Attached files
| file | filename |
|---|---|
| EX-32.1 - EX-32.1 - Astex Pharmaceuticals, Inc | a10-22648_1ex32d1.htm |
| EX-31.1 - EX-31.1 - Astex Pharmaceuticals, Inc | a10-22648_1ex31d1.htm |
| EX-31.2 - EX-31.2 - Astex Pharmaceuticals, Inc | a10-22648_1ex31d2.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K/A
(Amendment No. 1)
|
x |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
|
|
|
For the Fiscal Year Ended December 31, 2009 |
|
|
|
|
|
OR |
|
|
|
|
|
o |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission file number 0-27628
SUPERGEN, INC.
(Exact name of registrant as specified in its charter)
|
Delaware |
|
91-1841574 |
|
(State or other jurisdiction |
|
(IRS Employer |
|
of incorporation or organization) |
|
Identification Number) |
|
4140 Dublin Blvd., Suite 200, Dublin, CA |
|
94568 |
|
(Address of principal executive offices) |
|
(Zip Code) |
Registrant’s telephone number, including area code: (925) 560-0100
Securities registered pursuant to Section 12(b) of the Act:
|
Title of each class: |
|
Name of each exchange on which registered: |
|
Common Stock, $0.001 par value per share |
|
The NASDAQ Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports) and (2) has been subject to such filing requirements for the past 90 days. Yes x No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes o No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the Registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. o
Indicate by check mark whether registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
|
Large accelerated filer o |
|
Accelerated filer x |
|
|
|
|
|
Non-accelerated filer o |
|
Smaller reporting company o |
|
(do not check if a smaller reporting company) |
|
|
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes o No x.
The aggregate market value of the voting stock held by non-affiliates of the Registrant (based on the closing sale price of the Common Stock as reported on the Nasdaq Stock Market on June 30, 2009, the last business day of the Registrant’s most recently completed second fiscal quarter) was approximately $117,441,154. This determination of affiliate status is not necessarily a conclusive determination for other purposes. The number of outstanding shares of the Registrant’s Common Stock as of the close of business on March 5, 2010 was 60,215,632.
DOCUMENTS INCORPORATED BY REFERENCE
Items 10, 11, 12, 13 and 14 of Part III incorporate by reference information from the definitive proxy statement for the Registrant’s Annual Meeting of Stockholders.
EXPLANATORY NOTE
SuperGen, Inc. (“SuperGen,” the “Company,” “we,” “us,” or “our”) is filing this Amendment No. 1 on Form 10-K/A (“Amendment”) to its Annual Report on Form 10-K for the year ended December 31, 2009, as filed with the Securities and Exchange Commission on March 15, 2010 (“Form 10-K”).
We are filing this Amendment to re-file the complete text of those items for which we have included supplemental information, as follows:
· Part I, Item 1, Business — our “Patents and Proprietary Technology” section has been supplemented with additional information about our patents;
· Part II, Item 7, Management’s Discussion and Analysis of Financial Condition and Results of Operations — our “Results of Operations” section has been supplemented with additional information about our research and development expenses;
· Part II, Item 9A, Controls and Procedures, has been modified to include additional language;
· Part III, Item 11, Executive Compensation, has been modified to include supplemental information in the Compensation Discussion and Analysis section, as well as a revised Summary Compensation Table.
This Amendment contains currently dated certifications of our Chief Executive Officer and Chief Financial Officer.
This Amendment does not reflect events occurring after the filing of the Form 10-K, or after the filing of the definitive Proxy Statement for our 2010 Annual Meeting of Stockholders, as filed with the Securities and Exchange Commission on April 30, 2010 (the “Proxy Statement”), and does not update disclosures contained in either the Form 10-K or Proxy Statement, or modify or amend the Form 10-K or Proxy Statement except as specifically described in this explanatory note.
SUPERGEN, INC.
2009 ANNUAL REPORT ON FORM 10-K/A
AMENDMENT NO. 1
|
|
|
Page |
|
|
|
|
|
|
|
|
|
|
|
|
|
1 |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
17 |
|
|
27 |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
29 |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
46 |
||
|
|
|
|
|
S-1 |
||
Special Note Regarding Forward-Looking Statements
Our disclosure and analysis in this report contain forward-looking statements within the meaning of Section 27A of the Securities Act and Section 21E of the Exchange Act, and within the meaning of the Private Securities Litigation Reform Act of 1995. Forward-looking statements provide our current expectations or forecasts of future events. When we use the words “anticipate,” “estimate,” “project,” “intend,” “expect,” “plan,” “believe,” “should,” “likely” and similar expressions, we are making forward-looking statements. In particular, these statements include statements such as: our estimates about profitability; our forecasts regarding our revenues and research and development expenses; and our statements regarding the sufficiency of our cash to meet our operating needs. Our actual results could differ materially from those predicted in the forward-looking statements as a result of risks and uncertainties including, but not limited to, delays and risks associated with conducting and managing our clinical trials; the commercial success of Dacogen; developing products and obtaining regulatory approval; our ability to establish and maintain collaboration relationships; competition; our ability to protect our intellectual property; our expectations about the joint development program with GSK; our dependence on third party suppliers; risks associated with the hiring and loss of key personnel; adverse changes in the specific markets for our products; and our ability to launch and commercialize our products. Certain unknown or immaterial risks and uncertainties can also affect our forward-looking statements. Consequently, no forward-looking statement can be guaranteed and you should not rely on these forward-looking statements.
The forward-looking statements reflect our position as of the date of this report, and we undertake no obligation to update any forward-looking statements, whether as a result of new information, future events or otherwise. You are advised, however, to consult any further disclosures we make on related subjects in our Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, or other filings. Also note that we provide a cautionary discussion of risks and uncertainties relevant to our business under Item 1A—Risk Factors in this report. These are currently known and material risks that we believe could cause our actual results to differ materially from expected and historical results. Other unknown and immaterial risks besides those listed in this report could also adversely affect us.
We incorporated in March 1991 as a California corporation and changed our state of incorporation to Delaware in May 1997. Our executive offices are located at 4140 Dublin Blvd., Suite 200, Dublin, CA, 94568 and our telephone number at that address is (925) 560-0100. We maintain a website on the internet at www.supergen.com. This is a textual reference only. We do not incorporate the information on our website into this annual report on Form 10-K, and you should not consider any information on, or that can be accessed through, our website as part of this annual report on Form 10-K.
Overview
We are a pharmaceutical company dedicated primarily to the discovery and development of therapies to treat patients with cancer. Historically we acquired products that were developed by other companies and applied additional developmental effort to expand sales or advance these products clinically towards potential approval for marketing. In 2006, Dacogen® (decitabine) for Injection received approval for marketing in the United States, our commercial infrastructure and products were sold, and we acquired a discovery and development company to internally discover and develop our own products. These changes were implemented to mitigate the escalating risk of competitive in-licensing and maximize the return on both existing resources and our incoming royalty and milestone revenue.
Our new drug application (“NDA”) for Dacogen was approved by the United States Food and Drug Administration (“FDA”) in May 2006 for the treatment of patients with myelodysplastic syndromes (“MDS”). In August 2004, we had executed an agreement granting MGI PHARMA Inc. (“MGI”) exclusive worldwide rights to the development, manufacture, commercialization and distribution of Dacogen. In July 2006, MGI executed an agreement to sublicense Dacogen to Janssen-Cilag GmbH, a Johnson & Johnson company, granting exclusive development and commercialization rights in all territories outside North America. Janssen-Cilag companies are responsible for conducting regulatory and commercial activities related to Dacogen in all territories outside North America, while MGI retains all commercialization rights and responsibility for all activities in the United States, Canada and Mexico. MGI was acquired by Eisai Corporation of North America in January 2008.
Our current primary developmental efforts revolve around the products progressing out of our acquisition of Montigen Pharmaceuticals, Inc. (“Montigen”), a small-molecule drug discovery company, in 2006. We initiated Phase I in-human clinical trials in June 2007 and initiated Phase Ib clinical trials in late 2007 for the first Montigen product, amuvatinib (MP-470), a DNA repair suppressor. In early 2009, we initiated clinical trials for a second internally developed product, SGI-1776, a PIM kinase inhibitor.
In October 2009, we entered into a Commercial License and Research Agreement with GlaxoSmithKline (“GSK”). Pursuant to the terms of this agreement, we will collaborate with GSK over a period of five years to discover and develop specific epigenetic therapeutics. At the end of the research term, or earlier if GSK elects, GSK may exercise its option to license from us the compounds that are the result of the joint research effort, in order to continue the development and ultimately commercialize and sell the resulting products worldwide. Upon execution of the agreement, we received an upfront payment of $2 million from GSK, as well as a $3 million investment in shares of our common stock, sold at a 10% premium to market price. GSK is obligated to make certain additional payments to us if and when the compounds reach specified developmental milestones, as well as payments to us if and when the compounds that GSK has licensed achieve certain regulatory milestones. The agreement further provides that, if the licensed compounds derived from the joint research team become products, GSK will pay us sales milestone payments as well as royalties on annual net sales of such products. Total potential development and commercialization milestones payable to us could exceed $375 million. The tiered royalties, into double digit magnitudes, will be paid on a country-by-country and product-by-product basis.
Strategy
We are a pharmaceutical company dedicated to the discovery and development of therapies to treat patients with cancer. Our founding strategy was to acquire rights to late stage clinical products and commercialize these products by executing selective developmental and commercialization strategies that might allow these products to come into the market and be utilized by the widest possible patient populations. The competition for late-stage compounds that can be obtained through licensure or acquisition, that have shown initial efficacy in humans, has increased significantly with most major pharmaceutical companies taking positions in this market. The acquisition of Montigen mitigates the competitive risk of in-licensure and may allow us to out-license selective products to our licensing competitors or other pharmaceutical companies. Our primary objective is to become a leading developer and seller of therapies for patients suffering from cancer. Key elements of our strategy include the following:
Discover and advance into clinical trials at least one product about every twelve to eighteen months. Our drug discovery group has been optimizing our proprietary process called CLIMB™ that allows a small team of chemists and biologists to model difficult or previously unknown cancer targets for computerized drug creation and development. The flexibility and relatively low cost of both human and developmental capital for this type of discovery and development has allowed us to transition from being just a licensee to becoming a potential licensor.
Focus on drug targets that are difficult to screen by traditional methods. Most established pharmaceutical companies use some version of a high through-put screening. However, this methodology does not work well for a wide variety of complex targets. Our modeling process has demonstrated an ability to create lead candidates for these complex targets, including protein-to-protein interaction targets that might be disrupted by small molecules to be used as potential therapeutics.
Capitalize on our existing clinical expertise and regulatory development to maximize the commercial value of our products. Computer and animal models are only modestly predictive of how a product might work in humans. We have acquired significant expertise at planning, managing, and filing clinical data in both the United States and Europe. Proving the concept that a specific drug will translate into an approvable, commercially viable product in humans is a difficult task. Some drug candidates demonstrate this “proof of concept” very early in non-clinical development, while other drug candidates might need to be compared clinically to existing therapies to achieve such a proof of concept. Typically this proof of concept comes in Phase II trials where it is demonstrated that a drug candidate can destroy tumors in specific diseases through a specific process. As product candidates move from non-clinical into Phase I and Phase II clinical studies, the potential value of the drug candidates should increase as the proof of concept is achieved. Historically, products that are in Phase II trials command a higher in-licensing value than products that are still in Phase I trials. We believe our clinical and regulatory expertise facilitates efficient use of our resources to achieve appropriate proof of concept.
CLIMB Discovery Process
Traditional drug discovery processes may require five or more years before presenting a candidate suitable for the clinic. This lengthy timeline leads to high research and development costs. Utilizing our CLIMB platform, we have effectively streamlined the discovery and lead optimization process in order to get potentially life-saving therapeutics to the clinical testing stage of development faster and at a lower cost. CLIMB is SuperGen’s approach to small molecule drug discovery, which merges the rapid screening of compound libraries with computational chemistry and systems biology techniques to identify drug leads that bind to target proteins.
CLIMB is an iterative and evolving process, incorporating techniques from computational design to laboratory bench biology and chemistry, to yield targeted therapeutics for use in the clinic. In traditional small molecule screening, very large physical libraries of millions of compounds may be created and screened in order to identify the few that interact selectively with a disease-related protein target. This approach has worked fairly well for simple or very well characterized targets, but is very time and cost intensive. Since CLIMB works with a virtual library of compounds and compound fragments to screen against target models, we can screen up to three million virtual compounds per day against very complex targets.
CLIMB has been used to create models and identify products that have exhibited considerable activity while physically screening as few as several hundred rationally-selected compounds. This reduces the time from target identification to clinical candidate by several years, and decreases the cost of drug development. As part of CLIMB, our software development team is actively involved in the creation of algorithms to integrate computation, biochemistry, medicinal chemistry and systems biology to improve the predictive properties of our models and streamline the drug discovery process even further.
Products in Research and Development
The chart below lists our current products or projects in development:
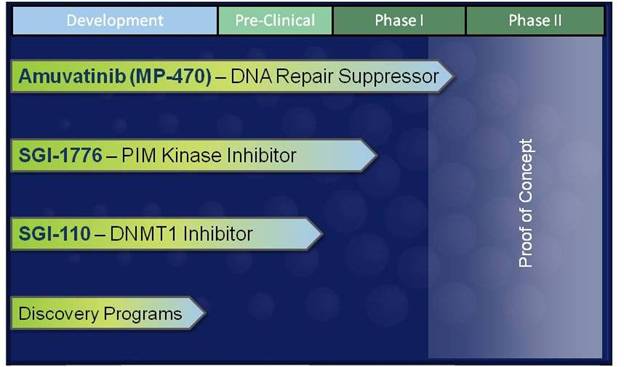
Amuvatinib (MP-470) — DNA Repair Suppressor
Amuvatinib is a multi-targeted Tyrosine Kinase Inhibitor that is specific for mutant forms of c-kit, PDGFRa, and FLT3. These protein kinase targets are involved in the growth and proliferation of cancer cells. Amuvatinib is also a suppressor of Rad51, a DNA repair protein which is involved in resistance to a variety of chemotherapy agents and radiation. We submitted an Investigational New Drug Application (“IND”) to the FDA in March 2007, and initiated a first-in-human Phase I single agent amuvatinib trial in June 2007.
Amuvatinib has a wide therapeutic window and shows minimal toxicity in the expected therapeutic dose range, despite suppressing several signaling pathways within cells. We have evaluated amuvatinib as a dry powder mix and as a lipid suspension formulation in multiple Phase I studies as a single agent in healthy volunteers and in cancer patients, as well as in combination with five standard of care chemotherapy regimens in different tumor types. Across these studies, over 170 patients/healthy volunteers received at least one dose of amuvatinib. As a single agent in cancer patients, gastrointestinal toxicity was the major adverse event noted at doses up to 1500 mg/day with the dry powder formulation. In the combination trial, preliminary data indicated twelve partial responses and numerous durable stable disease per RECIST criteria, mostly with the paclitaxel/carboplatin and carboplatin/etoposide standard of care chemotherapy regimens in combination with oral amuvatinib. Tumor types demonstrating clinical benefit include neuroendocrine, non-small cell lung, small cell lung, breast and endometrial carcinoma. The safety profile of amuvatinib in combination with standard of care was consistent with historical published data for each chemotherapeutic with no apparent increase in severity or prolongation of reported events.
We completed an additional Phase I pharmacokinetic study in healthy male volunteers to determine the relative bioavailability and define the safety profile of the lipid suspension capsules compared with the dry powder mix capsules. In a randomized, two-way crossover study, exposure levels of amuvatinib following administration of a single dose of the lipid suspension formulation (3 x 30 mg capsules) were higher than those observed following the administration of the single dose dry powder formulation (1 x 100 mg capsule). The relative bioavailability favored the lipid suspension formulation, adjusted for dose, with an overall increase in exposure by twofold.
We are also performing non-clinical studies with collaborators to further our understanding of the mechanisms of amuvatinib in DNA repair pathways, as well as the effects of the drug in combination studies in multiple solid tumor models. Pending full analysis of the responses in the Phase I studies and completion of the ongoing non-clinical programs, we intend to initiate one or more Phase II studies evaluating the safety and efficacy of amuvatinib lipid suspension formulation in tumor types that have demonstrated clinical benefit in the Phase I development program. The initial Phase II study is anticipated to be launched during the second half of 2010.
SGI-1776 — Pim Kinase Inhibitor
Pim kinases are proteins that play a pivotal survival role in cancer cells. Over-expression of Pim kinases in cells prevents programmed cell death that normally occurs when cells malfunction and can lead to unchecked cell survival, or cancer. Our Pim kinase inhibitor, SGI-1776, is a novel, orally administered, small molecule anticancer compound that effectively blocks the pro-survival activity of Pim kinases, allowing these potentially malignant cells to self-abort. Our IND for SGI-1776 received clearance from the FDA in November 2008, and we initiated a Phase I clinical trial to evaluate the safety, tolerability, and pharmacokinetic profile of SGI-1776 in the first half of 2009. The first in-human clinical trial program has enrolled patients with solid tumors with specific emphasis on hormone and docetaxel refractory prostate cancer and refractory non-Hodgkin’s lymphomas. These tumor types have been reported to over-express the Pim kinase family of proteins at a high frequency. Over-expression of Pim-1 kinase has been shown to be a marker of poor prognosis in these tumors. A second Phase I/II study is being planned in patients with refractory acute or chronic myeloid or lymphatic leukemias in which Pim kinases are also over-expressed, and correlated with poor prognosis and drug resistance. This study is anticipated to be launched during the first half of 2010.
SGI-110 — DNMT1 Inhibitor
In normal cells, silencing of unnecessary genes is commonly carried out by DNA methylation through the action of DNA Methyltransferase enzymes. However, this machinery can be usurped during the process of tumorigenesis, resulting in the inactivation of tumor suppressor genes and ultimately cancer. Inhibition of DNMT-1 activity in cancer cells causes the suppressed genes to become unmethylated and re-expressed. These re-expressed tumor suppressor genes interfere with the cancer cells proliferative pathways and lead to cell death. We have developed a compound called SGI-110 which targets and blocks the mechanism by which methylation occurs, thus allowing re-expression of tumor suppressor genes in tumors. SGI-110 is currently in the pre-clinical development stage with an anticipated IND filing in 2010. The initial Phase I/II study will evaluate multiple schedules in relapsed/refractory MDS and acute myeloid leukemia (AML).
Discovery Programs
JAK2 Inhibitor
Janus kinases (JAK) are a family of non-receptor intracellular tyrosine kinases that transduce signaling from type I and II cytokine receptors, which possess no catalytic kinase activity, to the signal transducers and activators of transcription (STAT) proteins which translocate into the nucleus to initiate the growth and
differentiation programs associated with the various receptor cytokine complexes. The JAK/STAT pathways play an important role in a diverse array of cellular processes, including cell survival, proliferation, differentiation and apoptosis. Activation of JAK kinases through mutation or aberrant signaling has been associated with disease progression in immune disorders, myeloproliferative disorders, and cancers.
Axl Inhibitor
Axl kinase is a receptor tyrosine kinase implicated in tumorigenesis. Over-expression of Axl is associated with increased cellular transformation, cell survival, proliferation, migration, angiogenesis, and adhesion. The oncogenic potential of Axl was first discovered in chronic myelogenous leukemia (CML) and has been shown to play a role in the development of acute myelogenous leukemia (AML) and myelodysplasia. Axl kinase is an exciting target for small molecule drug discovery. A series of small molecule inhibitors were discovered and are being developed for potency and selectivity against Axl.
ETK/BMX Inhibitors
Epithelial and endothelial tyrosine kinase (ETK), also known as bone marrow X kinase (BMX), is a nonreceptor tyrosine kinase that plays a central role in the proliferation, differentiation, apoptosis, adhesion, motility, and tumorigenicity of epithelial cells. ETK has been reported to be over-expressed in several aggressive metastatic carcinomas, such as hepatocellular carcinoma, cholangiocarcinoma, and prostate cancer. ETK kinase signals downstream of several important oncogenes, including Src, focal adhension kinase, and phosphatidylinositol 3-kinase and has been shown to be vital to the tumorigenic effects of such proteins. Taken together, these findings suggest ETK is an exciting potential therapeutic target in multiple tumor types. We have designed and developed a novel class of potent small molecule inhibitors with specificity toward ETK. Initial leads from this class of inhibitors have activity against ETK at low nM concentrations in a biochemical ETK kinase assay, and show good selectivity across a diverse panel of kinases.
Products Sublicensed or Sold
Dacogen
In September 2004, we executed an agreement granting MGI exclusive worldwide rights to the development, manufacture, commercialization and distribution of Dacogen. Under the terms of the agreement, MGI made a $40 million equity investment in us and agreed to pay up to $45 million in specific regulatory and commercialization milestones. To date, we have received $32.5 million of these milestones. The Dacogen license has also created for us a royalty income stream on worldwide net sales starting at 20% and escalating to a maximum of 30%.
In July 2006, MGI executed an agreement to sublicense Dacogen to Janssen-Cilag, a Johnson & Johnson company, granting exclusive development and commercialization rights in all territories outside North America. In accordance with our license agreement with MGI, we are entitled to receive 50% of certain payments MGI receives as a result of any sublicenses. We received $5 million, or 50% of the $10 million upfront payment MGI received, and, as a result of both the original agreement with MGI and this sublicense with Janssen-Cilag, we may receive up to $17.5 million in future milestone payments as they are achieved for Dacogen globally. Janssen-Cilag is responsible for conducting regulatory and commercial activities related to Dacogen in all territories outside North America, while MGI retains all commercialization rights and responsibility for all activities in the United States, Canada and Mexico. MGI was acquired by Eisai Corporation of North America in January 2008.
Nipent
We used to sell our drug Nipent in the United States and the European Union (“EU”) for the treatment of hairy cell leukemia, a type of B-lymphocytic leukemia, and it was our principal source of revenue from 1997-2006. We sold the North American rights to Nipent to Mayne Pharma (“Mayne”) in August 2006, and sold the remaining worldwide rights to Nipent to Mayne in April 2007. Mayne was acquired by Hospira, Inc. in February 2007.
Acquisition of New Products and Technologies
We are continually reviewing new product development opportunities in an effort to enhance and create a broader product pipeline for future development.
In 2006, we completed our acquisition of Montigen, a privately held, oncology-focused drug discovery and development company headquartered in Salt Lake City, Utah. Montigen’s assets included its research and development team, a proprietary drug discovery technology platform and optimization process, CLIMB, and late-stage non-clinical compounds. Pursuant to the terms of the merger agreement, we paid the Montigen stockholders a total of $17.9 million upon the closing of the transaction, consisting of $9.0 million in cash and $8.9 million in shares of SuperGen common stock. In April 2007, we paid the former Montigen stockholders a milestone payment of approximately $10.0 million, which was paid in shares of our common stock. In November 2008, we paid the former Montigen stockholders another milestone payment of approximately $5.2 million, which was paid in a combination of approximately $2.8 million in cash and 1.5 million shares of our common stock. We have an obligation to pay the Montigen stockholders an additional $6.8 million in shares of our stock, contingent upon achievement of one additional regulatory milestone.
Research and Development
Because of the stage of our development and the nature of our business, we expend significant resources on research and development activities. We expended $29.7 million in 2009, $32.7 million in 2008, and $23.4 million in 2007 on research and development activities. We conduct research internally and also through collaborations with third parties, and we intend to maintain our strong commitment to our research and development efforts in the future. Our major research and development projects are focused on our drug discovery and non-clinical activities as well as Phase I and Phase Ib clinical trials for amuvatinib and SGI-1776.
Sales and Marketing
We currently have no employees focused on sales, marketing, and sales support. Our marketing efforts are handled by our Corporate Communications and Business Development group.
Manufacturing
We currently outsource manufacturing of all our drug compounds to qualified United States and foreign suppliers. We expect to continue to outsource manufacturing in the near term. We believe our current suppliers will be able to efficiently manufacture our proprietary compounds in sufficient quantities and on a timely basis, maintaining product quality and compliance with FDA and foreign regulations. We maintain oversight of the quality of our third-party manufacturers through ongoing audits, rigorous review, control over documented operating procedures, and thorough analytical testing by qualified, contracted laboratories. We believe that our current strategy of outsourcing manufacturing is cost-effective because we avoid the high fixed costs of plant, equipment, and large manufacturing staffs.
The FDA must approve our drug manufacturing sites and deem a manufacturer acceptable under current good manufacturing practices (“GMPs”) before release of active pharmaceutical ingredients (“API”) and finished dosage forms for clinical testing.
We intend to continue evaluating our manufacturing requirements and may establish or acquire our own facilities to manufacture our products for distribution if doing so would be cost effective or improve control and flexibility of product supply.
Government Regulation: New Drug Development and Approval Process
Regulation by governmental authorities in the United States and other countries is a significant factor in the manufacture and marketing of pharmaceuticals and in our ongoing research and development activities. All of our drug products will require regulatory approval by governmental agencies prior to commercialization. In particular, human therapeutic products are subject to rigorous non-clinical testing, clinical trials, and other pre-marketing approval requirements by the FDA and regulatory authorities in other countries. In the United States, various federal, and in some cases state statutes and regulations, also govern or have an impact upon the manufacturing, safety, labeling, storage, record-keeping and marketing of such products. The lengthy process of seeking required approvals and the continuing need for compliance with applicable statutes and regulations require the expenditure of substantial resources. Regulatory approval, when and if obtained, may be limited in scope which may significantly limit the indicated uses for which a product may be marketed. Further, approved drugs, as well as their manufacturers, are subject to ongoing review and inspections which could reveal previously unknown problems with such products, which may result in restrictions on their manufacture, sale or use or in their withdrawal from the market.
The process for new drug approval has three major stages, discovery, non-clinical and clinical:
Drug discovery. In the initial stages of small molecule drug discovery, potential biological targets are identified, these targets are characterized, and then large numbers of potential compounds are screened for activity. This drug discovery process can take several years. Once a company defines a lead compound, the next steps are to conduct further preliminary studies on the mechanism of action, in vitro (test tube) screening against particular disease targets and some in vivo (animal) screening. If results are satisfactory, the compound progresses from discovery to non-clinical development.
Non-clinical testing. During the non-clinical testing stage, laboratory and animal studies are conducted to show biological activity of the compound against the disease target and the compound is evaluated for safety. These tests can take several years to complete and must be conducted in compliance with Good Laboratory Practice (“GLP”) regulations. If the compound passes these hurdles, animal toxicology studies are initiated. If the results demonstrate acceptable levels of toxicity, the compound emerges from non-clinical testing and moves into the clinical phase.
Clinical testing — The Investigational New Drug Application. After appropriate animal testing is evaluated and the candidate molecule is found to have an acceptable safety profile, we may decide to expand the development programs to a clinical setting. To accomplish this in the United States an IND is submitted to the FDA. IND applications include the known chemistry of the compound, how the compound is manufactured, the results of animal studies and other previous experiments, the method by which the drug is expected to work in the human body, a proposed clinical development plan and how, where and by whom the proposed new clinical studies will be conducted. Health authorities in Europe and the rest of the world require a similar clinical trial application. If the controlling authority does not object, we may initiate human testing. All clinical trials must be conducted in accordance with globally-accepted standards of good clinical practices (“GCPs”). This means we have specific obligations to protect trial subjects and potential patients, monitor the study, collect the data and prepare a report of the study. Clinical trial applications must be updated with new information obtained during the course of the trials.
Clinical protocols must be approved by independent reviewers, referred to as Institutional Review Boards (“IRB”) in the United States and Ethical Committees (“EC”) in Europe. The IRB/EC is charged with providing an independent assessment of the appropriateness of the study, particularly focusing on the safety of the patients that might enroll in the study. The IRB’s/EC’s responsibilities continue while the study is ongoing, focusing on protecting the rights and safety of those enrolled in the study.
We have an obligation to provide progress reports on clinical trials at least annually to the FDA. The FDA may, at any time during a clinical trial, impose a “clinical hold” if it has serious safety concerns about a trial. If this occurs, the clinical trial cannot continue until the FDA is satisfied that it is appropriate to proceed.
Clinical Development Plan. Clinical trials are typically conducted in three sequential phases, but the phases may overlap.
· Phase I clinical trials. After an IND becomes effective, Phase I human clinical trials can begin. These trials generally involve 20 to 40 heavily pre-treated cancer patients who may have a wide variety of cancers and typically take approximately one year to complete. These trials are designed to evaluate a drug’s safety profile and may include studies to assess the optimal safe dosage range. Phase I clinical studies may evaluate how a drug is absorbed, distributed, metabolized and excreted from the body. Phase I studies may be expanded to Phase Ib trials that test the research compound in combination with other agents to define the combined safety and dosing parameters.
· Phase II clinical trials. In Phase II clinical trials, studies are conducted in patients who have the specific targeted disease. The primary purpose of these trials is to demonstrate preliminary efficacy of the drug in the target patient population. These studies typically take a few years to complete. Once trial data is obtained that a specific dose and schedule is creating clinical efficacy that appears to be superior to other treatments, advancement to Phase III can begin.
· Phase III clinical trials. These trials are typically large, involving several hundred or even thousands of patients. Phase III trials typically compare an investigational agent against a control product or the standard of care, which could be a product or treatment already approved for use in that disease. The data generated in these studies are monitored regularly by clinical monitors as well as the participating physician. There are specific requirements for the reporting of any adverse reactions that may result from the use of the drug. Clinical monitors visit the sites regularly and transmit the data back to the company for analysis and ultimately for presentation to the FDA.
Marketing application. Companies have the opportunity to interact with health authorities during the course of a drug development program. Most companies take advantage of this access to gain further insights about the kind of data that will be expected in their marketing application. After completion of the clinical trial phase, a company must compile all of the chemistry, manufacturing, non-clinical and clinical data into a marketing application. In the United States, this is called an NDA; in the EU it is called a Marketing Authorization Application (“MAA”). This is a significant amount of information, often in excess of 100,000 pages, and it will be independently reviewed by these health authorities.
Both the FDA and the European Medicines Evaluation Agency (“EMEA”) review these submissions for overall content and completeness before accepting them for review and may request additional information. Once an application is accepted for filing, each agency independently begins its in-depth review. In both the United States and Europe, there are specified timeframes for the completion of review. This process may be extended if an agency requests additional information or clarification regarding the data provided in the submission.
In the United States, the FDA may refer the application to an appropriate advisory committee for a recommendation as to whether the application should be approved, but the FDA is not bound by this recommendation. The review process concludes with the issuance of a “complete response” letter from the FDA. If FDA evaluations of the NDA and the manufacturing facilities are favorable, the FDA will approve the application. If the FDA’s evaluation of the NDA submission or manufacturing facilities is not favorable, the FDA will reject the application in the complete response. This complete response will describe specific deficiencies, and, when possible, will outline recommended actions the applicant might take to get the application ready for approval. When and if any deficiencies are corrected, and actions are completed to the FDA’s satisfaction, the FDA will issue an approval letter authorizing commercialization of the drug for specific indications. The review and approval process in Europe has substantial similarities to that outlined for the United States.
Marketing approval. Once a health authority grants marketing approval for a drug, it can then be made available in that country or region. Periodic safety reports must be submitted to health authorities as a way to monitor the use of new drugs introduced to the market. Regulatory agencies around the world place great emphasis on pharmacovigilance, the process of monitoring the safety of a drug when it is released for general use, as the real world setting is very different from the controlled environment of clinical trials.
Phase IV clinical trials and post marketing studies. In addition to studies that might have been requested by health authorities as a condition of approval, clinical trials may be conducted to generate more information about the drug after initial approval of the product; including use for additional indications, the use of new dosage forms or new dosing regimens. These studies may generate approved label changes and publications that provide further information to patients and the medical community.
Fast Track. The FDA Modernization Act of 1997 specifies that the FDA can assign a fast track designation to a new drug or biologic product that is intended for the treatment of a serious or life-threatening condition and has the potential to address unmet medical needs for such a condition. Under this program, the sponsoring company may request this designation at any time during the development of the product. The FDA must determine whether the product qualifies within 60 days of receipt of the sponsoring company’s request. For a product designated as fast track, the FDA has the ability to define a faster review, including allowing the sponsor to provide the NDA in discrete sections. This process is called a “rolling” NDA and is intended to accelerate the review and approval process.
Priority Review. This is a designation by the FDA for a review period of 6 months, instead of the standard 10 months defined by federal regulation.
Accelerated Approval. This is a program intended to make promising products for life-threatening diseases available on the market on the basis of preliminary evidence prior to formal demonstration of patient benefit.
Approvals in the European Union. In 1993, the EU established a system for the registration of medicinal products in the EU whereby marketing authorization may be submitted at either a centralized or decentralized level. The centralized procedure is administered by the EMEA and is mandatory for the approval of biotechnology products and is available, at the applicant’s option, for other innovative products. The centralized procedure provides for the granting of a single marketing authorization that is valid in all EU member states. A mutual recognition procedure is available at the request of the applicant for all medicinal products that are not subject to the mandatory centralized procedure, under a decentralized procedure.
Approvals outside of the United States and European Union. Applications to market a new drug product must be made to virtually all countries prior to marketing. The approval procedure and the time required for approval vary from country to country and may involve additional testing and cost. There can be no assurance that approvals will be granted on a timely basis or at all. In addition, pricing approval is required in many countries and there can be no assurance that the resulting prices would be sufficient to generate an acceptable return on investment.
Off-Label Use. Drugs are approved for a specific use (“label use”) that is then set forth in the document (“label”) accompanying the dispensed drug. Physicians may prescribe drugs for uses that are not approved in the product’s label. Such “off-label” prescribing may be used by physicians across medical specialties. The FDA does not regulate the behavior of physicians in their choice of treatments but it does limit a manufacturer’s communications on the subject of off-label use. Companies cannot promote FDA approved drugs for off-label uses, nor can companies promote the use of a drug before it is approved.
Other Government Regulations
As a United States-based company, in addition to laws and regulations enforced by the FDA, we are also subject to regulation by other agencies under the Occupational Safety and Health Act, the Environmental Protection Act, the Toxic Substances Control Act, the Resource Conservation and Recovery Act and other present and potential future federal, state or local laws and regulations. These agencies have specialized responsibilities to monitor the controlled use of hazardous materials such as chemicals, viruses and various radioactive compounds.
Market Exclusivity
The commercial success of a product, once it is approved for marketing, will depend primarily on a company’s ability to create and sustain market share and exclusivity. Market exclusivity can be gained and maintained by a number of methods, including, but not limited to: patents, trade secrets, know-how, trademarks, branding and special market exclusivity provided by regulations.
Orphan Drug Designation
The United States, European Union, Japan and Australia have all enacted regulations to encourage the development of drugs intended to treat rare diseases. Orphan drug designation must be requested before submitting an application for marketing approval. After the granting of an orphan drug designation, the chemical identity of the therapeutic agent and its potential treatment use are disclosed publicly. If and when a product with orphan drug status receives marketing approval for the orphan indication, the product is entitled to marketing exclusivity, which means the regulatory authority may not approve any other applications to market the same drug for the same indication for seven years in the United States, ten years in Europe and Japan and four years in Australia.
Data Exclusivity and Generic Copies
There is an abbreviated regulatory review and approval process for a generic copy of an approved innovator drug product. The generic copy can be approved on the basis of an application that is usually limited to manufacturing and biologic equivalence data, by referring to the non-clinical and clinical data that were the bases of approval of the innovator product. The copy can be approved after expiration of relevant patents and any regulatory exclusivity afforded by special circumstances. A new chemical entity has 5 and 10 years of regulatory exclusivity in the United States and European Union, respectively, precluding approval of a generic copy. Additional exclusivity can be afforded in the United States by approval of a product or use that has orphan drug status (7 years), or that requires review of new clinical data (3 years), or that is an expansion of use to a pediatric population (6 months). These exclusivities are independent, and could run sequentially, effectively extending the period of regulatory exclusivity. There is no assurance that such special regulatory exclusivities are applicable for our compounds. Separate from regulatory data exclusivity is the exclusivity conferred by the Hatch Waxman Act based on patent protection of the drug. A company seeking to market a generic might, after the lapse of regulatory data exclusivity, successfully challenge the patent protection of the marketed drug, thereby shortening its exclusive marketing period.
Patents and Proprietary Technology
Patents are very important to us in establishing proprietary rights to the products we develop or license. The patent positions of pharmaceutical and biotechnology companies, including ours, can be uncertain and involve complex legal, scientific, and factual questions. See “Risk Factors—Our ability to protect our intellectual property rights will be critically important to the success of our business, and we may not be able to protect these rights in the United States or abroad.”
We actively pursue patent protection when applicable for our proprietary products and technologies, whether they are developed in-house or acquired from third parties. We attempt to protect our intellectual property position by filing United States and foreign patent applications related to our proprietary technology, inventions and improvements that are important to the development of our business. Importantly, we are prosecuting a number of patent applications directed to various compounds in our pipeline, including those from our Discovery group. Additionally, we have been granted patents and have received patent licenses relating to our proprietary formulation technology, non-oncology and non-core technologies.
There can be no assurance that the patents granted or licensed to us will afford adequate legal protection against competitors or provide significant proprietary protection or competitive advantage. The patents granted or licensed to us could be held invalid or unenforceable by a court, or infringed or circumvented by others. In addition, third parties could also obtain patents that we would need to license or circumvent. Competitors or potential competitors may have filed patent applications or received patents, and may obtain additional patents and proprietary rights relating to proteins, small molecules, compounds, or processes that are competitive with the products we are developing.
In general, we obtain licenses from various parties we deem necessary or desirable for the development, manufacture, use, or sale of our products or product candidates. Some of our proprietary products are dependent upon compliance with other licenses and agreements. These licenses and agreements may require us to make royalty and other payments, to reasonably exploit the underlying technology of applicable patents, and to comply with regulatory filings. If we fail to comply with these and other terms in these licenses and agreements, we could lose the underlying rights to one or more of these potential products, which would adversely affect our product development and harm our business.
We also have patents, licenses to patents and pending patent applications outside of the United States, such as in Europe, Australia, Japan, Canada, China, Israel and India. Limitations on patent protection in these countries, and the differences in what constitutes patentable subject matter in these countries outside the United States, may limit the protection we have on patents issued or licensed to us outside the United States. In addition, laws of foreign countries may not protect our intellectual property to the same extent as would laws in the United States. To minimize our costs and expenses and maintain effective protection, we focus our foreign patent and licensing activities primarily in the European Union, Canada, Australia and Japan. In determining whether or not to seek a patent or to license any patent in other specific foreign countries, we weigh the relevant costs and benefits, and consider, among other things, the market potential and profitability, the scope of patent protection afforded by the law of the jurisdiction and its enforceability, and the nature of terms with any potential licensees. Failure to obtain adequate patent protection for our proprietary drugs and technology would impair our ability to be commercially competitive in these markets.
SuperGen has 120 issued patents and 271 pending patent applications. Of the total patents and pending patent applications those that relate to each of our material commercial product or product candidate are described in further detail below:
· Decitabine, a product we have exclusively licensed to Eisai: there are 11 issued patents and 30 patent applications, having projected expiration dates ranging from June 5, 2022 to September 27, 2024, granted or pending in the jurisdictions of the U.S., Australia, Canada, Europe, Japan, China, India and Hong Kong.
· SGI-110, our DNMT1 inhibitor: there is one issued patent and seven patent applications, each having a projected expiration date of September 29, 2025, granted or pending in the jurisdictions of the U.S., the European Patent Office (the “EPO”), Israel, China, South Africa, Malaysia, Vietnam and New Zealand.
· Amuvatinib, our TK Inhibitor and DNA Repair Suppressor: there are four issued patents and eight patent applications, having projected expiration dates ranging from October 14, 2024 to March 1, 2027, granted or pending in the jurisdictions of the U.S., the EPO, Australia and Canada.
· SGI-1776, our PIM kinase inhibitor: there is one issued patent and 11 patent applications, each having a projected expiration date of November 6, 2027, granted or pending in the jurisdictions of the U.S., the EPO, Hong Kong, India, Malaysia, Brazil, Canada, Japan, Russia, Vietnam and China.
· Our discovery JAK2 inhibitor: there are 17 patent applications, having projected expiration dates ranging from February 29, 2028 to July 25, 2029, pending in the jurisdictions of the U.S., the EPO, Taiwan, Australia, Brazil, Canada, China, Indonesia, India, Japan, Korea, Mexico, New Zealand and Hong Kong.
· Our discovery AXL inhibitor: there are 13 patent applications, having projected expiration dates ranging from April 11, 2028 to February 8, 2030, pending in the jurisdictions of the U.S., the EPO, Australia, Canada, China, Japan, Korea, New Zealand and Taiwan.
· Relating to our GlaxoSmithKline discovery collaboration: there are three patent applications, having projected expiration dates of February 26, 2030, pending in the jurisdictions of the U.S., the Patent Cooperation Treaty participants and Taiwan.
Trade Secrets and Trademarks
We also rely on trade secret protection for certain proprietary technology. To protect our trade secrets and our other confidential information, we pursue a policy of having our employees and consultants execute proprietary information agreements upon commencement of employment or consulting relationships with us. These agreements provide that all confidential information developed or made known to the individual during the course of the relationship is confidential except in specified circumstances. Further, we minimize the dissemination of our trade secrets by limiting the knowledge of staff only to the specific knowledge of a trade secret to what they need to know, and protective sequestering of trade secrets behind, for example, locks and passwords.
Competition
The pharmaceutical industry in general and the oncology sector in particular is highly competitive and subject to significant and rapid technological change. There are many companies, both public and private, including well-known pharmaceutical companies that are engaged in the discovery and development of products for some of the applications that we are pursuing. Some of our competitors and probable competitors include ArQule, Array BioPharma, Astex Tx, Crystal Genomics, Exelixis, Infinity, Plexxikon, Vertex, Sanofi-Aventis, Bristol-Myers Squibb Company, Celgene, Eli Lilly & Co., GSK, Novartis AG, Pfizer, and others.
Many of our competitors have substantially greater financial, research and development, and manufacturing resources than we do and may represent substantial long-term competition for us. Some of our competitors have received regulatory approval for products or are developing or testing product candidates that compete directly with our product candidates. For example, amuvatinib faces competition from a multitude of other investigational drugs which are multi-targeted tyrosine kinase inhibitors and inhibitors of the DNA repair pathway. We also expect that there will be other inhibitors of Pim kinases that will emerge as competition for SGI-1776 as well as other investigational drugs progressing through our discovery pipeline. In addition, Dacogen faces competition from 5-aza-cytidine and other drugs in development to treat MDS.
Many of these competitors, either alone or together with their customers and partners, have significantly greater experience than we do in discovering products, undertaking non-clinical testing and clinical trials, obtaining FDA and other regulatory approvals, and manufacturing and marketing products. Accordingly, our competitors may succeed in obtaining patent protection, receiving FDA or other foreign marketing approval or commercializing products before we do. If we elect to commence commercial product sales of our product candidates, we could be at a disadvantage relative to many companies with greater marketing and manufacturing capabilities, areas in which we have limited or no experience.
Factors affecting competition in the pharmaceutical industry vary depending on the extent to which competitors are able to achieve an advantage based on superior differentiation of their products’ greater institutional knowledge or depth of resources. If we are able to establish and maintain a competitive advantage based on the ability of CLIMB to discover new drug candidates more quickly and against targets not accessible by many competitors, our advantage will likely depend primarily on the ability of our CLIMB technology to make accurate predictions about the effectiveness and safety of our drug candidates as well as our ability to effectively and rapidly develop investigational drugs.
Extensive research and development efforts and rapid technological progress characterize the industry in which we compete. Although we believe that our proprietary drug discovery capabilities afford us a competitive advantage relative to other discovery and development companies competing in oncology, we expect competitive intensity in this pharmaceutical segment to continue and increase over time. Discoveries by others may render CLIMB and our current and potential products noncompetitive. Our competitive position also depends on our ability to attract and retain qualified scientific and other personnel at all our geographic locations, develop effective proprietary products, implement development plans, obtain patent protection and secure adequate capital resources.
Employees
As of December 31, 2009, we had 80 full-time employees. We use consultants and temporary employees to complement our staffing. Our employees are not subject to any collective bargaining agreements, and we consider our relations with employees to be good.
Executive Officers
|
Name |
|
Age |
|
Position |
|
James S. J. Manuso, Ph.D. |
|
61 |
|
President, Chief Executive Officer and Director |
|
Mohammad Azab, M.D., M.Sc., MBA |
|
54 |
|
Chief Medical Officer |
|
Michael Molkentin |
|
55 |
|
Chief Financial Officer |
James S.J. Manuso, Ph.D., has served as our president and chief executive officer since January 1, 2004, as our chief executive officer-elect from September 2003 to December 2003 and as a director since February 2001. Dr. Manuso is co-founder and immediate past president and chief executive officer of Galenica Pharmaceuticals, Inc. Dr. Manuso co-founded and was general partner of PrimeTech Partners, a biotechnology venture management partnership, from 1998 to 2002, and co-founder and managing general partner of The Channel Group LLC, an international life sciences corporate advisory firm. He was also president of Manuso, Alexander & Associates, Inc., management consultants and financial advisors to pharmaceutical and
biotechnology companies. Dr. Manuso was a vice president and director of Health Care Planning and Development for The Equitable Companies (now Group Axa), where he also served as acting medical director. He currently serves on the boards of Novelos Therapeutics, Inc. (NVLT:OB) and privately-held KineMed, Inc. Previously, he served on the boards of Merrion Pharmaceuticals Ltd. (MERR:IEX; Dublin, Ireland), Inflazyme Pharmaceuticals, Inc., Symbiontics, Inc., Quark Biotech, Inc., Galenica Pharmaceuticals, Inc., and Supratek Pharma, Inc. Dr. Manuso earned a B.A. with Honors in Economics and Chemistry from New York University, a Ph.D. in Experimental Psychophysiology from the Graduate Faculty of The New School University, a Certificate in Health Systems Management from Harvard Business School, and an Executive M.B.A. from Columbia Business School. Dr. Manuso is the author of over 30 chapters, articles and books on topics including health care cost containment and biotechnology company management. He has taught and lectured at Columbia, New York University, Georgetown, Polytechnic University, and Waseda University (Japan). He has delivered invited addresses at meetings of the American Management Association, the American Medical Association, the Securities Industry Association, the Biotechnology Industry Organization, and many other professional associations. Dr. Manuso previously served as vice president and a member of the Board of Trustees of the Greater San Francisco Bay Area Leukemia & Lymphoma Society.
Mohammad Azab, M.D., M.Sc., MBA, joined SuperGen as chief medical officer in July 2009. He possesses more than 20 years of experience in worldwide drug development, clinical research, and medical affairs, resulting in eight approved drugs, including six in oncology. Most recently, he was president and chief executive officer of Intradigm Corporation, a privately held Palo Alto, CA company developing siRNA cancer therapeutics. Previously, Dr. Azab served as executive vice president of research and development, and chief medical officer, of Vancouver, British Columbia-based QLT Inc., where he led clinical development for now-approved drugs in oncology, gastro-intestinal, and ophthalmologic indications. Prior to this, he served as oncology drug team leader at UK-based Zeneca Pharmaceuticals, now Astra Zeneca, where he held responsibilities in global clinical development and regulatory submissions. In this capacity, he managed the approval of drugs for prostate, breast, colorectal, and lung cancer indications. Before Zeneca, Dr. Azab was an international medical manager in oncology at Sanofi Pharmaceuticals, now Sanofi-Aventis, in Gentilly, France. Dr. Azab received his medical degree in 1979 from Cairo University. He practiced as a medical oncologist and received post-graduate training and degrees in oncology research and statistics from the University of Paris-Sud and the University of Pierre and Marie Curie in France. He has published more than 100 medical papers and abstracts. He is an active member of the American Society of Clinical Oncology, the American Association of Cancer Research, and the European Society of Medical Oncology. Dr. Azab received an MBA, with Distinction, from the Richard Ivey School of Business, University of Western Ontario.
Michael Molkentin joined us as chief financial officer and corporate secretary in October 2003. Prior to joining us, Mr. Molkentin served as interim chief financial officer at Aradigm Corporation from May 2000 to September 2002. From January 1995 to April 2000, Mr. Molkentin served as division controller for Thermo Finnigan Corporation, a subsidiary of Thermo Electron. Mr. Molkentin served in a variety of financial management positions with technology companies, including field controller of Vanstar Corporation, controller of Republic Telcom Systems, Inc. and corporate controller of Computer Automation, Inc. Mr. Molkentin is a CPA and received a B.B.A. in accounting from Bernard M. Baruch College in New York City, New York.
Segment and Geographic Area Financial Information
We operate in one business segment — human therapeutics. We had no product revenue in 2009 or 2008. In 2007, 100% of our product revenue was from the EU.
Available Information
We are subject to the information requirements of the Securities Exchange Act of 1934 (the “Exchange Act”). Therefore, we file periodic reports, proxy statements, and other information with the Securities and Exchange Commission (“SEC”). Such reports, proxy statements, and other information may be obtained by visiting the Public Reference Room of the SEC at 100 F Street, NE, Washington, DC 20549 or by calling the SEC at 1-800-SEC-0330. In addition, the SEC maintains a website (www.sec.gov) that contains reports, proxy and information statements, and other information regarding issuers that file electronically.
Financial and other information about us is available on our website at www.supergen.com. We make available on our website, free of charge, copies of our annual report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act as soon as reasonably practicable after filing such material electronically or otherwise furnishing it to the SEC. Information on our website does not constitute a part of this annual report on Form 10-K.
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS.
You should read the following discussion together with our consolidated financial statements and related notes included elsewhere in this report. The results discussed below are not necessarily indicative of the results to be expected in any future periods. Our disclosure and analysis in this section of the report also contain forward-looking statements. When we use the words “anticipate,” “estimate,” “project,” “intend,” “expect,” “plan,” “believe,” “should,” “likely” and similar expressions, we are making forward-looking statements. Forward-looking statements provide our current expectations or forecasts of future events. In particular, these statements include statements such as: our estimates about profitability; the percentage of royalties we expect to earn on Dacogen sales under our agreement with MGI/Eisai; our forecasts regarding our research and development expenses; our expectations about the joint development program with GSK; and our statements regarding the sufficiency of our cash to meet our operating needs. Our actual results could differ materially from those predicted in the forward-looking statements as a result of risks and uncertainties including, but not limited to: the commercial success of Dacogen; delays and risks associated with conducting and managing our clinical trials; developing products and obtaining regulatory approval; ability to establish and maintain collaborative relationships; competition; ability to obtain funding; ability to protect our intellectual property; our dependence on third party suppliers; risks associated with the hiring and loss of key personnel; adverse changes in the specific markets for our products; and our ability to launch and commercialize products. Certain unknown or immaterial risks and uncertainties can also affect our forward-looking statements. Consequently, no forward-looking statement can be guaranteed and you should not rely on these forward-looking statements. For a discussion of the known and material risks that could affect our actual results, please see the “Risk Factors” section of this report. We undertake no obligation to update any forward-looking statements, whether as a result of new information, future events or otherwise. Readers should carefully review the Risk Factors section as well as other reports or documents we file from time to time with the Securities and Exchange Commission.
Overview
We are a pharmaceutical company dedicated to the discovery and development of novel cancer therapeutics in epigenetic and cell signaling modulation. We develop products through biochemical and clinical proof of concept to partner for further development and commercialization. We have a number of Aurora-A and Tyrosine Kinase inhibitors and DNA methyltransferase clinical and pre-clinical products under development. In 2006, Dacogen received approval for marketing in the United States, during 2006 and 2007 we sold the North American and remaining worldwide rights to Nipent to Mayne Pharma, in 2006 we acquired a drug discovery and development company to complement our ongoing licensing efforts, and in October 2009 we entered into a multi-year collaboration with GlaxoSmithKline to discover and develop cancer therapeutics based on epigenetic targets. These changes were implemented to mitigate the ongoing risk of competitive in-licensing and to maximize the return on both existing resources and our incoming royalty and milestone revenue.
Since our incorporation in 1991 we have devoted substantially all of our resources to our product development efforts. Our past development efforts have been focused primarily on the key compounds of Dacogen and Nipent.
Dacogen. Dacogen is approved by the FDA for the treatment of patients with MDS. In September 2004, we executed an agreement granting MGI/Eisai exclusive worldwide rights to the development, manufacture, commercialization and distribution of Dacogen. Under the terms of the agreement, MGI/Eisai made a $40 million equity investment in our company and agreed to pay up to $45 million in specific regulatory and
commercialization milestone payments. To date, we have received $32.5 million of these milestone payments, including $20 million upon first commercial sale of Dacogen in the U.S. in May 2006. In accordance with our agreement with MGI/Eisai, we are entitled to receive a royalty on worldwide net sales starting at 20% and escalating to a maximum of 30%. Our royalty revenues have increased from $22.3 million in 2007, to $38.4 million in 2008, and $41.2 million in 2009. We recognize royalty revenue when the royalty statement is received from MGI/Eisai because we do not have sufficient ability to accurately estimate Dacogen sales prior to that time.
In July 2006, MGI/Eisai executed an agreement to sublicense Dacogen to Janssen-Cilag, a Johnson & Johnson company, granting exclusive development and commercialization rights in all territories outside North America. In accordance with our agreement with MGI/Eisai, we are entitled to receive 50% of certain payments MGI/Eisai receives as a result of any sublicenses. We received $5 million, 50% of the $10 million upfront payment MGI/Eisai received, and, as a result of both the original agreement with MGI/Eisai and this sublicense with Janssen-Cilag, we may receive up to $17.5 million in future milestone payments as they are achieved for Dacogen globally. Janssen-Cilag companies will be responsible for conducting regulatory and commercial activities related to Dacogen in all territories outside North America, while MGI/Eisai retains all commercialization rights and responsibility for all activities in the United States, Canada and Mexico.
Nipent. Nipent is approved by the FDA and EMEA for the treatment of hairy cell leukemia. Nipent was marketed by us in the United States until August 2006, and distributed in Europe through March 2007.
On August 22, 2006, we closed a transaction with Mayne Pharma (USA), Inc., whereby Mayne acquired the North American rights to Nipent and our SurfaceSafe cleaning system. Pursuant to the Asset Acquisition Agreement, we received cash proceeds of $13.4 million upon closing of the transaction. In addition to the initial payment and holdbacks, we had the right to receive annual deferred payments totaling $14.1 million over the following five years based on achievement of specific sales targets. These annual deferred payments might be accelerated under certain circumstances, including a change of control of Mayne. Such a change of control occurred in February 2007 when Mayne was acquired by Hospira, Inc. and we received $10.3 million of the deferred payments. We continued to maintain our commercial operations organization to support the sales and marketing of Nipent during the six month transition period, which ended on February 21, 2007. We were reimbursed by Mayne/Hospira for all Nipent sales and marketing costs during the transition period, including employee salaries and overhead.
In April 2007, we closed another transaction with Mayne/Hospira, completing the sale of the remaining worldwide rights for Nipent for total consideration of up to $8.3 million. We received an initial up-front payment of $3.75 million as a condition of the closing, plus an additional $389,000 for the carrying value of the remaining Nipent inventory. The balance of the purchase price is payable in five annual installments on the anniversary of the closing date.
In September 2007, we received a sales milestone payment of $1.8 million from Mayne/Hospira and in December 2007 received $6 million in supply holdback payments. In February 2008, we received a $1 million indemnification holdback from Mayne/Hospira relating to the sale of the North American rights. In May 2008, we received $400,000, the first annual installment payment relating to the sale of the remaining worldwide rights, and in November 2008 we received the remaining $250,000 indemnification holdback. In March 2009, we received $500,000, the second annual installment payment relating to the sale of the remaining worldwide rights. We do not expect to receive any further payments from the sale of the North American rights.
Montigen Acquisition. In April 2006, we acquired Montigen, a privately-held oncology-focused drug discovery and development company headquartered in Salt Lake City, Utah. Montigen’s assets included its research and development team, a proprietary drug discovery technology platform and optimization process known as CLIMB, and late-stage non-clinical compounds targeting Aurora-A Kinase and members of the Tyrosine Kinase receptor family.
Pursuant to the terms of the merger agreement with Montigen, we paid the Montigen stockholders a total of $17.9 million upon the closing of the transaction in cash and shares of SuperGen common stock. The merger agreement also specified an additional $22 million due to the Montigen stockholders, payable in shares of SuperGen common stock, contingent upon achievement of specific regulatory milestones. The first Montigen compound was cleared in April 2007 by the FDA to begin Phase I clinical trials, which triggered the first milestone payment to the former Montigen stockholders of approximately $10 million which we paid in shares of our common stock. A second Montigen compound was cleared in November 2008 by the FDA to begin Phase I clinical trials, which triggered a milestone payment of $5.2 million, which we paid in a combination of cash and shares of our common stock.
GSK Collaboration. In October 2009, we entered into a multi-year collaboration with GSK to discover and develop cancer therapeutics based on epigenetic targets. Epigenetics refer to the regulation of genes with mechanisms other than changes to the underlying DNA sequence. Epigenetic processes are widely believed to play a central role in the development and progression of almost all cancers. Pursuant to the terms of the transaction, we will collaborate with GSK over a period of five years to discover and develop specific epigenetic therapeutics. At the end of the research term, or earlier if GSK elects, GSK may exercise its option to license from us the compounds that are the result of the joint research effort, in order to continue the development and ultimately commercialize and sell the products worldwide.
In connection with the transaction, we received $5 million upfront, inclusive of a $3 million purchase by GSK of shares of our common stock, priced at a premium to market. In addition, GSK is obligated to make certain payments to us if and when the compounds reach specified developmental milestones, as well as payments to us if and when the compounds that GSK has licensed achieve certain regulatory milestones. The agreement further provides that, if the licensed compounds derived from the joint research team become products, GSK will pay us sales milestone payments as well as royalties on annual net sales of such products. The royalties will be paid on a country-by-country and product-by-product basis. Total potential development and sales milestones payable to us could exceed $375 million. In addition, we may receive tiered royalties into double digit magnitudes, payable on net sales of any resulting products.
All of our current products are in the development or clinical trial stage, and will require substantial additional investments in research and development, clinical trials, regulatory and sales and marketing activities to commercialize these product candidates. Conducting clinical trials is a lengthy, time-consuming, and expensive process involving inherent uncertainties and risks, and our studies may be insufficient to demonstrate safety and efficacy to support FDA approval of any of our product candidates.
As a result of our substantial research and development expenditures and minimal product revenues, we have incurred cumulative losses of $356.6 million through December 31, 2009, and have not consistently generated enough funds through our operations to support our business. We expect to continue to incur operating losses over the next few years.
Ultimately, our ability to become profitable will depend upon a variety of factors, including regulatory approvals of our products, the timing of the introduction and market acceptance of our products and competing products, MGI/Eisai’s success in selling Dacogen, the success of our joint development program with GSK, the launch of new products and our ability to control our ongoing costs and operating expenses. If our drug discovery and research efforts are not successful, or if the results from our clinical trials are not positive, we may not be able to get sufficient funding to continue our trials or conduct new trials, and we would be forced to scale down or cease our business operations. Moreover, if our products are not approved or commercially accepted we will remain unprofitable for longer than we currently anticipate. Additionally, we might be forced to substantially scale down our operations or sell certain of our assets, and it is likely the price of our stock would decline precipitously.
Critical Accounting Policies
Our management discussion and analysis of our financial condition and results of operations is based upon our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses, and reported disclosures. On an on-going basis, we evaluate our estimates, including those related to revenue and gain recognition, the valuation of investments and stock-based compensation. We base our estimates on historical experience and on various other assumptions that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
Our significant accounting policies are more fully disclosed in Note 1 to our consolidated financial statements. However, some of our accounting policies are particularly important to the portrayal of our financial position and results of operations and require the application of significant judgment by our management. We believe the following critical accounting policies, among others, affect our more significant judgments and estimates used in the preparation of our consolidated financial statements.
Stock-Based Compensation
We account for stock-based compensation based upon the fair value estimated on the measurement date of our stock awards using the Black-Scholes option-pricing model based on assumptions for volatility, risk-free interest rates, expected life of the award, and dividends (if any). Expected volatility is determined based on a blend of historical volatility and implied volatility of our common stock based on the period of time corresponding to the expected life of the stock options. The expected life of our stock options is based on our historical data and represents the period of time that stock options granted are expected to be outstanding. The risk-free interest rate is based on the U.S. Treasury yield curve in effect at the time of grant commensurate with the expected life assumption.
We are using the straight-line attribution method to recognize stock-based compensation expense. The amount of stock-based compensation recognized during a period is based on the value of the portion of the awards that are ultimately expected to vest, including awards that vest based on certain performance criteria. For the awards that vest based on certain performance criteria we estimate the probability that the awards will vest as well as the time period over which they are expected to vest and refine these estimates as necessary each reporting period. We estimate forfeitures at the time of grant and revise them, if necessary, in subsequent periods if actual forfeitures differ from those estimates. The term “forfeitures” is distinct from “cancellations” or “expirations” and represents only the unvested portion of the surrendered option. This analysis is re-evaluated annually and the forfeiture rate is adjusted as necessary. Ultimately, the actual expense recognized over the vesting period will only be for those shares that vest.
As of December 31, 2009, there was $2.1 million of total unrecognized compensation cost related to unvested stock-based awards. This cost is expected to be recognized over a weighted average period of 2.06 years.
Revenue and Gain Recognition
Advance payments received in excess of amounts earned are classified as deferred revenue until earned. We enter into revenue arrangements with multiple deliverables, such as intellectual property rights and research and development services. For these arrangements, we generally do not meet the criteria to separate the deliverables for revenue recognition and we treat the deliverables as a combined unit of accounting. As such, non-refundable up-front payments received in connection with research and license agreements are deferred and recognized on a straight-line basis over the relevant estimated periods of continuing involvement, generally the research term. We re-evaluate the period of continuing involvement each reporting period and adjust our estimates accordingly. We recognize milestone fees upon completion of specified substantive at-risk milestones according to the related contract terms.
MGI/Eisai is required to pay us royalties starting at 20% and escalating to a maximum of 30% of net worldwide Dacogen sales within 45 days after the end of each calendar quarter. We recognize royalty revenue when we receive the royalty statement from MGI/Eisai because we do not have sufficient ability to accurately estimate Dacogen sales prior to this time.
The determination of the gain on sale of products from the sale of the rights to Nipent to Mayne/Hospira involves the estimation of our price protection exposure as we are obligated to reimburse Mayne/Hospira for three years for amounts paid to a new supplier of Nipent in excess of certain amounts. The remaining balance of the deferred gain on sale of products of $50,000 at December 31, 2009, reduced from $125,000 at December 31, 2008, consists of the estimated price protection exposure for the remaining year of the underlying agreement and is subject to management estimates and assumptions based on all available information including updated estimates of manufacturing yields and requirements and sales trends.
Impairment of Investments in Financial Instruments
Investments in financial instruments are carried at fair value based on quoted market prices, with unrealized gains and losses included in accumulated other comprehensive income or loss in stockholders’ equity. Our investment portfolio includes equity securities that could subject us to material equity market risk and corporate and U.S. government (or U.S. governmental agency) obligations that subject us to varying levels of credit risk. An other than temporary decline in fair value of a financial instrument may be subject to a write-down resulting in a charge against earnings. The determination of whether a decline in fair value is other than temporary requires significant judgment, and could have a material impact on our balance sheet and results of operations. Our management reviews the securities within our portfolio for other than temporary declines in value on a regular basis. As of December 31, 2009, the gross unrealized losses on available for sale investments was $59,000 (less than 0.1% of our portfolio value) and such losses were not attributed to changes in credit risk. The prices of some of our marketable equity securities are subject to considerable volatility. Currently we own 2,384,211 shares of AVI and recorded an other-than-temporary decline in value of $3.1 million related to this investment during the year ended December 31, 2008. As of December 31, 2009, the gross unrealized gain on our investment in AVI was approximately $811,000. Decreases in the fair value of our securities may continue to significantly impact our results of operations.
Investments in equity securities without readily determinable fair value are carried at cost. We periodically review those carried costs, amounting to $500,000 as of December 31, 2009 and evaluate whether an impairment has occurred. The determination of whether an impairment has occurred requires significant judgment, as each investment has unique market and development opportunities.
Recent Accounting Pronouncements
In October 2009, the FASB issued ASU 2009-13, which amends ASC Topic 605, Revenue Recognition, to change the existing criteria for separating consideration in multiple-deliverable arrangements and require companies to allocate revenue in multiple-element arrangements based on an element’s estimated selling price if vendor-specific or other third-party evidence of value is not available. ASU 2009-13 is effective for us beginning January 1, 2011. Earlier application is permitted. We are currently evaluating the impact of the pending adoption of the ASU on our consolidated financial statements and cannot estimate the impact of adoption at this time.
Results of Operations
|
Revenues (in thousands) |
|
2009 |
|
2008 |
|
2007 |
|
|||
|
Royalty revenue |
|
$ |
41,156 |
|
$ |
38,422 |
|
$ |
22,333 |
|
|
Development and license revenue |
|
97 |
|
— |
|
— |
|
|||
|
Net product revenue |
|
— |
|
— |
|
621 |
|
|||
|
Total revenues |
|
$ |
41,253 |
|
$ |
38,422 |
|
$ |
22,954 |
|
The increases in royalty revenue are due to higher Dacogen product sales by MGI/Eisai. MGI/Eisai is required to pay us royalties starting at 20% and escalating to a maximum of 30% of net Dacogen sales within 45 days after the end of each calendar quarter. We recognize royalty revenue when we receive the royalty statements from MGI/Eisai because we do not have sufficient ability to accurately estimate Dacogen sales prior to this time. Therefore, the royalty revenues recorded in 2009 represent MGI/Eisai’s Dacogen sales for the fourth quarter of 2008 and the first three quarters of 2009. The royalty revenues recorded in 2008 represent MGI/Eisai’s Dacogen sales for the fourth quarter of 2007 and the first three quarters of 2008. The royalty revenues recorded in 2007 represent MGI/Eisai’s Dacogen sales for the fourth quarter of 2006 and the first three quarters of 2007.
Development and license revenue relates to the agreements we entered into with GSK in October 2009. In connection with the agreements, we received an upfront payment of $2 million, in addition to a $3 million equity investment by GSK at above market price. As our substantive performance obligations under the agreements are estimated to be completed over a five year period, the $2 million upfront payment and the premium paid on the $3 million equity investment of $0.5 million are being recognized ratably over 60 months to development and license revenue.
The elimination of net product revenue is due to the disposal of our remaining commercial products in 2007. Our net product revenue had previously consisted of sales of Nipent and generic mitomycin. We sold the rights to mitomycin to Intas Pharmaceuticals in April 2007 and sold the remaining worldwide rights to Nipent to Mayne/Hospira in April 2007.
|
Costs and operating expenses (in thousands) |
|
2009 |
|
2008 |
|
2007 |
|
|||
|
Cost of product revenue |
|
$ |
— |
|
$ |
— |
|
$ |
221 |
|
|
Research and development |
|
29,689 |
|
32,685 |
|
23,423 |
|
|||
|
Selling, general and administrative |
|
8,994 |
|
11,119 |
|
13,520 |
|
|||
|
Acquired in-process research and development |
|
— |
|
5,185 |
|
9,967 |
|
|||
|
Gain on sale of products |
|
(595 |
) |
(2,236 |
) |
(33,677 |
) |
|||
|
Total costs and operating expenses |
|
$ |
38,088 |
|
$ |
46,753 |
|
$ |
13,454 |
|
The decrease in cost of product revenue is due to the disposal of our commercial products in 2006 and 2007 as noted in the discussion of the decline in our product revenue above.
The increase in research and development expenses from 2007 to 2008 was due to higher drug discovery research costs associated with the acquisition of Montigen in 2006, clinical trial costs related to the initiation of Phase I and Phase Ib studies for amuvatinib in 2007, and development and formulation costs for SGI-1776 in 2008. The decrease in research and development expenses from 2008 to 2009 is primarily due to lower contracted outside research and development services for several of our drug candidates and lower clinical trial costs related to our Phase I and Phase Ib clinical trials for amuvatinib.
We conduct research internally and through collaborations with third parties as we continue to maintain a strong commitment to our research and development efforts. Our research and development activities consist primarily of drug discovery, drug development, and pre-clinical and clinical development, as we advance our existing product candidates through clinical trials. Our research and development expenses consist primarily of salaries, employee benefits and other personnel related costs; stock-based compensation expense; laboratory equipment and supplies; third-party consultant fees and contract labor; costs for pre-clinical and clinical trials, including clinical research organizations; other outsourced research; depreciation expense; corporate overhead; and allocated facility costs.
We do not allocate certain of our internal research and development costs such as salaries and other personnel related costs, corporate overhead and facility costs to our development programs on a project-by-project basis. These costs are incurred across and contribute to many discovery, research and development programs including a broad range of scientific research projects, many of which fail in the early stages of discovery and development. We do allocate direct salaries and third party costs to certain major development programs that are generally in later stages of pre-clinical or clinical development. Our scientists record their time to such specific projects when appropriate; however, many activities simultaneously benefit multiple projects and cannot be readily attributed to a specific program. Accordingly, the accurate assignment of time and costs to a specific project is difficult and does not give a true indication of the actual costs of a particular project on a fully burdened basis. Below is a summary of third party costs and direct salaries that are identifiable for our major drug programs:
|
Third Party Costs and Direct |
|
2009 |
|
2008 |
|
2007 |
|
Cumulative To |
|
||||
|
Amuvatinib |
|
$ |
4,601 |
|
$ |
6,786 |
|
$ |
2,614 |
|
$ |
16,039 |
|
|
SGI-1776 |
|
3,243 |
|
4,093 |
|
324 |
|
7,703 |
|
||||
|
SGI-110 |
|
2,088 |
|
1,688 |
|
170 |
|
3,946 |
|
||||
|
All other projects |
|
3,216 |
|
3,530 |
|
4,861 |
|
16,302 |
|
||||
Conducting clinical trials is a lengthy, expensive and uncertain process. Completion of clinical trials may take several years or more. The length of time generally varies substantially according to the type, complexity, novelty and intended use of the product candidate. Our clinical trials may be suspended at any time if we or the FDA believe the patients participating in our studies are exposed to unacceptable health risks. We may encounter problems in our studies which will cause us or the FDA to delay or suspend the studies. Because of these uncertainties, we cannot predict when or whether we will successfully complete the development of our product candidates or the ultimate product development cost for any of our product candidates.
The decrease in selling, general and administrative expenses from 2007 to 2008 relates primarily to lower costs associated with sales and marketing programs due to the sale of our North American and worldwide rights to Nipent and Surface Safe to Mayne, lower costs related to our European operations, and lower stock-based compensation expense due to our lower stock price in recent years. We sold the rights to mitomycin to Intas Pharmaceuticals in April 2007 and sold the remaining worldwide rights to Nipent to Mayne Pharma in April 2007. The decrease in selling, general, and administrative expenses from 2008 to 2009 relates primarily to lower stock-based compensation and legal expenses, as well as the elimination of administrative costs related to our European operations, which were liquidated in October 2008.
Acquired in-process research and development expenses relate to our acquisition of Montigen Pharmaceuticals in April 2006. In April 2007, the first Montigen compound received clearance from the FDA to begin Phase I clinical trials, triggering the first milestone payment to the former Montigen stockholders of $9,967,000, which we paid through the issuance of 1,477,000 shares of our common stock, and which was recorded as acquired in-process research and development expense in the year ended December 31, 2007. In November 2008, a second Montigen compound received clearance from the FDA to begin Phase I clinical trials, triggering a second milestone payment of $5,185,000, which was paid through a combination of cash and the issuance of common stock. We had no similar transactions in 2009.
Gain on sale of products recorded in the year ended December 31, 2007 relates to the sale of our worldwide rights to Nipent and Surface Safe to Mayne/Hospira and the sale of mitomycin, paclitaxel, and etoposide to Intas Pharmaceuticals, as well as the first annual sales milestone payment of $1.8 million received from Mayne/Hospira in September 2007 relating to the achievement of certain Nipent sales targets and $6 million in supply holdbacks received in December 2007. During 2008 we received a $1 million indemnification holdback relating to the sale of the North American Nipent rights, a $250,000 indemnification holdback, and a $400,000 annual payment relating to the sale of the remaining worldwide Nipent rights. In addition, in 2008 we reduced our price protection reserve by $426,000 and reversed $160,000 of residual products return reserve for Nipent as the reserve was no longer required due to the expiration of the contractual return period. The sum of these 2008 transactions, $2,236,000, was recorded as a gain on sale of products during the year ended December 31. 2008. Gain on sale of products for the year ended December 31, 2009 represented the receipt of a $500,000 annual payment from Mayne/Hospira relating to the sale of the worldwide rights for Nipent, a $75,000 reduction of our price protection reserve, and $20,000 relating to the reversal of a residual products return reserve that was no longer required due to the expiration of the contractual return period.
|
Other income (expense) (in thousands) |
|
2009 |
|
2008 |
|
2007 |
|
|||
|
Interest income |
|
$ |
686 |
|
$ |
2,193 |
|
$ |
4,017 |
|
|
Other than temporary decline in value of investments |
|
— |
|
(3,055 |
) |
(65 |
) |
|||
|
Other income |
|
— |
|
34 |
|
40 |
|
|||
|
Income tax benefit (provision) |
|
886 |
|
48 |
|
(411 |
) |
|||
The declines in interest income have been due primarily to significant declines in interest rates.
During the years ended December 31, 2008 and 2007, we recorded write-downs of $3,055,000 and $65,000, respectively, related to other than temporary declines in the values of two of our equity investments. We had no similar write-downs in 2009.
Other income primarily represented transaction gains on foreign currency exchange relating to activities of our subsidiary EuroGen. EuroGen discontinued selling Nipent in 2007, and EuroGen was liquidated as of October 1, 2008.
In 2009, we recorded a tax benefit of $886,000, which was primarily due to the Worker, Home Ownership and Business Assistance Act of 2009, signed into law on November 6, 2009, that allowed for certain net operating losses to be used to eliminate or refund alternative minimum tax, as well as monetization of research credits and other state tax benefits. In 2008, we recorded a tax benefit of $48,000 due to estimated refundable research and development tax credits under a 2008 Housing Rescue bill. The tax provision recorded in 2007 reflected federal and state income taxes at statutory rates and the application of net operating loss carryforwards, offset by certain non-deductible expenses, including acquired in-process research and development expense.
Liquidity and Capital Resources
Our cash, cash equivalents, and current and non-current marketable securities totaled $100.8 million at December 31, 2009, compared to $88.3 million at December 31, 2008.
Net cash provided by operating activities was $8.7 million in 2009, and consisted primarily of the net income of $4.7 million, depreciation of $1.2 million, stock based compensation expense of $2.5 million, and an increase in deferred revenue from entering into the GSK agreements in 2009 of $2.4 million, offset in part by an
$818,000 increase in income tax receivable and a $1.1 million decrease in accounts payable and other liabilities. Net cash used in operating activities was $1.7 million in 2008, and consisted primarily of the net loss of $9.1 million and recognition of gain on sale of products of $1.6 million, offset in part by non-cash depreciation and amortization of $1.6 million, other than temporary declines in the value of investments of $3.1 million, stock based compensation expense of $2.8 million, and non-cash acquired in-process research and development expenses of $2.4 million. Net cash used in operating activities was $5.8 million in 2007, and consisted primarily of the net income of $13.1 million, plus the non-cash impacts of acquired in-process research and development expenses of $10 million, non-cash stock-based compensation expense of $4.3 million, and depreciation and amortization of $2 million. We also had a decrease in accounts receivable of $1.9 million due to the sale of our commercial operations, all more than offset by a decrease in accounts payable and other liabilities of $2.8 million, and the recognition of and change in the deferred gain on sale of products of $34.6 million.
Net cash used in investing activities was $52.6 million in 2009, and consisted primarily of $133.3 million for the purchase of marketable securities and $1 million for purchases of property and equipment, offset in part by $81.2 million in maturities of marketable securities. Net cash used in investing activities was $27.6 million in 2008, and consisted primarily of purchases of marketable securities of $46 million and property and equipment of $1.1 million, offset in part by proceeds from maturities of marketable securities of $17.9 million and the sale of products of $1.6 million. Net cash provided by investing activities was $12.4 million in 2007, and consisted primarily of milestone payments of $17.1 million from Mayne relating to the sale of the North American rights to Nipent, $5.1 million from Mayne for the sale of the remaining worldwide rights to Nipent, $1.5 million in maturities of marketable securities, and $1.2 million for the sale of products to Intas, partially offset by $10.9 million for purchases of marketable securities and $1.7 million for purchases of property and equipment.
Net cash provided by financing activities was $2.7 million in 2009, and related primarily to proceeds from the issuance of common stock to GSK, as well as proceeds from the issuance of common stock upon exercise of stock options and issuances under our employee stock purchase plan. Net cash provided by financing activities was $142,000 in 2008 and $3.8 million in 2007, and consisted of proceeds from the issuance of common stock upon exercise of stock options and warrants as well as issuances under our employee stock purchase plan.
Our contractual obligations as of December 31, 2009 are as follows (in thousands):
|
|
|
Payments Due by Period |
|
|||||||||||||
|
|
|
Total |
|
< 1 year |
|
1-3 years |
|
4-5 years |
|
After 5 years |
|
|||||
|
Operating leases, net of subleases |
|
$ |
2,801 |
|
$ |
2,279 |
|
$ |
522 |
|
$ |
— |
|
$ |
— |
|
|
Total contractual cash obligations |
|
$ |
2,801 |
|
$ |
2,279 |
|
$ |
522 |
|
$ |
— |
|
$ |
— |
|
Our principal administrative facility is currently located in leased general office space, containing approximately 50,000 square feet, in Dublin, California, under a lease that expires in November 2010. Our drug formulation laboratory operations are located in a 10,000 square foot industrial building that we own in Pleasanton, California. We are currently leasing 11,700 square feet of space for our drug discovery laboratory operations in Salt Lake City, Utah. The lease on this space expires in May 2012. We are currently exploring various options to address our space needs in all three locations, which would significantly increase our future lease obligations beyond those noted in the table above.
Avicine will require significant additional expenditures to complete the clinical development necessary to gain marketing approval from the FDA and equivalent foreign regulatory agencies. As part of our agreement with AVI BioPharma, we are obligated to make additional payments to AVI based on successful achievement of developmental, regulatory approval, and commercialization milestones over the next several years that could total $80 million. However, no significant development efforts have been incurred for Avicine since 2003 and none
are anticipated in the near future. We are unable to determine precisely when and if our payment obligations under our agreement with AVI will become due as these obligations are based on milestone events the achievement of which is subject to a significant number of risks and uncertainties. Because some of the milestone events are revenue-related and the payment obligation would not be triggered absent our receipt of revenues, we may be able to use funds generated from revenues to make the milestone payments if they become due.
We have financed our operations primarily through the issuance of equity and debt securities, the receipt of milestone, royalty and other payments in connection with collaborative agreements, and the sale of non-core assets. Based on our current forecasted product development activities, we believe that our current cash, cash equivalents, marketable securities and other investments will satisfy our cash requirements through at least December 31, 2011. We may pursue additional financing options, including the selling of additional shares of stock in public or private offerings.
We believe that our need for additional funding will increase in the future, especially if we acquire new product technologies for development and sale, and our ability to continue raising funds from external sources will be critical to our success. We continue to actively consider future contractual arrangements that would require significant financial commitments. If we experience currently unanticipated cash requirements, we could require additional capital much sooner than presently anticipated. We may raise money by the sale of our equity securities or debt, or the exercise of outstanding stock options by the holders of such options. However, given uncertain market conditions and the volatility of our stock price, we may not be able to sell our securities in public offerings or private placements at prices and on terms that are favorable to us, if at all. We may also choose to obtain funding through licensing and other contractual agreements. Such arrangements may require us to relinquish our rights to our technologies, products or marketing territories, or to grant licenses on terms that are not favorable to us. If we fail to obtain adequate funding in a timely manner, or at all, we will be forced to scale back our product development activities or our operations in a manner that will ensure we can discharge our obligations as they come due in the ordinary course of business.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements that are currently material or reasonably likely to be material to our consolidated financial position or results of operations.
Liquidation of EuroGen Pharmaceuticals
We established our European subsidiary, EuroGen Pharmaceuticals, Ltd. in 2001 to expand our commercial presence in Europe. Since that time, we changed our strategic focus due to the sublicense of Dacogen to MGI/Eisai in 2004 and the sale of Nipent to Mayne/Hospira in 2006 and 2007. As a result, in May 2008, we decided to discontinue our European operations and liquidate EuroGen. The liquidation was substantially completed by October 1, 2008. As part of this liquidation, we terminated the two employees at EuroGen as of September 30, 2008. As part of the employment agreements with these two employees, we were required to provide them with a twelve month notification period prior to termination. They continued to provide services from the May 2008 notification date through September 30, 2008, during which time they were compensated with their normal salary and benefits. At their termination date, we were obligated to pay these employees severance through the remainder of the twelve month notification period. During the year ended December 31, 2008, we recorded total severance costs of $420,000, which were recorded in selling, general and administrative expenses. We do not expect to incur any further severance costs related to this liquidation.
Income Taxes
As of December 31, 2009, we have net operating loss carryforwards for federal income tax purposes of approximately $264 million which expire in the years 2012 through 2029, net operating loss carryforwards for state income tax purposes of approximately $136 million which expire in the years 2010 through 2029, federal research and development credit carryforwards of approximately $11 million, which expire in the years 2010 through 2029, and state research and development carryforwards of approximately $10 million, some of which expire in 2022 and some of which have no expiration. The realization of these future tax benefits is dependent upon our ability to generate sufficient taxable income within the carryforward period. Because of our history of operating losses, we believe that these benefits are not currently likely to be realized, and, accordingly, the net deferred tax assets have been fully offset by a valuation allowance.
Utilization of our net operating loss carryforwards may be subject to substantial annual limitations due to ownership change limitations provided by the Internal Revenue Code and similar state provisions. Such an annual limitation could result in the expiration of the net operating losses before utilization.
We have no unrecognized tax benefits as of December 31, 2009. No interest and penalties expenses were recognized in the statements of operations for the years ended December 31, 2007, 2008 and 2009. We are subject to income tax examinations for U.S. Federal income taxes and state income taxes from 1994 forward due to net operating losses in tax years 1994 through 2005. We are subject to tax examinations in the United Kingdom from 2001 forward. We do not anticipate that total unrecognized tax benefits will significantly change prior to December 31, 2010.
ITEM 9A. CONTROLS AND PROCEDURES.
Evaluation of Disclosure Controls and Procedures.
We carried out an evaluation, under the supervision of and with the participation of our management, including our Chief Executive Officer and our Chief Financial Officer, of the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended) as of the end of the period covered by this amended report. Based on the evaluation, our Chief Executive Officer and Chief Financial Officer have concluded that as of December 31, 2009, our disclosure controls and procedures were effective.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining an adequate internal control structure and procedures for financial reporting as defined in Exchange Act Rules 13a-15(f) and 15d-15(f). Our internal control over financial reporting is a process designed by, or under the supervision of, our Chief Executive Officer and Chief Financial Officer and effected by our board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Our internal control over financial reporting includes those policies and procedures that:
· pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect our transactions and dispositions of assets;
· provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and our board of directors; and
· provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of our assets that could have a material effect on our financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions or that the degree of compliance with the policies or procedures may deteriorate.
Under the supervision of and with the participation of our management, including our Chief Executive Officer and our Chief Financial Officer, our management has assessed the effectiveness of internal control over financial reporting as of December 31, 2009. Our assessment was based on criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”) Internal Control-Integrated Framework. Based on using the COSO criteria, we believe our internal control over financial reporting as of December 31, 2009 was effective.
Ernst & Young LLP, our independent registered public accounting firm, has issued an attestation report that the Company’s internal control over financial reporting was effective as of December 31, 2009.
Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting that occurred during the fourth quarter of 2009 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
ITEM 11. EXECUTIVE COMPENSATION.
COMPENSATION DISCUSSION AND ANALYSIS
Overview of Compensation Program and Philosophy
Our executive compensation programs are designed to meet the following objectives:
· Attract and retain qualified executives with a view to the highly competitive nature of the marketplace in the San Francisco Bay Area biotechnology industry and other industries from which we may seek executive talent;
· Provide an executive compensation structure that is not only competitive in our geographic area and industry sector, but is internally equitable and consistent based on the level of responsibilities for each executive position;
· Motivate and reward our executives to perform to the best of their abilities;
· Align our financial results and compensation paid to executive officers with a goal to achieve our current year and longer-term strategic business goals and objectives; and
· Ensure that our executives are not motivated to incur excessive risk that could jeopardize the Company.
These objectives fit within our overall compensation philosophy by helping to continuously improve the Company’s performance, secure the future potential of our business, enhance stockholder value, and provide proper compliance with regulatory and related requirements.
To meet these objectives, we have implemented an executive compensation program based on the following policies:
· Pay executive base salaries that are competitive with the practices of other San Francisco Bay Area biotechnology companies and other relevant industries that are similar in size;
· Pay for performance through a management bonus plan that is based upon shorter-term incentives and through merit increases based on company and personal performance, as well as market data; and
· Pay for performance through equity compensation awards that provide a longer-term incentive to retain our executives.
The Compensation Committee is responsible for ensuring compliance with these objectives and policies and, accordingly, is empowered to review and approve the annual compensation arrangements of the Company’s executive officers, including annual base salary, annual incentive bonus, equity compensation, and other benefits or perquisites. The Company’s executive team was comprised of the following individuals during 2009:
· James S.J. Manuso, Ph.D. — President and Chief Executive Officer
· Mohammad Azab, M.D. — Chief Medical Officer
· Michael Molkentin, C.P.A. — Chief Financial Officer
· David J. Bearss, Ph.D. — Former Chief Scientific Officer (left the Company in October 2009)
· Gregory Berk, M.D. — Former Chief Medical Officer (left the Company in May 2009)
Throughout this Proxy Statement, our executive team may be referred to by name, title or referred to as the “executive officers” and comprises our “Named Executive Officers.”
Role of our Compensation Committee
Our Compensation Committee approves, administers and interprets our executive compensation program, and with respect to our Named Executive Officers, the 2003 Stock Plan. This Committee is appointed by the Board, and consists entirely of directors who are independent for purposes of the listing standards of the Nasdaq Stock Market, “outside directors” for purposes of Section 162(m) of the Internal Revenue Code of 1986, as amended, and “non-employee directors” for purposes of Rule 16b-3 under the Exchange Act. The Compensation Committee is comprised of Walter J. Lack (chairman) and Thomas V. Girardi and operates under a written charter that is periodically reviewed and revised by the Compensation Committee and the Board. A copy of this charter is available for review in the corporate governance section of our website at www.supergen.com. The Compensation Committee held one meeting in 2009 and approved several other matters by unanimous written consent.
Role of Executive Officers in Compensation Decisions
Dr. Manuso, our president and chief executive officer (CEO), reviews the performance of each executive officer, and presents his findings to the Compensation Committee, together with recommendations for compensation structures applicable to the fiscal year under consideration. The Compensation Committee considers these findings and recommendations, but makes its own final determinations. The Compensation Committee alone or in consultation with the full Board reviews Dr. Manuso’s performance.
Role of Compensation Consultants and Surveys
The Compensation Committee has adopted a policy where it has the sole authority to hire and fire outside compensation consultants to assist the Committee in analyzing executive compensation and discharging its related responsibilities. The Compensation Committee did not retain a compensation consultant to assist in determining or recommending the amount or form of executive compensation since it did not anticipate making substantial changes in the amounts and form of executive compensation for our executive officers based on the Company’s overall performance during 2009.
For 2009, the Compensation Committee primarily utilized two compensation surveys in evaluating the various elements of the compensation of our executive officers. The two surveys were the following:
· Radford Global Life Sciences Survey (“Radford Survey”) — uses peer group information from over 156 participants in the 50 to 149 employee size category regarding all elements of compensation, including base salary, bonus, and equity for new hires and existing employees of all levels, including executive officers. There are 48 participants in the San Francisco Bay Area in the 50 to 149 employee size category to provide comparative data for our Dublin/Pleasanton locations, and 39 participants in the mountain states to provide comparative data for our Salt Lake City location.
· BioWorld Executive Compensation Survey — analyzes information from proxy statements and annual reports of approximately 203 publicly traded biotechnology companies that were filed with the U.S. Securities and Exchange Commission for fiscal year 2008. This survey summarizes compensation data for up to seven officer level positions.
2009 and 2010 Performance Priorities
Executive incentive compensation decisions are primarily based upon the achievement or progress towards Company-wide performance objectives, the execution of operational and business specific strategic objectives, and the officer’s individual performance. Our performance objectives are both qualitative and quantitative. These performance objectives may be modified by the Compensation Committee based on changes
in market conditions or the business environment in which we operate. The Compensation Committee may include or exclude certain items related to the ongoing operations when calculating financial measures including gains or losses not anticipated or properly reflecting the cash flow or economic contribution of a transaction during the annual performance period under review. The Compensation Committee believes that our overall performance as a discovery-based drug development company is a more significant factor in our compensation decisions than our performance against any specific individual predetermined goal.
The Compensation Committee determines the amount payable, if any, to its named executive officers as an annual performance bonus relating to the 2009 and 2010 years based upon its determination as to the Company’s achievement of the 2009 and 2010 performance priorities. However, this determination is not mechanical. As noted above, the Compensation Committee also factors in, as the most significant factor in making its determination, the Company’s progress in its transition to a discovery-based drug development company. Thus, while there is no specific weighting accorded to each of the enumerated performance priorities for 2009 and 2010 or the officer’s individual performance, the Compensation Committee considers them of roughly equal weight, with the most weight given to the judgment regarding the Company’s progress and overall performance as a discovery-based drug development company. As described below, in 2009 the Company significantly exceeded the performance goals in each of the performance priority areas. Moreover, the Compensation Committee determined that the Company’s overall performance as a discovery-based drug development company in 2009 was outstanding.
2009 Performance Priorities
At the beginning of 2009, the Compensation Committee established performance priorities in four key areas for the Company:
1. Financial performance — Properly manage annual cash burn and year-end cash balance by targeting reasonable ranges for loss from operations, net loss, cash used in operations and cash on hand at the end of 2009. The target range for loss from operations and cash used in operations is plus or minus 20% from the approved financial targets, while the target range for cash, cash equivalents and marketable securities is plus or minus 10% of a targeted combined balance of $75 million at the end of the performance period.
The Company achieved the primary financial performance priorities for 2009. We recorded income from operations of $3.2 million, which was 125% better than the anticipated annual loss from operations of $12.5 million, which was the target. Our recorded net income of $4.7 million was 143% better than the targeted annual loss of $10.9 million. The cash provided by our operating activities of $8.7 million was 222% better than the targeted annual amount of cash used in operating activities of $7.1 million, and unrestricted cash, cash equivalents and marketable securities of $100.8 million exceeded the year-end target of $75 million by 34%.
2. Business development — Initiate and execute at least one business development transaction with terms, conditions and economic value consistent with other market transactions being executed for similar type products within the pharmaceutical industry sector in which we compete.
The Company achieved this business development priority for 2009 by executing a collaboration agreement with GlaxoSmithKline (“GSK”) to discover and develop specific epigenetic therapeutics. Total potential development and commercial milestones payable to the Company could exceed $375 million.
3. Product development — Advance to lead product candidacy, IND enabling research and/or IND filing one or more new product development candidates while advancing clinical-stage compounds.
The Company achieved the product development priority for 2009 with the advancement of several candidates in the clinical and pre-clinical stage and with multiple unannounced potential candidates in the discovery stage process.
4. Organizational development — Invest in essential infrastructure by recruiting additional talent for key operating positions with a focus on retaining, training and developing current organizational resources required to advance our drug research and business development initiatives. The Company is targeting headcount growth primarily in the research and development area with a focus on discovery, pre-clinical, manufacturing, and clinical operations.
The Company made progress with the organizational development priority with the addition of several key new hires in research and development and clinical operations areas. In addition, the Company assessed its future strategic organizational needs and initiated an organizational adjustment which was executed in early 2009 and reduced overall personnel by approximately 8% at that time.
The Compensation Committee believed the successful execution overall of our 2009 Performance Priorities, including the consideration of other actions not included in the specific priorities, would improve our overall performance and enhance stockholder value over the long term. The Compensation Committee continues to believe that the strategic shift from a specialty pharmaceutical company dependent on acquiring drugs from third parties into a discovery-based drug development company will not necessarily result in a short-term increase in stockholder value. In fact, the development of a novel and growing drug development pipeline, including the targeting, development and execution of new business development transactions and relationships, will require significant ongoing effort and achievement by our executive officers and other personnel during 2010 and beyond.
2010 Performance Priorities
At the beginning of 2010, the Compensation Committee established initial performance priorities in four key areas for the Company:
1. Financial performance — Properly manage annual cash burn and year-end cash balance by targeting reasonable ranges for loss from operations, net loss, cash used in operations and cash on hand at the end of 2010. The target range for income (loss) from operations and cash provided by (used in) operations is plus or minus 15% from the approved financial targets, while the target range for cash, cash equivalents and marketable securities is plus or minus 10% of a targeted combined balance of $85 million at the end of the performance period.
2. Business development - Initiate and execute at least one business development transaction with terms, conditions and economic value consistent with other market transactions being executed for similar type products within the pharmaceutical industry sector in which we compete.
3. Product development — Advance to lead product candidacy, IND enabling research and/or IND filing one or more new product development candidates while advancing clinical-stage compounds.
4. Organizational development — Invest in essential infrastructure by recruiting additional talent for key operating positions with a focus on retaining, training and developing current organizational resources required to advance our drug research and business development initiatives. The Company is targeting headcount growth primarily in the research and development area with a focus on discovery, pre-clinical, manufacturing, regulatory and clinical operations.
The Compensation Committee believes the 2010 Performance Priorities identified above are reasonably attainable, but will require significant and sustained effort on the part of all our Named Executive Officers and employees. The Compensation Committee believes that successful execution of our performance priorities will improve our overall performance and ultimately enhance stockholder value over the long term.
Chief Executive Officer Annual Bonuses
For 2009, our chief executive officer Dr. Manuso’s target annual bonus was $350,000 under a new executive employment agreement effective April 1, 2009. There was no threshold bonus or maximum bonus. The Compensation Committee assessed the 2009 performance of Dr. Manuso by conferring with the independent members of the Board of Directors. In addition to assessing Dr. Manuso’s contribution to the achievement of the 2009 performance priorities, the independent members of the Board of Directors and the Compensation Committee also considered other qualitative elements furthering the long-term success of the Company. Other elements discussed and considered include organizational leadership qualities, development and execution of business, product and operational development strategies and opportunities, development and execution of investor relations and other programs enhancing organizational visibility in the financial community, expanding the shareholder base, regulatory compliance, and overall financial stewardship of financial resources. After receiving this input, the Compensation Committee determined that the 2009 performance of Dr. Manuso was excellent. Due to the Company’s strong performance, with respect to both overall performance as a discovery-based drug development company and performance against the enumerated performance priorities, and based upon the Compensation Committee’s subjective appraisal of Dr. Manuso’s 2009 performance, Dr. Manuso was awarded his full target bonus of $350,000. Moreover, the Compensation Committee exercised its discretion to award Dr. Manuso an additional bonus payment of $150,000, based upon the Company’s and his superior 2009 performance.
In addition to the bonuses earned above, Dr. Manuso also earned a $150,000 guaranteed bonus on January 7, 2009 under terms of a prior executive compensation agreement that was superseded on April 1, 2009 with a new executive employment agreement.
For 2010, Dr. Manuso has a target annual bonus of $650,000. There is no specified threshold bonus or maximum bonus for Dr. Manuso for 2010.
Named Executive Officer Annual Bonuses
Our other named executive officers who received annual bonuses on account of the full 2009 fiscal year, Messrs. Azab and Molkentin had a 2009 target payout equal to 30% of their annual base salary and a maximum payout of 50% of their 2009 annual base salary. The Compensation Committee assessed the 2009 performance of Messrs. Azab and Molkentin by conferring with Dr. Manuso. The Compensation Committee and Dr. Manuso realize that the current size of the organization demands greater cross functional expertise and/or understanding with the multiple aspects of a drug discovery operation, and that many similar qualities and characteristics are required of its executives, and thus considered when assessing an officer’s contribution with the execution and achievement of any relevant annual performance or other priorities. Therefore, additional categories or areas were considered by the Compensation Committee when assessing the executive’s individual performance and organizational contributions.
Chief Medical Officer: In addition to assessing Dr. Azab’s contribution to the achievement of the 2009 performance priorities, the Compensation Committee also considered other qualitative elements furthering the future success of the Company. Other elements discussed and considered included Dr. Azab’s organizational leadership qualities, contribution to the development and execution of product development and clinical trial programs and strategies, development and execution of external relations and programs enhancing the organization’s visibility and interactions within the scientific community, and compliance and constructive interaction with the appropriate regulatory bodies.
Chief Financial Officer: In addition to assessing Mr. Molkentin’s contribution to the achievement of the 2009 performance priorities, the Compensation Committee also considered other qualitative elements furthering the long-term success of the organization. Other elements discussed and considered included organizational leadership qualities, development and execution of business and operational development strategies and opportunities, assistance in the development and execution of investor relations and other programs enhancing the organization’s visibility in the financial community and among its shareholder base, development and demonstration of overall financial stewardship and management of financial assets, and timely and accurate filing with all regulatory requirements and compliance associated with the management of multiple general and administrative functions.
After considering the organizational achievements of the various annual performance priorities, the above categories, and Dr. Manuso’s input, the Compensation Committee determined that the 2009 performance of Messrs. Azab and Molkentin was superb. There were no threshold-level bonuses for either Dr. Azab or Mr. Molkentin. Based upon the Company’s 2009 achievement both in the enumerated performance priority areas and upon the Company’s overall 2009 performance as a discovery-based drug development company, and based upon the Committee’s subjective appraisal of their individual performances, the Compensation Committee approved an annual bonus payout equal to 45% of Dr. Azab’s annual base salary and equal to 43% of Mr. Molkentin’s annual base salary. Dr. Azab’s 2009 annual bonus was pro-rated to reflect his partial year of service.
For 2010, Messrs. Azab and Molkentin have a target annual bonus equal to 30% of their annual base salary and a maximum payout equal to 50% of their annual base salary. The Compensation Committee, however, retains the discretion to pay outside of this range based upon its qualitative determinations.
Elements of Compensation for President and Chief Executive Officer
Original Employment Agreement
On September 22, 2006, the Company and James S.J. Manuso, Ph.D. executed an Executive Employment and Confidential Information and Invention Assignment Agreement (“Original Agreement”). Dr. Manuso is the Company’s current CEO and the agreement provided for the continuation of his service for the three year period or from January 1, 2007 through December 31, 2009. The prior three year employment agreement expired on December 31, 2006. We do not have employment agreements with any other executive officer.
The Original Agreement provided for (i) an initial base salary of $515,000, adjusted annually at twice the percentage increase in the Consumer Price Index, (ii) a guaranteed annual bonus of $150,000 provided Dr. Manuso remained continuously employed during the applicable year of the payment, (iii) a potential annual performance-based bonus of up to $350,000 to be paid no later than March 15 of the following year based on achievement of criteria established by the Compensation Committee in conjunction with the Board, (iv) relocation expenses not to exceed $75,000 in the event of an involuntary termination that would be subject to tax equalization adjustments, (v) an automobile allowance and related auto insurance of up to $29,000, and (vi) life insurance coverage of $2 million.
In addition, we provide the CEO with certain benefits that are available to all our employees. We do not provide pension arrangements, deferred compensation or other similar benefits to our CEO, except for certain termination benefits as described in detail under the section of this Proxy Statement entitled “Potential Payments Upon Involuntary Termination or a Change of Control.”
The Original Agreement also provided for grants of the following stock options:
· On January 1, 2007 (or the first business day following the effective date of the Original Agreement), and on each anniversary thereafter during the term of the agreement, an annual option to purchase 360,000 shares of the Company’s common stock at a per share exercise price equal to the fair market value on the date of grant. Each annual option vests as to 1/12th of the shares at the end of each month from the grant date.
· On January 3, 2007, an option to purchase 1,000,000 shares of the Company’s common stock at a per share price equal to the fair market value on the date of grant, which vests only upon achieving specified multiple performance milestones. Eight performance milestones are specified in the agreement that, if achieved, would trigger the vesting of the related portion of the option grant.
Pursuant to the Original Agreement, including all unvested performance options granted under the prior employment agreement which expired on December 31, 2006, in the event of an involuntary termination or a change of control of the Company prior to the granting of all of the annual options and any unvested performance options, the securities underlying all ungranted and unvested annual options and all unvested performance options will become fully vested in their entirety and any ungranted annual options shall immediately be granted and vested.
Amended Agreement
On October 28, 2008, the Company and Dr. Manuso executed an Amended and Restated Executive Employment and Confidential Information and Invention Assignment Agreement (the “Amended Agreement”). The Amended Agreement replaced in its entirety the Original Agreement that the Company entered into with Dr. Manuso effective January 1, 2007. The new and modified provisions of the Amended Agreement were intended to bring it into compliance with Section 409A of the Internal Revenue Code of 1986, as amended, and to effect certain other changes.
The changes primarily involved the timing of compensatory payments payable pursuant to the Amended Agreement, as well as a change to the definition of “involuntary termination” and the addition of a new section relating to Section 409A considerations. The substance of the bonus and stock option sections of the Amended Agreement remained unchanged. In addition, a proviso was added to the Amended Agreement to clarify that if Dr. Manuso is involuntarily terminated prior to the occurrence of a change of control transaction, he may be eligible for certain severance benefits as described in the section of this Proxy Statement entitled “Potential Payments Upon Involuntary Termination or a Change of Control.”
2009 Guaranteed Bonus and Performance-Based Bonuses: The guaranteed annual cash bonus of $150,000 was paid for the third year of the Amended Agreement on January 7, 2009. On February 9, 2009, the Compensation Committee approved a performance-based bonus award of $350,000 to Dr. Manuso, based on achievements and performance of the Company during 2008. In addition, on February 9, 2009, the Compensation Committee approved the vesting of 100,000 shares of the 150,000 share performance-based stock option granted in connection with the Amended Agreement.
On January 11, 2010, the Compensation Committee approved two performance-based cash bonus awards to Dr. Manuso. A bonus of $350,000 was granted in connection with the Amended Agreement, as well as an additional bonus of $150,000. The Compensation Committee determined to award both bonuses after reviewing the achievements and performance of the Company during 2009 and Dr. Manuso’s instrumental efforts in that regard. The cash bonuses were paid on January 21, 2010.
New Executive Employment and Confidential Information and Invention Assignment Agreement Effective April 1, 2009:
On April 1, 2009, the Company and Dr. Manuso executed an Executive Employment and Confidential Information and Invention Assignment Agreement (the “New Agreement”). The New Agreement, which supersedes and replaces the Amended Agreement, covers the period from April 1, 2009 through March 31, 2012.
The New Agreement provided for a base salary of $574,752 for the remainder of 2009, increasing to $625,000 on January 1, 2010. The base salary will be adjusted annually thereafter at twice the percentage increase in the Consumer Price Index. The New Agreement also provided for a performance-based bonus of up to $350,000 for 2009 (as described above), and performance bonuses of up to $650,000 per year for the remaining term of the agreement, with each performance bonus to be paid based on achieving criteria established by the Compensation Committee. The New Agreement does not include any guaranteed bonuses. In addition, the New Agreement provides for life insurance coverage of $2 million, an annual automobile allowance of $25,000 for 2009, increasing to $30,000 for each year thereafter, and relocation expenses not to exceed $100,000 in the event of an involuntary termination of employment.
The New Agreement also provides for grants of the following stock options:
· On April 1, 2009, concurrently with the execution of the agreement, Dr. Manuso was granted an option to purchase 1,200,000 shares of the Company’s common stock at a per share price equal to the fair market value the date of grant, with vesting subject to the achievement of specified performance milestones (the “Performance Option”). Ten performance milestone thresholds are specified in the agreement that, if achieved over the three-year term of the agreement, would trigger the vesting of the related portion of the Performance Option. The Performance Option represents approximately 63% of the total potential options that may be granted throughout the term of the New Agreement.
· On or about April 1, 2010 and on each anniversary thereafter during the term of the agreement, an option to purchase 360,000 shares of the Company’s common stock at a per share exercise price equal to the fair market value on the date of grant (the “Annual Options”). Each Annual Option will vest as to 1/12th of the shares at the end of each month from the grant date.
· The New Agreement also provides for the continued vesting of the performance options granted pursuant to Dr. Manuso’s previous employment agreements with the Company, provided the performance milestones are met while he remains employed with the Company.
Pursuant to the New Agreement, in the event of a change of control of the Company prior to: (i) the granting of all of the Annual Options, then all Annual Options yet to be granted will immediately be granted and 100% of the then-unvested shares subject to the Annual Options will vest and become exercisable and (ii) the vesting of the Performance Option, then 100% of the then-unvested shares will immediately vest and become exercisable. If Dr. Manuso’s employment terminates for specified reasons within one year following a change of control, he will receive the following benefits: (1) a lump sum payment equal to eighteen months of base salary; (2) a lump sum payment equal to any unpaid bonuses (not to exceed $1 million); and (3) full acceleration of the vesting of any then-unvested stock options.
Elements of Compensation for Other Executive Officers
The compensation for our other executive officers has three primary components:
· Base salary;
· Participation in the bonus plan; and
· Participation in annual equity compensation awards.
In addition, we provide our other executive officers with certain benefits that are available to all our employees. We do not provide pension arrangements, deferred compensation or other similar benefits to our executive officers, except for certain termination benefits as described in detail under the section of this Proxy Statement entitled “Potential Payments Upon Involuntary Termination or a Change of Control.”
We do not have any employment agreements with our other executive officers that provide for benefits upon involuntary termination or change of control, but we do have an Officer Severance Benefit Plan that describes the recommended severance in the event of an involuntary termination for the other executive officers. Any benefit paid under this plan is subject to approval by the Compensation Committee. We also have acceleration provisions relating to the vesting for option grants in the event of involuntary termination of service following a change of control transaction, as well as an extension of time to exercise such grants following such involuntary termination, to the sooner of twelve months from such termination or the original expiration date of the option.
We believe that this combination of compensation elements provides an appropriate mix of fixed and variable pay, balances short-term operational performance with long-term stockholder value, and facilitates executive retention and recruitment.
Base Salaries
Base salaries are designed to meet competitive norms and reward exemplary performance on an annual basis. In establishing base salaries for our executive officers, the Compensation Committee relies on data from the Radford Survey, the BioWorld Executive Compensation Survey as well as general market sources, to compare base salaries against those for companies with similar numbers of employees and located in similar geographic areas. Based on the competitive landscape, the Compensation Committee initially targets Named Executive Officers’ salaries to be in the 50th to 90th percentile of peer group companies.
For 2009, the Compensation Committee reviewed the base salaries to determine if annual merit increases were to be awarded to the other executive officers based on the achievement of our shorter-term objectives, progress and/or achievement of the Company’s 2009 Performance Priorities and the individual’s annual performance while considering changes in market conditions. The Compensation Committee determined that these goals were achieved and awarded a merit increase to Dr. Mohammad Azab, our Chief Medical Officer (“CMO”), and Mr. Michael Molkentin, our Chief Financial Officer (“CFO”). As discussed below, the merit increases became effective January 1, 2010.
Bonus Plan
We have a performance-based bonus plan that is intended to motivate and reward all employees, including our other executive officers, to perform well and contribute to the achievement of our shorter-term objectives. The amount of bonus is determined based on a target percentage of base salary of an executive officer’s position, the progress and/or achievement of the Company’s performance priorities, and the results of the officer’s individual annual performance review while also reflecting changes in market conditions. The bonus is paid in cash.
2009 Bonus Awards: For 2009, the Compensation Committee reviewed the bonus plan to determine if bonuses were to be awarded to the other executive officers based on the achievement of our shorter-term objectives, progress and/or achievement of the Company’s 2009 Performance Priorities and the executive officer’s individual performance. The Compensation Committee determined that these goals were achieved and awarded a bonus to our CMO and CFO. The initial bonus awards for 2009 were targeted between the 50th and
90th percentile of peer group companies using primarily Radford Survey data. The actual bonus awards for 2009 were calculated as a percent of the officer’s prior year annual base salary and were paid in 2010. The actual bonus awards for 2009 as a percent of base salary for the other executive officers were as follows:
|
|
|
Actual Bonus |
|
Bonus Award Target Range |
|
||
|
Name and Position |
|
Percentage (1) |
|
50th |
|
90th |
|
|
Mohammad Azab |
|
45 |
%(2) |
30 |
% |
50 |
% |
|
Michael Molkentin |
|
43 |
% |
30 |
% |
50 |
% |
(1) Calculated as a percent of prior year annual base salary
(2) The actual bonus award amount was prorated for the period of time the position was actually filled as CMO.
2010 Bonus Award Targets: The bonus awards for 2010 are initially targeted to be within the 50th to 90th percentile of peer group companies. The bonus awards are typically expressed as a percent of the executive officer’s base salary. Considering current year Radford Survey data, the modified bonus award as a percent of base salary for the other executive officers is as follows:
|
|
|
Bonus Award Target Range (Percentile) |
|
||
|
Name and Position |
|
50th |
|
90th |
|
|
Mohammad Azab |
|
30 |
% |
50 |
% |
|
Michael Molkentin |
|
30 |
% |
50 |
% |
After completion of the 2010 fiscal year, the Compensation Committee will determine if bonuses are to be awarded to the other executive officers and at what levels based on the achievement of the Company’s shorter-term objectives, progress and/or achievement of the 2010 Performance Priorities and the executive officer’s individual performance.
Summary of Grant Policies
Our Compensation Committee regularly monitors the environment in which we operate and makes changes to our equity compensation program to help us meet our goals, including the achievement of long-term stockholder value. We may use various forms of equity compensation to motivate and reward long-term performance and encourage our employees, including the executive officers, to participate in the ownership of the Company. Historically, we have granted equity awards to our executive officers in the form of stock options. In spite of the evolution of the accounting treatment of certain types of awards, which requires a company to recognize as an expense the fair value of stock options and other stock-based compensation granted to employees, the Compensation Committee has determined that it is in the best interests of the Company and our stockholders to continue this practice. The Compensation Committee utilizes a vesting schedule to encourage our executive officers to continue in the employ of SuperGen and to encourage executive officers to maintain a long-term perspective. With respect to the CEO, a substantial portion of his equity awards vest only upon achievement of specific performance milestones. In determining the size of stock option grants, the Compensation Committee considers information provided by the Radford Survey, as well as general market sources, and focuses on the executive officers’ current and expected future value to the Company and the competitive influence of peer organizations. The Compensation Committee also considers the number of granted and unvested options held by the executive officer.
The Board and the Compensation Committee have not adopted formal policies regarding the timing of granting equity compensation awards. For example, the Compensation Committee has not established a set date for equity compensation awards, but rather, has acted in a timely manner following the annual performance review process completed for all our employees, including the executive officers, which typically occurs during the first quarter of each fiscal year. Equity compensation grants are approved by the Compensation Committee at
scheduled meetings of the committee or by unanimous written consent. The timing of such actions is driven by the Compensation Committee’s need to conduct particular business, such as an equity compensation grant, and not by the Company’s stock price. The exercise price or calculation price used in connection with any equity compensation grant is determined as the closing price for the Company’s common stock on NASDAQ on the date of the meeting at which the grant is approved. The Compensation Committee has not granted, nor does it intend in the future to grant, equity compensation awards in anticipation of the release of material nonpublic information that is likely to result in changes to the price of our common stock, such as a significant positive or negative earnings announcement. Similarly, our Compensation Committee has not timed, nor does it intend in the future to time, the release of material nonpublic information based on equity award grant dates. We do not reprice or exchange underwater options without stockholder approval.
Equity Compensation
2009 Equity Awards: For the 2009 annual period, the Compensation Committee reviewed outstanding executive officer equity compensation to determine if equity awards were to be granted to the executive officers other than our CEO to motivate and reward longer-term performance, enhance retention and encourage participation in the ownership of the Company. The equity compensation grants were based on the achievement of our shorter-term objectives, progress and/or achievement of the Company’s 2009 Performance Priorities and the executive officer’s annual performance review. The Compensation Committee determined that these goals were achieved and awarded additional option grants to our CMO and CFO. The equity awards were originally targeted between the 50th and 90th percentile of peer group companies primarily using the Radford Survey. Equity compensation is made in the sole discretion of the Compensation Committee and is based on market information provided primarily by Radford Survey data, including recommendations by the CEO and other market considerations. The actual equity awards based on the 2009 period under review for the executive officers other than our CEO were as follows:
|
|
|
Actual Equity |
|
Equity Award Target Range (#) |
|
||
|
Name and Position |
|
Award (#) |
|
50th |
|
90th |
|
|
Mohammad Azab |
|
60,000 |
(1) |
50,000 |
|
150,000 |
|
|
Michael Molkentin |
|
110,000 |
|
50,000 |
|
150,000 |
|
(1) The actual equity award represents a prorated award for the period of time the position was actually filled as CMO.
2010 Equity Award Targets: The equity awards for 2010 are initially targeted to be within the 50th to 90th percentile of peer group companies. Considering current year Radford Survey data, the modified equity award target for the other executive officers is as follows:
|
|
|
Equity Award Target Range (#) |
|
||
|
Name and Position |
|
50th |
|
90th |
|
|
Mohammad Azab |
|
60,000 |
|
160,000 |
|
|
Michael Molkentin |
|
60,000 |
|
160,000 |
|
After completion of the 2010 fiscal year, the Compensation Committee will determine if equity awards are to be awarded to the executive officers other than our CEO and at what level based on the achievement of the Company’s shorter-term objectives, progress and/or achievement of the 2010 Performance Priorities and the executive officer’s individual performance.
Summary of Compensation Committee Actions for Other Executive Officers
On March 12, 2009, the Compensation Committee approved 2009 annual salaries, cash bonus awards, and granted options for the achievement of the Company’s 2008 Performance Priorities and annual performance to the following executive officers:
|
Name and Position |
|
Annual Salary |
|
Cash Bonus |
|
Stock Option |
|
||
|
David J. Bearss |
|
$ |
328,000 |
|
$ |
57,000 |
|
45,000 |
|
|
Gregory Berk |
|
383,000 |
|
112,000 |
|
80,000 |
|
||
|
Michael Molkentin |
|
327,000 |
|
119,000 |
|
80,000 |
|
||
(1) Annual salaries retroactive to January 1, 2009.
(2) Option grants are subject to terms and conditions of the 2003 Stock Plan, as amended, and will vest monthly over a period of 48 months.
On March 4, 2010, the Compensation Committee approved 2010 annual salaries, including cash bonus awards, and granted options for the achievement of the Company’s 2009 Performance Priorities and annual performance to the following executive officers:
|
Name and Position |
|
Annual Salary |
|
Cash Bonus |
|
Stock Option |
|
||
|
Mohammad Azab |
|
$ |
393,000 |
|
$ |
78,000 |
|
60,000 |
|
|
Michael Molkentin |
|
342,000 |
|
141,000 |
|
110,000 |
|
||
(1) Annual salaries are retroactive to January 1, 2010.
(2) Option grants are subject to terms and conditions of the 2003 Stock Plan, as amended, and will vest monthly over a period of 48 months.
Generally Available Benefit Programs
We also offer a number of other benefits to our employees and all executive officers including medical, prescription, dental and vision insurance, long-term and short-term disability insurance, life and accidental death and dismemberment insurance, health and dependent care flexible spending accounts, paid holidays, floating holidays, vacation, personal time off, and employee assistance programs.
We believe that these benefit programs generally enhance employee productivity and loyalty to the Company. The main objectives of our benefit programs are to give all our employees access to quality healthcare, financial protection from unforeseen events, assistance in achieving retirement financial goals, and enhanced health and productivity. These benefit programs typically do not specifically factor into decisions regarding an individual employee’s or executive officer’s total compensation or equity award package. The availability of these benefit programs are influenced more by competitive market considerations for biotech and other industries against whom we compete to either retain our current employees or attract new talent.
401(k) Plan
We also maintain a 401(k) Plan to provide retirement benefits through tax deferred salary deductions for all employees. We make matching employer contributions, at rates varying from 1% to 3%, up to a maximum of $6,000 annually, based on the rate of the employee’s 401(k) payroll contribution. Our matching contributions vest ratably over five years.
Internal Revenue Code Section 162(m) Implications for Executive Compensation
The Compensation Committee is responsible for addressing issues raised by Section 162(m) of the Internal Revenue Code. Section 162(m) limits the Company’s tax deduction for compensation paid to certain executive officers that does not qualify as “performance based” to $1 million per executive officer. The Compensation Committee believes that the stockholders’ interests are served by maintaining the discretion and flexibility in our executive compensation programs. Accordingly, the Compensation Committee may approve executive compensation that is not fully deductible. If our stockholders approve Proposal Two herein, that will enable us to continue to qualify certain awards under our 2003 Stock Plan as deductible performance-based compensation.
Compensation Committee Interlocks and Insider Participation
Our Compensation Committee was formed in January 1993. The Compensation Committee is composed of Mr. Lack (Chairman) and Mr. Girardi, who are independent directors of the Company. Neither of these persons was an employee of the Company or any of its subsidiaries, nor were there any Compensation Committee interlocks or other relationships during 2009 requiring disclosure under Item 407(e)(4) of Regulation S-K of the Securities Act of 1933, as amended.
Potential Payments Upon Involuntary Termination or Change of Control
Dr. Manuso’s New Agreement requires specific payments and/or benefits to be provided to Dr. Manuso in the event of an involuntary termination of employment without cause following a change of control of the Company. If an involuntary termination for cause occurs, Dr. Manuso will not receive any additional severance-type benefits under the New Agreement.
Dr. Manuso’s New Agreement defines “change of control” as the occurrence of any of the following events:
· Any “person” (as such term is used in Sections 13(d) and 14(d) of the Exchange Act) becomes the “beneficial owner” (as defined in Rule 13d-3 of the Exchange Act), directly or indirectly, of securities of the Company representing 50% or more of the total voting power represented by the Company’s then outstanding voting securities;
· The consummation of the sale or disposition by the Company of all or substantially all of the Company’s assets; or
· The consummation of a merger or consolidation of the Company with any other corporation, other than a merger or consolidation which would result in the voting securities of the Company outstanding immediately prior thereto continuing to represent (either by remaining outstanding or by being converted into voting securities of the surviving entity or its parent) at least 50% of the total voting power represented by the voting securities of the Company or such surviving entity or its parent outstanding immediately after such merger or consolidation.
“Involuntary Termination,” as used in the New Agreement, means the following:
· Without Dr. Manuso’s express written consent, a material diminution of his duties, position or responsibilities relative to his duties, position or responsibilities in effect immediately prior to such reduction;
· Without Dr. Manuso’s express written consent, a material diminution by the Company of his base salary as in effect immediately prior to such reduction;
· Any material breach by the Company of any of the terms of the New Agreement;
· Without Dr. Manuso’s express written consent, his relocation to a facility or a location more than fifty miles from the current location of the Company, which the Company and Dr. Manuso agree would constitute a material change in the geographic location at which he must perform services to the Company, or
· Any purported termination of Dr. Manuso other than for cause.
“Cause,” as used in the New Agreement, means the following:
· Any act of personal dishonesty taken by Dr. Manuso in connection with his employment which is intended to result in his personal enrichment;
· Dr. Manuso’s conviction of a felony;
· Any act by Dr. Manuso that constitutes material misconduct and is injurious to the Company; or
· Continued violations by Dr. Manuso of his obligations to the Company.
Under the New Agreement, Dr. Manuso may not resign for an Involuntary Termination without first providing the Company with:
· A written notice within ninety days of the event that he believes constitutes an Involuntary Termination specifically identifying the acts or omissions constituting the grounds for an Involuntary Termination, and
· A reasonable cure period of not less than thirty days following the date of such notice.
The New Agreement provides that if Dr. Manuso’s employment with the Company is terminated by the Company as a result of an Involuntary Termination and following a change of control, he would receive:
· A lump sum payment equivalent to eighteen months of his then current base salary;
· A lump sum payment equivalent to any unpaid amount of bonus due to him (up to a maximum of $1 million);
· Reimbursement for all reasonable relocation expenses to him or his family’s relocation from California to New York, including, but not limited to, short-term hotel costs or apartment rental for a period not to exceed six months, with the total relocation not to exceed $100,000. Additional cash compensation would be paid to fully offset taxes, or tax equalized, attributable to him as a result of payment of such reasonable relocation expenses;
· Full acceleration of the vesting of any then-unvested stock options held by him or any annual stock options not yet granted to him.
Estimated Value of Involuntary Termination or Change of Control Benefits for Other Executive Officers
Though the Company does not have employment agreements with any executive officer other than Dr. Manuso, we do have an Officer Severance Benefit Plan that describes the recommended severance in the event of certain involuntary terminations for all the Company’s executive officers, where applicable. Effective October 28, 2008, the Company revised its Officer Severance Benefit Plan, which provides for severance payments, career transition assistance and COBRA continuation coverage for the Company’s executive officers in the event they are involuntarily terminated, to bring it into compliance with Section 409A of the Internal Revenue Code.
Benefits under the Officer Severance Benefit Plan include the following:
· Cash Severance Benefit. Each eligible executive officer shall receive a cash severance benefit in an amount equal to the sum of (a) two weeks of such eligible executive officer’s base salary, which shall be paid in lieu of notice of termination of employment, (b) an additional thirty-nine weeks of such eligible executive officer’s base salary, (c) an additional two weeks of such eligible executive officer’s base salary for each full year of service completed, and (d) an additional one week of such eligible executive officer’s base salary for any partial year of service completed provided that such partial year of service is greater than six months in length.
· Career Transition Assistance. Following an eligible executive officer’s termination of employment, career transition services shall be provided through an outplacement service provider for a period of nine months. Outplacement services currently cost the Company approximately $8,500 per executive officer.
· COBRA Continuation Coverage. Each eligible executive officer who is enrolled in a health, dental, or vision plan sponsored by the Company may be eligible under COBRA to continue coverage under such health, dental, or vision plan (or to convert to an individual policy), at the time of his or her termination of employment. If COBRA is elected by an eligible executive officer, the Company shall pay COBRA premiums on behalf of the executive officer during the number of weeks of base salary in respect of which the amount paid to the eligible executive officer under the Cash Severance Benefit section, as described above, was calculated.
Eligible executive officers are required to sign and not revoke a release of claims in favor of the Company as a condition to receiving benefits. No benefits are payable upon any voluntary termination, upon any involuntary termination for misconduct or poor job performance or upon certain other terminations of employment. The Officer Severance Benefit Plan does not provide any income tax gross-ups for golden parachute excise taxes nor do we otherwise provide golden parachute excise tax gross-ups to our executive officers.
In addition to the Officer Severance Benefit Plan, our Named Executive Officers have double-trigger 100% vesting acceleration on their Company stock options. Specifically, if our Named Executive Officers are involuntarily terminated other than for cause following a change of control, then 100% of the shares subject to their outstanding stock options accelerate as to vesting. Additionally, in such event, their stock option post-termination exercise period is extended from three months after employment termination to twelve months after employment termination or, if earlier, the original maximum term of the option.
2009 Potential Payments Upon Termination Table
|
|
|
|
|
Severance |
|
Bonus |
|
Accelerated |
|
|
|
|
Name |
|
Termination Scenario |
|
($) (1) |
|
($) (2) |
|
Vesting ($) |
|
Other ($) |
|
|
James S.J. Manuso |
|
Change of control |
|
862,128 |
|
1,000,000 |
|
731,000 |
(3) |
144,118 |
(6) |
|
|
|
Involuntary (without cause) |
|
862,128 |
|
1,000,000 |
|
— |
|
144,118 |
(6) |
|
|
|
For cause |
|
— |
|
— |
|
— |
|
— |
|
|
Mohammad Azab |
|
Change of control |
|
303,558 |
|
— |
|
10,400 |
(4) |
10,407 |
(7) |
|
|
|
Involuntary (without cause) |
|
303,558 |
|
— |
|
— |
|
10,407 |
(7) |
|
|
|
For cause |
|
— |
|
— |
|
— |
|
— |
|
|
Michael Molkentin |
|
Change of control |
|
333,288 |
|
— |
|
77,630 |
(5) |
11,353 |
(7) |
|
|
|
Involuntary (without cause) |
|
333,288 |
|
— |
|
— |
|
11,353 |
(7) |
|
|
|
For cause |
|
— |
|
— |
|
— |
|
— |
|
|
(1) |
Assumes “severance” payment made to the Named Executive Officer as of the last business day of the fiscal year or December 31, 2009. |
|
(2) |
Represents bonus payout for remaining term of the employment agreement, not to exceed $1 million. |
|
(3) |
Represents accelerated vesting of 2,000,000 previously granted unvested performance-based stock options and the granting and vesting of 720,000 ungranted annual stock options remaining under the term of the New Agreement. The exercise price related to 850,000 of these options was under the fair market value of SuperGen’s stock as of December 31, 2009, and therefore, these options had an intrinsic value of $731,000 at December 31, 2009. |
|
(4) |
Represents accelerated vesting of 260,000 previously granted unvested stock options. The exercise price related to all of these options was under the fair market value of SuperGen’s stock as of December 31, 2009, and therefore, resulting in an intrinsic value of $10,400 at December 31, 2009. |
|
(5) |
Represents accelerated vesting of 136,109 previously granted unvested stock options. The exercise price of 119,000 of those options was under the fair market value of SuperGen’s stock as of December 31, 2009, resulting in an intrinsic value of $77,630 at December 31, 2009. |
|
(6) |
Represents reimbursement for relocation expenses not to exceed $100,000, which are subject to tax equalization adjustments, and employer-paid medical coverage for total estimated severance period. |
|
(7) |
Represents employer-paid medical coverage for total estimated severance period. |
The actual amount of the benefits paid to the Named Executive Officers in the event of an involuntary termination or a change of control can only be determined at the time of the executive’s actual termination from the Company.
2009 Summary Compensation Table
The following table presents the total compensation earned by each of the Named Executive Officers during the fiscal year ended December 31, 2009.
|
Name and |
|
Year |
|
Salary |
|
Bonus |
|
Option Awards |
|
All Other |
|
Total |
|
|
James S.J. Manuso |
|
2009 |
|
574,237 |
|
650,000 |
(2) |
1,670,653 |
|
36,644 |
(3) |
2,781,534 |
|
|
President and Chief Executive Officer |
|
2008 |
|
560,406 |
|
500,000 |
|
727,308 |
|
40,748 |
|
1,828,462 |
|
|
|
2007 |
|
512,731 |
|
500,000 |
|
3,888,340 |
|
33,721 |
|
4,834,692 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Mohammad Azab (4) |
|
2009 |
|
141,044 |
|
78,000 |
|
427,076 |
|
— |
|
568,120 |
|
|
Chief Medical Officer |
|
2008 |
|
— |
|
— |
|
— |
|
— |
|
— |
|
|
|
|
2007 |
|
— |
|
— |
|
— |
|
— |
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
David J. Bearss (5) |
|
2009 |
|
306,658 |
|
— |
|
45,594 |
|
6,000 |
(7) |
415,252 |
|
|
Former Chief Scientific Officer |
|
2008 |
|
236,486 |
|
57,000 |
|
102,366 |
|
6,000 |
|
389,732 |
|
|
|
2007 |
|
— |
|
44,880 |
|
— |
|
— |
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Gregory Berk (6) |
|
2009 |
|
175,476 |
|
— |
|
81,056 |
|
335,720 |
(8) |
704,252 |
|
|
Former Chief Medical Officer |
|
2008 |
|
370,125 |
|
112,000 |
|
150,380 |
|
6,000 |
|
617,505 |
|
|
|
2007 |
|
193,622 |
|
111,000 |
|
565,230 |
|
— |
|
778,852 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Michael Molkentin |
|
2009 |
|
326,375 |
|
141,000 |
|
81,056 |
|
6,000 |
(7) |
532,431 |
|
|
Chief Financial Officer |
|
2008 |
|
310,076 |
|
119,000 |
|
144,365 |
|
6,000 |
|
567,441 |
|
|
|
2007 |
|
265,398 |
|
107,000 |
|
128,502 |
|
6,000 |
|
476,580 |
|
|
(1) |
Reflects the aggregate grant date fair value using the Black-Scholes option pricing model for option awards granted during the year computed in accordance with ASC 718, Compensation-Stock Compensation. The value of the option awards reported for 2008 and 2007 have been revised from prior years’ proxy disclosure to reflect their grant date fair value in accordance with the revised SEC disclosure requirements relating to such awards. The assumptions used in the valuation of these awards are set forth in the notes to our consolidated financial statements, which are included in our Annual Report on Form 10-K for the year ended December 31, 2009, filed with the SEC on March 15, 2010. These amounts do not correspond to the actual value that could be realized by each Named Executive Officer. |
|
(2) |
Amount includes a $150,000 guaranteed bonus earned and paid on January 7, 2009 under terms of a prior executive employment agreement, in addition to a $350,000 annual bonus and a $150,000 discretionary bonus earned under the new executive employment agreement effective April 1, 2009. |
|
(3) |
Includes $26,804 for car allowances, $6,000 for 401(k) Company match, and $3,840 for life insurance premiums. |
|
(4) |
Dr. Azab joined the Company in July 2009. |
|
(5) |
Dr. Bearss was first appointed as an executive officer of the Company in November 2008. The prior year’s compensation is not shown as he was not an executive officer in 2007. Dr. Bearss left the Company in October 2009. |
|
(6) |
Dr. Berk left the Company in May 2009. |
|
(7) |
Represents 401(k) Company match. |
|
(8) |
Includes $4,278 for 401(k) Company match and $331,442 in severance pay. |
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES.
|
Exhibit |
|
Description of Document |
|
31.1 |
|
Certification of Chief Executive Officer under Section 302(a) of the Sarbanes-Oxley Act of 2002. |
|
31.2 |
|
Certification of Chief Financial Officer under Section 302(a) of the Sarbanes-Oxley Act of 2002. |
|
32.1 |
|
Certifications of Chief Executive Officer and Chief Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this Amendment No. 1 to the Report on Form 10-K/A to be signed on its behalf by the undersigned, thereunto duly authorized, on this 8th day of December 2010.
|
|
SUPERGEN, INC. |
|
|
|
|
|
|
|
By: |
/s/ JAMES S. J. MANUSO |
|
|
|
James S.J. Manuso |
POWER OF ATTORNEY
Pursuant to the requirements of the Securities Exchange Act of 1934, this Amendment No. 1 to the Report on Form 10-K/A has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated:
|
Signature |
|
Title |
|
Date |
|
|
|
|
|
|
|
/S/ JAMES S.J. MANUSO |
|
Chief Executive Officer, President and Director |
|
December 8, 2010 |
|
(James S.J. Manuso) |
|
(Principal Executive Officer) |
|
|
|
|
|
|
|
|
|
/S/ MICHAEL MOLKENTIN |
|
Chief Financial Officer |
|
December 8, 2010 |
|
(Michael Molkentin) |
|
(Principal Financial and Accounting Officer) |
|
|
|
|
|
|
|
|
|
* |
|
Director |
|
December 8, 2010 |
|
(Charles J. Casamento) |
|
|
|
|
|
|
|
|
|
|
|
* |
|
Director |
|
December 8, 2010 |
|
(Thomas V. Girardi) |
|
|
|
|
|
|
|
|
|
|
|
* |
|
Director |
|
December 8, 2010 |
|
(Allan R. Goldberg) |
|
|
|
|
|
|
|
|
|
|
|
* |
|
Director |
|
December 8, 2010 |
|
(Walter J. Lack) |
|
|
|
|
|
|
|
|
|
|
|
* |
|
Director |
|
December 8, 2010 |
|
(Michael D. Young) |
|
|
|
|
|
|
|
|
|
|
|
* By: /S/ JAMES S.J. MANUSO |
|
|
|
|
|
James S.J. Manuso |
|
|
|
|
|
Attorney-in-Fact |
|
|
|
|