Attached files
| file | filename |
|---|---|
| 8-K - 8-K - Innoviva, Inc. | a11-3522_18k.htm |
Exhibit 99.1
|
|
® ® 29th Annual J.P. Morgan Healthcare Conference Rick E Winningham January 11, 2011 |

|
|
Safe Harbor This presentation contains certain “forward-looking” statements as that term is defined in the Private Securities Litigation Reform Act of 1995 regarding, among other things, statements relating to goals, plans, objectives and future events. Theravance intends such forward-looking statements to be covered by the safe harbor provisions for forward-looking statements contained in Section 21E of the Exchange Act and the Private Securities Litigation Reform Act of 1995. The words “may”, “will”, “should”, “could”, “would”, “plan”, “anticipate”, “believe”, “estimate”, “intend”, “goal,” “project”, “potential”, “expect”, “consistent”, “supportive”, “target” and “promising” and similar expressions are intended to identify such forward-looking statements. Examples of such statements include statements relating to the goals and timing of clinical studies and product commercialization, statements regarding the potential benefits and mechanisms of action of drug candidates, statements concerning the timing of seeking regulatory approval of our product candidates, statements concerning enabling capabilities of Theravance's approach to drug discovery and its proprietary insights, statements concerning expectations for product candidates through development and commercialization and projections of revenue, expenses and other financial items. These statements are based on the current estimates and assumptions of the management of Theravance as of the date of this presentation and are subject to risks, uncertainties, changes in circumstances, assumptions and other factors that may cause the actual results of Theravance to be materially different from those reflected in its forward-looking statements. Important factors that could cause actual results to differ materially from those indicated by such forward-looking statements include, among others, risks related to delays or difficulties in commencing or completing clinical studies, risks related to the potential that results of clinical or preclinical studies indicate product candidates are unsafe or ineffective, our dependence on third parties in the conduct of our clinical studies, delays or failure to achieve regulatory approvals for product candidates, risks of relying on third-party manufacturers for the supply of our product candidates and risks of collaborating with third parties to develop and commercialize products. These and other risks are described in greater detail under the heading "Risk Factors" contained in Theravance’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission (SEC) on October 29, 2010 and the risks discussed in our other period filings with SEC. Given these uncertainties, you should not place undue reliance on these forward-looking statements. Theravance assumes no obligation to update its forward-looking statements. |

|
|
Theravance — Advancing Key Programs RELOVAIR is a trademark of Glaxo Group Limited and mark is intended for U.S. and subject to FDA approval. VIBATIV is a trademark of Astellas Pharma Inc. For full Prescribing Information and Medication Guide for VIBATIVTM please visit www.VIBATIV.com. PµMA MABA RELOVAIR™ In collaboration with GlaxoSmithKline Targeted to be next-generation combination LABA+ICS Large global Phase 3 programs in COPD and asthma ongoing In collaboration with GlaxoSmithKline Muscarinic antagonist and ß2-agonist pharmacology in a single molecule Phase 2b COPD study of GSK961081 initiated in December 2010 Targeted to be a once-daily, orally-administered therapy for OIC TD-1211 achieved positive Phase 2 Proof-of-Concept Plan to progress into further Phase 2 dose-ranging in 2011 Diverse Product Pipeline VIBATIV™ (telavancin) approved in U.S. and Canada for cSSSI Targeting “best-in-class” medicines in respiratory, bacterial infections, gastrointestinal disease, pain & cognitive disorders |

|
|
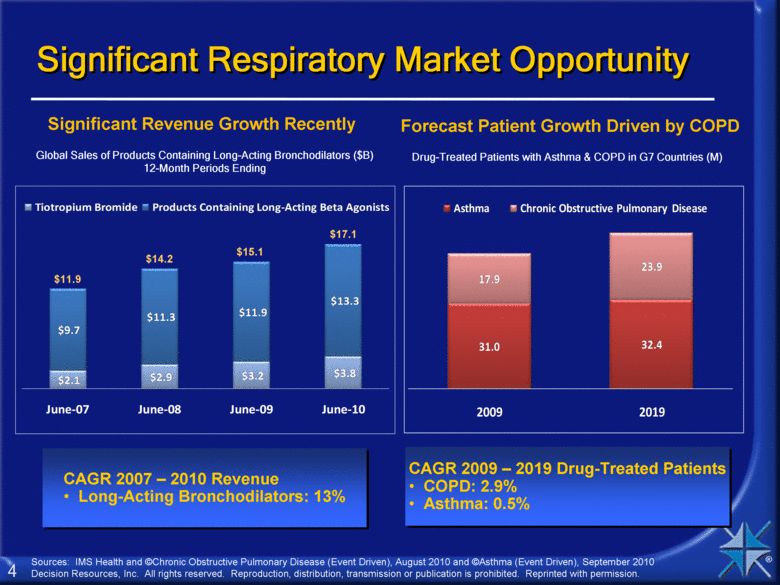
Significant Respiratory Market Opportunity Forecast Patient Growth Driven by COPD Significant Revenue Growth Recently CAGR 2007 – 2010 Revenue Long-Acting Bronchodilators: 13% Drug-Treated Patients with Asthma & COPD in G7 Countries (M) CAGR 2009 – 2019 Drug-Treated Patients COPD: 2.9% Asthma: 0.5% Global Sales of Products Containing Long-Acting Bronchodilators ($B) 12-Month Periods Ending $17.1 $15.1 $14.2 $11.9 Sources: IMS Health and ©Chronic Obstructive Pulmonary Disease (Event Driven), August 2010 and ©Asthma (Event Driven), September 2010 Decision Resources, Inc. All rights reserved. Reproduction, distribution, transmission or publication is prohibited. Reprinted with permission. $2.1 $2.9 $3.2 $3.8 $9.7 $11.3 $11.9 $13.3 June - 07 June - 08 June - 09 June - 10 Tiotropium Bromide Products Containing Long-Acting Beta Agonists 31.0 32.4 17.9 23.9 2009 2019 Asthma Chronic Obstructive Pulmonary Disease |
|
|
RELOVAIR™ with GSK Goal: Once-Daily LABA + ICS for COPD & Asthma Targeted to be next-generation combination COPD/asthma products Currently Seretide/Advair® is GSK’s largest product – ~£5.0B/~$7.8B in ‘09 ~50% COPD ~50% Asthma Initiated Phase 3 program in asthma in March ‘10 and in COPD in October ‘09 13 Phase 3 studies are progressing with the novel once-daily LABA, vilanterol trifenatate (VI), and the novel once-daily ICS, fluticasone furoate (FF) 5 studies in COPD 8 studies in Asthma Theravance would receive royalties of 15% on first $3B of annual net sales and 5% thereafter for approved LABA and LABA+ICS Theravance has no cost obligation through NDA/MAA RELOVAIR is a trademark of Glaxo Group Limited. Mark intended for U.S. and subject to FDA approval. Positive Phase 2b results in 3,000 COPD and asthma patients |

|
|
RELOVAIR™ Phase 3 Programs in COPD and Asthma Status of 13 Registrational Studies Phase 3a Study Status COPD Studies 12-month exacerbation study Enrollment complete; study ongoing 12-month exacerbation study Enrollment complete; study ongoing 6-month efficacy and safety study Enrollment complete; study ongoing 6-month efficacy and safety study Enrollment complete; study ongoing Detailed lung function profile study Completed Asthma Studies Exacerbation study Enrollment complete; study ongoing 12-month safety study Enrollment complete; study ongoing Hypothalamic-Pituitary-Adrenal (HPA) axis study Completed 24-week head-to-head study of RELOVAIR™ vs. Advair®/Seretide Recruiting 24-week high-dose combination efficacy study Recruiting 12-week low-dose combination efficacy study Recruiting 24-week fluticasone furoate vs. fluticasone propionate Recruiting 12-week vilanterol trifenatate vs. salmeterol Recruiting RELOVAIR™ Phase 3 programs in COPD and asthma have now enrolled over 9,000 patients RELOVAIR is a trademark of Glaxo Group Limited. Mark intended for U.S. and subject to FDA approval. |

|
|
MABA Advanced to Phase 2b Dual Pharmacology in a Single Molecule Muscarinic antagonist and ß2-agonist pharmacology in a single molecule Potential for triple therapy with an inhaled corticosteroid (ICS) in a single inhaler for the treatment of COPD Discovered through Theravance’s insights on secondary binding clefts on both the ß2 and muscarinic receptors Strategic Alliance with GSK Theravance has no cost obligation on the program Potential milestone payments Single-agent average percentage royalty rate low double-digits to mid-teens Positive topline Phase 2a clinical results with GSK961081 (‘081) |

|
|
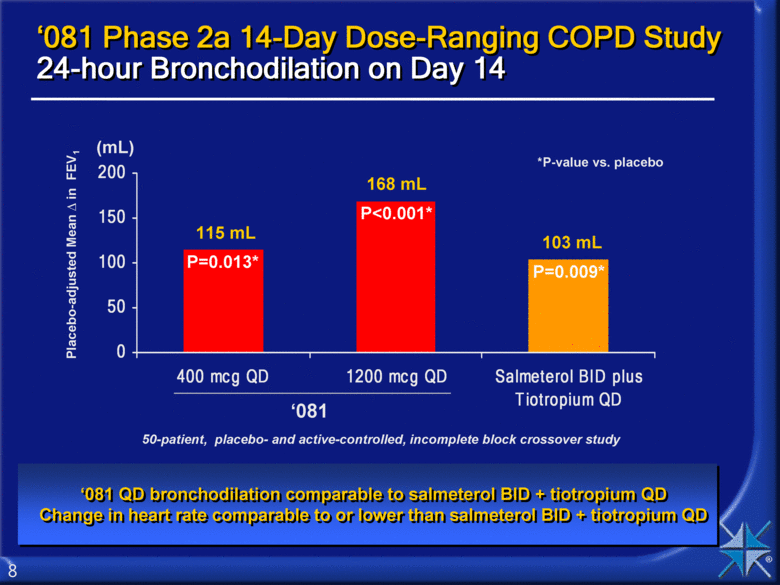
Placebo-adjusted Mean in FEV1 ‘081 ‘081 Phase 2a 14-Day Dose-Ranging COPD Study 24-hour Bronchodilation on Day 14 ‘081 QD bronchodilation comparable to salmeterol BID + tiotropium QD Change in heart rate comparable to or lower than salmeterol BID + tiotropium QD (mL) 115 mL 168 mL 103 mL *P-value vs. placebo P=0.013* P<0.001* P=0.009* 50-patient, placebo- and active-controlled, incomplete block crossover study 0 50 100 150 200 400 mcg QD 1200 mcg QD Salmeterol BID plus Tiotropium QD |
|
|
MABA Phase 2b Study in COPD Goal and Design Goal: To evaluate the dose response, dose interval, safety and efficacy of GSK961081 (‘081) administered QD and BID in COPD patients Design Multicenter, randomized, double-blind, double-dummy, parallel-group, placebo- and active-controlled 4-week treatment period Doses/Eight arms ‘081: 100 mcg, 400 mcg, and 800 mcg QD ‘081: 100 mcg, 200 mcg, and 400 mcg BID Salmeterol: 50 mcg BID Placebo 425 randomized patients Efficacy Endpoints Primary: Change from baseline in a.m. trough FEV1 on Day 29 Secondary: Includes serial FEV1 measures and use of albuterol rescue medication |

|
|
TD-1211 for OIC Goal: Once-Daily, Orally-Administered PµMA Targeted to be a best-in-class, once-a-day, orally-administered, peripherally selective, multivalent antagonist of the mu opioid receptor (PµMA), for the treatment of opioid-induced constipation (OIC) Theravance-discovered novel, multivalent compound designed to alleviate gastrointestinal side effects of opioid therapy without affecting analgesia Potential OIC Market Opportunity in U.S. could be >500M treatment days (TD) annually* 3.9B TD on non-injectable opioids* ~40% of patients on chronic opioids have constipation* 45% of OIC patients using laxatives still report <3 BM per week* TD-1211 achieved positive Phase 2 Proof-of-Concept *Sources: Theravance estimate based on IMS Health NPA; Kalso et al (2004) Pain 112:p372, Brown et al (2006) J of Opioid Mgmt 2:3 p137, Bell et al (2009) Pain Med 10:p35 |

|
|
TD-1211 Phase 2 Study in Patients Suffering from Opioid-Induced Constipation Double-blind, placebo-controlled, dose-escalation study, two U.S. sites 70 patients requiring chronic opioid therapy for non-cancer pain < 5 Spontaneous Bowel Movements (SBMs) during 2-week baseline At least one additional symptom of constipation 2-week baseline followed by 2-week treatment period and 1-week follow-up 3 days in unit after first dose (fasted for the first dose) Daily electronic Patient Reported Outcome (ePRO) diary to collect bowel movement (BM), symptom and quality-of-life metrics Primary endpoint: Change from baseline in average number of SBMs per week over 2-week treatment period Pre-Defined Proof-of-Concept: Lower bound of the 95% CI > 1 |

|
|
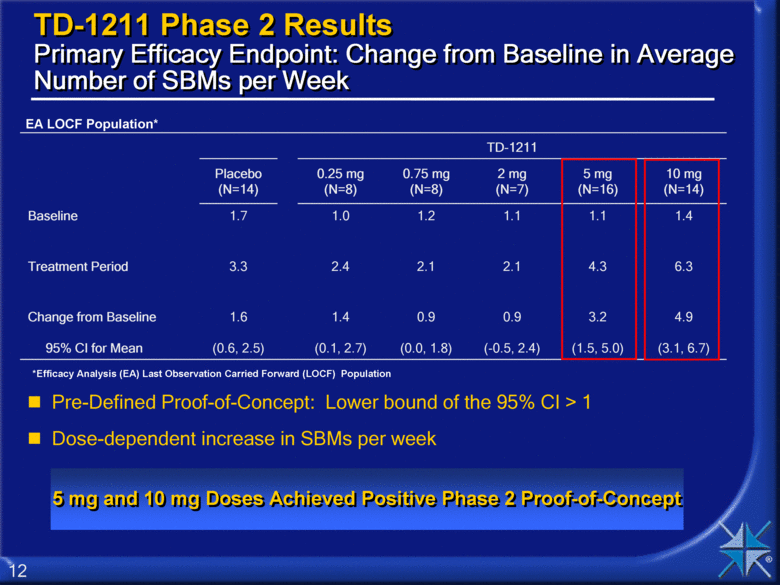
TD-1211 Phase 2 Results Primary Efficacy Endpoint: Change from Baseline in Average Number of SBMs per Week TD-1211 Placebo (N=14) 0.25 mg (N=8) 0.75 mg (N=8) 2 mg (N=7) 5 mg (N=16) 10 mg (N=14) Baseline 1.7 1.0 1.2 1.1 1.1 1.4 Treatment Period 3.3 2.4 2.1 2.1 4.3 6.3 Change from Baseline 95% CI for Mean 1.6 (0.6, 2.5) 1.4 (0.1, 2.7) 0.9 (0.0, 1.8) 0.9 (-0.5, 2.4) 3.2 (1.5, 5.0) 4.9 (3.1, 6.7) Pre-Defined Proof-of-Concept: Lower bound of the 95% CI > 1 Dose-dependent increase in SBMs per week 5 mg and 10 mg Doses Achieved Positive Phase 2 Proof-of-Concept EA LOCF Population* *Efficacy Analysis (EA) Last Observation Carried Forward (LOCF) Population |
|
|
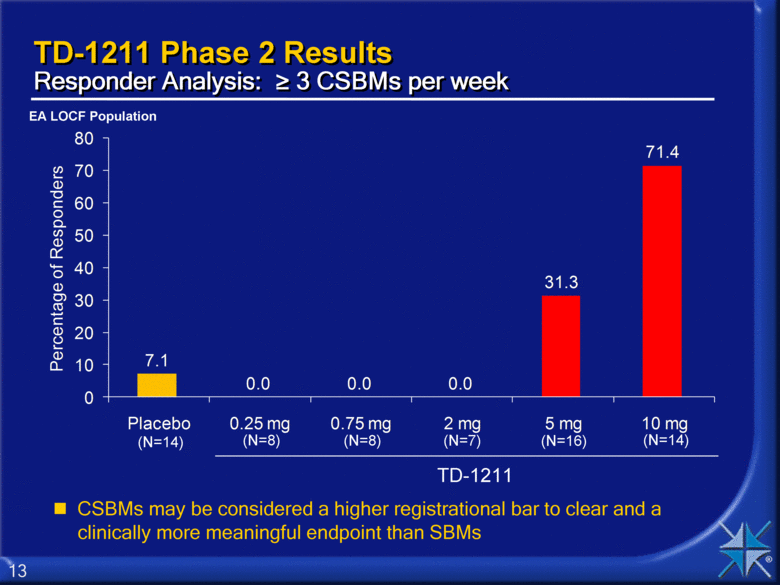
TD-1211 Phase 2 Results Responder Analysis: > 3 CSBMs per week CSBMs may be considered a higher registrational bar to clear and a clinically more meaningful endpoint than SBMs EA LOCF Population TD-1211 (N=8) (N=14) (N=8) (N=7) (N=16) (N=14) 7.1 0.0 0.0 0.0 31.3 71.4 0 10 20 30 40 50 60 70 80 Placebo 0.25 mg 0.75 mg 2 mg 5 mg 10 mg Percentage of Responders |
|
|
TD-1211 Phase 2 Results Safety Summary No Serious Adverse Events (SAEs) reported No evidence of CNS opioid withdrawal or analgesic interference Most adverse events (AEs) were mild/moderate Early onset (Day 1 or Day 2) Majority of GI-related AEs resolved within a few days No clinically significant changes in laboratory tests, ECGs, vital signs, physical exam Theravance retains sole ownership of TD-1211 and intends to progress it into further Phase 2 work in 2011 |

|
|
Launched in the U.S. for the treatment of cSSSI caused by susceptible Gram+ pathogens including Staphylococcus aureus/MRSA Dual mechanism of action Bactericidal, once-daily injectable antibiotic approved for adults Commercialized in partnership with Astellas Phase 3 results in nosocomial pneumonia (NP) published in Clinical Infectious Diseases in December 2010 Recent FDA communications: ATTAIN studies do not meet new NP draft guidance cSSTI and NP under review in Europe For full Prescribing Information, including Boxed Warning and Medication Guide for VIBATIV™, please visit www.VIBATIV.com. VIBATIVTM (telavancin) VIBATIV is a trademark of Astellas Pharma Inc. |

|
|
Financial Position Cash as of September 30, 2010: ~$193M GSK’s investment of ~$129M through a private placement in November ‘10 Projected 2010 expenses Approximately $80M to $85M for R&D + G&A Excludes SFAS 123(R) stock option expenses Access to development funding GSK pays all RELOVAIR™ and MABA program development costs Potential MABA milestone payments from GSK Potential VIBATIV™ milestone payments from Astellas Royalties from sales of VIBATIV™ RELOVAIR is a trademark of Glaxo Group Limited and mark is intended for U.S. and subject to FDA approval. VIBATIV is a trademark of Astellas Pharma Inc. For full Prescribing Information and Medication Guide for VIBATIVTM please visit www.VIBATIV.com. |

|
|
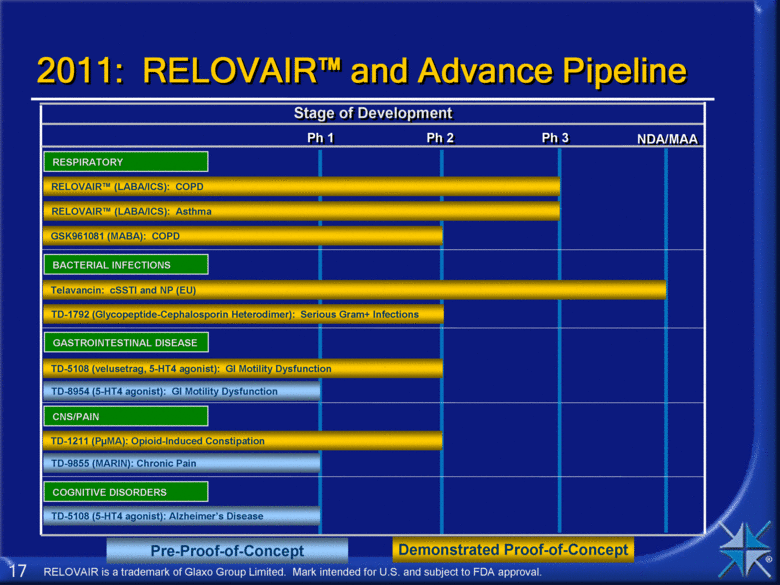
Ph 3 Stage of Development Ph 1 Ph 2 NDA/MAA BACTERIAL INFECTIONS Telavancin: cSSTI and NP (EU) Demonstrated Proof-of-Concept TD-1792 (Glycopeptide-Cephalosporin Heterodimer): Serious Gram+ Infections RESPIRATORY GASTROINTESTINAL DISEASE TD-5108 (velusetrag, 5-HT4 agonist): GI Motility Dysfunction COGNITIVE DISORDERS TD-8954 (5-HT4 agonist): GI Motility Dysfunction TD-1211 (PµMA): Opioid-Induced Constipation GSK961081 (MABA): COPD RELOVAIR™ (LABA/ICS): Asthma 2011: RELOVAIR™ and Advance Pipeline RELOVAIR™ (LABA/ICS): COPD TD-5108 (5-HT4 agonist): Alzheimer’s Disease Pre-Proof-of-Concept RELOVAIR is a trademark of Glaxo Group Limited. Mark intended for U.S. and subject to FDA approval. CNS/PAIN TD-9855 (MARIN): Chronic Pain |
|
|
® ® |

|
|
VIBATIVTM (telavancin) Important Safety Information VIBATIV is a trademark of Astellas Pharma Inc. Fetal Risk Women of childbearing potential should have a serum pregnancy test prior to administration of VIBATIV. Avoid use of VIBATIV during pregnancy unless the potential benefit to the patient outweighs the potential risk to the fetus. Adverse developmental outcomes observed in three animal species at clinically relevant doses raise concerns about potential adverse developmental outcomes in humans. If not already pregnant, women of childbearing potential should use effective contraception during VIBATIV treatment. Nephrotoxicity New onset or worsening renal impairment occurred in patients who received VIBATIV. Renal adverse events were more likely to occur in patients with baseline comorbidities known to predispose patients to kidney dysfunction and in patients who received concomitant medications known to affect kidney function. Monitor renal function in all patients receiving VIBATIV prior to initiation of treatment, during treatment, and at the end of therapy. If renal function decreases, the benefit of continuing VIBATIV versus discontinuing and initiating therapy with an alternative agent should be assessed. Clinical cure rates in telavancin-treated patients were lower in patients with baseline CrCl <50 mL/min compared to those with CrCl >50 mL/min. Consider these data when selecting antibacterial therapy for use in patients with baseline moderate/severe renal impairment. Geriatric Use Telavancin is substantially excreted by the kidney, and the risk of adverse reactions may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection in this age group. Infusion Related Reactions VIBATIV is a lipoglycopeptide antibacterial agent and should be administered over a period of 60 minutes to reduce the risk of infusion-related reactions. Rapid intravenous infusions of the glycopeptide class of antimicrobial agents can cause “Red-man Syndrome”-like reactions including: flushing of the upper body, urticaria, pruritus, or rash. Clostridium difficile-Associated Diarrhea Clostridium difficile-associated diarrhea (CDAD) has been reported with nearly all antibacterial agents and may range in severity from mild diarrhea to fatal colitis. CDAD must be considered in all patients who present with diarrhea following antibiotic use. Development of Drug Resistant Bacteria Prescribing VIBATIV in the absence of a proven or strongly suspected bacterial infection is unlikely to provide benefit to the patient and increases the risk of the development of drug-resistant bacteria. As with other antibacterial drugs, use of VIBATIV may result in overgrowth of nonsusceptible organisms, including fungi. QTc Prolongation Caution is warranted when prescribing VIBATIV to patients taking drugs known to prolong the QT interval. In a study involving healthy volunteers, VIBATIV prolonged the QTc interval. Use of VIBATIV should be avoided in patients with congenital long QT syndrome, known prolongation of the QTc interval, uncompensated heart failure, or severe left ventricular hypertrophy. Coagulation Test Interference VIBATIV does not interfere with coagulation, but does interfere with certain tests used to monitor coagulation such as prothrombin time, international normalized ratio, activated partial thromboplastin time, activated clotting time, and coagulation based factor Xa tests. Blood samples for these coagulation tests should be collected as close as possible prior to a patient’s next dose of VIBATIV. Adverse Reactions The most common adverse reactions (>10% of patients treated with VIBATIV) observed in the Phase III cSSSI clinical trials were taste disturbance, nausea, vomiting, and foamy urine. In the Phase III cSSSI clinical trials, serious adverse events were reported in 7% of patients treated with VIBATIV and most commonly included renal, respiratory, or cardiac events. Serious adverse events were reported in 5% of vancomycin-treated patients, and most commonly included cardiac, respiratory, or infectious events. For full Prescribing Information, including Boxed Warning and Medication Guide, please visit www.VIBATIV.com. |
