Attached files
| file | filename |
|---|---|
| EX-21 - EX-21 - Talecris Biotherapeutics Holdings Corp. | a11-1850_1ex21.htm |
| EX-31.2 - EX-31.2 - Talecris Biotherapeutics Holdings Corp. | a11-1850_1ex31d2.htm |
| EX-32.2 - EX-32.2 - Talecris Biotherapeutics Holdings Corp. | a11-1850_1ex32d2.htm |
| EX-23.1 - EX-23.1 - Talecris Biotherapeutics Holdings Corp. | a11-1850_1ex23d1.htm |
| EX-31.1 - EX-31.1 - Talecris Biotherapeutics Holdings Corp. | a11-1850_1ex31d1.htm |
| EX-32.1 - EX-32.1 - Talecris Biotherapeutics Holdings Corp. | a11-1850_1ex32d1.htm |
| EX-10.32 - EX-10.32 - Talecris Biotherapeutics Holdings Corp. | a11-1850_1ex10d32.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
Form 10-K
(Mark One)
|
x |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2010
or
|
o |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission file number: 001-34473
TALECRIS BIOTHERAPEUTICS HOLDINGS CORP.
(Exact name of registrant as specified in its charter)
|
Delaware |
|
20-2533768 |
|
(State or other jurisdiction of |
|
(IRS Employer Identification No.) |
P.O. Box 110526
4101 Research Commons
79 T.W. Alexander Drive
Research Triangle Park, North Carolina 27709
(Address of principal executive offices, including
Zip Code)
(919) 316-6300
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
|
Title of each class |
|
Name of each exchange on which |
|
Common Stock, $0.01 par value |
|
The NASDAQ Global Select Market |
Securities registered pursuant to section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files) Yes o No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
|
Large accelerated filer x |
|
Accelerated filer o |
|
|
|
|
|
Non- accelerated filer o |
|
Smaller reporting company o |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes o No x
The aggregate market value of the voting stock held by non-affiliates of the registrant, computed by reference to the closing price as of the last business day of the registrant’s most recently completed second fiscal quarter, June 30, 2010, was approximately $1.3 billion. The registrant has no non-voting common stock.
Indicate the number of shares outstanding of each of the registrant’s classes of common stock, as of the latest practicable date. 125,816,959 shares of Common Stock, $0.01 par value, as of February 21, 2011.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the Talecris Biotherapeutics Holdings Corp. Definitive Proxy Statement to the 2011 Annual Meeting of Stockholders to be filed with the Securities and Exchange Commission within 120 days after the close of the registrant’s fiscal year are incorporated by reference in Part III to the extent described therein.
Talecris Biotherapeutics Holdings Corp.
2010 Annual Report on Form 10-K
|
|
|
|
|
Page |
|
|
1 | |||
|
|
|
|
2 | |
|
|
|
2 | ||
|
|
|
18 | ||
|
|
|
42 | ||
|
|
|
42 | ||
|
|
|
43 | ||
|
|
|
|
|
|
|
|
|
|
46 | |
|
|
|
46 | ||
|
|
|
48 | ||
|
|
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
|
50 | |
|
|
|
91 | ||
|
|
|
93 | ||
|
|
Changes in and Disagreements With Accountants on Accounting and Financial Disclosure |
|
141 | |
|
|
|
141 | ||
|
|
|
141 | ||
|
|
|
|
|
|
|
|
|
|
142 | |
|
|
|
142 | ||
|
|
|
143 | ||
|
|
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
|
144 | |
|
|
Certain Relationships and Related Transactions, and Director Independence |
|
144 | |
|
|
|
144 | ||
|
|
|
|
|
|
|
|
|
|
145 | |
|
|
|
145 | ||
|
|
|
|
149 | |
Special Note Regarding Forward-Looking Statements
This Annual Report on Form 10-K (Annual Report) contains forward-looking statements that involve substantial risks and uncertainties. All statements, other than statements of historical facts, included in this Annual Report, regarding our strategy, future operations, future financial position, future revenue, projected costs, prospects, plans and objectives of management are forward-looking statements. Forward-looking statements may be identified by the use of forward-looking terms such as “may,” “will,” “would,” “expects,” “intends,” “believes,” “anticipates,” “plans,” “predicts,” “estimates,” “projects,” “targets,” “forecasts,” “seeks,” or the negative of such terms or other variations on such terms or comparable terminology. The forward-looking statements that we make are based upon assumptions about many important risk factors, many of which are beyond our control. Among the factors that could cause actual results to differ materially are the following:
· the impact of the announcement of our definitive merger agreement with Grifols and the potential impact of completion, termination, or delay of the proposed merger with Grifols, including, but not limited to, disruptions from the pending transaction, transaction costs, and the outcome of litigation and regulatory proceedings to which we may be a party;
· fluctuations in the balance between supply and demand with respect to the market for plasma-derived products;
· the unprecedented volatility in the global economy and fluctuations in financial markets;
· changes in economic conditions, political tensions, trade protection measures, licensing requirements, and tax matters in the countries in which we conduct business;
· the impact of competitive products and pricing;
· recently enacted and additional proposed U.S. healthcare legislation, regulatory action or legal proceedings affecting, among other things, the U.S. healthcare system, pharmaceutical pricing and reimbursement, including Medicaid, Medicare and the Public Health Service Program and additional legislation and regulatory action now under consideration;
· legislation or regulations in markets outside of the U.S. affecting product pricing, reimbursement, access, or distribution channels;
· our ability to procure adequate quantities of plasma and other materials which are acceptable for use in our manufacturing processes from our own plasma collection centers or from third-party vendors;
· our ability to maintain compliance with government regulations and licenses, including those related to plasma collection, production, and marketing;
· our ability to identify growth opportunities for existing products and our ability to identify and develop new product candidates through our research and development activities;
· the timing of, and our ability to, obtain and/or maintain regulatory approvals for new product candidates, the rate and degree of market acceptance, and the clinical utility of our products;
· unexpected shut-downs of our manufacturing and storage facilities or delays in opening new planned facilities;
· our and our suppliers’ ability to adhere to cGMP;
· our ability to manufacture at appropriate scale to meet the market’s demand for our products;
· our ability to resume or replace sales to countries affected by our Foreign Corrupt Practices Act (FCPA) investigation;
· potential sanctions, if any, that the Department of Justice (DOJ) or other federal agencies, may impose on us as a result of our internal FCPA investigation;
· the impact of the PCA judgment;
· the impact of geographic and product mix on our sales and gross profit;
· foreign currency exchange rate fluctuations in the international markets in which we operate;
· the impact of our substantial capital plan; and
· other factors identified elsewhere in this Annual Report.
No assurances can be provided as to any future financial results. Our forward-looking statements do not reflect the potential impact of any future acquisitions, mergers, dispositions, joint ventures, or investments we may make. Unless legally required, we do not undertake to update or revise any forward-looking statements, even if events make it clear that any projected results, expressed or implied, will not be realized.
Unless otherwise stated or the context otherwise requires, references in this Annual Report to “Talecris,” “we,” “us,” “our” and similar references refer to Talecris Biotherapeutics Holdings Corp. and its wholly-owned subsidiaries.
We are a biopharmaceutical company that, according to the Marketing Research Bureau (MRB), is one of the largest producers and marketers of plasma-derived protein therapies in the world. We develop, produce, market, and distribute therapies that extend and enhance the lives of people suffering from chronic and acute, often life-threatening, conditions, such as chronic inflammatory demyelinating polyneuropathy (CIDP), primary immune deficiencies (PI), alpha-1 antitrypsin deficiency, bleeding disorders, infectious diseases and severe trauma. Our products are derived from human plasma, the liquid component of blood, which is sourced from our plasma collection centers or purchased from third party plasma collection centers in the United States. Plasma contains many therapeutic proteins which we extract through a process known as fractionation at our Clayton, North Carolina and Melville, New York facilities. The fractionated intermediates are then purified, formulated into a final bulk, and aseptically filled into final containers for sale. We also sell fractionated intermediate products. Our manufacturing facilities currently have the capacity to fractionate approximately 4.2 million liters of human plasma per year. Purification, filling and finishing capacities are dependent on fraction mix.
We believe that many plasma-derived products are underutilized and have positive growth outlook. We believe worldwide unit volume demand for plasma-derived products will grow over the long term at a compound annual rate of approximately 5% to 8%, driven principally by the following factors:
· population growth;
· discovery and approval of new applications and indications for plasma-based products;
· growth of diagnosed cases;
· treatment of previously diagnosed, but untreated patients;
· increased patient compliance and appropriate dosing levels for diagnosed, treated patients; and
· increased utilization in developing countries.
Products
According to MRB, our largest product, Gamunex, Immune Globulin Intravenous (Human), 10% Caprylate/Chromatography Purified (Gamunex, Gamunex IGIV), is one of the leading products in the intravenous immune globulin (IGIV) segment, with a reputation as a premium product. Gamunex and its successor in the United States and Canada, Gamunex-C Immune Globulin Injection (Human) 10% Caprylate/Chromatography Purified, are the only IGIV products approved for the treatment of CIDP, a neurological indication, in the U.S. and Canada and, through a Mutual Recognition Procedure, in 16 European countries. The Gamunex IGIV share of sales was 23% in the U.S. in 2009 and 14% globally in 2008 based on data from MRB. Our second largest product, Prolastin Alpha-1 Proteinase Inhibitor (Human) and its successor in the United States and Canada, Prolastin-C Alpha 1 Proteinase Inhibitor (Human), had a 62% share of sales in the United States in 2009 and a 74% share of sales worldwide in 2008 and has a high degree of brand recognition within the alpha-1 proteinase inhibitor, or A1PI, category. Gamunex and Prolastin/Prolastin-C A1PI, together represented 76.4% of our net revenue in 2010. We also have a line of hyperimmune therapies that provide treatment for tetanus, rabies, hepatitis B, hepatitis A and Rh factor control during pregnancy and at birth. In addition, we provide plasma-derived therapies for critical care, including the treatment of hemophilia, an anti-coagulation factor, as well as albumin to expand blood volume. Although we sell our products worldwide, the majority of our sales are concentrated in the United States and Canada. Our products are primarily prescribed by specialty physicians, including neurologists, immunologists, pulmonologists, and hematologists. Our six key products categories and their indications are given in the table below:
|
Category and Talecris |
|
Talecris Indications |
|
Talecris |
|
Talecris Net |
|
IGIV Gamunex-C Gamunex IGIV |
|
U.S., Canada and EU—PI,ITP, CIDP. |
|
23%—U.S.(1) |
|
$660.0—U.S. |
|
|
Canada and EU—Post Bone Marrow Transplant, Pediatric HIV Infection. |
|
14%—Worldwide(2) |
|
$871.6(3)—Worldwide | |
|
|
|
EU only—Kawasaki Disease, Guillain Barre Syndrome, Chronic Lymphocytic Leukemia, Multiple Myeloma |
|
|
|
|
|
A1PI |
|
A1PI Deficiency related emphysema |
|
62%—U.S.(1) |
|
$231.7—U.S. |
|
|
|
|
74%—Worldwide(2) |
|
$351.5—Worldwide | |
|
Fraction V (Albumin and PPF) |
|
Plasma expanders, severe trauma, acute liver and kidney failures |
|
13%—U.S.(1) 5%—Worldwide(2) |
|
$46.7—U.S. $78.4(3)—Worldwide |
|
Factor VIII(4) |
|
Hemophilia A |
|
0.8%—U.S.(1) 1.3%—Worldwide(2) |
|
$17.6—U.S. $57.0—Worldwide |
|
Antithrombin III |
|
Heriditary antithrombin III deficiency |
|
92%—U.S.(1) 6%—Worldwide(2) |
|
$30.2—U.S. $30.2—Worldwide |
|
Hyperimmunes |
|
Hepatitis A, Hepatitis B, Rabies, RH Sensitization, Tetanus |
|
20%—U.S.(1) 7.5%—Worldwide(2) |
|
$53.3—U.S. $69.8—Worldwide |
(1) For the 2009 calendar year, according to MRB. The Plasma Fractions Market in the United States, 2009.
(2) For the 2008 calendar year, according to MRB. The Worldwide Fractions Market, 2008.
(3) Excludes contract fractionation revenues from the Canadian blood system operators.
(4) Sales include plasma-derived and recombinant Factor VIII products but exclude von Willenbrands.
The majority of our sales are concentrated in the therapeutic areas of: Immunology/Neurology, primarily through our IGIV product for the treatment of primary immune deficiency and CIDP, and Pulmonology, through our alpha-1 proteinase inhibitor (A1PI) product for the treatment of alpha-1 antitrypsin deficiency-related emphysema. These therapeutic areas are served by our branded products, Gamunex and Gamunex-C brands IGIV (Gamunex, Gamunex-C), Prolastin and Prolastin-C brands A1PI (Prolastin, Prolastin A1PI, Prolastin-C A1PI). Our six largest product categories and net revenues are included in the following table.
|
|
|
|
|
Net Revenue (in millions) |
| |||||||
|
Category |
|
|
|
2010 |
|
2009 |
|
2008 |
| |||
|
IGIV(1) |
|
|
|
$ |
871.6 |
|
$ |
826.4 |
|
$ |
677.7 |
|
|
A1PI |
|
|
|
$ |
351.5 |
|
$ |
319.1 |
|
$ |
316.5 |
|
|
Fraction V (Albumin and PPF) (1) |
|
|
|
$ |
78.4 |
|
$ |
84.8 |
|
$ |
61.1 |
|
|
Factor VIII |
|
|
|
$ |
57.0 |
|
$ |
46.5 |
|
$ |
40.2 |
|
|
Antithrombin III |
|
|
|
$ |
30.2 |
|
$ |
24.2 |
|
$ |
21.3 |
|
|
Hyperimmunes |
|
|
|
$ |
69.8 |
|
$ |
74.2 |
|
$ |
78.2 |
|
(1) Excludes contract fractionation revenues from the Canadian blood system operators.
We have a strong product portfolio with over ten licensed products focused on what we believe to be under diagnosed, underdeveloped markets. The following is a discussion of our key products or product classes:
IGIV—Gamunex-C/Gamunex IGIV
IGIV products are antibody-rich plasma therapies that have long been used in the treatment of immune related disorders such as primary immune deficiencies and certain autoimmune disorders, such as CIDP. For many indications, IGIV is thought to act as an immune modulator; however, in most cases formal regulatory approvals have not been obtained. We believe that the overall unit demand for IGIV is still significantly underdeveloped, due to under-diagnosis of conditions amenable to IGIV therapy, physician under-dosing for current indications and underutilization for many indications where it has demonstrated efficacy. We believe demand for IGIV products will generally increase as a result of new European Medicines Agency (EMA) and FDA approved indications, physician education on diagnosis and treatment options and development of consensus guidelines (to ensure appropriate therapeutic use and optimal dosing).
Our product, Gamunex-C, is a ready-to-use 10% liquid. Gamunex-C has the most approved indications of any liquid IGIV currently marketed in the U.S. Further, the FDA granted Gamunex-C/Gamunex IGIV orphan drug status, which provides marketing exclusivity for the CIDP indication in the U.S. until September 2015. According to an independent study by Harris Interactive, CIDP is the largest therapeutic use of IGIV volume, representing 29% of the total U.S. IGIV unit volume. We believe Gamunex-C/Gamunex IGIV indication for CIDP doubles our market access for licensed indications to 61% of total U.S. IGIV unit volume in the U.S. In an online survey conducted by Harris Interactive on our behalf during the first quarter of 2010, Gamunex was shown to be the preferred IGIV among neurologists who indicated a brand preference. The survey results showed that neurologists selected Gamunex over four times more often than all other available liquid IGIV therapies, with a statistically significant margin (p<0.05). In 2009, we submitted an sBLA with the FDA and an sNDS with Health Canada for subcutaneous route of administration for Gamunex IGIV for treatment of PI. In May 2010 and October 2010, Gamunex-C/Gamunex IGIV was approved for the subcutaneous route of administration for the PI indication in Canada and the U.S., respectively.
The approved indications for Gamunex-C/Gamunex IGIV in the U.S. and Canada and Gamunex in 17 countries in the European Union are Primary Humoral Immunodeficiency (PI), and Idiopathic Thrombocytopenic Purpura (ITP). Chronic Inflammatory Demyelinating Polyneuropathy (CIDP) is also an approved indication for Gamunex-C/Gamunex IGIV in the United States and Canada and for Gamunex in 16 European countries. Gamunex IGIV is also approved in the European Union for post bone marrow transplant and pediatric HIV infection as is Gamunex-C/Gamunex IGIV in Canada. Gamunex IGIV is approved in the European Union for Kawasaki Disease, Guillain Barre Syndrome, Chronic Lymphocytic Leukemia and Multiple Myeloma.
A1PI—Prolastin/Prolastin-C
A1PI is a naturally occurring, self-defensive protein produced in the liver. A1PI is used to treat congenital A1PI deficiency-related emphysema. This deficiency may predispose an individual to several illnesses but most commonly appears as emphysema in adults. U.S. sales of A1PI have experienced a compound annual growth rate of 15% between 1996 and 2009. Our Prolastin A1PI product represented 74% of worldwide A1PI sales in 2008 and 62% of U.S. A1PI sales in 2009 according to MRB.
Prolastin/Prolastin-C A1PI has the leading share of sales in the U.S., and approximately 87% share of sales in the European Union in 2008 according to MRB. From 1987 when our A1PI product, Prolastin A1PI, was granted orphan drug status, until 2003, when competitors began selling in the U.S., Prolastin had 100% share of A1PI sales in the U.S. Prolastin had a 62% share of U.S. sales in 2009 according to MRB. As a result of our first-mover advantage, pricing, brand strength and direct-to-patient distribution model, we have lost very few of our patients to competitors, which rely on identification of new patients to establish their market share. We consistently experience patient losses due to the nature of the disease. Globally, we continue to focus on access to new markets and patient identification efforts, including distribution of diagnostic kits as a way to increase sales of A1PI, and are seeking reimbursement in several European countries.
We believe there are approximately 11,000 individuals currently identified with A1PI deficiency in North America and Europe with 5,500 of those individuals currently undergoing A1PI treatment, based on internal estimates. There are an estimated 200,000 individuals with A1PI deficiency at high risk for development of emphysema in North America and Europe. Many individuals with symptoms are misdiagnosed before receiving a diagnosis of A1PI deficiency-related emphysema. Based on patient registries in many European countries, we believe that severe A1PI deficiency is also prevalent in Europe, and that European patients may represent approximately 30% of potential global sales.
Epidemiological surveys have demonstrated that there is significant latent demand for A1PI as only approximately 10% of all patients in need of treatment have been identified (source: Alpha-1-antitrypsin deficiency. High prevalence in the St. Louis area determined by direct population screening. Silverman EK, et al. Am Rev Respir Dis. 1989; 140:961-966). Even fewer patients are being treated using an A1PI product due to the limited number of countries with licensed product. This represents two distinct opportunities for market expansion—improved disease awareness leading to increased patient identification, and gaining licenses in new markets where there has not been access to A1PI.
We completed the conversion of our existing U.S. and Canadian Prolastin A1PI patients to our newer A1PI product Prolastin-C A1PI in 2010. Prolastin-C A1PI has improved yields, higher concentration, and significantly reduced infusion time. We believe Prolastin-C A1PI is differentiated in the United States by its unique direct-to-patient distribution and service model, Prolastin Direct, which provides easy enrollment, home infusion, access to insurance experts and patient-centered health management. Prolastin Direct health management provides better patient outcomes by reducing the frequency of respiratory exacerbations. Furthermore, Prolastin Direct results in high medication compliance, with over 94% of prescribed doses being administered annually and high patient loyalty, with an annual retention rate of over 96%. Unlike our competitors, our Prolastin-C A1PI product are primarily shipped in the U.S. directly to the patient through Centric Health Resources (Centric), a specialized pharmacy. We own 30% of Centric’s common stock as of December 31, 2010.
In 2006, we completed a Mutual Recognition Procedure to sell product in European countries with significant identified patient populations. Prolastin is approved in 15 European countries and we are currently established in six of these markets. We have been in reimbursement discussions with a number of these countries since late 2007 and these discussions must be concluded before we can expect to significantly increase sales in these countries. Competitors are currently only licensed in the U.S., Spain and France.
Hyperimmunes
Hyperimmunes are antibody rich preparations, the majority of which are used to provide antibodies to counter specific antigens. Other products, collectively referred to as hyperimmune globulins, are made from human plasma collected from donors with immunity to specific diseases. We have one of the broadest lines of FDA-approved hyperimmunes for hepatitis A, hepatitis B, rabies, tetanus and treatment of Rh negative women pregnant with Rh positive children.
We had the largest share of sales in the U.S. for HyperRab and HyperTet in 2009 and 2010 according to MRB.
Albumin and PPF
Albumin is the most abundant protein in human plasma. It is a protein synthesized by the liver and performs multiple functions, including the transport of many small molecules in the blood and the binding of toxins and heavy metals, which prevents damage they might otherwise cause.
Plasma Protein Factor (Human) (PPF) has similar therapeutic uses as albumin and both are derived from plasma Fraction V. We are licensed to produce and market albumin and PPF under the brand names: Plasbumin and Plasmanate.
Some of our indications for albumin are:
· Emergency treatment of Hypovolemic Shock
· Burn therapy
· Hypoproteinemia with or without edema
· Adult Respiratory Distress Syndrome (ARDS)
· Cardiopulmonary bypass
· Acute Liver Failure
· Neonatal Hemolytic Disease
· Acute Nephrosis
· Eythrocyte Resuspension
· Renal Dialysis
We are planning to expand our production capacity to support future growth.
Plasma-Derived Hematology Products
Plasma-derived hematology products are used to treat patients who either lack one of the necessary factors for blood clotting or suffer from conditions in which clotting occurs abnormally. There are 13 blood coagulation factors found in human blood.
· We produce plasma-derived Factor VIII, called Koate DVI. Factor VIII is the primary treatment for Hemophilia A, a congenital bleeding disorder caused by a deficiency of coagulation agents in the blood. Total sales of hemostasis products, including recombinant products, in 2009 were $2.3 billion in the U.S. Sales of plasma-derived hemostasis products in 2009 were $477.9 million in the U.S. In 2008, total sales were $7.0 billion worldwide (including plasma-derived and recombinant FVIII, FIX, ATIII, von Willenbrands, and FVII), while plasma-derived only product sales were $2.5 billion (includes plasma-derived Factor VIII, FIX, ATIII, and von Willebrands and FVII). We are expanding production capabilities in a phased approach to help meet demand.
· ATIII is an important anticoagulant and ATIII therapies are designed to treat and prevent thromboemboli, or spontaneous clotting within vital organs, in patients with congenital ATIII deficiency during high risk surgery, trauma, pregnancy, or childbirth. Our ATIII product, Thrombate III, represented 92% of sales in the U.S. in 2009 and 6% of worldwide sales in 2008 according to MRB, Thrombate III is currently produced for us by Bayer pursuant to a manufacturing agreement. We are currently validating a new production facility at our Clayton, North Carolina site with regulatory approval expected in 2012. We believe that we have sufficient inventory of intermediates and finished product to meet demand until the new facility is approved. The new facility will increase our capacity and will allow us to increase supply.
PPF-powder Intermediate Sales
Separately from our sales of PPF packaged for final use, we provide PPF powder to Bayer as an intermediate product for the fermentation of Kogenate, Bayer’s recombinant Factor VIII product. We will continue to provide PPF powder to the Bayer Kogenate business through 2012 pursuant to a supply agreement with potential extensions at Bayer’s option to 2015.
Manufacturing and Raw Materials
Our Clayton, North Carolina manufacturing site is one of the world’s largest fully integrated facilities for plasma-derived therapies. The site includes plasma receiving, fractionation, purification, filling/freeze-drying and packaging capabilities as well as freezer storage, testing laboratories and a cGMP pilot plant for clinical supply manufacture. In addition, we have a manufacturing facility in Melville, NewYork that provides additional fractionation capacity as well as capabilities for other contract manufacturing services. In addition to the on-site freezer storage, we also utilize a leased facility which allows for expanded inventory storage capacity.
Our manufacturing facilities currently have the capacity to fractionate approximately 4.2 million liters of human plasma per year. We processed approximately 3.8 million liters of plasma in 2010, which represents a utilization rate of approximately 90.5% of our fractionation capacity. The majority of the capacity is used for internal production requirements with a small amount utilized for contract fractionation, mainly for the Canadian blood system operators. We anticipate that we will reach our fractionation capacity in the near term depending upon the demand for our products, the availability of source plasma, the impact of variability in yield, potential inventory impairments, among other factors. To allow full fractionation capacity utilization of 4.2 million liters, harvest of albumin paste will be capped at 2.3 million liters.
We are planning to expand our fractionation capacity with a new facility that will be able to process annually, 6.0 million liters of Crypoprecipitate (Factor VIII) and II+III paste (IGIV) and 4.0 million liters for IV-I paste (Alpha-1). Fraction V paste (albumin) will continue to be fractionated in our existing facility in the mid-term at a capacity of 4.0 million liters. Purification, filling, and finishing capacities are dependent on fraction and vial size mix.
Talecris Plasma Resources
Plasma is the key raw material used in the production of plasma-derived biological products, representing greater than 50% of our cost of goods sold. Human plasma can be secured through internal or external collection networks or sources. As of December 31, 2010, we operated 69 plasma collection centers (67 FDA licensed, two unlicensed) with approximately 2,700 employees. Over the past four years, we have aggressively expanded our plasma supply through these collection centers under our wholly-owned subsidiary, TPR. These centers collectively represent substantially all of our currently planned collection center network for the next three years. We expect this network, once it fully matures, will provide in excess of 90% of our current plasma requirements. Our licensed centers collected approximately 69% of our plasma during the year ended December 31, 2010.
The rapid vertical integration of our plasma supply was accomplished through the development of an extensive infrastructure necessary to manage the multiple work streams to open and obtain FDA licenses at our centers, as well as the ramp up of their production. Prior to the execution of our vertical integration strategy, we relied exclusively on third parties for all of our plasma, a significant portion of which was provided to us through plasma collection centers owned or controlled by our competitors. Through the successful execution of our strategy, we have been able to reduce our reliance on third party suppliers, enhance our flexibility in procuring plasma, increase our operating efficiencies and improve our gross margin and profitability by reducing levels of under absorbed TPR infrastructure and start-up costs as a result of the maturation of our TPR plasma center platform.
We intend to continue to purchase some plasma from third parties through plasma supply contracts. On August 12, 2008, we signed a five-year plasma supply agreement with CSL Plasma Inc., a subsidiary of CSL Limited, a major competitor. This agreement provides us with minimum annual purchase commitments that decline from 550,000 liters in 2010 to 200,000 liters in 2013, the final year of the agreement. We have the ability to obtain additional volumes above the minimum purchase commitments under the terms of the agreement. CSL Plasma, Inc. is obligated to supply 300,000 liters of plasma to us in 2011 as we have not elected to take optional volumes for the 2011 contract year. In addition to the contract with CSL Plasma Inc., we have several other contracts to purchase minimum quantities of plasma with various third parties.
We plan to source our plasma supply through the continued growth of our plasma collection center platform and through our plasma supply contracts with third parties. In addition to the procurement of plasma on occasion, we have also purchased intermediate materials needed for the production of specific fractions. These materials are purchased from fractionators that have either excess capacity or do not have the processes to manufacture the final product.
We will need to significantly increase plasma collections generated from TPR in the near term to offset the expected decrease in plasma supplied by third parties and planned increases in our fractionation to meet anticipated demand. To meet our plasma requirements, we have increased donor fees, increased marketing expenses, and expanded plasma center hours of operations, among other initiatives, which may limit our ability to reduce our cost per liter of plasma.
Pathogen safety
Assuring the pathogen safety of our products is a priority for us. There are a number of steps used to help ensure the pathogen safety of the source plasma we use and the products we produce. The initial step is the application of donor qualification procedures by our plasma suppliers. We also test donated plasma for serum antibodies. The purpose of serological testing is to detect serum antibodies and other biological markers that appear specifically in association with certain diseases. Serological testing is provided under contract by qualified laboratories according to our specifications.
Prior to delivery of the source plasma from our suppliers to our manufacturing plant, we internally perform nucleic acid amplification testing, or NAT, for various viruses, including HBV, HCV, HIV, HAV, and B-19. Our ability to perform NAT testing internally provides us with a strategic advantage over competitors who do not have such facilities and must contract with a third party, the National Genetics Institute. We perform these tests in a 76,000 square foot testing facility, located in Raleigh, North Carolina. The facility is leased through September 2017, with an option to purchase through September 2011. The laboratory tested approximately 4.9 million samples in 2010 and is operated by 106 operations and quality employees.
Research and Development
As a result of our past and ongoing investment in research and development, we believe that we are positioned to continue as a leader in the plasma-derived therapies industry. Innovation by our research and development operations is critical to our future growth and ability to remain competitive in our industry. We have a strong commitment to science and technology with a track record of accomplishments and pipeline opportunities. As of December 31, 2010, we had 308 scientists and support staff engaged in research and development activities. We focus our research and development efforts in three key areas: continued enhancement of our process technologies (including pathogen safety), life cycle management for our existing products (including new indications), and development of new products. To the extent we wish to add new products to our research and development pipeline, we anticipate making opportunistic business acquisitions or partnering with other companies with projects that fit our expertise. Our research and development spending was $69.6 million, $71.2 million, and $66.0 million for the years ended December 31, 2010, 2009, and 2008, respectively.
The following table includes information regarding the clinical development stage of various product candidates currently in our development pipeline:
|
Product Candidate |
|
Therapeutic Area |
|
Product Type |
|
Use |
|
Development Phase |
|
|
|
|
|
|
|
|
|
|
|
Plasmin |
|
Thrombolytic |
|
Plasma-derived Plasmin |
|
aPAO |
|
Phase I completed; Phase II initiated |
|
|
|
|
|
|
|
|
|
|
|
Plasmin |
|
Thrombolytic |
|
Plasma-derived Plasmin |
|
Acute Ischemic Stroke |
|
Proof of concept clinical trial |
|
|
|
|
|
|
|
|
|
|
|
recPlasmin |
|
Thrombolytic |
|
Recombinant Plasmin |
|
Acute Ischemic Stroke |
|
Preclinical |
|
|
|
|
|
|
|
|
|
|
|
Prolastin-C A1PI |
|
Respiratory |
|
IVA1PI |
|
A1PI deficiency |
|
Phase IV commitment |
|
|
|
|
|
|
|
|
|
|
|
Recombinant FVIII |
|
Coagulation |
|
Intravenous |
|
Hemophilia A |
|
Preclinical |
|
|
|
|
|
|
|
|
|
|
|
Recombinant A1PI |
|
Respiratory |
|
Intravenous and/or Aerosolized |
|
A1PI deficiency, COPD, Cystic Fibrosis |
|
Preclinical |
The content of our development portfolio will change over time as new plasma products progress from pre-clinical to development to market, and as we discontinue testing of product candidates that do not prove to be promising or feasible to develop. Due to the uncertainties and difficulties of the development process, it is not unusual for protein therapeutics, especially those in the early stages of investigation, to be terminated or delayed as they progress through development.
While our current protein products are derived from human plasma, we expect that recombinant technologies will be a major source of new protein therapies commercialized in the future. We have initiated recombinant protein development programs for Plasmin, Factor VIII, and A1PI. Recombinant or recPlasmin is a patentable form of Plasmin that retains key properties of Plasmin that make it a candidate for development as a therapy for ischemic stroke. We have filed composition of matter patents covering recPlasmin. We are conducting pre-clinical development of recombinant Factor VIII and A1PI utilizing advanced protein production technology licensed from Crucell N.V.
Gamunex-C (TAL-05-0002)
We developed a subcutaneous route of administration option for Gamunex 10% IGIV to meet a growing demand for subcutaneous self-administration of immunoglobulin for PI patients. In October 2010, the FDA approved Gamunex-C for the subcutaneous route of administration for the PI indication. A required post-marketing study will be initiated in the second half of 2011. We received approval from Health Canada for Gamunex-C subcutaneous administration in Canada. We launched subcutaneous administration in Canada in the fourth quarter of 2010.
Plasmin (TAL-05-00013)
Plasmin, a direct-acting thrombolytic agent, is our most innovative pipeline product. Plasmin is purified from human plasma in its inactive, zymogen form, plasminogen. Historically, attempts to use plasminogen in clinical settings have been impeded by the inability to provide sufficient quantities of Plasmin at the site of clotting before it autodegraded or because such attempts required potentially toxic additives. We have avoided these difficulties by converting plasminogen to Plasmin, the active form of the enzyme, and stabilizing it by placing it in an acidified solution that allows us to administer it directly into patients at appropriate doses and low toxicity to dissolve blood clots as determined by pre-clinical investigation. Plasmin is being produced for clinical trials at our cGMP clinical manufacturing facilities in Clayton, North Carolina. Our intellectual property on Plasmin related to its method of use as a direct-acting thrombolytic, its formulation, and its manufacturing process is supported by multiple patents issued in the U.S., European Union, and elsewhere. In 2010, another broad claims method of use patent was allowed in the U.S.
Plasmin has an expected advantage over tissue plasminogen activator as it has a reduced likelihood of causing bleeding. As a natural human plasma enzyme, Plasmin plays a key role in maintaining hemostasis in human and animal physiology. Its main physiologic function is dissolution of blood clots. Plasmin is inactivated in less than a second when it is present in the bloodstream unless it is bound to a blood clot. To ensure that Plasmin remains active, it is delivered locally to a clot using a catheter. Once Plasmin is delivered to a clot, it binds and dissolves the clot. When the clot is dissolved and Plasmin is released into the blood stream it is immediately inactivated thereby preventing active Plasmin from circulating and causing bleeding at distal sites within the body. The balance between clot binding and systemic inhibition provides the basis for differentiation of our direct-acting Plasmin development product from any other thrombolytic agents, both licensed and those in development.
Plasmin’s formulation is designed for direct delivery into clots via state-of-the-art procedures (catheter-directed thrombolytic therapy) performed in catheterization laboratories of hospitals, rather than by the current method involving intravenous injection into the bloodstream. Potential applications of catheter delivered thrombolytics include aPAO, DVT (deep vein thrombosis), and ischemic stroke. Basically, wherever a vessel or device is occluded by a blood clot and is accessible by catheter, the potential exists for directed thrombolytic therapy. We have filed Investigational New Drug Applications (IND) for aPAO, DVT and HGO (hemodialysis graft occlusion). We believe Plasmin may have other significant indications as well. We are also evaluating the use of Plasmin for the treatment of clots in stroke. A proof of concept trial in acute ischemic stroke is ongoing in certain countries outside of the United States.
We filed an IND with the FDA in early 2003 and conducted a Phase I safety trial in hemodialysis patients who have clogged synthetic arterial-venous shunts. This study started in September 2003 and was completed in 2005. Plasmin was well-tolerated with no major bleeding events at all doses tested and there was a dose-dependent response in clot lysis with greater than 75% clot lysis in five of five patients at the highest dose of 24 mg.
Based on the encouraging outcome of this initial trial, we conducted a Phase I study in the U.S., EU and other countries to evaluate Plasmin in the treatment of aPAO, a condition in which arterial blood flow to extremities, usually the legs, becomes blocked by a clot. Affecting approximately 100,000 people in the U.S. each year, this condition is most common in people with underlying narrowing of arteries and gradual restriction of blood flow over time resulting from peripheral arterial disease (PAD). Without prompt intervention, aPAO can result in significant complications such as permanent nerve and muscle damage, and in the most severe cases, even amputation or death.
There is an unmet medical need for a proven safe direct-acting thrombolytic agent to treat aPAO. Current methods focus on pharmacologic, mechanical, or surgical removal of the blood clot, or bypass grafting to direct flow around the area of the clot. However, no clot-busting drugs currently are approved for this indication by regulatory authorities, and those currently used (plasminogen activators) may require a prolonged infusion averaging 24 to 36 hours and produce increased risk of bleeding complications. We received an orphan drug designation in the U.S. for the development of Plasmin for aPAO, which provides incentives for qualified clinical testing expenses and, if approved, market exclusivity for seven years. We are investigating Plasmin to assess its safety and efficacy in the treatment of aPAO. We completed our Phase I clinical trial in the second quarter of 2010 and initiated a Phase II clinical trial in several countries outside of the U.S. in the fourth quarter of 2010. In Phase I and preclinical trials, Plasmin appears able to rapidly dissolve blood clots without an elevated risk of bleeding. Pd-Plasmin delivered by catheter has dissolved blood clots over 30 cm long in two hours. We also have an open IND for Plasmin in deep vein thrombosis but currently have no plans to pursue an indication.
Prolastin-C A1PI
We completed pivotal clinical studies and submitted a sBLA with the FDA and a sNDS with Health Canada for the approval of Prolastin-C A1PI. Prolastin-C A1PI is a key product life cycle enhancement to Prolastin A1PI. Like Prolastin, Prolastin-C A1PI is intended to treat A1PI deficiency, an inherited disorder that causes a significant reduction in the naturally occurring protein A1PI. The modified production process of Prolastin-C A1PI will allow for increased yield and higher concentration versus our current Prolastin A1PI product, effectively increasing our capacity by 40%. The European Union regulatory authorities have indicated that a successful efficacy trial will be a prerequisite to applying for marketing authorization for Prolastin-C A1PI in the European Union. The sBLA has been approved by the FDA; and a post-approval clinical trial is required as a condition for approval. The sNDS has been approved by Health Canada. We completed the conversion of our existing U.S. and Canadian Prolastin A1PI patients to Prolastin-C A1PI in 2010.
Recombinant A1PI and Factor VIII
We are exploring the potential for human cell-line based protein production technology to produce A1PI and Factor VIII with human modifications. Current recombinant forms of A1PI and Factor VIII are produced in animal cell lines and therefore lack key protein modifications characteristic of the human plasma version of the two proteins. Human protein modifications have the potential to make the human recombinant forms physiologically similar to their plasma-derived counterparts. The availability of recombinant A1PI may facilitate the application of A1PI therapy to other respiratory diseases such as COPD or cystic fibrosis (CF).
Technical Support of Manufacturing Operations
Our R&D activities also support improvements in manufacturing and testing processes that enhance yield and product quality. In addition to the above initiatives, we are working to transfer production of our Thrombate III product from Bayer, with whom we currently have a supply agreement. We are currently validating a new production facility at our Clayton site with regulatory approval expected in early 2012. We believe that we have sufficient inventory of intermediates and finished product to meet demand until the new facility is approved.
Customers
FFF Enterprise Inc. and Amerisource Bergen collectively accounted for approximately 27% of our net revenue for both the years ended December 31, 2010 and 2009. Similarly, our accounts receivable balances have also been concentrated with a small number of customers. Amerisource Bergen accounted for approximately 13% of our accounts receivable, net, as of December 31, 2010 and FFF Enterprise Inc. accounted for approximately 15% of our accounts receivable, net, as of December 31, 2009. Additionally, we are the largest supplier of plasma-derived products to the Canadian blood system operators, Canadian Blood Services and Hema Quebec, and derive significant revenue and profits from these contracts. We have experienced and expect to continue to experience, annual volume declines in Canada, due to Canadian Blood Services objective to have multiple sources of supply which has and will continue to impact our overall IGIV growth. We expect that sales of our products to a limited number of customers will continue to represent a substantial amount of our revenues for the foreseeable future. For a discussion of certain risks related to our customer concentration, please see “Risk Factors—A substantial portion of our revenue is derived from a small number of customers, and the loss of one or more of these customers could have a material adverse effect on us.”
The following table includes a geographic breakdown of our net revenues:
|
|
|
Years Ended December 31, |
| |||||||
|
|
|
2010 |
|
2009 |
|
2008 |
| |||
|
United States |
|
$ |
1,098,969 |
|
$ |
1,011,468 |
|
$ |
906,376 |
|
|
Canada |
|
195,092 |
|
214,883 |
|
215,964 |
| |||
|
Europe |
|
196,571 |
|
185,297 |
|
168,081 |
| |||
|
Other |
|
110,987 |
|
121,561 |
|
83,871 |
| |||
|
Total net revenue |
|
$ |
1,601,619 |
|
$ |
1,533,209 |
|
$ |
1,374,292 |
|
Our international business is subject, in various degrees, to a number of risks inherent in conducting business in other countries. These include, but are not limited to, currency exchange rate fluctuations, capital and exchange control regulations, expropriation and other restrictive government actions. Our international business is also subject to government-imposed constraints, including laws on pricing, reimbursement, and access to our products.
Our worldwide net revenue does not reflect a significant degree of seasonality. We generally experience a favorable revenue and gross margin benefit during the second and third quarters due to higher sales of our hyperimmune therapies for the treatment of exposure to rabies.
Contract Services
Apart from contracts with the Canadian blood system operators, we also provide a variety of contract manufacturing or process services, including plasma fractionation, manufacturing, and analytical testing. These contracts are for a limited number of batches of product and development services during the clinical development phase of a project and then change to a guaranteed annual minimum purchase commitment for a term of typically three to five years during the commercial phase of each agreement.
Sales, Marketing and Distribution
We have sales and marketing teams in the U.S., Germany and Canada as well as a team dedicated to the development of other international markets. Within the U.S., our core market, we have developed a “push-pull” distribution network comprised of both direct and indirect channels. Our direct channel is comprised of three specialty sales teams: Immunology/Neurology, Pulmonary, and Hematology. Our specialty sales representatives are experienced professionals with a combination of extensive commercial and healthcare related experience calling on a variety of touch points including physicians, pharmacists, and homecare companies. For 2009, MRB estimates that 65% (including 10% sold to distributors) of the IGIV sold in the U.S. was purchased by hospitals for both in-patient and out-patient use; physician offices represented about 15% of IGIV volume; and home and specialty pharmacies represented 20% of the IGIV volume.
· The Immunology/Neurology team is primarily focused on promoting Gamunex-C for the use in PI, CIDP, and ITP. It is also responsible for promoting our portfolio of hyperimmune products and Plasbumin. This team calls on office-based and hospital-based specialty physicians including neurologists and immunologists. They also call on a variety of healthcare providers within the hospital and home-care settings (physicians, nurses, pharmacists).
· The Pulmonary team promotes Prolastin-C A1PI, focusing on identification of A1PI patients and driving brand choice for Prolastin-C A1PI, which is the most prescribed A1PI therapy in the U.S.
· The Hematology team promotes Koate-DVI and Thrombate III, calling on hematologists and other healthcare providers within the hospital and specialty treatment centers.
· In addition to our direct sales force, we have a managed markets sales team that manages relationships and contracting efforts with GPOs, distributors, home healthcare and specialty pharmacy providers and private commercial payors.
We sell, market and distribute our various products through both our direct sales personnel and our network of distributors. Our sales, marketing and distribution efforts focus on strengthening our relationships with physicians, pharmacists, nurses, patients, GPOs, distributors, home healthcare and specialty pharmacy providers. In the U.S., our Prolastin Direct Program has engaged a prescription fulfillment provider to ensure that Prolastin-C A1PI can be shipped directly to patients. This direct-to-patient distribution service is designed to enhance user convenience and ensure the patient of an uninterrupted supply of Prolastin-C A1PI, while providing us with valuable information about their usage patterns. We are the only A1PI provider to have completed a Mutual Recognition Procedure in the European Union. Prolastin A1PI is approved in 15 European countries and we are currently established in six of these markets. We have been in reimbursement discussions with a number of these countries since late 2007 and these discussions must be concluded before we can expect to significantly increase sales in these countries.
Packaging and Distribution Contracts
Since 2005, we significantly reduced the number of our distributors and simultaneously entered into long-term distribution agreements with major hospital group purchasing organizations, or GPOs, distributors, home care and specialty pharmacy providers and distributors which we consider favorable.
In addition, we have an agreement with Catalent Pharma Solutions pursuant to which they provide packaging, labeling and testing services for us in connection with the distribution of our products in Europe. The agreement expires in 2013, though we have the option to extend it twice, each time for a period of two years. We may terminate the agreement upon 12 months notice, and Catalent may terminate the agreement upon 24 months notice, beginning in 2011. Either party may terminate the agreement upon the bankruptcy of the other party or if there is a material breach which is not cured within 30 business days.
Patents and Other Intellectual Property Rights
Our success depends in part on our ability to obtain and maintain proprietary protection for our product candidates, technology and know-how, to operate without infringing the proprietary rights of others and to prevent others from infringing our proprietary rights. Our policy is to seek to protect our proprietary position by, among other methods, filing U.S. and foreign patent applications related to our proprietary technology, inventions and improvements that are important to the development of our business. We also rely on trade secrets, legal opinions, know-how, continuing technological innovation and in-licensing opportunities to develop and maintain our proprietary position.
Patents
As of December 31, 2010, we owned or licensed for uses within our field of business 117 U.S. patents and U.S. patent applications as well as numerous foreign counterparts to many of these patents and patent applications. At present, we do not consider any individual patent, patent application or patent family to be material to the operation of our business as a whole. Our patent portfolio includes patents and patent applications with claims directed to the composition of matter, manufacturing processes, pharmaceutical formulation and methods of use of many of our compounds and products, including a composition of matter patent application for a recombinant Plasmin molecule and a manufacturing process patent for Gamunex-C/Gamunex IGIV.
Trademarks
Given the importance of our name brands, particularly with respect to IGIV and A1PI products, we rely heavily on the protection of trade names and trademarks. We have registered trademarks for a number of our products, including Gamunex®, Gaminex 10%®, Prolastin®, Prolastin-C A1PI®, Plasbumin®, Plasmanate®, Koate®, Thrombate III®, GamaStan®, HyperHepB®, HyperRho®, HyperRab®, HyperTet®, Gamimune®, Talecris Direct®, and Prolastin Direct®.
Trade Secrets
We may rely, in some circumstances, on trade secrets to protect our technology. However, trade secrets can be difficult to protect. We seek to protect our proprietary technology and processes, in part, by maintaining a trade secret registry and by implementing confidentiality agreements with our employees, consultants, scientific advisors and contractors. We also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems. While we have confidence in these individuals, organizations and systems, agreements or security measures may be breached, and we may not have adequate remedies for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors.
In-License Agreements
We license from Bayer HealthCare LLC certain intellectual property rights. Under the licensing agreement with Bayer Healthcare, we were granted a royalty-free, worldwide and perpetual license covering certain intellectual properties not acquired by us in connection with our formation transaction. As amended on August 10, 2007, the agreement permits us, subject to specified limitations, to grant a sublicense to Baxter International Inc., Baxter Healthcare Corporation, and certain of their affiliates relating to certain Japanese patent rights not acquired by us in connection with our formation transaction. On August 1, 2006 we entered into a collaboration and development agreement with Activaero GmbH, which grants us exclusive rights to use the Akita™ inhalation device for the aerosol delivery of A1PI for any indication. Under that agreement, we are committed to a minimal level of spending each year during development and should the product reach commercialization we must pay a royalty on sales, with no milestone payments due to Activaero GmbH. The agreement is valid until the last to expire applicable patent right currently not expected until 2025. We have the right to terminate with up to 120 days notice, but must pay a progressively higher termination fee determined by considering both the reason for our termination and the number of years since the effective date of the agreement. On September 4, 2008 and December 17, 2008, we entered into exclusive commercial licenses with Crucell N.V. for two recombinant proteins, A1PI and Factor VIII (alone or combined with recombinant Von Willebrand Factor), respectively, utilizing advanced protein technology. We paid Crucell license fees and committed to pay them milestone payments when certain research and clinical targets have been met and royalty payments upon commercialization, while reserving the right for us to terminate the agreements without penalty upon 90 days notice.
Competition
The plasma products industry is highly competitive with changing competitive dynamics. We face, and will continue to face, competition from both U.S.-based and foreign producers of plasma-derived therapies, some of which have lower cost structure, greater capital, manufacturing facilities, resources for research and development, and marketing capabilities. In addition to competition from other large worldwide plasma products providers, we face competition in local areas from smaller entities. For example, in Europe, where the industry is highly regulated and health care systems vary from country to country, local entities may have greater knowledge of local health care systems, more established infrastructure and have existing regulatory approvals or a better understanding of the local regulatory process, allowing them to market their products more quickly. Moreover, plasma-derived therapies generally face competition from non-plasma products and other courses of treatment.
In 2008, we were ranked third along with Octapharma in global plasma product sales, each with an 11% share of worldwide sales. According to MRB, our principal plasma derivatives competitors are Baxter International, Inc., CSL Behring, Octapharma AG and Grifols, representing 22%, 18%, 11% and 8%, respectively, of worldwide plasma derivative sales in 2008. We also face competition in specific countries from local non-profit organizations. Currently, product production capacity may be limited by fractionation capacity or purification capacity. Our competitors and we have announced plans to invest in the development of additional fractionation and purification capacity, which should allow production of plasma-derived therapies to meet the pace of demand.
With regard to other large plasma product providers and certain key products of the plasma product business, the competition we face, and the principal bases on which we compete, vary from product class to product class.
IGIV
Since 2002, the majority of sales for IGIV products have shifted from lyophilized to liquid with liquids capturing 29% of the market in 2002 and 80% in 2008. Liquid IGIV products are viewed as preferable because they are ready-to-use (i.e., they require no reconstitution), with generally excellent efficacy and tolerability profiles. Product margin improvement opportunities are being driven by the transition from lower priced lyophilized products to higher priced liquids using manufacturing processes that deliver improved IgG (immune gamma globulin) yields.
Grifols has recently received FDA approval for and launched its liquid 10% IGIV Flebogamma 10% DIF. CSL received U.S. regulatory approval for its liquid 10% IGIV and launched its product in 2008. CSL launched Rhophylac for ITP in 2007 and in 2010 received FDA approval for HizentraTM, Immune Globulin subcutaneous (Human), 20% Liquid, for treating PI. In 2005, Baxter Bioscience launched Gammagard Liquid 10% soon after its purchase of the American Red Cross IGIV product line (Panglobulin and Polygam). Other Baxter efforts in subcutaneous formulation and delivery in flexible containers have also been announced. We expect additional producers of IGIV outside the U.S. to introduce products into the U.S. in the next few years. For example, Bio Products Laboratory, a not-for-profit organization wholly-owned by the U.K. government, received approval from the FDA for its 5 percent concentration IGIV for PI and Octapharma and Biotest have filed for approval of a 10% liquid IGIV. Octapharma’s IGIV products have been off the market during much of 2010 in the U.S. and other parts of the world. When Octapharma resumes sales of its products, it may discount prices to regain lost market share. Among producers of liquid IGIV products, we intend to compete primarily on the basis of our first-line positioning as a premium liquid IGIV brand with extensive clinical experience and well developed brand loyalty supplemented in the medium term with new indications and formulations. In addition, we believe our use of a caprylate purification process gives our product an advantage over competitors’ products that are purified by harsher solvent processes.
Gamunex also faces competition from non-plasma products and other courses of treatments. For example, two RhD hyperimmune globulins for intravenous administration, Cangene’s WinRho SDF and CSL Behring’s Rhophylac, are now approved for use to treat ITP, and GSK and Amgen launched thrombopoietin inhibitors targeting ITP patients in 2008 that may reduce the demand for IGIV to treat this immune disorder.
A1PI
Until 2003, our A1PI product, Prolastin A1PI, was the only plasma product licensed and sold for therapy of congenital A1PI deficiency-related emphysema in the U.S. Our share of units sold of Prolastin A1PI in the U.S. was approximately 62% of all A1PI product sales in 2009 according to MRB. We completed the conversion of our existing U.S. and Canadian Prolastin A1PI patients to our newer product, Prolastin-C A1PI, in 2010. We compete against Baxter’s Aralast and CSL Behring’s Zemaira, which were launched in 2003. In addition, Kamada Ltd. launched its liquid A1PI product, Glassia, in 2010 in the U.S. We expect to compete on the basis of our long clinical history as well as brand name recognition and customer acceptance. In that regard, we believe we have a significant advantage because of our existing direct-to-patient delivery program, Prolastin Direct, which is our primary distribution mode in the U.S. Prolastin Direct health management provides better patient outcomes by reducing the frequency of respiratory exacerbations. Furthermore, Prolastin Direct results in high medication compliance, with over 94% of prescribed doses being administered annually and high patient loyalty with, an annual retention rate of over 96%. Newer entrants will likely continue to gain some share of product sales for new patients (competitors have also invested in new patient identification efforts), but we have not incurred significant erosion of our existing customer base due to patients’ switching to alternative A1PI products and we have been successful in identifying new patients. We experience continual patient attrition due to the nature of the disease.
In the European Union, we had an 87% share of A1PI sales in 2008 according to MRB data, and have the only licensed A1PI product, other than Grifols, which has marketing authorization for Trypsone A1PI in Spain, and LFB, which sells Alfalastin in France. In 2006, we completed a Mutual Recognition Procedure in the European Union covering countries where over 1,300 symptomatic patients have been identified but have had no access to a licensed A1PI product. Prolastin A1PI is approved in 15 European countries and we are currently established in six of these markets. We have been in reimbursement discussions with a number of these countries since late 2007 and these discussions must be concluded before we can expect to significantly increase sales in these countries. Our competitors are currently pursuing licensing trials in Europe.
New products may reduce demand for plasma-derived A1PI. In addition to us, Arriva and GTC Biotherapeutics are in the early stages of development for a recombinant form of recA1PI. Although we are not aware of any active clinical trials for a recA1PI product or where the companies are in the development of these products, a successful recA1PI, prior to us developing a similar product, could gain first mover advantage and result in a loss of our A1PI market share. Similarly, several companies are attempting to develop products which would be substitution threats in the A1PI sector, including retinoic acid, oral synthetic elastase inhibitors and gene therapy.
Albumin
Among albumin products, competition is generally based on price, given that the products tend to be homogenous. In 2008, we had an approximate 5% share of global albumin and PPF sales. Most of our albumin sales are in the U.S.
Koate DVI
During the late 1970s and early 1980s, some plasma-derived Factor VIII patients were exposed to HIV and Hepatitis C through contaminated Factor VIII products. In an effort to eliminate the risk of pathogen transmission, scientists developed recombinant or synthetic alternatives to plasma-derived Factor VIII products for the treatment of Hemophilia A and Hemophilia B.
Recombinant products have taken a significant share of Factor VIII sales due to their near independence from human plasma. Of the 6.9 billion units of Factor VIII sold in 2008, approximately 58% were recombinant. On a per unit basis, recombinants are priced approximately 88% higher than plasma-derived products and have been adopted more rapidly in the U.S. and industrialized nations. Growth in the demand for plasma-derived Factor VIII is being driven by increased patient identification and treatment in developing countries. The high cost of recombinant products prevents their use in developing markets where plasma derivatives are the standard. We are conducting pre-clinical development of recombinant Factor VIII proteins utilizing advanced protein production technology.
Thrombate
We have the only plasma-derived brand ATIII licensed in the U.S. In 2009, a competing product—Atryn, transgenic ATIII (produced in transgenic goats) for the treatment of hereditary antithrombin deficiency was launched in the U.S.
Government Regulation
Government authorities in the U.S., at the federal, state and local level, and in other countries extensively regulate, among other things, the research, development, testing, approval, manufacturing, labeling, post-approval monitoring and reporting, packaging, promotion, storage, advertising, distribution, marketing and export and import of pharmaceutical products such as those we are developing. The process of obtaining regulatory approvals and the subsequent substantial compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources.
United States Government Regulation
In the U.S., the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act and implementing regulations. Failure to comply with the applicable FDA requirements at any time during the product development process, approval process or after approval may result in administrative or judicial sanctions. These sanctions could include the FDA’s imposition of a clinical hold on trials, refusal to approve pending applications, withdrawal of an approval, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties or criminal prosecution, or any combination of these sanctions. Any agency or judicial enforcement action could have a material adverse effect on us.
Drugs that are also biological products must also satisfy the requirements of the Public Health Services Act and its implementing regulations. In order for a biological drug product to be legally marketed in the U.S., the product must have a Biologic License Application (BLA), approved by the FDA.
Before approving a BLA, the FDA generally will inspect the facility or the facilities at which the product is manufactured. The FDA will not approve the product if it finds that the facility does not appear to be in cGMP compliance. If the FDA determines the application, manufacturing process or manufacturing facilities are not acceptable, it will either disapprove the application or issue an approvable letter in which it will outline the deficiencies in the BLA and provide the applicant an opportunity to meet with FDA representatives and subsequently to submit additional information or data to address the deficiencies. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
The testing and approval processes require substantial time, effort and financial resources, and each may take several years to complete. Data obtained from clinical activities are not always conclusive and may be susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. The FDA may not grant approval on a timely basis, or at all. We may encounter difficulties or unanticipated costs in our efforts to secure necessary governmental approvals, which could delay or preclude us from marketing our products. The FDA may limit the indications for use or place other conditions on any approvals that could restrict the commercial application of the products. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further testing requirements and FDA review and approval.
After regulatory approval of a product is obtained, we are required to comply with a number of post-approval requirements. For example, as a condition of approval of a BLA, the FDA may require post marketing testing and surveillance to monitor the product’s safety or efficacy. In addition, holders of an approved BLA are required to keep extensive records, to report certain adverse reactions and production problems to the FDA, to provide updated safety and efficacy information and to comply with requirements concerning advertising and promotional labeling for their products. Also, quality control and manufacturing procedures must continue to conform to cGMP regulations as well as the manufacturing conditions of approval set forth in the BLA. The FDA periodically inspects manufacturing facilities to assess compliance with cGMP regulations, which imposes certain procedural, substantive and recordkeeping requirements. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance.
Orphan Drug Designation
The FDA may grant orphan drug designation to drugs intended to treat a “rare disease or condition” that affects fewer than 200,000 individuals in the U.S., or more than 200,000 individuals in the U.S. and for which there is no reasonable expectation that the cost of developing and making available in the U.S. a drug for this type of disease or condition will be recovered from sales in the U.S. for that drug. Orphan drug designation must be requested before submitting an application for marketing approval. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. Orphan drug designation can provide opportunities for grant funding towards clinical trial costs, tax advantages and FDA user-fee benefits. In addition, if a product which has an orphan drug designation subsequently receives the first FDA approval for the indication for which it has such designation, the product is entitled to orphan drug exclusivity, which means the FDA may not approve any other application to market the same drug for the same indication for a period of seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity or a meaningfully different mode of administration. Competitors may receive approval of different drugs or biologics for the indications for which the orphan product has exclusivity. However, if a company with orphan drug exclusivity is not able to supply the market, the FDA could allow another company with the same drug a license to market for said indication.
Regulation Outside the United States
In addition to regulations in the U.S., we are subject to a variety of regulations in other jurisdictions governing clinical trials and commercial sales and distribution of our products. Whether or not we obtain FDA approval for a product, we must obtain approval of a product by the comparable regulatory authorities of countries outside the U.S. before we can commence clinical trials or marketing of the product in those countries. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country.
To obtain regulatory approval of a drug under European Union regulatory systems, we may submit marketing authorizations either under a centralized or decentralized procedure. The centralized procedure provides for the grant of a single marketing authorization that is valid for all European Union member states. All marketing authorizations for products designated as orphan drugs must be granted in accordance with the centralized procedure. The decentralized procedure provides for approval by one or more other, or concerned, member states of an assessment of an application performed by one member state, known as the reference member state. If a member state cannot approve the assessment report and related materials on the grounds of potential serious risk to the public health, the disputed points may eventually be referred to the European Commission, whose decision is binding on all member states.
The European Medicines Agency grants orphan drug designation to promote the development of products that may offer therapeutic benefits for life-threatening or chronically debilitating conditions affecting not more than five in 10,000 people in the European Union. In addition, orphan drug designation can be granted if the drug is intended for a life threatening, seriously debilitating or serious and chronic condition in the European Union and that without incentives it is unlikely that sales of the drug in the European Union would be sufficient to justify developing the drug. Orphan drug designation is only available if there is no other satisfactory method approved in the European Union of diagnosing, preventing or treating the condition, or if such a method exists, the proposed orphan drug will be of significant benefit to patients. Orphan drug designation provides opportunities for free protocol assistance, fee reductions for access to the centralized regulatory procedures before and during the first year after marketing authorization and 10 years of market exclusivity following drug approval. Fee reductions are not limited to the first year after authorization for small and medium enterprises. The exclusivity period may be reduced to six years if the designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity.
FDA-approved Talecris products are required to be registered and approved by local regulatory agencies in the majority of countries outside North America and Europe. There are a limited number of countries (Bahamas, Bermuda, Guam, Oman and Quatar) which do not require further local product registration and product may be distributed based on the existing FDA approval. In addition, any changes in the distributors supporting our export business could result in a loss of sales.
Pharmaceutical Pricing and Reimbursement
In the United States and markets in other countries, sales of any products for which we receive regulatory approval for commercial sale will depend in part on the availability of reimbursement from third-party payors. Third-party payors include government health programs, managed care providers, private health insurers and other organizations. These third-party payors are increasingly challenging the price and examining the cost-effectiveness of medical products and services. In addition, significant uncertainty exists as to the reimbursement status of newly approved healthcare products. Our products may not be considered cost effective. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development. In the U.S., Talecris’ products are reimbursed or purchased under several government programs, including Medicaid, Medicare Parts B and D, the 340B/Public Health Service (PHS) program, and pursuant to our contract with the Department of Veterans Affairs. Medicaid is a joint state and federal government health plan that provides covered outpatient prescription drugs for low-income individuals. Under Medicaid, drug manufacturers pay rebates to the states based on utilization data provided by the states.
The rebate amount for most branded drugs was previously equal to a minimum of 15.1% of the Average Manufacturer Price, which is referred to as AMP, or AMP less Best Price, which is referred to as AMP less BP, whichever is greater. The U.S. healthcare reform legislation that was signed into law in 2010 generally increased the size of the Medicaid rebates paid by drug manufacturers for single source and innovator multiple source (brand name) drugs from a minimum of 15.1% to 23.1% of the AMP, subject to certain exceptions, for example, for certain clotting factors the increase is limited to a minimum of 17.1% of the AMP. For non-innovator multiple source (generic) drugs, the rebate percentage is increased from a minimum of 11% of AMP to 13% of AMP. The legislation also extends the rebate obligation to prescription drugs covered by Medicaid managed care organizations. The increase in required rebates which became effective January 1, 2010 may adversely affect financial performance.
Medicare Part B reimburses providers for drugs provided in the outpatient setting based upon Average Sales Price (ASP). Beginning January 1, 2008 federal government reforms to Medicare Part B have reduced the reimbursement rates for IGIV. At that time CMS reduced the reimbursement for separately covered outpatient drugs and biologicals, including IGIV in the hospital outpatient setting, from ASP +6% to ASP +5% using 2006 Medicare claims data as a reference for this reduction. CMS reduced this reimbursement further in 2009 to ASP +4% using aggregate hospital cost report data as a reference for the reduction. Additional reductions in Medicare Part B reimbursement rates could restrict access to our products.
Medicare Part D is a partial, voluntary prescription drug benefit created by the federal government primarily for persons 65 years old and over. The Part D drug program is administered through private insurers that contract with CMS. Government payment for some of the costs of prescription drugs may increase demand for any products for which we receive marketing approval. However, to obtain payments under this program, we are required to negotiate prices with private insurers operating pursuant to federal program guidance. These prices may be lower than we might otherwise obtain.
Some payors, including Medicare Part D plans and some state Medicaid programs, reimburse providers for drugs based upon a discount off of the Average Wholesale Price (AWP). AWP is a list price determined by third party publishers, which does not reflect actual transactions in the distribution chain. We do not publish an AWP for any of our products. We may be at a competitive disadvantage where providers are reimbursed on an AWP basis and competitors’ products are reimbursed at higher rates than our corresponding product.
The availability of federal funds to pay for our products under the Medicaid and Medicare Part B programs requires that we extend discounts under the 340B/PHS drug pricing program. The 340B drug pricing program extends discounts to a variety of community health clinics and other entities that receive health services grants from the PHS, as well as hospitals that serve a disproportionate share of certain low income individuals. The PHS price or “ceiling price” cannot exceed the AMP (as reported to CMS under the Medicaid drug rebate program) less the Medicaid unit rebate amount. Talecris has entered into a Pharmaceutical Pricing Agreement (PPA) with the government in which the company has agreed to participate in the 340B/PHS program by charging eligible entities no more than the PHS ceiling price for drugs intended for outpatient use.
Beyond the changes made to the 340B program in the 2010 health care reform legislation, further reforms to the 340B program remain a possibility.
We also make our products available for purchase by authorized government users of the Federal Supply Schedule (FSS) pursuant to our FSS contract with the Department of Veterans Affairs. Under the Veterans Health Care Act of 1992, or the VHC Act, we are required to offer discounted FSS contract pricing to four Federal agencies — the Department of Veterans Affairs, the Department of Defense, the Coast Guard and the Public Health Service (including the Indian Health Service) — for federal funding to be made available for reimbursement of any of our products under the Medicaid program and for our products to be eligible to be purchased by those four Federal agencies. FSS pricing to those four Federal agencies must be equal to or less than the “Federal Ceiling Price,” which is, at a minimum, 24% off the Non-Federal Average Manufacturer Price, or “Non-FAMP”, for the prior fiscal year.
The marketability of any products for which we receive regulatory approval for commercial sale may suffer if the government and third-party payors fail to provide adequate coverage and reimbursement. Federal, state and local governments in the U.S. continue to consider legislation to limit the growth of healthcare costs, including the cost of prescription drugs. Future legislation could limit payments for pharmaceuticals such as the drug candidates that we are developing, including possibly permitting the federal government to negotiate prices directly with manufacturers. In addition, an increasing emphasis on managed care in the U.S. has increased and will continue to increase the pressure on pharmaceutical pricing. For a discussion of certain risks related to reimbursement and pricing, please see “Risk Factors—Risks Related to Healthcare Reform and Reimbursement—We could be adversely affected if government or private third-party payors decrease or otherwise limit the amount, price, scope or other eligibility requirements for reimbursement for the purchasers of our products.”
Other
Our operations and many of the products we manufacture or sell are subject to extensive regulation by numerous other governmental agencies, both within and outside the United States. In the United States, apart from the agencies discussed above, our facilities, operations, employees, products (their manufacture, sale, import and export) and services are regulated by the Drug Enforcement Agency, the Environmental Protection Agency, the Occupational Health & Safety Administration, the Department of Agriculture, the Department of Labor, Customs and Border Protection, the Department of Commerce, the Department of Treasury, the Department of Justice and others. Furthermore, because we supply products and services to healthcare providers that are reimbursed by federally funded programs such as Medicare, our activities are also subject to regulation by the Centers for Medicare and Medicaid Services and enforcement by the Office of the Inspector General within the Department of Health and Human Services. State agencies also regulate our facilities, operations, employees, products and services within their respective states. Government agencies outside the United States also regulate public health, product registration, manufacturing, environmental conditions, labor, exports, imports and other aspects of the company’s global operations. For further discussion of the impact of regulation on our business, see “Risk Factors—Risks Related to Our Business—Certain of our other business practices are subject to scrutiny by regulatory authorities.”
Employees
As of December 31, 2010, we had approximately 5,400 full-time employees, none of which are represented by labor unions or covered by collective bargaining agreements. We consider our relationships with our employees to be good.
Available Information
Our Internet website address is www.talecris.com. Our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, and amendments to those reports filed or furnished pursuant to section 13(a) or 15(d) of the Exchange Act are available free of charge through our website as soon as reasonably practicable after we electronically file with or furnish them to the Securities and Exchange Commission (SEC) and are available in print to any stockholder who requests a copy. Additionally, the charters of the standing committees of our board of directors are available on our website under “Investor Relations — Corporate Governance”. Information on our website shall not be deemed incorporated into, or to be a part of, this Annual Report on Form 10-K.
The public may also read and copy any materials that we file with the SEC at the SEC’s Public Reference Room at 100 F Street, NE, Washington, DC 20549. You may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. Additionally, the SEC maintains a website that contains reports, proxy statements, information statements and other information regarding issuers, including us, that file electronically with the SEC at www.sec.gov.
Risks Related to Proposed Merger
Termination of our proposed merger with Grifols could result in fluctuations in the price of our common stock.
On February 14, 2011, our shareholders approved our proposed merger transaction with Grifols. This transaction has not yet been consummated and may still be terminated, including for failure to obtain anti-trust clearance. Grifols agreed to provide written notice to the FTC staff at least thirty days prior to closing the transaction and, in any event, not to close the transaction until after 11:59 p.m. on March 20, 2011. There can be no assurance that Grifols will reach resolution with the FTC by March 20, 2011. Under the pending merger agreement, if this transaction is not closed by the current “outside date” of March 6, 2011, then under specified circumstances, either Grifols or we may elect to cause the “outside date” to be extended to a date not later than the expiration of Grifols financing for the transaction, or September 6, 2011, whichever is earlier. Any termination of the transaction could result in stockholders who purchased or held the stock in anticipation of receiving the merger consideration deciding to sell their stock. If a sufficient number of such stockholders seek to dispose of their stock in the near term, the increased number of shares for sale could materially depress our stock price.
Additionally, our stock price recently may have been impacted by the value of the expected merger consideration. If the merger is terminated, the market may reprice the value of our stock based on other factors. Such a repricing may result in increased volatility in our stock price and a decline in our stock price.
Any decline in our stock price resulting from a termination of the merger could also result in lawsuits by stockholders seeking to recover damages allegedly caused by the price decline or actions leading up to the merger termination. Any such lawsuits could seek material amounts in damages and could result in significant demands on management’s time and resources.
Our proposed merger with Grifols may adversely affect our operations and financial performance.
Our proposed merger with Grifols may result in the loss of key employees, suppliers, and customers. Employees, suppliers or customers who do not support the merger or do not want to work for or with the resulting entity may seek other opportunities as a result of our plans. Key employees may also decide to leave if the merger is subsequently not consummated. Additionally, the demand on management’s time and our resources relating to regulatory approvals and integration planning may interfere with management’s day-to-day oversight of operations. As a result, our operations and financial performance could be adversely affected while we prepare for the merger or in future periods should the merger not occur.
Lawsuits have been filed against us and certain of our officers and directors challenging the merger, and any adverse judgment for monetary damages could have a material adverse effect on the combined company’s operations after the transaction.
Four purported class action lawsuits have been filed by our stockholders challenging the proposed transaction. Two of the lawsuits were filed in the Court of Chancery of the State of Delaware and have been consolidated under the caption In re Talecris Biotherapeutics Holdings Shareholder Litigation, Consol. C.A. No. 5614-VCL. The other two lawsuits were filed in the Superior Court of the State of North Carolina and are captioned Rubin v. Charpie, et al., No. 10 CV 004507 (North Carolina Superior Court, Durham County), and Kovary v. Talecris Biotherapeutics Holdings Corp., et al., No. 10 CV 011638 (North Carolina Superior Court, Wake County). The lawsuits name as defendants Talecris, the members of the Talecris Board of Directors, Grifols, S.A. and its subsidiary, Grifols Inc., and, in the Delaware consolidated action, Talecris Holdings and Stream Merger Sub, Inc. The two North Carolina actions have been stayed.
All of the lawsuits allege that the individual defendants (and, in the consolidated Delaware action, Talecris Holdings) breached their fiduciary duties to our stockholders in connection with the proposed transaction with Grifols, and that Grifols (and, in one of the North Carolina cases, Talecris, and in the Delaware action, Grifols Inc.) aided and abetted those breaches. The Delaware complaint alleges, among other things, that the consideration offered to Talecris stockholders pursuant to the proposed transaction is inadequate; that our board of directors failed to take steps to maximize stockholder value; that our IPO and debt refinancing in 2009 were intended to facilitate a sale of Talecris; that Cerberus and Talecris Holdings arranged the proposed merger for the benefit of Cerberus, without regard to the interests of other stockholders; that the voting agreements impermissibly lock up the transaction; and that the merger agreement contains terms, including a termination fee, that favor Grifols and deter alternative bids. The Delaware complaint further alleges that the preliminary Form F-4 filed on August 10, 2010 contains material misstatements and/or omissions, including with respect to the availability of appraisal rights in the merger; the purpose and effects of the Virginia reincorporation merger; the antitrust risks of the proposed transaction; the financial advisors’ analyses regarding the Grifols’ non-voting stock to be issued in connection with the transaction; and the fees to be paid to Morgan Stanley by us
and Grifols in connection with the proposed transaction. The Delaware complaint also alleges that our stockholders are entitled to appraisal rights in connection with the transaction pursuant to Section 262 of the Delaware General Corporation Law, and that the transaction violates the Delaware General Corporation Law by failing to provide such rights. The Delaware action seeks equitable and injunctive relief, including a determination that the stockholders have appraisal rights in connection with the merger, and damages. On October 29, 2010, the parties to the Delaware litigation entered into a Memorandum of Understanding, or MOU, reflecting an agreement in principle to settle that litigation. The MOU provides, among other things, for the provision of appraisal rights in accordance with DGCL 262 in connection with the transaction as described at pages 135-137 of the MOU; for an increase in the merger consideration by an additional 500,000 shares of Grifols non-voting shares to holders of our common stock other than our specified affiliated stockholders as described at pages 144-145 of the MOU; and for certain additional disclosures provided herein. The MOU also provides for a dismissal of the action with prejudice and a release of claims. On January 21, 2011, the parties executed a formal Stipulation of Settlement documenting the agreement set forth in the MOU, and on January 25, 2011, the Delaware court entered an order preliminarily approving the settlement. The settlement remains subject, among other things, to notice to the class, final court approval and consummation of the transaction.
One of the conditions to the completion of the transaction is that no temporary restraining order, or preliminary or permanent injunction, or other judgment or order issued by a court or other governmental entity that prohibits or prevents the completion of the Talecris-Grifols merger shall be in effect. A preliminary injunction could delay or jeopardize the completion of the transaction, and an adverse judgment granting permanent injunctive relief could indefinitely enjoin completion of the transaction. An adverse judgment for monetary damages could have a material adverse effect on the operations of the combined company after the transaction.
Our Merger Agreement with Grifols may prevent us from consummating desirable business combinations.
As long as our merger agreement with Grifols is in place, we are prohibited from doing any material acquisition or business combinations without the consent Grifols. This may cause us to lose opportunities that would otherwise benefit our business.
Risks Related to the Healthcare Industry
We could be adversely affected by changes in the legal requirements for the market for medical care or healthcare coverage in the United States resulting from the implementation of 2010 healthcare reform legislation, regulatory rule making, or the enactment of additional legislation under consideration.
Substantial changes are being made to the current system for paying for healthcare in the U.S., including changes made in order to extend medical benefits to those who currently lack insurance coverage. Approximately 47 million Americans currently lack health insurance of any kind. While a provision in the health care reform legislation requiring those without insurance to pay a penalty was recently declared unconstitutional by two federal district courts, other federal district courts have upheld this provision, and other litigation relating to the legislation is pending. Provisions of the legislation that could increase coverage have not been affected. Extending coverage to such a large population could substantially change the structure of the health insurance system and the methodology for reimbursing medical services, drugs and devices. These structural changes could entail modifications to the existing system of private payors and government programs (Medicare, Medicaid and State Children’s Health Insurance Program), creation of state-sponsored healthcare insurance exchanges, as well as other changes. Restructuring the coverage of medical care in the U.S. could impact the reimbursement for prescribed drugs and biopharmaceuticals, such as those produced and marketed by us. If reimbursement for these products is substantially reduced in the future, or rebate obligations associated with them are substantially increased (discussed in more detail below), our business could be materially impacted.
Extending medical benefits to those who currently lack coverage will likely result in substantial cost to the federal government, which may force significant changes to the U.S. healthcare system. Much of the funding for expanded healthcare coverage may be sought through cost savings. While some of these savings may come from realizing greater efficiencies in delivering care, improving the effectiveness of preventive care and enhancing the overall quality of care, much of the cost savings may come from reducing the cost of care. Cost of care could be reduced by reducing the level of reimbursement for medical services or products (including those biopharmaceutical products produced and marketed by us), or by restricting coverage (and, thereby, utilization) of medical services or products. In either case, a reduction in the utilization of, or reimbursement for, our products could have a materially adverse impact on our financial performance.
All of the changes discussed above, and others passed in this legislation, are subject to rule-making and implementation timelines that extend for several years. This uncertainty limits our ability to forecast changes that may occur in the future and to manage our business accordingly. In addition, beginning in 2012, the new law may require us to issue Internal Revenue Service Form 1099 to plasma donors whose remuneration equals or exceeds six hundred dollars annually. The cost of implementing this requirement, as well as its potential impact on plasma donations, is unknown at this time.
We could be adversely affected by other provisions of recently passed United States healthcare reform legislation.
In the United States, our products are reimbursed or purchased under several government programs, including, Medicaid, Medicare Parts B and D, the 340B/PHS program, and pursuant to contracts with the Department of Veterans Affairs. In order for a drug manufacturer’s products to be reimbursed by federal funding under Medicaid, the manufacturer must enter into a Medicaid drug rebate agreement with the Secretary of the United States Department of Health and Human Services, and pay certain rebates to the states based on utilization data provided by each state to the manufacturer and to the Centers for Medicare & Medicaid Services, which is referred to as CMS, and pricing data provided by the manufacturer to the federal government. The states have been required to share this savings with the federal government. The rebate amount for most branded drugs was previously equal to a minimum of 15.1% of the Average Manufacturer Price, which is referred to as AMP, or AMP less Best Price, which is referred to as AMP less BP, whichever is greater. The recently enacted healthcare reform legislation generally increases the size of the Medicaid rebates paid by drug manufacturers for single source and innovator multiple source (brand name) drugs from a minimum of 15.1% to 23.1% of the AMP, subject to certain exceptions, for example, for certain clotting factors the increase is limited to a minimum of 17.1% of the AMP. For non-innovator multiple source (generic) drugs, the rebate percentage is increased from a minimum of 11% of AMP to 13% of AMP. The legislation also extends the rebate obligation to prescription drugs covered by Medicaid managed care organizations. The increase in required rebates, which became effective January 1, 2010, may adversely affect our financial performance.
In addition, many states have implemented special Medicaid rebate programs, approved by CMS. These programs often involve drug manufacturers paying supplemental rebates to the states pursuant to a supplemental rebate agreement between the drug manufacturer and the state. The supplemental rebates are typically a condition to the manufacturer obtaining “preferred status” for its drugs on the state’s Medicaid drug formularies and avoiding otherwise mandatory prior authorization by Medicaid officials as a condition to any Medicaid recipient using the manufacturer’s drugs. As with standard Medicaid rebates, the states have shared the savings from supplemental rebate programs with the federal government. However, under the recently enacted healthcare reform legislation, the savings realized from the increased rebate amounts, described above (e.g., the 8% increase in the minimum brand name drug rebate), will be retained entirely by the federal government, and not shared with the states. As a result, states with supplemental rebate programs that, for example, have already increased brand name drug rebates by up to 8%, may effectively lose the portion of the savings they previously shared with the federal government. While the increase in rebates from Medicaid managed care organizations may mitigate this issue for the states, this is not certain, and states with supplemental rebate arrangements may seek to increase supplemental rebate requirements in order to address this, thus affecting financial performance.
Medicare Part D is a partial, voluntary prescription drug benefit created by the United States federal government primarily for persons 65 years old and over. The Part D drug program is administered through private insurers that contract with CMS. To obtain payments under this program, we are required to negotiate prices with private insurers operating pursuant to federal program guidance. These prices may be lower than might otherwise be obtained. In addition, beginning in 2011, the recently enacted healthcare reform legislation generally requires drug manufacturers to provide 50% savings for brand name drugs and biologics provided to Medicare Part D beneficiaries who are in the “donut hole” (or a gap in Medicare Part D coverage for beneficiaries who have expended certain amounts for drugs). The rebate requirement could adversely affect financial performance, particularly if contracts with Part D plans cannot be favorably renegotiated.
The availability of federal funds to pay for our products under the United States Medicaid and Medicare Part B programs requires that we extend discounts under the 340B/PHS program. The 340B/PHS program extends discounts to a variety of community health clinics and other entities that receive health services grants from the PHS, as well as hospitals that serve a disproportionate share of certain low income individuals. The PHS price (or “ceiling price”) cannot exceed the AMP (as reported to CMS under the Medicaid drug rebate program) less the Medicaid unit rebate amount. We have entered into a Pharmaceutical Pricing Agreement with the government in which we have agreed to participate in the 340B/PHS program by charging eligible entities no more than the PHS ceiling price for drugs intended for outpatient use. Recently enacted healthcare reform legislation, as amended by a technical corrections bill signed into law on December 15, 2010, imposes a “must sell” obligation on manufacturers so that they must offer for sale their products to eligible entities at legally-mandated discount prices and expands the number of qualified 340B entities eligible to purchase products for outpatient use.
Beyond the changes made to the 340B program in the 2010 health care reform legislation, further reforms to the 340B program remain a possibility, which could have a material negative impact on our sales and margin given the significant price discount for 340B/PHS products as compared to our commercial prices.
The recently enacted United States healthcare reform legislation imposes a fee on manufacturers and importers of branded drugs and biologics based on their sales to United States government health programs. The fee will first be imposed for 2011 sales. The aggregate fee imposed on all covered entities is $2.5 billion for 2011, $2.8 billion for 2012, $2.8 billion for 2013, $3 billion for 2014, $3 billion for 2015, $3 billion for 2016, $4 billion for 2017, $4.1 billion for 2018 and $2.8 billion for 2019 and following years. The aggregate fee will be allocated among applicable manufacturers and importers based on their relative sales to government health programs, with the caveat that entities with lower sales will have their sales counted at less than 100% in allocating responsibility for the fee. This new fee will increase our costs. It is not clear that we will be able to pass this increased cost on to our customers.
We could be adversely affected if other government or private third-party payors decrease or otherwise limit the amount, price, scope or other eligibility requirements for reimbursement for the purchasers of our products.
We have experienced and expect to continue to experience pricing pressures on our current products and pipeline products from initiatives aimed at reducing healthcare costs by government and private third-party payors, the increasing influence of managed care organizations, and regulatory proposals, both in the United States and in foreign markets. Recently enacted healthcare reform in the United States is likely to increase the pressure. This pressure may include the effect of such healthcare reform changes as the introduction of a biosimilar pathway (which will permit companies to obtain FDA approval of generic versions of existing biologic based upon lessor showings of safety and efficacy than is required for the pioneer biologic), the redefinition of the term “single source” product, which plays a key role in determining reimbursements under the Medicare Part B program, and changes to the 340B Public Health Service (PHS) drug pricing program imposing a “must sell” obligation on manufacturers so that they must offer for sale their products to eligible entities at legally-mandated discount prices. Additional legislative changes to current pricing rules are possible. We cannot predict which additional changes, if any, will eventually be adopted, or their impact on us. Certain changes could have a materially adverse impact on our financial performance. In addition, certain pharmaceutical products, such as plasma-derived products, are subject to price controls in countries within the European Union. Price controls are expanding beyond those already imposed by governmental authorities in some of the countries where we operate. The existence of direct and indirect price controls over our products have affected, and may continue to materially adversely affect, our ability to maintain or increase gross margins.
In the United States, group purchasing organizations, which are referred to as GPOs, which are entities that act as purchasing intermediaries for hospitals and physicians, constitute the largest marketing channel. The United States GPO channel is dominated by a small number of companies. The GPOs’ large market position and their substantial purchasing volume provide them with significant negotiating power, resulting in price pressures for manufacturers like us. In addition, in the United States, health insurance providers have been setting a cap on the amounts that they will reimburse for certain products. This could have a negative effect on the price that we may be able to charge for our products and could function as an indirect nongovernmental price control.
If payors reduce the amount of reimbursement for a product, it may cause groups or individuals dispensing the product to discontinue administration of the product, to administer lower doses, to substitute lower cost products or to seek additional price related concessions. These actions could have a negative effect on our financial results, particularly in cases where we have a product that commands a premium price in the marketplace, or where changes in reimbursement induce a shift in the site of treatment. For example, beginning in 2005, the Medicare drug reimbursement methodology for physician and hospital outpatient payment schedules changed to Average Sales Price (ASP), which is referred to as ASP+6%. This payment was based on a volume-weighted average of all brands under a common billing code. Medicare payments to physicians between the fourth quarter of 2004 and the first quarter of 2005 dropped 14% for both the powder and liquid forms of intravenous immune globulin, which is referred to as IGIV. Medicare payments to hospitals fell 45% for powder IGIV and 30% for liquid IGIV between the fourth quarter of 2005 and the first quarter of 2006. The Medicare reimbursement changes resulted in the shift of a significant number of Medicare IGIV patients to hospitals from physicians’ offices beginning in 2005 as many physicians could no longer recover their costs of obtaining and administering IGIV in their offices and clinics. After 2006, some hospitals reportedly began to refuse providing IGIV to Medicare patients due to reimbursement rates that were below their acquisition costs. While subsequent changes have improved some of these Medicare reimbursement issues, on January 1, 2008, the CMS reduced the reimbursement for separately covered drugs and biologicals, including IGIV, in the hospital outpatient setting from ASP +6% to ASP +5% using 2006 Medicare claims data as a reference for this reduction. In addition, CMS reduced a hospital add-on payment from $75 to $38 per infusion. Beginning January 1, 2009, CMS further reduced the hospital outpatient reimbursement for separately covered outpatient drugs, including IGIV, to ASP +4%, and eliminated the add-on payment.
Physicians frequently prescribe legally available therapies for uses that are not described in the product’s labeling and that differ from those tested in clinical studies and approved by the FDA or similar regulatory authorities in other countries. These unapproved (also known as “off-label”) uses are common across medical specialties, and physicians may believe such off-label uses constitute the preferred treatment or treatment of last resort for many patients in varied circumstances. We believe that a significant portion of our IGIV volume may be used to fill physician prescriptions for indications not approved by the FDA or similar regulatory authorities. If reimbursement for off-label uses of our products, including IGIV, is reduced or eliminated by Medicare or other third-party payors, including those in the United States or the European Union, we could be adversely affected.
For example, CMS could initiate an administrative procedure known as a National Coverage Determination (NCD) by which the agency determines which uses of a therapeutic product would be reimbursable under Medicare and which uses would not. This determination process can be lengthy, thereby creating a long period during which the future reimbursement for a particular product may be uncertain. High levels of spending on IGIV products, along with increases in IGIV prices, increased IGIV utilization and the high proportion of off-label uses, may increase the risk of regulation of IGIV reimbursement by CMS. On the state level, similar limits could be proposed for therapeutic products covered under Medicaid. Moreover, the Deficit Reduction Act of 2005 incentivizes states to take innovative steps to control healthcare costs, which could include attempts to negotiate limits to, or reductions of, drug prices.
Healthcare reform legislation established and provided significant funding for a Patient-Centered Outcomes Research Institute to coordinate and fund Comparative Effectiveness Research (CER). While the stated intent of CER is to develop information to guide providers to the most efficacious therapies, outcomes of CER could influence the reimbursement or coverage for therapies that are determined to be less cost-effective than others. Should any of our products be determined to be less cost-effective than alternative therapies, the levels of reimbursement for these products, or the willingness to reimburse at all, could be impacted, which could materially impact our financial results.
For many payors, including private health insurers and self-insured health plans, as well as Medicare Part D plans and some state Medicaid programs, outpatient pharmaceuticals are often reimbursed based upon a discount calculated off of a pricing benchmark called “Average Wholesale Price,” which is referred to as AWP. AWP is a list price calculated and published by private third-party publishers (such as First DataBank, Thomson Reuters (Red Book) and Wolters Kluwer (Medi-Span)). AWP does not reflect actual transactions in the distribution chain (e.g., the publishers do not base the figure on actual transaction prices, including any prompt pay or other discounts, rebates or price reductions). Often, publishers calculate AWP based upon a standard markup of, for example, 20% over another list price which is reported by drug manufacturers to the publishers. This list price is called “Wholesale Acquisition Cost,” which is referred to as WAC. WAC is generally understood in the industry to be the list price drug manufacturers have for their drug wholesaler customers and, like AWP, is not calculated based on actual transaction prices, including any prompt pay or other discounts, rebates or price reductions. We do not publish an AWP for any of our products, reporting WAC for our products instead. We may be at a competitive disadvantage where providers are reimbursed on an AWP basis and competitors’ products are reimbursed based on a higher AWP than the corresponding AWP for our product.
The use of AWP and WAC as pricing benchmarks has been subject to legal challenge by both government officials and private citizens, often based on claims that the benchmarks were used in a misleading manner, thus defrauding consumers and third-party payors. It is possible that we, as a reporter of WAC, could be challenged on this basis. Additionally, the settlement of class action litigation against First DataBank and others has resulted in the downward revision of certain reported AWP listings (to a level of 20% over WAC). Issues regarding AWP have contributed to suggestions to eliminate its use as a drug pricing benchmark.
Risks Related to Our Business
Our business is highly concentrated on our two largest products, Gamunex-C/Gamunex IGIV and Prolastin/Prolastin-C A1PI, and our largest geographic region, the U.S. Any adverse market event with respect to either product or the U.S. region would have a material adverse effect on our business.
We rely heavily upon the sales of two of our products: Gamunex-C/Gamunex IGIV and Prolastin/Prolastin-C A1PI. Sales of Gamunex-C/Gamunex IGIV and Prolastin/Prolastin-C A1PI together comprised approximately 76% and 75% of our total net revenue for the years ended December 31, 2010 and 2009, respectively. Sales of Gamunex IGIV comprised approximately half of our total net revenue in each of these years. If either Gamunex-C/Gamunex IGIV or Prolastin/Prolastin-C A1PI lost significant sales, or were substantially or completely displaced in the market, we would lose a significant and material source of our net revenue. Similarly, if either Gamunex-C/Gamunex IGIV or Prolastin/Prolastin-C A1PI were to become the subject of litigation and/or an adverse governmental ruling requiring us to cease sales of either product, our business would be adversely affected.
A recent review of two previously conducted studies of the safety and effectiveness of alpha-1 antitrypsin, sold by us as Prolastin, was conducted by the Nordic Cochrane Centre at Rigshospitalet in Copenhagen, Denmark. The review of these older studies concluded that there was no statistically meaningful difference between treatment with intravenous alpha-1 antitrypsin or a placebo. While we believe that the Nordic Cochrane Centre review is flawed, the review could have an adverse affect on sales of Prolastin and the prospects of obtaining future reimbursement approvals in Europe. Additionally, the FDA recently objected to promotional claims in marketing materials for Prolastin as inconsistent with the product’s approved package insert because they were based on data which the FDA considered insufficient to demonstrate the long-term effects derived from chronic augmentation therapy of individuals with alpha-1 antitrypsin deficiency.
We rely heavily upon sales from the U.S. region, which comprised approximately 69% and 66% of our net revenue for the years ended December 31, 2010 and 2009, respectively. If our U.S. sales were significantly impacted by either material changes to government or private payor reimbursement, by other regulatory developments, or by competition, then our business would be adversely affected.
Our manufacturing processes are complex and involve biological intermediates that are susceptible to contamination and variations in yield.
Plasma is a raw material that is susceptible to damage and contamination and may contain human pathogens, any of which would render the plasma unsuitable as raw material for further manufacturing. For instance, improper storage of plasma, by us or third-party suppliers, may require us to destroy some of our raw material. If unsuitable plasma is not identified and discarded prior to the release of the plasma to our manufacturing process, it may be necessary to discard intermediate or finished product made from that plasma or to recall any finished product released to the market, resulting in a charge to cost of goods sold.
The manufacture of our plasma products is an extremely complex process of fractionation, purification, filling and finishing. Our products can become non-releasable or otherwise fail to meet our stringent specifications through a failure of one or more of our product testing, manufacturing, process controls, and quality assurance processes. We may detect instances in which an unreleased product was produced without adherence to our manufacturing procedures or plasma used in our production process was not collected or stored in a compliant manner consistent with our current Good Manufacturing Practices (cGMP) or other regulations. Such an event of non-compliance would likely result in our determination that the impacted products should not be released and therefore should be destroyed. For example, a malfunction of the Gamunex IGIV chromatography system just prior to our formation transaction in 2005 resulted in the processing of IGIV products containing elevated levels of antibodies for over one month. Our total cost related to this incident, including the costs of product loss, investigation, testing, disposal, and other remedial actions, was approximately $41.6 million. We subsequently recovered from Bayer $10.7 million through our 2005 working capital adjustment and $9.0 million in the first quarter of 2007 through a settlement. As an additional example, our 2010 inventory impairment provisions included $24.6 million of provisions for raw materials and work in process inventories related to a Gamunex-C/Gamunex IGIV production issue. We believe we have identified the cause of the issue and have implemented appropriate remediation steps.
Once we have manufactured our plasma-derived products, they must be handled carefully and kept at appropriate temperatures. Our failure, or the failure of third parties that supply, ship or distribute our products, to properly care for our products may require that those products be destroyed.
While we expect to write off small amounts of work-in-progress in the ordinary course of business due to the complex nature of plasma, our processes and our products, unanticipated events may lead to write-offs and other costs materially in excess of our expectations. We have in the past had issues with product quality and purity that have caused us to write off the value of the product. Such write-offs and other costs could cause material fluctuations in our profitability. Furthermore, contamination of our products could cause investors, consumers, or other third parties with whom we conduct business to lose confidence in the reliability of our manufacturing procedures, which could adversely affect our sales and profits. In addition, faulty or contaminated products that are unknowingly distributed could result in patient harm, threaten the reputation of our products and expose us to product liability damages and claims from companies for whom we do contract manufacturing.
Additionally, due to the nature of plasma there will be variations in the biologic properties of the plasma we collect or purchase for fractionation that may result in fluctuations in the obtainable yield of desired fractions, even if cGMP is followed. Lower yields may limit production of our plasma-derived products due to capacity constraints. If these batches of plasma with lower yields impact production for extended periods, it may reduce the total capacity of product that we could market and increase our cost of goods sold, thus reducing our profitability.
Our business requires substantial capital to operate and grow and to achieve our strategy of realizing increased operating leverage, including the completion of several large capital projects.
We intend to undertake several large capital projects to maintain compliance with cGMP and expand capacity. These projects are required for us to expand our production capabilities and achieve our strategy of realizing operating leverage. Capital projects of this magnitude involve technology and project management risks. Technologies that have worked well in a laboratory or in a pilot plant may cost more or not perform as well, or at all, in full scale operations. Projects may run over budget or be delayed. We cannot be certain that these projects will be completed in a timely manner or that we will maintain our compliance with cGMP, and we may need to spend additional amounts to achieve compliance. Additionally, by the time these multi-year projects are completed, market conditions may differ significantly from our assumptions regarding the number of competitors, customer demand, alternative therapies, reimbursement and public policy, and as a result capital returns might not be realized.
We began a spending program in 2010 to obtain FDA approval for new indications for existing products, to enhance the facilities in which and processes by which we manufacture existing products, to develop new product delivery mechanisms for existing products, to strengthen our plasma collection system and to develop innovative product additions. We anticipate spending substantial sums in capital and operating expense for this program over the next five years. We face a number of obstacles to successfully converting these efforts into profitable products including but not limited to the successful development of a experimental product for use in clinical trials, the design of clinical study protocols acceptable to FDA, the successful outcome of clinical trials, our ability to scale our manufacturing processes to produce commercial quantities or successfully transition technology, FDA approval of our product or process and our ability to successfully market an approved product with our new process or new indication.
We expect to operate at or near our fractionation capacity over the next few years depending upon the demand for our products, the availability of source plasma, the impact of yield variability, the potential impact of inventory impairments, and normal production shut-downs, among other factors. We plan to utilize most of our available fractionation capacity in the near term, which may result in increased inventory levels in order to attempt to maintain pace with projected future growth in product demand, although we have not been successful in building excess finished goods inventories to date as a result of the factors previously mentioned. Consequently, any disruption in meeting our fractionation and purification plans would most likely result in lower revenue, gross profit, net income, and operating cash flows as well as lower than planned growth given our fractionation and purification constraints. Our fractionation constraints would likely preclude us from participating in greater than estimated overall market demand or higher demand for Gamunex-C/Gamunex IGIV. In response to our capacity constraints, we have embarked on a substantial capital plan, which we currently estimate to be in the range of $750 million to $800 million on a cumulative basis over the next five years through 2015, excluding capitalized interest. The amount and timing of future capital spending is dependent upon a number of factors, including market conditions, regulatory requirements, and the extent and timing of particular projects, among other things. Given our expectations that we will need fractionation capacity in the near term, our ability to grow our business is dependent upon the timely completion of these facilities and obtaining the requisite regulatory approvals.
To finance these various activities, we may need to incur future debt or issue additional equity if our cash flows and capital resources are insufficient, and we may not be able to structure our debt obligations on favorable economic terms. A failure to fund these activities may harm our competitive position, quality compliance and financial condition.
Our ability to continue manufacturing and distributing our products depends on our and our suppliers’ continued adherence to cGMP regulations.
The manufacturing processes for our products are governed by detailed written procedures and federal regulations that set forth cGMP requirements for blood and blood products. Our Quality Operations unit monitors compliance with these procedures and regulations, and the conformance of materials, manufacturing intermediates, and final products to their specifications. Failure to adhere to established procedures or regulations, or to meet a specification, could require that a product or material be rejected and destroyed. There are relatively few opportunities for us to rework, reprocess or salvage nonconforming materials or products.
Our adherence to cGMP regulations and the effectiveness of our quality systems are periodically assessed through inspections of our facilities by the FDA in the U.S. and analogous regulatory authorities in other countries. We could be cited for deficiencies in the future. If deficiencies are noted during an inspection, we must take action to correct those deficiencies and to demonstrate to the regulatory authorities that our corrections have been effective. If serious deficiencies are noted or if we are unable to prevent recurrences, we may have to recall product or suspend operations until appropriate measures can be implemented. We are required to report some deviations from procedures to the FDA. Even if we determine that the deviations were not
material, the FDA could require us to take similar measures. Since cGMP reflects ever evolving standards, we regularly need to update our manufacturing processes and procedures to comply with cGMP. These changes may cause us to incur costs without improving our profitability or the safety of our products. For example, more sensitive testing assays may be required (if and when they become available) or existing procedures or processes may require revalidation, all of which may be costly and time-consuming and could delay or prevent the manufacturing of a product or launch of a new product.
Changes in manufacturing processes, including a change in the location where the product is manufactured or a change of a third-party manufacturer, may require prior FDA review and approval or revalidation of the manufacturing process and procedures in accordance with cGMP. There may be comparable foreign requirements. For example, we have completed the process of transferring the manufacture of our Thrombate III product from Bayer’s Berkeley, California, biologics manufacturing facility to our Clayton manufacturing facility that we are currently validating with regulatory approval expected in 2012. We cannot guarantee that we have a sufficient inventory of intermediates and finished product to meet demand until the new facility is approved and manufacturing can recommence. To validate our manufacturing processes and procedures following completion of upgraded facilities, we must demonstrate that the processes and procedures at the upgraded facilities are comparable to those currently in place at Bayer’s facilities. In order to provide such a comparative analysis, both the Bayer facility processes and the processes that we expect to be implemented at our upgraded facilities must comply with the regulatory standards prevailing at the time that our expected upgrade is completed. If the FDA does not approve the transfer, our ability to market our Thrombate III product will be seriously impaired or eliminated. In addition, regulatory requirements, including cGMP regulations, continually evolve. Failure to adjust our operations to conform to new standards as established and interpreted by applicable regulatory authorities would create a compliance risk that could impair our ability to sustain normal operations.
A number of inspections by the FDA and foreign control authorities, including the German Health Authority (GHA), have been conducted or are expected at our plasma collection centers in 2011. Some of these inspections are of licensed centers to assess ongoing compliance with cGMP, while others are of our currently unlicensed centers as a prerequisite to final approval of the centers’ license applications. If the FDA (or other authorities) finds these centers not to be in compliance, our ongoing operations and/or plans to expand plasma collections would be adversely affected.
We must continually monitor the performance of our products once approved and marketed for signs that their use may elicit serious and unexpected side effects, which could jeopardize our ability to continue marketing the products. We may also be required to conduct post-approval clinical trials as a condition to licensing a product.
As for all pharmaceutical products, the use of our products sometimes produces undesirable side effects or adverse reactions or events (referred to cumulatively as “adverse events”). For the most part, these adverse events are known, are expected to occur at some frequency and are described in the products’ labeling. Known side effects described in the labeling for our products are as follows: the use of Plasbumin 5%, 20%, 25% sometimes produce the following adverse events: allergic manifestations including urticaria, chills, fever and changes in respiration, pulse and blood pressure; the use of Plasmanate sometimes produces the following adverse events: hypotension, flushing, urticaria, back pain, nausea, headache; the use of Koate DVI sometimes produces the following adverse events: allergic type reactions; tingling in the arm, ear and face; blurred vision, headache, nausea, stomach ache and jittery feeling; the use of Gamunex sometimes produces the following adverse events: nausea, vomiting, asthenia, pyrexia, rigors, injection site reaction, allergic/anaphylactic reaction, aseptic meningitis, arthralgia, back pain, dizziness, headache, rash, pruritus, urticaria, hemolysis/hemolytic anemia; the use of Prolastin/Prolastin-C sometimes produces the following adverse events: dyspnea, tachycardia, rash, chest pain, chills, influenza-like symptons, hypersensitivity, hypotension, hypertension; the use of Thrombate III sometimes produces the following adverse events: dizziness, chest tightness, nausea, foul taste in mouth, chills, cramps, shortness of breath, chest pain, filmy vision, light-headedness, gastrointestinal fullness, hives, fever, hematoma formation; the use of HyperHEP B sometimes produces the following adverse events: local pain and tenderness at the injection site, urticaria, angioedema, anaphylactic reactions; the use of HyperRAB sometimes produces the following adverse events: soreness at injection site, mild temperature elevation, sensitivity to repeated injections in immunoglobulin-deficient patients, angioneurotic edema, skin rash, nephritic syndrome, anaphylactic shock; the use of HyperTET sometimes produces the following adverse events: soreness at injection site, sensitization to repeated injections of immunoglobulin, slight temperature elevation, angioneurotic edema, nephritic syndrome, anaphylactic shock; the use of HyperRHO sometimes produces the following adverse events: soreness at injection site, sensitization to repeated injections of immunoglobulin, slight temperature elevation, elevated bilirubin; and the use of GamaSTAN sometimes produces the following adverse events: local pain and injection site soreness, angioedema, anaphylaxis, and urticaria. When adverse events are reported to us, we investigate each event and circumstances surrounding it to determine whether it was caused by our product and whether it implies a previously unrecognized safety issue exists. Periodically, we report summaries of these events to the applicable regulatory authorities.
In addition, the use of our products may be associated with serious and unexpected adverse events, or with less serious reactions at a greater than expected frequency. This may be especially true when our products are used in critically ill patient populations. When these unexpected events are reported to us, we must make a thorough investigation to determine causality and implications for product safety. These events must also be specifically reported to the applicable regulatory authorities. If our evaluation concludes, or regulatory authorities perceive, that there is an unreasonable risk associated with the product, we would be obligated to withdraw the impacted lot(s) of that product. Furthermore, an unexpected adverse event of a new product could be recognized only after extensive use of the product, which could expose us to product liability risks, enforcement action by regulatory authorities and damage to our reputation and public image.
We have received reports that some Gamunex patients have experienced transient hemolysis and/or hemolytic anemia, which are known potential side effects for this class of drugs. Since 2005, a disproportionate number of these reports have been received from Canada, where our product accounted for 80% of all IGIV distributed in 2008. The Canadian product labeling was updated in 2005 after these hemolysis events were first reported to Health Canada.
Subsequently, Talecris provided annual updates on these events to Health Canada from 2006 to 2008, but no further action was recommended by the Canadian regulators. A serious adverse finding concerning the risk of hemolysis by any regulatory authority for intravenous immune globulin products in general, or Gamunex in particular, could adversely affect our business and financial results.
Once we produce a product, we rely on physicians to prescribe and administer them as we have directed and for the indications described on the labeling. It is not, however, unusual for physicians to prescribe our products for “off-label” uses or in a manner that is inconsistent with our directions. For example, a physician may prescribe an infusion rate for our Gamunex IGIV product that is greater than our directed infusion rate, which in turn may reduce its efficacy or result in some other adverse affect upon the patient. Similarly, a physician may prescribe a higher or lower dosage than the dosage we have indicated, which may also reduce our product’s efficacy or result in some other adverse affect upon the patient. To the extent such off-label uses and departures from our administration directions become pervasive and produce results such as reduced efficacy or other adverse effects, the reputation of our products in the marketplace may suffer.
When a new product is approved, the FDA or other regulatory authorities may require post-approval clinical trials, sometimes called Phase IV clinical trials. For example, FDA has required such trials for A1PI products, including our recently approved A1PI next generation product, Prolastin-C. If the results of such trials are unfavorable, this could result in the loss of the license to market the product, with a resulting loss of sales.
Our products face increased competition.
Recently, certain of our products have experienced increased competition. Until 2004, we were one of two North American suppliers with an approved liquid IGIV product. In 2004, Grifols launched Flebogamma® 5% liquid IGIV and Octapharma launched Octagam® 5% liquid IGIV. In 2005, Baxter’s Gammagard® 10% liquid IGIV was launched. In 2007 CSL Behring received approval for Privigen® 10% liquid IGIV. Privigen® was launched in the U.S. in 2008. In 2010, CSL Behring received FDA approval and launched Hizentra Immune Globulin Subcutaneous (Human) 20% liquid. Also in 2010, Grifols received FDA approval and launched its 10% liquid IGIV, Flebogamma 10% DIF.We expect Octapharma to launch a 10% liquid IGIV product in the U.S. Omrix and Biotest are both seeking approval for liquid IGIV products in the U.S., which, if approved, will further increase competition among liquid IGIV products. Additionally, Bio Products Laboratory received approval from the FDA for its 5% concentration IGIV for PI. As competition has increased, competitors have discounted the price of IGIV products. Furthermore, many customers are increasingly more price sensitive regarding IGIV products. Octapharma’s IGIV products have been off the market during much of 2010 in the U.S. and other parts of the world. When Octapharma resumes sales of its products, it may discount prices to regain lost market share. If customers demand lower priced products of competitors, we may lose sales or be forced to lower our prices.
In Canada, we have been the “supplier of record” since the 1980s. We have experienced, and expect to continue to experience, annual volume declines in Canada due to Canadian Blood Services objective to have multiple sources of supply, which has impacted and will continue to impact our overall IGIV growth. Canadian Blood Services may further reduce volumes to contract minimums and Hema Quebec may adopt a similar strategy.
Until December 2002, our A1PI product, Prolastin A1PI, was the only plasma product licensed and marketed for therapy of congenital A1PI deficiency-related emphysema in the U.S. Accordingly, until that time, Prolastin A1PI had virtually 100% market share in its category. In December 2002 and July 2003, predecessors of Baxter and CSL Behring received licenses for Aralast and Zemaira, respectively, which were launched in the U.S. in 2003, and Grifols received marketing authorization for Trypsone in Spain in 2003. Due in part to our inability to fully meet demand for A1PI product, as well as patient losses due to the nature of the disease, our share of sales has dropped to approximately 62% in 2009 in the United States and 74% in 2008 globally according to the Marketing Research Bureau, which is referred to as MRB. Competitors may increase their sales, lower their prices or change their distribution model, which may harm our product sales and financial condition. Also, if the attrition rate of our Prolastin/Prolastin-C A1PI patient base accelerates faster than we have forecast, we would have fewer patients and lower sales volume. In addition, Kamada Ltd. received approval of its BLA for its liquid A1PI product, Glassia, on July 1, 2010. In the European Union, we have an 87% share of A1PI sales in 2008 according to MRB data, and have the only licensed A1PI product, other than Grifols, which has marketing authorization for Trypsone A1PI in Spain, and LFB, which sells Alfalastin in France. Our competitors are currently pursuing licensing trials in Europe. Should our competitors receive approvals in the European Union sooner than expected, our unit volumes and share of sales will be impacted.
New products may reduce demand for plasma-derived A1PI. A recombinant form of A1PI (recA1PI) could gain market share through the elimination of the risk of plasma-borne pathogens, or through a reduced price permitted by significantly decreased costs (because the recA1PI would not be sourced from plasma). In addition to us, Arriva and GTC Biotherapeutics are in the early stages of development for a recombinant form of recA1PI. Although we are not aware of any active clinical trials for a recA1PI product, a successful recA1PI, prior to us developing a similar product, could gain first mover advantage and result in a loss of our A1PI market share. If a new formulation of A1PI is developed that has a significantly improved rate or method of administration, such as aerosol inhalation, the market share of Prolastin/Prolastin-C A1PI could be negatively impacted. Similarly, several companies are attempting to develop products which would be substitution threats in the A1PI sector, including retinoic acid, oral synthetic elastase inhibitors and gene therapy. While these products are all in early stages of development, the potential for successful product development and launch cannot be ruled out.
In addition, our plasma-derived therapeutics face competition from non-plasma products and other courses of treatments. For example, two RhD hyperimmune globulins for intravenous administration, Cangene’s WinRho SDF and CSL Behring’s Rhophylac, are now approved for use to treat ITP, and GSK and Amgen launched thrombopoietin inhibitors targeting ITP patients in 2008 that may reduce the demand for IGIV to treat this immune disorder. There is also a risk that indications for which our products are now used will be susceptible to new treatments, such as small molecules, monoclonal or recombinant products. Recombinant Factor VIII product competes with our own plasma-derived product in the treatment of Hemophilia A and is perceived by many experts to have lower risks of disease transmission. Additional recombinant products or the use of monoclonal antibodies, small molecules or stem cell transplantations could compete with our products and reduce the demand for our products. Crucell and Sanofi Pasteur have completed Phase II clinical trials for a monoclonal rabies product to compete with our rabies hyperimmune product. If successful, we estimate that the monoclonal product could take a significant portion of the rabies market in years subsequent to its introduction. Also, in February 2009, GTC Biotherapeutics obtained FDA approval of a competitive ATIII product for the treatment of hereditary antithrombin deficiency, which is derived from the milk of transgenic goats. This product now directly competes with our product, Thrombate III (Human), which had been the only FDA approved product.
We do not currently sell any recombinant products. Although we are attempting to develop recombinant versions of Plasmin, A1PI and Factor VIII, we cannot be certain that any of these products will ever be approved or commercialized. As a result, our product offerings may remain plasma-derived, even if our competitors offer recombinant products.
Our financial performance will suffer if we do not improve the cost efficiency of our plasma collection platform.
The opening of new plasma collection centers, which take up to several years to reach efficient production capacity, and the creation of a corporate infrastructure to support our vertical integration strategy have historically resulted in our per liter cost of plasma being higher than many of our larger competitors.
In order to continue to improve the cost per liter of plasma, we will need to significantly increase production levels to leverage fixed costs, improve operational efficiency, reduce overall fixed costs, reduce variable costs such as donor fees or a combination of some or all of the foregoing. Our inability to significantly reduce the cost per liter of plasma will result in higher costs of operations, lower margins and lower cash flow than our competitors. If by attempting to reduce these costs, we adversely affect compliance with cGMP, we may be required to write-off plasma and any intermediates and products manufactured with non-compliant plasma and we may face shortages of plasma needed to manufacture our products.
We would become supply-constrained and our financial performance would suffer if we could not obtain adequate quantities of FDA-approved source plasma.
In order for plasma to be used in the manufacturing of our products, the individual centers at which the plasma is collected must be licensed by the FDA, and approved by the regulatory authorities, such as the GHA, of those countries in which we sell our products. When a new plasma center is opened, and on an ongoing basis after licensure, it must be inspected by the FDA and GHA for compliance with cGMP and other regulatory requirements. An unsatisfactory inspection could prevent a new center from being licensed or risk the suspension or revocation of an existing license.
In order to maintain a plasma center’s license, its operations must continue to conform to cGMP and other regulatory requirements. In the event that we determine that plasma was not collected in compliance with cGMP, we may be unable to use and may ultimately destroy plasma collected from that center, which would be recorded as a charge to cost of goods. Additionally, if non-compliance in the plasma collection process is identified after the impacted plasma has been pooled with compliant plasma from other sources, entire plasma pools, in-process intermediate materials and final products could be impacted. Consequently, we could experience significant inventory impairment provisions and write-offs which could adversely affect our business and financial results. During 2008, we experienced such an event at one of our plasma collection centers, which resulted in a charge to cost of goods sold of $23.3 million, for which we subsequently recovered $19.4 million through December 31, 2010. In this particular instance, a portion of the impacted plasma had been released to manufacturing prior to our detection of the issue.
We plan to increase our supplies of plasma for use in our manufacturing processes through increased collections at our plasma collection centers and through selective remodeling or relocations of existing centers. This strategy is dependent upon our ability to successfully integrate new centers, to obtain FDA and GHA approval for the remaining unlicensed plasma centers, to maintain a cGMP compliant environment in all plasma centers, and to expand production and attract donors to our centers.
Our ability to expand production and increase our plasma collection centers to more efficient production levels may be affected by changes in the economic environment and population in selected regions where TPR operates plasma centers, by the entry of competitive plasma centers into regions where TPR operates, by misjudging the demographic potential of individual regions where TPR expects to expand production and attract new donors, by unexpected facility related challenges, or by unexpected management challenges at selected plasma centers. In addition, beginning in 2012, the recently enacted healthcare reform legislation may require us to issue Internal Revenue Service Form 1099 to plasma donors whose remuneration equals or exceeds six hundred dollars annually. The cost of implementing this requirement, as well as its potential impact on plasma donations, is unknown at this time.
Our financial performance is dependent upon third-party suppliers of FDA-approved source plasma.
For the years ended December 31, 2010 and 2009, we obtained 15.3% and 14.3% of our plasma under a five-year supply arrangement with CSL Plasma Inc., a subsidiary of CSL, a major competitor. The agreement with CSL Plasma Inc. provides that our minimum purchase obligations are: (i) 550,000 liters for calendar year 2010; (ii) 300,000 liters of plasma for each of calendar years 2011 and 2012; and (iii) 200,000 liters of plasma for calendar year 2013. Each quarter, CSL Plasma Inc. is obligated to deliver at least 20% of our minimum purchase obligation for that year. We notified CSL Plasma Inc that we will not elect to take optional volumes under the contract in 2011. Either we or CSL Plasma Inc. may terminate the agreement in the event of material nonperformance after a 30-day cure period. For the years ended December 31, 2010 and 2009, we obtained 9.9% and 8.9% of our plasma from Interstate Blood Bank, Inc. (IBBI). The agreement with IBBI requires us to make a minimum purchase of 330,000 liters of plasma for each year during the term of the agreement, which terminates at the end of 2016. We have a right of first refusal with respect to any material quantities of plasma that IBBI has available for sale in excess of this amount, as well as a right of first refusal with respect to the transfer of any asset, equity, or controlling interest of IBBI related to any of the centers which supply us. We also agreed to provide secured financing for additional centers approved by us and the relocation of IBBI’s plasma centers in a maximum amount of $1.0 million per center and $3.0 million in the aggregate. Either we or IBBI may terminate the agreement in the event of material nonperformance after a 30 day cure period. Were any dispute to arise or were CSL Plasma Inc. or IBBI to experience any plasma collection difficulties, it could be difficult or impossible for us to replace the shortfall, which would materially adversely affect our business.
Plasma volumes obtained under arrangements with independent third parties have not always met expectations. An inability of any of our suppliers to operate their business successfully and satisfy their obligations in a timely manner may cause a disruption in our plasma supply, which could materially adversely affect our business.
Industry-wide disruptions could reduce the availability of FDA-approved source plasma and our financial performance would suffer.
A number of other factors could disrupt our ability to increase source plasma collections, including but not limited to:
· A lack of alternative plasma supply sources. In recent years, there has been consolidation in the industry as several plasma derivatives manufacturers have acquired previously independent plasma collectors. As a result, it could be difficult or impossible to resolve any significant disruption in the supply of plasma or an increased demand for plasma with plasma from alternative sources.
· A reduction in the donor pool. Regulators in most of the large markets for plasma-derived products, including the United States, restrict the use of plasma collected from specific countries and regions in the manufacture of plasma-derived products. For example, the appearance of the variant Creutzfeldt-Jakob disease, commonly referred to as “mad cow” disease (which resulted in the suspension of the use of plasma collected from U.K. residents), and concern over the safety of blood products (which has led to increased domestic and foreign regulatory control over the collection and testing of plasma and the disqualification of certain segments of the population from the donor pool), have significantly reduced the potential donor pool.
Our products have historically been subject to supply-driven price fluctuations.
Our products, particularly IGIV, have historically been subject to price fluctuations as a result of changes in industry supply levels, the availability and pricing of plasma, development of competing products and the availability of alternative therapies. Higher prices for plasma-derived products have traditionally spurred increases in plasma production and collection capacity, resulting over time in increased product supply and lower prices. As demand continues to grow, if plasma supply and manufacturing capacity do not commensurately expand, prices tend to increase.
The demand for plasma derived products, particularly for IGIV, over the last few years has resulted in efforts on the part of companies, including ourselves, to increase manufacturing capacity and open new plasma collection centers to increase the availability of source plasma. Some of our competitors have announced plans to grow product supply at a rate above expected demand growth. The growth in demand for IGIV has been outpaced by the recent supply growth, as evidenced by increased supply in the distribution channel. We, or our competitors, may misjudge demand growth and over-invest in expanding plasma collection or manufacturing capacity, which ultimately may result in lower prices for, or inability to sell, our products.
Exchange rate fluctuations and our foreign currency hedges could adversely affect our financial results.
As a result of our international operations, currency exchange rate fluctuations may affect our results of operations, cash flows, and financial position. Our most significant foreign currency exposure is the euro. Although from time to time, we may enter into foreign currency exchange agreements with financial institutions to reduce our exposure to fluctuations in foreign currency values relative to our foreign receivables and forecasted sales transactions, these hedging transactions do not eliminate that risk entirely. These hedges may also serve to reduce any gain that we may have made based on favorable foreign currency fluctuations. Furthermore, these contracts have inherent levels of counterparty risk over which we have no control. We are exposed to potential losses if a counterparty fails to perform according to the terms of the agreement. We do not require collateral or other security to be furnished by counterparties to our derivative financial instruments. A number of financial institutions similar to those that serve or may serve as counterparties to our hedging arrangements were adversely affected by the global credit crisis. The failure of any of the counterparties to our hedging arrangements to fulfill their obligations to us could adversely affect our results of operations. At December 31, 2010, we were not a party to any foreign currency hedges. During the first quarter of 2011, we entered into approximately $49.5 (€37.1) million in notional value of fair value hedges against firm commitments and $38.4 (€28.2) million in notional value of cash flow hedges against anticipated future sales. The weighted average U.S. dollar to euro exchange rate on these foreign currency contracts is 1.3461.
We are investigating potential Foreign Corrupt Practices Act violations.
We are conducting an internal investigation into potential violations of the Foreign Corrupt Practices Act (FCPA) that we became aware of during the conduct of an unrelated review. The FCPA investigation is being conducted by outside counsel under the direction of a special committee of our board of directors. The investigation initially focused on sales to certain Eastern European and Middle Eastern countries, primarily Belarus, Russia and Iran, but we are also reviewing sales practices in Brazil, Bulgaria, China, Georgia, Libya, Poland, Turkey, Ukraine and other countries as deemed appropriate.
In July 2009, we voluntarily contacted the U.S. Department of Justice (DOJ) to advise them of the investigation and to offer our cooperation in any investigation that they want to conduct or they want us to conduct. The DOJ has not indicated what action it may take, if any, against us or any individual, or the extent to which it may conduct its own investigation. Even though we self-disclosed this matter to the DOJ, it or other federal agencies may seek to impose sanctions on us that may include, among other things, debarment, injunctive relief, disgorgement, fines, penalties, appointment of a monitor, appointment of new control staff, or enhancement of existing compliance and training programs. Other countries in which we do business may initiate their own investigations and impose similar penalties. As a result of this investigation, we suspended shipments to some of these countries while we put additional safeguards in place. In some cases, safeguards involved terminating consultants and suspending relations with or terminating distributors in countries under investigation as circumstances warranted. These actions unfavorably affected revenue from these countries in 2010 and 2009. We have resumed sales in countries where we believe we have appropriate safeguards in place and are reallocating product to other countries as necessary. To the extent that we conclude, or the DOJ concludes, that we cannot implement adequate safeguards or otherwise need to change our business practices, distributors, or consultants in affected countries or other countries, this may result in a permanent loss of business from those countries. We expect to complete our internal FCPA investigation and present our findings to the DOJ in 2011. The preliminary findings of our investigation indicate that it is probable that there were FCPA violations by persons associated with us that the DOJ or other regulators may assert are attributable to us. Given the preliminary nature of our findings, our continuing investigation and the uncertainties regarding this matter, we are unable to estimate the financial outcome. Any such sanctions or loss of business could have a material adverse effect on us or our results of operations financial condition, or cash flows.
A pending investigation relating to our compliance with the terms of the Pharmaceutical Pricing Agreement under the Public Health Service program may result in our being barred from allocating a fixed amount of IGIV as available for sale at the Public Health Service price.
In November 2009, we received a letter from the United States Attorney’s Office for the Eastern District of Pennsylvania (USAO). The USAO requested a meeting to review our compliance with the terms of the Pharmaceutical Pricing Agreement (PPA) under the Public Health Service program. Specifically, the USAO asked for information related to the sale of our IGIV product, Gamunex, under that program. In order to have federal financial participation apply to their products under the Medicaid program and to obtain Medicare Part B coverage, manufacturers are required to enter into a PPA. The PPA obligates manufacturers to charge covered entities the Public Health Service price for drugs intended for outpatient use. The Public Health Service price is based on the Medicaid rebate amount. We believe that we have complied with the terms of the PPA and federal law. If the USAO determines that our practices are inconsistent with the terms of the PPA, the USAO has stated that it may file a civil action against us under the Anti-fraud Injunction Act and seek a court order directing us to comply with the PPA or, potentially, proceed under some other legal theory. An adverse outcome in an Anti-fraud Injunction Act action could have a material adverse effect on us or our results of operation to the extent that we are barred from allocating a fixed amount of IGIV as available for sale at the Public Health Service price and we are forced to give a preference to those purchasers over all other customers. We could also be subject to fines, damages, penalties, appointment of a monitor, or enhancement of existing compliance and training programs as a result of government action. We are cooperating with the investigation and intend to respond to information requests from the USAO.
Our ability to export products to Iran requires annual export licenses and the use of intermediate or advisory banks.
In 2010, we had sales of $16.6 million, or approximately 1.0% of our net revenue, to customers located in Iran pursuant to an export license which must be renewed annually. Although the Office of Foreign Asset Control (OFAC) renewed our license to supply humanitarian products, tensions with Iran continue to impede our ability to conduct business in Iran. Our revenues related to our business in Iran have declined in 2010 compared to 2009 and are likely to continue to decline in the future.
Our ability to continue to produce safe and effective products depends on the safety of our plasma supply against transmittable diseases.
Despite overlapping safeguards, including the screening of donors and other steps to remove or inactivate viruses and other infectious disease causing agents, the risk of transmissible disease through plasma-derived products cannot be entirely eliminated. For example, since plasma-derived therapeutics involve the use and purification of human plasma, there has been concern raised about the risk of transmitting HIV, prions, West Nile virus, H1N1 virus (commonly known as the swine flu) and other blood-borne pathogens through plasma-derived products. There are also concerns about the future transmission of H5N1 virus (commonly known as the bird flu). In the 1980s, thousands of hemophiliacs worldwide were infected with HIV through the use of contaminated Factor VIII. Bayer and other producers of Factor VIII, though not us, are defendants in numerous lawsuits resulting from these infections.
New infectious diseases emerge in the human population from time to time. If a new infectious disease has a period during which time the causative agent is present in the bloodstream but symptoms are not present, it is possible that plasma donations could be contaminated by that infectious agent. Typically, early in an outbreak of a new disease, tests for the causative agent do not exist. During this early phase, we must rely on screening of donors (e.g., for behavioral risk factors or physical symptoms) to reduce the risk of plasma contamination. Screening methods are generally less sensitive and specific than a direct test as a means of identifying potentially contaminated plasma units.
During the early phase of an outbreak of a new infectious disease, our ability to manufacture safe products would depend on the manufacturing process’ capacity to inactivate or remove the infectious agent. To the extent that a product’s manufacturing process is inadequate to inactivate or remove an infectious agent, our ability to manufacture and distribute that product would be impaired.
If a new infectious disease were to emerge in the human population, the regulatory and public health authorities could impose precautions to limit the transmission of the disease that would impair our ability to procure plasma, manufacture our products or both. Such precautionary measures could be taken before there is conclusive medical or scientific evidence that a disease poses a risk for plasma-derived products.
In recent years, new testing and viral inactivation methods have been developed that more effectively detect and inactivate infectious viruses in collected plasma. There can be no assurance, however, that such new testing and inactivation methods will adequately screen for, and inactivate, infectious agents in the plasma used in the production of our products.
If our Clayton facility or other major facilities, or the facilities of our third-party suppliers, were to suffer a crippling accident, or a force majeure event materially affected our ability to operate and produce saleable products, a substantial part of our manufacturing capacity could be shut down for an extended period.
Substantially all of our revenues are derived from products manufactured, and services performed, at our plants located in Clayton, North Carolina and Melville, New York. In addition, a substantial portion of our plasma supply is stored at facilities in Benson, North Carolina, and our Clayton facility. If any of these facilities were to be impacted by an accident or a force majeure event such as an earthquake, major fire or explosion, major equipment failure or power failure lasting beyond the capabilities of our backup generators, our revenues would be materially adversely affected. In this situation, our manufacturing capacity could be shut down for an extended period and we could experience a loss of raw materials, work in process or finished goods inventory. Other force majeure events such as terrorist acts, influenza pandemic or similar events could also impede our ability to operate our business. In addition, in any such event, the reconstruction of our Clayton fractionation plant or our plasma storage facilities, the regulatory approval of the new facilities, and the replenishment of raw material plasma could be time-consuming. During this period, we would be unable to manufacture our products at other plants due to the need for FDA and foreign regulatory authority inspection and certification of such facilities and processes. While we maintain property damage and business interruption insurance with limits of $1 billion, these amounts may still be insufficient to mitigate the losses from any such event. We may also be unable to recover the value of the lost plasma or work-in-progress, as well as the sales opportunities from the products we would be unable to produce.
A significant number of our plasma collection centers are located near the U.S. border with Mexico. For the years ended December 31, 2010 and 2009, approximately 21% and 22%, respectively, of our internally sourced plasma came from collection centers located on the United States border with Mexico. Donations at these centers could be impacted by changes in U.S. visa rules and the recently enacted healthcare reform legislation, which may require us beginning in 2012 to issue Internal Revenue Service Form 1099 to plasma donors whose remuneration equals or exceeds six hundred dollars annually. The cost of implementing this requirement, as well as its potential impact on plasma donations, is unknown at this time. In addition, we have a number of plasma centers in regions of the southeast which could be affected by natural disasters such as hurricanes. A disruption in our source of plasma due to events arising in a geographic region where many of our collection centers are located would limit our ability to maintain our current production levels of plasma-derived products.
If we experience equipment difficulties or if the suppliers of our equipment or disposable goods fail to deliver key product components or supplies in a timely manner, our manufacturing ability would be impaired and our product sales could suffer.
We depend on a limited group of companies that supply and maintain our equipment and provide supplies such as chromatography resins, filter media, glass and stoppers used in the manufacture of our products. In some cases we have only one qualified supplier. If our equipment should malfunction, the repair or replacement of the machinery may require substantial time and cost, which could disrupt our production and other operations. Our plasma collection centers rely on disposable goods supplied by Haemonetics Corporation and information technology systems hosted by a subsidiary of Haemonetics Corporation. Our plasma collection centers cannot operate without an uninterrupted supply of these disposable goods and the operation of these systems. We have experienced periodic outages of these systems, but a material outage would affect our ability to operate our collection centers. Alternative sources for key component parts or disposable goods may not be immediately available. Any new equipment or change in supplied materials may require revalidation by us and/or review and approval by the FDA, or foreign regulatory authorities, including the German Health Authority, which may be time-consuming and require additional capital and other resources. We may not be able to find an adequate alternative supplier in a reasonable time period, or on commercially acceptable terms, if at all. As a result, shipments of affected products may be limited or delayed. Our inability to obtain our key source supplies for the manufacture of our products may require us to delay shipments of products, harm customer relationships and force us to curtail operations.
We purchase nearly all of our specialty plasma used for the production of hyperimmunes from a limited number of companies under short-term contracts.
We currently rely on three companies — Biotest Pharmaceuticals Corporation, Octapharma AG, and Advanced Bioservices, LLC, which is referred to as ABS, a subsidiary of Kedrion SpA, all of which are our direct competitors — to supply nearly all of our specialty plasma required for the production of our hyperimmunes, which represented $69.8 million, or 4.4%, of our net revenue for the year ended December 31, 2010 and $74.2 million, or 4.8%, of our net revenue for the year ended December 31, 2009. Specialty plasma is plasma that contains antibodies to specific diseases, usually because the donor has been vaccinated. Our contracts with suppliers of specialty plasma are usually on a short-term basis. We have entered into contracts with Octapharma and Biotest to secure our expected need for specialty plasma in 2011 and 2012. Depending upon these competitors’ production plans, it may be difficult to increase the amounts of plasma we purchase from them or to renew our contracts in the future. Our inability to replace the volumes provided by these suppliers through our own plasma collection efforts or through increased specialty plasma deliveries from other third parties would materially adversely affect our business. To the extent that we develop a supply of specialty plasma from our own collection centers, such specialty plasma may come at the expense of the plasma we use for our other products. It would also take significant time to obtain the necessary regulatory approvals and develop a sufficient donor base.
We rely in large part on third parties for the sale, distribution and delivery of our products.
In the U.S., we regularly enter into distribution, supply and fulfillment contracts with group purchasing organizations, home care companies, alternate infusion sites, hospital groups, and others. We are highly dependent on these contracts for the successful sale, distribution and delivery of our products. For example, we rely principally on group purchasing organizations and on our distributors to sell our IGIV product and on Centric Health Resources to fulfill prescriptions for Prolastin/Prolastin-C A1PI. If the parties with which we contract breach, terminate, or otherwise fail to perform under the agreements, our ability to effectively distribute our products will be impaired and our business may be materially and adversely affected. In addition, through circumstances outside of our control, such as general economic decline, market saturation, or increased competition, we may be unable to successfully renegotiate our contracts or secure terms which are as favorable to us. In addition, we rely on distributors for sales of our products outside the U.S. Disagreements or difficulties with our distributors supporting our export business could result in a loss of sales.
Product liability lawsuits against us could cause us to incur substantial liabilities, limit sales of our existing products and limit commercialization of any products that we may develop.
Our business exposes us to the risk of product liability claims that are inherent in the manufacturing, distribution, and sale of plasma-derived therapeutic protein products. We face an inherent risk of product liability exposure related to the testing of our product candidates in human clinical trials and an even greater risk when we commercially sell any products. If we cannot successfully defend ourselves against claims that our product candidates or products caused injuries, we could incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
· decreased demand for our products and any product candidates that we may develop;
· injury to our reputation;
· withdrawal of clinical trial participants;
· costs to defend the related litigation;
· substantial monetary awards to trial participants or patients;
· loss of revenue; and
· the inability to commercialize any products that we may develop.
Bayer is the defendant in continuing litigation alleging that use of products manufactured at our Clayton site in the 1980s, prior to our formation transaction and carve-out from Bayer, resulted in the transmission of Hepatitis C virus and HIV to patients. Bayer is also a defendant in litigation alleging that thimerosal, a preservative that was added to some intra muscular (hyperimmune) immune globulin products until 1996 (at which time its use was discontinued), was the cause of autism and other disorders in children who received these products. While we are not a party to any of these actions, and Bayer has agreed to fully indemnify us from any claims or losses arising out of these actions, we cannot assure you that our products or any of their constituents or additives may not someday give rise to similar product liability claims that we will be forced to defend and which may have a material adverse affect on our business.
We have a global insurance policy with limits of $100 million with a per claim deductible of $5 million and an aggregate deductible of $10 million. This amount of insurance may not be adequate to cover all liabilities that we may incur. We intend to expand our insurance coverage as our sales grow. Insurance coverage is, however, increasingly expensive. We may not be able to maintain insurance coverage at a reasonable cost and we may not be able to obtain insurance coverage that will be adequate to satisfy any liability that may arise.
Our products and manufacturing processes are subject to regulatory requirements and authority, including over our manufacturing practices and any product recalls.
Our business is heavily regulated in all jurisdictions where we collect plasma or manufacture or sell our products. In particular, plasma collection activities in the United States are regulated by the FDA, which requires a licensing and certification process for each plasma collection center prior to opening and conducts periodic inspections of facilities and processes. Many states also regulate plasma collection, imposing similar obligations and additional inspections and audits. In addition, the marketing and sale of a pharmaceutical product such as plasma derivatives and parenteral solutions are subject to the prior registrations, listings, licenses and approvals of such products with the competent authorities of the jurisdiction where the product is to be marketed and sold, including compliance with promotion, labeling and advertising requirements. Our manufacturing facilities located in Clayton, North Carolina, must meet strict European Union and FDA rules and our manufacturing facilities in Melville, New York, must comply with FDA rules. Our manufacturing facilities must also comply with applicable state laws. U.S. plasma centers collecting plasma for manufacture into products to be distributed in the European Union must also be approved by the competent European Health Authority.
Collection centers and manufacturing facilities are subject to periodic inspections by regulatory authorities. The consequences of adverse findings following inspections can be more serious, such as the temporary shutdown of such center or facility, the loss of that center’s or facility’s license because of alleged noncompliance with applicable requirements, a voluntary or mandatory recall of finished product released to the market, or the destruction of inventory. These more serious consequences are often highly public and may also prompt private products liability lawsuits, additional regulatory enforcement actions, the imposition of substantial fines or penalties by regulatory authorities, and damage to the reputation and public image of the collection or manufacturing facility.
With respect to product recalls, we have, from time to time, voluntarily recalled plasma products that had been released to the market in an effort to address drug safety issues and may do so again in the future. Since it’s formation in 2005, we have had four recalls of finished biological products. The products involved were: Plasma Protein Fraction (Human) 5% USP, Plasmanate®, Lot Number: 26N39N1; Antihemophilic Factor (Human), Koate DVI®; Lot Numbers: 26N7802, 26N6XW1, 26N6N01, 26N7H01; Rho(D) Immune Globulin (Human); HyperRHO S/D®, Mini-Dose, Lot Number: 26N7XX1; and Plasbumin-5®, Albumin (Human) 5%, USP, Lot Number: 26N9P21. In addition, plasma unit retrievals are routinely handled between Talecris Plasma Resources and its consignee, exclusively Talecris Biotherapeutics, when new information relevant to donor or plasma suitability is received after a donation is collected. Plasma unit retrievals are also triggered if units were distributed that should have been rejected by the plasma center. A minority of unit retrievals are required to be reported to the FDA as Biological Product Deviation Reports (“BPDRs”), and a relatively small number are classified by the FDA as recalls. There have been approximately 176 incidents that resulted in retrievals of plasma units by Talecris Plasma Resources from 2007 through December 2010 that have been classified as recalls by the FDA.
In addition, the FDA conducts ongoing monitoring and surveillance of advertising and promotional matter used by manufacturers to sell and promote their products. The FDA assesses these materials for compliance with the FDCA, regulations on misbranding and other requirements, for example, assessing if information about the risks and benefits of regulated products are communicated in a truthful, accurate, science-based, non-misleading and balanced manner. The FDA issued Untitled Letters on three occasions since 2008 requesting that we change advertising materials on the basis that they were inconsistent with the package insert for the product. We addressed these matters to the satisfaction of the FDA.
In particular, our manufacturing processes are governed by detailed and constantly evolving federal and sometimes state regulations that set forth cGMP for drugs and devices manufactured or distributed in the United States. We monitor compliance with these evolving procedures and regulations to help assure compliance, but failure to adhere to established procedures or regulations, or to meet a specification, could require that a product or material be rejected and destroyed, and could result in adverse regulatory actions against the companies. As a result of routine inspections by regulatory health authorities, we have been issued observations, for example, Form 483 FDA Inspection Observations, with regard to cGMP compliance. While these issues have been corrected, no assurances can be provided that we will avoid citation for deficiencies in the future. If serious deficiencies are noted or recur, compliance may be costly and difficult to achieve, and consequences may include the need to recall product or suspend operations until appropriate measures can be implemented. Also, certain deviations from procedures must be reported to the FDA, and even if we determine that the deviations were not material, the FDA could require us to take similar measures.
Later discovery of previously unknown problems with our products or failure by us or any third-party manufacturers, including Bayer, to comply with cGMP regulations, or failure to comply with regulatory requirements, may result in, among other things:
· restrictions on such products or manufacturing processes;
· withdrawal of products from the market;
· voluntary or mandatory recall;
· suspension or withdrawal of regulatory approvals and licenses;
· cessation of our manufacturing activities, which may be for an extended or indefinite period of time;
· product seizure; and
· injunctions or the imposition of civil or criminal penalties.
We could also be required to add warnings to our packaging or labeling that could negatively differentiate our product in the view of customers or patients.
For example, we settled a dispute with a customer in September 2007 regarding intermediate material manufactured by us, which is used by this customer in their manufacturing process. We recorded a charge to cost of goods sold of $7.9 million during the year ended December 31, 2007 for inventory impairment related to this material, which we recovered in its entirety during 2008 as the related material was determined to be saleable, converted into final product, and sold to other customers. Similarly, during 2008, we recorded an additional inventory impairment provision of $2.6 million related to this dispute for products held in Europe, for which we recovered $0.8 million and $1.8 million during the years ended December 31, 2009 and 2008, respectively, as the impacted material was determined to be saleable, converted into final product, and sold to other customers.
Separately, our plans to transition from Prolastin to our next generation therapy, Prolastin-C A1PI, in Europe have been delayed because we have yet to receive European regulatory approval. Presently additional clinical trials are being required by European regulators as a precursor to Prolastin-C A1PI approval. Additionally, we could face further delays with respect to launches in specific European countries. To the extent regulatory authorities do not act within the same time-frame, we will need to operate both new and old manufacturing processes in parallel with overlapping crews, higher costs, and lower yields.
Certain of our business practices are subject to scrutiny by regulatory authorities, as well as to lawsuits brought by private citizens under federal and state false claims laws. Failure to comply with applicable law or an adverse decision in lawsuits may result in adverse consequences to us.
The laws governing our conduct are enforceable by criminal, civil and administrative penalties. Violations of laws such as the Federal Food, Drug and Cosmetic Act, the False Claims Act and the Anti-Kickback Law, the Public Health Service Act, and any regulations promulgated under their authority, may result in jail sentences, fines, or exclusion from federal and state programs, as may be determined by Medicare, Medicaid and the Department of Defense and other regulatory authorities as well as by the courts. There can be no assurance that our activities will not come under the scrutiny of regulators and other government authorities or our practices will not be found to violate applicable laws, rules and regulations or prompt lawsuits by private citizen “relators” under federal or state false claims laws.
For example, under the Anti-Kickback Law, and similar state laws and regulations, even common business arrangements, such as discounted terms and volume incentives for customers in a position to recommend or choose drugs and devices for patients, such as physicians and hospitals, can result in substantial legal penalties, including, among others, exclusion from the Medicare and Medicaid programs, and arrangements with referral sources must be structured with care to comply with applicable requirements. Also, certain business practices, such as consulting fees to healthcare providers, sponsorship of educational or research grants, charitable donations, interactions with healthcare providers that prescribe products for uses not approved by the FDA, and financial support for continuing medical education programs, must be conducted within narrowly prescribed and controlled limits to avoid any possibility of wrongfully influencing healthcare providers to prescribe or purchase particular products or as a reward for past prescribing. Under the U.S. healthcare reform legislation, such payments by pharmaceutical manufacturers to United States healthcare practitioners and academic medical centers must be publicly disclosed starting with payments made in calendar year 2012. A number of states have similar laws in place. Additional and stricter prohibitions could be implemented by federal and state authorities. Where such practices have been found to be improper incentives to use such products, government investigations and assessments of penalties against manufacturers have resulted in substantial damages and fines. Many manufacturers have been required to enter into consent decrees or orders that prescribe allowable corporate conduct.
Failure to satisfy requirements under the Federal Food, Drug and Cosmetic Act can also result in penalties, as well as requirements to enter into consent decrees or orders that prescribe allowable corporate conduct.
Adverse consequences can also result from failure to comply with the requirements of the 340B/PHS program under the Public Health Service Act, which extends discounts to a variety of community health clinics and other entities that receive health services grants from the PHS. Although the recently passed health care reform legislation adds a “must sell” obligation to the terms of the program’s Pharmaceutical Pricing Agreement (PPA), under the current PPA no such obligation is stated, although some government regulators have suggested that a similar obligation exists. In November 2009, the United States Attorney’s Office for the Eastern District of Pennsylvania commenced an investigation with respect to our method of allocating our IGIV product, Gamunex, as available for sale at the PHS price to covered entities. We are cooperating with the investigation, and believe we have complied with the terms of the PPA and federal law, but an adverse outcome in this investigation could have a material adverse effect on us or our results of operation.
In addition, while regulatory authorities generally do not regulate physicians’ discretion in their choice of treatments for their patients, they do restrict communications by manufacturers on unapproved uses of approved drugs or on the potential safety and efficacy of unapproved products in development. Companies in the United States, Canada and European Union cannot promote approved products for other indications that are not specifically approved by the competent regulatory authorities (e.g., FDA in the United States), nor can companies promote unapproved products. In limited circumstances companies may disseminate to physicians information regarding unapproved uses of approved products or results of studies involving investigational products. If such activities fail to comply with applicable regulations and guidelines of the various regulatory authorities, we may be subject to warnings from, or enforcement action by, these authorities. Furthermore, if such activities are prohibited, it may harm demand for our products.
Promotion of unapproved drugs or devices or unapproved indications for a drug or device is a violation of the Federal Food, Drug and Cosmetic Act and subjects us to civil and criminal sanctions. Furthermore, sanctions under the Federal False Claims Act have recently been brought against companies accused of promoting off-label uses of drugs, because such promotion induces the use, and subsequent claims for reimbursement under Medicare and other federal programs. Similar actions for off-label promotion have been initiated by several states for Medicaid fraud. The U.S. healthcare reform legislation significantly strengthened provisions of the Federal False Claims Act, Medicare and Medicaid Anti-Kickback provisions, and other health care fraud provisions, leading to the possibility of greatly increased qui tam suits by relators for perceived violations. Violations or allegations of violations of the foregoing restrictions could materially and adversely impact our business.
To market and sell our products outside of the United States, we must obtain and maintain regulatory approvals and comply with regulatory requirements in such jurisdictions. The approval procedures vary among countries in complexity and timing. We may not obtain approvals from regulatory authorities outside the United States on a timely basis, if at all, which would preclude us from commercializing products in those markets.
In addition, some countries, particularly the countries of the European Union, regulate the pricing of prescription pharmaceuticals. In these countries, pricing discussions with governmental authorities can take considerable time after the receipt of marketing approval for a product. To obtain reimbursement or pricing approval in some countries, we may be required to conduct a clinical trial that compares the cost-effectiveness of their product candidate to other available therapies. Such trials may be time-consuming, expensive, and may not show an advantage in efficacy for our products. If reimbursement of our products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, in either the United States or the European Union, we could be adversely affected.
Our business involves the controlled use of hazardous materials, various biological compounds and chemicals. Although we believe that our safety procedures for handling and disposing of these materials comply with the standards prescribed by state and federal regulations, the risk of accidental contamination or injury from these materials cannot be eliminated. If an accident occurs, we could be held liable for resulting damages, which could be substantial. We are also subject to numerous environmental, health and workplace safety laws and regulations, including those governing laboratory procedures, exposure to blood-borne pathogens and the handling of biohazardous materials and chemicals. Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of these materials, this insurance may not provide adequate coverage against potential liabilities. We do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us. Additional federal, state, and local laws and regulations affecting our operations may be adopted in the future. We may incur substantial capital costs and operating expenses to comply with any of these laws or regulations and the terms and conditions of any permits required pursuant to such laws and regulations, including costs to install new or updated pollution control equipment, modify our operations or perform other corrective actions at our respective facilities. In addition, fines and penalties may be imposed for noncompliance with environmental and health and safety laws and regulations or for the failure to have or comply with the terms and conditions of required environmental permits.
On the international front, the Kyoto Protocol to the United Nations Framework Convention on Climate Change, which is commonly called the Kyoto Protocol, became effective in February 2005. Adopted by some of the countries in which we operate, the Kyoto Protocol requires the implementation of national programs to reduce emissions of certain gases, generally referred to as greenhouse gases, which contribute to global warming. Climate change-related legislation has also passed the U.S. House of Representatives, which, if enacted by the full Congress, would limit and reduce greenhouse gas emissions from large emitters of greenhouse gasses through a “cap-and-trade” system of allowances and credits and other provisions. Moreover, the Environmental Protection Agency, which is referred to as the EPA, issued a finding that the current and projected concentrations of certain greenhouse gases in the atmosphere, including carbon dioxide, which is referred to as CO2, threaten the public health and welfare of current and future generations. While this finding in itself does not impose any requirements on our industry, it authorizes the EPA to regulate directly greenhouse gas emissions through a rule-making process. Existing legislation and the future passage of climate control legislation or regulations that restricts emissions of greenhouse gases in the areas in which we operate could result in adverse financial and operational impacts on our business.
In addition, we export our products to a variety of countries whose legal regimes and business customs and practices differ significantly from those in the United States. A failure to comply with laws and regulations applicable to their international operations or export sales could expose them to significant penalties. These laws and regulations include data privacy requirements, labor relations laws, tax laws, competition regulations, anti-money laundering, import and trade restrictions, export requirements, including those of the U.S. Office of Foreign Assets Control, U.S. laws such as the Foreign Corrupt Practices Act, which is referred to as the FCPA, and local laws which also prohibit payments to corrupt governmental officials. While we require our employees to comply with applicable laws and we monitor legal compliance, we cannot be certain that our employees or agents will comply in all instances or that they will promptly identify violations. Violations of these laws and regulations could result in fines, criminal sanctions against us, our officers or our employees, and prohibitions on the conduct of our business. Any such violations could result in prohibitions on our ability to offer products in one or more countries, and could also materially damage our reputation, our products’ reputations, our international expansion efforts, our ability to attract and retain employees, our business and our operating results. We are currently conducting an internal investigation into violations of the FCPA of which we became aware during conduct of an unrelated review. As a result of the internal investigation, we suspended shipment to some countries while safeguards were implemented, and voluntarily contacted the U.S. Department of Justice. Any government sanctions or any continued loss of business from certain countries could have a material adverse effect on us or our operating results.
To enhance compliance with applicable health care laws, and mitigate potential liability in the event of noncompliance, regulatory authorities, such as the United States Health and Human Services Department Office of Inspector General (“OIG”), have recommended the adoption and implementation of a comprehensive health care compliance program that generally contains the elements of an effective compliance and ethics program described in Section 8B2.1 of the United States Sentencing Commission Guidelines Manual. Increasing numbers of United States-based pharmaceutical companies have such programs. While we have adopted U.S. healthcare compliance and ethics programs that generally incorporate the OIG’s recommendations, and train our U.S. employees in such compliance, having such a program can be no assurance that we will avoid any compliance issues.
We are required to provide accurate pricing information to the U.S. government for the purpose of calculating reimbursement levels by the Centers for Medicare and Medicaid Services (CMS) and for calculating certain federal prices and federal rebate obligations.
We are required to report detailed pricing information, net of included discounts, rebates and other concessions, to CMS for the purpose of calculating national reimbursement levels, certain federal prices, and certain federal rebate obligations. We have established a system for collecting and reporting this data accurately to CMS and have instituted a compliance program to assure that the information we collect is complete in all respects. If we report pricing information that is not accurate to the federal government, we could be subject to fines and other sanctions that could adversely affect our business. In addition, the government could change its calculation of reimbursement, federal prices, or federal rebate obligations which could negatively impact our financial results.
We seek to obtain and maintain protection for the intellectual property relating to our technology and products.
Our success depends in large part on our ability to obtain and maintain protection in the United States and other countries for the intellectual property covering or incorporated into our technology and products, especially intellectual property related to our purification processes. The patent situation in the field of biotechnology and pharmaceuticals generally is highly uncertain and involves complex legal and scientific questions. We may not be able to obtain additional issued patents relating to our technology or products. Even if issued, patents issued to us or our licensors may be challenged, narrowed, invalidated, held to be unenforceable or circumvented, which could limit our ability to stop competitors from marketing similar products or limit the length of term of patent protection we may have for our products. Additionally, most of our patents relate to the processes we use to produce our products, not the products themselves. In many cases, the plasma-derived products we produce or develop in the future will not, in and of themselves, be patentable. Since our patents relate to processes, if a competitor is able to design and utilize a process that does not rely on our protected intellectual property, that competitor could sell a plasma-derived product similar to one we developed or sell. Changes in either patent laws or in interpretations of patent laws in the United States and other countries may diminish the value of our intellectual property or narrow the scope of our patent protection. In addition, we are a party to a number of license agreements which may impose various obligations on us, including milestone and royalty payments. If we fail to comply with these obligations, the licensor may terminate the license, in which event we might not be able to market any product that is covered by the licensed patents.
Our patents also may not afford us protection against competitors with similar technology. Because patent applications in the United States and many other jurisdictions are typically not published until 18 months after filing, or in some cases not at all, and because publications of discoveries in the scientific literature often lag behind actual discoveries, neither we nor our licensors can be certain that we or they were the first to make the inventions claimed in our or their issued patents or pending patent applications, or that we or they were the first to file for protection of the inventions set forth in these patent applications. If a third party has also filed a U.S. patent application covering our product candidates or a similar invention, we may have to participate in an adversarial proceeding, known as an interference, declared by the U.S. Patent Office to determine priority of invention in the United States. The costs of these proceedings could be substantial and it is possible that our efforts could be unsuccessful, resulting in a loss of our anticipated U.S. patent position.
We also rely on unpatented technology, trade secrets, know-how and confidentiality agreements with our employees, consultants and third parties to protect our unpatented proprietary technology, processes and know-how. We require our officers, employees, consultants and advisors to execute proprietary information and invention and assignment agreements upon commencement of their relationships with us. There can be no assurance, however, that these agreements will provide meaningful protection for our inventions, trade secrets or other proprietary information in the event of unauthorized use or disclosure of such information. These agreements may be breached and we may not have adequate remedies for any such breach. In addition, our trade secrets may otherwise become known or be independently developed by competitors. If any trade secret, know-how or other technology not protected by a patent were to be disclosed to or independently developed by a competitor to develop alternative products, we could face increased competition and lose a competitive advantage.
We, like other companies in the pharmaceutical industry, may become aware of counterfeit versions of our products becoming available domestically and abroad. Counterfeit products may use different and possibly contaminated sources of plasma and other raw materials, and the purification process involved in the manufacture of counterfeit products may raise additional safety concerns, over which we have no control. Any reported adverse events involving counterfeit products that purport to be our products could harm our reputation and the sale of our products, in particular, and consumer willingness to use plasma-derived therapeutics generally.
We may infringe or be alleged to infringe intellectual property rights of third parties.
Our products or product candidates may infringe or be accused of infringing one or more claims of an issued patent or may fall within the scope of one or more claims in a published patent application that may be subsequently issued and to which we do not hold a license or other rights. Third parties may own or control these patents or patent applications in the United States and abroad. These third parties could bring claims against us or our collaborators that would cause us to incur substantial expenses and, if successful against us, could cause us to pay substantial damages. Further, if a patent infringement suit were brought against us or our collaborators, we or they could be forced to stop or delay research, development, manufacturing or sales of the product or product candidate that is the subject of the suit.
If we are found to infringe the patent rights of a third party, or in order to avoid potential claims, we or our collaborators may choose or be required to seek a license from a third party and be required to pay license fees or royalties or both. These licenses may not be available on acceptable terms, or at all. Even if we or our collaborators were able to obtain a license, the rights may be nonexclusive, which could result in our competitors gaining access to the same intellectual property. Ultimately, we could be prevented from commercializing a product, or be forced to cease some aspect of our business operations, if, as a result of actual or threatened patent infringement claims, we or our collaborators are unable to enter into licenses on acceptable terms.
There has been substantial litigation and other proceedings regarding patent and other intellectual property rights in the pharmaceutical and biotechnology industries. In addition to infringement claims against us, we may become a party to other patent litigation and other proceedings, including interference proceedings declared by the United States Patent and Trademark Office and opposition proceedings in the European Patent Office, regarding intellectual property rights with respect to our products. The cost to us of any patent litigation or other proceeding, even if resolved in our favor, could be substantial. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their substantially greater financial resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace. Patent litigation and other proceedings may also absorb significant management time.
Many of our employees were previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. We try to ensure that our employees do not use the proprietary information or know-how of others in their work for us. We may, however, be subject to claims that we or these employees have inadvertently or otherwise used or disclosed intellectual property, trade secrets or other proprietary information of any such employee’s former employer. Litigation may be necessary to defend against these claims and, even if we are successful in defending ourselves, could result in substantial costs to us or be distracting to our management. If we fail to defend any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel.
We may not be able to commercialize products in development.
Before obtaining regulatory approval for the sale of our product candidates or for marketing of existing products for new indicated uses, we must conduct, at our own expense, extensive preclinical tests to demonstrate the safety of our product candidates in animals and clinical trials to demonstrate the safety and efficacy of our product candidates in humans. Preclinical and clinical testing is expensive, difficult to design and implement, can take many years to complete and is uncertain as to outcome. A failure of one or more of our clinical trials can occur at any stage of testing. We may experience numerous unforeseen events during, or as a result of, preclinical testing and the clinical trial process that could delay or prevent our ability to receive regulatory approval or commercialize our product candidates, including:
· regulators or institutional review boards may not authorize us to commence a clinical trial or conduct a clinical trial within a country or at a prospective trial site respectively;
· the regulatory requirements for product approval may not be explicit, may evolve over time and may diverge by jurisdiction;
· our preclinical tests or clinical trials may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional preclinical testing or clinical trials or we may abandon projects that we had expected to be promising;
· the number of patients required for our clinical trials may be larger than we anticipate, enrollment in our clinical trials may be slower than we currently anticipate, or participants may drop out of our clinical trials at a higher rate than we anticipate, any of which would result in significant delays;
· our third-party contractors may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner;
· we might have to suspend or terminate our clinical trials if the participants are being exposed to unacceptable health risks or if any participant experiences an unexpected serious adverse event;
· regulators or institutional review boards may require that we hold, suspend or terminate clinical research for various reasons, including non-compliance with regulatory requirements;
· undetected or concealed fraudulent activity by a clinical researcher, if discovered, could preclude the submission of clinical data prepared by that researcher, lead to the suspension or substantive scientific review of one or more of our marketing applications by regulatory agencies, and result in the recall of any approved product distributed pursuant to data determined to be fraudulent;
· the cost of our clinical trials may be greater than we anticipate;
· the supply or quality of our product candidates or other materials necessary to conduct our clinical trials may be insufficient or inadequate because we do not currently have any agreements with third-party manufacturers for the long-term commercial supply of any of our product candidates;
· an audit of preclinical or clinical studies by the FDA or other regulatory authority may reveal non-compliance with applicable regulations, which could lead to disqualification of the results and the need to perform additional studies; and
· the effects of our product candidates may not be the desired effects or may include undesirable side effects or the product candidates may have other unexpected characteristics.
If we are required to conduct additional clinical trials or other testing of our product candidates beyond those that we currently contemplate, if we are unable to successfully complete our clinical trials or other testing, if the results of these trials or tests are not positive or are only modestly positive or if there are safety concerns, we may:
· be delayed in obtaining marketing approval for our product candidates;
· not be able to obtain marketing approval;
· not be able to obtain reimbursement for our products in some countries;
· obtain approval for indications that are not as broad as intended; or
· have the product removed from the market after obtaining marketing approval.
Our product development costs will also increase if we experience delays in testing or approvals. We do not know whether any preclinical tests or clinical trials will begin as planned, will need to be restructured or will be completed on schedule, if at all. Significant preclinical or clinical trial delays also could shorten the patent protection period during which we may have the exclusive right to commercialize our product candidates or allow our competitors to bring products to market before we do and impair our ability to commercialize our products or product candidates.
Even if clinical trials are successful, we may still be unable to commercialize the product due to difficulties in obtaining regulatory approval for the process or problems in scaling the engineering process to commercial production. Additionally, if produced, the product may not achieve an adequate level of market acceptance by physicians, patients, healthcare payors and others in the medical community to be profitable. The degree of market acceptance of our product candidates, if approved for commercial sale, will depend on a number of factors, some of which are beyond our control, including:
· the prevalence and severity of any side effects;
· the efficacy and potential advantages over alternative treatments;
· the ability to offer our product candidates for sale at competitive prices;
· relative convenience and ease of administration;
· the willingness of the target patient population to try new therapies and of physicians to prescribe these therapies;
· the strength of marketing and distribution support; and
· sufficient third-party coverage or reimbursement.
Therefore, we cannot guarantee that any products which we may seek to develop will ever be successfully commercialized, and to the extent they are not, such products could be a significant expense with no reward.
Our future success depends on our ability to retain members of our senior management and to attract, retain and motivate qualified personnel.
We are highly dependent on the principal members of our executive and scientific teams. The loss of the services of any of these persons might impede the achievement of our research, development, operational and commercialization objectives. In particular, we believe the loss of the services of Lawrence D. Stern, John M. Hanson, Mary J. Kuhn, Thomas J. Lynch, John R. Perkins, Joel E. Abelson, Stephen R. Petteway, John F. Gaither, Kari D. Heerdt, Daniel L. Menichella, James R. Engle and Bruce Nogales would significantly and negatively impact our business. Our risk of key employee turnover may increase due to the vesting of restricted shares in March 2010 and options in April 2010 and due to the announcement of our definitive merger agreement with Grifols.
Recruiting and retaining qualified operations, finance and accounting, scientific, clinical and sales and marketing personnel will be critical to our success. We may not be able to attract and retain these personnel on acceptable terms, given the competition among numerous pharmaceutical and biotechnology companies for similar personnel. We also experience competition for the hiring of scientific and clinical personnel from universities and research institutions. In addition, we rely on consultants and advisors, including scientific and clinical advisors, to assist us in formulating our research and development and commercialization strategy. Our consultants and advisors may be employed by employers other than us and may have commitments under consulting or advisory contracts with other entities that may limit their availability to us.
Federal cGMP regulations also require that the personnel we employ and hold responsible for the collection, processing, testing, storage or distribution of blood or blood components be adequate in number, educational background, training and experience, including professional training as necessary, or combination thereof, and have capabilities commensurate with their assigned functions, a thorough understanding of the procedures or control operations they perform, the necessary training or experience, and adequate information concerning the application of relevant cGMP requirements for their individual responsibilities. Our failure to attract, retain, and motivate qualified personnel may result in a regulatory violation, affect product quality, require recall or market withdrawal of affected product, or a suspension or termination of our license to market our products, or any combination thereof.
A substantial portion of our revenue is derived from a small number of customers, and the loss of one or more of these customers could have a material adverse effect on us.
FFF Enterprises Inc. and Amerisource Bergen collectively accounted for approximately 27% of our net revenue for both the years ended December 31, 2010 and 2009. Similarly, our accounts receivable balances have also been concentrated with a small number of customers. Amerisource Bergen accounted for approximately 13% of our accounts receivable, net, as of December 31, 2010 and FFF Enterprise, Inc. accounted for approximately 15% of our accounts receivable, net, as of December 31, 2009. In the event that any of these customers were to suffer an adverse downturn in their business or a downturn in their supply needs, our business could be materially adversely affected. We cannot guarantee that these customers will continue purchasing our products at past volumes, or, in the event that any of them were to cease doing business with us, that we could replace such customer on substantially similar terms or at all. Therefore, the loss of one or more of these customers could have a material adverse effect on our net sales, gross profit and financial condition. Under certain market conditions, our customers’ liquidity may worsen and they may demand longer payment terms, higher early payment discounts, volume rebates and other concessions which would have adverse financial consequences on us.
A significant amount of our U.S. Gamunex volume is contracted. As these contracts expire over the next few years, beginning in 2011, we may not be able to renew the commitments on as favorable terms, or at all.
Since the late 1980s we have been the “supplier of record” for the Canadian blood system. Under existing contracts, we are the largest supplier of plasma-derived products to the Canadian blood system operators, Canadian Blood Services and Hema Quebec. We transport plasma from Canadian Blood Services and Hema Quebec collection centers to our manufacturing facility in Clayton, North Carolina for manufacture, and return the finished product, along with commercial product, for sale to Canadian Blood Services and Hema Quebec. Pricing for our products and services is set at the beginning of the contract period, subject to adjustment for inflation. The U.S. dollar based contracts are terminable upon default, or the occurrence of certain events, including a third party obtaining Canadian regulatory approval to introduce a significantly superior product or fractionation service, our products or services becoming obsolete, or if we make certain nonrelated improvements and Canadian Blood Services or Hema Quebec do not accept the associated price increase. We were awarded new five year contracts in December 2007, which became effective April 1, 2008. The contracts may be extended for two one-year terms upon agreement of the parties. Under these contracts, we fractionate 100% of the Canadian plasma initially and a majority of the Canadian plasma throughout the contract period and supply a majority of the Canadian requirements for IGIV during the contract term as well. Canadian Blood Services has elected to pursue a multi-source strategy and although we will continue to be the primary supplier, we anticipate annual volume declines because of their strategy. Hema Quebec currently has a sole source strategy for fractionation of their plasma but could switch to a multi-source strategy. In 2010 we fractionated 71% of Canadian plasma and supplied 67% of Canadian requirements of IGIV. We derive significant revenue and profits under these contracts, and a failure to maintain contracts with the Canadian blood system operators or any diminution in the volume or price under future contracts could have a material adverse effect on our financial results.
Potential business combinations could require significant management attention and prove difficult to integrate with our business.
If we become aware of potential business combination candidates that are complementary to our business, we may decide to combine with such businesses or acquire their equity or assets. We have acquired businesses or product lines in the past. For example, in April 2005, we acquired Precision Pharma Services, Inc., a contract fractionator located in Melville, New York, and in November 2006 and June 2007 we acquired groups of plasma collection centers in varying stages of development and assumed certain liabilities from IBR, a supplier of source plasma. We have since acquired additional plasma collection centers on a case by case basis. Business combinations generally may involve a number of difficulties and risks to our business, including:
· failure to integrate management information systems, personnel, research and development, marketing, operations, sales and support;
· potential loss of key current employees or employees of the acquired company;
· disruption of our ongoing business and diversion of management’s attention from other business concerns;
· potential loss of the acquired company’s customers;
· failure to develop further the other company’s technology successfully;
· unanticipated costs and liabilities; and
· other accounting and operational consequences.
In addition, we may not realize the anticipated benefits from any business combination we may undertake in the future and any benefits we do realize may not justify the acquisition price. Any integration process would require significant time and resources, and we may not be able to manage the process successfully. If our customers are uncertain about our ability to operate on a combined basis, they could delay or cancel orders for our products. We may not successfully evaluate or utilize the acquired technology or accurately forecast the financial impact of a combination, including accounting charges or volatility in the stock price of the combined entity. If we fail to successfully integrate other companies with which we may combine in the future, our business and financial results could be harmed.
Talecris Holdings, LLC and its affiliated entities will continue to exercise significant control over us and could delay or prevent a change in corporate control.
As of December 31, 2010 Talecris Holdings, LLC owned approximately 48.7% of our outstanding common stock. Talecris Holdings, LLC is owned by (i) Cerberus-Plasma Holdings LLC, the managing member of which is Cerberus Partners, L.P., and (ii) limited partnerships affiliated with Ampersand Ventures. Substantially all rights of management and control of Talecris Holdings, LLC are held by Cerberus-Plasma Holdings LLC.
As long as Talecris Holdings, LLC owns or controls such a substantial portion of our outstanding voting power, it may have the ability to delay or prevent a change in control of us that may be favored by other stockholders and may otherwise exercise substantial control over all corporate actions requiring stockholder approval:
· the election and removal of directors and the size of our board;
· any amendment of our certificate of incorporation or bylaws;
· the approval of mergers and other significant corporate transactions, including a sale of substantially all of our assets; or
· the defeat of any non-negotiated takeover attempt that might otherwise benefit our other stockholders.
Our quarterly results of operations may fluctuate and this fluctuation may cause our stock price to decline, resulting in losses to our investors.
Our quarterly operating results are likely to fluctuate. A number of factors, many of which are not within our control, could cause variability in our operations and our operating results and may result in fluctuations in our stock price. These factors include the risks discussed elsewhere in this section, and may include:
· contamination of products or material that does not meet specifications in production or final product which could result in recalls, write-offs and other costs;
· changes in plasma procurement costs, yield, or other manufacturing costs that may increase our cost of goods sold for the period;
· non-capitalizable costs associated with our capital projects;
· seasonality of sales, particularly our hyperimmune products;
· competitor activities, including new product introductions;
· variations in product demand or price;
· regulatory developments in the United States and elsewhere;
· the departure of key personnel;
· interest rate fluctuations impacting our outstanding balances under our floating rate revolving credit facility and foreign currency exchange rate fluctuations in the international markets in which we operate; and
· general and industry-specific economic conditions that may affect our operations.
If our quarterly operating results fail to meet the expectations of stock market analysts and investors, the price of our common stock may rapidly decline, resulting in losses to our investors. Additional factors that could cause actual results to differ materially are included in “Special Note Regarding Forward Looking Statements” located elsewhere in this Annual Report.
We do not anticipate paying dividends in the foreseeable future.
We currently anticipate that we will retain all funds for use in the operation of our business, and we do not anticipate paying any cash dividends on our common stock for the foreseeable future. Therefore, any return on investment in our common stock is solely dependent upon the appreciation of the price of our common stock on the open market. We cannot guarantee that our common stock will appreciate in value. See the discussion contained elsewhere in this Annual Report under the heading “Dividend Policy.”
Despite our current indebtedness levels, we and our subsidiaries may be able to incur substantially more debt.
We and our subsidiaries may be able to incur substantially more additional indebtedness in the future, including by accessing approximately $322.6 million of unused available borrowing capacity under our existing revolving credit facility, based on our December 31, 2010 indebtedness. We are not fully restricted under the terms of the indenture governing the 7.75% Notes from incurring additional debt, securing existing or future debt, recapitalizing our debt or taking a number of other actions that are not prohibited by the terms of the indenture governing the 7.75% Notes, any of which actions could have the effect of diminishing our ability to make payments on the 7.75% Notes when due and further exacerbate the risks associated with our substantial indebtedness. Furthermore, the terms of the instruments governing our subsidiaries’ indebtedness may not fully prohibit us or our subsidiaries from taking such actions. Although the indenture for the 7.75% Notes and our revolving credit facility contain covenants limiting indebtedness, these covenants are subject to a number of significant exceptions and qualifications.
The indenture governing the 7.75% Notes and our revolving credit facility contain operating and financial restrictions on us that may limit our flexibility in operating our business.
Under the indenture governing the 7.75% Notes and under the revolving credit facility, we are required to satisfy a number of covenants that may restrict our ability to conduct our operations. For instance, these covenants limit or prohibit, among other things, our ability to incur additional debt, pay dividends on, redeem or repurchase capital stock, make certain investments, enter into certain types of transactions with affiliates, engage in unrelated businesses, incur certain liens; make prepayments of certain indebtedness and sell certain assets or merge with or into other’ companies. These covenants could adversely affect our operating results by significantly limiting our operating and financial flexibility.
Our ability to comply with these covenants may be affected by events beyond our control, and any breach could require us to seek waivers or amendments of covenants or alternative sources of financing, or to reduce expenditures. We cannot assure you that such waivers, amendments or alternative financing could be obtained or, if obtained, would be on terms favorable to us. In addition, under our revolving credit facility, as amended, we are required to satisfy a fixed charge coverage ratio of at least 1.10 to 1.00 if our borrowing availability based on eligible collateral is less than $48.75 million. If we were unable-to meet this fixed charge coverage ratio, the lenders could elect to terminate the facility and require us to repay outstanding borrowings. In such an event, unless we are able to refinance the indebtedness coming due and replace our revolving credit facility, we would likely not have sufficient liquidity for our business needs to service our debt or fund operations.
ITEM 1B. UNRESOLVED STAFF COMMENTS
Not applicable
Our primary facilities are described below. All of our owned real estate is pledged as security under our revolving credit facility.
· Clayton Site. A 175-acre site that we own, located in Clayton, North Carolina, which includes a 14-building complex of office space, lab space, warehouse, freezer storage, and biopharmaceutical manufacturing facilities consisting of 654,139 square feet. An additional 37,000 square feet of administrative office and 23,600 square feet of warehouse space are leased through December 2019 in a building located adjacent to our Clayton site. A 30,159 square foot climate controlled warehouse located next to the Clayton site is also leased through September 2014.
· Research Triangle Park. A leased three-building headquarters/administrative office facility consisting of 123,000 square feet. The main building housing our corporate headquarters and additional space in two other buildings are leased through May 2022. An expansion covering 45,359 square feet of office space is also leased through May 2022. We hold a five-year renewal option on the facility and a termination option exercisable effective 2018.
· Raleigh Test Lab. A laboratory space located in Raleigh, North Carolina consisting of 76,000 square feet leased through September 2017, with an option to purchase through September 2011.
· Melville Site. An 11-acre site that we own, located in Melville, New York consisting of 102,922 square feet of office space, lab space, warehouse, and biopharmaceutical manufacturing facilities.
· Research Triangle Park. An 18-acre site that we own, located in Research Triangle Park, North Carolina, on which is located a R&D building, consisting of 25,000 square feet of office space and 45,000 square feet of laboratory facilities.
· Benson Warehouse. A cold storage warehouse of 39,200 square feet used for plasma storage in Benson, North Carolina leased through December 2012.
· Centennial Campus North Carolina State University. A combined office and laboratory space in Raleigh, North Carolina consisting of 21,364 square feet leased through December 2011.
· Frankfurt. A 2,552 square meters office facility located in Frankfurt Germany, which serves as our European headquarters and which is leased until June 30, 2015.
· Canada. A 6,396 square foot office facility located in Mississauga, Ontario, which serves as our Canadian headquarters and which is leased until March 2012. We also lease a 2,356 square foot sales office in Ottawa, Ontario, which is leased until December 2012.
· Plasma Collection Centers. As of December 31, 2010, we operated 69 plasma collection centers of various sizes under non-cancellable lease agreements expiring at various dates.
We believe our properties are adequately maintained and suitable for their intended use. We continually evaluate our properties and believe that our current facilities plus any planned expansions and upgrades are generally sufficient to meet our expected needs and expected near-term growth. Expansion projects and facility closings are undertaken as necessary in response to market needs.
We are involved in a number of judicial, regulatory and arbitration proceedings (including those described below) concerning matters arising in connection with the conduct of our businesses. We believe, based on currently available information, that the results of such proceedings, in the aggregate, will not have a material adverse effect on our financial condition, but might be material to our operating results for any particular period, depending, in part, upon the operating results for such period.
National Genetics Institute/Baxter Healthcare Corporation Litigation
In May 2008, Baxter Healthcare Corporation (Baxter) and National Genetics Institute (NGI), a wholly-owned subsidiary of Laboratory Corporation of America, filed a complaint in the U.S. District Court for the Eastern District of North Carolina alleging that we infringed U.S. Patents Nos. 5,780,222, 6,063,563, and 6,566,052. The patents deal primarily with a method of screening large numbers of biological samples utilizing various pooling and matrix array strategies, and the complaint alleges that the patents are owned by Baxter and exclusively licensed to NGI. In November 2008, we filed our answer to their complaint, asserting anti-trust and other counterclaims, and filed a request for re-examination of the patents with the Patent and Trademark Office (PTO), which was subsequently granted. The case was settled effective October 1, 2010, whereby we paid $3.9 million to NGI and received a paid-up license to the technology subject to the disputed patents and the parties dismissed their claims and counterclaims.
Plasma Centers of America, LLC and G&M Crandall Limited Family Partnership Litigation
We had a three year Amended and Restated Plasma Sale/Purchase Agreement with Plasma Centers of America, LLC (PCA) under which we were required to purchase annual minimum quantities of plasma from plasma collection centers approved by us, including the prepayment of 90% for unlicensed plasma. We were also committed to finance the development of up to eight plasma collection centers, which were to be used to source plasma for us. Under the terms of the agreement, we had a conditional obligation to purchase such centers under certain conditions for a sum determined by a formula set forth in the agreement. We provided approximately $4.2 million in financing related to the development of such centers and advanced payments for unlicensed plasma. We recorded a provision within SG&A during 2008 related to loans and advances provided.
In August 2008, we notified PCA that they were in breach of the Amended and Restated Plasma Sale/Purchase Agreement. We terminated the agreement in September 2008. In November 2008, we filed suit in federal court in Raleigh, North Carolina, against the G&M Crandall Limited Family Partnership and its individual partners as guarantors of obligations of PCA. We were served in January 2009 in a parallel action by PCA, alleging breach of contract by TPR. The federal case has been stayed. On December 13, 2010, a jury in the state court case rendered a verdict in the amount of $37 million in favor of PCA against TPR in a breach of contract claim, which was confirmed by the court in post trial motions. We intend to appeal. Interest on the verdict, if sustained, will accrue at 8% simple interest from the date of breach, which is approximately $6.7 million at December 31, 2010.
Foreign Corrupt Practices Act Investigation
In July 2009, we voluntarily contacted the U.S. Department of Justice (DOJ) to advise them that we were conducting an internal investigation into potential violations of the Foreign Corrupt Practices Act (FCPA). The FCPA investigation is being conducted by outside counsel under the direction of a special committee of our Board of Directors. The investigation into possible improper payments to individuals and entities made after our formation initially focused on payments made in connection with sales in certain Eastern European and Middle Eastern countries, primarily Belarus, Russia, and Iran, but we are also reviewing sales practices in Brazil, Bulgaria, China, Georgia, Libya, Poland, Turkey, Ukraine, and other countries as deemed appropriate. The DOJ has not indicated what action it may take, if any, against us or any individual, or the extent to which it may conduct its own investigation. Even though we self-disclosed this matter to the DOJ, it or other federal agencies may seek to impose sanctions on us that may include, among other things, debarment, injunctive relief, disgorgement, fines, penalties, appointment of a monitor, appointment of new control staff, or enhancement of existing compliance and training programs. Other countries in which we do business may initiate their own investigations and impose similar penalties. We expect to complete our internal FCPA investigation and present our findings to the DOJ in 2011.
Compliance with Pharmaceutical Pricing Agreement
In November 2009, we received a letter from the United States Attorney’s Office for the Eastern District of Pennsylvania (USAO). The USAO requested a meeting to review the company’s compliance with the terms of the Pharmaceutical Pricing Agreement (PPA) under the Public Health Service program. Specifically, the USAO asked for information related to the sale of our IGIV product, Gamunex, under that program. In order to have federal financial participation apply to their products under the Medicaid program and to obtain Medicare Part B coverage, manufacturers are required to enter into a PPA. The PPA obligates manufacturers to charge covered entities the Public Health Service price for drugs intended for outpatient use. The Public Health Service price is based on the Medicaid rebate amount. We believe that we have complied with the terms of the PPA and federal law. If the USAO determines that our practices are inconsistent with the terms of the PPA, the USAO has stated that it may file a civil action against us under the Anti-fraud Injunction Act and seek a court order directing us to comply with the PPA or, potentially, proceed under some other legal theory. We could also be subject to fines, damages, penalties, appointment of a monitor, or enhancement of existing compliance and training programs as a result of government action. We are cooperating with the investigation and intend to respond to information requests from the USAO.
Talecris – Grifols Merger
Four purported class action lawsuits have been filed by our stockholders challenging the proposed merger transaction with Grifols. Two of the lawsuits were filed in the Court of Chancery of the State of Delaware and have been consolidated under the caption In re Talecris Biotherapeutics Holdings Shareholder Litigation, Consol. C.A. No. 5614-VCL. The other two lawsuits were filed in the Superior Court of the State of North Carolina and are captioned Rubin v. Charpie, et al., No. 10 CV 004507 (North Carolina Superior Court, Durham County), and Kovary v. Talecris Biotherapeutics Holdings Corp., et al., No. 10 CV 011638 (North Carolina Superior Court, Wake County). The lawsuits name as defendants Talecris, the members of the Talecris Board of Directors, Grifols, S.A. and its subsidiary, Grifols, Inc., and, in the Delaware consolidated action, Talecris Holdings and Stream Merger Sub, Inc. The two North Carolina actions have been stayed.
All of the lawsuits allege that the individual defendants (and, in the consolidated Delaware action, Talecris Holdings) breached their fiduciary duties to the Talecris stockholders in connection with the proposed transaction with Grifols, and that Grifols (and, in one of the North Carolina cases, Talecris, and in the Delaware action, Grifols, Inc.) aided and abetted those breaches. The Delaware complaint alleges, among other things, that the consideration offered to Talecris stockholders pursuant to the proposed transaction is inadequate; that the Talecris Board of Directors failed to take steps to maximize stockholder value; that Talecris’ IPO and debt refinancing in 2009 were intended to facilitate a sale of Talecris; that Cerberus and Talecris Holdings arranged the proposed merger for the benefit of Cerberus, without regard to the interests of other stockholders; that the voting agreements impermissibly lock up the transaction; and that the merger agreement contains terms, including a termination fee, that favor Grifols and deter alternative bids. The Delaware complaint further alleges that the preliminary Form F-4 filed on August 10, 2010 contains material misstatements and/or omissions, including with respect to the availability of appraisal rights in the merger; the purpose and effects of the Virginia reincorporation merger; the antitrust risks of the proposed transaction; the financial advisors’ analyses regarding the Grifols’ non-voting stock to be issued in connection with the transaction; and the fees to be paid to Morgan Stanley by Talecris and Grifols in connection with the proposed transaction. The Delaware complaint also alleges that Talecris stockholders are entitled to appraisal rights in connection with the transaction pursuant to Section 262 of the Delaware General Corporation Law, and that the transaction violates the Delaware General Corporation Law by failing to provide such rights. The Delaware action seeks equitable and injunctive relief, including a determination that the stockholders have appraisal rights in connection with the merger, and damages.
On October 29, 2010, the parties to the Delaware litigation entered into a MOU reflecting an agreement in principle to settle that litigation. The MOU provides, among other things, for the provision of appraisal rights in accordance with DGCL 262 in connection with the transaction as described at pages 135-137 of the MOU; for an increase in the merger consideration by an additional 500,000 shares of Grifols non-voting stock to holders of Talecris common stock other than the Talecris specified affiliated stockholders as described at pages 144-145 of the MOU; and for certain additional disclosures provided herein. The MOU also provides for a dismissal of the action with prejudice and a release of claims. On January 21, 2011, the parties executed a formal Stipulation of Settlement documenting the agreement set forth in the MOU, and on January 25, 2011, the Delaware court entered an order preliminarily approving the settlement. The settlement remains subject, among other things, to notice to the class, final court approval and consummation of the transaction.
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our common stock, par value $0.01, has been listed on The NASDAQ Global Select Market under the symbol “TLCR” since October 1, 2009. Prior to that time, there was no public market for our common stock. The initial public offering price of our common stock on October 1, 2009 was $19.00 per share. The following table sets forth the range of the high and low market prices of our common stock for the periods indicated as reported by The NASDAQ Global Select Market:
|
|
|
2009 |
|
2010 |
| |||||||||||
|
|
|
4th Quarter |
|
1st Quarter |
|
2nd Quarter |
|
3rd Quarter |
|
4th Quarter |
| |||||
|
High |
|
$ |
23.44 |
|
$ |
24.41 |
|
$ |
23.09 |
|
$ |
23.30 |
|
$ |
24.63 |
|
|
Low |
|
$ |
18.01 |
|
$ |
19.77 |
|
$ |
15.70 |
|
$ |
20.95 |
|
$ |
21.30 |
|
Stockholders
There were 43 registered accounts of record of our common stock as of the close of business on February 21, 2011. The number of holders of record is based upon the actual number of holders registered at such date and does not include holders of shares in “street names” or persons, partnerships, associates, corporations, or other entities identified in security position listings maintained by depositories.
Dividends
We did not declare or pay cash dividends on our common stock in either 2010 or 2009. We currently do not anticipate paying cash dividends on our common stock in the foreseeable future. Instead, we anticipate that all of our earnings, if any, in the foreseeable future will be used for working capital and to finance the growth and development of our business. Any future determination relating to dividend policy will be made at the discretion of our board of directors and will depend on a number of factors, including our outstanding indebtedness, earnings, capital requirements, financial condition and future prospects, applicable Delaware law, which provides that dividends are only payable out of surplus or net profit for the then current and immediately preceding fiscal years, and other factors that our board of directors may deem relevant.
Our revolving credit facility, as amended, permits the payment of cash dividends to holders of our common stock commencing with the first fiscal quarter of 2010, so long as (i) the Leverage Ratio determined as of the end of the immediately preceding fiscal quarter for the then most recently completed four fiscal quarters, is equal to or less than 2.00 to 1.00 and (ii) the minimum pro forma availability as of the date of such dividend (after giving effect to such cash dividend, the funding of all revolving loans, and the issuance of all letters of credit to be funded or issued as of such date) is not less than $48.75 million; provided that, the aggregate amount of restricted payments shall not exceed 50% of net income during the period from October 1, 2009 to the end of the most recently ended fiscal quarter as of the date of the restricted payment.
The indenture governing our 7.75% Notes permits us to make “Restricted Payments”, including the payment of dividends to holders of our common stock, only if (A) (i) there is no default or event of default under the indenture (and no default or event of default would occur as a result of the payment of dividends), (ii) we would be able to incur an additional dollar of indebtedness under the Fixed Charge Coverage Ratio test in the indenture, and (iii) the amount of such dividends along with all other Restricted Payments would not exceed our then existing Restricted Payment basket, or (B) we have availability under another specified basket to make a payment of dividends.
Recent Sales of Unregistered Securities
None.
Issuer Purchases of Equity Securities
During the fourth fiscal quarter ended December 31, 2010, we did not repurchase any of our outstanding common stock.
Performance Graph
We have presented below the cumulative total return to our stockholders during the period from October 1, 2009, the date of our initial public offering of common stock, through December 31, 2010 in comparison to the cumulative return on the NASDAQ Composite Index and a customized peer group of seventeen companies during that same period. Our peer group consisted of seventeen companies which are: Abbott Laboratories, Alcon Inc, Allergan Inc, Amgen Inc, Baxter International Inc, Bristol Myers Squibb Company, Covidien PLC, Edwards Lifesciences Corp., ELI Lilly & Company, Glaxosmithkline PLC, Hospira Inc, King Pharmaceuticals Inc, Medtronic Inc, Merck & Company Inc, Pfizer Inc, Sanofi-Aventis and Takeda Pharmaceutical Company. The results assume that $100 (with reinvestment of all dividends) was invested in our common stock, in the peer group, and in the index on October 1, 2009 and its relative performance tracked through December 31, 2010. The comparisons are based on historical data and are not indicative of, nor intended to forecast, the future performance of our common stock. The performance graph set forth below shall not be deemed incorporated by reference into any filing by us under the Securities Act of 1933 or the Securities Exchange Act of 1934 except to the extent that we specifically incorporate such information by reference, and shall not otherwise be deemed filed under such Acts.
COMPARISON OF CUMULATIVE TOTAL RETURN
Among Talecris Biotherapeutics Holdings Corp, The NASDAQ Composite Index
And a Peer Group
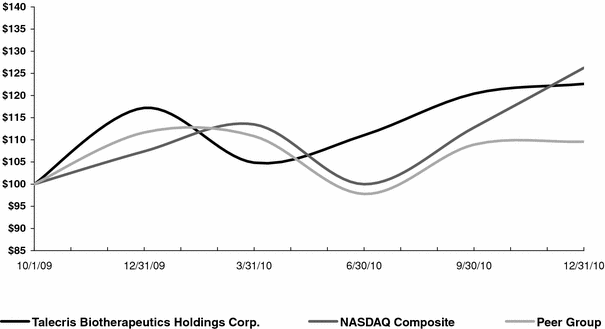
ITEM 6. SELECTED FINANCIAL DATA
The following is a summary of our historical consolidated financial data for the periods ended and at the dates indicated below. You are encouraged to read this information together with our audited consolidated financial statements and the related footnotes and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included elsewhere in this Annual Report.
The historical consolidated financial data for the years ended December 31, 2010, 2009, and 2008 and as of December 31, 2010 and 2009 has been derived from our audited consolidated financial statements, which are included elsewhere in this Annual Report. The historical consolidated financial data for the years ended December 31, 2007 and 2006 and as of December 31, 2008, 2007, and 2006 has been derived from our audited consolidated financial statements, which are not included in this Annual Report.
We believe that the comparability of our financial results between the periods presented in the table below is significantly impacted by the following items, many of which are more fully described in “Management’s Discussion and Analysis of Financial Condition and Results of Operations— Matters Affecting Comparability.”
· The financial impact related to our 2009 initial public offering (IPO) and refinancing transactions, including the repayment and termination of our First and Second Lien Term Loans; the issuance of our 7.75% Senior Notes, due November 15, 2016 (7.75% Notes), the write-off of previously deferred debt issuance costs, and charges related to the settlement and termination of our interest rate swap contracts;
· The increase in the number of shares of our common stock outstanding as a result of the issuance of new shares of our common stock in our IPO, to convert our Series A and B preferred stock, and to settle accrued dividends upon the conversion of our Series A and B preferred stock;
· Costs associated with our definitive merger agreement with Grifols;
· Costs associated with the judgment related to litigation with Plasma Centers of America, LLC (PCA);
· Costs and non-operating income associated with our terminated merger agreement with CSL Limited (CSL);
· Costs associated with our internal investigation into potential violations of the Foreign Corrupt Practices Act (FCPA);
· Costs associated with the development and vertical integration of our plasma collection center platform;
· Inventory impairment provisions, and subsequent recoveries, related to a plasma collection center cGMP issue;
· Inventory impairment provisions, and subsequent recoveries, related to a customer dispute settlement regarding intermediate material;
· Costs associated with share-based compensation awards and special recognition bonuses;
· Costs associated with transition-related activities to establish an independent company apart from Bayer;
· Non-operating income and costs related to a litigation settlement with Baxter; and
· Tax benefit due to the release of our deferred tax asset valuation allowance.
|
|
|
Years Ended December 31, |
| |||||||||||||
|
|
|
2010 |
|
2009 |
|
2008 |
|
2007 |
|
2006 |
| |||||
|
|
|
(in thousands, except share and per share amounts) |
| |||||||||||||
|
Income Statement Data: |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Net revenue: |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Product |
|
$ |
1,576,936 |
|
$ |
1,507,754 |
|
$ |
1,334,550 |
|
$ |
1,196,686 |
|
$ |
1,114,489 |
|
|
Other |
|
24,683 |
|
25,455 |
|
39,742 |
|
21,823 |
|
14,230 |
| |||||
|
Total |
|
1,601,619 |
|
1,533,209 |
|
1,374,292 |
|
1,218,509 |
|
1,128,719 |
| |||||
|
Cost of goods sold |
|
911,976 |
|
901,077 |
|
882,157 |
|
788,152 |
|
684,750 |
| |||||
|
Gross profit |
|
689,643 |
|
632,132 |
|
492,135 |
|
430,357 |
|
443,969 |
| |||||
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
| |||||
|
SG&A |
|
287,011 |
|
289,929 |
|
227,524 |
|
189,387 |
|
241,448 |
| |||||
|
R&D |
|
69,649 |
|
71,223 |
|
66,006 |
|
61,336 |
|
66,801 |
| |||||
|
Total |
|
356,660 |
|
361,152 |
|
293,530 |
|
250,723 |
|
308,249 |
| |||||
|
Income from operations |
|
332,983 |
|
270,980 |
|
198,605 |
|
179,634 |
|
135,720 |
| |||||
|
Other non-operating (expense) income: |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Interest expense, net |
|
(45,837 |
) |
(74,491 |
) |
(96,640 |
) |
(110,236 |
) |
(40,867 |
) | |||||
|
CSL merger termination fee |
|
— |
|
75,000 |
|
— |
|
— |
|
— |
| |||||
|
PCA judgment |
|
(43,690 |
) |
— |
|
— |
|
— |
|
— |
| |||||
|
Equity in earnings of affiliate |
|
991 |
|
441 |
|
426 |
|
436 |
|
684 |
| |||||
|
Loss on extinguishment of debt |
|
— |
|
(43,033 |
) |
— |
|
— |
|
(8,924 |
) | |||||
|
Litigation settlement |
|
— |
|
— |
|
— |
|
12,937 |
|
— |
| |||||
|
Income before income taxes and extraordinary items |
|
244,447 |
|
228,897 |
|
102,391 |
|
82,771 |
|
86,613 |
| |||||
|
(Provision) benefit for income taxes |
|
(78,379 |
) |
(75,008 |
) |
(36,594 |
) |
40,794 |
|
(2,222 |
) | |||||
|
Income before extraordinary items |
|
166,068 |
|
153,889 |
|
65,797 |
|
123,565 |
|
84,391 |
| |||||
|
Extraordinary items: |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Loss from unallocated negative goodwill |
|
— |
|
— |
|
— |
|
— |
|
(306 |
) | |||||
|
Gain from settlement of contingent consideration due Bayer |
|
— |
|
— |
|
— |
|
— |
|
3,300 |
| |||||
|
Net income |
|
$ |
166,068 |
|
$ |
153,889 |
|
$ |
65,797 |
|
$ |
123,565 |
|
$ |
87,385 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
|
Income (loss) before extraordinary items per common share: |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Basic |
|
$ |
1.35 |
|
$ |
4.56 |
|
$ |
39.01 |
|
$ |
65.58 |
|
$ |
(119.83 |
) |
|
Diluted |
|
$ |
1.29 |
|
$ |
1.50 |
|
$ |
0.71 |
|
$ |
1.36 |
|
$ |
(119.83 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
|
Cash dividends declared per common share: |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Basic |
|
— |
|
— |
|
— |
|
— |
|
$ |
132.82 |
| ||||
|
Diluted |
|
— |
|
— |
|
— |
|
— |
|
$ |
8.61 |
| ||||
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
|
Weighted average common shares outstanding: |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Basic |
|
123,323,722 |
|
31,166,613 |
|
1,310,448 |
|
1,685,784 |
|
5,679,456 |
| |||||
|
Diluted |
|
128,927,053 |
|
102,514,363 |
|
92,761,800 |
|
91,065,600 |
|
5,679,456 |
| |||||
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
|
Balance Sheet Data (at year end): |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Cash and cash equivalents |
|
$ |
197,876 |
|
$ |
65,239 |
|
$ |
16,979 |
|
$ |
73,467 |
|
$ |
11,042 |
|
|
Total assets |
|
$ |
1,738,453 |
|
$ |
1,445,005 |
|
$ |
1,307,399 |
|
$ |
1,142,322 |
|
$ |
903,474 |
|
|
Long-term debt and capital lease obligations |
|
$ |
605,301 |
|
$ |
605,267 |
|
$ |
1,194,205 |
|
$ |
1,129,692 |
|
$ |
1,102,920 |
|
|
Redeemable preferred stock |
|
— |
|
— |
|
$ |
110,535 |
|
$ |
110,535 |
|
$ |
110,535 |
| ||
|
Total stockholders’ equity (deficit) |
|
$ |
794,364 |
|
$ |
582,154 |
|
$ |
(316,725 |
) |
$ |
(390,757 |
) |
$ |
(528,980 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
|
Other Financial Data and Ratios (unaudited): |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Liters of plasma fractionated |
|
3,803 |
|
3,569 |
|
3,240 |
|
2,650 |
|
2,983 |
| |||||
|
Gross margin |
|
43.1 |
% |
41.2 |
% |
35.8 |
% |
35.3 |
% |
39.3 |
% | |||||
|
Operating margin |
|
20.8 |
% |
17.7 |
% |
14.5 |
% |
14.7 |
% |
12.0 |
% | |||||
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You are encouraged to read the following discussion and analysis of our financial condition and results of operations together with our audited consolidated financial statements and related footnotes included at the end of this Annual Report. This discussion and analysis contains forward-looking statements that involve risks and uncertainties. See “Risk Factors” included elsewhere in this Annual Report for a discussion of some of the important factors that could cause actual results to differ materially from those described or implied by the forward-looking statements contained in the following discussion and analysis. See “Special Note Regarding Forward-Looking Statements” included elsewhere in this Annual Report.
All tabular disclosures are presented in thousands, except share and per share amounts. Percentages and amounts presented herein may not calculate or sum precisely due to rounding.
A seven-for-one share dividend on our common stock was paid on September 10, 2009. All share and per share amounts have been retroactively adjusted for all periods to reflect the share dividend.
BUSINESS OVERVIEW
We are a biopharmaceutical company that researches, develops, manufactures, markets, and sells protein-based therapies that extend and enhance the lives of individuals who suffer from chronic and acute, often life-threatening, conditions, such as primary immune deficiencies, chronic inflammatory demyelinating polyneuropathy (CIDP), alpha-1 antitrypsin deficiency-related emphysema, bleeding disorders, infectious diseases, and severe trauma. Our primary products have orphan drug designation to serve populations with rare, chronic diseases. Our products are derived from human plasma, the liquid component of blood, which is sourced from our plasma collection centers or purchased from third parties with plasma collection centers located in the United States. Plasma contains many therapeutic proteins, which we extract through the process of fractionation at our Clayton, North Carolina and Melville, New York facilities. The fractionated intermediates are then purified, formulated into final bulk, and aseptically filled into final containers for sale. We also sell the fractionated intermediate products.
The majority of our sales are concentrated in the therapeutic areas of Immunology/Neurology and Pulmonology. Our largest product, representing 54.4%, 53.9%, and 49.3% of our net revenue for the years ended December 31, 2010, 2009, and 2008, respectively, Gamunex, Immune Globulin Intravenous (Human), 10% Caprylate/Chromatography Purified (Gamunex, Gamunex IGIV), provides a treatment for primary immunodeficiency (PI), idiopathic thrombocytopenic purpura (ITP), and autoimmune diseases, such as CIDP. In May 2010 and October 2010, Gamunex-C was approved for the subcutaneous route of administration for the PI indication in Canada and the U.S., respectively. Our second largest product, representing 22.0%, 20.8%, and 23.0% of our net revenue for the years ended December 31, 2010, 2009, and 2008, respectively, Prolastin Alpha-1 Proteinase Inhibitor (Human) (Prolastin, Prolastin A1PI, Prolastin-C A1PI), provides a treatment for alpha-1 antitrypsin deficiency-related emphysema. We completed the conversion of our existing U.S. and Canadian Prolastin A1PI patients to Prolastin-C A1PI in 2010.
We believe U.S. IGIV distribution increased between 6% and 8% during the year ended December 31, 2010. Despite solid demand growth for IGIV, there has been increased scrutiny and price sensitivity in the hospital segment. In addition, the increase in the number of hospitals qualifying for the 340B discounts has effectively reduced demand from GPO’s who are not permitted to service this discounted channel. This, among other factors, has led us to accept reduced volume tiers under certain of our GPO contracts. We have seen solid demand growth for Gamunex-C/Gamunex IGIV with most customer segments. We believe that U.S. and international IGIV demand will grow approximately 5% to 8% over the long-term, which is consistent with demand growth during the year ended December 31, 2010. However, IGIV demand can vary significantly on a quarter-to-quarter basis.
Our ability to expand our international business has been hampered by the effects of our internal Foreign Corrupt Practices Act (FCPA) investigation, our reliance on the tender process for generating business and increased price sensitivities of our customers. We expect to complete our internal FCPA investigation and present our findings to the Department of Justice (DOJ) in 2011. The preliminary findings of our investigation indicate that it is probable that there were FCPA violations by persons associated with us that the DOJ or other regulators may assert are attributable to us. We are unable to estimate the potential impact of any sanctions, that may be imposed. See “Management’s Discussion and Analysis of Financial Condition and Results of Operations— Matters Affecting Comparability—Foreign Corrupt Practices Act (FCPA)” for further discussion. Our business with an Iranian distributor, one of our major customers, has been in decline, which is likely to continue. Our profitability has and may continue to be negatively impacted by unfavorable euro/U.S. dollar exchange rates. We have experienced, and expect to continue to experience, annual volume declines in Canada due to Canadian Blood Services’ (CBS) objective to have multiple sources of supply, which has impacted and will continue to impact our overall IGIV growth. CBS may further reduce volumes to contract minimums and Hema Quebec may adopt a similar strategy.
We expect to operate at or near our fractionation capacity over the next few years depending upon the demand for our products, the availability of source plasma, the impact of yield variability, the potential impact of inventory impairments, and normal production shut-downs, among other factors. We plan to utilize most of our available fractionation capacity in the near term, which may result in increased inventory levels in order to attempt to maintain pace with projected future growth in product demand, although we have not been successful in building excess finished goods inventories to date as a result of the factors previously mentioned. Consequently, any disruption in meeting our fractionation and purification plans would most likely result in lower revenue, gross profit, net income, and operating cash flows as well as lower than planned growth given our fractionation and purification constraints. Our fractionation constraints would likely preclude us from participating in greater than estimated overall market demand or higher demand for Gamunex-C/Gamunex IGIV. In response to our capacity constraints, we have embarked on a substantial capital plan which we anticipate to be in the range of $750 million to $800 million on a cumulative basis from 2011 through 2015, excluding capitalized interest. Given the nature of our planned capital projects, we anticipate our capital spending to peak in a range of $250 million to $270 million in 2011, excluding capitalized interest. Our most significant capital project is the construction of our new fractionation facility, which we estimate will cost approximately $340 million, excluding capitalized interest. Through December 31, 2010, our capital spending on this project was approximately $90 million with estimated additional capital spending of $250 million to be incurred, excluding capitalized interest. Estimated costs related to the construction of our new purification facilities for Plasmin is $120 million with additional expenditures planned for Koate modernization and albumin purification expansion. The successful execution of this capital plan, which is discussed further in the section titled, “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Liquidity and Capital Resources—Access to Capital and Capital Resources,” will be necessary to support our projected future volume growth, particularly given our current fractionation and purification constraints, launch new products, and complete strategic initiatives.
As of December 31, 2010, we operated 69 plasma collection centers (67 FDA licensed, two unlicensed) with approximately 2,700 employees. Over the past four years, we have aggressively expanded our plasma supply through these collection centers under our wholly-owned subsidiary, Talecris Plasma Resources, Inc. (TPR). These centers collectively represent substantially all of our currently planned collection center network for the next three years. We expect this network, once it fully matures, will provide in excess of 90% of our current plasma requirements. Our licensed centers collected approximately 69% of our plasma during the year ended December 31, 2010. We intend to continue to purchase some plasma from third parties through plasma supply contracts. We have a five-year plasma supply contract with CSL Plasma, Inc., a subsidiary of CSL Limited, a major competitor. This agreement provides us with minimum annual purchase commitments that decline from 550,000 liters in 2010 to 200,000 liters in 2013, the final year of the agreement. We have the ability to obtain additional volumes above the minimum purchase commitments under the terms of the agreement. CSL Plasma, Inc. is obligated to supply 300,000 liters of plasma to us in 2011 as we have not elected to take optional volumes for the 2011 contract year. In addition to the contract with CSL Plasma, Inc., we have several other contracts to purchase minimum quantities of plasma with various third parties.
We will need to significantly increase plasma collections generated from TPR in the near term to offset the expected decrease in plasma supplied by third parties and planned increases in our fractionation to meet anticipated demand. To meet our plasma requirements, we have increased donor fees, increased marketing expenses, and expanded plasma collection center days and hours of operations, among other initiatives, which may limit our ability to reduce our cost per liter of plasma. Consequently, we expect to continue to produce plasma at a cost per liter which we believe is significantly higher than our competitors. However, TPR is in the process of upgrading its plasmapheresis machines to speed plasma collections as well as the installation of automated digital screening to improve compliance. These measures, in addition to reductions in infrastructure support as the platform matures, will improve costs as well as the donor experience.
Our historical results show a substantial reduction in both the collection cost per liter and the amount of excess period costs charged directly to cost of goods sold as a result of the maturation of our plasma collection center platform. Decreasing collection costs and the reduction of excess period costs, combined with leveraging our manufacturing facilities as a result of higher volumes, have contributed to improving our gross margins. Our cost of goods sold reflects $6.6 million, $44.0 million, and $98.5 million for the years ended December 31, 2010, 2009, and 2008, respectively, related to excess period costs associated with TPR. We believe that we have substantially eliminated unabsorbed TPR infrastructure and start-up costs. Consequently, future margin improvements will need to be derived from increases in product pricing and volumes, product mix, improvements in the cost per liter of plasma, manufacturing efficiencies, yield improvements or some combination thereof. We believe that we have limited opportunities to increase price as well as enhance product mix. We have recently experienced and expect to continue to experience higher costs of goods sold due to yield variability, inventory impairment provisions, less efficient utilization of each incremental liter of plasma fractionated as we increase Gamunex-C/Gamunex IGIV production, and higher non-capitalizable costs associated with our capital projects, particularly the construction of our new fractionation facility.
The combination of the factors mentioned above, particularly competitive pressures, slower than planned reductions in our cost per liter of plasma, yield variability as well as inefficient plasma utilization and the potential impact of inventory impairment provisions, among other factors, will most likely result in lower gross margins in future periods.
Our U.S. sales force is comprised of three specialty teams focused on Immunology/Neurology for the promotion of Gamunex-C for use in PI, ITP, and CIDP, as well as our portfolio of hyperimmune products and Plasbumin; Pulmonary for the promotion of Prolastin/Prolastin-C A1PI with an emphasis on patient identification; and Hematology which promotes Koate and Thrombate III. In addition to this direct sales force, we also have managed markets and national account sales teams that manage relationships and contracting efforts with GPO’s, distributors, home healthcare and specialty pharmacy providers and private commercial payors. In addition to our U.S. operations, we have operations located in Germany, Canada, as well as a team dedicated to the development of other international markets. We believe that we are well positioned in the IGIV market given the features and benefits of Gamunex-C/Gamunex IGIV. As a result of eliminating our plasma supply constraints, the attributes of Gamunex-C/Gamunex IGIV and its approval for CIDP have resulted in significant increases in our share of sales. Our fractionation constraints, however, will limit the supply of Gamunex-C/Gamunex IGIV and our ability to grow Gamunex-C/Gamunex IGIV volumes. In addition our current purification constraints related to albumin and Koate, our plasma-derived Factor VIII product, will continue.
HIGHLIGHTS
Our 2010 financial and business highlights are included below.
2010 Financial Highlights
The following summarizes our 2010 financial highlights. Additional information regarding our results of operations is included in the section entitled, “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Results of Operations.”
· Total net revenue increased 4.5% for the year ended December 31, 2010 to $1.602 billion as compared to $1.533 billion for the year ended December 31, 2009. We experienced year over year growth in our U.S. Gamunex-C and Prolastin-C A1PI net revenue of 10.0% and 12.4%, respectively, driven by both higher volumes and pricing. Pull-through sales growth by our distributors exceeded our ex-factory sales growth. In addition, we experienced a $6.8 million increase in international Prolastin/Prolastin-C A1PI net revenue, driven by higher volumes and pricing in Europe, including the impact of unfavorable foreign exchange. Despite strong European growth in Gamunex net revenue, international Gamunex-C/Gamunex IGIV net revenue declined 6.6% as a result of lower Canadian sales as a result of CBS’ multi-source strategy, as well as lower sales in other international regions.
· Gross margin improved approximately 200 basis points to 43.1% for the year ended December 31, 2010 as compared to 41.2% for the year ended December 31, 2009. Gross margin benefited primarily from a $37.4 million reduction in TPR unabsorbed infrastructure and start-up costs as a result of the maturation of our plasma collection center platform.
· Operating margin improved approximately 310 basis points to 20.8% for the year ended December 31, 2010 as compared to 17.7% for the year ended December 31, 2009.
· Net income was $166.1 million for the year ended December 31, 2010, as compared to $153.9 million for the year ended December 31, 2009. Diluted earnings per common share were $1.29 and $1.50 for the years ended December 31, 2010 and 2009, respectively. Our 2010 results include $27.7 million (approximately $17.3 million after tax) in transaction-related costs associated with our definitive merger agreement with Grifols as well as a charge, including accrued interest, of $43.7 million (approximately $26.6 million after tax) associated with the judgment in favor of Plasma Centers of America, LLC (PCA). Our 2009 results include the impact of the CSL merger termination income of $75.0 million (approximately $48.8 million after tax), transaction-related costs related to the terminated CSL merger agreement of $15.1 million (approximately $9.3 million after tax), and charges related to our refinancing transactions of $43.0 million (approximately $26.3 million after tax). We believe that a meaningful comparison of our results for the years presented is enhanced by a quantified presentation of the impact of theses items on our net income and diluted earnings per share, which is illustrated in the table below.
· Operating cash flows were $255.5 million and $234.2 million for the years ended December 31, 2010 and 2009, respectively. Capital expenditures were $152.8 million and $75.2 million for the years ended December 31, 2010 and 2009, respectively.
In addition, our 2010 diluted earnings per common share amounts reflect a significant increase in the number of weighted average common shares used in our computation of diluted earnings per share as discussed below and in the section entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations— Matters Affecting Comparability—Comparability of Outstanding Common Shares and Pro Forma Diluted Earnings Per Common Share.”
The adjusted net income and diluted earnings per share amounts in the table below are non-GAAP financial measures and should not be considered a substitute for any performance measure determined in accordance with U.S. GAAP. Additional information regarding the use of non-GAAP financial measures and their limitations are included in the section entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Non-GAAP Financial Measures.”
|
|
|
|
|
Income Tax |
|
|
|
Diluted Earnings |
| ||||
|
|
|
Pre- Tax |
|
Expense |
|
|
|
Per |
| ||||
|
|
|
Amount |
|
(Benefit) |
|
Net Income |
|
Common Share |
| ||||
|
Year Ended December 31, 2010 |
|
|
|
|
|
|
|
|
| ||||
|
U.S. GAAP |
|
$ |
244,447 |
|
$ |
(78,379 |
) |
$ |
166,068 |
|
$ |
1.29 |
|
|
Grifols merger-related expenses |
|
27,730 |
|
(10,454 |
) |
17,276 |
|
0.13 |
| ||||
|
PCA judgment |
|
43,690 |
|
(17,083 |
) |
26,607 |
|
0.21 |
| ||||
|
Excluding specific items |
|
$ |
315,867 |
|
$ |
(105,916 |
) |
$ |
209,951 |
|
$ |
1.63 |
|
|
|
|
|
|
|
|
|
|
|
| ||||
|
Year Ended December 31, 2009 |
|
|
|
|
|
|
|
|
| ||||
|
U.S. GAAP |
|
$ |
228,897 |
|
$ |
(75,008 |
) |
$ |
153,889 |
|
$ |
1.50 |
|
|
CSL merger termination fee |
|
(75,000 |
) |
26,250 |
|
(48,750 |
) |
(0.48 |
) | ||||
|
CSL merger-related expenses |
|
15,136 |
|
(5,873 |
) |
9,263 |
|
0.08 |
| ||||
|
Write-off of deferred debt issuance costs |
|
12,141 |
|
(4,711 |
) |
7,430 |
|
0.07 |
| ||||
|
Loss on extinguishment of interest rate swap contracts |
|
30,892 |
|
(11,986 |
) |
18,906 |
|
0.19 |
| ||||
|
Excluding specific items |
|
$ |
212,066 |
|
$ |
(71,328 |
) |
$ |
140,738 |
|
$ |
1.36 |
|
|
|
|
|
|
|
|
|
|
|
| ||||
|
As adjusted for pro forma weighted average number of shares (1) |
|
|
|
|
|
|
|
$ |
1.11 |
| |||
(1) As discussed further in the section entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Matters Affecting Comparability—Comparability of Outstanding Common Shares and Pro Forma Diluted Earnings Per Common Share,” we believe the comparability of our diluted earnings per share between the years presented is enhanced by the use of an adjusted share base to reflect the impact for the issuance of common shares to convert our Series A and B preferred stock, settle accrued dividends on the preferred stock, and complete our IPO as if these events occurred at the beginning of 2009.
Business Highlights
· During the fourth quarter of 2010, we initiated our Phase II clinical trial for our direct-acting thrombolytic Plasmin to treat acute Peripheral Arterial Occlusion.
· On December 13, 2010, a jury in the General Court of Justice, Superior Court Division, Wake County, North Carolina, rendered a verdict in the amount of $37.0 million in favor of PCA against TPR in a breach of contract claim, which was confirmed by the court in post trial motions. We intend to appeal. The jury verdict, if sustained, will bear simple interest at 8% per statute from the date of the breach, which totals approximately $6.7 million at December 31, 2010. We have included a charge of $43.7 million (approximately $26.6 million after tax) in our consolidated income statement for the year ended December 31, 2010 related to this judgment.
· During the fourth quarter of 2010, we initiated a clinical trial evaluating the safety and pharmacokinetic profile of two doses of Prolastin-C A1PI. The study will investigate the safety and pharmacokinetic profile of a higher dose, 120 mg/kg weekly, of Prolastin-C A1PI, versus the licensed dose of 60 mg/kg weekly.
· On October 13, 2010, we received approval from the FDA for Gamunex-C (Immune Globulin Injection [Human], 10% Caprylate/Chromatography Purified) for subcutaneous administration in the treatment of primary immunodeficiency (PI). Gamunex-C is the first and only immune globulin to provide both the intravenous route of administration and a new subcutaneous route of administration. The intravenous delivery mode is approved to treat PI, CIDP, and ITP. The subcutaneous mode is approved to treat only PI. A required post-marketing study will be initiated in the second half of 2011.
· In September 2010 and March 2010, we launched our next generation A1PI product, Prolastin-C, in Canada and the United States, respectively. We have completed the conversion of existing Canadian and U.S. Prolastin patients to Prolastin-C A1PI.
· On June 6, 2010, we entered into a definitive merger agreement with Grifols under which Grifols will acquire, through merger transactions, all of our common stock. On November 4, 2010, pursuant to a memorandum of understanding entered into in connection with the litigation described under “Legal Proceedings” included elsewhere in this Annual Report, the parties to the merger agreement entered into an amendment to the merger agreement, pursuant to which Grifols agreed to pay a per Talecris merger consideration of a combination of (1) $19.00 in cash and (2) subject to adjustment under limited circumstances, 0.6485 (or 0.641 for Talecris’ directors and Talecris Holdings, LLC) of a share of Grifols non-voting share.
· On May 13, 2010, we received approval from Health Canada to launch Gamunex for subcutaneous administration in Canada for the PI indication. We launched subcutaneous administration for Gamunex in Canada in the fourth quarter of 2010.
· During the first quarter of 2010, we received approval to proceed with the proof of concept trial for plasma-derived Plasmin to treat acute ischemic stroke in certain countries outside of the United States.
· In March 2010, we began constructing our new fractionation facility located in Clayton, North Carolina. The new fractionation facility, which is expected to be operational in 2015, will have the capacity to fractionate 6.0 million liters of human plasma annually.
· In February 2010, we were granted orphan drug designation by the U.S. FDA for the development of an aerosol formulation of A1PI to treat congenital alpha-1 antitrypsin (AAT) deficiency. AAT deficiency is a chronic, hereditary condition that increases the risk of certain diseases, particularly emphysema. Currently, there are no approved, inhaled treatments available for the treatment of AAT. We received a similar orphan drug designation for the aerosolized form of A1PI from the European Commission in June of 2008. We have decided not to initiate an aerosol trial with plasma-derived Prolastin-C A1PI.
SUBSEQUENT EVENTS
Special Meeting of Stockholders
On February 14, 2011, we held a special meeting at which holders of a majority of our outstanding common stock approved the adoption of the Agreement and Plan of Merger, dated as of June 6, 2010, among Grifols and Talecris Biotherapeutics Inc. The completion of the transaction is subject to obtaining certain regulatory approvals and other customary conditions. Grifols agreed to provide written notice to the FTC staff at least thirty days prior to closing the transaction and, in any event, not to close the transaction until after 11:59 p.m. on March 20, 2011. There can be no assurance that Grifols will reach resolution with the FTC by March 20, 2011. Under the pending merger agreement, if this transaction is not closed by the current “outside date” of March 6, 2011, then under specified circumstances, either Grifols or we may elect to cause the “outside date” to be extended to a date not later than the expiration of Grifols financing for the transaction, or September 6, 2011, whichever is earlier.
Foreign Currency Hedging Program
In order to reduce the impact of the volatility of foreign currency exchange rates and improve predictability, we initiated a foreign currency hedging program in the first quarter of 2011 as discussed further in the section titled “Quantitative and Qualitative Disclosures About Market Risk—Foreign Currency Risk,” included elsewhere in this Annual Report.
HEALTHCARE REFORM
In March 2010, healthcare reform legislation was enacted in the United States. This legislation contains several provisions that impact our business. Certain of these provisions are included below. Additional information regarding U.S. healthcare reform is included in the section titled, “Business—Pharmaceutical Pricing and Reimbursement”, located elsewhere in this Annual Report.
Although many provisions of the new legislation do not take effect immediately, several provisions became effective during 2010. These include (1) an increase in the minimum Medicaid rebate to states participating in the Medicaid program from 15.1% to 23.1% of the Average Manufacturer Price (AMP) on our branded prescription drugs, with a limitation of this increase on clotting factors to 17.1% of the AMP; (2) the extension of the Medicaid rebate to managed care organizations that dispense drugs to Medicaid beneficiaries; and (3) the expansion of the 340B Public Health Services (PHS) drug pricing program, which provides hospital outpatient drugs at reduced rates, to include additional hospitals, clinics, and healthcare centers. These new provisions did not have a material impact on our 2010 financial results.
Beginning in 2011, the new law requires that drug manufacturers provide a 50% discount to Medicare beneficiaries whose prescription drug costs cause them to be subject to the Medicare Part D coverage gap (commonly referred to as the donut hole). Also, beginning in 2011, we will be assessed our share of a new fee assessed on all branded prescription drug manufacturers and importers. This fee will be calculated based upon each organization’s percentage share of total branded prescription drug sales to U.S. government programs (such as Medicare, Medicaid and VA and PHS discount programs) made during the previous year. The aggregated industry wide fee is expected to range from $2.5 billion to $4.1 billion annually between 2011 and 2018 and remain at $2.8 billion in 2019 and subsequent years.
Beginning in 2012, the new law may require us to issue Internal Revenue Service Forms 1099 to plasma donors whose remuneration exceeds six hundred dollars annually. The cost of implementing this requirement, as well as its potential impact on plasma donations, is unknown at this time.
Presently, uncertainty exists as many of the specific determinations will be developed as regulatory bodies interpret the law and enact new regulations. For example, determination as to how the Medicare Part D coverage gap will operate and how the annual fee on branded prescriptions will be calculated and allocated remains to be clarified. As noted above, these programs will become effective in 2011.
BASIS OF PRESENTATION
Our consolidated financial statements include the accounts of Talecris Biotherapeutics Holdings Corp. and its wholly-owned subsidiaries. All significant intercompany transactions and balances have been eliminated upon consolidation. The effects of business acquisitions have been included in our consolidated financial statements from their respective date of acquisition.
The comparability of our financial results is impacted by significant events and transactions during the years presented as described in “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Matters Affecting Comparability.”
CRITICAL ACCOUNTING POLICIES AND ESTIMATES
The preparation of consolidated financial statements in accordance with accounting principles generally accepted in the United States of America (U.S. GAAP) requires us to make estimates and judgments in certain circumstances that affect the reported amounts of assets, liabilities, revenue and expenses, and the related disclosures of contingent assets and liabilities. A detailed description of our significant accounting policies is included in the footnotes to our audited consolidated financial statements included elsewhere in this Annual Report.
We believe that certain of our accounting policies are critical because they are the most important to the preparation of our consolidated financial statements. These policies require our most subjective and complex judgments, often requiring the use of estimates about the effects of matters that are inherently uncertain. We apply estimation methodologies consistently from year to year. Other than changes required due to the issuance of new accounting guidance, there have been no significant changes in our application of our critical accounting policies during 2010. We periodically review our critical accounting policies and estimates with the audit committee of our board of directors. The following is a summary of accounting policies that we consider critical to our consolidated financial statements.
Revenue Recognition and Gross-to-Net Revenue Adjustments
We recognize revenue when earned, which is generally at the time of delivery to the customer. Recognition of revenue also requires reasonable assurance of collection of sales proceeds, a fixed and determinable price, persuasive evidence that an arrangement exists, and completion of all other performance obligations. The recognition of revenue is deferred if there are significant post-delivery obligations, such as customer acceptance.
Allowances against revenues for estimated discounts, rebates, administrative fees, chargebacks, shelf-stock adjustments, and other items are established by us concurrently with the recognition of revenue. The standard terms and conditions under which products are shipped to our customers generally do not allow a right of return. In the rare instances in which we grant a right of return, revenue is reduced at the time of sale to reflect expected returns and deferred until all conditions of revenue recognition are met.
We have supply agreements with our major distributors, which require them to purchase minimum quantities of our products. We regularly review the supply levels of our products on hand at major distributors, primarily by analyzing inventory reports supplied by these distributors, available data regarding the sell-through of our products, our internal data, and other available information. When we believe distributor inventory levels have increased relative to underlying demand, we evaluate the need for sales return allowances. Factors that influence the allowance include historical sales return activity, levels of inventory in the distribution network, inventory turnover, demand history, demand projections, estimated product shelf-life, pricing, and competition. Sales returns have not been material during the years presented.
Revenue from milestone payments for which we have no continuing performance obligations is recognized upon achievement of the related milestone. When we have continuing performance obligations, the milestone payments are deferred and recognized as revenue over the term of the arrangement as we complete our performance obligations.
Gross product sales are subject to a variety of deductions that are generally estimated and recorded in the same period that the revenues are recognized, and primarily represent rebates to government agencies, chargebacks to wholesalers and distributors, and customer prompt pay discounts. These gross-to-net revenue adjustments are described below.
We offer rebates to some classes of trade, which we account for by establishing an accrual at the time the sale is recorded in an amount equal to our estimate of rebates attributable to each sale. We determine our estimate of the rebates primarily based on historical experience and current contract arrangements. We consider the sales performance of products subject to rebates and the levels of inventory in the distribution channel and adjust the accrual periodically to reflect actual experience. Rebates accrued upon sale are settled based on actual experience. Due to the limited classes of trade that participate in rebate programs and our visibility of inventories in the channel, adjustments for actual experience have not been material.
We participate in state government-managed Medicaid programs. We account for Medicaid rebates by establishing an accrual at the time the sale is recorded in an amount equal to our estimate of the Medicaid rebate claims attributable to such sale. We determine our estimate of the Medicaid rebates accrual primarily based on historical experience regarding Medicaid rebates, legal interpretations of the applicable laws related to the Medicaid program and any new information regarding changes in the Medicaid programs’ regulations and guidelines that would impact the amount of the rebates. We consider outstanding Medicaid claims, Medicaid payments, and levels of inventory in the distribution channel and adjust the accrual periodically to reflect actual experience. While these rebate payments to the states generally occur on a one to two quarter lag, any adjustments for actual experience have not been material.
As of December 31, 2010, our allowance for managed health care and Medicaid rebates and other items was $23.8 million. A hypothetical 10% change in payments made for managed health care and Medicaid rebates for the year ended December 31, 2010 would not have a material impact to our consolidated results of operations.
We enter into agreements with some customers to establish contract pricing for our products, which these entities purchase from the authorized wholesaler or distributor (collectively, wholesalers) of their choice. Consequently, when our products are purchased from wholesalers by these entities at the contract price which is less than the price charged by us to the wholesaler, we provide the wholesaler with a credit referred to as a chargeback. We record the chargeback accrual at the time of the sale. The allowance for chargebacks is based on our estimate of the wholesaler inventory levels, and the expected sell-through of our products by the wholesalers at the contract price based on historical chargeback experience and other factors. We periodically monitor the factors that influence our provision for chargebacks, and make adjustments when we believe that actual chargebacks may differ from established allowances. These adjustments occur in a relatively short period of time. As these chargebacks are typically settled within 30 to 45 days of the sale, adjustments for actual experience have not been material.
As of December 31, 2010, our allowance for chargebacks was $3.4 million. A hypothetical 10% change in credits issued for chargebacks for year ended December 31, 2010 would not have a material impact to our consolidated results of operations.
Sales allowances are established based upon consideration of a variety of factors, including, but not limited to, our sales terms, which generally provide for up to a 2% prompt pay discount on domestic and international sales, contractual agreements with customers, estimates of the amount of product in the pipeline and prescribing patterns. We believe that our sales allowance accruals are reasonably determinable and are based on the information available at the time to arrive at our best estimate of the accruals at the time of the sale. Actual sales allowances incurred are dependent upon future events. We periodically monitor the factors that influence sales allowances and make adjustments to these provisions when we believe that the actual sales allowances may differ from prior estimates. If conditions in future periods change, revisions to previous estimates may be required, potentially in significant amounts. As these prompt pay discounts are typically settled within 30 to 45 days of the sale, adjustments for actual experience have not been material.
As of December 31, 2010, our allowance for cash discounts was $1.2 million. A hypothetical 10% change in credits issued for cash discounts for the year ended December 31, 2010 would not have a material impact to our consolidated results of operations.
Shelf-stock adjustments are credits issued to customers to reflect decreases in the selling prices of products. Agreements to provide this form of price protection are customary in our industry and are intended to reduce a customer’s inventory cost to better reflect current market prices. Shelf-stock adjustments are based upon the amount of product that customers have remaining in their inventories at the time of a price reduction. Decreases in our selling prices are discretionary decisions made by us to reflect market conditions. Any amounts recorded for estimated price adjustments would be based upon the specific terms with customers, estimated declines in price, and estimates of inventory held by the customer. We have not experienced material shelf-stock adjustments during the years presented as a result of
the demand for plasma-derived products outpacing the supply due to our constraints in our industry. Recently, product supply and demand have become more balanced. We could experience material shelf-stock adjustments in the future in the event that the supply-demand dynamic become unbalanced and resulted in price declines.
We utilize information from external sources to estimate our significant gross-to-net revenue adjustments. Our estimates of inventory at wholesalers and distributors are based on written and oral information obtained from certain wholesalers and distributors with respect to their inventory levels and sell-through to customers. The inventory information received from wholesalers and distributors is a product of their record-keeping process. Our estimates are subject to inherent limitations of estimates that rely on third-party information, as certain third-party information was itself in the form of estimates, and reflect other limitations, including lags between the date as of which the third-party information is generated and the date on which we receive third-party information. We believe, based on our experience, that the information obtained from external sources provides a reasonable basis for our estimate.
The following table summarizes our gross-to-net revenue adjustments expressed in dollars and percentages:
|
|
|
Years Ended December 31, |
| |||||||
|
|
|
2010 |
|
2009 |
|
2008 |
| |||
|
Gross product revenue |
|
$ |
1,660,643 |
|
$ |
1,593,995 |
|
$ |
1,389,542 |
|
|
Chargebacks |
|
(28,013 |
) |
(24,380 |
) |
(13,927 |
) | |||
|
Cash discounts |
|
(20,186 |
) |
(18,710 |
) |
(15,147 |
) | |||
|
Rebates and other |
|
(35,130 |
) |
(42,397 |
) |
(24,008 |
) | |||
|
SG&A reimbursements |
|
(378 |
) |
(754 |
) |
(1,910 |
) | |||
|
Product net revenue |
|
$ |
1,576,936 |
|
$ |
1,507,754 |
|
$ |
1,334,550 |
|
|
|
|
Years Ended December 31, |
| ||||
|
|
|
2010 |
|
2009 |
|
2008 |
|
|
Gross product revenue |
|
100.0 |
% |
100.0 |
% |
100.0 |
% |
|
Chargebacks |
|
(1.7 |
)% |
(1.5 |
)% |
(1.0 |
)% |
|
Cash discounts |
|
(1.2 |
)% |
(1.2 |
)% |
(1.1 |
)% |
|
Rebates and other |
|
(2.1 |
)% |
(2.7 |
)% |
(1.7 |
)% |
|
SG&A reimbursements |
|
— |
|
— |
|
(0.1 |
)% |
|
Product net revenue |
|
95.0 |
% |
94.6 |
% |
96.1 |
% |
The following table provides a summary of activity with respect to our allowances:
|
|
|
|
|
Cash |
|
Rebates and |
|
|
| ||||
|
|
|
Chargebacks |
|
Discounts |
|
Other |
|
Total |
| ||||
|
Balance at December 31, 2007 |
|
$ |
2,688 |
|
$ |
1,074 |
|
$ |
11,432 |
|
$ |
15,194 |
|
|
Provision |
|
13,927 |
|
15,147 |
|
24,008 |
|
53,082 |
| ||||
|
Credits issued |
|
(12,752 |
) |
(14,727 |
) |
(23,029 |
) |
(50,508 |
) | ||||
|
Balance at December 31, 2008 |
|
3,863 |
|
1,494 |
|
12,411 |
|
17,768 |
| ||||
|
Provision |
|
24,380 |
|
18,710 |
|
42,397 |
|
85,487 |
| ||||
|
Credits issued |
|
(23,981 |
) |
(18,930 |
) |
(28,381 |
) |
(71,292 |
) | ||||
|
Balance at December 31, 2009 |
|
4,262 |
|
1,274 |
|
26,427 |
|
31,963 |
| ||||
|
Provision |
|
28,013 |
|
20,186 |
|
35,508 |
|
83,707 |
| ||||
|
Credits issued |
|
(28,850 |
) |
(20,280 |
) |
(38,118 |
) |
(87,248 |
) | ||||
|
Balance at December 31, 2010 |
|
$ |
3,425 |
|
$ |
1,180 |
|
$ |
23,817 |
|
$ |
28,422 |
|
As discussed elsewhere in this Annual Report, the recently enacted healthcare reform legislation increased the size of Medicaid rebates paid by drug manufacturers from 15.1% to 23.1% of the AMP, with a limitation of this increase on clotting factors to 17.1% of the AMP.
The increase in the provision for chargebacks during 2010 as compared to prior years was largely due to increased sales related to government contracts. Rebates and other adjustments were impacted by the reduction of channel inventories during 2010. The increase in the provision for rebates and other for the year ended December 31, 2009 as compared to the year ended December 31, 2008 was primarily due to higher GPO fees, Medicaid rebates, and the potential Canadian pricing adjustment, among others. The provision for chargebacks for the year ended December 31, 2009 as compared to the year ended December 31, 2008 was largely due to increased sales related to government contracts.
Concentrations of Credit Risk
Customer Concentrations
Our accounts receivable, net, includes amounts due from pharmaceutical wholesalers and distributors, buying groups, hospitals, physicians’ offices, patients, and others. Our concentrations with customers that represented more than 10% of our accounts receivable, net, were:
· At December 31, 2010: Amerisource Bergen- 12.8%
· At December 31, 2009: FFF Enterprise, Inc.- 14.6%
The following table summarizes our concentrations with customers that represented more than 10% of our total net revenue:
|
|
|
Years Ended December 31, |
| ||||
|
|
|
2010 |
|
2009 |
|
2008 |
|
|
FFF Enterprise, Inc. |
|
13.9 |
% |
14.4 |
% |
12.8 |
% |
|
Amerisource Bergen |
|
13.1 |
% |
12.3 |
% |
12.0 |
% |
|
Canadian Blood Services |
|
<10 |
% |
<10 |
% |
10.6 |
% |
In the event that any of these customers were to suffer an adverse downturn in their business or a downturn in their supply needs, our business could be materially adversely affected. Additional information regarding customer concentrations is included in the section titled “Risk Factors—Risks Related to our Business— A substantial portion of our revenue is derived from a small number of customers, and the loss of one or more customers could have a material adverse effect on us.”
Counterparty Risk
As discussed further in “Quantitative and Qualitative Disclosures About Market Risk—Foreign Currency Risk,” we initiated a foreign currency hedging program in the first quarter of 2011 for the purpose of managing the economic effects of the volatility associated with short-term changes in euro/U.S. dollar exchange rates on our earnings and cash flows. These derivative financial instruments present certain market and counterparty risks. We seek to manage the counterparty risks associated with these contracts by limiting transactions to counterparties with which we have established banking relationships and limit the duration of the contracts to less than one year. We are exposed to potential losses if a counterparty fails to perform according to the terms of the agreement. We do not require collateral or other security to be furnished by counterparties to our derivative financial instruments. There can be no assurance, however, that our practice effectively mitigates counterparty risk. A number of financial institutions similar to those that serve or may serve as counterparties to our hedging arrangements were adversely affected by the global credit crisis. The failure of any of the counterparties to our hedging arrangements to fulfill their obligations to us could adversely affect our results of operations and cash flows.
Research and Development
Our R&D expenses include the costs directly attributable to the conduct of research and development programs for new products and extensions or improvements of existing products and the related manufacturing processes. Such costs include salaries and related employee benefit costs, payroll taxes, materials (including the material required for clinical trials), supplies, depreciation on and maintenance of R&D equipment, services provided by outside contractors for clinical development and clinical trials, regulatory services, and fees. R&D also includes the allocable portion of facility costs such as rent, depreciation, utilities, insurance, and general support services. All costs associated with R&D activities are expensed as incurred.
We continue to invest in R&D to provide future sources of revenue through the development of new products, as well as through additional uses for existing products. We have a strong commitment to science and technology with a track record of accomplishments and pipeline opportunities. We focus our R&D efforts in three key areas: continued enhancement of our process technologies (including pathogen safety), life cycle management for our existing products (including new indications), and the development of new products. We expect overall R&D spending to increase in subsequent periods due to life-cycle management, new product projects, and licensure of technology or products.
The following table summarizes our significant R&D projects and expenses for the years presented:
|
|
|
Years Ended December 31, |
| |||||||
|
|
|
2010 |
|
2009 |
|
2008 |
| |||
|
Life Cycle Management: |
|
|
|
|
|
|
| |||
|
Gamunex CIDP |
|
$ |
50 |
|
$ |
200 |
|
$ |
600 |
|
|
Prolastin-C A1PI |
|
$ |
3,200 |
|
$ |
2,200 |
|
$ |
3,900 |
|
|
Prolastin Alpha-1 Aerosol |
|
$ |
1,900 |
|
$ |
8,900 |
|
$ |
6,100 |
|
|
Gamunex subcutaneous administration |
|
$ |
400 |
|
$ |
1,400 |
|
$ |
3,300 |
|
|
|
|
|
|
|
|
|
| |||
|
New Product Candidates: |
|
|
|
|
|
|
| |||
|
Plasmin and recombinant Plasmin |
|
$ |
26,500 |
|
$ |
25,500 |
|
$ |
18,500 |
|
|
Other recombinant products |
|
$ |
10,500 |
|
$ |
4,700 |
|
$ |
4,000 |
|
Additional information regarding the status of our life cycle management activities and new product candidates is included in “Business—Research and Development.”
Income Taxes
We calculate a provision for, or benefit from, income taxes using the asset and liability method, under which deferred tax assets and liabilities are recorded based on the difference between the financial statement and tax bases of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to reverse. A reduction in the carrying amounts of deferred tax assets by a valuation allowance is required, if, based on the available evidence, it is more likely than not that the assets will not be realized. Accordingly, we periodically assess the need to establish valuation allowances for deferred tax assets based on the more-likely-than-not realization threshold criterion. In assessing the need for a valuation allowance, we consider all available evidence, both positive and negative, including historical levels of income, expectations and risks associated with future taxable income, and ongoing prudent and feasible tax planning strategies.
We establish reserves for uncertain income tax positions, based on the technical support for the positions, our past audit experience with similar situations, and potential interest and penalties related to the matters. Our recorded reserves represent our best estimate of the amount, if any, that we will ultimately be required to pay to settle such matters. The resolution of our uncertain income tax positions is dependent on uncontrollable factors such as law changes, new case law and the willingness of the income tax authorities to settle, including the timing thereof and other factors. Although we do not anticipate significant changes to our uncertain income tax positions in the next twelve months, items outside of our control could cause our uncertain income tax positions to change in the future, which would be recorded within (provision) benefit for income taxes in our consolidated income statements. Interest and penalties related to unrecognized tax benefits are recognized as a component of our income tax provision.
Share-Based Compensation
We have long-term incentive plans which provide for the grant of awards in the form of incentive stock options, non-qualified stock options, share appreciation rights, restricted stock, restricted stock units (RSU’s), unrestricted shares of common stock, deferred share units, and performance share units (PSU’s) to eligible employees, directors, and consultants. We value share-based compensation at the grant date using a fair value model and recognize this value as expense over the employees’ requisite service period, typically the period over which the share-based compensation vests. We classify share-based compensation costs consistent with each grantee’s salary. We record income tax benefits which result from realizing a tax deduction in excess of previously recognized compensation expense as additional paid-in capital.
The fair value of our common stock on the grant date is a significant factor in determining the fair value of share-based compensation awards and the ultimate non-cash compensation cost that we will be required to record over the vesting period. Given the absence of a trading market for our common stock on grant dates prior to October 1, 2009, our board of directors, or special dividend committee or compensation committee designated by our board of directors, estimated the fair value of our common stock contemporaneously with each grant using numerous objective and subjective factors. These factors included: (i) our stage of development, our efforts to become independent from Bayer, and revenue growth; (ii) the timing of the anticipated launch of new products and new indications; (iii) business conditions and business challenges at the time; (iv) available market data, including observable market transactions, and valuations for comparable companies; (v) the illiquid nature of our stock options and stock grants; and (vi) the likelihood of achieving a liquidity event for the shares of common stock underlying the options, such as an initial public offering or sale of our company, given prevailing market conditions at the grant date. In making the assessment of common stock fair value on each award date, our board of directors or designated committee of our board of directors considered the guidance in American Institute of Certified Public Accountants Technical Practice Aid, “Valuation of Privately-Held Company Equity Securities Issued as Compensation.” The valuations were completed utilizing the market and/or an income approach and then the enterprise value was allocated using the “Probability-Weighted Expected Return Method,” which provides different probability weights of various likely scenarios (distressed; remain private; private sale; IPO), and develops valuations by determining the present value of the future expected common stock value under each of these scenarios. For option awards granted on October 1, 2009, the fair value of our common stock was determined to be the IPO price per share of $19.00. For option awards granted subsequent to our IPO, we consider the fair value of our common stock to be the closing share price as reported by The NASDAQ Global Select Market on the grant date.
We estimate the fair value of stock options at the grant date using a Black-Scholes option pricing model, which requires the use of a number of assumptions related to the risk-free interest rate, average life of options (expected term), expected volatility, and dividend yield. There was no trading market for our common stock or stock options on grant dates prior to October 1, 2009. Therefore, our application of the Black-Scholes pricing model incorporates historical volatility measures of similar public companies. A forfeiture rate based on historical attrition rates of award holders is used in estimating the granted awards not expected to vest. If actual forfeitures differ from the expected rate, we may be required to make additional adjustments to compensation expense in future periods. Our valuation utilized a dividend yield of zero. We believe that the valuation technique and the approach utilized to develop the underlying assumptions are appropriate in calculating the fair values of our stock options on their grant dates. Estimates of the values of these grants are not intended to predict actual future events or the value ultimately realized by employees who receive such awards.
Service-based awards vest annually in equal amounts over the vesting period. Performance-based awards vest annually upon the achievement of corporate performance objectives which are established by our board of directors. We make assessments as to whether the performance conditions related to the performance-based awards will be achieved. We record compensation cost for awards with performance conditions based on the probable outcome of the performance conditions.
The Black-Scholes option pricing model was developed for use in estimating the fair value of traded options that have no vesting restrictions and are fully transferable, characteristics that are not present in our option grants. If the model permitted consideration of the unique characteristics of employee stock options, the resulting estimate of the fair value of the stock options could be different. In addition, if we had made different assumptions and estimates than those described above, the amount of our recognized and to be recognized stock-based compensation expense, net income, and earnings per share amounts could have been materially different.
Additional information regarding the assumptions used in our accounting for share-based compensation awards is included in the footnotes to our audited consolidated financial statements included elsewhere in this Annual Report.
Accounts Receivable, net
Accounts receivable, net, consists of amounts owed to us by our customers on credit sales with terms generally ranging from 30 to 150 days from the date of invoice and are presented net of an allowance for doubtful accounts receivable on our consolidated balance sheets.
We maintain an allowance for doubtful accounts receivable for estimated losses resulting from our inability to collect from customers. In extending credit, we assess our customers’ creditworthiness by, among other factors, evaluating the customers’ financial condition, credit history, and the amount involved, both initially and on an ongoing basis. Collateral is generally not required. In evaluating the adequacy of our allowance for doubtful accounts receivable, we primarily analyze accounts receivable balances, the percentage of accounts receivable by aging category, and historical bad debts. We also consider, among other things, customer concentrations and changes in customer payment terms or payment patterns.
If the financial conditions of our customers were to deteriorate, resulting in an impairment of their ability to make payments or our ability to collect, an increase to the allowance may be required. Also, should actual collections of accounts receivable be different than our estimates included in determining the allowance, the allowance would be adjusted through charges or credits to SG&A in our consolidated income statement in the period in which such changes in collection become known. If conditions change in future periods, additional allowances or reversals may be required. Such allowances or reversals could be significant. While our credit losses have historically been within our expectations and the allowance established, we may not continue to experience the same credit loss rates that we have in the past.
Inventories
Inventories consist of raw material, work-in-process, and finished goods held for sale and are stated at the lower of cost or market, which approximates actual costs determined on a first-in, first-out basis and market being determined as the lower of replacement cost or estimated net realizable value. We establish inventory reserves through inventory impairment provision charges to cost of goods sold when conditions indicate that the selling price could be less than cost. These inventory impairment provisions establish a lower cost basis for the inventory.
Our raw materials, particularly plasma, are susceptible to damage and contamination and may contain human pathogens, any of which would render the plasma unsuitable as raw material for further manufacturing. In the event that we determine that the plasma was not collected in accordance with our standard operating procedures (SOP) or in a current Good Manufacturing Practices (cGMP) compliant fashion or that the collection center is unable to obtain FDA licensure, we may be unable to use and may ultimately destroy plasma collected from that center, which would be recorded as a charge to cost of goods sold during the period the plasma is determined to be unrealizable. From time to time, we have experienced significant impairment charges to cost of goods sold related to raw plasma that was collected or stored in a manner not consistent with our SOP or cGMP, such as the $23.3 million charge we recorded during the first half of 2008 as discussed further in the section titled, “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Matters Affecting Comparability—Plasma Center cGMP issue.”
The manufacture of our plasma-derived products is an extremely complex process of fractionation, purification, filling, and finishing. Although we attempt to maintain high standards for product testing, manufacturing, process controls, and quality assurance, our products can become non-releasable, or otherwise fail to meet our stringent specifications through a failure of one or more of these processes. Extensive testing is performed throughout the manufacturing process to ensure the safety and effectiveness of our products. We may, however, detect instances in which an unreleased product was produced without adherence to our SOP or cGMP. Such an event of noncompliance would likely result in our determination that the product should not be released and therefore would be destroyed, resulting in a charge to cost of goods sold. While we expect to write off small amounts of work-in-process inventory in the ordinary course of business, unanticipated events may lead to write-offs and other costs materially in excess of our expectations. Such write-offs and other costs could cause material fluctuations in our profitability.
Once we have manufactured our plasma-derived products, they must be handled carefully and kept at appropriate temperatures. Our failure, or the failure of third parties that supply, ship, or distribute our products, to properly care for our products may require those products be destroyed, resulting in a charge to cost of goods sold. Our finished goods are also subject to physical deterioration, obsolescence, reductions in estimated future demand, and reductions in selling prices. We generally record an inventory impairment provision for finished goods inventory six months prior to its expiry date when we do not reasonably expect to sell the product prior to expiration.
We capitalize the cost of unlicensed plasma when, based on our judgment, future economic benefit is probable. While unlicensed plasma cannot be sold to third parties or used in our manufacturing processes to make finished product until all regulatory approvals have been obtained, we have determined that it is probable that our plasma inventories are realizable. As part of the FDA licensing process for plasma collection centers, we are initially permitted to collect plasma utilizing the procedures and Quality Systems implemented and approved under our existing Biologics License Application (BLA) until such time as the FDA inspectors have conducted a pre-license inspection of the site and approved the site for inclusion in the BLA. At the conclusion of this process, we are permitted to sell or utilize previously collected plasma in the manufacturing of final product. We believe that our cumulative knowledge of the industry, standard industry practices, experience working with the FDA, established Quality Systems, and consistency with achieving licensure support our capitalization of unlicensed plasma. Total unlicensed plasma and related testing costs included in our raw material inventories was $2.6 million at December 31, 2010.
Impairment Reviews
We evaluate the recoverability of recorded goodwill and other indefinite-lived intangible asset amounts annually as of December 31 or when events or changes in circumstances indicate that evidence of potential impairment exists, using a fair value-based test. This test requires us to make estimates of factors that include, but are not limited to, projected future operating results and business plans, economic projections, anticipated future cash flows, comparable marketplace data from a consistent industry group and the cost of capital. Any applicable impairment loss is the amount, if any, by which the implied fair value is less than the carrying value.
We review the carrying amounts of other long-lived assets for potential impairment whenever events or changes in circumstances indicate that the carrying amount of the asset may not be recoverable. We periodically evaluate whether events or changes in circumstances have occurred that may warrant revision of the estimated useful lives of our long-lived assets or whether the remaining carrying amount of long-lived assets should be evaluated for possible impairment. An example of such a change in circumstances includes a significant adverse change in the extent or manner in which an asset is being used.
Recent Accounting Pronouncements Applicable to Our Company
In April 2010, the Financial Accounting Standards Board (FASB) issued new accounting guidance which clarifies questions surrounding the accounting implications of the different signing dates of the Health Care and Education Reconciliation Act (signed March 30, 2010) and the Patient Protection and Affordable Care Act (signed March 23, 2010). The new guidance states that the FASB and the Office of the Chief Accountant at the SEC would not be opposed to viewing the two Acts together for accounting purposes. The adoption of this guidance did not have a material impact on our consolidated financial statements.
In February 2010, the FASB issued new accounting guidance regarding disclosures related to subsequent events. An entity that is a U.S. Securities and Exchange Commission (SEC) filer is not required to disclose the date through which subsequent events have been evaluated. This change alleviates potential conflicts between the Accounting Standards Codification (ASC) and the SEC’s requirement. The adoption of this guidance did not have a material impact on our consolidated financial statements.
In January 2010, the FASB issued guidance that requires new disclosures for fair value measurements and provides clarity for existing disclosures. This update requires new disclosures for (1) transfers in and out of levels 1 and 2, and (2) activity in level 3, by requiring the reconciliation to present separate information about purchases, sales, issuance, and settlements. Also, this update clarifies the disclosures related to the fair value of each class of assets and liabilities and the input and valuation techniques for both recurring and nonrecurring fair value measurements in levels 2 and 3. The effective date for the disclosures and clarifications is for the interim and annual reporting periods beginning after December 15, 2009 except for the disclosures about purchases, sales, issuances and settlements, which is effective for fiscal years beginning after December 15, 2010. This update is not expected to have a material impact on our consolidated financial statements or related disclosures.
In October 2009, the FASB issued new accounting guidance regarding multiple-deliverable revenue arrangements. The new guidance addresses the accounting for multiple-deliverable arrangements to enable vendors to account for products or services (deliverables) separately rather than as a combined unit. This guidance establishes a hierarchy for determining the selling price of a deliverable, which is based on: (a) vendor-specific objective evidence; (b) third-party evidence; or (c) estimates. This guidance also eliminates the residual method of allocation and requires that arrangement consideration be allocated at the inception of the arrangement to all deliverables using the relative selling price method. In addition, this guidance significantly expands required disclosures related to a vendor’s multiple-deliverable revenue arrangements. The guidance is effective prospectively for revenue arrangements entered into or materially modified in fiscal years beginning on or after June 15, 2010 and early adoption is permitted. A company may elect, but will not be required, to adopt the guidance retrospectively for all prior periods. We do not anticipate that the adoption of this guidance will have a material impact on our consolidated financial statements or related disclosures.
In August 2009, the FASB released new accounting guidance concerning measuring liabilities at fair value. The new guidance provides clarification that in circumstances in which a quoted price in an active market for the identical liability is not available, a reporting entity is required to measure fair value using certain valuation techniques. Additionally, it clarifies that a reporting entity is not required to adjust the fair value of a liability for the existence of a restriction that prevents the transfer of the liability. This new guidance is effective for the first reporting period after its issuance; however, earlier application is permitted. The adoption of this guidance did not have a material impact on our consolidated financial statements or related disclosures.
On January 30, 2009, the SEC released the final rules requiring all registered companies to use eXtensible Business Reporting Language (XBRL) when submitting financial statements to the SEC. The new rules initially will require interactive data reporting only by domestic and foreign large accelerated filers that prepare their financial statements in accordance with U.S. GAAP and have a worldwide public common equity float above $5.0 billion for their first quarterly period ending after June 15, 2009 and all reporting periods thereafter. We will be required to file using XBRL beginning with our quarterly reporting period ending March 31, 2011.
MATTERS AFFECTING COMPARABILITY
We believe that the comparability of our financial results between the years presented is significantly impacted by the following items:
Plasma Center of America (PCA) Judgment
On December 13, 2010, a jury in the General Court of Justice, Superior Court Division, Wake County, North Carolina, rendered a verdict in the amount of $37.0 million in favor of PCA against TPR in a breach of contract claim, which was confirmed by the court in post trial motions. We intend to appeal. The jury verdict, if sustained, will bear simple interest at 8% per statute from the date of breach, which totals approximately $6.7 million at December 31, 2010. We have included a charge of $43.7 million (approximately $26.6 million after tax) in our consolidated income statement for the year ended December 31, 2010 related to this judgment.
Definitive Merger Agreement with Grifols
We have entered into agreements with investment bankers related to our definitive merger agreement with Grifols. We incurred fees totaling $2.5 million under these agreements during 2010. During 2010, we also incurred legal, accounting, and other fees of $18.3 million associated with the merger.
Under our agreements with investment bankers, we are obligated to pay additional fees totalling $21.3 million upon successful closing of the transaction.
Under the terms of the merger agreement with Grifols, we are permitted to offer retention amounts up to a total of $15.0 million to employees. As of December 31, 2010, we have offered retention amounts totaling approximately $10.2 million to employees, of which $2.9 million was paid during 2010 and the remaining amounts are expected to be paid in 2011, subject to the terms of the retention agreements. We incurred retention expenses, including fringe benefits, of $6.9 million during the year ended December 31, 2010. The remaining retention amounts will likely be recognized ratably through the second quarter of 2011, but we may offer additional retention packages.
Financial Impact of IPO and Refinancing Transactions
The following table summarizes the changes to our indebtedness during 2009, including the impact from the application of the net proceeds to us of $519.7 million from our IPO and the net proceeds to us of $583.9 million from our refinancing transactions:
|
|
|
December 31, |
|
2009 Net |
|
October 6, 2009 |
|
October 21, 2009 |
|
|
|
December 31, |
| ||||||
|
|
|
2008 |
|
Repayments |
|
IPO |
|
Refinancing |
|
Accretion |
|
2009 |
| ||||||
|
Revolving credit facility |
|
$ |
179,941 |
|
$ |
(124,348 |
) |
$ |
— |
|
$ |
(55,593 |
) |
$ |
— |
|
$ |
— |
|
|
First Lien Term Loan |
|
686,000 |
|
(5,250 |
) |
(389,812 |
) |
(290,938 |
) |
— |
|
— |
| ||||||
|
Second Lien Term Loan |
|
330,000 |
|
— |
|
(129,937 |
) |
(200,063 |
) |
— |
|
— |
| ||||||
|
7.75% Notes |
|
— |
|
— |
|
— |
|
600,000 |
|
— |
|
600,000 |
| ||||||
|
Discount on 7.75% Notes |
|
— |
|
— |
|
— |
|
(4,074 |
) |
120 |
|
(3,954 |
) | ||||||
|
Total indebtedness |
|
$ |
1,195,941 |
|
$ |
(129,598 |
) |
$ |
(519,749 |
) |
$ |
49,332 |
|
$ |
120 |
|
$ |
596,046 |
|
In addition to the debt principal repayments in the preceding table, we used $28.7 million of the net proceeds to us from the issuance of the 7.75% Notes to settle and terminate certain interest rate swap contracts with a notional amount of $390.0 million and $8.6 million to pay accrued interest associated with our then outstanding First and Second Lien Term Loans. In addition to the $4.1 million of discounts on the 7.75% Notes disclosed in the table above, approximately $12.0 million of commissions were deducted from the gross issuance proceeds. Subsequently, we settled and terminated our remaining interest rate swap contract with a notional amount of $50.0 million for $6.1 million. We incurred other costs related to our IPO of $3.9 million, of which $1.3 million is included within SG&A in our consolidated income statement for the year ended December 31, 2009 and $2.6 million is included as a reduction of additional paid-in capital on our December 31, 2009 consolidated balance sheet.
As a result of the IPO and refinancing transactions, we recognized a charge during the fourth quarter of 2009 of $12.1 million to write-off previously deferred debt issuance costs related to our First and Second Lien Term Loans and $30.9 million related to costs associated with the settlement and termination of our interest rate swap contracts. These charges, which totaled $43.0 million, are recorded within total other non-operating expense, net, in our consolidated income statement for the year ended December 31, 2009. We capitalized $14.9 million of debt issuance costs associated with the issuance of the 7.75% Notes and the revolving credit facility amendment during 2009.
The following table summarizes changes in deferred debt issuance costs during the year ended December 31, 2009:
|
|
|
|
|
|
|
Newly |
|
|
|
|
| |||||
|
|
|
|
|
|
|
Capitalized |
|
|
|
|
| |||||
|
|
|
December 31, |
|
|
|
Debt Issuance |
|
|
|
December 31, |
| |||||
|
|
|
2008 |
|
Charges |
|
Costs |
|
Amortization |
|
2009 |
| |||||
|
Revolving credit facility |
|
$ |
3,014 |
|
$ |
— |
|
$ |
1,545 |
|
$ |
(1,041 |
) |
$ |
3,518 |
|
|
First Lien Term Loan |
|
9,629 |
|
(8,054 |
) |
— |
|
(1,575 |
) |
— |
| |||||
|
Second Lien Term Loan |
|
4,744 |
|
(4,087 |
) |
— |
|
(657 |
) |
— |
| |||||
|
7.75% Notes |
|
— |
|
— |
|
13,334 |
|
(392 |
) |
12,942 |
| |||||
|
Total deferred debt issuance costs |
|
$ |
17,387 |
|
$ |
(12,141 |
) |
$ |
14,879 |
|
$ |
(3,665 |
) |
$ |
16,460 |
|
Comparability of Outstanding Common Shares and Pro Forma Diluted Earnings Per Common Share
As discussed elsewhere in this Annual Report, we completed our IPO and refinancing transactions during the fourth quarter of 2009. Our IPO consisted of 56,000,000 shares of our common stock of which 28,947,368 were newly issued and sold by us and 27,052,632 shares were sold by the selling stockholder, Talecris Holdings, LLC. We used the net primary proceeds to us of $519.7 million to repay amounts outstanding under our then existing First and Second Lien Term Loans. In connection with our IPO, we also converted our Series A and B preferred stock into 85,846,320 shares of our common stock and issued 2,381,548 shares of our common stock to settle $45.3 million of accrued dividends upon the conversion of our Series A and B preferred stock. The issuance of new common shares has resulted in a significant increase in the number of common shares used in our computation of diluted earnings per common share. The application of the net primary proceeds to us from our IPO to repay our then existing indebtedness has resulted in a significant reduction in interest expense in periods subsequent to our IPO.
We believe that the comparability of our financial results for the years presented is enhanced by the following pro forma presentation of our diluted earnings per common share. In the table below, the pro forma diluted earnings per common share computation for the year ended December 31, 2009 reflects an adjustment to net income for reduced interest expense as if the net primary proceeds to us from our IPO of $519.7 million had been applied to repay our debt at the beginning of 2009, net of interest rate differences from our 7.75% Notes issuance. The pro forma adjustments to the denominator reflects the impacts for the issuance of common shares to convert preferred stock, settle accrued dividends on the preferred stock and complete the IPO as if these events occurred at the beginning of 2009.
|
|
|
Years Ended December 31, |
| ||||||||||
|
|
|
2010 |
|
2009 |
|
2009 |
|
2008 |
| ||||
|
|
|
Actual |
|
Actual |
|
Pro forma |
|
Actual |
| ||||
|
Net income |
|
$ |
166,068 |
|
$ |
153,889 |
|
$ |
153,889 |
|
$ |
65,797 |
|
|
Interest expense reduction due to debt repayment |
|
|
|
|
|
5,555 |
|
— |
| ||||
|
Less accretion of common stock put option |
|
— |
|
— |
|
— |
|
(308 |
) | ||||
|
Net income available to common stockholders |
|
$ |
166,068 |
|
$ |
153,889 |
|
$ |
159,444 |
|
$ |
65,489 |
|
|
|
|
|
|
|
|
|
|
|
| ||||
|
Weighted average common shares outstanding |
|
123,323,722 |
|
31,166,613 |
|
31,166,613 |
|
1,310,448 |
| ||||
|
Plus incremental shares from assumed conversions: |
|
|
|
|
|
|
|
|
| ||||
|
Stock options and restricted shares |
|
5,603,331 |
|
7,374,601 |
|
7,374,601 |
|
5,605,032 |
| ||||
|
Series A preferred stock |
|
— |
|
53,654,795 |
|
53,654,795 |
|
72,000,000 |
| ||||
|
Series B preferred stock |
|
— |
|
10,318,354 |
|
10,318,354 |
|
13,846,320 |
| ||||
|
Shares issued for preferred stock dividend |
|
— |
|
— |
|
1,774,743 |
|
— |
| ||||
|
Newly issued shares for IPO |
|
— |
|
— |
|
22,047,585 |
|
— |
| ||||
|
Dilutive potential common shares |
|
128,927,053 |
|
102,514,363 |
|
126,336,691 |
|
92,761,800 |
| ||||
|
|
|
|
|
|
|
|
|
|
| ||||
|
Diluted net income per common share |
|
$ |
1.29 |
|
$ |
1.50 |
|
$ |
1.26 |
|
$ |
0.71 |
|
Definitive Merger Agreement with CSL Limited (CSL)
On August 12, 2008, we entered into a definitive merger agreement with CSL, the closing of which was subject to the receipt of certain regulatory approvals as well as other customary conditions. The U.S. Federal Trade Commission filed an administrative complaint challenging the merger and a complaint in federal district court seeking to enjoin the merger during the administrative process. On June 8, 2009, the merger parties agreed to terminate the definitive merger agreement. CSL paid Talecris a merger termination fee of $75.0 million (after tax amount of $48.8 million) during the 2009 second quarter, which is included as other non-operating income in our consolidated income statement for the year ended December 31, 2009. The U.S. Federal Trade Commission’s complaints were subsequently dismissed.
We recorded retention expense, including fringe benefits, of $9.2 million and $5.6 million for the years ended December 31, 2009 and 2008, respectively. We classified the cost of this retention program consistent with each recipient’s salary. We made payments of approximately $13.3 million under this retention program during 2009. All retention amounts were paid during 2009.
We incurred legal and other costs associated with the regulatory review process of $6.0 million and $8.3 million during the years ended December 31, 2009 and 2008, respectively, which are included in SG&A in our consolidated income statements.
Foreign Corrupt Practices Act (FCPA)
We are conducting an internal investigation into potential violations of the FCPA that we became aware of during the conduct of an unrelated review. The FCPA investigation is being conducted by outside counsel under the direction of a special committee of our board of directors. During the years ended December 31, 2010 and 2009, we incurred $5.0 million and $8.0 million, respectively, of legal costs associated with our internal investigation into this matter, which are recorded in SG&A in our consolidated income statement. We expect to complete our internal FCPA investigation and present our findings to the Department of Justice (DOJ) in 2011. The preliminary findings of our investigation indicate that it is probable that there were FCPA violations by persons associated with us that the DOJ or other regulators may assert are attributable to us. Even though we self-disclosed this matter to the DOJ, it or other federal agencies may seek to impose sanctions on us that may include, among other things, debarment, injunctive relief, disgorgement, fines, penalties, appointment of a monitor, appointment of a new control staff or enhancements of existing compliance and training programs. Any such sanctions or related loss of business could have a material adverse effect on us or our results of operations. It is possible, however, that any sanctions that DOJ or other federal agencies might otherwise consider imposing would be reduced, if not eliminated, in light of the comprehensive compliance measures that we have implemented. Given the preliminary nature of our findings, our continuing investigation and the uncertainties regarding this matter, we are unable to estimate the financial outcome. Additional information regarding our investigation into potential violations of the FCPA is included in the footnotes to our consolidated financial statements and in the section of this Annual Report entitled, “Risk Factors—Risks Related to Our Business—We are investigating potential Foreign Corrupt Practices Act violations.”
Unabsorbed TPR Infrastructure and Start-Up Costs
Our cost of goods sold includes $6.6 million, $44.0 million, and $98.5 million for the years ended December 31, 2010, 2009, and 2008, respectively, related to unabsorbed TPR infrastructure and start-up costs associated with the development of our plasma collection center platform. Until our plasma collection centers reach normal operating capacity, we charge unabsorbed overhead costs directly to cost of goods sold. The reduction in unabsorbed TPR infrastructure and start-up costs resulted primarily from the maturation of our plasma collection center platform. We believe we have substantially eliminated unabsorbed TPR infrastructure and start-up costs.
Plasma Center cGMP Issue
During the first and second quarters of 2008, we incurred charges to cost of goods sold of $16.3 million and $7.0 million, respectively, due to deviations from our SOP and cGMP at one of our plasma collection centers. Our preliminary investigations concluded that the deviations from our SOP and cGMP resulted in impairments to the related raw material and work-in-process inventories as we concluded there was no probable future economic benefit related to the impacted inventories. Subsequently, due to further investigations and new facts and circumstances, we determined that certain impacted inventories were saleable. We record recoveries directly to cost of goods sold after the impacted material is converted into final products and sold to third parties. During the years ended December 31, 2009 and 2008, we recorded recoveries of $1.9 million and $17.5 million, respectively. For the year ended December 31, 2008, recoveries totaled $17.5 million, resulting in a net provision of $5.8 million for 2008. No material recoveries were recorded during the year ended December 31, 2010.
Share-Based Compensation Awards
We have long-term incentive plans, which provide for the grant of awards in the form of incentive stock options, non-qualified stock options, share appreciation rights, restricted stock, restricted stock units (RSU’s), unrestricted shares of common stock, deferred share units, and performance share units, to eligible employees, directors, and consultants.
The following table summarizes our share-based compensation expense:
|
|
|
Years Ended December 31, |
| |||||||
|
|
|
2010 |
|
2009 |
|
2008 |
| |||
|
SG&A |
|
$ |
12,606 |
|
$ |
40,968 |
|
$ |
33,780 |
|
|
R&D |
|
1,024 |
|
2,303 |
|
2,361 |
| |||
|
Total operating expenses |
|
13,630 |
|
43,271 |
|
36,141 |
| |||
|
Cost of goods sold |
|
3,336 |
|
4,275 |
|
2,566 |
| |||
|
Total expense |
|
$ |
16,966 |
|
$ |
47,546 |
|
$ |
38,707 |
|
|
|
|
|
|
|
|
|
| |||
|
Capitalized in inventory |
|
$ |
2,265 |
|
$ |
3,574 |
|
$ |
3,233 |
|
In addition to incremental share-based compensation expense associated with share-based compensation awards granted during the years presented, the following items have impacted the comparability of our share-based compensation expense:
· The decrease in share-based compensation expense during 2010 was primarily driven by the final vesting of awards under our 2005 Stock Option and Incentive Plan on April 1, 2010 and the majority of the awards under our 2006 Restricted Stock Plan on March 31, 2010. In addition, a combination of estimate-to-actual true ups as these plans culminated during 2010 and the acceleration of certain options to our Chairman and Chief Executive Officer in 2009 (discussed further below) further impacted the comparability of share-based compensation expense between the years presented.
· During the third quarter of 2009, we entered into an amended and restated employment agreement with our Chairman and Chief Executive Officer which included accelerating the vesting of options to purchase 1,008,000 shares of our common stock at an exercise price of $21.25 per common share to August 19, 2009. The acceleration of these options resulted in the recognition of a non-cash charge of $11.8 million of compensation expense during 2009. Options to purchase these shares were previously scheduled to vest in April of 2010 (504,000 options) and April of 2011 (504,000 options).
· During the second quarter of 2008, the compensation committee of our board of directors amended the exercise price of 570,400 stock options outstanding to certain employees from $21.25 per share to $11.00 per share and also amended the exercise price of 17,152 stock options outstanding to certain members of our board of directors from $21.25 per share to $11.00 per share. The stock options that were re-priced were granted during 2007.
The following table summarizes the remaining unrecognized compensation cost related to our share-based compensation awards as of December 31, 2010 and the weighted average period over which non-cash compensation cost is expected to be recognized:
|
|
|
|
|
Weighted |
| |
|
|
|
Unrecognized |
|
Average |
| |
|
|
|
Compensation |
|
Period |
| |
|
|
|
Cost |
|
(Years) |
| |
|
Stock options |
|
$ |
3,965 |
|
2.10 |
|
|
Restricted share awards |
|
721 |
|
0.25 |
| |
|
RSU’s |
|
6,589 |
|
2.10 |
| |
|
PSU’s |
|
1,138 |
|
1.25 |
| |
|
Total |
|
$ |
12,413 |
|
|
|
In addition to the unrecognized compensation cost included in the table above, at December 31, 2010, $1.7 million of compensation cost was included in inventory on our consolidated balance sheet, which we expect to be recognized as non-cash compensation expense in our consolidated income statement primarily within the next twelve months. The amount of share-based compensation expense that we will ultimately be required to record could change in the future as a result of additional grants, changes in the fair value of shares for performance-based awards, differences between our anticipated forfeiture rate and the actual forfeiture rate, the probability of achieving targets established for performance award vesting, and other actions by our board of directors or its compensation committee. Additional information regarding our share-based compensation awards is included in “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Critical Accounting Policies— Share-Based Compensation” and in the footnotes to our audited consolidated financial statements included elsewhere in this Annual Report.
RESULTS OF OPERATIONS
We have included information regarding our results of operations in the following table. The subsequent discussion and analysis provides information which management believes is relevant to an assessment and understanding of our consolidated results of operations. You are encouraged to read the following discussion and analysis of our financial condition and results of operations together with our audited consolidated financial statements and related footnotes included elsewhere in this Annual Report. Additional information regarding significant matters affecting comparability of our results of operations is included in the section titled “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Matters Affecting Comparability.”
|
|
|
Years Ended December 31, |
| |||||||
|
|
|
2010 |
|
2009 |
|
2008 |
| |||
|
Net revenue: |
|
|
|
|
|
|
| |||
|
Product |
|
$ |
1,576,936 |
|
$ |
1,507,754 |
|
$ |
1,334,550 |
|
|
Other |
|
24,683 |
|
25,455 |
|
39,742 |
| |||
|
Total |
|
1,601,619 |
|
1,533,209 |
|
1,374,292 |
| |||
|
Cost of goods sold |
|
911,976 |
|
901,077 |
|
882,157 |
| |||
|
|
|
|
|
|
|
|
| |||
|
Gross profit |
|
689,643 |
|
632,132 |
|
492,135 |
| |||
|
Operating expenses: |
|
|
|
|
|
|
| |||
|
SG&A |
|
287,011 |
|
289,929 |
|
227,524 |
| |||
|
R&D |
|
69,649 |
|
71,223 |
|
66,006 |
| |||
|
Total |
|
356,660 |
|
361,152 |
|
293,530 |
| |||
|
|
|
|
|
|
|
|
| |||
|
Income from operations |
|
332,983 |
|
270,980 |
|
198,605 |
| |||
|
Other non-operating (expense) income: |
|
|
|
|
|
|
| |||
|
Interest expense, net |
|
(45,837 |
) |
(74,491 |
) |
(96,640 |
) | |||
|
PCA judgment |
|
(43,690 |
) |
— |
|
— |
| |||
|
CSL merger termination fee |
|
— |
|
75,000 |
|
— |
| |||
|
Loss on extinguishment of debt |
|
— |
|
(43,033 |
) |
— |
| |||
|
Equity in earnings of affiliate |
|
991 |
|
441 |
|
426 |
| |||
|
Total |
|
(88,536 |
) |
(42,083 |
) |
(96,214 |
) | |||
|
|
|
|
|
|
|
|
| |||
|
Income before income taxes |
|
244,447 |
|
228,897 |
|
102,391 |
| |||
|
Provision for income taxes |
|
(78,379 |
) |
(75,008 |
) |
(36,594 |
) | |||
|
|
|
|
|
|
|
|
| |||
|
Net income |
|
$ |
166,068 |
|
$ |
153,889 |
|
$ |
65,797 |
|
|
|
|
|
|
|
|
|
| |||
|
Earnings per common share: |
|
|
|
|
|
|
| |||
|
Basic |
|
$ |
1.35 |
|
$ |
4.56 |
|
$ |
39.01 |
|
|
Diluted |
|
$ |
1.29 |
|
$ |
1.50 |
|
$ |
0.71 |
|
|
|
|
|
|
|
|
|
| |||
|
Financial measures: |
|
|
|
|
|
|
| |||
|
Gross margin |
|
43.1 |
% |
41.2 |
% |
35.8 |
% | |||
|
Operating margin |
|
20.8 |
% |
17.7 |
% |
14.5 |
% | |||
|
Effective income tax rate |
|
32.1 |
% |
32.8 |
% |
35.7 |
% | |||
Primary Revenue and Expense Components
The following is a description of the primary components of our revenue and expenses:
· Product net revenue—Our product net revenue is presented net of allowances for estimated discounts, rebates, administrative fees, chargebacks, and sales allowances. Our product net revenue is also presented net of SG&A reimbursements to certain international distributors.
· Other net revenue—Our other net revenue primarily consists of royalties under our collaborative agreements, licensing fees, milestone revenues, and revenue related to contracted services performed for third parties at our Melville, New York facility.
· Cost of goods sold—Our cost of goods sold includes material costs for the products we sell, which primarily consists of plasma and other costs associated with the manufacturing process, such as personnel costs, utilities, consumables, and overhead. In addition, our cost of goods sold includes packaging costs and distribution costs. The most significant component of our cost of goods sold is plasma, which is the common raw material for our primary products. Due to our long manufacturing cycle times, which range from 100 days to in excess of 400 days for some specialty plasma, in addition to a required 60 day pre-production holding period for plasma, the cost of plasma is not expensed through cost of goods sold until a significant period of time subsequent to its acquisition.
· Gross profit—Our gross profit is impacted by the volume and pricing of our finished products, our raw material costs, production mix, yield, and cycle times, as well as our production capacities and normal production shut-downs, and the timing and amount of release of finished product. Our profitability is significantly impacted by the efficiency of our utilization of plasma, including, but not limited to, the production yields we obtain, the product reject rates that we experience, and the product through-put that we achieve.
· SG&A—Our SG&A consists primarily of salaries and related employee benefit costs for personnel in executive, sales and marketing, finance, information technology, human resources, and other administrative functions, as well as fees for professional services, facilities costs, and other general and administrative costs.
· R&D—Our R&D includes the costs directly attributable to the conduct of research and development programs for new products and life cycle management. Such costs include salaries and related employee benefit costs; materials (including the material required for clinical trials); supplies; depreciation on and maintenance of R&D equipment; various services provided by outside contractors related to clinical development, trials and regulatory services; and the allocable portion of facility costs such as rent, depreciation, utilities, insurance and general support services. R&D expenses are influenced by the number and timing of in-process projects and the nature of expenses associated with these projects.
· Interest expense, net—Our interest expense, net, consists of interest expense incurred on outstanding debt, capital lease obligations, derivative financial instruments, and amortization of debt issuance costs and debt discount, offset by interest income and capitalized interest associated with the construction of plant and equipment.
· Income tax provision—Our income tax provision includes United States Federal, state, local, and foreign income taxes, and is based on reported pre-tax income.
Year Ended December 31, 2010 as Compared to Year Ended December 31, 2009
The following table contains information regarding our results of operations for the year ended December 31, 2010 as compared to the year ended December 31, 2009:
|
|
|
Years Ended December 31, |
|
Change |
| |||||||
|
|
|
2010 |
|
2009 |
|
$ |
|
% |
| |||
|
Net revenue: |
|
|
|
|
|
|
|
|
| |||
|
Product |
|
$ |
1,576,936 |
|
$ |
1,507,754 |
|
$ |
69,182 |
|
4.6 |
% |
|
Other |
|
24,683 |
|
25,455 |
|
(772 |
) |
(3.0 |
)% | |||
|
Total |
|
1,601,619 |
|
1,533,209 |
|
68,410 |
|
4.5 |
% | |||
|
Cost of goods sold |
|
911,976 |
|
901,077 |
|
(10,899 |
) |
(1.2 |
)% | |||
|
|
|
|
|
|
|
|
|
|
| |||
|
Gross profit |
|
689,643 |
|
632,132 |
|
57,511 |
|
9.1 |
% | |||
|
Operating expenses: |
|
|
|
|
|
|
|
|
| |||
|
SG&A |
|
287,011 |
|
289,929 |
|
2,918 |
|
1.0 |
% | |||
|
R&D |
|
69,649 |
|
71,223 |
|
1,574 |
|
2.2 |
% | |||
|
Total |
|
356,660 |
|
361,152 |
|
4,492 |
|
1.2 |
% | |||
|
|
|
|
|
|
|
|
|
|
| |||
|
Income from operations |
|
332,983 |
|
270,980 |
|
62,003 |
|
22.9 |
% | |||
|
Other non-operating (expense) income: |
|
|
|
|
|
|
|
|
| |||
|
Interest expense, net |
|
(45,837 |
) |
(74,491 |
) |
28,654 |
|
38.5 |
% | |||
|
PCA judgment |
|
(43,690 |
) |
— |
|
(43,690 |
) |
nm |
| |||
|
CSL merger termination fee |
|
— |
|
75,000 |
|
(75,000 |
) |
nm |
| |||
|
Loss on extinguishment of debt |
|
— |
|
(43,033 |
) |
43,033 |
|
nm |
| |||
|
Equity in earnings of affiliate |
|
991 |
|
441 |
|
550 |
|
124.7 |
% | |||
|
Total |
|
(88,536 |
) |
(42,083 |
) |
(46,453 |
) |
(110.4 |
)% | |||
|
|
|
|
|
|
|
|
|
|
| |||
|
Income before income taxes |
|
244,447 |
|
228,897 |
|
15,550 |
|
6.8 |
% | |||
|
Provision for income taxes |
|
(78,379 |
) |
(75,008 |
) |
(3,371 |
) |
(4.5 |
)% | |||
|
|
|
|
|
|
|
|
|
|
| |||
|
Net income |
|
$ |
166,068 |
|
$ |
153,889 |
|
$ |
12,179 |
|
7.9 |
% |
|
|
|
|
|
|
|
|
|
|
| |||
|
Earnings per common share: |
|
|
|
|
|
|
|
|
| |||
|
Basic |
|
$ |
1.35 |
|
$ |
4.56 |
|
$ |
(3.21 |
) |
(70.4 |
)% |
|
Diluted |
|
$ |
1.29 |
|
$ |
1.50 |
|
$ |
(0.21 |
) |
(14.0 |
)% |
|
|
|
|
|
|
|
|
|
|
| |||
|
Financial measures: |
|
|
|
|
|
|
|
|
| |||
|
Gross profit margin |
|
43.1 |
% |
41.2 |
% |
|
|
|
| |||
|
Operating margin |
|
20.8 |
% |
17.7 |
% |
|
|
|
| |||
|
Effective income tax rate |
|
32.1 |
% |
32.8 |
% |
|
|
|
| |||
|
nm- not meaningful |
|
|
|
|
|
|
|
|
Net Revenue
The following table contains information regarding our net revenue
|
|
|
Years Ended December 31, |
|
Change |
| |||||||
|
|
|
2010 |
|
2009 |
|
$ |
|
% |
| |||
|
Product net revenue: |
|
|
|
|
|
|
|
|
| |||
|
Gamunex-C/Gamunex IGIV |
|
$ |
871,625 |
|
$ |
826,376 |
|
$ |
45,249 |
|
5.5 |
% |
|
Prolastin A1PI/ Prolastin-C A1PI |
|
351,499 |
|
319,080 |
|
32,419 |
|
10.2 |
% | |||
|
Fraction V (Albumin and Plasmanate) |
|
78,400 |
|
84,770 |
|
(6,370 |
) |
(7.5 |
)% | |||
|
Fraction VIII (Koate DVI) |
|
57,040 |
|
46,453 |
|
10,587 |
|
22.8 |
% | |||
|
Hyperimmunes |
|
69,800 |
|
74,203 |
|
(4,403 |
) |
(5.9 |
)% | |||
|
Other |
|
148,572 |
|
156,872 |
|
(8,300 |
) |
(5.3 |
)% | |||
|
Total product net revenue |
|
1,576,936 |
|
1,507,754 |
|
69,182 |
|
4.6 |
% | |||
|
Other net revenue |
|
24,683 |
|
25,455 |
|
(772 |
) |
(3.0 |
)% | |||
|
Total net revenue |
|
$ |
1,601,619 |
|
$ |
1,533,209 |
|
$ |
68,410 |
|
4.5 |
% |
|
|
|
|
|
|
|
|
|
|
| |||
|
U.S. product net revenue: |
|
|
|
|
|
|
|
|
| |||
|
Gamunex-C/Gamunex IGIV |
|
$ |
659,962 |
|
$ |
599,758 |
|
$ |
60,204 |
|
10.0 |
% |
|
Prolastin A1PI/ Prolastin-C A1PI |
|
231,712 |
|
206,099 |
|
25,613 |
|
12.4 |
% | |||
|
Fraction V (Albumin and Plasmanate) |
|
46,678 |
|
44,768 |
|
1,910 |
|
4.3 |
% | |||
|
Fraction VIII (Koate DVI) |
|
17,564 |
|
13,601 |
|
3,963 |
|
29.1 |
% | |||
|
Hyperimmunes |
|
53,259 |
|
59,500 |
|
(6,241 |
) |
(10.5 |
)% | |||
|
Other |
|
67,067 |
|
64,404 |
|
2,663 |
|
4.1 |
% | |||
|
Total U.S. product net revenue |
|
$ |
1,076,242 |
|
$ |
988,130 |
|
$ |
88,112 |
|
8.9 |
% |
|
|
|
|
|
|
|
|
|
|
| |||
|
International product net revenue: |
|
|
|
|
|
|
|
|
| |||
|
Gamunex-C/Gamunex IGIV |
|
$ |
211,663 |
|
$ |
226,618 |
|
$ |
(14,955 |
) |
(6.6 |
)% |
|
Prolastin A1PI/ Prolastin-C A1PI |
|
119,787 |
|
112,981 |
|
6,806 |
|
6.0 |
% | |||
|
Fraction V (Albumin and Plasmanate) |
|
31,722 |
|
40,002 |
|
(8,280 |
) |
(20.7 |
)% | |||
|
Fraction VIII (Koate DVI) |
|
39,476 |
|
32,852 |
|
6,624 |
|
20.2 |
% | |||
|
Hyperimmunes |
|
16,541 |
|
14,703 |
|
1,838 |
|
12.5 |
% | |||
|
Other |
|
81,505 |
|
92,468 |
|
(10,963 |
) |
(11.9 |
)% | |||
|
Total international product net revenue |
|
$ |
500,694 |
|
$ |
519,624 |
|
$ |
(18,930 |
) |
(3.6 |
)% |
Our product net revenue was $1,576.9 million and $1,507.8 million for the years ended December 31, 2010 and 2009, respectively, representing an increase of $69.2 million, or 4.6%. The increase consisted of higher volumes of $57.6 million and higher pricing of $11.6 million, including the effects of unfavorable foreign exchange of $6.9 million.
The $45.2 million increase in Gamunex-C/Gamunex IGIV net revenue was driven by higher volumes. We experienced higher Gamunex-C/Gamunex IGIV volumes of $74.9 million in the U.S. and Europe, which were partially offset by lower Gamunex-C/Gamunex IGIV volumes of $29.7 million in Canada and other international regions. Our 2010 Canadian Gamunex-C volumes were negatively impacted by lower commercial sales to CBS as discussed below. Our 2009 volumes in other international regions include opportunistic sales that did not recur during 2010. U.S. and Canadian pricing was $8.6 million higher during 2010 as compared to the prior year. The benefit of the higher pricing in the U.S. and Canada was offset by lower pricing of $8.6 million in Europe and other international regions as a result of country mix, competitive pressures, and the effects of unfavorable foreign exchange of $1.9 million. As indicated elsewhere in this Annual Report, the recently enacted healthcare reform legislation increased the size of the Medicaid rebates paid by drug manufacturers from 15.1% to 23.1% of the AMP, excluding clotting factors. We have also experienced higher Medicaid utilization and GPO administrative fees during 2010. We expect product pricing to be relatively flat given the current balance of supply and demand.
The $32.4 million increase in our Prolastin A1PI/ Prolastin-C A1PI net revenue consisted of higher volumes of $19.8 million and higher pricing of $12.6 million, including the effects of unfavorable foreign exchange of $5.0 million. The increase in volumes was driven by higher volumes of $18.8 million in the U.S. and Europe, where we experienced net patient gains during 2010. Prolastin volumes are largely a function of our ability to identify and enroll new patients as compared to the number of patients lost due to attrition and competition. Our ability to grow our European volumes will also depend on our ability to obtain appropriate reimbursement on a country by country basis. The increase in pricing was driven by higher pricing of $14.7 million in the U.S. as a result of a price increase implemented during the 2010 third quarter, as well as higher pricing of $2.9 million in Canada. The benefit of the higher pricing in the U.S. and Canada was partially offset by lower pricing in Europe, driven by unfavorable foreign exchange of $5.6 million.
Effective August 1, 2010, legislation was enacted in Germany to increase rebates paid by drug manufacturers from 6% to 16% in retail pharmacies, which impacts our Gamunex business. In addition, effective August 1, 2010, a price freeze on the basis of August 2009 prices became effective through 2013 for all products we sell in Germany.
Our Fraction V product category consists of albumin and Plasmanate, with albumin representing the majority of sales in the category. The $6.4 million decrease in our Fraction V net revenue resulted from lower volumes of $3.7 million and lower pricing of $2.7 million. We experienced lower Fraction V volumes of $11.0 million in other international regions due to opportunistic sales during 2009, which did not recur during 2010, partially offset by higher volumes of $7.3 million in the U.S., Canada, and Europe as a result of supply availability. We experienced declining albumin pricing, which has stabilized.
The $10.6 million increase in Factor VIII (Koate DVI) net revenue was primarily driven by higher volumes of $13.7 million in some international regions due to supply availability and opportunistic sales, as well as higher volumes of $2.1 million and higher pricing of $1.9 million in the U.S. These increases were partially offset by lower pricing of $7.1 million in other international regions. Pricing pressure for plasma-derived Factor VIII in international markets as we compete for tenders may continue.
The $4.4 million decrease in hyperimmune net revenue was driven by lower sales of $6.2 million in the U.S. primarily due to a customer systems issue, partially offset by higher sales of $1.5 million in Canada. We believe that the customer system issue is resolved and expect hyperimmune sales to recover to historical levels.
Our other product net revenue relates primarily to our contract manufacturing agreements for IGIV and Fraction V with the two Canadian blood system operators, CBS and Hema Quebec, intermediate products, Thrombate III (human), and contracted PPF powder. The $8.3 million decrease in other product revenue was driven by lower net revenue related to contract manufacturing of IGIV and Fraction V of $7.4 million due to CBS’ multi-source strategy as well as lower intermediate product sales of $3.0 million and lower PPF powder sales of $4.1 million, partially offset by higher Thrombate III revenues of $6.0 million.
We believe U.S. IGIV distribution increased between 6% and 8% during the year ended December 31, 2010. Despite solid demand growth for IGIV, there has been increased scrutiny and price sensitivity in the hospital segment. In addition, the increase in the number of hospitals qualifying for the 340B discounts has effectively reduced demand from GPO’s who are not permitted to service this discounted channel. This, among other factors, has led us to accept reduced volume tiers under certain of our GPO contracts. We have seen solid demand growth for Gamunex-C/Gamunex IGIV with most customer segments. We believe that U.S. and international IGIV demand will grow approximately 5% to 8% over the long-term, which is consistent with demand growth during the year ended December 31, 2010. However, IGIV demand can vary significantly on a quarter-to-quarter basis.
Our ability to expand our international business has been hampered by the effects of our internal FCPA investigation, our reliance on the tender process for generating business and increased price sensitivities of our customers. We expect to complete our internal FCPA investigation and present our findings to the Department of Justice (DOJ) in 2011. The preliminary findings of our investigation indicate that it is probable that there were FCPA violations by persons associated with us that the DOJ or other regulators may assert are attributable to us. Even though we self-disclosed this matter to the DOJ, it or other federal agencies may seek to impose sanctions on us that may include, among other things, disbarment, injunctive relief, disgorgement, fines, penalties, appointment of a monitor, appointment of a new control staff or enhancements of existing compliance and training programs. Any such sanctions or related loss of business could have a material adverse effect on us or our results of operations. It is possible, however, that any sanctions that DOJ or other federal agencies might otherwise consider imposing would be reduced, if not eliminated, in light of the comprehensive compliance measures that we have implemented. Given the preliminary nature of our findings, our continuing investigation and the uncertainties regarding this matter, we are unable to estimate the financial outcome. See “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Matters Affecting Comparability—Foreign Corrupt Practices Act (FCPA)” and “Risk Factors—Risks Related to Our Business—We are investigating potential Foreign Corrupt Practices Act violations” for further discussion. Our business with a distributor in Iran, one of our major international customers, has been in decline, which is likely to continue. Our profitability has been, and may continue to be, negatively impacted by unfavorable euro/U.S. dollar exchange rates.
We are the primary supplier of Canadian IGIV under our five year contracts with CBS and Hema Quebec, which became effective April 1, 2008. These five year contracts provide for escalated pricing, based on inflation, for contract fractionation services and our commercial products, including Gamunex-C, Plasbumin, and certain hyperimmune products, effective April 1 of each year throughout the term of the agreements. We have experienced, and expect to continue to experience, annual volume declines in Canada due to CBS’ objective to have multiple sources of supply, which has impacted and will continue to impact our overall IGIV growth. CBS may further reduce volumes to contract minimums and Hema Quebec may adopt a similar strategy. The combination of these factors, among others, may reduce our near term global IGIV growth, relative to our prior expectations.
We expect to operate at or near our fractionation capacity over the next few years depending upon the demand for our products, the availability of source plasma, the impact of variability in yield, the potential impact of inventory impairments, and normal production shut-downs, among other factors. We plan to utilize most of our available fractionation capacity in the near term, which may result in increased inventory levels in order to attempt to maintain pace with projected future growth in product demand, although we have not been successful in building excess finished goods inventories to date as a result of the factors previously mentioned. Consequently, any disruption in
meeting our fractionation and purification plans would most likely result in lower revenue, gross profit, net income, and operating cash flows as well as lower than planned growth given our fractionation and purification constraints. Our fractionation constraints would likely preclude us from participating in greater than estimated overall market demand or higher demand for Gamunex-C/Gamunex IGIV.
Cost of Goods Sold and Gross Profit
Our gross profit was $689.6 million and $632.1 million for the years ended December 31, 2010 and 2009, respectively, representing gross margins of 43.1% and 41.2%, respectively. In general, our gross margin and cost of goods sold are impacted by the volume and pricing of our finished products, our raw material costs, production mix, yield, inventory impairment provisions, and cycle times, as well as our production capacities and normal production shut-downs, and the timing and amount of release of finished product.
Our cost of goods sold was $912.0 million, or 56.9% of net revenue, for the year ended December 31, 2010, as compared to $901.1 million, or 58.8%, of net revenue for the year ended December 31, 2009. Our cost of goods sold benefited from lower TPR unabsorbed infrastructure and start-up costs of $37.4 million during the year ended December 31, 2010 as compared to the year ended December 31, 2009. Our costs of goods sold also benefited from lower costs of production of $16.9 million during 2010 primarily driven by production mix associated with the conversion to our next generation A1PI product, Prolastin-C A1PI, as well as source mix. Inventory impairment provisions, net of recoveries, were $60.6 million during the year ended December 31, 2010 as compared to $31.1 million during the year ended December 31, 2009, an increase of $29.5 million, primarily driven by higher provisions for raw material and work in process inventories mainly related to a Gamunex-C/Gamunex IGIV production issue. We believe that we have identified the cause of the issue and have implemented appropriate remediation steps. Non-capitalizable project spending during the year ended December 31, 2010 was $39.9 million as compared to $36.9 million during the prior year, driven by spending related to our new fractionation facility, Koate reengineering, and Thrombate III projects. We experienced higher costs of $32.6 million associated with increased volumes during the year ended December 31, 2010 as compared to the prior year.
The largest component of our cost of goods sold is the cost of source plasma, which represented greater than 50% of our cost of goods sold for each of the years presented. The overall cost of source plasma is impacted by the collection cost per liter, including donor fees, labor, soft goods, facility costs, testing, unabsorbed TPR infrastructure and start-up costs, the cost of plasma purchased from third parties and variability in protein yields, among other factors. Our internal cost per liter of plasma, including unabsorbed TPR infrastructure and start-up costs, decreased 5.9% for the year ended December 31, 2010, as compared to the prior year, primarily driven by lower unabsorbed TPR infrastructure and start-up costs and higher TPR plasma collections. Our acquisition cost per liter of third party plasma decreased 0.1% during the year ended December 31, 2010 as compared to the prior year. Our licensed centers collected approximately 69% of our plasma during 2010. Due to our long manufacturing cycle times, which range from 100 days to in excess of 400 days, the cost of plasma is not expensed through cost of goods sold until a significant period of time subsequent to its acquisition.
We believe we have substantially eliminated unabsorbed TPR infrastructure and start-up costs. Consequently, future margin improvements will need to be derived from increases in product pricing and volumes, product mix, improvements in the cost per liter of plasma, manufacturing efficiencies, yield improvements or some combination thereof. We believe that we have limited opportunities to increase price and product mix. We have recently experienced and expect to continue to experience higher costs of goods sold due to yield variability, inventory impairment provisions, less efficient utilization of each incremental liter of plasma fractionated as we increase Gamunex-C/Gamunex IGIV production, and higher non-capitalizable costs associated with our capital projects, particularly the construction of our new fractionation facility.
The combination of the factors mentioned above, particularly competitive pressures, slower than planned reductions in our cost per liter of plasma, the potential impact of inventory impairment provisions, and yield variability as well as inefficient plasma utilization, among other factors, will most likely result in lower gross margins in future periods.
Operating Expenses
Our SG&A was $287.0 million and $289.9 million for the years ended December 31, 2010 and 2009, respectively. The lower SG&A was driven by lower share-based compensation expense of $28.4 million, the absence of retention expenses (excluding fringe benefit) and legal fees related to our terminated merger agreement with CSL of $11.0 million, lower donations of $9.0 million, lower legal fees of $3.0 million related to our internal FCPA investigation, and the absence of $5.7 million of management fees to Talecris Holdings LLC as a result of the termination of the management agreement at the time of our initial public offering. The benefit of the aforementioned items was partially offset by $25.9 million in Grifols merger-related expenses and higher sales and marketing expenses of $23.3 million during 2010 as a result of our sales force expansion, marketing of our CIDP indication, Prolastin patient identification efforts, launch of Prolastin-C A1PI, as well as support for other products. In addition, our SG&A for the year ended December 31, 2010 included $4.7 million of unfavorable foreign exchange transactions as compared to $1.9 million of favorable foreign exchange transactions in the prior year.
We are in the process of expanding our sales forces to increase support for a number of our products both in the U.S. and internationally. We estimate that this sales force expansion and related sales and marketing expenses will increase SG&A by approximately $10 million in 2011.
SG&A in future periods will be impacted by additional Grifols merger-related transaction expenses such as legal and accounting costs as well as retention expenses as discussed further in the section titled “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Matters Affecting Comparability—Definitive Merger Agreement with Grifols,” included elsewhere in this Annual Report.
Our R&D was $69.6 million and $71.2 million for the years ended December 31, 2010 and 2009, respectively. As a percentage of net revenue, R&D was 4.3% and 4.6% for the years ended December 31, 2010 and 2009, respectively. R&D expenses are influenced by the timing of in-process projects and the nature and extent of expenses associated with these projects. Additional information regarding our R&D projects is included in “Business—Research and Development,” included elsewhere in this Annual Report. The decrease in R&D year over year was primarily driven by the timing of our Prolastin Alpha-1 aerosol trial and program, partially offset by increased Plasmin clinical trials expense related to aPAO and ischemic stroke. We anticipate that R&D will increase in subsequent periods primarily as a result of clinical trial activities related to Plasmin aPAO and ischemic stroke, and research related to recombinant product development. Additional information regarding R&D expenses by significant project is included in “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Critical Accounting Policies and Estimates—Research and Development,” included elsewhere in this Annual Report. Our focus on plasma-derived plasmin trials for aPAO and ischemic stroke, recombinant product develop and the assessment of new development candidates will likely increase R&D in the range of $10 million to $15 million in 2011.
Our operating margin improved 310 basis points to 20.8% for the year ended December 31, 2010 as compared to 17.7% for the year ended December 31, 2009, for the reasons described above.
Total Other Non-Operating Expenses, net
The primary recurring component of our other non-operating expense, net, is interest expense, which amounted to $47.3 million and $56.9 million for the years ended December 31, 2010 and 2009, respectively. The reduction in interest expense was driven by lower weighted average debt levels during 2010. Our weighted average annualized interest rates on our outstanding debt, excluding amortization of deferred debt issuance costs and debt discount, were 7.75% and 3.40% for the years ended December 31, 2010 and 2009, respectively.
Our total other non-operating expense, net, for the year ended December 31, 2010 also includes a $43.7 million charge, including accrued interest, related to a judgment in favor of PCA against TPR in a breach of contract claim. The judgment related to the PCA litigation is discussed further in the section titled “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Matters Affecting Comparability—Plasma Centers of America (PCA) Judgment.”
As a result of our IPO and refinancing transactions, we recognized a charge during 2009 of $12.1 million to write-off previously deferred debt issuance costs related to our First and Second Lien Term Loans and $30.9 million related to costs associated with the settlement and termination of our interest rate swap contracts. Additional information regarding our IPO and refinancing transactions is included in the section entitled, “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Matters Affecting Comparability—Financial Impact of IPO and Refinancing Transactions.”
Our total other non-operating expense, net, for the year ended December 31, 2009 also includes $75.0 million of non-operating income related to the CSL merger termination fee as discussed in the section entitled, “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Matters Affecting Comparability—Definitive Merger Agreement with CSL Limited (CSL).”
Provision for Income Taxes
Our income tax provisions were $78.4 million and $75.0 million for the years ended December 31, 2010 and 2009, respectively, resulting in effective income tax rates of 32.1% and 32.8%, respectively.
For the year ended December 31, 2010, our effective income tax rate was lower than the U.S. statutory Federal income tax rate of 35% primarily due to credits for Federal Research and Experimentation, orphan drug clinical testing expenditures and qualified production activities. Our tax rates were increased due to state income taxes and non-deductable transaction costs.
For the year ended December 31, 2009, our effective income tax rate was lower than the U.S. statutory Federal income tax rate primarily due to credits for Federal Research and Experimentation and orphan drug clinical testing expenditures and the deduction of previously capitalized transaction costs related to our terminated merger transaction with CSL, offset by state income taxes.
We have not provided for U.S. Federal income and foreign withholding taxes on our non-U.S. subsidiaries’ cumulative undistributed earnings of approximately $13.2 million as of December 31, 2010 as such earnings are intended to be reinvested outside of the U.S. indefinitely. It is not practicable to estimate the amount of tax that might be payable if some or all of such earnings were to be remitted, and foreign tax credits would be available to reduce or eliminate the resulting U.S. income tax liability.
At December 31, 2010, we had state tax credit carryforwards of $5.1 million that will start expiring in 2015. Our ability to offset future taxable income with tax credit carryforwards may be limited in certain circumstances, including changes in ownership.
As of December 31, 2010, our total gross unrecognized tax benefits were approximately $8.8 million, of which approximately $5.8 million would reduce our effective income tax rate if recognized. The IRS exam of the 2005, 2006, and 2007 tax years are effectively settled as the Joint Committee on Taxation has completed its review of our position and has agreed not to take exception to the refund approved by the Internal Revenue Service in its report dated June 2009. The favorable resolution of this matter resulted in a release of $4.7 million gross unrecognized tax benefits and a decrease in 2010 income tax expense of $4.9 million.
Net Income
Our net income was $166.1 million and $153.9 million for the years ended December 31, 2010 and 2009, respectively. The significant factors and events contributing to the increase in our net income are discussed above.
Year Ended December 31, 2009 as Compared to Year Ended December 31, 2008
The following table contains information regarding our results of operations for the year ended December 31, 2009 as compared to the year ended December 31, 2008:
|
|
|
Years Ended December 31, |
|
Change |
| |||||||
|
|
|
2009 |
|
2008 |
|
$ |
|
% |
| |||
|
Net revenue: |
|
|
|
|
|
|
|
|
| |||
|
Product |
|
$ |
1,507,754 |
|
$ |
1,334,550 |
|
$ |
173,204 |
|
13.0 |
% |
|
Other |
|
25,455 |
|
39,742 |
|
(14,287 |
) |
(35.9 |
)% | |||
|
Total |
|
1,533,209 |
|
1,374,292 |
|
158,917 |
|
11.6 |
% | |||
|
Cost of goods sold |
|
901,077 |
|
882,157 |
|
(18,920 |
) |
(2.1 |
)% | |||
|
|
|
|
|
|
|
|
|
|
| |||
|
Gross profit |
|
632,132 |
|
492,135 |
|
139,997 |
|
28.4 |
% | |||
|
Operating expenses: |
|
|
|
|
|
|
|
|
| |||
|
SG&A |
|
289,929 |
|
227,524 |
|
(62,405 |
) |
(27.4 |
)% | |||
|
R&D |
|
71,223 |
|
66,006 |
|
(5,217 |
) |
(7.9 |
)% | |||
|
Total |
|
361,152 |
|
293,530 |
|
(67,622 |
) |
(23.0 |
)% | |||
|
|
|
|
|
|
|
|
|
|
| |||
|
Income from operations |
|
270,980 |
|
198,605 |
|
72,375 |
|
36.4 |
% | |||
|
Other non-operating (expense) income: |
|
|
|
|
|
|
|
|
| |||
|
Interest expense, net |
|
(74,491 |
) |
(96,640 |
) |
22,149 |
|
22.9 |
% | |||
|
CSL merger termination fee |
|
75,000 |
|
— |
|
75,000 |
|
nm |
| |||
|
Loss on extinguishment of debt |
|
(43,033 |
) |
— |
|
(43,033 |
) |
nm |
| |||
|
Equity in earnings of affiliate |
|
441 |
|
426 |
|
15 |
|
3.5 |
% | |||
|
Total |
|
(42,083 |
) |
(96,214 |
) |
54,131 |
|
56.3 |
% | |||
|
|
|
|
|
|
|
|
|
|
| |||
|
Income before income taxes |
|
228,897 |
|
102,391 |
|
126,506 |
|
123.6 |
% | |||
|
Provision for income taxes |
|
(75,008 |
) |
(36,594 |
) |
(38,414 |
) |
(105.0 |
)% | |||
|
|
|
|
|
|
|
|
|
|
| |||
|
Net income |
|
$ |
153,889 |
|
$ |
65,797 |
|
$ |
88,092 |
|
133.9 |
% |
|
|
|
|
|
|
|
|
|
|
| |||
|
Earnings per common share: |
|
|
|
|
|
|
|
|
| |||
|
Basic |
|
$ |
4.56 |
|
$ |
39.01 |
|
$ |
(34.45 |
) |
(88.3 |
)% |
|
Diluted |
|
$ |
1.50 |
|
$ |
0.71 |
|
$ |
0.79 |
|
111.3 |
% |
|
|
|
|
|
|
|
|
|
|
| |||
|
Financial measures: |
|
|
|
|
|
|
|
|
| |||
|
Gross profit margin |
|
41.2 |
% |
35.8 |
% |
|
|
|
| |||
|
Operating margin |
|
17.7 |
% |
14.5 |
% |
|
|
|
| |||
|
Effective income tax rate |
|
32.8 |
% |
35.7 |
% |
|
|
|
| |||
nm- not meaningful
Net Revenue
The following table contains information regarding our net revenue:
|
|
|
Years Ended December 31, |
|
Change |
| |||||||
|
|
|
2009 |
|
2008 |
|
$ |
|
% |
| |||
|
Product net revenue: |
|
|
|
|
|
|
|
|
| |||
|
Gamunex |
|
$ |
826,376 |
|
$ |
677,737 |
|
$ |
148,639 |
|
21.9 |
% |
|
Prolastin A1PI |
|
319,080 |
|
316,495 |
|
2,585 |
|
0.8 |
% | |||
|
Fraction V (Albumin and Plasmanate) |
|
84,770 |
|
61,075 |
|
23,695 |
|
38.8 |
% | |||
|
Fraction VIII (Koate DVI) |
|
46,453 |
|
40,247 |
|
6,206 |
|
15.4 |
% | |||
|
Hyperimmunes |
|
74,203 |
|
78,178 |
|
(3,975 |
) |
(5.1 |
)% | |||
|
Other |
|
156,872 |
|
160,818 |
|
(3,946 |
) |
(2.5 |
)% | |||
|
Total product net revenue |
|
1,507,754 |
|
1,334,550 |
|
173,204 |
|
13.0 |
% | |||
|
Other net revenue |
|
25,455 |
|
39,742 |
|
(14,287 |
) |
(35.9 |
)% | |||
|
Total net revenue |
|
$ |
1,533,209 |
|
$ |
1,374,292 |
|
$ |
158,917 |
|
11.6 |
% |
|
|
|
|
|
|
|
|
|
|
| |||
|
U.S. product net revenue: |
|
|
|
|
|
|
|
|
| |||
|
Gamunex |
|
$ |
599,758 |
|
$ |
479,895 |
|
$ |
119,863 |
|
25.0 |
% |
|
Prolastin A1PI |
|
206,099 |
|
202,678 |
|
3,421 |
|
1.7 |
% | |||
|
Fraction V (Albumin and Plasmanate) |
|
44,768 |
|
38,701 |
|
6,067 |
|
15.7 |
% | |||
|
Fraction VIII (Koate DVI) |
|
13,601 |
|
8,574 |
|
5,027 |
|
58.6 |
% | |||
|
Hyperimmunes |
|
59,500 |
|
60,707 |
|
(1,207 |
) |
(2.0 |
)% | |||
|
Other |
|
64,404 |
|
77,378 |
|
(12,974 |
) |
(16.8 |
)% | |||
|
Total U.S. product net revenue |
|
$ |
988,130 |
|
$ |
867,933 |
|
$ |
120,197 |
|
13.8 |
% |
|
|
|
|
|
|
|
|
|
|
| |||
|
International product net revenue: |
|
|
|
|
|
|
|
|
| |||
|
Gamunex |
|
$ |
226,618 |
|
$ |
197,842 |
|
$ |
28,776 |
|
14.5 |
% |
|
Prolastin A1PI |
|
112,981 |
|
113,817 |
|
(836 |
) |
(0.7 |
)% | |||
|
Fraction V (Albumin and Plasmanate) |
|
40,002 |
|
22,374 |
|
17,628 |
|
78.8 |
% | |||
|
Fraction VIII (Koate DVI) |
|
32,852 |
|
31,673 |
|
1,179 |
|
3.7 |
% | |||
|
Hyperimmunes |
|
14,703 |
|
17,471 |
|
(2,768 |
) |
(15.8 |
)% | |||
|
Other |
|
92,468 |
|
83,440 |
|
9,028 |
|
10.8 |
% | |||
|
Total international product net revenue |
|
$ |
519,624 |
|
$ |
466,617 |
|
$ |
53,007 |
|
11.4 |
% |
Our product net revenue was $1,507.8 million and $1,334.6 million for the years ended December 31, 2009 and 2008, respectively, representing an increase of $173.2 million, or 13.0%. The increase consisted of higher volumes of $126.8 million and improved pricing of $46.4 million, net of the effects of unfavorable foreign exchange of $8.0 million.
Our other net revenue, which consists of royalties and licensing fees, milestones, and revenues related to contracted services performed for third parties at our Melville facility, decreased $14.3 million. Our other net revenue for the year ended December 31, 2008 included the recognition of $1.9 million of previously deferred revenue as a result of the termination of a licensed technology agreement with an unaffiliated third party and $2.6 million of a previously deferred upfront licensing fee as a result of the completion of a portion of our performance obligations under a licensed technology agreement with an unaffiliated third party. Our other net revenue for the year ended December 31, 2009 was negatively impacted by lower royalties and licensing fees of $0.7 million and lower Melville contracted services revenue of $7.3 million, as compared to 2008.
The $148.6 million increase in our Gamunex product net revenue consisted of higher volumes of $117.0 million and improved pricing of $31.6 million, net of the effects of unfavorable foreign exchange of $1.7 million. We experienced higher Gamunex volumes of $121.4 million in the United States, Europe, and other international regions, which were partially offset by lower volumes of $4.4 million in Canada. We experienced improved Gamunex pricing of $37.5 million in the United States and Canada, which was partially offset by lower pricing of $5.9 million in Europe and other international regions, including $1.7 million of unfavorable foreign exchange transactions.
In 2009, we experienced a significant increase in demand for Gamunex, driven primarily by supply availability, growth in our GPO and Specialty Pharmacy/Homecare business in the United States, and geographic expansion. As a result of the success of our plasma collection platform, as well as our plasma supply contract with CSL, we began to alleviate our plasma supply constraints in the second half of 2008, bringing significant additional IGIV volumes to the market, to meet the pent-up demand for Gamunex. We believe that this pent-up demand has largely been satisfied, and consequently, we would not expect to experience the same high level of accelerated IGIV volume growth that we experienced in the second half of 2008 and the full year 2009, which will affect our comparative growth in sales and margins in future periods. We expect that our volume growth rate will moderate substantially and effectively grow with the market (excluding Canada). The supply of IGIV inventory increased throughout the distribution channel as supply became available from the previous low levels.
The $2.6 million increase in our Prolastin product net revenue consisted of higher volumes of $3.7 million, partially offset by
lower pricing of $1.1 million. Our Prolastin net revenue for the year ended December 31, 2009 was negatively impacted by unfavorable foreign exchange of $6.1 million which offset price increases of $5.5 million in Europe. In 2009, we also experienced pricing adjustments of $3.4 million in Canada related to a pricing dispute. Prolastin volumes are largely a function of our ability to identify and enroll new patients as compared to the number of patients lost due to attrition and competition. Our ability to grow our European volumes will also depend upon our ability to obtain appropriate reimbursement on a country by country basis. We anticipate the launch of our next generation A1PI product, Prolastin-C, in the U.S. and Canada in the first half of 2010. We received FDA approval for our next generation A1PI product, Prolastin-C, in October 2009 and received Health Canada approval in February 2010. Additional clinical trials are being required by the European authorities as a precursor to Prolastin-C A1PI approval in Europe.
Our Fraction V product category consists of albumin and Plasmanate, with albumin representing the majority of sales in the category. The $23.7 million increase in our Fraction V product net revenue consisted of higher volumes of $19.5 million and improved pricing of $4.2 million. The increase in Fraction V volume was primarily driven by sales in the U.S. and other international regions (excluding Canada and Europe). The increase in Fraction V pricing was primarily driven by favorable pricing in other international regions (excluding Canada and Europe). Fraction V volumes during the year ended December 31, 2008 were negatively impacted by a change in production mix to contracted PPF powder from Fraction V as a result of the settlement of a customer dispute, which occurred during 2007. This change in production mix resulted in lower quantities of Fraction V available for sale during the year ended December 31, 2008.
Our other product net revenue consists primarily of revenue related to the Canadian blood system, where in addition to commercial sales of Gamunex, we have contract manufacturing contracts with the two national Canadian blood system operators, CBS and Hema Quebec, as well as sales of Koate DVI Factor VIII (human), hyperimmunes, intermediate products, Thrombate III (human) and contracted PPF powder.
Our other product net revenue was favorably impacted by improved sales of $8.6 million related to intermediate products, such as cryoprecipitate, and increased sales of Koate DVI Factor VIII (human) of $6.2 million as a result of higher volumes in the U.S. and improved pricing in the U.S. and other international regions (excluding Canada and Europe). Our other product net revenue was negatively impacted by lower contracted PPF powder sales of $14.3 million during the year ended December 31, 2009 as compared to the prior year. During 2008, we experienced higher contracted PPF powder sales as a result of the settlement of a customer dispute as previously discussed. During the year ended December 31, 2009, we recorded a sales adjustment of $3.2 million related to our terminated Bayer European distribution agreement, which we recorded as a reduction of other net product revenue.
We increased prices for several of our products in most of our geographic regions during 2009 as a result of higher costs and generally increasing demand. Our product net revenue was negatively impacted by $8.0 million, or 0.6%, as a result of unfavorable foreign exchange rate fluctuations in relation to the U.S. dollar during the year ended December 31, 2009 as compared to the prior year.
As a result of our internal investigation related to potential FCPA violations, we suspended shipments to affected countries while we put additional safeguards in place. We also terminated several consultants and suspended relations with or terminated some distributors in countries under investigation as circumstances warranted. These actions resulted in a decline in revenue from these countries during 2009. We resumed shipments to several countries during the fourth quarter of 2009.
Cost of Goods Sold
Our gross profit was $632.1 million and $492.1 million for the years ended December 31, 2009 and December 31, 2008, respectively, representing gross margin of 41.2% and 35.8%, respectively. In general, our gross margin and cost of goods sold are impacted by the volume and pricing of our finished products, our raw material costs, production mix, yield, and cycle times, as well as our production capacities and normal production shut-downs, and the timing and amount of release of finished product. The net impact of these items resulted in higher gross margin during the year ended December 31, 2009 as compared to the prior year.
Our cost of goods sold was $901.1 million, or 58.8% of net revenue, for the year ended December 31, 2009, as compared to $882.2 million, or 64.2% of net revenue, for the year ended December 31, 2008. The decrease in our cost of goods sold as a percentage of net revenue during 2009 was primarily attributable to lower TPR unabsorbed infrastructure and start-up costs of $54.5 million and lower inventory impairment provisions of $5.0 million. The beneficial effects of the foregoing were offset by higher costs of production and costs associated with an increase in production volumes, which aggregated $78.4 million. Due to the relatively fixed nature of our factory overhead and certain other production costs, we experienced operating leverage as production increased during 2009.
Our cost of goods sold for the year ended December 31, 2009 includes higher costs of production of $14.7 million, including foreign exchange, and higher costs associated with an increase in volumes of $52.8 million. The impact of foreign exchange in our cost of production for the year ended December 31, 2009 is not material. During 2009, we incurred non-capitalizable project and start-
up costs of $36.9 million, an increase of $10.9 million, as compared to the prior year, related to capital projects. The largest component of our cost of goods sold is the cost of source plasma, which represented greater than 50% of our cost of goods sold in both 2009 and 2008. The overall cost of source plasma is impacted by the fully-loaded collection cost per liter, including donor fees, labor, soft goods, facility costs, testing and unabsorbed TPR infrastructure and start-up costs, the cost of plasma purchased from third-parties and variability in protein yields, among other factors. Our internal cost per liter of plasma, including unabsorbed TPR infrastructure and start-up costs, declined by 21.4% during the year ended December 31, 2009, primarily driven by lower unabsorbed infrastructure and start-up costs. Our acquisition cost of plasma per liter of third party plasma increased slightly by 0.30% during the year ended December 31, 2009 as compared to the year ended December 31, 2008. We fractionated approximately 3.6 million liters of plasma during 2009, of which approximately 62% came from plasma collection centers we own and approximately 38% came from third-party plasma supply contracts. Due to our long manufacturing cycle times, which range from 100 days to in excess of 400 days, the cost of plasma is not expensed through cost of goods sold until a significant period of time subsequent to its acquisition.
Unabsorbed TPR infrastructure and start-up costs amounted to $44.0 million and $98.5 million for the years ended December 31, 2009 and 2008, respectively, representing approximately 2.9% and 7.2%, respectively, of our net revenue. Our cost of goods sold during 2009 benefited from lower unabsorbed TPR infrastructure and start-up costs, which resulted from higher plasma volumes collected at our plasma collection centers and improved labor efficiencies as well as lower consulting and management support costs. Unabsorbed TPR infrastructure and start-up costs during the year ended December 31, 2008 were negatively impacted by costs associated with remediation efforts at certain plasma collection centers.
Our inventory impairment provisions, net of recoveries, decreased $5.0 million during the year ended December 31, 2009 as compared to the prior year. During the year ended December 31, 2008, we recorded a net provision of $5.8 million due to the plasma center cGMP issue described in “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Matters Affecting Comparability—Plasma Center cGMP issue.” During the year ended December 31, 2009, we recorded recoveries of $1.9 million related to this issue. During the years ended December 31, 2009 and 2008, we recorded net recoveries of $0.8 million and $7.1 million related to a 2007 customer settlement. During the year ended December 31, 2008, we recorded an impairment charge of $3.6 million primarily within cost of goods sold related to capital lease assets and leasehold improvements at certain of our plasma collection centers which were closed or were under development and we no longer plan to open. During the year ended December 31, 2008, we also recorded a loss of $3.4 million within cost of goods sold related to two lease commitments associated with properties that we no longer plan to operate as plasma collection centers. During the year ended December 31, 2009, we experienced an incident in the restart of our manufacturing facility located in Melville, New York, following a scheduled maintenance shut-down, which resulted in an inventory impairment provision of $3.4 million. In addition, during 2009, we recorded provisions of $4.2 million related to short-dated finished goods inventories. During the year ended December 31, 2009, we experienced lower work-in-process inventory impairment provisions of $6.9 million as compared to the prior year.
We expect unabsorbed TPR infrastructure and start-up costs to decrease substantially in 2010, primarily due to increased collections as well as cost reductions. This benefit will be partially offset by increased cost of goods sold due to yield variability, less efficient utilization of each incremental liter of plasma fractionated as we increase Gamunex production, and uncapitalizable costs associated with our capital projects, particularly the construction of our new fractionation facility.
Operating Expenses
Our SG&A was $289.9 million and $227.5 million for the years ended December 31, 2009 and 2008, respectively, representing an increase of $62.4 million, or 27.4%. As a percentage of net revenue, SG&A was 18.9% and 16.6% for the years ended December 31, 2009 and 2008, respectively. Our share-based compensation expense recorded in SG&A increased $7.2 million during 2009 as compared to the prior year, driven primarily by the acceleration of the vesting of certain of our Chairman and Chief Executive Officer’s stock options. Our SG&A included legal expenses of $6.0 million and $8.3 million and retention expenses, excluding fringe benefits, of $5.0 million and $3.0 million related to our terminated merger agreement with CSL for the years ended December 31, 2009 and 2008 respectively. Our SG&A for the year ended December 31, 2009 was negatively impacted by $8.0 million of legal costs associated with our internal investigation into potential violations of the FCPA. We experienced higher sales and marketing expenses during 2009 as a result of costs associated with our Gamunex CIDP indication, Prolastin patient identification, as well as support for other products. We also experienced higher charitable donations of $14.9 million during the year ended December 31, 2009 as compared to the prior year. Our provision for uncollectible receivables and advances was $2.1 million higher during 2008 as compared to 2009 as a result of non-cash charges related to outstanding notes receivable and other advances made to one of our plasma suppliers due to uncertainty regarding collection. In order to grow revenues through leveraging our Gamunex brand and our CIDP indication, as well as our focus on A1PI patient identification, we began a significant expansion in the size of our U.S. sales force in the third quarter of 2009. We are also increasing our sales and marketing organization in Europe as well as in other international regions. We expect the sales force expansion to result in an annual increase in SG&A of approximately $10.0 million. Additionally, we expect that our share-based compensation expense will decline subsequent to the first quarter of 2010 when substantially all grants and awards under our 2005 Stock Option and Incentive Plan, our 2006 Restricted Stock Plan and our Special Recognition Bonus Plan are fully vested. Additional information regarding certain items impacting the comparability of our results of
operations is included in “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Matters Affecting Comparability.”
Our R&D was $71.2 million and $66.0 million for the years ended December 31, 2009 and 2008, respectively, representing an increase of $5.2 million, or 7.9%. As a percentage of net revenue, R&D was 4.6% and 4.8% for the years ended December 31, 2009 and 2008, respectively. R&D expenses are influenced by the timing of in-process projects and the nature and extent of expenses associated with these projects. The increase in R&D year over year is primarily attributable to increased spending during 2009 related to our Plasmin and recombinant Plasmin new product candidates, partially offset by lower spending attributable to our Prolastin-C A1PI and Gamunex subcutaneous administration life cycle management projects. Additional information regarding our R&D projects is included in “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Research and Development” and “Business—Research and Development.” We anticipate that R&D will increase in subsequent periods primarily as a result of increased Plasmin clinical trials related to aPAO and ischemic stroke as well as increased spending related to our development of recombinant A1PI and Factor VIII.
Total Other Non-Operating Expense, net
Our other non-operating expense, net, includes interest expense related to our indebtedness, which amounted to $56.9 million and $85.7 million for the years ended December 31, 2009 and 2008, respectively. The weighted average annualized interest rates on our outstanding indebtedness, excluding amortization of deferred debt issuance costs and debt discount, were 3.4% and 7.2% for the years ended December 31, 2009 and 2008, respectively. The benefit of the lower cost of borrowings during 2009 was partially mitigated by higher interest expense of $3.9 million related to our interest rate swaps as compared to 2008, as a result of falling three-month LIBOR as compared to our fixed interest rate swaps. The interest rate swap contracts were settled and terminated as discussed below.
As a result of our IPO and refinancing transactions, we recognized a charge during 2009 of $12.1 million to write-off previously deferred debt issuance costs related to our First and Second Lien Term Loans and $30.9 million related to costs associated with the settlement and termination of our interest rate swap contracts. Additional information regarding our IPO and refinancing transactions is included in “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Matters Affecting Comparability—Financial Impact of IPO and Refinancing Transactions.”
Our total other non-operating expense, net, for the year ended December 31, 2009 also includes $75.0 million of non-operating income related to the merger termination fee as discussed in “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Matters Affecting Comparability—Definitive Merger Agreement with CSL Limited (CSL).”
Provision for Income Taxes
Our income tax provision was $75.0 million and $36.6 million for the years ended December 31, 2009 and 2008, respectively, resulting in effective income tax rates of 32.8% and 35.7%, respectively. Our effective income tax rates differed from the U.S. statutory Federal income tax rate of 35% during each year due to the items discussed in the following paragraphs.
For the year ended December 31, 2009, our effective income tax rate is lower than the U.S. statutory Federal income tax rate primarily due to credits for Federal Research and Experimentation and Orphan Drug clinical testing expenditures and the deduction of previously capitalized transaction costs related to the terminated merger agreement with CSL.
For the year ended December 31, 2008, our effective income tax rate is higher than the U.S. statutory Federal income tax rate primarily because the benefit of the credits for Federal Research and Experimentation and Orphan Drug clinical testing expenditures aggregating $4.1 million offset by the effect of capitalizing transaction costs related to the CSL merger agreement, which was terminated in 2009.
At December 31, 2009, our gross unrecognized tax benefits were approximately $12.2 million, of which approximately $8.8 million would reduce our effective income tax rate if recognized.
We have not provided for U.S. Federal income and foreign withholding taxes on our non-U.S. subsidiaries’ cumulative undistributed earnings of approximately $9.7 million as of December 31, 2009 as such earnings are intended to be reinvested outside of the U.S. indefinitely. It is not practicable to estimate the amount of tax that might be payable if some or all of such earnings were to be remitted, and foreign tax credits would be available to reduce or eliminate the resulting U.S. income tax liability.
Net Income
Our net income was $153.9 million and $65.8 million for the years ended December 31, 2009 and 2008, respectively. Our
net income for the year ended December 31, 2009 reflects the impact of the CSL merger termination fee of $48.8 million, net of $26.2 million income tax effect, as well as the refinancing charges of $26.3 million, net of $16.7 million income tax effect. The significant factors and events contributing to the change in our net income are discussed above.
LIQUIDITY AND CAPITAL RESOURCES
Cash Flow Analysis
The following table and subsequent discussion and analysis contain information regarding our cash flows:
|
|
|
Years Ended December 31, |
| |||||||
|
|
|
2010 |
|
2009 |
|
2008 |
| |||
|
Operating activities: |
|
|
|
|
|
|
| |||
|
Net income |
|
$ |
166,068 |
|
$ |
153,889 |
|
$ |
65,797 |
|
|
Non-cash items |
|
61,888 |
|
92,375 |
|
67,272 |
| |||
|
Changes in operating assets and liabilities, excluding the effects of business acquisitions |
|
27,526 |
|
(12,109 |
) |
(100,055 |
) | |||
|
Net cash provided by operating activities |
|
$ |
255,482 |
|
$ |
234,155 |
|
$ |
33,014 |
|
|
|
|
|
|
|
|
|
| |||
|
Investing activities: |
|
|
|
|
|
|
| |||
|
Purchases of property, plant, and equipment |
|
$ |
(152,849 |
) |
$ |
(75,163 |
) |
$ |
(86,212 |
) |
|
Financing arrangements with third party suppliers, net of repayments |
|
— |
|
— |
|
(16,335 |
) | |||
|
Business acquisitions, net of cash acquired |
|
— |
|
(30,431 |
) |
(10,272 |
) | |||
|
Other |
|
765 |
|
976 |
|
880 |
| |||
|
Net cash used in investing activities |
|
$ |
(152,084 |
) |
$ |
(104,618 |
) |
$ |
(111,939 |
) |
|
|
|
|
|
|
|
|
| |||
|
Financing activities: |
|
|
|
|
|
|
| |||
|
(Repayments) borrowings under revolving credit facility, net |
|
$ |
— |
|
$ |
(179,941 |
) |
$ |
66,904 |
|
|
Repayments of borrowings under term loans |
|
— |
|
(1,016,000 |
) |
(7,000 |
) | |||
|
Repayments of capital lease obligations |
|
(751 |
) |
(574 |
) |
(1,192 |
) | |||
|
Proceeds from issuance of 7.75% Notes |
|
— |
|
600,000 |
|
— |
| |||
|
Discount on 7.75% Notes |
|
— |
|
(4,074 |
) |
— |
| |||
|
Financing transaction costs |
|
(394 |
) |
(14,879 |
) |
— |
| |||
|
Proceeds from initial public offering, net of issuance costs |
|
— |
|
519,749 |
|
— |
| |||
|
Costs related to initial public offering |
|
— |
|
(2,557 |
) |
— |
| |||
|
Repurchases of common stock |
|
(4,917 |
) |
(4,183 |
) |
(36,118 |
) | |||
|
Proceeds from exercises of stock options |
|
22,333 |
|
7,581 |
|
— |
| |||
|
Excess tax benefits from share-based payment arrangements |
|
13,481 |
|
13,406 |
|
— |
| |||
|
Net cash provided by (used in) financing activities |
|
$ |
29,752 |
|
$ |
(81,472 |
) |
$ |
22,594 |
|
|
|
|
|
|
|
|
|
| |||
|
Cash and cash equivalents (at end of year) |
|
$ |
197,876 |
|
$ |
65,239 |
|
$ |
16,979 |
|
We use our available cash balances to repay amounts outstanding under our revolving credit facility. We deposit any excess cash amounts into an overnight investment account. At December 31, 2010, we had $322.6 million of unused available borrowing capacity under our revolving credit facility.
We have financed our operations through a combination of equity funding and debt financing, and through internally generated funds. We expect our cash flows from operations combined with our cash balances and availability of our revolving credit facility to provide sufficient liquidity to fund our current obligations, projected working capital requirements, and capital expenditures for at least the next twelve months.
Cash Flows from Operating Activities
During the years ended December 31, 2010, 2009, and 2008, our net income was $166.1 million, $153.9 million, and $65.8 million, respectively. Our net income for the year ended December 31, 2010 includes a non-cash charge of $43.7 million (approximately $26.6 million after tax) related to the PCA judgment. Our net income for the year ended December 31, 2009 benefited from the $75.0 million (approximately $48.8 million after tax) payment we received from CSL as a result of the termination of the definitive merger agreement. The benefit of the CSL merger termination fee was partially offset by charges totaling $43.0 million (approximately $26.3 million after tax) as a result of the settlement and termination of our interest rate swap contracts and the write-off of deferred debt issuance costs associated with our First and Second Lien Term Loans. Additional information regarding our net income for the years presented is included in “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Results of Operations.”
Our non-cash operating items were $61.9 million, $92.4 million, and $67.3 million for the years ended December 31, 2010, 2009, and 2008, respectively. The following significant non-cash items impacted the comparability of the net cash provided by our operating activities during the years presented.
· Our depreciation and amortization expense for the years ended December 31, 2010, 2009, and 2008 was $36.0 million, $28.9 million, and $20.3 million, respectively. The increase in depreciation and amortization expense during the years presented reflects our cumulative capital investments primarily related to our manufacturing facilities and TPR.
· Our share-based compensation expense for the years ended December 31, 2010, 2009, and 2008 was $17.0 million, $47.5 million, and $38.7 million, respectively. The decrease in share-based compensation during 2010 was primarily driven by the final vesting of awards under the 2005 Stock Option and Incentive Plan on April 1, 2010 and the majority of the awards under the 2006 Restricted Stock Plan on March 31, 2010. In addition, a combination of estimate-to-actual true ups as these plans culminated during 2010 and the acceleration of certain options to our Chairman and Chief Executive Officer in 2009 further impacted the comparability of our share-based compensation expense between the years presented. The increase in share-based compensation during 2009 also resulted from the incremental expense associated with RSU and stock option awards granted in connection with our initial public offering on October 1, 2009.
· During the year ended December 31, 2009, we recognized a non-cash charge of $12.1 million related to the write-off of unamortized debt issuance costs associated with our First and Second Lien Term Loans as discussed in “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Matters Affecting Comparability—Financial Impact of IPO and Refinancing Transactions.”
· During the year ended December 31, 2008, we recognized previously deferred revenue of $4.8 million as discussed in “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Results of Operations.” No significant amounts were recognized during the years ended December 31, 2010 and 2009.
· During the year ended December 31, 2010, our deferred tax assets decreased $12.3 million, driven primarily by the effects of stock option exercises. During the year ended December 31, 2009, our deferred tax assets decreased $15.4 million, of which $1.2 million is included in operating activities and $14.2 million is included in non-cash financing activities related to the reclassification of the unrealized losses associated with our interest rate swap contracts to earnings upon their settlement and termination. During the year ended December 31, 2008, our deferred tax assets increased $5.5 million. Additional information regarding our net deferred tax assets is included in the footnotes to our audited consolidated financial statements included elsewhere in this Annual Report.
· Amortization of deferred compensation related to special recognition bonus awards was $2.0 million, $5.7 million, and $5.9 million for the years ended December 31, 2010, 2009, and 2008, respectively. We made the final special recognition bonus award payments during March 2010.
· During the years ended December 31, 2010 and 2009, we recognized excess tax benefits related to share-based compensation of $13.5 million and $13.4 million, respectively.
Our operating assets (excluding the effects of business acquisitions), net, decreased (increased) $27.5 million, $(12.1) million, and $(100.1) million for the years ended December 31, 2010, 2009, and 2008, respectively, and were driven by the following items.
· Our accounts receivable, net, (increased) decreased $(1.4) million, $8.6 million, and $(26.9) million for the years ended December 31, 2010, 2009, and 2008, respectively. Accounts receivable, net, balances are influenced by the timing of net revenue and customer collections. Our days sales outstanding (DSO) were 30 days, 32 days, and 34 days for the years ended December 31, 2010, 2009, and 2008, respectively. Our international sales terms generally range from 30 to 150 days due to industry and national practices outside of the U.S., which can impact our DSO results. We calculate DSO as our period end accounts receivable, net, divided by our prior three months’ net sales, multiplied by 90 days. Our calculation of DSO may not be consistent with similar calculations performed by other companies.
· Our inventories increased $52.0 million, $57.5 million, and $92.9 million for the years ended December 31, 2010, 2009, and 2008, respectively. Our inventories fluctuate based upon our plasma collections, production mix and cycle times, production capacities, normal production shut-downs, finished product releases, targeted safety stock levels, and demand for our products. Our biological manufacturing processes result in relatively long inventory cycle times ranging from 100 days to in excess of 400 days in addition to a required 60 day pre-production holding period for plasma. Specialty plasma, due to its nature, can often have cycle times in excess of one year. Consequently, we have significant investment in raw material and work-in-process inventories for extended periods. The increase in our inventories during 2010 was primarily driven by increased plasma collections as well as higher work in progress and finished goods inventories. Work in progress and finished goods inventories at December 31, 2009 were lower than at December 31, 2010 as a result of our Clayton turn around during 2009. The increase in our inventories during 2009 was primarily driven by Thrombate III inventory build in preparation of manufacturing transfer from Bayer, increased plasma collections as compared to the prior year, and higher hyperimmune inventory levels. During 2008, we repurchased inventories with a value of approximately $28.6 million from a Bayer affiliate in Germany, where we terminated our distribution agreement.
· Our prepaid expenses and other assets decreased (increased) $0.7 million, $8.0 million, and $(15.8) million for the years ended December 31, 2010, 2009, and 2008, respectively. The decrease in our prepaid expenses and other assets during 2009 was primarily driven by a reclassification of $10.1 million of prepaid plasma to raw material inventories as a result of plasma deliveries from IBR upon center licensures, for which we subsequently acquired the centers. This was partially offset by higher corporate prepaid amounts, including insurance and various service contracts. The increase in our prepaid expenses and other assets during 2008 was primarily driven by a $9.7 million increase in prepaid plasma and a $7.8 million increase in prepaid income taxes, partially offset by lower corporate prepaid amounts. Under the terms of our 2007 Supply Agreement with IBR, we were required to prepay 90% for unlicensed plasma. Upon center licensure, we were required to remit the remaining 10% to IBR, resulting in a reclassification of the prepaid amounts to raw material inventories.
· Our operating liabilities increased $80.3 million, $28.8 million, and $35.5 million for the years ended December 31, 2010, 2009, and 2008, respectively. Our liabilities fluctuate as a result of the varying due dates of accounts payable, accrued expenses, and other obligations. The increase in our liabilities in 2010 was primarily driven by accrued charges of $43.7 million related to the PCA judgment discussed previously, higher accrued goods and services of $35.7 million, and higher accrued payroll, bonuses and employee benefits of $6.3 million, partially offset by lower interest payable of $3.1 million and lower accounts payable obligations of $11.1 million. The increase in our liabilities in 2009 was primarily driven by higher accounts payable obligations of $16.1 million and higher Medicaid, commercial rebates, and chargebacks of $14.2 million, partially offset by lower interest payable and accrued goods and services. The increase in our liabilities in 2008 was primarily driven by higher accounts payable obligations of $16.6 million, higher accrued payroll, bonuses, and benefits of $13.7 million, higher Medicaid, commercial rebates, and chargebacks of $2.2 million, and generally higher liabilities for accrued goods, services, and other items, partially offset by lower taxes payable of $10.6 million.
Cash Flows from Investing Activities
Our capital expenditures were $152.8 million, $75.2 million, and $86.2 million for the years ended December 31, 2010, 2009, and 2008, respectively. Our capital expenditures reflect investments in our facilities to support a platform for future growth and efficiency improvements, including compliance enhancements, general infrastructure upgrades, capacity expansions, and new facilities. Our capital expenditures also reflect investments in our TPR infrastructure to support our plasma collection efforts.
Our capital expenditures for the years ended December 31, 2010 and 2009 reflect significantly lower spending related to our TPR infrastructure as compared to 2008 as a result of the maturation of our plasma collection platform, as well as the completion of several projects. Our 2010 and 2009 capital expenditures reflect higher spending related to reliability/compliance initiatives, as well as the initial investments in our new fractionation strategic program. In addition, significant investment in 2008 continued into 2009 for the new Thrombate III purification facility, which was mechanically complete in 2009. Two earlier investments, our Prolastin-C A1PI facility and our Koate purification expansion Phase I project, were approved by the FDA during 2009. Additional information regarding our currently planned capital programs is included in “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Liquidity and Capital Resources—Sources of Credit, Access to Capital and Cash Requirements, and Credit Ratings.”
Our cash flows used in investing activities in 2009 and 2008 also include various cash outflows for the development of our plasma collection center platform, including the purchase price of plasma collection centers acquired from IBR and loans and advances made to third-party plasma suppliers, net of repayments, for the development of plasma collection centers for which we had the option to purchase under certain conditions. We completed the acquisition of twelve plasma centers during 2009 as compared to three centers in 2008. The IBR center acquisition program was completed as of December 31, 2009.
Cash Flows from Financing Activities
We completed our IPO on October 6, 2009, which resulted in net proceeds to us of $519.7 million after deducting underwriters’ discounts and commissions. We used the net proceeds to us from the IPO to repay $389.8 million and $129.9 million of principal under our First and Second Lien Term Loans, respectively. We incurred legal and other costs related to our IPO of approximately $3.9 million, of which $2.6 million is included as a reduction of additional paid-in capital. On October 21, 2009, we completed a $600.0 million private placement of our 7.75% Notes at an issue price of 99.321% of par, which resulted in net proceeds to us of $583.9 million after deducting underwriters’ commissions and the discount. We used a portion of the net proceeds to us from the issuance of the 7.75% Notes to repay $290.9 million and $200.1 million under our First and Second Lien Term Loans, respectively, and $55.6 million of principal under our revolving credit facility. We incurred total debt issuance costs related to the issuance of the 7.75% Notes and the revolving credit facility amendment of $14.9 million. In addition to the term loan principal payments resulting from the application of net proceeds from our IPO and the 7.75% Notes issuance, we made contractual principal payments under our First Lien Term Loan during 2009 and 2008 as indicated in the table above. Outstanding amounts under our revolving credit facility fluctuate based upon our business needs.
During the year ended December 31, 2010, we received proceeds of $22.3 million from the exercise of 3,650,579 stock options. In addition, we withheld 246,823 shares of our common stock from employees for $4.9 million to settle their withholding tax obligations. During the year ended December 31, 2010, we recognized excess tax benefits related to share-based compensation of $13.5 million. During the year ended December 31, 2009, we received proceeds of $7.6 million from the exercise of 2,394,762 stock options. In addition, we withheld 251,108 shares of our common stock from employees for $4.2 million to settle their withholding tax obligations. During the year ended December 31, 2009, we recognized excess tax benefits related to share-based compensation of $13.4 million. During the year ended December 31, 2008, we repurchased 2,146,232 shares of our common stock from IBR for $33.5 million, plus accrued interest of $1.9 million, and 69,648 shares of our common stock from employees for $0.7 million to settle their withholding tax obligations.
Sources of Credit, Access to Capital and Cash Requirements, and Credit Ratings
Sources of Credit
Our sources of credit as of December 31, 2010 are summarized in the following table:
|
|
|
|
|
Reductions in |
|
|
|
|
| ||||
|
|
|
|
|
Available Credit |
|
|
|
|
| ||||
|
|
|
|
|
Facility for |
|
|
|
|
| ||||
|
|
|
Maximum |
|
Other |
|
|
| ||||||
|
|
|
Amounts |
|
Financial |
|
Amounts |
|
Amounts |
| ||||
|
Debt Instrument |
|
Available |
|
Instruments (1) |
|
Outstanding |
|
Available |
| ||||
|
7.75% Notes |
|
$ |
600,000 |
|
$ |
— |
|
$ |
600,000 |
|
$ |
— |
|
|
Revolving credit facility |
|
325,000 |
|
2,431 |
|
— |
|
322,569 |
| ||||
|
Total sources of credit |
|
$ |
925,000 |
|
$ |
2,431 |
|
$ |
600,000 |
|
$ |
322,569 |
|
(1) Amounts represent letters of credit used as security for utilities, insurance, and third party warehousing.
7.75% Unsecured Senior Notes, due November 15, 2016
On October 21, 2009, we completed the issuance of $600.0 million, 7.75% Senior Unsecured Notes, due November 15, 2016, at a price of 99.321% of par, in a private placement to certain qualified institutional buyers. The 7.75% Notes yield 7.875% to maturity and pay interest semi-annually on May 15 and November 15 to holders of record on the immediately preceding May 1 and November 1, respectively. The 7.75% Notes are guaranteed on a senior unsecured basis by our existing and future domestic subsidiaries. Except as described below, we will not be entitled to redeem the 7.75% Notes at our option prior to November 12, 2012.
We may redeem some or all of the 7.75% Notes, at our option, at any time on or after November 12, 2012, at the redemption prices (expressed as percentages of principal amount) set forth below plus accrued and unpaid interest and additional interest, if any, on the 7.75% Notes redeemed, to the applicable redemption date, if redeemed during the twelve-month period beginning on November 15 of the years indicated below:
|
Fiscal Year |
|
Percentage |
|
|
2012 |
|
103.875 |
% |
|
2013 |
|
102.583 |
% |
|
2014 |
|
101.292 |
% |
|
2015 and thereafter |
|
100.000 |
% |
In addition, at any time during each twelve-month period ending on November 15, 2010, 2011, and 2012, we may redeem up to 10% of the originally issued principal amount of the 7.75% Notes at a redemption price of 103% of the principal amount of the 7.75% Notes redeemed plus accrued and unpaid interest and additional interest, if any, to the redemption date, subject to the rights of the holders of the 7.75% Notes on the relevant record date to receive interest due on the relevant interest payment date. No principal amounts were redeemed during 2010.
At any time, or from time to time, on or prior to November 15, 2012, we may, at our option, redeem up to 35% of the aggregate principal amount of the 7.75% Notes issued under the indenture with the net cash proceeds to us of certain equity offerings at a redemption price equal to 107.75% of the principal amount of the 7.75% Notes plus accrued and unpaid interest and additional interest, if any, to the applicable redemption date, provided that at least 65% of the aggregate principal amount of the 7.75% Notes originally issued remains outstanding immediately after such redemption and the redemption occurs within 90 days of the date of the closing of such equity offering.
Under the Make-Whole redemption feature, we may redeem 100% of the principal plus a premium as defined under the indenture (computed using a discount rate equal to the U.S. Treasury rate as of such redemption date plus 0.50%), plus accrued and unpaid interest and additional interest, if any, prior to November 15, 2012, with respect to some or all of the 7.75% Notes, subject to the rights of the holders on the relevant record date to receive interest due on the relevant interest payment date.
We are not required to make mandatory redemption or sinking fund payments with respect to the 7.75% Notes.
Upon a change of control, the 7.75% Notes are puttable at 101% of principal plus accrued and unpaid interest and additional interest, if any.
We may incur additional indebtedness and our subsidiary guarantors may also incur additional indebtedness if our Fixed Charge Coverage Ratio for our most recently ended four full fiscal quarters immediately preceding the date on which such additional indebtedness is incurred would have been at least 2.00 to 1.00, determined on a pro forma basis.
The indenture contains certain covenants limiting, subject to exceptions, carve-outs and qualifications, our ability and our restricted subsidiaries to: (i) sell assets; (ii) pay distributions on, redeem or repurchase its capital stock or redeem or repurchase its subordinated debt; (iii) make certain investments; (iv) incur or guarantee additional indebtedness or issue preferred stock; (v) create or incur certain liens; (vi) enter into agreements that restrict distributions or other payments from our restricted subsidiaries to us; (vii) engage in certain sale and leaseback transactions; (viii) engage in certain transactions with affiliates; (ix) transfer or dispose of the capital stock of the restricted subsidiary to persons other than us or our restricted subsidiaries; and (x) create unrestricted subsidiaries. The indenture also contains certain customary events of default.
On July 19, 2010, we exchanged all of our then outstanding 7.75% Notes for similar 7.75% Notes that were registered under the Securities Act. This exchange did not impact our capitalization.
Revolving Credit Facility
We have a $325.0 million asset-based credit agreement administered by Wachovia Bank, N.A., an affiliate of Wells Fargo Securities, which was amended on October 15, 2009 as described below. We use our available cash balances to repay amounts outstanding under this revolving credit facility. We deposit any excess cash amounts into an overnight investment account. Outstanding principal under this facility is due and payable on the maturity date of December 6, 2011.
Borrowings under this facility bear interest at a rate based upon either ABR or LIBOR, at our option, plus applicable margins based upon borrowing availability. The ABR represents the greater of the Federal Funds Effective Rate plus 0.50% or the Prime Rate. Interest accrues on the revolving credit facility at the ABR plus 0.25-0.75% or LIBOR plus 1.50-2.00%. For the years ended December 31, 2010, 2009, and 2008, the weighted average interest rates of our revolving credit facility were 3.50%, 2.79%, and 4.79%, respectively. No amounts were outstanding under the revolving credit facility at December 31, 2010 and 2009.
The revolving credit facility is secured by a Pledge and Security Agreement dated December 6, 2006 under which substantially all of our personal property, including manufacturing equipment, accounts receivable, inventory, and stock are pledged as security, each as defined within the agreement.
The revolving credit facility contains default provisions, and, pursuant to the October 15, 2009 amendment described below, imposes restrictions on annual capital expenditures if our leverage ratio is 2.00 to 1.00 or less, and contains a financial covenant which requires us to maintain a fixed charge coverage ratio of at least 1.10 to 1.00 if our borrowing availability based on eligible collateral is less than $48.75 million. The revolving credit facility defines certain terms in calculating covenant ratios, including adjusted EBITDA and Indebtedness.
The borrowing base under our revolving credit facility is based on our accounts receivable and inventory, and is calculated as (i) 85% of our eligible accounts receivable plus (ii) the lesser of (a) 65% of our eligible inventory (valued on a first-in-first-out basis), (b) 85% of the net orderly liquidation value of our eligible inventory as determined by a recent appraisal, and (c) $300 million. Only up to $100 million may be advanced to us based on the value of our work-in-process inventory (with “filled-not-packed” and “packed-not-released” inventory being considered finished goods inventory). From time to time, the collateral agent under the revolving credit facility may modify our eligibility standards, establish or adjust reserves, or reduce one or more of the other elements used in computing the borrowing base.
On October 15, 2009, we entered into an amendment to the revolving credit facility dated as of October 12, 2009. The revolving credit facility, as amended, permitted the 7.75% Notes, described above, to be issued as long as the First and Second Lien Term Loan Credit Agreements were terminated in connection with the offering of the 7.75% Notes. The amendment also (i) increases the covenant baskets for permitted acquisitions to $250 million, (ii) permits the payment of cash dividends commencing with the first fiscal quarter of 2010 if certain conditions are met, and (iii) increases our capital expenditure baskets so that we will be permitted to make capital expenditures of up to $225 million in each of 2010 and 2011. Moreover, pursuant to the amendments, we are not subject to any limitation on our capital expenditures in any fiscal year if our leverage ratio, as defined, as of the end of the fiscal year most recently ended was less than or equal to 2.00 to 1.00. Minimum availability tests under the revolving credit facility were also increased from $32.5 million to $48.75 million in connection with the amendment.
Our revolving credit facility, as amended, permits the payment of cash dividends to holders of our common stock commencing with the first fiscal quarter of 2010, so long as (i) the ratio determined as of the end of the immediately preceding fiscal quarter for the then most recently completed four fiscal quarters, is equal to or less than 2.00 to 1.00 and (ii) the minimum pro forma availability as of the date of such dividend (after giving effect to such cash dividend, the funding of all revolving loans, and the issuance of all letters of credit to be funded or issued as of such date) is not less than $48.75 million; provided that, the aggregate amount of restricted payments shall not exceed 50% of Net Income during the period from October 1, 2009 to the end of the most recently ended fiscal quarter as of the date of the restricted payment.
First and Second Lien Term Loans
Our First and Second Lien Term Loans were repaid in full and terminated as a result of the application of the net proceeds to us from our October 6, 2009 IPO and the issuance of our 7.75% Notes on October 21, 2009. The weighted average annualized interest rates on the First Lien Term Loan were 4.66% and 6.60% for the years ended December 31, 2009 and 2008, respectively, and the weighted average annualized interest rates on the Second Lien Term Loan were 7.68% and 9.63% for the years ended December 31, 2009 and 2008, respectively. At December 31, 2008, the interest rate on the First and Second Lien Term Loans were 5.64% and 8.64%, respectively.
Interest Rate Swaps and Caps
In October 2009, we used $28.7 million of the net proceeds to us from the issuance of our 7.75% Notes to settle and terminate certain interest rate swap contracts with a notional amount of $390.0 million. Subsequently, we settled and terminated our remaining interest rate swap contract with a notional amount of $50.0 million for $6.1 million. As a result of the settlement and termination of these interest rate swap contracts, we recognized a charge of $30.9 million (approximately $18.9 million after tax) during the year ended December 31, 2009 within total other non-operating expense, net, in our consolidated income statement. At December 31, 2008, $23.3 million, net of taxes, was recorded in accumulated other comprehensive loss, related to our interest rate swap contracts. As a result of their settlement and termination, we reclassified $23.3 million out of accumulated other comprehensive loss to loss on extinguishment of debt within total other non-operating expense, net, in our consolidated income statement for the year ended December 31, 2009.
At December 31, 2009, we had two interest rate cap contracts with a notional principal amount of $175.0 million outstanding for which the cap rate of 6.00% was significantly higher than prevailing market interest rates; therefore, the fair market value was zero. The interest rate caps matured during February 2010.
Access to Capital and Cash Requirements
At December 31, 2010, our cash and cash equivalents totaled $197.9 million. We use our available cash balances to repay amounts outstanding under our revolving credit facility. We deposit any excess cash amounts into an overnight investment account. At December 31, 2010, no amounts were outstanding under our revolving credit facility and we had unused available borrowing capacity of $322.6 million.
We expect our cash flows from operations combined with our cash balances to provide sufficient liquidity to fund our current obligations, projected working capital requirements, and capital expenditures for at least the next twelve months. As of the date of this Annual Report, we believe that we are currently in compliance with all covenants or other requirements set forth in our credit facilities.
As noted above, our current revolving credit facility ends on December 6, 2011. We believe that we will be able to negotiate and enter into a new revolving credit facility with terms and conditions similar to or better than our current facility given our improved credit profile. A new revolving credit facility would require approval of Grifols under the terms of our definitive merger agreement which would impact the timing of our negotiation of a new credit agreement, if any.
Our working capital, which is driven primarily by our accounts receivable turnover and inventory production times, including plant turnarounds, can vary significantly period to period. Our capital requirements will depend on many factors, including our rate of sales growth, acceptance of our products, costs of securing access to adequate manufacturing capacities, maintaining cGMP compliant facilities, the timing and extent of research and development activities, and changes in operating expenses, including costs of production and sourcing of plasma, all of which are subject to uncertainty. We anticipate that our cash needs will be significant and that we may need to increase our borrowings under our credit facilities in order to fund our operations and strategic initiatives. We anticipate that our working capital will increase in order to grow our business. We expect to operate at or near our fractionation capacity over the next few years depending upon the demand for our products, the availability of source plasma, the impact of variability in yield, the potential impact of inventory impairments and normal production shut-downs, among other factors. We plan to utilize most of our available fractionation capacity in the near term, which may result in increased inventory levels in order to attempt to maintain pace with projected future growth in product demand, although we have not been successful in building excess finished goods inventories to date as a result of the factors previously mentioned. Consequently, any disruption in meeting our fractionation and purification plans would most likely result in lower revenue, gross profit, net income and operating cash flows.
We incurred significantly higher capital spending in 2010 compared to 2009 and 2008 and anticipate significantly higher capital spending in 2011. We expect that our cumulative capital spending from 2011 through 2015 to be in the range of $750 million to $800 million,excluding capitalized interest. Given the nature of our planned capital projects, we anticipate our capital spending to peak in a range of $250 million to $270 million in 2011, excluding capitalized interest. Most of the anticipated capital spending will be necessary to support our future volume growth, particularly with the anticipation that we will reach our fractionation capacity in the near term, launch new products, and complete strategic initiatives. Incremental capital spending will continue to be required to ensure ongoing maintenance and compliance of our facilities.
The amount and timing of future capital spending is dependent upon a number of factors, including market conditions, regulatory requirements, and the extent and timing of particular projects, among other things. Our most significant capital project is the construction of our new fractionation facility, which we estimate will cost approximately $340 million, excluding capitalized interest. Through December 31, 2010, our capital expenditures on this project were approximately $90 million, with estimated additional capital expenditures of $250 million to be incurred, excluding capitalized interest. We anticipate our new fractionation facility will be operational by 2015. The construction costs of new purification facilities for Plasmin are estimated at approximately $120 million; with additional expenditures planned for Koate modernization and albumin purification expansion and projects to support continued new product development, among others. To the extent we discontinue a capital project, we would write-off a portion or all previously capitalized amounts through a charge to our consolidated income statement. The first phase of our Factor VIII purification expansion project, as well as reliability improvements, have been completed. In addition, we have completed construction of our new Thrombate III purification facility as well as our new Prolastin-C A1PI purification facility.
On an ongoing basis, we expect to evaluate our capital spending. We may need to incur future debt or issue additional equity if our cash flows and capital resources are insufficient to finance these various activities. Additional funds may not be available on favorable terms to us, or at all.
Credit Ratings
Our credit ratings at December 31, 2010 were as follows:
|
|
|
|
|
Standard & |
|
|
|
|
Moody’s |
|
Poor’s |
|
|
7.75% Notes |
|
B1 |
|
BB |
|
|
Corporate Family Rating |
|
Ba3 |
|
BB |
|
Factors that can affect our credit ratings include changes in our operating performance, financial position, business strategy, and the overall economic environment for the plasma-derived products business. If a downgrade of our credit ratings were to occur, it could adversely impact, among other things, our future borrowing costs and access to capital markets.
CONTRACTUAL OBLIGATIONS
The following table summarizes our significant contractual obligations as of December 31, 2010:
|
|
|
Payments Due by Period |
| |||||||||||||
|
|
|
|
|
Less than |
|
|
|
|
|
More than |
| |||||
|
|
|
Total |
|
1 Year |
|
1-3 Years |
|
4-5 Years |
|
5 Years |
| |||||
|
Long-term debt (1) |
|
$ |
600,000 |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
600,000 |
|
|
Interest payments (2) |
|
279,000 |
|
46,500 |
|
139,500 |
|
93,000 |
|
— |
| |||||
|
Capital lease obligations |
|
13,916 |
|
1,814 |
|
5,528 |
|
3,508 |
|
3,066 |
| |||||
|
Operating lease obligations |
|
85,422 |
|
17,427 |
|
33,781 |
|
12,304 |
|
21,910 |
| |||||
|
Purchase commitments (3)(4) |
|
853,345 |
|
201,875 |
|
474,567 |
|
176,903 |
|
— |
| |||||
|
Total |
|
$ |
1,831,683 |
|
$ |
267,616 |
|
$ |
653,376 |
|
$ |
285,715 |
|
$ |
624,976 |
|
(1) Long-term debt in the table above consists of outstanding amounts under our 7.75% Notes. We also have a $325.0 million revolving credit facility, maturing on December 6, 2011, for which no amounts were outstanding at December 31, 2010. The 7.75% Notes are redeemable or puttable prior to their scheduled maturity of November 15, 2016 under certain circumstances as described further in “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Liquidity and Capital Resources.”
(2) Interest payments related to long-term debt in the table above consist of interest amounts under our 7.75% Notes. We also have a $325.0 million variable rate revolving credit facility, for which no amounts were outstanding at December 31, 2010.
(3) Includes material agreements to purchase goods and services that are enforceable and legally binding.
(4) We entered into a contract, effective January 1, 2011, for subcontract manufacturing services that is substantially similar to, and replaces, one to which we were a party that expired on December 31, 2010. The minimum purchase obligations under this contract, which expires on December 31, 2012, of approximately $30 million for both 2011 and 2012 are not included in the table above.
On January 6, 2011, we entered into a contract for subcontract manufacturing services. The minimum purchase obligations of approximately $4.0 million for each of the years 2012 through 2015 are not included in the table above.
In addition to the contractual obligations disclosed in the table above, we have other contractual obligations for which the timing and extent of future payments are not known. We have described these potential obligations in the following paragraphs.
We have employment agreements and offer letters with certain of our employees which require payments generally ranging from 100% to 150% of the employee’s annual compensation if employment is terminated not for cause by us, or by the employee, for good reason, as defined. Certain of these arrangements also include provisions for payments of bonuses under our annual incentive plan and the vesting of equity awards, as well as other customary payments, such as accrued personal days, bonuses, continuing benefits, and outplacement services. In the event of a change of control of the Company, our share-based compensation plans permit accelerated vesting of awards under defined circumstances.
We have two exclusive commercial license agreements for advanced protein production technology with Crucell. In consideration of the licenses that Crucell has granted us, we paid upfront license fees of $4.0 million during 2008 and additional milestones of $1.5 million and $0.5 million during 2010 and 2009, respectively, and could be required to pay up to $46.5 million of additional development milestones as certain activities are completed. Under the terms of both agreements, we may terminate either agreement by giving Crucell 90 days prior written notice and payment of all outstanding amounts owed to Crucell. If products developed under these agreements are sold, we would be required to pay royalties to Crucell ranging from 3% to 5% of net sales from recombinant Factor VIII and from 3.5% to 6% of net sales from recombinant A1PI. We currently anticipate paying milestones of $2.0 million during 2011 under these agreements.
At December 31, 2010, $8.8 million of unrecognized tax benefits have been recorded as liabilities for uncertain income tax positions. The ultimate resolution of our uncertain income tax positions is dependent on uncontrollable factors such as law changes, new case law, and the willingness of the income tax authorities to settle, including the timing thereof, and other factors. Although we do not anticipate significant changes to our uncertain income tax positions in the next twelve months, items outside of our control could cause our uncertain income tax positions to change in the future.
As indicated elsewhere in this Annual Report, we have embarked on a substantial capital plan, which we anticipate to be in the range of $750 million to $800 million on a cumulative basis from 2011 through 2015, excluding capitalized interest. Given the nature of our planned capital projects, we anticipate our capital spending to peak in a range of $250 million to $270 million in 2011, excluding capitalized interest. Actual spending will vary based upon changes to the timing and scope of planned projects, including project deferral or acceleration, as well as new opportunities. At December 31, 2010, we had commitments and open purchase orders for capital spending of approximately $214.5 million which are not included in the table above.
We have entered into agreements with investment bankers related to our definitive merger agreement with Grifols. Under the terms of the agreement, we are obligated to pay fees totaling $21.3 million upon successful closing of the merger.
Under the terms of the definitive merger agreement with Grifols, we are permitted to offer retention amounts up to a total of $15.0 million to employees. As of December 31, 2010, we have offered retention amounts totaling approximately $10.2 million to employees, of which $2.9 million was paid during 2010 and the remaining amounts are expected to be paid in 2011, subject to the terms of the retention agreement.
OFF-BALANCE SHEET ARRANGEMENTS
As of December 31, 2010, we do not have any off-balance sheet arrangements that are material or reasonably likely to be material to our consolidated financial position or results of operations.
NON-GAAP FINANCIAL MEASURES
In the section titled “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Highlights,” we have presented adjusted net income and diluted earnings per share amounts. Both of these measures are considered to be non-GAAP financial measures. In calculating adjusted net income and diluted earnings per share, we have presented a quantification of the impact to our financial results in 2009 for the CSL merger termination fee, CSL merger-related expenses, and charges related to our debt refinancing and in 2010 for the PCA judgment and Grifols merger-related expenses. We believe that we have further enhanced the comparability of our financial results between the years presented by using an adjusted share base in the computation of diluted earnings per share, reflecting the impact for the issuance of common shares to convert our Series A and B preferred stock, settle accrued dividends on the preferred stock, and completion our IPO as if these events occurred at the beginning of 2009.
We believe that a meaningful analysis of our historical operating performance is also enhanced by the use of adjusted EBITDA, as defined in our revolving credit facility, and Consolidated Cash Flow, as defined in our 7.75% Notes. Both adjusted EBITDA and Consolidated Cash Flow are financial measures that are not defined by U.S. GAAP. A non-GAAP financial measure is a numerical measure of a company’s financial performance that (i) excludes amounts, or is subject to adjustments that have the effect of excluding amounts, that are included in a comparable measure calculated and presented in accordance with U.S. GAAP in the statement of operations, such as net income, or the statement of cash flows, such as operating cash flow; or (ii) includes amounts, or is subject to adjustments that have the effect of including amounts, that are excluded from the comparable measure so calculated and presented. The non-GAAP financial measures that we use should not be considered a substitute for any performance measure determined in accordance with U.S. GAAP. We do not rely solely on these non-GAAP financial measures and also consider our U.S. GAAP results. Because the non-GAAP financial measures that we use are not calculated in the same manner by all companies, they may not be comparable to similarly titled measures used by other companies. To properly and prudently evaluate our business, we encourage you to also review our U.S. GAAP consolidated financial statements included elsewhere in this Annual Report, and not to rely on any single financial measure to evaluate our business. These non-GAAP financial measures have material limitations as analytical tools and you should not consider these measures in isolation, or as a substitute for analysis of our results as reported under U.S. GAAP.
Adjusted EBITDA and Consolidated Cash Flow are used by our management, our lenders, and the compensation committee of our board of directors as follows:
· Our management uses adjusted EBITDA as one of our primary financial performance measures in the day to day oversight of our business to, among other things, allocate financial and human resources across our organization, determine appropriate levels of capital investment and research and development spending, determine staffing needs and develop hiring plans, manage our plants’ production plans, and assess appropriate levels of sales and marketing initiatives. Our management uses adjusted EBITDA in its decision making because this supplemental operating performance measure facilitates internal comparisons to historical operating results and external comparisons to competitors’ historical operating results by eliminating various income and expense items which are either not part of operating income or may vary significantly when comparing our results among the years presented to our competitors or other companies.
· The compensation committee of our board of directors uses adjusted EBITDA as a financial performance objective because it is one of the primary financial performance measures used in the day-to-day oversight of our business to, among other things, allocate financial and human resources across our organization, determine appropriate staffing needs and develop hiring plans, manage our plants’ production plans, and assess appropriate levels of sales and marketing initiatives. In 2010, the compensation committee used revenue and net income in addition to adjusted EBITDA. In 2009 and 2008, the compensation committee used unlevered free cash flow because it measures management’s effectiveness in managing cash and the related impact on interest expense. In order to motivate top performance by our executives, we establish a target level for each of the various performance criteria that is high enough that there is no certainty it is achievable. The target level for any performance criterion changes from year to year. These target performance levels reflect challenges with respect to various factors such as sales volume and pricing, cost control, working capital management, plasma platform objectives, R&D objectives, and sales and marketing objectives, among others. Our compensation committee has discretion to adjust the actual results related to the performance targets positively or negatively for items which, in the opinion of the compensation committee, were not reasonably within management’s control. Our compensation committee also evaluates the manner in which actual results were achieved to determine if unusual actions or risks were taken that would impact or manipulate the results.
· Our lenders use adjusted EBITDA to determine compliance with the Leverage Ratio financial covenant under our revolving credit facility, which is calculated as debt less cash divided by the last twelve months’ adjusted EBITDA. The October 15, 2009 amendment to our revolving credit facility suspends restrictions on our annual capital expenditures if our Leverage Ratio is greater than 2.00 to 1.00. Our 7.75% Notes use a similar measure referred to as Consolidated Cash Flow to determine compliance with the Fixed Charge Coverage Ratio, which allows for the incurrence of indebtedness and issuance of qualified and preferred stock if this ratio is at least 2.00 to 1.00.
Certain items that we eliminate in calculating adjusted EBITDA and Consolidated Cash Flow have been, and we expect will continue to be, significant to our business. For example:
· Interest expense is a necessary element of our costs and is largely a function of our capital structure and reflects our debt levels;
· Depreciation and amortization primarily result from the allocation of resources relative to investment decisions by our management and board of directors;
· Income tax expense results from our performance and applying statutory tax rates in the jurisdictions in which we operate coupled with the application of income tax accounting guidance and tax planning strategies;
· Non-cash compensation expense is expected to be a recurring component of our costs, although we expect that we will not grant share-based compensation in the same magnitude in the future;
· Expenses related to our special recognition bonuses are significant, and although we do not expect to grant bonuses in this magnitude in the future, bonuses will continue to be a key component of compensation to retain and attract employees; and
· Expenses related to debt extinguishment represent a necessary element of our costs to the extent that we restructure our debt;
Although we currently believe other items such as management fees, the PCA judgment, and merger-related expenses will not recur in the future in the same magnitude that they have occurred in the past, we may incur similar charges in the future. Other items, such as impairment charges, are not predictable, and therefore, we could incur similar charges in the future.
In the following table, we have presented a reconciliation of adjusted EBITDA and Consolidated Cash Flow to the most comparable U.S. GAAP measure, net income:
|
|
|
Years Ended December 31, |
| |||||||
|
|
|
2010 |
|
2009 |
|
2008 |
| |||
|
Net income |
|
$ |
166,068 |
|
$ |
153,889 |
|
$ |
65,797 |
|
|
Interest expense, net (a) |
|
45,837 |
|
74,491 |
|
97,040 |
| |||
|
Income tax provision (b) |
|
78,379 |
|
75,008 |
|
36,594 |
| |||
|
Depreciation and amortization (c) |
|
36,030 |
|
28,936 |
|
20,269 |
| |||
|
EBITDA |
|
326,314 |
|
332,324 |
|
219,700 |
| |||
|
Management fees (d) |
|
— |
|
5,715 |
|
6,871 |
| |||
|
Non-cash share-based compensation expense (e) |
|
16,966 |
|
47,546 |
|
38,707 |
| |||
|
Special recognition bonus expense (f) |
|
2,091 |
|
6,310 |
|
6,622 |
| |||
|
Loss on extinguishment of debt (g) |
|
— |
|
43,033 |
|
— |
| |||
|
Equity in earnings of affiliate (h) |
|
(991 |
) |
(441 |
) |
(426 |
) | |||
|
Merger-related expenses (i) |
|
27,730 |
|
9,136 |
|
5,593 |
| |||
|
PCA judgment (j) |
|
43,690 |
|
— |
|
— |
| |||
|
Other (k) |
|
1,383 |
|
3,660 |
|
10,749 |
| |||
|
Adjusted EBITDA under revolving credit facility |
|
417,183 |
|
447,283 |
|
287,816 |
| |||
|
Merger termination fee (l) |
|
— |
|
(75,000 |
) |
— |
| |||
|
Consolidated Cash Flow under 7.75% Notes |
|
$ |
417,183 |
|
$ |
372,283 |
|
$ |
287,816 |
|
(a) Represents interest expense associated with our debt structure. Through the third quarter of 2009, our debt structure consisted of facilities totaling $1.355 billion, including our $700 million First Lien Term Loan, $330 million Second Lien Term Loan, and $325 million revolving credit facility, as well as our interest rate cap and swap contracts. As a result of our IPO and refinancing transactions during October 2009, we reduced our credit facilities to $925 million, consisting of our $600 million 7.75% Notes and $325 million revolving credit facility. We also settled and terminated our interest rate swap contracts.
(b) Represents our income tax provision as presented in our consolidated income statements.
(c) Represents depreciation and amortization expense associated with our property, plant, and equipment, and all other intangible assets.
(d) Represents the advisory fees paid to Talecris Holdings, LLC, under the Management Agreement, as amended. This agreement was terminated in connection with our IPO.
(e) Represents our non-cash share-based compensation expense associated with stock options, restricted stock, RSU’s, and PSU’s.
(f) Represents compensation expense associated with special recognition bonus awards granted to certain of our employees and senior executives to reward past performance. We made the final payments under the special recognition bonus awards during March 2010. We do not anticipate granting similar awards in the future.
(g) Represents charges to write-off previously capitalized financing charges associated with our First and Second Lien Term Loans as a result of their repayment and termination as well as costs associated with the settlement and termination of our interest rate swap contracts.
(h) Represents non-operating income associated with our investment in Centric, which we believe are not part of our core operations.
(i) Represents merger related retention expenses associated with our terminated merger agreement with CSL for 2009 and 2008, and merger-related expenses associated with our merger agreement with Grifols, including investment bankers, legal, accounting, and other costs, as well as retention expenses for 2010.
(j) Represents charges related to a judgment in the amount of $37.0 million in favor of PCA against TPR in a breach of contract claim and interest of $6.7 million from the date of the breach.
(k) For the year ended December 31, 2010, the amount represents charges/losses related to long-lived assets, net. For the year ended December 31, 2009, the amount represents $3.1 million of charges related primarily to capital lease assets and leasehold improvements, offset by recoveries of $1.9 million related to our 2008 plasma center cGMP issue. For the year ended December 31, 2009, the amount also includes $1.3 million of costs related to our October 6, 2009 IPO and $1.2 million of losses on disposals of our property, plant, and equipment. For the year ended December 31, 2008, the amount represents an inventory impairment charge, net of recoveries of $5.8 million due to our plasma center cGMP issue, an impairment charge of $3.6 million related primarily to capital lease assets and leasehold improvements, and other long-lived asset impairment charges of $0.7 million. For the year ended December 31, 2008, the amount also includes $0.9 million of costs related to the initial public offering that was discontinued during 2008, partially offset by insurance recoveries of $0.3 million.
(l) For the year ended December 31, 2009, the amount includes a $75.0 million merger termination fee that we received from CSL in connection with our terminated definitive merger agreement.
In addition to the adjustments we make in computing adjusted EBITDA and Consolidated Cash Flow, our management and compensation committee also consider the impact of other items when evaluating our operating performance. Certain of these items, which impact the comparability of our historical financial results, are included in the section titled, “Management’s Discussion and Analysis of Financial Condition and Results of Operations——Matters Affecting Comparability.”
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
We operate on a global basis, and are exposed to the risk that our earnings and cash flows could be adversely impacted by fluctuations in interest rates, foreign exchange, and commodity prices. The overall objective of our financial risk management program is to minimize the impact of these risks through operational means and by using various financial instruments. These practices may change as economic conditions change. At December 31, 2010, we were not a party to any derivative financial instruments. During the first quarter of 2011, we initiated a foreign currency hedging program as described below.
INTEREST RATE RISK
At December 31, 2010, our long-term debt consisted of our 7.75% Notes ($600.0 million outstanding), which bear a fixed interest rate, and our $325.0 million revolving credit facility, which bears interest at a rate based upon either ABR or LIBOR, at our option, plus applicable margins based upon borrowing availability. Our exposure to adverse movements in ABR or LIBOR during 2010 was not significant as a result of minimal average borrowings outstanding under the revolving credit facility during 2010. Assuming a fully drawn revolving credit facility and a 100 basis point increase in applicable interest rates, our interest expense, net, would increase by $3.25 million on an annual basis.
At December 31, 2010, we had cash and cash equivalents of $197.9 million. We use our available cash balances to repay amounts outstanding under our revolving credit facility. We deposit any excess amounts into an overnight investment account, which earns minimal interest. Because our cash and cash equivalents are short-term in duration, we believe that our exposure to interest rate risk is not significant and a 100 basis point movement in market interest rates would not have a significant impact on the carrying value of our cash and cash equivalents. We actively monitor changes in interest rates.
FOREIGN CURRENCY RISK
We operate internationally and enter into transactions denominated in foreign currencies. As such, our financial position, results of operations, cash flows, and competitive position are subject to the variability that arises from exchange rate movements in relation to the U.S. dollar. Our foreign currency exposures are primarily limited to the impact that fluctuations in the euro and the Canadian dollar have on our net revenue and the remeasurement of our euro-denominated accounts receivable.
Approximately 31% of our net revenue for the year ended December 31, 2010 was generated outside of the United States. Foreign currency exchange rate fluctuations in relation to the U.S. dollar unfavorably impacted our net revenue by $6.9 million, or 0.4%, for the year ended December 31, 2010. In addition, we incurred transaction losses, net, of $4.7 million during the year ended December 31, 2010, primarily related to the remeasurement of euro-denominated accounts receivable, which losses we recorded within SG&A in our consolidated income statement.
For the purpose of specific risk analysis, we used a sensitivity analysis to measure the potential impact to our consolidated income statement for a hypothetical 10% strengthening of the U.S. dollar compared with the euro and Canadian dollar for the year ended December 31, 2010. Assuming a 10% strengthening of the U.S. dollar, our product net revenue would have been negatively impacted by approximately $17.2 million for the year ended December 31, 2010. At December 31, 2010, we had approximately €36.1 million in receivables. An adverse movement in the value of the euro in relation to the U.S. dollar could have a significant impact on our profitability. We did not hedge our exposures to changes in foreign currency exchange rates during the years presented in this Annual Report.
In order to reduce the impact of volatility of foreign exchange rates on intercompany transactions, we initiated a foreign currency hedging program in the first quarter of 2011 related to both known and anticipated intercompany transactions of approximately one year in duration or less. The effective portion of the changes in fair value of these instruments is reported in other comprehensive income and reclassified into earnings in the same period or periods in which the hedged transactions affect earnings.
The changes in fair value of the hedges against firm commitments will be recognized in general and administrative expenses consistent with the underlying intercompany receivables being hedged. The changes in the fair value hedges against anticipated intercompany sales will be recognized as an adjustment to revenues when the hedged inventory sells through to third parties.
During the first quarter of 2011 we entered in approximately $49.5 (€37.1) million in notional value of fair value hedges against firm commitments and $38.4 (€28.2) million in notional value of cash flow hedges against anticipated future sales. The weighted average U.S. dollar to euro exchange rate on these foreign currency contracts is 1.3461.
COMMODITY RISK
Plasma is the key raw material used in the production of our products, which we obtain from our plasma collection centers, as well as third party plasma suppliers. As of December 31, 2010, our plasma collection center platform consisted of 69 operating plasma collection centers, of which 67 were FDA licensed and two were unlicensed. Our licensed centers collected approximately 69% of our plasma during the year ended December 31, 2010.
For the purpose of specific risk analysis, we used a sensitivity analysis to measure the potential impact to our consolidated income statement for a hypothetical 10% increase in the cost of plasma used to produce the products sold during the year ended December 31, 2010. Assuming this 10% increase in the cost of plasma, our cost of goods sold would have increased by $54.3 million for the year ended December 31, 2010, and our gross margin would have been negatively impacted by approximately 340 basis points. This sensitivity analysis assumes that we would not be able to pass the hypothetical cost increase to our customers in the form of price increases. This sensitivity analysis does not consider the fixed pricing of plasma purchased from our third-party plasma suppliers.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Index to Financial Statements and Financial Statement Schedule
|
|
|
Page |
|
|
|
|
|
Report of Management on Internal Control Over Financial Reporting |
|
94 |
|
|
|
|
|
|
95 | |
|
|
|
|
|
Consolidated Financial Statements: |
|
|
|
|
|
|
|
|
96 | |
|
|
|
|
|
|
97 | |
|
|
|
|
|
|
98 | |
|
|
|
|
|
|
99 | |
|
|
|
|
|
|
100 | |
|
|
|
|
|
Financial Statement Schedule: |
|
|
|
|
|
|
|
|
140 |
REPORT OF MANAGEMENT ON INTERNAL CONTROL OVER FINANCIAL REPORTING
Our management is responsible for establishing and maintaining adequate internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act). Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the United States of America. The Company’s internal control over financial reporting includes those policies and procedures that: (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the Company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with accounting principles generally accepted in the United States of America, and that receipts and expenditures of the Company are being made only in accordance with authorizations of management and directors of the Company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the Company’s assets that could have a material effect on the financial statements.
Our management conducted an evaluation of the effectiveness of our internal control over financial reporting as of the end of the period covered by this report based on the framework in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on this evaluation, management concluded that the Company’s internal control over financial reporting was effective as of the end of the period covered by this report.
Our independent registered public accounting firm, which has audited the financial statements included in Part II, Item 8 of this report, has also audited the effectiveness of the Company’s internal control over financial reporting as of December 31, 2010, as stated in their report.
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and Board of Directors of
Talecris Biotherapeutics Holdings Corp:
In our opinion, the consolidated financial statements listed in the accompanying index present fairly, in all material respects, the financial position of Talecris Biotherapeutics Holdings Corp. and its subsidiaries at December 31, 2010 and 2009, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2010 in conformity with accounting principles generally accepted in the United States of America. In addition, in our opinion, the financial statement schedule listed in the accompanying index presents fairly, in all material respects, the information set forth therein when read in conjunction with the related consolidated financial statements. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2010, based on the criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company’s management is responsible for these financial statements and financial statement schedule, for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Report of Management on Internal Control over Financial Reporting appearing in Item 8. Our responsibility is to express opinions on these financial statements, on the financial statement schedule, and on the Company’s internal control over financial reporting based on our audits, which was an integrated audit in 2010. We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
|
/s/ PricewaterhouseCoopers LLP |
|
|
|
|
|
PricewaterhouseCoopers LLP |
|
|
Raleigh, North Carolina |
|
February 23, 2011
Talecris Biotherapeutics Holdings Corp.
(in thousands, except share and per share amounts)
|
|
|
December 31, |
| ||||
|
|
|
2010 |
|
2009 |
| ||
|
Assets |
|
|
|
|
| ||
|
|
|
|
|
|
| ||
|
Current assets: |
|
|
|
|
| ||
|
Cash and cash equivalents |
|
$ |
197,876 |
|
$ |
65,239 |
|
|
Accounts receivable, net of allowances of $3,253 and $3,461, respectively |
|
134,842 |
|
136,978 |
| ||
|
Inventories |
|
694,499 |
|
644,054 |
| ||
|
Deferred income taxes |
|
96,593 |
|
88,652 |
| ||
|
Prepaid expenses and other |
|
29,662 |
|
31,466 |
| ||
|
Total current assets |
|
1,153,472 |
|
966,389 |
| ||
|
Property, plant, and equipment, net |
|
382,793 |
|
267,199 |
| ||
|
Investment in affiliate |
|
2,926 |
|
1,935 |
| ||
|
Intangible assets |
|
10,880 |
|
10,880 |
| ||
|
Goodwill |
|
172,860 |
|
172,860 |
| ||
|
Deferred income taxes |
|
— |
|
5,848 |
| ||
|
Other |
|
15,522 |
|
19,894 |
| ||
|
Total assets |
|
$ |
1,738,453 |
|
$ |
1,445,005 |
|
|
|
|
|
|
|
| ||
|
Liabilities and Stockholders’ Equity |
|
|
|
|
| ||
|
|
|
|
|
|
| ||
|
Current liabilities: |
|
|
|
|
| ||
|
Accounts payable |
|
$ |
59,975 |
|
$ |
71,046 |
|
|
Accrued expenses and other liabilities |
|
251,726 |
|
170,533 |
| ||
|
Current portion of capital lease obligations |
|
860 |
|
740 |
| ||
|
Total current liabilities |
|
312,561 |
|
242,319 |
| ||
|
Long-term debt and capital lease obligations |
|
605,301 |
|
605,267 |
| ||
|
Deferred income taxes |
|
14,432 |
|
— |
| ||
|
Other |
|
11,795 |
|
15,265 |
| ||
|
Total liabilities |
|
944,089 |
|
862,851 |
| ||
|
Commitments and contingencies (Note 14) |
|
|
|
|
| ||
|
Stockholders’ equity: |
|
|
|
|
| ||
|
Common stock, $0.01 par value; 400,000,000 shares authorized; 125,577,877 and 122,173,274 shares issued and outstanding, respectively |
|
1,253 |
|
1,212 |
| ||
|
Additional paid-in capital |
|
813,783 |
|
767,032 |
| ||
|
Accumulated deficit |
|
(20,378 |
) |
(186,446 |
) | ||
|
Accumulated other comprehensive (loss) income, net of tax |
|
(294 |
) |
356 |
| ||
|
Total stockholders’ equity |
|
794,364 |
|
582,154 |
| ||
|
Total liabilities and stockholders’ equity |
|
$ |
1,738,453 |
|
$ |
1,445,005 |
|
The accompanying Notes to Consolidated Financial Statements are an integral part of these consolidated financial statements.
Talecris Biotherapeutics Holdings Corp.
Consolidated Income Statements
(in thousands, except per share amounts)
|
|
|
Years Ended December 31, |
| |||||||
|
|
|
2010 |
|
2009 |
|
2008 |
| |||
|
Net revenue: |
|
|
|
|
|
|
| |||
|
Product |
|
$ |
1,576,936 |
|
$ |
1,507,754 |
|
$ |
1,334,550 |
|
|
Other |
|
24,683 |
|
25,455 |
|
39,742 |
| |||
|
Total |
|
1,601,619 |
|
1,533,209 |
|
1,374,292 |
| |||
|
Cost of goods sold |
|
911,976 |
|
901,077 |
|
882,157 |
| |||
|
Gross profit |
|
689,643 |
|
632,132 |
|
492,135 |
| |||
|
Operating expenses: |
|
|
|
|
|
|
| |||
|
Selling, general, and administrative |
|
287,011 |
|
289,929 |
|
227,524 |
| |||
|
Research and development |
|
69,649 |
|
71,223 |
|
66,006 |
| |||
|
Total |
|
356,660 |
|
361,152 |
|
293,530 |
| |||
|
Income from operations |
|
332,983 |
|
270,980 |
|
198,605 |
| |||
|
Other non-operating (expense) income |
|
|
|
|
|
|
| |||
|
Interest expense, net |
|
(45,837 |
) |
(74,491 |
) |
(96,640 |
) | |||
|
PCA judgment |
|
(43,690 |
) |
— |
|
— |
| |||
|
CSL merger termination fee |
|
— |
|
75,000 |
|
— |
| |||
|
Loss on extinguishment of debt |
|
— |
|
(43,033 |
) |
— |
| |||
|
Equity in earnings of affiliate |
|
991 |
|
441 |
|
426 |
| |||
|
Total |
|
(88,536 |
) |
(42,083 |
) |
(96,214 |
) | |||
|
Income before income taxes |
|
244,447 |
|
228,897 |
|
102,391 |
| |||
|
Provision for income taxes |
|
(78,379 |
) |
(75,008 |
) |
(36,594 |
) | |||
|
Net income |
|
166,068 |
|
153,889 |
|
65,797 |
| |||
|
Less dividends to preferred stockholders and other non-common stockholders’ charges |
|
— |
|
(11,744 |
) |
(14,672 |
) | |||
|
Net income available to common stockholders |
|
$ |
166,068 |
|
$ |
142,145 |
|
$ |
51,125 |
|
|
|
|
|
|
|
|
|
| |||
|
Net income per common share: |
|
|
|
|
|
|
| |||
|
|
|
|
|
|
|
|
| |||
|
Basic |
|
$ |
1.35 |
|
$ |
4.56 |
|
$ |
39.01 |
|
|
|
|
|
|
|
|
|
| |||
|
Diluted |
|
$ |
1.29 |
|
$ |
1.50 |
|
$ |
0.71 |
|
The accompanying Notes to Consolidated Financial Statements are an integral part of these consolidated financial statements.
Talecris Biotherapeutics Holdings Corp.
Consolidated Statements of Cash Flows
(in thousands)
|
|
|
Years Ended December 31, |
| |||||||
|
|
|
2010 |
|
2009 |
|
2008 |
| |||
|
Cash flows from operating activities: |
|
|
|
|
|
|
| |||
|
Net income |
|
$ |
166,068 |
|
$ |
153,889 |
|
$ |
65,797 |
|
|
Adjustments to reconcile net income to net cash provided by operating activities: |
|
|
|
|
|
|
| |||
|
Depreciation and amortization |
|
36,030 |
|
28,936 |
|
20,269 |
| |||
|
Amortization of deferred loan fees and debt discount |
|
4,262 |
|
3,785 |
|
3,764 |
| |||
|
Share-based compensation expense |
|
16,966 |
|
47,546 |
|
38,707 |
| |||
|
Amortization of deferred compensation |
|
1,983 |
|
5,714 |
|
5,922 |
| |||
|
Write-off of unamortized debt issuance costs |
|
— |
|
12,141 |
|
— |
| |||
|
Asset impairment |
|
595 |
|
3,061 |
|
4,282 |
| |||
|
Provision for doubtful receivables and advances |
|
3,519 |
|
2,858 |
|
4,978 |
| |||
|
Recognition of previously deferred revenue |
|
(230 |
) |
(230 |
) |
(4,784 |
) | |||
|
Equity in earnings of affiliate |
|
(991 |
) |
(441 |
) |
(426 |
) | |||
|
Loss on disposal of property, plant, and equipment |
|
896 |
|
1,196 |
|
48 |
| |||
|
Decrease (increase) in deferred tax assets |
|
12,339 |
|
1,215 |
|
(5,488 |
) | |||
|
Excess tax benefits from share-based payment arrangements |
|
(13,481 |
) |
(13,406 |
) |
— |
| |||
|
Changes in assets and liabilities, excluding the effects of business acquisitions |
|
27,526 |
|
(12,109 |
) |
(100,055 |
) | |||
|
Net cash provided by operating activities |
|
255,482 |
|
234,155 |
|
33,014 |
| |||
|
|
|
|
|
|
|
|
| |||
|
Cash flows from investing activities: |
|
|
|
|
|
|
| |||
|
Purchases of property, plant, and equipment |
|
(152,849 |
) |
(75,163 |
) |
(86,212 |
) | |||
|
Business acquisitions, net of cash acquired |
|
— |
|
(30,431 |
) |
(10,272 |
) | |||
|
Financing arrangements with third party suppliers, net of repayments |
|
— |
|
— |
|
(16,335 |
) | |||
|
Other |
|
765 |
|
976 |
|
880 |
| |||
|
Net cash used in investing activities |
|
(152,084 |
) |
(104,618 |
) |
(111,939 |
) | |||
|
|
|
|
|
|
|
|
| |||
|
Cash flows from financing activities: |
|
|
|
|
|
|
| |||
|
Borrowings under revolving credit facility |
|
915 |
|
1,201,749 |
|
1,430,092 |
| |||
|
Repayments of borrowings under revolving credit facility |
|
(915 |
) |
(1,381,690 |
) |
(1,363,188 |
) | |||
|
Repayments of borrowings under term loans |
|
— |
|
(1,016,000 |
) |
(7,000 |
) | |||
|
Repayments of capital lease obligations |
|
(751 |
) |
(574 |
) |
(1,192 |
) | |||
|
Proceeds from issuance of 7.75% Notes |
|
— |
|
600,000 |
|
— |
| |||
|
Discount on 7.75% Notes |
|
— |
|
(4,074 |
) |
— |
| |||
|
Financing transaction costs |
|
(394 |
) |
(14,879 |
) |
— |
| |||
|
Proceeds from initial public offering, net of issuance costs |
|
— |
|
519,749 |
|
— |
| |||
|
Costs related to initial public offering |
|
— |
|
(2,557 |
) |
— |
| |||
|
Repurchases of common stock |
|
(4,917 |
) |
(4,183 |
) |
(36,118 |
) | |||
|
Proceeds from exercises of stock options |
|
22,333 |
|
7,581 |
|
— |
| |||
|
Excess tax benefits from share-based payment arrangements |
|
13,481 |
|
13,406 |
|
— |
| |||
|
Net cash provided by (used in) financing activities |
|
29,752 |
|
(81,472 |
) |
22,594 |
| |||
|
Effect of exchange rate changes on cash and cash equivalents |
|
(513 |
) |
195 |
|
(157 |
) | |||
|
Net increase (decrease) in cash and cash equivalents |
|
132,637 |
|
48,260 |
|
(56,488 |
) | |||
|
Cash and cash equivalents at beginning of year |
|
65,239 |
|
16,979 |
|
73,467 |
| |||
|
Cash and cash equivalents at end of year |
|
$ |
197,876 |
|
$ |
65,239 |
|
$ |
16,979 |
|
The accompanying Notes to Consolidated Financial Statements are an integral part of these consolidated financial statements.
Talecris Biotherapeutics Holdings Corp.
Consolidated Statements of Stockholders’ Equity (Deficit)
(in thousands, except share amounts)
|
|
|
|
|
|
|
|
|
|
|
Accumulated |
|
|
| |||||
|
|
|
|
|
|
|
Additional |
|
|
|
Other |
|
|
| |||||
|
|
|
Common Stock |
|
Paid-in |
|
Accumulated |
|
Comprehensive |
|
|
| |||||||
|
|
|
Shares |
|
Amount |
|
Capital |
|
Deficit |
|
Income (Loss) |
|
Total |
| |||||
|
Balance at December 31, 2007 |
|
5,317,232 |
|
$ |
— |
|
$ |
27,010 |
|
$ |
(406,132 |
) |
$ |
(11,635 |
) |
$ |
(390,757 |
) |
|
Net income |
|
— |
|
— |
|
— |
|
65,797 |
|
— |
|
65,797 |
| |||||
|
Other comprehensive loss |
|
— |
|
— |
|
— |
|
— |
|
(11,772 |
) |
(11,772 |
) | |||||
|
Comprehensive income |
|
— |
|
— |
|
— |
|
— |
|
— |
|
54,025 |
| |||||
|
Share-based compensation cost |
|
— |
|
— |
|
29,258 |
|
— |
|
— |
|
29,258 |
| |||||
|
Issuance of restricted stock |
|
42,720 |
|
— |
|
— |
|
— |
|
— |
|
— |
| |||||
|
Forfeitures of restricted stock |
|
(287,784 |
) |
— |
|
— |
|
— |
|
— |
|
— |
| |||||
|
Repurchases and retirement of common stock |
|
(2,215,880 |
) |
— |
|
— |
|
— |
|
— |
|
— |
| |||||
|
Fair value adjustment on common stock with put/call feature |
|
— |
|
— |
|
(8,942 |
) |
— |
|
— |
|
(8,942 |
) | |||||
|
Interest accretion on IBR put option |
|
— |
|
— |
|
(309 |
) |
— |
|
— |
|
(309 |
) | |||||
|
Balance at December 31, 2008 |
|
2,856,288 |
|
— |
|
47,017 |
|
(340,335 |
) |
(23,407 |
) |
(316,725 |
) | |||||
|
Net income |
|
— |
|
— |
|
— |
|
153,889 |
|
— |
|
153,889 |
| |||||
|
Other comprehensive income |
|
— |
|
— |
|
— |
|
— |
|
476 |
|
476 |
| |||||
|
Reclassification of unrealized loss on derivatives to earnings |
|
— |
|
— |
|
— |
|
— |
|
23,287 |
|
23,287 |
| |||||
|
Comprehensive income |
|
— |
|
— |
|
— |
|
— |
|
— |
|
177,652 |
| |||||
|
Share-based compensation cost |
|
— |
|
— |
|
39,206 |
|
— |
|
— |
|
39,206 |
| |||||
|
Issuance of restricted stock |
|
14,464 |
|
— |
|
— |
|
— |
|
— |
|
— |
| |||||
|
Forfeitures of restricted stock |
|
(16,368 |
) |
— |
|
— |
|
— |
|
— |
|
— |
| |||||
|
Repurchases and retirement of common stock |
|
(251,108 |
) |
— |
|
(51 |
) |
— |
|
— |
|
(51 |
) | |||||
|
Series A and B preferred stock dividends declared |
|
— |
|
— |
|
(45,250 |
) |
— |
|
— |
|
(45,250 |
) | |||||
|
Conversion of Series A and B preferred stock to common stock |
|
88,227,868 |
|
882 |
|
154,903 |
|
— |
|
— |
|
155,785 |
| |||||
|
Initial public offering |
|
28,947,368 |
|
289 |
|
519,460 |
|
— |
|
— |
|
519,749 |
| |||||
|
Costs related to initial public offering |
|
— |
|
— |
|
(2,557 |
) |
— |
|
— |
|
(2,557 |
) | |||||
|
Fair value adjustment on common stock with put/call feature |
|
— |
|
— |
|
(6,585 |
) |
— |
|
— |
|
(6,585 |
) | |||||
|
Reclassification of mezzanine equity to permanent equity upon cancellation of |
|
|
|
|
|
|
|
|
|
|
|
— |
| |||||
|
common stock put/call feature |
|
— |
|
17 |
|
39,926 |
|
— |
|
— |
|
39,943 |
| |||||
|
Stock option exercises |
|
2,394,762 |
|
24 |
|
7,557 |
|
— |
|
— |
|
7,581 |
| |||||
|
Excess tax benefit from share-based compensation |
|
— |
|
— |
|
13,406 |
|
— |
|
— |
|
13,406 |
| |||||
|
Balance at December 31, 2009 |
|
122,173,274 |
|
1,212 |
|
767,032 |
|
(186,446 |
) |
356 |
|
582,154 |
| |||||
|
Net income |
|
— |
|
— |
|
— |
|
166,068 |
|
— |
|
166,068 |
| |||||
|
Other comprehensive loss |
|
— |
|
— |
|
— |
|
— |
|
(650 |
) |
(650 |
) | |||||
|
Comprehensive income |
|
— |
|
— |
|
— |
|
— |
|
— |
|
165,418 |
| |||||
|
Share-based compensation cost |
|
— |
|
— |
|
15,895 |
|
— |
|
— |
|
15,895 |
| |||||
|
Repurchases and retirement of common stock |
|
(246,823 |
) |
(3 |
) |
(4,914 |
) |
— |
|
— |
|
(4,917 |
) | |||||
|
Stock option exercises |
|
3,650,579 |
|
37 |
|
22,296 |
|
— |
|
— |
|
22,333 |
| |||||
|
Excess tax benefit from share-based compensation |
|
— |
|
— |
|
13,481 |
|
— |
|
— |
|
13,481 |
| |||||
|
Shares issued upon RSU vesting |
|
847 |
|
— |
|
— |
|
— |
|
— |
|
— |
| |||||
|
Vesting of restricted common stock |
|
— |
|
7 |
|
(7 |
) |
— |
|
— |
|
— |
| |||||
|
Balance at December 31, 2010 |
|
125,577,877 |
|
$ |
1,253 |
|
$ |
813,783 |
|
$ |
(20,378 |
) |
$ |
(294 |
) |
$ |
794,364 |
|
The accompanying Notes to Consolidated Financial Statements are an integral part of these consolidated financial statements.
Talecris Biotherapeutics Holdings Corp.
Notes to Consolidated Financial Statements
1. Description of Business
We are a biopharmaceutical company that researches, develops, manufactures, markets, and sells protein-based therapies that extend and enhance the lives of individuals who suffer from chronic and acute, often life-threatening, conditions, such as primary immune deficiencies, chronic inflammatory demyelinating polyneuropathy (CIDP), alpha-1 antitrypsin deficiency-related emphysema, bleeding disorders, infectious diseases, and severe trauma. Our primary products have orphan drug designation to serve populations with rare, chronic diseases. Our products are derived from human plasma, the liquid component of blood, which is sourced from our plasma collection centers or purchased from third parties, located in the United States. Plasma contains many therapeutic proteins, which we extract through the process of fractionation at our Clayton, North Carolina and Melville, New York facilities. The fractionated intermediates are then purified, formulated into final bulk, and aseptically filled into final containers for sale. We also sell the fractionated intermediate products.
The majority of our sales are concentrated in two key therapeutic areas of the plasma business: Immunology/Neurology, through our intravenous immune globulin (IGIV) product for the treatment of primary immune deficiency and autoimmune diseases, such as CIDP, and Pulmonology, through our alpha-1 proteinase inhibitor (A1PI) product for the treatment of alpha-1 antitrypsin deficiency-related emphysema. These therapeutic areas are served by our products, Gamunex, Immune Globulin Intravenous (Human), 10% Caprylate/Chromatography Purified (Gamunex, Gamunex IGIV) and Prolastin Alpha-1 Proteinase Inhibitor (Human) (Prolastin, Prolastin A1PI, Prolastin-C A1PI ). In March 2010, we launched Prolastin-C A1PI, our next generation A1PI product, in the United States, and in the third quarter of 2010, we launched Prolastin-C A1PI in Canada. As of December 31, 2010, we have completed the conversion of our existing U.S. and Canadian Prolastin patients to Prolastin-C A1PI. During 2010, Gamunex-C was approved for the subcutaneous route of administration for the PI indication in Canada and the U.S. Sales of Gamunex-C/Gamunex IGIV and Prolastin/Prolastin-C A1PI together comprised 76.4%, 74.7%, and 72.3% of our net revenue for the years ended December 31, 2010, 2009, and 2008, respectively. We also have a line of hyperimmune therapies that provides treatment for tetanus, rabies, hepatitis A, hepatitis B, and Rh factor control during pregnancy and at birth. In addition, we provide plasma-derived therapies for critical care/hemostasis, including the treatment of hemophilia, an anti-coagulation factor (Thrombate III), as well as albumin to expand blood volume. We sell our products worldwide, but 81% of our sales were in the United States and Canada in 2010.
We are headquartered in Research Triangle Park, North Carolina and our primary manufacturing facilities are a short distance away in Clayton, North Carolina. Our Clayton site is one of the world’s largest plasma protein processing facilities whose operations include fractionation, purification, filling, and finishing. We have an integrated plasma collection center platform, which as of December 31, 2010, consisted of 69 operating centers, of which 67 were FDA licensed and two were unlicensed. In addition to the United States, we have operations in Germany and Canada to support our international sales and marketing activities.
On October 6, 2009, we completed our initial public offering (IPO), which resulted in net proceeds to us of $519.7 million. In addition, during October 2009, we amended our revolving credit facility and completed the issuance of $600.0 million, 7.75% Unsecured Senior Notes, due November 15, 2016, at a price of 99.321% of par, in a private placement to certain qualified institutional buyers. The issuance of the 7.75% Notes resulted in net proceeds to us of $583.9 million. Proceeds from these transactions were used to repay and terminate our then existing First and Second Lien Term Loans, settle and terminate certain interest rate swap contracts, and repay amounts outstanding under our revolving credit facility. On July 19, 2010, we exchanged all of our then existing 7.75% Senior Notes due 2016 for 7.75% Senior Notes due 2016 that have been registered under the Securities Act of 1933, as amended. Additional information regarding our IPO and refinancing transactions are included in Note 4, “Initial Public Offering and Use of Proceeds,” and Note 12, “Long-Term Debt and Capital Lease Obligations,” respectively.
Until January 21, 2010, a majority of our outstanding common stock was owned by Talecris Holdings, LLC. Talecris Holdings, LLC is owned by (i) Cerberus-Plasma Holdings LLC, the managing member of which is Cerberus Partners, L.P., and (ii) limited partnerships affiliated with Ampersand Ventures. Substantially all rights of management and control of Talecris Holdings, LLC are held by Cerberus-Plasma Holdings LLC. As of December 31, 2010, Talecris Holdings, LLC owned approximately 48.7% of our outstanding common stock.
As discussed in Note 3, we entered into a definitive merger agreement with Grifols S.A. and Grifols, Inc. (Grifols) on June 6, 2010.
2. Summary of Significant Accounting Policies
Throughout our consolidated financial statements, references to “Talecris Biotherapeutics Holdings Corp.,” “Talecris,” “the Company,” “we,” “us,” and “our” are references to Talecris Biotherapeutics Holdings Corp. and its wholly-owned subsidiaries.
All tabular disclosures of dollar amounts are presented in thousands. All share and per share amounts are presented at their actual amounts.
A seven-for-one share dividend on our common stock was paid on September 10, 2009. All share and per-share amounts have been retroactively adjusted for all periods presented to reflect the share dividend.
Principles of Consolidation
The accompanying consolidated financial statements include the accounts of Talecris Biotherapeutics Holdings Corp. and its wholly-owned subsidiaries. All significant intercompany transactions and balances have been eliminated upon consolidation.
Use of Estimates
The preparation of financial statements in conformity with U.S. generally accepted accounting principles (U.S. GAAP) requires us to make estimates and judgments in certain circumstances that affect the reported amounts of assets, liabilities, revenues and expenses, and the related disclosures of contingent assets and liabilities. The most significant judgments we have made include, but are not limited to, estimates used in determining values of inventories, allowances for doubtful accounts and notes receivable, long-lived and indefinite-lived assets, litigation accruals and related settlements, losses under contractual obligations, leasehold impairments, deferred income taxes, income tax provisions, accruals for uncertain income tax positions, self-insurance accruals, share-based payment transactions, derivative instruments, and other operating allowances and accruals. We also use significant judgments in applying purchase accounting to business acquisitions.
We periodically evaluate estimates used in the preparation of the financial statements for reasonableness, including estimates provided by third parties. Appropriate adjustments to the estimates are made prospectively, as necessary, based on such periodic evaluations. We base our estimates on, among other things, currently available information, market conditions, and industry and historical experience, which collectively form the basis of making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Although we believe that our assumptions are reasonable under the circumstances, actual future results could differ materially. In addition, if we had used different estimates and assumptions, our financial position and results of operations could have differed materially from that which is presented.
Cash and Cash Equivalents
All highly liquid investments with original maturities of three months or less when purchased are considered cash equivalents and are carried at cost due to the short period of time to maturity.
Accounts Receivable, net
Accounts receivable, net, consists of amounts owed to us by our customers on credit sales with terms generally ranging from 30 to 150 days from date of invoice and are presented net of an allowance for doubtful accounts receivable on our consolidated balance sheets.
We maintain an allowance for doubtful accounts receivable for estimated losses resulting from our inability to collect from customers. In extending credit, we assess our customers’ creditworthiness by, among other factors, evaluating our customers’ financial condition, credit history, and the amount involved, both initially and on an ongoing basis. Collateral is generally not required. In evaluating the adequacy of our allowance for doubtful accounts receivable, we primarily analyze accounts receivable balances, the percentage of accounts receivable by aging category, and historical bad debts. We also consider, among other things, customer concentrations and changes in customer payment terms or payment patterns.
If the financial conditions of our customers were to deteriorate, resulting in an impairment of their ability to make payments or our ability to collect, an increase to the allowance may be required. Also, should actual collections of accounts receivable be different than our estimates included in determining the allowance, the allowance would be adjusted through charges or credits to selling, general, and administrative expenses (SG&A) in our consolidated income statements in the period in which such changes in collection become known. If conditions were to change in future periods, additional allowances or reversals may be required. Such allowances or reversals could be significant.
Concentrations of Credit Risk
Customer Concentration
Our accounts receivable, net, includes amounts due from pharmaceutical wholesalers and distributors, buying groups, hospitals, physicians’ offices, patients, and others. Our concentrations with customers that represented more than 10% of our accounts receivable, net, were:
· At December 31, 2010: Amerisource Bergen- 12.8%
· At December 31, 2009: FFF Enterprise, Inc.- 14.6%
The following table summarizes our concentrations with customers that represented more than 10% of our total net revenue:
|
|
|
Years Ended December 31, |
| ||||
|
|
|
2010 |
|
2009 |
|
2008 |
|
|
FFF Enterprise, Inc. |
|
13.9 |
% |
14.4 |
% |
12.8 |
% |
|
Amerisource Bergen |
|
13.1 |
% |
12.3 |
% |
12.0 |
% |
|
Canadian Blood Services |
|
<10 |
% |
<10 |
% |
10.6 |
% |
Counterparty Risk
As discussed further in Note 27, “Subsequent Events,” we initiated a foreign currency hedging program in the first quarter of 2011 for the purpose of managing the economic effects of the volatility associated with short-term changes in euro/U.S. dollar exchange rates on our earnings and cash flows. These derivative financial instruments present certain market and counterparty risks. We seek to manage the counterparty risks associated with these contracts by limiting transactions to counterparties with which we have established banking relationships and limit the duration of the contracts to less than one year. We are exposed to potential losses if a counterparty fails to perform according to the terms of the agreement. We do not require collateral or other security to be furnished by counterparties to our derivative financial instruments. There can be no assurance, however, that our practice effectively mitigates counterparty risk. A number of financial institutions similar to those that serve or may serve as counterparties to our hedging arrangements were adversely affected by the global credit crisis. The failure of any of the counterparties to our hedging arrangements to fulfill their obligations to us could adversely affect our results of operations and cash flows.
Inventories
Inventories consist of raw materials, work-in-process, and finished goods held for sale and are stated at the lower of cost or market, which approximates actual costs determined on the first-in, first-out method. In evaluating whether inventory is stated at the lower of cost or market, we consider such factors as the amount of inventory on hand and in the distribution channel, the estimated time required to sell such inventory, remaining shelf life, and current and expected market conditions, including levels of competition. As appropriate, provisions are recorded to reduce inventories to their net realizable value. We record provisions for work-in-process inventory when we believe the inventory does not meet all criteria to permit release to the market. Provisions are recorded for finished goods that do not have sufficient remaining shelf lives. We record recoveries directly to cost of goods sold after the impacted material is determined to be usable and is sold to third parties.
Property, Plant, and Equipment, net
Property, plant, and equipment are recorded at cost, less accumulated depreciation and amortization. Internal labor costs directly related to asset additions are capitalized. Major renewals and betterments are capitalized. All feasibility studies and maintenance and repair costs are expensed as incurred. Certain interest costs incurred by us during the construction period, based on our weighted average borrowing rates of debt, are capitalized and included in the cost of the related asset.
We generally depreciate and amortize property, plant, and equipment using the straight-line method over the useful lives presented in the following table:
|
Asset Type |
|
Useful Life (Years) |
|
Buildings |
|
10 to 45 |
|
Building improvements |
|
10 to 20 |
|
Machinery and equipment |
|
3 to 20 |
|
Furniture and fixtures |
|
5 to 10 |
|
Computer hardware and software |
|
3 to 7 |
|
Leasehold improvements |
|
the estimated useful life of the improvement or, if shorter, the life of the lease |
We lease various property and equipment. Leased property and equipment that meet certain criterion are capitalized and the present values of the related lease payments are recorded as liabilities. Capital lease payments are allocated between a reduction of the
lease obligation and interest expense using the interest rate implicit in the lease. All other leases are accounted for as operating leases and the related payments are expensed ratably over the rental period. Amortization of assets under capital leases is computed using the straight-line method over the shorter of the remaining lease term or the estimated useful life.
Business Acquisitions
Results of business acquisitions are included in our results of operations as of the respective acquisition dates. The purchase price of each acquisition is allocated to the net assets acquired based on estimates of their fair values at the date of the acquisition. Any purchase price in excess of the fair value of net assets acquired is recorded as goodwill. The accounting for business acquisitions requires us to make estimates and assumptions related to the estimated fair values of the net assets acquired. Significant judgments are used during this process, particularly with respect to intangible assets. Generally, definite-lived intangible assets are amortized over their estimated useful lives. Goodwill and other indefinite-lived intangible assets are not amortized, but are annually assessed for impairment. Therefore, the purchase price allocation to intangible assets and goodwill could have a significant impact on future operating results.
Identifiable Intangible Assets
Identifiable intangible assets are recorded at cost, or when acquired as part of a business acquisition, at estimated fair value. Definite-lived intangible assets are amortized over their useful lives. Indefinite-lived intangible assets, such as regulatory licenses, are not amortized, but are annually assessed for impairment.
Impairment Reviews
We evaluate the recoverability of recorded goodwill and other indefinite-lived intangible asset amounts annually as of December 31 or when events or changes in circumstances indicate that evidence of potential impairment exists, using a fair value based test. This test requires us to make estimates of factors that include, but are not limited to, projected future operating results and business plans, economic projections, anticipated future cash flows, comparable marketplace data from a consistent industry group, and the cost of capital. Any applicable impairment loss is the amount, if any, by which the implied fair value is less than the carrying value.
We review the carrying amounts of other long-lived assets for potential impairment whenever events or changes in circumstances indicate that the carrying amount of the asset may not be recoverable. We periodically evaluate whether events or changes in circumstances have occurred that may warrant revision of the estimated useful lives of our long-lived assets or whether the remaining carrying amount of long-lived assets should be evaluated for possible impairment. An example of such a change in circumstances includes a significant adverse change in the extent or manner in which an asset is being used.
Debt Issuance Costs and Debt Discount
We capitalize costs associated with the issuance of our debt and amortize these costs to interest expense, net, over the term of the related debt agreement using an effective yield amortization method, or similar method. Unamortized debt issuance costs are written off within total other non-operating expense, net, in our consolidated income statements when indebtedness under the related credit facility is repaid or restructured prior to maturity.
We record debt discounts as a reduction of the face amount of the related debt. Debt discounts are amortized to interest expense, net, over the term of the related debt agreement using an effective yield amortization method, or similar method.
Revenue Recognition and Gross-to-Net Revenue Adjustments
We recognize revenue when earned, which is generally at the time of delivery to the customer. Recognition of revenue also requires reasonable assurance of collection of sales proceeds, a fixed and determinable price, persuasive evidence that an arrangement exists, and completion of all other performance obligations. The recognition of revenue is deferred if there are significant post-delivery obligations, such as customer acceptance.
Allowances against revenue for estimated discounts, rebates, administrative fees, chargebacks, and shelf-stock adjustments are established by us concurrently with the recognition of revenue. The standard terms and conditions under which products are shipped to our customers generally do not allow a right of return. In the rare instances in which we grant a right of return, revenue is reduced at the time of sale to reflect expected returns and deferred until all conditions of revenue recognition are met.
We have supply agreements with our major distributors, which require them to purchase minimum quantities of our products. We regularly review the supply levels of our products on hand at major distributors, primarily by analyzing inventory reports supplied by these distributors, available data regarding the sell-through of our products, our internal data, and other available information. When we believe distributor inventory levels have increased relative to underlying demand, we evaluate the need for sales return allowances. Factors that influence the allowance include historical sales return activity, levels of inventory in the distribution network, inventory turnover, demand history, demand projections, estimated product shelf-life, pricing, and competition. Sales returns have not been material during the periods presented.
Revenue from milestone payments for which we have no continuing performance obligations is recognized upon achievement of the related milestone. When we have continuing performance obligations, the milestone payments are deferred and recognized as revenue over the term of the arrangement as we complete our performance obligations.
Gross product sales are subject to a variety of deductions that are generally estimated and recorded in the same period that the revenue is recognized, and primarily represent rebates to government agencies, chargebacks to wholesalers and distributors, and customer prompt payment discounts. These gross-to-net revenue adjustments are described below.
We offer rebates to certain classes of trade, which we account for by establishing an accrual at the time the sale is recorded in an amount equal to our estimate of rebates attributable to each sale. We determine our estimate of the rebates primarily based on historical experience and current contract arrangements. We consider the sales performance of products subject to rebates and the levels of inventory in the distribution channel and adjust the accrual periodically to reflect actual experience. Rebates accrued upon sale are settled based on actual experience. Due to the limited classes of trade that participate in rebate programs and our visibility of inventories in the channel, adjustments for actual experience have not been material.
We participate in state government-managed Medicaid programs. We account for Medicaid rebates by establishing an accrual at the time the sale is recorded in an amount equal to our estimate of the Medicaid rebate claims attributable to such sale. We determine our estimate of the Medicaid rebates accrual primarily based on historical experience regarding Medicaid rebates, legal interpretations of the applicable laws related to the Medicaid program and any new information regarding changes in the Medicaid programs’ regulations and guidelines that would impact the amount of the rebates. We consider outstanding Medicaid claims, Medicaid payments, and levels of inventory in the distribution channel and adjust the accrual periodically to reflect actual experience. Adjustments for actual experience have not been material.
Sales allowances are established based upon consideration of a variety of factors, including, but not limited to, our sales terms which generally provide for up to a 2% prompt pay discount on domestic and international sales, contractual agreements with customers, estimates of the amount of product in the pipeline, and prescribing patterns. We believe that our sales allowance accruals are reasonably determinable and are based on the information available at the time to arrive at our best estimate of the accruals. Actual sales allowances incurred are dependent upon future events. We periodically monitor the factors that influence sales allowances and make adjustments to these provisions when we believe that the actual sales allowances may differ from prior estimates. If conditions in future periods change, revisions to previous estimates may be required, potentially in significant amounts. As these prompt pay discounts are typically settled within 30 to 45 days of the sale, adjustments for actual experience have not been material.
Our estimates for discounts, customer and government rebates, and administrative fees are by their nature more predictable and less subjective. Estimates for chargebacks are more subjective and, consequently, may be more variable. We enter into agreements with certain customers to establish contract pricing for our products, which these entities purchase from the wholesaler or distributor (collectively, wholesalers) of their choice. Consequently, when our products are purchased from wholesalers by these entities at the contract price which is less than the price charged by us to the wholesaler, we provide the wholesaler with a credit referred to as a chargeback. The allowance for chargebacks is based on our estimate of the wholesaler inventory levels, and the expected sell-through of our products by the wholesalers at the contract price based on historical chargeback experience and other factors. Our estimates of inventory levels at the wholesalers are subject to inherent limitations, as our estimates rely on third party data, and their data may itself rely on estimates, and be subject to other limitations. We periodically monitor the factors that influence our provision for chargebacks, and make adjustments when we believe that actual chargebacks may differ from established allowances. These adjustments occur in a relatively short period of time.
Shelf-stock adjustments are credits issued to our customers to reflect decreases in the selling prices of our products. Agreements to provide this form of price protection are customary in our industry and are intended to reduce a customer’s inventory cost to better reflect current market prices. Shelf-stock adjustments are based upon the amount of product that our customers have remaining in their inventories at the time of the price reduction. Decreases in our selling prices are discretionary decisions made by us to reflect market conditions. Amounts recorded for estimated price adjustments are based upon specified terms with customers, estimated declines in market prices, and estimates of inventory held by customers. Our estimates of inventory levels at the customer are subject to inherent limitations, as our estimates may rely on third party data, and their data may itself rely on estimates, and be subject to other limitations. We regularly monitor these factors and evaluate our reserves for shelf-stock adjustments. We have not experienced significant shelf-stock adjustments during the periods presented.
Shipping and Handling
Shipping and handling costs incurred for inventory purchases are included in cost of goods sold in our consolidated income statements. Shipping and handling costs incurred to warehouse, pick, pack, and prepare inventory for delivery to customers are included in selling, general and administrative expenses (SG&A) in our consolidated income statements. Shipping and handling costs included in SG&A amounted to $3.7 million, $3.6 million, and $3.5 million for the years ended December 31, 2010, 2009, and 2008, respectively.
Advertising Costs
The costs of advertising are expensed as incurred within SG&A in our consolidated income statements. Our advertising costs consist primarily of product samples, print media, online advertising, and promotional material. We incurred advertising costs totaling $11.3 million, $10.2 million, and $10.5 million for the years ended December 31, 2010, 2009, and 2008, respectively.
Research and Development Expenses
Research and development (R&D) expenses include the costs directly attributable to the conduct of research and development programs for new products and extensions or improvements of existing products and the related manufacturing processes. Such costs include salaries and related employee benefit costs, payroll taxes, materials (including the material required for clinical trials), supplies, depreciation on and maintenance of R&D equipment, services provided by outside contractors for clinical development and clinical trials, regulatory services, and fees. R&D also includes the allocable portion of facility costs such as rent, depreciation, utilities, insurance, and general support services. All costs associated with R&D are expensed as incurred.
Share-Based Compensation
We value share-based compensation at the grant date using a fair value model and recognize this value as expense over the employees’ requisite service period, typically the period over which the share-based compensation vests. We classify share-based compensation costs consistent with each grantee’s salary. We record income tax benefits which result from realizing a tax deduction in excess of previously recognized compensation expense as additional paid-in capital.
The fair value of our common stock on the grant date is a significant factor in determining the fair value of share-based compensation awards and the ultimate non-cash compensation cost that we will be required to record over the vesting period. Given the absence of a trading market for our common stock on grant dates prior to October 1, 2009, our board of directors, or special dividend committee or compensation committee designated by our board of directors, estimated the fair value of our common stock contemporaneously with each grant using numerous objective and subjective factors. These factors included: (i) our stage of development, our efforts to become independent from Bayer, and revenue growth; (ii) the timing of the anticipated launch of new products and new indications; (iii) business conditions and business challenges at the time; (iv) available market data, including observable market transactions, and valuations for comparable companies; (v) the illiquid nature of our stock options and stock grants; and (vi) the likelihood of achieving a liquidity event for the shares of common stock underlying the options, such as an initial public offering or sale of our company, given prevailing market conditions at the grant date. In making the assessment of common stock fair value on each award date, our board of directors or designated committee of our board of directors considered the guidance in American Institute of Certified Public Accountants Technical Practice Aid, “Valuation of Privately-Held Company Equity Securities Issued as Compensation.” The valuations were completed utilizing the market and/or an income approach and then the enterprise value was allocated using the “Probability-Weighted Expected Return Method,” which provides different probability weights of various likely scenarios (distressed; remain private; private sale; IPO), and develops valuations by determining the present value of the future expected common stock value under each of these scenarios. For option awards granted on October 1, 2009, the fair value of our common stock was determined to be the IPO price per share of $19.00. For option awards granted subsequent to our IPO, we consider the fair value of our common stock to be the closing share price as reported by The NASDAQ Global Select Market on the grant date.
We estimate the fair value of stock options at the grant date using the Black-Scholes pricing model, which requires the use of a number of assumptions related to the risk-free interest rate, average life of options (expected term), expected volatility, and dividend yield. There was no trading market for our common stock or stock options on grant dates prior to October 1, 2009. Therefore, our application of the Black-Scholes pricing model incorporates historical volatility measures of similar public companies. A forfeiture rate based on historical attrition rates of award holders is used in estimating the granted awards not expected to vest. If actual forfeitures differ from the expected rate, we may be required to make additional adjustments to compensation expense in future periods. The Black-Scholes option pricing model was developed for use in estimating the fair value of traded options that have no
vesting restrictions and are fully transferable, characteristics that are not present in our option grants. If the model permitted consideration of the unique characteristics of employee stock options, the resulting estimate of the fair value of the stock options, and resulting compensation expense, could be different.
The stock options that we granted to employees typically have service-based and performance-based components. The performance stock unit (PSU) awards that we grant to employees vest based on the achievement of pre-established objective performance goals, which are generally financial in nature. The restricted stock and restricted stock unit (RSU) awards that we grant to employees are typically service-based only. Stock option grants, restricted stock, and RSU awards to non-employee directors are service-based only. Service-based awards vest annually in equal amounts over the vesting period. The performance-based component of the stock options vests annually upon the achievement of corporate performance objectives which are established by our board of directors. We make assessments as to whether the performance conditions related to the performance-based stock options will be achieved. We record compensation cost for awards with performance conditions based on the probable outcome of the performance conditions.
Litigation Accruals
We record an accrual for our exposures to our various litigation matters as a charge to our consolidated income statements when it becomes probable and can be reasonably estimated. The exposure to legal matters is evaluated and estimated, if possible, following consultation with legal counsel. Such estimates are based on currently available information and, given the subjective nature and complexities inherent in making these estimates, the ultimate outcome of our legal matters may be significantly different than the amounts estimated. Additional information regarding our possible litigation exposures is included in Note 14, “Commitments and Contingencies.”
Environmental Costs
We record liabilities when our environmental assessments indicate that remediation efforts are probable, and the costs can be reasonably estimated. We recognize a current period expense for the liability when clean-up efforts do not benefit future periods. We capitalize costs that benefit more than one accounting period. Estimates, when applicable, of our liabilities are based on currently available facts, existing technology, and presently enacted laws and environmental regulations taking into consideration the likely effects of inflation and other societal and economic factors, and include estimates of associated legal costs. The amounts also consider prior experience in remediating contaminated sites, other companies’ clean-up experience, and data released by the Environmental Protection Agency (EPA) or other organizations. The estimates are subject to revision in future periods based on actual costs or new circumstances. We evaluate recoveries from insurance coverage or government sponsored programs separately from our liability, and when recovery is assured, we record and report an asset separately from the associated liability. At December 31, 2010 and 2009, no environmental related assets or liabilities are reflected on our consolidated balance sheets as no amounts are probable or estimable.
Other Contingencies
We recognize liabilities for other contingencies when we have an exposure, that, when analyzed, indicates it is both probable that an asset has been impaired or a liability incurred, and the amount of impairment or loss can be reasonably estimated. Funds spent to remedy these contingencies are charged against the accrued liability, if one exists, or expensed, if no liability was previously established. When a range of probable loss can be estimated, we accrue the most likely amount within the range of probable losses.
Self-Insurance Programs
We maintain self-insured retentions and deductibles for some of our insurance programs and limit our exposure to claims by maintaining stop-loss and/or aggregate liability coverage under which the insurer is the primary obligor to the insured. The estimate of our claims liability is subject to inherent limitations as it relies on our judgment of the likely ultimate costs that will be incurred to settle reported claims and unreported claims for incidents incurred but not reported as of the balance sheet date. When estimating our liability for such claims, we consider a number of factors, including, but not limited to, self-insured retentions, deductibles, claim experience, demographic factors, severity factors, and maximum claims exposure. If actual claims exceed these estimates, additional charges may be required.
Income Taxes
We calculate a provision for, or benefit from, income taxes using the asset and liability method, under which deferred tax assets and liabilities are recorded based on the difference between the financial statement and tax bases of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to reverse. A reduction in the carrying amounts of deferred tax assets by a valuation allowance is required, if, based on the available evidence, it is more likely than not that the assets will not be realized. Accordingly, we periodically assess the need to establish valuation allowances for deferred tax assets based on
the more-likely-than-not realization threshold criterion. In assessing the need for a valuation allowance, we consider all available evidence, both positive and negative, including historical levels of income, expectations and risks associated with future taxable income, and ongoing prudent and feasible tax planning strategies.
We establish reserves for uncertain income tax positions, based on the technical support for the positions, our past audit experience with similar situations, and potential interest and penalties related to the matters. Our recorded reserves represent our best estimate of the amount, if any, that we will ultimately be required to pay to settle such matters. The resolution of our uncertain income tax positions is dependent on uncontrollable factors such as law changes, new case law and the willingness of the income tax authorities to settle, including the timing thereof and other factors. Although we do not anticipate significant changes to our uncertain income tax positions in the next twelve months, items outside of our control could cause our uncertain income tax positions to change in the future, which would be recorded within (provision) benefit for income taxes in our consolidated income statements. Interest and penalties related to unrecognized tax benefits are recognized as a component of our income tax provision.
Interest Costs
We capitalize a portion of the interest costs we incur during the construction of long-lived assets, primarily plant and equipment, as an additional cost of the related asset. The amount of interest capitalized is determined by applying our weighted average borrowing rate to the related capital spending during the construction period. We incurred interest costs related to our debt, including imputed interest on capital lease obligations, and interest rate swap contracts of $48.3 million, $72.8 million, and $97.2 million for the years ended December 31, 2010, 2009, and 2008, respectively, of which $5.9 million, $2.0 million, and $2.3 million, respectively, were capitalized related to the construction of property and equipment. Our interest rate swap contracts were settled and terminated during 2009.
Derivative Financial Instruments
All derivative financial instruments are recorded on our consolidated balance sheets as assets or liabilities and measured at fair value, which considers the instrument’s term, notional amount, discount rate, credit risk, and other factors. For derivatives designated as hedges of the fair value of assets or liabilities, the changes in fair values of both the derivatives and the hedged items are recorded in current earnings. For derivatives designated as cash flow hedges, the effective portion of the changes in fair value of the derivatives are recorded in other comprehensive income (loss) and subsequently recognized in earnings when the hedged items impact income. Changes in the fair value of derivatives not designated as hedges and the ineffective portion of cash flow hedges are recorded in current earnings. When determining the fair value of our derivative financial instruments, we analyze the instruments from a market participant’s perspective to determine a hypothetical exit price to the counterparty. At December 31, 2009, our derivative financial instruments consisted of two interest rate cap contracts with an aggregate notional amount of $175.0 million for which the cap rate of 6.00% was significantly higher than prevailing market interest rates; therefore, the fair market value was zero. At December 31, 2010, we did not have any derivative financial instruments. We initiated a foreign currency hedging program in the first quarter of 2011 as discussed in Note 27, “Subsequent Events.”
Fair Value of Financial Instruments
At December 31, 2010, we had no financial assets or liabilities which were required to be measured at fair value. At December 31, 2009, we had two interest rate cap contracts with an aggregate notional amount of $175.0 million for which the cap rate of 6.00% was significantly higher than prevailing market interest rates; therefore, the fair market value was zero.
At December 31, 2010 and 2009, the estimated fair value of our 7.75% Notes was $648.8 million and $607.9 million, which was calculated by reference to open bid/ask quotations of our 7.75% Notes. We had no amounts outstanding under our variable rate revolving credit facility at December 31, 2010 and 2009. At December 31, 2010 and 2009, we have notes receivable outstanding, which bear interest at market rates, and consequently, the recorded amounts approximate fair value. The recorded amounts of all other financial instruments, which consist of cash and cash equivalents, accounts receivable, net, accounts payable, accrued expenses and other liabilities, approximate fair value due to the short duration of the instruments.
Comprehensive Income
Comprehensive income is defined as the change in equity resulting from recognized transactions and other events and circumstances from non-owner sources. Comprehensive income includes net income as currently reported under U.S. GAAP and other comprehensive income (loss). Other comprehensive income (loss) considers the effect of additional economic events that are not required to be recorded in determining net income, but rather are reported as a separate component of stockholders’ equity (deficit).
The following table includes information regarding our other comprehensive income (loss):
|
|
|
Gross Amount |
|
Tax Effect |
|
Net Amount |
| |||
|
Year ended December 31, 2010 |
|
|
|
|
|
|
| |||
|
Foreign currency translation adjustments |
|
$ |
(576 |
) |
$ |
— |
|
$ |
(576 |
) |
|
Additional minimum pension liability |
|
(74 |
) |
— |
|
(74 |
) | |||
|
Other comprehensive loss |
|
$ |
(650 |
) |
$ |
— |
|
$ |
(650 |
) |
|
|
|
|
|
|
|
|
| |||
|
Year ended December 31, 2009 |
|
|
|
|
|
|
| |||
|
Foreign currency translation adjustments |
|
$ |
232 |
|
$ |
— |
|
$ |
232 |
|
|
Additional minimum pension liability |
|
244 |
|
— |
|
244 |
| |||
|
Reclassification of unrealized loss on derivative financial instruments |
|
37,513 |
|
(14,226 |
) |
23,287 |
| |||
|
Other comprehensive income |
|
$ |
37,989 |
|
$ |
(14,226 |
) |
$ |
23,763 |
|
|
|
|
|
|
|
|
|
| |||
|
Year ended December 31, 2008 |
|
|
|
|
|
|
| |||
|
Foreign currency translation adjustments |
|
$ |
(216 |
) |
$ |
— |
|
$ |
(216 |
) |
|
Net unrealized loss on derivative financial instruments |
|
(18,477 |
) |
6,973 |
|
(11,504 |
) | |||
|
Additional minimum pension liability |
|
(52 |
) |
— |
|
(52 |
) | |||
|
Other comprehensive loss |
|
$ |
(18,475 |
) |
$ |
6,973 |
|
$ |
(11,772 |
) |
The following table includes information regarding our accumulated other comprehensive (loss) income:
|
|
|
December 31, |
| ||||
|
|
|
2010 |
|
2009 |
| ||
|
Foreign currency translation adjustments |
|
$ |
(412 |
) |
$ |
164 |
|
|
Additional minimum pension liability |
|
118 |
|
192 |
| ||
|
Accumulated other comprehensive (loss) income |
|
$ |
(294 |
) |
$ |
356 |
|
During the year ended December 31, 2009, we settled and terminated our interest rate swap contracts, which resulted in a loss of $30.9 million. Our accumulated other comprehensive loss at December 31, 2008 included $23.3 million, net of taxes, related to unrealized losses associated with our interest rate swap contracts. As a result of their settlement and termination, we reclassified $23.3 million out of accumulated other comprehensive loss to loss on extinguishment of debt within total other non-operating expense, net, in our consolidated income statement for the year ended December 31, 2009.
Foreign Currency Translation
For our international operations, local currencies have been determined to be the functional currencies. We translate the financial statements of international subsidiaries to their U.S. dollar equivalents at end-of-period currency exchange rates for assets and liabilities and at average currency exchange rates for revenues and expenses. We record these translation adjustments as a component of other comprehensive income (loss) within stockholders’ equity (deficit). We recognize transaction gains and losses arising from fluctuations in currency exchange rates on transactions denominated in currencies other than the functional currency as incurred within SG&A in our consolidated income statements. We incurred foreign currency transaction (losses) gains of $(4.7) million, $1.9 million, and $(1.0) million for the years ended December 31, 2010, 2009, and 2008, respectively. As discussed in Note 27, “Subsequent Events,” we initiated a foreign currency hedging program in the first quarter of 2011 in order to reduce the impact of the volatility of foreign exchange rates and improve predictability.
Business Segments
We operate our plasma-derived protein therapeutics business as a single reportable business segment since all operating activities are directed from our North Carolina headquarters and all of our products result from a common manufacturing process based on a single feedstock.
Earnings per Share
We calculate basic earnings per share based upon the weighted average number of common shares outstanding. We calculate diluted earnings per share based upon the weighted average number of common shares outstanding plus the dilutive effect of common share equivalents calculated using the treasury stock method.
Recent Accounting Pronouncements
In April 2010, the Financial Accounting Standards Board (FASB) issued new accounting guidance which clarifies questions surrounding the accounting implications of the different signing dates of the Health Care and Education Reconciliation Act (signed March 30, 2010) and the Patient Protection and Affordable Care Act (signed March 23, 2010). The new guidance states that the FASB and the Office of the Chief Accountant at the SEC would not be opposed to viewing the two Acts together for accounting purposes. The adoption of this guidance did not have a material impact on our consolidated financial statements.
In February 2010, the FASB issued new accounting guidance regarding disclosures related to subsequent events. An entity that is a U.S. Securities and Exchange Commission (SEC) filer is not required to disclose the date through which subsequent events have been evaluated. This change alleviates potential conflicts between the Accounting Standards Codification (ASC) and the SEC’s requirement. The adoption of this guidance did not have a material impact on our consolidated financial statements.
In January 2010, the FASB issued guidance that requires new disclosures for fair value measurements and provides clarity for existing disclosures. This update requires new disclosures for (1) transfers in and out of levels 1 and 2, and (2) activity in level 3, by requiring the reconciliation to present separate information about purchases, sales, issuance, and settlements. Also, this update clarifies the disclosures related to the fair value of each class of assets and liabilities and the input and valuation techniques for both recurring and nonrecurring fair value measurements in levels 2 and 3. The effective date for the disclosures and clarifications is for the interim and annual reporting periods beginning after December 15, 2009 except for the disclosures about purchases, sales, issuances and settlements, which is effective for fiscal years beginning after December 15, 2010. This update is not expected to have a material impact on our consolidated financial statements or related disclosures.
In October 2009, the FASB issued new accounting guidance regarding multiple-deliverable revenue arrangements. The new guidance addresses the accounting for multiple-deliverable arrangements to enable vendors to account for products or services (deliverables) separately rather than as a combined unit. This guidance establishes a hierarchy for determining the selling price of a deliverable, which is based on: (a) vendor-specific objective evidence; (b) third-party evidence; or (c) estimates. This guidance also eliminates the residual method of allocation and requires that arrangement consideration be allocated at the inception of the arrangement to all deliverables using the relative selling price method. In addition, this guidance significantly expands required disclosures related to a vendor’s multiple-deliverable revenue arrangements. The guidance is effective prospectively for revenue arrangements entered into or materially modified in fiscal years beginning on or after June 15, 2010 and early adoption is permitted. A company may elect, but will not be required, to adopt the guidance retrospectively for all prior periods. We do not anticipate that the adoption of this guidance will have a material impact on our consolidated financial statements or related disclosures.
In August 2009, the FASB released new accounting guidance concerning measuring liabilities at fair value. The new guidance provides clarification that in circumstances in which a quoted price in an active market for the identical liability is not available, a reporting entity is required to measure fair value using certain valuation techniques. Additionally, it clarifies that a reporting entity is not required to adjust the fair value of a liability for the existence of a restriction that prevents the transfer of the liability. This new guidance is effective for the first reporting period after its issuance; however, earlier application is permitted. The adoption of this guidance did not have a material impact on our consolidated financial statements or related disclosures.
On January 30, 2009, the SEC released the final rules requiring all registered companies to use eXtensible Business Reporting Language (XBRL) when submitting financial statements to the SEC. The new rules initially will require interactive data reporting only by domestic and foreign large accelerated filers that prepare their financial statements in accordance with U.S. GAAP and have a worldwide public common equity float above $5.0 billion for their first quarterly period ending after June 15, 2009 and all reporting periods thereafter. We will be required to file using XBRL beginning with our quarterly reporting period ending March 31, 2011.
3. Definitive Merger Agreement with Grifols S.A. and Grifols, Inc. (Grifols)
We entered into a definitive merger agreement with Grifols on June 6, 2010, as amended by Amendment 1 on November 4, 2010. Under the terms of the agreement, Grifols will acquire, through merger transactions, all of the common stock of Talecris for a combination of $19.00 in cash and 0.6485 (or 0.641 for Talecris directors and Talecris Holdings, LLC) of a newly-issued non-voting Grifols’ (Class B) ordinary share for each outstanding Talecris share (the merger consideration). Under the terms of the agreement, completion of the transaction is subject to obtaining certain regulatory approvals, shareholder approvals, as well as other customary conditions. The 0.641 exchange ratio and the additional exchange ratio of 0.0075 (which together comprise the 0.6485 exchange ratio) are generally fixed but the 0.641 exchange ratio will be adjusted if the 0.641 exchange ratio would result in Grifols issuing in excess of 86.5 million Grifols non-voting shares and the additional 0.0075 exchange ratio will be adjusted if such additional exchange ratio would result in Grifols issuing in excess of 0.5 million Grifols non-voting shares. The Grifols non-voting shares will be listed on NASDAQ in the form of American Depositary Shares and the Madrid, Barcelona, Bilbao and Valencia stock exchanges and quoted on the Automated Quotation System of the Spanish Stock Exchanges. Grifols non-voting shares will carry the same economic rights as Grifols ordinary shares. Additionally, Talecris share-based compensation, whether vested or unvested, generally will be converted into the right to receive or acquire the merger consideration, or, in the case of employee stock options, the right to acquire the merger consideration, as described in the merger agreement in lieu of Talecris common stock. The merger agreement provides that if the merger agreement is terminated under specified circumstances Grifols will be required to pay Talecris a termination fee of either $100 million or $375 million, depending on the specified circumstances. If the merger agreement is terminated under other specified circumstances, Talecris will be required to pay Grifols a termination fee of $100 million. Generally, except as noted above, all fees and expenses incurred in connection with the merger agreement and the transactions contemplated by the merger agreement will be paid by the party incurring those expenses. We have incurred and will continue to incur significant costs related to investment banking, legal, and accounting activities, as well as retention expenses, related to this merger transaction. The leading shareholders of Grifols have entered into an agreement with us, subject to conditions, to vote their Grifols shares in favor of the transaction and, separately, Talecris Holdings, LLC, an affiliate of Cerberus Capital Management, L.P., which owns approximately 49% of the outstanding Talecris common stock, has entered into an agreement with Grifols, subject to conditions, to vote its Talecris shares in favor of the transaction.
Under the terms of the definitive merger agreement with Grifols, we are permitted to offer retention amounts up to a total of $15.0 million to employees. As of December 31, 2010, we have offered retention amounts totaling approximately $10.2 million to employees, of which $2.9 million was paid during 2010 and the remaining amounts are expected to be paid in 2011, subject to the terms of the retention agreements. We incurred retention expenses, including fringe benefits, of $6.9 million during the year ended December 31, 2010. The remaining retention amounts will likely be recognized ratably through the second quarter of 2011.
We have entered into agreements with investment bankers related to our definitive merger agreement with Grifols. We incurred fees totaling $2.5 million under these agreements during 2010. During the year ended December 31, 2010, we also incurred legal, accounting, and other fees of $18.3 million associated with the merger. We are obligated to pay additional fees totaling $21.3 million upon successful closing of the merger transaction.
4. Initial Public Offering and Use of Proceeds
On October 6, 2009, we completed our IPO of 56,000,000 shares of our common stock, par value $0.01 per share, at an offering price of $19.00 per share. Our IPO included 28,947,368 shares newly issued and sold by us and 27,052,632 shares sold by the selling stockholder, Talecris Holdings, LLC, including 6,000,000 shares sold by the selling stockholder pursuant to the underwriters’ option to purchase additional shares. After deducting the payment of underwriters’ discounts and commissions, the net primary proceeds to us from the sale of shares in our IPO were approximately $519.7 million, which we used to repay $389.8 million and $129.9 million of principal under our First and Second Lien Term Loans, respectively. We did not receive any proceeds from the sale of shares by the selling stockholder. In addition to the $30.3 million of underwriters’ discounts and commissions deducted from the offering proceeds, we incurred other offering costs of $3.9 million, of which $1.3 million is included in SG&A in our consolidated income statement for the year ended December 31, 2009 and $2.6 million is included as a reduction of additional paid-in capital on our December 31, 2009 consolidated balance sheet. At December 31, 2009, approximately $0.2 million of accrued offering expenses were payable to underwriters.
5. Definitive Merger Agreement with CSL Limited (CSL)
On August 12, 2008, we entered into a definitive merger agreement with CSL, under which CSL agreed to acquire us for cash consideration of $3.1 billion, less net debt, as defined. The closing of the transaction was subject to the receipt of certain regulatory approvals as well as other customary conditions. The U.S. Federal Trade Commission filed an administrative complaint before the Commission challenging the merger and a complaint in Federal district court seeking to enjoin the merger during the administrative process. On June 8, 2009, the merger parties agreed to terminate the definitive merger agreement. CSL paid us a merger termination fee of $75.0 million, which is included as other non-operating income in our consolidated income statement for the year ended December 31, 2009. The U.S. Federal Trade Commission’s complaints were subsequently dismissed.
In consideration of the definitive merger agreement with CSL, our board of directors approved a retention program in August 2008 for an amount up to $20.0 million. We recorded retention expense of $8.2 million and $5.1 million, excluding fringe benefit, during the years ended December 31, 2009 and 2008, respectively. We classified the cost of this retention program consistent with each recipient’s salary. We made payments of approximately $13.3 million under this retention program during 2009. No further payments are due.
6. Business Acquisitions
In November 2006, we entered into an Asset Purchase Agreement (APA) with International BioResources, L.L.C. and affiliated entities (IBR) pursuant to which we acquired certain assets and assumed certain liabilities from IBR. The APA was subsequently amended in June 2007 to provide for the acceleration of all milestones and other amounts owed to IBR under the contingent consideration provision of the APA, and as a result, we issued 2,146,232 shares of our common stock to IBR in June 2007. IBR had the right to put the shares of our common stock back to us for cash ($15.61 per common share) under certain circumstances prior to June 30, 2008. IBR was entitled to interest at a rate of 8% per annum from the issuance date of the shares through December 31, 2007. In January 2008, IBR exercised their put right, as amended, for 1,185,232 common shares, which we repurchased in February 2008. In March 2008, IBR exercised their put right, as amended, for the remaining 961,000 common shares, which we repurchased in April 2008. The repurchased shares were retired and the embedded put feature was cancelled.
The following table summarizes our purchase accounting for plasma collection centers acquired from IBR under the June 2007 Agreement. The plasma collection centers were acquired to support our plasma supply vertical integration strategy. The plasma collection centers’ results of operations have been included in our consolidated financial statements from their respective date of acquisition.
|
|
|
Years Ended December 31, |
| ||||
|
|
|
2009 |
|
2008 |
| ||
|
Payments at closing |
|
$ |
5,181 |
|
$ |
2,147 |
|
|
Notes receivable and other advances |
|
44,540 |
|
10,430 |
| ||
|
Performance incentive payments |
|
837 |
|
843 |
| ||
|
Allocable portion of accelerated contingent consideration |
|
6,020 |
|
2,580 |
| ||
|
Transaction costs |
|
— |
|
56 |
| ||
|
Total purchase price |
|
$ |
56,578 |
|
$ |
16,056 |
|
|
|
|
|
|
|
| ||
|
Cash and cash equivalents |
|
$ |
62 |
|
$ |
21 |
|
|
Inventory |
|
5,416 |
|
1,778 |
| ||
|
Other current assets |
|
183 |
|
— |
| ||
|
Property, plant, and equipment |
|
10,181 |
|
1,814 |
| ||
|
Intangible assets- regulatory licenses |
|
3,860 |
|
840 |
| ||
|
Goodwill |
|
37,060 |
|
11,643 |
| ||
|
Total assets acquired |
|
56,762 |
|
16,096 |
| ||
|
Current liabilities assumed |
|
(184 |
) |
(40 |
) | ||
|
Total purchase price |
|
$ |
56,578 |
|
$ |
16,056 |
|
|
|
|
|
|
|
| ||
|
Number of plasma collection centers acquired |
|
12 |
|
3 |
| ||
The purchase price for the plasma collection centers acquired from IBR during 2009 and 2008 consisted of various loans and advances made to IBR and performance incentive payments for achieving certain milestones related to the timing of plasma collection center openings. The purchase price also includes the allocable portion of accelerated contingent consideration due to IBR as discussed above. We have no further financing commitments to IBR under the terms of our June 2007 Agreement.
7. Goodwill and Intangible Assets
There were no changes to the carrying amount of goodwill for the year ended December 31, 2010. Changes to the carrying amount of goodwill for the year ended December 31, 2009 were as follows:
|
Balance at December 31, 2008 |
|
$ |
135,800 |
|
|
Acquisitions of plasma collection centers from IBR |
|
37,060 |
| |
|
Balance at December 31, 2009 |
|
$ |
172,860 |
|
Additional information regarding our business acquisitions is included in Note 6, “Business Acquisitions.”
We assess goodwill for impairment annually as of December 31, or more frequently if events and circumstances indicate that impairment may have occurred. The impairment test requires us to allocate goodwill to our reporting units and estimate the fair value of the reporting units that contain goodwill. If the carrying value of a reporting unit exceeds its fair value, the goodwill of that reporting unit is potentially impaired and we proceed to step two of the impairment analysis. In step two of the analysis, we would record an impairment loss determined by the excess of the carrying amount of the reporting unit’s goodwill over its implied fair value.
We have assessed goodwill at the reporting unit level. We allocated our Company’s enterprise value to our reporting units based upon their relative contributions to one of our principal operating performance measures, adjusted EBITDA. We determined that the allocated fair value of the reporting unit exceeded its carrying value, and as a result, no adjustment to our recorded goodwill was required at December 31, 2010. Additional information regarding the use of non-GAAP financial measures is included in Note 12, “Long-Term Debt and Capital Lease Obligations.”
At December 31, 2010 and 2009, we had $10.9 million of intangible assets recorded on our consolidated balance sheet, all of which were indefinite-lived regulatory licenses associated with our plasma collection centers. We performed our annual impairment testing of indefinite-lived intangible assets as of December 31, 2010, which resulted in no impairment of the recorded amounts.
8. Collaborative and Other Agreements
Supply and Service Agreement
We have a Supply and Service Agreement, as amended, through 2012 to provide albumin to an unaffiliated third party, which is used in conjunction with a proprietary product manufactured by them. We earn a commission on sales of the third party’s product at a fixed rate which depends on the territory where the product is sold, as defined in the agreement. We also provide regulatory support as required. We earned commissions of $6.6 million, $5.5 million, and $8.6 million under this agreement for the years ended December 31, 2010, 2009, and 2008, respectively, which have been recorded in other net revenue in our consolidated income statements.
Settlement Agreement
We were co-plaintiff along with Bayer Healthcare (Bayer) in patent litigation in the United States District Court for the District of Delaware against Baxter International Inc. and Baxter Healthcare (collectively, Baxter) in which, we, as exclusive licensee of Bayer’s U.S. Patent No. 6,686,191 (the ‘191 patent), alleged that Baxter by its manufacture and importation of its liquid IGIV product, Gammagard Liquid, had infringed the ‘191 patent. Pursuant to a Settlement Agreement with Baxter, Baxter will pay us an amount comprising 1.2% of Baxter’s net sales in the United States of Gammagard Liquid and any other product sold by Baxter or an affiliate in the United States under a different brand name that is a liquid intravenous immunoglobulin through August 2011. Thereafter, until expiration of the ‘191 patent, Baxter will continue to owe the same amount in royalties if Baxter continues to use the licensed technologies under a separate Sublicense Agreement. During the years ended December 31, 2010, 2009, and 2008, we recorded $10.0 million, $10.6 million, and $8.7 million, respectively, of fees from Baxter within other net revenue in our consolidated income statements.
Licensed Technology
We licensed certain technology related to the formulation of one of our products to an unaffiliated third party. As consideration for the technology transfer, we received an upfront licensing fee of $4.0 million during 2007, of which 33% is refundable under certain conditions. We recognized $2.6 million of the licensing fee during 2008 as a result of the completion of a portion of our performance obligations, which we recorded within other net revenue in our consolidated income statement. The remaining portion has been deferred on our consolidated balance sheets at December 31, 2010 and 2009. We will recognize the remaining portion of the deferred licensing fee once our remaining performance obligations have been completed. Under the terms of this agreement, we will also receive royalty payments from this third party, which escalates with volume. During the years ended December 31, 2010, 2009 and 2008, we recorded $2.0 million, $1.6 million and $1.1 million of royalties under this agreement within other net revenue in our consolidated income statements.
9. Inventories and Cost of Goods Sold
Inventories consisted of the following:
|
|
|
December 31, |
| ||||
|
|
|
2010 |
|
2009 |
| ||
|
Raw material |
|
$ |
184,664 |
|
$ |
171,866 |
|
|
Work-in-process |
|
346,086 |
|
312,178 |
| ||
|
Finished goods |
|
163,749 |
|
160,010 |
| ||
|
Total inventories |
|
$ |
694,499 |
|
$ |
644,054 |
|
Our raw material inventories include unlicensed plasma and related testing costs of $2.6 million and $7.6 million at December 31, 2010 and 2009, respectively, which we believe are realizable.
Unabsorbed Talecris Plasma Resources, Inc. (TPR) Infrastructure and Start-Up Costs
Our cost of goods sold includes $6.6 million, $44.0 million, and $98.5 million for the years ended December 31, 2010, 2009, and 2008, respectively, related to unabsorbed TPR infrastructure and start-up costs associated with the development of our plasma collection center platform. The reduction in unabsorbed TPR infrastructure and start-up costs resulted primarily from the maturation of our plasma collection center platform.
Plasma Center current Good Manufacturing Practices (cGMP) Issue
During the first and second quarters of 2008, we incurred charges to cost of goods sold of $16.3 million and $7.0 million, respectively, due to deviations from our standard operating procedures and cGMP at one of our plasma collection centers. Our preliminary investigations concluded that the deviations from our standard operating procedures and cGMP resulted in impairments to the related raw material and work-in-process inventories as we concluded there was no probable future economic benefit related to the impacted inventories. Subsequently, due to further investigations and new facts and circumstances, we determined that certain impacted inventories were saleable. We record recoveries directly to cost of goods sold after the impacted material is converted into final products and sold to third parties. During the years ended December 31, 2009 and 2008, we recorded recoveries of $1.9 million and $17.5 million, respectively. For the year ended December 31, 2008, recoveries totaled $17.5 million, resulting in a net provision of $5.8 million for 2008. During the year ended December 31, 2010, recoveries were not significant.
Customer Settlement
We settled a dispute with a customer in September 2007 regarding intermediate material manufactured by us, which is used by this customer in their manufacturing process. We recorded a charge to cost of goods sold of $7.9 million during the year ended December 31, 2007 for inventory impairment related to this material, which we recovered in its entirety during 2008 as the related material was determined to be saleable, converted into final product, and sold to other customers. During 2008, we recorded an additional inventory provision of $2.6 million related to this dispute for products held in Europe, for which we recovered $0.8 million and $1.8 million during 2009 and 2008, respectively, as the impacted material was determined to be saleable, converted into final product, and sold to other customers.
10. Property, Plant, and Equipment, net
Property, plant, and equipment, net, consisted of the following:
|
|
|
December 31, |
| ||||
|
|
|
2010 |
|
2009 |
| ||
|
Land |
|
$ |
4,136 |
|
$ |
4,136 |
|
|
Buildings and improvements |
|
88,652 |
|
68,417 |
| ||
|
Machinery and equipment |
|
143,813 |
|
102,887 |
| ||
|
Furniture and fixtures |
|
7,377 |
|
5,492 |
| ||
|
Computer hardware and software |
|
64,729 |
|
54,761 |
| ||
|
Capital leases of buildings |
|
8,704 |
|
8,374 |
| ||
|
|
|
317,411 |
|
244,067 |
| ||
|
Less: accumulated depreciation and amortization |
|
(95,319 |
) |
(62,463 |
) | ||
|
|
|
222,092 |
|
181,604 |
| ||
|
Construction in progress |
|
160,701 |
|
85,595 |
| ||
|
Total property, plant, and equipment, net |
|
$ |
382,793 |
|
$ |
267,199 |
|
Depreciation expense was $36.0 million, $28.8 million, and $20.1 million for the years ended December 31, 2010, 2009, and 2008, respectively.
During 2009 and 2008, we recorded impairment charges of $3.1 million and $3.6 million, respectively, primarily within cost of goods sold in our consolidated income statements related primarily to capital lease assets and leasehold improvements at certain of our plasma collection centers which were closed or were under development and we no longer plan to open. No material impairment charges related to property, plant, and equipment were recorded during 2010.
11. Investment in Affiliate
We have a 30% interest in the Class 1 common stock of Centric Health Resources, Inc. (Centric). Our investment in Centric is accounted for using the equity method of accounting based on our assessment that our interest allows us to exercise significant influence, but not control. Under the equity method, our investment, originally recorded at cost, is adjusted to recognize our share of net earnings or losses of Centric as they occur. Our recognition of losses is limited to the extent of our investment in, advances to, and commitments for the investment.
Centric provides services in the management of our Prolastin and Gamunex Direct programs. In this capacity, Centric provides warehousing, order fulfillment, distribution, home infusion, and customer relationship services for us primarily related to our U.S. sales of Prolastin/Prolastin-C A1PI. Centric maintains inventory on our behalf which they utilize to fill customer orders. Centric also provides services to us in collecting accounts receivable for sales made under the Prolastin and Gamunex Direct programs. We provide Centric a fee for each unit of product provided to patients which escalates with volume. The total fees for such services for the years ended December 31, 2010, 2009, and 2008 were $22.9 million, $20.3 million, $17.5 million, respectively. The majority of these fees are recorded within cost of goods sold in our consolidated income statements. The value of the finished goods inventories that Centric held on our behalf was $8.0 million and $7.1 million at December 31, 2010 and 2009, respectively.
12. Long-Term Debt and Capital Lease Obligations
We were obligated under the following debt instruments:
|
|
|
December 31, |
| ||||
|
|
|
2010 |
|
2009 |
| ||
|
7.75% Notes |
|
$ |
600,000 |
|
$ |
600,000 |
|
|
Discount on 7.75% Notes |
|
(3,379 |
) |
(3,954 |
) | ||
|
Revolving credit facility |
|
— |
|
— |
| ||
|
Capital lease obligations |
|
9,540 |
|
9,961 |
| ||
|
Total debt and capital lease obligations |
|
606,161 |
|
606,007 |
| ||
|
Less: current maturities |
|
(860 |
) |
(740 |
) | ||
|
Long-term debt and capital lease obligations, net of current maturities |
|
$ |
605,301 |
|
$ |
605,267 |
|
7.75% Unsecured Senior Notes, due November 15, 2016
On October 21, 2009, we completed the issuance of $600.0 million, 7.75% Senior Notes, due November 15, 2016, at a price of 99.321% of par, in a private placement to certain qualified institutional buyers. The 7.75% Notes yield 7.875% to maturity and pay interest semi-annually on May 15 and November 15 to holders of record on the immediately preceding May 1 and November 1, respectively. The 7.75% Notes are guaranteed on a senior unsecured basis by our existing and future domestic subsidiaries. Except as described below, we will not be entitled to redeem the 7.75% Notes at our option prior to November 12, 2012.
We may redeem some or all of the 7.75% Notes, at our option, at any time on or after November 12, 2012, at the redemption prices (expressed as percentages of principal amount) set forth below plus accrued and unpaid interest and additional interest, if any, on the 7.75% Notes redeemed, to the applicable redemption date, if redeemed during the twelve-month period beginning on November 15 of the years indicated below:
|
Fiscal Year |
|
Percentage |
|
|
2012 |
|
103.875 |
% |
|
2013 |
|
102.583 |
% |
|
2014 |
|
101.292 |
% |
|
2015 and thereafter |
|
100.000 |
% |
In addition, at any time during each twelve-month period ending on November 15, 2010, 2011, and 2012, we may redeem up to 10% of the originally issued principal amount of the 7.75% Notes at a redemption price of 103% of the principal amount of the 7.75% Notes redeemed plus accrued and unpaid interest and additional interest, if any, to the redemption date, subject to the rights of the holders of the 7.75% Notes on the relevant record date to receive interest due on the relevant interest payment date. No principal amounts were redeemed during 2010.
At any time, or from time to time, on or prior to November 15, 2012, we may, at our option, redeem up to 35% of the aggregate principal amount of the 7.75% Notes issued under the indenture with the net cash proceeds to us of certain equity offerings at a redemption price equal to 107.75% of the principal amount of the 7.75% Notes plus accrued and unpaid interest and additional interest, if any, to the applicable redemption date, provided that at least 65% of the aggregate principal amount of the 7.75% Notes originally issued remains outstanding immediately after such redemption and the redemption occurs within 90 days of the date of the closing of such equity offering.
Under the Make-Whole redemption feature, we may redeem 100% of the principal plus a premium as defined under the indenture (computed using a discount rate equal to the U.S. Treasury rate as of such redemption date plus 0.50%), plus accrued and unpaid interest and additional interest, if any, prior to November 15, 2012, with respect to some or all of the 7.75% Notes, subject to the rights of the holders on the relevant record date to receive interest due on the relevant interest payment date.
We are not required to make mandatory redemption or sinking fund payments with respect to the 7.75% Notes.
Upon a change of control, the 7.75% Notes are puttable at 101% of principal plus accrued and unpaid interest and additional interest, if any.
We may incur additional indebtedness and our subsidiary guarantors may also incur additional indebtedness if our Fixed Charge Coverage Ratio for our most recently ended four full fiscal quarters immediately preceding the date on which such additional indebtedness is incurred would have been at least 2.00 to 1.00, determined on a pro forma basis.
The indenture contains certain covenants limiting, subject to exceptions, carve-outs and qualifications, our ability and our restricted subsidiaries’ ability to: (i) sell assets; (ii) pay distributions on, redeem or repurchase its capital stock or redeem or repurchase its subordinated debt; (iii) make certain investments; (iv) incur or guarantee additional indebtedness or issue preferred stock; (v) create or incur certain liens; (vi) enter into agreements that restrict distributions or other payments from our restricted subsidiaries to us; (vii) engage in certain sale and leaseback transactions; (viii) engage in certain transactions with affiliates; (ix) transfer or dispose of the capital stock of the restricted subsidiary to persons other than us or our restricted subsidiaries; and (x) create unrestricted subsidiaries. The indenture also contains certain customary events of default.
On July 19, 2010, we exchanged all of our then outstanding 7.75% Notes for similar 7.75% Notes that were registered under the Securities Act. This exchange did not impact our capitalization.
Revolving Credit Facility
We have a $325.0 million asset-based credit agreement administered by Wachovia Bank, N.A., an affiliate of Wells Fargo Securities, which was amended on October 15, 2009 as described below. We use our available cash balances to repay amounts outstanding under our revolving credit facility. We deposit any excess amounts into an overnight investment account. Outstanding principal under this facility is due and payable on the maturity date of December 6, 2011. As such, any future outstanding balances will likely be recorded as current liabilities. At December 31, 2010, $2.4 million was being utilized for letters of credit and $322.6 million was unused and available. The letters of credit were used as security for utilities, insurance, and third party warehousing.
Borrowings under this facility bear interest at a rate based upon either the ABR or LIBOR, at our option, plus applicable margins based upon borrowing availability. The ABR represents the greater of the Federal Funds Effective Rate plus 0.50% or the Prime Rate. Interest accrues on the revolving credit facility at the ABR plus 0.25-0.75% or LIBOR plus 1.50-2.00%. For the years ended December 31, 2010, 2009, and 2008, the weighted average interest rates of our revolving credit facility were 3.50%, 2.79%, and 4.79%, respectively. No amounts were outstanding under the revolving credit facility at December 31, 2010 and 2009.
The revolving credit facility is secured by a Pledge and Security Agreement dated December 6, 2006 under which substantially all of our personal property, including real estate, manufacturing equipment, accounts receivable, inventory, and stock are pledged as security, each as defined within the agreement.
The revolving credit facility contains default provisions, and, pursuant to the October 15, 2009 amendment described below, imposes restrictions on annual capital expenditures if our leverage ratio is 2.00 to 1.00 or less, and contains a financial covenant which requires us to maintain a fixed charge coverage ratio of at least 1.10 to 1.00 if our borrowing availability based on eligible collateral is less than $48.75 million. The revolving credit facility defines certain terms in calculating covenant ratios, including adjusted EBITDA and Indebtedness.
The borrowing base under our revolving credit facility is based on our accounts receivable and inventory, and is calculated as
(i) 85% of our eligible accounts receivable plus (ii) the lesser of (a) 65% of our eligible inventory (valued on a first-in-first-out basis), (b) 85% of the net orderly liquidation value of our eligible inventory as determined by a recent appraisal, and (c) $300 million. Only up to $100 million may be advanced to us based on the value of our work-in-process inventory (with “filled-not-packed” and “packed-not-released” inventory being considered finished goods inventory). From time to time, the collateral agent under the revolving credit facility may modify our eligibility standards, establish or adjust reserves, or reduce one or more of the other elements used in computing the borrowing base.
On October 15, 2009, we entered into an amendment to the revolving credit facility dated as of October 12, 2009. The revolving credit facility, as amended, permitted the 7.75% Notes, described above, to be issued as long as the First and Second Lien Term Loan Credit Agreements were terminated in connection with the offering of the 7.75% Notes. The amendment also (i) increases the covenant baskets for permitted acquisitions to $250 million, (ii) permits the payment of cash dividends commencing with the first fiscal quarter of 2010 if certain conditions are met as described below, and (iii) increases our capital expenditure baskets so that we will be permitted to make capital expenditures of up to $225 million in each of 2010 and 2011. Moreover, pursuant to the amendments, we are not subject to any limitation on our capital expenditures in any fiscal year if our leverage ratio, as defined, as of the end of the fiscal year most recently ended was less than or equal to 2.00 to 1.00. Minimum availability tests under the revolving credit facility were also increased from $32.5 million to $48.75 million in connection with the amendment.
Our revolving credit facility, as amended, permits the payment of cash dividends to holders of our common stock commencing with the first fiscal quarter of 2010, so long as (i) the leverage ratio determined as of the end of the immediately preceding fiscal quarter for the then most recently completed four fiscal quarters, is equal to or less than 2.00 to 1.00 and (ii) the minimum pro forma availability as of the date of such dividend (after giving effect to such cash dividend, the funding of all revolving loans, and the issuance of all letters of credit to be funded or issued as of such date) is not less than $48.75 million; provided that, the aggregate amount of restricted payments shall not exceed 50% of Net Income during the period from October 1, 2009 to the end of the most recently ended fiscal quarter as of the date of the restricted payment.
First and Second Lien Term Loans
Our First and Second Lien Term Loans were repaid in full and terminated as a result of the application of the net proceeds to us from our October 6, 2009 IPO and the issuance of our 7.75% Notes on October 21, 2009. The weighted average annualized interest rates on the First Lien Term Loan were 4.66% and 6.60% for the years ended December 31, 2009 and 2008, respectively, and the weighted average annualized interest rates on the Second Lien Term Loan were 7.68% and 9.63% for the years ended December 31, 2009 and 2008, respectively.
Financial Impact of IPO and Refinancing Transactions
The following table summarizes the changes to our indebtedness during 2009, including the impact from the application of the net proceeds to us of $519.7 million from our IPO discussed in Note 4, “Initial Public Offering and Use of Proceeds,” and the net proceeds to us of $583.9 million from the refinancing transactions discussed below:
|
|
|
December 31, |
|
2009 Net |
|
October 6, 2009 |
|
October 21, 2009 |
|
|
|
December 31, |
| ||||||
|
|
|
2008 |
|
Repayments |
|
IPO |
|
Refinancing |
|
Amortization |
|
2009 |
| ||||||
|
Revolving Credit Facility |
|
$ |
179,941 |
|
$ |
(124,348 |
) |
$ |
— |
|
$ |
(55,593 |
) |
$ |
— |
|
$ |
— |
|
|
First Lien Term Loan |
|
686,000 |
|
(5,250 |
) |
(389,812 |
) |
(290,938 |
) |
— |
|
— |
| ||||||
|
Second Lien Term Loan |
|
330,000 |
|
— |
|
(129,937 |
) |
(200,063 |
) |
— |
|
— |
| ||||||
|
7.75% Notes |
|
— |
|
— |
|
— |
|
600,000 |
|
— |
|
600,000 |
| ||||||
|
Discount on 7.75% Notes |
|
— |
|
— |
|
— |
|
(4,074 |
) |
120 |
|
(3,954 |
) | ||||||
|
Total indebtedness |
|
$ |
1,195,941 |
|
$ |
(129,598 |
) |
$ |
(519,749 |
) |
$ |
49,332 |
|
$ |
120 |
|
$ |
596,046 |
|
In addition to the debt principal repayments in the preceding table, we used $28.7 million of the net proceeds to us from the issuance of the 7.75% Notes to settle and terminate certain interest rate swap contracts with a notional amount of $390.0 million and $8.6 million to pay accrued interest associated with our then outstanding First and Second Lien Term Loans. In addition to the $4.1 million of discounts on the 7.75% Notes disclosed in the table above, approximately $12.0 million of commissions were deducted from the gross issuance proceeds. Subsequently, we paid $6.1 million to settle and terminate our remaining interest rate swap contract with a notional amount of $50.0 million.
As a result of the IPO and refinancing transactions, we recognized a charge during the fourth quarter of 2009 of $12.1 million to write-off previously deferred debt issuance costs related to our First and Second Lien Term Loans and $30.9 million related to costs associated with the settlement and termination of our interest rate swap contracts. These charges, which totaled $43.0 million, were recorded within other non-operating expense, net, in our consolidated income statement for the year ended December 31, 2009. We
capitalized $14.9 million of debt issuance costs associated with the issuance of the 7.75% Notes and the revolving credit facility amendment. We incurred other costs related to our IPO of $3.9 million, of which $1.3 million is included within SG&A in our consolidated income statement for the year ended December 31, 2009 and $2.6 million is included as a reduction of additional paid-in capital on our December 31, 2009 consolidated balance sheet. The following table summarizes changes in deferred debt issuance costs during the year ended December 31, 2009:
|
|
|
|
|
|
|
Newly |
|
|
|
|
| |||||
|
|
|
|
|
|
|
Capitalized |
|
|
|
|
| |||||
|
|
|
December 31, |
|
|
|
Debt Issuance |
|
|
|
December 31, |
| |||||
|
|
|
2008 |
|
Charges |
|
Costs |
|
Amortization |
|
2009 |
| |||||
|
Revolving Credit Facility |
|
$ |
3,014 |
|
$ |
— |
|
$ |
1,545 |
|
$ |
(1,041 |
) |
$ |
3,518 |
|
|
First Lien Term Loan |
|
9,629 |
|
(8,054 |
) |
— |
|
(1,575 |
) |
— |
| |||||
|
Second Lien Term Loan |
|
4,744 |
|
(4,087 |
) |
— |
|
(657 |
) |
— |
| |||||
|
7.75% Notes |
|
— |
|
— |
|
13,334 |
|
(392 |
) |
12,942 |
| |||||
|
Total deferred debt issuance costs |
|
$ |
17,387 |
|
$ |
(12,141 |
) |
$ |
14,879 |
|
$ |
(3,665 |
) |
$ |
16,460 |
|
Deferred debt issuance costs are recorded within other long-term assets on our consolidated balance sheets and are amortized to interest expense, net, in our consolidated income statements on a straight-line basis, which approximates the effective yield amortization method, over the term of the related credit facility.
Interest Rate Swaps and Caps
During 2009, we used $28.7 million of the net proceeds to us from the issuance of our 7.75% Notes to settle and terminate certain interest rate swap contracts with a notional amount of $390.0 million. Subsequently, we paid $6.1 million to settle and terminate our remaining interest rate swap contract with a notional amount of $50.0 million. As a result of the settlement and termination of these interest rate swap contracts, we recognized a charge of $30.9 million (approximately $18.9 million after tax) during the year ended December 31, 2009 within total other non-operating expense, net, in our consolidated income statement. At December 31, 2008, approximately $23.3 million, net of taxes, was recorded in accumulated other comprehensive loss, related to our interest rate swap contracts. As a result of their settlement and termination, we reclassified $23.3 million out of accumulated other comprehensive loss to loss on extinguishment of debt within total other non-operating expense, net, in our consolidated income statement for the year ended December 31, 2009.
At December 31, 2009, we had two interest rate cap contracts with a notional principal amount of $175.0 million outstanding for which the cap rate of 6.00% was significantly higher than prevailing market interest rates; therefore, the fair market value was zero. The interest rate caps matured in February of 2010.
Additional Information Regarding Our Financial Covenants
The lenders under our revolving credit facility use adjusted EBITDA as the basis of calculation of our compliance with our Leverage Ratio (Total Debt divided by the last twelve months’ adjusted EBITDA) and Interest Coverage Ratio (last twelve months’ adjusted EBITDA divided by Cash Interest Expense). Both the Leverage Ratio and the Interest Coverage Ratio are measures our lenders use to monitor our performance and ability to generate positive cash flows.
Adjusted EBITDA is defined in our revolving credit facility as net income plus interest expense, depreciation and amortization, income taxes, and other adjustments. Other adjustments include, but are not limited to, the following to the extent that they are included in net income:
· Write offs, write-downs, asset revaluations and other non-cash charges, losses, and expenses, including non-cash equity compensation expense;
· Impairments of intangibles and goodwill;
· Extraordinary gains and losses;
· Fees paid pursuant to our Management Agreement, as amended, with Talecris Holdings, LLC, which was terminated in connection with our IPO;
· Fees and expenses incurred in connection with transactions and permitted acquisitions and investments;
· Extraordinary, unusual, or non-recurring charges and expenses including transition, restructuring, and “carve-out” expenses;
· Legal, accounting, consulting, and other expenses relating to potential or actual issuances of equity interests, including an initial public offering of common stock;
· Costs associated with our Special Recognition Bonuses; and
· Other items.
In addition to our lenders, adjusted EBITDA is used by our management and the compensation committee of our Board of Directors.
Our management uses adjusted EBITDA as one of our primary financial performance measures in the day-to-day oversight of our business to, among other things, allocate financial and human resources across our organization, determine appropriate levels of capital investment and research and development spending, determine staffing needs and develop hiring plans, manage our plants’ production plans, and assess appropriate levels of sales and marketing initiatives. Our management uses adjusted EBITDA in its decision making because this supplemental operating performance measure facilitates internal comparisons to historical operating results and external comparisons to competitors’ historical operating results by eliminating various income and expense items which are either not part of operating income or may vary significantly when comparing our results among the periods presented to our competitors or other companies.
The compensation committee of our Board of Directors uses adjusted EBITDA as a financial performance objective because it is one of our primary financial performance measures used in the day-to-day oversight of our business to, among other things, allocate financial and human resources across our organization, determine appropriate levels of capital investment and research and development spending, determine staffing needs and develop hiring plans, manage our plants’ production plans, and assess appropriate levels of sales and marketing initiatives. In order to motivate top performance by our executives, we establish a target level for each of the various performance criteria that is high enough that there is no certainty it is achievable. The target level for any performance criterion changes from year to year. These target performance levels reflect challenges with respect to various factors such as sales volume and pricing, cost control, working capital management, plasma platform objectives, R&D objectives and sales and marketing objectives, among others. Our compensation committee has discretion to adjust the actual results related to the performance targets positively or negatively for items which, in the opinion of the compensation committee, were not reasonably within management’s control. The compensation committee also evaluates the manner in which actual results were achieved to determine if unusual actions or risks were taken that would impact or manipulate the results.
13. Income Taxes
Components of our provision for income taxes are as follows:
|
|
|
Years Ended December 31, |
| |||||||
|
|
|
2010 |
|
2009 |
|
2008 |
| |||
|
Current provision: |
|
|
|
|
|
|
| |||
|
Federal |
|
$ |
58,907 |
|
$ |
68,960 |
|
$ |
28,639 |
|
|
State and local |
|
5,418 |
|
3,421 |
|
4,590 |
| |||
|
Foreign |
|
1,793 |
|
1,348 |
|
1,776 |
| |||
|
Total current provision |
|
66,118 |
|
73,729 |
|
35,005 |
| |||
|
Deferred provision: |
|
|
|
|
|
|
| |||
|
Federal |
|
12,366 |
|
69 |
|
7 |
| |||
|
State and local |
|
(105 |
) |
1,210 |
|
1,582 |
| |||
|
Total deferred provision |
|
12,261 |
|
1,279 |
|
1,589 |
| |||
|
Provision for income taxes |
|
$ |
78,379 |
|
$ |
75,008 |
|
$ |
36,594 |
|
A reconciliation of expected income tax expense at the U.S. Federal rate of 35% to actual income tax expense is as follows:
|
|
|
Years Ended December 31, |
| |||||||
|
|
|
2010 |
|
2009 |
|
2008 |
| |||
|
Amount computed at statutory rate |
|
$ |
85,556 |
|
$ |
80,114 |
|
$ |
35,837 |
|
|
State income taxes (net of Federal benefit) |
|
7,040 |
|
4,291 |
|
4,059 |
| |||
|
Research and development credits |
|
(11,426 |
) |
(7,732 |
) |
(4,052 |
) | |||
|
State tax credits (net of Federal benefit) |
|
(1,607 |
) |
(871 |
) |
(600 |
) | |||
|
Federal benefit of tax deductions for qualified production activities |
|
(6,080 |
) |
(2,764 |
) |
(2,037 |
) | |||
|
Capitalized transaction costs |
|
6,800 |
|
(2,352 |
) |
584 |
| |||
|
Nondeductible meals and entertainment expenses |
|
671 |
|
504 |
|
425 |
| |||
|
Other |
|
(2,575 |
) |
3,818 |
|
2,378 |
| |||
|
Provision for income taxes |
|
$ |
78,379 |
|
$ |
75,008 |
|
$ |
36,594 |
|
We calculate a provision for, or benefit from, income taxes using the asset and liability method. Deferred tax assets and liabilities are recognized for future consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carry-forwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. The major components of our deferred tax assets and liabilities are as follows:
|
|
|
December 31, |
| ||||
|
|
|
2010 |
|
2009 |
| ||
|
Current: |
|
|
|
|
| ||
|
Deferred income tax assets: |
|
|
|
|
| ||
|
Allowances on accounts receivable |
|
$ |
8,074 |
|
$ |
11,020 |
|
|
Inventories |
|
32,026 |
|
23,928 |
| ||
|
Revenue recognition |
|
1,455 |
|
7,857 |
| ||
|
Stock-based compensation |
|
23,855 |
|
30,952 |
| ||
|
Deferred bonuses |
|
4,200 |
|
4,617 |
| ||
|
Accrued expenses |
|
21,994 |
|
4,568 |
| ||
|
State tax credit carry-forward |
|
3,335 |
|
3,195 |
| ||
|
Other |
|
2,341 |
|
3,543 |
| ||
|
Total deferred income tax assets |
|
97,280 |
|
89,680 |
| ||
|
Deferred income tax liabilities: |
|
|
|
|
| ||
|
Other liabilities |
|
(687 |
) |
(1,028 |
) | ||
|
Total deferred income tax liabilities |
|
(687 |
) |
(1,028 |
) | ||
|
Net current deferred income tax assets |
|
$ |
96,593 |
|
$ |
88,652 |
|
|
|
|
|
|
|
| ||
|
Non-current: |
|
|
|
|
| ||
|
Deferred income tax assets: |
|
|
|
|
| ||
|
Property, plant, and equipment |
|
$ |
— |
|
$ |
14,170 |
|
|
Other |
|
459 |
|
252 |
| ||
|
Total deferred income tax assets |
|
459 |
|
14,422 |
| ||
|
Deferred income tax liabilities: |
|
|
|
|
| ||
|
Property, plant, and equipment |
|
(1,292 |
) |
— |
| ||
|
Intangibles |
|
(13,599 |
) |
(8,574 |
) | ||
|
Total deferred income tax liabilities |
|
(14,891 |
) |
(8,574 |
) | ||
|
Net non-current deferred income tax (liabilities) assets |
|
$ |
(14,432 |
) |
$ |
5,848 |
|
|
|
|
|
|
|
| ||
|
Net deferred income tax assets |
|
$ |
82,161 |
|
$ |
94,500 |
|
We record a valuation allowance to reduce our deferred tax assets to the amount that we believe is more likely than not to be realized. In assessing the need for a valuation allowance, we consider all available evidence, both positive and negative, including historical levels of income, expectations and risks associated with future taxable income, and ongoing prudent and feasible tax planning strategies.
We have not provided for U.S. Federal income and foreign withholding taxes on our non-U.S. subsidiaries’ cumulative undistributed earnings of approximately $13.2 million as of December 31, 2010 as such earnings are intended to be reinvested outside of the U.S. indefinitely. It is not practicable to estimate the amount of tax that might be payable if some or all of such earnings were to be remitted, and foreign tax credits would be available to reduce or eliminate the resulting U.S. income tax liability.
At December 31, 2010 we had state tax credit carryforwards of $5.1 million that will start expiring in 2015. Our ability to offset future taxable income with tax credit carryforwards may be limited in certain circumstances, including changes in ownership.
The following table summarizes activity related to our gross unrecognized tax positions:
|
Unrecognized tax benefits at December 31, 2007 |
|
$ |
6,885 |
|
|
Additions for tax positions taken in the current year |
|
3,626 |
| |
|
Reductions for tax positions taken in a prior year |
|
(521 |
) | |
|
Unrecognized tax benefits at December 31, 2008 |
|
9,990 |
| |
|
Additions for tax positions taken during a prior year |
|
3,899 |
| |
|
Reductions for tax positions taken in a prior year |
|
(1,642 |
) | |
|
Unrecognized tax benefits at December 31, 2009 |
|
12,247 |
| |
|
Additions for tax positions taken in the current year |
|
2,485 |
| |
|
Reductions for tax positions taken in a prior year |
|
(5,908 |
) | |
|
Unrecognized tax benefits at December 31, 2010 |
|
$ |
8,824 |
|
As of December 31, 2010, our total gross unrecognized tax benefits were approximately $8.8 million, of which approximately $5.8 million would reduce our effective income tax rate if recognized. Interest and penalties related to unrecognized tax benefits are included in income tax expense. No material interest or penalties were incurred during the years presented.
The IRS exam of the 2005, 2006, and 2007 tax years is effectively settled as the Joint Committee on Taxation has completed its review of our position and has agreed not to take exception to the refund approved by the Internal Revenue Service in its report dated June 2009. The favorable resolution of this matter resulted in a release of $4.7 million gross unrecognized tax benefits.
We have analyzed our filing positions for all open years in all significant Federal, state, and foreign jurisdictions where we are required to file income tax returns. The periods subject to examination by the major tax jurisdictions where we conduct business are tax periods 2005 through 2010.
14. Commitments and Contingencies
Leases
We lease office buildings, plasma collection centers, refrigerated storage, furniture, machinery, computer equipment, and miscellaneous equipment under leasing agreements. The majority of our leases are operating leases. In addition to rent, certain of our leases require us to pay directly for taxes, insurance, maintenance, and other operating expenses. Future minimum lease payments required under our capital leases and non-cancellable operating leases as of December 31, 2010 are as follows:
|
|
|
|
|
Non-cancellable |
| ||
|
|
|
|
|
Operating |
| ||
|
|
|
Capital |
|
Leases |
| ||
|
2011 |
|
$ |
1,814 |
|
$ |
17,427 |
|
|
2012 |
|
1,836 |
|
14,614 |
| ||
|
2013 |
|
1,862 |
|
10,246 |
| ||
|
2014 |
|
1,830 |
|
8,921 |
| ||
|
2015 |
|
1,729 |
|
7,016 |
| ||
|
Thereafter |
|
4,845 |
|
27,198 |
| ||
|
Total future minimum lease payments |
|
13,916 |
|
$ |
85,422 |
| |
|
Less: amounts representing interest |
|
(4,376 |
) |
|
| ||
|
Present value of net minimum lease payments |
|
9,540 |
|
|
| ||
|
Less: current portion of capital lease obligations |
|
(860 |
) |
|
| ||
|
Total |
|
$ |
8,680 |
|
|
| |
In the preceding table, the future minimum annual rentals payable under non-cancellable leases denominated in foreign currencies have been calculated based upon the December 31, 2010 foreign currency exchange rates. Rental cost was approximately $18.2 million, $16.6 million, and $14.8 million for the years ended December 31, 2010, 2009, and 2008, respectively.
Employment Agreements
We have employment agreements and offer letters with certain of our employees which require payments generally ranging from 100% to 150% of the employee’s annual compensation if employment is terminated not for cause by us, or by the employee, for good reason, as defined. Certain of these arrangements also include provisions for payments of bonuses under our annual incentive plan and the vesting of equity awards, as well as other customary payments, such as accrued personal days, bonuses, continuing benefits, and outplacement services. In the event of a change of control of the Company, our share-based compensation plans permit accelerated vesting of awards under defined circumstances.
Customer Commitments
We have supply agreements with some of our customers which require us to provide certain minimum quantities of our products for various periods. At December 31, 2010, we currently anticipate being able to fill these supply agreements in the foreseeable future and we do not consider our potential exposure for unfilled customer orders to be material.
Litigation
We are involved in various legal and regulatory proceedings that arise in the ordinary course of business. We record accruals for such contingencies to the extent that we conclude that their occurrence is both probable and estimable. We consider many factors in making these assessments, including the professional judgment of experienced members of management and our legal counsel. We have estimated the likelihood of unfavorable outcomes and the amounts of such potential losses. In our opinion, the ultimate outcome of these proceedings and claims is not anticipated to have a material adverse effect on our consolidated financial position, results of operations, or cash flows. However, the ultimate outcome of litigation is unpredictable and actual results could be materially different from our estimates. We record anticipated recoveries under applicable insurance contracts when we are assured of recovery.
National Genetics Institute/Baxter Healthcare Corporation Litigation
In May 2008, Baxter Healthcare Corporation (Baxter) and National Genetics Institute (NGI), a wholly-owned subsidiary of Laboratory Corporation of America, filed a complaint in the U.S. District Court for the Eastern District of North Carolina, alleging that we infringed U.S. Patent Nos. 5,780,222, 6,063,563, and 6,566,052. They subsequently withdrew and re-filed the case in November 2008. The patents deal primarily with a method of screening large numbers of biological samples utilizing various pooling and matrix array strategies, and the complaint alleges that the patents are owned by Baxter and exclusively licensed to NGI. In November 2008, we filed our answer to their complaint, asserting anti-trust and other counterclaims, and filed a request for re-examination of the patents with the Patent and Trademark Office (PTO), which was subsequently granted. The case was settled effective October 1, 2010, with us paying $3.9 million to NGI, which was accrued in 2009, and us receiving a paid-up license to the technology subject to the disputed patents and the parties dismissing their claims and counterclaims.
Plasma Centers of America, LLC and G&M Crandall Limited Family Partnership
We had a three year Amended and Restated Plasma Sale/Purchase Agreement with Plasma Centers of America, LLC (PCA) under which we were required to purchase annual minimum quantities of plasma from plasma collection centers approved by us, including the prepayment of 90% for unlicensed plasma. We were also committed to finance the development of up to eight plasma collection centers, which were to be used to source plasma for us. Under the terms of the agreement, we had a conditional obligation to purchase such centers for a sum determined by a formula set forth in the agreement. We provided $3.2 million in financing, including accrued interest, related to the development of such centers, and we advanced payment of $1.0 million for unlicensed plasma. We recorded a provision within SG&A during 2008 related to these loans and advances.
In August 2008, we notified PCA that they were in breach of the Amended and Restated Plasma Sale/Purchase Agreement. We terminated the agreement in September 2008. In November 2008, TPR filed suit in federal court in Raleigh against the G&M Crandall Limited Family Partnership and its individual partners as guarantors of obligations of PCA. We were served in January 2009 in parallel state action by PCA, alleging breach of contract by TPR. The federal case has been stayed. On December 13, 2010, a jury in the state court case rendered a verdict in the amount of $37.0 million in favor of PCA against TPR in a breach of contract claim, which was confirmed by the court in post trial motions. We intend to appeal. The jury verdict, if sustained, will bear simple interest at 8% per statute from the date of breach, which totals approximately $6.7 million at December 31, 2010. We have recorded a $43.7 million charge within other non-operating expense, net, in our consolidated income statement for the year ended December 31, 2010 as a result of the judgment.
Foreign Corrupt Practices Act
We are conducting an internal investigation into potential violations of the Foreign Corrupt Practices Act (FCPA) that we became aware of during the conduct of an unrelated review. The FCPA investigation is being conducted by outside counsel under the
direction of a special committee of our board of directors. The investigation initially focused on sales to certain Eastern European and Middle Eastern countries, primarily Belarus, Russia, and Iran, but we are also reviewing sales practices in Brazil, Bulgaria, China, Georgia, Libya, Poland, Turkey, Ukraine, and other countries as deemed appropriate.
In July 2009, we voluntarily contacted the U.S. Department of Justice (DOJ) to advise them of the investigation and to offer our cooperation in any investigation that they want to conduct or they want us to conduct. The DOJ has not indicated what action it may take, if any, against us or any individual, or the extent to which it may conduct its own investigation. Even though we self-disclosed this matter to the DOJ, it or other federal agencies may seek to impose sanctions on us that may include, among other things, debarment, injunctive relief, disgorgement, fines, penalties, appointment of a monitor, appointment of new control staff, or enhancement of existing compliance and training programs. Other countries in which we do business may initiate their own investigations and impose similar penalties. As a result of this investigation, we suspended shipments to some of these countries while we put additional safeguards in place. In some cases, safeguards involved terminating consultants and suspending relations with or terminating distributors in countries under investigation as circumstances warranted. These actions unfavorably affected revenue from these countries in 2010 and 2009. We have resumed sales in countries where we believe we have appropriate safeguards in place and are reallocating product to other countries as necessary. To the extent that we conclude, or the DOJ concludes, that we cannot implement adequate safeguards or otherwise need to change our business practices, distributors, or consultants in affected countries or other countries, this may result in a permanent loss of business from those countries. We expect to complete our internal FCPA investigation and present our findings to the DOJ in 2011. The preliminary findings of our investigation indicate that it is probable that there were FCPA violations by persons associated with us that the DOJ or other regulators may assert are attributable to us.
Any sanctions or related loss of business could have a material adverse effect on us or our results of operations. It is possible, however, that any sanctions that DOJ or other federal agencies might otherwise consider imposing would be reduced, if not eliminated, in light of the comprehensive compliance measures that we have implemented. Given the preliminary nature of our findings, our continuing investigation and the uncertainties regarding this matter, we are unable to estimate the financial outcome and consequently, we have not accrued any amounts related to the outcome of this matter.
Compliance with Pharmaceutical Pricing Agreement
In November 2009, we received a letter from the United States Attorney’s Office for the Eastern District of Pennsylvania (USAO). The USAO requested a meeting to review our compliance with the terms of the Pharmaceutical Pricing Agreement (PPA) under the Public Health Service program. Specifically, the USAO asked for information related to the sale of our IGIV product, Gamunex, under that program. In order to have federal financial participation apply to their products under the Medicaid program and to obtain Medicare Part B coverage, manufacturers are required to enter into a PPA. The PPA obligates manufacturers to charge covered entities the Public Health Service price for drugs intended for outpatient use. The Public Health Service price is based on the Medicaid rebate amount. We believe that we have complied with the terms of the PPA and federal law. If the USAO determines that our practices are inconsistent with the terms of the PPA, the USAO has stated that it may file a civil action against us under the Anti-fraud Injunction Act and seek a court order directing the company to comply with the PPA or, potentially, proceed under some other legal theory. We could also be subject to fines, damages, penalties, appointment of a monitor, or enhancement of existing compliance and training programs as a result of government action. We are cooperating with the investigation and intend to respond to information requests from the USAO. Based on the information obtained to date, we have not determined that any potential liability that may result is probable or can be reasonably estimated. Therefore, we have not made any accrual in our consolidated financial statements at December 31, 2010.
Exclusive License Agreements with Crucell N.V. (Crucell)
During September 2008, we entered into an exclusive commercial license agreement with Crucell for recombinant technology. In consideration of the license that Crucell granted us, we paid an upfront license fee of $2.5 million, which we recorded in R&D in our consolidated income statement during the year ended December 31, 2008. During the year ended December 31, 2010, we paid $1.5 million to Crucell, which we recorded in R&D in our consolidated income statement. We could be required to pay up to $28.0 million of additional development milestones as certain activities are completed. Upon commercialization of the product, we are required to pay a royalty at a tiered rate, ranging between 3.5% and 6%, based on the related net sales of the product.
During December 2008, we entered into another exclusive commercial license agreement with Crucell for recombinant technology. In consideration of the license that Crucell granted us, we paid an upfront license fee of $1.5 million, which we recorded in R&D in our consolidated income statement for the year ended December 31, 2008. During the year ended December 31, 2009, we paid $0.5 million to Crucell, which is included in R&D in our consolidated income statement. We could be required to pay up to $18.5 million of additional development milestones as certain activities are completed. Upon commercialization of the product, we are required to pay a royalty at a tiered rate, ranging between 3% and 5%, based on the related net sales of the product.
Under the terms of both exclusive license agreements with Crucell, we may terminate either agreement by giving Crucell 90 days prior written notice and payment of all outstanding amounts owed to Crucell.
Purchase Commitments
We have purchase agreements that require us to purchase minimum annual quantities of plasma, other raw materials, and associated subcontracted manufacturing services and other services. At December 31, 2010, our purchase commitments, generally subject to annual price negotiations, are as follows:
|
2011 |
|
$ |
201,875 |
|
|
2012 |
|
179,850 |
| |
|
2013 |
|
167,637 |
| |
|
2014 |
|
127,080 |
| |
|
2015 |
|
121,308 |
| |
|
Thereafter |
|
55,595 |
| |
|
Total |
|
$ |
853,345 |
|
An inability of any of our suppliers to satisfy their obligations in a timely manner could cause a disruption in our plasma supply, which could materially adversely affect our business.
We entered into a contract, effective January 1, 2011, for subcontract manufacturing services that is substantially similar to, and replaces, one to which we were a party that expired on December 31, 2010. The minimum purchase obligations under this contract, which expires on December 31, 2012, of approximately $30 million for both 2011 and 2012 are not included in the table above.
On January 6, 2011, we entered into a contract for subcontract manufacturing services. The minimum purchase obligations of approximately $4.0 million for each of the years 2012 through 2015 are not included in the table above.
In addition to the minimum purchase commitments in the table above, at December 31, 2010, we have commitments and open purchase orders for capital spending to be made of $214.5 million.
Environmental Matters
Our operations are subject to extensive and evolving federal, state, and local environmental laws and regulations. Compliance with such laws and regulations can be costly. Additionally, governmental authorities may enforce the laws and regulations with a variety of civil and criminal enforcement measures, including monetary penalties and remediation requirements. It is possible that new information or future developments could require us to reassess our potential exposure related to environmental matters. We may incur significant costs and liabilities in order to comply with existing environmental laws and regulations. It is also possible that other developments, such as increasingly strict environmental laws and regulations and claims for damages to property, employees, other persons, and the environment resulting from current or past operations, could result in substantial costs and liabilities in the future as this information becomes available, or other relevant developments occur. We establish accrued liabilities or adjust previously accrued amounts accordingly. While there are still uncertainties relating to the ultimate costs we may incur, based upon our evaluation and experience to date, we believe that compliance with all applicable laws and regulations will not have a material adverse impact on our financial position, operating results, or cash flows. At December 31, 2010 and 2009, no amounts have been accrued as we are not currently aware of any probable liabilities.
Other
All pharmaceutical companies, including us, are subject to periodic inspections by the FDA and other regulatory authorities of manufacturing and plasma collection facilities, procedures, and processes. If in the course of an inspection, the FDA or other regulatory authority notes conditions they believe are objectionable with respect to cGMP or other applicable regulations, we must implement effective corrective actions or face regulatory or enforcement sanctions.
15. Related Party Transactions
Until January 21, 2010, a majority of our outstanding common stock was owned by Talecris Holdings, LLC. Talecris Holdings, LLC is owned by (i) Cerberus-Plasma Holdings LLC, the managing member of which is Cerberus Partners, L.P., and (ii) limited partnerships affiliated with Ampersand Ventures. Substantially all rights of management and control of Talecris Holdings, LLC are held by Cerberus-Plasma Holdings LLC. As of December 31, 2010, Talecris Holdings, LLC owned approximately 48.7% of our outstanding common stock. We had a management agreement with Cerberus-Plasma Holdings LLC and an affiliate of Ampersand Ventures, which was terminated as of September 30, 2009 in connection with our IPO. We have a Master Consulting and Advisory Services Agreement with an affiliate of Cerberus to provide certain advisory services to us.
We have an equity investment in Centric as further discussed in Note 11, “Investment in Affiliate;” therefore, we consider Centric to be a related party during the periods presented.
The following table summarizes our related party transactions for the years ended December 31, 2010, 2009, and 2008 and our related party accounts payable balances at December 31, 2010 and 2009:
|
|
|
Years Ended December 31, |
| |||||||
|
Expenses |
|
2010 |
|
2009 |
|
2008 |
| |||
|
Centric (product distribution and other services) |
|
$ |
22,865 |
|
$ |
20,306 |
|
$ |
17,508 |
|
|
Cerberus/Ampersand (management fees) |
|
$ |
— |
|
$ |
5,715 |
|
$ |
6,871 |
|
|
Cerberus (operational support) |
|
$ |
— |
|
$ |
608 |
|
$ |
4,184 |
|
|
|
|
December 31, |
| ||||
|
Payable |
|
2010 |
|
2009 |
| ||
|
Centric (product distribution and other services) |
|
$ |
6,406 |
|
$ |
5,537 |
|
|
Cerberus (operational support) |
|
$ |
— |
|
$ |
349 |
|
16. Equity Transactions
On October 6, 2009, we completed our IPO of 56,000,000 shares of our common stock, par value $0.01 per share. Additional information regarding our IPO is included in Note 4, “Initial Public Offering and Use of Proceeds.”
A seven-for-one share dividend on our common stock was paid on September 10, 2009. All share and per-share amounts have been retroactively adjusted for all periods presented to reflect the share dividend.
On September 30, 2009, 1,000,000 shares of our Series A preferred stock and 192,310 shares of our Series B preferred stock were converted into an aggregate of 85,846,320 shares of our common stock in connection with our IPO. In addition, on September 30, 2009, 2,381,548 shares of our common stock were issued to settle $45.3 million of accrued dividends upon the conversion of our Series A and B preferred stock. Additional information regarding our Series A and B preferred stock is included in Note 17, “Redeemable Series A and B Senior Convertible Preferred Stock.”
During the year ended December 31, 2008, we repurchased 2,146,232 shares of our common stock from IBR for $35.4 million at a put value of $15.61 per share plus accrued charges. The shares were issued to IBR during the year ended December 31, 2007 as a result of the acceleration of the contingent consideration provision of our November 2006 Asset Purchase Agreement, as amended. Additional information regarding the shares repurchased from IBR is included in Note 6 “Business Acquisitions.”
During the year ended December 31, 2008, our board of directors approved the retirement of 2,212,640 shares of our common stock held in treasury, and approved that shares of our common stock repurchased in the future would be immediately retired by the Company.
Information regarding employee share-based compensation is included in Note 18, “Share-Based Compensation.”
17. Redeemable Series A and B Senior Convertible Preferred Stock
On September 30, 2009, 1,000,000 shares of our Series A preferred stock and 192,310 shares of our Series B preferred stock were converted into an aggregate of 85,846,320 shares of our common stock in connection with our IPO. In addition, on September 30, 2009, 2,381,548 shares of our common stock were issued to settle $45.3 million of accrued dividends upon the conversion of our Series A and B preferred stock.
18. Share-Based Compensation
In connection with our IPO, we ceased further grants under our 2005 Stock Option and Incentive Plan and 2006 Restricted Stock Plan. The Talecris Biotherapeutics Holdings Corp. 2009 Long-Term Incentive Plan (2009 Plan), which was adopted by our board of directors on August 7, 2009, became effective in connection with our IPO. The 2009 Plan provides for the grant of awards in the form of incentive stock options, nonqualified stock options, share appreciation rights, restricted stock, RSU’s, unrestricted shares of common stock, deferred share units, and performance awards. Our employees, directors, and consultants are eligible to receive awards under the 2009 Plan. The maximum number of shares that we may issue for all awards under the 2009 Plan is 7,200,000. As of December 31, 2010, 5,798,837 shares remain available for grant under the 2009 Plan.
We value share-based compensation at the grant date using a fair value model and recognize this value as expense over the employees’ requisite service period, typically the period over which the share-based compensation vests. We classify share-based compensation costs consistent with each grantee’s salary. In connection with stock option exercise and restricted share vesting, we recognized net tax benefits of $26.6 million, $20.4 million, and $3.3 million for the years ended December 31, 2010, 2009 and 2008, respectively. We record income tax benefits realized upon exercise or vesting of an award in excess of that previously recognized in earnings as additional paid-in-capital. We recognized excess tax benefits related to share-based compensation of $13.5 million and $13.4 million during the years ended December 31, 2010 and 2009, respectively.
Share-based compensation expense for the years ended December 31, 2010, 2009, and 2008 was as follows:
|
|
|
Years Ended December 31, |
| |||||||
|
|
|
2010 |
|
2009 |
|
2008 |
| |||
|
SG&A |
|
$ |
12,606 |
|
$ |
40,968 |
|
$ |
33,780 |
|
|
R&D |
|
1,024 |
|
2,303 |
|
2,361 |
| |||
|
Cost of goods sold |
|
3,336 |
|
4,275 |
|
2,566 |
| |||
|
Total expense |
|
$ |
16,966 |
|
$ |
47,546 |
|
$ |
38,707 |
|
|
|
|
|
|
|
|
|
| |||
|
Capitalized in inventory |
|
$ |
2,265 |
|
$ |
3,574 |
|
$ |
3,233 |
|
Amounts capitalized in inventory are recognized in cost of goods sold in our consolidated income statements primarily within twelve months.
The following table summarizes the remaining unrecognized compensation cost related to our share-based compensation awards as of December 31, 2010 and the weighted average period over which the non-cash compensation cost is expected to be recognized:
|
|
|
|
|
Weighted- |
| |
|
|
|
Unrecognized |
|
Average |
| |
|
|
|
Compensation |
|
Period |
| |
|
|
|
Cost |
|
(Years) |
| |
|
Stock options |
|
$ |
3,965 |
|
2.10 |
|
|
Restricted share awards |
|
721 |
|
0.25 |
| |
|
RSU’s |
|
6,589 |
|
2.10 |
| |
|
Performance share awards |
|
1,138 |
|
1.25 |
| |
|
Total |
|
$ |
12,413 |
|
|
|
In addition to the unrecognized compensation cost included in the table above, at December 31, 2010, $1.7 million of compensation cost was included in inventory on our consolidated balance sheet, which we expect to be recognized as non-cash compensation expense in our consolidated income statement primarily within the next twelve months. The amount of share-based compensation expense that we will ultimately be required to record could change in the future as a result of additional grants, changes in the fair value of shares for performance-based awards, differences between our anticipated forfeiture rate and the actual forfeiture rate, the probability of achieving targets established for performance award vesting, and other actions by our board of directors or its compensation committee.
Stock Options
Stock options, which entitle the holder to purchase, after the end of a vesting term, a specified number of shares of our common stock at a price per share equal to the exercise price, are accounted for at fair value at the date of the grant. Option awards are granted with an exercise price at least equal to the fair value of our common stock at the date of grant and generally vest over periods of three to five years. The exercise price of stock options is determined by our board of directors. The stock options that we granted to employees typically have service-based and performance-based components. The stock options granted to members of our board of directors are service-based only. Our stock options generally expire ten years after the date of grant, or earlier if an option holder ceases to be employed by the Company. Stock option exercises are settled with newly issued common stock previously authorized and available for issuance.
The following is a summary of stock option activity for the years ended December 31, 2010, 2009, and 2008:
|
|
|
|
|
|
|
Weighted |
|
|
| ||
|
|
|
|
|
|
|
Average |
|
|
| ||
|
|
|
|
|
Weighted |
|
Remaining |
|
|
| ||
|
|
|
|
|
Average |
|
Contractual |
|
Aggregate |
| ||
|
|
|
|
|
Exercise |
|
Term |
|
Intrinsic |
| ||
|
|
|
Shares |
|
Price |
|
(Years) |
|
Value |
| ||
|
Outstanding at December 31, 2007 |
|
12,932,344 |
|
$ |
6.42 |
|
|
|
|
| |
|
Granted |
|
2,291,304 |
|
$ |
10.98 |
|
|
|
|
| |
|
Forfeited |
|
(945,232 |
) |
$ |
2.98 |
|
|
|
|
| |
|
Outstanding at December 31, 2008 |
|
14,278,416 |
|
$ |
6.96 |
|
|
|
|
| |
|
Granted |
|
638,472 |
|
$ |
18.91 |
|
|
|
|
| |
|
Forfeited |
|
(392,688 |
) |
$ |
4.89 |
|
|
|
|
| |
|
Exercised |
|
(2,394,762 |
) |
$ |
3.17 |
|
|
|
|
| |
|
Outstanding at December 31, 2009 |
|
12,129,438 |
|
$ |
8.40 |
|
6.7 |
|
$ |
168,235 |
|
|
Granted |
|
73,593 |
|
$ |
20.39 |
|
|
|
|
| |
|
Forfeited |
|
(24,950 |
) |
$ |
19.05 |
|
|
|
|
| |
|
Exercised |
|
(3,650,579 |
) |
$ |
6.12 |
|
|
|
|
| |
|
Outstanding at December 31, 2010 |
|
8,527,502 |
|
$ |
9.45 |
|
5.7 |
|
$ |
118,090 |
|
|
|
|
|
|
|
|
|
|
|
| ||
|
Exercisable at December 31, 2010 |
|
7,882,452 |
|
$ |
8.66 |
|
4.8 |
|
$ |
115,419 |
|
|
|
|
|
|
|
|
|
|
|
| ||
|
Vested and expected to vest at December 31, 2010 |
|
8,488,004 |
|
$ |
9.36 |
|
5.7 |
|
$ |
118,296 |
|
At December 31, 2009 and 2008, stock options with a weighted average exercise price of $8.16 and $4.02 were exercisable for 8,711,838 shares and 6,950,872 shares, respectively. Our estimate of the stock options vested and expected to vest at December 31, 2010 considers an expected forfeiture rate of 3%.
The aggregate intrinsic value in the table above represents the difference between the $23.30 closing price of our common stock as reported by The NASDAQ Global Select Market on December 31, 2010 and the weighted average exercise price, multiplied by the number of options outstanding or exercisable. The total intrinsic value and net cash proceeds to us from stock option exercises during the year ended December 31, 2010 were $62.7 million and $22.3 million, respectively. We do not record the aggregate intrinsic value for financial accounting purposes and the value changes based upon changes in the fair value of our common stock. The total fair value of stock options that vested during the years ended December 31, 2010, 2009, and 2008 were $56.1 million, $72.5 million, and $24.7 million, respectively.
The weighted average grant date fair value of options granted during the years ended December 31, 2010, 2009, and 2008 were $9.99, $9.49, and $4.35, respectively. We estimated the fair value of stock options at their grant date using the Black-Scholes option pricing model. This model was developed for use in estimating the fair value of traded options that have no vesting restrictions and are fully transferable, characteristics that are not present in our option grants. If the model permitted consideration of the unique characteristics of employee stock options, the resulting estimate of the fair value of stock options could be different. The following weighted-average assumptions were used to estimate the fair value of stock options granted during the years ended December 31, 2010, 2009, and 2008:
|
|
|
Years Ended December 31, |
| ||||
|
|
|
2010 |
|
2009 |
|
2008 |
|
|
Risk-free interest rate |
|
2.66 |
% |
2.66 |
% |
2.65 |
% |
|
Expected term (years) |
|
5.66 |
|
5.97 |
|
5.20 |
|
|
Expected volatility |
|
50 |
% |
50 |
% |
50 |
% |
|
Expected dividend yield |
|
0 |
% |
0 |
% |
0 |
% |
The risk-free interest rate is based upon the U.S. Treasury yield curve at the time of grant. The expected life of options is reflective of our historical experience, vesting schedules, and contractual terms. There is limited trading history for our common stock; therefore, our application of the Black-Scholes option pricing model incorporated historical volatility measures of similar public companies. We currently do not expect to pay dividends in the future. We generally apply a 3% forfeiture rate to the options granted over the term of the award. This rate is calculated based upon historical attrition rates and represents an estimate of the granted options not expected to vest. If actual forfeitures differ from the expected rate, we may be required to make additional adjustments to compensation expense in future periods.
Restricted Stock Units
RSU’s, which entitle the holder to receive, at the end of a vesting term, a specified number of shares of our common stock, are accounted for at fair value at the date of grant. We granted 483,100 RSU’s in connection with our IPO. These RSU’s will vest one-third on each of April 1 of 2011, 2012, and 2013, subject to the award holder being employed on the vesting date. The aggregate fair value of the RSU’s was $8.4 million, which will be recognized as compensation expense ratably through April of 2013. The following is a summary of RSU activity for the years ended December 31, 2010 and 2009:
|
|
|
|
|
|
|
Weighted |
|
|
| ||
|
|
|
|
|
|
|
Average |
|
|
| ||
|
|
|
|
|
Weighted |
|
Remaining |
|
|
| ||
|
|
|
|
|
Average |
|
Contractual |
|
Aggregate |
| ||
|
|
|
|
|
Grant Date |
|
Term |
|
Intrinsic |
| ||
|
|
|
Shares |
|
Fair Value |
|
(Years) |
|
Value |
| ||
|
Outstanding at December 31, 2008 |
|
— |
|
— |
|
|
|
|
| ||
|
Granted |
|
483,100 |
|
$ |
19.00 |
|
|
|
|
| |
|
Forfeited |
|
(3,076 |
) |
$ |
19.00 |
|
|
|
|
| |
|
Outstanding at December 31, 2009 |
|
480,024 |
|
$ |
19.00 |
|
|
|
|
| |
|
Granted |
|
36,015 |
|
$ |
20.40 |
|
|
|
|
| |
|
Forfeited |
|
(30,299 |
) |
$ |
19.04 |
|
|
|
|
| |
|
Vested |
|
(1,182 |
) |
$ |
19.00 |
|
|
|
|
| |
|
Outstanding at December 31, 2010 |
|
484,558 |
|
$ |
19.10 |
|
2.16 |
|
$ |
11,290 |
|
Restricted Stock
Restricted shares of our common stock, which entitle the holder to receive, at the end of a vesting term, a specified number of shares of our common stock, are accounted for at fair value at the date of grant. Restricted share awards vest on terms determined by our board of directors or its compensation committee at the time of the grant. The majority of our restricted share awards currently outstanding vest annually over a four-year period from the date of grant unless accelerated by the compensation committee upon the event of a change in control, as defined. Any restricted share awards that have not vested at the time of termination of service are forfeited except in the event of death, disability, or a change in control. The restricted share awards are considered issued and outstanding and have full voting rights. Any dividends declared with respect to our common stock will vest at the same time as the underlying restricted stock award.
The following is a summary of restricted share activity for the years ended December 31, 2010, 2009, and 2008:
|
|
|
|
|
Weighted |
| |
|
|
|
|
|
Average |
| |
|
|
|
|
|
Grant Date |
| |
|
|
|
Shares |
|
Fair Value |
| |
|
December 31, 2007 unvested shares outstanding |
|
2,811,000 |
|
$ |
13.78 |
|
|
Granted |
|
42,720 |
|
$ |
9.88 |
|
|
Forfeited |
|
(287,784 |
) |
$ |
11.00 |
|
|
Vested |
|
(870,432 |
) |
$ |
13.45 |
|
|
December 31, 2008 unvested shares outstanding |
|
1,695,504 |
|
$ |
14.40 |
|
|
Granted |
|
14,464 |
|
$ |
16.63 |
|
|
Forfeited |
|
(16,368 |
) |
$ |
11.00 |
|
|
Vested |
|
(779,744 |
) |
$ |
13.50 |
|
|
December 31, 2009 unvested shares outstanding |
|
913,856 |
|
$ |
15.27 |
|
|
Vested |
|
(727,256 |
) |
$ |
13.74 |
|
|
December 31, 2010 unvested shares outstanding |
|
186,600 |
|
$ |
21.25 |
|
The total fair value of restricted shares that vested during the years ended December 31, 2010, 2009, and 2008 were $14.5 million, $13.0 million and $8.6 million, respectively.
Performance Share Units (PSU’s)
The following is a summary of performance share unit activity for the year ended December 31, 2010:
|
|
|
|
|
Weighted |
| |
|
|
|
|
|
Average |
| |
|
|
|
|
|
Grant Date |
| |
|
|
|
Shares |
|
Fair Value |
| |
|
Outstanding PSU’s outstanding at December 31, 2009 |
|
— |
|
$ |
— |
|
|
Granted |
|
261,327 |
|
$ |
21.51 |
|
|
Forfeited |
|
(1,627 |
) |
$ |
21.51 |
|
|
Unvested PSU’s outstanding at December 31, 2010 |
|
259,700 |
|
$ |
21.51 |
|
PSU’s are awards that vest based on the achievement of pre-established objective performance goals, which are generally financial in nature. For performance awards, the compensation committee establishes a performance period and the performance targets for each performance measure that must be achieved at the end of the performance period for awards to vest. The number of shares issued upon the vesting of the performance awards varies based on actual performance in a year relative to a defined minimum and maximum financial target for that year. The PSU’s granted on March 8, 2010 will vest annually over a three-year performance period with the potential for 0% to 125% payout, based on the achievement of annual earnings per share targets that were established at the time of grant.
Other Information about our Stock Option Plan
During the third quarter of 2009, we entered into an amended and restated employment agreement with our Chairman and Chief Executive Officer which included accelerating the vesting of options to purchase 1,008,000 shares of our common stock at an exercise price of $21.25 per common share to August 19, 2009. The acceleration of these options resulted in the recognition of a non-cash charge of $11.8 million of compensation expense during the third quarter of 2009. Options to purchase these shares were previously scheduled to vest in April of 2010 (504,000 options) and April of 2011 (504,000 options).
During the second quarter of 2008, the compensation committee of our board of directors amended the exercise price of 570,400 stock options outstanding to certain employees from $21.25 per share to $11.00 per share and during the second quarter of 2008, the compensation committee also amended the exercise price of 17,152 stock options outstanding to certain members of our board of directors from $21.25 per share to $11.00 per share. The stock options that were re-priced were granted during 2007.
During the first quarter of 2008, our board of directors revised the 2008 corporate objectives related to the performance-based component of stock options scheduled to vest on April 1, 2009. In addition, during the second quarter of 2008, we began recognizing compensation cost related to the performance-based component of stock options scheduled to vest on April 1, 2010 based on our probability assessment of achieving the related performance objectives.
19. Employee Benefit Plans
Savings Plan and Profit Sharing Plan
We have a defined contribution plan (Savings Plan), which qualifies as a deferred salary arrangement under Section 401(k) of the Internal Revenue Code (IRC). Once eligible, employees may elect to contribute a portion of their wages to the Savings Plan, subject to certain limitations. We match 100% of the first 3% of employee contributions and 50% of the next 2% of employee contributions. Our contributions and the employee contributions are fully vested when contributed. The plan assets are held in trust and invested as directed by the plan participants. Matching contribution cost for the Savings Plan was $8.9 million for the year ended December 31, 2010 and $7.8 million for both the years ended December 31, 2009 and 2008, and is recorded consistent with each participant’s salary.
Under the profit sharing portion of the Savings Plan, we may elect to contribute to eligible employees’ Savings Plan accounts up to 3% of their eligible earnings, as defined. The profit sharing portion of the plan is discretionary, with the percentage amount determined by the compensation committee of our board of directors, based upon the attainment of certain financial targets as established by the compensation committee. Our cost for the profit sharing portion of the Savings Plan was $7.2 million, $5.8 million, and $7.9 million for the years ended December 31, 2010, 2009, and 2008, respectively, and is recorded consistent with each participant’s salary.
Supplemental Savings Plan
We have a Supplemental Savings Plan, which is an unfunded nonqualified deferred compensation plan in which employees at certain executive levels are eligible to defer pre-tax earnings as well as to make additional contributions, subject to certain limitations. Our matching contribution is similar to the Savings Plan described above and is fully vested when contributed. Matching contribution cost for the periods presented were not material to our consolidated financial statements. At December 31, 2010 and 2009, we have recorded $6.5 million and $5.1 million, respectively, within accrued expenses and other liabilities on our consolidated balance sheets.
Other Plans
We provide an unfunded defined benefit pension plan to certain of our Talecris Biotherapeutics, GmbH employees in Germany as required by statutory law. Pension cost related to this plan was not material for the periods presented. At December 31, 2010 and 2009, no material obligations related to this plan were recorded on our consolidated balance sheets.
20. Deferred Compensation
In October of 2006, the compensation committee of our board of directors approved a Special Recognition Bonus Plan (Bonus Plan) as a vehicle to award certain employees, senior executives, and members of our board of directors for the financial success of our Company from its inception through the effective date of the Bonus Plan. We recorded compensation expense of $0.1 million, $0.6 million, and $0.7 million for the years ended December 31, 2010, 2009, and 2008, respectively, related to the bonus plan. We made payments of $0.9 million in both March of 2010 and 2009, and $1.2 million in March of 2008. No further payments are due under the Bonus Plan.
In December of 2006, the compensation committee of our board of directors approved a restricted share and cash recognition award to certain employees, senior executives, and members of our board of directors for the financial success of our Company from its inception through the effective date of the award. We funded an irrevocable trust for the cash installments under this award. We made cash payments of $5.8 million, $6.0 million, and $7.4 million in March of 2010, 2009, and 2008, respectively, related to the cash recognition portion of the award. No further cash payments are due under this plan. We recorded compensation expense of $2.0 million, $5.7 million, and $5.9 million for the years ended December 31, 2010, 2009, and 2008, respectively, related to this award.
21. Segment Reporting
We operate our plasma-derived protein therapeutics business as a single reportable business segment since all operating activities are directed from our North Carolina headquarters and all of our products are derived from a single source and result from a common manufacturing process. All products are manufactured from a single raw material source, human plasma, and are processed in whole, or in part, at our principal manufacturing facilities located in Clayton, North Carolina. Our Melville, New York, facility primarily supplies intermediate plasma fractions to our Clayton facilities. Gamunex-C/Gamunex IGIV and Prolastin/Prolastin-C A1PI constitute the majority of our net revenue. Although we sell our products worldwide, the majority of our net revenue was concentrated in the United States and Canada for the periods presented.
In the following table, we have presented our net revenue by significant product category. Our Immunology/Neurology product category includes the products that are used to provide antibodies to patients who have a genetic or acquired inability to produce these antibodies, as well as a treatment for CIDP, and also products that provide antibodies to counter specific antigens such as rabies. Our Pulmonology product category is comprised of our Prolastin/Prolastin-C A1PI product, which is used to treat patients with a genetic alpha-1 antitrypsin deficiency. Our Critical Care/Hemostasis product category includes products that are used to supplement, restore, or maintain normal plasma parameters such as volume or coagulation values. Other product net revenue primarily consists of sales of PPF powder and intermediate products, such as cryoprecipitate. Other net revenue consists of royalties and licensing fees, milestones, and revenues related to contracted services performed for third parties at our Melville, New York facility.
|
|
|
Years Ended December 31, |
| |||||||
|
|
|
2010 |
|
2009 |
|
2008 |
| |||
|
Product net revenue: |
|
|
|
|
|
|
| |||
|
Immunology/Neurology |
|
$ |
964,145 |
|
$ |
928,054 |
|
$ |
781,408 |
|
|
Pulmonology |
|
351,499 |
|
319,080 |
|
316,495 |
| |||
|
Critical Care/Hemostasis |
|
175,071 |
|
167,469 |
|
134,216 |
| |||
|
Other |
|
86,221 |
|
93,151 |
|
102,431 |
| |||
|
Total product net revenue |
|
1,576,936 |
|
1,507,754 |
|
1,334,550 |
| |||
|
Other revenue |
|
24,683 |
|
25,455 |
|
39,742 |
| |||
|
Total net revenue |
|
$ |
1,601,619 |
|
$ |
1,533,209 |
|
$ |
1,374,292 |
|
In the following table, we have presented our net revenue by geographic region. Net revenue for each region is based on the geographic location of the customer.
|
|
|
Years Ended December 31, |
| |||||||
|
|
|
2010 |
|
2009 |
|
2008 |
| |||
|
United States |
|
$ |
1,098,969 |
|
$ |
1,011,468 |
|
$ |
906,376 |
|
|
Canada |
|
195,092 |
|
214,883 |
|
215,964 |
| |||
|
Europe |
|
196,571 |
|
185,297 |
|
168,081 |
| |||
|
Other |
|
110,987 |
|
121,561 |
|
83,871 |
| |||
|
Total net revenue |
|
$ |
1,601,619 |
|
$ |
1,533,209 |
|
$ |
1,374,292 |
|
We did not maintain significant long-lived assets outside of the United States at December 31, 2010 and 2009.
22. Earnings per Share
The following table illustrates the calculation of our basic earnings per common share outstanding for the periods presented:
|
|
|
Years Ended December 31, |
| |||||||
|
|
|
2010 |
|
2009 |
|
2008 |
| |||
|
Net income |
|
$ |
166,068 |
|
$ |
153,889 |
|
$ |
65,797 |
|
|
Less: |
|
|
|
|
|
|
| |||
|
Series A preferred stock undeclared dividends |
|
— |
|
(9,602 |
) |
(11,745 |
) | |||
|
Series B preferred stock undeclared dividends |
|
— |
|
(2,142 |
) |
(2,619 |
) | |||
|
Accretion of common stock put option |
|
— |
|
— |
|
(308 |
) | |||
|
Net income available to common stockholders |
|
$ |
166,068 |
|
$ |
142,145 |
|
$ |
51,125 |
|
|
|
|
|
|
|
|
|
| |||
|
Weighted average common shares outstanding |
|
123,323,722 |
|
31,166,613 |
|
1,310,448 |
| |||
|
|
|
|
|
|
|
|
| |||
|
Basic net income per common share |
|
$ |
1.35 |
|
$ |
4.56 |
|
$ |
39.01 |
|
The following table illustrates the calculation of our diluted earnings per common share outstanding for the periods presented:
|
|
|
Years Ended December 31, |
| |||||||
|
|
|
2010 |
|
2009 |
|
2008 |
| |||
|
Net income |
|
$ |
166,068 |
|
$ |
153,889 |
|
$ |
65,797 |
|
|
Less accretion of common stock put option |
|
— |
|
— |
|
(308 |
) | |||
|
Net income available to common stockholders |
|
$ |
166,068 |
|
$ |
153,889 |
|
$ |
65,489 |
|
|
|
|
|
|
|
|
|
| |||
|
Weighted average common shares outstanding |
|
123,323,722 |
|
31,166,613 |
|
1,310,448 |
| |||
|
Plus incremental shares from assumed conversions: |
|
|
|
|
|
|
| |||
|
Series A preferred stock |
|
— |
|
53,654,795 |
|
72,000,000 |
| |||
|
Series B preferred stock |
|
— |
|
10,318,354 |
|
13,846,320 |
| |||
|
Stock options and restricted shares |
|
5,603,331 |
|
7,374,601 |
|
5,605,032 |
| |||
|
Dilutive potential common shares |
|
128,927,053 |
|
102,514,363 |
|
92,761,800 |
| |||
|
|
|
|
|
|
|
|
| |||
|
Diluted net income per common share |
|
$ |
1.29 |
|
$ |
1.50 |
|
$ |
0.71 |
|
For the years ended December 31, 2010, 2009, and 2008, 604,914, 2,168,730, and 2,016,000 stock options were excluded from the calculation of diluted earnings per share due to their anti-dilutive effect.
23. Other Consolidated Balance Sheet Information
Information regarding other accounts on our consolidated balance sheets is as follows:
|
|
|
December 31, |
| ||||
|
|
|
2010 |
|
2009 |
| ||
|
Accrued expenses and other liabilities: |
|
|
|
|
| ||
|
Accrued goods and services |
|
$ |
80,769 |
|
$ |
45,044 |
|
|
Accrued payroll, bonuses, and employee benefits |
|
80,317 |
|
73,983 |
| ||
|
Medicaid, commercial rebates, and chargebacks |
|
29,744 |
|
30,771 |
| ||
|
Interest payable |
|
6,011 |
|
9,111 |
| ||
|
PCA judgment |
|
43,690 |
|
— |
| ||
|
Other |
|
11,195 |
|
11,624 |
| ||
|
Total accrued expenses and other liabilities |
|
$ |
251,726 |
|
$ |
170,533 |
|
24. Cash Flow Supplemental Disclosures
Supplemental Disclosures of Cash Flow Information
Cash paid for:
|
|
|
Years Ended December 31, |
| |||||||
|
|
|
2010 |
|
2009 |
|
2008 |
| |||
|
Interest, net of amounts capitalized (1) |
|
$ |
44,546 |
|
$ |
55,131 |
|
$ |
87,213 |
|
|
Income taxes |
|
$ |
63,025 |
|
$ |
56,849 |
|
$ |
48,910 |
|
(1) Interest paid in the table above excludes payments related to our interest rate swap contracts, which amounted to $17.0 million and $9.2 million for the years ended December 31, 2009 and 2008, respectively. The interest rate swap contracts were terminated and settled during the fourth quarter of 2009 as discussed in Note 12, “Long-Term Debt and Capital Lease Obligations.”
Changes in assets and liabilities, excluding the effects of business acquisitions:
|
|
|
Years Ended December 31, |
| |||||||
|
|
|
2010 |
|
2009 |
|
2008 |
| |||
|
Changes in: |
|
|
|
|
|
|
| |||
|
Accounts receivable |
|
$ |
(1,382 |
) |
$ |
8,575 |
|
$ |
(26,894 |
) |
|
Inventories |
|
(52,034 |
) |
(57,452 |
) |
(92,856 |
) | |||
|
Prepaid expenses and other assets |
|
653 |
|
7,987 |
|
(15,823 |
) | |||
|
Accounts payable |
|
(11,071 |
) |
16,143 |
|
16,594 |
| |||
|
Interest payable |
|
(3,100 |
) |
(2,239 |
) |
(1,957 |
) | |||
|
Accrued expenses and other liabilities |
|
94,460 |
|
14,877 |
|
20,881 |
| |||
|
Total |
|
$ |
27,526 |
|
$ |
(12,109 |
) |
$ |
(100,055 |
) |
Supplemental Schedule of Non-Cash Investing and Financing Activities
For the Year Ended December 31, 2010
We entered into a capital lease agreement related to a building with an unaffiliated third party. We recorded $0.3 million directly to property, plant, and equipment, net, and capital lease obligations.
We retired 246,823 shares of our common stock, which were withheld from employees for payment of withholding taxes.
For the Year Ended December 31, 2009
We entered into a number of capital lease agreements related to buildings with an unaffiliated third party. We recorded $4.9 million directly to property, plant, and equipment, net and capital lease obligations.
The common shares that we have issued to employees and members of our board of directors under our share-based compensation plans had an embedded feature that permitted the participant (or designated beneficiary or estate) to sell, or “put,” the shares of our common stock back to us at fair market value in the event of the participant’s termination of service due to death or disability. In addition, we had the right to repurchase, or “call,” the shares of our common stock upon a participant’s termination of continuous service, as defined. We recorded a fair market value adjustment of $6.6 million related to the vested shares of our common stock to increase obligations under common stock put/call option and decrease additional paid-in capital on our consolidated balance sheet. Both our redemption rights and the participants’ put rights were terminated in connection with the closing of our IPO. As a result, we reclassified the fair value of vested common stock totaling $39.9 million from obligations under common stock put/call option to permanent equity on our consolidated balance sheet.
We declared a dividend of $45.3 million related to our Series A and B preferred stock. In connection with our IPO, 1,000,000 shares of our Series A preferred stock and 192,310 shares of our Series B preferred stock were converted into an aggregate of 85,846,320 shares of our common stock. In addition, 2,381,548 shares of our common stock were issued to settle the $45.3 million preferred stock dividend upon conversion of our Series A and B preferred stock.
We retired 251,108 shares of our common stock, which were withheld from employees for payment of withholding taxes.
We reclassified a previously unrealized loss related to our interest rate swap contracts of approximately $23.3 million, net of income tax benefit of $14.2 million, to earnings as a result of their settlement and termination. In addition, we recorded other comprehensive income of $0.5 million. Additional information regarding the components of our comprehensive income is included in Note 2, “Summary of Significant Accounting Policies.”
We entered into two plasma center development agreements related to buildings to be leased from an unaffiliated third party during 2008, for which we made the decision not to open as plasma collection centers during 2009. As a result, we recorded a loss on contract obligations of $3.4 million, which decreased our assets under capital leases.
For the Year Ended December 31, 2008
We entered into a number of capital lease agreements related to buildings with an unaffiliated third party. We recorded $6.0 million directly to property, plant, and equipment, net and capital lease obligations.
We reclassified $1.6 million of long-lived assets related to two plasma collection centers, net of impairment charges of $0.8 million, to assets held for sale within prepaid expenses and other on our consolidated balance sheet.
As a result of the put feature related to the common shares issued under our share-based compensation plans described above, we recorded a fair value adjustment of $8.9 million to increase obligations under common stock put/call option and decrease additional paid-in capital on our consolidated balance sheet.
We issued shares of our common stock, which had an embedded put option, to IBR in connection with our 2006 business acquisition as described below. We recorded accretion of $0.3 million related to the IBR put option directly to obligations under common stock put/call option and additional paid-in capital on our consolidated balance sheet.
We retired 2,215,880 shares of our common stock, of which 2,146,232 shares were repurchased from IBR and 69,648 shares were withheld from employees for payment of withholding taxes.
We recorded other comprehensive loss of $11.8 million, primarily related to unrealized losses associated with our interest rate swap contracts, net of taxes.
25. Selected Unaudited Quarterly Financial Data
The following table summarizes our unaudited quarterly financial results for the years ended December 31, 2010 and 2009. In our opinion, the quarterly financial results presented below have been prepared on the same basis as our annual audited consolidated financial statements.
|
|
|
2010 Quarter Ended |
| ||||||||||
|
|
|
March 31 |
|
June 30 |
|
September 30 |
|
December 31 |
| ||||
|
Net revenue |
|
$ |
380,961 |
|
$ |
402,826 |
|
$ |
407,001 |
|
$ |
410,831 |
|
|
Cost of goods sold |
|
217,351 |
|
223,217 |
|
229,908 |
|
241,500 |
| ||||
|
Gross profit |
|
163,610 |
|
179,609 |
|
177,093 |
|
169,331 |
| ||||
|
Operating expenses |
|
83,891 |
|
91,892 |
|
80,056 |
|
100,821 |
| ||||
|
Operating income |
|
79,719 |
|
87,717 |
|
97,037 |
|
68,510 |
| ||||
|
Total other non-operating expense, net |
|
(11,116 |
) |
(11,944 |
) |
(11,256 |
) |
(54,220 |
) | ||||
|
Income before income taxes |
|
68,603 |
|
75,773 |
|
85,781 |
|
14,290 |
| ||||
|
(Provision) benefit for income taxes |
|
(23,264 |
) |
(28,150 |
) |
(29,729 |
) |
2,764 |
| ||||
|
Net income |
|
$ |
45,339 |
|
$ |
47,623 |
|
$ |
56,052 |
|
$ |
17,054 |
|
|
|
|
|
|
|
|
|
|
|
| ||||
|
Earnings per share: |
|
|
|
|
|
|
|
|
| ||||
|
Basic |
|
$ |
0.37 |
|
$ |
0.39 |
|
$ |
0.45 |
|
$ |
0.14 |
|
|
Diluted |
|
$ |
0.35 |
|
$ |
0.37 |
|
$ |
0.43 |
|
$ |
0.13 |
|
|
|
|
2009 Quarter Ended |
| ||||||||||
|
|
|
March 31 |
|
June 30 |
|
September 30 |
|
December 31 |
| ||||
|
Net revenue |
|
$ |
371,795 |
|
$ |
375,570 |
|
$ |
395,731 |
|
$ |
390,113 |
|
|
Cost of goods sold |
|
209,201 |
|
224,008 |
|
230,666 |
|
237,202 |
| ||||
|
Gross profit |
|
162,594 |
|
151,562 |
|
165,065 |
|
152,911 |
| ||||
|
Operating expenses |
|
88,963 |
|
81,023 |
|
95,655 |
|
95,511 |
| ||||
|
Operating income |
|
73,631 |
|
70,539 |
|
69,410 |
|
57,400 |
| ||||
|
Total other non-operating (expense) income, net |
|
(21,256 |
) |
54,582 |
|
(19,475 |
) |
(55,934 |
) | ||||
|
Income before income taxes |
|
52,375 |
|
125,121 |
|
49,935 |
|
1,466 |
| ||||
|
Provision for income taxes |
|
(18,940 |
) |
(41,849 |
) |
(14,125 |
) |
(94 |
) | ||||
|
Net income |
|
$ |
33,435 |
|
$ |
83,272 |
|
$ |
35,810 |
|
$ |
1,372 |
|
|
|
|
|
|
|
|
|
|
|
| ||||
|
Earnings per share: |
|
|
|
|
|
|
|
|
| ||||
|
Basic |
|
$ |
25.09 |
|
$ |
47.42 |
|
$ |
12.01 |
|
$ |
0.01 |
|
|
Diluted |
|
$ |
0.36 |
|
$ |
0.89 |
|
$ |
0.38 |
|
$ |
0.01 |
|
Earnings per share amounts for the 2010 and 2009 quarters and full years have been computed separately. Accordingly, quarterly amounts may not add to the annual amounts because of differences in the weighted average shares outstanding during each quarter due to the effect of potentially dilutive securities only in the periods in which such effect would be dilutive.
Our net income for the fourth quarter of 2010 includes a $43.7 million charge (approximately $26.6 million after tax) related to the PCA judgment. Additional information regarding the PCA judgment is included in Note 14, “Commitments and Contingencies.”
Our net income for the second quarter of 2009 includes a $75.0 million (approximately $48.8 million after tax) payment we received from CSL as a result of the termination of the definitive merger agreement. Our net income for the fourth quarter of 2009 includes a charge of $43.0 million (approximately $26.3 million after tax) as a result of the settlement and termination of our interest rate swap contracts and the write-off of deferred debt issuance costs associated with our First and Second Lien Term Loans. Additional information regarding our terminated merger agreement with CSL is included in Note 5, “Definitive Merger Agreement with CSL Limited (CSL)” and additional information regarding our refinancing transactions is included in Note 12, “Long-Term Debt and Capital Lease Obligations.”
26. Condensed Consolidating Financial Information
In October 2009, we completed the issuance of our 7.75% Notes. The 7.75% Notes are guaranteed on a senior unsecured basis by our existing and future domestic subsidiaries. The accompanying condensed consolidating financial information has been prepared and presented pursuant to SEC Regulation S-X, Rule 3-10, “Financial Statements of Guarantors and Issuers of Guaranteed Securities Registered or Being Registered.” Each of the subsidiary guarantors are 100% owned, directly or indirectly, by us, and all guarantees are full and unconditional and joint and several. Our investments in our consolidated subsidiaries are presented under the equity method of accounting. No significant administrative costs are borne by the Parent.
Talecris Biotherapeutics Holdings Corp.
Condensed Consolidating Balance Sheets
December 31, 2010
|
|
|
Parent/ |
|
Guarantor |
|
Non-Guarantor |
|
Consolidating |
|
|
| |||||
|
|
|
Issuer |
|
Subsidiaries |
|
Subsidiaries |
|
Adjustments |
|
Consolidated |
| |||||
|
Assets: |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Current assets: |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Cash |
|
$ |
— |
|
$ |
183,737 |
|
$ |
14,139 |
|
$ |
— |
|
$ |
197,876 |
|
|
Accounts receivable, net |
|
— |
|
248,124 |
|
37,793 |
|
(151,075 |
) |
134,842 |
| |||||
|
Inventories |
|
— |
|
657,493 |
|
37,006 |
|
— |
|
694,499 |
| |||||
|
Other |
|
— |
|
126,684 |
|
(429 |
) |
— |
|
126,255 |
| |||||
|
Total current assets |
|
— |
|
1,216,038 |
|
88,509 |
|
(151,075 |
) |
1,153,472 |
| |||||
|
Property, plant, and equipment, net |
|
— |
|
381,707 |
|
1,086 |
|
— |
|
382,793 |
| |||||
|
Intangible assets |
|
— |
|
10,880 |
|
— |
|
— |
|
10,880 |
| |||||
|
Goodwill |
|
— |
|
172,860 |
|
— |
|
— |
|
172,860 |
| |||||
|
Investment in Subsidiaries |
|
826,782 |
|
(36,573 |
) |
— |
|
(790,209 |
) |
— |
| |||||
|
Advances and notes between Parent and Subsidiaries |
|
1,397,133 |
|
826,919 |
|
— |
|
(2,224,052 |
) |
— |
| |||||
|
Other |
|
— |
|
18,167 |
|
281 |
|
— |
|
18,448 |
| |||||
|
Total assets |
|
$ |
2,223,915 |
|
$ |
2,589,998 |
|
$ |
89,876 |
|
$ |
(3,165,336 |
) |
$ |
1,738,453 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
|
Liabilities and Stockholders’ Equity (Deficit): |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Current liabilities: |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Accounts payable |
|
$ |
— |
|
$ |
96,503 |
|
$ |
114,547 |
|
$ |
(151,075 |
) |
$ |
59,975 |
|
|
Accrued expenses and other liabilities |
|
6,011 |
|
234,730 |
|
10,985 |
|
— |
|
251,726 |
| |||||
|
Current portion of capital lease obligations |
|
— |
|
860 |
|
— |
|
— |
|
860 |
| |||||
|
Total current liabilities |
|
6,011 |
|
332,093 |
|
125,532 |
|
(151,075 |
) |
312,561 |
| |||||
|
Long-term debt and capital lease obligations |
|
596,621 |
|
8,680 |
|
— |
|
— |
|
605,301 |
| |||||
|
Advances and notes between Parent and Subsidiaries |
|
826,919 |
|
1,397,133 |
|
— |
|
— |
) |
— |
| |||||
|
Other |
|
— |
|
25,310 |
|
917 |
|
— |
|
26,227 |
| |||||
|
Total liabilities |
|
1,429,551 |
|
1,763,216 |
|
126,449 |
|
(2,375,127 |
) |
944,089 |
| |||||
|
Stockholders’ equity (deficit) |
|
794,364 |
|
826,782 |
|
(36,573 |
) |
(790,209 |
) |
794,364 |
| |||||
|
Total liabilities and stockholders’ equity (deficit) |
|
$ |
2,223,915 |
|
$ |
2,589,998 |
|
$ |
89,876 |
|
$ |
(3,165,336 |
) |
$ |
1,738,453 |
|
Talecris Biotherapeutics Holdings Corp.
Condensed Consolidating Balance Sheets
December 31, 2009
|
|
|
Parent/ |
|
Guarantor |
|
Non-Guarantor |
|
Consolidating |
|
|
| |||||
|
|
|
Issuer |
|
Subsidiaries |
|
Subsidiaries |
|
Adjustments |
|
Consolidated |
| |||||
|
Assets: |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Current assets: |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Cash |
|
$ |
— |
|
$ |
58,320 |
|
$ |
6,919 |
|
$ |
— |
|
$ |
65,239 |
|
|
Accounts receivable, net |
|
— |
|
222,007 |
|
64,454 |
|
(149,483 |
) |
136,978 |
| |||||
|
Inventories |
|
— |
|
605,324 |
|
38,730 |
|
— |
|
644,054 |
| |||||
|
Other |
|
— |
|
117,670 |
|
2,448 |
|
— |
|
120,118 |
| |||||
|
Total current assets |
|
— |
|
1,003,321 |
|
112,551 |
|
(149,483 |
) |
966,389 |
| |||||
|
Property, plant, and equipment, net |
|
— |
|
266,067 |
|
1,132 |
|
— |
|
267,199 |
| |||||
|
Intangible assets |
|
— |
|
10,880 |
|
— |
|
— |
|
10,880 |
| |||||
|
Goodwill |
|
— |
|
172,860 |
|
— |
|
— |
|
172,860 |
| |||||
|
Investment in Subsidiaries |
|
680,459 |
|
(27,925 |
) |
— |
|
(652,534 |
) |
— |
| |||||
|
Advances and notes between Parent and Subsidiaries |
|
1,355,631 |
|
862,406 |
|
— |
|
(2,218,037 |
) |
— |
| |||||
|
Other |
|
— |
|
27,054 |
|
623 |
|
— |
|
27,677 |
| |||||
|
Total assets |
|
$ |
2,036,090 |
|
$ |
2,314,663 |
|
$ |
114,306 |
|
$ |
(3,020,054 |
) |
$ |
1,445,005 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
|
Liabilities and Stockholders’ Equity (Deficit): |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Current liabilities: |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Accounts payable |
|
$ |
— |
|
$ |
103,460 |
|
$ |
117,069 |
|
$ |
(149,483 |
) |
$ |
71,046 |
|
|
Accrued expenses and other liabilities |
|
9,111 |
|
150,936 |
|
10,486 |
|
— |
|
170,533 |
| |||||
|
Current portion of capital lease obligations |
|
— |
|
740 |
|
— |
|
— |
|
740 |
| |||||
|
Total current liabilities |
|
9,111 |
|
255,136 |
|
127,555 |
|
(149,483 |
) |
242,319 |
| |||||
|
Long-term debt and capital lease obligations |
|
596,046 |
|
9,221 |
|
— |
|
— |
|
605,267 |
| |||||
|
Advances and notes between Parent and Subsidiaries |
|
848,779 |
|
1,355,626 |
|
13,632 |
|
(2,218,037 |
) |
— |
| |||||
|
Other |
|
— |
|
14,221 |
|
1,044 |
|
— |
|
15,265 |
| |||||
|
Total liabilities |
|
1,453,936 |
|
1,634,204 |
|
142,231 |
|
(2,367,520 |
) |
862,851 |
| |||||
|
Stockholders’ equity (deficit) |
|
582,154 |
|
680,459 |
|
(27,925 |
) |
(652,534 |
) |
582,154 |
| |||||
|
Total liabilities and stockholders’ equity (deficit) |
|
$ |
2,036,090 |
|
$ |
2,314,663 |
|
$ |
114,306 |
|
$ |
(3,020,054 |
) |
$ |
1,445,005 |
|
Talecris Biotherapeutics Holdings Corp.
Condensed Consolidating Income Statements
Year Ended December 31, 2010
|
|
|
Parent/ |
|
Guarantor |
|
Non-Guarantor |
|
Consolidating |
|
|
| |||||
|
|
|
Issuer |
|
Subsidiaries |
|
Subsidiaries |
|
Adjustments |
|
Consolidated |
| |||||
|
Net revenue |
|
$ |
— |
|
$ |
1,438,162 |
|
$ |
163,457 |
|
$ |
— |
|
$ |
1,601,619 |
|
|
Cost of goods sold |
|
— |
|
783,710 |
|
128,266 |
|
— |
|
911,976 |
| |||||
|
Gross profit |
|
— |
|
654,452 |
|
35,191 |
|
— |
|
689,643 |
| |||||
|
Operating expenses |
|
— |
|
315,350 |
|
41,310 |
|
— |
|
356,660 |
| |||||
|
Income from operations |
|
— |
|
339,102 |
|
(6,119 |
) |
— |
|
332,983 |
| |||||
|
Equity in earnings (losses) of Subsidiaries |
|
166,068 |
|
(7,998 |
) |
— |
|
(158,070 |
) |
— |
| |||||
|
Other non-operating (expense) income, net |
|
— |
|
(88,555 |
) |
19 |
|
— |
|
(88,536 |
) | |||||
|
Income (loss) before income taxes |
|
166,068 |
|
242,549 |
|
(6,100 |
) |
(158,070 |
) |
244,447 |
| |||||
|
Provision for income taxes |
|
— |
|
(76,481 |
) |
(1,898 |
) |
— |
|
(78,379 |
) | |||||
|
Net income (loss) |
|
$ |
166,068 |
|
$ |
166,068 |
|
$ |
(7,998 |
) |
$ |
(158,070 |
) |
$ |
166,068 |
|
Talecris Biotherapeutics Holdings Corp.
Condensed Consolidating Income Statements
Year Ended December 31, 2009
|
|
|
Parent/ |
|
Guarantor |
|
Non-Guarantor |
|
Consolidating |
|
|
| |||||
|
|
|
Issuer |
|
Subsidiaries |
|
Subsidiaries |
|
Adjustments |
|
Consolidated |
| |||||
|
Net revenue |
|
$ |
— |
|
$ |
1,389,172 |
|
$ |
144,037 |
|
$ |
— |
|
$ |
1,533,209 |
|
|
Cost of goods sold |
|
— |
|
784,635 |
|
116,442 |
|
— |
|
901,077 |
| |||||
|
Gross profit |
|
— |
|
604,537 |
|
27,595 |
|
— |
|
632,132 |
| |||||
|
Operating expenses |
|
5,715 |
|
321,525 |
|
33,912 |
|
— |
|
361,152 |
| |||||
|
Income (loss) from operations |
|
(5,715 |
) |
283,012 |
|
(6,317 |
) |
— |
|
270,980 |
| |||||
|
Equity in earnings (losses) of Subsidiaries |
|
108,854 |
|
(7,466 |
) |
— |
|
(101,388 |
) |
— |
| |||||
|
Other non-operating (expense) income, net |
|
75,000 |
|
(117,106 |
) |
23 |
|
— |
|
(42,083 |
) | |||||
|
Income (loss) before income taxes |
|
178,139 |
|
158,440 |
|
(6,294 |
) |
(101,388 |
) |
228,897 |
| |||||
|
(Provision) benefit for income taxes |
|
(24,250 |
) |
(49,586 |
) |
(1,172 |
) |
— |
|
(75,008 |
) | |||||
|
Net income (loss) |
|
$ |
153,889 |
|
$ |
108,854 |
|
$ |
(7,466 |
) |
$ |
(101,388 |
) |
$ |
153,889 |
|
Talecris Biotherapeutics Holdings Corp.
Condensed Consolidating Income Statements
Year Ended December 31, 2008
|
|
|
Parent/ |
|
Guarantor |
|
Non-Guarantor |
|
Consolidating |
|
|
| |||||
|
|
|
Issuer |
|
Subsidiaries |
|
Subsidiaries |
|
Adjustments |
|
Consolidated |
| |||||
|
Net revenue |
|
$ |
— |
|
$ |
1,232,366 |
|
$ |
141,926 |
|
$ |
— |
|
$ |
1,374,292 |
|
|
Cost of goods sold |
|
— |
|
764,952 |
|
117,205 |
|
— |
|
882,157 |
| |||||
|
Gross profit |
|
— |
|
467,414 |
|
24,721 |
|
— |
|
492,135 |
| |||||
|
Operating expenses |
|
6,871 |
|
257,281 |
|
29,378 |
|
— |
|
293,530 |
| |||||
|
Income (loss) from operations |
|
(6,871 |
) |
210,133 |
|
(4,657 |
) |
— |
|
198,605 |
| |||||
|
Equity in earnings (losses) of Subsidiaries |
|
70,263 |
|
(5,590 |
) |
— |
|
(64,673 |
) |
— |
| |||||
|
Other non-operating (expense) income, net |
|
— |
|
(96,832 |
) |
618 |
|
— |
|
(96,214 |
) | |||||
|
Income (loss) before income taxes |
|
63,392 |
|
107,711 |
|
(4,039 |
) |
(64,673 |
) |
102,391 |
| |||||
|
(Provision) benefit for income taxes |
|
2,405 |
|
(37,448 |
) |
(1,551 |
) |
— |
|
(36,594 |
) | |||||
|
Net income (loss) |
|
$ |
65,797 |
|
$ |
70,263 |
|
$ |
(5,590 |
) |
$ |
(64,673 |
) |
$ |
65,797 |
|
Talecris Biotherapeutics Holdings Corp.
Condensed Consolidating Statements of Cash Flows
Year Ended December 31, 2010
|
|
|
Parent/ |
|
Guarantor |
|
Non-Guarantor |
|
Consolidating |
|
|
| |||||
|
|
|
Issuer |
|
Subsidiaries |
|
Subsidiaries |
|
Adjustments |
|
Consolidated |
| |||||
|
Cash flows from operating activities: |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Net income (loss) |
|
$ |
166,068 |
|
$ |
166,068 |
|
$ |
(7,998 |
) |
$ |
(158,070 |
) |
$ |
166,068 |
|
|
Undistributed equity in (earnings) losses of Subsidiaries |
|
(166,068 |
) |
7,998 |
|
— |
|
158,070 |
|
— |
| |||||
|
Adjustments to reconcile net income (loss) to net cash flows provided by (used in) operating activities |
|
— |
|
39,946 |
|
29,728 |
|
19,740 |
|
89,414 |
| |||||
|
Net cash provided by (used in) operating activities |
|
— |
|
214,012 |
|
21,730 |
|
19,740 |
|
255,482 |
| |||||
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
|
Cash flows from investing activities: |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Purchases of property, plant, and equipment |
|
— |
|
(152,484 |
) |
(365 |
) |
— |
|
(152,849 |
) | |||||
|
Net advances and notes between Parent and Subsidiaries |
|
(30,897 |
) |
— |
|
— |
|
30,897 |
|
— |
| |||||
|
Other |
|
— |
|
14,397 |
|
(13,632 |
) |
— |
|
765 |
| |||||
|
Net cash (used in) provided by investing activities |
|
(30,897 |
) |
(138,087 |
) |
(13,997 |
) |
30,897 |
|
(152,084 |
) | |||||
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
|
Cash flows from financing activities: |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Net advances and notes between Parent and Subsidiaries |
|
— |
|
50,637 |
|
— |
|
(50,637 |
) |
— |
| |||||
|
Other |
|
30,897 |
|
(1,145 |
) |
— |
|
— |
|
29,752 |
| |||||
|
Net cash provided by (used in) financing activities |
|
30,897 |
|
49,492 |
|
— |
|
(50,637 |
) |
29,752 |
| |||||
|
Effect of exchange rate changes on cash and cash equivalents |
|
— |
|
— |
|
(513 |
) |
— |
|
(513 |
) | |||||
|
Net increase in cash and cash equivalents |
|
— |
|
125,417 |
|
7,220 |
|
— |
|
132,637 |
| |||||
|
Cash and cash equivalents at beginning of year |
|
— |
|
58,320 |
|
6,919 |
|
— |
|
65,239 |
| |||||
|
Cash and cash equivalents at end of year |
|
$ |
— |
|
$ |
183,737 |
|
$ |
14,139 |
|
$ |
— |
|
$ |
197,876 |
|
Talecris Biotherapeutics Holdings Corp.
Condensed Consolidating Statements of Cash Flows
Year Ended December 31, 2009
|
|
|
Parent/ |
|
Guarantor |
|
Non-Guarantor |
|
Consolidating |
|
|
| |||||
|
|
|
Issuer |
|
Subsidiaries |
|
Subsidiaries |
|
Adjustments |
|
Consolidated |
| |||||
|
Cash flows from operating activities: |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Net income (loss) |
|
$ |
153,889 |
|
$ |
108,854 |
|
$ |
(7,466 |
) |
$ |
(101,388 |
) |
$ |
153,889 |
|
|
Undistributed equity in (earnings) losses of Subsidiaries |
|
(108,854 |
) |
7,466 |
|
— |
|
101,388 |
|
— |
| |||||
|
Adjustments to reconcile net income (loss) to net cash flows provided by (used in) operating activities |
|
(2,007 |
) |
64,009 |
|
4,759 |
|
13,505 |
|
80,266 |
| |||||
|
Net cash provided by (used in) operating activities |
|
43,028 |
|
180,329 |
|
(2,707 |
) |
13,505 |
|
234,155 |
| |||||
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
|
Cash flows from investing activities: |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Purchases of property, plant, and equipment |
|
— |
|
(74,576 |
) |
(587 |
) |
— |
|
(75,163 |
) | |||||
|
Business acquisitions, net of cash acquired |
|
— |
|
(30,431 |
) |
— |
|
— |
|
(30,431 |
) | |||||
|
Net advances and notes between Parent and Subsidiaries |
|
(1,172,950 |
) |
— |
|
— |
|
1,172,950 |
|
— |
| |||||
|
Other |
|
— |
|
(2,788 |
) |
3,764 |
|
— |
|
976 |
| |||||
|
Net cash (used in) provided by investing activities |
|
(1,172,950 |
) |
(107,795 |
) |
3,177 |
|
1,172,950 |
|
(104,618 |
) | |||||
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
|
Cash flows from financing activities: |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Net repaments of borrowings |
|
— |
|
(1,196,515 |
) |
— |
|
— |
|
(1,196,515 |
) | |||||
|
Issuance of 7.75% Notes, net of discount |
|
595,926 |
|
— |
|
— |
|
— |
|
595,926 |
| |||||
|
Proceeds from initial public offering, net of issuance costs |
|
517,192 |
|
— |
|
— |
|
— |
|
517,192 |
| |||||
|
Net advances and notes between Parent and Subsidiaries |
|
— |
|
1,186,455 |
|
— |
|
(1,186,455 |
) |
— |
| |||||
|
Other |
|
16,804 |
|
(14,879 |
) |
— |
|
— |
|
1,925 |
| |||||
|
Net cash provided by (used in) financing activities |
|
1,129,922 |
|
(24,939 |
) |
— |
|
(1,186,455 |
) |
(81,472 |
) | |||||
|
Effect of exchange rate changes on cash and cash equivalents |
|
— |
|
— |
|
195 |
|
— |
|
195 |
| |||||
|
Net increase in cash and cash equivalents |
|
— |
|
47,595 |
|
665 |
|
— |
|
48,260 |
| |||||
|
Cash and cash equivalents at beginning of year |
|
— |
|
10,726 |
|
6,253 |
|
— |
|
16,979 |
| |||||
|
Cash and cash equivalents at end of year |
|
$ |
— |
|
$ |
58,321 |
|
$ |
6,918 |
|
$ |
— |
|
$ |
65,239 |
|
Talecris Biotherapeutics Holdings Corp.
Condensed Consolidating Statements of Cash Flows
Year Ended December 31, 2008
|
|
|
Parent/ |
|
Guarantor |
|
Non-Guarantor |
|
Consolidating |
|
|
| |||||
|
|
|
Issuer |
|
Subsidiaries |
|
Subsidiaries |
|
Adjustments |
|
Consolidated |
| |||||
|
Cash flows from operating activities: |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Net income (loss) |
|
$ |
65,797 |
|
$ |
70,263 |
|
$ |
(5,590 |
) |
$ |
(64,673 |
) |
$ |
65,797 |
|
|
Undistributed equity in (earnings) losses of Subsidiaries |
|
(70,263 |
) |
5,590 |
|
— |
|
64,673 |
|
— |
| |||||
|
Adjustments to reconcile net income (loss) to net cash flows provided by (used in) operating activities |
|
424 |
|
14,776 |
|
(63,115 |
) |
15,132 |
|
(32,783 |
) | |||||
|
Net cash provided by (used in) operating activities |
|
(4,042 |
) |
90,629 |
|
(68,705 |
) |
15,132 |
|
33,014 |
| |||||
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
|
Cash flows from investing activities: |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Purchases of property, plant, and equipment |
|
— |
|
(86,051 |
) |
(161 |
) |
— |
|
(86,212 |
) | |||||
|
Business acquisitions, net of cash acquired |
|
— |
|
(10,272 |
) |
— |
|
— |
|
(10,272 |
) | |||||
|
Net advances and notes between Parent and Subsidiaries |
|
40,160 |
|
— |
|
— |
|
(40,160 |
) |
— |
| |||||
|
Other |
|
— |
|
(15,455 |
) |
— |
|
— |
|
(15,455 |
) | |||||
|
Net cash (used in) provided by investing activities |
|
40,160 |
|
(111,778 |
) |
(161 |
) |
(40,160 |
) |
(111,939 |
) | |||||
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
|
Cash flows from financing activities: |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Net borrowings |
|
— |
|
58,712 |
|
— |
|
— |
|
58,712 |
| |||||
|
Net advances and notes between Parent and Subsidiaries |
|
— |
|
(34,896 |
) |
9,868 |
|
25,028 |
|
— |
| |||||
|
Other |
|
(36,118 |
) |
— |
|
— |
|
— |
|
(36,118 |
) | |||||
|
Net cash provided by (used in) financing activities |
|
(36,118 |
) |
23,816 |
|
9,868 |
|
25,028 |
|
22,594 |
| |||||
|
Effect of exchange rate changes on cash and cash equivalents |
|
— |
|
91 |
|
(248 |
) |
— |
|
(157 |
) | |||||
|
Net increase in cash and cash equivalents |
|
— |
|
2,758 |
|
(59,246 |
) |
— |
|
(56,488 |
) | |||||
|
Cash and cash equivalents at beginning of year |
|
— |
|
7,968 |
|
65,499 |
|
— |
|
73,467 |
| |||||
|
Cash and cash equivalents at end of year |
|
$ |
— |
|
$ |
10,726 |
|
$ |
6,253 |
|
$ |
— |
|
$ |
16,979 |
|
27. Subsequent Events
Special Meeting of Stockholders
On February 14, 2011, we held a special meeting at which holders of a majority of our outstanding common stock approved the adoption of the Agreement and Plan of Merger, dated as of June 6, 2010, among Grifols and Talecris Biotherapeutics Inc. The completion of the transaction is subject to obtaining certain regulatory approvals and other customary conditions. Grifols agreed to provide written notice to the FTC staff at least thirty days prior to closing the transaction and, in any event, not to close the transaction until after 11:59 p.m. on March 20, 2011. There can be no assurance that Grifols will reach resolution with the FTC by March 20, 2011. Under the pending merger agreement, if this transaction is not closed by the current “outside date” of March 6, 2011, then under specified circumstances, either Grifols or we may elect to cause the “outside date” to be extended to a date not later than the expiration of Grifols financing for the transaction, or September 6, 2011, whichever is earlier.
Foreign Currency Hedging Program
In order to reduce the impact of volatility of foreign exchange rates on intercompany transactions, we initiated a foreign currency hedging program in the first quarter of 2011 related to both known and anticipated intercompany transactions of approximately one year in duration or less. The effective portion of the changes in fair value of these instruments is reported in other comprehensive income and reclassified into earnings in the same period or periods in which the hedged transactions affect earnings.
The changes in fair value of the hedges against firm commitments will be recognized in general and administrative expenses consistent with the underlying intercompany receivables being hedged. The changes in the fair value hedges against anticipated intercompany sales will be recognized as an adjustment to revenues when the hedged inventory sells through to third parties.
During the first quarter of 2011 we entered into approximately $49.5 (€37.1) million in notional value of fair value hedges against firm commitments and $38.4 (€28.2) million in notional value of cash flow hedges against anticipated future sales. The weighted average U.S. dollar to euro exchange rate on these foreign currency contracts is 1.3461.
These derivative financial instruments present certain market and counterparty risks. We seek to manage the counterparty risks associated with these contracts by limiting transactions to counterparties with which we have established banking relationships and limit the duration of the contracts to less than one year. We are exposed to potential losses if a counterparty fails to perform according to the terms of the agreement. We do not require collateral or other security to be furnished by counterparties to our derivative financial instruments. There can be no assurance, however, that our practice effectively mitigates counterparty risk. A number of financial institutions similar to those that serve or may serve as counterparties to our hedging arrangements were adversely affected by the global credit crisis. The failure of any of the counterparties to our hedging arrangements to fulfill their obligations to us could adversely affect our results of operations and cash flows.
Talecris Biotherapeutics Holdings Corp.
Valuation and Qualifying Accounts
Years Ended December 31, 2010, 2009, and 2008
(in thousands)
|
|
|
Balance at |
|
Charges to |
|
Charges to |
|
|
|
Balance at |
| |||||
|
|
|
Beginning of |
|
Costs and |
|
Other |
|
|
|
End of |
| |||||
|
|
|
Period |
|
Expenses |
|
Accounts |
|
Deductions |
|
Period |
| |||||
|
Reserve for doubtful accounts receivable: |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Year ended December 31, 2010 |
|
$ |
3,461 |
|
$ |
3,519 |
|
$ |
— |
|
$ |
(3,727 |
)(1) |
$ |
3,253 |
|
|
Year ended December 31, 2009 |
|
$ |
2,020 |
|
$ |
2,858 |
|
$ |
— |
|
$ |
(1,417 |
)(1) |
$ |
3,461 |
|
|
Year ended December 31, 2008 |
|
$ |
2,631 |
|
$ |
728 |
|
$ |
— |
|
$ |
(1,339 |
)(1) |
$ |
2,020 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
|
Reserve for doubtful notes receivable and other advances: |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Year ended December 31, 2010 |
|
$ |
4,250 |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
4,250 |
|
|
Year ended December 31, 2009 |
|
$ |
4,250 |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
4,250 |
|
|
Year ended December 31, 2008 |
|
$ |
— |
|
$ |
4,250 |
(3) |
$ |
— |
|
|
|
$ |
4,250 |
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
|
Inventory reserves: |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Year ended December 31, 2010 |
|
$ |
44,377 |
|
$ |
55,419 |
|
$ |
— |
|
$ |
(39,796 |
)(2) |
$ |
60,000 |
|
|
Year ended December 31, 2009 |
|
$ |
49,766 |
|
$ |
21,758 |
|
$ |
— |
|
$ |
(27,147 |
)(2) |
$ |
44,377 |
|
|
Year ended December 31, 2008 |
|
$ |
47,534 |
|
$ |
36,840 |
|
$ |
— |
|
$ |
(34,608 |
)(2) |
$ |
49,766 |
|
(1) Includes write-offs of uncollectible accounts receivable and the effects of foreign exchange.
(2) Includes the net of write-offs, reversals of reserved inventory that was sold or recoverable for other purposes such as testing, and the effects of foreign exchange
(3) Includes a provision for $3.2 million related to notes receivables and $1.0 million related to advances for unlicensed plasma from a then existing third party supplier due to uncertainty regarding collection.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
There were no changes in or disagreements with our accountants on accounting and financial disclosure matters.
ITEM 9A. CONTROLS AND PROCEDURES
Role of Controls and Procedures
Our management, including our Chief Executive Officer and Chief Financial Officer, does not expect that our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act) or our internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act) will prevent all errors and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the control system’s objectives will be met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of the controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within a company have been detected. These inherent limitations include the realities that judgments in decision-making can be faulty and that breakdowns can occur because of simple error and mistake. Controls can also be circumvented by the individual acts of some persons, by collusion of two or more people, or by management override of the controls. The design of any system of controls is based in part on certain assumptions about the likelihood of future events. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected. Also projections of any evaluation of effectiveness of controls and procedures to future periods are subject to the risk that the controls and procedures may become inadequate because of changes in conditions, or that the degree of compliance with the controls and procedures may have deteriorated.
Report of Management on Internal Control Over Financial Reporting
The report of management on internal control over financial reporting is set forth above under Part II, Item 8, “Financial Statements and Supplementary Data—Report of Management on Internal Control Over Financial Reporting.”
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive Officer and our Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act) as of the end of the period covered by this report. Based upon that evaluation, our Chief Executive Officer and Chief Financial Officer concluded that at December 31, 2010, our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act) are effective to provide reasonable assurance that information that we are required to disclose in the reports that we file or submit to the SEC is recorded, processed, summarized, and reported within the time periods specified in the SEC’s rules, our disclosure controls and procedures are designed to ensure that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate to allow timely decisions regarding required disclosure.
Changes in Internal Control over Financial Reporting
There was no change in our internal control over financial reporting (as defined in Rule 13a-15(f) and 15d-15(f) of the Exchange Act) that occurred during the quarter ended December 31, 2010 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
None.
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
Executive Officers
Our executive officers and their respective ages and positions as of February 1, 2011 are as follows:
|
Name |
|
Age |
|
Position |
|
Lawrence D. Stern |
|
54 |
|
Chairman, Chief Executive Officer and Director |
|
John M. Hanson |
|
58 |
|
Executive Vice President and Chief Financial Officer |
|
John F. Gaither, Jr. |
|
61 |
|
Executive Vice President, General Counsel and Secretary |
|
Mary J. Kuhn |
|
52 |
|
Executive Vice President, Operations |
|
John R. Perkins |
|
40 |
|
Executive Vice President, Global Commercial |
|
Stephen R. Petteway, Jr. PhD |
|
65 |
|
Executive Vice President, Research & Development |
|
Joel E. Abelson |
|
52 |
|
Senior Vice President and General Manager, Portfolio Management and International Business |
|
James R. Engle |
|
55 |
|
Senior Vice President, Information Technology |
|
Kari D. Heerdt |
|
43 |
|
Senior Vice President, Human Resources |
|
Thomas J. Lynch, PhD |
|
60 |
|
Senior Vice President, Corporate Compliance, Regulatory and Public Policy |
|
Daniel L. Menichella |
|
51 |
|
Senior Vice President, Corporate Development and Strategy |
|
Bruce Nogales |
|
54 |
|
Senior Vice President and General Manager, Talecris Plasma Resources |
Lawrence D. Stern has served as our Chairman and Chief Executive Officer since June 2007, as a director since April 2005, and currently serves as chairman of the executive committee and as a member of our compliance committee. He was previously our Executive Chairman from April 2005 until June 2007. From December 2003 until March 2005, Mr. Stern worked primarily with Cerberus and Ampersand in connection with our formation transaction. From January through November 2003, Mr. Stern worked as an independent consultant supporting debt and equity transactions. Mr. Stern received his BS in Chemical Engineering from Cornell University and his MS from the Massachusetts Institute of Technology.
John M. Hanson has served as Executive Vice President and Chief Financial Officer for our company since October 2005. From April 2003 until he joined us in 2005, Mr. Hanson was employed at Andrx Corporation, a specialty pharmaceuticals company, where he ultimately served as Senior Vice President and Chief Financial Officer. Mr. Hanson took some time off from August 2001 to March 2003 to pursue personal interests. Mr. Hanson has an extensive history of executive experience in the pharmaceuticals industry. From November 2000 to July 2001, Mr. Hanson served as Chief Financial Officer of Mylan Laboratories and from September 1996 to October 2000, Mr. Hanson served as Chief Financial Officer of Zenith Goldline Pharmaceuticals, Inc., the U.S. generic products subsidiary of IVAX Corporation. Mr. Hanson was employed by Arthur Andersen LLP from 1984 to 1995. Mr. Hanson is a Certified Public Accountant and earned both his BS in Biology and his MPA from the University of South Dakota.
John F. Gaither, Jr. has been our Executive Vice President, General Counsel and Secretary since September 2006. Prior to joining us in 2006, Mr. Gaither served as Senior Vice President, General Counsel and Secretary of NeighborCare, Inc., one of the largest providers of institutional pharmacy services in the United States, from 2003 through 2005. Mr. Gaither earned both his BBA in Accounting and his JD from the University of Notre Dame.
Mary J. Kuhn became our Executive Vice President, Operations, in August 2009 after being our Senior Vice President, Operations since April 2005. Prior to joining us, Ms. Kuhn was a Senior Vice President of Operations at Bayer from October 2002 to April 2005. During the previous two decades, Ms. Kuhn held numerous positions with increasing responsibility within Bayer’s North American Pharmaceutical Division’s Headquarters, including Supervisor, Pharmaceutical Formulations Lab; Manager, Pharmaceutical Technical Services; Manager, Manufacturing Operations; Director, Manufacturing; and, Vice President, Technical Operations and Ethical Product Manufacturing; and Vice-President of Operations. Ms. Kuhn earned her BS in Pharmacy from Purdue University and her EMBA from the University of New Haven.
John R. Perkins joined our company in 2006 and since May 2010 has served as our Executive Vice President, Global Commercial. From April 2008 until May 2010, he served as our Vice President and General Manager, Portfolio Management and U.S. Business. Previously, he was Vice President and General Manager, U.S. Commercial Operations. Prior to joining our company, Mr. Perkins was a consultant to Cerberus Capital Management where he provided executive management since 2004 for several existing portfolio companies, including Talecris, and operational due diligence for potential investments. Mr. Perkins joined Cerberus from General Electric where he was an executive in its Corporate Business Development group since 2001. Mr. Perkins started his career with General Electric where he held a variety of additional positions including marketing/product, sales, operations, and corporate audit. Mr. Perkins is a graduate of General Electric’s Management Development Program. He received his BA from DePauw University and earned his MBA from Northwestern’s Kellogg School of Management.
Stephen R. Petteway, Jr., PhD became our Executive Vice President, Research and Development, in August 2009 after being our Senior Vice President, Research & Development since April 2005. Prior to our formation transaction, Dr. Petteway held various positions for Bayer Biological Products, including: Senior Director, Pathogen Safety & Research; Vice President, Pathogen Safety & Research; and Acting Head, Research & Technology. Dr. Petteway is the author of numerous scientific publications and patents related to diagnostics and therapeutics. Dr. Petteway earned his BA in Microbiology/Chemistry from the University of South Florida at Tampa, and his PhD in Molecular Cell Biology from the University of Alabama at Birmingham.
Joel E. Abelson has served as our Senior Vice President and General Manager, Portfolio Management and International Business since April 2008. Previously, he was Vice President and General Manager, International Commercial Operations from August 2007 to April 2008 and before that he was Vice President, Canada and Intercontinental Commercial Operations from March 2006 to August 2007. From April 2005 until March 2006, Mr. Abelson worked in a consulting role for Talecris supporting both the formation of the new company and the planning and establishment of our Canadian business. From July 2002 to March 2005, Mr. Abelson served as Vice President, Global Strategic Marketing for Bayer’s Biological Products Division. Mr. Abelson received his BA (Honours) degree from Carleton University and his MA degree in Public Administration from the University of Toronto.
James R. Engle has been our Senior Vice President, Information Technology since April 2005. Prior to joining us in 2005, Mr. Engle was with Aventis from 1988 to 2004. His most recent position at Aventis was Senior Vice President, IT for Aventis Behring (subsequently acquired by CSL Limited and renamed CSL Behring) from 1999 to 2004. Mr. Engle attended Delaware Valley College of Science and Agriculture, where he earned his BS in Business Administration.
Kari D. Heerdt has been our Senior Vice President, Human Resources since January 2008. Prior to joining Talecris, Ms. Heerdt served as Senior Vice President of Human Resources for Mervyns, a mid-market department store partially owned by Cerberus Capital Management, from 2004 to 2008. Prior to that, she was a consultant to Cerberus Capital Management, a leading investment management firm. From 2000 to 2004, Ms. Heerdt was a Senior Consultant for Hewitt Associates, a global Human Resources consulting firm. Ms. Heerdt earned a BA degree from Mount Holyoke College, and a MS from the Krannert Graduate School of Management at Purdue University.
Thomas J. Lynch, PhD began as our Vice President of Corporate Governance and Compliance in April 2005, and assumed his current position as Senior Vice President, Corporate Compliance, Regulatory and Public Affairs in early 2006. From 2003 to 2005, Dr. Lynch served as Vice President of Regulatory Affairs and Quality Assurance for CryoLife, Inc., a medical device and human tissue company. Prior to that, from 2000 to 2003, he served as Senior Vice President, Regulatory Affairs and Quality Assurance at Clearant, a biotechnology company engaged in pathogen inactivation research & development. From 1993 to 2000, Dr. Lynch was with the U.S. Food and Drug Administration, most recently as Deputy Director of the Division of Hematology. He holds a Ph.D. in biochemistry from Wayne State University and a JD from Georgetown University.
Daniel L. Menichella joined our company in October 2007 as Senior Vice President, Corporate Development and Strategy. Prior to joining our company, Mr. Menichella served in various capacities related to business development and strategic planning at Merck KGaA, the global pharmaceutical and chemical company, from December 2002 until October 2007. Over the course of his five years at Merck KGaA, Mr. Menichella served as Vice President, Business Development, Vice President, Corporate Strategic Planning and Vice President, Corporate Business Development and Alliance Management. Mr. Menichella earned his AB from Harvard University and his MBA from the University of North Carolina at Chapel Hill.
Bruce Nogales joined our company in 2005 as Vice President, International, responsible for our business transitions in Europe and Intercontinental Markets. He has also served as Vice President of both the Immunology and Pulmonary Commercial Development groups. In April 2007, Mr. Nogales was named Vice President and General Manager, International Commercial Operations and in September 2007, Mr. Nogales was named Senior Vice President and General Manager, Talecris Plasma Resources. Prior to joining us, Mr. Nogales had served from 1999 through 2004 as both Vice President and General Manager of Intercontinental Commercial Operations and of the Wound Healing Business Unit at Aventis Behring (subsequently acquired by CSL Limited and renamed CSL Behring), which is engaged in the global protein therapeutics business. Mr. Nogales earned his BS in Finance from California State University, Long Beach in 1978 and completed an Executive Development Program at the University of Pennsylvania’s Wharton School of Business.
The information under the sections entitled “Board of Directors”, “Corporate Governance” and “Section 16(a) Beneficial Ownership Reporting Compliance” from our definitive proxy statement for the 2011 Annual Meeting, which is to be filed with the SEC not later than 120 days after the close of our fiscal year ended December 31, 2010 (the 2011 Proxy Statement), is hereby incorporated by reference.
ITEM 11. EXECUTIVE COMPENSATION
The information under the sections entitled “Compensation Discussion & Analysis”, “Executive Compensation,” “Compensation Committee Interlocks and Insider Participation,” and “Compensation Committee Report” from the 2011 Proxy Statement is hereby incorporated by reference.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
Equity Compensation Plan Information
The following table sets forth certain information regarding the following equity compensation plans, pursuant to which we have made equity compensation available to eligible participants as of December 31, 2010: 2005 Stock Option and Incentive Plan (2005 Plan), 2006 Restricted Stock Plan (2006 Plan), and 2009 Long-Term Incentive Plan (2009 Plan). In connection with our IPO, we ceased further grants under our 2005 and 2006 Plans and the 2009 Plan became effective. The 2009 Plan provides for the grant of awards in the form of incentive stock options, nonqualified stock options, share appreciation rights, restricted stock, restricted stock units (RSU’s), unrestricted shares of common stock, deferred share units, and performance awards. All of our plans have been approved by our stockholders.
|
Plan Category |
|
(a) |
|
(b) |
|
(c) |
| |
|
|
|
|
|
|
|
|
| |
|
Equity compensation plans approved by shareholders |
|
9,271,760 |
|
$ |
10.29 |
|
5,798,837 |
|
|
Equity compensation plans not approved by shareholders |
|
— |
|
— |
|
— |
| |
|
Total |
|
9,271,760 |
|
$ |
10.29 |
|
5,798,837 |
|
|
(a) |
Number of securities to be issued upon exercise of outstanding options, warrants, and rights |
|
(b) |
Weighted-average exercise price of outstanding options, warrants, and rights |
|
(c) |
Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) |
The maximum number of shares that we may issue for all awards under the 2009 Plan is 7,200,000. At December 31, 2010, 8,527,502, 484,558 and 259,700 stock options, RSU’s, and PSU’s respectively, were outstanding, and 5,798,837 securities were remaining for future issuance under the 2009 Plan. PSU’s are awards that vest based on the achievement of pre-established performance goals, which are generally financial in nature. The number of shares issued upon vesting of the performance awards varies based on actual performance in a year relative to defined minimum and maximum financial targets for that year. The PSU’s outstanding on December 31, 2010 were granted on March 8, 2010 and will vest annually over a three year performance period with the potential for 0% to 125% payout, based on the achievement of annual earnings per share targets that were established at the time of the grant. The number of securities disclosed in column “(a)” above assumes a 100% payout.
The other information under the section entitled “Security Ownership of Certain Beneficial Holders and Management” from the 2011 Proxy Statement is hereby incorporated by reference.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information under the sections entitled “Board of Directors” and “Certain Relationships and Related Person Transactions” from the 2011 Proxy Statement is hereby incorporated by reference.
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
The information under the section entitled “Audit and Non-Audit Fees” from the 2011 Proxy Statement is hereby incorporated by reference.
ITEM 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES
1. Financial Statements. The following are included in Item 8, “Financial Statements and Supplementary Data:”
Report of Management on Internal Control Over Financial Reporting
Report of Independent Registered Public Accounting Firm
Consolidated Balance Sheets at December 31, 2010 and 2009
Consolidated Income Statements for the years ended December 31, 2010, 2009, and 2008
Consolidated Statements of Cash Flows for the years ended December 31, 2010, 2009, and 2008
Consolidated Statements of Stockholders’ Equity (Deficit) for the years ended December 31, 2010, 2009, and 2008
Notes to Consolidated Financial Statements
2. Financial Statement Schedules. The following is included in Item 8, “Financial Statements and Supplementary Data:”
Schedule II- Valuation and Qualifying Accounts for the years ended December 31, 2010, 2009, and 2008
All other schedules for which provision is made in the applicable accounting regulations of the SEC are not required under the related instructions or are inapplicable and therefore have been omitted.
3. Exhibits
|
Exhibit Number |
|
Exhibit Description |
|
|
|
|
|
2.1 |
|
Agreement and Plan of Merger, dated as of June 6, 2010, by and among Grifols, S.A., Grifols, Inc. and Talecris Biotherapeutics Holdings Corp. (incorporated by reference to Exhibit 2.1 to our Current Report on Form 8-K filed on June 10, 2010) |
|
|
|
|
|
2.2 |
|
Amendment No. 1 to the Agreement and Plan of Merger, dated as of November 4, 2010, by and among Grifols, S.A., Grifols, Inc. and Talecris Biotherapeutics Holdings Corp. (incorporated by reference to Exhibit 2.2 to our Current Report on Form 8-K filed on November 5, 2010) |
|
|
|
|
|
2.3 |
|
Appraisal Indemnity Agreement, dated as of November 4, 2010, by and among Talecris Biotherapeutics Holdings Corp., Grifols, S.A., and Talecris Holdings, LLC, and solely with respect to the provisions of Section 9, Cerberus Capital Management, L.P. (incorporated by reference to Exhibit 2.3 to our Current Report on Form 8-K filed on November 5, 2010) |
|
|
|
|
|
3.1 |
|
Amended and Restated Certificate of Incorporation of Talecris Biotherapeutics Holdings Corp. (incorporated by reference to Exhibit 3.1 of Amendment No. 9 to our Registration Statement on Form S-1 (File No. 333-144941) filed on September 11, 2009) |
|
|
|
|
|
3.2 |
|
Amended and Restated By-Laws of Talecris Biotherapeutics Holdings Corp. (incorporated by reference to Exhibit 3.2 of Amendment No. 8 to our Registration Statement on Form S-1 (File No. 333-144941) filed on August 19, 2009) |
|
|
|
|
|
4.1 |
|
Indenture dated as of October 21, 2009 among Talecris Biotherapeutics Holdings Corp., the Guarantors named therein, and The Bank of New York Mellon Trust Company, N.A. (incorporated by reference to Exhibit 4.1 to our Current Report on Form 8-K filed on October 21, 2009) |
|
|
|
|
|
10.1 |
|
Revolving Credit Agreement, dated as of December 6, 2006, by and among Talecris Biotherapeutics Holdings Corp., Talecris Biotherapeutics, Inc., Precision Pharma Services, Inc., Talecris Plasma Resources, Inc., Wells Fargo Foothill, Inc. and Wachovia Bank, N.A. (incorporated by reference to Exhibit 10.12 of Amendment No. 1 to our Registration Statement on Form S-1 (File No. 333-144941) filed on September 24, 2007) |
|
|
|
|
|
10.2 |
|
First Amendment to Revolving Credit Agreement, dated as of October 12, 2009 by and among Talecris Biotherapeutics Holdings Corp., Wachovia Bank, National Association, Wells Fargo Foothill, Inc., and other signatories thereto (incorporated by reference to Exhibit 10.1 to our Current Report on Form 8-K filed on October 16, 2009) |
|
|
|
|
|
10.3 |
|
Supply Agreement, dated as of March 31, 2005, by and between Bayer Healthcare LLC, Biological Products Division, and Talecris Biotherapeutics, Inc. (F/K/A NPS Biotherapeutics, Inc.) (incorporated by reference to Exhibit 10.31 of Amendment No. 9 to our Registration Statement on Form S-1 (File No. 333-144941) filed on September 11, 2009)+ |
|
|
|
|
|
10.4 |
|
Amendment to Supply Agreement, dated as of June 30, 2008, by and between Bayer Healthcare LLC and Talecris Biotherapeutics, Inc. (incorporated by reference to Exhibit 10.31.1 of Amendment No. 9 to our Registration Statement on Form S-1 (File No. 333-144941) filed on September 11, 2009)+ |
|
|
|
|
|
10.5 |
|
Amended and Restated Plasma Sale/Purchase Agreement, dated October 13, 2006, by and between |
|
|
|
Talecris Biotherapeutics, Inc. and Interstate Blood Bank, Inc. (incorporated by reference to Exhibit 10.32 of Amendment No. 8 to our Registration Statement on Form S-1 (File No. 333-144941) filed on August 19, 2009)+ |
|
|
|
|
|
10.6 |
|
Profit Sharing Agreement, dated November 18, 2006, by and between International BioResources, L.L.C. and Talecris Plasma Resources, Inc. (incorporated by reference to Exhibit 10.19 of Amendment No. 1 to our Registration Statement on Form S-1 (File No. 333-144941) filed on September 24, 2007) |
|
|
|
|
|
10.7 |
|
Fractionation Services and Commercial Products Agreement, dated as of April 1, 2008, between and amongst Canadian Blood Services/Societe Canadienne Du Sang, Talecris Biotherapeutics, Inc. and Talecris Biotherapeutics, Ltd. (incorporated by reference to Exhibit 10.29 of Amendment No. 9 to our Registration Statement on Form S-1 (File No. 333-144941) filed on September 11, 2009)+ |
|
|
|
|
|
10.8 |
|
Fractionation Services and Commercial Products Agreement, dated as of April 1, 2008, between and amongst Héma-Québec, Talecris Biotherapeutics, Ltd. and Talecris Biotherapeutics, Inc. (incorporated by reference to Exhibit 10.30.1 of Amendment No. 9 to our Registration Statement on Form S-1 (File No. 333-144941) filed on September 11, 2009)+ |
|
|
|
|
|
10.9 |
|
Amending Agreement No. 1, effective as of May 26, 2008, to Fractionation Services and Commercial Products, dated as of April 1, 2008, by and among Héma-Québec, Talecris Biotherapeutics, Ltd. and Talecris Biotherapeutics, Inc. (incorporated by reference to Exhibit 10.30.2 of Amendment No. 6 to our Registration Statement on Form S-1 (File No. 333-144941) filed on July 2, 2009)+ |
|
|
|
|
|
10.10 |
|
Toll Manufacturing Agreement for Testing and Packaging, dated April 4, 2008, by and between Talecris Biotherapeutics, GmbH and Catalent France Limoges SAS (incorporated by reference to Exhibit 10.35 of Amendment No. 8 to our Registration Statement on Form S-1 (File No. 333-144941) filed on August 19, 2009)+ |
|
|
|
|
|
10.11 |
|
Plasma Sale/Purchase Agreement, dated as of August 12, 2008, by and between CSL Plasma Inc. (f/k/a ZLB Bioplasma Inc.) and Talecris Biotherapeutics, Inc. (incorporated by reference to Exhibit 10.33 of Amendment No. 7 to our Registration Statement on Form S-1 (File No. 333-144941) filed on July 21, 2009)+ |
|
|
|
|
|
10.12 |
|
Amended and Restated Services Agreement, dated January 1, 2009, by and between Talecris Biotherapeutics, Inc. and Centric Health Resources, Inc. (incorporated by reference to Exhibit 10.34 of Amendment No. 9 to our Registration Statement on Form S-1 (File No. 333-144941) filed on September 11, 2009)+ |
|
|
|
|
|
10.13 |
|
Retained Intellectual Property License Agreement, dated as of March 31, 2005, by and between Bayer Healthcare LLC and Talecris Biotherapeutics, Inc. (f/k/a NPS Biotherapeutics, Inc.) (incorporated by reference to Exhibit 10.31.1 of Amendment No. 1 to our Registration Statement on Form S-1 (File No. 333-144941) filed on September 24, 2007) |
|
|
|
|
|
10.14 |
|
Amendment to Retained Intellectual Property Licensing Agreement, entered into as of August 10, 2007, by and between Bayer Healthcare LLC and Talecris Biotherapeutics, Inc. (incorporated by reference to Exhibit 10.31.2 of Amendment No. 1 to our Registration Statement on Form S-1 (File No. 333-144941) filed on September 24, 2007) |
|
|
|
|
|
10.15 |
|
Management Agreement, dated as of March 31, 2005, by and between Cerberus-Plasma Holdings LLC, Ampersand 2001 Limited Partnership, Talecris Biotherapeutics Holdings Corp., and Talecris Biotherapeutics, Inc. (incorporated by reference to Exhibit 10.27.1 of Amendment No. 1 to our Registration Statement on Form S-1 (File No. 333-144941) filed on September 24, 2007) |
|
|
|
|
|
10.16 |
|
Amendment to Management Agreement, dated as of December 6, 2006, by and between Cerberus-Plasma Holdings LLC, Ampersand 2001 Limited Partnership, Talecris Biotherapeutics Holdings Corp., and Talecris Biotherapeutics, Inc. (incorporated by reference to Exhibit 10.27.2 of Amendment No. 1 to our Registration Statement on Form S-1 (File No. 333-144941) filed on September 24, 2007) |
|
|
|
|
|
10.17 |
|
Second Amendment to Management Agreement, dated August 17, 2009, by and between Cerberus-Plasma Holdings LLC, Ampersand 2001 Limited Partnership, Talecris Biotherapeutics Holdings Corp., and Talecris Biotherapeutics, Inc. (incorporated by reference to Exhibit 10.27.3 of Amendment No. 9 to our Registration Statement on Form S-1 (File No. 333-144941) filed on September 11, 2009) |
|
|
|
|
|
10.18 |
|
Stockholders Agreement, dated March 31, 2005, by and among Talecris Biotherapeutics Holdings Corp., Talecris Holdings, LLC and Bayer Healthcare LLC and its affiliates party thereto (incorporated by reference to Exhibit 10.16.1 of Amendment No. 8 to our Registration Statement on Form S-1 (File No. 333-144941) |
|
|
|
filed on August 19, 2009) |
|
|
|
|
|
10.19 |
|
First Amendment to Stockholders Agreement, dated August 17, 2009, by and among Talecris Biotherapeutics Holdings Corp. and Talecris Holdings, LLC (incorporated by reference to Exhibit 10.15.2 of Amendment No. 9 to our Registration Statement on Form S-1 (File No. 333-144941) filed on September 11, 2009) |
|
|
|
|
|
10.20 |
|
Stockholders Agreement, dated December 7, 2006, by and among Talecris Biotherapeutics Holdings Corp., Talecris Holdings, LLC and certain other stockholders of Talecris Biotherapeutics Holdings Corp. party thereto (incorporated by reference to Exhibit 10.21 of Amendment No. 1 to our Registration Statement on Form S-1 (File No. 333-144941) filed on September 24, 2007) |
|
|
|
|
|
10.21 |
|
Voting Agreement, dated as of June 6, 2010, between Talecris Biotherapeutics Holdings Corp. and Deria, S.A. (incorporated by reference to Exhibit 10.1 to our Current Report on Form 8-K filed on June 10, 2010) |
|
|
|
|
|
10.22 |
|
Agreement for Domestic and Global Relocation Services, dated July 14, 2008, by and between Talecris Biotherapeutics, Inc. and GMAC Global Relocation Services, LLC (incorporated by reference to Exhibit 10.36.1 of Amendment No. 8 to our Registration Statement on Form S-1 (File No. 333-144941) filed on August 19, 2009) |
|
|
|
|
|
10.23 |
|
Amendment to Agreement for Domestic and Global Relocation Services, dated August 15, 2008, by and between Talecris Biotherapeutics, Inc. and GMAC Global Relocation Services, LLC (incorporated by reference to Exhibit 10.36.2 of Amendment No. 8 to our Registration Statement on Form S-1 (File No. 333-144941) filed on August 19, 2009) |
|
|
|
|
|
10.24 |
|
Master Consulting and Advisory Services Agreement, dated as of July 18, 2008, by and between Cerberus Operations and Advisory Company LLC and Talecris Biotherapeutics Holding Corp. (incorporated by reference to Exhibit 10.37 of Amendment No. 9 to our Registration Statement on Form S-1 (File No. 333-144941) filed on September 11, 2009)+ |
|
|
|
|
|
10.25 |
|
Talecris Biotherapeutics Holdings Corp. 2005 Stock Option and Incentive Plan (incorporated by reference to Exhibit 10.1 of Amendment No. 1 to our Registration Statement on Form S-1 (File No. 333-144941) filed on September 24, 2007)* |
|
|
|
|
|
10.26 |
|
Talecris Biotherapeutics Holdings Corp. Supplemental Savings Plan (incorporated by reference to Exhibit 10.4 of Amendment No. 1 to our Registration Statement on Form S-1 (File No. 333-144941) filed on September 24, 2007)* |
|
|
|
|
|
10.27 |
|
Talecris Biotherapeutics Holdings Corp. Irrevocable Trust Agreement, dated as of December 6, 2006, by and between Talecris Biotherapeutics Holdings Corp. and Wilmington Trust Company, as trustee (incorporated by reference to Exhibit 10.36 of Amendment No. 1 to our Registration Statement on Form S-1 (File No. 333-144941) filed on September 24, 2007)* |
|
|
|
|
|
10.28 |
|
Talecris Biotherapeutics Holdings Corp. Incentive Plan (Management Bonus Plan), as amended (incorporated by reference to Exhibit 10.6 of Amendment No. 1 to our Registration Statement on Form S-1 (File No. 333-144941) filed on September 24, 2007)* |
|
|
|
|
|
10.29 |
|
Talecris Biotherapeutics Holdings Corp. 2009 Long-Term Incentive Plan (incorporated by reference to Exhibit 10.7 of Amendment No. 9 to our Registration Statement on Form S-1 (File No. 333-144941) filed on September 11, 2009)* |
|
|
|
|
|
10.30 |
|
Form of Stock Option Award Agreement under the 2009 Long-Term Incentive Plan of Talecris Biotherapeutics Holdings Corp. (incorporated by reference to Exhibit 10.7.2 of Amendment No. 11 to our Registration Statement on Form S-1 (File No. 333-144941) filed on September 28, 2009)* |
|
|
|
|
|
10.31 |
|
Form of Restricted Share Unit Award Agreement under the 2009 Long-Term Incentive Plan of Talecris Biotherapeutics Holdings Corp. (incorporated by reference to Exhibit 10.7.3 of Amendment No. 11 to our Registration Statement on Form S-1 (File No. 333-144941) filed on September 28, 2009)* |
|
|
|
|
|
10.32 |
|
Form of Performance Share Award Agreement under the 2009 Long-Term Incentive Plan of Talecris Biotherapeutics Holdings Corp.* |
|
|
|
|
|
10.33 |
|
Third Restated Employment Agreement, dated as of April 1, 2009, by and between Talecris Biotherapeutics Holdings Corp. and Lawrence D. Stern (incorporated by reference to Exhibit 10.22 of Amendment No. 8 to our Registration Statement on Form S-1 (File No. 333-144941) filed on August 19, 2009)* |
|
|
|
|
|
10.34 |
|
Employment Agreement, as amended and restated as of October 10, 2008, by and between Talecris Biotherapeutics Holdings Corp. and John M. Hanson (incorporated by reference to Exhibit 10.23 of Amendment No. 6 to our Registration Statement on Form S-1 (File No. 333-144941) filed on July 2, 2009)* |
|
10.35 |
|
Letter Agreement, effective as of June 6, 2010, amending the terms of a certain Employment Agreement, dated September 14, 2005, as subsequently amended and restated on October 10, 2008, by and between Talecris Biotherapeutics Holdings Corp. and John M. Hanson (incorporated by reference to Exhibit 10.1 to our Current Report on Form 8-K filed on June 8, 2010)* |
|
|
|
|
|
10.36 |
|
Employment Agreement, as amended and restated as of November 6, 2008, by and between Talecris Biotherapeutics Holdings Corp. and John F. Gaither, Jr. (incorporated by reference to Exhibit 10.25 of Amendment No. 6 to our Registration Statement on Form S-1 (File No. 333-144941) filed on July 2, 2009)* |
|
|
|
|
|
10.37 |
|
Letter Agreement, effective as of June 4, 2010, amending the terms of a certain Employment Agreement, dated September 5, 2006, as subsequently amended and restated on September 5, 2008 and November 6, 2008, by and between Talecris Biotherapeutics Holdings Corp. and John F. Gaither, Jr. (incorporated by reference to Exhibit 10.2 to our Current Report on Form 8-K filed on June 8, 2010)* |
|
|
|
|
|
10.38 |
|
Employment Letter, dated November 19, 2004, by and among NPS LLC, Bayer Healthcare LLC and Mary J. Kuhn (incorporated by reference to Exhibit 10.24.1 of Amendment No. 6 to our Registration Statement on Form S-1 (File No. 333-144941) filed on July 2, 2009)* |
|
|
|
|
|
10.39 |
|
Employment Offer Letter and supplemental termination provisions, dated February 8, 2005, by and between Talecris Biotherapeutics, Inc. and Mary J. Kuhn (incorporated by reference to Exhibit 10.24 of Amendment No. 5 to our Registration Statement on Form S-1 (File No. 333-144941) filed on July 23, 2008)* |
|
|
|
|
|
10.40 |
|
Letter Agreement, dated December 19, 2008, amending the terms of a certain Employment Letter, dated November 19, 2004, by and between Talecris Biotherapeutics, Inc. and Mary J. Kuhn (incorporated by reference to Exhibit 10.24.3 of Amendment No. 6 to our Registration Statement on Form S-1 (File No. 333-144941) filed on July 2, 2009)* |
|
|
|
|
|
10.41 |
|
Employment Offer Letter, dated March 2, 2006, by and between Talecris Biotherapeutics, Inc. and John R. Perkins (incorporated by reference to Exhibit 10.26 of Amendment No. 5 to our Registration Statement on Form S-1 (File No. 333-144941) filed on July 23, 2008)* |
|
|
|
|
|
10.42 |
|
Letter Agreement, dated December 19, 2008, amending the terms of a certain Employment Letter, dated March 2, 2006, by and between Talecris Biotherapeutics, Inc. and John R. Perkins (incorporated by reference to Exhibit 10.26.2 of Amendment No. 6 to our Registration Statement on Form S-1 (File No. 333-144941) filed on July 2, 2009)* |
|
|
|
|
|
10.43 |
|
Letter Agreement, dated May 25, 2010, amending the terms of a certain Employment Letter, dated March 2, 2006, as subsequently amended on December 19, 2008, by and between Talecris Biotherapeutics Holdings Corp. and John R. Perkins (incorporated by reference to Exhibit 10.1 to our Current Report on Form 8-K filed on June 3, 2010)* |
|
|
|
|
|
10.44 |
|
Separation Pay Agreement, dated May 26, 2010, amending the terms of a certain Employment Letter, dated March 2, 2006, as subsequently amended on December 19, 2008, by and between Talecris Biotherapeutics Holdings Corp. and John R. Perkins (incorporated by reference to Exhibit 10.2 to our Current Report on Form 8-K filed on June 3, 2010)* |
|
|
|
|
|
10.45 |
|
Employment Agreement, as amended and restated as of November 26, 2008, by and between Talecris Biotherapeutics Holdings Corp. and Kari Heerdt (incorporated by reference to Exhibit 10.21 of Amendment No. 8 to our Registration Statement on Form S-1 (File No. 333-144941) filed on August 19, 2009)* |
|
|
|
|
|
10.46 |
|
Letter Agreement, dated February 24, 2010, amending the terms of an amended and restated Employment Agreement, dated November 26, 2008, by and between Talecris Biotherapeutics Holdings Corp. and Kari Heerdt (incorporated by reference to Exhibit 10.1 to our Current Report on Form 10-Q filed on April 28, 2010)* |
|
|
|
|
|
21 |
|
Subsidiaries of Talecris Biotherapeutics Holdings Corp. |
|
|
|
|
|
23.1 |
|
Consent of Independent Registered Public Accounting Firm |
|
|
|
|
|
31.1 |
|
Certification of Chief Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 |
|
|
|
|
|
31.2 |
|
Certification of Chief Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 |
|
|
|
|
|
32.1 |
|
Certification of Chief Executive Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002** |
|
|
|
|
|
32.2 |
|
Certification of Chief Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002** |
+ Portions of the exhibit have been omitted pursuant to an order granting confidential treatment dated September 30, 2009 by the Securities and Exchange Commission
* Management contract or compensatory plan or arrangement
** This certification shall not be deemed filed for purposes of Section 18 of the Exchange Act, or incorporated by reference in any filing under the Securities Act of 1933, as amended, or the Exchange Act, except as may be expressly set forth by specific reference in such filing
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, in the city of Research Triangle Park, State of North Carolina, on February 23, 2011.
|
|
TALECRIS BIOTHERAPEUTICS HOLDINGS CORP. | |
|
|
| |
|
|
By: |
/s/ Lawrence D. Stern |
|
|
| |
|
|
Lawrence D. Stern | |
|
|
Chairman and Chief Executive Officer | |
|
|
| |
|
|
By: |
/s/ John M. Hanson |
|
|
| |
|
|
John M. Hanson | |
|
|
Executive Vice President and Chief Financial Officer | |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons in the capacities and on the dates indicated:
|
Signature |
|
Title |
|
Date |
|
|
|
|
|
|
|
/s/ Lawrence D. Stern |
|
Chairman and Chief Executive Officer |
|
February 23, 2011 |
|
Lawrence D. Stern |
|
(principal executive officer) |
|
|
|
|
|
|
|
|
|
/s/ John M. Hanson |
|
Executive Vice President and Chief |
|
February 23, 2011 |
|
John M. Hanson |
|
Financial Officer (principal financial |
|
|
|
|
|
and accounting officer) |
|
|
|
|
|
|
|
|
|
/s/ Richard A. Charpie |
|
Director |
|
February 16, 2011 |
|
Richard A. Charpie |
|
|
|
|
|
|
|
|
|
|
|
/s/ Paul N. Clark |
|
Director |
|
February 17, 2011 |
|
Paul N. Clark |
|
|
|
|
|
|
|
|
|
|
|
/s/ W. Brett Ingersoll |
|
Director |
|
February 16, 2011 |
|
W. Brett Ingersoll |
|
|
|
|
|
|
|
|
|
|
|
/s/ James T. Lenehan |
|
Director |
|
February 16, 2011 |
|
James T. Lenehan |
|
|
|
|
|
|
|
|
|
|
|
/s/ Steven F. Mayer |
|
Director |
|
February 16, 2011 |
|
Steven F. Mayer |
|
|
|
|
|
|
|
|
|
|
|
/s/ Kenneth J. Martin |
|
Director |
|
February 21, 2011 |
|
Kenneth J. Martin |
|
|
|
|
|
|
|
|
|
|
|
/s/ Dean J. Mitchell |
|
Director |
|
February 21, 2011 |
|
Dean J. Mitchell |
|
|
|
|
|
|
|
|
|
|
|
/s/ Ruedi E. Waeger |
|
Director |
|
February 16, 2011 |
|
Ruedi E. Waeger |
|
|
|
|