be reviewed, approved and conducted under the auspices of an independent Institutional Review Board (“IRB”), and each trial, with limited exceptions, must include all subjects’ informed consent. The clinical evaluation phase typically involves the following sequential process:
51
Phase I clinical trials are conducted in a limited number of healthy subjects to determine the drug’s safety, tolerability, and biological performance. The total number of subjects in Phase I clinical trials varies, but is generally in the range of 20 to 80 people (or less in some cases, such as drugs with significant human experience).
Phase II clinical trials involve administering the drug to subjects suffering from the target disease or condition to evaluate the drug’s potential efficacy and appropriate dose. The number of subjects in Phase II trials is typically several hundred subjects or less. Generally, early Phase II or Phase II-A studies look at the absorption, metabolism and pharmacodynamics of a drug rather than at clinical benefit or safety. The second stage of Phase II development, Phase II-B studies look more like Phase III trials because they are looking for continued indications of clinical efficacy and safety in larger groups of patients. The Company, not the FDA, has designated its upcoming phase II clinical trial as a Phase II-B trial for the these reasons, plus the fact that VLN cigarettes made from the Company’s VLN tobacco have already demonstrated efficacy in other Phase II trials, specifically the Phase II trial at the University of Minnesota which had a very similar protocol compared to our Phase II-B trial.
Phase III clinical trials are performed after preliminary evidence suggesting effectiveness has been obtained and safety, tolerability, and appropriate dosing have been established. Phase III clinical trials are intended to gather additional data needed to evaluate the overall benefit-risk relationship of the drug and to provide adequate instructions for its use. Phase III trials usually include several hundred to several thousand subjects.
Throughout the clinical testing phase, samples of the product made in different batches are tested for stability to establish shelf life constraints. In addition, increasingly large-scale production protocols and written standard operating procedures must be developed for each aspect of commercial manufacturing and testing.
The clinical trial phase is both costly and time-consuming, and may not be completed successfully within any specified time period, if at all. The FDA closely monitors the progress of each of the three phases of clinical trials that are conducted under an IND and may, at its discretion, reevaluate, alter, suspend, or terminate the testing at any time for various reasons, including a finding that the subjects or patients are being exposed to an unacceptable health risk. The FDA can also request additional clinical testing as a condition to product approval. Additionally, new government requirements may be established that could delay or prevent regulatory approval of our products under development. Furthermore, institutional review boards, which are independent entities constituted to protect human subjects in the institutions in which clinical trials are being conducted, have the authority to suspend clinical trials in their respective institutions at any time for a variety of reasons, including safety issues.
New Drug Application and Review
After the completion of Phase III clinical trials, the sponsor of the new drug submits an NDA to the FDA requesting approval to market the product for one or more indications. An NDA is a comprehensive, multi-volume application that includes, among other things, the results of all preclinical and clinical studies, information about the drug’s composition, and the sponsor’s plans for producing, packaging, and labeling the drug. In most cases, the NDA must be accompanied by a substantial user fee. The FDA has 60 days after submission to review the completeness and organization of the application, and may refuse to accept it for continued review, or refuse to file, if the application is found deficient. After filing, the FDA reviews an NDA to determine, among other things, whether a product is safe and effective for its intended use. Drugs that successfully complete NDA review may be marketed in the United States, subject to all conditions imposed by the FDA.
Prior to granting approval, the FDA generally conducts an inspection of the facilities, including outsourced facilities that will be involved in the manufacture, production, packaging, testing and control of the drug for cGMP compliance. The FDA will not approve the application unless cGMP compliance is satisfactory. If the FDA determines that the marketing application, manufacturing process, or manufacturing facilities are not acceptable, it will outline the deficiencies in the submission and will often request additional testing or information. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the marketing application does not satisfy the regulatory criteria for approval and refuse to approve the application by issuing a “not approvable” letter.
The length of the FDA’s review can range from a few months to several years or more. Once an NDA is in effect, significant changes such as the addition of one or more new indications for use generally require prior approval of a supplemental NDA including additional clinical trials or other data required to demonstrate that the product as modified remains safe and effective.
52
Fast Track Development
The Food and Drug Administration Modernization Act of 1997 (the “Modernization Act”), establishes a statutory program for relatively streamlined approval of “Fast Track” products, which are defined under the Modernization Act as new drugs or biologics intended for the treatment of a serious or life-threatening condition that demonstrates the potential to address unmet medical needs for this condition. Fast Track status requires an official designation by the FDA. The Tobacco Control Act provides that products for smoking cessation be considered for “Fast Track” designation by the FDA.
We have submitted a request to the FDA for Fast Track designation in June 2011 and, although there can be no assurance, we believe that our X-22 smoking cessation aid will be granted Fast Track designation by the FDA. The FDA has informed us it will respond to this request by August 2011. A product that receives Fast Track designation is eligible for (i) more frequent meetings with the FDA to discuss the drug’s development plan and ensure collection of appropriate data needed to support drug approval, and (ii) more frequent written correspondence from the FDA about such things as the design of the proposed clinical trials. A Fast Track product is also eligible for Rolling Review, in which sections of the NDA can be submitted for review by the FDA before the entire application is completed. A Fast Track product would ordinarily meet FDA criteria for Priority Review. The FDA goal for reviewing a drug with Priority Review status is six months from the filing of the NDA. There is no guarantee that X-22 will qualify for “Fast Track” designation at the FDA.
Post-Approval Phase
Once the FDA has approved a new drug for marketing, the product becomes available for physicians to prescribe in the U.S. After approval, we must comply with post-approval requirements, including ongoing compliance with cGMP regulations, delivering periodic reports to the FDA, submitting descriptions of any adverse reactions reported, and complying with drug sampling and distribution requirements. We are required to maintain and provide updated safety and efficacy information to the FDA. We must also comply with requirements concerning advertising, product promotions, and labeling.
X-22 Clinical Trials
We have met with the FDA regarding the remaining X-22 clinical trials and, based on the FDA’s guidance, we plan to conduct a small Phase II-B trial and two larger and concurrent Phase III trials with the same protocols that entail measuring the quitting efficacy of the X-22 cigarette against a typical cigarette with conventional nicotine content that is visually indistinguishable from X-22 (the “active control”). The Phase II-B optimization trial will consist of approximately 200 participants over a six-week treatment period, and we expect the Phase III trials will use the same protocol with larger groups of participants. Accordingly, in all of the remaining clinical trials, half of the participants will smoke X-22 for six weeks and half of the participants will smoke the active control for six weeks, with all participants instructed to quit on the last day of the six-week treatment period. We expect Phase II-B to cost approximately $1.1 million. The Company will require additional funding of approximately $10 million to complete the Phase III clinical trials. We currently plan to seek to raise approximately $15 million in the fourth quarter 2011 through the sale of our common stock and/or securities convertible into common stock to fund Phase III trials, modified risk exposure studies and general working capital requirements of the Company. Before we can raise additional capital, the Company must satisfy the registration rights granted to investors who acquired shares of its common stock in the January 25, 2011 Private Placement and Merger. These registration rights provide that 22nd Century Group must have an effective registration statement covering the shares subject to the registration rights for a period of 90 days before it can sell any equity securities or securities convertible into equity securities.
Smokers who do not smoke over the four-week period immediately following the conclusion of the six-week treatment period (weeks 7 through 10) are considered abstinent. The abstinence (quit) rates of the X-22 group and the active control group will then be compared for statistical significance. With additional and adequate funding, we will be able to conduct our two Phase III clinical trials concurrently with the same protocols in order to expedite the FDA approval process. We have submitted our Pre-IND (PIND 103,589) and IND to the FDA, and we have initiated many activities for our Phase II-B clinical trial, including creating clinical trial related materials and training of personnel at our three clinical trial sites. Our IND contains all of the information and data of our PIND 103,589 plus standard tobacco industry smoke analyses of the X-22 clinical trial cigarette and the active control. Before Phase III trials, some additional information and testing of X-22 and its tobacco are required by the FDA, some of which we already have from our former licensee’s IND 69,185. All analyses that the FDA requires on the clinical trial materials are efficiently outsourced to Arista Laboratories which is one of the industry leaders in tobacco and tobacco smoke analyses with whom we have contracted since 2008. With additional and adequate funding, we intend to initiate our Phase III clinical trials in the fourth quarter of 2011 and to file our NDA with the FDA for X-22 in 2012. We expect the FDA to Fast Track the approval of X-22 and that we should receive FDA approval to commence the marketing and sales of X-22 in the U.S. as early as the fourth quarter of 2012. There is no guarantee that X-22 will qualify for “Fast Track” designation at the FDA.
53
Following FDA approval, we intend to register X-22 as a Medicinal Product (pharmacological) for smoking cessation with the European Medicines Agency (“EMA”) and other international FDA-equivalent agencies in targeted countries. Regulatory approval for X-22 as a smoking cessation aid is not required in some international markets since, unlike the FDA, some foreign drug regulatory agencies do not require approval to market a product as a smoking cessation aid if the product is allowed to be sold for other purposes.
Modified Risk Cigarettes
The Tobacco Control Act, which became law in June 2009, prohibits the FDA from banning cigarettes outright or mandating that nicotine levels be reduced to zero. However, among other things, it allows the FDA to require the reduction of nicotine or any other compound in cigarettes. In 2009, the Tobacco Control Act banned all sales in the United States of cigarettes with flavored tobacco (other than menthol). As of June 2010, all cigarette companies were required to cease using the terms “low tar,” “light” and “ultra light” in describing cigarettes sold in the United States. We believe this new regulatory environment represents a paradigm shift for the tobacco industry and will create opportunities for us in marketing BRAND A and BRAND B and in licensing our proprietary technology and/or tobaccos to larger competitors.
For the first time in history, a U.S. regulatory agency will scientifically evaluate cigarettes that may pose lower health risks as compared to conventional cigarettes. The Tobacco Control Act establishes procedures for the FDA to regulate the labeling and marketing of modified risk tobacco products, which includes cigarettes that (i) reduce exposure to tobacco smoke toxins and/or (ii) pose lower health risks, as compared to conventional cigarettes (“Modified Risk Cigarettes”). The Tobacco Control Act requires the FDA to issue specific regulations and guidance regarding applications that must be submitted to the FDA for the authorization to label and market Modified Risk Cigarettes. The FDA has yet to release its regulations regarding modified risk tobacco products. However, based in part on the timelines contained in the Tobacco Control Act, we expect the FDA to issue such regulations and guidance within the next year. Although we believe that BRAND A and BRAND B will qualify as Modified Risk Cigarettes, further research and exposure studies are needed for the FDA to make this determination for Brand A and Brand B. In addition, the Tobacco Control Act allows the FDA to mandate the use of reduced risk technologies in conventional tobacco products and cigarettes (e.g., Marlboro®) which could create opportunities for us to license our proprietary technology and/or our tobaccos to larger competitors.
We have begun to supply our cigarettes to researchers at the National Transdisciplinary Tobacco Use Research Centers in the U.S. so studies can be conducted to obtain additional information on our products. We expect this information will assist us, along with our own funded studies, in obtaining the necessary FDA authorizations to market BRAND A and BRAND B as Modified Risk Cigarettes and to obtain FDA approval for X-22 as a prescription smoking cessation aid.
In November 2010, as required by the Tobacco Control Act, the FDA issued a proposed rule to modify the required warnings that appear on cigarette packages and in cigarette advertisements. These warning were finalized on June 21, 2011 and consist of nine new textual warning statements accompanied by color graphics depicting the negative health consequences of smoking. The FDA selected nine images from the originally proposed 36 images after reviewing the relevant scientific literature, analyzing the results from an 18,000 person study and considering more than 1,700 comments from a variety of groups. The graphic health warnings will be located beneath the cellophane wrapping on cigarette packages, and will comprise the top 50 percent of the front and rear panels of cigarette packages. The graphic health warnings will occupy 20 percent of a cigarette advertisement and will be located at the top of the advertisement. Each warning is accompanied by a smoking cessation phone number, 1-800-QUIT-NOW. When implemented in September 2012, all cigarettes manufactured for sale or distribution in the United States will need to include the new graphic health warnings on their packages. Any reduction in the number smokers will probably reduce the demand for MAGIC and RED SUN, as well as X-22, Brand A and B, if and when approved/authorized by the FDA.
Biomass Products
Biomass Products are products such as ethanol made from the organic material, usually plants grown over a given area. We have funded extensive biomass field trials conducted by North Carolina State University (“NCSU”), and studies on feedstock digestibility and bioconversion at the National Renewable Energy Lab. Bioconversion is the conversion of organic matter into a source of energy, ethanol in our research, through the action of microorganisms. The results of our biomass studies have been summarized in a comprehensive feasibility study relating to our nicotine-free tobacco biomass crop (Verfola) to produce a variety of bioproducts. First, protein and other plant fractions are extracted, and then biofuels and other products are produced from the remaining cellulosic residue. In 2008, we put our biomass development projects on hold so that our management could focus its attention and resources on our modified risk cigarette business and our X-22 smoking cessation business. We do not plan to move forward with potential biomass business activities until some period of time after FDA approval of X-22 or FDA authorization to market Brand A or Brand B as a Modified Risk Cigarette. We currently are not spending any capital for such potential biomass business activities nor do we have any current plans to raise any capital for such potential biomass business activities.
54
Tobacco has a number of advantages as a starting point for development of novel bioproduct crop systems. Because tobacco is a widely cultivated crop, grown in over 100 countries throughout the world, tobacco agronomy is highly understood. For decades tobacco has been used as a model system for plant biology, and recently the tobacco genome has been mapped. Tobacco plants rapidly sprout back after each harvest and produce large amounts of leaf and total biomass. Tobacco grown for cigarettes yields about 3,000 pounds of cured leaf per acre (~20% moisture) per year from 7,500 tobacco plants. In our field trials in North Carolina, nicotine-free tobacco grown for biomass yields about 100,000 pounds of fresh weight per acre (which equals 10,000 pounds of dry weight) per year with multiple machine harvests from about 80,000 tobacco plants.
Research and Development
Most research and development (R&D) since our inception have been outsourced to highly qualified groups in their respective fields. Since 1998, 22nd Century has had multiple R&D agreements with North Carolina State University (“NCSU”) resulting in exclusive worldwide licenses to various patented technologies. We have utilized the model of many public-sector research organizations which entails obtaining an exclusive option or license agreement to any invention arising out of the funded research. In all cases, we fund and exclusively control all patent filings as the exclusive licensee. This model of contracting with public-sector researchers has enabled 22nd Century to control R&D costs while achieving our desired results, including obtaining exclusive intellectual property rights relating to all of our outsourced R&D.
Other R&D partners with the same arrangement have included the National Research Council of Canada, Plant Biotechnology Institute in Saskatoon, Canada (“NRC”), and the Nara Institute of Science and Technology in Nara, Japan (“NAIST”). Our R&D agreements with NCSU, NRC and NAIST have expired in 2009 and the majority these agreements have involved the biosynthesis of nicotine in plants. During the years ended December 31, 2010 and 2009, we incurred research and development expenses of approximately $364,000 and $540,000, respectively. In 2010, NAIST assigned all of their worldwide patents to us which were a result of our R&D at NAIST and that were previously licensed to 22nd Century on an exclusive basis. We did not have any outsourced R&D projects during 2010.
55
Finally, other than our planned clinical trials for X-22 and exposure studies for our Modified Risk Cigarette candidates, we have no other substantial third-party R&D commitments requiring funding. However, we do plan to carry out a minimal amount of other R&D not to exceed $250,000 per year, including the execution of more field trials from the inventory of hundreds of seed lots that resulted from our R&D at NCSU, NRC and NAIST.
Employees
We currently employ six people, none of whom are represented by a union, and we consider our employee relations to be good.
Description of Property
Our principal administrative offices are located in Williamsville, New York. We currently lease 1,302 square feet of office space at 8201 Main Street, Suite 6, Williamsville, New York 14221 at an aggregate cost of approximately $1,584 per month pursuant to a lease expires on October 31, 2011. This lease subject to automatic renewal for an additional one-year term absent notice of non-renewal from either party. We believe that our administrative office space is sufficient to support our current operations, although we may require additional space as the Company expands.
Legal Proceedings
From time to time we may be involved in claims arising in the ordinary course of business. To our knowledge, no legal proceedings, governmental actions, investigations or claims are currently pending against us or involve us that, in the opinion of our management, could reasonably be expected to have a material adverse effect on our business and financial condition.
56
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND
RESULTS OF OPERATIONS
The following discussion highlights the principal factors that have affected our financial condition and results of operations as well as our liquidity and capital resources for the periods described.
On January 25, 2011, 22nd Century Ltd completed a reverse merger transaction (the “Merger”) with 22nd Century Group. 22nd Century Ltd is a wholly owned subsidiary of 22nd Century Group, which continues to operate the business of 22nd Century Ltd. All reference to shareholders or common shares include the historical members and membership Units of 22nd Century Ltd because, in the Merger, such Units were exchanged for common shares on a one-for-one basis and from an accounting standpoint, they are equivalent. The Merger is being accounted for as a reverse acquisition and a recapitalization; 22nd Century Ltd is the acquirer for accounting purposes. Consequently, the assets and liabilities and the historical operations that are reflected in the financial statements prior to the Merger are those of 22nd Century Ltd and are recorded at the historical cost basis of 22nd Century Ltd, and the consolidated financial statements since completion of the Merger include the assets and liabilities of 22nd Century Ltd, historical operations of 22nd Century Ltd and operations of 22nd Century Group from the closing date of the Merger. For purposes of this Management’s Discussion and Analysis of Financial Condition and Results of Operations, references to the “Company,” “we,” us” or “our” refer to the operations of 22nd Century Group and its direct and indirect subsidiaries for the periods described herein.
This prospectus contains statements that the Company believes to be “forward-looking statements” within the meaning of the “safe harbor” provisions of the Private Securities Litigation Reform Act of 1995. Forward-looking statements include, without limitation, any statement that is not a statement of historical fact, including, without limitation, statements regarding the Company’s business strategy and plans and objectives of management for future operations or that may predict, forecast, indicate or imply future results, performance or achievements. The words “estimate,” “project,” “intend,” “forecast,” “anticipate,” “plan,” “planning,” “expect,” “believe,” “will,” “will likely,” “should,” “could,” “would,” “may” or the negative of such words or words or expressions of similar meaning are intended to identify forward-looking statements. These forward-looking statements are not guarantees of future performance, and all such forward-looking statements involve risks and uncertainties, many of which are beyond the Company’s ability to control. Actual results may differ materially from those expressed or implied by such forward-looking statements as a result of various factors including but not limited to the following and the risk factors set forth our S-1 registration statement filed with the SEC on April 8, 2011, including uncertainties regarding the process of obtaining Food & Drug Administration approval for our products, including time and expenses involved in seeking such approval, the Company’s inability to continue as a going concern, the Company’s inability to achieve or sustain profitability given a history of losses, the uncertainty of the Company’s ability to generate positive cash flow, the difficult of evaluating the Company’s current business and future prospects given the Company’s limited operating history, fluctuations in working capital requirements, continued instability in credit and financial market conditions, dependence on third parties to manufacture, market, sell and distribute the Company’s products, dependency on market acceptance of products among physicians, patients, third-party payers and the medical community, performance of competitors in the field, retention of key personnel, and other factors. All forward-looking statements and other information in this prospectus speak only as of the date of this prospectus. We do not undertake, and we disclaim, any obligation to update any forward-looking statements or to announce revisions to any of the forward-looking statements.
Our business activities are affected by many factors, including regulatory actions by the FDA and state governments, many of which are beyond the control of our management. Statements regarding the following subjects are forward-looking by their nature: management’s plans, FDA matters including matters such as obtaining “Fast Track” status and clinical trials including our Phase II-B clinical trial, expected future activities under our research cigarette program and future capital needs.
Please see “Risk Factors” as set forth in this S-1 registration statement.
Overview
We have operated at a loss since 2006, when we increased our research and development expenditures. Our license agreement with our former licensee was discontinued in 2007. In the first three months of 2011 we realized revenue of $117,456 from our research cigarette program; in 2010, we realized revenue of $49,784 from this program and in 2009 we realized sales of $27,612 from limited test marketing of our cigarettes. During 2009 and 2010 we transitioned from solely developing proprietary technology and tobacco to developing and commercializing our own products. In March 2011, our subsidiary, Goodrich Tobacco introduced two of our products, RED SUN and MAGIC, into customary market channels for tobacco products.
57
Our prospects depend on our ability to generate and sustain revenues from our X-22 smoking cessation aid and cigarettes made with our proprietary tobacco. Our ability to generate meaningful revenue from X-22, especially in the United States, depends on FDA approval, and our ability to generate meaningful revenue from our proprietary cigarette products depends in large part on obtaining FDA authorization to market these brands as modified risk cigarettes (as defined in the Overview of Management Discussion). Once our products are approved by the FDA, we must still meet the challenges to gain consumer acceptance, including successful marketing and distribution. We do not expect FDA approval of X-22 until the fourth quarter of 2012 at the earliest. We believe the FDA will issue regulations for modified risk tobacco products within the next year, and we therefore expect to submit applications in 2012 to the FDA to authorize the marketing and labeling of our proprietary cigarette products as modified risk cigarettes. This process is likely to take at least one year. Until these approvals and authorizations are received, sales of our proprietary cigarette products will be limited and will only include RED SUN, MAGIC and SPECTRUM. We intend to focus our marketing efforts for RED SUN and MAGIC on tobacconists, smoke shops and tobacco outlets. Accordingly, our cash flow from product sales will be limited and, in addition to the net proceeds from the Private Placement that closed on January 25, 2011, we will need cash from equity or debt financing to continue operations. As additional funds are required, we may raise such funds from time to time through public or private sales of equity or debt securities. Financing may not be available on acceptable terms, or at all, and our failure to raise capital when needed could materially adversely impact our growth plans and its financial condition and results of operations. Additional equity financing will be dilutive to the ownership interests of holders of the Common Stock, and debt financing, if available, may involve significant cash payment obligations and covenants that restrict our ability to operate our business.
In connection with our FDA activities we will incur substantial costs related to clinical trials and smoke exposure studies related to our modified risk product candidates. Our total budget for the Phase II-B project is approximately $1.1 million, and we paid approximately $350,000 of that amount through March 31, 2011.
At March 31, 2011, the Company had current assets of $1,961,651 (including cash of $872,401) and current liabilities of $1,458,997. On January 25, 2011, the Company completed the Private Placement of securities resulting in approximately $3.4 million in net cash proceeds and a reduction of debt obligations to shareholders that were on the balance sheet at December 31, 2010 of approximately $614,000, which were exchanged for equity interests in the Private Placement. These proceeds were sufficient to enable the Company to make substantial reductions in its outstanding current liabilities, which totaled $1,458,997 at March 31, 2011, as compared to $4,823,635 at December 31, 2010.
The 2009 Family Smoking Prevention and Tobacco Control Act (“Tobacco Control Act”) granted the FDA authority over the regulation of all tobacco products. While it prohibits the FDA from banning cigarettes outright, it allows the FDA to require the reduction of nicotine or any other compound in tobacco and cigarette smoke. The Tobacco Control Act also banned all sales in the U.S. of cigarettes with flavored tobacco (other than menthol). As of June 2010, all cigarette companies were required to cease the use of the terms “low tar,” “light” and “ultra light” in describing cigarettes sold in the U.S. Besides numerous other regulations, including certain marketing restrictions, for the first time in history, a U.S. regulatory agency will scientifically evaluate cigarettes that may pose lower health risks as compared to conventional cigarettes.
The Tobacco Control Act establishes procedures for the FDA to regulate the labeling and marketing of modified risk tobacco products, which includes cigarettes that (i) reduce exposure to tobacco smoke toxins and/or (ii) pose lower health risks, as compared to conventional cigarettes (“modified risk cigarettes”). The Tobacco Control Act requires the FDA to issue specific regulations and guidance regarding applications that must be submitted to the FDA for the authorization to label and market modified risk cigarettes. Based in part on the timelines contained in the Tobacco Control Act, we expect the FDA to issue such regulations and guidance within the next year. We believe that two of our cigarette products, which we refer to as BRAND A and BRAND B, will qualify as modified risk cigarettes. Compared to commercial cigarettes, we believe that the tobacco in BRAND A has approximately 95% less nicotine than tobacco in cigarettes previously marketed as “light” cigarettes, and BRAND B’s smoke contains the lowest amount of “tar” per milligram of nicotine.
58
Year Ended December 31, 2010 Compared to Year Ended December 31, 2009
Revenues. In 2010, we realized revenue of $49,784 from our research cigarette program and in 2009 we realized sales of $27,612 from limited test marketing our cigarettes in customary market channels for tobacco products.
Costs of goods sold. In 2010 costs of goods sold of $27,964 consisted mainly of product design costs related to our research cigarette program. These cigarettes are sold directly to researchers and do not enter the customary market channels for tobacco products. In 2009, costs of goods sold of $20,112 included federal excise taxes assessed at the manufacturer’s level on products sold in.
General and Administrative Expense. General and administrative expense was $590,826 in 2010, an increase of $310,117, or 110%, from $280,709 in 2009. Approximately, $167,000 of this increase was related to increased administrative payroll mainly due to adding two executive officers during 2010. In addition, approximately $92,000 of the increase in 2010 was for accounting, tax, and audit services. The balance was in various other expense categories such as supplies, printing and travel.
Research and Development Expense. Research and Development expense was $363,781 in 2010, a decrease of $176,519, or 32.6%, from $540,300 in 2009. The decrease was primarily due to a reduction compensation expense of approximately $168,000 as a result of an equity compensation award in 2009 that was nearly fully amortized to expense in that year.
Amortization Expense. Amortization expense relates solely to capitalized patent and trademark costs. Amortization expense increased 13.6% in 2010 to $164,456 from $144,792 in 2009. This increase of $19,644 is due to our investment in patents and trademarks in 2010 and 2009 of $147,912 and $227,942, respectively.
Interest Expense and Debt Expense. Interest expense and debt expense, which includes interest amortization of debt discount and debt issuance costs, increased in 2010 to $326,404 from $268,503 in 2009. This increase of $57,901 or 21.6% was mainly a result of additional borrowings and interest charges from a vendor offset by reduced amortization of debt discount.
Net Loss. We had a net loss in 2010 of $1,423,647 as compared to a net loss of $1,226,804 in 2009. The increase in the net loss of $196,843, or 16%, was mainly a result of higher total operating expenses in 2010 as compared to 2009.
Three Months Ended March 31, 2011 Compared to Three Months Ended March 31, 2010
Sales. In the three months ended March 31, 2011, we realized revenue of $117,456 from our research cigarette program. We had no sales in the first three months of 2010.
Other income. In the three months ended March 31, 2011, we recognized other income of $49,216 from a therapeutic grant award we received in fourth quarter of 2010; $174,324 remains to be recognized, which recognition is anticipated to occur in the second and third quarters of 2011 as we complete our Phase II-B clinical trial.
Costs of goods sold. In the first three months of 2011, costs of goods sold related to revenues under our research cigarette program was $47,002 or 40% of the related sales. There were no costs of goods sold in the first three months of 2010, because we had no sales during that period.
Research and Development Expense. Research and development expense was $216,291 in the three months ended March 31, 2011, an increase of $120,871, or 127%, from $95,420 in the three months ended March 31, 2010. This increase was primarily due to expenditures related to our Phase II-B clinical trials. We initiated this effort late in the fourth quarter of 2010 so there were no Phase II-B activities in the first three months of 2010.
General and Administrative Expense. General and administrative expense was $325,938 in the three months ended March 31, 2011, an increase of $229,830, or 239%, from $96,108 in three months ended March 31, 2010. Approximately $119,000 of this increase was related to increased legal and accounting and audit fees. In addition, approximately $75,000 of the increase was for payroll and directors’ fees due to increasing our administrative headcount from two to five people in the first quarter of 2011, as compared to the first quarter of 2010, and holding our first Board of Directors’ meeting since the Merger. The balance was in various other expense categories such as supplies, SEC filing fees and investor communications.
59
Sales and marketing costs. Sales and marketing costs were $171,525 in the three months ended 2011; we had no costs categorized as such in the three months ended March 31, 2010. The costs in 2011 related to product testing, product and packaging, and design, product branding, samples and advertising.
Amortization and Depreciation Expense. Amortization and depreciation expense relates almost entirely to capitalized patent and trademark costs. Amortization and depreciation expense increased 9% in the three months ended March 31, 2011 to $43,577, up from $39,860 in the three months ended March 31, 2010. This increase of $3,717 is mainly due to our additional investment in patents and trademarks in 2010 of $147,912.
Interest Expense and Debt Expense. Interest expense and debt expense, which include interest amortization of debt discount and debt issuance costs, decreased in the three months ended March 31, 2011 to $14,005, from $73,629 in the three months ended March 31, 2010. This decrease of $59,624 or 81% was mainly a result of reduced borrowings during the first quarter of 2011 compared to the first quarter of 2010. In January 2011, a substantial portion of our outstanding interest bearing debt was repaid or converted into shares of our common stock. New long term obligations in the amounts of $350,000 and $237,000 were entered at the end of the first quarter and had minimal impact on interest expense but will result in additional interest expense starting in the second quarter of 2011.
Net Loss. We had a net loss in the three months ended March 31, 2011 of $651,666 as compared to a net loss of $305,017 in the three months ended March 31, 2010. The increase in the net loss of $346,649, or 114%, was mainly a result of higher operating expenses and clinical trial expenses in the three months ended March 31, 2011, as compared to the three months ended March 31, 2010, offset by revenues and reduced interest and debt expense.
Liquidity and Capital Resources
Summary of Balances and Recent Sources and Uses
As of March 31, 2011, we had working capital of approximately $0.5 million compared to negative working capital of approximately $4.1 million at December 31, 2010. The improvement in our working capital position of $4.6 million was mainly a result of proceeds from the January 25, 2011 Private Placement and the transfer from accounts payable of $587,000 to notes payable as part of payment arrangements with two vendors for past due amounts we owed, offset by our net loss for the three months ended March 31, 2010.
Cash demands on operations
In 2009 and 2010, we operated at a loss and operating activities consumed more than $1,000,000 in cash during this two year period. In the three months ended March 31, 2011 we also operated at a loss while cash used in operating actives was approximately $1,653,000. Although the amount of cash used in operations for the first quarter of 2011 was impacted by seasonal purchases of inventory and payments on past due amounts we owed to vendors, cash used in operating activities will continue at higher than historical levels for the balance of 2011 due to increased headcount and expenditures related to the pursuit of FDA approval of our smoking cessation aid and modified risk cigarettes.
If we are unable to improve operations or raise funds during the next twelve months, there would be a material adverse effect on our ability to meet our working capital needs and the risk that we would be unable to continue operations would increase. We intend and expect to require additional funds before the end of 2011 to complete the FDA clinical trials, complete exposure studies for our potential modified risk tobacco products, launch X-22 and pay loan, note obligations as they become due. As additional funds are required, we may raise such funds from time to time through public or private sales of equity or debt securities. Financing may not be available on acceptable terms, or at all, and our failure to raise capital when needed could materially adversely impact our growth plans and its financial condition and results of operations. Additional equity financing will be dilutive to the ownership interests of holders of the Common Stock, and debt financing, if available, may involve significant cash payment obligations and covenants that restrict our ability to operate our business.
60
Net Cash used in Operating Activities.
In 2010, $909,939 of cash was used in operating activities compared to $165,213 of cash used in operating activities in 2009. This increase use of cash of $744,726 was due to the increase of approximately $490,000 in the cash portion of the net loss in 2010 as compared to 2009. The balance of the increase was a result of the net increase in working capital components related to operations.
In the first three months of 2011, $1,653,309 of cash was used in operating activities compared to $381,656 of cash used in operating activities in the first three months of 2010. This increased use of cash of $1,271,653 was due to the increase of $435,827 in the cash portion of the net loss in the 2011 three-month period as compared to the comparable 2010 period. The balance of the increase was a result of the net change in working capital components related to operations. This increase was in large part due to having the resources to pay past due amounts to third parties and to pay for increased tobacco leaf inventory, materials for RED SUN, MAGIC and SPECTRUM.
Net Cash used in Investing Activities.
In 2010, we used $108,116 of cash from the net activity related to third party costs incurred for patents and trademarks as compared to $6,840 used in 2009 because we had to pay a greater portion of current charges in 2010 than in 2009.
In the first three months of 2011, we used $523,200 of cash related to third party costs incurred for patents and trademarks and the acquisition of office furniture and fixtures as compared to $35,351 used in the first three months of 2010. This increase was partially attributable to our payment in the first three months of 2011 of $500,000 towards charges from the proceeds of our January 25, 2011 Private Placement that were deferred in prior periods.
Net Cash From Financing Activities.
During 2010, we generated $1,018,207 in our financing activities through the issuance of units, warrants, notes and advances from members with total proceeds of $1,275,411 offset by $120,028 in net repayments of advances from a related party, payment of private placement costs of $60,976 and repayments of debt of $76,200. During 2009, we generated net cash of approximately $159,000 from financing activities. Approximately $55,000 was generated by the issuance of notes and related warrants. We also received cash advances from our members and a related party of $105,000 and repaid $1,000 of bank demand loans.
During the first three months of 2011, we generated $3,048,600 in our financing activities primarily through the issuance of shares of 22nd Century Group’s common stock and warrants to purchase 22nd Century Group’s common stock in the January 25, 2011 Private Placement. This resulted in net proceeds of $3,461,708 offset by $393,275 in payments on notes to shareholders and $21,272 in net advances to a related party. During the first three months of 2010, we generated net cash of $417,000 from our financing activities; $450,000 was generated by the issuance of notes and warrants offset by the payment of $20,000 in deferred Private Placement costs and $13,000 in net advances to a related party.
We received approximately $3.4 million in net cash proceeds from a Private Placement that closed on January 25, 2011. We used approximately $1,400,000 of the proceeds to retire debt obligations that had matured or make payments on past due amounts owed to vendors and North Carolina State University (“NCSU”). We paid NCSU $400,000 in February 2011 out of proceeds from the Private Placement and to date we have paid NCSU approximately $53,000 for patent costs. The balance due to NCSU of approximately $728,000 was due in May 2011. We have requested NCSU to continue to defer payment of the balance owed until 2012 so that we would have sufficient funds in 2011 to complete our Phase II-B clinical trial, which we expect to cost approximately $1.1 million. We expect NCSU to comply with this request. As our licensor for 12 years, we believe NCSU has a vested interest in 22nd Century completing the Phase II-B clinical trials. Since 1998, we have funded approximately $6 million to NCSU in royalties, patent support payments and R&D, so we believe the amount we owe NCSU is not significant in light of the magnitude of the ongoing relationship between NCSU and the Company. If NCSU defers payment of the balance owed until 2012, we believe that the remaining proceeds from the January 25, 2011 Private Placement, together with cash receipts from our research cigarette program (approximately $680,000 from the purchase order we have received from RTI), will be sufficient to enable the Company to complete the Phase II-B clinical trial for X-22 and submit the results to the FDA later this year. However, we expect to require additional funds before the end of 2011 to complete the FDA Phase III clinical trials (which we expect to cost approximately $10 million), complete exposure studies for our potential modified risk tobacco products (which we expect to cost approximately $2 million), launch X-22, pay loan and note obligations as they become due, and continue to make payments to NCSU. We currently plan to seek to raise approximately $15 million in the fourth quarter 2011 through the sale of our common stock and/or securities convertible into common stock to fund Phase III trials, modified risk exposure studies and general working capital requirements of the Company. Before we can raise additional capital, we must satisfy our registration rights obligations with respect to investors who acquired shares of 22nd Century Group’s common stock in the January 25, 2011 Private Placement and Merger. These registration rights provide that the Company must have an effective registration statement covering the shares that are subject to these registration rights for a period of 90 days before it can sell any equity securities or securities convertible into equity securities. Our future capital requirements will depend on many factors, including the progress made in our X-22 clinical trials. As additional funds are required, we may raise such funds from time to time through public or private sales of equity or debt securities. Financing may not be available on acceptable terms, or at all, and our failure to raise capital when needed could materially adversely impact our growth plans and its financial condition and results of operations. Additional equity financing will be dilutive to the ownership interests of holders of common stock of 22nd Century Group, and debt financing, if available, may involve significant cash payment obligations and covenants that restrict our ability to operate our business.
61
Critical Accounting Policies and Estimates
Accounting principles generally accepted in the United States of America, or U.S. GAAP, require estimates and assumptions to be made that affect the reported amounts in our consolidated financial statements and accompanying notes. Some of these estimates require difficult, subjective and/or complex judgments about matters that are inherently uncertain and, as a result, actual results could differ from those estimates. Due to the estimation processes involved, the following summarized accounting policies and their application are considered to be critical to understanding our business operations, financial condition and results of operations.
Revenue Recognition
Revenue is recognized when tobacco products are shipped to customers and title passes. We also record appropriate provisions for rebates and discounts and credits for returns. These amounts are estimated based on information and historical experience. The Company was awarded a research cigarette program which includes three stages. Stages 1 and 2 are for product design, manufacture planning and material procurement for 9,000,000 cigarettes. The Company has completed each of Stages 1 and 2 and has recognized the revenue when the billing for each of these stages was approved by the customer. Stage 3 is the manufacture and shipment of the research cigarettes. Revenue under this stage will be recognized at the point when the cigarettes are shipped to the customer and title has transferred to the customer. In 2010, the Company received a grant as partial support for its Phase II-B clinical trial. This income will be recognized as a reduction of the costs of the clinical trial as such costs are incurred.
Impairment of Long-Lived Assets
We review the carrying value of amortizing long-lived assets whenever events or changes in circumstances indicate that the historical cost-carrying value of an asset may no longer be appropriate. We also assess recoverability of the asset by estimating the future undiscounted net cash flows expected to result from the asset, including eventual disposition. If the estimated future undiscounted net cash flows are less than the carrying value of the asset, an impairment loss is recorded equal to the difference between the asset’s carrying value and its fair value. Non-amortizing intangibles (trademarks) are reviewed annually for impairment. We have not recognized any impairment losses during the period ended March 31, 2011 or during the two years ended December 31, 2010.
Amortization Estimates
We generally determine amortization based on the estimated useful lives of the assets and record amortization expense on a straight-line method over such lives. The remaining life of the patent is generally used to determine the estimated useful life of the related patent costs.
Valuation of our Equity Securities
We have issued shares of 22nd Century Group’s common stock to satisfy obligations to vendors or employees that were due in cash. These shares have been valued based on the cash value of the obligation satisfied by their issuance. We have also issued warrants to purchase shares of 22nd Century Group’s stock in connection with the issuance of debt obligations. These warrants have been valued based on the value ascribed to the underlying shares issued in cash transactions or in settlement of cash obligations.
62
Income taxes
Prior to the closing of the Merger, 22nd Century Ltd was organized as a limited liability company and treated as a partnership for income tax purposes; accordingly, prior to the Merger, 22nd Century Ltd was not directly responsible for income taxes (income and loses passed through to its LLC members) and did not have to account for them. Since the closing of the Merger, our results of operations are subject to income taxes, and accounting for income taxes is now a critical accounting policy. In addition, we now account for deferred tax assets and liabilities, including the evaluation of the recoverability of deferred tax assets.
Derivative Financial Instruments
The warrants to purchase shares of 22nd Century Group stock that were issued in connection with the Merger are treated as derivative instruments for accounting purposes. Accordingly, upon issuance, these instruments are reported as liabilities rather than equity. We do not use derivative instruments to hedge exposures to cash flow, market or foreign currency risks. We evaluate all of our financial instruments to determine if such instruments are derivatives or contain features that qualify as embedded derivatives. For derivative financial instruments that are accounted for as liabilities, the derivative instrument is initially recorded at its fair market value and then is revalued at each reporting date, with changes in fair value reported in the consolidated statement of operations. We use a lattice model approach which for valuing our outstanding warrants classified as derivative instruments which includes probability weighted estimates of future events including volatility of our common stock. The classification of derivative instruments, including whether such instruments should be recorded as liabilities or equity, is evaluated at the end of each reporting period. Derivative instrument liabilities are classified in the balance sheet as current or non-current based on whether or not net-cash settlement of the derivative instrument could be required within twelve months of the balance sheet date.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements as defined by Item 303(a)(4) of Regulation S-K.
63
DIRECTORS AND EXECUTIVE OFFICERS
|
Name
|
Age
|
Position
|
||
|
Joseph Pandolfino
|
43
|
Chief Executive Officer and Director
|
||
|
Henry Sicignano, III
|
43
|
President, Secretary and Director
|
||
|
Michael R. Moynihan, Ph.D.
|
58
|
Vice President of R&D
|
||
|
C. Anthony Rider
|
59
|
Chief Financial Officer and Treasurer
|
||
|
Joseph Alexander Dunn, Ph.D.
|
57
|
Director
|
||
|
James W. Cornell
|
54
|
Director
|
||
|
Steven Katz
|
|
63
|
|
Director
|
Our directors and executive officers hold office until the earlier of their death, resignation, removal or until their successors have been duly elected and qualified. Our executive officers are appointed by the board of directors and serve at the discretion of the board. There are no family relationships among our directors and executive officers. In connection with the Merger, our Board was expanded to five members. The sole officer and sole member of the Board prior to the closing of the Merger, David Rector, resigned as an officer and a director after the closing of the Merger. The current members of our Board are Joseph Pandolfino, Henry Sicignano III, Joseph Alexander Dunn, Ph.D., James W. Cornell and Steven Katz. Our current executive officers are Joseph Pandolfino, Chief Executive Officer, Henry Sicignano III, President and Secretary, Michael R. Moynihan, Ph.D., Vice President of R&D, and C. Anthony Rider, Chief Financial Officer, Treasurer and Secretary. Each of Messrs. Pandolfino, Sicignano and Rider were executive officers of 22nd Century prior to the closing of the Merger.
Joseph Pandolfino, MBA, Chief Executive Officer and Director
Mr. Pandolfino has served as our Chief Executive Officer and as a director since the closing of the Merger. He founded 22nd Century in 1998 and has over 15 years experience in all aspects of the tobacco industry, including 12 years with genetically-engineered tobacco. He served as President of 22nd Century from its inception until April 2010 and as Chief Executive Officer of 22nd Century since April 2010. Mr. Pandolfino oversees our operations, strategy and product development. Mr. Pandolfino holds a B.S. Degree in Business Administration from Medaille College and an M.B.A. Degree from the State University of New York at Buffalo. Mr. Pandolfino’s significant experience in all aspect of the tobacco industry as well as his experience leading 22nd Century led to our conclusion that he should serve as a director of our Company.
Henry Sicignano, III, MBA, President and Director
Mr. Sicignano has served as our President and Secretary since the closing of the Merger, as a director since March 4, 2011, and as President of 22nd Century since April 2010. From August 2005 to April 2009, Mr. Sicignano served as a General Manager and as the Director of Corporate Marketing for NOCO Energy Corp., a petroleum products company; and from March 2003 to July 2005, as Vice President of Kittinger Furniture Company, Inc., a fine furniture manufacturer. From February 1997 through July 2002, he served as Vice President and Marketing Director of Santa Fe Natural Tobacco Company, a specialty tobacco company, prior to the sale of that company to R.J. Reynolds Tobacco Company in 2002. Mr. Sicignano holds a B.A. Degree in Government from Harvard College and a M.B.A. Degree from Harvard University. Mr. Sicignano’s extensive experience in management, including in the tobacco industry, led to our conclusion that he should serve as a director of our Company.
Michael R. Moynihan, Ph.D., Vice President of R&D
Dr. Moynihan has served as our Vice President of R&D since March 2011 and served as Vice President of R&D for 22nd Century since January, 2007. He has also been a consultant for 22nd Century since 1999. From 2001 to 2006 he served as Director of Biotechnology Development at Fundacion Chile and from 1995 to 2000 as Senior Project Director at InterLink Biotechnologies LLC. Dr. Moynihan holds a Bachelor of Science Degree in Biology from Brown University and a Master’s Degree and Ph.D. in Biology from Harvard University. He previously served as a Visiting Research Fellow at the Institute for Molecular and Cellular Biology, Osaka University, Japan; a Postdoctoral Associate in the Section of Plant Biology, Cornell University; and a Postdoctoral Associate at the Center for Agricultural Molecular Biology, Rutgers University.
64
C. Anthony Rider, CPA, Chief Financial Officer
Mr. Rider has served as our Chief Financial Officer and Treasurer since the closing of the Merger and served as the Chief Financial Officer of 22nd Century on a part-time basis since 2007. He has also served, since 2007, as Chief Financial Officer of Locke Acquisition Group LLC, which is unrelated to us. Mr. Rider served as the Chief Financial Officer of Astronics Corporation, a public company, and MOD-PAC Corp., a public company, each from 2000 to 2005, and as the Chief Financial Officer of IIMAK, a private-equity sponsored international manufacturing company, from 2005 to 2007. Mr. Rider holds a Bachelor of Science Degree from Canisius College. Mr. Rider is a member of the AICPA and the New York State Society of CPAs. From 1973 to 2000, Mr. Rider was employed by Ernst & Young.
Joseph Alexander Dunn, Ph.D., Director
Dr. Dunn has served as a director since March 4, 2011. Dr. Dunn is currently Associate Dean for Research and Professor of Pharmaceutical Sciences at D’Youville College of Pharmacy in Buffalo, New York and has served in this capacity since April 1, 2010. Dr. Dunn has also served as Chief Executive Officer of the National Center for Food and Agricultural Policy in Washington, D.C. since November 1, 2009 and as Chief Executive Officer and Director of Research at OmniPharm Research International, Inc., a drug company, and affiliated entities, Therex Technologies Inc., a drug company, and Therex LLC, a drug company, each located in Buffalo, New York since January, 1994. From May 1, 2008, until January 20, 2009, Dr. Dunn served as Deputy Under Secretary and from August 1, 2006, until April 30, 2008 Dr. Dunn served as Senior Scientific Advisor at the United States Department of Agriculture, Research, Education and Economics Mission Area in Washington, D.C. From December 1, 2006, until April 30, 2008 Dr. Dunn served as Executive Director of the United States Department of Agriculture NAREEE Advisory Board. From July, 1998 until July 1, 2006, Dr. Dunn served as Research Associate Professor in the Department of Oral Biology, School of Dental Medicine, at the State University of New York at Buffalo. Since June 1, 2010, Dr. Dunn has served as a member of the Board of Directors of Brothers of Mercy, Inc., a not-for-profit nursing and rehabilitation concern. Dr. Dunn holds a B.S. Degree in Medical Chemistry and a Ph.D. Degree in Pharmacology, both from the State University of New York at Buffalo School of Pharmacy. Dr. Dunn also served as a Postdoctoral Fellow in the Department of Pharmacology at Harvard Medical School and as a Staff Fellow at the National Institutes of Health, National Cancer Institute Laboratory of Cellular Carcinogenesis and Tumor Promotion. Dr. Dunn’s extensive scientific and regulatory background led to our conclusion that he should serve as a director of our Company.
James W. Cornell, Director
Mr. Cornell has served as a director since March 4, 2011. Mr. Cornell is currently the President and Chief Executive Officer of Praxiis, LLC, an enterprise that provides support for clients in organizational change, leadership development and transactional advisory services. He has served in this capacity since October, 1988. Mr. Cornell is also the current Manager of Larkin Center Management, LLC, a real estate development company, and has served in this capacity since October 2010. From September 2006 until September 2010, Mr. Cornell served as Managing Director of New York New Jersey Rail, LLC, which is part of the national transportation rail system and moves rail freight by rail barge across New York City Harbor, and he now continues to serve as principal business advisor to that firm. From March 2005 until September 2008, Mr. Cornell served as the Chairman of the Board of Directors of New York Regional Rail Corp., which operates as a short-haul regional trucking company. From April 2006, until February 2007, Mr. Cornell served as Chief Restructuring Officer of Regus Industries, a waste management firm, and from January 2001 until November 2004, he served as Special Advisor to Pinkerton Government Services, Inc. and Securitas Nuclear and Government Services Unit, security services providers to the energy industry and government. Mr. Cornell holds a B.S. Degree in Business, Management, and Economics and an M.B.A. Degree, both from the State University of New York, Empire College. Mr. Cornell’s extensive business management, strategy, and leadership experience led to our conclusion that he should serve as a director of our Company.
65
Steven Katz, Director
Mr. Katz has served as a director since March 4, 2011. Mr. Katz is currently the President of Steven Katz & Associates, Inc., a management consulting firm and has served in this capacity since January 1981. From April 2000 until March 9, 2007, Mr. Katz served on the Board of Directors, and as a member of the audit and compensation committees thereof, of Biophan Technologies, a technology development company. From November 1999 until May 13, 2010, Mr. Katz served on the Board of Directors, and as a member of the audit and compensation committees thereof, of USA Technologies, a cashless transactions solutions company. From July 2004 until July 20, 2007, Mr. Katz served on the Board of Directors, and as a member of the audit and compensation committees thereof, of Natural Nano, a nanomaterials company. From February 2005 until March 1, 2010, Mr. Katz served on the Board of Directors, and as a member of the audit and compensation committees thereof, of Health Systems Solutions, a technology and services company in the health care and mobile work force industries. From November 2006 until September 13, 2008, Mr. Katz served as Chairman of the Board of Directors and President of GammaCan International Inc., an immunotherapies products company; from September 2003 until May 4, 2006, he served on the Board of Directors of Nanoscience Technologies, a company previously engaged in the commercialization of third-party intellectual property; and from October 2004 until April 26, 2006, he served on the Board of Directors of Vivid Learning Systems, a company engaged in the providing computer-based compliance training products and services. From January 2000 until October 2001, Mr. Katz also served as a member of the Board of Directors, President, and Chief Operating Officer of Senesco Technologies, Inc., a company engaged in the identification and development of proprietary gene technology with application to human, animal and plant systems. Mr. Katz holds a B.A. Degree in Accounting from the City College of New York. Mr. Katz’s extensive experience in management consulting as well as his significant services on the boards of numerous public and private companies led to our conclusion that he should serve as a director of our Company.
Code of Ethics
In 2006, we adopted a Code of Ethics that applies to all of our employees. A copy of our Code of Ethics will be provided to any person requesting same without charge. To request a copy of our Code of Ethics, please make written request to our Chief Executive Officer c/o 22nd Century Group, Inc., 8201 Main Street, Suite 6, Williamsville, NY 14221.
Stockholder Communications
As of the date of this prospectus, we do not yet have a defined process for security holders to send communications to the Board. Security holders that wish to communicate with the Board are encouraged to contact the Company at its principal executive offices by letter or telephone.
Board Committees
Nominating Committee
At of the date of this prospectus, we do not have a nominating committee. We intend to adopt a nominating committee in the future.
As of the date of this prospectus, we do not have any defined policy or procedure requirements for stockholders to submit recommendations or nominations for directors. We do not currently have any specific or minimum criteria for the election of nominees to the Board, and does not have any specific process or procedure for evaluating such nominees. Our current Board assesses all candidates, whether submitted by management or stockholders, and makes recommendations for election or appointment.
Audit Committee
As of the date of this prospectus, the role of the audit committee is performed by the Board.
In this capacity, the Board is responsible for: (i) selection and oversight of our independent accountants; (ii) establishing procedures for the receipt, retention and treatment of complaints regarding accounting, internal controls and auditing matters; (iii) establishing procedures for the confidential, anonymous submission by our employees of concerns regarding accounting and auditing matters; (iv) engaging outside advisors; and (v) funding for the outside auditors and any outside advisors engaged by the Board.
We have determined that James W. Cornell qualifies as an “audit committee financial expert” as defined in Item 407(d)(5)(ii) of Regulation S-K.
66
From inception to present date, we believe that the members of our Board are collectively capable of analyzing and evaluating the Company’s financial statements and understanding internal controls and procedures for financial reporting.
Compensation Committee
We have determined that the functions ordinarily handled by such a committee should be handled by our entire Board.
Section 16(a) Beneficial Ownership Reporting Compliance
Section 16(a) of the Exchange Act requires our directors, executive officers, and stockholders holding more than 10% of our outstanding common stock to file with the SEC initial reports of ownership and reports of changes in beneficial ownership of our common stock. Executive officers, directors and greater-than-10% stockholders are required by SEC regulations to furnish us with copies of all Section 16(a) reports they file. We not have any information to report in this regard.
Director Compensation
Beginning in 2011, we are compensating each of the non-employee members of our board of directors at a rate of $10,000 per year, payable in equal quarterly installments. We expect to continue this arrangement going forward. On April 1, 2011, we also granted each non-employee director 25,000 shares of our common stock, subject only to certain restrictions on transfer. Additionally, we reimburse our non-employee directors for reasonable travel and other expenses incurred in connection with attending board of director and committee meetings. Our directors who are also employees are compensated for their service as employees and do not receive any additional compensation for their service on our board. Except with respect to the compensation described above, there is presently no compensation package being offered to members of our board of directors. We expect to establish long-term arrangements for our board members in the future. For the 2010 and 2009 calendar years and as of December 31, 2010, there were no non-employee directors of the Company. Information regarding compensation for those of our directors who are also employees is set forth in the Executive Compensation – Summary Compensation Table below.
EXECUTIVE COMPENSATION
The following table summarizes the compensation paid by us in each of the last two completed fiscal years ended December 31, 2010 for our principal executive officer and the two most highly compensated executive officers who received annual compensation in excess of $100,000. These officers are referred to herein as our “Named Executive Officers.”
Summary Compensation Table
|
Name and
Principal
Position
|
Year
|
Salary
($)
|
Bonus
($)
|
Stock
Awards
($)
|
Option
Awards
($)
|
Non-Equity
Incentive Plan
Compensation
($)
|
Nonqualified
Deferred
Compensation
Earnings
($)
|
All Other
Compensation
($)
|
Total
($)
|
|||||||||||||||||||||||||
|
Joseph Pandolfino,
|
2010
|
150,000 | (1) | - | - | - | - | - | - | 150,000 | ||||||||||||||||||||||||
|
Chief Executive Officer
|
2009
|
150,000 | (1) | - | - | - | - | - | - | 150,000 | ||||||||||||||||||||||||
|
Henry Sicignano III,
|
2010
|
85,000 | - | - | - | - | - | - | 85,000 | |||||||||||||||||||||||||
|
President
|
2009
|
- | - | - | - | - | - | - | - | |||||||||||||||||||||||||
|
Michael R. Moynihan, Ph.D.,
|
2010
|
114,019 | (2) | - | - | - | - | - | 114,019 | |||||||||||||||||||||||||
|
Vice President of R&D
|
2009
|
92,000 | (2) | - | 258,660 | (3) | - | - | - | 350,660 | ||||||||||||||||||||||||
|
David Rector (4)
|
2010
|
1,000 | - | - | - | - | - | - | 1,000 | |||||||||||||||||||||||||
|
2009
|
- | - | - | - | - | - | - | - | ||||||||||||||||||||||||||
|
Ronald Asirwatham (5)
|
2010
|
- | - | - | - | - | - | - | - | |||||||||||||||||||||||||
|
|
2009
|
- | - | - | - | - | - | - | - | |||||||||||||||||||||||||
|
Nanuk Warman (6)
|
2010
|
8,000 | - | - | - | - | - | 1,500 | 9,500 | |||||||||||||||||||||||||
|
|
2009
|
8,000 | - | - | - | - | - | - | 8,000 | |||||||||||||||||||||||||
67
(1) We paid a portion of Mr. Pandolfino’s salary in common stock in lieu of cash payment. During the year ended December 31, 2010, we issued to Mr. Pandolfino 236,909 shares of our common stock as compensation in lieu of $44,700 of salary. During the year ended December 31, 2009, we issued to Mr. Pandolfino’s 556,508 shares of our common stock as compensation in lieu of $137,500 of salary. The number of shares issued was determined by reference to the value of shares issued concurrently or recently in cash transactions to private investors who were not employees, officers or directors.
(2) We paid a portion of Dr. Moynihan’s salary in common stock in lieu of cash payment. During the year ended December 31, 2010, we issued to Dr. Moynihan’s 109,584 shares of our common stock as compensation in lieu of $23,537 of salary. Additionally, during 2010, we issued a short term promissory note payable to Dr. Moynihan in the amount of $9,537 as consideration for unpaid salary. Dr. Moynihan converted this note to 9,537 shares of our common stock during the Private Placement Offering. During the year ended December 31, 2009, we issued to Dr. Moynihan 74,201 shares of our common stock as compensation in lieu of $18,333 of salary and 4 membership interests in our subsidiary (100 units outstanding), Goodrich Tobacco Company, LLC (f/k/a Xodus, LLC) in lieu of $36,000 of salary. The number of shares issued was determined by reference to the value of shares issued concurrently or recently in cash transactions to private investors who were not employees, officers or directors.
(3) As an inducement to renew his employment agreement, on February 1, 2009, we granted Dr. Moynihan a warrant to purchase 445,207 shares of our common stock, vesting over a one year service period ending February 1, 2010.
(4) David Rector served as the Company’s sole executive officer and director from November 22, 2010 until January 25, 2011, at which time he resigned as an executive officer. Mr. Rector continued to serve as a director until March 4, 2011. Mr. Rector earned $1,000 for his services as an officer and director in 2010. He received no compensation from the Company in 2009.
(5) Ronald Asirwatham served as the Company’s sole executive officer and director from September 24,2010 through November 22, 2010. He received no compensation from the Company for either 2009 or 2010.
(6) Mr. Warman served as the Company’s sole executive officer and director from June 9, 2008 through September 24, 2010. In such capacity, he earned $8,000 in each of 2009 and 2010. In addition, subsequent to his September 24, 2010 resignation, Mr. Warman earned $1,500 for accounting services related to the preparation of the Company’s September 30, 2010 Annual Report on Form 10-K, and December 31, 2010 Quarterly Report on Form 10-Q.
Outstanding Equity Awards at Fiscal Year-End
As of December 31, 2010, there were no outstanding equity awards held by our Named Executive Officers or any other executive officers of either 22nd Century or the Company.
Agreements with Executive Officers
We have entered into employment agreements with each of Messrs. Pandolfino, Sicignano, and Rider and Dr. Moynihan that provide for annual compensation of $150,000, $150,000, $72,000, and $105,000 respectively, subject to increases as contained in such employment agreements and/or as decided by our board of directors. These employment agreements also contain non-compete covenants and change of control provisions.
68
The employment agreement of each of Messrs. Pandolfino, Sicignano and Rider provides that during the executive officer’s employment by us and for a period of two years after the executive officer ceases to be employed by us, the following non-compete covenants will apply: (i) the executive officer will not (except on behalf of us) provide or offer to provide any goods or services to any entity engaged in the United States in the making, offering, marketing, distributing and/or selling of products made from the tobacco (Nicotiana) plant, and/or providing or offering to provide the same or substantially similar services to any customer or prospective customer, (ii) the executive officer will not interfere with our relationships with any customer, prospective customer, supplier, distributer, farmer and/or manufacturer, and (iii) the executive will not induce or attempt to induce any persons employed by us to leave their employment with us, nor hire or employ, or attempt to hire or employ, any persons employed by us, nor assist or facilitate in any way any other person or entity in the hiring of any persons employed by us. The employment agreement of Dr. Moynihan provides that during Dr. Moynihan’s employment by us and for a period of three years after he ceases to be employed by us for any reason, Dr. Moynihan will be unable to engage in any business that offers the Same or Substantially Similar Services (as defined in his employment agreement) as the Company does to any Customer or Prospective Customer (as such terms are defined in his employment agreement). Furthermore, for a period of four years following Dr. Moynihan’s termination for any reason, Dr. Moynihan will undertake not to interfere with the Company’s relationship with any Customer, Prospective Customer, researcher, supplier, distributer, farmer and/or manufacturer. For a period of three years following Dr. Moynihan’s termination for any reason, he shall not solicit any of the Company’s employees, consultants or subcontractors.
The employment agreement of Mr. Rider provides that in the event of a change of control (as defined in his employment agreement) of our Company, Mr. Rider may resign his employment with the Company (or, if involuntarily terminated, give notice of his intention to collect benefits) and shall be entitled to receive the base salary set forth therein which remains unpaid for the remainder of the initial term of the employment agreement.
The employment agreements of Messers. Pandolfino and Sicignano provide that in the event of a change in control (as defined in the employment agreements) of our Company, then during the three-year period following such change in control if certain triggering events occur as defined in such employment agreements, such as if the executive is terminated other than for cause (as defined in each of the employment agreements), death or disability, or if the executive officer’s responsibilities are diminished after the change in control as compared to the executive officer’s responsibilities prior to the change in control, or if the executive officer’s base salary or benefits are reduced, or the executive is required to relocate more than 25 miles from his current place of employment, then in any such events the executive officer will have the option, exercisable within 90 days of the occurrence of such an event, to resign his employment with us, in which case the executive officer will be entitled to receive: (A) the greater of either his base salary for the then remaining portion of the initial 5-year term of the agreement or his base salary for three years thereafter; (B) reimbursement for eighteen (18) months of his reasonable costs for medical, dental, life, disability and other benefits and insurance coverage that the executive officer received during his employment; (C) outplacement services for two years; and (D) the immediate vesting of all options and/or restricted stock grants previously granted or to be granted to the executive officer. The employment agreement of Dr. Moynihan contains change in control provisions similar to the foregoing, with the exception that upon a change in control and upon exercising his option to resign his employment with us, Dr. Moynihan will be eligible to receive salary continuation until the expiration of the 90-day period only, plus eligibility to participate in benefits provided by the Company for 30 days following resignation.
Dr. Moynihan’s employment agreement contains a severance provision which provides that upon the termination of his employment without Cause (as defined in his employment agreement), Dr. Moynihan will receive severance compensation equal to the base salary then in effect beginning on the date of termination and continuing until the later of one year following termination or the expiration of the initial term of his employment agreement.
We also provide each of Messrs. Pandolfino, Sicignano, and Rider and Dr. Moynihan with health insurance and vacation benefits.
69
Equity Incentive Plan
On October 21, 2010, we established the 2010 Equity Incentive Plan (“EIP”) for officers, employees, directors, consultants and advisors to the Company and its affiliates, consisting of 4,250,000 shares of common stock. The EIP has a term of ten years and is administered by our Board or a committee to be established by our Board, to determine the various types of incentive awards that may be granted to recipients under this plan, such as stock grants, stock options, stock appreciation rights, performance share awards, restricted stock and restricted stock units, and the number of shares of common stock to underlie each such award under the EIP. The EIP also contains a provision which restricts the plan to granting awards relating to no more than 1,600,000 shares of common stock during the first twelve months following the effective date of the Merger or January 25, 2011. On April 1, 2011, under our EIP, the Board granted an aggregate of 1,150,000 shares of our common stock to our officers and directors and options to purchase an aggregate of 35,000 shares of our common stock to our employees.
70
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS
Immediately prior to the closing of the Merger, pursuant to the terms of the Split-Off Agreement, we transferred all of our pre-Merger operating assets and liabilities to the Split-Off Subsidiary. We then transferred all of the outstanding capital stock of the Split-Off Subsidiary to David Rector, our sole director and executive officer prior to the Merger, in exchange for $1, such consideration being deemed to be adequate by our Board prior to the Merger. Prior to the closing of the Merger, we paid Mr. Rector $1,500 in consideration for his service as our sole director and executive officer.
Prior to the closing of the Merger, we utilized office space located at 11923 SW 37 Terrace, Miami, Florida 33175 that was provided to us on a rent-free basis by Nanuk Warman, our former director and executive officer. Also, prior to the closing of the Merger, we cancelled 10,015,200 shares of our common stock held by Mr. Warman and entered into a mutual release agreement with Mr. Warman regarding such cancellation. In each of fiscal years 2009 and 2010, we paid Mr. Warman aggregate compensation of $8,000 in consideration for his services as our sole director and executive officer during those periods. We also paid Mr. Warman aggregate of $1,500 in consideration for his accounting services in preparation of our most recent Form 10-K and Form 10-Q filed prior to the closing of the Merger.
We have had numerous transactions with Alternative Cigarettes, Inc. (“AC”). AC is 95% owned by three holders of our common stock, including Joseph Pandolfino, our Chief Executive Officer, and Angelo Tomasello, who currently owns approximately 11.8% of our issued and outstanding common stock. We share office space and employee services with AC and AC reimburses us from time to time for the value of these activities. AC paid us $13,000 during fiscal year 2010 and approximately $52,000 during fiscal year 2009 for these services. AC has also advanced funds to us from time to time. Since January 1, 2009, the largest net amount due from us to AC was approximately $127,000. No interest has been accrued or paid on these amounts due to AC and there are no repayment terms between the parties.
In January 2008, we issued convertible promissory notes due and payable on January 15, 2011 to Messrs. Pandolfino and Tomasello in the principal amounts of $77,435 and $100,315, respectively, with 7% interest per annum accruing thereon. In December 2009, Mr. Pandolfino converted the principal balance and accrued interest under his note ($88,172) into 151,760 shares of our common stock. In May 2010, Mr. Tomasello agreed to amend his note to eliminate his right to convert the balance into shares of our common stock, and in January 2011, Mr. Tomasello’s note together with all accrued interest thereon was paid in full.
In November 2008, we issued a promissory note due and payable on November 11, 2010 to Mr. Tomasello in the principal amount of $325,000, with 10% interest per annum accruing thereon, and a warrant to purchase 371,006 shares of our common stock, which have since been exercised at a price of $.0001 per share. The note is guaranteed by Virgil Properties, LLC, which is jointly owned by Messrs. Pandolfino and Tomasello. Effective December 1, 2010, the $325,000 promissory note was amended to extend the maturity date until January 10, 2011 and to increase the interest rate to 15% during this extension period. On January 25, 2011, Mr. Tomasello converted the principal amount of this promissory note into 325,000 shares of our common stock through an investment in the Private Placement Offering and Mr. Tomasello was paid cash in the amount of $79,401 in January 2011, which represents the accrued interest on the original $325,000 promissory note. Mr. Tomasello has also made funds available to us in the form of cash advances. The largest net amount outstanding since January 1, 2009 was approximately $166,000. No interest was accrued or paid on such advances and there were no repayment terms between the parties. In December 2009, Mr. Tomasello was issued 504,553 shares of our common stock in lieu of repayment of $135,996 of such advances, and we issued him a promissory note that was exchanged for 204,639 shares of our common stock in June 2010.
71
Mr. Pandolfino has made funds available to us in the form of cash advances and deferred guaranteed payments due to him by us as consideration for his services as our Chief Executive Officer. The largest net amount of such advances and deferred guaranteed payments outstanding since January 1, 2009 was approximately $276,496. No interest was accrued or paid on such advances or deferrals and there are no repayment terms between the parties. In December 2009, we issued to Mr. Pandolfino 1,061,061 shares of our common stock in lieu of repayment of such amount; 556,508 of which shares were issued in lieu of deferred guaranteed payments. During the period between January 1, 2010 and October 5, 2010, we issued to Mr. Pandolfino 236,909 shares of our common stock in lieu of $44,700 due and payable to him for his services. On October 5, 2010, we issued to Mr. Pandolfino a promissory note, which Mr. Pandolfino then assigned to Mr. Sicignano, due and payable on January 31, 2011 in the principal amount of $58,873, with 15% interest per annum accruing thereon. In January 2011, we made payment in full to Mr. Sicignano on this assigned note together with all accrued interest thereon. In September 2010, Henry Sicignano III, our President and Secretary, loaned us $35,000, which amount was due and payable in November 2010, with 15% interest per annum accruing thereon. On December 16, 2010, Mr. Sicignano agreed to extend the maturity date of this loan until January 25, 2011. On December 28, 2010 we issued a promissory note to Mr. Sicignano due and payable on January 15, 2011 in the principal amount of $100,000, with 15% interest per annum accruing thereon. From time to time, Mr. Sicignano deferred guaranteed payments due to him by us as consideration for his services as our President with the largest net amount of such deferred guaranteed payments outstanding since January 1, 2009 being $85,000. On January 28, 2011 we made payment in full to Mr. Sicignano of all deferred guaranteed payments and all principal and accrued interest on all promissory notes then outstanding. Mr. Sicignano is also the managing member of Henry Sicignano III Group, LLC (“Sicignano Group”). On October 5, 2010, Sicignano Group purchased 112,396 shares of our common stock for $30,295 and, in a simultaneous related transaction, made a loan to the Company in the principal amount of $30,295, with 15% interest per annum accruing thereon, for which we issued Sicignano Group a promissory note due and payable on January 31, 2011. On January 25, 2011, Sicignano Group converted the principal amount of this promissory and the accrued interest thereon into 31,626 shares of our common stock through an investment in the Private Placement Offering.
Michael R. Moynihan, the Vice President of Research and Development, has deferred guaranteed payments due and payable to him as consideration for his services to us. The largest net balance of such amounts outstanding since January 1, 2009 was approximately $79,000. No interest was accrued or paid on such amounts owed and there were no repayment terms between the parties. In December 2009, we issued to Dr. Moynihan 74,201 shares of our common stock and 4 membership interests in our subsidiary (100 units outstanding), Goodrich Tobacco Company, LLC (f/k/a Xodus, LLC), in lieu of approximately $54,000 of such amount due and payable to him. During the period between January 1 and October 5, 2010, we issued to Dr. Moynihan 109,584 shares of our common stock in lieu of $23,537 of such amount due and payable to him for his services. We paid the remaining amounts owed to Dr. Moynihan as compensation for his services in cash.
On September 15 and October 15, 2009, we issued promissory notes payable to Clearwater Partners, LLC (“Clearwater”) in the amounts of $15,000 and $10,000, respectively. In conjunction with the $15,000 promissory note, a warrant to purchase 185,503 shares of our common stock, at a price per share of less than $.0001, was issued to Clearwater, and in conjunction with the $10,000 note, a warrant to purchase 92,751 shares of our common stock, at a price per share of less than $.0001, was issued to Clearwater. The promissory notes bear interest at a rate of 10%. These promissory notes had original maturity dates September 15, 2010 and October 15, 2010, respectively. On May 27, 2010, the maturity dates of both promissory notes were extended to January 31, 2012.
On March 1, 2010, we issued a four year warrant to purchase 1,706,626 shares of our common stock to Clearwater, which was exercised in full on May 27, 2010, at a price per share of $0.0001. On May 27, 2010, we further issued to Clearwater an additional four year warrant to purchase 1,409,821 shares of our common stock, which was immediately exercised in full at a price per share of $0.0001, and we issued to Clearwater a promissory note due and payable on January 31, 2012 in the principal amount of $45,000, with 10% interest per annum accruing thereon. These warrants and this promissory note were issued to Clearwater in lieu of repayment of $450,000 principal, and accrued interest thereon, of funds previously advanced to us by Clearwater. On October 5, 2010, Clearwater purchased 176,358 shares of our common stock for $47,535 and, in a simultaneous related transaction, made a loan to the Company in the principal amount of $47,535, with 15% interest per annum accruing thereon, for which we issued Clearwater a promissory note due and payable on January 31, 2011. On January 25, 2011, Clearwater converted the principal amount of this $47,535 promissory note and the accrued interest thereon, and the principal amount of the $45,000 promissory note and the accrued interest thereon, due and payable on January 31, 2012, into 97,544 shares of our common stock through an investment in the Private Placement Offering.
In February 2011, AC was paid $22,500 by 22nd Century for AC’s assignment of its MAGIC trademark to 22nd Century and other minor assets. On June 30, 2011, we issued a promissory note due and payable on August 30, 2011 to Mr. Sicignano in the principal amount of $150,000 with 12% interest per annum accruing thereon.
72
Lockup Agreements
All officers, directors, stockholders holding ten percent (10%) or more of our common stock after giving effect to the Merger, the Split-Off and the Private Placement Offering and our key employees (each a “Restricted Holder,” and collectively, the “Restricted Holders”), have entered into lock-up agreements with us for a term of eighteen (18) months following the date of the closing of the Merger during which time no Restricted Holder will offer or sell any shares of common stock owned by such Restricted Holder, except to another Restricted Holder. The lock-up agreements entered into by Clearwater Partners, LLC and Angelo Tomasello do not apply to any shares of our common stock or any Investor Warrant issued to Clearwater Partners, LLC (97,544 shares of our common stock and a warrant to purchase 48,772 shares of our common stock) or Mr. Tomasello (325,000 shares of our common stock and a warrant to purchase 167,500 shares of our common stock) upon consummation of the Merger in exchange for the Units of 22nd Century purchased by Clearwater Partners, LLC (97,544 limited liability company membership interests and a warrant to purchase 48,772 limited liability company membership interests) or Mr. Tomasello (325,000 limited liability company membership interests and a warrant to purchase 167,500 limited liability company membership interests) in the Private Placement Offering nor to any shares of our common stock issued to Clearwater Partners, LLC or Mr. Tomasello upon exercise of any Investor Warrant.
Policies and Procedures for Related Party Transactions
We do not currently have a formal written policy or procedure for the review and approval of related party transactions. However, effective as of the date ten (10) days following the date hereof, all future related party transactions will be reviewed and approved by a disinterested majority of the members of our Board.
Our Board intends to adopt a written related person transaction policy, which will set forth the policies and procedures for the review and approval or ratification of related person transactions. This policy will be administered by our Board and covers any transaction, arrangement or relationship, or any series of similar transactions, arrangements or relationships, in which we were or are to be a participant, where the amount involved exceeds $50,000 and a related person had or will have a direct or indirect material interest. While the policy covers related person transactions in which the amount involved exceeds $50,000, the policy states that related person transactions in which the amount involved exceeds $120,000 are required to be disclosed in applicable filings as required by the Securities Act of 1933, as amended (the “Securities Act”), the Exchange Act and related rules. Our Board set the $50,000 threshold for approval of related party transactions in the policy at an amount lower than that which is required to be disclosed under the Securities Act, the Exchange Act and related rules because we believe it is appropriate for our Board to review transactions or potential transactions in which the amount involved exceeds $50,000, as opposed to $120,000.
Pursuant to this policy, our Board will: (i) review the relevant facts and circumstances of each related person transaction, including if the transaction is on terms comparable to those that could be obtained in arm’s length dealings with an unrelated third party and the extent of the related person’s interest in the transaction, and (ii) take into account the conflicts of interest and corporate opportunity provisions of our code of business conduct and ethics. Management will present to our Board each proposed related person transaction, including all relevant facts and circumstances relating thereto, and will update the Board as to any material changes to any related person transaction. All related person transactions may only be consummated if our Board has approved or ratified such transactions in accordance with the guidelines set forth in the policy. Certain types of transactions have been pre-approved by our Board under the policy. These pre-approved transactions include: (i) certain compensation arrangements; (ii) transactions in the ordinary course of business where the related person’s interest arises only (a) from his or her position as a director of another entity that is party to the transaction, and/or (b) from an equity interest of less than 5% in another entity that is party to the transaction, or (c) from a limited partnership interest of less than 5%, subject to certain limitations; and (iii) transactions in the ordinary course of business where the interest of the related person arises solely from the ownership of a class of equity securities in our Company where all holders of such class of equity securities will receive the same benefit on a pro rata basis. No director may participate in the approval of a related person transaction for which he or she is a related person.
PLAN OF DISTRIBUTION
We are registering the shares offered by this prospectus on behalf of the selling stockholders. The selling stockholders, which as used herein includes donees, pledgees, transferees or other successors-in-interest selling shares of common stock or interests in shares of common stock received after the date of this prospectus from a selling stockholder as a gift, pledge, partnership distribution or other transfer, may, from time to time, sell, transfer or otherwise dispose of any or all of their shares of common stock or interests in shares of common stock on any stock exchange, market or trading facility on which the shares are traded or in private transactions. These dispositions may be at fixed prices, at prevailing market prices at the time of sale, at prices related to the prevailing market price, at varying prices determined at the time of sale, or at negotiated prices. To the extent any of the selling stockholders gift, pledge or otherwise transfer the shares offered hereby, such transferees may offer and sell the shares from time to time under this prospectus, provided that this prospectus has been amended under Rule 424(b)(3) or other applicable provision of the Securities Act to include the name of such transferee in the list of selling stockholders under this prospectus.
73
The selling stockholders may use any one or more of the following methods when disposing of shares or interests therein:
|
|
•
|
ordinary brokerage transactions and transactions in which the broker-dealer solicits purchasers;
|
|
|
•
|
block trades in which the broker-dealer will attempt to sell the shares as agent, but may position and resell a portion of the block as principal to facilitate the transaction;
|
|
|
•
|
purchases by a broker-dealer as principal and resale by the broker-dealer for its account;
|
|
|
•
|
an exchange distribution in accordance with the rules of the applicable exchange;
|
|
|
•
|
privately negotiated transactions;
|
|
|
•
|
short sales
|
|
|
•
|
through the writing or settlement of options or other hedging transactions, whether through an options exchange or otherwise;
|
|
|
•
|
broker-dealers may agree with the selling stockholders to sell a specified number of such shares at a stipulated price per share;
|
|
|
•
|
a combination of any such methods of sale; and
|
|
|
•
|
any other method permitted pursuant to applicable law.
|
The selling stockholders may, from time to time, pledge or grant a security interest in some or all of the shares of common stock owned by them and, if they default in the performance of their secured obligations, the pledgees or secured parties may offer and sell the shares of common stock, from time to time, under this prospectus, or under an amendment to this prospectus under Rule 424(b)(3) or other applicable provision of the Securities Act amending the list of selling stockholders to include the pledgee, transferee or other successors in interest as selling stockholders under this prospectus.
In connection with the sale of our common stock or interests therein, the selling stockholders may enter into hedging transactions with broker-dealers or other financial institutions, which may in turn engage in short sales of the common stock in the course of hedging the positions they assume. The selling stockholders may also sell shares of our common stock short and deliver these securities to close out their short positions, or loan or pledge the common stock to broker-dealers that in turn may sell these securities. The selling stockholders may also enter into option or other transactions with broker-dealers or other financial institutions or the creation of one or more derivative securities which require the delivery to such broker-dealer or other financial institution of shares offered by this prospectus, which shares such broker-dealer or other financial institution may resell pursuant to this prospectus (as supplemented or amended to reflect such transaction).
The aggregate proceeds to the selling stockholders from the sale of the common stock offered by them will be the purchase price of the common stock less discounts or commissions, if any. Each of the selling stockholders reserves the right to accept and, together with their agents from time to time, to reject, in whole or in part, any proposed purchase of common stock to be made directly or through agents.
The selling stockholders also may resell all or a portion of the shares in open market transactions in reliance upon Rule 144 under the Securities Act of 1933, provided that they meet the criteria and conform to the requirements of that rule.
The selling stockholders might be, and any broker-dealers that act in connection with the sale of securities will be, deemed to be “underwriters” within the meaning of Section 2(11) of the Securities Act, and any commissions received by such broker-dealers and any profit on the resale of the securities sold by them while acting as principals will be deemed to be underwriting discounts or commissions under the Securities Act. Each selling stockholder has represented and warranted to the company that it does not have any agreement or understanding, directly or indirectly with any Person to distribute shares.
To the extent required, the shares of our common stock to be sold, the names of the selling stockholders, the respective purchase prices and public offering prices, the names of any agents, dealer or underwriter, any applicable commissions or discounts with respect to a particular offer will be set forth in an accompanying prospectus supplement or, if appropriate, a post-effective amendment to the registration statement that includes this prospectus.
74
In order to comply with the securities laws of some states, if applicable, the common stock may be sold in these jurisdictions only through registered or licensed brokers or dealers. In addition, in some states the common stock may not be sold unless it has been registered or qualified for sale or an exemption from registration or qualification requirements is available and is complied with.
We have advised the selling stockholders that the anti-manipulation rules of Regulation M under the Exchange Act may apply to sales of shares in the market and to the activities of the selling stockholders and their affiliates. Regulation M's prohibition on purchases may include purchases to cover short positions by the selling stockholders, and a selling stockholder's failure to cover a short position at a lender's request and subsequent purchases by the lender in the open market of shares to cover such short positions, may be deemed to constitute an inducement to buy shares, which is prohibited by Regulation M.
We will make copies of this prospectus (as it may be supplemented or amended from time to time) available to the selling stockholders for the purpose of satisfying the prospectus delivery requirements of the Securities Act. The selling stockholders may indemnify any broker-dealer that participates in transactions involving the sale of the shares against certain liabilities, including liabilities arising under the Securities Act.
We have advised the selling stockholders that if a particular offer of shares is to be made on terms constituting a material change from the information described under a final prospectus, then, to the extent required, a supplement to the final prospectus must be distributed setting forth the terms and related information as required.
We have agreed to indemnify the selling stockholders against liabilities, including liabilities under the Securities Act and state securities laws, relating to the registration of the shares offered by this prospectus.
We agreed to keep this prospectus effective until the earlier of (i) the date that is two years following the date that the registration statement, of which this prospectus forms a part, is declared effective by the SEC; or (ii) the date on which the shares may be resold by the selling stockholders without registration and without regard to any volume limitations by reason of Rule 144(e) under the Securities Act or any other rule of similar effect. The resale shares will be sold only through registered or licensed brokers or dealers if required under applicable state securities laws. In addition, in certain states, the resale shares may not be sold unless they have been registered or qualified for sale in the applicable state or an exemption from the registration or qualification requirement is available and is complied with.
General
Our authorized capital stock consists of 300,000,000 shares of common stock and 10,000,000 shares of preferred stock, of which 27,909,646 shares of common stock are issued and outstanding. No shares of preferred stock are issued and outstanding. We will also have reserved 11,751,980 shares of common stock, which is comprised of (i) 8,151,980 shares reserved for issuance upon the exercise of the warrants issued in connection with the Merger, (ii) 500,000 shares reserved for issuance upon the exercise of the warrants that were issued to our financial advisor upon the closing of the Merger, and (iii) 3,100,000 shares underlying securities to be issued under our EIP.
The following summary of certain provisions of our capital stock does not purport to be complete and is subject to and is qualified in its entirety by our articles of incorporation and by-laws following the Merger and by the provisions of applicable law.
Common Stock
Holders of common stock are entitled to one vote per share with respect to each matter presented to our stockholders on which holders of common stock are entitled to vote. The common stock does not have cumulative voting rights. No share of common stock affords any preemptive rights or is convertible, redeemable, assessable or entitled to the benefits of any sinking or repurchase fund.
75
Subject to the prior rights of holders of preferred stock, if any, holders of common stock are entitled to receive dividends as may be lawfully declared from time to time by our board of directors. Upon our liquidation, dissolution or winding up, whether voluntary or involuntary, holders of common stock will be entitled to receive such assets as are available for distribution to our shareholders after there shall have been paid, or set aside for payment, the full amounts necessary to satisfy any preferential or participating rights to which the holders of each outstanding series of preferred stock are entitled by the express terms of the series.
The common stock is quoted on the OTC Bulletin Board under the symbol “XXII.OB.”
Preferred Stock
Our Board is authorized, without action by our stockholders, to designate and issue up to an aggregate of 10,000,000 shares of preferred stock in one or more series. Our board of directors has the discretion to determine the rights, preferences, privileges and restrictions, including voting rights, dividend rights, conversion rights, redemption privileges and liquidation preferences, of each series of preferred stock.
No shares of preferred stock are currently outstanding, and we have no current plans to issue preferred stock. The issuance of shares of preferred stock, or the issuance of rights to purchase preferred stock, could be used to discourage an unsolicited acquisition proposal. For example, a business combination could be impeded by the issuance of a series of preferred stock containing class voting rights that would enable the holder or holders of such series to block any such transaction. Alternatively, a business combination could be facilitated by the issuance of a series of preferred stock having sufficient voting rights to provide a required percentage vote of our stockholders. In addition, under some circumstances, the issuance of preferred stock could adversely affect the voting power and other rights of the holders of our common stock. Although prior to issuing any series of preferred stock our board is required to make a determination as to whether the issuance is in the best interests of our stockholders, our board could act in a manner that would discourage an acquisition attempt or other transaction that some, or a majority, of our stockholders might believe to be in their best interests or in which our stockholders might receive a premium for their stock over prevailing market prices of such stock. Our board of directors does not presently intend to seek stockholder approval prior to any issuance of currently authorized preferred stock, unless otherwise required by law or applicable stock exchange requirements.
Warrants
We issued five-year warrants to purchase 2,717,223 shares of our common stock, at an exercise price of $1.50 per share, in exchange for the warrants contained in the Securities purchased by investors in the Private Placement Offering (the “Investor Warrants”). These warrants contain among other things, a cashless exercise provision to become operative upon the later of: (A) February 1, 2012 if a registration statement pursuant to the Securities Act with regard to the shares of common stock issuable upon exercise of these warrants has not been filed by such date, and (B) 30 days following the date on which the earlier filed registration statement with regard to the shares of common stock received by the investors in the Private Placement Offering as a result of the Merger has been declared effective by the SEC, if a registration statement pursuant to the Securities Act with regard to the shares of common stock issuable upon exercise of these warrants has not been filed prior to the expiry of such 30 day period. A cashless exercise means that in lieu of paying the aggregate purchase price for the shares being purchased upon exercise of the warrants in cash, the holder will forfeit a number of shares underlying the warrants with a “fair market value” equal to the aggregate exercise price. We will not receive additional proceeds to the extent that warrants are exercised on a cashless basis. The exercise price and number of shares of our common stock issuable upon exercise of the warrants may be adjusted in certain circumstances, including in the event of a stock dividend, or our recapitalization, reorganization, merger or consolidation. These warrants also provide holders with weighted-average anti-dilution price protection. No fractional shares will be issued upon exercise of these warrants. If, upon exercise of these warrants, a holder would be entitled to receive a fractional interest in a share, we may, in our discretion, upon exercise, round up to the nearest whole number of shares of our common stock to be issued to the warrant holder or otherwise equitably adjust the exercise and exercise price per share.
76
We issued five-year warrants to purchase 5,000,000 shares of our common stock, at an exercise price of $3.00 per share, in exchange for the warrants held by the members of 22nd Century prior to the consummation of the Private Placement Offering (the “Century Warrants”). These warrants contain among other things, a cashless exercise provision to become operative upon the later of: (A) February 1, 2012 if a registration statement pursuant to the Securities Act with regard to the shares of common stock issuable upon exercise of these warrants has not been filed by such date, and (B) 30 days following the date on which the earlier filed registration statement with regard to the share of common stock received by the investors in the Private Placement Offering as a result of the Merger has been declared effective by the SEC, if a registration statement pursuant to the Securities Act with regard to the shares of common stock issuable upon exercise of the these warrants has not been filed prior to the expiry of such 30 day period. A cashless exercise means that in lieu paying the aggregate purchase price for the shares being purchased upon exercise of the warrants in cash, the holder will forfeit a number of shares underlying the warrants with a “fair market value” equal to the aggregate exercise price. We will not receive additional proceeds to the extent that warrants are exercised on a cashless basis. The exercise price and number of shares of our common stock issuable upon exercise of the warrants may be adjusted in certain circumstances, including in the event of a stock dividend, or our recapitalization, reorganization, merger or consolidation. These warrants also provide holders with weighted-average anti-dilution price protection. No fractional shares will be issued upon exercise of these warrants. If, upon exercise of these warrants, a holder would be entitled to receive a fractional interest in a share, we may, in our discretion, upon exercise, round up to the nearest whole number of shares of our common stock to be issued to the warrant holder or otherwise equitably adjust the exercise and exercise price per share.
We issued five-year warrants to purchase an aggregate of 434,755 shares of our common stock, at an exercise price of $1.50 per share, in exchange for the warrants issued to Rodman & Renshaw, LLC (the “Placement Agent”), who acted as placement agent in the Private Placement Offering, and Gottbetter Capital Markets, LLC (the “Sub-Agent”), who acted as sub-placement agent in the Private Placement Offering (the “Placement Agent Conversion Warrants”). These warrants contain among other things, a cashless exercise provision. A cashless exercise means that in lieu of paying the aggregate purchase price for the shares being purchased upon exercise of the warrants in cash, the holder will forfeit a number of shares underlying the warrants with a “fair market value” equal to the aggregate exercise price. We will not receive additional proceeds to the extent that warrants are exercised on a cashless basis. The exercise price and number of shares of our common stock issuable upon exercise of the warrants may be adjusted in certain circumstances, including in the event of a stock dividend, or our recapitalization, reorganization, merger or consolidation. These warrants also provide holders with weighted-average anti-dilution price protection. No fractional shares will be issued upon exercise of these warrants. If, upon exercise of these warrants, a holder would be entitled to receive a fractional interest in a share, we may, in our discretion, upon exercise, round up to the nearest whole number of shares of our common stock to be issued to the warrant holder or otherwise equitably adjust the exercise and exercise price per share.
We issued five-year warrants to purchase 500,000 shares of our common stock, at an exercise price of $1.50 per share, to the Placement Agent (the “Advisor Warrants”). These warrants contain among other things, a cashless exercise provision. A cashless exercise means that in lieu of paying the aggregate purchase price for the shares being purchased upon exercise of the warrants in cash, the holder will forfeit a number of shares underlying the warrants with a “fair market value” equal to the aggregate exercise price. We will not receive additional proceeds to the extent that warrants are exercised on a cashless basis. The exercise price and number of shares of our common stock issuable upon exercise of the warrants may be adjusted in certain circumstances, including in the event of a stock dividend, or our recapitalization, reorganization, merger or consolidation. These warrants also provide holders with weighted-average anti-dilution price protection. No fractional shares will be issued upon exercise of these warrants. If, upon exercise of these warrants, a holder would be entitled to receive a fractional interest in a share, we may, in our discretion, upon exercise, round up to the nearest whole number of shares of our common stock to be issued to the warrant holder or otherwise equitably adjust the exercise and exercise price per share.
Options
On April 1, 2011, under our EIP, the Board granted options to purchase an aggregate of 35,000 shares of our common stock to certain of our employees. These 5-year options vest in one year and have an exercise price of $1.20 and a cashless exercise provision.
Registration Rights
We agreed to a covenant in conjunction with the Private Placement Offering to use our commercially reasonable efforts to file with the SEC this registration statement, within seventy-five (75) days following the effective date of the Merger, which covers the resale of the common stock issued to the investors in the Private Placement as a result of the Merger in exchange for the Units contained in the Securities sold in the Private Placement Offering. We will use our commercially reasonable efforts to cause this registration statement to be declared effective by the SEC within 180 calendar days of filing with the SEC (240 days if the SEC reviews such registration statement). If we are late in filing this registration statement or if this registration statement is not declared effective within the prescribed time periods, then the holders of registrable common stock shall be entitled to monetary penalties payable by us at a rate equal to one-half percent (0.50%) of the offering price per Unit in the Private Placement Offering for each full month that (i) we are late in filing this registration statement or (ii) this registration statement is late in being declared effective by the SEC; provided, however, that in no event shall the aggregate of any such penalties exceed 5% of the offering price per Unit in the Private Placement Offering. Notwithstanding the foregoing, no penalties shall accrue with respect to any shares of common stock removed from the registration statement in response to a comment from the staff of the SEC limiting the number of shares of common stock which may be included in this registration statement (a “Cutback Comment”) or which may be resold by the holders of registrable common stock in accordance with Rule 144 under the Securities Act. We shall keep this registration statement effective and up to date for two years from the date it is declared effective by the SEC or until Rule 144 is available to the investors in the Private Placement Offering with respect to all of their shares of registrable common stock, whichever is earlier.
77
The holders of the Investor Warrants and the Placement Agent Conversion Warrants, as well as the holders of any shares of common stock removed from the registration statement described above as a result of a Cutback Comment (but not the Restricted Holders (as defined below)), shall have “piggyback” registration rights for the shares of common stock underlying such warrants with respect to any registration statement filed by us following the effectiveness of the registration statement described above, which would permit the inclusion of such underlying shares.
All officers, directors, stockholders holding 10% or more of our common stock after giving effect to the Merger, the Split-Off and the Private Placement Offering and our key employees (each a “Restricted Holder,” and collectively, the “Restricted Holders”), have entered into lock-up agreements with us for a term of eighteen (18) months following the date of the closing of the Merger during which time no Restricted Holder will offer or sell any shares of common stock owned by such Restricted Holder, except to another Restricted Holder. The lock-up agreements entered into by Clearwater Partners, LLC and Angelo Tomasello do not apply to any shares our common stock or any Investor Warrant issued to Clearwater Partners, LLC (97,544 shares of common stock and a warrant to purchase 48,772 shares of common stock) or Mr. Tomasello (325,000 shares of common stock and a warrant to purchase 167,500 shares of common stock) upon consummation of the Merger in exchange for the Units and warrant of 22nd Century contained in the PPO Securities purchased by Clearwater Partners, LLC (97,544 limited liability company membership interests and a warrant to purchase 48,772 limited liability company membership interests) or Mr. Tomasello (325,000 limited liability company membership interests and a warrant to purchase 167,500 limited liability company membership interests) in the Private Placement Offering nor to any shares of our common stock issued to Clearwater Partners, LLC or Mr. Tomasello upon exercise of any Investor Warrant.
In addition, for a period of eighteen (18) months following the closing of the Merger, we will not register or take any action to facilitate registration under the Securities Act of the shares of common stock issued pursuant to the Merger to the Restricted Holders.
Liability and Indemnification of Directors and Officers
Nevada Revised Statutes Sections 78.7502 and 78.751 provide us with the power to indemnify any of our directors and officers. The director and officer must have conducted himself or herself in good faith and reasonably believe that his or her conduct was in, or not opposed to, out best interests. In a criminal action, the director, officer, employee, or agent must not have had reasonable cause to believe that his or her conduct was unlawful.
Under Nevada Revised Statutes Section 78.751, advances for expenses may be made by agreement if the director or officer affirms in writing that he or she believes that he or she has met the statutory standards and will personally repay the expenses if it is determined that such officer or director did not meet the statutory standards.
Our amended and restated articles of incorporation allow for indemnification of directors and officers to the maximum extent permitted by the Nevada Revised Statutes.
Insofar as indemnification for liability under the Securities Act may be permitted for our directors, officers and controlling persons pursuant to the foregoing provisions, or otherwise, we have been advised that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable.
78
Future Stock Issuances
We intend to engage one or more third-parties to provide investors’ relations services to the Company. In addition to other consideration, such third-parties may be compensated with warrants to purchase up to 250,000 shares of our common stock.
Except as expressly set forth herein or pursuant to our equity incentive plan (“EIP”), we have no current plans to issue any additional shares of our capital stock.
Trading Information
Our common stock is quoted on the OTC Bulletin Board under the symbol “XXII.OB.”
Transfer Agent
The transfer agent and registrar for our common stock is Continental Stock Transfer & Trust Company, 17 Battery Place, 8th Floor, New York, NY 10004. We will serve as warrant agent for the outstanding warrants.
LEGAL MATTERS
We are being represented in connection with this registration statement by Foley & Lardner, LLP. On March 30, 2011, 22nd Century issued a promissory note to Foley & Lardner LLP in the principal amount of $350,000 with 4% interest per annum accruing thereon for fees for legal services due and payable to Foley & Lardner LLP by 22nd Century. We also executed this note to guarantee its payment by 22nd Century to Foley & Larder LLP. This note is due and payable on the earlier of: (a) July 1, 2012 or (b) the date on which we or any of our subsidiaries receive funding in the amount $5,000,000 or more. These fees were primarily incurred prior to December 31, 2010 and are included in accounts payable in the December 31, 2010 balance sheet.
EXPERTS
Freed Maxick & Battaglia CPAs, PC, an independent registered public accounting firm, has audited the financial statements of 22nd Century as of December 31, 2010 and 2009 and for each of the years then ended, as stated in their report appearing herein, and have been so included in reliance upon the report of the firm given upon their authority as experts in accounting and auditing.
WHERE YOU CAN FIND MORE INFORMATION
We file annual, quarterly and special reports, proxy statements and other information with the SEC under the Exchange Act (File No. 1-15997). We have also filed with the SEC a registration statement on Form S-1 under the Securities Act to register the shares offered by this prospectus. The term “registration statement” means the original registration statement and any and all amendments thereto, including the schedules and exhibits to the original registration statement or any amendment. This prospectus is part of that registration statement. This prospectus does not contain all of the information set forth in the registration statement or the exhibits to the registration statement. For further information with respect to us and the shares we are offering pursuant to this prospectus, you should refer to the registration statement and its exhibits. Statements contained in this prospectus as to the contents of any contract, agreement or other document referred to are not necessarily complete, and you should refer to the copy of that contract or other documents filed as an exhibit to the registration statement. You may read or obtain a copy of the registration statement at the SEC’s public reference facilities and Internet site referred to below.
You may read or obtain copies of these reports and other information filed with the SEC by us at the public reference facilities maintained by the SEC at 100 F Street, N.E., Washington, D.C. 20549. You can call the SEC at 1-800-SEC-0330 for information regarding the operations of its Public Reference Room and any copying charges assessed by the SEC. The SEC also maintains a website at http://www.sec.gov that contains registration statements, reports, proxy information statements and other information regarding registrants regarding registrants (including us) that file electronically. The information contained on the SEC’s website is not intended to be incorporated by reference in this prospectus and you should not consider that information a part of this prospectus.
79
CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS
ON ACCOUNTING AND FINANCIAL DISCLOSURE
On January 27, 2011, our Board approved the dismissal of Child, Van Wagoner & Bradshaw, PLLC (“Child”), as our independent registered public accounting firm and engaged Freed Maxick & Battaglia CPAs, PC, (“Freed”), as our independent registered public accounting firm, both effective as of January 27, 2011. Freed was the independent registered public accounting firm of 22nd Century prior to the Merger and, given that the business of 22nd Century is now our sole line of business, our Board concluded that Freed should serve as our independent registered public accounting firm.
Child’s report on our financial statements for each of 22nd Century Group, Inc.’s past two fiscal years ended September 30, 2010 and 2009 did not contain an adverse opinion or disclaimer of opinion, nor was it qualified or modified as to uncertainty, audit scope or accounting principles, except that the report was qualified as to 22nd Century Group, Inc.’s ability to continue as a going concern.
During the fiscal years ended September 30, 2010 and 2009 and the subsequent interim period through January 27, 2011, there were no: (i) disagreements with Child on any matter of accounting principles or practices, financial statement disclosure, or auditing scope of procedure which, if not resolved to the satisfaction of Child, would have caused Child to make reference to the matter in their report, or (ii) reportable events as defined in Item 304(a)(1)(v) of Regulation S-K.
During the fiscal years ended September 30, 2010 and 2009 and the subsequent interim period through January 27, 2011, neither 22nd Century Group, Inc. nor anyone acting on its behalf consulted Freed regarding either: (i) the application of accounting principles to a specific transaction, either completed or proposed, or the type of audit opinion that might be rendered on our financial statements; or (ii) any matter that was either the subject of a disagreement (as defined in Item 304(a)(1)(iv) of Regulation S-K) or a reportable event (as described in Item 304(a)(1)(v) of Regulation S-K).
80
22nd CENTURY LIMITED, LLC AND SUBSIDIARY
INDEX TO FINANCIAL STATEMENTS
|
Page
|
|
|
Report of Independent Registered Public Accounting Firm
|
F-2
|
|
Consolidated Financial Statements:
|
|
|
Consolidated Balance Sheets
|
|
|
December 31, 2010 and 2009
|
F-3
|
|
Consolidated Statements of Operations
|
|
|
For The Years Ended December 31, 2010 and 2009
|
F-4
|
|
Consolidated Statements of Members’ Deficit
|
|
|
For The Years Ended December 31, 2010 and 2009
|
F-5
|
|
Consolidated Statements of Cash Flows
|
|
|
For The Years Ended December 31, 2010 and 2009
|
F-6 |
|
Notes to the December 31, 2010 Consolidated Financial Statements
|
F7–F18
|
|
March 31, 2011 (unaudited) and December 31, 2010
|
F-19
|
|
Consolidated Statements of Operations
|
|
|
Three Months Ended March 31, 2011 and 2010 (unaudited)
|
F-20
|
|
Consolidated Statements of Cash Flows
|
|
|
Three Months Ended March 31, 2011 and 2010 (unaudited)
|
F-21
|
|
Consolidated Statement of Shareholders Deficit
|
|
|
Three Months Ended March 31, 2011
|
F-22
|
|
Notes to March 31, 2011 Consolidated Financial Statements
|
F-23
|
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Members
22nd Century Limited, LLC and Subsidiary
We have audited the accompanying consolidated balance sheets of 22nd Century Limited, LLC and Subsidiary as of December 31, 2010 and 2009, and the related consolidated statements of operations, members’ deficit, and cash flows for the years then ended. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of 22nd Century Limited, LLC as of December 31, 2010 and 2009, and the consolidated results of its operations and its cash flows for the years then ended in conformity with accounting principles generally accepted in the United States of America.
The accompanying consolidated financial statements have been prepared assuming that 22nd Century Limited, LLC will continue as a going concern. As discussed in Note 2 to the consolidated financial statements, since 2006 22nd Century Limited, LLC has suffered recurring losses from operations and as of December 31, 2010 has negative working capital of approximately $4.1 million. During January 2011, 22nd Century Limited, LLC raised $3.4 million of net cash proceeds in a private placement offering of its securities and reduced its current debt obligations on the balance sheet at December 31, 2010 by approximately $614,000. However, additional financing is expected to be required during 2011 in order to satisfy existing current obligations and finance working capital needs, as well as additional losses from operations that are expected in 2011. This raises substantial doubt about the Company's ability to continue as a going concern. Management's plans in regard to these matters are also described in Note 2. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ FREED MAXICK & BATTAGLIA, CPAs, PC
Buffalo, New York
March 21, 2011
F-2
|
22nd CENTURY LIMITED, LLC AND SUBSIDIARY
|
|
CONSOLIDATED BALANCE SHEETS
|
|
December 31,
|
|
2010
|
2009
|
|||||||
|
ASSETS
|
||||||||
|
Current assets:
|
||||||||
|
Cash
|
$ | 310 | $ | 158 | ||||
|
Grant receivable
|
223,540 | - | ||||||
|
Inventory
|
308,662 | 55,023 | ||||||
|
Prepaid expenses
|
211,717 | - | ||||||
|
Total current assets
|
744,229 | 55,181 | ||||||
|
Other assets:
|
||||||||
|
Patent and trademark costs, net
|
1,467,623 | 1,484,167 | ||||||
|
Debt issuance costs, net
|
- | 35,923 | ||||||
|
Deferred private placement costs
|
587,133 | - | ||||||
|
Deposits
|
1,535 | 1,535 | ||||||
|
Total other assets
|
2,056,291 | 1,521,625 | ||||||
|
Total assets
|
$ | 2,800,520 | $ | 1,576,806 | ||||
|
LIABILITIES AND MEMBERS' DEFICIT
|
||||||||
|
Current liabilities:
|
||||||||
|
Demand bank loans
|
$ | 174,925 | $ | 246,735 | ||||
|
Accounts payable
|
2,900,684 | 2,144,207 | ||||||
|
Accrued interest payable to members
|
190,977 | 80,188 | ||||||
|
Accrued expenses
|
227,724 | 36,500 | ||||||
|
Deferred grant revenue
|
223,540 | - | ||||||
|
Notes payable to members, net of unamortized discount
|
1,095,643 | 597,468 | ||||||
|
Due to related party
|
6,942 | 126,970 | ||||||
|
Due to member
|
3,200 | 930 | ||||||
|
Total current liabilities
|
4,823,635 | 3,232,998 | ||||||
|
Long-term notes to members, net of unamortized discount
|
65,557 | 141,551 | ||||||
|
Total liabilities
|
4,889,192 | 3,374,549 | ||||||
|
Commitments and contingencies (Note 9)
|
- | - | ||||||
|
Members' deficit:
|
||||||||
|
Contributed capital
|
3,598,856 | 2,466,138 | ||||||
|
Accumulated deficit
|
(5,687,394 | ) | (4,263,762 | ) | ||||
|
Non-controlling interest - consolidated subsidiary
|
(134 | ) | (119 | ) | ||||
|
Total members' deficit
|
(2,088,672 | ) | (1,797,743 | ) | ||||
|
Total liabilities and members' deficit
|
$ | 2,800,520 | $ | 1,576,806 | ||||
See accompanying notes to consolidated financial statements
F-3
|
22nd CENTURY LIMITED, LLC AND SUBSIDIARY
|
|
CONSOLIDATED STATEMENTS OF OPERATIONS
|
|
For the Years Ended December 31,
|
|
2010
|
2009
|
|||||||
|
Sales
|
$ | 49,784 | $ | 27,612 | ||||
|
Operating expenses:
|
||||||||
|
Costs of goods sold
|
27,964 | 20,112 | ||||||
|
Research and development
|
363,781 | 540,300 | ||||||
|
General and administrative
|
590,826 | 280,709 | ||||||
|
Amortization
|
164,456 | 144,792 | ||||||
| 1,147,027 | 985,913 | |||||||
|
Operating loss
|
(1,097,243 | ) | (958,301 | ) | ||||
|
Interest expense and debt expense:
|
||||||||
|
Members
|
(265,221 | ) | (256,803 | ) | ||||
|
Other
|
(61,183 | ) | (11,700 | ) | ||||
|
Net loss
|
(1,423,647 | ) | (1,226,804 | ) | ||||
|
Net loss attributable to non-controlling interest
|
15 | 119 | ||||||
|
Net loss attributed to members
|
$ | (1,423,632 | ) | $ | (1,226,685 | ) | ||
|
Loss per common unit - basic and diluted
|
$ | (0.11 | ) | $ | (0.23 | ) | ||
|
Units used in basic earnings per share calculation
|
12,437,983 | 5,304,423 | ||||||
See accompanying notes to consolidated financial statements
F-4
|
22nd CENTURY LIMITED, LLC AND SUBSIDIARY
|
|
CONSOLIDATED STATEMENTS OF MEMBERS’ DEFICIT
|
|
For the Years Ended December 31, 2010 and 2009
|
|
Member Units
|
||||||||||||||||||||
|
Outstanding
|
Contributed
|
Accumulated
|
Non-controlling
|
Members'
|
||||||||||||||||
|
(restated)
|
Capital
|
Deficit
|
Interest
|
Deficit
|
||||||||||||||||
|
Balance at December 31, 2008
|
5,238,176 | $ | 1,657,019 | $ | (3,037,077 | ) | $ | - | $ | (1,380,058 | ) | |||||||||
|
Membership Units issued in exchange for services
|
74,201 | 18,333 | - | - | 18,333 | |||||||||||||||
|
Warrants issued in exchange for services
|
- | 21,859 | - | - | 21,859 | |||||||||||||||
|
Membership Units issued as compensation in lieu of cash
|
630,710 | 155,833 | - | - | 155,833 | |||||||||||||||
|
Goodrich Tobacco Company units issued as compensation in lieu of cash
|
- | 36,000 | - | - | 36,000 | |||||||||||||||
|
Expensed portion of warrants issued as compensation
|
- | 215,554 | - | - | 215,554 | |||||||||||||||
|
Conversion of member advances to Membership Units
|
1,009,106 | 271,992 | - | - | 271,992 | |||||||||||||||
|
Conversion of member note and accrued interest to Membership Units
|
151,760 | 88,172 | - | - | 88,172 | |||||||||||||||
|
Warrants issued with debt
|
- | 36,395 | - | - | 36,395 | |||||||||||||||
|
Redemption of Membership Units
|
(51,637 | ) | (35,019 | ) | - | - | (35,019 | ) | ||||||||||||
|
Warrants exercised for Membership Units
|
37,624 | - | - | - | - | |||||||||||||||
|
Net loss
|
- | - | (1,226,685 | ) | (119 | ) | (1,226,804 | ) | ||||||||||||
|
Balance at December 31, 2009
|
7,089,940 | 2,466,138 | (4,263,762 | ) | (119 | ) | (1,797,743 | ) | ||||||||||||
|
Warrants issued
|
- | 405,000 | 405,000 | |||||||||||||||||
|
Units Issued
|
3,380,908 | 549,870 | 549,870 | |||||||||||||||||
|
Membership Units issued in exchange for services
|
8,132 | 2,192 | 2,192 | |||||||||||||||||
|
Membership Units issued as compensation in lieu of cash
|
346,493 | 68,237 | 68,237 | |||||||||||||||||
|
Membership Units issued as compensation
|
204,053 | 33,000 | 33,000 | |||||||||||||||||
|
Expensed portion of warrants issued as compensation
|
- | 43,108 | 43,108 | |||||||||||||||||
|
Conversion of member note and accrued interest to Membership Units
|
165,951 | 31,311 | 31,311 | |||||||||||||||||
|
Warrants exercised for Membership Units
|
4,804,523 | - | - | |||||||||||||||||
|
Net loss
|
- | - | (1,423,632 | ) | (15 | ) | (1,423,647 | ) | ||||||||||||
|
Balance at December 31, 2010
|
16,000,000 | $ | 3,598,856 | $ | (5,687,394 | ) | $ | (134 | ) | $ | (2,088,672 | ) | ||||||||
See accompanying notes to consolidated financial statements
F-5
|
22nd CENTURY LIMITED, LLC AND SUBSIDIARY
|
|
CONSOLIDATED STATEMENTS OF CASH FLOWS
|
|
For the Years Ended December 31,
|
|
2010
|
2009
|
|||||||
|
Cash flows from operating activities:
|
||||||||
|
Net loss
|
$ | (1,423,647 | ) | $ | (1,226,804 | ) | ||
|
Adjustments to reconcile net loss to cash used by operating activities:
|
||||||||
|
Amortization of intangible assets
|
164,456 | 144,792 | ||||||
|
Amortization of debt issuance costs
|
35,923 | 43,108 | ||||||
|
Amortization of warrants issued with notes payable
|
126,624 | 142,745 | ||||||
|
Equity based employee compensation expense
|
144,345 | 407,387 | ||||||
|
Equity based payments for outside services
|
2,192 | 40,192 | ||||||
|
Note issued to satisfy employee compensation obligation
|
9,537 | - | ||||||
|
Note issued to satisfy obligation for outside services
|
2,192 | - | ||||||
|
Increase in assets:
|
||||||||
|
Grant receivable
|
(223,540 | ) | - | |||||
|
Inventory
|
(253,639 | ) | (30,023 | ) | ||||
|
Prepaid expenses
|
(211,717 | ) | - | |||||
|
Increase in liabilities:
|
||||||||
|
Accounts payable
|
190,524 | 220,031 | ||||||
|
Accrued interest payable to members
|
112,047 | - | ||||||
|
Accrued expenses
|
191,224 | 93,359 | ||||||
|
Deferred grant revenue
|
223,540 | - | ||||||
|
Net cash used by operating activities
|
(909,939 | ) | (165,213 | ) | ||||
|
Cash flows from investing activities:
|
||||||||
|
Acquisition of patents and trademarks
|
(108,116 | ) | (6,840 | ) | ||||
|
Net cash used by investing activities
|
(108,116 | ) | (6,840 | ) | ||||
|
Cash flows from financing activities:
|
||||||||
|
Payment of deferred private placement costs
|
(60,976 | ) | - | |||||
|
Repayment of bank loans
|
(71,810 | ) | (1,371 | ) | ||||
|
Proceeds from issuance of notes and related warrants
|
- | 55,000 | ||||||
|
Payment on note payable to repurchase membership units
|
(4,390 | ) | - | |||||
|
Proceeds from issuance of notes
|
318,271 | - | ||||||
|
Proceeds from issuance of warrants
|
405,000 | - | ||||||
|
Proceeds from issuance of units
|
549,870 | - | ||||||
|
Net advances (repayments) from related party
|
(120,028 | ) | 69,161 | |||||
|
Net advances from members
|
2,270 | 35,860 | ||||||
|
Net cash provided by financing activities
|
1,018,207 | 158,650 | ||||||
|
Net increase (decrease) in cash
|
152 | (13,403 | ) | |||||
|
Cash - beginning of year
|
158 | 13,561 | ||||||
|
Cash - end of year
|
$ | 310 | $ | 158 | ||||
|
Cash paid during the year for:
|
||||||||
|
Interest
|
$ | 16,890 | $ | 5,661 | ||||
|
Supplemental disclosure of noncash investing and financing activities:
|
||||||||
|
Patent and trademark additions included in accounts payable
|
$ | 39,796 | $ | 221,102 | ||||
|
Deferred private placement cost additions included in accounts payable
|
$ | 526,157 | $ | - | ||||
|
Conversion of member note and accrued interest to Membership Units
|
$ | 31,311 | $ | 88,172 | ||||
|
Note payable issued to repurchase Membership Units
|
$ | - | $ | 35,019 | ||||
|
Debt discount related to warrants issued with notes payable
|
$ | - | $ | 36,395 | ||||
|
Conversion of member advances to Membership Units
|
$ | - | $ | 271,992 | ||||
See accompanying notes to consolidated financial statements
F-6
|
22nd CENTURY LIMITED, LLC AND SUBSIDIARY
|
|
NOTES TO CONSOLIDATED STATEMENTS
|
|
December 31, 2010
|
NOTE 1. - NATURE OF BUSINESS AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Nature of Business - 22nd Century Limited, LLC (“22nd Century”) is a plant biotechnology company founded in 1998. 22nd Century owns or has the exclusive right to use more than 97 issued patents in more than 79 countries related to modifying the content of nicotinic alkaloids in plants, specifically tobacco plants, through genetic engineering and plant breeding.
The overall objective of 22nd Century is to reduce smoking-related disease by increasing smoking cessation with, X-22, its botanical smoking cessation aid and reducing the harm to smokers with 22nd Century’s potential modified risk cigarettes, for smokers unwilling to quit.
22nd Century is primarily involved in the following activities:
|
|
·
|
The development of its botanical smoking cessation aid, X-22;
|
|
|
·
|
The development of its modified risk tobacco products;
|
|
|
·
|
The pursuit of necessary regulatory approvals at the FDA to market X-22 as a prescription smoking cessation aid and its proprietary cigarettes as modified risk tobacco products;
|
|
|
·
|
The manufacture, marketing and distribution of RED SUN and MAGIC proprietary cigarettes in traditional tobacco market channels in the U.S. through its subsidiary Goodrich Tobacco Company; and
|
|
|
·
|
The international licensing of 22nd Century’s trademarks, brands, proprietary tobaccos, and technology.
|
Principles of Consolidation - The accompanying consolidated financial statements include Goodrich Tobacco Company LLC (f/k/a Xodus, LLC), a subsidiary of 22nd Century (collectively, the “Company”). 22nd Century owns 96% of the outstanding Membership Units of Goodrich Tobacco Company. All intercompany accounts and transactions have been eliminated.
Inventory - Inventories are valued at the lower of cost or market. Cost is determined on the first-in, first-out (FIFO) method. The Company’s inventory consisted of the following categories:
|
2010
|
2009
|
|||||||
|
Materials mainly tobacco
|
$ | 292,480 | $ | 55,023 | ||||
|
Finished goods
|
16,182 | - | ||||||
|
Total
|
$ | 308,662 | $ | 55,023 | ||||
F-7
|
22nd CENTURY LIMITED, LLC AND SUBSIDIARY
|
|
NOTES TO CONSOLIDATED STATEMENTS
|
|
December 31, 2010
|
Intangible Assets - Intangible assets are recorded at cost and consist primarily of expenditures incurred with third parties related to the processing of patent claims and trademarks with government authorities. The Company also capitalized costs as a result of one of its exclusively licensed patent application being subject to an interference proceeding invoked by the U.S. Patent and Trademark Office, which favorably resulted in the Company obtaining rights to a third party’s issued patent. The amounts capitalized relate to patents the Company owns or has exclusive rights to and its trademarks, and exclude approximately $1.8 million recovered from a former licensee as direct reimbursements of costs incurred. These capitalized costs are amortized using the straight-line method over the remaining statutory life of the Company’s primary patent family, which expires in 2019 (the assets’ estimated lives). Periodic maintenance or renewal fees, which are generally due on an annual basis are expensed as incurred. Annual minimum license fees are charged to expense in the year the licenses are effective. Total patent and trademark costs capitalized and accumulated amortization amounted to $1,965,621 and $497,998 as of December 31, 2010 ($1,817,709 and $333,542 - 2009). Expected future annual amortization of patent costs and trademarks is approximately $173,000.
Impairment of Long-Lived Assets - The Company reviews the carrying value of its amortizing long-lived assets whenever events or changes in circumstances indicate that the historical cost-carrying value of an asset may no longer be recoverable.
The Company assesses recoverability of the asset by estimating the future undiscounted net cash flows expected to result from the asset, including eventual disposition. If the estimated future undiscounted net cash flows are less than the carrying value of the asset, an impairment loss is recorded equal to the difference between the asset’s carrying value and its fair value. There was no impairment loss recorded during the years ended December 31, 2010 or 2009.
Deferred Private Placement Costs - During 2010 the Company incurred costs related to the private placement that closed on January 25, 2011. Such costs were accumulated on the balance sheet and were charged against contributed capital upon closing of the offering. Deferred private placement costs amounted to $587,133 as of December 31, 2010 ($0 – 2009).
Income Taxes - The Company has elected to be treated as a Partnership for Federal and State income tax purposes. As a result, there is no corporate level tax because all taxable income, tax deductions and tax credits are passed through to the members of the Company. The Company’s Federal and State tax returns for the years ended December 31, 2007 to December 31, 2010 are currently open to audit under statutes of limitations.
Following the merger on January 25, 2011 (see Note 12) the Company’s operations will be subject to Federal and State income taxes.
Employee Equity-Based Compensation - The Company uses a fair-value based method to determine compensation for all arrangements under which Company members, employees and others receive Membership Units or warrants to purchase Membership Units of the Company.
Debt Discounts - The Company accounts for warrants issued to note holders as an inducement to provide financing for the Company in accordance with the FASB’s guidance on Accounting for Convertible Debt and Convertible Debt Issued with Stock Purchase Warrants. Fair value of the warrants is determined by Membership Unit price according to recent equity transactions since there is no vesting period and a negligible exercise price. The proceeds allocated to the warrant is recorded as a debt discount and amortized over the life of the corresponding financing as interest expense.
F-8
|
22nd CENTURY LIMITED, LLC AND SUBSIDIARY
|
|
NOTES TO CONSOLIDATED STATEMENTS
|
|
December 31, 2010
|
Unearned Grant Income - The Company received approval of a government grant as partial support for its next clinical trial with the FDA. This grant will be recognized as a reduction of the cost of the clinical trial as such costs are incurred. As of December 31, 2010, the Company recorded a grant receivable and corresponding deferred grant revenue amounting to $223,540. No unearned grant income existed as of December 31, 2009.
Revenue Recognition - The Company recognizes revenue at the point the product is shipped to a customer and title has transferred. Revenue from the sale of the Company’s products is recognized net of cash discounts, sales returns and allowances. Federal Excise Taxes are included in net sales and accounts receivable billed to customers.
Shipping Costs - Shipping costs are included in general and administrative expense and aggregated $2,290 in 2010 ($2,262 – 2009).
Advertising Costs - Advertising costs are expensed as incurred and are included in general and administrative expense. Advertising costs for the year ended December 31, 2010 amounted to $3,520 ($979 – 2009).
Research and Development - Research and development costs are expensed as incurred.
Loss Per Common Unit - Basic loss per common Membership Unit is computed using the weighted-average number of common Membership Units outstanding. Diluted loss per unit is computed assuming conversion of all potentially dilutive warrants. Potential common Membership Units outstanding are excluded from the computation if their effect is anti-dilutive.
Commitment and Contingency Accounting - The Company evaluates each commitment and/or contingency in accordance with the accounting standards, which state that if the item is more likely than not to become a direct liability, then the Company will record the liability in the financial statements. If not, the Company will disclose any material commitments or contingencies that may arise.
F-9
|
22nd CENTURY LIMITED, LLC AND SUBSIDIARY
|
|
NOTES TO CONSOLIDATED STATEMENTS
|
|
December 31, 2010
|
Use of Estimates - The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of income and expenses during the reporting period. Actual results could differ from those estimates.
Reclassifications - Certain 2009 amounts have been reclassified to conform to the 2010 presentation.
Membership Units Split - On October 5, 2010 the Company authorized a 37,100.5626 to 1 split of its Membership Units. The amounts shown for Membership Units, warrants and loss per unit amounts have been retroactively adjusted in all periods presented to reflect this split.
Subsequent Events - These statements have not been updated for subsequent events occurring after March 21, 2011, which is the date these financial statements were available to be issued.
NOTE 2. - LIQUIDITY AND MANAGEMENT’S PLANS
Since 2006, 22nd Century has experienced limited revenues and incurred substantial operating losses as it transitioned from being only a licensor of its proprietary technology and tobaccos to commercializing its own tobacco products. At December 31, 2010, the Company had current assets of $744,229 and current liabilities of $4,823,635. On January 25, 2011, the Company completed a private placement of securities resulting in approximately $3.4 million in net cash proceeds and a reduction of debt obligations to members that were on the balance sheet at December 31, 2010 of approximately $614,000, which were exchanged for equity interests in the offering. These proceeds are sufficient to enable the Company to make substantial reductions in its outstanding current liabilities, which management expects will allow the Company to generally be on satisfactory terms with its vendors. In addition, the Company will need to raise additional capital to further reduce outstanding current liabilities and complete the FDA-approval process for X-22. Before the Company can raise additional capital it must satisfy the registration rights granted to investors who acquired shares of its common stock in the January 25, 2011 private placement and Merger. These registration rights provide that the Company must have an effective registration statement covering the shares subject to the registration rights for a period of 90 days before it can sell any equity securities or securities convertible into equity securities. In addition, the ability to complete future financings on acceptable terms will depend on a number of factors, including the general performance of the capital markets, the Company’s progress in the FDA approval process and the manufacture, distribution and sale of its commercial products. Any future equity financings will be dilutive to the Company’s existing shareholders ownership percentages.
Prior to FDA approval of its prescription X-22 smoking cessation aid and FDA authorization of its two modified risk cigarettes, the Company expects to generate cash from the sale of its proprietary cigarettes in the U.S through its subsidiary, Goodrich Tobacco Company. Sales are expected in traditional tobacco market channels and to fulfill orders by the National Institute of Drug Abuse (“NIDA”) and researchers. In March 2011, the Company received a revised purchase order and authorization to bill $112,000 for manufacture planning and material procurement for 9 million cigarettes from RTI International (“RTI”). Payment was received on May 2, 2011. The Company received an additional purchase order from RTI for approximately $680,000 in May 2011 to ship the 9 million research cigarettes. These government research cigarettes will be distributed under the mark SPECTRUM.
The Company believes, but can offer no assurances that the above business plans will provide sufficient cash flow to fund the Company’s operations during 2011.
F-10
|
22nd CENTURY LIMITED, LLC AND SUBSIDIARY
|
|
NOTES TO CONSOLIDATED STATEMENTS
|
|
December 31, 2010
|
NOTE 3. - AMOUNTS OWED NORTH CAROLINA STATE UNIVERSITY (“NCSU”)
Pursuant to the terms of an exclusive license agreement with NCSU, the Company owes NCSU approximately $1,128,000 as of December 31, 2010 for patent costs ($1,045,000 – 2009). These amounts are included in accounts payable in the consolidated balance sheets. The Company was required to pay these amounts within thirty days of being invoiced and they are past due. NCSU has the right to claim interest on the balance. In February 2011 the Company made a payment to NCSU towards patent costs totaling $400,000 out of the net proceeds of the January 25, 2011 private placement and the Company and NCSU have entered into negotiations regarding the timing of payment of the balance owed. If the Company and NCSU cannot reach a satisfactory agreement, NCSU may have the right to terminate the exclusive license agreement, but can only do so with a 60-day prior written notice, including the opportunity to cure within this timeframe. As of December 31, 2010, patent costs associated with the exclusive license agreements that could potentially be terminated had a carrying value of approximately $783,000. Additionally, NCSU has not imposed interest charges on past due amounts invoiced to the Company and as such the Company has not recorded accrued interest or interest expense.
NOTE 4. - DEMAND BANK LOANS
The demand loan is payable to a commercial bank under a revolving credit agreement and is guaranteed by a member of the Company. This loan has a balance of $174,925 at both December 31, 2010 and 2009. The Company is required to pay interest monthly at 0.75% above the prime rate, or 4.00%, at December 31, 2010 (4.00% - 2009). The Company has met this interest payment obligation. The terms of the demand loan includes an annual “clean-up” provision, which require the Company to repay all principal amounts outstanding. The Company has not complied with this requirement; however, the bank has not demanded payment.
NOTE 5. - NOTES PAYABLE
Notes payable members, net of unamortized discount:
|
2010
|
2009
|
|||||||
|
Note dated October 28, 2008, net of unamortized discount
|
$ | 325,000 | $ | 271,041 | ||||
|
Note dated November 11, 2008, net of unamortized discount
|
325,000 | 271,041 | ||||||
|
Note dated May 20, 2009, net of unamortized discount
|
30,000 | 20,367 | ||||||
|
Note dated January 1, 2008
|
100,014 | - | ||||||
|
Note dated September 1, 2010
|
35,000 | - | ||||||
|
Notes dated October 4, 2010
|
150,000 | - | ||||||
|
Note dated December 31, 2010
|
100,000 | - | ||||||
|
Note payable to repurchase Membership Units
|
30,629 | 35,019 | ||||||
| $ | 1,095,643 | $ | 597,468 | |||||
F-11
|
22nd CENTURY LIMITED, LLC AND SUBSIDIARY
|
|
NOTES TO CONSOLIDATED STATEMENTS
|
|
December 31, 2010
|
Note Dated October 28, 2008 - On October 28, 2008, the Company issued a note payable to a third party in the amount of $325,000, and a warrant to purchase 371,006 Membership Units at less than $.0001 per unit. The warrant was valued at $129,500 and recorded as a discount to the note payable and is being amortized over the term of the note which significantly adjusts the effective interest rate. The weighted average annual effective rate on the note is 41%. The intrinsic value of the warrant at the time of issuance was determined to be $215,540; the debt discount recorded was based on allocating the $325,000 in transaction proceeds proportionally between the note and the warrant. The note bears interest at a rate of 10% and the outstanding principal and interest was due and payable on October 28, 2010, the maturity date. As of December 31, 2010, the outstanding principal and unamortized debt discount amounted to $325,000 and $0, ($325,000 and $53,959 – December 31, 2009), respectively. The warrant was exercised in 2010. The note is guaranteed by a related party, Virgil Properties, LLC (“Virgil”), which is owned by two members of the Company. The note is secured by a mortgage on property owned by Virgil. Virgil received 148,402 warrants as consideration for this guarantee. These warrants were valued at $86,216, recorded as a deferred financing and amortized over the term of the loan. On December 30, 2009, Virgil agreed to rescind these warrants. In consideration of the rescission of warrants, the Company agreed to convert certain cash advances totaling $271,992 from the two members of the Company that own Vigil into 1,009,106 Membership Units of the Company.
This note matured and remained outstanding at December 31, 2010; however, in January and February 2011 this note together with the $30,000 note dated May 20, 2009, and all the related accrued interest were satisfied by: (i) conversion of $150,000 into equity in connection with the January 25, 2011 private placement (ii) a cash payment of $142,300 and (iii) a new note for $140,000.
Note Dated November 11, 2008 - On November 11, 2008, the Company issued a note payable to a member in the amount of $325,000, and a warrant to purchase 371,006 Membership Units at less than $.0001 per unit. The warrant was valued at $129,500 and was recorded as a discount to the note payable and is being amortized over the term of the note which significantly adjusts the effective interest rate. The weighted average annual effective rate on the note is 41%. The intrinsic value of the warrant at the time of issuance was determined to be $215,540; the debt discount recorded was based on allocating the $325,000 in transaction proceeds proportionally between the note and the warrant. The note bears interest at a rate of 10% and the outstanding principal and interest was due and payable on November 11, 2010, the maturity date. As of December 31, 2010, the outstanding principal and unamortized debt discount amounted to $325,000 and $0, ($325,000 and $53,959 – December 31, 2009), respectively. The warrant was exercised in 2010. The note is guaranteed by a related party, Virgil, which is owned by two members of the Company.
The note was amended to extend the maturity sixty days to January 10, 2011 and increase the interest rate to 15% during the extension period. Following the maturity of this note in January 2011, the principal amount of $325,000 was converted to equity in connection with the January 25, 2011 private placement and the accrued interest was paid in cash.
Note Dated May 20, 2009 (unsecured) - On May 20, 2009, the Company issued a note payable in the amount of $30,000 and a warrant to purchase 185,503 Membership Units at less than $.0001 per unit to the same third party that was issued the October 28, 2008. The warrant was valued at $18,132 and recorded as a discount to the note payable and is being amortized over the term of the note, which significantly adjusts the effective interest rate. The weighted average annual effective rate on the note is 178%. The intrinsic value of the warrant at the time of issuance was determined to be $45,833; the debt discount recorded was based on allocating the $30,000 in transaction proceeds proportionally between the notes and the warrant. The note bears interest at a rate of 10% and the outstanding principal and interest was due and payable on May 19, 2010, the maturity date. The $30,000 in principal and accrued interest remained outstanding as of December 31, 2010. As of December 31, 2010, the outstanding principal and unamortized debt discount amounted to $30,000 and $0, ($30,000 and $6,233 – December 31, 2009) respectively. The warrant was exercised in 2010. This note was satisfied in January and February 2011 together with the October 28, 2008 note as described previously.
F-12
|
22nd CENTURY LIMITED, LLC AND SUBSIDIARY
|
|
NOTES TO CONSOLIDATED STATEMENTS
|
|
December 31, 2010
|
Note Dated January 1, 2008 (unsecured) - The Company issued a note to a member as of January 1, 2008 for $100,014. The note bears interest at a rate of 7%, and interest and principal are due on the maturity date of January 15, 2011 and was paid in cash in January 2011. The note is subordinated to senior debt, which consists of amounts payable on the bank demand loan.
Note Dated September 1, 2010 - The Company issued a note payable to a member in the amount of $35,000. The note bears interest at a rate of 15%, and interest and principal are due on the maturity date of November 1, 2010. The note was amended to extend the maturity to January 25, 2011. The note is guaranteed by a member of the Company. The note was paid in cash in January 2011.
Notes Dated October 4, 2010 (unsecured) -The Company issued notes payable to seven members in the aggregate amount of $150,000. The notes bears interest at a rate of 15%, and interest and principal are due on the maturity date of January 31, 2011. These notes were satisfied in January 2011 by conversion to equity in connection with the January 25, 2011 private placement and by payment of cash.
Note Dated December 29, 2010 (unsecured) - The Company issued a note payable to a member in the amount of $100,000. The note bears interest at a rate of 15%, and interest and principal are due on the maturity date of January 29, 2011. The note was paid in cash in January 2011.
Note Payable to Repurchase Membership Units (unsecured) - Prior to December 31, 2009, the Company agreed to repurchase 51,637 Membership Units previously issued to the member for $35,019 which remained unpaid as of December 31, 2009. Subsequently, the Company issued a note dated January 1, 2010 to evidence the obligation. The note bears interest at a rate of 7% and the outstanding principal and interest is due and payable on September 30, 2010, the maturity date. The note remained unpaid at the maturity date and was paid in February 2011. As of December 31, 2010 the outstanding principal amounted to $30,629 ($35,019 as of December 31, 2009).
Long term notes payable to members, net of unamortized discount:
|
2010
|
2009
|
|||||||
|
Notes dated September 15 and October 15, 2009, net of unamortized discount
|
$ | 20,557 | $ | 11,483 | ||||
|
Note dated May 27, 2010
|
45,000 | - | ||||||
|
Note dated January 1, 2008
|
- | 100,014 | ||||||
|
Note dated December 30, 2009
|
- | 30,054 | ||||||
| $ | 65,557 | $ | 141,551 | |||||
Notes Dated September 15 and October 15, 2009 (unsecured) - On September 15 and October 15, 2009, the Company issued two notes payable to the same third party in the amounts of $15,000 and $10,000, respectively. In conjunction with the $15,000 note, a warrant to purchase 185,503 membership units at less than $.0001 per unit was issued, and in conjunction with the $10,000 note, a warrant to purchase 92,751 Membership Units at less than $.0001 per unit was issued. The warrants were valued at $11,301 for the $15,000 note and $6,962 for the $10,000 and recorded as discounts to the respective notes payable and are being amortized over the term of each note which significantly adjusts the effective interest rate. The intrinsic value of the warrants at the time of issuance was determined to be $68,750; the debt discount recorded was based on allocating the $25,000 in transaction proceeds proportionally between the notes and the warrants. The notes bear interest at a rate of 10% and the outstanding principal and interest is due and payable at maturity - January 31, 2012. As of December 31, 2010, the total outstanding principal and unamortized debt discounts for the two notes amounted to $25,000 and $4,443 ($25,000 and $13,517 – December 31, 2009), respectively. The warrants were exercised in 2010.
F-13
|
22nd CENTURY LIMITED, LLC AND SUBSIDIARY
|
|
NOTES TO CONSOLIDATED STATEMENTS
|
|
December 31, 2010
|
Note Dated May 27, 2010 (unsecured) - During the first quarter of 2010 the holder of the Notes dated September 15 and October 15, 2009 advanced additional funds, totaling $450,000, to the Company and obtained conversion rights to warrants to acquire Membership Units. In March 2010, $225,000 was converted into warrants to acquire approximately 1,706,626 Membership Units and this amount was recorded as equity. Pursuant to an agreement effective on May 27, 2010, in exchange for the remaining $225,000 advanced, the Company issued warrants to acquire approximately 1,409,821 Membership Units and a note for $45,000. The note bears interest at 10%, which is due with the principal amount on January 31, 2012. The note was satisfied in January 2011 by conversion to equity in connection with the January 25, 2011 private placement.
Note Dated December 30, 2009 (unsecured) - On December 30, 2009, the Company issued a note to a member in exchange for advances the member previously made to the Company. The original amount of the note was $30,054 and, in June 2010, the Company agreed to allow the principal and accrued interest of $1,257 to be converted into 165,951 Membership Units.
NOTE 6. - DUE TO RELATED PARTY
The Company has conducted numerous transactions with a related party, Alternative Cigarettes, Inc. (“AC”). AC is entirely owned by certain members of the Company. AC shares office space and employee services with the Company for which the Company was reimbursed by AC in the amount of approximately $13,000 during 2010 ($32,000 – 2009). The net amount due to AC as a result of advances, repayments and expenses incurred and reimbursed amounted to $6,942 as of December 31, 2010 ($126,970 - 2009). No interest has been accrued or paid on amount due to AC and there are no repayment terms.
NOTE 7. - DUE TO MEMBERS
Amount due to members is a result of member advances to the Company for working capital purposes or services recorded as member advances. During 2009, two members accepted 504,553 Membership Units each in exchange for $135,996 of advances owed to each member by the Company. Also, during 2009, one member converted $30,054 of amounts due into a subordinated note payable as discussed in Note 5. As of December 31, 2010, the remaining unpaid amount due to a member for advances to the Company for working capital purposes was $3,200 ($930 - 2009). No interest has been accrued or paid on amount due to members and there are no repayment terms.
NOTE 8. - WARRANTS FOR MEMBERSHIP UNITS
The Company has granted warrants in connection with borrowings as an additional incentive for providing financing to the Company and as additional compensation to officers, consultants and advisors. The warrants are granted with a conversion price of less than $.0001, and the number of warrants issued has been negotiated based on the agreement at the time of the grant. The warrants have been issued for terms of two to five years.
F-14
|
22nd CENTURY LIMITED, LLC AND SUBSIDIARY
|
|
NOTES TO CONSOLIDATED STATEMENTS
|
|
December 31, 2010
|
Warrants issued and outstanding during the years ended December 31, 2010 and 2009 are as follows:
|
Number of
|
||||
|
Warrants
|
||||
|
Warrants outstanding at December 31, 2008
|
927,514 | |||
|
Warrants issued during 2009
|
946,064 | |||
|
Warrants exercised during 2009
|
(37,100 | ) | ||
|
Warrants forfeited during 2009
|
(148,402 | ) | ||
|
Warrants outstanding at December 31, 2009
|
1,688,076 | |||
|
Warrants issued during 2010
|
3,116,447 | |||
|
Warrants exercised during 2010
|
(4,804,523 | ) | ||
|
Warrants outstanding at December 31, 2010
|
- | |||
|
Warrants exercisable at December 31, 2010
|
- | |||
On February 1, 2009, the Company granted an award for service to an executive officer of 445,207 warrants, vesting over a one year service period ending February 1, 2010. The related compensation cost of $258,662 was determined by the intrinsic value of the underlying common Membership Units at the time of the award of $0.58 per unit and is being charged to expense on a straight line basis over the service period. For the year ended December 31, 2010, $43,108 ($215,554 -2009) was recorded as expense. There is no unrecognized compensation expense related to the grant of these warrants.
NOTE 9. - COMMITMENTS
License Agreements - Under its exclusive license agreement with NCSU the Company is required to pay minimum annual royalty payments, which are credited against running royalties on sales of licensed products. The annual minimum royalty for each of the calendar years 2010 through 2013 is $75,000, and in 2014 the annual minimum royalty increases to $200,000. The license agreement continues through the life of the last-to-expire patent. These minimum royalty payments are due each February following the end of the applicable calendar year reduced by any running royalties paid or payable for that year. The agreement also requires a milestone payment of $150,000 upon FDA approval of a product that uses the NCSU licensed technology. The Company is also responsible for reimbursing NCSU for actual third-party patent costs incurred. These costs vary from year to year and the Company has certain rights to direct the activities that result in these costs. During 2010, the costs incurred related to patent costs and patent maintenance amounted to $125,956 ($169,512 – 2009).
The Company has two other exclusive license agreements which require aggregate annual license fees of approximately $55,000, which are credited against running royalties on sales of licensed products. Each license agreement continues through the life of the last-to-expire patent.
Operating Leases - The Company leases office space under non-cancelable operating leases for $1,584 per month; expiring in October 2011. Rent expense under the operating lease was approximately $18,600 for the year ended December 31, 2010 ($18,600 – 2009). Future minimum payments due under the operating lease are approximately $15,800 in 2011.
NOTE 10. - FAIR VALUE OF FINANCIAL INSTRUMENTS
The estimated fair value of cash, advances from members and related party, demand bank loans and notes payable approximate the carrying value due to their short-term nature. In applying the accounting standards for fair value determination, the Company has taken into account what the Company would have to pay someone to take over its debt obligations. Considerable judgment is required in developing estimates of fair value. Therefore, the estimates presented herein are not necessarily indicative of the amounts that the Company would realize in a current market exchange.
F-15
|
22nd CENTURY LIMITED, LLC AND SUBSIDIARY
|
|
NOTES TO CONSOLIDATED STATEMENTS
|
|
December 31, 2010
|
The estimated fair value of long-term debt is summarized as follows:
|
2010
|
2009
|
|||||||||||||
|
Carrying
|
Estimated
|
Carrying
|
Estimated
|
|||||||||||
|
Amount
|
Fair Value
|
Amount
|
Fair Value
|
|||||||||||
| $ | 65,557 | $ | 63,000 | $ | 141,551 | $ | 92,000 | |||||||
Differences between fair value and carrying amount of long-term debt are primarily due to instruments that provide fixed interest or contain fixed interest rate elements. Inherently, such instruments are subject to fluctuations in fair value due to subsequent movements in interest rates that are available to the Company.
NOTE 11. - EARNINGS PER MEMBERSHIP UNIT
The following table sets forth the computation of basic and diluted earnings per share for the years ended December 31:
|
2010
|
2009
|
|||||||
|
Net loss attributed to members
|
$ | (1,423,632 | ) | $ | (1,226,685 | ) | ||
|
Denominator for basic earnings per share-weighted average units outstanding
|
12,437,983 | 5,304,423 | ||||||
|
Effect of dilutive securities:
|
||||||||
|
warrants outstanding
|
- | - | ||||||
|
Denominator for diluted earnings per unit - weighted average units adjusted for dilutive securities
|
12,437,983 | 5,304,423 | ||||||
| 2010 | 2009 | |||||||
|
Loss per common unit - basic
|
$ | (0.11 | ) | $ | (0.23 | ) | ||
|
Loss per common unit- diluted
|
$ | (0.11 | ) | $ | (0.23 | ) | ||
Securities outstanding that were excluded from the computation because they would have been anti-dilutive are as follows:
|
2010
|
2009
|
|||||||
|
Debt convertible into units (number of units)
|
- | 197,679 | ||||||
|
Warrants
|
- | 1,688,076 | ||||||
F-16
|
22nd CENTURY LIMITED, LLC AND SUBSIDIARY
|
|
NOTES TO CONSOLIDATED STATEMENTS
|
|
December 31, 2010
|
NOTE 12. - SUBSEQUENT EVENTS
Private Placement and Merger - On January 25, 2011, the Company completed a private placement offering (the “Private Placement”) of 5,434,446 units of its securities or “Units” at the purchase price of $1.00 per Unit, each Unit consisting of one (1) limited liability company membership interest and a five-year warrant to purchase one-half of one (1/2) limited liability company membership interest at an exercise price of $1.50 per whole limited liability company membership interest. In connection with the Private Placement, the Company approved a prorata distribution of 5,000,000 five-year warrants to purchase one limited liability company membership interest at an exercise price of $3.00 to its members immediately prior to the closing of the Private Placement. Private Placement proceeds included $614,070 from the conversion of Company indebtedness into Units and $395,376 from the conversion of Placement Agent Fees into Units, resulting in gross cash proceeds of $4,425,000. Private Placement offering expenses incurred included cash expenses of approximately $1,010,000 and non-cash expenses consisting of the Placement Agent fees of $395,376 and $390,000 for the estimated fair value of the Placement Agent and Advisor Warrants issued to the Placement Agent. 22nd Century received net cash proceeds of approximately $3,415,000 from the Private Placement and the Company reduced its debt obligations to members that were on the balance sheet at December 31, 2010 of approximately $614,000, which was exchanged for equity interests in the offering. Upon closing of the Private Placement, the Placement Agent was granted Placement Agent warrants to purchase 434,755 one half of one (1/2) limited liability company membership interest at an exercise price of $1.50 per whole limited liability company membership interest.
The Company agreed to a covenant in conjunction with the Private Placement Offering to use our best efforts to file, within 75 days following the effective date of the Merger, with the SEC a registration statement, which will cover the resale of the Common Stock issued to the investors in the Private Placement as a result of the Merger in exchange for the Units contained in the PPO Securities. The Company will use our best efforts to cause this registration statement to be declared effective by the SEC within one hundred eighty (180) calendar days of filing with the SEC (240 days if the SEC reviews such registration statement). If the Company is late in filing this registration statement or if this registration statement is not declared effective within the prescribed time periods, then the holders of Common Stock to be registered shall be entitled to monetary penalties at a rate equal to one-half percent (0.50%) of the offering price per Unit in the Private Placement Offering for each full month that (i) The Company are late in filing this registration statement or (ii) this registration statement is late in being declared effective by the SEC; provided, however, that in no event shall the aggregate of any such penalties exceed five percent (5%) of the offering price per Unit in the Private Placement Offering. In addition for a period of 90 days following the effective date of this registration statement the Company cannot sell any equity securities or securities convertible into equity securities.
Also, on January 25, 2011, the Company consummated a merger with 22nd Century Group, Inc., a public shell company with limited activity. In connection therewith, 22nd Century Limited, LLC became a wholly-owned subsidiary of the 22nd Century Group, Inc. and the pre-Merger holders of 22nd Century Limited, LLC Units and warrants were issued an aggregate of 21,434,446 shares of common stock of 22nd Century Group Inc. and warrants to purchase an aggregate of 8,151,980 shares of its common stock, respectively, in exchange for the securities of 22nd Century Limited LLC so held. Immediately following the consummation of the Merger, approximately 80.1% of the 26,759,646 shares of common stock issued and outstanding was held by pre-Merger holders of 22nd Century Limited LLC Units. Upon closing of the merger the Placement Agent was granted the Advisor Warrants to purchase 500,000 shares of common stock of 22nd Century Group, Inc. at an exercise price of $1.50 per common share
The merger is being accounted for as a reverse acquisition and recapitalization of the Company for financial accounting purposes whereby the Company is deemed to be the acquirer for accounting and financial reporting purposes. Consequently, the assets and liabilities and the historical operations that will be reflected in the financial statements prior to the merger will be those of the Company and will be recorded at the historical cost basis of the Company, and the consolidated financial statements after completion of the merger will include the assets and liabilities of 22nd Century Group, Inc. and the Company, historical operations of the Company and operations of 22nd Century Group, Inc. beginning on the closing date of the merger. No goodwill will be recorded as a result of the accounting for the merger. The Company will become an SEC reporting entity effective with the merger.
F-17
|
22nd CENTURY LIMITED, LLC AND SUBSIDIARY
|
|
NOTES TO CONSOLIDATED STATEMENTS
|
|
December 31, 2010
|
The unaudited supplemental pro forma revenue and net loss attributed to members of the Company if the acquisition had taken place as of January 1, 2009 are as follows:
|
2010
|
2009
|
|||||||
|
Revenue
|
$ | 49,784 | $ | 27,612 | ||||
|
Net loss attributed to members
|
$ | (1,473,632 | ) | $ | (1,276,685 | ) | ||
F-18
|
22nd CENTURY GROUP INC. AND SUBSIDIARIES
|
|
CONSOLIDATED BALANCE SHEETS
|
|
|
|
March 31, 2011
|
December 31, 2010
|
|||||||
|
(unaudited)
|
||||||||
|
ASSETS
|
||||||||
|
Current assets:
|
||||||||
|
Cash
|
$ | 872,401 | $ | 310 | ||||
|
Accounts receivable
|
112,056 | - | ||||||
|
Grant receivable
|
- | 223,540 | ||||||
|
Due from related party
|
14,330 | - | ||||||
|
Inventory
|
768,382 | 308,662 | ||||||
|
Prepaid expenses
|
194,482 | 211,717 | ||||||
|
Total current assets
|
1,961,651 | 744,229 | ||||||
|
Other assets:
|
||||||||
|
Patent and trademark costs, net
|
1,442,575 | 1,467,623 | ||||||
|
Office furniture and fixtures, net
|
4,670 | - | ||||||
|
Deferred private placement costs
|
- | 587,133 | ||||||
|
Deposits
|
1,535 | 1,535 | ||||||
|
Total other assets
|
1,448,780 | 2,056,291 | ||||||
|
Total assets
|
$ | 3,410,431 | $ | 2,800,520 | ||||
|
LIABILITIES AND SHAREHOLDERS' DEFICIT
|
||||||||
|
Current liabilities:
|
||||||||
|
Demand bank loan
|
$ | 174,925 | $ | 174,925 | ||||
|
Accounts payable
|
770,862 | 2,900,684 | ||||||
|
Accrued interest payable to shareholders
|
6,477 | 190,977 | ||||||
|
Accrued expenses
|
255,880 | 227,724 | ||||||
|
Deferred grant revenue
|
174,324 | 223,540 | ||||||
|
Notes payable to shareholders, net of unamortized discount
|
- | 1,095,643 | ||||||
|
Current portion of long-term debt
|
71,890 | - | ||||||
|
Due to related party
|
- | 6,942 | ||||||
|
Due to officer
|
4,639 | 3,200 | ||||||
|
Total current liabilities
|
1,458,997 | 4,823,635 | ||||||
|
Long-term notes
|
587,000 | - | ||||||
|
Long-term notes to shareholders, net of unamortized discount
|
161,890 | 65,557 | ||||||
|
Total long term debt
|
748,890 | 65,557 | ||||||
|
Less current portion
|
(71,890 | ) | - | |||||
| 677,000 | 65,557 | |||||||
|
Warrant derivative liability
|
3,061,750 | - | ||||||
|
Total liabilities
|
5,197,747 | 4,889,192 | ||||||
|
Commitments and contingencies (Note 10)
|
- | - | ||||||
|
Shareholders' deficit
|
||||||||
|
Capital stock:
|
||||||||
|
Authorized:
|
||||||||
|
10,000,000 preferred shares, $.00001 par value
|
||||||||
|
300,000,000 common shares, $.00001 par value
|
||||||||
|
Issued and outstanding:
|
||||||||
|
0 preferred shares
|
- | - | ||||||
|
26,759,646 common shares (16,000,000 at December 31, 2010)
|
268 | |||||||
|
Capital in excess of par value
|
4,551,610 | 3,598,856 | ||||||
|
Accumulated deficit
|
(6,336,694 | ) | (5,687,394 | ) | ||||
|
Non-controlling interest - consolidated subsidiary
|
(2,500 | ) | (134 | ) | ||||
|
Total sharehoders' deficit
|
(1,787,316 | ) | (2,088,672 | ) | ||||
|
Total liabilities and shareholders' deficit
|
$ | 3,410,431 | $ | 2,800,520 | ||||
See accompanying notes to financial statements.
F-19
|
CONSOLIDATED STATEMENTS OF OPERATIONS
|
|
Three Months Ended March 31, 2011 and 2010
|
|
(unaudited)
|
|
March 31, 2011
|
March 31, 2010
|
|||||||
|
Net Sales
|
$ | 117,456 | $ | - | ||||
|
Other Income - Therapeutic Grant Credit
|
49,216 | - | ||||||
|
Total gross revenue
|
166,672 | - | ||||||
|
Operating expenses:
|
||||||||
|
Costs of goods sold
|
47,002 | - | ||||||
|
Research and development
|
216,291 | 95,420 | ||||||
|
General and administrative
|
325,938 | 96,108 | ||||||
|
Sales and marketing costs
|
171,525 | - | ||||||
|
Amortization and depreciation
|
43,577 | 39,860 | ||||||
| 804,333 | 231,388 | |||||||
|
Operating loss
|
(637,661 | ) | (231,388 | ) | ||||
|
Interest expense and debt expense:
|
||||||||
|
Shareholders
|
(12,145 | ) | (70,982 | ) | ||||
|
Other
|
(1,860 | ) | (2,647 | ) | ||||
|
Net loss
|
(651,666 | ) | (305,017 | ) | ||||
|
Net loss attributable to non-controlling interest
|
2,366 | 4 | ||||||
|
Net loss attributed to common shareholders
|
$ | (649,300 | ) | $ | (305,013 | ) | ||
|
Loss per common share - basic and diluted
|
$ | (0.03 | ) | $ | (0.04 | ) | ||
|
Common shares used in basic earnings per share calculation
|
23,890,407 | 7,089,946 | ||||||
See accompanying notes to financial statements.
F-20
|
CONSOLIDATED STATEMENTS OF CASH FLOWS
|
|
Three Months Ended March 31, 2011 and 2010
|
|
(unaudited)
|
|
March 31, 2011
|
March 31, 2010
|
|||||||
|
Cash flows from operating activities:
|
||||||||
|
Net loss
|
$ | (651,666 | ) | $ | (305,017 | ) | ||
|
Adjustments to reconcile net loss to cash used by operating activities:
|
||||||||
|
Amortization and depreciation
|
43,577 | 39,860 | ||||||
|
Amortization of debt issuance costs
|
- | 10,777 | ||||||
|
Amortization of debt discount
|
1,334 | 40,341 | ||||||
|
Equity based employee compensation expense
|
- | 43,108 | ||||||
|
(Increase) decrease in assets:
|
||||||||
|
Accounts receivable
|
(112,056 | ) | - | |||||
|
Grant receivable
|
223,540 | - | ||||||
|
Inventory
|
(459,720 | ) | (206,737 | ) | ||||
|
Prepaid expenses
|
17,234 | (18,750 | ) | |||||
|
Increase (decrease) in liabilities:
|
||||||||
|
Accounts payable
|
(516,695 | ) | (82,090 | ) | ||||
|
Accrued interest payable to shareholders
|
(177,797 | ) | 19,865 | |||||
|
Accrued expenses
|
28,156 | 76,987 | ||||||
|
Deferred grant revenue
|
(49,216 | ) | - | |||||
|
Net cash used by operating activities
|
(1,653,309 | ) | (381,656 | ) | ||||
|
Cash flows from investing activities:
|
||||||||
|
Acquisition of patents and trademarks
|
(518,408 | ) | (35,351 | ) | ||||
|
Acquisition of office furniture and fixtures
|
(4,792 | ) | - | |||||
|
Net cash used by investing activities
|
(523,200 | ) | (35,351 | ) | ||||
|
Cash flows from financing activities:
|
||||||||
|
Payment of deferred private placement costs
|
- | (20,000 | ) | |||||
|
Proceeds from issuance of notes and warrants
|
- | 450,000 | ||||||
|
Payments on notes payable to shareholders
|
(393,275 | ) | - | |||||
|
Net proceeds from January 25, 2011 private placement
|
3,461,708 | - | ||||||
|
Net payments to from related party
|
(21,272 | ) | (13,000 | ) | ||||
|
Net advances from officers
|
1,439 | - | ||||||
|
Net cash provided by financing activities
|
3,048,600 | 417,000 | ||||||
|
Net increase (decrease) in cash
|
872,091 | (7 | ) | |||||
|
Cash - beginning of reporting period
|
310 | 158 | ||||||
|
Cash - end of reporting period
|
$ | 872,401 | $ | 151 | ||||
|
Cash paid during period for:
|
||||||||
|
Interest
|
$ | 189,736 | $ | 2,432 | ||||
|
Income tax
|
$ | - | $ | - | ||||
|
Supplemental disclosure of noncash investing and financing activities:
|
||||||||
|
Reduction of accounts payable not related to operating activities:
|
||||||||
|
Payment of accounts payable for patent costs
|
$ | 500,000 | $ | - | ||||
|
Payment of accounts payable for deferred private placement costs
|
526,127 | - | ||||||
|
Accounts payable converted to promissory notes
|
587,000 | - | ||||||
| $ | 1,613,127 | $ | - | |||||
|
Deferred private placement costs charged to contributed capital
|
$ | 587,133 | $ | - | ||||
|
Conversion of member notes and accrued interest to common shares and warrants
|
$ | 614,070 | $ | - | ||||
|
Issuance of derivative liability instrument and reduction of additional paid-in capital
|
$ | 3,061,750 | $ | - | ||||
|
Patent and trademark additions included in accounts payable
|
$ | - | $ | 22,255 | ||||
|
Deferred private placement cost additions included in accounts payable
|
$ | - | $ | 34,423 | ||||
See accompanying notes to financial statements.
F-21
|
22nd CENTURY GROUP INC. AND SUBSIDIARIES
|
|
CONSOLIDATED STATEMENT OF SHAREHOLDERS' DEFICIT
|
|
For the three months ended March 31, 2011
|
|
(unaudited)
|
|
Common
|
Par value
|
|||||||||||||||||||||||
|
Shares
|
of Common
|
Contributed
|
Accumulated
|
Non-controlling
|
Shareholders'
|
|||||||||||||||||||
|
Outstanding
|
Shares
|
Capital
|
Deficit
|
Interest
|
Deficit
|
|||||||||||||||||||
|
Balance at December 31, 2010
|
16,000,000 | $ | 3,598,856 | $ | (5,687,394 | ) | $ | (134 | ) | $ | (2,088,672 | ) | ||||||||||||
|
Distribution of 5,000,000 warrants for common stock, exercise price of $3.00 per share
|
(1,550,000 | ) | (1,550,000 | ) | ||||||||||||||||||||
|
Common Shares issued in January 25, 2011 private placement, net
|
5,434,446 | 2,503,022 | 2,503,022 | |||||||||||||||||||||
|
Merger of 22nd Century Limited and 22nd Century Group
|
5,325,200 | 268 | (268 | ) | - | |||||||||||||||||||
|
Net loss
|
(649,300 | ) | $ | (2,366 | ) | $ | (651,666 | ) | ||||||||||||||||
|
Balance at March 31, 2011
|
26,759,646 | $ | 268 | $ | 4,551,610 | $ | (6,336,694 | ) | $ | (2,500 | ) | $ | (1,787,316 | ) | ||||||||||
See accompanying notes to financial statements.
F-22
22nd CENTURY GROUP, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS MARCH 31, 2011
NOTE 1. - NATURE OF BUSINESS AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Introduction - These unaudited consolidated financial statements of 22nd Century Group Inc. (“22nd Century Group”) and its direct and indirect subsidiaries (collectively with 22nd Century Group, the “Company”) incorporate and reflect the reverse acquisition of 22nd Century Limited, LLC by 22nd Century Group, Inc. as described below.
On January 25, 2011, 22nd Century Limited, LLC (“22nd Century Ltd”) completed a reverse merger transaction (the “Merger”) with 22nd Century Group, Inc. (formerly Touchstone Mining Limited). As a result, 22nd Century Ltd became a wholly owned subsidiary of 22nd Century Group which continues to operate the business of 22nd Century Ltd. In connection with the Merger, 22nd Century Group issued 21,434,446 shares of its common stock to the holders of limited liability company membership interests of 22nd Century, which share amount represented 80.1% of the outstanding shares immediately following the Merger. All references contained herein to shareholders or common shares include the historical members and limited liability company membership interests of 22nd Century Ltd because, in the Merger, limited liability company membership interests were exchanged for common shares on a one-for-one basis and from an accounting standpoint they are equivalent.
The Merger is being accounted for as a reverse acquisition and a recapitalization; 22nd Century Ltd is the acquirer for accounting purposes. Consequently, the assets and liabilities and the historical operations that are reflected in the financial statements set forth herein for periods prior to the Merger are those of 22nd Century Ltd and are recorded at the historical cost basis of 22nd Century Ltd, and the consolidated financial statements set forth herein for periods beginning on and following completion of the Merger include the assets and liabilities of 22nd Century Ltd, historical operations of 22nd Century Ltd, and operations of 22nd Century Group from the closing date of the Merger.
Upon the closing of the Merger, 22nd Century Group transferred all of its operating assets and liabilities to Touchstone Split Corp. and split-off Touchstone Split Corp. through the sale of all of the outstanding capital stock of Touchstone Split Corp. (the “Split-Off”). After the completion of the Merger and Split Off, 22nd Century Group’s consolidated financial statements include only the assets and liabilities of 22nd Century Ltd. Refer to the Current Report on Form 8-K that the Company filed with the Securities and Exchange Commission on February 1, 2011 for further information on the Merger.
Immediately prior to the Merger on January 25, 2011, the 22nd Century Ltd completed a private placement offering (the “Private Placement”) of 5,434,446 securities (the “PPO Securities”) at the purchase price of $1.00 per Unit, each such Unit consisting of one (1) limited liability company membership interest of 22nd Century Ltd. and a five-year warrant to purchase one-half of one (1/2) limited liability company membership interest of 22nd Century Ltd. at an exercise price of $1.50 per whole common share. In connection with the Private Placement, 22nd Century Ltd approved a prorata distribution of 5,000,000 five-year warrants to purchase one limited liability company membership interest at an exercise price of $3.00. Private Placement proceeds included $614,070 from the conversion of 22nd Century Ltd indebtedness into PPO Securities and $395,376 from the conversion of placement agent fees into PPO Securities, resulting in gross cash proceeds of $4,425,000. Private Placement offering expenses incurred included cash expenses of approximately $1,025,000 and non-cash expenses consisting of the placement agent fees of $395,376 which were converted into PPO Securities and $390,000 for the estimated fair value of 934,755 placement agent and advisor warrants issued to the placement agent. 22nd Century Ltd/Group received net cash proceeds of approximately $3,462,000 from the Private Placement and the Company reduced its debt obligations to shareholders that were on the balance sheet at December 31, 2010 of approximately $614,000, which was exchanged for equity interests in the offering. A portion of the proceeds were allocated to the warrants issued, which are classified as liabilities (see note 9).
Basis of Presentation - The accompanying unaudited statements have been prepared in accordance with U.S. generally accepted accounting principles for interim financial information. Accordingly, they do not include all of the information and footnotes required by U.S. generally accepted accounting principles for complete financial statements. In the opinion of management, all adjustments, consisting of normal recurring accruals, considered necessary for a fair and non-misleading presentation of the financial statements have been included.
F-23
The results of operations for any interim period are not necessarily indicative of results for the full year. Operating results for the three month ended March 31, 2011 are not necessarily indicative of the results that may be expected for the year ending December 31, 2011. The balance sheet at December 31, 2010 has been derived from the audited financial statements at that date, but does not include all of the information and footnotes required by U.S. generally accepted accounting principles for complete financial statements.
Nature of Business - 22nd Century Group is a holding company and is the sole member of 22nd Century Ltd, which is a plant biotechnology company. 22nd Century Ltd owns or exclusively licenses 98 issued patents in 79 countries predominantly related to modifying the content of nicotinic alkaloids in plants, specifically tobacco plants, through genetic engineering and plant breeding.
The overall objectives of 22nd Century Ltd are to reduce smoking-related disease by increasing smoking cessation with, X-22, its botanical smoking cessation aid and reducing the harm to smokers with 22nd Century’s potential modified risk tobacco products (as defined in the Overview of Management Discussion), for smokers unwilling to quit. 22nd Century Ltd, including its subsidiary, Goodrich Tobacco Company Ltd. (“Goodrich Tobacco”) is primarily involved in the following activities:
|
|
·
|
The development of its botanical smoking cessation aid, X-22;
|
|
|
·
|
The development of its modified risk tobacco products;
|
|
|
·
|
The pursuit of necessary regulatory approvals at the U.S. Food and Drug Administration (the “FDA”) to market X-22 as a prescription smoking cessation aid and its proprietary cigarettes as modified risk tobacco products in the U.S.;
|
|
|
·
|
The manufacture, marketing and distribution of RED SUN and MAGIC proprietary cigarettes in traditional tobacco market channels in the U.S. through its subsidiary Goodrich Tobacco; and
|
|
|
·
|
The international licensing of 22nd Century’s trademarks, brands, proprietary tobaccos, and technology.
|
Principles of Consolidation - The accompanying consolidated financial statements include the accounts of 22nd Century Group, 22nd Century Ltd, and Goodrich Tobacco, a subsidiary of 22nd Century Ltd. 22nd Century Ltd owns 96% of the outstanding membership units of Goodrich Tobacco. All intercompany accounts and transactions have been eliminated.
Inventory - Inventories are valued at the lower of cost or market. Cost is determined on the first-in, first-out (FIFO) method. The Company’s inventory consisted of the following categories:
|
March 31,
|
December 31,
|
|||||||
|
2011
|
2010
|
|||||||
|
Materials, mainly tobacco
|
$ | 753,620 | $ | 292,480 | ||||
|
Finished goods
|
14,762 | 16,182 | ||||||
|
Total
|
$ | 768,382 | $ | 308,662 | ||||
F-24
Intangible Assets - Intangible assets are recorded at cost and consist primarily of expenditures incurred with third parties related to the processing of patent claims and trademarks with government authorities. The Company also capitalized costs as a result of one of its exclusively licensed patent application being subject to an interference proceeding invoked by the U.S. Patent and Trademark Office, which favorably resulted in the Company obtaining rights to a third party’s issued patent. The amounts capitalized relate to patents the Company owns or to which it has exclusive rights and its trademarks, and exclude approximately $1.8 million recovered from a former licensee as direct reimbursements of costs incurred. These capitalized costs are amortized using the straight-line method over the remaining statutory life of the Company’s primary patent family, which expires in 2019 (the assets’ estimated lives). Periodic maintenance or renewal fees, which are generally due on an annual basis, are expensed as incurred. Annual minimum license fees are charged to expense in the year the licenses are effective. Total patent and trademark costs capitalized and accumulated amortization amounted to $1,984,029 and $541,454 as of March 31, 2011 ($1,965,621 and $497,998 - as of December 31, 2010).
Impairment of Long-Lived Assets - The Company reviews the carrying value of its amortizing long-lived assets whenever events or changes in circumstances indicate that the historical cost-carrying value of an asset may no longer be recoverable.
The Company assesses recoverability of the asset by estimating the future undiscounted net cash flows expected to result from the asset, including eventual disposition. If the estimated future undiscounted net cash flows are less than the carrying value of the asset, an impairment loss is recorded equal to the difference between the asset’s carrying value and its fair value. There was no impairment loss recorded during the three month periods ended March 31, 2011 or 2010.
Deferred Private Placement Costs - During 2010, the Company incurred costs related to the Private Placement that closed on January 25, 2011. Such costs were accumulated on the balance sheet as of December 31, 2010 and were charged against contributed capital upon closing of the offering. Deferred Private Placement costs were $0 as of March 31, 2011 ($587,133 – December 31, 2010).
Income Taxes - Following the Merger on January 25, 2011, the Company’s operations are subject to Federal and State income taxes. Accordingly, since the merger date the Company is required to recognize deferred tax assets and liabilities for any differences in basis in assets and liabilities between tax and GAAP reporting. The corresponding asset that resulted has been fully offset by a valuation allowance. The Company incurred a net operating loss from the closing of the Merger to March 31, 2011 and, accordingly, has made no provision for current income taxes. The income tax asset arising from this net operating loss has been fully reserved because it is not probable that it will be realized.
Prior to the Merger, the Company had elected to be treated as a Partnership for Federal and State income tax purposes. As a result, there was no entity level income tax for periods prior to the Merger, because all taxable income, tax deductions and tax credits were passed through to the members of the Company. The Company’s Federal and State tax returns for the years ended December 31, 2007 to December 31, 2010 are currently open to audit under statutes of limitations. There are no pending audits as of March 31, 2011.
Employee Equity-Based Compensation - The Company uses a fair-value based method to determine compensation for all arrangements under which Company employees and others receive shares, options or warrants to purchase common shares of the 22nd Century Group.
Debt Discounts - The Company accounts for warrants issued to note holders as an inducement to provide financing for the Company in accordance with the FASB’s guidance on Accounting for Convertible Debt and Convertible Debt Issued with Stock Purchase Warrants. Fair value of the warrants is determined by common share price according to recent equity transactions, since there is no vesting period and a negligible exercise price. The proceeds allocated to each warrant is recorded as a debt discount and amortized over the life of the corresponding financing as interest expense.
Unearned Grant Revenue- The Company received approval of a government grant as partial support for its next clinical trial for the FDA. Costs associated with this grant are being recognized as such costs are incurred and included in research and development expense. As the costs are incurred, deferred revenue is also recognized into income. During the three months ended March 31, 2011, the Company recognized $49,216, which is included in other income on the statement of operations. Deferred grant revenue was $174,324 as of March 31, 2011 (as compared to $223,540 as of December 31, 2010).
F-25
Revenue Recognition - The Company recognizes revenue at the point the product is shipped to a customer and title has transferred. Revenue from the sale of the Company’s products is recognized net of cash discounts, sales returns and allowances. Federal Excise Taxes are included in net sales and accounts receivable billed to customers.
The Company was chosen to be a subcontractor for a 5-year government contract between RTI International (“RTI”) and the National Institute on Drug Abuse (“NIDA”) to supply NIDA research cigarettes which includes four stages. Stages 1 and 2 are for product design, stage 3 is for manufacture planning and material procurement and stage 4 is for the manufacture and shipping of the 9,000,000 cigarettes. The Company has completed each of stages 1 to 3 and recognized the revenue when billing was approved by the customer. Revenue under this stage 4 will be recognized at the point the cigarettes are shipped to the customer and title has transferred. These government research cigarettes will be distributed under the mark SPECTRUM.
Derivatives - We do not use derivative instruments to hedge exposures to cash flow, market or foreign currency risks. We evaluate all of our financial instruments to determine if such instruments are derivatives or contain features that qualify as embedded derivatives. For derivative financial instruments that are accounted for as liabilities, the derivative instrument is initially recorded at its fair market value and then is revalued at each reporting date, with changes in fair value reported in the consolidated statement of operations. The methodology for valuing our outstanding warrants classified as derivative instruments will use a lattice model approach which includes probability weighted estimates of future events including volatility of our common stock. The classification of derivative instruments, including whether such instruments should be recorded as liabilities or equity, is evaluated at the end of each reporting period. Derivative instrument liabilities are classified in the balance sheet as current or non-current based on whether or not net-cash settlement of the derivative instrument could be required within twelve months of the balance sheet date.
Research and Development - Research and development costs are expensed as incurred.
Loss Per Common Share - Basic loss per common share is computed using the weighted-average number of common shares outstanding. Diluted loss per share is computed assuming conversion of all potentially dilutive securities. Potential common shares outstanding are excluded from the computation if their effect is anti-dilutive.
Commitment and Contingency Accounting - The Company evaluates each commitment and/or contingency in accordance with the accounting standards, which state that if the item is more likely than not to become a direct liability then the Company will record the liability in the financial statements. If not, the Company will disclose any material commitments or contingencies that may arise.
Use of Estimates - The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of income and expenses during the reporting period. Actual results could differ from those estimates.
NOTE 2. - LIQUIDITY AND MANAGEMENT’S PLANS
Since 2006, 22nd Century Ltd has experienced limited revenues and incurred substantial operating losses as it transitioned from being only a licensor of its proprietary technology and tobaccos to commercializing its own tobacco products. At March 31, 2011, the Company had current assets of $1,961,651 (including cash of $872,401) and current liabilities of $1,458,997. On January 25, 2011, the Company completed the Private Placement of securities resulting in approximately $3.4 million in net cash proceeds and a reduction of debt and accrued interest obligations to shareholders that were on the balance sheet at December 31, 2010 of approximately $614,000, which were exchanged for equity interests in the Private Placement. These proceeds were sufficient to enable the Company to make substantial reductions in its outstanding current liabilities, which totaled $1,458,997 at March 31, 2011 as compared to $4,823,635 at December 31, 2010.
F-26
The Company will need to raise additional capital during the fourth quarter of 2011 to further reduce outstanding current liabilities, pay obligations that mature or begin to mature in 2012, and complete the FDA-approval process for X-22. Before the Company can raise additional capital, it must satisfy the registration rights granted to investors who acquired shares of its common stock in the January 25, 2011 Private Placement and Merger. These registration rights provide that 22nd Century Group must have an effective registration statement covering the shares subject to the registration rights for a period of 90 days before it can sell any equity securities or securities convertible into equity securities. In addition, the ability to complete future financings on acceptable terms will depend on a number of factors, including the general performance of the capital markets, the Company’s progress in the FDA approval process and the manufacture, distribution and sale of its products. Any future equity financings will be dilutive to 22nd Century Group’s existing shareholders’ respective ownership percentages.
Prior to FDA approval of its prescription X-22 smoking cessation aid and FDA authorization of its two potential modified risk cigarettes (as defined in the Overview of Management Discussion), the Company expects to generate cash from the sale of its proprietary cigarettes in the U.S. through its subsidiary, Goodrich Tobacco. Sales are expected in traditional tobacco market channels and to fulfill orders by the National Institute on Drug Abuse (“NIDA”). In March 2011, the Company received a revised purchase order and authorization to bill $112,000 for manufacture planning and material procurement for 9 million cigarettes from RTI International (“RTI”). Payment was received on May 2, 2011. The Company received an additional purchase order from RTI in May 2011 to ship the 9 million research cigarettes. We estimate the revenue from this purchase order will be approximately $680,000. These government research cigarettes will be distributed under the mark SPECTRUM.
The Company’s management believes, but can offer no assurances that, the above business plans will provide sufficient cash flow to fund the Company’s operations into the fourth quarter of 2011.
NOTE 3. - AMOUNTS OWED NORTH CAROLINA STATE UNIVERSITY (“NCSU”)
Pursuant to the terms of an exclusive license agreement with NCSU, the Company owes NCSU approximately $728,000 as of March 31, 2011 for patent costs (as compared to $1,128,000 owed as of December 31, 2010). These amounts are included in accounts payable in the consolidated balance sheets. The Company was required to pay these amounts within thirty days of being invoiced and they are past due. NCSU has the right to claim interest on the balance. In February 2011, the Company, after making a $400,000 payment to NCSU towards patent costs, proposed a deferred payment arrangement for the balance owed. If the Company and NCSU cannot reach a satisfactory agreement, NCSU may have the right to terminate the exclusive license agreement, but can only do so with a 60-day prior written notice, including the opportunity to cure within this timeframe. As of March 31, 2011, patent costs associated with the exclusive license agreements that could potentially be terminated had a carrying value of approximately $750,000. Additionally, NCSU has not imposed interest charges on past due amounts invoiced to the Company and as such the Company has not recorded accrued interest or interest expense.
NOTE 4. - DEMAND BANK LOANS
The demand loan that is among the Company’s short term liabilities is payable to a commercial bank under a revolving credit agreement and is guaranteed by a shareholder of the Company. This loan had a balance of $174,925 at March 31, 2011 and December 31, 2010, respectively. The Company is required to pay interest monthly at an annual rate of 0.75% above the prime rate, or 4.00% at March 31, 2010 (as compared to 4.00% at December 31, 2010). The Company is current in meeting this interest payment obligation. The terms of the demand loan includes an annual “clean-up” provision, which requires the Company to repay all principal amounts outstanding for a period of 30 consecutive days every year. The Company has not complied with this requirement; however, the bank has not demanded payment.
F-27
NOTE 5. - NOTES PAYABLE
Notes payable to shareholders consisted of the following:
|
March 31,
|
December 31,
|
|||||||
|
2011
|
2010
|
|||||||
|
Note dated October 28, 2008
|
$ | - | $ | 325,000 | ||||
|
Note dated November 11, 2008
|
- | 325,000 | ||||||
|
Note dated May 20, 2009
|
- | 30,000 | ||||||
|
Note dated January 1, 2008
|
- | 100,014 | ||||||
|
Note dated September 1, 2010
|
- | 35,000 | ||||||
|
Notes dated October 4, 2010
|
- | 150,000 | ||||||
|
Note dated December 31, 2010
|
- | 100,000 | ||||||
|
Note payable to repurchase common shares
|
- | 30,629 | ||||||
| $ | - | $ | 1,095,643 | |||||
Note Dated October 28, 2008 - On October 28, 2008, the Company issued a note payable to a third party in the amount of $325,000. The interest rate on the note was 10% and the outstanding principal and interest was due and payable on October 28, 2010, the maturity date.
This note matured and remained outstanding at December 31, 2010; however, in January and February 2011 this note together with the $30,000 note dated May 20, 2009 (described below), and all the related accrued interest of $77,300 were satisfied by: (i) conversion of $150,000 into equity in connection with the January 25, 2010 Private Placement; (ii) a cash payment of $142,300; and (iii) a new note dated January 25, 2011 (see Note 6) payable in the amount of $140,000.
Note Dated November 11, 2008 - On November 11, 2008, the Company issued a note payable to a shareholder in the amount of $325,000. The interest rate on the note was 10% and the outstanding principal and interest was due and payable on November 11, 2010, the maturity date. This note was extended as set forth below and remained outstanding at December 31, 2010.
The extended maturity date on the note was January 10, 2011 and the interest rate during the extension period was 15%. Following the maturity of this note on January 10, 2011, the principal amount of $325,000 was converted to equity in connection with the January 25, 2011 Private Placement and the accrued interest was paid in cash.
Note Dated May 20, 2009 (unsecured) - On May 20, 2009, the Company issued a note payable in the amount of $30,000 to the same third party that was issued the October 28, 2008 note. The interest rate on the note was 10% and the outstanding principal and interest was due and payable on May 19, 2010, the maturity date. The $30,000 in principal and accrued interest remained outstanding as of December 31, 2010. This note was satisfied in January and February 2011 together with the October 28, 2008 note as described previously.
Note Dated January 1, 2008 (unsecured) - The Company issued a note to a member on January 1, 2008 payable in the amount of $100,014. The interest rate on the note was 7%; the interest and principal were due on January 15, 2011, the maturity date, and were paid in cash in January 2011.
Note Dated September 1, 2010 - On September 1, 2010, the Company issued a note payable to a member in the amount of $35,000. The interest rate on the note was 15%, and, pursuant to an extension, the interest and principal were due on January 25, 2011, the extended the maturity date. The note was paid in cash in January 2011.
Notes Dated October 4, 2010 (unsecured) -The Company issued notes payable to seven shareholders in the aggregate amount of $150,000. The interest rate on each of these notes was 15%, and the interest and principal were due on the maturity date of January 31, 2011. These notes were satisfied in January 2011 by conversion of $87,367 to equity in connection with the January 25, 2011 Private Placement and by payment of $62,633 in cash.
F-28
Note Dated December 29, 2010 (unsecured) - 22nd Century Ltd issued a note payable to a member in the amount of $100,000. The interest rate on this note was 15%, and the interest and principal were due on the maturity date of January 29, 2011. The note was paid in cash in January 2011.
Note Payable to Repurchase Common Shares (unsecured) – On December 31, 2010, the Company agreed to repurchase 51,637 common shares previously issued to a member in exchange for a $35,019 note, which $30,629 remained unpaid as of December 31, 2010. The interest rate on the note was 7% and the outstanding principal and interest were due and payable on September 30, 2010, the maturity date. The note remained unpaid at the maturity date; the outstanding principal balance and accrued interest were paid in cash in February 2011.
NOTE 6. – LONG TERM NOTES PAYABLE
Long term notes payable, net of unamortized discount, consisted of the following as of the dates set forth below:
|
March 31,
|
December 31,
|
|||||||
|
2011
|
2010
|
|||||||
|
Convertible note dated March 31, 2011
|
$ | 237,000 | - | |||||
|
Note dated date March 30, 2011
|
350,000 | - | ||||||
|
Total long term notes
|
587,000 | - | ||||||
|
Notes dated September 15 and October 15, 2009, net of unamortized discount
|
$ | 21,890 | $ | 20,557 | ||||
|
Note dated May 27, 2010
|
- | 45,000 | ||||||
|
Note dated January 25, 2011
|
140,000 | - | ||||||
|
Total long term notes to shareholders
|
161, 890 | 65,557 | ||||||
|
Total long term debt
|
748,890 | 65,557 | ||||||
|
Less current portion
|
(71,890 | ) | - | |||||
| $ | 677,000 | $ | 65,557 | |||||
Convertible Note Dated March 31, 2011- On March 31, 2011, the Company issued a note to a vendor in the amount of $237,000 as satisfaction of past due invoices previously recorded by the Company in accounts payable. The note bears interest at an annual rate of 9%. Principal payments together with accrued interest are due as follows: $50,000 each January 1, 2012, May 1, 2012, August 1, 2012 and November 1, 2012; and $37,000 January 1, 2013, the maturity date. The note is convertible into common stock of 22nd Century Group at the option of the holder and the earliest the note can be exercised is 90 days after the Company’s registration statement covering 5,434,446 shares of common stock is declared effective by the Securities Exchange Commission. The conversion rate will be determined at the time of conversion based on the average of the closing price of the Company’s common stock on the day of and day after the conversion date.
Note Dated March 30, 2011- On March 30, 2011, the Company issued a note to a vendor in the amount of $350,000 as satisfaction of past due invoices previously recorded by the Company in accounts payable. The note bears interest at an annual rate of 4%. Principal and accrued interest are due on July 1, 2012. This maturity date is automatically accelerated if the Company receives funding of $5,000,000 or more prior to July 1, 2012.
F-29
Notes Dated September 15 and October 15, 2009 (unsecured) - On September 15, 2009 and October 15, 2009, the Company issued two notes payable to the same third party in the amounts of $15,000 and $10,000, respectively. In conjunction with the $15,000 note, a warrant to purchase 185,503 common shares of 22nd Century Ltd at less than $.0001 per unit was issued, and in conjunction with the $10,000 note, a warrant to purchase 92,751 common shares of 22nd Century Ltd at less than $.0001 per unit was issued. The warrants were valued at $11,301 for the $15,000 note and $6,962 for the $10,000 note, were recorded as discounts to the respective notes payable and are being amortized over the term of each note which significantly adjusts the effective interest rate. The intrinsic value of the warrants at the time of issuance was determined to be $68,750; the debt discount recorded was based on allocating the $25,000 in transaction proceeds proportionally between the notes and the warrants. The notes bear interest at a rate of 10% and the outstanding principal and interest is due and payable at maturity - January 31, 2012 in each case. As of March 31, 2011, the total outstanding principal and unamortized debt discounts for the two notes amounted to $25,000 and $3,110 (as compared to $25,000 and $4,442 at December 31, 2010), respectively. The warrants were exercised in 2010 prior to the Merger.
Note Dated May 27, 2010 - On May 27, 2010, the Company issued a note for $45,000. The note bears interest at an annual rate of 10%, which was due with the principal amount on January 31, 2012. The note was satisfied in January 2011 by conversion to equity in connection with the January 25, 2011 Private Placement.
Note Dated January 25, 2011- On January 25, 2011, the Company issued a note for $140,000 to a shareholder as satisfaction of the balance due for principal and interest on matured notes that were not paid in cash or converted to common stock of 22nd Century Group and warrants to purchase shares of common stock of 22nd Century Group. The note bears interest at 12% and is due on July 1, 2013 together with accrued interest.
NOTE 7. - DUE TO RELATED PARTY
The Company has conducted numerous transactions with a related party, Alternative Cigarettes, Inc. (“AC”). AC is entirely owned by certain shareholders of the Company, including the CEO. AC shares office space and employee services with the Company. During the three months ended March 31, 2011 the Company acquired its MAGIC trademark from AC for a purchase price of $22,500. There were no expense reimbursements from AC during each of the first three months of 2011 and 2010. The net amount due from AC as a result of advances, repayments and expenses incurred and reimbursed amounted to $14,330 as of March 31, 2011 (as compared with the Company owing AC $6,942 as of December 31, 2010). No interest has been accrued or paid on amount due to AC and there are no repayment terms.
NOTE 8. - DUE TO OFFICERS
Amount due to officers is a result of advances to the Company for working capital purposes and Company expenses paid directly by the officers net of advances or reimbursements. As of March 31, 2010, the net amount due to officers was $4,639 (as compared with $3,200 due to officers as of March 31, 2010). No interest has been accrued or paid on any amount due to officers and there are no repayment terms.
NOTE 9. - WARRANTS FOR COMMON STOCK
Prior to December 31, 2010, the Company granted warrants to purchase common shares of 22nd Century Group in connection with borrowings as an additional incentive for providing financing to the Company and as additional compensation to officers, consultants and advisors. The warrants were granted with a conversion price of less than $.0001, and the number of warrants issued was negotiated based on the agreement at the time of the grant. These warrants had been issued for terms of two to five years. All of these warrants were exercised prior to December 31, 2010.
In connection with the January 25, 2011 Private Placement and Merger, the Company issued 8,651,979 five year warrants to purchase shares of common stock of 22nd Century Group as follows: 5,000,000 with an exercise price of $3.00 per share, and 3,651,979 with an exercise price of $1.50 per share. These warrants contain “down round” provisions which provide for adjustments to the exercise price if the Company issues common shares of stock of 22nd Century Group at a price that is less than the respective warrant exercise prices. This provision is a guarantee of value which requires that these warrants be classified as derivatives for accounting purposes which means they are reported as a liability and marked to market at each balance sheet date. The value of this warrant liability as of March 31, 2011 was estimated to be the same as the value on January 25, 2011, the date of issuance. The amount of the warrant liability related to the 5,000,000 $3.00 warrants is $1,550,000 at March 31, 2011 and was recorded as a reduction of equity for the derivative liability issued to the existing stockholders; the amount of the warrant liability related to the 3,651,979 $1.50 warrants is $1,511,750, and, because it was recorded as a liability, the portion of proceeds from the Private Placement that was recorded as contributed capital was reduced accordingly.
F-30
The value of this warrant liability as of March 31, 2011 was estimated to be the same as the value on January 25, 2011, the date of issuance. The Company estimated the value of the warrant liability using the binomial lattice model to allocate total enterprise value to the warrants and other securities in the Company’s capital structure based upon the enterprise value implied in the January 25, 2011 Private Placement. Consideration was also given to the impact of expected future capital raises and issuances of securities under the Company’s 2010 Equity Investment Plan (“EIP”). Volatility was estimated based on historical observed equity volatilities and implied (forward) or expected volatilities for a sample group of guideline companies and consideration of recent market trends.
ASC 820 establishes a valuation hierarchy for disclosure of the inputs to valuation used to measure fair value. This hierarchy prioritizes the inputs into three broad levels as follows.
· Level 1 inputs are quoted prices (unadjusted) in active markets for identical assets or liabilities.
· Level 2 inputs are quoted prices for similar assets and liabilities in active markets or inputs that are observable for the asset or liability, either directly or indirectly through market corroboration, for substantially the full term of the financial instrument.
· Level 3 inputs are unobservable inputs based on the Company’s own assumptions used to measure assets and liabilities at fair value.
A financial asset or liability’s classification within the hierarchy is determined based on the lowest level input that is significant to the fair value measurement. The warrant liability is measured at fair value using certain estimated factors such as volatility and probability and are classified within Level 3 of the valuation hierarchy
|
Number of Warrants
|
||||
|
Warrants outstanding at December 31, 2009
|
1,688,076 | |||
|
Warrants issued during 2010
|
3,116,447 | |||
|
Warrants exercised during 2010
|
(4,804,523 | ) | ||
|
Warrants outstanding at December 31, 2010
|
- | |||
|
Warrants issued during 2011
|
8,651,979 | |||
|
Warrants exercised during 2011
|
-
|
|||
|
Warrants outstanding at March 31, 2011
|
- | |||
|
Warrants exercisable at March 31, 2011
|
8,651,979 | |||
On February 1, 2009, the Company granted an award for service to an executive officer of 445,207 warrants to purchase common shares, vesting over a one year service period ending February 1, 2010. The related compensation cost of $258,662 was determined by the intrinsic value of the underlying common shares at the time of the award of $0.58 per unit and was charged to expense on a straight line basis over the service period. The cost was fully amortized in the first quarter of 2010 with a charge of $43,090.
NOTE 10. - COMMITMENTS
License Agreements - Under its exclusive license agreement with NCSU, the Company is required to pay minimum annual royalty payments, which are credited against running royalties on sales of licensed products. The annual minimum royalty for each of the calendar years 2011 through 2013 is $75,000, and in 2014 the annual minimum royalty increases to $200,000. The license agreement continues through the life of the last-to-expire patent. These minimum royalty payments are due each February following the end of the applicable calendar year and are reduced by any running royalties paid or payable for that year. The agreement also requires a milestone payment of $150,000 upon FDA approval of a product that uses the NCSU licensed technology. The Company is also responsible for reimbursing NCSU for actual third-party patent costs incurred. These costs vary from year to year and the Company has certain rights to direct the activities that result in these costs. During the three months ended March 31, 2011, the costs incurred related to patent costs and patent maintenance amounted to $3,665 (as compared to $31,701 during the three months ended March 31, 2010).
F-31
The Company has two other exclusive license agreements which require aggregate annual license fees of approximately $55,000, which are credited against running royalties on sales of licensed products. Each license agreement continues through the life of the last-to-expire patent.
Registration Rights –Pursuant to a covenant in conjunction with the January 25, 2011 Private Placement the Company filed a registration statement with the Securities and Exchange Commission (“SEC”) on April 8, 2011 covering the resale of 5,434,446 shares of common stock of 22nd Century Group (“Common Stock”) issued to the investors in the Private Placement. The Company will use its best efforts to cause this registration statement to be declared effective by the SEC on or before December 4, 2011. If this registration statement is not declared effective by the SEC by December 4, 2011, then the holders of Common Stock to be registered shall be entitled to monetary penalties at a rate equal to $0.005 per common share for each full month that this registration statement is late in being declared effective by the SEC. However, in no event shall the aggregate of any such penalties exceed $0.05 per common share. In addition, for a period of 90 days following the effective date of this registration statement, the Company cannot sell any equity securities or securities convertible into equity securities.
NOTE 11. - FAIR VALUE OF FINANCIAL INSTRUMENTS
The estimated fair value of cash, advances from shareholders and related party, demand bank loans and notes payable approximate the carrying value due to their short-term nature. In applying the accounting standards for fair value determination, the Company has taken into account what the Company would have to pay someone to take over its debt obligations. Considerable judgment is required in developing estimates of fair value. Therefore, the estimates presented herein are not necessarily indicative of the amounts that the Company would realize in a current market exchange.
The estimated fair value of long-term debt is summarized as follows:
|
March 31, 2011
|
December 31, 2010
|
|||||||||||||
|
Carrying
|
Estimated
|
Carrying
|
Estimated
|
|||||||||||
|
Amount
|
Fair Value
|
Amount
|
Fair Value
|
|||||||||||
| $ | 748,890 | $ | 717,000 | $ | 65,557 | $ | 63,000 | |||||||
Differences between the fair value and carrying amount of long-term debt are primarily due to instruments that provide fixed interest or contain fixed interest rate elements. Inherently, such instruments are subject to fluctuations in fair value due to subsequent movements in interest rates that are available to the Company.
NOTE 12. - EARNINGS PER COMMON SHARE
The following table sets forth the computation of basic and diluted earnings per common share for the three months ended March 31:
|
2011
|
2010
|
|||||||
|
Net loss attributed to common shareholders
|
$ | (649,300 | ) | $ | (305,013 | ) | ||
|
Denominator for basic earnings per share-weighted average shares outstanding
|
23,890,407 | 7,089,946 | ||||||
|
Effect of dilutive securities:
|
||||||||
|
warrants outstanding
|
- | - | ||||||
|
Denominator for diluted earnings per common share - weighted average shares adjusted for dilutive securities
|
23,890,407 | 7,089,946 | ||||||
|
Loss per common share - basic
|
$ | (0.03 | ) | $ | (0.04 | ) | ||
|
Loss per common share- diluted
|
$ | (0.03 | ) | $ | (0.04 | ) | ||
F-32
Securities outstanding that were excluded from the computation because they would have been anti-dilutive are as follows:
|
2011
|
2010
|
|||||||
|
Warrants
|
8,651,979 | 4,804,523 | ||||||
NOTE 13 - EQUITY INCENTIVE PLAN
On October 21, 2010, the Company established the 2010 Equity Incentive Plan (“EIP”) for officers, employees, directors, consultants and advisors to the Company and its affiliates, consisting of 4,250,000 shares of common stock. The EIP has a term of ten years and is administered by our Board or a committee to be established by our Board (the “Administrator”), to determine the various types of incentive awards that may be granted to recipients under this plan and the number of shares of common stock to underlie each such award under the EIP. The EIP also contains a provision which restricts the plan to granting awards relating to no more than 1,600,000 shares of common stock of 22nd Century Group during the first twelve months following the effective date of the Merger. On March 31, 2011, the Company filed a registration statement with the SEC to register all of the shares of common stock of 22nd Century Group that it may issue under the EIP. On April 1, 2011, under our EIP, the Board granted an aggregate of 1,150,000 shares of common stock of 22nd Century Group to officers and directors and incentive stock options to purchase an aggregate of 35,000 shares of 22nd Century Group’s common stock to employees.
The number of shares of common stock of 22nd Century Group subject to the EIP, any number of shares subject to any numerical limit in the EIP, and the number of shares and terms of any incentive award will be adjusted in the event of any change in outstanding common stock of 22nd Century Group by reason of any stock dividend, spin-off, split-up, stock split, reverse stock split, recapitalization, reclassification, merger, consolidation, liquidation, business combination or exchange of shares or similar transaction.
The EIP authorizes the grant of incentive stock options, nonqualified stock options, stock appreciation rights, restricted stock, restricted stock units, performance shares, restricted stock or restricted stock units.
All awards made under the EIP may be subject to vesting and other contingencies as determined by the Administrator and will be evidenced by agreements approved by the Administrator which set forth the terms and conditions of each award.
F-33
5,434,446 Shares of Common Stock
22nd CENTURY GROUP, INC.
PROSPECTUS
July ___, 2011
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 13. Other Expenses of Issuance and Distribution.
Set forth below is an estimate (except for registration fees, which are actual) of the approximate amount of the fees and expenses payable by us in connection with the issuance and distribution of the shares of our common stock.
|
AMOUNT
|
||||
|
EXPENSE
|
||||
|
Registration Fees
|
$ | 788.68 | ||
|
Legal Fees and Expenses
|
85,000.00 | |||
|
Accounting Fees and Expenses
|
10,000.00 | |||
|
Miscellaneous Fees and Expenses
|
2,000.00 | |||
|
Total
|
$ | 97,788.68 | ||
Item 14. Indemnification of Directors and Officers.
Nevada Revised Statutes (NRS) Sections 78.7502 and 78.751 provide us with the power to indemnify any of our directors, officers, employees and agents. The person entitled to indemnification must have conducted himself in good faith, and must reasonably believe that his conduct was in, or not opposed to, our best interests. In a criminal action, the director, officer, employee or agent must not have had reasonable cause to believe that his conduct was unlawful.
Under NRS Section 78.751, advances for expenses may be made by agreement if the director or officer affirms in writing that he has met the standards for indemnification and will personally repay the expenses if it is determined that such officer or director did not meet those standards.
Our bylaws include an indemnification provision under which we have the power to indemnify, to the extent permitted under Nevada law, our current and former directors and officers, or any person who serves or served at our request for our benefit as a director or officer of another corporation or our representative in a partnership, joint venture, trust or other enterprise, against all expenses, liability and loss reasonably incurred by reason of being or having been a director, officer or representative of ours or any of our subsidiaries. We may make advances for expenses upon receipt of an undertaking by or on behalf of the director or officer to repay the amount if it is ultimately determined by a court of competent jurisdiction that he, she or it is not entitled to be indemnified by us.
Our articles of incorporation provide a limitation of liability such that no director or officer shall be personally liable to us or any of our stockholders for damages for breach of fiduciary duty as a director or officer, involving any act or omission of any such director or officer, provided there was no intentional misconduct, fraud or a knowing violation of the law, or payment of dividends in violation of NRS Section 78.300.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of ours under Nevada law or otherwise, we have been advised the opinion of the SEC is that such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event a claim for indemnification against such liabilities (other than payment by us for expenses incurred or paid by a director, officer or controlling person of ours in successful defense of any action, suit, or proceeding) is asserted by a director, officer or controlling person in connection with the securities being registered, we will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction, the question of whether such indemnification by it is against public policy in the Securities Act and will be governed by the final adjudication of such issue.
II-1
Item 15. Recent Sales of Unregistered Securities.
There have been no sales of unregistered securities within the last three years which would be required to be disclosed pursuant to Item 701 of Regulation S-K, except for the following:
Sales by the Company
Pre-Merger
On February 6, 2008, we sold 386,389 shares of common stock to one investor at a price of $0.13 per share, for an aggregate sale price of $50,000. The sale was made pursuant to the exception provided by Section 4(2) of the Securities Act since the issuance did not involve a public offering, the recipient had access to information that would be included in a registration statement, the recipient took the shares for investment and not resale, and the Company took appropriate measures to restrict resale.
In September 2007, we issued 8,346,000 shares of restricted common stock to Douglas Scheving, our then-current sole officer and director, in exchange for the forgiveness of $34,502 in indebtedness that was owed to him by us. The shares were issued pursuant to the exemption from registration provided by Section 4(2) of the Securities Act.
Merger
On January 25, 2011, we entered into an Agreement and Plan of Merger and Reorganization with Acquisition Sub, and 22nd Century. On that date, in consummation of the Merger contemplated under the Agreement and Plan of Merger and Reorganization, Acquisition Sub merged with and into 22nd Century, and 22nd Century, as the surviving entity, became our wholly-owned subsidiary.
Prior to the closing of the Merger, we transferred all of our pre-Merger operating assets and liabilities pursuant to the terms of a split-off agreement, to our wholly-owned subsidiary, Touchstone Split Corp., a Delaware corporation, or the Split-Off Subsidiary. Thereafter, pursuant to the split-off agreement, we transferred all of the outstanding capital stock of the Split-Off Subsidiary to our then-sole director in exchange for $1.00, such consideration being deemed to be adequate by our pre-Merger board of directors.
At the closing of the Merger, each membership interest of 22nd Century issued and outstanding immediately prior to the closing of the Merger was exchanged for one (1) share of our common stock, and each warrant to purchase limited liability company membership interests of 22nd Century was exchanged for one warrant of like tenor and term to purchase shares of our common stock. An aggregate of 21,434,446 shares of common stock and warrants to purchase an aggregate of 8,151,980 shares of common stock were issued to the holders of limited liability company membership interests and warrants, respectively, of 22nd Century, in the Merger. Immediately following the closing of the Merger, an aggregate of 26,759,646 shares of common stock were issued and outstanding and an aggregate of 8,651,980 shares of our common stock were reserved for issuance pursuant to the exercise of warrants to purchase shares of common stock. Of the 26,759,646 shares of common stock issued and outstanding after the closing of the Merger, approximately 59.8% of such issued and outstanding shares were held by individuals and entities that were holders of limited liability company membership interests of 22nd Century prior to consummation of the Private Placement Offering, approximately 20.3% were held by the investors in the Private Placement Offering, and approximately 19.9% were held by the pre-Merger stockholders of the Company.
The issuance of the securities in the Merger was not registered under the Securities Act, or the securities laws of any state, and were offered and sold in reliance on the exemption from registration afforded by Section 4(2) and Regulation D (Rule 506) under the Securities Act and corresponding provisions of state securities laws, which exempt transactions by an issuer not involving any public offering.
II-2
Item 16. Exhibits and Financial Statement Schedules.
|
Exhibit No.
|
Description
|
|
|
2.1(1)
|
Agreement and Plan of Merger and Reorganization dated as of January 25, 2011 by and among the Company, 22nd Century, and Acquisition Sub.
|
|
|
2.2(2)
|
Certificate of Merger dated as of January 25, 2011 Acquisition Sub. with and into 22nd Century
|
|
|
3.1(3)
|
Certificate of Incorporation of the Company
|
|
|
3.2(4)
|
Amended and Restate Certificate of Incorporation of the Company
|
|
|
3.3(3)
|
Bylaws of the Company
|
|
|
5.1*
|
Opinion of Foley & Lardner LLP
|
|
|
10.1(7)
|
2010 Equity Incentive Plan
|
|
|
10.2(8)
|
Form of Securities Purchase Agreement dated as of January 25, 2011 by and among 22nd Century, the purchaser(s) identified on the signature pages thereto and Parent, solely for the purposes of Section E and Section G thereof, as amended.
|
|
|
10.3(9)
|
Form of Conversion Agreement
|
|
|
10.4(10)
|
Form of Warrant dated as of January 25, 2011 issued to LLC members of 22nd Century prior to the consummation of the Private Placement Offering upon consummation of the Merger
|
|
|
10.5(11)
|
Form of Warrant dated as of January 25, 2011 issued to investors in the Private Placement Offering upon consummation of the Merger
|
|
|
10.6(12)
|
Form of Warrant dated as of January 25, 2011 issued to the Placement Agent and Sub-Agent upon consummation of the Merger
|
|
|
10.7(13)
|
Advisor Warrant dated as of January 25, 2011 issued to the Placement Agent in connection with that certain Advisory Agreement dated as of January 25, 2011 by and between the Company and the Placement Agent
|
|
|
10.8(14)
|
Advisory Agreement dated as of January 25, 2011 by and between the Company and the Placement Agent
|
|
|
10.9(15)
|
Placement Agency Agreement dated as of December 1, 2010 by and between 22nd Century and the Placement Agent
|
|
|
10.10(16)
|
Escrow Agreement dated as of December 2, 2010 by and among 22nd Century, the Placement Agent and Bank of America, National Association
|
|
|
10.11(17)
|
Split-Off Agreement dated as of January 25, 2011 by and among the Company, Touchstone Split. Corp and David Rector
|
|
|
10.12(18)
|
Letter from Paramount Strategy Corp dated as of December 21, 2010 regarding loan forgiveness
|
|
|
10.13(19)
|
Letter from Milestone Enhanced Fund Ltd. dated as of December 28, 2010 regarding loan forgiveness
|
II-3
|
10.14(20)
|
Letter from Mark Tompkins dated as of January 25, 2011 regarding loan forgiveness
|
|
|
10.15(21)†
|
Employment Agreement dated as of January 25, 2011 by and between the Company and Joseph Pandolfino
|
|
|
10.16(22)†
|
Employment Agreement dated as of January 25, 2011 by and between the Company and Henry Sicignano III
|
|
|
10.17(23)†
|
Employment Agreement dated as of January 25, 2011 by and between the Company and C. Anthony Rider
|
|
|
10.18†*
|
Employment Agreement dated as of March 15, 2011 by and between the Company and Michael R. Moynihan.
|
|
|
10.19(24)
|
Form of Lock-Up Agreement
|
|
| 10.20** |
Promissory Note dated June 30, 2011, payable by the Company to Henry Sicignano III in the principal amount of $150,000.
|
|
|
14.1(6)
|
Code of Ethics
|
|
|
16.1(25)
|
Letter from Child, Van Wagoner & Bradshaw, PLLC regarding change in independent registered public accountants.
|
|
|
17.1(26)
|
Letter from David Rector dated as of January 25, 2011 resigning as a director and officer of Parent
|
|
|
21.1*
|
Subsidiaries of the Company
|
|
|
23.1**
|
Consent of Freed Maxick & Battaglia, CPAs, PC
|
|
|
23.2**
|
Consent of Foley & Lardner LLP (included in Exhibit 5.1)
|
|
*
|
Previously filed.
|
|
**
|
Filed herewith.
|
|
†
|
Management contract or compensatory plan, contract or arrangement.
|
(1) Incorporated herein by reference to Exhibit 2.1 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(2) Incorporated herein by reference to Exhibit 2.2 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(3) Incorporated herein by reference to Exhibit 3.1 of the Company’s Registration Statement on Form SB-2 filed with the Commission on December 27, 2005.
(4) Incorporated herein by reference to Exhibit 3.2 of the Company’s Annual Report on Form 10-K for the year ended September 30, 2010 filed with the Commission on November 2, 2010.
(5) Incorporated herein by reference to Appendix B of the Company’s Definitive Information Statement on Schedule 14C filed with the Commission on November 2, 2010.
(6) Incorporated herein by reference to Exhibit 14.1 of the Company’s Annual Report on Form 10-KSB for the year ended September 30, 2006 filed with the Commission on December 20, 2006.
(7) Incorporated herein by reference to Exhibit 10.1 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
II-4
(8) Incorporated herein by reference to Exhibit 10.2 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(9) Incorporated herein by reference to Exhibit 10.3 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(10) Incorporated herein by reference to Exhibit 10.4 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(11) Incorporated herein by reference to Exhibit 10.5 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(12) Incorporated herein by reference to Exhibit 10.6 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(13) Incorporated herein by reference to Exhibit 10.7 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(14) Incorporated herein by reference to Exhibit 10.8 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(15) Incorporated herein by reference to Exhibit 10.9 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(16) Incorporated herein by reference to Exhibit 10.10 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(17) Incorporated herein by reference to Exhibit 10.11 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(18) Incorporated herein by reference to Exhibit 10.12 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(19) Incorporated herein by reference to Exhibit 10.13 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(20) Incorporated herein by reference to Exhibit 10.14 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(21) Incorporated herein by reference to Exhibit 10.15 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(22) Incorporated herein by reference to Exhibit 10.16 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(23) Incorporated herein by reference to Exhibit 10.17 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(24) Incorporated herein by reference to Exhibit 10.18 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(25) Incorporated herein by reference to Exhibit 16.1 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
II-5
(26) Incorporated herein by reference to Exhibit 17.1 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
II-6
Item 17. Undertakings.
The undersigned registrant hereby undertakes:
(A) The undersigned registrant hereby undertakes:
(1) To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement:
(i) To include any prospectus required by Section 10(a)(3) of the Securities Act of 1933;
(ii) To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than 20 percent change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement; and
(iii) To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement.
(2) That, for the purpose of determining any liability under the Securities Act of 1933, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered herein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
(3) To remove from registration by means of a post−effective amendment any of the securities being registered which remain unsold at the termination of the offering.
(4) That, for the purpose of determining liability under the Securities Act of 1933 to any purchaser:
(i) If the registrant is relying on Rule 430B:
(A) Each prospectus filed by the registrant pursuant to Rule 424(b)(3) shall be deemed to be part of the registration statement as of the date the filed prospectus was deemed part of and included in the registration statement; and
(B) Each prospectus required to be filed pursuant to Rule 424(b)(2), (b)(5), or (b)(7) as part of a registration statement in reliance on Rule 430B relating to an offering made pursuant to Rule 415(a)(1)(i), (vii), or (x) for the purpose of providing the information required by Section 10(a) of the Securities Act of 1933 shall be deemed to be part of and included in the registration statement as of the earlier of the date such form of prospectus is first used after effectiveness or the date of the first contract of sale of securities in the offering described in the prospectus. As provided in Rule 430B, for liability purposes of the issuer and any person that is at that date an underwriter, such date shall be deemed to be a new effective date of the registration statement relating to the securities in the registration statement to which that prospectus relates, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such effective date, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such effective date; or
II-7
(ii) If the registrant is subject to Rule 430C, each prospectus filed pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A, shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use.
(B) Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Act and will be governed by the final adjudication of such issue.
(C) The undersigned registrant hereby undertakes that:
(1) For purposes of determining any liability under the Securities Act of 1933, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant pursuant to Rule 424(b) (1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective.
(2) For the purpose of determining any liability under the Securities Act of 1933, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
II-8
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, the registrant has duly caused this registration statement to be signed on its behalf by the undersigned, in the City of Williamsville, State of New York on July 20, 2011.
|
By:
|
/s/ Joseph Pandolfino
|
|
|
Joseph Pandolfino
|
||
|
Title:
|
Chief Executive Officer
|
|
In accordance with the requirements of the Securities Act of 1933, as amended, this registration statement was signed by the following persons in the capacities and on the dates stated:
|
Signature
|
|
Title
|
|
Date
|
|
|
|
|
|
|
|
|
|
/s/ Joseph Pandolfino
|
|
|
|
|
|
|
Joseph Pandolfino
|
|
Chief Executive Officer and Director
(Principal Executive Officer)
|
|
July 20, 2011
|
|
|
|
|
|
|
|
|
|
/s/ C. Anthony Rider
|
|
|
|
|
|
|
C. Anthony Rider
|
|
Chief Financial Officer and Treasurer
(Principal Financial and Accounting Officer)
|
|
July 20, 2011
|
|
|
/s/ Henry Sicignano, III.
|
|
|
|
||
|
Henry Sicignano, III
|
|
President, Secretary and Director
|
|
July 20, 2011
|
|
|
/s/ Joseph A. Dunn, Ph.D.
|
|
|
|
|
|
|
Joseph A. Dunn, Ph.D.
|
|
Director
|
|
July 20, 2011
|
|
|
|
|
|
|
|
|
|
/s/ James W. Cornell
|
|
|
|
|
|
|
James W. Cornell
|
|
Director
|
|
July 20, 2011
|
|
|
|
|
|
|
|
|
|
/s/ Steven Katz
|
|
|
|
|
|
|
Steven Katz
|
|
Director
|
|
July 20, 2011
|
S-1
EXHIBIT INDEX
|
EXHIBIT
|
||
|
NUMBER
|
DESCRIPTION
|
|
|
2.1(1)
|
Agreement and Plan of Merger and Reorganization dated as of January 25, 2011 by and among the Company, 22nd Century, and Acquisition Sub.
|
|
|
2.2(2)
|
Certificate of Merger dated as of January 25, 2011 Acquisition Sub. with and into 22nd Century
|
|
|
3.1(3)
|
Certificate of Incorporation of the Company
|
|
|
3.2(4)
|
Amended and Restate Certificate of Incorporation of the Company
|
|
|
3.3(3)
|
Bylaws of the Company
|
|
|
5.1*
|
Opinion of Foley & Lardner LLP
|
|
|
10.1(7)
|
2010 Equity Incentive Plan
|
|
|
10.2(8)
|
Form of Securities Purchase Agreement dated as of January 25, 2011 by and among 22nd Century, the purchaser(s) identified on the signature pages thereto and Parent, solely for the purposes of Section E and Section G thereof, as amended.
|
|
|
10.3(9)
|
Form of Conversion Agreement
|
|
|
10.4(10)
|
Form of Warrant dated as of January 25, 2011 issued to LLC members of 22nd Century prior to the consummation of the Private Placement Offering upon consummation of the Merger
|
|
|
10.5(11)
|
Form of Warrant dated as of January 25, 2011 issued to investors in the Private Placement Offering upon consummation of the Merger
|
|
|
10.6(12)
|
Form of Warrant dated as of January 25, 2011 issued to the Placement Agent and Sub-Agent upon consummation of the Merger
|
|
|
10.7(13)
|
Advisor Warrant dated as of January 25, 2011 issued to the Placement Agent in connection with that certain Advisory Agreement dated as of January 25, 2011 by and between the Company and the Placement Agent
|
|
|
10.8(14)
|
Advisory Agreement dated as of January 25, 2011 by and between the Company and the Placement Agent
|
|
|
10.9(15)
|
Placement Agency Agreement dated as of December 1, 2010 by and between 22nd Century and the Placement Agent
|
|
|
10.10(16)
|
Escrow Agreement dated as of December 2, 2010 by and among 22nd Century, the Placement Agent and Bank of America, National Association
|
|
|
10.11(17)
|
Split-Off Agreement dated as of January 25, 2011 by and among the Company, Touchstone Split. Corp and David Rector
|
E-1
|
10.12(18)
|
Letter from Paramount Strategy Corp dated as of December 21, 2010 regarding loan forgiveness
|
|
|
10.13(19)
|
Letter from Milestone Enhanced Fund Ltd. dated as of December 28, 2010 regarding loan forgiveness
|
|
|
10.14(20)
|
Letter from Mark Tompkins dated as of January 25, 2011 regarding loan forgiveness
|
|
|
10.15(21)†
|
Employment Agreement dated as of January 25, 2011 by and between the Company and Joseph Pandolfino
|
|
|
10.16(22)†
|
Employment Agreement dated as of January 25, 2011 by and between the Company and Henry Sicignano III
|
|
|
10.17(23)†
|
Employment Agreement dated as of January 25, 2011 by and between the Company and C. Anthony Rider
|
|
|
10.18†*
|
Employment Agreement dated as of March 15, 2011 by and between the Company and Michael R. Moynihan.
|
|
|
10.19(24)
|
Form of Lock-Up Agreement
|
|
| 10.20** |
Promissory Note dated June 30, 2011, payable by the Company to Henry Sicignano III in the principal amount of $150,000.
|
|
|
14.1(6)
|
Code of Ethics
|
|
|
16.1(25)
|
Letter from Child, Van Wagoner & Bradshaw, PLLC regarding change in independent registered public accountants.
|
|
|
17.1(26)
|
Letter from David Rector dated as of January 25, 2011 resigning as a director and officer of Parent
|
|
|
21.1*
|
Subsidiaries of the Company
|
|
|
23.1**
|
Consent of Freed Maxick & Battaglia, CPAs, PC
|
|
|
23.2**
|
Consent of Foley & Lardner LLP (included in Exhibit 5.1)
|
|
*
|
Previously filed.
|
|
**
|
Filed herewith.
|
|
†
|
Management contract or compensatory plan, contract or arrangement.
|
(1) Incorporated herein by reference to Exhibit 2.1 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(2) Incorporated herein by reference to Exhibit 2.2 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(3) Incorporated herein by reference to Exhibit 3.1 of the Company’s Registration Statement on Form SB-2 filed with the Commission on December 27, 2005.
(4) Incorporated herein by reference to Exhibit 3.2 of the Company’s Annual Report on Form 10-K for the year ended September 30, 2010 filed with the Commission on November 2, 2010.
(5) Incorporated herein by reference to Appendix B of the Company’s Definitive Information Statement on Schedule 14C filed with the Commission on November 2, 2010.
E-2
(6) Incorporated herein by reference to Exhibit 14.1 of the Company’s Annual Report on Form 10-KSB for the year ended September 30, 2006 filed with the Commission on December 20, 2006.
(7) Incorporated herein by reference to Exhibit 10.1 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(8) Incorporated herein by reference to Exhibit 10.2 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(9) Incorporated herein by reference to Exhibit 10.3 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(10) Incorporated herein by reference to Exhibit 10.4 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(11) Incorporated herein by reference to Exhibit 10.5 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(12) Incorporated herein by reference to Exhibit 10.6 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(13) Incorporated herein by reference to Exhibit 10.7 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(14) Incorporated herein by reference to Exhibit 10.8 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(15) Incorporated herein by reference to Exhibit 10.9 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(16) Incorporated herein by reference to Exhibit 10.10 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(17) Incorporated herein by reference to Exhibit 10.11 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(18) Incorporated herein by reference to Exhibit 10.12 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(19) Incorporated herein by reference to Exhibit 10.13 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(20) Incorporated herein by reference to Exhibit 10.14 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(21) Incorporated herein by reference to Exhibit 10.15 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(22) Incorporated herein by reference to Exhibit 10.16 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(23) Incorporated herein by reference to Exhibit 10.17 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
E-3
(24) Incorporated herein by reference to Exhibit 10.18 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(25) Incorporated herein by reference to Exhibit 16.1 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
(26) Incorporated herein by reference to Exhibit 17.1 of the Company’s Current Report on Form 8-K filed with the Commission on February 1, 2011.
E-4