Attached files
Table of Contents
As filed with the Securities and Exchange Commission on May 26, 2011
Registration No. 333-
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
GenMark Diagnostics, Inc.
(Exact name of Registrant as specified in its charter)
| Delaware | 3841 | 27-2053069 | ||
| (State or other jurisdiction of incorporation or organization) |
(Primary Standard Industrial Classification Code Number) |
(I.R.S. Employer Identification Number) |
5964 La Place Court
Carlsbad, CA 92008
(760) 448-4300
(Address, including zip code, and telephone number, including area code, of Registrant’s principal executive offices)
Hany Massarany
Chief Executive Officer and President
GenMark Diagnostics, Inc.
5964 La Place Court
Carlsbad, CA 92008
(760) 448-4300
(Name, address, including zip code, and telephone number, including area code, of agent for service)
Copies to:
| Michael S. Kagnoff, Esq. DLA Piper LLP (US) 4365 Executive Drive, Suite 1100 San Diego, CA 92121 Tel: (858) 677-1400 Fax: (858) 677-1401 |
Timothy R. Curry, Esq. Jones Day 1755 Embarcadero Road Palo Alto, CA 94303 Tel: (650) 739-3939 Fax: (650) 739-3900 |
Approximate date of commencement of proposed sale to the public: As soon as practicable after the effective date of this registration statement.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, check the following box. ¨
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definitions of “large accelerated filer,” “accelerated filer,” and “smaller reporting company” in Rule 12b-2 of the Exchange Act (Check one):
| Large accelerated filer |
¨ |
Accelerated filer | ¨ | |||
| Non-accelerated filer |
x (Do not check if a smaller reporting company) |
Smaller reporting company | ¨ |
CALCULATION OF REGISTRATION FEE
| Title of Each Class of Securities to be Registered |
Proposed Aggregate Offering Price(1) |
Amount of Registration Fee | ||
| Common Stock, $0.0001 par value per share |
$28,750,000 | $3,338 | ||
| (1) | Estimated solely for the purpose of calculating the registration fee in accordance with Rule 457(o) under the Securities Act. Includes the offering price of additional shares of common stock that the underwriters have the option to purchase. |
The Registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this registration statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933 or until the registration statement shall become effective on such date as the Commission, acting pursuant to said Section 8(a), may determine.
Table of Contents
The information in this prospectus is not complete and may be changed. We may not sell these securities until the Securities and Exchange Commission declares our registration statement effective. This prospectus is not an offer to sell these securities and we are not soliciting an offer to buy these securities in any state where the offer or sale is not permitted.
Subject to completion, dated May 26, 2011

shares
GENMARK DIAGNOSTICS, INC.
Common Stock
We are offering shares of our common stock. Our common stock is traded on the NASDAQ Global Market under the symbol “GNMK.” On May 25, 2011, the last reported sale price of our common stock on the NASDAQ Global Market was $5.85 per share.
Investing in our common stock involves a high degree of risk. Please see the section entitled “Risk Factors” starting on page 9 of this prospectus to read about risks you should consider carefully before buying shares of our common stock.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or passed upon the accuracy or adequacy of this prospectus. Any representation to the contrary is a criminal offense.
| Per Share | Total | |||||||
| Public offering price |
$ | $ | ||||||
| Underwriting discount |
$ | $ | ||||||
| Proceeds, before expenses, to GenMark Diagnostics, Inc. |
$ | $ | ||||||
We have granted the underwriters a 30-day option to purchase up to an additional shares of our common stock at the public offering price, less the underwriting discount, to cover any over-allotments.
The underwriters expect to deliver the shares on or about , 2011.
Sole Book-Running Manager
Canaccord Genuity
Co-Lead Manager
William Blair & Company
The date of this prospectus is , 2011
Table of Contents

Table of Contents
| Page | ||||
| 1 | ||||
| 9 | ||||
| 30 | ||||
| 31 | ||||
| 31 | ||||
| 32 | ||||
| 35 | ||||
| MANAGEMENT’S DISCUSSION AND ANALYSIS OF RESULTS OF OPERATIONS AND FINANCIAL CONDITION |
37 | |||
| 50 | ||||
| 71 | ||||
| 77 | ||||
| 89 | ||||
| 91 | ||||
| 93 | ||||
| 96 | ||||
| CERTAIN MATERIAL U.S. FEDERAL INCOME AND ESTATE TAX CONSIDERATIONS TO NON-U.S. HOLDERS |
98 | |||
| 101 | ||||
| 104 | ||||
| 104 | ||||
| 104 | ||||
| F-1 | ||||
You should rely only on the information contained in this prospectus. We have not, and the underwriters have not, authorized any other person to provide you with different information. If anyone provides you with different or inconsistent information, you should not rely on it. This prospectus is not an offer to sell, nor are we or the underwriters seeking an offer to buy, these securities in any state where the offer or sale is not permitted. The information in this prospectus is complete and accurate as of the date on the front cover of this prospectus. Our business, financial condition, results of operations and prospects may have changed since that date.
No action is being taken in any jurisdiction outside of the United States to permit a public offering of the common stock or possession or distribution of this prospectus in that jurisdiction. Persons who come into possession of this prospectus in any jurisdiction outside of the United States are required to inform themselves about and to observe any restrictions as to this offering and the distribution of this prospectus applicable to that jurisdiction.
eSensor®, Osmetech®, GenMarkDx™ and our logo are our trademarks. This prospectus also includes trademarks, trade names and service marks that are the property of other organizations.
i
Table of Contents
The items in the following summary are described in more detail later in this prospectus. This summary provides an overview of selected information and does not contain all the information you should consider. Therefore, you should also read the more detailed information set out in this prospectus and the financial statements included elsewhere in this prospectus. In this prospectus, unless the context otherwise requires, references to “we,” “us” or “our” refer to GenMark Diagnostics, Inc.
Our Company
We are a molecular diagnostics company focused on developing and commercializing our proprietary eSensor detection technology. Our proprietary electrochemical technology enables fast, accurate and highly sensitive detection of up to 72 distinct biomarkers in a single sample. Our XT-8 system received 510(k) clearance from the Food and Drug Administration, or FDA, and is designed to support a broad range of molecular diagnostic tests with a compact and easy-to-use workstation and self-contained, disposable test cartridges. Within 30 minutes of receipt of an amplified DNA sample, our XT-8 system produces clear and accurate results. Our XT-8 system supports between one and three analyzers. Each analyzer holds up to eight independent test cartridges, resulting in the XT-8 system supporting up to 24 test cartridges, each of which can be run independently, resulting in a convenient and flexible workflow for our target customers, which are hospitals and reference laboratories. As of March 31, 2011, we had an installed base of 102 analyzers, or placements, with our customers.
We have developed four diagnostic tests for use with our XT-8 system and expect to expand this test menu by introducing two to four new tests annually. Three of our diagnostic tests have received FDA clearance, including our Cystic Fibrosis Genotyping Test, which detects pre-conception risks of cystic fibrosis, our Warfarin Sensitivity Test, which determines an individual’s ability to metabolize the oral anticoagulant warfarin, and our Thrombophilia Risk Test, which detects an individual’s increased risk of blood clots. We have demonstrated 100% accuracy in clinical studies compared to DNA sequencing in our Cystic Fibrosis Genotyping Test, our Warfarin Sensitivity Test and our Thrombophilia Risk Test. We have also developed a Respiratory Viral Panel Test, which detects the presence of major respiratory viruses and is labeled for investigational use only, or IUO. We intend to seek FDA clearance for our Respiratory Viral Panel Test in 2011. We also have a pipeline of several additional potential products in different stages of development or design, including diagnostic tests for an individual’s sensitivity to Plavix, a commonly prescribed anti-coagulant, and for mutations in a gene known as K-ras, which is predictive of an individual’s response rates to certain prescribed anti-cancer therapies.
We are also developing our next-generation platform, the NexGen system. We are designing the NexGen system (formerly referred to as the AD-8 system) to integrate automated nucleic acid extraction and amplification with our eSensor detection technology to enable technicians to place a raw or a minimally prepared patient sample into our test cartridge and obtain results without any additional steps. This sample-to-answer capability is enabled by the robust nature of our eSensor detection technology, which is not impaired by sample impurities that we believe hinder competing technologies. We are designing our NexGen system to further simplify workflow and provide powerful, cost-effective molecular diagnostics solutions to a significantly expanded group of hospitals and reference laboratories.
Our XT-8 system and planned menu of tests are intended to improve patient care and physician practices by providing high value, clinically useful information that aids in the diagnosis of disease and the selection of treatments tailored to an individual’s genetic profile. We believe that these improvements in patient care are economically attractive to our customers who are generally reimbursed for these tests by third-party payors and managed care providers through established reimbursement codes. Because the XT-8 system is designed to be flexible and easy-to-use, we believe that our customers will choose to perform a broad range of tests on our platform, in some cases providing our customers with the capability to perform diagnostic tests that they were
1
Table of Contents
not previously able to complete. By focusing our product development and commercialization efforts on high value, clinically useful opportunities in genetic and infectious diseases, cancer and personalized medicine, we believe we will drive widespread clinical adoption of our products.
Our Strategy
Our goal is to become the market leading provider of automated, multiplex molecular diagnostic testing systems. We intend to expand the use of our XT-8 system and diagnostic tests targeting mainly those reference laboratories and hospitals in the United States which perform a high volume of molecular diagnostic tests. To achieve this objective, we intend to:
| • | expand our menu of clinical diagnostic products; |
| • | grow our installed base of customers; |
| • | increase utilization of tests with our customers; |
| • | develop and commercialize our NexGen system; and |
| • | expand internationally and explore out-licensing opportunities. |
Market Opportunity and Limitations of Current Technologies
The U.S. market for molecular diagnostics was estimated to be $1.9 billion in 2009 and is anticipated to reach $3.4 billion in 2014 according to L.E.K., a market research firm. Many factors are driving growth of this market, including the expansion of genetic testing for disease predisposition, advances in personalized medicine, such as the tailoring of cancer therapies to those individuals most likely to respond, and increased demand for infectious disease diagnostics panels.
Commercially available molecular diagnostic testing systems, as well as “home-brew” or laboratory developed tests, or LDTs, are characterized by the following limitations:
Limited Menu of Diagnostic Tests. We believe LDTs are typically custom designed for one specific genetic biomarker or disease. In addition, we believe testing systems marketed as alternatives to LDTs currently offer only a limited number of tests for use with such systems.
Inability to Multiplex. Testing systems often lack the capacity to multiplex, or test for multiple biomarkers at the same time on a single patient sample. As a result, the laboratory must perform multiple, separate tests.
Poor Laboratory Workflow. Many LDTs and testing systems require significant sample preparation and washing steps, frequent calibration and time-consuming maintenance.
Risk of Human Error and Contamination. Many LDTs and testing systems require technicians to perform complex manual procedures, which may lead to contamination. In addition, LDTs and many testing systems require the operator to interpret results, which increases the potential for human error.
Intensive Resource Requirements. Laboratories need highly skilled technicians and dedicate significant capital, labor and laboratory space to conduct molecular diagnostic tests. Many multiplex tests currently used by national reference laboratories are so specialized that we believe only a limited number of their sites can perform these tests.
Shifting Regulatory Environment. Many LDTs and testing systems have not been submitted for FDA clearance. The FDA has imposed regulatory requirements on laboratories that use these tests. In the future, the FDA may further restrict use of non-FDA-cleared tests.
2
Table of Contents
Our Solution
Our XT-8 system is an automated molecular diagnostic system that enables reference laboratories and hospitals to perform fast, accurate and easy-to-use molecular diagnostic tests. The XT-8 system, which employs our proprietary electrochemical detection technology, consists of a compact bench-top workstation with an integrated touch screen computer and an analyzer module into which the self-contained, disposable test cartridges are inserted. The XT-8 system is user-friendly, intuitive, requires minimal maintenance and provides laboratories with the ability to perform multiplex molecular diagnostic tests in an efficient and cost-effective manner. Specifically, we believe that our XT-8 system and related diagnostic tests offer reference laboratories and hospitals the following benefits:
Versatile Platform for a Broad Menu. Our XT-8 system has broad application across a number of areas in molecular diagnostic testing. In addition to our FDA-cleared Cystic Fibrosis Genotyping Test, Warfarin Sensitivity Test and Thrombophilia Risk Test, and our Respiratory Viral Panel Test, which is labeled for IUO, we have a pipeline of several additional products in development or design in the fields of pharmacogenetics, genetic diseases, infectious diseases and cancer. We are currently developing a Plavix Sensitivity Test and a K-ras Mutation Test, and we have a pipeline of potential products in various stages of development or design. Laboratories using our system will be able to run our additional tests without any further capital investment or operator training.
FDA-Cleared Products. We have received FDA clearance for our Cystic Fibrosis Genotyping Test, Warfarin Sensitivity Test and Thrombophilia Risk Test, while our Respiratory Viral Panel Test is labeled for IUO. We intend to submit our Respiratory Viral Panel Test to the FDA for clearance in 2011. We intend to utilize IUO-labeled products in clinical studies within the broader process of seeking FDA clearance for our diagnostic tests.
Ease of Use. Our XT-8 system eliminates the need to use complex instrumentation to generate test results. Our XT-8 system minimizes manual processing steps and streamlines data analysis, making molecular diagnostic testing available to a broad spectrum of laboratories without the need for highly skilled technicians. As a result, our XT-8 system can provide national reference laboratories with the ability to perform our menu of molecular diagnostic tests across all of their locations. We also designed our XT-8 system to require minimal maintenance.
Accuracy and Reliability. Our XT-8 system provides accurate and reliable molecular diagnostic test results. We have demonstrated 100% accuracy in clinical studies compared to DNA sequencing in our Cystic Fibrosis Genotyping Test, our Warfarin Sensitivity Test and our Thrombophilia Risk Test. Our XT-8 system limits technician contact with a patient sample, thereby reducing contamination risk. It also provides clear reports, minimizing the risk of human error and increasing the repeatability of test results.
Enhanced Laboratory Work Flow. Our unique platform allows for random access, or the ability to initiate tests while other tests are in progress, resulting in a highly convenient and flexible workflow. Our XT-8 system provides random access for up to 24 independent test cartridges. In addition, our proprietary electrochemical detection technology streamlines the sample preparation process and eliminates the need for the additional washing steps required by some other detection methods, such as optical or fluorescent detection. Laboratories using our XT-8 system can expect to obtain test results within 30 minutes of receipt of the amplified DNA sample, resulting in a total turnaround time of generally under four hours.
Multiplex Capability. Our XT-8 system can detect up to 72 separate biomarkers in a single test cartridge. This allows laboratories to run multiple tests or panels on an individual patient sample in a one-step detection process. This capability reduces the time required for a laboratory to perform a diagnostic analysis that involves multiple genetic markers or infectious disease pathogens, which otherwise would require the laboratory to run multiple, separate molecular diagnostic tests.
3
Table of Contents
Our Diagnostic Tests
We currently offer four diagnostic tests for use with our XT-8 System, three of which have received 510(k) clearance from the FDA and one of which is currently labeled for IUO. We also have eight additional diagnostic tests in the development or design stage.
| Test |
Intended Application | |
| FDA-Cleared Tests |
||
| Cystic Fibrosis Genotyping |
Detects the most common mutations associated with cystic fibrosis | |
| Warfarin Sensitivity |
Identifies biomarkers associated with an individual’s ability to metabolize the oral anti-coagulant warfarin | |
| Thrombophilia Risk |
Detects the most common mutations associated with increased risk of blood clots | |
| IUO Test |
||
| Respiratory Viral Panel |
Detects major respiratory viruses and aids in the identification of respiratory infections | |
| Tests in Development or Design |
||
| Plavix Sensitivity |
Identifies biomarkers associated with the metabolism of Plavix, a commonly prescribed anti-coagulant | |
| K-ras Mutation |
Detects mutations in the K-ras gene associated with response to anti-epidermal growth factor receptor therapy, or anti-EGFR therapy, a type of cancer treatment that interferes with the growth of cancer cells | |
| Lower Respiratory Tract Infections |
Detects major viral and bacterial causes of lower respiratory tract infections | |
| Central Nervous System Infections |
Detects major infectious agents associated with meningitis and encephalitis | |
| Hepatitis C Virus Genotyping |
Identifies type and subtype of the hepatitis C virus | |
| 2D6 Tamoxifen Metabolism |
Identifies patients with altered 2D6 metabolism that can affect the effectiveness of tamoxifen, a drug used for the prevention and treatment of breast cancer | |
| EGFR Pathway |
Detects mutations in other genes besides K-ras involved in EGFR signaling | |
| Human Papillomavirus Genotyping |
Identifies human papillomavirus types associated with cervical cancer | |
Our NexGen System
We are developing our next-generation testing system to integrate automated nucleic acid extraction and amplification. We are designing the NexGen system (formerly referred to as our AD-8 system) to allow a technician to place a raw or minimally prepared patient sample into our test cartridge and then insert the cartridge into the NexGen system with no further user intervention. The NexGen system is designed to achieve full sample-to-answer capability. The NexGen system will provide the same customer benefits of the XT-8 system and further enhance workflow by significantly reducing or eliminating the level of sample processing required and incorporating amplification. We believe this advancement will make our eSensor technology attractive to the broad range of institutions that currently lack the technical or economic resources to perform molecular diagnostic testing. We believe the NexGen system may expand our target user base from approximately 1,000 to over 5,000 potential laboratories and hospitals in the United States.
The NexGen system is currently in development with substantial technical feasibility completed using diluted blood in our Warfarin Sensitivity Test. The NexGen system leverages the base technology and system hardware from our XT-8 system to reduce risk and accelerate the development of the automated sample preparation and amplification features. We believe our approach to a sample-to-answer system will achieve benefits over other competitive multiplex systems, which require extensive sample processing procedures in addition to other complex sample manipulations throughout their test process.
4
Table of Contents
Selected Risk Factors
Investing in our common stock involves substantial risk. Before participating in this offering, you should carefully consider all of the information in this prospectus, including risks discussed in “Risk Factors” beginning on page 9. Some of our most significant risks are:
| • | We have a history of net losses and we may never achieve or maintain profitability. |
| • | We may need to raise additional funds in the future, and such funds may not be available on a timely basis, or at all. |
| • | We are reliant on the commercial success of our XT-8 System and our FDA-cleared diagnostic tests. |
| • | We may fail to successfully expand the menu of diagnostic tests for our XT-8 System, obtain licenses to additional biomarkers on commercially reasonable terms or effectively predict the types of tests our existing customers want. |
| • | Our products could infringe patent rights of others, which may require costly litigation and, if we are not successful, could cause us to pay substantial damages or limit our ability to commercialize our products. |
| • | Our sales depend on third-party payors reimbursing our customers for the use of our products at levels sufficient for us to sell our products profitably. |
| • | We may not be successful in developing our NexGen system. |
Reorganization
GenMark Diagnostics, Inc., or GenMark, was formed by Osmetech plc, or Osmetech, in Delaware in February 2010 and had no operations prior to our initial public offering which was completed in June 2010. Immediately prior to the closing of our initial public offering, GenMark acquired all of the outstanding ordinary shares of Osmetech in a reorganization under the applicable laws of the United Kingdom. As a result of the reorganization, all of the issued ordinary shares in Osmetech were cancelled in consideration of (i) the issuance of common stock of GenMark to the former shareholders of Osmetech and (ii) the issuance of new shares in Osmetech to GenMark. Following the reorganization, Osmetech became a wholly-owned subsidiary of GenMark, and the former shareholders of Osmetech held shares of GenMark. Any historical discussion of GenMark prior to the reorganization relates to Osmetech and its consolidated subsidiaries.
Office Location
Our principal corporate offices are located at 5964 La Place Court, Carlsbad, California 92008 and our telephone number is (760) 448-4300.
5
Table of Contents
The Offering
| Common stock we are offering |
shares of common stock |
| Over-allotment option |
The underwriters have been granted an option to purchase up to additional shares of our common stock at the initial public offering price for a period of 30 days after the date of this prospectus. |
| Offering price |
The public offering price is $ per share of common stock |
| Common stock outstanding after the offering |
shares of common stock |
| Use of proceeds |
We intend to use the net proceeds from this offering to continue to develop a broad menu of tests, to fund our planned sales and marketing initiatives, to develop our NexGen System, to acquire complementary businesses, technologies or licenses, to fund working capital and for general corporate purposes. We have no commitment with respect to any future acquisitions or licenses. See “Use of Proceeds.” |
| NASDAQ Global Market Symbol |
GNMK |
The number of shares of our common stock outstanding immediately after this offering is based on 11,738,233 shares outstanding as of March 31, 2011, and assumes all of the shares offered hereby are sold. The number of shares of common stock excludes:
| • | 1,314,975 shares of common stock issuable upon exercise of options outstanding as of March 31, 2011, at a weighted average exercise price of $6.06 per share; |
| • | 88,317 shares of common stock issuable upon exercise of warrants outstanding as of March 31, 2011, at a weighted average exercise price of $9.98 per share; |
| • | 701,957 shares of common stock which will be available for future grant or issuance under our 2010 Equity Incentive Plan, or our 2010 Plan, and the annual increases in the number of shares authorized under this plan; and |
| • | shares of common stock to cover over-allotments. |
Unless otherwise indicated, all information in this prospectus assumes:
| • | that the underwriters do not exercise their option to purchase up to additional shares of our common stock to cover over-allotments, if any; and |
| • | no options, warrants or shares of common stock were issued after March 31, 2011, and no outstanding options or warrants were exercised after March 31, 2011. |
6
Table of Contents
SUMMARY CONSOLIDATED HISTORICAL FINANCIAL DATA
The following table summarizes our financial data. The summary consolidated historical financial data as of and for the year ended December 31, 2010, and as of and for the three months ended March 31, 2011 relate to GenMark. The summary consolidated historical financial data as of December 31, 2009 and for the three months ended March 31, 2010 and the years ended December 31, 2009 and 2008 relate to Osmetech and its consolidated subsidiaries, which became subsidiaries of GenMark in a reorganization under the applicable laws of the United Kingdom in 2010. We have derived the following summary consolidated statement of operations data for the years ended December 31, 2010, 2009 and 2008 and the consolidated balance sheet data as of December 31, 2010 and 2009 from our audited consolidated financial statements included elsewhere in this prospectus, which have been prepared in accordance with U.S. generally accepted accounting principles, or U.S. GAAP. The consolidated statements of operations data for the three months ended March 31, 2011 and 2010, and the consolidated balance sheet data as of March 31, 2011, are derived from our unaudited consolidated financial statements included elsewhere in this prospectus. We have prepared the unaudited financial information on the same basis as the audited consolidated financial statements and have included, in our opinion, all adjustments, consisting only of normal recurring adjustments, that we consider necessary for a fair presentation of the financial information set forth in those statements.
Our historical results are not necessarily indicative of the results that may be expected in the future, and our interim results are not necessarily indicative of the results to be expected for the full fiscal year. The summary consolidated historical financial data should be read together with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and the consolidated financial statements and unaudited condensed consolidated financial statements of GenMark and Osmetech and related notes included elsewhere in this prospectus.
| Three Months Ended March 31, | Year Ended December 31, | |||||||||||||||||||
| 2011 | 2010 | 2010 | 2009 | 2008 | ||||||||||||||||
| Consolidated Statements of Operations Data: |
||||||||||||||||||||
| Revenue: |
||||||||||||||||||||
| Product sales |
$ | 692,739 | $ | 384,249 | $ | 2,340,996 | $ | 910,527 | $ | 559,592 | ||||||||||
| License revenue and other |
71,664 | 15,015 | 163,872 | 87,889 | 87,500 | |||||||||||||||
| Total revenue |
764,403 | 399,264 | 2,504,868 | 998,416 | 647,092 | |||||||||||||||
| Cost of sales |
1,643,456 | 567,396 | 4,377,701 | 4,332,299 | 3,237,869 | |||||||||||||||
| Gross loss |
(879,053 | ) | (168,132 | ) | (1,872,833 | ) | (3,333,883 | ) | (2,590,777 | ) | ||||||||||
| Operating expenses: |
||||||||||||||||||||
| Sales and marketing |
1,130,389 | 1,058,285 | 4,282,521 | 3,181,762 | 3,393,665 | |||||||||||||||
| Research and development |
2,528,252 | 1,453,759 | 6,522,112 | 5,633,717 | 13,423,679 | |||||||||||||||
| General and administrative |
2,111,336 | 2,167,264 | 7,353,802 | 8,288,762 | 9,632,708 | |||||||||||||||
| Total operating expenses |
5,769,977 | 4,679,308 | 18,158,435 | 17,104,241 | 26,450,052 | |||||||||||||||
| Loss from operations |
(6,649,030 | ) | (4,847,440 | ) | (20,031,268 | ) | (20,438,124 | ) | (29,040,829 | ) | ||||||||||
| Other income: |
||||||||||||||||||||
| Foreign exchange gain (loss) |
11,899 | (1,110 | ) | (1,110 | ) | 303,523 | 504,921 | |||||||||||||
| Interest income |
6,258 | 4,654 | (582 | ) | 33,222 | 420,011 | ||||||||||||||
| Therapeutic discovery credit |
— | — | 1,645,292 | — | — | |||||||||||||||
| Total other income |
18,157 | 3,544 | 1,643,600 | 336,745 | 924,932 | |||||||||||||||
| Loss before income taxes |
(6,630,873 | ) | (4,843,896 | ) | (18,387,668 | ) | (20,101,379 | ) | (28,115,897 | ) | ||||||||||
| (Provision) benefit for income taxes |
(10,968 | ) | (5,049 | ) | (15,324 | ) | 138,770 | (246,736 | ) | |||||||||||
| Net loss from continuing operations |
$ | (6,641,841 | ) | $ | (4,848,945 | ) | $ | (18,402,992 | ) | $ | (19,962,609 | ) | $ | (28,362,633 | ) | |||||
| Net loss |
$ | (6,641,841 | ) | $ | (4,848,945 | ) | $ | (18,402,992 | ) | $ | (19,962,609 | ) | $ | (28,362,633 | ) | |||||
| Unaudited net loss from continuing operations per common share, basic and diluted |
$ | (0.56 | ) | $ | (0.68 | ) | $ | (1.88 | ) | $ | (4.41 | ) | $ | (28.13 | ) | |||||
| Weighted average shares used in unaudited per share amounts |
11,771,014 | 7,113,922 | 9,796,588 | 4,526,758 | 1,008,386 | |||||||||||||||
7
Table of Contents
| As of March 31, | As of December 31, | |||||||||||
| 2011 | 2010 | 2009 | ||||||||||
| Balance Sheet Data: |
||||||||||||
| Cash and cash equivalents |
$ | 17,054,095 | $ | 18,329,079 | $ | 16,482,818 | ||||||
| Total assets |
21,955,353 | 24,925,509 | 19,333,477 | |||||||||
| Long-term obligations |
2,622,644 | 612,932 | 795,334 | |||||||||
| Total liabilities |
7,055,185 | 3,858,091 | 4,008,659 | |||||||||
| Accumulated deficit |
(151,134,719 | ) | (144,492,881 | ) | (126,089,889 | ) | ||||||
| Total stockholders’ equity |
14,900,168 | 21,067,418 | 15,324,818 | |||||||||
8
Table of Contents
Investing in our common stock involves a high degree of risk. You should carefully consider the risks described below and all of the other information set forth in this prospectus, including our consolidated financial statements and the related notes and “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” before deciding to invest in our common stock. If any of the events or developments described below occurs, our business, financial condition or results of operations could be negatively affected. In that case, the market price of our common stock could decline, and you could lose all or part of your investment.
Risks Related to Our Business
We have a history of net losses, and we may never achieve or maintain profitability.
We have a history of significant net losses and a limited history commercializing our molecular diagnostic products. We obtained FDA clearance for our first generation molecular diagnostic system in 2006, and commenced a limited marketing effort for this system. We commenced offering our XT-8 system and our Warfarin Sensitivity Test in July 2008. We commenced offering our Cystic Fibrosis Genotyping Test in July 2009 and our Thrombophilia Risk Test in April 2010. Our Respiratory Viral Panel Test is currently labeled for IUO. Our net losses from continuing operations were approximately $6.6 million for the three months ended March 31, 2011, $18.4 million for the twelve months ended December 31, 2010, $20.0 million in 2009 and $28.4 million in 2008. At March 31, 2011, we had an accumulated deficit of approximately $151.1 million. We will continue to incur significant expenses for the foreseeable future for our sales and marketing, research and development and regulatory activities and maintaining our existing and obtaining additional intellectual property rights. We cannot provide you any assurance that we will ever achieve profitability and, even if we achieve profitability, that we will be able to sustain or increase profitability on a quarterly or annual basis. Further, because of our limited commercialization history and because the market for molecular diagnostic products is relatively new and rapidly evolving, we have limited insight into the trends that may emerge and affect our business. We may make errors in predicting and reacting to relevant business trends, which could harm our business and financial condition.
We will need to raise additional funds in the future, and such funds may not be available on a timely basis, or at all. If additional capital is not available, we may have to curtail or cease operations.
Until such time, if ever, as we can generate substantial product revenues, we will be required to finance our operations with our cash resources. We will need to raise additional funds in the future to support our operations. We cannot be certain that additional capital will be available as needed or on acceptable terms, or at all. If we require additional capital at a time when investment in our company, in molecular diagnostics companies or the marketplace in general is limited, we may not be able to raise such funds at the time that we desire, or at all. If we do raise additional funds through the issuance of equity or convertible securities, the percentage ownership of holders of our common stock could be significantly diluted and these newly issued securities may have rights, preferences or privileges senior to those of holders of our common stock. If we obtain debt financing, a substantial portion of our operating cash flow may be dedicated to the payment of principal and interest on such indebtedness, and the terms of the debt securities issued could impose significant restrictions on our operations. If we raise additional funds through collaborations and licensing arrangements, we could be required to relinquish significant rights to our technologies, and products or grant licenses on terms that are not favorable to us.
If our products do not perform as expected or the reliability of the technology on which our products are based is questioned, our operating results and business will suffer.
Our success depends on the market’s confidence that we can provide reliable, high-quality diagnostic systems and tests. We believe that customers in our target markets are likely to be particularly sensitive to product defects and errors. As a result, our reputation and the public image of our products or technologies will
9
Table of Contents
be significantly impaired if our products fail to perform as expected. Although our diagnostic systems are designed to be user-friendly, the functions they perform are quite complex, and our products may develop or contain undetected defects or errors.
If we experience a material defect or error, this could result in loss or delay of revenues, increased costs to produce our tests, delayed market acceptance, damaged reputation, diversion of development and management resources, legal claims, increased insurance costs or increased service and warranty costs, any of which could materially harm our business, financial condition and results of operations.
We also face the risk of product liability exposure related to the sale of our products. We currently carry product liability insurance that covers us against specific product liability claims up to an annual aggregate limit of $7.0 million. We also carry a separate general liability and umbrella policy that covers us against certain claims but excludes coverage for product liability. Any claim in excess of our insurance coverage, or for which we do not have insurance coverage, would have to be paid out of our cash reserves, which would harm our financial condition. We cannot assure you that we have obtained sufficient insurance or broad enough coverage to cover potential claims. Also, we cannot assure you that we can or will maintain our insurance policies on commercially acceptable terms, or at all. A product liability claim could significantly harm our business, financial condition and results of operations.
We may fail to successfully expand the menu of diagnostic tests for our XT-8 system or effectively predict the types of tests our existing and target customers want.
We currently market three FDA-cleared diagnostic tests and have developed one other diagnostic test currently labeled for IUO. In addition, we have several diagnostic tests in the development or design stage. Some hospital-based and reference laboratories may not consider adopting our XT-8 system until we offer a broader menu of diagnostic tests. Although we are developing additional tests to respond to the needs of these laboratories, we cannot guarantee that we will be able to license the appropriate technology, or develop and obtain required regulatory clearances or approvals, for enough additional tests quickly enough or in a manner that is cost-effective. The development of new or enhanced products is a complex and uncertain process requiring the accurate anticipation of technological and market trends, as well as precise technological execution. In addition, in order to commercialize our products, we are required to undertake time consuming and costly development activities, including clinical studies for which the outcome is uncertain. Products that appear promising during early development and preclinical studies may, nonetheless, fail to demonstrate the results needed to support regulatory approval or, if approved, may not generate the demand we expect. If we are unable to successfully develop and commercialize additional diagnostic tests for use with our XT-8 system, our revenues and our ability to achieve profitability will be significantly impaired.
We may not be able to correctly estimate or control our future operating expenses, which could lead to cash shortfalls.
Our operating expenses may fluctuate significantly in the future as a result of a variety of factors, many of which are outside of our control. These factors include:
| • | the time and resources required to develop, conduct clinical studies and obtain regulatory clearances for the additional diagnostic tests we develop; |
| • | the expenses we incur for research and development required to maintain and improve our technology, including developing our next-generation molecular diagnostic system; |
| • | the costs of preparing, filing, prosecuting, defending and enforcing patent claims and other patent related costs, including litigation costs and the results of such litigation. |
| • | the expenses we incur in connection with commercialization activities, including product marketing, sales and distribution; |
| • | the expenses we incur in licensing biomarkers from third parties to expand the menu of diagnostics tests we plan to offer; |
10
Table of Contents
| • | our sales strategy and whether the revenues from sales of our test cartridges or XT-8 system will be sufficient to offset our expenses; |
| • | the costs to attract and retain personnel with the skills required for effective operations; and |
| • | the costs associated with being a public company. |
Our budgeted expense levels are based in part on our expectations concerning future revenues from sales of our XT-8 system and diagnostic tests. We may be unable to reduce our expenditures in a timely manner to compensate for any unexpected shortfall in revenue. Accordingly, a significant shortfall in demand for our products could have an immediate and material impact on our business and financial condition.
We face intense competition from established and new companies in the molecular diagnostics field and expect to face increased competition in the future.
The markets for our technologies and products are very competitive, and we expect the intensity of competition to increase. We compete with many companies in the United States engaged in the development, commercialization and distribution of similar products intended for clinical molecular diagnostic applications. Categories of competitors include:
| • | companies developing and marketing multiplex molecular diagnostics systems, including Luminex Corporation; Nanosphere; Qiagen NV; Abbott Diagnostics; Hologic, Inc. and Innogenetics Inc.; |
| • | large hospital-based laboratories and reference laboratories who provide large-scale testing using their own proprietary testing methods including Quest Diagnostics and Laboratory Corporation of America; and |
| • | companies that manufacture laboratory-based tests and analyzers including Cephid; Gen-Probe, Inc.; Siemens; Hologic, Inc.; Qiagen NV; Roche Diagnostics; and Abbott Diagnostics. |
Our diagnostic tests also face competition with the laboratory-developed-tests, or LDTs, developed by national and regional reference laboratories and hospitals. Such laboratory-developed tests may not be subject to the same requirements for clinical trials and FDA submission requirements that may apply to our products.
We anticipate that we will face increased competition in the future as new companies enter the market with new technologies and our competitors improve their current products and expand their menu of diagnostic tests. Many of our current competitors, as well as many of our potential competitors, have greater name recognition, more substantial intellectual property portfolios, longer operating histories, significantly greater resources to invest in new technologies, more substantial experience in new product development, greater regulatory expertise, more extensive manufacturing capabilities and the distribution channels to deliver products to customers. The impact of these factors may result in our technologies and products becoming obsolete before we recover the expenses incurred to develop them or before they generate significant revenue.
We are reliant on the commercial success of our XT-8 system and our diagnostic tests.
We have primarily placed our XT-8 systems with customers at no initial charge through placement agreements, under which customers commit to purchasing minimum quantities of test cartridges over a period of one to three years, with a component of the reagent cartridge price allocated to recover the instrument cost. While we also offer our XT-8 systems for sale, we have sold only 12 of our systems. We expect sales of our diagnostic tests associated with our XT-8 system will account for the vast majority of our revenues for at least the next several years. We intend to dedicate a significant portion of our resources to the commercialization of our XT-8 system and our existing FDA-cleared diagnostic tests. Although we intend to develop a broad range of additional diagnostic tests for use with the XT-8 system and our NexGen system, we cannot assure you when or if we will obtain FDA clearance for the tests we develop in the future, or whether the market will accept such new products. As a result, to the extent that our XT-8 system and our existing and future FDA-cleared diagnostic tests are not commercially successful or are withdrawn from the market for any reason, our revenues will be harmed and our business, operating results and financial condition will be harmed.
11
Table of Contents
We may not be successful in developing our NexGen system.
We are developing a sample-to-answer platform, the NexGen system. We are designing this system to integrate automated nucleic acid extraction and amplification with our eSensor technology to allow technicians to be able to place a patient sample into our test cartridge and obtain results with significantly reduced or no processing. The development of the NexGen system is a complex process, and we may not be successful in completing the development of all the currently intended features and benefits of the system, which may limit its marketability. In addition, before commercializing the NexGen system we will be required to obtain regulatory approval for the system as well as each of the diagnostic tests to be used on the system, including those tests that previously received approval for use with our XT-8 system. If we are unable to successfully develop and obtain regulatory approval for our NexGen system and related diagnostic tests, our business plan will be impaired. Additionally, prior to or upon release of our NexGen System, sales of our XT-8 system may decrease as customers migrate over to our newer technology.
Our financial results will depend on the acceptance among reference laboratories and hospitals, third-party payors and the medical community of our molecular diagnostic technology and products.
Our future success depends on the acceptance by our target customers, third-party payors and the medical community that our molecular diagnostic products are a reliable, accurate and cost-effective replacement for other molecular diagnostic testing methods.
Medical offices and many hospitals outsource their molecular diagnostic testing needs to national or regional reference laboratories. Our business success depends on our ability to convince these target laboratories and hospitals to replace their current testing platforms and/or send-out tests, with our XT-8 system and related diagnostic tests. We must also continue to increase the number of available tests, and test sell-through, on our installed systems.
Many other factors may affect the market acceptance and commercial success of our molecular diagnostic technology and products, including:
| • | the relative convenience and ease of use of our diagnostic systems over competing products; |
| • | the introduction of new technologies and competing products that may make our technologies and products a less attractive solution for our target customers; |
| • | the breadth of our menu of available diagnostic tests relative to our competitors; |
| • | our success in training reference and hospital-based laboratories in the proper use of our products; |
| • | the acceptance in the medical community of our molecular diagnostic technology and products; |
| • | the extent and success of our marketing and sales efforts; and |
| • | general economic conditions. |
Manufacturing risks and inefficiencies may adversely affect our ability to produce products; we have a sole source of supply for our XT-8 System.
We must manufacture, or engage third parties to manufacture, components of our products in sufficient quantities and on a timely basis, while maintaining product quality, acceptable manufacturing costs and complying with regulatory requirements. In determining the required quantities of our products and the manufacturing schedule, we must make significant judgments and estimates based on inventory levels, current market trends and other related factors. Because of the inherent nature of estimates and our limited experience in marketing our products, there could be significant differences between our estimates and the actual amounts of products we require. This can result in shortages if we fail to anticipate demand, or excess inventory and write-offs if we order more than we need.
We currently manufacture our proprietary test cartridges at our Carlsbad, California manufacturing facility. We outsource manufacturing of our XT-8 system and much of the disposable component molding and
12
Table of Contents
component assembly for our test cartridges. Our XT-8 system is manufactured by Aubrey Group Inc., our single source supplier that specializes in contract design and manufacturing of electronic and electromechanical devices for medical use. While we work closely with Aubrey Group Inc. to try to ensure continuity of supply while maintaining high quality and reliability, we cannot guarantee that these efforts will be successful. Should Aubrey Group Inc. become unable or unwilling to continue to meet our supply needs, we may experience delays in qualifying a new source or may not obtain as favorable pricing or other terms, any of which could harm our business, financial condition or results of operation. In addition, our components are custom-made by only a few outside vendors. Reliance on third-party manufacturers entails risks to which we would not be subject if we manufactured these components ourselves, including:
| • | reliance on third parties for regulatory compliance and quality assurance; |
| • | possible breaches of manufacturing agreements by the third parties because of factors beyond our control; |
| • | possible regulatory violations or manufacturing problems experienced by our suppliers; and |
| • | possible termination or non-renewal of agreements by third parties, based on their own business priorities, at times that are costly or inconvenient for us. |
We may not be able to meet the demand for our products if one or more of these third-party manufacturers are not able or are unwilling to supply us with the necessary components that meet our specifications. It may be difficult to find alternate suppliers in a timely manner and on terms acceptable to us.
The manufacturing operations for our test cartridges in Carlsbad, California use highly technical processes involving unique, proprietary techniques. In addition, the manufacturing equipment we use would be costly to repair or replace and could require substantial lead time to repair or replace. Any interruption in our operations or decrease in the production capacity of our manufacturing facility or the facilities of any of our suppliers because of equipment failure, natural disasters such as earthquakes, tornadoes and fires or otherwise, would limit our ability to meet customer demand for the XT-8 system and tests and would have a material adverse effect on our business, financial condition and results of operations. Other possible disruptions may include power loss and telecommunications failures. In the event of a disruption, we may lose customers and we may be unable to regain those customers thereafter. Our insurance may not be sufficient to cover all of our potential losses and may not continue to be available to us on acceptable terms, or at all.
If we are unable to retain key members of our senior management and scientists or hire additional skilled employees, we may be unable to achieve our goals.
Our performance is substantially dependent on the performance of our senior management and key scientific and technical personnel. Our senior managers and other key employees can terminate their relationship with us at any time. We have a small number of senior managers, and the loss of services of any of these managers or our scientific or technical personnel could have a material adverse effect on our business, financial condition and results of operations. We do not maintain key-man life insurance on any of our employees.
In addition, our product development and marketing efforts could be delayed or curtailed if we are unable to attract, train and retain highly skilled employees and scientific advisors. To expand our research, product development and sales efforts, we will need to retain additional people skilled in areas such as electrochemical and molecular science, information technology, manufacturing, sales, marketing and technical support. Because of the complex and technical nature of our systems and the dynamic market in which we compete, any failure to attract and retain a sufficient number of qualified employees could materially harm our ability to develop and commercialize our technology. We may not be successful in hiring or retaining qualified personnel, and any failure to do so could have a material adverse effect on our business, financial condition and results of operations.
13
Table of Contents
Our success may depend upon how we and our competitors anticipate and adapt to market conditions.
The markets for our products are characterized by rapidly changing technology, evolving industry standards, changes in customer needs, emerging competition and new product introductions. New technologies, techniques or products could emerge with similar or better performance or may be perceived as providing better value than our systems and related tests and could exert pricing pressures on our products. It is critical to our success that we anticipate changes in technology and customer requirements and successfully introduce enhanced and competitive technology to meet our customers’ and prospective customers’ needs on a timely basis. We will need to respond to technological innovation in a rapidly changing industry and may not be able to maintain our technological advantages over emerging technologies in the future. If we fail to keep pace with emerging technologies, our systems and related tests will become uncompetitive and our market share will decline, which would harm our business, financial condition and results of operations.
We may be unsuccessful in our long-term goal of expanding sales of our product offerings outside the United States.
Assuming we receive the applicable regulatory approvals, we intend to market our diagnostic products outside the United States through third-party distributors. These distributors may not commit the necessary resources to market and sell our products to meet our expectations. If distributors do not perform adequately or in compliance with applicable laws and regulations in particular geographic areas, or if we are unable to locate distributors in particular geographic areas, our ability to realize long-term international revenue growth would be harmed.
In order to market our products in the European Union and many other foreign jurisdictions, we, or our distributors or partners, must obtain separate regulatory approvals and comply with numerous and varying regulatory requirements regarding safety and efficacy and governing, among other things, clinical studies and commercial sales and distribution of our products. The approval procedure varies among countries and can involve additional testing. The regulatory approval process outside the United States may include all of the risks associated with obtaining FDA approval, as well as additional risks. In addition, in many countries outside the United States, it is required that the product be approved for reimbursement before the product can be approved for sale in that country. We may not obtain approvals from regulatory authorities outside the United States on a timely basis, if at all, which could harm our ability to expand into markets outside the United States.
If we expand sales of our products outside the United States, our business will be susceptible to risks associated with international operations.
If we execute our plan to expand our operations outside the United States, our inexperience in operating in foreign countries increases the risk that our international expansion will not be successful. Conducting international operations would subject us to new risks that, generally, we have not faced in the United States, including:
| • | fluctuations in currency exchange rates; |
| • | unexpected changes in foreign regulatory requirements; |
| • | longer accounts receivable payment cycles and difficulties in collecting accounts receivable; |
| • | competition from companies located in the countries in which we offer our products, which may be a competitive disadvantage; |
| • | difficulties in managing and staffing international operations; |
| • | potentially adverse tax consequences, including the complexities of foreign value added tax systems, tax inefficiencies related to our corporate structure and restrictions on the repatriation of earnings; |
| • | the burdens of complying with a wide variety of foreign laws and different legal standards; |
| • | increased financial accounting and reporting burdens and complexities; |
| • | political, social and economic instability abroad, terrorist attacks and security concerns in general; and |
| • | reduced or varied protection for intellectual property rights in some countries. |
14
Table of Contents
The occurrence of any one of these risks could harm our business, results of operations and prospects. Additionally, operating internationally requires significant management attention and financial resources. We cannot be certain that the investment and additional resources required in establishing operations in other countries will produce desired levels of revenues or profitability.
Our Respiratory Viral Panel Test and other menu items that we develop in the future may have sales that fluctuate on a seasonal basis and, as a result, our results of operations for any particular quarter may not accurately reflect full-year trends.
Our Respiratory Viral Panel Test and other tests that we develop in the future may have sales that fluctuate on a seasonal basis. As a result, our results of operations for any particular quarter may not accurately reflect full-year trends. For example, we expect volume of testing for our Respiratory Viral Panel Test generally will decline during the spring and summer season and accelerate during the fall and winter season. As a result, comparison of our results from quarter-to-quarter may not accurately reflect trends or results for the full year.
We have limited experience in sales and marketing and may be unable to successfully commercialize our XT-8 system and related diagnostic tests.
We have limited marketing, sales and distribution experience and capabilities. In connection with our XT-8 system, we commenced offering our Warfarin Sensitivity Test in July 2008, our Cystic Fibrosis Genotyping Test in July 2009 and our Thrombophilia Risk Test in April 2010. We are currently in varying stages of development of 4 additional tests:
| • | Respiratory Viral Panel: A qualitative nucleic acid multiplex test designed for the simultaneous detection and identification of multiple respiratory virus nucleic acids and mutations; |
| • | Plavix Sensitivity: For the multiplexed detection and genotyping of the *2, *3, *4, *5, *6, *7, *8, *9, *10, *13 and *17 alleles of the cytochrome P450 (CYP450) 2C19 gene locus; |
| • | Kras-Mutation: Designed for the multiplexed detection and genotyping of 12 mutations in codons 12 and 13 of KRAS and the V600E mutation in BRAF; and |
| • | Hepatitis C Virus Genotyping: Designed to detect and subtype the different genotypes for the Hepatitis C Virus (HCV). |
As of March 31, 2011, we had 102 analyzers installed with customers. Our ability to achieve profitability depends on attracting customers for the XT-8 system, expanding the number of tests we offer, and building brand loyalty. To successfully perform sales, marketing, distribution and customer support functions ourselves, we face a number of risks, including:
| • | our ability to attract and retain the skilled support team, marketing staff and sales force necessary to commercialize and gain market acceptance for our technology and our products; |
| • | the ability of our sales and marketing team to identify and penetrate the potential customer base, including hospitals, national and regional reference laboratories; and |
| • | the difficulty of establishing brand recognition and loyalty for our products. |
In addition, we may seek to enlist one or more third parties to assist with sales, distribution and customer support globally or in certain regions of the world. If we do seek to enter into these arrangements, we may not be successful in attracting desirable sales and distribution partners, or we may not be able to enter into these arrangements on favorable terms, or at all. If our sales and marketing efforts, or those of any third-party sales and distribution partners, are not successful, our technologies and products may not gain market acceptance, which would harm our business operations.
Providing XT-8 systems to our customers through reagent rental agreements may harm our liquidity.
The majority of our XT-8 systems are sold to customers via “reagent rental” agreements, under which customers obtain the XT-8 System in return for a commitment to purchase minimum quantities of test cartridges over a period of one to three years. Accordingly, we must incur the expense of manufacturing XT-8
15
Table of Contents
Systems well in advance of receiving sufficient revenues from test cartridges to recover our manufacturing expenses. We also offer our XT-8 systems for sale. In 2010, we sold ten XT-8 systems to customers which included the sale of twelve analyzers. The amount of additional capital we may need to raise depends on the amount of our revenues from sales of test cartridges sold through these reagent rental agreements. We do not currently sell enough test cartridges to recover all of our fixed manufacturing expenses associated with the production of our systems and test cartridges, and therefore we currently have a high cost of sales relative to revenue, resulting in a gross loss. If we continue not to sell a sufficient number of test cartridges to offset our expenses associated with these reagent rental agreements, our liquidity will be adversely affected.
We use hazardous chemicals, biological materials and infectious agents in our business. Any claims relating to improper handling, storage or disposal of these materials could be time consuming and costly.
Our research, product development and manufacturing processes involve the controlled use of hazardous materials, including chemicals, biological materials and infectious disease agents. Our operations produce hazardous waste products. We cannot eliminate the risk of accidental contamination or discharge and any resultant injury from these materials. We may be sued for any injury or contamination that results from our use or the use by third parties of these materials, and our liability may exceed our insurance coverage and our total assets. Federal, state and local laws and regulations govern the use, manufacture, storage, handling and disposal of these hazardous materials and specified waste products, as well as the discharge of pollutants into the environment and human health and safety matters. Compliance with environmental laws and regulations may be expensive and may impair our research, development and production efforts. If we fail to comply with these requirements, we could incur substantial costs, including civil or criminal fines and penalties, clean-up costs or capital expenditures for control equipment or operational changes necessary to achieve and maintain compliance. In addition, we cannot predict the impact on our business of new or amended environmental laws or regulations or any changes in the way existing and future laws and regulations are interpreted and enforced.
Our corporate structure may create tax inefficiencies.
As a result our reorganization in 2010, Osmetech became a wholly-owned subsidiary of GenMark and a controlled foreign corporation for U.S. federal income tax purposes. This organizational structure may create inefficiencies, as certain types of income and investments of Osmetech that otherwise would not be currently taxable under general tax rules, may become taxable. In addition, conveyance of intellectual property rights from one subsidiary to another could create taxable income. Distributions from GenMark to its operating subsidiaries or amongst the U.S. operating subsidiaries of GenMark may be subject to additional U.S. and foreign income tax withholding and result in lower profits. It is our intention by the end of the first half of 2011 to streamline our corporate structure and, by doing so, we may lose some, if not most, of our tax loss carryforward benefits and/or certain activities of the restructuring could become taxable transactions in the United States. We cannot predict the outcome of such transactions and the impact such reorganization may have on U.S. and foreign tax liability and financial condition.
Our ability to use our net operating loss carryforwards might be limited.
As of December 31, 2010, we had net operating loss carryforwards of approximately $77.9 million for U.S. federal income tax purposes. These loss carryforwards will expire in varying amounts through 2030. To the extent these net operating loss carryforwards are available, we intend to use them to reduce the corporate income tax liability associated with our operations. Section 382 of the U.S. Internal Revenue Code generally imposes an annual limitation on the amount of net operating loss carryforwards that might be used to offset taxable income when a corporation has undergone significant changes in stock ownership. As a result, prior or future changes in ownership could put limitations on the availability of our net operating loss carryforwards. In addition, our ability to use the current net operating loss carryforwards might be further limited by the issuance of common stock in the future. To the extent our use of net operating loss carryforwards is significantly limited, our income could be subject to corporate income tax earlier than it would if we were able to use net operating loss carryforwards, which could result in lower profits.
16
Table of Contents
We have determined that we have experienced multiple ownership changes under Section 382. We have estimated that approximately $24.7 million of federal net operating losses may be utilized in the future based on limitations that we have calculated under Section 382. We are currently analyzing alternative positions and additional factual information that may increase the amount of net operating losses that could subsequently be utilized. To the extent that this additional information becomes available and could increase net operating losses available for use, we will adjust our deferred tax assets accordingly, with a corresponding adjustment to our valuation allowance. We also had non-U.S. net operating loss carryforwards of approximately $30.4 million as of December 31, 2010. Upon completion of our planned corporate restructuring, these non-U.S. net operating loss carryforwards will not be available for use.
Risks Related to Regulation
The regulatory clearance or approval process is expensive, time consuming and uncertain, and the failure to obtain and maintain required clearances or approvals could prevent us from commercializing our future products.
We are investing in the research and development of new diagnostic tests to expand our menu of testing options, as well as to develop our next-generation NexGen system, which we anticipate will reduce the need for sample preparation when using our system. Our products are subject to 510(k) clearance or pre-market approval by the FDA prior to their marketing for commercial use in the United States, and to any approvals required by foreign governmental entities prior to their marketing outside the United States. In addition, any changes or modifications to a device that has received regulatory clearance or approval that could significantly affect its safety or effectiveness, or would constitute a major change in its intended use, may require the submission of a new application for 510(k) clearance, pre-market approval or foreign regulatory approvals.
The 510(k) clearance and pre-market approval processes, as well as the process of obtaining foreign approvals, can be expensive, time consuming and uncertain. It generally takes from four to twelve months from submission to obtain 510(k) clearance, and from one to three years from submission to obtain pre-market approval; however, it may take longer, and 510(k) clearance or pre-market approval may never be obtained. Delays in receipt of, or failure to obtain, clearances or approvals for future products, including tests that are currently in design or development, would result in delayed, or no, realization of revenues from such products and in substantial additional costs which could decrease our profitability. We have limited experience in filing FDA applications for 510(k) clearance and pre-market approval. In addition, we are required to continue to comply with applicable FDA and other regulatory requirements once we have obtained clearance or approval for a product. There can be no assurance that we will obtain or maintain any required clearance or approval on a timely basis, or at all. Any failure to obtain or any material delay in obtaining FDA clearance or any failure to maintain compliance with FDA regulatory requirements could harm our business, financial condition and results of operations.
If third-party payors do not reimburse our customers for the use of our clinical diagnostic products or if reimbursement levels are set too low for us to sell our products at a profit, our ability to sell our products and our results of operations will be harmed.
We sell our products to hospital-based and reference laboratories, substantially all of which receive reimbursement for the health care services they provide to their patients from third-party payors, such as Medicare, Medicaid, other domestic and foreign government programs, private insurance plans and managed care programs. Reimbursement decisions by particular third-party payors depend upon a number of factors, including each third-party payor’s determination that use of a product is:
| • | a covered benefit under its health plan; |
| • | appropriate and medically necessary for the specific indication; |
| • | cost effective; and |
| • | neither experimental nor investigational. |
17
Table of Contents
Third-party payors may deny reimbursement for covered products if they determine that a medical product was not used in accordance with cost-effective diagnosis methods, as determined by the third-party payor, or was used for an unapproved indication. Third-party payors also may refuse to reimburse for procedures and devices deemed to be experimental.
Obtaining coverage and reimbursement approval for a product from each government or third-party payor is a time consuming and costly process that could require us to provide supporting scientific, clinical and cost-effectiveness data for the use of our product to each government or third-party payor. We may not be able to provide data sufficient to gain acceptance with respect to coverage and reimbursement. For example, Medicare and Medicaid generally do not reimburse providers who use our Warfarin Sensitivity Test. In addition, eligibility for coverage does not imply that any product will be covered and reimbursed in all cases or reimbursed at a rate that allows our potential customers to make a profit or even cover their costs.
In the United States, the American Medical Association assigns specific Current Procedural Terminology, or CPT, codes, which are necessary for reimbursement of diagnostic tests. Once the CPT code is established, the Centers for Medicare and Medicaid Services establish reimbursement payment levels and coverage rules under Medicaid and Medicare, and private payors establish rates and coverage rules independently. We cannot guarantee that any of our tests are or will be covered by the CPT codes that we believe may be applied to them or that any of our tests or other products will be approved for coverage or reimbursement by Medicare and Medicaid or any third-party payor. Third-party payors may nonetheless choose to reimburse our customers on a per test basis based on individual biomarker detection, rather than on the basis of the number of results given by the test. This may result in reference laboratories, public health institutions and hospitals electing to use separate tests to screen for each disease so that they can receive reimbursement for each test they conduct. In that event, these entities may purchase separate tests for each disease, rather than products, such as ours, that can be used to return multiple test results.
Third-party payors are increasingly attempting to contain health care costs by limiting both coverage and the level of reimbursement for medical products and services. Increasingly, Medicare, Medicaid and other third-party payors are challenging the prices charged for medical services, including clinical diagnostic tests. In addition, Medicare’s current freeze on its clinical laboratory fee schedule may harm the growth of the molecular diagnostics market for patients in the United States who are over 65 or have specific disabilities. Levels of reimbursement may decrease in the future, and future legislation, regulation or reimbursement policies of third-party payors may harm the demand for and reimbursement available for our products, which in turn, could harm pricing and sales. If our customers are not adequately reimbursed for our products, they may reduce or discontinue purchases of our products, which would cause our revenues to decline.
We and our suppliers, contract manufacturers and customers are subject to various governmental regulations, and we may incur significant expenses to comply with, and experience delays in our product commercialization as a result of, these regulations.
Our manufacturing processes and facilities, and those of some of our contract manufacturers, are required to comply with the federal Quality System Regulation, or the QSR, which covers the procedures and documentation of the design, testing, production, control, quality assurance, labeling, packaging, sterilization, storage and shipping of our devices. The FDA enforces the QSR through periodic announced and/or unannounced inspections of manufacturing facilities. We and our contract manufacturers have been, and anticipate in the future being, subject to such inspections, as well as to inspections by other federal and state regulatory agencies.
We must also file reports of device corrections and removals and adhere to the FDA’s rules on labeling and promotion. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability, including substantial monetary penalties and criminal prosecution.
18
Table of Contents
Failure to comply with applicable FDA requirements, or later discovery of previously unknown problems with our products or manufacturing processes, including our failure or the failure of one of our contract manufacturers to take satisfactory corrective action in response to an adverse QSR inspection, can result in, among other things:
| • | administrative or judicially imposed sanctions; |
| • | injunctions or the imposition of civil penalties; |
| • | recall or seizure of our products; |
| • | total or partial suspension of production or distribution; |
| • | the FDA’s refusal to grant pending future clearance or pre-market approval for our products; |
| • | withdrawal or suspension of marketing clearances or approvals; |
| • | clinical holds; |
| • | warning letters; |
| • | refusal to permit the import or export of our products; and |
| • | criminal prosecution. |
Any of these actions, in combination or alone, could prevent us from marketing, distributing or selling our products and would likely harm our business.
In addition, a product defect or regulatory violation could lead to a government-mandated or voluntary recall by us. We believe that the FDA would request that we initiate a voluntary recall if a product was defective or presented a risk of injury or gross deception. Regulatory agencies in other countries have similar authority to recall devices because of material deficiencies or defects in design or manufacture that could endanger health. Any recall would divert management attention and financial resources, could cause the price of our shares of common stock to decline and expose us to product liability or other claims, including contractual claims from parties to whom we sold products and harm our reputation with customers. A recall involving our XT-8 system or our FDA-cleared diagnostic tests would be particularly harmful to our business and financial results.
The use of our diagnostic products by our customers is also affected by the Clinical Laboratory Improvement Amendments of 1988, or CLIA, and related federal and state regulations that provide for regulation of laboratory testing. CLIA is intended to ensure the quality and reliability of clinical laboratories in the United States by mandating specific standards in the areas of personnel qualifications, administration, participation in proficiency testing, patient test management, quality assurance and quality control and inspections. Current or future CLIA requirements or the promulgation of additional regulations affecting laboratory testing may prevent some laboratories from using some or all of our diagnostic products.
Legislative or regulatory healthcare reforms may make it more difficult and costly for us to obtain regulatory clearance or approval of our products and to produce, market and distribute our products after clearance or approval is obtained.
From time to time, legislation is drafted and introduced in Congress that could significantly change the statutory provisions governing the regulatory clearance or approval, manufacture and marketing of regulated products or the reimbursement thereof. In addition, FDA regulations and guidance are often revised or reinterpreted by the FDA in ways that may significantly affect our business and our products. For example, in the future, the FDA may require more burdensome premarket approval of our system or diagnostic tests rather than the 510(k) clearance process we have used to date and anticipate primarily using in the future. Any new regulations or revisions or reinterpretations of existing regulations may impose additional costs or lengthen review times of our products. Delays in receipt of or failure to receive regulatory clearances or approvals for our new products would harm our business, financial condition and results of operations.
19
Table of Contents
Federal and state governments in the United States are also undertaking efforts to control growing health care costs through legislation, regulation and voluntary agreements with medical care providers and third-party payors. In March 2010, Congress enacted comprehensive health care reform legislation known as the Patient Protection and Affordable Care Act of 2010, or the PPACA. While the PPACA involves expanding coverage to more individuals, it includes new regulatory mandates and other measures designed to constrain medical costs. The PPACA also imposes significant new taxes on medical device manufacturers that are expected to cost the medical device industry up to $20 billion over the next decade. There are also stringent new reporting requirements of financial relationships between device manufacturers and physicians and teaching hospitals. Complying with PPACA could significantly increase our tax liabilities and costs, which could adversely affect our business and financial condition.
Our operations will also be impacted by the federal Patient Protection and Affordable Care Act of 2010, as modified by the Health Care and Education Reconciliation Act of 2010, or the Health Care Act. The Health Care Act imposes a 2.3% excise tax on sales of medical devices by manufacturers. Taxable devices include any medical device defined in Section 201(h) of the Federal Food, Drug and Cosmetic Act, or FDCA, and intended for use by humans, with limited exclusions for devices purchased by the general public at retail for individual use. There is no exemption for small companies, and we expect to begin paying the tax in 2013. The Health Care Act also requires manufacturers to report to the Department of Health and Human Services detailed information about financial arrangements with physicians and teaching hospitals. These reporting provisions preempt state laws that require reporting of the same information, but not those that require reports of different or additional information. Failure to comply subjects the manufacturer to significant civil monetary penalties. We expect compliance with the Health Care Act to impose significant administrative and financial burdens on us.
We are subject to various federal and state laws pertaining to health care fraud and abuse, including anti-kickback, self-referral, false claims and fraud laws, and any violations by us of such laws could result in fines or other penalties.
Our commercial, research, and other financial relationships with healthcare providers and institutions are subject to various federal and state laws intended to prevent health care fraud and abuse. The federal anti-kickback statute prohibits the knowing offer, receipt or payment of remuneration in exchange for or to induce the referral of patients or the use of products or services that would be paid for in whole or part by Medicare, Medicaid or other federal health care programs. Remuneration has been broadly defined to include anything of value, including cash, improper discounts, and free or reduced price items and services. Many states have similar laws that apply to their state health care programs as well as private payors. Violations of the anti-kickback laws can result in exclusion from federal health care programs and substantial civil and criminal penalties.
The federal False Claims Act, or the FCA, imposes liability on persons who, among other things, present or cause to be presented false or fraudulent claims for payment by a federal health care program. The FCA has been used to prosecute persons submitting claims for payment that are inaccurate or fraudulent, that are for services not provided as claimed, or for services that are not medically necessary. The FCA includes a whistleblower provision that allows individuals to bring actions on behalf of the federal government and share a portion of the recovery of successful claims. If our marketing or other arrangements were determined to violate anti-kickback or related laws, including the FCA, then our revenues could be adversely affected, which would likely harm on our business, financial condition and results of operations.
State and federal authorities have aggressively targeted medical device companies for alleged violations of these anti-fraud statutes, based on improper research or consulting contracts with doctors, certain marketing arrangements that rely on volume-based pricing, off-label marketing schemes and other improper promotional practices. Companies targeted in such prosecutions have paid substantial fines in the hundreds of millions of dollars or more, have been forced to implement extensive corrective action plans, and have often become subject to consent decrees severely restricting the manner in which they conduct their business. If we become
20
Table of Contents
the target of such an investigation or prosecution based on our contractual relationships with providers or institutions, or our marketing and promotional practices, we could face similar sanctions which would materially harm our business.
To the extent we commence commercial operations overseas, we will be subject to the U.S. Foreign Corrupt Practices Act, or the FCPA, and other countries’ anti-corruption/anti-bribery regimes, such as the U.K. Bribery Act. The FCPA prohibits improper payments or offers of payments to foreign governments and their officials for the purpose of obtaining or retaining business. Safeguards we implement to discourage improper payments or offers of payments by our employees, consultants, sales agents or distributors may be ineffective, and violations of the FCPA and similar laws may result in severe criminal or civil sanctions, or other liabilities or proceedings against us, any of which would likely harm our reputation, business, financial condition and result of operations.
Risks Related to Our Intellectual Property
We rely on third-party license agreements for patents and other technology related to our products. The termination of these agreements could delay or prevent us from being able to commercialize our products and the failure to negotiate new licenses could prevent us from expanding our menu of diagnostic products.
We depend on licenses to certain patents and patent applications that are related to electrochemical detection technology and other technology used in our molecular diagnostic systems and test cartridges. These licenses include both exclusive and non-exclusive arrangements. Many of these exclusive licenses obligate us to use commercially reasonable efforts to commercialize the subject inventions of the licensed patents, and if we fail to meet this obligation, we could lose one or more of those licenses. If, following such an event, any of our licensors were to provide a license to these patents to one or more of our competitors, our ability to compete in the market may be diminished. Furthermore, if we fail to comply with our material obligations under any of our patent license agreements, the licenses may be terminated and we could lose license rights that are important to our business.
The exclusive and non-exclusive licenses expire at various times, corresponding to the subject patents or patent applications, the expirations of which currently range from 2013 to 2028. We expect that we will need to license other technology or patents to commercialize future products, including licenses to additional biomarkers to expand our menu of diagnostic tests. These licenses may not be available to us on commercially reasonable terms, or at all, which could adversely affect our results of operations and growth prospects.
We may incur substantial costs as a result of litigation or other proceedings relating to the protection of our patents and other intellectual property rights and we may be unable to protect our rights to our technology.
If we or any of our licensors choose to go to court to stop a third party from using the inventions claimed in our owned or licensed patents, that third party may ask the court to rule that the patents are invalid and should not be enforced against that third party. These lawsuits are expensive and would consume time and other resources even if we were successful in stopping the infringement of these patents. In addition, there is a risk that the court will decide that these patents are not valid and that we do not have the right to stop others from using the inventions.
There is also the risk that, even if the validity of these patents is upheld, the court will refuse to stop the other party on the ground that such other party’s activities do not infringe our patents. In addition, the U.S. Supreme Court and the Court of Appeals for the Federal Circuit have recently changed certain tests regarding granting patents and assessing the validity of patent claims. As a consequence, issued patents may be found to contain invalid claims according to the newly revised and currently evolving standards. Some of our own or in-licensed patents may be subject to challenge and subsequent invalidation or significant narrowing of claim scope in a re-examination proceeding before the Patent and Trademark Office, or the PTO, or during litigation, under the revised criteria which make it more difficult to obtain patents.
We may also not be able to detect infringement against our own or in-licensed patents, which may be especially difficult for methods of use. While we intend to take actions reasonably necessary to enforce our patent rights, we depend, in part, on our licensors and collaborators to protect a substantial portion of our proprietary rights.
21
Table of Contents
Our products could infringe patent rights of others, which may require costly litigation and, if we are not successful, could cause us to pay substantial damages or limit our ability to commercialize our products.
Our commercial success depends on our ability to develop, manufacture and market our systems and tests and use our proprietary technology without infringing the patents and other proprietary rights of third parties. As the molecular diagnostic industry expands and more patents are issued, the risk increases that there may be patents issued to third parties that relate to our products and technology of which we are not aware or that we must challenge to continue our operations as currently contemplated. Our products may infringe or may be alleged to infringe these patents.
In addition, some patent applications in the United States may be maintained in secrecy until the patents are issued, because patent applications in the United States and many foreign jurisdictions are typically not published until eighteen months after filing and because publications in the scientific literature often lag behind actual discoveries, we cannot be certain that others have not filed patent applications for technology covered by our issued patents or our pending applications or that we were the first to invent the technology. Another party may have filed, and may in the future file, patent applications covering our products or technology similar to ours. Any such patent application may have priority over our patent applications or patents, which could further require us to obtain rights to issued patents covering such technologies. If another party has filed a U.S. patent application on inventions similar to ours, we may have to participate in an interference proceeding declared by the PTO to determine priority of invention in the United States. The costs of these proceedings could be substantial, and it is possible that such efforts would be unsuccessful if the other party had independently arrived at the same or similar invention prior to our own invention, resulting in a loss of our U.S. patent position with respect to such inventions.
There is a substantial amount of litigation involving patent and other intellectual property rights in the medical device, biotechnology and pharmaceutical industries generally. If a third party claims that we or any collaborator infringes its intellectual property rights, we may face a number of issues, including, but not limited to:
| • | infringement and other intellectual property claims which, regardless of merit, may be expensive and time-consuming to litigate and may divert our management’s attention from our core business; |
| • | substantial damages for infringement, which we may have to pay if a court decides that the product at issue infringes on or violates the third party’s rights, and if the court finds that the infringement was willful, we could be ordered to pay treble damages and the patent owner’s attorneys’ fees; |
| • | a court prohibiting us from selling or licensing our product unless the third party licenses its product rights to us, which it is not required to do; |
| • | if a license is available from a third party, we may have to pay substantial royalties, upfront fees or grant cross-licenses to intellectual property rights for our products; and |
| • | redesigning our products or processes so they do not infringe, which may not be possible or may require substantial monetary expenditures and time. |
Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. In addition, any uncertainties resulting from the initiation and continuation of any litigation could have a material adverse effect on our ability to raise the funds necessary to continue our operations.
We may be infringing on the patent rights of third parties, which could prevent us from selling our current or future products.
From time to time we may become engaged in litigation with third parties having patent or other intellectual property rights alleging that our products or proprietary technologies infringe their intellectual property rights. These third parties and others who may in the future threaten us with such litigation, are or may be better capitalized and have more resources than us. In addition, in order to commercialize certain new or
22
Table of Contents
existing tests including our Thrombophilia Risk Test, we may be required to license certain biomarkers or risk that a third party may claim that the use of certain biomarkers in our tests infringes their intellectual properly rights. We have received correspondence bringing to our attention certain patent rights held by third parties and offering to discuss licensing terms to the patents. Some of these letters relate to patents that are important to our products. Independently, we have also identified patents held by third parties that cover one or more of our products or planned products. Although we have taken licenses to numerous such third-party patents, we have also declined to license certain patents in instances where we do not believe our existing products infringe valid claims.
In May 2010, we received correspondence from Caliper Life Sciences, Inc., or Caliper, alleging that we infringe certain microfluidic patents held by Caliper relating to fluid handling technologies that we utilize in the cartridges used in all of our tests and demanding that we take a license to its patents or else Caliper would institute litigation against us. On November 10, 2010, we filed a complaint for declaratory judgment against Caliper in the United States District Court for the Northern District of California. In our complaint, we requested a declaration from the court that certain of Caliper’s microfluidic patents were invalid, and that we did not infringe on these patents. On March 1, 2011, we entered into a settlement agreement with Caliper pursuant to which we agreed to dismiss our action for declaratory judgment, without prejudice, and Caliper agreed not to assert infringement by us on these patents for a period of six months. Following the expiration of this six-month period, Caliper may again assert that we are infringing its patents and that we are required to take a license to its patents and could institute legal action. If one of Caliper’s patents or any other third-party patents were found to be valid and cover any of our products, proprietary technologies, including our fluid handling technologies used in our test cartridges, or their uses, we or any collaborator could be enjoined from using or selling our products by a court and/or required to pay damages and could be unable to commercialize our products or product candidates or use our proprietary technologies unless we or they obtained a license to the patent. A license may not be available to us or any collaborator on acceptable terms, or at all, which could potentially prevent us from selling our current products, using our fluid handling technologies used in our test cartridges or other core technologies or developing new tests. In addition, during litigation, the patent holder could obtain a preliminary injunction or other equitable relief that could prohibit us from making, using or selling our products, technologies or methods pending a trial on the merits, which could be years away. Furthermore, such litigation can be extremely costly and could significantly affect our results of operations and divert the attention of managerial and technical personnel.
If we are unable to obtain, maintain and enforce intellectual property protection covering our products, others may be able to make, use, or sell products substantially the same as ours, which could adversely affect our ability to compete in the market.
Our commercial success is dependent in part on obtaining, maintaining and enforcing intellectual property rights, including patents. If we are unable to obtain, maintain and enforce intellectual property protection covering our products, others may be able to make, use or sell products that are substantially the same as ours without incurring the sizeable development and licensing costs that we have incurred, which would adversely affect our ability to compete in the market.
We seek to obtain and maintain patents and other intellectual property rights to restrict the ability of others to market products that compete with our products. Currently, our patent portfolio is comprised, on a worldwide basis, of 100 issued U.S. patents, 50 issued foreign patents and 28 pending domestic and foreign patent applications, all of which we own directly or for which we are the exclusive licensee and that expire between 2013 and 2028. However, patents may not be issued based on any pending or future patent applications owned by or licensed to us and, moreover, issued patents owned or licensed to us now or in the future may be found by a court to be invalid or otherwise unenforceable. Also, even if our patents are determined by a court to be valid and enforceable, they may not be sufficiently broad to prevent others from marketing products similar to ours or designing around our patents, despite our patent rights, nor provide us with freedom to operate unimpeded by the patent rights of others.
We have also licensed certain intellectual property from third parties related to our products, and we rely on them to file and prosecute patent applications and maintain patents and otherwise protect the licensed
23
Table of Contents
intellectual property. We have not had and do not have primary control over these activities for certain of our patents or patent applications and other intellectual property rights. We cannot be certain that such activities by third parties have been or will be conducted in compliance with applicable laws and regulations or will result in valid and enforceable patents and other intellectual property rights. Pursuant to the terms of the license agreements with some of our licensors, the licensors may have the right to control enforcement of our licensed patents or defense of any claims asserting the invalidity of these patents and even if we are permitted to pursue such enforcement or defense, we will require the cooperation of our licensors. We cannot be certain that our licensors will allocate sufficient resources or prioritize their or our enforcement of such patents or defense of such claims to protect our interests in the licensed patents.
The patent positions of medical device companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. No consistent policy regarding the breadth of claims allowed in patents in these fields has emerged to date in the United States or in many foreign jurisdictions. Both the U.S. Supreme Court and the Court of Appeals for the Federal Circuit have made, and will likely continue to make, changes in how the patent laws of the U.S. are interpreted. In addition, Congress is currently considering legislation that would change provisions of the patent law. We cannot predict future changes in the interpretation of patent laws or changes to patent laws which might be enacted into law. Those changes may materially affect our patents, our ability to obtain patents or the patents and applications of our collaborators and licensors. The patent situation in the medical device and disease diagnostic fields outside the United States is even more uncertain.
Future protection for our proprietary rights is uncertain because legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage. For example:
| • | others may be able to make systems or devices that are similar to ours but that are not covered by the claims of our patents; |
| • | we may not be able to identify potential infringers of our technology due in part to the large number of competitors in the field; |
| • | we might not have been the first to make the inventions covered by our issued patents or pending patent applications; |
| • | we might not have been the first to file patent applications for these inventions; |
| • | our pending patent applications may not result in issued patents; |
| • | our issued patents may not provide us with any competitive advantages or may be held invalid or unenforceable as a result of legal challenges by third parties; |
| • | the claims of our issued patents or patent applications when issued may not cover our device or product candidates; |
| • | there may be dominating patents relevant to our product candidates of which we are not aware; |
| • | there may be prior public disclosures that could invalidate our inventions or parts of our inventions of which we are not aware; |
| • | the laws of foreign countries may not protect our proprietary rights to the same extent as the laws of the United States; and |
| • | we may not develop additional proprietary technologies that are patentable. |
We have a number of foreign patents and applications. However, the laws of some foreign jurisdictions do not protect intellectual property rights to the same extent as laws in the United States, and many companies have encountered significant difficulties in obtaining, protecting and defending such rights in foreign jurisdictions. If we encounter such difficulties or we are otherwise precluded from effectively protecting our intellectual property rights in foreign jurisdictions, our business prospects could be substantially harmed.
We also rely on trade-secret protection to protect our interests in proprietary know-how and for processes for which patents are difficult to obtain or enforce. We may not be able to protect our trade secrets adequately.
24
Table of Contents
We have limited control over the protection of trade secrets used by our licensors, collaborators and suppliers. Although we use reasonable efforts to protect our trade secrets, our employees, consultants, contractors, outside scientific collaborators and other advisors may unintentionally or willfully disclose our information to competitors. Enforcing a claim that a third-party illegally obtained and is using any of our trade secrets is expensive and time consuming, and the outcome is unpredictable. In addition, courts outside the United States are sometimes less willing to protect trade secrets. We rely, in part, on non-disclosure and confidentiality agreements with our employees, consultants and other parties to protect our trade secrets and other proprietary technology. These agreements may be breached and we may not have adequate remedies for any breach. Moreover, others may independently develop equivalent proprietary information, and third parties may otherwise gain access to our trade secrets and proprietary knowledge. Any disclosure of confidential data into the public domain or to third parties could allow our competitors to learn our trade secrets and use the information in competition against us.
We may be subject to claims that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
As is common in our industry, we employ individuals who were previously employed at other molecular diagnostics or medical device companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that these employees, or we, have used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
Risks Related to Our Common Stock and This Offering
The market price of our common stock may be volatile and fluctuate significantly, which could result in substantial losses for investors purchasing common stock in this offering and subject us to litigation.
The offering price for our common stock sold in this offering will be determined based upon negotiations with the underwriters and current market conditions. The public offering price for our common stock may vary from the market price of our common stock at the time of the offering. Among the factors that may cause the market price of our common stock to fluctuate are the risks described in this “Risk Factors” section and other factors, including:
| • | fluctuations in our operating results or the operating results of our competitors; |
| • | changes in estimates of our financial results or recommendations by securities analysts; |
| • | variance in our financial performance from the expectations of securities analysts; |
| • | changes in the estimates of the future size and growth rate of our markets; |
| • | changes in accounting principles or changes in interpretations of existing principles, which could affect our financial results; |
| • | failure of our products to achieve or maintain market acceptance or commercial success; |
| • | conditions and trends in the markets we serve; |
| • | changes in general economic, industry and market conditions; |
| • | success of competitive products and services; |
| • | changes in market valuations or earnings of our competitors; |
| • | changes in our pricing policies or the pricing policies of our competitors; |
| • | announcements of significant new products, contracts, acquisitions or strategic alliances by us or our competitors; |
| • | the timing and outcome of regulatory reviews and approvals of our products; |
| • | changes in legislation or regulatory policies, practices or actions; |
25
Table of Contents
| • | the commencement or outcome of litigation involving our company, our general industry or both; |
| • | recruitment or departure of key personnel; |
| • | changes in our capital structure, such as future issuances of securities or the incurrence of additional debt; |
| • | actual or expected sales of our common stock by the holders of our common stock; and |
| • | the trading volume of our common stock. |
In addition, the stock market in general, the NASDAQ Global Market and the market for diagnostics companies in particular may experience a loss of investor confidence. A loss of investor confidence may result in extreme price and volume fluctuations in our common stock that are unrelated or disproportionate to the operating performance of our business, our financial condition or results of operations. These broad market and industry factors may materially harm the market price of our common stock and expose us to securities class-action litigation. Class-action litigation, even if unsuccessful, could be costly to defend and divert management’s attention and resources, which could further materially harm our financial condition and results of operations.
Future sales of our common stock may depress our share price.
As of March 31, 2011, we had 11,738,233 shares of our common stock outstanding. Sales of a number shares of common stock in the public market, or the expectation of such sales, could cause the market price of our common stock to decline. In addition, our 2010 Plan provides for annual increases in the number of shares available for issuance under the plan. We may also sell additional common stock in subsequent public offerings, which may adversely affect the market price of our common stock.
We have broad discretion in the use of the net proceeds from this offering, and our investment of these proceeds may not yield a favorable return.
We cannot specify with certainty the particular uses of the net proceeds we will receive from this offering, and these uses may vary substantially from our current plans. Our management will have broad discretion in the application of the net proceeds, including for any of the purposes described in “Use of Proceeds.” Accordingly, you will have to rely upon the judgment of our management with respect to the use of the proceeds. Our management may spend a portion or all of the net proceeds from this offering in ways that holders of our common stock may not desire or that may not yield a significant return or any return at all. The failure by our management to apply these funds effectively could harm our business. Pending their use, we may also invest the net proceeds from this offering in a manner that does not produce income or that loses value.
We will incur costs and demands upon management as a result of complying with the laws and regulations affecting public companies in the United States, which may harm our operating results, and failure to achieve and maintain effective internal control over financial reporting in accordance with Section 404 of the Sarbanes-Oxley Act could cause investors to lose confidence in our operating results and in the accuracy of our financial reports and could harm our business and on the price of our common stock.
As a public company in the United States, we will be required, pursuant to Section 404 of the Sarbanes-Oxley Act of 2002, or Section 404, to furnish a report by management on, among other things, the effectiveness of our internal control over financial reporting. Our first report on compliance with Section 404 is expected to be in connection with our financial statements for the fiscal year ending December 31, 2011. The controls and other procedures are designed to ensure that information required to be disclosed by us in the reports that we file with the Securities and Exchange Commission, or SEC, is disclosed accurately and is recorded, processed, summarized and reported within the time periods specified in SEC rules and forms. We are in the early stages of conforming our internal control procedures to the requirements of Section 404 and we may not be able to complete our evaluation, testing and any required remediation needed to comply with Section 404 in a timely
26
Table of Contents
fashion. Our independent registered public accounting firm was not engaged for fiscal year 2010 to perform an audit of our internal control over financial reporting. Our independent registered public accounting firm’s audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of our internal control over financial reporting. Accordingly, no such opinion was expressed. Even if we develop effective controls, these new controls may become inadequate because of changes in conditions or the degree of compliance with these policies or procedures may deteriorate. Even after we develop these new procedures additional weaknesses in our internal control over financial reporting may be discovered. In order to fully comply with Section 404, we will need to retain additional employees to supplement our current finance staff and/or to engage a third party consulting firm to assist in risk assessment, documentation and testing of controls. In addition, in the process of evaluating our internal control over financial reporting we expect that certain of our internal control practices will need to be updated to comply with the requirements of Section 404 and the regulations promulgated thereunder, and we may not be able to do so on a timely basis, or at all. In the event that we are not able to demonstrate compliance with Section 404 in a timely manner, or are unable to produce timely or accurate financial statements, we may be subject to sanctions or investigations by regulatory authorities such as the SEC or the NASDAQ Global Market and investors may lose confidence in our operating results and the price of our common stock could decline. Furthermore, if we or our auditors are unable to certify that our internal control over financial reporting is effective and in compliance with Section 404 we may be subject to sanctions or investigations by regulatory authorities such as the SEC or the NASDAQ Global Market and we could lose investor confidence in the accuracy and completeness of our financial reports, which would materially harm our business and the price of our common stock and our ability to access the capital markets.
Furthermore, as a public company listed in the United States, we incur significant legal, accounting and other expenses. In addition, changing laws, regulations and standards relating to corporate governance and public disclosure, including regulations implemented by the SEC and the NASDAQ Global Market, may increase our legal and financial compliance costs and make some activities more time consuming. These laws, regulations and standards are subject to varying interpretations and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies. We intend to invest resources to comply with evolving laws, regulations and standards, and this investment may result in increased general and administrative expenses and a diversion of management’s time and attention from revenue-generating activities to compliance activities. If notwithstanding our efforts to comply with new laws, regulations and standards, we fail to comply, regulatory authorities may initiate legal proceedings against us and our business may be harmed.
Failure to comply with these rules might also make it more difficult or more expensive for us to obtain certain types of insurance, including director and officer liability insurance, and we might be forced to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. The impact of these events could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors, on committees of our board of directors or as members of senior management.
We do not currently intend to pay dividends on our common stock and, consequently, your ability to achieve a return on your investment will depend on appreciation in the price of our common stock.
We currently intend to invest our future earnings, if any, to fund the development and growth of our business. In addition, pursuant to our Loan and Security Agreement with Square 1 Bank, we are restricted from paying any dividends. The payment of dividends will be at the discretion of our board of directors and will depend on our results of operations, capital requirements, financial condition, future prospects, contractual arrangements, restrictions imposed by applicable law, any limitations on payments of dividends present in any debt agreements we may enter into and other factors our board of directors may deem relevant. If we do not pay dividends, your ability to achieve a return on your investment in our company will depend on future appreciation in the market price of our common stock. There is no guarantee that our common stock will appreciate in value or even maintain the price at which our holders have purchased their common stock.
27
Table of Contents
Provisions of our certificate of incorporation, our bylaws and Delaware law could make an acquisition of us, which may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove the current members of our board and management.
Certain provisions of our certificate of incorporation and bylaws could discourage, delay or prevent a merger, acquisition or other change of control that stockholders may consider favorable, including transactions in which you might otherwise receive a premium for your shares. Furthermore, these provisions could prevent or frustrate attempts by our stockholders to replace or remove members of our board of directors. These provisions also could limit the price that investors might be willing to pay in the future for our common stock, thereby depressing the market price of our common stock. Stockholders who wish to participate in these transactions may not have the opportunity to do so. These provisions:
| • | allow the authorized number of directors to be changed only by resolution of our board of directors; |
| • | provide that our stockholders may only remove our directors for cause; |
| • | establish a classified board of directors, such that not all members of the board of directors may be elected at one time; |
| • | authorize our board of directors to issue without stockholder approval up to 100,000,000 shares of common stock, that, if issued, would dilute our stock ownership and could operate as a “poison pill” to dilute the stock ownership of a potential hostile acquirer to prevent an acquisition that is not approved by our board of directors; |
| • | authorize our board of directors to issue without stockholder approval up to 5,000,000 shares of preferred stock, the rights of which will be determined at the discretion of the board of directors that, if issued, could operate as a “poison pill” to dilute the stock ownership of a potential hostile acquirer to prevent an acquisition that is not approved by our board of directors; |
| • | require that stockholder actions must be effected at a duly called stockholder meeting or by unanimous written consent; |
| • | establish advance notice requirements for stockholder nominations to our board of directors or for stockholder proposals that can be acted on at stockholder meetings; |
| • | limit who may call stockholder meetings; and |
| • | require the approval of the holders of 80% of the outstanding shares of our capital stock entitled to vote in order to amend certain provisions of our certificate of incorporation and bylaws. |
In addition, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which may, unless certain criteria are met, prohibit large stockholders, in particular those owning 15% or more of the voting rights on our common stock, from merging or combining with us for a prescribed period of time.
28
Table of Contents
SPECIAL NOTE REGARDING FORWARD LOOKING STATEMENTS
This prospectus contains forward-looking statements concerning our business, operations and financial performance and condition as well as our plans, objectives and expectations for our business operations and financial performance and condition. Any statements contained herein that are not of historical facts may be deemed to be forward-looking statements. You can identify these statements by words such as “aim,” “anticipate,” “assume,” “believe,” “could,” “due,” “estimate,” “expect,” “goal,” “intend,” “may,” “objective,” “plan,” “predict,” “potential,” “positioned,” “should,” “target,” “will,” “would” and other similar expressions that are predictions of or indicate future events and future trends. These forward-looking statements are based on current expectations, estimates, forecasts and projections about our business and the industry in which we operate and management’s beliefs and assumptions and are not guarantees of future performance or development and involve known and unknown risks, uncertainties and other factors that are in some cases beyond our control. As a result, any or all of our forward-looking statements in this prospectus may turn out to be inaccurate. Factors that may cause such differences include, but are not limited to, the risks described under “Risk Factors,” including:
| • | failure to obtain sufficient funding for the continued development and commercialization of our products; |
| • | failure to expand our menu of diagnostic tests, including the failure to obtain licenses to additional biomarkers on commercially reasonable terms; |
| • | increases in our projected expenditures on sales and marketing, research and development and administrative activities; |
| • | less than anticipated growth in the market for diagnostic testing generally and for the tests we are developing or may develop in the future; |
| • | failure of our products to gain market acceptance domestically or internationally; |
| • | inability to obtain regulatory clearance or approval for any of our products; |
| • | changes in the regulatory environment which may adversely impact the commercialization of our new products and result in significant additional capital expenditures; |
| • | failure to enter into or maintain successful strategic alliances, which may delay the development or commercialization of our products or may result in significant additional expenditures; |
| • | inability to attract or retain skilled personnel for our product development and commercialization efforts; |
| • | inability to protect our intellectual property and operate our business without infringing upon the intellectual rights of others, which could result in litigation and significant expenditures; |
| • | refusal of third-party payors to reimburse our customers for use of diagnostic systems and tests; and |
| • | failure to develop our NexGen System with the capabilities we intend to offer. |
Potential investors and other readers are urged to consider these factors carefully in evaluating the forward-looking statements and are cautioned not to place undue reliance on the forward-looking statements. These forward-looking statements speak only as of the date of this prospectus. Unless required by law, we do not intend to publicly update or revise any forward-looking statements to reflect new information or future events or otherwise. You should, however, review the factors and risks we describe in the reports we will file from time to time with the SEC after the date of this prospectus. See “Where You Can Find More Information.”
29
Table of Contents
MARKET, INDUSTRY AND OTHER DATA
Unless otherwise indicated, information contained in this prospectus concerning our industry and the market in which we operate, including our general expectations and market position, market opportunity and market size, is based on information from various sources including industry publications, third-party market research and publicly available information. This data involves a number of assumptions and limitations, and you are cautioned not to give undue weight to such estimates. While we believe the market position, market opportunity and market size information included in this prospectus is based on reasonable and sound assumptions, such information is inherently imprecise. The Company has paid for market research information provided by L.E.K. and Kalorama which appears in this prospectus.
30
Table of Contents
We expect to receive approximately $ million of net proceeds from the sale of our shares of common stock at the public offering price of $ per share, after deducting underwriting discounts and commissions and estimated offering expenses payable by us, or approximately $ million if the underwriters’ over-allotment option is exercised in full.
We intend to use the net proceeds of this offering for the following purposes:
| • | $ million for the development of a broad menu of tests; |
| • | $ million to fund our planned sales and marketing initiatives; and |
| • | $ million to continue to develop our NexGen system. |
We may also use net proceeds to fund acquisitions of complementary businesses, technologies or licenses and to fund working capital and other general corporate purposes. We have no commitments with respect to any future acquisitions or licenses.
The foregoing expected use of the net proceeds of this offering represents our current intentions based upon our present plans and business condition. The amounts and timing of our actual expenditures may vary significantly and will depend upon numerous factors, including cash flows from operations and the anticipated growth of our business. We will retain broad discretion in the allocation and use of our net proceeds. Pending the allocation of the net proceeds of this offering, we intend to invest the net proceeds of this offering in short-term, interest-bearing obligations, investment grade instruments, certificates of deposit or guaranteed obligations of the U.S. government.
We have never declared or paid any cash dividends on our capital stock and we do not currently anticipate declaring or paying cash dividends on our capital stock in the foreseeable future. In addition, pursuant to our Loan and Security Agreement with Square 1 Bank, we are restricted from paying any dividends. We currently intend to retain all of our future earnings, if any, to finance the operation and expansion of our business. Any future determination relating to our dividend policy will be made at the discretion of our board of directors and will depend on a number of factors, including future earnings, capital requirements, financial conditions, future prospects, contractual restrictions and covenants and other factors that our board of directors may deem relevant.
31
Table of Contents
The following table summarizes the capitalization as of March 31, 2011:
| • | on an actual historical basis for GenMark; and |
| • | on a pro forma basis to give effect to the sale of shares of our common stock in this offering at the public offering price of $ per share, after deducting underwriting discounts and commissions and estimated offering expenses payable by us, which total approximately $ million. |
You should read the following table in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” “Description of Capital Stock” and the financial statements of GenMark and Osmetech and related notes appearing elsewhere in this prospectus.
| GenMark Actual |
Pro Forma(1) | |||||||
| Stockholders’ equity: |
||||||||
| Common Stock $0.0001 par value; 100,000,000 authorized shares, actual and pro forma; 11,738,233 shares issued and outstanding, actual; shares issued and outstanding, pro forma |
$ | 1,172 | ||||||
| Preferred Stock, $0.0001 par value; 5,000,000 authorized, none issued, actual and pro forma |
— | |||||||
| Additional paid-in capital |
166,483,672 | |||||||
| Accumulated deficit |
(151,134,719 | ) | ||||||
| Accumulated other comprehensive loss |
(449,957 | ) | ||||||
| Total stockholders’ equity |
14,900,168 | |||||||
| Total capitalization |
$ | 14,900,168 | ||||||
| (1) | Pro forma reflects the sale of shares of GenMark common stock in this offering at the public offering price of $ per share, after deducting underwriting discounts and commissions and estimated offering expenses payable by us. |
The above table excludes the following:
| • | 1,314,975 shares of common stock issuable upon exercise of options outstanding as of March 31, 2011, at a weighted average exercise price of approximately $6.06 per share; |
| • | 88,317 shares of common stock issuable upon exercise of warrants outstanding as of March 31, 2011, at a weighted average exercise price of approximately $9.98 per share; and |
| • | 701,957 shares of common stock which will be available for future grant or issuance under our 2010 Equity Incentive Plan, and the annual increases in the number of shares authorized under this plan. |
32
Table of Contents
DILUTION
If you invest in our common stock, your interest in our net tangible book value will be diluted to the extent of the difference between the public offering price and the net tangible book value per share of our common stock immediately after the completion of this offering. Dilution results from the fact that the public offering price is substantially in excess of the book value per share attributable to the existing stockholders for the presently outstanding stock.
Our net tangible book value as of March 31, 2011 was approximately $14,832,126, or $1.26 per share. Net tangible book value per share is determined by dividing the amount of our total tangible assets less our total liabilities by the number of shares of common stock totaling 11,738,233 shares.
After giving effect to the sale of shares of common stock in this offering at the public offering price of $ per share, and after deducting underwriting discounts and commissions and our estimated offering expenses totaling approximately $ million, our pro forma net tangible book value as of March 31, 2011 would have been approximately $ million, or $ per share.
This amount represents an immediate increase in pro forma net tangible book value of $ per share and an immediate dilution of $ per share to new investors. The following table illustrates this calculation on a per share basis:
| Public offering price per share |
$ | |||||||
| Net tangible book value per share of common stock as of March 31, 2011 |
$ | |||||||
| Pro forma increase per share attributable to the offering |
||||||||
| Pro forma net tangible book value per share of common stock after this offering |
||||||||
| Dilution per share to new investors |
$ | |||||||
If the underwriters exercise an over-allotment option of shares in full, our pro forma net tangible book value will increase to $ per share, representing an increase to existing holders of $ per share, and there will be an immediate dilution of $ per share to new investors after giving effect to the underwriting discount of %.
The following table summarizes, on a pro forma basis, as of March 31, 2011, after giving effect to the sale of shares in this offering and the pro forma adjustments referred to above, the total number of shares of our common stock purchased from us and the total consideration and average price per share paid by existing stockholders and by new investors at the offering price of $ per share:
| Shares Purchased | Total Consideration | Average Price Per Share |
||||||||||||||||||
| Number | Percent | Amount | Percent | |||||||||||||||||
| Existing stockholders |
% | % | $ | |||||||||||||||||
| New investors |
||||||||||||||||||||
| Total |
100 | % | 100 | % | $ | |||||||||||||||
If the underwriters exercise an over-allotment option to purchase shares in full, the following will occur:
| • | the pro forma percentage of shares of our common stock held by existing stockholders will decrease to approximately % of the total number of pro forma shares of our common stock outstanding after this offering; and |
| • | the pro forma number of shares of our common stock held by new public investors will increase to , or approximately % of the total pro forma number of shares of our common stock outstanding after this offering. |
33
Table of Contents
The above discussion and tables exclude:
| • | 1,314,975 shares of common stock issuable upon exercise of options outstanding as of March 31, 2011, at a weighted average exercise price of $6.06 per share; |
| • | 88,317 shares of common stock issuable upon exercise of warrants outstanding as of March 31, 2011, at a weighted average exercise price of approximately $9.98 per share; and |
| • | 701,957 shares of common stock available for future grant or issuance under our 2010 Plan, and the annual increases in the number of shares authorized under this plan. |
The preceding discussion and tables assume no exercise of any options and warrants outstanding as of March 31, 2011. If all of our outstanding options and warrants as of March 31, 2011 were exercised, the pro forma net tangible book value per share after this offering would be $ per share, representing an increase to existing holders of $ per share, and there will be an immediate dilution of $ per share to new investors.
34
Table of Contents
SELECTED CONSOLIDATED FINANCIAL DATA
The selected consolidated financial data as of and for the year ended December 31, 2010, and as of and for the three months ended March 31, 2011, relate to GenMark and its consolidated subsidiaries. The selected consolidated financial data as of and for the years ended December 31, 2009, 2008, 2007 and 2006, and for the three months ended March 31, 2010, relate to Osmetech and its consolidated subsidiaries, which became subsidiaries of GenMark in a reorganization under the applicable laws of the United Kingdom in 2010. We have derived the following consolidated statement of operations data for the three years ended December 31, 2010 and the consolidated balance sheet data as of December 31, 2010 and 2009 from our audited consolidated financial statements included elsewhere in this prospectus. We have derived the consolidated statement of operations data for the year ended December 31, 2007, and the consolidated balance sheet data as of December 31, 2008 from the audited consolidated financial statements of Osmetech, which are not included in this prospectus. The consolidated statements of operations data for the three months ended March 31, 2011 and 2010, and the consolidated balance sheet data as of March 31, 2011, are derived from GenMark’s unaudited consolidated financial statements included elsewhere in this prospectus. We have prepared the unaudited financial information on the same basis as the audited consolidated financial statements and have included, in our opinion, all adjustments, consisting only of normal recurring adjustments, that we consider necessary for a fair presentation of the financial information set forth in those statements. Our historical results are not necessarily indicative of the results that may be expected in the future.
The selected consolidated financial statement of operations data of Osmetech presented below for the year ended December 2006 and the selected consolidated balance sheet data of Osmetech as of December 31, 2007 and 2006 have been derived from unaudited consolidated financial information of Osmetech, not included in this prospectus, and have been prepared by Osmetech in accordance with U.S. GAAP.
35
Table of Contents
The results for the periods shown below are not necessarily indicative of the results to be expected for any future periods, and our interim results are not necessarily indicative of the results to be expected for the full fiscal year. The selected consolidated financial data should be read together with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and with the consolidated financial statements and unaudited condensed consolidated financial statements of GenMark and Osmetech and related notes included elsewhere in this prospectus.
| Three months ended March 31, |
For the year ended December 31, | |||||||||||||||||||||||||||
| 2011 | 2010 | 2010 | 2009 | 2008 | 2007 | 2006 | ||||||||||||||||||||||
| Consolidated Statements of Operations Data: |
||||||||||||||||||||||||||||
| Revenue: |
||||||||||||||||||||||||||||
| Product sales |
$ | 692,739 | $ | 384,249 | $ | 2,340,996 | $ | 910,527 | $ | 559,592 | $ | 234,099 | $ | 50,500 | ||||||||||||||
| License revenue |
71,664 | 15,015 | 163,872 | 87,889 | 87,500 | 107,500 | 41,062 | |||||||||||||||||||||
| Total revenue |
764,403 | 399,264 | 2,504,868 | 998,416 | 647,092 | 341,599 | 91,562 | |||||||||||||||||||||
| Cost of sales |
1,643,456 | 567,396 | 4,377,701 | 4,332,299 | 3,237,869 | 2,624,589 | 2,331,430 | |||||||||||||||||||||
| Gross loss |
(879,053 | ) | (168,132 | ) | (1,872,833 | ) | (3,333,883 | ) | (2,590,777 | ) | (2,282,990 | ) | (2,239,868 | ) | ||||||||||||||
| Operating expenses: |
||||||||||||||||||||||||||||
| Sales and marketing |
1,130,389 | 1,058,285 | 4,282,521 | 3,181,762 | 3,393,665 | 2,220,098 | 905,962 | |||||||||||||||||||||
| Research and development |
2,528,252 | 1,453,759 | 6,522,112 | 5,633,717 | 13,423,679 | 12,554,236 | 10,606,562 | |||||||||||||||||||||
| General and administrative |
2,111,336 | 2,167,264 | 7,353,802 | 8,288,762 | 9,632,708 | 8,895,796 | 9,781,509 | |||||||||||||||||||||
| Total operating expenses |
5,769,977 | 4,679,308 | 18,158,435 | 17,104,241 | 26,450,052 | 23,670,130 | 21,294,033 | |||||||||||||||||||||
| Loss from operations |
(6,649,030 | ) | (4,847,440 | ) | (20,031,268 | ) | (20,438,124 | ) | (29,040,829 | ) | (25,953,120 | ) | (23,533,901 | ) | ||||||||||||||
| Other (expense) income: |
||||||||||||||||||||||||||||
| Foreign exchange gain (loss) |
11,899 | (1,110 | ) | (1,110 | ) | 303,523 | 504,921 | — | — | |||||||||||||||||||
| Interest income |
6,258 | 4,654 | (582 | ) | 33,222 | 420,011 | 1,715,211 | 522,293 | ||||||||||||||||||||
| Therapeutic discovery credit |
— | — | 1,645,292 | — | — | — | — | |||||||||||||||||||||
| Total other income |
18,157 | 3,544 | 1,643,600 | 336,745 | 924,932 | 1,715,211 | 522,293 | |||||||||||||||||||||
| Loss before income taxes |
(6,630,873 | ) | (4,843,896 | ) | (18,387,668 | ) | (20,101,379 | ) | (28,115,897 | ) | (24,237,909 | ) | (23,011,608 | ) | ||||||||||||||
| (Provision) benefit for income taxes |
(10,968 | ) | (5,049 | ) | (15,324 | ) | 138,770 | (246,736 | ) | 300,214 | 231,637 | |||||||||||||||||
| Net loss from continuing operations |
$ | (6,641,841 | ) | $ | (4,848,945 | ) | $ | (18,402,992 | ) | $ | (19,962,609 | ) | $ | (28,362,633 | ) | $ | (23,937,695 | ) | $ | (22,779,971 | ) | |||||||
| Net loss per common share from continuing operations (basic and diluted) |
$ | (0.56 | ) | $ | (0.68 | ) | $ | (1.88 | ) | $ | (0.02 | ) | $ | (0.12 | ) | $ | (0.12 | ) | $ | (0.14 | ) | |||||||
| Weighted average shares used in net loss per common share |
11,771,014 | 7,113,922 | 9,796,588 | 1,041,054,350 | 231,928,699 | 202,934,689 | 165,457,028 | |||||||||||||||||||||
| Unaudited pro forma net loss per common share from continuing operations, basic and diluted |
||||||||||||||||||||||||||||
| Weighted average shares used in unaudited pro forma per share amounts |
||||||||||||||||||||||||||||
| As of March 31, | As of December 31, | |||||||||||||||||||||||
| 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | |||||||||||||||||||
| Balance Sheet Data: |
||||||||||||||||||||||||
| Cash and cash equivalents |
$ | 17,054,095 | $ | 18,329,079 | $ | 16,482,818 | $ | 8,822,458 | $ | 27,619,715 | $ | 13,874,798 | ||||||||||||
| Total assets |
21,955,353 | 24,925,509 | 19,333,477 | 15,175,215 | 33,233,621 | 26,718,736 | ||||||||||||||||||
| Long-term obligations |
2,622,644 | 612,932 | 795,334 | 769,237 | 720,355 | 339,144 | ||||||||||||||||||
| Total liabilities |
7,055,185 | 3,858,091 | 4,008,659 | 5,237,946 | 3,265,933 | 8,359,361 | ||||||||||||||||||
| Accumulated deficit |
(151,134,719 | ) | (144,492,881 | ) | (126,089,889 | ) | (106,127,280 | ) | (77,764,647 | ) | (88,309,444 | ) | ||||||||||||
| Total stockholders’ equity |
14,900,168 | 21,067,418 | 15,324,818 | 9,937,269 | 29,967,688 | 18,359,375 | ||||||||||||||||||
36
Table of Contents
MANAGEMENT’S DISCUSSION AND ANALYSIS OF RESULTS OF OPERATIONS AND FINANCIAL CONDITION
You should read the following in conjunction with the “Selected Consolidated Financial Information” and the consolidated financial statements of GenMark and the related notes thereto that appear elsewhere in this prospectus. In addition to historical information, the following discussion and analysis includes forward-looking information that involves risks, uncertainties and assumptions. Actual results and the timing of events could differ materially from those anticipated by these forward-looking statements as a result of many factors, including those discussed under “Risk Factors” elsewhere in this prospectus. See also “Special Note Regarding Forward-Looking Statements” included elsewhere in this prospectus.
Overview
GenMark was formed by Osmetech in Delaware in February 2010 and had no operations prior to its initial public offering which was completed in June 2010. Immediately prior to the closing of the initial public offering, GenMark acquired all of the outstanding ordinary shares of Osmetech in a reorganization under the applicable laws of the United Kingdom. As a result of the reorganization, all of the issued ordinary shares in Osmetech were cancelled in consideration of (i) the issuance of common stock of GenMark to the former shareholders of Osmetech and (ii) the issuance of new shares in Osmetech to GenMark. Following the reorganization, Osmetech became a wholly-owned subsidiary of GenMark, and the former shareholders of Osmetech held shares of GenMark. Any historical discussion of GenMark relates to Osmetech and its consolidated subsidiaries prior to the reorganization.
We are a molecular diagnostics company focused on developing and commercializing our proprietary eSensor detection technology. Our proprietary electrochemical technology enables fast, accurate and highly sensitive detection of up to 72 distinct biomarkers in a single sample. Our XT-8 system received 510(k) clearance from the FDA and is designed to support a broad range of molecular diagnostic tests with a compact and easy-to-use workstation and self-contained, disposable test cartridges. Within 30 minutes of receipt of an amplified DNA sample, our XT-8 system produces clear and accurate results. Our XT-8 system supports between one and three analyzers. Each analyzer holds up to eight independent test cartridges, resulting in the XT-8 system supporting up to 24 test cartridges, each of which can be run independently, resulting in a convenient and flexible workflow for our target customers, which are hospitals and reference laboratories. As of March 31, 2011, we had an installed base of 102 analyzers, or placements, with our customers.
We have developed four diagnostic tests for use with our XT-8 system and expect to expand this test menu by introducing two to four new tests annually. Three of our diagnostic tests have received FDA clearance, including our Cystic Fibrosis Genotyping Test, which detects pre-conception risks of cystic fibrosis, our Warfarin Sensitivity Test, which determines an individual’s ability to metabolize the oral anticoagulant warfarin, and our Thrombophilia Risk Test, which detects an individual’s increased risk of blood clots. Our eSensor technology has demonstrated 100% accuracy in clinical studies compared to DNA sequencing in our Cystic Fibrosis Genotyping Test, our Warfarin Sensitivity Test and our Thrombophilia Risk Test. We have also developed a Respiratory Viral Panel Test, which detects the presence of major respiratory viruses and is labeled for IUO. We intend to seek FDA clearance for our Respiratory Viral Panel Test in 2011. We also have a pipeline of several additional potential products in different stages of development or design, including diagnostic tests for an individual’s sensitivity to Plavix, a commonly prescribed anti-coagulant, and for mutations in a gene known as K-ras, which is predictive of an individual’s response rates to certain prescribed anti-cancer therapies.
We are also developing our next-generation platform, the NexGen system. We are designing the NexGen system (formerly referred to as the AD-8 system) to integrate automated nucleic acid extraction and amplification with our eSensor detection technology to enable technicians using the NexGen system to be able to place a raw or a minimally prepared patient sample into our test cartridge and obtain results without any additional steps. This sample-to-answer capability is enabled by the robust nature of our eSensor detection
37
Table of Contents
technology, which is not impaired by sample impurities that we believe hinder competing technologies. We are designing our NexGen system to further simplify workflow and provide powerful, cost-effective molecular diagnostics solutions to a significantly expanded group of hospitals and reference laboratories.
Since inception, we have incurred net losses from continuing operations each year, and we expect to continue to incur losses for the foreseeable future. Our losses attributable to continuing operations for the three months ended March 31, 2011 and the fiscal years ended December 31, 2010, 2009 and 2008 were approximately $6.6 million, $18.4 million, $20.0 million and $28.4 million, respectively. As of March 31, 2011, we had an accumulated deficit of $151.1 million. Our operations to date have been funded principally through revenue, sales of capital stock and sales of our previous businesses. We expect to incur increasing expenses over the next several years, principally to develop additional diagnostic tests, as well as to further increase our spending to manufacture, sell and market our products.
Financial Results Overview
Revenue
Revenue from continuing operations includes product sales, principally of our eSensor Cystic Fibrosis Genotyping Test and, to a lesser extent, our Warfarin Sensitivity Test, for use with our XT-8 system and our predecessor eSensor 4800 System. We primarily place our XT-8 system with customers through a reagent rental agreement, under which customers commit to purchasing minimum quantities of test cartridges over a period of one to three years. We also offer our XT-8 system for sale, however, for the three months ended March 31, 2011 and the year ended December 31, 2010, we sold only one and ten XT-8 systems, respectively, to customers which included the sale of thirteen total analyzers, or placements.
Revenue also includes licensing revenue from the out-licensing of our electrochemical detection technology. In addition, revenue generated from service agreements recognized using the proportional performance method of accounting is included in this category. We may enter into additional sub-licenses of our technology generating additional revenue, but do not anticipate that this will provide a significant portion of our future revenue.
Our growth plans focus on both reagent rental agreements and system sales of our current XT-8 system and our next-generation NexGen system that is currently under development. We plan to expand our base of customers and systems as well as adding more tests for use with our systems. We believe these developments will drive accelerated use of our test cartridges, which we expect to be our primary source of revenue.
Cost of Sales
Cost of sales includes the cost of materials, direct labor and manufacturing overhead costs used in the manufacture of our consumable test kits for our XT-8 system and our predecessor eSensor 4800 System, including royalties on product sales. Cost of sales also includes depreciation on revenue generating systems that have been placed with our customers under a reagent rental agreement, and amortization of licenses related to our test cartridges.
Our XT-8 systems are procured from a contract manufacturer and generally capitalized as fixed assets and depreciated on a straight line basis over their useful life as a charge to cost of sales. We expect our cost of sales to increase as we place additional XT-8 systems and manufacture and sell an increasing menu of accompanying diagnostic tests.
We manufacture our test cartridges in our facility and have significant capacity for expansion. This underutilized capacity results in a high cost of sales relative to revenue, resulting in a gross loss. We believe cost of sales as a percentage of revenue will decrease as our sales of test cartridges grow.
38
Table of Contents
Sales and Marketing Expenses
Sales and marketing expenses include those costs associated with our direct sales force, sales management, marketing, technical support and business development departments. These expenses primarily consist of salaries, commissions, benefits, share-based compensation, travel, advertising and promotions. We expect sales and marketing costs to increase as we scale up our commercial efforts to increase our customer base.
Research and Development Expenses
Research and development expenses primarily include expenses related to the development of our XT-8 system and its predecessor eSensor 4800 System, including the detection system and the test cartridges. These expenses also include clinical study expenses incurred in the process of preparing for FDA clearance for these systems and test cartridges. The expenses primarily consist of salaries, benefits, share-based compensation costs, outside design and consulting services, laboratory supplies, contract research organizations, clinical study supplies and facility costs.
We expense all research and development costs in the periods in which they are incurred. We expect research and development costs to increase as we develop more advanced systems and increase the development of new tests for our XT-8 system.
General and Administrative Expenses
Our general and administrative expenses include our executive, accounting and finance, information technology, legal, intellectual property, human resource and investor relations departments. These expenses consist primarily of salaries, benefits, share-based compensation costs, independent auditor costs, legal fees, consultants, travel, insurance, relocation, and public company expenses such as stock transfer agent fees and listing fees for the Alternative Investment Market, or AIM, of the London Stock Exchange and the NASDAQ Global Market.
Foreign Exchange Gains and Losses
Transactions in currencies other than the functional currency are translated at the prevailing rates on the dates of the transaction. Foreign exchange gains and losses arise from differences in exchange rates during the period between the date a transaction denominated in a foreign currency is consummated and the date on which it is settled or translated. Exchange gains and losses also included those arising on cash balances held by Osmetech denominated in currencies other than its functional currency, the British pound. Since the initial public offering, the functional currency of GenMark has been the U.S. dollar.
Interest Income (expense)
Interest income (expense) includes interest earned on our cash and cash equivalents less interest accrued on other liabilities.
Benefit (Provision) for Income Taxes
We account for income taxes in accordance with ASC Topic 740, Income Taxes. Under ASC Topic 740, deferred taxes are provided on an asset and liability method whereby deferred tax assets are recognized for deductible temporary differences and operating loss carryforwards. Deferred tax liabilities are recognized for taxable temporary differences. Temporary differences are the differences between the reported amounts of assets and liabilities and the tax bases. Deferred tax assets are reduced by a valuation allowance when, in the opinion of management, it is more-likely-than-not that some portion or all of the deferred tax assets will not be realized. Deferred tax assets and liabilities are adjusted for the effects of changes in tax laws and rates on the date of enactment.
39
Table of Contents
Critical Accounting Polices and Significant Judgments and Estimates
Revenue
We recognize revenue from product sales and contract arrangements, net of discounts and sales related taxes. We recognize revenue from product sales when there is persuasive evidence that an arrangement exists, delivery has occurred, the price is fixed or determinable and collectability is reasonably assured. Where applicable, all revenue is stated net of sales taxes and trade discounts.
We offer customers the choice to either purchase a system outright or to receive a system free of charge in exchange for an annual minimum purchase commitment for test cartridges. When a system is sold, revenue is generally recognized upon shipment of the unit. When a system is placed free of charge under a “reagent rental” agreement, we retain title to the equipment and the system remains capitalized on the balance sheet under property and equipment. Under our reagent rental agreements, we retain the right to access or replace the systems at any time and our customers pay an additional system rental fee for each test cartridge purchased. The reagent rental fee varies based on the monthly volume of test cartridges purchased.
We sell our durable systems and disposable test cartridges through a direct sales force in the United States. Components are individually priced and can be purchased separately or together. Revenue on system and test cartridge sales is recognized upon shipment, which is when title and the risk of loss and rewards of ownership have been transferred to the customer and there are no other post-shipment obligations.
During the three months ended March 31, 2011 and the year ended December 31, 2010, we sold one and ten XT-8 systems, respectively, to customers which included the sale of thirteen total analyzers, or placements.
Revenue related to royalties received from licenses is recognized evenly over the contractual period to which the license relates. Revenue from service agreements is recognized using the proportional performance method of accounting.
Shipping and handling costs are expensed as incurred and included in cost of product sales. In those cases where we bill shipping and handling costs to customers, the amounts billed are classified as revenue.
Property and Equipment
Property, equipment and leasehold improvements are recorded at cost and depreciated using the straight-line method over the assets’ estimated useful lives, which are noted below. We generally capitalize our XT-8 systems, and previously the predecessor eSensor 4800 systems, and provide these to customers for no charge. Each category of property and equipment is analyzed to determine its useful life. We look at the manufacturers’ estimates of useful life and adjust these for actual experience in our operating environment. Useful lives are reviewed periodically and shortened if circumstances dictate a change.
| Machinery and laboratory equipment | 3 - 5 years | |
| Systems at customer location | 3 years | |
| Office equipment | 2 - 4 years | |
| Leasehold improvements | over the shorter period of the life of the lease or the useful economic life of the asset |
During 2009, our estimate of the useful life of our systems was changed from five years to three years. This estimate was revised due to a change in our strategy to accelerate the development of our next-generation system and did not have a significant impact on our results for the period.
Impairment of Long-Lived Assets
We assess the recoverability of long-lived assets, including intangible assets and systems at customer locations by periodically evaluating the carrying value of such assets whenever events or changes in circumstances indicate that the carrying amount of such assets may not be recoverable. If impairment is
40
Table of Contents
indicated, we write down the carrying value of the asset to the estimated fair value. This fair value is usually determined based on an estimate of future discounted cash flows. The primary cause for us to consider systems at customer locations for impairment is evidence that customers are not ordering the minimum quantities set forth in their reagent rental agreement. For impairment of systems at customers’ locations, which are assessed separately for each customer, we analyze the recoverability based on historical and estimated future sales of test cartridges to each customer. In the three months ended March 31, 2011 and the year ended December 31, 2010, no impairment charges were recorded. In the year ended December 31, 2009, we recorded an impairment against systems of $865,389, which was recorded within cost of sales ($665,718), sales and marketing ($129,712) and research and development ($69,959).
Share-Based Compensation
We have granted our options with an exercise price equal to the closing price of our common stock on the NASDAQ Global Market on each grant date. We use the Black-Scholes option-pricing model as the method for determining the estimated fair value of stock options. The Black-Scholes model requires the use of highly subjective and complex assumptions which determine the fair value of share-based awards, including the option’s expected term and the price volatility of the underlying stock. These assumptions include:
| • | Expected Term. Our expected term represents the period that our share-based awards are expected to be outstanding and is determined by evaluating past experience. |
| • | Expected Volatility. Expected volatility represents the volatility in our stock price expected over the expected term of the option. |
| • | Expected Dividend. The Black-Scholes valuation model calls for a single expected dividend yield as an input. We assumed no dividends as we have never paid dividends and have no current plans to do so. |
| • | Risk-Free Interest Rate. The risk-free interest rate used in the Black-Scholes valuation method is based on published government rates in effect at the time of grant for periods corresponding with the expected term of option. |
| • | Estimated Forfeitures. The estimated forfeiture rate is determined based on our historical forfeiture rates. We will monitor actual expenses and periodically update the estimate. |
| • | Valuation. Our board of directors determined the fair value of our common stock to be equivalent to the closing prices on the NASDAQ Global Market. GenMark’s shares trade on the NASDAQ Global Market on a daily basis and reflect prices that investors are willing to pay for GenMark’s shares. |
Income Taxes
Our income tax expense, deferred tax assets and liabilities and reserves for unrecognized tax benefits reflect management’s best assessment of estimated future taxes to be paid. We are subject to income taxes in both the United States and the United Kingdom. Significant judgments and estimates are required in determining the consolidated income tax expense.
We believe that it is more likely than not that the benefit from certain U.S. federal and U.S. state net operating loss carryforwards will not be realized. In recognition of this risk, we have provided a valuation allowance of approximately $13.1 million on the deferred tax assets relating to these net operating loss carryforwards and other deferred tax assets. If our assumptions change and we determine we will be able to realize these net operating losses, the tax benefits relating to any reversal of the valuation allowance on deferred tax assets at December 31, 2010 will be accounted for as a reduction of income tax expense.
Changes in tax laws and rates could also affect recorded deferred tax assets and liabilities in the future. Management is not aware of any such changes that would have a material effect on our results of operations, cash flows or financial position.
We recognize tax liabilities in accordance with ASC Topic 740 and we adjust these liabilities when our judgment changes as a result of the evaluation of new information not previously available. Due to the
41
Table of Contents
complexity of some of these uncertainties, the ultimate resolution may result in a payment that is materially different from our current estimate of the tax liabilities. These differences will be reflected as increases or decreases to income tax expense in the period in which they are determined.
Recent Accounting Pronouncements
In October 2009, authoritative guidance was provided on revenue arrangements with multiple deliverables. The guidance amended the accounting standards for multiple deliverable revenue arrangements to: (i) provide updated guidance on whether multiple deliverables exist, how the deliverables in an arrangement should be separated, and how the consideration should be allocated; (ii) require an entity to allocate revenue in an arrangement using estimated selling prices, or ESP of deliverables if a vendor does not have vendor-specific objective evidence of selling price, or VSOE or third-party evidence of selling price, or TPE; and (iii) eliminate the use of the residual method and require an entity to allocate revenue using the relative selling price method.
Arrangements that contain multiple deliverables include sales of systems and test cartridges. These are accounted for as separate units of accounting if the following criteria are met: (i) the delivered item or items have value to the customer on a standalone basis and (ii) for an arrangement that includes a general right of return relative to the delivered item(s), delivery or performance of the undelivered item(s) is considered probable and substantially in our control. We consider a deliverable to have standalone value if the item is sold separately or if the item could be resold by the customer. Our revenue arrangements generally do not include a right of return relative to delivered products. We sold our first systems in 2010. We elected to early adopt the new accounting guidance because we are able to meet the new separation criteria and have applied it to all applicable revenue arrangements entered into or materially modified beginning January 1, 2010.
Results of Operations—March 2011 compared to March 2010
Revenue
| March 31, | ||||||||||||||||
| 2011 | 2010 | $ Change | % Change | |||||||||||||
| Three months ended |
$ | 764,403 | $ | 399,264 | $ | 365,139 | 91 | % | ||||||||
The increase in revenue for the three month period ended March 31, 2011 as compared to the three month period ended March 31, 2010 was primarily due to a $331,000 increase in reagent revenue driven by the increase in number of our installed base of systems as well as an expanded menu of tests available for sale.
Cost of Sales and Gross Loss
| March 31, | ||||||||||||||||
| 2011 | 2010 | $ Change | % Change | |||||||||||||
| Cost of Sales-three months ended |
$ | 1,643,456 | $ | 567,396 | $ | 1,076,060 | 190 | % | ||||||||
| Gross Loss -three months ended |
$ | (879,053 | ) | $ | (168,132 | ) | $ | (710,921 | ) | 423 | % | |||||
The increase in cost of sales for the three months ended March 31, 2011 compared to the three months ended March 31, 2010 was due to $605,000 in increased expenses related directly to the increase in reagent and system shipments, as well as costs incurred in relocating our manufacturing facilities from Pasadena to our Carlsbad location in 2011, including $314,000 in higher payroll, benefits and temporary labor costs and $188,000 of additional facility-related charges. The increase in gross loss resulted primarily from costs associated with our expanded product offerings which will be reduced as a percentage of sales as our sales volume increases, and the one-time expense of relocating our manufacturing facility.
42
Table of Contents
Operating Expenses
Sales and Marketing
| March 31, | ||||||||||||||||
| 2011 | 2010 | $ Change | % Change | |||||||||||||
| Three months ended |
$ | 1,130,389 | $ | 1,058,285 | $ | 72,104 | 7 | % | ||||||||
The increase in sales and marketing expense was driven primarily by increased costs for product samples sent to prospective customers.
General and Administrative
| March 31, | ||||||||||||||||
| 2011 | 2010 | $ Change | % Change | |||||||||||||
| Three months ended |
$ | 2,111,336 | $ | 2,167,264 | $ | (55,928 | ) | (3 | )% | |||||||
General and administrative expense decreased for the three months ended March 31, 2011 compared to the three months ended March 31, 2010 due to $925,000 in lower payroll, severance and other headcount related costs associated with our former U.K. operations and lower facility-related costs. These reductions were offset by a $370,000 increase in professional services fees primarily related to corporate restructuring, and $550,000 in additional consulting, share-based compensation and recruiting costs for executive services.
Research and Development
| March 31, | ||||||||||||||||
| 2011 | 2010 | $ Change | % Change | |||||||||||||
| Three months ended |
$ | 2,528,252 | $ | 1,453,759 | $ | 1,074,493 | 74 | % | ||||||||
The increase in research and development expense for the three months ended March 31, 2011 was due to $150,000 in higher payroll expense due to increased headcount, severance related expenses of $239,000, increased development supplies and clinical trial costs of $160,000, a $133,000 increase in intellectual property related costs, costs of $250,000 incurred to obtain regulatory certification for our Carlsbad manufacturing facility and $117,000 of increased facility related costs in 2011 as compared to the same period in 2010. We expect research and development expenses to increase as we continue to focus on the development of our NexGen system.
Other Income, Net
| March 31, | ||||||||||||||||
| 2011 | 2010 | $ Change | % Change | |||||||||||||
| Three months ended |
$ | 18,157 | $ | 3,544 | $ | 14,613 | 412 | % | ||||||||
Interest and other income (expense) represent earnings on cash and cash equivalents and foreign currency gains or losses. The increase in revenue for the three months ended March 31, 2011 as compared to the same period in 2010 was due primarily to a foreign currency gain related to accounts receivable from the United Kingdom in 2010 that was received in 2011 at higher foreign currency translation rates. During the second quarter of 2010, we shut down our U.K. facility and changed our functional currency to the U.S. dollar. There are no remaining material operations in the United Kingdom.
Provision for Income Taxes
| March 31, | ||||||||||||||||
| 2011 | 2010 | $ Change | % Change | |||||||||||||
| Three months ended |
$ | 10,968 | $ | 5,049 | $ | 5,919 | 117 | % | ||||||||
43
Table of Contents
Due to our losses we have only recorded tax provisions or benefits related to interest on uncertain tax positions, minimum tax payments and refunds.
Results of Operations—2010 compared to 2009
Revenue
Revenue increased $1.5 million, or 151%, to $2.5 million for the year ended December 31, 2010 compared to $998,000 for the year ended December 31, 2009. Product sales increased $1.4 million, or 157%, to $2.3 million for the year ended December 31, 2010 compared to $911,000 for the year ended December 31, 2009. License and other revenue increased $76,000 to $164,000, or 86%, for the year ended December 31, 2010, due to increased service revenue, compared to $88,000 for the year ended December 31, 2009. The increase in product revenue was primarily driven by increased reagent revenues as well as system sales and other product revenue and was due to an increase in our installed base of systems and an expanded menu of tests available for sale. License revenue increased predominantly due to a collaboration agreement executed in conjunction with a clinical trial for Warfarin.
Cost of Sales and Gross Loss
Cost of sales increased $46,000, or 1%, to $4.4 million for the year ended December 31, 2010 compared to $4.3 million for the year ended December 31, 2009. The increase was primarily due to the increase in reagent and system shipments. Gross loss decreased $1.5 million or 44% to $1.9 million for the year ended December 31, 2010 compared to a gross loss of $3.3 million in 2009. The decrease was due to higher revenues in 2010 but not a corresponding increase in cost of sales due to increased capacity utilization.
Sales and Marketing
Sales and marketing expense increased $1.1 million, or 35% to $4.3 million for the year ended December 31, 2010, compared to $3.2 million for the year ended December 31, 2009. The increase was driven by higher payroll costs. We built our direct sales force during 2010 and expect these costs to increase during 2011 and beyond.
Research and Development
Research and development expense increased $888,000, or 16%, to $6.5 million for the year ended December 31, 2010 compared to $5.6 million for the year ended December 31, 2009. The increase was due to higher payroll costs, including relocation and recruiting fees and increased usage of project supplies.
General and Administrative
General and administrative expense decreased $935,000, or 11%, to $7.4 million for the year ended December 31, 2010 compared to $8.3 million for year ended December 31, 2009. The decline was due to reduced facility costs and professional fees offset by relocation costs related to our move from Pasadena to Carlsbad.
Foreign Exchange
We incurred a foreign exchange loss for the year ended December 31, 2010 of $1,000 as compared to a gain of $304,000 for the year ended December 31, 2009. The gain was due to the settlement of U.S. dollar liabilities during the year as the U.S. dollar weakened against the British pound combined with the benefit of maturing U.S. dollar forward contracts which were held by us during the period. There were few foreign exchange transactions during 2010.
Interest Income (Expense)
Interest income (expense), declined $34,000 to $1,000 net interest expense for the year ended December 31, 2010 compared to $33,000 net interest income for the year ended December 31, 2009, due to lower cash balances during the year as well as increased expense on a tax liability.
44
Table of Contents
Other Income (Therapeutic Discovery Credit)
We recorded other income related to the Therapeutic Discovery Credit of $1.6 million for the year ended December 31, 2010. In July 2010, we applied for certification of qualified investments eligible for credits and grants under the qualifying therapeutic discovery project program for the years ended December 31, 2009 and December 31, 2010. The $1.6 million in grant applications were for expenses incurred in 2010 and 2009. In February 2011, we received $561,000 for 2009 expenses and $1.1 million for 2010 expenses.
These development projects included the NexGen system (formerly referred to as the AD-8 system), K-ras mutation cancer treatment, Plavix Sensitivity Drug, Warfarin Sensitivity Test, Thrombophilia Risk Test, Respiratory Viral Panel and Cystic Fibrosis Genotyping. In November 2010, we were notified that we were awarded a total of $1.6 million under the program. As of December 31, 2010, We recorded the $1.6 million tax credit as an Other Current Assets on the Balance Sheet with a corresponding credit to Other Income on the Consolidated Statement of Operations.
Benefit (Provision) for Income Taxes
A tax provision of $15,000 was recorded for the year ended December 31, 2010, compared to a tax benefit of $139,000 for the year ended December 31, 2009. The amount of the 2010 tax provision consists primarily of state income taxes. During 2009, a benefit was recognized relating to a carry-back of tax losses to prior years following the enactment of the Worker, Homeownership and Business Assistance Act of 2009.
Results of Operations—2009 compared to 2008
Revenue
Revenue increased $351,000, or 54%, to $998,000 for the year ended December 31, 2009 compared to $647,000 for year ended December 31, 2008. Product sales increased $351,000 or 63% to $911,000 for the year ended December 31, 2009 compared to $560,000 for the year ended December 31, 2008. License revenue of $88,000 for the year ended December 31, 2009 was equivalent compared to the year ended December 31, 2008. License revenue was predominantly attributable to annual maintenance and minimum royalties from existing licensees.
Product sales consisted solely of test cartridge sales, which are only available for purchase through reagent rental agreements or through negotiated purchase orders following purchase of an XT-8 system. The increase in revenue for 2009 was driven by sales of our Cystic Fibrosis Genotyping Test which replaced the predecessor Cystic Fibrosis Carrier Detection Test following FDA clearance of the test in July 2009. Revenue growth was hampered during this period by the lack of sufficient capital and the use of a distributor-based sales effort instead of a direct sales force for a major portion of the year ended December 31, 2009. Distributors generally do not dedicate substantial time to educate customers and monitor the evaluation of high technology new products which we believe adversely impacted our sales.
Cost of Sales
Cost of sales increased $1.1 million, or 34%, to $4.3 million for the year ended December 31, 2009 compared to $3.2 million for the year ended December 31, 2008. The increase was due to $666,000 in impairment charges for systems, and $549,000 in impairment charges for intangibles, partially offset by lower expenses for manufacturing support and temporary labor as production processes improved.
Sales and Marketing
Sales and marketing expense decreased $212,000, or 6% to $3.2 million for the year ended December 31, 2009, compared to $3.4 million for the year ended December 31, 2008. The decrease was driven by lower salaries and travel expenses partially offset by $381,000 for a one-time market research study in 2009, relocation of the newly hired commercial team and increased depreciation of XT-8 systems used in marketing
45
Table of Contents
evaluations. During 2009, we changed our estimate of the useful life of systems used for marketing purposes from five years to three years, which increased our depreciation for 2009 compared to 2008 by $38,000, and we recorded an impairment charge of $130,000 for certain demonstration units.
Research and Development
Research and development expense declined $7.8 million, or 58%, to $5.6 million for the year ended December 31, 2009 compared to $13.4 million for the year ended December 31, 2008. The decline was due to a substantial reduction in research and development headcount and expenses in 2009 after the completion of the XT-8 system development. We also consolidated our Rockland, Massachusetts and Menlo Park, California research facilities into our headquarters in Pasadena, California.
General and Administrative
General and administrative expense decreased $1.3 million, or 14%, to $8.3 million for the year ended December 31, 2009 compared to $9.6 million for year ended December 31, 2008. The decline was due to costs during 2008 related to our fund raising activities.
Foreign Exchange
Foreign exchange gain declined $201,000, or 40%, to $304,000 for the year ended December 31, 2009 compared to $505,000 for the year ended December 31, 2008. The gain was due to the settlement of U.S. dollar liabilities during the year as the U.S. dollar weakened against the British pound combined with the benefit of maturing U.S. dollar forward contracts which were held by us during the period.
Interest Income
Interest income declined $387,000, or 92% to $33,000 for the year ended December 31, 2009 compared to $420,000 for the year ended December 31, 2008, due to lower cash balances and declining interest rates in 2009.
Benefit (Provision) for Income Taxes
A tax benefit of $139,000 was recorded for the year ended December 31, 2009, compared to a tax provision of $247,000 for the year ended December 31, 2008. During 2009, a benefit was recognized relating to a carry-back of tax losses to prior years following the enactment of the Worker, Homeownership and Business Assistance Act of 2009. During 2008, a tax provision was recorded due to amendments made to the research and development tax credit claimed in prior periods.
Liquidity and Capital Resources
To date we have funded our operations principally through revenue, sales of capital stock and sales of our previous businesses. We have incurred net losses from continuing operations each year and have not yet achieved profitability. At March 31, 2011, we had $14.7 million of working capital, including $17.1 million in cash and cash equivalents.
Cash Flows
The following table summarizes, for the periods indicated, selected items in our consolidated statements of cash flows:
| March 31, | ||||||||
| 2011 | 2010 | |||||||
| Three months ended: |
||||||||
| Cash used by operating activities |
$ | (3,090,451 | ) | $ | (4,982,165 | ) | ||
| Cash used by investing activities |
(184,533 | ) | (137,440 | ) | ||||
| Cash provided by financing activities |
2,000,000 | 4,734 | ||||||
| Decrease in cash and cash equivalents |
$ | (1,274,984 | ) | $ | (5,114,871 | ) | ||
46
Table of Contents
Cash flows used by operating activities
Net cash used in operating activities decreased $1.9 million to $3.1 million for the three months ended March 31, 2011 compared to $5.0 million for the three months ended March 31, 2010. The decreased use of cash was due primarily to collection of a $1.6 million therapeutic tax credit and $1.2 million of higher accounts payable and accrued liabilities in the current quarter, partially offset by accrued IPO costs at March 31, 2010 that were subsequently paid in 2010.
Cash flows used by investing activities
Net cash used in investing activities increased $48,000 to $185,000 for the three months ended March 31, 2011 compared to $137,000 for the three months ended March 31, 2010 primarily due to increased purchases of our XT-8 systems used for customer rentals which are included in property and equipment.
Cash flows provided by financing activities
Net cash provided by financing activities increased by $2.0 million for the three months ended March 31, 2011 compared to the three months ended March 31, 2010 resulting from proceeds of a loan payable drawn in March 2011 to finance equipment purchases and tenant improvements purchased in 2010.
In March 2010, we entered into a loan and security agreement with Square 1 Bank, pursuant to which we obtained a credit facility consisting of a revolving line of credit in the amount of up to $2 million and an equipment term loan in the amount of up to $2 million. Based upon certain financial covenants, interest on the revolving line of credit will be either (i) the greater of (a) the bank’s prime rate (3.25% as of March 31, 2011) plus 2.75%, or (b) 6%; or (ii) the greater of (a) the bank’s prime rate plus 3.75%, or (b) 7%. In addition, based upon certain financial covenants, interest on the equipment term loan will be either (i) the greater of (a) the bank’s prime rate plus 3.25%, or (b) 6.50%; or (ii) the greater of (a) the bank’s prime rate plus 4.25%, or (b) 7.50%. The revolving line matures in July 2011 and the term loan matures in July 2013. In March 2011, the loan and security agreement was amended, whereby the line of credit availability was increased to $3 million and the maturity was extended to July 2012. The term loan was modified to allow invoices up to 360 days to qualify to be submitted for credit extension. There were no other changes to these two loans.
In March 2011, an additional loan was made available under the amended loan and security agreement for up to $1 million to finance equipment purchases. Based upon certain financial covenants, interest on this equipment term loan will be either (i) the greater of (a) the bank’s prime rate plus 3.25%, or (b) 6.50%; or (ii) the greater of (a) the bank’s prime rate plus 4.25%, or (b) 7.50%. This term loan matures March 2014.
As of March 31, 2011, we had no outstanding loans on the line of credit or the 2011 equipment loan and had drawn $2.0 million to finance 2010 equipment purchases and tenant improvements to our Carlsbad facility against the original 2010 equipment term loan. The loan bears an interest rate of 6.5%.
Pursuant to the terms of the loan and security agreement, we are required to maintain a ratio of liquidity to bank indebtedness equal to at least 1.50 to 1.00. In addition, the loan and security agreement includes several restrictive covenants, including requirements that we obtain the consent of Square 1 Bank prior to entering into any change of control event unless all debt is repaid to Square 1 Bank prior to the change of control event, incurring other indebtedness or liens with respect to our property, making distributions to our stockholders, making certain investments or entering into certain transactions with affiliates and other restrictions on storing inventory and equipment with third parties. The agreement also limits the amount we can borrow under the term loan to license genetic biomarkers to $500,000. To secure the credit facility, we granted Square 1 Bank a first priority security interest in our assets and intellectual property rights. We are currently in compliance will all ratios and covenants.
Our management has prepared cash flow forecasts which indicate, based on the current cash resources available, the availability of unutilized credit facilities, and our ability to access the equity markets will be sufficient to fund our business for at least the next 12 months. We expect capital outlays and operating
47
Table of Contents
expenditures to increase over the next several years as we grow our customer base and revenues, expand our research and development, commercialization and manufacturing activities. The amount of additional capital we may need to raise in the future depends on many factors, including:
| • | the level of revenues and the rate of revenue growth; |
| • | the level of expenses required to expand our sales and marketing activities; |
| • | the number of systems placed on a reagent rental basis; |
| • | the level of research and development investment required to maintain and improve our technology; |
| • | the costs of filing, prosecuting, defending and enforcing patent claims and other intellectual property rights; |
| • | competing technological and market developments; |
| • | our need to acquire or license complementary technologies or acquire complementary businesses; and |
| • | changes in regulatory policies or laws that affect our operations. |
We cannot be certain that additional capital will be available when and as needed or that our actual cash requirements will not be greater than anticipated. If we require additional capital at a time when investment in diagnostics companies or in the marketplace in general is limited due to the then prevailing market or other conditions, we may not be able to raise such funds at the time that we desire, on acceptable terms, or at all. In addition, when we raise additional funds through the issuance of equity or convertible debt securities, the percentage ownership of our stockholders could be significantly diluted, and these newly issued securities may have rights, preferences or privileges senior to those of existing stockholders. When we obtain additional debt financing, a substantial portion of our operating cash flow may be dedicated to the payment of principal and interest on such indebtedness, and the terms of the debt securities issued could impose significant restrictions on our operations. If we raise additional funds through collaborations and licensing arrangements, we might be required to relinquish significant rights to our technologies or products, or grant licenses on terms that are not favorable to us.
Contractual Obligations
As of December 31, 2010, we had contractual obligations relating to our facilities leases as follows:
| Payments due by period | ||||||||||||||||||||
| Contractual Obligations |
Total | Less than 1 Year |
1-3 Years |
4-5 Years |
After 5 Years |
|||||||||||||||
| Operating lease obligations(1) |
$ | 4,703,084 | $ | 992,471 | $ | 1,746,464 | $ | 1,253,871 | $ | 710,278 | ||||||||||
| (1) | Included in these amounts are our facilities leases. We enter into operating leases in the ordinary course of business with respect to facilities. Our lease agreements have fixed payment terms based on the passage of time. Certain facility leases require payment of maintenance and real estate taxes. Our future operating lease obligations could change if we exit certain contracts or if we enter into additional operating leases. |
In addition to the obligations in the table above, we periodically purchase systems from a contract manufacturer. In order to guarantee delivery, we issue purchase orders each 90 day period for delivery of systems during that period. At December 31, 2010, we had outstanding purchase orders for $27,860 worth of systems. For the three months ended March 31, 2011, one new purchase agreement for instruments was completed.
Additionally, approximately $487,000 of unrecognized tax benefits, including accrued interest and penalties of $105,000, have been recorded as liabilities and we are uncertain as to if or when such amounts may be settled.
In March 2010, we entered into a loan and security agreement with Square 1 Bank, pursuant to which we obtained a credit facility consisting of a revolving line of credit in the amount of up to $2 million and an
48
Table of Contents
equipment term loan in the amount of up to $2 million. Based upon certain financial covenants, interest on the revolving line of credit will be either (i) the greater of (a) the bank’s prime rate (3.25% as of December 31, 2010) plus 2.75%, or (b) 6%; or (ii) the greater of (a) the bank’s prime rate plus 3.75%, or (b) 7%. In addition, based upon certain financial covenants, interest on the equipment term loan will be either (i) the greater of (a) the bank’s prime rate plus 3.25%, or (b) 6.50%; or (ii) the greater of (a) the bank’s prime rate plus 4.25%, or (b) 7.50%. The revolving line matures in July 2011 and the term loan matures in July 2013. As of December 31, 2010, we had not drawn any funds under this loan and security agreement.
In March 2011, the loan and security agreement was amended, whereby the line of credit availability was increased by $1 million to $3 million and the maturity was extended to July 2012. The term loan was modified to allow invoices up to 360 days to qualify to be submitted for credit extension. There were no other changes to these two loans.
An additional loan was made available under the amended loan and security agreement for up to $1 million to finance equipment purchases. Based upon certain financial covenants, interest on this equipment term loan will be either (i) the greater of (a) the bank’s prime rate plus 3.25%, or (b) 6.50%; or (ii) the greater of (a) the bank’s prime rate plus 4.25%, or (b) 7.50%. This term loan matures March 2014.
As of March 11, 2011, we had no outstanding loans on the line of credit and had drawn $2 million to finance 2010 equipment purchases and tenant improvements to its Carlsbad facility against the original term loan. The loan bears an annual interest rate of 7.5%.
In November 2009, we renegotiated our lease on our 25,000 square foot headquarters facility in Pasadena, California that lowered our rent and accelerated the termination of that lease to June 30, 2010.
In March 2008, we exercised our option to extend the operating lease of our approximately 8,400 square-foot former manufacturing facility in Pasadena, California, for a three-year period from August 1, 2008 until July 31, 2011 at a rental cost of $21,558 per month. On February 8, 2010, we entered into a seven-year and seven-month lease for a new 31,098 square foot facility in Carlsbad, California. The facility is part of a three-building office and research and development project located at 5964 La Place Court, Carlsbad, California, and the project totals 158,733 rentable square feet. Monthly rental payments are $48,260 and increases 3% annually. We also pay our pro-rata share of the building and project maintenance, property tax, management and other costs subject to certain limitations. We have paid a $55,000 security deposit and provided a $500,000 standby letter of credit as security for the future rent as well as for up to $2.0 million in landlord funded tenant improvements. The lease also provides for expansion rights and rights of first refusal for expansion within our building, subject to certain limitations.
Other Off-Balance Sheet Arrangements
We have no other off-balance sheet arrangements except for our unutilized credit facilities with Square 1 Bank that provides a revolving line of credit up to $2 million and an unutilized equipment term loan totaling $1 million at March 31, 2011.
49
Table of Contents
Overview
We are a molecular diagnostics company focused on developing and commercializing our proprietary eSensor detection technology. Our proprietary electrochemical technology enables fast, accurate and highly sensitive detection of up to 72 distinct biomarkers in a single sample. Our XT-8 system received 510(k) clearance from the FDA and is designed to support a broad range of molecular diagnostic tests with a compact and easy-to-use workstation and self-contained, disposable test cartridges. Within 30 minutes of receipt of an amplified DNA sample, our XT-8 system produces clear and accurate results. Our XT-8 system supports between one and three analyzers. Each analyzer holds up to eight independent test cartridges, resulting in the XT-8 system supporting up to 24 test cartridges, each of which can be run independently, resulting in a convenient and flexible workflow for our target customers, which are hospitals and reference laboratories. As of March 31, 2011, we had an installed base of 102 analyzers, or placements, with our customers.
We have developed four diagnostic tests for use with our XT-8 system and expect to expand this test menu by introducing two to four new tests annually. Three of our diagnostic tests have received FDA clearance, including our Cystic Fibrosis Genotyping Test, which detects pre-conception risks of cystic fibrosis, our Warfarin Sensitivity Test, which determines an individual’s ability to metabolize the oral anticoagulant warfarin, and our Thrombophilia Risk Test, which detects an individual’s increased risk of blood clots. Our eSensor technology has demonstrated 100% accuracy in clinical studies compared to DNA sequencing in our Cystic Fibrosis Genotyping Test, our Warfarin Sensitivity Test and our Thrombophilia Risk Test. We have also developed a Respiratory Viral Panel Test, which detects the presence of major respiratory viruses and is labeled for IUO. We intend to seek FDA clearance for our Respiratory Viral Panel Test in 2011. We also have a pipeline of several additional potential products in different stages of development or design, including diagnostic tests for an individual’s sensitivity to Plavix, a commonly prescribed anti-coagulant, and for mutations in a gene known as K-ras, which is predictive of an individual’s response rates to certain prescribed anti-cancer therapies.
We are also developing our next-generation platform, the NexGen system. We are designing the NexGen system (formerly referred to as the AD-8 system) to integrate automated nucleic acid extraction and amplification with our eSensor detection technology to enable technicians to place a raw or a minimally prepared patient sample into our test cartridge and obtain results without any additional steps. This sample-to-answer capability is enabled by the robust nature of our eSensor detection technology, which is not impaired by sample impurities that we believe hinder competing technologies. We are designing our NexGen system to further simplify workflow and provide powerful, cost-effective molecular diagnostics solutions to a significantly expanded group of hospitals and reference laboratories.
Our XT-8 system and planned menu of tests are intended to improve patient care and physician practices by providing high value, clinically useful information that aids in the diagnosis of disease and the selection of treatments tailored to an individual’s genetic profile. We believe that these improvements in patient care are economically attractive to our customers who are generally reimbursed for these tests by third-party payors and managed care providers through established reimbursement codes. Because the XT-8 system is designed to be flexible and easy-to-use, we believe that our customers will choose to perform a broad range of tests on our platform, in some cases providing our customers with the capability to perform diagnostic tests that they were not previously able to complete. By focusing our product development and commercialization efforts on high value, clinically useful opportunities in genetic and infectious diseases, cancer and personalized medicine, we believe we will drive widespread clinical adoption of our products.
Our Strategy
Our goal is to become the market leading provider of automated, multiplex molecular diagnostic testing systems. We intend to expand the use of our XT-8 system and diagnostic tests targeting mainly those reference laboratories and hospitals in the United States that perform a high volume of molecular diagnostic tests. To achieve this objective, we intend to:
Expand our Menu of Clinical Diagnostic Products. We intend to develop a broad menu of molecular diagnostic tests that we believe satisfy important medical needs and will be attractively reimbursed by third-
50
Table of Contents
party payors. We are pursuing and intend to continue to pursue FDA clearance or approval for our tests. We intend to explore tests that are either already in high demand or projected to experience rapid growth. Where required, we plan to gain access to these tests by in-licensing the appropriate biomarkers that have shown correlations to diseases or therapeutic response.
Grow our Installed Base of Customers. We have identified those laboratories and hospitals in the United States that we believe will be high volume customers and who will benefit from our eSensor technology. We intend to leverage our commercial organization to drive placements of our XT-8 system. We anticipate expansion of our installed base of customers will drive sales of our test cartridges, from which we anticipate generating the majority of our revenues.
Increase Utilization of Tests With Our Customers. We intend to increase the use of our diagnostic tests by developing and offering tools and support tailored to our products such as accredited physician education programs and seminars, product training for our customers and reimbursement support. These activities will aid in establishing the clinical utility of multiplex molecular diagnostic tests, which we believe will increase adoption of our products.
Develop and Commercialize our NexGen System. We are developing our NexGen system to provide a complete sample-to-answer solution for our customers. The NexGen system will retain all the customer benefits of our XT-8 system while also integrating automated nucleic acid extraction and amplification. These features will eliminate the need for time consuming and complex sample preparation steps and allow technicians to place a raw or minimally prepared patient sample into our test cartridge. We have already demonstrated feasibility of direct sample-to-answer on a NexGen system prototype using diluted blood. We believe this advancement will make our technology attractive to a broader range of hospitals and laboratories that lack the technical or economic resources to perform molecular diagnostic testing with existing products and technology. We believe such workflow enhancements may expand our target user base from approximately 1,000 customers to over 5,000 potential customers in the United States.
Expand Internationally and Explore Out-Licensing Opportunities. We plan to offer our molecular diagnostic products in European and other international markets in the future. We are currently developing a distribution strategy for these markets, and we anticipate using marketing partners and distributors as we expand internationally. We expect to supplement marketing partnerships with specialists who will train our partners’ sales forces and provide technical support. We also intend to explore opportunities to leverage our intellectual property position in detection technologies through out-licensing or the establishment of partnerships.
Our Market Opportunity
The U.S. market for molecular diagnostics was estimated to be $1.9 billion in 2009 and is anticipated to reach $3.4 billion in 2014 according to L.E.K., a market research firm. Molecular diagnostics generally refers to the detection and measurement of DNA or RNA biomarkers to diagnose disease and to optimize the treatment of patients. We believe that the following factors, among others, are contributing to the growth of this market:
Expansion of Genetic Testing for Disease Predisposition. Advances in the understanding of the relationship between an individual’s genetics and disease have led to increased reliance on molecular diagnostic testing for inherited diseases such as cystic fibrosis and thrombophilia. We expect new molecular diagnostic tests will be required as researchers continue to discover new relationships between genetics and disease, new medical treatments are developed, and as professional societies set guidelines regarding genetic disease and the role of genetic counseling in the interpretation of the results of these tests.
Adoption of FDA-Cleared Molecular Diagnostic Testing Methods. The FDA recommends that laboratories and hospitals use FDA-cleared molecular diagnostic tests when these tests are available, rather than
51
Table of Contents
tests known as “home-brew tests” or laboratory developed tests, or LDTs, that are not submitted to the FDA for approval. LDTs are broadly used by reference laboratories and hospitals to perform molecular diagnostic tests and are subject to strict regulatory requirements. As a result, we believe reference laboratories and hospitals will look to replace their existing LDTs and non-FDA-cleared molecular diagnostic tests with FDA-cleared tests as they become available.
Advances in Cancer Therapy. Tailoring treatments to an individual’s tumor type and genetics is an important trend in cancer therapy. Because cancer drugs can be expensive and are not effective in certain patients, the FDA has required or recommended that molecular diagnostic tests be performed before administration of certain drugs, such as Herceptin, Erbitux and Vectibix. We believe molecular diagnostic testing to determine an individual’s response to certain cancer therapies will drive demand for molecular diagnostics.
Increased Demand for Infectious Disease Diagnostic Panels. Different disease pathogens can produce similar symptoms, but with vastly distinct courses of disease progression and required medical treatment responses. For example, pneumonia caused by Mycoplasma may resolve without treatment, while pneumonia caused by Legionella will generally require aggressive medication and hospitalization. In order to improve patient care, we believe physicians are increasingly requesting infectious disease diagnostic panels to be performed. According to L.E.K., the market for molecular diagnostic testing of infectious diseases in the United States was estimated to be $1.1 billion in 2009. We intend to address the emerging multiplex diagnostic segment of this market which our management estimates will be in the range of $100 million to $150 million.
Advances in Personalized Medicine. Tailoring treatments to an individual’s genetic profile—called personalized medicine or pharmacogenetics—is emerging as an important trend and will drive demand for molecular diagnostic testing. Pharmaceutical companies, clinical researchers and pharmacy benefit managers are screening drugs for varied toxicity, dose response and efficacy among individuals with different genetic profiles. Additionally, regulating agencies continue to revise drug labels to improve safety and efficacy. Because these industry developments may improve clinical outcomes and reduce costs for third-party payors, we believe adoption of these tests will become more widespread in a managed care environment.
Limitations of Existing Diagnostic Products and Technologies
Scientists have developed a variety of genomic analysis methods, including DNA sequencing, gene expression analysis and genotyping, to measure genetic biomarkers and detect diseases. These analytical methods are performed using various molecular diagnostic testing technologies, the most common being polymerase chain reaction, or PCR, which involves amplifying, or generating exponential copies of, the DNA sequence in question and then detecting the DNA with the use of fluorescent dyes.
The first commercially used molecular diagnostic tests were “home brew tests” or LDTs developed by reference laboratories and large hospital-based laboratories. LDTs, which are still broadly used today, are generally single-purpose tests that involve a number of complex, manual procedures. To perform these tests, laboratories are required to employ highly skilled technicians and maintain specialized laboratory facilities and equipment. As the market for molecular diagnostic tests expanded, we believe a number of companies developed and began offering dedicated instrumentation and commercialized testing systems for specific genetic biomarkers and diseases. These commercialized testing systems, as well as LDTs, are characterized by the following limitations:
Limited Menu of Diagnostic Tests. We believe existing LDTs are typically custom designed for one specific genetic biomarker or disease. In addition, we believe commercialized testing systems currently offer only a limited number of molecular diagnostic tests for use with such systems. As a result, laboratories need numerous LDTs and commercialized testing systems to offer their physician and hospital clients with a range of molecular diagnostic testing options.
Inability to Multiplex. In many cases, testing for multiple genetic biomarkers may be necessary to diagnose a disease, differentially identify infectious agents or evaluate the appropriate treatment options for a
52
Table of Contents
patient. Many LDTs and commercialized testing systems lack the ability to multiplex, or test for multiple genetic biomarkers at the same time on a single patient sample. As a result, the laboratory must perform multiple, separate tests on a sample. Serial testing is time-consuming and expensive and significantly increases the amount of time and sample needed to complete a diagnostic analysis.
Poor Laboratory Workflow. LDTs and commercialized testing systems generally do not permit laboratories to initiate new tests while other tests are in progress. As a result, laboratories are required to batch process molecular diagnostic tests. To help control costs, laboratories may only run a particular molecular diagnostic test on an infrequent basis. In addition, LDTs and commercialized testing systems require significant sample preparation and additional washing steps, which adds to the complexity and time required to complete a molecular diagnostic test. Many of them also involve optical systems, robotics and complicated moving parts that must be frequently calibrated and are subject to maintenance and repair issues.
Risk of Human Error and Contamination. Many LDTs and commercialized testing systems generally require technicians to perform a series of complex manual procedures which may lead to contamination. Commercialized testing systems only automate certain steps in the testing process, and technicians are often required to use multiple instruments or manual processes in sequence to generate results. The handling of samples and the multiple manual procedures required by existing products can lead to increased risk of sample contamination and human error. In addition, LDTs and many commercialized testing systems require the operator to interpret results, which increases the potential for human error and leads to problems related to repeatability of results.
Intensive Resource Requirements. Laboratories are required to employ and train highly-skilled technicians and dedicate significant capital, labor and laboratory space to conduct molecular diagnostic tests. In fact, many of the multiplex LDTs and commercialized testing systems currently used by national reference laboratories are so specialized that we believe only a limited number of their sites have the capability to perform these tests. As a result, we believe national reference laboratories do not have the ability to perform their entire menu of multiplex tests across all of their locations, and smaller hospital-based laboratories face significant hurdles to initiate molecular diagnostic testing at limited volumes.
Shifting Regulatory Environment. A significant number of molecular diagnostic tests, including LDTs as well as commercialized testing systems, have not been submitted for FDA clearance. The FDA has imposed regulatory requirements on laboratories that use LDTs or other non-FDA-cleared commercialized testing systems, including the requirement to comply with CLIA standards. In the future, the FDA may restrict the use of LDTs and non-FDA-cleared molecular diagnostic tests unless the laboratories comply with medical device requirements, including the FDA’s Quality Systems Regulations and 510(k) clearance or premarket approval requirements.
These limitations have created a laboratory model for molecular diagnostic testing that is complex, inefficient and inaccessible to a large segment of reference laboratories and hospitals.
Our Solution
Our XT-8 system is an automated molecular diagnostic system that enables reference laboratories and hospitals to perform fast, accurate and easy-to-use molecular diagnostic tests. The XT-8 system, which employs our proprietary electrochemical detection technology, consists of a compact bench-top workstation with an integrated touch screen computer and an analyzer module into which the self-contained, disposable test cartridges are inserted. The XT-8 system is user-friendly, intuitive, requires minimal maintenance and provides laboratories with the ability to perform multiplex molecular diagnostic tests in an efficient and cost-effective manner. Specifically, we believe that our XT-8 system and related diagnostic tests offer reference laboratories and hospitals the following benefits:
Versatile Platform for a Broad Menu. Our XT-8 system has broad application across a number of areas in molecular diagnostic testing. In addition to our FDA-cleared Cystic Fibrosis Genotyping Test, Warfarin
53
Table of Contents
Sensitivity Test and Thrombophilia Risk Test, and our Respiratory Viral Panel Test, which is labeled for IUO, we have a pipeline of several additional products in development or design in the fields of pharmacogenetics, genetic diseases, infectious diseases and cancer. We are currently developing a Plavix Sensitivity Test and a K-ras Mutation Test, and we have a pipeline of potential products in various stages of development or design. Laboratories using our system will be able to run our additional tests without any further capital investment or operator training.
FDA-Cleared Products. We have received FDA clearance for our Cystic Fibrosis Genotyping Test, Warfarin Sensitivity Test and Thrombophilia Risk Test, while our Respiratory Viral Panel Test is labeled for IUO. We intend to submit our Respiratory Viral Panel Test to the FDA for clearance in 2011. We intend to utilize IUO-labeled products in clinical studies within the broader process of seeking FDA clearance for our diagnostic tests.
Ease of Use. Our XT-8 system eliminates the need to use complex instrumentation to generate test results. Our XT-8 system minimizes manual processing steps and streamlines data analysis, making molecular diagnostic testing available to a broad spectrum of laboratories without the need for highly skilled technicians. As a result, our XT-8 system can provide national reference laboratories with the ability to perform our menu of molecular diagnostic tests across all of their locations. We also designed our XT-8 system to require minimal maintenance.
Accuracy and Reliability. Our XT-8 system provides accurate and reliable molecular diagnostic test results. We have demonstrated 100% accuracy in clinical studies compared to DNA sequencing in our Cystic Fibrosis Genotyping Test, our Warfarin Sensitivity Test and our Thrombophilia Risk Test. Our XT-8 system limits technician contact with a patient sample, thereby reducing contamination risk. It also provides clear reports, minimizing the risk of human error and increasing the repeatability of test results.
Enhanced Laboratory Work Flow. Our unique platform allows for random access, or the ability to initiate tests while other tests are in progress, resulting in a highly convenient and flexible workflow. Our XT-8 system provides random access for up to 24 independent test cartridges. In addition, our proprietary electrochemical detection technology streamlines the sample preparation process and eliminates the need for the additional washing steps required by some other detection methods, such as optical or fluorescent detection. Laboratories using our XT-8 system can expect to obtain test results within 30 minutes of receipt of the amplified DNA sample, resulting in a total turnaround time of generally under four hours.
Multiplex Capability. Our XT-8 system can detect up to 72 separate biomarkers in a single test cartridge. This allows laboratories to run multiple tests or panels on an individual patient sample in a one-step detection process. This capability reduces the time required for a laboratory to perform a diagnostic analysis that involves multiple genetic markers or infectious disease pathogens, which otherwise would require the laboratory to run multiple, separate molecular diagnostic tests.
54
Table of Contents
Our Products
Our XT-8 System
Our FDA-cleared XT-8 System is an automated, multiplex molecular diagnostics workstation that provides a wide range of diagnostic testing capabilities in pharmacogenetics, genetic and infectious diseases and oncology. Our XT-8 system consists of a compact bench-top workstation with an integrated touch screen computer and an analyzer module into which the self-contained, disposable test cartridges are inserted. These features make the XT-8 system user-friendly, intuitive and virtually maintenance-free. With a footprint of approximately 16-by-16 inches in its standard configuration, the XT-8 system takes up less bench top space than most of our competitors’ systems, and its standalone design allows it to be installed and used without any required laboratory modifications.

Prior to performing a test, a laboratory technician takes isolated DNA from the patient sample and performs a DNA amplification step with materials supplied with our test cartridge. In some cases, the technician also performs a routine enzymatic treatment before adding our proprietary signal probes and transferring the solution into the sample compartment in our test cartridge. The technician enters sample identification and reagent information into our XT-8 system using the supplied bar code wand or on-screen keyboard and inserts the test cartridge into an open slot on the analyzer module. The on-board computer automatically assimilates input information and test cartridge information from the memory chip on the test cartridge and initiates the specified test protocol. The testing process typically takes under four hours to complete, and the test results can be viewed on the built-in touch screen monitor 30 minutes after the insertion of test cartridges into the XT-8. Test results can also be printed out or reported through the laboratory’s computer information system.
The key features of our XT-8 system include:
| Key Features |
Characteristics | |
| Ease of Use |
Intuitive touch-screen interface and clear reports | |
| Multiplex Capability |
Detects up to 72 distinct biomarkers in a single sample | |
| Accurate Results |
Our Cystic Fibrosis Genotyping Test, our Warfarin Sensitivity Test and our Thrombophilia Risk Test demonstrated 100% accuracy in clinical studies compared to DNA sequencing | |
| Fast Turnaround |
30 minutes to result from amplified DNA sample with minimal technician time needed | |
| Random Access |
Each of up to 24 test cartridge slots can be accessed independently | |
| Minimal Maintenance |
No routine maintenance or calibration required | |
| Small Footprint |
Approximately 16 inches in width and depth in its standard configuration |
55
Table of Contents
Our Test Menu
We have developed four diagnostic tests for use with our XT-8 system, three of which have received clearance from the FDA and one of which is currently labeled for IUO. During the three months ended March 31, 2011 and the fiscal year ended December 31, 2010, sales of our Cystic Fibrosis Genotyping Test represented approximately 45% and 43% of our revenues, respectively, and sales of our Warfarin Sensitivity Test represented approximately 5% and 13% of our revenues, respectively.
Cystic Fibrosis Genotyping. Our Cystic Fibrosis Genotyping Test is a multiplex genotyping test that detects a panel of mutations associated with cystic fibrosis based on guidelines published by the American College of Medical Genetics and the American College of Obstetricians and Gynecologists for screening of adult couples contemplating pregnancy. Our Cystic Fibrosis Genotyping Test demonstrated 100% accuracy in clinical studies compared to DNA sequencing and delivers results within 30 minutes of receipt of the amplified DNA sample. Test results are summarized in an easy-to-interpret report that includes a summary “carrier” or “non-carrier” determination as well as individual carrier status for each of the 23 recommended markers. Our Cystic Fibrosis Genotyping Test received FDA clearance in July 2009.
Our Cystic Fibrosis Genotyping Test addresses a market that was estimated in 2009 at over $70 million in the United States alone. More than 10 million Americans are carriers of one mutation of the cystic fibrosis gene. The American College of Obstetricians and Gynecologists suggests that all couples who are considering having a child, or those who are expecting a child, should have genetic carrier testing for cystic fibrosis. Much of current cystic fibrosis testing is performed by national reference laboratories. With the availability of highly accurate, easy to use cystic fibrosis tests, we expect that the market will continue to decentralize through regional reference laboratories and hospitals now capable of offering this test.
Warfarin Sensitivity. Our Warfarin Sensitivity Test is a multiplex pharmacogenetic test for the detection of three genetic markers that are known to play a critical role in metabolism of, and sensitivity to, warfarin. Warfarin, offered under the brand name Coumadin, is the most widely prescribed oral anticoagulant in North America and Europe and is used to prevent heart attacks, strokes, and blood clots in patients’ veins, arteries and lungs. Through detection of an individual’s sensitivity to warfarin, doctors are better able to accurately and efficiently determine the appropriate warfarin dosage level on an individual patient basis. Our Warfarin Sensitivity Test demonstrated 100% accuracy in clinical studies compared to DNA sequencing and delivers results within 30 minutes of receipt of the amplified DNA sample. Our Warfarin Sensitivity Test received FDA clearance in July 2008.
According to the Medco-Mayo Warfarin Effectiveness Study, there were approximately two million new patient prescriptions of warfarin in the United States in 2009. According to Biotechnology Healthcare 2008, a health-care focused journal, there were approximately 20 million patients on warfarin therapy in 2008. The FDA recently approved a labeling change that provides dose recommendations based on genetic test results.
Thrombophilia Risk. Thrombophilia is a condition where a person’s blood clots easily or excessively, placing them at risk of developing clots. Thrombophilia is a particular concern for high risk patients, including patients who are pregnant or undergoing certain surgeries. Our Thrombophilia Risk Test is a multiplex test for the detection of four common inherited genetic risk factors of thrombophilia: Factor V Leiden, Factor II prothrombin and two genetic markers in the methylenetetrahydrofolate reductase (MTHFR) gene. Our Thrombophilia Risk Test demonstrated 100% accuracy in clinical studies compared to DNA sequencing and delivers results within 30 minutes of receipt of the amplified DNA sample. Our Thrombophilia Risk Test received FDA clearance in April 2010.
Thrombophilia is one of the most common types of blood coagulation disorders affecting 1 in 1,000 individuals. We believe the U.S. market was approximately $55 million in 2008 based on statistics provided by Kalorama Information 2009, a market research firm.
56
Table of Contents
Respiratory Viral Panel (RVP). Our Respiratory Viral Panel Test, currently labeled for IUO, covers approximately 20 viruses, including influenza A (H1N1 and seasonal), influenza B, respiratory syncytial virus, or RSV, and numerous other upper respiratory viruses. We have initiated clinical studies on our RVP panel and currently plan to submit it for FDA clearance in 2011.
Respiratory pathogens are a major source of illness and can lead to hospitalizations and death. According to the Centers for Disease Control, each year in the United States on average, 5% to 20% of the population gets the flu; more than 200,000 people are hospitalized from flu-related complications; and about 36,000 people die from flu-related causes. RSV is the most common cause of bronchitis and pneumonia in infants and young children, with up to 125,000 children hospitalized annually in the United States. The challenge to the physician assessing a patient with a respiratory illness is determining what the underlying cause is so that an effective treatment plan can be determined.
Our Tests in Development and Design
We have a pipeline of potential products in various stages of development or design. We consider our diagnostic tests to be in the design phase once they have advanced beyond the conceptual stage. We perform market research, clinical publication reviews, customer interviews, technical feasibility and freedom to operate assessments to determine if a potential diagnostics test is a viable product candidate. We believe that all of our tests in the design stage have viable market potential and are technically feasible to develop using our eSensor technology. While we do not currently license biomarkers for all products in the design phase, we believe we will be able to obtain such licenses, if needed, on commercially reasonable terms.
We intend to introduce two to four new tests annually and currently expect that our Plavix Sensitivity Test, our Hepatitis C Viral Genotyping test and our K-ras test will be our next tests in development and design to be introduced. We have selected these tests based upon what we believe are clinically relevant products which address unmet market needs. Laboratories using our XT-8 system will be able to run our additional tests without any further capital investment or operator training. We are currently developing or designing the following diagnostic tests:
Plavix Sensitivity. Plavix is the most commonly prescribed anti-platelet drug with more than 25 million patients taking the drug in the United States each year. According to the Cheuvreux Sector Report, a market research report, over 1.6 million new patients were prescribed Plavix in 2009. In order for Plavix to be effective, it must be metabolized by the body using an enzyme referred to as 2C19. Patients with impaired 2C19 functionality will see reduced metabolism and therefore, reduced benefits from taking Plavix. We are currently in late stage development for a 2C19 multiplex genetic test that detects a panel of genetic markers associated with poor metabolism of Plavix. The FDA has recently revised the label for Plavix with a “black box” that warns of the reduced effectiveness of Plavix in patients who are poor metabolizers and informs physicians of the existence of genetic tests to identify these at-risk patients.
Plavix’s patents are expected to expire in late 2011, and we believe this expiration will lead to significant generic competition which will drive down the cost for Plavix and increase overall demand for the drug. According to the Plavix label, 2% to 14% of patients do not respond to Plavix. As a result, we believe there will be increased demand for the Plavix Sensitivity Test as third-party payors will have an added incentive to reimburse for a test that can reduce or avoid the use of these expensive therapies.
Infectious Disease Test Panels. The infectious disease diagnostics market is estimated to reach over $6 billion in the United States by 2012, with substantial growth expected in the molecular diagnostic segment. We are currently designing other infectious disease test panels that would align strategically with our existing respiratory viral panel test offering by leveraging our current and future XT-8 system placements in the acute care setting. The test panels we are designing fit into two categories: Genotyping tests for viruses such as hepatitis C virus (HCV) and human papillomavirus (HPV) or detection tests for panels of viruses, bacteria or fungi such as central nervous system infections or lower respiratory tract infections. Genotyping tests are run throughout the year whereas many detection tests have a seasonal component. In order to maximize the value of
57
Table of Contents
systems installed for infectious disease tests like our RVP product, we intend to develop a broad range of detection assays that have distinctly different seasonal peaks in prevalence to allow our customers to utilize our system for infectious disease testing throughout the year. Currently, several infectious disease panels and genotyping tests are in the design or development stage. These include: Lower Respiratory Tract Infections (LRTI); Central Nervous System Infections (CNS); and Hepatitis C Virus Genotyping (HCVg).
K-ras Mutation. Anti-EGFR therapy is a type of cancer treatment that interferes with the growth of cancer cells, slowing their growth and subsequent spread in the body. Anti-EGFR therapy is currently approved by the FDA to treat colorectal cancer as well as head and neck cancer. Scientific studies have demonstrated that patients whose tumors have genetic variations in the K-ras gene will not respond to anti-EGFR therapy. Currently approved anti-EGFR therapies are marketed under the brand names Erbitux and Vectibix. These therapies are approved for use in colorectal cancer and more recently head and neck cancer in the case of Erbitux.
According to the American Cancer Society, there are over one million new cases of colorectal cancer globally each year with approximately 150,000 cases in the United States alone. We are currently developing a multiplex K-ras test that detects a panel of common genetic markers in the K-ras gene. The FDA requires K-ras testing on the labels of the two approved anti-EGFR antibody therapeutics, Vectibix and Erbitux, for use in colorectal cancer.
Oncology and Personalized Medicine Tests. Given the trend in oncology towards tailoring treatment to an individual’s tumor type and the emerging interest in personalized medicine, we are currently researching and evaluating the development of test panels in these areas. Expanding our product offering into these two areas would align strategically with our existing products as well as development stage products by leveraging our current and future XT-8 system placements in these laboratories. Examples of tests panels that are under design include 2D6 for Tamoxifen Metabolism, which can affect the effectiveness of a drug used for the treatment and prevention of breast cancer, and EGFR Pathway, which detects mutations in genes other than K-ras involved in EGFR signaling.
Our NexGen System
We are developing our next-generation testing system to integrate automated nucleic acid extraction and amplification. We are designing the NexGen system (formerly referred to as our AD-8 system) to allow a technician to place a raw or minimally prepared patient sample into our test cartridge and then insert the cartridge into the NexGen system with no further user intervention. The NexGen system is designed to achieve full sample-to-answer capability. The NexGen system will provide the same customer benefits of the XT-8 system and further enhance workflow by eliminating or reducing the level of sample processing required and incorporating amplification. We believe this advancement will make our eSensor technology attractive to the broad range of institutions that currently lack the technical or economic resources to perform molecular diagnostic testing. We believe the NexGen system may expand our target user base from approximately 1,000 to over 5,000 potential laboratories and hospitals in the United States.
The NexGen system is currently in development with substantial technical feasibility completed using diluted blood in our Warfarin Sensitivity Test. The NexGen system leverages the base technology and system hardware from our XT-8 system to reduce risk and accelerate the development of the sample preparation and amplification features. We believe our approach to a sample-to-answer system will achieve benefits over other competitive multiplex systems, which require extensive sample processing procedures in addition to other complex sample manipulations throughout their test process.
Our Technology
Our eSensor Technology
Our proprietary eSensor technology is based on the principles of competitive DNA hybridization and electrochemical detection. DNA naturally forms a double-stranded structure, with each strand binding with high affinity, or hybridizing, only to a complementary strand. Our technology takes advantage of this highly specific
58
Table of Contents
binding by first creating two types of single-stranded DNA, the capture probe and the signal probe. The capture probe and signal probe are each complementary to a different segment of the target DNA, or biomarker, that is a focus of the diagnostic test. Using our proprietary technology and processes, we attach our capture probes to a proprietary monolayer on the surface of a gold electrode within our proprietary test cartridge. We separately attach ferrocene, an electrochemically active label, to our signal probes.
Before placing the sample into our test cartridge, the technician mixes the amplified DNA sample with our signal probe. If the target biomarker is present in the prepared patient sample, a segment of the biomarker DNA will hybridize with a solution containing our signal probe. This solution is then run past an electrode, against which our capture probes have been immobilized. The as-yet unbound segment of the target biomarker binds to our capture probe, creating a target DNA, signal probe, capture probe complex at the surface of the electrode. This complex produces an electrochemical signal analyzed and interpreted by the XT-8 system. Our test cartridges currently have 72 distinct electrodes, each of which can be configured to detect a different target biomarker, enabling multiplex testing.
Our eSensor technology is highly specific for the target biomarker, and is not based on optical or fluorescent detection. As a result, our diagnostic tests are less prone to sample contamination risk and do not require many of the time-consuming washing and preparation steps required by competing technologies. The only sample preparation step required before using our test cartridges is a PCR amplification, which involves amplifying, or generating billions of copies of, the target DNA molecules, followed by transfer of the sample to our test cartridge and insertion of the test cartridge into any open slot in our XT-8 system. In some tests, amplified DNA is subject to an additional enzymatic treatment to produce a single-stranded-DNA.
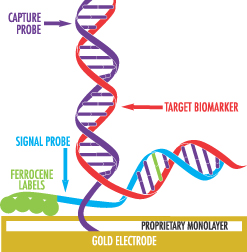
59
Table of Contents
Our Test Cartridges. Our test cartridges are self-contained devices specifically programmed and configured for a given diagnostic test. Each test cartridge includes a sample compartment and a plastic cover that forms a hybridization chamber. The test cartridge is fitted with a diaphragm pump and valves that circulate the hybridization solution, including the signal probe and prepared patient sample, when inserted into the XT-8 system. The test cartridge also includes a printed circuit board chip consisting of an array of 72 gold-plated working electrodes, a silver/silver chloride reference electrode, and two gold-plated auxiliary electrodes. Each electrode is customized with a proprietary monolayer that immobilizes the DNA capture probes specific for each target of a test panel. The test cartridge also contains an electrically erasable programmable read-only memory component that stores information related to the cartridge such as assay identifier, cartridge lot number and expiration date.

Our XT-8 System. Our XT-8 system is a multiplex workstation that has a modular design consisting of an integrated touch screen workstation and up to three analyzer modules each capable of analyzing eight individual test cartridges. The test cartridge slots operate independently of each other allowing up to 24 independent test cartridges to be loaded at one time, with the remaining slots available for use at any future time while the system is running. Each slot contains a test cartridge connector, a precision-controlled heater, an air pump and electronics. The air pumps drive the diaphragm pump and valve system in the test cartridge, eliminating fluid contact between the system and the cartridge. The pneumatic pumping enables recirculation of the hybridization solution allowing the target DNA and the signal probes to efficiently hybridize with the complementary capture probes on the electrodes. The diaphragm pump in the test cartridge is connected to a pneumatic source from the XT-8 system and provides unidirectional pumping of the hybridization mixture through the cartridge during hybridization.
The touch screen workstation controls each analyzer module, provides power and analyzes and stores data. Technicians can load patient identification numbers and reagent lot codes by the included bar code scanner, the touch screen or uploading a text file from a USB memory stick.
Advantages of Our eSensor Electrochemical Signal Detection
We believe our proprietary electrochemical signal detection technology has several advantages over other signal detection platforms:
Robust Signal. Our capture probes are highly target specific, reducing the binding of non-target DNA and, thereby, largely eliminating interference from other components in a patient’s sample, such as blood, saliva or urine. Similarly, constituents of blood that would normally interfere with fluorescence detection, such as hemoglobin or bilirubin, have no effect on the processed electronic signals produced by our eSensor technology. This robust functionality will, we believe, facilitate the development of integrated amplification and sample-to-answer systems for blood and other sample types.
60
Table of Contents
High Sensitivity and Accuracy. Our eSensor technology is highly sensitive in the detection of nucleic acids. Each electrode can routinely detect approximately 1 nanomolar of target DNA, and a sensitivity of 10 picomolar of target DNA has been achieved. Such concentrations are readily produced from patient samples using several commercially-validated amplification technologies such as PCR. Our eSensor technology has demonstrated 100% accuracy in clinical studies compared to DNA sequencing in our Cystic Fibrosis Genotyping Test, our Warfarin Sensitivity Test and our Thrombophilia Risk Test.
Streamlined Sample Preparation. Our technology directly detects the target DNA sequence with highly specific signal probes and electrode-bound capture probes. As a result, our test samples do not require many of the washing steps typically required to remove unbound target DNA and labels. We believe that our eSensor technology can minimize sample preparation requirements. We have already demonstrated direct PCR-based genotyping from diluted whole blood without the need for DNA sample preparation or washing out of interfering substances.
Efficient Multiplexing. Each of the 72 electrodes in our test cartridge configuration acts independently of the others and produces a comprehensive and informative signal. For example, a single eSensor electrode can measure the presence or absence of control DNA, which we use for quality control, and simultaneously indicate whether a patient sample contains zero, one or two copies of a particular sequence, corresponding to mutant, heterozygous or wild type genotypes. As a result, our eSensor technology eliminates the need for redundancy and the averaging of multiple measurements commonly required by competing technologies.
Small Footprint with Low Maintenance. Our eSensor technology enables users to perform hybridization and detection in a low-cost system with relatively few moving parts. In contrast, conventional microarray systems require robotic instrumentation to automate multistage fluidic handling processes. As a result, these systems are often bulky, complicated and expensive and require frequent calibration and maintenance. Our XT-8 system, for example, requires no calibration and virtually no maintenance and is self-contained in a small footprint of approximately 16-by-16 inches in its standard configuration.
Cost-Effective Development. The use of electrochemical technology allows our XT-8 system to leverage third-party advances in microelectronics such as miniaturization and manufacturing efficiencies. Many electronic components associated with our core processes are produced in large volumes at low cost and size for use in numerous fields including automotive, aerospace, information technology and medical devices. By avoiding the use of fluidic handling and optical or fluorescent detection, we believe our eSensor technology can be applied at low cost to numerous testing environments in addition to our current target markets, including field testing and point-of-care applications.
Straightforward Development of New Tests. Our eSensor technology is highly flexible, and we believe the main design consideration in developing new diagnostic tests for our XT-8 system is our ability to access and synthesize the appropriate capture and signal probes. Our versatile platform allows us to add new diagnostic tests to our menu or to add new content to existing diagnostic tests without modifying the XT-8 system. This ease of assay development and our versatile platform allows us to focus our research and development resources on developing new commercial test products.
Functionality Outside of Molecular Diagnostics. Our eSensor technology has broad applicability to detect a range of biomolecules. Independently, and through collaborative research with university and industry partners, we have demonstrated eSensor detection of proteins and small molecule drugs. This versatility opens the possibility of developing mixed analyte sensors, such as tests that can detect antibodies to a certain pathogen plus the pathogen itself, or genetic variations in drug metabolism plus monitoring of the drug level itself.
Research and Development
As of March 31, 2011, we had 17 employees focused on research and development. In 2010, we moved our research and development activities to our new approximately 31,000 square foot headquarters in Carlsbad,
61
Table of Contents
California. Our research and development expenditures were $2.5 million for the three months ended March 31, 2011, and $6.5 million, $5.6 million and $13.4 million for the years ended December 31, 2010, 2009 and 2008, respectively. The overall reduction from 2008 in research and development expenses was due to the completion of our XT-8 system in 2009. The increase from 2009 to 2010 was due to higher payroll costs, relocation and recruitment costs and higher facility allocations. Research and development expenses were $1.5 million for the three months ended March 31, 2010. The increase for the three months ended March 31, 2011 was due to increased headcount, severance related expenses, increased development supplies, clinical trial costs and intellectual property related costs, cost incurred to obtain regulatory certification for our Carlsbad manufacturing facility and increased facility related costs.
In addition to expanding the diagnostic test menu for our XT-8 system and developing our NexGen system, our research and development team is focused on the following initiatives:
Improving the Clinical and Practical Utility of our Tests. An important role of our research and development team is to help establish the clinical utility and value of our molecular diagnostic tests. We have and intend to continue to partner with academic and reference laboratories to perform validation and clinical studies on our tests. Key aspects of our efforts are aimed at improving workflow in the laboratory setting, positively comparing our tests to historical or “gold standard” tests and demonstrating that our tests can help improve patient care and lower diagnostic and medical treatment costs. We intend to publish the results from these clinical studies in peer-reviewed or trade journals, submit them to regulatory bodies and present them at industry conferences in support of our commercialization strategy.
Developing New Test Capabilities. We are developing capabilities for utilizing our eSensor technology in protein and small molecule detection, both independently and through research collaborations. These capabilities may enhance our future menu offerings or provide us with out-licensing opportunities. We are also exploring direct gene expression analysis opportunities through collaboration with oncology specialists in industry and academia. These opportunities may allow us to develop quantitative tests that are competitive with the “gold standard” real-time PCR tests but that are simple to perform in a multiplex manner with our XT-8 system.
Manufacturing
We manufacture our proprietary test cartridges and ancillary reagents at our approximately 31,000 square foot headquarters in Carlsbad, California after recently completing the move of our manufacturing operations from Pasadena, California. Our reagent formulation, test cartridge manufacturing and packaging of final components and cartridges are performed by us in accordance with applicable guidelines for medical device manufacturing. We outsource manufacturing of our XT-8 system, as well as the oligonucleotide raw materials and much of the disposable component molding and sub-component assembly for our test cartridges. In particular, our XT-8 system is manufactured by a single source supplier, Aubrey Group Inc., who specializes in contract design and manufacturing of electronic and electromechanical devices for medical use. We believe we can secure other suppliers on commercially reasonable terms for the products and parts we outsource.
We have implemented a quality management system designed to comply with FDA regulations and ISO standards governing diagnostic medical device products. These regulations carefully control the design, manufacture, testing and release of diagnostics products as well as raw material receipt and control. We also have controlled methods for the consistent manufacturing of our proprietary test cartridges and reagents at our facilities. All key outsourcing partners are generally ISO-certified to help assure a continual supply of high quality components.
We plan to continue to manufacture components that we determine are highly proprietary or highly custom to produce, while outsourcing more commodity-like components. We are likely to establish additional outsourcing partnerships as we manufacture more products. We believe our new facility in Carlsbad, California is adequate to meet our current and future manufacturing needs.
62
Table of Contents
Sales and Marketing
Our sales and marketing strategy is to expand the installed base and utilization of the XT-8 platform and consumables. Our products are sold in the United States through a geographically dispersed seven person direct sales force and four technical specialist service organization. They are supported by a centralized team of Product Managers, Marketing, Customer and Technical Support personnel.
Our sales representatives typically have extensive experience in molecular diagnostics and a network of laboratory contacts within their respective territories. We utilize our representatives’ knowledge along with market research databases to target and qualify our customers. We execute a variety of sales campaigns and strategies to meet the buying criteria of the different customer segments we serve. To support our expanding molecular test menu, growth in our customer base and launch plans of our next generation detection platform, we continue to make investments in these customer facing organizations.
We believe the XT-8 platform competes largely on the basis of strong performance and reliability, ease of use and streamlined laboratory workflow, a high value IVD menu with multiplexing capabilities, and a high return on investment. These and other advantages conferred by our chemistry are enabling us to provide clinicians and researchers with valuable molecular solutions. Our sales cycle typically includes customer evaluations and validations of our products. Upon successful validation, a customer can acquire our XT-8 system and consumables in the following ways:
| • | Reagent Rental: The reagent rental agreement requires a customer commitment to purchase a minimum number of cartridges over the term of the agreement, and a portion of the charge for each cartridge is a rental fee for the equipment. Our reagent rental agreements do not typically provide for any cancellation rights by the customer. The reagent rental agreements do allow us to remove, change or upgrade the XT-8 system at any time. |
| • | Capital Purchase: The XT-8 system is paid for upfront, and in its entirety, by the customer. Customers are also eligible to receive structured pricing incentives if they enter into an optional annual minimum cartridge commitment. For the three months ended March 31, 2011 and the year ended December 31, 2010, we sold one and ten XT-8 systems, respectively, to customers. This includes the sale of thirteen analyzers to customers, with each analyzer capable of analyzing eight test cartridges at one time. |
In the second half of 2011, we anticipate commencing planning for commercialization of our molecular diagnostic products in Europe and other international markets. We anticipate our sales and marketing strategy will involve a select network of partners and distributors. A distribution strategy is being developed for each relevant international market. It is expected that we will augment this effort with a team of our specialists who will enable our partners’ sales forces and provide technical support. We also intend to explore opportunities to leverage our intellectual property position in molecular diagnostics through out-licensing or the establishment of partnerships.
Customers
In 2010 and 2009, 28% and 38% of our revenues, respectively, were attributed to our three largest customers during the year. In 2010, one customer accounted for approximately 12% of our total revenues.
During 2010, we redefined the XT-8 system to be a product consisting of one control system and up to three analyzers, with each analyzer capable of analyzing eight test cartridges at one time. Placements are defined in terms of the number of analyzers sold to a customer, reflecting a direct correlation between the reagent test revenue opportunity and the number of test cartridges that can be analyzed at any one time. As of March 31, 2011, there were 102 analyzers at 76 unique customer sites, or approximately 1.3 analyzers per customer. This compares with 38 analyzers at 32 unique customer sites, or approximately 1.2 analyzers per customer as of March 31, 2010.
63
Table of Contents
The increase in analyzers and related revenue is due to an increase in the number of new customers buying our products and growth in additional tests from existing customers. We expect our clinical molecular diagnostic revenues to continue to increase in 2011.
Competition
We primarily face competition in the molecular diagnostic testing markets with testing products and systems developed by public and private companies such as Cepheid, Gen-Probe, Inc., Siemens, Hologic, Inc., Innogenetics, Inc, Luminex Corporation, Nanosphere, Inc., Qiagen NV, Roche Diagnostics and Abbott Diagnostics. Our diagnostic tests also face competition with the laboratory developed tests created by national and regional reference laboratories and hospitals. We believe that the XT-8 system competes largely on the basis of accuracy and reliability, enhanced laboratory workflow, multiplex capability, ease-of-use and return on investment for customers.
Many of our competitors have substantially greater financial, technical, research and other resources and larger, more established marketing, sales and distribution organizations than we do. Many of our competitors also offer broader product lines and have greater brand recognition than we do. Moreover, our existing and new competitors may make rapid technological developments that may result in our technologies and products becoming obsolete before we recover the expenses incurred to develop them or before they generate significant revenue.
Intellectual Property
To establish and protect our proprietary technologies and products, we rely on a combination of patent, copyright, trademark and trade-secret laws, as well as confidentiality provisions in our contracts. We have implemented a patent strategy designed to protect our technology and facilitate commercialization of our current and future products. Currently, our patent portfolio is comprised, on a worldwide basis, of 100 issued U.S. patents, 50 issued foreign patents (predominantly in Europe and Japan) and 28 pending domestic and foreign patent applications, all of which we own directly or for which we are the exclusive licensee. Our intellectual property portfolio for our core electrochemical technology was built through the combination of our acquisition of the Clinical Micro Sensors business from Motorola, licensing patents from third parties and the issuance of new patents to us to protect our ongoing development activities. Many of our issued and pending patents were exclusively licensed from the California Institute of Technology and Harvard University and generally cover our core technology relating to our XT-8 system.
We believe that our patent portfolio and licenses provide us with a robust intellectual property position for our electrochemical detection techniques, chemical insulators and attachment points on electrode surfaces and other technology that collectively form the staple of our eSensor platform.
In general, patents have a term of 20 years from the application filing date or earlier claimed priority date. Our issued and exclusively licensed patents will expire between 2013 and 2028 or later, with several of our pending applications having the potential to mature into patents that might expire in 2027, 2028 and 2029. Our success depends to a significant degree upon our ability to police infringement, derive licensing revenues and continue to develop proprietary products and technologies without infringing on the intellectual property rights of others.
We also rely in part on trade-secret protection of our intellectual property. We attempt to protect our trade secrets by entering into confidentiality agreements with third parties, employees and consultants. Our employees and consultants also sign agreements requiring that they assign to us their interests in intellectual property such as patents and copyrights arising from their work for us. All employees sign an agreement not to compete unfairly with us during their employment and upon termination of their employment through the misuse of confidential information, soliciting employees and soliciting customers.
64
Table of Contents
Government Regulation
The design, development, manufacture, testing and sale of our diagnostic products are subject to regulation by numerous governmental authorities, principally the FDA, and corresponding state and foreign regulatory agencies.
Regulation by the FDA
In the United States, the Federal Food, Drug, and Cosmetic Act, or FDCA, FDA regulations and other federal and state statutes and regulations govern, among other things, medical device design and development, preclinical and clinical testing, premarket clearance or approval, registration and listing, manufacturing, labeling, storage, advertising and promotion, sales and distribution, export and import, and post-market surveillance. The FDA regulates the design, manufacturing, servicing, sale and distribution of medical devices, including molecular diagnostic test kits and instrumentation systems. Failure to comply with applicable U.S. requirements may subject a company to a variety of administrative or judicial sanctions, such as FDA refusal to approve pending applications, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties and criminal prosecution.
Unless an exemption applies, each medical device we wish to distribute commercially in the United States will require marketing authorization from the FDA prior to distribution. The two primary types of FDA marketing authorization applicable to a device are premarket notification, also called 510(k) clearance, and premarket approval, also called PMA approval. The type of marketing authorization is generally linked to the classification of the device. The FDA classifies medical devices into one of three classes (Class I, II or III) based on the degree of risk the FDA determines to be associated with a device and the level of regulatory control deemed necessary to ensure the device’s safety and effectiveness. Devices requiring fewer controls because they are deemed to pose lower risk are placed in Class I or II. Class I devices are deemed to pose the least risk and are subject only to general controls applicable to all devices, such as requirements for device labeling, premarket notification and adherence to the FDA’s current Good Manufacturing Practices, or cGMP, and Quality System Requirements, as reflected in its QSR. Class II devices are intermediate risk devices that are subject to general controls and may also be subject to special controls such as performance standards, product-specific guidance documents, special labeling requirements, patient registries or postmarket surveillance. Class III devices are those for which insufficient information exists to assure safety and effectiveness solely through general or special controls and include life-sustaining, life-supporting or implantable devices, devices of substantial importance in preventing impairment of human health, or which present a potential, unreasonable risk of illness or injury.
Most Class I devices and some Class II devices are exempted by regulation from the 510(k) clearance requirement and can be marketed without prior authorization from the FDA. Some Class I devices that have not been so exempted and Class II devices are eligible for marketing through the 510(k) clearance pathway. By contrast, devices placed in Class III generally require PMA approval or 510(k) de novo clearance prior to commercial marketing. The PMA approval process is more stringent, time-consuming and expensive than the 510(k) clearance process, however, the 510(k) clearance process has also become increasingly stringent and expensive. The FDA has cleared our XT-8 system with our eSensor Warfarin Sensitivity Test, Cystic Fibrosis Genotyping Test and Thrombophilia Risk Test as Class II devices via the 510(k) clearance process.
510(k) Clearance. To obtain 510(k) clearance for a medical device, an applicant must submit a premarket notification to the FDA demonstrating that the device is “substantially equivalent” to a device legally marketed in the United States that is not subject to PMA approval, commonly known as the “predicate device.” A device is substantially equivalent if, with respect to the predicate device, it has the same intended use and has either (i) the same technological characteristics or (ii) different technological characteristics and the information submitted demonstrates that the device is as safe and effective as a legally marketed device and does not raise different questions of safety or effectiveness. A showing of substantial equivalence sometimes, but not always, requires clinical data. Generally, the 510(k) clearance process can exceed 90 days and may extend to a year or more.
65
Table of Contents
After a device has received 510(k) clearance for a specific intended use, any change or modification that significantly affects its safety or effectiveness, such as a significant change in the design, materials, method of manufacture or intended use, may require a new 510(k) clearance or PMA approval and payment of an FDA user fee. The determination as to whether or not a modification could significantly affect the device’s safety or effectiveness is initially left to the manufacturer using available FDA guidance; however, the FDA may review this determination to evaluate the regulatory status of the modified product at any time and may require the manufacturer to cease marketing and recall the modified device until 510(k) clearance or PMA approval is obtained. The manufacturer may also be subject to significant regulatory fines or penalties.
Before we can submit a medical device for 510(k) clearance, we may have to perform a series of generally short studies over a period of months, including method comparison, reproducibility, interference and stability studies to ensure that users can perform the test successfully. Some of these studies may take place in clinical environments, but are not usually considered clinical trials. For PMA submissions, we would generally be required to conduct a longer clinical trial over a period of years that supports the clinical utility of the device and how the device will be used.
Although clinical investigations of most devices are subject to the investigational device exemption, or IDE, requirements, clinical investigations of molecular diagnostic tests, including our products and products under development, are generally exempt from the IDE requirements. Thus, clinical investigations by intended users for intended uses of our products generally do not require the FDA’s prior approval, provided the clinical evaluation testing is non-invasive, does not require an invasive sampling procedure that presents a significant risk, does not intentionally introduce energy into the subject and is not used as a diagnostic procedure without confirmation by another medically established test or procedure. In addition, our products must be labeled per FDA regulations “for research use only-RUO” or “for investigational use only-IUO,” and distribution controls must be established to assure that our products distributed for research, method comparisons or clinical evaluation studies are used only for those purposes.
PMA Approval. A PMA application requires the payment of significant user fees. PMA applications must be supported by valid scientific evidence, which typically requires extensive data, including technical, preclinical, clinical and manufacturing data, to demonstrate to the FDA’s satisfaction the safety and effectiveness of the device. A PMA application must also include, among other things, a complete description of the device and its components, a detailed description of the methods, facilities and controls used to manufacture the device, and proposed labeling.
The FDA has 45 days from its receipt of a PMA to determine whether the application will be accepted for filing based on the agency’s threshold determination that it is sufficiently complete to permit substantive review. Once the submission is accepted for filing, the FDA begins an in-depth review. During this review period, the FDA may request additional information or clarification of information already provided. In addition, the FDA will conduct a pre-approval inspection of the manufacturing facility or facilities to ensure compliance with the QSR, which requires manufacturers to follow design, testing, control, documentation and other quality assurance procedures.
FDA review of an initial PMA application is required by statute to take between six to ten months, although the process typically takes significantly longer, and may require several years to complete. The FDA can delay, limit or deny approval of a PMA application for many reasons, including:
| • | it is not demonstrated that there is reasonable assurance that the device is safe or effective under the conditions of use prescribed, recommended, or suggested in the proposed labeling; |
| • | the data from preclinical studies and clinical trials may be insufficient to support approval; and |
| • | the manufacturing process, methods, controls or facilities used for the manufacture, processing, packing or installation of the device do not meet applicable requirements. |
66
Table of Contents
If the FDA evaluations of both the PMA application and the manufacturing facilities are favorable, the FDA will either issue an approval letter or an approvable letter, which usually contains a number of conditions that must be met in order to secure final approval of the PMA. If the FDA’s evaluation of the PMA or manufacturing facilities is not favorable, the FDA will deny approval of the PMA or issue a not approvable letter. A not approvable letter will outline the deficiencies in the application and, where practical, will identify what is necessary to make the PMA approvable. The FDA may also determine that additional clinical trials are necessary, in which case the PMA approval may be delayed for several months or years while the trials are conducted and then the data submitted in an amendment to the PMA. Once granted, PMA approval may be withdrawn by the FDA if compliance with post approval requirements, conditions of approval or other regulatory standards is not maintained or problems are identified following initial marketing.
Approval by the FDA of new PMA applications or PMA supplements may be required for modifications to the manufacturing process, labeling, device specifications, materials or design of a device that is approved through the PMA process. PMA supplements often require submission of the same type of information as an initial PMA application, except that the supplement is limited to information needed to support any changes from the device covered by the original PMA application and may not require as extensive clinical data or the convening of an advisory panel.
Regulation After FDA Clearance or Approval. Any devices we manufacture or distribute pursuant to clearance or approval by the FDA are subject to pervasive and continuing regulation by the FDA and certain state agencies. We are required to adhere to applicable regulations setting forth detailed cGMP requirements, as set forth in the QSR, which include, among other things, testing, control and documentation requirements. Non-compliance with these standards can result in, among other things, fines, injunctions, civil penalties, recalls or seizures of products, total or partial suspension of production, refusal of the government to grant 510(k) clearance or PMA approval of devices, withdrawal of marketing approvals and criminal prosecutions. We have designed and implemented our manufacturing facilities under the FDA’s cGMP requirements.
Because we are a manufacturer of medical devices, we must also comply with medical device reporting requirements by reviewing and reporting to the FDA whenever there is evidence that reasonably suggests that one of our products may have caused or contributed to a death or serious injury. We must also report any incident in which our product has malfunctioned if that malfunction would likely cause or contribute to a death or serious injury if it were to recur. Labeling and promotional activities are subject to scrutiny by the FDA and, in certain circumstances, by the Federal Trade Commission. Medical devices approved or cleared by the FDA may not be promoted for unapproved or uncleared uses, otherwise known as “off-label” promotion. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability, including substantial monetary penalties and criminal prosecution.
We are also subject to numerous federal, state and local laws relating to such matters as safe working conditions, manufacturing practices, environmental protection, fire hazard control and disposal of hazardous or potentially hazardous substances. Some of these laws require us to obtain licenses or permits to conduct our operations. We have numerous policies and procedures in place to ensure compliance with these laws and to minimize the risk of occupational exposure to hazardous materials. We do not expect the operations of our products to produce significant quantities of hazardous or toxic waste or radiation that would require use of extraordinary disposal practices. Although the costs to comply with these applicable laws and regulations have not been material, we cannot predict the impact on our business of new or amended laws or regulations or any changes in the way existing and future laws and regulations are interpreted or enforced, nor can we ensure we will be able to obtain or maintain any required licenses or permits.
Export of Our Products. Export of products subject to the 510(k) notification requirements, but not yet cleared to market, is permitted with FDA authorization provided certain requirements are met. Unapproved products subject to the PMA approval requirements may be exported if the exporting company and the device meet certain criteria, including, among other things, that the device complies with the laws of the receiving
67
Table of Contents
country and the company submits a “Simple Notification” to the FDA when the company begins to export. If the company or device does not comply with such criteria, FDA approval must be obtained for export. To obtain FDA export approval, if required, we must meet certain requirements, including, among other things and with some exceptions, documentation demonstrating that the product is approved for import into the country to which it is to be exported and, in some instances, safety data to demonstrate that export of the device will not be contrary to the public health or safety.
Clinical Laboratory Improvement Amendments of 1988. The use of our products is also affected by CLIA and related federal and state regulations, which provide for regulation of laboratory testing. Any customers using our products for clinical use in the United States will be regulated under CLIA, which is intended to ensure the quality and reliability of laboratory testing in the United States. In particular, these regulations mandate that clinical laboratories must be certified by the federal government or a federally approved accreditation agency, or must be located in a state that has been deemed exempt from CLIA requirements because the state has in effect laws that provide for requirements equal to or more stringent than CLIA requirements. Moreover, these laboratories must meet quality assurance, quality control and personnel standards, and they must undergo proficiency testing and inspections. The CLIA standards applicable to clinical laboratories are based on the complexity of the method of testing performed by the laboratory, which range from “waived” to “moderate complexity” to “high complexity.” We expect that most of our products will be categorized as “high complexity,” since most molecular diagnostic tests are currently FDA-cleared as CLIA “high complexity” devices.
Other Legislation. On September 27, 2007, the President signed the Food and Drug Administration Amendments Act of 2007, or FDAAA. Among other significant changes and requirements it imposes, the new legislation expands the federal government’s clinical trial registry and results databank maintained by the NIH to include all (with limited exceptions) medical device trials. In particular, it requires certain information about device trials, including a description of the trial, participation criteria, location of trial sites, and contact information, to be sent to NIH for inclusion in a publicly accessible database. In addition, the results of clinical trials that form the primary basis for efficacy claims or are conducted after a device is approved or cleared must be posted to the results databank. Under the FDAAA, companies that violate these and other provisions of the new law are subject to substantial civil monetary penalties.
Foreign Government Regulation. We intend to market our products in European and other selected international markets. Before doing so, we or our partners and distributors will need to receive regulatory approval. The regulatory review process for medical devices varies from country to country, and many countries also impose product standards, packaging requirements, labeling requirements and import restrictions on devices. Each country has its own tariff regulations, duties and tax requirements. Failure to comply with applicable foreign regulatory requirements may subject a company to fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Third-Party Payor Reimbursements
Obtaining reimbursement approval for a health care product or service from a government or other third-party payor is a time consuming and costly process that could require us to provide supporting scientific, clinical and health economic data for the use of our products to the payor. We may not be able to provide data sufficient to gain acceptance with respect to reimbursement. Even when a payor determines that a product or service is eligible for reimbursement, the payor may impose coverage limitations that preclude payment for some uses that are approved by the FDA or comparable authorities. In addition, there is a risk that full reimbursement may not be available for high-priced products. Moreover, eligibility for coverage does not imply that any product or service will be reimbursed in all cases or at a rate that allows our customers to make a profit or cover their costs. Initial or interim reimbursements for products and services, if available, may also not be sufficient to cover costs and may not be made permanent.
Successful sales of our products in the United States and other countries will depend on the availability of reimbursement from third-party payors such as private insurance plans, managed care organizations, and
68
Table of Contents
Medicare and Medicaid. Our customers have obtained reimbursement for our eSensor Cystic Fibrosis Genotyping Test and eSensor Thrombophilia Risk Test for the XT-8 system and we believe that each of our tests in development are covered by existing CPT codes and will be eligible for coverage by Medicare and Medicaid and most third-party payors. However, Medicare and Medicaid generally do not reimburse providers who use our Warfarin Sensitivity Test. In 2010, the American Medical Association formed a molecular pathology working group to provide recommendations for modernization of codes for molecular diagnostic and genetic tests. This group is expected to issue recommendations in 2011 that will be considered for implementation in 2012. Outside of the United States, health care reimbursement systems vary from country to country, and to the extent we begin to sell our products outside the United States, we may not be able to obtain adequate reimbursement coverage, if any, for our products.
In addition, we may develop tests in the future that do not relate to previously established CPT codes and we may need to obtain new CPT codes in order to obtain reimbursement. Reimbursement by a third-party payor depends on a number of factors, including applicable coverage policies and limitations, the level of demand by health care providers and the payor’s determination that the use of a new product is medically necessary and represents a clinical advance. In addition, both government and non-government third-party payors routinely limit reimbursement coverage and reimbursement amounts for diagnostic tests. If our customers cannot receive sufficient levels of reimbursement when using our products, our ability to sell them will be significantly constrained.
Fraud and Abuse Regulations
We are subject to numerous federal and state health care anti-fraud laws, including the federal anti-kickback statute and False Claims Act, that are intended to reduce waste, fraud and abuse in the health care industry. These laws are broad and subject to evolving interpretations. They prohibit many arrangements and practices that are lawful in industries other than health care, including certain payments for consulting and other personal services, some discounting arrangements, the provision of gifts and business courtesies, the furnishing of free supplies and services, and waivers of payments. In addition, many states have enacted or are considering laws that limit arrangements between medical device manufacturers and physicians and other health care providers and require significant public disclosure concerning permitted arrangements. These laws are vigorously enforced against medical device manufacturers and have resulted in manufacturers paying significant fines and penalties and being subject to stringent corrective action plans and reporting obligations. We must operate our business within the requirements of these laws and, if we were accused of violating them, could be forced to expend significant resources on investigation, remediation and monetary penalties. Companies targeted in such prosecutions have paid substantial fines in the hundreds of millions of dollars or more, have been forced to implement extensive corrective action plans, can be excluded from federal health care programs and become subject to substantial civil and criminal penalties, and have often become subject to consent decrees severely restricting the manner in which they conduct their business.
To the extent we commence commercial operations overseas, we will be subject to the FCPA and other countries’ anti-corruption/anti-bribery regimes, such as the U.K. Bribery Act. The FCPA prohibits improper payments or offers of payments to foreign governments and their officials for the purpose of obtaining or retaining business. Safeguards we implement to discourage improper payments or offers of payments by our employees, consultants, sales agents or distributors may be ineffective, and violations of the FCPA and similar laws may result in severe criminal or civil sanctions, or other liabilities or proceedings against us, any of which would likely harm our reputation, business, financial condition and result of operations.
Patient Protection and Affordable Care Act
Our operations will also be impacted by the federal Patient Protection and Affordable Care Act of 2010, as modified by the Health Care and Education Reconciliation Act of 2010, which we refer to as the Health Care Act. The Health Care Act imposes a 2.3% excise tax on sales of medical devices by manufacturers. Taxable devices include any medical device defined in Section 201(h) of the FDCA and intended for use by humans,
69
Table of Contents
with limited exclusions for devices purchased by the general public at retail for individual use. There is no exemption for small companies, and we expect to begin paying the tax in 2013. The Health Care Act also requires manufacturers to report to the Department of Health and Human Services detailed information about financial arrangements with physicians and teaching hospitals. These reporting provisions preempt state laws that require reporting of the same information, but not those that require reports of different or additional information. Failure to comply subjects the manufacturer to significant civil monetary penalties. We expect compliance with the Health Care Act to impose significant administrative and financial burdens on us.
Employees
As of March 31, 2011, we had 62 employees. Approximately 17 were involved in research and development, 22 in operations, manufacturing and quality assurance, 15 in sales and marketing, and 8 in finance, legal and other administrative functions. Our success will depend in large part upon our ability to attract and retain employees. We face competition in this regard from other companies, research and academic institutions, government entities and other organizations. None of our employees are covered by a collective bargaining agreement.
Legal Proceedings
We are from time to time subject to various claims and legal actions during the ordinary course of our business. We believe that there are currently no claims or legal actions that would reasonably be expected to have a material adverse effect on our results of operations or financial condition.
General Information
Our principal corporate offices are located at 5964 La Place Court, Carlsbad, California 92008 and our telephone number is (760) 448-4300. We were incorporated in Delaware in February 2010.
Our Internet address is www.genmarkdx.com. We make available on our website, free of charge, our annual report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and any amendments to those reports, as soon as reasonably practicable after we electronically file such material with, or furnish it to, the Securities and Exchange Commission, or SEC. We also make available on our Internet site public financial information for which a report is not required to be filed with or furnished to the SEC. Our SEC reports and other financial information can be accessed through the investor relations section of our Internet site. The information found on our Internet site is not part of this prospectus.
70
Table of Contents
Directors and Officers
The following table provides information regarding the directors and officers of GenMark Diagnostics, Inc., including their ages and positions, as of May 25, 2011:
| Name |
Age | Position | ||||
| Hany Massarany |
50 | Chief Executive Officer, President and Director | ||||
| Paul Ross |
37 | Chief Financial Officer, Treasurer and Secretary | ||||
| Jon Faiz Kayyem, Ph.D. |
47 | Chief Scientific Officer | ||||
| Jeffrey Hawkins |
33 | Sr. Vice President, Marketing and Business Development | ||||
| Jennifer Williams |
38 | Sr. Vice President, Global Operations | ||||
| Christopher Gleeson |
61 | Chairman of the Board | ||||
| Daryl J. Faulkner |
62 | Director | ||||
| James Fox, Ph.D. |
59 | Director | ||||
| Kevin C. O’Boyle |
55 | Director | ||||
The business address for our directors and senior management is c/o GenMark Diagnostics, Inc., 5964 La Place Court, Suite 100, Carlsbad, California.
Hany Massarany has served as President and Chief Executive Officer and Director of GenMark Diagnostics, Inc. since May 2011. From February 2009 to April 2011, Mr. Massarany served as President of Ventana Medical Systems and Head of Roche Tissue Diagnostics. From 1999 to 2009, he held various global leadership positions with Ventana, including Chief Operating Officer, Executive Vice President Worldwide Operations, Senior Vice President Corporate Strategy and Development, and Vice President North American Commercial Operations. Prior to Ventana, Mr. Massarany held executive management positions with Bayer Diagnostics and Chiron Diagnostics, working in both Asia Pacific and the United States. Mr. Massarany received a B.S. degree in Microbiology and Immunology from Monash University, Australia and an MBA from Melbourne University, Australia. We believe Mr. Massarany is qualified to serve on our board of directors based on his executive experience in the medical device and molecular diagnostics industries as described above.
Paul Ross has served as Chief Financial Officer, Treasurer and Secretary of GenMark Diagnostics, Inc. since April 2011. Mr. Ross previously served as the chief financial officer of Teledata Technology Solutions, a global provider of information technology consulting services, from October 2009 to March 2011. From March 2007 to April 2009, Mr. Ross served as Senior Vice President—Finance and Chief Financial Officer of Meade Instruments Corp., a NASDAQ-listed multinational consumer optics company. Mr. Ross also previously served as the Chief Financial Officer and Treasurer of Power-One, Inc., a NASDAQ-listed manufacturer of power supply products for use in communication, semiconductor, testing, medical, industrial and other electronic instruments, from May 2005 to March 2007. From April 2001 to May 2005, Mr. Ross held various positions with Power-One including Vice President Finance and Corporate Controller, Director of Corporate Finance, and Manager of Financial Planning and Reporting. From December 1998 to April 2001, Mr. Ross was the senior financial analyst of the external reporting group with BP/Atlantic Richfield Company (ARCO). From September 1996 to December 1998, Mr. Ross was an audit associate and then senior audit associate with PricewaterhouseCoopers LLP. Mr. Ross received his BA degree from UCLA and his MBA degree from USC, and is a Certified Public Accountant.
Jon Faiz Kayyem, Ph.D. has served as Chief Scientific Officer since August 2010. Previously, he served as President and Chief Executive Officer of GenMark Diagnostics, Inc. from March 2010 to August 2010 and as a Director from March 2010 to May 2011. Dr. Kayyem has served as President and Chief Executive Officer of Osmetech plc., our subsidiary following our reorganization, since August 2009 and Chairman of the board of directors of Osmetech plc from January 2009 to August 2009. Dr. Kayyem attended Yale University and
71
Table of Contents
received his combined Master and Bachelor of Sciences in Molecular Biophysics and Biochemistry in 1985. He received his Ph.D. in Molecular Biology in 1991 at The California Institute of Technology, or Caltech. Dr. Kayyem remained at Caltech as a Senior Research Fellow until 1995, when he founded Clinical Micro Sensors to commercialize technical innovations he developed while at Caltech. In 2000, Clinical Micro Sensors was sold to Motorola, Inc. for approximately $280 million, and subsequently purchased by Osmetech plc in 2005. In 2004, Dr. Kayyem left Clinical Micro Sensors and co-founded the biotechnology fund management company, Efficacy Capital Limited, where he served as managing partner until September 2009. We believe Dr. Kayyem is qualified to serve on our board of directors based on his executive experience at Clinical Micro Sensors where he led the development and growth of the company through its acquisition by Motorola, Inc.
Jeffrey Hawkins has served as Senior Vice President, Marketing and Business Development since November 2010. Prior to that, he held the position of Vice President of Business Development from May 2010 to November 2010 and in the same capacity for Osmetech Technology, Inc., a wholly-owned subsidiary of Osmetech plc, from March 2010 to May 2010 and as Vice President of Marketing from December 2009 to March 2010. From July 2008 until December 2009, Mr. Hawkins was Executive Director of Laboratory Marketing for Hologic, Inc., a developer, manufacturer and supplier of medical imaging systems and diagnostic and surgical products. From November 2006 until its acquisition by Hologic in June 2008, Mr. Hawkins served as Executive Director of Marketing of Third Wave Technologies Inc., a provider of DNA and RNA analysis products to clinical, research and agricultural customers. Prior to Third Wave, Mr. Hawkins has held various positions of increasing responsibility in the areas of Marketing, Product Development and Operations for Sysmex America and Abbott Laboratories. Mr. Hawkins is currently a member of the board of directors for Ohmx Corporation, a bioelectronic detection company developing a monitoring device to be used in all point-of-care (POC) settings. Mr. Hawkins received a B.A. in Chemistry with honors from Concordia University and an M.B.A from Keller Graduate School of Management.
Jennifer Williams was appointed Senior Vice President, Global Operations in November 2010, and is responsible for Manufacturing Operations, Human Resources and Asia Pacific Commercial Operations. Prior to joining GenMark, she held the position of Senior Human Resource Executive with Cerberus Operations and Advisory Company, a private equity firm, from February 2008 to May 2010, responsible for human resources oversight and transformation of global companies in the portfolio. From January 2005 to January 2008, she served as Vice President Human Resources at HD Supply, a wholesale distribution company serving the infrastructure, construction, and maintenance markets, initially as part of The Home Depot organization and subsequently spun off in 2007. Previous to that, she led Talent Management for The Home Depot including organization design, succession planning, leadership programs, and executive development. Ms. Williams began her career at Honeywell (formerly AlliedSignal) and held positions of increasing responsibility in Quality, Operations, Program Management, and Organization Effectiveness. Ms. Williams received her MBA from Case Western Reserve in Organizational Behavior and an undergraduate degree in Industrial and Operations Engineering from the University of Michigan. Ms. Williams holds a certification in Organization Design and is a Six Sigma greenbelt.
Christopher Gleeson has served as Chairman of the Board of GenMark Diagnostics, Inc. since March 2010. Mr. Gleeson served as Chief Executive Officer of GenMark Diagnostics, Inc. on an interim basis from August 2010 to May 2011, and has served as Chairman of the Board of Osmetech plc since July 2009. Mr. Gleeson was formerly President, Chief Executive Officer and a Director of Ventana Medical Systems, Inc., a leading supplier of automated diagnostic systems to the anatomical pathology market where he served from 1999 to February 2008. Following the acquisition of Ventana by Roche Diagnostics in February 2008 for $3.4 billion, Mr. Gleeson became a member of the board of directors of Roche Diagnostics. Prior to joining Ventana, Mr. Gleeson was Senior Vice-President of Bayer Diagnostics, the diagnostics division of Bayer Healthcare Pharmaceuticals and general manager of the U.S. commercial operations for Chiron Diagnostics, the diagnostics division of Chiron Corporation. Prior to that time, he was the founder, owner, and managing director of Australian Diagnostics Corporation. Mr. Gleeson attended the Pharmacy and Business Schools at Monash University in Australia. We believe Mr. Gleeson is qualified to serve on our board of directors based on his executive experience in the medical device and molecular diagnostics industries as described above.
72
Table of Contents
Daryl J. Faulkner has served on the board of directors of GenMark Diagnostics, Inc. since March 2010. Mr. Faulkner was appointed to the board of directors of Osmetech plc in August 2008, serving as Non-Executive Chairman until December 2008. Mr. Faulkner is currently a member of the board of directors of AspenBio Pharma, an emerging biotechnology company engaged in the research, development, manufacture, and licensing of novel diagnostics and drugs. He also served as Executive Chair and Interim Chief Executive Officer of AspenBio Pharma from February 2009 until March 2010. From August 2008 to January 2009, Mr. Faulkner served as a consultant to Qiagen NV, a leading provider of innovative sample and assay technologies and products, in connection with its integration of Digene Corp., a developer of gene-based diagnostic tests acquired by Qiagen in August 2007. Mr. Faulkner had served as President and Chief Executive Officer and a director of Digene from December 2006 until consummation of Qiagen’s acquisition of Digene. From 1998 until March 2006, Mr. Faulkner served in several executive roles at Invitrogen Corp., a life sciences company, including Senior Vice President, Business Segment Management from 2003 until March 2006. Mr. Faulkner received a B.S. in Industrial Relations from the University of North Carolina, Chapel Hill and an M.A. in Business Management from Webster University. We believe Mr. Faulkner is qualified to serve on our board of directors and as serve as Chair of our Corporate Governance and Nominating Committee based on his executive experience in the medical device and molecular diagnostics industries as described above.
James Fox, Ph.D., was appointed to the board of directors of GenMark Diagnostics, Inc. in September 2010. Dr. Fox has extensive experience in global technology and healthcare businesses. He led the start up of Invetech, an Australian contract research and development company that specializes in healthcare products and complex instruments for international markets. Invetech was merged with Australian Securities Exchange listed Vision Systems Limited in 1993, and Dr. Fox took over as Group Managing Director of the combined entity. In January 2007, Vision Systems Ltd., then a leading global cancer diagnostics company, was acquired by Danaher Corporation. Prior to Invetech, Dr. Fox spent seven years working as a consultant and director with PA Technology. Dr. Fox currently serves as Chairman of the Board of Biota Holdings Limited, a director of Air New Zealand Ltd., a director of TTP Group plc and as a director of MS Research Australia, a not-for-profit organization aimed at financing public multiple sclerosis research. Dr. Fox received his Bachelor’s, Master’s and Ph.D. degrees in engineering from the University of Melbourne. We believe Dr. Fox is qualified to serve on our board of directors and as serve as Chair of our Compensation Committee based on his executive experience in the medical device and molecular diagnostics industries as described above.
Kevin C. O’Boyle has served on the board of directors of GenMark Diagnostics, Inc. since March 2010. Mr. O’Boyle is currently a member of the board of directors of Tornier N.V., a publicly-held global orthopedics company, and serves as Senior Vice President and Chief Financial Officer at Advanced BioHealing, Inc. since December 2010. Previously, Mr. O’Boyle served as the Chief Financial Officer of NuVasive, Inc., a medical device company focused on the design, development and marketing of products for the surgical treatment of spine disorders, from January 2003 to December 2009 and the Executive Vice President of NuVasive from December 2004 to December 2009. Prior to that time, Mr. O’Boyle served in various positions during his seven years with ChromaVision Medical Systems, Inc., a publicly traded medical device firm specializing in the oncology market, including as its Chief Financial Officer and Chief Operating Officer. Also, Mr. O’Boyle held various positions during his six years with Albert Fisher North America, Inc., a publicly traded international food company, before it was sold in 1996, including Chief Financial Officer and Senior Vice President of Operations. Mr. O’Boyle is a CPA and received a B.S. in Accounting from the Rochester Institute of Technology and successfully completed the Executive Management Program at the University of California at Los Angeles, John E. Anderson Graduate Business School. We believe Mr. O’Boyle is qualified to serve on our board of directors and serve as Chair of our Audit Committee based on his executive experience in the medical device industry and his financial and accounting expertise as described above.
73
Table of Contents
Board Composition
Our certificate of incorporation and bylaws provide that the authorized number of directors may be changed only by resolution of the board of directors. We currently have five members serving on our board of directors. In accordance with our certificate of incorporation and bylaws, our board of directors is divided into three classes with staggered three-year terms. At each annual meeting of stockholders, the successors to the directors whose terms then expire will be elected to serve until the third annual meeting following the election. Our directors are divided among the three classes as follows:
| Director |
Class |
Expiration of Term | ||
| Daryl J. Faulkner |
Class I Director | 2014 Annual Meeting | ||
| James Fox, Ph.D. |
Class I Director | 2014 Annual Meeting | ||
| Hany Massarany |
Class II Director | 2012 Annual Meeting | ||
| Kevin C. O’Boyle |
Class II Director | 2012 Annual Meeting | ||
| Christopher Gleeson |
Class III Director | 2013 Annual Meeting |
Board Leadership Structure
In May 2011, our Board separated the positions of Chief Executive Officer and Chairman with the appointment of Hany Massarany as our Chief Executive Officer. Our Board believes that the separation of these positions strengthens the independence of our Board and allows us to have a Chairman focused on the leadership of the Board while allowing our Chief Executive Officer to focus more of his time and energy on company strategy and managing our operations.
Role of Board in Risk Oversight Process
The responsibility for the day-to-day management of risk lies with our management, while the Board is responsible for overseeing the risk management process to help ensure that it is properly designed, well-functioning and consistent with the our overall corporate strategy. Each year our management identifies what it believes are the top individual risks we face. These risks are then discussed and analyzed with the Board. This enables the Board to coordinate the risk oversight role, particularly with respect to risk interrelationships. However, in addition to the Board, the committees of the Board consider the risks within their areas of responsibility. The Audit Committee oversees the risks associated with the our financial reporting and internal controls, the Compensation Committee oversees the risks associated with our compensation practices, including an annual review of our risk assessment of its compensation policies and practices for its employees, and the Corporate Governance and Nominating Committee oversees the risks associated with our overall governance, corporate compliance policies (for example, policies addressing relationships with health care professionals and compliance with anti-kickback laws) and its succession planning process to understand that we have a slate of future, qualified candidates for key management positions.
Committees of the Board of Directors/Corporate Governance
Directors are expected to attend meetings of the Board and any Board committees on which they serve. The Board has three standing committees to facilitate and assist the Board in the execution of its responsibilities: the Audit Committee, the Compensation Committee and the Corporate Governance and Nominating Committee. Each of these committees has the responsibilities described in the committee charters which are available on our website at www.genmarkdx.com. Our Board may also establish other committees from time to time to assist in the discharge of its responsibilities.
Audit Committee. The Audit Committee currently consists of Daryl Faulkner, James Fox, Ph.D. and Kevin C. O’Boyle (Chair). The Board has determined that all members of the Audit Committee are independent directors under the NASDAQ Global Market listing standards and each of them is able to read and fundamentally understand financial statements. The Board has determined that Kevin C. O’Boyle qualifies as an “audit committee financial expert” as defined by the rules of the Securities and Exchange Commission. The purpose of the Audit Committee is to oversee both our accounting and financial reporting processes as well as
74
Table of Contents
audits of its financial statements. The responsibilities of the Audit Committee include appointing and approving the compensation of the independent registered public accounting firm selected to conduct the annual audit of our accounts, reviewing the scope and results of the independent audit, reviewing and evaluating internal accounting policies, and approving all professional services to be provided by our independent registered public accounting firm. The Audit Committee is governed by a written charter approved by the Board.
Compensation Committee. The Compensation Committee currently consists of Daryl Faulkner, James Fox, Ph.D. (Chair) and Kevin C. O’Boyle. The Board has determined that all members of the Compensation Committee are independent directors under the NASDAQ Global Market listing standards. The Compensation Committee administers our benefit and stock plans, reviews and administers all compensation arrangements for executive officers, and establishes and reviews general policies relating to the compensation and benefits of our officers and employees. The Compensation Committee meets several times a year to review, analyze and set compensation packages for our executive officers, which include our CEO, our Chief Scientific Officer, our Chief Financial Officer and each of our other senior officers. The Compensation Committee determines the CEO’s compensation and, as it deems appropriate, leverages industry benchmark compensation data. For the other executive officers, the CEO prepares and presents to the Compensation Committee performance assessments and compensation recommendations. Following consideration of the CEO’s presentation, the Compensation Committee may accept or adjust the CEO’s recommendations. The other executive officers are not present during this process. For more information, please see below under “Compensation Discussion and Analysis.” The Compensation Committee is governed by a written charter approved by the Board.
Our Compensation Committee reviews and evaluates potential risks related to our compensation policies and practices for employees and has determined that we have no compensation risks that are reasonably likely to have a material adverse effect on our company. We structure our compensation to address company-wide risk. We believe the combination of base salary, annual incentive bonuses and stock-based incentive awards with four-year vesting periods is balanced and serves to motivate our employees to accomplish our business plan without creating risks that are reasonably likely to have a material adverse effect on our company.
Corporate Governance and Nominating Committee. The Corporate Governance and Nominating Committee currently consists of Daryl Faulkner (Chair), James Fox, Ph.D. and Kevin C. O’Boyle, each of whom the Board has determined is an independent director under the NASDAQ Global Market listing standards. The Corporate Governance and Nominating Committee’s responsibilities include recommending to the Board nominees for possible election to the Board, ensuring that each of the committees of the Board have qualified and independent directors and providing oversight with respect to corporate governance and succession planning matters. The Corporate Governance and Nominating Committee is governed by a written charter approved by the Board.
Corporate Governance Guidelines
Our corporate governance guidelines are designed to ensure effective corporate governance. Our corporate governance guidelines cover topics including, but not limited to, director qualification criteria, director compensation, director orientation and continuing education, communications from stockholders to the Board, succession planning and the annual evaluations of the Board and its committees. Our corporate governance guidelines will be reviewed regularly by the Corporate Governance and Nominating Committee and revised when appropriate. The full text of our corporate governance guidelines is accessible to the public at www.genmarkdx.com. A printed copy may also be obtained by any stockholder upon request.
Code of Business Conduct and Ethics
Our Board adopted a code of business conduct and ethics to ensure that our business is conducted in a consistently legal and ethical manner. The code of business conduct and ethics establishes policies pertaining to, among other things, employee conduct in the workplace, securities trading, confidentiality, conflicts of interest, reporting violations and compliance procedures. All of our employees, including our executive officers, as well as members of our Board, are required to comply with our code of business conduct and ethics.
75
Table of Contents
The full text of code of business conduct and ethics is accessible to the public at www.genmarkdx.com. A printed copy may also be obtained by any stockholder upon request. Any waiver of the code of business conduct for our executive officers or directors must be approved by our Board after receiving a recommendation from our Audit Committee. We will disclose future amendments to our code of business conduct on our website, www.genmarkdx.com, within four business days following the date of the amendment or waiver.
Compensation Committee Interlocks and Insider Participation
As of December 31, 2010, the Compensation Committee consisted of Daryl Faulkner, James Fox, Ph.D. (Chair) and Kevin C. O’Boyle. During 2010, Christopher Gleeson served as Chairperson of the Compensation Committee from January 2010 until his resignation from the Compensation Committee in September 2010. Mr. Gleeson was replaced by James Fox, Ph.D. on September 12, 2010. All of the members of the Compensation Committee are non-employee directors. No members of the Compensation Committee have a relationship that would constitute an interlocking relationship as defined by SEC rules.
Director Independence
In accordance with our corporate governance principles, the majority of our Board members are independent directors. Our Board considers that a director is independent when the director is not an officer or employee of us or our subsidiaries, does not have any relationship which would, or could reasonably appear to, materially interfere with independent judgment, and otherwise meets the independence requirements under the rules of the NASDAQ Global Market and the SEC. Our Board has reviewed the materiality of any relationship that each of our directors has with us, either directly or indirectly. Based on this review, our Board has determined that Daryl Faulkner, James Fox, Ph.D. and Kevin C. O’Boyle are considered to be independent directors under the rules of the NASDAQ Global Market and the SEC.
No Family Relationship
There are no family relationships among our officers and directors, nor are there any arrangements or understandings between any of our directors or officers or any other person pursuant to which any officer or director was, or is, to be selected as an officer or director.
76
Table of Contents
Compensation Discussion and Analysis
Our compensation program is designed to attract and retain talented employees, to motivate them to achieve our key financial, operational and strategic goals and to reward them for superior performance. We believe that attracting and retaining high caliber employees and providing them with appropriate performance incentives are critical steps to helping us achieve our corporate goals and build long-term value for our stockholders.
The Compensation Committee of our board of directors oversees our executive compensation program. In this role, the Compensation Committee reviews and approves annually all compensation decisions relating to our executives, including our Chief Executive Officer.
Overview of Compensation Program
The elements of our compensation program are directed toward providing our executives with both short-term and long-term performance incentives, with the overall objective to motivate our executives to help us achieve our corporate goals and build long-term value for our stockholders. The elements of our compensation program include:
| • | base salary; |
| • | annual performance-based bonus awards; and |
| • | long-term stock-based incentive awards. |
We also provide our executives with insurance and a limited number of additional benefits that are typical for companies in our industry. Each of these compensation elements is described in more detail below.
In determining the relevant amounts for each of these compensation elements to be awarded to our executives, our Compensation Committee considers the following objectives:
A Substantial Portion of Executive Compensation Should Be Performance-Based. We believe that a substantial portion of the compensation received by each of our executives should be directly tied to, and contingent upon, the performance of our company as a whole and the executive’s individual contribution and performance. To support this objective, our Compensation Committee has established an Annual Bonus Incentive Plan, or the Bonus Plan. The Bonus Plan is designed to align each executive’s efforts with our key financial, operational and strategic goals by providing an opportunity for the executive to earn an annual cash or stock bonus with amounts determined by considering our success in achieving our corporate goals and the executive’s success in achieving individual performance goals set for that executive. Our coporate performance goals under the Bonus Plan include revenue and gross margin targets, new customer acquisition targets, XT-8 instrument and analyzer placement targets and new product development targets.
Stock-Based Incentive Awards Should Comprise a Substantial Portion of Executive Compensation. We believe that a substantial portion of executive compensation should be delivered in the form of stock-based incentive awards in order to align the long-term interests of our executives with those of our stockholders and to provide a retention incentive to our executives.
Our Executive Compensation Should Be Competitive and Fair. In order to help us attract and retain talented executives, we believe that our compensation programs should be competitive when compared to our peers as well as perceived as fair, when considered both externally as well as internally.
Compensation Process
Our Compensation Committee is responsible for establishing our compensation philosophy and setting the compensation levels for our executives, including base salaries, target performance-based awards under the Bonus Plan and stock-based incentive awards. The Compensation Committee is responsible for approving the
77
Table of Contents
corporate performance goals for purposes of the Bonus Plan. The Compensation Committee sets the compensation of our Chief Executive Officer utilizing industry benchmark compensation data. To assist the Compensation Committee, our Chief Executive Officer will prepare a report at the beginning of each fiscal year recommending base salaries, stock-based incentive awards, corporate performance goals for the fiscal year for all other executive officers. The Compensation Committee in its sole discretion may accept or adjust the compensation recommendations it is provided. In addition to this report, our Compensation Committee considers relevant market compensation data when seting the compensation for our other executive officers. No executive officer, including our Chief Executive Officer, is allowed to be present at the time his or her compensation is being discussed or determined by the Compensation Committee.
After the end of each fiscal year, our Compensation Committee also determines the performance-based bonus awards our executive officers should be paid under the Bonus Plan for the prior fiscal year. In making this determination, our Compensation Committee evaluates our success in achieving the corporate performance goals for purposes of awards under the Bonus Plan. To assist in this process, our Chief Executive Officer prepares a report for the Compensation Committee regarding the achievement of the applicable corporate performance goals. Based on this information, our Compensation Committee determines what percentage of the individual target awards under the Bonus Plan each of our executive officers should receive for the past fiscal year.
Determination of Executive Compensation
In setting the compensation for our executive officers, our Compensation Committee places significant emphasis on the recommendation of our Chief Executive Officer (other than with respect to determining his own compensation), considers our overall performance during the prior fiscal year and the executive’s individual contributions during the prior fiscal year, as well as relevant market data. With respect to new hires, the Compensation Committee considers an executive’s background and historical compensation in lieu of prior year performance. For 2010 and 2011, our Compensation Committee reviewed an analysis of competitive market data using Top 5 Data Services, Inc. Top 5 provides executive compensation data for companies in the medical device and diagnostic industries. Our Compensation Committee used this market data as one component of determining executive compensation for 2010 and 2011. Our Compensation Committee expects to retain an independent compensation consultant to assist it with the benchmarking process in future years.
Components of Executive Compensation
As indicated above, we compensate our executives through a combination of short-term and long-term incentives that are designed to motivate our executives to help us achieve our key corporate performance goals and build long-term value for our stockholders.
Base Salary. The Compensation Committee evaluates and sets the base salaries for our executives on an annual basis following annual performance reviews, as well as upon a promotion or other change in responsibility. In setting base salaries for our executive officers, our Compensation Committee considers the executive’s position, our success in achieving our prior year corporate performance goals, the individual’s contribution and performance during the prior fiscal year. The Compensation Committee also considers market survey data provided by Top 5 Data Services. The Compensation Committee also considers the evaluations and recommendations proposed by our Chief Executive Officer for our other executive officers. With respect to new hires, the Compensation Committee considers an executive’s background and historical compensation in lieu of prior year performance. Based on the determinations made by our Compensation Committee, base salaries for our executive officers in 2010 and 2011 are between the 25th and 75th percentiles as compared to our benchmark companies in the medical device and diagnostics industries.
78
Table of Contents
Our named executive officers were offered the following annualized base salaries for fiscal years 2010 and 2011:
| Name and Title |
Base Salary | |||||||
| 2010 | 2011 | |||||||
| Christopher Gleeson, Chairman(1) |
$ | — | $ | — | ||||
| Hany Massarany, Chief Executive Officer, President and Director(2) |
N/A | 450,000 | ||||||
| Paul Ross, Chief Financial Officer, Treasurer and Secretary(3) |
N/A | 240,000 | ||||||
| Jon Faiz Kayyem, Ph.D., Chief Scientific Officer |
230,000 | 230,000 | ||||||
| Jeffrey Hawkins, Senior Vice President, Marketing and Business Development |
190,000 | 210,000 | ||||||
| Jennifer Williams, Senior Vice President, Global Operations |
200,000 | 225,000 | ||||||
| (1) | Christopher Gleeson received an award of 109,375 shares of our restricted stock as compensation for his services as our Interim Chief Executive Officer for the period August 2010 to May 2011. |
| (2) | Mr. Massarany was appointed as our Chief Executive Officer and President in May 2011. |
| (3) | Mr. Ross was appointed as our Chief Financial Officer, Treasurer and Secretary in April 2011. |
Performance-Based Bonus Awards. Our Compensation Committee established a Bonus Plan for our executive officers. The Bonus Plan is designed to align each executive’s efforts with our annual financial, operational and strategic performance goals. Our Compensation Committee is responsible for administrating the Bonus Plan. Awards under the Bonus Plan may be paid in cash or stock.
Our Compensation Committee is responsible for setting the target bonus amounts for our executives under the Bonus Plan. The target bonus amounts for each executive is generally set at a percentage of his or her base salary. Executives will not become eligible for bonus payments unless we achieve certain revenue performance goals approved by the Compensation Committee. The bonuses are payable based on the weighted percentage assigned to each corporate performance goal by the Compensation Committee. For 2010, our corporate goals were weighted as follows: placement of XT-8 systems and analyzers (40%), product development milestones (40%) and achievement of our projected gross margin (20%).
After the end of each fiscal year, the Compensation Committee is responsible for awarding the actual bonus amounts under the Bonus Plan. To assist our Compensation Committee, each year our Chief Executive Officer provides the Compensation Committee with documentation regarding full or partial achievement of each of the corporate performance goals established by the Compensation Committee for that year.
The weighted average of each corporate goal is multiplied by the executive’s target bonus amount to determine the actual bonus amount paid in respect of each corporate performance goal. Actual amounts payable range from 0% to 150% of the target amounts, based upon achievement of the corporate performance goals. The total award paid is determined by adding up the sum of the weighted averages multiplied by the executive’s target bonus amounts. To reward exceptional performance in certain circumstances, the Compensation Committee may determine that a supplemental bonus in excess of the target bonus is appropriate and justified. However, individual incentive payments will not be an entitlement. Our Compensation Committee may terminate the Bonus Plan at any time, and may alter the terms and conditions under which the bonus awards are set, calculated or paid. The following sets forth the target bonus percentages and amounts for 2010 and 2011:
| Bonus | ||||||||||||||||
| 2010 | 2011 | |||||||||||||||
| Bonus % | $ at Target | Bonus % | $ at Target | |||||||||||||
| Executive Officer |
||||||||||||||||
| Christopher Gleeson, Chairman(1) |
— | — | — | — | ||||||||||||
| Hany Massarany, Chief Executive Officer, President and Director(2) |
— | — | 75 | % | 337,500 | |||||||||||
| Paul Ross, Chief Financial Officer, Treasurer and Secretary(3) |
— | — | 50 | % | 120,000 | |||||||||||
| Jon Faiz Kayyem, Ph.D., Chief Scientific Officer(4) |
30 | % | 69,000 | 50 | % | 115,000 | ||||||||||
| Jeffrey Hawkins, Senior Vice President, Marketing and Business Development |
25 | % | 47,500 | 50 | % | 105,000 | ||||||||||
| Jennifer Williams, Senior Vice President, Global Operations |
30 | % | 60,000 | 50 | % | 112,500 | ||||||||||
79
Table of Contents
| (1) | Christopher Gleeson received an award of 109,375 shares of our restricted stock as compensation for his services as our Interim Chief Executive Officer for the period August 2010 to May 2011. |
| (2) | Mr. Massarany was appointed as our Chief Executive Officer and President in May 2011. |
| (3) | Mr. Ross was appointed as our Chief Financial Officer, Treasurer and Secretary in April 2011. |
| (4) | Dr. Kayyem served as Chief Executive Officer from January 1, 2010 through August 9, 2010. His base pay was $275,000 and his bonus percentage was 50% during that period. |
Stock-Based Incentive Awards. In addition to our performance-based bonus awards, we provide long-term stock-based incentive awards to our executive officers. These stock-based incentive awards generally consist of options to purchase shares of our common stock, restricted stock grants or restricted stock units. We believe that equity awards help further our compensation objectives by encouraging our executives to remain with us through at least the vesting period for these awards and providing them with an incentive to continue to focus on our long-term financial performance and increasing stockholder value.
Our executive officers receive an equity award in connection with their initial hire, following promotions and on an annual basis. To assist the Compensation Committee, we have developed guidelines for initial and annual equity awards. The guidelines for initial grants are based on the executive’s position, market data and the guidelines for annual grants are primarily based on individual performance and contributions to the overall company performance. With respect to new hires, we also considered the executive’s background and historical compensation when determining the number of shares to grant to the executive. The actual number of shares for an executive may be higher or lower than these guidelines, based on their individual performance or extraordinary achievements.
Equity Grant Practices. Our Compensation Committee adopted a policy by which all stock and option awards to new and current employees, including our executive officers, are granted at pre-determined meeting dates of the Compensation Committee. Our Compensation Committee grants the equity awards in accordance with the dates fixed by this policy.
The exercise price of any option grant is determined by reference to the fair market value of such shares, which the 2010 Equity Incentive Plan defines as the daily volume-weighted average price of our common stock on the NASDAQ Global Market on the date of grant. Our equity grants generally vest 25% one year from the date of the grant, with the remaining 75% of the options vesting in annual installments over the subsequent three year period.
Other Benefits
In order to attract, retain and pay market levels of compensation, we provide our executives with the following benefits:
| • | Health Insurance. We provide each of our executives and their spouses and children the same health, dental and vision insurance coverage we make available to our other eligible employees. |
| • | Life and Disability Insurance. We provide each of our executives with the same disability and life insurance as we make available to our other eligible employees. |
| • | Pension Benefits. We do not provide pension arrangements or post-retirement health coverage for our executives or employees. Our executives and other eligible employees are eligible to participate in our 401(k) defined contribution plan. We do not currently make matching contributions to participants in the 401(k) plan, however we have previously made matching contributions, and we may opt to do so again in the future. |
| • | Nonqualified Deferred Compensation. We do not provide any nonqualified defined contribution or other deferred compensation plans to any of our employees. |
| • | Perquisites. We limit the perquisites that we make available to our executive officers. Our executives are entitled to relocation expenses on their initial hire and other benefits with de minimis value that are not otherwise available to all of our employees. |
80
Table of Contents
Employment Agreements
Christopher Gleeson
We entered into an executive consulting agreement with Mr. Gleeson, effective October 12, 2010 for his services as Interim Chief Executive Officer during the period from August 12, 2010 through May 2011. Pursuant to the consulting agreement, Mr. Gleeson was provided a restricted stock grant of 109,375 shares and a temporary housing benefit not to exceed $5,000 per month. Mr. Gleeson restricted stock grant is fully vested.
Hany Massarany
On April 5, 2011, we entered into an executive employment agreement with Hany Massarany, pursuant to which Mr. Massarany was appointed as our President and Chief Executive Officer, effective May 1, 2011. Mr. Massarany served as a consultant from April 5, 2011 to May 1, 2011. In addition, effective May 1, 2011, our board of directors appointed Mr. Massarany as a director. Mr. Massarany was designated as a Class II Director and will serve until our 2012 Annual Meeting of Stockholders, or until his successor has been duly elected and qualified.
Pursuant to the terms of the employment agreement, Mr. Massarany’s annual salary is $450,000, less applicable withholdings, and his target bonus is equal to 75% of base salary in 2011 and 100% of base salary in subsequent years. In addition, under the Bonus Plan, Mr. Massarany may earn up to 150% of his target bonus based on achievement of certain milestones and objectives established by the Compensation Committee. Mr. Massarany will be guaranteed a minimum bonus equal to 50% of his target bonus for 2011.
In connection with entering into the employment agreement, Mr. Massarany was awarded 275,000 stock options at an exercise price equal to the fair market value on the date of the grant and 176,739 restricted shares of common stock, in each case, pursuant to the terms of the 2010 Plan. The options will vest over four years, with 25% of the options vesting on April 5, 2012, and 75% of the options vesting in equal monthly installments thereafter, subject to acceleration upon a change of control of GenMark. The shares of restricted stock will vest over four years, with the shares vesting in equal quarterly installments beginning on July 5, 2011, subject to acceleration upon a change of control of GenMark. In the event of a change of control transaction in which our shareholders receive cash consideration, Mr. Massarany’s options shall be exchanged for (i) a cash payment equal to the number of shares subject to the options multiplied by (ii) the excess of the fair market value of each share over the exercise price.
We also reimburse Mr. Massarany for certain expenses, including relocation expenses which includes a temporary housing allowance of up to $5,000 per month through August 31, 2011.
Subject to the following paragraph, in the event Mr. Massarany is terminated without cause (as defined in the employment agreement) or Mr. Massarany terminates his employment for good reason (as defined in the employment agreement), Mr. Massarany will be entitled to receive (i) any accrued benefits during his time of service, (ii) a severance payment equal to his base salary at the time of termination, plus the last annual bonus paid to Mr. Massarany, (iii) immediate acceleration of the vesting of his outstanding options and shares of restricted stock, and (iv) during the one year period following his termination of employment, reimbursement for any payments made to continue his healthcare coverage, subject to certain limitations.
In the event Mr. Massarany’s employment is terminated without cause or Mr. Massarany terminates his employment for good reason within six month preceding or 24 months following a change in control (as defined in the 2010 Plan), in lieu of the benefits described above, Mr. Massarany will be entitled to receive (i) any accrued benefits during his time of service, (ii) a severance payment equal to the product of two multiplied by (a) his base salary at the time of termination, plus (b) the last annual bonus paid to Mr. Massarany, (iii) immediate acceleration of the vesting of his outstanding options and shares of restricted stock, and (iv) during the two year period following his termination of employment, reimbursement for any payments made to continue his healthcare coverage, subject to certain limitations.
81
Table of Contents
In the event Mr. Massarany’s employment is terminated for cause, except with respect to any obligations which accrued during his time of service, all other obligations under the employment agreement will automatically become terminated. In addition, in the event of Mr. Massarany’s death or disability, he will become entitled to receive (i) any accrued benefits during his time of service, (ii) a prorated portion of his annual bonus payable in accordance with the Bonus Plan, and (iii) immediate acceleration of the vesting of his outstanding options and shares of restricted stock.
Paul Ross
On March 11, 2011, we entered into an employment offer letter with Paul Ross, pursuant to which Mr. Ross was appointed our Chief Financial Officer, effective April 4, 2011.
Pursuant to the terms of the offer letter, Mr. Ross will earn a base salary initially set at $240,000 per year subject to our standard payroll practices and procedures. In addition, Mr. Ross will be eligible to earn a performance-based bonus of up to 50% of his base salary under our annual incentive bonus program. In the event Mr. Ross’ employment is terminated by us for any reason other than cause, Mr. Ross will entitled to receive six months base salary plus bonus consideration, the terms of which will be further detailed in an employment agreement to be entered into following the effectiveness of his appointment.
In connection with his appointment, Mr. Ross received an initial grant of 52,500 options to purchase shares of our common stock and an initial grant of 33,741 shares of restricted common stock. The options will vest over four years, with 25% of the options vesting on April 4, 2012, and 75% of the options vesting in equal monthly installments thereafter, subject to acceleration upon a change of control. The shares of restricted stock will vest over four years, with 25% of the shares vesting on April 4, 2012 and 25% of the shares vesting in equal quarterly installments thereafter, subject to acceleration upon a change of control.
Jon Faiz Kayyem, Ph.D.
We entered into an employment agreement, effective January 1, 2010, with Dr. Kayyem, pursuant to which he agreed to serve as our President and Chief Executive Officer. On August 9, 2010, Dr. Kayyem resigned as our President and Chief Executive Officer and simultaneous with his resignation was appointed as our Chief Scientific Officer. Dr. Kayyem’s current base salary is $230,000. He is also eligible to participate in the Bonus Plan of up to 30% variable pay based on his current base salary and he is eligible to participate in our 2010 Plan.
Jeffrey Hawkins
We entered into an employment agreement, effective March 1, 2010, with Jeffrey Hawkins. Pursuant to the terms of the agreement, Mr. Hawkins’ current base salary is $190,000. He is also eligible to participate in the Bonus Plan of up to 25% variable pay based on his current base salary and he is eligible to participate in our 2010 Plan.
Pankaj Singhal, Ph.D.
We entered into an employment agreement, effective January 1, 2010, with Pankaj Singhal. Pursuant to the terms of the agreement, Dr. Singhal’s current base salary is $220,000. He is also eligible to participate in the Bonus Plan of up to 30% variable pay based on his current base salary and he is eligible to participate in our 2010 Equity Incentive Plan.
On March 24, 2011, we entered into a separation agreement with Dr. Singhal, pursuant to which Dr. Singhal resigned effective April 29, 2011 (the “Separation Date”). Pursuant to the terms of the Separation Agreement, Dr. Singhal will receive separation payments at his current base salary through January 29, 2012, subject to applicable withholdings and taxes, and we will reimburse Dr. Singhal for payments made in connection with his participation in our medical plan through January 29, 2012. In addition, on the Separation Date, 47,938 shares of common stock will fully vest under Dr. Singhal’s outstanding stock options, after which
82
Table of Contents
Dr. Singhal will have until April 30, 2012 to exercise such options. Pursuant to the Separation Agreement, we also granted 4,000 restricted stock units to Dr. Singhal under our 2010 Equity Incentive Plan, which vested on May 31, 2011. The Separation Agreement also provides for a customary release of general claims by Dr. Singhal, as well as customary confidentiality, non-solicitation and non-disparagement restrictive covenants.
Tax and Accounting Considerations
To the extent possible, we attempt to provide compensation that is structured to maximize favorable accounting, tax and similar benefits for the Company.
Deductibility of Executive Compensation
Section 162(m) of the Internal Revenue Code of 1986, as amended, generally limits the deductibility of certain compensation in excess of $1,000,000 paid in any one year to any one named executive officer. Qualifying performance-based compensation will not be subject to this deduction limit if certain requirements are met.
The Compensation Committee periodically reviews and considers the deductibility of executive compensation under Section 162(m) in designing our compensation programs and arrangements. A portion of our annual cash incentive awards is determined based upon the achievement of certain predetermined financial performance goals of ours in order to permit us to deduct such amounts pursuant to Section 162(m). In addition, our equity incentive plans contain limits on the number of equity awards that can be granted to any one individual in any year for purposes of Section 162(m).
While we will continue to monitor our compensation programs in light of Section 162(m), the Compensation Committee considers it important to retain the flexibility to design compensation programs that are in the best long-term interests of our stockholders. As a result, the Compensation Committee may conclude that paying compensation at levels that are not deductible under Section 162(m) is nevertheless in the best interests of our stockholders.
83
Table of Contents
Summary Compensation Table
The following table sets forth information concerning compensation earned for services rendered by our “named executive officers” for the fiscal year ended December 31, 2010. The compensation described in this table does not include medical, group life insurance, or other benefits which are available generally to all of our salaried employees.
| Name and Principal Position |
Year | Salary | Bonuses ($) |
Stock Awards (2)(3) |
Option Awards (2) |
Non-Equity Incentive Plan Compensation ($) |
All Other Compensation |
Total | ||||||||||||||||||||||||
| Christopher Gleeson(4)(5) |
2010 | — | — | 487,813 | — | — | 18,344 | (10) | 506,157 | |||||||||||||||||||||||
| Chairman and Former Interim CEO |
2009 | — | — | — | — | — | — | — | ||||||||||||||||||||||||
| Jon Faiz Kayyem, Ph.D.(4) |
2010 | 251,923 | 77,360 | (1)(2) | — | — | — | 15,500 | (10) | 267,423 | ||||||||||||||||||||||
| Chief Scientific Officer |
2009 | 69,231 | — | — | 936,547 | — | — | 1,005,778 | ||||||||||||||||||||||||
| Steven J. Kemper(5) |
2010 | 230,000 | — | — | 81,303 | — | 172,500 | (10) | 483,801 | |||||||||||||||||||||||
| Former Chief Financial Officer |
2009 | 13,269 | — | — | 321,102 | — | — | 334,371 | ||||||||||||||||||||||||
| Jeffrey Hawkins(6) |
2010 | 190,000 | 33,725 | (1)(2) | 40,987 | 108,606 | — | 70,000 | (10) | 409,593 | ||||||||||||||||||||||
| Sr. Vice President, Marketing and Business Development |
2009 | 7,308 | — | — | — | — | — | 7,308 | ||||||||||||||||||||||||
| Pankaj Singhal, Ph. D.(7) |
2010 | 220,000 | — | — | — | — | 100,000 | (10) | 320,000 | |||||||||||||||||||||||
| Former Sr. Vice President, Product Development |
2009 | 220,000 | — | — | 321,102 | — | 6,600 | (11) | 547,702 | |||||||||||||||||||||||
| Jennifer Williams(8) |
2010 | 126,923 | 42,600 | (1)(2) | 272,791 | 84,630 | — | 100,000 | (10) | 584,344 | ||||||||||||||||||||||
| Sr. Vice President, Global Operations |
2009 | — | — | — | — | — | — | — | ||||||||||||||||||||||||
| John Bellano(9) |
2010 | 180,810 | 50,000 | — | — | — | 153,077 | (10) | 383,887 | |||||||||||||||||||||||
| Former Senior Vice President, Commercial Operations |
2009 | 7,692 | — | — | 321,102 | — | — | 328,794 | ||||||||||||||||||||||||
| (1) | Annual bonuses for services rendered in 2010 will be granted in the form of restricted stock. The grant date valuation is equal to the amount of the 2010 annual bonus. |
| (2) | Represents the grant date valuation of the awards computed in accordance with the Financial Accounting Standards Board (FASB) Accounting Standards Codification (ASC) Topic 718. For more information, see Note 6 in the Notes to Consolidated Financial Statements contained in our Annual Report on Form 10-K filed with the SEC on March 14, 2011. |
| (3) | Excludes restricted stock grants to named executive officers granted in lieu of 2010 annual bonuses which are reflected in the bonuses column. |
| (4) | Mr. Gleeson was appointed our Interim Chief Executive Officer and principal executive officer in August 2010. Prior to that time, Dr. Kayyem served as our Chief Executive Officer and principal executive officer. Mr. Gleeson resigned from his position as Interim Chief Executive Officer in May 2011. |
| (5) | Mr. Kemper resigned from the position of Chief Financial Officer and was replaced as the Company’s principal financial officer by Mr. Gleeson in November 2010. Mr. Kemper resigned from GenMark effective December 31, 2010. Mr. Ross was appointed to the position of Chief Financial Officer effective April 4, 2011. |
| (6) | Mr. Hawkins was appointed our Senior Vice President, Marketing and Business Development in November 2010. |
| (7) | Dr. Singhal resigned as our Senior Vice President, Product Development effective April 29, 2011. |
| (8) | Ms. Williams was appointed our Senior Vice President, Global Operations in November 2010. |
| (9) | Mr. Bellano resigned as our Senior Vice President, Commercial Operations in November 2010. |
| (10) | Other compensation consisted of a housing assistance for Mr. Gleeson and Dr. Kayyem, a severance payment for Mr. Kemper and Mr. Bellano, and a relocation allowance for Mr. Hawkins, Dr. Singhal, Ms. Williams and Mr. Bellano. |
| (11) | Other compensation consisted of a matching 401(k) contribution. |
84
Table of Contents
Grant of Plan-Based Awards
The following table sets forth information regarding grants of stock and option awards made to our named executive officers during the fiscal year ended December 31, 2010.
| Name |
Grant Date | All Other Stock Awards Number of Shares |
All Other Option Awards Number of Shares |
Exercise or Base Price of Option Awards ($/sh) |
Grant Date Fair Value of Stock and Option Awards |
|||||||||||||||
| Christopher Gleeson(1) |
5/28/10 | — | 24,050 | $ | 6.00 | $ | 86,669 | |||||||||||||
| Christopher Gleeson(1) |
8/12/10 | 109,375 | — | $ | — | $ | 487,813 | |||||||||||||
| Steven J. Kemper(2) |
5/28/10 | — | 4,633 | $ | 6.00 | $ | 17,684 | |||||||||||||
| Steven J. Kemper(2) |
5/28/10 | — | 16,667 | $ | 6.00 | $ | 63,617 | |||||||||||||
| Jeffrey Hawkins(1) |
5/28/10 | — | 16,667 | $ | 6.00 | $ | 63,513 | |||||||||||||
| Jeffrey Hawkins(1) |
5/28/10 | — | 11,833 | $ | 6.00 | $ | 45,092 | |||||||||||||
| Jeffrey Hawkins(1) |
11/3/10 | 9,444 | — | $ | — | $ | 40,987 | |||||||||||||
| Jennifer Williams(1) |
5/28/10 | — | 16,667 | $ | 6.00 | $ | 64,037 | |||||||||||||
| Jennifer Williams(1) |
5/28/10 | — | 54,333 | $ | 6.00 | $ | 208,755 | |||||||||||||
| Jennifer Williams(1) |
11/3/10 | 19,500 | — | $ | — | $ | 84,630 | |||||||||||||
| (1) | Mr. Gleeson was granted an award of restricted stock in connection with his appointment to the Interim CEO position in August 2010. Mr. Hawkins and Ms. Williams were granted an award of restricted stock in connection with their promotions in fiscal 2010. The value of the awards are calculated by multiplying the number of units awarded by the market closing price of our common stock on the date of grant, $4.46 on August 12, 2010 for Mr. Gleeson and $4.34 on November 3, 2010 for Mr. Hawkins and Ms. Williams. Mr. Gleeson resigned from his position as Interim Chief Executive Officer in May 2011. |
| (2) | Mr. Kemper resigned from the position of Chief Financial Officer in November 2010 and from GenMark effective December 31, 2010 and was replaced as our principal financial officer by Christopher Gleeson in November 2010. |
Outstanding Equity Awards at Fiscal Year-End
The following table sets forth information regarding outstanding equity awards held by our named executive officers as of December 31, 2010.
| Option Awards(1) | Stock Awards | |||||||||||||||||||||||||||
| Name |
Grant Date |
Number of Securities Underlying Unexercised Options (#) Exercisable |
Number of Securities Underlying Unexercised Options (#) Unexercisable |
Option Exercise Price ($) |
Option Expiration Date |
Number of shares that have not Vested |
Market Value of shares that have not Vested(13) |
|||||||||||||||||||||
| Christopher Gleeson(2) |
9/25/09 | 8,359 | 16,721 | $ | 6.93 | 9/25/19 | (3) | 109,375 | (10) | $ | 447,344 | |||||||||||||||||
| 12/23/09 | 4,821 | — | $ | 6.49 | 12/23/19 | (4) | — | — | ||||||||||||||||||||
| 12/23/09 | 9,405 | 15,675 | $ | 6.49 | 12/23/19 | (5) | — | — | ||||||||||||||||||||
| 5/28/10 | 16,033 | 8,017 | $ | 6.00 | 5/28/20 | (6) | — | — | ||||||||||||||||||||
| Jon Faiz Kayyem, Ph.D.(2) |
12/23/09 | 93,213 | 115,355 | $ | 6.49 | 12/23/19 | (5) | — | — | |||||||||||||||||||
| Steven J. Kemper (1) |
12/23/09 | 21,306 | — | $ | 6.49 | 12/31/11 | (7) | — | — | |||||||||||||||||||
| 5/28/10 | 2,778 | — | $ | 6.00 | 12/31/11 | (7) | — | — | ||||||||||||||||||||
| 5/28/10 | 773 | — | $ | 6.00 | 12/31/11 | (7) | — | — | ||||||||||||||||||||
| Jeffrey Hawkins |
12/23/09 | 10,653 | 31,959 | $ | 6.49 | 12/23/19 | (5) | 9,444 | (11) | 38,626 | ||||||||||||||||||
| 5/28/10 | 3,125 | 13,542 | $ | 6.00 | 5/28/20 | (8) | — | — | ||||||||||||||||||||
| 5/28/10 | 2,219 | 9,614 | $ | 6.00 | 5/28/20 | (8) | — | — | ||||||||||||||||||||
| Pankaj Singhal, Ph.D. |
12/23/09 | 40,836 | 44,388 | $ | 6.49 | 12/23/19 | (9) | — | — | |||||||||||||||||||
| Jennifer Williams |
5/28/10 | — | 16,667 | $ | 6.00 | 5/28/20 | (5) | 19,500 | (11) | 79,755 | ||||||||||||||||||
| 5/28/10 | — | 54,333 | $ | 6.00 | 5/28/20 | (5) | — | — | ||||||||||||||||||||
| John Bellano |
12/23/09 | 21,306 | — | $ | 6.49 | 11/11/11 | (12) | — | — | |||||||||||||||||||
85
Table of Contents
| (1) | Mr. Kemper resigned from the position of Chief Financial Officer and was replaced as our principal financial officer by Mr. Gleeson in November 2010. Mr. Kemper resigned from GenMark effective December 31, 2010. Mr. Kemper’s options remain exercisable until December 31, 2011. |
| (2) | Dr. Kayyem resigned from the position of President and Chief Executive Officer and was appointed Chief Scientific Officer on August 12, 2010. Mr. Gleeson was appointed Interim CEO on that date. Mr. Gleeson resigned from his position as Interim Chief Executive Officer in May 2011. |
| (3) | Options vest one third annually on the anniversary of the grant date and have a term of ten years. |
| (4) | Options vest 100% on grant date with a term of ten years. |
| (5) | Options vest 25% on the one year anniversary of the grant date, with the remaining shares vesting in 36 equal monthly installments thereafter. All option grants have a term of ten years. |
| (6) | Options vest in 12 equal monthly installments with a term of ten years. |
| (7) | Options are fully vested pursuant to the terms of a Separation Agreement between Mr. Kemper and GenMark and are exercisable through December 31, 2011. |
| (8) | Options vest 18.75% on December 7, 2010, with the remaining shares vesting in 39 equal monthly installments thereafter with a term of ten years. |
| (9) | Options in an aggregate total of 47,938 shares become fully vested upon Dr. Singhal’s termination on April 29, 2011. |
| (10) | Restricted stock vests 50% each on January 31, 2011 and July 30, 2011, respectively, contingent upon continued service to GenMark. |
| (11) | Restricted stock vests 25% on the one year anniversary of the grant date, with the remaining shares vesting in 36 equal monthly installments thereafter, contingent upon continued service to GenMark. |
| (12) | Options are fully vested and exercisable through November 11, 2011 pursuant to the terms of a Separation Agreement between Mr. Bellano and GenMark. |
| (13) | The market value was determined by multiplying the number of stock awards by the closing market price on December 31, 2010 of $4.09. |
Option Exercises
None of our named executive officers exercised options during the fiscal year ended December 31, 2010.
Potential Payments upon Termination or Change of Control
Steven Kemper
Steven Kemper resigned as our Chief Financial Officer in November 2010 and from GenMark effective December 31, 2010. Pursuant to the terms of his separation from GenMark, Mr. Kemper received severance of $172,500 and will receive reimbursement for health, dental and vision insurance coverage through December 31, 2011 expected to total approximately $23,000.
John Bellano
John Bellano resigned as our Senior Vice President, Commercial Operations effective November 12, 2010. Pursuant to the terms of his separation from the company, Mr. Bellano received severance of $50,000 and the Company agreed to continue group health coverage through January 31, 2011 at a cost of approximately $4,000.
86
Table of Contents
DIRECTOR COMPENSATION
Non-employee directors receive cash and equity consideration for their services as members of the Board and the Audit Committee. The following table sets forth the annual compensation offered to our non-employee directors for fiscal 2010 and 2011.
| Position |
Annual Compensation | |||||||
| 2010 | 2011 | |||||||
| Board |
$ | 60,000 | $ | 60,000 | ||||
| Chairperson of the Board |
$ | 40,000 | $ | 40,000 | ||||
| Chairperson of Audit Committee |
$ | 15,000 | $ | 15,000 | ||||
No compensation is paid to any director who is also an employee of the Company. Compensation is pro rated based on length of service for independent directors serving for only a portion of the year. The Board has discretion to allocate the percent of the compensation to paid in cash and the percent of the compensation payable in equity. The equity compensation will consist of stock options or restricted stock awards, or a combination thereof. In addition, each director is also entitled to be reimbursed for reasonable travel and other expenses incurred in connection with attending meetings of the board of directors and any committee on which he or she serves.
In connection with our initial public offering, we also granted additional options to the non-employee members of our board of directors in May 2010 in addition to their respective annual grants and cash retainer.
Director Summary Compensation Table
The following table summarizes director compensation during the fiscal year ended December 31, 2010.
| Name |
Fees Earned or Paid in Cash ($) |
Stock Awards ($)(1) |
Option Awards ($)(1) |
Totals ($) |
||||||||||||
| Christopher Gleeson(2) |
25,000 | — | 86,669 | 111,669 | ||||||||||||
| Daryl Faulkner(3) |
15,000 | — | 52,074 | 67,074 | ||||||||||||
| James Fox, Ph.D.(4) |
5,000 | 209,375 | — | 214,375 | ||||||||||||
| Kevin C. O’Boyle(5) |
16,042 | — | 160,111 | 176,153 | ||||||||||||
| (1) | Represents the grant date valuation of the awards computed in accordance with the FASB ASC Topic 718. For more information, see Note 4 in the Notes to Consolidated Financial Statements contained in our Annual Report on Form 10-K filed with the SEC on March 14, 2011. |
| (2) | Mr. Gleeson served as a non-executive director until August 2010. As of December 31, 2010, Christopher Gleeson directly held 109,375 shares of restricted common stock, of which 54,687 shares were unvested. |
| (3) | As of December 31, 2010, Mr. Faulkner held 45,315 options to purchase shares of common stock and no shares of restricted stock. |
| (4) | Dr. Fox was appointed to the Board in September 2010. As of December 31, 2010, Dr. Fox held no options to purchase shares of common stock and 51,954 shares of restricted stock, 42,852 shares of which were unvested. |
| (5) | Mr. O’Boyle was appointed to the Board of GenMark in March 2010. As of December 31, 2010, Mr. O’Boyle held 43,050 options to purchase shares of common stock and no shares of restricted stock. |
87
Table of Contents
During fiscal 2010, our non-employee directors were issued restricted stock and options to purchase shares of our common stock as set forth in the following table.
| Name |
Date of Grants |
Restricted Stock Granted |
Options Granted |
Vesting Terms | ||||||||||
| Christopher Gleeson |
5/28/2010 | — | 24,050 | Vests in 12 equal monthly installments starting May 16, 2010. | ||||||||||
| James Fox, Ph.D. |
9/1/2010 | 41,032 | — | 25% vests one year from grant date, remainder vests in 12 quarterly installments thereafter. | ||||||||||
| James Fox, Ph.D. |
9/1/2010 | 10,922 | — | One-third vested September 30, 2010, 25% vested on January 30, 2011 and the remainder vests on July 30, 2011. | ||||||||||
| Daryl Faulkner |
5/28/2010 | — | 14,450 | 25% vests on April 16, 2011, remainder vests in 36 monthly installments thereafter. | ||||||||||
| Kevin C. O’Boyle |
5/28/2010 | — | 25,000 | 25% vested on March 1, 2011, remainder vests in 36 monthly installments thereafter. | ||||||||||
| Kevin C. O’Boyle |
5/28/2010 | — | 18,050 | Vested in 12 equal monthly installments starting March 1, 2010. | ||||||||||
88
Table of Contents
The following table sets forth information regarding ownership of our common stock as of March 31, 2011 (or such other date as provided below) based on information available to us and filings with the Securities and Exchange Commission by (a) each person known to us to own more than 5% of the outstanding shares of our common stock, (b) each of our directors, (c) our Chief Executive Officer and each other named executive officer and (d) all directors and executive officers as a group. Each stockholder’s percentage ownership is based on 11,738,233 shares of our common stock outstanding as of March 31, 2011. The information in this table is based solely on statements in filings with the Securities and Exchange Commission (the “SEC” ) or other reliable information.
| Name and Address of Beneficial Owner(1) |
Amount and Nature of Beneficial Ownership(2) |
Percent of Class |
||||||
| Principal Stockholders |
||||||||
| FMR LLC(3) |
1,188,150 | 10.1 | % | |||||
| 82 Devonshire Street Boston, MA 02109 |
||||||||
| Gartmore Investment Limited, et. al.(4) |
1,165,694 | 9.9 | % | |||||
| Walker House, 87 Mary Street George Town, Grand Cayman KYI-9005 Cayman Islands |
||||||||
| Ronin Capital, LLC(5) |
1,102,704 | 9.4 | % | |||||
| 350 N. Orleans Street, Suite 2N Chicago, IL 60654 |
||||||||
| Efficacy Capital, Ltd.(6) |
738,986 | 6.3 | % | |||||
| 11622 El Camino Real, Ste. 100 San Diego, CA 92130 |
||||||||
| Visium Balanced Master Fund, Ltd.(7) |
678,217 | 5.8 | % | |||||
| 950 Third Avenue New York, NY 10022 |
||||||||
| Directors and Named Executive Officers |
||||||||
| Christopher Gleeson(8) |
1,863,529 | 15.9 | % | |||||
| Daryl Faulkner(9) |
53,622 | * | ||||||
| James Fox, Ph.D.(10) |
88,229 | * | ||||||
| Jon Faiz Kayyem, Ph.D.(11) |
880,820 | 7.5 | % | |||||
| Kevin C. O’Boyle(12) |
25,342 | * | ||||||
| Jeffrey Hawkins(13) |
55,248 | * | ||||||
| Jennifer Williams(14) |
149,983 | 1.3 | % | |||||
| Pankaj Singhal, Ph.D.(15) |
51,938 | * | ||||||
| Steven Kemper(16) |
29,857 | * | ||||||
| John Bellano(17) |
29,638 | * | ||||||
| All directors and executive officers as a group (10 persons)(18) |
3,228,207 | 27.5 | % | |||||
| * | Represents beneficial ownership of less than 1% of the outstanding shares of our common stock. |
| (1) | Unless otherwise indicated, the address of each beneficial owner is c/o GenMark Diagnostics, Inc., 5964 La Place Court, Carlsbad, CA 92008. |
| (2) | Beneficial ownership of shares and percentage ownership are determined in accordance with the rules of the SEC. In calculating the number of shares beneficially owned by an individual or entity and the percentage ownership of that individual or entity, shares underlying options or warrants held by that individual or entity that are either currently exercisable or exercisable within 60 days from March 31, 2011 are deemed outstanding. These shares, however, are not deemed outstanding for the purpose of computing the percentage |
89
Table of Contents
| ownership of any other individual or entity. Unless otherwise indicated and subject to community property laws where applicable, the individuals and entities named in the table above have sole voting and investment power with respect to all shares of our common stock shown as beneficially owned by them. |
| (3) | Based solely upon Appendix A to Schedule 13G jointly filed on February 14, 2011 by FMR LLC and Edward C. Johnson III (the “FMR Reporting Persons”) containing information as of December 31, 2010. Fidelity Management & Research Company (“Fidelity”), wholly-owned subsidiary of FMR LLC and a registered investment adviser, is the beneficial owner of 1,188,150 shares as a result of acting as investment adviser to various investment companies. Each of the FMR Reporting Persons, through its control of Fidelity, has sole power to dispose of the 1,188,150 shares, but neither FMR Reporting Person has the sole power to vote or direct the voting of the shares owned directly by the Fidelity funds; such power resides with the individual funds’ boards of trustees. Fidelity carries out the voting of the shares under written guidelines established by the funds’ boards of trustees. |
| (4) | Based solely upon reporting provided by Gartmore Investments Limited as of March 25, 2011 and upon a Schedule 13G filed on July 16, 2010 by Gartmore Group Limited containing information as of June 3, 2010, 489,275 of the shares of common stock are held directly by Alphagen Volantis Fund Limited, 318,520 of the shares of common stock are held directly by Gartmore Fund Managers Limited A/C Gartmore UK & Irish Smaller Companies and 357,899 of the shares of common stock are held directly by Strathclyde Pension Fund. Gartmore Investment Limited is the investment manager for each of these funds. Gartmore Investment Limited is a wholly owned subsidiary of Gartmore Investment Management Limited, which is a wholly owned subsidiary of Gartmore Group Ltd. Voting and dispositive power over the shares resides with the board of directors of Gartmore Group Ltd. The board of directors of Gartmore Group Ltd consists of Andrew Skirton, Jeffrey Meyer, Keith Starling, Patrick Healy, Blake Kleinman, David Barclay and David Lindsell. |
| (5) | Based solely upon a Schedule 13G jointly filed on February 14, 2011 by Ronin Capital LLC and Ronin Trading U.K. LLP, 927,582 shares are held directly by Ronin Capital LLC and 175,122 of the shares of common stock are held by Ronin Trading U.K. LLP. Ronin Capital LLC is the Designated Member with a controlling interest in Ronin Trading U.K. LLP and has power to vote and direct the shares and power to dispose or direct the disposition of the shares. Ronin Capital, LLC is a registered broker dealer and its wholly owned subsidiary DART Executions, LLC is a FINRA member. |
| (6) | Mark Lappe, the general partner of Efficacy Capital, Ltd., has voting and dispositive power with respect to the shares. Mr. Lappe disclaims beneficial ownership except to the extent of his pecuniary interest therein. |
| (7) | Based solely upon a Schedule 13G jointly filed on February 11, 2011 by Visium Balanced Master Fund, Ltd., Visium Asset Management, LP, JG Asset, LLC and J.J. Jacob Gottlieb containing information as of December 31, 2010. By virtue of its position as investment manager to pooled funds, Visium Asset Management, LP may be deemed to beneficially own the 678,217 shares of our common stock owned by the pooled investment vehicles. By virtue of its position as general partner to Visium Asset Management, LP, JG Asset, LLC may be deemed to beneficially own the 678,217 shares of our common stock owned by Visium Asset Management, LP. By virtue of his position as the Managing Director of JG Asset, LLC, J.J. Gottlieb may be deemed to beneficially own the 678,217 shares of our common stock owned by JG Asset, LLC. Visium Asset Management, LP, JG Asset, LLC and J.J. Jacob Gottlieb all disclaim beneficial ownership to the securities, except to the extent of his or its pecuniary interests therein. |
| (8) | Includes warrants to purchase 88,317 shares, 57,087 options to purchase shares currently exercisable or exercisable within 60 days of March 31, 2011 and 54,687 shares of unvested restricted stock at March 31, 2011. Also includes 1,562,565 shares held by the Gleeson Family Trust. Mr. Gleeson is the trustee of the Gleeson Family Trust and may be deemed to have beneficial ownership of these shares. |
| (9) | Includes 36,956 options to purchase shares currently exercisable or exercisable within 60 days of March 31, 2011. |
| (10) | Includes 42,852 unvested shares of restricted stock at March 31, 2011. Also includes 36,275 shares held by Penashe Holdings Propriety Limited. Dr. Fox is an executive director of Penashe Holdings Propriety Limited and may be deemed to have beneficial ownership of these securities, except to the extent of any indirect pecuniary interest in his distributive shares therein. |
| (11) | Includes 61,651 shares of common stock held by HI Charitable Remainder Uni Trust, 124,934 shares of common stock held by The Jon Faiz Kayyem and Paige N. Gates Family Trust, dated April 1, 2000, and 569,308 shares of common stock held by IFIN LP. Dr. Kayyem is trustee of the HI Charitable Remainder Uni Trust, trustee of The Jon Faiz Kayyem and Paige N. Gates Family Trust, dated April 1, 2000, and President of In-Motion LLC, the general partner of IFIN LP. Dr. Kayyem may be deemed to have beneficial ownership of the shares held by these entities. Also includes 113,927 shares subject to options currently exercisable or exercisable within 60 days of March 31, 2011 and 11,000 unvested shares of restricted stock at March 31, 2011. |
| (12) | Consists of 25,342 options to purchase shares currently exercisable or exercisable within 60 days of March 31, 2011. |
| (13) | Includes 23,404 options to purchase shares currently exercisable or exercisable within 60 days of March 31, 2011 and 31,844 unvested shares of restricted stock at March 31, 2011. |
| (14) | Includes 17,750 options to purchase shares currently exercisable or exercisable within 60 days of March 31, 2011 and 43,400 unvested shares of restricted stock at March 31, 2011. |
| (15) | Includes 47,938 options to purchase shares currently exercisable or exercisable within 60 days of March 31, 2011 and 4,000 unvested shares of restricted stock at March 31, 2011. |
| (16) | Includes 24,857 options to purchase shares currently exercisable. Mr. Kemper resigned from GenMark as Chief Financial Officer in November 2010 and from GenMark effective December 31, 2010. |
| (17) | Includes 21,306 options to purchase shares currently exercisable. Mr. Bellano resigned from GenMark as Senior Vice President, Commercial Operations on November 10, 2010. |
| (18) | Includes 368,567 options to purchase shares currently exercisable or exercisable within 60 days of March 31, 2011 and 187,783 unvested shares of restricted stock at March 31, 2011. |
90
Table of Contents
Since January 1, 2008, except as set forth below, there has not been nor are there currently proposed any transactions or series of similar transactions to which we were or are to be a party in which the amount involved exceeds $120,000 and in which any director, executive officer, holder of more than 5% of our common stock or any member of the immediate family of any of the foregoing persons had or will have a direct or indirect material interest.
Issuances of Ordinary Shares by Osmetech
In 2008, Osmetech completed two private financing transactions, in which it issued 1,050,813 ordinary shares at $3.36 per share for net proceeds of $3.5 million on December 5, 2008 and 1,942,624 ordinary shares at $3.36 per share for net proceeds of $6.5 million on December 21, 2008. Efficacy Capital Limited purchased 396,533 ordinary shares in the December 5, 2008 financing and 733,066 ordinary shares in the December 21, 2008 financing. Dr. Kayyem, our Chief Executive Officer and one of our directors, served as a managing partner of Efficacy Capital Limited at the time of these financings.
In 2009, Osmetech completed two private financing transactions, in which it issued 1,139,285 ordinary shares at $7.59 per share for net proceeds of $8.6 million on June 25, 2009 and 2,086,090 ordinary shares at $7.60 per share for net proceeds of $15.8 million on December 21, 2009. The securities purchased by investors in these financings included the following:
| • | Mr. Gleeson, one of our directors, purchased 132,475 ordinary shares in the June 25, 2009 financing and 127,942 ordinary shares in the December 21, 2009 financing. |
| • | Efficacy Capital Limited purchased 33,119 ordinary shares in the June 25, 2009 financing and 31,985 ordinary shares in the December 21, 2009 financing. Dr. Kayyem, our Chief Executive Officer and one of our directors, served as a managing partner of Efficacy Capital Limited at the time of these financings. |
Issuance of Shares in our Initial Public Offering
Certain of our directors, officers and beneficial owners of 5% or more of our common stock purchased shares in our initial public offering which closed on June 3, 2010 at the public offering price of $6.00 per share as follows:
| • | Mr. Christopher Gleeson, our then Chairman, and current Chairman and former Interim Chief Executive Officer, purchased an aggregate of 1,333,333 shares of common stock with an aggregate public offering price of $8,000,000; |
| • | Dr. Jon Faiz Kayyem, our then President, Chief Executive Officer and director, and our current Scientific Officer and director, purchased an aggregate of 250,000 shares of common stock with an aggregate public offering price of up to $1,500,000; and |
| • | Ronin Capital L.L.C., a greater than 5% stockholder, purchased an aggregate of 332,238 shares of common stock with an aggregate public offering price of up to $1,500,000. |
Recruitment Services
In 2010, we made payments to Novasyte, LLC for recruitment services and related expenses totaling $126,675. Christopher Gleeson, our Chairman and former Interim CEO, is a 33% shareholder in Novasyte, LLC.
Employment
Michael Gleeson, who is an immediate family member of our Chairman and former Interim CEO Christopher Gleeson, serves as our Vice President, U.S. Sales. Michael Gleeson was compensated $150,028 for base salary, bonus and commissions in 2010.
91
Table of Contents
Other Agreements with Management
We have entered into employment agreements with certain of our executive officers, which contain compensation terms and vesting acceleration or severance benefits upon termination of employment or a change of control. We previously entered into a consulting agreement with Christopher Gleeson, our Chairman, during his service as Interim Chief Executive Officer from August 2010 to May 2011. See “Executive Compensation—Employment Arrangements” for a description of these agreements. Please see “Principal Stockholders” for a description of the option and stock holdings of our directors and executive officers.
Indemnification Agreements
We have entered into indemnification agreements with our directors and executive officers for the indemnification of and advancement of expenses to these persons. We also intend to enter into these agreements with our future directors and executive officers. The indemnification agreements provide, among other things, that subject to certain procedures and conditions, we will, to the fullest extent permitted by Delaware law, indemnify the directors and officers against all liabilities and expenses, actually or reasonably incurred by a director or officer in connection with the investigation, defense, settlement or appeal of a proceeding if, by reason of the indemnitee’s status as a director or officer, the indemnitee was or is a party or is threatened to be made a party to the proceeding. In addition, the indemnification agreements provide for the advancement of expenses incurred by the indemnitee, subject to certain conditions and exceptions, in connection with any proceeding covered by the indemnification agreements. The indemnification agreements also require us to maintain directors’ and officers’ liability insurance in a reasonable amount from established and reputable insurers.
Policy for Approval of Related Party Transactions
Pursuant to the written charter of our Audit Committee, the Audit Committee is responsible for reviewing and approving all transactions in which we are a participant and in which any parties related to us, including our executive officers, our directors, beneficial owners of more than 5% of our securities, immediate family members of the foregoing persons and any other persons whom our Board determines may be considered related parties, has or will have a direct or indirect material interest. If advanced approval is not feasible, the Audit Committee has the authority to ratify a related party transaction at the next Audit Committee meeting. For purposes of our Audit Committee charter, a material interest is deemed to be any consideration received by such a party in excess of $120,000 per year.
In reviewing and approving such transactions, the Audit Committee shall obtain, or shall direct our management to obtain on its behalf, all information that the committee believes to be relevant and important to a review of the transaction prior to its approval. Following receipt of the necessary information, a discussion shall be held of the relevant factors if deemed to be necessary by the committee prior to approval. If a discussion is not deemed to be necessary, approval may be given by written consent of the committee. This approval authority may also be delegated to the Chairman of the Audit Committee in respect of any transaction in which the expected amount is less than $250,000. No related party transaction may be entered into prior to the completion of these procedures.
The Audit Committee or its Chairman, as the case may be, shall approve only those related party transactions that are determined to be in, or not inconsistent with, the best interests of us and our stockholders, taking into account all available facts and circumstances as the committee or the Chairman determines in good faith to be necessary. These facts and circumstances will typically include, but not be limited to, the material terms of the transaction, the nature of the related party’s interest in the transaction, the significance of the transaction to the related party and the nature of our relationship with the related party, the significance of the transaction to us, and whether the transaction would be likely to impair (or create an appearance of impairing) the judgment of a director or executive officer to act in our best interest. No member of the Audit Committee may participate in any review, consideration or approval of any related party transaction with respect to which the member or any of his or her immediate family members is the related party, except that such member of the Audit Committee will be required to provide all material information concerning the related party transaction to the Audit Committee.
92
Table of Contents
The following description of the capital stock and provisions of our certificate of incorporation and bylaws are summaries and are qualified by reference to our certificate of incorporation and bylaws. For the complete terms of our capital stock, please refer to our certificate of incorporation and our bylaws.
General
Pursuant to our certificate of incorporation, we are authorized to issue 100,000,000 shares of common stock, $0.0001 par value per share, and 5,000,000 shares of preferred stock, $0.0001 par value per share. Our board of directors may establish the rights and preferences of the preferred stock from time to time.
Common Stock
As of March 31, 2011, we had 11,738,233 shares of common stock outstanding and no shares of preferred stock outstanding. As of March 31, 2011, there were 9,128 holders of record of common stock.
Based on the number of shares outstanding as of March 31, 2011, we will have approximately shares of common stock outstanding immediately following this offering, and approximately shares outstanding if the underwriters exercise their over-allotment option in full.
Holders of common stock are entitled to one vote for each share held of record on all matters submitted to a vote of the stockholders, and do not have cumulative voting rights. Subject to preferences that may be applicable to any outstanding shares of preferred stock, holders of common stock are entitled to receive ratably such dividends, if any, as may be declared from time to time by our board of directors out of funds legally available for dividend payments. The holders of common stock have no preferences or rights of conversion, exchange, pre-emption or other subscription rights. There are no redemption or sinking fund provisions applicable to the common stock. In the event of any liquidation, dissolution or winding-up of our affairs, holders of common stock will be entitled to share ratably in our assets that are remaining after payment or provision for payment of all of our debts and obligations and after liquidation payments to holders of outstanding shares of preferred stock, if any.
Preferred Stock
We have no shares of preferred stock outstanding. Our board of directors has the authority, without further stockholder authorization, to issue from time to time up to 5,000,000 shares of preferred stock in one or more series and to fix the terms, limitations, voting rights, relative rights and preferences and variations of each series. The rights of the holders of common stock will be subject to, and may be adversely affected by, the rights of holders of any preferred stock that may be issued in the future. Although we have no present plans to issue any other shares of preferred stock, the issuance of shares of preferred stock, or the issuance of rights to purchase such shares, could decrease the amount of earnings and assets available for distribution to the holders of common stock, could adversely affect the rights and powers, including voting rights, of the common stock, and could have the effect of delaying, deterring or preventing a change of control of us or an unsolicited acquisition proposal.
Stock Options
We reserved an aggregate of 2,000,000 shares of common stock for issuance under the 2010 Equity Incentive Plan, which is subject to increase on an annual basis pursuant to the terms of the plan. As of March 31, 2011, we had outstanding options to purchase an aggregate of 1,314,975 shares of common stock. All outstanding options are governed by the terms of the 2010 Plan. Of this aggregate amount, the outstanding options have a weighted-average exercise price of $6.06 per share, and options to purchase 516,596 shares have been vested as of March 31, 2011.
93
Table of Contents
Warrants
As of March 31, 2011, there were outstanding warrants to purchase 88,317 shares of our common stock at a weighted-average exercise price of $9.98 per share.
Anti-Takeover Provisions of Delaware Law, Our Certificate of Incorporation and Our Bylaws
Provisions of the DGCL and our certificate of incorporation and by-laws could make it more difficult to acquire us by means of a tender offer, a proxy contest or otherwise, or to remove incumbent officers and directors. These provisions, summarized below, are expected to discourage certain types of coercive takeover practices and takeover bids that our board of directors may consider inadequate and to encourage persons seeking to acquire control of us to first negotiate with our board of directors. We believe that the benefits of increased protection of our ability to negotiate with the proponent of an unfriendly or unsolicited proposal to acquire or restructure us outweigh the disadvantages of discouraging takeover or acquisition proposals because, among other things, negotiation of these proposals could result in an improvement of their terms.
Delaware Anti-Takeover Statute
We are subject to Section 203 of the DGCL, an anti-takeover statute. In general, Section 203 of the DGCL prohibits a publicly held Delaware corporation from engaging in a “business combination” with an “interested stockholder” for a period of three years following the time the person became an interested stockholder, unless the business combination or the acquisition of shares that resulted in a stockholder becoming an interested stockholder is approved in a prescribed manner. Generally, a “business combination” includes a merger, asset or stock sale, or other transaction resulting in a financial benefit to the interested stockholder. Generally, an “interested stockholder” is a person who, together with affiliates and associates, owns (or within three years prior to the determination of interested stockholder status did own) 15% or more of a corporation’s voting stock. The existence of this provision would be expected to have an anti-takeover effect with respect to transactions not approved in advance by our board of directors, including discouraging attempts that might result in a premium over the market price for the shares of common stock held by our stockholders.
Classified Board
Our certificate of incorporation and our bylaws provide that our board of directors is divided into three classes, each comprised of three directors. The directors designated as a Class I directors have a term expiring at our annual meeting of stockholders in May 2014. The directors designated as a Class II directors have a term expiring at our annual meeting of stockholders in 2012, and the directors designated as Class III directors have a term expiring at our annual meeting of stockholders in 2013. Directors for each class will be elected at the annual meeting of stockholders held in the year in which the term for that class expires and thereafter will serve for a term of three years. At any meeting of stockholders for the election of directors at which a quorum is present, the election will be determined by a plurality of the votes cast by the stockholders entitled to vote at the election. Under the classified board provisions, it will take at least two elections of directors for any individual or group to gain control of our board. Accordingly, these provisions could discourage a third party from initiating a proxy contest, making a tender offer or otherwise attempting to gain control of us.
Removal of Directors
Our bylaws provide that our stockholders may only remove our directors with cause.
Amendment
Our certificate of incorporation and our bylaws provide that the affirmative vote of the holders of at least 80% of our voting stock then outstanding is required to amend certain provisions relating to the number, term, election and removal of our directors, the filling of our board vacancies, stockholder notice procedures, the calling of special meetings of stockholders and the indemnification of directors.
94
Table of Contents
Size of Board and Vacancies
Our bylaws provide that the number of directors on our board of directors is fixed exclusively by our board of directors. Newly created directorships resulting from any increase in our authorized number of directors will be filled by a majority of our board of directors then in office, provided that a majority of the entire board of directors, or a quorum, is present and any vacancies in our board of directors resulting from death, resignation, retirement, disqualification, removal from office or other cause will be filled generally by the majority vote of our remaining directors in office, even if less than a quorum is present.
Special Stockholder Meetings
Our certificate of incorporation provides that only the Chairman of our board of directors, our Chief Executive Officer or our board of directors pursuant to a resolution adopted by a majority of the entire board of directors may call special meetings of our stockholders.
Stockholder Action by Unanimous Written Consent
Our certificate of incorporation expressly eliminates the right of our stockholders to act by written consent other than by unanimous written consent. Stockholder action must take place at the annual or a special meeting of our stockholders or be effected by unanimous written consent.
Requirements for Advance Notification of Stockholder Nominations and Proposals
Our bylaws establish advance notice procedures with respect to stockholder proposals and nomination of candidates for election as directors other than nominations made by or at the direction of our board of directors or a committee of our board of directors.
No Cumulative Voting
The DGCL provides that stockholders are denied the right to cumulate votes in the election of directors unless our certificate of incorporation provides otherwise. Our certificate of incorporation does not provide for cumulative voting.
Undesignated Preferred Stock
The authority that will be possessed by our board of directors to issue preferred stock could potentially be used to discourage attempts by third parties to obtain control of our company through a merger, tender offer, proxy contest or otherwise by making such attempts more difficult or more costly. Our board of directors may issue preferred stock with voting rights or conversion rights that, if exercised, could adversely affect the voting power of the holders of common stock.
Authorized but Unissued Shares
Our authorized but unissued shares of common stock and preferred stock will be available for future issuance without stockholder approval. We may use additional shares for a variety of purposes, including future public offerings to raise additional capital, to fund acquisitions and as employee compensation. The existence of authorized but unissued shares of common stock and preferred stock could render more difficult or discourage an attempt to obtain control of us by means of a proxy contest, tender offer, merger or otherwise.
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is American Stock Transfer and Trust Company.
Stock Exchange
Our common stock is listed on the NASDAQ Global Market under the symbol “GNMK.”
95
Table of Contents
SHARES ELIGIBLE FOR FUTURE SALES
We cannot assure you that a significant public market for our common stock will be sustained after this offering. Future sales of substantial amounts of our common stock, including shares of common stock issuable upon exercise of outstanding options and warrants, in the public market after this offering, or the perception that these sales could occur, could adversely affect the prevailing market price of our common stock and could impair our future ability to raise capital through the sale of equity securities.
Sale of Outstanding Shares
Based on the number of shares outstanding as of March 31, 2011, we will have approximately shares of common stock outstanding after the completion of this offering, and approximately shares if the underwriters exercise their over-allotment option in full. Of these shares, the shares of common stock sold in this offering, plus the additional shares if the underwriters exercise their over-allotment option in full, will be freely transferable without restriction, unless purchased by our affiliates, as that term is defined under Rule 144 of the Securities Act.
The remaining shares of common stock to be outstanding immediately following the completion of this offering, will be freely transferable without restriction, unless such shares are held by our affiliates as defined under Rule 144 of the Securities Act or such shares are subject to the terms and conditions of a lock-up agreement as discussed further below.
In addition, of the 1,314,975 shares of our common stock that were subject to stock options outstanding as of March 31, 2011, options to purchase 516,596 shares of common stock were vested as of March 31, 2011. Shares received upon exercise of these stock options will be eligible for sale subject to the lock-up agreements described below and the requirements of Rule 144 under the Securities Act for shares held by our affiliates.
Lock-Up Agreements
Pursuant to certain “lock-up” agreements, shares of common stock held by our executive officers and directors are subject to lock-up agreements with the underwriters. These individuals have agreed, subject to certain limited exceptions, not to, for a period from the date of the final prospectus for this offering through and including the 90th day after the date of the final prospectus, offer, pledge, announce the intention to sell, contract to sell, sell any option or contract to purchase, purchase any option or contract to sell, grant any options, right or warrant to purchase or otherwise dispose of, directly or indirectly, any shares of our common stock or securities convertible into or exchangeable or exercisable for shares of our common stock or to file or request the filing of any registration statement under the Securities Act with respect of such shares. The 90-day restricted period will be automatically extended if (i) during the last 17 days before the last day of the 90-day restricted period we issue an earnings release or material news or a material event relating to us occurs or (ii) prior to the expiration of the 90-day restricted period, we announce that we will release earnings results during the 16-day period beginning on the last day of the 90-day restricted period, in either of which case the restrictions described above will continue to apply until the expiration of the date that is 18 days after the issuance of the earnings release or the material news or material event occurs. These lock-up restrictions apply to our shares of our common stock and to securities convertible into or exchangeable or exercisable for or repayable with shares of our common stock. The lock-up restrictions will not apply to certain transfers not involving a disposition for value, provided that the recipient agrees to be bound by these lock-up restrictions. The lock-up restrictions will not apply to grants we may make under our existing stock options plans or to exercises of stock options under our stock options plans, but will apply to shares acquired upon exercise of these options. Canaccord Genuity may agree, at any time or from time to time and without notice, to release for sale in the public market all or any portion of the securities subject to these restrictions.
96
Table of Contents
Stock Options
Assuming no options to purchase shares of our common stock are issued after March 31, 2011, we will have outstanding options to purchase an aggregate of 1,314,975 shares of common stock immediately following this offering with a weighted-average exercise price of $6.06 per share. All of the outstanding options at the time of closing of this offering will be governed by the terms of the 2010 Plan. We have registered all of the common shares issued or reserved for future issuance under the 2010 Plan by filing a Form S-8 registration statement under the Securities Act, and as a result, all common shares purchased upon exercise of our outstanding options generally will be available for resale in the public market, subject to the terms of the lock-up agreements and the requirements of Rule 144 under the Securities Act for shares held by our affiliates.
Warrants
Assuming no warrants to purchase shares of our common stock are issued after March 31, 2011, we will have outstanding warrants to purchase an aggregate of 88,317 shares of common stock immediately following this offering with a weighted-average exercise price of $9.98 per share. Upon exercise of the warrants, the holder may sell the shares of common stock in the market in accordance with Rule 144 under the Securities Act.
Affiliate Sales
Our affiliates, as defined under Rule 144 of the Securities Act, are entitled to sell within any three-month period a number of shares that does not exceed the greater of:
| • | one percent of the number of shares of our common stock then outstanding, which will equal approximately shares immediately after this offering; and |
| • | the average weekly trading volume of our common stock on the NASDAQ Global Market during the four calendar weeks preceding the filing of a notice on Form 144 with respect to the sale, or if no such notice is required, the date of receipt of the order to execute the sale. |
Sales of restricted shares under Rule 144 by our affiliates are also subject to requirements regarding the manner of sale, notice and the availability of current public information about us.
Rule 144
Under Rule 144 of the Securities Act, as in effect on the date of this prospectus, a person who holds restricted shares of our common stock and is not one of our affiliates at any time during the three months preceding a sale, and who has beneficially owned these restricted shares for at least six months, would be entitled to sell an unlimited number of shares of our common stock, provided current public information about us is available. In addition, under Rule 144, a person who holds restricted shares of our common stock and is not one of our affiliates at any time during the three months preceding a sale, and who has beneficially owned these restricted shares for at least one year, would be entitled to sell an unlimited number of shares immediately upon the closing of this offering without regard to whether current public information about us is available.
97
Table of Contents
CERTAIN MATERIAL U.S. FEDERAL INCOME AND ESTATE TAX CONSIDERATIONS TO NON-U.S. HOLDERS
The following is a discussion of the material U.S. federal income and estate tax considerations with respect to the ownership and disposition of our common stock that may be relevant to a non-U.S. holder that acquires our common stock pursuant to this offering. The discussion is based on provisions of the Internal Revenue Code of 1986, as amended, or the Code, applicable U.S. Treasury regulations promulgated thereunder and U.S. Internal Revenue Service, or IRS, rulings and pronouncements and judicial decisions, all as in effect on the date of this prospectus and all of which are subject to change (possibly on a retroactive basis) or to differing interpretations so as to result in tax considerations different from those summarized below. We cannot assure you that a change in law will not alter significantly the tax considerations that we describe in this summary.
The discussion is limited to non-U.S. holders that hold our common stock as a “capital asset” within the meaning of Section 1221 of the Code (generally, property held for investment). As used in this discussion, the term “non-U.S. holder” means a beneficial owner of our common stock that is not, for U.S. federal income tax purposes:
| • | an individual who is a citizen or resident of the United States; |
| • | a corporation including any entity treated as a corporation for U.S. federal income tax purposes created or organized in or under the laws of the United States or any political subdivision thereof; |
| • | a partnership including any entity treated as a partnership for U.S. federal income tax purposes; |
| • | an estate, the income of which includes gross income for U.S. federal income tax purposes regardless of its source; or |
| • | a trust (1) if a U.S. court is able to exercise primary supervision over the administration of the trust and one or more U.S. persons have authority to control all substantial decisions of the trust or (2) that has made a valid election to be treated as a U.S. person for such purposes. |
This discussion does not address the U.S. federal income and estate tax rules applicable to any person who holds our common stock through entities treated as partnerships for U.S. federal income tax purposes or to such entities themselves. If a partnership (including any entity or arrangement treated as a partnership for U.S. federal income tax purposes) owns our common stock, the tax treatment of a partner in that partnership will depend upon the status of the partner and the activities of the partnership. A holder that is a partnership or a holder of interests in a partnership should consult such holder’s tax advisor regarding the tax consequences of the purchase, ownership and disposition of our common stock.
This discussion does not consider:
| • | any state, local or foreign tax consequences; |
| • | any tax consequences or computation of the alternative minimum tax; |
| • | any U.S. federal gift tax consequences; or |
| • | any U.S. federal tax considerations that may be relevant to a non-U.S. holder in light of its particular circumstances or to non-U.S. holders that may be subject to special treatment under U.S. federal tax laws, including without limitation, banks or other financial institutions, insurance companies, tax-exempt organizations, certain trusts, hybrid entities, “controlled foreign corporations,” “passive foreign investment companies,” certain former citizens or residents of the U.S., holders subject to U.S. federal alternative minimum tax, broker-dealers, dealers or traders in securities or currencies and holders that hold our common stock as part of a “straddle,” “hedge,” “conversion transaction,” “synthetic security” or other integrated investment. |
Prospective investors are urged to consult their tax advisors regarding the application of the U.S. federal income and estate tax laws to their particular situations and the consequences under U.S. federal gift tax laws, as well as foreign, state and local laws and tax treaties.
98
Table of Contents
Dividends
As previously discussed, we do not anticipate paying dividends on our common stock in the foreseeable future. If we pay dividends on our common stock, however, those payments will constitute dividends for U.S. federal income tax purposes to the extent paid from our current or accumulated earnings and profits, as determined under U.S. federal income tax principles. To the extent those distributions exceed our current and accumulated earnings and profits, the distributions will constitute a return of capital and first reduce the non-U.S. holder’s adjusted tax basis, but not below zero, and then will be treated as gain from the sale of stock, as described in the section of this prospectus entitled “Gain on Disposition of Common Stock.”
A dividend paid to a non-U.S. holder generally will be subject to withholding of U.S. federal income tax at a 30% rate, or a lower rate under an applicable income tax treaty, unless the dividend is effectively connected with the conduct of a trade or business of the non-U.S. holder within the U.S. (and, if an applicable income tax treaty so requires, is attributable to a permanent establishment of the non-U.S. holder within the U.S.). Non-U.S. holders (generally on a properly executed IRS Form W-8 BEN) will be required to satisfy certain certification and disclosure requirements in order to claim a reduced rate of withholding pursuant to an applicable income tax treaty. These forms must be periodically updated. Non-U.S. holders should consult their tax advisors regarding their entitlement to benefits under a relevant income tax treaty. Special rules apply in the case of common stock held by certain non-U.S. holders that are entities rather than individuals.
Dividends that are effectively connected with a non-U.S. holder’s conduct of a trade or business in the United States and, if an applicable income tax treaty so requires, attributable to a permanent establishment in the United States will be taxed on a net income basis at the regular graduated U.S. federal income tax rates in the same manner as if the non-U.S. holder were a resident of the United States. In such cases, we will not have to withhold U.S. federal income tax if the non-U.S. holder complies with applicable certification and disclosure requirements. In addition, a “branch profits tax” may be imposed at a 30% rate, or a lower rate under an applicable income tax treaty, on dividends received by a foreign corporation that are effectively connected with the conduct of a trade or business in the United States.
A non-U.S. holder may obtain a refund or credit of any excess amounts withheld by filing an appropriate claim for a refund together with the required information with the IRS.
Gain on Disposition of Common Stock
A non-U.S. holder generally will not be subject to U.S. federal income tax with respect to gain realized on a sale or other disposition of our common stock unless one of the following applies:
| • | the gain is effectively connected with the non-U.S. holder’s conduct of a trade or business in the U.S. and, if an applicable income tax treaty so requires, is attributable to a permanent establishment maintained by the non-U.S. holder in the United States; in these cases, the non-U.S. holder generally will be taxed on its net gain derived from the disposition at the regular graduated rates and in the manner applicable to United States persons and, if the non-U.S. holder is a foreign corporation, the “branch profits tax” described above may also apply; |
| • | the non-U.S. holder is an individual present in the United States for 183 days or more in the taxable year of the disposition and certain other conditions are met; in this case, the non-U.S. holder will be subject to a 30% tax on the amount by which the gain derived from the sale or other disposition of our common stock and any other U.S.-source capital gains realized by the non-U.S. holder in the same taxable year exceed the U.S.-source capital losses realized by the non-U.S. holder in that taxable year unless an applicable income tax treaty provides an exemption or a lower rate; or |
| • | we are or have been a “U.S. real property holding corporation” for U.S. federal income tax purposes at any time within the shorter of the five year period ending on the date of disposition or the period that the non-U.S. holder held our common stock. We do not believe that we have been, are, or will become, a U.S. real property holding corporation, although there can be no assurance in this regard. If we are, |
99
Table of Contents
| or were to become, a U.S. real property holding corporation at any time during the applicable period, however, any gain recognized on a disposition of our common stock by a non-U.S. holder that did not own (directly, indirectly or constructively) more than 5% of our common stock during the applicable period generally would not be subject to U.S. federal income tax, provided that our common stock is “regularly traded on an established securities market” (within the meaning of Section 897(c)(3) of the Code). |
Federal Estate Tax
Common stock owned or treated as owned by an individual who is not a citizen or resident of the United States (as specifically defined for U.S. federal estate tax purposes) at the time of death are considered U.S. situs assets includible in the individual’s gross estate for U.S. federal estate tax purposes and therefore may be subject to U.S. federal estate tax, unless an applicable estate tax treaty provides otherwise.
Information Reporting and Backup Withholding Tax
Dividends and proceeds from the sale or other taxable disposition of our common stock are potentially subject to backup withholding. In general, backup withholding will not apply to dividends on our common stock paid by us or our paying agents, in their capacities as such, to a non-U.S. holder if the holder has provided the required certification that it is a non-U.S. holder.
Generally, we must report to the IRS the amount of dividends paid, the name and address of the recipient, and the amount, if any, of tax withheld. Pursuant to income tax treaties or some other agreements, the IRS may make its reports available to tax authorities in the recipient’s country of residence.
In general, backup withholding and information reporting will not apply to proceeds from the disposition of our common stock paid to a non-U.S. holder within the United States or conducted through certain U.S.-related financial intermediaries the holder has provided the required certification that it is a non-U.S. holder.
Backup withholding is not an additional tax. Any amount withheld may be refunded or credited against the holder’s U.S. federal income tax liability, if any, provided that the required information is furnished to the IRS in a timely manner.
Non-U.S. holders should consult their tax advisors regarding the application of the information reporting and backup withholding rules to them.
Prospective non-U.S. holders of our common stock should consult their tax advisors with respect to the particular tax consequences to them of owning and disposing of our common stock, including the consequences under the laws of any state, local or foreign jurisdiction or under any applicable tax treaty.
Recent Legislation Affecting Taxation of Our Common Stock Held By or Through Foreign Entities
Recent legislation generally will impose a U.S. federal withholding tax of 30% on dividends and the gross proceeds of a disposition of our common stock paid after December 31, 2012 to certain foreign entities (including, financial institutions), unless various U.S. information reporting and due diligence requirements (that are different from, and in addition to, the certification requirements described above) have been satisfied that generally relate to ownership by United States persons of interests in or accounts with those entities. Prospective investors should consult their own tax advisors regarding the possible implications of this legislation on their investment in our common stock.
100
Table of Contents
We are offering the shares of common stock described in this prospectus through a number of underwriters. Canaccord Genuity Inc. is acting as sole book-running manager of the offering and as representative of the underwriters. William Blair & Company, L.L.C. is acting as co-lead manager for this offering. Subject to the terms and conditions of the underwriting agreement, we have agreed to sell to the underwriters, and each underwriter has agreed, severally and not jointly, to purchase, the number of shares indicated next to its name in the following table:
| Underwriters |
Number of Shares |
|||
| Canaccord Genuity Inc. |
||||
| William Blair & Company, L.L.C. |
||||
| Total |
||||
The underwriters are offering the common stock subject to their acceptance of the shares from us and subject to prior sale. The underwriting agreement provides that the obligations of the underwriters to pay for and accept delivery of the common stock offered by this prospectus are subject to the approval of certain legal matters by their counsel and to certain other conditions. The underwriting agreement provides that the underwriters are obligated to take and pay for all of the common stock if any such shares are purchased, other than those shares covered by the overallotment option described below.
The underwriters have advised us that they propose to offer the shares of common stock directly to the public at the public offering price set forth on the cover page of this prospectus, and to selected dealers at the public offering price less a selling concession not in excess of $ per share. The underwriters also may allow, and dealers may reallow, a concession not in excess of $ per share to brokers and dealers. After the public offering of the shares, the underwriters may change the offering price and other selling terms.
Overallotment Option
We have granted to the underwriters an option to purchase up to an aggregate of additional shares of common stock from us at the public offering price less the underwriting discount. The underwriters may exercise this option solely for the purpose of covering over-allotments, if any, made in connection with the offering of the shares of common stock offered by this prospectus. The underwriters have up to 30 days from the date of this prospectus to exercise this over-allotment option. If any shares are purchased with this over-allotment option, the underwriters will purchase shares in approximately the same proportion as shown in the table above. If any additional shares of common stock are purchased, the underwriters will offer the additional shares on the same terms as those on which the shares are being offered.
Discounts and Expenses
The following table shows the public offering price, the total underwriting discounts to be paid to the underwriters by us and the proceeds, before expenses, to us, both on a per share basis and in total. These amounts are shown assuming both no exercise and full exercise of the over-allotment option.
| Per Share |
Total | |||||||||||
| Without Overallotment Exercise |
With Overallotment Exercise |
|||||||||||
| Public offering price |
$ | $ | $ | |||||||||
| Underwriting discounts paid by us |
||||||||||||
| Proceeds, before expenses, to us |
||||||||||||
101
Table of Contents
We estimate expenses payable by us in connection with the offering of common stock, other than the underwriting discounts referred to above, will be approximately $ .
Indemnification
We have agreed to indemnify the underwriters against certain liabilities, including liabilities under the Securities Act, or to contribute to payments that the underwriters may be required to make in respect of those liabilities.
Lock-up Agreements
We and our executive officers and directors have entered into lock-up agreements with the underwriters. Under these agreements, we and each of these persons may not, without the prior written approval of Canaccord Genuity Inc., subject to limited exceptions, offer, sell, assign, transfer, contract to sell, or otherwise dispose of, or announce the intention to otherwise dispose of, or enter into any swap or other arrangement that transfers any economic consequences of ownership of our common stock or securities convertible into or exercisable or exchangeable for our common stock. These restrictions will be in effect for a period of 90 days after the date of this prospectus. Notwithstanding the termination of the lock-up period outlined above, and subject to certain exceptions, in the event that either (i) during the last 17 days of the lock-up period, we issue an earnings release or material news or a material event relating to us occurs, or (ii) prior to the expiration of the lock-up period, we announce that we will release earnings results during the 16-day period beginning on the last day of the lock-up period, then the expiration of the lock-up period will be extended until the expiration of the 18-day period beginning on the date of the issuance of an earnings release or the occurrence of the material news or material event, as applicable, unless the underwriter waives, in writing, such extension. At any time and without public notice, the underwriter may in its sole discretion release all or some of the securities from these lock-up agreements.
Price Stabilization, Short Positions and Penalty Bids
Until distribution of the shares of our common stock is completed, SEC rules may limit the underwriters from bidding for and purchasing shares of our common stock. However, the underwriters may engage in transactions that stabilize the price of the shares of our common stock, such as bids or purchases to peg, fix or maintain that price.
If the underwriters create a short position in our common stock in connection with this offering (i.e., if they sell more shares of our common stock than are listed on the cover page of this prospectus), the underwriters may reduce that short position by purchasing shares of our common stock in the open market. The underwriters may also elect to reduce any short position by exercising all or part of the over-allotment option described above. Purchases of shares of our common stock to stabilize its price or to reduce a short position may cause the price of shares of our common stock to be higher than it might be in the absence of such purchases.
The underwriters also may impose a penalty bid, whereby the underwriters may reclaim selling concessions allowed to other broker-dealers in respect of the common stock sold in the offering for their account if the underwriters repurchase the shares in stabilizing or covering transactions. These activities may stabilize, maintain or otherwise affect the market price of the common stock, which may be higher than the price that might otherwise prevail in the open market. The imposition of a penalty bid may also affect the price of the shares of our common stock in that it discourages resales of those shares of our common stock. The underwriters have advised us that these transactions may be effected on the NASDAQ Global Market or otherwise. Neither we nor the underwriters make any representation or prediction as to the direction or magnitude of any effect that the transactions described above may have on the price of shares of our common stock. In addition, neither we nor the underwriters make any representation that the underwriters will engage in these transactions or that these transactions, once commenced, will not be discontinued without notice.
102
Table of Contents
Electronic Distribution
This prospectus may be made available in electronic format on websites or through other online services maintained by the underwriters of the offering, or by their affiliates. Other than the prospectus in electronic format, the information on such websites and any information contained in any other website maintained by the underwriters or any of their affiliates is not part of the prospectus or the registration statement of which this prospectus forms a part, has not been approved or endorsed by us or the underwriters in their capacity as underwriters and should not be relied upon by investors.
Other Relationships
Certain of the underwriters and their affiliates have provided in the past to us and our affiliates and may provide from time to time in the future certain commercial banking, financial advisory, investment banking and other services for us and such affiliates in the ordinary course of their business, for which they have received and may continue to receive customary fees and commissions. Canaccord Genuity Inc. has also provided various financial advisory and investment banking services to Osmetech, our predecessor company, for which it received customary fees and expenses. In addition, from time to time, certain of the underwriters and their affiliates may effect transactions for their own account or the account of customers, and hold on behalf of themselves or their customers, long or short positions in our debt or equity securities or loans, and may do so in the future.
103
Table of Contents
The validity of the shares of common stock we are offering will be passed upon for us by DLA Piper LLP (US). Jones Day is counsel to the underwriters in connection with the offering.
The financial statements of GenMark as of December 31, 2010 included in this prospectus have been audited by Deloitte & Touche LLP (US), an independent registered public accounting firm, as stated in their report appearing herein. Such financial statements are included in reliance upon the report of such firm given upon their authority as experts in accounting and auditing.
The financial statements of Osmetech as of December 31, 2009 and for each of the two years in the period ended December 31, 2009 included in this prospectus have been audited by Deloitte LLP (UK), an independent registered public accounting firm, as stated in their report appearing herein. Such financial statements are included in reliance upon the report of such firm given upon their authority as experts in accounting and auditing.
WHERE YOU CAN FIND MORE INFORMATION ABOUT US
We have filed with the Securities and Exchange Commission a registration statement on Form S-1 under the Securities Act with respect to the common stock offered by this prospectus. This prospectus, which is part of the registration statement, omits certain information, exhibits, schedules, and undertakings set forth in the registration statement. For further information pertaining to us and our common stock, reference is made to the registration statement and the exhibits and schedules to the registration statement. Statements contained in this prospectus as to the contents or provisions of any documents referred to in this prospectus are not necessarily complete, and in each instance where a copy of the document has been filed as an exhibit to the registration statement on Form S-1 of which this prospectus forms a part, reference is made to the exhibit for a more complete description of the matters involved. We are required to file annual, quarterly and current reports, proxy statements and other information with the Securities and Exchange Commission. We make these documents publicly available, free of charge, on our website at www.genmarkdx.com as soon as practicable after filing such documents with the Securities and Exchange Commission.
You may read and copy any document that we file at the public reference room of the Securities and Exchange Commission at 100 F Street, N.E., Washington, D.C. 20549. Copies of the registration statement may be obtained from the Securities and Exchange Commission at prescribed rates from the public reference room at such address. You may obtain information regarding the operation of the public reference room by calling 1-800-SEC-0330. In addition, this registration statement and our future filings filed electronically with the Securities and Exchange Commission are publicly available through its website at www.sec.gov.
104
Table of Contents
F-1
Table of Contents
FINANCIAL STATEMENTS
CONDENSED CONSOLIDATED BALANCE SHEETS
(Unaudited)
| As of March 31, 2011 |
As of December 31, 2010 |
|||||||
| Current assets |
||||||||
| Cash and cash equivalents |
$ | 17,054,095 | $ | 18,329,079 | ||||
| Accounts receivable, net |
762,272 | 677,648 | ||||||
| Inventories, net |
871,033 | 896,809 | ||||||
| Other current assets |
382,194 | 2,193,160 | ||||||
| Total current assets |
19,069,594 | 22,096,696 | ||||||
| Property and equipment, net |
2,762,362 | 2,702,478 | ||||||
| Intangible assets, net |
68,042 | 70,980 | ||||||
| Other long-term assets |
55,355 | 55,355 | ||||||
| Total assets |
$ | 21,955,353 | $ | 24,925,509 | ||||
| Current liabilities |
||||||||
| Accounts payable |
$ | 1,651,985 | $ | 823,242 | ||||
| Accrued compensation |
1,019,545 | 1,171,989 | ||||||
| Other current liabilities |
1,761,011 | 1,249,928 | ||||||
| Total current liabilities |
4,432,541 | 3,245,159 | ||||||
| Long-term liabilities |
||||||||
| Loan payable |
2,000,000 | — | ||||||
| Other non-current liabilities |
622,644 | 612,932 | ||||||
| Total liabilities |
$ | 7,055,185 | $ | 3,858,091 | ||||
| Stockholders’ equity |
||||||||
| Common stock, $.0001 par value; 100,000,000 authorized; 11,738,233 and 11,723,512 issued and outstanding as of March 31, 2011 and December 31, 2010, respectively |
1,172 | 1,172 | ||||||
| Preferred stock, $0.0001 par value; 5,000,000 authorized, none issued |
— | — | ||||||
| Additional paid-in capital |
166,483,672 | 166,009,084 | ||||||
| Accumulated deficit |
(151,134,719 | ) | (144,492,881 | ) | ||||
| Accumulated other comprehensive loss |
(449,957 | ) | (449,957 | ) | ||||
| Total stockholders’ equity |
14,900,168 | 21,067,418 | ||||||
| Total liabilities and stockholders’ equity |
$ | 21,955,353 | $ | 24,925,509 | ||||
See accompanying notes to unaudited condensed consolidated financial statements.
F-2
Table of Contents
CONDENSED CONSOLIDATED STATEMENTS OF OPERATIONS
(Unaudited)
| Three Months Ended March 31, |
||||||||
| 2011 | 2010 | |||||||
| Product Revenue |
$ | 692,739 | $ | 384,249 | ||||
| License and other revenue |
71,664 | 15,015 | ||||||
| Total revenue |
764,403 | 399,264 | ||||||
| Cost of sales |
1,643,456 | 567,396 | ||||||
| Gross loss |
(879,053 | ) | (168,132 | ) | ||||
| Operating expenses |
||||||||
| Sales and marketing |
1,130,389 | 1,058,285 | ||||||
| General and administrative |
2,111,336 | 2,167,264 | ||||||
| Research and development |
2,528,252 | 1,453,759 | ||||||
| Total operating expenses |
5,769,977 | 4,679,308 | ||||||
| Loss from operations |
(6,649,030 | ) | (4,847,440 | ) | ||||
| Other income |
||||||||
| Other income (expense) |
11,899 | (1,110 | ) | |||||
| Interest income |
6,258 | 4,654 | ||||||
| Total other income |
18,157 | 3,544 | ||||||
| Loss before income taxes |
(6,630,873 | ) | (4,843,896 | ) | ||||
| Provision for income taxes |
(10,968 | ) | (5,049 | ) | ||||
| Net loss |
$ | (6,641,841 | ) | $ | (4,848,945 | ) | ||
| Net loss per share, basic and diluted |
$ | (0.56 | ) | $ | (0.68 | ) | ||
| Weighted average number of shares outstanding |
11,771,014 | 7,113,922 | ||||||
| Condensed consolidated statements of comprehensive loss three and three months ended March 31, 2011 and 2010 |
||||||||
| Net loss |
$ | (6,641,841 | ) | $ | (4,848,945 | ) | ||
| Foreign currency translation adjustment |
— | (34,647 | ) | |||||
| Comprehensive loss |
$ | (6,641,841 | ) | $ | (4,883,592 | ) | ||
See accompanying notes to unaudited condensed consolidated financial statements.
F-3
Table of Contents
CONDENSED CONSOLIDATED STATEMENTS OF CASH FLOWS
(Unaudited)
| Three Months
Ended March 31, |
||||||||
| 2011 | 2010 | |||||||
| Cash flows from operating activities: |
||||||||
| Net loss |
$ | (6,641,841 | ) | $ | (4,848,945 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||
| Depreciation and amortization |
288,771 | 318,369 | ||||||
| Change in allowance for doubtful accounts, net of write-offs |
(47,785 | ) | — | |||||
| Change in allowance for excess and obsolete inventory |
(546 | ) | — | |||||
| Share-based compensation |
474,588 | 347,530 | ||||||
| Changes in operating assets and liabilities: |
||||||||
| Trade accounts receivable |
(36,839 | ) | (71,316 | ) | ||||
| Inventories |
26,322 | (65,138 | ) | |||||
| Other current assets |
1,810,966 | (469,561 | ) | |||||
| Accounts payable |
667,559 | (316,564 | ) | |||||
| Accrued compensation |
(152,444 | ) | 279,184 | |||||
| Accrued and other liabilities |
520,798 | (155,724 | ) | |||||
| Net cash used in operating activities |
(3,090,451 | ) | (4,982,165 | ) | ||||
| Investing activities: |
||||||||
| Purchases of property and equipment |
(184,533 | ) | (137,440 | ) | ||||
| Net cash used in investing activities |
(184,533 | ) | (137,440 | ) | ||||
| Financing activities: |
||||||||
| Proceeds of loan payable |
2,000,000 | — | ||||||
| Proceeds from stock option exercises |
— | 4,734 | ||||||
| Net cash provided by financing activities |
2,000,000 | 4,734 | ||||||
| Effect of foreign exchange rate changes |
— | (46,935 | ) | |||||
| Net decrease in cash and cash equivalents |
(1,274,984 | ) | (5,114,871 | ) | ||||
| Cash and cash equivalents at beginning of period |
18,329,079 | 16,482,818 | ||||||
| Cash and cash equivalents at end of period |
$ | 17,054,095 | $ | 11,321,012 | ||||
| Noncash investing and financing activities: |
||||||||
| Reclassification of deposits on systems in other current assets |
— | $ | 285,284 | |||||
| IPO costs incurred but not paid included in accounts payable |
— | 1,537,192 | ||||||
| Property and equipment costs incurred but not paid included in accounts payable |
$ | 161,184 | — | |||||
See accompanying notes to unaudited condensed consolidated financial statements.
F-4
Table of Contents
Notes to Unaudited Condensed Consolidated Financial Statements
1. Organization and basis of presentation
Genmark Diagnostics, Inc. (the “Company” or “GenMark”) is a molecular diagnostics company focused on developing and commercializing the Company’s proprietary e-sensor technology. On February 12, 2010, the Company was established to serve as the parent company of Osmetech plc (“Osmetech”) upon a corporate reorganization and initial public offering (“IPO”). On June 3, 2010, the Company completed an IPO for 4,600,000 shares. Immediately prior to the completion of the IPO, the Company underwent a corporate reorganization whereby the ordinary shares of Osmetech were exchanged by its shareholders for the common stock of the Company on a 230 for 1 basis.
As the reorganization is deemed to be a transaction under common control, GenMark accounted for the reorganization in a manner similar to a pooling-of-interests, meaning:
(i) assets and liabilities were carried over at their respective carrying values;
(ii) common stock was carried over at the nominal value of the shares issued by GenMark;
(iii) additional paid-in capital represents the difference between the nominal value of the shares issued by GenMark, and the total of the additional paid-in capital and nominal value of Osmetech’s shares cancelled pursuant to the described reorganization; and
(iv) the accumulated deficit represents the aggregate of the accumulated deficit of Osmetech and the Company.
Once the reorganization became effective, all stock options granted under the Osmetech plc 2003 U.S. Equity Compensation Plan, Long Term Incentive Awards and all warrants issued were exchanged for options and warrants exercisable for the common stock of the Company.
The preferred stock may be issued from time to time in one or more series.
In these consolidated financial statements, the Company means Osmetech when referring to periods prior to the corporate reorganization and IPO.
The Company evaluated subsequent events through May 13, 2011, being the date of issuance of the unaudited condensed consolidated financial statements.
The accompanying financial statements have been prepared on a going-concern basis, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. The Company has incurred net losses from operations since its inception and has an accumulated deficit of $151,134,719 at March 31, 2011. Cash and cash equivalents at March 31, 2011 were $17,054,095.
Management expects operating losses to continue through the foreseeable future until the Company has expanded its product offerings and increased its product revenues to an extent covers the fixed cost base of the business. The Company’s management has prepared cash flow forecasts which indicate, based on the current cash resources available and the availability of unutilized credit facilities, that the Company has sufficient capital to fund its operations for at least the next twelve months.
The Company has prepared the accompanying unaudited condensed consolidated financial statements in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”) for interim financial information and Article 10 of Regulation S-X. Accordingly, they do not include all of the information and disclosures required by U.S. GAAP for audited financial statements. In the opinion of management, all adjustments, which include only normal recurring adjustments considered necessary for a fair
F-5
Table of Contents
Genmark Diagnostics, Inc.
Notes to Unaudited Condensed Consolidated Financial Statements—(Continued)
presentation, have been included. Operating results for the three months ended March 31, 2011 are not necessarily indicative of the results that may be expected for the year ending December 31, 2011. The information presented in the condensed consolidated financial statements and related footnotes at March 31, 2011, and for the three months ended March 31, 2011 and 2010, is unaudited and the condensed consolidated balance sheet amounts and related footnotes at December 31, 2010 have been derived from our audited financial statements. For further information, refer to the consolidated financial statements and accompanying footnotes included in our annual report Form 10-K filed with the Securities and Exchange Commission (“SEC”) on March 14, 2011.
The Company operates in one reportable segment, and substantially all of the Company’s operations and assets are in the United States of America.
Principles of Consolidation-The unaudited condensed consolidated financial statements include the accounts of the Company and its wholly owned subsidiaries. All intercompany balances and transactions have been eliminated in consolidation.
Recent Accounting Pronouncements
From time to time, new accounting pronouncements are issued by the Financial Accounting Standards Board (FASB) or other standard setting bodies that are adopted by the Company as of the specified effective date. We believe that the impact of recently issued standards that are not yet effective will not have a material impact on our financial position or results of operations upon adoption.
Fair Value of Financial Instruments
The Company’s financial instruments consist of cash equivalents, accounts receivable, accounts payable and loan payable. The carrying amounts of accounts receivable, accounts payable and the loan payable are considered reasonable estimates of their fair value, due to the short maturity of these instruments. There were no significant financial instruments requiring one-time or recurring measurements of fair value during the three months ended March 31, 2011.
Accounting literature provides a fair value hierarchy, which classifies fair value measurements based on the inputs used in measuring fair value. These inputs include: Level 1, defined as observable inputs such as quoted prices for identical instruments in active markets; Level 2, defined as inputs other than quoted prices in active markets that are either directly or indirectly observable; and Level 3, defined as unobservable inputs for which little or no market data exists, therefore requiring an entity to develop its own assumptions. There were no transfers of items between Levels 1, 2 or 3.
Cash and cash equivalents: The carrying amounts reported in the balance sheets for cash and cash equivalents are stated at their fair market value. Cash and cash equivalents are classified as Level 1.
Loan payable: The carrying amount reported in the balance sheets for the loan payable is considered a reasonable estimate of fair value, based on the short maturity and comparable terms for similar credit facilities. The loan payable is classified as Level 2.
Non-recurring measurements: The Company measures the fair value of its long-lived assets on a periodic basis when it appears that there may be requirement to do so, such as an indication of impairment. There was no impairment recorded for the three months ended March 31, 2011.
F-6
Table of Contents
Genmark Diagnostics, Inc.
Notes to Unaudited Condensed Consolidated Financial Statements—(Continued)
Income Taxes
Current income tax expense is the amount of income taxes expected to be payable for the current year. A deferred income tax liability or asset is established for the expected future tax consequences resulting from the differences in financial reporting and tax bases of assets and liabilities. A valuation allowance is provided if it is more likely than not that some or all of the deferred tax assets will not be realized. A full valuation allowance has been recorded against the Company’s deferred tax assets due to the uncertainty surrounding the Company’s ability to utilize these assets in the future. The Company provides for uncertain tax positions when such tax positions do not meet the recognition thresholds or measurement standards prescribed by the authoritative guidance on income taxes. Amounts for uncertain tax positions are adjusted in periods when new information becomes available or when positions are effectively settled. The Company recognizes accrued interest and penalties related to uncertain tax positions as a component of income tax expense.
2. Share-Based Compensation
The Company recognizes share-based compensation expense related to share options, warrants and restricted stock issued to employees, directors and consultants in exchange for services. The compensation expense is based on the fair value of the awards, which are determined by utilizing various assumptions regarding the underlying attributes of the options and shares. The estimated fair value of options granted and restricted stock, net of forfeitures expected to occur during the vesting period, is amortized as compensation expense on a straight line basis over the period the vesting occurs. The share-based compensation expense is recorded in cost of sales, sales and marketing, research and development and general and administrative expenses based on the employee’s or consultant’s respective function. The option and warrant-related expense is derived from the Black-Scholes Option Pricing Model that uses several judgment based variables to calculate the expense. The inputs include the expected life of the option or warrant, the expected volatility and other factors. The compensation expense related to the restricted stock is calculated as the difference between the fair market value of the stock on the date of grant, less the cost to acquire the shares, which is $0.0001 per share.
On June 3, 2010, the Company exchanged all of the outstanding options under the Osmetech plc 2003 U.S. Equity Compensation Plan (the “U.S. Plan”) for options under the 2010 Equity Incentive Plan (the “Plan”). The options were exchanged using an exchange ratio of 230 options to purchase shares of Osmetech plc to one share of the Company and was accounted for as a modification of the share-based payment arrangement. There was no additional compensation cost recorded related to the exchange as there was no change in the economic value of the options exchanged.
Employee participation in the Plan is at the discretion of the compensation committee or senior management of the Company. All options granted since June 3, 2010 are exercisable at a price equal to the average closing quoted market price of the Company’s shares on the NASDAQ on the date of grant. Options granted prior to June 3, 2010 under the Osmetech plc 2003 U.S. Equity Compensation Plan were exercisable at a price equal to the average closing quoted market price of the Osmetech plc’s shares on the Alternative Investment Market of the London Stock Exchange on the date of the grant as adjusted for the exchange ratio to the Company’s shares as described above. Options generally vest between 1 and 4 years.
Options are generally exercisable for a period up to 10 years after grant and are forfeited if the employee leaves the Company before the options vest. As of March 31, 2011, 701,957 shares remained available for future grant of awards under the Plan. Restricted stock grants reduce the amount of stock options available for grant under the 2010 Plan and are excluded from the table below.
F-7
Table of Contents
Genmark Diagnostics, Inc.
Notes to Unaudited Condensed Consolidated Financial Statements—(Continued)
The following table summarizes stock option activity during the three months ended March 31, 2011:
| Number of Share options |
Weighted average exercise price |
|||||||
| Outstanding at December 31, 2010 |
1,107,920 | $ | 6.40 | |||||
| Granted |
226,500 | 4.37 | ||||||
| Exercised |
— | — | ||||||
| Cancelled |
(19,445 | ) | (0.37 | ) | ||||
| Outstanding at March 31, 2011 |
1,314,975 | $ | 6.06 | |||||
| Exercisable at March 31, 2011 |
516,596 | $ | 7.12 | |||||
As of March 31, 2011, there were 1,314,975 options that are vested or expected to vest and these options have a remaining weighted average contractual term of 8.63 years, and an aggregate intrinsic value of $0.
During the three months ended March 31, 2011, the Company granted 130,800 shares of restricted stock to senior management employees and 10,000 shares of restricted stock to an outside consultant. The restricted stock granted to senior management employees vests over a four year period except for 4,000 shares of restricted stock issued to one employee as part of a separation agreement that vests May 31, 2011. The restricted stock granted to the outside consultant vested on March 1, 2011 commensurate with the period of service rendered to the Company.
Valuation of Share-Based Awards—The Black-Scholes option pricing model was used for estimating the grant date fair value of stock options granted during the three months ended March 31, 2011 with the following assumptions:
| Expected volatility (%) |
70.0 | |||
| Expected life (years) |
6.08 | |||
| Risk free rate (%) |
2.51 | |||
| Expected dividend yield (%) |
0 |
3. Net Loss Per Common Share
Basic net loss per share is computed by dividing loss available to common shareholders (the numerator) by the weighted average number of common shares outstanding during the period (the denominator). Shares issued during the period and shares reacquired during the period are weighted for the portion of the period that they were outstanding. Diluted loss per share is calculated in a similar way to basic loss per share except that the denominator is increased to include the number of additional shares that would have been outstanding if the dilutive potential shares had been issued unless the effect would be anti-dilutive. As the Company had a net loss in each of the periods presented, basic and diluted net loss per ordinary share are the same.
The computations of diluted net loss per share did not include the effects of the following securities as the inclusion of these items would have been anti-dilutive:
| Three months ended March 31, |
||||||||
| 2011 | 2010 | |||||||
| Share options |
1,314,975 | 960,624 | ||||||
| Warrants |
88,317 | 88,317 | ||||||
| Restricted Stock—vested; not issued or outstanding |
61,057 | — | ||||||
| 1,464,349 | 1,048,941 | |||||||
F-8
Table of Contents
Genmark Diagnostics, Inc.
Notes to Unaudited Condensed Consolidated Financial Statements—(Continued)
Common Stock Warrants—During 2009, the Company issued warrants to purchase 132,475 of Osmetech’s ordinary shares with an exercise price of £4.60 per share, and warrants to purchase 88,317 of Osmetech’s ordinary shares with an exercise price of £6.90 per share to a director for services to the Company in connection with the share offering completed in 2009. Pursuant to the terms of the warrant, the warrant to purchase 132,475 was cancelled upon the closing of the IPO in June 2010. At the same time, the warrant to purchase 88,317 of Osmetech’s ordinary shares was converted to warrants to purchase 88,317 shares of the Company’s common stock at an exercise price of $9.98. These warrants were fully vested and exercisable upon issue, and shall continue to be exercisable up to and including the earlier to occur of (i) 60 days after the director leaving the Company’s board of directors (for whatever reason) and (ii) June 30, 2012.
4. Property and Equipment, net
Property and equipment was comprised of the following as of March 31, 2011 and December 31, 2010:
| March 31, 2011 |
December 31, 2010 |
|||||||
| Property and equipment—at cost: |
||||||||
| Plant and machinery |
$ | 2,473,579 | $ | 2,451,775 | ||||
| Rental systems |
3,125,399 | 2,821,665 | ||||||
| Office equipment |
1,548,235 | 1,541,544 | ||||||
| Leasehold improvements |
611,021 | 597,523 | ||||||
| Total property and equipment—at cost |
7,758,234 | 7,412,506 | ||||||
| Less accumulated depreciation |
(4,995,872 | ) | (4,710,029 | ) | ||||
| Net property and equipment |
$ | 2,762,362 | $ | 2,702,478 | ||||
Depreciation expense amounted to $285,833 and $198,687 for the three months ended March 31, 2011 and 2010, respectively.
5. Loan payable
In March 2010, the Company entered into a loan and security agreement with Square 1 Bank, pursuant to obtaining a credit facility consisting of a revolving line of credit in the amount of up to $2 million and an equipment term loan in the amount of up to $2 million. Based upon certain financial covenants, interest on the revolving line of credit will be either (i) the greater of (a) the bank’s prime rate (3.25% as of March 31, 2011) plus 2.75%, or (b) 6%; or (ii) the greater of (a) the bank’s prime rate plus 3.75%, or (b) 7%. In addition, based upon certain financial covenants, interest on the equipment term loan will be either (i) the greater of (a) the bank’s prime rate plus 3.25%, or (b) 6.50%; or (ii) the greater of (a) the bank’s prime rate plus 4.25%, or (b) 7.50%. The revolving line matures in July 2011 and the term loan matures in July 2013. In March 2011, the loan and security agreement was amended, whereby the line of credit availability was increased to $3 million and the maturity was extended to July 2012. The term loan was modified to allow invoices up to 360 days to qualify to be submitted for credit extension. There were no other changes to these two loans.
In March 2011, an additional loan was made available under the amended loan and security agreement for up to $1.0 million to finance equipment purchases. Based upon certain financial covenants, interest on this equipment term loan will be either (i) the greater of (a) the bank’s prime rate plus 3.25%, or (b) 6.50%; or (ii) the greater of (a) the bank’s prime rate plus 4.25%, or (b) 7.50%. This term loan matures March 2014.
As of March 31, 2011, the Company had no outstanding loans on the line of credit or the 2011 equipment loan and had drawn $2.0 million to finance 2010 equipment purchases and tenant improvements to its Carlsbad facility against the original 2010 equipment term loan. The loan bears an interest rate of 6.5%.
F-9
Table of Contents
Genmark Diagnostics, Inc.
Notes to Unaudited Condensed Consolidated Financial Statements—(Continued)
Pursuant to the terms of the loan and security agreement, we are required to maintain a ratio of liquidity to bank indebtedness equal to at least 1.50 to 1.00. In addition, the loan and security agreement includes several restrictive covenants, including requirements that we obtain the consent of Square 1 Bank prior to entering into any change of control event unless all debt is repaid to Square 1 Bank prior to the change of control event, incurring other indebtedness or liens with respect to our property, making distributions to our stockholders, making certain investments or entering into certain transactions with affiliates and other restrictions on storing inventory and equipment with third parties. The agreement also limits the amount we can borrow under the term loan to license genetic biomarkers to $500,000. To secure the credit facility, we granted Square 1 Bank a first priority security interest in our assets and intellectual property rights. We are currently in compliance will all ratios and covenants.
6. Income taxes
The Company uses an estimated annual effective tax rate, which is based on expected annual income, statutory tax rates and tax planning opportunities available in the various jurisdictions in which the Company operates, to determine its quarterly provision for income taxes. Certain significant or unusual items are separately recognized in the quarter in which they occur and can be a source of variability in the effective tax rates from quarter to quarter.
As of March 31, 2011, the Company has recorded a full valuation allowance against its deferred tax assets due to the uncertainty surrounding the Company’s ability to utilize these assets in the future. Provision for income tax was $11,000 and $5,000 for the three months ended March 31, 2011 and 2010, respectively. Due to the Company’s losses it only records tax provision or benefit related to minimum tax payments or refunds and interest and penalties related to its uncertain tax positions.
The total amount of unrecognized tax benefits was $382,000 as of March 31, 2011 which would impact the effective tax rate if recognized. The gross liability for income taxes related to unrecognized tax benefits is included in other long-term liabilities in the Company’s condensed consolidated balance sheets.
The total balance of accrued interest related to uncertain tax positions was $104,770 as of March 31, 2011. The Company recognizes interest and penalties related to uncertain tax positions as a component of income tax expense. The Company does not expect its unrecognized tax benefits to change significantly over the next twelve months.
The Company is subject to taxation in the U.S., UK based on its legacy operations, and in various state jurisdictions. As of March 31, 2011 the Company’s tax years after 2007 are subject to examination by the UK tax authorities. Except for net operating losses generated in prior years carrying forward to the current year, as of March 31, 2011, the Company is no longer subject to U.S. federal, state, local or foreign examinations by tax authorities for years before 2006.
F-10
Table of Contents
REPORTS OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRMS
To the Board of Directors and Stockholders of
GenMark Diagnostics, Inc.
We have audited the accompanying consolidated balance sheet of GenMark Diagnostics, Inc. and subsidiaries (the “Company”) (formerly Osmetech plc and subsidiaries) as of December 31, 2010, and the related consolidated statements of operations, comprehensive loss, stockholders’ equity, and cash flows for the year then ended. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the consolidated financial statements based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audit provides a reasonable basis for our opinion.
In our opinion, such consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2010, and the results of its operations and cash flows for the year then ended, in conformity with accounting principles generally accepted in the United States of America.
/s/ DELOITTE & TOUCHE, LLP
San Diego, CA
March 11, 2011
F-11
Table of Contents
To the Board of Directors and Stockholders of
Osmetech plc
London, United Kingdom
We have audited the accompanying consolidated balance sheet of Osmetech plc and subsidiaries (the “Company”) (the predecessor entity of GenMark Diagnostics, Inc.) as of December 31, 2009, and the related consolidated statements of operations, comprehensive income (loss), stockholders’ equity, and cash flows for each of the two years in the period ended December 31, 2009. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, such consolidated financial statements present fairly, in all material respects, the financial position of Osmetech plc and subsidiaries as of December 31, 2009, and the results of their operations and their cash flows for each of the two years in the period ended December 31, 2009, in conformity with accounting principles generally accepted in the United States of America.
/s/ DELOITTE LLP
St. Albans, United Kingdom
March 19, 2010
F-12
Table of Contents
Consolidated Balance Sheets as of December 31, 2010 and 2009
| As of December 31, | ||||||||
| 2010 | 2009 | |||||||
| Current assets |
||||||||
| Cash and cash equivalents |
$ | 18,329,079 | $ | 16,482,818 | ||||
| Accounts receivable—net |
677,648 | 169,842 | ||||||
| Inventories—net |
896,809 | 136,967 | ||||||
| Other current assets |
2,193,160 | 992,181 | ||||||
| Total current assets |
22,096,696 | 17,781,808 | ||||||
| Property and equipment—net |
2,702,478 | 1,381,618 | ||||||
| Intangible assets—net |
70,980 | 170,051 | ||||||
| Other long-term assets |
55,355 | |||||||
| Total assets |
$ | 24,925,509 | $ | 19,333,477 | ||||
| Current liabilities |
||||||||
| Accounts payable |
$ | 823,242 | $ | 1,504,905 | ||||
| Accrued compensation |
1,171,989 | 822,388 | ||||||
| Other current liabilities |
1,249,928 | 886,032 | ||||||
| Total current liabilities |
3,245,159 | 3,213,325 | ||||||
| Other non-current liabilities |
612,932 | 795,334 | ||||||
| Total liabilities |
3,858,091 | 4,008,659 | ||||||
| Commitments and contingencies—See note 6 |
||||||||
| Stockholders’ equity |
||||||||
| Ordinary shares, £0.23 ($0.3634 as of December 31, 2009) par value; -0- and 7,101,928 shares issued and outstanding as of December 31, 2010 and December 31, 2009, respectively |
— | 2,573,857 | ||||||
| Deferred shares, £0.0099 ($0.01709 as of December 31, 2009) par value; -0- and 689,478,300 shares issued and outstanding as of December 31, 2010 and December 31, 2009, respectively |
— | 11,780,709 | ||||||
| Common stock, $.0001 par value; 100,000,000 authorized; 11,728,233 and -0- issued and outstanding as of December 31, 2010 and December 31, 2009, respectively |
1,172 | — | ||||||
| Preferred stock, $0.0001 par value; 5,000,000 authorized, none issued |
— | — | ||||||
| Additional paid-in capital |
166,009,084 | 127,475,450 | ||||||
| Accumulated deficit |
(144,492,881 | ) | (126,089,889 | ) | ||||
| Accumulated other comprehensive loss |
(449,957 | ) | (415,309 | ) | ||||
| Total stockholders’ equity |
21,067,418 | 15,324,818 | ||||||
| Total liabilities and stockholders’ equity |
$ | 24,925,509 | $ | 19,333,477 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
F-13
Table of Contents
Consolidated Statements of Operations
For the Years ended December 31, 2010, 2009 and 2008
| Year ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Revenue |
||||||||||||
| Product revenue |
$ | 2,340,996 | $ | 910,527 | $ | 559,592 | ||||||
| License and other revenue |
163,872 | 87,889 | 87,500 | |||||||||
| Total revenue |
2,504,868 | 998,416 | 647,092 | |||||||||
| Cost of sales |
4,377,701 | 4,332,299 | 3,237,869 | |||||||||
| Gross loss |
(1,872,833 | ) | (3,333,883 | ) | (2,590,777 | ) | ||||||
| Operating expenses |
||||||||||||
| Sales and marketing |
4,282,521 | 3,181,762 | 3,393,665 | |||||||||
| Research and development |
6,522,112 | 5,633,717 | 13,423,679 | |||||||||
| General and administrative |
7,353,802 | 8,288,762 | 9,632,708 | |||||||||
| Total operating expenses |
18,158,435 | 17,104,241 | 26,450,052 | |||||||||
| Loss from operations |
(20,031,268 | ) | (20,438,124 | ) | (29,040,829 | ) | ||||||
| Other income |
||||||||||||
| Foreign exchange gain (loss) |
(1,110 | ) | 303,523 | 504,921 | ||||||||
| Interest income (expense) |
(582 | ) | 33,222 | 420,011 | ||||||||
| Therapeutic discovery credit |
1,645,292 | — | — | |||||||||
| Total other income |
1,643,600 | 336,745 | 924,932 | |||||||||
| Loss before income taxes |
(18,387,668 | ) | (20,101,379 | ) | (28,115,897 | ) | ||||||
| (Provision) benefit for income taxes |
(15,324 | ) | 138,770 | (246,736 | ) | |||||||
| Net loss |
$ | (18,402,992 | ) | $ | (19,962,609 | ) | $ | (28,362,633 | ) | |||
| Net loss per share, basic and diluted |
$ | (1.88 | ) | $ | (4.41 | ) | $ | (28.13 | ) | |||
| Weighted average number of shares outstanding |
9,796,588 | 4,526,758 | 1,008,386 | |||||||||
| Consolidated Statements of Comprehensive Loss For the Years ended December 31, 2010, 2009 and 2008 |
||||||||||||
| Net loss |
$ | (18,402,992 | ) | $ | (19,962,609 | ) | $ | (28,362,633 | ) | |||
| Foreign currency translation adjustment |
(34,648 | ) | (93,682 | ) | (1,157,707 | ) | ||||||
| Comprehensive loss |
$ | (18,437,640 | ) | $ | (20,056,291 | ) | $ | (29,520,340 | ) | |||
The accompanying notes are an integral part of these consolidated financial statements.
F-14
Table of Contents
Consolidated Statements of Stockholders’ Equity
For the Years ended December 31, 2010, 2009 and 2008
| Ordinary Shares | Deferred Stock | Common Stock Shares |
Par Value |
Additional paid-in Capital |
Accumulated other compreh- ensive income (loss) |
Accumulated Deficit |
Total | |||||||||||||||||||||||||||||||||
| Shares | Par value | Shares | Par value | |||||||||||||||||||||||||||||||||||||
| Balance—January 1, 2008 |
203,056,639 | $ | 360,439 | 689,478,300 | $ | 11,780,709 | — | $ | — | 94,755,107 | $ | 836,080 | $ | (77,764,647 | ) | $ | 29,967,688 | |||||||||||||||||||||||
| Share-based compensation related to stock options |
— | — | — | — | — | — | (256,219 | ) | — | — | (256,219 | ) | ||||||||||||||||||||||||||||
| Exercise of share options |
60,000 | 119 | — | — | — | — | 22,840 | — | — | 22,959 | ||||||||||||||||||||||||||||||
| Issuance of ordinary shares, net of offering expenses |
688,490,518 | 1,006,504 | — | — | — | — | 8,716,677 | — | — | 9,723,181 | ||||||||||||||||||||||||||||||
| Foreign currency translation adjustment |
— | — | — | — | — | — | — | (1,157,707 | ) | — | (1,157,707 | ) | ||||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | (28,362,633 | ) | (28,362,633 | ) | ||||||||||||||||||||||||||||
| Balance—December 31, 2008 |
891,607,157 | $ | 1,367,062 | 689,478,300 | $ | 11,780,709 | — | $ | — | 103,238,405 | $ | (321,627 | ) | $ | (106,127,280 | ) | $ | 9,937,269 | ||||||||||||||||||||||
| Share-based compensation related to share options |
— | — | — | — | — | — | 1,311,033 | — | — | 1,311,033 | ||||||||||||||||||||||||||||||
| Issuance of ordinary shares, net of offering expenses |
741,836,194 | 1,206,795 | — | — | — | — | 22,926,012 | — | — | 24,132,807 | ||||||||||||||||||||||||||||||
| Foreign currency translation adjustment |
— | — | — | — | — | — | — | (93,682 | ) | — | (93,682 | ) | ||||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | (19,962,609 | ) | (19,962,609 | ) | ||||||||||||||||||||||||||||
| Balance—December 31, 2009 |
1,633,443,351 | $ | 2,573,857 | 689,478,300 | $ | 11,780,709 | — | $ | — | 127,475,450 | $ | (415,309 | ) | $ | (126,089,889 | ) | $ | 15,324,818 | ||||||||||||||||||||||
| Share-based compensation related to share options |
— | — | — | — | — | — | 1,552,871 | — | — | 1,552,871 | ||||||||||||||||||||||||||||||
| Exercise of share options |
4,964,403 | 7,482 | — | — | — | — | — | — | — | 7,482 | ||||||||||||||||||||||||||||||
| Reorganization |
(1,638,407,754 | ) | (2,581,339 | ) | (689,478,300 | ) | (11,780,709 | ) | 7,128,233 | 712 | 14,361,336 | — | — | — | ||||||||||||||||||||||||||
| Issuance of common stock, net of offering expenses |
— | — | — | — | 4,600,000 | 460 | 22,619,427 | — | — | 22,619,887 | ||||||||||||||||||||||||||||||
| Foreign currency translation adjustment |
— | — | — | — | — | — | — | (34,648 | ) | — | (34,648 | ) | ||||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | (18,402,992 | ) | (18,402,992 | ) | ||||||||||||||||||||||||||||
| Balance—December 31, 2010 |
— | $ | — | — | $ | — | 11,728,233 | $ | 1,172 | 166,009,084 | $ | (449,957 | ) | $ | (144,492,881 | ) | $ | 21,067,418 | ||||||||||||||||||||||
The accompanying notes are an integral part of these financial statements.
F-15
Table of Contents
Consolidated Statements of Cash Flows
For the Years Ended December 31, 2010, 2009 and 2008
| Year ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Cash flows from operating activities |
||||||||||||
| Net loss |
$ | (18,402,992 | ) | $ | (19,962,609 | ) | $ | (28,362,633 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities |
||||||||||||
| Depreciation and amortization |
1,063,311 | 1,569,074 | 1,157,655 | |||||||||
| Loss from disposal of property and equipment |
— | 8,462 | 31,335 | |||||||||
| Impairment losses |
— | 1,505,642 | — | |||||||||
| Share-based compensation |
1,552,871 | 1,311,033 | (256,219 | ) | ||||||||
| Changes in operating assets and liabilities: |
||||||||||||
| Accounts receivable |
(507,806 | ) | (51,068 | ) | (21,056 | ) | ||||||
| Inventories |
(651,130 | ) | 1,227,383 | (736,121 | ) | |||||||
| Other current assets |
(1,404,305 | ) | 315,985 | (172,491 | ) | |||||||
| Accounts payable |
(1,058,342 | ) | (857,307 | ) | 1,365,330 | |||||||
| Accrued and other current liabilities |
547,670 | (510,168 | ) | 825,595 | ||||||||
| Net cash used in operating activities |
(18,860,723 | ) | (15,443,573 | ) | (26,168,605 | ) | ||||||
| Cash flows from investing activities |
||||||||||||
| Proceeds from the sale of property and equipment and intangible assets |
— | 10,000 | 160,000 | |||||||||
| Purchases of property and equipment |
(1,859,877 | ) | (1,068,671 | ) | (1,592,715 | ) | ||||||
| Net cash used in investing activities |
(1,859,877 | ) | (1,058,671 | ) | (1,432,715 | ) | ||||||
| Cash flows from financing activities |
||||||||||||
| Proceeds from the issuance of ordinary shares and common stock |
27,600,000 | 24,132,807 | 9,723,182 | |||||||||
| Costs incurred in conjunction with initial public offering |
(4,990,937 | ) | — | — | ||||||||
| Proceeds from stock option exercises |
4,734 | — | 22,959 | |||||||||
| Net cash provided by financing activities |
22,613,797 | 24,132,807 | 9,746,141 | |||||||||
| Effect of foreign exchange rate changes |
(46,936 | ) | 29,797 | (942,078 | ) | |||||||
| Net increase (decrease) in cash and cash equivalents |
1,846,261 | 7,660,360 | (18,797,257 | ) | ||||||||
| Cash and cash equivalents—Beginning of year |
16,482,818 | 8,822,458 | 27,619,715 | |||||||||
| Cash and cash equivalents—End of year |
$ | 18,329,079 | 16,482,818 | $ | 8,822,458 | |||||||
| Supplemental cash flow disclosures: |
||||||||||||
| Cash received for income taxes |
$ | 5,049 | $ | 181,162 | $ | 391,086 | ||||||
| Cash received for interest |
$ | 25,025 | $ | 33,222 | $ | 420,011 | ||||||
| Noncash investing and financing activities: |
||||||||||||
| Reclassification of deposits on systems in other current assets, and inventory to property and equipment in 2010 and 2009, respectively |
$ | 288,962 | $ | 256,909 | — | |||||||
| IPO Costs incurred but not paid |
$ | 103,626 | — | — | ||||||||
| VAT tax refund related to IPO costs recorded but not received |
$ | 114,450 | — | — | ||||||||
| Transfer of systems from property and equipment into inventory |
$ | 108,712 | — | — | ||||||||
| Fixed asset acquisitions included in accounts payable |
$ | 275,799 | — | — | ||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-16
Table of Contents
Notes to Consolidated Financial Statements
1. Organization and basis of presentation
GenMark Diagnostics, Inc. (the “Company” or “GenMark”) is a molecular diagnostics company focused on developing and commercializing the Company’s proprietary e-sensor technology. On February 12, 2010, the Company was established to serve as the parent company of Osmetech plc (“Osmetech”) upon a corporate reorganization and initial public offering (“IPO”). On June 3, 2010, the Company completed an IPO for 4,600,000 shares. Immediately prior to the completion of the IPO, the Company underwent a corporate reorganization whereby the ordinary shares of Osmetech were exchanged by its shareholders for the common stock of the Company on a 230 for 1 basis.
As the reorganization was deemed to be a transaction under common control, GenMark accounted for the reorganization in a manner similar to a pooling-of-interests, meaning:
| (i) | assets and liabilities were carried over at their respective carrying values; |
| (ii) | common stock was carried over at the nominal value of the shares issued by GenMark; |
| (iii) | additional paid-in capital represented the difference between the nominal value of the shares issued by GenMark, and the total of the additional paid-in capital and nominal value of Osmetech’s shares cancelled pursuant to the reorganization; and |
| (iv) | the accumulated deficit represented the aggregate of the accumulated deficit of Osmetech and GenMark. |
Once the reorganization became effective, all stock options granted under the Osmetech plc 2003 U.S. Equity Compensation Plan, Long Term Incentive Awards and all warrants issued were exchanged for options and warrants exercisable for the common stock of the Company.
The preferred stock may be issued from time to time in one or more series.
In these consolidated financial statements, the Company means Osmetech when referring to periods prior to the IPO.
Subsequent events have been evaluated through March 11, 2011, being the date that the financial statements were available to be issued.
The accompanying financial statements have been prepared on a going-concern basis, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. The Company has incurred net losses from operations since its inception and has an accumulated deficit of $144,492,881 at December 31, 2010. Cash and cash equivalents at December 31, 2010 were $18,329,079.
Management expects operating losses to continue through the foreseeable future until the Company has expanded its product offering and consequently increased its product revenues to an extent to cover the fixed cost base of the business. The Company’s management has prepared cash flow forecasts which indicate, based on the current cash resources available and the availability of credit facilities of up to $4,000,000, that the Company has sufficient capital to fund its operations for at least the next twelve months.
The accompanying consolidated financial statements have been prepared in accordance with United States generally accepted accounting principles and applicable regulations of the Securities and Exchange Commission (“SEC”). The Company’s operating results for the year ended December 31, 2010 are not necessarily indicative of the results that may be expected for any future periods.
F-17
Table of Contents
GenMark Diagnostics, Inc.
Notes to Consolidated Financial Statements—(Continued)
Principles of Consolidation—The consolidated financial statements include the accounts of the Company and its wholly owned subsidiaries. All intercompany accounts and transactions have been eliminated in consolidation.
2. Summary of Significant Accounting Policies
Cash and Cash Equivalents
The Company considers all highly liquid investments with an original maturity of three months or less, at date of purchase, to be cash equivalents. The majority of these funds are held in interest-bearing money market and bank checking accounts. Interest income is recorded on the accrual basis as earned.
Receivables
Accounts receivable consists of amounts due to the Company for sales to customers and are recorded net of an allowance for doubtful accounts. Prior to 2010, the Company did not reserve or write-off any receivables.
Inventories
Inventories are stated at the lower of cost (first-in, first-out) or market and include direct labor, materials, and manufacturing overhead. The Company periodically reviews inventory for evidence of slow-moving or obsolete parts, and writes inventory down to market. This write down is based on management’s reviews of inventories on hand, compared to estimated future usage and sales, shelf-life assumptions, and assumptions about the likelihood of obsolescence. During 2009, due to a change in business strategy, the Company changed the intention to sell its systems, and determined that the systems would be placed at customer sites pursuant to reagent rental agreements. Therefore, $256,909 was transferred from inventory to property and equipment-net.
Property and Equipment-net
Property, equipment and leasehold improvements are recorded at cost and depreciated using the straight-line method over the assets’ estimated useful lives, which are:
| Machinery and laboratory equipment |
- | 3 – 5 years | ||||
| Systems at customer locations |
- | 3 years | ||||
| Office equipment |
- | 2 – 4 years | ||||
| Leasehold improvements |
- | over the shorter period of the life of the lease or the useful economic life of the asset |
Maintenance and repair costs are expensed as incurred.
Intangible Assets
Intangible assets are comprised of licenses or sublicenses to technology covered by patents owned by third parties, and are amortized on a straight-line basis over the expected useful lives of these assets, generally five years. Amortization of licenses begins upon the Company obtaining FDA clearance to sell products containing the licensed technology and is recorded in cost of sales.
Impairment of Long-Lived Assets
The Company assesses the recoverability of long-lived assets, including intangible assets, by periodically evaluating the carrying value whenever events or changes in circumstances indicate that the carrying amount
F-18
Table of Contents
GenMark Diagnostics, Inc.
Notes to Consolidated Financial Statements—(Continued)
may not be recoverable. If impairment is indicated, the Company writes down the carrying value of the asset to its estimated fair value. This fair value is primarily determined based on estimated discounted cash flows.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the amounts reported in the financial statements and the notes thereto. The Company’s significant estimates included in the preparation of the financial statements are related to inventories, plant and equipment, intangible assets, certain accrued liabilities related to the Company’s former facilities and share-based compensation. Actual results could differ from those estimates.
Revenue Recognition
The Company recognizes revenue from product sales and contract arrangements, net of discounts and sales related taxes. The Company recognizes revenue from product sales when there is persuasive evidence that an arrangement exists, delivery has occurred, the price is fixed or determinable and collectability is reasonably assured. Where applicable, all revenue is stated net of sales taxes and trade discounts.
The Company’s XT-8 systems are placed free of charge with customers in exchange for an annual minimum purchase commitment of products from the customer, while the Company retains the right to access or replace the systems at any time. Therefore, the systems remain capitalized on the balance sheet. Revenue from sales of the test cartridges and related products are recognized when the risks and rewards of ownership are transferred to the customer, which is generally at the time of product shipment.
Revenues related to royalties received from licenses are recognized evenly over the contractual period to which the license relates. Services provided are recognized evenly over the contractual period to which the services relate.
Shipping and handling costs are expensed as incurred and included in cost of product sales. In those cases where the Company bills shipping and handling costs to customers, the amounts billed are classified as revenue.
Product Warranties
The Company generally offers a one-year warranty for its systems sold to customers and provides for the estimated cost of the product warranty at the time the system sale is recognized. Factors that affect the Company’s warranty reserves include the number of units sold, historical and anticipated rates of warranty repairs and the cost per repair. The Company periodically assesses the adequacy of the warranty reserve and adjusts the amount as necessary. Because there were no system sales in 2009 or 2008, the product warranty reserve has been zero prior to 2010.
Product warranty reserve activity for the year ended December 31, 2010 is as follows:
| 2010 | ||||
| Beginning balance |
$ | — | ||
| Provisions |
25,000 | |||
| Ending balance |
$ | 25,000 | ||
Research and Development Costs
Research and development costs are expensed as incurred.
F-19
Table of Contents
GenMark Diagnostics, Inc.
Notes to Consolidated Financial Statements—(Continued)
Income Taxes
The Company accounts for deferred tax assets and liabilities for the expected future tax consequences of events that have been included in the financial statements or tax returns. Deferred tax assets and liabilities are determined based on the differences between the financial reporting and tax bases of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to reverse. A valuation allowance is provided to reduce deferred tax assets to the amount management believes will, more likely than not, be recovered.
A tax position that is more likely than not to be realized is measured at the largest amount of tax benefit that is greater than 50% likely of being realized upon settlement with the taxing authority that has full knowledge of all relevant information. Measurement of a tax position that meets the more likely than not threshold considers the amounts and probabilities of the outcomes that could be realized upon settlement using the facts, circumstances and information available at the reporting date.
Share-Based Compensation
The Company recognizes share-based compensation expense related to share options and warrants issued to employees and directors in exchange for services. The compensation expense is based on the fair value of the share-based compensation utilizing various assumptions regarding the underlying attributes of the options and shares. The estimated fair value of options granted, net of forfeitures expected to occur during the vesting period, is amortized as compensation expense on an accelerated basis to reflect the vesting as it occurs. The share-based compensation expense is recorded in cost of sales, sales and marketing, research and development and general and administrative expenses based on the employee’s respective function. The expense is derived from the Black-Scholes Option Pricing Model that uses several judgment based variables to calculate the expense. The inputs include the expected life of the option or warrant, the expected volatility and other factors.
Fair Value of Financial Instruments
The carrying amount of the Company’s financial instruments, including cash and cash equivalents, accounts receivable and accounts payable approximate their fair values.
Foreign Currency Translation
During 2010, the Company changed its functional currency from the British Pound to the U.S. Dollar. Prior to this change, monetary assets and liabilities of the Company’s entities outside of the U.S. were translated into U.S. dollars based on foreign currency exchange rates in effect at the end of each period, and revenues and expenses were translated at weighted average exchange rates during the periods. Gains or losses resulting from these foreign currency translations of the Company’s assets and liabilities were recorded in accumulated other comprehensive income in the consolidated balance sheets.
Transactions in foreign currencies were translated into the relevant functional currency at the rate of exchange prevailing at the date of the transaction. Foreign currency transaction gains (losses), which are included in the results of operations, totaled $(1,110), $303,523 and $504,921, for the years ended December 31, 2010, 2009, and 2008, respectively, and relate primarily to transactions denominated in U.S. dollars which were undertaken by Osmetech.
Derivative Financial Instruments
In 2008, derivative financial instruments were used principally in the management of foreign currency and interest rate exposures and were recorded in the consolidated balance sheets at fair value. Derivative
F-20
Table of Contents
GenMark Diagnostics, Inc.
Notes to Consolidated Financial Statements—(Continued)
instruments not designated as hedges were marked-to-market at the end of 2008 with the results included in results of operations. The effect on earnings was not material. The Company did not use derivative financial instruments in 2010 or 2009.
Net Loss Per Common Share
Basic net loss per share is computed by dividing loss available to common shareholders (the numerator) by the weighted average number of common shares outstanding during the period (the denominator). Shares issued during the period and shares reacquired during the period are weighted for the portion of the period that they were outstanding. Diluted loss per share is calculated in a similar way to basic loss per share except that the denominator is increased to include the number of additional shares that would have been outstanding if the dilutive potential shares had been issued unless the effect would be anti-dilutive. As the Company had a net loss in each of the periods presented, basic and diluted net loss per ordinary share are the same.
The computations of diluted net loss per share for the years ended December 31, 2010, 2009 and 2008 did not include the effects of the following options and warrants to acquire ordinary stock which were outstanding as of the end of each year as the inclusion of these securities would have been anti-dilutive.
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Share options |
1,107,920 | 993,214 | 108,590 | |||||||||
| Warrants |
88,317 | 220,791 | — | |||||||||
| Restricted Stock |
204,115 | — | — | |||||||||
| 1,400,352 | 1,214,005 | 108,590 | ||||||||||
Segment Information
The Company operates in one reportable segment, and substantially all of the Company’s operations and assets are in the United States of America.
Concentration of Risk
The Company had sales to customers representing greater than 10% of the total as follows:
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Customer A |
12 | % | — | — | ||||||||
| Customer B |
— | 15 | % | 13 | % | |||||||
| Customer C |
— | 12 | % | 23 | % | |||||||
| Customer D |
— | 11 | % | — | ||||||||
| Customer E |
— | — | 18 | % | ||||||||
The Company’s XT-8 system is manufactured by a single source supplier that specializes in contract design and manufacturing of electronic and electromechanical devices for medical use.
Comprehensive Income (Loss)
U.S. GAAP requires that all components of comprehensive income (loss), including net income (loss), be reported in the financial statements in the period in which they are recognized. Comprehensive income (loss) is defined as the change in equity during a period from transactions and other events and circumstances from non-owner sources, including accumulated translation adjustments. The Company reports comprehensive income (loss) as a separate component of stockholders’ equity.
F-21
Table of Contents
GenMark Diagnostics, Inc.
Notes to Consolidated Financial Statements—(Continued)
Recent Accounting Pronouncements
In October 2009, authoritative guidance was provided on revenue arrangements with multiple deliverables. The guidance amended the accounting standards for multiple deliverable revenue arrangements to: (i) provide updated guidance on whether multiple deliverables exist, how the deliverables in an arrangement should be separated, and how the consideration should be allocated; (ii) require an entity to allocate revenue in an arrangement using estimated selling prices (“ESP”) of deliverables if a vendor does not have vendor-specific objective evidence of selling price (“VSOE”) or third-party evidence of selling price (“TPE”); and (iii) eliminate the use of the residual method and require an entity to allocate revenue using the relative selling price method.
Arrangements that contain multiple deliverables include sales of systems and test cartridges. These are accounted for as separate units of accounting if the following criteria are met: (i) the delivered item or items have value to the customer on a standalone basis and (ii) for an arrangement that includes a general right of return relative to the delivered item(s), delivery or performance of the undelivered item(s) is considered probable and substantially in our control. The Company considers a deliverable to have standalone value if the item is sold separately or if the item could be resold by the customer. The Company’s revenue arrangements generally do not include a right of return for delivered products.
The Company sold its first systems in the year ended December 31, 2010. The Company elected to early adopt the new accounting guidance because it is able to meet the new separation criteria and has applied it to all applicable revenue arrangements entered into or materially modified beginning January 1, 2010. The adoption of the new guidance had an immaterial effect on the financial statements and on loss per share for the year ended December 31, 2010.
The adoption of this guidance did not result in a change in the Company’s units of accounting or in how the Company allocates arrangement consideration to its units of accounting, as the arrangements to which the new accounting guidance is applicable were first entered into during the year ended December 31, 2010.
3. Intangible assets
Intangible assets, consisting of purchased intellectual property, as of December 31, 2010 and 2009 comprise the following:
| December 31, 2010 | December 31, 2009 | |||||||||||||||||||||||
| Gross carrying amount |
Accumulated amortization |
Net carrying amount |
Gross carrying amount |
Accumulated amortization |
Net carrying amount |
|||||||||||||||||||
| Patents and trademarks |
$ | 438,032 | $ | (438,032 | ) | $ | — | $ | 438,032 | $ | (438,032 | ) | $ | — | ||||||||||
| Intellectual property |
877,140 | (877,140 | ) | — | 877,140 | (877,140 | ) | — | ||||||||||||||||
| Licenses |
1,251,518 | (1,180,538 | ) | 70,980 | 1,251,518 | (1,081,467 | ) | 170,051 | ||||||||||||||||
| $ | 2,566,690 | $ | (2,495,710 | ) | $ | 70,980 | $ | 2,566,690 | $ | (2,396,639 | ) | $ | 170,051 | |||||||||||
F-22
Table of Contents
GenMark Diagnostics, Inc.
Notes to Consolidated Financial Statements—(Continued)
Licenses have a weighted average remaining amortization period of 7.5 months as of December 31, 2010. Amortization expense for intangible assets amounted to $68,247, $164,662 and $105,455 for the years ended December 31, 2010, 2009, and 2008, respectively. Additionally, during 2009, licenses that were used for the manufacture of certain of the Company’s consumables were impaired due to the Company outsourcing this manufacturing process. This resulted in an impairment charge of $549,148 charged to cost of sales. In addition, an impairment of $91,105 was recorded as a general and administrative expense. Estimated future amortization expense for these licenses is as follows:
| Years Ending December 31, |
||||
| 2011 |
$ | 61,749 | ||
| 2012 |
9,231 | |||
| Total |
$ | 70,980 | ||
4. Share-based compensation
The Company recognizes share-based compensation expense related to share options, warrants and restricted stock issued to employees and directors in exchange for services. The compensation expense is based on the fair value of the awards, which are determined by utilizing various assumptions regarding the underlying attributes of the options and shares. The estimated fair value of options granted and restricted stock, net of forfeitures expected to occur during the vesting period, is amortized as compensation expense on a straight line basis over the period the vesting occurs. The share-based compensation expense is recorded in cost of sales, sales and marketing, research and development and general and administrative expenses based on the employee’s respective function. The option and warrant-related expense is derived from the Black-Scholes Option Pricing Model that uses several judgment based variables to calculate the expense. The inputs include the expected life of the option or warrant, the expected volatility and other factors. The compensation expense related to the restricted stock is calculated as the difference between the fair market value of the stock on the date of grant, less the cost to acquire the shares, which is $0.0001 per share.
Employee participation is at the discretion of the compensation committee or senior management of the Company. All options are exercisable at a price equal to the average closing quoted market price of the Company’s shares on the NASDAQ on the date of grant and generally vest between 1 and 4 years.
Options are generally exercisable for a period up to 10 years after grant and are forfeited if the employee leaves the Company before the options vest. As of December 31, 2010, 687,965 shares remained available for future grant of awards under the Plan. Restricted stock grants reduce the amount of stock options available for grant under the 2010 Plan and are excluded from the table below.
The following table summarizes stock option activity during the year ended December 31, 2010:
| Number of shares |
Weighted average exercise price (translated to dollars) |
|||||||
| Outstanding at December 31, 2009 |
993,214 | $ | 6.96 | |||||
| Granted |
429,300 | 5.29 | ||||||
| Exercised |
(21,589 | ) | 0.37 | |||||
| Cancelled |
(293,005 | ) | (5.48 | ) | ||||
| Outstanding at December 31, 2010 |
1,107,920 | $ | 6.40 | |||||
| Exercisable at December 31, 2010 |
437,399 | $ | 7.16 | |||||
F-23
Table of Contents
GenMark Diagnostics, Inc.
Notes to Consolidated Financial Statements—(Continued)
The weighted average fair value of options granted during 2010, 2009 and 2008 was $5.29, $3.68 and $27.37, respectively. The intrinsic value of options exercised in 2010, 2009 and 2008 was $136,157, $0 and $3,116, respectively. No options were exercised in 2009. As of December 31, 2010, there were 992,565 options that are vested or expected to vest and these options have a remaining weighted average contractual term of 8.56 years, and an aggregate intrinsic value of $0. Options that are exercisable as of December 31, 2010 have a remaining weighted average contractual term of 7.52 years, and an aggregate intrinsic value of $0.
Valuation of Share-Based Awards—The Black-Scholes option pricing model was used for estimating the grant date fair value of stock options granted during the years ended December 31, 2010, 2009 and 2008 with the following assumptions:
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Expected volatility (%) |
70.0 | 66.7 | 49.0 | |||||||||
| Expected life (years) |
5.91 | 0.4 | 3.0 | |||||||||
| Risk free rate (%) |
2.1 | 2.2 | 4.6 | |||||||||
| Expected dividend yield (%) |
0 | 0 | 0 | |||||||||
Share Warrants—During 2009, the Company issued warrants to purchase 132,475 of Osmetech’s ordinary shares with an exercise price of £4.60 per share, and warrants to purchase 88,317 of Osmetech’s ordinary shares with an exercise price of £6.90 per share to a director for services to the Company in connection with the share offering completed in 2009. Pursuant to the terms of the warrant, the warrant to purchase 132,475 was cancelled upon the closing of the IPO. At the same time, the warrant to purchase 88,317 of Osmetech’s ordinary shares was converted to a warrant to purchase 88,317 shares of the Company’s common stock at an exercise price of $9.98. These warrants were fully vested and exercisable upon issue, and shall continue to be exercisable up to and including the earlier to occur of (i) 60 days after the director leaving the Company’s board of directors (for whatever reason) and (ii) June 30, 2012.
Additionally, Osmetech’s deferred shares, which were created at the time of a 10-for-1 consolidation of ordinary shares on September 30, 2005 are excluded from basic and diluted net loss per ordinary share. Management considers these shares to be of minimal value. The deferred shares do not entitle the holder to payment of any dividend or other distribution or to receive notice or attend or vote at any general meeting of Osmetech. The deferred shares are non transferable. In the event of a return of assets on winding up of Osmetech, the deferred shareholders receive 1 pence in respect of their shareholding in its entirety.
During the year ended December 31, 2010, the company granted 161,329 shares of restricted stock to two board members.
The restricted stock granted to the Interim Chief Executive Officer vests over the twelve month period ending July 2011 and the restricted stock granted to our new board member vests over the four year period of his board of director’s duties and over the twelve month period ending August 2011, for his initial and annual board compensation grants, respectively.
F-24
Table of Contents
GenMark Diagnostics, Inc.
Notes to Consolidated Financial Statements—(Continued)
Share-Based Compensation—Share-based compensation, was recognized in the consolidated statements of operations as follows:
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Cost of sales |
$ | 18,916 | $ | 19,364 | $ | 23,243 | ||||||
| Sales and marketing |
260,823 | 37,344 | 44,826 | |||||||||
| Research and development |
162,065 | 48,409 | 58,107 | |||||||||
| General and administrative |
1,111,067 | 1,205,916 | (382,395 | ) | ||||||||
| $ | 1,552,871 | $ | 1,311,033 | $ | (256,219 | ) | ||||||
No share-based compensation was capitalized during the periods presented, and there was no unrecognized tax benefit related to share-based compensation for the years ended December 31, 2010, 2009 and 2008. During 2008, the Company determined that certain performance based criteria for options previously issued to certain executives would not be met. Accordingly, all expenses that had previously been recognized were reversed. No other options with performance based conditions have been outstanding during the periods presented. At December 31, 2010, the estimated total remaining unamortized compensation expense, net of forfeitures, associated with share-based awards was $2,728,305 which is expected to be recognized over a weighted-average period of 1.42 years.
5. Income Taxes
The components of loss before income taxes were as follows:
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Domestic (U.S. Entities) |
$ | (18,387,668 | ) | $ | (18,332,641 | ) | $ | (25,585,488 | ) | |||
| Foreign (Non U.S. Entities) |
0 | (1,768,738 | ) | (2,530,409 | ) | |||||||
| $ | (18,387,668 | ) | $ | (20,101,379 | ) | $ | (28,115,897 | ) | ||||
The components of the income tax expense (benefit) for continuing operations are as follows for the years ended December 31:
| 2010 | 2009 | 2008 | ||||||||||
| Current expense (benefit): |
||||||||||||
| U.S. Provision |
$ | — | $ | (165,339 | ) | $ | — | |||||
| State |
15,324 | 2,872 | 8,583 | |||||||||
| Foreign (Non-U.S. entities) |
— | — | 180,023 | |||||||||
| Total Current |
15,324 | (162,467 | ) | 188,606 | ||||||||
| Non-current expense |
||||||||||||
| U.S. Provision |
— | — | — | |||||||||
| State |
— | 23,697 | 58,130 | |||||||||
| Foreign (Non-U.S. Entities) |
— | — | — | |||||||||
| Total Non-current expense |
— | 23,697 | 58,130 | |||||||||
| Deferred expense |
||||||||||||
| U.S. Provision |
— | — | — | |||||||||
| State |
— | — | — | |||||||||
| Foreign (Non-U.S. Entities) |
— | — | — | |||||||||
| Total Deferred Expense |
— | — | — | |||||||||
| Total Expense |
$ | 15,324 | $ | (138,770 | ) | $ | 246,736 | |||||
F-25
Table of Contents
GenMark Diagnostics, Inc.
Notes to Consolidated Financial Statements—(Continued)
The components of net deferred income taxes consist of the following as of December 31:
| 2010 | 2009 | 2008 | ||||||||||
| Current deferred income tax assets (liabilities): |
||||||||||||
| Compensation Accruals |
$ | 676,350 | $ | 82,262 | $ | 95,135 | ||||||
| Accruals and Reserves |
304,704 | 568,189 | 837,665 | |||||||||
| State Tax Provision |
10,587 | 1,251 | 8,099 | |||||||||
| Federal Benefit of State UTP |
165,580 | — | — | |||||||||
| Valuation allowance |
(1,157,221 | ) | (651,702 | ) | (940,899 | ) | ||||||
| Total current deferred income taxes |
— | — | — | |||||||||
| Noncurrent deferred income tax assets (liabilities): |
||||||||||||
| Depreciation and Amortization |
960,808 | 1,068,097 | (98,712 | ) | ||||||||
| Intercompany Interest Expense |
1,980,233 | 2,140,075 | 2,006,305 | |||||||||
| NOL and Credits |
8,996,736 | 34,941,648 | 26,949,147 | |||||||||
| Valuation allowance |
(11,937,777 | ) | (38,149,820 | ) | (28,856,740 | ) | ||||||
| Total noncurrent deferred income taxes |
— | — | — | |||||||||
| Net deferred income taxes |
$ | — | $ | — | $ | — | ||||||
A reconciliation of income tax (expense) / benefit for continuing operations to the amount computed by applying the statutory federal income tax rate ( the federal rate has been utilized as the Company’s main operation are taxed at the federal rate) to the loss from continuing operations is summarized as follows:
| 2010 | 2009 | 2008 | ||||||||||
| U.S. Federal statutory income tax rate |
34.0 | % | 34.0 | % | 34.0 | % | ||||||
| Permanent Differences |
0.4 | % | (0.1 | %) | (1.8 | %) | ||||||
| State Taxes |
(0.1 | %) | (0.1 | %) | (0.2 | %) | ||||||
| Effect of non-U.S. Operations |
(0.0 | %) | (0.5 | %) | (0.5 | %) | ||||||
| Effective Rate Change non- U.S. |
(0.0 | %) | (0.7 | %) | (2.1 | %) | ||||||
| Valuation allowance |
(34.4 | %) | (31.9 | %) | (30.3 | %) | ||||||
| Total tax provision |
(0.1 | %) | 0.7 | % | (0.9 | %) | ||||||
As of December 31 2010, the Company had net operating loss carryforwards of approximately $77.9 million and $72.6 million for federal and state income tax purposes, respectively. These may be used to offset future taxable income and will begin to expire in varying amounts through 2030. In addition, the Company has non-U.S. net operating loss carryforwards of $30.4 million. Because the Company intends to restructure its operations during 2011, the non-U.S. net operating losses and other deferred tax assets have been removed from the Company’s table of deferred income taxes above.
Internal Revenue Code Section 382 places a limitation on future utilization of the federal and state net operating losses, to the extent that the Company incurs an ownership change as defined by Section 382. The Company has determined that it has experienced multiple ownership changes under Section 382. Management has estimated that approximately $24.7 million and $9.5 million of federal and state net operating losses, respectively, can be utilized in the future based on limitations that it has calculated under Section 382. As of December 31, 2010 approximately $10 million of federal net operating losses are available immediately. Additionally, federal net operating losses ranging from $0.2 million to $2.3 million become available each year. Management is currently analyzing alternative positions and additional factual information that may increase
F-26
Table of Contents
GenMark Diagnostics, Inc.
Notes to Consolidated Financial Statements—(Continued)
the amount of net operating losses that could subsequently be utilized up to $41.6 million and $24.7 million of federal and state net operating losses, respectively. To the extent that this additional information becomes available and could increase net operating losses available for use, the Company would adjust its deferred tax assets accordingly, with a corresponding adjustment to its valuation allowance. Utilization of net operating losses is also dependent upon sufficient taxable income generated within the appropriate carryforward periods.
The Company has established a full valuation allowance for its deferred tax assets due to uncertainties that preclude it from determining that it is more likely than not that the Company will be able to generate sufficient taxable income to realize such assets. Management assesses the available positive and negative evidence to estimate if sufficient future taxable income will be generated to utilize the existing deferred tax assets. A significant piece of objective negative evidence evaluated was the cumulative loss incurred over the three year period ended December 31, 2010. Such objective evidence limits the ability to consider other subjective evidence such as our projections for future growth. Based on this evaluation, as of December 31, 2010, a valuation allowance of $13.1 million has been recorded in order to measure only the portion of the deferred tax asset that more likely than not will be realized. The amount of the deferred tax asset considered realizable, however, could be adjusted if estimates of future taxable income during the carryforward period are reduced or if objective negative evidence in the form of cumulative losses is no longer present and additional weight may be given to subjective evidence such as our projections for growth.
The Company adopted certain provisions of ASC 740, “Income Taxes” (previously reported as Interpretation No. 48 “Accounting for Uncertainty in Income Taxes—an interpretation of FASB Statement No. 109), which contains a two-step approach to recognizing and measuring uncertain tax positions. The first step is to evaluate the tax position for recognition by determining if the weight of available evidence indicates it is more likely than not, that the position will be sustained on audit, including resolution of related appeals or litigation processes, if any. The second step is to measure the tax benefit as the largest amount, which is more than 50% likely of being realized upon ultimate settlement. Income tax positions must meet a more likely than not recognition threshold at the effective date to be recognized upon the adoption of ASC 740 and in subsequent periods. This interpretation also provides guidance on measurement, derecognition, classification, interest and penalties, accounting in interim periods, disclosure and transition.
Upon adoption of ASC 740 on January 1, 2007, the Company did not have any unrecognized tax benefits. In accordance with the adoption, a reconciliation of the beginning and ending amount of unrecognized tax benefits, exclusive of accrued interest and penalties, is as follows:
| 2010 | 2009 | 2008 | ||||||||||
| Balance at January 1 |
$ | 382,000 | $ | 382,000 | $ | 382,000 | ||||||
| Additions based on tax positions related to the current year |
— | — | — | |||||||||
| Additions for tax positions of prior years |
— | — | — | |||||||||
| Reductions for tax positions of prior years |
— | — | ||||||||||
| Lapse of statute |
— | — | — | |||||||||
| Settlements |
— | — | — | |||||||||
| Balance at December 31 |
$ | 382,000 | $ | 382,000 | $ | 382,000 | ||||||
At December 31, 2010 and 2009, the Company classified $486,770 and $463,000, respectively, of total unrecognized tax benefits, which includes accrued interest and penalties of $104,770 and $81,000 for 2010 and 2009, respectively, as a component of other long-term liabilities. This represents the amount of unrecognized tax benefits that would, if recognized, reduce the Company’s effective income tax rate in any future periods. The Company does not expect its unrecognized tax benefits to change significantly over the next 12 months. The Company recognizes interest and penalties related to uncertain tax positions in income tax expense.
F-27
Table of Contents
GenMark Diagnostics, Inc.
Notes to Consolidated Financial Statements—(Continued)
The Company is subject to taxation in the UK, US and various states jurisdictions. As of December 31, 2010 the Company’s tax years after 2007 are subject to examination by the UK tax authorities. Except for net operating losses generated in prior years carrying forward to the current year, as of December 31, 2010, the Company is no longer subject to U.S. federal, state, local or foreign examinations by tax authorities for years before 2006.
6. Commitments and Contingencies
The Company has various operating lease agreements for its office, manufacturing, warehousing and laboratory space. Rent and operating expenses charged were $958,607, $1,124,655 and $1,228,173 for the years ended December 31, 2010, 2009, and 2008, respectively. Pursuant to the Company’s lease agreements, a portion of the monthly rental has been deferred. The balance deferred as at December 31, 2010 and 2009 was $133,542 and $186,949, respectively.
Annual future minimum obligations for operating leases as of December 31, 2010 are as follows:
| Years Ending December 31, |
Operating leases |
|||
| 2011 |
$ | 992,471 | ||
| 2012 |
565,362 | |||
| 2013 |
582,155 | |||
| 2014 |
598,947 | |||
| 2015 |
617,606 | |||
| Thereafter |
1,346,543 | |||
| Total minimum lease payments |
$ | 4,703,084 | ||
7. Inventory
Inventory on hand as of December 31, 2010 and 2009 was comprised of the following:
| 2010 | 2009 | |||||||
| Raw materials |
$ | 396,956 | $ | 38,973 | ||||
| Work-in-process |
103,013 | 31,062 | ||||||
| Finished goods |
396,840 | 66,932 | ||||||
| $ | 896,809 | $ | 136,967 | |||||
The increase in raw materials and finished goods inventory is due to incremental revenue growth and inventory build-up in anticipation of the move of the manufacturing facility from Pasadena, California, to Carlsbad, California.
8. Property and Equipment, net
Property and equipment was comprised of the following as of December 31, 2010 and 2009:
| 2010 | 2009 | |||||||
| Property and equipment—at cost: |
||||||||
| Plant and machinery |
$ | 2,451,775 | $ | 2,201,033 | ||||
| Rental systems |
2,821,665 | 2,073,082 | ||||||
| Office equipment |
1,541,544 | 1,079,214 | ||||||
| Leasehold improvements |
597,523 | 74,394 | ||||||
| Total property and equipment—at cost |
7,412,506 | 5,427,723 | ||||||
| Less accumulated depreciation |
(4,710,029 | ) | (4,046,105 | ) | ||||
| Net property and equipment |
$ | 2,702,478 | $ | 1,381,618 | ||||
F-28
Table of Contents
GenMark Diagnostics, Inc.
Notes to Consolidated Financial Statements—(Continued)
The depreciation expense amounted to $995,064, $1,404,412 and $1,052,200 for the years ended December 31, 2010, 2009 and 2008 respectively.
During 2010, $288,962 of deposits on systems were transferred from other current assets to property and equipment, net. During 2009, $256,909 of systems were transferred out of finished goods inventory into property and equipment, net. These transfers were as a result of a change in the Company’s strategy from outright sales of systems to placing systems with customers for no initial charge and recovering that cost through the sale of test cartridges pursuant to reagent rental agreements.
In 2009, due to the anticipated acceleration of the release of future generations of the Company’s products, in particular the NexGen system, the Company assessed all systems for impairment. For systems placed with customers the carrying amount was written down to fair value based on the projected discounted net cash flows to be generated from the sale of test cartridges. Systems that were not expected to generate any future revenues were impaired to $0. The Company recorded an aggregate impairment charge of $865,389 of which $665,718 was charged to cost of sales in respect of systems placed with customers, $69,959 was charged to research and development expenses in respect of systems being used for research purposes, and $129,712 was charged to sales and marketing expenses in respect of systems being used for demonstration purposes only. Additionally in 2009, the Company revised the estimated useful life of systems from 5 to 3 years, although this did not result in a material increase in the depreciation charge during the year.
9. Employee Benefit Plan
The Company has a 401(k) tax-deferred savings plan, whereby eligible employees may contribute a percentage of their eligible compensation. Company contributions are discretionary. Including administrative fees, the expense was $78,572, $172,668 and $304,449 for the years ended December 31, 2010, 2009 and 2008, respectively. Additionally, the Company has made contributions to other defined contribution plans on behalf of its employees amounting to $0, $58,004 and $98,325 for the years ended December 31, 2010, 2009 and 2008, respectively. These other defined contribution plans were terminated in 2010.
10. Fair Value of Financial Instruments
The Company’s financial instruments consist of cash equivalents, accounts receivable, and accounts payable. The carrying amounts of accounts receivable and accounts payable are considered reasonable estimates of their fair value, due to the short maturity of these instruments.
Accounting literature provides a fair value hierarchy, which classifies fair value measurements based on the inputs used in measuring fair value. These inputs include: Level 1, defined as observable inputs such as quoted prices for identical instruments in active markets; Level 2, defined as inputs other than quoted prices in active markets that are either directly or indirectly observable; and Level 3, defined as unobservable inputs for which little or no market data exists, therefore requiring an entity to develop its own assumptions.
Cash and cash equivalents: The carrying amounts reported in the balance sheets for cash and cash equivalents are stated at their fair market value. Cash and cash equivalents are classified as Level 1.
Foreign exchange contracts: The Company does not use derivative financial instruments for speculative or trading purposes. Prior to 2009, the Company entered into foreign exchange forward contracts to hedge certain balance sheet exposures and intercompany balances against movement in foreign exchange rates. Gains and losses on the foreign exchange contracts were included in interest and other income, net, which offset foreign exchange gains or losses from revaluation of foreign currency-denominated balance sheet items and intercompany balances.
F-29
Table of Contents
GenMark Diagnostics, Inc.
Notes to Consolidated Financial Statements—(Continued)
The foreign exchange forward contracts required the Company to exchange foreign currencies to U.S. dollars or vice versa, and generally mature in one month or less. As of December 31, 2010, 2009 and 2008, the Company had outstanding foreign exchange forward contracts with aggregate notional amounts of $0, $0 and $6.0 million, respectively, which had remaining maturities of less than six months. The fair value recorded on the consolidated balance sheets for foreign exchange contracts is not material.
Non-recurring measurements: The Company measures the fair value of its long-lived assets on a periodic basis when it appears that there may be requirement to do so, such as an indication of impairment. During the year ended December 31, 2009, impairment indicators required that an assessment of the fair value of certain intangible assets and systems. These fair value measurements were done on the basis of unobservable Level 3 inputs, for which little or no market data exists. These inputs included the assumptions of future cash flows related to the items, and a discount rate applied to these cash flows. The assumed cash flows were projected based on management’s best estimates for the remaining net cash flows for each item over its the estimated remaining useful life. Due to the relatively short-term period of future cash flows on these items, the use of a discount rate did not have a material impact on the valuation of these items. Impairments recorded during the period as a result of these fair value measurements were $640,253 for intangible assets (note 3), and $865,389 on the laboratory systems (note 8).
There were no transfers of items between Levels 1, 2 or 3.
11. Other current assets and liabilities, and other non-current liabilities consisted of the following as of December 31, 2010 and 2009:
| 2010 | 2009 | |||||||
| Other current assets |
||||||||
| Therapeutic discovery credit receivable |
$ | 1,645,292 | $ | — | ||||
| Deposits and prepaid expenses |
290,920 | 344,558 | ||||||
| Tax receivable |
256,948 | — | ||||||
| Other |
— | 647,623 | ||||||
| Total |
$ | 2,193,160 | $ | 992,181 | ||||
| Other non-current assets |
||||||||
| Deposit |
$ | 55,355 | $ | — | ||||
| Total |
$ | 55,355 | $ | — | ||||
| Other current liabilities |
||||||||
| Accrued professional fees |
$ | 350,097 | $ | 544,524 | ||||
| Rental related liabilities |
330,424 | 188,070 | ||||||
| Accrued warranties |
179,594 | — | ||||||
| Other |
389,813 | 153,438 | ||||||
| Total |
$ | 1,249,928 | $ | 886,032 | ||||
| Other non-current liabilities |
||||||||
| Liability pertaining to uncertain tax position |
$ | 486,770 | $ | 463,000 | ||||
| Tax payable |
10,516 | — | ||||||
| Rental related liabilities |
115,646 | 332,334 | ||||||
| Total |
$ | 612,932 | $ | 795,334 | ||||
F-30
Table of Contents
GenMark Diagnostics, Inc.
Notes to Consolidated Financial Statements—(Continued)
In July 2010, the Company applied for certification of qualified investments eligible for credits and grants under the qualifying therapeutic discovery project program for the years ending December 31, 2009 and December 31, 2010. The $1.6 million in grant applications were for $561,000 of expenditures in 2009 and $1.1 million of expenditures in 2010.
These development projects included the NexGen System (formerly the “AD-8 system”), K-ras mutation cancer treatment, Plavix Sensitivity Drug, Warfarin Sensitivity Test, Thrombophilia Risk Test, Respiratory Viral Panel and Cystic Fibrosis Genotyping. In November 2010, the company was notified that it had been awarded a total of $1.6 million under the program. As of December 31, 2010, the Company recorded the $1.6 million tax credit as an Other Current Assets on the Balance Sheet with a corresponding credit to Other Income on the Consolidated Statement of Operations.
In February 2011, the Company requested payment from the U.S. Department of Treasury, and $1.6 million in cash was received.
12. Selected Quarterly Financial Data (Unaudited)
| 2010 Quarters | ||||||||||||||||
| (in thousands, except per share data) | ||||||||||||||||
| First | Second | Third | Fourth | |||||||||||||
| Total revenue |
$ | 399 | $ | 651 | $ | 667 | $ | 787 | ||||||||
| Gross loss |
$ | (168 | ) | $ | (212 | ) | $ | (554 | ) | $ | (939 | ) | ||||
| Loss from operations |
$ | (4,847 | ) | $ | (5,142 | ) | $ | (4,924 | ) | $ | (5,118 | ) | ||||
| Net loss |
$ | (4,849 | ) | $ | (5,137 | ) | $ | (4,917 | ) | $ | (3,500 | ) | ||||
| Per share data: |
||||||||||||||||
| Net loss per common share—basic and diluted |
$ | (0.68 | ) | $ | (0.60 | ) | $ | (0.42 | ) | $ | (0.30 | ) | ||||
| 2009 Quarters | ||||||||||||||||
| (in thousands, except per share data) | ||||||||||||||||
| First | Second | Third | Fourth | |||||||||||||
| Total revenue |
$ | 188 | $ | 249 | $ | 255 | $ | 306 | ||||||||
| Gross loss |
$ | (1,186 | ) | $ | (484 | ) | $ | (506 | ) | $ | (1,158 | ) | ||||
| Loss from operations |
$ | (4,628 | ) | $ | (4,264 | ) | $ | (5,648 | ) | $ | (5,898 | ) | ||||
| Net loss |
$ | (4,144 | ) | $ | (4,267 | ) | $ | (5,653 | ) | $ | (5,628 | ) | ||||
| Per share data: |
||||||||||||||||
| Net loss per common share—basic and diluted |
$ | (1.14 | ) | $ | (1.08 | ) | $ | (1.13 | ) | $ | (1.07 | ) | ||||
13. Subsequent Events
In March 2010, the Company entered into a loan and security agreement with Square 1 Bank, pursuant to which the Company obtained a credit facility consisting of a revolving line of credit in the amount of up to $2 million and an equipment term loan in the amount of up to $2 million. Based upon certain financial covenants, interest on the revolving line of credit will be either (i) the greater of (a) the bank’s prime rate (3.25% as of December 31, 2010) plus 2.75%, or (b) 6%; or (ii) the greater of (a) the bank’s prime rate plus 3.75%, or (b) 7%. In addition, based upon certain financial covenants, interest on the equipment term loan will be either (i) the greater of (a) the bank’s prime rate plus 3.25%, or (b) 6.50%; or (ii) the greater of (a) the bank’s prime rate plus 4.25%, or (b) 7.50%. The revolving line matures in July 2011 and the term loan matures in July 2013. As of December 31, 2010, the Company had not drawn any funds under this loan and security agreement.
F-31
Table of Contents
GenMark Diagnostics, Inc.
Notes to Consolidated Financial Statements—(Continued)
In March 2011, the loan and security agreement was amended, whereby the line of credit availability was increased by $1 million to $3 million and the maturity was extended to July 2012. The term loan was modified to allow invoices up to 360 days to qualify to be submitted for credit extension. There were no other changes to these two loans.
An additional loan was made available under the amended loan and security agreement for up to $1 million to finance equipment purchases. Based upon certain financial covenants, interest on this equipment term loan will be either (i) the greater of (a) the bank’s prime rate plus 3.25%, or (b) 6.50%; or (ii) the greater of (a) the bank’s prime rate plus 4.25%, or (b) 7.50%. This term loan matures March 2014.
As of March 11, 2011, the Company had no outstanding loans on the line of credit and had drawn $2 million to finance 2010 equipment purchases and tenant improvements to its Carlsbad facility against the original term loan. The loan bears an interest rate of 7.5%.
F-32
Table of Contents
Shares
GENMARK DIAGNOSTICS, INC.
Common Stock
PROSPECTUS
Canaccord Genuity
William Blair & Company
, 2011
Table of Contents
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 13. Other Expenses of Issuance and Distribution.
The following table indicates the expenses to be incurred in connection with the offering described in this registration statement all of which will be paid by us. All of the amounts are estimated except for the SEC registration fee, the FINRA filing fee and the Nasdaq filing fee.
| Amount to be paid |
||||
| SEC registration fee |
$ | 3,338 | ||
| Printing and mailing |
$ | * | ||
| FINRA filing fee |
$ | * | ||
| Nasdaq filing fee |
$ | * | ||
| Legal fees and expenses |
$ | * | ||
| Accounting fees and expenses |
$ | * | ||
| Transfer agent and registrar |
$ | * | ||
| Miscellaneous |
$ | * | ||
| Total |
$ | * | ||
| * | Estimated expenses only include information that is known at the time of filing this registration statement. Estimated expenses are subject to future contingencies, including additional expenses for future offerings. |
Item 14. Indemnification of Directors and Officers.
Our certificate of incorporation and bylaws that will be effective upon completion of the offering provide that each person who was or is made a party or is threatened to be made a party to or is otherwise involved (including, without limitation, as a witness) in any action, suit or proceeding, whether civil, criminal, administrative or investigative, by reason of the fact that he or she is or was one of our directors or officers or is or was serving at our request as a director, officer, or trustee of another corporation, or of a partnership, joint venture, trust or other enterprise, including service with respect to an employee benefit plan, whether the basis of such proceeding is alleged action in an official capacity as a director, officer or trustee or in any other capacity while serving as a director, officer or trustee, shall be indemnified and held harmless by us to the fullest extent authorized by the Delaware General Corporation Law against all expense, liability and loss (including attorneys’ fees, judgments, fines, ERISA excise taxes or penalties and amounts paid in settlement) reasonably incurred or suffered by such.
Section 145 of the Delaware General Corporation Law permits a corporation to indemnify any director or officer of the corporation against expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred in connection with any action, suit or proceeding brought by reason of the fact that such person is or was a director or officer of the corporation, if such person acted in good faith and in a manner that he or she reasonably believed to be in, or not opposed to, the best interests of the corporation, and, with respect to any criminal action or proceeding, if he or she had no reason to believe his or her conduct was unlawful. In a derivative action, or an action brought by or on behalf of the corporation, indemnification may be provided only for expenses actually and reasonably incurred by any director or officer in connection with the defense or settlement of such an action or suit if such person acted in good faith and in a manner that he or she reasonably believed to be in, or not opposed to, the best interests of the corporation, except that no indemnification shall be provided if such person shall have been adjudged to be liable to the corporation, unless and only to the extent that the court in which the action or suit was brought shall determine that the defendant is fairly and reasonably entitled to indemnity for such expenses despite such adjudication of liability.
II-1
Table of Contents
Pursuant to Section 102(b)(7) of the Delaware General Corporation Law, Article 12 of our certificate of incorporation eliminates the liability of a director to us or our stockholders for monetary damages for such a breach of fiduciary duty as a director, except for liabilities arising:
from any breach of the director’s duty of loyalty to us or our stockholders;
from acts or omissions not in good faith or which involve intentional misconduct or a knowing violation of law;
under Section 174 of the Delaware General Corporation Law; and
from any transaction from which the director derived an improper personal benefit.
The foregoing discussion of our certificate of incorporation, bylaws, indemnification agreements, and Delaware law is not intended to be exhaustive and is qualified in its entirety by such certificate of incorporation, bylaws, indemnification agreements, or law.
Reference is made to Item 17 of our undertakings with respect to liabilities arising under the Securities Act. Reference is also made to the form of underwriting agreement filed as Exhibit 1.1 to this registration statement for the indemnification agreements between us and the underwriters.
Item 15. Recent Sales of Unregistered Securities
Stock Options Grants
Since May 1, 2008, GenMark granted stock options, which were not registered under the Securities Act to employees and directors pursuant to which the optionees may purchase up to an aggregate of 1,613,925 shares of common stock at a weighted average exercise price of $6.05 per share. Of the options granted during this period, no options to purchase shares have been exercised, a total of 303,885 options to purchase shares were forfeited and a total of 77,375 options expired. The sale and issuance of these securities were exempt from registration under Rule 701 under the Securities Act.
Issuances of Ordinary Shares and Warrants by Osmetech
On December 5, 2008, Osmetech issued 1,050,813 ordinary shares at $3.36 per share to “accredited investors” as defined in Regulation D under the Securities Act of 1933, or Regulation D, for net proceeds of approximately $3.5 million. On December 21, 2008, Osmetech issued 1,942,624 ordinary shares to “accredited investors” as defined in Regulation D at $3.36 per share for net proceeds of approximately $6.5 million.
On June 25, 2009, Osmetech issued 1,139,285 ordinary shares to “accredited investors” as defined in Regulation D at $7.59 per share for net proceeds of approximately $8.6 million. On December 21, 2009, Osmetech issued 2,086,090 ordinary shares to “accredited investors” as defined in Regulation D at $7.60 per share for net proceeds of approximately $15.8 million.
Effective July 1, 2009, Osmetech issued a warrant to purchase 132,475 ordinary shares at an exercise price of $6.94 and a warrant to purchase 88,317 ordinary shares at an exercise price of $10.40. Each of the warrants was issued to an “accredited investor” as defined in Regulation D.
Each of the foregoing share numbers gives effect to the reorganization, as a result of which Osmetech became a subsidiary of GenMark. No underwriters were involved in the foregoing sales of securities. To the extent an exemption from registration was required, the securities were sold and issued to accredited investors in reliance upon the exemption from the registration requirements of the Securities Act, as set forth in Section 4(2) under the Securities Act and, with respect to the sales of ordinary shares, Rule 506 of Regulation D under the Securities Act. The purchasers of shares and warrants represented to Osmetech in connection with their purchase that they were accredited investors and were acquiring the securities for investment and not distribution, that they could bear the risks of the investment and could hold the securities for an indefinite
II-2
Table of Contents
period of time. The purchasers received written disclosures that the securities had not been registered under the Securities Act and that any resale must be made pursuant to a registration or an available exemption from such registration. The sales of these securities were made without general solicitation or advertising.
Issuances of Common Stock by GenMark Diagnostics, Inc.
Since our incorporation in February 2010, we have issued and sold the following securities that were not registered under the Securities Act:
In connection with our incorporation and initial organization, we issued 1,000 shares of common stock at par value to Osmetech plc for a total consideration of $0.10, which was exempt from registration pursuant to Regulation S under the Securities Act.
Concurrent with our initial public offering in June 2010, we issued shares of our common stock to the existing shareholders of Osmetech plc in exchange for the cancellation of their outstanding Osmetech ordinary shares pursuant to a scheme of arrangement under Part 26 of the U.K. Companies Act of 2006. Based on the number of ordinary shares of Osmetech outstanding and the exchange ratio set forth in the scheme of arrangement, we issued 7,123,512 shares of common stock to the existing shareholders of Osmetech plc. The scheme of arrangement was approved by the courts in the United Kingdom and by the Osmetech shareholders. The issuances were exempt from registration pursuant to Section 3(a)(10) of the Securities Act.
In connection with the scheme of arrangement, the holders of outstanding options and warrants to purchase ordinary shares in Osmetech were given the opportunity to surrender their options and warrants for options and warrants to purchase shares of common stock in GenMark. Based on the options and warrants of Osmetech outstanding and the exchange ratio set forth in the scheme of arrangement, we issued options and warrants exercisable for 1,184,305 shares of GenMark common stock to the existing option and warrant holders of Osmetech. The issuance of these options and warrants are exempt from registration under the Securities Act in reliance on Section 4(2) of the Securities Act or Rule 701 promulgated under Section 3(b) of the Securities Act as transactions not involving a public offering or transactions under compensatory benefit plans and contracts relating to compensation as provided under Rule 701.
Item 16. Exhibits and Financial Statement Schedules.
(a) See the Exhibit Index on the page immediately preceding the exhibits for a list of exhibits filed as part of this registration statement on Form S-1, which Exhibit Index is incorporated herein by reference.
(b) Financial Statement Schedules: All schedules have been omitted because they are not required or are not applicable or the required information is shown in the financial statements or notes thereto.
Item 17. Undertakings
(a) The undersigned registrant hereby undertakes to provide to the underwriters at the closing specified in the underwriting agreement, certificates in such denominations and registered in such names as required by the underwriters to permit prompt delivery to each purchaser.
(b) Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the registrant pursuant to the provisions described under Item 14 above, or otherwise, the registrant has been informed that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled
II-3
Table of Contents
by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
(c) The undersigned Registrant hereby undertakes that:
(1) For purposes of determining any liability under the Securities Act of 1933, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective.
(2) For the purpose of determining any liability under the Securities Act of 1933, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
II-4
Table of Contents
SIGNATURES
Pursuant to the requirements of the Securities Act, the Registrant certifies that it has reasonable grounds to believe that it meets all of the requirements for filing on Form S-1 and has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized, in Carlsbad, California, on May 26, 2011.
| GENMARK DIAGNOSTICS, INC. | ||
| By: |
/s/ Hany Massarany | |
| Name: Hany Massarany | ||
| Title: Chief Executive Officer and President | ||
We, the undersigned officers and directors of GenMark Diagnostics, Inc., hereby severally constitute and appoint Hany Massarany and Paul Ross, and both or any one of them, our true and lawful attorneys-in-fact and agents, with full power of substitution and re-substitution in each of them for him and in his name, place and stead, and in any and all capacities, to sign any and all amendments (including post-effective amendments) to this registration statement, and any registration statement for the same offering that is to be effective upon filing pursuant to Rule 462(b) under the Securities Act of 1933, as amended, and to file the same, with all exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing necessary or desirable to be done in and about the premises, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that each of said attorneys-in-fact or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Act of 1933, this registration statement has been signed below by the following persons in the capacities and on the dates indicated below.
| Signature |
Title |
Date | ||
| /s/ Hany Massarany Hany Massarany |
President and Chief Executive Officer and Director (Principal Executive Officer) | May 26, 2011 | ||
| /s/ Paul Ross Paul Ross |
Chief Financial Officer (Principal Financial Officer and Principal Accounting Officer) | May 26, 2011 | ||
| /s/ Christopher Gleeson Christopher Gleeson |
Chairman of the Board | May 26, 2011 | ||
| /s/ Daryl J. Faulkner Daryl J. Faulkner |
Director | May 26, 2011 | ||
| /s/ James Fox, Ph.D. James Fox, Ph.D. |
Director | May 26, 2011 | ||
| /s/ Kevin C. O’Boyle Kevin C. O’Boyle |
Director | May 26, 2011 | ||
Table of Contents
INDEX TO EXHIBITS
| Exhibit |
Description of Exhibits | |||
| 1.1 | Form of Underwriting Agreement.** | |||
| 3.1 | Certificate of Incorporation (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on March 19, 2010). | |||
| 3.2 | By-Laws (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on March 19, 2010). | |||
| 4.1 | Form of Warrant (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on May 13, 2010). | |||
| 5.1 | Opinion of DLA Piper LLP (US) regarding the legality of the securities being registered.* | |||
| 10.1 | Lease between The Campus Carlsbad, LLC and Clinical Micro Sensors, Inc. dba Osmetech Molecular Diagnostics, dated February 8, 2010 (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on March 19, 2010). | |||
| 10.2 | Commercial Lease Agreement between Collis P. and Howard Huntington Memorial Hospital Trust and Osmetech Technology Inc., dated March 24, 2008 (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on March 19, 2010). | |||
| 10.3 | First Amendment to Commercial Lease Agreement between Collis P. and Howard Huntington Memorial Hospital Trust and Osmetech Technology Inc., dated February 1, 2009 (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on April 20, 2010). | |||
| 10.4 | Second Amendment and Termination of Commercial Lease Agreement between Collis P. and Howard Huntington Memorial Hospital Trust and Osmetech Technology Inc., dated November 1, 2009 (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on April 20, 2010). | |||
| 10.5 | Commercial Lease Agreement between Kandamerica, Inc., and Osmetech Inc., dated August 1, 2005 (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on April 20, 2010). | |||
| 10.6 | Amendment to Commercial Lease Agreement between Kandamerica, Inc., and Osmetech Inc., dated March 12, 2008 (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on April 20, 2010). | |||
| 10.7 | License Agreement by and between California Institute of Technology and Clinical Micro Sensors, Inc. dba Osmetech Molecular Diagnostics, dated February 8, 1995 (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on May 21, 2010). ++ | |||
| 10.8 | Amended and Restated License Agreement by and between President and Fellows of Harvard College and Clinical Micro Sensors, Inc. dba Osmetech Molecular Diagnostics, dated July 14, 1997 (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on May 21, 2010). ++ | |||
| 10.9 | Exclusive License Agreement by and between Marshfield Clinic and Clinical Micro Sensors, Inc. dba Osmetech Molecular Diagnostics, dated October 15, 2007 (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on May 25, 2010). ++ | |||
Table of Contents
| Exhibit |
Description of Exhibits | |||
| 10.10 | Non-Exclusive Patent License Agreement by and between the University of Washington and Clinical Micro Sensors, Inc. dba Osmetech Molecular Diagnostics, dated February 28, 2007 (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on May 21, 2010). ++ | |||
| 10.11 | Amended and Restated Chemically Modified Enzymes Kit Patent License Agreement by and between Roche Molecular Systems, Inc., F. Hoffman-La Roche Ltd., and Clinical Micro Sensors, Inc. dba Osmetech Molecular Diagnostics, dated February 27, 2008 (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on May 21, 2010). ++ | |||
| 10.12 | Non-Exclusive License Agreement by and between The Johns Hopkins University and Clinical Micro Sensors, Inc. dba Osmetech Molecular Diagnostics, dated December 29, 2006 (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on May 25, 2010). ++ | |||
| 10.13 | License Agreement by and between the Regents of the University of Michigan, HSC Research and Development Limited Partnership and Clinical Micro Sensors, Inc. dba Osmetech Molecular Diagnostics, dated March 15, 2006 (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on May 21, 2010). ++ | |||
| 10.14 | License Agreement by and between HSC Research and Development Limited Partnership and Clinical Micro Sensors, Inc. dba Osmetech Molecular Diagnostics, dated March 15, 2006 (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on May 25, 2010). ++ | |||
| 10.15 | 2010 Equity Incentive Plan (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on April 20, 2010). + | |||
| 10.16 | Form of Stock Option Agreement (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on April 20, 2010). + | |||
| 10.17 | Form of Director and Officer Indemnification Agreement (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on March 19, 2010). + | |||
| 10.18 | Executive Employment Agreement, dated January 1, 2010, by and between Osmetech Technology Inc. and Jon Faiz Kayyem (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on March 19, 2010). + | |||
| 10.19 | Executive Employment Agreement, dated November 30, 2009, by and between Clinical Micro Sensors, Inc. dba Osmetech Molecular Diagnostics and Steven Kemper (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on March 19, 2010). + | |||
| 10.20 | Executive Employment Agreement, dated January 1, 2010, by and between Osmetech Technology Inc., and Pankaj Singhal (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on March 19, 2010). + | |||
| 10.21 | Executive Employment Agreement, dated March 1, 2010, by and between Osmetech Technology Inc. and John Bellano (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on March 19, 2010). + | |||
| 10.22 | Compromise Agreement, dated August 10, 2009, by and between Osmetech plc and James White (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on March 19, 2010). + | |||
Table of Contents
| Exhibit |
Description of Exhibits | |||
| 10.23 | Compromise Agreement, dated March 10, 2010, by and between Osmetech plc and David Sandilands (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on March 19, 2010). + | |||
| 10.24 | Loan and Security Agreement, dated March 12, 2010, by and among Square 1 Bank and Osmetech Technology Inc., Clinical Micro Sensors, Inc. dba Osmetech Molecular Diagnostics, and GenMark Diagnostics, Inc. (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on March 19, 2010). | |||
| 10.25 | First Amendment to Loan and Security Agreement, dated August 17, 2010, by and among Square 1 Bank and Osmetech Technology, Inc., Clinical Micro Sensors, Inc. dba Osmetech Molecular Diagnostics, and GenMark Diagnostics, Inc. (incorporated by reference to our Annual Report on Form 10-K filed with the Commission on March 14, 2011). | |||
| 10.26 | Second Amendment to Loan and Security Agreement, dated August 17, 2010, by and among Square 1 Bank and Osmetech Technology, Inc., Clinical Micro Sensors, Inc. dba Osmetech Molecular Diagnostics, and GenMark Diagnostics, Inc. (incorporated by reference to our Annual Report on Form 10-K filed with the Commission on March 14, 2011). | |||
| 10.27 | Third Amendment to Loan and Security Agreement, dated August 17, 2010, by and among Square 1 Bank and Osmetech Technology, Inc., Clinical Micro Sensors, Inc. dba Osmetech Molecular Diagnostics, and GenMark Diagnostics, Inc. (incorporated by reference to our Annual Report on Form 10-K filed with the Commission on March 14, 2011). | |||
| 10.28 | Manufacturing Services Agreement, dated February 1, 2007, by and between Aubrey Group, Inc. and Clinical Micro Sensors, Inc. dba Osmetech Molecular Diagnostics (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on April 20, 2010). ++ | |||
| 10.29 | First Amendment to Manufacturing Services Agreement, dated May 7, 2009, by and between Aubrey Group, Inc. and Clinical Micro Sensors, Inc. dba Osmetech Molecular Diagnostics (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on April 20, 2010). | |||
| 10.30 | Separation Agreement and General Release, dated March 24, 2011, by and between Clinical Micro Sensors, Inc. d.b.a. GenMark Diagnostics, Inc. and Pankaj Singhal (incorporated by reference to our Form 8-K filed with the Commission on March 28, 2010). + | |||
| 10.31 | Employee Offer Letter, dated March 11, 2011, by and between Clinical Micro Sensors, Inc. d.b.a. GenMark Diagnostics, Inc. and Paul Ross (incorporated by reference to our Form 8-K/A filed with the Commission on April 7, 2011). + | |||
| 10.32 | Executive Employment Agreement, dated March 1, 2010, by and between Clinical Micro Sensors, Inc. d.b.a. GenMark Diagnostics, Inc. and Jeffrey Hawkins (incorporated by reference to our Quarterly Report on Form 10-Q filed with the Commission on May 13, 2011). + | |||
| 10.33 | Executive Consulting Agreement, dated October 12, 2010, by and between Clinical Micro Sensors, Inc. d.b.a. GenMark Diagnostics, Inc. and Kuranda Partners, LLC (incorporated by reference to our Quarterly Report on Form 10-Q filed with the Commission on May 13, 2011) + | |||
| 10.34 | Executive Employment Agreement, dated April 5, 2011, by and between GenMark Diaganostics, Inc. and Hany Massarany (incorporated by reference to our Quarterly Report on Form 10-Q filed with the Commission on May 13, 2011) + | |||
| 21.1 | List of Subsidiaries (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on May 13, 2010). | |||
Table of Contents
| Exhibit |
Description of Exhibits | |||
| 23.1 | Consent of DLA Piper LLP (US) (included in Exhibit 5.1).* | |||
| 23.2 | Consent of Deloitte & Touche LLP (US).* | |||
| 23.3 | Consent of Deloitte LLP (UK).* | |||
| 24.1 | Powers of Attorney (included in the signature page).* | |||
| + | Management Compensation Plan |
| ++ | Confidential Treatment Granted |
| * | Filed herewith. |
| ** | To be filed. |