Attached files
| file | filename |
|---|---|
| EX-23 - EXHIBIT 23.1 - POLYMEDIX, INC | exhibit23.htm |
| EX-32 - EXHIBIT 32 - POLYMEDIX, INC | exhibit32_123110.htm |
| EX-31 - EXHIBIT 31.2 - POLYMEDIX, INC | exhibit312_123110.htm |
| EX-31 - EXHIBIT 31.1 - POLYMEDIX, INC | exhibit311_123110.htm |
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
x Annual Report Pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934.
For the Fiscal Year Ended December 31, 2010
o Transition Report Pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934
for The Transition Period From __________To __________
Commission file number: 000-51895
![]()
POLYMEDIX, INC.
(Exact name of Registrant as specified in its charter)
|
Delaware |
|
27-0125925 |
|
(State or other jurisdiction of incorporation or organization) |
|
(I.R.S. Employer Identification No.) |
|
170 N. Radnor-Chester Road; Suite 300 Radnor, PA 19087 (Address of principal executive offices including zip code) |
|
|
|
(484) 598-2332 (Registrant’s telephone number, including area code) |
Securities registered pursuant to Section 12(b) of the Act: None
Securities registered pursuant to Section 12(g) of the Act: Common Stock, $0.001 par value
Indicate by check mark if the Registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. o Yes x No
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. o Yes x No
Indicate by check whether the registrant: (1) filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirement for the past 90 days. xYes o No
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). oYes o No
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of Registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendments to this Form 10-K. o
Indicate by check mark whether the Registrant is a large accelerated filer, an accelerated filer, or a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer,” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer o Accelerated filer x Non-accelerated filer o Smaller reporting company o
(Do not check if a smaller reporting company)
Indicate by check mark whether the Registrant is a shell company (as defined in Rule 12b-2 of the Act). o Yes x No
The aggregate market value of the registrant’s Common Stock, $0.001 par value per share, held by non-affiliates of the registrant on June 30, 2010 was approximately $77,135,980 (based on the closing sales price of the registrant’s Common Stock on that date ($1.00)).
The number of shares of the issuer’s Common Stock outstanding as of March 7, 2011 was 80,999,610.
DOCUMENTS INCORPORATED BY REFERENCE
None.
TABLE OF CONTENTS
Page
-1-
Note Regarding Forward-Looking Statements.
This report contains forward-looking statements within the meaning of Section 27A of the Securities Act, as amended, Section 21E of the Securities Exchange Act of 1934, as amended, and the Private Securities Litigation Reform Act of 1995, that are subject to risks and uncertainties. All statements other than statements of historical facts contained in this report, including statements regarding our financial condition, operations, plans, objectives, goals, business strategies, future events, capital expenditures, future results, our competitive strengths, and the trends in our industry are forward-looking statements. The words “believe,” “may,” “could,” “estimate,” “continue,” “anticipate,” “intend,” “should,” “plan,” “expect,” “appear,” “future,” “likely,” “probably,” “suggest,” “goal,” “potential” and similar expressions, as they relate to us, are intended to identify forward-looking statements.
Forward-looking statements reflect only our current expectations. In any forward-looking statement, where we express an expectation or belief as to future results or events, such expectation or belief is expressed in good faith as of the date of such statement and believed to have a reasonable basis, but there can be no assurance that the statement of expectation or belief will be achieved or accomplished. Our actual results, performance or achievements could differ materially from those expressed in or inferred from the forward-looking statements due to a number of uncertainties, many of which are unforeseen, including:
Ø our need for, and the availability of, substantial capital in the future to fund our operations and planned clinical trials;
Ø the conditions in the capital markets and the biopharmaceutical industry that make raising capital or entering into strategic arrangements difficult and expensive;
Ø the timing of our product development and evaluation;
Ø the timing and magnitude of expenditures we may incur in connection with our ongoing research and development activities;
Ø the results of our preclinical and clinical trials, including regulatory approvals necessary for advancement and continuation of our development programs;
Ø the maintenance of our existing licenses with the University of Pennsylvania (Penn) and the University of Massachusetts (UMass);
Ø the success, timing and financial consequences of our formation of new business relationships and alliances; and
Ø the timing and volume of sales of products for which we may obtain marketing approval.
In addition, you should refer to the “Risk Factors” section of this report for a discussion of other factors that may cause our actual results to differ materially from those implied by our forward-looking statements. Because of these factors or others, the forward-looking statements in this report may not prove to be accurate. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time frame, if at all. Accordingly, you should not place undue reliance on these forward-looking statements. All subsequent written and oral forward looking statements attributable to us or the persons acting on our behalf are expressly qualified in their entirety by the applicable cautionary statements. We undertake no obligation to update any forward looking statements, whether as a result of new information, future events or otherwise, except as may be required by applicable law or regulation.
-2-
PART I
In this annual report on Form 10-K, "PolyMedix," "we," "us" and "our" refer to PolyMedix, Inc. and its wholly owned subsidiary PolyMedix Pharmaceuticals, Inc. and "Common Stock" refers to PolyMedix’s Common Stock, par value $0.001 per share.
This Business section outlines both how we perceive the current status of our business initiatives as well as our plans to further develop our business and product portfolio. We do not currently have the funding resources necessary to carry out all of our planned operating activities. We believe that our current cash and investment balances will fund our ongoing Phase 2 studies for PMX-30063 and PMX-60056 and can fund our operations for at least the next twelve months. We plan to seek additional funding in one or more equity or debt financings, among other sources. However, if we are unable to secure adequate additional funding, we will delay, scale-back or eliminate certain of our future research, drug discovery or development activities or certain other aspects of our operations and our business until such time as we are successful in securing adequate additional funding. In the absence of adequate additional financing, we believe that we have the ability to scale our operations such that our current cash and investment balances can fund our operations for at least the next twelve months.
SUMMARY:
We are a biotechnology company focused on developing small synthetic molecule compounds that mimic the activity of large natural protein molecules, for the treatment of infectious diseases and acute cardiovascular disorders. Using our proprietary computational drug design technology, we have created novel defensin mimetic antibiotic compounds and heparin antagonist compounds (or “heptagonists”), and other drug compounds intended for human therapeutic use. The following chart illustrates the current stage of development of our internally developed pipeline along with planned development timings for our two lead programs that are in clinical development:
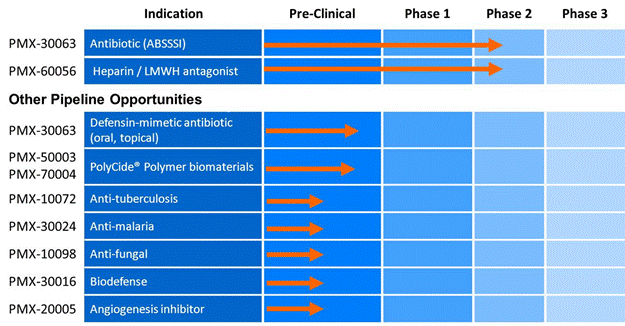

Note: Other pipeline opportunities are primarily funded through government grants and contracts.
1
CLINICAL PROGRAMS:
Defensin Mimetic Antibiotic: PMX-30063
Market Opportunity
Worldwide approximately 49 million patients are treated for infections annually, with more than 7 million patients hospitalized for bacterial infections. According to DataMonitor, a provider of healthcare market analysis data, in 2008 the global and U.S. markets for systemic anti-bacterials were approximately $38.1 billion and $9.7 billion, respectively. A 2007 Public Health Report issued by the Centers for Disease Control (CDC) estimates that in 2002 there were approximately 1.7 million healthcare acquired infections (HAI’s) in U.S. hospitals (adjusted to include federal facilities) which accounted for 99,000 deaths. Direct expenditures for healthcare associated infections cost U.S. hospitals approximately $45 billion in 2007, according to a 2009 analysis done by the CDC.
The public health threat posed by the increasing spread of bacterial resistance to mainstay antibiotics, including antibiotics that until recently were thought of as treatments of last resort, continues to be a major worldwide concern. The World Health Organization (WHO) has identified antibiotic resistance as “one of the three greatest threats to human health.” A February 2008 report from the Association for Professionals in Infection Control and Epidemiology estimates that 70% of infections may now be resistant to antibiotics. A recent analysis of antibiotic-resistant infection data conducted at Chicago Cook County Hospital suggested that the direct and indirect economic costs of antibiotic resistance to the U.S. health care system are in excess of $20 billion annually, more than $35 billion in societal costs. As drug‑resistance continues to spread, the need for potent novel antibiotic drugs increases.
There is a significant need for new antibiotics to treat serious drug-resistant Gram-positive infections, such as methicillin-resistant Staphylococcus aureus (MRSA). A recent CDC-supported study published in the Journal of the American Medical Association estimated that invasive MRSA infects more than 94,000 people and kills nearly 19,000 people annually in the U.S. Invasive MRSA infections represent only one segment of the greater MRSA problem in the U.S. The market for MRSA antibiotics is growing at a compound annual growth rate of 18% with U.S. sales nearly doubling from $778 million in 2005 to $1.5 billion in 2009.
The portion of the antibiotic market that we initially intend to target is the North American acute‑care hospital market. This market includes intravenous or subcutaneously infused products that are administered or prescribed in the hospital. According to DataMonitor, the size of this market in 2006 was valued at $3.7 billion.
Host Defense Proteins
Our small molecule antibiotics mimic the activity of host defense proteins. Host defense proteins are part of the innate immune system. In the human body, host defense proteins represent a first line of defense against bacterial attack and primarily exist in the respiratory tract, the urogenital tract, the gastrointestinal track and the epidermal tissues under the skin, all locations where microbial pathogens first enter the human body. Host defense proteins act rapidly against bacteria, unlike other parts of the immune system that take longer to work.
Host defense proteins use a simple, but effective method for killing bacteria by targeting bacterial membranes and creating an instability of the cellular contents and membrane, which results in the killing of the bacterial cell.
Antibiotics on the market today generally act on specific molecular targets in bacteria and many must enter the bacteria cell to work. Bacterial cells can become resistant to antibiotics through:
Ø genetic mutations that modify the molecular targets themselves, rendering them invulnerable to the antibiotic in question; or
Ø metabolic responses that cause the cell to pump out foreign agents, preventing the antibiotics from accessing the molecular targets.
By contrast, host defense proteins physically disrupt the cell from the outside. The mechanism of action of the host defense proteins makes it difficult for bacteria to develop resistance because of several reasons:
Ø they do not have to enter the bacterial cell to work;
Ø they act quickly, killing bacteria within minutes of exposure, thereby limiting the bacterial response time; and
Ø in order to develop effective defenses, the bacteria would have to alter the structure of its cell membrane, which is a highly complex multi‑step response that would likely reduce the ability of the newly mutated bacteria to grow and survive in a natural environment due to changes in membrane transport of essential nutrients and wastes.
2
It has been documented in many studies published by the American Society of Microbiology and others that susceptible bacteria do not readily develop resistance to host defense proteins under experimental conditions where resistance readily develops against conventional antibiotics. Furthermore, bacteria remain sensitive to the host defense proteins despite hundreds of millions of years of evolution in which bacteria have been exposed to host defense proteins’ antimicrobial mechanism of action.
Another favorable attribute of host defense proteins is that they selectively target bacteria and not mammalian cells, by recognizing the differences in the composition of bacterial and mammalian cell membranes. The outer surfaces of bacterial cell membranes are more negatively charged than mammalian cells. Bacterial cell membranes also lack cholesterol, an essential component of all mammalian membranes. Host defense proteins specifically target membranes that lack cholesterol and have a high degree of negative electrical charge. Therefore, they selectively attack bacterial cell membranes while mitigating harm to mammalian cells.
We believe that the host defense proteins provide an attractive mechanistic approach for the development of a new type of antibiotic therapeutic drug due to their strong antimicrobial activity, unique mechanism of action for which bacterial resistance appears less likely to develop, and selective targeting of bacteria but not mammalian cells. Attempts were made in the past by other companies to develop natural host defense proteins as novel antibiotics. Those products lacked robust activity in models of systemic infection, and were studied for niche topical applications, likely because of their limited systemic availability. None of them has been successfully commercialized to date because of difficulties relating to some of the inherent complexities associated with protein drugs: a lack of systemic bioavailability and stability. For systemic applications, all protein drugs have a number of limitations, including often not being bioavailable with either injectable or oral administration, often expensive to produce, often unstable even with intravenous administration, and at risk of eliciting immunological reactions which can neutralize their activity.
Our Approach
We have developed PMX‑30063, our lead antibiotic, and other novel small molecule antibiotics that mimic the activity of host defense proteins. We are developing these defensin mimetic antibiotics in a variety of forms to combat drug‑resistant bacterial infections. Our current focus is developing PMX-30063 for the treatment of Acute Bacterial Skin and Skin Structure Infections (ABSSSI), caused by drug susceptible and drug resistant strains of Staph aureus bacteria. PMX‑30063 and our other defensin mimetic antibiotics are completely synthetic, which make them easier and less expensive to produce than proteins. Importantly, our small molecule compounds demonstrate activity in animal models of systemic infection, activity that was lacking in others’ past attempts to develop natural host defense proteins. These defensin mimetic antibiotics may be developed in a variety of formulations, including injectable, tablet and topical, for a wide range of antibiotic applications. The results of our preclinical experiments for PMX‑30063 and our other defensin mimetic antibiotics suggest several advantages compared to marketed antibiotics. Such advantages include:
Ø Targeted spectrum of activity, including against resistant bacterial strains. Because they act on bacterial cell membranes, we believe the structure activity relationship of our antibiotics can be optimized for activity against Gram‑positive and Gram‑negative pathogens. In our in vitro efficacy studies and animal models of systemic infections, our defensin mimetic antibiotics as a class demonstrated potent antibacterial activity on hundreds of common bacterial pathogens. Our lead defensin mimetic antibiotic, PMX-30063, has been optimized for Gram-positive pathogens to support our initial clinical target of ABSSSI.
Ø Lower likelihood for the development of drug‑resistance. It is very difficult for bacteria to develop drug resistance to natural antimicrobial host defense proteins because these proteins act on bacterial membranes rather than on a single, mutable molecular target, such as an enzyme. Because our defensin mimetic antibiotics are designed to have the same mechanism of action as host defense proteins, we anticipate they will have a similar lower likelihood of developing bacterial resistance compared to conventional antibiotics. This reduced likelihood of bacterial resistance has been demonstrated in laboratory serial passage experimental studies.
Ø Potentially faster acting than other antibiotics. In time‑kill studies, our defensin mimetic antibiotics act quickly when bacteria are exposed to them. We observed bactericidal activity in a matter of minutes after exposure to our defensin mimetic antibiotics. In contrast, many currently marketed drugs can take hours or even several days to show effect.
3
PMX-30063 Clinical Development
Our first product candidate is an intravenous (or “IV”) formulation initially intended for the treatment of ABSSSI, caused by drug susceptible and drug resistant strains of Staph aureus bacteria. In the future, we also hope to expand indications to potentially include other infections and bacterial strains.
Clinical Studies to Date
In December 2008, we completed and announced positive results from our single dose escalation clinical study of healthy subjects receiving PMX-30063 at various dose levels (Phase 1A). This ascending single-dose intravenous pharmacokinetic and safety study met the study endpoints of defining both a limiting single dose and the plasma distribution/elimination kinetics. In this study, the dose was not limited by any measurable clinical or laboratory parameters. A subjective syndrome of paresthesias was identified, appearing only at the higher dosages and consisting of abnormal numbness and/or tingling. These effects were graded as mild to moderate by investigators or subjects, but their reproducibility and dose-proportionality allowed dose-escalation to be successfully concluded after achieving levels well in excess of that which we expect, based on other studies, to be the therapeutic range. The effects were temporary and resolved on their own. The same study provided detailed information on the time course of the drug during and after dosing. These pharmacokinetics appear favorable for therapeutic use of the drug. The half-life for elimination from the plasma was approximately 12 to 15 hours, allowing for flexibility in dosing to obtain optimal peak and trough drug levels.
In March 2010, we completed and announced positive results from our ascending multi-dose intravenous pharmacokinetic and safety study of healthy subjects receiving PMX-30063 at various dose levels (Phase 1B). The Phase 1B blinded, randomized, placebo-controlled ascending multiple-dose study was designed to find the limiting tolerable dosage for PMX-30063 when administered as five daily doses, and to define the resulting plasma distribution/elimination kinetics. In this study, the limiting effects of the dose were found to be the same subjective syndrome of paresthesias identified in the previously completed Phase 1A single-dose study, which appeared only at the higher dosages and consisted of abnormal numbness and/or tingling. In all cases the effects were temporary and resolved without treatment shortly after discontinuation of PMX-30063 administration. Transient changes in liver enzymes (ALT and AST) were also observed in some subjects at higher doses. In most cases these changes were less than twice the upper limit of normal, and in all cases resolved without treatment after completion of the study. These changes were not considered clinically significant by the study physicians, and were similar to those often seen with many other marketed drugs.
Antimicrobial assays were also performed with blood samples drawn from the study subjects. In these assays, blood samples were taken from the subjects after they had received a single dose of PMX-30063. Staphylococcus aureus bacteria was then added to these blood samples, to determine if, and at what concentrations, the PMX-30063 in the subjects blood would have antimicrobial activity. The bacteriostatic and bactericidal activity against MSSA (methicilin-sensitive Staphylococcus aureus) and MRSA (methicillin-resistant Staphylococcus aureus, or drug-resistant Staph) strains were achieved at doses between 0.1 mg/kg and 0.3 mg/kg, and largely correlated with those established in normal medium and in experimental animal studies (as seen in data presented by PolyMedix at the American Society of Microbiology ICAAC 2009 meeting). These data suggest that antimicrobial activity and bacteriostatic and bactericidal concentrations can be achieved in human subjects following multiple administrations of PMX-30063 at doses below the identified limiting dosage. Based upon the doses of PMX-30063 administered in the study, and blood levels of drug observed in subjects in this study, together with our pre-clinical studies, we believe that a beneficial therapy may exist.
In February 2011, we completed and announced positive results from a randomized, double-blind, placebo-controlled Phase 1 exposure-escalation study of twenty healthy male and female subjects receiving PMX-30063. PMX-30063 met the study safety endpoints regarding high dosing beyond five days of administration, and the pharmacokinetic profiles in male subjects were very similar to those in female subjects. Similar to results from two previously completed Phase 1 clinical studies with PMX-30063, the most frequently reported side effects included transient sensations of paraesthesia affecting the lips, face, and fingers. These sensations did not worsen over the course of the study and resolved on their own without treatment shortly after discontinuation of PMX-30063 administration. There were no significant clinical neurosensory abnormalities noted. One subject discontinued from the study due to paraesthesia. Several subjects were discontinued by the investigator after receiving between nine and thirteen days of dosing due to increased blood pressure and heart rate, for which there were no associated clinical signs or symptoms. After nine days of dosing, there was one reportable adverse event of atrial fibrillation that was fully reversed within a few hours following dosing. As seen in previous Phase 1 clinical studies, there were a few minor and transient increases in liver enzymes that were not considered clinically significant. All subjects demonstrated complete return to normal and reported no adverse effects by the time of the last follow up visit. The doses of PMX-30063 administered in this study were identical to the highest dosing level being administered for five days in our current Phase 2 efficacy study in Staph infections.
4
Ongoing Clinical Studies
In September 2010, we initiated a Phase 2 clinical trial with PMX-30063. This randomized, blinded, active controlled Phase 2 clinical trial is being conducted at multiple sites in Canada and Europe. The study objective is to evaluate the safety and efficacy of PMX-30063 as treatment for ABSSSI caused by Staphylococcus aureus. The trial will enroll up to 200 patients that have ABSSSI due to either MSSA or MRSA. Patients will be randomized to receive one of three dosing regimens of PMX-30063 or daptomycin active control, and will have an early treatment assessment at 48 and 72 hours for clinical and microbiologic response. Following the treatment assessment, patients will be re-evaluated at day 10 to 15 for test of cure and again for safety at four weeks. Clinical studies conducted outside the United States have and are being conducted to United States standards are intended to support regulatory approval in the United States.
Heptagonist: PMX-60056
Unfractionated Heparin (or “UFH”) and Low Molecular Weight Heparin (or “LMWH”) are blood clot prevention drugs, which are commonly used in numerous post‑surgical applications as anti‑coagulants, as well as during cardiac surgery with extracorporeal bypass. Overdoses of UFH or LMWH are dangerous due to potentially life‑threatening bleeding. While widely used, anti-coagulation with UFH and LMWH carries the risk of adverse bleeding events, a certain percentage of which are life-threatening or clinically significant. These bleeding events require medical intervention and cause significant direct costs.
Market Opportunity
Unfractionated Heparin (UFH)
Protamine is the only available reversing agent for UFH. Protamine is used both as an antidote in the event of overdose, and more commonly in standard treatment as a reversing agent following coronary artery bypass grafts (CABG, or “bypass” procedures), in which standard practice involves administering UFH while the patient is on a heart‑lung bypass machine to prevent blood clots from forming in the machine. However, protamine has many significant potential problems with its use, including:
Ø difficult to predict efficacy, resulting in variable inter‑patient activity;
Ø anticoagulant activity of protamine can actually increase and worsen the anticoagulant activity of UFH, and which requires complex dose‑titration and careful administration of often several doses;
Ø allergic anaphylactic reactions in some patients due to the fact that protamine is a biological product derived from fish sperm;
Ø inferior fibrokinetics, or clot formation, can result in leaky clots and in more serious cases rebound bleeding may occur; and
Ø immune reactions with neutralizing antibodies formed after sensitization to protamine in patients who have previously received protamine can impact efficacy and result in severe reaction; some patients, such as diabetic patients receiving zinc insulin, or males who have undergone vasectomy, may have pre‑existing antibodies that could pre‑dispose them to sensitivity to protamine.
As a result, we believe there is a significant medical and market need for a safer UFH antagonist. Worldwide, we estimate that approximately two million extracorporeal blood procedures, including cardiothoracic procedures, are performed that utilize heparin and may require UFH reversal. According to IMS Health, six to eight million doses of protamine are used annually worldwide. We believe our heptagonists have the potential to be a safer alternative to protamine.
5
Low Molecular Weight Heparin (LMWH)
The LMWHs are often used for longer term prevention of blood clots (thromboprophylaxis), in indications such as deep vein thrombosis (DVT), in patients who have had heart attacks (post Myocardial Infarction, or MI), and in cancer patients. All of these patient groups are at risk of developing potentially life‑threatening blood clots, and are given anti‑clotting drugs including LMWHs to prevent dangerous clot formation. Based on a study by the Agency for Healthcare Research and Quality, a division of the Department of Health and Human Services, most hospitalized patients have at least one risk factor for deep vein thrombosis and should receive prophylaxis with UFH or LMWH. This includes a significant number of the 23 million people in the US who undergo surgery as well as medical patients. According to IMS data, sales for LMWHs in the US and Canada were approximately $3 billion annually, corresponding to approximately 100 million individual doses of LMWH dispensed. In Europe, where LMWH is used more widely, the number of doses is estimated to be twice as high. Based on IMS sales data we estimate that more than 20 million patients receive LMWH annually.
While having favorable risk/benefit characteristics, clinical studies have shown that there is a significant incidence of bleeding in patients who receive LMWHs. Most of these studies generally have shown that 1%‑4% of patients experience serious, life‑threatening bleeding, and up to 20% may experience clinically significant bleeding. Neither protamine nor any other drug is approved for reversal of LMWH. In patients who experience bleeding problems while on LMWH, current care may involve hospitalization, blood transfusions, and surgery, which can be expensive and unreliable.
LMWHs are not currently used in acute surgical settings, such as cardiothoracic procedures, because of the lack of a reversing agent. The long half‑life of LMWH products of up to 24 hours, while an advantage for longer‑term administration, can be a major disadvantage if a patient has a major bleeding episode or must be re‑operated on shortly after surgery. If clotting time is prolonged due to systemic LMWH, re‑operation may not be possible. The availability of a reversing agent for LMWHs could create an opportunity for these drugs to be used in cardiothoracic surgical procedures. Thus, an approved LMWH reversing agent could have the potential to substantially increase the market and sales of LMWHs, thereby increasing the need and market for that same reversing agent.
Our Approach
UFH and LMWH are composed of sulfated polysaccharides, which provide an attractive target for the structure based design of novel molecules. Based on a model for the three‑dimensional structure of these molecules, we have produced a series of compounds that bind very tightly to the critical pentasaccharide site found on both UFH and LMWH. In clinical experiments, our compounds demonstrated efficacy in safely reversing the effect of both UFH and LMWH.
Our objectives in this program are to develop a product that:
Ø is as effective as protamine in reversing the anticoagulant effect of UFH;
Ø is safer than protamine;
Ø is easy to use;
Ø has superior fibrokinetic activity (less deleterious impact on clot formation than protamine);
Ø is less sensitizing than protamine; and
Ø is effective in reversing the anticoagulant effect of LMWH.
We believe that a heptagonist with these attributes could be an attractive alternative to protamine, expand the heparin reversal market by introducing a reversing agent for LMWHs, provide a new treatment for patients receiving LMWHs and who experience bleeding complications and require reversal, and further expand the LMWH market by allowing physicians to more widely use LMWHs.
6
PMX-60056 Clinical Development
Clinical Experiments to Date
In October 2009, we successfully completed a double-blind, placebo-controlled, crossover, proof-of-concept UFH reversal study (Phase 1B/2) of PMX-60056, which was conducted at a single site in the United States. PMX-60056 met the study safety and efficacy endpoints in reversing the anticoagulant activity of UFH. In all subjects receiving PMX-60056, there was a rapid and complete reversal of the anticoagulant action of UFH, as measured by activated clotting time (ACT) and activated partial thromboplastin time (aPTT). The UFH reversal appeared to occur at or before the end of the 10-minute infusion of PMX-60056, suggesting that the dose may have been in excess of requirements for reversal of the administered UFH dose. Furthermore, the reversal was permanent: there was no return of anticoagulation during the hours required for heparin’s effects to decline naturally. PMX-60056 was well tolerated in this study, with no serious or reportable adverse events occurring. Subjects in the study experienced minimal side effects, which consisted of mild itching or warmth lasting only minutes, and brief reductions in blood pressure, which were transient and not clinically significant.
In June 2010, we successfully completed a double-blind, placebo-controlled, crossover, proof-of-concept tinzaparin (a LMWH) reversal study (Phase 1B/2) of PMX-60056, which was conducted at a single site in the United States. PMX-60056 met the study safety and efficacy endpoints in reversing the anticoagulant activity of tinzaparin. PMX-60056, administered as a ten-minute infusion, reduced both anti-Xa activity (a critical clotting factor in the blood) and aPTT. After the first administration of PMX-60056, the tinzaparin continued to enter the subjects’ systems from the subcutaneous injection site, and as it did so, anticoagulation returned or continued. A second ten-minute infusion of PMX-60056 was administered three hours later, which showed similar results. PMX-60056 was well tolerated in this study, with no serious or reportable adverse events occurring. Subjects in the study experienced minimal side effects, which consisted of mild itching or warmth lasting only minutes, and brief reductions in blood pressure, which were transient and not clinically significant when LMWH was present.
In August 2010, we successfully completed an open label, dose titration, UFH reversal study (Phase 1B/2) of PMX-60056, which was conducted at a single site in the United States. PMX-60056 met the study safety and efficacy endpoints in both the reversal of surgical levels of UFH and in subsequent re-anticoagulation with UFH and re-reversal with PMX-60056. PMX-60056 was well tolerated in this study, with no serious or reportable adverse events occurring. Subjects in the study experienced minimal side effects, which consisted of transient reductions in blood pressure, which were not clinically significant and were seen only at the end of some reversals when ACT was already nearing baseline after the last dose of PMX-60056.
Ongoing Clinical Studies
In February 2011, we initiated a Phase 2 clinical trial to assess the safety and efficacy of PMX-60056, in reversing UFH in patients undergoing Percutaneous Coronary Intervention (PCI) procedures. This multi-center, open-label Phase 2 clinical study is intended to enroll up to 40 patients undergoing PCI in the United States. All patients will receive PMX-60056 by intravenous infusion, in a dose calculated to reduce the post-procedure ACT to less than 20 seconds above the baseline level. The primary endpoint of the study is to evaluate the safety and efficacy of PMX-60056 in reversing UFH in a surgical setting. Data collected from this study are intended to support further development of PMX-60056 in larger and more diverse patient populations.
In October 2007, we received a Small Business Innovation Research (SBIR) grant of $100,000 from the National Institute of Health (NIH) to support the development of biomimetic compounds, such as PMX-60056, as anti-coagulant antagonists. The research under this grant has been completed and, in June 2009, we received a Phase II SBIR grant in the amount of $923,080, to be disbursed over two years, to support the development of LMWH anticoagulant reversing agents. As of December 31, 2010, we had approximately $132,000 still available for research under this grant.
PRECLINICAL PROGRAMS:
In addition to our clinical programs for PMX‑30063 and PMX‑60056, we have conducted preclinical studies for other product candidates that are described below. These preclinical programs are either currently on hold or are being supported by grants or research contracts for the NIH or US Department of Defense. Further, we may consider expanding or initiating other development activities if and when we are able to generate additional funding and resources.
7
Defensin Mimetic Antibiotics
We also hope to conduct pre-clinical development of PMX-30063 and other defensin mimetic antibiotic compounds for topical inhaled and oral antibiotic applications. We are presently investigating the development of defensin mimetics for the treatment of oral candidiasis, a fungal infection in the mouth of immune-compromised patients caused by the yeast, Candida albicans. This work is being supported by a Phase 2 SBIR grant from the National Institute of Dental and Craniofacial Research (NIDCR) of the NIH, and is an extension of work initiated in a Phase 1 grant from the same Institute. Other programs that are currently on hold until we are able to obtain and allocate sufficient resources include the development of an ophthalmic topical antibiotic for the treatment of eye infections, and the development of a non-absorbed oral antibiotic for the treatment of gastrointestinal infections such as Clostridium difficile. With sufficient resources, we plan to advance pre-clinical development and file additional Investigational New Drug applications (IND) for the ophthalmic topical and non-absorbed oral antibiotic indications in the future.
We are also evaluating our defensin mimetic antibiotic compounds for a number of other topical and local applications, including topical antibiotic use for skin structure infections, oral healthcare applications for treatment of periodontal disease, a type of gum disease, topical treatment for ear infections, topical treatment of fungal infections, topical treatment of acne, and a variety of non‑therapeutic applications in personal care and materials applications.
We are also exploring the potential use of our defensin-mimetic antibiotics for the treatment of other infectious diseases. In particular, recent pre-clinical studies by PolyMedix and our academic collaborators have shown activity of certain of our compounds for potential use against tuberculosis and malaria. We have obtained a fast track SBIR grant from the National Institute of Infectious and Allergic Diseases (NIAID) of the NIH for development of a defensin mimetic to treat malaria. In addition, we have a NIH U01 grant for development of defensin mimetics to treat illnesses caused by foodborne pathogens and a Phase 2 grant from the US Army to develop defensin mimetics for treatment of bacterial infections associated with biofilm formation.
Medical Device, Industrial and Consumer Applications
We have conducted preliminary experiments which demonstrate that polymer derivatives of certain of our compounds, such as PMX-70004 and PMX‑50003, which we refer to as our PolyCides®, may be used as effective antimicrobial agents as additives to materials, such as paints, plastics and textiles.
For example, when our PolyCides are incorporated into the plastic of intravenous catheter tubing or sutures and then exposed to a high inoculum of bacteria, significant anti‑bacterial activity is observed compared to untreated plastics.
We do not currently plan to conduct advanced development of any medical device, industrial or consumer application of our PolyCides, but rather intend to focus on generating a limited core of basic enabling data to support our efforts to license these PolyCides to partners who will continue research and development efforts in developing products.
In August 2010, we received a Phase 1 grant from the National Science Foundation for $150,000 in support of development of antimicrobial sutures formulated with PolyCides to combat MRSA and other pathogens associated with surgical site infections. As of December 31, 2010, we had approximately $92,000 still available for research under this grant.
Biodefense
Over the past decade, improving our nation’s defense against bioterrorism has become a greater priority. Antibiotic activity against anthrax and other biowarfare pathogens, with a mechanism by which resistance is unlikely to develop, has commercial, medical and national security value. Preliminary data from our preclinical experiments indicate that our product candidates have potent activity against biowarfare agents that cause anthrax, tularemia and plague.
As a result of this preliminary data, we are pursuing grants and research contracts from government sources, such as the Department of Defense, the Defense Advanced Research Projects Agency and other military and security agencies to further test and advance our product candidates for indications important to national security. To-date we have been awarded the following grants and contracts in support of biodefense applications:
8
Ø In June 2009, we were awarded a $1.6 million contract with the Defense Threat Reduction Agency to develop new defensin mimetic antibiotic compounds to combat biowarfare pathogens. As of December 31, 2010, we had approximately $270,000 still available for research under this contract.
Ø In July 2009, we, and our academic collaborators at the University of Massachusetts (or UMass), received a contract for $70,000 from the Office of Naval Research to develop antibacterial compounds against pathogens of military interest. As of December 31, 2010, research under this contract has been completed.
Ø In July 2009, we, and our academic collaborators at UMass, received a grant for up to $6.6 million from the NIH to develop new compounds for biodefense and emerging food-borne infectious diseases. As of December 31, 2010, we had approximately $6,100,000 still available for research under this grant.
Ø In August 2009, we and our academic collaborators at UMass, received a Phase 1 contract for $100,000 from the Army Research Office to develop compounds against drug-resistant bacterial biofilms. As of December 31, 2010, research under this contract was completed.
Ø In October 2010, we were awarded a $750,000 Phase 2 contract from the Army Research Office (ARO) to continue our research on treating biofilm-associated infections. As of December 31, 2010, we had approximately $702,000 still available for research under this grant.
Anti-Tuberculosis
Tuberculosis is a highly contagious disease that affects one-third of the world’s population today. Approximately, 1.7 billion people are infected worldwide, including 15 million cases in the United States. There are 8 million newly reported cases each year and 3.1 million people die from the disease annually. Tuberculosis is the leading cause of death of women, AIDs patients and the young in the world and there are more deaths from tuberculosis than any other single infectious disease. Mycobacterium tuberculosis is the primary infectious agent for tuberculosis and drug resistance has become a paramount issue, accounting for over 50 million infections. The drug-resistant forms of the disease are significantly more difficult to treat, leading to higher mortality rates and escalating costs of care. PolyMedix compounds, including PMX-10070, have demonstrated encouraging in vitro specificity for tuberculosis bacteria versus mammalian cells.
Anti-Malaria Agents
Recent pre-clinical studies with PolyMedix’s defensin mimetic antibiotics show encouraging activity for the potential treatment of the malaria parasite, Plasmodium falciparum, the infectious agent for the most prevalent and deadly forms of malaria. P. falciparum accounts for 80% of all human malarial infections and 90% of deaths from such infections. More than 120 million clinical cases of malaria and between 1 to 1.5 million deaths occur worldwide every year. Current therapies for malaria are plagued by rapid resistance, which has become endemic in certain regions of the world. Several of our compounds, including PMX-70008 and PMX-30024, have demonstrated encouraging in vitro specificity for the parasite versus mammalian cells.
As a result of these preliminary data, we are pursuing grants and research contracts from government sources, such as the National Institutes of Health to further test and advance our product candidates. In July 2010, we received a Fast Track Advanced Technology SBIR grant from the NIAID to support the development of defensin-mimetic antimicrobial compounds for the treatment of malaria. The initial phase of this grant provides PolyMedix with $0.5 million over the first year, with the opportunity for an additional $0.5 million in the second year, to validate and pursue defensin-mimetic antimicrobial compounds for therapeutic development. The goal of the first phase of research is to generate proof-of-concept through in vitro and in vivo efficacy testing. Subject to satisfactory performance of the initial phase and availability of funds, the grant provides for funding of up to $1 million per year for each of an additional three years, which brings the potential value of the grant up to an aggregate of $4 million over the five-year period. As of December 31, 2010, we had approximately $410,000 still available for research under the first year of this grant.
Antifungal Agents
Our compounds, such as PMX-10098 and PMX-70004, have demonstrated promising activity against fungus strains that often cause human infectious diseases. In preclinical experiments, certain of our compounds have demonstrated effectiveness at inhibiting fungal growth and for certain strains of fungus, effectiveness was achieved at lower concentrations than that of fluconazole, a commonly used antifungal agent. We intend to continue to investigate the potential of our compounds as novel treatments for human fungal infections.
9
As a result of these preliminary data, we are pursuing grants and research contracts from government sources, such as the National Institutes of Health to further test and advance our product candidates. In May 2008, we received a Phase 1 SBIR grant of $126,000 from the NDICR to support preliminary research for antifungal defensin mimetic applications. A Phase 2 grant totaling $1,000,000 was awarded in October, 2010 from the same institute to continue our anti-fungal research for development of defensin mimetics to treat oral candidiasis. As of December 31, 2010, the Phase 1 grant was completed, and we had approximately $885,000 still available for use under the Phase 2 grant.
Angiogenesis Inhibitor
Our angiogenesis inhibitor compounds, such as PMX‑20005, have demonstrated promising activity in in vitro and animal models of inhibition of angiogenesis, the abnormal growth of blood vessels. An angiogenesis inhibitor may be effective in treating age related macular degeneration (or AMD); a common form of blindness caused by excessive and abnormal growth of blood vessels in the back of the eye. With adequate additional financing and resources, we hope to continue to investigate the potential of our compounds as novel treatments of AMD and other angiogenesis related diseases.
Our Strategy
Our primary goal is to discover and develop novel agents for the treatment of infectious diseases and patients with cardiovascular disorders. To achieve this objective, we are implementing the following strategies:
Advance the Development of our Lead Product Candidates. We believe our current financial resources provide us with sufficient funding to support the ongoing Phase 2 clinical trials for each PMX-30063 and PMX-60056. Further studies for current and future potential indications will require additional financial resources.
Retain Rights to Market Hospital‑Based Products in North America. We have exclusive commercial rights to all of our current programs and in addition have either exclusive ownership rights or options to obtain exclusive license rights to any new products that result from our academic relationships. Our objective is to generate maximum value from sales of our product candidates if regulatory approval is achieved. To achieve this goal, we intend to build our own specialty sales force to market hospital‑based therapeutics, such as PMX‑30063 and PMX‑60056, in North America. Additionally, we plan to collaborate with third parties to commercialize our products in primary care, international and other markets.
Pursue Near‑Term Revenue Opportunities through Out‑Licensing and Partnership Agreements. We intend to out‑license selected product candidates from internal programs, such as our PolyCides, that are not part of our strategic focus. Such collaborations could generate multiple sources of revenue, such as up‑front fees, research funding, milestone payments and royalties.
Utilize our Technology to Pursue Additional Drug Development Programs for Other Therapeutic Areas. Using our computational drug design tools, we have already identified potential product candidates for other therapeutic areas. If sufficient resources become available, we plan to pursue development in these areas through partnerships or on our own in order to expand our product pipeline.
Strengthen Collaborations with Existing Partners and Enter into Agreements with Potential New Partners. We have entered into collaborations and agreements with leading academic institutions. We are actively pursuing additional agreements that would provide for license fees, research funding and milestone payments and royalties to PolyMedix from research results and subsequent product development and commercialization.
Research and Development
We incurred research and development expenses of $10,951,000, $7,374,000, and $7,401,000 for the years ended December 31, 2010, 2009 and 2008, respectively.
10
Proprietary Rights
University of Pennsylvania
In January 2003, we entered into a Patent License Agreement with the University of Pennsylvania, or Penn. Under the terms of the agreement, as amended, we have been granted an exclusive, worldwide royalty‑bearing license to use, make and sell products utilizing seven of Penn’s issued patents or pending patent applications for the life of such patents. These patents and applications cover our clinical and preclinical stage product candidates, as well as future product candidates utilizing the licensed patents and applications. The licensed patents and applications include:
Ø three issued U.S. patents and three pending U.S. patent applications covering the composition of matter on antimicrobial compounds, including small molecules, oligomers and polymers. The first issued patent expires in 2017, the second issued patent expires in 2022, and the third issued patent expires in 2023. The other patents, if issued, will expire at varying times from 2022 to 2025. There are corresponding foreign applications to one of the issued U.S. patents and to each of the three pending U.S. patent applications.
Ø one U.S. patent application and two corresponding foreign patent applications covering polycationic compounds and their use for treating cancer. The patent, if issued, will expire in 2026.
Penn may terminate the licenses if:
Ø we are more than 60 days late in paying to Penn royalties, expenses, or any other undisputed amounts due under the agreement and we do not pay such amounts within 30 days’ written notice of such delinquency;
Ø we become insolvent, enter into bankruptcy or a similar proceeding or call a meeting of our creditors in order to arrange adjustment of our debts; or
Ø we otherwise materially breach the agreement.
If this license agreement is properly terminated by Penn, we may not be able to execute our strategy to develop and commercialize our IV antibiotic, PMX-30063, our heptagonist, PMX-60056, our oral antibiotic product candidates, our PolyCides for biomaterials applications (PolyCides®), or future product candidates utilizing the licensed patents.
We also entered into a Software License Agreement with Penn in May 2003. Under the terms of the agreement, Penn granted us a non‑exclusive, royalty‑free license to use three software programs and an exclusive, royalty‑free license to three patent applications relating to such software programs. The software programs and patents covered by the agreement include a suite of proprietary computational algorithms that we use in the development, refinement and testing of our product candidates. The licenses expire contemporaneously with our Patent License Agreement with Penn. The patents relating to the software licenses, if issued, will expire at varying times in 2023 and 2024. Penn may terminate the agreement if:
Ø we are more than 60 days late in paying to Penn any undisputed amounts due under the agreement and we do not pay such amounts within 30 days’ written notice of such delinquency;
Ø we become insolvent, enter into bankruptcy or a similar proceeding or call a meeting of our creditors in order to arrange adjustment of our debts; or
Ø we otherwise materially breach the agreement.
If this license agreement is properly terminated by Penn, our ability to advance our current product candidates or develop new product candidates may be adversely affected.
We also retain as a paid consultant Dr. William DeGrado, a Professor of Biochemistry and Biophysics at Penn, who provides these consulting services on our premises. Any inventions made under this arrangement are intended to be covered by our existing intellectual property, our existing Patent License Agreement with Penn, or a new license agreement with Penn containing substantially the same terms. Dr. DeGrado’s current consulting services for us focuses on further development of our antimicrobial and heptagonist applications, and other general matters of pharmaceutical research and development relating to our programs. For these services, we pay Dr. DeGrado a consulting fee of $5,000 per month. In addition, we have granted stock options to Dr. DeGrado. Dr. DeGrado also serves as Chairman of our Scientific Advisory Board. Either we or Dr. DeGrado may terminate his consulting arrangement at any time.
11
University of Massachusetts
In January 2004, we entered into a Sponsored Research Agreement with the University of Massachusetts, or UMass. Under the terms of this agreement, as amended, we have agreed to sponsor certain research of Dr. Gregory Tew, an Assistant Professor in the Polymer Science and Engineering Department at UMass, through March 2009, and have the exclusive option to license any intellectual property generated by such research. We may exercise this option by issuing 7,500 shares of our Common Stock to UMass for each $100,000 of research conducted by Dr. Tew. We are in the process of negotiating an extension to the term of this Sponsored Research Agreement. During 2007, we issued 12,500 shares to UMass in connection with this agreement. Dr. Tew’s research focuses on methods to add our antibiotic agents to biomaterials, testing the physical properties of our antibiotic agents and safety evaluation of our antimicrobial agents. The agreement will terminate if Dr. Tew leaves UMass or ceases work in the antimicrobial field of research. In addition, UMass may terminate the agreement including any licenses granted thereunder if we materially breach the agreement, including by nonpayment of amounts due under the agreement. If the Sponsored Research Agreement or any license thereunder is properly terminated by UMass, our ability to develop new product candidates may be adversely affected.
In January 2005, we entered into an Exclusive License Agreement with UMass, pursuant to which UMass granted us an exclusive, worldwide license to use, make and sell products under one U.S. patent application and seven corresponding foreign patent applications covering polynorborene co‑polymers and methods of use for the life of such patents. The patents, if issued, will expire in 2025. UMass may terminate the license agreement if we materially breach the agreement, including by nonpayment of amounts due under the agreement or in the case of a change in our ownership or control. If the license agreement is properly terminated by UMass, we may not be able to execute our strategy to develop and commercialize our PolyCides for biomaterials applications or to develop and commercialize future product candidates utilizing the licensed patents.
We also retain as a paid consultant Dr. Gregory Tew, Assistant Professor in the Polymer Science and Engineering Department at UMass, who provides these consulting services for biomaterials applications for our antimicrobial agents. Any inventions made under this arrangement are intended to be covered by our existing intellectual property, our existing Exclusive License Agreement with UMass, or a new license agreement with UMass containing substantially the same terms. For these services, we pay Dr. Tew a consulting fee of $2,500 per month. In addition, we have granted stock options to Dr. Tew. Dr. Tew also serves on our Scientific Advisory Board. Either we or Dr. Tew may terminate his consulting arrangement at any time.
Our Technology
We initiate the design of our product candidates using our proprietary computational drug design technology that is licensed from Penn. There are several computational methods used to assist the drug design process, which includes the ability to:
Ø model molecular interactions in the presence of solvent, such as an aqueous environment, rather than in a vacuum, which more closely resembles the real‑life characteristics of how molecules interact with each other;
Ø simulate molecular interactions for hundreds of microseconds which are time frames much longer than possible with previous molecular dynamics technologies; and
Ø design compounds which target the bacterial cell membrane, instead of a biochemical cell target.
Our innovative and proprietary de novo drug design approach starts with protein targets with well‑understood physical structures and biological activity, and designs small molecule compounds that mimic or regulate the activity of these targets. This biomimetic approach of regulating biological activities by mimicking nature itself with synthetic small molecule compounds allows us to rationally design product candidates that we believe greatly improve the efficiency of new drug discovery.
12
Intellectual Property
We rely on a combination of patents and trade secrets, as well as confidentiality and non‑use agreements to protect our intellectual property. Our patent strategy is designed to facilitate commercialization of our current and future product candidates; and create barriers to entry. Our intellectual property portfolio currently consists of three provisional applications, two patent applications (U.S. and foreign) owned by us, two issued U.S. patents we exclusively license from Penn and eight U.S. patent applications we exclusively license from Penn and/or UMass (as well as foreign counterparts thereof). Our patent applications and the ten issued patents and pending patent applications we exclusively license from Penn and/or UMass include:
Ø seven U.S. patent applications that have been filed (as well as foreign counterparts thereof) covering the composition of matter on 11 classes of antimicrobial compounds, including small molecules, oligomers and polymers. Two U.S. patents have been granted on two of these applications;
Ø one patent application covering the use of claimed oligomers for treating ophthalmic and otic infections;
Ø one patent application covering the use of claimed oligomers for treating mycobacterium
Ø one patent application covering the use of claimed oligomers for treating malarial infections
Ø one patent application covering synthetic mimetics of host defense proteins and their use
Ø one patent application covering the combination of claimed compounds and polymers with sesquiterpenoid enhancing agents;
Ø one patent application covering novel angiogenesis inhibitors with a wide range of therapeutic uses;
Ø one patent application covering the use of claimed oligomers for treating cancer;
Ø one patent application covering processes for preparing a polymeric compound;
Ø two patent applications covering anti-heparin compounds;
Ø one patent application covering methods of immune modulation;
Ø one patent application covering facially amphiphilic oligomers and uses thereof; and
Ø three patent applications covering our proprietary, computational algorithms and models, such as the coarse grain model and a new force field. These patent applications do not disclose the specific computer code, which either will be copyrighted or kept as a proprietary trade secret. One U.S. patent has been granted on one of these applications.
We expect to continue to expand our intellectual property portfolio with additional filings of both composition of matter and method of use patent applications. A number of new patent applications are currently in process.
We attempt to protect our trade secrets by entering into confidentiality agreements with third parties, employees and consultants. Our employees and consultants also sign agreements requiring that they assign to us their interests in patents and other intellectual property arising from their work for us. All employees sign an agreement not to engage in any conflicting employment or activity during their employment with us and not to disclose or misuse confidential information.
In addition, we have received trademark protection for “PolyMedix®” and “PolyCide®.”
13
COMPETITION:
Defensin Mimetic Antibiotic Competition
The pharmaceutical industry is highly competitive. We face significant competition from pharmaceutical companies and biotechnology companies that are researching and selling products designed to treat bacterial infections. Many major pharmaceutical companies, such as GlaxoSmithKline, Pfizer, Bayer, Merck, Novartis, Johnson & Johnson, and Sanofi Aventis have already established significant positions in the antibiotic market. Additionally, many mid-sized and smaller companies, such as Astellas Pharma, Forest Laboratories, Cubist Pharmaceuticals, Basilea Pharmaceutica, Theravance, The Medicines Company, Trius Therapeutics, NovaBay Pharmaceuticals, Inc., Paratek, and Inhibitex have either marketed antibiotics and/or are attempting to enter this market by developing novel and more potent antibiotics that are intended to be effective against drug‑resistant bacterial strains.
These companies, as well as other potential entrants into the antibiotic market, have longer operating histories, larger customer or user bases, greater brand recognition and significantly greater financial, marketing and other resources than we do. Many of these current or potential competitors can devote substantially greater resources to the development and promotion of their products than we can.
Additionally, there has been consolidation within the pharmaceutical industry and larger, well‑established and well‑financed entities may continue to acquire, invest in or form joint ventures to gain access to additional technology or products. Any of these trends would increase the competition we face and could adversely affect our business and operating results.
Heptagonist Competition
There are currently no products available in the marketplace or approved as an antidote for and antagonist to LWMH, while protamine is the only available antidote for and antagonist to UFH and, as such, protamine currently dominates this market. Because of protamine’s potentially problematic side effect profile, we believe that our heptagonist products may penetrate into the UFH antagonist market.
Government Regulation
Government authorities in the U.S., at the federal, state, and local level, and foreign countries extensively regulate, among other things, the following areas relating to our products candidates:
Ø research and development;
Ø testing, manufacture, labeling and distribution;
Ø advertising, promotion, sampling and marketing; and
Ø import and export.
All of our product candidates will require regulatory approvals by government agencies prior to commercialization, but none of our product candidates has received these approvals. In particular, human therapeutic products are subject to rigorous preclinical and clinical trials to demonstrate safety and efficacy and other approval procedures of the FDA and similar regulatory authorities in foreign countries. Various federal, state, local, and foreign statutes and regulations also govern testing, manufacturing, labeling, distribution, storage and record keeping related to such products and their promotion and marketing. The process of obtaining these approvals and the compliance with federal, state, local, and foreign statutes and regulations require the expenditure of substantial time and financial resources. In addition, the current regulatory and political environment at the FDA could lead to increased testing and data requirements that could impact regulatory timelines and costs.
U.S. Government Regulation
In the U.S., the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act and implementing regulations. If we fail to comply with the applicable requirements at any time during the product development process, approval process, or after approval, we may become subject to administrative or judicial sanctions. These sanctions could include:
14
Ø refusal to approve pending applications;
Ø withdrawals of approvals;
Ø clinical holds;
Ø warning letters;
Ø product recalls and product seizures; and
Ø total or partial suspension of our operations, injunctions, fines, civil penalties or criminal prosecution.
Any agency enforcement action could have a material adverse effect on us.
Currently there is a substantial amount of congressional and administration review of the FDA and the regulatory approval process for drug candidates in the U.S. As a result, there may be significant changes made to the regulatory approval process in the U.S.
The steps required before a drug may be marketed in the U.S. include:
Ø preclinical laboratory tests, animal studies and formulation studies;
Ø submission to the FDA of an IND, which must become effective before human clinical trials may begin;
Ø execution of adequate and well‑controlled clinical trials to establish the safety and efficacy of the product for each indication for which approval is sought;
Ø submission to the FDA of a New Drug Application (NDA), or biologics license application, or BLA;
Ø satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance with current Good Manufacturing Practices (cGMP); and
Ø FDA review and approval of the NDA or BLA, or any supplements thereto, including, if applicable, a determination of its controlled substance schedule.
Preclinical studies generally are conducted in laboratory animals to evaluate the potential safety and activity of a product. Violation of the FDA’s good laboratory practices regulations can, in some cases, lead to invalidation of the studies, requiring these studies to be replicated. In the U.S., drug developers submit the results of preclinical trials, together with manufacturing information and analytical and stability data, to the FDA as part of the IND, which must become effective before clinical trials can begin in the U.S. An IND becomes effective 30 days after receipt by the FDA unless before that time the FDA raises concerns or questions about the proposed clinical trials outlined in the IND. In that case, the IND sponsor and the FDA must resolve any outstanding FDA concerns or questions before clinical trials can proceed. If these concerns or questions are unresolved, the FDA may not allow the clinical trials to commence.
Clinical trials involve the administration of the investigational product candidate or approved products to human subjects under the supervision of qualified investigators. Clinical trials are conducted under protocols detailing, among other things, the objectives of the study and the parameters to be used in assessing the safety and the effectiveness of the drug. Typically, clinical evaluation involves a time‑consuming and costly three‑phase sequential process, but the phases may overlap. Each trial must be reviewed, approved and conducted under the auspices of an independent institutional review board, and each trial must include each patient’s informed consent. The following sets forth a brief description of the typical phases of clinical trials:
Phase 1 Refers typically to closely monitored clinical trials and includes the initial introduction of an investigational new drug into human patients or healthy volunteer subjects. Phase 1 clinical trials are designed to determine the safety, metabolism and pharmacologic actions of a drug in humans, the potential side effects of the product candidates associated with increasing drug doses and, if possible, to gain early evidence of the product candidate’s effectiveness. Phase 1 trials also include the study of structure‑activity relationships and mechanism of action in humans, as well as studies in which investigational drugs are used as research tools to explore biological phenomena or disease processes. During Phase 1 clinical trials, sufficient information about a drug’s pharmacokinetics and pharmacological effects should be obtained to permit the design of well‑controlled, scientifically valid Phase 2 studies. The total number of subjects and patients included in Phase 1 clinical trials varies, but is generally in the range of 20 to 80 people.
15
Phase 2 Refers to controlled clinical trials conducted to evaluate appropriate dosage and the effectiveness of a drug for a particular indication or indications in patients with a disease or condition under study and to determine the common short‑term side effects and risks associated with the drug. These clinical trials are typically well controlled, closely monitored and conducted in a relatively small number of patients, usually involving no more than several hundred subjects.
Phase 3 Refers to expanded controlled and uncontrolled clinical trials. These clinical trials are performed after preliminary evidence suggesting effectiveness of a drug has been obtained. Phase 3 clinical trials are intended to gather additional information about the effectiveness and safety that is needed to evaluate the overall benefit‑risk relationship of the drug and to provide an adequate basis for physician labeling. Phase 3 trials usually include from several hundred to several thousand subjects.
Phase 4 Refers to trials conducted after approval of a new drug and which explore approved uses and approved doses of the product. These trials must also be approved and conducted under the auspices of an institutional review board. Phase 4 studies may be required as a condition of approval.
Clinical testing may not be completed successfully within any specified time period, if at all. The FDA closely monitors the progress of each of the first three phases of clinical trials that are conducted in the U.S. and may, at its discretion, reevaluate, alter, suspend or terminate the testing based upon the data accumulated to that point and the FDA’s assessment of the risk/benefit ratio to the patient. The FDA can also provide specific guidance on the acceptability of protocol design for clinical trials. The FDA or we may suspend or terminate clinical trials at any time for various reasons, including a finding that the subjects or patients are being exposed to an unacceptable health risk. The FDA can also request additional clinical trials be conducted as a condition to product approval. During all clinical trials, physicians monitor the patients to determine effectiveness and to observe and report any reactions or other safety risks that may result from use of the drug candidate.
Assuming successful completion of the required clinical trial, drug developers submit the results of preclinical studies and clinical trials, together with other detailed information including information on the chemistry, manufacture and control of the product, to the FDA, in the form of an NDA or BLA, requesting approval to market the product for one or more indications. In most cases, the NDA/BLA must be accompanied by a substantial user fee. The FDA reviews an NDA/BLA to determine, among other things, whether a product is safe and effective for its intended use.
Before approving an application, the FDA will inspect the facility or facilities where the product is manufactured. The FDA will not approve the application unless cGMP compliance is satisfactory. The FDA will issue an approval letter if it determines that the application, manufacturing process and manufacturing facilities are acceptable. If the FDA determines that the application, manufacturing process or manufacturing facilities are not acceptable, it will outline the deficiencies in the submission and will often request additional testing or information. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval and refuse to approve the application by issuing a “not approvable” letter.
The testing and approval process requires substantial time, effort and financial resources, which may take several years to complete. The FDA may not grant approval on a timely basis, or at all. We may encounter difficulties or unanticipated costs in our efforts to secure necessary governmental approvals, which could delay or preclude us from marketing our products. Furthermore, the FDA may prevent a drug developer from marketing a product under a label for its desired indications or place other conditions, including restrictive labeling, on distribution as a condition of any approvals, which may impair commercialization of the product. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further FDA review and approval.
If the FDA approves the NDA/BLA, the drug can be marketed to physicians to prescribe in the U.S. After approval, the drug developer must comply with a number of post‑approval requirements, including delivering periodic reports to the FDA, submitting descriptions of any adverse reactions reported, biological product deviation reporting, and complying with drug sampling and distribution requirements. The holder of an approved NDA/BLA is required to provide updated safety and efficacy information and to comply with requirements concerning advertising and promotional labeling. Also, quality control and manufacturing procedures must continue to conform to cGMP after approval. Drug manufacturers and their subcontractors are required to register their facilities and are subject to periodic unannounced inspections by the FDA to assess compliance with cGMP, which imposes procedural, and documentation requirements relating to manufacturing, quality assurance and quality control. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance. The FDA may require post‑market testing and surveillance to monitor the product’s safety or efficacy, including additional studies to evaluate long‑term effects.
16
In addition to studies requested by the FDA after approval, a drug developer may conduct other trials and studies to explore use of the approved drug for treatment of new indications, which require submission of a supplemental or new NDA and FDA approval of the new labeling claims. The purpose of these trials and studies is to broaden the application and use of the drug and its acceptance in the medical community.
Foreign Regulation
Whether or not we obtain FDA approval for a product, we must obtain approval of a clinical trial or product by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement also vary greatly from country to country. Although governed by the applicable country’s regulations, clinical trials conducted outside of the U.S. typically are administered with the three‑phase sequential process that is discussed above under “U.S. Government Regulation.” However, the foreign equivalent of an IND is generally not a prerequisite to performing pilot studies or Phase 1 clinical trials.
Furthermore, regulatory approval of prices is required in most countries other than the U.S. We face the risk that the resulting prices would be insufficient to generate an acceptable return to us or our collaborators.
Business History
PolyMedix, Inc. (formerly known as BTHC II Acquisition Corp.) was originally organized in the State of Texas as BTHC II LLC, a public shell company. On September 29, 2004, BTHC II LLC and its sister companies filed an amended petition under Chapter 11 of the United States Bankruptcy Code. On November 22, 2004, the court approved BTHC II LLC's Amended Plan of Reorganization. On March 24, 2005, and in accordance with its Amended Plan of Reorganization, BTHC II LLC changed its state of organization from Texas to Delaware by merging with and into BTHC II Acquisition Corp., a Delaware corporation formed solely for the purpose of effecting the reincorporation. After this merger, BTHC II Acquisition Corp. became the public shell company.
PolyMedix Pharmaceuticals, Inc. (formerly known as PolyMedix, Inc.) was incorporated on August 8, 2002 in the State of Delaware as PolyMedix, Inc. to serve as a biotechnology company focused on treating infectious diseases. On October 6, 2005, the former BTHC II Acquisition Corp. entered into an Agreement and Plan of Merger and Reorganization with the former PolyMedix, Inc., pursuant to which the stockholders of the former PolyMedix, Inc. exchanged their equity ownership interests in the former PolyMedix, Inc. for an aggregate 94% equity ownership interest in the former BTHC II Acquisition Corp. In November 2005, pursuant to the Agreement and Plan of Merger and Reorganization, the former PolyMedix, Inc. merged with a wholly-owned subsidiary of the former BTHC II Acquisition Corp. and, as such, became a wholly-owned subsidiary of the former BTHC II Acquisition Corp.
At the time of the merger, the former BTHC II Acquisition Corp. had no operations and no assets or liabilities. Because the stockholders of the former PolyMedix, Inc. exchanged their equity ownership interests in the former PolyMedix, Inc. for an aggregate 94% equity ownership interest in the former BTHC II Acquisition Corp., the former PolyMedix, Inc. was, for accounting purposes, the surviving entity of the merger. Accordingly, all financial information included in this report or incorporated herein by reference for the periods prior to the merger is for the former PolyMedix, Inc.
17
On February 24, 2006, the former BTHC II Acquisition Corp. changed its corporate name to PolyMedix, Inc., and the former PolyMedix, Inc. changed its corporate name to PolyMedix Pharmaceuticals, Inc.
Employees
We currently have 29 full-time employees, including one M.D. and 15 employees with Ph.D. degrees. Twenty-one of our employees are focused on research and development and eight are focused on general administration. We also utilize a number of consultants to assist with research and development activities.
Securities and Exchange Commission Filings
We file annual reports on Form 10-K, quarterly reports on Form 10-Q and current reports on Form 8-K and other information with the SEC. Members of the public may read and copy materials that we file with the SEC at the SEC’s Public Reference Room at 100 F Street, N.E., Washington, D.C. 20549. Members of the public may also obtain information on the Public Reference Room by calling the SEC at 1-800-732-0330. The SEC also maintains an Internet web site that contains reports, proxy and information statements and other information regarding issuers, including us, that file electronically with the SEC. The address of that site is www.sec.gov.
Risks Related to Our Business
We have incurred losses since inception and anticipate that we will continue to incur losses for the foreseeable future.
We are a development stage company with a limited operating history. As of December 31, 2010, we had an accumulated deficit of approximately $64,204,000. We expect to continue to incur significant and increasing operating losses, in the aggregate and on a per share basis, for the foreseeable future. These losses, among other things, have had and will continue to have an adverse effect on our results of operations, financial condition, stockholders’ equity, net current assets and working capital.
Because of the numerous risks and uncertainties associated with developing new drugs, we are unable to predict the extent of any future losses or when we will become profitable, if at all. Currently, we have no products available for commercial sale, and, to date, we have not generated any product revenue. We have financed our operations and internal growth primarily through equity and debt financings. We have devoted substantially all of our efforts to research and development, including current cGMP manufacturing and current Good Laboratory Practices (cGLP) compliant toxicology, safety pharmacology, genotoxicity studies and clinical studies for our lead clinical product candidates.
If we are unable to meet our needs for additional funding in the future, we will be required to limit, scale back or cease operations.
We do not currently have the funding resources necessary to carry out all of our proposed operating activities. We will need to obtain additional financing in the future in order to fully fund these product candidates through the regulatory approval process. Costs of our proposed activities unrelated to our two lead product candidates would be in addition to these estimates.
We believe that our current cash and investment balances as of the date of this filing will be sufficient to fund the completion of our ongoing Phase 2 studies for each PMX-30063 and PMX-60056, as well as fund our operations for at least the next 12 months. If we are unable to secure adequate additional funding in the future, we may delay, scale back or eliminate certain of our planned research, drug discovery and development activities and certain other aspects of our operations and our business until such time as we are successful in securing adequate additional funding. As a result, our business may be materially and adversely affected.
Our future capital requirements will depend on many factors, including:
Ø the timing and success of our clinical trials for PMX-30063 and PMX-60056;
Ø continued progress of and increased spending related to our research and development activities, including our plan to hire additional research and development employees;
18
Ø the conditions in the capital markets and the biopharmaceutical industry that make raising capital or entering into strategic arrangements difficult and expensive;
Ø progress with preclinical experiments and clinical trials, including regulatory approvals necessary for advancement and continuation of our development programs;
Ø changes in regulatory requirements and guidance of the FDA and other regulatory authorities, which may require additional clinical trials to evaluate safety and/or efficacy, and thus have significant impacts on our timelines, cost projections, and financial requirements;
Ø ongoing general and administrative expenses related to our being a reporting company;
Ø the cost, timing, and results of regulatory reviews and approvals;
Ø the maintenance of our existing licenses with the Penn and UMass;
Ø the success, timing, and financial consequences of any future collaborative, licensing and other arrangements that we may establish;
Ø the cost of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights;
Ø the costs of commercializing any of our other product candidates;
Ø technological and market developments;
Ø the cost of manufacturing development; and
Ø timing and volume of sales of products for which we obtain marketing approval.
These factors could result in variations from our currently projected operating and liquidity requirements. Additional funds may not be available when needed, or, if available, we may not be able to obtain such funds on terms acceptable to us. If adequate funds are unavailable, we may be required, among other things, to:
Ø delay, reduce the scope of or eliminate one or more of our research or development programs;
Ø license rights to technologies, product candidates or products at an earlier stage than otherwise would be desirable or on terms that are less favorable to us than might otherwise be available; or
Ø obtain funds through arrangements that may require us to relinquish rights to product candidates or products that we would otherwise seek to develop or commercialize by ourselves.
Funding, especially on terms acceptable to us, may not be available to meet our future capital needs because of the state of the credit and capital markets.
Global market and economic conditions have been, and continue to be, disruptive and volatile. The debt and equity capital markets have been impacted by significant write-offs in the financial services sector and the re-pricing of credit risk in the broadly syndicated market, among other things. These events have negatively affected general economic conditions and it is uncertain when the credit and capital markets will stabilize or improve.
In particular, the cost of raising money in the debt and equity capital markets has increased substantially while the availability of funds from those markets has diminished significantly. Also, as a result of concern about the stability of financial markets generally and the solvency of counterparties specifically, the cost of obtaining money from the credit markets has increased as many lenders and institutional investors have increased interest rates, enacted tighter lending standards and reduced and, in some cases, ceased to provide funding to borrowers. Low valuations and decreased appetite for equity investments, among other factors, may make the equity markets difficult to access on acceptable terms or unavailable altogether.
If funding is not available when needed, or is available only on unfavorable terms, meeting our capital needs or otherwise taking advantage of business opportunities may become challenging, which could have a material adverse effect on our business plans.
19
We have a Loan and Security Agreement with Hercules Technology II, L.P., who is a secured creditor of ours and has certain remedies available to it in the event we default on our loan.
If we become unable to make our principal and interest payments to Hercules on a timely basis, fail to uphold any of the covenants set forth in the loan and security agreement, experience a material adverse effect, or experience any other event of default under the loan and security agreement, we will be in default of our loan obligations. We have granted Hercules a security interest in our personal property other than intellectual property. Hercules, as a secured creditor, has certain remedies available to it in the event of our default which could result in the acceleration of any of our obligations under the agreement, our inability to request further advances, and seizure of some or all of our assets to satisfy our loan obligations.
We may not be able to access sufficient funds under our Equity Line with Dutchess Opportunity Fund, II, when needed.
Our ability to put shares to Dutchess and obtain funds under our Equity Line is limited by the terms and conditions in the Investment Agreement with Dutchess, including restrictions on when we may exercise our put rights, restrictions on the amount we may put to Dutchess at any one time, which is determined in part by the trading volume of our common stock, and a limitation on our ability to put shares to Dutchess to the extent that it would cause Dutchess to beneficially own more than 4.99% of our outstanding shares. In addition, we do not expect the Equity Line to satisfy all of our funding needs, even if we are able and choose to take full advantage of the Equity Line.
Development of our product candidates is a lengthy, expensive and uncertain process that requires investment of substantial amounts of time and money that may not yield viable products, which may cause our business and results of operations to suffer.
We face the risks of failure inherent in developing drugs based on new technologies and particularly those with novel mechanisms of action. Product candidates with novel mechanisms of action may receive increased scrutiny by the FDA and other regulatory bodies due to the greater uncertainty and questions regarding potential novel toxicities, which could result in requirements for additional safety and other studies, which, if required, would increase the time and cost of development. None of our product candidates have received regulatory approval for commercial sale and our product candidates may never be commercialized. In addition, all of our product candidates are in the early stages of development and certain of our programs are on hold until we are able to obtain and allocate sufficient resources, including financing. The progress and results of any ongoing or future clinical trials or future pre-clinical testing are uncertain, and if our product candidates do not receive regulatory approvals, our business, operating results, financial condition and cash flows will be materially adversely affected. Our product candidates are not expected to be commercially available for several years, if at all. Our development programs require a significant amount of cash to support the development of product candidates. We will need to obtain additional financing in the future. We may not be able to obtain such additional financing on terms acceptable to us or at all.
In addition, a number of potential drugs, including drugs intended to be utilized for similar indications as our product candidates, have shown promising results in early studies, but failed in subsequent clinical trials and/or failed to obtain necessary regulatory approvals. Data obtained from such studies are susceptible to varying interpretations, which may delay, limit or prevent regulatory approval. Regulatory authorities may refuse or delay approval as a result of many other factors, including changes in regulatory policy during the period of product development.
The FDA is actively working to revise certain of its guidance and in August 2010 issued draft guidance for clinical trial conduct, evaluation, clinical endpoints, and requirements for regulatory approval for antibiotic product candidates intended for use in acute bacterial infections, including skin and skin structure infections (ABSSSI), which has been the intended first clinical indication for PMX-30063. Based on the potential impacts of revised final FDA guidance, there may be delays in the conduct of clinical trials in the United States. Furthermore, there has not been a new product candidate for heparin reversal considered by the FDA in recent years, and there has never to date been a product candidate for reversal of LMWH considered by the FDA. The FDA and/or other foreign regulatory authorities may in the future request clinical trial parameters that result in clinical trials that are too expensive or too difficult to demonstrate pharmacoeconomic and therapeutic benefits compared to current therapy. As a result, our present plans for clinical development, including the cost and timing for development of our product candidates, may ultimately be substantially different than our present expectations, and there is no way of predicting what impacts on our timelines, cost projections, and clinical trials requirements may result from these changes in policy, requirements, standards, and guidance of the FDA and other regulatory authorities.
20
Completion of clinical trials may take many years. The length of time required varies substantially according to the type, complexity, novelty and intended use of the product candidate. The FDA and/or other foreign regulatory authorities monitor the progress of each phase of testing, and may require the modification, suspension, or termination of a trial if it is determined to present excessive risks to patients. The clinical trial process may also be accompanied by substantial delay and expense and the data generated in these studies ultimately may not be sufficient for marketing approval by the FDA and/or other foreign regulatory authorities. Our rate of commencement and completion of clinical trials may be delayed by many factors, including:
Ø our inability to manufacture or obtain sufficient quantities of materials for use in clinical trials;
Ø variability in the number and types of patients available for each study;
Ø difficulty in maintaining contact with patients after treatment, resulting in incomplete data;
Ø safety issues or side effects;
Ø poor or unanticipated effectiveness of products during the clinical trials;
Ø government or regulatory delays; and/or
Ø changes in regulatory requirements and guidance of the FDA and other regulatory authorities, which may require additional clinical trials to evaluate safety and/or efficacy, and thus have significant impacts on our timelines, cost projections, and financial requirements.
Notwithstanding our efforts during the application process and the submission of any requested additional information, the FDA and/or other foreign regulatory authorities ultimately may decide that the application does not satisfy the regulatory criteria for approval and refuse to approve the application. Furthermore, the FDA and/or other foreign regulatory authorities may prevent us from marketing a product candidate under a label for its desired indications or place other conditions, including restrictive labeling, on distribution as a condition of any approvals, which may impair commercialization of the product.
We are a development stage company, which makes it difficult to evaluate our existing business and business prospects and increases the risk that the value of any investment in our company may decline.
We are a development stage company and to date, our only revenues have been from research grants and contracts. We will not be able to generate revenue from product sales or royalties unless and until we receive regulatory approval and begin commercialization of our product candidates or otherwise out‑license our compounds. We are not certain of when, if ever, that will occur. Although we intend to introduce products, we may not do so. Because the expected markets for our products are not established, uncertain and evolving, and because we are a development stage company, it is difficult to assess or predict the growth rate, if any, and the size of this market. We may not develop additional product candidates or be able to introduce products, a market for our products may not develop, and/or our products may not achieve market acceptance.
If our product candidates are not demonstrated to be sufficiently safe and effective in clinical trials, they will not receive regulatory approval and we will be unable to commercialize them and our business and results of operations will suffer.
Our product candidates must satisfy rigorous standards of safety and efficacy before they can be approved by the FDA and/or other foreign regulatory authorities for commercial use. The FDA and foreign regulatory authorities have full discretion over this approval process. We will need to conduct significant additional research, involving testing in animals and in humans, before we can file applications for product approval. Typically, in the pharmaceutical industry there is a high rate of attrition for product candidates in pre‑clinical testing and clinical trials. Also, satisfaction of regulatory requirements typically takes many years, is dependent upon the type, complexity and novelty of the product and requires the expenditure of substantial resources. Success in pre‑clinical testing and early clinical trials does not ensure that later clinical trials will be successful. For example, a number of companies in the pharmaceutical industry, including biotechnology companies, have suffered significant setbacks in advanced clinical trials, even after promising results in earlier trials and in interim analyses. In addition, delays or rejections may be encountered based upon additional government regulation, including any changes in regulatory policy, during the process of product development, clinical trials and regulatory approvals.
21
If we are not able to retain our current management and advisory team and attract and retain qualified scientific, technical and business personnel, our business will suffer.
We are highly dependent on our executive officers and other key management and technical personnel, including Nicholas Landekic, Richard Scott, Ph.D., Edward Smith, and Bozena Korczak, Ph.D. The loss of any of them could have a material adverse effect on our future operations. We presently do not maintain “key person” life insurance policies on any of our personnel.
Our success is also dependent on our ability to attract, retain and motivate highly trained technical, marketing, sales and management personnel for the development, maintenance and expansion of our activities. We may not be able to retain our existing personnel or attract additional qualified employees. The loss of key personnel or the inability to hire and retain additional qualified personnel in the future could have a material adverse effect on our business, financial condition and results of operation.
Our success is dependent on the continuation of certain licensing arrangements and other strategic relationships with third parties. These arrangements and relationships may not continue and we may not be successful in entering into other similar arrangements and relationships.
All of our product candidates are licensed from or based upon licenses from either Penn or UMass. If either or any of these license agreements, or the related consulting arrangements, are properly terminated, our ability to advance our current product candidates or develop new product candidates will be materially adversely affected.
We depend, and will continue to depend, on these arrangements, and potentially on other licensing arrangements and/or strategic relationships with third parties for the research, development, manufacturing and commercialization of our product candidates. If any of our licenses or relationships are terminated or breached, we may:
Ø lose our rights to develop and market our product candidates;
Ø lose patent and/or trade secret protection for our product candidates;
Ø experience significant delays in the development or commercialization of our product candidates;
Ø not be able to obtain any other licenses on acceptable terms, if at all; and/or
Ø incur liability for damages.
Licensing arrangements and strategic relationships in the pharmaceutical and biotechnology industries can be very complex, particularly with respect to intellectual property rights. Disputes may arise in the future regarding ownership rights to technology developed by or with other parties. These and other possible disagreements between us and third parties with respect to our licenses or our strategic relationships could lead to delays in the research, development, manufacture and commercialization of our product candidates. These third parties may also pursue alternative technologies or product candidates either on their own or in strategic relationships with others in direct competition with us. These disputes could also result in litigation or arbitration, both of which are time consuming and expensive, and could require significant time and attention from our management.
Even if regulatory authorities approve our product candidates, they may not be commercially successful.
Our product candidates may not be commercially successful because physicians, government agencies and other third party payors may not accept them, or the potential markets may prove smaller than we estimate. Third parties may develop superior products or have proprietary rights that preclude us from marketing our products. We also expect that most of our product candidates will be very expensive, if approved. If we do obtain regulatory approval for any of our product candidates, we will need to achieve patient acceptance and demand in order to be commercially successful. Patient acceptance of and demand for any product candidates will depend upon many factors, including but not limited to, the extent, if any, of reimbursement of drug and treatment costs by government agencies and other third party payers, pricing, the safety and effectiveness of alternative products, and the prevalence and severity of side effects associated with our products. If we are unable to demonstrate pharmacoeconomic and therapeutic benefits, we will not achieve product acceptance and sufficient demand, and our operating results and financial condition will be materially adversely affected.
We do not currently have sales, marketing or distribution capabilities. If we fail to effectively sell, market and distribute any product candidate for which we receive regulatory approval, our business and results of operations will suffer.
If we are unable to create sales, marketing and distribution capabilities or enter into agreements with third parties to perform these functions, we will not be able to successfully commercialize any of our product candidates that may receive regulatory approval in the future. We currently have no sales, marketing or distribution capabilities. In order to successfully commercialize any of our product candidates, we must either internally develop sales, marketing and distribution capabilities or make arrangements with third parties to perform these services.
22
If we do not develop a marketing and sales force with technical expertise and supporting distribution capabilities, we will be unable to market any of our products directly. To promote any of our products through third parties, we will have to locate acceptable third parties for these functions and enter into agreements with them on acceptable terms and we may not be able to do so. In addition, any third‑party arrangements we are able to enter into may result in lower revenues than we could have achieved by directly marketing and selling our products.
We may suffer losses from product liability claims.
Any of our product candidates could cause adverse events to patients, such as immunologic or allergic reactions. These reactions may not be observed in clinical trials, but may nonetheless occur after commercialization. If any of these reactions occur, they may render our product candidates ineffective in some patients and our sales would suffer.
We may be susceptible to product liability lawsuits from events arising out of the use of product candidates in clinical studies. If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of any products or product candidates. Our business exposes us to potential product liability risks, which are inherent in the testing, manufacturing, marketing and sale of pharmaceutical products. We may not be able to avoid product liability claims. Product liability insurance for the pharmaceutical and biotechnology industries is generally expensive, if available at all. If we are unable to protect against potential product liability claims, we may be unable to commercialize our product candidates. A successful product liability claim brought against us in excess of our insurance coverage may cause us to incur substantial liabilities and, as a result, our business may fail.
Due to our reliance on third party manufacturers, suppliers and research organizations, we may be unable to implement our manufacturing, supply and clinical operations strategies, which would materially harm our business.
If our current and future licensing, manufacturing and supply strategies are unsuccessful, then we may be unable to complete any preclinical or clinical trials and/or commercialize our product candidates in a timely manner, if at all. Completion of any preclinical or clinical trials and/or commercialization of our product candidates will require access to, or development of, facilities to manufacture a sufficient supply of our product candidates, or the ability to license them to other companies to perform these functions. We do not have the resources, facilities or experience to manufacture our product candidates on our own and do not intend to develop or acquire facilities for the manufacture of product candidates for pre‑clinical trials, clinical trials or commercial purposes in the foreseeable future. We intend to continue to license technology and/or rely on contract manufacturers to produce sufficient quantities of our product candidates necessary for any pre‑clinical or clinical testing we undertake. Such contract manufacturers may be the sole source of production and may have limited experience at manufacturing, formulating, analyzing, filling and finishing our types of product candidates.
We also intend to rely on third parties to supply the components that we will need to develop, test and commercialize all of our product candidates. There may be a limited supply of these components. We might not be able to enter into agreements that assure us of the availability of such components in the future from any supplier. Our potential suppliers may not be able to adequately supply us with the components necessary to successfully conduct our pre‑clinical and clinical trials and/or to commercialize our product candidates. If we cannot acquire an acceptable supply of components to produce our product candidates, we will not be able to complete pre‑clinical and clinical trials and will not be able to commercialize our product candidates.
In addition, we rely on contract research organizations in conducting clinical trials for our product candidates. We do not have the resources, facilities or experience to conduct clinical studies for our product candidates on our own and do not intend to develop or acquire such resources, facilities or experience in the foreseeable future. The quality, cost and timing of work performed by our contracted contract research organizations has a significant impact on our clinical programs and our business.
23
If we make technology or product acquisitions, we may incur a number of costs, may have integration difficulties and may experience other risks that could harm our business and results of operations.
All of our product candidates are licensed from, or based upon technologies licensed from, third parties. We may acquire and/or license additional product candidates and/or technologies in the future. Any product candidate or technology we license or acquire will likely require additional development efforts prior to commercial sale, including extensive clinical testing and approval by the FDA and applicable foreign regulatory authorities, if any. All product candidates are prone to risks of failure inherent in biotechnology product development, including the possibility that the product candidate or product developed based on licensed technology will not be shown to be sufficiently safe and effective for approval by regulatory authorities. In addition, we cannot assure you that any product candidate that we develop based on acquired or licensed technology that is granted regulatory approval will be manufactured or produced economically, successfully commercialized or widely accepted in the marketplace. Moreover, integrating any newly acquired product could be expensive and time‑consuming. If we cannot effectively manage these aspects of our business strategy, our business may not succeed.
Furthermore, proposing, negotiating and implementing an economically viable acquisition or license can be a lengthy, costly and complex process. Other companies, including those with substantially greater financial, marketing and sales resources, may compete with us for the acquisition or license of product candidates and/or technologies. We may not be able to acquire the rights to alternative product candidates and/or technologies on terms that we find acceptable, or at all. Our failure to acquire or license alternative products and/or technologies could have a material adverse effect on our business, prospects and financial condition.
Failure to effectively manage our growth may have a material adverse effect on our business, results of operations and financial condition.
In November 2009 and March 2010, we obtained significant additional financing to fund the continued clinical development of our two lead drug candidates, PMX-30063 and PMX-60056. As a result, we have begun to expand our operations, including hiring of additional personnel. However, we may not be able to effectively grow and expand our business. Successful implementation of our business plan will require management of growth, which will result in an increase in the level of responsibility for management personnel. To manage growth effectively, we will be required to continue to implement and improve our operating and financial systems and controls to expand, train and manage our employee base. The management, systems and controls currently in place or to be implemented may not be adequate for such growth, and the steps taken to hire personnel and to improve such systems and controls might not be sufficient. If we are unable to manage our growth effectively, it will have a material adverse effect on our business, results of operations and financial condition.
Our executive officers and directors have the ability to significantly influence matters submitted to our stockholders for approval.
As of December 31, 2010, our executive officers and directors, in the aggregate, beneficially owned shares representing approximately 15% of our common stock. Beneficial ownership includes shares over which an individual or entity has investment or voting power and includes shares that could be issued upon the exercise of options and warrants within 60 days after the date of determination. On matters submitted to our stockholders for approval, holders of our common stock are entitled to one vote per share. If our executive officers and directors choose to act together, they would have significant influence over all matters submitted to our stockholders for approval, as well as our management and affairs. For example, these individuals, if they chose to act together, would have significant influence on the election of directors and approval of any merger, consolidation or sale of all or substantially all of our assets. This concentration of voting power could delay or prevent an acquisition of our company on terms that other stockholders may desire.
Risks Related to our Intellectual Property
We rely on third parties to provide software necessary to the future success of our business.
Currently, we rely on a non‑exclusive license from Penn to use, copy, perform, display, distribute, modify and prepare derivative works based on three software packages, which include a suite of proprietary computational algorithms that we use in the development, refinement, and testing of our product candidates. If this license agreement is properly terminated by Penn, our ability to advance our current product candidates or develop new product candidates may be adversely affected.
In the future, we expect to rely upon the software programs licensed from Penn, as well as software licensed from other third parties, including software that might be integrated with our software and used to perform key functions. If we license such third‑party software, it is likely that certain of these licenses may not contain favorable terms for us, including duration for limited terms, can be renewed only by mutual consent and may be terminated if we breach the terms of the license and fail to cure the breach within a specified period of time. Such licenses may not be available to us on commercially reasonable terms, if at all. The loss of or inability to maintain or obtain licenses on such third party software could result in the discontinuation of, or delays or reductions in, product availability unless and until equivalent technology is identified, licensed and integrated with our software. Any such discontinuation, delay or reduction would harm our business, results of operations and financial condition. In addition, financial or other difficulties that may be experienced by such third‑party vendors may have a material adverse effect upon the technologies that may be incorporated into our products. If such technologies become unavailable, we may not be able to find suitable alternatives, which could harm our business, operating results, and financial condition.
24
The obstacles to procurement and enforcement of our intellectual property and proprietary rights could harm our competitive position by allowing competitors access to our proprietary technology and to introduce competing products.
We regard our product candidates as proprietary and rely primarily on a combination of patents, trademarks, copyrights, and trade secrets and other methods to protect our proprietary rights. We maintain confidentiality agreements with our employees, consultants and current and potential affiliates, customers and business partners.
If we fail to secure and then enforce patents and other intellectual property rights underlying our product candidates and technologies, we may be unable to compete effectively. The pharmaceutical industry places considerable importance on obtaining patent and trade secret protection for new technologies, products and processes. Our success will depend, in part, on our ability, and the ability of our licensors, to obtain and to keep protection for our products and technologies under the patent laws of the U.S. and other countries, so that we can stop others from using our inventions. Our success also will depend on our ability to prevent others from using our trade secrets.
Our pending U.S. and foreign patent applications may not result in the issuance of patents to us or may not result in the issuance of patents that will be advantageous to us. If we do not receive patents for these applications or do not receive adequate protections, our developments will not have any proprietary protection and other entities will be able to make the products and compete with us. Also, any patents we have obtained or do obtain may be challenged by reexamination, opposition or other administrative proceeding, or may be challenged in litigation, and such challenges could result in a determination that the patent is invalid or unenforceable. In addition, competitors may be able to design alternative methods or devices that avoid infringement of our patents. To the extent our intellectual property protection offers inadequate protection, or is found to be invalid, we are exposed to a greater risk of direct competition. Both the patent application process and the process of managing patent disputes can be time consuming and expensive and may require significant time and attention from our management. Furthermore, the laws of some foreign countries may not protect our intellectual property rights to the same extent as do the laws of the United States.
In addition, the standards that the United States Patent and Trademark Office, (U.S. PTO), uses to grant patents can change. Consequently, we may be unable to determine the type and extent of patent claims that will be issued to us or to our licensors in the future, if any patent claims are issued at all. In addition, if the U.S. PTO and/or other patent offices where we file our patent applications increase the fees associated with filing and prosecuting patent applications we would incur higher expenses and our intellectual property strategy could be adversely affected.
The confidentiality agreements we require of our employees and those which we enter into with other parties may not provide adequate protection for our trade secrets, know‑how or other confidential information or prevent any unauthorized use or disclosure or the unlawful development by others. If any of our confidential intellectual property is disclosed, our business may suffer. Such agreements may not be enforceable or may not provide meaningful protection for our trade secrets or other proprietary information in the event of unauthorized use or disclosure or other breaches of the agreements, and we may not be able to prevent such unauthorized disclosure. Monitoring unauthorized disclosure is difficult, and we do not know whether the steps we have taken to prevent such disclosure are, or will be, adequate.
We may have to engage in costly litigation to enforce or protect our proprietary technology, which may harm our business, results of operations, financial condition and cash flows.
The pharmaceutical field is characterized by a large number of patent filings involving complex legal and factual questions, and, therefore, we cannot predict with certainty whether any patents or in‑licensed patents will be enforceable. Additionally, we may not be aware of all of the patents potentially adverse to our interests that may have been issued to others. Should third parties file patent applications, or be issued patents, claiming technology also claimed by us in pending applications, we may be required to participate in interference proceedings in the U.S. PTO to determine priority of invention. We, or our licensors, also could be required to participate in interference proceedings involving our issued patents and pending applications of another entity.
25
In the event a competitor infringes upon our patent or other intellectual property rights, litigation to enforce our intellectual property rights or to defend our patents against challenge, even if successful, could be expensive and time consuming and could require significant time and attention from our management. We may not have sufficient resources to enforce our intellectual property rights or to defend any patents against challenges from others. If our intellectual property does not provide adequate protection against our competitors’ products, our competitive position and business could be adversely affected.
Our commercial success depends significantly on our ability to develop and commercialize our products without infringing the intellectual property rights of third parties.
Our commercial success will depend, in part, on our not infringing the patents or proprietary rights of third parties. Third parties that believe we are infringing on their rights could bring actions against us claiming damages and seeking to enjoin clinical testing, manufacturing and marketing of the affected product or products.
If we become involved in any litigation, it could consume a substantial portion of our resources, regardless of the outcome of the litigation. If any of these actions are successful, we could be required, in addition to any potential liability for damages, to obtain a license to continue to manufacture or market the affected product, in which case we may be required to pay substantial royalties or grant cross‑licenses to our patents. However, any such license may not be available on acceptable terms or at all. Ultimately, we could be prevented from commercializing a product, or forced to cease some aspect of our business operations, as a result of patent infringement claims, which would harm our business.
We may enter into licensing agreements with third party intellectual property owners for use of their property in connection with our products in order to ensure that such third party’s rights are not infringed. Although we are not aware that any of our intended products would materially infringe the rights of others, a claim of infringement may be asserted against us and any such assertion may result in costly litigation or may require us to obtain a license in order to make, use, or sell our products. Third parties may assert infringement claims against us in the future with respect to current or future products. Any such claims or litigation, with or without merit, could be costly and a diversion of management’s attention, which could have a material adverse effect on our business, operating results and financial condition. Adverse determinations in such claims or litigation could harm our business, operating results and financial condition.
We may be unable to protect the intellectual property rights of the third parties from whom we license certain of our intellectual property or with whom we have entered into other strategic relationships, which may harm our business.
We are, and will continue to be, reliant upon such third parties to protect their intellectual property rights to this technology. Such third parties may determine not to protect the intellectual property rights that we license from them and we may be unable defend such intellectual property rights on our own or we may have to undertake costly litigation to defend the intellectual property rights of such third parties. We may not continue to have proprietary rights to the intellectual property that we license from such third parties or otherwise have the right to use through similar strategic relationships. Any loss or limitations on use with respect to our right to use such intellectual property licensed from third parties or otherwise obtained from third parties with whom we have entered into strategic relationships could have a material adverse effect on our business, operating results and financial condition.
Many countries, including certain countries in Europe, have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In addition, most countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may be limited to monetary relief and may be unable to enjoin infringement, which could materially diminish the value of the patent. Compulsory licensing of life‑saving products is also becoming increasingly popular in developing countries, through either direct legislation or international initiatives. Such compulsory licenses could be extended to include some of our product candidates, which may limit our potential revenue opportunities.
26
International patent protection is particularly uncertain, and if we are involved in opposition proceedings in foreign countries, we may have to expend substantial sums and management resources.
Patent law outside the U.S. is even more uncertain than in the U.S. and is periodically reviewed and revised in many countries. Further, the laws of some foreign countries may not protect our intellectual property rights to the same extent as U.S. laws. For example, certain countries do not grant patent claims that are related to the treatment of humans. We may participate in opposition proceedings to determine the validity of our foreign patents or our competitors’ foreign patents, which could result in substantial costs and diversion of our management’s efforts.
The government maintains certain rights to data and government use of our technology that we develop using government grant and contract money and we may lose the revenues from such technology if we do not commercialize and utilize the technology pursuant to established government guidelines.
Some of our research has been or are being funded in part by government grants and contracts. As a result of such funding, the U.S. Government has established guidelines and have certain rights in the technology developed with the grant and contract. If we fail to meet these guidelines, we would lose our exclusive rights to these products, and we would lose potential revenue derived from the sale of these products.
Risks Related to our Industry
We may experience delays in obtaining or we may not obtain required regulatory approvals in the U.S. to market our proposed product candidates.
Delays in regulatory approval, limitations in regulatory approval and withdrawals of regulatory approval may have a negative impact on our results of operations. If we experience significant delays in testing or approvals, or in our ability to license product candidates, our product development costs may increase. If the FDA grants regulatory approval of a product, this approval will be limited to those disease states and conditions for which the product has been demonstrated through clinical trials to be safe and effective. Any product approvals that we receive in the future could also include significant restrictions on the use or marketing of our products. Product approvals, if granted, can be withdrawn for failure to comply with regulatory requirements or upon the occurrence of adverse events following commercial introduction of the products. Failure to comply with applicable FDA or other applicable regulatory requirements may result in criminal prosecution, civil penalties, recall or seizure of products, total or partial suspension of production or injunction, as well as other regulatory action against our product candidates or us. If approvals are withdrawn for a product, or if a product were seized or recalled, we would be unable to sell or license that product and our revenues would suffer. In addition, outside the U.S., our ability to market any of our potential products is contingent upon receiving market application authorizations from the appropriate regulatory authorities and these foreign regulatory approval processes include all of the risks associated with the FDA approval process described above.
The FDA is actively working to revise certain of its guidance and in August 2010 issued draft guidance for clinical trial conduct, evaluation, clinical endpoints, and requirements for regulatory approval for antibiotic product candidates intended for use in acute bacterial infections, including skin and skin structure infections (ABSSSI), which has been the intended first clinical indication for PMX-30063. Based on the potential impacts of revised final FDA guidance, there may be delays in the conduct of clinical trials in the United States. Furthermore, there has not been a new product candidate for heparin reversal considered by the FDA in recent years, and there has never to date been a product candidate for reversal of LMWH considered by the FDA. The FDA and/or other foreign regulatory authorities may in the future request clinical trial parameters that result in clinical trials that are too expensive or too difficult to demonstrate pharmacoeconomic and therapeutic benefits compared to current therapy. As a result, our present plans for clinical development, including the cost and timing for development of our product candidates, may ultimately be substantially different than our present expectations, and there is no way of predicting what impacts on our timelines, cost projections, and clinical trials requirements may result from these changes in policy, requirements, standards, and guidance of the FDA and other regulatory authorities.
If competitors develop and market products that offer advantages as compared to our product candidates, our commercial opportunities will be limited.
Other companies have products and product candidates in development to treat the conditions we are seeking to ultimately treat. If these competitors are able to develop products that are more effective, have fewer side effects, are less expensive or offer other advantages as compared to our product candidates, our commercial opportunities will be limited. Furthermore, if our competitors commercialize competing products before we do, then our ability to penetrate the market and sell our products may be impaired.
27
Our competitors include fully integrated pharmaceutical companies and biotechnology companies, universities and public and private research institutions. Many of the organizations competing with us have substantially greater capital resources, larger research and development staffs and facilities, greater experience in drug development and in obtaining regulatory approvals, and greater manufacturing and marketing capabilities than we do. These organizations also compete with us to:
Ø attract parties for acquisitions, joint ventures or other collaborations;
Ø license proprietary technology that is competitive with the technology we are practicing;
Ø attract funding; and
Ø attract and hire scientific talent.
In the antibiotic market, many major pharmaceutical companies, such as GlaxoSmithKline, Pfizer, Bayer, Merck and Sanofi Aventis have already established significant positions. Additionally, many smaller companies, such as Astellas Pharma, Forest Laboratories, Cubist Pharmaceuticals, Basilea Pharmaceutica, Theravance, The Medicines Company, Trius Therapeutics, NovaBay Pharmaceuticals, Inc., Paratek, and Inhibitex either have marketed, or are attempting to enter this market by developing, novel and more potent antibiotics that are intended to be effective against drug‑resistant bacterial strains. In the UFH antagonist market, protamine is the only available antidote for and antagonist to UFH and, as such, protamine currently dominates this market. Because of protamine’s virtual monopoly of the UFH antidote/antagonist market, we believe that it may be difficult for our future UFH antidote/antagonist products to penetrate this market. There may be additional competitive products about which we are not aware.
Recently enacted health care reform legislation may increase our costs, impair our ability to match our pricing with any such increased costs, and therefore could materially and adversely affect our business, financial condition and results of operations.
The Patient Protection and Affordable Care Act (“PPACA”) was signed into law on March 23, 2010. The PPACA was subsequently amended on March 30, 2010 by the Health Care and Education Reconciliation Act of 2010 (the “Reconciliation Act”). The PPACA and Reconciliation Act (collectively the “Act”) entail sweeping health care reforms with staggered effective dates from 2010 through 2018, and many provisions in the Act require the issuance of additional guidance from the U.S. Department of Labor, the Internal Revenue Service, the U.S. Department of Health & Human Services, and the states. This reform includes, but is not limited to: the implementation of a small business tax credit; required changes in the design of our healthcare policy including providing insurance coverage to part-time workers working thirty or more hours per week; “grandfathering” provisions for existing policies; state insurance exchanges; “pay or play” requirements; and a “Cadillac plan” excise tax. We are currently unable to determine the long-term impact of such legislation on our business. Since many provisions of the Act do not become operative until future years, we do not expect the Act to have a material adverse impact on our results of operations in 2011. However, health care reform as mandated and implemented under the Act and any future federal or state mandated health care reform could materially and adversely affect our financial position and results of operations by increasing our costs, hindering our ability to effectively match our cost of providing health insurance with our pricing and impeding our ability to attract and retain customers as well as potentially changing our business model or causing us to lose certain current competitive advantages.
Healthcare reform measures could adversely affect our business.
The business and financial condition of pharmaceutical companies is affected by the efforts of governmental and third party payors to contain or reduce the costs of healthcare. In the U.S. and in foreign jurisdictions there have been, and we expect that there will continue to be, a number of legislative and regulatory proposals aimed at changing the healthcare system. For example, in some countries other than the U.S., pricing of prescription drugs is subject to government control, and we expect proposals to implement similar controls in the U.S. to continue. The implementation of such additional controls could have the effect of reducing the prices that we are able to charge for any products we develop and sell through these plans. Prescription drug legislation and related amendments or regulations could also cause third party payors other than the federal government, including the states under the Medicaid program, to discontinue coverage for any products we develop or to lower reimbursement amounts that they pay.
Further federal, state and foreign healthcare proposals and reforms are likely. While we cannot predict the legislative or regulatory proposals that will be adopted or what effect those proposals may have on our business, including the future reimbursement status of any of our product candidates, the pendency or approval of such proposals could result in a decrease in our stock price or limit our ability to raise capital or to obtain strategic partnerships or licenses.
28
Because our activities may involve the use of hazardous materials, we may be subject to claims relating to improper handling, storage or disposal of these materials that could be time consuming and costly.
If we use biological and hazardous materials in a manner that causes injury, we may be liable for damages. Our research and development activities may involve the controlled use of potentially harmful biological materials as well as hazardous materials, chemicals and various radioactive compounds. We cannot completely eliminate the risk of accidental contamination or injury from the use, storage, handling or disposal of these materials. In the event of contamination or injury, we could be held liable for damages that result, and any liability could exceed our resources. We are subject to federal, state and local laws and regulations governing the use, storage, handling and disposal of these materials and specified waste products. The cost of compliance with these laws and regulations could be significant.
Risks Related to our Capital Stock
Our common stock price is volatile, our stock is highly illiquid, and any investment in our securities could decline substantially in value.
Our common stock is currently quoted on the OTC Bulletin Board. Trading of our stock through the OTC Bulletin Board is frequently thin and highly volatile. In light of our small size and limited resources, as well as the uncertainties and risks that can affect our business and industry, our stock price is expected to continue to be highly volatile and can be subject to substantial drops, with or even in the absence of news affecting our business. The following factors, in addition to the other risk factors described in this Form 10-K and the potentially low volume of trades in our common stock, may have a significant impact on the market price of our common stock, some of which are beyond our control:
• results and timing for achievements of preclinical and clinical milestones;
• commercial success of approved products;
• corporate partnerships;
• technological innovations by us or competitors;
• changes in laws and government regulations both in the U.S. and overseas;
• changes in key personnel at our company;
• developments concerning proprietary rights, including patents and litigation matters;
• public perception relating to the commercial value or safety of any of our product candidates;
• future sales of our common stock;
• future issuance of our common stock causing dilution;
• anticipated or unanticipated changes in our financial performance or future prospects;
• general trends related to the biopharmaceutical and biotechnological industries; and
• general conditions in the stock market
The stock market in general has experienced relatively large price and volume fluctuations. In particular, the market prices of securities of smaller biotechnology companies have experienced dramatic fluctuations that often have been unrelated or disproportionate to the operating results of these companies. Continued market fluctuations could result in extreme volatility in the price of our common stock, which could cause a decline in its value. You should also be aware that price volatility may be worse if the trading volume of the common stock remains limited or declines
A decline in the price of our common stock could affect our ability to raise further working capital and adversely impact our ability to continue operations.
A prolonged decline in the price of our common stock could result in a reduction in the liquidity of our common stock and a reduction in our ability to raise capital. Because a significant portion of our operations has been and will continue to be financed through the sale of equity securities, a decline in the price of our common stock could be especially detrimental to our liquidity and our operations. Such reductions may force us to reallocate funds from other planned uses and may have a significant negative effect on our business plans and operations, including our ability to develop our product candidates and continue our current operations. If our stock price declines, it may be more difficult to raise additional capital. If we are unable to raise sufficient capital in the future, and we are unable to generate funds from operations sufficient to meet our obligations, we will not be able to have the resources to continue our normal operations.
29
The market price for our common stock may also be affected by our ability to meet or exceed expectations of analysts or investors. Any failure to meet these expectations, even if minor, may have a material adverse effect on the market price of our common stock.
When we issue additional shares in the future, it will likely result in the dilution of our existing stockholders.
Our certificate of incorporation authorizes the issuance of up to 250,000,000 shares of common stock with a $0.001 par value and 10,000,000 preferred shares with a par value of $0.001, of which approximately 81,000,000 common shares were issued and outstanding as of December 31, 2010. We may need to increase our authorized share count in order to issue additional shares of common stock in the future. From time to time we also will increase the number of shares available for issuance in connection with our equity compensation plans and we may issue awards to our employees outside the terms of our stock plans. As of December 31, 2010, we had approximately 11,002,000 additional shares of common stock reserved for future issuance under our stock plans. Our board of directors may fix and determine the designations, rights, preferences or other variations of each class or series within each class of preferred stock and may choose to issue some or all of such shares to provide additional financing or acquire more businesses in the future.
Moreover, in the past, we issued warrants and options to acquire shares of common stock. As of December 31, 2010, we had warrants and options to purchase an aggregate of approximately 53,779,000 shares of our common stock outstanding. In addition, our Equity Line with Dutchess contemplates the issuance of up to 12,000,000 shares of our common stock to Dutchess, subject to certain restrictions and obligations.
The issuance of any securities for acquisition, licensing or financing efforts, upon conversion of any preferred stock or exercise of warrants and options, pursuant to our equity compensation plans, or otherwise may result in a reduction of the book value and market price of the outstanding shares of our common stock. If we issue any such additional shares, such issuance will cause a reduction in the proportionate ownership and voting power of all current stockholders. Further, such issuance may result in a change in control of our corporation.
Financial Industry Regulatory Authority (FINRA) sales practice requirements may also limit a stockholder’s ability to buy and sell our Common Stock.
FINRA has adopted rules that require that in recommending an investment to a customer, a broker‑dealer must have reasonable grounds for believing that the investment is suitable for that customer. Prior to recommending speculative low priced securities to their non‑institutional customers, broker‑dealers must make reasonable efforts to obtain information about the customer’s financial status, tax status, investment objectives and other information. Under interpretations of these rules, FINRA believes that there is a high probability that speculative low priced securities will not be suitable for at least some customers. FINRA requirements make it more difficult for broker‑dealers to recommend that their customers buy our common stock, which may limit your ability to buy and sell our stock and have an adverse effect on the market for our shares.
We have never paid dividends on our common stock and do not anticipate paying any in the foreseeable future.
We have never declared or paid a cash dividend on our common stock and we do not expect to pay cash dividends in the foreseeable future. If we do have available cash, we intend to use it to grow our business.
Sales of a substantial number of shares of our common stock into the public market, including shares of common stock that we may issue to Dutchess pursuant to our Equity Line, may result in significant downward pressure on the price of our common stock and could affect your ability to realize the current trading price of our common stock.
We have registered the resale by Dutchess of a maximum of 12,000,000 shares of common stock that may be issued to Dutchess pursuant to our Equity Line. The common stock to be issued to Dutchess pursuant to the Investment Agreement with Dutchess will be sold at a 5% discount to the volume weighted average price (VWAP) of our common stock during the five consecutive trading day period beginning on the trading day immediately following the date of delivery of a put notice by us to Dutchess, subject to certain exceptions. Dutchess has a financial incentive to sell our common stock upon receiving the shares to realize the profit equal to the difference between the discounted price and the market price.
Sales of a substantial number of shares of our common stock in the public market, including shares of common stock that we may issue to Dutchess pursuant to our Equity Line, could cause a reduction in the market price of our common stock. To the extent stockholders sell shares of common stock, the price of our common stock may decrease due to the additional shares of common stock in the market.
30
Any significant downward pressure on the price of our common stock as stockholders sell their shares could encourage short sales by our stockholders or others. Any such short sales could place further downward pressure on the price of our common stock.
Delaware law, our stockholder rights plan, and our amended and restated certificate of incorporation and amended and restated bylaws contain provisions that could delay and discourage takeover attempts that stockholders may consider favorable.
Certain provisions of our amended and restated certificate of incorporation and our amended and restated bylaws and applicable provisions of Delaware corporate law may make it more difficult for or prevent a third party from acquiring control of us or changing our board of directors and management. These provisions include:
Ø the ability of our board of directors to issue preferred stock with voting or other rights or preferences;
Ø limitations on the ability of stockholders to amend our charter documents, including stockholder supermajority voting requirements;
Ø requirements that special meetings of our stockholders may only be called by the chairman of our board of directors, our president, or upon a resolution adopted by, or an affirmative vote of, a majority of our board of directors; and
Ø advance notice procedures our stockholders must comply with in order to nominate candidates for election to our board of directors or to place stockholders’ proposals on the agenda for consideration at meetings of stockholders.
We will also be afforded the protections of Section 203 of the Delaware General Corporation Law, which will prevent us from engaging in a business combination with a person who acquires at least 15% of our common stock for a period of three years from the date such person acquired such common stock, unless board or stockholder approval were obtained.
We review these provisions from time to time. Any delay or prevention of a change in control transaction or changes in our board of directors or management could deter potential acquirers or prevent the completion of a transaction in which our stockholders could receive a premium over the then current market price for their shares.
In addition, we have a stockholder rights plan pursuant to which we distributed rights to purchase shares of our Series C Preferred Stock. The rights become exercisable upon the earlier of ten business days after a public announcement that a person or group of affiliated or associated persons has acquired, or obtained the right to acquire, 15% or more of our common stock then outstanding or ten business days after the commencement of a tender or exchange offer that would result in a person or group beneficially owning 15% or more of our common stock then outstanding. These rights may cause substantial dilution to a person or group that attempts to acquire us (or which otherwise acquires 15% or more of our common stock) on terms or in a manner not approved by our board of directors, except pursuant to an offer conditioned upon the negation, purchase or redemption of these rights. Accordingly, these rights could delay or discourage someone from acquiring our business, even if doing so would benefit our stockholders.
Item 1B. Unresolved Staff Comments.
None.
We currently lease 24,223 square feet of office and laboratory facilities at 170 N. Radnor-Chester Road; Suite 300 in Radnor, Pennsylvania, pursuant to a twelve-year lease. All of our tangible personal property, consisting mainly of computers, office furniture and lab equipment, is located at our leased office and laboratory facilities and is in good operating condition and repair (subject to normal wear and tear).
To our knowledge, we are not a party to any material pending or threatened legal proceedings. To our knowledge, no governmental authority is contemplating commencing a legal proceeding in which we would be named as a party.
31
PART II
Item 5. Market for Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
Market Information
Since August 18, 2006, our Common Stock has been traded on the OTC Bulletin Board under the symbol “PYMX”. The market for our Common Stock is limited and volatile. The following table sets forth the range of high and low sales prices for our Common Stock for each of the periods indicated as reported by the OTC Bulletin Board. The prices quoted on the OTC Bulletin Board reflect inter-dealer prices, without retail mark-up, markdown or commissions. The OTC Bulletin Board prices listed below may not represent actual transaction prices.
|
|
Year Ended December 31, 2010 |
|
|
|
High |
Low |
|
First Quarter |
$ 1.38 |
$ 0.99 |
|
Second Quarter |
$ 1.18 |
$ 0.91 |
|
Third Quarter |
$ 1.04 |
$ 0.74 |
|
Fourth Quarter |
$ 1.08 |
$ 0.80 |
|
|
Year Ended December 31, 2009 |
|
|
|
High |
Low |
|
First Quarter |
$ 1.24 |
$ 0.63 |
|
Second Quarter |
$ 0.85 |
$ 0.64 |
|
Third Quarter |
$ 0.98 |
$ 0.63 |
|
Fourth Quarter |
$ 1.45 |
$ 0.93 |
Holders of Record
As of March 7, 2011, there were approximately 700 holders of record of shares of our Common Stock.
Dividends
Since our reincorporation in Delaware in March 24, 2005, we have not paid or declared any cash dividends on our Common Stock. We currently intend to retain any earnings for future growth and, therefore, do not expect to pay cash dividends on our Common Stock in the foreseeable future.
Recent Sales of Unregistered Securities
We have not made any sales of unregistered securities over the past three years that have not been previously disclosed in our filings with the Securities and Exchange Commission.
33
Item 6. Selected Financial Data
The following selected financial data is derived from our audited financial statements and should be read in conjunction with, and is qualified in its entirety by, Item 7 “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and Item 8 “Financial Statements and Supplementary Data.”
|
YEAR ENDED DECEMBER 31, |
||||||||||
|
(in thousands) |
2010 |
2009 |
2008 |
2007 |
2006 |
|||||
|
|
|
|
|
|
||||||
|
Operations data: |
|
|
|
|
|
|
|
|
|
|
|
Revenues |
$ 2,144 |
$ 1,048 |
$ 1,066 |
$ 1,126 |
$ 821 |
|||||
|
Operating expenses |
|
16,718 |
|
12,931 |
|
12,276 |
|
13,801 |
|
7,480 |
|
Net loss |
|
(15,516) |
|
(11,829) |
|
(10,986) |
|
(12,164) |
|
(5,966) |
|
Beneficial conversion and conversion inducement on Preferred Stock |
- |
- |
(5,845) |
(2,247) |
(2,899) |
|||||
|
Net loss attributable to common shareholders |
|
(15,516) |
|
(11,829) |
|
(16,831) |
|
(14,411) |
|
(8,865) |
|
Net loss per share - basic and diluted |
|
$ (0.19) |
|
$ (0.19) |
|
$ (0.40) |
|
$ (0.61) |
|
$ (0.72) |
|
Balance sheet data: |
|
|
|
|
|
|
|
|
|
|
|
Cash and investments |
$ 20,821 |
$ 23,974 |
$ 15,106 |
$ 8,903 |
$ 14,529 |
|||||
|
Working capital |
|
14,281 |
|
22,864 |
|
13,404 |
|
5,101 |
|
13,587 |
|
Total assets |
22,445 |
25,392 |
16,077 |
9,722 |
15,142 |
|||||
|
Long-term debt and capital lease obligations |
|
6,550 |
|
- |
|
- |
|
104 |
|
69 |
|
Shareholders’ equity |
9,203 |
22,319 |
13,143 |
5,134 |
13,746 |
|||||
34
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
The following discussion and analysis should be read in conjunction with the consolidated financial statements including the notes thereto. This discussion and analysis may contain forward-looking statements based upon current expectations that involve risks and uncertainties. Our actual results may differ materially as a result of various factors, including those set forth under “Risk Factors” or elsewhere in this Form 10-K.
Overview
We are a development stage biotechnology company focused on developing treatments for patients with infectious diseases and acute cardiovascular disorders with synthetic small molecule compounds referred to as biomimetics. Using our proprietary computational drug design technology, we have created novel defensin mimetic antibiotic compounds, heparin antagonist compounds, and other drug compounds intended for human therapeutic use. Since 2002, we have been a development stage enterprise, and accordingly, our operations have been directed primarily toward developing business strategies, raising capital, research and development activities, conducting pre-clinical testing and human clinical trials of our product candidates, exploring marketing channels and recruiting personnel.
We have incurred operating losses since inception, have not generated any product sales revenues and have not achieved profitable operations. Our deficit accumulated during the development stage through December 31, 2010 was $64,204,000, and we expect to continue to incur substantial losses in future periods.
None of our product candidates have received regulatory approval for commercial sale and our product candidates may never be commercialized. In addition, all of our product candidates are in the early stages of development and all but two of our programs are either on hold pending additional funding or being developed only to the extent they are substantially funded by targeted grants or research contracts. The progress and results of our current and any future clinical trials or future pre-clinical testing are uncertain, and if our product candidates do not receive regulatory approvals, our business, operating results, financial condition and cash flows will be materially adversely affected. Our development programs require a significant amount of cash to support the development of product candidates.
We are highly dependent on the success of our research, development and licensing efforts and, ultimately, upon regulatory approval and market acceptance of our products under development. Our short and long-term capital requirements depend upon a variety of factors, including market acceptance for our technologies and product candidates and various other factors. We believe that our current cash and investment balances as of the date of this filing will be sufficient to fund our operations for at least the next 12 months.
In March 2010, we successfully completed a Phase 1B clinical study with our novel defensin-mimetic antibiotic, PMX-30063. The data from the study suggest that multiple doses of PMX-30063 can be administered at dose levels which showed bactericidal activity in blood samples drawn from subjects in the study. PMX-30063, a small-molecule mimetic of host defense proteins, has a novel mechanism of action distinct from other antibiotics, which is believed to make bacterial resistance unlikely to develop. In September 2010, we initiated a Phase 2 trial for PMX-30063.
In June 2010, we completed a Phase 1B/2 clinical study to evaluate the safety and efficacy of PMX-60056 in reversing the anticoagulant activity of the LMWH tinzaparin. In August 2010, we completed a Phase 1B/2 dose-ranging clinical study evaluating UFH reversal with PMX-60056. PMX-60056 met the study endpoints for each of these studies. In February 2011, we initiated a Phase 2 clinical trial to assess the safety and efficacy of PMX-60056, in reversing UFH in patients undergoing PCI procedures.
The following discussion and analysis provides information that we believe is relevant to an assessment and understanding of our results of operations for the years ended December 31, 2010, 2009, and 2008, and financial condition as of December 31, 2010 and 2009.
Critical Accounting Policies and Practices
The preparation of our consolidated financial statements in conformity with accounting principles generally accepted in the United States requires management to adopt critical accounting policies and to make estimates and assumptions that affect the amounts reported in our consolidated financial statements and accompanying notes. These critical accounting policies and estimates have been reviewed by our audit committee of our board of directors. The principal items in our consolidated financial statements reflecting critical accounting policies or requiring significant estimates and judgments are as follows:
35
Stock-based compensation
Since inception, we have used the Black-Scholes Merton formula to estimate the fair value of stock options and warrants and have elected to continue to estimate the fair value of stock options and warrants using the Black-Scholes Merton formula. The volatility and expected term assumptions have the most significant effect on the results obtained from the Black-Scholes Merton formula. We have to date assumed that stock options and warrants have an expected life of five years, representing about half of their contractual life, and assumed Common Stock volatility of between 41% and 75%. Higher estimates of volatility and expected life of the option increase the value of an option and the resulting expense. Given the absence of an active market for our Common Stock in prior periods, the fair value of our Common Stock has periodically been estimated using several criteria, including progress and milestones achieved in our research activities along with the price per share of our preferred and Common Stock offerings.
Results of Operations
Year ended December 31, 2010 compared to year ended December 31, 2009
Grant and contract revenues were $2,144,000 and $1,048,000 for the years ended December 31, 2010 and 2009, respectively. The increase was due to several new grants and contracts that commenced during the third quarter of 2009 and continued into the third quarter of 2010, as well as new grants that commenced in the fourth quarter of 2010.
Research and development expenses were $10,951,000 and $7,374,000 for the years ended December 31, 2010 and 2009, respectively. The increase was due to the additional Phase 1B activities completed in the second half of 2010 for PMX-60056, and drug manufacturing and other activities in preparation for and commencing Phase 2 clinical trials for PMX-30063 and PMX-60056, as well as increased costs associated with our research grants and contracts.
General and administrative expenses were $5,767,000 and $5,557,000 for the years ended December 31, 2010 and 2009, respectively. General and administrative expenses remained relatively consistent with the prior year’s level.
Interest income was $96,000 and $55,000 for the years ended December 31, 2010 and 2009, respectively. The increase was due to higher average cash and investment balances and higher interest rates in 2010.
Interest expense was $1,038,000 and $2,000 for the years ended December 31, 2010 and 2009, respectively. The increase was due to interest on our $10,000,000 long-term debt, which commenced on March 31, 2010 resulting in significantly higher interest expense than was incurred solely from our capital lease obligations in 2009.
Cash Flows
Operating Activities. Cash used in operating activities during the year ended December 31, 2010 increased to $12,414,000 as compared to $10,248,000 used in the year ended December 31, 2009. The increase was driven primarily by an increase in research and development costs to advance our lead product candidates through Phase 1B clinical trials and preparations for and commencement of Phase 2.
Investing Activities. Cash used for investing activities represents cash paid for purchases of investments and property and equipment, net of maturities of investments. During the year ended December 31, 2010 and 2009, we purchased $5,448,000 and $3,798,000 of investments, respectively. During the year ended December 31, 2010 and 2009, maturities of our investments were $3,800,000 and $11,703,000, respectively. During the years ended December 31, 2010 and 2009, property and equipment purchases were $474,000 and $38,000, respectively.
Financing Activities. To date, we have financed our operating and investing activities primarily from the proceeds from the sale of equity securities and issuance of debt. During the year ended December 31, 2010, we received net proceeds of $9,839,000 from the issuance of debt, and paid $106,000 in financing costs associated with our November 2009 equity financing and the filing of a shelf registration statement in 2010. During the year ended December 31, 2009, we received $18,753,000 in net proceeds from our November 2009 financing, $500,000 in proceeds from the exercise of outstanding warrants, and paid $104,000 in principal associated with our then outstanding capital lease obligations.
36
Year ended December 31, 2009 compared to year ended December 31, 2008
Grant and contract revenues were $1,048,000 and $1,066,000 for the years ended December 31, 2009 and 2008, respectively. The slight decrease in grant and contract revenues was the result of the timing of our expenditures and subsequent reimbursement related to our grant and contract activities, as well as several new grants and contracts which commenced during the third quarter of 2009. The grants and contracts, which commenced in the third quarter 2009, were not fully utilized as of December 31, 2009 due to the timing and extent of the activities commenced under each grant or contract.
Research and development expenses were $7,374,000 and $7,401,000 for the years ended December 31, 2009 and 2008, respectively. The slight decrease was due to the successful completion of Phase 1B clinical trials of our PMX-60056 product candidate at a significantly lower cost than Phase 1A, offset by our ongoing Phase 1B clinical trials of PMX-30063, as well as increased research activities which we have performed using grant and contract revenues.
General and administrative expenses were $5,557,000 and $4,875,000 for the years ended December 31, 2009 and 2008, respectively. The increase in general and administrative costs is primarily associated with increased legal fees and equity compensation expense.
Interest income and other expenses were $54,000 and $224,000 for the years ended December 31, 2009 and 2008, respectively. The decrease in interest income and other expenses was primarily the result of lower interest rates and decreased average cash and investment balances during 2009, resulting in less interest income.
Cash Flows
Operating Activities. Cash used in operating activities during the year ended December 31, 2009 decreased to $10,248,000 as compared to $10,926,000 used for the year ended December 31, 2008. The decrease was primarily due to increased grant and contract receivables slightly offset by increased current liabilities.
Investing Activities. Cash used for investing activities represents cash paid for purchases of investments and property and equipment, net of maturities of investments. During the year ended December 31, 2009 and 2008, we purchased $3,798,000 and $9,553,000 of investments, respectively. During the year ended December 31, 2009 and 2008, maturities of our investments were $11,703,000 and $5,700,000, respectively. During the years ended December 31, 2009 and 2008, property and equipment purchases were $38,000 and $0, respectively.
Financing Activities. We have financed our operating and investing activities primarily from the proceeds from the sale of equity securities. During the years ended December 31, 2009 and 2008, we received $18,753,000 and $16,966,000, respectively, in net proceeds of such issuances of equity securities. Additionally, during 2009 and 2008 we received $500,000 and $214,000, respectively from the exercise of warrants and stock options.
Financial Condition, Liquidity and Capital Resources
In March 2010, we closed a debt financing for initial gross proceeds of $10,000,000, and in November 2009, we closed an equity financing for gross proceeds of $20,700,000. Proceeds from these financing activities are being used to fund our ongoing clinical development of PMX-30063 and PMX-60056 and for general corporate purposes. We will seek additional funds through equity or debt financing, among other sources, or through government grants and contracts. However, we may not be able to obtain additional funding on favorable terms, if at all. If we are unable to secure adequate additional funding in the future, we may delay, scale-back or eliminate certain of our planned research, drug discovery and development activities and certain other aspects of our operations and our business until such time as we are successful in securing adequate additional funding. As a result, our business may be materially and adversely affected.
As of December 31, 2010 and 2009, we had cash and investment balances of approximately $20,821,000 and $23,974,000, respectively, and total liabilities of approximately $13,257,000 and $3,073,000, respectively. The decrease in our cash and investment balances was primarily attributable to the funding of our ongoing operations and clinical trials, offset by the proceeds received from our March 2010 debt financing. The increase in our total liabilities was primarily attributable to our March 2010 debt financing.
We presently have an Investment Agreement (Investment Agreement) with Dutchess Opportunity Fund, II, L.P. (formerly known as Dutchess Equity Fund, L.P.) (Dutchess), pursuant to which Dutchess committed to purchase up to $10,000,000 (but not to exceed 12,000,000 shares) of our common stock, subject to the terms and conditions of the Investment Agreement. As of December 31, 2010, we have not issued any shares pursuant to this Investment Agreement. This Investment Agreement expires in July 2012.
37
We are a development stage company and have not experienced significant revenue generating activities since our formation. We have incurred operating losses for each year since our inception in 2002. In the near term, we expect to continue to incur significant and increasing operating losses as a result of the research and development expenses we anticipate incurring in developing our product candidates and the general and administrative expenses we incur as a reporting company under the Securities Exchange Act of 1934, as amended. Additionally, we do not expect to generate any revenues from sources other than research grants and contracts for the foreseeable future.
We are subject to many risks associated with development-stage businesses, including the above-discussed risks associated with the ability to raise capital. Please see the section entitled “Risk Factors” in this Form 10-K for more information regarding risks associated with our business.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements as defined in Item 303(a)(4) of Regulation S-K.
Commitments and Contingencies
As described above, we believe our current cash and investment balances are adequate to fund operations, including the following commitments and contingencies, at least for the next twelve months. If we are unable to secure adequate additional funding during 2011, we may delay, scale back or eliminate certain of our future research, drug discovery or development activities or certain other aspects of our operations and our business until such time as we are successful in securing adequate additional funding, if at all.
Operating Lease
In June 2006, we entered into an operating lease agreement for 24,223 square feet of combined office and laboratory space located in Radnor, Pennsylvania. The initial term of the lease is 12 years. Payments under the lease commenced on December 1, 2006. Our annual future minimum lease payments under this non-cancelable operating lease are as follows (in thousands):
|
|
Operating Leases |
|
2011 |
$ 667 |
|
2012 |
686 |
|
2013 |
705 |
|
2014 |
724 |
|
2015 |
743 |
|
Thereafter |
2,144 |
|
Total minimum lease payments |
$ 5,669 |
Prior to the commencement of our current operating lease for our Radnor facility, we leased approximately 3,500 square feet of combined office and laboratory space on a month-to-month basis in Philadelphia, Pennsylvania. Rent expense was $598,000 for each of the three years ended December 31, 2010, and $3,073,000 for the period from August 8, 2002 (Inception) to December 31, 2010.
Contractual Obligations
On March 31, 2010, we entered into a Loan and Security Agreement (LSA) with Hercules Technology II, L.P. (HTGC) whereby we borrowed $10,000,000. This loan bears an interest rate at the greater of 12.35%, or the Federal Reserve Prime Rate plus 7.10%, not to exceed 14%, and has a term of 42 months with interest-only payments for the first nine months. In connection with this loan, we paid the following (in thousands):
|
|
Years Ended December 31, |
Period from August 8, 2002 (Inception) to |
|
|
|
2010 |
2009 |
December 31, 2010 |
|
Principal |
$ - |
$ - |
$ - |
|
Interest |
841 |
- |
841 |
|
Total principal and interest |
$ 841 |
$ - |
$ 841 |
38
Our annual future payments under this loan obligation are as follows (in thousands):
|
|
Principal |
Interest |
Total |
|
2011 |
$ 3,237 |
$ 1,070 |
$ 4,307 |
|
2012 |
3,664 |
643 |
4,307 |
|
2013 |
3,099 |
165 |
3,264 |
|
Total minimum loan payments |
$ 10,000 |
$ 1,878 |
$ 11,878 |
Patent License Agreements
University of Pennsylvania. In January 2003, we entered into a Patent License Agreement with the University of Pennsylvania, (Penn). Under the terms of the agreement, we were granted an exclusive, worldwide royalty-bearing license to make and sell products utilizing seven of Penn’s issued or pending patents for the life of such patents. One issued patent and five patent applications cover the composition of matter on antimicrobial compounds, including small molecules, oligomers and polymers. One patent application covers the composition and use of polycationic compounds for treating cancer. If a change-of-control event occurs, in which we transfer the license to these patents to a third party, we are acquired by another company, or we conduct an initial public offering of our securities, we are required to pay a 3% royalty on the gross sales for licensed products that are sold as pharmaceuticals and a 1.5% royalty on products sold as coatings for use in medical devices. We are permitted to sublicense the patents provided that (a) the sublicensee is prohibited from further licensing of the patents and (b) the sublicensee is subject to all of the terms of the original license granted to us. In addition, we are required to share with Penn any consideration we receive from sublicensing our patents to a third party.
University of Massachusetts. In January 2004, we entered into a sponsored research agreement with the University of Massachusetts (UMass) which, as amended, continued through March 2009. We are currently negotiating an extension of this agreement with UMass. Under the terms of this agreement, we have the exclusive option to license any intellectual property that may be generated by Dr. Gregory Tew pursuant to research sponsored under the agreement. We may exercise this option by issuing 7,500 shares of our Common Stock to UMass for each $100,000 of research conducted by Dr. Tew. If we exercise this option, we are also required to reimburse UMass for direct patent costs incurred by it for the patents licensed by us. During 2007, we issued 12,500 shares to UMass in connection with this agreement.
In January 2005, we entered into an Exclusive License Agreement with UMass, pursuant to which UMass granted us an exclusive, worldwide license to use, make and sell products under one U.S. patent application and seven corresponding foreign patent applications covering polynorborene co‑polymers and methods of use for the life of such patents. The patents, if issued, will expire in 2025. UMass may terminate the license agreement if we materially breach the agreement, including by nonpayment of amounts due under the agreement or in the case of a change in our ownership or control. If the license agreement is properly terminated by UMass, we may not be able to execute our strategy to develop and commercialize our PolyCides® for biomaterials applications or to develop and commercialize future product candidates utilizing the licensed patents.
Other
Agreements with Employees. We have entered into employment agreements with various executives. These agreements provide for severance arrangements and accelerated vesting of equity compensation awards in the event that the executive is terminated by us other than for cause or disability or if the executive resigns for good reason.
Credit Line. In November 2010, we terminated our then outstanding line and letter of credit and entered into a similar letter of credit agreement with a new financial institution to secure our payment obligations under our facility operating lease. This letter of credit is for $1,400,000, expires on December 1, 2011, and is secured by certificates of deposits held at the same financial institution.
Recently Issued Accounting Pronouncements
In January 2010, the Financial Accounting Standards Board (FASB) issued an Accounting Standards Update (ASU) relating to fair value disclosures, to add new requirements for disclosures about transfers into and out of Levels 1 and 2, and separate disclosures about purchases, sales, issuances, and settlements relating to Level 3 measurements. It also clarifies existing fair value disclosures about the level of disaggregation and about inputs and valuation techniques used to measure fair value. The guidance in this ASU is effective for the first reporting period (including interim periods) beginning after December 15, 2009, except for the requirement to provide the Level 3 activity of purchases, sales, issuances, and settlements on a gross basis, which will be effective for fiscal years beginning after December 15, 2010, and for interim periods within those fiscal years. The adoption of this guidance as it relates to Levels 1 and 2 investments did not have a material impact on our consolidated financial statements. We do not expect the adoption of this guidance as it relates to Level 3 investments to have a material impact on our consolidated financial statements.
39
In February 2010, the FASB issued an ASU which amends existing guidance regarding an entity’s requirement to perform and disclose subsequent events procedures. The amendments add definitions for “SEC filer” and “revised financial statements,” and remove the requirement for SEC filers to disclose the date through which subsequent events have been evaluated, and clarify that an SEC filer is to evaluate subsequent events through the date the financial statements are issued. The guidance in this ASU was effective immediately. The adoption of this guidance did not have a material impact on our consolidated financial statements.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk.
Interest Rate Risk
Our investment assets consist solely of certificates of deposit with a financial institution. The interest rates are fixed over the duration of the certificate, and are backed by the full faith and credit of the financial institution. We will hold such certificates to maturity at which time the certificate will be redeemed at face value, and all accrued interest will be paid. Due to the backing of the financial institution, we do not believe that we have a material exposure to interest rate risk related to our investment portfolio.
Our long-term debt carries a variable interest rate indexed to the prime rate, subject to a cap. The prime rate in the U.S. has remained at 3.25% since December of 2008. While we cannot predict when, if at all, this rate will be increased, the prime rate would have to increase by more than 2% to increase our current interest rate on our long-term debt, and our interest rate can not increase by more than 1.65%. We believe the stability of the prime rate over the past two years and our interest rate cap sufficiently mitigate interest rate risk related to our debt portfolio.
Foreign Exchange Risk
We have entered into some agreements denominated, wholly or partly, in Canadian Dollars, Euros or other foreign currencies, and, in the future, we may enter into additional, significant agreements denominated in foreign currencies. If the values of these currencies increase against the dollar, our costs would increase. To date, we have not entered into any contracts to reduce the risk of fluctuations in currency exchange rates. In the future, depending upon the amounts payable under any such agreements, we may enter into forward foreign exchange contracts to reduce the risk of unpredictable changes in these costs. However, due to the variability of timing and amount of payments under any such agreements, foreign exchange contracts may not mitigate the potential adverse impact on our financial results.
Item 8. Financial Statements and Supplementary Data.
Our consolidated financial statements, accompanying notes and Report of Independent Registered Public Accounting Firm are attached to this Annual Report on Form 10-K beginning on page F-1.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure.
None.
Item 9A. Controls and Procedures.
Conclusions regarding disclosure controls and procedures. An evaluation was performed under the supervision and with the participation of our management, including our Chief Executive Officer, or CEO, and Chief Financial Officer, or CFO, of the effectiveness of our disclosure controls and procedures, as such term is defined under Rule 13a-15(e) or 15d-15(e) promulgated under the Securities Exchange Act of 1934, as amended (the “Exchange Act”) as of the end of the period covered by this report. Based on that evaluation, our management, including the CEO and CFO, concluded that our disclosure controls and procedures are effective to ensure that information required to be disclosed by us in reports that we file or submit under the Exchange Act, is recorded, processed, summarized and reported, and also accumulated and communicated to our management, including the CEO and CFO, to allow timely decisions regarding required disclosure as specified in Securities and Exchange Commission rules and forms.
40
Management’s Report on Financial Statements. Our management is responsible for the preparation, integrity and fair presentation of information in our consolidated financial statements, including estimates and judgments. The consolidated financial statements presented in this report have been prepared in accordance with accounting principles generally accepted in the United States. Our management believes the consolidated financial statements and other financial information included in this report fairly present, in all material respects, our financial condition, results of operations and cash flows as of and for the periods presented in this report. The consolidated financial statements have been audited by Deloitte & Touche LLP, an independent registered public accounting firm, as stated in their report, which is included herein.
Management’s Report on Internal Control Over Financial Reporting. Our management is responsible for establishing and maintaining an adequate system of internal control over financial reporting. Our system of internal control over financial reporting is designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the United States.
Our internal control over financial reporting includes those policies and procedures that:
|
• |
|
Pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect our transactions and dispositions of our assets. |
|
• |
|
Provide reasonable assurance that our transactions are recorded as necessary to permit preparation of our financial statements in accordance with accounting principles generally accepted in the United States, and that our receipts and expenditures are being made only in accordance with authorizations of our management and our directors. |
|
• |
|
Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements. |
Because of its inherent limitations, a system of internal control over financial reporting can provide only reasonable assurance and may not prevent or detect misstatements. Further, because of changes in conditions, effectiveness of internal control over financial reporting may vary over time. Our system contains self-monitoring mechanisms, and actions are taken to correct deficiencies as they are identified.
Our management conducted an evaluation of the effectiveness of the system of internal control over financial reporting based on the framework in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on this evaluation, our management concluded that our system of internal control over financial reporting was effective as of December 31, 2010. The effectiveness of our internal controls over financial reporting has been audited by Deloitte & Touche LLP, an independent registered public accounting firm, as stated in their report, which is included herein.
Audit Committee Oversight. The Audit Committee of the Board of Directors, which is comprised solely of independent directors, has oversight responsibility for our financial reporting process, including our internal control over financial reporting, and the audits of our consolidated financial statements. The Audit Committee meets regularly with management and our independent registered public accounting firm to review matters related to the quality and integrity of our financial reporting, internal control over financial reporting (including compliance matters related to our Code of Ethics and Business Conduct), and the nature, extent, and results of internal and external audits. Our independent registered public accounting firm has full and free access and reports directly to the Audit Committee. The Audit Committee recommended, and the Board of Directors approved, that the audited consolidated financial statements be included in this Form 10-K.
Changes in internal control over financial reporting. There were no changes in our internal control over financial reporting identified in connection with the evaluation required by paragraph (d) of Exchange Act Rules 13a-15 or 15d-15 that occurred during our last fiscal quarter that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
41
PART III
Item 10. Directors and Executive Officers and Corporate Governance.
The following table sets forth the respective names, ages and positions of our executive officers and directors as of March 7, 2011:
|
Name |
|
Age |
|
Position(s) |
|
|
|
|
|
|
|
Nicholas Landekic |
|
52 |
|
President, Chief Executive Officer and Director |
|
Bozena Korczak |
|
58 |
|
Sr. Vice President, Drug Development and Chief Development Officer |
|
Richard W. Scott, Ph.D. |
|
57 |
|
Vice President, Research |
|
Edward F. Smith |
|
39 |
|
Vice President, Finance, Chief Financial Officer and Secretary |
|
Frank P. Slattery, Jr. |
|
73 |
|
Chairman of the Board of Directors |
|
Brian Anderson |
|
64 |
|
Director |
|
Richard W. Bank, M.D. |
|
77 |
|
Director |
|
Michael E. Lewis, Ph.D. |
|
59 |
|
Director |
|
Stefan Loren, Ph.D. |
|
47 |
|
Director |
|
Shaun F. O’Malley |
|
75 |
|
Director |
|
Douglas J. Swirsky |
|
41 |
|
Director |
|
|
|
|
|
|
Executive Officers and Directors
Nicholas Landekic, has served as President, Chief Executive Officer and Director of PolyMedix, Inc. since November 2005 and of PolyMedix Pharmaceuticals, Inc. where he served in the same capacity since inception in August 2002. Mr. Landekic has more than 20 years of pharmaceutical experience. From 2000 to 2002, he was President and Chief Executive Officer of Locus Discovery. From 1995 to 2000, Mr. Landekic was Senior Vice President of Corporate Development & Investor Relations at Guilford Pharmaceuticals. From 1991 to 1995, Mr. Landekic served as Senior Director of Business Development at Cephalon and, from 1988 to 1991, served as Senior Manager for Strategic Marketing at Bristol-Myers Squibb. He also held positions in Finance and Business Development at Johnson & Johnson Corporation (McNeil Pharmaceutical) from 1985 to 1988 and positions in the research laboratories at the Mt. Sinai Medical Center from 1982 to 1983. Mr. Landekic received an M.B.A. from the State University of New York at Albany, an M.A. in Biology from Indiana University and a B.S. in Biology from Marist College.
Bozena Korczak, Ph.D., has served as Senior Vice President, Drug Development and Chief Development Officer of PolyMedix, Inc. since January 2011 and prior to that Vice President, Drug Development of PolyMedix, Inc. since November 2007. Dr. Korczak has over 20 years of experience in discovery, pre‑clinical and clinical research at burgeoning biotechnology organizations. Prior to joining PolyMedix, Dr. Korczak was a principal for PharmaReach, a private drug development consulting company, from October 2005 to November 2007. From September 2001 to October 2005, Dr. Korczak was Vice President of Research and Development of Cytochroma, Inc., a private biotechnology company. Prior to Cytochroma, Inc., Dr. Korczak served in various positions of increasing responsibility at Glycodesign, Inc., Allelix Biopharmaceutical, Inc. and Mount Sinai Hospital. She is an author of thirty‑five peer review scientific papers and holds a Ph.D. in biochemistry from the Polish Academy of Science.
Richard W. Scott, Ph.D., has served as Vice President, Research of PolyMedix, Inc. since November 2005 and of PolyMedix Pharmaceuticals, Inc. since November 2002. Dr. Scott has approximately 20 years of biopharmaceutical industry experience. Most recently, he was Vice President of Biology at Cephalon, Inc. where he held positions of increasing responsibility from 1991 to 2001. From 1985 to 1990, Dr. Scott worked at the research laboratories of DuPont and Company. Dr. Scott has authored more than 45 papers and book chapters, and is named on six patents. Dr. Scott holds a Ph.D. in Microbiology from the University of Pennsylvania and a B.S. in Biology from Muhlenberg College.
43
Edward F. Smith, has served as Vice President, Finance and Chief Financial Officer of PolyMedix, Inc. since January 2006. Mr. Smith has approximately 15 years of combined biopharmaceutical industry and financial management experience. From 2000 to 2005, he was Executive Director of Finance at InKine Pharmaceutical Company, Inc. (acquired by Salix Pharmaceuticals, Ltd. in 2005). From 1993 to 1999, Mr. Smith held various positions of increasing responsibility in public accounting, most recently as a manager in the audit practice at Deloitte & Touche LLP. Mr. Smith is licensed as a Certified Public Accountant in Pennsylvania and holds a B.S. degree in Business Administration from the University of Hartford.
Frank Slattery, Jr., has served as Chairman of the Board of Directors of PolyMedix, Inc. since November 2005 and was a co‑founder of PolyMedix Pharmaceuticals, Inc. where he served in the same capacity since inception in August 2002. Frank Slattery was the President, Chief Executive Officer and Director of LFC Financial Corp. from 1969 to 1994. Mr. Slattery has founded and currently serves as the Chairman of the Board of Directors of several privately held companies, GelMed Inc., Probaris Technologies, Inc., MinusNine Inc., Knite, Inc., and NanoSelect, Inc. He also currently serves as a director of Clarient, Inc., a publicly held company, and as Vice Chairman of the Jefferson Health System. For the past year he was the Executive Director and CEO of The Philadelphia Orchestra Association, retiring in January 2010. Mr. Slattery holds an A.B. from Princeton University and a J.D. from the Law School of the University of Pennsylvania. We believe that Mr. Slattery’s business expertise, in particular his experience as an executive officer and as a board member of various technology-based companies, give him the qualifications and skills to serve as a Director of PolyMedix.
Brian Anderson, has served as a Director of PolyMedix, Inc. since January 2008. Since March 2007 he has served as an advisor to companies involved in the health care industry. From January 2006 until March 2007 Mr. Anderson was employed by Alkermes, Inc., in a market development capacity. Prior to that, from April 2004 to October 2005 Mr. Anderson was Chief Business officer for MediciNova,Inc., a publicly traded specialty pharmaceutical company. Mr. Anderson was an advisor to Montridge, Inc., a boutique investor relations firm from September 2002 to January 2004. He was President and CEO of Cognetix, Inc., a biotechnology company, from 1998 to 2002. Prior to that, from 1995 to 1998, Mr. Anderson was Senior Vice President for Commercial Development at Indevus (formerly Interneuron) Pharmaceuticals, a publicly traded biopharmaceutical company. Mr. Anderson has held various senior level positions in sales, marketing and business development at Bristol‑Myers Squibb and The Upjohn Company of Canada (which later became Pharmacia and Upjohn and is now a part of Pfizer). Mr. Anderson graduated from the University of Manitoba where he received his Bachelors degree. We believe that Mr. Anderson’s business expertise, in particular his experience as an executive officer in various capacities with both large and small life science organizations, give him the qualifications and skills to serve as a Director of PolyMedix.
Richard W. Bank, M.D., has served as a Director of PolyMedix, Inc. since July 2008. Dr. Bank is currently the President of BioVest Advisors, a consulting company, since July 2006. Dr. Bank has served as Senior Portfolio Manager, Managing Director, and Senior Vice President of the Liberty View Health Sciences Fund, a division of Liberty View Capital Management, a Lehman Brothers company, from July 2004 to July 2006. Dr. Bank has served as President and Managing Director of First-Tier Biotechnology Partners from February 1995 until it acquisition by Lehman Brothers in July 2004. From February 1995 through April 1996, Dr. Bank served as President and Secretary of Biomedical Sciences, Incorporated. He has also served as President and Secretary of BioVest Health Sciences, Incorporated since its organization in April 1996 to July 2004. Dr. Bank was Senior Research Analyst Director/Biotechnology SBC Warburg Dillon Read from 1998 to 1999. He was also Entrepreneur-In-Residence in Life Sciences for Tucker Anthony Sutro for 2000 through 2001. Dr. Bank received his B.S. from Washington & Lee University and his M.D. from Finch University of Health Sciences, The Chicago Medical School. We believe that Dr. Bank’s business and medical experience, in particular his experience as a portfolio manager and doctor of medicine, give him the qualifications and skills to serve as a Director of PolyMedix.
Michael E. Lewis, Ph.D., has served as a Director of PolyMedix, Inc. since November 2005 and of PolyMedix Pharmaceuticals, Inc. since its inception. Dr. Lewis has more than 30 years of scientific experience in academic and government laboratories and in several major pharmaceutical and biotechnology companies. Since 1994, Dr. Lewis has served as President of BioDiligence Partners, Inc., where he co‑founded three other biotechnology companies, Cara Therapeutics, Inc., Arena Pharmaceuticals, Inc. and Adolor Corporation. Dr. Lewis has served as Chief Scientific Advisor and a director of Cara Therapeutics, Inc. since its inception in 2004. He has also served as a director of Aeolus Pharmaceuticals, Inc. since 2004. After serving as Chief Scientific Advisor of Adolor Corporation from 1994 to 1997, Dr. Lewis served as Chief Scientific Advisor of Arena Pharmaceuticals, Inc. from 1997 to 2003, and as a director from 1997 to 2000. Dr. Lewis is a co‑founder of Cephalon, Inc. and served at Cephalon as Senior Scientist, Director of Pharmacology, and then as Senior Director of Scientific Affairs from 1988 to 1993. He supervised a molecular pharmacology research laboratory at the E.I. DuPont Co. from 1985 to 1987, and received postdoctoral training in pharmacology at the National Institutes of Health, the University of Michigan, and at the University of Cambridge. He received a Ph.D. from Clark University in 1977. We believe that Dr. Lewis’s business and scientific experience, in particular his experience as a founder, executive and director of life science organizations, give him the qualifications and skills to serve as a Director of PolyMedix.
44
Stefan D. Loren, Ph.D., has served as a Director of PolyMedix, Inc. since July 2008. Dr. Loren is currently a Managing Director of Westwicke Partners, a consulting company and a consultant to MTB Investment Advisors, a family of equity funds, and has held these positions since August, 2008. Prior to that, Dr. Loren was Analyst/Portfolio Manager with Perceptive Advisors, a health care hedge fund, from May 2007 to August, 2008 and MTB Investment Advisors, a family of equity funds, from August 2005 to May 2007. From July 1997 to August 2005, Dr. Loren was a Managing Director in the healthcare group at Legg Mason. Prior to that, Dr. Loren was a Research Chemist at the advanced technologies division of Abbott Laboratories and a research fellow at the Scripps Research Institute. Dr. Loren received his B.A. from the University of California, San Diego, and his Ph.D. from University of California, Berkeley. We believe that Dr. Loren’s business and scientific experience, in particular his experience as a sell-side and buy-side analyst and research chemist, give him the qualifications and skills to serve as a Director of PolyMedix.
Shaun F. O’Malley, has served as a Director of PolyMedix, Inc. since January 2006. Mr. O’Malley is currently the Chairman Emeritus of Price Waterhouse LLP, a title he has held since July 1995. Prior to 1995, he served as Chairman and Senior Partner of Price Waterhouse LLP. He currently serves as a member of the Board of Directors of the Finance Company of Pennsylvania. Mr. O’Malley holds a B.S. in Economics from the Wharton School of the University of Pennsylvania. We believe that Mr. O’Malley’s business and accounting experience, in particular his experience in public accounting and as a board member, give him the qualifications and skills to serve as a Director of PolyMedix.
Douglas J. Swirsky has served as a Director of PolyMedix, Inc. since June 30, 2009. Mr. Swirsky presently serves as Senior Vice President, Chief Financial Officer, Treasurer, and Corporate Secretary of GenVec, Inc., a biopharmaceutical company, a position he has held since 2006. Prior to joining GenVec, Mr. Swirsky was a Managing Director and the Head of Life Sciences Investment Banking at Stifel Nicolaus and held the same position at Legg Mason prior to Stifel Financial’s acquisition of the Legg Mason Capital Markets business in 2005. Mr. Swirsky, a Certified Public Accountant and a CFA charterholder, has also previously held investment banking positions at UBS, PaineWebber, and Morgan Stanley. His experience also includes positions in public accounting and consulting. Mr. Swirsky received his B.S. in Business in Administration from Boston University and his M.B.A. from the Kellogg School of Management at Northwestern University. We believe that Mr. Swirsky’s business and accounting experience, in particular his experience as an executive officer, investment banker and accountant, give him the qualifications and skills to serve as a Director of PolyMedix.
Executive officers of PolyMedix, Inc. and PolyMedix Pharmaceuticals, Inc. are appointed by the board of directors of PolyMedix, Inc. and PolyMedix Pharmaceuticals, Inc., respectively. The Chief Executive Officer, Treasurer and Secretary of PolyMedix, Inc. and PolyMedix Pharmaceuticals, Inc. are elected annually by the board of directors of PolyMedix, Inc. and PolyMedix Pharmaceuticals, Inc., respectively, at its first meeting following the annual meeting of stockholders. Other officers of PolyMedix, Inc. and PolyMedix Pharmaceuticals, Inc. may be appointed by the board of directors of PolyMedix, Inc. and PolyMedix Pharmaceuticals, Inc., respectively, at any meeting. Such officers shall hold office until his or her successor is elected and qualified, unless a different term is specified in the resolution electing or appointing such officer, or until such officer’s earlier death, resignation or removal.
All directors of PolyMedix, Inc. and PolyMedix Pharmaceuticals, Inc. are elected at each annual meeting of the stockholders of the PolyMedix, Inc. and PolyMedix Pharmaceuticals, Inc., respectively, to hold office until the next annual meeting of stockholders and until their successor is elected and qualified, or until such director’s earlier death, resignation or removal.
Corporate Governance
Our business, property and affairs are managed by, or under the direction of, our Board, in accordance with the General Corporation Law of the State of Delaware and our By‑Laws. Members of the Board are kept informed of our business through discussions with the Chief Executive Officer and other key members of management, by reviewing materials provided to them by management, and by participating in meetings of the Board and its Committees.
45
We continue to review our corporate governance policies and practices by comparing our policies and practices with those suggested by various groups or authorities active in evaluating or setting best practices for corporate governance of public companies. Based on this review, we have adopted, and will continue to adopt, changes that the Board believes are the appropriate corporate governance policies and practices for our Company. We have adopted changes and will continue to adopt changes, as appropriate, to comply with the Sarbanes‑Oxley Act of 2002 and subsequent rule changes made by the SEC and any applicable securities exchange.
Independence of Directors
The Corporate Governance Committee operates pursuant to a charter, which can be viewed on our website at www.polymedix.com (under “Investors/Governance”). In determining the independence of our directors, we apply the definition of “independent director” provided under the listing rules of The Nasdaq Stock Market LLC (NASDAQ). Pursuant to these applicable NASDAQ rules, the Board concluded its annual review of director independence in March 2010. After considering all relevant facts and circumstances, the Board affirmatively determined that all of the directors then serving on the Board, including those nominated for election at the Annual Meeting held on June 30, 2010, are independent of us under NASDAQ rules, with the exception of Nicholas Landekic, who is employed by us.
Committees of our Board of Directors
The Board has three committees: the Audit Committee (which was established in accordance with Section 3(a)(58)(A) of the Exchange Act; all Audit Committee members satisfy the independence standards of the Exchange Act and NASDAQ rules), the Compensation Committee, and the Governance Committee. Shaun O’Malley (Chairman), Stefan Loren, Doug Swirsky, and Frank Slattery are the current members of the Audit Committee. Brian Anderson (Chairman), Richard Bank, and Michael Lewis are the current members of the Compensation Committee. Stefan Loren (Chairman), Shaun O’Malley and Frank Slattery are the current members of the Governance Committee. Charters have been adopted for all committees.
Audit Committee
The Audit Committee consists of four non‑employee directors, all of whom are “independent” as defined under the rules of the SEC. In addition, the Board has determined that Shaun O’Malley, the Chairman of our Audit Committee, qualifies as an “audit committee financial expert” as defined in the rules of the SEC. The Audit Committee operates pursuant to a charter, which can be viewed on our website at www.polymedix.com (under “Investors/Governance”). The Audit Committee met four times during 2010 and no member missed any such meetings of the Audit Committee. The role of the Audit Committee is to:
Ø oversee management’s preparation of our financial statements and management’s conduct of the accounting and financial reporting processes;
Ø oversee management’s maintenance of internal controls and procedures for financial reporting;
Ø oversee our compliance with applicable legal and regulatory requirements, including without limitation, those requirements relating to financial controls and reporting;
Ø oversee the independent auditor’s qualifications and independence;
Ø oversee the performance of the independent auditors, including the annual independent audit of our financial statements;
Ø prepare the report required by the rules of the SEC to be included in our proxy statement; and
Ø discharge such duties and responsibilities as may be required of the Committee by the provisions of applicable law or rule or regulation of the Sarbanes‑Oxley Act of 2002.
Compensation Committee
The Compensation Committee consists of three non‑employee directors, all of whom are “independent” as defined in applicable NASDAQ rules. The Compensation Committee met three times during 2010 and no members missed any such meeting of the Compensation Committee. The role of the Compensation Committee is to:
46
Ø develop and recommend to the independent directors of the Board the annual compensation (base salary, bonus, stock options and other benefits) for our President/Chief Executive Officer;
Ø review, approve and recommend to the independent directors of the Board the annual compensation (base salary, bonus and other benefits) for all of our executives (Vice Presidents and above);
Ø review, approve and recommend to the Board the aggregate number of stock options to be granted to employees below the level of Vice President;
Ø ensure that a significant portion of executive compensation is reasonably related to the long‑term interest of our stockholders; and
Ø prepare certain portions of our annual proxy statement, including an annual report on executive compensation.
A copy of the charter of the Compensation Committee is available on our website at www.polymedix.com (under “Investors/Governance”).
The Compensation Committee may form and delegate a subcommittee consisting of one or more members to perform the functions of the Compensation Committee. The Compensation Committee may engage outside advisers, including outside auditors, attorneys and consultants, as it deems necessary to discharge its responsibilities. The Compensation Committee has sole authority to retain and terminate any compensation expert or consultant to be used to provide advice on compensation levels or assist in the evaluation of director, President/Chief Executive Officer or senior executive compensation, including sole authority to approve the search firm’s fees and other retention terms. During 2010, the Compensation Committee engaged Compensation Resources, Inc., or “Compensation Resources”, to provide salary, bonus, equity and other compensation data for executives for similarly sized companies in our industry. In connection with the engagement of Compensation Resources, the Compensation Committee instructed Compensation Resources to determine, and conduct a study of, a peer group based on other publicly traded companies in the same industry and similar size as us to analyze our position in the marketplace with respect to total compensation packages for our executive officers and non-employee directors. Compensation Resources was further instructed to clarify our executive compensation philosophy and address our competitive position in the marketplace, the appropriate mix of compensation elements, the extent to which our executives will be paid for performance, and the degree to which non-traditional compensation programs will be utilized. In addition, the Compensation Committee considers, but is not bound by, the recommendations of our Chief Executive Officer with respect to the compensation packages of our other executive officers.
Nominating and Corporate Governance Committee
The Nominating and Corporate Governance Committee, or the “Governance Committee”, consists of three independent directors, as that term is defined in applicable NASDAQ rules. The Governance Committee met three times during 2010 and no members missed any such meeting of the Governance Committee. The role of the Governance Committee is to:
Ø evaluate from time to time the appropriate size (number of members) of the Board and recommend any increase or decrease;
Ø determine the desired skills and attributes of members of the Board, taking into account the needs of the business and listing standards;
Ø establish criteria for prospective members, conduct candidate searches, interview prospective candidates, and oversee programs to introduce the candidate to us, our management, and operations;
Ø to review on an annual basis and recommend to the Board one member of the Board to serve as Chair;
Ø annually recommend to the Board persons to be nominated for election as directors;
Ø recommend to the Board the members of all standing Committees;
47
Ø adopt or develop for Board consideration corporate governance principles and policies; and
Ø provide oversight to the strategic planning process conducted annually by our management.
A copy of the charter of the Governance Committee is available on our website at www.polymedix.com (under “Investors/Governance”).
Director Qualifications and Diversity
The board seeks independent directors who represent a diversity of backgrounds and experiences that will enhance the quality of the board’s deliberations and decisions. Candidates shall have substantial experience with one or more publicly traded companies or shall have achieved a high level of distinction in their chosen fields. The board is particularly interested in maintaining a mix that includes individuals who are active or retired executive officers and senior executives, particularly those with experience in drug discovery and development; science and medicine; finance, accounting and banking; or marketing and sales.
In evaluating nominations to the Board of Directors, the Governance Committee also looks for certain personal attributes, such as integrity, ability and willingness to apply sound and independent business judgment, comprehensive understanding of a director’s role in corporate governance, availability for meetings and consultation on Company matters, and the willingness to assume and carry out fiduciary responsibilities. Qualified candidates for membership on the Board will be considered without regard to race, color, religion, sex, ancestry, national origin or disability.
Compensation Committee Interlocks and Insider Participation
During 2010, Brian Anderson, Richard Bank, and Michael Lewis served on the Compensation Committee. None of these individuals has ever been an officer or employee of ours. In addition, none of our executive officers serves as a member of the board of directors or compensation committee of any entity that has one or more executive officers serving as a member of our Board of Directors or the Compensation Committee.
Section 16(a) Beneficial Ownership Reporting Compliance
Based solely upon a review of reports of stock ownership (and changes in stock ownership) and written representations received by us, we believe that our directors and executive officers met all of their filing requirements under Section 16(a) of the Exchange Act during the year ended December 31, 2010.
Code of Conduct
We adopted a Code of Business Conduct and Ethics (“Code of Ethics”) applicable to our principal executive officer and principal financial and accounting officer and any persons performing similar functions. In addition, the Code of Ethics applies to our employees, officers, directors, agents and representatives. The Code of Ethics requires, among other things, that our employees avoid conflicts of interest, comply with all laws and other legal requirements, conduct business in an honest and ethical manner, and otherwise act with integrity and in our best interest.
The Code of Ethics includes procedures for reporting violations of the Code of Ethics. In addition, the Sarbanes-Oxley Act of 2002 requires companies to have procedures to receive, retain and treat complaints received regarding accounting, internal accounting controls or auditing matters and to allow for the confidential and anonymous submission by employees of concerns regarding questionable accounting or auditing matters. The Code of Ethics is intended to comply with the rules of the SEC and includes these required procedures. The Code of Ethics is available on our website at www.polymedix.com (under “Investor Relations”).
Risk Oversight
Enterprise risks are identified and prioritized by management and each prioritized risk is assigned to a board committee or the full board for oversight as follows:
Full Board - Risks and exposures associated with strategic, financial and execution risks and other current matters that may present material risk to our operations, plans, prospects or reputation.
48
Audit Committee - Risks and exposures associated with financial matters, particularly financial reporting, tax, accounting, disclosure, internal control over financial reporting, financial policies, investment guidelines and credit and liquidity matters.
Governance Committee - Risks and exposures relating to corporate governance; and management and director succession planning.
Compensation Committee - Risks and exposures associated with leadership assessment, and compensation programs and arrangements, including incentive plans.
Board Leadership Structure
The Chairman of the Board presides at all meetings of the Board. The Chairman is appointed on an annual basis by at least a majority vote of the remaining directors. Currently, the offices of Chairman of the Board and Chief Executive Officer are separated. The company has no fixed policy with respect to the separation of the offices of the Chairman of the Board and Chief Executive Officer. The Board believes that the separation of the offices of the Chairman of the Board and Chief Executive Officer is part of the succession planning process and that it is in the best interests of the company to make this determination from time to time.
49
Item 11. Executive Compensation.
The following summary compensation table sets forth information concerning compensation for services rendered in all capacities for the years ended December 31, 2010, 2009 and 2008 awarded to, earned by, or paid to: (i) Nicholas Landekic, who served as our President and Chief Executive Officer (our CEO) during 2010, 2009 and 2008, (ii) Edward F. Smith, who served as our Vice President, Finance, Chief Financial Officer and Corporate Secretary (our CFO) during 2010, 2009, and 2008 and (iii) our three most highly paid executive officers (as determined based on total compensation) other than our CEO and CFO as of December 31, 2010. These individuals are referred to in this report as the Named Executive Officers (or “NEOs”).
|
Name and Principal Position |
Year |
Salary ($) |
Bonus ($) (1) |
Stock Awards ($) (2) |
Option Awards ($) (2) |
All Other Compensation ($) |
Total ($) |
|
Nicholas Landekic President, Chief Executive Officer and Director |
2010 2009 2008 |
370,000 370,000 370,000 |
150,550 240,000 101,750 |
— — — |
1,210,861 (3) — 1,118,702 (4) |
— — — |
1,731,411 610,000 1,590,452 |
|
Edward F. Smith Vice President, Finance, Chief Financial Officer and Secretary |
2010 2009 2008 |
245,000 245,000 225,000 |
76,500 75,000 56,250 |
— — — |
206,672 (5) — 317,108 (6) |
— — — |
528,172 320,000 598,358 |
|
Bozena Korczak, Ph.D. Vice President, Drug Development |
2010 2009 2008 |
258,333 245,000 230,000 |
85,500 75,000 51,750 |
— — — |
317,957 (7) — 360,196 (8) |
— — — |
661,790 320,000 641,946 |
|
R. Eric McAllister, M.D., Ph.D. (13) Vice President, Clinical Development, Chief Medical Officer |
2010 2009 2008 |
280,000 280,000 280,000 |
36,000 50,000 63,000 |
— — — |
248,706 (9) — 492,966 (10) |
— — — |
538,570 330,000 835,966 |
|
Richard W. Scott, Ph.D. Vice President, Research |
2010 2009 2008 |
265,000 265,000 265,000 |
81,100 132,500 46,375 |
— — — |
256,883 (11) — 195,281 (12) |
— — — |
602,983 397,500 506,656 |
(1) Represents performance bonus awards. The 2008 bonus award was paid in 2009, the 2009 bonus award was paid in 2010, and the 2010 bonus award was paid in 2011.
(2) This column reflects the total grant date fair value for all stock options or awards granted in the applicable fiscal year pursuant to our equity compensation plans. The assumptions used in the calculation of these amounts are described in footnote 6 to our audited financial statements for the year ended December 31, 2010 and our discussion of stock‑based compensation in this annual report on Form 10‑K filed under “Management’s Discussion and Analysis Of Financial Condition and Results of Operations—Critical Accounting Policies and Practices” for the year ended December 31, 2010.
(3) This amount reflects the total grant date fair value of options to purchase common stock granted in 2010 to Mr. Landekic. 1,750,000 options were granted on February 24, 2010, and 187,500 options were granted on July 30, 2010, pursuant to our 2005 Omnibus Equity Compensation Plan
(4) This amount reflects the total grant date fair value of options to purchase common stock granted in 2008 to Mr. Landekic. 600,000 options were granted on January 23, 2008 and 1,000,000 options were granted on December 30, 2008, pursuant to our 2005 Omnibus Equity Compensation Plan except that 350,000 options from the January 23, 2008 were deemed to have been issued outside of the 2005 Omnibus Equity Compensation Plan.
(5) This amount reflects the total grant date fair value of 325,000 options to purchase common stock granted on February 24, 2010 to Mr. Smith, pursuant to our 2005 Omnibus Equity Compensation Plan.
(6) This amount reflects the total grant date fair value of options to purchase common stock granted in 2008 to Mr. Smith. 125,000 options were granted on January 23, 2008 and 325,000 options were granted on December 30, 2008, pursuant to our 2005 Omnibus Equity Compensation Plan.
(7) This amount reflects the total grant date fair value of 500,000 options to purchase common stock granted on February 24, 2010 to Dr. Korczak, pursuant to our 2005 Omnibus Equity Compensation Plan.
50
(8) This amount reflects the total grant date fair value of 500,000 options to purchase common stock granted on December 30, 2008 to Dr. Korczak, pursuant to our 2005 Omnibus Equity Compensation Plan.
(9) This amount reflects the total grant date fair value of options to purchase common stock granted in 2010 to Dr. McAllister. 350,000 options were granted on February 24, 2010, and 50,000 options were granted on July 30, 2010, pursuant to our 2005 Omnibus Equity Compensation Plan.
(10) This amount reflects the total grant date fair value of options to purchase common stock granted in 2008 to Dr. McAllister. 200,000 options were granted on January 23, 2008 and 500,000 options were granted on December 30, 2008, pursuant to our 2005 Omnibus Equity Compensation Plan.
(11) This amount reflects the total grant date fair value of options to purchase common stock granted in 2010 to Dr. Scott. 200,000 options were granted on February 14, 2010 and 225,000 options were granted on May 14, 2010, pursuant to our 2005 Omnibus Equity Compensation Plan.
(12) This amount reflects the total grant date fair value of options to purchase common stock granted in 2008 to Dr. Scott. 50,000 options were granted on January 23, 2008 and 225,000 options were granted on December 30, 2008, pursuant to our 2005 Omnibus Equity Compensation Plan.
(13) In January 2011, Dr. McAllister ceased to serve as an NEO in the position of Vice President, Clinical Development and Chief Medical Officer and assumed a new role as Vice President of Cardiovascular Clinical Development.
Narrative Disclosure to Summary Compensation Table
In 2010, the Compensation Committee of the Board awarded cash bonuses to each of our NEOs. These awards are reflected in the column titled “Bonus” in the Summary Compensation Table above. Such awards were made by the Compensation Committee in its sole discretion at the end of the year after reviewing the recommendations of our Chief Executive Officer for each NEO other than himself, individual goals and targets, the performance of each NEO in the applicable fiscal year, evaluating other components of each NEO’s total compensation package, including the balance of equity to non-equity compensation, each NEO’s total compensation package relative to executives in benchmark peer group companies holding similar positions, the report of the Compensation Committee’s consultant, and the Company’s cash position. For benchmarking executive compensation, the Compensation Committee engaged a compensation consulting firm which assisted in establishing a peer group of companies, analyzing peer company compensation data and comparing our compensation programs with the practices of the companies represented in the compensation data reviewed. For 2010, the Compensation Committee’s consulting firm, Compensation Resources, Inc. established a peer group of 20 publicly traded biotechnology companies of similar size to us. The Compensation Committee’s primary consideration in awarding the cash bonuses for 2010 was to award each NEO’s for their respective performance in the achievement of individual goals and objectives.
51
Outstanding Equity Awards
The following table provides information on all stock option awards held by our NEOs as of December 31, 2010. All outstanding equity awards are in shares of our common stock.
Outstanding Equity Awards at 2010 Fiscal Year‑End
|
|
Option Awards (1) | ||||
|
Name |
Number of Securities Underlying Unexercised Options Exercisable (#) |
Number of Securities Underlying Unexercised Options Unexercisable (#) |
Option Exercise Price ($) |
Option Expiration Date |
|
|
Nicholas Landekic President, Chief Executive Officer and Director |
1,000,000 (2) 500,000 (3) 231,000 (3) 583,333 (3) 666,667 (3) 486,111 (3) 26,042 (3)
|
— — — 16,667 333,333 1,263,889 161,458 |
1.50 1.50 2.85 1.10 1.18 1.05 0.90 |
08/10/2015 12/01/2015 02/04/2016 01/22/2018 12/29/2018 02/23/2020 07/29/2020 |
|
|
Edward F. Smith Vice President, Finance, Chief Financial Officer and Secretary |
250,000 (3) 30,000 (3) 121,528 (3) 216,667 (3) 90,278 (3)
|
— — 3,472 108,333 234,722 |
1.50 2.85 1.10 1.18 1.05 |
01/01/2016 02/04/2016 01/22/2018 12/29/2018 02/23/2020 |
|
|
Bozena Korczak, Ph.D. Vice President, Drug Development |
250,000 (3) 333,333 (3) 138,889 (3) |
— 166,667 361,111 |
0.98 1.18 1.05 |
11/12/2017 12/29/2018 02/23/2020 |
|
|
R. Eric McAllister, M.D., Ph.D. (5) Vice President, Clinical Development, Chief Medical Officer |
400,000 (4) 194,444 (3) 333,333 (3) 97,222 (3) 6,944 (3)
|
— 5,556 166,667 252,778 43,056 |
3.50 1.10 1.18 1.05 0.90 |
11/12/2016 01/22/2018 12/29/2018 02/23/2020 07/29/2020 |
|
|
Richard W. Scott, Ph.D. Vice President, Research |
250,000 (2) 40,000 (3) 48,611 (3) 150,000 (3) 55,556 (3) 43,750 (3)
|
— — 1,389 75,000 144,444 181,250 |
1.50 2.85 1.10 1.18 1.05 0.97 |
08/10/2015 02/04/2016 01/22/2018 12/29/2018 02/23/2020 05/13/2020 |
|
––––––––––––
(1) There are no restricted stock awards outstanding for any of the NEOs.
(2) Grants with an expiration date of August 10, 2015 have a stated term of ten years and vest 50% on the date of grant and 50% on the one‑year anniversary of the grant. If a “change in control” (as defined in our 2002 Equity Compensation Plan) were to occur, these options would become immediately exercisable in full.
(3) Grants with expiration dates of December 1, 2015, January 1, 2016, February 4, 2016, January 22, 2018, November 12, 2017, December 29, 2018, February 23, 2020, May 13, 2020 and July 29, 2020 have a stated term of ten years and vest in monthly installments over a three‑year period beginning after the date of grant. If a “change in control” (as defined in our 2005 Omnibus Equity Compensation Plan) were to occur, these options would become immediately exercisable in full.
(4) Grants with expiration dates of November 12, 2016 have a stated term of ten years and vest in 50% on the two‑year anniversary of the grant and the remaining 50% vests in monthly installments over a two‑year period beginning after the two‑year anniversary of the grant.
52
(5) In January 2011, Dr. McAllister ceased to serve as an NEO in the position of Vice President, Clinical Development and Chief Medical Officer and assumed a new role as Vice President of Cardiovascular Clinical Development.
Employment Contracts and Termination of Employment and Change‑in‑Control Arrangements
Nicholas Landekic
We extended the offer of employment to Nicholas Landekic for the position of President and Chief Executive Officer pursuant to an offer letter dated July 30, 2002, which provided that his annual salary will be at least $250,000 per year. Mr. Landekic’s base salary was increased to $350,000, 370,000 and $400,000 as of January, 2006, February 2007 and January 2011, respectively. Mr. Landekic is eligible to receive additional compensation depending upon achievement of performance goals as established by the Board of Directors. As an “at‑will” employee, Mr. Landekic’s employment can be terminated by us or by him, at any time and for any reason. In the event Mr. Landekic is terminated by us other than for “cause” or other than by reason of his “disability,” or he resigns for “good reason” (each as defined in his offer letter), Mr. Landekic will be entitled to full vesting of all unvested stock options and restricted stock previously granted to him and a cash payment equal to two‑years of his then current base salary.
Edward Smith
We extended the offer of employment to Edward Smith for the position of Vice President Finance, Chief Financial Officer and Corporate Secretary pursuant to an offer letter dated December 5, 2005, which provides that his annual salary will be at least $200,000 per year. Mr. Smith’s base salary was increased to $225,000, $245,000 and $267,500 as of February 2007, January 2009 and January 2011, respectively. Mr. Smith is eligible to receive additional compensation depending upon achievement of performance goals as established by the Board. As an “at‑will” employee, Mr. Smith’s employment can be terminated by us or by him, at any time and for any reason. In the event Mr. Smith is terminated by us other than for “cause” or other than by reason of his “disability” (each as defined in his offer letter), Mr. Smith will be entitled to full vesting of all unvested stock options previously granted to him and a cash payment equal to one–year of his then current base salary.
Bozena Korczak, Ph.D
We extended the offer of employment to Bozena Korczak for the position of Vice President, Drug Development pursuant to an offer letter dated November 5, 2007, which provides that her annual salary will be at least $230,000 per year. Dr. Korczak’s base salary increased to $245,000, $285,000, and $310,000 effective January 2009, September 2010 and January 2011, respectively. Dr. Korczak is eligible to receive additional compensation depending upon achievement of performance goals as established by the Board. As an “at‑will” employee, Dr. Korczak’s employment can be terminated by us or by her, at any time and for any reason. In the event Ms. Korczak is terminated by us other than for “cause” or other than by reason of his “disability” (each as defined in her offer letter), Dr. Korczak will be entitled to full vesting of all unvested stock options previously granted to her and a cash payment equal to one‑year of her then current base salary.
R. Eric McAllister, M.D., Ph.,D
We extended the offer of employment to R. Eric McAllister, M.D., Ph.D., for the position of Vice President, Clinical Development and Chief Medical Officer pursuant to an offer letter dated October 19, 2006, provides that his annual salary will be at least $280,000 per year. In January 2011, Dr. McAllister ceased to serve as an executive officer in the position of Vice President, Clinical Development and Chief Medical Officer and assumed a new role as Vice President of Cardiovascular Clinical Development. Dr. McAllister is eligible to receive a discretionary cash bonus based on his performance and our company’s performance. Dr. McAllister is eligible to receive additional compensation depending upon achievement of performance goals as established by the Board. As an “at‑will” employee, Dr. McAllister’s employment can be terminated by us or by him, at any time and for any reason. If Dr. McAllister is terminated by the Company following a Change in Control (as defined in his offer letter), other than by reason of his “Disability” (as defined in his offer letter), Dr. McAllister will be entitled to full vesting of all unvested stock options previously granted to him and a cash payment equal to twelve months of his then current base salary.
53
Richard Scott, Ph.D.
We extended the offer of employment to Richard Scott for the position of Vice President, Research pursuant to an offer letter dated September 23, 2002, which provides that his annual salary will be at least $150,000 per year. Dr. Scott’s base salary was increased to $250,000, $265,000 and $292,000 effective January 2006, February 2007 and January 2011, respectively. Dr. Scott is eligible to receive additional compensation depending upon achievement of performance goals as established by the Board. As an “at‑will” employee, Dr. Scott’s employment can be terminated by us or by him, at any time and for any reason. In the event Dr. Scott is terminated by us other than for “cause” or other than by reason of his “disability” (each as defined in his offer letter), Dr. Scott will be entitled to full vesting of all unvested stock options and restricted stock previously granted to him and a cash payment equal to one‑year of his then current base salary.
Director Compensation for 2010
The following Director Compensation table sets forth information concerning compensation for services rendered by our independent directors for fiscal year 2010:
|
Name |
Fees Earned or Paid in Cash ($) |
Stock Awards ($) |
Option Awards ($) (1) |
Total ($) |
|
Frank P. Slattery, Jr. Chairman of the Board |
40,000 |
— |
21,840 (2) |
61,840 |
|
Shaun F. O’Malley Chairman, Audit Committee |
35,000 |
— |
21,840 (2) |
56,840 |
|
Brian Anderson Chairman, Compensation Committee |
29,000 |
— |
21,840 (3) |
50,840 |
|
Richard W. Bank, M.D. |
27,000 |
— |
21,840 (3) |
48,840 |
|
Michael E. Lewis, Ph.D. |
25,500 |
— |
21,840 (2) |
47,340 |
|
Stefan D. Loren, Ph.D. Chairman, Governance Committee |
34,000 |
— |
21,840 (3) |
55,840 |
|
Douglas J. Swirsky |
29,000 |
— |
21,840 (3) |
50,840 |
––––––––––––
(1) This column reflects the total grant date fair value for all stock options or awards granted in the applicable fiscal year pursuant to our equity compensation plans. The assumptions used in the calculation of these amounts are described in footnote 6 to our audited financial statements for the year ended December 31, 2010 and our discussion of stock‑based compensation in this annual report on Form 10‑K under “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Critical Accounting Policies and Practices” for the year ended December 31, 2010.
(2) Represents the total grant date fair value of options to purchase 50,000 shares of common stock granted to this director on September 2, 2010. As of December 31, 2010, this director holds options to purchase 418,500 shares of common stock.
(3) Represents the total grant date fair value of options to purchase 50,000 shares of common stock granted to this director on September 2, 2010. As of December 31, 2010, this director holds options to purchase 200,000 shares of common stock.
Compensation of Directors
Directors who are also our employees receive no additional compensation for serving as a director or as a member of any Committee of the Board. All non‑employee directors receive a fee of $1,500 and $1,000 per Board meeting and Committee meeting, respectively, and are reimbursed for expenses incurred in connection with attending Board and Committee meetings. In addition, our Chairman of the Board receives an annual retainer of $20,000, the Chairman of the Audit Committee receives an annual retainer of $16,000, the Chairmen of the Compensation and Nominating and Corporate Governance Committees receive annual retainers of $14,000 and all other non‑employee directors receive an annual retainer of $12,000.
54
All new non‑employee directors receive an initial grant of options to purchase shares of common stock upon first becoming a member of the Board. In addition, each non‑employee director may, at the discretion of the Board or Compensation Committee of the Board, receive additional equity compensation awards. See the Director Compensation Table above for more details.
Continuing Education of Directors
We are committed to supporting the continuing education of our directors on relevant matters. The Governance Committee of the Board will decide on a case‑by‑case basis the appropriate level and frequency of support to provide.
55
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
The following table shows information known to us about beneficial ownership of our common stock by:
Ø each of our directors;
Ø each of our current named executive officers as well as any additional individuals identified as Named Executive Officers in the section of this report titled “Executive Compensation”;
Ø all of our directors and executive officers as a group; and
Ø each person known by us to beneficially own 5% or more of our common stock.
Beneficial ownership and percentage ownership are determined in accordance with the rules of the SEC. Under these rules, beneficial ownership generally includes any shares as to which the individual or entity has sole or shared voting power or investment power and includes any shares that an individual or entity has the right to acquire beneficial ownership of within 60 days of February 28, 2011, through the exercise of any option, warrant, conversion privilege or similar right. In computing the number of shares beneficially owned by a person and the percentage ownership of that person, shares of our common stock that could be issued upon the exercise of outstanding options and warrants that are exercisable within 60 days of February 28, 2011 are considered to be outstanding. These shares, however, are not considered outstanding as of February 28, 2011 when computing the percentage ownership of each other person.
To our knowledge, except as indicated in the footnotes to the following table and subject to state community property laws where applicable, all beneficial owners named in this table have sole voting and investment power with respect to all shares shown as beneficially owned by them. Percentage of ownership is based on 80,999,610 shares of common stock outstanding as of February 28, 2011.
|
Name and Address of Beneficial Owner (1) |
|
Amount and Nature of Beneficial Ownership Of Common Stock |
|
Percent of Common Stock |
|
||
|
|
|
|
|
|
|
||
|
Directors and Executive Officers |
|
|
|
|
|
||
|
Nicholas Landekic |
|
4,775,986 |
(2) |
|
5.6 |
% |
|
|
Frank P. Slattery, Jr. |
|
2,519,850 |
(3) |
|
3.1 |
% |
|
|
Richard W. Scott, Ph.D. |
|
1,205,100 |
(4) |
|
1.5 |
% |
|
|
R. Eric McAllister, M.D., Ph.D. |
|
1,125,000 |
(5) |
|
1.4 |
% |
|
|
Michael Lewis, Ph.D. |
|
879,750 |
(6) |
|
1.1 |
% |
|
|
Bozena Korczak, Ph.D. |
|
844,583 |
(7) |
|
1.0 |
% |
|
|
Edward Smith |
|
790,833 |
(8) |
|
1.0 |
% |
|
|
Shaun F. O’Malley |
|
442,610 |
(9) |
|
* |
|
|
|
Stefan D. Loren, Ph.D. |
|
224,250 |
(10) |
|
* |
|
|
|
Brian Anderson |
|
181,250 |
(11) |
|
* |
|
|
|
Richard W. Bank, M.D. |
|
181,250 |
(11) |
|
* |
|
|
|
Douglas J. Swirsky |
|
162,500 |
(12) |
|
* |
|
|
|
|
|
|
|
|
|
|
|
|
All Directors and Executive Officers as a Group (12 persons): |
|
13,332,963 |
(13) |
|
14. |
7% |
|
|
|
|
|
|
|
|
|
|
|
Five Percent Stockholders: |
|
|
|
|
|
|
|
|
Act Capital Management, LLLP 2 Radnor Corporate Center, Suite 111 Radnor, PA 19087 (12) |
|
5,948,100 |
(14) |
|
7. |
3% |
|
––––––––––––
* Less than 1%.
(1) Unless otherwise indicated, the address of all individuals and entities listed below is PolyMedix, Inc., 170 N. Radnor Chester Rd., Suite 300, Radnor, Pennsylvania 19087.
(2) Includes 3,603,986 shares of common stock issuable upon exercise of options.
56
(3) Includes 399,750 shares of common stock issuable upon exercise of options and 489,000 shares of common stock issuable upon exercise of warrants. The warrants and 677,000 shares of common stock are held in the name of Kate Partners XX, L.P. (“Kate Partners”). Mr. Slattery is a general partner and owns a controlling interest in Kate Partners.
(4) Includes 667,500 shares of common stock issuable upon exercise of options.
(5) Includes 1,125,000 shares of common stock issuable upon exercise of options.
(6) Includes 399,750 shares of common stock issuable upon exercise of options.
(7) Includes 844,583 shares of common stock issuable upon exercise of options.
(8) Includes 790,833 shares of common stock issuable upon exercise of options.
(9) Includes 399,750 shares of common stock issuable upon exercise of options and 21,430 shares of common stock issuable upon exercise of warrants.
(10) Includes 181,250 shares of common stock issuable upon exercise of options and 21,500 shares of common stock issuable upon exercise of warrants.
(11) Includes 181,250 shares of common stock issuable upon exercise of options.
(12) Includes 162,500 shares of common stock issuable upon exercise of options.
(13) Includes 8,937,403 shares of common stock issuable upon exercise of options and 531,930 shares of common stock issuable upon exercise of warrants.
(14) Based on the filing of a Schedule 13G on January 31, 2011 with the SEC, we understand that Amir L. Ecker and Carol G. Frankenfield are the General Partners of ACT Capital Management, LLLP and Act Capital Partners, LP and that voting and investment decisions made on behalf of ACT Capital Management, LLLP are made primarily by its General Partners. This figure does not include 3,380,000 shares of common stock issuable upon exercise of outstanding Series A warrants at an exercise price of $1.00 per share, 1,180,000 shares of common stock issuable upon exercise of outstanding Series B Warrants at an exercise price of $1.00 per share, or 379,500 shares of common stock issuable upon exercise of outstanding Series C Warrants at an exercise price of $1.25 per share, which are not currently exercisable as a result of aggregate holdings limitations set forth in the warrants. Also includes 46,000 shares of common stock held in various accounts of which Mr. Ecker personally has sole investment and voting power, 22,500 shares of common stock held by Ms. Frankenfield, 35,000 shares of common stock held jointly by Ms. Frankenfield and her husband, and 2,100 shares of common stock held by Ms. Frakenfield’s husband.
Equity Compensation Plan Information
|
Plan Category |
|
(a) |
|
(b) |
|
(c) |
|
|
|
Equity compensation plans approved by security holders (1) |
|
13,272,000 |
|
$1.27 |
|
11,002,000 |
|
|
|
Equity compensation plans not approved by security holders (2) |
|
2,320,000 |
|
$1.38 |
|
— |
|
|
|
Total: |
|
15,592,000 |
|
$1.28 |
|
11,002,000 |
|
|
57
______________
(1) Represents 918,000 and 12,354,000 shares of our Common Stock issuable under our 2002 Equity Compensation Plan and our 2005 Omnibus Equity Compensation Plan, respectively, as of December 31, 2010.
(2) An aggregate of 2,320,000 shares of our Common Stock represent portions of prior grants that exceeded the aggregate individual grant limit under our 2002 Equity Compensation Plan or 2005 Omnibus Equity Compensation Plan, as applicable, and are considered to have occurred outside such plans.
Item 13. Certain Relationships and Related Transactions and Director Independence.
Other than compensation agreements and other arrangements with our executive officers and directors and the transactions described below, during our last two fiscal years, there has not been, and there is not currently proposed, any transaction or series of similar transactions to which we were or will be a party in which the amount involved exceeded or will exceed the lesser of $120,000 or one percent of the average of our total assets at year-end for the last two completed fiscal years and in which any of our directors, nominees for director, executive officers, holders of more than five percent of any class of our voting securities or any member of the immediate family of the foregoing persons had or will have a direct or indirect material interest.
Private Placement
On November 16, 2009, we closed on a registered offering selling 20,700,000 units at a purchase price of $1.00 per unit. Each unit sold included one share of our common stock and a warrant to purchase up to 0.3 shares of our common stock at an exercise price of $1.25. In connection, with the offering, an entity controlled by Frank Slattery Jr., a director of ours, purchased 200,000 units at an aggregate offering price of $200,000.
The information contained in Item 10 under the heading Independence of Directors is incorporated herein by reference.
Item 14. Principal Accountants Fees and Services.
The Audit Committee of the Board has appointed Deloitte & Touche LLP as our independent registered public accounting firm to audit our financial statements for the fiscal year ending December 31, 2010. Deloitte & Touche LLP has served as our independent registered public accounting firm since February 2006. Prior to 2006, we did not have an independent registered public accounting firm audit our financial statements or provide other audit services.
The Audit Committee pre-approved the audit fees, audit-related fees, tax fees and all other fees described below in accordance with our pre-approval policy and believe such fees are compatible with the independence of Deloitte & Touche LLP, the member firms of Deloitte Touche Tohmatsu Limited, and their respective affiliates.
|
|
2010 |
2009 |
|
Audit Fees |
$190,000 |
$194,660 |
|
Audit Related Fees |
$25,862 |
$118,944 |
|
Tax Fees |
$0 |
$0 |
|
All Other Fees |
$0 |
$0 |
Audit Fees. The “Audit Fees” are the aggregate fees of Deloitte & Touche LLP attributable to professional services rendered in 2010 and 2009 for the audit of our annual financial statements and for review of financial statements included in our quarterly reports on Form 10-Q or for services that are normally provided by Deloitte & Touche LLP in connection with statutory and regulatory filings or engagements for those fiscal years.
Audit-Related Fees. The “Audit Related Fees” are the aggregate fees paid to Deloitte & Touche LLP for their consent to the incorporation by reference of our financial statements in various registration statements in 2010 and 2009.
Tax Fees. There were no fees billed in 2010 or 2009 for tax compliance, tax advice or tax planning services.
58
All Other Fees. There were no fees billed in 2010 or 2009 for other products or services provided by Deloitte & Touche LLP besides the services reported above under “Audit Fees” or “Audit-Related Fees.”
Pre-approval Policies and Procedures.
The Audit Committee is required to review and approve in advance the retention of the independent auditors for the performance of all audit and lawfully permitted non-audit services and the fees for such services. The Audit Committee may delegate to one or more of its members the authority to grant pre-approvals for the performance of non-audit services, and any such Audit Committee member who pre-approves a non-audit service must report the pre-approval to the full Audit Committee at its next scheduled meeting. The Audit Committee is required to periodically notify the Board of their approvals. The required pre-approval policies and procedures were complied with during 2010 and 2009.
Item 15. Exhibits and Financial Statement Schedules.
(a) Documents filed as part of this report:
1. Financial Statements. The financial statements as set forth under Item 8 of this Annual Report on Form 10-K are incorporated herein.
2. Financial Statement Schedules. All financial statement schedules have been omitted because they are not applicable, not required, or the information is shown in the financial statements or related notes.
3. Exhibits. See (b) below.
(b) Exhibits:
|
Exhibit No. |
Description of Exhibit |
|
2.1 |
Agreement and Plan of Merger and Reorganization, dated October 6, 2005, among the Registrant, PolyMedix Merger Sub, Inc., PolyMedix Pharmaceuticals, Inc. and those stockholders of Registrant identified on Exhibit A thereto.(1) |
|
3.1 |
Amended and Restated Certificate of Incorporation of Registrant.(1) |
|
3.2 |
Amendment to the Amended and Restated Certificate of Incorporation of Registrant.(1) |
|
3.3 |
Amended and Restated By‑Laws of Registrant.(1) |
|
3.4 |
Amendment to Amended and Restated By‑Laws of Registrant.(1) |
|
3.5 |
Amendment to the Amended and Restated Certificate of Incorporation of Registrant, as amended.(2) |
|
3.6 |
Certificate of Amendment to Amended and Restated Certificate of Incorporation of Registrant, as amended. (3) |
|
3.7 |
Certificate of Designation, Preferences and Rights of Series C Preferred Stock. (4) |
|
4.1 |
Form of common stock Purchase Warrant issued to Fordham Financial Management, Inc. on November 8, 2005, December 8, 2005, January 10, 2006 and February 15, 2006.(1) |
|
4.2 |
Form of common stock Purchase Warrant. (5) |
|
4.3 |
Form of Registration Rights Substitution Agreement.(6) |
|
4.4 |
Form of Amendment No. 1 to Registration Rights Substitution Agreement.(7) |
|
4.5 |
Form of Amendment No. 2 to Registration Rights Substitution Agreement.(7) |
|
4.6 |
Form of Series A Warrant Agreement (including Series A Warrant certificate).(8) |
|
4.7 |
Form of Placement Agent Warrant.(8) |
|
4.8 |
Form of Securities Purchase Agreement as executed by the Registrant and the Investors. (9) |
|
4.9 |
Form of Series B Warrant to Purchase Capital Stock. (9) |
|
4.10 |
Rights Agreement, by and between PolyMedix, Inc. and American Stock Transfer & Trust Company, LLC, as Rights Agent, including exhibits thereto, dated as of May 12, 2009.(4) |
|
4.11 |
Form of Purchase Agreement in connection with the November 2009 offering.(10) |
|
4.12 |
Form of Series C Warrant to Purchase Common Stock issued in connection with the November 2009 offering.(10) |
|
4.13 |
Form of Placement Agent Warrant issued in connection with the November 2009 offering.(10) |
|
10.1 |
Patent License Agreement, dated January 3, 2003, between the Registrant and the University of Pennsylvania.(1) |
|
10.2 |
Letter Agreement, dated December 23, 2003, amending the Patent License Agreement, dated January 3, 2003, between the Registrant and the University of Pennsylvania.(1) |
|
10.3 |
Software License Agreement, dated May 30, 2003, between the Registrant and the University of Pennsylvania.(1) |
|
10.4 |
Exclusive License Agreement, dated January 2, 2005, between the Registrant and the University of Massachusetts.(1) |
|
10.5** |
Employment Agreement, dated July 30, 2002, between Nicholas Landekic and the Registrant.(1) |
|
10.6** |
Employment Agreement, dated December 5, 2005, between Edward Smith and the Registrant.(1) |
|
10.7** |
Employment Agreement, dated March 28, 2003, between Richard Scott, Ph.D. and the Registrant.(1) |
|
10.8 |
Letter Agreement, dated February 25, 2004, between Dr. William DeGrado and the Registrant.(1) |
|
10.9 |
Lab/Office Space License Agreement for 3701 Market Street, dated February 22, 2006, between the Registrant and the University City Science Center.(1) |
|
10.10 |
Pennsylvania Full‑Service Lease Agreement for 170 N. Radnor‑Chester Road; Suite 300, Radnor, PA 19087, dated May 26, 2006, between the Registrant and the Radnor Properties — SDC, L.P.(11) |
|
10.11** |
Amended and Restated 2005 Omnibus Equity Compensation Plan of the Registrant.(12) |
|
10.12** |
2002 Equity Compensation Plan of the Registrant.(13) |
|
10.13** |
Form of Incentive Stock Option Agreement.(1) |
|
10.14** |
Form of Nonqualified Stock Option Agreement.(1) |
|
10.15 |
Sponsored Research Agreement, dated January 5, 2004, between the Registrant and the University of Massachusetts.(14) |
|
10.16 |
Merrill Lynch Loan Management Account Agreement, dated April 13, 2006, between the Registrant and Merrill Lynch Bank USA.(11) |
|
10.17** |
Employment Agreement, dated October 19, 2006, between R. Eric McAllister and the Registrant.(14) |
|
10.18** |
Offer Letter to Bozena Korczak, Ph.D.(15) |
|
10.19 |
Form of Amended and Restated Co‑Placement Agent Agreement by and among the Registrant, Carter Securities, LLC and Fordham Financial Management, Inc. (8) |
|
10.20** |
Offer Letter to J. Gregory Ford.(16) |
|
10.21 |
Investment Agreement by and between PolyMedix, Inc. and Dutchess Equity Fund, LP dated May 20, 2009.(17) |
|
10.22 |
Registration Rights Agreement by and between PolyMedix, Inc. and Dutchess Equity Fund, LP dated May 20, 2009.(17) |
|
10.23 |
Amendment No. 1 to Investment Agreement dated July 8, 2009, between the Registrant and Dutchess Equity Fund, LP.(18) |
|
10.24 |
Loan and Security Agreement dated March 31, 2010 by and among PolyMedix, Inc., PolyMedix Pharmaceuticals, Inc. and Hercules Technology II, L.P. (19) |
|
10.25 |
Common Stock Purchase Warrant dated March 31, 2010(19) |
|
10.26 |
Registration Rights Agreement dated March 31, 2010 by and between PolyMedix, Inc. and Hercules Technology II, L.P. (19) |
|
10.27 |
Offer Letter, dated as of January 18, 2011, by and between the Company and R. Eric McAllister(20) |
|
21 |
List of Subsidiaries(14) |
|
23.1* |
Consent of Deloitte & Touche LLP |
|
24* |
Power of Attorney (Included on signature page) |
|
31.1* |
Certification Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
|
31.2* |
Certification Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
|
32* |
Certification Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
59
* Filed herewith.
** Management contract or compensatory arrangement.
(1) Filed as an Exhibit to the Registration Statement on Form 10‑SB (File No. 000‑51895) filed on April 5, 2006 and incorporated herein by reference.
60
(2) Filed as an Exhibit to the Current Report on Form 8‑K filed on June 5, 2007 and incorporated herein by reference.
(3) Filed as an Exhibit to the Current Report on Form 8-K filed on December 12, 2008 and incorporated herein by reference.
(4) Filed as an Exhibit to the Current Report on Form 8-K filed on May 14, 2009 and incorporated herein by reference.
(5) Filed as an Exhibit to Amendment No. 5 to the Registration Statement on Form SB‑2 (File No. 333‑146180) on November 30, 2007 and incorporated herein by reference.
(6) Filed as an Exhibit to the Registration Statement on Form SB‑2 (File No. 333‑146180) on September 19, 2007 and incorporated herein by reference.
(7) Filed as an Exhibit to Amendment No. 2 to the Registration Statement on Form SB‑2 (File No. 333‑146180) on October 16, 2007 and incorporated herein by reference.
(8) Filed as an Exhibit to Amendment No. 1 to the Registration Statement on Form S-1 (File No. 333-151084) on June 27, 2008 and incorporated herein by reference.
(9) Filed as an Exhibit to the Current Report on Form 8-K filed on September 24, 2008 and incorporated herein by reference.
(10) Filed as an Exhibit to Amendment No. 2 to the Registration Statement on Form S-1 (File No. 333-160833) on November 6, 2009 and incorporated herein by reference.
(11) Filed as an Exhibit to Amendment No. 2 to the Registration Statement on Form 10‑SB/A (File No. 000‑51895) filed on June 19, 2006 and incorporated herein by reference.
(12) Filed as Appendix A to the Registrant’s Definitive Proxy Statement filed on May 15, 2009 and incorporated herein by reference.
(13) Filed as an Exhibit to the Registration Statement on Form S‑8 (File No. 333‑139686) filed on December 26, 2006 and incorporated herein by reference.
(14) Filed as an Exhibit to the Annual Report on Form 10‑KSB filed on March 19, 2007 and incorporated herein by reference.
(15) Filed as an Exhibit to the Current Report on Form 8‑K filed on November 16, 2007 and incorporated herein by reference.
(16) Filed as an Exhibit to the Current Report on Form 8-K filed on December 4, 2008 and incorporated herein by reference.
(17) Filed as an Exhibit to the Current Report on Form 8-K filed on May 22, 2009 and incorporated herein by reference.
(18) Filed as an Exhibit to the Registration Statement on Form S-1 (File No. 333-160470) on July 8, 2009 and incorporated herein by reference.
(19) Filed as an Exhibit to the Current Report on Form 8-K filed on April 2, 2010 and incorporated herein by reference.
(20) Filed as an Exhibit to the Current Report on Form 8-K filed on January 24, 2011 and incorporated herein by reference.
(c) None.
61
SIGNATURES
In accordance with the requirements of the Exchange Act, the registrant caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, this registrant caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
PolyMedix, Inc.
March 7, 2011 By: /s/ Nicholas Landekic
Date Nicholas Landekic
President & Chief Executive Officer
March 7, 2011 By: /s/ Edward F. Smith
Date Edward F. Smith
Vice President & Chief Financial Officer
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints each of Nicholas Landekic and Edward F. Smith as his attorney-in-fact, with the full power of substitution, for him or her in any and all capacities, to sign any amendments to this Report, and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that said attorney-in-fact, or his substitute or substitutes, may do or cause to be done by virtue hereof.
Pursuant to the requirements of the Exchange Act, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated:
|
Name |
Capacity |
Date |
|
/s/ Nicholas Landekic Nicholas Landekic |
President & Chief Executive Officer and Director (Principal Executive Officer) |
March 7, 2011 |
|
/s/ Edward F. Smith Edward F. Smith |
Vice President, Finance & Chief Financial Officer (Principal Financial and Accounting Officer) |
March 7, 2011 |
|
/s/ Frank Slattery, Jr. Frank Slattery, Jr. |
Chairman of the Board of Directors |
March 7, 2011 |
|
/s/ Brian Anderson Brian Anderson |
Director |
March 7, 2011 |
|
/s/ Richard W. Bank, M.D. Richard W. Bank, M.D. |
Director |
March 7, 2011 |
|
/s/ Michael E. Lewis, Ph.D. Michael E. Lewis, Ph.D. |
Director |
March 7, 2011 |
|
/s/ Stefan D. Loren, Ph.D. Stefan D. Loren, Ph.D. |
Director |
March 7, 2011 |
|
/s/ Shaun F. O’Malley Shaun F. O’Malley |
Director |
March 7, 2011 |
|
/s/ Douglas J. Swirsky Douglas J. Swirsky |
Director |
March 7, 2011 |
62
FINANCIAL STATEMENTS
POLYMEDIX, INC.
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED FINANCIAL STATEMENTS
AS OF DECEMBER 31, 2010 AND 2009 AND
FOR THE YEARS ENDED DECEMBER 31, 2010, 2009 AND 2008
AND FOR THE PERIOD
AUGUST 8, 2002 (INCEPTION) TO DECEMBER 31, 2010
CONTENTS
Page
|
F-2 |
|
|
|
|
|
PolyMedix, Inc. Consolidated Balance Sheets as of December 31, 2010 and 2009 |
F-4 |
|
|
|
|
F-5 |
|
|
|
|
|
F-6 |
|
|
|
|
|
F-8 |
|
|
|
|
|
F-9 |
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
PolyMedix, Inc.
Radnor, Pennsylvania
We have audited the accompanying consolidated balance sheets of PolyMedix, Inc. and its subsidiary (a development stage company) (the "Company") as of December 31, 2010 and 2009, and the related consolidated statements of operations, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2010, and for the period from August 8, 2002 (date of inception) to December 31, 2010. We also have audited the Company's internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission. The Company's management is responsible for these financial statements, for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on these financial statements and an opinion on the Company's internal control over financial reporting based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
A company's internal control over financial reporting is a process designed by, or under the supervision of, the company's principal executive and principal financial officers, or persons performing similar functions, and effected by the company's board of directors, management, and other personnel to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company's internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company's assets that could have a material effect on the financial statements.
Because of the inherent limitations of internal control over financial reporting, including the possibility of collusion or improper management override of controls, material misstatements due to error or fraud may not be prevented or detected on a timely basis. Also, projections of any evaluation of the effectiveness of the internal control over financial reporting to future periods are subject to the risk that the controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
F-2
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of PolyMedix, Inc. and its subsidiary as of December 31, 2010 and 2009, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2010, and for the period from August 8, 2002 (date of inception) to December 31, 2010, in conformity with accounting principles generally accepted in the United States of America. Also, in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2010, based on the criteria established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission.
|
/s/ DELOITTE & TOUCHE LLP |
|
|
|
|
|
|
|
|
|
March 7, 2011 |
|
|
|
F-3
POLYMEDIX,
INC.
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED BALANCE SHEETS
DECEMBER 31, 2010 AND 2009
(IN THOUSANDS, EXCEPT PER SHARE AMOUNTS)
|
December 31, |
December 31, |
||||
|
2010 |
2009 |
||||
|
ASSETS |
|
|
|
|
|
|
Current Assets: |
|||||
|
Cash and cash equivalents |
|
|
$ 19,171 |
|
$ 23,974 |
|
Prepaid expenses and other current assets |
166 |
258 |
|||
|
Grant and contract receivable |
|
|
701 |
|
778 |
|
Total current assets |
20,038 |
25,010 |
|||
|
|
|
|
|
|
|
|
Property and Equipment: |
|||||
|
Computers |
|
|
616 |
|
214 |
|
Office furniture and lab equipment |
734 |
725 |
|||
|
Accumulated depreciation |
|
|
(677) |
|
(557) |
|
Total property and equipment |
673 |
382 |
|||
|
|
|
||||
|
Long-term investments |
|
|
1,650 |
|
- |
|
Long-term assets - other |
84 |
- |
|||
|
|
|
|
|
|
|
|
TOTAL ASSETS |
$ 22,445 |
$ 25,392 |
|||
|
|
|
|
|
|
|
|
LIABILITIES AND STOCKHOLDERS' EQUITY |
|||||
|
Current Liabilities: |
|
|
|
|
|
|
Accounts payable |
$ 1,112 |
$ 892 |
|||
|
Accrued expenses |
|
|
1,530 |
|
1,254 |
|
Current portion of long-term debt |
3,115 |
- |
|||
|
Total current liabilities |
|
|
5,757 |
|
2,146 |
|
Long-term debt, less current portion |
6,550 |
- |
|||
|
Deferred rent |
|
|
935 |
|
927 |
|
Total liabilities |
13,242 |
3,073 |
|||
|
|
|
|
|
|
|
|
Commitments and contingencies |
- |
- |
|||
|
|
|
|
|
|
|
|
Stockholders' Equity: |
|||||
|
Preferred Stock ($0.001 par value; 10,000 shares authorized; 0 issued and outstanding at December 31, 2010 and December 31, 2009) |
|
|
- |
|
- |
|
Common Stock ($0.001 par value; 250,000 shares authorized; 81,000 issued and outstanding at December 31, 2010 and December 31, 2009) |
81 |
81 |
|||
|
Additional paid-in capital |
|
|
73,326 |
|
70,926 |
|
Deficit accumulated during the development stage |
(64,204) |
(48,688) |
|||
|
Total stockholders' equity |
|
|
9,203 |
|
22,319 |
|
|
|
||||
|
TOTAL LIABILITIES AND STOCKHOLDERS' EQUITY |
$ 22,445 |
$ 25,392 |
|||
The accompanying notes are an integral part of these consolidated financial statements.
F-4
POLYMEDIX, INC.
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED STATEMENTS OF OPERATIONS
YEARS ENDED DECEMBER 31, 2010, 2009 AND 2008 AND
PERIOD FROM AUGUST 8, 2002 (INCEPTION) THROUGH DECEMBER 31, 2010
(IN THOUSANDS, EXCEPT PER SHARE AMOUNTS)
|
Years Ended December 31, |
For the period August 8, 2002 (Inception) to December 31, |
|||||||
|
2010 |
2009 |
2008 |
2010 |
|||||
|
Revenues: |
|
|
|
|
|
|
|
|
|
Grant and research revenues |
$ 2,144 |
$ 1,048 |
$ 1,066 |
$ 6,705 |
||||
|
Total revenues |
|
2,144 |
|
1,048 |
|
1,066 |
|
6,705 |
|
Operating Expenses: |
|
|
|
|
|
|
|
|
|
Research and development |
$ 10,951 |
7,374 |
7,401 |
42,916 |
||||
|
General and administrative |
|
5,767 |
|
5,557 |
|
4,875 |
|
28,598 |
|
Total operating expenses |
16,718 |
12,931 |
12,276 |
71,514 |
||||
|
|
|
|
|
|
|
|
|
|
|
Other Income and Expenses: |
||||||||
|
Interest income |
|
96 |
|
55 |
|
243 |
|
1,691 |
|
Interest expense |
(1,038) |
(1) |
(19) |
(1,086) |
||||
|
Total other income (expense) |
|
(942) |
|
54 |
|
224 |
|
605 |
|
|
|
|
|
|||||
|
Net loss |
|
$ (15,516) |
|
$ (11,829) |
|
$ (10,986) |
|
$ (64,204) |
|
Beneficial conversion feature on Series 2008 preferred stock, conversion inducement and dividends on Series 1 preferred stock |
- |
- |
(5,845) |
(11,118) |
||||
|
|
|
|
|
|
|
|
|
|
|
Net loss attributable to common stockholders |
$ (15,516) |
$ (11,829) |
$ (16,831) |
$ (75,322) |
||||
|
|
|
|
|
|
|
|
|
|
|
Net loss per common share - basic and diluted |
$ (0.19) |
$ (0.19) |
$ (0.40) |
|||||
|
|
|
|
|
|
|
|
|
|
|
Weighted average common shares outstanding - basic and diluted |
81,000 |
62,437 |
42,007 |
|||||
The accompanying notes are an integral part of these consolidated financial statements.
F-5
POLYMEDIX,
INC.
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED STATEMENT OF CHANGES IN STOCKHOLDERS’ EQUITY
FROM AUGUST 8, 2002 (INCEPTION) THROUGH DECEMBER 31, 2010
(IN THOUSANDS)
The accompanying notes are an integral part of these consolidated financial statements.
F-6
POLYMEDIX,
INC.
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED STATEMENT OF CHANGES IN STOCKHOLDERS’ EQUITY
FROM AUGUST 8, 2002 (INCEPTION) THROUGH DECEMBER 31, 2010 - Continued
(IN THOUSANDS)
The accompanying notes are an integral part of these consolidated financial statements.
F-7
POLYMEDIX, INC.
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED STATEMENTS OF CASH FLOWS
YEARS ENDED DECEMBER 31, 2010, 2009 AND 2008 AND
PERIOD FROM AUGUST 8, 2002 (INCEPTION) THROUGH DECEMBER 31, 2010
(IN THOUSANDS)
|
Years Ended December 31, |
For the period August 8, 2002 (Inception) to |
|||||||
|
2010 |
2009 |
2008 |
December 31, 2010 |
|||||
|
Cash Flows from Operating Activities: |
|
|
|
|
|
|
|
|
|
Net loss |
$ (15,516) |
$ (11,829) |
$ (10,986) |
$ (64,204) |
||||
|
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
|
|
|
Depreciation |
182 |
205 |
213 |
910 |
||||
|
Amortization |
|
35 |
|
- |
|
- |
|
35 |
|
Accretion of discount on investment securities |
(2) |
(17) |
(70) |
(461) |
||||
|
Stock-based compensation |
|
2,005 |
|
1,786 |
|
1,404 |
|
9,635 |
|
Non-cash interest expense |
91 |
- |
- |
91 |
||||
|
(Increase) decrease in prepaid expenses and other current assets |
|
214 |
|
(629) |
|
(365) |
|
(822) |
|
Increase (decrease ) in accounts payable, accrued expenses and deferred rent |
577 |
236 |
(1,122) |
3,562 |
||||
|
Loss on disposal of fixed assets |
|
- |
|
- |
|
- |
|
23 |
|
Net cash used in operating activities |
(12,414) |
(10,248) |
(10,926) |
(51,231) |
||||
|
|
|
|
|
|
|
|
|
|
|
Cash Flows from Investing Activities: |
||||||||
|
Cash paid for property and equipment |
|
(474) |
|
(38) |
|
- |
|
(1,263) |
|
Purchases of investments |
(5,448) |
(3,798) |
(9,553) |
(39,489) |
||||
|
Proceeds from maturities of investments |
|
3,800 |
|
11,703 |
|
5,700 |
|
38,301 |
|
Net cash provided by (used in) investing activities |
(2,122) |
7,867 |
(3,853) |
(2,451) |
||||
|
|
|
|
|
|
|
|
|
|
|
Cash Flows from Financing Activities: |
||||||||
|
Proceeds from issuance of stock, net of financing costs |
|
(106) |
|
18,753 |
|
16,966 |
|
62,447 |
|
Proceeds from warrant and stock option exercises |
- |
500 |
214 |
898 |
||||
|
Principal payments on capital lease obligations |
|
- |
|
(104) |
|
(131) |
|
(331) |
|
Proceeds from issuance of debt, net of issue costs |
9,839 |
- |
- |
9,839 |
||||
|
Net cash provided by financing activities |
|
9,733 |
|
19,149 |
|
17,049 |
|
72,853 |
|
|
|
|
||||||
|
Net increase (decrease) in cash and cash equivalents |
|
(4,803) |
|
16,768 |
|
2,270 |
|
19,171 |
|
Cash and cash equivalents - beginning of period |
|
23,974 |
|
7,206 |
|
4,936 |
|
- |
|
|
|
|
||||||
|
Cash and cash equivalents - end of period |
|
$ 19,171 |
|
$ 23,974 |
|
$ 7,206 |
|
$ 19,171 |
|
Cash Paid for Interest |
|
$ 841 |
|
$ 2 |
|
$ 19 |
|
$ 889 |
|
Non-Cash Investing Activities: |
|
|
|
|
|
|
|
|
|
Capital expenditures acquired on account |
$ - |
$ - |
$ - |
$ 15 |
||||
|
|
|
|
|
|
|
|
|
|
|
Non-Cash Financing Activities: |
||||||||
|
Conversion inducement and dividends on Series 1 Preferred Stock and Beneficial conversion feature on Series 2008 Preferred Stock |
|
$ - |
|
$ - |
|
$ 5,845 |
|
$ 11,118 |
|
Warrants issued to creditor and placement agent |
$ 426 |
$ 119 |
$ 783 |
$ 3,059 |
||||
|
Accrued financing costs |
|
$ - |
|
$ 73 |
|
$ 51 |
|
$ - |
The accompanying notes are an integral part of these consolidated financial statements.
F-8
POLYMEDIX, INC.
(A DEVELOPMENT STAGE COMPANY)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
AS OF DECEMBER 31, 2010 AND 2009 AND
FOR THE YEARS ENDED DECEMBER 31, 2010, 2009 AND 2008
AND FOR THE PERIOD
AUGUST 8, 2002 (INCEPTION) TO DECEMBER 31, 2010
In these consolidated financial statements, "PolyMedix," "we," "us" and "our" refer to PolyMedix, Inc. and its wholly owned subsidiary PolyMedix Pharmaceuticals, Inc. and "Common Stock" refers to PolyMedix’s Common Stock, par value $0.001 per share
NOTE 1 – ORGANIZATION AND BUSINESS ACTIVITIES
We are a development stage biotechnology company focused on developing treatments for patients with infectious diseases and acute cardiovascular disorders with synthetic small molecule compounds that mimic the activity of large natural protein molecules, compounds referred to as biomimetics. Using our proprietary computational drug design technology, we have created novel defensin mimetic antibiotic compounds, novel heparin antagonist compounds, and other drug compounds intended for human therapeutic use. Since 2002, we have been a development stage enterprise, and accordingly, our operations have been directed primarily toward developing business strategies, raising capital, research and development activities, conducting pre-clinical testing and human clinical trials of our product candidates, exploring marketing channels and recruiting personnel.
We have incurred operating losses since inception, have not generated any product sales revenues and have not achieved profitable operations. Our deficit accumulated during the development stage through December 31, 2010 was $64,204,000, and we expect to continue to incur substantial losses in future periods. None of our product candidates have received regulatory approval for commercial sale and our product candidates may never be commercialized. In addition, all of our product candidates are in the early stages of development and several programs are on hold pending additional financing. Our development programs require a significant amount of cash to support the development of product candidates. The progress and results of our current and any future clinical trials or future pre‑clinical testing are uncertain, and if our product candidates do not receive regulatory approvals, our business, operating results, financial condition and cash flows will be materially adversely affected.
We are highly dependent on the success of our research, development and licensing efforts and, ultimately, upon regulatory approval and market acceptance of our products under development. Our short and long-term capital requirements depend upon a variety of factors, including the timing and results of our clinical trials, market acceptance for our technologies and product candidates and various other factors. We believe that our current cash and investment balances as of the date of this filing will be sufficient to fund our operations for at least the next 12 months. Our ability to advance the clinical development of our drug candidates and ultimately seek regulatory approval depends upon our ability to meet the regulatory requirements of the United States Food and Drug Administration (FDA) and other regulatory authorities on an ongoing basis, many of which develop and change over time and are subject to interpretation by such regulatory authorities. Any changes in regulatory requirements may have significant impacts on the timing and cost of clinical trials, and our ability to complete such trials, if at all.
In March 2010, we closed a debt financing for gross proceeds of $10,000,000, and in November 2009, we closed an equity financing for gross proceeds of $20,700,000. Proceeds from these financing activities are being used to fund our ongoing clinical development of PMX-30063 and PMX-60056 and for general corporate purposes. We may seek additional funds through equity or debt financing, among other sources, or through government grants and contracts. However, we may not be able to obtain additional funding on favorable terms, if at all. If we are unable to secure adequate additional funding in the future, we may delay, scale-back or eliminate certain of our planned research, drug discovery and development activities and certain other aspects of our operations and our business until such time as we are successful in securing adequate additional funding. As a result, our business, operating results, financial condition and cash flows may be materially and adversely affected.
F-9
NOTE 2 – SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Principles of Consolidation
The consolidated financial statements include the accounts of PolyMedix, Inc. and its wholly owned subsidiary PolyMedix Pharmaceuticals, Inc. All intercompany accounts and transactions have been eliminated in consolidation.
Development Stage Company
We are considered to be in the development stage as defined by accounting principles generally accepted in the United States (“GAAP”). We have devoted substantially all of our efforts to business planning, research and development, recruiting management and technical staff, acquiring operating assets and raising capital.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts in the financial statements and accompanying notes. Actual results could differ from those estimates. Significant estimates include the value of our stock options and warrants.
Cash and Cash Equivalents
We consider all highly liquid investments purchased with a maturity of three months or less at the time of purchase to be cash equivalents. Cash and cash equivalents consist of cash in banks and money market funds.
Property and Equipment
Property and equipment are stated at cost. Depreciation is computed using the straight-line method over the estimated useful life of the assets; three years for computer equipment and related software, seven years for office furniture and five years for lab equipment. When property and equipment are sold or otherwise disposed of, the cost and related accumulated depreciation are removed from the accounts and the resulting gain or loss is included in operating expenses. Depreciation expense was $182,000, $205,000, $213,000 and $910,000 for the years ended December 31, 2010, 2009 and 2008 and for the period from August 8, 2002 (Inception) to December 31, 2010 respectively.
Impairment of Long-Lived Assets
Long-lived assets are reviewed for impairment whenever events or changes in circumstances indicate that the basis of an asset may not be recoverable. In accordance with GAAP, impairment is assessed by measuring the carrying amount of the assets against the estimated undiscounted future cash flows associated with them. If the expected future cash flows are less than the carrying value, an impairment loss is recognized for the amount by which the carrying value exceeds the fair value of the assets.
Long-term Investments
Investments purchased with a maturity of more than 12 months are classified as long-term investments. We generally hold investments to maturity and have classified these investments as such as defined by GAAP.
Grant and Contract Revenue
Sponsored research grant and contract revenues are recognized pursuant to the terms of the related agreements as work is performed.
Research and Development Expense
Research and development costs are expensed as incurred.
F-10
Income Taxes
We recognize deferred tax assets and liabilities for temporary differences between the financial reporting basis and the tax basis of our assets and liabilities and the expected benefits of using net operating loss carryforwards. The impact of changes in tax rates and laws on deferred taxes, if any, applied during the years in which temporary differences are expected to be settled, is reflected in the consolidated financial statements in the period of enactment. The measurement of deferred tax assets is reduced, if necessary, if, based on weight of the evidence, it is more likely than not that some, or all, of the deferred tax assets will not be realized. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in the period that such tax rate changes are enacted. At December 31, 2010 and 2009, we have concluded that a full valuation allowance is necessary for deferred tax assets (See Note 5).
We apply the provisions of GAAP as it relates to the recognition threshold and measurement attribute for the financial statement recognition and measurement of uncertain tax positions taken or expected to be taken in a tax return. We had no material amounts recorded for uncertain tax positions, interest or penalties in the accompanying consolidated financial statements. See Note 5 for additional information.
Loss per Share of Common Stock
We calculate our loss per share in accordance with GAAP, which requires a dual presentation of “basic” and “diluted” loss per share on the face of the income statement. Basic loss per share is computed by dividing loss by the weighted average number of shares of common stock outstanding during each period. Diluted loss per share includes the effect, if any, from the potential exercise or conversion of securities, such as unvested restricted stock, convertible preferred stock, stock options and warrants, which would result in the issuance of incremental shares of common stock. In computing the basic and diluted net loss per share allocable to common stockholders, the weighted average number of shares remains the same for both calculations due to the fact that when a net loss exists, dilutive shares are not included in the calculation. Potentially dilutive securities include options and warrants to purchase our common stock. These potentially dilutive securities are more fully described in Note 6.
Comprehensive Loss
Comprehensive loss consists of reported net income or loss and unrealized gains or losses on available for sale securities. The comprehensive loss for each of the periods presented approximates the net loss in the consolidated statements of operations.
Segment Information
In accordance with the definitions provided by GAAP, we have one reportable segment operating within the United States.
Reclassification
Certain prior year amounts have been reclassified to conform to current year presentation. Specifically, interest income and expense have been disclosed separately on our consolidated statement of operations.
Fair Value of Financial Instruments and Credit Risk
The carrying amounts reported in the consolidated balance sheets for cash and cash equivalents, accounts payable and accrued expenses approximate fair value because of the immediate or short-term maturity of these financial instruments.
We invest our cash in accordance with a policy objective that seeks to ensure both liquidity and safety of principal. The policy limits investments to instruments issued by the U.S. government and commercial institutions with strong investment grade credit ratings and places restrictions on maturity terms and concentrations by type and issuer.
Stock-Based Compensation
We currently sponsor equity compensation plans under which we have granted options to purchase our common stock to employees, directors, and key consultants. We have also granted awards outside of these plans. Compensation expense associated with equity awards is based on the fair value estimated at the date of grant, recognized ratably over the period in which the related services are provided. The total stock-based compensation expense was $2,005,000, $1,786,000, $1,404,000, and $9,635,000 for the years ended December 31, 2010, 2009 and 2008 and for the period from August 8, 2002 (Inception) to December 31, 2010, respectively.
F-11
Recently Issued and Adopted Accounting Standards
In January 2010, the Financial Accounting Standards Board (FASB) issued an Accounting Standards Update (ASU) relating to fair value disclosures, to add new requirements for disclosures about transfers into and out of Levels 1 and 2, and separate disclosures about purchases, sales, issuances, and settlements relating to Level 3 measurements. It also clarifies existing fair value disclosures about the level of disaggregation and about inputs and valuation techniques used to measure fair value. The guidance in this ASU is effective for the first reporting period (including interim periods) beginning after December 15, 2009, except for the requirement to provide the Level 3 activity of purchases, sales, issuances, and settlements on a gross basis, which will be effective for fiscal years beginning after December 15, 2010, and for interim periods within those fiscal years. The adoption of this guidance as it relates to Levels 1 and 2 investments did not have a material impact on our consolidated financial statements. We do not expect the adoption of this guidance as it relates to Level 3 investments to have a material impact on our consolidated financial statements.
In February 2010, the FASB issued an ASU which amends existing guidance regarding an entity’s requirement to perform and disclose subsequent events procedures. The amendments add definitions for “SEC filer” and “revised financial statements,” and remove the requirement for SEC filers to disclose the date through which subsequent events have been evaluated, and clarify that an SEC filer is to evaluate subsequent events through the date the financial statements are issued. The guidance in this ASU was effective immediately. The adoption of this guidance did not have a material impact on our consolidated financial statements.
NOTE 3 – LONG-TERM INVESTMENTS
Long-term investments consist of investments with maturities greater than one year. All long-term investments represent certificates of deposit, classified as “held to maturity,” and are recorded at cost. As discussed in Note 8, these certificates of deposit secure our letter of credit.
NOTE 4 – ACCRUED EXPENSES
At December 31, 2010 and 2009, accrued expenses consisted of the following (in thousands):
|
|
|
2010 |
|
|
2009 |
|
||
|
Payroll related costs |
|
$ |
939 |
|
|
$ |
1,014 |
|
|
Research and development related costs |
|
|
158 |
|
|
|
84 |
|
|
Accrued interest expense |
|
|
106 |
|
|
|
-- |
|
|
Other costs |
|
|
327 |
|
|
|
156 |
|
|
Total accrued expenses |
|
$ |
1,530 |
|
|
$ |
1,254 |
|
NOTE 5 – PROVISION FOR INCOME TAXES
We account for income taxes using the asset and liability approach as required by GAAP. Deferred income taxes are provided for the differences between the tax bases of assets or liabilities and their reported amounts in the financial statements. This method also requires the recognition of future tax benefits, such as net operating loss carryforwards and research and development credits, to the extent that realization of such benefits is more likely than not. At December 31, 2010 and 2009, deferred tax assets consisted of the following (in thousands):
|
|
|
2010 |
|
|
2009 |
|
||
|
Deferred tax assets: |
|
|
|
|
|
|
|
|
|
Net operating loss carryforward |
|
$ |
20,871 |
|
|
$ |
15,595 |
|
|
Stock compensation expense |
|
|
1,900 |
|
|
|
1,809 |
|
|
Contribution carryforward |
|
|
- |
|
|
|
33 |
|
|
R & D credit carryforward |
|
|
2,026 |
|
|
|
1,754 |
|
|
Straight-line rent |
|
|
380 |
|
|
|
376 |
|
|
Other |
|
|
151 |
|
|
|
82 |
|
|
Gross deferred tax assets |
|
|
25,328 |
|
|
|
19,649 |
|
|
Less: Valuation allowance |
|
|
(25,292 |
) |
|
|
(19,591 |
) |
|
Net deferred tax assets |
|
|
36 |
|
|
|
58 |
|
|
Deferred tax liability |
|
|
(36 |
) |
|
|
(58 |
) |
|
Net deferred tax |
|
|
- |
|
|
|
- |
|
F-12
We believe there is not sufficient evidence that future taxable income will be adequate to permit the realization of the future tax deductions embedded in this asset. As such, in accordance with GAAP, we have established a valuation allowance to reduce the deferred tax asset since it is more likely than not that the deferred tax asset will not be realized.
The difference between the provision for (benefit from) income taxes and the amount computed by applying the federal statutory income tax rate to income (loss) before provision for (benefit from) income taxes is explained below (in thousands):
|
|
|
|
Period from |
|
||||||||||||
|
|
|
|||||||||||||||
|
|
Years ended December 31, |
|||||||||||||||
|
|
|
2010 |
|
|
2009 |
|
|
2008 |
|
2010 |
|
|
||||
|
Loss before provision for income taxes |
$ |
(15,516 |
) |
$ |
(11,829 |
) |
$ |
(10,986 |
) |
$ |
(64,204 |
) |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
Tax benefit at federal statutory rate |
|
(5,275 |
) |
|
(4,022 |
) |
|
(3,735 |
) |
|
(21,830 |
) |
|
|||
|
State tax benefits, net of federal |
|
(1,023 |
) |
|
(506 |
) |
|
(883 |
) |
|
(4,221 |
) |
|
|||
|
Research and development credits |
|
(322 |
) |
|
(253 |
) |
|
(290 |
) |
|
(1,534 |
) |
|
|||
|
Other permanent differences |
|
728 |
|
|
432 |
|
|
113 |
|
|
1,370 |
|
|
|||
|
Other |
|
191 |
|
|
2 |
|
|
205 |
|
|
923 |
|
|
|||
|
Change in valuation allowance |
|
5,701 |
|
|
4,347 |
|
|
4,590 |
|
|
25,292 |
|
|
|||
|
Provision for income taxes |
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
|
|||
We have available at December 31, 2010, approximately $51,000,000 in unused federal and state operating loss carryforwards that expire between 2022 and 2030. We also have available at December 31, 2010, approximately $2,000,000 unused federal research and development credit carryforwards that may provide future tax benefits and expire between 2025 and 2030.
The Tax Reform Act of 1986 contains provisions that may limit the NOL and research and development credit carryforwards available to be used in any given year upon the occurrence of certain events, including significant changes in ownership interest. Generally, a change in ownership of a company of greater than 50% within a three-year period results in an annual limitation on that company’s ability to utilize its NOL carryforwards and tax credits from the tax periods prior to the ownership change. A study needs to be performed to determine if we have undergone a change in ownership, however, we believe that we have undergone an ownership change and are subject to an annual limitation on the use of our NOL carryforwards pursuant to these provisions.
No adjustments were made to retained earnings for unrecognized tax benefits after applying the “more likely than not” threshold to the initial recognition and derecognition of our tax positions.
We file income tax returns in the U.S. federal jurisdiction and Pennsylvania. Our policy is to record interest and penalties on uncertain tax positions as income tax expense. The tax years back to 2002 remain open to examination by the major taxing jurisdictions where we file. Net operating loss and credit carryforwards from earlier periods also remain open to examination by taxing authorities, and will for a period post utilization. We do not reasonably estimate that the unrecognized tax benefit will change significantly within the next twelve months. Any changes in the future would also have no impact on our consolidated financial statements due to the existence of a full valuation allowance. As of December 31, 2010, we had no material amounts recorded for uncertain tax positions, interest or penalties in the accompanying consolidated financial statements.
F-13
NOTE 6 – STOCKHOLDERS’ EQUITY
Common Stock
We are authorized to issue 250,000,000 shares of common stock, with a par value of $0.001, of which approximately 81,000,000 were issued and outstanding at December 31, 2010 and 2009, respectively.
In November 2009, we completed a registered offering of 20,700,000 units. Each unit consisted of one share of our common stock and a Series C Warrant to purchase three-tenths of a share of our common stock. Each unit was priced at $1.00 per unit with the exercise price of the Series C Warrants issued at $1.25 per share. We received gross proceeds of $20,700,000 in the registered offering before fees and expenses. In connection with this registered offering, we incurred commission fees and expenses of approximately $1,904,000.
Preferred Stock
We are authorized to issue 10,000,000 shares of preferred stock, with a par value of $0.001, of which no shares were issued and outstanding at December 31, 2010 and 2009, respectively.
Warrants
We presently have the following warrants outstanding to purchase shares of our common stock (number of warrants in thousands):
|
Warrant Series |
Number of Warrants Outstanding |
Exercise Price |
Expiration Date |
|
2007 |
2,489 |
$1.10 |
December 2012 |
|
Series A |
22,285 |
$1.00 |
July 2013 |
|
Series B |
6,368 |
$1.00 |
September 2013 |
|
Series C |
6,417 |
$1.25 |
November 2014 |
|
HTGC |
628 |
$1.16 |
March 2015 |
|
Total |
38,187 |
|
|
In November 2010, approximately 4,372,000 warrants with an exercise price of $1.16 expired unexercised. All warrants are exercisable beginning five years prior to their expiration date.
In March 2010, we issued to Hercules Technology II, L.P. (HTGC) a warrant to purchase 627,586 shares of our common stock in connection with the signing of a Loan Security Agreement (LSA) with HTGC (included in table above). The fair value of this warrant at the time of issuance was $445,000, which was estimated using the Black-Scholes-Merton formula with the following assumptions: no dividends; risk-free interest rate of 2.55%; estimated life of five years and volatility of 72%. This warrant expires on March 30, 2015. As discussed in Note 7, this value was used to determine the relative fair value of the warrants of $426,000, which was recorded to additional paid-in capital.
F-14
Stock-Based Compensation
We maintain equity compensation plans under which grants of incentive stock options, nonqualified stock options, stock appreciation rights, stock awards, stock units and other stock-based awards are granted to selected employees, non-employee directors and key advisors. Since our inception on August 8, 2002, we have recognized equity compensation expense over the requisite service period using the Black-Scholes-Merton formula to estimate the fair value of stock options. The following table summarizes the total stock-based compensation expense included in our Consolidated Statements of Operations (in thousands):
|
|
Years Ended December 31, |
Period from August 8, 2002 (Inception) to December 31, | ||
|
|
2010 |
2009 |
2008 |
2010 |
|
Research and development |
$ 950 |
$ 704 |
$ 440 |
$ 3,455 |
|
General and administrative |
1,055 |
1,082 |
964 |
6,180 |
|
Total stock-based compensation expense |
$ 2,005 |
$ 1,786 |
$ 1,404 |
$ 9,635 |
During the year ended December 31, 2010, approximately 4,963,000 stock options were awarded to non-employee directors, officers, employees and consultants for services performed. Options awarded to our non-employee consultants are granted for their continuing services under our consulting agreements which have no definitive end date, and therefore, we apply a similar approach in identifying the grant date and vesting period for these awards as we do for our employees. As of December 31, 2010, there were 15,592,000 shares of common stock issuable upon exercise of outstanding stock options and 11,002,000 shares of Common Stock available for issuance of future equity compensation awards in connection with our equity compensation plans.
Stock Options
As of December 31, 2010, there was approximately $3,051,000 of total unrecognized compensation cost related to non-vested stock options, which will be amortized over the weighted average remaining service period of approximately 0.99 years. This expected cost does not include the impact of any future stock option awards. Options granted are generally exercisable for a period of up to ten years from the date of grant at an exercise price which is not less than the fair value on the date of grant and generally vest over terms ranging from immediately to four years. A summary of the status of our stock options as of December 31, 2010 and changes during the year ended December 31, 2010 is presented below (in thousands, except per share amounts):
|
|
Options |
Weighted Average Exercise Price per share |
Weighted Average Remaining Contractual Life (in years) |
Aggregate Intrinsic Value |
|
Outstanding at beginning of period |
10,629 |
$1.41 |
|
|
|
Granted |
4,963 |
$1.01 |
|
|
|
Exercised |
- |
- |
|
|
|
Expired |
- |
- |
|
|
|
Forfeited or cancelled |
- |
- |
|
|
|
Outstanding at end of period |
15,592 |
$1.28 |
7.43 |
$377 |
|
Exercisable at end of period |
10,509 |
$1.41 |
6.68 |
$192 |
F-15
The aggregate intrinsic value in the preceding tables represent the total pretax intrinsic value that would have been received by the option holders had all option holders exercised their options on the last day of 2010. Intrinsic value is determined by calculating the difference between the value of our stock on the last day of the year and the exercise price, multiplied by the number of options. For options where our stock price on December 31, 2010 was below the exercise price, the intrinsic value was given as $0.
The following table summarizes the fair value of options granted during the years ended December 31, 2010, 2009 and 2008 and for the period from August 8, 2002 (Inception) to December 31, 2010, respectively (in thousands, except per share amounts):
|
|
Years Ended December 31, |
Period from August 8, 2002 (Inception) to December 31, | ||
|
|
2010 |
2009 |
2008 |
2010 |
|
Fair value of options granted |
$ 2,994 |
$ 417 |
$ 3,210 |
$ 12,179 |
|
Fair value of options granted (per share) |
$ 0.60 |
$ 0.49 |
$ 0.67 |
$ 0.73 |
The fair value of options granted is amortized over the requisite service period. The fair value of each option grant is estimated on the date of grant using the Black-Scholes-Merton formula with the following weighted average assumptions:
|
|
Year Ended December 31, 2010 |
Year Ended December 31, 2009 |
Year Ended December 31, 2008 |
|
Range of risk free interest rates |
1.11% – 2.40% |
1.65% - 2.51% |
1.37% - 3.25% |
|
Dividend yield |
0% |
0% |
0% |
|
Expected volatility |
69% – 72% |
72% - 74% |
69% - 75% |
|
Expected life of options (in years) |
5 |
5 |
5 |
|
Forfeitures |
0% |
0% |
0% |
Expected volatility and expected life for the years ended December 31, 2010, 2009, and 2008 were estimated based upon historical activity. The risk-free interest rate is calculated using the U.S. Treasury yield curves in effect at the time of grant. We currently estimate that all of our outstanding options will vest.
Restricted Common Stock Grants
We have also issued shares of restricted Common Stock to selected employees, non-employee directors and key advisors. During the three-year period ended December 31, 2010, there were no grants of restricted stock. As of December 31, 2010, there was no recognized or unrecognized compensation cost related to non-vested restricted stock grants. The restriction period for restricted stock awards was for a four year vesting period, commencing from the grant date.
Since inception, August 8, 2002, we granted 1,464,000 shares of restricted stock . No shares of restricted stock vested during the three-year period ended December 31, 2010. The total grant date fair value of these restricted stock awards was $34,000. All restricted shares awarded have grant date fair values of $0.025 per share.
Common Stock Grants
We also issued Common Stock grants to selected employees, non-employee directors and key advisors. For the period from August 8, 2002 (Inception) to December 31, 2010, we issued Common Stock grants of 1,244,000 shares. The total grant date fair value of these common stock grants and expense recognized for the period from August 8, 2002 (inception) to December 31, 2010 was $834,000. We did not grant or recognize any expense related to these awards for the three year period ended December 31, 2010.
F-16
Additionally, during January 2008, and December, 2007, in connection with agreements with certain terminated employees, we granted 50,000 and 25,000 shares, respectively, of Common Stock as a one-time termination benefit to certain employees. These shares were issued ratably over the first five-months following the grant date. During 2007, we also issued 12,500 shares to UMass in connection with our license agreement. We recognized $0, $0, and $26,000, for the years ended December 31, 2010, 2009, and 2008, respectively, and $96,000 for the period from August 8, 2002 (inception) to December 31, 2010, in equity based compensation costs associated with these shares, which is included in research and development costs on our Consolidated Statements of Operations.
Equity Line
In May 2009, we signed an Investment Agreement with Dutchess Opportunity Fund, II (Dutchess) which allows us to put shares of our common stock at an aggregate value of up to $10,000,000, but not to exceed 12,000,000 shares, to Dutchess, subject to the terms and conditions of the agreement. The purchase price will be determined on a volume-weighted average price calculated in accordance with the terms of the agreement. As of December 31, 2010, we have not put any shares of our common stock to Dutchess under this Investment Agreement.
NOTE 7 – DEBT
On March 31, 2010, we entered into a Loan and Security Agreement (LSA) with Hercules Technology II, L.P. (HTGC) whereby we borrowed $10,000,000. This loan bears an interest rate at the greater of 12.35%, or the Federal Reserve Prime Rate plus 7.10%, not to exceed 14%, and has a term of 42 months with interest-only payments for the first nine months. We have also granted HTGC a security interest in our personal property other than intellectual property. In connection with this loan, we also issued to HTGC a detachable warrant to purchase 627,586 shares of our common stock which expires on March 30, 2015 (see Note 6 for additional information on the warrant). The following table summarizes the debt included on our consolidated balance sheet as of December 31, 2010 (in thousands):
|
|
Principal |
Unamortized Discount |
|
Current portion |
$ 3,237 |
$ 122 |
|
Long-term portion |
6,763 |
213 |
|
Total |
$ 10,000 |
$ 335 |
We allocated the $10,000,000 of proceeds received to both the debt and the detachable warrant based on the relative fair value of the debt instrument without the warrant and the warrant itself. The relative fair value at the time of issuance was $9,574,000 and $426,000 for the debt and warrant, respectively. The carrying value of the Company's long-term debt approximates fair value because the interest rate approximates current market rates available to the company. The portion allocated to the warrant was recorded to additional paid-in capital, and is amortized to interest expense as a discount to the principal balance of the debt. As of December 31, 2010, we had not repaid any portion of the principal balance and had accrued interest of $106,000. We are not required to maintain any financial covenants. As of December 31, 2010, we were in compliance with all nonfinancial covenants.
We incurred $161,000 in debt issue costs in connection with securing the LSA, which we have recorded as an asset to be amortized over the life of the loan.
NOTE 8 – COMMITMENTS AND CONTINGENCIES
Operating Lease
In June 2006, we entered into an operating lease agreement for 24,223 square feet of combined office and laboratory space located in Radnor, Pennsylvania. The initial term of the lease is 12 years. Payments under the lease commenced on December 1, 2006. Our annual future minimum lease payments under this non-cancelable operating lease are as follows (in thousands):
|
|
Operating Leases |
|
2011 |
$ 667 |
|
2012 |
686 |
|
2013 |
705 |
|
2014 |
724 |
|
2015 |
743 |
|
Thereafter |
2,144 |
|
Total minimum lease payments |
$ 5,669 |
F-17
Prior to the commencement of our current operating lease for our Radnor facility, we leased approximately 3,500 square feet of combined office and laboratory space on a month-to-month basis in Philadelphia, Pennsylvania. Rent expense was $598,000 for each of the three years ended December 31, 2010, and $3,073,000 for the period from August 8, 2002 (Inception) to December 31, 2010.
Patent License Agreements
University of Pennsylvania. In January 2003, we entered into a Patent License Agreement with the University of Pennsylvania, (Penn). Under the terms of the agreement, we were granted an exclusive, worldwide royalty-bearing license to make and sell products utilizing seven of Penn’s issued or pending patents for the life of such patents. One issued patent and five patent applications cover the composition of matter on antimicrobial compounds, including small molecules, oligomers and polymers. One patent application covers the composition and use of polycationic compounds for treating cancer. If a change-of-control event occurs, in which we transfer the license to these patents to a third party, we are acquired by another company, or we conduct an initial public offering of our securities, we are required to pay a 3% royalty on the gross sales for licensed products that are sold as pharmaceuticals and a 1.5% royalty on products sold as coatings for use in medical devices. We are permitted to sublicense the patents provided that (a) the sublicensee is prohibited from further licensing of the patents and (b) the sublicensee is subject to all of the terms of the original license granted to us. In addition, we are required to share with Penn any consideration we receive from sublicensing our patents to a third party.
University of Massachusetts. In January 2004, we entered into a sponsored research agreement with the University of Massachusetts (UMass) which, as amended, continued through March 2009. We are currently negotiating an extension of this agreement with UMass. Under the terms of this agreement, we have the exclusive option to license any intellectual property that may be generated by Dr. Gregory Tew pursuant to research sponsored under the agreement. We may exercise this option by issuing 7,500 shares of our Common Stock to UMass for each $100,000 of research conducted by Dr. Tew. If we exercise this option, we are also required to reimburse UMass for direct patent costs incurred by it for the patents licensed by us. During 2007, we issued 12,500 shares to UMass in connection with this agreement.
In January 2005, we entered into an Exclusive License Agreement with UMass, pursuant to which UMass granted us an exclusive, worldwide license to use, make and sell products under one U.S. patent application and seven corresponding foreign patent applications covering polynorborene co‑polymers and methods of use for the life of such patents. The patents, if issued, will expire in 2025. UMass may terminate the license agreement if we materially breach the agreement, including by nonpayment of amounts due under the agreement or in the case of a change in our ownership or control. If the license agreement is properly terminated by UMass, we may not be able to execute our strategy to develop and commercialize our PolyCides® for biomaterials applications or to develop and commercialize future product candidates utilizing the licensed patents.
Other
Agreements with Employees. We have entered into employment agreements with various executives. These agreements provide for severance arrangements and accelerated vesting of equity compensation awards in the event that the executive is terminated by us other than for cause or disability or if the executive resigns for good reason.
Credit Line. In November 2010, we terminated our then outstanding line and letter of credit and entered into a similar letter of credit agreement with a new financial institution to secure our payment obligations under our facility operating lease. This letter of credit is for $1,400,000, expires on December 1, 2011, and is secured by certificates of deposits held at the same financial institution.
F-18
NOTE 9 – QUARTERLY FINANCIAL INFORMATION (UNAUDITED)
The following tables set forth certain unaudited financial information for each of the quarters in the years ended December 31, 2010 and 2009. This unaudited quarterly information has been prepared on the same basis as the audited financial statements and includes all necessary adjustments, consisting only of normal recurring adjustments that we consider necessary for a fair presentation of the unaudited quarterly results when read in conjunction with the audited financial statements and notes. We believe that quarter-to-quarter comparisons of financial results are not necessarily meaningful and should not be relied upon as an indication of future performance (in thousands, except per share amounts).
*includes adjustment of $355,000, which resulted from our increased estimated bonus accrual
F-19
|
EXHIBIT INDEX
| |
|
Exhibit No. |
Description of Exhibit |
|
2.1 |
Agreement and Plan of Merger and Reorganization, dated October 6, 2005, among the Registrant, PolyMedix Merger Sub, Inc., PolyMedix Pharmaceuticals, Inc. and those stockholders of Registrant identified on Exhibit A thereto.(1) |
|
3.1 |
Amended and Restated Certificate of Incorporation of Registrant.(1) |
|
3.2 |
Amendment to the Amended and Restated Certificate of Incorporation of Registrant.(1) |
|
3.3 |
Amended and Restated By‑Laws of Registrant.(1) |
|
3.4 |
Amendment to Amended and Restated By‑Laws of Registrant.(1) |
|
3.5 |
Amendment to the Amended and Restated Certificate of Incorporation of Registrant, as amended.(2) |
|
3.6 |
Certificate of Amendment to Amended and Restated Certificate of Incorporation of Registrant, as amended. (3) |
|
3.7 |
Certificate of Designation, Preferences and Rights of Series C Preferred Stock. (4) |
|
4.1 |
Form of common stock Purchase Warrant issued to Fordham Financial Management, Inc. on November 8, 2005, December 8, 2005, January 10, 2006 and February 15, 2006.(1) |
|
4.2 |
Form of common stock Purchase Warrant. (5) |
|
4.3 |
Form of Registration Rights Substitution Agreement.(6) |
|
4.4 |
Form of Amendment No. 1 to Registration Rights Substitution Agreement.(7) |
|
4.5 |
Form of Amendment No. 2 to Registration Rights Substitution Agreement.(7) |
|
4.6 |
Form of Series A Warrant Agreement (including Series A Warrant certificate).(8) |
|
4.7 |
Form of Placement Agent Warrant.(8) |
|
4.8 |
Form of Securities Purchase Agreement as executed by the Registrant and the Investors. (9) |
|
4.9 |
Form of Series B Warrant to Purchase Capital Stock. (9) |
|
4.10 |
Rights Agreement, by and between PolyMedix, Inc. and American Stock Transfer & Trust Company, LLC, as Rights Agent, including exhibits thereto, dated as of May 12, 2009.(4) |
|
4.11 |
Form of Purchase Agreement in connection with the November 2009 offering.(10) |
|
4.12 |
Form of Series C Warrant to Purchase Common Stock issued in connection with the November 2009 offering.(10) |
|
4.13 |
Form of Placement Agent Warrant issued in connection with the November 2009 offering.(10) |
|
10.1 |
Patent License Agreement, dated January 3, 2003, between the Registrant and the University of Pennsylvania.(1) |
|
10.2 |
Letter Agreement, dated December 23, 2003, amending the Patent License Agreement, dated January 3, 2003, between the Registrant and the University of Pennsylvania.(1) |
|
10.3 |
Software License Agreement, dated May 30, 2003, between the Registrant and the University of Pennsylvania.(1) |
|
10.4 |
Exclusive License Agreement, dated January 2, 2005, between the Registrant and the University of Massachusetts.(1) |
|
10.5** |
Employment Agreement, dated July 30, 2002, between Nicholas Landekic and the Registrant.(1) |
|
10.6** |
Employment Agreement, dated December 5, 2005, between Edward Smith and the Registrant.(1) |
|
10.7** |
Employment Agreement, dated March 28, 2003, between Richard Scott, Ph.D. and the Registrant.(1) |
|
10.8 |
Letter Agreement, dated February 25, 2004, between Dr. William DeGrado and the Registrant.(1) |
|
10.9 |
Lab/Office Space License Agreement for 3701 Market Street, dated February 22, 2006, between the Registrant and the University City Science Center.(1) |
|
10.10 |
Pennsylvania Full‑Service Lease Agreement for 170 N. Radnor‑Chester Road; Suite 300, Radnor, PA 19087, dated May 26, 2006, between the Registrant and the Radnor Properties — SDC, L.P.(11) |
|
10.11** |
Amended and Restated 2005 Omnibus Equity Compensation Plan of the Registrant.(12) |
|
10.12** |
2002 Equity Compensation Plan of the Registrant.(13) |
|
10.13** |
Form of Incentive Stock Option Agreement.(1) |
|
10.14** |
Form of Nonqualified Stock Option Agreement.(1) |
|
10.15 |
Sponsored Research Agreement, dated January 5, 2004, between the Registrant and the University of Massachusetts.(14) |
|
10.16 |
Merrill Lynch Loan Management Account Agreement, dated April 13, 2006, between the Registrant and Merrill Lynch Bank USA.(11) |
|
10.17** |
Employment Agreement, dated October 19, 2006, between R. Eric McAllister and the Registrant.(14) |
|
10.18** |
Offer Letter to Bozena Korczak, Ph.D.(15) |
|
10.19 |
Form of Amended and Restated Co‑Placement Agent Agreement by and among the Registrant, Carter Securities, LLC and Fordham Financial Management, Inc. (8) |
|
10.20** |
Offer Letter to J. Gregory Ford.(16) |
|
10.21 |
Investment Agreement by and between PolyMedix, Inc. and Dutchess Equity Fund, LP dated May 20, 2009.(17) |
|
10.22 |
Registration Rights Agreement by and between PolyMedix, Inc. and Dutchess Equity Fund, LP dated May 20, 2009.(17) |
|
10.23 |
Amendment No. 1 to Investment Agreement dated July 8, 2009, between the Registrant and Dutchess Equity Fund, LP.(18) |
|
10.24 |
Loan and Security Agreement dated March 31, 2010 by and among PolyMedix, Inc., PolyMedix Pharmaceuticals, Inc. and Hercules Technology II, L.P. (19) |
|
10.25 |
Common Stock Purchase Warrant dated March 31, 2010(19) |
|
10.26 |
Registration Rights Agreement dated March 31, 2010 by and between PolyMedix, Inc. and Hercules Technology II, L.P. (19) |
|
10.27 |
Offer Letter, dated as of January 18, 2011, by and between the Company and R. Eric McAllister(20) |
|
21 |
List of Subsidiaries(14) |
|
23.1* |
Consent of Deloitte & Touche LLP |
|
24* |
Power of Attorney (Included on signature page) |
|
31.1* |
Certification Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
|
31.2* |
Certification Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
|
32* |
Certification Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
F-20
* Filed herewith.
** Management contract or compensatory arrangement.
(1) Filed as an Exhibit to the Registration Statement on Form 10‑SB (File No. 000‑51895) filed on April 5, 2006 and incorporated herein by reference.
(2) Filed as an Exhibit to the Current Report on Form 8‑K filed on June 5, 2007 and incorporated herein by reference.
(3) Filed as an Exhibit to the Current Report on Form 8-K filed on December 12, 2008 and incorporated herein by reference.
(4) Filed as an Exhibit to the Current Report on Form 8-K filed on May 14, 2009 and incorporated herein by reference.
(5) Filed as an Exhibit to Amendment No. 5 to the Registration Statement on Form SB‑2 (File No. 333‑146180) on November 30, 2007 and incorporated herein by reference.
(6) Filed as an Exhibit to the Registration Statement on Form SB‑2 (File No. 333‑146180) on September 19, 2007 and incorporated herein by reference.
(7) Filed as an Exhibit to Amendment No. 2 to the Registration Statement on Form SB‑2 (File No. 333‑146180) on October 16, 2007 and incorporated herein by reference.
(8) Filed as an Exhibit to Amendment No. 1 to the Registration Statement on Form S-1 (File No. 333-151084) on June 27, 2008 and incorporated herein by reference.
F-21
(9) Filed as an Exhibit to the Current Report on Form 8-K filed on September 24, 2008 and incorporated herein by reference.
(10) Filed as an Exhibit to Amendment No. 2 to the Registration Statement on Form S-1 (File No. 333-160833) on November 6, 2009 and incorporated herein by reference.
(11) Filed as an Exhibit to Amendment No. 2 to the Registration Statement on Form 10‑SB/A (File No. 000‑51895) filed on June 19, 2006 and incorporated herein by reference.
(12) Filed as Appendix A to the Registrant’s Definitive Proxy Statement filed on May 15, 2009 and incorporated herein by reference.
(13) Filed as an Exhibit to the Registration Statement on Form S‑8 (File No. 333‑139686) filed on December 26, 2006 and incorporated herein by reference.
(14) Filed as an Exhibit to the Annual Report on Form 10‑KSB filed on March 19, 2007 and incorporated herein by reference.
(15) Filed as an Exhibit to the Current Report on Form 8‑K filed on November 16, 2007 and incorporated herein by reference.
(16) Filed as an Exhibit to the Current Report on Form 8-K filed on December 4, 2008 and incorporated herein by reference.
(17) Filed as an Exhibit to the Current Report on Form 8-K filed on May 22, 2009 and incorporated herein by reference.
(18) Filed as an Exhibit to the Registration Statement on Form S-1 (File No. 333-160470) on July 8, 2009 and incorporated herein by reference.
(19) Filed as an Exhibit to the Current Report on Form 8-K filed on April 2, 2010 and incorporated herein by reference.
(20) Filed as an Exhibit to the Current Report on Form 8-K filed on January 24, 2011 and incorporated herein by reference.
F-22