Attached files
| file | filename |
|---|---|
| EX-32.1 - CERTIFICATION - ENDOLOGIX INC /DE/ | elgxex32120101231q4.htm |
| EX-23.1 - CONSENT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM - ENDOLOGIX INC /DE/ | elgxex23120101231q4.htm |
| EX-32.2 - CERTIFICATION - ENDOLOGIX INC /DE/ | elgxex32220101231q4.htm |
| EX-31.2 - CERTIFICATION - ENDOLOGIX INC /DE/ | elgxex31220101231q4.htm |
| EX-31.1 - CERTIFICATION - ENDOLOGIX INC /DE/ | elgxex31120101231q4.htm |
| EX-21.1 - LIST OF SUBSIDIARIES - ENDOLOGIX INC /DE/ | elgxex21120101231q4.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
_______________________________
Form 10-K
_______________________________
(Mark One)
x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2010
or
o | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transaction period from to .
Commission file number: 000-28440
_______________________________
Endologix, Inc.
(Exact name of registrant as specified in its charter)
_______________________________
Delaware | 68-0328265 | |
(State or other jurisdiction of incorporation or organization) | (IRS Employer Identification No.) |
11 Studebaker, Irvine, California 92618
(Address of principal executive offices, including zip code)
Registrant’s telephone number, including area code: (949) 595-7200
_______________________________
Securities registered pursuant to Section 12(b) of the Act:
Title of each class | Name of each exchange on which registered | |
Common Stock, $0.001 par value | The NASDAQ Stock Market, LLC |
Securities registered pursuant to Section 12(g) of the Act: None
_______________________________
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes o No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K.
o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer | o | Accelerated filer | x | ||
Non-accelerated filer | o (Do not check if a smaller reporting company) | Smaller reporting company | o |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes o No x
As of June 30, 2010, the aggregate market value of the voting stock held by non-affiliates of the Registrant was approximately $146,296.509 (based upon the closing price for shares of the Registrant’s Common Stock as reported by the NASDAQ Global Market on June 30, 2010, the last trading date of the Registrant’s most recently completed second fiscal quarter).
On February 25, 2011, approximately 56,048,849 shares of the Registrant’s Common Stock, $0.001 par value, were outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of Part III of this Annual Report on Form 10-K are incorporated by reference into the Registrant’s Proxy Statement for its Annual Meeting of Stockholders to be held on May 25, 2011.
ENDOLOGIX, INC.
ANNUAL REPORT ON
Form 10-K
For the Fiscal Year Ended December 31, 2010
TABLE OF CONTENTS
Page | ||
PART I | ||
Item 1 | ||
Item 1A | ||
Item 1B | ||
Item 2 | ||
Item 3 | ||
Item 4 | (Removed and Reserved) | |
PART II | ||
Item 5 | ||
Item 6 | ||
Item 7 | ||
Item 7A | ||
Item 8 | ||
Item 9 | ||
Item 9A | ||
Item 9B | ||
PART III | ||
Item 10 | ||
Item 11 | ||
Item 12 | ||
Item 13 | ||
Item 14 | ||
PART IV | ||
Item 15 | ||
This Annual Report on Form 10-K contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. You can identify forward-looking statements generally by the use of forward-looking terminology such as “believes,” “expects,” “may,” “will,” “intends,” “plans,” “should,” “could,” “seeks,” “pro forma,” “anticipates,” “estimates,” “continues,” or other variations thereof, including their use in the negative, or by discussions of strategies, opportunities, plans or intentions. In addition, any statements that refer to projections of our future financial performance, trends in our businesses, or other characterizations of future events or circumstances are forward-looking statements. We have based these forward-looking statements largely on our current expectations based on information currently available to us and projections about future events and trends affecting the financial condition of our business. These forward-looking statements are subject to a number of risks, uncertainties, and assumptions including, among other things:
• | continued market acceptance of our Endologix ELG device; |
• | our ability to successfully incorporate the operations of Nellix and Nellix's products and technology into our business; |
• | the level and availability of third party payor reimbursement for our products; |
• | our ability to effectively manage our anticipated growth; |
• | our ability to protect our intellectual property rights and proprietary technology; |
• | our ability to operate without infringing the intellectual property rights and proprietary technology of third parties; |
• | our ability to effectively develop new or complementary technologies; |
• | development and management of our business and anticipated trends of our business; |
• | our ability to attract, retain and motivate qualified personnel; |
• | our ability to manufacture product to meet demand; |
• | the nature of regulatory requirements that apply to us, our suppliers and competitors and our ability to obtain and maintain any required regulatory approvals; |
• | our future capital expenditures and needs; |
• | our ability to effectively compete; |
• | general economic and business conditions; and |
• | other risks set forth under “Risk Factors” in Item 1A of this Annual Report on Form 10-K. |
The forward-looking statements involve known and unknown risks, uncertainties and other factors that may cause actual results to differ in significant ways from any future results expressed or implied by the forward-looking statements. Unless otherwise required by law, we undertake no obligation to publicly update or revise any forward-looking statements, either as a result of new information, future events or otherwise after the date of this Annual Report on Form 10-K.
The industry and market data contained in this Annual Report on Form 10-K are based either on our management’s own estimates or on independent industry publications, reports by market research firms or other published independent sources. Although we believe these sources are reliable, we have not independently verified the information and cannot guarantee its accuracy and completeness, as industry and market data are subject to change and cannot always be verified with complete certainty due to limits on the availability and reliability of raw data, the voluntary nature of the data gathering process and other limitations and uncertainties inherent in any statistical survey of market shares. Accordingly, you should be aware that the industry and market data contained in this Annual Report on Form 10-K, and estimates and beliefs based on such data, may not be reliable. Unless otherwise indicated, all information contained in this Annual Report on Form 10-K concerning our industry in general or any segment thereof, including information regarding our general expectations and market opportunity, is based on management’s estimates using internal data, data from industry related publications, consumer research and marketing studies and other externally obtained data.
Endologix® and Powerlink® are registered trademarks of Endologix, Inc. IntuiTrak, IntuiTrak Express, PowerFit, Powerlink XL, Nellix and our logos are trademarks of Endologix, Inc. Other trademarks used in this Annual Report on Form 10-K are the property of their respective holders.
1
PART I
Item 1. | Business |
Introduction
We develop, manufacture, market and sell innovative treatments for aortic disorders. Our principal product is an endoluminal graft, or ELG, for the treatment of abdominal aortic aneurysm, or AAA. AAA is a weakening of the wall of the aorta, the largest artery of the body. Once AAA develops, it continues to enlarge and if left untreated becomes increasingly susceptible to rupture. The overall patient mortality rate for ruptured AAAs is between 50% and 80%, making it a leading cause of death in the United States today.
The Endologix ELG device consists of a self-expanding cobalt chromium alloy stent covered by high density expanded polytetrafluoroethylene, or ePTFE, graft material. The device is loaded in a delivery catheter and implanted in the abdominal aorta, which is accessed through the femoral artery. Once the Endologix ELG is deployed into its proper position, blood flow is shunted away from the weakened or “aneurysmal” section of the aorta, reducing pressure and the potential for the aorta to rupture. This procedure is known as endovascular aneurysm repair, or EVAR. Our clinical trials demonstrated that implantation of our products reduces the mortality and morbidity rates associated with conventional AAA surgery, as well as provides a clinical alternative for many patients who could not undergo conventional surgery. Sales of the Endologix ELG in the United States, Europe, Asia, and South America are the sole source of our reported revenue.
Merger and Private Placement Transaction
On December 10, 2010, we completed our acquisition of Nellix, Inc., or Nellix, by way of a merger of a wholly-owned subsidiary of our company with and into Nellix. As a result of the merger, Nellix became, and is, a wholly-owned subsidiary of our company. Upon the closing of the merger, we issued an aggregate of 2,935,227 unregistered shares of our common stock to certain former securityholders of Nellix in exchange for the shares of Nellix capital stock held by them immediately prior to the closing of the merger. We also delivered an aggregate of 264,214 unregistered shares of our common stock to Wells Fargo Bank N.A., in its capacity as escrow agent, to secure our rights to indemnification as provided in the merger agreement. In addition, we may be required to issue to the former securityholders of Nellix as contingent consideration additional shares of our common stock upon our achievement of certain performance milestones related to the Nellix technology, or other events.
Also on December 10, 2010, concurrent with the closing of our acquisition of Nellix, we issued and sold to Essex Woodlands Health Ventures Fund VII, L.P., or Essex Woodlands Fund VII, and Essex Woodlands Fund VII purchased from us, an aggregate of 3,170,577 unregistered shares of our common stock, at purchase price of $4.731 per share, resulting in gross proceeds to us of $15,000,000.
Industry Background
Atherosclerosis is the thickening and hardening of arteries. Some hardening of arteries occurs naturally as people grow older. Atherosclerosis involves deposits of fatty substances, cholesterol, cellular waste products, calcium and other substances on the inner lining of an artery. Atherosclerosis is a slow, complex disease that starts in childhood and often progresses with age.
Atherosclerosis also can reduce the integrity and strength of the blood vessel wall, causing the vessel to expand or balloon out, which is known as an aneurysm. Aneurysms are commonly diagnosed in the aorta, which is the body’s largest artery. The highest incidence of aortic aneurysms occurs in the infrarenal aorta, the segment between the renal arteries and the area where the aorta divides into the two iliac arteries that travel down the legs. Once diagnosed, patients with AAA require non-invasive monitoring, or depending on the size of the AAA, must undergo a procedure to repair the aneurysm.
Traditional AAA repair is a highly invasive, open surgical procedure requiring a large incision in the patient’s abdomen, withdrawal of the patient’s intestines to provide access to the aneurysm, and the cross clamping of the aorta to stop blood flow. This procedure typically lasts two to four hours and is performed under general anesthesia. The complication rates for the open surgical procedure, as well as ELG systems, vary depending upon patient anatomy and risk classification.
An article published in the New England Journal of Medicine on January 31, 2008 compared the results of open surgical repair and EVAR on more than 45,000 patients over a three year period. Among the findings discussed in the article were:
• | The mortality rate of all patients in the study undergoing EVAR was approximately 1.2% as compared to 4.8% for open surgical repair. Importantly, these findings are based on a patient population that typically has a significantly higher co-morbidity rate compared with those patients treated by open surgery. |
• | Patients treated by EVAR were three times as likely to be discharged to their homes rather than another |
2
rehabilitation facility as compared to patients treated with open repair. This results in substantial clinical and economic benefits for patients and payors alike.
• | The average hospital stay for patients in the study undergoing EVAR was 3.4 days versus 9.3 days for patients undergoing open repair. |
Market Opportunity
In the United States alone, it is estimated that between 1.2 million and 2 million people have an AAA. Although AAA is one of the most serious cardiovascular diseases, many AAAs are never detected. Approximately 75% of AAA patients do not have symptoms at the time of their initial diagnosis, and AAAs generally are discovered inadvertently during procedures to diagnose unrelated medical conditions. Once an AAA develops, it continues to enlarge and if left untreated, becomes increasingly susceptible to rupture. The overall patient mortality rate for ruptured aneurysms is between 50% and 80%.
We estimate that in 2010 over 200,000 people were diagnosed with AAA in the United States and that approximately 72,000 of those diagnosed underwent aneurysm repair, either via EVAR or open surgery.
AAAs are generally more prevalent in people over the age of 65 and are more common in men than in women. In addition to the current pool of potential patients, we expect that the number of persons seeking treatment for their condition will increase based on demographic factors. In 2010, the age 65 and over population in the United States numbered approximately 40 million, or 13% of the total population, and is expected to be 75 million by 2030. It is growing at a higher rate than the overall United States population.
Over the next several years, we estimate that the United States ELG market will increase by at least 8% per year, and we estimate that up to 75% of AAA procedures will be performed using ELGs by 2015. We estimate that the current total worldwide AAA market is approximately $885 million, with approximately $535 million of the market in the United States alone, and the total worldwide AAA market is expected to grow to approximately $1.3 billion by 2015. We expect that the total aortic stent graft market (including thoracic stent grafts, for which we have future product development plans, but do not currently have an approved product) will grow to $1.7 billion by 2015.
Our Strategy
Our objective is to become a leader in the development and commercialization of innovative products for the treatment of aortic disorders. Key elements of our strategy to accomplish this objective are as follows:
• | Focus exclusively on the aorta and become the industry leader in the development, manufacture and sale of minimally invasive devices for the treatment of aortic disorders. |
• | Provide innovative, easy to use devices that enable physicians to treat more patients and achieve excellent clinical outcomes. |
• | Provide excellent clinical and technical support to physicians worldwide by building an experienced and knowledgeable sales and marketing organization. |
Our Products
Endologix ELG System
Our current ELG device consists of a self-expanding cobalt chromium alloy stent covered with high density ePTFE graft material. The device is loaded in a delivery catheter and implanted in the abdominal aorta, gaining access through a small incision into the femoral artery. Once the Endologix ELG is deployed into its proper position, blood flow is shunted away from the weakened, or aneurysmal, section of the aorta, reducing pressure and the potential for the aorta to rupture.
We believe the Endologix ELG is a superior design that overcomes the inherent limitations of other AAA devices and offers the following advantages:
• | Anatomical Fixation. The Endologix ELG is unique in that the placement of the main body stent graft is directly on the patient's aortoiliac bifurcation. This provides a solid foundation and long-term stability for the device. Alternative ELG's rely on hooks, barbs and radial force to anchor into the aorta near the renal arteries. These fixation methods are less reliable and can sometimes lead to device migration. |
• | Fully Supported. The main body and limbs of the Endologix ELG are fully supported by a cobalt chromium alloy stent. The cobalt chromium stent greatly reduces or eliminates the risk of kinking of the stent graft in even tortuous anatomies, eliminating the need for additional procedures or costly peripheral |
3
stents. Kinking may result in reduced blood flow and limb thrombosis.
• | Unique, Minimally Invasive Delivery System. The Endologix ELG requires only a small surgical incision in one leg. The other leg needs only placement of a non-surgical introducer sheath, three millimeters in diameter. Other ELGs typically need surgical exposure of the femoral artery in both legs to introduce the multiple components. Our unique delivery system permits our technology to be used in patients having small or very tortuous access vessels. |
Endologix ELG Components
Variations in patient anatomies require an adaptive technology. We designed our ELG device with multiple proximal aortic extensions, limb extensions, bifurcated main body lengths and diameters to simplify procedures, improve clinical results, and drive product adoption by offering physicians a full line of products that are adaptable for treatment of the majority of patients with AAA disease.
Powerlink Infrarenal Bifurcated Systems. The Powerlink Infrarenal Bifurcated System is our foundational stent graft and is available in multiple diameters and lengths and can treat patients that have an aortic neck up to 32 millimeters in diameter. The infrarenal device is made of a self-expanding cobalt chromium alloy stent covered by high density ePTFE graft material for placement below the renal arteries. We commenced commercial sales of this product in the United States in late 2004 and in Japan in February 2008 through Cosmotec, our exclusive distributor in that country.
Powerlink Aortic Cuffs and Limb Extensions. The Powerlink Proximal Aortic Extensions and Limb Extensions permit the physician to treat a greater number of patients. Proximal Extensions extend the aortal stent graft and are available in 25, 28 and 34 millimeter diameters and various lengths. They also are available in both infrarenal and suprarenal (i.e., the segment above the renal arteries) configurations. In addition, in the third quarter of 2010 we introduced the PowerFit Aortic Extension which improves proximal seal by increasing the number of contact points the stent makes in the aortic neck.
Limb Extensions are available in 16, 20 and 25 millimeter diameters and various lengths, allowing the physician to customize the implant to treat a wide range of patient anatomies. We market our large diameter 34 millimeter Proximal Extensions under the trademark Powerlink XL.
IntuiTrak. In October 2008, we received FDA approval for a new system to deliver and deploy the Powerlink System. The new system, IntuiTrak, further simplifies the implant procedure and provides a lower profile advantage over many competitive devices. We completed full market introduction for the product in the United States in the second quarter of 2009.
IntuiTrak Express. In March 2009, we received FDA approval for a new system to deliver the Powerlink XL aortic extension. This completes the application of IntuiTrak technology to the full range of sizes of the Powerlink System. We introduced the IntuiTrak Express to the United States market at the Society for Vascular Surgery meeting in June 2009.
Nellix Products
We acquired Nellix in order to expand our product line, to treat more patients and to achieve better long-term outcomes than those available with other EVAR devices. It is estimated that up to 40% of infrarenal AAA are not suitable for EVAR using currently available devices due to unfavorable neck anatomy (e.g., highly angulated, dilated or short) or distal anatomy. These issues can increase the risk of graft migration, or Type 1 endoleaks, which are caused by an incomplete proximal seal, and resultant secondary intervention. In addition, Type II endoleaks, which are caused by retrograde flow into the aneurysmal sac from collateral vessels, following EVAR, is an issue for all currently available endovascular devices. The Nellix system uses endobags that are filled with a biostable polymer that conforms to the patient's specific anatomy. Through this process, the Nellix endograft completely fills and stabilizes the aneurysm, reducing the risk of Type II endoleaks, while providing a blood conduit through the endoframes. In addition, the implant uniquely conforms to the patient's anatomy, mitigating the risk of graft migration and the potential loss of seal which can result in a Type III endoleak, which is caused by device separation. Thus, in addition to its advantages in treating varying anatomies, the Nellix system may mitigate Type I, II and III endoleaks associated with currently available modular endograft devices. Because the Nellix system fills the aneurysm sac, it does not need hooks or barbs to attach to the aortic wall. In addition, the Nellix system does not apply continuous radial force on the aorta like all other EVAR devices.
Clinical Trials
We continue to conduct clinical trials for other products related to the Endologix ELG system. In November 2009, we received an Investigational Device Exemption from the FDA to begin a prospective, multicenter, randomized clinical trial for a bilateral percutaneous approach to AAA repair. We initiated this trial in the first quarter of 2010. The total cost of the trial is
4
expected to be approximately $1.5 million, with the majority of costs incurred in 2011. We expect to conclude the enrollment of 150 patients at 20 domestic clinical sites during 2011.
Nellix began its feasibility clinical trial in 2008. To date, the clinical results have been positive, with 34 patients being treated with the Nellix technology with an average follow up time of 15 months. Nearly half of these patients had anatomical features that would have made them unsuitable for treatment with other EVAR devices. The data demonstrates exceptional clinical results as evidenced by complete freedom from AAA-related mortality, no aneurysm ruptures and no stent graft migration for up to two years post-procedure.
Research and Development
We spent $11.2 million in 2010, $6.6 million in 2009, and $6.1 million in 2008, on research and development, including clinical studies. Our focus is to continually develop innovative and cost effective medical device technology for the treatment of aortic disorders. We believe that our ability to develop new technologies is a key to our future growth and success. Our research and development activities have focused on technology that makes our existing products easier to use, developing products to treat more AAA patients, and new technologies to address other aortic disorders, such as stent grafts for the thoracic region of the aorta. Historically, we have focused on developing devices for infrarenal AAA. However, we expect to devote more resources in the future to developing new technologies to treat juxtarenal and thoracic aneurysms, in order to continue growing our business. Completing these new product development activities will likely require significant cash resources and could take many years to complete, if at all.
Marketing and Sales
We sell and market products both in the United States and internationally. We sell our products in the United States through a direct sales force and internationally through exclusive independent distributors. As of February 25, 2011, we marketed our products in 31 countries outside of the United States through 14 independent distributors.
United States. We market and sell our products in the United States through a direct sales force consisting of 63 sales territories as of December 31, 2010 and we plan to add 15% more territories by the end of 2011. The primary customer and decision maker for these devices in the United States is the vascular surgeon. Through our direct sales force, we provide clinical support and service to many of the approximately 1,800 hospitals in the United States which perform EVAR. We devote significant resources to the training of our sales representatives, including educational programs on their interactions with physicians. Approximately 82% of our revenues for the year ended December 31, 2010 were generated from product sales in the United States.
Europe. The market for ELGs in Europe is influenced by vascular surgeons, interventional radiologists and, to a lesser extent, interventional cardiologists who perform EVAR. We have obtained the right to affix a CE mark to our products. Europe represents a smaller market opportunity due to capitated hospital budgets and a selling price that is typically less than in the United States. Approximately 6% of our revenues for the year ended December 31, 2010 were generated from product sales in Europe. In addition, we have obtained regulatory approval but have not initiated the distribution process in several other countries, including Norway, Poland, Portugal, and Spain. We may or may not pursue these markets depending on the availability of a suitable distribution partner.
Asia. We commenced commercial sales in Japan in February 2008 after receipt of Shonin approval. We also commenced commercial sales in China in 2010 after receiving regulatory approval. Approximately 5% of our revenues for the year ended December 31, 2010 were generated from product sales in Asia.
South America and Mexico. We have obtained regulatory approval and have active distribution partners in a number of countries, including Argentina, Brazil, Chile, Colombia, and Mexico. Approximately 6% of our revenues for the year ended December 31, 2010 were generated from product sales in South America and Mexico.
Manufacturing and Supply
All of our commercial products are manufactured, assembled, packaged and sterilized at our 30,200 square foot leased facility in Irvine, California. Our current manufacturing process is labor intensive and involves shaping and forming a cobalt chromium stent, producing the high density ePTFE graft material to form the outside of the device, suturing the graft material on to the stent, and loading the device into a delivery catheter.
In April 2007, we received FDA approval to manufacture the high density ePTFE graft material used in our products. Since 2008, we have manufactured all of our requirements for this graft material. Our cost to manufacture this graft material is
5
significantly lower than our cost previously paid to a third party supplier of the graft material, which positively impacted our gross margins.
We rely on third parties for the supply of certain components used in our products, such as wire used to form our cobalt chromium alloy stent. We outsource the manufacturing of these components as it allows us have relationships with suppliers who have appropriate competencies while minimizing our capital investment and costs. Our third party manufacturers are required to meet FDA and/or ISO 13485 and other quality standards. While we obtain some of these components from single source suppliers, we believe there are alternative vendors for the supply of these components.
Patents and Proprietary Information
We believe that our intellectual property and proprietary information is key to protecting our technology. We are building a portfolio of apparatus and method patents covering various aspects of our current and future technology. In the AAA area, we have 18 United States patents issued, including 393 claims, and 21 pending United States patent applications. Our current AAA related patents begin expiring in 2017 and the last patent expires in 2019. We intend to continue to file for patent protection to strengthen our intellectual property position as we continue to develop our technology. As a result of our acquisition of Nellix, we acquired an additional nine issued United States patents and six issued foreign patents, with expiration dates beginning in 2015.
In addition to our AAA intellectual property, we own or have the rights to 36 issued United States patents, one issued European patent, one issued Japanese patent, and two pending United States patent applications relating to intravascular radiation, stents, and various catheter or intravascular technologies. The non-AAA patents begin expiring in 2012 and the last patent expires in 2018.
Our policy is to protect our proprietary position by, among other methods, filing United States and foreign patent applications to protect technology, inventions and improvements that are important to the development of our business. We also own trademarks to protect the names of our products. In addition to patents and trademarks, we rely on trade secrets and proprietary know-how. We seek protection of these trade secrets and proprietary know-how, in part, through confidentiality and proprietary information agreements. We make diligent efforts to require our employees, directors, consultants and advisors, other advisors and other individuals and entities to execute confidentiality agreements upon the start of employment, consulting or other contractual relationships with us. These agreements provide that all confidential information developed or made known to the individual or entity during the course of the relationship is to be kept confidential and not be disclosed to third parties, except in specific circumstances. In the case of employees and some other parties, the agreements provide that all inventions conceived by the individual will be our exclusive property.
Competition
The medical device industry is marked by intense competition. Any product we develop that gains regulatory clearance or approval will have to compete for market acceptance and market share. We believe that the primary competitive factors in the market for AAA devices are:
• | clinical effectiveness; |
• | product safety, reliability and durability; |
• | ease of use; |
• | sales force experience and relationships; and |
• | price. |
We experience significant competition in the endovascular graft market and we expect that the intensity of competition will increase over time. Three manufacturers, Medtronic Inc., W.L. Gore Inc., and Cook Medical Products, Inc. have obtained FDA marketing approval for their endovascular stent grafts in the U.S. However, we believe that our technology offers clinical advantages over these other currently available technologies. The cardiovascular device industry is marked by rapid technological improvements and, as a result, physicians are open to improved designs. Significant market share and revenue can be captured by designs demonstrating superior clinical outcomes. We believe that ease of use, the dependability of clinical results and the durability of the product design are the most important product characteristics. The Endologix ELG is the only available EVAR device that provides anatomical fixation and gives physicians the choice of either infrarenal or suprarenal aortic extensions.
6
The following chart details the stent graft characteristics of the EVAR stent grafts being sold in the United States.
FDA Approved
Stent Graft Characteristics
Manufacturer/Product Name | Design | Fixation | ||
Endologix/ Powerlink | Long main body, short limbs | Anatomical fixation | ||
Medtronic/ Endurant® & AneuRx® | Short main body, long modular limbs | Radial force, suprarenal stent and barbs | ||
Cook/ Zenith® | Short main body, long modular limbs | Radial force, suprarenal stent and barbs | ||
WL Gore/ Excluder® | Short main body, long modular limbs | Radial force and barbs |
In addition to the competitors mentioned above, Terumo-Vascutek, Trivascular, Aptus, Attura, Cordis, Jotec, Bolton, St. Jude Medical and Lombard Medical are believed to have active development programs.
Most of our existing competitors have substantially greater capital resources than we do and also have greater resources and expertise in the areas of research and development, obtaining regulatory approvals, manufacturing, marketing, and sales. In addition, many of our competitors have multiple product offerings, which some physicians may find more convenient. We also compete with other medical device companies for clinical sites and for the hiring of qualified personnel, including sales representatives.
Third-Party Reimbursement
In the United States, hospitals are the primary purchasers of our products. Hospitals then bill various third-party payors, such as Medicare, Medicaid, and other government programs and private insurance plans, for the healthcare services and products provided to patients. Government agencies, private insurers and other payors determine whether to provide coverage for a particular procedure and reimburse hospitals for medical treatment at a fixed rate based on the diagnosis-related group established by the United States Centers for Medicare and Medicaid Services, or CMS. The fixed rate of reimbursement is based on the procedure performed, and is unrelated to the specific devices used in that procedure.
Reimbursement of procedures utilizing our products currently is covered under a diagnosis-related group. Some payors may deny reimbursement if they determine that the device used in a treatment was unnecessary, inappropriate or not cost-effective, experimental or used for a non-approved indication. Therefore, we cannot assure you that reimbursement for any new product we develop will be available to hospitals and other users, or that future reimbursement policies of payors will not hamper our ability to sell current or new products on a profitable basis.
In October 2000, the CMS issued a guideline regarding the proper coding of our procedures for billing purposes. CMS instructed that code 39.71, for endovascular graft repair of aneurysm, be utilized. For purposes of hospital reimbursement, the majority of patients using the Endologix ELG will be classified under DRG 237, Major Cardiovascular Procedures with Complication/ Comorbidity. In the latest data published by CMS, the national average reimbursement for DRG 237, which includes hospital costs, approximated $28,500.
Outside the United States, market acceptance of products depends partly upon the availability of reimbursement within the prevailing healthcare payment system. Reimbursement systems vary significantly by country, and by region within some countries, and reimbursement approvals must be obtained on a country-by-country basis. Reimbursement is obtained from a variety of sources, including government sponsored healthcare and private health insurance plans. In Europe, reimbursement for the procedure, including the device, typically comes from the hospital's general fund and is usually about three-quarters of the reimbursement available in the United States.
Some countries have centrally organized healthcare systems, but in most cases there is a degree of regional autonomy either in deciding whether to pay for a particular procedure or in setting the reimbursement level. The manner in which new devices enter the healthcare market depends on the system. There may be a national appraisal process leading to a new procedure or product coding, or it may be a local decision made by the relevant hospital department. The latter is particularly the case where a global payment is made that does not detail specific technologies used in the treatment of a patient. Most foreign countries also have private insurance plans that may reimburse patients for alternative therapies.
7
Government Regulation
United States
Our products are regulated in the United States as medical devices by the FDA under the Federal Food, Drug and Cosmetic Act. The FDA classifies medical devices into one of three classes based upon controls the FDA considers necessary to reasonably ensure their safety and effectiveness. Class I devices are subject to general controls such as labeling, adherence to good manufacturing practices and maintenance of product complaint records, but are usually exempt from premarket notification requirements. Class II devices are subject to the same general controls and also are subject to special controls such as performance standards, and FDA guidelines, and may also require clinical testing prior to approval. Class III devices are subject to the highest level of controls because they are used in life-sustaining or life-supporting implantable devices. Class III devices require rigorous clinical testing prior to their approval and generally require a PMA or PMA supplement approval prior to their sale.
If a medical device manufacturer can establish that a device is “substantially equivalent” to a legally marketed Class I or Class II device, or to an unclassified device, or to a Class III device for which the FDA has not called for PMAs, the manufacturer may seek clearance from the FDA to market the device by filing a 510(k) premarket notification. The 510(k) notification must be supported by appropriate data establishing the claim of substantial equivalence to the satisfaction of the FDA. Following submission of the 510(k) notification, the manufacturer may not place the device into commercial distribution in the United States until an order is issued by the FDA.
Manufacturers must file an investigational device exemption, or IDE, application if human clinical studies of a device are required and if the FDA considers experimental use of the device to represent significant risk to the patient. The IDE application must be supported by data, typically including the results of animal and mechanical testing of the device. If the IDE application is approved by the FDA, human clinical studies may begin at a specific number of investigational sites with a maximum number of patients, as approved by the FDA. The clinical studies must be conducted under the review of an independent institutional review board to ensure the protection of the patients’ rights.
Generally, upon completion of these human clinical studies, a manufacturer seeks approval of a Class III medical device from the FDA by submitting a PMA application. A PMA application must be supported by extensive data, including the results of the clinical studies, as well as testing and literature to establish the safety and effectiveness of the device. The Endologix ELG was approved through this PMA process and we anticipate that the Nellix system will likewise go through the PMA process.
FDA regulations require us to register as a medical device manufacturer with the FDA. Additionally, the California Department of Health Services, or CDHS, requires us to register as a medical device manufacturer within the state. Because of this, the FDA and the CDHS inspect us on a routine basis for compliance with Quality System Records, or QSR, regulations. These regulations require that we manufacture our products and maintain related documentation in a prescribed manner with respect to manufacturing, testing and control activities. We have undergone and expect to continue to undergo regular QSR inspections in connection with the manufacture of our products at our facility. Further, the FDA requires us to comply with various FDA regulations regarding labeling. The Medical Device Reporting laws and regulations require us to provide information to the FDA on deaths or serious injuries alleged to have been associated with the use of our devices, as well as product malfunctions that likely would cause or contribute to death or serious injury if the malfunction were to recur. In addition, the FDA prohibits an approved device from being marketed for “off-label” purposes.
We are subject to other federal, state and local laws, regulations and recommendations relating to safe working conditions, laboratory and manufacturing practices.
International
Our international sales are subject to regulatory requirements in the countries in which our products are sold. The regulatory review process varies from country to country and may in some cases require the submission of clinical data. In addition, the FDA must approve the export to certain countries of devices that require a PMA but are not yet approved domestically.
In Europe, we need to comply with the requirements of the Medical Devices Directive, or MDD, and affix the CE mark on our products to attest to such compliance. To achieve compliance, our products must meet the “Essential Requirements” of the MDD relating to safety and performance and we must successfully undergo verification of our regulatory compliance, or conformity assessment, by a Notified Body selected by us. The level of scrutiny of such assessment depends on the regulatory class of the product.
In December 1998, we received ISO 9001:1994/ EN46001:1996 certification from our Notified Body with respect to the
8
manufacturing of all of our products in our facilities. In September 2002, we received ISO 9001:1994/ EN46001:1996 and ISO 13485:1996 certification. In December 2005, we received ISO13485:2003 certification. We are subject to continued surveillance by our Notified Body and will be required to report any serious adverse incidents to the appropriate authorities. We also must comply with additional requirements of individual countries in which our products are marketed.
Fraud and Abuse; HIPPA
We may directly or indirectly be subject to various federal and state laws pertaining to healthcare fraud and abuse, including anti-kickback laws. In particular, the federal healthcare program anti-kickback statute prohibits persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual, or the furnishing, arranging for or recommending a good or service, for which payment may be made in whole or part under federal healthcare programs, such as the Medicare and Medicaid programs. The anti-kickback statute is broad and prohibits many arrangements and practices that are lawful in businesses outside of the healthcare industry. In implementing the statute, the Office of Inspector General, or OIG, has issued a series of regulations, known as the “safe harbors,” which began in July 1991. These safe harbors set forth provisions that, if all their applicable requirements are met, will assure healthcare providers and other parties that they will not be prosecuted under the anti-kickback statute. The failure of a transaction or arrangement to fit precisely within one or more safe harbors does not necessarily mean that it is illegal or that prosecution will be pursued. However, conduct and business arrangements that do not fully satisfy all requirements of an applicable safe harbor may result in increased scrutiny by government enforcement authorities such as the OIG. Penalties for violations of the federal anti-kickback statute include criminal penalties and civil sanctions such as fines, imprisonment and possible exclusion from Medicare, Medicaid and other federal healthcare programs. Various states have also enacted laws regulating the interactions of medical device companies with healthcare providers to prevent fraud and abuse.
The federal False Claims Act prohibits persons from knowingly filing or causing to be filed a false claim to, or the knowing use of false statements to obtain payment from, the federal government. Suits filed under the False Claims Act, known as “qui tam” actions, can be brought by any individual on behalf of the government. These individuals, sometimes known as “relators” or, more commonly, as “whistleblowers”, may share in any amounts paid by the entity to the government in fines or settlement. The number of filings of qui tam actions has increased significantly in recent years, causing more healthcare companies to have to defend a False Claim action. If an entity is determined to have violated the federal False Claims Act, it may be required to pay up to three times the actual damages sustained by the government, plus civil penalties for each separate false claim. Various states have also enacted similar laws modeled after the federal False Claims Act which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor.
The Health Insurance Portability and Accountability Act of 1996, or HIPAA, created two new federal crimes: healthcare fraud and false statements relating to healthcare matters. The healthcare fraud statute prohibits knowingly and willfully executing a scheme to defraud any healthcare benefit program, including private payors. A violation of this statute is a felony and may result in fines, imprisonment or exclusion from government sponsored programs. The false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items, or services. A violation of this statute is a felony and may result in fines or imprisonment.
We have implemented and maintain a comprehensive program of oversight and training of our employees to ensure compliance with the foregoing laws and regulations.
Product Liability
The manufacture and marketing of medical devices carries the risk of financial exposure to product liability claims. Our products are used in situations in which there is a high risk of serious injury or death. Such risks will exist even with respect to those products that have received, or in the future may receive, regulatory approval for commercial sale. We are currently covered under a product liability insurance policy with coverage limits of $10 million per occurrence and $10 million per year in the aggregate subject to usual self insured retention amounts.
Employees
As of December 31, 2010, we had 297 employees, including 173 in manufacturing, 13 in research and development, eight in regulatory and clinical affairs, 83 in sales, marketing and customer service and 20 in administration. We believe that the success of our business will depend, in part, on our ability to attract and retain qualified personnel. Our employees are not subject to a collective bargaining agreement, and we believe we have good relations with our employees.
9
General Information
We were incorporated in California in March 1992 under the name Cardiovascular Dynamics, Inc. and reincorporated in Delaware in June 1993. In January 1999, Cardiovascular Dynamics, Inc. merged with privately held Radiance Medical Systems, Inc., and we changed our name to Radiance Medical Systems, Inc. In May 2002, we merged with privately held Endologix, Inc., and we changed our name to Endologix, Inc. On December 10, 2010, we completed the merger of a wholly-owned subsidiary of our company with and into Nellix. As a result of the merger, Nellix became a wholly-owned subsidiary of our company.
Our principal executive office is located at 11 Studebaker, Irvine, California and our telephone number is (949) 595-7200. Our website is located at www.endologix.com. The information on, or that can be accessed through, our website is not incorporated by reference into this Annual Report on Form 10-K and should not be considered to be a part hereof.
We make our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to these reports available on our website, at www.endologix.com, free of charge as soon as practicable after filing with the U.S. Securities and Exchange Commission, or SEC.
All such reports are also available free of charge via EDGAR through the SEC website at www.sec.gov. In addition, the public may read and copy materials filed by us with the SEC at the SEC’s public reference room located at 100 F Street, NE, Washington, D.C., 20549. Information regarding operation of the SEC’s public reference room can be obtained by calling the SEC at 1-800-SEC-0330.
Item 1A. | Risk Factors |
The following risks could affect our business, financial results and results of operations. These risk factors should be considered in connection with evaluating the forward-looking statements contained in this Annual Report on Form 10-K because these factors could cause actual results and conditions to differ materially from those projected in the forward-looking statements.
All of our revenue is generated from a limited number of products, and any declines in the sales of these products will negatively impact our business.
We have focused heavily on the development and commercialization of a limited number of products for the treatment of AAA because of limited resources. If we are unable to continue to achieve market acceptance of these products and do not achieve sustained positive cash flow from operations, we will be constrained in our ability to fund development and commercialization of improvements and other product lines. In addition, if we are unable to market our products as a result of failure to maintain regulatory approvals, we would lose our only source of revenue and our business would be negatively affected.
Our success depends on the growth in the number of AAA patients treated with endovascular devices.
In the United States, over 200,000 new diagnoses of AAA are made each year. In 2010, approximately 72,000 AAA patients were treated by either endovascular repair or by open surgery. Our success with our ELG's will depend on an increasing percentage of patients with AAA being diagnosed, and an increasing percentage of those diagnosed receiving endovascular, as opposed to open surgical procedures. Initiatives to increase screening for AAA are underway but are out of our control and such general screening programs may never gain wide acceptance. The failure to diagnose more patients with AAA could negatively impact our sales.
Our success depends on convincing physicians to use our products in more endovascular AAA procedures.
Our AAA products utilize a different fixation approach than the competitive products. Based upon our favorable clinical results, product improvements and increasing the size of our sales force, we have been able to increase sales at a rate higher than the market growth. However, if we are unable to continue convincing physicians to use our products, our business could be negatively impacted.
Our international operations subject us to certain operating risks, which could adversely impact our net sales, results of operations and financial condition.
Sales of our products outside the United States represented approximately 18% of our revenue in 2010. During 2010, we sold our products through fourteen distributors located in the following countries outside of the United States: Argentina, Brazil, Chile, Colombia, Germany, Greece, Ireland, Italy, Japan, Mexico, New Zealand, China, and Turkey. The sales territories authorized within these various distribution agreements cover a total of thirty-one countries. The sale and shipment of our products across international borders, as well as the purchase of components and products from international sources, subject us
10
to extensive U.S. and foreign governmental trade, import and export, and custom regulations and laws. Compliance with these regulations is costly and exposes us to penalties for non-compliance. Other laws and regulations that can significantly impact us include various anti-bribery laws, including the U.S. Foreign Corrupt Practices Act and anti-boycott laws. Any failure to comply with applicable legal and regulatory obligations could impact us in a variety of ways that include, but are not limited to, significant criminal, civil and administrative penalties, including imprisonment of individuals, fines and penalties, denial of export privileges, seizure of shipments, restrictions on certain business activities, and exclusion or debarment from government contracting. Also, the failure to comply with applicable legal and regulatory obligations could result in the disruption of our shipping and sales activities.
In addition, many of the countries in which we sell our products are, to some degree, subject to political, economic or social instability. Our international operations expose us and our distributors to risks inherent in operating in foreign jurisdictions. These risks include:
• | difficulties in enforcing or defending intellectual property rights. |
• | pricing pressure that we may experience internationally; |
• | a shortage of high-quality sales people and distributors; |
• | changes in third-party reimbursement policies that may require some of the patients who receive our products to directly absorb medical costs or that may necessitate the reduction of the selling prices of our products; |
• | the imposition of additional U.S. and foreign governmental controls or regulations; |
• | economic instability; |
• | changes in duties and tariffs, license obligations and other non-tariff barriers to trade; |
• | the imposition of restrictions on the activities of foreign agents, representatives and distributors; |
• | scrutiny of foreign tax authorities which could result in significant fines, penalties and additional taxes being imposed on us; |
• | laws and business practices favoring local companies; |
• | longer payment cycles; |
• | difficulties in maintaining consistency with our internal guidelines; |
• | difficulties in enforcing agreements and collecting receivables through certain foreign legal systems; |
• | the imposition of costly and lengthy new export licensing requirements; |
• | the imposition of U.S. or international sanctions against a country, company, person or entity with whom we do business that would restrict or prohibit continued business with the sanctioned country, company, person or entity; and |
• | the imposition of new trade restrictions. |
If we experience any of these risks, our sales in international countries may be harmed and our results of operations would suffer.
If our products or processes infringe upon the intellectual property of third parties, the sale of our products may be challenged and we may have to defend costly and time-consuming infringement claims.
We may need to engage in expensive and prolonged litigation to assert or defend any of our intellectual property rights or to determine the scope and validity of rights claimed by other parties. With no certainty as to the outcome, litigation could be too expensive for us to pursue. Our failure to prevail in such litigation or our failure to pursue litigation could result in the loss of our rights that could hurt our business substantially. In addition, the laws of some foreign countries do not protect our intellectual property rights to the same extent as the laws of the United States, if at all.
Our failure to obtain rights to intellectual property of third parties or the potential for intellectual property litigation could force us to do one or more of the following:
• | stop selling, making or using products that use the disputed intellectual property; |
• | obtain a license from the intellectual property owner to continue selling, making, licensing or using products, which license may not be available on reasonable terms, or at all; |
• | redesign our products, processes or services; or |
• | subject us to significant liabilities to third parties. |
11
If any of the foregoing occurs, we may be unable to manufacture and sell our products and may suffer severe financial harm. Whether or not an intellectual property claim is valid, the cost of responding to it, in terms of legal fees and expenses and the diversion of management resources, could harm our business.
If third-party payors do not provide reimbursement for the use of our products, our revenues may be negatively impacted.
Our success in marketing our products depends in large part on whether domestic and international government health administrative authorities, private health insurers and other organizations will reimburse customers for the cost of our product. Reimbursement systems in international markets vary significantly by country and by region within some countries, and reimbursement approvals must be obtained on a country-by-country basis. Further, many international markets have government managed healthcare systems that control reimbursement for new devices and procedures. In most markets there are private insurance systems as well as government-managed systems. If sufficient reimbursement is not available for our current or future products, in either the United States or internationally, the demand for our products will be adversely affected.
Healthcare policy changes, including recent federal legislation to reform the U.S. healthcare system, may have a material adverse effect on us.
In response to perceived increases in health care costs in recent years, there have been and continue to be proposals by the federal government, state governments, regulators and third-party payors to control these costs and, more generally, to reform the U.S. healthcare system. Certain of these proposals could limit the prices we are able to charge for our products or the amounts of reimbursement available for our products and could limit the acceptance and availability of our products. Moreover, as discussed below, recent federal legislation would impose significant new taxes on medical device makers such as us. The adoption of some or all of these proposals, including the recent federal legislation, could have a material adverse effect on our financial position and results of operations.
On March 23, 2010, President Obama signed the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or the PPACA. The total cost imposed on the medical device industry by the PPACA may be up to approximately $20 billion over ten years. The PPACA includes, among other things, a deductible 2.3% excise tax on any entity that manufactures or imports medical devices offered for sale in the U.S., with limited exceptions, effective 2013. This excise tax will result in a significant increase in the tax burden on our industry, and if any efforts we undertake to offset the excise tax are unsuccessful, the increased tax burden could have a material adverse affect on our results of operations and cash flows. Other elements of the PPACA, including comparative effectiveness research, an independent payment advisory board, payment system reforms including shared savings pilots and other provisions, may significantly affect the payment for, and the availability of, healthcare services and result in fundamental changes to federal healthcare reimbursement programs, any of which may materially affect numerous aspects of our business.
Current challenges in the commercial and credit environment may adversely affect our business and financial condition.
The global financial markets have experienced unprecedented levels of volatility. Our ability to generate cash flows from operations or enter into or maintain existing financing arrangements on acceptable terms could be adversely affected if there is a material decline in the demand for our products or in the solvency of our customers, deterioration in our key financial ratios, maintenance of compliance with financial covenants in existing credit agreements, or credit ratings, or other significantly unfavorable changes in conditions. While these conditions and current economic instability have not meaningfully impaired our ability to access credit markets or meaningfully adversely affected our operations to date, continuing volatility in the global financial markets could increase borrowing costs or affect our ability to access the capital markets. Current or worsening economic conditions may also adversely affect the business of our customers, including their ability to pay for our products and services, and the amount spent on healthcare generally. This could result in a decrease in the demand for our products and services, longer sales cycles, slower adoption of new technologies and increased price competition.
Our operating results may vary significantly from quarter to quarter, which may negatively impact our stock price in the future.
Our quarterly revenues and results of operations may fluctuate due to, among others, the following reasons:
• | physician acceptance of our products; |
• | the conduct and results of clinical trials; |
• | the timing and expense of obtaining future regulatory approvals; |
• | fluctuations in our expenses associated with expanding our operations; |
• | the introduction of new products by our competitors; |
• | supplier, manufacturing or quality problems with our devices; |
12
• | the timing of stocking orders from our distributors; |
• | changes in our pricing policies or in the pricing policies of our competitors or suppliers; and |
• | changes in third-party payors' reimbursement policies. |
Because of these and possibly other factors, it is likely that in some future period our operating results will not meet investor expectations or those of public market analysts.
Any unanticipated change in revenues or operating results is likely to cause our stock price to fluctuate since such changes reflect new information available to investors and analysts. New information may cause investors and analysts to revalue our stock, which could cause a decline in the trading price of our stock.
We have limited resources to invest in research and development and to grow our business and may need to raise additional funds in the future for these activities.
We believe that our growth will depend, in significant part, on our ability to develop new technologies for the treatment of AAA and other aortic disorders and technology complementary to our current products. Our existing resources may not allow us to conduct all of the research and development activities that we believe would be beneficial for our future growth. As a result, we may need to seek funds in the future to finance these activities. If we are unable to raise funds on favorable terms, or at all, we may not be able to increase our research and development activities and the growth of our business may be negatively impacted.
Our future success depends on our ability to develop, receive regulatory clearance or approval for, and introduce new products or product enhancements that will be accepted by the market in a timely manner.
It is important to our business that we continue to build a more complete product offering for treatment of AAA and other aortic disorders. As such, our success will depend in part on our ability to develop and introduce new products. However, we may not be able to successfully develop and obtain regulatory clearance or approval for product enhancements, or new products, or these products may not be accepted by physicians or the payors who financially support many of the procedures performed with our products.
The success of any new product offering or enhancement to an existing product will depend on several factors, including our ability to:
• | properly identify and anticipate physicians and patient needs; |
• | develop and introduce new products or product enhancements in a timely manner; |
• | avoid infringing upon the intellectual property rights of third parties; |
• | demonstrate, if required, the safety and efficacy of new products with data from preclinical studies and clinical trials; |
• | obtain the necessary regulatory clearances or approvals for new products or product enhancements; |
• | be fully FDA-compliant with marketing of new devices or modified products; |
• | provide adequate training to potential users of our products; |
• | receive adequate coverage and reimbursement for procedures performed with our products; and |
• | develop an effective and FDA-compliant, dedicated marketing and distribution network. |
If we do not develop new products or product enhancements in time to meet market demand or if there is insufficient demand for these products or enhancements, our results of operations will suffer.
If clinical trials of our current or future products do not produce results necessary to support regulatory clearance or approval in the United States or elsewhere, we will be unable to commercialize these products.
We are currently conducting clinical trials and will likely need to conduct additional clinical trials in the future in support of new product approvals or approval for new indications for use. Clinical testing is expensive, typically takes many years and has an uncertain outcome. The initiation and completion of any of these studies may be prevented, delayed or halted for numerous reasons, including, but not limited to, the following:
• | the FDA, institutional review boards or other regulatory authorities do not approve a clinical study protocol, force us to modify a previously approved protocol, or place a clinical study on hold; |
• | patients do not enroll in, or enroll at the expected rate, or complete a clinical study; |
13
• | patients or investigators do not comply with study protocols; |
• | patients do not return for post-treatment follow-up at the expected rate; |
• | patients experience serious or unexpected adverse side effects for a variety of reasons that may or may not be related to our products such as the advanced stage of co-morbidities that may exist at the time of treatment, causing a clinical study to be put on hold; |
• | sites participating in an ongoing clinical study may withdraw, requiring us to engage new sites; |
• | difficulties or delays associated with bringing additional clinical sites on-line; |
• | third-party clinical investigators decline to participate in our clinical studies, do not perform the clinical studies on the anticipated schedule or consistent with the investigator agreement, clinical study protocol, good clinical practices, and other FDA and Institutional Review Board requirements; |
• | third-party organizations do not perform data collection and analysis in a timely or accurate manner; |
• | regulatory inspections of our clinical studies require us to undertake corrective action or suspend or terminate our clinical studies; |
• | changes in U.S. federal, state, or foreign governmental statutes, regulations or policies; |
• | interim results are inconclusive or unfavorable as to immediate and long-term safety or efficacy; or |
• | the study design is inadequate to demonstrate safety and efficacy. |
Clinical failure can occur at any stage of the testing. Our clinical trials may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical and/or non-clinical testing in addition to those we have planned. Our failure to adequately demonstrate the efficacy and safety of any of our devices would prevent receipt of regulatory clearance or approval and, ultimately, the commercialization of that device or indication for use.
We may not realize all of the anticipated benefits of our acquisition of Nellix.
The success of our acquisition of Nellix will depend, in part, on our ability to realize the anticipated growth opportunities and synergies from combining the businesses of our company and Nellix. Our ability to realize these benefits, and the timing of this realization, depend upon a number of factors and future events, many of which we cannot control. These factors and events include:
• | the results of future clinical trials of the Nellix Product, which consists of a stent-graft that employs an endoframe and endobags, which are filled with a polyethylene glycol polymer; |
• | the receipt of CE Mark approval of the Nellix Product from its European Union notified body; |
• | the receipt of approval from the FDA to sell the Nellix Product in the United States; |
• | obtaining and maintaining patent rights relating to the Nellix technology; |
• | effectively consolidating research and development operations; |
• | retaining and attracting key employees; |
• | consolidating corporate and administrative functions; |
• | building an effective direct sales and marketing organization in Europe; |
• | preserving our and Nellix's important business relationships; and |
• | minimizing the diversion of management's attention from ongoing business concerns. |
We may be obligated to issue additional shares of our common stock to the former stockholders of Nellix upon our satisfaction of certain milestones set forth in the merger agreement with Nellix and the other parties thereto. Any future issuances of our common stock as contingent merger consideration will dilute the ownership interests of our stockholders, and such dilution may not be commensurate with the benefit our stockholders realize from the merger.
Under the terms of the merger agreement with Nellix and the other parties thereto, we agreed to issue additional shares of our common stock to the former stockholders of Nellix as contingent consideration upon our satisfaction of one or both of two milestones related to the Nellix Product and described in the merger agreement, or upon a change of control of our company prior to our completion of one or both milestones. The maximum aggregate number of shares of our common stock issuable to the Nellix stockholders upon our achievement of both milestones, or upon a change of control of our company prior to our achievement of both milestones, is 10,190,475 shares. Issuing additional shares of our common stock to the former
14
stockholders as contingent consideration will dilute the ownership interests of holders of our common stock on the dates of such issuances. If we are unable to realize the strategic, operational and financial benefits anticipated from our acquisition of Nellix, our stockholders may experience dilution of their ownership interests in our company upon any such future issuances of shares of our common stock without receiving any commensurate benefit.
If any future acquisitions or business development efforts are unsuccessful, our business may be harmed.
As part of our business strategy to be an innovative leader in the treatment of aortic disorders, we may need to acquire other companies, technologies and product lines in the future. Acquisitions involve numerous risks, including the following:
• | the possibility that we will pay more than the value we derive from the acquisition, which could result in future non-cash impairment charges; |
• | difficulties in integration of the operations, technologies, and products of the acquired companies, which may require significant attention of our management that otherwise would be available for the ongoing development of our business; |
• | the assumption of certain known and unknown liabilities of the acquired companies; and |
• | difficulties in retaining key relationships with employees, customers, partners and suppliers of the acquired company. |
In addition, we may invest in new technologies that may not succeed in the marketplace. If they are not successful, we may be unable to recover our initial investment, which could include the cost of acquiring the license, funding development efforts, acquiring products or purchasing inventory. Any of these would negatively impact our future growth and cash reserves.
Our business is subject to extensive governmental regulation that could make it more expensive and time consuming for us to introduce new or improved products.
Our products must comply with regulatory requirements imposed by the FDA and similar agencies in foreign countries. These requirements involve lengthy and detailed laboratory and clinical testing procedures, sampling activities, an extensive FDA review process, and other costly and time-consuming procedures. It often takes several years to satisfy these requirements, depending on the complexity and novelty of the product. We also are subject to numerous additional licensing and regulatory requirements relating to safe working conditions, manufacturing practices, environmental protection, fire hazard control and disposal of hazardous or potentially hazardous substances. Some of the most important requirements we face include:
FDA approval process;
California Department of Health Services requirements;
ISO 9001:1994 and ENISO 13485:2003; and
European Union CE mark requirements.
Government regulation may impede our ability to conduct continuing clinical trials and to manufacture our existing and future products. Government regulation also could delay our marketing of new products for a considerable period of time and impose costly procedures on our activities. The FDA and other regulatory agencies may not approve any of our future products on a timely basis, if at all. Any delay in obtaining, or failure to obtain, such approvals could negatively impact our marketing of any proposed products and reduce our product revenues.
Our products remain subject to strict regulatory controls on manufacturing, marketing and use. We may be forced to modify or recall our product after release in response to regulatory action or unanticipated difficulties encountered in general use. Any such action could have a material effect on the reputation of our products and on our business and financial position.
Further, regulations may change, and any additional regulation could limit or restrict our ability to use any of our technologies, which could harm our business. We could also be subject to new federal, state or local regulations that could affect our research and development programs and harm our business in unforeseen ways. If this happens, we may have to incur significant costs to comply with such laws and regulations, which will harm our results of operations.
The use, misuse or off-label use of our products may harm our image in the marketplace or result in injuries that lead to product liability suits, which could be costly to our business or result in FDA sanctions if we are deemed to have engaged in such promotion.
Our currently marketed products have been cleared by the FDA for specific treatments and anatomies. We cannot, however, prevent a physician from using our products outside of those indications cleared for use, known as off-label use.
15
There may be increased risk of injury if physicians attempt to use our products off-label. We train our sales force not to promote our products for off-label uses. Furthermore, the use of our products for indications other than those indications for which our products have been cleared by the FDA may not effectively treat such conditions, which could harm our reputation in the marketplace among physicians and patients.
Physicians may also misuse our products or use improper techniques if they are not adequately trained, potentially leading to injury and an increased risk of product liability. If our products are misused or used with improper technique, we may become subject to costly litigation by our customers or their patients. Product liability claims could divert management's attention from our core business, be expensive to defend and result in sizable damage awards against us that may not be covered by insurance. If we are deemed by FDA to have engaged in the promotion of our products for off-label use, we could be subject to FDA prohibitions on the sale or marketing of our products or significant fines and penalties, and the imposition of these sanctions could also affect our reputation and position within the industry. Any of these events could harm our business and results of operations and cause our stock price to decline.
If we fail to develop and grow our direct sales force, our business could suffer.
We have a nationally staffed direct sales force and we utilize a network of third-party distributors for sales outside of the United States. As we launch new products and increase our marketing efforts with respect to existing products, we will need to retain and develop our direct sales personnel to build upon their experience, tenure with our products, and their relationships with customers. There is significant competition for sales personnel experienced in relevant medical device sales. If we are unable to attract, motivate, develop, and retain qualified sales personnel and thereby grow our sales force, we may not be able to maintain or increase our revenues.
Our third-party distributors may not effectively distribute our products.
We depend on medical device distributors and strategic relationships for the marketing and selling of our products internationally. We depend on these distributors' efforts to market our product, yet we are unable to control their efforts completely. In addition, we are unable to ensure that our distributors comply with all applicable laws regarding the sales of our products. If our distributors fail to market and sell our products effectively and in compliance with applicable laws, our operating results and business may suffer substantially, or we may have to make significant additional expenditures or concessions to market our products.
Our commercialization strategy with respect to the Nellix product line may adversely impact the efforts of our distributors who sell our other products.
Our proposed commercialization strategy with respect to the Nellix product line will involve developing a direct sales force in certain countries in Europe. As a result, it may be difficult for us to maintain relationships with some of our European distributors. If we are unable to maintain or build relationships with our European distributors, our operating results and business may suffer, or we may be required to make significant additional expenditures or concessions to market our products.
We are in a highly competitive market segment, which is subject to rapid technological change. If our competitors are better able to develop and market products that are safer, more effective, less costly, easier to use, or otherwise more attractive than any products that we may develop, our ability to generate revenue will be reduced.
Our industry is highly competitive and subject to rapid and profound technological change. Our success depends, in part, upon our ability to maintain a competitive position in the development of technologies and products for use in the treatment of AAA and other aortic disorders. We face competition from both established and development stage companies. Many of the companies developing or marketing competing products enjoy several advantages, including:
• | greater financial and human resources for product development, sales and marketing and patent litigation; |
• | significantly greater name recognition; |
• | established relationships with physicians, customers and third-party payors; |
• | additional lines of products, and the ability to offer rebates or bundle products to offer greater discounts or incentives to gain a competitive advantage; |
• | established sales and marketing, and distribution networks; and |
• | greater experience in conducting research and development, manufacturing, clinical trials, preparing regulatory submissions and obtaining regulatory clearance or approval for products and marketing approved products. |
Our competitors may develop and patent processes or products earlier than us, obtain regulatory clearance or approvals for competing products more rapidly than us, and develop more effective or less expensive products or technologies that render
16
our technology or products obsolete or non-competitive. We also compete with our competitors in recruiting and retaining qualified scientific, sales, and management personnel, establishing clinical trial sites and patient enrollment in clinical trials, as well as in acquiring technologies and technology licenses complementary to our products or advantageous to our business. If our competitors are more successful than us in these matters, our business may be harmed.
Our dependence upon key personnel to operate our business puts us at risk of a loss of expertise if key personnel were to leave us.
We depend upon the experience and expertise of our executive management team. The competition for executives, as well as for skilled product development and technical personnel and sales representatives in the medical device industry is intense and we may not be able to retain or recruit the personnel we need. If we are not able to attract and retain existing and additional highly qualified management, sales, regulatory, clinical and technical personnel, we may not be able to successfully execute our business strategy.
If we fail to properly manage our anticipated growth, our business could suffer.
We may experience periods of rapid growth and expansion, which could place a significant strain on our limited personnel, information technology systems, and other resources. In particular, the increase in our direct sales force requires significant management and other supporting resources. Any failure by us to manage our growth effectively could have an adverse effect on our ability to achieve our development and commercialization goals. To achieve our revenue goals, we must successfully increase production output as required by customer demand. We may in the future experience difficulties in increasing production, including problems with production yields and quality control and assurance, component supply, and shortages of qualified personnel. These problems could result in delays in product availability and increases in expenses. Any such delay or increased expense could adversely affect our ability to generate revenues. Future growth will also impose significant added responsibilities on management, including the need to identify, recruit, train and integrate additional employees. In addition, rapid and significant growth will place a strain on our administrative and operational infrastructure. In order to manage our operations and growth we will need to continue to improve our operational and management controls, reporting and information technology systems, and financial internal control procedures. If we are unable to manage our growth effectively, it may be difficult for us to execute our business strategy and our operating results and business could suffer.
We have a history of operating losses and may be required to obtain additional funds.
We have a history of operating losses and may need to seek additional capital in the future. Our cash requirements in the future may be significantly different from our current estimates and depend on many factors, including:
• | the results of our commercialization efforts for our existing and future products; |
• | the need for additional capital to fund future development programs; |
• | the costs involved in obtaining and enforcing patents or any litigation by third parties regarding intellectual property; |
• | the establishment of high volume manufacturing and increased sales and marketing capabilities; and |
• | our success in entering into collaborative relationships with other parties. |
To finance these activities, we may seek funds through borrowings or through additional rounds of financing, including private or public equity or debt offerings and collaborative arrangements with corporate partners. We may be unable to raise funds on favorable terms, or at all. During the recent economic instability, it has been difficult for many companies, particularly small cap medical device companies, to obtain financing in the public markets or to obtain debt financing on commercially reasonable terms, if at all. In addition, the sale of additional equity or convertible debt securities could result in additional dilution to our stockholders. If we borrow additional funds or issue debt securities, these securities could have rights superior to holders of our common stock, and could contain covenants that will restrict our operations. We might have to obtain funds through arrangements with collaborative partners or others that may require us to relinquish rights to our technologies, product candidates or products that we otherwise would not relinquish. If we do not obtain additional resources, our ability to capitalize on business opportunities will be limited, and the growth of our business will be harmed.
The surviving corporation of the Nellix acquisition, which is a wholly-owned subsidiary of our company, possesses not only all of the assets, but also all of the liabilities of Nellix. Discovery of previously undisclosed or unknown liabilities could have an adverse effect on our business, operating results and financial condition.
Acquisitions involve risks, including inaccurate assessment of undisclosed, contingent or other liabilities or problems. As a result of our acquisition of Nellix, we acquired not only all of the assets, but also all of the liabilities of Nellix. Although we conducted a due diligence investigation of Nellix and its known and potential liabilities and obligations prior to completing the acquisition, it is possible that undisclosed, contingent or other liabilities or problems may arise which we were previously
17
unaware. These undisclosed liabilities could have an adverse effect on our business, operating results and financial condition. The amount of such liabilities may be in excess of the value of the shares of our common stock that we placed into the escrow fund for fifteen months following the closing of the Nellix acquisition to secure our rights to indemnification under the merger agreement, or such liabilities may not be uncovered until after the shares of our common stock that were placed into the escrow fund have been released from the escrow fund.
We rely solely on an in-house process to manufacture our graft material, and any disruption in our ability to produce this material could delay or prevent us from producing products for sale.
Currently, we rely solely on an in-house manufacturing process to produce graft material, which is a primary component for our AAA products. Our reliance on a sole source exposes our operations to disruptions in supply caused by:
• | failure to comply with quality or regulatory requirements; |
• | fire, flood or earthquake, or other natural disaster; and |
• | a supply interruption in the underlying raw material for the process. |
Although we attempt to retain a significant stock of the graft material, the occurrence of any of the above disruptions in supply or other unforeseen events that could cause a disruption in our process to manufacture graft material may cause us to halt or experience a disruption in manufacturing the Powerlink System. Because we do not have alternative suppliers, our sales and operating results would be harmed in the event of a disruption.
We rely on a single vendor to supply certain components for our products, and any disruption in our supply could delay or prevent us from producing products for sale.
Currently, we rely on certain vendors as a sole source to supply us with certain primary components for our products. Our reliance on a sole source supplier exposes our operations to disruptions in supply caused by:
• | failure to comply with regulatory requirements; |
• | any strike or work stoppage; |
• | disruptions in shipping; |
• | a natural disaster caused by fire, floods or earthquakes; |
• | a supply shortage experienced by our sole source suppliers and; |
• | the fiscal health and manufacturing strength of our sole source suppliers. |
Although we retain significant stock in sole source components, the occurrence of any of the above disruptions in supply or other unforeseen events that could cause a disruption in supply from our sole source suppliers could prevent us from manufacturing our products and harm our business.
If we are unable to protect our intellectual property, our business may be negatively affected.
The market for medical devices is subject to frequent litigation regarding patent and other intellectual property rights. It is possible that our patents or licenses may not withstand challenges made by others or protect our rights adequately.
Our success depends in large part on our ability to secure effective patent protection for our products and processes in the United States and internationally. We have filed and intend to continue to file patent applications for various aspects of our technology. However, we face the risks that:
• | we may fail to secure necessary patents prior to or after obtaining regulatory clearances, thereby permitting competitors to market competing products; and |
• | our already-granted patents may be re-examined, re-issued or invalidated. |
We also own trade secrets and confidential information that we try to protect by entering into confidentiality agreements with other parties. However, the confidentiality agreements may not be honored or, if breached, we may not have sufficient remedies to protect our confidential information. Further, our competitors may independently learn our trade secrets or develop similar or superior technologies. To the extent that our consultants, key employees or others apply technological information to our projects that they develop independently or others develop, disputes may arise regarding the ownership of proprietary rights to such information, and such disputes may not be resolved in our favor. If we are unable to protect our intellectual property adequately, our business and commercial prospects likely will suffer.
If we are unable to effectively manage our inventory held on consignment by our intended customers, we will not achieve
18
our expected results.
Our current products are sold on a consignment basis to certain hospitals which purchase our product as they use it. In these consignment locations, we do not have physical possession of our products. We therefore must rely on information from our customers as well as periodic inspections by our sales personnel to determine when our products have been used. Our efforts to strengthen our monitoring and management of consigned inventory may not be adequate to meaningfully reduce the risk of inventory loss. If we are not able to effectively manage appropriate consigned inventory levels, we may suffer inventory losses which will reduce our operating results.
We may face product liability claims that could result in costly litigation and significant liabilities.
Manufacturing and marketing of our commercial products, and clinical testing of our products under development, may expose us to product liability claims. Although we have, and intend to maintain, product liability insurance, the coverage limits of our insurance policies may not be adequate and one or more successful claims brought against us may have a material adverse effect on our business and results of operations. Additionally, adverse product liability actions could negatively affect the reputation and sale of our products, our ability to obtain and maintain regulatory approval for our products and may divert management's attention from other matters.
Our operations for the Endologix ELG are currently conducted at a single location that may be at risk from earthquakes or other natural disasters.
We currently conduct all of our manufacturing, development and management activities at a single location in Irvine, California, near known earthquake fault zones. We have taken precautions to safeguard our facilities, including insurance, health and safety protocols, and off-site storage of computer data. However, any future natural disaster, such as an earthquake, could cause substantial delays in our operations, damage or destroy our equipment or inventory, and cause us to incur additional expenses. A disaster could seriously harm our business and results of operations. The insurance coverage we maintain may not be adequate to cover our losses in any particular case.
The price of our stock may fluctuate unpredictably in response to factors unrelated to our operating performance.
The stock market periodically experiences significant price and volume fluctuations that are unrelated to the operating performance of particular companies. These broad market fluctuations may cause the market price of our common stock to drop. In particular, the market price of securities of small medical device companies, like ours, has been very unpredictable and may vary in response to:
• | announcements by us or our competitors concerning technological innovations; |
• | introductions of new products; |
• | FDA and foreign regulatory actions; |
• | developments or disputes relating to patents or proprietary rights; |
• | failure of our results of operations to meet the expectations of stock market analysts and investors; |
• | changes in stock market analyst recommendations regarding our common stock; |
• | changes in healthcare policy in the United States or other countries; and |
• | general stock market conditions and other factors unrelated to our operating performance. |
Trading in our stock over the last twelve months has been limited, so investors may not be able to sell as much stock as they want at prevailing prices.
The average daily trading volume in our common stock for the year ended December 31, 2010 was approximately 226,000 shares. If limited trading in our stock continues, it may be difficult for investors to sell their shares in the public market at any given time at prevailing prices. Moreover, the market price for shares of our common stock may be made more volatile because of the relatively low volume of trading in our common stock. When trading volume is low, significant price movement can be caused by the trading of a relatively small number of shares. Volatility in our common stock could cause stockholders to incur substantial losses.
Some provisions of our charter documents and Delaware law may make takeover attempts difficult, which could depress the price of our stock and inhibit your ability to receive a premium price for your shares.
Provisions of our amended and restated certificate of incorporation could make it more difficult for a third party to acquire control of our business, even if such change in control would be beneficial to our stockholders. Our amended and restated certificate of incorporation allows our board of directors to issue up to five million shares of preferred stock and to fix
19
the rights and preferences of such shares without stockholder approval. Any such issuance could make it more difficult for a third party to acquire our business and may adversely affect the rights of our stockholders. In addition, our board of directors is divided into three classes for staggered terms of three years. We are also subject to anti-takeover provisions under Delaware law, each of which could delay or prevent a change of control. Together these provisions may delay, deter or prevent a change in control of us, adversely affecting the market price of our common stock.
We do not anticipate declaring any cash dividends on our common stock.
We have never declared or paid cash dividends on our common stock and do not plan to pay any cash dividends in the near future. Our current policy is to retain all funds and any earnings for use in the operation and expansion of our business. The payment of cash dividends by us is restricted by our revolving credit facility, which contains restrictions prohibiting us from paying any cash dividends without the lender's prior approval. If we do not pay dividends, our stock may be less valuable to you because a return on your investment will only occur if our stock price appreciates.
Item 1B. | Unresolved Staff Comments |
None.
Item 2. | Properties |
Currently, we lease two adjacent facilities aggregating approximately 49,800 square feet in Irvine, California under separate lease agreements that expire in August 2011 and August 2012, both of which may be renewed for two additional twelve month periods, at our option. Additionally, through Nellix we lease approximately 7,500 square feet in Palo Alto, California under a lease agreement that expires in April 2012. We believe that our current facilities will be adequate and suitable for our operations through the current lease term.
Item 3. | Legal Proceedings |
We are currently involved in litigation with Cook Medical Incorporated, or Cook. Cook has alleged that we infringed two of Cook's patents, granted in 1991 and 1998, respectively. The lawsuit was filed by Cook in the United States District Court for the Southern District of Indiana, on October 8, 2009. In December 2009, the United States Patent and Trademark Office, or PTO, granted our request for a reexamination of the two patents asserted by Cook in the lawsuit. In January 2010, the Court ordered that the lawsuit be stayed pending the outcome of the patent reexaminations. In February 2010, the PTO completed its initial reexamination process and confirmed the patentability of one of the two patents, and on March 31, 2010 issued a reexamination certificate to that effect. As to the second patent, the PTO rejected as unpatentable those patent claims asserted by Cook against us. On April 14, 2010, the PTO indicated its intent to issue a reexamination certificate confirming the patentability of the amended and new claims and issued the certificate on July 21, 2010. On June 2, 2010, the stay of the court proceedings was lifted and discovery is underway. We are raising numerous defenses in the case, one of which is that Cook's lawsuit is barred by a prior judgment in an earlier case between the same parties. A hearing on the construction of the asserted claims of the patents is scheduled for April, 2011. We intend to continue our vigorous defense against these claims and believe our defenses are meritorious.
We are also involved in litigation with Bard Peripheral Vascular, Inc., or Bard, in which Bard alleges that we infringe one of Bard's patents that issued in 2002. Bard filed the lawsuit against us and another defendant, Atrium Medical Corp., on August 10, 2010 in the United States District Court for the District of Arizona. Bard alleged in its complaint that the ePTFE material used in our Endologix ELG infringes U.S. Patent No. 6,436,135, entitled “Prosthetic Vascular Graft”, and seeks damages for the infringement. Bard also alleges that our infringement was willful and seeks treble damages, prejudgment interest and its attorney fees as well as a permanent injunction. Bard served us the complaint on November 24, 2010.We intend to vigorously defend ourselves against these claims and believe that our defenses are meritorious.
At this time, we are unable to predict the outcomes of these matters. We currently believe that the outcome of these matters will not have a material adverse effect on our financial position, results of operations, or cash flow. However, as these matters are ongoing, there is no assurance they will be resolved favorably by us or will not result in a material loss.
Item 4. | (Removed and Reserved) |
20
PART II
Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Our common stock trades on the NASDAQ Global Market under the symbol “ELGX.” The following table sets forth the high and low sale prices for our common stock as reported on the NASDAQ Global Market for the periods indicated.
High | Low | ||||||
Year Ended December 31, 2009 | |||||||
First Quarter | $ | 2.25 | $ | 1.08 | |||
Second Quarter | 3.51 | 2.05 | |||||
Third Quarter | 6.25 | 3.28 | |||||
Fourth Quarter | 6.27 | 4.05 | |||||
Year Ended December 31, 2010 | |||||||
First Quarter | $ | 5.89 | $ | 3.30 | |||
Second Quarter | 5.60 | 3.95 | |||||
Third Quarter | 5.01 | 3.68 | |||||
Fourth Quarter | 7.55 | 4.42 | |||||
On February 25, 2011, the closing sale price of our common stock on the NASDAQ Global Market was $5.87 per share and there were 262 record holders of our common stock.
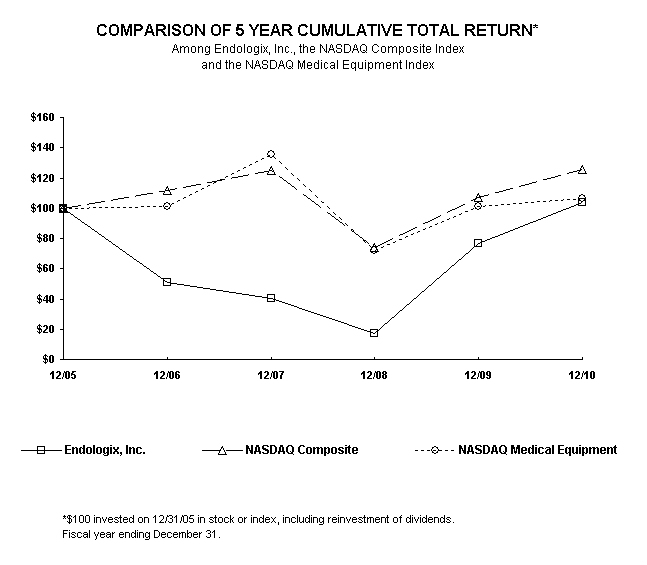
21
Dividend Policy
We have never paid any dividends. We currently intend to retain all earnings, if any, for use in the expansion of our business and therefore do not anticipate paying any dividends in the foreseeable future. Additionally, the terms of our credit facility prohibit us from paying cash dividends without the lender’s consent.
22
Item 6. | Selected Financial Data |
The following selected consolidated financial data has been derived from our audited consolidated financial statements. The audited consolidated financial statements for the fiscal years ended December 31, 2010, 2009, and 2008 are included elsewhere in this Annual Report on Form 10-K. The information set forth below should be read in conjunction with Management’s Discussion and Analysis of Financial Condition and Results of Operations and consolidated financial statements and notes thereto included herein.
Year Ended December 31, | |||||||||||||||||||
2010 | 2009 | 2008 | 2007 | 2006 | |||||||||||||||
(In thousands, except per share data) | |||||||||||||||||||
Consolidated Statement of Operations Data: | |||||||||||||||||||
Total revenue | $ | 67,251 | $ | 52,441 | $ | 37,664 | $ | 27,771 | $ | 14,672 | |||||||||
Cost of revenue | 15,030 | 13,181 | 10,380 | 10,539 | 6,330 | ||||||||||||||
Gross profit | 52,221 | 39,260 | 27,284 | 17,232 | 8,342 | ||||||||||||||
Operating costs and expenses: | |||||||||||||||||||
Research and development | 11,166 | 6,569 | 6,082 | 6,381 | 6,765 | ||||||||||||||
Marketing and sales | 31,869 | 26,483 | 23,794 | 20,142 | 14,579 | ||||||||||||||
General and administrative | 13,410 | 8,550 | 9,455 | 6,371 | 5,585 | ||||||||||||||
Termination of supply agreement | — | — | — | 550 | — | ||||||||||||||
Total operating costs and expenses | 56,445 | 41,602 | 39,331 | 33,444 | 26,929 | ||||||||||||||
Loss from operations | (4,224 | ) | (2,342 | ) | (12,047 | ) | (16,212 | ) | (18,587 | ) | |||||||||
Total other income(expense) | (160 | ) | (92 | ) | 55 | 1,137 | 1,044 | ||||||||||||
Net loss before income tax | (4,384 | ) | (2,434 | ) | (11,992 | ) | (15,075 | ) | (17,543 | ) | |||||||||
Income tax benefit | 15,037 | — | — | — | — | ||||||||||||||
Net income (loss) | $ | 10,653 | $ | (2,434 | ) | (11,992 | ) | (15,075 | ) | $ | (17,543 | ) | |||||||
Basic net earnings (loss) per share | $ | 0.22 | $ | (0.05 | ) | $ | (0.28 | ) | $ | (0.35 | ) | $ | (0.44 | ) | |||||
Shares used in computing basic net earnings (loss) per share | 48,902 | 45,194 | 43,045 | 42,796 | 40,010 | ||||||||||||||
Diluted net earnings (loss) per share | $ | 0.21 | $ | (0.05 | ) | $ | (0.28 | ) | $ | (0.35 | ) | $ | (0.44 | ) | |||||
Shares used in computing diluted net earnings (loss) per share | 50,544 | 45,194 | 43,045 | 42,796 | 40,010 | ||||||||||||||
December 31, | |||||||||||||||||||
2010 | 2009 | 2008 | 2007 | 2006 | |||||||||||||||
(In thousands) | |||||||||||||||||||
Consolidated Balance Sheet Data: | |||||||||||||||||||
Cash, restricted cash and cash equivalents | $ | 38,191 | $ | 24,065 | $ | 8,111 | $ | 9,228 | $ | 6,771 | |||||||||
Marketable securities available-for-sale | — | — | — | — | 13,417 | ||||||||||||||
Working capital | 48,585 | 31,061 | 15,951 | 18,365 | 26,933 | ||||||||||||||
Total assets | 134,375 | 51,292 | 37,263 | 40,043 | 52,686 | ||||||||||||||
Long term debt | — | 83 | 4,250 | — | — | ||||||||||||||
Accumulated deficit | (135,510 | ) | (146,164 | ) | (143,730 | ) | (131,738 | ) | (116,663 | ) | |||||||||
Total stockholders’ equity | 93,903 | 42,880 | 25,892 | 34,675 | 46,505 | ||||||||||||||
23
Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
The following discussion and analysis should be read in conjunction with “Selected Financial Data” and our consolidated financial statements and the related notes included in this Annual Report on Form 10-K. This discussion contains forward-looking statements that involve risks and uncertainties. Our actual results could differ materially from those anticipated in the forward-looking statements as a result of various factors including the risks we discuss in Item 1A of Part I, “Risk Factors” and elsewhere in this Annual Report on Form 10-K.
Overview
We are develop, manufacture, market and sell innovative treatments for aortic disorders. Our primary product is an ELG for the treatment of AAA. Since 2009, our sole source of revenue has been sales of the Powerlink System. Prior to 2009, we also generated license revenue from the licensing of our technology from a previous line of business.
For the year ended December 31, 2010, we had net sales of $67.3 million, which was an increase of approximately 28% from the year ended December 31, 2009, and we had net earnings of $10.7 million. As of December 31, 2010, we had an accumulated deficit of approximately $135.5 million. We have experienced year over year sales growth since the commercial launch of the Powerlink System in the United States in 2004. We now sell our products in the United States, Europe, Asia and South America.
As a result of our history of operating losses, we had limited resources with which to develop additional products beyond the Powerlink System. However, in 2009 we generated approximately $5 million of cash flow from operations and received approximately $14.8 million in net proceeds from a public offering of our common stock, as well as entered into a new long term credit facility whereby we may borrow up to $10 million. In 2010, we received approximately $15.0 million in net proceeds from a private placement offering of our common stock to Essex Woodlands in connection with our acquisition of Nellix. As a result of these increases in our capital resources, we increased our research and development efforts in order to develop new technologies for the treatment of aortic disorders and we expanded the size of our direct sales force in the United States, both of which we believe are vital to the future growth of our business.
Summary of Accounting Policies and Use of Estimates
The preparation of financial statements in conformity with generally accepted accounting principles requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, revenues and expenses, and related disclosure of contingent assets and liabilities. On an on-going basis, we evaluate our estimates, including those related to collectibility of customer accounts, whether the cost of inventories can be recovered, the value assigned to and estimated useful life of intangible assets, the realization of tax assets and estimates of tax liabilities, contingent liabilities and the potential outcome of litigation. We base our estimates on historical experience and on various other assumptions that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities. Actual results may differ from these estimates under different assumptions or conditions.
The following critical accounting policies and estimates were used in the preparation of the consolidated financial statements:
Revenue Recognition and Accounts Receivable
We comply with the revenue recognition guidelines in SEC Staff Accounting Bulletin No. 104, “Revenue Recognition.” We recognize revenue when all of the following criteria are met:
• | Persuasive evidence of an arrangement exists; |
• | The sales price is fixed or determinable; |
• | Collection of the relevant receivable is probable at the time of sale; and |
• | Products have been shipped or used and the customer has taken ownership and assumed risk of loss. |
For domestic sales, we generally recognize revenue upon completion of a procedure, when our product is implanted in a patient. For international sales, we recognize revenue at the time of shipment of our products to a distributor.We do not offer rights of return and we have no post delivery obligations other than our specified warranty.
We maintain allowances for doubtful accounts for estimated losses resulting from the inability of our customers to make
24
required payments. These estimates are based on our review of the aging of customer balances, correspondence with the customer, and the customer’s payment history. If additional information becomes available to us indicating the financial condition of the customer is deteriorating, additional allowances may be required.
Inventories
We write down our inventory for estimated obsolescence or unmarketable inventory equal to the difference between the cost of inventory and the estimated realizable value based upon assumptions about future demand, as driven by economic and market conditions, and the product’s shelf life. If actual demand, or economic or market conditions are less favorable than those projected by management, additional inventory write-downs may be required.
Goodwill, Intangible Assets and Long-Lived Assets
We record an impairment charge, or expense, for long-lived assets whenever events or changes in circumstances indicate that the value recorded for the asset may not be recoverable. Future changes in operations could cause us to write down the asset value and record an expense to better reflect our current estimate of its value. Goodwill and indefinite-lived intangible assets are tested for impairment annually, or more frequently if events or changes in circumstances indicate that the goodwill or indefinite-lived intangible assets are impaired. Factors that may impact whether there is potential goodwill impairment include a significant decrease in our stock price and our evaluation of a control premium that may be used when estimating our total fair value. Our stock price may decline, or other factors may arise, which could result in goodwill impairment in future periods. Factors that may impact whether there is a potential impairment to our indefinite-lived intangible assets include legal and regulatory considerations.
In-process research and development, or IPR&D, acquired as part of the Nellix acquisition will be amortized over its useful life once commercial sales of the Nellix product begin.
In determining the value of IPR&D, we used the excess earnings method. The excess earnings method reflects the present value of the operating cash flows generated by the IPR&D, after taking into account the cost to realize the revenue and an appropriate discount rate to reflect the time value and risk associated with the invested capital.
The key drivers, which require significant judgment, are:
•Projected revenue and earnings generated by the project;
•Estimated timing of and expected costs to complete the in-process projects;
•Projecting regulatory approvals;
•The expected life of the asset;
•The contributory asset charges that would be paid to the requisite operating assets; and
•A discount rate that reflects the level of the risk associated with receiving future cash flows.
In our valuation, we projected that the Nellix product will be launched internationally in the second half of 2011, and domestically in 2015. The discount rate we used was 26.0 percent.
If the Nellix product is not successfully developed, our sales and profitability will be adversely affected in future periods. Additionally, the value of the acquired IPR&D assets may become impaired. Our annual assessment will include a comparison of the fair value of IPR&D to our existing carrying value. We will recognize an impairment for amounts greater than the determined fair value.
In the judgment of management, the value of the Nellix IPR&D was $40.1 million at the time of the acquisition. As of the time of acquisition, we expect that all acquired IPR&D will reach technological feasibility, but there can be no assurance that the commercial viability will actually be achieved. The nature of the efforts to develop the acquired technologies into commercially viable products consists principally of planning, designing and conducting clinical trials necessary to obtain regulatory approvals. The risks associated with achieving commercialization include, but are not limited to, delay or failure to obtain regulatory approvals to conduct clinical trials, failure of clinical trials, delay or failure to obtain required market clearances, and patent litigation. If commercial viability is not achieved, we will not realize the original estimated financial benefits.
25
Valuation of Contingent Acquisition Consideration Payable
In connection with the purchase price allocations for acquisitions, we estimate the fair value of contingent consideration payments utilizing a probability-based income approach inclusive of an estimated discount rate.
Although we believe the assumptions and estimates made are reasonable, they are based in part on historical experience and information obtained from the management of the acquired businesses and are inherently uncertain. Examples of critical estimates in valuing certain of the intangible assets and any contingent consideration we have acquired or may acquire in the future include but are not limited to:
• | the feasibility and timing of achievement of development, regulatory and commercial milestones; |
• | expected costs to develop the in-process research and development into commercially viable products; and |
• | future expected cash flows from product sales. |
Unanticipated events and circumstances may occur which may affect the accuracy or validity of such assumptions, estimates or actual results.
As part of our acquisition of Nellix, we recorded a contingent acquisition consideration payable related to potential future payments based on certain milestones being met. As of the closing date of the acquisition, we assessed the fair value of the contingent acquisition consideration payable using significant judgment in the timing of when milestones will be achieved and assumed discount periods and rates. Each period we will reassess the fair value of the contingent acquisition consideration payable and record increases in the fair value as contingent consideration expense and record decreases in the fair value as a reduction of contingent consideration expense. Increases or decreases in the fair value of the contingent acquisition consideration payable can result from changes in assumed probability adjustments with respect to regulatory approval, changes in the assumed timing of when milestones will be achieved and changes in assumed discount periods and rates. Significant judgment is employed in determining the appropriateness of these assumptions each period. Accordingly, future business and economic conditions, as well as changes in any of the assumptions described in the accounting for business combinations above can materially impact the amount of contingent consideration expense that we record in any given period
We included two contingent consideration payments in the purchase price. The contingent consideration payments are subject to certain regulatory approvals and a revenue target, which are described below.
OUS Milestone
The outside the U.S., or OUS, milestone represents a contingency which concerns generating revenue in excess of $10 million outside the U.S. within a certain time period following our receipt of a CE Mark for the Nellix product. The CE Mark is a mandatory conformance mark on any products placed on the single market in the European Economic Area.
The right to receive the OUS milestone payment extends through the expiration date of one of Nellix' significant patents, which is estimated to expire in May 2026. Furthermore, if we experience a change of control event during the term of the OUS milestone, the contingent consideration shall be paid in full, even if not achieved.
The OUS milestone is subject to a payment table that provides for higher payments the earlier the milestone is met. The set of payments range from a low of $10 million should the milestone be reached more than 6 years after receiving the CE Mark, and a high of $24 million should the milestone be reached within 8 months after receiving the CE Mark. Payment of the milestone will be made in shares of our common stock, and is bound by a share price floor of $3.50 through expiration and a price ceiling of $7.50 for 24 months following receipt of a CE Mark. We have valued the OUS milestone using a probability weighted methodology to capture the probability of milestones being achieved, a change in control event, and no event occurring. Based on our judgment, we generated a set of probabilities over an applicable term of the OUS milestone.
After applying the applicable probabilities and payments with the respective payment periods, we applied an appropriate discount rate. We have estimated the value of the OUS milestone to be approximately $19.2 million.
PMA Milestone
The premarket approval, or PMA, milestone represents the contingency which concerns receiving PMA for the Nellix product. PMA is the FDA process of scientific and regulatory review to evaluate the safety and effectiveness of Class III medical devices.
26
The right to receive the PMA milestone payment extends through the expiration date of one of Nellix' significant patents. Furthermore, if we experience a change in control event during the term of the PMA milestone, the contingent consideration shall be paid in full, even if not achieved. If we achieve the PMA milestone, we will make a payment of $15 million (less the dollar value of certain cash payments and other deductions) in shares of our common stock. Furthermore, the shares issuable upon achievement of the PMA milestone are bound by a share price floor of $4.50 through expiration.
We have valued the PMA milestone using a probability weighted methodology to capture the probability of milestones being achieved, a change in control event, and no event occurring. After applying the applicable probabilities and payments within the respective payment periods, we applied an appropriate discount rate. We have estimated the value of the PMA milestone to be $9.0 million.
Income Taxes
Our consolidated balance sheets reflect net deferred tax assets that primarily represent the tax benefit of net operating loss carryforwards and credits and timing differences between book and tax recognition of certain revenue and expense items, net of a valuation allowance. When it is more likely than not that all or some portion of deferred tax assets may not be realized, we establish a valuation allowance for the amount that may not be realized. Each quarter, we evaluate the need to retain all or a portion of the valuation allowance on our net deferred tax assets. Our evaluation considers historical earnings, estimated future taxable income and ongoing prudent and feasible tax planning strategies. Adjustments to the valuation allowance increase or decrease net income/(loss) in the period such adjustments are made. If our estimates require adjustments, it could have a significant impact on our consolidated financial statements.
We continually review the adequacy and necessity of the valuation allowance. If it is more likely than not that we would not realize the deferred tax benefits, then all or a portion of the valuation allowance may need to be re-established. Changes in tax laws and rates could also affect recorded deferred tax assets in the future. Management is not aware of any such changes that would have a material effect on our consolidated financial statements.
Stock-based compensation
We recognize compensation expense over a stock option award vesting period based on the fair value of the award at the date of grant. We use the Black-Scholes option pricing model to value stock option grants. The fair value for awards that are expected to vest is then amortized on a straight-line basis over the requisite service period of the award, which is generally the option vesting term. The amount of expense attributed is net of an estimated forfeiture rate, which is updated as appropriate. This option pricing model requires the input of highly subjective assumptions, including the expected volatility of our common stock, pre-vesting forfeiture rate and the option’s expected life. The financial statements include such amounts based on our best estimates and judgments.
We amortize restricted stock grants on a straight line basis over the vesting period of the grant. If vesting is dependent on reaching certain milestones, or adjusted based on the timing of reaching milestones, we use significant judgment in estimating the likelihood and timing of achieving the milestones. Each period, we will reassess the likelihood and estimate the timing of reaching the milestones, and will adjust expense accordingly.
Results of Operations
Comparison of Years Ended December 31, 2010 and 2009
Sales. Sales increased 28% to $67.3 million in 2010 from $52.4 million in 2009. Domestic sales increased from $43.7 million to $55.4 million. The increase in domestic sales was primarily due to the expanded size and increased productivity of our sales force, and the market introduction of new Powerlink sizes and Powerfit aortic extensions.
Sales to distributors outside the United States increased from $8.8 million in 2009 to $11.8 million in 2010. This increase was driven primarily by the release of the IntuiTrak delivery system to most of our international distributors.
We expect that product sales will increase in 2011 by an estimated 16% to 22% from 2010, to $78 million to $82 million. Based on the timing of new product launches and continued improvements in sales force productivity, we expect that the majority of the revenue growth will be weighted in the second half of the year. Outside the United States, we expect growth in each of our major markets of Europe, Asia, and South America.
Cost of Revenue. Cost of revenue, which includes labor, overhead, materials and parts, rent, depreciation, small tools and supplies, samples for destructive testing, and utilities, among other items, increased 14% from $13.2 million in 2009 to $15.0 million in 2010, due to an increase in the volume of Endologix ELG sales. As a percentage of revenue, cost of revenue decreased to 22% in 2010 from 25% in 2009. The percentage decline in the cost of revenue was due to a more favorable product mix, volume related efficiencies, and utilization of our in-house ePTFE graft material for products sold to our
27
distributor in Japan.
Gross Profit. Gross profit increased 33% to $52.2 million in 2010 from $39.3 million in 2009. The increase in gross profit resulted from higher sales in 2010 as compared to 2009 and an increase in gross profit as a percentage of revenue from 75% in 2009 to 78% in 2010. The increase in gross profit percentage was due to the introduction of new Powerlink sizes and Powerfit aortic extensions into the domestic market and cost efficiencies due to higher volume.
We believe that gross profit dollars will increase in 2011 due to higher commercial sales of our Powerlink System both in and outside of the United States, a full year of impact from the new products launched in mid-2010, and due to the introduction of additional new products in the second half of 2011. We expect that gross profit as a percentage of revenues in 2011 will be largely unchanged from 2010.
Research, Development and Clinical. Research, development and clinical expenses increased by 70% to $11.2 million from $6.6 million in 2009. The increase primarily resulted from costs associated with enhancing and expanding our Endologix ELG product line, our acquisition of balloon expandable stent technology, our acquisition of the Nellix technology, and costs associated with our percutaneous endovascular aneurysm repair, or PEVAR, clinical trial.
We expect that these expenses will continue to significantly increase in 2011 as we pursue opportunities to develop additional new products for the treatment of aortic disorders, particularly the development of the Nellix technology, and costs associated with the PEVAR clinical trial.
Marketing and Sales. Marketing and sales expenses increased by 20% to $31.9 million from $26.5 million in 2009. This increase was due to higher variable compensation expense on the 28% growth in domestic sales revenue and an increase in the number of staffed sales territories.
We expect that marketing and sales expenses will increase in 2011 due to higher commission expense on the expected increase in sales, higher compensation costs associated with the expected increase in the number of domestic sales territories, and costs associated with the development of a direct sales organization in Europe during 2011.
General and Administrative. General and administrative expenses increased by 57% to $13.4 million from $8.6 million in 2009. The increase was due to $3.75 million of acquisition and diligence costs associated with our acquisition of Nellix, and $1.55 million in legal costs associated with patent disputes. We expect that general and administrative expenses in 2011 will decrease from 2010. Legal costs associated with patent disputes are expected to be greater in 2011 than in 2010, but the costs related to our acquisition of Nellix are non-recurring.
Other Expense. Other expense increased 74% to $160,000 in 2010 from $92,000 in 2009. The increase in other expense was primarily due to losses related to foreign currency exchange offset by lower interest expense due to lower debt. We do not expect to incur any significant other income or expense in 2011.
Income Tax Benefit. We recorded an income tax benefit of $15.0 million in the year ended December 31, 2010. The income tax benefit is related to the release of valuation allowances of approximately $15.0 million primarily due to additional deferred tax liabilities related to the IPR&D we recorded from the Nellix acquisition.
Comparison of Years Ended December 31, 2009 and 2008
Sales. Sales increased 39% to $52.4 million in 2009 from $37.6 million in 2008 primarily due to the increased productivity of our sales force, the introduction of new products, including Powerlink XL, our suprarenal proximal extension, and the IntuiTrak delivery system, and increased physician acceptance of the Powerlink System. Domestic sales increased from $31.9 million to $43.7 million, and sales to distributors outside the United States increased from $5.7 million in 2008 to $8.8 million in 2009. This increase was driven primarily by the introduction of IntuiTrak to most of our international distributors. Additionally, we had higher sales to our distributors in South America and Japan and an initial sale generating stocking order from our distributor in China.
Cost of Revenue. The cost of revenue, which includes labor, overhead, materials and parts, rent, depreciation, small tools and supplies, samples for destructive testing, and utilities, among other items, increased 27% from $10.4 million in 2008 to $13.2 million in 2009. The increase was primarily attributable to an increase in the volume of Powerlink System sales, partially offset by efficiencies in our manufacturing process.
Gross Profit. Gross profit increased 44% to $39.3 million in 2009 from $27.3 million in 2008. The increase in gross profit resulted from higher sales in 2009 as compared to 2008 and from a favorable product mix due to new product introductions, which have a higher average selling price. Gross profit as a percentage of revenue increased to 75% in 2009 from 72% in 2008
28
for these reasons.
Research, Development and Clinical. Research, development and clinical expenses increased by 8% to $6.6 million from $6.1 million in 2008. The increase primarily resulted from costs associated with the development of new products for the treatment of aortic disorders.
Marketing and Sales. Marketing and sales expenses increased by 11% to $26.5 million from $23.8 million in 2008. This increase was due to higher sales commission payouts on the 37% growth in domestic sales revenue and an increase in marketing efforts related to our product launches in 2009.
General and Administrative. General and administrative expenses decreased by 10% to $8.6 million from $9.5 million in 2008. The decrease was primarily due to $700,000 in costs associated with our chief executive officer succession, which occurred in May 2008, and significant legal fees that occurred in 2008. These reductions were partially offset by higher stock based compensation charges and incentive compensation accruals based on performance metrics in 2009.
Other Income/(expense). Other income/(expense) decreased 267% to ($92,000) in 2009 from $55,000 in 2008. An increase in interest income was offset by expenses related to interest expense incurred in the first three quarters of 2009.
Liquidity and Capital Resources
For the years ended December 31, 2010 and 2009, we incurred net earnings (losses) of $10.7 million and $(2.4) million, respectively. As of December 31, 2010, we had an accumulated deficit of approximately $135.5 million. Historically, we have relied on the sale and issuance of equity securities to provide a significant portion of funding for our operations. In August 2009, we completed a sale of our common stock that resulted in net proceeds of approximately $14.8 million. During 2009, we generated positive cash flows from operations for the first time in our history.
In October 2009, we entered into a revolving credit facility with Wells Fargo Bank, National Association, or Wells Fargo, whereby we may borrow up to $10.0 million. All outstanding amounts under the credit facility bear interest at a variable rate equal to the greater of 90 day LIBOR, the federal funds rate, or the lender’s prime rate, plus 1.25%, which is payable on a monthly basis. The unused portion is subject to an unused revolving line facility fee, payable quarterly, in arrears, on a calendar year basis, in an amount equal to 0.2% per annum of the average unused portion of the revolving line, as determined by Wells Fargo. The credit facility also contains customary covenants regarding operations of our business and financial covenants relating to ratios of current assets to current liabilities and tangible net worth during any calendar quarter and is collateralized by all of our assets with the exception of our intellectual property. All amounts owing under the credit facility will become due and payable on April 30, 2012. As of December 31, 2010, we did not have any outstanding borrowings under this credit facility; however, the Company is reporting a tangible net worth of $22 million and, therefore, is not in compliance with one covenant. We have obtained a waiver from our lender whereby the lender has agreed to forbear from enforcing their default rights under the agreement for this specified instance. The waiver does not apply to any subsequent breaches of the same provision nor any breach of any other provision specified within the agreement.
On December 10, 2010, we issued and sold to Essex Woodlands Fund VII, and Essex Woodlands Fund VII purchased from us, an aggregate of 3,170,577 unregistered shares of our common stock, at purchase price of $4.731 per share, resulting in gross proceeds to us of $15.0 million.
At December 31, 2010, we had cash and cash equivalents of $38.2 million. We used $4.1 million of operating cash flow in 2010. We believe that our current cash balance, and borrowings available under our credit facility, will be sufficient to meet anticipated cash needs for operating and capital expenditures through at least December 31, 2011. If we do not realize expected revenue and gross profit margin levels, are unable to manage our operating expenses in line with our revenues, or cannot maintain our days sales outstanding accounts receivable level, we may need to obtain additional financing.
We believe that the future growth of our business will depend upon our ability to successfully develop new technologies for the treatment of aortic disorders and bring these technologies to market, and to increase the size and productivity of our direct sales force. In order to achieve these objectives, we may need to seek additional sources of financing. In the event that we require additional funding, we will attempt to raise the required capital through either debt or equity arrangements.
The timing and amount of our future capital requirements will depend on many factors, including:
• | the need for additional capital to fund future development programs or sales force expansion; |
• | the need for additional capital to fund business development acquisition(s); |
• | our requirements for additional facility space or manufacturing capacity; |
• | our requirements for additional information technology infrastructure and systems; and |
29
• | adverse outcome(s) from current or future litigation and the cost to defend such litigation. |
If we are required to obtain additional financing, we may not be able to do so on acceptable terms, if at all. Even if we are able to obtain such financing it may cause substantial dilution for our stockholders, in the case of an equity financing, or may contain burdensome restrictions on the operations of our business, in the case of debt financing.
Accounts Receivable. Trade accounts receivable, net, increased 46% to $12.2 million at December 31, 2010 from $8.3 million at December 31, 2009. The increase was due to the 41% increase in sales in the fourth quarter of 2010 as compared to the fourth quarter of 2009.
Inventories. Inventories increased 51% to $8.4 million at December 31, 2010 from $5.5 million at December 31, 2009. The increase was primarily the result of the increased number of product codes available for sale.
Accounts Payable and Accrued Expenses. Accounts payable and accrued expenses increased 56% to $11.2 million at December 31, 2010 from $7.3 million at December 31, 2009. The increase is primarily attributable higher accruals in 2010 related to incentive bonus and commission programs and accruals related to our acquisition of Nellix.
Cash Provided by/(Used in) Operations. In 2010, cash used in operations was $4.1 million as compared to cash provided by operations of $5.0 million in 2009. The change was primarily attributed to the increases noted above in both operating expenses and working capital.
Cash Used in Investing Activities. Cash used in investing activities increased to $99,000 for the year ended December 31, 2010 from $98,000 for the year ended December 31, 2009. The increase was due to the additional property and equipment purchases in 2010 and a decrease in restricted cash in 2009.
Cash Provided by Financing Activities. Cash provided by financing activities increased to $18.4 million for the year ended December 31, 2010 from $11.5 million for the year ended December 31, 2009 The difference was primarily as a result of a $5.0 million loan being paid off in 2009. In addition, there were more proceeds from the sale of common stock under the employee stock purchase plan and from the exercise of stock options.Off-Balance Sheet Arrangements
We do not maintain any off-balance sheet arrangements.
Commitments. As of December 31, 2010, expected future cash payments related to contractual obligations were as follows:
Total | 2011 | 2012 | 2013 | 2014 | 2015 | ||||||||||||||||||
(In thousands) | |||||||||||||||||||||||
Contractual Obligations: | |||||||||||||||||||||||
Operating lease obligations | $ | 1,019 | $ | 675 | $ | 261 | $ | 64 | $ | 19 | $ | — | |||||||||||
Short term debt | 83 | 83 | — | — | — | — | |||||||||||||||||
Interest expense on borrowings | 2 | 2 | — | — | — | — | |||||||||||||||||
Total | $ | 1,104 | $ | 760 | $ | 261 | $ | 64 | $ | 19 | $ | — | |||||||||||
Item 7A. | Quantitative and Qualitative Disclosures About Market Risk |
We do not believe that we currently have material exposure to interest rate, foreign currency exchange rate or other relevant market risks.
Interest Rate and Market Risk. Our exposure to market risk for changes in interest rates relates primarily to our revolving credit facility. Under our revolving credit facility all outstanding amounts bear interest at a variable rate equal to the greater of 90 day LIBOR, the federal funds rate, or the lender’s prime rate, plus 1.25%. As of December 31, 2010, we had no amounts outstanding under the revolving line of credit. We may be exposed to market risk with respect to the revolving line of credit due to changes in interest rates.
We do not use derivative financial instruments in our investment portfolio. We place our investments with high credit quality issuers and, by policy, limit the amount of credit exposure to any one issuer. We are averse to principal loss and try to ensure the safety and preservation of our invested funds by limiting default risk, market risk, and reinvestment risk. We attempt to mitigate default risk by investing in only the safest and highest credit quality securities and by constantly positioning our portfolio to respond appropriately to a significant reduction in a credit rating of any investment issuer or guarantor. At
30
December 31, 2010, our investment portfolio consisted of money market instruments.
Foreign Currency Transaction Risk. We do not currently have material foreign currency exposure as the majority of our assets are denominated in U.S. currency and our foreign-currency based transaction exchange risk is not material. For the years ended December 31, 2010, 2009, and 2008, we recorded $0, ($49,000), and $194,000, respectively, of foreign currency transaction gains (losses).
Item 8. | Financial Statements and Selected Quarterly Financial Data |
The financial statements required by this Item 8 are set forth at the pages indicated at Item 15(a)(1).
Summarized Quarterly Data
(Unaudited)
March 31 | June 30 | September 30 | December 31 | ||||||||||||
(in thousands, except per share amounts) | |||||||||||||||
2010: | |||||||||||||||
Total revenues | $ | 14,480 | $ | 15,654 | $ | 17,874 | $ | 19,243 | |||||||
Gross profit | 11,119 | 12,042 | 14,052 | 15,007 | |||||||||||
Net earnings (loss) | (225 | ) | (380 | ) | (466 | ) | 11,724 | ||||||||
Basic net earnings (loss) per share | — | (0.01 | ) | (0.01 | ) | 0.23 | |||||||||
Diluted net earnings (loss) per share | — | (0.01 | ) | (0.01 | ) | 0.22 | |||||||||
2009: | |||||||||||||||
Total revenues | $ | 11,834 | $ | 13,168 | $ | 13,777 | $ | 13,662 | |||||||
Gross profit | 8,929 | 9,912 | 10,118 | 10,301 | |||||||||||
Net loss | (1,177 | ) | (425 | ) | (156 | ) | (676 | ) | |||||||
Basic and diluted net loss per share | (0.03 | ) | (0.01 | ) | — | (0.01 | ) | ||||||||
Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
None.
Item 9A. | Controls and Procedures |
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rules 13a-15(f) and 15d-15(f) under the Securities Exchange Act of 1934, as amended, or the Exchange Act. Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of the financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. This process includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of our assets; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures are being made only in accordance with authorizations of our management and directors; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on our financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of the internal control over financial reporting to future periods are subject to risk that the internal control may become inadequate because of changes in conditions, or that the degree of compliance with policies or procedures may deteriorate.
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2010. In making this assessment, it used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control-Integrated Framework. Based on our assessment, we have concluded that, as of December 31, 2010, our internal control over financial reporting was effective based on those criteria.
31
The effectiveness of our internal control over financial reporting as of December 31, 2010 has been audited by PricewaterhouseCoopers LLP, an independent registered public accounting firm, as stated in its report, which is included herein.
Disclosure controls and procedures
We carried out an evaluation, under the supervision and with the participation of our management, including our chief executive officer and chief financial officer, of the effectiveness of our disclosure controls and procedures as of December 31, 2010, pursuant to Rules 13a-15(e) and 15d-15(e) under the Exchange Act. Based on that evaluation, our chief executive officer and chief financial officer have concluded that our disclosure controls and procedures, as of such date, were effective.
Changes in internal control over financial reporting
There has been no change in our internal control over financial reporting during the fourth fiscal quarter that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Item 9B. | Other Information |
None.
32
PART III
Item 10. | Directors, Executive Officers and Corporate Governance |
The information required hereunder is incorporated herein by reference to our Proxy Statement to be filed within 120 days of December 31, 2010 and delivered to stockholders in connection with our Annual Meeting of Stockholders to be held on May 25, 2011.
Item 11. | Executive Compensation |
The information required hereunder is incorporated herein by reference to our Proxy Statement to be filed within 120 days of December 31, 2010 and delivered to stockholders in connection with our Annual Meeting of Stockholders to be held on May 25, 2011.
Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
Certain information required hereunder is incorporated herein by reference to our Proxy Statement to be filed within 120 days of December 31, 2010 and delivered to stockholders in connection with our Annual Meeting of Stockholders to be held on May 25, 2011.
Equity Compensation Plan Information
The following table sets forth information regarding outstanding options and rights and shares reserved for future issuance under our existing equity compensation plans as of December 31, 2010:
Plan Category | Number of Securities to be Issued upon Exercise of Outstanding Options (a) | Weighted Average Exercise Price of Outstanding Options (b) | Number of Securities Remaining Available for Future Issuance Under Equity Compensation Plans (Excluding Securities Reflected in Column A) (c) | ||||||
Equity compensation plans approved by security holders: | |||||||||
2006 Stock Incentive Plan | 5,040,797 | $ | 3.55 | 1,347,972 | |||||
1996 Stock Option/ Stock Issuance Plan | 985,542 | $ | 5.29 | — | |||||
2010 Stock Acquisition Plan | 480,000 | $ | — | 34,000 | |||||
2006 Employee Stock Purchase Plan | — | — | 738,613 | ||||||
Total | 6,506,339 | $ | 3.56 | 2,120,585 | |||||
Item 13. | Certain Relationships and Related Transactions, and Director Independence |
The information required hereunder is incorporated herein by reference to our Proxy Statement to be filed within 120 days of December 31, 2010 and delivered to stockholders in connection with our Annual Meeting of Stockholders to be held on May 25, 2011.
Item 14. | Principal Accountant Fees and Services |
The information required hereunder is incorporated herein by reference to our Proxy Statement to be filed within 120 days of December 31, 2000 and delivered to stockholders in connection with our Annual Meeting of Stockholders to be held on May 25, 2011.
33
PART IV
Item 15. | Exhibits, Financial Statement Schedules |
(a) | The following documents are filed as a part of this Annual Report on Form 10-K: |
1. | Financial Statements. |
Report of Independent Registered Public Accounting Firm
Consolidated Balance Sheets — December 31, 2010 and 2009
Consolidated Statements of Operations for the years ended December 31, 2010, 2009 and 2008
Consolidated Statements of Stockholders’ Equity and Comprehensive Loss for the years ended December 31, 2010, 2009 and 2007
Consolidated Statements of Cash Flows for the years ended December 31, 2010, 2009 and 2008
Notes to Consolidated Financial Statements for the years ended December 31, 2010, 2009 and 2008
2. | Financial Statement Schedule. |
Schedule II — Valuation and Qualifying Accounts
Schedules not listed above have been omitted because they are not applicable or are not required to be set forth herein as such information is included in the Consolidated Financial Statements or the notes thereto.
34
3. | Exhibits. |
The following exhibits are filed as part of this Annual Report on Form 10-K:
Exhibit | ||||
Number | Description | |||
2.1 | Agreement and Plan of Merger and Reorganization, dated October 27, 2010, by and among Endologix, Nellix Acquisition Corporation, Nellix, Inc., certain of Nellix, Inc.’s stockholders listed therein and Essex Woodlands Health Ventures, Inc., as representative of Nellix, Inc.’s stockholders (Incorporated by reference to Exhibit 2.1 to Endologix Current Report on Form 8-K, filed with the SEC on October 27, 2010). | |||
3.1 | Amended and Restated Certificate of Incorporation (Incorporated by reference to Exhibit 3.1 to Endologix Quarterly Report on Form 10-Q, filed with the SEC on July 28, 2009). | |||
3.2 | Amended and Restated Bylaws, as amended (Incorporated by reference to Exhibit 3.1 to Endologix Current Report on Form 8-K, filed with the SEC on December 14, 2010). | |||
4.1 | Specimen Certificate of Common Stock (Incorporated by reference to Exhibit 4.1 to Amendment No. 2 to Endologix Registration Statement on Form S-1, No. 333-04560, filed with the SEC on June 10, 1996). | |||
10.1 | (2) | 1997 Supplemental Stock Option Plan (Incorporated by reference to Exhibit 99.1 to Endologix Registration Statement on Form S-8, No. 333-42161, filed with the SEC on December 12, 1997). | ||
10.2 | (2) | 1996 Stock Option/Stock Issuance Plan (Incorporated by reference to Exhibit 4.1 to Endologix Registration Statement on Form S-8, No. 333-122491, filed with the SEC on February 2, 2005). | ||
10.3 | (2) | 2006 Stock Incentive Plan (Incorporated by reference to Exhibit 10.1 to Endologix Current Report on Form 8-K, filed with the SEC on May 26, 2010). | ||
10.4 | (2) | Form of Stock Option Agreement under 2006 Stock Incentive Plan (Incorporated by reference to Exhibit 10.1 to Endologix Quarterly Report on Form 10-Q, filed with the SEC on November 9, 2006). | ||
10.5 | (2) | Form of Restricted Stock Award Agreement under 2006 Stock Incentive Plan (Incorporated by reference to Exhibit 10.2 to Endologix Quarterly Report on Form 10-Q, filed with the SEC on November 9, 2006). | ||
10.6 | (2) | 2006 Employee Stock Purchase Plan (Incorporated by reference to Exhibit 10.1 to Endologix Current Report on Form 8-K, filed with the SEC on June 17, 2009). | ||
10.7 | Form of Indemnification Agreement entered into with Endologix officers and directors (Incorporated by reference to Exhibit 10.41 to Endologix Quarterly Report on Form 10-Q, No. 000-28440, filed with the SEC on November 13, 2002). | |||
10.8 | Standard Industrial/Commercial Single-Tenant Lease — Net, dated November 2, 2004, by and between Endologix and Del Monico Investments, Inc. (Incorporated by reference to Exhibit 10.46 to Endologix Current Report on Form 8-K, No. 000-28440, filed with the SEC on November 24, 2004). | |||
35
Exhibit | |||||
Number | Description | ||||
10.8.1 | Addendum No. 2 to Standard Industrial/Commercial Single-Tenant Lease — Net, by and between Endologix, Inc. and Del Monico Investments, Inc., dated June 9, 2009 (Incorporated by reference to Exhibit 10.1 to Endologix Quarterly Report on Form 10-Q, filed with the SEC on November 2, 2009). | ||||
10.9 | (2) | Offer Letter, dated April 28, 2008, between Endologix and John McDermott (Incorporated by reference to Exhibit 10.1 to Endologix Current Report on Form 8-K, filed with the SEC on May 16, 2008). | |||
10.1 | (2) | Employment Agreement, dated as of December 29, 2008, by and between Endologix and John McDermott (Incorporated by reference to Exhibit 10.1 to Endologix Current Report on Form 8-K, filed with the SEC on January 2, 2009). | |||
10.11 | (2) | Employment Agreement, dated as of December 29, 2008, by and between Endologix and Robert J. Krist (Incorporated by reference to Exhibit 10.2 to Endologix Current Report on Form 8-K, filed with the SEC on January 2, 2009). | |||
10.12 | (2) | Employment Agreement, dated as of December 29, 2008, by and between Endologix and Stefan G. Schreck, Ph.D. (Incorporated by reference to Exhibit 10.3 to Endologix Current Report on Form 8-K, filed with the SEC on January 2, 2009). | |||
10.13 | (2) | Employment Agreement, dated as of December 29, 2008, by and between Endologix and Janet Fauls (Incorporated by reference to Exhibit 10.4 to Endologix Current Report on Form 8-K, filed with the SEC on January 2, 2009). | |||
10.14 | Standard Industrial/Commercial Multi -Tenant Lease — Net, by and between Endologix, Inc. and Four-In-One Associates, dated August 28, 2009 (Incorporated by reference to Exhibit 10.2 to Endologix Quarterly Report on Form 10-Q, filed with the SEC on November 2, 2009). | ||||
10.15 | Credit Agreement, dated October 30, 2009, by and between Endologix and Wells Fargo Bank, National Association (Incorporated by reference to Exhibit 10.16 to Endologix Annual Report on Form 10-K, filed with the SEC on March 5, 2010). | ||||
10.16 | (2) | Employment Agreement, dated as of April 13, 2009, by and between Endologix, Inc. and Joseph DeJohn (Incorporated by reference to Exhibit 10.17 to Endologix Annual Report on Form 10-K, filed with the SEC on March 5, 2010). | |||
10.17 | Securities Purchase Agreement, dated as of October 27, 2010, by and between Endologix and Essex Woodlands Health Ventures Fund VII, L.P. (Incorporated by reference to Exhibit 10.1 to Endologix Current Report on Form 8-K, filed with the SEC on October 27, 2010). | ||||
10.17.1 | Amendment to Securities Purchase Agreement, dated as of December 9, 2010, by and between Endologix and Essex Woodlands Health Ventures Fund VII, L.P. (Incorporated by reference to Exhibit 10.1 to Endologix Current Report on Form 8-K, filed with the SEC on December 14, 2010). | ||||
10.18 | (2 | ) | Employment Agreement, dated as of December 10, 2010, by and between Endologix and Robert D. Mitchell (Incorporated by reference to Exhibit 10.2 to Endologix Current Report on Form 8-K, filed with the SEC on December 14, 2010). | ||
14 | Code of Ethics for Chief Executive Officer and Principal Financial Officers (Incorporated by reference to Exhibit 14 to Endologix Annual Report on Form 10-K, No. 000-28440, filed with the SEC on March 26, 2004). | ||||
21.1 | List of Subsidiaries. | ||||
23.1 | Consent of Independent Registered Public Accounting Firm. | ||||
24.1 | Power of Attorney (included on signature page hereto). | ||||
31.1 | Certification of Chief Executive Officer Pursuant to Rule 13a-14(a)/15d-14(a) under the Securities Exchange Act of 1934. | ||||
31.2 | Certification of Chief Financial Officer Pursuant to Rule 13a-14(a)/15d-14(a) under the Securities Exchange Act of 1934. | ||||
32.1 | Certification of Chief Executive Officer Pursuant to Rule 13a-14(b)/15d-14(b) under the Securities Exchange Act of 1934 and 18 U.S.C. Section 1350. | ||||
32.2 | Certification of Chief Financial Officer Pursuant to Rule 13a-14(b)/15d-14(b) under the Securities Exchange Act of 1934 and 18 U.S.C. Section 1350. | ||||
(1) | Portions of this exhibit are omitted and were filed separately with the Securities and Exchange Commission pursuant to Endologix application requesting confidential treatment under Rule 24b-2 of the Securities Exchange Act of 1934. |
(2) | These exhibits are identified as management contracts or compensatory plans or arrangements of Endologix pursuant to Item 15(a)(3) of Form 10-K. |
36
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this amendment to be signed on its behalf by the undersigned, thereunto duly authorized.
ENDOLOGIX, INC. | ||
By: | /S/ JOHN MCDERMOTT | |
John McDermott Chief Executive Officer and Director (Principal Executive Officer) | ||
Date: March 16, 2011
POWER OF ATTORNEY
We, the undersigned directors and officers of Endologix, Inc., do hereby constitute and appoint John McDermott and Robert J. Krist, and each of them, as our true and lawful attorneys-in-fact and agents with power of substitution, to do any and all acts and things in our name and behalf in our capacities as directors and officers and to execute any and all instruments for us and in our names in the capacities indicated below, which said attorney-in-fact and agent may deem necessary or advisable to enable said corporation to comply with the Securities Exchange Act of 1934, as amended, and any rules, regulations and requirements of the Securities and Exchange Commission, in connection with this Annual Report on Form 10-K, including specifically but without limitation, power and authority to sign for us or any of us in our names in the capacities indicated below, any and all amendments (including post-effective amendments) hereto; and we do hereby ratify and confirm all that said attorney-in-fact and agent, shall do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Signature | Title | Date | ||
/s/ JOHN MCDERMOTT | Chief Executive Officer and Director | March 16, 2011 | ||
(John McDermott) | (Principal Executive Officer) | |||
/S/ ROBERT J. KRIST | Chief Financial Officer and Secretary | March 16, 2011 | ||
(Robert J. Krist) | (Principal Financial and Accounting Officer) | |||
/S/ FRANKLIN D. BROWN | Chairman of the Board | March 16, 2011 | ||
(Franklin D. Brown) | ||||
/S/ RODERICK DE GREEF | Director | March 16, 2011 | ||
(Roderick de Greef) | ||||
/S/ DAN LEMAITRE | Director | March 16, 2011 | ||
(Dan Lemaitre) | ||||
/s/ THOMAS C. WILDER | Director | March 16, 2011 | ||
(Thomas C. Wilder) | ||||
/S/ JEFFREY F. O’DONNELL | Director | March 16, 2011 | ||
(Jeffrey F. O’Donnell) | ||||
/s/ GUIDO J. NEELS | Director | March 16, 2011 | ||
(Guido J. Neels) | ||||
/s/ GREGORY D. WALLER | Director | March 16, 2011 | ||
(Gregory D. Waller) | ||||
37
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of Endologix, Inc.:
In our opinion, the consolidated financial statements listed in the index appearing under Item 15(a)(1) present fairly, in all material respects, the financial position of Endologix, Inc. and its subsidiaries at December 31, 2010 and 2009, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2010 in conformity with accounting principles generally accepted in the United States of America. In addition, in our opinion, the financial statement schedule listed in the index appearing under Item 15(a)(2), presents fairly, in all material respects, the information set forth therein when read in conjunction with the related consolidated financial statements. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company's management is responsible for these financial statements and financial statement schedule, for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in Management's Report on Internal Control over Financial Reporting appearing under Item 9A. Our responsibility is to express opinions on these financial statements, on the financial statement schedule, and on the Company's internal control over financial reporting based on our integrated audits. We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ PricewaterhouseCoopers LLP
Orange County, California
March 16, 2011
F-1
ENDOLOGIX, INC.
CONSOLIDATED BALANCE SHEETS
(In thousands, except share and per share amounts)
December 31, | |||||||
2010 | 2009 | ||||||
ASSETS | |||||||
Current assets: | |||||||
Cash and cash equivalents | $ | 38,191 | $ | 24,065 | |||
Accounts receivable, net of allowance for doubtful accounts of $118 and $97 | 12,212 | 8,342 | |||||
Other receivables | 515 | 3 | |||||
Inventories | 8,350 | 5,540 | |||||
Other current assets | 560 | 389 | |||||
Total current assets | 59,828 | 38,339 | |||||
Property and equipment, net | 2,429 | 2,089 | |||||
Goodwill | 27,073 | 4,631 | |||||
Intangibles, net | 44,863 | 6,104 | |||||
Other assets | 182 | 129 | |||||
Total assets | $ | 134,375 | $ | 51,292 | |||
LIABILITIES AND STOCKHOLDERS’ EQUITY | |||||||
Current liabilities: | |||||||
Accounts payable and accrued expenses | $ | 11,160 | $ | 7,199 | |||
Current portion of long term debt | 83 | 79 | |||||
Total current liabilities | 11,243 | 7,278 | |||||
Long-term liabilities: | |||||||
Long term debt | — | 83 | |||||
Other long-term liabilities | 29,229 | 1,051 | |||||
Total long-term liabilities | 29,229 | 1,134 | |||||
Total liabilities | 40,472 | 8,412 | |||||
Commitments and contingencies (Note 8) | |||||||
Stockholders’ equity: | |||||||
Preferred stock, $0.001 par value; 5,000,000 shares authorized, no shares issued and outstanding | — | — | |||||
Common stock, $0.001 par value; 75,000,000 shares authorized, 56,896,000 and 49,152,000 shares issued, and 56,401,000 and 48,657,000 outstanding | 57 | 49 | |||||
Additional paid-in capital | 230,017 | 189,656 | |||||
Accumulated deficit | (135,510 | ) | (146,164 | ) | |||
Treasury stock, at cost, 495,000 shares | (661 | ) | (661 | ) | |||
Total stockholders’ equity | 93,903 | 42,880 | |||||
Total liabilities and stockholders’ equity | $ | 134,375 | $ | 51,292 | |||
The accompanying notes are an integral part of these Consolidated Financial Statements.
F-2
ENDOLOGIX, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
Year Ended December 31, | |||||||||||
2010 | 2009 | 2008 | |||||||||
(In thousands, except per share amounts) | |||||||||||
Total revenue | $ | 67,251 | $ | 52,441 | $ | 37,664 | |||||
Cost of revenue | 15,030 | 13,181 | 10,380 | ||||||||
Gross profit | 52,221 | 39,260 | 27,284 | ||||||||
Operating costs and expenses: | |||||||||||
Research and development | 11,166 | 6,569 | 6,082 | ||||||||
Marketing and sales | 31,869 | 26,483 | 23,794 | ||||||||
General and administrative | 13,410 | 8,550 | 9,455 | ||||||||
Total operating costs and expenses | 56,445 | 41,602 | 39,331 | ||||||||
Loss from operations | (4,224 | ) | (2,342 | ) | (12,047 | ) | |||||
Other income(expense): | |||||||||||
Interest income | 30 | 48 | 170 | ||||||||
Interest expense | (16 | ) | (192 | ) | (106 | ) | |||||
Other income(expense), net | (174 | ) | 52 | (9 | ) | ||||||
Total other income(expense) | (160 | ) | (92 | ) | 55 | ||||||
Net loss before income tax | (4,384 | ) | (2,434 | ) | (11,992 | ) | |||||
Income tax benefit | 15,037 | — | — | ||||||||
Net earnings (loss) | $ | 10,653 | $ | (2,434 | ) | (11,992 | ) | ||||
Basic net earnings (loss) per share | $ | 0.22 | $ | (0.05 | ) | $ | (0.28 | ) | |||
Shares used in computing basic net (earnings) loss per share | 48,902 | 45,194 | 43,045 | ||||||||
Diluted net earnings (loss) per share | $ | 0.21 | $ | (0.05 | ) | $ | (0.28 | ) | |||
Shares used in computing diluted net earnings (loss) per share | 50,544 | 45,194 | 43,045 | ||||||||
The accompanying notes are an integral part of these Consolidated Financial Statements.
F-3
ENDOLOGIX, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY AND COMPREHENSIVE INCOME (LOSS)
Common Stock | Additional Paid-In Capital | Accumulated Deficit | Treasury Stock | Stockholders’ Equity | Comprehensive Income (Loss) | |||||||||||||||||||||
Shares | Amount | |||||||||||||||||||||||||
(In thousands) | ||||||||||||||||||||||||||
Balance at December 31, 2007 | 43,453 | $ | 43 | $ | 166,912 | $ | (131,738 | ) | $ | (661 | ) | $ | 34,556 | — | ||||||||||||
Exercise of common stock options | 30 | — | 29 | — | — | 29 | ||||||||||||||||||||
Employee stock purchase plan | 357 | — | 480 | — | — | 480 | ||||||||||||||||||||
Amortization of stock compensation expense | — | — | 2,381 | — | — | 2,381 | ||||||||||||||||||||
Grant of restricted stock | 525 | 1 | 5 | — | — | 6 | ||||||||||||||||||||
Amortization expense of restricted stock | — | — | 432 | — | — | 432 | ||||||||||||||||||||
Net loss | — | — | — | (11,992 | ) | — | (11,992 | ) | (11,992 | ) | ||||||||||||||||
Balance at December 31, 2008 | 44,365 | $ | 44 | $ | 170,239 | $ | (143,730 | ) | $ | (661 | ) | $ | 25,892 | $ | (11,992 | ) | ||||||||||
Exercise of common stock options | 238 | — | 782 | — | — | 782 | ||||||||||||||||||||
Employee stock purchase plan | 489 | 1 | 785 | — | — | 786 | ||||||||||||||||||||
Sale of Common Stock | 3,900 | 4 | 14,773 | — | — | 14,777 | ||||||||||||||||||||
Amortization of stock compensation expense | — | — | 2,272 | — | — | 2,272 | ||||||||||||||||||||
Grant of restricted stock | 160 | — | — | — | — | — | ||||||||||||||||||||
Amortization expense of restricted stock | — | — | 772 | — | — | 772 | ||||||||||||||||||||
Amortization expense of non-employee stock options | — | — | 33 | — | — | 33 | ||||||||||||||||||||
Net loss | — | — | — | (2,434 | ) | — | (2,434 | ) | (2,434 | ) | ||||||||||||||||
Balance at December 31, 2009 | 49,152 | $ | 49 | $ | 189,656 | $ | (146,164 | ) | $ | (661 | ) | $ | 42,880 | $ | (2,434 | ) | ||||||||||
Exercise of common stock options | 676 | 1 | 2,242 | — | — | 2,243 | ||||||||||||||||||||
Employee stock purchase plan | 293 | — | 1,117 | — | — | 1,117 | ||||||||||||||||||||
Sale of Common Stock | 3,171 | 3 | 14,987 | — | — | 14,990 | ||||||||||||||||||||
Issuance of Common Stock for acquisition | 3,199 | 3 | 19,385 | — | — | 19,388 | ||||||||||||||||||||
Amortization of stock compensation expense | — | — | 2,215 | — | — | 2,215 | ||||||||||||||||||||
Grant of restricted stock | 405 | 1 | — | — | — | 1 | ||||||||||||||||||||
Amortization expense of restricted stock | — | — | 422 | — | — | 422 | ||||||||||||||||||||
Compensation expense of non-employee stock options | — | — | (7 | ) | — | — | (7 | ) | ||||||||||||||||||
Net earnings | — | — | — | 10,653 | — | 10,653 | 10,653 | |||||||||||||||||||
Balance at December 31, 2010 | 56,896 | $ | 57 | $ | 230,017 | $ | (135,510 | ) | $ | (661 | ) | $ | 93,902 | $ | 10,653 | |||||||||||
The accompanying notes are an integral part of these Consolidated Financial Statements.
F-4
ENDOLOGIX, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
Year Ended December 31, | |||||||||||
2010 | 2009 | 2008 | |||||||||
(In thousands) | |||||||||||
Operating activities: | |||||||||||
Net Income (loss) | $ | 10,653 | $ | (2,434 | ) | $ | (11,992 | ) | |||
Adjustments to reconcile net income (loss) to net cash provided by (used in) operating activities: | |||||||||||
Income tax benefit | (15,067 | ) | |||||||||
Depreciation and amortization | 2,444 | 2,765 | 2,483 | ||||||||
Stock-based compensation | 2,546 | 3,092 | 2,900 | ||||||||
Loss on disposal of assets | — | — | 23 | ||||||||
Changes: | |||||||||||
Accounts receivable | (3,870 | ) | (1,971 | ) | (1,844 | ) | |||||
Inventories | (2,964 | ) | 1,686 | 980 | |||||||
Other receivables and other assets | (672 | ) | 29 | 369 | |||||||
Accounts payable, accrued expenses and long term liabilities | 2,843 | 1,878 | 884 | ||||||||
Net cash provided by (used in) operating activities | (4,087 | ) | 5,045 | (6,197 | ) | ||||||
Investing activities: | |||||||||||
Decrease in restricted cash equivalents | — | 500 | — | ||||||||
Cash acquired in Nellix acquisition | 698 | — | — | ||||||||
Capital expenditures for property and equipment | (861 | ) | (598 | ) | (447 | ) | |||||
Net cash used in investing activities | (163 | ) | (98 | ) | (447 | ) | |||||
Financing activities: | |||||||||||
Proceeds from sale of common stock, net of expenses | 14,990 | 14,777 | — | ||||||||
Proceeds from sale of common stock under employee stock purchase plan | 1,118 | 786 | 497 | ||||||||
Proceeds from exercise of stock options | 2,347 | 782 | 30 | ||||||||
Proceeds (repayments) for capital purchase | (79 | ) | 162 | — | |||||||
Proceeds (repayment) under term loan and line of credit facilities | — | (5,000 | ) | 5,000 | |||||||
Net cash provided by financing activities | 18,376 | 11,507 | 5,527 | ||||||||
Net increase (decrease) in cash and cash equivalents | 14,126 | 16,454 | (1,117 | ) | |||||||
Cash and cash equivalents, beginning of year | 24,065 | 7,611 | 8,728 | ||||||||
Cash and cash equivalents, end of year | $ | 38,191 | $ | 24,065 | $ | 7,611 | |||||
Supplemental Disclosure of Cash Flow Activities: | |||||||||||
Cash paid during the year for interest | $ | 16 | $ | 192 | $ | 106 | |||||
Non-cash investing and financing activities: | |||||||||||
Shares issued for acquisition | $ | 19,385 | $ | — | $ | — | |||||
The accompanying notes are an integral part of these Consolidated Financial Statements.
F-5
ENDOLOGIX, INC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(in thousands, except share or performance unit and per share amounts)
1. Business, Basis of Presentation and Summary of Critical Accounting Policies
Business and Basis of Presentation
Endologix, Inc. (the “Company”) was incorporated in California in March 1992 and reincorporated in Delaware in June 1993. In January 1999, the Company merged with privately held Radiance Medical Systems, Inc. (“former Radiance”), and changed its name to Radiance Medical Systems, Inc. In May 2002, the Company merged with privately held Endologix, Inc., and changed its name to Endologix, Inc.
Since the merger in May 2002, the Company has been engaged in the development, manufacture, sales and marketing of minimally invasive therapies for the treatment of vascular disease. The Company’s primary product is the Powerlink System, a catheter-based alternative treatment for abdominal aortic aneurysms, or AAA. AAA is a weakening of the wall of the aorta, the largest artery of the body.
The consolidated financial statements include the accounts of the Company and its wholly-owned subsidiaries. Intercompany transactions have been eliminated in consolidation. The Company operates in a single business segment.
Summary of Accounting Policies and Use of Estimates
The preparation of financial statements in conformity with generally accepted accounting principles requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, revenues and expenses, and related disclosure of contingent assets and liabilities. On an on-going basis, the Company evaluates its estimates, including those related to collectibility of customer accounts, whether the cost of inventories can be recovered, the value assigned to and estimated useful life of intangible assets, the realization of tax assets and estimates of tax liabilities, contingent liabilities and the potential outcome of litigation. The Company bases its estimates on historical experience and on various other assumptions that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities. Actual results may differ from these estimates under different assumptions or conditions.
Cash and Cash Equivalents
Cash and cash equivalents includes cash on hand, demand deposits and money market funds with original maturities of three months or less from the date of purchase.
Accounts Receivables
Trade accounts receivable are recorded at the invoiced amount and do not bear interest. The allowance for doubtful accounts is the Company’s best estimate of the amount of probable credit losses in existing accounts receivable. The Company determines the allowance based on historical write-off experience. The Company reviews the allowance for doubtful accounts monthly. Past due balances over 90 days and over a specified amount are reviewed individually for collectibility. Account balances are charged off against the allowance when the Company believes it is probable the receivable will not be recovered.
Inventories
The Company values inventory at the lower of the actual cost to purchase or manufacture the inventory or the market value for such inventory. Cost is determined on the first-in, first-out method. The Company regularly reviews inventory quantities in process and on hand and records a provision for obsolete inventory based on actual loss experience and a forecast of product demand compared to the remaining shelf life.
Property and Equipment
Property and equipment are stated at cost and depreciated on a straight-line basis over the estimated useful lives of the assets, with the exception of the Company’s in-house ePTFE manufacturing equipment, which is depreciated by a per unit produced basis and approximates a six year useful life. Leasehold improvements are amortized over the term of the lease or the estimated useful life of the asset, whichever is shorter. Maintenance and repairs are expensed as incurred while renewals or betterments are capitalized. Upon sale or disposition of property and equipment, any gain or loss is included in the statement of operations. The estimated useful lives for furniture and equipment range from three to seven years and the estimated useful life for leasehold improvements is five years.
F-6
ENDOLOGIX, INC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—Continued
(in thousands, except share and per share amounts)
Intangible Assets
Goodwill and other intangible assets with indefinite lives are not subject to amortization but are tested for impairment annually or whenever events or changes in circumstances indicate that the asset might be impaired. The Company most recently performed its annual impairment analysis as of June 30, 2010 and will continue to test for impairment annually as of June 30. No impairment was indicated.
Developed technology is being amortized over its estimated useful life of 10 years. In-process research and development will be amortized upon commencement of commercial sales and it is expected to be amortized over its useful life.
Long-Lived Assets
Long-lived assets and intangible assets with determinate lives are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. The Company evaluates potential impairment by comparing the carrying amount of the asset with the estimated undiscounted future cash flows associated with the use of the asset and its eventual disposition. Should the review indicate that the asset is not recoverable, the Company’s carrying value of the asset would be reduced to its estimated fair value, which is measured by future discounted cash flows.
Contingent Acquisition Consideration Payable
The Company determines the fair value of contingent acquisition consideration payable on the acquisition date using a probability-based income approach utilizing an appropriate discount rate. Changes in the fair value of the contingent acquisition consideration payable are determined each period end and recorded in the intangible asset amortization and contingent consideration on the consolidated statements of operations.
Fair Value of Financial Instruments
The carrying amount of all financial instruments approximates fair value because of the short maturities of the instruments.
Concentrations of Credit Risk and Significant Customers
The Company maintains its cash and cash equivalents in deposit accounts and in money market securities administered by a major financial institution.
The Company sells its products primarily to hospitals and distributors worldwide. The Company performs credit evaluations of its customers’ financial condition and generally does not require collateral from customers. Management believes that an adequate allowance for doubtful accounts has been provided.
No single customer accounted for more than 10% of the Company’s total revenue in 2010, 2009, and 2008.
As of December 31, 2010 and December 31, 2009, no single customer accounted for more than 10% of the Company’s accounts receivable balance.
Product Sales by Geographic Region
The Company had product sales by region, based on where the Company ships the product, as follows:
Year Ended December 31, | |||||||||||
2010 | 2009 | 2008 | |||||||||
United States | $ | 55,443 | $ | 43,682 | $ | 31,950 | |||||
Europe | 4,054 | 2,964 | 3,095 | ||||||||
South America | 3,653 | 2,717 | 1,033 | ||||||||
Asia | 3,334 | 2,870 | 1,370 | ||||||||
Other | 767 | 208 | 183 | ||||||||
$ | 67,251 | $ | 52,441 | $ | 37,631 | ||||||
F-7
ENDOLOGIX, INC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—Continued
(in thousands, except share and per share amounts)
Revenue Recognition
The Company recognizes revenue when all of the following criteria are met:
• | Persuasive evidence of an arrangement exists; |
• | The sales price is fixed or determinable; |
• | Collection of the relevant receivable is probable at the time of sale; and |
• | Products have been shipped or used and the customer has taken ownership and assumed risk of loss. |
For domestic sales, the Company is generally able to recognize revenue upon completion of a procedure, when the product is implanted in a patient, as this represents when the product has been used For international sales, the Company is able to recognize revenue at the time of shipment of the products to a distributor, as this represents the period that the customer has taken ownership and assumed risk of loss.
The Company does not offer rights of return or price protection and has no post delivery obligations other than its specified warranty.
Shipping Costs
Shipping costs billed to customers are included in revenue with the related costs in costs of goods sold.
Foreign Currency
The assets and liabilities of foreign subsidiaries are translated at the rates of exchange at the balance sheet date. The income and expense items of these subsidiaries are translated at average monthly rates of exchange. Gains and losses resulting from foreign currency transactions, which are denominated in a currency other than the respective entity’s functional currency are included in the consolidated statement of operations.
Income Taxes
The Company records the estimated future tax effects of temporary differences between the tax basis of assets and liabilities and amounts reported in the financial statements, as well as operating losses and tax credit carry forwards. It has recorded a full valuation allowance to reduce its deferred tax assets to zero, because the Company believes that, based upon a number of factors, it is more likely than not that the deferred tax assets will not be realized. If the Company were to determine that it would be able to realize their deferred tax assets in the future, an adjustment to the valuation allowance on its deferred tax assets would increase net income in the period such determination was made.
Net Earnings(Loss) Per Share
Net earnings(loss) per common share is computed using the weighted average number of common shares outstanding during the periods presented. Because of the net losses during the years ended December 31, 2009, and 2008, options to purchase the common stock of the Company were excluded from the computation of net loss per share because the effect would have been antidilutive.
If anti-dilutive stock options were included, the number of shares used to compute net earnings(loss) per share would have been increased by approximately 2,761,000 shares, 3,389,000 shares, and 4.802,000 shares, for the years ended December 31, 2010, 2009, and 2008, respectively. Of these amounts, 1,119,000, 2,422,000, and 4,746,000 shares had an exercise price above the average closing price for the years ended December 31, 2010, 2009, and 2008, respectively.
Research and Development Costs
Research and development costs are expensed as incurred.
Product Warranty
Within six months of shipment, certain customers may request replacement of products they receive that do not meet the manufacturer’s product specifications. No other warranties are offered and the Company disclaims responsibility for any consequential or incidental damages associated with the use of the products. Historically, the Company has not experienced a significant amount of costs as a result of its product warranty policy.
F-8
ENDOLOGIX, INC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—Continued
(in thousands, except share and per share amounts)
2. Acquisitions
On December 10, 2010, the Company completed the merger acquisition of Nellix Inc. The total consideration given related to the acquisition follows.
Market value ($6.06 per share at purchase date) | $ | 19,389 | ||
Contingent acquisition consideration payable | 28,200 | |||
Total consideration | $ | 47,589 | ||
The consideration given to Nellix is 3,199,441 shares issued to the former securityholders of Nellix at the market value of $6.06 on the date of the acquisition, December 10, 2010.
The Company also agreed to pay Nellix stockholders additional consideration in future periods if certain sales and development milestones are met. The fair value of the contingent acquisition consideration payments on the acquisition date was $28.2 million and was estimated by applying a probability-based income approach utilizing an appropriate discount rate. This estimation was based on significant inputs that are not observable in the market, referred to as level 3 inputs. As of December 31, 2010, the range of outcomes and assumptions used to develop these estimates have not changed. The contingent payable of $28,200 will be evaluated on a quarterly basis starting in 2011, with adjustments made based upon management's assessment of the probability of meeting performance milestones as set-forth within the Agreement.
One milestone payment referred to above relates to receiving a CE mark and generating a revenue of $10 million outside of the United States, prior to the expiration of a key Nellix patent expected to occur in 2026. The amount owed is dependent on the time it takes to reach the revenue milestone after the CE mark is approved, and is payable in Endologix common stock. The total amount of payment, if the milestone is achieved, can range from $10,000 to $24,000, and the shares granted in this milestone are bound by a share price floor of $3.50 through expiration and a price ceiling of $7.50 for 24 months from receiving a CE Mark. The estimated value of this contingency, after taking into account applicable probabilities and discount rates, is $19.2 million.
The other milestone payment referred to above represents the contingency which concerns receiving PMA. PMA is the FDA process of scientific and regulatory review to evaluate the safety and effectiveness of Class III medical devices. The right to receive the PMA milestone extends through the expiration date of a key Nellix patent, expected to occur in 2026. If the PMA milestone is achieved, payment of $15,000 will be made in Endologix common shares, bound by a share price floor of $4.50. The estimated value of this contingency, after taking into account applicable probabilities and discount rates, is $9.0 million.
Purchase Price Allocation | ||||
Cash | $ | 698 | ||
Receivables and other assets | 65 | |||
Property, plant, and equipment | 382 | |||
Patent | 65 | |||
IPR&D | 40,100 | |||
Accounts payable and other liabilities | (1,096 | ) | ||
Deferred tax liability | (15,067 | ) | ||
Net assets acquired | $ | 25,147 | ||
Consideration given | $ | 47,589 | ||
Less: Net assets acquired | 25,147 | |||
Goodwill | $ | 22,442 | ||
Goodwill resulting from the merger agreement is largely attributable to the unique, patented technology which will enable the Company to reach patients with anatomies that could only be repaired through open surgery. Purchased identifiable
F-9
ENDOLOGIX, INC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—Continued
(in thousands, except share and per share amounts)
intangible assets includes $40.1 million of in-process research and development which will be amortized on a straight-line basis upon commencement of commercial sales for the Nellix product.
Nellix has been recorded in the Company's consolidated financial statements since the date of acquisition. For the period from acquisition, December 10, 2010 to December 31, 2010, Nellix did not have revenue and incurred a net loss of approximately $0.8 million. If Nellix had been included in the Company's results of operations since January 1, 2009, the unaudited consolidated revenue, net loss, and earnings per share would be as follows:
December 31, | |||||||
2010 | 2009 | ||||||
Revenue | $ | 67,251 | $ | 52,441 | |||
Net Loss | (3,388 | ) | (13,684 | ) | |||
Basic Loss per Share | (0.07 | ) | (0.30 | ) | |||
These unaudited pro forma amounts are not necessarily indicative of the results that would have occurred if the acquisition had been completed at the beginning of 2009, nor are they indicative of the future operating results from the combined companies.
3. Inventories
Inventories consisted of the following:
December 31, | |||||||
2010 | 2009 | ||||||
Raw materials | $ | 2,051 | $ | 1,866 | |||
Work in process | 1,851 | 1,414 | |||||
Finished goods | 4,448 | 2,260 | |||||
$ | 8,350 | $ | 5,540 | ||||
4. Property and Equipment
Property and equipment consisted of the following:
December 31, | |||||||
2010 | 2009 | ||||||
Furniture and equipment | $ | 6,017 | $ | 5,321 | |||
Leasehold improvements | 2,281 | 2,127 | |||||
Contruction in Progress | 19 | — | |||||
8,317 | 7,448 | ||||||
Less: accumulated depreciation | (5,888 | ) | (5,359 | ) | |||
$ | 2,429 | $ | 2,089 | ||||
Depreciation expense for property and equipment for the years ended December 31, 2010, 2009, and 2008 was $904, $1,361 and $1,078, respectively.
F-10
ENDOLOGIX, INC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—Continued
(in thousands, except share and per share amounts)
5. Goodwill and Intangibles
Goodwill resulting from the acquisition of Nellix totaled $22.4 million. Existing goodwill totaled $4.6 million.
Intangible assets other than goodwill consisted of the following:
December 31, | |||||
2010 | 2009 | ||||
Developed technology | 14,050 | 14,050 | |||
Accumulated amortization | (12,060 | ) | (10,654 | ) | |
Developed technology, net | 1,990 | 3,396 | |||
Patent | 100 | — | |||
Accumulated amortization | (35 | ) | — | ||
Patent, net | 65 | ||||
In-process research and development | 40,100 | — | |||
Trademarks and trade names | 2,708 | 2,708 | |||
Intangible assets, net | 44,863 | 6,104 | |||
On December 10, 2010, the Company acquired $40.1 million of in-process research and development as part of the Nellix acquisition. Amortization of the in-process research and development will start once the Nellix product has commercial sales. Amortization expense for intangible assets for the years ended December 31, 2010, 2009, and 2008 was $1,406, $1,404 and $1,405, respectively. Estimated amortization in future years is as follows:
Year Ending December 31, | |||
2011 | $ | 1,425 | |
2012 | 605 | ||
2013 | 20 | ||
Thereafter | 5 | ||
$ | 2,055 | ||
6. Accounts Payable and Accrued Expenses
Accounts payable and accrued expenses consisted of the following:
December 31, | |||||||
2010 | 2009 | ||||||
Accounts payable | $ | 5,370 | $ | 2,150 | |||
Accrued payroll and related expenses | 4,974 | 4,629 | |||||
Accrued performance units | 336 | 183 | |||||
Accrued clinical expenses | 248 | 79 | |||||
Customer deposits | 212 | 122 | |||||
Other accrued expenses | 20 | 36 | |||||
$ | 11,160 | $ | 7,199 | ||||
F-11
ENDOLOGIX, INC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—Continued
(in thousands, except share and per share amounts)
7. Long Term Liabilities
Long term liabilities consisted of the following:
December 31, | |||||||
2010 | 2009 | ||||||
Contingent acquisition consideration payable | $ | 28,200 | $ | — | |||
Deferred income taxes | 1,029 | 1,029 | |||||
Other | 83 | 184 | |||||
Total long-term liabilities | 29,312 | 1,213 | |||||
Current portion of long-term debt | (83 | ) | (79 | ) | |||
Long term portion | 29,229 | 1,134 | |||||
In October 2009, the Company entered into a revolving credit facility with Wells Fargo Bank, National Association (“Wells Fargo”), whereby the Company may borrow up to $10.0 million. All outstanding amounts under the credit facility bear interest at a variable rate equal to the greater of 90 day LIBOR, the federal funds rate, or lender’s prime rate, plus 1.25%, which is payable on a monthly basis. The unused portion is subject to an unused revolving line facility fee, payable quarterly, in arrears, on a calendar year basis, in an amount equal to 0.2% per annum of the average unused portion of the revolving line, as determined by Wells Fargo. The credit facility also contains customary covenants regarding operations of the business and financial covenants, including requiring the Company to maintain a tangible net worth of $23 million, and is collateralized by all of its assets with the exception of its intellectual property. All amounts owing under the credit facility will become due and payable on April 30, 2012. As of December 31, 2010, the Company did not have any outstanding borrowings under this credit facility; however, the Company is reporting a tangible net worth of $22 million and, therefore, is not in compliance with one covenant. We have obtained a waiver from our lender whereby the lender has agreed to forbear from enforcing their default rights under the agreement for this specified instance. The waiver does not apply to any subsequent breaches of the same provision nor any breach of any other provision specified within the agreement.
Contingent Acquisition Consideration Payable
As further disclosed in Note 2 regarding the acquisition of Nellix, the Company recorded a contingent acquisition consideration payable related to potential future payments based on certain milestones being met.
8. Commitments and Contingencies
Operating Leases
The Company leases its administrative, research and manufacturing facility and certain equipment under long-term, non-cancelable lease agreements that have been accounted for as operating leases. Certain of these leases include renewal options and require the Company to pay operating costs, including property taxes, insurance and maintenance as proscribed by the agreements.
Future minimum payments by year under non-cancelable operating leases with initial terms in excess of one year were as follows as of December 31, 2010:
Year Ending December 31, | |||
2011 | 760 | ||
2012 | 261 | ||
2013 | 64 | ||
2014 | 19 | ||
2015 | — | ||
Thereafter | — | ||
$ | 1,104 | ||
F-12
ENDOLOGIX, INC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—Continued
(in thousands, except share and per share amounts)
Rental expense charged to operations for all operating leases during the years ended December 31, 2010, 2009 and 2008, was $549, $414, and $312, respectively.
Employment Agreements and Retention Plan
The Company has entered into employment agreements with its officers and “key employees” under which payment and benefits would become payable in the event of termination by the Company for any reason other than cause; or upon a change in control or corporate transaction, by the key employee for good reason, as such terms are defined in the agreement. If due, the payment will generally be equal to six months of the key employee’s then current salary for termination by the Company without cause, and generally be equal to twelve months of salary if by the key employee for good reason upon a change in control or corporate transaction.
9. Stockholders’ Equity
Authorized Shares of Common Stock
In October 2003, shareholders approved an increase in the number of authorized shares of common stock from 30,000,000 to 50,000,000. In May 2006, shareholders approved an amendment, which increased the number of authorized shares of common stock from 50,000,000 to 60,000,000. In May 2009, shareholders approved an amendment that increased the number of authorized shares of common stock from 60,000,000 to 75,000,000.
Sale of Common Stock
In August 2009, the Company completed a public offering of 3,900,000 shares of its common stock at a purchase price of $4.10 per share, which resulted in net proceeds of approximately $14,777 after deducting the offering expenses. In addition, in December 2010, concurrent with the close of the Merger, the Company issued and sold 3,170,577 shares to Essex Woodlands Fund VII at a purchase price of $4.731, resulting in gross proceeds of $15 million.
Stock Options
Pursuant to the Company’s 1996 Stock Option/Issuance Plan (the “1996 Plan”), the 1997 Supplemental Stock Option Plan (the “1997 Plan”), and the Company’s 2006 Stock Incentive Plan (the “2006 Plan” and together with the 1996 Plan and 1997 Plan, the “Plans”), either incentive stock options, non-qualified options, restricted stock, or awards may be granted. Under the Plans, options are granted at a price not less than 100% of the value of the Company’s common stock on the date of grant and are exercisable over a maximum term of ten years from the date of grant and generally vest over a four-year period.
At December 31, 2010 and 2009, there were approximately 1,382,000 and 717,000 shares of common stock available for future stock grants. The stock option activity under the plans is summarized below:
2010 | 2009 | 2008 | ||||||||||||||||||
Number of Shares | Weighted Average Exercise Price | Number of Shares | Weighted Average Exercise Price | Number of Shares | Weighted Average Exercise Price | |||||||||||||||
Outstanding — Beginning of Year | 5,466,144 | $ | 3.71 | 4,985,261 | $ | 3.69 | 4,124,739 | $ | 4.34 | |||||||||||
Granted | 1,424,376 | 4.53 | 1,096,460 | 3.56 | 2,025,000 | 2.57 | ||||||||||||||
Exercised | 598,840 | 3.92 | 238,058 | 3.28 | 30,000 | 2.82 | ||||||||||||||
Forfeited | 357,196 | 3.18 | 315,019 | 3.21 | 891,978 | 4.00 | ||||||||||||||
Expired | 19,600 | 7.01 | 62,500 | 3.40 | 242,500 | 4.63 | ||||||||||||||
Outstanding — End of Year | 5,914,884 | $ | 3.91 | 5,466,144 | $ | 3.71 | 4,985,261 | $ | 3.69 | |||||||||||
Exercisable — End of Year | 3,437,992 | $ | 3.81 | 3,168,307 | $ | 4.07 | 2,266,879 | $ | 4.52 | |||||||||||
Weighted Average Fair Value of Options Granted During Year | $ | 4.53 | $ | 3.56 | $ | 2.57 | ||||||||||||||
Under the Plans, the total intrinsic value for shares outstanding was approximately $11,798, $9,172, and $11 as of December 31, 2010, 2009, and 2008, respectively. The total intrinsic value for shares exercisable was approximately $6,571,
F-13
ENDOLOGIX, INC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—Continued
(in thousands, except share and per share amounts)
$4,337, and $5 as December 31, 2010, 2009, and 2008, respectively. The total intrinsic value of options exercised was approximately $1,359, $322, and $56 in 2010, 2009, and 2008, respectively.
As of December 31, 2010, there was $3,491 of total unrecognized compensation cost related to unvested stock options granted. This unrecognized compensation cost is expected to be recognized over a weighted average period of 2.7 years.
The following table summarizes information regarding stock grants outstanding at December 31, 2010:
Outstanding | Exercisable | |||||||||||||||
Range of Exercise Prices | Granted Shares Outstanding | Weighted- Average Remaining Contractual Life (Years) | Weighted- Average Exercise Price | Granted Shares Exercisable | Weighted- Average Exercise Price | |||||||||||
$1.07 — 2.25 | 308,771 | 7.2 | $ | 2.02 | 179,398 | $ | 1.98 | |||||||||
2.26 — 2.62 | 521,753 | 6.9 | 2.55 | 389,296 | 2.55 | |||||||||||
2.67 — 2.69 | 520,312 | 7.4 | 2.67 | 332,916 | 2.67 | |||||||||||
2.75 — 3.16 | 446,318 | 7.1 | 2.85 | 351,944 | 2.84 | |||||||||||
3.35 — 3.45 | 534,845 | 5.3 | 3.41 | 464,747 | 3.41 | |||||||||||
3.46 — 3.90 | 519,998 | 7.1 | 3.57 | 259,979 | 3.63 | |||||||||||
3.92 — 4.31 | 465,921 | 7.5 | 4.13 | 227,064 | 4.10 | |||||||||||
4.32 — 4.51 | 1,445,832 | 8.2 | 4.39 | 478,815 | 4.38 | |||||||||||
4.56 — 5.68 | 541,048 | 6.3 | 5.24 | 305,333 | 5.16 | |||||||||||
5.72 — 7.12 | 610,086 | 5.3 | 6.10 | 448,500 | 6.12 | |||||||||||
$0.95 — 8.75 | 5,914,884 | 7.0 | $ | 3.91 | 3,437,992 | $ | 3.81 | |||||||||
The weighted-average grant-date fair value of stock granted during 2010, 2009, and 2008 where the exercise price on the date of grant was equal to the stock price on that date, was $4.53, $3.56, and $2.57, respectively.
Expense related to non-employee stock options is being amortized over the vesting period, which is generally four years. During the years ended December 31, 2010, 2009, and 2008, $(7), $33, and $(8), respectively, was recorded as compensation expense for the change in the fair value of unvested non-employee option grants. During the years ended December 31, 2010 and 2009, the Company did not grant any options to non-employees. For the year ended December 31, 2008, the Company granted 10,000 options to non-employees. As of December 31, 2010, 2009, and 2008, a total of 68,750, 83,350, and 95,000 non-employee stock options, respectively, were outstanding. As of December 31, 2010, 2009, and 2008, a total of 68,750, 83,350, and 76,667, non-employee stock options, respectively, were fully vested.
F-14
ENDOLOGIX, INC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—Continued
(in thousands, except share and per share amounts)
Restricted Stock
The following table summarizes activity and related information for our restricted stock awards:
Number of Shares | Weighted Average Grant-Date Fair Value | |||||
Nonvested as of December 31, 2007 | — | $ | 2.65 | |||
Granted | 525,000 | 2.65 | ||||
Vested | — | — | ||||
Nonvested as of December 31, 2008 | 525,000 | 2.65 | ||||
Granted | 160,000 | 3.25 | ||||
Vested | — | — | ||||
Nonvested as of December 31, 2009 | 685,000 | $ | 5.90 | |||
Granted | 518,045 | 5.95 | ||||
Canceled | (35,965 | ) | 2.88 | |||
Vested | (575,625 | ) | 2.72 | |||
Nonvested as of December 31, 2010 | 591,455 | $ | 5.62 | |||
During the years ended December 31, 2010, 2009, and 2008, we granted 518,045, 160,000, and 525,000 shares of restricted stock, respectively. Restricted stock is granted subject to restrictions as to sale or other disposition of shares and to restrictions that require continuous employment with the Company. The restrictions generally expire in either two or four years from the date of grant. Grants with a two year expiration completely vest after two years. Grants with a four year term vest 25% after one year, with the remainder vesting in equal monthly amounts over the remaining three years. The grant-date fair value of shares granted during the years ended December 31, 2010, 2009, and 2008, was $3,083, $520, and $1,389, respectively. The weighted-average grant-date fair value per share for restricted stock granted was based upon the closing market price of the Company’s common stock on the grant dates of the awards and was $5.95, $3.25, and $2.65, per share for the years ended December 31, 2010, 2009, and 2008, respectively. During the year ended December 31, 2010, 35,965 restricted shares canceled and 575,625 restricted shares vested. The Company recorded stock-based compensation related to restricted stock of $422, $772, and $432 for the years ended December 31, 2010, 2009, and 2008, respectively. As of December 31, 2010, the unrecorded stock-based compensation balance related to restricted stock awards was $2,645, and will be recognized over an estimated weighted average amortization period of 2.5 years.
Included in the table are 100,000 restricted shares granted to non-employees in connection with Nellix acquisition. These shares are unregistered and issued outside of the plans. For the years ended December 31, 2008 and 2007, the Company did not grant restricted shares to non-employees. As of December 31, 2010, a total of 100,000 non-employee restricted shares were outstanding. As of December 31, 2010, there were no non-employee restricted shares fully vested nor exercised and no non-employee restricted shares were canceled.
Employee Stock Purchase Plan
Under the terms of the Company’s 2006 Employee Stock Purchase Plan (the “Purchase Plan”), eligible employees can purchase common stock through payroll deductions at a price equal to the lower of 85% of the fair market value of the Company’s common stock at the beginning or end of the applicable offering period. In 2009, an additional 1,500,000 shares of common stock were approved. No additional shares of common stock were approved for issuance under the Purchase Plan in 2010. During the years ended December 31, 2010, 2009, and 2008, $468, $335, and $169, respectively, was recorded as stock based compensation expense under the Purchase Plan. During 2010, 2009, and 2008, a total of approximately 293,000, 489,000, and 357,000, shares of common stock, respectively, were purchased at an average price of $3.81, $1.61, and $1.39, respectively.
Stock Based Compensation
The Company uses the Black-Scholes option pricing model as the valuation model to calculate the fair value of stock
F-15
ENDOLOGIX, INC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—Continued
(in thousands, except share and per share amounts)
based compensation. The model requires extensive use of financial estimates and accounting judgment, including estimates of the expected period of time employees will retain their vested stock options before exercising them, the expected volatility of the Company’s common stock over the expected term, and the number of shares that are expected to be forfeited before they are vested. Application of alternative assumptions could produce significantly different estimates of the fair value of the stock-based compensation and as a result, significantly different results recognized in the consolidated statements of operations.
The weighted average of the assumptions used to estimate the fair value of stock options granted using the Black-Scholes valuation method was as follows:
2010 | 2009 | 2008 | ||||||
Expected Life (in years) (1) | 6.0 | 5.5 | 5.5 | |||||
Expected Volatility (2) | 56.4 | % | 55.8 | % | 56.1 | % | ||
Risk Free Interest Rate (3) | 2.4 | % | 2.5 | % | 3.1 | % | ||
Dividend Yield (4) | — | % | — | % | — | % | ||
1. | Estimated based on historical experience. |
2. | Volatility based on historical experience over a period equivalent to the expected life in years. |
3. | Based on the US Treasury constant maturity interest rate with a term consistent with the expected life of the options granted. |
4. | The Company does not pay dividends on its common stock and the Company currently does not have any plans to pay or declare any cash dividends. |
Stock compensation expense was as follows:
2010 | 2009 | 2008 | |||||||||
General and Administrative | $ | 1,168 | $ | 1,592 | $ | 1,413 | |||||
Marketing and Sales | 919 | 989 | 1,018 | ||||||||
Research, Development, and Clinical | 377 | 299 | 236 | ||||||||
Cost of Sales | 188 | 177 | 245 | ||||||||
Total Stock Based Compensation | $ | 2,652 | $ | 3,057 | $ | 2,912 | |||||
In addition, the Company had $43, $63, and $78 of stock based compensation capitalized in inventory as of December 31, 2010, 2009, and 2008, respectively.
10. Related Party Transactions
A director of a hospital facility from which the Company contracts for physician training and clinical research services also served as a member of the board of directors of the Company until the expiration of his term on June 11, 2009. Payments totaling $0, $23, and $97 for the periods ended December 31, 2010, June 11, 2009 December 31, 2008, respectively, were made to this hospital. In addition, this hospital purchased products from the Company totaling $0, $508, and $816 for the periods ended December 31, 2010, June 11, 2009, and December 31, 2008, respectively. All transactions were in accordance with normal commercial terms and conditions.
11. Income Taxes
Income tax expense (benefit) consists of the following:
F-16
ENDOLOGIX, INC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—Continued
(in thousands, except share and per share amounts)
2010 | 2009 | 2008 | |||||||||
Current | |||||||||||
Federal | $ | 49 | $ | 1 | $ | (30 | ) | ||||
State | 20 | 20 | 2 | ||||||||
Foreign | — | — | — | ||||||||
69 | 21 | (28 | ) | ||||||||
Deferred | |||||||||||
Federal | (13,634 | ) | — | — | |||||||
State | (1,472 | ) | — | — | |||||||
Total tax expense (benefit) | $ | (15,037 | ) | $ | 21 | $ | (28 | ) | |||
For the years ended December 31, 2009 and 2008, income tax expense (benefit) is included in other income/(expense) on the Consolidated Statement of Operations.
Income taxes for 2010, 2009 and 2008 differ from income taxes for those years computed by applying the U.S. federal statutory rate of 34% to income/(loss) before taxes for those years as follows:
2010 | 2009 | 2008 | |||||||||
Tax benefit at U.S. statutory rate | $ | (1,490 | ) | $ | (827 | ) | $ | (4,077 | ) | ||
State tax expense (benefit) net of federal benefit | (1,458 | ) | 20 | (372 | ) | ||||||
Meals & Entertainment (50% addback) | 182 | 114 | 128 | ||||||||
Research & Development Credits | (327 | ) | (117 | ) | (110 | ) | |||||
Stock based compensation | 684 | 471 | 500 | ||||||||
Net change in valuation allowance | (13,974 | ) | 361 | 3,898 | |||||||
Other, net | 1,346 | (1 | ) | 5 | |||||||
$ | (15,037 | ) | $ | 21 | $ | (28 | ) | ||||
Significant components of the Company’s deferred tax assets and (liabilities) are as follows at December 31:
2010 | 2009 | ||||||
Net operating loss carryforwards | $ | 48,976 | $ | 39,484 | |||
Accrued expenses | 503 | 172 | |||||
Tax credits | 8,673 | 6,453 | |||||
Bad debt | 44 | 37 | |||||
Depreciation and amortization | 672 | 699 | |||||
Inventory | 85 | 146 | |||||
Capitalized research and development | 89 | 172 | |||||
Developed technology and trademark | (15,855 | ) | (1,279 | ) | |||
Trademarks and tradenames | (1,029 | ) | (1,029 | ) | |||
Deferred compensation | 2,210 | 2,007 | |||||
Other | 226 | 99 | |||||
Net deferred tax assets | 44,594 | 46,961 | |||||
Valuation allowance | (45,623 | ) | (47,990 | ) | |||
Net deferred tax liability | $ | (1,029 | ) | $ | (1,029 | ) | |
Based upon the Company’s history of operating losses, realization of its deferred tax assets does not appear more likely than not. The Company recorded a valuation allowance of $45,623. In determining the net asset subject to a valuation allowance, the Company recorded a deferred tax liability related to its indefinite-lived other intangible assets that are not
F-17
ENDOLOGIX, INC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—Continued
(in thousands, except share and per share amounts)
expected to reverse in the foreseeable future resulting in a net deferred tax liability of approximately $1,029 after application of the valuation allowance.
The valuation allowance decreased by $2,357 in 2010 and increased by $425, and $4,973, in 2009 and 2008, respectively. The principal reason for decrease in valuation allowance in 2010 was due to recording of deferred tax liability associated with the Nellix acquisition.
At December 31, 2010, the Company had net operating loss carryforwards for federal and state income tax purposes of approximately $133,932 and $93,731, respectively. Included in the net operating loss carryfoward balances are federal and state net operating losses of $30,145 and $29,750 respectively in connection with the Nellix acquisition. The acquired federal and state net operating losses will begin expiring in 2021 and 2017, respectively. The federal net operating loss carryforward will begin expiring in 2017. The majority of the state net operating losses are attributable to the state of California and will begin expiring in 2016. In addition, the Company had research and development and other tax credits for federal and state income tax purposes of approximately $4,420 and $4,155, respectively, which will begin to expire in 2018. The state research and development credits do not expire for California purposes.
The table of deferred tax assets and liabilities shown above does not include certain deferred tax assets at December 31, 2010 and 2009 that arose directly from (or the use of which was postponed by) tax deductions related to equity compensation in excess of compensation recognized for financial reporting. Those deferred tax assets include federal and state net operating losses. Equity will be increased by $0.2 million if and when such deferred tax assets are ultimately realized. The Company uses SFAS 109 ordering for purposes of determining when excess tax benefits have been realized.
Utilization of the NOL and R&D credit carryforwards may be subject to a substantial annual limitation due to an “ownership change” (as defined) that may have occurred or that could occur in the future, as required by Section 382 of the Internal Revenue Code of 1986, as amended (the “Code”), as well as similar state provisions. These ownership changes may limit the amount of NOL and R&D credit carryforwards, and other tax attributes, that can be utilized annually to offset future taxable income and tax, respectively. In general, an “ownership change” as defined by Section 382 of the Code results from a transaction or series of transactions over a three-year period resulting in an ownership change of more than 50 percentage points of the outstanding stock of a company by certain stockholders or public groups. Since the Company’s formation, the Company has raised capital through the issuance of capital stock on several occasions which, combined with the purchasing stockholders’ subsequent disposition of those shares, may have resulted in such an ownership change, or could result in an ownership change in the future upon subsequent disposition.
The Company is in the process of completing a study to assess whether an ownership change has occurred or whether there have been multiple ownership changes since the Company's formation. Due to the complexity and cost associated with such a study, this study is still in process. If the Company has experienced an ownership change at any time since its formation, utilization of the NOL, R&D credit carryforwards, and other tax attributes, would be subject to an annual limitation under Section 382 of the Code. In general, the annual limitation, which is determined by first multiplying the value of the Company’s stock at the time of the ownership change by the applicable long-term, tax-exempt rate, could further be subject to additional adjustments, as required. Any limitation may result in the expiration of a portion of the NOL or R&D credit carryforwards before utilization. Further, until a study is completed and any limitation is known, no amounts are being considered as an uncertain tax position or disclosed as an unrecognized tax benefit under FIN 48. Due to the existence of the valuation allowance, future changes in the Company’s unrecognized tax benefits will not impact its effective tax rate. Any carryforwards that will expire prior to utilization as a result of a limitation under Section 382 will be removed from deferred tax assets with a corresponding reduction of the valuation allowance.
The Company has not recognized any additional liability for unrecognized tax benefits. The Company expects any resolution of unrecognized tax benefits, if created, would occur while the full valuation allowance of deferred tax assets is maintained; therefore, the Company does not expect to have any unrecognized tax benefits that, if recognized, would affect the effective tax rate.
The Company is subject to taxation in the U.S. and various states. The Company’s tax years for 2007, 2008, and 2009 are subject to examination by the taxing authorities. With few exceptions, the Company is no longer subject to U.S. federal, state, local or foreign examinations by taxing authorities for years before 2007.
12. Employee Benefit Plan
The Company provides a 401(k) Plan for all employees 21 years of age or older. Under the 401(k) Plan, eligible
F-18
ENDOLOGIX, INC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—Continued
(in thousands, except share and per share amounts)
employees voluntarily contribute to the Plan up to 100% of their salary through payroll deductions, subject to statutory limitations. Employer contributions may be made by the Company at its discretion based upon matching employee contributions, within limits, and profit sharing provided for in the Plan. No employer contributions were made in 2010, 2009, or 2008.
13. Legal Matters
The Company is involved from time to time in various claims and legal proceedings of a nature considered normal and incidental to its business, including product liability, intellectual property, employment and other matters. The Company accrues for contingent liabilities when it is probable that a liability has been incurred and the amount can be reasonably estimated. The accruals are adjusted periodically as assessments change or as additional information becomes available.
The Company is currently involved in litigation with Cook Medical Incorporated (“Cook”), in which Cook alleges that the Company infringed two of Cook's patents, granted in 1991 and 1998, respectively. The lawsuit was filed by Cook in the United States District Court, Southern District of Indiana (“Court”), on October 8, 2009. In December 2009, the United States Patent and Trademark Office (“PTO”) granted the Company's requests for a reexamination of the two patents asserted by Cook in the lawsuit. In January 2010, the Court ordered that the lawsuit be stayed pending the outcome of the patent reexaminations. In February 2010, the PTO completed its initial reexamination process and confirmed the patentability of one of the two patents (the '706 patent), and on March 31, 2010 issued a reexamination certificate to that effect. As to the second patent (the '777 patent), the PTO rejected as unpatentable those patent claims asserted by Cook against the Company. Cook subsequently amended the '777 patent and added certain new claims. On April 14, 2010 the PTO indicated its intent to issue a reexamination certificate confirming the patentability of these amended and new claims and issued the certificate on July 21, 2010. On June 2, 2010, the stay of the court proceedings was lifted and discovery is underway. The Company is raising numerous defenses in the case, one of which is that Cook's lawsuit is barred by a prior judgment in an earlier case between the same parties. A hearing on the construction of the asserted claims of the '706 and '777 patents is scheduled for April 2011. The Company intends to continue its vigorous defense against these claims and believes its defenses are meritorious.
The Company is also involved in litigation with Bard Peripheral Vascular, Inc. (“Bard”), in which Bard alleges that the Company infringes one of Bard's patents that issued in 2002. Bard filed the lawsuit against the Company and another defendant, Atrium Medical Corp. on August 10, 2010 alleging that the Company infringes U.S. Patent No. 6,436,135 (“'135 patent”) entitled “Prosthetic Vascular Graft.” Bard alleged in the complaint that the ePTFE material used in the Company's Powerlink System infringes the '135 patent and seeks damages for the infringement. Bard also alleges that the Company's infringement was willful and seeks treble damages, prejudgment interest, and its attorney fees as well as a permanent injunction. The Complaint was served on the Company by Bard on November 24, 2010. The Company intends to vigorously contest the case.
At December 31, 2010, the Company had not accrued for any contingent losses in connection with the Cook or the Bard suits because an unfavorable outcome with respect to these matters is not probable. However, as these matters are ongoing, there is no assurance they will be resolved favorably by the Company or will not result in a material loss.
No other matters require disclosure.
(2) Financial Statement Schedule
F-19
ENDOLOGIX, INC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—Continued
(in thousands, except share and per share amounts)
SCHEDULE II — VALUATION AND QUALIFYING ACCOUNTS
Years Ended December 31, 2010, 2009 and 2008
Column A | Column B | Column C | Column D | Column E | |||||||||||||||
Additions (Reductions) | |||||||||||||||||||
Description | Balance at Beginning of Period | Charges to Costs and Expenses | Charged to Other Accounts (b) | Deductions(a),(b) | Balance at End of Period | ||||||||||||||
(In thousands) | |||||||||||||||||||
Year ended December 31, 2010 | |||||||||||||||||||
Allowance for doubtful accounts | $ | 97 | $ | 20 | $ | — | $ | 117 | |||||||||||
Income tax valuation allowance | $ | 47,990 | $ | — | $ | 15,106 | $ | (17,473 | ) | $ | 45,623 | ||||||||
Year ended December 31, 2009 | |||||||||||||||||||
Allowance for doubtful accounts | $ | 72 | $ | 103 | $ | — | $ | (78 | ) | $ | 97 | ||||||||
Income tax valuation allowance | $ | 47,565 | $ | 425 | $ | — | $ | — | $ | 47,990 | |||||||||
Year ended December 31, 2008 | |||||||||||||||||||
Allowance for doubtful accounts | $ | 100 | $ | 89 | $ | — | $ | (117 | ) | $ | 72 | ||||||||
Income tax valuation allowance | $ | 42,592 | $ | 4,973 | $ | — | $ | — | $ | 47,565 | |||||||||
(a) | Deductions represent the actual write-off of accounts receivable balances or the disposal of inventory. |
(b) | Represents recording of deferred tax liability that is charged to Goodwill related to the Nellix acquisition and associated release of valuation allowance. |
F-20
EXHIBIT INDEX
Exhibit Number | Description | |
2.1 | Agreement and Plan of Merger and Reorganization, dated October 27, 2010, by and among Endologix, Nellix Acquisition Corporation, Nellix, Inc., certain of Nellix, Inc.'s stockholders listed therein and Essex Woodlands Health Ventures, Inc., as representative of Nellix, Inc.'s stockholders (Incorporated by reference to Exhibit 2.1 to Endologix Current Report on Form 8-K, filed with the SEC on October 27, 2010). | |
3.1 | Amended and Restated Certificate of Incorporation (Incorporated by reference to Exhibit 3.1 to Endologix Quarterly Report on Form 10-Q, filed with the SEC on July 28, 2009). | |
3.2 | Amended and Restated Bylaws (Incorporated by reference to Exhibit 3.2 to Endologix Annual Report on Form 10-K filed with the SEC on March 29, 2001). | |
4.1 | Specimen Certificate of Common Stock (Incorporated by reference to Exhibit 4.1 to Amendment No. 2 to Endologix Registration Statement on Form S-1, No. 333-04560, filed with the SEC on June 10, 1996). | |
10.1(2) | 1997 Supplemental Stock Option Plan (Incorporated by reference to Exhibit 99.1 to Endologix Registration Statement on Form S-8, No. 333-42161, filed with the SEC on December 12, 1997). | |
10.2(2) | 1996 Stock Option/Stock Issuance Plan (Incorporated by reference to Exhibit 4.1 to Endologix Registration Statement on Form S-8, No. 333-122491, filed with the SEC on February 2, 2005). | |
10.3(2) | 2006 Stock Incentive Plan (Incorporated by reference to Exhibit 10.1 to Endologix Current Report on Form 8-K, filed with the SEC on May 26, 2006). | |
10.4(2) | Form of Stock Option Agreement under 2006 Stock Incentive Plan (Incorporated by reference to Exhibit 10.1 to Endologix Quarterly Report on Form 10-Q, filed with the SEC on November 9, 2006). | |
10.5(2) | Form of Restricted Stock Award Agreement under 2006 Stock Incentive Plan (Incorporated by reference to Exhibit 10.2 to Endologix Quarterly Report on Form 10-Q, filed with the SEC on November 9, 2006). | |
10.6(2) | 2006 Employee Stock Purchase Plan (Incorporated by reference to Exhibit 10.1 to Endologix Current Report on Form 8-K, filed with the SEC on June 17, 2009). | |
10.7 | Form of Indemnification Agreement entered into with Endologix officers and directors (Incorporated by reference to Exhibit 10.41 to Endologix Quarterly Report on Form 10-Q, filed with the SEC on November 13, 2002). | |
10.8 | Standard Industrial/Commercial Single-Tenant Lease — Net, dated November 2, 2004, by and between Endologix and Del Monico Investments, Inc. (Incorporated by reference to Exhibit 10.46 to Endologix Current Report on Form 8-K, filed with the SEC on November 24, 2004). | |
10.8.1 | Addendum No. 2 to Standard Industrial/Commercial Single-Tenant Lease — Net, by and between Endologix, Inc. and Del Monico Investments, Inc., dated June 9, 2009 (Incorporated by reference to Exhibit 10.1 to Endologix Quarterly Report on Form 10-Q, filed with the SEC on November 2, 2009). | |
10.9(2) | Offer Letter, dated April 28, 2008, between Endologix and John McDermott (Incorporated by reference to Exhibit 10.1 to Endologix Current Report on Form 8-K, filed with the SEC on May 16, 2008). | |
10.10(2) | Employment Agreement, dated as of December 29, 2008, by and between Endologix and John McDermott (Incorporated by reference to Exhibit 10.1 to Endologix Current Report on Form 8-K, filed with the SEC on January 2, 2009). | |
10.11(2) | Employment Agreement, dated as of December 29, 2008, by and between Endologix and Robert J. Krist (Incorporated by reference to Exhibit 10.2 to Endologix Current Report on Form 8-K, filed with the SEC on January 2, 2009). | |
F-21
Exhibit Number | Description | |
10.12(2) | Employment Agreement, dated as of December 29, 2008, by and between Endologix and Stefan G. Schreck, Ph.D. (Incorporated by reference to Exhibit 10.3 to Endologix Current Report on Form 8-K, filed with the SEC on January 2, 2009). | |
10.13(2) | Employment Agreement, dated as of December 29, 2008, by and between Endologix and Janet Fauls (Incorporated by reference to Exhibit 10.4 to Endologix Current Report on Form 8-K, filed with the SEC on January 2, 2009). | |
10.14 | Standard Industrial/Commercial Multi -Tenant Lease — Net, by and between Endologix, Inc. and Four-In-One Associates, dated August 28, 2009 (Incorporated by reference to Exhibit 10.2 to Endologix Quarterly Report on Form 10-Q, filed with the SEC on November 2, 2009). | |
10.15 | Credit Agreement, dated October 30, 2009, by and between Endologix and Wells Fargo Bank, National Association. | |
10.16(2) | Employment Agreement, dated as of April 13, 2009, by and between Endologix, Inc. and Joseph DeJohn. | |
10.17 | Securities Purchase Agreement, dated as of October 27, 2010, by and between Endologix and Essex Woodlands Health Ventures Fund VII, L.P. (Incorporated by reference to Exhibit 10.1 to Endologix Current report on form 8-K, filed with the SEC on October 27, 2010). | |
10.17.1 | Amendment to Securities Purchase Agreement, dated as of December 9, 2010, by and between Endologix and Essex Woodlands Health Ventures Fund VII, L.P. (Incorporated by reference to Exhibit 10.1 to Endologix Current Report on Form 8-K, filed with the SEC on December 14, 2010). | |
10.18(2) | Employment Agreement, dated as of December 10, 2010, by and between Endologix and Robert D. Mitchell (Incorporated by reference to Exhibit 10.2 to Endologix Current Report on Form 8-K, filed with the SEC on December 14, 2010). | |
14 | Code of Ethics for Chief Executive Officer and Principal Financial Officers (Incorporated by reference to Exhibit 14 to Endologix Annual Report on Form 10-K filed with the SEC on March 26, 2004). | |
21.1 | List of Subsidiaries. | |
23.1 | Consent of Independent Registered Public Accounting Firm. | |
24.1 | Power of Attorney (included on signature page hereto). | |
31.1 | Certification of Chief Executive Officer Pursuant to Rule 13a-14(a)/15d-14(a) under the Securities Exchange Act of 1934. | |
31.2 | Certification of Chief Financial Officer Pursuant to Rule 13a-14(a)/15d-14(a) under the Securities Exchange Act of 1934. | |
32.1 | Certification of Chief Executive Officer Pursuant to Rule 13a-14(b)/15d-14(b) under the Securities Exchange Act of 1934 and 18 U.S.C. Section 1350. | |
32.2 | Certification of Chief Financial Officer Pursuant to Rule 13a-14(b)/15d-14(b) under the Securities Exchange Act of 1934 and 18 U.S.C. Section 1350. | |
_______________________
(1) | Portions of this exhibit are omitted and were filed separately with the Securities and Exchange Commission pursuant to Endologix application requesting confidential treatment under Rule 24b-2 of the Securities Exchange Act of 1934. |
(2) | These exhibits are identified as management contracts or compensatory plans or arrangements of Endologix pursuant to Item 15(a)(3) of Form 10-K. |
F-22