Attached files
| file | filename |
|---|---|
| EX-23 - CONSENT OF AUDITOR - PALATIN TECHNOLOGIES INC | ex23-02.htm |
As filed with the Securities and Exchange Commission on February 23, 2011
Registration No. 333-170227
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
______________________
FORM S-1
Amendment No. 2
REGISTRATION STATEMENT UNDER THE SECURITIES ACT OF 1933
______________________
PALATIN TECHNOLOGIES, INC.
(Exact name of registrant as specified in its charter)
|
Delaware
|
2834
|
95-4078884
|
|
(State or other jurisdiction of
incorporation or organization)
|
(Primary Standard Industrial
Classification Code Number)
|
(I.R.S. Employer Identification
Number)
|
4C Cedar Brook Drive
Cranbury, New Jersey 08512
(609) 495-2200
(Address, including zip code, and telephone number,
including area code, of Registrant’s principal executive offices)
______________________
Stephen T. Wills, Chief Financial Officer
4C Cedar Brook Drive
Cranbury, New Jersey 08512
(609) 495-2200
(Name, address, including zip code, and telephone number,
including area code, of agent for service)
______________________
Please send copies of all communications to:
|
Faith L. Charles, Esq.
Thompson Hine LLP
335 Madison Avenue, 12th Floor
New York, NY 10017
(212) 344-5680
|
Stephen A. Slusher, Esq.
Chief Legal Officer
4C Cedar Brook Drive
Cranbury, NJ 08512
(609) 495-2200
|
John D. Hogoboom, Esq.
Lowenstein Sandler PC
65 Livingston Avenue
Roseland, NJ 07068
(973) 597-2500
|
______________________
As soon as practicable after the effective date of this Registration Statement.
(Approximate date of commencement of proposed sale to the public)
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933 check the following box: þ
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer o Accelerated filer o Non-accelerated filer o Smaller reporting company þ
(Do not check if a smaller reporting company)
Calculation of Registration Fee
|
Title of Each Class of Securities to be Registered
|
Proposed Maximum Aggregate Offering Price (1) (2)
|
Amount of Registration Fee
|
|||
|
Units, each unit consisting of (i) one share of Common Stock, par value $0.01 per share, (ii) a Series A Warrant to purchase 0.087 shares of Common Stock and (iii) a Series B Warrant to purchase 0.913 shares of Common Stock
|
$23,000,000
|
$2,670.30
|
|||
|
Common Stock, par value $0.01 per share, included in Units (3)
|
-
|
-
|
|||
|
Series A Warrants to purchase Common Stock, included in Units (3)
|
-
|
-
|
|||
|
Series B Warrants to purchase Common Stock, included in Units (3)
|
-
|
-
|
|||
|
Common Stock issuable upon exercise of Series A Warrants
|
$2,000,000
|
$232.20
|
|||
|
Underwriter Warrants
|
$575,000
|
$66.76
|
|||
|
Total
|
$ 25,575,000
|
$ 2,969.26
|
(4)
|
NOTES TO FEE TABLE:
(1) Estimated solely for the purpose of calculating the registration fee pursuant to Rule 457(o) under the Securities Act of 1933, as amended (the “Securities Act of 1933”).
(2) Pursuant to Rule 416, this registration statement shall be deemed to cover additional securities that may be offered or issued to prevent dilution resulting from stock splits, stock dividends or similar transactions.
(3) No fee is required pursuant to Rule 457(g) under the Securities Act of 1933.
(4) Previously paid.
The registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the registrant shall file a further amendment which specifically states that this registration statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933, as
amended, or until the registration statement shall become effective on such date as the Securities and Exchange Commission, acting pursuant to said Section 8(a), may determine.
|
The information in this prospectus is not complete and may be changed. We may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This prospectus is not an offer to sell these securities and it is not soliciting an offer to buy these securities in any state where the offer or sale is not permitted.
|
|
PROSPECTUS
|
SUBJECT TO COMPLETION
|
February 23, 2011
|
|
|
PALATIN TECHNOLOGIES, INC.
Units to Purchase
23,000,000 Shares of Common Stock,
Series A Warrants to Purchase up to 2,000,000 Shares of Common Stock and
Series B Warrants to Purchase up to 21,000,000 Shares of Common Stock
Up to 2,000,000 Shares of Common Stock Issuable Upon Exercise of Series A Warrants
We are offering 23,000,000 shares of our common stock, Series A Warrants to purchase up to 2,000,000 shares of our common stock and Series B Warrants to purchase up to 21,000,000 shares of our common stock in a firm commitment public offering. This prospectus also covers up to 2,000,000 shares of our common stock issuable upon exercise of the Series A Warrants. We are not registering the 21,000,000 shares of common stock issuable upon the exercise of the Series B Warrants. We refer to the Series A Warrants and the Series B Warrants as the warrants.
The common stock and the warrants will be sold in units, with each unit consisting of one share of common stock, a Series A Warrant exercisable for 0.087 shares of our common stock and a Series B Warrant exercisable for 0.913 shares of common stock. Units will not be issued or certificated. The shares of common stock and the warrants are immediately separable and will be issued separately. The Series A Warrants are exercisable immediately upon issuance and expire on the fifth anniversary of the date of issuance. The Series B Warrants are exercisable beginning one year and one day from the date of issuance, but only if our stockholders increase the number of our authorized shares of common stock, and expire on the fifth anniversary of the date they first become exercisable. For a more detailed description of the securities offered by this prospectus, see the section entitled “Description of the Securities” beginning on page 52 of this prospectus.
Our common stock is quoted on the NYSE Amex under the symbol “PTN.” On February 22, 2011, the closing price of the common stock was $1.12.
Investing in our securities involves a high degree of risk. You should purchase these units only if you can afford a complete loss of your investment. See “Risk Factors” beginning on page 7 of this prospectus.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or passed upon the adequacy or accuracy of this prospectus. Any representation to the contrary is a criminal offense.
|
Per Unit
|
Total
|
|
|
Public offering price
|
$ _.__
|
$ [ * ]
|
|
Underwriting discounts and commissions
|
$ _.__
|
$ [ * ]
|
|
Proceeds to us, before expenses
|
$ _.__
|
$ [ * ]
|
The underwriters have reserved for sale at the public offering price [ * ] units offered by this prospectus for Carl Spana, Ph.D., our President, Chief Executive Officer, and Director, and Stephen T. Wills, our Executive Vice President – Operations and Chief Financial Officer. Messrs. Spana and Wills have entered into lock-up agreements. See “Underwriting.”
In addition to the discounts and commissions listed above, we have agreed to issue to the underwriters or their designees underwriter warrants to purchase shares of common stock equal to 2.5% of the total number of shares included in the Units. We also have agreed to reimburse the underwriters for certain of their out-of-pocket expenses. See “Underwriting.”
The underwriters expect to deliver the shares of our common stock, the Series A Warrants and the Series B Warrants on or about February [ * ], 2011 through the book-entry facilities of The Depository Trust Company.
Sole Book-Running Manager
Roth Capital Partners
Co-Manager
Madison Williams and Company
The date of this prospectus is February __, 2011
You should rely only on the information contained in this prospectus and any free writing prospectus prepared by us or on our behalf. We have not authorized anyone to provide you with different or additional information. If anyone provides you with different or additional information, you should not rely on it. The information in this prospectus is accurate only as of the date on the front of this prospectus. Our business, financial condition, results of operations and prospects may have changed since the date of this prospectus. This prospectus is not an offer or solicitation relating to the securities in any jurisdiction in which such an offer or solicitation relating to the securities is not authorized. You should not consider this prospectus to be an offer or solicitation relating to the securities if the person making the offer or solicitation is not qualified to do so, or if it is unlawful for you to receive such an offer or solicitation.
We are not making any representation to you regarding the legality of an investment in us under any legal investment or similar laws or regulations. You should not consider any information in this prospectus to be legal, business, tax or other advice. You should consult your own attorney, business advisor and tax advisor for legal, business and tax advice regarding an investment in us.
TABLE OF CONTENTS
|
Prospectus
|
|
|
Page
|
|
|
3
|
|
|
7
|
|
|
18
|
|
|
19
|
|
|
20
|
|
|
21
|
|
|
21
|
|
|
22
|
|
|
29
|
|
|
39
|
|
|
39
|
|
|
40
|
|
|
42
|
|
|
49
|
|
|
51
|
|
|
52
|
|
|
59
|
|
|
62
|
|
|
62
|
|
|
62
|
|
|
F-1
|
|
PROSPECTUS SUMMARY
This summary highlights certain information appearing elsewhere in this prospectus. This summary is not complete and does not contain all of the information you should consider prior to investing. After you read this summary, you should read and consider carefully the more detailed information and financial statements and related notes that we include in this prospectus, especially the sections entitled “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operation.” If you invest in our securities, you are assuming a high degree of risk.
Unless we have indicated otherwise or the context otherwise requires, references in the prospectus to “Palatin,” the “Company,” “we,” “us” and “our” or similar terms are to Palatin Technologies, Inc. and its subsidiary.
Our Company
We are a biopharmaceutical company dedicated to the development of peptide, peptide mimetic and small molecule agonist compounds with a focus on melanocortin and natriuretic peptide receptor systems. We have a pipeline of development programs targeting melanocortin and natriuretic receptors, including development of proposed products for treatment of sexual dysfunction, acute asthma, heart failure, hypertension, obesity, diabetes and metabolic syndrome.
Our Product Candidates
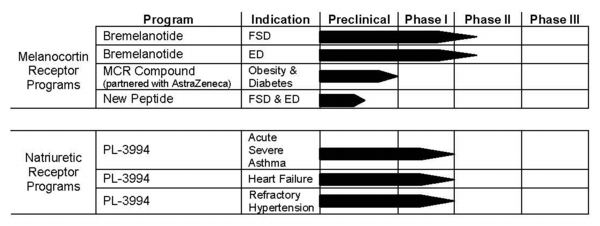
We currently have the following active drug development programs:
|
|
·
|
Bremelanotide, a peptide melanocortin receptor agonist, for treatment of sexual dysfunction, targeting female sexual dysfunction (FSD) and erectile dysfunction (ED) in patients non-responsive to current therapies.
|
|
|
·
|
Peptide melanocortin receptor agonists for treatment of FSD and ED.
|
|
|
·
|
PL-3994, a peptide mimetic natriuretic peptide receptor A (NPR-A) agonist, for treatment of acute exacerbations of asthma, heart failure and refractory or difficult-to-control hypertension.
|
We have licensed several families of melanocortin receptor-based compounds for treatment of obesity, diabetes and related metabolic syndrome to AstraZeneca AB (AstraZeneca) pursuant to our research collaboration and license agreement with AstraZeneca.
Recent Events
Reverse Stock Split. On September 24, 2010, we announced that we were implementing a one-for-ten reverse stock split of our common stock, which had been authorized by our stockholders at our annual meeting held on May 13, 2010. The reverse stock split, which became effective on September 27, 2010, reduced the number of shares of our common stock issued and outstanding from approximately 118.2 million to approximately 11.8 million. All share and per share amounts in this prospectus, including shares of common stock issuable upon exercise, vesting or conversion of all outstanding options, warrants and convertible preferred stock, are presented on a post-reverse-split basis.
Realignment of Resources. On September 24, 2010, we announced our strategic decision to focus resources and efforts on clinical trials for bremelanotide and PL-3994 and preclinical development of an inhaled formulation of PL-3994 and a new peptide drug candidate for sexual dysfunction. As part of this decision, we suspended further research and development efforts on new product candidates and implemented a reduction in staffing levels. We now have 17 full-time employees.
Strategy
Key elements of our business strategy include: using our technology and expertise to develop and commercialize products in our active drug development programs; entering into alliances and partnerships with pharmaceutical companies to facilitate the development, manufacture, marketing, sale and distribution of product candidates we are developing; and, partially funding our product development programs with the cash flow from our AstraZeneca research collaboration and license agreement and any future agreements with other companies.
Summary Financial Information
The following tables summarize our financial data. We have derived this summary for the fiscal years ended June 30, 2010 and 2009 and the three and six month periods ended December 31, 2010 and 2009 from our audited and unaudited consolidated financial statements appearing elsewhere in this prospectus. This summary of our financial data should be read together with our audited and unaudited consolidated financial statements and related notes and the section entitled “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operation” in this prospectus.
|
Three Months Ended
December 31,
|
Six Months Ended
December 31,
|
Year Ended
June 30,
|
|||||||||
|
2010
|
2009
|
2010
|
2009
|
2010
|
2009
|
||||||
|
Statement of Operations Data:
|
|||||||||||
|
Revenues
|
$ 1,042,176
|
$ 7,283,299
|
$ 1,258,323
|
$ 10,945,918
|
$ 14,180,727
|
$ 11,351,774
|
|||||
|
Operating expenses
|
2,873,916
|
3,847,834
|
7,708,454
|
7,671,129
|
17,195,113
|
18,653,610
|
|||||
|
Other income and tax benefit
|
731,238
|
1,066,410
|
749,309
|
1,190,021
|
1,221,878
|
2,499,604
|
|||||
|
Net income (loss)
|
$ (1,100,502)
|
$ 4,501,875
|
$ (5,700,822)
|
$ 4,464,810
|
$ (1,792,508) )
|
$ (4,802,232)
|
|||||
|
December 31,
|
June 30,
|
||||
|
2010
|
2010
|
2009
|
|||
|
Balance Sheet Data:
|
|||||
|
Cash and available-for-sale investments
|
$ 3,684,281
|
$ 8,867,619
|
$ 7,818,312
|
||
|
Current assets
|
3,929,305
|
9,263,811
|
8,819,664
|
||
|
Total assets
|
6,469,856
|
12,388,877
|
13,199,811
|
||
|
Current liabilities
|
2,043,794
|
2,394,931
|
8,670,332
|
||
|
Total liabilities
|
2,438,771
|
3,070,604
|
9,886,312
|
||
Company Information
We incorporated in Delaware in 1986 and commenced operations in the biopharmaceutical area in 1996. Our corporate offices and research and development facility are located at 4C Cedar Brook Drive, Cranbury, New Jersey 08512 and our telephone number is (609) 495-2200.
This prospectus contains trademarks of others, including Viagra®, Levitra®, Cialis®, Caverject Impulse®, MUSE® and Natrecor®. Viagra® is a registered trademark of Pfizer Inc., Levitra® is a registered trademark of Bayer Aktiengesellschaft, Cialis® is a registered trademark of Eli Lilly and Company, Caverject Impulse® is a registered trademark of Pharmacia & Upjohn Company LLC, MUSE® is a registered trademark of VIVUS, Inc. and Natrecor® is a registered trademark of Scios Inc.; we claim no rights to these drugs or these trademarks.
Palatin Technologies® and NeutroSpec® are our trademarks.
THE OFFERING
|
Securities offered
|
23,000,000 shares of our common stock, Series A Warrants to purchase up to 2,000,000 shares of our common stock and Series B Warrants to purchase up to 21,000,000 shares of our common stock. The common stock and warrants will be sold in units, with each unit consisting of one share of common stock, a Series A Warrant exercisable for 0.087 shares of our common stock and a Series B Warrant exercisable for 0.913 shares of common stock. Units will not be issued or certificated. The shares of common stock and the warrants are immediately separable and will be issued separately.
|
|
|
Offering price
|
$[ * ] per unit.
|
|
|
Description of warrants
|
The Series A Warrants will be exercisable immediately upon issuance until the fifth anniversary of the issuance date at an initial exercise price per share equal to 100% of the public offering price per unit set forth on the cover page of this prospectus. The Series B Warrants will be exercisable one year and one day from the date of issuance, but only if our stockholders increase the number of our authorized shares of common stock, until the fifth anniversary of the initial exercise date at an initial exercise price per share equal to 100% of the public offering price per unit set forth on the cover page of this prospectus. We do not have a sufficient number of authorized shares to permit exercise of the Series B Warrants. Thus, we may be unable to issue shares upon exercise thereof unless we obtain stockholder approval to effect an amendment to our certificate of incorporation to increase our authorized shares to an amount sufficient to permit such exercise. In the event that we are unable to increase our authorized shares by the date on which the Series B Warrants initially become exercisable, we will pay holders of Series B Warrants liquidated damages in an aggregate of $2,500,000. See “Description of the Securities–Series A and Series B Warrants–Stockholder Approval; Payment of Liquidated Damages; Registration of Series B Warrant Shares.”
|
|
|
Common stock outstanding before this offering
|
11,854,028 shares.
|
|
|
Common stock to be outstanding after this offering
|
34,854,028 shares, excluding 23,575,000 shares of common stock issuable upon exercise of the warrants (assuming, with respect to Series B Warrants and underwriter warrants, our stockholders approve an increase in the number of our authorized shares of common stock).
|
|
|
Use of proceeds
|
We plan to use the net proceeds of this offering to further develop our product candidates, primarily a Phase 2 clinical trial with subcutaneously administered bremelanotide for female sexual dysfunction, and secondarily for our PL-3994 development programs for asthma and a development program for new peptides for sexual dysfunction, and for general working capital purposes. For a more complete description of our intended use of proceeds from this offering, see “Use of Proceeds.”
|
|
|
Risk factors
|
See “Risk Factors” beginning on page 7 and the other information set forth in this prospectus for a discussion of factors you should consider before deciding to invest in our securities.
|
|
|
NYSE Amex symbol
|
|
“PTN.”
|
The number of shares of our common stock to be outstanding after the closing of this offering is based on 11,854,028 shares of our common stock outstanding as of February 22, 2011 and excludes:
|
|
·
|
821,918 shares of common stock issuable upon exercise of options outstanding and having a weighted average exercise price of $11.77 per share;
|
|
|
·
|
1,551,748 shares of common stock issuable upon exercise of warrants outstanding and having a weighted average exercise price of $8.46;
|
|
|
·
|
2,000,000 shares of common stock issuable upon exercise of the Series A Warrants;
|
|
|
·
|
21,000,000 shares of common stock issuable upon exercise of the Series B Warrants (assuming our stockholders approve an increase in the number of our authorized shares of common stock);
|
|
|
·
|
575,000 shares of common stock issuable upon exercise of the underwriters’ warrants (assuming our stockholders approve an increase in the number of our authorized shares of common stock);
|
|
|
·
|
54,500 shares of common stock issuable under restricted stock units that vest no later than March 15, 2011, subject to the fulfillment of service conditions;
|
|
|
·
|
360,336 shares of common stock reserved for future issuance under our 2005 Stock Plan; and
|
|
|
·
|
26,865 shares of common stock issuable upon conversion of immediately convertible Series A Convertible Preferred Stock outstanding.
|
RISK FACTORS
You should carefully consider the risks described below and other information in this prospectus, including the financial statements and related notes that appear at the end of this prospectus, before deciding to invest in our securities. These risks should be considered in conjunction with any other information included herein, including in conjunction with forward-looking statements made herein. If any of the following risks actually occur, they could materially adversely affect our business, financial condition, operating results or prospects. Additional risks and uncertainties that we do not presently know or that we currently deem immaterial may also impair our business, financial condition, operating results and prospects.
Risks Relating to our Company
We will continue to incur substantial losses over the next few years and we may never become profitable.
We have never been profitable and we may never become profitable. As of December 31, 2010, we had an accumulated deficit of $214.9 million. We expect to incur additional losses as we continue our development of bremelanotide, PL-3994 and other product candidates. Unless and until we receive approval from the U. S. Food and Drug Administration (FDA) or other equivalent regulatory authorities outside the United States, we cannot sell our products and will not have product revenues from them. Therefore, for the foreseeable future, we will have to fund all of our operations and capital expenditures from reimbursements and other contract revenue under collaborative development agreements, existing cash balances and outside sources of financing, which may not be available on acceptable terms, if at all.
We need to raise additional funds and will need to continue to raise funds in the future, and funds may not be available on acceptable terms, or at all.
As of December 31, 2010, we had cash and cash equivalents of $1.7 million and available-for-sale investments of $2.0 million, with current liabilities of $2.0 million. We have curtailed our operations significantly, including suspending early stage research and discovery programs and implementing a reduction in our workforce. However, our currently available working capital will not fund our currently planned operations for the next twelve months. We will also need additional funds to continue development of bremelanotide and PL-3994, including planned clinical trials and preclinical development efforts.
We expect the net proceeds from this offering will be sufficient to fund our planned operations for the next eighteen months, but will likely not be sufficient to complete required clinical trials for any of our product candidates. We will need additional funding to complete required clinical trials and, assuming those clinical trials are successful, as to which there can be no assurance, complete submission of required regulatory applications to the FDA for any of our product candidates. We may raise additional funds through public or private equity financings, debt financings, collaborative arrangements on our product candidates or other sources. However, additional funding may not be available on acceptable terms, or at all. To obtain additional funding, we may need to enter into arrangements that require us to develop only certain of our product candidates or relinquish rights to certain technologies, product candidates and/or potential markets.
If we are unable to raise sufficient additional funds, we will implement plans for the orderly wind down of our business operations, including curtailing operations significantly and further decreasing staffing levels, and will seek to license, sell or otherwise dispose of our product candidates, technologies and contractual rights, including rights under our research collaboration and license agreement with AstraZeneca, on the best possible terms available. Even if we are able to license, sell or otherwise dispose of our product candidates, technologies and contractual rights, it is likely to be on unfavorable terms and for less value than if we had the financial resources to develop or otherwise advance our product candidates, technologies and contractual rights ourselves.
Our independent registered public accounting firm has expressed doubt about our ability to continue as a going concern, which may hinder our ability to obtain future financing.
Our consolidated financial statements as of June 30, 2010 and December 31, 2010 have been prepared under the assumption that we will continue as a going concern. Our independent registered public accounting firm has issued a report dated September 27, 2010 that included an explanatory paragraph referring to our recurring net losses and negative cash flows from operations and expressing substantial doubt in our ability to continue as a going concern. Our ability to continue as a going concern is dependent upon our ability to obtain additional equity financing or other capital, attain further operating efficiencies, reduce expenditures, and, ultimately, to generate revenue. The consolidated financial statements do not include any adjustments that might result from the outcome of
this uncertainty. We are continually evaluating opportunities to raise additional funds through public or private equity financings, as well as evaluating prospective business partners, and will continue to do so. However, if adequate funds are not available to us when we need it, and we are unable to enter into some form of strategic relationship that will give us access to additional cash resources, we will be required to even further curtail our operations which would, in turn, further raise substantial doubt about our ability to continue as a going concern.
We are not in compliance with continued listing standards of NYSE Amex, and our common stock may be delisted, making it difficult to trade shares of our common stock.
Our common stock trades on NYSE Amex. On November 26, 2010, we received a letter from NYSE Amex advising us that, based on our Quarterly Report on Form 10-Q for the period ended September 30, 2010, we are not in compliance with certain continued listing standards under Section 1003 of the NYSE Amex Company Guide. Specifically, NYSE Amex stated that we are not in compliance with Section 1003(a)(iii) of the Company Guide because our stockholders’ equity is less than the required $6,000,000 and we have losses from continuing operations and net losses in our five most recent fiscal years, and Section 1003(a)(iv) of the Company Guide because we have sustained losses which are so substantial in relation to our overall operations or existing financial resources, or our financial condition has become so impaired that it appears questionable, in the opinion of the NYSE Amex, as to whether we will be able to continue operations and/or meet our obligations as they mature.
In order to maintain our listing on NYSE Amex, we submitted a plan addressing how we intend to regain compliance with Section 1003(a)(iv) by February 28, 2011 and Section 1003(a)(iii) by May 26, 2011. On January 31, 2011, NYSE Amex notified us that it had accepted our plan for regaining compliance, and that our listing was being continued pursuant to an extension. We may be able to continue our listing during the plan period through February 28, 2011 with respect to Section 1003(a)(iv) and May 26, 2011 with respect to Section 1003(a)(iii), subject to periodic review by NYSE Amex to determine if we are making progress consistent with the plan. Failure to make progress consistent with the plan or to regain compliance with continued listing standards by the relevant extension periods could result in our common stock being delisted from NYSE Amex.
If we are delisted from NYSE Amex, then our common stock will trade, if at all, only on the over-the-counter market, and then only if one or more registered broker-dealer market makers comply with quotation requirements. Delisting of our common stock could also further depress our stock price, substantially limit liquidity of our common stock and materially adversely affect our ability to raise capital on terms acceptable to us, or at all. Delisting from NYSE Amex could also have other negative results, including the potential loss of confidence by suppliers and employees, the loss of institutional investor interest and fewer business development opportunities.
We have a limited operating history upon which to base an investment decision.
Our operations are primarily focused on acquiring, developing and securing our proprietary technology, conducting preclinical and clinical studies and formulating and manufacturing on a small-scale basis our principal product candidates. These operations provide a limited basis for stockholders to assess our ability to commercialize our product candidates.
We have not yet demonstrated our ability to perform the functions necessary for the successful commercialization of any of our current product candidates. The successful commercialization of our product candidates will require us to perform a variety of functions, including:
|
|
·
|
continuing to conduct preclinical development and clinical trials;
|
|
|
·
|
participating in regulatory approval processes;
|
|
|
·
|
formulating and manufacturing products, or having third parties formulate and manufacture products;
|
|
|
·
|
post-approval monitoring and surveillance of our products;
|
|
|
·
|
conducting sales and marketing activities, either alone or with a partner; and
|
|
|
·
|
obtaining additional capital.
|
If we are unable to obtain regulatory approval of any of our product candidates, to successfully commercialize any products for which we receive regulatory approval or to obtain additional capital, we may not be able to recover our investment in our development efforts.
Budget constraints negatively impact our research and development, forcing us to delay our efforts to develop certain product candidates in favor of developing others, which may prevent us from commercializing our product candidates as quickly as possible.
Research and development is an expensive process. As part of our plan to realign resources, we have decided to focus resources and efforts on clinical trials for bremelanotide and PL-3994 and preclinical development of an inhaled formulation of PL-3994 and a new peptide drug candidate for sexual dysfunction. As part of this decision, we have suspended further research and development efforts on new product candidates. We do not currently have sufficient funds to progress the programs we have prioritized, and clinical trial and development priorities may change depending on terms required by investors in our company, including investors in this offering. Because we have had to prioritize our development candidates as a result of budget constraints, and because these priorities may change, we may not be able to fully realize the value of our product candidates in a timely manner, if at all.
Development and commercialization of our product candidates involves a lengthy, complex and costly process, and we may never successfully develop or commercialize any product.
Our product candidates are at various stages of research and development, will require regulatory approval, and may never be successfully developed or commercialized. Our product candidates will require significant further research, development and testing before we can seek regulatory approval to market and sell them.
We must demonstrate that our product candidates are safe and effective for use in patients in order to receive regulatory approval for commercial sale. Preclinical studies in animals, using various doses and formulations, must be performed before we can begin human clinical trials. Even if we obtain favorable results in the preclinical studies, the results in humans may be different. Numerous small-scale human clinical trials may be necessary to obtain initial data on a product candidate’s safety and efficacy in humans before advancing to large-scale human clinical trials. We face the risk that the results of our trials in later phases of clinical trials may be inconsistent with those obtained in earlier phases. Adverse or inconclusive results could delay the progress of our development programs and may prevent us from filing for regulatory approval of our product candidates. Additional factors that can cause delay or termination of our human clinical trials include:
|
|
·
|
the availability of sufficient capital to sustain operations and clinical trials;
|
|
|
·
|
timely completion of clinical site protocol approval and obtaining informed consent from subjects;
|
|
|
·
|
the rate of patient enrollment in clinical studies;
|
|
|
·
|
adverse medical events or side effects in treated patients; and
|
|
|
·
|
lack of effectiveness of the product being tested.
|
You should evaluate us in light of these uncertainties, delays, difficulties and expenses commonly experienced by early stage biopharmaceutical companies, as well as unanticipated problems and additional costs relating to:
|
|
·
|
product approval or clearance;
|
|
|
·
|
regulatory compliance;
|
|
|
·
|
good manufacturing practices;
|
|
|
·
|
intellectual property rights;
|
|
|
·
|
product introduction; and
|
|
|
·
|
marketing and competition.
|
The regulatory approval process is lengthy, expensive and uncertain, and may prevent us from obtaining the approvals we require.
Government authorities in the United States and other countries extensively regulate the advertising, labeling, storage, record-keeping, safety, efficacy, research, development, testing, manufacture, promotion, marketing and distribution of drug products. Drugs are subject to rigorous regulation by the FDA and similar regulatory bodies in other countries. The steps ordinarily required by the FDA before a new drug may be marketed in the United States include:
|
|
·
|
completion of non-clinical tests including preclinical laboratory and formulation studies and animal testing and toxicology;
|
|
|
·
|
submission to the FDA of an Investigational New Drug (IND) application, which must become effective before clinical trials may begin;
|
|
|
·
|
performance of adequate and well-controlled Phase 1, 2 and 3 human clinical trials to establish the safety and efficacy of the drug for each proposed indication;
|
|
|
·
|
submission to the FDA of a New Drug Application (NDA); and
|
|
|
·
|
FDA review and approval of the NDA before any commercial marketing or sale.
|
Satisfaction of FDA pre-market approval requirements for new drugs typically takes a number of years and the actual time required for approval may vary substantially based upon the type, complexity and novelty of the product or disease. The results of product development, preclinical studies and clinical trials are submitted to the FDA as part of an NDA. The NDA also must contain extensive manufacturing information. Once the submission has been accepted for filing, the FDA generally has ten months to review the application and respond to the applicant. The review process is often significantly extended by FDA requests for additional information or clarification. Success in early stage clinical trials does not assure success in later stage clinical trials. Data obtained from clinical trials is not always conclusive and may be susceptible to varying interpretations that could delay, limit or prevent regulatory approval. The FDA may refer the NDA to an advisory committee for review, evaluation and recommendation as to whether the application should be approved, but the FDA is not bound by the recommendation of the advisory committee. The FDA may deny or delay approval of applications that do not meet applicable regulatory criteria or if the FDA determines that the clinical data do not adequately establish the safety and efficacy of the drug. Therefore, our proposed products could take a significantly longer time than we expect or may never gain approval. If regulatory approval is delayed or never obtained, our business and our liquidity would be adversely affected.
Upon approval, a product candidate may be marketed only in those dosage forms and for those indications approved by the FDA. Once approved, the FDA may withdraw the product approval if compliance with regulatory requirements is not maintained or if problems occur after the product reaches the marketplace. In addition, the FDA may require post-marketing studies, referred to as Phase 4 studies, to monitor the approved products in a larger number of patients than were required for product approval and may limit further marketing of the product based on the results of these post-market studies. The FDA has broad post-market regulatory and enforcement powers, including the ability to seek injunctions, levy fines and civil penalties, criminal prosecution, withdraw approvals and seize products or request recalls.
If regulatory approval of any of our product candidates is granted, it will be limited to certain disease states or conditions. Adverse experiences with the product must be reported to the FDA and could result in the imposition of market restriction through labeling changes or in product removal. Product approvals may be withdrawn if compliance with regulatory requirements is not maintained or if problems concerning safety or efficacy of the product occur following approval.
Outside the United States, our ability to market our product candidates will also depend on receiving marketing authorizations from the appropriate regulatory authorities. The foreign regulatory approval process generally includes all of the risks associated with FDA approval described above. The requirements governing the conduct of clinical trials and marketing authorization vary widely from country to country. At present, foreign marketing authorizations are applied for at a national level, although within the European Community, or EC, registration procedures are available to companies wishing to market a product to more than one EC member state. If the regulatory authority is satisfied that adequate evidence of safety, quality and efficiency has been presented, a marketing authorization will be granted.
If any approved product does not achieve market acceptance, our business will suffer.
Regulatory approval for the marketing and sale of any of our product candidates does not assure the product’s commercial success. Any approved product will compete with other products manufactured and marketed by major pharmaceutical and other biotechnology companies. The degree of market acceptance of any such product will depend on a number of factors, including:
|
|
·
|
perceptions by members of the healthcare community, including physicians, about its safety and effectiveness;
|
|
|
·
|
cost-effectiveness relative to competing products and technologies;
|
|
|
·
|
availability of reimbursement for our products from third party payors such as health insurers, health maintenance organizations and government programs such as Medicare and Medicaid; and
|
|
|
·
|
advantages over alternative treatment methods.
|
If any approved product does not achieve adequate market acceptance, our business, financial condition and results of operations will be adversely affected.
We rely on third parties to conduct clinical trials for our product candidates and their failure to timely perform their obligations could significantly harm our product development.
We rely on outside scientific collaborators such as researchers at clinical research organizations and universities in certain areas that are particularly relevant to our research and product development plans, such as the conduct of clinical trials and non-clinical tests. There is competition for these relationships, and we may not be able to maintain our relationships with them on acceptable terms. These outside collaborators generally may terminate their engagements with us at any time. As a result, we can control their activities only within certain limits, and they will devote only a certain amount of their time to conduct research on our product candidates and develop them. If they do not successfully carry out their duties under their agreements with us, fail to inform us if these trials fail to comply with clinical trial protocols or fail to meet expected deadlines, our ability to develop our product candidates and obtain regulatory approval on a timely basis, if at all, may be adversely affected.
Production and supply of our product candidates depend on contract manufacturers over whom we have no control.
We do not have the facilities to manufacture bremelanotide, PL-3994, melanocortin receptor agonist compounds or our other potential products. Our contract manufacturers must perform these manufacturing activities in a manner that complies with FDA regulations. Our ability to control third-party compliance with FDA requirements will be limited to contractual remedies and rights of inspection. The manufacturers of approved products and their manufacturing facilities will be subject to continual review and periodic inspections by the FDA and other authorities where applicable, and must comply with ongoing regulatory requirements, including the FDA’s current good manufacturing practices (GMPs) regulations. Failure of third-party manufacturers to comply with GMPs or other FDA requirements may result in enforcement action by the FDA. Failure to conduct their activities in compliance with FDA regulations could delay our development programs or negatively impact our ability to receive FDA approval of our potential products or continue marketing if they are approved. Establishing relationships with new suppliers, who must be FDA-approved, is a time-consuming and costly process.
We are subject to extensive regulation in connection with the laboratory practices and the hazardous materials we use.
We are subject to various laws and regulations regarding laboratory practices, the experimental use of animals and the use and disposal of hazardous or potentially hazardous substances in connection with our research. In each of these areas, as noted above, the FDA and other regulatory authorities have broad regulatory and enforcement powers, including the ability to levy fines and civil penalties, suspend or delay issuance of approvals, seize or recall products and withdraw approvals, any one or more of which could have a material adverse effect on us. We are also subject to numerous federal, state and local laws relating to such matters as safe working conditions, manufacturing practices, environmental protection, fire hazard control and disposal of hazardous or potentially hazardous substances. Even though we have suspended research and development efforts on new product candidates, we are maintaining selected laboratory capabilities, and will be subject to regulations in connection with decommissioning animal facilities, disposal of chemicals and hazardous or potentially hazardous substances, and decommissioning and disposing of laboratory equipment. We may incur significant costs to comply with such laws and regulations now or in the future.
Contamination or injury from hazardous materials used in the development of our products could result in a liability exceeding our financial resources.
Our research and development has involved the use of hazardous materials and chemicals, including radioactive compounds. We cannot completely eliminate the risk of contamination or injury from these materials. In the event of contamination or injury, we may be responsible for any resulting damages. Damages could be significant and could exceed our financial resources, including the limits of our insurance.
We have no experience in marketing, distributing and selling products and will substantially rely on our marketing partners to provide these capabilities.
We are developing bremelanotide and melanocortin receptor agonist compounds for sexual dysfunction and PL-3994 for the treatment of asthma, heart failure and related indications. We do not have marketing partners for any of these products. If any of these products are approved by the FDA or other regulatory authorities, we must either develop marketing, distribution and selling capacity and expertise, which will be costly and time consuming, or enter into agreements with other companies to provide these capabilities. We may not be able to enter into suitable agreements on acceptable terms, if at all.
We do not control the development of compounds licensed to third parties and, as a result, we may not realize a significant portion of the potential value of any such license arrangements.
Under our research collaboration and license agreement with AstraZeneca for our melanocortin-based therapeutic compounds for obesity, diabetes and related metabolic syndrome, we have no direct control over the development of compounds and have only limited, if any, input on the direction of development efforts. If the results of development efforts are negative or inconclusive, AstraZeneca may decide to abandon further development of this program, including terminating the agreement, by giving us notice of termination. Because much of the potential value of the license arrangement with AstraZeneca is contingent upon the successful development and commercialization of the licensed technology, the ultimate value of this license will depend on the efforts of AstraZeneca. If AstraZeneca does not succeed in developing the licensed technology for any reason, or elects to discontinue the development of this program, we may be unable to realize the potential value of this arrangement.
Competing products and technologies may make our proposed products noncompetitive.
There are a number of other products being developed for FSD and ED. In addition to three oral FDA-approved phosphodiesterase-5 (PDE-5) inhibitor drugs for the treatment of ED, there are other approved products and devices for ED, and other products are being developed for ED and FSD and are in clinical trials. There is competition to develop drugs for ED in patients non-responsive to PDE-5 inhibitor drugs, and to develop drugs for treatment of FSD.
There are a large number of products approved for use in asthma, and a number of other products being developed for treatment of acute exacerbations of asthma, including products in clinical trials. There is intense competition to develop drugs for treatment of acute exacerbations of asthma.
We are aware of one recombinant natriuretic peptide product for acutely decompensated congestive heart failure approved and marketed in the United States, and another recombinant natriuretic peptide product approved and marketed in Japan. Clinical trials on other natriuretic peptide products are being conducted in the United States. In addition, other products for treatment of heart failure are either currently being marketed or in development.
The biopharmaceutical industry is highly competitive. We are likely to encounter significant competition with respect to bremelanotide, other melanocortin receptor agonist compounds and PL-3994. Most of our competitors have substantially greater financial and technological resources than we do. Many of them also have significantly greater experience in research and development, marketing, distribution and sales than we do. Accordingly, our competitors may succeed in developing, marketing, distributing and selling products and underlying technologies more rapidly than we can. These competitive products or technologies may be more effective and useful or less costly than bremelanotide, other melanocortin receptor agonist compounds or PL-3994. In addition, academic institutions, hospitals, governmental agencies and other public and private research organizations are also conducting research and may develop competing products or technologies on their own or through strategic alliances or collaborative arrangements.
Our ability to achieve revenues from the sale of our products in development will depend, in part, on our ability to obtain adequate reimbursement from Medicare, Medicaid, private insurers and other healthcare payers.
Our ability to successfully commercialize our products in development will depend, in significant part, on the extent to which we or our marketing partners can obtain reimbursement for our products and also reimbursement at appropriate levels for the cost of our products and related treatment. Obtaining reimbursement from governmental payers, insurance companies, health maintenance organizations (HMOs) and other third-party payers of healthcare costs is a time-consuming and expensive process. There is no guarantee that our products will ultimately be reimbursed. If we are able to obtain reimbursement, continuing efforts by governmental and third party payers to contain or reduce costs of healthcare may adversely affect our future revenues and ability to achieve profitability. Third-party payers are increasingly challenging the prices charged for diagnostic and therapeutic products and related services. Reimbursement from governmental payers is subject to statutory and regulatory changes, retroactive rate adjustments, administrative rulings and other policy changes, all of which could materially decrease the range of products for which we are reimbursed or the rates of reimbursement by government payers. In addition,
recent legislation reforming the healthcare system may result in lower prices or the actual inability of prospective customers to purchase our products in development. The cost containment measures that healthcare payers and providers are instituting and the effect of any healthcare reform could materially and adversely affect our ability to operate profitably. Furthermore, even if reimbursement is available, it may not be available at price levels sufficient for us to realize a positive return on our investment.
If we fail to adequately protect or enforce our intellectual property rights or secure rights to patents of others, the value of our intellectual property rights would diminish.
Our success, competitive position and future revenues will depend in part on our ability and the abilities of our licensors to obtain and maintain patent protection for our products, methods, processes and other technologies, to preserve our trade secrets, to prevent third parties from infringing on our proprietary rights and to operate without infringing the proprietary rights of third parties. We cannot predict:
|
|
·
|
the degree and range of protection any patents will afford us against competitors, including whether third parties will find ways to invalidate or otherwise circumvent our patents;
|
|
|
·
|
if and when patents will be issued;
|
|
|
·
|
whether or not others will obtain patents claiming aspects similar to those covered by our patents and patent applications; and
|
|
|
·
|
whether we will need to initiate litigation or administrative proceedings, which may be costly whether we win or lose.
|
If our products, methods, processes and other technologies infringe the proprietary rights of other parties, we could incur substantial costs and we may have to:
|
|
·
|
obtain licenses, which may not be available on commercially reasonable terms, if at all;
|
|
|
·
|
redesign our products or processes to avoid infringement;
|
|
|
·
|
stop using the subject matter claimed in the patents held by others;
|
|
|
·
|
pay damages; or
|
|
|
·
|
defend litigation or administrative proceedings, which may be costly whether we win or lose, and which could result in a substantial diversion of our management resources.
|
If we are unable to keep our trade secrets confidential, our technologies and other proprietary information may be used by others to compete against us.
In addition to our reliance on patents, we attempt to protect our proprietary technologies and processes by relying on trade secret laws and agreements with our employees and other persons who have access to our proprietary information. These agreements and arrangements may not provide meaningful protection for our proprietary technologies and processes in the event of unauthorized use or disclosure of such information. In addition, our competitors may independently develop substantially equivalent technologies and processes or gain access to our trade secrets or technology, either of which could materially and adversely affect our competitive position.
We may incur substantial liabilities and may be required to limit commercialization of our products in response to product liability lawsuits.
The testing and marketing of medical products entails an inherent risk of product liability. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our products or cease clinical trials. Our inability to obtain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of pharmaceutical products we develop, alone or with corporate collaborators. We currently carry liability insurance as to certain clinical trial risks. We, or any corporate collaborators, may not in the future be able to obtain insurance at a reasonable cost or in sufficient amounts, if at all. Even if our agreements with any future corporate collaborators entitle us to indemnification against losses, such indemnification may not be available or adequate should any claim arise.
We are highly dependent on our management team, senior research professionals and third-party contractors and consultants, and the loss of their services could materially adversely affect our business.
We rely on our management team, our employees and various contractors and consultants to provide critical services. Our ability to execute our bremelanotide and PL-3994 clinical programs and our preclinical programs on an inhaled formulation of PL-3994 and a new peptide drug candidate for sexual dysfunction depends on our continued retention and motivation of our management and scientific personnel, including executive officers and senior members of development and management who possess significant technical expertise and experience and oversee our development programs. Our success also depends on our ability to develop and maintain relationships with contractors, consultants and scientific advisors. If we lose the services of existing personnel or fail to attract new personnel, our development programs could be adversely affected. Competition for personnel is intense. In addition, because of our reduction in staffing levels we anticipate we will need to hire consultants or contractors for development activities previously undertaken by our employees.
Anti-takeover provisions of Delaware law and our charter documents may make potential acquisitions more difficult and could result in the entrenchment of management.
We are incorporated in Delaware. Anti-takeover provisions of Delaware law and our charter documents may make a change in control or efforts to remove management more difficult. Also, under Delaware law, our board of directors may adopt additional anti-takeover measures. Under Section 203 of the Delaware General Corporation Law, a corporation may not engage in a business combination with an “interested stockholder” for a period of three years after the date of the transaction in which the person first becomes an “interested stockholder,” unless the business combination is approved in a prescribed manner.
Pursuant to approvals by our stockholders at the annual meeting of stockholders held on May 13, 2010, effective July 23, 2010 we increased our authorized common stock from 150,000,000 to 400,000,000, and on September 27, 2010 we implemented a one-for-ten reverse stock split, which reduced our authorized common stock to 40,000,000 shares. This could have the effect of making it more difficult for a third party to acquire a majority of our outstanding voting stock.
Our charter authorizes us to issue up to 10,000,000 shares of preferred stock and to determine the terms of those shares of stock without any further action by our stockholders. If we exercise this right, it could be more difficult for a third party to acquire a majority of our outstanding voting stock.
In addition, our equity incentive plans generally permit us to accelerate the vesting of options and other stock rights granted under these plans in the event of a change of control. If we accelerate the vesting of options or other stock rights, this action could make an acquisition more costly.
The application of these provisions could have the effect of delaying or preventing a change of control, which could adversely affect the market price of our common stock.
Risks Relating to Owning our Common Stock
Our stock price is volatile and we expect it to remain volatile, which could limit investors’ ability to sell stock at a profit.
The volatile price of our stock makes it difficult for investors to predict the value of their investment, to sell shares at a profit at any given time, or to plan purchases and sales in advance. A variety of factors may affect the market price of our common stock. These include, but are not limited to:
|
|
·
|
publicity regarding actual or potential clinical results relating to products under development by our competitors or us;
|
|
|
·
|
delay or failure in initiating, completing or analyzing preclinical or clinical trials or unsatisfactory designs or results of these trials;
|
|
|
·
|
interim decisions by regulatory agencies, including the FDA, as to clinical trial designs, acceptable safety profiles and the benefit/risk ratio of products under development;
|
|
|
·
|
achievement or rejection of regulatory approvals by our competitors or by us;
|
|
|
·
|
announcements of technological innovations or new commercial products by our competitors or by us;
|
|
|
·
|
developments concerning proprietary rights, including patents;
|
|
|
·
|
developments concerning our collaborations;
|
|
|
·
|
regulatory developments in the United States and foreign countries;
|
|
|
·
|
economic or other crises and other external factors;
|
|
|
·
|
period-to-period fluctuations in our revenue and other results of operations;
|
|
|
·
|
changes in financial estimates by securities analysts; and
|
|
|
·
|
sales of our common stock.
|
We will not be able to control many of these factors, and we believe that period-to-period comparisons of our financial results will not necessarily be indicative of our future performance. If our revenues, if any, in any particular period do not meet expectations, we may not be able to adjust our expenditures in that period, which could cause our operating results to suffer further. If our operating results in any future period fall below the expectations of securities analysts or investors, our stock price may fall by a significant amount.
For the 12 month period ended January 31, 2011, the price of our stock has been volatile, ranging from a high of $3.50 per share to a low of $0.84 per share.
In addition, the stock market in general, and the market for biotechnology companies in particular, has experienced extreme price and volume fluctuations that may have been unrelated or disproportionate to the operating performance of individual companies. These broad market and industry factors may seriously harm the market price of our common stock, regardless of our operating performance.
We have implemented a reverse stock split, which has reduced our trading volume and may result in a decrease in our market capitalization.
Effective September 27, 2010, we implemented a one-for-ten reverse stock split. This reverse stock split was implemented because we had received notice that the NYSE Amex deemed it appropriate for us to effect a reverse stock split because of the low selling price of our common stock. At our annual meeting of stockholders held on May 13, 2010, the stockholders authorized a reverse stock split. We cannot guarantee that the price increase of our common stock price resulting from the reverse split will:
|
|
·
|
be proportionate to the reverse split ratio;
|
|
|
·
|
last in the marketplace for any length of time;
|
|
|
·
|
remain at a price sufficient to meet the listing requirements of the NYSE Amex; or
|
|
|
·
|
be sufficient to facilitate raising capital.
|
We do not intend to pay cash dividends in the foreseeable future.
We do not anticipate paying any cash dividends in the foreseeable future and intend to retain future earnings, if any, for the development and expansion of our business. In addition, the terms of existing or future agreements may limit our ability to pay dividends. Therefore, our stockholders will not receive a return on their shares unless the value of their shares increases.
Risks Related to this Offering
You will experience immediate and substantial dilution as a result of this offering.
As of December 31, 2010, we had a net tangible book value of approximately $4.0 million, or $0.34 per share of common stock, assuming the conversion of all then convertible preferred stock and no exercise of any warrants or options. Based on the public offering price of $[ * ] per unit, and attributing no value to the warrants included in the units, investors in this offering will experience immediate and substantial dilution of $[ * ] per share in the net tangible book value of the common stock. See “Dilution.”
As of February 22, 2011 , there were 2,455,031 shares of common stock underlying outstanding convertible preferred stock, options, warrants and restricted stock units, and you may experience dilution from the conversion of preferred stock, exercise of outstanding options and warrants and the vesting of restricted stock units.
As of February 22, 2011 , holders of our outstanding dilutive securities had the right to acquire the following amounts of underlying common stock:
|
|
·
|
26,865 shares issuable on the conversion of immediately convertible Series A Convertible preferred stock, subject to adjustment, for no further consideration;
|
|
|
·
|
1,551,748 shares issuable on the exercise of warrants, at exercise prices ranging from $2.00 to $28.80 per share;
|
|
|
·
|
821,918 shares issuable on the exercise of stock options, at exercise prices ranging from $1.30 to $47.00 per share; and
|
|
|
·
|
54,500 shares issuable under restricted stock units that vest no later than March 15, 2011, subject to the fulfillment of service conditions.
|
If the holders convert, exercise or receive those securities, or similar dilutive securities we may issue in the future, stockholders may experience dilution in the net tangible book value of their common stock. In addition, the sale or availability for sale of the underlying shares in the marketplace could depress our stock price. We have registered or agreed to register for resale substantially all of the underlying shares listed above. Holders of registered underlying shares could resell the shares immediately upon issuance, resulting in significant downward pressure on our stock.
We will have broad discretion over the use of the proceeds of this offering and may not realize a return.
We will have considerable discretion in the application of the net proceeds of this offering. We intend to use the net proceeds to further develop our product candidates and for general corporate purposes. We may use the net proceeds for purposes that do not yield a significant return, if any, for our stockholders.
There is no public market for the warrants to purchase common stock in this offering.
There is no established public trading market for the warrants included in the units, and we do not expect a market to develop. In addition, we do not intend to apply for listing the warrants on any securities exchange. Without an active market, the liquidity of the warrants will be limited.
If the registration statement covering the shares issuable upon exercise of the warrants contained in the units is no longer effective, such shares will be issued with restrictive legends unless such shares are eligible for sale under Rule 144 or another exemption under the Securities Act of 1933.
We are required to hold a stockholders’ meeting no later than June 30, 2011 to vote on a proposal related to this offering, and if we fail to obtain such approval, we are required to pay $2,500,000 to holders of Series B Warrants.
We have agreed to hold a stockholders’ meeting no later than June 30, 2011 to approve an increase in the authorized shares of our common stock to permit the exercise of the Series B Warrants. If we are unable to obtain the required stockholder approval by February [ * ], 2012, we will be required to pay holders of Series B Warrants an aggregate of $2,500,000, which could have a negative effect on our business and harm the market price of our common stock.
The warrants may not have any value.
The Series A Warrants have an initial exercise price per share equal to 100% of the public offering price per unit set forth on the cover page of this prospectus and can be exercised during the five-year period beginning on the date of issuance. The Series B Warrants have an initial exercise price per share equal to 100% of the public offering price per unit set forth on the cover page of this prospectus and can be exercised during the five-year period beginning one year and one day after the date of issuance, but only if our stockholders increase the number of our authorized shares of common stock. In the event our common stock price does not exceed the exercise price of the warrants during the period when the warrants are exercisable, or if, in the case of the Series B Warrants, our stockholders fail to approve an increase in our authorized shares of common stock, the warrants may not have any value.
Holders of our warrants will have no rights as a common stockholder until they acquire our common stock.
Until you acquire shares of our common stock upon exercise of your warrants, you will have no rights with respect to our common stock. Upon exercise of your warrants, you will be entitled to exercise the rights of a common stockholder only as to matters for which the record date occurs after the exercise date.
You will be unable to exercise the warrants under certain circumstances.
If we are unable to issue the shares of common stock upon exercise of the warrants because the registration statement covering the shares is subject to a stop order or has had its effectiveness suspended or withdrawn or if we
are otherwise unable to issue the shares, and no exemption from registration is available by virtue of a cashless exercise or otherwise, the warrants will not be exercisable. In such event, the warrants will not expire until five days after the date we are first able to issue the shares of common stock. In addition, if our stockholders fail to approve an increase in our authorized shares of common stock, the Series B Warrants will not be exercisable. In no event will the warrants be net cash settled.
NOTE CONCERNING FORWARD-LOOKING STATEMENTS
This prospectus contains forward-looking statements, within the meaning of Section 27A of the Securities Act of 1933 and Section 21E of the Securities Exchange Act of 1934, that involve risk and uncertainties. Any statements contained in this prospectus that are not statements of historical fact may be forward-looking statements. When we use the words “anticipates,” “plans,” “expects” and similar expressions, we are identifying forward-looking statements. Forward-looking statements involve risks and uncertainties which may cause our actual results, performance or achievements to be materially different from those expressed or implied by forward-looking statements. These factors include, among others:
|
•
|
current or future financial performance,
|
|||
|
•
|
management’s plans and objectives for future operations,
|
|||
|
•
|
uncertainties associated with product research and development,
|
|||
|
•
|
clinical trials and results,
|
|||
|
•
|
uncertainties associated with dependence upon the actions of our collaborators and of government regulatory agencies,
|
|||
|
•
|
product plans and performance,
|
|||
|
•
|
management’s assessment of market factors, and
|
|||
|
•
|
statements regarding our strategy and plans and those of our strategic partners.
|
|||
These forward-looking statements involve known and unknown risks, uncertainties and other factors that could cause our actual results to be materially different from our historical results or from any results expressed or implied by forward-looking statements. Our future operating results are subject to risks and uncertainties and are dependent upon many factors, including, without limitation, the risks identified under the caption “Risk Factors,” and in our other Securities and Exchange Commission (SEC) filings. The statements we make in this prospectus are as of the date of this prospectus.
Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee future results, levels of activity, performance or achievements. Except as may be required by law, we do not intend to update any of the forward-looking statements for any reason after the date of this prospectus to conform such statements to actual results or if new information becomes available.
All forward-looking statements attributable to us, or to persons acting on our behalf, are expressly qualified in their entirety by these cautionary statements.
USE OF PROCEEDS
We estimate that the net proceeds from this offering will be approximately $[ * ] million, after deducting the underwriting discounts and commissions and estimated offering expenses payable by us. This amount does not include the proceeds which we may receive in connection with the exercise of the warrants. We cannot predict when or if the warrants will be exercised, and it is possible that the warrants may expire and never be exercised.
We intend to use the net proceeds of this offering for general corporate purposes and working capital, including our clinical trial programs with bremelanotide for female sexual dysfunction, and secondarily for our PL-3994 development programs for asthma and a development program for new peptides for sexual dysfunction. Pending use of the net proceeds, we intend to invest these net proceeds in interest-bearing, investment-grade securities.
The amounts actually expended for each purpose may vary significantly depending upon numerous factors, including the amount and timing of the proceeds from this offering. Expenditures will also depend upon the availability of additional financing, whether we are able to enter into an agreement with a development and marketing partner for either bremelanotide for erectile dysfunction or PL-3994, and if so, the terms and conditions of such agreement, and other factors. Investors will be relying on the judgment of our management regarding the application of the proceeds of any sale of securities.
The proceeds from this offering will likely not be sufficient to complete clinical trials and other studies required for the approval of any product by the FDA, and we will need significant additional funds in the future. See the sections entitled “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operation.”
Our net tangible book value as of December 31, 2010, was approximately $4.0 million, or $0.34 per share of common stock. Net tangible book value per share is calculated by subtracting our total liabilities from our total tangible assets, which is total assets less intangible assets, and dividing this amount by the number of shares of common stock outstanding as of December 31, 2010.
After giving effect to the sale by us of 23,000,000 units at a public offering price of $[ * ] per unit, and after deducting the underwriting discounts and commissions and estimated offering expenses payable by us, and attributing no value to the warrants, our as adjusted net tangible book value as of December 31, 2010, would have been approximately $[ * ] million, or $[ * ] per share of common stock. This represents an immediate increase in net tangible book value of $[ * ] per share to our existing stockholders and an immediate and substantial dilution of $[ * ] per share to purchasers of units in this offering, as illustrated in the following table:
|
Offering price per unit
|
$
|
[ * ]
|
||||||
|
Net tangible book value per share
|
$
|
0.34
|
||||||
|
Increase in net tangible book value per share attributable to new investors
|
$
|
[ * ]
|
||||||
|
Net tangible book value per share after this offering
|
$
|
[ * ]
|
||||||
|
Dilution per share to new investors
|
$
|
[ * ]
|
||||||
The foregoing table does not take into account both (a) the initial classification of Series B Warrants and underwriter warrants as a liability until such time as stockholders approve an increase in our authorized common shares in an amount sufficient to permit exercise of Series B Warrants and underwriter warrants and (b) the effect of the exercise of any of the warrants included within the units.
The foregoing table further does not take into account further dilution to purchasers of units in this offering that could occur upon the exercise of outstanding options and warrants having a per share exercise price less than the per share offering price to the public in this offering. As of December 31, 2010, there were 11,854,028 shares of common stock outstanding, which does not include:
|
•
|
864,166 shares of common stock issuable upon exercise of options outstanding as of December 31, 2010, at a weighted average exercise price of $12.18 per share;
|
||
|
•
|
1,551,748 shares of common stock issuable upon exercise of warrants outstanding as of December 31, 2010, at a weighted average exercise price of $8.46;
|
||
|
•
|
332,413 shares of common stock reserved for future issuance under our 2005 Stock Plan; and
|
||
|
•
|
26,865 shares of common stock issuable upon conversion of immediately convertible Series A Convertible Preferred Stock outstanding as of December 31, 2010.
|
||
MARKET PRICE OF COMMON STOCK
Our common stock has been listed on NYSE Amex under the symbol “PTN” since December 21, 1999. It previously traded on The Nasdaq SmallCap Market under the symbol “PLTN.” The table below provides, for the fiscal quarters indicated, the reported high and low sales prices for our common stock on the NYSE Amex since July 1, 2008. Prices per share of our common stock have been adjusted for the one-for-ten reverse stock split on September 27, 2010 on a retroactive basis.
|
FISCAL YEAR ENDING JUNE 30, 2011
|
HIGH
|
LOW
|
|
Third Quarter (through February 22, 2011 )
|
$1.45
|
$1.01
|
|
Second Quarter
|
1.90
|
0.84
|
|
First Quarter
|
2.40
|
1.26
|
|
FISCAL YEAR ENDED JUNE 30, 2010
|
HIGH
|
LOW
|
|
Fourth Quarter
|
$3.50
|
$1.70
|
|
Third Quarter
|
3.70
|
2.50
|
|
Second Quarter
|
4.40
|
2.30
|
|
First Quarter
|
4.80
|
2.20
|
|
FISCAL YEAR ENDED JUNE 30, 2009
|
HIGH
|
LOW
|
|
Fourth Quarter
|
$3.70
|
$1.00
|
|
Third Quarter
|
1.40
|
0.60
|
|
Second Quarter
|
10.50
|
0.60
|
|
First Quarter
|
3.40
|
1.10
|
On February 22, 2011, the closing price as reported on NYSE Amex of our common stock was $1.12 per share. As of February 22, 2011, we had 229 record holders of our common stock. This number does not include stockholders for whom shares were held in “nominee” or “street” name.
DIVIDEND POLICY
We have never declared or paid any dividends. We currently intend to retain earnings, if any, for use in our business. We do not anticipate paying dividends in the foreseeable future. Any future determination as to the declaration and payment of dividends, if any, will be at the discretion of our board of directors and will depend on then existing conditions, including our financial condition, operating results, contractual restrictions, capital requirements, business prospects and other factors our board of directors may deem relevant. Our outstanding Series A Preferred Stock, consisting of 4,997 shares on February 22, 2011, provides that we may not pay a dividend or make any distribution to holders of any class of stock unless we first pay a special dividend or distribution of $100 per share to the holders of the Series A Preferred Stock.
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION
AND RESULTS OF OPERATIONS
The following discussion and analysis should be read in conjunction with the consolidated financial statements and notes to the consolidated financial statements filed as part of this prospectus.
Critical Accounting Policies.
Our significant accounting policies are described in Note 2 to our audited consolidated financial statements and Note 3 to our unaudited interim consolidated financial statements for the three and six months ended December 31, 2010 and 2009, both of which are included in this prospectus. We believe that our accounting policies and estimates relating to revenue recognition, accrued expenses and stock-based compensation charges are the most critical.
Revenue Recognition
Revenue from corporate collaborations and licensing agreements consists of up-front fees, research and development funding and milestone payments. Non-refundable up-front fees are deferred and amortized to revenue on a straight-line basis over the related performance period. We estimate the performance period as the period in which we perform certain development activities under the applicable agreement. Reimbursements for research and development activities are recorded in the period that we perform the related activities under the terms of the applicable agreements. Revenue resulting from the achievement of milestone events stipulated in the applicable agreements is recognized when the milestone is achieved, provided that such milestone is substantive in nature. Revenue from grants is recognized as we provide the services stipulated in the underlying grants based on the time and materials incurred.
The $10.0 million upfront payment received in January 2007 under the research collaboration and license agreement with AstraZeneca and the additional $5.0 million received pursuant to the September 2009 amendment has been recognized as revenue over the period ended January 2010, the completion of the research collaboration portion of the agreement.
In 2004, we entered into a collaborative development and marketing agreement with King Pharmaceuticals, Inc. (King) to jointly develop and commercialize bremelanotide, which agreement was terminated effective December 2007. Deferred revenue related to the King agreement had been recognized as revenue over the estimated period of our performance during the initial development term of this agreement. In connection with the termination of the agreement, we recognized as revenue in our fiscal year ended June 30, 2008 all remaining deferred up-front license fees received from King, together with associated deferred costs, in the amounts of $6.5 million and $0.8 million, respectively.
Accrued Expenses
Third parties perform a significant portion of our development activities. We review the activities performed under significant contracts each quarter and accrue expenses and the amount of any reimbursement to be received from our collaborators based upon the estimated amount of work completed. Estimating the value or stage of completion of certain services requires judgment based on available information. If we do not identify services performed for us but not billed by the service-provider, or if we underestimate or overestimate the value of services performed as of a given date, reported expenses will be understated or overstated.
Stock-based Compensation
The fair value of stock options granted has been calculated using the Black-Scholes option pricing model, which requires us to make estimates of expected volatility and expected option lives. We estimate these factors at the time of grant based on our own prior experience, public sources of information and information for comparable companies. The amount of recorded compensation related to an option grant is not adjusted for subsequent changes in these estimates or for actual experience. The amount of our recorded compensation is also dependent on our estimates of future option forfeitures and the probability of achievement of performance conditions. If we initially over-estimate future forfeitures, our reported expenses will be understated until such time as we adjust our estimate. Changes in estimated forfeitures will affect our reported expenses in the period of change and future periods.
The amount and timing of compensation expense to be recorded in future periods related to grants of restricted stock units may be affected by employment terminations. As a result, stock-based compensation charges may vary significantly from period to period.
Results of Operations
Three and Six Months Ended December 31, 2010 Compared to the Three and Six Months Ended December 31, 2009
Revenue – For the three and six months ended December 31, 2010, we recognized $0.2 million and $0.4 million, respectively, in revenue compared to $7.3 million and $10.9 million, respectively, for three and six months ended December 31, 2009 pursuant to our license agreement with AstraZeneca.
Revenue for the three and six months ended December 31, 2010 consisted entirely of reimbursement of development costs and per-employee compensation, earned at the contractual rate. Revenue for the three and six months ended December 31, 2009 consisted of $1.1 million and $2.0 million, respectively, related to our research services performed during those periods, and $6.2 million and $8.9 million, respectively, of revenue related to AstraZeneca’s up-front license fee. In connection with the completion of the research collaboration portion of the research collaboration and license agreement, we recognized as revenue in fiscal 2010 all remaining deferred up-front license fees received from AstraZeneca. Future contract revenue from AstraZeneca, in the form of reimbursement of development costs, will fluctuate based on development activities in our obesity program. We may also earn contract revenue based on the attainment of development milestones.
Research and Development – Research and development expenses for the three months ended December 31, 2010 decreased to $2.0 million from $2.7 million for the three months ended December 31, 2009. This decrease is the result of reducing staffing levels pursuant to our strategic decision to focus resources and efforts on clinical trials of bremelanotide and PL-3994 and preclinical development of an inhaled formula of PL-3994 and a new peptide drug candidate for sexual dysfunction announced in September 2010. Research and development expenses remained fairly constant at $5.4 million for the six months ended December 31, 2010 and for the six months ended December 31, 2009.
Research and development expenses related to our bremelanotide, other melanocortin receptor agonists, PL-3994, obesity and other preclinical programs were $0.2 million and $1.0 million, respectively, for the three and six months ended December 31, 2010 compared to $0.6 million and $1.1 million, respectively, for the three and six months ended December 31, 2009. Spending to date has been primarily related to the identification and optimization of lead compounds and pre-clinical development, and secondarily to a study of the effects of melanocortin receptor-specific compounds on food intake, obesity and other metabolic parameters and a study of subcutaneously administered bremelanotide. The amount of such spending and the nature of future development activities are dependent on a number of factors, including primarily the availability of funds to support future development activities, success of our clinical trials and preclinical and discovery programs, and our ability to progress compounds in addition to bremelanotide and PL-3994 into human clinical trials.
The historical amounts of project spending above exclude general research and development spending, which decreased $0.3 million to $1.8 million for the three months ended December 31, 2010, from $2.1 million for the three months ended December 31, 2009. This decrease is the result of reducing staffing levels pursuant to our strategic decision to focus resources and efforts on clinical trials of bremelanotide and PL-3994 and preclinical development of an inhaled formula of PL-3994 and a new peptide drug candidate for sexual dysfunction announced in September 2010. General research and development spending increased to $4.4 million for the six months ended December 31, 2010 compared to $4.3 million for the six months ended December 31, 2009, primarily related to the recognition of severance related expenses of $0.6 million in the three months ended September 30, 2010.
Cumulative spending from inception to December 31, 2010 on our bremelanotide, NeutroSpec (a previously marketed imaging product on which all work is suspended) and other programs (which include PL-3994, other melanocortin receptor agonists, obesity, and other discovery programs) amounts to approximately $136.8 million, $55.6 million and $58.6 million, respectively. Due to various risk factors described in our periodic reports filed with the SEC, including the difficulty in currently estimating the costs and timing of future Phase 1 clinical trials and larger-scale Phase 2 and Phase 3 clinical trials for any product under development, we cannot predict with reasonable certainty when, if ever, a program will advance to the next stage of development or be successfully completed, or when, if ever, net cash inflows will be generated.
General and Administrative – General and administrative expenses decreased to $0.9 million for the three months ended December 31, 2010 compared to $1.1 million for the three months ended December 31, 2009. The decrease is primarily related to reducing staffing levels pursuant to our strategic decision to focus resources and efforts on clinical trials of bremelanotide and PL-3994 and preclinical development of an inhaled formula of PL-3994 and a new peptide drug candidate for sexual dysfunction. General and administrative expenses remained fairly constant at $2.3 million for the six months ended December 31, 2010 and for the six months ended December 31, 2009.
Year Ended June 30, 2010 Compared to the Year Ended June 30, 2009
Revenue – For the fiscal year ended June 30, 2010 (fiscal 2010), we recognized $14.2 million in revenue compared to $11.4 million for the fiscal year ended June 30, 2009 (fiscal 2009) pursuant to our research collaboration and license agreement with AstraZeneca.
Revenue from AstraZeneca for fiscal 2010 and fiscal 2009 consists of $3.2 million and $9.7 million, respectively, of revenue related to our research services performed during those periods, and $11.0 million and $1.7 million, respectively, of revenue related to AstraZeneca’s up-front license fee. In connection with the completion of the research collaboration portion of the research collaboration and license agreement, we recognized as revenue in fiscal 2010 all remaining deferred up-front license fees received from AstraZeneca. Future contract revenue from AstraZeneca, in the form of reimbursement of development costs, will fluctuate based on development activities in our obesity program. We may also earn contract revenue based on the attainment of development milestones.
Research and Development – Research and development expenses decreased to $12.3 million for fiscal 2010 compared to $13.4 million for fiscal 2009. The decrease is the result of the restructuring of our clinical-stage product portfolio and development programs.
Research and development expenses related to our bremelanotide, other melanocortin receptor agonists, PL-3994, obesity and other preclinical programs were $4.1 million in each of fiscal years 2010 and 2009. Spending to date has been primarily related to the identification and optimization of lead compounds, and secondarily to study the effects of melanocortin receptor-specific compounds on food intake, obesity and other metabolic parameters and preclinical studies and a Phase 1 trial with subcutaneously administered bremelanotide. The amount of such spending and the nature of future development activities are dependent on a number of factors, including primarily the availability of funds to support future development activities, success of our clinical trials and preclinical and discovery programs, and our ability to progress compounds in addition to bremelanotide and PL-3994 into human clinical trials.
The historical amounts of project spending above exclude general research and development spending, which decreased to $8.2 million for fiscal 2010 compared to $9.3 million for fiscal 2009. The decrease is primarily related to management’s refinement of operations and expense control.
Cumulative spending from inception to June 30, 2010 on our bremelanotide, NeutroSpec (a previously marketed imaging product on which all work is suspended) and other programs (which include PL-3994, other melanocortin receptor agonists, obesity and other discovery programs) amounts to $133.2 million, $55.5 million and $56.8 million, respectively. Due to various risk factors described in this prospectus, including the difficulty in currently estimating the costs and timing of future Phase 1 clinical trials and larger-scale Phase 2 and Phase 3 clinical trials for any product under development, we cannot predict with reasonable certainty when, if ever, a program will advance to the next stage of development or be successfully completed, or when, if ever, related net cash inflows will be generated. See the section entitled “Risk Factors.”
General and Administrative – General and administrative expenses decreased to $4.9 million for fiscal 2010 compared to $5.3 million for fiscal 2009. The decrease is primarily related to management’s refinement of operations and expense control.
Income Tax Benefit – Income tax benefits of $1.0 million in fiscal 2010 and $1.7 million in fiscal 2009 relate to the sale of New Jersey state net operating loss carryforwards. The amount of such losses and tax credits that we are able to sell depends on annual pools and allocations established by the state of New Jersey.
Year Ended June 30, 2009 Compared to the Year Ended June 30, 2008
Revenue – For fiscal 2009, we recognized $11.4 million in revenue compared to $11.5 million for the fiscal year ended June 30, 2008 (fiscal 2008). Revenue consisted of the following:
|
Fiscal 2009
|
Fiscal 2008
|
Revenue related to:
|
|
|
$11.4 million
|
$3.0 million
|
our license agreement with AstraZeneca
|
|
|
-
|
$8.2 million
|
bremelanotide for ED and FSD pursuant to our collaboration agreement with King, which was terminated effective December 2007
|
|
Fiscal 2009
|
Fiscal 2008
|
Revenue related to:
|
|
|
-
|
$0.3 million
|
NeutroSpec, pursuant to our collaboration agreement with Mallinckrodt.
|
Revenue from AstraZeneca for fiscal 2009 and fiscal 2008 consists of $9.7 million and $1.3 million, respectively, of revenue related to our research services performed during those periods, and $1.7 million and $1.7 million, respectively, of revenue related to AstraZeneca’s up-front license fee. As discussed under “Revenue Recognition” above, in fiscal 2008 we recognized remaining deferred up-front license fees and associated deferred costs on termination of our collaboration agreement with King. Contract revenue from Mallinckrodt, with whom we have a strategic collaboration agreement to develop NeutroSpec, primarily reflects Mallinckrodt’s share of the costs incurred in NeutroSpec development activities. There were no substantive development activities on NeutroSpec in fiscal 2009 or fiscal 2010, though the agreement with Mallinckrodt has not been terminated.
Research and Development – Research and development expenses decreased to $13.4 million for fiscal 2009 compared to $21.2 million for fiscal 2008. The decrease is the result of the restructuring of our clinical-stage product portfolio and development programs and the reduction in workforce initiated in May 2008.
Research and development expenses related to our bremelanotide, other melanocortin receptor agonists, PL-3994, obesity, NeutroSpec and other preclinical programs were $4.1 million for fiscal 2009 and $7.1 million for fiscal 2008. Spending to date has been primarily related to the identification and optimization of lead compounds, and secondarily to a study of the effects of melanocortin receptor-specific compounds on food intake, obesity and other metabolic parameters and preclinical studies, a Phase 1 and a Phase 2A trial with PL-3994 and additional preclinical studies and a Phase 1 trial with subcutaneously administered bremelanotide. The amount of such spending and the nature of future development activities are dependent on a number of factors, including primarily the availability of funds to support future development activities, success of our clinical trial, preclinical and discovery programs, and our ability to progress compounds in addition to bremelanotide and PL-3994 into human clinical trials.
The historical amounts of project spending above exclude general research and development spending, which decreased to $9.3 million for fiscal 2009 compared to $14.1 million for fiscal 2008. The decrease is primarily related to the reduction in workforce initiated in May 2008.
Cumulative spending from inception to June 30, 2009 on our bremelanotide, NeutroSpec and other programs (which includes PL-3994, PL-6983, obesity, and other discovery programs) amounts to approximately $126.8 million, $55.5 million and $51.0 million, respectively. Due to various risk factors described in this prospectus, including the difficulty in currently estimating the costs and timing of future Phase 1 clinical trials and larger-scale Phase 2 and Phase 3 clinical trials for any product under development, we cannot predict with reasonable certainty when, if ever, a program will advance to the next stage of development or be successfully completed, or when, if ever, related net cash inflows will be generated. See the section entitled “Risk Factors.”
General and Administrative – General and administrative expenses decreased to $5.3 million for fiscal 2009 compared to $6.9 million for fiscal 2008. The decrease is primarily related to the reduction in workforce initiated in May 2008.
Income Tax Benefit – Income tax benefits of $1.7 million in fiscal 2009 and $1.3 million in fiscal 2008 relate to the sale of New Jersey state net operating loss carryforwards. The amount of such losses and tax credits that we are able to sell depends on annual pools and allocations established by the state of New Jersey.
Liquidity and Capital Resources
Since inception, we have incurred net operating losses, primarily related to spending on our research and development programs. We have financed our net operating losses primarily through equity financings and amounts received under collaborative agreements.
Our product candidates are at various stages of development and will require significant further research, development and testing and may never be successfully developed or commercialized. We may experience uncertainties, delays, difficulties and expenses commonly experienced by early stage biopharmaceutical companies, which may include unanticipated problems and additional costs relating to:
|
|
·
|
the development and testing of products in animals and humans;
|
|
|
·
|
product approval or clearance;
|
|
|
·
|
regulatory compliance;
|
|
|
·
|
good manufacturing practices;
|
|
|
·
|
intellectual property rights;
|
|
|
·
|
product introduction;
|
|
|
·
|
marketing, sales and competition; and
|
|
|
·
|
obtaining sufficient capital.
|
Failure to obtain timely regulatory approval for our product candidates and indications would impact our ability to increase revenues and could make it more difficult to attract investment capital for funding our operations. Any of these possibilities could materially and adversely affect our operations and require us to curtail or cease certain programs.
During the six months ended December 31, 2010, we used $5.2 million of cash for our operating activities, compared to $3.2 million used in the six months ended December 31, 2009. Higher net cash outflows from operations in the six months ended December 31, 2010 resulted primarily from lower revenues. During fiscal 2010, we used $5.7 million of cash for our operating activities, compared to $5.4 million used in fiscal 2009. Net cash outflows from operations in fiscal 2010 were favorably impacted by the decrease in research and development expenses and the receipt of $5.0 million in additional payments from AstraZeneca. Net cash outflows from operations in fiscal 2009 were favorably impacted by the receipt of $6.6 million in additional payments from AstraZeneca. Our periodic accounts receivable balances will continue to be highly dependent on the timing of receipts from collaboration partners and the division of development responsibilities between us and our collaboration partners.
During the six months ended December 31, 2010, cash provided by investing activities was $1.5 million from the sale of available-for-sale investments. During the six months ended December 31, 2009, cash provided by investing activities of $0.1 million consisted solely of the sale of supplies. In fiscal 2010, net cash provided by investing activities was $38,000 consisting mainly of the sale of property compared to $0.7 million provided by investing activities in fiscal 2009, which consisted mainly of the sale of property and equipment.
During the six months ended December 31, 2010, cash provided by financing activities of $36,000 consisted primarily from the exercise of warrants during the period, offset by cash used for payment of withholding taxes related to restricted stock. During the six months ended December 31, 2009, cash provided by financing activities was $2.6 million, consisting of approximately $2.8 million from the sale of equity units in a registered direct offering offset by payments on capital lease obligations. For fiscal 2010, net cash provided by financing activities was $6.7 million, primarily reflecting the aggregate net proceeds of approximately $7.0 million from the sales in August 2009, February 2010 and June 2010 of 948,485 units, 962,963 units and 1,000,000 units, respectively, in registered direct offerings. Each unit from the August 2009 offering consisted of one share of common stock and a five-year warrant to purchase 0.35 shares of common stock at an exercise price of $3.30 per share. Each unit from the February 2010 offering consisted of one share of common stock, a Series A warrant exercisable for 0.33 shares of our common stock at an exercise price of $3.00 per share of common stock and a Series B warrant exercisable for 0.33 shares of common stock at an exercise price of $2.70 per share of common stock. The Series A warrant is exercisable 181 days from the date of issuance and expires three years thereafter, the Series B warrant was exercisable immediately upon issuance and originally expired 180 days from the date of issuance. Management extended the expiration date of the Series B warrants an additional 180 days. Each unit from the June 2010 offering consisted of one share of common stock and a one-year warrant to purchase 0.14 shares of common stock at an exercise price of $2.00 per share. During fiscal 2009, net cash used in financing activities was $0.3 million, consisting entirely of payments on capital lease obligations.
We have incurred cumulative negative cash flows from operations since our inception, and have expended, and expect to continue to expend in the future, substantial funds to complete our planned product development efforts. As of December 31, 2010, our cash and cash equivalents were $1.7 million, our available-for-sale investments were $2.0 million, and our current liabilities were $2.0 million.
We believe that our cash, cash equivalents and available-for-sale investments prior to this offering are not sufficient to fund our planned operations for the next twelve months. This raises substantial doubt about our ability to continue as a going concern. We have made the strategic decision to focus resources and efforts on clinical trials for bremelanotide and PL-3994 and preclinical development of an inhaled formulation of PL-3994 and a new peptide drug candidate for sexual dysfunction, and have ceased research and development efforts on new product
candidates. As part of this decision, we implemented reductions in staffing levels, and now have seventeen employees. The accompanying consolidated financial statements have been prepared assuming that we continue as a going concern.
We expect that the net proceeds from this offering will be sufficient to fund our planned operations for the next eighteen months, during which time we will focus resources and efforts primarily on clinical trials with bremelanotide for female sexual dysfunction and secondarily on our PL-3994 inhaled formulation development program and development program for new peptides for sexual dysfunction. We intend to seek additional capital through collaborative arrangements on PL-3994 and bremelanotide for erectile dysfunction and through other sources. However, sufficient additional funding to support clinical trials with either PL-3994 or bremelanotide for erectile dysfunction, or both, may not be available on acceptable terms or at all. In any event, the net proceeds from this offering will likely not be sufficient to complete all of the required clinical trials for any of our clinical products, but should allow us to complete planned Phase 2 clinical trials with bremelanotide for female sexual dysfunction. Within the next eighteen months we will need to obtain additional capital, through public or private equity or debt financings, collaborative arrangements on our product candidates, or other sources, in order to complete required clinical trials, including Phase 3 clinical trials with bremelanotide for female sexual dysfunction, and complete submission of an NDA to the FDA for any of our product candidates. Sufficient funds may not be available on acceptable terms or at all. We may be required to seek collaborators for our product candidates at an earlier stage than otherwise would be desirable and on terms that are less favorable than might otherwise be available, and relinquish, license or otherwise dispose of rights on unfavorable terms to technologies and product candidates that we would otherwise seek to develop or commercialize ourselves. The nature and timing of our development activities are highly dependent on our financing activities.
If we are unable to raise sufficient additional funds to advance at least one of our product candidates, we will implement plans for the orderly wind down of our business operations, including curtailing operations significantly and further decreasing staffing levels, and will seek to license, sell or otherwise dispose of our product candidates, technologies and contractual rights, including rights under our research collaboration and license agreement with AstraZeneca, on the best possible terms available. Even if we are able to license, sell or otherwise dispose of our product candidates, technologies and contractual rights, it is likely to be on unfavorable terms and for less value than if we had the financial resources to develop or otherwise advance our product candidates, technologies and contractual rights ourselves.
We anticipate incurring additional losses over at least the next few years. To achieve profitability, we, alone or with others, must successfully develop and commercialize our technologies and proposed products, conduct preclinical studies and clinical trials, obtain required regulatory approvals and successfully manufacture and market such technologies and proposed products. The time required to reach profitability is highly uncertain, and we do not know whether we will be able to achieve profitability on a sustained basis, if at all.
Off-Balance Sheet Arrangements
None.
Contractual Obligations
We have entered into various contractual obligations and commercial commitments. The following table summarizes our most significant contractual obligations as of June 30, 2010:
|
Payments due by Period
|
|||||
|
Total
|
Less than 1 Year
|
1 - 3 Years
|
3 - 5 Years
|
More than 5 Years
|
|
|
Facility operating leases
|
$ 4,948,401
|
$ 2,196,655
|
$ 2,290,236
|
$ 461,510
|
$ -
|
|
Capital lease obligations
|
37,107
|
22,264
|
14,843
|
-
|
-
|
|
License agreements
|
210,000
|
15,000
|
30,000
|
30,000
|
135,000
|
|
Total contractual obligations
|
$ 5,195,508
|
$ 2,233,919
|
$ 2,335,079
|
$ 491,510
|
$ 135,000
|
Our license agreement related to NeutroSpec requires royalty payments on commercial net sales and payments of up to $2.25 million contingent on the achievement of specified cumulative net margins on sales by
Mallinckrodt. No contingent amounts will be payable related to NeutroSpec unless we recommence sales and marketing of NeutroSpec. We do not expect to make any such contingent payments during the next twelve months.
We are a biopharmaceutical company dedicated to the development of peptide, peptide mimetic and small molecule agonist compounds with a focus on melanocortin and natriuretic peptide receptor systems. We have a pipeline of development programs targeting melanocortin and natriuretic receptors, including development of proposed products for treatment of sexual dysfunction, acute asthma, heart failure, hypertension, obesity, diabetes and metabolic syndrome.
Our Product Candidates
We currently have the following active drug development programs:
|
|
·
|
Bremelanotide, a peptide melanocortin receptor agonist, for treatment of sexual dysfunction, targeting female sexual dysfunction (FSD) and erectile dysfunction (ED) in patients non-responsive to current therapies.
|
|
|
·
|
Peptide melanocortin receptor agonists for treatment of FSD and ED.
|
|
|
·
|
PL-3994, a peptide mimetic natriuretic peptide receptor A (NPR-A) agonist, for treatment of acute exacerbations of asthma, heart failure and refractory or difficult-to-control hypertension.
|
We have licensed several families of melanocortin receptor-based compounds for treatment of obesity, diabetes and related metabolic syndrome to AstraZeneca AB (AstraZeneca) pursuant to our research collaboration and license agreement with AstraZeneca.
Melanocortin Receptor-Specific Programs
The melanocortin system is involved in a large and diverse number of physiologic functions, and therapeutic agents modulating this system may have the potential to treat a variety of conditions and diseases, including sexual dysfunction, obesity and related disorders, cachexia (wasting syndrome) and inflammation-related diseases.
Bremelanotide for Sexual Dysfunction. We are developing subcutaneously administered bremelanotide for the treatment of FSD and ED in patients non-responsive to current therapies. Bremelanotide, which is a melanocortin agonist (a compound which binds to a cell receptor and triggers a response) drug candidate, is a synthetic peptide analog of the naturally occurring hormone alpha-MSH (melanocyte-stimulating hormone).
Medical Need - FSD. FSD is a multifactorial condition that has anatomical, physiological, medical, psychological and social components. FSD includes four disorders, hypoactive sexual desire disorder, female sexual arousal disorder, sexual pain disorder and orgasmic disorder. To establish a diagnosis of FSD, these syndromes must be associated with personal distress, as determined by the affected women. Approximately 40 million American women are affected by FSD. The National Health and Social Life Survey, a probability sample study of sexual behavior in a demographically representative cohort of United States adults ages 18 to 59, found that approximately 43% of women suffer from some form of FSD.
There are no drugs in the United States approved for FSD indications.
Medical Need - ED. ED is the consistent inability to attain and maintain an erection sufficient for sexual intercourse. The condition is correlated with increasing age, cardiovascular disease, hypertension, diabetes,
hyperlipidemia and smoking. In addition, certain prescription drugs and psychogenic issues may contribute to ED. According to the Massachusetts Male Aging Study, more than 50% of men aged 40-70 report episodes of ED and more than 30 million men in the United States may be afflicted with some form of ED, with less than 20% seeking treatment. The incidence of ED increases with age. Studies show that chronic ED affects about 5% of men in their 40s and 15% to 25% of men by the age of 65. The current market size for ED is more than $4 billion per year.
PDE-5 inhibitors such as sildenafil (Viagra®), vardenafil (Levitra®) and tadalafil (Cialis®) are used to treat ED, but an estimated 35% of ED patients are non-responsive to PDE-5 inhibitor therapy. There are limited therapeutic options for ED patients non-responsive or inadequately responsive to PDE-5 inhibitor therapy, including alprostadil for direct penis injection or urethral suppositories, surgical penile implants and various devices.
Mechanisms of Action with Bremelanotide. Bremelanotide is believed to act through activation of melanocortin receptors in the central nervous system, which is a different mechanism of action from currently marketed PDE-5 inhibitor ED therapies that act directly on the vascular system. Studies have demonstrated efficacy with bremelanotide in ED patients non-responsive to PDE-5 inhibitor therapies. Studies have also demonstrated an additive effect in ED patients co-administered both bremelanotide and a PDE-5 inhibitor.
Clinical Trials with Intranasal Formulations. We extensively studied bremelanotide for sexual dysfunction in nasal formulations, administered as a single spray in one nostril. Increases in blood pressure were observed in some patients receiving nasally administered bremelanotide, and this observed increase was a significant factor leading us to discontinue work on nasally administered bremelanotide as a first-line therapy for sexual dysfunction. We believe that the amount of increase in blood pressure, as well as the rate of nausea and emesis (vomiting), was due, at least partially, to high doses resulting from variability in drug uptake with nasal administration. Studies showed wide variation in plasma levels of bremelanotide in patients receiving nasally administered bremelanotide.
While we are no longer developing intranasal formulations of bremelanotide for commercialization, trials with intranasal formulations of bremelanotide did demonstrate potential utility of bremelanotide. Phase 2B double blind, placebo-controlled, parallel doses clinical trials evaluating nasal bremelanotide for ED, conducted in 726 non-diabetic and 294 diabetic patients, showed that over 30% of ED patients were restored to a normal level of function. Phase 2A clinical trials of post-menopausal FSD patients showed a statistically significant increase in the level of sexual desire and genital arousal in subjects receiving nasal bremelanotide compared to subjects receiving placebo and, in pre-menopausal FSD patents, a trend to increases in the level of sexual desire and genital arousal in subjects receiving nasal bremelanotide compared to subjects receiving placebo. In trials conducted to date, almost 2,000 patients received at least one dose of bremelanotide, with about 1,500 receiving multiple doses.
Subcutaneous Administration of Bremelanotide. In a Phase 1 clinical trial designed to evaluate the blood pressure effects of subcutaneously administered bremelanotide in pre-menopausal women, no statistically significant difference in mean changes in blood pressure was seen in subjects receiving bremelanotide compared to placebo. No subject discontinued participation in the trial as a result of protocol stopping rules based on blood pressure changes. In addition, there was no difference in the incidence of emesis in subjects receiving bremelanotide compared to placebo. This Phase 1 trial was a two-week, randomized, double-blind, placebo-controlled study in subjects who received 45 repeat doses of bremelanotide or placebo subcutaneously. Each administered dose of bremelanotide achieved plasma levels shown to be efficacious for improving erectile function in multiple previous Phase 1 and Phase 2 erectile dysfunction studies. With subcutaneous administration of bremelanotide variability in plasma exposure was significantly decreased.
We have completed a placebo-controlled, randomized, double-blind, cross over safety study evaluating blood pressure effects of subcutaneous bremelanotide in healthy male volunteers between 45 and 65 years old. The study also evaluated dose-to-dose consistency of plasma exposure of bremelanotide. A total of 49 subjects were dosed in the safety study; 19 of the subjects were enrolled in a sub-study and completed a graded exercise treadmill test as a surrogate for the cardiovascular effects of sexual activity. The results show that subcutaneous administration of bremelanotide provides better control of both plasma drug levels and blood pressure effects.
Next Clinical Trial Steps. We submitted a protocol and held a meeting with the FDA on initiation of an at-home Phase 2 clinical trial of subcutaneously administered bremelanotide for pre-menopausal women with FSD. At that meeting we reached agreement with the FDA on the protocol and clinical trial design. We will be submitting a revised protocol to the FDA reflecting that agreement, and assuming the sale of the units offered in this prospectus, intend to start this Phase 2 at home clinical trial for women with FSD in the second quarter of calendar 2011.
We are seeking a development and marketing partner for subcutaneously administered bremelanotide for men with ED who are non-responsive or inadequately responsive to PDE-5 inhibitor therapies. The partner would fund, in whole or in part, an in-clinic Phase 2 clinical trial, as either monotherapy or a combination therapy with a
PDE-5 inhibitor such as sildenafil. We have not yet submitted a protocol to the FDA for this trial, and do not presently intend to do so unless and until we reach agreement with a development and marketing partner.
Delivery of Bremelanotide. Injection sites for subcutaneous injection of bremelanotide include the abdomen, thigh and upper arms. We are exploring various delivery devices for subcutaneous administration. We believe that fine needle devices, pen injectors and needle-free injector systems can be used for subcutaneous administration of bremelanotide, and are evaluating various delivery devices for potential commercialization. If Phase 2 clinical trials for ED or FSD are successful, we anticipate that Phase 3 clinical trials will be conducted with a delivery device intended for commercialization.
Peptide and Small Molecule Melanocortin Receptor Agonists for Treatment of Sexual Dysfunction. We developed a series of lead alternative melanocortin receptor-specific peptides for treatment of sexual dysfunction, and have demonstrated efficacy with certain of these peptides in inducing erections in animal models.
In developing these peptides, we used a novel screening platform that examined the effectiveness of peptides in animal models of sexual response and also determined cardiovascular effects, primarily looking at changes in blood pressure. In these animal models, certain of these peptides resulted in significantly smaller increases in blood pressure at doses effective for a sexual response than increases in blood pressure in the same models seen with comparably effective doses of bremelanotide. Additionally, many of these peptides are highly selective for the specific melanocortin receptor believed to be involved in sexual response, and thus may have an improved side effect and safety profile.
We have suspended further discovery work on our alternative melanocortin receptor-specific peptides, but intend to advance one or more of the peptides we have developed to preclinical toxicology and other studies required by the FDA prior to initiating human clinical trials.
We have suspended our development program for small molecule melanocortin receptor-specific compounds for treatment of sexual dysfunction.
Obesity. In 2007, we entered into an exclusive research collaboration and license agreement with AstraZeneca to discover, develop and commercialize compounds that target melanocortin receptors for the treatment of obesity, diabetes and related metabolic syndrome. In June and December 2008 and in September 2009, the research collaboration and license agreement was amended to include additional compounds and associated intellectual property we developed and to modify royalty rates and milestone payments. Active work under the collaboration portion of the agreement concluded in January 2010, but we are still providing certain clinical trial related and other services to AstraZeneca.
Obesity is a multifactorial condition with significant biochemical components relating to satiety (feeling full), energy utilization and homeostasis. A number of different metabolic and hormonal pathways are being evaluated by companies around the world in efforts to develop better treatments for obesity. Scientific research has established that melanocortin receptors have a role in eating behavior and energy homeostasis, and that some melanocortin receptor agonists decrease food intake and induce weight loss.
Obesity is a significant healthcare issue, often correlated with a variety of cardiovascular and other diseases, including diabetes. More than 1.1 billion adults and over 150 million children worldwide are overweight, with over 300 million adults categorized as obese. According to the American Obesity Association, obesity is the second leading cause of preventable death after smoking and nearly one-third of adults in the United States are obese. Increased mortality, high blood pressure, diabetes and other substantial health risks are associated with being overweight and obese. Over 2.6 million deaths are attributed to diabetes each year worldwide and almost $120 billion is spent on related costs of obesity, according to the U.S. Surgeon General.
We developed classes of small molecule and peptide compounds targeting melanocortin receptors which are effective in the treatment of obesity in animal models. Certain of these compounds have been demonstrated to be effective in normal diet-induced obese and genetically obese animal models for decreasing food intake and body weight, without an increase in sexual response in normal animals at the same or higher dose levels. Pursuant to clinical trial agreements with AstraZeneca, we have conducted proof-of-principle clinical trials on the effects of a melanocortin receptor-specific compound on food intake, obesity and other metabolic parameters, and have agreed to conduct additional related studies at a negotiated rate.
Our agreement with AstraZeneca remains in effect as long as AstraZeneca is developing a compound covered by the agreement or commercializing a product for which a royalty is owed. The agreement may be terminated by AstraZeneca upon notice to us, or by either party upon notice in the event of a material breach. Upon termination by AstraZeneca without cause or by us for cause, all rights and licenses we granted to AstraZeneca
terminate, but AstraZeneca remains obligated to pay royalties and milestones on compounds developed during the collaboration portion of the agreement. In the event AstraZeneca terminates the agreement because we breached the agreement, rights and licenses we granted under the agreement become permanent, with financial terms, including royalties, to be determined by arbitration.
We have received up-front and other licensing payments totaling $15 million from AstraZeneca under the agreement. We are eligible for milestone payments totaling up to $145 million, with up to $85 million contingent upon development and regulatory milestones and the balance on achievement of sales targets, plus mid to high single-digit royalties on sales of approved products. AstraZeneca has responsibility for product commercialization, product discovery and development costs.
Other Melanocortin Programs. We have suspended work on our other early stage research and discovery programs exploring additional indications and targets. These programs include development of highly selective melanocortin-1 and melanocortin-3 receptor agonists for treatment of inflammation-related diseases and disorders and melanocortin-4 receptor antagonists for treatment of cachexia. We do not anticipate that any significant effort will be devoted to these programs during the next twelve months.
Natriuretic Peptide Receptor-Specific Programs
The natriuretic peptide receptor system has numerous cardiovascular functions, and therapeutic agents modulating this system may be useful in treatment of heart failure, hypertension, acute asthma and other cardiovascular diseases.
PL-3994. PL-3994 is an NPR-A agonist compound in development for treatment of acute exacerbations of asthma, heart failure and refractory hypertension. PL-3994 activates NPR-A, a receptor known to play a role in cardiovascular homeostasis. PL-3994 increases plasma cyclic guanosine monophosphate (cGMP) levels, a pharmacological response consistent with the effects of endogenous (naturally produced) natriuretic peptides on cardiovascular function and smooth muscle relaxation. PL-3994 also decreases activity of the renin-angiotensin-aldosterone system (RAAS), a hormone system that regulates blood pressure and fluid balance. The RAAS system is frequently over-activated in heart failure patients, leading to worsening of cardiovascular function.
PL-3994 is one of a number of natriuretic peptide receptor agonist compounds we have developed. PL-3994 is a synthetic molecule incorporating a novel and proprietary amino acid mimetic structure. It has an extended half-life, with reduced affinity for the endogenous natriuretic peptide clearance receptor and significantly increased resistance to neutral endopeptidase, an endogenous enzyme that degrades natriuretic peptides.
PL-3994 for Acute Exacerbations of Asthma. Acute exacerbations of asthma, also called acute severe asthma, is an ongoing asthma episode in which asthma symptoms do not adequately respond to initial bronchodilator or corticosteroid therapy. Inhaled beta-2 adrenergic receptor agonists, such as albuterol, and inhaled corticosteroids are primary treatments for asthma episodes. Some patients with acute exacerbations of asthma become unresponsive to beta-2 adrenergic receptor agonists, significantly limiting treatment options and increasing risk.
In 2006, the most recent year reported, there were almost 1.7 million emergency room visits due to asthma, with 440,000 hospitalizations attributed to asthma. In 2008, approximately 23.3 million Americans had asthma, with a projected 2010 economic cost in the United States of $20.7 billion, of which the largest single direct medical expenditure, $5.9 billion, is for prescription drugs.
Existing therapies for acute exacerbations of asthma in patients unresponsive to beta-2 adrenergic receptor agonists have limitations, including typically taking several hours for significant patient improvement. Existing therapies include oxygen, systemic steroids and anticholinergic drugs. PL-3994, which works through a different pathway than beta-2 adrenergic receptor agonists and other approved bronchodilators, is intended to address this unmet medical need.
Research over the past two decades has demonstrated potent bronchodilator effects with both systemic and inhalation administration of natriuretic peptides. NPR-A agonism is known to relax smooth muscles in airways and works through a pathway independent of the beta-2 adrenergic receptor. Preclinical testing demonstrated potent airway smooth muscle relaxation in rat, guinea pig and human tissues using PL-3994, and animal studies in sensitized guinea pigs has demonstrated a bronchodilator effect with PL-3994 using both subcutaneous and inhalation administration.
Endogenous natriuretic peptides have a very short half-life, due primarily to degradation by neutral endopeptidase and clearance through the natriuretic peptide clearance receptor. PL-3994 is resistant to neutral
endopeptidase and clears from the body much more slowly than endogenous natriuretic peptides. PL-3994 has a blood-plasma half-life of at least three hours in humans when administered by subcutaneous injection, with biological effects seen for over eight hours post-administration.
We have submitted an IND application with a clinical trial protocol to the FDA for a proof-of-concept human trial for asthma using a subcutaneously administered formulation of PL-3994. The FDA has reviewed the IND application and protocol, and this trial is allowed to proceed at any time. We also have commenced development of an inhalation formulation of PL-3994. We are seeking a development and marketing partner for PL-3994, which would include both the proof-of-concept human trial for asthma using a subcutaneously administered formulation and development of an inhalation formulation. We do not intend to initiate either the proof-of-concept human trial or preclinical inhalation toxicity studies unless and until we reach agreement with a development and marketing partner or receive funding to support the proof-of-concept human trial or preclinical inhalation toxicity studies from a third party, such as grant funding from an agency of the federal government.
PL-3994 for Heart Failure. Heart failure is an illness in which the heart is unable to pump blood efficiently, and includes acutely decompensated heart failure with dyspnea (shortness of breath) at rest or with minimal activity. Endogenous natriuretic peptides have a number of beneficial effects, including vasodilation (relaxation of blood vessels), natriuresis (excretion of sodium), and diuresis (excretion of fluids).
Patients who have been admitted to the hospital with an episode of worsening heart failure have an increased risk of either death or hospital readmission in the three months following discharge. Up to 15% of patients die in this period and as many as 30% need to be readmitted to the hospital. We believe that decreasing mortality and hospital readmission in patients discharged following hospitalization for worsening heart failure is a large unmet medical need for which PL-3994 may be effective. PL-3994 would be utilized as an adjunct to existing heart failure medications, and may, if successfully developed, be self-administered by patients as a subcutaneous injection following hospital discharge. We believe that PL-3994, through activation of NPR-A, may, if successful, reduce cardiac hypertrophy (increase in heart size due to disease), which is an independent risk factor for cardiovascular morbidity and mortality.
Over 5.7 million Americans suffer from heart failure, with 670,000 new cases of heart failure diagnosed each year, with disease incidence expected to increase with the aging of the American population. Despite the treatment of heart failure with multiple drugs, almost all heart failure patients will experience at least one episode of acute heart failure that requires treatment with intravenous medications in the hospital. Heart failure has tremendous human and financial costs. Estimated direct costs in the United States for heart failure are $37.2 billion in 2009, with heart failure constituting the leading cause of hospitalization in people over 65 years of age, with over 1.1 million hospital discharges for heart failure in 2006. Heart failure is also a high mortality disease, with approximately one-half of heart failure patients dying within five years of initial diagnosis.
We have planned a repeat dose Phase 2 clinical trial in patients hospitalized with heart failure, which will evaluate safety profiles in patients given repeat doses of PL-3994 as well as pharmacokinetic (period to metabolize or excrete the drug) and pharmacodynamic (period of action or effect of the drug) endpoints, but have not determined whether or when this trial will commence.
PL-3994 for Refractory Hypertension. PL-3994 may potentially also be used for treatment of refractory or difficult-to-control hypertension, which is high blood pressure despite a three-drug regimen that includes a diuretic. Refractory hypertension is commonly found in patients with congestive heart failure or renal disease. While there are a large number of approved drugs for treatment of hypertension, there are no approved drugs for hypertension that are active through the NPR-A system. Refractory and other difficult-to-control hypertension can be caused by increased aldosterone levels. PL-3994 is believed to act through the NPR-A system on the RAAS to decrease renin and aldosterone secretion and thereby decrease blood pressure. In a Phase 2A study of subjects with controlled hypertension, the data suggested an increased effect of PL-3994 in reducing systemic blood pressure when taken with an angiotensin-converting enzyme (ACE) inhibitor, a common class of drugs for controlling hypertension. PL-3994 thus may be suitable for use as an adjunct therapy to one or more existing hypertension drugs, including an ACE inhibitor.
Clinical Studies with PL-3994. Preclinical studies in animals established a dose-dependent effect on blood pressure and diuresis, and in animal models of heart failure showed improved kidney function and prevention of cardiac hypertrophy. Human clinical studies of PL-3994 commenced with a Phase 1 trial which concluded in the first quarter of calendar year 2008. This was a randomized, double-blind, placebo-controlled, study in 26 healthy volunteers who received either PL-3994 or a placebo subcutaneously. The evaluations included safety, tolerability, pharmacokinetics and several pharmacodynamic endpoints, including levels of cGMP, a natural messenger
nucleotide. Dosing concluded with the successful achievement of the primary endpoint of the study, a prespecified reduction in systemic blood pressure. No volunteer experienced a serious or severe adverse event. Elevations in plasma cGMP levels, increased diuresis and increased natriuresis were all observed for several hours after single subcutaneous doses.
In the second quarter of calendar year 2008, we conducted a Phase 2A trial in volunteers with controlled hypertension who were receiving one or more conventional antihypertensive medications. In this trial, which was a randomized, double-blind, placebo-controlled, single ascending dose study in 21 volunteers, the objective was to demonstrate that PL-3994 can be given safely to patients taking antihypertensive medications commonly used in heart failure and hypertension patients. Dosing concluded with the successful achievement of the primary endpoint of the study, a prespecified reduction in systemic blood pressure. No volunteer experienced a serious or severe adverse event. Elevations in plasma cGMP levels were observed for several hours after single subcutaneous doses.
Administration of PL-3994. We are developing PL-3994 for acute exacerbations of asthma indications as either a subcutaneously administered drug or as an inhaled drug. For asthma indications we believe that inhalation administration may be preferable to subcutaneous or other systemic administration.
We are developing PL-3994 for heart failure and refractory hypertension indications as a subcutaneously administered drug. PL-3994 is well absorbed through this route of administration. In human studies, the pharmacokinetic and pharmacodynamic half-lives were on the order of hours, significantly longer than the comparable half-lives of endogenous natriuretic peptides. We believe that subcutaneous PL-3994, if successful, will be amenable to self-administration by patients, similar to insulin and other self-administered drugs.
Other Natriuretic Peptide Receptor-Specific Programs. We have suspended work on our early stage discovery and development programs in the natriuretic peptide receptor field. We do not anticipate that any significant effort will be devoted to these programs during the next twelve months.
Other Programs
We previously marketed NeutroSpec®, a radiolabeled monoclonal antibody product for imaging and diagnosing infection, which is the subject of a strategic collaboration agreement with the Mallinckrodt division of Covidien Ltd. We have suspended marketing, clinical trials and securing regulatory approvals of NeutroSpec, and do not anticipate conducting any substantive work or incurring substantial expenditures on NeutroSpec over the next twelve months.
Technologies We Use
We used a rational drug design approach to discover and develop proprietary peptide, peptide mimetic and small molecule agonist compounds, focusing on melanocortin and natriuretic peptide receptor systems. Computer-aided drug design models of receptors are optimized based on experimental results obtained with peptides and small molecules we develop, supported by conformational analyses of peptides in solution utilizing nuclear magnetic resonance spectroscopy. By integrating both technologies, we believe we are developing an advanced understanding of the factors which drive agonism.
We have developed a series of proprietary technologies used in our drug development programs. One technology employs novel amino acid mimetics in place of selected amino acids. These mimetics provide the receptor-binding functions of conventional amino acids, while providing structural, functional and physiochemical advantages. The amino acid mimetic technology is employed in PL-3994, our compound in development for treatment of acute exacerbations of asthma, heart failure and refractory hypertension.
Some compound series have been derived using our proprietary and patented platform technology, called MIDAS™ (Metal Ion-induced Distinctive Array of Structures). This technology employs metal ions to fix the three-dimensional configuration of peptides, forming conformationally rigid molecules that remain folded specifically in their active state. These MIDAS molecules are generally simple to synthesize, are chemically and proteolytically stable, and have the potential to be orally bioavailable. In addition, MIDAS molecules are information-rich and provide data on structure-activity relationships that may be used to design small molecule, non-peptide drugs.
Estimate of Amount Spent on Research and Development Activities
Research and development expenses were $12.3 million for the fiscal year ended June 30, 2010 (fiscal 2010) and $13.4 million for the fiscal year ended June 30, 2009 (fiscal 2009), of which $3.2 million and $4.7 million of our research and development expenses for fiscal 2010 and fiscal 2009, respectively, were borne by AstraZeneca pursuant to the research collaboration and license agreement.
Competition
General. Our products under development will compete on the basis of quality, performance, cost effectiveness and application suitability with numerous established products and technologies. We have many competitors, including pharmaceutical, biopharmaceutical and biotechnology companies. Furthermore, there are several well-established products in our target markets that we will have to compete against. Products using new technologies which may be competitive with our proposed products may also be introduced by others. Most of the companies selling or developing competitive products have financial, technological, manufacturing and distribution resources significantly greater than ours and may represent significant competition for us.
The pharmaceutical and biotechnology industries are characterized by extensive research efforts and rapid technological change. Many biopharmaceutical companies have developed or are working to develop products similar to ours or that address the same markets. Such companies may succeed in developing technologies and products that are more effective or less costly than any of those that we may develop. Such companies may be more successful than us in developing, manufacturing and marketing products.
We cannot guarantee that we will be able to compete successfully in the future or that developments by others will not render our proposed products under development or any future product candidates obsolete or non-competitive or that our collaborators or customers will not choose to use competing technologies or products.
Bremelanotide and Other Melanocortin Receptor Agonists for Treatment of Sexual Dysfunction. There is competition and financial incentive to develop, market and sell drugs for the treatment of ED and FSD. Leading drugs approved for ED indications are PDE-5 inhibitors which target the vascular system, such as sildenafil (sold under the trade name Viagra®), vardenafil (sold under the trade name Levitra®) and tadalafil (sold under the trade name Cialis®). In addition, we are aware of other PDE-5 inhibitors under development. Other drugs approved for ED indications include alprostadil for injection (sold under the trade name Caverject Impulse® among others), which is injected directly into the penis, and alprostadil in urethral suppository format (sold under the trade name MUSE®). In addition, a variety of devices, including vacuum devices and surgical penile implants, have been approved for ED indications. We are aware of a number of companies developing new drugs for ED indications, including at least one company developing a new drug for treatment of ED not sufficiently responsive to PDE-5 inhibitors, some of which are in clinical trials in the United States and elsewhere. We are not aware of any company actively developing a melanocortin receptor agonist drug for ED.
There are no products specifically approved for an FSD indication in the United States. A number of hormonal therapies have been commercialized for other indications, including progestin, androgen and localized estrogen therapies, but none have been approved by the FDA for FSD indications. A number of drugs, including hormonal drugs, are in various stages of research or development for FSD. We are not aware of any company actively developing a melanocortin receptor agonist drug for FSD.
PL-3994 for Acute Exacerbations of Asthma Indications. The asthma market is intensively competitive, with substantial competition and financial incentive to develop, market and sell drugs for treatment of asthma, with projected costs of prescription drugs of $5.9 billion in the United States in 2010. We are aware of companies developing drugs for the specific indications of either acute exacerbations of asthma or acute severe asthma, including at least one company with a drug reported to be currently in clinical trials. In addition, a number of clinical trials are conducted by hospitals, research institutes and others exploring various methods and combinations of drugs to treat acute exacerbations of asthma. There are a number of drugs and therapies currently used to treat acute exacerbations of asthma, including administration of oral or intravenous systemic steroids, use of oxygen or heliox, a mixture of helium and oxygen, nebulized short-acting beta-2 adrenergic receptor agonists, intravenous or nebulized anticholinergic agents and, for patients in or approaching respiratory arrest, intubation and mechanical ventilation. However, each of these drugs or therapies has recognized limitations or liabilities, and we believe that there remains an unmet medical need for a safe and effective treatment for acute exacerbations of asthma. We are not aware of any company actively developing a drug to relax smooth muscles in airways through a natriuretic peptide receptor pathway.
PL-3994 for Heart Failure Indications. Nesiritide (sold under the trade name Natrecor®), a recombinant human B-type natriuretic peptide drug, is marketed in the United States by Scios Inc., a Johnson & Johnson company. Nesiritide is approved for treatment of acutely decompensated congestive heart failure patients who have dyspnea at rest or with minimal activity. Carperitide, a recombinant human atrial natriuretic peptide drug, is marketed in Japan and is reported to be available for licensing in other countries. Ularitide, a synthetic form of urodilatin, a naturally occurring human natriuretic peptide related to atrial natriuretic peptide, is reported to be in clinical trials. Nesiritide, carperitide and ularitide are administered by intravenous infusion. Because of the very
short half-lives of nesiritide, carperitide and ularitide, we believe these drugs are unlikely to be suitable for subcutaneous administration or for long-term treatment of heart failure. We are aware of other companies developing intravenously administered natriuretic peptide drugs, with at least one reported to be in Phase 2 clinical trials for acute heart failure. In addition, there are a number of approved drugs and drugs in development for treatment of heart failure through mechanisms or pathways other than agonism of NPR-A.
Obesity. There are several FDA-approved drugs for the treatment of obesity, and a large number of products in clinical development by other companies, including products which target melanocortin receptors. Clinical trials for obesity are lengthy, time-consuming and expensive, and we may not be able to proceed if AstraZeneca discontinues work under or terminates our research collaboration and license agreement. See the discussion under the heading “We do not control the development of compounds licensed to third parties and, as a result, we may not realize a significant portion of the potential value of any such license arrangements” in “Risk Factors” in this prospectus.
Patents and Proprietary Information
Patent Protection. Our success will depend in substantial part on our ability to obtain, defend and enforce patents, maintain trade secrets and operate without infringing upon the proprietary rights of others, both in the United States and abroad. We own a number of issued United States patents and have pending United States patent applications, many with issued or pending counterpart patents in selected foreign countries. We seek patent protection for our technologies and products in the United States and those foreign countries where we believe patent protection is commercially important.
We own two issued United States patents claiming the bremelanotide substance, issued patents claiming the bremelanotide substance in Japan, Mexico, Austria, Belgium, Cyprus, Denmark, Finland, France, Germany, Greece, Ireland, Luxembourg, Monaco, Netherlands, Portugal, Spain, Sweden, Switzerland, United Kingdom, Italy, Australia and New Zealand, and pending patent applications claiming the bremelanotide substance in Korea, Brazil, Canada and Mexico. The issued United States patents have a term until 2020, which term may be subject to extension for a maximum period of up to five years as compensation for patent term lost during drug development and the FDA regulatory review process, pursuant to the Drug Price Competition and Patent Term Restoration Act of 1984 (the Hatch-Waxman Amendments). Whether we will be able to obtain patent term extensions under the Hatch-Waxman Amendments and the length of the extension to which we may be entitled cannot be determined until the FDA approves for marketing, if ever, a product in which bremelanotide is the active ingredient. In addition, the claims of issued patents covering bremelanotide may not provide meaningful protection. Further, third parties may challenge the validity or scope of any issued patent.
We also own an issued United States patent claiming non-oral administration of bremelanotide in combination with oral administration of a PDE-5 inhibitor. This patent has a term until 2025. However, this patent would apply only if we develop bremelanotide for use in combination therapy with a PDE-5 inhibitor. If we obtain regulatory approval for bremelanotide for use in combination therapy with a PDE-5 inhibitor, which may never occur, then the patent term may be subject to extension under the Hatch-Waxman Amendments, but we cannot presently evaluate the duration of any potential patent term extension.
We own patent applications on one class of alternative melanocortin receptor-specific peptides for treatment of sexual dysfunction which are pending in the United States, Australia, Brazil, Canada, China, India, Israel, Japan, Korea, Mexico and South Africa and before the European and Eurasian patent offices. If any patent issues in the United States, the presumptive term will be until 2029. We also own a patent application under the Patent Cooperation Treaty for a second class of alternative melanocortin receptor-specific peptides for treatment of sexual dysfunction. We will be required to enter national stage prosecution on this application, including filing the application in countries we select, by November 2011. If we enter national stage prosecution in the United States, and if any patent issues, the presumptive term will be until 2030. Until one or more product candidates covered by a claim of one of these patent applications are developed for commercialization, which may never occur, we cannot evaluate the duration of any potential patent term extension under the Hatch-Waxman Amendments.
We own an issued United States patent claiming the PL-3994 substance and other natriuretic peptide receptor agonist compounds we have developed, which has a term until 2027, and two pending related United States patent applications, one claiming a precursor molecule and other claiming related compounds. Patent applications claiming the PL-3994 substance and other compounds, including precursor molecules, are pending in Australia, Brazil, Canada, China, India, Israel, Japan, Korea, Mexico, Philippines and South Africa and before the European and Eurasian patent offices. We do not know the full scope of patent coverage we will obtain, or whether any patents will issue other than the United States patent claiming PL-3994. Until one or more product candidates
covered by a claim of one of these patent applications are developed for commercialization, which may never occur, we cannot evaluate the duration of any potential patent term extension under the Hatch-Waxman Amendments.
We additionally have 25 issued United States patents and 9 patent applications, including 3 patent applications under the Patent Cooperation Treaty which have not yet entered national stage prosecution, on melanocortin receptor specific peptides and small molecules, but we are not actively developing any product candidate covered by a claim of one of these patents or applications. Most of the pending applications have not yet been examined, and we do not know the scope of patent claims that will be allowed, or whether any patents will issue.
We own a number of United States and foreign patent applications that are licensed to AstraZeneca under our research collaboration and license agreement relating to our obesity program. Under the agreement, AstraZeneca is responsible for prosecution of these patent applications in the United States and other countries. Additionally, AstraZeneca is prosecuting patent applications in its name resulting from its collaboration with us, including patent applications naming our employees as inventors, on which royalties would be payable under our agreement with AstraZeneca if a compound covered by a claim of one of these applications is developed for commercialization. Many of these patent applications have not yet been examined, and we do not know the scope of patent claims that will be allowed, or whether any patents will issue. Additionally, until one or more compounds subject to the agreement with AstraZeneca are developed for commercialization, which may never occur, we cannot evaluate the duration of patents or their effect on the program.
In the event that a third party has also filed a patent application relating to an invention we claimed in a patent application, we may be required to participate in an interference proceeding adjudicated by the United States Patent and Trademark Office to determine priority of invention. The possibility of an interference proceeding could result in substantial uncertainties and cost, even if the eventual outcome is favorable to us. An adverse outcome could result in the loss of patent protection for the subject of the interference, subjecting us to significant liabilities to third parties, the need to obtain licenses from third parties at undetermined cost, or requiring us to cease using the technology.
Future Patent Infringement. We do not know for certain that our commercial activities will not infringe upon patents or patent applications of third parties, some of which may not even have been issued. Although we are not aware of any valid United States patents which are infringed by bremelanotide or PL-3994, we cannot exclude the possibility that such patents might exist or arise in the future. We may be unable to avoid infringement of any such patents and may have to seek a license, defend an infringement action, or challenge the validity of such patents in court. Patent litigation is costly and time consuming. If such patents are valid and we do not obtain a license under any such patents, or we are found liable for infringement, we may be liable for significant monetary damages, may encounter significant delays in bringing products to market, or may be precluded from participating in the manufacture, use or sale of products or methods of treatment covered by such patents.
Proprietary Information. We rely on proprietary information, such as trade secrets and know-how, which is not patented. We have taken steps to protect our unpatented trade secrets and know-how, in part through the use of confidentiality and intellectual property agreements with our employees, consultants and certain contractors. If our employees, scientific consultants, collaborators or licensees develop inventions or processes independently that may be applicable to our product candidates, disputes may arise about the ownership of proprietary rights to those inventions and processes. Such inventions and processes will not necessarily become our property, but may remain the property of those persons or their employers. Protracted and costly litigation could be necessary to enforce and determine the scope of our proprietary rights.
If trade secrets are breached, our recourse will be solely against the person who caused the secrecy breach. This might not be an adequate remedy to us, because third parties other than the person who causes the breach will be free to use the information without accountability to us. This is an inherent limitation of the law of trade secret protection.
Governmental Regulation
The FDA, comparable agencies in other countries and state regulatory authorities have established regulations and guidelines which apply to, among other things, the clinical testing, manufacturing, safety, efficacy, labeling, storage, record keeping, advertising, promotion, marketing and distribution of our proposed products. Noncompliance with applicable requirements can result in fines, recalls or seizures of products, total or partial suspension of production, refusal of the regulatory authorities to approve marketing applications, withdrawal of approvals and criminal prosecution.
Before a drug product is approved by the FDA for commercial marketing, three phases of human clinical trials are usually conducted to test the safety and effectiveness of the product. Phase 1 clinical trials most typically involve testing the drug on a small number of healthy volunteers to assess the safety profile of the drug at different dosage levels. Phase 2 clinical trials, which may also enroll a relatively small number of patient volunteers, are designed to further evaluate the drug’s safety profile and to provide preliminary data as to the drug’s effectiveness in humans. Phase 3 clinical trials consist of larger, well-controlled studies that may involve several hundred or thousand patient volunteers representing the drug’s targeted population. During any of these phases, the clinical trial can be placed on clinical hold, or temporarily or permanently stopped for a variety of reasons, principally for safety concerns.
After approving a product for marketing, the FDA may require post-marketing testing, including extensive Phase 4 studies, and surveillance to monitor the safety and effectiveness of the product in general use. The FDA may withdraw product approvals if compliance with regulatory standards is not maintained or if problems occur following initial marketing. In addition, the FDA may impose restrictions on the use of a drug that may limit its marketing potential. The failure to comply with applicable regulatory requirements in the United States and in other countries in which we conduct development activities could result in a variety of fines and sanctions, such as warning letters, product recalls, product seizures, suspension of operations, fines and civil penalties or criminal prosecution.
In addition to obtaining approval of an NDA from the FDA for any of our proposed products, any facility that manufactures such a product must comply with GMPs. This means, among other things, that the drug manufacturing establishment must be registered with, and subject to inspection by, the FDA. Foreign manufacturing establishments must also comply with GMPs and are subject to periodic inspection by the FDA or by corresponding regulatory agencies in such other countries under reciprocal agreements with the FDA. In complying with standards established by the FDA, manufacturing establishments must continue to expend time, money and effort in the areas of production and quality control to ensure full technical compliance. We will use contract manufacturing establishments, in the United States or in foreign countries, to manufacture our proposed products, and will depend on those establishments to comply with GMPs and other regulatory requirements.
Third-Party Reimbursements
Successful sales of our proposed products in the United States and other countries will depend, in large part, on the availability of adequate reimbursement from third-party payors such as governmental entities, managed care organizations, HMOs and private insurance plans. Reimbursement by a third-party payor may depend on a number of factors, including the payor’s determination that the product has been approved by the FDA for the indication for which the claim is being made, that it is neither experimental nor investigational, and that the use of the product is safe and efficacious, medically necessary, appropriate for the specific patient and cost effective. Since reimbursement by one payor does not guarantee reimbursement by another, we or our licensees may be required to seek approval from each payor individually. Seeking such approvals is a time-consuming and costly process. Third-party payors routinely limit the products that they will cover and the amount of money that they will pay and, in many instances, are exerting significant pressure on medical suppliers to lower their prices. There is significant uncertainty concerning third-party reimbursement for the use of any pharmaceutical product incorporating new technology and we are not sure whether third-party reimbursement will be available for our proposed products once approved, or that the reimbursement, if obtained, will be adequate. There is also significant uncertainty concerning third-party reimbursement for products treating FSD and ED. Less than full reimbursement by governmental and other third-party payors for our proposed products would adversely affect the market acceptance of these proposed products. Further, healthcare reimbursement systems vary from country to country, and we are not sure whether third-party reimbursement will be made available for our proposed products under any other reimbursement system.
Manufacturing and Marketing
To be successful, our proposed products will need to be manufactured in commercial quantities under GMPs prescribed by the FDA and at acceptable costs. We do not have the facilities to manufacture any of our proposed products under GMPs. We intend to rely on collaborators, licensees or contract manufacturers for the commercial manufacture of our proposed products.
Our bremelanotide product candidate is a synthetic peptide. While the production process involves well-established technology, there are few manufacturers capable of scaling up to commercial quantities under GMPs at acceptable costs. We have identified one third-party manufacturer for the production of bremelanotide, and have validated manufacturing of the bremelanotide drug substance under GMPs with that manufacturer. However, we
have not negotiated a long-term supply agreement with the third-party manufacturer, and may not be able to enter into a supply agreement on acceptable terms, if at all.
Our PL-3994 product candidate is a peptide mimetic molecule, incorporating a proprietary amino acid mimetic structure and amino acids. We have identified a manufacturer which made the product in quantities sufficient for Phase 1 and some anticipated Phase 2 clinical trials, and are in the process of evaluating commercial-scale manufacturers. Scaling up to commercial quantities may involve production, purification, formulation and other problems not present in the scale of manufacturing done to date.
Certain of our melanocortin receptor agonist product candidate are synthetic peptides, which we have primarily manufactured in-house. We have not contracted with a third-party manufacturer to produce these synthetic peptides for either clinical trials or commercial purposes. While the production process involves well-established technology, there are few manufacturers capable of scaling up to commercial quantities under GMPs at acceptable costs. Additionally, scaling up to commercial quantities may involve production, purification, formulation and other problems not present in the scale of manufacturing done to date.
The failure of any manufacturer or supplier to comply with FDA GMPs or to supply the drug substance and services as agreed, would force us to seek alternative sources of supply and could interfere with our ability to deliver product on a timely and cost effective basis or at all. Establishing relationships with new manufacturers or suppliers, any of whom must be FDA-approved, is a time-consuming and costly process.
Product Liability and Insurance
Our business may be affected by potential product liability risks which are inherent in the testing, manufacturing, marketing and use of our proposed products. We have liability insurance providing up to $10 million coverage in the aggregate as to certain clinical trial risks.
Employees
As of February 22, 2011, we employed 17 persons full time, of whom 9 are engaged in research and development activities and 8 are engaged in administration and management. While we have been successful in attracting skilled and experienced scientific personnel, competition for personnel in our industry is intense. None of our employees are covered by a collective bargaining agreement. All of our employees have executed confidentiality and intellectual property agreements. We consider relations with our employees to be good.
From time to time, we hire scientific consultants to work on specific research and development programs. We also rely on independent organizations, advisors and consultants to provide services, including aspects of manufacturing, clinical management, regulatory strategy and market research. Our independent advisors and consultants sign agreements that provide for confidentiality of our proprietary information and rights to any intellectual property developed while working for us.
Our corporate offices and research and development facilities are located at 4C Cedar Brook Drive, Cedar Brook Corporate Center, Cranbury, NJ 08512, where we lease approximately 28,000 square feet under a lease which expires July 17, 2012. We also lease 10,000 square feet of additional office space and 12,000 square feet of laboratory space in two other buildings in the same center under leases that expire in 2015 and February 28, 2012, respectively. The 10,000 square feet of additional office space is subleased to a third party under a sublease that expires February 28, 2012. We have ceased using the 12,000 square feet of laboratory space under the lease that expires February 28, 2012, and are seeking to sublease or otherwise terminate our lease as to this property. The leased properties are in good condition.
We are involved, from time to time, in various claims and legal proceedings arising in the ordinary course of our business. We are not currently a party to any claim or legal proceeding.
Identification of Directors
The following table sets forth the names, ages, positions and committee memberships of our directors. All directors hold office until the next annual meeting of stockholders or until their successors have been elected and qualified. All current directors were re-elected at our annual stockholders’ meeting on May 13, 2010.
|
Name (1)
|
Age
|
Position with Palatin
|
|
Carl Spana, Ph.D.
|
48
|
Chief executive officer, president and a director
|
|
John K.A. Prendergast, Ph.D.
|
57
|
Director, chairman of the board of directors
|
|
Perry B. Molinoff, M.D.
|
70
|
Director
|
|
Robert K. deVeer, Jr. (2) (3) (4)
|
64
|
Director
|
|
Zola P. Horovitz, Ph.D. (2) (3) (4)
|
75
|
Director
|
|
Robert I. Taber, Ph.D. (2) (3)
|
74
|
Director
|
|
J. Stanley Hull
|
58
|
Director
|
|
(1) Errol De Souza, Ph.D., resigned as a director and member of the Compensation Committee and Nominating and Corporate Governance Committee effective December 31, 2010. The board has not yet determined whether to fill the vacancy created by Dr. De Souza’s resignation or fix the number of directors at seven.
(2) Member of the Audit Committee.
(3) Member of the Compensation Committee.
(4) Member of the Nominating and Corporate Governance Committee.
|
||
CARL SPANA, Ph.D., co-founder of Palatin, has been our chief executive officer and president since June 14, 2000. He has been a director of Palatin since June 1996 and has been a director of our wholly-owned subsidiary, RhoMed Incorporated, since July 1995. From June 1996 through June 14, 2000, Dr. Spana served as an executive vice president and our chief technical officer. From June 1993 to June 1996, Dr. Spana was vice president of Paramount Capital Investments, LLC, a biotechnology and biopharmaceutical merchant banking firm, and of The Castle Group Ltd., a medical venture capital firm. Through his work at Paramount Capital Investments and The Castle Group, Dr. Spana co-founded and acquired several private biotechnology firms. From July 1991 to June 1993, Dr. Spana was a Research Associate at Bristol-Myers Squibb, a publicly-held pharmaceutical company, where he was involved in scientific research in the field of immunology. Dr. Spana is a director of AVAX Technologies, Inc., a life science company. Dr. Spana received his Ph.D. in molecular biology from The Johns Hopkins University and his B.S. in biochemistry from Rutgers University.
Dr. Spana’s qualifications for our board include his leadership experience, business judgment and industry experience. As a senior executive of Palatin for almost fifteen years, he provides in-depth knowledge of our company, our drug products under development and the competitive and corporate partnering landscape.
JOHN K.A. PRENDERGAST, Ph.D., co-founder of Palatin, has been chairman of the board since June 14, 2000, and a director since August 1996. Dr. Prendergast has been president and sole stockholder of Summercloud Bay, Inc., an independent consulting firm providing services to the biotechnology industry, since 1993. He is a member of the board of AVAX Technologies, Inc. and MediciNova, Inc., life science companies, and was a member of the board of Avigen, Inc. until its acquisition by MediciNova in 2009. Currently, he is the chairman and chief executive officer of AVAX Technologies, Inc. and executive chairman of the board of directors of Antyra, Inc., a privately-held biopharmaceutical firm. From October 1991 through December 1997, Dr. Prendergast was a managing director of The Castle Group Ltd., a medical venture capital firm. Dr. Prendergast received his M.Sc. and Ph.D. from the University of New South Wales, Sydney, Australia and a C.S.S. in administration and management from Harvard University.
Dr. Prendergast is a co-founder of Palatin, and brings a historical perspective to our board coupled with extensive industry experience in corporate development and finance in the life sciences field. His service on other publicly traded company boards provides experience relevant to good corporate governance practices.
PERRY B. MOLINOFF, M.D. has been a director since November 2001. He served as our executive vice president for research and development from September 2001 until November 3, 2003, when he resigned to accept a position as Vice Provost for Research at the University of Pennsylvania, which he held from November 2003 through September 2006. He is also a director of Cypress Bioscience, Inc., a publicly-held life science company. Dr. Molinoff has more than 30 years of experience in both the industrial and educational sectors. From 1981 to 1994, he was a professor of pharmacology and chairman of the Department of Pharmacology at the University of Pennsylvania School of Medicine in Philadelphia. From January 1995 until March 2001, he was vice president of neuroscience and genitourinary drug discovery for the Bristol-Myers Squibb Pharmaceutical Research Institute, where he was responsible for directing and implementing the Institute’s research efforts. Dr. Molinoff earned his medical degree from Harvard Medical School.
Dr. Molinoff has extensive academic and pharmaceutical company experience, with scientific knowledge that makes him a resource to our executive officers and other board members. As a former officer of Palatin, Dr. Molinoff has significant knowledge of our technologies and drug products under development, as well as the markets potentially addressed by our drug products under development.
ROBERT K. deVEER, Jr. has been a director since November 1998. Since January 1997, Mr. deVeer has been the president of deVeer Capital LLC, a private investment company. He is also a director of Solutia Inc., a publicly-held chemical-based materials company. From 1995 until his retirement in 1996, Mr. deVeer served as Managing Director, Head of Industrial Group, at New York-based Lehman Brothers. From 1973 to 1995, he held increasingly responsible positions at New York-based CS First Boston, including Head of Project Finance, Head of Industrials and Head of Natural Resources. He was a managing director, member of the investment banking committee and a trustee of the First Boston Foundation. He received a B.A. in economics from Yale University and an M.B.A. in finance from Stanford Graduate School of Business.
Mr. deVeer has extensive experience in investment banking and corporate finance, including the financing of life sciences companies, and serves as the Audit Committee’s financial expert.
ZOLA P. HOROVITZ, Ph.D. has been a director since February 2001. Before he retired from Bristol-Myers Squibb in 1994, Dr. Horovitz spent 34 years in various positions, including associate director of the Squibb Institute for Medical Research, vice president of development, vice president, scientific liaison, vice president of licensing, and vice president of business development and planning for the pharmaceutical division of Bristol-Myers Squibb. He held advisory positions at the University of Pittsburgh, Rutgers College of Pharmacy and Princeton University. He is also currently a director of BioCryst Pharmaceuticals, Inc. and GenVec, Inc., publicly-held life science companies. Within the past five years, Dr. Horovitz also served on the board of directors of Genaera Corp., Immunicon Corp., NitroMed, Inc., Avigen, Inc. and DOV Pharmaceutical, Inc. Dr. Horovitz earned his Ph.D. in pharmacology from the University of Pittsburgh.
Dr. Horovitz has extensive experience in development of pharmaceutical drugs, business development and licensing, and has served on the board of directors of a number of publicly-held life science companies.
ROBERT I. TABER, Ph.D. has been a director since May 2001. Dr. Taber began his career in the pharmaceutical industry in 1962, holding a succession of positions within Schering Corporation’s biological research group before leaving in 1982 as director of biological research. He has also held a number of increasingly important positions with DuPont Pharmaceuticals and the DuPont Merck Pharmaceutical Company, including director of pharmaceutical research, director of pharmaceutical and biotechnology research, vice president of pharmaceutical research and vice president of extramural research and development. From 1994 to 1998, Dr. Taber held the position of senior vice president of research and development at Synaptic Pharmaceuticals Corporation before founding Message Pharmaceuticals, Inc. in 1998, serving as president and chief executive officer until 2000. Dr. Taber earned his Ph.D. in pharmacology from the Medical College of Virginia.
Dr. Tabor has extensive experience in pharmaceutical research and development both in large pharmaceutical companies and in smaller biotechnology and biopharmaceutical companies.
J. STANLEY HULL has been a director since September 2005. Mr. Hull has over three decades of experience in the field of sales and marketing. Mr. Hull joined GlaxoSmithKline, a research-based pharmaceutical company, in October 1987 and retired as Senior Vice President, Pharmaceuticals in May 2010, having previously served in the R&D organization of GlaxoSmithKline as Vice President and Worldwide Director of Therapeutic Development and Product Strategy – Neurology and Psychiatry. Prior to that, he was Vice President of Marketing – Infectious Diseases and Gastroenterology for Glaxo Wellcome Inc. Mr. Hull started his career in the pharmaceutical industry with SmithKline and French Laboratories in 1978. Mr. Hull received his B.S. in business administration from the University of North Carolina at Greensboro.
Mr. Hull has extensive experience in commercial operations, development and marketing of pharmaceutical drugs and corporate alliances between pharmaceutical companies and biotechnology companies.
Director Independence
The board of directors has determined that all of the directors except for Dr. Spana (our chief executive officer and president) and Dr. Prendergast (our chairman) are independent directors, as defined in the NYSE Amex listing standards.
Executive Officers
Executive officers are appointed by the board and serve at the discretion of the board. Each officer holds his position until his successor is appointed and qualified. The current executive officers hold office under employment agreements.
|
Name (1)
|
Age
|
Position with Palatin
|
|
Carl Spana, Ph.D.
|
48
|
Chief executive officer, president and director
|
|
Stephen T. Wills, MST, CPA
|
54
|
Chief financial officer and executive vice president of operations, secretary and treasurer
|
_______________________________
|
(1)
|
Trevor Hallam, Ph.D., served as executive vice president of research and development from May 2005 through December 31, 2010, the effective date of his resignation.
|
Additional information about Dr. Spana is included above under the heading “Identification of Directors.”
STEPHEN T. WILLS, MST, CPA, has been vice president, secretary, treasurer and chief financial officer since 1997 and has been executive vice president of operations since 2005. From July 1997 to August 2000, Mr. Wills was also a vice president and the chief financial officer of Derma Sciences, Inc., a publicly-held company which provides wound and skin care products, and currently serves as lead director of Derma. Mr. Wills is also a director of U.S. Helicopter Corp., a publicly-held company. From 1991 to August 2000, he was the president and chief operating officer of Golomb, Wills & Company, P.C., a public accounting firm. Mr. Wills, a certified public accountant, received his B.S. in accounting from West Chester University, and an M.S. in taxation from Temple University.
EXECUTIVE COMPENSATION
Summary Compensation Table
The following table summarizes the compensation earned by or paid to our principal executive officer, principal financial officer and our one other executive officer (our named executive officers) for our fiscal years ended June 30, 2010 and 2009. We have no non-equity incentive plan, no defined benefit or actuarial pension plan, and no deferred compensation plan.
|
Name and Principal Position
|
Fiscal
Year
|
Salary
($)
|
Bonus (1)
($)
|
Stock
awards (2)
($)
|
Option
awards (2)
($)
|
All
other
compen-
sation (3)
($)
|
Total
($)
|
|
Carl Spana, Ph.D., chief executive officer and president
|
2010
|
390,000
|
0
|
0
|
62,305
|
12,250
|
464,555
|
|
2009
|
390,000
|
25,000
|
22,500
|
38,455
|
9,750
|
485,705
|
|
|
Stephen T. Wills, MST, CPA, chief financial officer and executive vice president of operations
|
2010
|
321,000
|
0
|
0
|
49,844
|
12,250
|
383,094
|
|
2009
|
321,000
|
25,000
|
22,500
|
30,764
|
11,500
|
410,764
|
|
|
Name and Principal Position
|
Fiscal
Year
|
Salary
($)
|
Bonus (1)
($)
|
Stock
awards (2)
($)
|
Option
awards (2)
($)
|
All
other
compen-
sation (3)
($)
|
Total
($)
|
|
Trevor Hallam, Ph.D., executive vice president of research and development (4)
|
2010
|
321,000
|
0
|
0
|
49,844
|
12,250
|
383,094
|
|
2009
|
321,000
|
25,000
|
22,500
|
30,764
|
11,500
|
410,764
|
|
(1)
|
Performance based bonus amounts for fiscal 2009 were paid on December 31, 2008. There were no bonuses awarded to any of our executive officers for fiscal 2010.
|
|
(2)
|
Amounts in these columns represent the aggregate grant date fair value for stock awards and option awards computed in accordance with FASB ASC Topic 718. For a description of the assumptions we used to calculate these amounts, see Note 9 to the consolidated financial statements included in this prospectus and Note 9 to the consolidated financial statements included in our Annual Report on Form 10-K for the fiscal year ended June 30, 2009.
|
|
(3)
|
Consists of matching contributions to 401(k) plan accounts.
|
|
(4)
|
Dr. Hallam resigned effective December 31, 2011.
|
Employment Agreements
Effective July 1, 2010, we entered into employment agreements with Dr. Spana, Mr. Wills and Dr. Hallam, which continue through June 30, 2013 unless terminated earlier. On November 15, 2010, we entered into a separation agreement with Dr. Hallam, pursuant to which he resigned as an officer and employee effective December 31, 2010.
Under the employment agreements, which replace substantially similar agreements that expired on June 30, 2010, Dr. Spana is serving as chief executive officer and president at a base salary of $390,000 per year and Mr. Wills is serving as chief financial officer and executive vice president of operations at a base salary of $321,000 per year. Each agreement also provides for:
|
|
·
|
annual discretionary bonus compensation, in an amount to be decided by the Compensation Committee and approved by the board, based on achievement of yearly objectives; and
|
|
|
·
|
participation in all benefit programs that we establish, to the extent the executive’s position, tenure, salary, age, health and other qualifications make him eligible to participate.
|
Each agreement allows us or the executive to terminate the agreement upon written notice, and contains other provisions for termination by us for “cause,” or by the employee for “good reason” or due to a “change in control” (as these terms are defined in the employment agreements and set forth below). Early termination may, in some circumstances, result in severance pay at the salary then in effect, plus continuation of medical and dental benefits then in effect for a period of two years (Dr. Spana) or 18 months (Mr. Wills). In addition, the agreements provide that options and restricted stock units granted to these officers accelerate upon termination of employment except for voluntary resignation by the officer or termination for cause. In the event of retirement, termination by the officer for good reason, or termination by us other than for “cause”, options may be exercised until the earlier of twenty-four months following termination or expiration of the option term. Arrangements with our named executive officers in connection with a termination following a change in control are described below. Each agreement includes non-competition, non-solicitation and confidentiality covenants.
Under the separation agreement with Dr. Hallam, his resignation was a termination at the election of the employee, and options and restricted stock units not vested expired on December 31, 2010, and vested options expire three months after that date. The separation agreement with Dr. Hallam provides we will provide him with salary continuation for a period of six months following the date of termination of employment, and Dr. Hallam will provide consulting services to us during that period. Non-competition, non-solicitation and confidentiality covenants of the employment agreement with Dr. Hallam, which employment agreement is substantially identical to that of Mr. Wills, survive Dr. Hallam’s termination of employment.
The Compensation Committee determined not to award any discretionary bonuses to our named executive officers or to authorize any increase in our named executive officers’ salaries for fiscal 2010, based on results of operations during fiscal 2009, including our financial condition and our common stock price.
Stock Option and Restricted Stock Unit Grants
In October 2006, we granted 37,500, 30,000 and 30,000 restricted stock units to Dr. Spana, Mr. Wills and Dr. Hallam, respectively, which vested on March 26, 2010. The terms of these restricted stock units require that each executive retain ownership of at least 33% of the vested stock for the duration of the executive’s employment with us unless there is a change in control or for hardship as determined by the board of directors. In connection with the grant of the restricted stock units to our executive officers in October 2006, we determined at that time that the executive officers would not receive any further stock options or stock awards during the remainder of fiscal 2007 or the next three fiscal years thereafter, subject, however, to annual review by the Compensation Committee, which is authorized to make additional grants if warranted based on market conditions, our common stock price, the need to retain our executive officers and the interests of our stockholders.
In fiscal 2008, the Compensation Committee determined that additional stock option grants were necessary in order to motivate and retain our executive officers, and on March 26, 2008, Dr. Spana, Mr. Wills and Dr. Hallam were granted options to purchase 37,500, 30,000 and 30,000 shares of common stock, respectively, vesting over four years. Twenty-five percent of the shares underlying each option were granted at an exercise price in excess of the fair market value on the date of grant in order to incentivize the executive to improve our financial condition.
In each of fiscal 2009 and 2010, the Compensation Committee determined that additional equity grants were necessary in order to motivate and retain our executive officers. Effective on each of July 1, 2008 and July 1, 2009, Dr. Spana, Mr. Wills and Dr. Hallam were granted options to purchase 25,000, 20,000 and 20,000 shares of common stock, respectively, vesting over four years with an exercise price equal to the closing price of our common stock on the respective date of grant. In addition, on December 10, 2008, we granted restricted stock units as to 25,000 shares of common stock to each of Dr. Spana, Mr. Wills and Dr. Hallam, which vested on December 31, 2009.
On July 21, 2010, we granted 25,000, 20,000 and 20,000 restricted stock units to Dr. Spana, Mr. Wills and Dr. Hallam, respectively, which vested as to 50% on September 15, 2010 and the remaining 50% will vest on March 15, 2011, provided that the executive remains employed by us through such dates, subject to earlier vesting in the event of a change in control or termination of employment other than a voluntary termination or termination for cause. Under the separation agreement with Dr. Hallam, his restricted stock units, which would otherwise have vested on March 15, 2011, were terminated without vesting.
Outstanding Equity Awards at 2010 Fiscal Year-End
The following table summarizes all of the outstanding equity awards granted to our named executive officers as of June 30, 2010, the end of our fiscal year. No stock awards were outstanding as of June 30, 2010.
|
Option awards (1)
|
|||||
|
Name
|
Option
grant
date
|
Number of
securities
underlying
unexercised
options
(#)
exercisable
|
Number of
securities
underlying
unexercised
options
(#)
unexercisable
|
Option
exercise
price
($)
|
Option
expiration
date
|
|
Carl Spana
|
08/01/00
|
14,000
|
0
|
51.25
|
08/01/10
|
|
10/01/01
|
10,000
|
0
|
31.90
|
10/01/11
|
|
|
12/11/02
|
10,000
|
0
|
20.00
|
12/11/12
|
|
|
07/16/03
|
10,000
|
0
|
32.40
|
07/16/13
|
|
|
07/01/05
|
7,500
|
0
|
37.50
|
07/01/15
|
|
|
07/01/05
|
8,300
|
0
|
17.50
|
07/01/15
|
|
|
Option awards (1)
|
|||||
|
Name
|
Option
grant
date
|
Number of
securities
underlying
unexercised
options
(#)
exercisable
|
Number of
securities
underlying
unexercised
options
(#)
unexercisable
|
Option
exercise
price
($)
|
Option
expiration
date
|
|
10/06/06
|
9,375
|
3,125
|
24.90
|
10/06/16
|
|
|
03/26/08
|
14,062
|
14,062
|
2.80
|
03/26/18
|
|
|
03/26/08
|
2,344
|
2,344
|
5.00
|
03/26/18
|
|
|
03/26/08
|
2,344
|
2,344
|
6.60
|
03/26/18
|
|
|
07/01/08
|
6,250
|
18,750
|
1.80
|
07/01/18
|
|
|
07/01/09
|
0
|
25,000
|
2.80
|
07/01/19
|
|
|
Stephen T. Wills
|
08/01/00
|
6,500
|
0
|
51.25
|
08/01/10
|
|
10/01/01
|
7,000
|
0
|
31.90
|
10/01/11
|
|
|
12/11/02
|
8,000
|
0
|
20.00
|
12/11/12
|
|
|
07/16/03
|
8,000
|
0
|
32.40
|
07/16/13
|
|
|
07/01/05
|
5,000
|
0
|
37.50
|
07/01/15
|
|
|
07/01/05
|
7,300
|
0
|
17.50
|
07/01/15
|
|
|
10/06/06
|
7,500
|
2,500
|
24.90
|
10/06/16
|
|
|
03/26/08
|
11,250
|
11,250
|
2.80
|
03/26/18
|
|
|
03/26/08
|
1,875
|
1,875
|
5.00
|
03/26/18
|
|
|
03/26/08
|
1,875
|
1,875
|
6.60
|
03/26/18
|
|
|
07/01/08
|
5,000
|
15,000
|
1.80
|
07/01/18
|
|
|
07/01/09
|
0
|
20,000
|
2.80
|
07/01/19
|
|
|
Trevor Hallam (2)
|
05/09/05
|
35,000
|
0
|
19.90
|
05/09/15
|
|
10/06/06
|
7,500
|
2,500
|
24.90
|
10/06/16
|
|
|
03/26/08
|
11,250
|
11,250
|
2.80
|
03/26/18
|
|
|
03/26/08
|
1,875
|
1,875
|
5.00
|
03/26/18
|
|
|
03/26/08
|
1,875
|
1,875
|
6.60
|
03/26/18
|
|
|
07/01/08
|
5,000
|
15,000
|
1.80
|
07/01/18
|
|
|
07/01/09
|
0
|
20,000
|
2.80
|
07/01/19
|
|
|
(1)
|
Stock option vesting schedules: All options granted before October 6, 2006 have fully vested. Options granted on or after October 6, 2006 vest over four years with 1/4 of the shares vesting per year starting on the first anniversary of the grant date.
|
|
(2)
|
Options granted to Dr. Hallam: Under the separation agreement with Dr. Hallam, he ceased to be an employee as of December 31, 2010. All stock options that had not vested terminated as of that date, and all vested options expire three months following termination, that is on March 31, 2011.
|
Termination and Change-In-Control Arrangements
The employment agreements and restricted stock unit agreements with Dr. Spana and Mr. Wills contain the following provisions concerning severance compensation and the vesting of stock options and restricted stock units upon termination of employment or upon a change in control. The executive’s entitlement to severance, payment of health benefits and accelerated vesting of options is contingent on the executive executing a general release of claims against us.
Termination Without Severance Compensation. Regardless of whether there has been a change in control, if we terminate employment for cause or the executive terminates employment without good reason (as those terms are defined in the employment agreement and set forth below), then the executive receives only his accrued salary and vacation benefits through the date of termination. He may also elect to receive medical and dental benefits pursuant to COBRA for up to two years (Dr. Spana) or 18 months (Mr. Wills), but must remit the cost of coverage to us. Under the terms of our outstanding options and restricted stock units, all unvested options and restricted stock units would terminate immediately, and vested options would be exercisable for three months after termination.
Severance Compensation Without a Change in Control. If we terminate or fail to extend the employment agreement without cause, or the executive terminates employment with good reason, then the executive will receive as severance pay his salary then in effect, paid in a lump sum, plus medical and dental benefits at our expense, for a period of two years (Dr. Spana) or 18 months (Mr. Wills) after the termination date. In addition, upon such event all unvested options would immediately vest and be exercisable for two years after the termination date or, if earlier, the expiration of the option term, and all unvested restricted stock units would accelerate and become fully vested.
Severance Compensation After a Change in Control. If, within one year after a change in control, we terminate employment or the executive terminates employment with good reason, then the executive will receive as severance pay 200% (Dr. Spana) or 150% (Mr. Wills) of his salary then in effect, paid in a lump sum, plus medical and dental benefits at our expense, for a period of two years (Dr. Spana) or 18 months (Mr. Wills) after the termination date. We would also reimburse the executive for up to $25,000 in fees and expenses during the six months following termination, for locating employment. We would also reimburse the executive for any excise tax he might incur on “excess parachute payments” (as defined in Section 280G(b) of the Internal Revenue Code). All unvested options would immediately vest and be exercisable for two years after the termination date or, if earlier, the expiration of the option term. All unvested restricted stock units would vest upon a change in control, without regard to whether the executive’s employment is terminated.
Option Vesting Upon a Change in Control. A change in control by itself does not change compensation or benefits while the employment agreement remains in effect. However, if any options are to be terminated in connection with a change in control, those options will vest in full immediately before the change in control.
Definitions. Under the employment agreements, a “change in control,” “cause” and “good reason” are defined as follows:
A “change in control” occurs when:
|
|
(a)
|
some person or entity acquires more than 50% of the voting power of our outstanding securities;
|
|
|
(b)
|
the individuals who, during any twelve month period, constitute our board of directors cease to constitute at least a majority of the board of directors;
|
|
|
(c)
|
we enter into a merger or consolidation; or
|
(d) we sell substantially all our assets.
|
|
The term “cause” means:
|
|
|
(a)
|
the occurrence of (i) the executive’s material breach of, or habitual neglect or failure to perform the material duties which he is required to perform under, the terms of his employment agreement; (ii) the executive’s material failure to follow the reasonable directives or policies established by or
|
|
|
at the direction of our board of directors; or (iii) the executive’s engaging in conduct that is materially detrimental to our interests such that we sustain a material loss or injury as a result thereof, provided that the breach or failure of performance is not cured, to the extent cure is possible, within ten days of the delivery to the executive of written notice thereof;
|
|
|
(b)
|
the willful breach by the executive of his obligations to us with respect to confidentiality, invention and non-disclosure, non-competition or non-solicitation; or
|
|
|
(c)
|
the conviction of the executive of, or the entry of a pleading of guilty or nolo contendere by the executive to, any crime involving moral turpitude or any felony.
|
The term “good reason” means the occurrence of any of the following, with our failure to cure such circumstances within 30 days of the delivery to us of written notice by the executive of such circumstances:
|
|
(a)
|
any material adverse change in the executive’s duties, authority or responsibilities, which causes the executive’s position with us to become of significantly less responsibility, or assignment of duties and responsibilities inconsistent with the executive’s position;
|
|
|
(b)
|
a material reduction in the executive’s salary;
|
|
|
(c)
|
our failure to continue in effect any material compensation or benefit plan in which the executive participates, unless an equitable arrangement has been made with respect to such plan, or our failure to continue the executive’s participation therein (or in a substitute or alternative plan) on a basis not materially less favorable, both in terms of the amount of benefits provided and the level of the executive’s participation relative to other participants;
|
|
|
(d)
|
our failure to continue to provide the executive with benefits substantially similar to those enjoyed by the executive under any of our health and welfare insurance, retirement and other fringe-benefit plans, the taking of any action by us which would directly or indirectly materially reduce any of such benefits, or our failure to provide the executive with the number of paid vacation days to which he is entitled; or
|
|
|
(e)
|
the relocation of the executive to a location which is a material distance from Cranbury, New Jersey.
|
Director Compensation
The following table sets forth the compensation we paid to all directors during fiscal 2010, except for Dr. Spana, whose compensation is set forth above in the Summary Compensation Table and related disclosure. Dr. Spana did not receive any separate compensation for his services as a director.
|
Name
|
Fees earned or paid in cash ($)
|
Option awards
($) (1) (2)
|
Total ($)
|
|
John K.A. Prendergast, Ph.D.
|
60,000
|
14,953
|
74,953
|
|
Perry B. Molinoff, M.D.
|
30,000
|
9,969
|
39,969
|
|
Robert K. deVeer, Jr.
|
34,000
|
9,969
|
43,969
|
|
Zola P. Horovitz, Ph.D.
|
30,000
|
9,969
|
39,969
|
|
Robert I. Taber, Ph.D.
|
32,000
|
9,969
|
41,969
|
|
Errol De Souza, Ph.D.
|
30,000
|
9,969
|
39,969
|
|
J. Stanley Hull
|
30,000
|
9,969
|
39,969
|
|
(1)
|
Amounts in this column represent the aggregate grant date fair value for option awards granted in fiscal 2010 computed in accordance with FASB ASC Topic 718. For a description of the assumptions we used to calculate these amounts, see Note 9 to the consolidated financial statements included in this prospectus.
|
|
(2)
|
The aggregate number of shares underlying option awards outstanding at June 30, 2010 for each director was:
|
| Dr. Prendergast | 76,100 | ||
| Dr. Molinoff | 56,458 | ||
| Mr. deVeer | 45,500 | ||
| Dr. Horovitz | 39,500 | ||
| Dr. Taber | 39,000 | ||
| Dr. De Souza | 34,875 | ||
| Mr. Hull | 30,666 |
On July 21, 2010, as the annual option grant for the current fiscal year, the chairman of the board received an option to purchase 6,000 shares of common stock and each other non-employee director received an option to purchase 4,000 shares of common stock. All of these options have an exercise price of $1.70 per share, the closing price of our common stock on the date of grant, vest in twelve monthly installments beginning on July 31, 2010, expire ten years from the date of grant and provide for accelerated vesting in the event of involuntarily termination as a director following a change in control, with exercise permitted following accelerated vesting for up to the earlier of one year after termination or the expiration date of the option.
Following the resignation of Dr. De Souza effective December 31, 2010, the Compensation Committee determined to amend the options previously granted to him, such that vested options are exercisable until the earlier of June 30, 2012 or the expiration of the option.
Non-Employee Directors’ Cash Compensation. Dr. Prendergast serves as chairman of the board and receives an annual retainer of $60,000, payable quarterly. Other non-employee directors receive an annual retainer of $30,000, payable on a quarterly basis, with the Audit Committee chairperson and Compensation Committee chairperson receiving an additional $4,000 and $2,000, respectively, payable on a quarterly basis.
Non-Employee Directors’ Expenses. Non-employee directors are reimbursed for expenses incurred in performing their duties as directors, including attending all meetings of the board and any committees on which they serve.
Employee Directors. Employee directors are not separately compensated for services as directors, but are reimbursed for expenses incurred in performing their duties as directors, including attending all meetings of the board and any committees on which they serve.
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT
The tables below show the beneficial stock ownership and voting power, as of February 22, 2011, of:
|
|
·
|
each director, each of the named executive officers, and all current directors and officers as a group; and
|
|
|
·
|
all persons who, to our knowledge, beneficially own more than five percent of the common stock or Series A preferred stock.
|
“Beneficial ownership” here means direct or indirect voting or investment power over outstanding stock and stock which a person has the right to acquire now or within 60 days after February 22, 2011. See the footnotes for more detailed explanations of the holdings. To our knowledge, the persons named in the tables beneficially own and have sole voting and investment power over all shares listed.
The common stock has one vote per share and the Series A preferred stock has approximately 5.38 votes per share. Total voting power is the sum of common stock and Series A preferred stock outstanding as of February 22, 2011, on which date 11,854,028 shares of common stock and 4,997 shares of Series A preferred stock were outstanding.
The address for all members of our management is c/o Palatin Technologies, Inc., 4C Cedar Brook Drive, Cranbury, NJ 08512. Addresses of other beneficial owners are in the table.
MANAGEMENT:
|
Class
|
Name of beneficial owner
|
Amount and nature of beneficial ownership
|
Percent of
class
|
Percent of total voting
power
|
|
Common
|
Carl Spana, Ph.D.
|
159,981 (1)
|
1.3%
|
*
|
|
Common
|
Stephen T. Wills
|
130,824 (2)
|
1.1%
|
*
|
|
Common
|
Trevor Hallam, Ph.D.
|
120,448 (3)
|
1.0%
|
*
|
|
Common
|
John K.A. Prendergast, Ph.D.
|
59,867 (4)
|
*
|
*
|
|
Common
|
Perry B. Molinoff, M.D.
|
52,958 (5)
|
*
|
*
|
|
Common
|
Robert K. deVeer, Jr.
|
34,475 (6)
|
*
|
*
|
|
Common
|
Zola P. Horovitz, Ph.D.
|
33,000 (7)
|
*
|
*
|
|
Common
|
Robert I. Taber, Ph.D.
|
35,000 (8)
|
*
|
*
|
|
Common
|
J. Stanley Hull
|
26,166 (9)
|
*
|
*
|
|
All current directors and executive officers as a group (eight persons)(10)
|
532,271(11)
|
4.3%
|
*
|
*Less than one percent.
|
(1)
|
Includes 105,175 shares which Dr. Spana has the right to acquire under options.
|
|
(2)
|
Includes 82,800 shares which Mr. Wills has the right to acquire under options.
|
|
(3)
|
Includes 75,000 shares which Dr. Hallam has the right to acquire under options. Dr. Hallam resigned effective December 31, 2010.
|
|
(4)
|
Includes 58,100 shares which Dr. Prendergast has the right to acquire under options.
|
|
(5)
|
Includes 51,625 shares which Dr. Molinoff has the right to acquire under options.
|
|
(6)
|
Includes 34,375 shares which Mr. deVeer has the right to acquire under options.
|
|
(7)
|
Includes 32,500 shares which Dr. Horovitz has the right to acquire under options.
|
|
(8)
|
Includes 34,500 shares which Dr. Taber has the right to acquire under options.
|
|
(9)
|
Represents shares which Mr. Hull has the right to acquire under options.
|
|
(10)
|
Excludes Dr. Hallam, who resigned effective December 31, 2010.
|
|
(11)
|
Includes 425,574 shares which directors and officers have the right to acquire under options.
|
MORE THAN 5% BENEFICIAL OWNERS:
|
Class
|
Name and address of beneficial owner
|
Amount and nature of beneficial ownership
|
Percent of
class
|
Percent of total voting
power
|
|
Common
|
Michael David Marcus
PO Box 9330
Richmond Heights, MO 63117
|
780,000
|
6.6%
|
6.6%
|
|
Series A
Preferred
|
Tokenhouse PTE LTD
9 – 11 Reitergasse
Zurich 8027, Switzerland
|
667
|
13.3%
|
*
|
|
Series A
Preferred
|
Steven N. Ostrovsky
43 Nikki Ct.
Morganville, NJ 07751
|
500
|
10.0%
|
*
|
|
Series A
Preferred
|
Thomas L. Cassidy IRA Rollover
38 Canaan Close
New Canaan, CT 06840
|
500
|
10.0%
|
*
|
|
Series A
Preferred
|
Jonathan E. Rothschild
300 Mercer St., #28F
New York, NY 10003
|
500
|
10.0%
|
*
|
|
Series A
Preferred
|
103336 Canada Inc.
168 Forest Hill Rd.
Toronto, Ontario, M5P2M9
|
300
|
6.0%
|
*
|
|
Series A
Preferred
|
Arthur J. Nagle
19 Garden Avenue
Bronxville, NY 10708
|
250
|
5.0%
|
*
|
|
Series A
Preferred
|
Thomas P. and Mary E. Heiser, JTWROS
10 Ridge Road
Hopkinton, MA 01748
|
250
|
5.0%
|
*
|
|
Series A
Preferred
|
Carl F. Schwartz
31 West 87th St.
New York, NY 10016
|
250
|
5.0%
|
*
|
|
Series A
Preferred
|
Michael J. Wrubel
3650 N. 36 Avenue, #39
Hollywood, FL 33021
|
250
|
5.0%
|
*
|
|
Series A
Preferred
|
Myron M. Teitelbaum, M.D.
175 Burton Lane
Lawrence, NY 11559
|
250
|
5.0%
|
*
|
|
Series A
Preferred
|
Laura Gold Galleries Ltd. Profit Sharing Trust Park South Gallery at Carnegie Hall
154 West 57th Street, Suite 114
New York, NY 10019-3321
|
250
|
5.0%
|
*
|
|
Class
|
Name and address of beneficial owner
|
Amount and nature of beneficial ownership
|
Percent of
class
|
Percent of total voting
power
|
|
Series A
Preferred
|
Laura Gold
180 W. 58th Street
New York, NY 10019
|
250
|
5.0%
|
*
|
|
|
*Less than one percent.
|
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS
As a condition of employment, we require all employees to disclose in writing actual or potential conflicts of interest, including related party transactions. Our code of corporate conduct and ethics, which applies to employees, officers and directors, requires that the Audit Committee review and approve related party transactions. Since July 1, 2007, there have been no transactions or proposed transactions in which we were or are to be a participant, in which any related person had or will have a direct or indirect material interest.
DESCRIPTION OF SECURITIES
General
The following description of our capital stock is intended as a summary only and is qualified in its entirety by reference to our amended and restated certificate of incorporation and bylaws, which are filed as exhibits to the registration statement of which this prospectus forms a part.
Our authorized capital stock consists of
|
|
·
|
40,000,000 shares of common stock, par value $0.01 per share, and
|
|
|
·
|
10,000,000 shares of preferred stock, par value $0.01 per share, of which 9,736,000 shares are undesignated.
|
As of December 31, 2010, we had outstanding:
|
|
·
|
11,854,028 shares of our common stock;
|
|
|
·
|
options to purchase 864,166 shares of our common stock under our stock plans, at a weighted average exercise price of $12.18 per share, with options for 655,507 shares vested and exercisable, at a weighted average exercise price of $14.95;
|
|
|
·
|
warrants to purchase 1,551,748 shares of our common stock at a weighted average exercise price $8.46;
|
|
|
·
|
55,500 shares issuable under restricted stock units that vest no later than March 15, 2011, subject to the fulfillment of service conditions; and
|
|
|
·
|
4,997 shares of Series A Convertible Preferred Stock, convertible into 26,865 shares of common stock.
|
Common Stock
Holders of our common stock are entitled to one vote per share for the election of directors and on all other matters that require stockholder approval. Holders of shares of common stock do not have any cumulative voting rights. Subject to any preferential rights of any outstanding preferred stock, in the event of our liquidation, dissolution or winding up, holders of our common stock are entitled to share ratably in the assets remaining after payment of liabilities and the liquidation preferences of any outstanding preferred stock. Our common stock does not carry any redemption rights or any preemptive or preferential rights enabling a holder to subscribe for, or receive shares of, any class of our common stock or any other securities convertible into shares of any class of our common stock.
Preferred Stock
We have the authority to issue 10,000,000 shares of preferred stock. As of December 31, 2010, 264,000 shares of our preferred stock were designated as a single class, Series A Convertible Preferred Stock, of which 4,997 shares were outstanding (see “Series A Convertible Preferred Stock” below). The description of preferred stock provisions set forth below is not complete and is subject to and qualified in its entirety by reference to our amended and restated certificate of incorporation and the certificate of designations relating to the Series A Convertible Preferred Stock.
The board of directors has the right, without the consent of holders of common stock, to designate and issue one or more series of preferred stock, which may be convertible into common stock at a ratio determined by the board. A series of preferred stock may bear rights superior to common stock as to voting, dividends, redemption, distributions in liquidation, dissolution, or winding up, and other relative rights and preferences. The board may set the following terms of any series preferred stock:
|
|
·
|
the number of shares constituting the series and the distinctive designation of the series;
|
|
|
·
|
dividend rates, whether dividends are cumulative, and, if so, from what date and the relative rights of priority of payment of dividends;
|
|
|
·
|
voting rights and the terms of the voting rights;
|
|
|
·
|
conversion privileges and the terms and conditions of conversion, including provision for adjustment of the conversion rate;
|
|
|
·
|
redemption rights and the terms and conditions of redemption, including the date or dates upon or after which shares may be redeemable, and the amount per share payable in case of redemption, which may vary under different conditions and at different redemption dates;
|
|
|
·
|
sinking fund provisions for the redemption or purchase of shares;
|
|
|
·
|
rights in the event of voluntary or involuntary liquidation, dissolution or winding up of the corporation, and the relative rights of priority of payment; and
|
|
|
·
|
any other relative powers, preferences, rights, privileges, qualifications, limitations and restrictions of the series.
|
Dividends on outstanding shares of preferred stock will be paid or declared and set apart for payment before any dividends may be paid or declared and set apart for payment on the common stock with respect to the same dividend period.
If upon any voluntary or involuntary liquidation, dissolution or winding up of the corporation, the assets available for distribution to holders of preferred stock are insufficient to pay the full preferential amount to which the holders are entitled, then the available assets will be distributed ratably among the shares of all series of preferred stock in accordance with the respective preferential amounts (including unpaid cumulative dividends, if any) payable with respect to each series.
Holders of preferred stock will not be entitled to preemptive rights to purchase or subscribe for any shares of any class of capital stock of the corporation. The preferred stock will, when issued, be fully paid and nonassessable. The rights of the holders of preferred stock will be subordinate to those of our general creditors.
Series A Convertible Preferred Stock
The board of directors established a series of 264,000 shares of preferred stock, designated Series A Convertible Preferred Stock, par value $0.01 per share (the “Series A”). We issued 137,780 shares of Series A in 1997, of which 4,997 shares remain outstanding as of February 22, 2011 , the rest having been converted into common stock. The Series A has the following rights and preferences.
Optional conversion. Each share of Series A is convertible at any time, at the option of the holder, into the number of shares of common stock equal to $100 divided by the conversion price, as defined in the Series A certificate of designations. The current conversion price is $18.60, so each share of Series A is currently convertible into approximately 5 shares of common stock.
Mandatory conversion. We may, at our option, cause the conversion of the Series A, in whole or in part, on a pro rata basis, into common stock, if the closing bid price of the common stock has exceeded 200% of the conversion price for at least 20 trading days in any 30 consecutive trading day period, ending three days prior to the date of mandatory conversion.
Price protection provisions. The conversion price decreases if we sell common stock (or equivalents) for a price per share less than the conversion price or less than the market price of the common stock. The conversion price is also subject to adjustment upon the occurrence of a merger, reorganization, consolidation, reclassification, stock dividend or stock split which results in an increase or decrease in the number of shares of common stock outstanding.
Dividend and distribution preference. We may not pay a dividend or make any distribution to holders of any other capital stock unless and until we first pay a special dividend or distribution of $100 per share to the holders of Series A.
Liquidation preference. Upon (i) liquidation, dissolution or winding up of the Company, whether voluntary or involuntary, (ii) sale or other disposition of all or substantially all of the assets of the Company, or (iii) any consolidation, merger, combination, reorganization or other transaction in which Palatin is not the surviving entity or in which the shares of common stock constituting in excess of 50% of the voting power of the Company are exchanged for or changed into other stock or securities, cash and/or any other property, after payment or provision for payment of the debts and other liabilities of the Company, the holders of Series A will be entitled to receive, pro rata and in preference to the holders of any other capital stock, an amount per share equal to $100 plus accrued but unpaid dividends, if any.
Voting rights. Each holder of Series A has the number of votes equal to the number of shares of common stock issuable upon conversion of the holder’s Series A at the record date for determination of the stockholders
entitled to vote or, if no record date is established, at the date a vote is taken. Except as provided above or as required by applicable law, the holders of the Series A are entitled to vote together with the holders of the common stock and not as a separate class.
Warrants
As of December 31, 2010, warrants for the issuance of 1,551,748 shares of our common stock were outstanding, which are exercisable at a weighted average exercise price of $8.46, all of which are exercisable through various dates expiring between February 28, 2011 and August 12, 2014. This description of the warrants is only a summary, and is qualified in its entirety by the provisions of the forms of the warrants, which are attached as exhibits to the registration statement of which this prospectus forms a part.
Series A and Series B Warrants
The following description of the warrants is a summary only, is not intended to be complete and is qualified in its entirety by reference to the warrant agreement and the forms of the Series A Warrant and the Series B Warrant, all of which have been filed as exhibits to the registration statement of which this prospectus is a part. See “Where You Can Find More Information.”
Number of Warrants; Warrant Agent. After giving effect to this offering, 2,000,000 Series A Warrants and 21,000,000 Series B Warrants will be issued and outstanding. The warrants are being issued pursuant to a Warrant Agreement entered into between us and American Stock Transfer & Trust Company, LLC, as warrant agent. The warrants will be issued separately from the common stock included in the units offered hereby and may be transferred separately immediately thereafter. Warrants may be in certificated form or represented by one or more book-entry certificates.
Exercise Price and Duration of Warrants. Series A Warrants will be exercisable immediately upon issuance and at any time up to the fifth anniversary of the date of issuance at an initial exercise price per share equal to 100% of the public offering price per unit set forth on the cover page of this prospectus. Series B Warrants will be exercisable commencing one year and one day from the date of issuance, but only if our stockholders increase the number of authorized shares of common stock, until the fifth anniversary of the initial exercise date at an initial exercise price per share equal to 100% of the public offering price per unit set forth on the cover page of this prospectus. Warrants may be exercised in whole or in part by delivering, not later than 5:00 P.M., New York time, on any business day during the exercise period to the warrant agent the certificate representing the warrant or, in the case of book-entry warrants, the warrants being exercised free on the records of the Depositary Trust Company (DTC) to an account of the warrant agent at DTC along with a completed election to purchase and the payment of the exercise price for each warrant to be exercised by certified or official bank check or by bank wire transfer in immediately available funds.
If we are unable to issue the shares of common stock upon exercise of the warrants because the registration statement covering the shares is subject to a stop order or has had its effectiveness suspended or withdrawn or if we are otherwise unable to issue the shares, and no exemption from registration is available by virtue of a cashless exercise as described below or otherwise, the warrants will not be exercisable. In such event, the warrants will not expire until five days after the date we are first able to issue the shares of common stock. In no event may the warrants be net cash settled.
Cashless Exercise. If a registration statement covering the issuance of the shares underlying the warrants is not available, the warrants may also be exercised on a cashless basis pursuant to which the holder will receive a net number of shares of common stock determined according to the following formula:
Net number of shares = (A x B) - (A x C)
B
where:
A = the total number of shares with respect to which the warrant is then being exercised;
B = the arithmetic average of the closing sale prices of the shares of common stock for the fifteen consecutive trading days ending on the date immediately preceding the date of exercise; and
C = the exercise price then in effect.
Delivery of Shares Upon Exercise. Shares of common stock issuable upon exercise of the warrants will be issued to the holder no later than 5:00 P.M., New York time, on the second business day after the date of proper exercise of the warrants. In lieu of delivering physical certificates representing shares of common stock issuable
upon the exercise of warrants, if our transfer agent is participating in DTC’s Fast Automated Securities Transfer program, we will use our reasonable best efforts to cause the transfer agent to electronically transmit the shares by crediting the account of the registered holder’s prime broker with DTC or of a participant through DTC’s Deposit Withdrawal Agent Commission system.
Stockholder Approval; Payment of Liquidated Damages; Registration of Series B Warrant Shares. We have agreed to hold a stockholders meeting no later than June 30, 2011 in order to seek stockholder approval for an amendment to our certificate of incorporation to increase in the authorized number of shares of our common stock from 40,000,000 to 100,000,000 shares. In the event that we do not increase the authorized number of shares of our common stock on or prior to the initial exercise date of the Series B Warrants, we will be required to pay the holders of the Series B Warrants liquidated damages in the aggregate amount of $2,500,000.
After the increase in the authorized number of shares of common stock, we have agreed to register under the Securities Act the shares of our common stock issuable upon exercise of the Series B Warrants and to list those shares on the NYSE Amex. We will not be required to register the shares of our common stock issuable upon exercise of the Series B Warrants if we deliver an opinion of counsel reasonably satisfactory to the representative of the underwriters that registration is not required because of either the cashless exercise rights described above or because an exemption from registration is available. If we deliver the opinion of counsel, we will publicly announce that no registration statement will be filed and explain how holders may exercise their Series B Warrants.
Certain Adjustments. The exercise price and number of shares of common stock issuable on exercise of the warrants is subject to adjustment in the event of any stock split, reverse stock split, stock dividend, recapitalization, reorganization or similar transaction. However, the warrants will not be adjusted for issuances of shares of common stock at a price below their respective exercise prices.
Fundamental Transactions. In the event of a fundamental transaction involving our consolidation or merger with or into another entity where we are not the surviving entity, the sale or all or substantially all of our properties or assets or the reorganization, recapitalization or reclassification of our common stock, the issuance of a specified amount of new common stock or our liquidation, it is a condition to such fundamental transaction that any successor to us whose common stock is traded on an eligible market assume or remain bound by the warrants to deliver in exchange for the warrants a written instrument substantially similar to the warrants entitling the holder to acquire the successor’s capital stock at an exercise price that reflects the terms of the transaction. In the event that the successor does not have common stock traded on an eligible market, a holder of warrants will be entitled to receive an instrument substantially similar to the warrants exercisable for the consideration that would have been issuable in the fundamental transaction had the warrants been exercised immediately prior thereto.
At least thirty days prior to the consummation of any fundamental transaction, we are obligated to notify the holders that a fundamental transaction will occur. For a period until five days before such fundamental transaction, a warrant holder may require us to redeem all or part of its warrants upon notice to us. We will redeem the warrants covered by the notice for cash at a price equal to the Black-Scholes value of the warrants to be redeemed, in the case of an all cash transaction, a transaction consisting of cash and other consideration (to the extent of the percentage of the cash consideration received, in a going private transaction subject to Rule 13e-3 under the Exchange Act or certain transactions not involving an entity trading on an eligible market, or in the case of any other fundamental transaction, in a number of shares of Common Stock having a value equal to the Black-Scholes value of the warrants to be redeemed divided by 95% of the closing price of our common stock on the trading day immediately preceding the date on which the applicable fundamental transaction is consummated. In the event that a warrant holder gives a notice of redemption, we will be obligated to escrow the payments to be made to the warrant holder prior to effecting a fundamental transaction. Until the payment of the redemption price, the warrants to be redeemed may be exercised at any time.
Limitations on Exercise. Unless the initial holder advises the representative in writing that it does not want to be bound by this limitation at the time it purchases units from the underwriters, or unless otherwise agreed to by the holder, the warrant agent and us on or prior to the initial issuance of the warrants, the number of warrant shares that may be acquired by the registered holder upon any exercise of warrants will be limited to the extent necessary to insure that, following such exercise, the total number of shares of common stock then beneficially owned by such holder and its affiliates and any other persons whose beneficial ownership of common stock would be aggregated with the holder’s for purposes of Section 13(d) of the Exchange Act, does not exceed 4.9% (which may be increased by the holder to up to 9.9% upon not less than 61 days prior notice) of the total number of issued and outstanding shares of common stock (including for such purpose the shares of Common Stock issuable upon such exercise). This restriction may not be waived.
No Rights as Shareholders. Warrant holders do not have the rights or privileges of holders of common stock, including voting rights, until they exercise their warrants and receive shares of common stock. After the issuance of shares of common stock upon exercise of the warrants, each holder will be entitled to one vote for each share held of record on all matters to be voted on by stockholders.
Amendments. The warrant agreement provides that any amendment, modification or waiver of the warrants requires the written consent of the representative of the underwriters and the holders of a majority of the then outstanding warrants.
Fractional Shares. No fractional shares will be issued upon exercise of the warrants. If a holder exercises warrants and would be entitled to receive a fractional interest of a share, we will round up or down the number of common stock to be issued to the warrant holder to the nearest whole number of shares.
Transfers. The warrants may be transferred at the option of the warrant holder upon surrender of the warrants with the appropriate instruments of transfer. We will not pay any stamp or other tax or governmental charge required to be paid in connection with any transfer involved in the issue of shares of common stock issuable upon the exercise of warrants. In the event of any such transfer, we will not issue or deliver any shares until such tax or other charge shall have been paid or it has been established to our satisfaction that no such tax or other charge is due.
Trading. We do not intend to make an application to list the warrants on any exchange or other market and do not expect a market for the warrants to develop.
Underwriter Warrants
In addition, we will issue warrants to the underwriters covering an aggregate of 575,000 common shares on substantially the same terms as the Series B Warrants offered in this offering as part of their compensation in connection with this offering, except that the underwriter warrants will comply with FINRA Rule 5110(g)(1) in that for a period of 180 days after the issuance date of the underwriter warrants (which shall not be earlier than the applicable closing date of this offering), neither the underwriter warrants nor any shares of our common stock issued upon exercise of the underwriter warrants may be sold, transferred, assigned, pledged, or hypothecated, or be the subject of any hedging, short sale, derivative, put, or call transaction that would result in the effective economic disposition of such securities by any person for a period of 180 days immediately following the date of effectiveness or commencement of sales of the offering pursuant to which the underwriter warrants are being issued, except the transfer of any security:
• by operation of law or by reason of reorganization of the Company;
• to any FINRA member firm participating in this offering and the officers or partners thereof, if all securities so transferred remain subject to the lock-up restriction described above for the remainder of the time period;
• if the aggregate amount of securities of the Company held by either an underwriter or a related person do not exceed 1% of the securities being offered;
• that is beneficially owned on a pro-rata basis by all equity owners of an investment fund, provided that no participating member manages or otherwise directs investments by the fund, and participating members in the aggregate do not own more than 10% of the equity in the fund; or
• the exercise or conversion of any security, if all securities received remain subject to the lock-up restriction set forth above for the remainder of the time period.
In addition, pursuant to FINRA Rule 5110(f)(2)(H), the underwriter warrants may not contain certain terms.
Anti-Takeover Effects of Provisions of Delaware Law and Our Charter Documents
Amended and Restated Certificate of Incorporation. Our amended and restated certificate of incorporation authorizes the issuance of up to 10,000,000 shares of preferred stock, par value $.01 per share, of which 264,000 shares are currently designated as Series A Convertible Preferred Stock. The board of directors has the authority, without further approval of the stockholders, to issue and determine the rights and preferences of other series of preferred stock, except as limited by the certificate of designation for the Series A. The board could issue one or more series of preferred stock with voting, conversion, dividend, liquidation, or other rights which would adversely affect the voting power and ownership interest of holders of common stock. This authority may have the effect of
deterring hostile takeovers, delaying or preventing a change in control, and discouraging bids for our common stock at a premium over the market price.
Section 203 of the Delaware General Corporation Law. We are subject to Section 203 of the Delaware General Corporation Law, which, subject to certain exceptions, prohibits a Delaware corporation from engaging in any business combination with any interested stockholder for a period of three years following the time that such stockholder became an interested stockholder, unless:
|
|
·
|
prior to such time, the board of directors approved either the business combination or the transaction that resulted in the stockholder becoming an interested holder;
|
|
|
·
|
upon consummation of the transaction that resulted in the stockholder becoming an interested stockholder, the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction commenced, excluding for purposes of determining the number of shares outstanding those shares owned (a) by persons who are directors and also officers and (b) by employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; or
|
|
|
·
|
at or subsequent to such time, the business combination is approved by the board of directors and authorized at an annual or special meeting of stockholders, and not by written consent, by the affirmative vote of at least two thirds of the outstanding voting stock which is not owned by the interested stockholder.
|
In general, Section 203 defines “business combination” to include the following:
|
|
·
|
any merger or consolidation involving the corporation and the interested stockholder;
|
|
|
·
|
any sale, transfer, pledge or other disposition of 10% or more of the assets of the corporation involving the interested stockholder;
|
|
|
·
|
subject to certain exceptions, any transaction that results in the issuance or transfer by the corporation of any stock of the corporation to the interested stockholder;
|
|
|
·
|
any transaction involving the corporation that has the effect of increasing the proportionate share of the stock or any class or series of the corporation beneficially owned by the interested stockholder; or
|
|
|
·
|
the receipt by the interested stockholder of the benefit of any loans, advances, guarantees, pledges or other financial benefits provided by or through the corporation.
|
In general, Section 203 defines “interested stockholder” as an entity or person beneficially owning 15% or more of the outstanding voting stock of the corporation and any entity or person affiliated with or controlling or controlled by such entity or person.
Indemnification and Limitation of Liability. Our amended and restated certificate of incorporation and bylaws require us to indemnify our directors, officers, employees and agents against the costs (including fines, judgments and attorney fees) from involvement in legal proceedings arising from their position or service, provided that the person seeking indemnification acted:
|
|
·
|
in good faith;
|
|
|
·
|
in a manner he or she reasonably believed to be in, or not opposed to, the best interests of the corporation; and,
|
|
|
·
|
with respect to any criminal action or proceeding, had no reasonable cause to believe his or her conduct was unlawful.
|
The amended and restated certificate of incorporation and bylaws allow us to buy indemnification insurance for this purpose.
Our certificate of incorporation provides that, to the fullest extent permissible under Delaware law, no director shall be personally liable to the corporation or its stockholders for monetary damages for breach of a fiduciary duty as a director. However, this provision does not eliminate the duty of care, and in appropriate circumstances, equitable remedies such as injunctive or other forms of non-monetary relief that will remain available under Delaware law. In addition, each director will continue to be subject to liability for (a) breach of the director’s duty of loyalty to us or our stockholders, (b) acts or omissions not in good faith or which involve
intentional misconduct or a knowing violation of law, (c) violating Section 174 of the Delaware General Corporation Law, or (d) any transaction from which the director derived an improper personal benefit. The provision also does not affect a director’s responsibilities under any other law, such as the federal securities laws or state or federal environmental laws.
Market Information
Our common stock is quoted on the NYSE Amex under the symbol “PTN.” On February 22, 2011, the closing price of the common stock was $1.12.
Transfer Agent and Registrar
The transfer agent for our common stock is American Stock Transfer & Trust Company, located at 59 Maiden Lane, Plaza Level, New York, New York 10038.
We have entered into an underwriting agreement with the underwriters named herein, for whom Roth Capital Partners, LLC is acting as the representative and the sole book-running manager, with respect to the units subject to this offering. Subject to certain conditions, we have agreed to sell to the underwriters, and the underwriters have severally agreed to purchase, the number of units set forth opposite their respective names in the table below.
| Underwriter | Number of Units | |
| Roth Capital Partners, LLC | [ * ] | |
| Madison Williams and Company LLC | [ * ] | |
| Total | [ * ] |
The underwriters are offering the units subject to their acceptance of the units from us and subject to prior sale. The underwriting agreement provides that the obligation of the underwriters to pay for and accept delivery of the units offered by this prospectus is subject to the approval of certain legal matters by their counsel and to certain other conditions. The underwriters are obligated to take and pay for all of the units if any such units are taken.
Commission and Expenses
The underwriters have advised us that they propose to offer the units to the public at the public offering price set forth on the cover page of this prospectus and to certain dealers at that price less a concession not in excess of $[ * ] per unit. The underwriters may allow, and certain dealers may reallow, a discount from the concession not in excess of $[ * ] per unit to certain brokers and dealers. After this offering, the public offering price, concession and reallowance to brokers and dealers may be changed by the underwriters. No such change shall change the amount of proceeds to be received by us as set forth on the cover page of this prospectus. The units are offered by the underwriters as stated herein, subject to receipt and acceptance by them and subject to their right to reject any order in whole or in part. The underwriters have informed us that they do not intend to confirm sales to any accounts over which they exercise discretionary authority.
The following table shows the underwriting discounts and commissions payable to the underwriters by us in connection with this offering.
| Per Unit | Total | ||
| Underwriting discounts and commissions payable by us | $[ * ] | $[ * ] |
We estimate that expenses payable by us in connection with the offering of units, other than the underwriting discounts and commissions referred to above, will be approximately $250,000 . We have agreed to reimburse the underwriters for certain out-of-pocket expenses not to exceed $50,000 in the aggregate.
Reserved Shares
The underwriters have reserved for sale at the public offering price [ * ] units offered by this prospectus for Carl Spana, Ph.D., our President, Chief Executive Officer, and Director, and Stephen T. Wills, our Executive Vice President – Operations and Chief Financial Officer. Messrs. Spana and Wills have entered into lock-up agreements, as described below.
Indemnification
We have agreed to indemnify the underwriters against certain liabilities, including liabilities under the Securities Act of 1933, as amended, or the Securities Act, and liabilities arising from breaches of representations and warranties contained in the underwriting agreement, or to contribute to payments that the underwriters may be required to make in respect of those liabilities.
Lock-up Agreements
We, our officers and our directors have agreed, subject to limited exceptions, for a period of 30 days after the date of the underwriting agreement, not to offer, sell, contract to sell, pledge, grant any option to purchase, make any short sale or otherwise dispose of, directly or indirectly, any shares of common stock or any securities convertible into or exchangeable for our common stock either owned as of the date of the underwriting agreement or thereafter acquired without the prior written consent of the underwriter. This 30-day period may be extended if (1) during the last 17 days of the 30-day period, we issue an earnings release or material news or a material event
regarding us occurs or (2) prior to the expiration of the 30-day period, we announce that we will release earnings results during the 16-day period beginning on the last day of the 30-day period, then the period of such extension will be 18-days, beginning on the issuance of the earnings release or the occurrence of the material news or material event. If after any announcement described in clause (2) of the preceding sentence, we announce that we will not release earnings results during the 16-day period, the lock-up period shall expire the later of the expiration of the 30-day period and the end of any extension of such period made pursuant to clause (1) of the preceding sentence. The representative may, in its sole discretion and at any time or from time to time before the termination of the lock-up period, without notice, release all or any portion of the securities subject to the lock-up.
Electronic Distribution
This prospectus may be made available in electronic format on websites or through other online services maintained by the underwriters, or by their affiliates. Other than this prospectus in electronic format, the information on the underwriters’ website and any information contained in any other website maintained by the underwriters is not part of this prospectus or the registration statement of which this prospectus forms a part, has not been approved and/or endorsed by us or the underwriters in their capacity as underwriters, and should not be relied upon by investors.
Over-Allotment and Price Stabilization
The underwriters have advised us that they will not over-allot or engage in any stabilization transactions in connection with the sale of the units offered hereby.
Other
The underwriters and/or their respective affiliates have provided, and may in the future provide, various investment banking and other financial services for us for which services they have received and, may in the future receive, customary fees. Except for services provided in connection with this offering, no underwriter has provided any investment banking or other financial services to us during the 180-day period preceding the date of this prospectus and we do not expect to retain any of the underwriters to perform any investment banking or other financial services for at least 90 days after the date of this prospectus.
Notice to Investors in the United Kingdom
In relation to each Member State of the European Economic Area which has implemented the Prospectus Directive (each, a “Relevant Member State”) an offer to the public of any shares and warrants which are the subject of the offering contemplated by this prospectus may not be made in that Relevant Member State except that an offer to the public in that Relevant Member State of any shares or warrants may be made at any time under the following exemptions under the Prospectus Directive, if they have been implemented in that Relevant Member State:
(a) to legal entities which are authorized or regulated to operate in the financial markets or, if not so authorized or regulated, whose corporate purpose is solely to invest in securities;
(b) to any legal entity which has two or more of (1) an average of at least 250 employees during the last financial year; (2) a total balance sheet of more than €43,000,000 and (3) an annual net turnover of more than €50,000,000, as shown in its last annual or consolidated accounts;
(c) by the underwriters to fewer than 100 natural or legal persons (other than qualified investors as defined in the Prospectus Directive); or
(d) in any other circumstances falling within Article 3(2) of the Prospectus Directive, provided that no such offer of shares or warrants shall result in a requirement for the publication by the issuer or the underwriters of a prospectus pursuant to Article 3 of the Prospectus Directive.
For the purposes of this provision, the expression an “offer to the public” in relation to any shares or warrants in any Relevant Member State means the communication in any form and by any means of sufficient information on the terms of the offer and any shares and warrants to be offered so as to enable an investor to decide to purchase any shares or warrants, as the same may be varied in that Member State by any measure implementing the Prospectus Directive in that Member State and the expression “Prospectus Directive” means Directive 2003/71/EC and includes any relevant implementing measure in each Relevant Member State.
Each underwriter has represented, warranted and agreed that:
(a) it has only communicated or caused to be communicated and will only communicate or cause to be communicated any invitation or inducement to engage in investment activity (within the meaning of section 21 of
the Financial Services and Markets Act 2000 (the FSMA)) received by it in connection with the issue or sale of any of the shares and warrants in circumstances in which section 21(1) of the FSMA does not apply to the issuer; and
(b) it has complied with and will comply with all applicable provisions of the FSMA with respect to anything done by it in relation to the shares and warrants in, from or otherwise involving the United Kingdom.
European Economic Area
In particular, this document does not constitute an approved prospectus in accordance with European Commission’s Regulation on Prospectuses no. 809/2004 and no such prospectus is to be prepared and approved in connection with this offering. Accordingly, in relation to each Member State of the European Economic Area which has implemented the Prospectus Directive (being the Directive of the European Parliament and of the Council 2003/71/EC and including any relevant implementing measure in each Relevant Member State) (each, a Relevant Member State), with effect from and including the date on which the Prospectus Directive is implemented in that Relevant Member State (the Relevant Implementation Date) an offer of securities to the public may not be made in that Relevant Member State prior to the publication of a prospectus in relation to the shares and the warrants which has been approved by the competent authority in that Relevant Member State or, where appropriate, approved in another Relevant Member State and notified to the competent authority in that Relevant Member State, all in accordance with the Prospectus Directive, except that it may, with effect from and including the Relative Implementation Date, make an offer of securities to the public in that Relevant Member State at any time:
• to legal entities which are authorized or regulated to operate in the financial markets or, if not so authorized or regulated, whose corporate purpose is solely to invest in securities;
• to any legal entity which has two or more of (1) an average of at least 250 employees during the last financial year; (2) a total balance sheet of more than 43,000,000 euros; and (3) an annual net turnover of more than 50,000,000 euros, as shown in the last annual or consolidated accounts; or
• in any other circumstances which do not require the publication by the Issuer of a prospectus pursuant to Article 3 of the Prospectus Directive.
LEGAL MATTERS
The validity of the issuance of the securities offered by this prospectus will be passed upon for us by Thompson Hine LLP, New York, New York. Lowenstein Sandler PC, Roseland, New Jersey, is acting as counsel for the underwriters in this offering.
The consolidated financial statements of Palatin Technologies, Inc. as of June 30, 2009 and 2010, and for each of the years in the three-year period ended June 30, 2010, have been included herein and in this prospectus in reliance upon the report of KPMG LLP, independent registered public accounting firm, appearing elsewhere herein, and upon the authority of said firm as experts in accounting and auditing. The audit report covering the June 30, 2010 consolidated financial statements contains an explanatory paragraph that states that Palatin Technologies, Inc. has incurred recurring losses and negative cash flows from operations and will require substantial additional financing to continue to fund its planned development activities, which conditions raise substantial doubt about the ability of the Company to continue as a going concern. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
WHERE YOU CAN FIND MORE INFORMATION
We have filed a registration statement on Form S-1 under the Securities Act of 1933 with the SEC with respect to the shares of common stock and warrants we are offering by this prospectus. This prospectus does not contain all of the information included in the registration statement. For further information pertaining to us and our common stock, you should refer to the registration statement and the exhibits and schedules filed with the registration statement. Whenever we make reference in this prospectus to any of our contracts, agreements or other documents, the references are not necessarily complete, and you should refer to the exhibits attached to the registration statement for copies of the actual contract, agreement or other document.
We file annual, quarterly and special reports and other information with the SEC (Commission File Number 001-15543). These filings contain important information that does not appear in this prospectus. For further information about us, you may read and copy any reports, statements and other information filed by us at the SEC’s Public Reference Room at 100 F Street, N.E., Washington, D.C. 20549-0102. You may obtain further information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. Our SEC filings are also available on the SEC Internet site at http://www.sec.gov, which contains periodic reports and other information regarding issuers that file electronically. You can find information about Palatin on our website at http://www.palatin.com. Information found on our website is not part of this prospectus. You may also request a copy of any of our periodic reports filed with the SEC by writing or telephoning us at the following address:
Stephen T. Wills
Chief Financial Officer
Palatin Technologies, Inc.
4C Cedar Brook Drive
Cranbury, New Jersey 08512
Telephone (609) 495-2200
Table of Contents
Consolidated Financial Statements
|
Page
|
||
|
Audited Consolidated Financial Statements
|
||
|
F-2
|
||
|
F-3
|
||
|
F-4
|
||
|
F-5
|
||
|
F-6
|
||
|
F-7
|
||
|
Interim Consolidated Financial Statements (Unaudited)
|
||
|
F-19
|
||
|
F-20
|
||
|
F-21
|
||
|
F-22
|
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Palatin Technologies, Inc.:
We have audited the accompanying consolidated balance sheets of Palatin Technologies, Inc. and subsidiary (the Company) as of June 30, 2010 and 2009, and the related consolidated statements of operations, cash flows, and stockholders’ equity and comprehensive loss for each of the years in the three-year period ended June 30, 2010. These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Palatin Technologies, Inc. and subsidiary as of June 30, 2010 and 2009, and the results of their operations and their cash flows for each of the years in the three-year period ended June 30, 2010, in conformity with U.S. generally accepted accounting principles.
The accompanying consolidated financial statements have been prepared assuming the Company will continue as a going concern. As discussed in note 1 to the consolidated financial statements, the Company has incurred recurring net losses and negative cash flows from operations and will require substantial additional financing to continue to fund its planned development activities. These conditions raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ KPMG LLP
Philadelphia, Pennsylvania
September 27, 2010
PALATIN TECHNOLOGIES, INC.
and Subsidiary
Consolidated Balance Sheets
|
June 30, 2010
|
June 30, 2009
|
|||
|
ASSETS
|
||||
|
Current assets:
|
||||
|
Cash and cash equivalents
|
$ 5,405,430
|
$ 4,378,662
|
||
|
Available-for-sale investments
|
3,462,189
|
3,439,650
|
||
|
Accounts receivable
|
2,879
|
508,528
|
||
|
Prepaid expenses and other current assets
|
393,313
|
492,824
|
||
|
Total current assets
|
9,263,811
|
8,819,664
|
||
|
Property and equipment, net
|
2,388,365
|
3,650,783
|
||
|
Restricted cash
|
475,000
|
475,000
|
||
|
Other assets
|
261,701
|
254,364
|
||
|
Total assets
|
$ 12,388,877
|
$ 13,199,811
|
||
|
LIABILITIES AND STOCKHOLDERS’ EQUITY
|
||||
|
Current liabilities:
|
||||
|
Capital lease obligations
|
$ 19,670
|
$ 87,675
|
||
|
Accounts payable
|
155,795
|
206,363
|
||
|
Accrued expenses
|
2,219,466
|
1,420,741
|
||
|
Deferred revenue
|
-
|
6,955,553
|
||
|
Total current liabilities
|
2,394,931
|
8,670,332
|
||
|
Capital lease obligations
|
14,284
|
33,954
|
||
|
Deferred rent
|
661,389
|
1,182,026
|
||
|
Total liabilities
|
3,070,604
|
9,886,312
|
||
|
Commitments and contingencies (Note 8)
|
||||
|
Stockholders’ equity:
|
||||
|
Preferred stock of $0.01 par value – authorized 10,000,000 shares;
|
||||
|
Series A Convertible; issued and outstanding 4,997 shares as of June 30, 2010 and 2009, respectively
|
50
|
50
|
||
|
Common stock of $0.01 par value – authorized 40,000,000 shares; issued and outstanding 11,702,818 and 8,666,290 shares as of June 30, 2010 and 2009, respectively
|
117,028
|
86,663
|
||
|
Additional paid-in capital
|
218,236,723
|
210,492,345
|
||
|
Accumulated other comprehensive income
|
138,650
|
116,111
|
||
|
Accumulated deficit
|
(209,174,178)
|
(207,381,670)
|
||
|
Total stockholders’ equity
|
9,318,273
|
3,313,499
|
||
|
Total liabilities and stockholders’ equity
|
$ 12,388,877
|
$ 13,199,811
|
The accompanying notes are an integral part of these consolidated financial statements.
PALATIN TECHNOLOGIES, INC.
and Subsidiary
Consolidated Statements of Operations
|
Year Ended June 30,
|
|||||
|
2010
|
2009
|
2008
|
|||
|
REVENUES
|
$ 14,180,727
|
$ 11,351,774
|
$ 11,483,287
|
||
|
OPERATING EXPENSES:
|
|||||
|
Research and development
|
12,293,910
|
13,356,751
|
21,187,762
|
||
|
General and administrative
|
4,901,203
|
5,296,859
|
6,928,295
|
||
|
Total operating expenses
|
17,195,113
|
18,653,610
|
28,116,057
|
||
|
Loss from operations
|
(3,014,386)
|
(7,301,836)
|
(16,632,770)
|
||
|
OTHER INCOME (EXPENSE):
|
|||||
|
Investment income
|
141,635
|
233,319
|
1,030,452
|
||
|
Interest expense
|
(13,165)
|
(26,159)
|
(73,495)
|
||
|
Gain on sale of supplies and equipment
|
95,000
|
550,968
|
-
|
||
|
Total other income, net
|
223,470
|
758,128
|
956,957
|
||
|
Loss before income taxes
|
(2,790,916)
|
(6,543,708)
|
(15,675,813)
|
||
|
Income tax benefit
|
998,408
|
1,741,476
|
1,291,444
|
||
|
NET LOSS
|
$ (1,792,508)
|
$ (4,802,232)
|
$(14,384,369)
|
||
|
Basic and diluted net loss per common share
|
$ (0.18)
|
$ (0.56)
|
$ (1.69)
|
||
|
Weighted average number of common shares outstanding used in computing basic and diluted net loss per common share
|
9,861,215
|
8,637,030
|
8,522,057
|
||
The accompanying notes are an integral part of these consolidated financial statements.
PALATIN TECHNOLOGIES, INC.
and Subsidiary
Consolidated Statements of Stockholders’ Equity and Comprehensive Loss
|
Accumulated
|
|||||||||
|
Additional
|
Other
|
||||||||
|
Preferred Stock
|
Common Stock
|
Paid-in
|
Comprehensive
|
Accumulated
|
|||||
|
Shares
|
Amount
|
Shares
|
Amount
|
Capital
|
Income
|
Deficit
|
Total
|
||
|
Balance, July 1, 2007
|
4,997
|
$ 50
|
8,512,692
|
$ 85,127
|
$ 206,641,580
|
$ -
|
$ (188,195,069)
|
$ 18,531,688
|
|
|
Exercise of options and warrants
|
-
|
-
|
7,725
|
77
|
110,152
|
-
|
-
|
110,229
|
|
|
Stock-based compensation
|
-
|
-
|
31,991
|
320
|
2,265,179
|
-
|
-
|
2,265,499
|
|
|
Comprehensive loss:
|
|||||||||
|
Unrealized gain on investments
|
-
|
-
|
-
|
-
|
-
|
29,117
|
-
|
29,117
|
|
|
Net loss
|
-
|
-
|
-
|
-
|
-
|
-
|
(14,384,369)
|
(14,384,369)
|
|
|
Total comprehensive loss
|
(14,355,252)
|
||||||||
|
Balance, June 30, 2008
|
4,997
|
50
|
8,552,408
|
85,524
|
209,016,911
|
29,117
|
(202,579,438)
|
6,552,164
|
|
|
Stock-based compensation
|
-
|
-
|
113,882
|
1,139
|
1,475,434
|
-
|
-
|
1,476,573
|
|
|
Comprehensive loss:
|
|||||||||
|
Unrealized gain on investments
|
-
|
-
|
-
|
-
|
-
|
86,994
|
-
|
86,994
|
|
|
Net loss
|
-
|
-
|
-
|
-
|
-
|
-
|
(4,802,232)
|
(4,802,232)
|
|
|
Total comprehensive loss
|
(4,715,238)
|
||||||||
|
Balance, June 30, 2009
|
4,997
|
50
|
8,666,290
|
86,663
|
210,492,345
|
116,111
|
(207,381,670)
|
3,313,499
|
|
|
Sale of common stock units, net of costs
|
-
|
-
|
2,911,448
|
29,114
|
6,931,491
|
-
|
-
|
6,960,605
|
|
|
Exercise of options
|
-
|
-
|
6,725
|
67
|
11,371
|
-
|
-
|
11,438
|
|
|
Stock-based compensation
|
-
|
-
|
172,500
|
1,725
|
966,836
|
-
|
-
|
968,561
|
|
|
Payment of withholding taxes related
|
|
|
|||||||
|
to restricted stock units
|
- | - | (54,145) | (541) | (165,320) | - | - | (165,861) | |
|
Comprehensive loss:
|
|||||||||
|
Unrealized gain on investments
|
-
|
-
|
-
|
-
|
-
|
22,539
|
-
|
22,539
|
|
|
Net loss
|
-
|
-
|
-
|
-
|
-
|
-
|
(1,792,508)
|
(1,792,508)
|
|
|
Total comprehensive loss
|
(1,769,969)
|
||||||||
|
Balance, June 30, 2010
|
4,997
|
$ 50
|
11,702,818
|
$ 117,028
|
$ 218,236,723
|
$ 138,650
|
$ (209,174,178)
|
$ 9,318,273
|
|
The accompanying notes are an integral part of these consolidated financial statements.
PALATIN TECHNOLOGIES, INC.
and Subsidiary
Consolidated Statements of Cash Flows
|
Year Ended June 30,
|
|||||
|
2010
|
2009
|
2008
|
|||
|
CASH FLOWS FROM OPERATING ACTIVITIES:
|
|||||
|
Net loss
|
$ (1,792,508)
|
$ (4,802,232)
|
$ (14,384,369)
|
||
|
Adjustments to reconcile net loss to net cash
|
|||||
|
used in operating activities:
|
|||||
|
Depreciation and amortization
|
1,269,413
|
1,364,644
|
1,393,077
|
||
|
Gain on sale of supplies and equipment
|
(95,000)
|
(550,968)
|
-
|
||
|
Stock-based compensation
|
968,561
|
1,476,573
|
2,265,499
|
||
|
Amortization of deferred revenue
|
(11,905,553)
|
(683,336)
|
(9,669,031)
|
||
|
Changes in operating assets and liabilities:
|
|||||
|
Accounts receivable
|
505,649
|
(502,781)
|
602,094
|
||
|
Prepaid expenses and other assets
|
92,174
|
(5,513)
|
1,115,350
|
||
|
Accounts payable
|
(50,568)
|
(428,820)
|
(485,711)
|
||
|
Accrued expenses and other liabilities
|
278,088
|
(1,311,164)
|
(1,414,834)
|
||
|
Deferred revenues
|
5,000,000
|
-
|
-
|
||
|
Net cash used in operating activities
|
(5,729,744)
|
(5,443,597)
|
(20,577,925)
|
||
|
CASH FLOWS FROM INVESTING ACTIVITIES:
|
|||||
|
Purchase of available-for-sale investments
|
-
|
-
|
(1,000,012)
|
||
|
Sale of supplies and equipment
|
45,000
|
700,000
|
-
|
||
|
Purchases of property and equipment
|
(6,995)
|
(36,383)
|
(263,938)
|
||
|
Net cash provided by (used in) investing activities
|
38,005
|
663,617
|
(1,263,950)
|
||
|
CASH FLOWS FROM FINANCING ACTIVITIES:
|
|||||
|
Payments on capital lease obligations
|
(87,675)
|
(263,128)
|
(294,199)
|
||
|
Payment of withholding taxes related to restricted
|
|||||
|
stock units
|
(165,861)
|
-
|
-
|
||
|
Proceeds from common stock, stock option
|
|||||
|
and warrant issuances
|
6,972,043
|
-
|
110,229
|
||
|
Net cash provided by (used in) financing
activities
|
6,718,507
|
(263,128)
|
(183,970)
|
||
|
NET INCREASE (DECREASE) IN CASH
|
|||||
|
AND CASH EQUIVALENTS
|
1,026,768
|
(5,043,108)
|
(22,025,845)
|
||
|
CASH AND CASH EQUIVALENTS, beginning
|
|||||
|
of year
|
4,378,662
|
9,421,770
|
31,447,615
|
||
|
CASH AND CASH EQUIVALENTS, end of year
|
$ 5,405,430
|
$ 4,378,662
|
$ 9,421,770
|
||
|
SUPPLEMENTAL CASH FLOW INFORMATION:
|
|||||
|
Cash paid for interest
|
$ 13,165
|
$ 36,959
|
$ 58,495
|
||
|
Unrealized gain on available-for-sale
investments
|
22,539
|
86,994
|
29,117
|
||
|
Equipment acquired under financing arrangements
|
-
|
-
|
186,989
|
||
The accompanying notes are an integral part of these consolidated financial statements.
PALATIN TECHNOLOGIES, INC.
and Subsidiary
Notes to Consolidated Financial Statements
(1) ORGANIZATION:
Nature of Business – Palatin Technologies, Inc. (Palatin or the Company) is a biopharmaceutical company dedicated to the development of peptide, peptide mimetic and small molecule agonist compounds with a focus on melanocortin and natriuretic peptide receptor systems. Palatin has a diverse pipeline of active development programs targeting melanocortin and natriuretic receptors. The melanocortin system is involved in a large and diverse number of physiologic functions, and therapeutic agents modulating this system may have the potential to treat a variety of conditions and diseases, including sexual dysfunction, obesity and related disorders, cachexia (wasting syndrome) and inflammation-related diseases. The natriuretic peptide receptor system has numerous cardiovascular functions, and therapeutic agents modulating this system may be useful in treatment of acute asthma, heart failure, hypertension and other cardiovascular diseases.
The Company’s active drug development programs consist of bremelanotide for treatment of sexual dysfunction, other peptide melanocortin receptor agonists for treatment of sexual dysfunction, and PL-3994, an agonist peptide mimetic which binds to natriuretic peptide receptor A, for treatment of acute asthma and heart failure. The Company has an exclusive global research collaboration and license agreement with AstraZeneca AB (AstraZeneca) to commercialize compounds that target melanocortin receptors for the treatment of obesity, diabetes and related metabolic syndrome.
Key elements of the Company’s business strategy include using its technology and expertise to develop and commercialize therapeutic products; entering into alliances and partnerships with pharmaceutical companies to facilitate the development, manufacture, marketing, sale and distribution of product candidates the Company is developing; and, partially funding its product candidate development programs with the cash flow from the Company’s AstraZeneca collaboration agreement and any future agreements with other companies.
Reverse Stock Split – Effective on September 27, 2010, the Company implemented a one-for-ten reverse stock split of its common stock, which reduced the number of shares of its common stock issued and outstanding from approximately 118,200,000 to approximately 11,800,000. All share and per share amounts in these consolidated financial statements, including shares of common stock issuable upon exercise, vesting or conversion of all outstanding options, warrants and convertible preferred stock, are presented on a post-reverse-split basis.
Business Risk and Liquidity – The Company has incurred negative cash flows from operations since its inception, and has expended, and expects to continue to expend in the future, substantial funds to complete its planned product development efforts. As shown in the accompanying consolidated financial statements, the Company has an accumulated deficit as of June 30, 2010 and incurred a net loss for fiscal 2010. The Company anticipates incurring additional losses in the future as a result of spending on its development programs. To achieve profitability, the Company, alone or with others, must successfully develop and commercialize its technologies and proposed products, conduct successful preclinical studies and clinical trials, obtain required regulatory approvals and successfully manufacture and market such technologies and proposed products. The time required to reach profitability is highly uncertain, and there can be no assurance that the Company will be able to achieve profitability on a sustained basis, if at all.
As of June 30, 2010, the Company’s cash and cash equivalents were $5.4 million and its available-for-sale investments were $3.5 million. The Company has made the strategic decision to focus resources and efforts on clinical trials for bremelanotide and PL-3994 and preclinical development of an inhaled formulation of PL-3994 and a new peptide drug candidate for sexual dysfunction, and has ceased research and development efforts on new product candidates. As part of this decision, the Company plans to reduce staffing levels, and anticipates having no more than twenty employees by December 31, 2010, and intends to seek additional capital. Management does not believe that the Company’s existing capital resources, together with expected revenues, will be adequate to fund its currently planned operations for the next twelve months. The accompanying consolidated financial statements have been prepared assuming that the Company continues as a going concern, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. The consolidated financial statements do not include any adjustments relating to the recoverability and classification of recorded asset amounts or the amounts and classification of liabilities that might results from the outcome of this uncertainty.
The Company intends to seek additional capital through public or private equity or debt financings, collaborative arrangements on our product candidates, or other sources. However, sufficient additional funding to support projected operations, including clinical trials with either bremelanotide or PL-3994, or both, may not be available on acceptable terms or at all. These matters raise substantial doubt over the Company’s ability to continue as a going concern.
If the Company is unable to raise sufficient additional funds to advance at least one of its product candidates, management will implement plans for the orderly wind down of its business operations, including curtailing operations significantly and further decreasing staffing levels, and will seek to license, sell or otherwise dispose of the Company’s product candidates, technologies and contractual rights, including rights under the research collaboration and license agreement with AstraZeneca, on the best possible terms available.
The nature and timing of the Company’s development activities are highly dependent on its financing activities. There can be no assurance that the Company will be able to obtain financing when required, or that financing efforts will be successful. Additionally, the Company may be required to seek collaborators for its product candidates at an earlier stage than otherwise would be desirable and on terms that are less favorable than might otherwise be available, and relinquish, license or otherwise dispose of rights on unfavorable terms to technologies and product candidates that the Company would otherwise seek to develop or commercialize itself.
Concentrations – Concentrations in the Company’s assets and operations subject it to certain related risks. Financial instruments that subject the Company to concentrations of credit risk primarily consist of cash and cash equivalents, available-for-sale investments and accounts receivable. The Company’s cash and cash equivalents are primarily invested in one money market fund sponsored by a large financial institution. Revenues from collaboration partners as a percentage of total revenues were as follows:
|
Year Ended June 30,
|
|||||
|
2010
|
2009
|
2008
|
|||
|
AstraZeneca
|
100%
|
100%
|
26%
|
||
|
King Pharmaceuticals, Inc.
|
-
|
-
|
71%
|
||
|
Mallinckrodt
|
-
|
-
|
3%
|
||
(2) SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES:
Principles of Consolidation – The consolidated financial statements include the accounts of Palatin and its wholly-owned inactive subsidiary. All intercompany accounts and transactions have been eliminated in consolidation.
Use of Estimates – The preparation of consolidated financial statements in conformity with U.S. generally accepted accounting principles requires management to make estimates and assumptions that affect the reported amount of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Cash and Cash Equivalents – Cash and cash equivalents include cash on hand, cash in banks and all highly liquid investments with a purchased maturity of less than three months. Cash equivalents consist of $4,111,051 and $3,250,191 in a money market fund at June 30, 2010 and 2009, respectively. Restricted cash secures letters of credit for security deposits on leases.
Investments – The Company classifies its investments as available-for-sale investments and all such investments are recorded at fair value based on quoted market prices. Unrealized holding gains and losses are generally excluded from earnings and are reported in accumulated other comprehensive income/loss until realized. Interest and dividends on securities classified as available-for-sale are included in investment income. Gains and losses are recorded in the statement of operations when realized or when unrealized holding losses are determined to be other than temporary, on a specific-identification basis.
Fair Value of Financial Instruments – The Company’s financial instruments consist primarily of cash equivalents, available-for-sale investments, accounts receivable, accounts payable and capital lease obligations. Management believes that the carrying values of these assets and liabilities are representative of their respective fair values based on quoted market prices for investments and the short-term nature of the other instruments.
Property and Equipment – Property and equipment consists of office and laboratory equipment, office furniture and leasehold improvements and includes assets acquired under capital leases. Property and equipment are recorded at cost. Depreciation is recognized using the straight-line method over the estimated useful lives of the
related assets, generally five years for laboratory and computer equipment, seven years for office furniture and equipment and the lesser of the term of the lease or the useful life for leasehold improvements. Amortization of assets acquired under capital leases is included in depreciation expense. Maintenance and repairs are expensed as incurred while expenditures that extend the useful life of an asset are capitalized.
Impairment of Long-Lived Assets – The Company reviews its long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of the assets may not be fully recoverable. To determine recoverability of a long-lived asset, management evaluates whether the estimated future undiscounted net cash flows from the asset are less than its carrying amount. If impairment is indicated, the long-lived asset would be written down to its fair value. Fair value is determined by an evaluation of available price information at which assets could be bought or sold, including quoted market prices, if available, or the present value of the estimated future cash flows based on reasonable and supportable assumptions.
Deferred Rent – The Company’s operating leases provide for rent increases over the terms of the leases. Deferred rent consists of the difference between periodic rent payments and the amount recognized as rent expense on a straight-line basis, as well as tenant allowances for leasehold improvements. Rent expenses are being recognized ratably over the terms of the leases.
Revenue Recognition – Revenue from corporate collaborations and licensing agreements consists of up-front fees, research and development funding, and milestone payments. Non-refundable up-front fees are deferred and amortized to revenue over the related performance period. The Company estimates the performance period as the period in which it performs certain development activities under the applicable agreement. Reimbursements for research and development activities are recorded in the period that the Company performs the related activities under the terms of the applicable agreements. Revenue resulting from the achievement of milestone events stipulated in the applicable agreements is recognized when the milestone is achieved, provided that such milestone is substantive in nature.
Research and Development Costs – The costs of research and development activities are charged to expense as incurred, including the cost of equipment for which there is no alternative future use.
Stock-Based Compensation – The Company charges to expense the fair value of stock options and other equity awards granted. The Company determines the fair value of stock options utilizing the Black-Scholes option pricing model. Compensation costs for share-based awards with pro-rata vesting are allocated to periods on a straight-line basis
Income Taxes – The Company and its subsidiary file consolidated federal and separate-company state income tax returns. Income taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of assets and liabilities and their respective tax basis and operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences or operating loss and tax credit carryforwards are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in the period that includes the enactment date. The Company has recorded a valuation allowance against its deferred tax assets based on the history of losses incurred.
During the years ended June 30, 2010, 2009 and 2008, the Company sold New Jersey state net operating loss carryforwards, which resulted in the recognition of $998,408, $1,741,476 and $1,291,444, respectively, in tax benefits.
Net Loss per Common Share – Basic and diluted earnings per common share (EPS) are calculated in accordance with the provisions of Financial Accounting Standards Board (FASB) Accounting Standards Codification (ASC) Topic 260, “Earnings per Share.” In June 2008, the FASB issued guidance stating that non-vested share-based payment awards that include non-forfeitable rights to dividends or dividend equivalents, whether paid or unpaid, are considered participating securities, and the two-class method of computing EPS is required for all periods presented. The Company adopted the provisions of ASC Topic 260 relating to the two-class method of computing EPS effective July 1, 2009.
The Company’s outstanding shares of Series A Convertible Preferred stock contain rights that entitle the holder to a special dividend or distribution of $100 per share before the Company can pay dividends or make distributions to the common stockholders. The outstanding share-based compensation awards do not include non-forfeitable rights to dividends. Accordingly, only the outstanding Series A Convertible Preferred stock is considered a participating security and must be included in the computation of EPS. The adoption of the provisions of ASC
Topic 260 relating to the two-class method of computing EPS did not impact the basic and diluted EPS for the years ended June 30, 2010, 2009 or 2008, respectively, as the Company incurred a net loss in each period.
As of June 30, 2010, 2009 and 2008, common shares issuable upon conversion of Series A Convertible Preferred Stock, the exercise of outstanding options and warrants and the vesting of restricted stock units amounted to an aggregate of 2,569,695, 1,407,660 and 1,394,159, respectively.
Recently Issued Accounting Pronouncements – In June 2009, the FASB issued ASC 105-10 (formerly Statement of Financial Accounting Standards 168), “The FASB Accounting Standards Codification and the Hierarchy of Generally Accepted Accounting Principles,” which was effective for the Company beginning July 1, 2009. The FASB Accounting Standards Codification (the Codification) officially became the single source of authoritative nongovernmental generally accepted accounting principles (GAAP), superseding existing FASB, American Institute of Certified Public Accountants, Emerging Issues Task Force and related accounting literature. After that date, only one level of authoritative GAAP exists. All other accounting literature is considered non-authoritative. The Codification reorganizes the thousands of GAAP pronouncements into roughly 90 accounting topics and displays them using a consistent structure. Also included in the Codification is relevant SEC guidance organized using the same topical structure in separate sections within the Codification. The Company adopted this statement and has updated its existing GAAP references to the new codification.
In September 2009, the FASB issued Accounting Standards Update (ASU) 2009-13, Revenue Recognition (Topic 605), “Multiple-Deliverable Revenue Arrangements (ASU 2009-13)”, which requires companies to allocate revenue in arrangements involving multiple deliverables based on the estimated selling price of each deliverable when such deliverables are not sold separately either by the company or other vendors. ASU 2009-13 eliminates the requirement that all undelivered elements must have objective and reliable evidence of fair value before a company can recognize the portion of the overall arrangement fee that is attributable to items that already have been delivered. As a result, the new guidance may allow some companies to recognize revenue on transactions that involve multiple deliverables earlier than under current requirements. ASU 2009-13 is effective for revenue arrangements entered into or materially modified in fiscal years beginning on or after June 15, 2010. Early adoption is permitted at the beginning of a company’s fiscal year. The Company adopted ASU 2009-13 on July 1, 2010 and does not expect ASU 2009-13 to have a material impact on its consolidated financial statements.
In January 2010, the FASB issued ASU 2010-06, Fair Value Measurements and Disclosures (Topic 820), “Improving Disclosures about Fair Value Measurements (ASU 2010-06)”, which amends the existing fair value measurement and disclosure guidance currently included in ASC Topic 820, “Fair Value Measurements and Disclosures”, to require additional disclosures regarding fair value measurements. Specifically, ASU 2010-06 requires companies to disclose the amounts of significant transfers between Level 1 and Level 2 of the fair value hierarchy and the reasons for these transfers, the reasons for any transfer in or out of Level 3 and information in the reconciliation of recurring Level 3 measurements about purchases, sales, issuances and settlements on a gross basis. In addition, ASU 2010-06 also clarifies the requirements for companies to disclose information about both the valuation techniques and inputs used in estimating Level 2 and Level 3 fair value measurements. ASU 2010-06 is effective for interim and annual reporting periods beginning after December 15, 2009, except for additional disclosures related to Level 3 fair value measurements, which are effective for fiscal years beginning after December 15, 2010. The adoption of ASU 2010-06 did not impact the Company’s consolidated financial statements or results of operations.
In April 2010, the FASB issued ASU No. 2010-17, Revenue Recognition – Milestone Method (ASU 2010-17). ASU 2010-17 provides guidance on applying the milestone method to milestone payments for achieving specified performance measures when those payments are related to uncertain future events. Under ASU 2010-17, entities can make an accounting policy election to recognize arrangement consideration received for achieving specified performance measures during the period in which the milestones are achieved, provided certain criteria are met. This ASU is effective for fiscal years beginning January 1, 2011, with early adoption permitted. The Company does not believe adoption will have a material impact on its consolidated financial position and results of operations.
(3) AGREEMENT WITH ASTRAZENECA
In January 2007, the Company entered into an exclusive global research collaboration and license agreement with AstraZeneca to discover, develop and commercialize compounds that target melanocortin receptors for the treatment of obesity, diabetes and related metabolic syndrome. In June 2008, the collaboration agreement was amended to include additional compounds and associated intellectual property developed by the Company. In December 2008, the collaboration agreement was further amended to include additional compounds and associated intellectual property developed by the Company and extended the research collaboration for an additional year through January 2010. In September 2009, the collaboration agreement was further amended to modify royalty rates
and milestone payments. The collaboration is based on the Company’s melanocortin receptor obesity program and includes access to compound libraries, core technologies and expertise in melanocortin receptor drug discovery and development. As part of the September 2009 amendment to the research collaboration and license agreement, the Company agreed to conduct additional studies on the effects of melanocortin receptor specific compounds on food intake, obesity and other metabolic parameters
In December 2009 and 2008, the Company also entered into clinical trial sponsored research agreements with AstraZeneca, under which the Company agreed to conduct studies of the effects of melanocortin receptor specific compounds on food intake, obesity and other metabolic parameters. Under the terms of these clinical trial agreements, AstraZeneca paid $5,000,000 as of March 31, 2009 upon achieving certain objectives and pays all costs associated with these studies. The Company recognized $1,082,762 and $7,632,136, respectively, as revenue in the years ended June 30, 2010 and 2009 under these clinical trial sponsored research agreements.
The Company received an up-front payment of $10,000,000 from AstraZeneca on execution of the research collaboration and license agreement. Under the September 2009 amendment the Company was paid an additional $5,000,000 in consideration of reduction of future milestones and royalties and providing specific materials to AstraZeneca. The Company is now eligible for milestone payments totaling up to $145,250,000, with up to $85,250,000 contingent on development and regulatory milestones and the balance contingent on achievement of sales targets. In addition, the Company will receive royalties on sales of any approved products. AstraZeneca assumed responsibility for product commercialization, product discovery and development costs, with both companies contributing scientific expertise in the research collaboration. The Company provided research services to AstraZeneca through January 2010, the expiration of the research collaboration portion of the research collaboration and license agreement, at a contractual rate per full-time-equivalent employee.
The Company has determined that the license portion of the agreement and research services should be evaluated together as a single unit for purposes of revenue recognition. Accordingly, the aggregate payments of $15,000,000 have been recognized as revenue over the period ended January 2010. For the years ended June 30, 2010, 2009 and 2008, the Company recognized as revenue $10,972,219, $1,666,667 and $1,666,667, respectively, related to these aggregate payments. Per-employee compensation from AstraZeneca for research services was recognized as earned at the contractual rate, which approximates the fair value of such services. Revenue recognized for research services for the years ended June 30, 2010, 2009 and 2008 were $2,125,746, $2,052,968 and $1,250,000, respectively. Payments received upon the attainment of substantive milestones are recognized as revenue when earned.
(4) AGREEMENT WITH KING
King Pharmaceuticals, Inc. (King) terminated, effective December 2007, a collaborative development and marketing agreement between the Company and King entered into in August 2004, relating to development and commercialization of bremelanotide for treatment of sexual dysfunction. As a result of the termination, Palatin solely owns all rights to bremelanotide. In connection with the termination of the agreement, for the year ended June 30, 2008, the Company recognized as revenue all remaining deferred up-front license fees received from King, together with associated deferred costs, in the amounts of $6,499,796 and $815,561, respectively. King retains Company common stock obtained upon entering into the agreement in August 2004 and pursuant to a September 2005 agreement.
(5) INVESTMENTS AND FAIR VALUE MEASUREMENTS
The following is a summary of available-for-sale investments:
|
June 30,
|
June 30,
|
||||
|
2010
|
2009
|
||||
|
Cost
|
$
|
3,323,539
|
$
|
3,323,539
|
|
|
Gross unrealized gains
|
173,658
|
116,170
|
|||
|
Gross unrealized losses
|
(35,008)
|
(59)
|
|||
|
Total available-for-sale investments
|
$
|
3,462,189
|
$
|
3,439,650
|
|
The fair value of investments and cash equivalents are classified using a hierarchy prioritized based on inputs. Level 1 inputs are quoted prices (unadjusted) in active markets for identical assets or liabilities. Level 2 inputs are quoted prices for similar assets and liabilities in active markets or inputs that are observable for the asset or liability, either directly or indirectly through market corroboration, for substantially the full term of the financial
instrument. Level 3 inputs are unobservable inputs based on management’s own assumptions used to measure assets and liabilities at fair value. A financial asset or liability’s classification within the hierarchy is determined based on the lowest level input that is significant to the fair value measurement.
The following table provides the assets carried at fair value as of June 30, 2010 and 2009:
|
Fair Value
|
Quoted prices in active markets (Level 1)
|
Quoted prices in active markets (Level 2)
|
Quoted prices in active markets (Level 3)
|
|||||
|
June 30, 2010 –
|
||||||||
|
Money Market Fund
|
$
|
4,111,051
|
$
|
4,111,051
|
$
|
-
|
$
|
-
|
|
Mutual Funds
|
3,462,189
|
3,462,189
|
-
|
-
|
||||
|
June 30, 2009 –
|
||||||||
|
Money Market Fund
|
$
|
3,250,191
|
$
|
3,250,191
|
$
|
-
|
$
|
-
|
|
Mutual Funds
|
3,439,650
|
3,439,650
|
-
|
-
|
(6) PROPERTY AND EQUIPMENT, NET
Property and equipment, net, consists of the following:
|
June 30,
|
June 30,
|
|||||||
|
2010
|
2009
|
|||||||
|
Office equipment
|
$ 1,662,830
|
$ 1,662,830
|
||||||
|
Laboratory equipment
|
4,137,242
|
4,130,247
|
||||||
|
Leasehold improvements
|
7,088,462
|
7,088,462
|
||||||
|
12,888,534
|
12,881,539
|
|||||||
|
Less: Accumulated depreciation and amortization
|
(10,500,169)
|
(9,230,756)
|
||||||
|
$ 2,388,365
|
$ 3,650,783
|
|||||||
The cost of assets acquired under capital leases was $941,974 as of June 30, 2010 and 2009, respectively. Accumulated amortization associated with assets acquired under capital leases was $728,868 and $552,157 as of June 30, 2010 and 2009, respectively.
(7) ACCRUED EXPENSES
Accrued expenses consist of the following:
|
June 30,
|
June 30,
|
|||||||
|
2010
|
2009
|
|||||||
|
Clinical study costs
|
$ 798,744
|
$ 300,776
|
||||||
|
Other research related expenses
|
315,439
|
263,731
|
||||||
|
Deferred rent, current portion
|
421,443
|
356,012
|
||||||
|
Other
|
683,840
|
500,222
|
||||||
|
$ 2,219,466
|
$ 1,420,741
|
(8) COMMITMENTS AND CONTINGENCIES
Leases – The Company currently leases facilities under three non-cancelable operating leases. Future minimum lease payments under these leases are as follows:
|
Year Ending June 30,
|
|
|
2011
|
$ 2,196,655
|
|
2012
|
1,995,860
|
|
2013
|
294,376
|
|
2014
|
236,335
|
|
2015
|
225,175
|
|
$ 4,948,401
|
For the years ended June 30, 2010, 2009 and 2008, rent expense was $1,520,807, $1,613,534 and $1,650,273, respectively.
Capital Leases – The Company has acquired certain of its laboratory equipment under leases classified as capital leases. Scheduled future payments related to capital leases as of June 30, 2010 are as follows:
|
Year Ending June 30,
|
|
|
2011
|
$ 22,264
|
|
2012
|
14,843
|
|
37,107
|
|
|
Amount representing interest
|
(3,153)
|
|
Net
|
$ 33,954
|
Employment Agreements – The Company has employment agreements with three executive officers which provide a stated annual compensation amount, subject to annual increases, and annual bonus compensation in an amount to be approved by the Company’s Board of Directors. Each agreement allows the Company or the employee to terminate the agreement in certain circumstances. In some circumstances, early termination by the Company may result in severance pay to the employee for a period of 18 to 24 months at the salary then in effect, continuation of health insurance premiums over the severance period and immediate vesting of all stock options and restricted stock units. Termination following a change in control will result in a lump sum payment of one and one-half to two times the salary then in effect and immediate vesting of all stock options and restricted stock units.
License Agreements – The Company has license agreements related to NeutroSpec, a radiolabeled monoclonal antibody product for which the Company has suspended marketing, clinical trials and securing regulatory approvals, that require minimum annual payments of $15,000, royalty payments on commercial net sales and payments of up to $2,250,000 contingent on the achievement of specified cumulative net margins on sales. No royalty payments or other contingent amounts will be payable under these agreements unless the Company recommences sales and marketing of NeutroSpec. The Company does not expect to make any such contingent payments during the next twelve months.
Employee Retirement Savings Plan – The Company maintains a defined contribution 401(k) plan for the benefit of its employees. The Company currently matches a portion of employee contributions to the plan. For the years ended June 30, 2010, 2009 and 2008, Company contributions were $221,599, $254,127 and $341,997, respectively.
Contingencies – The Company accounts for litigation losses in accordance with ASC 450-20, “Loss Contingencies.” Under ASC 450-20, loss contingency provisions are recorded for probable losses when management is able to reasonably estimate the loss. Any outcome upon settlement that deviates from the Company’s best estimate may result in additional expense or in a reduction in expense in a future accounting period. The Company records legal expenses associated with such contingencies as incurred.
On January 21, 2008, the Company entered into a settlement agreement and release with Competitive Technologies, Inc. (CTI), resolving all outstanding disputes between the Company and CTI. The license agreement between CTI and the Company was terminated, with the Company retaining all rights to bremelanotide and CTI retaining all rights to a peptide called variously MT-II or PT-14. The settlement agreement and release also includes mutual covenants not to sue and releases of all claims by either party against the other based on, arising out of or in any way involving the subject matter of the license agreement. As part of the settlement, the Company remitted a one-time payment to CTI of $800,000 that was charged to general and administrative expense in the year ended June 30, 2008.
(9) STOCKHOLDERS’ EQUITY
Series A Convertible Preferred Stock – As of June 30, 2010, 4,997 shares of Series A Convertible Preferred Stock were outstanding. Each share of Series A Convertible Preferred Stock is convertible at any time, at the option
of the holder, into the number of shares of common stock equal to $100 divided by the Series A Conversion Price. As of June 30, 2010, the Series A Conversion Price was $18.60, so each share of Series A Convertible Preferred Stock is currently convertible into approximately 5 shares of common stock. The Series A Conversion Price is subject to adjustment, under certain circumstances, upon the sale or issuance of common stock for consideration per share less than either (i) the Series A Conversion Price in effect on the date of such sale or issuance, or (ii) the market price of the common stock as of the date of such sale or issuance. The Series A Conversion Price is also subject to adjustment upon the occurrence of a merger, reorganization, consolidation, reclassification, stock dividend or stock split which will result in an increase or decrease in the number of shares of common stock outstanding. Shares of Series A Convertible Preferred Stock have a preference in liquidation, including certain merger transactions, of $100 per share, or $499,700 in the aggregate as of June 30, 2010. Additionally, the Company may not pay a dividend or make any distribution to holders of any class of stock unless the Company first pays a special dividend or distribution of $100 per share to holders of the Series A Convertible Preferred Stock.
Common Stock Transactions – In August 2009, the Company sold 948,485 units in a registered direct offering for gross proceeds of $3,100,000. Each unit consisted of one share of common stock and a five-year warrant to purchase 0.35 shares of common stock at an exercise price of $3.30 per share. Net proceeds to the Company, after costs of the offering, were approximately $2,800,000. In addition, the Company issued to the placement agent warrants to purchase 47,424 shares of common stock at an exercise price of $4.10 per share.
In February 2010, the Company sold 962,963 units in a registered direct offering for gross proceeds of $2,600,000. Each unit consisted of one share of common stock, a Series A warrant exercisable for 0.33 shares of common stock at an exercise price of $3.00 per share and a Series B warrant exercisable for 0.33 shares of common stock at an exercise price of $2.70 per share. The Series A warrant became exercisable on August 30, 2010 and expires on August 30, 2013. The Series B warrant was exercisable immediately upon issuance and initially expired August 29, 2010, but the Company extended the expiration date through February 28, 2011. Net proceeds to the Company, after costs of the offering, were approximately $2,300,000. In addition, the Company issued to the placement agent warrants to purchase 48,148 shares of common stock at an exercise price of $3.40 per share.
In June 2010, the Company sold 1,000,000 units in a registered direct offering for gross proceeds of $2,000,000. Each unit consisted of one share of common stock and a one-year warrant to purchase 0.14 shares of common stock at an exercise price of $2.00 per share. Net proceeds to the Company, after costs of the offering, were approximately $1,800,000. In addition, the Company issued to the placement agent warrants to purchase 50,000 shares of common stock at an exercise price of $2.50 per share.
At the annual meeting of stockholders held on May 13, 2010, the stockholders authorized, at the discretion of the Company’s Board of Directors, an amendment to the Company’s restated certificate of incorporation to increase the number of authorized shares of common stock, $0.01 par value per share, from 150,000,000 to 400,000,000, and separately an amendment to the Company’s restated certificate of incorporation to effect a reverse stock split of common stock at a ratio of between one-for-two and one-for-fifteen (Note 1). At the direction of the Board of Directors, the amendment increasing the number of authorized shares of common stock, $0.01 par value per share, to 400,000,000 was filed effective July 23, 2010. Pursuant to the reverse stock split effective September 27, 2010, both the outstanding common stock and number of authorized shares of common stock were reduced proportionately.
Outstanding Stock Purchase Warrants – As of June 30, 2010, the Company had outstanding warrants exercisable for shares of common stock as follows:
|
Shares of Common Stock
|
Exercise Price per Share
|
Latest Termination Date
|
|
140,000
|
$ 2.00
|
June 29, 2011
|
|
50,000
|
2.50
|
November 26, 2012
|
|
317,777
|
2.70
|
February 28, 2011
|
|
317,777
|
3.00
|
August 30, 2013
|
|
331,969
|
3.30
|
August 12, 2014
|
|
48,148
|
3.40
|
November 26, 2012
|
|
47,424
|
4.10
|
November 26, 2012
|
|
1,500
|
28.20
|
December 11, 2012
|
|
329,359
|
28.80
|
April 17, 2011
|
|
1,500
|
40.00
|
December 15, 2010
|
|
1,585,454
|
Stock Plan – The Company’s 2005 Stock Plan was initially approved by the Company’s stockholders in June 2005 and provides for incentive and nonqualified stock option grants and other stock-based awards to employees, non-employee directors and consultants for up to 500,000 shares of common stock. On December 7, 2007, the Company received stockholder approval to increase the number of authorized shares available for grant to 1,000,000, and on May 13, 2009 the Company received stockholder approval to increase the number of authorized shares available for grant to 1,500,000. The 2005 Stock Plan is administered under the direction of the Board of Directors, which may specify grant terms and recipients. Options granted by the Company generally expire ten years from the date of grant and generally vest over three to four years. As of June 30, 2010, 459,006 shares were available for grant under the 2005 Stock Plan.
The Company also has outstanding options that were granted under previous plans. The Company expects to settle option exercises under any of its plans with authorized but currently unissued shares.
The following table summarizes option activity for the years ended June 30, 2010, 2009 and 2008:
|
2010
|
2009
|
2008
|
||||||
|
Number of Shares
|
Weighted Average Exercise Price
|
Number of Shares
|
Weighted Average Exercise Price
|
Number of Shares
|
Weighted Average Exercise Price
|
|||
|
Outstanding at beginning of year
|
882,862
|
$16.60
|
654,345
|
$24.00
|
639,472
|
$28.90
|
||
|
Granted
|
174,276
|
2.60
|
287,455
|
1.70
|
178,745
|
10.40
|
||
|
Forfeited
|
(34,303)
|
16.00
|
(27,097)
|
19.70
|
(138,154)
|
23.20
|
||
|
Exercised
|
(6,725)
|
1.70
|
-
|
-
|
-
|
-
|
||
|
Expired
|
(58,736)
|
34.10
|
(31,841)
|
31.90
|
(25,718)
|
48.20
|
||
|
Outstanding at end of year
|
957,374
|
13.20
|
882,862
|
16.60
|
654,345
|
24.00
|
||
|
Exercisable at end of year
|
631,313
|
18.00
|
546,380
|
23.10
|
439,285
|
29.30
|
||
|
Weighted average grant-date fair value of options granted during the year
|
$2.20
|
$1.40
|
$7.30
|
|||||
The following table summarizes options outstanding as of June 30, 2010:
|
Number of Shares
|
Weighted Average Exercise Price
|
Weighted Average Remaining Term in Years
|
Aggregate Intrinsic Value
|
||||||
|
Options outstanding at end of year
|
957,374
|
$ 13.20
|
6.2
|
$ 15,599
|
|||||
|
Options vested and exercisable at end of year
|
631,313
|
18.00
|
5.3
|
4,084
|
|||||
|
Unvested options expected to vest
|
308,382
|
4.00
|
8.0
|
10,289
|
The intrinsic value of options exercised during the year ended June 30, 2010 was $7,998.
The fair value of option grants is estimated at the grant date using the Black-Scholes model. For grants during the year ended June 30, 2010, the Company’s weighted average assumptions for expected volatility, dividends, term and risk-free interest rate were 96%, 0%, 8.1 years and 3.2%, respectively. For grants during the year ended June 30, 2009, the Company’s weighted average assumptions for expected volatility, dividends, term and risk-free interest rate were 85%, 0%, 8.8 years and 3.8%, respectively. For grants during the year ended June 30, 2008, the Company’s weighted average assumptions for expected volatility, dividends, term and risk-free interest rate were 80%, 0%, 6.2 years and 3.7%, respectively. Expected volatilities are based primarily on the Company’s historical volatility. The expected term of options is based upon the simplified method, which represents the average
of the vesting term and the contractual term. The risk-free interest rate is based on U.S. Treasury yields for securities with terms approximating the expected term of the option.
For the years ended June 30, 2010, 2009 and 2008 the Company recorded stock-based compensation related to stock options of $633,532, $700,618 and $1,016,579, respectively. The Company did not record a tax benefit related to stock-based compensation expense. As of June 30, 2010, there was $549,292 of total unrecognized compensation cost related to unvested options, which is expected to be recognized over a weighted-average period of 1.03 years.
In July 2010, the Company granted 30,000 options to its non-employee directors under the Company’s 2005 stock plan.
Restricted Stock Units – In October 2006, the Company made grants of restricted stock units to three executive officers for an aggregate of 97,500 shares of common stock. Under the original vesting conditions, 32,500 shares vested if the quoted market price of Palatin’s common stock was $40.00 or more for 20 consecutive trading days, an additional 32,500 shares vested if the quoted market price of Palatin’s common stock was $60.00 or more for 20 consecutive trading days and the remaining 32,500 shares vested if the quoted market price of Palatin’s common stock was $80.00 or more for 20 consecutive trading days. The fair value of the restricted stock units was estimated at the grant date using a lattice-type model. The Company’s assumptions for expected volatility, dividends and risk-free rate were 80%, 0% and 4.56%, respectively. The expected volatility was based on the Company’s historical volatility and the risk-free rate was based on U.S. Treasury yields for securities with terms approximating the contractual term of the units. The aggregate fair value of the units at the date of grant was $1,846,000, which was recognized over a weighted-average period ended December 31, 2009. For the years ended June 30, 2010, 2009 and 2008 the Company recognized $201,500, $470,031 and $671,125, respectively, of stock-based compensation expense related to these restricted stock units.
In March 2008, the Company’s Compensation Committee revised the vesting conditions of the above restricted stock units granted to the three executive officers. Under the revised conditions, the restricted stock units granted to each of the executive officers became fully vested on March 26, 2010 and on such date, after adjusting for withholding taxes, 66,160 shares of common stock were issued. The restricted stock unit agreements require that each executive officer retain ownership of at least 33% of the stock received for the duration of the executive’s employment with the Company unless there is a change in control or for hardship as determined by the Board of Directors. In addition to the original grant-date fair value of these awards, the Company recognized an incremental fair value adjustment to these restricted stock units, totaling $273,000, on a straight-line basis through March 26, 2010. For the years ended June 30, 2010, 2009 and 2008, the Company recognized $102,375, $136,500 and $34,125, respectively, of stock-based compensation expense related to these restricted stock units.
On December 10, 2008, the Company granted restricted stock units to its executive officers under the Company’s 2005 Stock Plan totaling 75,000 shares of common stock. The restricted stock units vested on December 31, 2009 and on such date, after adjusting for withholding taxes, 52,195 shares of common stock were issued. The Company amortized the fair value of these restricted stock units, totaling $67,500, on a straight-line basis through December 31, 2009. For the years ended June 30, 2010, and 2009, the Company recognized $31,154 and $36,346, respectively, as stock-based compensation expense related to these restricted stock units.
In September 2007, the Company issued 157,391 restricted stock units under the Company’s 2005 Stock Plan as retention bonuses to its employees, other than the executive officers, that were not affected by the September 2007 reduction in workforce. On September 30, 2008, after adjusting for forfeitures and early vesting due to involuntary position elimination, 113,882 shares of common stock vested. The Company amortized the fair value of these restricted stock units of $676,748 on a straight-line basis over a one-year period. For the years ended June 30, 2009 and 2008, the Company recognized $133,078 and $543,670, respectively, of stock-based compensation expense related to these restricted stock units.
In July 2010, the Company granted 205,000 restricted stock units to its employees under the Company’s 2005 stock plan. On September 15, 2010, 99,500 shares of common stock vested. The Company will amortize the fair value of these restricted stock units of approximately $340,000 over the nine months ending March 31, 2010.
(10) INCOME TAXES
The Company has had no income tax expense or benefit since inception because of operating losses, except for amounts recognized for sales of New Jersey state net operating loss carryforwards. Deferred tax assets and liabilities are determined based on the estimated future tax effect of differences between the financial statement and tax reporting basis of assets and liabilities, as well as for net operating loss carryforwards and research and development credit carryforwards, given the provisions of existing tax laws.
As of June 30, 2010, the Company had federal and state net operating loss carryforwards of approximately $194,000,000 and $83,000,000, respectively, which expire between 2011 and 2030 if not utilized. As of June 30, 2010, the Company had federal research and development credits of approximately $5,400,000 that will begin to expire in 2012, if not utilized.
The Tax Reform Act of 1986 (the Act) provides for limitation on the use of net operating loss and research and development tax credit carryforwards following certain ownership changes (as defined by the Act) that could limit the Company’s ability to utilize these carryforwards. The Company may have experienced various ownership changes, as defined by the Act, as a result of past financings. Accordingly, the Company’s ability to utilize the aforementioned carryforwards may be limited. Additionally, U.S. tax laws limit the time during which these carryforwards may be applied against future taxes; therefore the Company may not be able to take full advantage of these carryforwards for federal income tax purposes.
The Company’s net deferred tax assets are as follows:
|
June 30,
|
June 30,
|
||
|
2010
|
2009
|
||
|
Net operating loss carryforwards
|
$ 72,603,000
|
$ 70,810,000
|
|
|
Research and development tax credits
|
5,390,000
|
5,288,000
|
|
|
Accrued expenses, deferred revenue and other
|
2,911,000
|
5,768,000
|
|
|
80,904,000
|
81,866,000
|
||
|
Valuation allowance
|
(80,904,000)
|
(81,866,000)
|
|
|
Net deferred tax assets
|
$ -
|
$ -
|
In assessing the realizability of deferred tax assets, the Company considers whether it is more likely than not that some portion or all of the deferred tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income and the application of loss limitation provisions related to ownership changes. Due to the Company’s history of losses, the deferred tax assets are fully offset by a valuation allowance as of June 30, 2010 and 2009. The valuation allowance for the year ended June 30, 2010 decreased by $962,000 due to the tax treatment of certain deferred revenue.
During the years ended June 30, 2010, 2009 and 2008, the Company sold New Jersey state net operating loss carryforwards, which resulted in the recognition of $998,408, $1,741,476 and $1,291,444, respectively, in tax benefits.
(11) CONSOLIDATED QUARTERLY FINANCIAL DATA – UNAUDITED
The following tables provide quarterly data for the years ended June 30, 2010 and 2009:
|
Three Months Ended
|
|||||||
|
June 30,
2010
|
March 31, 2010
|
December 31, 2009
|
September 30, 2009
|
||||
|
(amounts in thousands, except per share data)
|
|||||||
|
Total revenues
|
$ 675
|
$ 2,560
|
$ 7,283
|
$ 3,663
|
|||
|
Total operating expenses
|
4,929
|
4,594
|
3,848
|
3,824
|
|||
|
Total other income, net
|
17
|
14
|
68
|
124
|
|||
|
Income/(loss) before income taxes
|
(4,237)
|
(2,020)
|
3,503
|
(37)
|
|||
|
Income tax benefit
|
-
|
-
|
998
|
-
|
|||
|
Net income (loss)
|
$ (4,237)
|
$ (2,020)
|
$ 4,501
|
$ (37)
|
|||
|
Basic net income/(loss) per common share
|
$ (0.40)
|
$ (0.20)
|
$ 0.41
|
$ (0.00)
|
|||
|
Weighted average number of common shares outstanding used in computing basic net income/(loss) per common share
|
10,722,061
|
9,987,323
|
9,616,954
|
9,130,622
|
|||
|
Diluted net income/(loss) per common share
|
$ (0.40)
|
$ (0.20)
|
$ 0.41
|
$ (0.00)
|
|||
|
Weighted average number of common shares outstanding used in computing diluted net income/(loss) per common share
|
10,722,061
|
9,987,323
|
9,664,507
|
9,130,622
|
|||
|
Three Months Ended
|
|||||||
|
June 30,
2009
|
March 31, 2009
|
December 31, 2008
|
September 30, 2008
|
||||
|
(amounts in thousands, except per share data)
|
|||||||
|
Total revenues
|
$ 4,228
|
$ 5,159
|
$ 1,211
|
$ 754
|
|||
|
Total operating expenses
|
4,461
|
5,087
|
3,991
|
5,115
|
|||
|
Total other income, net
|
32
|
26
|
622
|
78
|
|||
|
Income/(loss) before income taxes
|
(201)
|
98
|
(2,158)
|
(4,283)
|
|||
|
Income tax benefit
|
-
|
-
|
1,741
|
-
|
|||
|
Net income/(loss)
|
$ (201)
|
$ 98
|
$ (417)
|
$ (4,283)
|
|||
|
Basic and diluted net income/(loss) per common share
|
$ (0.02)
|
$ 0.01
|
$ (0.05)
|
$ (0.50)
|
|||
|
Weighted average number of common shares outstanding used in computing basic and diluted net income/(loss) per common share
|
8,666,290
|
8,666,290
|
8,664,064
|
8,552,431
|
|||
PALATIN TECHNOLOGIES, INC.
and Subsidiary
Consolidated Balance Sheets
(unaudited)
|
December 31,
2010
|
June 30,
2010
|
|||
|
ASSETS
|
||||
|
Current assets:
|
||||
|
Cash and cash equivalents
|
$ 1,731,615
|
$ 5,405,430
|
||
|
Available-for-sale investments
|
1,952,666
|
3,462,189
|
||
|
Accounts receivable
|
-
|
2,879
|
||
|
Prepaid expenses and other current assets
|
245,024
|
393,313
|
||
|
Total current assets
|
3,929,305
|
9,263,811
|
||
|
Property and equipment, net
|
1,794,970
|
2,388,365
|
||
|
Restricted cash
|
475,000
|
475,000
|
||
|
Other assets
|
270,581
|
261,701
|
||
|
Total assets
|
$ 6,469,856
|
$ 12,388,877
|
||
|
LIABILITIES AND STOCKHOLDERS' EQUITY
|
||||
|
Current liabilities:
|
||||
|
Capital lease obligations
|
$ 20,708
|
$ 19,670
|
||
|
Accounts payable
|
502,125
|
155,795
|
||
|
Accrued compensation
|
564,887
|
-
|
||
|
Unearned revenue
|
132,090
|
-
|
||
|
Accrued expenses
|
823,984
|
2,219,466
|
||
|
Total current liabilities
|
2,043,794
|
2,394,931
|
||
|
Capital lease obligations
|
3,663
|
14,284
|
||
|
Deferred rent
|
391,314
|
661,389
|
||
|
Total liabilities
|
2,438,771
|
3,070,604
|
||
|
Stockholders' equity:
|
||||
|
Preferred stock of $.01 par value – authorized 10,000,000 shares;
|
||||
|
Series A Convertible; issued and outstanding 4,997 shares as of December 31, 2010 and June 30, 2010, respectively
|
50
|
50
|
||
|
Common stock of $.01 par value – authorized 40,000,000 shares; issued and outstanding 11,854,028 and 11,702,818 shares as of December 31, 2010 and June 30, 2010, respectively
|
118,540
|
117,028
|
||
|
Additional paid-in capital
|
218,718,757
|
218,236,723
|
||
|
Accumulated other comprehensive income
|
68,738
|
138,650
|
||
|
Accumulated deficit
|
(214,875,000)
|
(209,174,178)
|
||
|
Total stockholders’ equity
|
4,031,085
|
9,318,273
|
||
|
Total liabilities and stockholders’ equity
|
$ 6,469,856
|
$ 12,388,877
|
The accompanying notes are an integral part of these consolidated financial statements.
F-19
PALATIN TECHNOLOGIES, INC.
and Subsidiary
Consolidated Statements of Operations
(unaudited)
|
Three Months Ended December 31,
|
Six Months Ended December 31,
|
|||||||
|
2010
|
2009
|
2010
|
2009
|
|||||
|
REVENUES:
|
||||||||
|
License and contract
|
$ 195,408
|
$ 7,283,299
|
$ 411,555
|
$ 10,945,918
|
||||
|
Grant
|
846,768
|
-
|
846,768
|
-
|
||||
|
Total revenues
|
1,042,176
|
7,283,299
|
1,258,323
|
10,945,918
|
||||
|
OPERATING EXPENSES:
|
||||||||
|
Research and development
|
1,984,440
|
2,712,871
|
5,437,202
|
5,382,435
|
||||
|
General and administrative
|
889,476
|
1,134,963
|
2,271,252
|
2,288,694
|
||||
|
Total operating expenses
|
2,873,916
|
3,847,834
|
7,708,454
|
7,671,129
|
||||
|
Income (loss) from operations
|
(1,831,740)
|
3,435,465
|
(6,450,131)
|
3,274,789
|
||||
|
OTHER INCOME (EXPENSE):
|
||||||||
|
Investment income
|
32,987
|
70,317
|
53,362
|
103,629
|
||||
|
Interest expense
|
(1,329)
|
(2,315)
|
(3,633)
|
(7,016)
|
||||
|
Gain on sale of securities
|
60,389
|
-
|
60,389
|
-
|
||||
|
Gain on sale of supplies and equipment
|
1,800
|
-
|
1,800
|
95,000
|
||||
|
Total other income, net
|
93,847
|
68,002
|
111,918
|
191,613
|
||||
|
Income (loss) before income taxes
|
(1,737,893)
|
3,503,467
|
(6,338,213)
|
3,466,402
|
||||
|
Income tax benefit
|
637,391
|
998,408
|
637,391
|
998,408
|
||||
|
NET INCOME (LOSS)
|
$ (1,100,502)
|
$ 4,501,875
|
$ (5,700,822)
|
$ 4,464,810
|
||||
|
Basic net income (loss) per common share
|
$ (0.09)
|
$ 0.42
|
$ (0.48)
|
$ 0.42
|
||||
|
Diluted net income (loss) per common share
|
$ (0.09)
|
$ 0.42
|
$ (0.48)
|
$ 0.42
|
||||
|
Weighted average number of common shares outstanding used in computing basic net income (loss) per common share
|
11,839,309
|
9,616,954
|
11,785,470
|
9,373,788
|
||||
|
Weighted average number of common shares outstanding used in computing diluted net income (loss) per common share
|
11,839,309
|
9,664,507
|
11,785,470
|
9,417,662
|
||||
The accompanying notes are an integral part of these consolidated financial statements.
F-20
PALATIN TECHNOLOGIES, INC.
and Subsidiary
Consolidated Statements of Cash Flows
(unaudited)
|
Six Months Ended December 31,
|
||||
|
2010
|
2009
|
|||
|
CASH FLOWS FROM OPERATING ACTIVITIES:
|
||||
|
Net income (loss)
|
$ (5,700,822)
|
$ 4,464,810
|
||
|
Adjustments to reconcile net loss to net cash
|
||||
|
used in operating activities:
|
||||
|
Depreciation and amortization
|
593,395
|
666,789
|
||
|
Gain on sale of supplies and equipment
|
(1,800)
|
(95,000)
|
||
|
Gain on sale of available-for-sale investments
|
(60,389)
|
-
|
||
|
Stock-based compensation
|
438,139
|
635,108
|
||
|
Amortization of deferred revenue
|
-
|
(7,276,736)
|
||
|
Changes in operating assets and liabilities:
|
||||
|
Accounts receivable
|
2,879
|
(1,382,467)
|
||
|
Prepaid expenses and other assets
|
139,409
|
202,595
|
||
|
Accounts payable
|
346,330
|
98,810
|
||
|
Accrued expenses and compensation
|
(1,100,670)
|
(506,545)
|
||
|
Unearned revenues
|
132,090
|
-
|
||
|
Net cash used in operating activities
|
(5,211,439)
|
(3,192,636)
|
||
|
CASH FLOWS FROM INVESTING ACTIVITIES:
|
||||
|
Proceeds from sale of supplies and equipment
|
1,800
|
95,000
|
||
|
Proceeds from sale of available-for-sale investments
|
1,500,000
|
-
|
||
|
Net cash provided by investing activities
|
1,501,800
|
95,000
|
||
|
CASH FLOWS FROM FINANCING ACTIVITIES:
|
||||
|
Payments on capital lease obligations
|
(9,583)
|
(76,950)
|
||
|
Payment of withholding taxes related to restricted stock units
|
(18,993)
|
(84,379)
|
||
|
Proceeds from sale of common stock units and warrant and
|
||||
|
exercise of common stock options
|
64,400
|
2,802,988
|
||
|
Net cash provided by financing activities
|
35,824
|
2,641,659
|
||
|
NET DECREASE IN CASH AND
|
||||
|
CASH EQUIVALENTS
|
(3,673,815)
|
(455,977)
|
||
|
CASH AND CASH EQUIVALENTS, beginning
|
||||
|
of period
|
5,405,430
|
4,378,662
|
||
|
CASH AND CASH EQUIVALENTS, end of period
|
$ 1,731,615
|
$ 3,922,685
|
||
|
SUPPLEMENTAL CASH FLOW INFORMATION:
|
||||
|
Cash paid for interest
|
$ 3,633
|
$ 7,016
|
||
|
Unrealized loss on available-for-sale investments
|
$ (9,523)
|
$ (7,926)
|
||
The accompanying notes are an integral part of these consolidated financial statements.
F-21
PALATIN TECHNOLOGIES, INC.
and Subsidiary
Notes to Consolidated Financial Statements
(unaudited)
(1) ORGANIZATION:
Nature of Business – Palatin Technologies, Inc. (Palatin or the Company) is a biopharmaceutical company dedicated to the development of peptide, peptide mimetic and small molecule agonist compounds with a focus on melanocortin and natriuretic peptide receptor systems. Palatin has a diverse pipeline of active development programs targeting melanocortin and natriuretic receptors. The melanocortin system is involved in a large and diverse number of physiologic functions, and therapeutic agents modulating this system may have the potential to treat a variety of conditions and diseases, including sexual dysfunction, obesity and related disorders, cachexia (wasting syndrome) and inflammation-related diseases. The natriuretic peptide receptor system has numerous cardiovascular functions, and therapeutic agents modulating this system may be useful in treatment of acute asthma, heart failure, hypertension and other cardiovascular diseases.
The Company’s active drug development programs consist of bremelanotide for treatment of sexual dysfunction, other peptide melanocortin receptor agonists for treatment of sexual dysfunction, and PL-3994, an agonist peptide mimetic which binds to natriuretic peptide receptor A, for treatment of acute asthma and heart failure. The Company has an exclusive global research collaboration and license agreement with AstraZeneca AB (AstraZeneca) to commercialize compounds that target melanocortin receptors for the treatment of obesity, diabetes and related metabolic syndrome.
Key elements of the Company’s business strategy include using its technology and expertise to develop and commercialize therapeutic products; entering into alliances and partnerships with pharmaceutical companies to facilitate the development, manufacture, marketing, sale and distribution of product candidates the Company is developing; partially funding its product candidate development programs with the cash flow from the Company’s AstraZeneca collaboration agreement and any future agreements with other companies.
Business Risk and Liquidity – The Company has incurred negative cash flows from operations since its inception, and has expended, and expects to continue to expend in the future, substantial funds to complete its planned product development efforts. As shown in the accompanying consolidated financial statements, the Company has an accumulated deficit as of December 31, 2010 and incurred a net loss for the three and six months ended December 31, 2010. The Company anticipates incurring additional losses in the future as a result of spending on its development programs. To achieve profitability, the Company, alone or with others, must successfully develop and commercialize its technologies and proposed products, conduct successful preclinical studies and clinical trials, obtain required regulatory approvals and successfully manufacture and market such technologies and proposed products. The time required to reach profitability is highly uncertain, and there can be no assurance that the Company will be able to achieve profitability on a sustained basis, if at all.
As of December 31, 2010, the Company’s cash and cash equivalents were $1.7 million and its available-for-sale investments were $2.0 million. The Company has made the strategic decision to focus resources and efforts on clinical trials for bremelanotide and PL-3994 and preclinical development of an inhaled formulation of PL-3994 and a new peptide drug candidate for sexual dysfunction, and has ceased research and development efforts on new product candidates. As part of this decision, the Company reduced staffing levels to 18 employees as of December 31, 2010. Management does not believe that the Company’s existing capital resources, together with expected revenues, will be adequate to fund its currently planned operations for the next twelve months and intends to seek additional capital. The accompanying consolidated financial statements have been prepared assuming that the Company continues as a going concern, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. The consolidated financial statements do not include any adjustments relating to the recoverability and classification of recorded asset amounts or the amounts and classification of liabilities that might results from the outcome of this uncertainty.
The Company intends to seek additional capital through public or private equity or debt financings, collaborative arrangements on our product candidates, or other sources. However, sufficient additional funding to support projected operations, including clinical trials with either bremelanotide or PL-3994, or both, may not be
available on acceptable terms, or at all. These matters raise substantial doubt over the Company’s ability to continue as a going concern.
If the Company is unable to raise sufficient additional funds to advance at least one of its product candidates, management will implement plans for the orderly wind down of its business operations, including curtailing operations significantly and further decreasing staffing levels, and will seek to license, sell or otherwise dispose of the Company’s product candidates, technologies and contractual rights, including rights under the research collaboration and license agreement with AstraZeneca, on the best possible terms available.
The nature and timing of the Company’s development activities are highly dependent on its financing activities. There can be no assurance that the Company will be able to obtain financing when required, or that financing efforts will be successful. Additionally, the Company may be required to seek collaborators for its product candidates at an earlier stage than otherwise would be desirable and on terms that are less favorable than might otherwise be available, and relinquish, license or otherwise dispose of rights on unfavorable terms to technologies and product candidates that the Company would otherwise seek to develop or commercialize itself.
Concentrations – Concentrations in the Company’s assets and operations subject it to certain related risks. Financial instruments that subject the Company to concentrations of credit risk primarily consist of cash and cash equivalents, available-for-sale investments and accounts receivable. The Company’s cash and cash equivalents are primarily invested in one money market fund sponsored by a large financial institution. For the three and six months ended December 31, 2010 and 2009, 100% of license and contract revenues were from AstraZeneca.
(2) BASIS OF PRESENTATION:
The accompanying unaudited consolidated financial statements have been prepared in accordance with U.S. generally accepted accounting principles for interim financial information and with the instructions to Form 10-Q. Accordingly, they do not include all of the information and footnote disclosures required to be presented for complete financial statements. In the opinion of management, these consolidated financial statements contain all adjustments (consisting of normal recurring adjustments) considered necessary to present fairly the Company’s financial position as of December 31, 2010, and its results of operations and its cash flows for the three and six months ended December 31, 2010 and 2009. The results of operations for the three and six months ended December 31, 2010 may not necessarily be indicative of the results of operations expected for the full year, except that the Company expects to incur a significant loss for the fiscal year ending June 30, 2011.
The accompanying consolidated financial statements should be read in conjunction with the audited consolidated financial statements and notes thereto included in the Company’s annual report on Form 10-K for the year ended June 30, 2010, filed with the Securities and Exchange Commission (SEC), which includes consolidated financial statements as of June 30, 2010 and 2009 and for each of the fiscal years in the three-year period ended June 30, 2010.
(3) SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES:
Principles of Consolidation – The consolidated financial statements include the accounts of Palatin and its wholly-owned inactive subsidiary. All significant intercompany accounts and transactions have been eliminated in consolidation.
Use of Estimates – The preparation of consolidated financial statements in conformity with U.S. generally accepted accounting principles requires management to make estimates and assumptions that affect the reported amount of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Cash and Cash Equivalents – Cash and cash equivalents include cash on hand, cash in banks and all highly liquid investments with a purchased maturity of less than three months. Cash equivalents consist of $1,409,820 and $4,111,051 in a money market fund at December 31, 2010 and June 30, 2010, respectively. Restricted cash secures letters of credit for security deposits on leases.
Investments – The Company classifies its investments as available-for-sale investments and all such investments are recorded at fair value based on quoted market prices. Unrealized holding gains and losses are
generally excluded from earnings and are reported in accumulated other comprehensive income/loss until realized. Interest and dividends on securities classified as available-for-sale are included in investment income. Gains and losses are recorded in the statement of operations when realized or when unrealized holding losses are determined to be other than temporary, on a specific-identification basis.
Fair Value of Financial Instruments – The Company’s financial instruments consist primarily of cash equivalents, available-for-sale investments, accounts receivable, accounts payable, and capital lease obligations. Management believes that the carrying value of these assets and liabilities are representative of their respective fair values based on quoted market prices for investments and the short-term nature of the other instruments.
Property and Equipment – Property and equipment consists of office and laboratory equipment, office furniture and leasehold improvements and includes assets acquired under capital leases. Property and equipment are recorded at cost. Depreciation is recognized using the straight-line method over the estimated useful lives of the related assets, generally five years for laboratory and computer equipment, seven years for office furniture and equipment and the lesser of the term of the lease or the useful life for leasehold improvements. Amortization of assets acquired under capital leases is included in depreciation expense. Maintenance and repairs are expensed as incurred while expenditures that extend the useful life of an asset are capitalized.
Impairment of Long-Lived Assets – The Company reviews its long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of the assets may not be fully recoverable. To determine recoverability of a long-lived asset, management evaluates whether the estimated future undiscounted net cash flows from the asset are less than its carrying amount. If impairment is indicated, the long-lived asset would be written down to fair value. Fair value is determined by an evaluation of available price information at which assets could be bought or sold, including quoted market prices if available, or the present value of the estimated future cash flows based on reasonable and supportable assumptions.
Deferred Rent – The Company’s operating leases provide for rent increases over the terms of the leases. Deferred rent consists of the difference between periodic rent payments and the amount recognized as rent expense on a straight-line basis, as well as tenant allowances for leasehold improvements. Rent expenses are being recognized ratably over the terms of the leases.
Revenue Recognition – Revenue from corporate collaborations and licensing agreements consists of up-front fees, research and development funding, and milestone payments. Non-refundable up-front fees are deferred and amortized to revenue over the related performance period. The Company estimates the performance period as the period in which it performs certain development activities under the applicable agreement. Reimbursements for research and development activities are recorded in the period that the Company performs the related activities under the terms of the applicable agreements. Revenue resulting from the achievement of milestone events stipulated in the applicable agreements is recognized when the milestone is achieved, provided that such milestone is substantive in nature. Revenue from grants is recognized as the Company provides the services stipulated in the underlying grants based on the time and materials incurred.
Research and Development Costs – The costs of research and development activities are charged to expense as incurred, including the cost of equipment for which there is no alternative future use.
Stock-Based Compensation – The Company charges to expense the fair value of stock options and other equity awards granted. The Company determines the value of stock options utilizing the Black-Scholes option pricing model. Compensation costs for share-based awards with pro rata vesting are allocated to periods on a straight-line basis.
Income Taxes – The Company and its subsidiary file consolidated federal and separate-company state income tax returns. Income taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of assets and liabilities and their respective tax basis and operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences or operating loss and tax credit carryforwards are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in the period that includes the enactment date. The Company has recorded a valuation allowance against its deferred tax assets based on the history of losses incurred.
During the three months ended December 31, 2010 and 2009, the Company sold New Jersey state net operating loss carryforwards, which resulted in the recognition of $637,391 and $998,408, respectively, in tax benefits.
Net Income (Loss) per Common Share – Basic and diluted earnings per common share (EPS) are calculated in accordance with the provisions of Financial Accounting Standards Board (FASB) Accounting Standards Codification (ASC) Topic 260, “Earnings per Share.” In June 2008, the FASB issued guidance stating that non-vested share-based payment awards that include non-forfeitable rights to dividends or dividend equivalents, whether paid or unpaid, are considered participating securities, and the two-class method of computing EPS is required for all periods presented. The Company adopted the provisions of ASC Topic 260 relating to the two-class method of computing EPS effective July 1, 2009.
The Company’s outstanding shares of Series A Convertible Preferred stock contain rights that entitle the holder to a special dividend or distribution of $100 per share before the Company can pay dividends or make distributions to the common stockholders. The other outstanding share-based compensation awards do not include non-forfeitable rights to dividends. Accordingly, only the outstanding Series A Convertible Preferred stock is considered a participating security and must be included in the computation of EPS. The adoption of the provisions of ASC Topic 260 relating to the two-class method of computing EPS reduced the basic EPS by $0.05 and $0.06, respectively for the three and six month period ended December 31, 2009 and diluted EPS by $0.05 for the three and six month period ended December 31, 2009.
The following table sets forth the computation of basic and diluted EPS:
|
Three months ended December 31,
|
Six months ended December 31,
|
||||||
|
2010
|
2009
|
2010
|
2009
|
||||
|
Net income (loss) per common share – Basic:
|
|||||||
|
Net income (loss)
|
$ (1,100,502)
|
$ 4,501,875
|
$ (5,700,822)
|
$ 4,464,810
|
|||
|
Net income allocated to Series A Preferred Shares
|
-
|
(508,876)
|
-
|
(508,791)
|
|||
|
Net income (loss) available to common stockholders
|
$ (1,100,502)
|
$ 3,992,999
|
$ (5,700,822)
|
$ 3,956,019
|
|||
|
Weighted average common shares outstanding
|
11,839,309
|
9,616,954
|
11,785,470
|
9,373,788
|
|||
|
Net income (loss) per common share - Basic
|
$ (0.09)
|
$ 0.42
|
$ (0.48)
|
$ 0.42
|
|||
|
Net income (loss) per common share – Diluted:
|
|||||||
|
Net income (loss)
|
$ (1,100,502)
|
$ 4,501,875
|
$ (5,700,822)
|
$ 4,464,810
|
|||
|
Net income allocated to Series A Preferred Shares
|
-
|
(508,876)
|
-
|
(508,791)
|
|||
|
Net income (loss) available to common stockholders
|
$ (1,100,502)
|
$ 3,992,999
|
$ (5,700,822)
|
$ 3,956,019
|
|||
|
Weighted average common shares outstanding
|
11,839,309
|
9,616,954
|
11,785,470
|
9,373,788
|
|||
|
Dilutive securities
|
-
|
47,553
|
-
|
43,874
|
|||
|
Weighted average common and dilutive shares outstanding
|
11,839,309
|
9,664,507
|
11,785,470
|
9,417,662
|
|||
|
Net income (loss) per common share - Diluted
|
$ (0.09)
|
$ 0.42
|
$ (0.48)
|
$ 0.42
|
As of December 31, 2010 and 2009, common shares issuable upon conversion of Series A Convertible Preferred Stock, the exercise of outstanding options and warrants and the vesting of restricted stock units amounted to an aggregate of 2,498,279 and 1,799,390, respectively.
Recently Issued Accounting Pronouncements – In September 2009, the FASB issued Accounting Standards Update (ASU) 2009-13, Revenue Recognition (Topic 605), “Multiple-Deliverable Revenue Arrangements (ASU 2009-13)”, which requires companies to allocate revenue in arrangements involving multiple deliverables based on the estimated selling price of each deliverable when such deliverables are not sold separately either by the company or other vendors. ASU 2009-13 eliminates the requirement that all undelivered elements must have objective and reliable evidence of fair value before a company can recognize the portion of the overall arrangement fee that is attributable to items that already have been delivered. As a result, the new guidance may allow some companies to recognize revenue on transactions that involve multiple deliverables earlier than under current requirements. ASU 2009-13 is effective for revenue arrangements entered into or materially modified in fiscal years beginning on or after June 15, 2010. Early adoption is permitted at the beginning of a company’s fiscal year. The adoption of ASU 2009-13 on July 1, 2010 had no impact on the Company’s consolidated financial statements.
In April 2010, the FASB issued ASU No. 2010-17, “Revenue Recognition – Milestone Method (ASU 2010-17).” ASU 2010-17 provides guidance on applying the milestone method to milestone payments for achieving specified performance measures when those payments are related to uncertain future events. Under ASU 2010-17, entities can make an accounting policy election to recognize arrangement consideration received for achieving specified performance measures during the period in which the milestones are achieved, provided certain criteria are met. This ASU is effective for fiscal years beginning January 1, 2011, with early adoption permitted. The Company does not believe adoption will have a material impact on its consolidated financial position and results of operations.
(4) AGREEMENT WITH ASTRAZENECA:
In January 2007, the Company entered into an exclusive global research collaboration and license agreement with AstraZeneca to discover, develop and commercialize compounds that target melanocortin receptors for the treatment of obesity, diabetes and related metabolic syndrome. In June 2008, the collaboration agreement was amended to include additional compounds and associated intellectual property developed by the Company. In December 2008, the collaboration agreement was further amended to include additional compounds and associated intellectual property developed by the Company and extended the research collaboration for an additional year through January 2010. In September 2009, the collaboration agreement was further amended to modify royalty rates and milestone payments. The collaboration is based on the Company’s melanocortin receptor obesity program and includes access to compound libraries, core technologies and expertise in melanocortin receptor drug discovery and development. As part of the September 2009 amendment to the research collaboration and license agreement, the Company agreed to conduct additional studies on the effects of melanocortin receptor specific compounds on food intake, obesity and other metabolic parameters.
In December 2009 and 2008, the Company also entered into clinical trial sponsored research agreements with AstraZeneca, under which the Company agreed to conduct studies of the effects of melanocortin receptor specific compounds on food intake, obesity and other metabolic parameters. Under the terms of these clinical trial agreements, AstraZeneca paid $5,000,000 as of March 31, 2009 upon achieving certain objectives and pays all costs associated with these studies. The Company recognized $195,408 and $411,555, respectively, as revenue in the three and six months ended December 31, 2010 and $121,284 and $243,375, respectively, as revenue in the three months ended December 31, 2009 under these clinical trial sponsored research agreements.
The Company received an up-front payment of $10,000,000 from AstraZeneca on execution of the research collaboration and license agreement. Under the September 2009 amendment the Company was paid an additional $5,000,000 in consideration of reduction of future milestones and royalties and providing specific materials to
AstraZeneca. The Company is now eligible for milestone payments totaling up to $145,250,000, with up to $85,250,000 contingent on development and regulatory milestones and the balance contingent on achievement of sales targets. In addition, the Company will receive royalties on sales of any approved products. AstraZeneca assumed responsibility for product commercialization, product discovery and development costs, with both companies contributing scientific expertise in the research collaboration. The Company provided research services to AstraZeneca through January 2010, the expiration of the research collaboration portion of the research collaboration and license agreement, at a contractual rate per full-time-equivalent employee.
The Company has determined that the license portion of the agreement and research services should be evaluated together as a single unit for purposes of revenue recognition. Accordingly, the aggregate payments of $15,000,000 have been recognized as revenue over the period ended January 2010. For the three and six months ended December 31, 2009, the Company recognized as revenue $6,232,599, and $8,926,736, respectively, related to these aggregate payments. Per-employee compensation from AstraZeneca for research services was recognized as earned at the contractual rate, which approximates the fair value of such services. Revenue recognized for research services for the three and six months ended December 31, 2009 were $929,416 and $1,775,807, respectively. Payments received upon the attainment of substantive milestones are recognized as revenue when earned.
(5) INVESTMENTS AND FAIR VALUE MEASUREMENTS:
The following is a summary of available-for-sale investments:
|
December 31,
|
June 30,
|
|||
|
2010
|
2010
|
|||
|
Cost
|
$ 1,883,928
|
$ 3,323,539
|
||
|
Gross unrealized gains
|
123,279
|
173,658
|
||
|
Gross unrealized losses
|
(54,541)
|
(35,008)
|
||
|
Total available-for-sale investments
|
$ 1,952,666
|
$ 3,462,189
|
||
The fair value of investments and cash equivalents are classified using a hierarchy prioritized based on inputs. Level 1 inputs are quoted prices (unadjusted) in active markets for identical assets or liabilities. Level 2 inputs are quoted prices for similar assets and liabilities in active markets or inputs that are observable for the asset or liability, either directly or indirectly through market corroboration, for substantially the full term of the financial instrument. Level 3 inputs are unobservable inputs based on management’s own assumptions used to measure assets and liabilities at fair value. A financial asset or liability’s classification within the hierarchy is determined based on the lowest level input that is significant to the fair value measurement.
The following table provides the assets carried at fair value:
|
Fair Value
|
Quoted prices in active markets (Level 1)
|
Quoted prices in active markets (Level 2)
|
Quoted prices in active markets (Level 3)
|
|
|
December 31, 2010;
|
||||
|
Money Market Fund
|
$ 1,409,820
|
$ 1,409,820
|
$ -
|
$ -
|
|
Mutual Funds
|
$ 1,952,666
|
$ 1,952,666
|
$ -
|
$ -
|
|
June 30, 2010;
|
||||
|
Money Market Fund
|
$ 4,111,051
|
$ 4,111,051
|
$ -
|
$ -
|
|
Mutual Funds
|
$ 3,462,189
|
$ 3,462,189
|
$ -
|
$ -
|
(6) COMPREHENSIVE LOSS:
Comprehensive loss consists of the following:
|
Three months ended December 31,
|
Six months ended December 31,
|
|||||||
|
2010
|
2009
|
2010
|
2009
|
|||||
|
Net income (loss)
|
$ (1,100,502)
|
$ 4,501,875
|
$ (5,700,822)
|
$ 4,464,810
|
||||
|
Unrealized loss on available-for-sale investments
|
(19,533)
|
(34,949)
|
(9,523))
|
(7,926)
|
||||
|
Comprehensive income (loss)
|
$ (1,120,035)
|
$ 4,466,926
|
$ (5,710,345)
|
$ 4,456,884
|
||||
(7) GRANT REVENUE:
In October 2010, the Company was awarded $977,917 in grants under the Patient Protection and Affordable Care Act of 2010 (PPACA). The grants relate to four of the Company’s projects: melanocortin agonists for sexual dysfunction; melanocortin agonists for obesity and related metabolic syndrome; natriuretic peptide mimetic PL-3994 for acute asthma; and, subcutaneously-delivered natriuretic peptide mimetic PL-3994 for cardiovascular disease. For the three and six months ended December 31, 2010, the Company received and recorded grant revenue of $846,768. The remainder of the grant of $131,149 will be available no later than 30 days after the Company’s fiscal year ending June 30, 2011, provided that the Company incurs appropriate project expenditures.
(8) STOCKHOLDERS’ EQUITY:
Restricted Stock Units – In July 2010, the Company granted 205,000 restricted stock units to its employees under the Company’s 2005 Stock Plan. On September 15, 2010, October 15, 2010 and November 30, 2010, respectively, 99,500, 14,500 and 15,000 shares of common stock vested. The Company is amortizing the grant-date fair value of these restricted stock units of $331,000 over the nine month vesting period ending March 31, 2011. The Company recognized $72,994 and $282,519, respectively, of stock-based compensation expense related to these restricted stock units during the three and six months ended December 31, 2010.
Stock-based compensation costs for the three and six months ended December 31, 2010 for stock options and equity-based instruments issued other than the restricted stock units described above was $54,702 and $155,620, respectively, and $317,576 and $635,108, respectively, for the three and six months ended December 31, 2009.
[BACK COVER]
This prospectus is part of a registration statement we filed with the SEC. You should rely only on the information or representations contained in this prospectus. We have not authorized anyone to provide information other than that provided in this prospectus. We are not making an offer of these securities in any jurisdiction or state where the offer is not permitted. You should not assume that the information in this prospectus is accurate as of any date other than the date on the front of the document.
|
|
PALATIN TECHNOLOGIES, INC.
Units to Purchase
23,000,000 Shares of Common Stock,
Series A Warrants to Purchase up to 2,000,000 Shares of Common Stock and
Series B Warrants to Purchase up to 21,000,000 Shares of Common Stock
Up to 2,000,000 Shares of Common Stock Issuable Upon Exercise of Series A Warrants
PROSPECTUS
Sole Book-Running Manager
Roth Capital Partners
Co-Manager
Madison Williams and Company
February __, 2011
PART II – INFORMATION NOT REQUIRED IN PROSPECTUS
Item 13. Other Expenses of Issuance and Distribution.
The following table sets forth expenses (estimated except for the SEC registration fees) in connection with the offering described in the registration statement:
|
SEC registration fees
|
$2,969
|
|
Costs of printing
|
$5,000
|
|
Legal fees and expenses
|
$100,000
|
|
Accountants fees and expenses
|
$75,000
|
|
Exchange fees
|
$45,000
|
|
Miscellaneous
|
$22,031
|
|
TOTAL
|
$250,000
|
Item 14. Indemnification of Directors and Officers.
Section 102(b)(7) of the Delaware General Corporation Law (DGCL) allows a corporation to provide in its certificate of incorporation for the elimination or limitation of personal liability of a director to the corporation or its stockholders for monetary damages for breach of fiduciary duty as a director, with some exceptions. Article V, Section 3 of our certificate of incorporation provides that to the fullest extent permitted by the DGCL, no director shall be personally liable to us or our stockholders for monetary damages for breach of a fiduciary duty as a director.
Section 145 of the DGCL provides that a corporation may indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative by reason of the fact that he is or was a director, officer, employee or agent of the corporation, or serving at the request of the corporation in similar capacities, against expenses (including attorneys’ fees), judgments, fines, and amounts paid in settlement actually and reasonably incurred by him in connection with such action, suit or proceeding if he acted in good faith and in a manner he reasonably believed to be in or not opposed to the best interests of the corporation, and, with respect to any criminal action or proceeding, had no reasonable cause to believe his conduct was unlawful. In the case of an action or suit by or in the right of the corporation, no indemnification shall be made with respect to any claim, issue or matter as to which such person shall have been adjudged to be liable to the corporation unless and only to the extent that the court having jurisdiction shall determine that such person is fairly and reasonably entitled to indemnity.
Article VI of our certificate of incorporation and Article IX of our bylaws provide that we shall make the indemnification permitted under the DGCL, as summarized above, but only (unless ordered by a court) upon a determination by a majority of a quorum of disinterested directors, by independent legal counsel in a written opinion, or by the stockholders, that the indemnified person has met the applicable standard of conduct.
Article VI of our certificate of incorporation and Article IX of our bylaws further provide that we may advance expenses for defending actions, suits or proceedings upon such terms and conditions as our Board of Directors deems appropriate, and that we may purchase insurance on behalf of indemnified persons whether or not we would have the power to indemnify such persons under Section 145 of the DGCL. We have obtained a directors’ and officers’ liability insurance policy which covers, among other things, certain liabilities arising under the Securities Act of 1933.
We also refer you to Section 7(b) of the underwriting agreement attached to this registration statement as Exhibit 1.01, which provides that the underwriters will indemnify our directors and officers for certain liabilities arising in connection with this offering.
Item 15. Recent Sales of Unregistered Securities.
In the three years preceding the filing of this registration statement, we have sold and issued the following unregistered securities in reliance on the exemptions from the registration statement requirements under Section 4(2) of the Securities Act of 1933, as transactions not involving a public offering:
On August 17, 2009, in connection with the closing of our registered direct offering of common stock and warrants to purchase common stock, we issued to Rodman & Renshaw, LLC or its permitted designees, as partial consideration for its services as placement agent, warrants to purchase up to 47,424 shares of our common stock at
an exercise price of $4.12 per share. The warrants are exercisable at the option of the holder at any time beginning on issuance through and including November 27, 2012.
On March 2, 2010, in connection with the closing of our registered direct offering of common stock and warrants to purchase common stock, we issued to Rodman & Renshaw, LLC or its permitted designees, as partial consideration for its services as placement agent, warrants to purchase up to 48,148 shares of our common stock at an exercise price of $3.37 per share. The warrants are exercisable at the option of the holder at any time beginning on issuance through and including November 26, 2012.
On June 29, 2010, in connection with the closing of our registered direct offering of common stock and warrants to purchase common stock, we issued to Rodman & Renshaw, LLC or its permitted designees, as partial consideration for its services as placement agent, warrants to purchase up to 50,000 shares of our common stock at an exercise price of $2.50 per share. The warrants are exercisable at the option of the holder at any time beginning on issuance through and including November 26, 2012.
Item 16. Exhibits and Financial Statement Schedules.
The following exhibits are filed with this registration statement:
|
No.
|
Description
|
|
1.01
|
Underwriting agreement.*
|
|
3.01
|
Restated certificate of incorporation. Incorporated by reference to Exhibit 3.01 of our Form 10-K for the year ended June 30, 2010, filed with the SEC on September 27, 2010.
|
|
3.02
|
Bylaws. Incorporated by reference to Exhibit 3.1 of our Form 10-Q for the quarter ended December 31, 2007, filed with the SEC on February 8, 2008.
|
|
4.01
|
Form of warrant agreement.*
|
|
4.02
|
Form of Series A Warrant certificate.*
|
|
4.03
|
Form of Series B Warrant certificate.*
|
|
4.04
|
Form of underwriter warrant.*
|
|
5.01
|
Opinion of Thompson Hine LLP, counsel to the registrant, regarding legality.*
|
|
10.01
|
1996 Stock Option Plan, as amended. Incorporated by reference to Exhibit 10.01 of our Annual Report on Form 10-K for the year ended June 30, 2009, filed with the SEC on September 28, 2009.+
|
|
10.02
|
Strategic Collaboration Agreement dated as of August 17, 1999, between Palatin and Mallinckrodt, Inc. Incorporated by reference to Exhibit 10.21 of our amended Annual Report on Form 10-KSB/A for the year ended June 30, 1999, filed with the SEC on December 28, 1999.
|
|
10.03
|
Amendment To Strategic Collaboration Agreement dated as of May 13, 2002 between Palatin and Mallinckrodt, Inc. Incorporated by reference to Exhibit 10.1 of our Quarterly Report on Form 10-Q for the quarter ended March 31, 2002, filed with the SEC on May 15, 2002. We have obtained confidential treatment of certain provisions contained in this exhibit. The copy filed as an exhibit omits the information subject to the confidentiality request.
|
|
10.04
|
Amendment to Strategic Collaboration Agreement dated as of October 1, 2005, between Palatin and Mallinckrodt, Inc. Incorporated by reference to Exhibit 10.32 of our Quarterly Report on Form 10-Q for the quarter ended September 30, 2005, filed with the SEC on November 8, 2005. We have requested confidential treatment of certain provisions contained in this exhibit. The copy filed as an exhibit omits the information subject to the confidentiality request.
|
|
10.05
|
Form of Option Certificate (incentive option) under the 2005 Stock Plan. Incorporated by reference to Exhibit 10.1 of our Current Report on Form 8-K, filed with the SEC on September 21, 2005.+
|
|
10.06
|
Form of Incentive Stock Option Agreement – Standard under the 2005 Stock Plan. Incorporated by reference to Exhibit 10.2 of our Current Report on Form 8-K, filed with the SEC on September 21, 2005.+
|
|
10.07
|
Form of Option Certificate (non-qualified option) under the 2005 Stock Plan. Incorporated by reference to Exhibit 10.3 of our Current Report on Form 8-K, filed with the SEC on September 21, 2005.+
|
|
10.08
|
Form of Non-Qualified Stock Option Agreement under the 2005 Stock Plan. Incorporated by reference to Exhibit 10.4 of our Current Report on Form 8-K, filed with the SEC on September 21, 2005.+
|
|
10.09
|
Form of stock purchase agreement for our April 2006 private placement. Incorporated by reference to Exhibit 10.2 of our Current Report on Form 8-K, filed with the SEC on April 12, 2006.
|
|
10.10
|
Research Collaboration and License Agreement dated January 30, 2007, between Palatin and AstraZeneca AB. Incorporated by reference to Exhibit 10.2 of our Quarterly Report on Form 10-Q for the quarter ended December 31, 2006, filed with the SEC on February 8, 2007. We have requested confidential treatment of certain provisions contained in this exhibit. The copy filed as an exhibit omits the information subject to the confidentiality request.
|
|
10.11
|
Palatin Technologies, Inc. 2007 Change in Control Severance Plan. Incorporated by reference to Exhibit 10.4 of our Quarterly Report on Form 10-Q for the quarter ended December 31, 2007, filed with the SEC on February 8, 2008.+
|
|
10.12
|
2005 Stock Plan, as amended effective December 7, 2007, March 10, 2009 and May 13, 2009. Incorporated by reference to Exhibit 10.1 of our Quarterly Report on Form 10-Q for the quarter ended December 31, 2009, filed with the SEC on May 15, 2009.+
|
|
10.13
|
Form of Executive Officer Option Certificate. Incorporated by reference to Exhibit 10.1 of our Quarterly Report on Form 10-Q for the quarter ended March 31, 2008, filed with the SEC on May 14, 2008.+
|
|
10.14
|
Form of Amended Restricted Stock Unit Agreement. Incorporated by reference to Exhibit 10.2 of our Quarterly Report on Form 10-Q for the quarter ended March 31, 2008, filed with the SEC on May 14, 2008.+
|
|
10.15
|
Form of Amended Option Certificate (incentive option) under the 2005 Stock Plan. Incorporated by reference to Exhibit 10.3 of our Quarterly Report on Form 10-Q for the quarter ended March 31, 2008, filed with the SEC on May 14, 2008.+
|
|
10.16
|
First Amendment dated June 27, 2008 to the Research Collaboration and License Agreement between Palatin and AstraZeneca AB. Incorporated by reference to Exhibit 10.28 of our Annual Report on Form 10-K for the year ended June 30, 2008, filed with the SEC on September 29, 2008. We have requested confidential treatment of certain provisions contained in this exhibit. The copy filed as an exhibit omits the information subject to the confidentiality request.
|
|
10.17
|
Second Amendment dated December 5, 2008 to the Research Collaboration and License Agreement between Palatin and AstraZeneca AB. Incorporated by reference to Exhibit 10.2 of our Quarterly Report on Form 10-Q for the quarter ended December 31, 2008, filed with the SEC on February 17, 2009. We have requested confidential treatment of certain provisions contained in this exhibit. The copy filed as an exhibit omits the information subject to the confidentiality request.
|
|
10.18
|
Clinical Trial Sponsored Research Agreement dated December 5, 2008 to the Research Collaboration and License Agreement between Palatin and AstraZeneca AB. Incorporated by reference to Exhibit 10.3 of our Quarterly Report on Form 10-Q for the quarter ended December 31, 2008, filed with the SEC on February 17, 2009. We have requested confidential treatment of certain provisions contained in this exhibit. The copy filed as an exhibit omits the information subject to the confidentiality request.
|
|
10.19
|
Form of Executive Officer Restricted Stock Unit Agreement. Incorporated by reference to Exhibit 10.19 of our Form 10-K for the year ended June 30, 2010, filed with the SEC on September 27, 2010.+
|
|
10.20
|
Form of securities purchase agreement for our August 2009 registered direct offering. Incorporated by reference to Exhibit 10.1 of our Current Report on Form 8-K, filed with the SEC on August 13, 2009.
|
|
10.21
|
Form of securities purchase agreement for our February 2010 registered direct offering. Incorporated by reference to Exhibit 10.1 of our Current Report on Form 8-K, filed with the SEC on March 1, 2010.
|
|
10.22
|
Form of securities purchase agreement for our June 2010 registered direct offering. Incorporated by reference to Exhibit 10.1 of our Current Report on Form 8-K, filed with the SEC on June 28, 2010.
|
|
10.23
|
Employment Agreement effective as of July 1, 2010 between Palatin and Carl Spana. Incorporated by reference to Exhibit 10.23 of our Form 10-K for the year ended June 30, 2010, filed with the SEC on September 27, 2010.+
|
|
10.24
|
Employment Agreement effective as of July 1, 2010 between Palatin and Stephen T. Wills. Incorporated by reference to Exhibit 10.24 of our Form 10-K for the year ended June 30, 2010, filed with the SEC on September 27, 2010.+
|
|
10.25
|
Employment Agreement effective as of July 1, 2010, between Palatin and Trevor Hallam. Incorporated by reference to Exhibit 10.25 of our Form 10-K for the year ended June 30, 2010, filed with the SEC on September 27, 2010.+
|
|
10.26
|
Third Amendment dated September 24, 2009 to the Research Collaboration and License Agreement between Palatin and AstraZeneca AB. Incorporated by reference to Exhibit 10.1 of our Quarterly Report on Form 10-Q for the quarter ended September 30, 2009, filed with the SEC on November 13, 2009. We have requested confidential treatment of certain provisions contained in this exhibit. The copy filed as an exhibit omits the information subject to the confidentiality request.
|
|
10.27
|
Separation Agreement, Waiver and Release by and between Palatin and Trevor Hallam, dated November 15, 2010. Incorporated by reference to Exhibit 10.1 of our Current Report on Form 8-K, filed with the SEC on November 19, 2010.+
|
|
21
|
Subsidiaries of the registrant. Incorporated by reference to Exhibit 21 of our Form 10-K for the year ended June 30, 2010, filed with the SEC on September 27, 2010.
|
|
23.01
|
Consent of Thompson Hine LLP, included in Exhibit 5.01.*
|
|
23.02
|
Consent of KPMG LLP, independent registered public accounting firm.**
|
|
24.01
|
Power of attorney. Previously filed.
|
*Previously filed.
**Filed with this registration statement.
+Management contract or compensatory plan or arrangement.
Item 17. Undertakings.
(a) The undersigned registrant hereby undertakes:
(1) To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement:
i. To include any prospectus required by Section 10(a)(3) of the Securities Act of 1933;
ii. To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the Securities and Exchange Commission (the “Commission”) pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than 20 percent change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement;
iii. To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement;
provided, however, that paragraphs (a)(1)(i), (a)(1)(ii) and (a)(1)(iii) of this section do not apply if the registration statement is on Form S-3 and the information required to be included in a post-effective amendment by those paragraphs is contained in reports filed with or furnished to the Commission by the registrant pursuant to Section 13 or Section 15(d) of the Exchange Act that are incorporated by reference in the registration statement, or is contained in a form of prospectus filed pursuant to Rule 424(b) that is part of the registration statement.
(2) That, for the purpose of determining any liability under the Securities Act of 1933, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
(3) To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering.
(4) That, for the purpose of determining liability under the Securities Act of 1933 in a primary offering of securities of the undersigned registrant pursuant to this registration statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the undersigned registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser:
i. Any preliminary prospectus or prospectus of the undersigned registrant relating to the offering required to be filed pursuant to Rule 424;
ii. Any free writing prospectus relating to the offering prepared by or on behalf of the undersigned registrant or used or referred to by the undersigned registrant;
iii. The portion of any other free writing prospectus relating to the offering containing material information about the undersigned registrant or its securities provided by or on behalf of the undersigned registrant; and
iv. Any other communication that is an offer in the offering made by the undersigned registrant to the purchaser.
(b) Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Commission such indemnification is against public policy as expressed in the Securities Act of 1933 and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate
jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act of 1933 and will be governed by the final adjudication of such issue.
(c) If the registrant is subject to Rule 430C, each prospectus filed pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A, shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use.
(d) The undersigned registrant hereby undertakes that:
(1) For purposes of determining any liability under the Securities Act of 1933, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act of 1933 shall be deemed to be part of this registration statement as of the time it was declared effective.
(2) For the purpose of determining any liability under the Securities Act of 1933, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, the registrant has duly caused this Amendment No. 1 to registration statement on Form S-1 to be signed on its behalf by the undersigned, thereunto duly authorized, in the Township of Cranbury, State of New Jersey, on February 23, 2011.
|
PALATIN TECHNOLOGIES, INC.
|
|
|
By:
|
/s/ CARL SPANA
Carl Spana, Ph.D.
President and Chief Executive Officer
|
Pursuant to the requirements of the Securities Act of 1933, this Amendment No. 1 to registration statement has been signed by the following persons in the capacities and on the dates indicated.
|
Signature
|
Title
|
Date
|
|
|
/s/ CARL SPANA
|
President, Chief Executive Officer and Director
|
February 23, 2011
|
|
|
Carl Spana
|
(principal executive officer)
|
||
|
/s/ STEPHEN T. WILLS
|
Executive Vice President and Chief Financial Officer
|
February 23, 2011
|
|
|
Stephen T. Wills
|
(principal financial and accounting officer)
|
||
|
*
|
Chairman and Director
|
February 23, 2011
|
|
|
John K.A. Prendergast
|
|||
|
*
|
Director
|
February 23, 2011
|
|
|
Perry B. Molinoff
|
|||
|
*
|
Director
|
February 23, 2011
|
|
|
Robert K. deVeer, Jr.
|
|||
|
*
|
Director
|
February 23, 2011
|
|
|
Zola P. Horovitz
|
|||
|
*
|
Director
|
February 23, 2011
|
|
|
Robert I. Taber
|
|||
|
*
|
Director
|
February 23, 2011
|
|
|
J. Stanley Hull
|
EXHIBIT INDEX
|
No.
|
Description
|
|
1.01
|
Underwriting agreement.*
|
|
3.01
|
Restated certificate of incorporation. Incorporated by reference to Exhibit 3.01 of our Form 10-K for the year ended June 30, 2010, filed with the SEC on September 27, 2010.
|
|
3.02
|
Bylaws. Incorporated by reference to Exhibit 3.1 of our Form 10-Q for the quarter ended December 31, 2007, filed with the SEC on February 8, 2008.
|
|
4.01
|
Form of warrant agreement.*
|
|
4.02
|
Form of Series A Warrant certificate.*
|
|
4.03
|
Form of Series B Warrant certificate.*
|
|
4.04
|
Form of underwriter warrant.*
|
|
5.01
|
Opinion of Thompson Hine LLP, counsel to the registrant, regarding legality.*
|
|
10.01
|
1996 Stock Option Plan, as amended. Incorporated by reference to Exhibit 10.01 of our Annual Report on Form 10-K for the year ended June 30, 2009, filed with the SEC on September 28, 2009.+
|
|
10.02
|
Strategic Collaboration Agreement dated as of August 17, 1999, between Palatin and Mallinckrodt, Inc. Incorporated by reference to Exhibit 10.21 of our amended Annual Report on Form 10-KSB/A for the year ended June 30, 1999, filed with the SEC on December 28, 1999.
|
|
10.03
|
Amendment To Strategic Collaboration Agreement dated as of May 13, 2002 between Palatin and Mallinckrodt, Inc. Incorporated by reference to Exhibit 10.1 of our Quarterly Report on Form 10-Q for the quarter ended March 31, 2002, filed with the SEC on May 15, 2002. We have obtained confidential treatment of certain provisions contained in this exhibit. The copy filed as an exhibit omits the information subject to the confidentiality request.
|
|
10.04
|
Amendment to Strategic Collaboration Agreement dated as of October 1, 2005, between Palatin and Mallinckrodt, Inc. Incorporated by reference to Exhibit 10.32 of our Quarterly Report on Form 10-Q for the quarter ended September 30, 2005, filed with the SEC on November 8, 2005. We have requested confidential treatment of certain provisions contained in this exhibit. The copy filed as an exhibit omits the information subject to the confidentiality request.
|
|
10.05
|
Form of Option Certificate (incentive option) under the 2005 Stock Plan. Incorporated by reference to Exhibit 10.1 of our Current Report on Form 8-K, filed with the SEC on September 21, 2005.+
|
|
10.06
|
Form of Incentive Stock Option Agreement – Standard under the 2005 Stock Plan. Incorporated by reference to Exhibit 10.2 of our Current Report on Form 8-K, filed with the SEC on September 21, 2005.+
|
|
10.07
|
Form of Option Certificate (non-qualified option) under the 2005 Stock Plan. Incorporated by reference to Exhibit 10.3 of our Current Report on Form 8-K, filed with the SEC on September 21, 2005.+
|
|
10.08
|
Form of Non-Qualified Stock Option Agreement under the 2005 Stock Plan. Incorporated by reference to Exhibit 10.4 of our Current Report on Form 8-K, filed with the SEC on September 21, 2005.+
|
|
10.09
|
Form of stock purchase agreement for our April 2006 private placement. Incorporated by reference to Exhibit 10.2 of our Current Report on Form 8-K, filed with the SEC on April 12, 2006.
|
|
10.10
|
Research Collaboration and License Agreement dated January 30, 2007, between Palatin and AstraZeneca AB. Incorporated by reference to Exhibit 10.2 of our Quarterly Report on Form 10-Q for the quarter ended December 31, 2006, filed with the SEC on February 8, 2007. We have requested confidential treatment of certain provisions contained in this exhibit. The copy filed as an exhibit omits the information subject to the confidentiality request.
|
|
10.11
|
Palatin Technologies, Inc. 2007 Change in Control Severance Plan. Incorporated by reference to Exhibit 10.4 of our Quarterly Report on Form 10-Q for the quarter ended December 31, 2007, filed with the SEC on February 8, 2008.+
|
|
10.12
|
2005 Stock Plan, as amended effective December 7, 2007, March 10, 2009 and May 13, 2009. Incorporated by reference to Exhibit 10.1 of our Quarterly Report on Form 10-Q for the quarter ended December 31, 2009, filed with the SEC on May 15, 2009.+
|
|
10.13
|
Form of Executive Officer Option Certificate. Incorporated by reference to Exhibit 10.1 of our Quarterly Report on Form 10-Q for the quarter ended March 31, 2008, filed with the SEC on May 14, 2008.+
|
|
10.14
|
Form of Amended Restricted Stock Unit Agreement. Incorporated by reference to Exhibit 10.2 of our Quarterly Report on Form 10-Q for the quarter ended March 31, 2008, filed with the SEC on May 14, 2008.+
|
|
10.15
|
Form of Amended Option Certificate (incentive option) under the 2005 Stock Plan. Incorporated by reference to Exhibit 10.3 of our Quarterly Report on Form 10-Q for the quarter ended March 31, 2008, filed with the SEC on May 14, 2008.+
|
|
10.16
|
First Amendment dated June 27, 2008 to the Research Collaboration and License Agreement between Palatin and AstraZeneca AB. Incorporated by reference to Exhibit 10.28 of our Annual Report on Form 10-K for the year ended June 30, 2008, filed with the SEC on September 29, 2008. We have requested confidential treatment of certain provisions contained in this exhibit. The copy filed as an exhibit omits the information subject to the confidentiality request.
|
|
10.17
|
Second Amendment dated December 5, 2008 to the Research Collaboration and License Agreement between Palatin and AstraZeneca AB. Incorporated by reference to Exhibit 10.2 of our Quarterly Report on Form 10-Q for the quarter ended December 31, 2008, filed with the SEC on February 17, 2009. We have requested confidential treatment of certain provisions contained in this exhibit. The copy filed as an exhibit omits the information subject to the confidentiality request.
|
|
10.18
|
Clinical Trial Sponsored Research Agreement dated December 5, 2008 to the Research Collaboration and License Agreement between Palatin and AstraZeneca AB. Incorporated by reference to Exhibit 10.3 of our Quarterly Report on Form 10-Q for the quarter ended December 31, 2008, filed with the SEC on February 17, 2009. We have requested confidential treatment of certain provisions contained in this exhibit. The copy filed as an exhibit omits the information subject to the confidentiality request.
|
|
10.19
|
Form of Executive Officer Restricted Stock Unit Agreement. Incorporated by reference to Exhibit 10.19 of our Form 10-K for the year ended June 30, 2010, filed with the SEC on September 27, 2010.+
|
|
10.20
|
Form of securities purchase agreement for our August 2009 registered direct offering. Incorporated by reference to Exhibit 10.1 of our Current Report on Form 8-K, filed with the SEC on August 13, 2009.
|
|
10.21
|
Form of securities purchase agreement for our February 2010 registered direct offering. Incorporated by reference to Exhibit 10.1 of our Current Report on Form 8-K, filed with the SEC on March 1, 2010.
|
|
10.22
|
Form of securities purchase agreement for our June 2010 registered direct offering. Incorporated by reference to Exhibit 10.1 of our Current Report on Form 8-K, filed with the SEC on June 28, 2010.
|
|
10.23
|
Employment Agreement effective as of July 1, 2010 between Palatin and Carl Spana. Incorporated by reference to Exhibit 10.23 of our Form 10-K for the year ended June 30, 2010, filed with the SEC on September 27, 2010.+
|
|
10.24
|
Employment Agreement effective as of July 1, 2010 between Palatin and Stephen T. Wills. Incorporated by reference to Exhibit 10.24 of our Form 10-K for the year ended June 30, 2010, filed with the SEC on September 27, 2010.+
|
|
10.25
|
Employment Agreement effective as of July 1, 2010, between Palatin and Trevor Hallam. Incorporated by reference to Exhibit 10.25 of our Form 10-K for the year ended June 30, 2010, filed with the SEC on September 27, 2010.+
|
|
10.26
|
Third Amendment dated September 24, 2009 to the Research Collaboration and License Agreement between Palatin and AstraZeneca AB. Incorporated by reference to Exhibit 10.1 of our Quarterly Report on Form 10-Q for the quarter ended September 30, 2009, filed with the SEC on November 13, 2009. We have requested confidential treatment of certain provisions contained in this exhibit. The copy filed as an exhibit omits the information subject to the confidentiality request.
|
|
10.27
|
Separation Agreement, Waiver and Release by and between Palatin and Trevor Hallam, dated November 15, 2010. Incorporated by reference to Exhibit 10.1 of our Current Report on Form 8-K, filed with the SEC on November 19, 2010.+
|
|
21
|
Subsidiaries of the registrant. Incorporated by reference to Exhibit 21 of our Form 10-K for the year ended June 30, 2010, filed with the SEC on September 27, 2010.
|
|
23.01
|
Consent of Thompson Hine LLP, included in Exhibit 5.01.*
|
|
23.02
|
Consent of KPMG LLP, independent registered public accounting firm.**
|
|
24.01
|
Power of attorney. Previously filed.
|
*Previously filed.
**Filed with this registration statement.
+Management contract or compensatory plan or arrangement.