Attached files
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
| For the Fiscal Year ended December 31, 2010 or |
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
| For the Transition Period from to |
Commission File Number: 000-23265
Salix Pharmaceuticals, Ltd.
(Exact name of Registrant as specified in its charter)
| Delaware | 94-3267443 | |
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) |
1700 Perimeter Park Drive
Morrisville, North Carolina 27560
(Address of principal executive offices, including zip code)
(919) 862-1000
(Registrant’s telephone number, including area code)
Securities Registered Pursuant to Section 12(b) of the Act:
| Title of Each Class |
Name of Exchange on which Registered | |
| Common Stock, $0.001 Par Value Preferred Share Purchase Rights |
Nasdaq Global Market Nasdaq Global Market |
Securities Registered Pursuant to Section 12(g) of the Act: None
Indicate by check mark if the Registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. YES x NO ¨
Indicate by check mark if the Registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. YES ¨ NO x
Indicate by check mark whether the Registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. YES x NO ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). YES ¨ NO ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the Registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer,” and “smaller reporting company” in Rule 12b-2 of the Act (Check one):
Large accelerated filer x Accelerated filer ¨ Non-accelerated filer ¨ Smaller reporting company ¨
Indicate by check mark whether registrant is a shell company (as defined) in Rule 12b-2 of the Exchange Act. YES ¨ NO x
The aggregate market value of the Registrant’s common stock held by non-affiliates of the Registrant on June 30, 2010 (based on the closing sale price of U.S. $39.03 of the Registrant’s common stock, as reported on The Nasdaq Global Market on such date) was approximately U.S. $1,886,965,222. Common stock held by each officer and director and by each person known to the Company who owned 10% or more of the outstanding common stock have been excluded in that such persons may be deemed to be affiliates. This determination of affiliate status is not necessarily a conclusive determination for other purposes.
The number of shares of the Registrant’s common stock outstanding at February 25, 2011 was 58,171,363.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the Registrant’s definitive Proxy Statement to be filed for its 2011 Annual Meeting of Stockholders currently scheduled to be held June 16, 2011 are incorporated by reference into Part III of this report.
Table of Contents
ANNUAL REPORT ON FORM 10-K
TABLE OF CONTENTS
| Page | ||||||
| PART I | ||||||
| Item 1. |
1 | |||||
| Item 1A. |
22 | |||||
| Item 1B. |
30 | |||||
| Item 2. |
30 | |||||
| Item 3. |
31 | |||||
| Item 4. |
32 | |||||
| 32 | ||||||
| PART II | ||||||
| Item 5. |
34 | |||||
| Item 6. |
36 | |||||
| Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
37 | ||||
| Item 7A. |
53 | |||||
| Item 8. |
53 | |||||
| Item 9. |
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
53 | ||||
| Item 9A. |
54 | |||||
| Item 9B. |
54 | |||||
| PART III | ||||||
| Item 10. |
55 | |||||
| Item 11. |
55 | |||||
| Item 12. |
Security Ownership of Certain Beneficial Owners and Management and |
55 | ||||
| Item 13. |
Certain Relationships and Related Transactions, and Director Independence |
56 | ||||
| Item 14. |
56 | |||||
| PART IV | ||||||
| Item 15. |
57 | |||||
| 63 | ||||||
i
Table of Contents
This report contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933 and Section 21E of the Securities Exchange Act of 1934. These forward-looking statements are subject to risks and uncertainties, including those set forth under “Item 1A. Risk Factors” and “Cautionary Statement” included in “Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations” and elsewhere in this report, that could cause actual results to differ materially from historical results or anticipated results. Unless otherwise indicated or required by the context, the terms “we,” “our,” “us” and the “Company” refer to Salix Pharmaceuticals, Ltd. and all of its subsidiaries.
Salix Pharmaceuticals, Ltd., a Delaware corporation, is a specialty pharmaceutical company dedicated to acquiring, developing and commercializing prescription drugs used in the treatment of a variety of gastrointestinal diseases, which are those affecting the digestive tract. Our website address is www.salix.com. Information on our website is not incorporated herein by reference. We make available free of charge through our website our press releases, Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and all amendments to those reports as soon as reasonably practicable after electronically filed with or furnished to the Securities and Exchange Commission.
OVERVIEW
We are a specialty pharmaceutical company dedicated to acquiring, developing and commercializing prescription drugs used in the treatment of a variety of gastrointestinal disorders, which are those affecting the digestive tract. Our strategy is to:
| • | identify and acquire rights to products that we believe have potential for near-term regulatory approval or are already approved; |
| • | apply our regulatory, product development, and sales and marketing expertise to commercialize these products; and |
| • | use our approximately 250-member specialty sales and marketing team focused on high-prescribing U.S. gastroenterologists, who are doctors who specialize in gastrointestinal disorders, to sell our products. |
Our current products demonstrate our ability to execute this strategy. As of December 31, 2010, our products were:
| • | XIFAXAN® (rifaximin) Tablets 200 mg, indicated for travelers’ diarrhea; |
| • | XIFAXAN® (rifaximin) Tablets 550 mg, indicated for hepatic encephalopathy, which we began selling in the second quarter of 2010; |
| • | MOVIPREP® (PEG 3350, Sodium Sulfate, Sodium Chloride, Potassium Chloride, Sodium Ascorbate and Ascorbic Acid for Oral Solution); |
| • | OSMOPREP™ (sodium phosphate monobasic monohydrate, USP and sodium phosphate dibasic anhydrous, USP) Tablets; |
| • | VISICOL® (sodium phosphate monobasic monohydrate, USP, and sodium phosphate dibasic anhydrous, USP) Tablets; |
| • | APRISO™ (mesalamine) extended-release capsules 0.375g; |
| • | METOZOLV™ ODT (metoclopramide HCl) 5mg and 10mg orally disintegrating tablets; |
| • | AZASAN® Azathioprine Tablets, USP, 75mg and 100mg; |
| • | ANUSOL-HC® 2.5% (Hydrocortisone Cream, USP), ANUSOL-HC® 25 mg Suppository (Hydrocortisone Acetate); |
1
Table of Contents
| • | PROCTOCORT® Cream (Hydrocortisone Cream, USP) 1% and PROCTOCORT® Suppository (Hydrocortisone Acetate Rectal Suppositories) 30 mg; |
| • | PEPCID® (famotidine) for Oral Suspension; |
| • | Oral Suspension DIURIL® (Chlorothiazide); and |
| • | COLAZAL® (balsalazide disodium) Capsules 750 mg. |
We generate revenue primarily by selling our products, namely prescription drugs, to pharmaceutical wholesalers. These direct customers resell and distribute our products to and through pharmacies to patients who have had our products prescribed by doctors. We currently market our products, and intend to market future products, if approved by the U.S. Food and Drug Administration, or FDA, to U.S. gastroenterologists and other physicians through our own direct sales force. In December 2000, we established our own field sales force to market Colazal in the United States. As of December 31, 2010, this sales force had approximately 160 sales representatives in the field marketing our approved products. Although the creation of an independent sales organization involved substantial costs, we believe that the financial returns from our direct product sales have been and will continue to be more favorable to us than those from the indirect sale of products through marketing partners. We enter into distribution or licensing relationships outside the United States and in certain markets in the U.S. where a larger sales organization is appropriate. As of December 31, 2010, our sales and marketing staff, including our sales representatives, consisted of approximately 250 people.
Because demand for our products originates with doctors, our sales force calls on high-prescribing specialists, primarily gastroenterologists, and we monitor new and total prescriptions for our products as key performance indicators for our business. Prescriptions result in our products being used by patients, requiring our direct customers to purchase more products to replenish their inventory. However, our revenue might fluctuate from quarter to quarter due to other factors, such as increased buying by wholesalers in anticipation of a price increase or because of the introduction of new products. Revenue could be less than anticipated in subsequent quarters as wholesalers’ increased inventory is used up.
Our primary product candidates currently under development and their status are as follows:
| Compound |
Indication |
Status | ||
| Rifaximin |
Irritable bowel syndrome, or IBS | New Drug Applications, or NDA, submitted June 7, 2010; Prescription Drug User Fee Action, or PDUFA, date March 7, 2011; Complete Response Letter anticipated on or before March 7, 2011 | ||
| Crofelemer |
HIV-associated diarrhea | Phase III completed | ||
| Balsalazide disodium tablet |
Ulcerative colitis | Complete response letter received April 27, 2010 | ||
| Budesonide foam |
Ulcerative proctitis | Phase III | ||
PRODUCTS
Xifaxan® (rifaximin) tablets
Xifaxan is a gastrointestinal-specific oral antibiotic. The FDA approved Xifaxan 200mg in May 2004 for the treatment of patients 12 years of age and older with travelers’ diarrhea caused by noninvasive strains of E coli. According to the Centers for Disease Control, each year between 30% and 50% of international travelers, an estimated 20.0 million people, develop diarrhea, with approximately 80% of the cases caused by bacteria. Approximately 5.8 million people sought treatment in the United States for infectious diarrhea in 2010 and approximately 3.6 million of those patients were prescribed a drug.
2
Table of Contents
Xifaxan 550mg was approved by the FDA in March 2010 for reduction in risk of overt hepatic encephalopathy, or HE, recurrence in patients 18 years of age or older. There are reported to be approximately 200,000 patients in the United States who suffer from episodic overt HE.
We believe the advantages of Xifaxan to treat these conditions are two-fold: (1) site-targeted antibiotic delivery; and (2) improved tolerability compared to other treatments. Less than 0.4% of the drug is absorbed into the bloodstream when it is taken orally. In addition, the drug might also cause fewer side effects or discomforts such as nausea, headache or dizziness than observed with currently available, more highly-absorbed antibiotics. We believe Xifaxan is also less likely to cause harmful interaction with other drugs a patient may be taking. We believe Xifaxan is unique because there is no other U.S.-approved oral antibiotic with its potential lack of systemic absorption and safety profile.
We launched Xifaxan 200mg in the United States in July 2004 and Xifaxan 550mg in May 2010 using our own direct sales force. We are exploring potential additional indications, formulations, clinical trials and co-promotion arrangements to capitalize on the potential for Xifaxan, including our development program in irritable bowel syndrome. Based on this potential indication, we believe Xifaxan can potentially compete in an annual U.S. market in excess of approximately $9.5 billion. While the potential market for Xifaxan is large, we expect to capture only a portion of each market due to competition, ability to capture share in each market, market acceptance and other factors.
The patents for the rifaximin composition of matter (also covering a process of making rifaximin and using rifaximin to treat gastrointestinal infectious diseases) expired in May 2001 in the United States and Canada. Rifaximin is a new chemical entity and was granted a five-year new chemical exclusivity by the FDA when it was approved in May 2004. Rifaximin, therefore, had data exclusivity to May 2009. In May 2006, U.S. Patent No. 7,045,620 (the ‘620 patent, which is a composition of matter and process patent that covers several physical states of rifaximin) was issued. We believe this patent extends the protection of the current form of rifaximin until June 2024. In November 2009, U.S. Patent No. 7,612,199 (the ‘199 patent) issued and should provide protection until June 2024. This patent provides further protection relating to U.S. Patent No. 7,045,620 that covers several physical states, or polymorphous forms, of rifaximin and provides protection for all indications currently marketed and being assessed. Alfa Wasserman S.p.a., the owner of the ‘620 and the ‘199 patents, has licensed the rights to Salix in the United States. In July 2006, Salix entered into an agreement with Cedars-Sinai Medical Center, or CSMC, for the right to use its patent and patent applications relating to methods of diagnosis and treating irritable bowel syndrome and other disorders caused by small intestinal bacterial overgrowth. The CSMC agreement provides Salix the right to use U.S. Patent No. 7,452,857, which issued in November 2008, providing protection relating to rifaximin for treating irritable bowel syndrome, or IBS, caused by small intestinal bacterial overgrowth; US 7,718,608, which issued May of 2010 and provides protection relating to rifaximin for treating IBS; and U.S. Patent No. 7,605,240, which issued in October 2009, providing protection relating to rifaximin for treating bloating caused by small intestinal bacterial overgrowth related to IBS, each of with provide protection until August 2019. Below is a tabular summary of issued U.S. patents related to rifaximin that we own or have licensed.
| U.S. Patent No. |
Issue Date |
Expiration |
Subject | |||||
| 7,045,620* | May 2006 |
June 2024 |
Composition of matter and process patent covering several physical states of rifaximin | |||||
| 7,452,857** | November 2008 |
August 2019 |
Use of rifaximin for treating irritable bowel syndrome | |||||
| 7,605,240** | October 2009 |
August 2019 |
Treatment of bloating caused by small intestinal bacterial overgrowth associated with irritable bowel syndrome | |||||
| 7,718,608** | May 2010 |
August 2019 |
Use of rifaximin for treating irritable bowel syndrome | |||||
| 7,612,199* | November 2009 |
June 2024 |
Covers several physical states, or polymorphous forms of rifaximin | |||||
| * | Licensed from Alfa Wasserman S.p.a. |
| ** | Licensed from Cedars-Sinai Medical Center |
3
Table of Contents
In addition, we have filed applications for patents relating to additional indications using rifaximin and related chemical substances. In September 2009, Lupin Ltd. granted Salix the exclusive right in the United States to its bioadhesive drug delivery technology for use with rifaximin. In October 2009, Cipla, Limited granted Salix the exclusive rights in the United States to its amorphous rifaximin application PCT Patent Application No. PCT/GB2007/003629; WO 2008/035109.
MoviPrep® (PEG 3350, sodium sulfate, sodium chloride, potassium chloride, sodium ascorbate and ascorbic acid) oral solution
In December 2005, we acquired exclusive rights to sell MoviPrep in the United States from Norgine B.V. MoviPrep is a patent-protected, liquid polyethylene glycol-salt , or PEG, bowel cleansing product that was approved by the FDA in August 2006 and competes with a number of liquid PEG bowel cleansing products. MoviPrep is differentiated from other liquid PEG bowel cleansing products by the inclusion of ascorbic acid in its formulation. MoviPrep is indicated for bowel cleansing prior to colonoscopy, intestinal surgery and barium enema X-ray examinations.
Norgine, B.V. and Norgine Europe, B. V. own U.S. Patent No. 7,169,381, or the ‘381 patent, which is listed with the FDA as protecting our MoviPrep product to September 2024. Norgine licensed MoviPrep and the ‘381 patent to us for commercialization in the United States. On February 9, 2010 U.S. Patent No. 7,658,914 was issued and listed in the Orange Book for MoviPrep. In August 2010 we entered into a Sublicense Agreement that granted Novel Laboratories, Inc. a license to the patents covering MoviPrep such that Novel is permitted to launch a generic MoviPrep on September 24, 2018.
OsmoPrep® and Visicol® (sodium phosphate monobasic monohydrate, USP, sodium phosphate dibasic anhydrous, USP) tablets
In September 2005, we acquired Visicol with the completion of the acquisition of InKine Pharmaceutical Company, Inc. Visicol and OsmoPrep tablets are indicated for cleansing of the colon as a preparation for colonoscopy in adults 18 years of age or older. Visicol was the first, and it and OsmoPrep are the only, tablet bowel cleansing products approved by the FDA and marketed in the United States. OsmoPrep is a patented, second-generation tablet bowel cleansing product approved by the FDA in March 2006. OsmoPrep offers potential benefits compared to Visicol such as its lack of microcrystalline cellulose, smaller tablet size and possible lower dose administration.
CDC, LLC, owns U.S. Patent No. 5,616,346, or the ‘346 patent, for the formulation and use of OsmoPrep, which CDC licensed to us for commercialization in the United States. The ‘346 patent is listed with the FDA as protecting our OsmoPrep product to 2013. US 7,687,075, which issued in March 2010 provides protection until June 2028. In September 2010 we entered into a Sublicense Agreement which granted Novel Laboratories, Inc. a license under the patents covering OsmoPrep such that Novel is permitted to launch a generic OsmoPrep on November 16, 2019.
On December 11, 2008, the FDA announced a proposed boxed warning for OsmoPrep and Visicol that addresses the potential risk of acute kidney injury. During 2009, working with the FDA, we revised the labels to include the boxed warning, and developed a risk evaluation and mitigation strategy, or REMS, including a medication guide. We plan to conduct post–marketing clinical trials as part of this strategy.
Apriso™ (mesalamine) extended-release capsules 0.375g
In July 2002 we acquired the exclusive development rights in the United States to a granulated mesalamine product from Dr. Falk Pharma GmbH, one of the most recognized gastroenterology companies worldwide. On October 31, 2008, the FDA granted marketing approval for Apriso for the maintenance of remission of ulcerative colitis in adults. Apriso is a locally-acting aminosalicylate and is the first and only mesalamine product approved
4
Table of Contents
by the FDA for once-a-day dosing for the maintenance of remission of ulcerative colitis. Apriso is designed to provide for the distribution of the active ingredient beginning in the small bowel and continuing throughout the colon. The product’s unique prolonged release mechanism might allow us to expand the range of treatment options for ulcerative colitis. We shipped Apriso to wholesalers in the fourth quarter of 2008 and launched Apriso to physicians in March 2009. Apriso is patent protected until 2018.
Metozolv™ ODT (metoclopramide HCl) 5mg and 10mg orally disintegrating tablets
In September 2007, we acquired exclusive, worldwide rights to metoclopramide-Zydis® from Wilmington Pharmaceuticals LLC. Wilmington submitted an NDA seeking approval to market Metozolv ODT and on February 26, 2009, Wilmington received a complete response letter from the FDA, indicating that it requires a REMS for Metozolv prior to approval of the NDA. In a separate action on February 26, 2009, the FDA issued a class-wide requirement for all manufacturers of metoclopramide in the United States to provide a REMS for their products. On September 8, 2009 the FDA granted marketing approval for METOZOLV™ ODT (metoclopramide HCl) 5 mg and 10 mg orally disintegrating tablets. METOZOLV ODT is indicated for the relief of symptomatic gastroesophageal reflux or short-term (4-12 weeks) therapy for adults with symptomatic, documented gastroesophageal reflux who fail to respond to conventional therapy and diabetic gastroparesis or the relief of symptoms in adults associated with acute and recurrent diabetic gastroparesis. METOZOLV ODT is a fast-dissolve formulation of metoclopramide that has patent protection until 2017 and additional patent protection pending that, if issued, will provide intellectual property protection until 2023. On November 3, 2010, we received a paragraph IV notification from Novel Laboratories, Inc. stating that Novel had filed an ANDA application to seek approval to market a generic version of Metoclopramide Hydrochloride ODT, 5 mg and 10 mg. The notification letter asserted non-infringement of U.S. Patent No. 6,413,549 (“the ‘549 patent”). Upon examination of the relevant sections of the ANDA, we concluded that the ‘549 patent would not be enforced against Novel Laboratories.
Azasan® (azathioprine) tablets
In November 2003, we acquired from aaiPharma LLC the exclusive right to sell 25, 75 and 100 milligram dosage strengths of azathioprine tablets in North America under the brand name Azasan. Azasan is an FDA-approved drug that suppresses immune system responses and is indicated for preventing rejection of kidney transplants and treatment of severe arthritis. In February 2004, we launched the 75 and 100 milligram dosage strengths of Azasan in the United States. The patents and data exclusivity for Azasan have expired.
Anusol-HC® and Proctocort® (hydrocortisone) creams and suppositories
In June 2004, we acquired the exclusive right to sell Anusol-HC 2.5% (hydrocortisone USP) cream, Anusol-HC 25 mg (hydrocortisone acetate) rectal suppositories, Proctocort 1% (hydrocortisone USP) cream and Proctocort 30 mg (hydrocortisone acetate) rectal suppositories from King Pharmaceuticals, Inc. The two cream products are topical corticosteroids indicated for relief of the inflammatory and pruritic, or itching, manifestations of corticosteroid-responsive dermatoses. The two suppository products are indicated for use in inflamed hemorrhoids and postirradiation proctitis, as well as an adjunct in the treatment of chronic ulcerative colitis and other inflammatory conditions. The patents and data exclusivity for Anusol-HC and Proctocort have expired.
Pepcid® (famotidine) for Oral Suspension and Oral Suspension Diuril® (Chlorothiazide)
In February 2007, we purchased the U.S. prescription pharmaceutical product rights to Pepcid Oral Suspension and Diuril Oral Suspension from Merck & Co., Inc. Pepcid Oral Suspension is a widely known prescription pharmaceutical product indicated for several gastrointestinal indications, including the treatment of duodenal ulcer, benign gastric ulcer and gastro-esophageal reflux disease. Pepcid Oral Suspension and Diuril Oral Suspension, both liquid formulations of their solid dosage form counterparts, compete in a combined annual
5
Table of Contents
U.S. market of approximately $183 million, concentrated in pediatric and hospitalized patient populations. The patents and data exclusivity for Pepcid Oral Suspension and Diuril Oral Suspension have expired. In May 2010, the FDA approved a generic famotidine oral suspension product, and we launched an authorized generic famotidine product. In June 2010 the FDA approved another generic famotidine oral suspension product.
Colazal® (balsalazide disodium) capsules
Our first drug, Colazal, was approved by the FDA in 2000 for the treatment of mildly to moderately active ulcerative colitis. We launched Colazal to physicians in the United States in January 2001. In December 2006, the FDA approved Colazal for use in pediatric patients between 5 to 17 years of age with ulcerative colitis. The pediatric use of Colazal has been granted orphan drug designation. On December 28, 2007, the Office of Generic Drugs, or OGD, approved three generic balsalazide capsule products, and we announced that we had entered into an agreement with Watson Pharma, Inc. to market and sell an authorized generic of Colazal. We do not anticipate significant Colazal sales in future periods.
DEVELOPMENT PROGRAMS
Xifaxan® (rifaximin) tablets
Irritable bowel syndrome, characterized by abdominal pain, bloating and altered bowel habits, is one of the most common chronic medical conditions and is associated with substantial medical costs. In September 2009, we announced the successful completion and outcome of our two identical pivotal Phase III trials to evaluate the efficacy and safety of rifaximin 550 mg dosed three-a-day in the treatment of non-constipation irritable bowel syndrome, or non-C IBS. In each trial rifaximin versus placebo treated patients demonstrated a statistically significant improvement for the primary endpoint of the adequate relief of IBS symptoms as assessed over a one-month period (weeks 3, 4, 5 and 6) following completion of a 14-day course of therapy (weeks 1 and 2). Consistent with the primary endpoint in each trial, the key secondary endpoint of relief of IBS-related bloating also demonstrated statistical significance of rifaximin versus placebo in each trial. In June 2010 we submitted an efficacy supplement to NDA 21–361 for Xifaxan 550 mg tablets for the proposed indication of treatment of non–constipation irritable bowel syndrome and IBS–related bloating. In August 2010 the FDA accepted this filing for Priority Review and issued a PDUFA goal date of December 7, 2010. In October 2010 the FDA extended the PDUFA goal date to March 7, 2010. On February 24, 2011, we announced that based on a telephone conversation with the FDA we anticipate receiving a Complete Response Letter on or before March 7, 2011. The FDA issues a Complete Response Letter to indicate that the review cycle for an application is complete and that the application is not ready for approval. Based on this telephone conversation, we are of the understanding that the FDA deems that the Xifaxan 550 mg sNDA is not ready for approval, primarily due to a newly expressed need for retreatment information. We will consider next steps with respect to the Xifaxan 550 mg sNDA for non-C-IBS following receipt of a Complete Response Letter.
Crofelemer
In December 2008 we acquired rights to crofelemer from Napo Pharmaceuticals, Inc. We are investigating crofelemer as an anti-secretory anti-diarrheal agent for the treatment of chronic diarrhea in people living with HIV, or HIV-associated diarrhea. Crofelemer is a first-in-class, naturally occurring, gut-targeted, oral anti-secretory, anti-diarrheal agent that has minimal absorption. In August 2009, we completed Stage 1 (dose selection stage involving approximately 200 patients) and initiated Stage 2 (final stage involving 150 patients) of our Phase III trial. The FDA reviewed and granted the protocol for this trial as a Special Protocol Assessment, or SPA, and granted the crofelemer IND fast track designation. In November 2010 we announced the successful completion and outcome of our Phase III trial to evaluate the efficacy, safety and tolerability of crofelemer in treating HIV-associated diarrhea. In this study, crofelemer provided relief of diarrhea for a highly statistically significant proportion of patients compared to placebo. The study utilized an adaptive design consisting of two stages. In Stage 1, subjects received 125 mg, 250 mg or 500 mg crofelemer or placebo. Based on Stage 1 results, an independent panel of experts chose a dose of 125 mg crofelemer for Stage 2. Stage 2 allowed for additional subjects to be enrolled into the 125 mg and placebo groups. The 125 mg dose performed very similarly to the 500
6
Table of Contents
mg maximum dose in regards to the primary endpoint. Due to the adaptive design of the trial, the trial prespecified a treatment difference at a p-value target of 0.025. The actual p-value achieved in the study was 0.0096. In this trial, the overall safety profile of crofelemer was balanced across all groups, including treatment and placebo.
Patents for crofelemer provide intellectual property protection to 2018. If crofelemer receives marketing approval, it should be eligible for five years of marketing exclusivity as a new molecular entity. The product also might be entitled to patent term restoration.
Balsalazide Disodium tablets
We have developed an 1100 mg tablet formulation of balsalazide disodium. We believe the convenience the balsalazide tablet formulation is designed to provide, by means of twice-a-day dosing and a reduced number of pills, demonstrates our ongoing commitment to bring products to market that better serve the needs of gastroenterologists and their patients. On July 17, 2007 we submitted an NDA to the FDA seeking approval to market an 1100 mg tablet formulation of balsalazide disodium. On May 16, 2008 we received an approvable letter from the FDA. We submitted a complete response to the approvable letter on June 30, 2008. On December 22, 2008, the FDA issued a complete response letter. We met with the FDA on March 16, 2009 to discuss the NDA, and on October 26, 2009 we submitted a complete response to the December 22, 2008 FDA complete response letter. On April 27, 2010 the FDA issued a complete response letter. In this complete response letter there are no requests for new pre–clinical or clinical trials. The sole issue raised in this letter concerns a site deficiency, not directly related to balsalazide tablet production, at the manufacturing facility identified in this NDA. The manufacturer has responded to the FDA and continues to work with the FDA to resolve the matter. We do not intend to conduct additional clinical investigation of balsalazide tablets in this indication. The patent for balsalazide disodium tablets will expire in 2018.
Budesonide
In March 2008, we acquired a license from Dr. Falk Pharma GmbH to a family of budesonide products, including a budesonide rectal foam, in the United States. In November 2009, we initiated two Phase III trials to evaluate the effectiveness and safety of budesonide rectal foam for the treatment of mild to moderate ulcerative proctitis or proctosigmoiditis. The rectal foam product has patent coverage in the United States until 2015.
COLLABORATIVE AND PRODUCT ACQUISITION AGREEMENTS
We have and plan to continue to enter into various collaborations and product acquisition agreements with licensors, licensees and others. To date, we have entered into the following agreements:
Product Acquisitions and In-License Agreements
aaiPharma LLC
In November 2003, we acquired from aaiPharma LLC for $2.0 million the exclusive right to sell 25, 75 and 100 milligram dosage strengths of azathioprine tablets in North America under the name Azasan. In addition, the agreement provides that Salix is to pay aaiPharma, on a quarterly basis, a low double-digit percentage royalty payment based on Salix’s net sales of Azasan in exchange for aaiPharma supplying us with drug product. Because the amount of the royalty payment is based on net sales during a quarter, with no minimum royalty amount, Salix is unable to prospectively disclose the absolute amount of such royalty payments. Royalties are only incurred if there is associated revenue, and then are included in “Cost of products sold” in the Statements of Operations. The license agreement and royalty obligations do not have a fixed expiration date, but may be terminated by either party if the other materially breaches the agreement and fails to cure the breach after notice. In addition, aaiPharma has the right to terminate the agreement if Salix is adjudged bankrupt, and Salix has the right to terminate the agreement at any time upon six months’ written notice to aaiPharma.
7
Table of Contents
Alfa Wassermann S.P.A.
In June 1996, Salix entered into a license agreement with Alfa Wassermann S.p.A, a privately held pharmaceutical company headquartered in Italy, pursuant to which Alfa Wassermann licensed to Salix the exclusive rights to make, use and sell rifaximin (Xifaxan) in the United States and Canada for the treatment of gastrointestinal and respiratory tract diseases. Pursuant to the license agreement, we agreed to pay Alfa Wassermann a net sales-based single- digit percentage royalty, as well as milestone payments. Salix made annual milestone payments in varying amounts to Alfa Wassermann until the commercial launch of Xifaxan in July 2004. No more milestone payments remain under this agreement. Our obligation to pay royalties commenced upon the commercial launch of the product and continues until the later of (1) the expiration of the period in which the manufacture, use or sale of the products by an unlicensed third party would constitute an infringement on the patent covering the product or (2) 10 years from commercial launch. The last patent is currently scheduled to expire in 2024. Thereafter, the licenses granted to us shall continue as irrevocable royalty-free paid-up licenses. However, we would remain obligated to pay a net sales based royalty for use of the product trademark if we choose to continue using it after the other licenses expired. Because the amount of the royalty payment is based on net sales during a quarter, with no minimum royalty amount, Salix is unable to prospectively disclose the absolute amount of such royalty payments. Royalties are only incurred if there is associated revenue, and then are included in “Cost of products sold” in the Statements of Operations.
Alfa Wassermann has agreed separately to supply us with bulk active ingredient rifaximin at a fixed price. Salix is committed to purchase a percentage of its rolling 12-month forecast that is updated monthly, until July 2014 or introduction of a generic product, whichever occurs first, and these amounts are included in the “Purchase Commitments” line of its contractual commitments table in its Management’s Discussion and Analysis of Financial Condition and Results of Operations.
The license agreement does not have a fixed expiration date, but continues until the earlier of 10 years from the commercial launch of the first non-orphan indication or the launch of a generic rifaximin product. After the occurrence of the earlier of those two events, the agreement may continue year to year at the option of both parties. There is a six-month notice period to cancel thereafter. Either party to the agreement may terminate it following a material breach by the other party and the failure of the breaching party to remedy the breach within 60 days. In addition, Alfa Wassermann has the right to terminate the agreement on three months’ written notice in the event that we fail to sell the product for a period of six consecutive months after commercial launch. In addition, Alfa Wassermann may terminate the agreement if we become involved in bankruptcy, liquidation or similar proceedings. We may terminate the agreement in respect of any indication or any part of the territory covered on 90 days’ notice, at which point our rights with respect to that indication or territory shall cease.
Biorex Laboratories Limited
Pursuant to an agreement entered into between us and Biorex in 1992, Biorex granted us the exclusive worldwide right (other than Japan, Taiwan, Korea and the United States) to develop, manufacture and sell balsalazide for all disease indications for a period of 15 years from the date of commercial launch, subject to early termination in certain circumstances, including upon the material breach by either party and, in the case of Biorex, in the event of our bankruptcy or if a sub-licensee of ours terminates or becomes entitled to terminate its sublicense as a result of actions by us. Pursuant to this agreement, Salix must pay to Biorex a percentage of any gross profits realized by Salix, plus a percentage of fees payable to Salix in connection with any sublicense by Salix of the rights under the agreement. Under a separate agreement, Biorex granted us the exclusive right to develop, manufacture and sell balsalazide for all disease indications in the United States for a period of nine years from the date of commercial launch or the term of the applicable patent, whichever is longer. Under these agreements, we paid Biorex fees upon entering into the agreements and are obligated to make additional milestone and royalty payments for the drug. The royalty payments to be made by us pursuant to the agreement governing the United States are based on net sales, subject to minimum royalty payments for the first five years following commercial launch. Under the agreement governing territories other than the United States, we are obligated to pay to Biorex a portion of any gross profit on sales of balsalazide outside the United States. Under
8
Table of Contents
these agreements, we undertook to complete preclinical testing, perform clinical trials and obtain regulatory approvals for balsalazide. During 2001, we acquired from Biorex the exclusive right and license to develop, manufacture and sell balsalazide in Japan, Korea and Taiwan. We did not have to pay any fees to Biorex upon entering into this agreement, but we are obligated to pay Biorex a portion of any upfront payments, milestone payments and gross profit on sales of balsalazide in Japan, Korea and Taiwan as well. In December 2009 we terminated these agreements with Biorex and have no additional future payments due to Biorex.
Cedars-Sinai Medical Center
On June 28, 2006, Salix entered into a license agreement with Cedars-Sinai Medical Center, or CSMC, for the right to use a patent and a patent application relating to methods of diagnosing and treating irritable bowel syndrome and other disorders caused by small intestinal bacterial overgrowth. Under the agreement, CSMC grants Salix the right to use its patent and patent application relating to methods of treating irritable bowel syndrome and other disorders caused by small intestinal bacterial overgrowth. CSMC also grants Salix a nonexclusive license to use any unpublished research and development information, know-how and technical data of CSMC as necessary to exploit all rights granted to Salix with respect to rifaximin, with a right to sublicense. In November 2008, the U.S. Patent and Trademark Office, or USPTO, issued a patent to Cedars-Sinai Medical Center providing protection relating to rifaximin for treating irritable bowel syndrome caused by small intestinal bacterial overgrowth until August 2019. In October 2009, the USPTO issued a patent to Cedars-Sinai Medical Center providing protection relating to rifaximin for treating bloating caused by small intestinal overgrowth related to IBS. In May 2010, the USPTO issued a patent to Cedars-Sinai Medical Center providing protection relating to rifaximin for treating irritable bowel syndrome until August 2019. Salix has an exclusive license to these patents from Cedars–Sinai Medical Center to make, have made, use, sell and have sold and import licensed products related to the use of rifaximin. As of December 31, 2010, Salix had paid the aggregate $1.2 million license fee. A portion of the $1.2 million was considered an up-front, non-refundable and irrevocable licensing fee. The balance was considered a prepaid, non-refundable and irrevocable royalty applicable as credit towards royalty amounts due and payable to CSMC, if any, under the agreement. At such time as the use of rifaximin is approved by the FDA as a treatment for irritable bowel syndrome, Salix will be required to pay CSMC low single digit percentage royalties on net sales of licensed products. An additional term of the license agreement provides that Salix will expend a minimum amount per calendar year to seek and obtain regulatory approval and develop and commercialize licensed products. Because the license agreement provides the ability for Salix to terminate the agreement upon giving written notice of not less than 90 days, Salix does not include amounts payable under the license agreement as a purchase obligation in its contractual commitments table in Management’s Discussion and Analysis of Financial Condition and Results of Operations. The license agreement does not have a fixed expiration date, but continues until terminated in accordance with its terms or until the last patent expires, which is currently in 2019. Royalty obligations terminate with the related patents on a country-by-country basis and when the license agreement terminates. The agreement will terminate automatically if Salix is declared insolvent. The agreement and royalty obligations may be terminated by CSMC if Salix materially breaches the agreement and fails to cure the breach after notice.
Clinical Development Capital Partnership
In connection with Salix’s acquisition of InKine in September 2005, Salix assumed a license agreement with ALW Partnership for the worldwide rights, in perpetuity, to develop, use, market, sell, manufacture, have manufactured and sub-license Visicol and improvements, including OsmoPrep, in the field of colonic purgatives, along with ALW Partnership’s body of proprietary technical information, trade secrets and related know-how. Pursuant to this license agreement, Salix pays to Clinical Development Capital, or CDC, ALW’s successor, on a quarterly basis, a single-digit percentage royalty payment based on Salix’s net sales of these products. Because the amounts of the royalty payments are based on net sales during a quarter, Salix is unable to prospectively disclose the amount of such royalty payments. The agreement requires a minimum annual royalty payment of $0.1 million. Additional royalties are only incurred if there is associated revenue, and then are included in “Cost of products sold” in the Statements of Operations. The license agreement does not have a fixed expiration date,
9
Table of Contents
but continues until terminated in accordance with its terms or until the last patent expires, which is currently in 2028. The agreement and royalty obligations may be terminated by either party if the other materially breaches the agreement and fails to cure the breach after notice. Salix may terminate the agreement with 60 days’ written notice to CDC. CDC has the right to terminate the agreement in the event Salix is declared insolvent.
Dr. Falk Pharma GmbH
Pursuant to Salix’s license agreement, as amended, with Dr. Falk Pharma GmbH, Salix acquired the rights to develop and market a granulated formulation of mesalamine. The agreement provides that Salix make milestone payments in an aggregate amount of up to $11.0 million to Dr. Falk Pharma upon certain events prior to the commercial launch of the product, and quarterly low double-digit percentage royalty payments thereafter. As of December 31, 2010 Salix had made all of these milestone payments. Royalties are only incurred if there is associated revenue, and then are included in “Cost of products sold” in the Statements of Operations. The agreement and our obligation to pay royalties continue until the later of expiration of the last patent, which is currently scheduled in 2018, or 15 years from commercial launch, which would be December 2023 because we launched the product in December 2008. The agreement and royalty obligations may be terminated by either party if the other materially breaches the agreement and fails to cure the breach after notice. Dr. Falk Pharma may terminate the agreement if Salix sells all or substantially all of its assets or stock without notifying Dr. Falk Pharma and making a non-termination payment to Dr. Falk Pharma.
In March 2008 we acquired a license from Dr. Falk Pharma to a family of budesonide products, including a budesonide rectal foam in the United States. The rectal foam product has patent coverage in the U.S. until 2015. The agreement requires Salix to make an upfront payment and regulatory milestone payments that could total up to $9.5 million to Dr. Falk Pharma, with the majority contingent upon achievement of U.S. regulatory approval. At such time as the use of this product is approved by the FDA, Salix will be required to pay Dr. Falk Pharma low double-digit percentage royalties on net sales of licensed products. As of December 31, 2010, $1.5 million of upfront and milestone payments had been made. The remaining milestone payments are contingent upon filing an NDA and achievement of regulatory approvals. Because these milestone payments are conditioned upon events that might never occur, we do not consider the potential milestone payments as purchase obligations nor a commitment to be reported in our contractual commitments table in Management’s Discussion and Analysis of Financial Condition and Results of Operations. The agreement term continues until the later of expiration of the last patent or 17 years from commercial launch. The last patent is currently scheduled to expire in 2015, and we have yet to launch this product. The agreement may be terminated if either party materially breaches the agreement and fails to cure the breach after notice. Salix may terminate the agreement if development milestones are not achieved. Dr. Falk Pharma may terminate the agreement if development milestones are not achieved or if Salix sells all or substantially all of its assets or stock without notifying Dr. Falk Pharma and making a non-termination payment to Dr. Falk Pharma.
King Pharmaceuticals, Inc.
In June 2004, we acquired the exclusive right to sell Anusol-HC® 2.5% (hydrocortisone USP) cream, Anusol-HC® 25 mg (hydrocortisone acetate) rectal suppositories, Proctocort® 1% (hydrocortisone USP) cream and Proctocort® 30 mg (hydrocortisone acetate) rectal suppositories from King Pharmaceuticals, Inc. We paid $13.0 million cash for the four products, and entered into a supply agreement for the suppository products and the Anusol-HC cream product with King Pharmaceuticals; we established an alternate supply arrangement with a contract manufacturer for the Proctocort cream product. Once payment amounts under this and other supply agreements are known and are non-cancelable, Salix includes them in its contractual commitments table in Management’s Discussion and Analysis of Financial Condition and Results of Operations.
Lupin, Ltd.
In September 2009, we entered into a Development, Commercialization and License Agreement with Lupin Ltd for Lupin’s proprietary drug delivery technology for rifaximin. The agreement provides that we are obligated
10
Table of Contents
to make upfront and milestone payments to Lupin that could total up to $53.0 million over the term of the agreement. As of December 31, 2010, we had paid $5.0 million of milestone payments. The remaining milestone payments are contingent upon achievement of certain clinical and regulatory milestones. Because these milestone payments are conditioned upon events that might never occur, we do not consider the potential milestone payments as purchase obligations nor a commitment to be reported in our contractual commitments table in Management’s Discussion and Analysis of Financial Condition and Results of Operations. The agreement terminates upon the termination of the low double-digit royalty obligations which is the earlier of expiration of the last patent, or 10 years from commercial launch. Currently, if issued, the last patent would be scheduled to expire in June 2028, or later if patent term adjustments are granted. The agreement may be terminated by either party if the other materially breaches the agreement and fails to cure the breach after notice or if either party is declared insolvent. Salix may terminate the agreement if we determine development of the product is not commercially feasible. Lupin may terminate the agreement if the Supply Agreement described below is terminated.
In September 2009, we also entered into a Rifaximin Manufacturing and Supply Agreement. Under the this Supply Agreement, Lupin agrees to manufacture and supply us with rifaximin at a set price pursuant to rolling monthly forecasts and quarterly firm forecasts. Effective January 1, 2010, we must take or pay for rifaximin in an amount equal to not less than 50% of our requirements of rifaximin, and these amounts are included in the “Purchase Commitments” line of our contractual commitments table in our Management’s Discussion and Analysis of Financial Condition and Results of Operations. The agreement terminates 10 years from commercial launch unless extended for additional periods at Salix’s option and upon notice to Lupin. The agreement may be terminated by Salix immediately upon notice to Lupin in the event of product withdrawal by regulatory authorities. Salix may also terminate the agreement after the commercial sale or distribution of a generic version of the product by a third party. The agreement may be terminated by either party if the other materially breaches the agreement, including declaration of insolvency by either party, and fails to cure the breach after notice.
Merck & Co., Inc.
In February 2007, we entered into a Master Purchase and Sale and License Agreement with Merck & Co., Inc., to purchase the U.S. prescription pharmaceutical product rights to Pepcid Oral Suspension and Diuril Oral Suspension. Pursuant to the Agreement, Salix paid Merck $55.0 million at the closing of the transaction. In addition, Salix will make additional payments to Merck up to an aggregate of $6.0 million upon the achievement of certain annual gross sales targets for the acquired products during any of the five calendar years beginning in 2007 and ending in 2011. Because these payments are conditioned upon events that might never occur, we do not consider these payments as purchase obligations nor a commitment to be reported in our contractual commitments table in Management’s Discussion and Analysis of Financial Condition and Results of Operations.
In return for these payments, Salix obtained (1) all rights to the U. S. regulatory approvals and related data, open purchase orders, inventory and customer lists related to the acquired oral suspension products, (2) an exclusive license to the Pepcid Oral Suspension and Diuril Oral Suspension trademarks for the use of prescription sale of the acquired oral suspension products in the United States, and (3) an exclusive license to certain know-how related to the manufacture of the acquired oral suspension products in the United States. In the event that Salix is acquired by another party or if Salix sells all or substantially all of the rights to the acquired products, and Merck determines in its reasonable judgment that such transaction will result in material harm to the Pepcid Oral Suspension name or the licensed trademark, Merck has the right to terminate one or more of the above licenses and the supply obligation. In May 2010, the FDA approved a generic famotidine oral suspension product, and we launched an authorized generic famotidine product. In June 2010 the FDA approved another generic famotidine oral suspension product.
Napo Pharmaceuticals, Inc.
In December 2008 we acquired rights to crofelemer from Napo Pharmaceuticals, Inc. Patents for crofelemer provide intellectual property protection to 2018. We have completed a Phase III study of crofelemer as an anti-
11
Table of Contents
secretory anti-diarrheal agent for the treatment of chronic diarrhea in people living with HIV, or HIV-associated diarrhea. We made an initial payment of $5.0 million, consisting of $4.5 million in an upfront license fee, and a $0.5 million equity investment in Napo. In addition, we will make up to $50.0 million in milestone payments to Napo contingent on regulatory approvals and up to $250.0 million in milestone payments contingent on reaching certain sales thresholds. We will be responsible for development costs of crofelemer, but costs exceeding $12.0 million for development of crofelemer for the HIV-associated diarrhea indication will be credited towards regulatory milestones and thereafter against sales milestones. Additionally, Salix will pay tiered royalties, ranging from lower double-digits to 20 percent, depending on annual sales levels, on net sales of crofelemer, if approved. Because these milestone payments are conditioned upon events that might never occur, we do not consider the potential milestone payments as purchase obligations nor a commitment to be reported in our contractual commitments table in Management’s Discussion and Analysis of Financial Condition and Results of Operations. The license agreement and royalty obligations do not have a fixed expiration date, but continue until terminated in accordance with its terms or until the last patent expires. The last patent is currently scheduled to expire in 2018. The agreement and royalty obligations may be terminated by either party if the other materially breaches the agreement and fails to cure the breach after notice or is declared insolvent. Salix may terminate the agreement if we determine development of the product is not commercially feasible.
Norgine B.V.
In December 2005, we acquired from Norgine B.V. the exclusive rights to sell NRL944 (now marketed by us under the trade name MoviPrep), a proprietary, liquid PEG bowel cleansing product in the United States. The agreement provides that Salix make an upfront payment and milestone payments to Norgine that could total up to $37.0 million. As of December 31, 2010, Salix had made $27.0 million of upfront and milestone payments. The remaining milestone payment is contingent upon reaching a sales threshold. Because this milestone payment is conditioned upon an event that might never occur, we do not consider the potential milestone payment as a purchase obligation nor a commitment to be reported in our contractual commitments table in Management’s Discussion and Analysis of Financial Condition and Results of Operations. We pay Norgine a royalty in the teens as a percentage of net sales, and we purchase finished product from Norgine. The royalty obligations terminate upon the earlier of expiration of the last patent or approval of an Abbreviated New Drug Application for the product. The last patent is currently scheduled to expire in 2024. The license agreement does not have a fixed expiration date, but continues until terminated in accordance with its terms. The agreement may be terminated by either party if the other materially breaches the agreement and fails to cure the breach after notice or becomes insolvent. Salix may terminate the agreement with 12 months’ written notice. In August 2010 we entered into the First Amendment to License and Supply Agreement in which we and Norgine agreed to modify effective as of October 1, 2010, our obligation to source from Norgine, and Norgine’s obligation to supply, our requirements for MoviPrep. We and Norgine also agreed to a reduction in our royalty obligations to Norgine. The First Amendment to License and Supply Agreement also provides for Norgine to reimburse us for one-half of the facilities improvement and expansion payments that we are to make to Novel, up to a specified amount.
Photocure ASA
In October 2010, we acquired from Photocure ASA the worldwide exclusive rights, excluding the Nordic region, to develop and commercialize LumacanTM for diagnosing, staging or monitoring gastrointestinal dysplasia or cancer. We made an initial payment of $4.0 million to Photocure. We will be responsible for development costs of Lumacan, but Photocure will reimburse us up to $3.0 million for certain out-of-pocket costs incurred by Salix. In addition, we will make up to $76.5 million in milestone payments to Photocure contingent on development and regulatory milestones, and up to $50.0 million in milestone payments contingent on reaching certain sales thresholds. Additionally, Salix will pay tiered royalties, ranging from lower to middle double-digits, depending on annual sales levels, on net sales of Lumacan, if approved. Because these milestone payments are conditioned upon events that might never occur, we do not consider the potential milestone payments as purchase obligations nor a commitment to be reported in our contractual commitments table in Management’s Discussion and Analysis of Financial Condition and Results of Operations. Salix may terminate the License Agreement at any time at its sole discretion after the completion of screening procedures sufficient to develop adequate
12
Table of Contents
information to allow Salix to reach an informed judgment as to the scientific, regulatory and commercial potential of Lumacan products. Either party may terminate the License Agreement due to the insolvency of, or a material breach of the agreement by, the other party. Photocure may terminate the entire License Agreement if Salix or its sublicensees institutes or participates in a challenge of any of the patents licensed by Photocure to Salix under the License Agreement or if Salix fails to meet a specified clinical milestone. Photocure may also terminate the License Agreement in respect of either or both of Europe or Asia if in respect of the relevant region Salix fails to meet a specified commercialization milestone. If termination of the License Agreement occurs after initial marketing approval of a Lumacan product, Salix is entitled to royalties at a rate in the low-to-mid single digits on net sales by Photocure and its licensees of Lumacan products. If termination of the License Agreement in respect of Europe or Asia occurs after initial marketing approval of a Lumacan product in a specified major market country in the relevant region, then Salix is entitled to royalties at a rate in the low single digits on net sales by Photocure and its licensees of the Lumacan products in the region in respect of which the termination has occurred.
Wilmington Pharmaceuticals, LLC
In September 2007, we acquired the exclusive, worldwide right to sell metoclopramide–Zydis® (trade name Metozolv) from Wilmington Pharmaceuticals, LLC. The agreement provides that Salix make an upfront payment and milestone payments that could total up to $8.0 million. The Company also loaned Wilmington $2.8 million, which we netted against the payment of the approval milestone as a result of FDA approval on September 8, 2009. As of December 31, 2010, we had paid all upfront and milestone payments under this agreement. Additionally, Salix will pay mid-teen percentage royalties on net sales of Metozolv. Royalties are only incurred if there is associated revenue, and then are included in “Cost of products sold” in the Statements of Operations. The agreement terminates upon the termination of the royalty obligations, which is the earlier of expiration of the last patent or 10 years from commercial launch. We launched this product in 2009, but the last patent is currently scheduled to expire in June 2017. Salix may terminate the agreement at any time with 180 days’ written notice to Wilmington. Wilmington has the right to terminate the agreement in the event Salix opposes the grant of a patent on any patent application, or disputes or directly assists a third party to dispute the validity of any patent covered by the agreement. The agreement may be terminated by either party if the other materially breaches the agreement and fails to cure the breach after notice or if either party is declared insolvent.
Product Out-License Collaborations
Dr. Falk Pharma GmbH
In April 2007, we licensed to Dr. Falk exclusive rights to market Diacol™ 1500 mg Tablets in 28 territories in Europe. We sell Diacol, or sodium phosphate monobasic monohydrate, USP, and sodium phosphate dibasic anhydrous, tablets, USP, in the United States under the trade name OsmoPrep™ Tablets. As part of the agreement, Dr. Falk also has a non–exclusive option to market Diacol in Italy and France. Under the terms of the agreement, we could receive up to $4.0 million in milestone payments, as well as royalty payments based on product sales. Dr. Falk made the first milestone payment of $1.5 million upon execution of the agreement. Dr. Falk is obligated to use all reasonable efforts to obtain Marketing Authorization by means of the Mutual Recognition Procedure in the territories and option countries.
Menarini Pharmaceutical Industries S.R.L.
Menarini, headquartered in Italy, is the largest manufacturer and distributor of pharmaceuticals in Southern Europe. Menarini also has extensive experience developing and marketing therapies for gastrointestinal disease in its markets. Under our agreements with Menarini, we granted Menarini certain manufacturing rights and exclusive distribution rights with respect to balsalazide in Italy, Spain, Portugal and Greece. The agreement calls for additional milestone revenues to be paid to us relating to additional European marketing approvals, if any, in the Menarini territories. Under the terms of the agreements, we will sell the bulk active ingredient balsalazide to
13
Table of Contents
Menarini for marketing and distribution in its territories at cost plus a sales-based royalty. Menarini did not purchase any bulk active ingredient balsalazide from us during 2010. During 2009 and 2008, we sold Menarini approximately $0.1 million and $0.4 million of bulk active ingredient balsalazide, respectively.
Unless terminated sooner in accordance with its terms, the agreement with Menarini continues until the earlier of the expiration of (1) the patents relating to the product or (2) 15 years from the date of the agreement, provided however that in any case the agreement shall continue for a period of 10 years from the date of first launch. Either party may terminate the agreement upon a material breach by the other party and the failure to remedy such breach within 30 days in the case of a payment breach or 90 days in the case of any other material breach or if a party enters liquidation, bankruptcy or similar proceedings.
Mayoly-Spindler S.A.S.
In October 2007, we licensed exclusive rights to market OsmoPrep™ (sodium phosphate monobasic monohydrate, USP and sodium phosphate dibasic anhydrous, USP) Tablets in France to Mayoly–Spindler S.A.S of Chatou, France. Under the terms of the agreement, we may receive up to $1.0 million in milestone payments, as well as royalty payments based on product sales.
Pharmatel PTY Limited.
In June 2004, we granted Pharmatel certain manufacturing rights and exclusive distribution rights with respect to balsalazide in Australia and New Zealand. Under the terms of the agreements, Pharmatel pays us royalties based on product sales.
Zeria Pharmaceutical Co. Ltd.
In August 2001, InKine licensed exclusive commercial rights in Japan to Visiclear® Tablets for colon cleansing to Zeria Pharmaceutical Co., Ltd. of Tokyo, Japan. Zeria launched Visiclear in June 2007. We market Visiclear, or sodium phosphate monobasic monohydrate and sodium phosphate dibasic anhydrous, tablets in the United States under the trade name Visicol®. Under the terms of the agreements, Zeria pays us royalty payments based on product sales.
Supply and Distribution Agreement with Watson Pharma, Inc.
On December 28, 2007, we announced that we entered into a Supply and Distribution Agreement with Watson Pharma, Inc., pursuant to which Watson is Salix’s exclusive distributor to market and sell an authorized generic of Colazal (balsalazide disodium) Capsules 750 mg, Salix’s anti-inflammatory drug approved for the treatment of mildly to moderately active ulcerative colitis, in the United States. Watson agrees to use commercially reasonable efforts to sell authorized generic Colazal, and has sole discretion to establish prices and terms. Watson will pay Salix a portion of its profits for sales under the agreement, and Salix will supply Watson with all its requirements for the product. The agreement terminates in October 2011, provided that either party may terminate immediately upon bankruptcy of the other, or for uncured breaches of the other party. Salix may also terminate on 30 days notice if the agreement has become commercially unviable, if it obtains the right to prohibit other generics from being sold or if it ceases distribution of branded Colazal. In May 2010 we terminated this agreement.
MANUFACTURING
We own no manufacturing facilities. We have in the past used and plan to continue to use third-party vendors to produce material for use in clinical trials and for commercial product. This manufacturing strategy enables us to direct our financial resources to product in-licensing and acquisition, product development, and sales and marketing efforts, without devoting resources to the time and cost associated with building and maintaining manufacturing or packaging facilities.
14
Table of Contents
Under our supply agreement with Alfa Wassermann, Alfa Wassermann is obligated to supply us with bulk rifaximin drug substance, the active pharmaceutical ingredient in Xifaxan 200mg rifaximin tablets and Xifaxan 550mg rifaximin tablets, until July 2014 or introduction of a generic product, whichever occurs first. Our supply of rifaximin drug substance supplied by Alfa Wassermann is manufactured by ZaCh Systems in Lonigo, Italy, and Sanofi-Aventis in Brindisi, Italy. Under our supply agreement with Lupin, we are obligated to purchase 50% of our annual requirements of bulk rifaximin drug substance from Lupin. Under a long-term supply agreement, rifaximin is converted into Xifaxan drug product for us by Patheon, Inc. in Whitby, Ontario. Bulk Xifaxan tablets are packaged into finished Xifaxan commercial bottles by Patheon and packaged into Xifaxan commercial blister packs by Pharma Packaging Solutions in Norris, Tennessee.
Under our long-term supply agreement with aaiPharma in Wilmington, North Carolina, aaiPharma manufactures and packages our commercial supply of 75mg and 100mg Azasan finished product.
Under our long-term supply agreement with Paddock Laboratories, Inc. in Minneapolis, Minnesota, Paddock produces our commercial supply of finished product of Anusol-HC Cream, Anusol-HC Suppositories, Proctocort Suppositories, Pepcid Oral Suspension and Diuril Oral Suspension. In addition, through prior supply arrangements between King Pharmaceuticals and Crown Laboratories, Inc. in Johnson City, Tennessee, Crown continues to produce our commercial supply of Proctocort Cream finished product.
Under our supply agreement with WellSpring Pharmaceutical Corporation in Oakville, Ontario, WellSpring will produce our commercial supply of OsmoPrep finished product through October 2011. In September 2010 we entered into a supply agreement with Novel Laboratories, Inc. and Actavis Inc. Under this supply agreement we agreed to purchase from Actavis, all of our requirements of OsmoPrep beginning in October 2011.
Under our supply agreement with Norgine in Hengoed, Wales, Norgine will produce our commercial supply of MoviPrep finished kits through 2011. In August 2010 we entered into a supply agreement with Novel Laboratories, Inc. and Actavis Inc. Under this supply agreement we agreed to purchase from Actavis, all of our requirements in excess of a certain amount of MoviPrep in 2011 and all of our requirements of MoviPrep beginning in 2012.
Bayer AG in Wuppertal, Germany supplies us with bulk mesalamine active ingredient. Under a long-term supply agreement with Catalent Pharma Solutions in Winchester, Kentucky, Catalent converts this mesalamine into our commercial supply of bulk Apriso, 375mg mesalamine capsules. Bulk Apriso capsules are packaged into finished Apriso commercial bottles by Pharma Packaging Solutions in Norris, Tennessee.
Cosma S.P.A. in Bergamo, Italy supplies us with bulk metoclopramide active ingredient. Under a long-term supply agreement with Catalent in Swindon, United Kingdom, Catalent converts this metoclopramide into our commercial supply of Metozolv, 5mg and 10mg tablets in blister packaging. The Metozolv blister packs are then packaged into finished cartons by Pharma Packaging Solutions in Norris, Tennessee.
Under long-term supply agreements, we use balsalazide drug substance, the active pharmaceutical ingredient in Colazal capsules, manufactured by OmniChem s.a., a subsidiary of Ajinomoto in Belgium, and PharmaZell in Raubling, Germany. Also, under a long-term supply agreement, balsalazide is encapsulated into Colazal drug product for us by Nexgen Pharma, Inc. in Irvine, California. Bulk Colazal capsules are packaged into finished Colazal commercial bottles by Nexgen and Pharma Packaging Solutions in Norris, Tennessee.
With respect to our 1100mg balsalazide tablet formulation which is currently under development, we plan to negotiate commercial supply agreements with the same manufacturers who supplied the drug substance and drug product for the supplies of the Phase III clinical trial material, if approved.
15
Table of Contents
Under our supply agreement with Glenmark Pharmaceuticals, Ltd. in Mumbai, India, Glenmark supplies us with crofelemer drug substance. With respect to our crofelemer tablet formulation, which is currently under development, and our 2mg budesonide foam formulation, which is currently under development, we plan to negotiate commercial supply agreements with the same manufacturers who supplied the drug substance and drug product for the supplies of the Phase III clinical trial material, if approved.
SALES AND MARKETING
We currently market our products and intend, if approved by the FDA, to market future products to U.S. gastroenterologists and other physicians through our own direct sales force. We enter into distribution relationships outside the United States and in markets where a larger sales organization is appropriate. As of December 31, 2010, our sales and marketing staff consisted of approximately 250 people, and we plan to hire additional people to sell our products for additional indications or sell additional products, if and when acquired and/or approved for U.S. marketing. Because there are a relatively small number of gastroenterologists that write a majority of the prescriptions in our indications, we believe that the size of our sales force is appropriate to reach our target physicians. As of December 31, 2010, our sales force consisted of approximately 160 employees who regularly call on approximately 21,000 healthcare professionals. We also had approximately ten national account managers who regularly call on major drug wholesalers, managed care organizations, large retail chains, formularies and related organizations. We believe we have created an attractive incentive program for our sales force that is based upon goals in prescription growth, market share achievement and customer service.
We cultivate relationships of trust and confidence with the high prescribing gastroenterologists in the United States. We use a variety of marketing techniques to promote our products including sampling, journal advertising, promotional materials, specialty publications, coupons, money-back or product replacement guarantees, educational conferences and informational websites.
16
Table of Contents
PATENTS AND PROPRIETARY RIGHTS
General
The patents for the rifaximin composition of matter (also covering a process of making rifaximin and using rifaximin to treat gastrointestinal infectious diseases) expired in May 2001 in the United States and Canada. Rifaximin was a new chemical entity and was granted a five-year new chemical exclusivity by the FDA when it was approved in May 2004. Rifaximin, therefore, had data exclusivity to May 2009. In May 2006, U.S. Patent No. 7,045,620 (the’620 patent, which is a composition of matter and process patent that covers several physical states of rifaximin) was issued. We believe this patent extends the protection of the current form of rifaximin until June 2024. In November 2009, U.S. Patent No. 7,612,199 (the ‘199 patent) issued and should provide protection until June 2024. This patent provides further protection relating to U.S. Patent No. 7,045,620 that covers several physical states, or polymorphous forms, of rifaximin and provides protection for all indications currently marketed and being assessed. Alfa Wasserman S.p.a., the owner of the ‘620 and the ‘199 patents, has licensed the rights to Salix in the United States. In July 2006, Salix entered into an agreement with Cedars-Sinai Medical Center, or CSMC, for the right to use its patent and patent applications relating to methods of diagnosis and treating irritable bowel syndrome and other disorders caused by small intestinal bacterial overgrowth. The CSMC agreement provides Salix the right to use U.S. Patent No. 7,452,857, which issued in November 2008, providing protection relating to rifaximin for treating irritable bowel syndrome, or IBS, caused by small intestinal bacterial overgrowth; US 7,718,608, which issued May of 2010 and provides protection relating to rifaximin for treating IBS; and U.S. Patent No. 7,605,240, which issued in October 2009, providing protection relating to rifaximin for treating bloating caused by small intestinal bacterial overgrowth related to IBS, each of which provide protection until August 2019. Below is a tabular summary of issued U.S. patents related to rifaximin that we own or have licensed.
| U.S. Patent No. |
Issue Date |
Expiration |
Subject | |||||
| 7,045,620* | May 2006 |
June 2024 |
Composition of matter and process patent covering several physical states of rifaximin | |||||
| 7,452,857** | November 2008 |
August 2019 |
Use of rifaximin for treating irritable bowel syndrome | |||||
| 7,605,240** | October 2009 |
August 2019 |
Treatment of bloating caused by small intestinal bacterial overgrowth associated with irritable bowel syndrome | |||||
| 7,718,608** | May 2010 |
August 2019 |
Use of rifaximin for treating irritable bowel syndrome | |||||
| 7,612,199* | November 2009 |
June 2024 |
Covers several physical states, or polymorphous forms of rifaximin | |||||
| * | Licensed from Alfa Wasserman S.p.a. |
| ** | Licensed from Cedars-Sinai Medical Center |
In addition, we have filed applications for patents relating to additional indications using rifaximin and related chemical substances. In September 2009, Lupin Ltd. granted Salix the exclusive right in the United States to its bioadhesive drug delivery technology for use with rifaximin. In October 2009, Cipla, Limited granted Salix the exclusive rights in the United States to its amorphous rifaximin application PCT Patent Application No. PCT/GB2007/003629; WO 2008/035109.
The patent for the treatment of the intestinal tract with Apriso, the granulated mesalamine product, provides patent coverage to 2018. In June 2009, U.S. Patent No. 7,547,451 issued, which relates to methods of producing Apriso and provides further protection.
The patent for balsalazide 1100 mg tablets provides patent coverage to 2018.
17
Table of Contents
The patent for balsalazide 1100 mg tablets relating to the increase of bioavailability provides coverage until November 2027 and in December 2009 a related patent issued, U.S. Patent No. 7,625,884, which also provides protection until November 2027.
The patent for Visicol provides patent coverage to 2013.
The patent for OsmoPrep provides patent coverage to 2013. We are seeking additional patent protection on OsmoPrep that, if approved, will provide patent coverage to 2024. In September 2010 we entered into a sublicense agreement which granted Novel Laboratories, Inc. a license under the patents covering OsmoPrep permitting Novel to launch a generic OsmoPrep on November 16, 2019.
The patent for MoviPrep provides patent coverage to 2024. This patent was issued by the USPTO in January 2007 and it contains composition of matter and kit claims. A second patent which covers the MoviPrep product, U.S. Patent No. 7,658,914, issued on February 9, 2010 and has been listed in the Orange Book. In August 2010 we entered into a sublicense agreement that granted Novel Laboratories, Inc. a license to the patents covering MoviPrep permitting Novel to launch a generic MoviPrep on September 24, 2018.
The patent for Metozolv is a formulation patent for its fast-dissolve formulation of metoclopramide that has patent protection until 2017 and additional patent protection pending that, if issued, will provide intellectual property protection until 2023. On November 3, 2010, we received a paragraph IV notification from Novel stating that Novel had filed an ANDA application to seek approval to market a generic version of Metoclopramide Hydrochloride ODT, 5 mg and 10 mg. The notification letter asserted non-infringement of U.S. Patent No. 6,413,549 (“the ‘549 patent”). Upon examination of the relevant sections of the ANDA, we concluded that the 549 patent would not be enforced against Novel Laboratories.
The patents for crofelemer provide protection to 2018. We are seeking applications for patents relating to additional indications using crofelemer and related chemical substances.
The budesonide rectal foam product has patent coverage in the U.S. until 2015.
The patents for the balsalazide composition of matter and method of treating ulcerative colitis with balsalazide expired in July 2001 in the United States; however, we were granted five years of new chemical entity data exclusivity for balsalazide until July 2005 and an extension of such patent under the Waxman-Hatch Act through July 2006. We also obtained patent extensions for the composition of balsalazide in Italy and the United Kingdom until July 2006. We have filed applications for patents relating to additional indications using balsalazide and related chemical substances. In November of 2008, the United States Patent and Trademark Office issued a patent covering methods for increasing the bioavailability of balsalazide; and in December 2009 a related patent issued, U.S. Patent No. 7,625,884, which also provides protection until November 2027.
The patents for Lumacan including, US 6,034,267 and US 7,247,655 provide protection until March 2016.
Data Exclusivity
Rifaximin was a new chemical entity, therefore, the FDA granted us five-year new chemical entity exclusivity when it was approved for the treatment of travelers’ diarrhea in May 2004. Therefore, rifaximin had data exclusivity for the travelers’ diarrhea indication through May 2009. Rifaximin 550mg, which is approved for the reduction in risk of overt hepatic encephalopathy (HE) recurrence in patients > 18 years of age was granted orphan exclusivity through March 2017.
Apriso, the granulated mesalamine product, is not a new chemical entity, but is entitled to three years of exclusivity from its approval based on the new clinical investigations that have been required during the approval process. The exclusivity prevents the FDA from approving an ANDA for a granulated mesalamine product which relied upon the new clinical investigation in our NDA for three years from October 31, 2008. The patent for the granulated mesalamine protects the product until April 2018.
18
Table of Contents
Metoclopramide, which is not a new chemical entity, was not entitled to three years of data exclusivity from the date of its approval. Our metoclopramide product is a fast-dissolve formulation that has patent protection until 2017 and additional patent protection pending that, if issued, will provide intellectual property protection until 2023. On November 3, 2010, we received a paragraph IV notification from Novel stating that Novel had filed an ANDA application to seek approval to market a generic version of Metoclopramide Hydrochloride ODT, 5 mg and 10 mg. The notification letter asserted non-infringement of U.S. Patent No. 6,413,549 (the ‘549 patent). Upon examination of the relevant sections of the ANDA, we concluded that the ‘549 patent would not be enforced against Novel Laboratories.
Azasan and the Anusol-HC and Proctocort product lines are mature products, thus, there are no available patent or exclusivity rights.
The Pepcid product line is a mature product line, thus, there are no available patent or exclusivity rights.
Crofelemer, which is a new chemical entity, should be eligible for market exclusivity for five years in the United States. As a new molecular entity, we believe crofelemer may be entitled to patent term restoration. The patents for crofelemer provide protection until 2018.
Lumacan, which is not a new chemical entity, will only be eligible for three years of data exclusivity. The patents that may cover a Lumacan product provide protection until March 2016.
GOVERNMENT REGULATION
The research, testing, manufacture, marketing and distribution of drug products are extensively regulated by governmental authorities in the United States and other countries. In the United States, drugs are subject to rigorous regulation by the FDA. The Federal Food, Drug and Cosmetic Act, as amended, and the regulations promulgated thereunder, and other federal and state statutes and regulations, govern, among other things, the research, development, testing, manufacture, storage, record keeping, labeling, promotion and marketing and distribution of pharmaceutical products. Failure to comply with applicable regulatory requirements may subject a company to administrative sanctions or judicially imposed sanctions such as civil penalties, criminal prosecution, injunctions, product seizure or detention, product recalls, and total or partial suspension of product marketing and/or approvals. In addition, non-compliance may result in the FDA’s refusal to approve pending NDAs or supplements to approved NDAs or in the withdrawal of an NDA. Any such sanction could result in adverse publicity, which could have a material adverse effect on our business, financial conditions, and results of operation.
The steps ordinarily required before a new pharmaceutical product containing a new chemical entity may be marketed in the United States include: (1) preclinical laboratory tests, preclinical studies in animals and formulation studies; (2) the submission to the FDA of a notice of claimed investigational exemption for a new drug, which must become effective before clinical testing may commence; (3) adequate and well-controlled clinical human trials to establish the safety and efficacy of the drug for each indication; (4) the submission of an NDA to the FDA; and (5) FDA review and approval of the NDA prior to any commercial sale or shipment of the drug. Preclinical tests include laboratory evaluation of product chemistry and formulation, as well as animal studies to assess the potential safety and efficacy of the product. Preclinical tests must be conducted in compliance with Good Laboratory Practice regulations. The results of preclinical testing are submitted to the FDA as part of an IND. A 30-day waiting period after the filing of each IND is required prior to the commencement of clinical testing in humans. In addition, the FDA may, at any time during this 30-day period or at any time thereafter, impose a clinical hold on proposed or ongoing clinical trials. If the FDA imposes a clinical hold, clinical trials cannot commence or recommence without FDA authorization and then only under terms authorized by the FDA. In some instances, the IND application process can result in substantial delay and expense.
19
Table of Contents
Clinical trials to support NDAs are typically conducted in three sequential phases, but the phases may overlap. In Phase I, the initial introduction of the drug into healthy human subjects or patients, the drug is tested to assess metabolism, pharmacokinetics and pharmacological actions and safety, including side effects associated with increasing doses. Phase II usually involves studies in a limited patient population to (1) assess the efficacy of the drug in specific, targeted indications, (2) assess dosage tolerance and optimal dosage and (3) identify possible adverse effects and safety risks. If a compound is found to be potentially effective and to have an acceptable safety profile in Phase II evaluations, Phase III trials are undertaken to further demonstrate clinical efficacy and to further test for safety within an expanded patient population at geographically dispersed clinical study sites. There can be no assurance that Phase I, Phase II or Phase III testing will be completed successfully within any specified time period, if at all, with respect to any of our products subject to such testing.
After successful completion of the required clinical testing, generally an NDA is submitted. FDA approval of the NDA is required before marketing may begin in the United States. The FDA reviews all NDAs submitted before it accepts them for filing and may request additional information rather than accepting an NDA for filing. In such an event, the NDA must be resubmitted with the additional information and, again, is subject to review before filing. Once the submission is accepted for filing, the FDA begins an in-depth review of the NDA. The FDA generally has 10 months in which to review the NDA and respond to the applicant. The review process is often significantly extended by FDA requests for additional information or clarification regarding information already provided in the submission. In the last few years, FDA review times have lengthened. The FDA may refer the application to an appropriate advisory committee, typically a panel of clinicians, for review, evaluation and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee. If the FDA’s evaluation of the NDA submission or manufacturing facilities is not favorable, the FDA may refuse to approve the NDA or issue a not approvable letter, outlining the deficiencies in the submission and often requiring additional testing or information. If FDA evaluations of the NDA and the manufacturing facilities are favorable, the FDA may issue either an approval letter or an complete response letter, which usually contains a number of conditions that must be met in order to secure final approval of the NDA. When and if those conditions have been met to the FDA’s satisfaction, the FDA will issue an approval letter, authorizing commercial marketing of the drug for certain indications. Furthermore, approval may entail ongoing requirements for post-marketing studies, and marketed products, manufacturers and manufacturing facilities are subject to continual review and periodic inspections. In addition, identification of certain side effects after a drug is on the market or the occurrence of manufacturing problems could cause subsequent withdrawal of approval, reformulation of the drug, additional preclinical testing or clinical trials and changes in labeling of the product.
Under the Orphan Drug Act, the FDA may designate a product as an orphan drug if it is a drug intended to treat a disease or condition that affects populations of fewer than 200,000 individuals in the United States or a disease whose incidence rates number more than 200,000 where the sponsor establishes that it does not realistically anticipate that its product sales will be sufficient to recover its costs. The sponsor that obtains the first marketing approval for a designated orphan drug for a given rare disease is eligible to receive marketing exclusivity for use of that drug for the orphan indication for a period of seven years. Rifaximin for the treatment of hepatic encephalopathy and Colazal for the treatment of mildly to moderately active ulcerative colitis in pediatric patients between 5 to 17 years of age have been granted orphan drug status.
Regulation of Drug Compounds Outside of the United States
Outside the United States, the ability to market a drug is contingent upon receiving marketing authorizations from the appropriate regulatory authorities. The requirements governing the conduct of clinical trials and marketing authorization vary widely from country to country. Currently, foreign marketing authorizations are applied for at a national level, although within the European Union procedures are available to companies wishing to market a product in more than one European Union member state. The foreign regulatory approval process includes all of the risks associated with FDA approval set forth above.
20
Table of Contents
If and when necessary, we will choose the appropriate route of European regulatory filing to accomplish the most rapid regulatory approvals. However, the chosen regulatory strategy might not secure regulatory approvals or approvals of our chosen product indications. Furthermore, we must obtain pricing approval in addition to regulatory approval prior to launching the product in the approving country. Failure to obtain pricing approval in a timely manner or approval of pricing which would support an adequate return on investment or generate a sufficient margin to justify the economic risk might delay or prohibit the commercial launch of the product in those countries.
COMPETITION
Competition in our business is intense and characterized by extensive research efforts, rapid technological progress and an increasing rate of generic product approvals. Technological developments by competitors, earlier regulatory approval for marketing competitive products, including generic versions of our products, such as those launched against Colazal in December 2007, and Pepcid in 2010, or superior marketing capabilities possessed by competitors could adversely affect the commercial potential of our products and could have a material adverse effect on our revenue and results of operations. We believe that there are numerous pharmaceutical and biotechnology companies, including large well known pharmaceutical companies and generic manufacturers, as well as academic research groups throughout the world, engaged in research and development efforts with respect to pharmaceutical products targeted at gastrointestinal diseases and conditions addressed by our current and potential products. In particular, we are aware of products in research or development by competitors that address the diseases being targeted by our products. Developments by others might render our current and potential products obsolete or non-competitive. Competitors might be able to complete the development and regulatory approval process sooner and, therefore, market their products earlier than us. Many of our competitors have substantially greater financial, marketing and personnel resources and development capabilities than we do.
For example, lactulose, a drug that is used to treat hepatic encephalopathy and competes with Xifaxan 550mg, is offered by various pharmaceutical manufacturers, including several generic manufacturers. Many large, well capitalized companies already offer products in the United States and Europe that target the indications for balsalazide and our mesalamine extended-release capsule product, including mesalamine (GlaxoSmithKline plc, Giuliani S.p.A., Axcan Pharma, Inc., Abbott Laboratories, Warner Chilcott plc and Shire Pharmaceuticals Group plc), sulfasalazine (Pharmacia & Upjohn, Inc.), and olsalazine (Alaven Pharmaceutical LLC). Asacol, marketed by Warner Chilcott, is currently the most prescribed product for the treatment of ulcerative colitis in the United States, and Shire introduced once-a-day Lialda in 2007. In addition, on December 28, 2007, the Office of Generic Drugs approved three generic balsalazide capsule products.
Several prescription liquid PEG products compete with Visicol, OsmoPrep and MoviPrep in the bowel cleansing market. These prescription products include Colyte, Golytely, Halflytely, SuPrep and Nulytely (Braintree) and Trilyte (Alaven Pharmaceutical LLC). Generic prescription, liquid PEG products are also available.
The most frequently prescribed product for treatment of travelers’ diarrhea in the United States currently is ciprofloxacin, commonly known as “Cipro®” and marketed by Bayer AG. The most frequently prescribed products that compete with Azasan are Imuran®, marketed by Prometheus Laboratories, Inc., and its various generics and Purinethol®, marketed by GATE Pharmaceuticals, and it’s various generics. The most frequently prescribed products that compete with Anusol-HC and Proctocort are AnaMantle HC, marketed by Nycomed; Analpram HC, marketed by Ferndale Laboratories; Proctofoam-HC and Proctocream-HC, marketed by Alaven Pharmaceutical LLC; Procto-Kit, marketed by Ranbaxy Pharmaceuticals; and various generics. The most frequently prescribed products that compete with Metozolv are Reglan®, marketed by Alaven Pharmaceuticals LLC, and various generics.
EMPLOYEES
As of December 31, 2010, we had approximately 390 full-time employees. We believe that our future success will depend in part on our continued ability to attract, hire, and retain qualified personnel, including sales
21
Table of Contents
and marketing personnel in particular. Competition for such personnel is intense, and there can be no assurance that we will be able to identify, attract, and retain such personnel in the future. None of our employees are represented by a labor union. We have not experienced any work stoppages and consider our relations with our employees to be good.
This report contains forward-looking statements that involve risks and uncertainties. Our actual results could differ materially from those discussed in this report. Factors that could cause or contribute to these differences include, but are not limited to, those discussed below and elsewhere in this report and in any documents incorporated in this report by reference.
If any of the following risks, or other risks not presently known to us or that we currently believe to not be significant, develop into actual events, then our business, financial condition, results of operations or prospects could be materially adversely affected. If that happens, the market price of our common stock could decline, and stockholders may lose all or part of their investment.
Future sales of Xifaxan and our other marketed products might be less than expected.
We currently market and sell twelve primary products, with a majority of our historical revenue derived from sales of Colazal prior to 2008. We expect Xifaxan, which was launched in mid-2004 for the treatment of traveler’s diarrhea, and approved and launched in March 2010 for the treatment of hepatic encephalopathy to be our most significant source of revenue in the future. If sales of our marketed products decline or if we experience product returns significantly in excess of estimated amounts recorded, particularly with respect to Xifaxan, it would have a material adverse effect on our business, financial condition and results of operations.
The degree of market acceptance of our products among physicians, patients, healthcare payors and the medical community will depend upon a number of factors including:
| • | the timing of regulatory approvals and product launches by us or competitors, and including any generic or over-the-counter competitors; |
| • | perceptions by physicians and other members of the healthcare community regarding the safety and efficacy of the products; |
| • | price increases, and the price of our products relative to other drugs or competing treatments; |
| • | patient and physician demand; |
| • | adverse side effects or unfavorable publicity concerning our products or other drugs in our class; |
| • | the results of product development efforts for new indications; |
| • | the scope and timing of additional marketing approvals and favorable reimbursement programs for expanded uses; |
| • | availability of sufficient commercial quantities of the products; and |
| • | our success in getting other companies to distribute our products outside of the U.S. gastroenterology market. |
Regulatory approval of our product candidates is time-consuming, expensive and uncertain, and could result in unexpectedly high expenses and delay our ability to sell our products.
Development of our products is subject to extensive regulation by governmental authorities in the United States and other countries. This regulation could require us to incur significant unexpected expenses or delay or
22
Table of Contents
limit our ability to sell our product candidates, including specifically rifaximin for irritable bowel syndrome, or IBS. In August 2010, the FDA accepted our NDA for rifaximin for IBS, and gave us an action date of December 7, 2010. In October 2010 the FDA informed us they were extending the action date by three months to provide for a full review and extended our action date to March 7, 2011. There is no assurance that the FDA will approve rifaximin for IBS in a timely manner, or at all. In early 2008, the FDA announced that because of its large workload it might not meet its target dates to respond to NDA submissions, and since then we have experienced delays in FDA review of Metozolv, balsalazide tablets and rifaximin for HE.
Our clinical studies might be delayed or halted, or additional studies might be required, for various reasons, including:
| • | the drug is not effective; |
| • | patients experience severe side effects during treatment; |
| • | appropriate patients do not enroll in the studies at the rate expected, as was the case with our Xifaxan Phase III trial in Thailand for the prevention of travelers’ diarrhea; |
| • | drug supplies are not sufficient to treat the patients in the studies; or |
| • | we decide to modify the drug during testing. |
If regulatory approval of any product is granted, it will be limited to those indications for which the product has been shown to be safe and effective, as demonstrated to the FDA’s satisfaction through clinical studies. For example, rifaximin has been approved for treatment of travelers’ diarrhea and HE, but we are developing rifaximin for IBS and other indications. The FDA might not ever approve any of our compounds in the indications we are pursuing, which would mean we cannot market these compounds for use in these indications.
Regulatory approval, even if granted, might entail ongoing requirements or restrictions on marketing that could increase our expenses and limit revenue.
Approval might entail ongoing requirements for post-marketing studies, or limit how or to whom we can sell our products. Even if we obtain regulatory approval, labeling and promotional activities are subject to continual scrutiny by the FDA and state regulatory agencies and, in some circumstances, the Federal Trade Commission. For example, in 2008 the FDA required us to put a “black box” warning on the OsmoPrep and Visicol labels regarding potential kidney damage that could result from their use, and a “black box” warning for Metozolv regarding tardive dyskensia which could result from its use. These warnings could limit future sales of those products. In addition, FDA enforcement policy prohibits the marketing of approved products for unapproved, or off-label, uses. These regulations and the FDA’s interpretation of them might increase our expenses, impair our ability to effectively market our products, and limit our revenue.
Our intellectual property rights might not afford us with meaningful protection.
The intellectual property rights protecting our products might not afford us with meaningful protection from generic and other competition. In addition, because our strategy is to in-license or acquire pharmaceutical products which typically have been discovered and initially researched by others, future products might have limited or no remaining patent protection due to the time elapsed since their discovery. Competitors could also design around any of our intellectual property or otherwise design competitive products that do not infringe our intellectual property.
Any litigation that we become involved in to enforce intellectual property rights could result in substantial cost to us. In addition, claims by others that we infringe their intellectual property could be costly. Our patent or other proprietary rights related to our products might conflict with the current or future intellectual property rights of others. Litigation or patent interference proceedings, either of which could result in substantial cost to
23
Table of Contents
us, might be necessary to defend any patents to which we have rights and our other proprietary rights or to determine the scope and validity of other parties’ proprietary rights. The defense of patent and intellectual property claims is both costly and time-consuming, even if the outcome is favorable. Any adverse outcome could subject us to significant liabilities to third parties, require disputed rights to be licensed from third parties, or require us to cease selling one or more of our products. We might not be able to obtain a license to any third-party technology that we require to conduct our business, or, if obtainable, that technology might not be available at a reasonable cost.
Upon patent expiration, our drugs could be subject to generic competition, which could negatively affect our pricing and sales volume. As previously disclosed, this has already happened to Colazal, which had been our largest selling drug prior to 2008.
Patent applications relating to rifaximin compositions and related chemical substances were filed together with Alfa Wasserman and issued in May 2006. This patent extends patent coverage to 2024 and in 2009 a related patent, U.S. Patent No. 7,612,199 issued to provide further coverage of rifaximin. In November of 2008 and in October 2009, the United States Patent and Trademark Office issued a patent covering the use of rifaximin for treating irritable bowel syndrome (IBS) and bloating caused by small intestinal bacterial overgrowth, respectively. The patent for the treatment of the intestinal tract with the Apriso will provide patent coverage to 2018. In June 2009, U.S. Patent No. 7,547,451 issued, which relates to methods of producing Apriso and provides further protection. The patent application relating to the dosage form for metoclopramide protects the product until July 2017. The patent for crofelemer, relating to enteric formulations and uses thereof provide protection until 2018 and should be entitled to patent term restoration as a new molecular entity. There is no assurance that these patents or the patent term restorations will be issued or granted, respectively. Patent expiration dates listed herein, unless otherwise noted, are for U.S. patents and assume there are no patent term adjustments, extensions or other adverse events that could affect the term or scope of a patent. Dates provided herein for the expiration of patent applications are merely estimates based on knowledge at this time and could be altered, for example, by terminal disclaimer or if patent term extensions or adjustments are available. The patents for Lumacan including, US 6,034,267 and US 7,247,655 provide protection until March 2016.
In January 2007, the United States Patent and Trademark Office issued a patent covering composition of matter and kit claims for MoviPrep. The MoviPrep patent provides coverage to September 2024. Norgine, B.V. and Norgine Europe, B.V., which we refer to collectively as Norgine, own U.S. Patent No. 7,169,381 (the ‘381 patent). The ‘381 patent is listed with the FDA as protecting our MoviPrep product. Norgine licensed MoviPrep and the ‘381 patent to the Company for commercialization in the United States. Novel filed an Abbreviated New Drug Application, or ANDA, with the FDA seeking approval to market a generic version of MoviPrep in the United States prior to the September 2024 expiration of the ‘381 patent. On May 14, 2008, we and Norgine filed a lawsuit in the United States District Court for the District of New Jersey against Novel for infringement of the ‘381 patent. Upon entry by the court on August 30, 2010 of a Consent Judgment included as an exhibit to the Settlement Agreement among the parties and Actavis, Inc., the settlement will resolve all of the parties’ outstanding claims and defenses in the lawsuit. The Consent Judgment provides that Novel’s proposed generic product, absent a license as granted to Novel under the settlement, would infringe the ‘381 patent and further provides that Novel acknowledges and agrees not to contest the validity and enforceability of the ‘381 patent. In addition to the Settlement Agreement, we have entered into a Sublicense Agreement with Norgine and Novel, as well as a Supply Agreement with Novel and Actavis and a First Amendment to License and Supply Agreement with Norgine. Under the terms of the Sublicense Agreement, we and Norgine have granted Novel a fully paid up license under the MoviPrep patents such that it is permitted to launch a generic MoviPrep product on September 24, 2018.
The patent for Visicol and OsmoPrep will expire in 2013. US 7,687,075 issued 30 March 2010 and provide protection for OsmoPrep until June 2028. CDC III, LLC, owns U.S. Patent No. 5,616,346, or the ‘346 patent. The ‘346 patent is listed with the United States Food and Drug Administration as protecting our OsmoPrep product. CDC licensed OsmoPrep and the ‘346 patent to us for commercialization in the United States. In addition, we
24
Table of Contents
own U.S. Patent No. 7,687,075, or the ‘075 patent, protecting OsmoPrep. Novel Laboratories, Inc., filed an ANDA with the FDA seeking approval to market a generic version of OsmoPrep in the United States prior to the May 2013 expiration of the ‘346 patent and the 2024 expiration of the ‘075 patent. On September 8, 2008, we filed a lawsuit in the United States District Court for the District of New Jersey against Novel for the infringement of the ‘346 patent and seeking a declaratory judgment confirming the validity of the patent. The lawsuit also joined CDC as a party. With the entry by the court of a Consent Judgment, the settlement will resolve all of the parties’ outstanding claims and defenses in the lawsuit. The Consent Judgment provides that Novel’s proposed generic product, absent a license as granted to Novel under the Sublicense Agreement described below, would infringe the ‘346 patent and further provides that Novel acknowledges and agrees not to contest the validity and enforceability of the ‘346 patent. In connection with the settlement, we entered into: a Settlement Agreement with CDC, Novel, Actavis Inc. and the general partnership of Craig Aronchick, William H. Lipshutz and Scott H. Wright (the “General Partnership”) that was the initial licensor of the ‘346 patent to Salix; a Sublicense Agreement with CDC, the General Partnership and Novel; a Supply Agreement with Novel; and, a Second Amendment to Supply Agreement with CDC and the General Partnership. Under the terms of the Sublicense Agreement, we, CDC and the General Partnership have granted Novel a fully paid up, non-exclusive license under the OsmoPrep patents such that it is permitted to launch a generic OsmoPrep product on November 16, 2019.
Rifaximin is a new chemical entity and was granted five-year new chemical entity exclusivity by the FDA when it was approved in May 2004. Rifaximin, therefore, had data exclusivity until May 2009. Accordingly, the Office of Generic Drugs would have been able to accept an ANDA for Xifaxan tablets on or any time subsequent to May 2008, if the applicant made certifications of patent non-infringement or invalidity. If this occurred, a Paragraph IV notification would have to be provided to us by the applicant. Although we do not possess any specific knowledge of any such filing at the current time, the expiration of data exclusivity could result in a challenge to the related intellectual property rights of Xifaxan 200mg tablets at any time in the future. In May 2008 we submitted a Citizen Petition, requesting the director of the Office of Generic Drugs of the Food and Drug Administration impose scientifically appropriate standards for the demonstration of bioequivalence for abbreviated new drug applications citing Xifaxan as the reference listed drug. Rifaximin 550mg, which is approved for the reduction in risk of overt hepatic encephalopathy (HE) recurrence in patients > 18 years of age was granted orphan exclusivity through March 2017. Accordingly, the Office of Generic Drugs, or OGD, would have been able to accept an ANDA for Xifaxan 550mg tablets on or any time subsequent to March 2010, if the applicant made certifications of patent non-infringement or invalidity. If this occurred, a Paragraph IV notification would have to be provided to us by the applicant. Although we do not possess any specific knowledge of any such filing at the current time, the orphan exclusivity period does not prohibit the filing of an ANDA and thus, an ANDA filing could result in a challenge to the related intellectual property rights of Xifaxan 550mg tablets at any time in the future. The OGD would be unable to finally approve an ANDA until the expiration of the orphan exclusivity in March 2017.
On November 3, 2010, we received a paragraph IV notification from Novel Laboratories, Inc. stating that Novel had filed an ANDA application to seek approval to market a generic version of Metoclopramide Hydrochloride ODT, 5 mg and 10 mg. The notification letter asserted non-infringement of U.S. Patent No. 6,413,549 (“the ‘549 patent”). Upon examination of the relevant sections of the ANDA, we concluded that the ‘549 patent would not be enforced against Novel Laboratories.
Because Azasan, Anusol-HC, Pepcid and Proctocort are mature products, there are no patents or data exclusivity rights available which subjects us to greater risk of generic competition for those products.
We also rely on trade secrets, proprietary know-how and technological advances, which we seek to protect, in part, through confidentiality agreements with collaborative partners, employees and consultants. These agreements might be breached and we might not have adequate remedies for any such breach. In addition, our trade secrets and proprietary know-how might otherwise become known or be independently developed by others.
25
Table of Contents
Intense competition might render our products noncompetitive or obsolete.
Competition in our business is intense and characterized by extensive research efforts and rapid technological progress. Technological developments by competitors, regulatory approval for marketing competitive products, including potential generic or over-the-counter products, or superior marketing resources possessed by competitors could adversely affect the commercial potential of our products and could have a material adverse effect on our revenue and results of operations. Generic competition is an increasing risk, as we have experienced with Colazal and Pepcid, and with challenges to our bowel-cleansing products’ intellectual property noted above. We believe that there are numerous pharmaceutical and biotechnology companies, including large well-known pharmaceutical companies, as well as academic research groups throughout the world, engaged in research and development efforts with respect to pharmaceutical products targeted at gastrointestinal diseases and conditions addressed by our current and potential products. In particular, we are aware of products in research or development by competitors that address the diseases being targeted by our products. Developments by others might render our current and potential products obsolete or noncompetitive. Competitors might be able to complete the development and regulatory approval process sooner and, therefore, market their products earlier than we can.
Many of our competitors have substantially greater financial, marketing and personnel resources and development capabilities than we do. For example, many large, well-capitalized companies already offer products in the United States and Europe that target the indications for:
| • | Xifaxan for HE, including lactulose (various manufacturers); |
| • | Xifaxan for travelers’ diarrhea, including ciprofloxacin, commonly known as Cipro (Bayer AG); |
| • | Visicol, OsmoPrep and MoviPrep, including Colyte, Golytely, Halflytely, SuPrep, and Nulytely (Braintree) and Trilyte (Alaven Pharmaceutical LLC), as well as potential generics from Novel Laboratories or others; |
| • | Apriso, including Asacol (Warner Chilcott), sulfasalazine (Pfizer), Dipentum (Alaven Pharmaceutical LLC), Pentasa (Shire Pharmaceuticals Group, plc), once-a-day Lialda (Shire), and three generic balsalazide capsule products; |
| • | Xifaxan under development, including Lotronex® (Prometheus), Amitiza (Sucampo Pharmaceuticals, Inc.) and Zelnorm® (Novartis), which is no longer available on the market in the United States, for IBS; and |
| • | Metozolv ODT, including Reglan• (Alaven Pharmaceutical LLC), and various generics. |
In addition, other products are in research or development by competitors that address the diseases and diagnostic procedures being targeted by these and our other products.
We could be exposed to significant product liability claims that could prevent or interfere with our product commercialization efforts.
We have been in the past and might continue to be subjected to product liability claims that arise through the testing, manufacturing, marketing and sale of our products. For example, we are currently and might continue to be subject to a number of product liability claims relating to OsmoPrep and Visicol in connection with their “box” label warning. We intend to defend these claims vigorously but we are currently unable to predict the outcome or to reasonably estimate the range of potential expenses or loss, if any. We currently maintain liability coverage for both clinical trials and the commercialization of our products but it is possible that this coverage, and any future coverage, will be insufficient to satisfy any liabilities that arise. We would have to assume defense of the lawsuits and be responsible for damages, fees and expenses, if any, that are awarded against us or for amounts in excess of our product liability coverage. These claims could expose us to significant liabilities that could prevent or interfere with our product commercialization efforts. Product liability claims could require us to spend significant time and money in litigation or to pay significant damages. In the future, we might not be able to obtain adequate coverage at an acceptable cost or might be unable to obtain adequate coverage at all.
26
Table of Contents
If government and other third-party payors do not provide coverage or reimburse patients for our products, our ability to derive revenues might suffer.
Our success will depend in part on the extent to which government and health administration authorities, private health insurers and other third-party payors will pay for our products. Reimbursement for newly approved healthcare products is uncertain. In the United States and elsewhere, third-party payors, such as Medicaid, are increasingly challenging the prices charged for medical products and services. Government and other third-party payors are increasingly attempting to contain healthcare costs by limiting both coverage and the level of reimbursement for new therapeutic products. In the United States, a number of legislative and regulatory proposals aimed at changing the healthcare system have been passed in recent years, including the Patient Protection and Affordable Care Act. Many significant changes in this legislation do not take effect until 2014, and various legal challenges and ongoing political opposition create uncertainty as to the final form of this Legislation. These changes to the healthcare system could increase our costs and reduce the amount we can charge for our drugs. In addition, an increasing emphasis on managed care in the United States has and will continue to increase pressure on pharmaceutical pricing. While we cannot predict whether legislative or regulatory proposals will be adopted or what effect those proposals or managed care efforts, including those relating to Medicaid payments, might have on our business, the announcement and/or adoption of such proposals or efforts could increase costs and reduce or eliminate profit margins. Third-party insurance coverage might not be available to patients for our products. If government and other third-party payors do not provide adequate coverage and reimbursement levels for our products, the market acceptance of these products might be reduced.
We expect to be profitable and have positive cash flow during 2011, assuming we receive FDA approval for rifaximin in irritable bowel syndrome, but we might need additional capital.
We expect to be profitable and have positive cash flow during 2011, assuming we receive FDA approval for rifaximin in irritable bowel syndrome. We believe that our current cash and cash equivalents together with cash generated from the sale of our products will be sufficient to fund our operations for 2011 and beyond, but that might not be the case. Our future capital requirements will depend on many factors, including but not limited to:
| • | the results, costs and timing of our research and development activities, regulatory approvals and product launches; |
| • | the status of competitive products, including current and potential generics; |
| • | the cost and number of products we acquire or in-license; |
| • | any impact on us of current conditions and uncertainties in the economy generally and the financial markets; |
| • | patient and physician demand for our products; and |
| • | our ability to reduce our costs in the event product demand is less than expected, or regulatory approvals are delayed or more expensive than expected. |
If we need additional capital, we might seek additional debt or equity financing or both to fund our operations or acquisitions. If we incur more debt, we might be restricted in our ability to raise additional capital and might be subject to financial and restrictive covenants. If we issued additional equity, our stockholders could suffer dilution. We might also enter into additional collaborative arrangements that could provide us with additional funding in the form of equity, debt, licensing, milestone and/or royalty payments. We might not be able to enter into such arrangements or raise any additional funds on terms favorable to us or at all, especially in the current economic environment. Our common stock is likely to decrease in value if the market believes that we will be unable to return to profitability, or that we will be required to raise additional capital.
Our ability to increase revenue in the future will depend in part on our success in in-licensing or acquiring additional pharmaceutical products.
We currently intend to in-license or acquire additional pharmaceutical products, as we did with crofelemer and budesonide, that have been developed beyond the initial discovery phase and for which late-stage human
27
Table of Contents
clinical data is already available, or as we did with Pepcid Oral Suspension, that has already received regulatory approval. These kinds of pharmaceutical products might not be available to us on attractive terms or at all. To the extent we acquire rights to additional products, we might incur significant additional expense in connection with the development and, if approved by the FDA, marketing of these products.
We are dependent on third parties to supply us with products.
We rely entirely on third parties to supply us with our commercially marketed products and our products under development.
For example, Glenmark Pharmaceuticals, Ltd., a corporation organized and located in India, manufactures and supplies us with drug substance for crofelemer, an anti-secretory agent that we are developing for the treatment of HIV-associated diarrhea. The raw material used in production of the crofelemer drug substance grows in select countries in South America. The amount of resources Glenmark devotes to these activities and its ability to successfully obtain raw material is not within our control. Failure by Glenmark to manufacture and supply us with crofelemer drug substance, whether due to international, political or economic conditions or otherwise, could delay development, increase expenses, delay regulatory approval, or eventually prevent us from generating revenue from crofelemer, if approved, any of which could have a material adverse effect on our business. Likewise, interruption of supply of any of our other products, whether for clinical use or commercial use, could have a material adverse effect on our business.
We are dependent on third parties to manufacture our products.
We own no manufacturing facilities, and we have limited capabilities in manufacturing pharmaceutical products. We do not generally expect to engage directly in the manufacturing of products, but instead contract with and rely on third-party vendors for these services. A limited number of contract manufacturers exist which are capable of manufacturing our marketed products and our product candidates. We might fail to contract with the necessary manufacturers or might contract with manufacturers on terms that may not be entirely acceptable to us. For example, we filed an NDA for balsalazide tablets in July 2007, and received a complete response letter from the FDA in April 2010. The sole issue raised in this letter concerns a deficiency of the manufacturing facility for this application. Given our ongoing dependence on third party vendors for supply of material for use in clinical trials and for commercial product, our manufacturing strategy presents the following risks:
| • | the manufacture of products might be difficult to scale up when required and result in delays, inefficiencies and poor or low yields of quality products; |
| • | some of our contracts contain purchase commitments that require us to make minimum purchases that might exceed our needs or limit our ability to negotiate with other manufacturers, which might increase costs; |
| • | the cost of manufacturing certain products might make them prohibitively expensive; |
| • | delays in scale-up to commercial quantities and any change in manufacturers could delay clinical studies, regulatory submissions and commercialization of our products; |
| • | manufacturers are subject to the FDA’s cGMP regulations and similar foreign standards, and we do not have control over compliance with these regulations by the third-party manufacturers; |
| • | if we need to change manufacturers, transfers of technical expertise would be required which would include educating the new manufacturer in the processes necessary for the production of our products, which may not be successful; and |
| • | if we need to change manufacturers, FDA and comparable foreign regulators may require additional testing and compliance inspections prior to the new manufacturer being qualified for the production of our products. |
28
Table of Contents
Failure to comply with manufacturing regulation could harm us financially and could hurt our reputation.
We and our third-party manufacturers, such as Glenmark Pharmaceuticals, Ltd. that produces crofelemer drug substance, are also required to comply with the applicable FDA current Good Manufacturing Practices (cGMP) regulations which include requirements relating to manufacturing, packaging, documentation, quality control, and quality assurance. Further, manufacturing facilities must be approved by the FDA before they can be used to manufacture our products. For example, in April 2010 we received a complete response letter from the FDA regarding our NDA for Giazo. In the complete response letter there were no requests for new pre–clinical or clinical trials. The sole issue raised in this letter concerned a deficiency of the manufacturing facility for this application. The manufacturer has responded to the FDA and continues to work with the FDA to resolve the matter, however the manufacturer has not resolved these issues and we have been unable to receive approval or launch Giazo. Such facilities are subject to periodic FDA inspection. Manufacturing regulations can increase our expenses and delay production, either of which could reduce our margins. In addition, if we fail to comply with any of FDA’s continuing regulations, we could be subject to reputational harm and sanctions, including:
| • | delays, warning letters and fines; |
| • | product recalls or seizures and injunctions on sales; |
| • | refusal of the FDA to review pending applications; |
| • | total or partial suspension of production; |
| • | withdrawals of previously approved marketing applications; and |
| • | civil penalties and criminal prosecutions. |
In addition, the occurrence of manufacturing-related compliance issues could cause subsequent withdrawal of the drug approval, reformulation of the drug product, additional testing or changes in labeling of the finished product.
Our results of operations might fluctuate from period to period, and a failure to meet the expectations of investors or the financial community at large could result in a decline in our stock price.
As they have in the past, our results of operations might fluctuate significantly on a quarterly and annual basis due to, among other factors:
| • | the timing of regulatory approvals and product launches by us or competitors, including potential generic or over-the-counter competitors; |
| • | the level of revenue generated by commercialized products, including potential increased purchases of inventory by wholesalers in anticipation of potential price increases or introductions of new dosages or bottle sizes, and subsequent lower than expected revenue as the inventory is used; |
| • | the timing of any up-front payments that might be required in connection with any future acquisition of product rights; |
| • | the timing of milestone payments that might be required to our current or future licensors; |
| • | fluctuations in our development and other costs in connection with ongoing product development programs; |
| • | the level of marketing and other expenses required in connection with product launches and ongoing product growth; |
| • | the timing of the acquisition and integration of businesses, assets, products and technologies; and |
| • | general and industry-specific business and economic conditions. |
29
Table of Contents
Our stock price is volatile.
Our stock price has been extremely volatile and might continue to be, making owning our stock risky. Between January 1, 2007 and February 25, 2011, the price of a share of our common stock varied from a low of $5.07 to a high of $49.05. In 2010, there were thirteen days on which our stock price increased or decreased by 5% or more.
The securities markets have experienced significant price and volume fluctuations unrelated to the performance of particular companies, including as a result of the current credit and economic crisis. In addition, the market prices of the common stock of many publicly traded pharmaceutical and biotechnology companies have in the past been and can in the future be expected to be especially volatile. Announcements of prescription trends, technological innovations or new products by us or our competitors, generic approvals, developments or disputes concerning proprietary rights, publicity regarding actual or potential medical results relating to products under development by us or our competitors, regulatory developments in both the United States and other countries, public concern as to the safety of pharmaceutical products, and economic and other external factors, as well as period-to-period fluctuations in financial results, might have a significant impact on the market price of our common stock.
Antitakeover provisions could discourage a takeover that stockholders consider to be in their best interests or prevent the removal of our current directors and management.
We have adopted a number of provisions that could have antitakeover effects or prevent the removal of our current directors and management. We have adopted a stockholder protection rights plan, commonly referred to as a poison pill. The rights plan is intended to deter an attempt to acquire us in a manner or on terms not approved by our board of directors. The rights plan will not prevent an acquisition that is approved by our board of directors. We believe our rights plan assisted in our successful defense against a hostile takeover bid earlier in 2003. Our charter authorizes our board of directors to determine the terms of up to 5,000,000 shares of undesignated preferred stock and issue them without stockholder approval. The issuance of preferred stock could make it more difficult for a third party to acquire, or discourage a third party from acquiring, voting control in order to remove our current directors and management. Our bylaws also eliminate the ability of the stockholders to act by written consent without a meeting or make proposals at stockholder meetings without giving us advance written notice, which could hinder the ability of stockholders to quickly take action that might be opposed by management. These provisions could make more difficult the removal of current directors and management or a takeover of Salix, even if these events could be beneficial to stockholders. These provisions could also limit the price that investors might be willing to pay for our common stock.
Item 1B. Unresolved Staff Comments
None.
Due to the growth of the Company since we moved to our current Morrisville, NC location in 2005, in February 2011 we committed to relocate our corporate headquarters to its former location in Raleigh, North Carolina. At that time, we entered into a lease with a target commencement date of September 2011 for approximately 127,000 square feet of office space, including office space that will become a part of the premises upon the termination of existing leases between the landlord and third parties. The lease expires in April 2023. At the same time, we amended our lease for office space also in Raleigh, currently being subleased to a third party. These premises were our former headquarters prior to May 2005 and consist of approximately 26,000 square feet of office space. The amendment extended the term of the lease until July 2018. In connection with our relocation to Raleigh, we terminated our current lease in Morrisville, North Carolina, under the terms of the lease. We have until October 2011 to vacate the current premises and will pay a termination fee of approximately $1.7 million. We also lease a small amount of additional space in Palo Alto, California. We believe our existing facilities are adequate for our current needs and that suitable additional or alternative space will be available in the future on commercially reasonable terms as needed.
30
Table of Contents
From time to time, we are party to various legal proceedings or claims, either asserted or unasserted, which arise in the ordinary course of business. Management has reviewed pending legal matters and believes that the resolution of such matters will not have a significant adverse effect on our financial condition or results of operations.
Regulatory data exclusivity for Xifaxan 200mg tablets ended on or about May 24, 2009. Accordingly, the Office of Generic Drugs would have been able to accept an ANDA for Xifaxan tablets on or any time subsequent to May 24, 2008, if the applicant certified that its generic rifaximin does not infringe Salix’s patent. If this occurred, a Paragraph IV notification would have to be provided to us by the applicant. Although we do not know of any such filing at the current time, the expiration of data exclusivity could result in a challenge to the related intellectual property rights of Xifaxan 200mg tablets at any time in the future. We intend to vigorously enforce the patent rights for Xifaxan.
On or about July 14, 2008, Strides Arcolab Limited filed a Suitability Petition with the FDA seeking permission to submit an ANDA for change of dosage form from tablet to capsule as suitable for a 200mg generic version of Xifaxan. We intend to vigorously enforce the regulatory and intellectual property rights regarding Xifaxan. We are unable to predict the outcome of any ensuing regulatory action or litigation at the present time.
We are currently and might continue to be subject to product liability claims that arise through the testing, manufacturing, marketing and sale of our products, including a number of claims relating to OsmoPrep, Visicol and Metozolv ODT in connection with their respective “box” label warning. We intend to defend these claims vigorously but are currently unable to predict the outcome or to reasonably estimate the range of total potential expenses or losses, if any. We currently maintain liability coverage for our products but it is possible that this coverage, and any future coverage, will be insufficient to satisfy any liabilities that arise. We would have to assume defense of the lawsuits and be responsible for damages, fees and expenses, if any, that are awarded against us or for amounts in excess of our product liability coverage.
We filed a lawsuit against Novel Laboratories, Inc. because Novel was seeking FDA approval to market a generic version of our MoviPrep product. Norgine, B.V. and Norgine Europe, B.V. own U.S. Patent No. 7,169,381 (the ‘381 patent). The ‘381 patent is listed with the FDA as protecting our MoviPrep product. Norgine licensed MoviPrep and the ‘381 patent to us for commercialization in the United States. Novel filed an Abbreviated New Drug Application, or ANDA, with the FDA seeking approval to market a generic version of MoviPrep in the United States prior to the September 2024 expiration of the ‘381 patent. On May 14, 2008, we and Norgine filed a lawsuit in the United States District Court for the District of New Jersey against Novel for infringement of the ‘381 patent. Novel filed an Answer and Counterclaims on June 20, 2008. On June 25, 2009 we and Norgine amended the complaint to add a claim for correction of inventorship for the ‘381 patent. Novel filed an Answer and Counterclaims to the first Amended Complaint on July 10, 2009. Novel denied infringement and asserted various affirmative defenses, including defenses of patent invalidity and unenforceability. On February 9, 2010, an additional patent, U.S. Patent No. 7,658,914, was issued and listed in the Orange Book for MoviPrep.
On August 26, 2010 the parties agreed to a confidential settlement of the MoviPrep lawsuit which was approved by the Court on August 30, 2010 and resolved all of the parties’ outstanding claims and defenses in the lawsuit. The Consent Judgment provides that Novel’s proposed generic product, absent a license as granted to Novel under the settlement, would infringe the ‘381 patent, and further provides that Novel acknowledges and agrees not to contest the validity and enforceability of the ‘381 patent. Under the terms of the settlement, Novel was granted a fully paid up license under the MoviPrep patents such that it is permitted to launch a generic MoviPrep product on September 24, 2018, or earlier under certain circumstances.
We filed a second lawsuit against Novel because Novel was also seeking FDA approval to market a generic version of our OsmoPrep product. CDC, LLC, owns U.S. Patent No. 5,616,346 (the ‘346 patent). The ‘346 patent
31
Table of Contents
is listed with the FDA as protecting our OsmoPrep product. CDC, by its predecessor, licensed OsmoPrep and the ‘346 patent to us for commercialization in the United States. Novel filed an ANDA with the FDA seeking approval to market a generic version of OsmoPrep in the United States prior to the May 2013 expiration of the ‘346 patent. On September 8, 2008, we filed a lawsuit in the United States District Court for the District of New Jersey against Novel for the infringement of the ‘346 patent. The lawsuit also joined CDC as a party. Novel filed an Answer and Counterclaims on December 16, 2008. Novel denied infringement and asserted a defense of patent invalidity. On February 4, 2010 the Court conducted a patent claim construction, or Markman, hearing. On March 30, 2010, an additional patent, U.S. Patent No. 7,687,075 was issued and listed in the Orange Book for OsmoPrep.
On September 30, 2010 the parties agreed to a confidential settlement of the OsmoPrep lawsuit which was approved by the Court on the same day and resolved all of the parties’ outstanding claims and defenses in the lawsuit. The Consent Judgment provides that Novel’s proposed generic product, absent a license as granted to Novel under the settlement, would infringe the ‘346 patent and further provides that Novel acknowledges and agrees not to contest the validity and enforceability of the ‘346 patent. Under the terms of the settlement, Novel was granted a fully paid up license under the OsmoPrep patents such that it is permitted to launch a generic OsmoPrep product on November 16, 2019, or earlier under certain circumstances.
On or about March 19, 2010, we received a Warning Letter from the Division of Marketing, Advertising, and Communications of the FDA, or DDMAC, alleging that certain Metozolv ODT promotional material did not comply with applicable regulatory and legal parameters. We promptly ceased use of such material as an accommodation and implemented a corrective messaging plan. On or about December 27, 2010, DDMAC advised us in writing that, in light of the actions taken by the Company, DDMAC considers this matter closed. We are not aware of any other enforcement action concerning this matter.
Executive Officers of the Registrant
The following table sets forth information concerning our executive officers as of March 1, 2010:
| Name |
Age | Position | ||||
| Carolyn J. Logan |
62 | President, Chief Executive Officer, and Director | ||||
| Adam C. Derbyshire |
45 | Executive Vice President, Finance and Administration, and Chief Financial Officer | ||||
| William P. Forbes |
49 | Executive Vice President, Research and Development and Chief Development Officer | ||||
Carolyn J. Logan has served as President and Chief Executive Officer and as a member of the Board of Directors since July 2002. She previously served as Senior Vice President, Sales and Marketing from June 2000 to July 2002. Prior to joining us, Ms. Logan served as Vice President, Sales and Marketing of the Oclassen Dermatologics division of Watson Pharmaceuticals, Inc. from May 1997 to June 2000, and as Vice President, Sales from February 1997 to May 1997. Prior to that date, she served as Director, Sales of Oclassen Pharmaceuticals, Inc. from January 1993 to February 1997. Prior to joining Oclassen, Ms. Logan held various sales and marketing positions with Galderma Laboratories, Ulmer Pharmacal and Westwood Pharmaceuticals. Ms. Logan received a B.S. degree in Biology and Dental Hygiene from the University of North Carolina at Chapel Hill.
Adam C. Derbyshire has served as Executive Vice President, Finance and Administration and Chief Financial Officer since January 2009. Mr. Derbyshire previously served as Senior Vice President, Finance and Administration and Chief Financial Officer from June 2003 to January 2009 and as Vice President, Finance and Administration and Chief Financial Officer from June 2000 to June 2003. From June 1999 to June 2000, Mr. Derbyshire was Vice President, Corporate Controller and Secretary of Medco Research, Inc., acquired by
32
Table of Contents
King Pharmaceuticals, Inc. in February 2000, Corporate Controller and Secretary of Medco from September 1995 to June 1999 and Assistant Controller of Medco from October 1993 to September 1995. Mr. Derbyshire received his B.S. degree from the University of North Carolina at Wilmington and his MBA from the University of North Carolina at Charlotte.
William P. Forbes has served as Executive Vice President, Research and Development and Chief Medical Officer since January 2010. Dr. Forbes previously served as Senior Vice President, Research and Development and Chief Medical Officer from January 2009 to January 2010. Dr. Forbes previously served as Vice President, Research and Development, and Chief Medical Officer from January 2005 to January 2009. From 2002 through 2004, Dr. Forbes was Vice President, Clinical Development and Regulatory Affairs of Metabasis Therapeutics, Inc. He has also worked for Otsuka America Pharmaceutical, Inc. in a variety of roles of increasing responsibility from 1991 to 2002 and Glaxo, Inc. from 1989 through 1991. He has extensive experience in clinical development, regulatory affairs and project management. Dr. Forbes received his Doctor of Pharmacy degree from Creighton University.
33
Table of Contents
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Our common stock is traded on the Nasdaq Global Market under the symbol “SLXP”. The following table sets forth the high and low sales prices of our common stock, as reported on the Nasdaq Global Market for the eight quarters ended December 31, 2010.
| High | Low | |||||||
| Fiscal year ended December 31, 2009 |
||||||||
| First quarter |
$ | 10.00 | $ | 6.14 | ||||
| Second quarter |
12.64 | 8.78 | ||||||
| Third quarter |
21.47 | 9.35 | ||||||
| Fourth quarter |
25.86 | 18.18 | ||||||
| Fiscal year ended December 31, 2010 |
||||||||
| First quarter |
$ | 38.00 | $ | 23.53 | ||||
| Second quarter |
43.02 | 33.15 | ||||||
| Third quarter |
45.00 | 36.75 | ||||||
| Fourth quarter |
49.05 | 35.64 | ||||||
On February 25, 2011 the closing price for the common stock as reported on the Nasdaq Global Market was $33.49. As of February 25, 2011 there were 239 stockholders of record, which excludes stockholders whose shares were held in nominee or street name by brokers.
The securities markets have from time to time experienced significant price and volume fluctuations unrelated to the operating performance of particular companies. Our stock has been particularly volatile, including for example the approximate 21.8% single-day drop on December 28, 2007, when three generic versions of our drug Colazal were approved, the approximate 51.1% single-day increase on September 14, 2009 when we announced data from our Phase III IBS trials, and the approximate 20.5% single-day increase on February 24, 2010, the day after the Gastrointestinal Drugs Advisory Committee of the FDA recommended by a vote of 14 to 4 in favor of the approval of Xifaxan 550 mg for the maintenance of remission of hepatic encephalopathy. The market prices of the common stock of Salix and many publicly traded pharmaceutical and biotechnology companies have in the past and can in the future be expected to be especially volatile. Announcements of technological innovations or new products by us or our competitors, developments or disputes concerning proprietary rights, publicity regarding actual or potential medical results relating to products under development by us or our competitors, regulatory developments in both the United States and other countries, public concern as to the safety of pharmaceutical products and economic and other external factors, as well as period-to-period fluctuations in our financial results, might have a significant impact on the market price of our common stock.
34
Table of Contents
Performance Graph
The following graph compares our cumulative total stockholder return from December 31, 2005 with those of the Nasdaq Composite Index and the Nasdaq Biotech Index and assumes that all dividends were reinvested. The graph assumes that U.S. $100 was invested on December 31, 2005 in (1) our common stock, (2) the Nasdaq Composite Index and (3) the Nasdaq Biotech Index. The measurement points utilized in the graph consist of the last trading day in each calendar year, which closely approximates the last day of the respective fiscal year of the Company. The historical stock performance presented below is not intended to and may not be indicative of future stock performance.
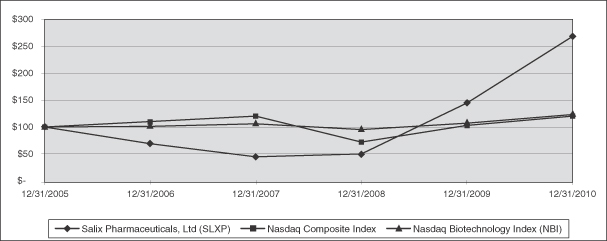
| 12/31/05 | 12/31/06 | 12/31/07 | 12/31/08 | 12/31/09 | 12/31/10 | |||||||||||||||||||
| SLXP |
$ | 100 | $ | 69 | $ | 45 | $ | 50 | $ | 144 | $ | 267 | ||||||||||||
| Nasdaq Composite Index |
$ | 100 | $ | 110 | $ | 120 | $ | 72 | $ | 103 | $ | 120 | ||||||||||||
| Nasdaq Biotech Index |
$ | 100 | $ | 101 | $ | 106 | $ | 95 | $ | 107 | $ | 123 | ||||||||||||
Dividend Policy
We have never declared or paid cash dividends on our common stock. We currently expect to retain future earnings, if any, for use in the operation and expansion of business and do not anticipate paying any cash dividends in the foreseeable future.
Equity Compensation Plans
The information required by Item 5 of Form 10-K regarding equity compensation plans is incorporated herein by reference to “Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.”
35
Table of Contents
Item 6. Selected Financial Data
The following selected consolidated financial data should be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and the Consolidated Financial Statements and the Notes thereto included elsewhere in this report.
Consolidated Statements of Operations Data:
| Year Ended December 31, | ||||||||||||||||||||
| 2010 | 2009 | 2008 | 2007 | 2006 | ||||||||||||||||
| (U.S. dollars, in thousands, except per share data) | ||||||||||||||||||||
| Revenues: |
||||||||||||||||||||
| Net product revenues |
$ | 336,973 | $ | 232,890 | $ | 178,766 | $ | 235,792 | $ | 208,533 | ||||||||||
| Costs and expenses: |
||||||||||||||||||||
| Cost of products sold |
68,677 | 52,025 | 36,710 | 55,024 | 41,443 | |||||||||||||||
| Amortization of product rights and intangible assets |
10,370 | 11,485 | 9,891 | 8,627 | 4,907 | |||||||||||||||
| Intangible impairment charge |
34,656 | — | — | — | — | |||||||||||||||
| Research and development |
73,346 | 89,466 | 83,735 | 73,797 | 49,213 | |||||||||||||||
| Selling, general and administrative |
156,101 | 120,020 | 95,088 | 86,492 | 82,636 | |||||||||||||||
| Total costs and expenses |
343,150 | 272,996 | 225,424 | 223,940 | 178,199 | |||||||||||||||
| Income (loss) from operations |
(6,177 | ) | (40,106 | ) | (46,658 | ) | 11,852 | 30,334 | ||||||||||||
| Interest expense |
(20,652 | ) | (6,746 | ) | (3,755 | ) | (1,336 | ) | — | |||||||||||
| Interest and other income |
2,626 | 1,221 | 2,690 | 4,662 | 2,552 | |||||||||||||||
| Income (loss) before income tax expense |
(24,203 | ) | (45,631 | ) | (47,723 | ) | 15,178 | 32,886 | ||||||||||||
| Income tax (expense) benefit |
(2,858 | ) | 2,012 | 116 | (6,953 | ) | (1,376 | ) | ||||||||||||
| Net income (loss) |
$ | (27,061 | ) | $ | (43,619 | ) | $ | (47,607 | ) | $ | 8,225 | $ | 31,510 | |||||||
| Net income (loss) per share, basic(1) |
$ | (0.47 | ) | $ | (0.88 | ) | $ | (0.99 | ) | $ | 0.17 | $ | 0.68 | |||||||
| Net income (loss) per share, diluted(1) |
$ | (0.47 | ) | $ | (0.88 | ) | $ | (0.99 | ) | $ | 0.17 | $ | 0.65 | |||||||
| Shares used in computing net income (loss) per share, basic(1) |
57,300 | 49,350 | 47,898 | 47,329 | 46,634 | |||||||||||||||
| Shares used in computing net income (loss) per share, diluted(1) |
57,300 | 49,350 | 47,898 | 48,678 | 48,369 | |||||||||||||||
Consolidated Balance Sheet Data:
| As of December 31, | ||||||||||||||||||||
| 2010 | 2009 | 2008 | 2007 | 2006 | ||||||||||||||||
| Cash and cash equivalents |
$ | 518,030 | $ | 192,512 | $ | 120,153 | $ | 111,272 | $ | 76,465 | ||||||||||
| Working capital |
560,110 | 217,537 | 113,795 | 108,891 | 126,216 | |||||||||||||||
| Total assets |
851,543 | 543,040 | 400,484 | 397,102 | 323,123 | |||||||||||||||
| Borrowings under credit facility |
— | 15,000 | 15,000 | 15,000 | — | |||||||||||||||
| Convertible senior notes |
323,005 | 47,299 | 44,759 | — | — | |||||||||||||||
| Long term portion of capital lease obligations |
90 | 499 | 791 | 612 | — | |||||||||||||||
| Accumulated deficit |
(223,025 | ) | (195,964 | ) | (152,345 | ) | (104,738 | ) | (112,963 | ) | ||||||||||
| Stockholders’ equity |
401,919 | 370,024 | 265,401 | 292,570 | 277,551 | |||||||||||||||
| (1) | See Note 2 of Notes to Consolidated Financial Statements for an explanation of shares used in computing net income (loss) per share. |
36
Table of Contents
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
OVERVIEW
We are a specialty pharmaceutical company dedicated to acquiring, developing and commercializing prescription drugs used in the treatment of a variety of gastrointestinal disorders, which are those affecting the digestive tract. Our strategy is to:
| • | identify and acquire rights to products that we believe have potential for near-term regulatory approval or are already approved; |
| • | apply our regulatory, product development, and sales and marketing expertise to commercialize these products; and |
| • | use our approximately 250-member specialty sales and marketing team focused on high-prescribing U.S. gastroenterologists, who are doctors who specialize in gastrointestinal disorders, to sell our products. |
Our current products demonstrate our ability to execute this strategy. As of December 31, 2010, our products were:
| • | XIFAXAN® (rifaximin) Tablets 200 mg, indicated for travelers’ diarrhea; |
| • | XIFAXAN® (rifaximin) Tablets 550 mg, indicated for hepatic encephalopathy, which we began selling in the second quarter of 2010; |
| • | MOVIPREP® (PEG 3350, Sodium Sulfate, Sodium Chloride, Potassium Chloride, Sodium Ascorbate and Ascorbic Acid for Oral Solution); |
| • | OSMOPREP™ (sodium phosphate monobasic monohydrate, USP and sodium phosphate dibasic anhydrous, USP) Tablets; |
| • | VISICOL® (sodium phosphate monobasic monohydrate, USP, and sodium phosphate dibasic anhydrous, USP) Tablets; |
| • | APRISO™ (mesalamine) extended-release capsules 0.375g; |
| • | METOZOLV™ ODT (metoclopramide HCl) 5mg and 10mg orally disintegrating tablets; |
| • | AZASAN® Azathioprine Tablets, USP, 75mg and 100 mg; |
| • | ANUSOL-HC® 2.5% (Hydrocortisone Cream, USP), ANUSOL-HC® 25 mg Suppository (Hydrocortisone Acetate); |
| • | PROCTOCORT® Cream (Hydrocortisone Cream, USP) 1% and PROCTOCORT® Suppository (Hydrocortisone Acetate Rectal Suppositories) 30 mg; |
| • | PEPCID® (famotidine) for Oral Suspension; |
| • | Oral Suspension DIURIL® (Chlorothiazide); and |
| • | COLAZAL® (balsalazide disodium) Capsules 750 mg. |
We generate revenue primarily by selling our products, namely prescription drugs, to pharmaceutical wholesalers. These direct customers resell and distribute our products to and through pharmacies to patients who have had our products prescribed by doctors. We currently market our products, and intend to market future products, if approved by the U.S. Food and Drug Administration, or FDA, to U.S. gastroenterologists and other physicians through our own direct sales force. In December 2000, we established our own field sales force to market Colazal in the United States. As of December 31, 2010, this sales force had approximately 160 sales representatives in the field and markets our approved products. Although the creation of an independent sales organization involved substantial costs, we believe that the financial returns from our direct product sales have
37
Table of Contents
been and will continue to be more favorable to us than those from the indirect sale of products through marketing partners. We enter into distribution or licensing relationships outside the United States and in certain markets in the U.S. where a larger sales organization is appropriate. As of December 31, 2010, our sales and marketing staff, including our sales representatives, consisted of approximately 250 people.
Because demand for our products originates with doctors, our sales force calls on high-prescribing specialists, primarily gastroenterologists, and we monitor new and total prescriptions for our products as key performance indicators for our business. Prescriptions result in our products being used by patients, requiring our direct customers to purchase more products to replenish their inventory. However, our revenue might fluctuate from quarter to quarter due to other factors, such as increased buying by wholesalers in anticipation of a price increase or because of the introduction of new products. Revenue could be less than anticipated in subsequent quarters as wholesalers’ increased inventory is used up.
Our primary product candidates currently under development and their status are as follows:
| Compound |
Indication |
Status | ||
| Rifaximin |
Irritable bowel syndrome, or IBS | New Drug Applications, or NDA, submitted June 7, 2010; Prescription Drug User Fee Action, or PDUFA, date March 7, 2011; Complete Response Letter anticipated on or before March 7, 2011 | ||
| Crofelemer |
HIV-associated diarrhea | Phase III | ||
| Balsalazide disodium tablet |
Ulcerative colitis | Complete response letter received April 27, 2010 | ||
| Budesonide foam |
Ulcerative proctitis | Phase III | ||
CRITICAL ACCOUNTING POLICIES
General
Our discussion and analysis of our financial condition and results of operations are based upon our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States of America. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses, and related disclosure of contingent assets and liabilities. On an on-going basis, we evaluate our estimates, including those related to sales of our products, bad debts, inventories, investments, intangible assets and legal issues. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results might differ materially from these estimates under different assumptions or conditions.
Methodologies used and assumptions selected by management in making these estimates, as well as the related disclosures, have been reviewed by and discussed with the Audit Committee of our Board of Directors.
We believe the following critical accounting policies affect our more significant judgments and estimates used in the preparation of our consolidated financial statements.
38
Table of Contents
Revenue Recognition
We recognize revenue when it is realized or realizable and earned. Revenue is realized or realizable and earned when all of the following criteria are met: (a) persuasive evidence of an arrangement exists; (b) delivery has occurred or services have been rendered; (c) the seller’s price to the buyer is fixed and determinable; and (d) collectibility is reasonably assured.
We recognize revenue from sales transactions where the buyer has the right to return the product at the time of sale only if (1) our price to the buyer is substantially fixed or determinable at the date of sale, (2) the buyer has paid us, or the buyer is obligated to pay us and the obligation is not contingent on resale of the product, (3) the buyer’s obligation to us would not be changed in the event of theft or physical destruction or damage of the product, (4) the buyer acquiring the product for resale has economic substance apart from any provided by us, (5) we do not have significant obligations for future performance to directly bring about resale of the product by the buyer, and (6) the amount of future returns can be reasonably estimated. We recognize revenues for product sales at the time title and risk of loss are transferred to the customer, which is generally at the time products are shipped. Our net product revenue represents our total revenues less allowances for customer credits, including estimated discounts, rebates, chargebacks, and product returns.
We establish allowances for estimated rebates, chargebacks and product returns based on numerous quantitative and qualitative factors, including:
| • | the number of and specific contractual terms of agreements with customers; |
| • | estimated levels of inventory in the distribution channel; |
| • | historical rebates, chargebacks and returns of products; |
| • | direct communication with customers; |
| • | anticipated introduction of competitive products or generics; |
| • | anticipated pricing strategy changes by us and/or our competitors; |
| • | analysis of prescription data gathered by a third-party prescription data provider; |
| • | the impact of changes in state and federal regulations; and |
| • | estimated remaining shelf life of products. |
In our analyses, we use prescription data purchased from a third-party data provider to develop estimates of historical inventory channel pull-through. We utilize an internal analysis to compare historical net product shipments to estimated historical prescriptions written. Based on that analysis, we develop an estimate of the quantity of product in the channel that might be subject to various rebate, chargeback and product return exposures. At least quarterly for each product line, we prepare an internal estimate of ending inventory units in the distribution channel by adding estimated inventory in the channel at the beginning of the period, plus net product shipments for the period, less estimated prescriptions written for the period. Based on that analysis, we develop an estimate of the quantity of product in the channel that might be subject to various rebate, chargeback and product return exposures. This is done for each product line by applying a rate of historical activity for rebates, chargebacks and product returns, adjusted for relevant quantitative and qualitative factors discussed above, to the potential exposed product estimated to be in the distribution channel. Internal forecasts that are utilized to calculate the estimated number of months in the channel are regularly adjusted based on input from members of our sales, marketing and operations groups. The adjusted forecasts take into account numerous factors including, but not limited to, new product introductions, direct communication with customers and potential product expiry issues. Adjustments to estimates are recorded in the period when significant events or changes in trends are identified.
Consistent with industry practice, we periodically offer promotional discounts to our existing customers. These discounts are calculated as a percentage of the current published list price and are treated as off-invoice allowances. Accordingly, the discounts are recorded as a reduction of revenue in the period that the program is
39
Table of Contents
offered. In addition to promotional discounts, at the time that we implement a price increase, we generally offer our existing customers an opportunity to purchase a limited quantity of product at the previous list price. Shipments resulting from these programs generally are not in excess of ordinary levels, therefore, we recognize the related revenue upon shipment and include the shipments in estimating our various product related allowances. In the event we determine that these shipments represent purchases of inventory in excess of ordinary levels for a given wholesaler, the potential impact on product returns exposure would be specifically evaluated and reflected as a reduction in revenue at the time of such shipments.
Allowances for estimated rebates and chargebacks were $40.8 million and $17.2 million as of December 31, 2010 and 2009, respectively. These balances exclude amounts related to Colazal, which are included in the reserve discussed below. These allowances reflect an estimate of our liability for items such as rebates due to various governmental organizations under the Medicare/Medicaid regulations, rebates due to managed care organizations under specific contracts and chargebacks due to various organizations purchasing certain of our products through federal contracts and/or group purchasing agreements. We estimate our liability for rebates and chargebacks at each reporting period based on a methodology of applying the relevant quantitative and qualitative assumptions discussed above. Due to the subjectivity of our accrual estimates for rebates and chargebacks, we prepare various sensitivity analyses to ensure our final estimate is within a reasonable range as well as review prior period activity to ensure that our methodology is still reasonable. Had a change in one or more variables in the analyses (utilization rates, contract modifications, etc.) resulted in an additional percentage point change in the trailing average of estimated chargeback and rebate activity for the year ended December 31, 2010, we would have recorded an adjustment to revenues of approximately $4.4 million, or less than 1.3%, for the year.
Allowances for product returns were $18.2 million and $11.3 million as of December 31, 2010 and 2009, respectively. These allowances reflect an estimate of our liability for product that may be returned by the original purchaser in accordance with our stated return policy. We estimate our liability for product returns at each reporting period based on historical return rates, the estimated inventory in the channel, and the other factors discussed above. Due to the subjectivity of our accrual estimates for product returns, we prepare various sensitivity analyses as well as review prior period activity to ensure that our methodology is still reasonable.
Colazal, our balsalazide disodium capsule, accounted for a majority of the Company’s revenue prior to 2008. On December 28, 2007, the Office of Generic Drugs, or OGD, approved three generic balsalazide capsule products. As a result of these generic approvals, sales of this product significantly decreased in 2008, 2009 and 2010 and we expect the future sales of Colazal to be insignificant. At December 31, 2010 and 2009, respectively, $1.5 million and $3.5 million were recorded as a liability to reflect the Company’s estimate of the Company’s liability for Colazal that may be returned or charged back by the original purchaser in accordance with the Company’s stated policies as a result of these generic approvals. We developed this estimate based on the following:
| • | our estimate of the quantity and expiration dates of Colazal inventory in the distribution channel based on historical net product shipments less estimated historical prescriptions written; |
| • | our estimate of future demand for Colazal based on the actual erosion of product demand for several comparable products that were previously genericized, and the most recent demand for Colazal prior to the generic approvals; |
| • | the actual demand for Colazal experienced during 2008, 2009 and 2010 subsequent to the generic approvals; |
| • | our estimate of chargeback and rebate activity based on price erosion as a result of the generic approvals; and |
| • | other relevant factors. |
40
Table of Contents
During 2010 we adjusted our estimate of Colazal returns based on our review of current trends for returns, chargebacks and purchases of Colazal, and recorded approximately $7.0 million as an increase to the reserve and a reduction of revenue during the period.
For the years ended December 31, 2010, 2009 and 2008, our absolute exposure for rebates, chargebacks and product returns has grown primarily as a result of increased sales of our existing products, the approval of new products and the acquisition of products, and also as a result of the approval of generic balsalazide capsule products. Accordingly, reductions to revenue and corresponding increases to allowance accounts have likewise increased. The estimated exposure to these revenue-reducing items as a percentage of gross product revenue in the years ended December 31, 2010, 2009 and 2008 was 14.9%, 9.8% and 6.3% for rebates, chargebacks and discounts and was 2.9%, 5.5% and 7.2% for product returns excluding the Colazal return reserve, respectively.
During the fourth quarter of 2009 we shipped Metozolv to wholesalers and began detailing this drug to physicians. Because we have limited experience with Metozolv’s indication we do not currently have the ability to estimate returns for Metozolv. We will recognize revenue for Metozolv based upon prescription pull-through until we have enough historical information to estimate returns.
During the second quarter of 2010 we recognized product revenue related to initial shipments to wholesalers of Xifaxan 550mg, which was approved by the FDA on March 24, 2010 for reduction in risk of overt hepatic encephalopathy, or HE, recurrence in patients 18 years of age or older, and launched to physicians in May 2010. Based on our historical experience with Xifaxan 200mg, which we distribute through the same distribution channels and is prescribed by the same physicians as Xifaxan 550mg, we have the ability to estimate returns for Xifaxan 550mg and therefore recognized revenue upon shipment to the wholesalers.
Inventories
Raw materials, work-in-process and finished goods inventories are stated at the lower of cost (which approximates actual cost on a first-in, first-out cost method) or market value. In evaluating whether inventory is stated at the lower of cost or market, management considers such factors as the amount of inventory on hand and in the distribution channel, estimated time required to sell such inventory, remaining shelf life, and current and expected market conditions, including levels of competition, including generic competition.
We expense pre-approval inventory unless we believe it is probable that the inventory will be saleable. We capitalize inventory costs associated with marketed products and certain products prior to regulatory approval and product launch, based on management’s judgment of probable future commercial use and net realizable value. Capitalization of this inventory does not begin until the product candidate is considered to have a high probability of regulatory approval, which is generally after we have analyzed Phase III data or filed an New Drug Application, or NDA. If we are aware of any specific risks or contingencies that are likely to impact the expected regulatory approval process or if there are any specific issues identified during the research process relating to safety, efficacy, manufacturing, marketing or labeling of the product candidate, we do not capitalize the related inventory. Once we capitalize inventory for a product candidate that is not yet approved, we monitor, on a quarterly basis, the status of this candidate within the regulatory approval process. We could be required to expense previously capitalized costs related to pre-approval inventory upon a change in our judgment of future commercial use and net realizable value, due to a denial or delay of approval by regulatory bodies, a delay in the timeline for commercialization or other potential factors. On a quarterly basis, we evaluate all inventory, including inventory capitalized for which regulatory approval has not yet been obtained, to determine if any lower of cost or market adjustment is required. As it relates to pre-approval inventory, we consider several factors including expected timing of FDA approval, projected sales volume and estimated selling price. At December 31, 2010 and 2009, there were no amounts included in inventory related to pre-approval inventory.
Inventory at December 31, 2010 consisted of $9.6 million of raw materials, $5.1 million of work-in-process and $1.4 million of finished goods. Inventory at December 31, 2009 consisted of $10.9 million of raw materials,
41
Table of Contents
$8.4 million of work-in-process and $5.0 million of finished goods. As of December 31, 2010, we had recorded inventory reserves totaling $6.6 million, compared to $2.8 million as of December 31, 2009, to reduce inventories to their net realizable value.
Intangible Assets and Goodwill
Our intangible assets consist of license agreements, product rights and other identifiable intangible assets, which result from product and business acquisitions. Goodwill represents the excess purchase price over the fair value of assets acquired and liabilities assumed in a business combination.
When we make product acquisitions that include license agreements, product rights and other identifiable intangible assets, we record the purchase price of such intangibles, along with the value of the product-related liabilities that we assume, as intangible assets. We allocate the aggregate purchase price to the fair value of the various tangible and intangible assets in order to determine the appropriate carrying value of the acquired assets and then amortize the cost of the intangible assets as an expense in the consolidated statements of operations over the estimated economic useful life of the related assets. We assess the impairment of identifiable intangible assets whenever events or changes in circumstances indicate that the carrying value might not be recoverable. We believe the following factors could trigger an impairment review: significant underperformance relative to expected historical or projected future operating results; significant changes in the manner of use of the acquired assets or the strategy for our overall business; approval of generic products; and significant negative industry or economic trends.
In assessing the recoverability of our intangible assets, we must make assumptions regarding estimated future cash flows and other factors. If the estimated undiscounted future cash flows do not exceed the carrying value of the intangible assets, we must determine the fair value of the intangible assets. If the fair value of the intangible assets is less than the carrying value, we will recognize an impairment loss equal to the difference. We review goodwill for impairment on an annual basis, and goodwill and other intangible assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable.
In November 2003, we acquired from aaiPharma LLC for $2.0 million the exclusive right to sell 25, 75 and 100 milligram dosage strengths of azathioprine tablets in North America under the name Azasan. The purchase price was fully allocated to product rights and related intangibles and is being amortized over a period of ten years. Although Azasan does not have any patent protection, we believe ten years is an appropriate amortization period based on established product history and management’s experience. At December 31, 2010 and 2009, accumulated amortization for the Azasan intangible was $1.4 million and $1.2 million, respectively.
In June 2004, we acquired the exclusive U.S. rights to Anusol-HC 2.5% (hydrocortisone Cream USP), Anusol-HC 25 mg Suppository (Hydrocortisone Acetate), Proctocort Cream (Hydrocortisone Cream USP) 1% and Proctocort Suppositories (Hydrocortisone Acetate Rectal Suppositories, 30 mg) from King Pharmaceuticals, Inc. for $13.0 million. The purchase price was fully allocated to product rights and related intangibles and is being amortized over a period of ten years. Although Anusol-HC and Proctocort do not have any patent protection, we believe ten years is an appropriate amortization period based on established product sales history and management’s experience. At December 31, 2010 and 2009, accumulated amortization for the King product intangibles was $8.4 million and $7.1 million, respectively.
In September 2005, we acquired InKine Pharmaceutical Company, Inc. for $210.0 million. We allocated $74.0 million of the purchase price to in-process research and development, $9.3 million to net assets acquired and $37.0 million to specifically identifiable product rights and related intangibles with an ongoing economic benefit to us. We allocated the remaining $89.7 million to goodwill, which is not being amortized. The InKine product rights and related intangibles are being amortized over an average period of 14 years, which we believed was an appropriate amortization period due to the product’s patent protection and the estimated economic lives of the product rights and related intangibles. In September 2010 we entered into a Sublicense Agreement which
42
Table of Contents
granted Novel Laboratories, Inc. a license under the patents covering OsmoPrep such that Novel is permitted to launch a generic OsmoPrep on November 16, 2019. As a result of this agreement the amortization period was adjusted prospectively, and the remaining net book value of the intangible asset is being amortized through November 16, 2019, which is our revised estimate of its remaining economic life. As a result of this agreement, we assessed whether there was an impairment to the carrying value of the related intangible asset due to its reduced economic life and determined that there was no impairment. At December 31, 2010 and 2009, accumulated amortization for the InKine intangibles was $15.7 million and $12.8 million, respectively. There is no future research or development planned for the products purchased from InKine.
In December 2005, we entered into a License and Supply Agreement with Norgine B.V., granting Salix the exclusive right to sell a patented-protected, liquid PEG bowel cleansing product, NRL 944, in the United States. In August 2006, we received Food and Drug Administration marketing approval for NRL 944 under the branded name of MoviPrep. In January 2007 the United States Patent Office issued a patent providing coverage to September 1, 2024. Pursuant to the terms of the Agreement, Salix paid Norgine milestone payments of $15.0 million in August 2006, $5.0 million in December 2008 and $5.0 million in December 2009. We were amortizing these milestone payments over a period of 17.3 years through 2022, which we believed was an appropriate amortization period due to the product’s patent protection and the estimated economic life of the related intangible. In August 2010 we entered into a Sublicense Agreement that granted Novel Laboratories, Inc. a license to the patents covering MoviPrep such that Novel is permitted to launch a generic MoviPrep on September 24, 2018. As a result of this agreement we adjusted the amortization period prospectively, and we are now amortizing the remaining net book value of the intangible asset through September 24, 2018, which is our revised estimate of its remaining economic life. As a result of this agreement, we assessed whether there was an impairment to the carrying value of the related intangible asset due to its reduced economic life and determined that there was no impairment. At December 31, 2010 and 2009, accumulated amortization for the MoviPrep intangible was $6.8 million and $5.0 million, respectively.
In February 2007, we entered into a Master Purchase and Sale and License Agreement with Merck & Co. Inc., to purchase the U.S prescription pharmaceutical product rights to Pepcid Oral Suspension and Diuril Oral Suspension from Merck. We paid Merck $55.0 million at the closing of this transaction. The purchase price was fully allocated to product rights and related intangibles, and is being amortized over a period of 15 years. Although Pepcid and Diuril do not have patent protection, we believe 15 years was an appropriate amortization period based on established product history and management experience. In May 2010, the FDA approved a generic famotidine oral suspension product, and we launched an authorized generic famotidine product. In June 2010 the FDA approved another generic famotidine oral suspension product. As a result of these events, we assessed whether there was an impairment to the carrying value of the related intangible asset. Based on this analysis, we recorded a $30 million impairment charge to reduce the carrying value of the intangible asset to its estimated fair value during the three-month period ended June 30, 2010. At December 31, 2010 and 2009, accumulated amortization for the Merck products was $12.9 million and $10.5 million, respectively.
In July 2002, we acquired the rights to develop and market a granulated formulation of mesalamine from Dr. Falk Pharma GmbH. On October 31, 2008, the FDA granted marketing approval for Apriso for the maintenance of remission of ulcerative colitis in adults. In November 2008, we made a $6.0 million milestone payment to Dr. Falk. In December 2009, we made a $2.0 million milestone payment to Dr. Falk. We are amortizing these milestone payments over a period of 9.5 years, which we believe is an appropriate amortization period due to the product’s patent protection and the estimated economic life of the related intangible. At December 31, 2010 and 2009, accumulated amortization for the Apriso intangible was $1.8 million and $1.0 million, respectively.
In September 2007, we acquired the exclusive, worldwide right to sell metoclopramide–Zydis® (trade name Metozolv) from Wilmington Pharmaceuticals, LLC. On September 8, 2009 the FDA granted marketing approval for Metozolv™ ODT (metoclopramide HCl) 5 mg and 10 mg orally disintegrating tablets. Metozolv ODT is indicated for the relief of symptomatic gastroesophageal reflux or short-term (4-12 weeks) therapy for adults with symptomatic, documented gastroesophageal reflux who fail to respond to conventional therapy and diabetic
43
Table of Contents
gastroparesis or the relief of symptoms in adults associated with acute and recurrent diabetic gastroparesis. In October 2009, we made a $7.3 million milestone payment to Wilmington. We are amortizing this milestone payment over a period of 8 years, which we believe is an appropriate amortization period due to the product’s patent protection and the estimated economic life of the related intangible. On November 3, 2010, we received a paragraph IV notification from Novel stating that Novel had filed an ANDA application to seek approval to market a generic version of Metoclopramide Hydrochloride ODT, 5 mg and 10 mg. The notification letter asserted non-infringement of U.S. Patent No. 6,413,549 (“the ‘549 patent”). Upon examination of the relevant sections of the ANDA, we concluded that the ‘549 patent would not be enforced against Novel Laboratories. As a result of this event, we assessed whether there was an impairment to the carrying value of the related intangible asset. Based on this analysis, we recorded a $4.6 million impairment charge to reduce the carrying value of the intangible asset to its estimated fair value during the three-month period ended December 31, 2010. At December 31, 2010 and 2009 accumulated amortization for the Metozolv intangible was $1.2 million and $0.2 million, respectively.
Allowance for Uncollectible Accounts
Based on a review of specific customer balances, industry experience and the current economic environment, we currently reserve for specific past due accounts that may represent collection concerns plus a percentage of our outstanding trade accounts receivable balance as an allowance for uncollectible accounts, which at December 31, 2010 and 2009 was approximately $0.5 million and $0.5 million, respectively. Refer to “Schedule II—Valuation and Qualifying Accounts” for a roll-forward of the allowance for uncollectible accounts.
Cash and Cash Equivalents
We consider all highly liquid investments with maturities from date of purchase of three months or less to be cash equivalents. We maintain our cash and cash equivalents in several different financial instruments with various banks and brokerage houses. This diversification of risk is consistent with our policy to maintain liquidity and ensure the safety of principal. At December 31, 2010 and 2009, cash and cash equivalents consisted primarily of demand deposits, overnight investments in Eurodollars, certificates of deposit and money market funds at reputable financial institutions and did not include any auction rate securities. We have not experienced any loss of principal or liquidity in any of our cash and cash equivalents.
Research and Development
We expense research and development costs, both internal and externally contracted, as incurred. For nonrefundable advance payments for goods or services that will be used or rendered for future research and development activities, we initially capitalize the advance payment. We then recognize such amounts as an expense as the related goods are delivered or the related services are performed. At December 31, 2010 and 2009, the net liability related to on-going research and development activities was $10.2 million and $14.5 million, respectively.
Due to the significant risks and uncertainties inherent in the clinical development and regulatory approval processes, the cost to complete projects and development timelines for their completion cannot be reasonably estimated. Enrollment in clinical trials might be delayed for reasons beyond our control, requiring additional cost and time. Results from clinical trials might not be favorable, or might require us to perform additional unplanned clinical trials, requiring additional cost and time, or resulting in termination of the project. Further, data from clinical trials is subject to varying interpretation, and might be deemed insufficient by the regulatory bodies reviewing applications for marketing approvals, requiring additional cost and time, or resulting in termination of the project. Process development and manufacturing scale-up for production of clinical and commercial product supplies might take longer and cost more than our forecasts. As such, clinical development and regulatory programs are subject to risks and changes that might significantly impact cost projections and timelines.
44
Table of Contents
The following table summarizes costs incurred for our significant projects, in thousands. We consider a project significant if expected spend for any year exceeds 10% of our development project budget for that year.
| Project |
Year ended December 31, | Cumulative through |
||||||||||||||
| 2010 | 2009 | 2008 | December 31, 2010 |
|||||||||||||
| Rifaximin for hepatic encephalopathy, or HE |
$ | 6,037 | $ | 7,411 | $ | 12,489 | $ | 35,498 | ||||||||
| Rifaximin for irritable bowel syndrome, or IBS |
93 | 21,100 | 15,459 | 46,404 | ||||||||||||
| Crofelemer for HIV-associated diarrhea |
10,979 | 9,229 | 2,375 | 22,583 | ||||||||||||
| Balsalazide disodium tablet for ulcerative colitis |
— | 250 | 6,150 | 40,030 | ||||||||||||
| Budesonide foam for ulcerative proctitis |
9,707 | 5,886 | 210 | 15,803 | ||||||||||||
| Granulated mesalamine for ulcerative colitis—Apriso |
— | 1,063 | 7,535 | 27,540 | ||||||||||||
| Other non-significant rifaximin clinical projects (6 in 2010, 9 in 2009, 4 in 2008) |
4,362 | 4,063 | 3,762 | N/A | ||||||||||||
| All other clinical programs (8 in 2010, 5 in 2009 and 2008) |
5,924 | 1,956 | 1,224 | N/A | ||||||||||||
RESULTS OF OPERATIONS
Years Ended December 31, 2010, 2009 and 2008
Revenues
The following table summarizes net product revenues, in thousands, for the years ended December 31, 2010, 2009 and 2008:
| Year ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Xifaxan |
$ | 250,459 | $ | 117,939 | $ | 79,928 | ||||||
| % of net product revenues |
74 | % | 51 | % | 45 | % | ||||||
| Purgatives—MoviPrep/OsmoPrep/Visicol |
54,207 | 76,287 | 62,862 | |||||||||
| % of net product revenues |
16 | % | 33 | % | 35 | % | ||||||
| Inflammatory Bowel Disease—Colazal/Apriso |
24,459 | 5,782 | 10,545 | |||||||||
| % of net product revenues |
7 | % | 2 | % | 6 | % | ||||||
| Other—Anusol/Azasan/Diuril/Pepcid/Proctocort/Metozolv |
7,848 | 32,882 | 25,431 | |||||||||
| % of net product revenues |
3 | % | 14 | % | 14 | % | ||||||
| Net product revenues |
$ | 336,973 | $ | 232,890 | $ | 178,766 | ||||||
Net product revenues for 2010 were $337.0 million, compared to $232.9 million for 2009. The net product revenue increase from 2009 to 2010 was primarily due to:
| • | increased unit sales of Xifaxan, primarily due to the approval of Xifaxan 550mg for hepatic encephalopathy, which we began selling in the second quarter of 2010; |
| • | increased unit sales of Apriso; and |
| • | price increases on our products. |
These increases were partially offset by decreased unit sales of OsmoPrep, MoviPrep and Pepcid and a change in the estimate in the reserve for Colazal returns of approximately $7.0 million.
Total milligrams of Xifaxan prescribed during 2010 increased 49% compared to 2009. Prescription growth for 2010 compared to 2009 was a 11% increase for our purgatives. Prescriptions for MoviPrep increased 23% for
45
Table of Contents
2010 compared to prescriptions for 2009. Prescriptions for OsmoPrep for 2010 declined 27% compared to prescriptions for 2009 due to the boxed label warning announced by the FDA on December 11, 2008.
Net product revenues for 2009 were $232.9 million, compared to $178.8 million for 2008. The net product revenue increase from 2008 to 2009 was primarily due to:
| • | increased unit sales of Xifaxan; |
| • | increased unit sales of MoviPrep; |
| • | increased unit sales of Apriso; and |
| • | price increases on our products. |
These increases were partially offset by decreased unit sales of OsmoPrep and Visicol, and an adjustment in the reserve for Colazal returns.
Prescription growth for 2009 compared to 2008 was 14% for Xifaxan and 16% for our purgative franchise. Prescriptions for MoviPrep increased 71% for 2009 compared to prescriptions for 2008. Prescriptions for OsmoPrep for 2009 declined 39% compared to prescriptions for 2008 due to the boxed label warning announced by the FDA on December 11, 2008.
Costs and Expenses
Total costs and expenses were $343.2 million, $273.0 million and $225.4 million for 2010, 2009 and 2008, respectively. Higher operating expenses in absolute terms for 2010 compared to 2009 were due primarily to increased cost of products sold related to the corresponding increase in product revenue, intangible impairment charges, and increased selling, general and administrative expenses due to the expansion of our sales force from 96 sales representatives to 160 sales representatives in mid 2009, partially offset by reduced research and development expenses. Higher operating expenses in absolute terms for 2009 compared to 2008 were due primarily to increased research and development activities, increased cost of products sold related to the corresponding increase in product revenue, and increased selling, general and administrative expenses due to the expansion of our sales force from 96 sales representatives to 160 sales representatives in mid 2009.
Cost of Products Sold
Cost of products sold were $68.7 million, $52.0 million and $36.7 million for 2010, 2009 and 2008, respectively. Gross margin on total product revenue, excluding $10.4 million, $11.5 million and $9.9 million in amortization of product rights and intangible assets for 2010, 2009 and 2008, respectively, was 80%, 78% and 79% in 2010, 2009 and 2008, respectively. Included in cost of products sold for 2010 are $6.2 million of costs related to Metozolv that was expensed. The increase in cost of products sold in absolute dollars from 2009 to 2010 was primarily due to increased sales of Xifaxan and Apriso, partially offset by decreased sales of OsmoPrep and MoviPrep. The increase in cost of products sold in absolute dollars from 2008 to 2009 was primarily due to increased sales of Xifaxan, Apriso and MoviPrep, partially offset by decreased sales of OsmoPrep and Visicol. The lower gross margin in 2009 compared to 2008 is due to the product revenue mix in the respective periods. Cost of products sold does not include amortization of product rights and intangibles. Refer to “Critical Accounting Policies—Intangible Assets and Goodwill” above.
Intangible Impairment Charges
Intangible impairment charges consists of a $30.0 million impairment charge recorded during the three-month period ended June 30, 2010 related to Pepcid OS, and a $4.6 million impairment charge recorded during the three-month period ended December 31, 2010 related to Metozolv. The Pepcid charge was a result of FDA approval of two generic famotidine oral suspension products in May 2010 and June 2010. The Metozolv charge was a result of Novel filing an ANDA application to seek approval to market a generic version of Metoclopramide Hydrochloride ODT, 5 mg and 10 mg, and our determination that the applicable patent could not be enforced against Novel.
46
Table of Contents
Research and Development
Research and development expense was $73.3million, $89.5 million and $83.7 million for 2010, 2009 and 2008, respectively.
The decrease in research and development expenses from 2009 to 2010 was due primarily to:
| • | reduced expenses related to our Phase III studies of rifaximin for IBS, which were completed in late 2009; |
| • | the $5.0 million upfront collaboration payment to Lupin in 2009 that did not recur in 2010; and |
| • | reduced expenses related to our hepatic encephalopathy development program, which was substantially completed in 2009. |
This decrease was partially offset by:
| • | increased expenses related to our development programs for crofelemer and budesonide; and |
| • | increased headcount costs. |
The increase in research and development expenses from 2008 to 2009 was due primarily to:
| • | increased expenses related to our Phase III studies of rifaximin for IBS; |
| • | increased expenses related to the continuation of our development program for crofelemer, which we acquired from Napo in December 2008; |
| • | increased expenses related to our development program for budesonide; |
| • | a $5.0 million upfront payment in connection with our collaboration with Lupin; |
| • | increased expenses related to investigator-initiated studies; and |
| • | increased headcount costs. |
These increases were partially offset by:
| • | reduced expenses related to our development program for our 1100mg balsalazide tablet; |
| • | reduced expenses related to our development program for granulated mesalamine, or Apriso, which the FDA approved in October 2008; |
| • | a $5.0 million upfront payment to Napo Pharmaceuticals in connection with the acquisition of the rights to crofelemer made in 2008 that did not recur in 2009; and |
| • | reduced expenses related to our hepatic encephalopathy development program for rifaximin, because our Phase III trials were completed in mid 2009. |
Through December 31, 2010, we had incurred research and development expense of approximately $77.6 million for balsalazide, $149.9 million for rifaximin, $41.5 million for granulated mesalamine, $20.3 million for crofelemer, and $8.7 million for budesonide.
Due to the significant risks and uncertainties inherent in the clinical development and regulatory approval processes, the cost to complete projects and development timelines for their completion cannot be reasonably estimated. Enrollment in clinical trials might be delayed for reasons beyond our control, requiring additional cost and time. Results from clinical trials might not be favorable, or might require us to perform additional unplanned clinical trials, requiring additional cost and time, or resulting in termination of the project. Further, data from clinical trials is subject to varying interpretation, and might be deemed insufficient by the regulatory bodies reviewing applications for marketing approvals, requiring additional cost and time, or resulting in termination of the project. Process development and manufacturing scale-up for production of clinical and commercial product supplies might take longer and cost more than our forecasts. As such, clinical development and regulatory
47
Table of Contents
programs are subject to risks and changes that might significantly impact cost projections and timelines. We generally expect research and development costs to increase in absolute terms in future periods as we pursue additional indications and formulations for rifaximin, continue development for our budesonide product candidate and crofelemer, and if and when we acquire new products.
Selling, General and Administrative
Selling, general and administrative expenses were $156.1 million, $120.0 million and $95.1 million for 2010, 2009 and 2008, respectively. The increase from 2009 to 2010 was primarily due to:
| • | increased headcount costs, primarily due to the expansion of our sales force from 96 sales representatives to 160 sales representatives in mid 2009; |
| • | payments made to Novel in 2010 under our agreement related to MoviPrep and OsmoPrep; and |
| • | marketing expenses related to the launch of Xifaxan 550mg for hepatic encephalopathy. |
The increase from 2008 to 2009 was primarily due to:
| • | increased headcount costs, primarily due to the expansion of our sales force from 96 sales representatives to 160 sales representatives in mid 2009; |
| • | increased marketing expenses related to Apriso and MoviPrep; and |
| • | increased legal costs for to the patent litigation related to MoviPrep and OsmoPrep; |
| • | partially offset by decreased marketing costs related to balsalazide tablets and OsmoPrep. |
We expect selling, general and administrative expenses to increase in absolute terms as we expand our sales and marketing efforts for our current products, and the launch of other indications for rifaximin, if approved.
Interest Expense
Interest expense was $20.6 million, $6.7 million and $4.2 million in 2010, 2009 and 2008, respectively. Interest expense for 2010 consisted of:
| • | $0.3 million of interest expense on our credit facility, which we paid off in May 2010; and |
| • | $6.6 million of interest expense on our convertible notes issued in August 2008, including $2.9 million of amortization of debt discount; and |
| • | $13.7 million of interest expense on our 2015 convertible notes issued in June 2010, including $7.2 million of amortization of debt discount. |
Interest expense for 2009 consisted of:
| • | $0.4 million of interest expense on our credit facility; and |
| • | $6.3 million of interest expense on our convertible notes issued in August 2008, including $2.5 million of amortization of debt discount. |
Interest expense for 2008 consisted of:
| • | $1.1 million of interest expense on our credit facility; |
| • | $2.0 million of interest expense on our convertible notes issued in August 2008, including $0.6 million of amortization of debt discount; and |
| • | a $1.1 million non-cash charge to expense a portion of the unamortized costs related to the credit facility. |
48
Table of Contents
Interest and Other Income
Interest and other income was $2.6 million, $1.2 million and $3.1 million for 2010, 2009 and 2008, respectively.
Due to the current economic climate, we expect 2011 interest rates paid to us on our cash and cash equivalents will be equal to or lower than we experienced during 2010.
Provision for Income Tax
Income tax (benefit) expense was $2.9 million, ($2.0) million and ($0.1) million in 2010, 2009 and 2008, respectively. Our effective tax rate was (11.6)%, 4.4% and 0.2% in 2010, 2009 and 2008, respectively. Tax expense in 2010 was primarily a result of state income taxes.
At December 31, 2010, 2009 and 2008, we had U.S. federal net operating loss carryforwards of approximately $90.3 million, $134.3 million and $108.7 million, respectively. These carryforwards will expire on various dates beginning in 2020 through 2025 if not utilized. Utilization of the federal net operating loss and credit carryforwards might be subject to a substantial annual limitation due to the “change in ownership” provisions of the Internal Revenue Code. The annual limitation might result in the expiration of net operating losses and credits before utilization.
Quarterly Results of Operations
See Note 12 of Notes to Consolidated Financial Statements for a presentation of our quarterly results of operations for the years ended December 31, 2010 and 2009.
LIQUIDITY AND CAPITAL RESOURCES
From inception until first achieving profitability in the third quarter of 2004, we financed product development, operations and capital expenditures primarily from public and private sales of equity securities and from funding arrangements with collaborative partners. Since launching Colazal in January 2001, net product revenue has been a growing source of cash. In August 2008 we closed an offering of $60.0 million in convertible senior notes due 2028, with net proceeds of $57.3 million. In November 2009 we closed an offering of 6.3 million shares of our common stock, with net proceeds of $128.4 million. On June 3, 2010 the Company closed an offering of $345.0 million in convertible senior notes due May 15, 2015 (“2015 Notes”), with net proceeds of approximately $334.2 million. As of December 31, 2010, we had $518.0 million in cash and cash equivalents, compared to $192.5 million as of December 31, 2009.
The decline in the stock market during 2008 and 2009, and the related lack of credit availability and financial institution difficulties had a limited effect on our business. We believe our cash and cash equivalent balances should be sufficient to satisfy our cash requirements for the foreseeable future. At December 31, 2010, our cash and cash equivalents consisted primarily of demand deposits, certificates of deposit, overnight investments in Eurodollars and money market funds at reputable financial institutions, and did not include any auction rate securities. We have not realized any material loss in principal or liquidity in any of our investments to date. However, declines in the stock market and deterioration in the overall economy could lead to a decrease in demand for our marketed products, which could have an adverse effect on our business, financial condition and results of operations.
Net cash provided by operating activities was $25.5 million in 2010 and was primarily attributable to our net loss for the period, net of non-cash charges including the intangible impairment charges, offset by an increase in accounts receivable due to increased sales in the fourth quarter of 2010. Net cash used by operating activities was $56.3 million in 2009 and was primarily attributable to our net loss for the period, and an increase in accounts receivable due to increased sales in the fourth quarter of 2009. Net cash used by operating activities was $20.9 million in 2008 and was primarily attributable to our net loss for the period, and product returns and chargebacks for Colazal, partially offset by collection of accounts receivable for product revenue recognized in the fourth quarter of 2007.
49
Table of Contents
Net cash used in investing activities was $9.4 million in 2010 and was primarily due to the decrease in restricted cash as a result of our repayment of our credit facility, net of purchases of property and equipment. Net cash used in investing activities was $12.1 million in 2009 and was primarily attributable to the approval milestone payment to Wilmington Pharmaceuticals for Metozolv, and purchases of property and equipment. Net cash used in investing activities was $26.9 million in 2008 and was primarily attributable to the transfer of $15.0 million of cash to restricted cash as a result of the amendment of our credit facility, a $6.0 million milestone payment to Dr. Falk in connection with the approval of Apriso, and a $5.0 million sales milestone payment to Norgine for MoviPrep.
Net cash provided by financing activities of $290.6 million in 2010 consisted primarily of the net proceeds from the closing of our offering of 2015 Notes in June 2010 and proceeds from the exercise of stock options. Net cash provided by financing activities of $140.8 million in 2009 consisted primarily of the proceeds of our public offering of common stock closed in November 2009 and proceeds from the issuance common stock upon the exercise of stock options. Net cash provided by financing activities of $56.6 million in 2008 consisted primarily of the proceeds of our convertible debt offering closed in August 2008.
As of December 31, 2010, we had non-cancelable purchase order commitments for inventory purchases of approximately $87.3 million, which includes any minimum purchase commitments under our manufacturing agreements. We anticipate significant expenditures related to our on-going sales, marketing, product launch efforts and our on-going development efforts for rifaximin, our budesonide product candidates, crofelemer and other existing product candidates. To the extent we acquire rights to additional compounds, we will incur additional expenditures.
Our contractual commitments for non-cancelable purchase commitments of inventory, minimum lease obligations for all non-cancelable operating leases, debt and minimum capital lease obligations (including interest) as of December 31, 2010 were as follows (in thousands), reflecting the future obligations under the office leases signed in February 2011:
| Total | < 1 year | 1-3 years | 3-5 years | > 5 years | ||||||||||||||||
| Operating leases |
$ | 36,642 | $ | 2,899 | $ | 3,117 | $ | 6,554 | $ | 24,072 | ||||||||||
| Purchase commitments |
87,278 | 79,363 | 7,915 | — | — | |||||||||||||||
| 2028 convertible senior notes(1) |
118,300 | 3,300 | 6,600 | 6,600 | 101,800 | |||||||||||||||
| 2015 convertible senior notes(2) |
386,508 | 9,488 | 18,975 | 358,045 | — | |||||||||||||||
| Capital lease obligations |
501 | 410 | 91 | — | — | |||||||||||||||
| Total |
$ | 629,229 | $ | 95,460 | $ | 36,698 | $ | 371,199 | $ | 125,872 | ||||||||||
| (1) | Contractual interest and principal obligations related to our 2028 convertible senior notes total $118.3 million at December 31, 2010, including $3.3 million, $6.6 million, $6.6 million and $101.8 million due in one year or less, two to three years, four to five years, and greater than five years, respectively. If these notes had been converted at December 31, 2010 based on the closing price of our stock of $46.96 per share on that date and we chose to settle them in cash, the settlement amount would have been approximately $304.5 million. |
| (2) | Contractual interest and principal obligations related to our 2015 convertible senior notes total $386.5 million at December 31, 2010, including $9.5 million, $18.9 million, and $358.0 million due in one year or less, two to three years, and four to five years, respectively. |
We enter into license agreements with third parties that may require us to make royalty, milestone or other payments that are contingent upon the occurrence of certain future events linked to the successful development and commercialization of pharmaceutical products. Certain of the payments are contingent upon the successful achievement of an important event in the development life cycle of these pharmaceutical products, which might or might not occur. If required by the agreements, we will make royalty payments based upon a percentage of the sales of a pharmaceutical product if regulatory approval to market this product is obtained and the product is
50
Table of Contents
commercialized. Because of the contingent nature of these payments, we have not attempted to predict the amount or period in which such payments would possibly be made and thus they are not included in the table of contractual obligations.
In February 2007, we entered into a $100.0 million revolving credit facility that was to mature in February 2012. On August 4, 2008 we amended the credit facility to waive defaults that may have arisen as a result of the approval of three generic balsalazide capsule products by the Office of Generic Drugs on December 28, 2007 and reduced the credit facility to $20.0 million. On August 22, 2008 we further amended the credit facility to allow us to issue the 2028 Notes described below. On May 6, 2010, the Company repaid the $15.0 million then drawn under our credit facility and terminated the facility.
On August 22, 2008 we closed an offering of $60.0 million in convertible senior notes due 2028 (“2028 Notes”). Net proceeds from the offering were $57.3 million. The 2028 Notes are governed by an indenture, dated as of August 22, 2008, between us and U.S. Bank National Association, as trustee. The 2028 Notes bear interest at a rate of 5.5% per year, payable semiannually in arrears on February 15 and August 15 of each year, beginning on February 15, 2009. The 2028 Notes will mature on August 15, 2028, unless previously converted or repurchased in accordance with their terms prior to such date. The 2028 Notes are senior unsecured obligations, and rank (i) equally to any of our existing and future unsecured senior debt, (ii) senior to any of our future indebtedness that is expressly subordinated to these 2028 Notes, and (iii) effectively junior to any secured indebtedness to the extent of the value of the assets securing such indebtedness. We may redeem the 2028 Notes, in whole or in part, at any time after August 15, 2013 for cash equal to the principal amount of the 2028 Notes to be redeemed, plus any accrued and unpaid interest. On August 15, 2013, August 15, 2018 and August 15, 2023 or upon the occurrence of a “fundamental change”, as defined in the Indenture, the holders may require us to repurchase all or a portion of the 2028 Notes for cash at 100% of the principal amount of the 2028 Notes being purchased, plus any accrued and unpaid interest.
The 2028 Notes are convertible into approximately 6,486,000 shares of our common stock under certain circumstances prior to maturity at a conversion rate of 108.0847 shares per $1,000 principal amount of 2028 Notes, which represents a conversion price of approximately $9.25 per share, subject to adjustment under certain conditions. Holders of the 2028 Notes may convert their 2028 Notes at their option on any day prior to the close of business on the business day immediately preceding the maturity date of August 15, 2028 only if one or more of the following conditions is satisfied: (1) during any fiscal quarter commencing after September 30, 2008, if the last reported sale price of our common stock for at least 20 trading days in the period of 30 consecutive trading days ending on the last trading day of the immediately preceding fiscal quarter is equal to or more than 130% of the conversion price of the 2028 Notes on the last day of such preceding fiscal quarter; (2) during the five business day period following any five consecutive trading day period in which the trading price for the 2028 Notes, per $1,000 principal amount of the 2028 Notes, for each such trading day was less than 98% of the product of the last reported sale price of our common stock and the conversion rate of the 2028 Notes on such date; (3) if we enter into specified corporate transactions; or (4) upon a redemption notice. The first of these conditions was met as of December 31, 2010. The 2028 Notes will be convertible, regardless of whether any of the foregoing conditions has been satisfied, on or after March 15, 2028 at any time prior to the close of business on the business day immediately preceding the stated maturity date of August 15, 2028. Upon conversion, we will pay cash, shares of our common stock or a combination of cash and stock, as determined by us in our discretion.
As long as the 2028 Notes are outstanding, we are prohibited from incurring any debt other than “permitted debt”, as defined in the Indenture, except that we may incur debt in certain circumstances, including meeting a consolidated leverage ratio test and a consolidated fixed charge coverage ratio test. The 2015 Notes described below are “permitted debt” under the Indenture.
On June 3, 2010 we closed an offering of $345.0 million in convertible senior notes due 2015 (“2015 Notes”). Net proceeds from the offering were $334.2 million. The 2015 Notes are governed by an indenture, dated as of June 3, 2010, between us and U.S. Bank National Association, as trustee. The 2015 Notes bear
51
Table of Contents
interest at a rate of 2.75% per year, payable semiannually in arrears on May 15 and November 15 of each year, beginning on November 15, 2010. The 2015 Notes will mature on May 15, 2015, unless previously converted or repurchased in accordance with their terms prior to such date. The 2015 Notes are senior unsecured obligations, and rank (i) equally to any of our existing and future unsecured senior debt, (ii) senior to any of our future indebtedness that is expressly subordinated to these 2015 Notes, and (iii) effectively junior to any secured indebtedness to the extent of the value of the assets securing such indebtedness.
The 2015 Notes are convertible into approximately 7,439,000 shares of our common stock under certain circumstances prior to maturity at a conversion rate of 21.5592 shares per $1,000 principal amount of 2015 Notes, which represents a conversion price of approximately $46.38 per share, subject to adjustment under certain conditions. Holders may convert their 2015 Notes at their option on any day prior to the close of business on the business day immediately preceding the maturity date of May 15, 2015 only if one or more of the following conditions is satisfied: (1) during any fiscal quarter commencing after June 30, 2010, if the last reported sale price of our common stock for at least 20 trading days in the period of 30 consecutive trading days ending on the last trading day of the immediately preceding fiscal quarter is equal to or more than 130% of the conversion price of the 2015 Notes on the last day of such preceding fiscal quarter; (2) during the five business day period following any five consecutive trading day period in which the trading price for the 2015 Notes, per $1,000 principal amount of the 2015 Notes, for each such trading day was less than 98% of the product of the last reported sale price of our common stock and the conversion rate of the 2015 Notes on such date; or (3) if we enter into specified corporate transactions. None of these conditions had been met as of December 31, 2010. The 2015 Notes will be convertible, regardless of whether any of the foregoing conditions have been satisfied, on or after January 13, 2015 at any time prior to the close of business on the second scheduled trading day immediately preceding the stated maturity date of May 15, 2015. Upon conversion, we may pay cash, shares of our common stock or a combination of cash and stock, as determined by us at our discretion.
In connection with the issuance of the 2015 Notes, we entered into capped call transactions with certain counterparties covering approximately 7,439,000 shares of our common stock. The capped call transactions have a strike price of $46.38 and a cap price of $62.44, and are exercisable when and if the 2015 Notes are converted. If upon conversion of the 2015 Notes, the price of our common stock is above the strike price of the capped calls, the counterparties will deliver shares of our common stock and/or cash with an aggregate value approximately equal to the difference between the price of our common stock at the conversion date (as defined, with a maximum price for purposes of this calculation equal to the cap price) and the strike price, multiplied by the number of shares of our common stock related to the capped call transactions being exercised. We paid $44.3 million for these capped calls, and charged that amount to additional paid-in capital.
As of December 31, 2010, we had an accumulated deficit of $223.0 million, and cash and cash equivalent balances of $518.0 million. At December 31, 2010, cash and cash equivalents consisted primarily of demand deposits, certificates of deposit, overnight investments in Eurodollars and money market funds at reputable financial institutions, and did not include any auction rate securities. We believe our cash and cash equivalent balances should be sufficient to satisfy our cash requirements for at least the next twelve months. Based on our current projections, we believe that we will return to a positive cash flow position without requiring additional capital. However, we might seek additional debt or equity financing or both to fund our operations or acquisitions, and our actual cash needs might vary materially from those now planned because of a number of factors including: FDA and foreign regulatory processes; the status of competitive products, including potential generics; intellectual property risks; our success selling products; the results of research and development activities; establishment of and change in collaborative relationships; general economic conditions; technological advances by us and other pharmaceutical companies; whether we acquire rights to additional products; and risk associated with acquisitions. If we incur more debt, we might be restricted in our ability to raise additional capital and might be subject to financial and restrictive covenants. If we issue additional equity, our stockholders could suffer dilution. We might also enter into additional collaborative arrangements that could provide us with additional funding in the form of equity, debt, licensing, milestone and/or royalty payments. We might not be able to enter into such arrangements or raise any additional funds on terms favorable to us or at all.
52
Table of Contents
RECENTLY ISSUED ACCOUNTING PRONOUNCEMENTS
In April 2010, the FASB issued authoritative guidance on defining a milestone and determining when it may be appropriate to apply the milestone method of revenue recognition for research or development transactions. Consideration that is contingent on achievement of a milestone in its entirety may be recognized as revenue in the period in which the milestone is achieved only if the milestone is judged to meet certain criteria to be considered substantive. Milestones should be considered substantive in their entirety and may not be bifurcated. An arrangement may contain both substantive and non substantive milestones, and each milestone should be evaluated individually to determine if it is substantive. The guidance is effective on a prospective basis for milestones achieved in fiscal years, and interim periods within those years, beginning on or after June 15, 2010, with early adoption permitted. We do not expect the adoption of this guidance to have a material impact on our consolidated financial statements.
CAUTIONARY STATEMENT
We operate in a highly competitive environment that involves a number of risks, some of which are beyond our control. The following statement highlights some of these risks. For more detail, see “Item 1A . Risk Factors”.
Statements contained in this Form 10-K that are not historical facts are or might constitute forward-looking statements under the safe harbor provisions of the Private Securities Litigation Reform Act of 1995. Although we believe the expectations reflected in such forward-looking statements are based on reasonable assumptions, our expectations might not be attained. Forward-looking statements involve known and unknown risks that could cause actual results to differ materially from expected results. Factors that could cause actual results to differ materially from our expectations expressed in the report include, among others: intense competition, including from generics; the high cost and uncertainty of the research, clinical trials and other development activities involving pharmaceutical products; the unpredictability of the duration and results of regulatory review of New Drug Applications and Investigational New Drug Applications; the possible impairment of, or inability to obtain intellectual property rights and the costs of obtaining such rights from third parties; our dependence on our first nine pharmaceutical products, particularly Xifaxan and MoviPrep, and the uncertainty of market acceptance of our products; general economic conditions; our need to return to profitability; the uncertainty of obtaining, and our dependence on, third parties to manufacture and sell our products; and results of future litigation and other risk factors detailed from time to time in our other SEC filings.
Item 7A. Quantitative and Qualitative Disclosures about Market Risk
Our purchases of raw materials and finished goods are denominated primarily in U.S. dollars, and our purchases denominated in currencies other than the U.S. dollar are insignificant. Additionally, our net assets denominated in currencies other than the U.S. dollar are insignificant and have not historically exposed us to material risk associated with fluctuations in currency rates. Given these facts, we have not considered it necessary to use foreign currency contracts or other derivative instruments to manage changes in currency rates. However, these circumstances might change.
Item 8. Financial Statements and Supplementary Data
The information required by this Item is set forth in the Consolidated Financial Statements and Notes thereto beginning at page F-1 of this Report.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
53
Table of Contents
Item 9A. Control and Procedures
Conclusion Regarding the Effectiveness of Disclosure Controls and Procedures
Disclosure controls and procedures (as defined in Exchange Act Rule 13a-15(e)) are designed only to provide reasonable assurance that information to be disclosed in our Exchange Act Reports is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms. As of the end of the period covered by this report, the Company carried out an evaluation, under the supervision and with the participation of the Company’s management, including the Company’s President and Chief Executive Officer and Executive Vice President, Finance and Administration and Chief Financial Officer, of the effectiveness of the Company’s disclosure controls and procedures pursuant to Exchange Act Rule 13a-15(e). Based upon this evaluation, the Company’s President and Chief Executive Officer and Executive Vice President, Finance and Administration and Chief Financial Officer have concluded that our disclosure controls and procedures are effective to provide the reasonable assurance discussed above.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rule 13a-15(f). Our internal control over financial reporting is designed to provide reasonable assurance to our management and board of directors regarding the preparation and fair presentation of published financial statements. A control system, no matter how well designed and operated, can only provide reasonable, not absolute, assurance that the objectives of the control system are met. Because of these inherent limitations, management does not expect that our internal controls over financial reporting will prevent all error and all fraud. Under the supervision and with the participation of our management, including our CEO and our Executive Vice President, Finance and Administration and Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on our evaluation under the framework in Internal Control—Integrated Framework, our management concluded that our internal control over financial reporting was effective as of December 31, 2010.
The effectiveness of our internal control over financial reporting as of December 31, 2010, has been audited by PricewaterhouseCoopers LLP, an independent registered public accounting firm, as stated in their report which is included on page F-2 herein.
Changes in Internal Control over Financial Reporting
There was no change in our internal controls over financial reporting during the fourth quarter of the period covered by this Annual Report that has materially affected, or is reasonably likely to materially affect, our internal controls over financial reporting.
Not applicable.
54
Table of Contents
Item 10. Directors, Executive Officers and Corporate Governance
Information required by this Item concerning our directors is incorporated by reference from the section captioned “Proposal One—Election of Directors” contained in our proxy statement related to the 2011 Annual Meeting of Stockholders scheduled to be held on June 16, 2011 which we intend to file with the SEC within 120 days of the end of our fiscal year pursuant to General Instruction G(3) of Form 10-K.
The Board of Directors has determined that the members of the Audit Committee are independent as defined in Rule 4200(a)(15) of the National Association of Securities Dealers’ listing standards. The Board of Directors has also determined that John F. Chappell, Thomas W. D’Alonzo and William P. Keane are “audit committee financial experts” as defined in Item 407 (d)(5) of Regulation S-K.
Our Board of Directors adopted a code of conduct that applies to all of our directors and employees. Our Board also adopted a separate code of ethics for our Chief Executive Officer, Chief Financial Officer, Chief Accounting Officer and Controller, or persons performing similar functions. We will provide copies of our code of conduct and code of ethics without charge upon request. To obtain a copy of our code of conduct and code of ethics, please send your written request to Salix Pharmaceuticals, Ltd., 1700 Perimeter Park Drive, Morrisville, NC 27560, Attn: General Counsel. In addition, you can find those codes on our website at www.salix.com/pdf/Salix_Code_Bus_Cond.pdf.
The information required by this Item concerning executive officers of the Registrant is set forth at the end of Part I of this report.
The information required by this Item concerning compliance with Section 16(a) of the United States Securities Exchange Act of 1934, as amended, is incorporated by reference from the section of the proxy statement captioned “—Section 16(a) Beneficial Ownership Reporting Compliance.”
Item 11. Executive Compensation
The information required by this Item is incorporated by reference to the information under the sections captioned “—Grant of Plan Based Awards for 2010,” “—Outstanding Equity Awards at 2010 Fiscal Year End,” “—Option Exercises and Stock Vested in 2010,” “—Director Compensation For 2010,” “—Compensation Discussion and Analysis,” “—Summary Compensation Table,” “—Compensation Committee Report,” and “—Compensation Committee Interlocks and Insider Participation” contained in the proxy statement.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The following table sets forth the indicated information as of December 31, 2010 with respect to our equity compensation plans:
| Plan Category |
(a) Number of Securities to be issued Upon Exercise of Outstanding Options, Warrants and Rights, or Vesting of Restricted Shares |
(b) Weighted Average Exercise Price of Outstanding Options, Warrants, Rights and Restricted Shares |
(c) Number of Securities Remaining Available for Future Issuance Under Equity Compensation Plans (Excluding Securities Related in Column (a)) |
|||||||||
| Equity Compensation Plans Approved by Security Holders |
3,752,211 | $ | 16.30 | 1,551,571 | ||||||||
| Equity Compensation Plans Not Approved by Security Holders |
— | — | — | |||||||||
| Total |
3,752,211 | $ | 16.30 | 1,551,571 | ||||||||
55
Table of Contents
The other information required by this Item is incorporated by reference to the information under the section captioned “—Security Ownership of Management and Certain Beneficial Owners” contained in the proxy statement.
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information required by this Item is incorporated by reference to the information under the section captioned “—Transactions with Related Persons” and “Proposal One—Election of Directors—Corporate Governance Matters” contained in the proxy statement.
Item 14. Principal Accountant Fees and Services
The information required by this Item is incorporated by reference to the information under the section captioned “—Audit Committee Report” contained in the proxy statement.
56
Table of Contents
Item 15. Exhibits and Financial Statement Schedules
(a) 1. Financial Statements
The following statements are filed as part of this report:
| Page | ||||
| F-2 | ||||
| F-3 | ||||
| F-4 | ||||
| F-5 | ||||
| F-6 | ||||
| F-7 | ||||
2. Financial Statement Schedules
| F-32 |
Schedules not listed above have been omitted because the information required to be set forth therein is not applicable or is shown in the financial statements or notes thereto.
3. Exhibits
| Exhibit |
Description of Document |
Registrant’s Form |
Dated | Exhibit Number |
Filed Herewith |
|||||||||||||
| 1.1 | Purchase Agreement dated August 18, 2008 by and between Salix Pharmaceuticals, Ltd. and Banc of America Securities LLC. | 8-K | 8/22/08 | 1.1 | ||||||||||||||
| 2.1 | Agreement and Plan of Merger dated June 23, 2005 among Salix Pharmaceuticals, Ltd., InKine Pharmaceutical Company, Inc. and Metal Acquisition Corp. | 8-K | 6/24/05 | 2.1 | ||||||||||||||
| 2.2 | Certificate of Domestication. | S-3 | 02/12/02 | 2.1 | ||||||||||||||
| 2.3* | Asset Purchase Agreement dated June 30, 2004 between King Pharmaceuticals, Inc., Monarch Pharmaceuticals, Inc., Parkedale Pharmaceuticals, Inc., Salix Pharmaceuticals, Inc. and Salix Pharmaceuticals, Ltd. | 10-Q | 08/09/04 | 2.2 | ||||||||||||||
| 3.1 | Certificate of Incorporation, as amended. | X | ||||||||||||||||
| 3.2 | Amended and Restated Bylaws. | 8-K | 09/02/03 | 3.2 | ||||||||||||||
| 4.1 | Indenture dated August 22, 2008 by and between Salix Pharmaceuticals, Ltd. and U.S Bank, National Association. | 8-K | 08/22/08 | 4.1 | ||||||||||||||
| 4.2 | Form of 5.5% Convertible Senior Note due 2028 (included in Exhibit 4.1). | 8-K | 08/22/08 | 4.2 | ||||||||||||||
| 10.3 | Form of 1996 Stock Plan for Salix Holdings, Ltd., as amended September 2000 and form of Notice of Stock Option Grant and Stock Option Agreement thereunder, as amended March 12, 2001. | 10-Q | 08/09/04 | 10.3 | ||||||||||||||
57
Table of Contents
| Exhibit |
Description of Document |
Registrant’s Form |
Dated | Exhibit Number |
Filed Herewith |
|||||||||||||
| 10.4* | Amendment Agreement effective as of September 17, 1992 by and among Glycyx Pharmaceuticals, Ltd., Salix Pharmaceuticals, Inc. and Biorex. | S-1 | 08/15/97 | 10.4 | ||||||||||||||
| 10.5* | License Agreement, dated September 17, 1992 between Biorex Laboratories Limited and Glycyx Pharmaceuticals, Ltd. and letter agreement amendments thereto. | S-1 | 08/15/97 | 10.5 | ||||||||||||||
| 10.6* | Research and Development Agreement dated September 21, 1992 between Glycyx Pharmaceuticals, Ltd. and AB Astra and letter agreement amendments thereto. | S-1 | 08/15/97 | 10.6 | ||||||||||||||
| 10.7* | Distribution Agreement dated September 21, 1992 between Glycyx Pharmaceuticals, Ltd. and AB Astra. | S-1 | 08/15/97 | 10.7 | ||||||||||||||
| 10.8* | Amended and Restated License Agreement by and between Salix Pharmaceuticals, Inc. and Biorex Laboratories, Limited, dated April 16, 1993. | S-1 | 08/15/97 | 10.8 | ||||||||||||||
| 10.9* | Co-Participation Agreement, dated April 30, 1993 between Salix Pharmaceuticals, Inc. and AB Astra as amended by Amendment No. 1 thereto effective September 30, 1993. | S-1 | 08/15/97 | 10.9 | ||||||||||||||
| 10.9.1 | Letter Agreement dated October 16, 1998 to Co-Participation Agreement dated April 30, 1993 by and between Salix Pharmaceuticals, Inc. and AB Astra. | 10-Q | 11/16/98 | 10.9.1 | ||||||||||||||
| 10.11* | Distribution Agreement, dated September 23, 1994 between Glycyx Pharmaceuticals, Ltd. and Menarini International Operations Luxembourg SA and amendments thereto. | S-1 | 08/15/97 | 10.11 | ||||||||||||||
| 10.12* | License Agreement, dated June 24, 1996, between Alfa Wassermann S.p.A. and Salix Pharmaceuticals, Ltd. | S-1 | 08/15/97 | 10.12 | ||||||||||||||
| 10.13* | Supply Agreement, dated June 24, 1996, between Alfa Wassermann S.p.A. and Salix Pharmaceuticals, Ltd. | S-1 | 08/15/97 | 10.13 | ||||||||||||||
| 10.22 | Termination and Settlement Agreement dated as of December 22, 1999, by and between Astra AB and Salix Pharmaceuticals Inc. (a wholly owned subsidiary of Salix Pharmaceuticals, Ltd.). | 8-K | 12/28/99 | 10.22 | ||||||||||||||
| 10.23 | Agreement dated December 22, 1999, between Glycyx Pharmaceuticals, Ltd. and Astra AB. | 8-K | 12/28/99 | 10.23 | ||||||||||||||
| 10.25* | Agreement dated May 17, 2000 between Glycyx Pharmaceuticals, Ltd. and Shire Pharmaceuticals Group plc. | 10-Q | 08/14/00 | 10.25 | ||||||||||||||
| 10.26* | Agreement dated May 17, 2000 between Biorex Laboratories Limited and Glycyx Pharmaceuticals, Ltd. | 10-Q | 08/14/00 | 10.26 | ||||||||||||||
58
Table of Contents
| Exhibit |
Description of Document |
Registrant’s Form |
Dated | Exhibit Number |
Filed Herewith |
|||||||||||||
| 10.29 | Lease Agreement dated June 30, 2000 by and between Colonnade Development, LLC and Salix Pharmaceuticals, Inc. | 10-Q | 08/14/01 | 10.29 | ||||||||||||||
| 10.30* | License Agreement between Biorex Laboratories Limited and Glycyx Pharmaceuticals, Ltd. dated August 22, 2001. | 10-Q | 11/14/01 | 10.30 | ||||||||||||||
| 10.31 | Form of Employment Agreement for executive officers. | 10-Q | 11/14/01 | 10.31 | ||||||||||||||
| 10.32* | License Agreement by and between Salix Pharmaceuticals, Inc. and Dr. Falk Pharma GmbH dated July 15, 2002. | 10-Q | 11/14/02 | 10.32 | ||||||||||||||
| 10.36 | Rights Agreement, dated as of January 10, 2003, between Salix Pharmaceuticals, Ltd. and Computershare Investor Services LLC, as Rights Agent. | 8-K | 01/10/03 | 10.36 | ||||||||||||||
| 10.37 | Common Stock Purchase Agreement dated November 6, 2003 among Salix Pharmaceuticals, Ltd. and the investors listed therein. | 8-K | 11/10/03 | 10.37 | ||||||||||||||
| 10.39* | License Agreement dated October 17, 2003, between Glycyx Pharmaceuticals, Ltd (a wholly owned subsidiary of Salix Pharmaceuticals, Ltd.) and Chong Kun Dang Pharmaceutical Corporation. | 10-Q | 11/14/03 | 10.39 | ||||||||||||||
| 10.40* | Amendment Agreement dated November 24, 2003 between Salix Pharmaceuticals, Inc. and Dr. Falk Pharma Gmbh. | 10-K | 03/12/04 | 10.40 | ||||||||||||||
| 10.41* | License Agreement dated October 31, 2003 between aaiPharma LLC, aaiPharma Inc. and Salix Pharmaceuticals, Ltd. | 10-K | 03/12/04 | 10.41 | ||||||||||||||
| 10.44* | Supply Agreement dated June 30, 2004 between King Pharmaceuticals, Inc., Parkedale Pharmaceuticals, Inc., Salix Pharmaceuticals, Inc. and Salix Pharmaceuticals, Ltd. | 10-Q | 08/09/04 | 10.44 | ||||||||||||||
| 10.45 | License Assignment and Consent Agreement dated June 30, 2004 between Parkedale Pharmaceuticals, Inc., King Pharmaceuticals, Inc., Salix Pharmaceuticals, Inc., Salix Pharmaceuticals, Ltd., Warner-Lambert Company LLC and Parke, Davis & Company LLC. | 10-Q | 08/09/04 | 10.45 | ||||||||||||||
| 10.46 | Assignment of Trademarks Agreement dated June 30, 2004 between Parkedale Pharmaceuticals, Inc. and Salix Pharmaceuticals, Inc. | 10-Q | 08/09/04 | 10.46 | ||||||||||||||
| 10.47 | License Agreement dated June 30, 2004 between Monarch Pharmaceuticals, Inc., Parkedale Pharmaceuticals, Inc., King Pharmaceuticals, Inc., Salix Pharmaceuticals, Inc. and Salix Pharmaceuticals, Ltd. | 10-Q | 08/09/04 | 10.47 | ||||||||||||||
59
Table of Contents
| Exhibit |
Description of Document |
Registrant’s Form |
Dated | Exhibit Number |
Filed Herewith |
|||||||||||||
| 10.48 | Office lease dated as of November 24, 2004 between Salix Pharmaceuticals, Ltd. And Duke Realty Limited Partnership. | 8-K | 12/13/04 | 10.48 | ||||||||||||||
| 10.49 | Co-Promotion Agreement dated March 2, 2005 between Salix Pharmaceuticals, Inc. and Altana Pharma US, Inc. | 10-Q/A | 08/23/05 | 10.49 | ||||||||||||||
| 10.50 | 2005 Stock Plan and forms of Notice of Option Grant and Stock Option Agreement. | S-8 | 06/30/05 | 10.50 | ||||||||||||||
| 10.51 | Termination Agreement dated August 19, 2005 between Salix Pharmaceuticals, Inc. and Altana Pharma US, Inc. | 8-K | 08/22/05 | 10.51 | ||||||||||||||
| 10.53 | License and Supply Agreement dated as of December 7, 2005 between Salix Pharmaceuticals, Inc. and Norgine B.V. | 8-K | 12/13/05 | 10.53 | ||||||||||||||
| 10.54 | Form of Restricted Stock Grant to be granted pursuant to the 2005 Stock Plan. | 10-K | 03/16/06 | 10.54 | ||||||||||||||
| 10.55 | License Agreement entered into on June 18, 2006 between Cedars-Sinai Medical Center and Salix Pharmaceuticals, Inc. | 8-K | 07/05/06 | 10.55 | ||||||||||||||
| 10.56 | Development and License Agreement, dated September 5, 2006 with Debiovision Inc. | 10-Q | 11/09/06 | 10.56 | ||||||||||||||
| 10.57 | $100.0 million Credit Facility by and among Salix Pharmaceuticals, Ltd., Bank of America, N.A., as Administrative Agent, Swing Line Lender and L/C Issuer, Wachovia Bank, National Association, as Syndication Agent, and RBC Centura Bank, as Documentation Agent, and the Other Lenders Party thereto, and Banc of America Securities LLC, as Sole Lead Arranger and Sole Book Manager, dated February 22, 2007. | 8-K | 02/28/06 | 10.57 | ||||||||||||||
| 10.58* | Master Purchase and Sale and License Agreement dated February 22, 2007 between Merck & Co., Inc. and Salix Pharmaceuticals, Ltd. | 10-Q | 5/10/07 | 10.58 | ||||||||||||||
| 10.59* | License Agreement dated April 16, 2007 between Salix Pharmaceuticals, Inc. and Dr. Falk Pharma GmbH. | 10-Q | 5/10/07 | 10.59 | ||||||||||||||
| 10.60* | Co-Promotion Agreement dated September 4, 2007, with Eisai Inc. | 10-Q | 8/09/07 | 10.60 | ||||||||||||||
| 10.61** | Supply and Distribution Agreement between Salix Pharmaceuticals, Inc. and Watson Pharma, Inc. | 10-K | 3/14/08 | 10.61 | ||||||||||||||
| 10.62* | License Agreement dated March 13, 2008 between Dr. Falk Pharma GmbH and Salix Pharmaceuticals, Inc. | 10-Q | 5/07/08 | 10.62 | ||||||||||||||
60
Table of Contents
| Exhibit |
Description of Document |
Registrant’s Form |
Dated | Exhibit Number |
Filed Herewith |
|||||||||||||
| 10.63 | Waiver and First Amendment to Credit Agreement, by and among Salix Pharmaceuticals, Ltd., Bank of America, N.A., as Administrative Agent, and the lender parties thereto, dated as of August 4, 2008 | 10-Q | 8/05/08 | 10.63 | ||||||||||||||
| 10.64** | Collaboration Agreement between Napo Pharmaceuticals, Inc. and Salix Pharmaceuticals, Inc. | 10-K | 3/11/09 | 10.64 | ||||||||||||||
| 10.65** | Manufacturing and Supply Agreement between Salix Pharmaceuticals, Inc. and Glenmark Pharmaceuticals Ltd. | 10-K | 3/11/09 | 10.65 | ||||||||||||||
| 10.66* | Commercial Manufacturing Agreement between Salix Pharmaceuticals, Inc. and Catalent Pharma Solutions, LLC | 10-Q | 8/17/09 | 10.66 | ||||||||||||||
| 10.67* | Development, Commercialization and License Agreement Between Lupin Ltd. and Salix Pharmaceuticals, Inc. | 10-Q | 11/09/09 | 10.67 | ||||||||||||||
| 10.68* | Rifaximin Manufacturing and Supply Agreement Between Salix Pharmaceuticals, Inc. and Lupin Ltd. | 10-Q | 11/09/09 | 10.68 | ||||||||||||||
| 10.69 | Confirmation for base capped call transaction dated as of May 27, 2010, between Bank of America, N.A. and the Company. | 8-K | 6/03/10 | 10.69 | ||||||||||||||
| 10.70 | Confirmation for additional capped call transaction dated as of June 2, 2010, between Bank of America N.A. and the Company. | 8-K | 6/03/10 | 10.70 | ||||||||||||||
| 10.71** | Settlement Agreement dated August 27, 2010, by and among Salix Pharmaceuticals, Inc., Norgine B.V., Norgine Europe B.V., Novel Laboratories, Inc., and Actavis Inc. | 10-Q | 11/09/10 | 10.71 | ||||||||||||||
| 10.72** | Sublicense Agreement dated August 27, 2010, by and among Salix Pharmaceuticals, Inc., Norgine B.V., Norgine Europe B.V., and Novel Laboratories, Inc. | 10-Q | 11/09/10 | 10.72 | ||||||||||||||
| 10.73** | Supply Agreement dated August 27, 2010, by and among Salix Pharmaceuticals, Inc, Actavis Inc., and Novel Laboratories, Inc. | 10-Q | 11/09/10 | 10.73 | ||||||||||||||
| 10.74** | First Amendment to License and Supply Agreement dated August 27, 2010, by and between Norgine B.V. and Salix Pharmaceuticals, Inc. | 10-Q | 11/09/10 | 10.74 | ||||||||||||||
| 10.75** | Settlement Agreement dated September 29, 2010, by and among Salix Pharmaceuticals, Inc., CDC III, LLC, a general partnership of Craig Aronchick, William H. Lipshutz and Scott H. Wright, Novel Laboratories, Inc., and Actavis Inc. | 10-Q | 11/09/10 | 10.75 | ||||||||||||||
61
Table of Contents
| Exhibit |
Description of Document |
Registrant’s Form |
Dated | Exhibit Number |
Filed Herewith |
|||||||||||||
| 10.76** | Sublicense Agreement dated September 29, 2010, by and among Salix Pharmaceuticals, Inc., CDC III, LLC, a general partnership of Craig Aronchick, William H. Lipshutz and Scott H. Wright, and Novel Laboratories, Inc. | 10-Q | 11/09/10 | 10.76 | ||||||||||||||
| 10.77** | Supply Agreement dated September 29, 2010, by and between Salix Pharmaceuticals, Inc. and Novel Laboratories, Inc. | 10-Q | 11/09/10 | 10.77 | ||||||||||||||
| 10.78** | Second Amendment to License Agreement dated September 29, 2010, by and among CDC III, LLC, a general partnership of Craig Aronchick, William H. Lipshutz and Scott H. Wright, and Salix Pharmaceuticals, Inc. | 10-Q | 11/09/10 | 10.78 | ||||||||||||||
| 10.79** | License Agreement dated October 19, 2010 by and between Photocure ASA, a Norwegian corporation and Salix Pharmaceuticals, Inc. | X | ||||||||||||||||
| 21.1 | Subsidiaries of the Registrant. | X | ||||||||||||||||
| 23.1 | Consent of Independent Registered Public Accounting Firm. | X | ||||||||||||||||
| 31.1 | Certification by the Chief Executive Officer pursuant to Section 240.13a-14 or Section 240.15d-14 of the Securities and Exchange Act of 1934, as amended. | X | ||||||||||||||||
| 31.2 | Certification by the Chief Financial Officer pursuant to Section 240.13a-14 or Section 240.15d-14 of the Securities and Exchange Act of 1934, as amended. | X | ||||||||||||||||
| 32.1 | Certification by the Chief Executive Officer pursuant to 18 U.S.C. 1350 as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | X | ||||||||||||||||
| 32.2 | Certification by the Chief Financial Officer pursuant to 18 U.S.C. 1350 as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | X | ||||||||||||||||
| * | The registrant has received confidential treatment with respect to portions of this exhibit. Those portions have been omitted from this exhibit and filed separately with the U.S. Securities and Exchange Commission. |
| ** | The registrant has requested confidential treatment with respect to portions of this exhibit. Those portions have been omitted from the exhibit and filed separately with the U.S. Securities and Exchange Commission. |
(b) Exhibits
See Item 15(a)(3) above.
(c) Financial Statement Schedules
See Item 15(a)(1) above.
62
Table of Contents
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this Form 10-K to be signed on its behalf by the undersigned, thereunto duly authorized.
| SALIX PHARMACEUTICALS, LTD. |
| /s/ CAROLYN J. LOGAN |
| Carolyn J. Logan President and Chief Executive Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, this Form 10-K has been signed below by the following persons on behalf of the Registrant and on the dates indicated.
| Date: February 28, 2011 |
/s/ CAROLYN J. LOGAN | |||
| Carolyn J. Logan President, Chief Executive Officer (Principal Executive Officer) and Director | ||||
| Date: February 28, 2011 |
/s/ ADAM C. DERBYSHIRE | |||
| Adam C. Derbyshire Executive Vice President, Finance & Administration and Chief Financial Officer (Principal Financial and Accounting Officer) | ||||
| Date: February 28, 2011 |
/s/ JOHN F. CHAPPELL | |||
| John F. Chappell Chairman of the Board | ||||
| Date: February 28, 2011 |
/s/ THOMAS W. D’ALONZO | |||
| Thomas W. D’Alonzo Director | ||||
| Date: February 28, 2011 |
/s/ RICHARD A. FRANCO | |||
| Richard A. Franco Director | ||||
| Date: February 28, 2011 |
/s/ WILLIAM P. KEANE | |||
| William P. Keane Director | ||||
| Date: February 28, 2011 |
/s/ MARK A. SIRGO | |||
| Mark A. Sirgo Director | ||||
63
Table of Contents
Index to Consolidated Financial Statements
| PAGE | ||||
| AUDITED CONSOLIDATED FINANCIAL STATEMENTS |
||||
| F-2 | ||||
| F-3 | ||||
| F-4 | ||||
| F-5 | ||||
| F-6 | ||||
| F-7 | ||||
| CONSOLIDATED FINANCIAL STATEMENT SCHEDULE |
||||
| F-31 | ||||
F-1
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To Board of Directors and Stockholders of
Salix Pharmaceuticals, Ltd.
In our opinion, the accompanying consolidated balance sheet and the related consolidated statements of operations, stockholders’ equity and cash flows present fairly, in all material respects, the financial position of Salix Pharmaceuticals, Ltd. at December 31, 2010 and 2009, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2010 in conformity with accounting principles generally accepted in the United States of America. In addition, in our opinion, the financial statement schedule listed in the index appearing under Item 15(a)(2) presents fairly, in all material respects, the information set forth therein when read in conjunction with the related consolidated financial statements. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company’s management is responsible for these financial statements and financial statement schedule, for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in Management’s Report on Internal Control over Financial Reporting appearing in Item 9A. Our responsibility is to express opinions on these financial statements, on the financial statement schedule and on the Company’s internal control over financial reporting based on our integrated audits. We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ PricewaterhouseCoopers LLP
Raleigh, North Carolina
March 1, 2011
F-2
Table of Contents
Consolidated Balance Sheets
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| (U.S. dollars, in thousands, except share and per share amounts) |
||||||||
| A S S E T S |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 518,030 | $ | 192,512 | ||||
| Accounts receivable, net |
140,177 | 98,248 | ||||||
| Inventory, net |
16,021 | 24,341 | ||||||
| Prepaid and other current assets |
11,054 | 10,918 | ||||||
| Total current assets |
685,282 | 326,019 | ||||||
| Property and equipment, net |
7,897 | 5,349 | ||||||
| Restricted cash |
— | 15,000 | ||||||
| Goodwill |
85,257 | 85,257 | ||||||
| Product rights and intangibles, net |
64,577 | 109,603 | ||||||
| Other assets |
8,530 | 1,812 | ||||||
| Total assets |
$ | 851,543 | $ | 543,040 | ||||
|
L I A B I L I T I E S A N D S T O C K H O L D E R S’ E Q U I T Y |
||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 18,475 | $ | 15,645 | ||||
| Accrued liabilities |
45,759 | 59,985 | ||||||
| Reserve for product returns, rebates and chargebacks |
60,538 | 32,041 | ||||||
| Current portion of capital lease obligations |
400 | 811 | ||||||
| Total current liabilities |
125,172 | 108,482 | ||||||
| Long-term liabilities: |
||||||||
| Convertible senior notes |
323,005 | 47,299 | ||||||
| Borrowings under credit facility |
— | 15,000 | ||||||
| Lease incentive obligation |
1,357 | 1,736 | ||||||
| Long term portion of capital lease obligations |
90 | 499 | ||||||
| Total long-term liabilities |
324,452 | 64,534 | ||||||
| Stockholders’ equity: |
||||||||
| Preferred stock, $0.001 par value; 5,000,000 shares authorized, issuable in series, none outstanding |
— | — | ||||||
| Common stock, $0.001 par value; 150,000,000 shares authorized, 58,139,941 and 56,245,759 shares issued and outstanding at December 31, 2010 and 2009, respectively |
58 | 56 | ||||||
| Additional paid-in-capital |
624,886 | 565,932 | ||||||
| Accumulated deficit |
(223,025 | ) | (195,964 | ) | ||||
| Total stockholders’ equity |
401,919 | 370,024 | ||||||
| Total liabilities and stockholders’ equity |
$ | 851,543 | $ | 543,040 | ||||
The accompanying notes are an integral part of these consolidated financial statements
F-3
Table of Contents
Consolidated Statements of Operations
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| (U.S. dollars, in thousands, except per share data) |
||||||||||||
| Revenues: |
||||||||||||
| Net product revenues |
$ | 336,973 | $ | 232,890 | $ | 178,766 | ||||||
| Costs and expenses: |
||||||||||||
| Cost of products sold (excluding $10,370, $11,485 and $9,891 in amortization of product rights and intangible assets for the years ended December 31, 2010, 2009 and 2008, respectively) |
68,677 | 52,025 | 36,710 | |||||||||
| Amortization of product rights and intangible assets |
10,370 | 11,485 | 9,891 | |||||||||
| Intangible impairment charges |
34,656 | — | — | |||||||||
| Research and development |
73,346 | 89,466 | 83,735 | |||||||||
| Selling, general and administrative |
156,101 | 120,020 | 95,088 | |||||||||
| Total costs and expenses |
343,150 | 272,996 | 225,424 | |||||||||
| Loss from operations |
(6,177 | ) | (40,106 | ) | (46,658 | ) | ||||||
| Interest expense |
(20,652 | ) | (6,746 | ) | (3,755 | ) | ||||||
| Interest and other income |
2,626 | 1,221 | 2,690 | |||||||||
| Loss before income tax |
(24,203 | ) | (45,631 | ) | (47,723 | ) | ||||||
| Income tax (expense) benefit |
(2,858 | ) | 2,012 | 116 | ||||||||
| Net loss |
$ | (27,061 | ) | $ | (43,619 | ) | $ | (47,607 | ) | |||
| Net loss per share, basic |
$ | (0.47 | ) | $ | (0.88 | ) | $ | (0.99 | ) | |||
| Net loss per share, diluted |
$ | (0.47 | ) | $ | (0.88 | ) | $ | (0.99 | ) | |||
| Shares used in computing net loss per share, basic |
57,300 | 49,350 | 47,898 | |||||||||
| Shares used in computing net loss per share, diluted |
57,300 | 49,350 | 47,898 | |||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-4
Table of Contents
Consolidated Statements of Stockholders’ Equity
| Common Stock | Additional Paid-in- capital |
Accumulated Deficit |
Total Stockholders’ Equity |
|||||||||||||||||
| Shares | Amount | |||||||||||||||||||
| (U.S. dollars, in thousands, except share amounts) | ||||||||||||||||||||
| Balance at December 31, 2007 |
47,708,985 | $ | 47 | $ | 397,261 | $ | (104,738 | ) | $ | 292,570 | ||||||||||
| Issuance of common stock upon exercise of stock options |
67,713 | — | 443 | — | 443 | |||||||||||||||
| Payments related to net settlement of stock-based awards |
— | — | (195 | ) | — | (195 | ) | |||||||||||||
| Issuance of common stock upon vesting of restricted stock |
301,502 | 1 | — | — | 1 | |||||||||||||||
| Income tax benefit from non-qualified stock option exercises |
— | — | 285 | — | 285 | |||||||||||||||
| Issuance of convertible debt |
— | — | 15,148 | — | 15,148 | |||||||||||||||
| Compensation expense related to restricted stock awards |
— | — | 4,756 | — | 4,756 | |||||||||||||||
| Net loss |
— | — | — | (47,607 | ) | (47,607 | ) | |||||||||||||
| Balance at December 31, 2008 |
48,078,200 | 48 | 417,698 | (152,345 | ) | 265,401 | ||||||||||||||
| Issuance of common stock upon exercise of stock options |
1,284,846 | 1 | 14,205 | — | 14,206 | |||||||||||||||
| Payments related to net settlements of stock-based awards |
— | — | (566 | ) | — | (566 | ) | |||||||||||||
| Issuance of common stock upon vesting of restricted stock |
557,713 | 1 | — | — | 1 | |||||||||||||||
| Issuance of common stock in public offering |
6,325,000 | 6 | 128,411 | — | 128,417 | |||||||||||||||
| Income tax expense from non-qualified stock option exercises |
— | — | (252 | ) | — | (252 | ) | |||||||||||||
| Compensation expense related to restricted stock awards |
— | — | 6,436 | — | 6,436 | |||||||||||||||
| Net loss |
— | — | — | (43,619 | ) | (43,619 | ) | |||||||||||||
| Balance at December 31, 2009 |
56,245,759 | 56 | 565,932 | (195,964 | ) | 370,024 | ||||||||||||||
| Issuance of common stock upon exercise of stock options |
1,140,862 | 1 | 17,120 | — | 17,121 | |||||||||||||||
| Payments related to net settlements of stock-based awards |
— | — | (2,952 | ) | — | (2,952 | ) | |||||||||||||
| Issuance of common stock upon vesting of restricted stock |
753,320 | 1 | (1 | ) | — | — | ||||||||||||||
| Purchase of call options |
— | — | (44,333 | ) | — | (44,333 | ) | |||||||||||||
| Issuance of convertible debt |
— | — | 76,923 | — | 76,923 | |||||||||||||||
| Income tax expense from non-qualified stock option exercises |
— | — | 2,257 | — | 2,257 | |||||||||||||||
| Compensation expense related to restricted stock awards |
— | — | 9,940 | — | 9,940 | |||||||||||||||
| Net loss |
— | — | — | (27,061 | ) | (27,061 | ) | |||||||||||||
| Balance at December 31, 2010 |
58,139,941 | $ | 58 | $ | 624,886 | $ | (223,025 | ) | $ | 401,919 | ||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-5
Table of Contents
Consolidated Statements of Cash Flows
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| (U.S. dollars, in thousands) | ||||||||||||
| Cash Flows from Operating Activities |
||||||||||||
| Net income loss |
$ | (27,061 | ) | $ | (43,619 | ) | $ | (47,607 | ) | |||
| Adjustments to reconcile net loss to net cash provided (used) by operating activities: |
||||||||||||
| Loss/(gain) on disposal of property and equipment |
(59 | ) | (4 | ) | — | |||||||
| Depreciation and amortization |
13,315 | 14,500 | 12,973 | |||||||||
| Intangible impairment charge |
34,656 | — | — | |||||||||
| Amortization of debt discount |
10,124 | 2,540 | 630 | |||||||||
| Stock-based compensation expense |
9,940 | 6,436 | 4,756 | |||||||||
| Changes in operating assets and liabilities: |
||||||||||||
| Accounts receivable, inventory, prepaid expenses and other assets |
(32,119 | ) | (66,916 | ) | 22,591 | |||||||
| Accounts payable and accrued liabilities |
(11,775 | ) | 32,717 | 4,397 | ||||||||
| Reserve for product returns, rebates and chargebacks |
28,497 | (1,993 | ) | (18,623 | ) | |||||||
| Net cash provided (used) by operating activities |
25,518 | (56,339 | ) | (20,883 | ) | |||||||
| Cash Flows from Investing Activities |
||||||||||||
| Purchases of property and equipment |
(5,572 | ) | (2,860 | ) | (856 | ) | ||||||
| Purchase of product rights, intangibles and other assets |
— | (9,266 | ) | (11,000 | ) | |||||||
| Decrease (increase) in restricted cash |
15,000 | — | (15,000 | ) | ||||||||
| Net cash provided (used) by investing activities |
9,428 | (12,126 | ) | (26,856 | ) | |||||||
| Cash Flows from Financing Activities |
||||||||||||
| Principal payments on credit facility |
(15,000 | ) | — | — | ||||||||
| Net proceeds from convertible senior note offering |
345,000 | — | 57,266 | |||||||||
| Debt issuance costs |
(10,839 | ) | — | — | ||||||||
| Purchase of call options |
(44,333 | ) | — | — | ||||||||
| Proceeds from offering of common stock |
— | 128,417 | — | |||||||||
| Principal payments on capital lease obligations |
(682 | ) | (981 | ) | (1,179 | ) | ||||||
| Excess tax benefit (expense) from stock-based compensation |
2,257 | (252 | ) | 285 | ||||||||
| Payments related to net settlement of stock-based awards |
(2,952 | ) | (566 | ) | (195 | ) | ||||||
| Proceeds from issuance of common stock upon exercise of stock awards |
17,121 | 14,206 | 443 | |||||||||
| Net cash provided by financing activities |
290,572 | 140,824 | 56,620 | |||||||||
| Net increase in cash and cash equivalents |
325,518 | 72,359 | 8,881 | |||||||||
| Cash and cash equivalents at beginning of year |
192,512 | 120,153 | 111,272 | |||||||||
| Cash and cash equivalents at end of year |
$ | 518,030 | $ | 192,512 | $ | 120,153 | ||||||
| Supplemental Disclosure of Cash Flow Information |
||||||||||||
| Cash paid (refunded) for income taxes |
$ | (1,587 | ) | $ | 1,822 | $ | 591 | |||||
| Cash paid for interest |
$ | 7,655 | $ | 3,572 | $ | 875 | ||||||
Supplemental Non-Cash Disclosure
At December 31, 2009, $5.0 million is included as an accrued liability that was paid to Norgine in January 2010. This related to the milestone payment for achievement of a sales milestone.
The accompanying notes are an integral part of these consolidated financial statements.
F-6
Table of Contents
Notes to Consolidated Financial Statements
December 31, 2010
(1) ORGANIZATION AND BASIS OF PRESENTATION
Salix Pharmaceuticals, Ltd., a Delaware corporation (“Salix” or the “Company”), is a specialty pharmaceutical company dedicated to acquiring, developing and commercializing prescription drugs used in the treatment of a variety of gastrointestinal diseases, which are those affecting the digestive tract.
These consolidated financial statements are stated in U.S. dollars and are prepared under accounting principles generally accepted in the United States. The accompanying consolidated financial statements include the accounts of the Company and its wholly owned subsidiaries. All significant inter-company balances and transactions have been eliminated in the consolidation.
The accompanying consolidated financial statements include all adjustments that, in the opinion of management, are necessary for a fair presentation of financial position, results of operations, and cash flows.
(2) SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Use of Estimates
The preparation of consolidated financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the amounts of assets and liabilities reported at the date of the financial statements, the disclosure of contingent assets and liabilities, and the reported amounts of revenues and expenses recognized during the reporting periods. On an ongoing basis, we evaluate our estimates, including but not limited to those related to product returns, rebates, chargebacks, collectability of receivables, inventory, intangible assets, income taxes and contingencies and litigation. Actual results could differ materially from those estimates.
Revenue Recognition
The Company recognizes revenue when it is realized or realizable and earned. Revenue is realized or realizable and earned when all of the following criteria are met: (a) persuasive evidence of an arrangement exists; (b) delivery has occurred or services have been rendered; (c) the Company’s price to the buyer is fixed or determinable; and (d) collectibility is reasonably assured.
The Company recognizes revenue from sales transactions where the buyer has the right to return the product at the time of sale only if (1) the Company’s price to the buyer is substantially fixed or determinable at the date of sale, (2) the buyer has paid the Company, or the buyer is obligated to pay the Company and the obligation is not contingent on resale of the product, (3) the buyer’s obligation to the Company would not be changed in the event of theft or physical destruction or damage of the product, (4) the buyer acquiring the product for resale has economic substance apart from any provided by the Company, (5) the Company does not have significant obligations for future performance to directly bring about resale of the product by the buyer, and (6) the amount of future returns can be reasonably estimated. The Company recognizes revenues for product sales at the time title and risk of loss are transferred to the customer, which is generally at the time products are shipped. The Company’s net product revenue represents the Company’s total revenues less allowances for customer credits, including estimated discounts, rebates, chargebacks, and product returns.
The Company establishes allowances for estimated rebates, chargebacks and product returns based on numerous qualitative and quantitative factors, including:
| • | the number of and specific contractual terms of agreements with customers; |
| • | estimated levels of inventory in the distribution channel; |
F-7
Table of Contents
SALIX PHARMACEUTICALS, LTD.
Notes to Consolidated Financial Statements — Continued
| • | historical rebates, chargebacks and returns of products; |
| • | direct communication with customers; |
| • | anticipated introduction of competitive products or generics; |
| • | anticipated pricing strategy changes by the Company and/or its competitors; |
| • | analysis of prescription data gathered by a third-party prescription data provider; |
| • | the impact of changes in state and federal regulations; and |
| • | estimated remaining shelf life of products. |
In its analyses, the Company uses prescription data purchased from a third-party data provider to develop estimates of historical inventory channel pull-through. The Company utilizes an internal analysis to compare historical net product shipments to estimated historical prescriptions written. Based on that analysis, it develops an estimate of the quantity of product in the channel which may be subject to various rebate, chargeback and product return exposures. At least quarterly for each product line, the Company prepares an internal estimate of ending inventory units in the distribution channel by adding estimated inventory in the channel at the beginning of the period, plus net product shipments for the period, less estimated prescriptions written for the period. Based on that analysis, the Company develops an estimate of the quantity of product in the channel that might be subject to various rebate, chargeback and product return exposures. This is done for each product line by applying a rate of historical activity for rebates, chargebacks and product returns, adjusted for relevant quantitative and qualitative factors discussed above, to the potential exposed product estimated to be in the distribution channel. The Company regularly adjusts internal forecasts that are utilized to calculate the estimated number of months in the channel based on input from members of the Company’s sales, marketing and operations groups. The adjusted forecasts take into account numerous factors including, but not limited to, new product introductions, direct communication with customers and potential product expiry issues. Adjustments to estimates are recorded in the period when significant events or changes in trends are identified.
The Company periodically offers promotional discounts to the Company’s existing customer base. These discounts are calculated as a percentage of the current published list price and are treated as off-invoice allowances. Accordingly, the discounts are recorded as a reduction of revenue in the period that the program is offered. In addition to promotional discounts, at the time that the Company implements a price increase, it generally offers its existing customer base an opportunity to purchase a limited quantity of product at the previous list price. Shipments resulting from these programs generally are not in excess of ordinary levels, therefore, the Company recognizes the related revenue upon shipment and includes the shipments in estimating various product related allowances. In the event the Company determines that these shipments represent purchases of inventory in excess of ordinary levels for a given wholesaler, the potential impact on product returns exposure would be specifically evaluated and reflected as a reduction in revenue at the time of such shipments.
Allowances for estimated rebates and chargebacks were $40.8 million and $17.2 million as of December 31, 2010 and 2009, respectively. These balances exclude amounts related to Colazal, which are included in the reserves discussed below. These allowances reflect an estimate of the Company’s liability for items such as rebates due to various governmental organizations under the Medicare/Medicaid regulations, rebates due to managed care organizations under specific contracts and chargebacks due to various organizations purchasing our products through federal contracts and/or group purchasing agreements. The Company estimates its liability for rebates and chargebacks at each reporting period based on a methodology of applying quantitative and qualitative assumptions discussed above. Due to the subjectivity of the Company’s accrual estimates for rebates and chargebacks, the Company prepares various sensitivity analyses to ensure the Company’s final estimate is within a reasonable range as well as review prior period activity to ensure that the Company’s methodology continues to be appropriate.
Allowances for product returns were $18.2 million and $11.3 million as of December 31, 2010 and 2009, respectively. These allowances reflect an estimate of the Company’s liability for product that may be returned by
F-8
Table of Contents
SALIX PHARMACEUTICALS, LTD.
Notes to Consolidated Financial Statements — Continued
the original purchaser in accordance with the Company’s stated return policy. The Company estimates its liability for product returns at each reporting period based on historical return rates, estimated inventory in the channel and the other factors discussed above. Due to the subjectivity of the Company’s accrual estimates for product returns, the Company prepares various sensitivity analyses and also reviews prior period activity to ensure that the Company’s methodology is still reasonable.
Colazal, the Company’s balsalazide disodium capsule, accounted for a majority of the Company’s revenue prior to 2008. On December 28, 2007, the Office of Generic Drugs, or OGD, approved three generic balsalazide capsule products. As a result of these generic approvals, sales of this product decreased significantly in 2008, 2009 and 2010 and the Company expects the future sales of Colazal to be insignificant. At December 31, 2010 and 2009, respectively, $1.5 million and $3.5 million were recorded as a liability to reflect an estimate of the Company’s liability for Colazal that may be returned or charged back by the original purchaser in accordance with the Company’s stated policies as a result of these generic approvals. The Company based this estimate on an estimate of Colazal inventory in the channel and related expiration dates of this inventory, estimated erosion of Colazal demand based on the generic approvals and the resulting estimated pull-through of Colazal, and other factors. During 2010 the Company adjusted its estimate of Colazal returns based on its review of current trends for returns, chargebacks and purchases of Colazal and recorded approximately $7.0 million as an increase to the reserve and a reduction in revenue during the period.
The Company’s provision for revenue-reducing items such as rebates, chargebacks, and product returns as a percentage of gross product revenue in the years ended December 31, 2010, 2009 and 2008 was 14.9%, 9.8% and 6.3% for rebates, chargebacks and discounts and was 2.9%, 5.5% and 7.2% for product returns, respectively, excluding the Colazal return reserve.
During the fourth quarter of 2008 the Company recognized $9.1 million of net product revenue related to shipments to wholesalers of Apriso, which was approved by the FDA on October 31, 2008, and was launched to physicians in late February 2009. Based on the Company’s historical experience with Colazal, which has the same indication, is distributed through the same distribution channels and prescribed by the same physicians as Apriso, the Company has the ability to estimate returns for Apriso and therefore recognized revenue.
During the fourth quarter of 2009 the Company shipped Metozolv to wholesalers and began detailing this drug to physicians. Because the Company has limited experience with Metozolv’s indication, the Company does not currently have the ability to estimate returns for Metozolv. The Company will recognize revenue for Metozolv based upon prescription pull-through until enough historical information is obtained to estimate returns.
During the second quarter of 2010 we recognized product revenue related to initial shipments to wholesalers of Xifaxan 550mg, which was approved by the FDA on March 24, 2010 for reduction in risk of overt hepatic encephalopathy, or HE, recurrence in patients 18 years of age or older, and launched to physicians in May 2010. Based on our historical experience with Xifaxan 200mg, which the Company distributes through the same distribution channels and is prescribed by the same physicians as Xifaxan 550mg, we have the ability to estimate returns for Xifaxan 550mg and therefore recognized revenue upon shipment to the wholesalers.
Research and Development
The Company expenses research and development costs, both internal and externally contracted, as incurred. For nonrefundable advance payments for goods or services that will be used or rendered for future research and development activities, the Company initially capitalizes the advance payment. The Company then recognizes such amounts as an expense as the related goods are delivered or the related services are performed. At December 31, 2010 and 2009, the net liability related to on-going research and development activities was $10.2 million and $14.5 million, respectively.
F-9
Table of Contents
SALIX PHARMACEUTICALS, LTD.
Notes to Consolidated Financial Statements — Continued
Cash and Cash Equivalents
The Company considers all highly liquid investments with maturities from date of purchase of three months or less to be cash equivalents. The Company maintains its cash and cash equivalents in several different financial instruments with various banks and brokerage houses. This diversification of risk is consistent with Company policy to maintain liquidity and ensure the safety of principal. At December 31, 2010, cash and cash equivalents consisted primarily of demand deposits, overnight investments in Eurodollars, certificates of deposit and money market funds at reputable financial institutions and did not include any auction rate securities.
Accounts Receivable
The Company extends credit on an uncollateralized basis primarily to wholesale drug distributors and retail pharmacy chains throughout the United States. The Company is required to estimate the level of accounts receivable which ultimately will be uncollectible. The Company calculates this estimate based on a review of specific customer balances, industry experience and the current economic environment. Currently, the Company reserves for specific accounts plus a percentage of the Company’s outstanding trade accounts receivable balance as an allowance for uncollectible accounts. Our allowance for uncollectible accounts at December 31, 2010 and 2009 was $0.5 million and $0.5 million, respectively.
Fair Value of Financial Instruments
The carrying amounts of the Company’s financial instruments, which include cash and cash equivalents, accounts receivable, accounts payable, accrued liabilities and capital lease obligations, approximated their fair values as of December 31, 2010 and 2009 due to the short-term nature of these financial instruments. The carrying amount of the Company’s credit facility, which we paid off in May 2010, approximated its fair value at December 31, 2009 due to the fact that interest rate was determined based on prevalent market rates.
Inventories
Raw materials, work-in-process and finished goods inventories are stated at the lower of cost (which approximates actual cost on a first-in, first-out cost method) or market value. In evaluating whether inventory is stated at the lower of cost or market, management considers such factors as the amount of inventory on hand and in the distribution channel, estimated time required to sell such inventory, remaining shelf life, and current and expected market conditions, including levels of competition, including generic competition.
The Company expenses pre-approval inventory unless the Company believes it is probable that the inventory will be saleable. The Company capitalizes inventory costs associated with marketed products and certain products prior to regulatory approval and product launch, based on management’s judgment of probable future commercial use and net realizable value. Capitalization of this inventory does not begin until the product candidate is considered to have a high probability of regulatory approval, which is generally after the Company has analyzed Phase III data or filed an NDA. If the Company is aware of any specific risks or contingencies that are likely to impact the expected regulatory approval process or if there are any specific issues identified during the research process relating to safety, efficacy, manufacturing, marketing or labeling of the product candidate, the Company does not capitalize the related inventory. Once the Company capitalizes inventory for a product candidate that is not yet approved, the Company monitors, on a quarterly basis, the status of this candidate within the regulatory approval process. The Company could be required to expense previously capitalized costs related to pre-approval inventory upon a change in its judgment of future commercial use and net realizable value, due to a denial or delay of approval by regulatory bodies, a delay in the timeline for commercialization or other potential factors. On a quarterly basis, the Company evaluates all inventory, including inventory capitalized for which regulatory approval has not yet been obtained, to determine if any lower of cost or market adjustment is required. As it relates to pre-approval inventory, the Company considers several factors including expected timing of FDA approval, projected sales volume and estimated selling price. At December 31, 2010 and 2009, there were no amounts included in inventory related to pre-approval inventory.
F-10
Table of Contents
SALIX PHARMACEUTICALS, LTD.
Notes to Consolidated Financial Statements — Continued
Inventory at December 31, 2010 consisted of $9.6 million of raw materials, $5.1 million of work-in-process, and $1.4 million of finished goods. Inventory at December 31, 2009 consisted of $10.9 million of raw materials, $8.4 million of work-in-process and $5.0 million of finished goods. As of December 31, 2010, inventory reserves totaling $6.6 million, compared to $2.8 million as of December 31, 2009, have been recorded to reduce inventories to their estimated net realizable value.
Property and Equipment
Property and equipment are stated at cost and depreciated over the estimated useful lives of the assets, generally three to five years, using the straight-line method.
Intangible Assets and Goodwill
The Company’s intangible assets consist of license agreements, product rights and other identifiable intangible assets, which result from product and business acquisitions. Goodwill represents the excess purchase price over the fair value of assets acquired and liabilities assumed in a business combination.
When the Company makes product acquisitions that include license agreements, product rights and other identifiable intangible assets, it records the purchase price of such intangibles, along with the value of the product related liabilities that it assumes, as intangible assets. The Company allocates the aggregate purchase price to the fair value of the various tangible and intangible assets in order to determine the appropriate carrying value of the acquired assets and then amortizes the cost of the intangible assets as an expense in its consolidated statement of operations over the estimated economic useful life of the related assets. The Company assesses the impairment of identifiable intangible assets whenever events or changes in circumstances indicate that the carrying value may not be recoverable. The Company believes that the following factors could trigger an impairment review: significant underperformance relative to expected historical or projected future operating results; significant changes in the manner of the Company’s use of the acquired assets or the strategy for the Company’s overall business; approval of generic products; and significant negative industry or economic trends.
In assessing the recoverability of its intangible assets, the Company must make assumptions regarding estimated future cash flows and other factors. If the estimated undiscounted future cash flows do not exceed the carrying value of the intangible assets, the Company must determine the fair value of the intangible assets. If the fair value of the intangible assets is less than the carrying value, the Company will recognize an impairment loss in an amount equal to the difference. The Company reviews goodwill for impairment on an annual basis in the fourth quarter, and goodwill and other intangible assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. At December 31, 2010 there is no impairment to goodwill.
The following table reflects the components of all specifically identifiable intangible assets as of December 31 (in thousands):
| 2010 | 2009 | |||||||||||||||||||||||
| Gross Amount |
Accumulated Amortization |
Net Carrying Value |
Gross Amount |
Accumulated Amortization |
Net Carrying Value |
|||||||||||||||||||
| Goodwill |
$ | 85,257 | $ | — | $ | 85,257 | $ | 85,257 | $ | — | $ | 85,257 | ||||||||||||
| Finite lived intangible assets |
112,997 | 48,420 | 64,577 | 147,653 | 38,050 | 109,603 | ||||||||||||||||||
| Indefinite lived intangible assets |
— | — | — | — | — | — | ||||||||||||||||||
| Total |
$ | 198,254 | $ | 48,420 | $ | 149,834 | $ | 232,910 | $ | 38,050 | $ | 194,860 | ||||||||||||
The average remaining life of our finite lived intangible assets was seven years and nine years at December 31, 2010 and 2009, respectively.
F-11
Table of Contents
SALIX PHARMACEUTICALS, LTD.
Notes to Consolidated Financial Statements — Continued
Amortization expense is calculated on a straight-line basis over the estimated useful life of the asset. Amortization expense for the years ended December 31, 2010, 2009 and 2008 was $10.4 million, $11.5 million and $9.9 million, respectively. Estimated amortization expense related to intangible assets existing as of December 31, 2010 is $9.7 million annually for each of the succeeding five years.
In November 2003, the Company acquired from aaiPharma LLC for $2.0 million the exclusive right to sell 25, 75 and 100 milligram dosage strengths of azathioprine tablets in North America under the name Azasan. The purchase price was fully allocated to product rights and related intangibles and is being amortized over a period of ten years. Although Azasan does not have any patent protection, the Company believes ten years is an appropriate amortization period based on established product sales history and management’s experience. At December 31, 2010 and 2009, accumulated amortization for the Azasan intangible was $1.4 million and $1.2 million, respectively.
In June 2004, the Company acquired the exclusive U.S. rights to Anusol-HC 2.5% (hydrocortisone Cream USP), Anusol-HC 25 mg Suppository (Hydrocortisone Acetate), Proctocort Cream (Hydrocortisone Cream USP) 1% and Proctocort Suppositories (Hydrocortisone Acetate Rectal Suppositories, 30 mg) from King Pharmaceuticals, Inc. for $13.0 million. The purchase price was fully allocated to product rights and related intangibles and is being amortized over a period of ten years. Although Anusol-HC and Proctocort do not have any patent protection, the Company believes ten years is an appropriate amortization period based on established product sales history and management’s experience. At December 31, 2010 and 2009, accumulated amortization for the King product intangibles was $8.4 million and $7.1 million, respectively.
In September 2005, the Company acquired InKine Pharmaceutical Company, Inc. for $210.0 million. The Company allocated $74.0 million of the purchase price to in-process research and development, $9.3 million to net assets acquired and $37.0 million to specifically identifiable product rights and related intangibles with an ongoing economic benefit to the Company. The Company allocated the remaining $89.7 million to goodwill, which is not being amortized. The InKine product rights and related intangibles were being amortized over an average period of 14 years, which the Company believed was an appropriate amortization period due to the product’s patent protection and the estimated economic lives of the product rights and related intangibles. In September 2010, the Company entered into a Sublicense Agreement which granted Novel Laboratories, Inc. a license under the patents covering OsmoPrep such permitting Novel to launch a generic OsmoPrep on November 16, 2019. As a result of this agreement the amortization period was adjusted prospectively, and the remaining net book value of the intangible asset will be amortized through November 16, 2019, which is the Company’s revised estimate of its remaining economic life. The Company assessed whether there was an impairment to the carrying value of the related intangible asset due to its reduced economic life and determined that there was no impairment. At December 31, 2010 and 2009, accumulated amortization for the InKine intangibles was $15.7 million and $12.8 million, respectively.
In December 2005, the Company entered into a License and Supply Agreement with Norgine B.V., granting Salix the exclusive right to sell a patented-protected, liquid PEG bowel cleansing product, NRL 944, in the United States. In August 2006, the Company received Food and Drug Administration marketing approval for NRL 944 under the branded name of MoviPrep. In January 2007 the United States Patent Office issued a patent providing coverage to September 1, 2024. Pursuant to the terms of the Agreement, Salix paid Norgine milestone payments of $15.0 million in August 2006, $5.0 million in December 2008 and $5.0 million in December 2009. The Company was amortizing these milestone payments over a period of 17.3 years through 2022, which the Company believed was an appropriate amortization period due to the product’s patent protection and the estimated economic life of the related intangible. In August 2010 the Company entered into a Sublicense Agreement that granted Novel Laboratories, Inc. a license to the patents covering MoviPrep permitting Novel to launch a generic MoviPrep on September 24, 2018. As a result of this agreement the amortization period was adjusted prospectively, and the remaining net book value of the intangible asset will be amortized through September 24, 2018, which is the Company’s revised estimate of its remaining economic life. The Company assessed whether there was an impairment to the carrying value of the related intangible asset due to its reduced economic life and determined that there was no impairment. At December 31, 2010 and 2009, accumulated amortization for the MoviPrep intangible was $6.8 million and $5.0 million, respectively.
F-12
Table of Contents
SALIX PHARMACEUTICALS, LTD.
Notes to Consolidated Financial Statements — Continued
In February 2007, the Company entered into a Master Purchase and Sale and License Agreement with Merck & Co. Inc., to purchase the U.S prescription pharmaceutical product rights to Pepcid Oral Suspension and Diuril Oral Suspension from Merck. The Company paid Merck $55.0 million at the closing of this transaction. The Company fully allocated the purchase price to product rights and related intangibles, and it is being amortized over a period of 15 years. Although Pepcid and Diuril do not have patent protection, the Company believes 15 years was an appropriate amortization period based on established product history and management experience. In May 2010, the FDA approved a generic famotidine oral suspension product, and the Company launched an authorized generic famotidine product. In June 2010 the FDA approved another generic famotidine oral suspension product. As a result of these events, the Company assessed whether there was an impairment to the carrying value of the related intangible asset. Based on this analysis, the Company recorded a $30 million impairment charge to reduce the carrying value of the intangible asset to its estimated fair value during the three-month period ended June 30, 2010. At December 31, 2010 and 2009, accumulated amortization for the Merck products was $12.9 million and $10.5 million, respectively and the carrying value was $12.1 million at December 31, 2010.
In July 2002, the Company acquired the rights to develop and market a granulated formulation of mesalamine from Dr. Falk Pharma GmbH. On October 31, 2008, the FDA granted marketing approval for Apriso for the maintenance of remission of ulcerative colitis in adults. In November 2008, the Company made a $6.0 million milestone payment to Dr. Falk. The Company is amortizing this milestone payment over a period of 9.5 years, which the Company believes is an appropriate amortization period due to the product’s patent protection and the estimated economic life of the related intangible. At December 31, 2010 and 2009, accumulated amortization for the Apriso intangible was $1.8 million and $1.0 million, respectively.
In September 2007, the Company acquired the exclusive, worldwide right to sell metoclopramide–Zydis® (trade name Metozolv) from Wilmington Pharmaceuticals, LLC. On September 8, 2009 the FDA granted marketing approval for Metozolv™ ODT (metoclopramide HCl) 5 mg and 10 mg orally disintegrating tablets. Metozolv ODT is indicated for the relief of symptomatic gastroesophageal reflux or short-term (4-12 weeks) therapy for adults with symptomatic, documented gastroesophageal reflux who fail to respond to conventional therapy and diabetic gastroparesis or the relief of symptoms in adults associated with acute and recurrent diabetic gastroparesis. In October 2009, the Company made a $7.3 million milestone payment to Wilmington. The Company was amortizing this milestone payment over a period of 8 years, which the Company believed was an appropriate amortization period due to the product’s patent protection and the estimated economic life of the related intangible. On November 3, 2010, the Company received a paragraph IV notification from Novel stating that Novel had filed an ANDA application to seek approval to market a generic version of Metoclopramide Hydrochloride ODT, 5 mg and 10 mg. The notification letter asserted non-infringement of U.S. Patent No. 6,413,549 (the ‘549 patent). Upon examination of the relevant sections of the ANDA, the Company concluded that the ‘549 patent would not be enforced against Novel Laboratories. As a result of this event, the Company assessed whether there was an impairment to the carrying value of the related intangible asset. Based on this analysis, the Company recorded a $4.6 million impairment charge to reduce the carrying value of the intangible asset to its estimated fair value during the three-month period ended December 31, 2010. At December 31, 2010 and 2009 accumulated amortization for the Metozolv intangible was $1.2 million and $0.2 million, respectively and the carrying value was $1.5 million at December 31, 2010.
F-13
Table of Contents
SALIX PHARMACEUTICALS, LTD.
Notes to Consolidated Financial Statements — Continued
Restricted Cash
At December 31, 2009, restricted cash of $15.0 million represents the collateral on deposit with the administrative agent related to the Company’s credit facility. On May 6, 2010, the Company repaid the $15.0 million then drawn under our credit facility and terminated the facility.
Shipping and Handling Costs
The Company does not charge its customers for freight costs. The amounts of such costs are included in selling, general and administrative expenses and are not material.
Advertising Costs
The Company charges advertising costs to expense as incurred. Advertising expenses were approximately $14.3 million, $6.7 million and $8.3 million for the years ended December 31, 2010, 2009 and 2008, respectively.
Segment Reporting
The Company operates in a single industry and segment acquiring, developing and commercializing prescription drugs used in the treatment of a variety of gastrointestinal diseases, which are those affecting the digestive tract. Accordingly, the Company’s business is classified as a single reportable segment.
The following table presents net product revenues by product (in thousands):
| Year ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Xifaxan |
$ | 250,459 | $ | 117,939 | $ | 79,928 | ||||||
| Purgatives—Visicol/OsmoPrep/MoviPrep |
54,207 | 76,287 | 62,862 | |||||||||
| Inflammatory Bowel Disease—Colazal/Apriso |
24,459 | 5,782 | 10,545 | |||||||||
| Other—Anusol/Azasan/Diuril/Pepcid/Proctocort/Metozolv |
7,848 | 32,882 | 25,431 | |||||||||
| Net product revenues |
$ | 336,973 | $ | 232,890 | $ | 178,766 | ||||||
Comprehensive Income (Loss)
For the periods presented, comprehensive income (loss) equaled reported net income (loss).
Stock-Based Compensation
At December 31, 2010, the Company had one active share-based compensation plan, the 2005 Stock Plan, allowing for the issuance of stock options and restricted stock. The Company estimates the fair value of share-based payment awards on the date of the grant. The cost is to be recognized over the period during which an employee is required to provide service in exchange for the award.
Income Taxes
The Company provides for income taxes under the liability method. This approach requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of differences between the tax basis of assets or liabilities and their carrying amounts in the consolidated financial statements. The Company provides a valuation allowance for deferred tax assets if it is more likely than not that these items will either expire before the Company is able to realize their benefit or if future deductibility is uncertain.
The Company applies the provisions of ASC 740-10 with respect to accounting for uncertainty in income taxes. The unrecognized tax benefits as of December 31, 2010 and 2009 relate to federal tax credit carryforwards. The Company does not expect the unrecognized tax benefits to change significantly over the next 12 months.
F-14
Table of Contents
SALIX PHARMACEUTICALS, LTD.
Notes to Consolidated Financial Statements — Continued
The Company continues to fully recognize its tax benefits that are offset by a valuation allowance to the extent that it is more likely than not that the deferred tax assets will not be realized.
The Company files a consolidated U.S. federal income tax return and consolidated and separate company income tax returns in many U.S. state jurisdictions. Generally, the Company is no longer subject to federal and state income tax examinations by U.S. tax authorities for years prior to 1993.
The Company recognizes any interest accrued related to unrecognized tax benefits in interest expense and penalties in operating expenses. During the twelve-month period ended December 31, 2010 and 2009 there was no such interest or penalties.
Net Income (Loss) Per Share
The Company computes basic net income (loss) per share by dividing net income (loss) by the weighted average number of common shares outstanding. Diluted net income (loss) per share is computed by dividing net income (loss) by the weighted average number of common shares and dilutive common share equivalents then outstanding. Common share equivalents consist of the incremental common shares issuable upon the exercise of stock options and the impact of unvested restricted stock grants. The Company accounts for the effect of the convertible notes on diluted net income (loss) per share using the treasury stock method. As a result, the convertible notes have no effect on diluted net income (loss) per share until the Company’s stock price exceeds the conversion price of $9.25 per share for the 2028 Notes, and $46.38 for the 2015 Notes. For the years ended December 31, 2009 and 2010, the effect of the approximately 6,486,000 shares issuable upon conversion of the 2028 Notes and the approximately 7,439,000 shares issuable upon conversion of the 2015 Notes were excluded from the diluted net income per share calculation, because their inclusion would have an anti-dilutive effect due to the net loss during those periods.
For the year ended December 31, 2010, weighted average common shares, diluted were equal to weighted average common shares, basic, because inclusion of the 1,438,241 and 1,527,186 shares of restricted stock and stock options, respectively, would have an anti-dilutive effect due to the net loss during that period. For the year ended December 31, 2009, weighted average common shares, diluted were equal to weighted average common shares, basic, because inclusion of the 756,699 and 922,011 shares of restricted stock and stock options, respectively, would have an anti-dilutive effect due to the net loss during that period. For the year ended December 31, 2008, weighted average common shares, diluted were equal to weighted average common shares, basic, because inclusion of the 133,053 and 578,386 shares of restricted stock and stock options, respectively, would have an anti-dilutive effect due to the net loss during that period. For the years ended 2010, 2009 and 2008, there were 113,957, 1,761,782, and 4,434,835, respectively, potential common shares outstanding that were excluded from the diluted net income (loss) per share calculation because their effect would have been anti-dilutive. For the years ended 2010 and 2009 there were 4,890,188 and 2,131,889, potential common shares outstanding, respectively, as a result of our convertible debt that were excluded from the diluted net income (loss) per share calculation because their effect would have been anti-dilutive.
The following table reconciles the numerator and denominator used to calculate diluted net income (loss) per share (in thousands):
| Year ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Numerator: |
||||||||||||
| Net income (loss) |
$ | (27,061 | ) | $ | (43,619 | ) | $ | (47,607 | ) | |||
| Denominator: |
||||||||||||
| Weighted average common shares, basic |
57,300 | 49,350 | 47,898 | |||||||||
| Dilutive effect of stock options and restricted stock awards |
— | — | — | |||||||||
| Weighted average common shares, diluted |
57,300 | 49,350 | 47,898 | |||||||||
F-15
Table of Contents
SALIX PHARMACEUTICALS, LTD.
Notes to Consolidated Financial Statements — Continued
Recently Issued Accounting Pronouncements
In April 2010, the FASB issued authoritative guidance on defining a milestone and determining when it may be appropriate to apply the milestone method of revenue recognition for research or development transactions. Consideration that is contingent on achievement of a milestone in its entirety may be recognized as revenue in the period in which the milestone is achieved only if the milestone is judged to meet certain criteria to be considered substantive. Milestones should be considered substantive in their entirety and may not be bifurcated. An arrangement may contain both substantive and non substantive milestones, and each milestone should be evaluated individually to determine if it is substantive. The guidance is effective on a prospective basis for milestones achieved in fiscal years, and interim periods within those years, beginning on or after June 15, 2010, with early adoption permitted. The Company does not expect the adoption of this guidance to have a material impact on its consolidated financial statements.
(3) PROPERTY AND EQUIPMENT
Property and equipment consisted of the following at December 31 (in thousands):
| 2010 | 2009 | |||||||
| Cost: |
||||||||
| Furniture and equipment |
$ | 9,479 | $ | 5,838 | ||||
| Computer equipment |
8,411 | 7,694 | ||||||
| Assets under capital lease |
3,453 | 3,523 | ||||||
| 21,343 | 17,055 | |||||||
| Accumulated depreciation: |
||||||||
| Furniture and equipment |
(5,407 | ) | (4,838 | ) | ||||
| Computer equipment |
(5,078 | ) | (4,649 | ) | ||||
| Assets under capital lease |
(2,961 | ) | (2,219 | ) | ||||
| (13,446 | ) | (11,706 | ) | |||||
| Net property and equipment |
$ | 7,897 | $ | 5,349 | ||||
Depreciation expense was approximately $2.9 million, $3.0 million and $3.1 million in 2010, 2009 and 2008, respectively.
(4) ACCRUED LIABILITIES
Accrued liabilities consisted of the following at December 31 (in thousands):
| 2010 | 2009 | |||||||
| Accrued expenses |
$ | 35,524 | $ | 43,744 | ||||
| Deferred revenue |
2,717 | 10,231 | ||||||
| Accrued royalties |
7,518 | 6,010 | ||||||
| Total accrued liabilities |
$ | 45,759 | $ | 59,985 | ||||
(5) CREDIT FACILITY
In February 2007, the Company entered into a $100.0 million revolving credit facility that was to mature in February 2012. On August 4, 2008 the credit facility was amended to waive defaults that may have arisen as a result of the approval of three generic balsalazide capsule products by the Office of Generic Drugs on December 28, 2007 and
F-16
Table of Contents
SALIX PHARMACEUTICALS, LTD.
Notes to Consolidated Financial Statements — Continued
the credit facility was reduced to $20.0 million. On August 22, 2008 the credit facility was further amended to allow the Company to issue the 2028 notes described in Note 6 below. As a result of the execution of the amendment to our credit facility on August 4, 2008, the Company recorded a $1.1 million non-cash charge to expense a portion of the unamortized costs related to the credit facility to interest expense. On May 6, 2010, the Company repaid the $15.0 million then drawn under our credit facility and terminated the facility.
(6) CONVERTIBLE SENIOR NOTES
Convertible Senior Notes Due 2028
On August 22, 2008 the Company closed an offering of $60.0 million in Convertible Senior Notes due 2028 (“2028 Notes”). Net proceeds from the offering were $57.3 million. The 2028 Notes are governed by an indenture, dated as of August 22, 2008, between the Company and U.S. Bank National Association, as trustee.
The 2028 Notes bear interest at a rate of 5.5% per year, payable semiannually in arrears on February 15 and August 15 of each year, beginning on February 15, 2009. The 2028 Notes will mature on August 15, 2028, unless previously converted or repurchased in accordance with their terms prior to such date.
The 2028 Notes are senior unsecured obligations, and rank (i) equally to any of the Company’s existing and future unsecured senior debt, (ii) senior to any of the Company’s future indebtedness that is expressly subordinated to these 2028 Notes, and (iii) effectively junior to any secured indebtedness to the extent of the value of the assets securing such indebtedness.
The Company may redeem the 2028 Notes, in whole or in part, at any time after August 15, 2013 for cash equal to the principal amount of the Notes to be redeemed, plus any accrued and unpaid interest.
On August 15, 2013, August 15, 2018 and August 15, 2023 or upon the occurrence of a “fundamental change”, as defined in the indenture, the holders may require the Company to repurchase all or a portion of the 2028 Notes for cash at 100% of the principal amount of the Notes being purchased, plus any accrued and unpaid interest.
The 2028 Notes are convertible into approximately 6,486,000 shares of the Company’s common stock under certain circumstances prior to maturity at a conversion rate of 108.0847 shares per $1,000 principal amount of 2028 Notes, which represents a conversion price of approximately $9.25 per share, subject to adjustment under certain conditions. Holders may convert their 2028 Notes at their option on any day prior to the close of business on the business day immediately preceding the maturity date of August 15, 2028 only if one or more of the following conditions is satisfied: (1) during any fiscal quarter commencing after September 30, 2008, if the last reported sale price of the Company’s common stock for at least 20 trading days in the period of 30 consecutive trading days ending on the last trading day of the immediately preceding fiscal quarter is equal to or more than 130% of the conversion price of the 2028 Notes on the last day of such preceding fiscal quarter; (2) during the five business day period following any five consecutive trading day period in which the trading price for the 2028 Notes, per $1,000 principal amount of the 2028 Notes, for each such trading day was less than 98% of the product of the last reported sale price of the Company’s common stock and the conversion rate of the Notes on such date; (3) if the Company enters into specified corporate transactions; or (4) upon a redemption notice. The first of these conditions was met as of December 31, 2010. The 2028 Notes will be convertible, regardless of whether any of the foregoing conditions have been satisfied, on or after March 15, 2028 at any time prior to the close of business on the business day immediately preceding the stated maturity date of August 15, 2028. Upon conversion, the Company may pay cash, shares of the Company’s common stock or a combination of cash and stock, as determined by the Company in its discretion.
As long as the 2028 Notes are outstanding, the Company and its subsidiaries are prohibited from incurring any debt other than “permitted debt”, as defined in the indenture, except that the Company and its subsidiaries may incur debt in certain circumstances, including meeting a consolidated leverage ratio test and a consolidated fixed charge coverage ratio test. The 2015 Notes described below are “permitted debt” under the indenture.
F-17
Table of Contents
SALIX PHARMACEUTICALS, LTD.
Notes to Consolidated Financial Statements — Continued
In connection with the issuance of the 2028 Notes, the Company incurred $2.7 million of issuance costs, which primarily consisted of investment banker, legal and other professional fees. These costs are being amortized and are recorded as additional interest expense through August 2013, the first scheduled date on which holders have the option to require the Company to repurchase the 2028 Notes.
The Company has separately accounted for the liability and equity components of the convertible debt instrument by allocating the proceeds from issuance of the 2028 Notes between the liability component and the embedded conversion option, or equity component. This allocation was done by first estimating an interest rate at the time of issuance for similar notes that do not include the embedded conversion option. This interest rate of 12.5% was used to compute the initial fair value of the liability component of $44.1 million. The excess of the initial proceeds received from the convertible 2028 Notes over the initial amount allocated to the liability component, of $15.9 million, is allocated to the embedded conversion option, or equity component. This excess is reported as a debt discount and subsequently amortized as interest cost, using the interest method, through August 2013, the first scheduled date on which the holders have the option to require the Company to repurchase the 2028 Notes.
The carrying value of the equity component at December 31, 2010 and 2009 was $15.2 million. The effective interest rate on the liability component for the years ended December 31, 2010, 2009 and 2008 was 12.6%. Total interest cost of $6.6 million, $5.8 million and $1.8 million was recognized during the years ended December 31, 2010, 2009 and 2008, respectively, including 2.9 million, $2.5 million and $0.6 million of amortization of debt discount, respectively. The fair value of the 2028 Notes was approximately $300.7 million at December 31, 2010.
Convertible Senior Notes Due 2015
On June 3, 2010 the Company closed an offering of $345.0 million in Convertible Senior Notes due May 15, 2015 (“2015 Notes”). Net proceeds from the offering were approximately $334.2 million. The 2015 Notes are governed by an indenture, dated as of June 3, 2010 between the Company and U.S. Bank National Association, as trustee.
The 2015 Notes bear interest at a rate of 2.75% per year, payable semiannually in arrears on May 15 and November 15 of each year, beginning on November 15, 2010. The 2015 Notes will mature on May 15, 2015, unless earlier converted or repurchased in accordance with their terms prior to such date.
The 2015 Notes are senior unsecured obligations, and rank (i) equally to any of the Company’s existing and future unsecured senior debt, (ii) senior to any of the Company’s future indebtedness that is expressly subordinated to these 2015 Notes, and (iii) effectively junior to any secured indebtedness to the extent of the value of the assets securing such indebtedness.
The 2015 Notes are convertible into approximately 7,439,000 shares of the Company’s common stock under certain circumstances prior to maturity at a conversion rate of 21.5592 shares per $1,000 principal amount of 2015 Notes, which represents a conversion price of approximately $46.38 per share, subject to adjustment under certain conditions. Holders may convert their 2015 Notes at their option on any day prior to the close of business on the business day immediately preceding the maturity date of May 15, 2015 only if one or more of the following conditions is satisfied: (1) during any fiscal quarter commencing after June 30, 2010, if the last reported sale price of the Company’s common stock for at least 20 trading days in the period of 30 consecutive trading days ending on the last trading day of the immediately preceding fiscal quarter is equal to or more than 130% of the conversion price of the 2015 Notes on the last day of such preceding fiscal quarter; (2) during the five business day period following any five consecutive trading day period in which the trading price for the 2015 Notes, per $1,000 principal amount of the 2015 Notes, for each such trading day was less than 98% of the product of the last reported sale price of the Company’s common stock and the conversion rate of the 2015 Notes on such date; or (3) if the Company enters into specified corporate transactions. None of these conditions had been met as of December 31, 2010. The 2015 Notes will be convertible, regardless of whether any of the foregoing conditions have been satisfied, on or after January 13, 2015 at any time prior to the close of business on the second scheduled trading day immediately preceding the stated
F-18
Table of Contents
SALIX PHARMACEUTICALS, LTD.
Notes to Consolidated Financial Statements — Continued
maturity date of May 15, 2015. Upon conversion, the Company may pay cash, shares of the Company’s common stock or a combination of cash and stock, as determined by the Company in its discretion.
The Company is required to separately account for the liability and equity components of the convertible debt instrument by allocating the proceeds from issuance of the 2015 Notes between the liability component and the embedded conversion option, or equity component. This allocation was done by first estimating an interest rate at the time of issuance for similar notes that do not include the embedded conversion option. This interest rate of 8.35% was used to compute the initial fair value of the liability component of $265.6 million. The excess of the initial proceeds received from the convertible 2015 Notes over the initial amount allocated to the liability component, of $79.4 million, is allocated to the embedded conversion option, or equity component. This excess is reported as a debt discount and subsequently amortized as interest cost, using the interest method, through May 2015, the maturity date of the 2015 Notes.
In connection with the issuance of the 2015 Notes, the Company incurred $10.8 million of issuance costs, which primarily consisted of investment banker, legal and other professional fees. The portion of these costs related to the equity component of $2.5 million was charged to additional paid-in capital. The portion of these costs related to the debt component of $8.3 million is being amortized and are recorded as additional interest expense through May 2015, the maturity date of the 2015 Notes.
In connection with the issuance of the 2015 Notes, the Company entered into capped call transactions with certain counterparties covering approximately 7,439,000 shares of the Company’s common stock. The capped call transactions have a strike price of $46.38 and a cap price of $62.44, and are exercisable when and if the 2015 Notes are converted. If upon conversion of the 2015 Notes, the price of the Company’s common stock is above the strike price of the capped calls, the counterparties will deliver shares of the Company’s common stock and/or cash with an aggregate value approximately equal to the difference between the price of the Company’s common stock at the conversion date (as defined, with a maximum price for purposes of this calculation equal to the cap price) and the strike price, multiplied by the number of shares of the Company’s common stock related to the capped call transactions being exercised. The Company paid $44.3 million for these capped calls and charged this to additional paid-in capital.
The carrying value of the equity component related to the 2015 Notes at December 31, 2010 was $79.4 million. The effective interest rate on the liability component for the year ended December 31, 2010 was 8.35%. Total interest cost of $13.7 million was recognized during the year ended December 31, 2010 including $7.2 million of amortization of debt discount. The fair value of the 2015 Notes is approximately $444.1 million at December 31, 2010.
The following table summarizes information on our convertible debt at December 31(in thousands):
| 2010 | 2009 | |||||||
| Convertible Notes due 2028: |
||||||||
| Principal amount of the liability component |
$ | 60,000 | $ | 60,000 | ||||
| Unamortized discount |
(9,824 | ) | (12,701 | ) | ||||
| Net carrying amount |
$ | 50,176 | $ | 47,299 | ||||
| Convertible Notes due 2015: |
||||||||
| Principal amount of the liability component |
$ | 345,000 | $ | — | ||||
| Unamortized discount |
(72,171 | ) | — | |||||
| Net carrying amount |
$ | 272,829 | $ | — | ||||
| Total Convertible Senior Notes |
||||||||
| Principal amount of the liability component |
$ | 405,000 | $ | 60,000 | ||||
| Unamortized discount |
(81,995 | ) | (12,701 | ) | ||||
| Net carrying amount |
$ | 323,005 | $ | 47,299 | ||||
F-19
Table of Contents
SALIX PHARMACEUTICALS, LTD.
Notes to Consolidated Financial Statements — Continued
(7) STOCKHOLDERS’ EQUITY
Preferred Stock
A total of 5,000,000 shares of preferred stock are authorized and issuable. No shares of preferred stock were issued or outstanding as of December 31, 2010 or 2009.
Common Stock
As of December 31, 2010 the Company was authorized to issue up to 150,000,000 shares of $0.001 par value common stock. As of December 31, 2010 and 2009, there were 58,139,941 and 56,245,759 shares of common stock issued and outstanding, respectively.
Stockholder Rights Plan
On January 9, 2003, the Company’s board of directors adopted an updated stockholder rights plan. Consequently, the Board authorized the redemption, effective on January 20, 2003, of rights under its existing stockholder rights plan for $0.0001 per right. Pursuant to the updated plan, stock purchase rights were distributed to stockholders at the rate of one right with respect to each share of common stock held of record as of January 20, 2003. The rights plan is designed to enhance the Board’s ability to prevent an acquirer from depriving stockholders of the long-term value of their investment and to protect stockholders against attempts to acquire the Company by means of unfair or abusive takeover tactics. The rights become exercisable based upon certain limited conditions related to acquisitions of stock, tender offers and business combinations involving the Company.
Stock Plans
The Company’s 1994 Stock Plan (the “1994 Plan”) was adopted by the Board of Directors in March 1994 and approved by the stockholders in March 1995. The Company’s 1996 Stock Plan (the “1996 Plan”) was adopted by the Board of Directors and approved by the Company’s stockholders in February 1996. The Company’s 2005 Stock Plan (the “2005 Plan”) was adopted by the Board of Directors in April 2005 and approved by the stockholders in June 2005. The stock granted under the 1994 Plan, the 1996 Plan and the 2005 Plan may be either stock options or restricted shares. Stock options expire no later than ten years from the date of grant.
Option exercise prices must be at least 100% of the fair market value on the date of grant. If, at the time the Company grants an option, the optionee directly or by attribution owns stock possessing more than 10% of the total combined voting power of all classes of stock of the Company, the exercise price for an incentive stock option must be at least 110% of the fair market value and the option may not be exercisable more than five years after the date of grant. The options generally become exercisable in increments of 1/48th per month over a period of 48 months from the date of grant. Options may be granted with different vesting terms as determined by the board of directors. Since inception of the Company’s 1994 Plan, 1996 Plan and 2005 Plan, the Company’s stock option grants were all at fair market value. Starting in 2006, the Company began issuing restricted shares to employees, executives and directors of the Company.
Stock-Based Compensation
At December 31, 2010, the Company had one active share-based compensation plan, the 2005 Stock Plan, allowing for the issuance of stock options and restricted stock. The Company estimates the fair value of share-based payment awards on the date of the grant. The Company recognizes cost over the period during which an employee is required to provide service in exchange for the award.
Starting in 2006, the Company began issuing restricted shares to employees, executives and directors of the Company. The restrictions on the restricted stock lapse according to one of two schedules. For employees and executives of the Company, restrictions lapse 25% annually over four years or 33% over 3 years. For Board members
F-20
Table of Contents
SALIX PHARMACEUTICALS, LTD.
Notes to Consolidated Financial Statements — Continued
of the Company, restrictions lapse 100% after one year. The fair value of the restricted stock was estimated using an assumed forfeiture rate of 9.2% and is being expensed on a straight-line basis over the period during which the restrictions lapse. For the years ended December 31, 2010, 2009 and 2008, the Company recognized $9.9 million, $7.0 million and $4.8 million in share based compensation expense related to the restricted shares, respectively. As of December 31, 2010, the total amount of unrecognized compensation cost related to nonvested restricted stock awards, to be recognized as expense subsequent to December 31, 2010, was approximately $21.8 million, and the related weighted-average period over which it is expected to be recognized is approximately 3 years.
Aggregate stock plan activity is as follows:
| Total Shares Available For Grant |
Stock Options | Restricted Shares | Stock Options and Restricted Shares |
|||||||||||||||||||||||||
| Number | Weighted Average Price |
Number Subject to Issuance |
Weighted Average Price |
Number | Weighted Average Price |
|||||||||||||||||||||||
| Balance at December 31, 2007 |
611,253 | 5,214,529 | $ | 14.03 | 1,123,199 | $ | 12.68 | 6,337,728 | $ | 13.80 | ||||||||||||||||||
| Additional shares authorized |
837,311 | — | — | — | — | — | — | |||||||||||||||||||||
| Granted |
(1,009,597 | ) | — | — | 1,009,597 | $ | 7.10 | 1,009,597 | $ | 7.10 | ||||||||||||||||||
| Exercised |
— | (67,713 | ) | $ | 3.66 | — | — | (67,713 | ) | $ | 3.66 | |||||||||||||||||
| Vested |
— | — | — | (301,502 | ) | $ | 12.65 | (301,502 | ) | $ | 12.65 | |||||||||||||||||
| Canceled/forfeited |
397,655 | (528,983 | ) | $ | 18.55 | (182,439 | ) | $ | 11.54 | (711,422 | ) | $ | 16.75 | |||||||||||||||
| Balance at December 31, 2008 |
836,622 | 4,617,833 | $ | 13.67 | 1,648,855 | $ | 9.39 | 6,266,688 | $ | 12.55 | ||||||||||||||||||
| Additional shares authorized |
2,000,000 | — | — | — | — | — | — | |||||||||||||||||||||
| Granted |
(1,228,471 | ) | — | — | 1,228,471 | $ | 10.66 | 1,228,471 | $ | 10.66 | ||||||||||||||||||
| Exercised |
— | (1,284,846 | ) | $ | 10.65 | — | — | (1,284,846 | ) | $ | 10.65 | |||||||||||||||||
| Vested |
— | — | — | (611,974 | ) | $ | 9.43 | (611,974 | ) | $ | 9.43 | |||||||||||||||||
| Canceled/forfeited |
251,717 | (151,353 | ) | $ | 18.11 | (161,935 | ) | $ | 9.85 | (313,288 | ) | $ | 13.84 | |||||||||||||||
| Balance at December 31, 2009 |
1,859,868 | 3,181,634 | $ | 14.68 | 2,103,417 | $ | 10.08 | 5,285,051 | $ | 12.85 | ||||||||||||||||||
| Granted |
(590,208 | ) | — | — | 590,208 | $ | 36.78 | 590,208 | $ | 36.78 | ||||||||||||||||||
| Exercised |
— | (1,140,862 | ) | $ | 14.96 | — | — | (1,140,862 | ) | $ | 14.96 | |||||||||||||||||
| Vested |
— | — | — | (699,065 | ) | $ | 11.22 | (699,065 | ) | $ | 11.22 | |||||||||||||||||
| Canceled/forfeited |
281,911 | (32,260 | ) | $ | 40.29 | (250,861 | ) | $ | 9.03 | (283,121 | ) | $ | 12.59 | |||||||||||||||
| Balance at December 31, 2010 |
1,551,571 | 2,008,512 | $ | 14.11 | 1,743,699 | $ | 18.81 | 3,752,211 | $ | 16.30 | ||||||||||||||||||
Exercise prices for options outstanding as of December 31, 2010 ranged from $1.13 to $48.58 per share.
| Options Outstanding and Exercisable | ||||||||||||
| Exercise Price |
Number Outstanding | Weighted Average Remaining Contractual Life (Yrs) |
Weighted Average Exercise Price |
|||||||||
| $ 1.13 – 4.92 |
422,125 | 1.58 | $ | 4.07 | ||||||||
| $ 5.40 – 14.46 |
370,967 | 2.73 | 9.54 | |||||||||
| $15.02 – 17.94 |
563,541 | 4.15 | 17.27 | |||||||||
| $18.14 – 20.89 |
497,796 | 3.59 | 18.99 | |||||||||
| $21.07 – 48.58 |
154,083 | 3.44 | 25.47 | |||||||||
| 2,008,512 | 3.18 | $ | 14.12 | |||||||||
At December 31, 2010, there were 2,008,512 exercisable options with a weighted average exercise price of $14.12. At December 31, 2009, there were 3,181,634 exercisable options with a weighted average exercise price of $14.68. At December 31, 2008, there were 4,617,833 exercisable options with a weighted average exercise price of $13.67.
F-21
Table of Contents
SALIX PHARMACEUTICALS, LTD.
Notes to Consolidated Financial Statements — Continued
For the year ended December 31, 2010, 1.1 million shares of the Company’s outstanding stock with a market value of $41.4 million were issued upon the exercise of stock options. For the year ended December 31, 2009, 1.3 million shares of the Company’s outstanding stock with a market value of $25.3 million were issued upon the exercise of stock options. For the year ended December 31, 2008, 0.1 million shares of the Company’s outstanding stock with a market value of $0.5 million were issued upon the exercise of stock options. The Company recognized no share-based compensation expense related to stock options for the years ended December 31, 2010, 2009 or 2008, nor any income tax benefit. The total intrinsic value of options exercised for the years ended December 31, 2010, 2009 and 2008 was $24.3 million, $11.6 million, and $0.3 million, respectively. As of December 31, 2010, there was no unrecognized compensation cost for stock options due to the fact that all stock options were fully vested as noted above. For the years ended December 31, 2010, 2009 and 2008, the Company received $17.1 million, $14.2 million and $0.4 million in cash from stock option exercises, respectively.
The following table summarizes stock-based compensation expense incurred for the years ended December 31, in thousands:
| 2010 | 2009 | 2008 | ||||||||||
| Research and development |
$ | 2,470 | $ | 1,773 | $ | 1,136 | ||||||
| Selling, general and administrative |
7,470 | 4,663 | 3,620 | |||||||||
| Total |
$ | 9,940 | $ | 6,436 | $ | 4,756 | ||||||
(8) INCOME TAXES
As of December 31, 2010, the Company had U.S. federal net operating loss carryforwards of $90.3 million. Of this amount, approximately $21.7 million is limited by section 382 of the Internal Revenue Code. Of the Company’s remaining net operating loss carryforwards of approximately $68.6 million, approximately $51.2 million relates to excess stock option benefit which, if and when realized, will credit additional paid-in capital. The Company also has gross North Carolina net operating loss carryforwards in the amount of $53.8 million.
The provision for income taxes in the accompanying consolidated statements of operations for the years ended December 31 consisted of the following (in thousands):
| 2010 | 2009 | 2008 | ||||||||||
| Current: |
||||||||||||
| Federal |
$ | 10 | $ | (1,885 | ) | $ | (541 | ) | ||||
| State |
2,848 | (127 | ) | (701 | ) | |||||||
| Total current taxes |
2,858 | (2,012 | ) | (1,242 | ) | |||||||
| Deferred: |
||||||||||||
| Federal |
— | — | 1,126 | |||||||||
| State |
— | — | — | |||||||||
| Total deferred taxes |
— | — | 1,126 | |||||||||
| Total tax expense (benefit) |
$ | 2,858 | $ | (2,012 | ) | $ | (116 | ) | ||||
The “Worker, Homeownership, and Business Assistance Act of 2009”, passed in November 2009, extended the general carryback period for 2008 and 2009 applicable net operating losses from two years to up to five years. An applicable net operating loss is a net operating loss that arises in a tax year either beginning or ending in 2008 or 2009. These provisions of the Act also suspend the 90% limit on the utilization of alternative tax net operating losses against alternative minimum taxable income for all years in the carryback period. In the case of an election to carry back the applicable net operating loss to the fifth preceding year, the amount of the net operating loss carryback that may be
F-22
Table of Contents
SALIX PHARMACEUTICALS, LTD.
Notes to Consolidated Financial Statements — Continued
applied in the fifth carryback year is limited to 50% of taxable income of that fifth preceding year. The Company plans to carry back applicable net operating losses from 2008 three years to recapture the benefit due to the suspension of the 90% limit on alternative tax net operating losses. The Company received a benefit of $1.6 million related to this extended carryback.
The tax effects of temporary differences that give rise to significant portions of the deferred tax asset at December 31 are as follows (in thousands):
| 2010 | 2009 | |||||||
| Net operating loss carryforwards |
$ | 9,187 | $ | 40,759 | ||||
| Capitalized research and development expenses |
533 | 1,145 | ||||||
| Research and development credits |
20,629 | 18,915 | ||||||
| Other credits |
40 | 40 | ||||||
| Intangible impairment charges |
15,969 | — | ||||||
| Charitable contribution carryforward |
5,982 | — | ||||||
| Timing differences, including reserves, accruals, and writeoffs |
34,587 | 25,998 | ||||||
| Total deferred tax assets |
86,927 | 86,857 | ||||||
| Valuation allowance |
(48,727 | ) | (74,497 | ) | ||||
| Net deferred tax assets |
38,200 | 12,360 | ||||||
| Intangible assets |
— | (9,731 | ) | |||||
| Property, plant and equipment |
(684 | ) | — | |||||
| Debt discount and issuance costs |
(33,125 | ) | — | |||||
| Accrued return allowance |
— | (2,629 | ) | |||||
| 481 (a) adjustments |
(4,391 | ) | — | |||||
| Net deferred tax liabilities |
(38,200 | ) | (12,360 | ) | ||||
| Net deferred taxes |
$ | — | $ | — | ||||
The Company has provided a valuation allowance for the gross deferred tax asset due to the uncertainty regarding the Company’s ability to realize the entire asset. The valuation allowance has decreased by approximately $25.8 million during the year ended December 31, 2010, and increased by approximately, $14.1 million and $18.0 million during the years ended December 31, 2009 and 2008, respectively. The decrease for 2010 primarily relates to a decrease of $38.5 million related to our 2015 convertible notes issued in June 2010.
Utilization of the federal net operating loss carryforwards might be subject to a substantial annual limitation due to the change in ownership provisions of the Internal Revenue Code of 1986. If this limitation applies, the ultimate ability for the Company to use existing net operating loss carryovers and tax credit carryovers to offset future income may be limited. The Company’s net operating loss carryforwards begin to expire at the end of the taxable year ending December 31, 2021.
A reconciliation of the statutory income tax rate to the effective income tax rate is as follows:
| 2010 | 2009 | 2008 | ||||||||||
| Federal statutory income tax rate |
35.0 | % | 35.0 | % | 35.0 | % | ||||||
| State income taxes, net of federal income tax benefit |
6.8 | % | 1.9 | % | (0.5 | )% | ||||||
| Federal research and development credit |
6.4 | % | 2.2 | % | 14.8 | % | ||||||
| Meals and entertainment |
(6.5 | )% | (2.5 | )% | (1.6 | )% | ||||||
| Change in deferred tax estimate |
(0.6 | )% | (3.4 | )% | — | |||||||
| ASC 740-10 reserve |
(1.2 | )% | (2.0 | )% | — | |||||||
| Change in valuation allowance |
(50.3 | )% | (30.9 | )% | (40.6 | )% | ||||||
| Tax credit, non-deductible expenses and other |
(1.2 | )% | 4.1 | % | (6.9 | )% | ||||||
| (11.6 | )% | 4.4 | % | 0.2 | % | |||||||
F-23
Table of Contents
SALIX PHARMACEUTICALS, LTD.
Notes to Consolidated Financial Statements — Continued
The Company applies the provisions of ASC 740-10 with respect to accounting for uncertainty in income taxes. The unrecognized tax benefits as of December 31, 2010 and 2009 relate to federal tax credit carryforwards. The Company does not expect its unrecognized tax benefits to change significantly over the next 12 months.
The following table summarizes the activity related to our unrecognized tax benefits for the years ended December 31 (in thousands):
| 2010 | 2009 | 2008 | ||||||||||
| Balance at January 1 |
$ | 3,338 | $ | 2,448 | $ | 2,448 | ||||||
| Increases related to prior year tax positions |
593 | 890 | 18,413 | |||||||||
| Decreases related to prior year tax positions |
(290 | ) | — | (18,413 | ) | |||||||
| Balance at December 31 |
$ | 3,641 | $ | 3,338 | $ | 2,448 | ||||||
If these unrecognized tax benefits of $3.6 million at December 31, 2010 were recognized, there would have been no impact to the Company’s annual effective tax rate. No expense has been recorded for interest and penalties related to these tax positions.
The 1993 through 2009 tax years remain subject to examination by federal and state taxing authorities. The audit of the 2005 tax year by the Internal Revenue Service was closed during 2008.
(9) SIGNIFICANT CONCENTRATIONS
The Company operates in a single industry acquiring, developing and commercializing prescription drugs used in the treatment of a variety of gastrointestinal diseases, which are those affecting the digestive tract. The Company’s principal financial instruments subject to potential concentration of credit risk are accounts receivable, which are unsecured, and cash equivalents. The Company’s cash equivalents consist primarily of money market funds. The amount of bank deposits might at times exceed the FDIC insurance limits.
The Company’s primary customers are wholesale pharmaceutical distributors and retail pharmacy chains in the United States.
Total revenues from customers representing 10% or more of total revenues for the respective years, are summarized as follows:
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Customer 1 |
30 | % | 35 | % | 42 | % | ||||||
| Customer 2 |
30 | % | 28 | % | 24 | % | ||||||
| Customer 3 |
18 | % | 14 | % | 12 | % | ||||||
Additionally, 88% and 76% of the Company’s accounts receivable balances were due from these three customers at December 31, 2010 and 2009, respectively.
Under the Company’s supply agreement with Alfa Wassermann, the Company is obligated to purchase from Alfa Wassermann bulk rifaximin drug substance, the active pharmaceutical ingredient in Xifaxan 200mg rifaximin tablets and Xifaxan 550mg rifaximin tablets, until July 2014 or introduction of a generic product, whichever occurs first. The Company’s supply of rifaximin drug substance supplied by Alfa Wassermann is manufactured by ZaCh Systems in Lonigo, Italy, and Aventis in Brindisi, Italy. Under a supply agreement with Lupin, the Company is obligated to purchase 50% of its annual requirements of bulk rifaximin drug substance from Lupin. Under a long-term supply agreement, rifaximin is converted into Xifaxan drug product by Patheon, Inc. in Whitby, Ontario. Bulk Xifaxan tablets are packaged into finished Xifaxan commercial bottles by Pathoen and packaged into Xifaxan commercial blister packs by Pharma Packaging Solutions in Norris, Tennessee.
F-24
Table of Contents
SALIX PHARMACEUTICALS, LTD.
Notes to Consolidated Financial Statements — Continued
Under the Company’s long-term supply agreement with aaiPharma, aaiPharma produces the Company’s commercial supply of 25 mg, 75 mg and 100 mg Azasan finished product.
Under the Company’s long-term supply agreement with Paddock Laboratories in Minneapolis, Minnesota, Paddock produces the Company’s commercial supply of finished product of Anusol-HC Cream, Anusol-HC Suppositories, Proctocort Suppositories, Pepcid Oral Suspension and Diuril Oral Suspension. In addition, through prior supply arrangements between King Pharmaceuticals and Crown Laboratories in Johnson City, Tennessee, Crown continues to produce the Company’s commercial supply of Proctocort Cream finished product.
Under the Company’s supply agreement with WellSpring Pharmaceuticals in Oakville, Ontario, WellSpring will produce commercial supply of OsmoPrep finished product through October 2011. In September 2010 the Company entered into a supply agreement with Novel Laboratories, Inc. and Actavis Inc. Under this supply agreement the Company agreed to purchase from Actavis, all of its requirements of OsmoPrep beginning in October 2011.
Under the Company’s long-term supply agreement with Norgine in Hengoed, Wales, Norgine will produce commercial supply of MoviPrep finished kits through 2011. In August 2010 the Company entered into a supply agreement with Novel Laboratories, Inc. and Actavis Inc. Under this supply agreement the Company agreed to purchase from Actavis all of its requirements in excess of a certain amount of MoviPrep in 2011 and all of its requirements of MoviPrep beginning in 2012.
Bayer AG in Wuppertal, Germany supplies the Company with bulk mesalamine active ingredient. Under a long-term supply agreement with Catalent Pharma Solutions in Winchester, Kentucky, Catalent converts this mesalamine into the Company’s commercial supply of bulk Apriso, 375mg mesalamine capsules. The bulk Apriso capsules are then packaged into finished Apriso commercial bottles by Pharma Packaging Solutions in Norris, Tennessee.
Cosma S.P.A. in Bergamo, Italy supplies the Company with bulk metoclopramide active ingredient. Under a long-term supply agreement with Catalent in Swindon, United Kingdom, Catalent converts this metoclopramide into the Company’s commercial supply of Metozolv, 5mg and 10mg tablets in blister packaging. The Metozolv blister packs are then packaged into finished cartons by Pharma Packaging Solutions in Norris, Tennessee.
Under long-term supply agreements, the Company uses balsalazide drug substance, the active pharmaceutical ingredient in Colazal capsules, manufactured by Omnichem s.a., a subsidiary of Ajinomoto in Belgium, and Pharmazell (formerly Noveon Pharma, GmbH) in Raubling, Germany. Also, under long-term supply agreements, balsalazide is encapsulated into Colazal drug product by Nexgen Pharma, inc. (formerly Anabolic Laboratories) in Irvine, California. Bulk Colazal capsules are packaged into finished Colazal commercial bottles by Nexgen and Pharma Packaging Solutions in Norris, Tennessee.
With respect to the Company’s 1100 mg balsalazide tablet formulation currently under development, the Company plans to negotiate commercial supply agreements with the same manufacturers who supplied the drug substance and drug product for the supplies of the Phase III clinical trial material, if approved.
(10) 401(k) PLAN
In 1996, the Company adopted the Salix Pharmaceuticals, Inc. 401(k) Retirement Plan. Eligible participants may elect to defer a percentage of their compensation. From inception through June 2006, the Company matched up to 50% of participant deferrals up to 6% of the participant’s compensation. Effective July 2006, the Company matches up to 75% of participant deferrals up to 6% of the participant’s compensation. The Company’s total matching contributions for all participants were approximately $1.5 million, $1.2 million and $1.1 million in 2010, 2009 and 2008, respectively. Additional discretionary employer contributions may be made on an annual basis.
F-25
Table of Contents
SALIX PHARMACEUTICALS, LTD.
Notes to Consolidated Financial Statements — Continued
(11) COMMITMENTS
At December 31, 2010, the Company had binding purchase order commitments for inventory purchases aggregating approximately $87.3 million over the next five years, which includes any minimum purchase commitments under our manufacturing agreements.
Lease Agreements
The Company leases fleet vehicles for its domestic sales force under a four-year lease agreement. The vehicles were capitalized at $1.5 million, which approximates the present value of the minimum lease payments.
The Company leases office facilities under various non-cancelable operating leases, the last of which expires on April 30, 2015. Certain of these leases contain future payment obligations that escalate over time. Rent expense was approximately $1.5 million, $1.6 million and $1.5 million for the years ended December 31, 2010, 2009 and 2008, respectively. The Company sub-leased its former corporate headquarters of approximately 26,000 square feet of office space under a lease expiring in 2011, total future minimum sublease payments due under this sublease were $ 0.2 million as of December 31, 2010.
As of December 31, 2010, future minimum payments for leases were as follows (in thousands):
| Years ending December 31, |
Operating Leases |
Capital Leases |
||||||
| 2011 |
$ | 1,735 | $ | 409 | ||||
| 2012 |
1,490 | 92 | ||||||
| 2012 |
1,461 | — | ||||||
| 2014 |
1,490 | — | ||||||
| 2015 |
500 | — | ||||||
| Thereafter |
0 | — | ||||||
| Total future minimum payments required |
$ | 6,676 | 501 | |||||
| Less: Amount representing interest |
11 | |||||||
| Present value of net minimum lease payments |
490 | |||||||
| Less: Current portion of capital lease obligations |
400 | |||||||
| Long term portion of capital lease obligations |
$ | 90 | ||||||
Potential Milestone Payments
The Company has entered into collaborative agreements with licensors, licensees and others. Pursuant to the terms of these collaborative agreements, the Company is obligated to make one or more payments upon the occurrence of certain milestones. The following is a summary of the material payments that the Company might be required to make under its collaborative agreements if certain milestones are satisfied.
License Agreement with Dr. Falk Pharma GmbH for budesonide—In March 2008, the Company entered into a license agreement with Dr. Falk Pharma GmbH. The agreement provides the Company with an exclusive license to develop and commercialize in the United States Dr. Falk Pharma’s budesonide products. The products covered in the license agreement include U.S. patent-protected budesonide rectal foam and budesonide gastro-resistant capsule, patents for which expire in 2015 and 2016, respectively. Pursuant to the license agreement the Company is obligated to make an upfront payment and regulatory milestone payments that could total up to $23.0 million to Dr. Falk Pharma, with the majority contingent upon achievement of U.S. regulatory approval. As of December 31, 2010, the Company had paid $1.0 million of these milestone payments.
F-26
Table of Contents
SALIX PHARMACEUTICALS, LTD.
Notes to Consolidated Financial Statements — Continued
Development, Commercialization and License Agreement with Lupin Ltd—In September 2009, the Company entered into a development, commercialization and license Agreement with Lupin Ltd for Lupin’s proprietary drug delivery technology for rifaximin. The agreement provides that the Company is obligated to make upfront and milestone payments to Lupin that could total up to $53.0 million over the term of the agreement. As of December 31, 2010, we had paid $5.0 million of milestone payments. The remaining milestone payments are contingent upon achievement of certain clinical and regulatory milestones.
License Agreement with Merck & Co, Inc.—In February 2007, the Company entered into a master purchase and sale and license agreement with Merck, paying Merck $55.0 million to purchase the U.S. prescription pharmaceutical product rights to Pepcid® Oral Suspension and Diuril® Oral Suspension. Pursuant to the license agreement, the Company is obligated to make additional milestone payments to Merck up to an aggregate of $6.0 million contingent upon reaching certain sales thresholds during any of the five calendar years beginning in 2007 and ending in 2011. None of these sales thresholds had been met at December 31, 2010.
License Agreement with Napo Pharmaceuticals, Inc.—In December 2008 the Company entered into a collaboration agreement with Napo. Pursuant to the agreement, the Company has an exclusive, royalty-bearing license to crofelemer for the treatment of HIV-associated diarrhea and additional indications of pediatric diarrhea and acute infectious diarrhea in a certain territory. The Company also has a non-exclusive, worldwide, royalty-bearing license to use Napo-controlled trademarks associated with crofelemer. The Company has made an initial payment of $5.0 million to Napo and will make up to $50 million in milestone payments to Napo contingent on regulatory approvals and up to $250.0 million in milestone payments contingent on reaching certain sales thresholds. The Company is responsible for development costs of crofelemer, but costs exceeding $12.0 million for development of crofelemer used for the HIV-associated diarrhea indication will be credited towards regulatory milestones and thereafter against sales milestones. No milestone payments had been made as of December 31, 2010.
License and Supply Agreement with Norgine B.V.—In December 2005, the Company entered into a license and supply agreement with Norgine for the rights to sell NRL944, a bowel cleansing product the Company now markets in the United States under the trade name MoviPrep. Pursuant to the terms of this agreement, the Company is obligated to make upfront and milestone payments to Norgine that could total up to $37.0 million over the term of the agreement. As of December 31, 2010, the Company had paid $27.0 million of milestone payments. The remaining milestone payments are contingent upon reaching sales thresholds.
License Agreement with Photocure ASA—In October 2010, the Company entered into a license agreement with Photocure for the worldwide exclusive rights, excluding the Nordic region, to develop and commercialize LumacanTM for diagnosing, staging or monitoring gastrointestinal dysplasia or cancer. The Company made an initial payment of $4.0 million to Photocure, and will be responsible for development costs of Lumacan, but Photocure will reimburse us up to $3 million for certain out-of-pocket costs incurred by Salix. In addition, the Company is obligated to make up to $76.5 million in milestone payments to Photocure contingent on development and regulatory milestones, and up to $50.0 million in milestone payments contingent on reaching certain sales thresholds over the term of the agreement. No milestone payments had been made as of December 31, 2010.
License Agreement with Wilmington Pharmaceuticals, LLC—In September 2007, the Company entered into an Exclusive Sublicense Agreement with Wilmington Pharmaceuticals. The agreement provides that the Company is obligated to make upfront and milestone payments up to an aggregate amount of $8.0 million to Wilmington. As of December 31, 2010, the Company had paid these milestone payments in full. The Company also loaned Wilmington $2.0 million which was netted against the payment of the approval milestone as a result of FDA approval on September 8, 2009.
F-27
Table of Contents
SALIX PHARMACEUTICALS, LTD.
Notes to Consolidated Financial Statements — Continued
(12) QUARTERLY RESULTS OF OPERATIONS (UNAUDITED)
The following is a summary of the unaudited quarterly results of operations for the years ended December 31, 2010 and 2009:
| Mar 31 | June 30 | Sept 30 | Dec 31 | |||||||||||||
| (in thousands, except per share amounts) | (unaudited) | |||||||||||||||
| 2010 |
||||||||||||||||
| Net product revenue |
$ | 44,077 | $ | 93,773 | $ | 80,625 | $ | 118,498 | ||||||||
| Cost of products sold, excluding amortization of product rights and intangible assets |
9,799 | 17,160 | 14,497 | 27,221 | ||||||||||||
| Net income (loss) |
(25,204 | ) | (23,607 | ) | (2,721 | ) | 24,471 | |||||||||
| Net income (loss) per share, basic(1) |
(0.45 | ) | (0.41 | ) | (0.05 | ) | 0.42 | |||||||||
| Net income (loss) per share, diluted(1) |
(0.45 | ) | (0.41 | ) | (0.05 | ) | 0.40 | |||||||||
| 2009 |
||||||||||||||||
| Net product revenue |
$ | 44,774 | $ | 52,234 | $ | 65,658 | $ | 70,224 | ||||||||
| Cost of products sold, excluding amortization of product rights and intangible assets |
9,905 | 11,411 | 13,207 | 17,502 | ||||||||||||
| Net loss |
(13,966 | ) | (15,317 | ) | (7,315 | ) | (7,021 | ) | ||||||||
| Net loss per share, basic(1) |
(0.29 | ) | (0.32 | ) | (0.15 | ) | (0.13 | ) | ||||||||
| Net loss per share, diluted(1) |
(0.29 | ) | (0.32 | ) | (0.15 | ) | (0.13 | ) | ||||||||
| (1) | The sum of per share earnings by quarter may not equal earnings per share for the year due to the changes in average share calculations. This is in accordance with prescribed reporting requirements. |
(13) LEGAL PROCEEDINGS
From time to time, the Company is party to various legal proceedings or claims, either asserted or unasserted, which arise in the ordinary course of business. Management has reviewed pending legal matters and believes that the resolution of such matters will not have a significant adverse effect on the Company’s financial condition or results of operations.
Regulatory data exclusivity for Xifaxan 200mg tablets ended on or about May 24, 2009. Accordingly, the Office of Generic Drugs would have been able to accept an ANDA for Xifaxan tablets on or any time subsequent to May 24, 2008, if the applicant certified that its generic rifaximin does not infringe Salix’s patent. If this occurred, a Paragraph IV notification would have to be provided to the Company by the applicant. Although the Company does not know of any such filing at the current time, the expiration of data exclusivity could result in a challenge to the related intellectual property rights of Xifaxan 200mg tablets at any time in the future. The Company intends to vigorously enforce the patent rights for Xifaxan.
On or about July 14, 2008, Strides Arcolab Limited filed a Suitability Petition with the FDA seeking permission to submit an ANDA for change of dosage form from tablet to capsule as suitable for a 200mg generic version of Xifaxan. The Company intends to vigorously enforce the regulatory and intellectual property rights regarding Xifaxan. The Company is unable to predict the outcome of any ensuing regulatory action or litigation at the present time.
The Company is currently and might continue to be subject to product liability claims that arise through the testing, manufacturing, marketing and sale of its products, including a number of claims relating to OsmoPrep, Visicol and Metozolv ODT in connection with their respective “box” label warning. The Company intends to defend these claims vigorously but is currently unable to predict the outcome or to reasonably estimate the range of total potential expenses or losses, if any. The Company currently maintains liability coverage for its products but it is possible that this coverage, and any future coverage, will be insufficient to satisfy any liabilities that arise. The Company would have to assume defense of the lawsuits and be responsible for damages, fees and expenses, if any, that are awarded against it or for amounts in excess of the Company’s product liability coverage.
F-28
Table of Contents
SALIX PHARMACEUTICALS, LTD.
Notes to Consolidated Financial Statements — Continued
The Company filed a lawsuit against Novel Laboratories, Inc. because Novel was seeking FDA approval to market a generic version of the Company’s MoviPrep product. Norgine, B.V. and Norgine Europe, B.V. own U.S. Patent No. 7,169,381 (the ‘381 patent). The ‘381 patent is listed with the FDA as protecting the Company’s MoviPrep product. Norgine licensed MoviPrep and the ‘381 patent to the Company for commercialization in the United States. Novel filed an Abbreviated New Drug Application, or ANDA, with the FDA seeking approval to market a generic version of MoviPrep in the United States prior to the September 2024 expiration of the ‘381 patent. On May 14, 2008, the Company and Norgine filed a lawsuit in the United States District Court for the District of New Jersey against Novel for infringement of the ‘381 patent. Novel filed an Answer and Counterclaims on June 20, 2008. On June 25, 2009 the Company and Norgine amended the complaint to add a claim for correction of inventorship for the ‘381 patent. Novel filed an Answer and Counterclaims to the first Amended Complaint on July 10, 2009. Novel denied infringement and asserted various affirmative defenses, including defenses of patent invalidity and unenforceability. On February 9, 2010, an additional patent, U.S. Patent No. 7,658,914, was issued and listed in the Orange Book for MoviPrep.
On August 26, 2010 the parties agreed to a confidential settlement of the MoviPrep lawsuit which was approved by the Court on August 30, 2010 that resolved all of the parties’ outstanding claims and defenses in the lawsuit. The Consent Judgment provides that Novel’s proposed generic product, absent a license as granted to Novel under the settlement, would infringe the ‘381 patent, and further provides that Novel acknowledges and agrees not to contest the validity and enforceability of the ‘381 patent. Under the terms of the settlement, Novel was granted a fully paid up license under the MoviPrep patents such that it is permitted to launch a generic MoviPrep product on September 24, 2018, or earlier under certain circumstances.
The Company filed a second lawsuit against Novel because Novel was also seeking FDA approval to market a generic version of the Company’s OsmoPrep product. CDC, LLC, owns U.S. Patent No. 5,616,346 (the ‘346 patent). The ‘346 patent is listed with the FDA as protecting the Company’s OsmoPrep product. CDC, by its predecessor, licensed OsmoPrep and the ‘346 patent to the Company for commercialization in the United States. Novel filed an ANDA with the FDA seeking approval to market a generic version of OsmoPrep in the United States prior to the May 2013 expiration of the ‘346 patent. On September 8, 2008, the Company filed a lawsuit in the United States District Court for the District of New Jersey against Novel for the infringement of the ‘346 patent. The lawsuit also joined CDC as a party. Novel filed an Answer and Counterclaims on December 16, 2008. Novel denied infringement and asserted a defense of patent invalidity. On February 4, 2010 the Court conducted a patent claim construction, or Markman, hearing. On March 30, 2010, an additional patent, U.S. Patent No. 7,687,075 was issued and listed in the Orange Book for OsmoPrep.
On September 30, 2010 the parties agreed to a confidential settlement of the OsmoPrep lawsuit which was approved by the Court on the same day and resolved all of the parties’ outstanding claims and defenses in the lawsuit. The Consent Judgment provides that Novel’s proposed generic product, absent a license as granted to Novel under the settlement, would infringe the ‘346 patent and further provides that Novel acknowledges and agrees not to contest the validity and enforceability of the ‘346 patent. Under the terms of the settlement, Novel was granted a fully paid up license under the OsmoPrep patents such that it is permitted to launch a generic OsmoPrep product on November 16, 2019, or earlier under certain circumstances.
On or about March 19, 2010, the Company received a Warning Letter from the Division of Marketing, Advertising, and Communications of the FDA, or DDMAC, alleging that certain Metozolv ODT promotional material did not comply with applicable regulatory and legal parameters. The Company promptly ceased use of such material as an accommodation and implemented a corrective messaging plan. On or about December 27, 2010, DDMAC advised the Company in writing that, in light of the actions taken by the Company, DDMAC considers this matter closed. The Company is not aware of any other enforcement action concerning this matter.
F-29
Table of Contents
SALIX PHARMACEUTICALS, LTD.
Notes to Consolidated Financial Statements — Continued
(14) SUBSEQUENT EVENTS
On February 7, 2011 the Company announced that it had entered into an exclusive worldwide (except Japan) agreement by which the Company licensed rights to Relistor® (methylnaltrexone bromide). Relistor Subcutaneous Injection is indicated for the treatment of opioid–induced constipation (OIC) in patients with advanced illness who are receiving palliative care, when response to laxative therapy has not been sufficient. Relistor is a peripherally acting mu–opioid receptor antagonist that counteracts the constipating effects of opioid pain medications in the gastrointestinal tract without affecting their ability to relieve pain. The methylnaltrexone license includes intellectual property from the University of Chicago, Progenics Pharmaceuticals, and Wyeth Pharmaceuticals, including patents and applications with expiration dates that will range from 2017 through 2031. Relistor was approved in the United States in 2008, and currently the drug is approved for use in over 50 countries worldwide. In 2010, Relistor single–use, pre–filled syringes were approved for use in the United States, Canada and the European Union. Worldwide net sales of Relistor totaled $16 million in 2010. Financial terms of the transaction include a $60 million up–front payment and development milestones totaling $90 million, contingent upon the achievement of certain U.S. regulatory milestones. The Company will pay sales–based milestones of up to $200 million plus royalties on product sales in the U.S., as well as 60% of all revenue received from non–U.S. sublicensees. The Company will fund all development, registration and commercialization activities for Relistor in markets worldwide other than in Japan.
In February 2011 we gave notice to the landlord for our current corporate headquarters in Morrisville, NC under the terms of the lease that we are terminating this lease as of October 2011. In February 2011 we entered into a lease for approximately 127,000 square feet for a corporate headquarters in Raleigh, North Carolina, which begins in September 2011 and expires in 2023. We currently sub-lease our former corporate headquarters located in Raleigh, North Carolina, of approximately 26,000 square feet of office space under a lease expiring in 2011. In February 2011we extended the lease on this space through 2018, and we plan to re-occupy the space in 2011.
F-30
Table of Contents
SCHEDULE II—VALUATION AND QUALIFYING ACCOUNTS
Allowance for Rebates, and Chargebacks
| Beginning Balance |
Additions | Deductions | Ending Balance |
|||||||||||||||||
| Year ended December 31, |
Provision Related to Current Period Sales |
Rebates, Chargebacks and Discounts or Credits Related to Current Period Sales |
Rebates, Chargebacks and Discounts or Credits Related to Prior Period Sales |
|||||||||||||||||
| (in thousands) |
||||||||||||||||||||
| 2010 |
$ | 17,158 | $ | 77,877 | * | $ | 44,323 | $ | 9,879 | $ | 40,833 | |||||||||
| 2009 |
$ | 7,437 | $ | 28,815 | $ | 12,615 | $ | 6,479 | $ | 17,158 | ||||||||||
| 2008 |
$ | 9,676 | $ | 13,576 | $ | 10,333 | $ | 5,482 | $ | 7,437 | ||||||||||
Allowance for Returns
| Beginning Balance |
Additions | Deductions | Ending Balance |
|||||||||||||
| Year ended December 31, |
Provision Related to Current Period Sales |
Returns or Credits Related to Prior Period Sales |
||||||||||||||
| (in thousands) |
||||||||||||||||
| 2010 |
$ | 14,883 | $ | 15,646 | * | $ | 10,824 | $ | 19,705 | |||||||
| 2009 |
$ | 26,597 | $ | 4,599 | $ | 16,313 | $ | 14,883 | ||||||||
| 2008 |
$ | 43,322 | $ | 4,074 | $ | 20,799 | $ | 26,597 | ||||||||
| * | 2010 activity includes an aggregate of approximately $7.0 million of changes in estimates for Colazal. |
Allowance for Uncollectible Accounts
| Beginning Balance |
Additions | Deductions | Ending Balance |
|||||||||||||
| Year ended December 31, |
Charged to Costs and Expenses |
Accounts Written Off During Period |
||||||||||||||
| (in thousands) |
||||||||||||||||
| 2010 |
$ | 508 | — | — | $ | 508 | ||||||||||
| 2009 |
$ | 565 | — | $ | 57 | $ | 508 | |||||||||
| 2008 |
$ | 3,112 | — | $ | 2,547 | $ | 565 | |||||||||
Inventory Allowance
| Beginning Balance |
Additions | Deductions | Ending Balance |
|||||||||||||
| Year ended December 31, |
Charged to Costs and Expenses |
Amounts Recovered During Period |
||||||||||||||
| (in thousands) |
||||||||||||||||
| 2010 |
$ | 2,783 | $ | 4,339 | $ | 491 | $ | 6,631 | ||||||||
| 2009 |
$ | 3,768 | — | $ | 985 | $ | 2,783 | |||||||||
| 2008 |
$ | 789 | $ | 3,297 | $ | 318 | $ | 3,768 | ||||||||
Valuation Allowance on Deferred Tax Assets
| Beginning Balance |
Additions | Deductions | Ending Balance |
|||||||||||||
| Year ended December 31, |
Provisions for Valuation Allowance |
Release of Valuation Allowance /Other |
||||||||||||||
| (in thousands) |
||||||||||||||||
| 2010 |
$ | 74,497 | — | $ | 25,771 | $ | 48,726 | |||||||||
| 2009 |
$ | 60,378 | $ | 14,119 | — | $ | 74,497 | |||||||||
| 2008 |
$ | 42,345 | $ | 18,033 | — | $ | 60,378 | |||||||||
F-31