Attached files
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2010
OR
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Commission File No. 000-30334
Angiotech Pharmaceuticals, Inc.
(Exact Name of Registrant as Specified in its Charter)
| British Columbia, Canada | 98-0226269 | |
| (State or Other Jurisdiction of Incorporation or Organization) |
(I.R.S. Employer Identification No.) | |
| 1618 Station Street, Vancouver, BC, Canada | V6A 1B6 | |
| (Address of principal executive offices) | (Zip Code) | |
Registrant’s telephone number, including area code: (604) 221-7676
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class |
Name of each exchange on which registered | |
| Common shares, without par value | OTC Bulletin Board |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the Registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the Registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ¨ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ¨ | Accelerated filer | x | |||
| Non-accelerated filer | ¨ (Do not check if a smaller reporting company) | Smaller reporting company | ¨ | |||
Indicate by check mark whether the Registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
The aggregate market value of the common shares held by non-affiliates of the registrant as of June 30, 2010 (based on the last reported sale price of the common shares of U.S. $0.75, as reported on The NASDAQ Global Select Market) was approximately U.S. $63,877,707.
As of March 1, 2011 the registrant had 85,185,497 outstanding common shares.
DOCUMENTS INCORPORATED BY REFERENCE
None.
Table of Contents
Angiotech Pharmaceuticals, Inc.
Table of Contents
Annual Report on Form 10-K for the year ended December 31, 2010
Table of Contents
In this Annual Report on Form 10-K, references to “the Company”, “Angiotech”, “us” or “we” are to Angiotech Pharmaceuticals, Inc. and all of its subsidiaries as a whole, except where it is clear that these terms only refer to Angiotech Pharmaceuticals, Inc.
Accounting Standards
In this Annual Report on Form 10-K, all dollar amounts are in U.S. dollars, except where otherwise stated and financial reporting is made in accordance with U.S. generally accepted accounting principles (“U.S. GAAP”).
Currency and Exchange Rates
The Company’s consolidated functional and reporting currency is the U.S. dollar. The Canadian dollar is the functional currency for the parent company Angiotech Pharmaceuticals, Inc. (on a non-consolidated basis) and its Canadian subsidiaries, the U.K pound is the functional currency of our U.K subsidiary, Pearsalls Limited, the Danish Kroner is the functional currency of our Danish subsidiary, PBN Medicals Denmark A/S, and the U.S. dollar is the functional currency for all other subsidiaries.
NOTICE REGARDING WEBSITE ACCESS TO COMPANY REPORTS
We file electronically with the Securities and Exchange Commission (“SEC”) our annual reports on Form 10-K, quarterly reports on Form 10-Q and current reports on Form 8-K pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934 (“the Exchange Act”). You may obtain a free copy of our annual reports on Form 10-K, quarterly reports on Form 10-Q and current reports on Form 8-K, and any amendments to those reports, on the day of filing with the SEC and on our website at: www.angiotech.com. Our website and the information on our website is not part of this Annual Report on Form 10-K.
You may also read and copy any materials we file with the SEC at the SEC’s Public Reference Room at 100 F Street, NE, Washington, DC 20549. You may also obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC maintains an Internet site at www.sec.gov that contains reports, proxy and information statements, and other information regarding the issuers such as us that file electronically.
1
Table of Contents
| Item 1. | BUSINESS |
Summary
We are a medical device company that discovers, develops and markets innovative technologies primarily focused on acute and surgical applications. We currently operate in two segments: Medical Products and Pharmaceutical Technologies. Our Medical Products segment manufactures and markets a wide range of single-use specialty medical products and medical device components. These products are sold directly to end users or other third-party medical device manufacturers. Our Pharmaceutical Technologies segment focuses primarily on establishing product development and marketing collaborations with major medical device, pharmaceutical or biomaterials companies, and to date has derived the majority of its revenue from royalties due from partners that develop, market and sell products incorporating our technologies. Our principal revenues in this segment are from royalties derived from sales by Boston Scientific Corporation (“BSC”) of TAXUS® coronary stent systems incorporating the drug paclitaxel (“TAXUS”). For the year ended December 31, 2010, we recorded $211.8 million in direct sales of our various medical products and $34.7 million in royalties and license fees received from our partners. For additional financial information about our two segments and the geographic areas in which we operate, refer to note 21 of the audited consolidated financial statements included in this Annual Report on Form 10-K.
Our research and development efforts focus on understanding and characterizing biological conditions that often occur concurrent with medical device implantation, surgery or acute trauma, including scar formation and inflammation, cell proliferation, bleeding and coagulation, infection, and tumor tissue overgrowth. Our strategy is to utilize our various technologies in the areas of medical devices, drugs, drug delivery, surface modification and biomaterials to create and commercialize novel, proprietary medical products that reduce surgical procedure side effects, improve surgical outcomes, shorten hospital stays, or are easier or safer for a physician to use. We have patent protected, or have filed patent applications for, certain of our technology and many of our products and potential product candidates.
Over the last three years, revenue in our Pharmaceutical Technologies segment has declined significantly, primarily due to lower royalties derived from sales by BSC of TAXUS. The decline in sales of TAXUS is primarily due to continued and increasing competitive pressure in the market for drug-eluting stents. This rapid decline in royalty revenue has negatively and materially impacted our liquidity, capital resources and results of operations. As a result of this and other factors impacting our business.
On October 29, 2010, the Company and certain of its subsidiaries entered into the Recapitalization Support Agreement (as amended, the “RSA”) with holders of approximately 73% of the aggregate principal amount outstanding of our 7.75% Senior Subordinated Notes due 2014 (the “Subordinated Notes” and such holders, together with all holders of Subordinated Notes who executed a consent agreement with the Company and certain of its subsidiaries prior to November 30, 2010, the “Consenting Noteholders”) to effectuate a recapitalization that would eliminate $250 million of our total outstanding indebtedness and related interest obligations by exchanging Subordinated Notes for new common shares in the Company (the “Recapitalization Transaction”).
On October 29, 2010, the Company also entered into a support agreement (as amended, the FRN Support Agreement”) with holders of approximately 51% of the aggregate principal amount outstanding of the Company’s senior floating rates notes due 2013 (the “Existing Floating Rate Notes”) pursuant to which such holders agreed to support and participate in the Company’s offer to exchange new senior floating rate notes (the “New Floating Rate Notes”) for its Existing Floating Rate Notes (the “FRN Exchange Offer”).
2
Table of Contents
In accordance with the terms of the RSA, on January 28, 2011 we and certain of our subsidiaries (the “Angiotech Entities”) filed for creditor protection (the “CCAA Proceedings”) under the Companies’ Creditors Arrangement Act, (Canada) (the “CCAA”) and an initial order (the “Initial Order” was granted by the Supreme Court of British Columbia (the “Canadian Court”). The CCAA Proceedings were commenced in order to implement the Recapitalization Transaction through a plan of compromise or arrangement (as amended, supplemented or restated from time to time, the “CCAA Plan”). In addition, in order to have the CCAA Proceedings and the relief granted therein recognized in the United States, on January 30, 2011, we and certain of our subsidiaries also commenced proceedings under Chapter 15 of Title 11 of the United States Code (the “U.S. Bankruptcy Code”) in the United States Bankruptcy Court for the District of Delaware (the “Chapter 15 Cases”). Under the terms of the CCAA Plan, all of our existing common shares and options to acquire common shares will be cancelled without payment or consideration therefore. For more information on the CCAA Proceedings and the Recapitalization Transaction, refer to Management’s Discussion and Analysis of Financial Condition and Results of Operations, included in this Annual Report on Form 10-K and notes 1, 16 and 26 in the audited consolidated financial statements included in this Annual Report on Form 10-K. Throughout 2011, we will continue to evaluate our various financial and strategic alternatives.
Business Strategy
Our strategy is to apply our various technologies to develop and commercialize novel, proprietary medical products that address the most frequent problems or complications observed in connection with medical device use or surgical procedures. Specific tactical elements of our strategy include:
| • | Identifying and Prioritizing Market Opportunities. We begin our product development process by identifying medical devices or surgical procedures where problems or complications arise soon after device implantation or the initial procedure, and where re-intervention may be expensive, potentially harmful for patients or difficult to perform. We target areas where we have previously developed successful technologies, such as the treatment of scar formation and cell proliferation with paclitaxel and its analogues or derivatives, or where we have particular scientific focus or expertise. |
| • | Developing Novel, Proprietary Product Candidates. After prioritizing opportunities, we identify the biology that may contribute to complications or failures of a device or procedure. We may then engineer a novel medical device product design, or screen and select a potential drug or drugs to combine with our biomaterial or drug delivery technologies to create proprietary drug-device combinations or other novel product candidates. |
| • | Establishing and Developing Our Intellectual Property Portfolio. After identifying potentially useful medical technologies or developing novel product candidates, we incorporate these elements into new patent filings or our existing patent portfolio, and we attempt to develop and establish new intellectual property in jurisdictions throughout the world. |
| • | Selecting Commercialization Path. Once we reach an advanced stage with a technology or product candidate, we select a development and commercialization path. Where we have a distribution channel, we may elect to fully develop a product ourselves or, in certain cases where product opportunities may not fit our commercial capabilities, we will license certain of our products, technologies or product candidates to be developed, manufactured or commercialized by partners. |
| • | Pursuing Selective Strategic Acquisitions and Licenses. To support our product development and commercialization activities, we have pursued, and will continue to selectively pursue, acquisitions or licenses to obtain proprietary pharmaceutical compounds or compound classes, formulation technologies, medical device technologies and intellectual property or other commercial assets. |
3
Table of Contents
Business Overview
Medical Products
Our Medical Products segment manufactures and markets a wide range of single-use specialty medical products and medical device components. These products are sold directly to end users or other third-party medical device manufacturers. This segment contains specialized direct sales and distribution capabilities as well as significant manufacturing capabilities. Many of our medical products are made using our proprietary manufacturing processes and/or are protected by our intellectual property.
Certain of our product lines, referred to as our Proprietary Medical Products, are marketed and sold by our direct sales group. We believe these product lines contain technology advantages that may have the potential for more substantial revenue growth as compared to our overall product portfolio. We also may invest significant capital and resources in sales and marketing and research and development with respect to existing and potential new Proprietary Medical Products.
Certain of our product lines, referred to as our Base Medical Products, represent more mature finished goods product lines. These product lines are supported by a small group of direct sales personnel, as well as a network of independent sales representatives and medical product distributors. These sales tend to exhibit greater volatility or slower growth when compared to sales of our Proprietary Medical Products. This is particularly the case with our sales of components to third-party medical device manufacturers, which may be impacted by customer concentration and business issues that certain of our large customers may face, as well as to a more limited extent by economic and credit market conditions.
Proprietary Medical Products
Our most significant Proprietary Medical Products have included:
| • | Quill™ Knotless Tissue-Closure Device. Our Quill™ Knotless Tissue-Closure Device (“Quill”) product line is a surface modified suture material that contains proprietary patterns of tiny barbs or other patterns, which can eliminate the need for surgeons to tie knots when closing certain wound types. We believe that use of Quill may lead to faster surgical times, improved wound cosmesis and lower wound infection and complication rates. Quill may also provide for stronger wound closure due to its ability to distribute tension across the entire length of a wound over large numbers of barbs or anchors, as opposed to a far fewer number of knots or anchor points with traditional sutures or surgical staples. Due to this ability to distribute tension, we believe Quill may enable certain tissue types to be repaired that are not addressable by other wound closure technologies. |
| • | SKATER™ Drainage Catheters. Our SKATER drainage catheter product (“SKATER”) is used to facilitate drainage of fluid from wounds, infectious tissues or surgical sites. SKATER features larger lumens and drainage holes, kink resistance, resistance to encrustation and high radiopacity. SKATER is offered in multiple sizes and configurations to address the most typical drainage applications including specific designs for centesis, or small volume drainage procedures, drainage from the biliary duct and nephrostomy procedures. |
| • | BioPince™ Full Core Biopsy Devices. Our BioPince biopsy instrument (“BioPince”) product line is used to obtain tissue samples from patients for analysis and diagnosis of disease. BioPince features a proprietary tri-axial “Cut and Trap” cannula system, which allows the device to deliver cylindrical, full-length biopsy specimens that are complete and largely undamaged, which we believe significantly improves the diagnostic value of the sample. |
| • | 5-FU-Eluting Medical Device Product Candidates. Our proprietary 5-flourouracil (“5-FU”) technology is designed to be combined with selected implantable medical devices in order to reduce infections which may occur concurrently with the use of such devices. Through our proprietary drug identification strategy, we elected to evaluate 5-FU, a drug previously approved by the FDA for treatment of various types of cancer, as a compound that may help to prevent certain types of infection that can occur with medical device implants, including for example, central venous catheters (“CVC”). CVCs are usually inserted into critically ill patients for extended periods of time to administer fluids, drugs, and nutrition, as well as |
4
Table of Contents
| facilitate frequent blood draws. To evaluate the safety and efficacy of our 5-FU CVC, we conducted a randomized, single-blind, 960-patient, 25-center study to evaluate whether our 5-FU-eluting CVC prevents bacterial colonization at least as well as the market-leading anti-infective CVC. In October 2007, we announced this study had met its primary statistical endpoint of non-inferiority as compared to the market leading anti-infective CVC (a chlorhexidine / silver sulfadiazine (“CH-SS”) coated CVC) and indicated an excellent safety profile. There were no statistically significant differences in the rate of adverse events related to the study devices, or in the rates of catheter-related bloodstream infections. Additionally, there was no evidence for acquired resistance to 5-FU in clinical isolates exposed to the drug for a second time. Based on the positive results achieved in the study, in December 2007, we filed a request for 510(k) clearance from the FDA to market and sell the CVC in the U.S. and in April 2008, we announced that we had received 510(k) clearance from the FDA to market our 5-FU CVC in the U.S. As a result of our significantly constrained liquidity and capital resources, we have to date not been able to pursue commercialization activities for this product. Development of future launch plans and commercialization of this product, as well as related product line extensions, will depend on our liquidity, capital resources, or our ability to secure future financing. |
Base Medical Products
Our Base Medical Products are comprised of four primary product groups, including:
| • | Biopsy Products. We manufacture, market and sell a wide range of soft tissue biopsy products, both disposable and re-usable for use in different types of breast, lung, spinal and bone marrow biopsies. Some key features of our soft tissue biopsy products include echogenic tips for placement under ultrasound guidance, numerically ordered centimeter markings to facilitate precise depth placement, and adjustable needle stops allowing the restriction of forward movement and localizing the needle to the biopsy site. We also offer a range of bone marrow biopsy products. The most significant of these product lines, the T-Lok™, has an ergonomically designed twist-lock handle, which improves a clinician’s ability to penetrate hard bone to obtain the biopsy sample. An adjustable needle stop to control the depth of penetration allows the clinician to safely aspirate at the sternum of the patient. Other selected products in our biopsy group include fixation pins, breast localization markers, tunneling stylets and surgical tunnelers, and isotope seed spacers and needles for prostate cancer treatment. |
| • | Ophthalmic Products. We manufacture, market and sell a wide range of surgical blades, marketed under the SharpPoint™ and SharpGuard™ brands, which are used primarily in cataract or other ophthalmic surgery where very small incisions are required. Our products include clear corneal knives, standard implant knives, pilot tip implant knives, precision depth knives, Sharptome™ crescent knives, spoon knives, stab knives, vitrectomy knives to create small, precise incisions, sub-2mm series knives, and a variety of slit and specialty knives. Other selected products in our ophthalmic group include a variety of cannula needles for the administration of anesthetic or for irrigation or aspiration in ophthalmic surgery, a full line of absorbable and non-absorbable microsurgical ophthalmic sutures, our UltraPlug™ punctum plugs for treatment of dry eye symptoms and our Ultrafit™ trephine blades for penetrating keratoplasty surgery and the Tan EndoGlide for the precise transplantation of human corneal endothelial cells. |
| • | Surgery Products. We manufacture, market and sell a wide range of traditional surgical wound closure products, sold primarily under our Sharpoint and Look™ brand names. These products are used by surgeons in a wide variety of applications in hospitals, surgery centers and physician offices. We also sell a significant amount of our general suture products for use in dental surgery. Our suture configurations vary in diameter, length, and material. Our product lines also include a full line of bio-absorbable and non-absorbable suture materials, including PGA, Monoderm™, polypropylene, poly-ethylene-terephthatate, glycolide and e-caprolactone, polyglycolic acid, plain and chromic gut, nylon and silk. This wide range of materials allows us to offer a range of handling characteristics and bio-absorption times for our product line. Our product lines also include a wide variety of needle types, including drilled-end and channel |
5
Table of Contents
| surgical needles. Drilled-end suture needles have a precisely drilled butt end for maximum suture holding strength and are adequately tempered for securing strong attachment of the suture. Channel needles have a channel with an underlying receptacle for attachment of the suture. |
| • | Medical Device Component Manufacturing. We manufacture, market and sell a wide range of medical device components for other medical device manufacturers, who then use the provided components as part of assembling their own finished medical device products. The components we manufacture and sell are often similar to selected components used in our own Base Medical Product offerings, or are custom components produced for certain customers by utilizing specific proprietary capabilities or expertise. Our most significant manufacturing capabilities include metal grinding and polishing, plasma welding, chemical etching, electropolishing and electro-chemical marking, plastics injection molding, plastics and polymer extrusion and silk suture formation, weaving of medical grade textiles and packaging and assembly. In addition, our manufacturing facilities each maintain engineering and machining capabilities in order to support production orders and new product opportunities for our key customers. |
Pharmaceutical Technologies
Our Pharmaceutical Technologies segment focuses primarily on establishing product development and marketing collaborations with major medical device, pharmaceutical or biomaterials companies, and to date has derived the majority of its revenue from royalties due from partners that develop, market and sell products incorporating our technologies. Our principal revenues in this segment are from royalties derived from sales by BSC of TAXUS. We also have a partnership with Cook Medical Inc. (“Cook”) whereby Cook is utilizing our proprietary paclitaxel technology in peripheral vascular stent applications, specifically in connection with its Zilver® PTX® paclitaxel-eluting peripheral vascular stent, which is currently approved for sale in the European Union, and which is awaiting approval for sale in the U.S. If Cook were to receive approval to market and sell Zilver-PTX in the U.S., we may begin to receive more substantive royalties derived from sales by Cook of Zilver-PTX peripheral stent systems in 2011.
TAXUS Paclitaxel-Eluting Coronary Stents
The most significant commercial products containing technology developed and licensed by our Pharmaceutical Technologies segment are the TAXUS Express, TAXUS Liberté® and TAXUS Element coronary stent systems. These products are sold by BSC, our exclusive licensee in the coronary paclitaxel-eluting stent field. We are entitled to receive royalties based on the commercial sale of all of BSC’s various TAXUS products. BSC has received approval in various countries throughout the world and commenced commercial sales of a broad portfolio of coronary stent products utilizing our proprietary paclitaxel technology, including:
| • | January 2003. BSC commenced sales of the TAXUS Express paclitaxel-eluting coronary stent system in the European Union and other countries outside of the United States. |
| • | March 2004. BSC commenced sales of the TAXUS Express paclitaxel-eluting coronary stent system in the United States. |
| • | January 2005. BSC commenced sales of the TAXUS Liberté paclitaxel-eluting coronary stent system in 18 countries outside of the European Union and the U.S. The TAXUS Liberté stent system represents BSC’s second generation commercial product incorporating our research, technology and intellectual property related to the use of paclitaxel to treat restenosis and other local inflammatory diseases. |
| • | September 2005. BSC commenced sales of the TAXUS Liberté paclitaxel-eluting coronary stent system in Europe. |
| • | September 2008. BSC received approval to market and sell the Taxus Express2 Atom™ Paclitaxel-Eluting Coronary Stent System in the United States. The TAXUS Atom stent systems are the only drug-eluting stents available that are specifically designed to treat lesions with diameters as small as 2.25 millimeters. |
6
Table of Contents
| • | October 2008. BSC commenced sales of the TAXUS Liberté paclitaxel-eluting coronary stent system in the U.S. |
| • | May 2009. BSC commenced sales of the TAXUS Liberté Atom Paclitaxel-Eluting Coronary Stent System in the U.S. |
| • | July 2009. BSC commenced sales of the TAXUS Liberté Long Stent in the U.S., which at 38 millimeters is the longest available drug-eluting stent. |
| • | June 2010. BSC commenced sales of the TAXUS Element paclitaxel-eluting coronary stent in the Europe. The TAXUS Element stent system represents BSC’s third-generation commercial product incorporating our research, technology and intellectual property related to the use of paclitaxel to treat restenosis and other local inflammatory diseases. |
BSC has concluded several clinical trials of paclitaxel-eluting coronary stents. These various studies have universally indicated that paclitaxel-eluting coronary stents significantly reduce the incidence of restenosis and the need for repeat surgical procedures at the treated lesion site in coronary artery disease patients. In addition, there have been independent clinical trials of paclitaxel-eluting coronary stents that have demonstrated similarly positive results.
Certain clinicians suggested in 2006 that the use of drug-eluting stents may cause an increased rate of late thrombosis (the formation of blood clots in the stent long after the initial stent implantation) and in turn, potentially lead to an increased rate of myocardial infarctions (heart attacks) or deaths as compared to bare metal stents. These suggestions led to a significant decrease in the use of drug-eluting stents, beginning in the second half of 2006 and continuing through 2008. While subsequent analysis of clinical data suggest that these concerns may have been unreasonable and use of drug-eluting stents began to recover in 2009, usage has yet to reach to the levels observed in 2005 prior to the initial reporting of physician concerns.
In addition, starting in mid 2008 and early 2009, several new competitive drug-eluting stents were made commercially available in the United States, Europe and Japan. The reduction in drug-eluting stent usage, combined with the entry of new competitors, has had significant negative impacts on the levels of royalty revenue we receive from BSC in 2008, 2009 and 2010. Royalty revenues derived from sales of stent systems from BSC declined by 32% from 2008 to 2009 and 47% from 2009 to 2010.
Zilver-PTX Paclitaxel-Eluting Peripheral Vascular Stents
The Zilver-PTX stent, which is under evaluation in clinical trials being conducted by our partner Cook, is a specialized stent product incorporating our proprietary paclitaxel technology and is designed for placement in diseased arteries in the limbs to restore blood flow. We license Cook the rights to use paclitaxel with peripheral stents and certain other non-coronary medical devices. Subject to the terms of our license agreement, as amended, with Cook, we are entitled to receive royalty payments upon the commercial sale of paclitaxel-eluting peripheral vascular stent products, including the Zilver-PTX stent.
In August 2009, we announced that our partner Cook had reported CE Mark approval and commercial launch of the Zilver-PTX paclitaxel-eluting peripheral vascular stent in the European Union (“E.U.”). In June 2010, we announced Cook had submitted its Pre-Market Approval (PMA) application to the U.S. Food and Drug Administration (FDA) to request approval to market and sell Zilver –PTX in the U.S.
Cook has conducted several clinical trials of Zilver-PTX, These various studies, including Cook’s pivotal clinical trial designed for submission to the FDA to seek approval to market and sell Zilver-PTX in the U.S., have indicated that Zilver-PTX significantly reduces the incidence of restenosis at the treated lesion site in peripheral vascular disease patients as compared to balloon angioplasty, a traditional treatment for peripheral vascular disease.
Research and Product Development
We maintain significant research and development capabilities and personnel. Our capabilities include medical device and mechanical engineering, drug formulation, analytical chemistry, drug delivery and carrier manipulation and formulation. We also employ personnel with expertise in regulatory affairs and quality control that are responsible for regulatory and quality control activities with respect to certain of our existing and new product candidates.
7
Table of Contents
We, or our partners, are evaluating several new product candidates, including certain product candidates undergoing human clinical testing conducted both by us and our partners. We also maintain certain pre-clinical or research stage programs and conduct various product and process improvement initiatives related to our currently marketed products and our manufacturing facilities. The following discussion outlines our most advanced product candidates and their stage of development, and summarizes our most recently concluded or terminated clinical development programs.
8
Table of Contents
Ongoing Clinical Programs
The following discussion describes our product candidates, or certain of our partners’ product candidates, that are being evaluated in ongoing human clinical trials and their stage of development:
Partner Clinical Programs
Independent TAXUS Clinical Data
a) In May 2009 we announced that BSC had disclosed the publication of an article in the Journal of the American College of Cardiology (“JACC”) reviewing data on more than 19,000 patients from the Swedish national registry who were evaluated for restenosis, or the re-narrowing of arteries, after percutaneous coronary intervention (“PCI”). The Swedish Coronary Angiography and Angioplasty Registry holds data on all patients undergoing PCI in Sweden. The objective of this independent study was to evaluate restenosis rates of drug-eluting stents (“DES”) in patients with and without diabetes in a real-world setting. The article reported that patients who received a TAXUS Liberté® paclitaxel-eluting stent had numerically lower incidences of repeat procedures to treat restenosis at two years as compared to patients treated with ‘olimus-based DES, including Johnson & Johnson, Inc.’s Cypher® stent and Medtronic Inc.’s Endeavor® stent.
In the patients with diabetes, the TAXUS Liberté demonstrated a statistically significant lower restenosis rate compared to the Endeavor, which had more than two times the risk of repeat procedures. The JACC article reported that both the TAXUS Liberté stent and BSC’s first-generation paclitaxel-eluting stent, the TAXUS Express®, were the only stents in the study showing no increased risk of restenosis for patients with diabetes as compared to those without diabetes. Data for both the Cypher and Endeavor stents indicated significant increased risk of restenosis in patients with diabetes. In addition, the study showed that the TAXUS Liberté had an approximately 23 percent lower restenosis rate at two years compared to the prior-generation TAXUS Express.
The Swedish registry study included four DES brands: TAXUS Liberté, TAXUS Express, Cypher and Endeavor. In total, the registry included 35,478 DES implants during 22,962 procedures in 19,004 patients, with 1,807 restenoses reported over a mean 29-month follow-up period. For the entire study population, the repeat revascularization rate per stent was 3.5 percent after one year and 4.9 percent after two years. Overall, the adjusted risk of restenosis was 1.23 times higher in patients with diabetes than in patients without diabetes. In patients with diabetes, restenosis was higher in the non-TAXUS stents. The sirolimus-eluting Cypher and the zotorolimus-eluting Endeavor had higher restenosis rates in patients with diabetes compared with those in patients without diabetes (1.25 times and 1.77 times, respectively).
b) On March 16, 2010, we announced BSC’s results from an analysis of one-year subset data from the HORIZONS acute myocardial infarction trial assessing the impact of diabetes on clinical and angiographic outcomes in heart attack patients treated with the TAXUS Express2 Paclitaxel-Eluting Stent System or the Express bare-metal stent. The results demonstrated that the TAXUS Express Stent significantly reduced ischemia-driven target lesion revascularization (“TLR”) at one year and binary in-stent restenosis at 13 months in diabetic patients experiencing an acute myocardial infarction compared to an otherwise identical bare-metal control stent. Analysis of the data was presented on March 16, 2010 at the American College of Cardiology Annual Scientific Sessions.
On September 25, 2010 we further announced that BSC released three-year follow-up data from the HORIZONS-AMI trial. The trial, sponsored by the Cardiovascular Research Foundation (“CRF”) with grant support from Boston Scientific and The Medicines Company, is designed to determine the safety and efficacy of the TAXUS® Express2™ Paclitaxel-Eluting Coronary Stent System compared to bare-metal stenting in patients experiencing an acute myocardial infarction (“AMI”), or heart attack. With 3,006 patients enrolled worldwide, HORIZONS-AMI is the largest randomized trial to compare the use of drug-eluting stents to bare-metal stents for AMI patients. Analysis of the data was presented by Gregg W. Stone, M.D., Professor of Medicine and the Director of Research and Education at the Center for Interventional Vascular Therapy at the Columbia University Medical Center/New York-Presbyterian Hospital and Principal Investigator of the trial, at CRF’s annual Transcatheter Cardiovascular Therapeutics (“TCT”) scientific symposium in Washington, D.C. HORIZONS-AMI demonstrated that the TAXUS Express Stent
9
Table of Contents
significantly reduced clinical and angiographic restenosis compared to an otherwise identical bare-metal Express® control stent. After three years follow-up, the primary efficacy endpoint of ischemia-driven TLR was 9.4 percent for patients treated with TAXUS Express vs. 15.1 percent for bare-metal Express (p<0.001), a relative reduction of 40 percent. The secondary efficacy endpoint of ischemia-driven target vessel revascularization was 12.4 percent for TAXUS Express vs. 17.6 percent for bare-metal Express (p<0.001), a relative reduction of 32 percent. The primary safety endpoint of major adverse cardiac events (“MACE”) at three years was comparable for TAXUS Express and bare-metal Express patients (13.6 percent vs. 12.9 percent, respectively, p=0.66), which is consistent with findings at one and two years. Individual rates of death, repeat heart attack, stroke, and stent thrombosis between the two groups through three years of follow-up were also comparable.
On September 22, 2010 we announced that BSC had released comprehensive data from the TAXUS ATLAS clinical program, a series of global, prospective, single-arm trials evaluating the TAXUS® Liberté® Paclitaxel-Eluting Stent System in a variety of lesions and patient groups. Five-year results from the TAXUS ATLAS trial and four-year results from the TAXUS ATLAS Small Vessel and Long Lesion trials continue to show significant advantages for the thin-strut TAXUS Liberté Stent when compared to the first-generation TAXUS® Express® Stent. Analysis of the data was presented by the Co-Principal Investigators of the TAXUS ATLAS trials, Mark A. Turco, M.D., Director of the Center for Cardiac and Vascular Research, Washington Adventist Hospital, and John A. Ormiston, M.D., Mercy Angiography Unit, Mercy Hospital, Auckland, New Zealand, at CRF’s annual TCT scientific symposium in Washington, D.C.
TAXUS Element™ Paclitaxel-Eluting Coronary Stent System.
The platinum chromium TAXUS Element paclitaxel-eluting coronary stent system (“TAXUS Element Stent System”) is the third-generation BSC coronary stent platform that incorporates our research, technology and intellectual property related to the use of paclitaxel. The TAXUS Element stent features BSC’s proprietary platinum chromium alloy, which is designed to enable thinner stent struts, increased flexibility and a lower stent profile while improving radial strength, recoil and radiopacity. In addition, the TAXUS Element stent platform incorporates new balloon technology intended to improve upon BSC’s market-leading Maverick® balloon catheter technology.
The platinum chromium TAXUS Element Stent System is currently being evaluated by BSC in its PERSEUS clinical trial, which commenced in July 2007. On October 8, 2008, we announced that BSC had completed enrollment in the PERSEUS clinical trial. The PERSEUS clinical program compared the TAXUS Element stent to prior-generation stents in more than 1,600 patients in two parallel trials at 90 centers worldwide. This clinical program evaluated the safety and efficacy of the TAXUS Element stent in two studies and is designed for submission to the FDA in an application for approval to market and sell the TAXUS Element Stent System in the U.S. On March 15, 2010, we announced BSC’s results for this clinical trial. The first study evaluated the safety and efficacy of the TAXUS Element stent compared to the TAXUS Express2 stent in 1,264 patients with “workhorse” lesions from 2.75 to 4.0 mm. This prospective, randomized (3:1) trial met its primary endpoint of non-inferiority for target lesion failure (“TLF”) at 12 months with rates of 5.6 percent for the TAXUS Element Stent and 6.1 percent for the TAXUS Express2 stent. The secondary endpoint of in-segment percent diameter stenosis at nine months, as measured by quantitative coronary angiography, was also met. The second study compared the TAXUS Element stent to a historic control (TAXUS V de novo bare-metal Express coronary stent system) in 224 patients with lesions from 2.25 to 2.75 mm. The trial met its primary endpoint of superiority for in-stent late loss at nine months with unadjusted values of 0.38 mm for the TAXUS Element stent and 0.80 mm for the Express stent, which was statistically significant. The trial also met its secondary endpoint of superiority for TLF at 12 months, showing a reduction, which was statistically significant, with an unadjusted rate of 7.3 percent for the TAXUS Element stent compared to a pre-specified performance goal of 19.5 percent based on historical outcomes for the Express control stent.
On September 22, 2010 we announced BSC released results from an analysis of 1,166 patients from the PERSEUS clinical program comparing the performance of the TAXUS Element Stent System in diabetic versus non-diabetic patients. Results demonstrated that despite the known increased risk of restenosis for diabetics versus non-diabetics in patients undergoing coronary revascularization, the TAXUS Element Stent had comparable levels of TLR and late loss in both diabetic and non-diabetic patients. Analysis of the
10
Table of Contents
data was presented by Louis A. Cannon, M.D., of the Cardiac and Vascular Research Center of Northern Michigan in Petoskey, Michigan, and Co-Principal Investigator of the PERSEUS clinical program, at CRF’s annual TCT scientific symposium in Washington, D.C. The PERSEUS diabetic analysis included clinical outcomes at one year among 314 diabetic patients and 852 non-diabetic patients treated with the TAXUS Element Stent from the PERSEUS Workhorse and Small Vessel clinical trials. Due to significant disparity in baseline characteristics between diabetic and non-diabetic patients, propensity score analysis was used to allow for adjustment of baseline differences (other than the presence of diabetes) between the two groups. Results showed that the TAXUS Element Stent maintained comparable rates of TLR at one year, whether adjusted or unadjusted, in the diabetic and non-diabetic patient populations (5.5 percent vs. 4.1 percent, p=0.43, adjusted). The adjusted and unadjusted rates of TLF at one year (defined as ischemia-driven TLR, or myocardial infarction (“MI”)/cardiac death related to the target vessel) were also similar between the patient groups (7.5 percent vs. 5.4 percent, p=0.31, adjusted). Adjusted one-year rates of MACE, cardiac death, MI and Academic Research Consortium definite/probable stent thrombosis showed no differences between the two populations (p-values of 0.14, 0.12, 0.38, and 0.77, respectively). Nine-month adjusted angiographic outcomes showed similar in-segment late loss in diabetics and non-diabetics (0.23 mm vs. 0.19 mm, p=0.52). Rates of late loss for the TAXUS Element Stent were numerically lower than rates in prior studies for the TAXUS Express® and TAXUS Liberté® Stents.
In May 2010, we announced that BSC received CE Mark approval for the TAXUS Element Stent System, which included approval for a specific indication for the treatment of diabetic patients. In June 2010, we further announced that BSC had commercially launched and implanted its first TAXUS Element Stent System in the European Union and other CE Mark countries.
In the U.S. BSC expects FDA approval for the TAXUS Element Stent System in mid 2011. In Japan, BSC expects approval for the TAXUS Element Stent System in late 2011 or early 2012.
Zilver-PTX Paclitaxel-Eluting Peripheral Vascular Stent System
The Zilver PTX paclitaxel –eluting peripheral vascular stent (“Zilver PTX Stent System”) is a specialized stent, which incorporates our proprietary paclitaxel technology, and is designed for placement in diseased arteries in the limbs to restore blood flow by expanding and holding open the arteries upon deployment. The Zilver PTX Stent achieves targeted drug delivery without using a polymer to adhere the drug to the stent body, which eliminates the potential patient risks associated with polymer-coated devices, including clot formation and inflammation. Cook is a co-exclusive licensee, together with BSC, of our proprietary paclitaxel technology to reduce restenosis following stent placement in patients suffering from peripheral artery disease (“PAD”). The Zilver PTX stent is currently undergoing multiple human clinical trials in the E.U., U.S., Japan, and selected other countries to assess product safety and efficacy.
It is estimated that approximately 27 million people suffer from PAD in Europe and North America. Of patients with PAD, approximately only one quarter indicate any substantive symptoms. The “silent” nature of this condition can result in a number of patients being diagnosed only when their condition has progressed to the severe stage. In patients with severe PAD whose condition is not improving with risk-factor modification, exercise programs or pharmacological therapy, invasive surgical procedures may be necessary. These procedures may include angioplasty, stenting or surgery. To date, stent procedures for PAD in the limbs have been limited due to high observed rates of restenosis or other failure of the procedure requiring a subsequent surgical procedure, as well as risk of stent fracture with existing products, as these stents are exposed and not protected by the patient’s anatomy as with coronary stents.
In August 2009, we announced that our partner Cook had reported CE Mark approval and commercial launch of the Zilver PTX stent in the E.U. and in June 2010, we announced that Cook had submitted its Pre-Market Approval (“PMA”) application to the FDA for Zilver PTX. We believe the data from the clinical trial described below supports this PMA application.
In January 2007, Cook released nine-month data from its E.U. clinical study. The preliminary data presented by Cook on the first 60 patients in the randomized trial, which is examining the safety of using Cook’s Zilver PTX stent to treat blockages, or lesions, of the superficial femoral artery (“SFA”) above the knee, indicated that the Zilver PTX stent showed an equal adverse event rate to conventional angioplasty for treating SFA lesions. The Zilver PTX stent also displayed a zero percent fracture rate for 41 lesions at six months and 18 lesions at one year.
11
Table of Contents
In July 2007, Cook announced that the first U.S. patients in a randomized pivotal human clinical trial of the Zilver PTX stent were treated at Tri-City Medical Center in Oceanside, CA. The trial is designed to randomize patients to receive either the Zilver PTX stent or balloon angioplasty. Data from this clinical trial is intended to be used to support submission to the FDA for approval in the U.S. to market the device. In addition, data collected on Japanese and U.S. patients is expected to be combined for the final evaluation of the device and used for regulatory submissions in both markets for approval.
In June 2008, Cook reported positive interim results from the registry arm of a clinical study designed to measure the efficacy of the Zilver PTX stent in treating PAD patients, specifically in the treatment of blockages in the femoropopliteal artery.
The data published in the Zilver PTX registry involved 791 patients from Europe, Russia, Canada and Korea demonstrated highly positive results. Only 8 percent of all patients with de novo (new) lesions required a reintervention to reopen the artery in the first 12 months — a rate significantly surpassing existing treatments for PAD in the SFA, such as balloon angioplasty and bare metal (non-drug-eluting) stents. In addition, specific patient groups that are often very hard to treat, such as diabetics and patients with in-stent restenosis (those treated previously with a noncoated stent), were shown in the trial to benefit from the Zilver PTX. As indicated by the trial data, the superior results achieved in the first year have been largely maintained throughout 24 months, which is an important clinical milestone. In comparison with other trial data obtained, the Zilver PTX stent demonstrated a reduction in reintervention of between 50 percent and 75 percent, which is an important patient benefit.
The results were reported by trial investigators at the 2008 SVS Vascular Annual Meeting, and revealed clinical improvement, excellent durability and fracture resistance, high rates of event-free survival (“EFS”) and freedom from TLR. Interim data was compiled at six and 12 months using 435 patients and 200 patients, respectively. The corresponding EFS rates were 94% and 84%, and freedom from TLR was 96% and 88%. Evaluation of stent x-rays is ongoing, and currently suggests stent fractures in approximately one percent of cases at six months and less than two percent of cases at 12 months. In addition, the Zilver PTX stent exhibited no safety concerns and results were better than expected for Trans-Atlantic Inter-Society Consensus class C and D lesions, occlusions, in-stent restenosis and lesions greater than seven centimeters. Follow-up to the registry arm of the study will continue through two years.
In September 2008, Cook announced it had completed enrollment in its pivotal human clinical trial for the Zilver PTX. The 420 patients enrolled in Cook’s randomized trial include PAD patients treated in Germany, the U.S. and Japan. On the same date, Cook announced that it had enrolled an additional 780 patients in the E.U., Canada and Korea in a clinical registry to evaluate the safety of the Zilver PTX stent. Those data were used to obtain CE Mark approval in the E.U. and launch the device there, with additional regulatory submissions pending in additional markets.
In April 2009, we announced that Cook had reported data that showed that 82 percent of patients who were treated with Cook’s Zilver PTX stent were free from reintervention at two-year follow up. The Zilver PTX Registry study, involving 792 patients globally, is assessing the safety and efficacy of the Zilver PTX in treating peripheral artery disease. The most recent results were reported at the 31st International Symposium Charing Cross Controversies Challenges Consensus. Data was compiled at 12 and 24 months for 593 patients and 177 patients respectively. The registry, which enrolled a broad spectrum of patients, includes patients with complex lesions (e.g., long lesions, total occlusions, in-stent restenosis). The corresponding event-free survival rates were 87 percent and 78 percent, respectively, and freedom from target lesion revascularization was 89 percent and 82 percent, respectively. Clinical measures that included ankle-brachial index, Rutherford score, and walking distance and speed scores showed significant improvement at six and 12 months and were maintained through 24 months. Detailed evaluation of stent x-rays demonstrated excellent stent integrity through 12 months, confirming previously published results showing 99 percent completely intact stents (less than 1 percent stent fracture rates observed) with a mean follow up of 2.4 years in the challenging superior femoral artery and popliteal arteries, including behind the knee locations.
12
Table of Contents
In April 2010, we announced that Cook had enrolled its first patient in its landmark Formula™ PTX® clinical trial. The trial is the first of its kind to evaluate the safety and effectiveness of a paclitaxel-eluting stent to treat renal artery disease, the narrowing of the arteries that supply blood to the kidneys. The multi-center, randomized trial plans to enroll 120 patients at sites across Europe. The trial utilizes Cook’s Formula renal stent, which is designed with a very low profile that may help it cross tightly blocked vessels for placement into diseased renal arteries.
In May 2010, we announced that Cook presented one-year data at Euro PCR that confirmed sustained clinical outcomes with Zilver PTX. According to data presented, 86.2% of all patient subgroups treated with Zilver PTX demonstrated vessel patency at 12 months without the requirement for an additional intervention. The data was collected as part of the world’s largest clinical study of endovascular treatment for PAD in the SFA. The trial is based on a group of 787 patients, including symptomatic patients, diabetics, and those with the most complex lesions, including long lesions, total occlusions and in-stent restenosis.
On September 24, 2010 we announced that a summary of the final clinical trial results for the randomized study of Cook’s Zilver PTX Stent for use in patients with peripheral arterial disease in the SFA was presented at CRF’s annual TCT symposium in Washington D.C. The study met its 12-month primary endpoint showing non-inferior EFS and superior patency for the Zilver PTX as compared to balloon angioplasty. Cook Medical, a license holder of our proprietary paclitaxel technology, has developed Zilver-PTX, a self-expanding, highly durable nitinol stent that uses a proprietary technology to deliver a locally therapeutic dose of paclitaxel, a proven anti-restenotic drug, to the target lesion.
This multicenter, multinational, prospective, randomized study compared the safety and effectiveness of the Zilver PTX to balloon angioplasty and bare metal stenting. Patients with de novo or restenotic SFA lesions were randomized to balloon angioplasty or Zilver PTX stent placement. Balloon angioplasty patients experiencing acute failure (> 30% residual stenosis) underwent secondary randomization to provisional stenting with Zilver BMS (bare metal stent) or Zilver PTX. Endpoints included EFS, stent integrity by radiographic core laboratory analysis and primary patency by Duplex ultrasound core laboratory analysis (peak systolic velocity ratio < 2.0). The study enrolled 479 patients at 55 institutions in the United States, Japan and Germany, with 241 patients randomized to the Zilver PTX group and 238 to the balloon angioplasty group. Demographics and lesion characteristics were similar for both groups. Approximately half the balloon angioplasty group experienced acute failure and underwent secondary randomization, assigning 59 and 61 patients to provisional stenting with Zilver BMS and Zilver PTX, respectively.
Angiotech Clinical Programs
MultiStem® Stem Cell Therapy
The MultiStem stem cell therapy is under evaluation in clinical trials being conducted together with our partner Athersys, Inc. (“Athersys”) for the treatment of acute myocardial infarction. MultiStem stem cells are proprietary adult stem cells derived from bone marrow that have demonstrated the ability in laboratory experiments to form a wide range of cell types. MultiStem may work through several mechanisms, but a primary mechanism appears to be the production of multiple therapeutic molecules produced in response to inflammation and tissue damage. We and Athersys believe that MultiStem may represent a unique “off the shelf” stem cell product candidate, based on its potential ability to be used without tissue matching or immunosupression, and its potential capacity for large scale production. We entered into an agreement with Athersys in May 2006 to co-develop and commercialize MultiStem for use in the indications of acute myocardial infarction and peripheral vascular disease. On December 20, 2007, we and Athersys announced we had received authorization from the FDA to commence a phase I human clinical trial to evaluate the safety of MultiStem in the treatment of acute myocardial infarction.
In February 2010, Athersys announced that enrollment for the phase I clinical trial had been completed. The phase I clinical trial is an open label, multi-center dose escalation trial evaluating the safety and maximum tolerated dose of a single administration of allogeneic MultiStem cells following an acute myocardial infarction. Following standard treatment, enrolled patients receive MultiStem delivered via a catheter that enables rapid and efficient delivery of MultiStem into the region of damage in the heart. The
13
Table of Contents
study is being conducted at multiple cardiovascular treatment centers in the United States, including the Cleveland Clinic, Columbia University Medical Center and Henry Ford Health System, and includes patients in three treatment cohorts or dose groups, as well as a non-treated registry group. In preclinical studies conducted by Athersys and independent cardiovascular research teams, administration of MultiStem following an ischemic injury to the heart has been associated with a number of benefits, including an increase in ejection fraction, or volume of blood pumped from the heart, a reduction of inflammation in the region of ischemic injury and increased angiogenesis, each believed to help augment recovery and healing.
In July 2010, we announced that Athersys had announced positive results from its phase I clinical trial of MultiStem®. The study results, which represented at least four months of post-treatment patient data, demonstrated that MultiStem was well tolerated at all dose levels and also suggested improvement in heart function in treated patients.
In September 2010, updated study results were presented at the TCT conference held in Washington, D.C. Dr. Marc Penn, M.D., Ph.D., co-principal investigator of this study and Director of Cardiovascular Cell Therapy at the Cleveland Clinic, and Director of the Skirball Laboratory for Cardiovascular Cellular Therapeutics, presented the results at the Symposium “Strategies for Cardiovascular Repair: Stem Cell Therapy and Beyond.”
New data presented by Dr. Penn included additional information about the nature and incidence of adverse events (“AEs”) over the first four months of the trial, demonstrating that the AEs were generally mild-to-moderate in nature, there was no dose dependent effect of MultiStem on AEs, and overall, MultiStem had a favorable safety profile. Further, Dr. Penn shared the results from additional analysis of echocardiogram data collected over the first four months of the study, which suggests that MultiStem administration may also provide improvements in other measures of heart function. Patients receiving MultiStem, for instance, demonstrated a meaningful improvement in mean wall motion score at four months compared to baseline, though this improvement was not statistically significant. Among those patients with more severe heart attacks (i.e. left ventricular ejection fraction, a measure of heart function, £ 45), the mean wall motion score for treated patients improved over the four-month period, while for registry patients it worsened over this time.
On September 23, 2010 we, along with our partner Athersys, announced updated results from the phase I clinical trial of MultiStem®, an allogeneic cell therapy product, administered to individuals following acute myocardial infarction, more commonly referred to as a heart attack. The phase I clinical trial was an open label, multi-center dose escalation trial evaluating the safety and maximum tolerated dose of a single administration of allogeneic MultiStem cells following an acute myocardial infarction. Enrolled patients received MultiStem delivered via a catheter into the damaged region of the heart 2-5 days following percutaneous coronary intervention, a standard treatment for heart attack. The study included patients in three treatment cohorts or dose groups (20 million, 50 million and 100 million cells per patient) and a registry group where patients received only standard of care. Nineteen treated- and 6 registry subjects were enrolled in the study. The trial was conducted at multiple cardiovascular treatment centers in the United States, including the Cleveland Clinic, Columbia University Medical Center and Henry Ford Health System.
Upon completion of the phase I human clinical trial which is currently being conducted by Athersys, we may, at our option, assume lead responsibility for further clinical development. We currently own marketing and commercialization rights with respect to this product candidate.
Completed or Suspended Clinical Programs
Bio-Seal Lung Biopsy Tract Plug System.
Bio-Seal is a novel technology designed to prevent air leaks in patients having lung biopsies by plugging the biopsy track with an expanding hydrogel plug. On contact with moist tissue, the hydrogel plug absorbs fluids and expands to fill the void created by the biopsy needle puncture. The plug is absorbed into the body after healing of the puncture site has occurred. We are the worldwide manufacturer and distributor of Bio-Seal, which has received CE Mark approval and is currently marketed and sold in the E.U.
Bio-Seal has undergone a human clinical trial in the U.S. which was designed to assess the safety and efficacy of Bio-Seal, with the primary endpoint being reduction in rates of pneumothorax in patients
14
Table of Contents
undergoing lung biopsy procedures. The study was designed to provide a basis for FDA clearance for the commercialization of Bio-Seal. The prospective, randomized, controlled clinical trial enrolled and randomized 339 patients at 15 different investigational sites. Inspiratory upright chest x-rays were performed at 30 to 60 minutes, 24 hours and 30 days after treatment. The trial enrolled its first patient in October 2005 and completed enrollment in June 2008. In March 2009, we announced positive results from this clinical trial at the Society of Interventional Radiologists Annual Scientific Meeting in San Diego, CA. The trial demonstrated a statistically significant clinical benefit in the group receiving Bio-Seal. The Bio-Seal treatment arm hit the primary end point of clinical success for rate of absence of pneumothorax at each time period as compared to traditional treatment. Based on the per-protocol population, the clinical success rate was 85% using Bio-Seal and 69% in the control group. This difference was statistically significant (p=0.002). Although not powered for statistical analysis, positive trends were also observed for Bio-Seal subjects as compared to the control group in various secondary endpoints, including fewer Bio-Seal subjects admitted to the hospital for pneumothoraces (9.4% vs. 13.6%), fewer chest tube placements in Bio-Seal patients (3.5% vs. 10.7%), and fewer additional chest x-rays required in Bio-Seal patients (0.6% vs. 5.3%).
Data from this clinical trial was submitted to the FDA, with the objective of receiving 510(k) clearance to market Bio-Seal in the U.S. Subsequent to this submission, the FDA asked us various questions about the study and our submission and we responded to all of the questions from the FDA. In October 2009, we announced that we had received correspondence from the FDA regarding our 510(k) submission for Bio-Seal, stating the FDA’s view that Bio-Seal is a class III device that requires Pre-Market Approval (“PMA”) and therefore a more extensive clinical study in order to obtain FDA marketing clearance. As a result, we are reviewing our options with respect to this product candidate, including possibly appealing this FDA decision, and we are discussing the possible preparation of a PMA submission with our partner, Biopsy Sciences, LLC. We have not incurred material expense to date with respect to activities regarding this product candidate. Should we elect to continue to pursue development or regulatory approvals for this product candidate that require us to incur material expense, we will provide further updates in our public disclosure.
5-FU-Eluting Central Venous Catheter
CVCs are usually inserted into critically ill patients for extended periods of time to administer fluids, drugs, and nutrition, as well as facilitate frequent blood draws. Through our proprietary drug identification strategy, we have elected to evaluate 5-FU, a drug previously approved by the FDA for treatment of various types of cancer, as a compound that may help to prevent certain types of infection in patients receiving a CVC. We completed a human clinical trial in the U.S. designed to assess the safety and efficacy of our 5-FU-eluting CVC in preventing various types of catheter related infections. The study was a randomized, single-blind, 960-patient, 25-center study and was designed to evaluate whether our 5-FU-eluting CVC prevents bacterial colonization at least as well as the market-leading anti-infective CVC. On July 10, 2007, we announced that we had completed enrollment of the study, and on October 9, 2007, we announced this study had met its primary statistical endpoint of non-inferiority as compared to the market leading anti-infective CVC (a CH-SS coated CVC) and indicated an excellent safety profile. In March 2008, we presented the clinical trial data at the 28th International Symposium on Intensive Care and Emergency Medicine in Brussels, Belgium. Based on the clinical trial data, the investigators concluded that our 5-FU CVC met the primary endpoint of the study; specifically that our 5-FU CVC product candidate was non-inferior in its ability to prevent bacterial colonization of the catheter tip when compared to catheters coated with CH-SS. There were no statistically significant differences in the rate of adverse events related to the study devices, or in the rates of catheter-related bloodstream infections. Additionally, there was no evidence for acquired resistance to 5-FU in clinical isolates exposed to the drug for a second time. Based on the positive results achieved in the study, in December 2007 we filed a request for 510(k) clearance from the FDA to market and sell the CVC in the U.S. and on April 17, 2008, we announced that we had received 510(k) clearance from the FDA to market our 5-FU CVC in the U.S.
As a result of our significantly constrained liquidity and capital resources, we have elected to date not to pursue commercialization activities for this product. Development of future launch plans and commercialization of this product, as well as related product line extensions, will depend on our liquidity, capital resources, or our ability to secure future financing.
15
Table of Contents
Patents, Proprietary Rights and Licenses
Patents and other proprietary rights are essential to our business. We file patent applications to protect technology, inventions and improvements to inventions that are considered important to our business. We also rely upon trade secrets, continuing technological innovations and licensing opportunities to develop and maintain our competitive position. Furthermore, we endeavor to extend our intellectual property portfolio by licensing patents and patent applications from others and initiating research collaborations with outside sources.
Our patents and patent applications relate to, among other things:
| • | the use of paclitaxel and other agents for the treatment of diseases, such as restenosis; |
| • | compositions of and use of pharmacologic agents as medical device coatings and drug-eluting biomaterials; |
| • | hybrid polymer systems designed for localized delivery of bioactive agents and controlled elution of drugs from medical device surfaces; |
| • | non-reactive hydrophilic/hydrophobic polymer matrix, which can be varied to optimize performance characteristics such as lubricity, flexibility and hydration; |
| • | hemostatic and sealant compositions, as well as broad intellectual property rights relating to collagen; |
| • | drug delivery compositions, such as our proprietary systemic formulation, medical device coatings and other local drug delivery systems; |
| • | self-anchoring devices, including those for wound closure; and |
| • | methods of making and using the above. |
As part of our patent strategy, we have filed a variety of patent applications internationally. Oppositions have been filed against various granted patents that we either own or license and which are related to certain of our technologies. See “— Legal Proceedings” elsewhere in this Annual Report on Form 10-K for a discussion of the proceedings related to certain of such oppositions. An adverse decision by an Opposition Division in any country, or subsequently, by a Board of Appeal, could result in revocation of our patent or a narrowing of the scope of protection afforded by the patent. The ultimate outcomes of the pending oppositions, including appeals, are uncertain at this time. We do not know whether the patents that we have received or licensed, or those we may be able to obtain or license in the future, would be held valid or enforceable by a court or whether our patents would be found to infringe a competitor’s patents related to its technology or product. Further, we have no assurance that third parties will not (whether pursuant to or in breach of the terms) modify or terminate any license they have granted to us.
Regulatory
The research and development, manufacturing, labeling, advertising, sale and marketing of our drug and medical device products and those of our collaborators are subject to extensive regulation by the FDA and comparable regulatory agencies in state and local jurisdictions and in foreign countries. Because in some cases we may focus on combining pharmaceutical compounds with medical devices and biomaterials, we may be subject to both drug and device regulations depending upon the categorization of the commercial product or potential product candidate.
Drug and device licensing laws require registration of manufacturing facilities, carefully controlled research and testing of products, government review and approval or clearance of results prior to marketing of products, adherence to current Good Manufacturing Practices (“cGMPs”), during production and processing of products, and compliance with comprehensive post-marketing requirements including restrictions on advertising and promotion and requirements for reporting adverse events to the FDA in the United States and other regulatory authorities abroad. In the United States, these activities are subject to rigorous regulation by the FDA. Similar regulations apply in the European Union and other markets.
Our success is ultimately dependent upon our and our collaborators obtaining marketing approval or clearance for potential product candidates currently under development and will depend on our and our
16
Table of Contents
collaborators’ ability to comply with FDA and other market regulations governing the manufacturing, quality control, pre-clinical evaluation, and clinical testing of those candidates. If we or our present or future collaborators are not able to comply with the continuing FDA regulations that accompany FDA approval, the FDA may halt our clinical trials, withdraw approval of the IND (investigational new drug application) or IDE (investigational device exemption), require us to recall a drug or device from distribution, withdraw approval for the relevant drug, the 510(k) or Premarket Approval for the relevant device. Depending on the circumstances surrounding the clinical evaluation of a product, we may sponsor clinical trials, contract clinical trial activities to contract research organizations (“CROs”) or rely upon our corporate collaborators to sponsor such clinical development. There can be no assurance that any of our and our collaborators’ clinical trials or product development activities will provide favorable data or that the data will be acceptable to any government regulatory agency to provide us with approval to market and sell our products or product candidates. Moreover, even if we and our collaborators believe the results of our product development activities are favorable, regulatory authorities may interpret the results differently than we do.
We have significant medical device component engineering and manufacturing, as well as finished goods and packaging manufacturing operations. The manufacturing processes used in the production of finished medical devices are subject to FDA regulatory inspection, and must comply with FDA regulations, including its Quality Systems Regulation, which sets forth the cGMP requirements for medical devices. The Quality Systems Regulation requires manufacturers of finished medical devices to follow elaborate design, testing, control, documentation and other quality assurance procedures during the finished device manufacturing process. Our FDA registered facilities are subject to FDA inspection at any time for compliance with the Quality Systems Regulation and other FDA regulatory requirements. Failure to comply with these regulatory requirements and similar foreign requirements may result in civil and criminal enforcement actions, including financial penalties, seizures, injunctions or other measures. In some cases, failure to comply with the Quality System Regulation could prevent us or our customers from marketing products or gaining clearance to market additional products. Our products must also comply with state and foreign regulatory requirements.
Manufacturing and Distribution
We have significant manufacturing capabilities, with 11 medical device manufacturing facilities located in the United States, Europe and Puerto Rico. Our medical device manufacturing capabilities include polymer handling and extrusion, metals, plastics, textiles and packaging and assembly. Our suture manufacturing capabilities include the ability to make and handle various approved polymer materials in multiple lengths, diameters and configurations. Our metal manufacturing capabilities include bending, grinding, flattening, drilling, polishing, chemical etching, plasma welding, thermal curing and wire winding. Our plastics manufacturing capabilities include injection molding, plastic extrusion, insert molding and wire coating. Our textile manufacturing capabilities include braiding and embroidery of selected suture materials, including silk. Our assembly and packaging capabilities include small lot assembly, suture attachment and kit assembly. We also maintain manufacturing capabilities for certain of our biomaterials and coating technologies, including dedicated resources and technology to process and manufacture certain types of drug-eluting medical devices. Our established distribution network, allows us to reach customers in all of our established markets in an efficient and cost effective way.
We devote resources to develop procedures and process controls for our products and product candidates to ensure successful technology transfer for commercial scale manufacturing. We expect that process and assay development performed during pre-clinical and clinical stages of manufacturing will result in products with the desired use and stability. These activities include scaling up production methods, developing quality control systems, optimizing batch-to-batch reproducibility, testing sterilization methods and establishing reliable sources of raw materials for synthesizing proprietary products.
Sales and Marketing
We market and sell our selected Proprietary Medical Products through a specialty direct sales organization comprised of approximately 70 direct representatives as at December 31, 2010. In the case of our Base Medical Products, we employ a small group of direct sales representatives, a larger group of independent sales representatives and independent medical products distributors to market and sell these products. The majority of our direct sales teams are based in the United States.
17
Table of Contents
Our Proprietary Medical Products’ direct sales team targets hospital-based physician customers and alternate site health care providers such as ambulatory surgery centers and urgent care centers. The primary goal of our direct sales teams is to increase market share of our Proprietary Medical Products that have the highest margins or the most substantial technological advantages.
Our Base Medical Products’ sales team, sales representatives and independent distributors target hospital customers, alternate site health care providers; such as ambulatory surgery centers and urgent care centers; physician offices, dental offices and other third party medical products manufacturers or distributors. The primary goal of this group is to maintain our market share and revenue levels for our Base Medical Products.
Competition
We expect to face competition from companies marketing, selling and developing technologies that target the same diseases, clinical indications and customers that our technologies target, as well as competition from other manufacturers of medical device components who sell to similar third party medical device manufacturers to whom we do business with. Some of these companies have substantially greater product development, sales and marketing or manufacturing capabilities or experience, as well as more substantive financial, resources than we do.
The medical device, biotechnology and pharmaceutical industries are subject to rapid and substantial technological change. There can be no assurance that developments by others will not render obsolete our products or technologies or that we will be able to keep pace with technological developments. Some competitive products have an entirely different approach or means to accomplish the desired therapeutic effect as compared to our product candidates. These competitive products, including any enhancements or modifications made to such products in the future, could be more effective and/or less costly than our products, technologies or our product candidates.
There are a number of companies that are marketing or developing competing drug-eluting coronary stents or other treatments for restenosis that may be considered to be directly competitive with our technology. Certain of our competitors have developed and commercialized coronary drug-eluting stents that compete with BSC’s TAXUS stents and which have been approved for use in the United States, European Union and certain other countries. The launch of these competing products has had a significant impact on BSC’s sales of TAXUS, and has resulted in a decline in our royalty revenue received from BSC. The continued successful commercialization of any of these or other technologies to treat restenosis following angioplasty or stent placement could have a material adverse impact on our business.
We compete with a number of additional large companies across various of our product lines. These competitors have substantially greater business and financial resources than we do. These competitors include, but are not limited to, the Ethicon division of Johnson & Johnson and Covidien, Inc. in surgical sutures and wound closure devices; C.R. Bard Inc., the Allegiance division of Cardinal Health, Inc. and the Sherwood, Davis & Geck division of Covidien, Inc. in biopsy needle devices; Alcon, Inc., Allergan, Inc. and the Allegiance division of Cardinal Health, Inc. in ophthalmology products, and Accelent Inc., Teleflex Incorporated and Symmetry Medical Inc.. in the manufacture of medical devices and device components for other third party medical device manufacturers.
Employees
As of December 31, 2010, on a worldwide basis, we had approximately 1,368 employees, including 995 in manufacturing, 45 in R&D, 100 in Quality, 140 in sales and marketing and 87 in administration. In addition, we have contractual arrangements with scientists at various research and academic institutions. All personnel or their management companies execute confidentiality agreements with us. While we will continue to seek to hire highly qualified employees, we believe that we have employed a sufficient number of qualified personnel.
18
Table of Contents
Reimbursement
In the United States, health care providers generally rely on third-party payers, including both private health insurance plans and governmental payers, such as Medicare and Medicaid, to cover and reimburse all or part of the cost of a medical device and the procedures in medical devices that are used. Our ability to commercialize human therapeutic products and product candidates successfully will depend in part on the extent to which coverage and reimbursement for such products and related treatments will be available from government health administration authorities, private health insurers and other third-party payers or supported by the market for these products. Third-party payers are increasingly challenging the price of medical products and services and instituting cost containment measures to control or significantly influence the purchase of medical products and services. These cost containment measures, if instituted in a manner affecting the coverage for or payment of our products, could have a material adverse effect on our ability to operate profitably.
There can be no assurance that third-party payers’ coverage and reimbursement will be available or sufficient for our current products or the products we might develop. We believe that recommendations and endorsements by physicians are essential for market acceptance of our products, and we do not know whether these recommendations or endorsements will be obtained. We also believe that surgeons will not use these products unless they determine, based on clinical data and other factors, that the clinical benefits to patients and cost savings achieved through use of these products outweigh their cost. Acceptance among physicians may also depend upon the ability to train surgeons and other potential users of our products and the willingness of such users to learn new techniques.
In the United States, there have been and we expect there will continue to be a number of legislative and regulatory proposals to change the healthcare system in ways that could significantly affect our business. Efforts by governmental and third-party payers to reduce healthcare costs or the announcement of legislative proposals or reforms to implement government controls could cause a reduction in sales or in the selling price of our products, which could seriously harm our business. We cannot predict the impact on our business of any legislation or regulations related to the healthcare system that may be enacted or adopted in the future.
Environmental
Our business is subject to a broad range of government regulation and requirements, including federal, state, local and foreign environmental laws and regulations governing, among other matters, air emissions, wastewater discharges, workplace health and safety as well as the use, handling, storage and disposal of hazardous materials and remediation of contamination associated with the release of these materials at or from our facilities or off-site disposal locations. Some of these laws can impose liability for remediation costs on a party regardless of fault or the lawfulness of its conduct. Many of our manufacturing processes involve the use and subsequent regulated disposal of hazardous materials. To date, such matters have not had a material adverse impact on our business or financial condition. We cannot assure you, however, that such matters will not have such an effect in the future.
International Operations
The Company’s medical devices are manufactured and sold worldwide. Approximately 39% of the Company’s revenues are generated outside of the United States and geographic expansion remains a core component of the Company’s strategy. The Company currently sells its products in the following international territories: Europe, Asia-Pacific, Latin America and Canada. The Company is subject to certain risks inherent in conducting business outside the United States. For more information regarding these risks, see the risk factors under the captions “We are subject to risks associated with doing business globally” and “We may incur losses associated with foreign currency fluctuations” in Item 1A of this Annual Report on Form 10-K, all of which information is incorporated herein by reference.
For financial information about foreign and domestic operations and geographic information, see note 21 in the audited consolidated financial statements for the year ended December 31, 2010. In addition, for more information regarding foreign currency exchange risk, refer to the discussion under “Foreign Currency Risk” under Item 7A of this Annual Report on Form 10-K.
19
Table of Contents
| Item 1A. | RISK FACTORS |
You should consider carefully the following information about these risks, together with all of the other information contained within this document. Additional risks and uncertainties not currently known to us or that we currently deem immaterial may impair our business operations. If any of the following risks actually occur, our business, results of operations and financial condition could be harmed.
Risks Related to the Recapitalization Transaction
We may not be successful in implementing the Recapitalization Transaction.
We have been under CCAA protection since January 28, 2011 during which time the Company’s management, with input from the Company’s board of directors (the “Board of Directors”) and a committee of independent directors of the Board of Directors formed on July 29, 2010 (the “Independent Committee”), have negotiated the terms of the CCAA Plan and related agreements.
If the CCAA Plan is not implemented before April 30, 2011 and another plan is not proposed that meets the approval requirements of the Canadian Court, we may remain under CCAA protection for an indefinite period of time and our businesses could substantially erode or an insolvency proceeding involving the liquidation of our assets with a view to recovering the amounts owing to the creditors could result.
If the Recapitalization Transaction is not completed, there is no assurance that we will be able to complete a recapitalization or restructuring of our businesses or that any such recapitalization or restructuring will be on terms that provide equivalent value to our creditors compared to the consideration to be received by creditors pursuant to the Recapitalization Transaction and the CCAA Plan.
If we are not able to consummate the Recapitalization Transaction by April 30, 2011, the RSA may be terminated and the FRN Support Agreement may be terminated, and we will be required to pursue other alternatives that could have a more negative effect on the Company and its stakeholders.
Pursuant to the terms of the RSA, if the Recapitalization Transaction is not implemented on or before April 30, 2011 the RSA may be terminated by the holders of a majority of the aggregate principal amount outstanding of the Subordinated Notes party to the RSA. Similarly, under the terms of the FRN Support Agreement, if the Recapitalization Transaction is not implemented on or before April 30, 2011 the FRN Support Agreement may be terminated by holders of a majority of the aggregate principal amount outstanding of Existing Floating Rate Notes party to the FRN Support Agreement.
It is a condition to the FRN Support Agreement that the transactions contemplated by the RSA are concurrently implemented with the transactions contemplated by the FRN Support Agreement. Further, the implementation of the transactions under the FRN Support Agreement is a condition of the RSA. As a result, the termination of one of either the RSA or the FRN Support Agreement will result in a condition of the other that cannot be satisfied.
If either or both of the RSA or the FRN Support Agreement are terminated and the CCAA Plan is not implemented, we will have an immediate need to pursue other alternatives to manage our liquidity needs, including potentially filing for protection under the insolvency laws of various jurisdictions on terms other than as contemplated by the CCAA Plan. We do not expect, and there can be no assurance, that any alternative to such filings would be found.
20
Table of Contents
If we are unable to consummate the CCAA Plan, the Company will face an immediate liquidity crisis that will likely result in the Company filing for protection under the insolvency laws of various jurisdictions on terms other than as contemplated by the CCAA Plan, which could materially adversely affect the relationships between us and our existing and potential customers, employees, partners and other stakeholders.
We believe that seeking relief under the CCAA, Chapter 15 of the U.S. Bankruptcy Code and/or other similar insolvency laws and seeking to implement a restructuring on terms other than as contemplated by the CCAA Plan could materially adversely affect the relationships between us and our existing and potential customers, employees, partners and other stakeholders. For example:
| • | such bankruptcy filings could erode our customers’ confidence in our ability to provide our products and services and, as a result, there could be a significant and precipitous decline in our revenues, profitability and cash flow; |
| • | our employees could be distracted from performance of their duties, or more easily attracted to other career opportunities; |
| • | it may be more difficult to attract or replace key employees; |
| • | our lenders and other partners could seek to terminate their relationship with us, require financial assurances or enhanced performance, or refuse to provide credit on the same terms as prior to the reorganization case; |
| • | we could be forced to operate in bankruptcy for an extended period of time while we tried to develop a reorganization plan that could be confirmed, which we believe may impair our business and prospects; or |
| • | our suppliers, vendors, and service providers could terminate their relationship with us or require financial assurances or enhanced performance. |
In addition, any distributions in respect of, Affected Claims (as defined in the CCAA Plan,) under a liquidation or under a protracted reorganization case or cases under insolvency laws, other than in connection with the CCAA Plan, would likely be substantially delayed and the value of any potential recovery would likely be adversely impacted by such delay.
Furthermore, in the event of any foreclosure, dissolution, winding-up, liquidation or reorganization, or other insolvency proceeding other than in connection with the CCAA Plan, there can be no assurance as to the value, if any, that would be available to Affected Creditors (as defined in the CCAA Plan). The Affected Claims (including claims in respect of the Subordinated Notes) are unsecured obligations of the Company and rank junior to the secured obligations of the Company under the credit agreement with Wells Fargo Capital Finance, LLC (f/k/a Wells Fargo Foothill, LLC, “Wells Fargo”) dated February 27, 2009 (as amended, the “Existing Credit Facility”). Accordingly, upon any distribution to our creditors in any foreclosure, dissolution, winding-up, liquidation or reorganization, or other insolvency proceeding relating to us or our property other than in connection with the CCAA Plan, the indebtedness under our Existing Credit Facility will be entitled to be paid in full before any payment may be made with respect to the Affected Claims. If the CCAA Plan is not approved, Affected Creditors may receive less value for their Affected Claims in the event of any foreclosure, dissolution, winding-up, liquidation or reorganization, or other insolvency proceeding other than the CCAA Plan than they would under the Recapitalization Transaction.
In the event the Recapitalization Transaction is not implemented and creditor protection is no longer available under the CCAA, we will be required to pursue other alternatives that could have a more negative effect on the Company and its stakeholders, including the sale of core assets or non-consensual proceedings under insolvency statutes.
Should creditor protection no longer be available under the CCAA, then:
| • | the Company’s long-term debt will not be reduced by approximately $250 million and the associated net reduction in debt service costs would not be achieved; |
21
Table of Contents
| • | the holders of Existing Floating Rate Notes and Subordinated Notes may enforce their remedies against the Company pursuant to the indentures governing such notes as a result of the defaults caused by the missed interest payment under the terms of those agreements; and |
| • | the Company believes its cash flow from operations and available liquidity would be insufficient to provide adequate funds to finance its operations, and the Company would be unable to meet its obligations as they generally become due. |
In addition, any distributions in respect of Affected Claims under a liquidation or under a protracted reorganization case or cases under insolvency laws, other than in connection with the CCAA Plan, would likely be substantially delayed and the value of any potential recovery would likely be adversely impacted by such delay.
Furthermore, in the event of any foreclosure, dissolution, winding-up, liquidation or reorganization, or other insolvency proceeding other than in connection with the CCAA Plan, there can be no assurance as to the value, if any, that would be available to Affected Creditors. The Affected Claims including the Subordinated Notes are unsecured obligations and rank junior to the secured obligations under the Existing Credit Facility. Accordingly, upon any distribution to our creditors in any foreclosure, dissolution, winding-up, liquidation or reorganization, or other insolvency proceeding relating to us or our property other than in connection with the CCAA Plan, the indebtedness under our Existing Credit Facility will be entitled to be paid in full before any payment may be made with respect to the Affected Claims. If the CCAA Plan is not approved, Affected Creditors may receive less value for their Affected Claims in the event of any foreclosure, dissolution, winding-up, liquidation or reorganization, or other insolvency proceeding other than the CCAA Plan than they would under the Recapitalization Transaction.
The conditions to the CCAA Plan may not be satisfied or waived.
Implementation of the CCAA Plan is subject to various conditions, including the granting of the order of the Canadian Court sanctioning and approving the CCAA Plan (the “Sanction Order”), which must be fulfilled prior to implementation and effectiveness of the CCAA Plan. As of the date hereof, there can be no assurance that any or all of the conditions in the CCAA Plan or in the agreements pertaining to the CCAA Proceedings, such as the RSA and the FRN Support Agreement, will be satisfied (or waived, if applicable). Accordingly, there can be no assurance that the CCAA Plan will be consummated even if approved by the Affected Creditors.
The consummation of the Recapitalization Transaction may be delayed.
We have incurred significant costs and expenses to date in connection with our ongoing restructuring efforts. Even if the Recapitalization Transaction is completed, it may not be completed on the schedule described in this Annual Report on Form 10-K or on or prior to April 30, 2011. Accordingly, Affected Creditors participating in the Recapitalization Transaction may have to wait longer than expected to receive consideration, if any, for their Affected Claims. In addition, if the Recapitalization Transaction is not completed on the schedule described in this Annual Report on Form 10-K, we may incur additional expenses.
There is uncertainty regarding the financial condition of our business.
Adverse publicity or news coverage relating to the Company’s CCAA Proceedings could have an adverse effect on all or parts of our businesses. Following the implementation of the CCAA Plan, there can be no assurance that negative publicity may not adversely affect our results from operations or have a long-term negative effect on our businesses.
In addition, such uncertainty may adversely affect the Company’s relationships with its suppliers and customers. Following the implementation of the CCAA Plan, suppliers and customers may continue to be
22
Table of Contents
concerned about the financial condition of the Company’s businesses and, as a result, they may demand faster payment terms or not extend normal trade credit, both of which could adversely affect the Company’s working capital position. The Company may not be successful in obtaining alternative suppliers and customers if the need arises and this would adversely affect the Company’s results from operations and our ability to conduct our business.
We have filed under the U.S. Bankruptcy Code to effectuate the CCAA Proceedings in the United States as set forth in the CCAA Plan.
We cannot assure you that any plan of reorganization ultimately confirmed in the Chapter 15 Cases will have the same terms as set forth in the CCAA Plan. Claims may be asserted in Chapter 15 Cases of which the Company is presently unaware and for which the Company may ultimately be liable. There can be no assurances that we will have sufficient excess liquidity upon the implementation of the CCAA Plan to satisfy obligations owing under our debtor-in-possession credit agreement, dated as of February 7, 2011, among Angiotech, certain of its subsidiaries, the lenders party thereto and Wells Fargo, as arranger and administrative agent (the “DIP Facility”).
Even if the Recapitalization Transaction is successful, we will continue to face risks.
The Recapitalization Transaction is generally designed to reduce the amount of our indebtedness and cash interest expense and improve our liquidity as well as our financial and operational flexibility in order to generate long-term growth. Even if the Recapitalization Transaction is consummated, we will continue to face a number of risks, including certain risks that are beyond our control, such as changes in economic conditions, changes in our industry, regulatory changes and changes in consumer demand for our products. As a result of these risks and others, there is no guarantee that the Recapitalization Transaction will achieve our stated goals (see “—Risks Related to Our Business”).
Change-of-control, consent or notice provisions in agreements may be triggered upon the effectiveness of the CCAA Plan.
We may be a party to agreements that contain change-of-control, consent or notice provisions that may be triggered following the issuance of the new common shares pursuant to the CCAA Plan. Such provisions may also be triggered in connection with the Chapter 15 Cases. These change-of-control, consent or notice provisions, if triggered and not waived by any beneficiaries of those provisions, could result in termination of an agreement or in unanticipated expenses following the Recapitalization Transaction and could adversely affect our results of operations and financial condition.
The Recapitalization Transaction may not improve the financial condition of our business.
Management believes that the consummation of the Recapitalization Transaction will enhance the Company’s liquidity and provide it with improved operating flexibility. However, such belief is based on certain assumptions, including, without limitation, that the Company’s consolidated sales and relationships with suppliers, customers and competitors will not be materially adversely affected while the Recapitalization Transaction is underway and that they will be stable or will improve following the completion of the Recapitalization Transaction , that general economic conditions and the markets for the Company’s products or for the products of its partners will remain stable or improve, as well as the Company’s continued ability to manage costs. Should any of those assumptions prove false, the financial position of the Company may be materially adversely affected and the Company may not be able to pay its debts as they become due.
23
Table of Contents
Risks Related to Our Business
Our business activities involve various elements of risk. The risks described below and incorporated herein by reference are not the only ones facing us. Additional risks that are presently unknown to us or that we currently deem immaterial may also impact our business. We consider the following issues to be the most critical risks to the success of our business:
The Company has historically been unprofitable and may not be able to achieve and maintain profitability.
We began operations in 1992 and have incurred a loss from operations in a majority of the years in which we have been operating. Our ability to become profitable and maintain profitability will depend on, among other things, our ability to expediently complete the Recapitalization Transaction and thereby implement our proposed restructuring and reduce our outstanding indebtedness; our ability to maintain and improve sales of our existing product lines; our ability to successfully market and sell certain new products and technologies; the amount of royalty revenue we receive from our corporate partners; our ability to research, develop and successfully launch new products and technologies; our ability to improve our profit margins through lower manufacturing costs and efficiencies or improved product sales mix; and our ability to effectively control our various operating costs.
Our working capital and funding needs may vary significantly depending upon a number of factors including, but not limited to, our levels of sales and gross profit; costs associated with our manufacturing operations, including capital expenditures, labor and raw materials costs, and our ability to realize manufacturing efficiencies from our various operations; fluctuations in certain working capital items, including inventory and accounts receivable, that may be necessary to support the growth of our business or new product introductions; the level of royalty revenue we receive from corporate partners; progress of our research and development programs and costs associated with completing clinical studies and the regulatory process; collaborative and license arrangements with third parties; the cost of filing, prosecuting and enforcing our patent claims and other intellectual property rights; expenses associated with litigation; opportunities to in-license complementary technologies or potential acquisitions; potential milestone or other payments we may make to licensors or corporate partners; and technological and market developments that impact our royalty revenue, sales levels or competitive position in the marketplace.
The significant decline in royalty payments we receive from BSC, the large amount of our outstanding indebtedness and the related cash interest payments due on such indebtedness, as well as the working capital, capital and other expenditures required to develop the medical products segment of our business, among other factors, have adversely affected our operations and liquidity. Under the terms of the Existing Credit Agreement and our proposed new credit facility to be implemented in connection with the implementation of the Recapitalization Transaction (the “Exit Facility”), will replace our Existing Credit Facility, financing may not be available when it is needed, and financing in addition to such agreements may not be available, if at all, on attractive or acceptable terms due to credit market conditions and other factors. We have been pursuing options to improve our capital structure and address our potential working capital needs, including the Recapitalization Transaction. If we do not expediently consummate the Recapitalization Transaction, due to numerous factors that may impact our future cash position, working capital and liquidity, as discussed below, and given the significant cash that will be necessary to continue to service our current level of debt obligations, there can be no assurances that we will have adequate liquidity and capital resources to satisfy our financial obligations beyond Q2 2011. If we do not consummate the Recapitalization Transaction and our cash flows are worse than expected, we may require additional funds in order to fulfill the funding requirements of our commercial operations for our research and development programs, to satisfy certain contractual obligations, to meet other operating and capital requirements, to make milestone or other payment obligations we may owe to licensors or corporate partners, to repay or refinance our indebtedness or for potential acquisitions or in-licensing of technologies.
24
Table of Contents
Our success depends on the successful commercialization of our existing products and new product candidates.
The continued and successful commercialization of our medical products and technology is crucial for our success. Successful product commercialization and product development in the medical device and pharmaceutical industries are highly uncertain, and very few product commercialization initiatives or research and development projects produce a significant or successful commercial product. Our products and product candidates are in various stages of commercial and clinical development and face a variety of risks and uncertainties. Principally, these risks and uncertainties include the following:
| • | Commercialization risk. Even if our products or product candidates are successfully developed, receive all necessary regulatory approvals and are commercially produced, there is no guarantee that there will be market acceptance of them or that they will generate or sustain any significant revenues. Our ability to achieve sustainable market acceptance for any of our products will depend on a number of factors, including whether competitors develop technologies which are superior to, or less costly than, our products or product candidates, whether our products or product candidates lead to or directly cause unwanted side effects, and whether government and private third-party payers provide adequate coverage and reimbursement for our products or product candidates. |
| • | Clinical development risk. Future clinical trial results may show that some or all of our technology, or the technology of our strategic collaborators that incorporates our technology, is not safe or effective or is not as safe or effective as similar technologies produced by our competitors. |
| • | Manufacturing and scale-up risk. We and our strategic collaborators may face significant or unforeseen difficulties in manufacturing our products or the products that use certain aspects of our technology. These difficulties may become apparent when we or our strategic collaborators manufacture the medical devices or surgical implants on a small scale for clinical trials and regulatory approval or may only become apparent when scaling up the manufacturing to commercial scale. |
| • | Liquidity and working capital risk. We may not have the working capital and financial resources necessary to effectively market or distribute our products to customers or potential customers. Our available working capital to market and distribute our products and product candidates is in part dependent on our ability to expediently complete the Recapitalization Transaction and thereby implement our proposed restructuring and reduce our outstanding indebtedness. The Recapitalization Transaction and related proceedings may also increase uncertainty about our ability to market and distribute our products or product candidates, thereby impacting our ability to sustain or increase our revenues. |
If we are unsuccessful in dealing with any of these risks, or if we are unable to successfully commercialize our technology for some other reason, it would likely seriously harm our ability to sustain or to generate additional revenue.
We may be unsuccessful in marketing, selling and distributing certain of our products.
We distribute a number of our products worldwide. In order to achieve commercial success for our approved products, we have established sales and marketing resources in the United States, Europe and other parts of the world. If our distribution personnel or methods are not sufficient to achieve market acceptance of our products, to maintain our current customer relationships or to ensure we have supply to meet demand for our products, it could harm our prospects, business, financial condition and results of operations.
To the extent that we enter into co-promotion or other marketing and sales arrangements with other companies, any revenues received will be dependent on the efforts of others, and we do not know whether these efforts will be successful. Failure to develop an adequate sales and marketing operation or to enter into appropriate arrangements with other companies to market and sell our products will reduce our ability to sustain or generate revenues.
25
Table of Contents
Many of the products we are depending on to grow our business are not yet ready for sale or have only recently been introduced for sale.
Many of the products we are depending on to produce future growth are not yet ready for sale, or have only recently been introduced for sale. Introduction of a new product to the marketplace requires significant financial expenditures that we may not be able to fund. If any of our products are not approved for sale or do not achieve market acceptance, our ability to generate new revenues will be adversely affected.
If our products are alleged to be harmful, we may not be able to sell them, we may be subject to product liability claims not covered by insurance and our reputation could be damaged.
The nature of our business exposes us to potential liability risks inherent in the testing, manufacturing and marketing of pharmaceutical products and medical devices. Using our medical device or drug product candidates in clinical trials may expose us to product liability claims. These risks will expand with respect to medical devices or drugs, if any, that receive regulatory approval for commercial sale. In addition, some of the products we manufacture and sell are designed to be implanted in the human body for varying periods of time. Even if a medical device or drug were approved for commercial use by an appropriate governmental agency, there can be no assurance that users will not claim that effects other than those intended may have resulted from our products. Component failures, manufacturing flaws, quality system failures, design defects, inadequate disclosure of product-related risks or product-related information or other safety issues with respect to these or other products we manufacture or sell could result in an unsafe condition or injury to, or death of, a patient. In addition, although many of our products are subject to review and approval by the U.S. Food and Drug Administration (the “FDA”) or other regulatory agencies, under the current state of the law, any approval of our products by such agencies will not prohibit product liability lawsuits from being brought against us in the event that our products are alleged to be defective, even if such products have been used for their approved indications and appropriate labels have been included.
In the event that anyone alleges that any of our products are harmful, we may experience reduced consumer demand for our products or our products may be recalled from the market. In addition, we may be forced to defend individual or class action lawsuits and, if unsuccessful, to pay a substantial amount in damages. A recall of some of our products could result in exposure to additional product liability claims, lost sales and significant expense to perform the recall. The outcome of litigation, particularly class action lawsuits, is difficult to assess or quantify. Plaintiffs in these types of lawsuits often seek recovery of very large or indeterminate amounts, including not only actual damages, but also punitive damages. The magnitude of the potential loss relating to these types of lawsuits may remain unknown for substantial periods of time. In addition, the cost to defend against any future litigation may be significant.
We do not have insurance covering our costs and losses as a result of any recall of products or devices incorporating our technologies, whether such recall is instituted by a device manufacturer or us as required by a regulatory agency. Insurance to cover costs and losses associated with product recalls is expensive. If we seek insurance covering product recalls in the future, it may not be available on acceptable terms. Even if obtained, insurance may not fully protect us against potential liability or cover our losses. Some manufacturers that suffered such claims in the past have been forced to cease operations or declare bankruptcy.
We do have insurance covering product liability. However, our insurance may not fully protect us from potential product liability claims. If a product liability claim or a series of claims is brought against us in excess of our insurance coverage, our business could suffer. Some manufacturers that suffered such claims in the past have been forced to cease operations or declare bankruptcy.
Our royalty revenues we receive from BSC have declined significantly, and may continue to decline, thereby negatively impacting our cash flows and liquidity.
We do not have control over the sales and marketing efforts, stent pricing, production volumes, distribution or regulatory environment related to BSC’s paclitaxel-eluting coronary stent program. Our involvement is limited to the terms of our 1997 License Agreement (as amended) with BSC and Cook (the “1977 License Agreement”), which provides for the receipt of royalty revenue based on the net sales of TAXUS®.
Royalty revenue from BSC has declined significantly, as described in “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included in this Annual Report on Form 10-K.
26
Table of Contents
BSC has attributed this significant decline in royalties primarily to competition in the drug-eluting stent market, price pressure in the drug-eluting stent market and a decline in the overall number of drug-eluting stent procedures. Our royalties may decline further due to these and other factors in the future.
In addition, if BSC is impaired in its ability to market and distribute TAXUS®, whether for these reasons or due to a failure to comply with applicable regulatory requirements, discovery of a defect in the device, increased incidence of adverse events or identification of other safety issues, or previously unknown problems with the manufacturing operations for TAXUS® (any of which could, under certain circumstances, result in a manufacturing injunction), our revenues could be further reduced. BSC’s failure to resolve these issues in a timely manner and to the satisfaction of the FDA and other regulatory authorities, or the occurrence of similar problems in the future, could delay the launch of additional TAXUS® product lines in the United States and could have a significant impact on our royalty revenue from sales of TAXUS®.
Additionally, BSC may terminate our 1997 License Agreement under certain circumstances, including if BSC is unable to acquire a supply of paclitaxel at a commercially reasonable price; if BSC reasonably determines that the paclitaxel-eluting coronary stent is no longer commercially viable; or if our exclusive license agreement with the Public Health Service of the U.S. through the National Institutes of Health (the “NIH”), whereby the NIH granted the Company an exclusive, worldwide license to certain technologies of the NIH for the use of paclitaxel (the “the NIH License Agreement”), certain rights under which are sublicensed to BSC, terminates.
The amounts payable by BSC to us vary from 1% to 9% of net sales depending on various factors, including the volume of sales in each quarterly period and patent protection laws in the country of sale. From these amounts, we must pay certain royalties to our licensors, including the University of British Columbia (“UBC”), under license agreements. The royalty rates paid by BSC to us are described in our 2009 MD&A. There is no guarantee that royalty payments under our 1997 License Agreement will continue, and demand for BSC’s paclitaxel-eluting coronary stent products could continue to decline as a result of the factors stated above, as well as technological change, changes in reimbursement rates or other factors. Also, the royalty rate payable by BSC could decline if and when patent protection expires, or no longer exists (as defined by our 1997 License Agreement) in certain jurisdictions.
Our royalty revenue derived from the sale of paclitaxel-eluting coronary stents depends on BSC’s ability to continue to sell its various TAXUS® paclitaxel-eluting stent products and to launch next generation paclitaxel-eluting stents in the United States and in other countries. Historically, stent manufacture and sale have been subject to a substantial amount of patent litigation, and BSC and others may become involved in material legal proceedings related to paclitaxel-eluting stents that could impact BSC’s ability to sell TAXUS®, thereby negatively impacting our royalty revenue derived from the sale of paclitaxel-eluting coronary stents.
We face and will continue to face significant competition.
Competition from pharmaceutical companies, medical device companies, biotechnology companies and academic and research institutions is significant. Many of our competitors and potential competitors have substantially greater product development capabilities, experience conducting clinical trials and financial, scientific, manufacturing, sales and marketing resources and experience than our company. We also face competition from non-medical device companies, such as pharmaceutical companies, which may offer non-surgical alternative therapies for disease states that are currently treated or are intended to be treated using our products. Other companies may:
| • | develop and obtain patent protection for products earlier than we do; |
| • | design around patented technology developed by us; |
| • | obtain regulatory approvals for such products more rapidly; |
27
Table of Contents
| • | have greater manufacturing capabilities and other resources; |
| • | have larger or more experienced sales forces; |
| • | develop more effective or less expensive products; or |
| • | have greater success in obtaining adequate third-party payer coverage and reimbursement for their competing products. |
While we intend to expand our technological capabilities in order to remain competitive, there is a risk that:
| • | research and development by others will render our technology or product candidates obsolete or non-competitive; |
| • | treatments or cures developed by others will be superior to any therapy developed by us; and |
| • | any therapy developed by us will not be preferred to any existing or newly developed technologies. |
If physicians do not recommend and endorse our products or products that use our technology, or if our working relationships with physicians deteriorate, our products or products that use our technology may not be accepted in the marketplace, which could adversely affect our sales and royalty revenues.
In order for us to sell our products or continue to receive royalty revenues from the sale of products that use our technologies, physicians must recommend and endorse them. We believe that recommendations and endorsements by physicians are and will be essential for market acceptance of our products, and we do not know whether we will obtain the necessary recommendations or endorsements from physicians. Acceptance of our products or of products that use our technology depends on educating the medical community as to the distinctive characteristics, perceived benefits, safety, clinical efficacy and cost-effectiveness of these products compared to products of competitors, and on training physicians in the proper application of these products. If we are not successful in obtaining the recommendations or endorsements of physicians for our products, or our collaborators are not successful in doing the same for their products that use our technology, our sales and royalty revenues may not increase or may decline.
In addition, if we fail to maintain our working relationships with physicians, many of our products may not be developed and marketed in line with the needs and expectations of professionals who use and support our products. The research, development, marketing and sales of many of our new and improved products are dependent upon our maintaining working relationships with physicians. We rely on these professionals to provide us with considerable knowledge and experience regarding our products and the marketing of our products. Physicians assist us as researchers, marketing consultants, product consultants, inventors and public speakers. If we are unable to maintain our strong relationships with these professionals while continuing to receive their advice and input, the development and marketing of our products could suffer, which could adversely affect the acceptance of our products in the marketplace and our sales.
The commercial potential of our products and product candidates will be significantly limited if we are not able to obtain adequate levels of reimbursement for them.
Our ability to commercialize our medical products and product candidates successfully will depend in part on the extent to which coverage and reimbursement for such products and related treatments will be available from government health administration authorities, private health insurers and other third-party payers or otherwise supported by the market for these products. There can be no assurance that third-party payers’ coverage and reimbursement will be available or sufficient for the products we might develop.
Third-party payers are increasingly challenging the price of medical products and services and instituting cost-containment measures to control or significantly influence the purchase of medical products and services. These cost containment measures, if instituted in a manner affecting the coverage of or payment for our products, could have a material adverse effect on our ability to operate profitably. In some countries in the E.U. and in the United States, significant uncertainty exists as to the reimbursement status of newly approved health care products, and we do not know whether adequate third-party coverage and
28
Table of Contents
reimbursement will be available for us to realize an appropriate return on our investment in product development, which could seriously harm our business. In the United States, while reimbursement amounts previously approved appear to have provided a reasonable rate of return, there can be no assurance that our products will continue to be reimbursed at current rates or that third-party payers will continue to consider our products cost-effective and provide coverage and reimbursement for our products, in whole or in part.
We cannot be certain that our products will gain commercial acceptance among physicians, patients and third-party payers, even if necessary international and United States marketing approvals are maintained. We believe that recommendations and endorsements by physicians will be essential for market acceptance of our products, and we do not know whether these recommendations or endorsements will be obtained. We also believe that surgeons will not use these products unless they determine, based on clinical data and other factors, that the clinical benefits to patients and cost savings achieved through use of these products outweigh their cost. Acceptance among physicians may also depend upon the ability to train surgeons and other potential users of our products and the willingness of such users to learn these relatively new techniques.
If we are unable to license new technologies, or our existing license agreements are terminated, our ability to maintain our competitive advantage in our existing products and to develop future products may be adversely affected.
We have entered into, and we expect that we will continue to enter into, licensing agreements with third parties to give us access to technologies that we may use to develop products, or that we may use to gain access to new products for us to further develop, market and sell through our commercial sales organizations. The technologies governed by these license agreements may be critical to our ability to maintain our competitive advantage in our existing products and to develop future products. For example, through licenses with the NIH and UBC, we have been granted access to technologies that have contributed to the developments of the TAXUS® paclitaxel-eluting coronary stent.
Pursuant to terms of our existing license agreements, licensors have the right under certain specified circumstances to terminate their respective licenses. Events that may allow licensors to exercise these termination provisions include:
| • | sub-licensing without the licensor’s consent; |
| • | a transaction which results in a change of control of us; |
| • | our failure to use the required level of diligence to develop, market and sell products based on the licensed technology; |
| • | our failure to maintain adequate levels of insurance with respect to the licensed technologies; |
| • | our bankruptcy; or |
| • | other acts or omissions that may constitute a breach by us of our license agreement. |
In addition, any failure to continue to have access to these technologies may materially adversely affect the benefits that we currently derive from our collaboration and partnership arrangements and may adversely affect our results and operations.
We may depend on certain strategic collaborators for the development, regulatory approval, testing, manufacturing and potential commercialization of our products.
Historically, our strategy has been to enter into various arrangements with corporate and academic collaborators, licensors, licensees and others for the research, development, clinical testing, regulatory approval, manufacturing, marketing and commercialization of our product candidates. For instance, we collaborate with BSC and Cook to develop and market paclitaxel-eluting coronary and peripheral stents. Strategic collaborators, both existing (particularly BSC) and those that we may collaborate with in the future, are or may be essential to the development of our technology and potential revenue and we have little control over or access to information regarding our collaborators’ activities with respect to our products.
29
Table of Contents
Our strategic collaborators may fail to successfully develop or commercialize our technology to which they have rights for a number of reasons, including:
| • | failure of a strategic collaborator to continue, or delays in, its funding, research, development and commercialization activities; |
| • | the pursuit or development by a strategic collaborator of alternative technologies, either on its own or with others, including our competitors, as a means for developing treatments for the diseases targeted by our programs; |
| • | the preclusion of a strategic collaborator from developing or commercializing any product, through, for example, litigation or other legal action; and |
| • | the failure of a strategic collaborator to make required milestone payments, meet contractual milestone obligations or exercise options which may result in our terminating applicable licensing arrangements. |
If our process related to product development does not result in an approved and commercially successful product, our business could be adversely affected.
We focus our research and development activities on areas in which we have particular strengths. The outcome of any development program is highly uncertain, notwithstanding how promising a particular program may seem. Success in pre-clinical and early-stage clinical trials may not necessarily translate into success in large-scale clinical trials. Further, clinical trials are expensive and require increased investment, which will adversely affect our short-term profitability.
In addition, we will need to obtain and maintain regulatory approval in order to market new products. Notwithstanding the outcome of clinical trials for new products, regulatory approval may not be achieved. The results of clinical trials are susceptible to varying interpretations that may delay, limit or prevent approval or result in the need for post-marketing studies. In addition, changes in regulatory policy for product approval during the period of product development and review by regulators of a new application may cause delays or rejection. Even if we receive regulatory approval, this approval may include limitations on the indications for which we can market the product. There is no guarantee that we will be able to satisfy the applicable regulatory requirements, and we may suffer a significant variation from planned revenue as a result.
Our planned clinical trials may not begin on time, or at all, and our planned and ongoing clinical trials may not be completed on schedule, or at all.
The commencement or completion of any of our clinical trials may be delayed or halted for numerous reasons, including, but not limited to, the following:
| • | the FDA or other regulatory authorities do not approve a clinical trial protocol or a clinical trial, or place a clinical trial on hold; |
| • | the data and safety monitoring committee of a clinical trial recommends that a trial be placed on hold or suspended; |
| • | patients do not enroll in clinical trials at the rate we expect; |
| • | patients are not followed up at the rate we expect; |
| • | patients experience adverse side effects or events related to our products; |
30
Table of Contents
| • | patients die or suffer adverse medical effects during a clinical trial for a variety of reasons, including the advanced stage of their disease and medical problems, which may or may not be related to our product candidates; |
| • | regulatory inspections of our clinical trials or manufacturing facilities, which may, among other things, require us to undertake corrective action or suspend or terminate our clinical trials if investigators find us not to be in compliance with regulatory requirements; |
| • | the failure of our manufacturing process to produce finished products which conform to design and performance specifications; |
| • | changes in governmental regulations or administrative actions; |
| • | the interim results of the clinical trial are inconclusive or negative; |
| • | pre-clinical or clinical data is interpreted by third parties in different ways; |
| • | our clinical trial expenditures are constrained by our budgetary considerations; |
| • | our trial design, although approved, is inadequate to demonstrate safety and/or efficacy; or |
| • | our financial resources are not sufficient to commence or complete a trial. |
Clinical trials may require the enrollment of large numbers of patients, and suitable patients may be difficult to identify and recruit. Patient enrollment in clinical trials and completion of patient follow-up in clinical trials depend on many factors, including the size of the patient population, the nature of the trial protocol, the proximity of patients to clinical sites and the eligibility criteria for the study and patient compliance. For example, patients may be discouraged from enrolling in our clinical trials if the trial protocol requires them to undergo extensive post-treatment procedures to assess the safety and effectiveness of our products, or they may be persuaded to participate in contemporaneous trials of competitive products. Delays in patient enrollment or failure of patients to continue to participate in a study may cause an increase in costs and delays or may result in the failure of the trial.
Our clinical trial costs will increase if we have material delays in our clinical trials or if we need to perform more or larger clinical trials than planned. Adverse events during a clinical trial could cause us to repeat a trial, terminate a trial or cancel an entire program.
Pre-clinical development is a long, expensive and uncertain process, and we may terminate one or more of our ongoing pre-clinical development programs.
We may determine that certain of our pre-clinical product candidates or research programs do not have sufficient potential to warrant the allocation of resources. In addition, we may not have or be able to obtain adequate funding to sustain the development of any or all of our pre-clinical product candidates or research programs. Accordingly, we may elect to terminate programs for such product candidates or programs. If we terminate a pre-clinical product candidate or program in which we have invested significant resources, our prospects may suffer, as we will have expended resources on a program that will not provide a return on our investment and we will have missed the opportunity to have allocated those resources to potentially more productive uses. For example, as part of our cost reduction measures we have terminated several of our pre-clinical development programs.
We may not be able to protect our intellectual property or obtain necessary intellectual property rights from third parties, which could adversely affect our business.
Our success depends, in part, on ensuring that our intellectual property rights are covered by valid and enforceable patents or effectively maintained as trade secrets and on our ability to detect violations of our intellectual property rights and enforce such rights against others.
31
Table of Contents
The validity of our patent claims depends, in part, on whether pre-existing public information described or rendered obvious our inventions as of the filing date of our patent applications. We may not have identified all prior art, such as U.S. and foreign patents, published applications or published scientific literature that could adversely affect the validity of our issued patents or the patentability of our pending patent applications. For example, patent applications in the U.S. are maintained in confidence for up to 18 months after their filing. In some cases, however, patent applications remain confidential in the U.S. Patent and Trademark Office (the “USPTO”) for the entire time prior to their issuance as a U.S. patent. Patent applications filed in countries outside the U.S. are not typically published until at least 18 months from their first filing date. Similarly, publication of discoveries in scientific or patent literature often lags behind actual discoveries. Therefore, we cannot be certain that we were the first to invent, or the first to file patent applications related to, our technology. In the event that a third party has also filed a U.S. patent application covering a similar invention, we may have to participate in an adversarial proceeding, known as an interference, declared by the USPTO to determine priority of invention in the U.S. It is possible that we may be unsuccessful in the interference, resulting in a loss of some portion or all of our U.S. patent positions. The laws in some foreign jurisdictions do not protect intellectual property rights to the same extent as in the U.S., and many companies have encountered significant difficulties in protecting and defending such rights in foreign jurisdictions. If we encounter such difficulties or we are otherwise precluded from effectively protecting our intellectual property rights in foreign jurisdictions, our business prospects could be substantially harmed.
We frequently seek patents to protect our intellectual property. It should be recognized that we may not be able to obtain patent protection for key elements of our technology, as the patent positions of medical device, pharmaceutical and biotechnology companies are uncertain and involve complex legal and factual questions for which important legal issues are largely unresolved. For example, no consistent policy has emerged regarding the scope of health-related patent claims that are granted by the USPTO or enforced by the U.S. federal courts. Rights under any of our issued patents may not provide commercially meaningful protection for our products or afford us a commercial advantage against our competitors or their competitive products or processes. In addition, even if a patent is issued, the coverage claimed in a patent application may be significantly reduced in the patent as granted.
There can be no assurance that:
| • | patent applications will result in the issuance of patents; |
| • | additional proprietary products developed will be patentable; |
| • | licenses we have obtained from third parties that we use in connection with our technology will not be terminated; |
| • | patents issued will provide adequate protection or any competitive advantages; |
| • | patents will not be successfully challenged by any third parties; or |
| • | the patents of others will not impede our or our collaborators’ ability to commercialize our technology. |
For example, the drug paclitaxel is itself not covered by composition of matter patents. Therefore, although we have developed and are developing an intellectual property portfolio around the use of paclitaxel for intended commercial applications, others may be able to engage in off-label use of paclitaxel for the same indications, causing us to lose potential revenue. Furthermore, others may independently develop similar products or technologies or, if patents are issued to us, design around any patented technology developed by us, which could affect our potential to generate revenues and harm our results of operations.
Patent protection for our technology may not be available based on prior art. The publication of discoveries in scientific or patent literature often lags behind actual discoveries. As a consequence, there may be uncertainty as to whether we or a third party were the first creator of inventions covered by issued patents or pending patent applications or that we or a third party were the first to file patent applications for
32
Table of Contents
such inventions. Moreover, we might have to participate in interference proceedings declared by the USPTO, or other proceedings outside the U.S., including oppositions, to determine priority of invention or patentability, which could result in substantial cost to us even if the outcome were favorable. An unfavorable outcome in an interference or opposition proceeding could preclude us, our collaborators and our licensees from making, using or selling products using the technology or require us to obtain license rights from prevailing third parties. We do not know whether any prevailing party would offer us a license on commercially acceptable terms, if at all. We may also be forced to pay damages or royalties for our past use of such intellectual property rights, as well as royalties for any continued usage.
As part of our patent strategy, we have filed a variety of patent applications internationally. Oppositions have been filed against various granted patents that we either own or license and which are related to certain of our technologies. For a summary of certain of our material current legal proceedings, including those relating to our patents and intellectual property, see the section entitled “Legal Proceedings” under Part I, Item 3 of this Annual Report on Form 10-K.
Our future success and competitive position depend in part on our ability to obtain and maintain certain proprietary intellectual property rights used in our approved products and principal product candidates. Any such success depends in part on effectively instituting claims against others who we believe are infringing our rights and by effectively defending claims of intellectual property infringement brought by our competitors and others. The stent-related markets have experienced rapid technological change and obsolescence in the recent past, and our competitors have strong incentives to attempt to stop or delay us from introducing new products and technologies. See “—We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights.”
We do not know whether the patents that we have obtained or licensed, or may be able to obtain or license in the future, would be held valid or enforceable by a court or whether a competitor’s technology or product would be found to infringe such patents. Further, we have no assurance that third parties will not properly or improperly modify or terminate any license they have granted to us.
We have obtained licenses from third parties with respect to their intellectual property that we use in connection with certain aspects of our technology or products. However, we may need to obtain additional licenses for the development of our current or future products. Licenses may not be available on satisfactory terms or at all. If available, these licenses may obligate us to exercise diligence in bringing our technology to market and may obligate us to make minimum guarantee or milestone payments. This diligence and these milestone payments may be costly and could adversely affect our business. We may also be obligated to make royalty payments on the sales, if any, of products resulting from licensed technology and may be responsible for the costs of filing and prosecuting patent applications. These costs could affect our results of operations and decrease our earnings.
Certain of our key technologies include trade secrets and know-how that may not be protected by patents. There can be no assurance that we will be able to protect our trade secrets. To help protect our rights, we require employees, consultants, advisors and collaborators to enter into confidentiality agreements. We cannot assure you that all employees, consultants, advisors and collaborators have signed such agreements, or that these agreements will adequately protect our trade secrets, know-how or other proprietary information in the event of any unauthorized use or disclosure. Furthermore, we may not have adequate remedies for any such breach. Any disclosure of confidential data into the public domain or to third parties could allow our competitors to learn our trade secrets and use the information in competition against us.
If certain single-source suppliers fail to deliver key product components in a timely manner, our manufacturing ability would be impaired and our product sales could suffer.
We depend on certain single-source suppliers that supply components used in the manufacture of certain of our products. If we need alternative sources for key component parts for any reason, these component parts may not be immediately available to us. If alternative suppliers are not immediately available, we will have to identify and qualify alternative suppliers, and production of these components may be delayed. We may not be able to find an adequate alternative supplier in a reasonable time period or
33
Table of Contents
on commercially acceptable terms, if at all. Shipments of affected products have been limited or delayed as a result of such problems in the past, and similar problems could occur in the future. Our inability to obtain our key source supplies for the manufacture of our products may require us to delay shipments of products, harm customer relationships or force us to curtail or cease operations.
Compulsory licensing and/or generic competition may affect our business in certain countries.
In a number of countries, governmental authorities have the right to grant licenses to others to use a pharmaceutical or medical device company’s patents or other intellectual property without the consent of the owner of the patent or other intellectual property. Governmental authorities could use this right to grant licenses under our patents or other intellectual property to others or could require us to grant licenses under our patents or other intellectual property to others, thereby allowing our competitors to manufacture and sell their own versions of our products. In other circumstances, governmental authorities could use this right to require us or our licensees to reduce the prices of our products. In all of these situations, our sales or the sales of our licensee(s), and the results of our operations, in these countries could be adversely affected.
We are subject to litigation that could strain our resources and distract management.
From time to time, we are involved in a variety of claims, lawsuits and other disputes. These suits may concern issues including product liability, contract disputes, employee-related matters, intellectual property and personal injury matters. It is not feasible to predict the outcome of all pending suits and claims, and the ultimate resolution of these matters as well as future lawsuits could have a material adverse effect on our business, financial condition, results of operations or cash flows or reputation.
For a summary of certain of our material current legal proceedings, see the section entitled “Legal Proceedings” under Part I, Item 3 of this Annual Report on Form 10-K.
We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights.
In connection with maintaining the value of our various intellectual property and exclusivity rights, we regularly evaluate the activities of others worldwide. Our success will depend, in part, on our ability to obtain patents, or licenses to patents, maintain trade secret protection and enforce our rights against others. Should it become necessary to protect those rights, we intend to pursue all cost-efficient strategies, including, when appropriate, negotiation or litigation in any relevant jurisdiction. For a summary of certain of our current legal proceedings, including proceedings in respect of patent infringement and intellectual property matters, see the section entitled “Legal Proceedings” under Part I, Item 3 of this Annual Report on Form 10-K.
We intend to pursue and to defend vigorously any and all actions of third parties related to our material patents and technology. Any failure to obtain and protect intellectual property could adversely affect our business and our ability to operate could be hindered by the proprietary rights of others.
Our involvement in intellectual property litigation could result in significant expense, adversely affecting the development of product candidates or sales of the challenged product or intellectual property and diverting the efforts of our technical and management personnel, whether or not such litigation is resolved in our favor. Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources, and intellectual property litigation may be used against us as a means of gaining a competitive advantage. Competing parties frequently file multiple suits to leverage patent portfolios across product lines, technologies and geographies and to balance risk and exposure between the parties. Uncertainties resulting from the initiation and continuation of any litigation could affect our ability to continue our operations. In the event of an adverse outcome as a defendant in any such litigation, we may, among other things, be required to:
| • | pay substantial damages or back royalties; |
34
Table of Contents
| • | cease the development, manufacture, use or sale of product candidates or products that infringe upon the intellectual property of others; |
| • | expend significant resources to design around a patent or to develop or acquire non-infringing intellectual property; |
| • | discontinue processes incorporating infringing technology; or |
| • | obtain licenses to the infringed intellectual property. |
We cannot assure you that we will be successful in developing or acquiring non-infringing intellectual property or that necessary licenses will be available upon reasonable terms, if at all. Any such development, acquisition or license could require the expenditure of substantial time and other resources and could adversely affect our business and financial results. If we cannot develop or acquire such intellectual property or obtain such licenses, we could encounter delays in any introduction of products or could find that the development, manufacture or sale of products requiring such licenses could be prohibited.
If third parties file patent applications, or are issued patents claiming technology also claimed by us in pending applications, we may be required to participate in interference proceedings with the USPTO, or other proceedings outside the United States, including oppositions, to determine priority of invention or patentability, which could result in substantial cost to us even if the eventual outcome were favorable.
Our ability to operate could be hindered by the proprietary rights of others.
A number of pharmaceutical, biotechnology and medical device companies as well as research and academic institutions have developed technologies, filed patent applications or received patents on various technologies that may be related to our business. Some of these technologies, applications or patents may conflict with or adversely affect our technologies or intellectual property rights, including those that we license from others. We are aware of other parties holding intellectual property rights that may represent prior art or other potentially conflicting intellectual property. Any conflicts with the intellectual property of others could limit the scope of the patents, if any, that we may be able to obtain or result in the denial of our current or future patent applications altogether.
If patents that cover our activities are issued to other persons or companies, we could be charged with infringement. In the event that other parties’ patents cover any portion of our activities, we may be forced to develop alternatives or negotiate a license for such technology. We do not know whether we would be successful in either developing alternative technologies or acquiring licenses upon reasonable terms, if at all. Obtaining any such licenses could require the expenditure of substantial time and other resources and could harm our business and decrease our earnings. If we do not obtain such licenses, we could encounter delays in the introduction of our products or could find that the development, manufacture or sale of products requiring such licenses is prohibited.
Technological advances and evolving industry standards could reduce our future product sales, which could cause our revenues to grow more slowly or decline.
The markets for our products are characterized by rapidly changing technology, changing customer needs, evolving industry standards and frequent new product introductions and enhancements. The emergence of new industry standards in related fields may adversely affect the demand for our products. This could happen, for example, if new standards and technologies emerged that were incompatible with customer deployments of our applications. In addition, any compounds, products or processes that we develop may become obsolete or uneconomical before we recover any of the expenses incurred in connection with their development. We cannot assure you that we will succeed in developing and marketing product enhancements or new products that respond to technological change, new industry standards, changed customer requirements or competitive products on a timely and cost-effective basis. Additionally, even if we are able to develop new products and product enhancements, we cannot assure you that they will achieve market acceptance.
35
Table of Contents
Future legislation or regulatory changes to, or consolidation in, the health care system may affect our ability to sell our products profitably.
There have been, and we expect there will continue to be, a number of legislative and regulatory proposals to change the health care system, and some could involve changes that could significantly affect our business. For example, the recent health care reform bill signed into law in the United States includes a 2.3% excise tax on a wide range of medical devices. Efforts by governmental and third-party payers to reduce health care costs or the announcement of legislative proposals or reforms to implement government controls could cause a reduction in sales or in the selling price of our products, which would seriously harm our business. Additionally, initiatives to reduce the cost of health care have resulted in a consolidation trend in the health care industry, including hospitals. This in turn has resulted in greater pricing pressures and the exclusion of certain suppliers from certain market segments as consolidated groups such as group purchasing organizations, independent delivery networks and large single accounts continue to consolidate purchasing decisions for some of our hospital customers. We expect that market demand, government regulation, and third-party reimbursement policies will continue to change the worldwide health care industry, resulting in further business consolidations and alliances among our customers and competitors, which may reduce competition, exert further downward pressure on the prices of our products and may adversely impact our business, financial condition or results of operations.
We may be subject to damages resulting from claims that we or our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
Many of our employees were previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although no such claims against us are currently pending, we may be subject to claims that these employees or we have used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management. If we fail in defending such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. A loss of key research personnel or their work product could hamper or prevent our ability to commercialize certain product candidates, which could severely harm our business.
We may incur significant costs complying with environmental laws and regulations.
Our research and development processes and manufacturing operations involve the use of hazardous materials. In the countries in which we operate or sell our products, we are subject to federal, state, provincial, local and other laws and regulations that govern the use, manufacture, storage, handling and disposal of such materials and certain waste products. The risk of accidental contamination or injury from these materials cannot be completely eliminated. In the event of an accident or the discovery of pre-existing contamination at one or more of our facilities, we could be held liable for any damages that result, and any such liability could exceed our resources. In addition, our manufacturing facilities may become subject to new or increased legislation or regulation as a result of climate control initiatives. We may not be specifically insured with respect to these liabilities, and we do not know whether we will be required to incur significant costs to comply with environmental laws and regulations in the future, or whether our operations, business or assets will be harmed by current or future environmental laws or regulations.
We must receive regulatory approval for each of our product candidates before they can be sold commercially in Canada, the United States or internationally, which can take significant time and be very costly.
The development, manufacture and sale of medical devices and human therapeutic products in Canada, the United States and internationally is governed by a variety of statutes and regulations. These laws require, among other things:
| • | regulatory approval of manufacturing facilities and practices; |
| • | adequate and well-controlled research and testing of products in pre-clinical and clinical trials; |
36
Table of Contents
| • | review and approval of submissions containing manufacturing, pre-clinical and clinical data in order to obtain marketing approval based on establishing the safety and efficacy of the product for each use sought, including adherence to current cGMPs during production and storage; and |
| • | control of marketing activities, including advertising and labeling. |
The product candidates currently under development by us or our collaborators will require significant research, development, pre-clinical and clinical testing, pre-market review and approval, and investment of significant funds prior to their commercialization. In many instances, we are dependent on our collaborators for regulatory approval and compliance, and have little or no control over these matters. The process of completing clinical testing and obtaining such approvals is likely to take many years and require the expenditure of substantial resources, and we do not know whether any clinical studies by us or our collaborators will be successful, whether regulatory approvals will be received, or whether regulatory approvals will be obtained in a timely manner. Despite the time and resources expended by us, regulatory approval is never guaranteed. Even if regulatory approval is obtained, regulatory agencies may limit the approval to certain diseases, conditions or categories of patients who can use them.
If any of our development programs are not successfully completed in a timely fashion, required regulatory approvals are not obtained in a timely fashion, or products for which approvals are obtained are not commercially successful, it could seriously harm our business.
Our products and manufacturing facilities that have, or may receive, regulatory approval, are or will be subject to ongoing regulation. In addition, we have little or no control over the manufacturing facilities of our collaborators in which certain of our products are manufactured.
Our products and manufacturing operations are subject to extensive regulation in the United States by the FDA and by similar regulatory agencies abroad. Ongoing regulation includes compliance with an array of manufacturing and design controls and testing, quality control, storage and documentation procedures. Regulatory agencies may also require expensive post-approval studies. Any adverse events associated with our products must also be reported to regulatory authorities. If deficiencies in our or our collaborators’ manufacturing and laboratory facilities are discovered, or we or our collaborators fail to comply with applicable post-market regulatory requirements, a regulatory agency may close the facility or suspend manufacturing.
With respect to products manufactured by third-party contractors, we are, and we expect to continue to be, dependent on our collaborators for continuing regulatory compliance, and we may have little or no control over these matters. Our ability to control third-party compliance with the FDA and other regulatory requirements will be limited to contractual remedies and rights of inspection. Our failure or the failure of third-party manufacturers to comply with regulatory requirements applicable to our products may result in legal or regulatory action by those regulatory authorities. There can be no assurance that our or our collaborators’ manufacturing processes will satisfy regulatory, cGMPs or International Standards Organization requirements.
In addition, there may be uncertainty as to whether or not we or others who are involved in the manufacturing process will be able to make the transition to commercial production of some of our newly developed products. A failure to achieve regulatory approval for manufacturing facilities or a failure to make the transition to commercial production for our products will adversely affect our prospects, business, financial condition and results of operations.
If we are unable to fully comply with federal and state “fraud and abuse laws”, we could face substantial penalties, which may adversely affect our business, financial condition and results of operations.
We are subject to various laws pertaining to health care fraud and abuse, including the U.S. Anti-Kickback Statute, physician self-referral laws, the U.S. False Claims Act, the U.S. Health Insurance Portability and Accountability Act of 1996, the U.S. False Statements Statute, and state law equivalents to these U.S. federal laws, which may not be limited to government-reimbursed items and may not contain identical exceptions. Violations of these laws are punishable by criminal and civil sanctions, including, in some instances, civil and criminal penalties, damages, fines, exclusion from participation in U.S. federal
37
Table of Contents
and state health care programs, including Medicare and Medicaid, and the curtailment or restructuring of operations. Any action against us for violation of these laws could have a significant impact on our business. In addition, we are subject to the U.S. Foreign Corrupt Practices Act. Any action against us for violation by us or our agents or distributors of this act could have a significant impact on our business.
Acquisition of companies or technologies may result in disruptions to our business.
As part of our business strategy, we may acquire additional assets and businesses principally relating to or complementary to our current operations. Any acquisitions or mergers by us will be accompanied by the risks commonly encountered in acquisitions of companies. These risks include, among other things, higher than anticipated acquisition costs and expenses, the difficulty and expense of integrating the operations and personnel of the companies and the loss of key employees and customers as a result of changes in management.
In addition, geographic distances may make integration of acquired businesses more difficult. We may not be successful in overcoming these risks or any other problems encountered in connection with any acquisitions.
If significant acquisitions are made for cash consideration, we may be required to use a substantial portion of our available cash, cash equivalents and short-term investments. Future acquisitions by us may cause large one-time expenses or create goodwill or other intangible assets that could result in significant asset impairment charges in the future. Acquisition financing may not be available on acceptable terms, if at all.
If we fail to hire and retain key management, scientific and technical personnel, we may be unable to successfully implement our business plan.
We are highly dependent on our senior management and scientific and technical personnel. The competition for qualified personnel in the health care field is intense, and we rely heavily on our ability to attract and retain qualified managerial, scientific and technical personnel. Our ability to manage growth effectively will require continued implementation and improvement of our management systems and the ability to recruit and train new employees. We may not be able to successfully attract and retain skilled and experienced personnel, which could harm our ability to develop our product candidates and generate revenues.
Risks Relating to Our Indebtedness, Organization and Financial Position
There is significant doubt regarding our ability to remain a going concern.
There are material uncertainties regarding our ability to continue as a going concern, and such ability will depend upon our ability to: (i) complete the Recapitalization Transaction, (ii) restructure, refinance or amend the terms of our other existing debt obligations including the Existing Floating Rate Notes; (iii) remain in compliance with our debt covenants and maintain access to our Existing Credit Facility and the DIP Facility; (iv) obtain additional equity and debt financing (including the Exit Facility); (v) achieve revenue growth and improvement of gross margins; and (vi) achieve greater operating efficiencies or a reduction of expenses. We reported net losses for our fiscal quarters ending March 31, 2010, June 30, 2010, September 30, 2010 and December 31, 2010. Our cumulative losses, high level of outstanding indebtedness and inability to comply with related debt covenants, in addition to our current liquidity situation, lead to uncertainty as to our ability to continue as a going concern for a period longer than twelve months from December 31, 2010. Our ability to continue as a going concern depends on, among other factors, a return to profitable operations and a successful restructuring of our obligations related to the Subordinated Notes and Existing Floating Rate Notes. Until the completion of the Recapitalization Transaction, our future remains uncertain, and there can be no assurance that our efforts in this regard will be successful.
38
Table of Contents
As a result of filing for creditor protection, our business and operations will be subject to certain risks.
The CCAA Proceedings (together with the Chapter 15 Cases, as applicable) by Angiotech and certain of our subsidiaries have subjected our business and operations to various risks, including but not limited to, the following:
| • | The CCAA and/or U.S. ancillary filings may adversely affect our business prospects and our ability to operate during the restructuring process; |
| • | The coordination of a CCAA and/or U.S. ancillary filing and operating under the protection of an insolvency proceeding involve significant costs, including expenses of legal counsel and other professional advisors; |
| • | We may have difficulty continuing to obtain and maintain contracts necessary to continue our operations and at affordable rates with competitive terms; |
| • | We may have difficulty maintaining existing and building new customer and supplier relationships; |
| • | Transactions outside the ordinary course of business would be subject to the prior approval of the courts, which may limit our ability to respond to certain events or take advantage of certain opportunities in a timely manner; |
| • | We may not be able to obtain court approval or such approval may be delayed with respect to motions made in the restructuring proceedings; |
| • | We may be unable to retain and motivate key executives and associates through the process of restructuring, and we may have difficulty attracting new employees; |
| • | There can be no assurance as to our ability to maintain or obtain sufficient financing sources for operations or to fund any restructuring plan and meet future obligations; and |
| • | There can be no assurance that we will be able to successfully develop, prosecute, confirm and consummate one or more plans of compromise or arrangement that are acceptable to the court and our creditors and other parties in interest. |
We may not be able to service our debt or obtain future financing and we may be limited operationally.
We may incur additional debt from time to time to finance our operations, capital expenditures or for other purposes if we comply with the restrictions in the indenture governing the New Floating Rate Notes (the “New Notes Indenture”) and those anticipated to be included in our proposed Exit Facility.
The debt that we carry may have important consequences to us, including the following:
| • | our ability to obtain additional financing, if necessary, for working capital, capital expenditures, acquisitions or other purposes may be impaired or additional financing may not be available on favorable terms; |
| • | we may be required to use a portion of our operating cash flow to pay the interest or principal on our debt. These payments reduce the funds that would otherwise be available for our operations and future business opportunities; |
| • | a substantial decrease in our net operating cash flows could make it difficult for us to meet our debt service requirements and force us to further modify our operations; |
| • | we may be placed at a competitive disadvantage compared to our less leveraged competitors; and |
| • | we may be more vulnerable to a downturn in our business or the economy generally. |
39
Table of Contents
If we cannot service our debt, we will be forced to take actions such as reducing or delaying acquisitions and/or capital expenditures, selling assets, restructuring or refinancing our debt, or seeking additional equity capital. We can give no assurance that we can do any of these things on satisfactory terms or at all.
We have effected reductions in our operating costs and, as a result, our ability to cut costs further and sustain our business initiatives may be limited.
Beginning in late 2008 and continuing into 2009, 2010 and 2011, we have implemented various initiatives to reduce operating costs across all functions of the Company and focus our business efforts on our most promising near-term product opportunities. As a result of these cost-cutting initiatives, we may have a more limited ability to further reduce costs to increase our liquidity should such a measure become necessary. Any further reductions may have a materially negative impact on our business.
Restrictive covenants in our existing and future credit agreements may adversely affect us.
We must comply with operating and financing restrictions in our Existing Credit Facility, the DIP Facility, the indentures governing our notes and those anticipated to be included in our proposed Exit Facility. These restrictions affect, and in many respects limit or prohibit, our ability to:
| • | incur additional indebtedness; |
| • | make any distributions, declare or pay any dividends on or acquire any of our capital stock; |
| • | incur liens; |
| • | make investments, including joint venture investments; |
| • | sell assets; |
| • | repurchase certain debt; and |
| • | merge or consolidate with or into other companies or sell substantially all of our assets. |
We may also have similar restrictions with any future debt.
We expect that our proposed Exit Facility will require us to make mandatory payments upon the occurrence of certain events, including the sale of assets, in each case subject to certain limitations and conditions. We also expect that our proposed Exit Facility will contain financial covenants, including maintaining certain levels of EBITDA and interest coverage ratios. These restrictions could limit our ability to plan for or react to market conditions or to meet extraordinary capital needs or otherwise could restrict our activities. These restrictions could also adversely affect our ability to finance our future operations or capital needs or to engage in other business activities that would be in our interest.
Our obligation to pay cash interest on our notes has had, and may continue to have, an adverse effect on our liquidity.
We are obligated to make periodic cash interest payments on the Existing Floating Rate Notes and will be similarly obligated under the Subordinated Notes if we do not effect the Recapitalization Transaction. Upon consummation of the Recapitalization Transaction, the Subordinated Notes will no longer be outstanding and our only cash interest payments will be those under our Existing Floating Rate Notes, if any, and under the New Floating Rate Notes. As a result of these required cash interest payments, combined with the significant decline in royalty payments we receive from BSC, we have had significant liquidity issues. If our cash flows are worse than expected, we may be unable to access additional sources of liquidity to fund our cash needs, which could further adversely affect our financial condition or results of operations and our ability to make payments on our debt.
40
Table of Contents
The current global credit and financial market conditions may exacerbate certain risks affecting our business.
The current significantly challenging global credit and financial market conditions may make obtaining additional financing for our business difficult or impossible, or may make it more difficult to pursue a restructuring or other transaction with respect to our existing indebtedness.
In addition, the recent tightening of global credit may lead to a disruption in the performance of our third-party contractors, suppliers or collaborators. We rely on third parties for several important aspects of our business, including royalty revenue, portions of our product manufacturing, clinical development of future collaboration products, conduct of clinical trials and raw materials. If such third parties are unable to satisfy their commitments to us, our business would be adversely affected.
Certain of our products are used in elective medical procedures which are not covered by insurance. Adverse changes in the economy or other conditions or events have had and may continue to have an adverse effect on consumer spending and may reduce the demand for these procedures. Any such changes, conditions or events could have an adverse effect on our sales and results of operations.
We and our subsidiaries are permitted to incur substantially more debt, which could further exacerbate the risks associated with our leverage.
The respective terms of the indenture governing the Existing Floating Rate Notes (the “FRN Indenture”), the New Notes Indenture and our proposed Exit Facility expressly permit, or are anticipated to permit, as applicable, the incurrence of additional amounts of debt for specified purposes. Moreover, the FRN Indenture does not, and it is anticipated that the New Notes Indenture and our proposed Exit Facility will not, impose any limitation on our incurrence of liabilities that are not defined as “Indebtedness” under such indentures or facility (such as trade payables). If we are unable to expediently complete the Recapitalization Transaction or otherwise restructure our existing indebtedness, we may be forced to incur additional debt or other liabilities, exacerbating the related risks that we now face.
We are subject to risks associated with doing business globally.
As a medical device company with significant operations in Canada, the United States and other countries, we are subject to political, economic, operational, legal, regulatory and other risks that are inherent in conducting business globally. These risks include foreign exchange fluctuations, exchange controls, capital controls, new laws or regulations or changes in the interpretation or enforcement of existing laws or regulations, political instability, macroeconomic changes, including recessions and inflationary or deflationary pressures, increases in prevailing interest rates by central banks or financial services companies, economic uncertainty, which may reduce the demand for our products or reduce the prices that our customers are willing to pay for our products, import or export restrictions, tariff increases, price controls, nationalization and expropriation, changes in taxation, diminished or insufficient protection of intellectual property, lack of access to impartial court systems, violations of law, including the U.S. Foreign Corrupt Practices Act and the U.K. Bribery Act, disruption or destruction of operations or changes to the Company’s business position, regardless of cause, including war, terrorism, riot, civil insurrection, social unrest, strikes and natural or man-made disasters, including famine, flood, fire, earthquake, storm or disease. The impact of any of these developments, either individually or cumulatively, could have a material adverse effect on our business, financial condition and results of operations.
We may incur losses associated with foreign currency fluctuations.
We report our operating results and financial position in U.S. dollars in order to more accurately represent the currency of the economic environment in which we operate.
Our operations are in some instances conducted in currencies other than the U.S. dollar, and fluctuations in the value of foreign currencies relative to the U.S. dollar could cause us to incur currency
41
Table of Contents
exchange losses. In addition to the U.S. dollar, we currently conduct operations in Canadian dollars, Euros, Swiss francs, Danish kroner, Swedish kroner, Norwegian kroner and U.K. pounds sterling. Exchange rate fluctuations may reduce our future operating results and comprehensive income. For a description of the effects of exchange rate fluctuations on our results, see “Quantitative and Qualitative Disclosures about Market Risk” in our Annual Report on Form 10-K for the year ended December 31, 2010.
We have not entered into any forward currency contracts or other financial derivatives to hedge foreign exchange risk, and therefore we are subject to foreign currency transaction and translation gains and losses. We purchase goods and services in U.S. and Canadian dollars, Euros, Swiss francs, Danish kroner, and U.K. pounds sterling, and earn a significant portion of our license and milestone revenues in U.S. dollars. We primarily manage our foreign exchange risk by satisfying foreign-denominated expenditures with cash flows or assets denominated in the same currency.
We are not currently paying dividends on our common shares and have no current plans to do so in the future.
We cannot pay dividends on our common shares as the Company is insolvent. We have no current plans to pay dividends in the future with respect to the new common shares to be issued upon implementation of the CCAA Plan. Further, the terms of the FRN Indenture and our Existing Credit Facility restrict our ability to do so, and we expect the Exit Facility and the New Notes Indenture to contain similar restrictions.
Nasdaq and the TSX have recently delisted our common shares from quotation on their respective exchanges.
The Nasdaq Stock Market (the “NASDAQ”) and the Toronto Stock Exchange (the “TSX”) have recently delisted our common shares from quotation on their exchanges, in each case for failure to continue to meet their respective continued listing requirements. The delisting of our common shares from NASDAQ and the TSX could have material adverse effects by, among other things:
| • | reducing the liquidity and market price of our common shares; |
| • | reducing the number of investors willing to hold or acquire our common shares, thereby further restricting our ability to obtain equity financing; |
| • | reducing the amount of news and analyst coverage of the Company; and |
| • | reducing our ability to retain, attract and motivate our directors, officers and employees. |
United States investors may not be able to obtain enforcement of civil liabilities against us.
We were formed under the laws of British Columbia, Canada. A substantial portion of our assets are located outside the United States. In addition, a majority of the members of our board of directors and our officers are residents of countries other than the United States. As a result, it may be impossible for United States investors to effect service of process within the United States upon us or these persons or to enforce against us or these persons any judgments in civil and commercial matters, including judgments under United States federal or state securities laws. In addition, judgments of U.S. courts will not necessarily be recognized by courts in non-U.S. jurisdictions. Accordingly, even if you obtain a favorable judgment in a U.S. court, you may be required to re-litigate your claim in other jurisdictions. In addition, in certain jurisdictions in which certain of our subsidiaries are organized, it is questionable whether a court would accept jurisdiction and impose civil liability if proceedings were commenced in an original action predicated only upon U.S. federal securities laws.
| Item 1B. | UNRESOLVED STAFF COMMENTS |
None.
42
Table of Contents
| Item 2. | PROPERTIES |
As at December 31, 2010, we have 20 facilities located in six different countries, which include Canada, the United States, Puerto Rico, the United Kingdom, Denmark and Switzerland. Of the 20 facilities, 10 are primarily used to manufacture and distribute medical devices or materials for medical device products. Our other 10 facilities are primarily used for research, development and administrative activities. Collectively, these facilities have over 540,000 square feet of modern technical manufacturing, research and administrative operations. The following chart summarizes the facilities where our domestic and international operations occur:
| Location 1 |
Primary Purpose |
Segment |
Owned or Leased | |||
| St. Gregoire, France |
Administration | Medical Products | Leased | |||
| Lausanne, Switzerland |
Administration | Medical Products | Leased | |||
| Stockholm, Sweden |
Administration | Medical Products | Leased | |||
| Taunton, United Kingdom |
Manufacturing | Medical Products | Owned | |||
| Stenlose, Denmark (3 facilities) |
Manufacturing | Medical Products | 2 Leased, 1 Owned | |||
| Sarasota, FL |
Administration and Research | Pharmaceutical Technologies | Leased | |||
| Chicago, IL |
Administration | Medical Products | Leased | |||
| Wheeling, IL |
Manufacturing | Medical Products | Owned | |||
| Gainesville, FL |
Manufacturing | Medical Products | Owned | |||
| Henrietta, NY |
Manufacturing | Medical Products | Leased | |||
| Seattle, WA |
Administration | Pharmaceutical Technologies | Leased | |||
| Reading, PA |
Manufacturing | Medical Products | Owned | |||
| Aguadilla, Puerto Rico |
Manufacturing | Medical Products | Leased | |||
| Rincon, Puerto Rico |
Manufacturing | Medical Products | Leased | |||
| Sao Paulo, Brazil |
Administration | Medical Products | Leased | |||
| Montreal, Q.C. |
Administration and Research |
Pharmaceutical Technologies | Leased | |||
| Vancouver, B.C. (2 facilities) |
Administration and Research |
Pharmaceutical Technologies |
Leased |
| 1 | Excluded from this list are our two properties that are vacant and are held for sale. One facility is in Syracuse, NY with 77,000 square feet and the other is a 1.6 acre parcel of bare land adjacent to our corporate headquarters in Vancouver, B.C. |
We have several important, and in some cases proprietary, manufacturing capabilities and processes. These include specific capabilities in the areas of various metal bending, grinding and chemical etching, electroplating and electropolishing, plastic injection molding, wire coating, braiding and weaving of surgical textiles and kit assembly and packaging.
| Item 3. | LEGAL PROCEEDINGS |
On April 4, 2005, together with BSC, the Company commenced a legal action in the Netherlands against Sahajanand Medical Technologies Pvt. Ltd. (“SMT”) for patent infringement. On May 3, 2006, the Dutch trial court ruled in the Company’s favor, finding that our EP (NL) 0 706 376 patent was valid, and that SMT’s Infinnium™ stent infringed the patent. On March 13, 2008, a Dutch Court of Appeal held a hearing to review the correctness of the trial court’s decision. The Court of Appeal released their judgment on January 27, 2009, ruling against SMT (finding the Company’s patent novel, inventive, and sufficiently disclosed). The Court of Appeal however requested amendment of claim 12 before rendering their decision on infringement. The Company has filed a response to the Court of Appeal’s decision, and have additionally filed a further writ against SMT seeking to prevent SMT from, among other things, using its CE mark for SMT’s Infinnium stent. Any decision of the Court of Appeal is appealable to the Supreme
43
Table of Contents
Court of the Netherlands. On October 22, 2009, Angiotech and BSC settled this litigation with SMT on favorable terms. The Company has continued this matter before the Court of Appeal with respect to the requested amendment to claim 12. A hearing was held on April 22, 2010 and on August 31, 2010 this court rendered a decision favorable to Angiotech. This matter is therefore concluded.
In July 2004, Dr. Gregory Baran initiated legal action, alleging infringement by the Company’s subsidiary Medical Device Technologies Inc. (“MDT”), of two U.S. patents owned by Dr. Baran. These patents allegedly cover MDT’s BioPince™ automated biopsy device. On September 25, 2007, the judge issued her decision pursuant to the Markman hearing of December 2005. The Company submitted a Motion for Summary Judgment to the court based upon the Markman decision and on September 30, 2009, the judge ruled in MDT’s favor, finding that the BioPince device did not infringe a key claim of Dr. Baran’s patents. Dr. Baran appealed this decision. A hearing on Dr. Baran’s appeal was held before the U.S. Court of Appeals for the Federal Circuit on June 9, 2010; on August 12, 2010, the court ruled in our favor. Dr. Baran petitioned for a rehearing, but this has been denied. Dr. Baran has filed a Writ of Certiorari with the U.S. Supreme Court seeking review of the CAFC decision. We filed a Brief in Opposition on March 7, 2011. As the Supreme Court denied Certiorari, this matter is therefore concluded.
On October 4, 2010, the Company was notified that QSR Holdings, Inc. (“QSR”), as a representative for former stockholders of Quill Medical, Inc. (“QMI”), had made a formal demand (the “Arbitration Demand”) to the American Arbitration Association naming the Company, QMI and Angiotech Pharmaceuticals (US), Inc. (“Angiotech US”) as respondents (collectively, the “Respondents”). The Arbitration Demand alleged that the Respondents failed to satisfy certain obligations under the Agreement and Plan of Merger, dated May 25, 2006, by and among Angiotech, Angiotech US, Quaich Acquisition, Inc. and QMI (the “Merger Agreement”), notably including the obligations to make certain earn out payments and the orthopedic milestone payment. The Arbitration Demand sought either direct monetary damages or, in the alternative, extension of the earn out period for a sixth year as more fully set forth in the Merger Agreement. In addition, QSR commenced an action in the United States District Court for the Middle District of North Carolina on October 1, 2010 against the Respondents, entitled QSR Holdings, Inc. v. Angiotech Pharmaceuticals, Inc., Angiotech Pharmaceuticals (US), Inc. and Quill Medical, Inc. (the “Federal Litigation”). The Complaint in the Federal Litigation alleged, among other items, that the Respondents: (a) breached certain contractual obligations under the Merger Agreement; (b) made certain misrepresentations or omissions during the initial negotiation of the Merger Agreement; and (c) tortiously interfered with the Merger Agreement. QSR sought damages in an unstated amount together with punitive damages and/or attorneys fees to the extent allowed by law. On October 15, 2010, the Respondents moved to dismiss the Complaint in part and to transfer venue to federal court in Washington State.
Subsequent to the initiation of the Federal Litigation and the delivery of the Arbitration Demand, representatives of Angiotech and QSR engaged in substantial settlement discussions. Those discussions culminated in an agreement (the “Settlement Agreement”), dated as of January 27, 2011, resolving, among other things, any and all claims that the parties may have arising out of the Merger Agreement (including, without limitation, all claims, counterclaims or defenses that were or could have been asserted in the Arbitration Demand and the Federal Litigation) in exchange for a payment of $6 million (the “Settlement Amount”) by Angiotech US. $2.0 million of the Settlement Amount is payable on the earlier of (i) 10 business days after the implementation of the Recapitalization Transaction contemplated by the RSA (the “Implementation Date”) and (ii) May 31, 2011. The remainder of the Settlement Amount is payable in 24 equal monthly installments of $166,667 each beginning on the earlier of (i) 30 days after the Implementation Date; and (ii) July 31, 2011 (subject to reduction in the event of pre-payment as more fully set forth in the Settlement Agreement). The Settlement Agreement further provides for complete and mutual releases between the parties, as more fully set forth therein. In accordance with its obligations under the Settlement Agreement, on February 24, 2011, QSR irrevocably dismissed the Federal Litigation and the Arbitration Demand with prejudice.
In light of the Company’s recent liquidity and capital constraints, on November 11, 2010, Angiotech US informed Rex Medical, LP (“Rex”) that it would not be able to continue selling the Option IVC Filter under the existing terms of the License, Supply, Marketing and Distribution Agreement, dated as of March 13, 2008, by and between Angiotech US and Rex (as amended, the “Option Agreement”). Subsequently, on November 18, 2010, Rex initiated an arbitration (the “Rex Arbitration”) against Angiotech US alleging that
44
Table of Contents
Angiotech US had breached the Option Agreement. Rex sought monetary damages in excess of $3.0 million, including costs, fees and expenses incurred in connection with the Rex Arbitration. On December 2, 2010, Rex obtained a temporary restraining order and preliminary injunction against Angiotech US from the United States District Court for the Southern District of New York (such action, the “SDNY Action”) requiring Angiotech US to honor its obligations under the Option Agreement for a period of 180 days or until the Rex Arbitration was concluded. On January 24, 2011, Angiotech US received a termination notice from Rex indicating that the Option Agreement would cease on April 24, 2011.
On February 16, 2011, Angiotech US entered into a Settlement and License Termination Agreement with Rex (the “Rex Settlement Agreement”) which provides for the full and final settlement and/or dismissal of all claims arising under the Option Agreement. Pursuant to the Rex Settlement Agreement, the Company and Rex agreed to withdraw any claims that have been or could have made in the Rex Arbitration, as well as to dismiss the SDNY Action with prejudice. The Rex Settlement Agreement further provides that: (i) the Option Agreement will terminate on March 31, 2011, or at an earlier date if so elected by Rex (the “Termination Date”), upon which Angiotech US will no longer market, sell or distribute the Option IVC Filter; (ii) Angiotech US will make a payment in the amount of $1.5 million to Rex within five business days of the effective date of the Rex Settlement Agreement in full and final payment and settlement of all claims and royalties due under the Option Agreement relating to the sales of the Option IVC Filter recorded by Angiotech US prior to January 1, 2011, and all milestone payments due or that may come due to Rex under the Option Agreement now or in the future; (iii) within five business days of the effective date, Angiotech US will make a royalty payment to Rex based upon actual cash receipts collected from customers attributable to sales of the Option IVC Filter that are recorded and accrued between January 1, 2011 and the effective date; (iv) from the effective date through the Termination Date, Angiotech US will make ongoing royalty payments to Rex on a weekly basis based upon actual cash receipts collected during each previous calendar week in respect of sales of the Option IVC Filter; and (v) Angiotech US will deliver to Rex certain materials relating to the marketing and sale of the Option IVC Filter. The Rex Settlement Agreement became effective on March 10, 2011 upon the Canadian Court granting an order authorizing Angiotech US to enter into the Rex Settlement Agreement and approving the terms thereof.
As contemplated by the RSA, on January 28, 2011, the Angiotech Entities filed for protection under the CCAA and the Initial Order was granted by the Canadian Court. The Initial Order imposed a stay of proceedings against the Angiotech Entities preventing any party from commencing or continuing any action, suit or proceeding against the Angiotech Entities or from exercising other enforcement rights that could arise as a result of the commencement of proceedings under the CCAA. The stay of proceedings provided for in the Initial Order generally precludes parties from taking any action against the Angiotech Entities for breach of contractual or other obligations. The stay period currently is set to terminate on April 15, 2011 and may be extended by further order of the Canadian Court. In order to have the CCAA Proceedings recognized in the United States, on January 30, 2011 the Angiotech Entities commenced the Chapter 15 Cases. The purpose of the Chapter 15 Cases was to obtain recognition and enforcement in the United States of certain relief granted in the CCAA Proceedings, and to obtain the assistance of the U.S. court in effectuating the Recapitalization Transaction. On February 22, 2011, the U.S. court granted an order that among other things, recognized the CCAA Proceedings as a foreign main proceeding and provided that the Initial Order would be enforced on a final basis and given full force and effect in the United States.
For more information on the CCAA Proceedings and Recapitalization Transaction, refer to Management’s Discussion and Analysis of Financial Condition and Results of Operations, along with notes 1, 16 and 26 in the audited consolidated financial statements in this Annual Report on Form 10-K.
At the European Patent Office (“EPO”), various patents either owned or licensed by or to the Company are in opposition proceedings including the following:
| • | In EP_1_155_689, the EPO decided to revoke the patent as a result of the oral hearing held on November 25, 2010, and the Company has until March 21, 2011 to file an appeal. |
| • | In EP_1_159_974, the EPO decided to revoke the patent as a result of Oral Proceedings held on June 9, 2010. Angiotech filed a Notice of Appeal on August 27, 2010. A Statement of Grounds of Appeal was filed on November 2, 2010. |
| • | In EP_1_159_975, a Notice of Opposition was filed on June 5, 2009. On February 4, 2010, Angiotech requested that the EPO set oral proceedings. On April 20, 2010, Angiotech filed a response to the Opposition, and on December 2, 2010, the opponents filed their response. |
45
Table of Contents
| • | In EP_0_876_165, the EPO revoked the patent as an outcome of an oral hearing held on June 24, 2009. On August 20, 2009, the Company filed a Notice of Appeal. On April 8, 2010, the opponent filed their response to the appeal. |
| • | In EP_1_857_236, a Notice of Opposition was filed against Quill Medical, Inc. by ITV Denkendorf Produkservice GmbH. Quill Medical has until January 29, 2011, to file its response. |
| • | In EP_1_858_243, a Notice of Opposition was filed against Quill Medical, Inc. by ITV Denkendorf Produkservice GmbH. Quill Medical has until January 22, 2011, to file its response. |
| • | In EP_1_867_288, a Notice of Opposition was filed against Quill Medical, Inc. by ITV Denkendorf Produkservice GmbH. |
| Item 4. | [REMOVED AND RESERVED.] |
46
Table of Contents
| Item 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Trading Price and Volume
Our common shares were publicly traded on the Toronto Stock Exchange (“TSX”) under the trading symbol “ANP” and on the NASDAQ Global Select Market under the trading symbol “ANPI”. As previously announced, we were delisted from the NASDAQ Global Select Market on January 13, 2011 and from the Toronto Stock Exchange on March 3, 2011. Our common shares are now quoted on the OTC Bulletin Board under the symbol “ANPIQ”. For more details regarding our delistings, refer to note 26 of the audited consolidated financial statements in this Annual Report on Form 10-K. The following table summarizes the high and low prices for our common shares during 2010 and 2009 on a quarterly basis. The prices listed below are in Canadian dollars for trading on the TSX and in U.S dollars for trading on the NASDAQ Global Select Market.
| Quarter |
Canadian
Dollars (TSX) |
U.S. Dollars (NASDAQ) |
||||||||||||||
| 2010: |
High | Low | High | Low | ||||||||||||
| Q1 |
$ | 1.41 | $ | 0.99 | $ | 1.37 | $ | 0.95 | ||||||||
| Q2 |
1.23 | 0.77 | 1.21 | 0.75 | ||||||||||||
| Q3 |
0.80 | 0.43 | 0.78 | 0.41 | ||||||||||||
| Q4 |
0.62 | 0.18 | 0.60 | 0.18 | ||||||||||||
| Quarter |
Canadian
Dollars (TSX) |
U.S. Dollars (NASDAQ) |
||||||||||||||
| 2009: |
High | Low | High | Low | ||||||||||||
| Q1 |
$ | 0.68 | $ | 0.33 | $ | 0.56 | $ | 0.26 | ||||||||
| Q2 |
3.10 | 0.55 | 2.94 | 0.44 | ||||||||||||
| Q3 |
2.41 | 1.39 | 2.17 | 1.20 | ||||||||||||
| Q4 |
1.88 | 1.27 | 1.77 | 1.20 | ||||||||||||
47
Table of Contents
Comparative Shareholder Return Performance Graph
The following graph compares cumulative total Shareholder return on $100 invested in Common shares of the Company on December 31, 2004 with the cumulative total return of the S&P/TSX Composite Index (“S&P/TSX”) and the NASDAQ Biotechnology Index, assuming the re-investment of dividends. The Common shares began trading on the TSX on December 18, 1997, and were delisted from the NASDAQ on January 13, 2011 and from the TSX on March 3, 2011 (refer to note 26 of the audited consolidated financial statements for details of our delistings in early 2011).
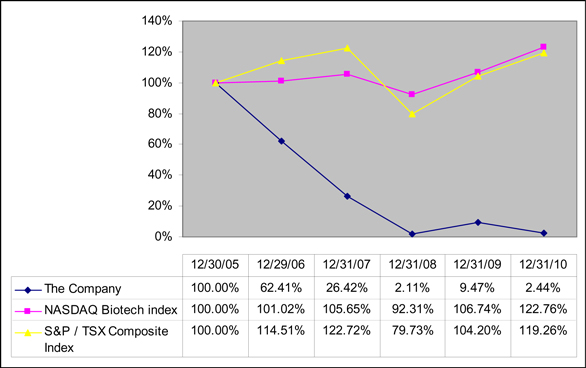
Based on information provided by our transfer agent, as of March 9, 2011, we had 502 shareholders of record.
Dividend Policy
We have not declared or paid any dividends on our common shares since inception. The declaration of dividend payments is at the sole discretion of our Board of Directors. The Board of Directors may declare dividends in the future depending upon numerous factors that ordinarily affect dividend policy, including the results of our operations, our financial position and general business conditions.
48
Table of Contents
Securities Authorized for Issuance under Equity Compensation Plan
The following table provides information as of December 31, 2010 with respect to the shares of the Company’s common stock that may be issued under existing equity compensation plans:
| Plan Category |
(a) Number of securities to be issued upon exercise of outstanding options, warrants and rights |
(b) Weighted average of exercise price of outstanding options, warrants and rights |
(c) Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) |
|||||||||
| Common shares, without par value, pursuant to the Angiotech Pharmaceuticals Inc. 2006 Stock Incentive Plan |
9,165,715 | $ | 2.86 | 3,422,173 | ||||||||
| Common shares, without par value, pursuant to the American Medical Instruments Holdings Inc. 2003 Stock Option Plan |
300,480 | $ | 15.44 | Nil | ||||||||
Sale of Unregistered Securities Within the Past Twelve Months
None.
Purchases of Equity Securities by the Company and its Affiliates Made in the Fourth Quarter
None.
49
Table of Contents
| Item 6. | SELECTED FINANCIAL DATA |
The following table sets forth selected consolidated financial information for each of our five most recently completed financial years. This data should be read in conjunction with our Management’s Discussion and Analysis of Financial Condition and Results of Operations, the audited consolidated financial statements, including the notes to the financial statements, and the risk factors set out in this Annual Report on Form 10-K.
CONSOLIDATED STATEMENTS OF INCOME
| (in thousands of U.S. $) |
Year
ended December 31, 2010 |
Year
ended December 31, 2009 |
Year
ended December 31, 2008 |
Year
ended December 31, 2007 |
Year
ended December 31, 2006 |
|||||||||||||||
| REVENUE |
||||||||||||||||||||
| Product sales, net (1) |
$ | 211,495 | $ | 191,951 | $ | 190,816 | $ | 170,193 | $ | 138,590 | ||||||||||
| Royalty revenue |
34,461 | 62,171 | 91,546 | 116,659 | 175,254 | |||||||||||||||
| License fees (2) |
286 | 25,556 | 910 | 842 | 1,231 | |||||||||||||||
| 246,242 | 279,678 | 283,272 | 287,694 | 315,075 | ||||||||||||||||
| EXPENSES |
||||||||||||||||||||
| Cost of products sold |
106,304 | 104,616 | 101,052 | 94,949 | 69,543 | |||||||||||||||
| License and royalty fees |
5,889 | 10,431 | 14,258 | 18,652 | 25,986 | |||||||||||||||
| Research and development |
26,790 | 23,701 | 53,192 | 53,963 | 45,393 | |||||||||||||||
| Selling, general and administration |
89,238 | 81,504 | 98,483 | 99,315 | 78,933 | |||||||||||||||
| Depreciation and amortization |
33,745 | 33,251 | 33,998 | 33,429 | 36,014 | |||||||||||||||
| In-process research and development |
— | — | 2,500 | 8,125 | 1,042 | |||||||||||||||
| Write-down of assets held for sale (3) |
1,450 | 3,090 | 1,283 | — | — | |||||||||||||||
| Write-down of property, plant and equipment (4) |
4,779 | — | — | — | — | |||||||||||||||
| Write-down of intangible assets (5) |
2,814 | — | — | — | — | |||||||||||||||
| Escrow settlement recovery (6) |
(4,710 | ) | — | — | — | — | ||||||||||||||
| Write-down of goodwill (7) |
— | — | 649,685 | — | — | |||||||||||||||
| 266,299 | 256,593 | 954,451 | 308,433 | 256,911 | ||||||||||||||||
| Operating income (loss) |
(20,057 | ) | 23,085 | (671,179 | ) | (20,739 | ) | 58,164 | ||||||||||||
| Other (expenses) income: |
||||||||||||||||||||
| Foreign exchange (loss) gain |
1,011 | (1,612 | ) | 540 | (341 | ) | 515 | |||||||||||||
| Investment and other income |
(571 | ) | 378 | 1,192 | 10,393 | 6,235 | ||||||||||||||
| Debt restructuring costs (8) |
(9,277 | ) | ||||||||||||||||||
| Interest expense on long-term debt |
(40,258 | ) | (38,039 | ) | (44,490 | ) | (51,748 | ) | (35,502 | ) | ||||||||||
| Write-down and other deferred financing charges (9) |
(291 | ) | (643 | ) | (16,544 | ) | — | (9,297 | ) | |||||||||||
| Write-down / loss on redemption of investments (10) |
(1,297 | ) | — | (23,587 | ) | (8,157 | ) | — | ||||||||||||
| Loan settlement gain (11) |
1,880 | |||||||||||||||||||
| Gain on disposal of Laguna Hills manufacturing facility (12) |
2,005 | |||||||||||||||||||
| Total other (expenses) income |
(46,798 | ) | (39,916 | ) | (82,889 | ) | (49,853 | ) | (38,049 | ) | ||||||||||
| (Loss) income from continuing operations before income taxes and cumulative effect of change in accounting policy |
(66,855 | ) | (16,831 | ) | (754,068 | ) | (70,592 | ) | 20,115 | |||||||||||
| Income tax (recovery) expense |
(44 | ) | 6,037 | (12,892 | ) | (14,545 | ) | 2,092 | ||||||||||||
| (Loss) income from continuing operations before cumulative effect of change in accounting policy |
(66,811 | ) | (22,868 | ) | (741,176 | ) | (56,047 | ) | 18,023 | |||||||||||
| Loss from discontinued operations, net of income taxes (13) |
— | — | — | (9,893 | ) | (7,708 | ) | |||||||||||||
| Cumulative effect of change in accounting policy |
— | — | — | — | 399 | |||||||||||||||
| Net (loss) income |
(66,811 | ) | (22,868 | ) | $ | (741,176 | ) | $ | (65,940 | ) | $ | 10,714 | ||||||||
50
Table of Contents
| (in thousands of U.S. $) |
Year
ended December 31, 2010 |
Year
ended December 31, 2009 |
Year
ended December 31, 2008 |
Year
ended December 31, 2007 |
Year
ended December 31, 2006 |
|||||||||||||||
| Basic net income (loss) per common share: |
||||||||||||||||||||
| Continuing operations |
$ | (0.78 | ) | $ | (0.27 | ) | $ | (8.71 | ) | $ | (0.66 | ) | $ | 0.21 | ||||||
| Discontinued operations |
— | — | — | (0.12 | ) | (0.09 | ) | |||||||||||||
| Total |
$ | (0.78 | ) | $ | (0.27 | ) | $ | (8.71 | ) | $ | (0.78 | ) | $ | 0.12 | ||||||
| Diluted net income (loss) per common share: |
||||||||||||||||||||
| Continuing operations |
$ | (0.78 | ) | $ | (0.27 | ) | $ | (8.71 | ) | $ | (0.66 | ) | $ | 0.21 | ||||||
| Discontinued operations |
— | — | (0.12 | ) | (0.09 | ) | ||||||||||||||
| Total |
$ | (0.78 | ) | $ | (0.27 | ) | $ | (8.71 | ) | $ | (0.78 | ) | $ | 0.12 | ||||||
| Basic weighted average number of common shares outstanding (in thousands) |
85,168 | 85,130 | 85,118 | 85,015 | 84,752 | |||||||||||||||
| Diluted weighted average number of common shares outstanding (in thousands) |
85,168 | 85,130 | 85,118 | 85,015 | 85,437 | |||||||||||||||
BALANCE SHEET INFORMATION
| As at | December 31, | |||||||||||||||||||
| (in thousands of U.S.$) |
2010 | 2009 | 2008 | 2007 | 2006 | |||||||||||||||
| Cash, cash equivalents and short-term investments |
$ | 37,968 | $ | 57,322 | $ | 39,800 | $ | 91,326 | $ | 108,617 | ||||||||||
| Working capital |
(518,739 | ) | 66,512 | 51,767 | 97,745 | 115,892 | ||||||||||||||
| Total assets |
305,998 | 370,059 | 385,197 | 1,150,108 | 1,224,624 | |||||||||||||||
| Total long-term obligations |
50,211 | 620,160 | 623,655 | 645,096 | 658,366 | |||||||||||||||
| Deficit |
(933,352 | ) | (866,541 | ) | (843,673 | ) | (102,497 | ) | (34,893 | ) | ||||||||||
| Total shareholders’ (deficit) equity |
(384,076 | ) | (313,287 | ) | (299,873 | ) | 442,072 | 498,692 | ||||||||||||
QUARTERLY RESULTS
The following tables present our unaudited consolidated quarterly results of operations for each of our last eight quarters:
| Quarter ended | ||||||||||||||||
| (in thousands of U.S.$, except per share data) |
December 31, 2010 |
September 30, 2010 |
June 30, 2010 |
March 31, 2010 |
||||||||||||
| Revenue |
||||||||||||||||
| Product sales |
$ | 55,699 | $ | 51,868 | $ | 52,948 | $ | 50,980 | ||||||||
| Royalty and license revenue |
6,332 | 7,116 | 8,938 | 12,361 | ||||||||||||
| Total revenues |
62,031 | 58,984 | 61,886 | 63,341 | ||||||||||||
| Gross Margin: |
||||||||||||||||
| Medical Products |
29,278 | 25,060 | 25,077 | 25,776 | ||||||||||||
| Pharmaceutical Technologies |
5,327 | 5,946 | 7,461 | 10,124 | ||||||||||||
| Total Gross Margin |
34,605 | 31,006 | 32,538 | 35,900 | ||||||||||||
| Operating (loss) income |
(11,792 | ) | (6,020 | ) | (5,376 | ) | 3,131 | |||||||||
| Net loss |
(27,542 | ) | (18,500 | ) | (14,074 | ) | (6,695 | ) | ||||||||
| Basic and diluted loss per share |
$ | (0.32 | ) | $ | (0.22 | ) | $ | (0.17 | ) | $ | (0.08 | ) | ||||
51
Table of Contents
| Quarter ended | ||||||||||||||||
| (in thousands of U.S.$, except per share data) |
December 31, 2009 |
September 30, 2009 |
June 30, 2009 |
March 31, 2009 |
||||||||||||
| Revenue |
||||||||||||||||
| Product sales |
$ | 49,976 | $ | 48,660 | $ | 47,179 | $ | 46,136 | ||||||||
| Royalty and license revenue |
13,585 | 14,584 | 17,394 | 42,164 | ||||||||||||
| Total revenues |
63,561 | 63,244 | $ | 64,573 | 88,300 | |||||||||||
| Gross Margin: |
||||||||||||||||
| Medical Products |
20,965 | 22,702 | 21,497 | 22,170 | ||||||||||||
| Pharmaceutical Technologies |
11,091 | 12,121 | 14,826 | 39,259 | ||||||||||||
| Total Gross Margin |
32,056 | 34,823 | 36,323 | 61,429 | ||||||||||||
| Operating (loss) income |
(8,016 | ) | 4,018 | (412 | ) | 27,495 | ||||||||||
| Net (loss) income |
$ | (15,633 | ) | $ | (7,802 | ) | $ | (11,877 | ) | $ | 12,444 | |||||
| Basic (loss) income per share |
$ | (0.18 | ) | $ | (0.09 | ) | $ | (0.14 | ) | $ | 0.15 | |||||
| Diluted loss per share |
$ | (0.18 | ) | $ | (0.09 | ) | $ | (0.14 | ) | $ | 0.14 | |||||
| 1) | During the year ended December 31, 2007, we recorded a credit of $2.6 million against our product sales related to our decision to accept returns of our Contour Threads product as part of our consolidation and discontinuation of the Contour Threads brand name, which was concurrent with our launch of our Quill Knotless Tissue-Closure Device product line (formerly know as our Quill SRS brand name). |
| 2) | In the first quarter of 2009, we received a one-time up-front payment of $25.0 million from Baxter International Inc. (“Baxter”) under the terms of our Amended and Restated Distribution and Licence Agreement between Angiotech US, Angiotech International GmbH, Baxter Healthcare Corporation and Baxter Healthcare, S.A., dated as of January 1, 2009 (the “Baxter Distribution and License Agreement”). The payment was received in lieu and settlement of future royalty and milestone obligations relating to existing formulations of CoSeal and any future products. |
| 3) | During the year ended December 31, 2009, we recorded impairment charges totaling $1.3 million on two properties, located in Syracuse and Puerto Rico, which were classified as held for sale. In 2009, we recorded further impairment charges of $3.1 million on the Syracuse property and a Vancouver based property, which were both classified as assets held for sale. In 2010 the Puerto Rico property was sold for net proceeds of $0.7 million with no resulting gain or loss and we recorded further write-downs of $1.5 million on the Vancouver and Syracuse properties. In addition, the $3.1 million carrying values of the Vancouver and Syracuse properties were reclassified back into Property, Plant and Equipment given that they no longer meet the held-for-sale criteria specified under ASC No. 360-10 – Property, Plant and Equipment. The carrying values of the remaining properties were measured at the lower of (a) the carrying values of the properties prior to their classification as held for sale, adjusted for depreciation that would have been recognized if the properties had remained classified as held and used; and (b) the fair values of the properties at the date of the subsequent decision to not sell the properties. |
| 4) | During the year ended December 31, 2010, we recorded impairment write-downs of $4.8 million on certain of our property, plant and equipment. The impairment charges recorded in 2010 are primarily due to restructuring changes that we have initiated in response to our recent liquidity constraints and recapitalization plans. |
| 5) | During the year ended December 31, 2010, we recorded a $1.7 million write-down on the intellectual property acquired from Haemacure Corporation (“Haemacure”) in connection with our decision to indefinitely suspend research and development activities related to the Haemacure fibrin and thrombin technologies. In addition, in connection with the termination of our 2008 License, Supply, Marketing and Distribution Agreement with Rex Medical LP (“Rex”), we recorded a $1.1 million write-down on the intellectual property associated with the Option IVC Filter. |
| 6) | The $4.7 million recovery represents settlement funds that were received in connection with the resolution of our dispute with RoundTable Healthcare Partners, LP (“Roundtable”) over $13.5 million of escrow funds that were being held by LaSalle Bank in connection with our 2006 acquisition of American Medical Instruments Holding, Inc. All legal proceedings have since been dismissed. |
52
Table of Contents
| 7) | At the end of the third quarter of 2008, we wrote-down the goodwill carrying value associated with our Medical Products segment by $599.4 million. The remaining balance of $26.8 million was subsequently written off in the fourth quarter of 2008. We also determined that the goodwill associated with our Pharmaceuticals Technology segment was impaired as at December 31, 2008 and accordingly, wrote off the remaining balance of $23.5 million. As a result, our goodwill balance for the Medical Products and Pharmaceutical Technology segments was $nil as at December 31, 2008 and 2009. |
| 8) | During the year ended December 31, 2010, we incurred debt restructuring costs of $9.3 million for fees and expenses related to the Recapitalization Transaction. These fees and expenses represent professional fees paid to both our and the Consenting Noteholders’ financial and legal advisors to assist in the analysis of financial and strategic alternatives. |
| 9) | In the third quarter of 2008, we expensed deferred financing charges of $13.5 million related to the suspension of a note purchase agreement. A further $3.0 million was expensed in the fourth quarter of 2008 related to this matter. In connection with the early termination of the term loan portion of our credit facility with Wells Fargo Foothill LLC in the second quarter of 2009, we wrote off $0.6 million of deferred financing charges. During the year ended December 31, 2010, we wrote-off $0.3 million of deferred financing costs related to our shelf registration statement that was filed with the SEC in 2009. |
| 10) | In 2007, we wrote-down and realized a loss on investments of $8.2 million. In 2008, we wrote-down and realized a loss on investments of $10.7 million, $1.9 million and $11.0 million in the second, third and fourth quarters respectively. In early 2010, we wrote off one of our long term investments totaling $0.3 million. During the year ended December 31, 2010, we recorded $1.3 million of write-offs related to our long term investments in three private biotechnology companies that were determined to be irrecoverable. |
| 11) | During the year ended December 31, 2010, we recorded a $1.9 million loan settlement gain relating to the settlement of our loan to Haemacure in connection with our acquisition of certain product candidates and technology assets from Haemacure. |
| 12) | During the year ended December 31, 2010, we recorded a $2.0 million gain from the sale of our Laguna Hills manufacturing facility and related long-lived assets. |
| 13) | In the first quarter of 2007, we recorded impairment charges of $8.9 million related to the discontinuance of the following subsidiaries: American Medical Instruments Inc. (Dartmouth), Point Technologies, Inc and Point Technologies SA. |
53
Table of Contents
| Item 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
ANGIOTECH PHARMACEUTICALS, INC.
For the year ended December 31, 2010
(All amounts following are expressed in U.S. dollars unless otherwise indicated.)
The following management’s discussion and analysis (“MD&A”), dated December 31, 2010, should be read in conjunction with our audited consolidated financial statements for the year ended December 31, 2010 prepared in accordance with United States generally accepted accounting principles (“U.S. GAAP”) and the applicable rules and regulations of the U.S. Securities and Exchange Commission (“SEC”) for the presentation of annual financial information. Additional information relating to our company is available by accessing the SEDAR website at www.sedar.com or the SEC’s EDGAR website at www.sec.gov/edgar.shtml.
Forward-Looking Statements and Cautionary Factors That May Affect Future Results
Statements contained in this Annual Report on Form 10-K that are not based on historical fact, including without limitation statements containing the words “believes,” “may,” “plans,” “will,” “estimates,” “continues,” “anticipates,” “intends,” “expects” and similar expressions, constitute “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995 and constitute “forward-looking information” within the meaning of applicable Canadian securities laws. All such statements are made pursuant to the “safe harbor” provisions of applicable securities legislation. Forward-looking statements may involve, but are not limited to, comments with respect to our objectives and priorities for 2011 and beyond, our strategies or future actions, our targets, expectations for our financial condition and the results of, or outlook for, our operations, research and development and product and drug development. Such forward-looking statements involve known and unknown risks, uncertainties and other factors that may cause the actual results, events or developments to be materially different from any future results, events or developments expressed or implied by such forward-looking statements.
Many such known risks, uncertainties and other factors are taken into account as part of our assumptions underlying these forward-looking statements and include, among others, the following: general economic and business conditions in the U.S., Canada and the other regions in which we operate; uncertainty involved in the CCAA Proceedings, the Chapter 15 Cases and the implementation of the CCAA Plan; market demand; technological changes that could impact our existing products or our ability to develop and commercialize future products; competition; existing governmental legislation and regulations and changes in, or the failure to comply with, governmental legislation and regulations; availability of financial reimbursement coverage from governmental and third-party payers for products and related treatments; adverse results or unexpected delays in pre-clinical and clinical product development processes; adverse findings related to the safety and/or efficacy of our products or products sold by our partners; decisions, and the timing of decisions, made by health regulatory agencies regarding approval of our technology and products; the requirement for substantial funding to conduct research and development, to expand manufacturing and commercialization activities; and any other factors that may affect our performance.
In addition, our business is subject to certain operating risks that may cause any results expressed or implied by the forward-looking statements in this Annual Report on Form 10-K to differ materially from our actual results. These operating risks include: our ability to attract and retain qualified personnel; our ability to successfully complete pre-clinical and clinical development of our products; changes in our business strategy or development plans; our failure to obtain patent protection for discoveries; loss of patent protection resulting from third-party challenges to our patents; commercialization limitations imposed by patents owned or controlled by third parties; our ability to obtain rights to technology from licensors; liability for patent claims and other claims asserted
54
Table of Contents
against us; our ability to obtain and enforce timely patent and other intellectual property protection for our technology and products; the ability to enter into, and to maintain, corporate alliances relating to the development and commercialization of our technology and products; market acceptance of our technology and products; our ability to successfully manufacture, market and sell our products; the availability of capital to finance our activities; our ability to restructure and to service our debt obligations; and any other factors referenced in our other filings with the applicable Canadian securities regulatory authorities or the SEC.
For a more thorough discussion of the risks associated with our business, see the section entitled “Risk Factors” in this Annual Report on Form 10-K.
Given these uncertainties, assumptions and risk factors, investors are cautioned not to place undue reliance on such forward-looking statements. Except as required by law, we disclaim any obligation to update any such factors or to publicly announce the result of any revisions to any of the forward-looking statements contained in this Annual Report on Form 10-K to reflect future results, events or developments.
This Annual Report on Form 10-K contains forward-looking information that constitutes “financial outlooks” within the meaning of applicable Canadian securities laws. We have provided this information to give shareholders general guidance on management’s current expectations of certain factors affecting our business, including our future financial results. Given the uncertainties, assumptions and risk factors associated with this type of information, including those described above, investors are cautioned that the information may not be appropriate for other purposes.
Business Overview
The Company discovers, develops and markets innovative medical technologies primarily focused on acute and surgical applications. We generate our revenue through our sales of medical products and components, as well as from royalties derived from sales by our partners of products utilizing certain of our proprietary technologies. For the year ended December 31, 2010, we recorded $211.5 million in direct sales of our various medical products and $34.7 million in royalties and license fees received from our partners.
We currently operate in two segments: Pharmaceutical Technologies and Medical Products.
Our Pharmaceutical Technologies segment focuses primarily on establishing product development and marketing collaborations with major medical device, pharmaceutical or biomaterials companies, and to date has derived the majority of its revenue from royalties due from partners that develop, market and sell products incorporating our technologies. Our principal revenues in this segment have been royalties derived from sales by BSC of TAXUS®.
Our Medical Products segment manufactures and markets a wide range of single-use specialty medical products and medical device components. These products are sold directly to end users or other third-party medical device manufacturers. This segment contains two specialized direct sales and distribution organizations as well as significant manufacturing capabilities. Many of our medical products are made using our proprietary manufacturing processes or are protected by our intellectual property.
Certain of our product lines, referred to as our Proprietary Medical Products, are marketed and sold by our direct sales groups. We believe certain of these product lines may contain technology advantages that have the potential for more substantial revenue growth compared to our overall product portfolio. Our most significant commercialized Proprietary Medical Products in 2010 included our Quill™ Knotless Tissue-Closure Device product line, SKATER™ line of drainage catheters, and BioPince™ full core biopsy device.
Certain of our product lines, referred to as our Base Medical Products, represent more mature finished goods product lines in the ophthalmology, biopsy and general surgery areas, as well as medical device components manufactured by us and sold to other third-party medical device manufacturers who assemble these components into finished medical devices. Sales of our Base Medical Products are supported by a small group of direct sales personnel, as well as a network of independent sales representatives and medical product distributors. These sales tend to exhibit greater volatility or slower growth when compared to sales of our Proprietary Medical Products. This is particularly the case with our sales of components to third- party medical device manufacturers, which may be impacted by customer concentration, business issues that certain of our large customers may face, and to a more limited extent economic and credit market conditions.
55
Table of Contents
Our research and development efforts focus on understanding and characterizing biological conditions that often occur concurrently with medical device implantation, surgery or acute trauma, including scar formation and inflammation, cell proliferation, bleeding and coagulation, infection and tumor tissue overgrowth. Our strategy is to utilize our various technologies in the areas of medical devices, drugs, drug delivery, surface modification and biomaterials to create and commercialize novel, proprietary medical products that reduce surgical procedure side effects, improve surgical outcomes, shorten hospital stays, or are easier or safer for a physician to use. We have patent protected, or have filed patent applications for, certain of our technologies, products and potential product candidates.
Significant Recent Developments
Recapitalization Transaction, CCAA and Related Proceedings
Over the past several years, revenues in our Pharmaceutical Technologies Segment, specifically our royalties derived from sales of TAXUS by BSC, have experienced significant declines. These declines have led to significant constraints on our liquidity, working capital and capital resources. Accordingly, our ability to continue to support our business initiatives and our debt obligations is uncertain. As a result, we have explored a range of financial and strategic alternatives in order to address these declines. After extensively exploring various possible solutions, we recently announced a Recapitalization Transaction, negotiated with a significant percentage of the holders of our outstanding indebtedness, to effectuate a significant reduction in the Company’s total indebtedness that would improve our liquidity, working capital and financial position.
On October 1, 2010, we did not make the $9.7 million interest payment due on our Subordinated Notes in connection with discussions being conducted with certain of the holders of our Subordinated Notes (the “Subordinated Noteholders”). Pursuant to the terms of the indenture governing the Subordinated Notes (as amended, the “SSN Indenture”), we had a 30-day grace period from October 1, 2010 to make the required interest payment before an event of default occurred and Subordinated Noteholders could exercise their right to demand repayment of all outstanding obligations thereunder. Pursuant to the terms of the supplemental indenture entered into by Angiotech and U.S. Bank National Association, as successor trustee under the SSN Indenture, at the direction of the holders of a majority in aggregate principal amount outstanding of the Subordinated Notes, on January 27, 2011, the grace period applicable to interest payments on the Subordinated Notes was ultimately extended to 211 days, or April 30, 2011 for purposes of the interest payments due on October 1, 2010.
In connection with the Company’s decision not to make the interest payment due to holders of the Subordinated Notes on October 1, 2010, we entered into a Forbearance Agreement (the “Forbearance Agreement”) with Wells Fargo on September 30, 2010 to preserve our Existing Credit Facility and avoid an event of default under the Existing Credit Facility that would have otherwise resulted from such non-payment while we completed negotiations with our Subordinated Noteholders.
On October 29, 2010, we entered into the RSA with the holders of approximately 73% of the aggregate principal amount of our outstanding Subordinated Notes pursuant to which such Consenting Noteholder agreed to, among other things, support the Recapitalization Transaction that would eliminate $250 million of our total outstanding indebtedness and related interest obligations by exchanging Subordinated Notes for new common shares in the Company, thereby providing significant improvements to our credit ratios, liquidity and financial flexibility. As at March 14, 2011, holders of approximately 85% of the aggregate principal amount of our outstanding Subordinated Notes have agreed to the terms of the RSA. In accordance with the terms of the RSA, (i) the Subordinated Noteholders will receive their pro rata share of approximately 91.5% of the new common shares of the Company to be issued and outstanding upon completion of the Recapitalization Transaction and (ii) the Consenting Noteholders will receive an additional amount of the new common shares equal to their pro rata share of approximately 3.5% of the new common shares of the Company to be issued and outstanding upon completion of the Recapitalization Transaction, in each case subject to potential dilution.
56
Table of Contents
On October 29, 2010, we also entered into the FRN Support Agreement with holders (the “Consenting FRN Holders”) of approximately 51% of the aggregate principal amount outstanding of our Existing Floating Rate Notes pursuant to which the Consenting FRN Holders agreed to support and tender to an offer to exchange their Existing Floating Rate Notes for New Floating Rate Notes in the FRN Exchange Offer. The New Floating Rate Notes will be issued on substantially the same terms and conditions as the Existing Floating Rate Notes, except that, among other things: (i) subject to certain exceptions, the New Floating Rate Notes will be secured by second-priority liens over substantially all of the assets, property and undertaking of Angiotech and certain of its subsidiaries; (ii) the New Floating Rate Notes will accrue interest at LIBOR plus 3.75%, subject to a LIBOR floor of 1.25%; and (iii) certain covenants related to the incurrence of additional indebtedness and asset sales and the definitions of permitted liens, asset sales and change of control will be modified in the indenture that governs the New Floating Rate Notes. The assets, property and undertakings of the Company and certain of its subsidiaries that secure the New Floating Rate Notes will be subject to a first priority lien under the proposed Exit Facility. Pursuant to the terms of the RSA, the obligation of the Consenting Noteholders to complete the Recapitalization Transaction is conditioned on the completion of the transactions contemplated by the FRN Support Agreement. Under the terms of the FRN Support Agreement, the obligation of all parties thereto to consummate the FRN Exchange Offer is conditioned upon the concurrent implementation of the Recapitalization Transaction. As at March 14, 2011, holders of approximately 89% of the aggregate principal amount outstanding of the Existing Floating Rate Notes have agreed to the terms of the FRN Exchange Offer.
The completion of the Recapitalization Transaction is subject to the establishment of a Key Employee Incentive Plan (“KEIP”) that is satisfactory to us and the Consenting Noteholders. The KEIP will provide payments to the named beneficiaries totaling no more than $1.0 million, subject to the completion of the Recapitalization Transaction. Two-thirds will be paid on the completion date with the remaining one third to be paid three months after the completion date.
In addition, the completion of the Recapitalization Transaction is subject to the establishment of a Management Incentive Plan (“MIP”) that is satisfactory to us and the Consenting Noteholders. On the completion date of the Recapitalization Transaction, the MIP will provide the beneficiaries with that number of new common shares that is equal to 4% of the new common shares to be issued in connection with the implementation of the CCAA Plan and options to acquire that number of new common shares that is equal to 6% of the new common shares on a fully-diluted basis to be issued in connection with the implementation of the CCAA Plan. Options to acquire an additional 5% of the new common shares, on a fully-diluted basis, as of the completion date, will be reserved for issuance at a later date at the discretion of the board of directors in place on the completion date of the Recapitalization Transaction.
On November 4, 2010, we entered into an amended and restated forbearance agreement with the lenders party thereto and Wells Fargo, as arranger and administrative agent, which amended and restated in its entirety the Forbearance Agreement (the “Amended and Restated Forbearance Agreement”). Under the Amended and Restated Forbearance Agreement, Wells Fargo agreed not to immediately exercise any rights or remedies it has related to: (a) our failure to make our $9.7 million interest payment due to the Subordinated Noteholders on October 1, 2010; (b) the litigation and arbitration relating to our dispute with QSR Holdings, Inc.; or (c) any failure to deliver audited financial statements with an unqualified audit opinion with respect to the going concern assumption for the fiscal year ending December 31, 2010. While the Amended and Restated Forbearance Agreement has not been terminated, Wells Fargo’s obligations thereunder, including its agreement to not exercise certain remedies, ceased to be of effect upon the Company’s commencement of the CCAA Proceedings Wells Fargo’s obligations, however, were nonetheless stayed in connection with such proceedings, as well as our proceedings under Chapter 15 of the U.S. Bankruptcy Code.
57
Table of Contents
In conjunction with the Amended and Restated Forbearance Agreement, on November 4, 2010, we entered into an amendment to our Existing Credit Facility with Wells Fargo that provides for, among other things:
| (i) | an amendment of financial covenants pertaining to minimum EBITDA levels and fixed charge coverage ratios; |
| (ii) | an increase the borrowing base available to secure loans by including in such borrowing base the value of certain real property, securities and intellectual property owned by the Company and its subsidiaries, in addition to accounts receivable and inventory, in each case subject to reserves, eligibility standards and advance rate formulas. As a result of such amendment, the amount of financing available to us under our Existing Credit Facility immediately increased to approximately $25 million, which excludes $2.8 million relating to outstanding secured letters of credit under the Existing Credit Facility; |
| (iii) | an amendment to the specified use of proceeds provisions to allow for certain secured and unsecured intercompany loans by our U.S. entities to our Canadian entities; and |
| (iv) | an amendment to permit the sale of certain assets of the Company. |
On January 28, 2011, in connection with the provisions of the RSA, Angiotech, 0741693 B.C. Ltd., Angiotech International Holdings, Corp., Angiotech Pharmaceuticals (US), Inc., American Medical Instruments Holdings, Inc., NeuColl, Inc., Angiotech BioCoatings Corp., Afmedica, Inc., Quill Medical, Inc., Angiotech America, Inc., Angiotech Florida Holdings, Inc., B.G. Sulzle, Inc., Surgical Specialties Corporation, Angiotech Delaware, Inc., Medical Device Technologies, Inc., Manan Medical Products, Inc. and Surgical Specialties Puerto Rico, Inc. (collectively, the “Angiotech Entities”) initiated the CCAA Proceedings and an initial order (the “Initial Order”) was granted by the Canadian Court. The CCAA Proceedings were commenced in order to implement the Recapitalization Transaction through the CCAA Plan. In order to have the CCAA Proceedings recognized in the United States, on January 30, 2011, the Angiotech Entities commenced the Chapter 15 Cases (together with the CCAA Proceedings, the “Creditor Protection Proceedings”). On February 22, 2011, the U.S. court granted an order that, among other things, recognized the CCAA Proceedings as a foreign main proceeding and provided that the Initial Order would be enforced on a final basis and given full force and effect in the United States.
Under the terms of the proposed CCAA Plan, upon implementation of the CCAA Plan, the Subordinated Notes will be exchanged for equity interests in Angiotech and retired in full, thereby eliminating approximately $250.0 million of our outstanding indebtedness, plus the accrued and unpaid interest related thereto. In addition, all of our existing common shares and options to acquire common shares will be cancelled on such date without any payment or consideration therefor. The Initial Order, among other things, provides for a general stay of proceedings against the Angiotech Entities preventing creditors and certain other parties from exercising rights to recover amounts owning to them as of January 28, 2011 or to exercise other rights that could arise as a result of the commencement of proceedings under the CCAA. The stay of proceedings was initially scheduled to expire on February 17, 2011 and was subsequently extended to April 15, 2011 and may be extended further by order of the Canadian Court (the “Stay Period”).
On February 17, 2011, the Canadian Court granted an order (the “Claims Procedure Order”) establishing a procedure for the adjudication, resolution and determination of claims of the Affected Creditors (as defined in the CCAA Plan) for voting and distribution purposes under the CCAA Plan. Creditors who do not file a proof of claim in accordance with the Claims Procedure Order will be barred from receiving distributions under the CCAA Plan and from asserting their claim in the future. As at January 28, 2011, the claims of Affected Creditors totalled approximately $3.6 million. In accordance with the terms of the fourth amendment to the RSA, the maximum amount of permitted claims, excluding the Company’s obligations under the Subordinated Notes and Existing Floating Rate Notes, is $30.0 million. Under the terms of the Claims Procedure Order, for the purposes of voting and distributions under the CCAA Plan, the amount of the claims in respect of the Subordinated Notes will only include the accrued amounts owing directly by Angiotech under the SSN
58
Table of Contents
Indenture and by the other Angiotech Entities under the guarantees executed by such other Angiotech Entities in respect of the Subordinated Notes up to the date of the Initial Order, January 28, 2011.
On February 17, 2011, the Canadian Court also granted an order (the “Meeting Order”) authorizing the holding of a meeting (the “Meeting”) of the Affected Creditors on April 4, 2011. The purpose of the Meeting is for the Affected Creditors to consider, and if deemed advisable, vote for or against the resolution approving the CCAA Plan. If the CCAA Plan is approved by a majority in number of Affected Creditors representing at least two-thirds in value of all such Affected Creditors’ claims that have been accepted for purposes of voting at the Meeting, in accordance with the provisions of the Claims Procedure Order, the Meeting Order and the CCAA, in each case, present and voting in person or by proxy at the Meeting, the Angiotech Entities intend to bring a further motion before the Canadian Court on or about April 6, 2011 seeking an order sanctioning the CCAA Plan.
The Canadian Court has appointed Alvarez & Marsal Canada, Inc. as the monitor (the “Monitor”) of the Angiotech Entities’ business and financial affairs, with the powers set forth in the CCAA and orders of the Canadian Court, during the CCAA Proceedings. The Monitor will report to the Canadian Court on a regular basis on our financial and operational position and other relevant matters. In addition, the Monitor may advise us on certain restructuring matters throughout our CCAA Proceedings.
In accordance with the terms of the Initial Order, on February 7, 2011, we and certain of our subsidiaries entered into a definitive agreement with Wells Fargo to secure a debtor-in-possession credit facility (the “DIP Facility”). On February 25, 2011, following entry of the February 22, 2011 recognition order of the CCAA Proceedings under the U.S. Bankruptcy Code (the “Recognition Order”) the loans under the DIP Facility were made available to the Company for borrowing. The DIP Facility will provide liquidity for working capital, general corporate purposes and expenses while we implement the CCAA Plan. The DIP Facility permits us to make revolving credit loans and provide letters of credit in an aggregate principal amount (including the face amount of any letters of credit) of up to $28.0 million. The DIP Facility is required to be repaid and terminated in connection with the implementation of the CCAA Plan. Upon implementation of the CCAA Plan, the Company expects to have entered into the proposed Exit Facility which is expected to provide for sufficient borrowing capacity to, among other things, repay all amounts outstanding under the DIP Facility and the Existing Credit Facility, as amended. For more information, refer to the Liquidity and Capital Resources and Long Term Debt sections of our Management, Discussion and Analysis and notes 1, 16 and 26 in our audited consolidated financial statements for the year ended December 31, 2010. Given that the initiation of the Creditor Protection Proceedings qualifies as an immediate event of default under the terms of our outstanding indebtedness, all obligations under (i) the Existing Credit Facility, (ii) the SSN Indenture, and (iii) the indenture governing the Existing Floating Rate Notes between the Company and Deutsche Bank National Trust Company in its capacity as successor trustee, dated December 11, 2006 (as amended, the “FRN Indenture”), have become due and payable under the terms of the respective agreements. The stay of proceedings granted under the Initial Order effectively prohibits these creditors from taking action to remedy or enforce their rights thereunder. The enforcement rights of these creditors are also subject to the applicable provisions of the CCAA and U.S. Bankruptcy Code.
The terms of the Initial Order have created a number of new charges against substantially all of our current and future assets. These charges, include: (i) an administration charge to secure amounts owing to the Monitor and certain restructuring and financial advisors, up to a maximum of $2,500,000 (the “Administrative Charge”); (ii) a directors’ charge to secure the indemnity created under the Initial Order in favor of the directors and officers of the Angiotech Entities, up to a maximum of $2,000,000; (iii) a DIP charge to the extent of any obligations outstanding under the DIP Facility (the “DIP Charge”); and (iv) charges to secure amounts owing by our Canadian entities to our U.S. entities and by our U.S. entities to our Canadian entities pursuant to the intercompany advances made in accordance with intercompany transaction practices existing as of the date of the Initial Order, to the extent of any obligations outstanding under those advances (the “Inter-Company Charges”). These new charges rank in the following priority:
| (i) | the Administration Charge (to the maximum amount of $1,500,000); |
59
Table of Contents
| (ii) | the Directors’ Charge (to the maximum amount of $1,000,000); |
| (iii) | any existing liens, charges, security interests or other encumbrances securing the obligations under the Existing Credit Facility; |
| (iv) | the DIP Charge; |
| (v) | the Inter-Company Charges, which rank pari passu with each other; |
| (vi) | the Administration Charge (to the maximum amount of $1,000,000); and |
| (vii) | the Director’s Charge (to the maximum amount of $1,000,000). |
Under bankruptcy legislation, certain payroll and commodity tax obligations also qualify as priority claims.
On February 10, 2011, we commenced the FRN Exchange Offer pursuant to the terms of the FRN Support Agreement. The FRN Exchange Offer is open to all holders of the Existing Floating Rate Notes. Upon consummation of the FRN Exchange Offer and the consent of holders owning at least a majority in aggregate principal amount outstanding of the Existing Floating Rate Notes, those holders of Existing Floating Rate Notes who do not participate in the FRN Exchange Offer will continue to hold Existing Floating Rate Notes under the FRN Indenture, as modified by certain amendments providing for, among other things, the elimination of most of the restrictive covenants and events of default contained therein. As of March 14, 2011, holders of approximately 89% of the aggregate principal amount outstanding of our Existing Floating Rate Notes support the terms of the FRN Support Agreement and have agreed to consent and tender their Existing Floating Rate Notes for New Floating Rate Notes in the FRN Exchange Offer.
For the year ended December 31, 2010, we incurred approximately $9.3 million of fees and expenses relating to the Recapitalization Transaction. These fees and expenses represent professional fees paid to our and the Consenting Noteholders’ financial and legal advisors to assist in the analysis of financial and strategic alternatives.
For more information pertaining to our Creditor Protection Proceedings, including the Monitor’s reports, refer to our Monitor’s website at www.alvarezandmarsal.com/angiotech and our website at www.angiotech.com. With the exception of the filings specifically incorporated by reference in this Annual Report on Form 10-K, material contained on or accessible through our website or the Monitor’s website is specifically not incorporated into this Annual Report on Form 10-K.
NASDAQ and TSX Delisting
On July 6, 2010, we received a notice from The NASDAQ Stock Market (“NASDAQ”) stating that the bid price for our common shares had closed below the required minimum of $1.00 per share for the previous 30 consecutive business days, and that in accordance with the applicable NASDAQ listing rule, we had a grace period of 180 calendar days, until January 3, 2011, to regain compliance with the applicable NASDAQ listing rule. On January 4, 2011, we received a letter from NASDAQ indicating that our common shares would be delisted from NASDAQ as a result of our failure to regain compliance with the applicable NASDAQ listing rule. We did not appeal NASDAQ’s decision to delist our common shares, and accordingly, our common shares were delisted from NASDAQ at the opening of business on January 13, 2011. A Form 25-NSE was filed with the Securities and Exchange Commission (the “SEC”) on January 21, 2011, notifying the SEC of the removal of our common shares from listing on NASDAQ.
On January 28, 2011 we received notice from the Toronto Stock Exchange (the “TSX”) stating that the TSX would be reviewing the eligibility for continued listing on TSX of our securities pursuant to Part VII of The Toronto Stock Exchange Company Manual. In addition, the TSX advised that our securities would be suspended from trading on the TSX until further notice from the TSX. On February 1, 2011, we received
60
Table of Contents
further notice from the TSX that our common shares would be delisted from the TSX effective at the close of market on March 3, 2011. The delisting was imposed as a result of our not meeting the continued listing requirements of the TSX. The TSX also advised that trading in our common shares would remain suspended until the time of delisting. We did not appeal the TSX’s decision to delist our common shares and our common shares were delisted from the TSX at the close of market on March 3, 2011.
National Institutes of Health
In November 1997, we entered into an exclusive license agreement with the Public Health Service of the U.S., through the National Institutes of Health (the “NIH”), whereby the NIH granted us an exclusive, worldwide license to certain technologies of the NIH for the use of paclitaxel (the “NIH License Agreement”). Pursuant to the NIH License Agreement, we agreed to pay to the NIH certain milestone payments upon achievement of specified clinical and commercial development milestones and to pay royalties on net TAXUS® sales by BSC and Zilver-PTX sales by Cook Medical Inc. and its affiliates (“Cook”). At December 31, 2010 and December 31, 2009, we accrued royalty fees and interest of $7.2 million and $7.3 million, respectively, payable to the NIH under the NIH License Agreement relating to royalties on net TAXUS® sales for the years 2010 and 2009, respectively. On December 29, 2010, we entered into an amendment to the NIH License Agreement whereby NIH agreed to eliminate: (i) approximately $7.2 million of unpaid royalties and interest due on sales of TAXUS® by BSC and (ii) future royalties payable on licensed products sold by BSC going forward, in exchange for a 0.25% increase of existing royalty rates for licensed products sold by Cook and an extension of the term for payment of such royalties of approximately two years. Given that the unpaid royalties have not been extinguished and will effectively be paid out through the incurrence of future royalty fees for licensed products sold by Cook, which were indeterminable and could not be forecasted at the time of our assessment, the $7.2 million of royalties remains accrued for as at December 31, 2010 in accordance with Accounting Standards Codification (“ASC”) No. 470-60-35 on Troubled Debt Restructuring.
QSR Holdings, Inc. Arbitration and Litigation
On October 4, 2010, the Company was notified that QSR Holdings, Inc. (“QSR”), as a representative for the former stockholders of Quill Medical, Inc. (“QMI”), had made a formal demand (the “Arbitration Demand”) to the American Arbitration Association naming the Company, QMI and Angiotech Pharmaceuticals (US), Inc. (“Angiotech US”) as respondents (collectively, the “Respondents”). The Arbitration Demand alleged that the Respondents failed to satisfy certain obligations under the Agreement and Plan of Merger, dated May 25, 2006, by and among Angiotech, Angiotech US, Quaich Acquisition, Inc. and QMI (the “Merger Agreement”), notably including the obligations to make certain earn out payments and the orthopedic milestone payment. The Arbitration Demand sought either direct monetary damages or, in the alternative, extension of the earn out period for a sixth year as more fully set forth in the Merger Agreement. In addition, QSR commenced an action in the United States District Court for the Middle District of North Carolina on October 1, 2010 against the Respondents, entitled QSR Holdings, Inc. v. Angiotech Pharmaceuticals, Inc., Angiotech Pharmaceuticals (US), Inc. and Quill Medical, Inc. (the “Federal Litigation”). The Complaint in the Federal Litigation alleged, among other items, that the Respondents: (a) breached certain contractual obligations under the Merger Agreement; (b) made certain misrepresentations or omissions during the initial negotiation of the Merger Agreement; and (c) tortiously interfered with the Merger Agreement. QSR sought damages in an unstated amount together with punitive damages and/or attorneys fees to the extent allowed by law. On October 15, 2010, the Respondents moved to dismiss the Complaint in part and to transfer venue to federal court in Washington State.
Subsequent to the initiation of the Federal Litigation and the delivery of the Arbitration Demand, representatives of Angiotech and QSR engaged in substantial settlement discussions. Those discussions culminated in an agreement (the “Settlement Agreement”), dated as of January 27, 2011, resolving, among other things, any and all claims that the parties may have arising out of the Merger Agreement (including, without limitation, all claims, counterclaims or defenses that were or could have been asserted in the Arbitration Demand and the Federal Litigation) in exchange for a payment of $6 million (the “Settlement Amount”) by Angiotech US. $2.0 million of the Settlement Amount is payable on the earlier of (i) 10 business days after the implementation of the Recapitalization Transaction contemplated by the RSA (the
61
Table of Contents
“Implementation Date”) and (ii) May 31, 2011. The remainder of the Settlement Amount is payable in 24 equal monthly installments of $166,667 each beginning on the earlier of (i) 30 days after the Implementation Date; and (ii) July 31, 2011 (subject to reduction in the event of pre-payment as more fully set forth in the Settlement Agreement). The Settlement Agreement further provides for complete and mutual releases between the parties, as more fully set forth therein. Pursuant to the Settlement Agreement, QSR has irrevocably dismissed the Federal Litigation and the Arbitration Demand with prejudice. As at December 31, 2010, we have recorded $0.6 million of litigation expense, related to these legal proceedings. In accordance with the guidance on contingent consideration under Financial Accounting Standard (“FAS”) 141, which pertains to business combinations arising prior to December 15, 2008, the remaining Settlement Amount is expected to be recognized as goodwill in our 2011 first quarter results.
Rex Medical, LP Arbitration and Litigation
In light of the Company’s recent liquidity and capital constraints, on November 11, 2010, Angiotech US informed Rex Medical, LP (“Rex”) that it would not be able to continue selling the Option IVC Filter under the existing terms of the License, Supply, Marketing and Distribution Agreement, dated as of March 13, 2008, by and between Angiotech US and Rex (as amended, the “Option Agreement”). Subsequently, on November 18, 2010, Rex initiated an arbitration (the “Rex Arbitration”) against Angiotech US alleging that Angiotech US had breached the Option Agreement. Rex sought monetary damages in excess of $3.0 million, including costs, fees and expenses incurred in connection with the Rex Arbitration. On December 2, 2010, Rex obtained a temporary restraining order and preliminary injunction against Angiotech US from the United States District Court for the Southern District of New York (such action, the “SDNY Action”) requiring Angiotech US to honor its obligations under the Option Agreement for a period of 180 days or until the Rex Arbitration was concluded. On January 24, 2011, Angiotech US received a termination notice from Rex indicating that the Option Agreement would cease on April 24, 2011.
On February 16, 2011, Angiotech US entered into a Settlement and License Termination Agreement with Rex (the “Rex Settlement Agreement”) which provides for the full and final settlement and/or dismissal of all claims arising under the Option Agreement. Pursuant to the Rex Settlement Agreement, the Company and Rex agreed to withdraw any claims that have been or could have made in the Rex Arbitration, as well as to dismiss the SDNY Action with prejudice. The Rex Settlement Agreement further provides that: (i) the Option Agreement will terminate on March 31, 2011, or at an earlier date if so elected by Rex (the “Termination Date”), upon which Angiotech US will no longer market, sell or distribute the Option IVC Filter; (ii) Angiotech US will make a payment in the amount of $1.5 million to Rex within five business days of the effective date of the Rex Settlement Agreement in full and final payment and settlement of all claims and royalties due under the Option Agreement relating to the sales of the Option IVC Filter recorded by Angiotech US prior to January 1, 2011, and all milestone payments due or that may come due to Rex under the Option Agreement now or in the future; (iii) within five business days of the effective date, Angiotech US will make a royalty payment to Rex based upon actual cash receipts collected from customers attributable to sales of the Option IVC Filter that are recorded and accrued between January 1, 2011 and the effective date; (iv) from the effective date through the Termination Date, Angiotech US will make ongoing royalty payments to Rex on a weekly basis based upon actual cash receipts collected during each previous calendar week in respect of sales of the Option IVC Filter; and (v) Angiotech US will deliver to Rex certain materials relating to the marketing and sale of the Option IVC Filter. The Rex Settlement Agreement became effective on March 10, 2011 upon the Canadian Court granting an order authorizing Angiotech US to enter into the Rex Settlement Agreement and approving the terms thereof.
As at December 31, 2010, we recorded a $0.9 million inventory write-down and a $1.1 million intangible asset write-down in connection with the termination of the Option Agreement. In accordance with ASC No. 855 on Subsequent Events, we are currently assessing the impact of the Rex Settlement Agreement on our 2011 first quarter results.
62
Table of Contents
Ongoing Clinical Programs
The following discussion describes our product candidates, or certain of our partners’ product candidates, that are being evaluated in ongoing human clinical trials and their stage of development:
Partner Clinical Programs
Independent TAXUS Clinical Data
a) In May 2009 we announced that BSC had disclosed the publication of an article in the Journal of the American College of Cardiology (“JACC”) reviewing data on more than 19,000 patients from the Swedish national registry who were evaluated for restenosis, or the re-narrowing of arteries, after percutaneous coronary intervention (“PCI”). The Swedish Coronary Angiography and Angioplasty Registry holds data on all patients undergoing PCI in Sweden. The objective of this independent study was to evaluate restenosis rates of drug-eluting stents (“DES”) in patients with and without diabetes in a real-world setting. The article reported that patients who received a TAXUS Liberté® paclitaxel-eluting stent had numerically lower incidences of repeat procedures to treat restenosis at two years as compared to patients treated with ‘olimus-based DES, including Johnson & Johnson, Inc.’s Cypher® stent and Medtronic Inc.’s Endeavor® stent.
In the patients with diabetes, the TAXUS Liberté demonstrated a statistically significant lower restenosis rate compared to the Endeavor, which had more than two times the risk of repeat procedures. The JACC article reported that both the TAXUS Liberté stent and BSC’s first-generation paclitaxel-eluting stent, the TAXUS Express®, were the only stents in the study showing no increased risk of restenosis for patients with diabetes as compared to those without diabetes. Data for both the Cypher and Endeavor stents indicated significant increased risk of restenosis in patients with diabetes. In addition, the study showed that the TAXUS Liberté had an approximately 23 percent lower restenosis rate at two years compared to the prior-generation TAXUS Express.
The Swedish registry study included four DES brands: TAXUS Liberté, TAXUS Express, Cypher and Endeavor. In total, the registry included 35,478 DES implants during 22,962 procedures in 19,004 patients, with 1,807 restenoses reported over a mean 29-month follow-up period. For the entire study population, the repeat revascularization rate per stent was 3.5 percent after one year and 4.9 percent after two years. Overall, the adjusted risk of restenosis was 1.23 times higher in patients with diabetes than in patients without diabetes. In patients with diabetes, restenosis was higher in the non-TAXUS stents. The sirolimus-eluting Cypher and the zotorolimus-eluting Endeavor had higher restenosis rates in patients with diabetes compared with those in patients without diabetes (1.25 times and 1.77 times, respectively).
b) On March 16, 2010, we announced BSC’s results from an analysis of one-year subset data from the HORIZONS acute myocardial infarction trial assessing the impact of diabetes on clinical and angiographic outcomes in heart attack patients treated with the TAXUS Express2 Paclitaxel-Eluting Stent System or the Express bare-metal stent. The results demonstrated that the TAXUS Express Stent significantly reduced ischemia-driven target lesion revascularization (“TLR”) at one year and binary in-stent restenosis at 13 months in diabetic patients experiencing an acute myocardial infarction compared to an otherwise identical bare-metal control stent. Analysis of the data was presented on March 16, 2010 at the American College of Cardiology Annual Scientific Sessions.
On September 25, 2010 we further announced that BSC released three-year follow-up data from the HORIZONS-AMI trial. The trial, sponsored by the Cardiovascular Research Foundation (“CRF”) with grant support from Boston Scientific and The Medicines Company, is designed to determine the safety and efficacy of the TAXUS® Express2™ Paclitaxel-Eluting Coronary Stent System compared to bare-metal stenting in patients experiencing an acute myocardial infarction (“AMI”), or heart attack. With 3,006 patients enrolled worldwide, HORIZONS-AMI is the largest randomized trial to compare the use of drug-eluting stents to bare-metal stents for AMI patients. Analysis of the data was presented by Gregg W. Stone, M.D., Professor of Medicine and the Director of Research and Education at the Center for Interventional Vascular Therapy at the Columbia University Medical Center/New York-Presbyterian Hospital and Principal Investigator of the trial, at CRF’s annual Transcatheter Cardiovascular Therapeutics (“TCT”) scientific symposium in Washington, D.C. HORIZONS-AMI demonstrated that the TAXUS Express Stent significantly reduced clinical and angiographic restenosis compared to an otherwise identical bare-metal
63
Table of Contents
Express® control stent. After three years follow-up, the primary efficacy endpoint of ischemia-driven TLR was 9.4 percent for patients treated with TAXUS Express vs. 15.1 percent for bare-metal Express (p<0.001), a relative reduction of 40 percent. The secondary efficacy endpoint of ischemia-driven target vessel revascularization was 12.4 percent for TAXUS Express vs. 17.6 percent for bare-metal Express (p<0.001), a relative reduction of 32 percent. The primary safety endpoint of major adverse cardiac events (“MACE”) at three years was comparable for TAXUS Express and bare-metal Express patients (13.6 percent vs. 12.9 percent, respectively, p=0.66), which is consistent with findings at one and two years. Individual rates of death, repeat heart attack, stroke, and stent thrombosis between the two groups through three years of follow-up were also comparable.
On September 22, 2010 we announced that BSC had released comprehensive data from the TAXUS ATLAS clinical program, a series of global, prospective, single-arm trials evaluating the TAXUS® Liberté® Paclitaxel-Eluting Stent System in a variety of lesions and patient groups. Five-year results from the TAXUS ATLAS trial and four-year results from the TAXUS ATLAS Small Vessel and Long Lesion trials continue to show significant advantages for the thin-strut TAXUS Liberté Stent when compared to the first-generation TAXUS® Express® Stent. Analysis of the data was presented by the Co-Principal Investigators of the TAXUS ATLAS trials, Mark A. Turco, M.D., Director of the Center for Cardiac and Vascular Research, Washington Adventist Hospital, and John A. Ormiston, M.D., Mercy Angiography Unit, Mercy Hospital, Auckland, New Zealand, at CRF’s annual TCT scientific symposium in Washington, D.C.
TAXUS Element™ Paclitaxel-Eluting Coronary Stent System.
The platinum chromium TAXUS Element paclitaxel-eluting coronary stent system (“TAXUS Element Stent System”) is the third-generation BSC coronary stent platform that incorporates our research, technology and intellectual property related to the use of paclitaxel. The TAXUS Element stent features BSC’s proprietary platinum chromium alloy, which is designed to enable thinner stent struts, increased flexibility and a lower stent profile while improving radial strength, recoil and radiopacity. In addition, the TAXUS Element stent platform incorporates new balloon technology intended to improve upon BSC’s market-leading Maverick® balloon catheter technology.
The platinum chromium TAXUS Element Stent System is currently being evaluated by BSC in its PERSEUS clinical trial, which commenced in July 2007. On October 8, 2008, we announced that BSC had completed enrollment in the PERSEUS clinical trial. The PERSEUS clinical program compared the TAXUS Element stent to prior-generation stents in more than 1,600 patients in two parallel trials at 90 centers worldwide. This clinical program evaluated the safety and efficacy of the TAXUS Element stent in two studies and is designed for submission to the FDA in an application for approval to market and sell the TAXUS Element Stent System in the U.S. On March 15, 2010, we announced BSC’s results for this clinical trial. The first study evaluated the safety and efficacy of the TAXUS Element stent compared to the TAXUS Express2 stent in 1,264 patients with “workhorse” lesions from 2.75 to 4.0 mm. This prospective, randomized (3:1) trial met its primary endpoint of non-inferiority for target lesion failure (“TLF”) at 12 months with rates of 5.6 percent for the TAXUS Element Stent and 6.1 percent for the TAXUS Express2 stent. The secondary endpoint of in-segment percent diameter stenosis at nine months, as measured by quantitative coronary angiography, was also met. The second study compared the TAXUS Element stent to a historic control (TAXUS V de novo bare-metal Express coronary stent system) in 224 patients with lesions from 2.25 to 2.75 mm. The trial met its primary endpoint of superiority for in-stent late loss at nine months with unadjusted values of 0.38 mm for the TAXUS Element stent and 0.80 mm for the Express stent, which was statistically significant. The trial also met its secondary endpoint of superiority for TLF at 12 months, showing a reduction, which was statistically significant, with an unadjusted rate of 7.3 percent for the TAXUS Element stent compared to a pre-specified performance goal of 19.5 percent based on historical outcomes for the Express control stent.
On September 22, 2010 we announced BSC released results from an analysis of 1,166 patients from the PERSEUS clinical program comparing the performance of the TAXUS Element Stent System in diabetic versus non-diabetic patients. Results demonstrated that despite the known increased risk of restenosis for diabetics versus non-diabetics in patients undergoing coronary revascularization, the TAXUS Element Stent had comparable levels of TLR and late loss in both diabetic and non-diabetic patients. Analysis of the data was presented by Louis A. Cannon, M.D., of the Cardiac and Vascular Research Center of Northern
64
Table of Contents
Michigan in Petoskey, Michigan, and Co-Principal Investigator of the PERSEUS clinical program, at CRF’s annual TCT scientific symposium in Washington, D.C. The PERSEUS diabetic analysis included clinical outcomes at one year among 314 diabetic patients and 852 non-diabetic patients treated with the TAXUS Element Stent from the PERSEUS Workhorse and Small Vessel clinical trials. Due to significant disparity in baseline characteristics between diabetic and non-diabetic patients, propensity score analysis was used to allow for adjustment of baseline differences (other than the presence of diabetes) between the two groups. Results showed that the TAXUS Element Stent maintained comparable rates of TLR at one year, whether adjusted or unadjusted, in the diabetic and non-diabetic patient populations (5.5 percent vs. 4.1 percent, p=0.43, adjusted). The adjusted and unadjusted rates of TLF at one year (defined as ischemia-driven TLR, or myocardial infarction (“MI”)/cardiac death related to the target vessel) were also similar between the patient groups (7.5 percent vs. 5.4 percent, p=0.31, adjusted). Adjusted one-year rates of MACE, cardiac death, MI and Academic Research Consortium definite/probable stent thrombosis showed no differences between the two populations (p-values of 0.14, 0.12, 0.38, and 0.77, respectively). Nine-month adjusted angiographic outcomes showed similar in-segment late loss in diabetics and non-diabetics (0.23 mm vs. 0.19 mm, p=0.52). Rates of late loss for the TAXUS Element Stent were numerically lower than rates in prior studies for the TAXUS Express® and TAXUS Liberté® Stents.
In May 2010, we announced that BSC received CE Mark approval for the TAXUS Element Stent System, which included approval for a specific indication for the treatment of diabetic patients. In June 2010, we further announced that BSC had commercially launched and implanted its first TAXUS Element Stent System in the European Union and other CE Mark countries.
In the U.S. BSC expects FDA approval for the TAXUS Element Stent System in mid 2011. In Japan, BSC expects approval for the TAXUS Element Stent System in late 2011 or early 2012.
Zilver-PTX Paclitaxel-Eluting Peripheral Vascular Stent System
The Zilver PTX paclitaxel –eluting peripheral vascular stent (“Zilver PTX Stent System”) is a specialized stent, which incorporates our proprietary paclitaxel technology, and is designed for placement in diseased arteries in the limbs to restore blood flow by expanding and holding open the arteries upon deployment. The Zilver PTX Stent achieves targeted drug delivery without using a polymer to adhere the drug to the stent body, which eliminates the potential patient risks associated with polymer-coated devices, including clot formation and inflammation. Cook is a co-exclusive licensee, together with BSC, of our proprietary paclitaxel technology to reduce restenosis following stent placement in patients suffering from peripheral artery disease (“PAD”). The Zilver PTX stent is currently undergoing multiple human clinical trials in the E.U., U.S., Japan, and selected other countries to assess product safety and efficacy.
It is estimated that approximately 27 million people suffer from PAD in Europe and North America. Of patients with PAD, approximately only one quarter indicate any substantive symptoms. The “silent” nature of this condition can result in a number of patients being diagnosed only when their condition has progressed to the severe stage. In patients with severe PAD whose condition is not improving with risk-factor modification, exercise programs or pharmacological therapy, invasive surgical procedures may be necessary. These procedures may include angioplasty, stenting or surgery. To date, stent procedures for PAD in the limbs have been limited due to high observed rates of restenosis or other failure of the procedure requiring a subsequent surgical procedure, as well as risk of stent fracture with existing products, as these stents are exposed and not protected by the patient’s anatomy as with coronary stents.
In August 2009, we announced that our partner Cook had reported CE Mark approval and commercial launch of the Zilver PTX stent in the E.U. and in June 2010, we announced that Cook had submitted its Pre-Market Approval (“PMA”) application to the FDA for Zilver PTX. We believe the data from the clinical trial described below supports this PMA application.
In January 2007, Cook released nine-month data from its E.U. clinical study. The preliminary data presented by Cook on the first 60 patients in the randomized trial, which is examining the safety of using Cook’s Zilver PTX stent to treat blockages, or lesions, of the superficial femoral artery (“SFA”) above the knee, indicated that the Zilver PTX stent showed an equal adverse event rate to conventional angioplasty for treating SFA lesions. The Zilver PTX stent also displayed a zero percent fracture rate for 41 lesions at six months and 18 lesions at one year.
65
Table of Contents
In July 2007, Cook announced that the first U.S. patients in a randomized pivotal human clinical trial of the Zilver PTX stent were treated at Tri-City Medical Center in Oceanside, CA. The trial is designed to randomize patients to receive either the Zilver PTX stent or balloon angioplasty. Data from this clinical trial is intended to be used to support submission to the FDA for approval in the U.S. to market the device. In addition, data collected on Japanese and U.S. patients is expected to be combined for the final evaluation of the device and used for regulatory submissions in both markets for approval.
In June 2008, Cook reported positive interim results from the registry arm of a clinical study designed to measure the efficacy of the Zilver PTX stent in treating PAD patients, specifically in the treatment of blockages in the femoropopliteal artery.
The data published in the Zilver PTX registry involved 791 patients from Europe, Russia, Canada and Korea demonstrated highly positive results. Only 8 percent of all patients with de novo (new) lesions required a reintervention to reopen the artery in the first 12 months — a rate significantly surpassing existing treatments for PAD in the SFA, such as balloon angioplasty and bare metal (non-drug-eluting) stents. In addition, specific patient groups that are often very hard to treat, such as diabetics and patients with in-stent restenosis (those treated previously with a noncoated stent), were shown in the trial to benefit from the Zilver PTX. As indicated by the trial data, the superior results achieved in the first year have been largely maintained throughout 24 months, which is an important clinical milestone. In comparison with other trial data obtained, the Zilver PTX stent demonstrated a reduction in reintervention of between 50 percent and 75 percent, which is an important patient benefit.
The results were reported by trial investigators at the 2008 SVS Vascular Annual Meeting, and revealed clinical improvement, excellent durability and fracture resistance, high rates of event-free survival (“EFS”) and freedom from TLR. Interim data was compiled at six and 12 months using 435 patients and 200 patients, respectively. The corresponding EFS rates were 94% and 84%, and freedom from TLR was 96% and 88%. Evaluation of stent x-rays is ongoing, and currently suggests stent fractures in approximately one percent of cases at six months and less than two percent of cases at 12 months. In addition, the Zilver PTX stent exhibited no safety concerns and results were better than expected for Trans-Atlantic Inter-Society Consensus class C and D lesions, occlusions, in-stent restenosis and lesions greater than seven centimeters. Follow-up to the registry arm of the study will continue through two years.
In September 2008, Cook announced it had completed enrollment in its pivotal human clinical trial for the Zilver PTX. The 420 patients enrolled in Cook’s randomized trial include PAD patients treated in Germany, the U.S. and Japan. On the same date, Cook announced that it had enrolled an additional 780 patients in the E.U., Canada and Korea in a clinical registry to evaluate the safety of the Zilver PTX stent. Those data were used to obtain CE Mark approval in the E.U. and launch the device there, with additional regulatory submissions pending in additional markets.
In April 2009, we announced that Cook had reported data that showed that 82 percent of patients who were treated with Cook’s Zilver PTX stent were free from reintervention at two-year follow up. The Zilver PTX Registry study, involving 792 patients globally, is assessing the safety and efficacy of the Zilver PTX in treating peripheral artery disease. The most recent results were reported at the 31st International Symposium Charing Cross Controversies Challenges Consensus. Data was compiled at 12 and 24 months for 593 patients and 177 patients respectively. The registry, which enrolled a broad spectrum of patients, includes patients with complex lesions (e.g., long lesions, total occlusions, in-stent restenosis). The corresponding event-free survival rates were 87 percent and 78 percent, respectively, and freedom from target lesion revascularization was 89 percent and 82 percent, respectively. Clinical measures that included ankle-brachial index, Rutherford score, and walking distance and speed scores showed significant improvement at six and 12 months and were maintained through 24 months. Detailed evaluation of stent x-rays demonstrated excellent stent integrity through 12 months, confirming previously published results showing 99 percent completely intact stents (less than 1 percent stent fracture rates observed) with a mean follow up of 2.4 years in the challenging superior femoral artery and popliteal arteries, including behind the knee locations.
66
Table of Contents
In April 2010, we announced that Cook had enrolled its first patient in its landmark Formula™ PTX® clinical trial. The trial is the first of its kind to evaluate the safety and effectiveness of a paclitaxel-eluting stent to treat renal artery disease, the narrowing of the arteries that supply blood to the kidneys. The multi-center, randomized trial plans to enroll 120 patients at sites across Europe. The trial utilizes Cook’s Formula renal stent, which is designed with a very low profile that may help it cross tightly blocked vessels for placement into diseased renal arteries.
In May 2010, we announced that Cook presented one-year data at Euro PCR that confirmed sustained clinical outcomes with Zilver PTX. According to data presented, 86.2% of all patient subgroups treated with Zilver PTX demonstrated vessel patency at 12 months without the requirement for an additional intervention. The data was collected as part of the world’s largest clinical study of endovascular treatment for PAD in the SFA. The trial is based on a group of 787 patients, including symptomatic patients, diabetics, and those with the most complex lesions, including long lesions, total occlusions and in-stent restenosis.
On September 24, 2010 we announced that a summary of the final clinical trial results for the randomized study of Cook’s Zilver PTX Stent for use in patients with peripheral arterial disease in the SFA was presented at CRF’s annual TCT symposium in Washington D.C. The study met its 12-month primary endpoint showing non-inferior EFS and superior patency for the Zilver PTX as compared to balloon angioplasty. Cook Medical, a license holder of our proprietary paclitaxel technology, has developed Zilver-PTX, a self-expanding, highly durable nitinol stent that uses a proprietary technology to deliver a locally therapeutic dose of paclitaxel, a proven anti-restenotic drug, to the target lesion.
This multicenter, multinational, prospective, randomized study compared the safety and effectiveness of the Zilver PTX to balloon angioplasty and bare metal stenting. Patients with de novo or restenotic SFA lesions were randomized to balloon angioplasty or Zilver PTX stent placement. Balloon angioplasty patients experiencing acute failure (> 30% residual stenosis) underwent secondary randomization to provisional stenting with Zilver BMS (bare metal stent) or Zilver PTX. Endpoints included EFS, stent integrity by radiographic core laboratory analysis and primary patency by Duplex ultrasound core laboratory analysis (peak systolic velocity ratio < 2.0). The study enrolled 479 patients at 55 institutions in the United States, Japan and Germany, with 241 patients randomized to the Zilver PTX group and 238 to the balloon angioplasty group. Demographics and lesion characteristics were similar for both groups. Approximately half the balloon angioplasty group experienced acute failure and underwent secondary randomization, assigning 59 and 61 patients to provisional stenting with Zilver BMS and Zilver PTX, respectively.
Angiotech Clinical Programs
MultiStem® Stem Cell Therapy
The MultiStem stem cell therapy is under evaluation in clinical trials being conducted together with our partner Athersys, Inc. (“Athersys”) for the treatment of acute myocardial infarction. MultiStem stem cells are proprietary adult stem cells derived from bone marrow that have demonstrated the ability in laboratory experiments to form a wide range of cell types. MultiStem may work through several mechanisms, but a primary mechanism appears to be the production of multiple therapeutic molecules produced in response to inflammation and tissue damage. We and Athersys believe that MultiStem may represent a unique “off the shelf” stem cell product candidate, based on its potential ability to be used without tissue matching or immunosupression, and its potential capacity for large scale production. We entered into an agreement with Athersys in May 2006 to co-develop and commercialize MultiStem for use in the indications of acute myocardial infarction and peripheral vascular disease. On December 20, 2007, we and Athersys announced we had received authorization from the FDA to commence a phase I human clinical trial to evaluate the safety of MultiStem in the treatment of acute myocardial infarction.
In February 2010, Athersys announced that enrollment for the phase I clinical trial had been completed. The phase I clinical trial is an open label, multi-center dose escalation trial evaluating the safety and maximum tolerated dose of a single administration of allogeneic MultiStem cells following an acute myocardial infarction. Following standard treatment, enrolled patients receive MultiStem delivered via a catheter that enables rapid and efficient delivery of MultiStem into the region of damage in the heart. The
67
Table of Contents
study is being conducted at multiple cardiovascular treatment centers in the United States, including the Cleveland Clinic, Columbia University Medical Center and Henry Ford Health System, and includes patients in three treatment cohorts or dose groups, as well as a non-treated registry group. In preclinical studies conducted by Athersys and independent cardiovascular research teams, administration of MultiStem following an ischemic injury to the heart has been associated with a number of benefits, including an increase in ejection fraction, or volume of blood pumped from the heart, a reduction of inflammation in the region of ischemic injury and increased angiogenesis, each believed to help augment recovery and healing.
In July 2010 we announced that Athersys had announced positive results from its phase I clinical trial of MultiStem®. The study results, which represented at least four months of post-treatment patient data, demonstrated that MultiStem was well tolerated at all dose levels and also suggested improvement in heart function in treated patients.
In September 2010, updated study results were presented at the TCT conference held in Washington, D.C. Dr. Marc Penn, M.D., Ph.D., co-principal investigator of this study and Director of Cardiovascular Cell Therapy at the Cleveland Clinic, and Director of the Skirball Laboratory for Cardiovascular Cellular Therapeutics, presented the results at the Symposium “Strategies for Cardiovascular Repair: Stem Cell Therapy and Beyond.”
New data presented by Dr. Penn included additional information about the nature and incidence of adverse events (“AEs”) over the first four months of the trial, demonstrating that the AEs were generally mild-to-moderate in nature, there was no dose dependent effect of MultiStem on AEs, and overall, MultiStem had a favorable safety profile. Further, Dr. Penn shared the results from additional analysis of echocardiogram data collected over the first four months of the study, which suggests that MultiStem administration may also provide improvements in other measures of heart function. Patients receiving MultiStem, for instance, demonstrated a meaningful improvement in mean wall motion score at four months compared to baseline, though this improvement was not statistically significant. Among those patients with more severe heart attacks (i.e. left ventricular ejection fraction, a measure of heart function, £ 45), the mean wall motion score for treated patients improved over the four-month period, while for registry patients it worsened over this time.
On September 23, 2010 we, together with Athersys, announced updated results from the phase I clinical trial of MultiStem®, an allogeneic cell therapy product, administered to individuals following acute myocardial infarction, more commonly referred to as a heart attack. The phase I clinical trial was an open label, multi-center dose escalation trial evaluating the safety and maximum tolerated dose of a single administration of allogeneic MultiStem cells following an acute myocardial infarction. Enrolled patients received MultiStem delivered via a catheter into the damaged region of the heart 2-5 days following percutaneous coronary intervention, a standard treatment for heart attack. The study included patients in three treatment cohorts or dose groups (20 million, 50 million and 100 million cells per patient) and a registry group where patients received only standard of care. Nineteen treated- and 6 registry subjects were enrolled in the study. The trial was conducted at multiple cardiovascular treatment centers in the United States, including the Cleveland Clinic, Columbia University Medical Center and Henry Ford Health System.
Upon completion of the phase I human clinical trial which is currently being conducted by Athersys, we may, at our option, assume lead responsibility for further clinical development. We currently own marketing and commercialization rights with respect to this product candidate.
Completed or Suspended Clinical Programs
Bio-Seal Lung Biopsy Tract Plug System.
Bio-Seal is a novel technology designed to prevent air leaks in patients having lung biopsies by plugging the biopsy track with an expanding hydrogel plug. On contact with moist tissue, the hydrogel plug absorbs fluids and expands to fill the void created by the biopsy needle puncture. The plug is absorbed into the body after healing of the puncture site has occurred. We are the worldwide manufacturer and distributor of Bio-Seal, which has received CE Mark approval and is currently marketed and sold in the E.U.
Bio-Seal has undergone a human clinical trial in the U.S. which was designed to assess the safety and efficacy of Bio-Seal, with the primary endpoint being reduction in rates of pneumothorax in patients
68
Table of Contents
undergoing lung biopsy procedures. The study was designed to provide a basis for FDA clearance for the commercialization of Bio-Seal. The prospective, randomized, controlled clinical trial enrolled and randomized 339 patients at 15 different investigational sites. Inspiratory upright chest x-rays were performed at 30 to 60 minutes, 24 hours and 30 days after treatment. The trial enrolled its first patient in October 2005 and completed enrollment in June 2008. In March 2009, we announced positive results from this clinical trial at the Society of Interventional Radiologists Annual Scientific Meeting in San Diego, CA. The trial demonstrated a statistically significant clinical benefit in the group receiving Bio-Seal. The Bio-Seal treatment arm hit the primary end point of clinical success for rate of absence of pneumothorax at each time period as compared to traditional treatment. Based on the per-protocol population, the clinical success rate was 85% using Bio-Seal and 69% in the control group. This difference was statistically significant (p=0.002). Although not powered for statistical analysis, positive trends were also observed for Bio-Seal subjects as compared to the control group in various secondary endpoints, including fewer Bio-Seal subjects admitted to the hospital for pneumothoraces (9.4% vs. 13.6%), fewer chest tube placements in Bio-Seal patients (3.5% vs. 10.7%), and fewer additional chest x-rays required in Bio-Seal patients (0.6% vs. 5.3%).
Data from this clinical trial was submitted to the FDA, with the objective of receiving 510(k) clearance to market Bio-Seal in the U.S. Subsequent to this submission, the FDA asked us various questions about the study and our submission and we responded to all of the questions from the FDA. In October 2009, we announced that we had received correspondence from the FDA regarding our 510(k) submission for Bio-Seal, stating the FDA’s view that Bio-Seal is a class III device that requires Pre-Market Approval (“PMA”) and therefore a more extensive clinical study in order to obtain FDA marketing clearance. As a result, we are reviewing our options with respect to this product candidate, including possibly appealing this FDA decision, and we are discussing the possible preparation of a PMA submission with our partner, Biopsy Sciences, LLC. We have not incurred material expense to date with respect to activities regarding this product candidate. Should we elect to continue to pursue development or regulatory approvals for this product candidate that require us to incur material expense, we will provide further updates in our public disclosure.
5-FU-Eluting Central Venous Catheter
Central venous catheters (“CVC”) are usually inserted into critically ill patients for extended periods of time to administer fluids, drugs, and nutrition, as well as facilitate frequent blood draws. Through our proprietary drug identification strategy, we have elected to evaluate 5-Fluorouracil (“5-FU”), a drug previously approved by the FDA for treatment of various types of cancer, as a compound that may help to prevent certain types of infection in patients receiving a CVC. We completed a human clinical trial in the U.S. designed to assess the safety and efficacy of our 5-FU-eluting CVC in preventing various types of catheter related infections. The study was a randomized, single-blind, 960-patient, 25-center study and was designed to evaluate whether our 5-FU-eluting CVC prevents bacterial colonization at least as well as the market-leading anti-infective CVC. On July 10, 2007, we announced that we had completed enrollment of the study, and on October 9, 2007, we announced this study had met its primary statistical endpoint of non-inferiority as compared to the market leading anti-infective CVC (a chlorhexidine / silver sulfadiazine (“CH-SS”) coated CVC) and indicated an excellent safety profile. In March 2008, we presented the clinical trial data at the 28th International Symposium on Intensive Care and Emergency Medicine in Brussels, Belgium. Based on the clinical trial data, the investigators concluded that our 5-FU CVC met the primary endpoint of the study; specifically that our 5-FU CVC product candidate was non-inferior in its ability to prevent bacterial colonization of the catheter tip when compared to catheters coated with CH-SS. There were no statistically significant differences in the rate of adverse events related to the study devices, or in the rates of catheter-related bloodstream infections. Additionally, there was no evidence for acquired resistance to 5-FU in clinical isolates exposed to the drug for a second time. Based on the positive results achieved in the study, in December 2007 we filed a request for 510(k) clearance from the FDA to market and sell the CVC in the U.S. and on April 17, 2008, we announced that we had received 510(k) clearance from the FDA to market our 5-FU CVC in the U.S.
As a result of our significantly constrained liquidity and capital resources, we have elected to date not to pursue commercialization activities for this product. Development of future launch plans and commercialization of this product, as well as related product line extensions, will depend on our liquidity, capital resources, or our ability to secure future financing.
69
Table of Contents
Acquisitions
As part of our business development efforts, we completed several significant acquisitions. Terms of certain of these acquisitions may require us to make future milestone or contingent payments upon achievement of certain product development and commercialization objectives, as discussed under “Contractual Obligations”.
On April 6, 2010, we acquired certain product candidates and technology assets of Haemacure Corporation (“Haemacure”) for approximately $3.9 million. Haemacure had been involved in proceedings under the Bankruptcy and Insolvency Act (Canada) and Chapter 11 of the U.S. Bankruptcy Code. Through an asset sale transaction, we acquired all of the relevant research and development activities, manufacturing operations, key personnel, and intellectual property rights necessary to pursue further clinical development of Haemacure’s human biomaterial product candidates, specifically fibrin sealant and thrombin hemostat. Under ASC No. 805, the transaction qualified as a business combination and the acquisition method was used to account for the transaction.
Prior to the acquisition, we entered into various license, distribution, development and supply agreements for fibrin and thrombin technologies. The collaboration was intended to provide us with access to technology for certain of its surgical product candidates which are currently in preclinical development. As these agreements did not contain any settlement provisions and were neither favorable nor unfavorable when compared to current market pricing for similar transactions, no settlement gains or losses were recognized.
As part of the collaboration, we provided Haemacure with a senior secured bridge financing facility (the “loan”) of $2.5 million in multiple draw-downs throughout 2009. The loan was for a two- year term, bore an annual interest rate of 10%, compounded quarterly, and was senior to all of Haemacure’s other indebtedness, subject to certain exceptions. The loan was secured by substantially all of Haemacure’s assets. As at December 31, 2009, the loans were recognized at their fair value of $1.6 million with the remainder being amortized on account of an arrangement for prepaid services. On January 11, 2010 Haemacure announced it filed a notice of intention to make a proposal to its creditors under the Bankruptcy and Insolvency Act (Canada) and its wholly -owned U.S. subsidiary sought court protection under Chapter 11 in the U.S. These actions were taken in light of Haemacure’s financial condition and inability to raise additional financing. We subsequently provided an additional $1.0 million of financing to fund Haemacure’s insolvency proceedings and day-to-day operations. The loan and accrued interest was effectively settled in exchange for certain assets acquired from Haemacure. In addition, we paid an additional $0.2 million in cash to settle certain obligations owed by Haemacure.
As at April 5, 2010, the carrying value of the loan owed by Haemacure was $2.5 million compared to the contractual amount of $3.7 million. In accordance with ASC No. 805-10-55-21, we recognized a $1.2 million gain on the effective settlement of the pre-existing debt arrangement, which reflects the difference between the contractual settlement amount of the loan and its carrying value.
The estimated fair values of the net assets acquired at the date of acquisition are summarized as follows:
| April 6, 2010 | ||||
| Property, plant and equipment |
$ | 2,906 | ||
| Intangible assets |
1,700 | |||
| Deferred income tax liability |
(720 | ) | ||
| Estimated fair value of net assets acquired |
3,886 | |||
| Consideration: |
||||
| Face value of the loan receivable, including accrued interest |
3,686 | |||
| Cash paid |
200 | |||
| Total consideration |
$ | 3,886 | ||
70
Table of Contents
We used the income approach to determine the fair value of the intangible assets acquired. The fair values of the property, plant and equipment were estimated using a cost-based approach which considers replacement values as well as the values ascribed to the assets for their highest and best use as defined by U.S. GAAP.
As the acquisition qualified as a business combination, transaction-related expenses of $0.6 million were charged to selling, general and administration costs during the year ended December 31, 2010. The assets acquired from Haemacure have been included in the consolidated financial statements since the date of acquisition. Given our current liquidity constraints and the Recapitalization Transaction being pursued through the Creditor Protection Proceedings; we have temporarily suspended research and development activities related to the Haemacure fibrin and thrombin technologies. As a result, as at December 31, 2010, we wrote-off the $3.9 million of net assets acquired from Haemacure during the year. This impairment charge triggered a corresponding reduction of the $0.7 million deferred income tax liability and expense, thereby increasing the loan settlement gain to $1.9 million as at December 31, 2010.
Pro forma information (unaudited)
The following unaudited pro forma information is provided for the business combination assuming that it occurred at the beginning of the earliest period presented, January 1, 2009. The pro forma information combines the financial results of the Company with the financial results of Haemacure for the year ended December 31, 2010 and December 31, 2009.
| Year
ended December 31, 2010 |
Year
ended December 31, 2009 |
|||||||
| Net loss |
$ | (68,997 | ) | $ | (27,505 | ) | ||
| Net loss per share |
$ | (0.81 | ) | $ | (0.32 | ) | ||
The information presented above is for illustrative purposes only and is not indicative of the results that would have been achieved had the business combination taken place at the beginning of the earliest period presented. For comparative purposes, the pro forma information above excludes the impact of the $1.9 million non-recurring loan settlement gain and $0.6 million of transaction costs which were incurred in connection with the business combination during the year ended December 31, 2010. The pro forma impact on revenue was not material for any of the periods presented.
Collaboration, License and Sales and Distribution Agreements
In connection with our research and development efforts, we have entered into various arrangements with corporate and academic collaborators, licensors, licensees and others for the research, development, clinical testing, regulatory approval, manufacturing, marketing and commercialization of our product candidates. Terms of the various license agreements may require us, or our collaborators, to make milestone payments upon achievement of certain product development and commercialization objectives and pay royalties on future sales of commercial products, if any, resulting from the collaborations.
As discussed above, in April 2010, we announced the closing of the acquisition of certain product candidates and technology assets of Haemacure. Prior to the acquisition and purchase of the assets, we entered into various license, distribution, development and supply agreements for fibrin and thrombin technologies. The collaboration was intended to provide us with access to technology for certain of its surgical product candidates, which are currently in preclinical development. We funded approximately $1.5 million in expenses prior to the acquisition, which include the funding of Haemacure’s insolvency proceedings and day-to-day operations, legal fees and expenses. Given our current liquidity constraints and the Recapitalization Transaction being pursued through the Creditor Protection Proceedings, we have suspended research and development activities related to the Haemacure fibrin and thrombin technologies. As a result, as at December 31, 2010, we wrote-off the $3.9 million of net assets acquired from Haemacure during the year.
Agreements relating to our material collaborations, licenses, sales and distribution arrangements are listed in the exhibits index to this Annual Report on Form 10-K and may be found at the locations specified therein.
71
Table of Contents
Critical Accounting Policies and Estimates
Our consolidated financial statements are prepared in accordance with U.S. GAAP. These accounting principles require management to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenue and expenses. We believe that the estimates and assumptions upon which we rely are reasonable and are based upon information available to us at the time the estimates and assumptions were made. Actual results could differ materially from our estimates.
We believe the following policies to be critical to understanding our financial condition, results of operations, and expectations for 2011 because these policies require management to make significant estimates, assumptions and judgments about matters that are inherently uncertain.
Revenue Recognition
Product Sales
We recognize revenue from product sales, including shipments to distributors, when the product is shipped from our facilities to the customer provided that we have not retained any significant risks of ownership or future obligations with respect to products shipped. We recognize revenue from product sales net of provisions for future returns. These provisions are established in the same period as the related product sales are recorded and are based on estimates derived from our historical experience. The estimates are adjusted to reflect actual returns.
We consider revenue to be realized or realizable and earned when all of the following criteria are met: persuasive evidence of a sales arrangement exists; delivery has occurred or services have been rendered; the price is fixed or determinable; and collection is reasonably assured. These criteria are generally met at the time of shipment when the risk of loss and title pass to the customer or distributor.
We include amounts billed to customers for shipping and handling in revenue. The corresponding costs for shipping and handling are included in cost of products sold.
Royalty Revenue
We recognize royalty revenue when we have fulfilled the terms in accordance with the contractual agreement, have no future obligations, the amount of the royalty fee is determinable and collection is reasonably assured. We record royalty revenue from BSC on a cash basis due to the difficulty of accurately estimating the BSC royalty before we receive the reports and payments from BSC. This results in a one quarter lag between the time we record royalty revenue and the time the associated sales were recorded by BSC.
License Fees
License fees are comprised of initial fees and milestone payments derived from collaborative and other licensing arrangements. License fees are recognized as revenue when persuasive evidence of an arrangement exists, the contracted fee is fixed or determinable, the intellectual property is delivered to the customer, collection is reasonably assured and the Company has substantially completed its performance obligations. Non-refundable milestone payments are recognized upon the achievement of specified milestones when the milestone payment is substantive in nature, achievement of the milestone was not reasonably assured at the inception of the agreement and the Company has no further significant performance obligations to fulfill under the arrangement. Initial fees and non-refundable milestone payments received which require the ongoing involvement of the Company are deferred and amortized into income on a straight-line basis over the period of ongoing involvement if there is no other method under which performance can be objectively measured.
Income Tax Expense
Income taxes are accounted for using the liability method. Deferred income tax assets and liabilities result from the temporary differences between the amount of assets and liabilities recognized for financial statement and income tax purposes, and for operating losses, capital losses and tax credit carry forwards,
72
Table of Contents
using tax rates expected to be in effect for the years in which the differences are expected to reverse. The carrying value of our net deferred income tax assets assumes that we will be able to generate sufficient future taxable income in certain tax jurisdictions to realize the value of these assets. Management evaluates the realizability of the deferred tax assets and assesses the need for any valuation allowance adjustment on a periodic basis. A valuation allowance is provided to reduce the net deferred income tax assets if, based upon the available evidence, it is more likely than not that some or all of the deferred income tax assets will not be realized.
Significant estimates are required in determining our provision for income taxes including, but not limited to, reserves for uncertain tax positions and valuation allowances for deferred income tax assets. Some of these estimates are based on interpretations of existing tax laws or regulations. Our effective tax rate may change from period to period based on the mix of income among the different foreign jurisdictions in which we operate, changes in tax laws in these jurisdictions, and changes in the amount of valuation allowance recorded.
The Company uses a two-step approach for recognizing and measuring the income tax benefits of uncertain tax positions taken or expected to be taken in a tax return. A tax benefit from an uncertain tax position is recognized if it is more-likely-than-not that the position will be sustained upon examination by the tax authority based on the technical merits of the position. The tax benefit of an uncertain tax position that meets the more-likely-than-not recognition threshold is measured as the largest amount that is greater than 50% likely to be realized upon settlement with the tax authority. To the extent a full benefit is not expected to be realized, an income tax liability is established. Any change in judgment related to the expected resolution of uncertain tax positions are recognized in the year of such a change. Accrued interest and penalties related to unrecognized tax benefits are recorded in income tax expense in the current year.
Stock-based Compensation
We account for stock-based compensation in accordance with ASC No. 718 — Compensation-Stock Compensation. ASC No. 718 requires us to recognize the grant date fair value of share-based compensation awards granted to employees over the requisite service period. We use the Black-Scholes option pricing model to calculate stock option values, which requires certain assumptions including the future stock price volatility and expected time to exercise. Changes to any of these assumptions, or the use of a different option pricing model (such as the binomial model), could produce a different fair value for stock-based compensation, which could have a material impact on our earnings.
Cash Equivalents and Investments
We may invest our excess cash balances in short-term securities, principally investment grade commercial debt and government agency notes. At December 31, 2010, we held one equity security which was classified as available-for-sale, and accordingly, was recorded at fair market value.
As part of our strategic product development efforts, we also invest in equity securities of certain companies with which we have collaborative agreements. The equity securities of some of these companies do not have a readily determinable fair value. These investments are recorded using the cost method of accounting and are tested for impairment by reference to anticipated undiscounted cash flows expected to result from the investment, the results of operations and financial position of the investee, and other evidence supporting the net realizable value of the investment.
Goodwill
We test goodwill for possible impairment at least annually and whenever changes in circumstances occur that would indicate impairment in the value of goodwill. When the carrying value of a reporting unit’s goodwill exceeds the implied fair value of the goodwill, an impairment loss is recognized in an amount equal to the excess. Circumstances that could trigger impairment include adverse changes or outcomes in legal or regulatory matters, technological advances, decreases in anticipated demand, unanticipated competition, and significant declines in our share price. We estimate fair value based on a discounted projection of future cash flows which are subject to significant uncertainty and estimates. If future cash flows are less than those projected, an impairment charge may become necessary that could have a material impact on our financial position and results of operations.
73
Table of Contents
During the year ended December 31, 2008 we tested our goodwill for impairment and determined that, due to a significant and sustained decline in our public stock market capitalization during 2008, our goodwill was impaired and as a consequence, we recorded an impairment charge of $649.7 million during year ended December 31, 2008. Accordingly, we had no goodwill remaining on our balance sheet as at December 31, 2008, December 31, 2009 and December 31, 2010.
Intangible Assets
Our identifiable intangible assets are primarily comprised of technologies acquired through our business combinations. Intangible assets also include in-licensed proven medical technologies. We amortize intangible assets to reflect the pattern in which the economic benefits of the intangible asset are consumed over the estimated life of the technologies, which is generally on a straight-line basis and ranges from two to twelve years depending on the circumstances and the intended use of the technology. We determine the estimated useful lives for intangible assets based on a number of factors such as legal, regulatory or contractual limitations; known technological advances; anticipated demand for our products; and the existence or absence of competition.
We review long-lived assets subject to amortization to determine if any adverse conditions exist or a change in circumstances has occurred that would indicate impairment or a change in the remaining useful life. Conditions that may indicate impairment include, but are not limited to, a significant adverse change in legal factors or business climate that could affect the value of an asset, a product recall, or an adverse action or assessment by a regulator. If an impairment indicator exists, we test the asset (asset group) for recoverability. For purposes of the recoverability test, intangible assets are grouped with other assets and liabilities at the lowest level of identifiable cash flows if the intangible asset does not generate cash flows independent of other assets and liabilities. If the carrying value of the asset (asset group) exceeds the undiscounted cash flows expected to result from the use and eventual disposition of the asset (asset group), an impairment charge is recorded to write the carrying value down to the fair value in the period identified.
We generally calculate fair value of assets (asset groups) as the present value of estimated future cash flows expected to generate from the asset using a risk-adjusted discount rate. In determining the estimated future cash flows associated with our assets (asset groups), assumptions about future revenue contributions, cost structures and remaining useful lives of the asset (asset group) are used. The use of alternative assumptions, including estimated cash flows, discount rates, and alternative estimated remaining useful lives could result in different calculations of impairment.
During the year ended December 31, 2010, we recorded impairment write-downs on our intangible assets totaling $2.8 million. Approximately $1.1 million of the intangible asset write-downs relates to the termination of our 2008 License, Supply, Marketing and Distribution Agreement with Rex described under the Recent Significant Developments section above. The remaining $1.7 million write-down was recorded as a result of our temporary suspension of our research and development activities on our Haemacure fibrin and thrombin technologies discussed under the Acquisitions section above. No intangible asset impairment charges were recorded for the year ended December 31, 2009. During the year ended December 31, 2008, we recorded an intangible asset write-down of $0.1 million.
74
Table of Contents
Results of Operations
Overview
| Years ended December 31, | ||||||||||||
| (in thousands of U.S.$, except per share data) |
2010 | 2009 | 2008 | |||||||||
| Revenues |
||||||||||||
| Medical Products |
$ | 211,495 | $ | 191,951 | $ | 190,816 | ||||||
| Pharmaceutical Technologies |
34,747 | 87,727 | 92,456 | |||||||||
| Total revenues |
246,242 | 279,678 | 283,272 | |||||||||
| Operating income (loss) |
(20,057 | ) | 23,085 | (671,179 | ) | |||||||
| Other (expense) income |
(46,798 | ) | (39,916 | ) | (82,889 | ) | ||||||
| Net loss before income taxes |
(66,855 | ) | (16,831 | ) | (754,068 | ) | ||||||
| Income tax expense (recovery) |
(44 | ) | 6,037 | (12,892 | ) | |||||||
| Net loss |
$ | (66,811 | ) | $ | (22,868 | ) | $ | (741,176 | ) | |||
| Basic and diluted net loss per common share |
$ | (0.78 | ) | $ | (0.27 | ) | $ | (8.71 | ) | |||
For the year ended December 31, 2010, we recorded a net loss of $66.8 million ($0.78 basic and diluted net loss per common share), compared to a net loss of $22.9 million ($0.27 basic and diluted net loss per common share) for the year ended December 31, 2009. The $43.9 million increase in net loss is due to several factors, including: (i) the receipt of a $25.0 million one-time payment from Baxter International, Inc. (“Baxter”) in 2009, which did not recur in 2010; (ii) a $27.8 million decline in royalty revenue related to the lower sales of TAXUS by BSC, primarily as a result of increased competition in the stent market; (iii) a $1.6 million increase in research and development salary and benefit costs associated with new hires recruited to conduct research and development activities on certain of our Proprietary Medical Products; (iv) a $7.1 million increase in selling, general and administrative costs, primarily due to the addition of sales and marketing staff required to support our Quill Knotless Tissue-Closure Device product line; (v) $0.6 million of transaction expenses related to our 2010 acquisition of certain Haemacure assets, and $1.1 million of research and development costs incurred in connection with research and development activities performed on the related fibrin and thrombin technologies; (vi) $9.3 million of professional fees and other debt restructuring costs incurred in connection with the Recapitalization Transaction; (vii) $2.8 million of intangible asset impairment write-downs; (viii) an additional $3.2 million of property, plant and equipment impairment write-downs compared to 2009; (ix) $1.3 million of write-downs on our long term investments; and (x) a $3.2 million increase in the amortization of our deferred financing costs related to a change in the estimated useful lives of the expected interest period for the Subordinated Notes and a change in the expected life of the Existing Credit Facility, which has effectively been replaced and superseded by the DIP Facility. The unfavorable impact of the factors described above were partially offset by: (i) a $17.9 million increase in gross margin due to an increase in sales of our higher gross margin products as well as improvements achieved with respect to certain manufacturing costs, including reduced labor costs and more efficient utilization of our fixed overhead costs; (ii) a $4.5 million reduction in license and royalty fees associated with the simultaneous drop in royalty revenue noted above; (iii) a $4.7 million recovery from a litigation settlement, and a corresponding $3.8 million drop in related litigation expenses; (iv) $1.0 million of foreign exchange gains; (v) a $1.9 million gain on the settlement of the Haemacure loan upon completion of our acquisition of certain of their assets; (vi) a $2.0 million gain recognized from the sale of a manufacturing facility; and (vii) a $6.0 million tax recovery.
For the year ended December 31, 2009, we recorded a net loss of $22.9 million ($0.27 basic and diluted net loss per common share), compared to a net loss of $741.2 million ($8.71 basic and diluted net loss per common share) for the year ended December 31, 2008. The decrease in net loss of $718.3 million is primarily due to the write-down of goodwill of $649.7 million recorded in 2008. The remaining decrease in net loss of $68.6 million is due to several factors, including: (i) a $29.5 million decrease in research and development costs; (ii) a $17.0 million decrease in selling, general and administrative expenses, (iii) a $23.6 million decrease in write-downs of various investments; (iv) a $15.9 million decrease in deferred financing charges related primarily to the write-down of certain transaction costs incurred in 2008 totaling $16.5 million; (v) a $6.5 million decrease in interest expense due primarily to lower interest expense
75
Table of Contents
incurred on our Existing Floating Rate Notes”), and (vi) a $25.0 million payment received from our partner Baxter in the first quarter of 2009. These factors were offset by a $22.8 million net reduction in royalty revenue derived from BSC’s sales of paclitaxel-eluting coronary stent systems as compared to the comparable period in 2008 (net of license and royalty fees), and an income tax expense of $6.0 million recorded in 2009 as compared to an income tax recovery of $12.9 million recorded in 2008.
For the year ended December 31, 2008, we recorded a net loss from continuing operations of $741.2 million ($8.71 basic and diluted net loss per share) compared to net loss from continuing operations of $56.0 million ($0.66 basic and diluted net loss per share) for the year ended December 31, 2007. The increase in the net loss of $685.2 million is due primarily to the write-down of an aggregate of $649.7 million of goodwill recorded in 2008. Also increasing the net loss were (i) a $25.1 million reduction in royalty revenue, as BSC’s sales of paclitaxel-eluting coronary stent systems declined substantially in 2008 as compared to prior comparable periods; (ii) $16.5 million of transaction costs accrued related to the abandoned proposed financing transaction; (iii) an $18.6 million write-down or loss on redemption of two available-for-sale equity securities, one of which we disposed of, and the other of which was trading below cost for an extended period of time (as compared to $8.2 million in 2007); (iv) a $5.0 million write-down of a long term investment for which we could not obtain sufficient assurance we would be able to recover our carrying value; and (v) a $2.5 million payment for in-process research and development expense made during the first quarter of 2008 (as compared to $8.0 million in 2007). Partially offsetting the increase in the net loss was a $14.5 million increase in gross profit relating to higher medical products sales and a $7.2 million reduction in interest expense, as the interest rate applied to our Existing Floating Rate Notes declined.
Revenues
| Years ended December 31, | ||||||||||||
| (in thousands of U.S.$) |
2010 | 2009 | 2008 | |||||||||
| Medical Products: | ||||||||||||
| Product sales |
$ | 211,495 | $ | 191,951 | $ | 190,816 | ||||||
| Pharmaceutical Technologies: | ||||||||||||
| Royalty revenue — paclitaxel-eluting stents |
30,501 | 57,420 | 84,079 | |||||||||
| Royalty revenue — other |
3,960 | 4,751 | 7,467 | |||||||||
| License fees |
286 | 25,556 | 910 | |||||||||
| 34,747 | 87,727 | 92,456 | ||||||||||
| Total revenues |
$ | 246,242 | $ | 279,678 | $ | 283,272 | ||||||
We operate in two reportable segments:
Medical Products
Our Medical Products segment manufactures and markets a wide range of single-use specialty medical products and medical device components. These products are sold directly to end users or other third-party medical products manufacturers. This segment contains specialized direct sales and distribution capabilities as well as significant manufacturing capabilities. Many of our medical products are made using our proprietary manufacturing processes or are protected by our intellectual property.
Revenue from our Medical Products segment for the year ended December 31, 2010 was $211.5 million, compared to $192.0 million for the year ended December 31, 2009. The $19.5 million increase is attributable to a 17% increase in sales of our Proprietary Medical Products and a 7% increase in sales of our Base Medical Products.
Sales of our Proprietary Medical Products increased to $69.7 million during the year ended December 31, 2010 from $59.8 million during the year ended December 31, 2009. The 17% increase was primarily due to sales growth of certain of our newer product lines, which include our Quill Knotless Tissue-Closure Device product line and our Option IVC Filter. The revenue increase was partly offset by the elimination of revenue from our EnSnareTM product line at the beginning of 2010 in connection with the discontinuation
76
Table of Contents
of our licensing agreement with Hatch Medical, LLC. In 2009, we elected not to match a competing offer to purchase the distribution rights and as a result, we no longer sell EnSnare. Foreign currency changes, sales did not have a significant impact on the sales growth of our Proprietary Medical Products during the year ended December 31, 2010 as compared to the prior year.
Sales of our Base Medical Products increased to $141.8 million during the year ended December 31, 2010 from $132.1 million during the year ended December 31, 2009. The 7% increase is primarily due to sales growth of certain of our biopsy, dental suture and microsurgical knife product lines, as well as modest sales growth of medical device components that are manufactured by us and subsequently sold to third-party medical device manufacturers. Excluding the impact of foreign currency changes, sales of our Base Medical Products would have increased by 8% during the year ended December 31, 2010 as compared to the prior year.
Revenue from our Medical Products segment for the year ended December 31, 2009 was $192.0 million, an increase of 1% from the $190.8 million recorded for the same period in 2008. This slight increase was attributable to a 32% increase in sales of our Proprietary Medical Products, partly offset by a 9% decline in sales of our Base Medical Products.
Sales of our Proprietary Medical Products increased to $59.8 million during the year ended December 31, 2009 from $45.2 million during the year ended December 31, 2008, due primarily to higher sales of certain of our product lines, most significantly our Quill Knotless Tissue-Closure Device product line, SKATER product line, Option IVC Filter and Hemostream dialysis catheter. Sales of our Proprietary Medical Products during the year ended December 31, 2009 as compared to the same period in 2008 were also negatively impacted by foreign currency fluctuations. Excluding the impact of foreign currency changes, sales of our Proprietary Medical Products would have increased by 35% during the year ended December 31, 2009 as compared to the prior year.
The increase in sales of our Proprietary Medical Products was offset by a 9% decline in sales of our Base Medical Products to $132.1 million during the year ended December 31, 2009 from $145.6 million during the year ended December 31, 2008, due primarily to lower sales of medical device components to other third-party medical device manufacturers. During the year, certain of our customers elected to postpone or cancel orders, or implemented inventory reduction programs in response to changing economic and credit market conditions. In November 2008, we began the transfer of the manufacturing of surgical needles to our facility in Aguadilla, Puerto Rico from our facility in Syracuse, New York. We believe that the closure of our Syracuse production facility in November 2008 and the finalization of our move of surgical needle production to Aguadilla, combined with the difficult economic and credit market environment, negatively impacted our Base Medical Product sales during the year ended December 31, 2009 as compared to the prior year. Sales of our Base Medical Products were also impacted to a lesser extent by foreign currency fluctuations. Excluding the impact of foreign currency changes, sales of our Base Medical Products would have decreased by 8% during the year ended December 31, 2009 as compared to the prior year.
We believe that certain of our Proprietary Medical Products have a high potential for growth and expect that revenue in our Medical Products segment will continue to increase during 2011 as compared to 2010. This expected growth may be mitigated or offset by more limited growth of our Base Medical Products, in particular if our sales of medical devices and device components to third-party customers continue at lower than expected levels.
At the end of 2010, we terminated our licensing and distribution agreement related to our sale of the Hemostream dialysis catheter. We do not expect the elimination of the EnSnare and Hemostream product lines to have a material impact on our 2011 product sales growth as compared to 2010. Also, as of March 2011 we will conclude our licensing and distribution agreement related to our sale of the Option IVC Filter, as part of our settlement of litigation and arbitration proceedings with our partner Rex. The elimination of the Option IVC Filter product line is expected to negatively impact revenue growth of our Proprietary Medical Products in 2011.
77
Table of Contents
Pharmaceutical Technologies
Royalty revenue derived from sales of TAXUS by BSC for the year ended December 31, 2010 decreased by 47% compared to the year ended December 31, 2009. The decrease in royalty revenues is due to lower sales of TAXUS by BSC, primarily as a result of continuing competitive pressures in the market for drug-eluting coronary stents. Royalty revenue for the year ended December 31, 2010 was based on BSC’s net sales of $539 million for the period October 1, 2009 to September 30, 2010, of which $271 million was in the U.S., compared to net sales of $926 million for the same period in 2009, of which $411 million was in the U.S. The average gross royalty rate earned on BSC’s net sales for the year ended December 31, 2010 was 6.0% for sales in the U.S. and 5.3% for sales in other countries, compared to an average rate of 6.4% for sales in the U.S. and 6.1% for sales in other countries for corresponding period in 2009. Our average gross royalty rates are dictated by our tiered royalty rate structure for sales in certain territories including the U.S., E.U. and Japan. Our current year average gross royalty rates have therefore declined in connection with lower sales volumes of TAXUS in these territories.
Royalty revenue derived from sales of TAXUS by BSC for the year ended December 31, 2009 decreased by 32% as compared to the year ended December 31, 2008. The decrease in royalty revenues was a result of lower sales of TAXUS by BSC. Royalty revenue for 2009 was based on BSC’s net sales of $926 million for the period October 1, 2008 to September 30, 2009, of which $411 million was in the U.S., compared to net sales of $1.2 billion for the same period in 2008, of which $637 million was in the U.S. The average gross royalty rate earned in the year ended December 31, 2009 on BSC’s net sales was 6.4% for sales in the U.S. and 6.1% for sales in other countries, compared to an average rate of 7.1% for sales in the U.S. and 6.4% for sales in other countries for year ended December 31, 2008. Our average royalty rates declined during the year ended December 31, 2009 as compared to the same period in 2008 as a result of our tiered royalty rate structure for sales in the U.S., E.U. and Japan. In addition, the average gross royalty rate relating to sales in countries other than the U.S. declined in 2009 primarily due to an additional royalty payment we received during the first quarter of 2008 relating to royalties on sales outside of the U.S. that were owed from prior periods in 2007.
For the year ended December 31, 2010, other royalty revenue decreased by $0.8 million as compared to the year ended December 31, 2009 due to the elimination of royalty payments from Baxter in connection with our entry into the Baxter Distribution and License Agreement which was completed in 2009. The entry into the the Baxter Distribution and License Agreement also explains the $2.7 million decrease in other royalty revenue for the year ended December 31, 2009 as compared to the year ended December 31, 2008.
License fees decreased by $25.3 million for the year ended December 31, 2010 compared to the year ended December 31, 2009 primarily due to a $25.0 million one-time payment received in the first quarter of 2009 in connection with the the Baxter Distribution and License Agreement discussed above. The payment was recorded as revenue in 2009 because the Company had fulfilled its performance obligations and had no further ongoing involvement in the collaboration. Similarly, license fees increased by $24.6 million for the year ended December 31, 2009 as compared to the year ended December 31, 2008 due to the $25.0 million one-time payment discussed above.
Revenues in our Pharmaceutical Technologies segment will continue to decrease in 2011 as compared to revenues recorded in 2010, 2009 and 2008, primarily as a result of the continued competitive pressures in the market for drug-eluting coronary stent systems and the related potential decline in royalty revenues we derive from sales by BSC of TAXUS. These declines may be offset by royalty revenues we may receive from our partner Cook, should Cook receive approval to market and sell its Zilver-PTX stent in the U.S.
Expenditures
| Years ended December 31, | ||||||||||||
| (in thousands of U.S.$) |
2010 | 2009 | 2008 | |||||||||
| Cost of products sold |
$ | 106,304 | $ | 104,616 | $ | 101,052 | ||||||
| License and royalty fees |
5,889 | 10,431 | 14,258 | |||||||||
| Research and development |
26,790 | 23,701 | 53,192 | |||||||||
78
Table of Contents
| Years ended December 31, | ||||||||||||
| (in thousands of U.S.$) |
2010 | 2009 | 2008 | |||||||||
| Selling, general and administrative |
89,238 | 81,504 | 98,483 | |||||||||
| Depreciation and amortization |
33,745 | 33,251 | 33,998 | |||||||||
| In-process research and development |
— | — | 2,500 | |||||||||
| Write-down of assets held for sale |
1,450 | 3,090 | 1,283 | |||||||||
| Write-down of property, plant and equipment |
4,779 | — | — | |||||||||
| Write-down of intangible assets |
2,814 | — | — | |||||||||
| Write-down of goodwill |
— | — | 649,685 | |||||||||
| Escrow settlement recovery |
(4,710 | ) | — | — | ||||||||
| $ | 266,299 | $ | 256,593 | $ | 954,451 | |||||||
Cost of Products Sold
Cost of products sold is comprised of costs and expenses related to the production of our various medical devices, device components and biomaterial products and technologies, including direct labor, raw materials, depreciation and certain fixed overhead costs related to our various manufacturing facilities and operations.
Cost of products sold increased by $1.7 million to $106.3 million for the year ended December 31, 2010 compared to $104.6 million for the year ended December 31, 2009. The increase was due primarily to the higher aggregate sales in our Medical Products segment leading to higher direct manufacturing costs, as well as increases in royalty expenses relating to increased sales of certain of our Proprietary Medical Products, most specifically our Option IVC filter. These factors were partially offset by the positive impact of favorable manufacturing variances. In 2009, we incurred significant non-recurring costs due to manufacturing inefficiencies related to the transfer of certain manufacturing operations from our facilities in Syracuse, New York and Reading, Pennsylvania to our Aguadilla, Puerto Rico facility.
Consolidated gross margin for our Medical Products segment sales was 49.7% for the year ended December 31, 2010 compared to 45.5% for the year ended December 31, 2009. The increase in gross margins is primarily due to sales growth of certain higher margin product lines, labor cost savings resulting from our relocation of certain manufacturing operations to lower cost jurisdictions, and improved absorption of fixed manufacturing overhead costs resulting from higher sales volumes. These increases were partially offset by the impact of royalty expenses relating to increased sales of certain of our Proprietary Medical Products, most specifically our Option IVC filter.
Cost of products sold increased by $3.5 million to $104.6 million for the year ended December 31, 2009 compared to $101.1 million for the year ended December 31, 2008. The increase was due primarily (i) excess labor and other costs incurred during the first half of 2009 relating to surgical needle orders expected from a large customer that were subsequently cancelled; (ii) increases in royalty expense relating to increased sales of certain of our Proprietary Medical Products; specifically our Option IVC filter and Hemostream dialysis catheter; (iii) changes in our mix of product sales among our Base Medical Products to certain items that have higher production costs relative to the revenue they produced; (iv) added costs incurred to re-work certain product inventory due to packaging errors during the fourth quarter in our facilities in Stenlose, Denmark and Gainsville, FL, and (v) modest increases in distribution costs relating to the move of our E.U. product distribution activities to a new central location in Belgium from our previous location in Denmark. These factors were partially offset by lower labor and other production costs for surgical needles in our new facility in Aguadilla, Puerto Rico as compared to our old production facility in Syracuse, NY.
Gross margins for our Medical Products segment sales were 45.5% for year ended December 31, 2009 as compared to 47.1% for year ended December 31, 2008. Gross margins during 2009 as compared to 2008 were negatively impacted by the factors noted above. In addition, certain product sales strategies for our Base Medical Products, which are designed to increase sales volume and improve our utilization of fixed production capacity, negatively impacted gross margin percentage during 2009 most significantly during the fourth quarter of 2009. Partly offsetting the impact of these negative factors was a relative increase in sales of our higher gross margin Proprietary Medical Products.
79
Table of Contents
We expect that cost of products sold will continue to be significant and that gross margins may improve in 2011, primarily as a result of improved sales mix and specifically due to potential increases in sales of certain of our Proprietary Medical Products, in particular our Quill product line, that provide higher relative gross margins. These improvements may be offset by the impact of certain sales and pricing initiatives for our Base Medical Products designed to improve sales volume, market share, fixed manufacturing capacity utilization and overall operating cash flow.
At the end of 2010, we terminated our licensing and distribution agreement related to our sale of the Hemostream dialysis catheter. We do not expect the elimination of the Hemostream product line to have a material impact on our 2011 gross margin as compared to 2010. Also, as of March 2011 we will conclude our licensing and distribution agreement related to our sale of the Option IVC Filter, as part of our settlement of litigation and arbitration proceedings with our partner Rex. The elimination of the Option IVC Filter product line is expected to positively impact our gross profit margin due to the elimination of high product acquisition and royalty expense costs relating to this product.
License and Royalty Fees on Royalty Revenue
License and royalty fee expenses include license and royalty payments due to certain of our licensors, primarily relating to paclitaxel-eluting coronary stent system royalty revenue received from BSC. License and royalty fees decreased by $4.5 million to $5.9 million for the year ended December 31, 2010 compared to $10.4 million for year ended December 31, 2009. The decreases in 2010, 2009 and 2008 reflect the reduction in royalty revenue discussed above. We expect that license and royalty fees expense will decrease significantly in 2011, due to the elimination of future license and royalty fees payable to the NIH relating to future sales of TAXUS by BSC, per the amended NIH License Agreement. These decreases may be offset by license and royalty fees that would be due to NIH should Cook receive approval to market and sell its Zilver-PTX stent in the U.S. and record significant sales thereof in 2011.
Research and Development
Our research and development expense is comprised of costs incurred in performing research and development activities, including salaries and benefits, clinical trial and related clinical manufacturing costs, contract research costs, patent procurement costs, materials and supplies, and operating and occupancy costs. Our research and development activities occur in two main areas:
(i) Discovery and pre-clinical research. Our discovery and pre-clinical research efforts are divided into several distinct areas of activity, including screening and pre-clinical evaluation of pharmaceuticals and various biomaterials and drug delivery technologies, evaluation of mechanism of action of pharmaceuticals, mechanical engineering and pursuing patent protection for our discoveries.
(ii) Clinical research and development. Clinical research and development refers to internal and external activities associated with clinical studies of product candidates in humans, and advancing clinical product candidates towards a goal of obtaining regulatory approval to manufacture and market these product candidates in various geographies.
Research and development expenditures increased by $3.1 million to $26.8 million for the year ended December 31, 2010 as compared to $23.7 million for the same period in 2009. The increase is primarily due to the following factors: (i) higher salaries and benefit costs incurred by personnel conducting research and development activities on certain of our Proprietary Medical Products; and (ii) $1.1 million of costs related to new personnel retained, and occupancy costs incurred, in connection with our acquisition of assets from Haemacure (see the Acquisitions section above).
Research and development expenditures decreased significantly to $23.7 million for the year ended December 31, 2009 as compared to $53.2 million for the year ended December 31, 2008. The decrease was primarily due to the suspension of enrollment in our Vascular Wrap clinical trial, a reduction of staffing within our clinical and research departments and reduction in other direct costs in the second half of 2008 relating to the postponement or elimination of other selected research programs and activities.
80
Table of Contents
We expect that our research and development expenditures in 2011 will be lower as compared to 2010, as we conclude or cancel certain programs, and due to reduction in certain research and development headcount. Although we expect the level of research and development expenditures to be lower in 2011 as compared to 2010, we anticipate we may continue to incur significant research and development expenditures in 2011 and in future periods.
Selling, General and Administrative Expenses
Our selling, general and administrative expenses are comprised of direct selling and marketing costs related to the sale of our various medical products, including salaries, benefits and sales commissions, and our various management and administrative support functions, including salaries, commissions, benefits and other operating and occupancy costs.
Selling, general and administrative expenditures increased by $7.7 million to $89.2 million for the year ended December 31, 2010 compared to $81.5 million for the year ended December 31, 2009. The increase is primarily due to the following factors: (i) a $6.1 million rise in salaries and benefits related to new sales and marketing personnel hired to expand sales of certain of our Proprietary Medical Products, most specifically relating to our Quill product line; (ii) a $1.3 million increase in travel costs attributed to the increased personnel; and (iii) a $3.8 million increase in operating costs relating to the increased personnel. The net $11.2 million increase in these costs was offset by an overall $3.8 million decrease in legal expenses, primarily related lower litigation costs due to a settlement of certain litigation as compared to 2009.
Selling, general and administrative expenditures were $81.5 million for the year ended December 31, 2009 as compared to $98.5 million for the same period in 2008. The lower expenditures during 2009 were primarily due to reduced severance costs and salaries and benefits as a result of the reduction in staff in selling, general and administrative departments completed in the first and third quarters of 2008, lower professional fees as well as reduced severance and other restructuring charges related to the closure and consolidation of our Syracuse, NY manufacturing facility completed in the fourth quarter of 2008.
We expect that selling, general and administrative expenses in 2011 will be consistent with the levels of such expenses incurred in 2010. These expenditures could fluctuate depending on product sales levels, the timing of the launch of certain new products, growth of new product sales, unforeseen litigation or other legal expenses that may be incurred to support and defend our intellectual property portfolio or other aspects of our business.
Depreciation and Amortization
Depreciation and amortization is comprised of depreciation expense of property, plant and equipment and amortization expense of licensed technologies and identifiable assets purchased through business combinations.
Depreciation and amortization expense of $33.7 million for the year ended December 31, 2010 was comparable to $33.3 million in 2009 and $34.0 million for the year ended December 31, 2008.
We expect depreciation and amortization expense during 2011 to be lower than 2010 due to impairment write-downs recorded on our intangible assets and property, plant and equipment during the year ended December 31, 2010.
In-process Research and Development (“IPR&D”)
We record IPR&D expense relating to acquired or in-licensed technologies that are at an early stage of development and have no alternative future use. We incurred no IPR&D expense during the years ended December 31, 2010 and December 31, 2009. For the year ended December 31, 2008, we recorded IPR&D expense of $2.5 million for an initial license payment made to Rex Medical to obtain the marketing rights for the Option IVC Filter.
We may incur further IPR&D expenditures in future periods in the event we in-license or acquire additional early stage technologies.
81
Table of Contents
Write-down of Assets Held For Sale; Property, Plant and Equipment and Intangible Assets
During the year ended December 31, 2010, we recorded intangible asset impairment write-downs of $2.8 million (December 31, 2009 – nil, December 31, 2008 – nil) related to (i) the termination of our 2008 License, Supply, Marketing and Distribution a licensing agreement with Rex (see Significant Recent Developments for additional information) and (ii) our temporary suspension of research and development activities related to our fibrin and thrombin technologies which were acquired from Haemacure in 2010 (see Acquisitions for additional information).
During the year ended December 31, 2010, we recorded property, plant and equipment impairment write-downs of $4.8 million related to our suspension of certain research and development activities, including those related to our fibrin and thrombin technologies as discussed above (December 31, 2009 – nil, December 31, 2008 – nil).
During the year ended December 31, 2008 and in connection with the Company’s capacity rationalization and consolidation plans, the Company reclassified three of its properties as held for sale. Based on the Company’s annual asset impairment assessment, the Company recorded impairment charges of $1.5 million, $3.1 million and $1.3 million for the years ended December 31, 2010, December 31, 2009 and December 31, 2008, respectively, to adjust the carrying values of the properties to the lower of cost and fair value less estimated selling costs. In early 2010, one of these properties was sold for gross proceeds of $0.8 million with no resulting gain or loss. As at December 31, 2010, the two remaining properties failed to meet the held-for-sale classification criteria defined under ASC No. 360-10 – Property, Plant and Equipment. As a result, the $3.0 million carrying value of the two remaining properties was reclassified back into Property, Plant and Equipment.
Escrow Settlement Recovery
As noted above, during the year ended December 31, 2010 we received $4.7 million in full settlement of an outstanding litigation matter with RoundTable. The proceeds were recorded as a non-recurring escrow settlement recovery in 2010.
Write-down of Goodwill
As a result of a significant and sustained decline in our public stock market capitalization during 2008, we determined that our goodwill was impaired and recorded an impairment charge of $649.7 million during year ended December 31, 2008. Accordingly, we had no goodwill remaining on our balance sheet as of December 31, 2008, 2009 and 2010. We utilized the income approach in determining the implied fair value of the goodwill, which requires estimates of future operating results and cash flows discounted using estimated discount rates.
Other Income (Expense)
| Years ended December 31, | ||||||||||||
| (in thousands of U.S.$) |
2010 | 2009 | 2008 | |||||||||
| Foreign exchange (loss) gain |
$ | 1,011 | $ | (1,612 | ) | $ | 540 | |||||
| Investment and other expense (income) |
(571 | ) | 378 | 1,192 | ||||||||
| Debt restructuring costs |
(9,277 | ) | — | — | ||||||||
| Interest expense on long term-debt |
(40,258 | ) | (38,039 | ) | (44,490 | ) | ||||||
| Write-down of deferred financing charges |
(291 | ) | (643 | ) | (16,544 | ) | ||||||
| Write-down or net losses on redemption of investments |
(1,297 | ) | — | (23,587 | ) | |||||||
| Loan settlement gain |
1,880 | — | — | |||||||||
| Gain on sale of assets |
2,005 | — | — | |||||||||
| $ | (46,798 | ) | $ | (39,916 | ) | $ | (82,889 | ) | ||||
82
Table of Contents
Net foreign exchange gains and losses were primarily the result of changes in the relationship of the U.S. to Canadian dollar and other foreign currency exchange rates when translating our foreign currency denominated cash, cash equivalents and short-term investments to U.S. dollars for reporting purposes at period end. We continue to hold foreign currency denominated cash and cash equivalents to meet our anticipated operating and capital expenditure needs in future periods in jurisdictions outside of the U.S. We do not use derivatives to hedge against exposures to foreign currency arising from our balance sheet financial instruments and therefore are exposed to future fluctuations in the U.S. dollar to foreign currency exchange rates.
Investment and other income for the year ended December 31, 2010 decreased by $1.0 million when compared to the year ended December 31, 2009. The decrease is primarily due to a $0.3 million write-off of leasehold improvements for a leased property that is no longer in use, a $0.2 million increase in losses on asset disposals, and lower interest income received in 2010 due to lower excess cash balances available for investment. Investment and other income for the year ended December 31, 2009 of $0.4 million represented a decrease of $0.8 million when compared to investment and other income of $1.2 million for the same period in 2008, primarily as a result of lower interest income received in 2009 due to lower excess cash balances available for investment.
During the year ended December 31, 2010, we incurred debt restructuring costs of $9.3 million for fees and expenses related to the Recapitalization Transaction. These fees and expenses represent professional fees paid to both our, and the Consenting Noteholders’ financial and legal advisors to assist in the analysis of financial and strategic alternatives. Over the next three to six months, we expect to continue to incur significant costs related to the execution and completion of the proposed restructuring transaction as well as the exploration of other financial and strategic alternatives. For more information see notes 1, 3, 17 and 27 under the Notes to the Consolidated Financial Statements included in Part IV of the 2010 10-K.
We incurred interest expense of $40.3 million and $38.0 million for the years ended December 31, 2010 and December 31, 2009, respectively. For the year ended December 31, 2010, interest expense consists of $32.9 million of interest incurred; related to our Existing Floating Rate Notes, Subordinated Notes and Existing Credit Facility; $0.7 million of interest expense incurred from royalties owing to NIH; and $6.7 million of amortization of deferred financing costs. Similarly, interest expense for the year ended December 31, 2009 consists of $35.2 million of interest incurred; related to our Existing Floating Rate Notes and Subordinated Notes; and $2.8 million of amortization of deferred financing costs. The $2.3 million decline in interest expense on our long term debt obligations is primarily due to a decline in LIBOR rates, which are used to derive the interest rate applicable to our Existing Floating Rate Notes. The average interest rate for the year ended December 31, 2010 was 4.1% compared to 4.7% for the year ended December 31, 2009.
During the year ended December 31, 2009, we incurred interest expense of $38.0 million on our outstanding long-term debt obligations, as compared to $44.5 million for the year ended December 31, 2008. The decrease resulted from a decline in the interest rate applicable on our Existing Floating Rate Notes due to lower LIBOR rates. The interest rate decline resulted in an average interest rate of 4.7% on our Existing Floating Rate Notes for the year ended December 31, 2009 as compared to 7.7% for the year ended December 31, 2008. Interest expense also includes $2.8 million and $2.3 million for amortization of deferred financing costs for the year ended December 31, 2009 and 2008, respectively.
During the year ended December 31, 2010, we recorded a $0.3 million impairment write-down of deferred financing costs related to our 2009 shelf registration statement filed with the SEC. Write-down and other deferred financing charges for the year ended December 31, 2009 related to a charge of $0.6 million resulting from the write-off of the portion of the deferred financing charges related to our term loan facility with Wells Fargo that terminated on May 29, 2009. During the year ended December 31, 2009, we wrote-down $16.5 million of deferred financing costs related to the exploration of certain proposed financing and strategic alternatives that were subsequently abandoned.
During the year ended December 31, 2010, we recorded $1.3 million of write-offs related to our long term investments in three private biotechnology companies that were determined to be irrecoverable. During the year ended December 31, 2009, there were no write-downs of long term investments.
83
Table of Contents
The write-down or net loss on redemption of investments for the year ended December 31, 2008 of $23.6 million is comprised of a loss of $8.8 million realized on the sale of our common stock holdings in CombinatoRx, Inc., an unrealized loss of $9.8 million on an available-for-sale equity security that had been trading below carrying value for an extended period and for which management does not intend to hold for a sufficient period of time to reasonably expect a recovery to initial carrying value and an unrealized loss of $5.0 million on the write-down in carrying value of shares held in a private biotechnology company classified as long term investments. The write-down or net loss on redemption of investments for the year ended December 31, 2007 of $8.2 million is comprised of a loss of $9.6 million realized on the sale of our common stock holdings in Orthovita, Inc., partially offset by a gain of $1.4 million realized on the sale of our common stock holdings in NuVasive, Inc.
During the year ended December 31, 2010, we recorded a $1.9 million loan settlement gain relating to the settlement of the loan with Haemacure in connection with the acquisition of Haemacure’s assets as described under “Acquisitions” above.
During the year ended December 31, 2010, we sold our Laguna Hills manufacturing operations, which is part of our Medical Products operating segment. The sale included certain assets and liabilities for total net proceeds of $2.8 million. The Company recorded a $2.0 million gain from the sale of the net assets under consideration.
Income Tax
Income tax recovery for the year ended December 31, 2010 was $0.04 million compared to income tax expense of $6.0 million for the year ended December 31, 2009. The income tax recovery for 2010 is primarily due to a recovery on the amortization of identifiable intangible assets.
The effective tax rate for the current period differs from the statutory Canadian tax rate of 28.5% (30% in 2009) and is primarily due to valuation allowances on net operating losses, the net effect of lower tax rates on earnings in foreign jurisdictions, and permanent differences not subject to tax.
For the year ended December 31, 2010, income tax recovery of $0.04 million consisted of a current and deferred income tax recovery of $0.16 million on a net loss from Canadian operations and a current and deferred income tax expense of $0.12 million on net income from U.S. and foreign operations.
Income tax expense for the year ended December 31, 2009 was $6.0 million compared to income tax recovery of $12.9 million for the year ended December 31, 2008. The income tax expense for 2009 is primarily due to a net income from U.S. and foreign operations and includes the recognition of a $6.1 million deferred tax charge relating to taxes paid in prior years in connection with an inter-company transfer of the intellectual property underlying the transaction with Baxter.
For the year ended December 31, 2009, income tax expense of $6.0 million consisted of a current and deferred income tax expense of $0.6 million on a net loss from Canadian operations and a current and deferred income tax expense of $5.4 million on net income from U.S. and foreign operations.
Liquidity and Capital Resources
At December 31, 2010, we had a working capital deficiency of $518.7 million and cash and cash equivalents of $33.3 million. For the year ended December 31, 2010, our working capital decreased by $585.3 million as compared to December 31, 2009, primarily due to the reclassification of $575.0 million of long-term debt to current (see note 17 of our audited consolidated financial statements for the year ended December 31, 2010), decreases in cash and cash equivalents, short-term investments, accounts payable and accrued liabilities, and income taxes payable, partly offset by increases in accounts receivable and prepaid expenses and other assets.
84
Table of Contents
Our most significant financial liabilities are our Existing Floating Rate Notes and our Subordinated Notes, which together comprise $575.0 million in aggregate principal amount of our outstanding indebtedness. As discussed, these debt obligations were originally supported in substantive part by royalty revenue derived from sales of TAXUS coronary stent systems by BSC. The sustained and significant decline in royalty revenues received from BSC over the past several years has had a significant negative impact on our liquidity and capital resources, and therefore on our ability to service our long term debt obligations. As a result, we have, over the past several years, explored a range of financial and strategic alternatives in order to address these circumstances. After extensive exploration of various possible solutions, we recently announced a Recapitalization Transaction, negotiated with a significant majority of the holders of our outstanding indebtedness, to effectuate a significant reduction in our total debt and thereby substantially improve our liquidity, working capital and financial position.
We did not make our $9.7 million interest payment due to holders of the Subordinated Notes on October 1, 2010. Subsequently, on October 29, 2010, we entered into the RSA with the Consenting Noteholders to, among other things, eliminate $250.0 million of our outstanding indebtedness and the related interest expenses associated therewith by exchanging Subordinated Notes for new common shares in the Company. As at March 14, 2011, holders of approximately 85% of the Subordinated Notes had agreed to the terms of the RSA.
On October 29, 2010, we also entered into the FRN Support Agreement, and, in accordance with the terms thereof, we commenced the FRN Exchange Offer on February 10, 2011, pursuant to which we offered to exchange, our Existing Floating Rate Notes for New Floating Rate Notes. The New Floating Rate Notes will be issued on substantially the same terms and conditions as the Existing Floating Rate Notes, except that, among other things: (i) subject to certain exceptions, the New Floating Rate Notes will be secured by second-priority liens over substantially all of the assets, property and undertakings of Angiotech and certain of its subsidiaries; (ii) the New Floating Rate Notes will accrue interest at LIBOR plus 3.75%, subject to a LIBOR floor of 1.25%; and (iii) certain covenants related to the incurrence of additional indebtedness and asset sales and the definitions of permitted liens, asset sales and change of control will be modified in the New Notes Indenture. If the FRN Exchange Offer is consummated, any Existing Floating Rate Notes not tendered in the FRN Exchange Offer will remain outstanding and subject to the terms of the FRN Indenture; as amended by the terms of the FRN Support Agreement. As at March 14, 2011, approximately 89% of the Existing Floating Rate Noteholders have agreed to the terms of the FRN Support Agreement, including the FRN Exchange Offer.
The Company obtained approval from the Consenting Noteholders to implement the Recapitalization Transaction through the CCAA Plan and the Angiotech Entities voluntarily commenced the CCAA Proceedings and Chapter 15 Cases on January 28, 2011 and January 30, 2011, respectively (for more information, refer to the Significant Recent Developments section above).
Pursuant to the terms of the SSN Indenture, the Company initially had a 30-day grace period from October 1, 2010 to make the required interest payment before an event of default occurred and the Subordinated Noteholders could exercise their right under the SSN Indenture to demand repayment of all obligations. Pursuant to the terms of the supplemental indenture entered into by Angiotech and U.S. Bank National Association, as successor trustee under the SSN Indenture, at the direction of the holders of a majority in aggregate principal amount outstanding of the Subordinated Notes, on January 27, 2011, the grace period applicable to interest payments on the Subordinated Notes was extended to 211 days. However, given that the initiation of the Creditor Protection Proceedings qualifies as an immediate event of default under the terms of our outstanding indebtedness, all obligations under (i) the Existing Credit Facility, (ii) the SSN Indenture and (iii) the FRN Indenture, have become due and payable under the terms of the respective agreements. The stay of proceedings granted in connection with the Creditor Protection Proceedings effectively prohibits these creditors from taking action to remedy or enforce their rights thereunder. The enforcement rights of these creditors are also subject to the applicable provisions of Chapter 15 and the CCAA. Without the applicable extensions or protection granted by the stay of proceedings, an event of default under the SSN Indenture would permit the holders of 25% or more of the aggregate outstanding principal amount of the Subordinated Notes to demand immediate repayment of amounts outstanding under the Subordinated Notes, other than for certain events of default resulting from bankruptcy matters, which would accelerate amounts outstanding immediately and without further action. Similarly, an event of default under the FRN Indenture arising from a cross-default failure to make the interest payment on the Subordinated Notes within the applicable grace period would permit the holders of 25% or more of the aggregate outstanding principal amount of the Existing Floating Rate Notes to demand immediate repayment of amounts outstanding under the Existing Floating Rate Notes.
85
Table of Contents
As at December 31, 2010, our Existing Credit Facility provided for borrowings in an amount up to $35.0 million, which includes an additional $10.0 million of temporary supplemental borrowing provided under the Fourth Amendment (as defined below). The amount we are able to borrow under the Existing Credit Facility at any given time is limited by a borrowing base formula, derived from the value of certain of our subsidiaries’ finished good inventory, accounts receivable, certain real property, securities and intellectual property owned by the Company and its subsidiaries, and therefore fluctuates from month to month. As of December 31, 2010, the amount of financing available under the Existing Credit Facility was approximately $14.8 million, which is net of $2.8 million in letters of credit issued under the Existing Credit Facility. As of December 31, 2010, $10.0 million of advances were outstanding under the Existing Credit Facility.
Borrowings outstanding under the Existing Credit Facility bear interest at an annual rate ranging from LIBOR, for a period selected by the Company of 1, 2 or 3 months, plus 3.25% to LIBOR plus 3.75%, with a minimum LIBOR of 2.25%. As the minimum LIBOR under the Existing Credit Facility is 2.25% and LIBOR for periods of 1, 2 and 3 months was, in each case, less than 0.50% on December 31, 2010, a 0.50% increase or decrease in the LIBOR rate as of December 31, 2010 would have no impact on interest payable under the Existing Credit Facility.
The utilization of the Existing Credit Facility is subject to, among other terms and conditions, certain restrictive covenants pertaining to operations and the maintenance of minimum levels of adjusted earnings before interest, taxes, depreciation and amortization (“Adjusted EBITDA”) and fixed charge coverage ratios. Under the terms of the Existing Credit Facility, a breach of any of these covenants may result in our inability to draw on the facility or the immediate repayment of any then outstanding principal and interest. In April 2010 we completed an amendment to the Existing Credit Facility, which included, among other items, amendments to financial covenants pertaining to minimum EBITDA levels, fixed charge coverage ratios and the definition of Adjusted EBITDA. This amendment allow us continued access to funds per the terms of the Existing Credit Facility, and are primarily intended to reflect the continued decline and uncertainty of sales of TAXUS by BSC and the related potential impact on our Adjusted EBITDA.
In connection with the Company’s failure to make the $9.7 million interest payment owing to the Subordinated Noteholders on October 1, 2010, the Company entered into the Forbearance Agreement to preserve the Existing Credit Facility. On November 4, 2010, the Company entered into the Amended and Restated Forbearance Agreement pursuant to which Wells Fargo agreed, subject to certain terms and conditions, to continue to forbear from exercising its rights and remedies under the Existing Credit Facility in order to allow us an opportunity to implement the Recapitalization Transaction. Under the terms of the Amended and Restated Forbearance Agreement, Wells Fargo, among other things, agreed not to immediately exercise any rights or remedies it has related to:
| (a) | the Company’s failure to make its interest payment due to the Subordinated Noteholders on October 1, 2010; |
| (b) | the Company’s litigation with QSR Holdings; or |
| (c) | any future failure to deliver an unqualified audit opinion with respect to the going concern assumption for the fiscal year ending December 31, 2010. |
While the Amended and Restated Forbearance Agreement has not been terminated, Wells Fargo’s obligations thereunder, including its agreement not to exercise certain remedies, ceased to be of effect upon our commencement of the CCAA Proceedings. Wells Fargo’s rights and remedies under the Existing Credit Facility, however, were nonetheless stayed in connection with the Creditor Protection Proceedings.
On November 4, 2010, we entered into an amendment to our Existing Credit Facility (the “Fourth Amendment”) to provide Angiotech with increased liquidity to facilitate the completion of the
86
Table of Contents
Recapitalization Transaction. In accordance with the terms of our Creditor Protection Proceedings, on February 7, 2011, we and certain of our subsidiaries (the “Borrowers”) entered into a definitive agreement with Wells Fargo to secure the DIP Facility. On February 25, 2011, following the entry of the February 22, 2011 Recognition Order the loans under the DIP Facility were made available to the Company for borrowing. The DIP Facility provides for up to $28.0 million in aggregate principal amount (subject to a borrowing base and certain reserves) of revolving loans to fund working capital requirements and implementation costs during the Recapitalization Transaction. The DIP Facility effectively supersedes and terminates use of the Existing Credit Facility beyond February 7, 2011. Under the terms of the DIP Facility, we are required to apply our domestic post-petition cash collections of the Angiotech Entities to the pre-petition Existing Credit Facility until all obligations outstanding under the Existing Credit Facility are paid in full. In order to satisfy this requirement, we are required to implement control agreements over bank accounts comprising a substantial portion of our cash balance. These control agreements require the Company to periodically sweep receipts from certain of our deposit accounts into Wells Fargo’s account until such time as the pre-petition obligations owing under the Existing Credit Facility have been fully repaid. The DIP Facility was issued on substantially the same terms as the Existing Credit Facility, subject to certain changes to reflect our filing for protection under the Creditor Protection Proceedings and will be used to fund daily cash needs prior to the repayment in full of the pre-petition Existing Credit Facility.
Borrowings under the DIP Facility are also subject to a borrowing base formula based on certain balances of: eligible inventory, accounts receivable, real property, securities and intellectual property, net of applicable reserves. In accordance with the agreed upon budget provided to Wells Fargo, the proceeds of the DIP Facility may only be used to pay for working capital requirements, capital expenditures and transaction fees and expenses incurred in connection with the Recapitalization Transactionre and the establishment of the DIP Facility. The proceeds may not be used to pay any debt outstanding prior to the date of the Initial Order, January 28, 2011, including those arising under the Existing Credit Facility.
Borrowings under the DIP Facility are secured by certain assets held by certain of our subsidiaries (the “DIP Guarantors”). Under the terms of the DIP Facility, our DIP Guarantors irrevocably and unconditionally jointly and severally guarantee: (a) the punctual payment; whether at stated maturity, by acceleration or otherwise; of all borrowings; including principal, interest, expenses or otherwise; when due and payable; and (b) the fulfillment of and compliance with all terms, conditions and covenants under the DIP Facility and all other related loan documents. Our DIP Guarantors also agreed to pay any and all expenses that Wells Fargo may incur in enforcing its rights under the guarantee.
The principal amount of the loans outstanding under the DIP Facility, plus interest accrued and unpaid thereon will be due and payable in full at maturity, which is defined as the earliest of:
| (a) | June 30, 2011; |
| (b) | the date of implementation of the CCAA Plan that has been sanctioned by an order of the Canadian Court and recognized by a recognition order of the US Bankruptcy Court; |
| (c) | the date immediately preceding the date on which any distributions under any the CCAA Plan commence; |
| (d) | the date of a sale of substantially all of the assets of Borrowers and the DIP Guarantors (which may take the form of an asset sale, stock sale or otherwise as approved by Wells Fargo); |
| (e) | the date that is 28 days after the Filing Date, if the Recognition Order has not been entered by the US Bankruptcy Court; and |
| (f) | such earlier date on which all advances and other obligations for the payment of money shall become due and payable in accordance with the terms of the DIP Facility and the other Loan Documents (as defined in the DIP Facility). |
Borrowings under the DIP Facility bear interest at a rate equal to, at our option, either (a) the Base Rate (as defined in the DIP Facility) plus either 3.25% or 3.50%, depending on the availability we have under the DIP Credit Agreement on the date of determination or (b) LIBOR plus either 3.50% or 3.75%, depending on the availability we have under the DIP Facility on the date of determination. If we default on our obligations under the DIP Facility, Wells Fargo may choose to implement a default rate of an additional 2%
87
Table of Contents
above the already applicable interest rate. In addition to interest, we are also required to pay an unused line fee of 0.5% per annum in respect to the unutilized commitments. In addition to certain customary affirmative and negative covenants, the DIP Facility requires us to comply with a specified minimum Adjusted EBIDTA and a minimum fixed charge coverage ratio. In addition to certain customary events of default, the DIP Facility includes defined events of default related to the progress of the Creditor Protection Proceedings. While we are currently in compliance with the Adjusted EBITDA and fixed charge covenants specified under the Existing Credit Facility and DIP Facility, we cannot guarantee that we will be able to comply with these covenants and related conditions throughout the remainder of 2011 and beyond. A breach of these covenants, our failure to obtain waivers to cure any further breaches of the covenants and the restrictions set forth under the DIP Facility, our failure to complete the Recapitalization Transaction or the FRN Exchange Offer or our failure to obtain required extensions of the Creditor Protection Proceedings and the stay of proceedings granted in connection therewith, may limit our ability to obtain additional advances or result in demands for accelerated repayment, thus further straining our working capital position and ability to continue operations.
Our cash resources and any borrowings available under the Existing Credit Facility and DIP Facility, in addition to cash generated from operations or cash available per commitments of certain of our creditors, are used to support our continuing sales and marketing initiatives, working capital requirements, debt servicing requirements, clinical studies, research and development initiatives and for general corporate purposes.
In accordance with the terms of the RSA, upon consummation of the Recapitalization Transaction, we will replace our Existing Credit Facility and our DIP Facility with the proposed Exit Facility to support ongoing liquidity needs. As at March 14, 2011, the terms of the Exit Facility have not yet been determined.
Due to numerous factors that may impact our future cash position, working capital and liquidity as discussed below, and the significant cash that will be necessary to continue to execute our business plan, including selected growth and new product development initiatives in our Medical Products segment, initiatives to maintain sales of our Base Medical Products and to service our debt obligations, there can be no assurance that we will have adequate liquidity and capital resources to satisfy our future financial obligations, including obligations under the New Floating Rate Notes.
Our cash flows, cash balances and liquidity position are subject to numerous uncertainties, including but not limited to our ability to complete the Recapitalization Transaction and the FRN Exchange Offer, changes in drug-eluting coronary stent markets, including the impact of increased competition in such markets and research relating to the efficacy of drug-eluting stents and the sales achieved in such markets by BSC, the timing and success of product sales and marketing initiatives and new product launches, the timing and success of our research, product development and clinical trial activities, the timing of completing certain operational initiatives including but not limited to facility closures, our ability to effect reductions in certain aspects of our budgets in an efficient and timely manner, changes in interest rates, fluctuations in foreign exchange rates and regulatory or legislative changes.
In particular, as our royalties received from BSC have continued to decline significantly as compared to prior periods, and given that the level of such royalties significantly impact our operating cash flows, liquidity position and cash balances immediately in the period they are recorded, our current liquidity and capital resources have been materially adversely affected by the decline in royalties.
As a result of our recent Creditor Protection Proceedings and liquidity concerns, there is substantial doubt about our ability to continue operations as a going concern. Our ability to continue operations is dependent on our ability to: (i) execute and complete the Recapitalization Transaction and the FRN Exchange Offer as described above and in note 16 of the audited consolidated financial statements; (ii) continue servicing our debt obligations, including future obligations under the New Floating Rate Notes and the Exit Facility; (iii) remain in compliance with our debt covenants with respect to our DIP Facility and subsequent Exit Facility; (iv) obtain additional equity and debt financing; (v) achieve revenue growth and improvement in our operating cash flows and liquidity; and (vi) achieve greater operating efficiencies or a reduction of our expenses.
88
Table of Contents
Our current initiatives and future plans to manage our operating, going concern and liquidity risks include, but are not limited to the following:
| • | execution and completion of the Recapitalization Transaction and the FRN Exchange Offer to reduce the burden of existing debt levels and interest expense; |
| • | maintenance and utilization of the DIP Facility with Wells Fargo and negotiation and finalization of the Exit Facility; |
| • | continued exploration of various other strategic and financial alternatives, including but not limited to raising new equity or debt capital or exploring sales of various assets or businesses; |
| • | continued evaluation of budgets and forecasts for certain sales and marketing and research and development programs; |
| • | selected gross margin improvement initiatives; |
| • | potential sale of non-productive property, plant and equipment assets; and |
| • | optimization of day-to-day operating cash flows. |
We continue to closely monitor and manage liquidity by regularly preparing rolling cash flow forecasts, monitoring the condition and value of assets available to be used as security in financing arrangements, seeking flexibility in financing arrangements and establishing programs to maintain compliance with the terms of financing agreements.
Although we have entered into various agreements with the intention to recapitalize the Company, there is no assurance that we will be able to successfully execute and complete the Recapitalization Transaction. The Recapitalization Transaction is subject to governmental, court, regulatory, shareholder and third party approvals. In the event that we are unable to successfully complete the Recapitalization Transaction prior to the expiration of the Stay Period, the Subordinated Noteholders, holders of the Company’s Existing Floating Rate Notes and Wells Fargo may elect to exercise provisions under their respective indentures or credit agreements allowing them to accelerate repayment and, in the case of Wells Fargo under the Company’s Existing Credit Facility and DIP Facility, refuse to extend further credit to the Company. In this case, the Company’s current cash and credit capacity would not be sufficient to service its principal and interest obligations under its outstanding indebtedness. In addition, restrictions on the Company’s ability to borrow and the possible accelerated demand of repayment of existing or future obligations by any of the Company’s creditors might jeopardize the Company’s working capital position and ability to sustain its operations.
While we believe that we have developed planned courses of action and identified opportunities to mitigate the operating, going concern and liquidity risks outlined above, there is no assurance that we will be able to complete any or all of the plans or initiatives that have been identified or obtain sufficient liquidity to execute our business plan. Furthermore, there may be other material risks and uncertainties that may impact our liquidity position that have not yet been identified.
89
Table of Contents
Cash Flow Highlights
| (in thousands of U.S.$) |
Years ended December 31, | |||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Cash and cash equivalents, beginning of year |
$ | 49,542 | $ | 38,952 | $ | 91,326 | ||||||
| Cash (used in) provided by operating activities |
(21,609 | ) | 21,971 | (38,007 | ) | |||||||
| Cash (used in) investing activities |
(3,695 | ) | (8,187 | ) | (8,239 | ) | ||||||
| Cash provided by (used in) financing activities |
9,342 | (3,400 | ) | (4,378 | ) | |||||||
| Effect of exchange rate changes on cash |
(265 | ) | 206 | (1,750 | ) | |||||||
| Net increase (decrease) in cash and cash equivalents |
(16,227 | ) | 10,590 | (52,374 | ) | |||||||
| Cash and cash equivalents, end of year |
$ | 33,315 | $ | 49,542 | $ | 38,952 | ||||||
Cash Flows from Operating Activities
Cash used in operating activities for the year ended December 31, 2010 was $21.6 million compared to cash provided by operating activities of $22.0 million for the year ended December 31, 2009. The $43.5 million decrease in cash provided by operating activities is primarily due to the following factors: (i) a $25.0 million cash payment received from Baxter in the first quarter of 2009; (ii) a $27.8 million decline in royalty revenue related to lower sales of TAXUS by BSC; (iii) $7.2 million paid for transaction and professional fees and expenses related to our Recapitalization Transaction; and (iv) a $10.3 million increase in net working capital requirements, offset by: (i) improved gross margins of $17.9 million; (ii) $4.7 million received in settlement of a litigation matter; (iii) $2.1 million received from a government grant; and (iv) $2.0 million of proceeds received in connection with the sale of our Laguna Hills manufacturing facility. Working capital changes during the year ended December 31, 2010 included net cash outflows of $5.6 million related to the increase in accounts receivable associated with sales growth of certain of our higher margin Proprietary Products and net cash outflows $2.3 million from accounts payable and accrued liabilities due to payments made during the period as well as the reclassification of an accrual for certain license and royalty fees owing to one of our licensors from current to long term due to an amendment of the contractual payments terms . Other working capital changes include net cash inflows of $9.8 million related to our decision to defer our October 1, 2010 interest payment in connection with the Recapitalization Transaction, and net cash outflows of $8.6 million relating to income taxes payable.
Cash provided by operating activities for the year ended December 31, 2009 was $22.0 million compared to cash used in operating activities of $38.0 million for the year ended December 31, 2008. The increase in cash provided by operating activities of $60.0 million was due primarily to: (i) the one-time cash payment of $25.0 million received from Baxter in the first quarter of 2009; (ii) a lower net loss resulting from reduced research and development and selling, general and administrative costs, offset by a reduction of royalty revenue; and (iii) a $25.7 million improvement in cash from changes in working capital. Working capital changes during the year ended December 31, 2009 included net cash outflows from accounts receivable of $2.1 million due primarily to sales growth of our Quill SRS product line and our Option IVC Filter during the year, net cash outflows from accounts payable and accrued liabilities of $5.4 million due to payments made during the period, net cash inflows resulting from lower inventory of $3.2 million, net cash inflows of $1.7 million relating to income taxes payable and net cash inflows of $7.6 million relating to prepaid expenses and other assets due to non-cash tax expense.
Cash Flows from Investing Activities
Net cash used in investing activities for the year ended December 31, 2010 was $3.7 million compared to $8.2 million of net cash used for the year ended December 31, 2009. For 2010, the net cash used in investing activities primarily consists of $5.7 million of capital expenditures, $3.2 million of proceeds related to our disposition of certain assets and our Laguna Hills manufacturing facility, and $1.0 million advanced to Haemacure in connection with our subsequent purchase of certain fibrin and thrombin product candidates and the related technology.
Net cash used in investing activities for the year ended December 31, 2009 was $8.2 million compared to net cash used of $8.2 million for the year ended December 31, 2008. For 2009, the net cash used in investing activities was primarily for capital expenditures of $4.4 million. In addition, payments were made to our partners Rex Medical and Haemacure of $2.5 million and $1.6 million, respectively during 2009. For 2008, the net cash used in investing activities consisted of $7.7 million of capital expenditures, a $0.8 million licensing milestone payment to a partner upon our successful completion of enrollment in our Bio-Seal human clinical trial, a $2.5 million investment in IPR&D for an initial license payment to Rex Medical and $2.8 million of proceeds from the sale of certain short-term investments.
Depending on the level of our cash portfolio, we may invest our excess cash in short-term marketable securities, principally investment grade commercial debt and government agency notes. Investments are made with the secondary objective of achieving the highest rate of return while meeting our primary objectives of liquidity and safety of principal. Our investment policy limits investments to certain types of instruments issued by institutions with investment grade credit ratings and places restrictions on maturities and concentration by type and issuer.
90
Table of Contents
At December 31, 2010 and December 31, 2009, we retained the following cash and cash equivalents denominated in foreign currencies in order to meet our anticipated foreign operating and capital expenditures in future periods.
| (in thousands of U.S.$) |
December 31, 2010 |
December 31, 2009 |
||||||
| Canadian dollars |
$ | 933 | $ | 5,542 | ||||
| Swiss franc |
575 | 986 | ||||||
| Euro |
1,447 | 5,680 | ||||||
| Danish krone |
1,090 | 412 | ||||||
| Other |
1,491 | 2,587 | ||||||
Cash Flows from Financing Activities
Net cash used in financing activities for the year ended December 31, 2010 was $9.3 million, which consisted of $0.7 million of fees incurred related to the completion of the third and fourth amendments to our Existing Credit Facility and $10.0 million of funds drawn under our Existing Credit Facility to support working capital and liquidity needs (see “Liquidity and Capital Resources” above and note 1, 3, 17 and 27 to the audited consolidated financial statements included in this Annual Report on Form 10-K).
Net cash used in financing activities for the year ended December 31, 2009 was $3.4 million, primarily for expenditures related to our Existing Credit Facility (see “Liquidity and Capital Resources” above and note 14(f) to the audited consolidated financial statements for the year ended December 31, 2009).
Net cash used in financing activities for the year ended December 31, 2008 was $4.4 million, primarily for expenditures related to an abandoned financing transaction, as well as expenditures related to our Existing Credit Facility as announced on March 2, 2009 (see “Liquidity and Capital Resources” above and note 14(f) to the audited consolidated financial statements for the year ended December 31, 2009).
Debt
(a) Subordinated Notes
On March 23, 2006, we issued Subordinated Notes in the aggregate principal amount of $250.0 million. These Subordinated Notes bear interest at an annual rate of 7.75% payable semi-annually in arrears on April 1 and October 1 of each year through to maturity. The Subordinated Notes are unsecured obligations. The Subordinated Notes and related note guarantees provided by us and certain of our subsidiaries are subordinated to our Existing Floating Rate Notes described below, the Existing Credit Facility and other existing and future senior indebtedness.
In connection with recent liquidity and capital resource constraints, on October 1, 2010 we did not make our $9.7 million interest payment due to the Subordinated Noteholders and, on October 29, 2010, we entered into the RSA in an effort to eliminate all outstanding indebtedness under the Subordinated Notes by exchanging Subordinated Notes for new common shares in the Company. On January 28, 2011, the Angiotech Entities commenced the CCAA Proceedings. On January 30, 2011 the Angiotech Entities commenced the Chapter 15 Proceedings to obtain recognition of the CCAA Proceedings and the relief granted therein in the United States. The Recapitalization Transaction is expected to significantly reduce our debt and interest obligations, thereby improving our liquidity, credit ratios and financial flexibility. Including the interest due on October 1, 2010, we accrued $14.5 million for our total interest obligation on the Subordinated Notes as at December 31, 2010. Upon the implementation of the CCAA Plan, the Subordinated Notes and all outstanding principal and interest thereunder will be cancelled.
(b) Existing Floating Rate Notes
On December 11, 2006, we issued the Existing Floating Rate Notes in the aggregate principal amount of $325 million. The Existing Floating Rate Notes bear interest at an annual rate of LIBOR plus 3.75%, which is reset quarterly. Interest is payable quarterly in arrears on March 1, June 1, September 1, and December 1 of each year through to maturity. On March 1, 2011, the Company paid $3.3 million in interest to the holders of its Existing Floating Rate Notes. The Existing Floating Rate Notes are unsecured senior obligations, are guaranteed by certain of our subsidiaries and rank equally in right of payment to all of our existing and future senior indebtedness. At December 31, 2010, the interest rate on these notes was 4.1%. Under the terms of the Existing Floating Rate Notes, we have the option to redeem all or a part of the notes at the redemption prices (expressed as percentages of principal amount) set forth below plus accrued and unpaid interest, if any, on the notes redeemed, to the applicable redemption date, if redeemed during the twelve-month period beginning on the dates indicated below:
91
Table of Contents
| Year |
Percentage (%) | |||
| December 1, 2010 |
101 | |||
| December 1, 2011 |
100 | |||
In addition, under certain change of control situations, we would be required to make an offer to purchase the then-outstanding Existing Floating Rate Notes at a price equal to 101% of their stated principal amount, plus accrued and unpaid interest to the applicable repurchase date, if any. On October 29, 2010, we entered into the FRN Support Agreement with holders of approximately 51% of the aggregate principal amount outstanding of our Existing Floating Rate Notes pursuant to which we commenced the FRN Exchange Offer. The FRN Exchange Offer is expected to close concurrent with the implementation of the CCAA Plan. The New Floating Rate Notes would be issued on substantially the same terms and conditions as the Existing Floating Rates Notes, except that, among other things: (i) subject to certain exceptions, the New Floating Rate Notes will be secured by second-priority liens over substantially all of the assets, property and undertakings of the Company and certain of its subsidiaries; (ii) the New Floating Rate Notes will accrue interest at LIBOR plus 3.75%, subject to a LIBOR floor of 1.25%; and (iii) certain covenants related to the incurrence of additional indebtedness and asset sales and the definitions of permitted liens, asset sales and change of control will be modified in the New Notes Indenture.
If the FRN Exchange Offer is consummated, any Existing Floating Rate Notes not tendered in the FRN Exchange Offer will remain outstanding and subject to the terms of the FRN indenture, as amended by the terms of the FRN Support Agreement.
As discussed above, in the event that we are unable to successfully complete the Recapitalization Transaction and the FRN Exchange Offer, the Subordinated Noteholders and holders of our Existing Floating Rate Notes may elect to exercise provisions under the respective indentures allowing them to accelerate amounts owing thereunder. An event of default under the FRN Indenture, arising from a cross-default for failure to make the interest payment on the Subordinated Notes within the applicable grace period would permit the holders of 25% or more of the aggregate outstanding principal amount of the Existing Floating Rate Notes to demand immediate repayment of our debt obligations owing under the Existing Floating Rate Notes. In this case, our current cash and credit capacity would not be sufficient to service principal debt and interest obligations. In addition, restrictions on our ability to borrow and the possible accelerated demand of repayment of existing or future obligations by any of our creditors may jeopardize our working capital position and ability to sustain its operations.
(c) Proforma Interest Expense
The following table (unaudited) shows the estimated interest payable for both the Existing Floating Rate Notes and the Subordinated Notes over the next four years without reflecting the potential impact of the Recapitalization Transaction or the FRN Exchange Offer discussed above. The estimate assumes a December 31, 2010 base LIBOR rate of 0.25% as well as the impact on the estimated interest payable of a 0.5% increase or a 0.25% decrease in the base LIBOR rate:
| Year |
(unaudited) | |||||||||||
| Base LIBOR | 0.5% Increase in base LIBOR |
0.25% Decrease in base LIBOR |
||||||||||
| 2011 |
$ | 32,720 | $ | 34,368 | $ | 31,732 | ||||||
| 2012 |
32,720 | 34,368 | 31,732 | |||||||||
| 2013 |
31,587 | 33,095 | 30,682 | |||||||||
| 2014 |
4,844 | 4,844 | 4,844 | |||||||||
Assuming that the Recapitalization Transaction and the FRN Exchange Offer are successfully completed and that the Subordinated Notes are retired in accordance with the CCAA Plan, we have also calculated the pro forma estimated interest payable for the New Floating Rate Notes over the next three years. Our estimates assume a December 31, 2010 base LIBOR rate of 0.3% from January 1, 2011 to April 30, 2011 and a LIBOR floor of 1.25% effective May 1, 2011 onwards. We have also calculated the impact on the estimated interest payable of a 0.5% increase in the base LIBOR rate:
| (unaudited) | ||||||||
| Year |
Base LIBOR | 0.5% Increase in base LIBOR * |
||||||
| 2011 |
$ | 14,047 | $ | 15,555 | ||||
| 2012 |
15,076 | 16,584 | ||||||
| 2013 |
15,076 | 16,584 | ||||||
92
Table of Contents
| * | Given that the interest on the New Floating Rate Notes is subject to a fixed LIBOR floor of 1.25%, we did not calculate the impact of a decrease in the LIBOR rate. |
(d) Debt Covenants
The terms of the SSN Indenture and the FRN Indenture include various covenants that impose restrictions on the operation of our business and the business of our subsidiaries, including the incurrence of certain liens and other indebtedness.
In addition, our Existing Credit Facility and DIP Facility include and the Exit Facility is expected to include a number of customary financial covenants, including a requirement to maintain certain levels of Adjusted EBITDA and interest coverage ratios, as well as covenants that limit our ability to, among other things, incur indebtedness, create liens, merge or consolidate, sell or dispose of assets, change the nature of our business, make distributions or make advances, loans or investments.
Material covenants under the SSN Indenture and FRN Indenture specify maximum or permitted amounts for certain types of capital transactions. In addition, these indentures contain certain restrictions on asset sales, the use of proceeds and the payment of dividends by the Company. Under the terms of the indentures, all outstanding principal amounts and interest accrued and unpaid become due and payable upon the occurrence of a defined event of default.
Upon the closing of the FRN Exchange Offer, which will occur concurrently with the effective date of the CCAA Plan, the FRN Indenture will be amended to provide for the elimination of most of the covenants and events of default therein. However, the indenture governing the New Floating Rate Notes will contain substantially identical covenants and events of default as the FRN Indenture in its present form, except with respect to certain modifications to the covenants relating to the incurrence of additional indebtedness, liens and asset sales.
Contractual Obligations
At December 31, 2010, our significant contractual obligations for the next five years and thereafter include the following, which does not give effect to the Recapitalization Transaction, the FRN Exchange Offer or the transactions contemplated thereby, which would eliminate all principal and interest obligations under the Subordinated Notes and provide for the issuance of New Floating Rate Notes in exchange for Existing Floating Rate Notes:
| (in thousands of U.S.$) |
Payments due by period | |||||||||||||||||||
| Total | Less than 1 year | 1 to 3 years | 3 to 5 years | More than 5 years | ||||||||||||||||
| Long-term debt repayments |
575,000 | 575,000 | — | — | — | |||||||||||||||
| Existing credit facility repayments |
10,000 | 10,000 | — | — | — | |||||||||||||||
| Long-term debt interest obligations |
101,871 | 32,720 | 64,307 | 4,844 | — | |||||||||||||||
| Operating leases |
28,511 | 3,843 | 7,308 | 7,127 | 10,233 | |||||||||||||||
| Other tax liability |
4,368 | — | — | — | 4,368 | |||||||||||||||
| Total obligations |
$ | 719,750 | $ | 621,563 | $ | 71,615 | $ | 11,971 | $ | 14,601 | ||||||||||
Long-term debt includes $325.0 million of Existing Floating Rate Notes and $250.0 million of Subordinated Notes. Repayments are based on contractual commitments as defined in the indentures governing the notes. Long-term debt interest obligations on variable (floating) rate debt are estimated using the current interest rates in effect at December 31, 2010. Long-term debt repayments and interest obligations assume no early repayment of principal.
We have entered into operating leases in the ordinary course of business for office and laboratory space with various third parties. The longest term of these leases will expire in 2019.
The table above does not include any cost sharing or milestone payments in connection with research and development collaborations with third parties as these payments are contingent on the achievement of specific developmental, regulatory or commercial activities and milestones as described in note 21 to the audited consolidated financial statements included in this Annual Report on Form 10-K. In addition, we may have to make royalty payments based on a percentage of future sales of certain products in the event regulatory approval for marketing is obtained.
93
Table of Contents
Contingencies
We are party to various legal proceedings, including patent infringement litigation and other matters. See Part I, Item 3 and Note 18(b) “Contingencies”, in the Notes to the Consolidated Financial Statements of Part II, Item 8 of this Annual Report on Form 10-K for more information.
Inflation
The effects of inflation or changing prices have not had a material impact on our net sales, revenues or income from continuing operations for the last three years.
Off-Balance Sheet Arrangements
As of December 31, 2010, we do not have any off-balance sheet arrangements, as defined by applicable securities regulators in Canada and the U.S., that have, or are reasonably likely to have, a current or future effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources that would be material to investors.
Recently adopted accounting policies
In June 2009, the FASB issued a new standard under ASC No. 810-10, Consolidation which changes the consolidation model for Variable Interest Entities (“VIE”). The revised model increases the qualitative analysis required when identifying which entity is the primary beneficiary that has (i) the power to direct the activities of a VIE that most significantly impact the entity’s economic performance and (ii) the obligation to absorb losses or the right to receive benefits from the VIE. The new standard eliminates the QSPE exemption, requires ongoing reconsideration of the primary beneficiary and amends the events which trigger reassessment of whether an entity is a VIE. The Company adopted the new guidance effective January 1, 2010. Adoption of this standard did not have a material impact on the Company’s financial results for the year ended December 31, 2010.
In January 2010, the FASB issued ASU No. 2010-02, Consolidation: Accounting and Reporting for Decreases in Ownership of a Subsidiary – a Scope Clarification, under ASC No. 810. The update clarifies the scope of decreases in ownership provisions and expands required disclosures for subsidiaries that are deconsolidated or group of assets that are derecognized. ASU No. 2010-02 is effective beginning in the first interim or annual reporting period ending on or after December 15, 2009, however, the amendments must be applied retrospectively to the first period that an entity adopted ASC No. 810-10. The adoption of the update did not have any impact on the Company’s financial statements.
In January 2010, the FASB issued ASU No. 2010-06, Fair Value Measurements and Disclosures: Improving Disclosures about Fair Value Measurements, under ASC No. 820-10. The update requires new disclosures about significant transfers in and out of Level 1 and Level 2 fair value measurements. ASU No. 2010-06 also clarifies disclosures required about inputs, valuation techniques and the level of disaggregation applied to each class of assets and liabilities. These updates are effective for interim and annual reporting periods beginning after December 15, 2009. These amendments have had no material impact on the Company’s financial results given that they relate to disclosure and presentation only.
In February 2010, the FASB issued ASU No. 2010-09, Subsequent Events: Amendments to Certain Recognition and Disclosure Requirements, The amendment removes the requirement for an SEC filer to disclose the date through which subsequent events have been evaluated in both issued and revised financial statements. SEC filers are still required to evaluate subsequent events through the date that the financial statements are issued. ASU No. 2010-09 was effective upon issuance and had no material impact on the Company’s financial statements or disclosures.
In February 2010, the FASB issued ASU No. 2010-08, Technical Corrections to Various Topics. The amendments effectively eliminate certain inconsistencies and outdated provisions in various topics under US GAAP to provide further clarification on the FASB’s original intent. ASU No. 2010-08 is effective for year’s beginning after December 15, 2009. The amendments are generally non-substantive in nature and have not resulted in significant or pervasive changes to our previous accounting assessments of the relevant accounting standards which are considered under ASU No. 2010-08.
94
Table of Contents
In April 2010, the FASB issued ASU No. 2010-17, Revenue Recognition – Milestone Method of Revenue Recognition, under ASC No. 605. The new guidance defines specific criteria for evaluating whether the milestone method is appropriate for the purposes of assessing revenue recognition. ASU No. 2010-17 stipulates that consideration tied to the achievement of a milestone may only be recognized if it meets all of the defined criteria for the milestone to be considered substantive. The guidance also requires expanded disclosures about the overall arrangement, the nature of the milestones, the consideration and the assessment of whether the milestones are substantive. ASU No. 2010-17 is effective on a prospective basis for milestones achieved in fiscal years and interim periods beginning on or after June 15, 2010. The adoption of this standard had no material impact on the Company’s recognition of revenue from current collaboration and revenue contracts.
In July 2010, the FASB issued ASU No. 2010-20, Disclosures about the Credit Quality of Financing Receivables and the Allowance for Credit Losses under ASC No. 310. The new guidance requires entities to provide a greater level of disaggregated information about the credit quality of its financing receivables and its allowance for credit losses. Disclosures may include information about credit quality indicators, overdue accounts, and modifications of financing receivables. The guidance is effective for public entities with interim and annual reporting periods ending on or after December 15, 2010. This guidance had no material impact on the Company’s financial results given that it relates to disclosure and presentation only.
Future accounting pronouncements
In October 2009, the FASB issued ASU No. 2009-13, Multiple-Deliverable Revenue Arrangements, under ASC No. 605. The new guidance provides a more flexible alternative to identify and allocate consideration among multiple elements in a bundled arrangement when vendor-specific objective evidence or third-party evidence of selling price is not available. ASU No. 2009-13 requires the use of the relative selling price method and eliminates the residual method to allocation arrangement consideration. Additional expanded qualitative and quantitative disclosures are also required. The guidance is effective prospectively for revenue arrangements entered into or materially modified in years beginning on or after June 15, 2010. The Company is still assessing the potential impact of ASU No. 2009-13, however, given that the Company does not currently have any bundled revenue arrangements; adoption of this guidance is not expected to have a significant impact on financial results.
In January 2010, the FASB issued ASU No. 2010-06, Fair Value Measurements and Disclosures: Improving Disclosures about Fair Value Measurements, under ASC No. 820-10. The update requires new disclosures about roll forward activity affecting Level 3 fair value measurements. ASU No. 2010-06 also clarifies disclosures required about inputs, valuation techniques and the level of disaggregation applied to each class of assets and liabilities. New disclosures about Level 3 fair value measurements are effective for interim and annual reporting periods beginning after December 15, 2010. The Company is assessing the potential impact that the adoption of ASU No. 2010-13 will have on its financial statement disclosures.
In April 2010, the FASB issued ASU No. 2010-13, Effect of Denominating the Exercise Price of a Share-Based Payment Award in the Currency of the Market in Which the Underlying Equity Security Trades. ASC No. 718, Compensation – Stock Compensation, provides guidance on whether share-based payments should be classified as either equity or a liability. Under this section, share-based payment awards that contain conditions which are not market, performance or service conditions are classified as liabilities. The updated guidance clarifies that an employee share-based payment award, with an exercise price denominated in the currency of a market in which substantial portion of the entity’s equity securities trade and which differs from the functional currency of the employer entity or payroll currency of the employee, are not considered to contain a condition that is not a market, performance or service condition. As such, these awards are classified as equity. ASU No. 2010-13 is effective for fiscal years and interim periods beginning on or after December 15, 2010. The cumulative effect of adopting this guidance should be applied to the opening balance of retained earnings. The Company is assessing the potential impact of adopting ASU No. 2010-13.
In December 2010, the FASB issues ASU No. 2010-29, Disclosure of Supplementary Pro Forma Information for Business Combinations. The new guidance clarifies that if a public entity presents comparative financial statements, the entity should disclose revenue and earnings of the combined entity as if the business combination occurred as of the beginning of the comparable prior annual reporting period only. ASU No. 2010-29 also requires additional disclosures about the nature and amount of material, non-recurring pro forma adjustments, which are directly attributable to the business combination and are included in the pro forma revenue and earnings estimates. The guidance is effective for all business combinations beginning on first annual reporting period beginning on or after December 15, 2010. The Company is currently assessing whether ASU No. 2010-29 will impact any of its future disclosures.
In December 2010, the FASB issues ASU No. 2010-28, Intangibles—Goodwill and Other (Topic 350). The amendments in this Update modify Step 1 of the goodwill impairment test for reporting units with zero or negative carrying amounts. For those reporting units, an entity is required to perform Step 2 of the goodwill impairment test if it is more likely than not that a goodwill impairment exists. In determining whether it is more likely than not that a goodwill impairment exists, an entity should consider whether there are any adverse qualitative factors indicating that an impairment may exist. The qualitative factors are consistent with the existing guidance and examples in paragraph 350-20-35-30, which requires that goodwill of a reporting unit be tested for impairment between annual tests if an event occurs or circumstances change that would more likely than not reduce the fair value of a reporting unit below its carrying amount. For public entities, the amendments in this Update are effective for fiscal years, and interim periods within those years, beginning after December 15, 2010. As the Company had no goodwill as of December, 31, 2010, it does not believe there will be any impact on its financial statements or disclosures.
95
Table of Contents
In January 2011, the FASB issues ASU No. 2011-01, Deferral of the Effective Date of Disclosures about Troubled Debt Restructurings in Update No. 2010-20. The new guidance temporarily delays the effective date of the disclosures about troubled debt restructurings in Update 2010-20 for public entities. The delay is intended to allow the Board time to complete its deliberations on what constitutes a troubled debt restructuring. The effective date of the new disclosures about troubled debt restructurings for public entities and the guidance for determining what constitutes a troubled debt restructuring will then be coordinated. Currently, that guidance is anticipated to be effective for interim and annual periods ending after June 15, 2011. The Company is currently assessing whether ASU No. 2011-01 will impact any of its future disclosures.
In January 2011, the Company filed for voluntary creditor protection under the CCAA and the U.S. Bankruptcy Code to effectuate an in-court Recapitalization Transaction as contemplated by the provisions under the RSA. Upon emergence from Creditor Protection Proceedings, the Company will likely be required to adopt fresh-start accounting. Under fresh-starting accounting, further adjustments will likely be required that may materially impact the current carrying values and classifications of the Company’s assets, liabilities, long-term debt and shareholder’s equity. The Company is still assessing the provisions of reorganization accounting under U.S. GAAP to determine whether it qualifies for fresh-start accounting. During the period of Creditor Protection Proceedings, the Company will continue to apply its current accounting policies described in note 2 of the audited consolidated financial statements for the year ended December 31, 2010.
Outstanding Share Data
As of December 31, 2010, there were 85,180,377 common shares issued and outstanding for a total of $472.8 million in share capital. At December 31, 2010, we had 5,275,184 CDN dollar stock options, and stock options with tandem stock appreciation rights, which we refer to as awards, outstanding under the Angiotech Pharmaceuticals, Inc. stock option plan (of which 3,049,990 were exercisable) at a weighted average price of CDN$3.83. We also had 3,890,999 U.S. dollar awards outstanding under this plan at December 31, 2010 (of which 1,846,660 were exercisable), at a weighted average exercise price of US$1.51. Pursuant to the 2006 Stock Incentive Plan, holders of each CDN dollar award and U.S. dollar award issued on or after October 1, 2002 may exercise their tandem stock appreciation right instead of the underlying stock option. The holder exercising the underlying stock option receives one common share of Angiotech Pharmaceuticals, Inc. for each option exercised. The holder exercising the tandem stock appreciation right portion of an award receives a portion of a common share of Angiotech Pharmaceuticals, Inc. with a fair value equal to the excess of the fair value of the common share subject to the underlying option at the time of exercise over the option price as set forth in the option agreement. Holders of each CDN dollar stock option issued prior to October 1, 2002 may exercise each option for one common share of Angiotech Pharmaceuticals, Inc.
As of December 31, 2010, there were 78 stock options outstanding in the AMI stock option plan (of which 48 were exercisable). Each AMI stock option is exercisable for approximately 3,852 common shares of Angiotech Pharmaceuticals, Inc. at a weighted average exercise price of US$15.44 per option.
In accordance with the Company’s Recapitalization Transaction and the terms of the CCAA Plan filed with the Canadian Court on February 17, 2011, all existing common shares and stock options will be cancelled without further payment or consideration.
96
Table of Contents
| Item 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
The primary objective of our investment activities is to preserve our capital to fund operations. We also seek to maximize income from our investments without assuming significant risk. To achieve our objectives, we maintain a portfolio of cash equivalents and investments in a variety of securities of high credit quality. As of December 31, 2010, we had cash and cash equivalents of $33.3 million.
Interest Rate Risk
As the issuer of the Existing Floating Rate Notes, we are exposed to interest rate risk. The interest rate on the Existing Floating Rate Notes is reset quarterly to 3-month LIBOR plus 3.75%. The aggregate principal amount of the Existing Floating Rate Notes is $325 million and the notes bear interest at a rate of approximately 4.05% at December 31, 2010 (4.01% at December 31, 2009). Based upon the average floating rate debt levels of the Company during the year, a 100 basis point increase in interest rates would have impacted our interest expense by approximately $3.3 million for the years ended December 31, 2010 and 2009, respectively. We do not use derivatives to hedge against interest rate risk.
Foreign Currency Risk
We operate internationally and enter into transactions denominated in foreign currencies. As such, our financial results are subject to the variability that arises from exchange rate movements in relation to the U.S. dollar. Our foreign currency exposures are primarily limited to the Canadian dollar, the Swiss franc, the Danish kroner, the Euro and the U.K. pound sterling. We incurred foreign exchange gain of $1.0 million for the year ended December 31, 2010 (December 31, 2009 - $1.6 million foreign exchange loss) primarily as a result of changes in the relationship of the U.S. to Canadian dollar and other foreign currency exchange rates when translating our foreign currency-denominated cash, cash equivalents and short-term investments to U.S. dollars for reporting purposes at period end. We have not entered into any forward currency contracts or other financial derivatives to hedge foreign exchange risk, and therefore we are subject to foreign currency transaction and translation gains and losses. We purchase goods and services in U.S. and Canadian dollars, Swiss francs, Danish kroner, Euros, and U.K. pounds sterling, and earn a significant portion of our license and milestone revenues in U.S. dollars. Foreign exchange risk is managed primarily by satisfying foreign denominated expenditures with cash flows or assets denominated in the same currency.
Since we operate internationally and approximately 13% of our net revenue for the year ended December 31, 2010 (December 31, 2009 – 11.4%) was generated in other than the U.S. dollar, foreign currency exchange rate fluctuations could significantly impact our financial position, results of operations, cash flows and competitive position.
For purposes of specific risk analysis, we used a sensitivity analysis to measure the potential impact to our consolidated financial statements for a hypothetical 10% strengthening of the U.S. dollar compared within the other currencies which we denominate product sales in for the year ended December 31, 2010. Assuming a 10% strengthening of the U.S. dollar, our product net revenue would have been negatively impacted by approximately $2.8 million (December 31, 2009 - $2.9 million).
| Item 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
The consolidated financial statements and financial statement schedules in Part IV, Item 15(a) (1) and (2) of this Annual Report on Form 10-K are incorporated by reference into this Item 8.
Management’s Report on Internal Control over Financial Reporting
Refer to Part II, Item 9A(b) of this Annual Report on Form 10-K.
97
Table of Contents
| Item 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
None.
| Item 9A. | CONTROLS AND PROCEDURES |
(a) Disclosure Controls and Procedures.
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed by us in reports we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our Chief Executive Officer (“CEO”) and Chief Financial Officer (“CFO”), as appropriate, to allow timely decisions regarding required disclosure. In designing and evaluating disclosure controls and procedures, management recognizes that any control and procedure, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and management necessarily applies its judgment in evaluating the cost-benefit relationship regarding the potential utilization of certain controls and procedures.
As required by Rules 13(a)-15(b) under the Exchange Act, our management, with the participation of our CEO and CFO, evaluated the design and operation of our disclosure controls and procedures (as defined in Rules 13a-15(e) or 15d-15(e) under the Exchange Act). Based on such evaluation, our CEO and CFO have concluded that, as of December 31, 2010, our disclosure controls and procedures were effective and were operating at a reasonable assurance level.
(b) Management Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting (as defined in Rule 13a-15(f) under the Exchange Act) to provide reasonable assurance regarding the reliability of our financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles.
Due to its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate due to changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Under the supervision and with the participation of our management, including our CEO and CFO, we conducted an evaluation of the effectiveness of our internal control over financial reporting using the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control — Integrated Framework.
Based on our assessment, management has concluded that our internal control over financial reporting was effective as of December 31, 2010 to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external reporting purposes in accordance with U.S. GAAP.
The effectiveness of our internal control over financial reporting as of December 31, 2010 has been audited by PricewaterhouseCoopers LLP, an independent registered public accounting firm, as stated in its report which appears herein.
(c) Changes in Internal Control over Financial Reporting
No change was made to our internal control over financial reporting during the fiscal quarter ended December 31, 2010 that has materially affected, or is reasonably likely to materially affect, such internal control over financial reporting.
| Item 9B. | OTHER INFORMATION |
None.
98
Table of Contents
| Item 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE |
Directors, Executive Officers, Promoters and Control Persons
Directors of the Company
Each of the following individuals is currently serving as a director of the Company. Information concerning such persons, as furnished by the individual directors, is as listed in the chart below. In connection with the implementation of the CCAA Plan, the current Board of Directors will be replaced by the board of directors designated in the Sanction Order approving the CCAA Plan.
| Name, Age, Province or State and Country of Residence and Position |
Principal occupation during the past 5 years |
Director Since |
||||
| William L. Hunter, MD, MSc. British Columbia, Canada President, Chief Executive Officer and Director Age: 48 |
September 2002 to present — President & Chief Executive Officer, the Company | 1992 | ||||
| David T. Howard British Columbia, Canada Chair of the Board of Directors, Director (1)(2) Age: 61 |
September 2002 to present — Chair of the Board of Directors, the Company
May 2000 to July 2003 – CEO of SCOLR, Inc. August 2003 to August 2005 – Chair of SCOLR, Inc. |
2000 | ||||
| Edward M. Brown, MBA California, U.S.A. Director (1)(2) Age: 47 |
June 2010 to present — Senior Adviser, Health Evolution Partners June 2007 to December 2009 — Managing Director, TPG Growth
June 2004 to June 2007 — Managing Director & Co-founder of Healthcare Investment Partners |
2004 | ||||
| Arthur H. Willms British Columbia, Canada Director (1)(2) Age: 71 |
1999 to present — Independent Company Director | 2004 | ||||
| Laura Brege, MBA California, U.S.A. Director (1)(2) Age: 53 |
2006 to present — Executive Vice President & Chief Operating Officer, Onyx Pharmaceuticals, Inc.
1999 to 2007 — General Partner, Red Rock Management |
2007 | ||||
| Henry A. McKinnell Jr., MBA, Ph.D. Wyoming, U.S.A. Director (1)(2) Age: 68 |
January 2011 to present — Board of Optimer Pharmaceuticals Chair, Science and Technology Committee & Member of Governance Committee
June 2002 to present — Chairman, Accordia Global Health Foundation
2001 to 2006 — Chief Executive Officer & Chairman of Pfizer Inc. |
2008 | ||||
Notes:
| (1) | Member of the Company’s Audit Committee. |
| (2) | Member of the Company’s Governance, Nominating and Compensation Committee. |
99
Table of Contents
The following are brief biographies of the Company’s directors. The information presented includes information each director has given about his or her age, all positions held, his or her principal occupation and business experience for the last five years, and the names of other publicly-held companies of which he or she currently serves as a director or has served as a director during the past five years. Also included is a brief discussion of the specific experience, qualifications, attributes, and skills of each nominee that lead the Board of Directors to the conclusions that the nominee should serve as a director.
William L. Hunter, MD, MSc ., President & Chief Executive Officer, Director . Dr. Hunter is a founder of the Company, has been Chief Executive Officer of the Company since 1997, and has been a member of the Company’s scientific and management teams since its inception. He serves as a director of Cardiome Pharma, Corp. and CombinatoRx Incorporated. Dr. Hunter has been honored with many awards including, most recently, the 2005 BC Innovation Council’s Cecil Green Award for Science and Technology Entrepreneurship and the 2005 Canadian Venture Capital Association’s Entrepreneur of the Year. He received his Bachelor of Science Degree from McGill University, Montreal in 1985 and his Masters of Science and Doctor of Medicine Degree from the University of British Columbia in 1989 and 1992, respectively. Dr. Hunter’s medical knowledge, research and development experience, commercial acumen and involvement with the Company as a founder and CEO, as well as his experience serving as a director of other companies, are invaluable to the discussions of the Board of Directors.
David T. Howard, Chair of the Board of Directors, Director . David Howard joined the Company’s Board of Directors in March 2000 and became Chair of the Board of Directors in September 2002. Mr. Howard is a director of Via Pharmaceuticals, Inc., and has served as a director of SemBioSys Genetics, Inc. and SCOLR Inc. Previously he was CEO, President and Chairman of the Board of SCOLR, Inc., a biopharmaceutical company located in Redmond, WA. Prior to this, he has served as President and Chief Operating Officer of Novopharm International, Inc. of Toronto, Ontario and President of Novopharm USA, Inc. Mr. Howard’s industry experience includes operational and strategic positions with Boehringer Mannheim Canada, where he was Vice President Pharmaceuticals, and Rhône-Poulenc Pharma in Montreal and Paris, where he was Vice-President Sales and Marketing. Mr. Howard’s strategic and operational experience as an executive of a biopharmaceutical company, as well as his substantial prior experience in the pharmaceutical industry, provide the Board of Directors valuable insight into the Company’s industry.
Edward M. Brown, MBA, Director . Edward (Ned) Brown is a Senior Advisor at health Evolution Partners, where he focuses on private equity investments in healthcare companies. Prior to joining Health Evolution Partners, Mr. Brown was a Managing Director of TPG Growth, a private equity firm, where he focused on private equity investments in healthcare companies. Prior to joining TPG Growth, Mr. Brown was a co-founder of Healthcare Investment Partners (“HIP”), a private equity firm focused exclusively on healthcare investments. Before HIP, Mr. Brown was a Managing Director in the Healthcare Group of Credit Suisse First Boston, an investment bank, where he led the firm’s West Coast healthcare effort and was one of the senior partners responsible for the firm’s global life sciences practice. Mr. Brown has over 23 years of experience as a financial advisor and investor in the healthcare industry. Mr. Brown also serves on the Board of Directors of Cardiovascular Systems, Inc. Mr. Brown graduated Phi Beta Kappa, Magna Cum Laude with High Honors from Middlebury College with a B.A. Degree in English and received his MBA degree from Anderson Graduate School of Management at UCLA. Mr. Brown brings to the Board of Directors years of experience in advising and providing financing for companies within the healthcare industry, and provides insight into the Company’s strategic and business development activities.
Arthur H. Willms, MA, Director . Arthur Willms is a Director and retired President and Chief Operating Officer of Westcoast Energy, Inc. where he held senior executive positions for 24 years. Mr. Willms’ other directorships are Pacific Northern Gas Ltd., and BC Lotteries Corp. He is also the Chair of the Board for the Vancouver Symphony Orchestra. Mr. Willms has been a lecturer in Economics at the University of Calgary and holds a Bachelor of Arts Degree in Education, a Bachelor of Science Degree in Mathematics, and a Masters Degree in Economics from the University of Calgary. Mr. Willms is a member of two audit committees and is the chair of one of these committees. Mr. Willms’ past operational experience as a senior executive, his experience as a director of companies in a variety of industries, and in particular his service on several audit committees, provide the Board of Directors financial and accounting expertise.
Laura Brege, MBA, Director . Ms. Brege is currently Executive Vice President and Chief Operating Officer at Onyx Pharmaceuticals, Inc., where she is responsible for Sales and Marketing, Medical and Scientific Affairs, Program Leadership, Technical Operations, Manufacturing, Commercial Operations and Strategy. Onyx Pharmaceuticals is focused on developing and commercializing innovative cancer treatments. Prior to joining Onyx Pharmaceuticals, Ms. Brege was a General Partner at Red Rock Management, a venture capital firm specializing in early stage financing for technology companies. Prior to her work at Red Rock Management, she was the Senior Vice President and Chief Financial Officer at COR Therapeutics, a biotechnology company that specialized in drug therapies for cardiovascular disease. While at COR, Ms. Brege was instrumental in helping the company grow from a research and development start-up to an operations-based enterprise with a commercialized product. Before joining COR, Ms. Brege was Vice President and Chief Financial Officer at Flextronics, a multi-billion dollar electronics manufacturer. She was also Vice President and Treasurer of The Cooper Companies, a Fortune 500 diversified company with operations worldwide. Ms. Brege is also a member of the Board of Directors of Acadia Pharmaceuticals, Inc. Ms. Brege earned her undergraduate degree from Ohio University and has an MBA from the University of Chicago. Ms. Brege’s breadth of operational, financial and sales and marketing experience as both a venture capitalist and in corporate operations provide a valuable sales and marketing, financial and operational
100
Table of Contents
perspective to the Board of Directors with respect to the Company’s own operations, as well as with respect to established companies within the Company’s industry and to venture-stage companies in which the Company may invest. Additionally, her experience in growth-stage biotechnology firms provides valuable insight into implementing research and development strategies for companies within the Company’s industry.
Henry A. McKinnell Jr., Director . Henry A. McKinnell Jr. is the Chairman of the Governance, Nominating and Compensation Committee, is a member of the Audit Committee and serves as the lead independent director of the Board of Directors of Moody’s Corporation and Optimer Pharmaceuticals, Inc. Dr. McKinnell served as Chairman of the Board of Pfizer Inc., a pharmaceutical company, from May 2001 until his retirement in December 2006 and Chief Executive Officer from January 2001 to July 2006. He served as President of Pfizer Inc. from May 1999 to December 2000 and as Executive Vice president from 1992 to 1999. Dr. McKinnell serves a Chairman of the Board of the Accordia Global Health Foundation. He is Chairman of the Science and Technology Committee and member of the Governance Committee of Optimer Pharmaceuticals, Inc. He is Chairman Emeritus of the Connecticut Science Center, and is a member of the Academic Alliance for AIDS Care and Prevention in Africa. He served as director of Pfizer Inc. and ExxonMobil Corporation until 2007 and John Wiley & Sons until 2005. Dr. McKinnell’s strategic and operational experience as an executive of Pfizer Inc., a public pharmaceuticals company, his experiences as a public company director, as well as his substantial involvement with business and civic organizations, allow the Board of Directors to draw on his years of experience with publicly traded companies with global operations in evaluating corporate best practices and strategic decision-making.
The following sets out the identity of our independent and non-independent directors together with a list of the other reporting issuers of which our directors are also a director.
| Name of Director |
Independent/ |
Other Reporting Issuers Of Which Director Is Also A Director Or Has Been a Director In The Last Five Years | ||
| William L. Hunter | Non-Independent | Cardiome Pharma, Corp; CombinatoRx, Incorporated (f/k/a Neuromed Pharmaceuticals, Inc.) | ||
| David T. Howard (Chair) | Independent | SemBioSys Genetics, Inc.; Via Pharmaceuticals, Inc., SCOLR, Inc.; Bioasis Technologies Inc. | ||
| Edward M. Brown | Independent | Cardiovascular Systems Inc. | ||
| Laura Brege | Independent | Acadia Pharmaceuticals, Inc. | ||
| Arthur H. Willms | Independent | Pacific Northern Gas Ltd.; BC Lotteries Corp., Pristine Power Inc. | ||
| Henry A. McKinnell Jr. | Independent | Moody’s Corporation, Pfizer Inc., ExxonMobil Corporation, John Wiley & Sons, Optimer Pharmaceuticals, Inc. |
The Board of Directors held a total of thirty-one meetings in the year ended December 31, 2010, inclusive of committee meetings. During 2010, Mr. Brown attended fewer than 75% of the aggregate meetings of the Board of Directors and the committees of which he is a member as a result of being required to recuse himself from certain Board of Directors meetings and committee meetings relating to the exploration of certain financial and strategic alternatives during 2010due to a potential conflict of interest. Excluding such meetings from which Mr. Brown was recused, his attendance exceeds the 75% attendance requirement.
101
Table of Contents
Executive Officers
The following sets forth certain information regarding the Company’s executive officers as of December 31, 2010 (ages are as of the date hereof):
| Name, Age, Province or State and Country and Position |
Position with the Company |
Principal Occupation within the Past Five Years | ||
| William L. Hunter, MD, MSc. Age: 48 |
President, Chief Executive Officer, and Director | September 2002 to present — President & CEO, the Company November 1992 to present — Director, the Company | ||
| K. Thomas Bailey Washington, Age: 42 |
Chief Financial Officer | December 2005 to present — Chief Financial Officer, the Company January 2004 to December 2005 — Vice President, Business Development, the Company | ||
| David D. McMasters, ESQ Age: 51 |
Senior Vice President, Legal and General Counsel | January 2004 to present — Senior Vice President, Legal & General Counsel, the Company
| ||
| Rui Avelar, MD (2) British Columbia, Canada Age: 49 |
Chief Medical Officer | December 2005 to February 2011 — Chief Medical Officer, the Company January 2004 to December 2005 — Senior Vice President, Medical Affairs & Communications, the Company | ||
| Jonathan W. Chen(1) British
Columbia, Canada Age: 36 |
Senior Vice President, Business Development & Financial Strategy | January 2008 to January 2011 — Senior Vice President, Business Development & Financial Strategy, the Company September 2005 to January 2008 — Vice President, Business Development & Financial Strategy, the Company September 2000 to May 2005 — Investment Banker, Credit Suisse First Boston | ||
| Steven Bryant South
Carolina, U.S.A. Age: 60 |
Senior Vice President, Sales & Marketing, Medical Device Technologies | June 2009 to present — Senior Vice President, Sales & Marketing, the Company 2001 to June 2009 — Vice President, Sales & Marketing, the Company | ||
| Sean Cunliffe Pennsylvania, U.S.A. Age: 54 |
Senior Vice President, Sales & Marketing | September 2009 to present — Senior Vice President, Sales & Marketing, the Company May 2008 to September 2009 — Chief Commercial Officer, Neuromed Pharmaceuticals, Inc. March 2005 to May 2008 — Vice President, Commercial Development, Neuromed Pharmaceuticals, Inc. | ||
| Jeffrey Gross British
Columbia, Canada Age: 47 |
Senior Vice President, Research and Development | Oct 2008 — present, Senior Vice President, Research and Development, the Company July 2007 — Sep 2008, Vice President, Engineering, Research and Development, the Company 2004 — 2007, Group Director, Biologics Research & Development, Medtronic Spinal & Biologics | ||
102
Table of Contents
| Name, Age, Province or State and |
Position with the Company |
Principal Occupation within the Past Five Years | ||
| Jay Dent British Columbia, Canada Age: 52 |
Senior Vice President, Finance | December 2006 to present — Senior Vice President, Finance, the Company October 2005 to December 2006 — Vice President, Finance & Accounting, the Company May 2001 to September 2005, Controller, Ballard Power Systems, Inc. | ||
| Victor Diaz Illinois, U.S.A. Age: 50 |
Senior Vice President, Global Manufacturing and Supply Chain Management | 2007 to present — Senior Vice President, Global Manufacturing & Supply Chain, the Company 2003 to 2006 — Vice President, Global Operations, Teleflex Medical | ||
| Tammy Neske British Columbia, Canada Age: 40 |
Senior Vice President, Human Resources | January 2008 to present — Senior Vice President, Human Resources, the Company January 2007 to January 2008 — Vice President, Human Resources, the Company July 2005 to January 2007 — Director, Corporate Development, the Company 2000 to 2005 — Manager, Employment and Compensation, Ballard Power Systems, Inc. | ||
Notes:
| (1) | Mr. Chen ceased employment on February 1, 2011. |
| (2) | Mr. Avelar ceased employment on February 28, 2011. |
There are no family relationships among the directors and executive officers of the Company.
For each of the past ten years, none or our directors or executive officers has been a director or officer of another issuer which, while that person was acting in that capacity or within one year of that person ceasing to act in that capacity, became bankrupt or made a proposal under any legislation relating to bankruptcy or insolvency or was subject to or instituted any proceedings, arrangements or compromises with creditors or had a receiver, manager or trustee appointed to hold its assets.
Code of Ethics
The Company has adopted a code of ethics for our Chief Executive Officer, Chief Financial Officer and Senior Vice President of Finance. In addition, the Company has also adopted a guide of business conduct for our employees. The current code of ethics and the guide of business conduct are available on the Company’s website at www.angiotech.com. Management reports violations of the codes and any remedial actions it has taken to the Board of Directors.
Audit Committee
The charter for the Audit Committee of the Board of Directors requires that it be comprised of at least three unrelated and independent directors. The Audit Committee is currently comprised of five members — Arthur Willms (Chair), David Howard, Edward Brown, Laura Brege and Henry McKinnell. All of the members of the Audit Committee are independent of management (see “— Directors, Executive Officers, Promoters, and Control Persons” for a description of the relationship each member has to the Company). The Audit Committee held four meetings during the year ended December 31, 2010. All Audit Committee members are required to be financially literate and at least one member is required to have accounting or related financial management expertise.
Our Board of Directors has determined that it has at least one audit committee financial expert serving on its Audit Committee. Mr. Arthur Willms has been determined by the Board of Directors to meet the “audit committee financial expert” criteria prescribed by the SEC and is independent, as that term is defined by the Canadian and U.S. securities laws applicable to the Company. The SEC has indicated that the designation of Mr. Willms as an audit committee financial expert does not make him an “expert” for any purpose, impose any duties, obligations or liability on him that are greater than those imposed on other members of the Audit Committee and the Board of Directors who do not carry this designation, or affect the duties, obligations or liability of any other member of the Audit Committee.
The Audit Committee Charter is available on the Company’s website at www.angiotech.com.
103
Table of Contents
Special Committee
The Board of Directors formed an independent committee (the “Independent Committee”) on July 29, 2010, originally consisting of Laura Brege and Messrs. Howard, McKinnell and Willms. On September 24, 2010 Mr. Brown was appointed to the Independent Committee. The Independent Committee’s responsibility included, among other things: (a) to explore and consider strategic alternatives available to Angiotech to maximize value in light of the financial condition of Angiotech and the state of the economy and capital markets generally; (b) to work as necessary with management of Angiotech and external advisors in connection with the foregoing; (c) to make recommendations to the Board of Directors in respect of the foregoing; and (d) to take all such other steps as the Independent Committee considered to be necessary or appropriate and in the best interests of Angiotech with respect to the foregoing. The Independent Committee met frequently to consider the alternatives available to Angiotech, including the terms of the Recapitalization Transaction. It received written and oral advice from management, the Angiotech Entities’ counsel and financial advisor, and from the Independent Committee’s counsel, Stikeman Elliott LLP, and its financial advisor, Lazard LLP.
104
Table of Contents
| Item 11. | EXECUTIVE COMPENSATION |
Compensation Discussion & Analysis
Overview of Compensation Program
The Company competes in a global market for executives, management, scientists, engineers, sales and marketing, manufacturing and other professionals. Many of our professional and management candidates are sought by other attractive employers, such as multi-national medical device companies, technology companies, multi-national pharmaceutical companies and university research institutions. Our overall compensation philosophy reflects our need to motivate, recruit and retain executive talent with the diversity of experience necessary to articulate and execute the critical elements of our corporate strategy and manage our operating businesses. Specifically, our approach is designed to motivate executives and facilitate the recruitment and retention of talent with the range of competencies necessary to deliver the strategy and operational goals of:
| i. | our Proprietary Medical Products business, which involves managing new technologies, substantive investments in research and development and sales and marketing and more rapid revenue growth, and |
| ii. | our Base Medical Products business, which involves managing more mature technologies with slower revenue growth and higher near-term operating profit, well established sales and marketing channels, including significant third party distribution arrangements, customer relationships with third-party medical device manufacturers that buy medical devices and medical device components made by us, and substantive medical device manufacturing operations. |
The Governance, Nominating and Compensation Committee (for purposes of this discussion, the “Committee”) of the Board of Directors is charged, on behalf of the Board of Directors, to develop, review and approve compensation policies and practices applicable to executive officers, including determining the benchmarks, criteria and performance metrics upon which executive compensation such as base salary, annual bonuses, equity compensation and other benefits are based. The Committee held six meetings during the year ended December 31, 2010. The Committee Charter is available on the Company’s website at www.angiotech.com.
As described below, the Committee approves the compensation of each of our Named Executive Officers (“NEO’s”). In 2010, our NEOs were: Dr. Hunter, Mr. Bailey, Mr. McMasters, Dr. Avelar and Mr. Bryant.
Compensation Philosophy and Objectives
The Company’s objectives for its executive compensation policies, programs, and practices are to:
| • | Attract and retain experienced and talented executive officers; |
| • | Pay for performance; |
| • | Inspire excellence in the performance of executive officers; and |
| • | Align shareholder and executive officer interests. |
To further these objectives, the Committee:
| • | Designs pay and performance systems that reflect the level of job responsibility, and incumbent specific considerations; |
| • | Aligns the Company’s compensation programs with those of similarly complex specialty pharmaceutical, biotechnology and medical device companies; |
| • | Motivates executive performance by aligning pay-for-performance-based compensation programs to the achievement of objectives that will drive future success and enhance shareholder value; and |
| • | Links a significant portion of annual awards to overall corporate performance and attainment of specific value enhancing goals. |
The Committee periodically evaluates compensation to ensure that the Company maintains its ability to attract and retain high caliber talented employees in key positions and that compensation provided to our executives remains competitive relative to the compensation paid to executives of peer companies. To that end, the Committee believes executive compensation packages provided by the Company should include salary, which provides base compensation, an annual cash bonus and long-term stock-based compensation, which reward performance as measured against established goals on a short-term and long-term basis.
In the fall of 2009, the Committee developed a formal benchmarking and pay mix approach to be used in setting 2010 executive compensation levels which, together with our overall compensation philosophy are designed to promote our strategy and operational goals described above. We target base salary and annual bonus opportunity at the 50th percentile of companies in our peer group, with the potential to earn above market median when individual performance or the business results exceed annual targets. We target equity compensation at or above the 75th percentile of companies in our comparator group, measured as annual grant in units as a percentage
105
Table of Contents
common shares outstanding, which we use because traditional methods of valuing equity grants such as fair market value or Black-Scholes, combined with current share price, create the appearance that our executive officers are under-equitized measured by the aggregate value of their equity. Using these methods of benchmarking cash and equity, we will be able to extrapolate whether our compensations levels and our pay mix are relatively consistent with those of our peer group, in any given year.
Role of Executive Officers in Compensation Decisions
The Committee makes all decisions pertaining to the compensation of the Chief Executive Officer. The Committee reviews and approves recommendations for compensation awards to other executive officers presented by the Chief Executive Officer.
The Chair of the Board of Directors and the Committee annually review the performance of the Chief Executive Officer. The Chief Executive Officer annually reviews the performance of the other executive officers. The conclusions reached and recommendations based on these reviews, including compensation awards, are presented to the Committee for review and approval. The Committee may exercise its discretion in modifying any recommended adjustments or awards to executive officers.
Setting Executive Compensation
Based on the foregoing objectives, the Committee has structured the Company’s compensation programs to motivate executives to achieve the short- and long-term business goals of the Company, and to reward executives for achieving such goals. When making compensation decisions, the Committee seeks the advice and recommendations of the Chief Executive Officer and Senior Vice President, Human Resources, and also retains independent compensation consultants as and when it deems appropriate to provide advice and data related to appropriate industry benchmarks and compensation trends. In the fall of 2009, the Committee undertook an executive compensation benchmarking analysis of industry peers for purposes of determining 2010 compensation levels.
For purposes of establishing 2010 compensation, the peer group used to benchmark executive compensation is made up of the following companies:
| Proprietary Business Comparators |
Base Business Comparators | |
| Onyx Pharmaceuticals Inc. |
Becton, Dickinson and Company | |
| Alexion Pharmaceuticals, Inc. |
CR Bard Inc. | |
| Cubist Pharmaceuticals Inc. |
Edwards Lifesciences Corp. | |
| Intuitive Surgical, Inc. |
ev3, Inc. | |
| Medicines Co. |
Integra LifeSciences Holdings Corp. | |
| Medicis Pharmaceutical Corp. |
Kinetic Concepts Inc. | |
| NuVasive, Inc. |
Merit Medical Systems Inc. | |
| OSI Pharmaceuticals Inc. |
Symmetry Medical, Inc. | |
| United Therapeutics Corporation |
Teleflex Inc. | |
| Vertex Pharmaceuticals Incorporated |
Wright Medical Group Inc. |
As mentioned previously, we target base salary and annual bonus opportunity at the 50th percentile of companies in our peer group, with the potential to earn above market median when individual performance or the business results exceed annual targets. We target equity compensation at or above the 75th percentile of companies in our comparator group, measured as annual grant in units as a percentage of common shares outstanding.
Stock Ownership Guidelines and Stock Retention Requirements
In March 2010, ownership guidelines were implemented to coincide with anticipated shareholder approval of the 2010 Stock Incentive Plan which would have facilitated compliance with the guidelines. The proposed plan was defeated by shareholders. Under the guideline, the Chief Executive Officer is expected to hold five times his base salary in shares, RSUs and DSUs, and other executive officers are expected to hold one times base salary in shares, RSUs and DSUs.
Adjustment or Recovery of Awards upon Restatement of Financial Results
The Company implemented a policy in the fall of 2009 to take effect for 2010. The Company will, in all circumstances deemed appropriate by the Board of Directors and to the extent permitted by governing law, require reimbursement of any bonus to an executive after January 1, 2010 where:
| i. | the payment was predicated upon achieving certain financial results that were subsequently the subject of a material restatement of Company financial statements filed with the SEC; |
| ii. | in the Board of Directors’ view the executive engaged in misconduct that caused or substantially caused the need for the material restatement; and |
| iii. | a lower payment would have been made to the executive based upon the restated financial results. |
106
Table of Contents
In each such instance, the Company will, to the extent practicable, seek to recover from the individual executive the amount by which the individual executive’s incentive payments for the relevant periods exceeded the lower payment that would have been made based on the restated financial results. Further, following a restatement of the Company’s financial statements, the Company will recover any bonuses paid to the Chief Executive Officer and the Chief Financial Officer that are required to be recovered by Section 304 of the Sarbanes-Oxley Act of 2002 (“SOX”).
107
Table of Contents
Accounting of Stock-Based Compensation
The Company accounts for stock-based compensation under its long-term incentive plans in accordance with the requirements of FASB ASC Topic 718.
2010 Executive Compensation Elements
For the fiscal year ended December 31, 2010, the principal components of compensation for NEOs were:
| • | Annual base salary; |
| • | Annual bonus program; |
| • | Long-term incentive program; |
| • | Retirement savings plan; and |
| • | Benefits programs including health coverage, insurance plans and other perquisites. |
Annual Base Salary
NEOs and other employees receive a salary which provides them with a base level of income. The base salary of each NEO is reviewed annually. The level of base salary is based on the executive’s responsibilities, performance, peer group data, experience and the general economic climate. Based on a combination of these factors the Committee awarded salary increases of 6% for Mr. Bailey, 9% for Dr. Avelar, 9% for Mr. Bryant and 0% for Dr. Hunter and Mr. McMasters. Annual base salary earned by NEOs during 2010 is set out in the Summary Compensation Table for the Fiscal Year Ending December 31, 2010.
Annual Bonus Program
The annual bonus program generally provides a performance-based cash incentive to NEOs and other employees. The program awards for individual performance that leads to the achievement of annual corporate objectives. The NEO bonus targets established by the Committee were not changed for 2010 with the exception of Dr. Hunter’s bonus target. Based on the peer group data, the Committee increased Dr. Hunter’s bonus target to 100% of base salary. Mr. Bailey’s bonus target was 50% of base salary. Mr. McMasters’, Dr. Avelar’s and Mr. Bryant’s bonus target was 40% of base salary.
At the beginning of 2010, key corporate objectives were set by the Board of Directors to represent a broad range of activities that span the Company’s global and complex business. The 2010 corporate objectives are categorized into goal themes: commercial, research & product development, operations and people. The commercial objectives included internal sales revenue targets, the research and product development objectives included new product launch and new product development activities, the operational and financial objectives included internal earnings, expense and margin goals and the people objective was related to the engagement of the workforce. Commercial objectives were weighted at 20%, research and product development objectives at 40%, operational and financial objectives at 30% and people objectives at 10%. The weights are intended to reflect the relative priority, importance and impact of each objective on the Company’s successes. Objectives are set with targets that represent significant “stretch” goals for the organization. Each objective is also assigned a minimum achievement target and a maximum achievement target.
After the year-end results are compiled, performance relative to the corporate objectives is measured to determine a “corporate performance result.” If an objective is achieved on target, the performance result is 100% and the annual bonus pool is funded at 100% for that objective. If the minimum achievement target is not met for an objective, the annual bonus pool will not be funded for that objective. If the minimum achievement target is met, the annual bonus pool will be funded at 50% of the target bonus pool for that objective. If the maximum achievement level is met, the annual bonus pool will be funded up to 150% of the target bonus pool for that objective. The Board of Directors gives consideration to how challenging each goal was and considers other business, commercial and organizational factors that may have impacted achievement of the goals in arriving at its final assessment of the corporate performance result.
The formula for determining the amount each NEO received under the annual bonus program is as follows:
Base Salary x Bonus Target % x Individual Performance Result x Corporate Performance Result
The individual performance result is based on each NEO’s achievement of his functional and individual goals (set at the beginning of each year) and demonstrated leadership competencies as assessed by the Chief Executive Officer and Committee. The CEO’s individual goals are generally the same as the corporate objectives. The other NEOs functional and individual goals contain a spectrum of commercial, financial, technical, scientific and special project goals specific to the NEO’s area of responsibility. The Committee and CEO do not use quantitative targets and formulas to assess individual executive performance. The Committee determines the individual performance result of the Chief Executive Officer and approves the recommendations made by the Chief Executive Officer for the individual performance result of the other NEOs.
108
Table of Contents
For 2010, the themes, objectives and relative achievements of those objectives were:
| Area |
Goal |
Weight | Target | Result | ||||||||
| Commercial |
Product Revenue (excluding royalty revenue) | 20 | % | $220mm | 16 | % | ||||||
| Research & Product Dev. |
Commercial launch of a drug-device product Commercial launch of a medical device product Advance pipeline of a drug device product |
40 | % | Internal milestones |
30 | % | ||||||
| Operations |
Achieve key operational and financial metrics | 30 | % | Internal metrics |
39 | % | ||||||
| People |
Improve employee engagement | 10 | % | Internal metrics |
5 | % | ||||||
The 2010 corporate performance result was determined by the Committee. In determining the corporate performance result for 2010, the Committee reviewed results for each 2010 objective. Results related to the commercial objective which consisted of internal sales goals were slightly underachieved. Results for the research and product development objective were mixed. While the Company made progress on targeted research and product development projects and successfully launched several new products and line extensions, it did not meet all internally targeted timelines. Results related to the operational and financial objective were also mixed. The internal earnings goal was over-achieved, the internal expense target was over-achieved and the internal margin target was over-achieved. The people goal related to employee engagement under-achieved but still fell within the performance range. The Committee determined that a 90% score reflected the Company’s corporate performance result against its targeted achievement level.
For 2010, the Committee awarded an individual performance result of 100% of target to all NEO’s which was then multiplied by the corporate performance result of 90% to award a bonus at 90% of their respective annual bonus targets. Information about the dollar value of bonus awards granted for 2010 is set forth below in the Summary Compensation Table.
Long-Term Incentive Program
The purpose of the Company’s long-term incentive awards is to promote the long-term success of the Company by aligning the interests of the Company’s employees with the interests of its shareholders. The awards provide employees and executive officers an equity-based interest in the Company and thereby encourage those people to perform their duties to the best of their abilities, and to devote their time and effort to further the long-term growth and development of the Company. The awards are also intended to assist the Company in attracting and retaining individuals with superior experience and ability and providing competitive levels of compensation to such individuals. Subject to the approval of the Board of Directors, the plan allows for grants of options and tandem SARs. 2010 awards for NEOs were granted at levels reduced from 2008 and 2009 grants. Awards vary among participants based on their position, performance and relative contribution. Information about the awards granted in 2010 is set forth below in the Grant of Plan-Based Awards table.
Options/tandem SARs granted in 2010 were awarded at the TSX (for Canadian executives) or NASDAQ (for U.S. executives) closing price of the Company’s common stock on the business day immediately preceding the date of grant. Typically, options/tandem SARs vest 1/48th per month over the first four years of the five-year option term. Vesting and exercise rights cease immediately upon termination of employment for cause. In the case of termination without cause and voluntary termination, vesting rights cease upon termination of employment and exercise of previously vested options/tandem SARs cease after 30 days or expiration of the option, whichever is earlier. In the event of death, disability or retirement, vesting rights cease upon death, disability or retirement and exercise of previously vested cease after 365 days or the expiration of the option, whichever is earlier.
Long-term equity incentive awards are typically granted early spring following the Company’s release of year-end financial results.
Evaluation of Risk in Compensation Plans
The Board of Directors reviews the Company’s compensation plans as they relate to risk management practices and risk-taking incentives. In the judgment of the Board of Directors, the compensation plans currently in place are not likely to create risks that are reasonably likely to create a material adverse effect to the Company.
109
Table of Contents
Angiotech Stock Option Plans
In June 2006, the shareholders approved the adoption of the 2006 Stock Incentive Plan (the “2006 Stock Incentive Plan”) which superseded the Company’s previous stock option plans. The 2006 Stock Incentive Plan incorporated all of the options granted under the previous stock option plans and, in total, provides for the issuance of non-transferable stock options and stock-based awards (as defined below) to purchase up to 13,937,756 common shares to employees, officers, directors of the Company, and persons providing ongoing management or consulting services to the Company. The 2006 Stock Incentive Plan provides for, but does not require, the granting of tandem stock appreciation rights that at the option of the holder may be exercised instead of the underlying option. A stock option with a tandem stock appreciation right is referred to as an award. When the tandem stock appreciation right portion of an award is exercised, the underlying option is cancelled. The tandem stock appreciation rights are settled in equity and the optionee receives common shares with a fair market value equal to the excess of the fair value of the shares subject to the option at the time of exercise (or the portion thereof so exercised) over the aggregate option price of the shares set forth in the option agreement. The exercise price of the options and awards is fixed by the Board of Directors, but will generally be at least equal to the market price of the common shares at the date of grant, and for options issued under the 2006 Stock Incentive Plan and the Company’s 2004 Stock Option Plan, the term may not exceed five years. For options grandfathered from the stock option plans prior to the 2004 Stock Option Plan, the term did not exceed 10 years. Options and awards granted are also subject to certain vesting provisions. Options and awards generally vest monthly after being granted over varying terms from two to four years.
A summary of CDN$ stock option and award activity is as follows:
| No. of Stock Options and Awards |
Weighted average exercise price (in CDN$) |
Weighted
average remaining contractual term (years) |
Aggregate intrinsic value (in CDN$) |
|||||||||||||
| Outstanding at December 31, 2008 |
7,644,632 | $ | 12.82 | 2.67 | $ | 196 | ||||||||||
| Granted |
1,469,000 | 0.41 | ||||||||||||||
| Exercised |
(10,229 | ) | 0.21 | |||||||||||||
| Forfeited |
(1,266,694 | ) | 16.06 | |||||||||||||
| Cancelled |
(2,665,000 | ) | 17.73 | |||||||||||||
| Outstanding at December 31, 2009 |
5,171,709 | 5.99 | 3.18 | 2,800 | ||||||||||||
| Granted |
919,500 | 0.41 | ||||||||||||||
| Exercised |
(10,000 | ) | 0.21 | |||||||||||||
| Forfeited |
(806,025 | ) | 16.06 | |||||||||||||
| Outstanding at December 31, 2010 |
5,275,184 | $ | 3.83 | 2.75 | $ | 132 | ||||||||||
| Exercisable at December 31, 2010 |
3,049,990 | $ | 5.91 | 2.26 | $ | 67 | ||||||||||
| Exercisable and expected to vest at December 31, 2010 |
3,049,990 | $ | 5.91 | 2.26 | $ | 67 | ||||||||||
Notes:
| (1) | Exercisable and expected to vest at December 31, 2010 equals only those options exercisable at December 31, 2010 as no further options are expected to vest as all stock options under existing plans are to be cancelled as part of the recapitalization process. |
110
Table of Contents
A summary of U.S.$ award activity is as follows:
| No. of Stock Options and Awards |
Weighted average exercise price (in U.S.$) |
Weighted
average remaining contractual term (years) |
Aggregate intrinsic value (in U.S.$) |
|||||||||||||
| Outstanding at December 31, 2008 |
1,790,968 | $ | 3.19 | 4.19 | $ | 64 | ||||||||||
| Granted |
1,386,000 | 0.40 | ||||||||||||||
| Exercised |
(6,970 | ) | 0.26 | |||||||||||||
| Forfeited |
(125,250 | ) | 2.33 | |||||||||||||
| Cancelled |
(60,000 | ) | 18.00 | |||||||||||||
| Outstanding at December 31, 2009 |
2,984,748 | 1.64 | 3.74 | $ | 2,278 | |||||||||||
| Granted |
1,194,000 | 1.05 | ||||||||||||||
| Exercised |
(52,480 | ) | 0.26 | |||||||||||||
| Forfeited |
(235,269 | ) | 1.07 | |||||||||||||
| Outstanding at December 31, 2010 |
3,890,999 | $ | 1.51 | 3.14 | $ | 179 | ||||||||||
| Exercisable at December 31, 2010 |
1,846,660 | $ | 2.40 | 2.66 | $ | 92 | ||||||||||
| Exercisable and expected to vest at December 31, 2010 |
1,846,660 | $ | 2.40 | 2.66 | $ | 92 | ||||||||||
In connection with the implementation of the CCAA Plan, any options, warrants or other rights or entitlements to acquire or purchase common shares of Angiotech that are existing or issued and outstanding immediately prior to the implementation of the CCAA Plan, including any options to acquire common shares of Angiotech issued under the Company’s 2006 Stock Incentive Plan or the Company’s previous stock option plans, will be cancelled and terminated without any liability, payment or other compensation in respect thereof and the Company’s 2006 Stock Incentive Plan or the Company’s previous stock option plans will be terminated. It is anticipated that the Company will adopt a new stock incentive plan concurrent with the implementation of the CCAA Plan.
American Medical Instruments Stock Option Plan
On March 23, 2006, the Company acquired all of the issued and outstanding share capital of American Medical Instruments Holdings, Inc. (“AMI”). On March 9, 2006, AMI granted 304 stock options under AMI’s 2003 Stock Option Plan (the “AMI Stock Option Plan”), each of which is exercisable for approximately 3,852 common shares of the Company upon exercise. All outstanding options and warrants granted prior to the March 9, 2006 grant were settled and cancelled upon the acquisition of AMI. No further options to acquire common shares of the Company can be issued pursuant to the AMI Stock Option Plan. Approximately 1,171,092 of the Company’s common shares were reserved in March 2006 to accommodate future exercises of the AMI options.
The AMI options included in the above are all non-transferable and were granted to employees, officers and directors of AMI and persons providing ongoing management and consulting services to AMI. The options are subject to graded vesting over a four-year period after the second anniversary date of the grant date. Each option will expire on the earliest of (i) ten years from the date it is granted; (ii) 12 months after the participant dies; (iii) six months after the participant is disabled; (iv) 90 days after the participant retires or the participant’s employment is terminated, other than for cause; and (v) immediately upon the participant being terminated for cause. AMI options to purchase 300,480 of the Company’s common shares were outstanding as of December 31, 2010.
In connection with the implementation of the CCAA Plan, any options, warrants or other rights or entitlements to acquire or purchase common shares of Angiotech that are existing or issued and outstanding immediately prior to the implementation of the CCAA Plan, including any options to acquire common shares of Angiotech issued under the AMI Stock Option Plan, will be cancelled and terminated without any liability, payment or other compensation in respect thereof and the AMI Stock Option Plan (as well as the Company’s other stock option plans) will be terminated. It is anticipated that the Company will adopt a new stock incentive plan concurrent with the implementation of the CCAA Plan.
Retirement Savings Plans
NEOs participate in the retirement plans made available for all employees. In Canada, NEOs receive a Company contribution to an individual Group Registered Retirement Savings Plan (the “RRSP”) (a Canadian tax-qualified plan) in an amount equal to 5% of their base salary, up to the maximums allowable by Canada Revenue Agency. NEOs Dr. Hunter and Dr. Avelar received Company contributions of CDN$21,396 and CDN$20,137, respectively, to their RRSP. In the United States, NEOs receive a Company match of
111
Table of Contents
100% on their contributions up to the first 3% and a 50% match on their contributions up to the next 2% for a total potential Company match of 4% to the Company’s 401(k) plan (a U.S. tax-qualified plan). During 2010, Mr. Bryant received Company contributions of $10,704 and Mr. Bailey and Mr. McMasters did not participate in the U.S. employee 401(k) plan. All contributions to the retirement savings plans are fully vested.
The Company does not have a defined benefit pension plan for any of its employees, including executive officers.
Benefits Programs
The Company provides benefits to all employees including the NEOs. Benefits provided at the Company’s expense include:
| • | Medical and extended health care insurance; |
| • | Dental insurance; |
| • | Vision care insurance; |
| • | Employee and dependent life insurance; |
| • | AD&D insurance; and |
| • | Short- and long-term disability insurance. |
NEOs are also eligible to participate in an executive perquisite program. The program allows NEOs to be reimbursed up to a maximum of $15,000 per year for expenses associated with a car lease, financial planning, tax advice and preparation, and health-related services.
Key Employee Incentive Program
On September 30, 2010, the Company adopted a key employee incentive program (as subsequently amended, the “KEIP”), which is designed to incentivize certain of the Company’s key employees, including the NEOs, to successfully and promptly address certain of the Company’s outstanding financial obligations by giving these key employees an incentive payment tied to the accomplishment of these goals. Pursuant to the KEIP, the cash incentive awards payable thereunder will be paid in two installments upon the occurrence of each of the following performance milestones, subject to a participant’s continued employment by the Company or one of its subsidiaries through each applicable milestone date:
| • | Two-thirds (2/3) of the award will be paid upon the consummation of the Recapitalization Transaction; and |
| • | One-third (1/3) of the Award will be paid on the date that is 90 days following such consummation. |
Although Dr. William Hunter, our Chief Executive Officer, was granted an award under the KEIP, he elected not to receive his award in recognition of the Company’s current business needs. Other NEO awards are as follows: Bailey $289,380, McMasters $194,876, Avelar $119,306 and Bryant $60,005.
Employment Agreements
The Company maintains employment agreements with each of the NEOs. The employment agreements are generally effective until terminated by the Company or the executive and provide for minimum base salary and participation by each of the NEOs in the Company’s annual bonus program, the Company’s benefit plans and the applicable retirement plans maintained by the Company. The terms of the executive employment agreements that are applicable upon termination of an NEO’s employment with the Company are summarized below in the section of this Item 11 titled “— Potential Payments Upon Termination or Change of Control”.
112
Table of Contents
Summary Compensation Table for the Fiscal Year Ended December 31, 2010
The following tables contain certain information about the compensation and benefits paid or provided to each of the Chief Executive Officer, the Chief Financial Officer and the other three most highly compensated executive officers of the Company (determined based on total compensation) as at December 31, 2010 (collectively the “Named Executive Officers” or “NEOs”).
| Name and Principal |
Year | Salary ($) (1) |
Bonus ($) (9) |
Stock Awards ($) |
Option Awards ($) (2) |
Non-equity incentive plan compensation ($) (1)(3) |
Change in pension value and nonqualified deferred compensation earnings ($) |
All other compensation ($) (4)(5)(6)(7)(8) |
Total ($) | |||||||||||||||||||||||||||
| William L. Hunter, |
2010 | $ | 766,990 | Nil | Nil | $ | 240,902 | $ | 690,291 | Nil | $ | 96,549 | $ | 1,794,732 | ||||||||||||||||||||||
| MD, MSc President, |
2009 | $ | 691,769 | Nil | Nil | $ | 102,843 | $ | 363,200 | Nil | $ | 100,954 | $ | 1,258,766 | ||||||||||||||||||||||
| Chief Executive Officer and Director |
2008 | $ | 741,088 | Nil | Nil | $ | 60,970 | Nil | Nil | $ | 87,769 | $ | 889,827 | |||||||||||||||||||||||
| K. Thomas Bailey |
2010 | $ | 385,840 | Nil | Nil | $ | 125,004 | $ | 173,628 | Nil | $ | 90,820 | $ | 775,292 | ||||||||||||||||||||||
| Chief Financial |
2009 | $ | 364,000 | Nil | Nil | $ | 45,000 | $ | 127,400 | Nil | $ | 194,866 | $ | 731,266 | ||||||||||||||||||||||
| Officer |
2008 | $ | 350,000 | Nil | Nil | $ | 22,500 | Nil | Nil | $ | 218,087 | $ | 590,587 | |||||||||||||||||||||||
| Rui Avelar, |
2010 | $ | 399,432 | Nil | Nil | $ | 108,406 | $ | 143,796 | Nil | $ | 33,096 | $ | 684,730 | ||||||||||||||||||||||
| MD Chief Medical |
2009 | $ | 329,895 | Nil | Nil | $ | 41,137 | $ | 106,226 | Nil | $ | 28,711 | $ | 505,969 | ||||||||||||||||||||||
| Officer |
2008 | $ | 350,016 | Nil | Nil | $ | 24,388 | $ | 142,780 | Nil | $ | 38,276 | $ | 555,460 | ||||||||||||||||||||||
| David McMasters, |
2010 | $ | 487,190 | Nil | Nil | $ | 105,472 | $ | 175,388 | Nil | $ | 38,447 | $ | 806,497 | ||||||||||||||||||||||
| Esq., Senior Vice |
2009 | $ | 487,190 | Nil | Nil | $ | 36,000 | $ | 156,875 | Nil | $ | 35,535 | $ | 715,600 | ||||||||||||||||||||||
| President, Legal & General Counsel |
2008 | $ | 483,643 | Nil | Nil | $ | 18,000 | $ | 196,825 | Nil | $ | 44,915 | $ | 743,383 | ||||||||||||||||||||||
| Steve Bryant, |
2010 | $ | 293,288 | $ | 102,835 | Nil | $ | 39,064 | $ | 108,009 | Nil | $ | 21,552 | $ | 564,748 | |||||||||||||||||||||
| Senior Vice President, |
2009 | $ | 243,002 | Nil | Nil | $ | 3,960 | $ | 165,835 | Nil | $ | 14,084 | $ | 426,881 | ||||||||||||||||||||||
| Sales & Marketing, Medical Device Technologies |
2008 | $ | 204,173 | Nil | Nil | $ | 3,500 | $ | 72,704 | Nil | $ | 8,970 | $ | 289,347 | ||||||||||||||||||||||
Notes:
| (1) | Amounts paid to Dr. Hunter and Dr. Avelar were paid in Canadian dollars and, for the purposes of this table, converted to U.S. dollars using the average exchange rate of 0.971, 0.876 and 0.938 for each of the years ended December 31, 2010, December 31, 2009 and December 31, 2008, respectively. |
| (2) | Represents the fair value of option awards granted during the respective year calculated using the Black-Scholes option pricing model. |
| (3) | Non-equity incentive plan compensation for the NEOs includes annual bonus accrued at December 31 but paid subsequent to December 31. |
| (4) | Amounts included in all other compensation for 2010 for Dr. Hunter include: $33,842 of costs related to reimbursement of family travel costs (2009 — $52,488; 2008 — $41,204); $22,915 in auto lease payments paid by the Company on behalf of the NEO (2009 — $20,665; 2008 — $21,695); $20,775 of RRSP contributions made by the Company on behalf of the NEO (2009 — $17,294; 2008 — $18,527; 2007 — $18,783); and $96,549 of other miscellaneous perquisites (2009 — $10,507; 2008 — $6,343; 2007 — $5,991). |
| (5) | Amounts included in all other compensation for 2010 for Mr. Bailey include: $nil of relocation allowance (2009 — $118,508; 2008 — $182,875); $28,531 of rent paid by the Company on behalf of the NEO (2009 – $25,597; 2008 – $26,615); $50,757 in personal tax make-whole payments (2009 — $38,819; 2008 — $nil); $11,532 in auto lease payments paid by the Company on behalf of the NEO (2009 — $11,942; 2008 — $nil; 2007 — $nil); and $nil of other miscellaneous perquisites (2009 — $nil; 2008 — $8,593). |
| (6) | Amounts included in all other compensation for 2010 for Mr. McMasters include: $38,447 of rent paid by the Company on behalf of the NEO (2009 — $33,625; 2008 — $36,023); and $nil of other miscellaneous perquisites (2009 — $1,910; 2008 — $8,893). |
| (7) | Amounts included in all other compensation for 2010 for Dr. Avelar include: $13,545 in auto lease payments paid by the Company on behalf of the NEO (2009 — $12,216; 2008 — $13,087); $19,551 of RRSP contributions made by the Company on behalf of the NEO (2009 — $16,495; 2008 — $17,501); and $nil of other miscellaneous perquisites (2009 — $nil; 2008 — $7,688). |
| (8) | Amounts included in all other compensation for 2010 for Mr. Bryant include: $10,848 in auto lease payments paid by the Company on behalf of the NEO (2009 — $4,520; 2008 — $nil); and $10,704 of 401K contributions made by the company on behalf of the NEO (2009 — $9,564; 2008 — $8,970). |
| (9) | The amount paid to Mr. Bryant in 2010 under “Bonus” relates to a retention bonus paid to Mr. Bryant in that year. |
113
Table of Contents
Grant of Plan-Based Awards
All amounts are expressed in U.S. Dollars unless otherwise noted.
| Name |
Grant Date | Estimated Future Payouts Under Non-Equity Incentive Plans |
All Other Option Awards; Number of Securities Underlying Options (#) |
Exercise or Base Price of Option Awards ($/Sh) |
Grant Date Fair Value of Stock and Option Awards (1) |
|||||||||||||||||||||||
| Threshold ($) |
Target ($) |
Maximum ($) |
||||||||||||||||||||||||||
| William L. Hunter |
Mar 8, 2010 | Nil | Nil | Nil | 300,000 | CDN$ | 1.09 | CDN$ | 0.83 | |||||||||||||||||||
| K. Thomas Bailey |
Mar 8, 2010 | Nil | Nil | Nil | 160,000 | $ | 1.05 | $ | 0.78 | |||||||||||||||||||
| Rui Avelar |
Mar 8, 2010 | Nil | Nil | Nil | 135,000 | CDN$ | 1.09 | CDN$ | 0.83 | |||||||||||||||||||
| David McMasters |
Mar 8, 2010 | Nil | Nil | Nil | 135,000 | $ | 1.05 | $ | 0.78 | |||||||||||||||||||
| Steve Bryant |
Mar 8, 2010 | Nil | Nil | Nil | 50,000 | $ | 1.05 | $ | 0.78 | |||||||||||||||||||
Note:
| (1) | Represents the grant date fair value of option awards granted during the respective year calculated using the Black-Scholes option pricing model. |
114
Table of Contents
Outstanding Equity Awards at Fiscal Year-End
The table below shows option awards to NEOs outstanding as of December 31, 2010. All exercise prices are denoted in Canadian dollars unless otherwise specified.
| Option Awards | ||||||||||||||||||||
| Name |
Number of securities underlying unexercised options (#) exercisable |
Number of securities underlying unexercised options (#) unexercisable |
Equity incentive plan awards: number of securities underlying unexercised unearned options (#) |
Option exercise price ($) |
Option Expiration date |
|||||||||||||||
| William L. Hunter |
400,000 | — | — | $ | 13.468 | 12/17/2012 | ||||||||||||||
| 335,416 | 14,584 | (2) | — | $ | 8.900 | 2/4/2012 | ||||||||||||||
| 250,000 | 250,000 | (1) | — | $ | 0.210 | 12/7/2013 | ||||||||||||||
| 218,749 | 281,251 | (3) | — | $ | 0.380 | 9/03/2014 | ||||||||||||||
| 56,249 | 243,751 | (5) | — | $ | 1.090 | 7/03/2015 | ||||||||||||||
| K. Thomas Bailey |
191,666 | 8,334 | (2) | — | U.S.$ | 7.650 | 2/4/2012 | |||||||||||||
| 125,000 | 125,000 | (1) | — | U.S.$ | 0.160 | 12/7/2013 | ||||||||||||||
| 109,374 | 140,626 | (3) | — | U.S.$ | 0.270 | 9/03/2014 | ||||||||||||||
| 29,997 | 130,003 | (5) | — | U.S.$ | 1.060 | 7/03/2015 | ||||||||||||||
| Rui Avelar |
70,000 | — | — | $ | 13.468 | 12/17/2012 | ||||||||||||||
| 177,291 | 7,709 | (2) | — | $ | 8.900 | 2/4/2012 | ||||||||||||||
| 100,000 | 100,000 | (1) | — | $ | 0.210 | 12/7/2013 | ||||||||||||||
| 87,499 | 112,501 | (3) | — | $ | 0.380 | 9/03/2014 | ||||||||||||||
| 25,308 | 109,692 | (5) | — | $ | 1.090 | 7/03/2015 | ||||||||||||||
| David McMasters |
50,000 | — | — | $ | 13.468 | 12/17/2012 | ||||||||||||||
| 167,707 | 7,293 | (2) | — | U.S.$ | 7.650 | 2/4/2012 | ||||||||||||||
| 100,000 | 100,000 | (1) | — | U.S.$ | 0.160 | 12/7/2013 | ||||||||||||||
| 87,499 | 112,501 | (3) | — | U.S.$ | 0.270 | 9/3/2014 | ||||||||||||||
| 25,308 | 109,692 | (5) | — | U.S.$ | 1.060 | 7/03/2015 | ||||||||||||||
| Steve Bryant |
14,375 | 625 | (2) | — | U.S.$ | 7.650 | 2/4/2012 | |||||||||||||
| 13,020 | 11,980 | (6) | — | U.S.$ | 0.260 | 10/11/2013 | ||||||||||||||
| 9,623 | 12,377 | (3) | — | U.S.$ | 0.270 | 9/3/2014 | ||||||||||||||
| 9,369 | 40,631 | (5) | — | U.S.$ | 1.060 | 7/03/2015 | ||||||||||||||
Notes:
| (1) | Vests monthly over 48 months to December 2012. |
| (2) | Vests monthly over 48 months to February 2011. |
| (3) | Vests monthly over 48 months to March 2013. |
| (4) | Vests monthly over 48 months to July 2011. |
| (5) | Vests monthly over 48 months to March 2014. |
| (6) | Vests monthly over 48 months to November 2012. |
Option Exercises and Stock Vested in 2010
There were no options exercised by NEOs during the last completed fiscal year.
Potential Payments Upon Termination or Change of Control
The tables below reflect the amount of compensation payable to each of the NEOs in the event of termination of such executive’s employment. The amount of compensation payable to each NEO upon voluntary termination, involuntary without-cause termination, termination by executive for good reason, termination following change of control and in the event of death of the executive is also shown below. The amounts shown assume that such termination was effective as of December 31, 2010, and thus includes amounts earned through such time and are estimates of the amounts which would be paid out to the executives upon their termination. The
115
Table of Contents
amounts shown below assume a share price of $0.32 based on the December 31, 2010 closing price of the common shares on the NASDAQ. The actual amounts to be paid out can only be determined at the time of the executive’s separation from the Company.
Dr. Hunter
Termination Without Cause or Termination by Dr. Hunter for Good Reason:
| • | Under the terms of his employment agreement, if Dr. Hunter’s employment is terminated without cause or Dr. Hunter terminates his employment for good reason, and Dr. Hunter does not enter into a Consulting Agreement (described below), Dr. Hunter will receive a lump sum severance payment equivalent to 12 months’ salary, plus an amount equal to the average annual bonus payments received in the previous three years; and monthly payments equivalent to one-half of monthly salary for 24 months, plus an additional amount equivalent to the average annual bonus payments received in the previous three years paid in 24 equal monthly installments. Health and certain other benefits will also be continued for 24 months, or Dr. Hunter will receive an amount equal to the cost to the Company of providing those benefits. The estimated total of the foregoing payments is approximately $2.85 million. The Company’s obligation to provide the foregoing payments and benefits is conditional on Dr. Hunter’s compliance with a Confidentiality, Inventions and Non Competition Agreement, which, among other things, restricts competitive activity for one year following the termination date. |
| • | Under the terms of his employment agreement, outstanding vested stock options as of the termination date may be exercised within 30 days of the termination date, and, if not exercised, will be cancelled. |
Termination after Change of Control:
| • | Under the terms of his change of control agreement, if Dr. Hunter is terminated for any reason or resigns for good reason within 12 months after a change of control, Dr. Hunter will receive a lump sum payment equivalent to three years’ salary, plus an amount equal to three times the average annual bonus payments received in the previous three years. Health and certain other benefits will also be continued for 36 months. The estimated total of the foregoing payments is approximately $3.6 million. |
Consulting Agreement:
| • | If Dr. Hunter retires or resigns from his position as President and Chief Executive Officer, resigns for good reason, or is terminated without cause, the Company will offer Dr. Hunter a Consulting Agreement to provide services to the Company as a management consultant on a part-time basis for 24 months. |
| • | If Dr. Hunter enters into a Consulting Agreement, he will receive a lump sum payment equivalent to 12 months’ salary, plus an amount equal to the average annual bonus payments received in the previous three years; and monthly payments equivalent to one-half of monthly salary for 24 months, plus an additional amount equivalent to the average annual bonus payments received in the previous three years paid in 24 equal monthly installments. Health and certain other benefits will also be continued for 24 months, unless ongoing coverage is not permitted under any applicable group insurance plan, in which case the Company will provide, or reimburse Dr. Hunter for the cost of maintaining, comparable individual coverage. Outstanding but unvested stock options will also be deemed to be vested immediately to the extent those options would have vested within 24 months if Dr. Hunter had continued to be employed by the Company. |
| • | The Company’s obligations under the Consulting Agreement are conditional on Dr. Hunter’s compliance with a Confidentiality, Inventions and Non Competition Agreement, which, among other things, restricts his competitive activity with the Company for one year following termination of employment. |
| • | If Dr. Hunter enters into a Consulting Agreement following his retirement or resignation, except resignation for good reason, Dr. Hunter is also restricted from entering into other gainful employment for two years. |
| Executive Payments Upon Termination (in U.S. Dollars) |
Voluntary Termination by Executive Without Good Reason or For Cause Termination |
Without Cause Termination or Termination by Executive With Good Reason |
Without Cause Termination or Termination by Executive With Good Reason Within 12 Months After a Change of Control |
|||||||||
| Lump sum severance |
— | 2,260,585 | 2,298,900 | |||||||||
| Bonus |
— | 363,993 | 1,091,978 | |||||||||
| Lump sum compensation for loss of benefits |
— | 206,157 | 58,387 | |||||||||
| Exercise of options (1) |
— | 25,463 | (1) | 50,925 | (2) | |||||||
| TOTAL |
— | 2,856,198 | 3,500,190 | |||||||||
| (1) | Represents the intrinsic value of vested options held as of December 31, 2010. |
| (2) | Represents the intrinsic value of vested and unvested options held as of December 31, 2010 as a result of the accelerated vesting provision in a change of control situation. |
116
Table of Contents
Mr. Bailey, Mr. McMasters, Dr. Avelar, and Dr. Bryant
Under the terms of the executive employment agreements of Mr. Bailey, Mr. McMasters, Dr. Avelar and Dr. Gross, if the executive is terminated without cause or resigns for good reason he will receive:
| • | 12 months of base salary plus an additional two months of base salary for each full year of employment completed up to a combined maximum of 24 months of base salary; |
| • | Lump sum amount as compensation for loss of any benefits made available during employment in the amount of $24,000 plus an additional $2,000 for each full year of employment completed up to a combined maximum of $48,000; |
| • | The balance of any payment due under the bonus plan, including, if applicable, a prorated payment under the bonus plan earned in respect of the fiscal year of termination; |
| • | Outstanding vested stock options as of the termination date may be exercised within 30 days of the termination date, and, if not, will be cancelled; and |
| • | The Company’s obligation to provide the foregoing payments and benefits is conditional on the executive’s ongoing compliance with all applicable post-employment obligations which, among other things, restricts competitive activity for one year following termination date. |
Under the terms of the executive employment agreements, if Mr. Bailey, Mr. McMasters, Dr. Avelar and Mr. Bryant are Terminated Without Cause or Resign for Good Reason following a Change of Control they will receive:
| • | 24 months of base salary plus an additional two months of base salary for each full year of employment completed up to a combined maximum of 36 months of base salary; |
| • | a lump sum amount as compensation for loss of benefits made available during employment in the amount of $48,000 plus an additional $2,000 for each full year of employment completed up to a combined maximum of $72,000; |
| • | the balance of any payment due under the bonus plan, including, if applicable, a prorated payment under the bonus plan earned in respect of the fiscal year of termination; |
| • | a lump sum amount equal to two times the greater of the average of the payments made to the executive under the bonus plan in each of the two preceding fiscal years and the amount of executive’s target bonus opportunity; |
| • | a lump sum amount equivalent to the amount the executive would have received if he had been able to exercise equity-based incentives in the event that the vesting of such incentives does not accelerate under the provisions of such plans; |
| • | in the event that any payment made by the Company to executive is subject to excise tax under Section 4999 of the Code and the reduction of the amounts payable to the maximum amount that could be paid by the Company without triggering the excise tax would provide executive with a greater after-tax amount than if such amounts were not reduced, then the amounts payable to executive will be reduced to the safe harbor cap; and |
| • | in the event that the Change of Control is an unapproved Change of Control, payment subject to excise tax under Section 4999 of the Code will be grossed up by the sum of the excise tax and the product of any deductions disallowed because of the inclusion of the gross-up payment in executive’s adjusted gross income and the highest applicable marginal rate of federal income tax for the calendar year in which the gross-up payment is made. |
The Company’s obligation to provide the foregoing payments and benefits is conditional on the executive’s ongoing compliance with all applicable post-employment obligations which, among other things, restricts competitive activity for one year following termination date.
117
Table of Contents
The following table shows the potential payments upon termination for Mr. Bailey, Mr. McMasters, Dr. Avelar and Mr. Bryant. Payments upon termination will be calculated in accordance with each executive’s employment agreement.
| Name |
Voluntary Termination by Executive Without Good Reason or For Cause Termination |
Without Cause Termination or Termination by Executive With Good Reason |
Without Cause Termination or Termination by Executive With Good Reason Within 24 Months After a Change of Control |
Without Cause Termination or Termination by Executive With Good Reason Within 24 Months After an Unapproved Change of Control |
Death | Termination Resulting from Loss of Eligibility to Work in Canada/ U.S. |
||||||||||||||||||
| K. Thomas Bailey: |
||||||||||||||||||||||||
| Lump sum severance |
— | 771,680 | 1,157,520 | 1,157,520 | — | 771,680 | ||||||||||||||||||
| Bonus for fiscal year of termination (2010) |
— | 173,628 | 173,628 | 173,628 | 173,628 | 173,628 | ||||||||||||||||||
| Lump sum compensation for loss of benefits |
— | 36,000 | 60,000 | 60,000 | — | 60,000 | ||||||||||||||||||
| Further lump sum |
— | — | 385,840 | 385,840 | — | — | ||||||||||||||||||
| Exercise of options |
— | — | 25,469 | 25,469 | — | — | ||||||||||||||||||
| Safe harbor cap adjustment |
— | — | (158,530 | ) | — | — | — | |||||||||||||||||
| Gross up payment |
— | — | — | 517.939 | — | — | ||||||||||||||||||
| Total |
— | 981,308 | 1,643,927 | 2,320,396 | 127,400 | 981,308 | ||||||||||||||||||
| Rui Avelar: |
||||||||||||||||||||||||
| Lump sum severance |
— | 798,145 | 1,197,218 | 1,197,218 | — | 798,145 | ||||||||||||||||||
| Bonus for fiscal year of termination (2010) |
— | 143,666 | 143,666 | 143,666 | 143,666 | 143,666 | ||||||||||||||||||
| Lump sum compensation for loss of benefits |
— | 38,800 | 62,080 | 62,080 | — | 38,800 | ||||||||||||||||||
| Further lump sum |
— | — | 319,258 | 319,258 | — | — | ||||||||||||||||||
| Exercise of options |
— | — | 10,185 | 10,185 | — | — | ||||||||||||||||||
| Total |
— | 980,611 | 1,732,407 | 1,732,407 | 143,666 | 980,611 | ||||||||||||||||||
| Steve Bryant: |
||||||||||||||||||||||||
| Lump sum severance |
— | 600,050 | 900,075 | 900,075 | — | 600,050 | ||||||||||||||||||
| Bonus for fiscal year of termination (2010) |
— | 108,009 | 108,009 | 108,009 | 108,009 | 108,009 | ||||||||||||||||||
| Lump sum compensation for loss of benefits |
— | 44,000 | 68,000 | 68,000 | — | 44,000 | ||||||||||||||||||
| Further lump sum |
— | — | 240,020 | 240,020 | — | — | ||||||||||||||||||
| Exercise of options |
— | — | 1,262 | 1,262 | — | — | ||||||||||||||||||
| Safe harbor cap adjustment |
— | — | — | — | — | — | ||||||||||||||||||
| Gross up payment |
— | — | — | — | — | — | ||||||||||||||||||
| Total |
— | 752,059 | 1,317,366 | 1,317,366 | 108,009 | 752,059 | ||||||||||||||||||
| David McMasters: |
||||||||||||||||||||||||
| Lump sum severance |
— | 974,380 | 1,461,570 | 1,461,570 | — | 974,380 | ||||||||||||||||||
| Bonus for fiscal year of termination (2010) |
— | 175,388 | 175,388 | 175,388 | 175,388 | 175,388 | ||||||||||||||||||
| Lump sum compensation for loss of benefits |
— | 44,000 | 68,000 | 68,000 | — | 44,000 | ||||||||||||||||||
| Further lump sum |
— | — | 389,752 | 389,752 | — | — | ||||||||||||||||||
| Exercise of options |
— | — | 20,375 | 20,375 | — | — | ||||||||||||||||||
| Safe harbor cap adjustment |
— | — | — | — | — | — | ||||||||||||||||||
| Gross up payment |
— | — | — | — | — | — | ||||||||||||||||||
| Total |
— | 1,193,768 | 2,115,085 | 2,115,085 | 175,388 | 1,193,768 | ||||||||||||||||||
Amendments to Employment Agreements as a Condition of CCAA Plan
As a condition to the implementation of the CCAA Plan, Dr Hunter is expected to enter into an amendment to his exsting employement agreement (the “Hunter Amendment”). The Hunter Amendment, which will become effective upon implementation of the CCAA Plan, provides, among other things, the following: (i) the transactions contemplated by the RSA will not constitute a “change of control” for purposes of the Hunter Executive Employment Agreement or the Hunter Executive Change of Control Agreement; (ii) in the event of a “change of control” (as defined in the Hunter Change of Control Agreement), all of Dr. Hunter’s equity-based incentive awards will vest and, if applicable, will remain exercisable through the expiration of their original terms; and (iii) in the event Dr. Hunter is terminated for any reason or resigns for “good reason” (as is defined in the Hunter Employment Agreement) upon or within the 12 months following a “change of control,” he will be entitled to the same severance payments and benefits to which he would have been entitled had such termination occurred prior to the “change of control.” The Hunter Amendment also provides that in the event Dr. Hunter is terminated without “cause” or resigns for “good reason,” all of his equity-based incentive awards will vest and, if applicable, will remain exercisable until the earlier of: (i) the first anniversary of his termination date; and (ii) the expiration of the original terms of the awards; provided, that if the stock underlying the awards is not publicly traded at the time of termination, the awards will remain exercisable until the earlier of: (x) the first anniversary of the date
118
Table of Contents
the stock becomes publicly traded, and (y) the expiration of the original terms of the awards. Pursuant to the Hunter Amendment, Dr. Hunter will also agree not to exercise his rights to enter into a consulting agreement for at least one year following the implementation of the CCAA Plan.
Also as a condition to the implementation of the CCAA Plan, executive officers including NEO’s will enter into an amendment to their existing employment agreement (the “Executive Amendments”). The Executive Amendments, which will become effective upon the Plan Implementation Date, provide, among other things, the following: (i) the transactions contemplated by the RSA will not constitute a “change of control” for purposes of the executive employment agreements; (ii) in the event of a “change of control” (as defined in the executive employment agreements), all of the executives’ equity-based incentive awards will vest and, if applicable, will remain exercisable through the expiration of their original terms; and (iii) in the event the executives are terminated without “cause” or resign for “good reason,” they will be entitled to the same severance payments, regardless of whether such termination occurs before or after a “change of control.” The Executive Amendments also provide that in the event the executives are terminated without “cause” or resign for “good reason,” all of their equity-based incentive awards will vest and, if applicable, will remain exercisable until the earlier of: (i) the first anniversary of his termination date; and (ii) the expiration of the original terms of the awards; provided, that if the stock underlying the awards is not publicly traded at the time of termination, the awards will remain exercisable until the earlier of: (x) the first anniversary of the date the stock becomes publicly traded, and (y) the expiration of the original terms of the awards.
119
Table of Contents
Compensation of Directors
Amounts paid to non-executive directors during the year ended December 31, 2010 are presented in the table below.
| Name |
Fees earned or paid in cash ($) (1) |
Stock awards ($) |
Option awards ($) |
Non equity incentive plan compensation ($) |
Change in pension value and nonqualified deferred compensation earnings |
All other compensation ($) |
Total ($) | |||||||||||||||||||||
| David T. Howard |
$ | 137,000 | — | — | — | — | — | $ | 137,000 | |||||||||||||||||||
| Arthur H. Willms |
$ | 117,000 | — | — | — | — | — | $ | 117,000 | |||||||||||||||||||
| Edward M. Brown |
$ | 71,000 | — | — | — | — | — | $ | 71,000 | |||||||||||||||||||
| Henry McKinnell |
$ | 113,000 | — | — | — | — | — | $ | 113,000 | |||||||||||||||||||
| Laura Brege |
$ | 83,000 | — | — | — | — | — | $ | 83,000 | |||||||||||||||||||
Notes:
| (1) | Includes fees for Board of Directors and committee membership. |
For the 12-month period ended December 31, 2010, each director who was not an employee (“a non-employee director”) was paid in quarterly installments as follows:
| Chair of the Board of Directors retainer |
$ | 75,000 per annum | ||
| Board of Directors member retainer |
$ | 35,000 per annum | ||
| Committee Chair retainer |
$ | 20,000 per annum | ||
| Board of Directors and Committee attendance fee |
$ | 2,000 per meeting |
Upon initially being elected or appointed to the Board of Directors, each independent director will be granted an option to purchase 25,000 common shares of the Company. On an annual basis thereafter, occurring at the time of the annual general meeting of the Company and December 1 of each year, if he or she continues to be an independent director of the Company, he or she will be granted a further option to purchase 12,500 common shares. Options granted are typically exercisable for a period of five years from the grant date, are granted at market price and typically vest over a period of six or 12 months.
For year ended December 31, 2010, the independent directors of the Company were paid approximately $521,000 in aggregate for their service on the Board of Directors. Independent directors of the Company were not issued stock options during the year ended December 31, 2010.
The compensation of independent directors is reviewed annually. The Governance, Nominating and Compensation Committee reviewed data on the amount of directors’ compensation and the market rates for comparable companies with a similar size capitalization and complexity in the U.S. and Canadian medical device and specialty pharmaceutical industry. Based on this information, the Governance, Nominating and Compensation Committee provides a report to the Board of Directors recommending any changes to compensation.
Compensation Committee Interlocks And Insider Participation
No member of the Compensation Committee is or has been a former or current executive officer or employee of the Company. None of our executive officers served as a director or member of a compensation committee (or other committee serving an equivalent function) of any other entity whose executive officers served as a director or member of our Compensation Committee during the fiscal year ended December 31, 2010.
120
Table of Contents
Report of the Governance, Nominating and Compensation Committee
The Company’s Compensation Discussion and Analysis is included in this Annual Report on Form 10-K. The Company has the primary responsibility for the Compensation Discussion and Analysis.
On behalf of the Board of Directors, the Governance, Nominating and Compensation Committee oversees the development, review and approval of the Company’s compensation program for executive officers. As part of this responsibility, the Governance, Nominating and Compensation Committee has reviewed and discussed with the Company’s management the contents of the Compensation Discussion and Analysis. Based on its review and discussion, and subject to the limitations on the role and responsibility of the Governance, Nominating and Compensation Committee, the Committee recommended to the Board of Directors that the Compensation Discussion and Analysis be included in this Form 10-K and incorporated into the Company’s Proxy Statement for the year ended December 31, 2010.
THE GOVERNANCE, NOMINATING AND COMPENSATION COMMITTEE
Henry McKinnell (Chair)
David Howard
Arthur Willms
Edward Brown
Laura Brege
121
Table of Contents
| Item 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
Equity Compensation Plan Information
The following table sets out information relating to the Company’s equity compensation plans in place at December 31, 2010. The only equity compensation plans the Company has are the 2006 Stock Incentive Plan, pursuant to which the Company grants options in Canadian or U.S. dollars and the AMI Stock Option Plan. The 2006 Stock Incentive Plan and the AMI Stock Option Plan are described above under “Executive Compensation — Angiotech Stock Option Plans.”
In connection with the implementation of the CCAA Plan, all outstanding any options, warrants or other rights or entitlements to acquire or purchase common shares of Angiotech that are existing or issued and outstanding immediately prior to the implementation of the CCAA Plan, including any options to acquire common shares of Angiotech issued under the Company’s equity compensation plans, will be cancelled and terminated without any liability, payment or other compensation in respect thereof and the Company’s equity compensation plans will be terminated. It is anticipated that the Company will adopt a new stock incentive plan concurrent with the implementation of the CCAA Plan.
| Plan Category |
Number of securities to be issued upon exercise of outstanding options and rights (a) |
Weighted-average exercise price of outstanding options and rights (b) |
Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) (c) |
|||||||||
| Equity compensation plans approved by securityholders: |
||||||||||||
| 2006 Stock Incentive Plan — CDN$ grants |
5,275,184 | CDN$ | 3.83 | 4,242,866 | (1) | |||||||
| 2006 Stock Incentive Plan — U.S.$ grants |
3,890,999 | U.S.$ | 1.51 | |||||||||
| Equity compensation plans not approved by securityholders: |
||||||||||||
| AMI Stock Option Plan |
300,480 | (2) | U.S.$ | 15.44 | Nil | |||||||
| Total |
9,466,663 | 4,242,866 | ||||||||||
Notes:
| (1) | Available for future issuance under the 2006 Stock Incentive Plan in either Canadian or U.S. dollars. |
| (2) | Represents the number of Angiotech common shares issuable upon the exercise of AMI options. |
Security Ownership of Certain Beneficial Owners and Management
The authorized capital of the Company consists of 250,000,000 shares without par value divided into 200,000,000 common shares and 50,000,000 Class I Preference shares.
As of March 1, 2011, there were 85,185,497 common shares and no Class I Preference shares issued and outstanding.
Beneficial Owners of More than Five Percent
As of March 1, 2011, to the knowledge of the directors and executive officers of the Company, no person or group beneficially owns, directly or indirectly, or exercises control or direction over, greater than 5% of any class of voting securities of the Company other than the following:
| Title of Class |
Name and Address of Beneficial Holder |
Amount and Nature of Beneficial Ownership (owned or controlled or directed, directly or indirectly) |
% of | |||||
| Common |
William L. Hunter, MD, MSc. (2) | 7,296,822 | 8.4 % | |||||
Notes:
| (1) | In accordance with Rule 13d-3(d)(1) under the Exchange Act, the applicable percentage of ownership for each person is based on 85,190,317 common shares outstanding as of March 1, 2011, plus any securities held by such person exercisable for or convertible into common shares within 60 days after the date of this Annual Report on Form 10-K. |
| (2) | (i) The address of William L. Hunter and Cathryn Hunter is 1618 Station Street, Vancouver, BC V6A 1B6, Canada and (ii) the address of |
122
Table of Contents
| Hunter Limited Partnership and K-Bunny Ventures, LTD. is c/o Lawson Lundell LLP, 1600 — 925 Georgia St. W., Vancouver, BC V6C 3L2, Canada. As of March 1, 2011, (a) William L. Hunter has sole voting power and sole dispositive power over 80,396 shares and shared voting power and shared dispositive power over 7,216,426 shares, (b) Cathryn Hunter has shared voting power and shared dispositive power over 7,216,426 shares and (c) Hunter Limited Partnership and K-Bunny Ventures, LTD. have sole voting power and sole dispositive power over 80,396 shares. |
Beneficial Ownership of Directors and Named Executive Officers
As at March 1, 2011, the number of common shares beneficially owned by each director and Named Executive Officer (defined below) of the Company, or over which each director and Named Executive Officer exercises control or direction, directly or indirectly, is set out in the following table:
| Title of Class |
Name and Address of Beneficial Holder |
Amount and Nature of Beneficial Ownership (owned or controlled or directed, directly or indirectly) |
% of
Class (1) |
|||||||
| Common |
William L. Hunter, MD, MSc. British Columbia, Canada |
7,296,822 | (2) | 8.4 | % | |||||
| Common |
David T. Howard British Columbia, Canada |
275,717 | 0.3 | % | ||||||
| Common |
Edward M. Brown California, U.S.A. |
42,500 | 0.1 | % | ||||||
| Common |
Arthur H. Willms British Columbia, Canada |
128,260 | (3) | 0.2 | % | |||||
| Common |
Laura Brege California, U.S.A. |
42,500 | 0.1 | % | ||||||
| Common |
Henry A. McKinnell Jr. Wyoming, U.S.A. |
87,500 | 0.1 | % | ||||||
| Common |
K. Thomas Bailey Washington, U.S.A. |
549,370 | 0.6 | % | ||||||
| Common |
Rui Avelar, MD British Columbia, Canada |
9,800 | (4) | 0.0 | % | |||||
| Common |
David D. McMasters, ESQ Washington, U.S.A. |
435,188 | (5) | 0.5 | % | |||||
| Common |
Steve Bryant South Carolina, U.S.A. |
88,801 | 0.1 | % | ||||||
Notes:
| (1) | In accordance with Rule 13d-3(d)(1) under the Exchange Act, the applicable percentage of ownership for each person is based on 85,190,317 common shares outstanding as of March 1, 2011, plus any securities held by such person exercisable for or convertible into common shares within 60 days after the date of this Annual Report on Form 10-K. |
| (2) |
Dr. Hunter’s holdings include 5,495,435 common shares held indirectly through his wife. |
| (3) |
Mr. Willms’ holdings include 260 common shares held indirectly through his wife. |
| (4) |
Dr. Avelar’s holdings include 3,684 common shares held indirectly through his wife. |
| (5) |
Mr. McMasters’ holdings include 2,000 common shares held indirectly through his wife. |
As of March 1, 2011, directors and executive officers of the Company beneficially owned or exercised control or direction over, directly or indirectly, 9,984,647 common shares representing approximately 11.7% of the outstanding common shares in the aggregate.
123
Table of Contents
| Item 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
Related-Party Transactions Policy
Pursuant to our Audit Committee Charter, all transactions between us and any of our directors, executive officers or other related parties are subject to review by our audit committee. Our audit committee, in its sole discretion, is permitted to consider any factors it deems appropriate in approving any such related-party transactions.
Certain Relationships and Related Party Transactions
Since January 1, 2009, there has not been, nor is there currently proposed, any transaction or series of similar transactions to which we were or are a party in which the amount involved exceeds $120,000 and in which any director, executive officer or beneficial holder of more than 5% of any class of our voting securities or members of such person’s immediate family had or will have a direct or indirect material interest.
As of March 1, 2011 the Company is not aware of any proceeding or action involving a director, executive officer or nominee which would be adverse to the Company.
Director Independence
Our Board of Directors currently consists of five independent members and one executive member. The following sets out the identity of our independent and non-independent directors.
| Name of Director |
Independent/ Non-Independent | |
| William L. Hunter |
Non-Independent | |
| David T. Howard (Chair) |
Independent | |
| Edward M. Brown |
Independent | |
| Laura Brege |
Independent | |
| Arthur H. Willms |
Independent | |
| Henry A. McKinnell Jr. |
Independent |
Under both SOX and the NASDAQ rules, the independence of a director is determined based upon certain criteria which evaluate whether the director has a relationship with the Company that would interfere with the exercise of the director’s independent business judgment. At present, five out of six of the Company’s directors would be considered independent under these rules.
The charters of both the Audit Committee and the Governance, Nominating, and Compensation Committee of the Board of Directors stipulate that each be comprised of at least three directors, each of whom is unrelated and independent. The Audit Committee is currently comprised of five members — Arthur Willms (Chair), David Howard, Edward Brown, Laura Brege and Henry McKinnell. All of the members of the Audit Committee are independent of management. The Governance, Nominating and Compensation Committee is currently comprised of five members — Henry McKinnell (Chair), Edward Brown, David Howard, Laura Brege and Arthur Willms. Each of the members of the Governance, Nominating and Compensation Committee is independent of management.
124
Table of Contents
| Item 14. | PRINCIPAL ACCOUNTANT FEES AND SERVICES |
The firm of PricewaterhouseCoopers LLP, Chartered Accountants, of 250 Howe Street, British Columbia (“PWC”) served as independent auditors for the Company for the year ended December 31, 2009. PWC was first appointed as independent auditors of the Company on June 8, 2006. The Audit Committee pre-approves all services provided by PWC.
The aggregate fees for professional services rendered by the independent auditors, PWC, for the Company for the years ending December 31, 2010 and December 31, 2009 totaled $1,835,643 and $1,485,186, respectively, as detailed in the following table. All amounts are in U.S. dollars and have been converted from Canadian dollars using an average foreign exchange rate for the period indicated:
| (in U.S. dollars) |
Year Ended December 31, 2010 |
Year Ended December 31, 2009 |
||||||
| Audit Fees |
$ | 1,611,746 | $ | 1,377,038 | ||||
| Tax Fees |
$ | 69,519 | $ | 54,293 | ||||
| All Other Fees |
$ | 154,378 | $ | 53,855 | ||||
| TOTAL |
$ | 1,835,643 | $ | 1,485,186 | ||||
The nature of the services provided by PWC under each of the categories indicated in the foregoing table is described below.
Audit Fees
Audit fees were for professional services rendered for the audit of the Company’s annual financial statements (including internal control over financial reporting requirements under Section 404 of SOX), quarterly reviews of interim financial statements and services provided in connection with regulatory filings or engagements.
Tax Fees
Tax fees were for tax compliance, tax advice and tax planning professional services. These services consisted of tax compliance, including the review of tax returns and tax planning and advisory services relating to common forms of domestic and international taxation (i.e. income tax, capital tax, goods and services tax, value added tax and payroll tax).
All Other Fees
In 2010, “All Other Fees” related to services in regards to the Company’s proxy circular. In 2009, “All Other Fees” related primarily to services in regards to auditor consents required for the Company’s S-3 and S-8 filings.
Pre-approval Policies and Procedures
The Audit Committee must pre-approve any audit or any permissible non-audit services to be provided by the independent auditors and has established pre-approval policies and procedures. All audit and permissible non-audit fees are pre-approved by the Audit Committee on a project-by-project basis. Before approving the services described under “Tax Fees” above, the Audit Committee reviewed whether the independent auditors could provide those services and maintain its independence. The Audit Committee approved 100% of the audit-related and tax fees for 2010 and 2009.
125
Table of Contents
| Item 15. | EXHIBITS AND FINANCIAL STATEMENT SCHEDULES |
| (a) | The following documents are included as part of this Annual Report on Form 10-K. |
| (1) | Financial Statements: |
| † | Page numbers refer to Exhibit 13 of this Annual Report on Form 10-K. |
| (2) | Financial Statement Schedule: |
| Page † | ||
| Schedule II — Valuation of Qualifying Accounts for the years ended December
31, 2010, 2009 and |
F-53 |
| † | Page numbers refer to Item F of this Annual Report on Form 10-K. |
All other schedules are omitted as the information required is inapplicable or the information is presented in the consolidated financial statements or related notes.
| (3) | Exhibits: |
The exhibits listed below are filed as part of this Annual Report on Form 10-K. References under the caption “Location” to exhibits or other filings indicate that the exhibit or other filing has been filed, that the indexed exhibit and the exhibit or other filing referred to are the same and that the exhibit or other filing referred to is incorporated by reference. Management contracts and compensatory plans or arrangements filed as exhibits to this Annual Report on Form 10-K are identified by an asterisk. The Commission file number for our Exchange Act filings referenced below is 000-30334.
| Exhibit |
Description |
Location | ||
| 3.1 | Notice of Articles of Angiotech Pharmaceuticals, Inc. | Exhibit 4.1, Form S-3 filed July 23, 2009 | ||
| 3.2 | Articles of Angiotech Pharmaceuticals, Inc. | Exhibit 3, Form 6-K filed March 31, 2005 | ||
| 3.3 | Amendment to the Articles of Angiotech Pharmaceuticals, Inc. | Exhibit 4.3, Form S-3 filed July 23, 2009 | ||
| 4.1 | Indenture dated March 23, 2006 among Angiotech Pharmaceuticals, Inc., certain of its subsidiaries and Wells Fargo Bank N.A., for 7.75% Senior Subordinated Notes due 2014 | Exhibit 99.2, Form 6-K filed April 3, 2006 | ||
| 4.2 | Indenture dated December 11, 2006 among Angiotech Pharmaceuticals, Inc., certain of its subsidiaries and Wells Fargo Bank N.A., for Senior Floating Rate Notes due 2013 | Exhibit 2, Form 6-K filed December 15, 2006 | ||
| 4.3 | Supplemental Indenture dated June 8, 2010 among Angiotech America, Inc., Angiotech Florida Holdings, Inc., Angiotech Pharmaceuticals, Inc., certain of its subsidiaries and Deutsche Bank National Trust Company, for 7.75% Senior Subordinated Notes due 2014. | Exhibit 10.2, Form 10-Q filed August 9, 2010 | ||
| 4.4 | Supplemental Indenture dated June 8, 2010 among Angiotech America, Inc., Angiotech Florida Holdings, Inc., Angiotech Pharmaceuticals, Inc., certain of its subsidiaries and Deutsche Bank National Trust Company, for Senior Floating Rate Notes due 2013. | Exhibit 10.3, Form 10-Q filed August 9, 2010 | ||
126
Table of Contents
| Exhibit |
Description |
Location | ||
| 4.5 | Supplemental Indenture, dated October 29, 2010, between Angiotech Pharmaceuticals, Inc. and U.S. Bank National Association, relating to the Company’s 7.75% Subordinated Notes. | Exhibit 10.3, Form 8-K filed November 2, 2010 | ||
| 4.6 | Supplemental Indenture, dated November 29, 2010, between Angiotech Pharmaceuticals, Inc. and U.S. Bank National Association, relating to the Company’s 7.75% Subordinated Notes. | Exhibit 10.3, Form 8-K filed December 2, 2010 | ||
| 4.7 | Supplemental Indenture, dated December 21, 2010, between Angiotech Pharmaceuticals, Inc. and U.S. Bank National Association, relating to the Company’s 7.75% Subordinated Notes. | Exhibit 10.1, Form 8-K filed December 23, 2010 | ||
| 4.8 | Supplemental Indenture, dated January 27, 2011, between Angiotech Pharmaceuticals, Inc. and U.S. Bank National Association, relating to the Company’s 7.75% Subordinated Notes. | Exhibit 10.3, Form 8-K filed January 28, 2011 | ||
| 4.9 | Shareholder Rights Plan Agreement between Angiotech Pharmaceuticals, Inc. and Computershare Trust Company of Canada as Rights Agent, Amended and Restated as of October 30, 2008 | Exhibit 4.3, Form 10-K filed March 16, 2009 | ||
| 10.1 | Credit Agreement, dated as of February 27, 2009, by and among Angiotech Pharmaceuticals, Inc., as Parent, the subsidiaries of Parent listed as borrowers on the signature pages thereto, as Borrowers, the lenders signatory thereto, as Lenders, and Wells Fargo Foothill, LLC, as Arranger and Administrative Agent | Exhibit 10.1, Form 8-K filed March 5, 2009 | ||
| 10.2 | First Amendment, dated as of May 29, 2009, to Credit Agreement, dated as of February 27, 2009, by and among Angiotech Pharmaceuticals, Inc., as Parent, the subsidiaries of Parent listed as borrowers on the signature pages thereto, as Borrowers, the lenders signatory thereto, as Lenders, and Wells Fargo Foothill, LLC, as Arranger and Administrative Agent | Exhibit 10.1, Form 8-K filed June 3, 2009 | ||
| 10.3 | Second Amendment, dated as of November 23, 2009, to Credit Agreement, dated as of February 27, 2009, by and among Angiotech Pharmaceuticals, Inc., as Parent, the subsidiaries of Parent listed as borrowers on the signature pages thereto, as Borrowers, the lenders signatory thereto, as Lenders, and Wells Fargo Foothill, LLC, as Arranger and Administrative Agent | Exhibit 10.3, Form 10-K/A filed April 29, 2010 | ||
| 10.4 | Third Amendment to Credit Agreement, dated as of April 21, 2010, by and among Angiotech, as Parent, the subsidiaries of Parent listed as borrowers on the signature pages thereto, as borrowers, the lenders signatory thereto, as lenders, and Wells Fargo Capital Finance, LLC (f/k/a Wells Fargo Foothill, LLC), as arranger and administrative agent. | Exhibit 10.1, Form 8-K filed April 27, 2010 | ||
| 10.5 | Fourth Amendment to Credit Agreement, dated as of November 4, 2010, by and among Angiotech Pharmaceuticals, Inc., as Parent, the subsidiaries of Parent listed as borrowers on the signature pages thereto, as borrowers, the lenders signatory thereto, as lenders, and Wells Fargo Capital Finance, LLC (f/k/a Wells Fargo Foothill, LLC), as arranger and administrative agent. | Exhibit 10.1, Form 8-K filed November 9, 2010 | ||
| 10.6 | Credit Agreement, dated as of February 7, 2011, among Angiotech Pharmaceuticals, Inc., certain of its subsidiaries, the lenders signatory thereto and Wells Fargo Capital Finance, LLC, as arranger and administrative agent. | Exhibit 10.3, Form 8-K filed February 10, 2011 | ||
| 10.7 | License Agreement by and among Angiotech Pharmaceuticals, Inc., Boston Scientific Corporation and Cook Incorporated, dated as of July 9, 1997 | Exhibit, Form 20-FR filed October 5, 1999 | ||
| 10.8 | September 24, 2004 Amendment Between Angiotech Pharmaceuticals, Inc. and Cook Incorporated Modifying July 9, 1997 License Agreement Among Angiotech Pharmaceuticals, Inc., Boston Scientific Corporation, and Cook Incorporated** | Exhibit 10.3, Form 10-K filed March 16, 2009 | ||
| 10.9 | May 20, 2010 Amendment between Angiotech Pharmaceuticals, Inc. and Cook Incorporated to the July 9, 1997 License Agreement among Angiotech Pharmaceuticals, Inc., Boston Scientific Corporation, and Cook Incorporated.** | Exhibit 10.1, Form 10-Q filed August 9, 2010 | ||
127
Table of Contents
| Exhibit |
Description |
Location | ||
| 10.10 | Distribution and License Agreement by and among Angiodevice International GmbH (as assignee of Angiotech Pharmaceuticals, Inc., Angiotech International, GmbH, and Cohesion Technologies, Inc.), Baxter Healthcare Corporation and Baxter Healthcare, S.A., dated April 1, 2003** | Exhibit 10.5, Form 10-K filed March 16, 2009 | ||
| 10.11 | Manufacturing and Supply Agreement, by and among Angiodevice International GmbH (as assignee of Angiotech Pharmaceuticals, Inc., Angiotech International, GmbH, and Cohesion Technologies, Inc.), Baxter Healthcare Corporation and Baxter Healthcare, S.A., dated April 1, 2003** | Exhibit 10.6, Form 10-K filed March 16, 2009 | ||
| 10.12 | Amendment No. 1 dated December 23, 2004 to Distribution and License Agreement, by and among Angiodevice International GmbH (as assignee of Angiotech Pharmaceuticals, Inc., Angiotech International, GmbH, and Cohesion Technologies, Inc.), Baxter Healthcare Corporation and Baxter Healthcare, S.A., dated April 1, 2003 | Exhibit 10.7, Form 10-K filed March 16, 2009 | ||
| 10.13 | Amendment No. 1 dated January 21, 2005 to Manufacturing and Supply Agreement, by and among Angiodevice International GmbH (as assignee of Angiotech Pharmaceuticals, Inc., Angiotech International, GmbH, and Cohesion Technologies, Inc.), Baxter Healthcare Corporation and Baxter Healthcare, S.A., dated April 1, 2003 | Exhibit 10.8, Form 10-K filed March 16, 2009 | ||
| 10.14 | Amendment No. 2 dated October 8, 2007 to Distribution and License Agreement, by and among Angiodevice International GmbH (as assignee of Angiotech Pharmaceuticals, Inc., Angiotech International, GmbH, and Cohesion Technologies, Inc.), Baxter Healthcare Corporation and Baxter Healthcare, S.A., dated April 1, 2003** | Exhibit 10.9, Form 10-K filed March 16, 2009 | ||
| 10.15 | Amended and Restated Distribution and License Agreement by and among Angiotech Pharmaceuticals (U.S.) Inc., Angiodevice International GmbH, Baxter Healthcare Corporation and Baxter Healthcare, S.A, dated as of January 1, 2009** | Exhibit 10.1, Form 10-Q filed May 8, 2009 | ||
| 10.16 | License Agreement between Public Health Service (through the Office of Technology Transfer, National Institutes of Health) and Angiotech Pharmaceuticals, Inc., dated November 19, 1997** | Exhibit 10.11, Form 10-K filed March 16, 2009 | ||
| 10.17 | First Amendment between Public Health Service (through the Office of Technology Transfer, National Institutes of Health) and Angiotech Pharmaceuticals, Inc., dated March 28, 2002 modifying the License Agreement between Public Health Service (through the Office of Technology Transfer, National Institutes of Health) and Angiotech Pharmaceuticals, Inc., dated November 19, 1997** | Exhibit 10.12, Form 10-K filed March 16, 2009 | ||
| 10.18 | Second Amendment between Public Health Service (through the Office of Technology Transfer, National Institutes of Health) and Angiotech Pharmaceuticals, Inc., dated May 23, 2007 modifying the License Agreement between Public Health Service (through the Office of Technology Transfer, National Institutes of Health) and Angiotech Pharmaceuticals, Inc., dated November 19, 1997** | Exhibit 10.13, Form 10-K filed March 16, 2009 | ||
| 10.19 | License Agreement between Angiotech Pharmaceuticals, Inc. and The University of British Columbia, dated August 1, 1997** | Exhibit 10.14, Form 10-K filed March 16, 2009 | ||
| 10.20 | Amendment to License Agreement between Angiotech Pharmaceuticals, Inc. and The University of British Columbia, dated February 27, 2004, modifying the License Agreement between Angiotech Pharmaceuticals, Inc. and The University of British Columbia, dated August 1, 1997** | Exhibit 10.15, Form 10-K filed March 16, 2009 | ||
| 10.21* | Form of U.S. Director Indemnification Agreement | Exhibit 10.2, Form 8-K filed October 4, 2010 | ||
| 10.22* | Form of Canadian Director Indemnification Agreement | Exhibit 10.3, Form 8-K filed October 4, 2010 | ||
| 10.23* | Form of U.S. Executive Indemnification Agreement | Exhibit 10.4, Form 8-K filed October 4, 2010 | ||
128
Table of Contents
| Exhibit |
Description |
Location | ||
| 10.24* | Form of Canadian Executive Indemnification Agreement | Exhibit 10.5, Form 8-K filed October 4, 2010 | ||
| 10.25* | Form of William L. Hunter Indemnification Agreement | Exhibit 10.6, Form 8-K filed October 4, 2010 | ||
| 10.26* | Executive Employment Agreement between Angiotech Pharmaceuticals, Inc. and William L. Hunter, dated April 23, 2004 | Exhibit 10.19, Form 10-K filed March 16, 2009 | ||
| 10.27* | Executive Employment Agreement between Angiotech Pharmaceuticals, Inc. and K. Thomas Bailey, dated August 8, 2007** | Exhibit 10.20, Form 10-K filed March 16, 2009 | ||
| 10.28* | Executive Employment Agreement between Angiotech Pharmaceuticals, Inc. and David D. McMasters, dated October 30, 2007** | Exhibit 10.21, Form 10-K filed March 16, 2009 | ||
| 10.29* | Executive Employment Agreement between Angiotech Pharmaceuticals, Inc. and Rui Avelar, dated October 15, 2007 | Exhibit 10.22, Form 10-K filed March 16, 2009 | ||
| 10.30* | Executive Employment Agreement between Angiotech Pharmaceuticals, Inc. and Steve Bryant, dated June 11, 2009** | Filed herewith | ||
| 10.31* | Angiotech Pharmaceuticals, Inc. 2001 Stock Option Plan | Exhibit to Form S-8, filed September 27, 2001 | ||
| 10.32* | Angiotech Pharmaceuticals, Inc. 2004 Stock Option Plan | Exhibit 10.27, Form 10-K filed March 16, 2009 | ||
| 10.33* | Angiotech Pharmaceuticals, Inc. 2006 Stock Option Plan | Exhibit 10.28, Form 10-K filed March 16, 2009 | ||
| 10.34* | American Medical Instruments Holdings, Inc. 2003 Stock Option Plan | Exhibit 10.29, Form 10-K filed March 16, 2009 | ||
| 10.35 | Agreement and Plan of Merger, dated as of May 25, 2006, by and among Angiotech Pharmaceuticals, Inc., Angiotech Pharmaceuticals (US), Inc., Quaich Acquisition, Inc. and Quill Medical, Inc. | Exhibit 10.4, Form 8-K filed November 2, 2010 | ||
| 10.36 | Settlement Agreement, dated January 27, 2011, among Angiotech Pharmaceuticals, Inc., certain of its subsidiaries, QSR Holdings, Inc. and the other signatories thereto. | Exhibit 10.4, Form 8-K filed January 28, 2011 | ||
| 10.37 | License, Supply, Marketing and Distribution Agreement by and between Angiotech Pharmaceuticals (US), Inc. and Rex Medical, LP, dated as of March 13, 2008. | Exhibit 10.1, Form 10-Q filed November 9, 2010 | ||
| 10.38 | Amendment dated August 9, 2009 to License, Supply, Marketing and Distribution Agreement by and between Angiotech Pharmaceuticals (US), Inc. and Rex Medical, LP, dated as of March 13, 2008. | Exhibit 10.2, Form 10-Q filed November 9, 2010 | ||
| 10.39 | Settlement and License Termination Agreement, dated as of February 16, 2011, by and between Angiotech Pharmaceuticals (US), Inc. and Rex Medical, LP. | Exhibit 10.1, Form 8-K filed February 18, 2011 | ||
| 10.40 | Forbearance Agreement, dated as of September 30, 2010, by and among Angiotech Pharmaceuticals, Inc. (“Parent”), the subsidiaries of Parent listed as borrowers on the signature pages thereto, the lenders signatory thereto and Wells Fargo Capital Finance, LLC (f/k/a Wells Fargo Foothill, LLC), as arranger and administrative agent. | Exhibit 10.1, Form 8-K filed October 4, 2010 | ||
| 10.41 | Amended and Restated Forbearance Agreement, dated as of November 4, 2010, by and among Angiotech Pharmaceuticals, Inc., as Parent, the subsidiaries of Parent listed as borrowers on the signature pages thereto, the lenders signatory thereto and Wells Fargo Capital Finance, LLC (f/k/a Wells Fargo Foothill, LLC), as arranger and administrative agent. | Exhibit 10.2, Form 8-K filed November 9, 2010 | ||
| 10.42 | Recapitalization Support Agreement, dated as of October 29, 2010, among Angiotech Pharmaceuticals, Inc., and the holders of senior subordinated notes signatory thereto. | Exhibit 10.1, Form 8-K filed November 2, 2010 | ||
129
Table of Contents
| Exhibit |
Description |
Location | ||
| 10.43 | Agreement to Amend the Recapitalization Support Agreement, dated November 29, 2010, among Angiotech Pharmaceuticals, Inc., and the holders of senior subordinated notes signatory thereto. | Exhibit 10.1, Form 8-K filed December 2, 2010 | ||
| 10.44 | Second Agreement to Amend the Recapitalization Support Agreement, dated December 15, 2010, among Angiotech Pharmaceuticals, Inc. and the holders of senior subordinated notes signatory thereto. | Exhibit 10.1, Form 8-K filed December 17, 2010 | ||
| 10.45 | Third Agreement to Amend the Recapitalization Support Agreement, dated January 11, 2011, among Angiotech Pharmaceuticals, Inc. and the holders of senior subordinated notes signatory thereto. | Exhibit 10.1, Form 8-K filed January 13, 2011 | ||
| 10.46 | Fourth Agreement to Amend the Recapitalization Support Agreement, dated January 27, 2011, among Angiotech Pharmaceuticals, Inc., and the holders of senior subordinated notes signatory thereto. | Exhibit 10.1, Form 8-K filed January 28, 2011 | ||
| 10.47 | Fifth Agreement to Amend the Recapitalization Support Agreement, dated February 7, 2011, among Angiotech Pharmaceuticals, Inc. and the holders of senior subordinated notes signatory thereto. | Exhibit 10.2, Form 8-K filed February 10, 2011 | ||
| 10.48 | Floating Rate Note Support Agreement, dated as of October 29, 2010, among Angiotech Pharmaceuticals, Inc., and the holders of floating rate notes signatory thereto. | Exhibit 10.2, Form 8-K filed November 2, 2010 | ||
| 10.49 | Agreement to Amend the Floating Rate Note Support Agreement, dated as of November 29, 2010, among Angiotech Pharmaceuticals, Inc., and the holders of floating rate notes signatory thereto. | Exhibit 10.2, Form 8-K filed December 2, 2010 | ||
| 10.50 | Second Agreement to Amend the Floating Rate Note Support Agreement, dated as of December 15, 2010, among Angiotech Pharmaceuticals, Inc., and the holders of floating rate notes signatory thereto. | Exhibit 10.2, Form 8-K filed December 17, 2010 | ||
| 10.51 | Third Agreement to Amend the Floating Rate Note Support Agreement, dated as of January 11, 2011, among Angiotech Pharmaceuticals, Inc., and the holders of floating rate notes signatory thereto. | Exhibit 10.2, Form 8-K filed January 13, 2011 | ||
| 10.52 | Fourth Agreement to Amend the Floating Rate Note Support Agreement, dated as of January 27, 2011, among Angiotech Pharmaceuticals, Inc., and the holders of floating rate notes signatory thereto. | Exhibit 10.2, Form 8-K filed January 28, 2011 | ||
| 10.53 | Fifth Agreement to Amend the Floating Rate Note Support Agreement, dated as of February 7, 2011, among Angiotech Pharmaceuticals, Inc., and the holders of floating rate notes signatory thereto. | Exhibit 10.1, Form 8-K filed February 10, 2011 | ||
| 10.54 | Sixth Agreement to Amend the Floating Rate Note Support Agreement, dated as of February 22, 2011, among Angiotech Pharmaceuticals, Inc., and the holders of floating rate notes signatory thereto. | Exhibit 10.1, Form 8-K filed February 24, 2011 | ||
| 12 | Statement re computation of ratios | Filed herewith | ||
| 14.1 | Code of Ethics for Chief Executive Officer, Chief Financial Officer and SVP Finance | Exhibit 14.1, Form 10-K filed March 16, 2010 | ||
| 14.2 | Guide of Business Conduct | Exhibit 14.2, Form 10-K filed March 16, 2010 | ||
| 21 | List of Worldwide Subsidiaries | Filed herewith | ||
| 23.1 | Consent of PricewaterhouseCoopers LLP | Filed herewith | ||
| 31.1 | Certification of CEO Pursuant to Securities Exchange Act Rules 13a-14 and 15d-14 as Adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | Filed herewith | ||
130
Table of Contents
| Exhibit |
Description |
Location | ||
| 31.2 | Certification of CFO Pursuant to Securities Exchange Act Rules 13a-14 and 15d-14 as Adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | Filed herewith | ||
| 32.1 | Certification of CEO Pursuant to 18 U.S.C. Section 1350 as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | Filed herewith | ||
| 32.2 | Certification of CFO Pursuant to 18 U.S.C. Section 1350 as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | Filed herewith | ||
| ** | portions of this exhibit have been omitted and filed separately with the Commission pursuant to a request for confidential treatment |
131
Table of Contents
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized.
| Angiotech Pharmaceuticals, Inc. | ||||
| Date: March 16, 2011 | By: | /S/ K. THOMAS BAILEY | ||
| K. Thomas Bailey Chief Financial Officer | ||||
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS that each person whose signature appears below constitute and appoint William L. Hunter, Chief Executive Officer, and K. Thomas Bailey, Chief Financial Officer, and each of them, the lawful attorney and agent or attorneys and agents with power and authority to do any and all acts and things and to execute all instruments which said attorneys and agents, or either of them, determine may be necessary or advisable or required to enable Angiotech Pharmaceuticals, Inc. to comply with the Securities Exchange Act of 1934, as amended, and any rules or regulations or requirements of the Securities and Exchange Commission in connection with this Annual Report on Form 10-K. Without limiting the generality of the foregoing power and authority, the powers granted include the power and authority to sign the names of the undersigned officers and directors in the capacities indicated below to this Annual Report on Form 10-K or amendments or supplements thereto, and each of the undersigned hereby ratifies and confirms all that said attorneys and agents or either of them, shall do or cause to be done by virtue hereof. This Power of Attorney may be signed in several counterparts.
Pursuant to the requirements of the Securities Exchange Act of 1934, this Report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signatures |
Title |
Date | ||
| /S/ WILLIAM L. HUNTER William L. Hunter, M.D. |
President, Chief Executive Officer and |
March 16, 2011 | ||
| /S/ K. THOMAS BAILEY K. Thomas Bailey |
Chief Financial Officer |
March 16, 2011 | ||
| /S/ JAY DENT Jay Dent |
Senior Vice President, Finance |
March 16, 2011 | ||
| /S/ DAVID HOWARD David Howard |
Chairman of the Board of Directors |
March 16, 2011 | ||
| /S/ LAURA BREGE Laura Brege |
Director |
March 16, 2011 | ||
| /S/ EDWARD BROWN Edward Brown |
Director |
March 16, 2011 | ||
| /S/ ARTHUR H. WILLMS Arthur Willms |
Director |
March 16, 2011 | ||
| /S/ HENRY A, MCKINNELL JR. Henry A, McKinnell Jr. |
Director |
March 16, 2011 | ||
132
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Shareholders and Board of Directors of Angiotech Pharmaceuticals, Inc.
We have audited the accompanying consolidated balance sheets of Angiotech Pharmaceuticals, Inc. and its subsidiaries as of December 31, 2010 and December 31, 2009, and the related consolidated statements of operations, shareholders’ (deficit) equity and cash flows for each of the years in the three-year period ended December 31, 2010. In addition, we audited the financial statement schedule listed in the index appearing under Item 15(a)(2) of the Annual Report on Form 10-K. We also have audited Angiotech Pharmaceuticals, Inc.’s internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control - Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Management is responsible for these financial statements and the financial statements schedule, for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on these consolidated financial statements and an opinion on the company’s internal control over financial reporting based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the consolidated financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of consolidated financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Angiotech Pharmaceuticals, Inc. and its subsidiaries as of December 31, 2010 and December 31, 2009, and the results of its operations and its cash flows for each of the years in the three-year period ended December 31, 2010 in conformity with accounting principles generally accepted in the United States of America. In addition, in our opinion, the financial statement schedule presents fairly, in all material aspects, the information set forth therein when read in conjunction with the related consolidated financial statements. Also in our opinion, Angiotech Pharmaceuticals, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control - Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO).
The accompanying financial statements have been prepared assuming that Angiotech Pharmaceuticals, Inc. and its subsidiaries will continue as a going concern. As discussed in Note 1 to the consolidated financial statements, there are material uncertainties related to the company’s financial condition that raise substantial doubt about its ability to continue as a going concern. In addition, as discussed in Notes 1 and 26, Angiotech Pharmaceuticals, Inc. and certain of its subsidiaries filed an application on January 28, 2011 with the Supreme Court of British Columbia for a reorganization under the provisions of the Companies’ Creditors Arrangement Act. Further, on January 30, 2011, ancillary proceedings were commenced in the United States Bankruptcy Court for the District of Delaware under Chapter 15 of Title 11 of the United States Code. Management’s plans in regard to these matters are also described in Notes 1 and 26 to the consolidated financial statements. The consolidated financial statements do not include any adjustments that might result from the outcome of these uncertainties.
| /s/ PricewaterhouseCoopers LLP |
| Vancouver, Canada |
| March 16, 2011 |
F-1
Table of Contents
Angiotech Pharmaceuticals, Inc.
CONSOLIDATED BALANCE SHEETS
(All amounts expressed in thousands of U.S. dollars)
| December 31, 2010 |
December 31, 2009 |
|||||||
| ASSETS |
||||||||
| Current assets |
||||||||
| Cash and cash equivalents [note 5] |
$ | 33,315 | $ | 49,542 | ||||
| Short-term investments [note 6] |
4,653 | 7,780 | ||||||
| Accounts receivable |
33,494 | 28,167 | ||||||
| Income taxes receivable |
669 | 1,090 | ||||||
| Inventories [note 7] |
34,631 | 35,541 | ||||||
| Deferred income taxes, current portion [note 14] |
4,102 | 4,284 | ||||||
| Deferred financing costs [note 16(e)] |
5,611 | — | ||||||
| Prepaid expenses and other current assets |
4,649 | 3,294 | ||||||
| Total current assets |
121,124 | 129,698 | ||||||
| Assets held for sale [note 8] |
— | 5,300 | ||||||
| Property, plant and equipment [note 9] |
46,261 | 46,879 | ||||||
| Intangible assets [note 10(a)] |
137,973 | 173,019 | ||||||
| Deferred financing costs [note 16(e)] |
— | 11,409 | ||||||
| Other assets |
640 | 3,754 | ||||||
| Total assets |
$ | 305,998 | $ | 370,059 | ||||
| LIABILITIES AND SHAREHOLDERS’ DEFICIT |
||||||||
| Current liabilities |
||||||||
| Accounts payable and accrued liabilities [note 12] |
$ | 36,847 | $ | 46,324 | ||||
| Income taxes payable |
2,255 | 10,858 | ||||||
| Interest payable on long-term debt |
15,761 | 6,004 | ||||||
| Revolving credit facility [note 16(f)] |
10,000 | — | ||||||
| Long-term debt [note 16 (a) & (b)] |
575,000 | — | ||||||
| Total current liabilities |
639,863 | 63,186 | ||||||
| Deferred leasehold inducement [note 13] |
4,785 | 2,888 | ||||||
| Deferred income taxes [note 14] |
31,702 | 36,778 | ||||||
| Other tax liability [note 15] |
4,367 | 3,898 | ||||||
| Other liabilities [note 20 (a)(i)-(ii))] |
9,357 | 1,596 | ||||||
| Long-term debt [note 16 (a) & (b)] |
— | 575,000 | ||||||
| Total non-current liabilities |
$ | 50,211 | $ | 620,160 | ||||
| Commitments and contingencies [note 20] |
||||||||
| Shareholders’ deficit |
||||||||
| Share capital [note 17] |
||||||||
| Authorized: |
||||||||
| 200,000,000 Common shares, without par value 50,000,000 Class I Preference shares, without par value Common shares issued and outstanding: |
||||||||
| December 31, 2010 — 85,180,377 |
||||||||
| December 31, 2009 — 85,138,081 |
472,753 | 472,742 | ||||||
| Additional paid-in capital |
35,470 | 33,687 | ||||||
| Accumulated deficit |
(933,352 | ) | (866,541 | ) | ||||
| Accumulated other comprehensive income |
41,053 | 46,825 | ||||||
| Total shareholders’ deficit |
(384,076 | ) | (313,287 | ) | ||||
| Total liabilities and shareholders’ deficit |
$ | 305,998 | $ | 370,059 | ||||
Going concern and liquidity [note 1]
Subsequent events [note 26]
See accompanying notes to the consolidated financial statements
F-2
Table of Contents
Angiotech Pharmaceuticals, Inc.
CONSOLIDATED STATEMENTS OF OPERATIONS
(All amounts expressed in thousands of U.S. dollars, except share and per share data)
| Year ended December 31, 2010 |
Year ended December 31, 2009 |
Year ended December 31, 2008 |
||||||||||
| REVENUE |
||||||||||||
| Product sales, net |
$ | 211,495 | $ | 191,951 | $ | 190,816 | ||||||
| Royalty revenue |
34,461 | 62,171 | 91,546 | |||||||||
| License fees [note 18] |
286 | 25,556 | 910 | |||||||||
| 246,242 | 279,678 | 283,272 | ||||||||||
| EXPENSES |
||||||||||||
| Cost of products sold |
106,304 | 104,616 | 101,052 | |||||||||
| License and royalty fees |
5,889 | 10,431 | 14,258 | |||||||||
| Research and development |
26,790 | 23,701 | 53,192 | |||||||||
| Selling, general and administration |
89,238 | 81,504 | 98,483 | |||||||||
| Depreciation and amortization |
33,745 | 33,251 | 33,998 | |||||||||
| In-process research and development |
— | — | 2,500 | |||||||||
| Write-down of assets held for sale [note 8] |
1,450 | 3,090 | 1,283 | |||||||||
| Write-down of property, plant and equipment [note 9] |
4,779 | — | — | |||||||||
| Write-down of intangible assets [note 10(a)] |
2,814 | — | — | |||||||||
| Escrow settlement recovery [note 19] |
(4,710 | ) | — | — | ||||||||
| Write-down of goodwill [note 10(b)] |
— | — | 649,685 | |||||||||
| 266,299 | 256,593 | 954,451 | ||||||||||
| Operating (loss) income |
(20,057 | ) | 23,085 | (671,179 | ) | |||||||
| Other (expenses) income: |
||||||||||||
| Foreign exchange gain (loss) |
1,011 | (1,612 | ) | 540 | ||||||||
| Other (expense) income |
(571 | ) | 378 | 1,192 | ||||||||
| Debt restructuring costs [note 16 (a)] |
(9,277 | ) | — | — | ||||||||
| Interest expense on long-term debt |
(40,258 | ) | (38,039 | ) | (44,490 | ) | ||||||
| Write-down of deferred financing charges [note 16(e)] |
(291 | ) | (643 | ) | (16,544 | ) | ||||||
| Write-downs and losses on redemption of investments [note 8] |
(1,297 | ) | — | (23,587 | ) | |||||||
| Loan settlement gain [note 4] |
1,880 | — | — | |||||||||
| Gain on disposal of Laguna Hills manufacturing facility [note 11] |
2,005 | — | — | |||||||||
| Total other expenses |
(46,798 | ) | (39,916 | ) | (82,889 | ) | ||||||
| Loss before income taxes |
(66,855 | ) | (16,831 | ) | (754,068 | ) | ||||||
| Income tax expense (recovery) [note 14] |
(44 | ) | 6,037 | (12,892 | ) | |||||||
| Net loss |
$ | (66,811 | ) | $ | (22,868 | ) | $ | (741,176 | ) | |||
| Basic and diluted net loss per common share [note 22] |
$ | (0.78 | ) | $ | (0.27 | ) | $ | (8.71 | ) | |||
| Basic and diluted weighted average number of common shares outstanding (in thousands) [note 22] |
85,168 | 85,130 | 85,118 | |||||||||
See accompanying notes to the consolidated financial statements
F-3
Table of Contents
Angiotech Pharmaceuticals, Inc.
CONSOLIDATED STATEMENTS OF SHAREHOLDERS’ (DEFICIT) EQUITY
(All amounts expressed in thousands of U.S. dollars, except share data)
| Common Shares | Additional paid-in capital |
Accumulated deficit |
Accumulated other comprehensive income |
Comprehensive income (loss) |
Total shareholders’ equity(deficit) |
|||||||||||||||||||||||
| Shares | Amount | |||||||||||||||||||||||||||
| Balance at December 31, 2007 |
85,073,983 | $ | 472,618 | $ | 29,669 | $ | (102,497 | ) | $ | 42,282 | $ | 442,072 | ||||||||||||||||
| Exercise of stock options for cash |
48,000 | 121 | 121 | |||||||||||||||||||||||||
| Stock-based compensation |
2,438 | 2,438 | ||||||||||||||||||||||||||
| Net unrealized loss on available-for-sale securities, net of taxes (nil) |
(9,572 | ) | $ | (9,572 | ) | (9,572 | ) | |||||||||||||||||||||
| Reclassification of realized loss on available-for-sale securities, net of taxes (nil) |
13,860 | 13,860 | 13,860 | |||||||||||||||||||||||||
| Cumulative translation adjustment |
(7,616 | ) | (7,616 | ) | (7,616 | ) | ||||||||||||||||||||||
| Net loss |
(741,176 | ) | (741,176 | ) | (741,176 | ) | ||||||||||||||||||||||
| Comprehensive loss |
$ | (744,504 | ) | |||||||||||||||||||||||||
| Balance at December 31, 2008 |
85,121,983 | $ | 472,739 | $ | 32,107 | $ | (843,673 | ) | $ | 38,954 | $ | (299,873 | ) | |||||||||||||||
| Exercise of stock options for cash |
16,098 | 3 | 3 | |||||||||||||||||||||||||
| Stock-based compensation |
1,580 | 1,580 | ||||||||||||||||||||||||||
| Net unrealized gain on available-for-sale securities, net of taxes (nil) |
6,932 | 6,932 | 6,932 | |||||||||||||||||||||||||
| Cumulative translation adjustment |
939 | 939 | 939 | |||||||||||||||||||||||||
| Net loss |
(22,868 | ) | (22,868 | ) | (22,868 | ) | ||||||||||||||||||||||
| Comprehensive loss |
$ | (14,997 | ) | |||||||||||||||||||||||||
| Balance at December 31, 2009 |
85,138,081 | $ | 472,742 | $ | 33,687 | $ | (866,541 | ) | $ | 46,825 | $ | (313,287 | ) | |||||||||||||||
| Exercise of stock options for cash |
42,296 | 11 | 11 | |||||||||||||||||||||||||
| Stock-based compensation |
1,783 | 1,783 | ||||||||||||||||||||||||||
| Net unrealized loss on available-for-sale securities, net of taxes (nil) |
(3,127 | ) | (3,127 | ) | (3,127 | ) | ||||||||||||||||||||||
| Cumulative translation adjustment |
(2,645 | ) | (2,645 | ) | (2,645 | ) | ||||||||||||||||||||||
| Net loss |
(66,811 | ) | (66,811 | ) | (66,811 | ) | ||||||||||||||||||||||
| Comprehensive loss |
$ | (72,583 | ) | |||||||||||||||||||||||||
| Balance at December 31, 2010 |
85,180,377 | $ | 472,753 | $ | 35,470 | $ | (933,352 | ) | $ | 41,053 | $ | (384,076 | ) | |||||||||||||||
See accompanying notes to the consolidated financial statements
F-4
Table of Contents
Angiotech Pharmaceuticals, Inc.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(All amounts expressed in thousands of U.S. dollars)
| Year ended December 31, 2010 |
Year ended December 31, 2009 |
Year ended December 31, 2008 |
||||||||||
| OPERATING ACTIVITIES |
||||||||||||
| Net loss |
$ | (66,811 | ) | $ | (22,868 | ) | $ | (741,176 | ) | |||
| Adjustments to reconcile net income to cash provided by operating activities: |
||||||||||||
| Depreciation and amortization |
37,625 | 37,026 | 38,171 | |||||||||
| Loss on disposition of property, plant and equipment |
615 | 369 | — | |||||||||
| Gain on disposal of Laguna Hills manufacturing facility [note 11] |
(2,005 | ) | — | — | ||||||||
| Write-down of intangible assets [note 10(a)] |
2,814 | — | 113 | |||||||||
| Write-down of assets held for sale [note 8] |
1,450 | 3,090 | 1,837 | |||||||||
| Write-down of property, plant and equipment [note 9] |
4,779 | — | — | |||||||||
| Write-downs, disposals and losses on redemption of investments |
1,297 | — | 23,584 | |||||||||
| Write-down of deferred financing charges [note 16 (e)] |
291 | 643 | 16,544 | |||||||||
| Write-down of goodwill |
— | — | 649,685 | |||||||||
| Loan settlement gain [note 4] |
(1,880 | ) | — | — | ||||||||
| Deferred income taxes |
(3,837 | ) | (4,321 | ) | (12,464 | ) | ||||||
| Stock-based compensation expense |
1,783 | 1,579 | 2,438 | |||||||||
| Non-cash interest expense |
6,175 | 2,788 | 2,237 | |||||||||
| In-process research and development |
— | — | 2,500 | |||||||||
| Deferred leasehold inducements [note 13] |
2,700 | — | — | |||||||||
| Other |
(750 | ) | (850 | ) | (335 | ) | ||||||
| Net change in non-cash working capital items relating to operating activities [note 23] |
(5,855 | ) | 4,515 | (21,141 | ) | |||||||
| Cash provided by (used in) operating activities |
(21,609 | ) | 21,971 | (38,007 | ) | |||||||
| INVESTING ACTIVITIES |
||||||||||||
| Proceeds from short-term investments |
— | — | 2,756 | |||||||||
| Purchase of property, plant and equipment |
(5,726 | ) | (4,366 | ) | (7,669 | ) | ||||||
| Proceeds from disposition of property, plant and equipment and Laguna Hills manufacturing facility |
1,272 | — | — | |||||||||
| Purchase of intangible assets |
— | (2,500 | ) | (1,000 | ) | |||||||
| Proceeds from disposition of Laguna Hills manufacturing facility |
2,005 | — | — | |||||||||
| Loans advanced |
(1,015 | ) | (1,573 | ) | — | |||||||
| Asset acquisition costs |
(231 | ) | ||||||||||
| In-process research and development |
— | — | (2,500 | ) | ||||||||
| Other |
— | 252 | 174 | |||||||||
| Cash used in investing activities |
(3,695 | ) | (8,187 | ) | (8,239 | ) | ||||||
| FINANCING ACTIVITIES |
||||||||||||
| Deferred financing charges and costs |
(669 | ) | (3,403 | ) | (4,499 | ) | ||||||
| Proceeds from stock options exercised |
11 | 3 | 121 | |||||||||
| Advances under credit facility |
10,000 | — | — | |||||||||
| Cash provided by (used in) financing activities |
9,342 | (3,400 | ) | (4,378 | ) | |||||||
| Effect of exchange rate changes on cash |
(265 | ) | 206 | (1,750 | ) | |||||||
| Net increase (decrease) in cash and cash equivalents |
(16,227 | ) | 10,590 | (52,374 | ) | |||||||
| Cash and cash equivalents, beginning of year |
49,542 | 38,952 | 91,326 | |||||||||
| Cash and cash equivalents, end of year |
$ | 33,315 | $ | 49,542 | $ | 38,952 | ||||||
Supplemental note disclosure [note 23]
See accompanying notes to the consolidated financial statements
F-5
Table of Contents
Angiotech Pharmaceuticals, Inc.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
Angiotech Pharmaceuticals, Inc. (“Angiotech” or the “Company”), is incorporated under the Business Corporations Act (British Columbia). Angiotech is a medical technology company that discovers develops and markets innovative technologies primarily focused on acute and surgical applications. The Company generates its revenue through sales of medical products and components, as well as from royalties derived from sales by its partners of products utilizing certain of its proprietary technologies.
The Company’s research and development efforts focus on understanding and characterizing biological conditions that often occur concurrent with medical device implantation, surgery or acute trauma, including scar formation and inflammation, cell proliferation, bleeding and coagulation, infection, and tumor tissue overgrowth. Its strategy is to utilize its various technologies in the areas of medical device engineering, drugs, drug delivery, surface modification and biomaterials to create and commercialize novel, proprietary medical products that reduce surgical procedure side effects, improve surgical outcomes, shorten hospital stays, or are easier or safer for a physician to use.
The Company has patent protected, or has filed patent applications for, certain of its technology and many of its products and potential product candidates.
1. BASIS OF PRESENTATION
These audited consolidated financial statements have been prepared in accordance with generally accepted accounting principles in the United States of America (“U.S. GAAP”) and pursuant to the rules and regulations of the U.S. Securities and Exchange Commission (“SEC”). In management’s opinion, all adjustments (which include reclassification and normal recurring adjustments), necessary to present fairly the consolidated balance sheets, consolidated statements of operations, consolidated statements of stockholders’ deficit and consolidated statements of cash flows at December 31, 2010 and for all periods presented, have been made. All amounts herein are expressed in U.S. dollars and all tabular amounts are expressed in thousands of U.S. dollars, except share and per share data, unless otherwise noted.
CREDITOR PROTECTION PROCEEDINGS AND RECAPITALIZATION TRANSACTION
Over the past several years, revenues in the Company’s Pharmaceutical Technologies Segment, specifically its royalties derived from sales of TAXUS® coronary stent systems (“TAXUS”) by Boston Scientific Corporation (“BSC”), have experienced significant declines. These declines have led to significant constraints on the Company’s liquidity, working capital and capital resources and accordingly, its ability to continue to support its business initiatives and service its debt obligations is uncertain. As a result, over the past several years the Company has explored a range of financial and strategic alternatives to address these circumstances. After extensive exploration of various possible solutions, the Company recently announced a proposed recapitalization transaction that would eliminate $250 million of its total outstanding indebtedness and related interest obligations by exchanging its 7.75% Senior Subordinated Notes due 2014 (the “Subordinated Notes”) for new common shares in the Company (the “Recapitalization Transaction”) and thereby substantially improve its liquidity, working capital and financial position.
As part of the Company’s recapitalization plan, on October 1, 2010, it did not make its $9.7 million interest payment due on its $250 million 7.75% Senior Subordinated Notes (the “Subordinated Notes”) in connection with discussions being conducted with certain of the holders of its Subordinated Notes (the “Subordinated Noteholders”). Pursuant to the terms of the indenture governing the Subordinated Notes (as amended, the “SSN Indenture”), the Company had a 30-day grace period from October 1, 2010 to make the required interest payment before an event of default occurred and Subordinated Noteholders could exercise their right to demand repayment of all outstanding obligations thereunder. Pursuant to the terms of the supplemental indenture entered into by Angiotech and U.S. Bank National Association, as successor trustee under the SSN Indenture, at the direction of the holders of a majority in aggregate principal amount outstanding of the Subordinated Notes, on January 27, 2011, the grace period applicable to interest payments on the Subordinated Notes was ultimately extended to 211 days. Including the $9.7 million of interest due on October 1, 2010, the Company has accrued $14.5 million for its total interest obligation on the Subordinated Notes as at December 31, 2010.
In connection with the Company’s decision not to make the interest payment due to holders of the Subordinated Notes on October 1, 2010, it entered into a forbearance agreement (the “Forbearance Agreement”) with Wells Fargo Capital Finance, LLC (“Wells Fargo”) on September 30, 2010 to preserve its existing revolving credit facility (as amended, the “Existing Credit Facility”) and avoid an event of default under the Existing Credit Facility that would have otherwise resulted from such non-payment while the Company completed negotiations with its Subordinated Noteholders.
F-6
Table of Contents
On October 29, 2010, the Company entered into a Recapitalization Support Agreement (as amended, the “RSA”) with the holders of approximately 73% of the aggregate principal amount of its outstanding Subordinated Notes (together with all holders of the Subordinated Notes who executed a consent agreement with the Company and certain of its subsidiaries prior to November 30, 2010, the “Consenting Noteholders”) to, among other things, effectuate the Recapitalization Transaction thereby eliminating $250 million of its total outstanding indebtedness and related interest obligations by exchanging Subordinated Notes for new common shares in the Company thereby providing significant improvements to its credit ratios, liquidity and financial flexibility. As of March 14, 2011, holders of approximately 85% of the aggregate principal amount of the Company’s outstanding Subordinated Notes had agreed to the terms of the RSA. In accordance with the terms of the RSA, (i) the Subordinated Noteholders will receive their pro rata share of approximately 91.5% of the new common shares to be issued and outstanding upon completion of the Recapitalization Transaction and (ii) the Consenting Noteholders will receive an additional amount of the new common shares equal to their pro rata share of approximately 3.5% of the new common shares of the Company to be issued and outstanding upon completion of the Recapitalization Transaction, in each case subject to potential dilution.
On October 29, 2010, the Company also entered into a support agreement (as amended, the “FRN Support Agreement”) with holders (the “Consenting FRN Holders”) of approximately 51% of the aggregate principal amount outstanding of our existing senior floating rate notes (the “Existing Floating Rate Notes”) pursuant to which the Consenting FRN Holders agreed to support and tender to an offer to exchange their Existing Floating Rate Notes for new senior floating rate notes (the “New Floating Rate Notes” and such exchange, the “FRN Exchange Offer”). The New Floating Rate Notes will be issued on substantially the same terms and conditions as the Existing Floating Rate Notes, except that, among other things: (i) subject to certain exceptions, the New Floating Rate Notes will be secured by second-priority liens over substantially all of the assets, property and undertakings of the Company and certain of its subsidiaries; (ii) the New Floating Rate Notes will accrue interest at LIBOR plus 3.75%, subject to a LIBOR floor of 1.25%; and (iii) certain covenants related to the incurrence of additional indebtedness and asset sales and the definitions of permitted liens, asset sales and change of control will be modified in the indenture that governs the New Floating Rate Notes. The assets, property and undertakings of the Company and certain of its subsidiaries that secure the New Floating Rate Notes will be subject to a first priority lien under an exit credit facility that we expect will be in place upon completion of the Recapitalization Transaction. Pursuant to the terms of the RSA, the obligation of the Consenting Noteholders to complete the Recapitalization Transaction is conditioned on the completion of the transactions contemplated by the FRN Support Agreement. Under the terms of the FRN Support Agreement, the obligation of all parties thereto to consummate the FRN Exchange Offer is conditioned upon the concurrent implementation of the Recapitalization Transaction. As of March 14, 2011, approximately 89% holders of the aggregate principal amount of the outstanding Existing Floating Rate Notes have agreed to the terms of the FRN Exchange Offer.
On November 4, 2010, the Company entered into an amended and restated forbearance agreement with the lenders party thereto and Wells Fargo, as arranger and administrative agent, which amended and restated in its entirety the Forbearance Agreement (the “Amended and Restated Forbearance Agreement”). Under the Amended and Restated Forbearance Agreement, Wells Fargo agreed not to immediately exercise any rights or remedies it has related to: (a) the Company’s failure to make its $9.7 million interest payment due to the Subordinated Noteholders on October 1, 2010; (b) the litigation and arbitration relating to the Company’s dispute with QSR Holdings, Inc. (“QSR”); or (c) any failure to deliver audited financial statements with an unqualified audit opinion with respect to the going concern assumption for the fiscal year ending December 31, 2010. While the Amended and Restated Forbearance Agreement has not been terminated, Wells Fargo’s obligations thereunder, including its agreement not to exercise certain remedies, ceased to be of effect upon the Company’s commencement of the CCAA Proceedings (as defined below). Wells Fargo’s obligations, however, were nonetheless stayed in connection with such proceedings, as well as our proceedings under Chapter 15 of the U.S. Bankruptcy Code.
In conjunction with the Amended and Restated Forbearance Agreement, on November 4, 2010, the Company entered into an amendment to its Existing Credit Facility with Wells Fargo that provides for, among other changes:
| (i) | an amendment of financial covenants pertaining to minimum EBITDA levels and fixed charge coverage ratios; |
| (ii) | an increase the borrowing base available to secure loans by including in such borrowing base the value of certain real property, securities and intellectual property owned by the Company and its subsidiaries, in addition to accounts receivable and inventory, in each case subject to reserves, eligibility standards and advance rate formulas. As a result of such amendment, the amount of financing available to us under our Existing Credit Facility immediately increased to approximately $25 million, which excludes $2.8 million relating to outstanding secured letters of credit under the Existing Credit Facility; |
| (iii) | an amendment to the specified use of proceeds provisions to allow for certain secured and unsecured intercompany loans by the Company’s U.S. entities to its Canadian entities; and |
| (iv) | an amendment to permit the sale of certain assets of the Company. |
The Company incurred $0.1 million to complete both the Forbearance Agreement and the Amended and Restated Forbearance Agreement and $0.3 million to complete the Fourth Amendment.
F-7
Table of Contents
On January 28, 2011, in connection with the provisions under the RSA, Angiotech, 0741693 B.C. Ltd., Angiotech International Holdings, Corp., Angiotech Pharmaceuticals (US), Inc., American Medical Instruments Holdings, Inc., NeuColl, Inc., Angiotech BioCoatings Corp., Afmedica, Inc., Quill Medical, Inc., Angiotech America, Inc., Angiotech Florida Holdings, Inc., B.G. Sulzle, Inc., Surgical Specialties Corporation, Angiotech Delaware, Inc., Medical Device Technologies, Inc., Manan Medical Products, Inc. and Surgical Specialties Puerto Rico, Inc. (collectively, the “Angiotech Entities”) voluntarily filed for creditor protection (the “CCAA Proceedings”) under the Companies’ Creditors Arrangement Act (Canada) (the “CCAA”) and an initial order (the “Initial Order”) was granted by the Supreme Court of British Columbia (the “Canadian Court”). The CCAA Proceedings were commenced in order to implement the Recapitalization Transaction through a plan of compromise and arrangement (as amended, supplemented or restated from time to time, the “CCAA Plan”) under the CCAA. In order to have the CCAA Proceedings recognized in the United States, on January 30, 2011, the Angiotech Entities commenced proceedings under Chapter 15 of Title 11 of the United States Code (the “U.S. Bankruptcy Code”) in the United States Bankruptcy Court for the District of Delaware (together with the CCAA Proceedings, the “Creditor Protection Proceedings”).
Under the terms of the proposed CCAA Plan, the Subordinated Notes will be exchanged for equity interests in Angiotech and retired in full, thereby eliminating approximately $250.0 million of our outstanding indebtedness, plus the accrued and unpaid interest related thereto. In addition, all of the existing common shares and options to acquire common shares will be cancelled without any payment or consideration therefor. The CCAA Plan will also address certain other unsecured obligations which are expected to be limited. The Initial Order, among other things, provided for a general stay of proceedings against the Angiotech Entities preventing any party from commencing or continuing any action, suit or proceeding against the Angiotech Entities or from exercising other rights that could arise as a result of the commencement of proceedings under the CCAA. The stay of proceedings was initially scheduled to expire on February 17, 2011 and was subsequently extended to April 15, 2011 and may be extended further by order of the Canadian Court (the “Stay Period”). On February 22, 2011, the U.S. court granted an order that, among other things, recognized the CCAA Proceeding as a foreign main proceeding and provided that the Initial Order would be enforced on a final basis and given full force and effect in the United States.
The Canadian Court has appointed Alvarez & Marsal Canada, Inc. as the monitor (the “Monitor”) of the Angiotech Entities’ business and financial affairs, with the powers set forth in the CCAA and orders of the Canadian Court, during the CCAA Proceedings. The Monitor will report to the Canadian Court on a regular basis on our financial and operational position and other relevant matters. In addition, the Monitor may advise the Company on certain restructuring matters throughout its CCAA Proceedings.
On February 17, 2011, the Canadian Court granted an order (the “Claims Procedure Order”) establishing a procedure for the adjudication, resolution and determination of claims of the Affected Creditors (as defined in the CCAA Plan) for voting and distribution purposes under the CCAA Plan. Creditors who do not file a proof of claim in accordance with the Claims Procedure Order will be barred from receiving distributions under the CCAA Plan and from asserting their claim in the future. As of March 7, 2011, the claims of Affected Creditors totalled approximately $3.6 million. In accordance with the terms of the fourth amendment to the RSA, the maximum amount of permitted claims, excluding the Company’s obligations under the Subordinated Notes and Existing Floating Rate Notes, is $30.0 million. Under the terms of the Claims Procedure Order, for the purposes of voting and distributions under the CCAA Plan, the amount of the claims in respect of the Subordinated Notes will only include the accrued amounts owing directly by Angiotech under the SSN Indenture and by the other Angiotech Entities under the guarantees executed by such other Angiotech Entities in respect of the Subordinated Notes up to the date of the Initial Order, January 28, 2011.
On February 17, 2011, the Canadian Court also granted an order (the “Meeting Order”) authorizing the holding of a meeting (the “Meeting”) of the Affected Creditors on April 4, 2011. The purpose of the Meeting is for the Affected Creditors to consider, and if deemed advisable, vote for or against the resolution approving the CCAA Plan. If the CCAA Plan is approved by a majority in number of Affected Creditors representing at least two-thirds in value of all such Affected Creditors’ claims that have been accepted for purposes of voting at the Meeting, in accordance with the provisions of the Claims Procedure Order, the Meeting Order and the CCAA, in each case, present and voting in person or by proxy at the Meeting, the Angiotech Entities intend to bring a further motion before the Canadian Court on or about April 6, 2011 seeking an order sanctioning the CCAA Plan.
In accordance with the terms of the Initial Order, on February 7, 2011, the Company and certain of its subsidiaries entered into a definitive agreement with Wells Fargo to secure a debtor-in-possession credit facility (the “DIP Facility”). On February 25, 2011, following entry of the February 22, 2011 recognition order of the CCAA Proceedings under the U.S. Bankruptcy Code (the “Recognition Order”),the loans under the DIP Facility were made available to the Company for borrowing. The DIP Facility will provide liquidity for working capital, general corporate purposes and expenses while the Company implements the CCAA Plan. The DIP Facility permits the Company to make revolving credit loans and provide letters of credit in an aggregate principal amount (including the face amount of any letters of credit) of up to $28.0 million. The DIP Facility is required to be repaid and terminated in connection with the implementation of the CCAA Plan. The Company expects to have a new credit facility in place upon implementation of the CCAA Plan in order to provide for, among other things, the repayment of any borrowings outstanding under the DIP Facility and the Existing Credit Facility, as amended. The Company incurred $1.2 million in fees to obtain the DIP Facility and expects additional fees of approximately $0.3 million to complete the DIP Facility. For more information, refer to notes 1, 16 and 26.
F-8
Table of Contents
Given that the initiation of the Creditor Protection Proceedings qualifies as an immediate event of default under the terms of the Company’s outstanding indebtedness, all obligations under (i) the Existing Credit Facility, (ii) the SSN Indenture, and (iii) the indenture governing the Existing Floating Rate Notes between the Company and Deutsche Bank National Trust Company in its capacity as successor trustee, dated December 11, 2006 (as amended, the “FRN Indenture”), have become due and payable under the terms of the respective agreements. The stay of proceedings granted under the Initial Order effectively prohibits these creditors from taking action to remedy or enforce their rights thereunder. The enforcement rights of these creditors are also subject to the applicable provisions of the CCAA and U.S. Bankruptcy Code.
The terms of the Initial Order have created a number of new charges against substantially all of the Company’s current and future assets. These charges, include (i) an administration charge to secure amounts owing to the Monitor and certain restructuring and financial advisors, up to a maximum of $2,500,000 (the “Administrative Charge”); (ii) a directors’ charge to secure the indemnity created under the Initial Order in favor of the directors and officers of the Angiotech Entities, up to a maximum of $2,000,000; (iii) a DIP charge to the extent of any obligations outstanding under the DIP Facility (the “DIP Charge”); and (iv) charges to secure amounts owing by the Company’s Canadian entities to its U.S. entities and by its U.S. entities to its Canadian entities pursuant to the intercompany advances made in accordance with intercompany transaction practices existing as of the date of the Initial Order, to the extent of any obligations outstanding under those advances (the “Inter-Company Charges”). These new charges rank in the following priority:
| (i) | the Administration Charge (to the maximum amount of $1,500,000); |
| (ii) | the Directors’ Charge (to the maximum amount of $1,000,000); |
| (iii) | any existing liens, charges, security interests or other encumbrances securing the obligations under the Existing Credit Facility; |
| (iv) | the DIP Charge; |
| (v) | the Inter-Company Charges, which rank pari passu with each other; |
| (vi) | the Administration Charge (to the maximum amount of $1,000,000); and |
| (vii) | the Director’s Charge (to the maximum amount of $1,000,000). |
Under bankruptcy legislation, certain payroll and commodity tax obligations also qualify as priority claims.
On February 10, 2011, the Company commenced the FRN Exchange Offer pursuant to the terms of the FRN Support Agreement. The FRN Exchange Offer is open to all holders of the Existing Floating Rate Notes. If the FRN Exchange Offer is consummated because holders owning at least a majority in aggregate principal amount outstanding of the Existing Floating Rate Notes tender their notes and consent, those holders of Existing Floating Rate Notes who do not participate in the FRN Exchange Offer will continue to hold Existing Floating Rate Notes under the FRN Indenture as modified by certain amendments providing for, among other things, elimination of most of the restrictive covenants and events of default contained therein. As of March 14, 2011, holders of approximately 89% of the aggregate principal amount of the outstanding Existing Floating Rate Notes support the terms of the FRN Support Agreement and have agreed to consent and tender their Existing Floating Rate Notes for New Floating Rate Notes in the FRN Exchange Offer. The closing of the FRN Exchange Offer is expected to occur concurrently with the implementation of the CCAA Plan.
For the year ended December 31, 2010, the Company incurred approximately $9.3 million of fees and expenses relating to the Recapitalization Transaction. These fees and expenses represent professional fees paid to the Company and the Consenting Noteholders’ financial and legal advisors to assist in the analysis of financial and strategic alternatives.
On January 13, 2011 and March 3, 2011, respectively, the Company’s common shares were delisted from the NASDAQ Stock Market (“NASDAQ”) and the Toronto Stock Exchange (“TSX”). For more information, refer to note 26.
GOING CONCERN AND LIQUIDITY
The Creditor Protection Proceedings and DIP Facility have provided the Company with temporary relief and financial support to enable it to continue to operate with minimal disruption while the Recapitalization Transaction is implemented. As a result, the audited consolidated financial statements have been prepared on a going concern basis, which assumes that the Company will continue to realize certain assets and discharge certain liabilities while operations are ongoing during the recapitalization period. In addition to the Creditor Protection Proceedings, there are several material uncertainties, which are described below, that cast substantial doubt about the Company’s ability to continue its operations as a going concern.
F-9
Table of Contents
The financial statements do not reflect any adjustments that may result from the impact of the Recapitalization Transaction and Creditor Protection Proceedings including: (i) the realizable value and availability of the Company’s assets on a liquidation basis to satisfy liabilities; (ii) the amounts, status and priority of pre-petition liabilities that may be allowed for claims or contingencies; (iii) the impact of any changes to the Company’s equity accounts that may result from recapitalization; or (iv) the impact on the Company’s Consolidated Statements of Operation that may result from any future restructuring changes made to the business. Upon emergence from Creditor Protection Proceedings, the Company will likely be required to adopt fresh-start accounting. Under fresh-starting accounting, further adjustments will likely be required that may materially impact the current carrying values and classifications of the Company’s assets, liabilities, long-term debt and shareholder’s equity. The Company is still assessing the provisions of reorganization accounting under U.S. GAAP to determine whether it qualifies for fresh-start accounting. During the period of Creditor Protection Proceedings, the Company will continue to apply its current accounting policies described in note 2 – Summary of Significant Accounting Policies.
| • | The Company’s most significant financial liabilities are its $325 million Existing Floating Rate Notes due December 1, 2013 and its $250 million Subordinated Notes due April 1, 2014. Interest payments related to this debt were historically supported in part by the Company’s license revenue derived from sales of TAXUS by BSC. However, the sustained and sharp decline in royalty revenues derived from sales of TAXUS over the last several years has significantly strained the Company’s liquidity and ability to service its long term debt obligations. As a result of the decline in TAXUS and other liquidity constraints, on January 28, 2011 the Angiotech Entities commenced Creditor Protection Proceedings to effectuate an in-court Recapitalization Transaction as contemplated by provisions under the RSA (see the Creditor Protection Proceedings and Recapitalization Transaction section above and note 16 for more information). The Recapitalization Transaction would eliminate the Company’s $250 million Subordinated Notes and related interest obligations in exchange for new common shares in the Company thereby providing significant improvements to its credit ratios, liquidity and financial flexibility. In accordance with the CCAA Plan, key elements of the Recapitalization Transaction are as follows: |
| i. | Under the terms of the RSA, the Consenting Noteholders have agreed to, among other things; exchange their Subordinated Notes for new common shares issued by the Company. All holders of the Subordinated Notes will be affected by the CCAA Plan. Under the terms of the CCAA Plan, Affected Creditors (including the Subordinated Noteholders) will receive their pro rata share of approximately 92.5% of the new common shares of the Company to be issued and outstanding upon completion of the Recapitalization Transaction as implemented by the CCAA Plan and Consenting Noteholders will receive, as additional consideration for their Subordinated Notes, their pro rata share of approximately 3.5% of the new common shares to be issued and outstanding immediately following the implementation of the CCAA Plan, in each case, subject to potential dilution. Upon implementation of the CCAA Plan, the Subordinated Notes, the SSN Indenture and all obligations thereunder will be deemed to be irrevocably and finally cancelled and eliminated. The RSA includes provisions pursuant to which the Consenting Noteholders have agreed to support the Recapitalization Transaction. As described in note 1, Creditor Protection Proceedings were commenced on January 28, 2011. |
| ii. | Under the terms of the FRN Support Agreement, the Consenting FRN Holders have agreed to, among other things; exchange their Existing Floating Rate Notes for new senior floating rate notes (the “New Floating Rates Notes”) through an Exchange Offer (the “FNR Exchange Offer”). The New Floating Rate Notes will be issued on substantially the same terms and conditions as the Existing Floating Rate Notes, except that, among other things, (i) subject to certain exceptions, the New Floating Rate Notes will be secured by second-priority liens over substantially all of the assets, property and undertaking of Angiotech and certain of its subsidiaries; (ii) the New Floating Rate Notes will accrue interest at LIBOR plus 3.75%, subject to a LIBOR floor of 1.25%; and (iii) certain covenants related to the incurrence of additional indebtedness and assets sales and the definition of permitted liens, asset sales and change of control will be modified in the indenture that governs the New Floating Rate Notes (the “New Notes Indenture”). |
| ii. | Under the terms of the CCAA Plan, existing common shares will be cancelled without further payment or compensation. |
| iii. | Under the terms of the CCAA Plan, all existing options, warrants or other rights to purchase common shares will be cancelled without further payment or compensation. |
| iv. | Upon implementation of the Recapitalization Transaction, the RSA required that the Company’s Existing Credit Facility with Wells Fargo be amended, refinanced or replaced to provide not less than $25 million and not more than $35 million in liquidity. On February 7, 2011, the Company entered into a $28 million DIP Facility with Wells Fargo (see note 1 and 16 (f) below for further information), which loans became available for borrowing on February 25, 2011. The Company expects to enter into a new credit facility in connection with the implementation of the Recapitalization Transaction. |
| v. | The completion of the Recapitalization Transaction is subject to the establishment of a stock-based incentive plan that is satisfactory to the Company and the Consenting Noteholders. The stock-based incentive plan may provide, over the life of the plan, for the grant of common stock and options of up to 15% of the new common shares, subject to potential dilution, as well as vesting and other considerations consistent with such stock-based incentive plans. |
F-10
Table of Contents
| vi. | The completion of the Recapitalization Transaction is subject to the establishment of a Key Employee Incentive Plan (“KEIP”) that is satisfactory to the Company and the Consenting Noteholders. The KEIP will provide payments to the named beneficiaries totaling no more than $1.0 million, subject to the completion of the Recapitalization Transaction. Two thirds will be paid on the completion date with the remaining one third to be paid three months after the completion date. |
| vii. | The completion of the Recapitalization Transaction is subject to the establishment of a Management Incentive Plan (“MIP”) that is satisfactory to the Company and the Consenting Noteholders. On the completion date of the Recapitalization Transaction, the MIP will provide the beneficiaries with 4% of the new common shares issued and options to acquire 6% of the new common shares issued, all subject to potential dilution and vesting considerations. These options are expected to expire on the seventh anniversary of the completion date of the Recapitalization Transaction. Options to acquire an additional 5% of the new common shares will be reserved for issuance at a later date at the discretion of the board of directors in place on the completion date of the Recapitalization Transaction. |
| vii. | The RSA requires completion of the Recapitalization Transaction no later than April 30, 2011. |
| • | As discussed above, for the year ended December 31, 2010, the Company incurred approximately $9.3 million of professional fees and expenses relating to the Recapitalization Transaction. The Company expects that it will continue to incur substantial costs related to the pursuit of the Recapitalization Transaction . In addition, upon successful completion of the Recapitalization Transaction certain of the Company’s financial advisors would be entitled to receive participation fees of approximately $5.3 million. These costs may further negatively impact the Company’s cash and liquidity position. |
| • | As discussed in note 16 (f), utilization of the Existing Credit Facility and DIP Facility are subject to a borrowing base formula based on the value of certain finished goods inventory and accounts receivable as well as certain restrictive covenants pertaining to operations and the maintenance of minimum levels of Adjusted EBITDA and fixed charge coverage ratios. While the Company complied with the Adjusted EBITDA and fixed charge coverage covenants specified under the Existing Credit Facility as at December 31, 2010, there are no assurances that the Company will continue to comply with the covenants specified under the DIP Facility and future borrowing arrangements in 2011 and beyond. A breach of these covenants, failure to obtain waivers to cure any further breaches of the covenants and restrictions set forth under the DIP Facility, failure to complete the proposed Recapitalization Transaction and failure to obtain required extensions of the Stay Period and Creditor Protection Proceedings, may limit the Company’s ability to obtain additional advances or result in demands for accelerated repayment, thus further straining the Company’s working capital position and ability to continue operations. As at March 10, 2011, approximately $11.0 million of financing is available under the DIP Facility, which is net of $2.8 million of secured letters of credit. The Company currently has $3.3 million outstanding under the Existing Credit Facility and $6.7 million outstanding under the DIP Facility other than such letters of credit. |
| • | For the year ended December 31, 2010, the Company reported a significant net loss of $66.8 million. Furthermore, as at December 31, 2010, the Company reported a stockholder’s deficit totaling $384.1 million and a net decrease in cash resources of $16.2 million as compared to December 31, 2009. During the year ended December 31, 2010, $21.6 million was used in operating activities; $3.7 million was used to fund investing activities and $9.3 million was provided by financing activities. Further operating losses and negative operating cash flows would continue to have an adverse impact on the Company’s liquidity position and ability to meet future obligations. |
| • | As discussed above, the Company’s long term-debt interest payments continue to be significant and strain the Company’s liquidity. During the year ended December 31, 2010, the Company incurred interest expense of $32.9 million on the Existing Floating Rate Notes and Subordinated Notes, as compared to $34.9 million for 2009. If the Company is able to successfully eliminate its $250 million aggregate principal amount outstanding of Subordinated Notes through the Recapitalization Transaction, the Company’s future interest obligations with respect to its total outstanding indebtedness are expected to be significantly reduced. The Existing Floating Rate Notes reset quarterly to an interest rate of 3-month London Interbank Offered Rate (“LIBOR”) plus 3.75%. As LIBOR may fluctuate from period to period, such volatility affects quarterly interest rates on the Existing Floating Rate Notes, thereby affecting the magnitude of interest costs within a given period. As December 31, 2010, the Existing Floating Rate Notes bore interest rates of 4.1% (December 31, 2009 - 4.0%). Under the FRN Support Agreement, the New Floating Rate Notes will accrue interest at LIBOR plus 3.75%, subject to a LIBOR floor of 1.25%. Given that the Company has drawn upon the Existing Credit Facility described above, additional interest expense of $0.1 million was incurred during 2010. The Company expects to incur further interest charges as it continues to borrow under the DIP Facility. |
| • | During the year ended December 31, 2010, the Company’s Pharmaceutical Technologies segment experienced a significant and sharp decline of $53.0 million in revenues. Revenue from the Company’s Pharmaceutical Technologies segment for the year ended December 31, 2010 was $34.7 million compared to $87.7 million in the prior year. The decline is primarily attributable to (a) the receipt of a $25.0 million one-time license fee received from Baxter International, Inc. (“Baxter”) under an Amended and Restated Distribution and License Agreement in 2009 and (b) a decrease in royalty revenues based on declining sales of TAXUS by BSC. The decline in sales is primarily due to the entry of several new competitors into the drug-eluting stent market primarily over the past two years. Under its license agreement with BSC, the Company does not |
F-11
Table of Contents
| have the ability to control direct or indirect sales of TAXUS products. The Company expects that competitive products will continue to adversely impact TAXUS related revenues and cash flows in 2011 and subsequent years. |
| • | As described in note 20, on January 27, 2011 the Company entered into a Settlement Agreement with QSR Holdings, Inc. (“QSR”), resolving among other things, any and all claims arising out of the Agreement of Plan and Merger dated May 25, 2006 related to the Company’s 2006 acquisition of Quill Medical Inc. Under the terms of the Settlement Agreement, Angiotech Pharmaceuticals (US), Inc. (“Angiotech US”) is obligated to pay QSR $6.0 million (the “Settlement Amount”) over a two-year period. The Settlement Amount is expected to negatively impact the Company’s cash flow and liquidity position. |
| • | As reported in note 20, the Company has entered into various research and development collaboration agreements, under which it may be required to participate in certain cost sharing or funding arrangements. The Company may also be obligated to make certain royalty and milestone payments contingent upon the achievement of contractual development, collaboration, regulatory and/or commercial milestones, which are not necessarily within the Company’s control. As such, acceleration of research and development activities under collaboration agreements by counterparties and any unexpected outcomes may trigger contingent payments earlier than are currently expected, thus negatively impacting the Company’s cash flows and liquidity position. |
The Company’s ability to continue as a going concern will depend upon its ability to: (i) execute and complete the Recapitalization Transaction as described above and in note 16, (ii) restructure, refinance or amend terms of its other existing debt obligations; (iii) remain in compliance with its debt covenants to maintain access to its DIP Facility and the proposed new credit facility to be implemented in connection with the implementation of the Recapitalization Transaction (the “Exit Facility”); (iv) obtain additional equity or debt financing when needed; (v) achieve revenue growth and improvement of gross margins; and (vi) achieve greater operating efficiencies or a reduction of expenses.
The Company’s future plans and current initiatives to manage its operating, going concern and liquidity risks include, but are not limited to the following:
| • | execution and completion of the Recapitalization Transaction and the FRN Exchange Offer to reduce the burden of existing debt levels and interest expense; |
| • | maintenance and utilization of the DIP Facility with Wells Fargo and negotiation and finalization of the Exit Facility; |
| • | continued exploration of various other strategic and financial alternatives, including but not limited to raising new equity or debt capital or exploring sales of various assets or businesses; |
| • | continued evaluation of budgets and forecasts for certain sales and marketing and research and development programs; |
| • | selected gross margin improvement initiatives; |
| • | potential sale of non-productive property, plant and equipment assets; and |
| • | optimization of day-to-day operating cash flows. |
Management continues to closely monitor and manage liquidity by regularly preparing rolling cash flow forecasts, monitoring the condition and value of assets available to be used as security in financing arrangements, seeking flexibility in financing arrangements and establishing programs to maintain compliance with the terms of financing agreements.
Although the Company has entered into agreements with the intent to recapitalize the Company, there is no assurance that the Company will be able to successfully execute and complete the Recapitalization Transaction All transactions contemplated under the terms of the RSA are subject to approval by the Canadian Court, the Consenting Noteholders, as well as other Affected Creditors. The completion of the Recapitalization Transaction may also be subject to approval from certain third parties, government bodies and regulatory bodies. In the event that the Company is unable to successfully complete the Recapitalization Transaction within the applicable Stay Period; the Subordinated Noteholders, holders of the Company’s Existing or Wells Fargo may elect to exercise rights under the respective indentures or credit agreements allowing them to accelerate repayment and, in the case of the Wells Fargo under the Company’s Existing Credit Facility and DIP Facility, refuse to extend further credit to the Company. In this case, the Company’s current cash and credit capacity would not be sufficient to service principal and interest obligations under our outstanding debt. In addition, restrictions on the Company’s ability to borrow and the possible accelerated demand of repayment of existing or future obligations by any of the Company’s creditors might jeopardize the Company’s working capital position and ability to sustain its operations.
F-12
Table of Contents
While the Company believes that it has developed planned courses of action and identified other opportunities to mitigate the operating, going concern and liquidity risks outlined above, there is no assurance that it will be able to complete any or all of the plans or initiatives that have been identified or obtain sufficient liquidity to execute its business plan. Furthermore, there may be other material risks and uncertainties that may impact the Company’s liquidity position that have not yet been identified.
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
(a) Consolidation
These consolidated financial statements include the accounts of the Company and its wholly-owned subsidiaries. All intercompany transactions and balances have been eliminated on consolidation.
(b) Use of estimates
The Company’s preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amount of assets and liabilities and disclosures of contingent assets and liabilities at the dates of the financial statements and the reported amounts of revenue and expenses during the reported periods. Actual results could differ materially from these estimates.
(c) Foreign currency translation
The Company’s functional and reporting currency is the U.S. dollar. Foreign currency denominated revenues and expenses are translated using average rates of exchange during the year. Foreign currency denominated monetary assets and liabilities are translated at the rate of exchange in effect at the balance sheet date with foreign exchange gains (losses) included in the Statement of Operations, except for the translation gains (losses) arising from available-for-sale instruments which are recorded through Other Comprehensive Income until realized through disposal or impairment. Foreign currency denominated non monetary assets and liabilities are re-translated using historical exchange rates.
The assets and liabilities of foreign subsidiaries that have a functional currency different from the reporting currency are translated into the reporting currency with all resulting exchange differences recognized in other comprehensive income. Assets and liabilities are translated at each balance sheet date at the closing rate at the date of the balance sheet. Income and expenses are translated at average exchange rates. The Company has certain subsidiaries which also have a U.S. dollar denominated functional and reporting currency and therefore it does not record additional translation gains (losses) on consolidation since the functional and reporting currency of the subsidiary is the same as the parent Company.
(d) Cash equivalents
The Company considers all highly liquid financial instruments purchased with an original maturity of three months or less to be cash equivalents. Cash equivalents are recorded at cost plus accrued interest. The carrying values of these cash equivalents approximate their fair values.
(e) Investments
The Company classifies marketable equity securities as available-for-sale. Investments that are classified as available-for-sale are carried at fair value with unrealized gains or losses, net of tax, reflected in accumulated other comprehensive income (loss). Realized losses are determined on a specific identification method and are included in write-downs/losses on redemption of investments. When the Company believes that the unrealized losses are other-than-temporary, these losses are included in the statement of operations and reclassified out of accumulated other comprehensive income (loss).
The Company classifies its investments in equity securities as long-term investments, unless the Company expects to sell the investment in less than one year. Long-term investments for which fair value is not readily determinable are recorded at cost. The Company regularly reviews its long-term investments for impairment indicators by reference to quoted market prices, the results of operations, financial position of the investee and other evidence supporting the net realizable value of the investment. If an impairment indicator is identified which signals that the carrying amount may not be recoverable and the impact is other-than-temporary, the investment is written down to its estimated fair value and the resulting losses are included in the determination of income for the period. Where fair value information is not readily available, the fair value of investments may be estimated using inputs which are in compliance with ASC No. 810 — Fair Value Measurements and Disclosures but may not be readily observable in active markets. A valuation model is developed for each investment using key assumptions about future cash flows and discount rates. These assumptions are based on historical experience, market trends and the anticipated return for each investment.
(f) Allowance for doubtful accounts
Accounts receivable are presented net of an allowance for doubtful accounts. In determining the allowance for doubtful accounts, which include specific reserves, the Company reviews accounts receivable aging, customer financial strength, credit standing and
F-13
Table of Contents
payment history to assess the probability of collection. The Company continually monitors the collectability of the receivables. Receivables are written off when management determines they are uncollectible.
Prior to the write-off of any specific account, the Company utilizes the services of approved third party collection agencies to assist in the collection of accounts deemed uncollectible. If the efforts of the collection agency are deemed to be unsuccessful, the account is written off upon final approval by one or more duly authorized persons.
(g) Inventories
Raw materials are recorded at the lower of cost, determined on a specific item basis, and net realizable value. Work-in-process, which includes inventory stored at a stage preceding final assembly and packaging, and finished goods are recorded at the lower of cost, determined on a standard cost basis which approximates average cost and net realizable value.
(h) Property, plant and equipment
Property, plant and equipment are recorded at cost less accumulated depreciation. Depreciation is provided using the straight-line method over the following terms:
| Buildings |
40 years | |
| Leasehold improvements |
Term of the lease | |
| Manufacturing equipment |
3 – 10 years | |
| Research equipment |
5 years | |
| Office furniture and equipment |
3 – 10 years | |
| Computer equipment |
3 – 5 years |
(i) Goodwill and intangible assets
As at December 31, 2008, the Company’s goodwill balance of $649.7 million was determined to be impaired and was therefore written off (see note 10 (b)). Goodwill represents the difference between the purchase price and the estimated fair value of the net assets acquired under the purchase method of accounting. In accordance with Accounting Standards Codification (“ASC”) No. 350, Intangibles — Goodwill and Other, goodwill was not amortized and was pushed-down to the reporting entities.
Where goodwill is recorded, it is tested for impairment at least annually in all of the Company’s reporting units and whenever changes in circumstances occur that would indicate impairment in the value of goodwill. When the carrying value of a reporting unit’s goodwill exceeds the implied fair value of the goodwill, an impairment loss is recognized which is equal to the excess. Examples of circumstances that could trigger impairment include adverse changes or outcomes in legal or regulatory matters, technological advances, decreases in anticipated demand, unanticipated competition, and significant declines in the Company’s share price. The Company estimates fair value based on a discounted projection of future cash flows which are subject to significant uncertainty and estimates.
Intangible assets with finite lives are amortized based on their estimated useful lives. The amortization method is selected to best reflect the pattern in which the economic benefits are derived from the intangible asset. If the pattern cannot be reliably or reasonably determined, straight line amortization is applied. Amortization is generally determined using the straight line method over the following terms:
| Acquired technologies |
2 - 10 years | |
| Customer relationships |
10 years | |
| In-licensed technologies |
5 - 10 years | |
| Trade name and other |
2 - 12 years |
(j) Impairment of long-lived assets
The Company reviews long-lived assets subject to amortization to determine if any adverse conditions exist or a change in circumstances has occurred that would indicate impairment or a change in the remaining useful life. Conditions that may indicate impairment include, but are not limited to, a significant adverse change in legal factors or business climate that could affect the value of an asset, a product recall, or an adverse action or assessment by a regulator. If an impairment indicator exists, the Company tests the asset (asset group) for recoverability. For purposes of the recoverability test, long-lived assets are grouped with other assets and liabilities at the lowest level of identifiable cash flows if the long-lived asset does not generate cash flows independent of other assets and liabilities. If the carrying value of the asset (asset group) exceeds the undiscounted cash flows expected to result from the use and eventual disposition of the asset (asset group), an impairment charge is recorded to write the carrying value down to the fair value in the period identified.
F-14
Table of Contents
The Company estimates the fair value of assets (asset groups) by calculating the present value of estimated future cash flows expected to be generated from the asset using a risk-adjusted discount rate. In determining the estimated future cash flows associated with our assets (asset groups), certain assumptions are made about future revenue contributions, cost structures, discount rates and the remaining useful lives of the asset (asset group). Variation in the assumptions used could result could result in materially differences when estimating impairment write-downs.
(k) Revenue recognition
(i) Product sales
Revenue from product sales, including shipments to distributors, is recognized when the product is shipped from the Company’s facilities to the customer provided that the Company has not retained any significant risks of ownership or future obligations with respect to products shipped. Revenue from product sales is recognized net of provisions for future returns. These provisions are established in the same period as the related product sales are recorded and are based on estimates derived from historical experience and adjusted to actual returns when determinable.
Revenue is considered to be realized or realizable and earned when all of the following criteria are met: persuasive evidence of a sales arrangement exists; delivery has occurred or services have been rendered; the price is fixed or determinable; and collectability is reasonably assured. These criteria are generally met at the time of shipment when the risk of loss and title passes to the customer or distributor.
Amounts billed to customers for shipping and handlings are included in revenue. Where applicable, revenue is recorded net of sales taxes. The corresponding costs for shipping and handling are included in cost of products sold.
(ii) Royalty revenue
Royalty revenue is recognized when the Company has substantially fulfilled its performance obligations under the terms of the contractual agreement, no significant future obligations remain, the amount of the royalty fee is determinable, and collection is reasonably assured. The Company records royalty revenue from BSC on a cash basis due to the difficulty in accurately estimating the BSC royalty before the reports and payments are received by the Company.
(iii) License fees
License fees are comprised of initial fees and milestone payments derived from collaborative and other licensing arrangements. License fees are recognized as revenue when persuasive evidence of an arrangement exists, the contracted fee is fixed or determinable, the intellectual property is delivered to the customer, collection is reasonably assured, and the Company has substantially completed its performance obligations. Non-refundable milestone payments are recognized upon the achievement of specified milestones when the milestone payment is substantive in nature, achievement of the milestone was not reasonably assured at the inception of the agreement, and the Company has no further significant performance obligations to fulfill under the arrangement. Initial fees and non-refundable milestone payments received which require the ongoing involvement of the Company are deferred and amortized into income on a straight-line basis over the period of ongoing involvement if there is no other method under which performance can be objectively measured.
(l) Income taxes
Income taxes are accounted for using the liability method. Deferred income tax assets and liabilities result from the temporary differences between the amount of assets and liabilities recognized for financial statement and income tax purposes, and for operating losses, capital losses and tax credit carry forwards, using tax rates expected to be in effect for the years in which the differences are expected to reverse. A valuation allowance is provided to reduce the deferred income tax assets if, based upon the available evidence, it is more likely than not that some or all of the deferred income tax assets will not be realized.
(m) Accounting for Uncertainty in Income Taxes
The Company accounts for uncertainty related to income tax positions in accordance with ASC No. 740-10 — Income taxes. ASC No. 740-10 requires the use of a two-step approach for recognizing and measuring the income tax benefits of uncertain tax positions taken or expected to be taken in a tax return, as well as enhanced disclosures regarding uncertain tax positions. A tax benefit from an uncertain tax position may only be recognized if it is more-likely-than-not that the position will be sustained upon examination by the tax authority based on the technical merits of the position. The tax benefit of an uncertain tax position that meets the more-likely-than-not recognition threshold is measured as the largest amount that is greater than 50% likely to be realized upon settlement with the tax authority. To the extent a full benefit is not expected to be realized, an income tax liability is established. Any change in judgment related to the expected resolution of uncertain tax positions are recognized in the year of such a change. Accrued interest and penalties related to unrecognized tax benefits are recorded in income tax expense in the current year.
F-15
Table of Contents
(n) Research and development costs
Research and development expenses are comprised of costs incurred in performing research and development activities including salaries and benefits, clinical trial and related clinical manufacturing costs, contract research costs, patent procurement costs, materials and supplies, and other operating and occupancy costs. Amounts paid for medical technologies used solely in research and development activities and with no alternative future use are expensed in the year incurred.
Advance payments for non-refundable portions of research and development activities are deferred and capitalized until the related goods are delivered or services are performed.
(o) In-process research and development costs
In-process research and development costs, including upfront fees and milestones paid to collaborators, are expensed in the year incurred if the technology has not demonstrated technological feasibility and does not have any alternative future use.
(p) Net loss per common share
Net loss per common share is calculated using the weighted average number of common shares outstanding during the period. Diluted net loss per common share is calculated using the treasury stock method which uses the weighted average number of common shares outstanding during the period and also includes the dilutive effect of potentially issuable common shares from outstanding stock options.
(q) Stock-based compensation
The Company accounts for stock based compensation expense in accordance with ASC No. 718 — Compensation: Stock Compensation and ASC No. 505-50 — Equity: Equity Based Payments to Non-Employees. These standards require the Company to recognize the grant date fair value of stock-based compensation awards granted to employees over the requisite service period. The compensation expense recognized reflects estimates of award forfeitures at the time of grant and revised in subsequent periods, if necessary when forfeitures rates are expected to change. The Company uses yield rates on U.S. Treasury or Canadian Government securities for a period approximating the expected term of the award to estimate the risk-free interest rate in the grant-date fair value assessment. The Company used its historical volatility as a basis to estimate the expected volatility assumption used in the Black-Scholes model. The Company has not paid any dividends on common stock since its inception and does not anticipate paying dividends on its common stock in the foreseeable future. Generally, the stock options granted have a maximum term of five years and vest over a four-year period from the date of the grant. When an employee ceases employment at the Company, any unexercised vested options granted will expire either immediately, within 365 days from the last date of service or on the original expiration date at the time the option was granted, as defined in the Company’s Stock Incentive Plan. The expected life of employee stock options is based on a number of factors, including historic exercise patterns, cancellations and forfeiture rates, vesting periods and contractual terms of the options.
(r) Deferred leasehold inducement
Leasehold inducements are deferred and amortized to reduce rent expense on a straight line basis over the term of the lease.
(s) Deferred financing costs
Financing costs for long-term debt are capitalized and amortized on a straight-line basis, which approximates the effective-interest rate method to interest expense over the life of the debt instruments. The Company revisits the life of debt instruments as events warrant.
(t) Costs for patent litigation and legal proceedings
Costs for patent litigation or other legal proceedings are expensed as incurred and included in selling, general and administrative expenses.
(u) Recently adopted accounting policies
In June 2009, the FASB issued a new standard under ASC No. 810-10, Consolidation which changes the consolidation model for Variable Interest Entities (“VIE”). The revised model increases the qualitative analysis required when identifying which entity is the primary beneficiary that has (i) the power to direct the activities of a VIE that most significantly impact the entity’s economic performance and (ii) the obligation to absorb losses or the right to receive benefits from the VIE. The new standard eliminates the QSPE exemption, requires ongoing reconsideration of the primary beneficiary and amends the events which trigger reassessment of whether an entity is a VIE. The Company adopted the new guidance effective January 1, 2010. Adoption of this standard did not have a material impact on the Company’s financial results for the year ended December 31, 2010.
In January 2010, the FASB issued ASU No. 2010-02, Consolidation: Accounting and Reporting for Decreases in Ownership of a Subsidiary – a Scope Clarification, under ASC No. 810. The update clarifies the scope of decreases in ownership provisions and
F-16
Table of Contents
expands required disclosures for subsidiaries that are deconsolidated or group of assets that are derecognized. ASU No. 2010-02 is effective beginning in the first interim or annual reporting period ending on or after December 15, 2009, however, the amendments must be applied retrospectively to the first period that an entity adopted ASC No. 810-10. The adoption of the update did not have any impact on the Company’s financial statements.
In January 2010, the FASB issued ASU No. 2010-06, Fair Value Measurements and Disclosures: Improving Disclosures about Fair Value Measurements, under ASC No. 820-10. The update requires new disclosures about significant transfers in and out of Level 1 and Level 2 fair value measurements. ASU No. 2010-06 also clarifies disclosures required about inputs, valuation techniques and the level of disaggregation applied to each class of assets and liabilities. These updates are effective for interim and annual reporting periods beginning after December 15, 2009. These amendments have had no material impact on the Company’s financial results given that they relate to disclosure and presentation only.
In February 2010, the FASB issued ASU No. 2010-09, Subsequent Events: Amendments to Certain Recognition and Disclosure Requirements, The amendment removes the requirement for an SEC filer to disclose the date through which subsequent events have been evaluated in both issued and revised financial statements. SEC filers are still required to evaluate subsequent events through the date that the financial statements are issued. ASU No. 2010-09 was effective upon issuance and had no material impact on the Company’s financial statements or disclosures.
In February 2010, the FASB issued ASU No. 2010-08, Technical Corrections to Various Topics. The amendments effectively eliminate certain inconsistencies and outdated provisions in various topics under US GAAP to provide further clarification on the FASB’s original intent. ASU No. 2010-08 is effective for year’s beginning after December 15, 2009. The amendments are generally non-substantive in nature and have not resulted in significant or pervasive changes to our previous accounting assessments of the relevant accounting standards which are considered under ASU No. 2010-08.
In April 2010, the FASB issued ASU No. 2010-17, Revenue Recognition – Milestone Method of Revenue Recognition, under ASC No. 605. The new guidance defines specific criteria for evaluating whether the milestone method is appropriate for the purposes of assessing revenue recognition. ASU No. 2010-17 stipulates that consideration tied to the achievement of a milestone may only be recognized if it meets all of the defined criteria for the milestone to be considered substantive. The guidance also requires expanded disclosures about the overall arrangement, the nature of the milestones, the consideration and the assessment of whether the milestones are substantive. ASU No. 2010-17 is effective on a prospective basis for milestones achieved in fiscal years and interim periods beginning on or after June 15, 2010. The adoption of this standard had no material impact on the Company’s recognition of revenue from current collaboration and revenue contracts.
In July 2010, the FASB issued ASU No. 2010-20, Disclosures about the Credit Quality of Financing Receivables and the Allowance for Credit Losses under ASC No. 310. The new guidance requires entities to provide a greater level of disaggregated information about the credit quality of its financing receivables and its allowance for credit losses. Disclosures may include information about credit quality indicators, overdue accounts, and modifications of financing receivables. The guidance is effective for public entities with interim and annual reporting periods ending on or after December 15, 2010. This guidance had no material impact on the Company’s financial results given that it relates to disclosure and presentation only.
(v) Future accounting pronouncements
In October 2009, the FASB issued ASU No. 2009-13, Multiple-Deliverable Revenue Arrangements, under ASC No. 605. The new guidance provides a more flexible alternative to identify and allocate consideration among multiple elements in a bundled arrangement when vendor-specific objective evidence or third-party evidence of selling price is not available. ASU No. 2009-13 requires the use of the relative selling price method and eliminates the residual method to allocation arrangement consideration. Additional expanded qualitative and quantitative disclosures are also required. The guidance is effective prospectively for revenue arrangements entered into or materially modified in years beginning on or after June 15, 2010. The Company is still assessing the potential impact of ASU No. 2009-13, however, given that the Company does not currently have any bundled revenue arrangements; adoption of this guidance is not expected to have a significant impact on financial results.
In January 2010, the FASB issued ASU No. 2010-06, Fair Value Measurements and Disclosures: Improving Disclosures about Fair Value Measurements, under ASC No. 820-10. The update requires new disclosures about roll forward activity affecting Level 3 fair value measurements. ASU No. 2010-06 also clarifies disclosures required about inputs, valuation techniques and the level of disaggregation applied to each class of assets and liabilities. New disclosures about Level 3 fair value measurements are effective for interim and annual reporting periods beginning after December 15, 2010. The Company is assessing the potential impact that the adoption of ASU No. 2010-13 will have on its financial statement disclosures.
In April 2010, the FASB issued ASU No. 2010-13, Effect of Denominating the Exercise Price of a Share-Based Payment Award in the Currency of the Market in Which the Underlying Equity Security Trades. ASC No. 718, Compensation – Stock Compensation, provides guidance on whether share-based payments should be classified as either equity or a liability. Under this section, share-based payment awards that contain conditions which are not market, performance or service conditions are classified as liabilities. The updated guidance clarifies that an employee share-based payment award, with an exercise price denominated in the currency of a market in which substantial portion of the entity’s equity securities trade and which differs from the functional currency of the employer entity or payroll currency of the employee, are not considered to contain a condition that is not a market, performance or service condition. As such, these awards are classified as equity. ASU No. 2010-13 is effective for fiscal years and interim periods beginning on or after December 15, 2010. The cumulative effect of adopting this guidance should be applied to the opening balance of retained earnings. The Company is assessing the potential impact of adopting ASU No. 2010-13.
F-17
Table of Contents
In December 2010, the FASB issues ASU No. 2010-29, Disclosure of Supplementary Pro Forma Information for Business Combinations. The new guidance clarifies that if a public entity presents comparative financial statements, the entity should disclose revenue and earnings of the combined entity as if the business combination occurred as of the beginning of the comparable prior annual reporting period only. ASU No. 2010-29 also requires additional disclosures about the nature and amount of material, non-recurring pro forma adjustments, which are directly attributable to the business combination and are included in the pro forma revenue and earnings estimates. The guidance is effective for all business combinations beginning on first annual reporting period beginning on or after December 15, 2010. The Company is currently assessing whether ASU No. 2010-29 will impact any of its future disclosures.
In December 2010, the FASB issues ASU No. 2010-28, Intangibles—Goodwill and Other (Topic 350). The amendments in this Update modify Step 1 of the goodwill impairment test for reporting units with zero or negative carrying amounts. For those reporting units, an entity is required to perform Step 2 of the goodwill impairment test if it is more likely than not that a goodwill impairment exists. In determining whether it is more likely than not that a goodwill impairment exists, an entity should consider whether there are any adverse qualitative factors indicating that an impairment may exist. The qualitative factors are consistent with the existing guidance and examples in paragraph 350-20-35-30, which requires that goodwill of a reporting unit be tested for impairment between annual tests if an event occurs or circumstances change that would more likely than not reduce the fair value of a reporting unit below its carrying amount. For public entities, the amendments in this Update are effective for fiscal years, and interim periods within those years, beginning after December 15, 2010. As the Company had no goodwill as of December, 31, 2010, it does not believe there will be any impact on its financial statements or disclosures.
In January 2011, the FASB issues ASU No. 2011-01, Deferral of the Effective Date of Disclosures about Troubled Debt Restructurings in Update No. 2010-20. The new guidance temporarily delays the effective date of the disclosures about troubled debt restructurings in Update 2010-20 for public entities. The delay is intended to allow the Board time to complete its deliberations on what constitutes a troubled debt restructuring. The effective date of the new disclosures about troubled debt restructurings for public entities and the guidance for determining what constitutes a troubled debt restructuring will then be coordinated. Currently, that guidance is anticipated to be effective for interim and annual periods ending after June 15, 2011. The Company is currently assessing whether ASU No. 2011-01 will impact any of its future disclosures.
3. FINANCIAL INSTRUMENTS AND FINANCIAL INSTRUMENT RISK
For certain of the Company’s financial assets and liabilities, including cash and cash equivalents, accounts receivable, accounts payable and accrued liabilities, the carrying amounts approximate fair value due to their short-term nature. The fair value of short-term investments approximates $4.7 million as at December 31, 2010 (December 31, 2009 - $7.8 million). The total fair value of long-term debt and accrued interest approximates $418.6 million (December 31, 2009 - $436.8 million) and has a carrying value of $590.8 million as at December 31, 2010 (December 31, 2009 - $581.0 million). The fair values of short-term investments and long-term debt, including accrued interest, is based on quoted market prices in active markets for identical assets as at December 31, 2010 and 2009.
Financial risk includes interest rate risk, exchange rate risk and credit risk.
| • | Interest rate risk arises due to the Company’s long-term debt bearing fixed and variable interest rates. The interest rate on the Existing Floating Rate Notes is reset quarterly to 3-month LIBOR plus 3.75%. The Existing Floating Rate Notes currently bear interest at a rate of 4.1% (December 31, 2009 – 4.0%). The Company does not use derivatives to hedge against interest rate risks. |
| • | Foreign exchange rate risk arises as a portion of the Company’s investments, revenues and expenses are denominated in other than U.S. dollars. The Company’s financial results are subject to the variability that arises from exchange rate movements in relation to the U.S. dollar, and is primarily limited to the Canadian dollar, the Swiss franc, the Euro, the Danish kroner and the UK pound sterling. Foreign exchange risk is primarily managed by satisfying foreign denominated expenditures with cash flows or assets denominated in the same currency. |
| • | Credit risk arises as the Company provides credit to its customers in the normal course of business. The Company performs credit evaluations of its customers on a continuing basis and the majority of its trade receivables are unsecured. The maximum credit risk loss that the Company could face is limited to the carrying amount of accounts receivable. Concentrations of credit risk with respect to trade accounts receivable are limited due to the large number of customers and their dispersion across many geographic areas. However, a significant amount of trade accounts receivable is with the national healthcare systems of several countries. Efforts by governmental and third-party payers to reduce health care costs or the announcement of legislative proposals or reforms to implement government controls could impact the Company’s credit risk. Although the Company does not currently foresee a credit risk associated with these receivables, collection is dependent upon the financial stability of those countries’ national economies. At December 31, 2010, accounts receivable is net of an allowance for uncollectible accounts of $2.4 million (December 31, 2009 -$0.5 million). |
F-18
Table of Contents
4. ACQUISITION OF CERTAIN ASSETS OF HAEMACURE CORPORATION
On April 6, 2010, the Company acquired certain Haemacure Corporation (“Haemacure”) assets for consideration of $3.9 million. Under ASC No. 805, the transaction qualified as a business combination and the acquisition method was used to account for the transaction.
Prior to the acquisition, the Company entered into various license, distribution, development and supply agreements for fibrin and thrombin technologies. The collaboration was intended to provide the Company with access to technology for certain of its surgical product candidates which are currently in preclinical development. As these agreements did not contain any settlement provisions and were neither favorable nor unfavorable when compared to current market pricing for similar transactions, no settlement gains or losses were recognized.
As part of the collaboration, the Company provided Haemacure with a senior secured bridge financing facility (the “loan”) of $2.5 million in multiple draw-downs throughout 2009. The loan was for a two year term, bore an annual interest rate of 10%, compounded quarterly, and was senior to all of Haemacure’s indebtedness, subject to certain exceptions. The loan was secured by substantially all of Haemacure’s assets. As at December 31, 2009, the loan was recognized at its fair value of $1.6 million with the remainder being amortized on account of an arrangement for prepaid services. On January 11, 2010 Haemacure announced it filed a notice of intention to make a proposal to its creditors under the Bankruptcy and Insolvency Act (Canada) and its wholly-owned U.S. subsidiary sought court protection under Chapter 11 of the U.S. Bankruptcy Code. These actions were taken in light of Haemacure’s financial condition and inability to raise additional financing. The Company subsequently provided an additional $1.0 million of financing to fund Haemacure’s insolvency proceedings and day-to-day operations. The loan and accrued interest was effectively settled in exchange for certain assets acquired from Haemacure. In addition, the Company paid an additional $0.2 million in cash to settle certain obligations owed by Haemacure.
As at April 5, 2010, the carrying value of the loan owed by Haemacure was $2.5 million compared to the contractual amount of $3.7 million. In accordance with ASC No. 805-10-55-21, the Company recognized a $1.2 million gain on the effective settlement of the pre-existing debt arrangement, which reflects the difference between the contractual settlement amount of the loan and its carrying value.
The estimated fair values of the net assets acquired at the date of acquisition are summarized as follows:
| April 6, 2010 | ||||
| Property, plant and equipment |
$ | 2,906 | ||
| Intangible assets |
1,700 | |||
| Deferred income tax liability |
(720 | ) | ||
| Estimated fair value of net assets acquired |
3,886 | |||
| Consideration: |
||||
| Face value of the loan receivable, including accrued interest |
3,686 | |||
| Cash paid |
200 | |||
| Total consideration |
$ | 3,886 | ||
The Company used the income approach to determine the fair value of the intangible assets acquired. The fair values of the property, plant and equipment were estimated using a cost-based approach which considers replacement values as well as the values ascribed to the assets for their highest and best use as defined by U.S. GAAP.
As the acquisition qualified as a business combination, transaction-related expenses of $0.6 million were charged to selling, general and administration costs during the year ended December 31, 2010. The assets acquired from Haemacure have been included in the consolidated financial statements since the date of acquisition. Given our current liquidity constraints and recapitalization plans being pursued through the Creditor Protection Proceedings; we have indefinitely suspended research and development activities related to the Haemacure fibrin and thrombin technologies. As a result, as at December 31, 2010, we wrote-off the $3.9 million of net assets acquired from Haemacure during the year. This impairment charge triggered a corresponding reduction of the $0.7 million deferred income tax liability and expense, thereby increasing the loan settlement gain to $1.9 million as at December 31, 2010.
Pro forma information (unaudited)
The following unaudited pro forma information is provided for the business combination assuming that it occurred at the beginning of the earliest period presented, January 1, 2009. The pro forma information combines the financial results of the Company with the financial results of Haemacure for the year ended December 31, 2010 and December 31, 2009.
F-19
Table of Contents
| Year ended December 31, 2010 |
Year ended December 31, 2009 |
|||||||
| Net loss |
$ | (68,997 | ) | $ | (27,505 | ) | ||
| Net loss per share |
$ | (0.81 | ) | $ | (0.32 | ) | ||
The information presented above is for illustrative purposes only and is not indicative of the results that would have been achieved had the business combination taken place at the beginning of the earliest period presented. For comparative purposes, the pro forma information above excludes the impact of the $1.9 million non-recurring loan settlement gain and $0.6 million of transaction costs which were incurred in connection with the business combination during the year ended December 31, 2010. The pro forma impact on revenue was not material for any of the periods presented.
5. CASH AND CASH EQUIVALENTS
Cash and cash equivalents includes the following:
| December 31, 2010 |
December 31, 2009 |
|||||||
| U.S. dollars |
$ | 27,779 | $ | 34,335 | ||||
| Canadian dollars |
933 | 5,542 | ||||||
| Swiss francs |
575 | 986 | ||||||
| Euros |
1,447 | 5,680 | ||||||
| Danish kroner |
1,090 | 412 | ||||||
| Other |
1,491 | 2,587 | ||||||
| $ | 33,315 | $ | 49,542 | |||||
All cash and cash equivalents are held in interest and non-interest bearing bank accounts.
6. SHORT TERM INVESTMENTS
| December 31, 2010 |
Cost | Net Unrealized Gains |
Carrying value | |||||||||
| Short-term investments: |
||||||||||||
| Available-for-sale equity securities |
$ | 848 | $ | 3,805 | $ | 4,653 | ||||||
| December 31, 2009 |
Cost | Net Unrealized Gains |
Carrying value | |||||||||
| Short-term investments: |
||||||||||||
| Available-for-sale equity securities |
$ | 848 | $ | 6,932 | $ | 7,780 | ||||||
Short term investments as at December 31, 2010 and 2009 include investments in a publicly traded biotechnology company.
During the year ended December 31, 2010, the Company recorded $1.3 million of write-offs related to three of its long term investments in private biotechnology companies that were determined to be irrecoverable. During the year ended December 31, 2009, there were no write-downs of long term investments. During the year ended December 31, 2008, the Company recorded a $14.8 million other-than-temporary impairment on certain investments and realized an $8.8 million loss on the disposition of a short-term available-for-sale investment.
7. INVENTORIES
| December 31, 2010 |
December 31, 2009 |
|||||||
| Raw materials |
$ | 9,760 | $ | 10,352 | ||||
| Work in process |
11,770 | 12,283 | ||||||
| Finished goods |
13,101 | 12,906 | ||||||
| $ | 34,631 | $ | 35,541 | |||||
As at December 31, 2010, the inventory allowance was $3.4 million (December 31, 2009 - $3.0 million). For the year ended December 31, 2010, inventory write-downs and obsolescence charges were $3.0 million (December 31, 2009 - $1.8 million, December 31, 2008 - $1.0 million).
F-20
Table of Contents
8. ASSETS HELD FOR SALE
During the year ended December 31, 2008 and in connection with the Company’s capacity rationalization and consolidation plans, the Company reclassified three of its properties as held for sale. Based on the Company’s annual asset impairment assessment, the Company recorded impairment charges of $1.5 million, $3.1 million and $1.3 million for the years ended December 31, 2010, December 31, 2009 and December 31, 2008, respectively, to adjust the carrying values of the properties to the lower of cost and fair value less estimated selling costs. In early 2010, one of these properties was sold for net proceeds of $0.7 million with no resulting gain or loss. As at December 31, 2010, the two remaining properties failed to meet the held-for-sale classification criteria defined under ASC No. 360-10 – Property, Plant and Equipment. As a result, the $3.1 million carrying value of the two remaining properties was reclassified back into Property, Plant and Equipment. The carrying values of the remaining properties were measured at the lower of (a) the carrying values of the properties prior to their classification as held for sale, adjusted for depreciation that would have been recognized if the properties had remained classified as held and used; and (b) the fair values of the properties at the date of the subsequent decision to not sell the properties.
9. PROPERTY, PLANT AND EQUIPMENT
| December 31, 2010 |
Cost | Accumulated Depreciation |
Net Book Value |
|||||||||
| Land |
$ | 7,345 | $ | — | $ | 7,345 | ||||||
| Buildings |
14,614 | 2,394 | 12,220 | |||||||||
| Leasehold improvements |
17,296 | 7,259 | 10,037 | |||||||||
| Manufacturing equipment |
25,949 | 11,367 | 14,582 | |||||||||
| Research equipment |
631 | 429 | 202 | |||||||||
| Office furniture and equipment |
4,252 | 3,812 | 440 | |||||||||
| Computer equipment |
8,445 | 7,010 | 1,435 | |||||||||
| $ | 78,532 | $ | 32,271 | $ | 46,261 | |||||||
| December 31, 2009 |
Cost | Accumulated Depreciation |
Net Book Value |
|||||||||
| Land |
$ | 4,796 | $ | — | $ | 4,796 | ||||||
| Buildings |
14,736 | 2,248 | 12,488 | |||||||||
| Leasehold improvements |
19,709 | 6,292 | 13,417 | |||||||||
| Manufacturing equipment |
20,396 | 9,175 | 11,221 | |||||||||
| Research equipment |
8,197 | 5,235 | 2,962 | |||||||||
| Office furniture and equipment |
4,398 | 3,577 | 821 | |||||||||
| Computer equipment |
8,183 | 7,009 | 1,174 | |||||||||
| $ | 80,415 | $ | 33,536 | $ | 46,879 | |||||||
Depreciation expense, including depreciation expense allocated to cost of goods sold was $6.8 million for the year ended December 31, 2010 (December 31, 2009 - $7.1 million, December 31, 2008 - $7.9 million).
During the year ended December 31, 2010, the Company recorded impairment write-downs of $4.8 million (December 31, 2009 – nil, December 31, 2008 – nil) to its Medical Products segment relating to: (i) certain lab and manufacturing equipment held at its Haemacure operations due to the Company’s decision to indefinitely suspend further research and development activities for that operation; and (ii) certain lab equipment related to its head office lab facilities due to the restructuring changes that the Company initiated in response to its recent liquidity constraints and recapitalization plans.
10. INTANGIBLE ASSETS AND GOODWILL
(a) Intangible Assets
| December 31, 2010 |
Cost | Accumulated Amortization |
Net Book Value |
|||||||||
| Acquired technologies |
$ | 131,947 | $ | 78,731 | $ | 53,216 | ||||||
| Customer relationships |
110,176 | 50,886 | 59,290 | |||||||||
| In-licensed technologies |
53,100 | 34,426 | 18,674 | |||||||||
| Trade names and other |
14,288 | 7,495 | 6,793 | |||||||||
| $ | 309,511 | $ | 171,538 | $ | 137,973 | |||||||
F-21
Table of Contents
| December 31, 2009 |
Cost | Accumulated Amortization |
Net Book Value |
|||||||||
| Acquired technologies |
$ | 135,060 | $ | 64,935 | $ | 70,125 | ||||||
| Customer relationships |
110,778 | 41,530 | 69,248 | |||||||||
| In-licensed technologies |
53,882 | 28,710 | 25,172 | |||||||||
| Trade names and other |
14,511 | 6,037 | 8,474 | |||||||||
| $ | 314,231 | $ | 141,212 | $ | 173,019 | |||||||
Amortization expense for the year ended December 31, 2010 was $30.8 million (December 31, 2009 - $30.0 million, December 31, 2008 -$30.2 million).
During the year ended December 31, 2010, the Company recorded an impairment write-downs of $2.8million (December 31, 2009 – nil) to its Medical Products segment relating to: (i) a write-down on the intellectual property acquired from Haemacure in connection with the Company’s decision to indefinitely suspend further research and development activities for that operation; and (ii) a write-down on the intellectual property associated with the Option IVC Filter in connection with the termination of the Company’s 2008 License, Supply, Marketing and Distribution Agreement with Rex.
The following table summarizes the estimated amortization expense for each of the five succeeding fiscal years for intangible assets held as of December 31, 2010:
| 2011 |
$ | 31,167 | ||
| 2012 |
28,930 | |||
| 2013 |
28,930 | |||
| 2014 |
27,440 | |||
| 2015 |
20,713 |
(b) Goodwill
During the year ended December 31, 2008, the Company tested goodwill for impairment and determined that goodwill was impaired due to a significant and sustained decline in the Company’s public share price and market capitalization, reorganization activities, uncertainties about future cash flows and implied risk premiums used by market participants. As a result, the Company recorded a $649.7 million write-down of goodwill, which consisted of a $626.2 million write-down to the Medical Products segment and a $23.5 million write-down to the Pharmaceutical Technologies segment. The carrying value of goodwill is therefore $nil for the years ended December 31, 2010 and 2009.
11. DISPOSAL OF LAGUNA HILLS MANUFACTURING FACILITY
During the year ended December 31, 2010, the Company sold its Laguna Hills manufacturing operations under its Medical Products operating segment. The sale included certain assets and liabilities for total net proceeds of $2.8 million. The Company recorded a $2.0 million gain from the sale of the net assets under consideration.
F-22
Table of Contents
12. ACCOUNTS PAYABLE AND ACCRUED LIABILITIES
| December 31, 2010 |
December 31, 2009 |
|||||||
| Trade accounts payable |
$ | 6,016 | $ | 5,110 | ||||
| Accrued license and royalty fees |
3,350 | 13,738 | ||||||
| Employee-related accruals |
14,470 | 14,054 | ||||||
| Accrued professional fees |
5,898 | 4,141 | ||||||
| Accrued contract research |
559 | 704 | ||||||
| Other accrued liabilities |
6,554 | 8,577 | ||||||
| $ | 36,847 | $ | 46,324 | |||||
13. DEFERRED LEASEHOLD INDUCEMENT
As at December 31, 2010, the Company had $4.8 million of deferred leasehold inducements related to its Canadian and Puerto Rican subsidiaries, Angiotech Pharmaceuticals Inc. and Surgical Specialties Puerto Rico, Inc, respectively. The increase in the deferred leasehold inducements for the year ended December 31, 2010 is primarily due to the receipt of a $2.1 million government grant by Surgical Specialties Puerto Rico. The government grant was received for the construction of a clean room and completion of certain tenant improvements. Given that title and ownership of the clean room and tenant improvements ultimately resides with the landlord of the leased property, the grant has been recorded as a deferred leasehold inducement. The deferred leasehold inducements are being amortized over the relevant lease terms with a corresponding reduction to rent expense.
14. INCOME TAXES
(a) The components of the provision for (recovery of) income taxes from continuing operations are as follows:
| Year ended December 31, 2010 |
Year ended December 31, 2009 |
Year ended December 31, 2008 |
||||||||||
| Current income tax expense (recovery): |
||||||||||||
| Canada |
$ | 376 | $ | 523 | $ | (4,281 | ) | |||||
| Foreign |
4,714 | 4,505 | 5,138 | |||||||||
| 5,090 | 5,028 | 857 | ||||||||||
| Deferred income tax expense (recovery): |
||||||||||||
| Canada |
(533 | ) | 63 | (1,897 | ) | |||||||
| Foreign |
(4,601 | ) | 946 | (11,852 | ) | |||||||
| (5,134 | ) | 1,009 | (13,749 | ) | ||||||||
| Income tax expense (recovery) |
$ | (44 | ) | $ | 6,037 | $ | (12,892 | ) | ||||
(b) The provision for income taxes is based on net income (loss) from continuing operations before income taxes as follows:
| Year ended December 31, 2010 |
Year ended December 31, 2009 |
Year ended December 31, 2008 |
||||||||||
| Canada |
$ | (24,740 | ) | $ | (27,984 | ) | $ | (20,510 | ) | |||
| Foreign |
(42,115 | ) | 11,153 | (733,558 | ) | |||||||
| $ | (66,855 | ) | $ | (16,831 | ) | $ | (754,068 | ) | ||||
(c) The reconciliation between the combined Canadian federal and provincial income taxes at the statutory rate and the provision for income taxes from continuing operations, is as follows:
| Year
ended December 31, 2010 |
Year
ended December 31, 2009 |
Year
ended December 31, 2008 |
||||||||||
| Net income (loss) before income taxes |
$ | (66,855 | ) | $ | (16,831 | ) | $ | (754,068 | ) | |||
F-23
Table of Contents
| Statutory tax rate |
28.5% | 30.0% | 31.0% | |||||||||
| Expected income tax expense (recovery) |
(19,053 | ) | (5,049 | ) | (233,761 | ) | ||||||
| Tax rate differential on deferred tax assets and liabilities |
5,939 | (426 | ) | 2,143 | ||||||||
| Foreign tax rate differences |
(2,278 | ) | (5,464 | ) | 4,200 | |||||||
| Foreign exchange losses |
24 | (640 | ) | (14,049 | ) | |||||||
| Change in valuation allowance |
11,191 | 3,091 | 64,001 | |||||||||
| Reassessment of prior years’ taxes [see paragraph (g)] |
— | 1,057 | (3,845 | ) | ||||||||
| Non-deductible portion of the capital (gains) losses |
— | — | (5,484 | ) | ||||||||
| Capital losses |
(2,311 | ) | — | (45,045 | ) | |||||||
| Non-deductible stock based compensation |
378 | 323 | 583 | |||||||||
| Goodwill impairment |
— | — | 198,369 | |||||||||
| Uncertain tax positions |
721 | 740 | 6,082 | |||||||||
| Taxable dividends from subsidiaries |
5,070 | 1,719 | 4,208 | |||||||||
| Cost capitalized to investment in subsidiaries |
(1,193 | ) | 1,124 | — | ||||||||
| Release of prepaid taxes |
84 | 6,211 | 422 | |||||||||
| Permanent differences and other |
1,384 | 3,351 | 9,284 | |||||||||
| Income tax expense (recovery) |
$ | (44 | ) | $ | 6,037 | $ | (12,892 | ) | ||||
(d) Deferred income tax assets and liabilities result from the temporary differences between the amount of assets and liabilities recognized for financial statement and income tax purposes. The significant components of the net deferred income tax assets and liabilities are as follows:
| December 31, 2010 |
December 31, 2009 |
|||||||
| Deferred income tax assets |
||||||||
| Property, plant and equipment |
$ | 1,822 | $ | 1,538 | ||||
| Intangible assets |
7,240 | 9,478 | ||||||
| Other assets |
5,828 | 5,532 | ||||||
| Accrued liabilities |
5,559 | 5,742 | ||||||
| Unrealized foreign exchange losses |
4,126 | 3,239 | ||||||
| Operating loss carry forwards |
28,314 | 31,037 | ||||||
| Capital loss carry forwards |
56,491 | 51,943 | ||||||
| Tax credits |
22,989 | 18,987 | ||||||
| Total gross deferred income tax assets |
132,369 | 127,496 | ||||||
| Less: valuation allowance |
(106,594 | ) | (95,159 | ) | ||||
| Total deferred income tax assets |
$ | 25,775 | $ | 32,337 | ||||
| Deferred income tax liabilities |
||||||||
| Property, plant and equipment |
$ | 444 | $ | 1,503 | ||||
| Identifiable intangible assets |
50,921 | 62,093 | ||||||
| Other liabilities |
95 | 98 | ||||||
| Undistributed earnings of foreign subsidiaries [see paragraph (f)] |
1,950 | 1,138 | ||||||
| Total deferred tax liabilities |
53,410 | 64,832 | ||||||
| Net deferred income tax assets (liabilities) |
$ | (27,635 | ) | $ | (32,495 | ) | ||
The realization of deferred income tax assets is dependent upon the generation of sufficient taxable income during future periods in which the temporary differences are expected to reverse. The valuation allowance is reviewed on a quarterly basis and if the assessment of the “more likely than not” criteria changes, the valuation allowance is adjusted accordingly. The valuation allowance continues to be applied against certain deferred income tax assets where the Company has assessed that the realization of such assets does not meet the “more likely than not” criteria. During 2010, the Company increased the valuation allowance by $11.4 million related primarily to the Company’s Canadian operations.
(e) The Company has unclaimed U.S. federal and state research and development investment tax credits of approximately $4.4 million (December 31, 2009 - $4.4 million) available to reduce future U.S. income taxes otherwise payable. The Company also has Canadian federal and provincial investment tax credits of approximately $15.5 million (December 31, 2009 - $14.1 million) available.
F-24
Table of Contents
The Company has a net operating loss carry forward balance of approximately $214.8 million (December 31, 2009 - $206.1 million) available to offset future taxable income in Canada ($73.4 million, December 31, 2009 - $35.9 million), the U.S. ($30.0 million, December 31, 2009 - $53.3 million), Switzerland ($111.4 million, December 31, 2009 - $116.9 million) and other European countries (nil, December 31, 2009 - nil). A portion of the losses in the U.S. are subjected to limitation but the Company does not expect the limitation will impair the use of any of the losses.
The Company has foreign tax credits in the U.S. of approximately $2.1 million (December 31, 2009 - $2.1 million) available to use against foreign-source income.
The Company has a capital loss carry forward balance of approximately $445.5 million (December 31, 2009 - $406.6 million) available to offset future capital gains in the U.S. ($3.1 million, December 31, 2009 - $4.4 million) and Canada ($442.4 million, December 31, 2009 - $402.2 million). The capital losses can be carried forward indefinitely for Canada and five years for the U.S.
The investment tax credits and loss carry forwards expire as follows:
| Federal tax credits |
Provincial/state tax credits |
Operating Loss Carry forwards |
||||||||||
| 2010 |
— | — | — | |||||||||
| 2011 |
— | — | 15,370 | |||||||||
| 2012 |
926 | — | 16,100 | |||||||||
| 2013 |
2,093 | 72 | 6,618 | |||||||||
| 2014 |
1,412 | — | 2,938 | |||||||||
| 2015 |
1,519 | — | 16,098 | |||||||||
| 2016 |
— | — | 46,502 | |||||||||
| 2017 |
— | — | 8,095 | |||||||||
| 2018 |
436 | 27 | — | |||||||||
| 2019 |
573 | — | — | |||||||||
| 2020 |
464 | 19 | — | |||||||||
| 2021 |
278 | — | — | |||||||||
| 2022 |
189 | — | — | |||||||||
| 2023 |
232 | — | — | |||||||||
| 2024 |
457 | — | — | |||||||||
| 2025 |
276 | — | — | |||||||||
| 2026 |
2,405 | — | 4,347 | |||||||||
| 2027 (or later) |
6,284 | 2,252 | 98,765 | |||||||||
| $ | 17,544 | $ | 2,370 | $ | 214,833 | |||||||
(f) The Company has not recognized a deferred income tax liability for the undistributed earnings of certain foreign subsidiaries which are essentially investments in those foreign subsidiaries and are permanent in duration. It is not practical to determine the amount of these liabilities.
(g) In September 2006, the Quebec National Assembly enacted legislation (Bill 15) that retroactively changed certain tax laws that subject the Company to additional taxes for the 2004 and 2005 taxation years. As a result of the Quebec income tax assessment, a total of $14.4 million had been accrued as of December 31, 2007 of which $1.1 million was accrued in 2007. During 2008, the Company settled with the Canada Revenue Agency and Revenue Quebec and recognized a recovery of $3.8 million.
15. OTHER TAX LIABILITY
The Company accounts for income taxes in accordance with ASC No. 740, Income Taxes. As of December 31, 2010 and December 31, 2009, the total amount of the Company’s unrecognized tax benefits were $12.5 million and $11.4 million, respectively. If recognized in future periods, the unrecognized tax benefits would affect our effective tax rate.
F-25
Table of Contents
The reserve for uncertain tax positions includes accrued interest and penalties of $0.9 million (December 31, 2009 - $0.7 million). In accordance with the Company’s accounting policies, accrued interest and penalties, if incurred, relating to unrecognized tax benefits are recognized as a component of income tax expense.
The taxation years 2006 - 2009 remain open to examination by the Canada Revenue Agency, taxation years 2003 – 2009 remain open to examination by the Internal Revenue Service, and various years remain open in the foreign jurisdictions. The Company files income tax returns in Canada, the U.S. and various foreign jurisdictions.
A reconciliation of the change in the reserves for an uncertain tax positions from January 1, 2009 – December 31, 2010 is as follows:
| 2010 | 2009 | |||||||
| Balance as of January 1 |
$ | 11,367 | $ | 10,416 | ||||
| Tax positions related to current year: |
||||||||
| Additions |
1,600 | 1,049 | ||||||
| Reductions |
— | — | ||||||
| Tax positions related to prior years: |
||||||||
| Additions |
371 | 294 | ||||||
| Reductions |
(700 | ) | (300 | ) | ||||
| Settlements |
— | (92 | ) | |||||
| Lapses in statutes of limitations |
— | — | ||||||
| Balance as of December 31 |
$ | 12,638 | $ | 11,367 | ||||
16. DEBT
The Company has the following debt issued and outstanding:
| December 31, 2010 | December 31, 2009 | |||||||
| 7.75% Senior Subordinated Notes |
$ | 250,000 | $ | 250,000 | ||||
| Senior Floating Rate Notes |
325,000 | 325,000 | ||||||
| Revolving credit facility |
10,000 | — | ||||||
| $ | 585,000 | $ | 575,000 | |||||
(a) Senior Subordinated Notes
On March 23, 2006, the Company issued the Subordinated Notes due on April 1, 2014 in the aggregate principal amount of $250 million. The Subordinated Notes are unsecured obligations and bear interest at an annual rate of 7.75% payable semi-annually in arrears on April 1 and October 1 of each year through to maturity. The Subordinated Notes and related guarantees provided by the Company and certain of its subsidiaries are subordinated in right of payment to the Existing Floating Rate Notes described below and the Company’s existing and future senior indebtedness. Prior to the Creditor Protection Proceedings described in note 1, the Company had the option to redeem all or a part of the Subordinated Notes at the redemption prices stipulated below (expressed as percentages of principal amount) plus accrued and unpaid interest, if any, on the notes redeemed, to the applicable redemption date if redeemed during the twelve-month period beginning on the dates indicated below:
| Year |
Percentage (%) | |||
| April 1, 2010 |
103.875 | |||
| April 1, 2011 |
101.938 | |||
| April 1, 2012 and thereafter |
100.000 | |||
In addition, under certain change of control situations, the Company would have been required to make an offer to purchase the then-outstanding Subordinated Notes at a price equal to 101% of their stated principal amount, plus accrued and unpaid interest, if any, to the applicable repurchase date.
On October 1, 2010 the Company did not make its $9.7 million interest payment due to holders of the Subordinated Notes and on January 28, 2011 the Angiotech Entities commenced the Creditor Protection Proceedings to effectuate an in-court
F-26
Table of Contents
Recapitalization Transaction as contemplated under the RSA (see note 1 and 26 for more information). The Recapitalization Transaction would eliminate the Company’s $250 million aggregate principal amount outstanding of Subordinated Notes and related interest obligations in exchange for new common shares in the Company thereby providing significant improvements to its credit ratios, liquidity and financial flexibility. For more information on key elements of the Recapitalization Transaction, refer to note 1. For the year ended December 31, 2010, the Company incurred approximately $9.3 million of professional fees and expenses relating to the Recapitalization Transaction.
(b) Existing Floating Rate Notes
The Existing Floating Rate Notes were issued on December 11, 2006 and are due on December 1, 2013. The Existing Floating Rate Notes bear interest at an annual rate of LIBOR plus 3.75%, which is reset quarterly. Interest is payable quarterly in arrears on March 1, June 1, September 1, and December 1 of each year through to maturity. The Existing Floating Rate Notes are unsecured senior obligations, which are guaranteed by certain of the Company’s subsidiaries, and rank equally in right of payment to all of the Company’s existing and future senior indebtedness. The guarantees of its guarantor subsidiaries are unconditional, joint and several. The Company has provided condensed consolidating guarantor financial information as at December 31, 2010 and 2009 and for the years ended December 31, 2010, 2009 and 2008 (note 25). As at December 31, 2010, the interest rate on the Existing Floating Rate Notes was 4.1% (December 31, 2009 – 4.0%).
The Company has the option to redeem all or a part of the Existing Floating Rate Notes at the redemption prices (expressed as percentages of principal amount) set forth below plus accrued and unpaid interest, if any, on the notes redeemed, to the applicable redemption date, if redeemed during the twelve-month period beginning on the dates indicated below:
| Year |
Percentage (%) | |||
| December 1, 2010 |
101 | |||
| December 1, 2011 |
100 | |||
In addition, under certain change of control situations, the Company is required to make an offer to purchase the then-outstanding Senior Floating Rate Notes at a price equal to 101% of their stated principal amount, plus accrued and unpaid interest to the applicable repurchase date, if any.
On October 29, 2010 the Company entered into the FRN Support Agreement with the Consenting FRN Holders to exchange their Existing Floating Rate Notes for New Floating Rate Notes pursuant to which the Company commenced the FRN Exchange Offer on February 10, 2011. The FRN Exchange Offer is open to all qualifying holders of Existing Floating Rate Notes. The New Floating Rate Notes will be issued on substantially the same terms and conditions as the Existing Floating Rates Notes, except that, among other things, the indenture governing the New Floating Rate Notes will differ from the FRN Indenture as follows:
| • | Changes to covenants pertaining to the incurrence of additional indebtedness, asset sales and the definitions of asset sales, change of control and permitted liens, including a reduction in the allowance from $100 million to $50 million for the Company to obtain new senior secured indebtedness having seniority to the New Floating Rate Notes; |
| • | The New Floating Rate Notes would accrue interest at LIBOR plus 3.75%, subject to a LIBOR floor of 1.25%; and |
| • | The provision of additional security for holders of the Existing Floating Rate Notes by providing a second lien over the property, assets and undertakings of the Company and certain of its subsidiaries, that secure the Existing Credit Facility and successive DIP Facility, subject to customary carve-outs and certain permitted liens. |
The FRN Exchange Offer is expected to close concurrently with the implementation of the CCAA Plan.
In the event that the Company is unable to successfully complete the Recapitalization Transaction and the FRN Exchange Offer, the Subordinated Noteholders or holders of our Existing Floating Rate Notes may elect to exercise rights under the respective indentures allowing them to accelerate amounts owing thereunder. An event of default under the FRN Indenture, arising from the failure to make the interest payment on the Subordinated Notes within the applicable grace period, would permit the holders of 25% or more of the aggregate outstanding principal amount of the Existing Floating Rate Notes to demand immediate repayment of our debt obligations owing under the Subordinated Notes. In this case, the Company’s current cash and credit capacity would not be sufficient to service principal debt and interest obligations. In addition, restrictions on the Company’s ability to borrow and the possible accelerated demand of repayment of existing or future obligations by any of its creditors may jeopardize its working capital position and ability to sustain operations.
F-27
Table of Contents
(c) Covenants
The terms of the SSN Indenture and the FRN Indenture include various covenants that impose restrictions on the operation of the Company’s business and the business of its subsidiaries, including the incurrence of certain liens and other indebtedness.
In addition, the Existing Credit Facility and DIP Facility include a number of customary financial covenants, including a requirement to maintain certain levels of Adjusted EBITDA and interest coverage ratios, as well as covenants that limit the Company’s ability to, among other things, incur indebtedness, create liens, merge or consolidate, sell or dispose of assets, change the nature of its business, make distributions or make advances, loans or investments.
Material covenants under the SSN Indenture and FRN Indenture specify maximum or permitted amounts for certain types of capital transactions. In addition, these indentures contain certain restrictions on asset sales, the use of proceeds and the payment of dividends by the Company. Under the terms of the indentures, all outstanding principal amounts and interest accrued and unpaid become due and payable upon the occurrence of a defined event of default.
Upon the closing of the FRN Exchange Offer, which will occur concurrently with the effective date of the CCAA Plan, the FRN Indenture will be amended to provide for the elimination of most of the covenants and events of default therein. However, the indenture governing the New Floating Rate Notes will contain substantially identical covenants and events of default as the FRN Indenture in its present form, except with respect to certain modifications to the covenants relating to the incurrence of additional indebtedness, liens and asset sales.
(d) Interest
The Existing Floating Rate Notes bear interest at an annual rate of LIBOR (London Interbank Offered Rate), which is reset quarterly, plus 3.75%. The effective interest rate on these notes for the year ended December 31, 2010 was 4.2% (December 31, 2009 – 4.0%) and the rate in effect on December 31, 2010 was 4.1% (December 31, 2009 – 4.01%). Interest is payable quarterly in arrears on March 1, June 1, September 1, and December 1 of each year through to maturity. On March 1, 2011, the Company paid $3.3 million in interest to the holders of its Existing Floating Rate Notes. As discussed under note 16 (b) above, the New Floating Rate Notes which will be issued pursuant to the FRN Exchange Offer, will accrue interest at LIBOR plus 3.75%, subject to a LIBOR floor of 1.25%. For the year ended December 31, 2010, the Company incurred $13.5 million of interest expense on the Existing Floating Rate Notes.
The Subordinated Notes bear interest at 7.75% annually payable in arrears on April 1 and October 1 of each year through to maturity. For the year ended December 31, 2010, the Company incurred $19.4 million of interest expense on the Subordinated Notes.
e) Deferred Financing Costs
Deferred financing costs are capitalized and amortized on a straight-line basis, which approximates the effective interest rate method, to interest expense over the life of the debt instruments.
| December 31, 2010 |
Cost | Accumulated amortization |
Net book value |
|||||||||
| Debt issuance costs relating to: |
||||||||||||
| Senior Floating Rate Notes |
$ | 8,000 | $ | 4,654 | $ | 3,346 | ||||||
| 7.75% Senior Subordinated Notes |
8,718 | 7,765 | 953 | |||||||||
| Revolving Credit Facility (Note 16 (f)) |
3,213 | 1,901 | 1,312 | |||||||||
| $ | 19,931 | $ | 14,320 | $ | 5,611 | |||||||
| December 31, 2009 |
Cost | Accumulated amortization |
Net book value |
|||||||||
| Debt issuance costs relating to: |
||||||||||||
| Senior Floating Rate Notes |
$ | 8,000 | $ | 3,506 | $ | 4,494 | ||||||
| 7.75% Senior Subordinated Notes |
8,718 | 4,087 | 4,631 | |||||||||
| Revolving Credit Facility (Note 16(f)) |
2,563 | 551 | 2,012 | |||||||||
| Other |
272 | — | 272 | |||||||||
| $ | 19,553 | $ | 8,144 | $ | 11,409 | |||||||
F-28
Table of Contents
During the year ended December 31, 2010, the Company recorded $6.2 million of non-cash interest expense related to the amortization of the deferred financing costs. This includes a $3.2 million adjustment resulting from a change in the estimated useful lives of the deferred financing costs related to the Subordinated Notes and Existing Credit Facility as a result of the Creditor Protection Proceedings and the proposed timeline for the execution of the Recapitalization Transaction. In addition, the Company recorded a $0.3 million impairment write-down of deferred financing costs related to its shelf registration statement which was filed in July 2009.
During the year ended December 31, 2008, the Company recorded a $16.5 million write-down of deferred financing costs, in connection with the Company’s 2008 termination of its tender offer for its outstanding Existing Floating Rate Notes and Subordinated Notes.
f) Existing Credit Facility and DIP Facility
As at December 31, 2010, the Company had an Existing Credit Facility with Wells Fargo with an original maturity date of February 27, 2013, for up to $35.0 million which includes an additional $10.0 million of temporary supplemental borrowing base that was added through the Fourth Amendment (see details below) to the Existing Credit Facility. The purpose of this facility was to provide additional liquidity for working capital and general corporate purposes.
The amount the Company is able to borrow under the Existing Credit Facility at any given time is limited by a borrowing base formula, derived from the value of certain finished good inventory, accounts receivable, certain real property, securities and intellectual property owned by the Company and certain of its subsidiaries, and therefore fluctuates from month to month. As of December 31, 2010, the amount of financing available under the Existing Credit Facility was approximately $14.8 million, which is net of $2.8 million in letters of credit issued under the Existing Credit Facility. As of December 31, 2010, $10.0 million of advances were outstanding under the Existing Credit Facility.
Borrowings outstanding under the Existing Credit Facility bear interest at an annual rate ranging from LIBOR, for a period selected by the Company of 1, 2 or 3 months, plus 3.25% to LIBOR plus 3.75%, with a minimum LIBOR of 2.25%. As the minimum LIBOR under the Existing Credit Facility is 2.25% and LIBOR for periods of 1, 2 and 3 months was, in each case, less than 0.50% on December 31, 2010, a 0.50% increase or decrease in the LIBOR rate as of December 31, 2010 would have no impact on interest payable under the Existing Credit Facility.
The utilization of the Existing Credit Facility is subject to, among other terms and conditions, certain restrictive covenants pertaining to operations and the maintenance of minimum levels of adjusted earnings before interest, taxes, depreciation and amortization (“Adjusted EBITDA”) and fixed charge coverage ratios. Under the terms of the Existing Credit Facility, a breach of any of these covenants may result in the Company’s inability to draw on the facility or the immediate repayment of any then outstanding principal and interest. In April 2010, the Company completed the Third Amendment to the Existing Credit Facility, which included, among other items, amendments to financial covenants pertaining to minimum EBITDA levels, fixed charge coverage ratios and the definition of Adjusted EBITDA. This amendment enabled the Company to continue to access to funds per the terms of the Existing Credit Facility, and was primarily intended to reflect the continued decline and uncertainty of sales of TAXUS by BSC and the related potential impact on its Adjusted EBITDA.
Borrowings under the Existing Credit Facility were secured by certain assets held by the Company’s North American operations. In addition, the Canadian parent company, Angiotech Pharmaceuticals Inc. and certain of its subsidiaries; were named as the guarantor. Under the General Continuing Guaranty agreement, the guarantor irrevocably and unconditionally guaranteed (a) the due and punctual payment of all borrowings, including interest, when due and payable and (b) the fulfillment of and compliance with all terms, conditions and covenants under the Existing Credit Facility agreement and all other related loan documents.
In connection with the Company’s failure to make the $9.7 million interest payment owing to the Subordinated Noteholders on October 1, 2010, the Company entered into the Forbearance Agreement on September 30, 2010 to preserve the Existing Credit Facility. On November 4, 2010, the Company entered into the Amended and Restated Forbearance Agreement pursuant to which Wells Fargo agreed, subject to certain terms and conditions, to continue to forbear from exercising its rights and remedies under the Existing Credit Facility in order to allow the Company to implement the Recapitalization Transaction. Under the Amended and Restated Forbearance Agreement, Wells Fargo, among other things, agreed to not immediately exercise any rights or remedies it has with respect to:
| (a) | the Company’s failure to make its $9.7 million interest payment due to the Subordinated Noteholders on October 1, 2010; |
F-29
Table of Contents
| (b) | the litigation and the arbitration relating to the dispute with QSR; or |
| (c) | any failure to deliver audited financial statements with an unqualified audit opinion with respect to the going concern assumption for the fiscal year ending December 31, 2010. |
The Company incurred $0.1 million to complete both the Forbearance Agreement and Amended and Restated Forbearance Agreement. The Amended and Restated Forbearance Agreement was scheduled to terminate on the earliest to occur of:
| (i) | the occurrence of another event constituting an Event of Default under the Existing Credit Facility; |
| (ii) | the occurrence of a material adverse change as defined under the Existing Credit Facility, subject to exclusions for changes relating to the Recapitalization Transaction; |
| (iii) | failure to attain certain intermediate milestones with respect to the Recapitalization Transaction; |
| (iv) | termination of the RSA or the FRN Support Agreement; |
| (v) | consummation of the Recapitalization Transaction on terms and conditions not reasonably satisfactory to Wells Fargo under the Existing Credit Facility; |
| (vi) | failure to remain within the applicable grace period with respect to the non-payment of the interest payment due to the Subordinated Noteholders on October 1, 2010; and |
| (vii) | April 30, 2011. |
While the Amended and Restated Forbearance Agreement has not been terminated, Wells Fargo’s obligations thereunder, including its agreement not to exercise certain remedies, ceased to be of effect upon our commencement of the CCAA Proceedings. Wells Fargo’s rights and remedies under the Existing Credit Facility, however, were nonetheless stayed in connection with the Creditor Protection Proceedings.
On November 4, 2010, the Company entered into the Fourth Amendment to the Existing Credit Facility to, among other changes:
| • | Amend the existing financial covenants pertaining to minimum EBITDA levels and fixed charge coverage ratios; |
| • | Increase the borrowing base available to secure loans by including in such borrowing base the value of certain real property, securities and intellectual property owned by the Company and its subsidiaries, in addition to accounts receivable and inventory, in each case subject to reserves, eligibility standards and advance rate formulas. As a result, amount of financing available to the Company under the Existing Credit Facility as at November 4, 2010 increased to approximately $25 million, which excluded $2.8 million outstanding secured letters of credit; |
| • | Amend the specified use of proceeds provisions to allow for certain secured and unsecured intercompany loans by US entities to Canadian entities; and |
| • | Permit the sale of certain assets of the Company. |
In accordance with the terms of our Creditor Protection Proceedings, on February 7, 2011, the Company and certain of its subsidiaries (the “Borrowers”) entered into a definitive agreement with Wells Fargo to secure a DIP Facility. On February 25, 2011, following the entry of the February 22, 2011 recognition order the loans under the DIP Facility were made available for borrowing by the Company. The DIP Facility will provide the Company with up to $28.0 million in aggregate principal amount (subject to a borrowing base and certain reserves) of revolving loans to fund working capital requirements and implementation costs during the Recapitalization Transaction. The DIP Facility effectively supersedes and terminates use of the Existing Credit Facility beyond February 7, 2011. Under the terms of the DIP Facility, the Company is required to apply its domestic post-petition cash collections of the Angiotech Entities to the pre-petition Existing Credit Facility until all obligations outstanding under the Existing Credit Facility are paid in full. In order to satisfy this requirement, the Company is required to implement control agreements over bank accounts comprising a substantial portion of its cash balance. These control agreements require the Company to periodically sweep receipts from certain of our deposit accounts into Wells Fargo’s account until such time as the pre-petition obligations owing under the Existing Credit Facility have been fully repaid. The DIP Facility was issued on substantially the same terms as the Existing Credit Facility, subject to certain changes to reflect the Company’s filing for protection under the Creditor Protection Proceedings and will be used to fund daily cash needs prior to the repayment in full of the pre-petition Existing Credit Facility.
Borrowings under the DIP Facility are also subject to a borrowing base formula based on certain balances of: eligible inventory, accounts receivable, real property, securities and intellectual property, net of applicable reserves. In accordance with the agreed upon budget provided to Wells Fargo, the proceeds of the DIP Facility may only be used to pay for working capital requirements, capital expenditures and transaction fees and expenses incurred in connection with the
F-30
Table of Contents
Recapitalization Transaction and the establishment of the DIP Facility. The proceeds may not be used to pay any debt outstanding prior to the date of the Initial Order (i.e. January 28, 2011), including those arising under the Existing Credit Facility.
Borrowings under the DIP Facility are secured by certain assets held by certain of the Company’s subsidiaries (the “DIP Guarantors”). Under the terms of the DIP Facility, the Company’s DIP Guarantors irrevocably and unconditionally jointly and severally guarantee: (a) the punctual payment; whether at stated maturity, by acceleration or otherwise; of all borrowings; including principal, interest, expenses or otherwise; when due and payable; and (b) the fulfillment of and compliance with all terms, conditions and covenants under the DIP Facility and all other related loan documents. The Company’s DIP Guarantors also agree to pay any and all expenses that Wells Fargo may incur in enforcing its rights under the guarantee.
The principal amount of the loans outstanding under the DIP Facility, plus interest accrued and unpaid thereon will be due and payable in full at maturity, which is defined as the earliest of:
(a) June 30, 2011;
| (b) | the date of implementation of the CCAA Plan that has been sanctioned by an order of the Canadian Court and recognized by a recognition order of the US Bankruptcy Court; |
| (c) | the date immediately preceding the date on which any distributions under any the CCAA Plan commence; |
| (d) | the date of a sale of substantially all of the assets of Borrowers and the DIP Guarantors (which may take the form of an asset sale, stock sale or otherwise as approved by Wells Fargo); |
| (e) | the date that is 28 days after the Filing Date, if the Recognition Order has not been entered by the US Bankruptcy Court; and |
| (f) | such earlier date on which all advances and other obligations for the payment of money shall become due and payable in accordance with the terms of the DIP Facility and the other Loan Documents (as defined n the DIP Facility). |
Borrowings under the DIP Facility bear interest at a rate equal to, at the Company’s option, either (a) the Base Rate (as defined in the DIP Facility) plus either 3.25% or 3.50%, depending on the availability we have under the DIP Facility on the date of determination or (b) LIBOR plus either 3.50% or 3.75%, depending on the availability the Company has under the DIP Facility on the date of determination. If the Company defaults on its obligations under the DIP Facility, Wells Fargo may choose to implement a default rate of an additional 2% above the already applicable interest rate. In addition to interest, the Company is required to pay an unused line fee of 0.5% per annum in respect to the unutilized commitments. In addition to certain customary affirmative and negative covenants, the DIP Facility requires the Company to comply with a specified minimum Adjusted EBIDTA and a minimum fixed charge coverage ratio. In addition to certain customary events of default, the DIP Facility includes defined events of default related to the progress of the Creditor Protection Proceedings. While the Company is currently in compliance with the Adjusted EBITDA and fixed charge covenants specified under the Existing Credit Facility and DIP Facility, there are no guarantees that the Company will be able to comply with these covenants and related conditions throughout the remainder of 2011 and beyond. A breach of these covenants, failure to obtain waivers to cure any further breaches of the covenants and the restrictions set forth under the DIP Facility, failure to complete the Recapitalization Transaction or the FRN Exchange Offer or failure to obtain required extensions of the Creditor Protection Proceedings and the stay granted in connection therewith, may limit the Company’s ability to obtain additional advances or result in demands for accelerated repayment, thus further straining its working capital position and ability to continue operations. The completion of the Recapitalization Transaction is also subject to completion of definitive documentation and satisfaction of any conditions contained therein. As a result, management is still in the process of assessing the impact of the transaction on the Company’s financial results.
In accordance with the terms of the RSA, upon consummation of the Recapitalization Transaction, the Company will be required to replace its Existing Credit Facility and DIP Facility with a new revolving credit facility (the “Exit Facility”) to support ongoing liquidity needs. As at March 14, 2011, the terms of the Exit Facility have not yet been determined.
The Company incurred $1.2 million in fees to obtain the DIP Facility and expects additional fees of approximately $0.3 million to complete the DIP Facility. In addition, the Company incurred $0.3 million to complete the Fourth Amendment and is obligated to pay certain agency fees and letter of credit fees when issued. As of December 31, 2010, the amount of financing available under the Existing Credit Facility was approximately $14.8 million, which is net of $2.8 million in letters of credit secured under the Existing Credit Facility and the $10.0 million of outstanding advances. As at March 16, 2011, approximately $11.0 million of financing is available under the DIP Facility, which is net of $2.8 million of secured letters of credit, $3.3 million is outstanding under the Existing Credit Facility and $6.7 million is outstanding under the DIP Facility.
F-31
Table of Contents
17. SHARE CAPITAL
(a) Authorized
200,000,000 Common shares without par value
50,000,000 Class I Preference shares without par value
The Class I Preference shares are issuable in Series. The directors may, by resolution, fix the number of shares in a series of Class I Preference shares and create, define and attach special rights and restrictions as required. None of these shares are currently issued and outstanding.
During the year ended December 31, 2010, the Company issued 42,296 common shares upon exercises of stock options (December 31, 2009 – 16,098, December 31, 2008 – 48,000). The Company issues new shares to satisfy stock option and award exercises.
(b) Stock Options
Angiotech Pharmaceuticals, Inc.
In June 2006, the shareholders approved the adoption of the 2006 Stock Incentive Plan (the “2006 Plan”) which superseded the Company’s previous stock option plans. The 2006 Plan incorporated all of the options granted under the previous stock option plans and, in total, provides for the issuance of non-transferable stock options and stock-based awards (as defined below) to purchase up to 13,937,756 common shares to employees, officers, directors of the Company, and persons providing ongoing management or consulting services to the Company. The 2006 Plan provides for, but does not require, the granting of tandem stock appreciation rights that at the option of the holder may be exercised instead of the underlying option. A stock option with a tandem stock appreciation right is referred to as an award. When the tandem stock appreciation right portion of an award is exercised, the underlying option is cancelled. The tandem stock appreciation rights are settled in equity and the optionee receives common shares with a fair market value equal to the excess of the fair value of the shares subject to the option at the time of exercise (or the portion thereof so exercised) over the aggregate option price of the shares set forth in the option agreement. The exercise price of the options and awards is fixed by the Board of Directors, but will generally be at least equal to the market price of the common shares at the date of grant, and for options issued under the 2006 Plan and the Company’s 2004 Stock Option Plan (the “2004 Plan”), the term may not exceed five years. For options grandfathered from the stock option plans prior to the 2004 Plan, the term did not exceed 10 years. Options and awards granted are also subject to certain vesting provisions. Options and awards generally vest monthly after being granted over varying terms from 2 to 4 years. Any one person is permitted, subject to the approval of the Company’s Board of Directors, to receive options and awards to acquire up to 5% of its issued and outstanding common shares.
A summary of CDN$ stock option and award activity is as follows:
| No. of Stock Options and Awards |
Weighted average exercise price (in CDN$) |
Weighted average remaining contractual term (years) |
Aggregate intrinsic value (in CDN$) |
|||||||||||||
| Outstanding at December 31, 2008 |
7,644,632 | 12.82 | 2.67 | 196 | ||||||||||||
| Granted |
1,469,000 | 0.41 | ||||||||||||||
| Exercised |
(10,229 | ) | 0.21 | |||||||||||||
| Forfeited |
(1,266,694 | ) | 16.06 | |||||||||||||
| Cancelled |
(2,665,000 | ) | 17.73 | |||||||||||||
| Outstanding at December 31, 2009 |
5,171,709 | $ | 5.99 | 3.18 | $ | 2,800 | ||||||||||
| Granted |
919,500 | 1.09 | ||||||||||||||
| Exercised |
(10,000 | ) | 0.31 | |||||||||||||
| Forfeited |
(806,025 | ) | 14.63 | |||||||||||||
| Outstanding at December 31, 2010 |
5,275,184 | $ | 3.83 | 2.75 | $ | 132 | ||||||||||
| Exercisable at December 31, 2010 |
3,049,990 | $ | 5.91 | 2.26 | $ | 67 | ||||||||||
A summary of U.S.$ award activity is as follows:
| No. of Awards |
Weighted average exercise price (in U.S.$) |
Weighted average remaining contractual term (years) |
Aggregate intrinsic value (in U.S.$) |
|||||||||||||
| Outstanding at December 31, 2008 |
1,790,968 | 3.19 | 4.19 | 64 | ||||||||||||
| Granted |
1,386,000 | 0.40 | ||||||||||||||
| Exercised |
(6,970 | ) | 0.26 | |||||||||||||
F-32
Table of Contents
| No. of Awards |
Weighted average exercise price (in U.S.$) |
Weighted average remaining contractual term (years) |
Aggregate intrinsic value (in U.S.$) |
|||||||||||||
| Forfeited |
(125,250 | ) | 2.33 | |||||||||||||
| Cancelled |
(60,000 | ) | 18.00 | |||||||||||||
| Outstanding at December 31, 2009 |
2,984,748 | $ | 1.64 | 3.74 | $ | 2,278 | ||||||||||
| Granted |
1,194,000 | 1.05 | ||||||||||||||
| Exercised |
(52,480 | ) | 0.26 | |||||||||||||
| Forfeited |
(235,269 | ) | 1.07 | |||||||||||||
| Outstanding at December 31, 2010 |
3,890,999 | $ | 1.51 | 3.14 | $ | 179 | ||||||||||
| Exercisable at December 31, 2010 |
1,846,660 | $ | 2.40 | 2.66 | $ | 92 | ||||||||||
As described under note 1, all stock options under existing plans are to be cancelled in connection with the implementation of the CCAA Plan.
American Medical Instruments Holdings, Inc. (“AMI”)
On March 9, 2006, AMI granted 304 stock options under AMI’s 2003 Stock Option Plan (the “AMI Stock Option Plan”), each of which is exercisable for approximately 3,852 common shares of the Company upon exercise. All outstanding options and warrants granted prior to the March 9, 2006 grant were settled and cancelled upon the acquisition of AMI. No further options to acquire common shares of the Company can be issued pursuant to the AMI Stock Option Plan. Approximately 1,171,092 of the Company’s common shares were reserved in March 2006 to accommodate future exercises of the AMI options.
| No. of Shares Underlying Options |
Weighted average exercise price (in U.S.$) |
Weighted average remaining contractual term (years) |
Aggregate intrinsic value (in U.S.$) |
|||||||||||||
| Outstanding at December 31, 2008 |
363,077 | 15.44 | 7.19 | — | ||||||||||||
| Forfeited |
(62,597 | ) | 15.44 | |||||||||||||
| Outstanding at December 31, 2009 |
300,480 | 15.44 | 6.19 | — | ||||||||||||
| Outstanding at December 31, 2010 |
300,480 | 15.44 | 5.19 | — | ||||||||||||
| Exercisable at December 31, 2010 |
187,797 | $ | 15.44 | 5.19 | — | |||||||||||
As described under note 1, all stock options under existing plans are to be cancelled in connection with the implementation of the CCAA Plan.
(c) Stock-based compensation expense
For the year ended December 31, 2010, the Company recorded stock-based compensation expense of $1.8 million (December 31, 2009 - $1.6 million, December 31, 2008 - $2.4 million) relating to awards granted under its stock option plan, modified or settled subsequent to October 1, 2002. The Company expenses the compensation cost of stock-based payments over the service period using the straight-line method over the vesting period and is estimated using the Black-Scholes option pricing model using the following weighted average assumptions for grants in the respective periods:
| Year Ended December 31, 2010 |
Year Ended December 31, 2009 |
Year Ended December 31, 2008 |
||||||||||
| Dividend Yield |
Nil | Nil | Nil | |||||||||
| Expected Volatility |
99.6% - 101.7 | % | 108.0% -126.8 | % | 81.8% - 95.9 | % | ||||||
| Weighted Average Volatility |
100.7 | % | 113.9 | % | 90.6 | % | ||||||
| Risk-free Interest Rate |
1.8 | % | 0.37% -1.31 | % | 1.11% - 2.25 | % | ||||||
| Expected Term (Years) |
5 | 3 | 3 | |||||||||
The weighted average fair value of awards granted in the years ended December 31, 2010, 2009 and 2008 are presented below:
F-33
Table of Contents
| Year Ended December 31, 2010 |
Year Ended December 31, 2009 |
Year Ended December 31, 2008 |
||||||||||
| CDN$ awards |
CDN$ | 0.83 | CDN$ | 0.28 | CDN$ | 0.14 | ||||||
| U.S.$ awards |
$ | 0.78 | $ | 0.27 | $ | 0.11 | ||||||
A summary of the status of the Company’s nonvested awards as of December 31, 2010 and changes during the year ended December 31, 2010, is presented below:
| Nonvested CDN$ Awards |
No. of Awards |
Weighted average grant-date fair value (in CDN$) |
||||||
| Nonvested at December 31, 2009 |
2,449,492 | $ | 0.53 | |||||
| Granted |
919,500 | 0.83 | ||||||
| Vested |
(1,093,796 | ) | 0.89 | |||||
| Forfeited |
(50,002 | ) | 1.32 | |||||
| Nonvested at December 31, 2010 |
2,225,194 | $ | 0.46 | |||||
| Nonvested U.S.$ Awards |
No. of Awards |
Weighted average grant-date fair value (in U.S.$) |
||||||
| Nonvested at December 31, 2009 |
2,061,569 | $ | 0.37 | |||||
| Granted |
1,194,000 | 0.78 | ||||||
| Vested |
(1,002,257 | ) | 0.62 | |||||
| Forfeited |
(208,973 | ) | 0.45 | |||||
| Nonvested at December 31, 2010 |
2,044,339 | $ | 0.48 | |||||
A summary of the status of the Company’s common shares underlying nonvested AMI stock options as of December 31, 2010 and changes during the year ended December 31, 2010, is presented below:
| Company Shares Underlying Nonvested AMI Options |
No. of Shares Underlying Options |
Weighted average grant-date fair value (in U.S.$) |
||||||
| Nonvested at December 31, 2009 |
187,801 | $ | 6.51 | |||||
| Vested |
(75,118 | ) | 6.51 | |||||
| Nonvested at December 31, 2010 |
112,683 | $ | 6.51 | |||||
As of December 31, 2010, there was $1.6 million of total unrecognized compensation cost related to nonvested stock awards granted under the 2006 Plan.
As of December 31, 2010, there was $0.4 million of total unrecognized compensation cost related to the nonvested AMI stock options. The total fair value of options vested during the year ended December 31, 2010 was $0.5 million (December 31, 2009 - $0.5 million).
During the years ended December 31, 2010, 2009 and 2008 the following activity occurred:
| (in thousands) |
Year Ended December 31, 2010 |
Year Ended December 31, 2009 |
Year Ended December 31, 2008 |
|||||||||
| Total intrinsic value of stock options exercised: |
||||||||||||
| CDN dollar options |
$ | 7 | $ | 17 | $ | 33 | ||||||
| U.S. dollar options |
$ | 33 | $ | 10 | $ | n/a | ||||||
| Total fair value of stock awards vested |
$ | 1,565 | $ | 1,559 | $ | 2,367 | ||||||
Cash received from stock option exercises for the years ended December 31, 2010, 2009, and 2008 were $11,000, $3,000 and $121,000 respectively. The tax benefit realized from the option exercises of the share-based payment arrangements totaled $nil for the year ended December 31, 2010, $7,900 for the year ended December 31, 2009, and $nil for the year ended December 31, 2008.
The Black-Scholes pricing model was developed for use in estimating the fair value of traded options which have no vesting restrictions and are fully transferable. In addition, option valuation models require the input of highly subjective assumptions including the expected stock price volatility. The Company’s employee stock options have characteristics significantly different from those of traded options and changes in the subjective input assumptions can materially affect the fair value estimate.
F-34
Table of Contents
(d) Shareholder rights plan
The Company adopted a shareholder’s rights plan (“the Plan”) on February 10, 1999, amended and restated March 5, 2002, June 9, 2005 and October 30, 2008. According to the Plan, each shareholder is issued one right. Each right will entitle the holder to acquire common shares of the Company at a 50% discount to the market upon certain specified events, including upon a person or group of persons acquiring 20% or more of the total common shares of the Company. The rights are not exercisable in the event of a Permitted Bid as defined in the Plan. The Plan must be reconfirmed by the Company’s shareholders every three years. In connection with the implementation of the CCAA Plan, the Plan and all rights issued thereunder will be cancelled.
18. LICENCE FEE REVENUE
Under an Amended and Restated Distribution and Licence Agreement with our partner Baxter, in 2009 the Company received a one-time payment of $25.0 million, which was received in lieu and settlement of future royalty and milestone obligations related to existing formulations of CoSeal surgical sealant or any future products. All deliverables were provided to Baxter and the Company’s performance obligations were substantially complete when the payment was recognized as licence fee revenue.
19. ESCROW SETTLEMENT RECOVERY
In connection with the 2006 acquisition of AMI, RoundTable Healthcare Partners, LP (“RoundTable”) and Angiotech entered into an Escrow Agreement on March 23, 2006. Under this agreement, Angiotech deposited $20.0 million with LaSalle Bank (“LaSalle”), which was designated as the Escrow Agent. On April 4, 2007, Angiotech issued an Escrow Claim Notice to LaSalle to return the $20.0 million, which was subsequently disputed by RoundTable. After various legal proceedings, hearings and discussions, a Joint Letter of Direction was executed and $6.5 million was released to RoundTable, thereby leaving the amount in dispute at approximately $13.5 million. On January 14, 2010, a settlement agreement was signed whereby Angiotech received $4.7 million from the escrow account. On January 15, 2010, the Company received the settlement funds, which were recorded as a recovery. All legal proceedings have now been dismissed.
20. COMMITMENTS AND CONTINGENCIES
(a) Commitments
i) Lease commitments
The Company has entered into operating lease agreements for office, laboratory and manufacturing space which expire through July 2019. Future minimum annual lease payments under these leases are as follows:
| 2011 |
3,843 | |||
| 2012 |
3,731 | |||
| 2013 |
3,578 | |||
| 2014 |
3,570 | |||
| 2015 |
3,557 | |||
| Thereafter |
10,233 | |||
| $ | 28,512 | |||
Rent expense for the year ended December 31, 2010 amounted to $3.3 million (December 31, 2009 - $3.2 million, December 31, 2008 - $3.5 million).
Other liabilities include a $0.9 million accrual for estimated exit costs associated with a leased property which is no longer in use.
ii) Commitments
We have entered into research and development collaboration agreements that involve joint research efforts. Certain collaboration costs and any eventual profits will be shared as per terms provided for in the agreements. We may also be required to make milestone, royalty, and/or other research and development funding payments under research and development collaboration and other agreements with third parties. These payments are contingent upon the achievement of specific development, regulatory and/or commercial milestones. We accrue for these payments when it is probable that a liability has been incurred and the amount can be reasonably estimated. We do not accrue for these payments if the outcome of achieving these milestones is not determinable. Our significant contingent milestone, royalty and other research and development commitments are as follows:
F-35
Table of Contents
Quill Medical, Inc. (“Quill”)
In connection with the acquisition of Quill in June 2006, the Company was required to make additional contingent payments of up to $150 million upon the achievement of certain revenue growth and a development milestone. These payments were primarily contingent upon the achievement of significant incremental revenue growth over a five-year period beginning on July 1, 2006, subject to certain conditions. In connection with the litigation with QSR Holdings, Inc. (“QSR”) as described under the Contingencies section below, on January 27, 2011 the Company entered into a settlement agreement for the full and final settlement of all claims, including all potential contingent payments, under the Agreement and Plan of Merger dated May 25, 2006. Under the terms of the settlement agreement, the Company is required to pay QSR $6.0 million. The first $2.0 million is due and payable at the earlier of the completion of the CCAA proceedings or May 31, 2011, with the remainder being payable in equal monthly installments over a 24 month period. As at December 31, 2010, the Company recorded $0.6 million of litigation expense, related to these legal proceedings. In accordance with the guidance on contingent consideration under Financial Accounting Standard (“FAS”) 141, which pertains to business combinations arising prior to December 15, 2008, the Company is currently assessing the impact of the remaining Settlement Amount on its 2011 first quarter results.
National Institutes of Health (“NIH”)
In November 1997, the Company entered into an exclusive license agreement with the Public Health Service of the U.S., through the National Institutes of Health (the “NIH”), whereby the NIH granted the Company an exclusive, worldwide license to certain technologies of the NIH for the use of paclitaxel (the “NIH License Agreement”). Pursuant to the NIH License Agreement, the Company agreed to pay to the NIH certain milestone payments upon achievement of specified clinical and commercial development milestones and to pay royalties on net TAXUS® sales by BSC and Zilver-PTX sales by Cook Medical Inc. and its affiliates (“Cook”). At December 31, 2010 and December 31, 2009, the Company had accrued royalty fees and interest of $7.2 million and $7.3 million, respectively, payable to the NIH under the NIH License Agreement relating to royalties on net TAXUS® sales for the years 2010 and 2009, respectively. On December 29, 2010, the Company and the NIH entered into an amendment to the NIH License Agreement whereby the parties agreed to eliminate: (i) approximately $7.2 million of unpaid royalties and interest due on sales of TAXUS® by BSC and (ii) future royalties payable on licensed products sold by BSC going forward, in exchange for a 0.25% increase of existing royalty rates for licensed products sold by Cook and an extension of the term for payment of such royalties of approximately two years. Given that the unpaid royalties have not been extinguished and will effectively be paid out through the incurrence of future royalty fees, which were indeterminable and could not be forecasted at the time of the Company’s assessment, the $7.2 million of royalties remains accrued for as at December 31, 2010 in accordance with ASC No. 470-60-35 on Troubled Debt Restructuring. In addition, based on this amendment, the $7.2 million accrual was reclassified from current accounts payable and accrued liabilities to non-current other liabilities.
Biopsy Sciences LLC (“Biopsy Sciences”)
In connection with the acquisition of certain assets from Biopsy Sciences in January 2007, the Company may be required to make certain contingent payments of up to $2.7 million upon the achievement of certain clinical and regulatory milestones and up to $10.7 million for achieving certain commercialization milestones. No clinical or regulatory milestones were achieved in 2010. As a result, no payments were made to Biopsy Sciences in 2010.
Rex Medical L.P. (“Rex”)
In March 2008, the Company entered into a License, Supply, Marketing and Distribution Agreement (the “Option Agreement”) with Rex to manufacture and distribute the Option Inferior Vena Cava Filter (“Option IVC Filter”). During 2008 the Company made an upfront payment of $2.5 million to Rex to obtain the marketing rights for the Option IVC Filter. This payment was recorded as an in-process and research development cost. During 2009, the Company made milestone payments of $2.5 million, in connection with the achievement of U.S. Food and Drug Administration (“FDA”) 510(k) clearance, and $0.5 million, in connection with the achievement of the first sales target. During the year ended December 31, 2010, the Company further achieved the second and third sales targets which triggered milestone payments of $1.0 million and $1.5 million respectively. All milestone payments were capitalized to intangible assets. The Company is also committed to making escalating royalty payments of 30.0% to 47.5% based on annual net sales of these products, which are recognized as cost of products sold. On January 24, 2011, the Company received a Notice from Rex indicating that the Option Agreement would terminate on April 24, 2011 and on February 16, 2011, in connection with the its ongoing litigation with Rex, the Company entered into a Settlement and License Termination Agreement with Rex (the “Rex Settlement Agreement”), which provides for the full and final settlement and dismissal of all claims arising under the Option Agreement. The Rex Settlement Agreement included, among other things, a $1.5 million settlement payment for the full and final settlement of all claims, and royalty and milestone obligations incurred prior to January 1, 2011 under the Option Agreement. As at December 31, 2010, the Company recorded a $0.9 million inventory write-down and a $1.1 million intangible asset write-down in connection with the termination of the Option Agreement. In accordance with ASC No. 855 on Subsequent Events, the Company is currently assessing the impact of the Rex Settlement Agreement on its 2011 first quarter results.
F-36
Table of Contents
(b) Contingencies
From time to time, we may be subject to claims and legal proceedings brought against us in the normal course of business. Such matters are subject to many uncertainties. We believe that adequate provisions have been made in the accounts where required. However, we are not able to determine the outcome or estimate potential losses of the pending legal proceedings listed below, or other legal proceedings, to which we may become subject in the normal course of business or estimate the amount or range of any possible loss we might incur if it does not prevail in the final, non-appealable determinations of such matters. We cannot provide assurance that the legal proceedings listed here, or other legal proceedings not listed here, will not have a material adverse impact on our financial condition or results of operations.
BSC, a licensee, is often involved in legal proceedings (to which we are not a party) concerning challenges to its stent business. If a party opposing BSC is successful, and if a court were to issue an injunction against BSC, royalty revenue would likely be significantly reduced. The ultimate outcome of any such proceedings is uncertain at this time. BSC has recently settled two significant litigation matters with Johnson & Johnson involving its stent business in exchange for payment to Johnson & Johnson of over $2.4 billion.
On April 4, 2005, together with BSC, the Company commenced a legal action in the Netherlands against Sahajanand Medical Technologies Pvt. Ltd. (“SMT”) for patent infringement. On May 3, 2006, the Dutch trial court ruled in the Company’s favor, finding that our EP (NL) 0 706 376 patent was valid, and that SMT’s Infinnium™ stent infringed the patent. On March 13, 2008, a Dutch Court of Appeal held a hearing to review the correctness of the trial court’s decision. The Court of Appeal released their judgment on January 27, 2009, ruling against SMT (finding the Company’s patent novel, inventive, and sufficiently disclosed). The Court of Appeal however requested amendment of claim 12 before rendering their decision on infringement. The Company has filed a response to the Court of Appeal’s decision, and has additionally filed a further writ against SMT seeking to prevent SMT from, among other things, using its CE mark for SMT’s Infinnium stent. Any decision of the Court of Appeal is appealable to the Supreme Court of the Netherlands. On October 22, 2009, Angiotech and BSC settled this litigation with SMT on favorable terms. The Company has continued this matter before the Court of Appeal with respect to the requested amendment to claim 12. A hearing was held on April 22, 2010 and on August 31, 2010, this court rendered a decision favorable to Angiotech. This matter is therefore concluded.
In July 2004, Dr. Gregory Baran initiated legal action, alleging infringement by the Company’s subsidiary Medical Device Technologies Inc. (“MDT”) of two U.S. patents owned by Dr. Baran. These patents allegedly cover MDT’s BioPince™ automated biopsy device. On September 25, 2007, the judge issued her decision pursuant to the Markman hearing of December 2005. The Company submitted a Motion for Summary Judgment to the court based upon the Markman decision and on September 30, 2009, the judge ruled in MDT’s favor, finding that the BioPince device did not infringe a key claim of Dr. Baran’s patents. Dr. Baran appealed this decision. A hearing on Dr. Baran’s appeal was held before the U.S. Court of Appeals for the Federal Circuit on June 9, 2010 and on August 12, 2010, this court decided in our favor. Dr. Baran petitioned for a rehearing, but this has been denied. Dr. Baran has filed a Writ of Certiorari with the U.S. Supreme Court seeking review of the CAFC decision. The Company filed a Brief in Opposition on March 7, 2011. As the Supreme Court denied Certiorari, this matter is therefore concluded.
On October 4, 2010, the Company was notified that QSR Holdings, Inc. (“QSR”), as a representative for former stockholders of Quill Medical, Inc. (“QMI”), had made a formal demand (the “Arbitration Demand”) to the American Arbitration Association naming the Company, QMI and Angiotech Pharmaceuticals (US), Inc. as respondents (collectively, the “Respondents”). The Arbitration Demand alleged that the Respondents failed to satisfy certain obligations under the Agreement and Plan of Merger, dated May 25, 2006, by and among Angiotech, Angiotech US, Quaich Acquisition, Inc. and QMI (the “Merger Agreement”), notably including the obligations to make certain earn out payments and the orthopedic milestone payment. The Arbitration Demand sought either direct monetary damages or, in the alternative, extension of the earn out period for a sixth year as more fully set forth in the Merger Agreement. In addition, QSR commenced an action in the United States District Court for the Middle District of North Carolina on October 1, 2010 against the Respondents, entitled QSR Holdings, Inc. v. Angiotech Pharmaceuticals, Inc., Angiotech Pharmaceuticals (US), Inc. and Quill Medical, Inc. (the “Federal Litigation”). The Complaint in the Federal Litigation alleged, among other items, that the Respondents: (a) breached certain contractual obligations under the Merger Agreement; (b) made certain misrepresentations or omissions during the initial negotiation of the Merger Agreement; and (c) tortiously interfered with the Merger Agreement. QSR sought damages in an unstated amount together with punitive damages and/or attorneys fees to the extent allowed by law. On October 15, 2010, the Respondents moved to dismiss the Complaint in part and to transfer venue to federal court in Washington State.
Subsequent to the initiation of the Federal Litigation and the delivery of the Arbitration Demand, representatives of Angiotech and QSR engaged in substantial settlement discussions. Those discussions culminated in an agreement (the “Settlement Agreement”), dated as of January 27, 2011, resolving, among other things, any and all claims that the parties may have arising out of the Merger Agreement (including, without limitation, all claims, counterclaims or defenses that were or could have been asserted in the Arbitration Demand and the Federal Litigation) in exchange for a payment of $6 million (the “Settlement Amount”) by Angiotech US. $2.0 million of the Settlement Amount is payable on the earlier of (i) 10 business days after the implementation of the Recapitalization Transaction contemplated by the RSA (the “Implementation Date”) and (ii) May 31, 2011. The remainder of the Settlement Amount is payable in 24 equal monthly installments of $166,667 each beginning on the earlier of (i) 30 days after the Implementation Date; and (ii) July 31, 2011 (subject to reduction in the event of pre-payment as more fully set forth in the Settlement Agreement). The Settlement Agreement further provides for complete and mutual releases between the parties, as more fully set forth therein. Pursuant to the Settlement Agreement, QSR irrevocably dismissed the Federal Litigation and the Arbitration Demand with prejudice.
F-37
Table of Contents
In light of the Company’s recent liquidity and capital constraints, on November 11, 2010, Angiotech US informed Rex Medical, LP (“Rex”) that it would not be able to continue selling the Option IVC Filter under the existing terms of the License, Supply, Marketing and Distribution Agreement, dated as of March 13, 2008, by and between Angiotech US and Rex (as amended, the “Option Agreement”). Subsequently, on November 18, 2010, Rex initiated an arbitration (the “Rex Arbitration”) against Angiotech US alleging that Angiotech US had breached the Option Agreement. Rex sought monetary damages in excess of $3.0 million, including costs, fees and expenses incurred in connection with the Rex Arbitration. On December 2, 2010, Rex obtained a temporary restraining order and preliminary injunction against Angiotech US from the United States District Court for the Southern District of New York (such action, the “SDNY Action”) requiring Angiotech US to honor its obligations under the Option Agreement for a period of 180 days or until the Rex Arbitration was concluded. On January 24, 2011, Angiotech US received a termination notice from Rex indicating that the Option Agreement would cease on April 24, 2011.
On February 16, 2011, Angiotech US entered into a Settlement and License Termination Agreement with Rex (the “Rex Settlement Agreement”) which provides for the full and final settlement and/or dismissal of all claims arising under the Option Agreement. Pursuant to the Rex Settlement Agreement, the Company and Rex agreed to withdraw any claims that have been or could have made in the Rex Arbitration, as well as to dismiss the SDNY Action with prejudice. The Rex Settlement Agreement further provides that: (i) the Option Agreement will terminate on March 31, 2011, or at an earlier date if so elected by Rex (the “Termination Date”), upon which Angiotech US will no longer market, sell or distribute the Option IVC Filter; (ii) Angiotech US will make a payment in the amount of $1.5 million to Rex within five business days of the effective date of the Rex Settlement Agreement in full and final payment and settlement of all claims and royalties due under the Option Agreement relating to the sales of the Option IVC Filter recorded by Angiotech US prior to January 1, 2011, and all milestone payments due or that may come due to Rex under the Option Agreement now or in the future; (iii) within five business days of the effective date, Angiotech US will make a royalty payment to Rex based upon actual cash receipts collected from customers attributable to sales of the Option IVC Filter that are recorded and accrued between January 1, 2011 and the effective date; (iv) from the effective date through the Termination Date, Angiotech US will make ongoing royalty payments to Rex on a weekly basis based upon actual cash receipts collected during each previous calendar week in respect of sales of the Option IVC Filter; and (v) Angiotech US will deliver to Rex certain materials relating to the marketing and sale of the Option IVC Filter. The Rex Settlement Agreement became effective on March 10, 2011 upon the Canadian Court granting an order approving the authority of Angiotech US to enter into the Rex Settlement Agreement. The Canadian Court is overseeing the financial restructuring of the Company and certain of its subsidiaries pursuant to the CCAA.
At the European Patent Office (“EPO”), various patents either owned or licensed by or to the Company are in opposition proceedings including the following:
| • | In EP_1_155_689, the EPO decided to revoke the patent as a result of the oral hearing held on November 25, 2010, and the Company has until March 21, 2011 to file an appeal. |
| • | In EP_1_159_974, the EPO decided to revoke the patent as a result of Oral Proceedings held on June 9, 2010. Angiotech filed a Notice of Appeal on August 27, 2010. A Statement of Grounds of Appeal was filed on November 2, 2010. |
| • | In EP_1_159_975, a Notice of Opposition was filed on June 5, 2009. On February 4, 2010, Angiotech requested that the EPO set oral proceedings. On April 20, 2010, Angiotech filed a response to the Opposition, and on December 2, 2010, the opponents filed their response. |
| • | In EP_0_876_165, the EPO revoked the patent as an outcome of an oral hearing held on June 24, 2009. On August 20, 2009, the Company filed a Notice of Appeal. On April 8, 2010, the opponent filed their response to the appeal. |
| • | In EP_1_857_236, a Notice of Opposition was filed against Quill Medical, Inc. by ITV Denkendorf Produkservice GmbH. Quill Medical has until January 29, 2011, to file its response. |
| • | In EP_1_858_243, a Notice of Opposition was filed against Quill Medical, Inc. by ITV Denkendorf Produkservice GmbH. Quill Medical has until January 22, 2011, to file its response. |
| • | In EP_1_867_288, a Notice of Opposition was filed against Quill Medical, Inc. by ITV Denkendorf Produkservice GmbH. |
21. SEGMENTED INFORMATION
The Company operates in two reportable segments: (i) Pharmaceutical Technologies and (ii) Medical Products.
The Pharmaceutical Technologies segment includes royalty revenue generated from out-licensing technology related to the drug-eluting stent and other technologies.
The Medical Products segment includes revenues and gross margins of single use, specialty medical devices including suture needles, biopsy needles / devices, micro surgical ophthalmic knives, drainage catheters, self-anchoring sutures, other specialty devices, biomaterials and other technologies.
F-38
Table of Contents
The Company reports segmented information on each of these segments to the gross margin level. All other income and expenses are not allocated to segments as they are not considered in evaluating the segment’s operating performance. The following tables represent reportable segment information for the years ended December 31, 2010, 2009 and 2008:
| Year ended December 31, 2010 |
Year ended December 31, 2009 |
Year ended December 31, 2008 |
||||||||||
| Revenue |
||||||||||||
| Medical Products |
$ | 211,495 | $ | 191,951 | $ | 190,816 | ||||||
| Pharmaceutical Technologies |
34,747 | 87,727 | 92,456 | |||||||||
| Total revenue |
246,242 | $ | 279,678 | $ | 283,272 | |||||||
| Cost of products sold — Medical Products |
106,304 | 104,616 | 101,052 | |||||||||
| License and royalty fees — Pharmaceutical Technologies |
5,889 | 10,431 | 14,258 | |||||||||
| Gross margin |
||||||||||||
| Medical Products |
105,191 | 87,335 | 89,764 | |||||||||
| Pharmaceutical Technologies |
28,858 | 77,296 | 78,198 | |||||||||
| Total gross margin |
134,049 | $ | 164,631 | $ | 167,962 | |||||||
| Research and development |
26,790 | 23,701 | 53,192 | |||||||||
| Selling, general and administration |
89,238 | 81,504 | 98,483 | |||||||||
| Depreciation and amortization |
33,745 | 33,251 | 33,998 | |||||||||
| In-process research and development |
— | — | 2,500 | |||||||||
| Write-down of assets held for sale |
1,450 | 3,090 | 1,283 | |||||||||
| Write-down of property, plant and equipment |
4,779 | — | — | |||||||||
| Write-down of intangible assets |
2,814 | — | — | |||||||||
| Escrow settlement recovery |
(4,710 | ) | — | — | ||||||||
| Write-down of goodwill |
— | — | 649,685 | |||||||||
| Operating (loss) income |
$ | (20,057 | ) | $ | 23,085 | $ | (671,179 | ) | ||||
| Other (expense) income |
(46,798 | ) | (39,916 | ) | (82,889 | ) | ||||||
| Loss from continuing operations before income taxes |
$ | (66,855 | ) | $ | (16,831 | ) | $ | (754,068 | ) | |||
The Company allocates its assets to the two reportable segments; however, as noted above, depreciation, income taxes and other expense and income are not allocated to segment operating units.
During the year ended December 31, 2010, revenue from one licensee represented approximately 12% of total revenue. During the year ended December 31, 2009, revenue from two licensees represented 29% of total revenue (December 31, 2008 – 32%).
The following tables represent total assets and capital expenditures for each reportable segment at December 31, 2010 and 2009:
| December 31, 2010 |
December 31, 2009 |
|||||||
| Total assets |
||||||||
| Pharmaceutical Technologies |
$ | 35,860 | $ | 70,848 | ||||
| Medical Products |
270,138 | 299,211 | ||||||
| Total assets |
$ | 305,998 | $ | 370,059 | ||||
| Capital expenditures |
||||||||
| Pharmaceutical Technologies |
$ | — | $ | 1,188 | ||||
| Medical Products |
5,726 | 3,178 | ||||||
| Total capital expenditures |
5,726 | $ | 4,366 | |||||
Geographic information
Revenues are attributable to countries based on the location of the Company’s customers or, for revenue from collaborators, the location of the collaborator’s customers.
| December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| United States |
$ | 150,428 | $ | 172,318 | $ | 169,181 | ||||||
| Europe |
49,420 | 60,001 | 65,166 | |||||||||
| Others |
46,394 | 47,359 | 48,925 | |||||||||
F-39
Table of Contents
| December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Total |
$ | 246,242 | $ | 279,678 | $ | 283,272 | ||||||
During the year ended December 31, 2009, the Company classified $5.3 million of land and buildings as held for sale. As at December 31, 2010, the remaining $3.1 million carrying value of land and buildings was reclassified back to property, plant and equipment given that they did not meet the held-for-sale classification criteria defined under ASC No. 360-10 – Property, Plant and Equipment (see note 8). The remaining carrying values of these assets are included in the table below for net long lived assets by country:
| For the year ended December 31, |
||||||||
| 2010 | 2009 | |||||||
| United States |
$ | 30,002 | $ | 34,655 | ||||
| Canada |
11,435 | 11,807 | ||||||
| Other |
4,824 | 5,717 | ||||||
| Total |
$ | 46,261 | $ | 52,179 | ||||
22. LOSS PER SHARE
Loss per share was calculated as follows:
| Year ended December 31, 2010 |
Year ended December 31, 2009 |
Year ended December 31, 2008 |
||||||||||
| Numerator: |
||||||||||||
| Net loss |
$ | (66,811 | ) | $ | (22,868 | ) | $ | (741,176 | ) | |||
| Denominator: |
||||||||||||
| Basic weighted average common shares outstanding |
85,168 | 85,130 | 85,118 | |||||||||
| Basic and diluted net loss per common share: |
$ | (0.78 | ) | $ | (0.27 | ) | $ | (8.71 | ) | |||
For the year ended December 31, 2010 6,045,830 stock options were excluded from the calculation of diluted net loss per common share, as the effect of including them would have been anti-dilutive (December 31, 2009 – 3,411,357, December 31, 2008 – 9,798,677).
23. CHANGE IN NON-CASH WORKING CAPITAL ITEMS RELATING TO OPERATING ACTIVITIES AND SUPPLEMENTAL CASH FLOW INFORMATION
The change in non-cash working capital items relating to operations was as follows:
| Year ended December 31, 2010 |
Year ended December 31, 2009 |
Year ended December 31, 2008 |
||||||||||
| Accounts receivable |
$ | (5,638 | ) | $ | (2,122 | ) | $ | (5,524 | ) | |||
| Inventories |
768 | 3,244 | (5,156 | ) | ||||||||
| Prepaid expenses and other current assets |
(223 | ) | 7,623 | 2,038 | ||||||||
| Income taxes receivable |
420 | |||||||||||
| Accounts payable and accrued liabilities |
(2,337 | ) | (5,417 | ) | (11,843 | ) | ||||||
| Income taxes payable |
(8,603 | ) | 1,698 | 156 | ||||||||
| Interest payable |
9,758 | (511 | ) | (812 | ) | |||||||
| $ | (5,855 | ) | $ | 4,515 | $ | (21,141 | ) | |||||
F-40
Table of Contents
Supplemental disclosure
| Year ended December 31, 2010 |
Year ended December 31, 2009 |
Year ended December 31, 2008 |
||||||||||
| Interest paid |
$ | 23,523 | $ | 35,671 | $ | 43,066 | ||||||
| Income taxes paid |
13,904 | 8,748 | 3,720 | |||||||||
| Income tax refund |
634 | 5,179 | 7,373 | |||||||||
| Financing activities not yet paid |
2,090 | — | 7,745 | |||||||||
24. COMPARATIVE FIGURES
Certain prior period figures have been reclassified to conform to current period presentation.
25. CONDENSED CONSOLIDATING GUARANTOR FINANCIAL INFORMATION
The following presents the condensed consolidating guarantor financial information as of December 31, 2010 and 2009, and for the years ended December 31, 2010, 2009 and 2008 for the direct and indirect subsidiaries of the Company that serve as guarantors of the Senior Subordinated Notes and the Senior Floating Rate Notes, and for the Company’s subsidiaries that do not serve as guarantors. Non-guarantor subsidiaries include the Swiss subsidiaries and a Canadian Trust that cannot guarantee the debt of the Company. All of the Company’s subsidiaries are 100% owned, and all guarantees are full and unconditional, joint and several.
F-41
Table of Contents
Condensed Consolidating Balance Sheet
Year end December 31, 2010
USD (in ‘000s)
| Parent
Company Angiotech Pharmaceuticals, Inc. |
Guarantor Subsidiaries |
Non
Guarantor Subsidiaries |
Consolidating Adjustments |
Consolidated Total |
||||||||||||||||
| ASSETS |
||||||||||||||||||||
| Current assets |
||||||||||||||||||||
| Cash and cash equivalents |
$ | 11,180 | $ | 15,340 | $ | 6,795 | $ | — | $ | 33,315 | ||||||||||
| Short term investments |
4,653 | — | — | — | 4,653 | |||||||||||||||
| Accounts receivable |
356 | 21,279 | 11,859 | — | 33,494 | |||||||||||||||
| Income taxes receivable |
669 | — | 0 | — | 669 | |||||||||||||||
| Inventories |
— | 29,177 | 7,176 | (1,722 | ) | 34,631 | ||||||||||||||
| Deferred income taxes |
— | 4,102 | — | — | 4,102 | |||||||||||||||
| Deferred financing costs |
4,300 | 1,311 | — | — | 5,611 | |||||||||||||||
| Prepaid expenses and other current assets |
2,311 | 1,933 | 405 | — | 4,649 | |||||||||||||||
| Total Current Assets |
23,469 | 73,142 | 26,235 | (1,722 | ) | 121,124 | ||||||||||||||
| Investment in subsidiaries |
182,887 | 151,710 | — | (334,597 | ) | — | ||||||||||||||
| Assets held for sale |
— | — | — | — | — | |||||||||||||||
| Property, plant and equipment |
11,544 | 29,893 | 4,824 | — | 46,261 | |||||||||||||||
| Intangible assets |
9,931 | 125,548 | 2,494 | — | 137,973 | |||||||||||||||
| Deferred financing costs |
— | — | — | — | — | |||||||||||||||
| Deferred income taxes |
— | — | — | — | — | |||||||||||||||
| Other assets |
80 | 375 | 185 | — | 640 | |||||||||||||||
| Total Assets |
227,911 | 380,668 | 33,738 | (336,319 | ) | 305,998 | ||||||||||||||
| LIABILITIES AND SHAREHOLDERS’ (DEFICIT) EQUITY |
||||||||||||||||||||
| Current liabilities |
||||||||||||||||||||
| Accounts payable and accrued liabilities |
10,719 | 20,596 | 5,527 | 5 | 36,847 | |||||||||||||||
| Income taxes payable |
669 | (480 | ) | 2,066 | — | 2,255 | ||||||||||||||
| Interest payable on long-term debt |
15,665 | 96 | — | — | 15,761 | |||||||||||||||
| Revolving credit facility |
— | 10,000 | — | — | 10,000 | |||||||||||||||
| Long-term debt |
575,000 | — | — | — | 575,000 | |||||||||||||||
| Total Current Liabilities |
602,053 | 30,212 | 7,593 | 5 | 639,863 | |||||||||||||||
| Deferred leasehold inducement |
2,247 | 2,538 | — | — | 4,785 | |||||||||||||||
| Deferred income taxes |
(1,783 | ) | 32,991 | 494 | — | 31,702 | ||||||||||||||
| Other tax liabilities |
2,205 | 961 | 1,201 | — | 4,367 | |||||||||||||||
| Other liabilities |
7,265 | 1,111 | 981 | — | 9,357 | |||||||||||||||
| Long-term debt |
— | — | — | — | — | |||||||||||||||
| Total Non-current Liabilities |
9,934 | 37,601 | 2,676 | — | 50,211 | |||||||||||||||
| Total Shareholders’ (Deficit) Equity |
(384,076 | ) | 312,855 | 23,469 | (336,324 | ) | (384,076 | ) | ||||||||||||
| Total Liabilities and Shareholders’ (Deficit) Equity |
$ | 227,911 | $ | 380,668 | $ | 33,738 | $ | (336,324 | ) | $ | 305,998 | |||||||||
F-42
Table of Contents
Condensed Consolidating Balance Sheet
Year end December 31, 2009
USD (in ‘000s)
| Parent Company Angiotech Pharmaceuticals, Inc. |
Guarantors Subsidiaries |
Non Guarantors Subsidiaries |
Consolidating Adjustments |
Consolidated Total |
||||||||||||||||
| ASSETS |
||||||||||||||||||||
| Current assets |
||||||||||||||||||||
| Cash and cash equivalents |
$ | 20,602 | $ | 16,912 | $ | 12,028 | $ | — | $ | 49,542 | ||||||||||
| Short term investments |
7,780 | — | — | — | 7,780 | |||||||||||||||
| Accounts receivable |
— | 18,272 | 9,826 | 69 | 28,167 | |||||||||||||||
| Income tax receivable |
1,090 | — | — | — | 1,090 | |||||||||||||||
| Inventories |
— | 29,776 | 6,776 | (1,011 | ) | 35,541 | ||||||||||||||
| Deferred income taxes, current portion |
1,692 | 6,918 | — | (4,326 | ) | 4,284 | ||||||||||||||
| Prepaid expenses and other current assets |
1,554 | 1,329 | 411 | — | 3,294 | |||||||||||||||
| Total Current Assets |
32,718 | 73,207 | 29,041 | (5,268 | ) | 129,698 | ||||||||||||||
| Investment in subsidiaries |
231,026 | 407,344 | — | (638,370 | ) | — | ||||||||||||||
| Assets held for sale |
3,000 | 2,300 | — | — | 5,300 | |||||||||||||||
| Property, plant and equipment |
8,807 | 30,719 | 7,353 | — | 46,879 | |||||||||||||||
| Intangible assets |
12,490 | 140,798 | 19,731 | 173,019 | ||||||||||||||||
| Deferred financing costs |
9,398 | 2,011 | — | — | 11,409 | |||||||||||||||
| Other assets |
2,479 | 459 | 816 | — | 3,754 | |||||||||||||||
| Total Assets |
299,918 | 656,838 | 56,941 | (643,638 | ) | 370,059 | ||||||||||||||
| LIABILITIES AND SHAREHOLDERS’ (DEFICIT) EQUITY |
||||||||||||||||||||
| Current liabilities |
||||||||||||||||||||
| Accounts payable and accrued liabilities |
17,974 | 22,205 | 6,292 | (147 | ) | 46,324 | ||||||||||||||
| Income taxes payable |
9,749 | 714 | 395 | — | 10,858 | |||||||||||||||
| Interest payable on long-term debt |
5,965 | 39 | — | — | 6,004 | |||||||||||||||
| Total Current Liabilities |
33,688 | 22,958 | 6,687 | (147 | ) | 63,186 | ||||||||||||||
| Deferred leasehold inducement |
2,506 | 382 | — | — | 2,888 | |||||||||||||||
| Deferred income taxes |
— | 40,668 | 439 | (4,329 | ) | 36,778 | ||||||||||||||
| Other tax liabilities |
2,011 | 756 | 1,131 | — | 3,898 | |||||||||||||||
| Long-term debt |
575,000 | — | — | — | 575,000 | |||||||||||||||
| Other liabilities |
— | — | 1,596 | — | 1,596 | |||||||||||||||
| Total Non-current Liabilities |
579,517 | 41,806 | 3,166 | (4,329 | ) | 620,160 | ||||||||||||||
| Total Shareholders’ (Deficit) Equity |
(313,287 | ) | 592,074 | 47,088 | (639,161 | ) | (313,287 | ) | ||||||||||||
| Equity |
$ | 299,918 | $ | 656,838 | $ | 56,941 | $ | (643,638 | ) | $ | 370,059 | |||||||||
F-43
Table of Contents
Condensed Consolidated Statement of Operations
Year ended December 31, 2010
USD (in ‘000s)
| Parent
Company Angiotech Pharmaceuticals, Inc. |
Guarantors Subsidiaries |
Non
Guarantors Subsidiaries |
Consolidating Adjustments |
Consolidated Total |
||||||||||||||||
| REVENUE |
||||||||||||||||||||
| Product sales, net |
$ | 22 | $ | 160,479 | $ | 69,822 | $ | (18,828 | ) | $ | 211,495 | |||||||||
| Royalty revenue |
30,691 | 3,770 | — | — | 34,461 | |||||||||||||||
| License fees |
— | (24 | ) | 310 | — | 286 | ||||||||||||||
| 30,713 | 164,225 | 70,132 | (18,828 | ) | 246,242 | |||||||||||||||
| EXPENSES |
||||||||||||||||||||
| Cost of products sold |
(26 | ) | 74,568 | 49,530 | (17,768 | ) | 106,304 | |||||||||||||
| License and royalty fees |
5,889 | — | — | — | 5,889 | |||||||||||||||
| Research & development |
15,952 | 10,477 | 361 | — | 26,790 | |||||||||||||||
| Selling, general and administration |
15,645 | 61,101 | 12,492 | — | 89,238 | |||||||||||||||
| Depreciation and amortization |
4,186 | 28,530 | 1,029 | — | 33,745 | |||||||||||||||
| Write-down of assets held for sale |
700 | 750 | — | — | 1,450 | |||||||||||||||
| Write-down of property, plant and equipment |
1,734 | 3,045 | — | — | 4,779 | |||||||||||||||
| Write-down of intangible assets |
— | 2,814 | — | — | 2,814 | |||||||||||||||
| Escrow settlement recovery |
— | (4,710 | ) | — | — | (4,710 | ) | |||||||||||||
| 44,080 | 176,575 | 63,412 | (17,768 | ) | 266,299 | |||||||||||||||
| Operating income (loss) |
(13,367 | ) | (12,350 | ) | 6,720 | (1,060 | ) | (20,057 | ) | |||||||||||
| Other income (expense) |
||||||||||||||||||||
| Debt restructuring charges |
(9,277 | ) | — | — | — | (9,277 | ) | |||||||||||||
| Foreign exchange gain (loss) |
56,840 | (57,633 | ) | 1,804 | — | 1,011 | ||||||||||||||
| Other (expense) income |
(246,320 | ) | (21,578 | ) | 6,249 | 261,078 | (571 | ) | ||||||||||||
| Interest expense on long-term debt |
(38,439 | ) | (1,635 | ) | (184 | ) | — | (40,258 | ) | |||||||||||
| Write-down of deferred financing |
(291 | ) | — | — | — | (291 | ) | |||||||||||||
| Write-down of investment |
(561 | ) | — | (736 | ) | — | (1,297 | ) | ||||||||||||
| Gain on loan settlement |
(34,401 | ) | (5,619 | ) | 41,900 | — | 1,880 | |||||||||||||
| Gain on disposal of Laguna Hills manufacturing facility |
— | 2,005 | — | — | 2,005 | |||||||||||||||
| Total other expenses |
(272,449 | ) | (84,460 | ) | 49,033 | 261,078 | (46,798 | ) | ||||||||||||
| Income (loss) before income taxes |
(285,816 | ) | (96,810 | ) | 55,753 | 260,018 | (66,855 | ) | ||||||||||||
| Income tax (recovery) expense |
(171 | ) | (3,388 | ) | 3,515 | — | (44 | ) | ||||||||||||
| Income (loss) from operations |
(285,645 | ) | (93,422 | ) | 52,238 | 260,018 | (66,811 | ) | ||||||||||||
| Equity in subsidiaries |
218,834 | 12,479 | — | (231,313 | ) | — | ||||||||||||||
| Net income (loss) |
$ | (66,811 | ) | $ | (80,943 | ) | $ | 52,238 | $ | 28,705 | $ | (66,811 | ) | |||||||
F-44
Table of Contents
Condensed Consolidated Statement of Operations
Year ended December 31, 2009
USD (in ‘000s)
| Parent Company Angiotech Pharmaceuticals, Inc. |
Guarantors Subsidiaries |
Non Guarantors Subsidiaries |
Consolidating Adjustments |
Consolidated Total |
||||||||||||||||
| REVENUE |
||||||||||||||||||||
| Product sales, net |
$ | — | $ | 153,364 | $ | 58,887 | $ | (20,300 | ) | $ | 191,951 | |||||||||
| Royalty revenue |
57,420 | 3,414 | 1,337 | — | 62,171 | |||||||||||||||
| License fees |
— | 356 | 25,200 | — | $ | 25,556 | ||||||||||||||
| 57,420 | 157,134 | 85,424 | (20,300 | ) | 279,678 | |||||||||||||||
| EXPENSES |
||||||||||||||||||||
| Cost of products sold |
85 | 82,905 | 42,488 | (20,862 | ) | 104,616 | ||||||||||||||
| License and royalty fees |
10,421 | — | 10 | — | 10,431 | |||||||||||||||
| Research & development |
14,491 | 8,827 | 383 | — | 23,701 | |||||||||||||||
| Intercompany R&D charges |
2,078 | (2,078 | ) | — | — | — | ||||||||||||||
| Selling, general and administration |
16,375 | 53,334 | 11,795 | — | 81,504 | |||||||||||||||
| Depreciation and amortization |
4,535 | 24,527 | 4,189 | — | 33,251 | |||||||||||||||
| Write-down of assets held for sale |
2,290 | 800 | — | — | 3,090 | |||||||||||||||
| 50,275 | 168,315 | 58,865 | (20,862 | ) | 256,593 | |||||||||||||||
| Operating income (loss) |
7,145 | (11,181 | ) | 26,560 | 562 | 23,085 | ||||||||||||||
| Other income (expense) |
||||||||||||||||||||
| Foreign exchange gain (loss) |
7,393 | (2,100 | ) | (6,905 | ) | — | (1,612 | ) | ||||||||||||
| Investment and other income |
(78,446 | ) | (245 | ) | 111 | 78,958 | 378 | |||||||||||||
| Interest expense on long-term debt |
58,755 | (96,039 | ) | (755 | ) | — | (38,039 | ) | ||||||||||||
| Write-down of deferred financing charges |
— | (643 | ) | — | — | (643 | ) | |||||||||||||
| Management fees |
(47 | ) | 47 | — | — | — | ||||||||||||||
| Total other expenses |
(12,345 | ) | (98,980 | ) | (7,549 | ) | 78,958 | (39,916 | ) | |||||||||||
| Income (loss) before income taxes |
(5,200 | ) | (110,161 | ) | 19,011 | 79,520 | (16,831 | ) | ||||||||||||
| Income tax (recovery) expense |
129 | (4,099 | ) | 3,975 | 6,032 | 6,037 | ||||||||||||||
| Income (loss) from operations |
(5,329 | ) | (106,062 | ) | 15,036 | 73,488 | (22,868 | ) | ||||||||||||
| Equity in subsidiaries |
(17,539 | ) | 257 | — | 17,282 | — | ||||||||||||||
| Net income (loss) |
$ | (22,868 | ) | $ | (105,805 | ) | $ | 15,036 | $ | 90,769 | $ | (22,868 | ) | |||||||
F-45
Table of Contents
Condensed Consolidated Statement of Operations
Year ended December 31, 2008
USD (in ‘000s)
| Parent Company Angiotech Pharmaceuticals, Inc. |
Guarantors Subsidiaries |
Non Guarantors Subsidiaries |
Consolidating Adjustments |
Consolidated Total |
||||||||||||||||
| REVENUE |
||||||||||||||||||||
| Royalty revenue |
$ | 84,079 | $ | 2,426 | $ | 5,041 | $ | — | $ | 91,546 | ||||||||||
| Product sales, net |
— | 143,578 | 79,012 | (31,774 | ) | 190,816 | ||||||||||||||
| License fees |
— | 389 | 1,000 | (479 | ) | 910 | ||||||||||||||
| 84,079 | 146,393 | 85,053 | (32,253 | ) | 283,272 | |||||||||||||||
| EXPENSES |
||||||||||||||||||||
| License and royalty fees |
14,250 | 8 | — | — | 14,258 | |||||||||||||||
| Cost of products sold |
— | 75,631 | 56,505 | (31,084 | ) | 101,052 | ||||||||||||||
| Research & development |
32,442 | 19,621 | 1,129 | — | 53,192 | |||||||||||||||
| Intercompany R&D charges |
5,329 | (6,405 | ) | 753 | 323 | — | ||||||||||||||
| Selling, general and administration |
22,969 | 57,764 | 17,750 | — | 98,483 | |||||||||||||||
| Depreciation and amortization |
4,806 | 25,445 | 3,747 | — | 33,998 | |||||||||||||||
| In-process research and development |
— | 2,500 | — | — | 2,500 | |||||||||||||||
| Write-down of assets held for sale |
— | 1,283 | — | — | 1,283 | |||||||||||||||
| Write-down of goodwill |
— | 537,531 | 112,154 | — | 649,685 | |||||||||||||||
| 79,796 | 713,378 | 192,038 | (30,761 | ) | 954,451 | |||||||||||||||
| Operating loss |
4,283 | (566,984 | ) | (106,985 | ) | (1,492 | ) | (671,179 | ) | |||||||||||
| Other income (expense) |
||||||||||||||||||||
| Foreign exchange gain (loss) |
49,985 | 2,284 | (51,729 | ) | — | 540 | ||||||||||||||
| Investment and other income |
(9,419 | ) | (438 | ) | 11,049 | — | 1,192 | |||||||||||||
| Interest expense on long-term debt |
(32,251 | ) | (20,302 | ) | 8,063 | — | (44,490 | ) | ||||||||||||
| Write-down and other deferred financing charges |
(16,531 | ) | — | (13 | ) | — | (16,544 | ) | ||||||||||||
| Write-down/loss on redemption of investments |
(23,583 | ) | — | (4 | ) | — | (23,587 | ) | ||||||||||||
| Management fees |
(2,597 | ) | 2,384 | (109 | ) | 322 | — | |||||||||||||
| Total other expenses |
(34,396 | ) | (16,072 | ) | (32,743 | ) | 322 | (82,889 | ) | |||||||||||
| Loss from continuing operations before income taxes |
(30,113 | ) | (583,057 | ) | (139,728 | ) | (1,170 | ) | (754,068 | ) | ||||||||||
| Income tax expense (recovery) |
(3,176 | ) | (11,088 | ) | 1,498 | (126 | ) | (12,892 | ) | |||||||||||
| Loss from continuing operations |
(26,937 | ) | (571,968 | ) | (141,226 | ) | (1,044 | ) | (741,176 | ) | ||||||||||
| Loss from discontinued operations, net of income taxes |
— | — | — | — | — | |||||||||||||||
| Equity in subsidiaries |
(714,239 | ) | (51,062 | ) | — | 765,301 | — | |||||||||||||
| Net income (loss) |
$ | (741,176 | ) | $ | (623,030 | ) | $ | (141,226 | ) | $ | 764,257 | $ | (741,176 | ) | ||||||
F-46
Table of Contents
Condensed Consolidating Statement of Cash Flows
Year end December 31, 2010
USD (in ‘000s)
| Parent Company Angiotech Pharmaceuticals, Inc. |
Guarantors Subsidiaries |
Non Guarantors Subsidiaries |
Consolidating Adjustments |
Consolidated Total |
||||||||||||||||
| OPERATING ACTIVITIES: |
||||||||||||||||||||
| Cash used in operating activities |
$ | (15,931 | ) | $ | (13,920 | ) | $ | 8,242 | $ | — | (21,609 | ) | ||||||||
| INVESTING ACTIVITIES: |
||||||||||||||||||||
| Purchase of property, plant and equipment |
(3,588 | ) | (2,099 | ) | (39 | ) | — | (5,726 | ) | |||||||||||
| Proceeds from disposition of property, plant and equipment |
— | 1,272 | — | — | 1,272 | |||||||||||||||
| Proceeds from disposition of intangible assets |
— | 2,005 | — | — | 2,005 | |||||||||||||||
| Loans advanced |
(1,015 | ) | — | — | — | (1,015 | ) | |||||||||||||
| Asset acquisition costs |
(231 | ) | — | — | — | (231 | ) | |||||||||||||
| Cash used in investing activities |
(4,834 | ) | 1,178 | (39 | ) | — | (3,695 | ) | ||||||||||||
| FINANCING ACTIVITIES: |
||||||||||||||||||||
| Deferred financing charges and costs |
(669 | ) | — | — | — | (669 | ) | |||||||||||||
| Dividends received / (paid) |
13,170 | (13,170 | ) | — | — | |||||||||||||||
| Proceeds from stock options exercised |
11 | — | — | — | 11 | |||||||||||||||
| Advances under credit facility |
— | 10,000 | — | — | 10,000 | |||||||||||||||
| Intercompany notes payable/receivable |
12,001 | (12,001 | ) | — | — | |||||||||||||||
| Cash provided by (used in) financing activities |
11,343 | 11,169 | (13,170 | ) | — | 9,342 | ||||||||||||||
| Effect of exchange rate changes on cash and cash equivalents |
— | 1 | (266 | ) | — | (265 | ) | |||||||||||||
| Net (decrease) increase in cash and cash equivalents |
(9,422 | ) | (1,572 | ) | (5,233 | ) | — | (16,227 | ) | |||||||||||
| Cash and cash equivalents, beginning of period |
20,602 | 16,912 | 12,028 | — | 49,542 | |||||||||||||||
| Cash and cash equivalents, end of period |
$ | 11,180 | $ | 15,340 | $ | 6,795 | $ | — | $ | 33,315 | ||||||||||
F-47
Table of Contents
Condensed Consolidating Statement of Cash Flows
Year end December 31, 2009
USD (in ‘000s)
| Parent Company Angiotech Pharmaceuticals, Inc. |
Guarantors Subsidiaries |
Non Guarantors Subsidiaries |
Consolidating Adjustments |
Consolidated Total |
||||||||||||||||
| OPERATING ACTIVITIES: |
||||||||||||||||||||
| Cash used in operating activities |
$ | (1,379 | ) | $ | 43,966 | $ | (20,891 | ) | $ | 21,696 | ||||||||||
| INVESTING ACTIVITIES: |
||||||||||||||||||||
| Purchase of property, plant and equipment |
(1,581 | ) | (2,258 | ) | (625 | ) | — | (4,464 | ) | |||||||||||
| Purchase of intangible assets |
— | (2,500 | ) | — | — | (2,500 | ) | |||||||||||||
| Loans advanced |
(1,200 | ) | (1,200 | ) | ||||||||||||||||
| Other |
12 | 334 | (94 | ) | — | 252 | ||||||||||||||
| Cash used in investing activities |
(2,769 | ) | (4,424 | ) | (719 | ) | — | (7,912 | ) | |||||||||||
| FINANCING ACTIVITIES: |
||||||||||||||||||||
| Deferred financing charges and costs |
(3,403 | ) | — | — | — | (3,403 | ) | |||||||||||||
| Dividends received / (paid) |
24,800 | (21,800 | ) | (3,000 | ) | — | — | |||||||||||||
| Intercompany notes payable/receivable |
(6,830 | ) | (7,418 | ) | 14,248 | — | — | |||||||||||||
| Proceeds from stock options exercised |
3 | — | — | — | 3 | |||||||||||||||
| Cash provided by (used in) financing activities |
14,570 | (29,218 | ) | 11,248 | — | (3,400 | ) | |||||||||||||
| Effect of exchange rate changes on cash and cash equivalents |
— | 16 | 190 | — | 206 | |||||||||||||||
| Net (decrease) increase in cash and cash equivalents |
10,422 | 10,340 | (10,172 | ) | — | 10,590 | ||||||||||||||
| Cash and cash equivalents, beginning of period |
10,180 | 6,572 | 22,200 | — | 38,952 | |||||||||||||||
| Cash and cash equivalents, end of period |
$ | 20,602 | $ | 16,912 | $ | 12,028 | $ | — | $ | 49,542 | ||||||||||
F-48
Table of Contents
Condensed Consolidating Statement of Cash Flows
Year ended December 31, 2008
USD (in ‘000s)
| Parent Company Angiotech Pharmaceuticals, Inc. |
Guarantors Subsidiaries |
Non Guarantors Subsidiaries |
Consolidating Adjustments |
Consolidated Total |
||||||||||||||||
| OPERATING ACTIVITIES: |
||||||||||||||||||||
| Cash (used in) provided by operating activities |
$ | (55,123 | ) | $ | (15,046 | ) | $ | 32,162 | $ | — | $ | (38,007 | ) | |||||||
| INVESTING ACTIVITIES: |
||||||||||||||||||||
| Proceeds from short-term investments |
2,756 | — | — | — | 2,756 | |||||||||||||||
| Purchase of property, plant and equipment |
(1,005 | ) | (4,942 | ) | (1,722 | ) | — | (7,669 | ) | |||||||||||
| Purchase of intangible assets |
— | (1,000 | ) | — | — | (1,000 | ) | |||||||||||||
| In-process research and development |
— | (2,500 | ) | — | — | (2,500 | ) | |||||||||||||
| Other |
236 | 28 | (90 | ) | — | 174 | ||||||||||||||
| Cash provided by (used in) investing activities |
1,987 | (8,414 | ) | (1,812 | ) | — | (8,239 | ) | ||||||||||||
| FINANCING ACTIVITIES: |
||||||||||||||||||||
| Deferred financing costs on long-term obligations |
(4,485 | ) | — | (14 | ) | — | (4,499 | ) | ||||||||||||
| Proceeds from stock options exercised |
121 | — | — | — | 121 | |||||||||||||||
| Dividends received / (paid) |
20,000 | 12,344 | (32,344 | ) | — | — | ||||||||||||||
| Intercompany notes payable/receivable |
23,891 | (2,596 | ) | (21,295 | ) | — | — | |||||||||||||
| Cash provided by (used in) financing activities |
39,527 | 9,748 | (53,653 | ) | — | (4,378 | ) | |||||||||||||
| Effect of exchange rate changes on cash and cash equivalents |
— | (51 | ) | (1,699 | ) | — | (1,750 | ) | ||||||||||||
| Net increase (decrease) in cash and cash equivalents |
(13,609 | ) | (13,763 | ) | (25,002 | ) | — | (52,374 | ) | |||||||||||
| Cash and cash equivalents, beginning of year |
23,790 | 20,334 | 47,202 | — | 91,326 | |||||||||||||||
| Cash and cash equivalents, end of year |
$ | 10,181 | $ | 6,571 | $ | 22,200 | $ | — | $ | 38,952 | ||||||||||
F-49
Table of Contents
26. SUBSEQUENT EVENTS
Creditor Protection Proceedings
In connection with the Company’s obligations under the RSA, on January 28, 2011 the Angiotech Entities commenced Creditor Protection Proceedings to effectuate an in-court Recapitalization Transaction as contemplated by the RSA (see note 1 and 16 above for more information). The Recapitalization Transaction would eliminate the $250 million Subordinated Notes and related interest obligations in exchange for new common shares in the Company thereby providing significant improvements to the Company’s credit ratios, liquidity and financial flexibility. In addition, all of the Company’s existing common shares and options to acquire common shares would be cancelled without any payment as consideration therefor.
Amendments to the RSA and FRN Support Agreement
Fifth Amendment to the FRN Support Agreement
On February 7, 2011, the Company, certain of its subsidiaries and the Consenting FRN Holders entered into the Fifth Agreement (the “Fifth FRN Extension Agreement”) to amend the FRN Support Agreement. The Fifth FRN Extension Agreement extends a deadline outlined in the previously announced FRN Support Agreement dated October 29, 2010. The Fifth FRN Extension Agreement extended to February 10, 2011 the deadline by which the Company must commence the FRN Exchange Offer to exchange the Existing Floating Rate Notes for New Floating Rate Notes.
Fifth Amendment to the RSA
On February 7, 2011, the Company and certain of its subsidiaries entered into the fifth agreement to amend the RSA (the “Fifth RSA Extension Agreement”) with the Consenting Noteholders. The Fifth RSA Extension Agreement extends certain deadlines outlined in the previously announced RSA dated October 29, 2010. Under the Fifth RSA Extension Agreement, the deadline by which the Company must commence the FRN Exchange Offer was extended to February 10, 2011 and the deadline by which the Company must obtain the Meeting Order and the Claims Procedure Order was extended to February 17, 2011.
Sixth Amendment to the FRN Support Agreement
On February 22, 2011, the Company entered into the sixth amendment (“Sixth Amendment”) to the FRN Support Agreement in connection with the proposed FRN Exchange Offer launched on February 10, 2011. Under the terms of the Sixth Amendment, the Company agreed to amend the FRN Support Agreement to provide that, among other things: (i) the New Floating Rate Notes to be issued in connection with the FRN Exchange Offer would accrue interest subject to a LIBOR floor of 1.25%; (ii) the FRN Indenture would impose restrictions on the Company’s ability to apply net proceeds received from certain asset sales; and (iii) the Consenting FRN Holders party to the FRN Support Agreement would not oppose the Recapitalization Transaction currently contemplated by the Company. Concurrent with the execution of the Sixth Amendment, additional holders of Existing Floating Rate Notes executed joinders to the FRN Support Agreement, thereby bringing the aggregate level of support for the FRN Exchange Offer to approximately 89% of the outstanding Existing Floating Rate Notes.
Launch of the FRN Exchange Offer
In connection with the Recapitalization Transaction, on February 10, 2011 the Company commenced the FRN Exchange Offer pursuant to the terms of the FRN Support Agreement. Refer to note 1 and 16 for more information.
On March 4, 2011 the Company announced that it had extended its FRN Exchange Offer offer to 11:59 p.m. New York City time on March 30, 2011. The FRN Exchange Offer was originally scheduled to expire at 11:59 p.m. New York City time on March 17, 2011.
The Company also announced that in connection with the Sixth Amendment it delivered a supplement (the “Supplement”) to the offering memorandum relating to the FRN Exchange Offer, dated February 10, 2011, to holders of Existing Floating Rate Notes. The Supplement amends certain provisions of the FRN Exchange Offer to give effect to the Sixth Amendment and provides that, among other things: (i) the New Floating Rate Notes will accrue interest at the London Interbank Offered Rate (“LIBOR”) plus 3.75%, subject to a LIBOR floor of 1.25% and (ii) certain covenants relating to asset sales and the definition of asset sale will be modified in the indenture that governs the New Floating Rate Notes.
As of March 14, 2011, approximately 89% of the aggregate principal amount outstanding of the Existing Floating Rate Notes have agreed to the terms of the FRN Exchange Offer.
F-50
Table of Contents
DIP Facility
In connection with the RSA and the Company’s Creditor Protection Proceedings, on February 7, 2011, the Company and certain of its subsidiaries entered into a definitive agreement with Wells Fargo to secure a DIP Facility. On February 25, 2011, following entry of the February 22, 2011 Recognition Order the loans under the DIP Facility were made available to the Company for borrowing. The DIP Facility will provide liquidity for working capital, general corporate purposes and expenses while it implements the CCAA Plan. The DIP Facility permits the Company to make revolving credit loans and provide letters of credit in an aggregate principal amount (including the face amount of any letters of credit) of up to $28.0 million (refer to note 1 and 16 for more information).
QSR Holdings, Inc. Arbitration and Litigation
On October 4, 2010, the Company was notified that QSR filed an Arbitration Demand with the American Arbitration Association naming the Company, QMI and Angiotech US as respondents (collectively, the “Respondents”). The Arbitration Demand alleged that the Respondents failed to satisfy certain obligations under the Merger Agreement (notably including the obligations to pay earn out payments and the orthopedic milestone payment), and sought either direct monetary damages or, in the alternative, extension of the Earn Out period for a sixth year as more fully set forth in the Merger Agreement. In addition, QSR commenced the “Federal Litigation. The Complaint in the Federal Litigation alleged, among other items, that the Respondents: (a) breached certain contractual obligations under the Merger Agreement; (b) made certain misrepresentations or omissions during the initial negotiation of the Merger Agreement; and (c) tortiously interfered with the Merger Agreement. QSR sought damages in an unstated amount together with punitive damages and/or attorneys fees to the extent allowed by law. On October 15, 2010, the Respondents moved to dismiss the Complaint in part and to transfer venue to federal court in Washington State.
Subsequent to the initiation of the Federal Litigation and the delivery of the Arbitration Demand, representatives of Angiotech and QSR engaged in substantial settlement discussions. Those discussions culminated the Settlement Agreement, dated as of January 27, 2011, resolving, among other things, any and all claims that the parties may have arising out of the Merger Agreement (including, without limitation, all claims, counterclaims or defenses that were or could have been asserted in the Arbitration Demand and the Federal Litigation) in exchange for a payment of $6.0 million (the “Settlement Amount”) by Angiotech US. $2.0 million of the Settlement Amount is payable on the earliest of (i) 10 business days after the implementation of the Recapitalization Transaction contemplated by the RSA (the “Implementation Date”) and (ii) May 31, 2011. The remainder of the Settlement Amount is payable in 24 equal monthly installments beginning on the earlier of (i) 30 days after the Implementation Date; and (ii) July 31, 2011 (subject to reduction in the event of pre-payment as more fully set forth in the Settlement Agreement). The Settlement Agreement further provides for complete and mutual releases between the parties, as more fully set forth therein. Pursuant to the Settlement Agreement QSR has irrevocably dismissed the Federal Litigation and the Arbitration Demand with prejudice. As at December 31, 2010, the Company recorded $0.6 million of litigation expense, related to these legal proceedings. In accordance with the guidance on contingent consideration under FAS 141, which pertains to business combinations arising prior to December 15, 2008, the Company is currently assessing the impact of the remaining Settlement Amount on its 2011 first quarter results.
Rex Arbitration and Litigation Settlement and Agreement Termination
In light of the Company’s recent liquidity and capital constraints, on November 11, 2010, the Company informed Rex that it would not be able to continue selling the Option IVC Filter under the existing terms of the Option Agreement. Subsequently, on November 18, 2010, Rex initiated an arbitration hearing against the Company alleging that it breached the Agreement. Rex sought monetary damages in excess of $3.0 million, including costs, fees and expenses incurred in connection with the arbitration proceeding. On December 2, 2010, Rex obtained a temporary restraining order and preliminary injunction against the Company from the United States District Court for the Southern District of New York requiring the Company to honor its obligations under the Option Agreement for a period of 180 days or until the arbitration was concluded. On January 24, 2011, the Company received a termination notice from Rex indicating that the contract would cease on April 24, 2011.
On February 16, 2011, the Company entered into the Rex Settlement Agreement, which provides for the full and final settlement and dismissal of all claims arising under the Option Agreement. The Rex Settlement Agreement includes the following principal terms: (i) the termination of the Option Agreement at March 31, 2011 or at an earlier date if so elected by Rex, upon which Angiotech US will no longer market, sell and distribute the Option IVC Filter; (ii) a payment in the amount of $1.5 million to be made to Rex within five business date of the of the effective date of the Rex Settlement Agreement, which is the first business day following receipt of approval of the Canadian Court to make such payment in respect of Angiotech’s ongoing CCAA proceedings, in full and final settlement of all claims, all royalties due under the Option Agreement related to sales of the Option IVC Filter recorded by Angiotech US prior to January 1, 2011, and all milestone payments due or that may come due to Rex under the Rex Agreement now or in the future; and (iii) the delivery to Rex of certain materials relating to the marketing and sale of the Option IVC Filter. The Rex Settlement Agreement became effective on March 10, 2011 upon the Canadian Court granting an order approving the authority of Angiotech US to enter into the Rex Settlement Agreement. As at December 31, 2010, the Company recorded a $0.9 million inventory write-down and a $1.1 million intangible asset write-down in connection with the termination of the Option Agreement. In accordance with ASC No. 855 on Subsequent Events, the Company is currently assessing the impact of the Rex Settlement Agreement on its 2011 first quarter results.
F-51
Table of Contents
NASDAQ and TSX Delisting
On July 6, 2010, the Company received a notice from NASDAQ stating that the bid price for the Company’s common shares had closed below the required minimum of $1.00 per share for the previous 30 consecutive business days, and that in accordance with the applicable NASDAQ listing rule, Angiotech had a grace period of 180 calendar days, until January 3, 2011, to regain compliance with the applicable NASDAQ listing rule. On January 4, 2011, the Company received a letter from NASDAQ confirming that the Company’s common shares would be delisted from NASDAQ as a result of its failure to regain compliance with the applicable NASDAQ listing rule. Accordingly, the Company’s common shares were delisted from NASDAQ at the opening of business on January 13, 2011. A Form 25-NSE was filed with the SEC on January 21, 2011, removing the Company’s common shares from listing on NASDAQ.
On January 28, 2011 we received notice from the Toronto Stock Exchange (the “TSX”) stating that the TSX would be reviewing the eligibility for continued listing on TSX of our securities pursuant to Part VII of The Toronto Stock Exchange Company Manual. In addition, the TSX advised that our securities would be suspended from trading on the TSX until further notice from the TSX. On February 1, 2011, we received further notice from the TSX that our common shares would be delisted from the TSX effective at the close of market on March 3, 2011. The delisting was imposed as a result of our not meeting the continued listing requirements of the TSX. The TSX also advised that trading in our common shares would remain suspended until the time of delisting. We did not appeal the TSX’s decision to delist our common shares and our common shares were delisted from the TSX at the close of market on March 3, 2011.
F-52
Table of Contents
VALUATION AND QUALIFYING ACCOUNTS
Years Ended December 31, 2010, 2009 and 2008
(in thousands of U.S. dollars)
| Balance at Beginning of Year |
Additions | Deductions | Balance at End of Year |
|||||||||||||||||
| Charged to Costs and Expenses |
Charged to Other accounts |
|||||||||||||||||||
| Deferred Income Tax Valuation Allowance: |
||||||||||||||||||||
| Year ended December 31, 2010 |
$ | 95,159 | 11,435 | — | — | $ | 106,594 | |||||||||||||
| Year ended December 31, 2009 |
91,986 | — | — | $ | 3,173 | $ | 95,159 | |||||||||||||
| Year ended December 31, 2008 |
42,005 | 55,514 | (579 | ) | — | 96,940 | ||||||||||||||
| Balance at Beginning of Year |
Balance at End of Year |
|||||||
| Accounts Receivable Allowance: |
||||||||
| Year ended December 31, 2010 |
$ | 471 | $ | 2,435 | ||||
| Year ended December 31, 2009 |
$ | 270 | $ | 471 | ||||
| Year ended December 31, 2008 |
214 | 270 | ||||||
F-53