Attached files
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, DC 20549
FORM 10-K
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended June 30, 2010
or
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission File Number 000-51122
PSIVIDA CORP.
(Exact name of registrant as specified in Its charter)
| Delaware | 26-2774444 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) |
| 400 Pleasant Street Watertown, MA |
02472 | |
| (Address of principal executive offices) | (Zip Code) |
Registrant’s telephone number, including area code: (617) 926-5000
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class |
Name of each exchange on which registered | |
| Common Stock, $.001 par value per share | The NASDAQ Stock Market LLC (NASDAQ Global Market) |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ¨ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer,” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer ¨ |
Accelerated filer ¨ | |
| Non-accelerated filer ¨ |
Smaller reporting company x | |
| (Do not check if a smaller reporting company) |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ¨ No x
The aggregate market value of the common stock held by non-affiliates of the registrant, computed by reference to the closing price of the common stock on the NASDAQ Global Market on December 31, 2009, the last trading day of the registrant’s most recently completed second fiscal quarter, was approximately $57,457,000.
There were 18,531,392 shares of the registrant’s common stock, $0.001 par value, outstanding as of September 22, 2010.
DOCUMENTS INCORPORATED BY REFERENCE
Specified portions of the registrant’s definitive proxy statement, to be filed in connection with the Annual Meeting of Stockholders to be held on December 9, 2010, are incorporated by reference into Part III of this Annual Report on Form 10-K.
Table of Contents
PSIVIDA CORP.
Form 10-K
For the Fiscal Year Ended June 30, 2010
Table of Contents
Preliminary Note Regarding Forward-Looking Statements
This Form 10-K and our 2010 Annual Report contain forward-looking statements, within the meaning of Section 27A of the Securities Act of 1933, as amended (Securities Act) and Section 21E of the Securities Exchange Act of 1934, as amended (Exchange Act). Forward-looking statements are inherently subject to risks, uncertainties and potentially inaccurate assumptions. Such statements give our current expectations or forecasts of future events; they do not relate strictly to historical or current facts. All statements other than statements of historical fact could be deemed forward-looking statements, including, without limitation, any expectations of revenue, expenses, cash flows, earnings or losses from operations, capital or other financial items; any statements of the plans, strategies and objectives of management for future operations; any statements concerning product research, development and commercialization timelines; any statements of expectations or belief; and any statements of assumptions underlying any of the foregoing. We often, although not always, identify forward-looking statements by using words or phrases such as the following: “likely”, “expect”, “intend”, “anticipate”, “believe”, “estimate”, “plan”, “project”, “forecast” and “outlook”.
We cannot guarantee that the results and other expectations expressed, anticipated or implied in any forward-looking statement will be realized. The risks set forth under Item 1A of this Form 10-K describe major risks to our business, and you should read and interpret any forward-looking statements together with these risks. A variety of factors, including these risks, could cause our actual results and other expectations to differ materially from the anticipated results or other expectations expressed, anticipated or implied in our forward-looking statements. Should known or unknown risks materialize, or should our underlying assumptions prove inaccurate, actual results could differ materially from past results and those anticipated, estimated or projected in the forward-looking statements. You should bear this in mind as you consider any forward-looking statements.
Our forward-looking statements speak only as of the dates on which they are made. We do not undertake any obligation to update any forward-looking statement, whether to reflect new information, future events or otherwise. You are advised, however, to consult any further disclosures we may make in our future reports to the SEC, on our website, www.psivida.com, or otherwise.
| ITEM 1. | BUSINESS |
Introduction
We develop tiny, sustained release, drug delivery products that are administered by implantation, injection or insertion. Once administered, a drug is released on a controlled and level basis for months or years. We have two core technology systems, Durasert™ and BioSilicon™. Utilizing three generations of our Durasert technology system, we have one product candidate for chronic eye disease that has been given Priority Review by the U.S. Food and Drug Administration (FDA) and two of the only three products approved by the FDA for the long-term, sustained release delivery of drug to treat chronic eye disease. We have a collaboration with Pfizer, Inc. (Pfizer), our largest shareholder, to develop additional ophthalmic products.
Iluvien™, the product candidiate with Priority Review, is designed to provide sustained release treatment for Diabetic Macula Edema (DME). DME is a leading cause of vision loss for people under the age of 65 and has been estimated to affect over 1,000,000 people in the United States. Using the third-generation of our Durasert technology system, Iluvien is injected into the eye and delivers the corticosteroid fluocinolone acetonide (FA) over a period of up to 3 years.
Iluvien is licensed to Alimera Sciences, Inc (Alimera), which is completing fully-recruited Phase III clinical trials. Based on 24-month data released in December 2009, Alimera filed a New Drug Application (NDA) with the FDA in June 2010 and registration filings in various European countries in July 2010. On August 30, 2010, the FDA granted Priority Review status and, as a result, Alimera could receive a response to its NDA from the
1
Table of Contents
FDA by the end of calendar year 2010. If approved, Alimera has indicated that it expects to commercialize Iluvien as early as the first calendar quarter of 2011. Under our collaboration agreement with Alimera, Iluvien is also being studied in investigator-sponsored pilot clinical trials designed to assess the safety and efficacy of Iluvien in both wet and dry Age-Related Macular Degeneration (AMD) and Retinal Vein Occlusion (RVO).
Our two FDA-approved products utilize earlier generations of our Durasert technology system, second-generation Retisert® for the treatment of posterior uveitis, and first-generation Vitrasert® for the treatment of AIDS-related cytomegalovirus (CMV) retinitis. We have licensed both of these products and the technologies underlying them to Bausch & Lomb Incorporated (Bausch & Lomb). Retisert provides sustained release treatment for approximately two and a half years, and Vitrasert provides sustained release treatment for six to nine months.
Under our worldwide collaborative research and license agreement with Pfizer, we are working together on a joint research program aimed at developing certain ophthalmic applications of our sustained drug delivery technologies not licensed to others.
BioSilicon, our other principal technology system, is a fully-erodible, nanostructured, porous silicon designed to provide sustained delivery of various therapeutics, including small drug molecules, proteins and peptides. Based on results of our preliminary studies, we are currently targeting BioSilicon as a key second prong of our drug delivery technology platform.
Medidur™, Durasert™, BioSilicon™, BrachySil™ and CODRUG™ are our trademarks. Retisert® and Vitrasert® are Bausch & Lomb’s trademarks. Iluvien™ is Alimera’s trademark. This Report also contains trademarks, trade names and service marks of other companies, which are the property of their respective owners.
Market Overview
Drug Delivery Generally
The therapeutic value of a drug depends on its distribution throughout the body, reaction with the targeted site, reaction with other tissues and organs in the body and clearance from the body. In an ideal treatment, the appropriate amount of drug is delivered to the intended site at an adequate concentration and maintained there for a sufficient period of time without adverse effect to other tissues and organs. Accordingly, the manner in which a drug is delivered can be as important to the ultimate therapeutic value of the treatment as the intrinsic properties of the drug itself.
Drugs are typically administered systemically by oral dosing or by injection, and are subsequently dispersed throughout the body via the circulatory system. In many cases, systemic administration does not deliver drugs to the intended site at an adequate concentration for a sufficient period of time or fails to achieve the maximum potential therapeutic benefit.
Because systemically delivered drugs disperse throughout the body, they often must be administered at high dosage levels in order to achieve sufficient concentrations at the intended site. Some areas of the body, such as the eyes, joints, brain and nervous system, have natural barriers that impede the movement of drugs to those areas, requiring the administration of even higher systemic doses. These high dosage levels can cause harmful side effects when the drug interacts with other tissues and organs.
Timely and repeated administration of drugs is often necessary to maintain therapeutic drug levels over an extended period of time. However, patients often fail to take drugs as prescribed or fail to attend follow-up visits and, as a result, they do not receive the potential therapeutic benefit. The risk of patient noncompliance increases if multiple drugs are required, if the dosing regimen is complicated or if the patient is elderly or cognitively impaired.
2
Table of Contents
Due to the drawbacks of traditional systemic drug delivery, the development of methods to deliver drugs to patients in a more precise, controlled fashion over sustained periods of time has become a multi-billion dollar industry. Such methods include oral and injectable controlled-release products and skin patches. These methods seek to improve the consistency of the dosage over time and extend the duration of delivery. However, most of these methods still cannot provide constant, controlled dosage or deliver drugs for a sufficiently long duration. This reduces their effectiveness for diseases that are chronic or require precise dosing. In addition, most of these methods still deliver drugs systemically, and, as a result, can still cause adverse side effects throughout the body.
Ophthalmic Drug Delivery
Delivery of drugs to treat back-of-the-eye diseases is a significant issue in ophthalmology. Due to the effectiveness of the blood/eye barrier, it is difficult for systemically administered drugs to reach the eye in sufficient quantities to have a beneficial effect without adverse side effects to other parts of the body. There is a need for drug delivery inside the eye in a manner that is safe, effective and practical for long-term use. While there are currently many approaches to delivering medications to the eye, most do not achieve sufficient and consistent concentrations within the eye for the appropriate period of time.
Injecting drugs in solution directly into the back of the eye can achieve effective, but often transient, drug levels in the eye, requiring repeated injections. Examples include Macugen® (pegaptanib sodium) and Lucentis® (ranibizumab, formerly RhuFab V2), both of which may be injected into the eye as frequently as approximately every four to six weeks. Apart from inconvenience and cost, repeated intravitreal injections carry risks, including intraocular infection, perforated sclera, vitreous hemorrhage and cataract formation.
Technologies and Products
Our primary technology systems are Durasert and BioSilicon.
Durasert Technology System
Iluvien, Retisert and Vitrasert, as well as some of our other product candidates, use our proprietary Durasert technology system, which delivers specific quantities of drugs directly to a target site in the body at controlled rates for predetermined periods of time ranging from days to years. The Durasert technology system is designed to provide the benefits of direct delivery of appropriate quantities of drug over an extended period, while addressing the drawbacks of systemic drug delivery, including adverse side effects characteristic of high dosing levels and reduced treatment benefits due to variations in drug levels at the target site. The Durasert technology system has three principal attributes designed to deliver these advantages:
| • | Localized Delivery. The Durasert technology system permits implantation, injection or other application of a drug directly at the target site. This administration allows the natural barriers of the body to isolate and assist in maintaining appropriate concentrations of the drug at the target site in an effort to achieve the maximum therapeutic effect of a drug while minimizing unwanted systemic effects. |
| • | Controlled Release Rate. The Durasert technology system releases drugs at a constant, controlled rate. We believe that this feature allows our products and product candidates to deliver and maintain optimal drug concentrations at a target site and eliminate variability in dosing over time. |
| • | Extended Delivery. The Durasert technology system delivers drugs for predetermined periods of time ranging from days to years. We believe that uninterrupted, sustained delivery offers the opportunity to develop products that reduce the need for repeat applications, eliminate the risk of patient noncompliance and provide more effective treatment. |
The Durasert technology system uses a drug core with one or more surrounding polymer layers. The drug release is controlled by the permeability of the polymer layers. By changing the design of the Durasert technology system, we can control both the rate and duration of release to meet different therapeutic needs. We believe that the Durasert technology system can be used to deliver a wide variety of different drugs.
3
Table of Contents
Our portfolio of Durasert products and product candidates includes:
| Product |
Disease |
Stage of Development |
Licensee | |||
| Vitrasert |
CMV Retinitis | FDA-approved; commercialized since 1996 | Bausch & Lomb | |||
| Retisert |
Posterior uveitis | FDA-approved; commercialized since 2005 | Bausch & Lomb | |||
| Iluvien |
Diabetic macular edema (DME) | Phase III clincial trials; NDA filed June 2010 | Alimera | |||
| Iluvien |
Wet age-related macular degeneration (Wet AMD) | Investigator-sponsored pilot clinical trial | Alimera | |||
| Iluvien |
Dry age-related macular degeneration (Dry AMD) | Investigator-sponsored pilot clinical trial | Alimera | |||
| Iluvien |
Retinal vein occlusion | Investigator-sponsored pilot clinical trial | Alimera | |||
| TBD |
Glaucoma | Pre-clinical | ||||
| TBD |
Retinitis pigmentosa | Pre-clinical | ||||
| TBD |
Dry AMD | Pre-clinical |
Iluvien
Iluvien is designed to treat DME, a disease that causes swelling in the macula, the most sensitive part of the retina. DME is a major cause of vision loss in diabetics and a leading cause of vision loss for Americans under 65, and has been estimated to affect over 1,000,000 people in the United States. Iluvien, which is inserted via a 25-gauge, transconjunctival delivery system to the back of the eye in an in-office procedure, is designed to deliver FA on a sustained basis for up to 36 months. There is currently no FDA-approved drug treatment for DME. The only FDA-approved method for treating DME is laser photocoagulation therapy, which has only modest efficacy and can leave irreversible blind spots.
Alimera is also studying Iluvien in three investigator-sponsored pilot clinical trials with respect to other chronic eye diseases. One trial is designed to assess the safety and efficacy of Iluvien in conjunction with Lucentis in patients with exudative age-related macular degeneration (wet AMD) to provide information on the potential of Iluvien to maintain the efficacy of Lucentis while reducing the overall number of Lucentis treatments. A second trial is designed to assess the safety and efficacy of Iluvien in patients with bilateral geographic atrophy secondary to dry-AMD. The third trial is designed to assess the safety and efficacy of Iluvien in patients with macular edema secondary to retinal vein occlusion.
Development Program for Iluvien for the Treatment of DME
Alimera is currently completing the FAME Study for Iluvien involving 956 patients in sites across the United States, Canada, Europe and India to assess the efficacy and safety of Iluvien in the treatment of DME. Combined enrollment was completed in October 2007, and the 24-month clinical readout from the FAME Study was received in December 2009. Alimera submitted an NDA in the United States for the low dose of Iluvien to the FDA in June 2010 based on the 24-month clinical data and the following month submitted a Marketing Authorization Application (MAA) for low dose Iluvien to the Medicines and Healthcare Products Regulatory Agency (MHRA) in the United Kingdom. The MAA is being submitted through the Decentralized Procedure with the UK MHRA as the Reference Member State (RMS). Applications have also been submitted to the following other Concerned Member States (CMS) in the European Union: Austria, France, Germany, Italy, Portugal and Spain. In August 2010, Alimera was notified by the FDA that its NDA was accepted for review and granted Priority Review status. Under Priority Review, a decision from the FDA could be received by the end of calendar year 2010. Alimera has indicated that the Iluvien injection system will not require a separate device application, but it must meet the safety and regulatory requirements of the applicable regulatory authorities when evaluated as part of the drug product marketing application. Alimera has indicated that it plans to follow its MAA submission with a registration filing in Canada in the near future.
4
Table of Contents
Consistent with the FDA requirement for registration and approval of drugs being developed for diabetic retinopathy, including DME, the primary efficacy endpoint for the FAME Study is the difference in the percentage of patients whose best corrected visual acuity (BCVA) improved from baseline by 15 or more letters on the Early Treatment Diabetic Retinopathy Study (ETDRS) eye chart between the treatment and control groups at month 24. The ETDRS eye chart is the standard used in clinical trials for measuring sharpness of sight as established by the National Eye Institute’s Early Treatment Diabetic Retinopathy Study. In addition, the FDA requires a numerical comparison of the percentage of patients with BCVA improvement of 15 or more letters between the month 24 and month 18 data to determine if the month 24 results are equal to or greater than the month 18 results. Patients enrolled in the FAME Study will be followed by Alimera for 36 months. Although Alimera has reported that it will submit the additional 12 months of clinical data to applicable regulatory authorities, the approval of Iluvien by regulatory authorities, including the FDA, will be based on the month 24 clinical data from the FAME Study.
FAME Study
The FAME Study, initiated by Alimera in September 2005, is divided into Trial A and Trial B (each having identical protocols) and completed enrollment in October 2007 of 956 patients across 101 academic and private practice centers. Trial A drew patients from sites located in the northern regions of the United States, Europe and India and all sites in Canada, while sites in the southern regions of the United States, India and Europe comprise Trial B.
The FAME Study was designed to assess the safety and efficacy of Iluvien in patients with DME involving the center of the macula, and who had at least one prior macular laser treatment 12 weeks or more before study entry. The inclusion criteria for the FAME Study were designed to select DME patients with BCVA between 20/50 (68 letters on the ETDRS eye chart) and 20/400 (19 letters on the ETDRS eye chart) in the study eye and no worse than 20/400 in the non-study eye. Patients who had received steroid drug treatments for DME within three months of screening, or anti-VEGF injections within two months of screening, and patients with glaucoma, ocular hypertension, IOP greater than 21mmHg or concurrent therapy with IOP-lowering agents in the study eye at screening were not eligible to participate in the trial.
Patient characteristics, such as age, gender and baseline BCVA, were balanced across the treatment and control groups. As part of randomization, the patients were divided into two separate groups, those with a baseline BCVA score greater than or equal to 49 letters on the ETDRS eye chart and those with a baseline BCVA score of less than 49 letters on the ETDRS eye chart.
Patients participating in the FAME Study were randomly assigned to one of three groups at a ratio of 2:2:1. The first two of these groups were assigned to an active drug formulation and the third group serves as the control group, undergoing a sham insertion procedure designed to mimic an intravitreal insertion. The treatment groups consist of one group receiving a low dose of Iluvien and another group receiving a high dose of Iluvien. To reduce potential bias, these trials use a randomized, double-masked study design so that neither the patient nor the investigational staff involved with assessing the patient knows to which group the patient belongs. In order to simulate an insertion and help to maintain proper patient masking, the sham insertion procedure includes all steps involved in the insertion procedure, except that a blunt inserter without a needle is used to apply pressure to the anesthetized eye.
As part of the FAME Study, investigators were able to re-treat each patient with Iluvien following their month 12 follow-up visit. Through month 24, 24.5% of patients had been treated with more than one Iluvien insert and 2.5% of patients had been treated with more than two Iluvien inserts.
Primary Efficacy Endpoint. The primary efficacy endpoint for the FAME Study is the difference in the percentage of patients with improved BCVA from baseline of 15 or more letters on the ETDRS eye chart at month 24 between the treatment and control groups. In December 2009, Alimera received the month 24 clinical
5
Table of Contents
readout for the FAME Study and analyzed the full data set consistent with the recommendations regarding the appropriate population for primary analysis as described in the FDA-adopted International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Guidance E9, “Statistical Principles for Clinical Trials”. ICH is a joint venture involving regulatory authorities and pharmaceutical industry representatives from Europe, Japan and the United States who discuss scientific and technical aspects of product registration.
The full data set includes all 956 patients randomized into the FAME Study, with data imputation employed, using “last observation carried forward” (LOCF), for data missing because of patients who discontinued the trial or are unavailable for follow-up (the Full Analysis Set). As part of Alimera’s analyses, it determined statistical significance based on the Hochberg-Bonferroni procedure (H-B procedure), which is a procedure employed to control for multiple comparisons. Alimera also made a target p-value adjustment of 0.0001 to account for each of the nine instances that the independent data safety monitoring board reviewed unmasked interim clinical data. These adjustments resulted in a required p-value of 0.0491 or lower for each of Trial A and Trial B to demonstrate statistical significance for both the low dose and high dose of Iluvien. Based upon the H-B procedure, if either dose of Iluvien in a trial did not meet statistical significance, the alternate dose was required to achieve a p-value of 0.02455 or lower in that trial to demonstrate statistical significance.
In the Full Analysis Set, the primary efficacy endpoint was met with statistical significance for both the low dose and the high dose of Iluvien in Trial A and Trial B, as well as on a combined basis. The table below summarizes the primary efficacy variable results.
| Patients Gaining At Least 15 Letters At Month 24 | |||||||||||||||||||||
| Trial A | Trial B | Combined | |||||||||||||||||||
| Study Group |
Individuals | % | p-value | Individuals | % | p-value | Individuals | % | p-value | ||||||||||||
| Control |
14/95 | 14.7 | % | — | 16/90 | 17.8 | % | — | 30/185 | 16.2 | % | — | |||||||||
| Low Dose |
51/190 | 26.8 | % | 0.029 | 57/186 | 30.6 | % | 0.030 | 108/376 | 28.7 | % | 0.002 | |||||||||
| High Dose |
51/196 | 26.0 | % | 0.034 | 62/199 | 31.2 | % | 0.027 | 113/395 | 28.6 | % | 0.002 | |||||||||
Additionally, as required by the FDA, a numerical comparison of the responder rates at month 18 and month 24 in the Full Analysis Set demonstrated that the responder rates for both the low dose and high dose of Iluvien at month 24 were numerically greater than the month 18 responder rates in both Trial A and Trial B.
The FAME Study protocol provides for analysis of the all-randomized and treated data set, which includes 953 patients randomized into the FAME Study and treated, with data imputation employed, using the LOCF method, for data missing because of patients who discontinued the trial or are unavailable for follow-up (the ART Data Set). Three patients who were randomized, but not treated, are included in the Full Data Set and excluded from the ART Data Set. In the ART Data Set, the primary efficacy endpoint was met with statistical significance for both doses of Iluvien in both Trial A and Trial B. The percentage of patients in the ART Data Set achieving improved BCVA of 15 or more letters at month 24 for Trial A is 14.7% for the control group, 26.8% for the low dose (p-value 0.029) and 26.2% for the high dose (p-value 0.032). The percentage of patients in the ART Data Set achieving improved BCVA of 15 or more letters at month 24 for Trial B is 17.8% for the control group, 30.8% for the low dose (p-value 0.028) and 31.3% for the high dose (p-value 0.026).
The protocol also provides for analysis of the modified ART Data Set, which includes all 953 patients included in the ART Data Set and excludes data collected subsequent to the use of treatments prohibited by the protocol, such as Avastin®, Lucentis, triamcinolone acetonide or vitrectomy (the Modified ART Data Set). In instances when a treatment prohibited by the FAME Study protocol was used, the last observation prior to the protocol violation was imputed forward to month 24 using the LOCF method. The percentage of patients in the Modified ART Data Set achieving improved BCVA of 15 or more letters for Trial A is 12.6% for the control group, 22.6% for the low dose (p-value 0.057) and 24.1% for the high dose (p-value 0.026). Neither dose of
6
Table of Contents
Iluvien for Trial A was statistically significant based on the H-B procedure. The percentage of patients in the Modified ART Data Set achieving improved BCVA of 15 or more letters at month 24 for Trial B is 13.3% for the control group, 29.7% for the low dose (p-value 0.004) and 29.3% for the high dose (p-value 0.005). Both doses of Iluvien for Trial B were statistically significant.
The FAME Study protocol provides that the primary assessment of efficacy is based on the Modified ART Data Set and that other data sets are considered secondary. The protocol did not specify the Full Analysis Set as a data set for analyzing the study; however, consistent with the recommendations regarding the appropriate population for primary analysis as described in the FDA-adopted ICH Guidance E9, Alimera has reported that it believes that the FDA will consider the Full Analysis Set to be the most relevant data set for determining the safety and efficacy of Iluvien in Trials A and B.
Additional Clinical Observations. In addition to the primary efficacy variable, Alimera also reported that it observed a number of other clinically relevant results in the month 24 clinical data from the FAME Study. These observations included, among others, the following:
| • | patients with improved BCVA of 15 or more letters at each follow up visit; |
| • | BCVA improvement of 15 or more letters relative to baseline BCVA; |
| • | Mean change in BCVA letter score; and |
| • | decrease in excess foveal thickness. |
The analyses of these Full Analysis Set observations set forth below are presented for Trial A and Trial B on a combined basis for patients who received the low dose of Iluvien in comparison to the control group. Statements regarding statistical significance do not reflect any adjustments to the p-values calculated for multiple comparisons and analyses.
Patients With Improved BCVA of 15 Letters or More at Each Follow Up Visit. Alimera’s analysis of the results of the FAME Study through month 24 indicates that the low dose of Iluvien provides an improvement in BCVA as early as three weeks after insertion. The low dose of Iluvien was statistically significantly better than the control group in the FAME Study by week 3 of patient follow up, and maintained a statistically significant advantage over the control through month 24. The chart below demonstrates the treatment effect of the low dose of Iluvien versus the control group, as measured by an improvement in BCVA of 15 letters or more, at each scheduled follow up visit during the FAME Study.
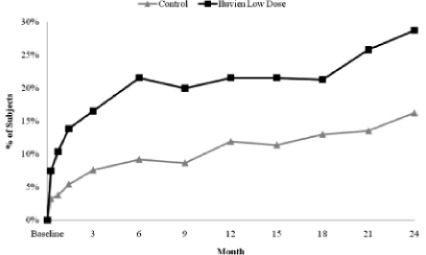
7
Table of Contents
BCVA Improvement of 15 or More Letters Relative to Baseline BCVA. Alimera’s analysis of the results of the FAME Study at month 24 indicates that Iluvien has a statistically significant advantage over the control group irrespective of the severity of a patient’s baseline BCVA. The table below demonstrates the statistically significant treatment effect of Iluvien versus the control group in patients with baseline BCVA of more than 49 letters on the EDTRS eye chart, and patients with BCVA of 49 letters or less on the EDTRS eye chart at baseline.
| Trial A & Trial B Combined | ||||||||
| Baseline BCVA |
Control | Low Dose | p-value | |||||
| Greater than 49 Letters |
11.8 | % | 21.1 | % | 0.027 | |||
| 49 Letters or Less |
28.6 | % | 46.1 | % | 0.039 | |||
Mean Change in BCVA Letter Score. Alimera’s analysis of the results of the FAME Study through month 24 indicates that the low dose of Iluvien provided a more beneficial improvement in visual acuity than the control group as analyzed by the mean change in the BCVA letter score from baseline. As demonstrated in the graph below, the mean change in BCVA for the patients receiving the low dose of Iluvien was an increase of 4.4 letters at month 24, peaking at an increase of 6.0 letters at month 6, compared to an increase of 1.7 letters in the control group, peaking at an increase of 2.6 letters at week 6. The low dose of Iluvien was statistically significantly better than the control group at month 24 (p-value 0.020)
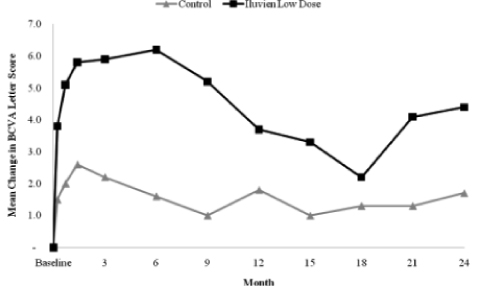
During the first 24 months of follow up in the FAME Study, Alimera reported that, of patients who were phakic (had a natural lens and no prior cataract surgery) at baseline, 50 of 121, or 41.3% of the control group and 182 of 235, or 77.4% of the low dose, had cataract formation reported as an adverse event through month 24. For these same phakic patients, Alimera reported that 19.8% of the control group and 66.0% of the low dose group underwent cataract surgery through month 24. For the patients in the low dose group the median time to reporting cataract formation as an adverse event was approximately 12 months from randomization into the FAME Study. The median time to cataract surgery was approximately 18 months. This interval between the report of cataract formation as an adverse event and cataract surgery may account for the decrease in the mean change in BCVA in patients receiving the low dose of Iluvien from the month 6 follow up visit to the month 18 follow up visit.
The effect of cataract progression on BCVA reported by Alimera is illustrated by comparing the mean change in BCVA of the 140 low dose patients that were pseudophakic (had an artificial lens) to the 235 that were phakic (natural lens and no prior cataract surgery) at baseline. The chart below shows the pseudophakic subset (those who would not have vision affected by a cataract) achieved a mean change in BCVA of more than 7 letters
8
Table of Contents
by month 6 and maintained this mean change through month 24 while the phakic subset experienced a decrease in the mean change in BCVA from the month 6 follow up visit to the month 18 follow up visit. The temporary decrease in mean change in BCVA in the phakic population is consistent with the total low dose population.
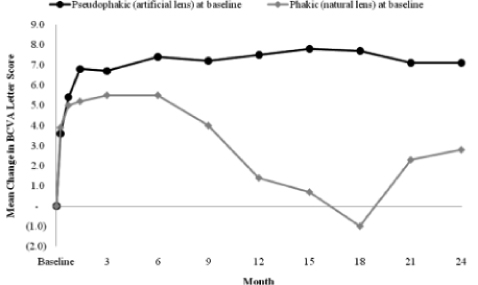
Decrease In Excess Foveal Thickness. In addition to the functional measures of BCVA, Alimera assessed the ability of Iluvien to effect a decrease in excess foveal thickness, an anatomic outcome, as measured by optical coherence tomography. Excess foveal thickness is a measurement of the swelling of the macula at its center point (known as the fovea). Alimera reported that it considers any measurement above 180 microns to represent excess foveal thickness. Based on a review of the month 24 clinical readout as summarized in the chart below, Alimera reported that patients receiving the low dose of Iluvien demonstrated a statistically significant difference versus the control group in decreasing excess foveal thickness by week 1 of patient follow up of the FAME Study, and maintained a statistically significant advantage through month 24. At month 24, patients receiving the low dose of Iluvien demonstrated a mean decrease in excess foveal thickness of 156.1 microns versus 100.5 microns for the control group.
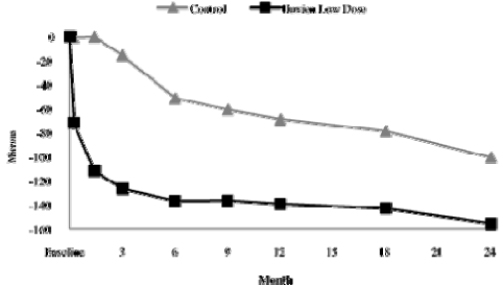
9
Table of Contents
Safety. Alimera reported that its safety assessment in connection with the month 24 clinical readout of the FAME Study included all reported adverse events at that time, regardless of a patient’s progression in the FAME Study. Some reported adverse events occurred beyond patients’ month 24 follow up visits. Alimera reported that Iluvien was well tolerated through this readout in both the low and high dose patient populations. Alimera’s preliminary assessment of adverse event data indicates that there is no apparent risk of systemic adverse events to patients as a result of the use of Iluvien. The use of corticosteroids in the eye is primarily associated with two undesirable side effects: increased IOP, which may increase the risk of glaucoma and require additional procedures to manage, and cataract formation. Excluding IOP-related side effects and cataracts, Alimera reported that it observed no significant eye related adverse events when comparing both the low dose and high dose patient populations to control. Thus, Alimera believes that the adverse events associated with the use of Iluvien are within the acceptable limits of a drug for the treatment of DME.
The table below summarizes the IOP related adverse events occurring in all patients randomized and treated in the FAME Study.
| Trial A & Trial B Combined | |||||||||
| Control N=185 |
Low Dose N=375 |
High Dose N=393 |
|||||||
| IOP > 30 mmHg (1) |
2.7 | % | 16.3 | % | 21.6 | % | |||
| Trabeculoplasty |
0.0 | % | 1.3 | % | 2.5 | % | |||
| IOP-Lowering Surgeries |
|||||||||
| Trabeculectomy (filtration) |
0.0 | % | 2.1 | % | 5.1 | % | |||
| Vitrectomy |
0.0 | % | 0.3 | % | 0.5 | % | |||
| Other Surgery Performed |
0.5 | % | 1.6 | % | 2.5 | % | |||
| Percentage of Patients Requiring One or More IOP-Lowering Surgeries |
0.5 | % | 3.7 | % | 7.4 | % | |||
| (1) | An IOP of 30 mmHg is a clinically significant level that Alimera used in assessing adverse events. |
According to the CDC, diabetic individuals aged 50 or older are 1.5 times more likely to develop cataracts than non-diabetic individuals. A review of the baseline characteristics of the FAME patient population reflects this increased risk of cataracts for diabetic patients, with 34.8% of the patients treated in the FAME Study having previously undergone a cataract surgery in the study eye. Alimera reported that the month 24 clinical readout from the FAME study (which includes reported adverse events that occurred beyond patients’ month 24 follow-up visits) indicated that, of patients who had a natural lens (no prior cataract surgery) at baseline, 46.3% of the control group, 80.0% of the low dose and 87.5% of the high dose had cataract formation reported as an adverse event through month 24. Additionally, of the patients who had a natural lens at baseline, 23.1% of the control group, 74.9% of the low dose and 84.5% of the high dose underwent cataract surgery.
PK Study
Alimera initiated an open-label Phase 2 human pharmacokinetic clinical study (PK Study) in August 2007 to assess the systemic exposure of FA by measuring plasma levels of FA. Analysis of plasma levels through month 18 in September 2009 demonstrated no systemic exposure of FA (plasma levels were below the limit of detection of 100 picograms per milliliter). Based on these results, Alimera reported that it filed a carcinogenicity waiver with the applicable regulatory authorities, including with the FDA, in connection with its NDA submission.
A total of 37 patients were enrolled in the PK Study, 17 patients on the high dose of Iluvien and 20 patients on the low dose of Iluvien. The last patient was enrolled in the study at the end of February 2008. Data from the PK Study are being evaluated on an ongoing basis with interim evaluations at months 3, 6, 12, 18, 24, 30 and 36.
Current Durasert Products
Retisert. Retisert is the only product approved by the FDA for the treatment of posterior uveitis, an autoimmune condition characterized by inflammation of the inside of the eye that can cause sudden or gradual
10
Table of Contents
vision loss. In the United States, this disease has been estimated to affect 175,000 people and to have resulted in blindness in approximately 30,000 people. Retisert, which is about the size of a grain of rice, is surgically implanted through a 3-4 mm incision and delivers sustained levels of the anti-inflammatory corticosteroid FA for 30 months. Although there are off-label treatments for posterior uveitis, those treatments generally only slow the progression of the disease and can have more serious side effects than Retisert. Clinical trials have shown that many patients treated with Retisert experience improved vision. Retisert was approved as an orphan drug in 2005, which provided for seven-year exclusive marketing rights. Retisert is marketed and sold in the United States by Bausch & Lomb.
Vitrasert. Vitrasert treats CMV retinitis, a blinding eye disease that occurs in individuals with advanced AIDS. Vitrasert, which is surgically implanted through a 5-6 mm incision, provides sustained treatment for six to eight months through the intravitreal delivery of the anti-viral drug ganciclovir. Studies have shown that Vitrasert is one of the most effective approved treatments for CMV retinitis. Vitrasert has been sold since 1996 in the United States and abroad, first by Chiron Corporation, which was subsequently acquired by Bausch & Lomb. Although CMV retinitis was common in the early 1990s, improvements in the treatment of AIDS/HIV have since significantly decreased the incidence of the disease in more developed countries. Accordingly, sales of Vitrasert have decreased significantly over time.
Other Durasert Research
Under our worldwide collaborative research and license agreement with Pfizer, Pfizer is licensed to develop certain ophthalmic applications of Durasert and BioSilicon. We are working together on a joint research program aimed at developing such products.
We are also conducting pre-clinical studies utilizing a bioerodible version of our Durasert technology designed to treat glaucoma, dry-AMD and retinitis pigmentosa.
BioSilicon Technology System
Our proprietary BioSilicon technology system utilizes a “honeycomb” structure of nano-porous, elemental silicon to deliver therapeutics. BioSilicon is both biocompatible and biodegradable. We believe BioSilicon can be used to deliver a wide variety of drugs, including small chemical entities, peptides, proteins and other therapeutics.
Based on results of our preliminary studies, we currently are targeting BioSilicon as a key second prong of our drug delivery technology platform. We are presently evaluating a form of BioSilicon technology designed to treat both retinal vein occlusion and age-related macular degeneration. We have previously out-licensed certain non-core field of use applications of BioSilicon in the areas of nutraceuticals and food science and diagnostics.
CODRUG Technology System
Our proprietary CODRUG system allows for the simultaneous release of two or more drugs from the same product at the same controlled rate over a predetermined period of time. Using this technology, we chemically link two or more identical or different drugs. CODRUGs can be administered by virtually any delivery method and dissolve into the body at a predetermined rate, and then separate into the original active drug(s) when the chemical bond breaks apart.
Strategic Collaborations
We have entered into a number of collaboration/license agreements to develop and commercialize our product candidates and technologies. In all of our collaboration agreements, we retain the right to use and develop the underlying technologies outside of the scope of the exclusive licenses granted.
11
Table of Contents
Alimera
Under a collaboration agreement with Alimera, as amended in March 2008 (the “Alimera Agreement”), we licensed Alimera the rights to develop, market and sell Iluvien. Alimera also has a worldwide non-exclusive license to make and sell certain additional Durasert-based products for the treatment and prevention of eye diseases other than uveitis that (i) are not exclusively licensed to Bausch & Lomb, (ii) have a drug core within a polymer layer and (iii) are approved or designed to be approved (a) to deliver a corticosteroid and no other active ingredient by implantation, injection or other direct delivery method to the posterior portion of the eye or (b) to treat DME by delivering a compound or formulation through implantation, injection or other direct delivery method other than through an incision smaller than that required for a 25 gauge needle. Other than the licenses to Bausch & Lomb, we are not permitted to use, or grant a license to any third party to use, such technologies to make or sell any products subject to the non-exclusive license granted to Alimera.
Alimera is completing fully-enrolled Phase III trials for Iluvien in DME. Based on 24-month data released in December 2009, Alimera filed an NDA with the FDA in June 2010. On August 30, 2010, the FDA granted Priority Review status for the NDA. Under Priority Review, Alimera could receive a response to the NDA from the FDA by the end of calendar year 2010.
Upon execution of the Alimera Agreement, we received consideration of $12.0 million in cash and Alimera cancelled $5.7 million of accrued development cost liabilities, including related penalties and accrued interest, owed by us to Alimera as of March 14, 2008. In addition, we received a $15.0 million conditional note providing for aggregate principal and interest payments of up to $21.3 million through September 2012, Alimera agreed to pay us a $25.0 million milestone payment upon FDA approval of Iluvien for DME; and Alimera assumed all financial responsibility for the development of licensed products under the Alimera Agreement, which had previously been shared equally, including reimbursement of approved development costs incurred by us in support of the ongoing clinical studies of Iluvien and anticipated regulatory submissions. In exchange, we decreased our share in any future profits, as defined, on sales of Iluvien by Alimera from 50% to 20%, subject to an offset of 20% of pre-profitability commercialization costs, as defined, incurred by Alimera. In the event Alimera sublicenses commercialization, we are entitled to receive 20% of royalties and 33% of non-royalty consideration received by Alimera, less certain permitted deductions. Alimera has indicated that it intends to commercialize Iluvien, if approved, through a direct sales force in the United States and to seek marketing collaboration partners for the commercialization of Iluvien outside of the United States.
The scheduled payment terms on the $15.0 million conditional note consisted of (i) interest only at an annual rate of 8% payable quarterly through March 2010 and (ii) principal payments of $500,000 per month commencing April 30, 2010 together with interest payable quarterly at an annual rate of 20%. Upon the occurrence of certain defined liquidity events (such as an initial public offering of Alimera, other sales of capital stock of Alimera and/or the sale or other disposition of substantially all of Alimera’s assets) that resulted in aggregate cash and/or noncash proceeds to Alimera in excess of $75 million, the note became immediately due and payable. Failure by Alimera to pay the note upon the occurrence of a defined liquidity event constituted an event of default under the note. If no liquidity event occurred on or before September 30, 2012, the note would automatically be cancelled. Based upon the terms of the note, payment was within the control of Alimera unless there was a liquidity event or an event of default. Through March 31, 2010, we received total interest payments of approximately $2.5 million under the terms of the note. On April 27, 2010, following consummation of its initial public offering, Alimera paid the $15.0 million conditional note in full, together with $225,000 of accrued and unpaid interest.
For fiscal 2010, fiscal 2009 and fiscal 2008 we derived revenues of approximately $22.3 million, $11.8 million and $3.3 million, respectively, under the Alimera Agreement.
Either party may terminate the agreement for the other party’s uncured material breach. We may terminate the agreement with respect to a particular product if Alimera notifies us that it is abandoning or has abandoned such product, in which case the agreement provides for specific, exclusive remedies.
12
Table of Contents
Pfizer
In April 2007, we entered into an exclusive worldwide collaborative research and license agreement with Pfizer for the use of certain of our technologies in ophthalmic applications that are not licensed to others.
Under the terms of the agreement, we are eligible to receive up to $153.5 million in development and sales-related milestones. We are working together on a joint research program aimed at developing ophthalmic products using our sustained drug delivery technology. Beginning with the first calendar quarter of 2008, under the agreement, Pfizer has paid us $500,000 per quarter and is required to continue to make quarterly payments of at least $500,000 until a first Phase III clinical trial commences. Pfizer will have an exclusive license to market any products developed under the agreement, and will pay us a royalty on net sales of those products. Pfizer may terminate the agreement without cause and without penalty on 60 days notice.
Pfizer has made equity investments totaling $11.5 million in pSivida, making Pfizer our largest shareholder, owning approximately 10.0% of total shares outstanding as of August 31, 2010.
Bausch & Lomb
Retisert and Vitrasert were developed and commercialized under a 1992 licensing and development agreement with Bausch & Lomb. Bausch & Lomb has a worldwide exclusive license to make and sell Vitrasert and our first-generation products (as defined in the agreement, including the Retisert device) in return for royalties based on sales. Bausch & Lomb can terminate its agreement with us without penalty at any time upon 90 days’ written notice.
Intrinsiq
In connection with a January 2008 exclusive field of use license with Intrinsiq Materials Cayman Limited (Intrinsiq) for nutraceutical and food science applications of BioSilicon, we received license fee payments of $730,000 in fiscal 2009 and $500,000 in fiscal 2008. During fiscal 2010, we received the first contractual minimum royalty payment of $450,000. Subject to continuation of the license agreement, which is cancellable by Intrinsiq on 90 days prior written notice, we are entitled to receive additional scheduled minimum royalties totaling approximately $3.1 million through April 2014, creditable against quarterly royalties earned, if any. The next scheduled minimum royalty payment of $630,000 is due in January 2012.
Research and Development
Our primary activity is the development of products based on our Durasert and BioSilicon technology systems. Our research and development expenses were $7.0 million in fiscal 2010, $8.0 million in fiscal 2009 and $14.4 million in fiscal 2008. Of these amounts, approximately $3.4 million in fiscal 2010, $4.4 million in fiscal 2009 and $10.2 million in fiscal 2008 were incurred for costs of research and development personnel, clinical trials, contract services, testing and laboratory facilities, and included approximately $4.7 million in fiscal 2008 of Iluvien co-development costs incurred prior to the amendment and restatement of the 2005 collaboration agreement with Alimera. Such costs were charged to operations as incurred. The remaining expense of $3.6 million in each of fiscal 2010 and fiscal 2009, and $4.2 million in fiscal 2008 consisted of non-cash charges for amortization of intangible assets, depreciation of property, plant and equipment and stock-based compensation expense specifically allocated to research and development personnel.
13
Table of Contents
Intellectual property
Our intellectual property rights are crucial to our business. We hold or are licensed patents relating to our core technology systems in the United States and international markets. The following table provides general details relating to our owned and licensed patents (including both patents that have been issued and applications that have been accepted for issuance) and patent applications as of August 31, 2010.
| Technology |
United States Patents |
United States Applications |
Foreign Patents |
Foreign Applications |
Patent Families | |||||
| Durasert |
10 | 17 | 67 | 87 | 19 | |||||
| BioSilicon |
12 | 9 | 64 | 33 | 22 | |||||
| CODRUG |
3 | 9 | 12 | 18 | 12 | |||||
| Other |
— | 2 | — | 4 | 3 | |||||
| Total |
25 | 37 | 143 | 142 | 56 | |||||
Employees
We had 23 employees as of August 31, 2010. None of our employees is covered by a collective bargaining agreement.
Sales and Marketing
We have no marketing or sales staff. We depend on collaborative partners to market our products. Significant additional expenditures would be required for us to develop an independent sales and marketing organization.
Reimbursement
The successful commercialization of our current products depends, and of any future products will depend, in significant part on the extent to which reimbursement of the cost of the products and the related administration procedures will be available from government health administration authorities, private health insurers and other organizations. Medicaid and Medicare, most major health maintenance organizations and most health insurance carriers reimburse $4,240 for the cost of the Vitrasert implant, with associated surgical fees reimbursed separately. The Centers for Medicare and Medicaid Services designated Retisert as eligible for Medicare reimbursement at the rate of $19,345, with associated surgical fees reimbursed separately.
Competition
We are engaged in healthcare product development, an industry that is characterized by extensive research efforts and rapid technological progress. We believe that pharmaceutical, drug delivery and biotechnology companies, research organizations, governmental entities, universities, hospitals, other nonprofit organizations and individual scientists are seeking to develop drugs, therapies and novel delivery methods to treat our targeted diseases.
Retisert is the only FDA-approved treatment for posterior uveitis, although steroids and other existing drugs approved for other uses are commonly administered systemically or by local injection to treat this condition in off-label use. Vitrasert primarily competes with treatments involving the systemic delivery of ganciclovir, a Roche Holdings AG product, and other drugs.
Many companies are pursuing products to treat back of the eye diseases. These include the following:
| • | Genentech Inc.’s products Lucentis (ranibizumab injection) and Avastin (bevacizumab), both antibodies that block all isoforms of VEGF, are being studied for the treatment of DME. However, only Lucentis is currently enrolled in Phase III clinical trials for the treatment of DME. Lucentis is currently |
14
Table of Contents
| approved in the United States for the treatment of patients with neovascular wet AMD. Avastin is currently marketed as an oncology product. Neither product is indicated for the treatment of DME, dry AMD or RVO. Genentech is a wholly-owned member of the Roche Group. |
| • | Allergan, Inc.’s product Ozurdex® (dexamethasone intraveal implant), is a bioerodible extended release implant that delivers the corticosteroid dexamethasone. Ozurdex was approved in 2009 for macular edema following branch or central RVO and showed a duration of therapy of three to five months. In addition, Allergan’s product Trivaris (triamcinolone acetonide injectable suspension) is approved for sympathetic ophthalmia, temporal arteritis, uveitis and other inflammatory conditions unresponsive to topical corticosteroids. Trivaris is not indicated for the treatment of DME, dry AMD, wet AMD or RVO. |
| • | Eyetech, Inc.’s product Macugen (pegaptanib sodium injection) is an anti-VEGF aptamer against VEGF 165. It has been FDA-approved for treatment of all subtypes of choroidal neovascularization in patients with AMD. Macugen is not indicated for the treatment of DME, dry AMD or RVO. |
| • | Regeneron Pharmaceuticals, Inc. is developing a drug for wet-AMD and DME. This drug (VEGF-trap) is designed, like Lucentis and Macugen, to be injected directly into the vitreous on a regular basis. Regeneron is currently conducting Phase III clinical trials for wet-AMD and Phase II trials for DME. |
We believe that competition for treatments of back-of-the-eye diseases is based upon the effectiveness of the treatment, potential side effects, timing to market, reimbursement and price.
Revenues
We operate in one segment. The following table summarizes our revenues by type and by geographical location. Revenue is allocated geographically by the location of the subsidiary that earns the revenue. For more detailed information regarding our operations, see our Consolidated Financial Statements commencing on page F-1.
| Year Ended June 30, | |||||||||||||||||||||||||||
| 2010 | 2009 | 2008 | |||||||||||||||||||||||||
| United States |
United Kingdom |
Total | United States |
United Kingdom |
Total | United States |
United Kingdom |
Total | |||||||||||||||||||
| (In thousands) | |||||||||||||||||||||||||||
| Revenue: |
|||||||||||||||||||||||||||
| Collaborative research and development |
$ | 22,449 | $ | 121 | $ | 22,570 | $ | 11,925 | $ | 77 | $ | 12,002 | $ | 3,328 | $ | — | $ | 3,328 | |||||||||
| Royalty income |
483 | — | 483 | 160 | — | 160 | 148 | — | 148 | ||||||||||||||||||
| $ | 22,932 | $ | 121 | $ | 23,053 | $ | 12,085 | $ | 77 | $ | 12,162 | $ | 3,476 | $ | — | $ | 3,476 | ||||||||||
Government Regulation
The FDA and comparable regulatory agencies in foreign countries impose substantial requirements upon the clinical development, manufacture and marketing of pharmaceutical and radiological products. These agencies regulate, among other things, the research, development, testing, manufacture, quality control, labeling, storage, record-keeping, approval, distribution, advertising and promotion of our drug delivery products. The process required by the FDA under the new drug provisions of the Federal Food, Drug, and Cosmetic Act before our products may be marketed in the United States generally involves the following:
| • | pre-clinical laboratory and animal tests; |
| • | submission to the FDA of an investigational new drug application (IND), which must become effective before clinical trials may begin; |
15
Table of Contents
| • | adequate and well-controlled human clinical trials to establish the safety and efficacy of the proposed pharmaceutical for its intended use; |
| • | submission to the FDA of an NDA; and |
| • | FDA review and approval of the NDA. |
The testing and approval process requires substantial time, effort and financial resources, and we cannot be certain that any approval will be granted on a timely basis, if at all.
Pre-clinical tests include laboratory evaluation of the product, its chemistry, formulation and stability, as well as animal studies to assess the potential safety and efficacy of the product. The results of the pre-clinical tests, together with manufacturing information, analytical data and protocols for proposed human clinical trials, are submitted to the FDA as part of an IND, which must become effective before the IND sponsor may begin human clinical trials. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises concerns or questions about the conduct of the proposed clinical trials as outlined in the IND, and imposes a clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before clinical trials can begin. There is no certainty that pre-clinical trials will result in the submission of an IND, or that submission of an IND will result in FDA authorization to commence clinical trials.
Clinical trials involve the administration of the investigational product to human subjects under the supervision of qualified investigators. Clinical trials are conducted in accordance with protocols that detail the objectives of the study, the parameters to be used to monitor safety and any efficacy criteria to be evaluated. Each protocol must be submitted to the FDA as part of the IND. Further, each clinical study must be conducted under the auspices of an independent institutional review board (IRB) at the institution where the study will be conducted. The IRB will consider, among other things, ethical factors, safety of human subjects and possible liability of the institution. Some clinical trials, called “investigator-sponsored” clinical trials, are conducted by third-party investigators. The results of these trials may be used as supporting data by a company in its application for FDA approval, provided that the company has contractual rights to use the results.
Human clinical trials are typically conducted in three sequential phases which may overlap:
| • | Phase I: The drug is initially introduced into healthy human subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion. |
| • | Phase II: Studies are conducted in a limited patient population to identify possible adverse effects and safety risks, to determine the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage. |
| • | Phase III: Phase III trials are undertaken to further evaluate clinical efficacy and to further test for safety in an expanded patient population, often at geographically dispersed clinical study sites. |
In the case of products for life-threatening diseases such as cancer, or severe conditions such as blinding eye disease, or for products that require invasive delivery, the initial human testing is often conducted in patients with the disease rather than in healthy volunteers. Since these patients already have the targeted disease or condition, these studies may provide initial evidence of efficacy traditionally obtained in Phase II trials, and so these trials are frequently referred to as Phase I/II or IIa trials.
We cannot be certain that we or our collaborative partners will successfully complete Phase I, Phase II or Phase III testing of our product candidates within any specific time period, if at all. Furthermore, we, our collaborative partners, the FDA, the IRB or the sponsor, if any, may suspend clinical trials at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk.
The Food and Drug Administration Amendments Act of 2007 (FDAAA) is designed to provide the public with more easily accessible information about the safety and efficacy of marketed drugs and the FDA with
16
Table of Contents
increased authority to ensure drug safety. The FDAAA requires that we register each controlled clinical trial, aside from a Phase I trial, on a website administered by National Institutes of Health (NIH), including descriptive information (e.g., a summary in lay terms of the study design, type and desired outcome), recruitment information (e.g., target number of participants and whether healthy volunteers are accepted), location and contact information and administrative data (e.g., FDA identification numbers). Within one year of a trial’s completion, information about the trial, including characteristics of the patient sample, primary and secondary outcomes, trial results written in lay and technical terms and the full trial protocol must be submitted to the FDA. The information is then posted to the website unless the drug has not yet been approved, in which case the FDA posts the information shortly after approval. An NDA supplement and certain other submissions to the FDA require certification of compliance with the FDAAA clinical trials reporting requirements.
The results of product development, pre-clinical studies and clinical studies are submitted to the FDA as part of an NDA for approval of the marketing and commercial shipment of the product. The FDA may deny an NDA if the applicable regulatory criteria are not satisfied, or may require additional clinical data. Even if the additional data are submitted, the FDA ultimately may decide that the NDA does not satisfy the criteria for approval. As a condition of approval, the FDA may require post-marketing “Phase IV” clinical trials to confirm that the drug is safe and effective for its intended uses. Once issued, the FDA may withdraw product approval for non-compliance with regulatory requirements or if safety or efficacy problems occur after the product reaches the market. The FDA may also require surveillance programs to monitor approved products which have been commercialized. The FDA also has the power to require changes in labeling or to prevent further marketing of a product based on the results of these post-marketing programs.
Satisfaction of FDA requirements or similar requirements of foreign regulatory agencies typically takes several years, and the actual time required may vary substantially based upon various factors, including the type, complexity and novelty of the pharmaceutical product. Such government regulation may delay or prevent marketing of potential products for a considerable period of time, and may impose costly procedures upon our activities. Success in pre-clinical or early stage clinical trials does not assure success in later stage clinical trials. Data from pre-clinical and clinical activities is not always conclusive, and may be susceptible to varying interpretations which could delay or prevent regulatory approval. Even if a product receives regulatory approval, the approval may be subject to significant limitations based on data from pre-clinical and clinical activities. Further, discovery of previously unknown problems in connection with a product’s use may result in restrictions on the product, or even complete withdrawal of the product from the market.
Any product manufactured or distributed under FDA approval is subject to pervasive and continuing regulation by the FDA, including requirements for record-keeping and reporting adverse experiences with the product. Drug manufacturers and their subcontractors are required to register with the FDA and state agencies. Drug manufacturers and their subcontractors are also subject to periodic unannounced inspections by the FDA and state agencies for compliance with good manufacturing practices, which impose procedural and documentation requirements upon us and our third-party manufacturers.
The passage of the FDAAA significantly enhanced the FDA’s authority to regulate drugs post-approval. For certain drugs that the FDA determines pose risks that outweigh the benefits, FDA approval may be subject to the manufacturers’ continued adherence to a Risk Evaluation Mitigation Strategy (REMS). REMS, which are tailored to specifically address the risks of a given drug, may contain elements that restrict distribution of the drug to certain physicians, pharmacists and patients or that require the use of communication tools such as letters to healthcare providers and patients detailing the risks associated with the drug. In addition to REMS, the FDAAA also provides the FDA with increased authority to require the manufacturer to conduct post-approval clinical trials and to submit drug advertisements to the FDA for review before dissemination.
We are also subject to numerous other federal, state and local laws relating to such matters as safe working conditions, manufacturing practices, environmental protection, fire hazard control and disposal of hazardous or
17
Table of Contents
potentially hazardous substances. We may incur significant costs to comply with such laws and regulations now or in the future. In addition, we cannot predict what adverse governmental regulations may arise from future U.S. or foreign governmental action.
We and our collaborative partners are also subject to foreign regulatory requirements governing human clinical trials and marketing approval for pharmaceutical products sold in their countries. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary widely by country. Whether or not FDA approval is obtained, we or our collaborative partners must obtain approval of a product by the comparable regulatory authorities of foreign countries before manufacturing or marketing the product in those countries. The approval process varies from country to country, and the time required for these approvals may differ substantially from that required for FDA approval. There is no assurance that clinical trials conducted in one country will be accepted by other countries, or that approval in one country will result in approval in any other country. For clinical trials conducted outside the U.S., the clinical stages generally are comparable to the phases of clinical development established by the FDA.
The time and expense required to perform the clinical testing necessary to obtain FDA clearance or approval for regulated products can frequently exceed the time and expense of the research and development initially required to create the product. Even after initial FDA approval has been obtained, we or our collaborative partners could be required to conduct further studies to provide additional data on safety or efficacy or, should we desire, to gain approval for the use of a product as a treatment for additional clinical indications. In addition, use of a product during testing and after marketing approval has been obtained could reveal side effects which, if serious, could limit uses, or in the most serious cases, result in a market withdrawal of the product or expose us to product liability claims.
Corporate Information
pSivida Corp. was organized as a Delaware corporation in March 2008. Its predecessor (pSivida Limited) was formed in December 2000 as an Australian company incorporated in Western Australia. On June 19, 2008, we reincorporated from Western Australia to the United States (the Reincorporation). Except as otherwise indicated, references in this Annual Report to “pSivida”, “the Company”, “we”, “us”, “our” or similar terms refer to pSivida Limited, a West Australia corporation, and its subsidiaries prior to June 19, 2008, and refer to pSivida Corp., a Delaware corporation, and its subsidiaries from such date. All share amounts and all information relating to options and warrants in this Annual Report have been retroactively adjusted to reflect the Reincorporation share exchange ratio, unless otherwise stated. Our principal executive office is located at 400 Pleasant Street, Watertown, Massachusetts 02472 and our telephone number is (617) 926-5000.
Additional Information
Our website address is http://www.psivida.com. Information contained on, or connected to, our website is not incorporated by reference into this Annual Report on Form 10-K. Copies of our annual reports on Form 10-K, proxy statements, quarterly reports on Form 10-Q, current reports on Form 8-K and, if applicable, amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934, are available free of charge through our website under “SEC Filings” as soon as reasonably practicable after we electronically file these materials with, or otherwise furnish them to, the Securities and Exchange Commission (SEC).
18
Table of Contents
| ITEM 1A. | RISK FACTORS |
RISKS RELATED TO OUR COMPANY AND OUR BUSINESS
We may be required to seek additional capital in order to fund our operations, and our ability to obtain additional capital is uncertain.
Our cash, cash equivalents and marketable securities totaled approximately $17.6 million at June 30, 2010. We believe we can fund our operations as currently conducted into at least calendar year 2012. Whether we will require additional capital will be influenced by many factors, including, but not limited to:
| • | the continuation of our existing collaborations with Pfizer and Alimera, including their continued funding of our programs and our receipt of milestone, royalty and other payments; |
| • | the timely development, regulatory approval and commercialization of Iluvien; |
| • | sales of Retisert, on which we receive royalty payments from Bausch & Lomb; |
| • | the scope and extent of our internally funded operations and programs, any new product candidates and any new business opportunities; |
| • | our ability to establish and maintain strategic arrangements for product candidates for research, development, clinical testing, manufacturing and marketing; |
| • | the success of our products and product candidates, including the timing and costs of regulatory approvals and the commercial success of approved products; |
| • | the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims; and |
| • | changes in our operating plan, including the pursuit of any new business opportunities, which may affect our need for capital. |
In particular, our future cash position depends significantly on the timing of FDA approval and the initiation and success of marketing of Iluvien. Alimera has agreed to pay us $25 million upon FDA approval of Iluvien for DME. In addition, we will be entitled to 20% of any future profits, as defined, on sales of Iluvien by Alimera, subject to an offset of 20% of defined pre-profitability commercialization costs incurred by Alimera. In the event Alimera sublicenses commercialization, we would receive 20% of royalties and 33% of non-royalty consideration received by Alimera, less certain permitted deductions. However, there is no assurance that the FDA will approve Iluvien or that Iluvien will achieve market acceptance even if it is approved by the FDA.
The downturn in the economy and the disruptions in the financial and credit markets have made it significantly more difficult and more expensive to obtain financing. If we determine that it is desirable or necessary to raise additional capital in the future, we do not know if it will be available when needed or on terms favorable to us or our stockholders. If available, additional equity financing may be dilutive to stockholders, debt financing may involve restrictive covenants or other unfavorable terms and potential dilutive equity, and funding through collaboration agreements may be on unfavorable terms, including requiring us to relinquish rights to certain of our technologies or products. If adequate financing is not available if and when needed, we may be required to delay, reduce the scope of or eliminate one or more of our research or development programs, postpone the pursuit of product candidates and new business opportunities, or otherwise reduce our cash requirements.
We have a history of losses and may incur losses in the future.
With the exception of fiscal 2010, we have incurred operating losses since our inception in 2000. For fiscal 2010, we recorded net income of $8.8 million, primarily due to the accelerated payment in full by Alimera of a $15.0 million conditional note. For fiscal 2009 and fiscal 2008 we incurred net losses of $2.5 million and $75.7 million, respectively. We may incur losses for the foreseeable future if our Iluvien product candidate is not
19
Table of Contents
timely approved and successfully commercialized. Even if Iluvien is approved and marketed, our profit share on sales of Iluvien, combined with royalty income from our current products, and any other sources of revenue, may not be sufficient to result in profitability.
Our results could be adversely affected as a result of the impact of impairment of our intangible assets, which could adversely affect the price of our securities.
Impairment charges on our intangible assets could have a material effect on our results of operations, which could in turn adversely affect the price of our securities. We have recorded significant amounts of intangible assets in connection with earlier acquisitions. We took a $60.1 million impairment charge on goodwill as of June 30, 2008 (which reduced the carrying value of our goodwill to zero), and a $45.3 million impairment charge on the recorded value of our Durasert intangible asset as of June 30, 2007. We still have $23.9 million of intangible assets on our balance sheet as of June 30, 2010, of which approximately $16.0 million relates to our BioSilicon technology and approximately $7.9 million relates to our Durasert technology. We will continue to conduct impairment analyses of our intangible assets as required, and may be required to take significant impairment charges in the future.
Our results could be adversely affected by non-cash charges due to fluctuations in the fair values of certain of our outstanding warrants, which could adversely affect the price of our securities.
In connection with certain capital raising transactions during the years ended June 30, 2008 and 2007, we issued detachable warrants denominated in Australian dollars (A$). The fair values of the warrants have been recorded as derivative liabilities on our balance sheet. We are required to assess the fair value of these warrants at each subsequent balance sheet date, and changes in their fair values will result in adjustments to our recorded derivative liabilities, and a corresponding gain or loss on our statement of operations. The fair values of these warrants are sensitive to changes in our share price, among other factors, and are measured using the Black-Scholes valuation model. Fluctuations in the fair values of these warrants could be substantial and will continue to affect our operating results until the last-to-expire of these warrants in July 2012.
Our operating results may fluctuate significantly from period to period.
Our operating results have fluctuated significantly from period to period in the past and may continue to do so in the future due to many factors, including:
| • | the timing, receipt and amount of payments, if any, from current and potential future collaboration partners and the revenue recognition policies related thereto; |
| • | changes in accounting estimates, policies or principles; |
| • | the entry into, or termination of, collaboration agreements; |
| • | the scope, duration and effectiveness of our collaboration arrangements; |
| • | the quarterly income or expense amounts recorded from the revaluation of our derivative liabilities; |
| • | the amount of research and development costs, including pre-clinical studies and clinical trials, that are funded internally; |
| • | general and industry-specific adverse economic conditions that may affect, among other things, our and our collaborators’ operations and financial results; and |
| • | impairment write-downs of one or more of our intangible assets. |
Due to fluctuations in our operating results, quarterly comparisons of our financial results may not necessarily be meaningful, and investors should not rely upon such results as an indication of future performance.
20
Table of Contents
In addition, investors may react adversely if our reported operating results are less favorable than in a prior period or are less favorable than those anticipated by investors in the financial community, which may result in a decrease in our stock price.
Our Retisert royalty income may never become a material source of our revenues.
In consideration of a June 2005 royalty advance of $3.0 million, we agreed that Bausch & Lomb would retain $6.25 million of future Retisert royalties that otherwise would be payable to us. In the quarter ended June 30 2010, the last remaining $60,000 was retained by Bausch & Lomb to complete that agreement, and a royalty payment of $342,000 was subsequently received by the Company. Although we have now resumed receiving Retisert royalties, the annual trend of these royalties (including the historical amounts retained by Bausch & Lomb) has been declining, and there is no assurance that our Retisert royalty payments will increase in the future or become a material source of revenue.
RISKS RELATED TO THE DEVELOPMENT AND COMMERCIALIZATION OF OUR PRODUCTS AND PRODUCT CANDIDATES
Our ability to generate significant revenues in the near term is heavily dependent on the success of the partnered product candidate Iluvien, for which an NDA has been accepted and granted a Priority Review by the FDA. If our collaborative partner is unable to obtain regulatory approval for and successfully commercialize Iluvien, or experiences significant delays in doing so, our business will be materially harmed.
In the near term, our ability to generate significant revenues depends on the ability of Alimera to obtain regulatory approval for and successfully commercialize Iluvien. Based on Alimera’s analysis of the month 24 clinical readout from its Phase III pivotal clinical trials for the use of Iluvien in the treatment of DME (collectively, its “FAME Study”), Alimera filed an NDA for approval of the low dose of Iluvien in the United States in June 2010, followed by registration filings in Austria, France, Germany, Italy, Portugal and Spain in July 2010. Although the FDA accepted Alimera’s submission and granted a Priority Review, they may request additional information from Alimera, including data from additional clinical trials, and ultimately may not grant marketing approval for Iluvien. Furthermore, although Alimera and we believe the month 24 clinical readout from Alimera’s FAME Study demonstrates Iluvien’s efficacy in the treatment of DME, clinical data often is susceptible to varying interpretations, and many companies that believed their products performed satisfactorily in clinical trials have nonetheless failed to obtain FDA approval for their products.
If Alimera is not successful in obtaining regulatory approval for and commercializing Iluvien, or is significantly delayed in doing so, our business will be materially harmed. Alimera’s ability to successfully obtain regulatory approval for and commercialize Iluvien will depend on, among other things, its ability to:
| • | receive marketing approval from the FDA and similar foreign regulatory authorities; |
| • | maintain commercial manufacturing arrangements with third-party manufacturers; |
| • | produce, or have its third-party manufacturers produce, sufficient quantities of Iluvien in a validated process to permit successful commercialization; |
| • | launch commercial sales of Iluvien; and |
| • | secure acceptance of Iluvien in the medical community and with third-party payors. |
Both we and Alimera believe the FDA will consider the most relevant population for determining the safety and efficacy of Iluvien to be the full data set of all 956 patients randomized into Alimera’s FAME Study, with data imputation employed using “last observation carried forward” for data missing because of patients who discontinued the trial or were unavailable for follow-up (the “Full Analysis Set”). The primary efficacy endpoint was met with statistical significance for both the low dose and the high dose of Iluvien in both trials using the Full Analysis Set, and Alimera intends to submit to the FDA an analysis based on this data set for the low dose of
21
Table of Contents
Iluvien. However, Alimera’s FAME Study protocol did not include the Full Analysis Set. The FAME Study protocol provides that the primary assessment of efficacy will be based on another data set that excludes from the Full Analysis Set three patients who were enrolled but never treated, excludes data collected for patients subsequent to their use of treatments prohibited by Alimera’s FAME Study protocol and imputes the last observation prior to the protocol violation forward to month 24 using the LOCF method (the “Modified ART Data Set”). Statistical significance was not achieved for either the low dose or the high dose of Iluvien in one trial using the Modified ART Data Set. There is no assurance that the FDA will utilize the Full Analysis Set and not the Modified ART Data Set or another data set in determining whether Iluvien is safe and effective, which could result in the FDA not granting marketing approval for Iluvien.
Alimera reports that it expects to obtain a regulatory agency waiver from the requirement to perform carcinogenicity studies of Iluvien in animals. Alimera’s month 18 readouts from its open-label Phase II human pharmacokinetic clinical trial (the “PK Study”) indicated to Alimera that there is negligible systemic absorption of FA in patients being treated with Iluvien. However, Alimera may be unable to demonstrate negligible systemic absorption of FA in its PK Study beyond month 18, or may not obtain a regulatory agency waiver from the requirement to perform carcinogenicity studies of Iluvien in animals regardless. Alimera reports that if it is required to perform carcinogenicity studies of Iluvien in animals, the approval of Iluvien could be delayed by up to 36 months.
Iluvien utilizes FA, a corticosteroid that has demonstrated undesirable side effects in the eye, and the success of Iluvien, therefore, will be dependent upon achieving an acceptable risk/benefit profile.
Iluvien utilizes FA, a corticosteroid whose use in the eye has been associated with undesirable side effects such as increased incidence of intraocular pressure (“IOP”), which may increase the risk of glaucoma and cataract formation. Alimera has performed a full analysis of only the month 24 clinical data from its FAME Study, and the extent of Iluvien’s long-term side-effect profile is not yet known. Upon review of Alimera’s NDA for the low dose of Iluvien in the treatment of DME, the FDA may conclude that Alimera’s FAME Study did not demonstrate that Iluvien has sufficient levels of efficacy to outweigh the risks associated with its side-effect profile. Conversely, the FDA may conclude that Iluvien’s side-effect profile does not demonstrate an acceptable risk/benefit relationship in line with Iluvien’s demonstrated efficacy. In the event of such conclusions, Alimera may not receive regulatory approval from the FDA or from similar regulatory agencies in other countries.
Although Alimera’s NDA was accepted with Priority Review, Iluvien may not be approved in a timely manner.
Although the FDA has granted Priority Review under FDA procedures, that designation does not necessarily mean a faster regulatory review process, nor does it necessarily confer any advantage with respect to approval as compared to conventional FDA procedures. Receiving Priority Review from the FDA does not guarantee approval within the six-month review/approval cycle.
Even if Alimera receives regulatory approval for Iluvien, the FDA and other regulatory agencies may impose limitations on the indicated uses for which Iluvien may be marketed, may subsequently withdraw approval for Iluvien or may take other actions against Iluvien that would be adverse to our business.
Regulatory agencies generally approve products for particular indications. If any regulatory agency approves Iluvien for a limited indication, the size of the potential market for Iluvien will be reduced. For example, the potential market for Iluvien would be reduced if the FDA limited the indications of use to only those patients diagnosed with clinically significant DME as opposed to all DME, or restricted the use to patients exhibiting IOP below a certain level at the time of treatment.
Additionally, product approvals, once granted, may be withdrawn if problems occur after initial marketing. If and when Iluvien does receive regulatory approval or clearance, the marketing, distribution and manufacture of Iluvien will be subject to regulation by the FDA in the United States and by similar entities in other countries. Alimera will need to comply with facility registration and product listing requirements of the FDA and similar
22
Table of Contents
entities in other countries, and will need to adhere to the FDA’s Quality System Regulations. Noncompliance with applicable FDA and similar entities’ requirements could result in warning letters, fines, injunctions, civil penalties, recall or seizure of Iluvien, total or partial suspension of production, refusal of regulatory agencies to grant approvals, withdrawal of approvals by regulatory agencies or criminal prosecution. Alimera also will need to maintain compliance with federal, state and foreign laws regarding sales incentives, referrals and other programs.
If we or our licensees are unable to complete clinical trials for our product candidates or do not receive the necessary regulatory approvals, we or our licensees will be unable to commercialize our product candidates.
Our current and future activities are and will be subject to stringent regulation by governmental authorities both in the United States and in any other country in which our products are marketed. Before we or our licensees can manufacture, market and sell any of our product candidates, approval from the FDA and/or foreign regulatory authorities is required. Generally, in order to obtain these approvals, pre-clinical studies and clinical trials must demonstrate that each of these product candidates is safe for human use and effective for its targeted disease or condition.
Our product candidates, other than Iluvien for DME, are in early stages of development. Product development involves a high degree of risk, and generally only a small proportion of research and development programs result in an approved product. If clinical trials for any of our product candidates do not provide the necessary evidence of safety and effectiveness, those product candidates could not be manufactured and sold, and would not generate revenues. Clinical trials initiated by us or our collaborative partners for product candidates may fail or be delayed by many factors, including the following:
| • | our (or our licensees’) lack of sufficient funding to pursue trials rapidly or at all; |
| • | our (or our licensees’) inability to attract clinical investigators for trials; |
| • | our (or our licensees’ ) inability to recruit patients in sufficient numbers or at the expected rate; |
| • | our inability to reach agreement with a licensee to undertake the clinical trials; |
| • | adverse side effects; |
| • | failure of the trials to demonstrate a product’s safety or efficacy; |
| • | our (or our licensees’) failure to meet FDA or other regulatory agency requirements for clinical trial design; |
| • | our (or our licensees’) inability to follow patients adequately after treatment; |
| • | changes in the design or manufacture of a product; |
| • | failures by, or changes in our (or our licensees’) relationship with, contract research organizations, third-party vendors and investigators responsible for pre-clinical testing and clinical trials; |
| • | our (or our licensees’) inability to manufacture sufficient quantities of materials for use in clinical trials; |
| • | governmental or regulatory agency assessments of pre-clinical or clinical testing that differs from our (or our licensees’) interpretations or conclusions that product candidates meet quality standards for stability, quality, purity and potency; and |
| • | governmental or regulatory delays, or changes in approval policies or regulations. |
Results from pre-clinical testing and early clinical trials often do not accurately predict results of later clinical trials. Data obtained from pre-clinical and clinical activities are susceptible to varying interpretations, which may delay, limit or prevent regulatory approval. Data from pre-clinical studies, early clinical trials and interim periods in multi-year trials are preliminary and may change, and final data from pivotal trials for such
23
Table of Contents
products may differ significantly. Adverse side effects may develop that delay, limit or prevent the regulatory approval of products, or cause such regulatory approvals to be limited or even rescinded.
Additional trials necessary for approval may not be undertaken or may ultimately fail to establish the safety and efficacy of our product candidates. The FDA or other relevant regulatory agencies may not approve our product candidates for manufacture and sale, and any approval by the FDA does not ensure approval by other regulatory agencies (which could require us to comply with numerous and varying regulatory requirements, possibly including additional clinical testing). Any product approvals we or our licensees achieve could also be withdrawn for failure to comply with regulatory standards or due to unforeseen problems after the products’ marketing approval. In either case, marketing efforts with respect to the affected product would have to cease. In addition, the FDA or other regulatory agencies may impose limitations on the indicated uses for which a product may be marketed.
In addition to testing, regulatory agencies impose various requirements on manufacturers and sellers of products under their jurisdiction, such as packaging, labeling, manufacturing practices, record keeping and reporting. Regulatory agencies may also require post-marketing testing and surveillance programs to monitor a product’s effects. Furthermore, changes in existing regulations or the adoption of new regulations could prevent us from obtaining, or affect the timing of, future regulatory approvals.
We have a limited ability to develop and market products ourselves. If we are unable to find marketing or commercialization partners, or our marketing or commercialization partners do not successfully develop or market our products, we may be unable to effectively develop and market products on our own.
We have limited product development capability and no marketing or sales staff. Developing products and achieving market acceptance for them will require extensive and substantial efforts by experienced personnel as well as expenditure of significant funds. We may not be able to establish sufficient capabilities necessary to develop products and achieve market penetration ourselves.
Our business strategy includes entering into collaborative and licensing arrangements for the development and commercialization of our product candidates, and we currently have collaboration and licensing arrangements with Alimera, Pfizer, Bausch & Lomb and Intrinsiq. The curtailment or termination of any of these arrangements could adversely affect our business, our ability to develop and commercialize our products, product candidates and proposed products and our ability to fund operations.
The success of these and future collaborative and licensing arrangements will depend heavily on the experience, resources, efforts and activities of our licensees. Our licensees have, and are expected to have, significant discretion in making decisions related to the development of product candidates and the commercialization of products under these collaboration agreements. Risks that we face in connection with our collaboration and licensing strategy include the following:
| • | our collaborative and licensing arrangements are, and are expected to be, subject to termination under various circumstances, including on short notice and without cause; |
| • | we are required, and expect to be required, under our collaborative and licensing arrangements not to conduct specified types of research and development in the field that is the subject of the arrangement, limiting the areas of research and development that we can pursue; |
| • | our licensees may develop and commercialize, either alone or with others, products that are similar to or competitive with our products; |
| • | our licensees, consistent with other pharmaceutical and biotechnology companies that have historically acted similarly, may for a variety of reasons change the focus of their development and commercialization efforts or decrease or fail to increase spending related to our products or product candidates, thereby limiting the ability of these products to reach their potential; |
24
Table of Contents
| • | our licensees may lack the funding, personnel or experience to develop and commercialize our products successfully or may otherwise fail to do so; and |
| • | our licensees may not perform their obligations, in whole or in part. |
To the extent that we choose not to, or we are unable to, enter into future license agreements with marketing and sales partners and, alternatively, seek to market and sell products ourselves, we would experience increased capital requirements to develop the ability to manufacture, market and sell future products. We may not be able to manufacture, market or sell our products or future products independently in the absence of such agreements.
Our current licensees may terminate their agreements with us at any time, and if they do, we may not be able to effectively develop and sell products currently licensed to them.
Our licensees have rights of termination under our agreements with them. Exercise of termination rights by one or more of our licensees may leave us, at least temporarily, without development, marketing or sales resources, which may have an adverse effect on our business, financial condition and results of operations. Additionally, our interests may not continue to coincide with those of our partners, and our partners may develop, independently or with third parties, products or technologies that could compete with our products. Further, disagreements over rights or technologies or other proprietary interests may occur.
We have exclusively licensed certain of our controlled drug delivery technologies to Pfizer for certain ophthalmic applications. Pfizer is currently funding early stage research and pre-clinical development of potential product candidates under our worldwide collaborative research and license agreement with it. Pfizer may terminate the agreement without penalty at any time and for any reason upon 60 days’ written notice. We have exclusively licensed our technology underlying Vitrasert and Retisert to Bausch & Lomb, which can terminate its agreement with us without penalty at any time upon 90 days’ written notice. We have licensed the technology underlying Iluvien and certain ophthalmic applications to Alimera. Alimera has the financial responsibility for the development of Iluvien and any other licensed products developed under our collaboration agreement, along with sole responsibility for the commercialization of such licensed products. Alimera may abandon the development and commercialization of any licensed product at any time.
Any of Pfizer, Alimera or Bausch & Lomb may decide not to continue with or commercialize any or all of the licensed products, change strategic focus, pursue alternative technologies or develop competing products. While Pfizer and Bausch & Lomb have significant experience in the ophthalmic field and have substantial resources, there is no assurance whether, and to what extent, that experience and those resources will be devoted to our technologies. Because we do not currently have sufficient funding or internal capabilities to develop and commercialize our products and product candidates, decisions, actions, breach or termination of these agreements by Pfizer, Bausch & Lomb or Alimera could delay or stop the development or commercialization of Retisert, Iluvien or other potential future product candidates licensed to such entities.
If our competitors and potential competitors develop products that receive regulatory approval before our product candidates are approved or reach the market prior to our product candidates, are more effective or have fewer side effects than our products or product candidates or are more effectively marketed or cost less, our products or product candidates may not achieve the sales we anticipate and could be rendered obsolete.
We believe that pharmaceutical, drug delivery and biotechnology companies, research organizations, governmental entities, universities, hospitals, other nonprofit organizations and individual scientists are seeking to develop the drugs, therapies, products, approaches or methods to treat our targeted diseases or their underlying causes. For many of our targeted diseases, competitors have alternate therapies that are already commercialized or are in various stages of development ranging from discovery to advanced clinical trials. Any of these drugs, therapies, products, approaches or methods may receive government approval or gain market acceptance more rapidly than our products and product candidates, may offer therapeutic or cost advantages, or may cure our targeted diseases or their underlying causes completely, which could reduce demand for our products and product
25
Table of Contents
candidates and could render them noncompetitive or obsolete. For example, sales of Vitrasert for the treatment of CMV retinitis, a disease that affects people with late-stage AIDS, declined significantly because of treatments that delay the onset of late-stage AIDS.
Many of our competitors and potential competitors have substantially greater financial, technological, research and development, marketing and personnel resources than us. Our competitors may succeed in developing alternate technologies and products that, in comparison to the products we have and are seeking to develop:
| • | are more effective and easier to use; |
| • | are more economical; |
| • | have fewer side effects; or |
| • | may otherwise render our products less competitive or obsolete. |
Many of these competitors have greater experience in developing products, conducting clinical trials, obtaining regulatory approvals or clearances and manufacturing and marketing products.
Our products and product candidates may not achieve and maintain market acceptance, and may never generate significant revenues.
In both domestic and foreign markets, the commercial success of our products and product candidates will require not only obtaining regulatory approvals but also obtaining market acceptance by retinal specialists and other doctors, patients, government health administration authorities and other third-party payors. Whether and to what extent our products and product candidates achieve and maintain market acceptance will depend on a number of factors, including: demonstrated safety and efficacy, cost-effectiveness, potential advantages over other therapies, our and our collaborative partners’ marketing and distribution efforts and the reimbursement policies of government and other third-party payors. In particular, if government and other third-party payors do not provide adequate coverage and reimbursement levels for our products and product candidates, the market acceptance of our products and product candidates will be limited. Both government and other third-party payors attempt to contain healthcare costs by limiting coverage and the level of reimbursement for products and, accordingly, they might challenge the price and cost-effectiveness of our products, or refuse to provide coverage for uses of our products for certain disease indications. If our products and product candidates fail to achieve and maintain market acceptance, they may fail to generate significant revenues and our business may be significantly harmed.
Guidelines, recommendations and studies published by various organizations could reduce the use of our products and product candidates.
Government agencies, professional societies, practice management groups, private health and science foundations and organizations focused on various diseases may publish guidelines, recommendations or studies related to our products and product candidates or our competitors’ products. Any such guidelines, recommendations or studies that reflect negatively on our products or product candidates could result in decreased use, sales of, and revenues from, one or more of our products and product candidates. Furthermore, our success depends in part on our and our partners’ ability to educate healthcare providers and patients about our products and product candidates, and these education efforts could be rendered ineffective by, among other things, third-parties’ guidelines, recommendations or studies.
26
Table of Contents
RISKS RELATED TO OUR INTELLECTUAL PROPERTY
We rely heavily upon patents and trade secrets to protect our proprietary technologies. If we fail to protect our intellectual property or infringe on others’ technologies, our ability to develop and market our products and product candidates may be compromised.
Our success is dependent on whether we can obtain patents, defend our existing patents and operate without infringing on the proprietary rights of third parties. As of August 31, 2010, we had 168 patents and 179 pending patent applications, including patents and pending applications covering our Durasert, BioSilicon and CODRUG technologies. Intellectual property protection of our technologies is uncertain. We expect to seek to patent and protect our proprietary technologies. However, there is no assurance that any additional patents will be issued to us as a result of our pending or future patent applications or that any of our patents will withstand challenges by others. In addition, we may not have sufficient funds to patent and protect our proprietary technologies to the extent that we would desire, or at all. If we were determined to be infringing any third party patent, we could be required to pay damages, alter our products or processes, obtain licenses, pay royalties or cease certain operations. We may not be able to obtain any required licenses on commercially favorable terms, if at all. In addition, many foreign country laws may treat the protection of proprietary rights differently from, and may not protect our proprietary rights to the same extent as, laws in the United States and Patent Co-operation Treaty countries.
Prior art may reduce the scope or protection of, or invalidate, patents. Previously conducted research or published discoveries may prevent patents from being granted, invalidate issued patents or narrow the scope of any protection obtained. Reduction in scope of protection or invalidation of our licensed or owned patents, or our inability to obtain patents, may enable other companies to develop products that compete with our products and product candidates on the basis of the same or similar technology. As a result, our patents and those of our licensors may not provide any or sufficient protection against competitors. While we have not been, and are not currently involved in, any litigation over intellectual property, such litigation may be necessary to enforce any patents issued or licensed to us or to determine the scope and validity of third party proprietary rights. We may also be sued by one or more third parties alleging that we infringe their intellectual property rights. Any intellectual property litigation would be likely to result in substantial costs to us and diversion of our efforts, and could prevent or delay our discovery or development of product candidates. If our competitors claim technology also claimed by us, and if they prepare and file patent applications in the U.S. or other jurisdictions, we may have to participate in interference proceedings declared by the U.S. Patent and Trademark office or the appropriate foreign patent office to determine priority of invention, which could result in substantial cost to us and diversion of our efforts. Any such litigation or interference proceedings, regardless of the outcome, could be expensive and time consuming. Litigation could subject us to significant liabilities to third parties, requiring disputed rights to be licensed from third parties and/or requiring us to cease using certain technologies.
We also rely on trade secrets, know-how and technology that are not protected by patents to maintain our competitive position. We try to protect this information by entering into confidentiality agreements with parties that have access to it, such as our corporate partners, collaborators, employees, and consultants. Any of these parties could breach these agreements and disclose our confidential information, or our competitors may learn of the information in some other way. If any material trade secret, know-how or other technology not protected by a patent were to be disclosed to or independently developed by a competitor, our competitive position could be materially harmed.
RISKS RELATED TO OUR BUSINESS, INDUSTRY, STRATEGY AND OPERATIONS
If we fail to retain some or all of our key personnel, our business could suffer.
We are dependent upon the principal members of our management, administrative and scientific staff. In addition, we believe that our future success in developing our products and achieving a competitive position will depend to a large extent on whether we can attract and retain additional qualified management and scientific personnel. There is strong competition for such personnel within the industry in which we operate and we may
27
Table of Contents
not be able to continue to attract such personnel either to Massachusetts, where much of our research and development is conducted, or to Malvern in the U.K. As we do not have large numbers of employees and our products are unique and highly specialized, the loss of the services of one or more of the senior management or scientific staff, or the inability to attract and retain additional personnel and develop expertise as needed, could have a material adverse effect on our results of operations and financial condition.
If we are subject to product liability suits, we may not have sufficient insurance to cover damages.
The testing, manufacturing, and marketing and sale of the products utilizing our technologies involves risks that product liability claims may be asserted against us and/or our licensees. Our current clinical trial and product liability insurance may not be adequate to cover damages resulting from product liability claims. Regardless of their merit or eventual outcome, product liability claims could require us to spend significant time, money and other resources to defend such claims, could result in decreased demand for our products and product candidates or result in reputational harm and could result in the payment of a significant damage award. Our product liability insurance coverage is subject to deductibles and coverage limitations and may not be adequate in scope to protect us in the event of a successful product liability claim. Further, we may not be able to acquire sufficient clinical trial or product liability insurance in the future on reasonable commercial terms, if at all.
The trend towards consolidation in the pharmaceutical and biotechnology industries may adversely affect us.
There is an ongoing trend of consolidation in the pharmaceutical and biotechnology industries. This consolidation trend could result in the remaining companies having greater financial resources and technological capabilities, thus intensifying competition. This trend could also result in fewer potential collaboration partners or licensees for our product candidates. In addition, if a consolidating company is already doing business with our competitors, we could lose existing or potential future licensees or collaboration partners as a result of such consolidation.
If we fail to comply with environmental laws and regulations, our ability to manufacture and commercialize products may be adversely affected.
Medical and biopharmaceutical research and development involves the controlled use of hazardous materials, such as radioactive compounds and chemical solvents. We are subject to federal, state and local laws and regulations in the U.S. and abroad governing the use, manufacture, storage, handling and disposal of such materials and waste products. We could be subject to both criminal liability and civil damages in the event of an improper or unauthorized release of, or exposure of individuals to, hazardous materials. In addition, claimants may sue us for injury or contamination that results from our use or the use by third parties of these materials, and our liability may exceed our total assets. Compliance with environmental laws and regulations is expensive, and current or future environmental regulations may impair our research, development or production efforts or harm our operating results.
If we encounter problems with product manufacturing, we could experience delays in product development and commercialization, which would adversely affect our future profitability.
Our ability to conduct timely pre-clinical and clinical research and development programs, obtain regulatory approvals, develop and commercialize our product candidates and fulfill our contract manufacturing obligations to others will depend, in part, upon our and our collaborative partners’ ability to manufacture our products and product candidates, either directly or through third parties, in accordance with FDA and other regulatory requirements. The manufacture, packaging and testing of our products and product candidates are regulated by the FDA and similar foreign regulatory entities and must be conducted in accordance with applicable current good manufacturing practices, or cGMP. Any change in a manufacturing process or procedure used for one of our products or product candidates, including a change in the location at which a product or product candidate is being manufactured or in the third-party manufacturer being used, may require the FDA’s and similar foreign
28
Table of Contents
regulatory entities’ prior review and/or approval in accordance with applicable cGMP regulations. Additionally, the FDA and similar foreign regulatory entities may implement new standards, or change their interpretation and enforcement of existing standards, for the manufacture, packaging and testing of products at any time.
There are a limited number of manufacturers that operate under cGMP regulations that are both capable of manufacturing our products and product candidates and are willing to do so. Failure by us, our collaborative partners, or our or their third-party manufacturers, to comply with applicable manufacturing requirements could result in sanctions being imposed on us, including fines, injunctions, civil penalties, failure of regulatory authorities to grant marketing approval of our product candidates, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of product, operating restrictions and criminal prosecutions. In addition, we or our collaborative partners may not be able to manufacture our product candidates successfully or have a third party manufacture them in a cost-effective manner. If we or our collaborative partners are unable to develop our own manufacturing facilities or to obtain or retain third-party manufacturing on acceptable terms, we may not be able to conduct certain future pre-clinical and clinical testing or to supply commercial quantities of our products.
We manufacture clinical supplies in connection with pre-clinical or clinical studies conducted by us or our collaboration partners. Under our collaboration agreements with Alimera, Pfizer and Bausch & Lomb, we have provided our licensees the exclusive rights to manufacture commercial quantities of products, once approved for marketing. Our current reliance on third-party manufacturers entails risks, including:
| • | the possibility that third parties may not comply with the FDA’s cGMP regulations, other regulatory requirements, and those of similar foreign regulatory bodies, and may not employ adequate quality assurance practices; |
| • | supply disruption, deterioration in product quality or breach of a manufacturing or license agreement by the third party because of factors beyond our control; |
| • | the possible termination or non-renewal of a manufacturing or licensing agreement with a third party at a time that is costly or inconvenient to us; and |
| • | our inability to identify or qualify an alternative manufacturer in a timely manner, even if contractually permitted to do so. |
Alimera reports that it currently intends to rely on a single third-party manufacturer of the Iluvien insert (Alliance Medical Products, Inc. (“Alliance”)), a single third-party manufacturer of the Iluvien inserter (Flextronics International, Ltd. or an affiliate of Flextronics International, Ltd. (“Flextronics”)) and a single third-party manufacturer of Iluvien’s active pharmaceutical ingredient (FARMABIOS S.R.L./Byron Chemical Company Inc. (“FARMABIOS”)). Alimera reported that it finalized a long-term agreement with Alliance for the manufacture of the Iluvien insert, but has not reported that it finalized a long-term agreement with Flextronics for the manufacture of the Iluvien inserter or with FARMABIOS for the manufacture of Iluvien’s active pharmaceutical ingredient. Our business could be significantly harmed if these third parties are not able to satisfy demand for Iluvien and alternative sources are not available. In addition, the materials necessary to produce Iluvien or to formulate the active pharmaceutical ingredient may not be available on commercially reasonable terms, or at all, which could affect the development and commercialization of Iluvien.
Problems associated with international business operations could affect our ability to manufacture and sell our products. If we encounter such problems, our costs could increase and our development of products could be delayed.
We currently maintain offices and research and development facilities in the U.S. and the U.K., and we intend to license products for sale and/or sell products in most major world healthcare markets. A number of risks are inherent in our international strategy. In order for us to license and manufacture our products, we must obtain country and jurisdiction-specific regulatory approvals or clearances to comply with regulations regarding safety and quality. We may not be able to obtain or maintain regulatory approvals or clearances in such countries,
29
Table of Contents
and we may be required to incur significant costs in obtaining or maintaining foreign regulatory approvals or clearances. In addition, our operations and revenues may be subject to a number of risks associated with foreign commerce, including the following:
| • | staffing and managing foreign operations; |
| • | political and economic instability; |
| • | foreign currency exchange fluctuations; |
| • | foreign tax laws, tariffs and freight rates and charges; |
| • | timing and availability of export licenses; |
| • | inadequate protection of intellectual property rights in some countries; and |
| • | obtaining required governmental approvals. |
Credit and financial market conditions may exacerbate certain risks affecting our business.
Sales of our products depend on the availability and extent of reimbursement from government and other third-party payors. Difficult credit and financial market conditions may increase the risk that government and other third-party payors will reduce the availability or extent of reimbursement for our products, and the risk that third-party payors will delay or default on reimbursement obligations.
Development and sales of our products and product candidates also heavily depend on collaborative partners and third-party suppliers. Difficult credit and financial market conditions may increase the risk that there are delays, disruptions or defaults in the performance of these third parties’ obligations to us.
Legislative or regulatory changes may adversely affect our business, operations and financial results.
Our industry is highly regulated and new laws, regulations and judicial decisions, and new interpretations of existing laws, regulations and judicial decisions, may adversely affect our business, operations and financial results.
The Patient Protection and Affordable Care Act of 2010, as amended by the Health Care and Education Reconciliation Act of 2010 (the “PPACA”), is intended to expand U.S. healthcare coverage primarily through the imposition of health insurance mandates on employers and individuals and expansion of the Medicaid program. Several provisions of this new law could significantly reduce payments from Medicare and Medicaid for our products and product candidates over the next 10 years, resulting in potentially significant reductions of our revenues. The PPACA’s effects cannot be fully known until its provisions are implemented, and the Centers for Medicare & Medicaid Services, and other federal and state agencies, issue applicable regulations or guidance. Proposed U.S. state healthcare reforms, and any foreign healthcare reforms, also could alter the availability, methods and rates of reimbursements from the government and other third-party payors for our products and product candidates, and could adversely affect our business strategy, operations and financial results.
The U.S. Food and Drug Administration Amendment Act of 2007 has granted the FDA enhanced authority over products already approved for sale, including authority to require post-marketing studies and clinical trials, labeling changes based on new safety information and compliance with risk evaluations and mitigation strategies approved by the FDA. The FDA’s exercise of this relatively new authority could result in delays and increased costs during product development, clinical trials and regulatory review and approval, increased costs following regulatory approval to assure compliance with new post-approval regulatory requirements, and potential restrictions on the sale or distribution of approved products following regulatory approval.
30
Table of Contents
RISKS RELATED TO OUR COMMON STOCK
The price of our common stock may be volatile.
The price of our common stock (including common stock represented by CHESS Depositary Interests (CDIs)) may be affected by developments directly affecting our business and by developments out of our control or unrelated to us. The biotechnology sector, in particular, and the stock market generally, are vulnerable to abrupt changes in investor sentiment. Prices of securities and trading volume of companies in the biotechnology industry, including ours, can swing dramatically in ways unrelated to, or that bear a disproportionate relationship to, operating performance. The price of our stock (and CDIs) and their trading volumes may fluctuate based on a number of factors including, but not limited to:
| • | clinical trial results and other product and technological developments and innovations; |
| • | FDA and other governmental regulatory actions, receipt and timing of approvals of our product candidates, and any denials and withdrawals of approvals; |
| • | competitive factors, including the commercialization of new products in our markets by our competitors; |
| • | advancements with respect to treatment of the diseases targeted by our product candidates; |
| • | developments relating to collaborative partners, including execution and termination of agreements, achievement of milestones and receipt of payments; |
| • | the success of our collaborative partners in marketing any approved products and the amount and timing of the royalties payable to us; |
| • | availability and cost of capital and our financial and operating results; |
| • | changes in reimbursement policies or other practices relating to our product candidates or the pharmaceutical industry generally; |
| • | meeting, exceeding or failing to meet analysts’ or investors’ expectations, and changes in evaluations and recommendations by securities analysts; |
| • | economic, industry and market conditions, changes or trends; and |
| • | other factors unrelated to us or the biotechnology industry. |
In addition, low trading volume in our common stock or our CDIs may increase their price volatility. As of August 31, 2010, we had approximately 18.5 million shares of common stock outstanding. The combined daily trading volume in the common stock (and CDIs) on the exchanges in which our common stock are listed averaged approximately 31,000 shares during the period May to August 2010. Holders of our common stock and CDIs may not be able to liquidate their positions at the desired time or price.
If the holders of our outstanding warrants and stock options exercise their warrants and options, ownership of our common stock holders may be diluted, and our stock price may decline.
The issuance of shares of our common stock upon exercise of our outstanding warrants and stock options would result in dilution to the interests of other holders of our common stock and could adversely affect our stock price. As of September 15, 2010, we had outstanding approximately 11.0 million warrants and 2.9 million options to acquire shares of our common stock, or approximately 42.8% of our shares on a fully diluted basis. Certain of the options are subject to performance conditions and certain of the options are subject to shareholder approval. Although the exercise prices of all of these warrants and some of the stock options are substantially above the market price at that date, the overhang of such warrants and options may adversely affect our stock price. The warrant exercise prices may be adjusted under certain circumstances, including, among others, in the event we issue securities in a rights offering at a lower price than the exercise price.
31
Table of Contents
Pfizer owns a significant percentage of our common stock and is a collaborative partner and therefore may be able to influence our business in ways that are not beneficial to you.
Pfizer owned approximately 10.0% of our outstanding shares as of August 31, 2010 and is a collaborative partner. As a result, Pfizer may be able to exert significant influence over our board of directors and how we operate our business. The concentration of ownership may also have the effect of delaying or preventing a change in control of our company.
We have paid penalties pursuant to registration agreements with securities holders relating to resale registration statements, and any requirement to pay such penalties in the future may have a material adverse effect on our financial condition.
We have registration rights agreements that require us to file and maintain the effectiveness of registration statements for the resale of our common stock, which provide for monetary penalties in the event of our failure to do so. During the year ended June 30, 2007, we paid registration delay penalties of approximately $2.3 million in connection with our then outstanding Sandell convertible promissory note and Absolute subordinated convertible notes. Our failure or inability to maintain the effectiveness of any of our required registration statements or to adequately update information in the related prospectuses may subject us to additional penalties under our current registration rights agreements. Payment of additional penalties may have a material adverse effect on our financial condition and may require us to suspend, curtail or terminate our operations or delay, reduce the scope of or eliminate one or more of our research and development programs, any of which could have a material adverse effect on our business.
We do not currently intend to pay dividends on our common stock, and any return to investors will come, if at all, only from potential increases in the price of our common stock.
At the present time, we intend to use available funds to finance our operations. Accordingly, while payment of dividends rests within the discretion of our board of directors, no cash dividends on our common shares have been declared or paid by us and we have no intention of paying any such dividends in the foreseeable future.
| ITEM 1B. | UNRESOLVED STAFF COMMENTS |
None.
| ITEM 2. | PROPERTIES |
We do not own any real property. We lease the following:
| • | 3,940 square feet of laboratory space, 1,582 square feet of clean room space and 7,890 square feet of office space in Watertown, Massachusetts under a lease agreement that expires in April 2011, with a three-year option to renew; and |
| • | 1,500 square feet of laboratory space and 1,800 square feet of office space in Malvern, United Kingdom under lease agreements that expire in June 2012, subject to a six month advance notice of termination by either party. |
| ITEM 3. | LEGAL PROCEEDINGS |
None.
| ITEM 4. | [REMOVED AND RESERVED] |
32
Table of Contents
| ITEM 4A. | EXECUTIVE OFFICERS OF THE REGISTRANT |
Each of our officers holds office until the first meeting of the board of directors following the next annual meeting of stockholders and until such officer’s respective successor is chosen and qualified, unless a shorter period shall have been specified by the terms of such officer’s election or appointment. Our current officers are listed below.
Paul Ashton, 49
President and Chief Executive Officer
Dr. Ashton has served as President and Chief Executive Officer since January 2009 and was previously the Managing Director of the Company from January 2007 and its Executive Director of Strategy from December 2005 to January 2007. From 1996 until its acquisition by the Company in December 2005, Dr. Ashton was the President and Chief Executive Officer of Control Delivery Systems, Inc. (CDS), a drug delivery company that he co-founded in 1991. Dr. Ashton was previously a joint faculty member in the Departments of Ophthalmology and Surgery at the University of Kentucky, served on the faculty of Tufts University and worked as a pharmaceutical scientist at Hoffman-LaRoche.
Lori Freedman, 43
Vice President of Corporate Affairs, General Counsel and Company Secretary
Ms. Freedman has served as Vice President of Corporate Affairs, General Counsel and Company Secretary since May 2006, and of Control Delivery Systems, Inc. (CDS) from 2001 to May 2006. Prior to that, Ms. Freedman served as Vice President, Business Development, and Counsel of Macromedia, Inc., a provider of software for creating Internet content and business applications, from March 2001 through September 2001. Ms. Freedman has also served as Vice President, General Counsel, and Secretary of Allaire Corporation, a provider of Internet infrastructure for building business applications, from 1999 until Allaire’s acquisition by Macromedia in 2001, as Corporate Counsel of Polaroid Corporation from May 1998 to December 1998 and with the law firm of McDermott, Will & Emery.
Leonard S. Ross, 60
Vice President, Finance and Principal Financial Officer
Mr. Ross has served as Vice President, Finance since November 2009 and previously as Corporate Controller from October 2006. Mr. Ross was designated as the Company’s principal financial officer in March 2009. From 2001 through April 2006, Mr. Ross served as Corporate Controller for NMT Medical, Inc., a medical device company. From 1990 to 1999, Mr. Ross was employed by JetForm Corporation, a developer of workflow software solutions, where he served in various capacities, including Vice President, Finance and Vice President, International Operations.
33
Table of Contents
| ITEM 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Market Information, Holders and Dividends
Our common stock is traded on the NASDAQ Global Market under the trading symbol “PSDV”. The following table sets forth the high and low prices per share of our common stock as reported on the NASDAQ Global Market for the periods indicated:
| High | Low | |||||
| Fiscal year ended June 30, 2010: |
||||||
| First Quarter |
$ | 6.25 | $ | 1.51 | ||
| Second Quarter |
6.06 | 2.86 | ||||
| Third Quarter |
4.72 | 3.08 | ||||
| Fourth Quarter |
5.14 | 3.26 | ||||
| Fiscal year ended June 30, 2009: |
||||||
| First Quarter |
$ | 3.65 | $ | 1.35 | ||
| Second Quarter |
2.49 | 0.51 | ||||
| Third Quarter |
1.20 | 0.60 | ||||
| Fourth Quarter |
2.22 | 0.81 | ||||
On September 22, 2010, the last reported sale price of our common stock on the NASDAQ Global Market was $4.28. As of that date, we had approximately 49 holders of record of our common stock and, according to our estimates, approximately 3,075 beneficial owners of our common stock. In addition, as of that date, there were approximately 2,700 registered owners of our CDIs.
We have never paid cash dividends, and we do not anticipate paying cash dividends in the foreseeable future.
Equity Compensation Plan Information
The following table provides information about the securities authorized for issuance under the Company’s equity compensation plans as of June 30, 2010:
| Plan category |
Number of securities to be issued upon exercise of outstanding options, warrants and rights (a) |
Weighted-average exercise price of outstanding options, warrants and rights (*) (b) |
Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in Column a) (c) | ||||
| Equity Compensation plans approved by security holders |
2,151,312 | $ | 3.26 | 614,953 | |||
| Equity Compensation plans not approved by security holders |
— | — | — | ||||
| Total |
2,151,312 | $ | 3.26 | 614,953 | |||
| (*) | Of the total outstanding options, 185,312 are denominated in A$ and were translated at the June 30, 2010 exchange rate of A$1.00 = US$0.8567. |
Beginning on July 1, 2010 and on each subsequent anniversary date through July 1, 2017, the number of shares reserved for issuance under the Company’s 2008 Incentive Plan will be increased by the least of
34
Table of Contents
(i) 750,000 shares; (ii) 4% of the then outstanding shares of common stock; and (iii) any such lesser amount of shares as is determined by the Compensation Committee of the Board of Directors. On July 1, 2010, the number of shares issuable under the 2008 Incentive Plan increased by 741,255 shares, representing 4% of the outstanding shares at June 30, 2010.
Recent Sales of Unregistered Securities; Uses of Proceeds from Registered Securities; Issuer Repurchases of Equity Securities
None
| ITEM 6. | SELECTED FINANCIAL DATA |
The selected historical financial data set forth below as of June 30, 2010 and 2009 and for the years ended June 30, 2010, 2009 and 2008 have been derived from our audited consolidated financial statements, which are included elsewhere in this Annual Report on Form 10-K. The selected historical financial data as of June 30, 2008 and 2007 and for the years ended June 30, 2007 and 2006 have been derived from our audited consolidated financial statements contained in our Form 10-K filed with the SEC on September 26, 2008. The selected consolidated balance sheet data as of June 30, 2006 has been derived from our audited consolidated financial statements contained in our Form 8-K filed with the SEC on June 20, 2008.
The following selected consolidated financial data should be read in conjunction with Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations”, and the audited consolidated financial statements, and the notes thereto, and other financial information included elsewhere in this Annual Report on Form 10-K. The historical results are not necessarily indicative of the results to be expected for any future period.
| Year Ended June 30, | |||||||||||||||||||
| 2010 (1) | 2009 (1) | 2008 (1,2) | 2007 (3,4) | 2006 (5) | |||||||||||||||
| (In thousands except per share data) | |||||||||||||||||||
| Consolidated Statements of Operations Data: |
|||||||||||||||||||
| Revenues |
$ | 23,053 | $ | 12,162 | $ | 3,476 | $ | 1,785 | $ | 1,036 | |||||||||
| Income (loss) from continuing operations |
8,753 | (2,511 | ) | (75,670 | ) | (83,525 | ) | (45,312 | ) | ||||||||||
| Net income (loss) |
8,753 | (2,511 | ) | (75,670 | ) | (81,203 | ) | (46,957 | ) | ||||||||||
| Income (loss) per share—basic |
|||||||||||||||||||
| Income (loss) from continuing operations |
$ | 0.48 | $ | (0.14 | ) | $ | (4.17 | ) | $ | (7.57 | ) | $ | (6.02 | ) | |||||
| Net income (loss) |
$ | 0.48 | $ | (0.14 | ) | $ | (4.17 | ) | $ | (7.36 | ) | $ | (6.24 | ) | |||||
| Income (loss) per share—diluted |
|||||||||||||||||||
| Income (loss) from continuing operations |
$ | 0.46 | $ | (0.14 | ) | $ | (4.17 | ) | $ | (7.57 | ) | $ | (6.02 | ) | |||||
| Net income (loss) |
$ | 0.46 | $ | (0.14 | ) | $ | (4.17 | ) | $ | (7.36 | ) | $ | (6.24 | ) | |||||
| Weighted average shares outstanding: |
|||||||||||||||||||
| Basic |
18,405 | 18,263 | 18,166 | 11,038 | 7,521 | ||||||||||||||
| Diluted |
18,895 | 18,263 | 18,166 | 11,038 | 7,521 | ||||||||||||||
| As of June 30, | |||||||||||||||
| 2010 | 2009 | 2008 | 2007 | 2006 | |||||||||||
| (In thousands) | |||||||||||||||
| Consolidated Balance Sheet Data: |
|||||||||||||||
| Cash and cash equivalents |
$ | 15,514 | $ | 6,899 | $ | 15,609 | $ | 2,670 | $ | 6,692 | |||||
| Marketable securities |
2,051 | — | — | — | — | ||||||||||
| Total assets |
43,014 | 37,104 | 55,784 | 107,220 | 165,504 | ||||||||||
| Total deferred revenue |
6,896 | 10,534 | 18,590 | 1,702 | 1,948 | ||||||||||
| Long-term debt |
— | — | — | — | 2,912 | ||||||||||
| Total stockholders’ equity |
33,041 | 23,541 | 30,078 | 88,265 | 130,747 | ||||||||||
35
Table of Contents
| (1) | During the years ended June 30, 2010, 2009 and 2008, we recognized $22.3 million, $11.8 million and $3.3 million, respectively, of collaborative research and development revenue under our collaboration agreement with Alimera. See Note 3 to the accompanying audited consolidated financial statements for additional information. |
| (2) | At June 30, 2008, in connection with our annual review of goodwill, we incurred a $60.1 million goodwill impairment charge. See Note 4 to the accompanying consolidated financial statements for additional information. |
| (3) | At June 30, 2007, we recorded a $45.3 million impairment charge related to our Durasert intangible asset. |
| (4) | In April 2007, we sold the stock of our AION Diagnostics, Inc. subsidiary for a pre-tax and after-tax gain of $3.6 million. |
| (5) | In December 2005, we completed the acquisition of Control Delivery Systems, Inc. (CDS) for aggregate consideration of $108.2 million. For the years ended June 30, 2010, 2009, 2008, 2007 and 2006, substantially all of our collaborative research and development revenues and royalty income were attributable to the operations of CDS (renamed pSivida US, Inc.). Approximately $25.0 million of the purchase price was allocated to our Iluvien product candidate and was charged to acquired in-process research and development expense for the year ended June 30, 2006. |
36
Table of Contents
| ITEM 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
The following discussion and analysis of financial condition and results of operations should be read in conjunction with our audited consolidated financial statements and related notes beginning on page F-1 of this Annual Report on Form 10-K. This discussion contains forward-looking statements, based on current expectations and related to future events and our future financial performance, that involve risks and uncertainties. Our actual results may differ significantly from those anticipated or implied in these forward-looking statements as a result of many important factors, including, but not limited to, those set forth under Item 1A, “Risk Factors”, and elsewhere in this report.
Overview
We develop tiny, sustained release, drug delivery products that are administered by implantation, injection or insertion. Once administered, a drug is released on a controlled and level basis for months or years. We have two core technology systems, Durasert and BioSilicon. Utilizing three generations of our Durasert technology system, we have one product candidate for chronic eye disease that has been given Priority Review by the FDA and two of the only three products approved by the FDA for the long-term, sustained release delivery of drug to treat chronic eye disease. We have a collaboration agreement with Pfizer, our largest shareholder, to develop additional ophthalmic products.
Iluvien, the product candidate with Priority Review, is designed to provide sustained release treatment for DME. DME is a leading cause of vision loss for people under the age of 65 and has been estimated to affect over 1,000,000 people in the United States. Using the third-generation of our Durasert technology system, Iluvien is injected into the eye and delivers the corticosteroid FA over a period of up to 3 years.
Iluvien is licensed to Alimera, which is completing fully-recruited Phase III clinical trials. Based on 24-month data released in December 2009, Alimera filed an NDA with the FDA in June 2010 and registration filings in various European countries in July 2010. On August 30, 2010, the FDA granted Priority Review status and, as a result, Alimera could receive a response to its NDA from the FDA by the end of calendar year 2010. If approved, Alimera has indicated that it expects to commercialize Iluvien as early as the first calendar quarter of 2011. Under our collaboration agreement with Alimera, Iluvien is also being studied in investigator-sponsored pilot clinical trials designed to assess the safety and efficacy of Iluvien in both wet and dry AMD and RVO.
Our two FDA-approved sustained release products utilize earlier generations of our Durasert technology system, second-generation Retisert for the treatment of posterior uveitis and our first-generation Vitrasert for the treatment of AIDS-related CMV retinitis. We have licensed both of these products and the technologies underlying them to Bausch & Lomb.
Under our worldwide collaborative research and license agreement with Pfizer, we are working together on a joint research program aimed at developing ophthalmic products using our sustained release drug delivery technologies not licensed to others. Retisert provides sustained release treatment for approximately two and a half years, and Vitrasert provides sustained release treatment for six to nine months.
BioSilicon, our other principal technology system, is a fully-erodible, nanostructured, porous silicon designed to provide sustained delivery of various therapeutics, including small drug molecules, proteins and peptides. Based on results of our preliminary studies, we are currently targeting BioSilicon as a key second prong of our drug delivery technology platform.
Effective June 19, 2008, we reincorporated from Western Australia to the United States (the Reincorporation).
37
Table of Contents
Equity Financing
In July 2007, we sold 3,600,500 units at a price of $5.00 per unit for gross proceeds of $18.0 million. Each unit consisted of (i) one share of common stock; and (ii) one warrant to purchase 0.4 share of common stock at $6.60 per share. In addition, we simultaneously completed a sale of 513,699 units for gross proceeds of approximately $2.6 million.
License and Collaboration Agreements
Alimera
Upon execution of the Alimera Agreement in March 2008, we received consideration of $12.0 million in cash and Alimera cancelled $5.7 million of accrued development cost liabilities, including related penalties and accrued interest, owed by us to Alimera as of March 14, 2008. In addition, we received a $15.0 million conditional note providing for aggregate principal and interest payments of up to approximately $21.3 million through September 2012, Alimera agreed to pay us a $25.0 million milestone payment upon FDA approval of Iluvien for DME and Alimera assumed all financial responsibility for the development of licensed products under the Alimera Agreement, which had previously been shared equally, including reimbursement of approved development costs incurred by us in support of the ongoing clinical studies of Iluvien and anticipated regulatory submissions. In exchange, we decreased our share in any future profits, as defined, on sales of Iluvien by Alimera from 50% to 20%, subject to an offset of 20% of pre-profitability commercialization costs, as defined, incurred by Alimera. In the event Alimera sublicenses commercialization, we are entitled to receive 20% of royalties and 33% of non-royalty consideration received by Alimera, less certain permitted deductions. Alimera has indicated that it intends to commercialize Iluvien, if approved, through a direct sales force in the United States and to seek marketing collaboration partners for the commercialization of Iluvien outside of the United States.
The scheduled payment terms on the $15.0 million conditional note consisted of (i) interest only at an annual rate of 8% payable quarterly through March 2010 and (ii) principal payments of $500,000 per month commencing April 30, 2010 together with interest payable quarterly at an annual rate of 20%. Upon the occurrence of certain defined liquidity events (such as an initial public offering of Alimera, other sales of capital stock of Alimera and/or the sale or other disposition of substantially all of Alimera’s assets) that resulted in aggregate cash and/or noncash proceeds to Alimera in excess of $75 million, the note became immediately due and payable. Failure by Alimera to repay the note upon the occurrence of a defined liquidity event constituted an event of default under the note. If no liquidity event occurred on or before September 30, 2012, the note would automatically be cancelled. Based upon the terms of the note, payment was within the control of Alimera unless there was a liquidity event or an event of default.
Pursuant to the Alimera Agreement, a total of $18.3 million of deferred revenue was recognized as revenue on a straight-line basis over the 21.5 month performance obligation period from the amendment effective date through December 31, 2009. Following consummation of the Alimera Agreement, we received conditional note interest payments and reimbursements of approved development costs totaling approximately $1.5 million, $1.9 million and $437,000 during the years ended June 30, 2010, 2009 and 2008, respectively. In addition, on April 27, 2010, following consummation of its initial public offering, Alimera paid the $15.0 million conditional note in full. Cash consideration received from Alimera during the performance period was recognized as revenue ratably over the performance period, including immediate revenue recognition catch-up for the pro rata period from the amendment effective date to the date of each receipt. Cash consideration received subsequent to December 31, 2009 is being recognized as revenue upon receipt or at such earlier date, if applicable, on which any such amount is both fixed and determinable and reasonably assured of collection.
Pfizer
Under our worldwide collaborative research and license agreement with Pfizer, beginning in calendar year 2008 and continuing until commencement of the first Phase III clinical trial, Pfizer has agreed to provide us
38
Table of Contents
with a minimum of $500,000 per quarter in research funding. To date, the joint research program has consisted of pre-clinical studies. Under the agreement, we are also entitled to receive clinical development milestone payments and, following commercialization of any products, sales-based milestones and royalties. Because we have been unable to define the period of our performance obligations under this agreement, none of the cash payments received from Pfizer to date has been recognized as revenue. At June 30, 2010, approximately $5.8 million was recorded as non-current deferred revenue on our consolidated balance sheet.
Bausch & Lomb
Bausch & Lomb sells Vitrasert and Retisert. Our collaboration agreement with Bausch & Lomb provides for royalties on such sales. In June 2005 we received a $3.0 million advance from Bausch & Lomb in consideration of $6.25 million of future Retisert royalties that otherwise would be payable to us. Bausch & Lomb retained $1.2 million in fiscal 2010, $1.6 million in fiscal 2009 and $1.8 million in fiscal 2008 of Retisert royalties that otherwise would have been payable to us. During the quarter ended June 30, 2010, Bausch & Lomb retained the final portion of these royalties otherwise payable and we recorded an incremental $342,000 of royalty income, which was paid by Bausch & Lomb . Subsequent to June 30, 2010, we are entitled to receive 100% of the Retisert royalties pursuant to the collaboration agreement. Vitrasert royalties were $141,000 in fiscal 2010, $160,000 in fiscal 2009 and $148,000 in fiscal 2008.
Intrinsiq
In connection with a January 2008 exclusive field of use license with Intrinsiq for nutraceutical and food science applications of BioSilicon, we received license fee payments of $730,000 and $500,000 during fiscal years 2009 and 2008, respectively. During fiscal 2010, we received the first contractual minimum royalty payment of $450,000. Subject to continuation of the license agreement, which is cancellable by Intrinsiq on 90 days notice, we are entitled to receive additional scheduled minimum royalty payments totaling approximately $3.1 million through April 2014, creditable against quarterly royalties earned, if any. The next scheduled minimum royalty payment of $630,000 is due in January 2012.
Summary of Critical Accounting Policies and Estimates
Our discussion and analysis of the Company’s financial condition and results of operations are based upon our consolidated financial statements, which have been prepared in accordance with United States generally accepted accounting principles, or U.S. GAAP. The preparation of these financial statements requires that we make certain estimates, judgments and assumptions that affect the reported amounts of assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting periods. We base our estimates on historical experience, anticipated results and trends and various other factors believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily available from other sources. By their nature, these estimates, judgments and assumptions are subject to an inherent degree of uncertainty. Actual results may differ from our estimates under different assumptions or conditions.
Our significant accounting policies are more fully described in Note 2 to the accompanying consolidated financial statements. We believe that a few of these accounting policies and related estimates used in the preparation of our financial statements are most critical to an understanding of our historical and future performance because these specific areas require us to make judgments and estimates about matters that are uncertain at the time, and different estimates—which also could have been reasonable—might have produced different financial results. It is important that the discussion of our operating results that follows be read in conjunction with the critical accounting policies discussed below.
Revenue Recognition
Our business strategy includes entering into collaborative license and development agreements for the development and commercialization of product candidates utilizing our Durasert and BioSilicon technology
39
Table of Contents
systems. The terms of these arrangements typically include multiple deliverables by us (for example, granting of license rights, providing research and development services and manufacturing of clinical materials, participating on joint research committee) in exchange for consideration to us of some combination of non-refundable license fees, funding of research and development activities, payments based upon achievement of clinical development milestones and royalties in the form of a designated percentage of product sales or profits.
License Fees and Multiple Element Arrangements. We recognize non-refundable license fees as revenue when we have a contractual right to receive such payment, the contract price is fixed or determinable, the collection of the resulting receivable is reasonably assured and we have no further performance obligations under the license agreement. We analyze multiple element arrangements, such as license and development arrangements, to determine whether the deliverables can be separated or whether they must be accounted for as a single unit of accounting. We recognize up-front license payments as revenue upon delivery of the license only if the license has stand-alone value and the fair value of the undelivered performance obligations can be determined. If the fair value of the undelivered performance obligations can be determined, such obligations would then be accounted for separately as performed. If we determine the license either (i) does not have stand-alone value or (ii) has stand-alone value but the fair value of any of the undelivered performance obligations cannot be determined, the arrangement is accounted for as a single unit of accounting. Whenever we determine that an arrangement should be accounted for as a single unit of accounting, we must assess the period over which our performance obligations will be completed. We then recognize collaborative research and development revenue on a straight-line basis over the expected performance period using the cumulative catch-up method. Under this method, the portion of any such payment (except payments contingent upon achievement of substantive milestones) represented by the time elapsed from the commencement of the performance period to the payment date as a percentage of the total performance period is recognized immediately as revenue, with the remainder amortized on a straight-line basis over the remaining performance period. We limit the cumulative amount of revenue recognized under an arrangement to the cumulative amount of payments received as of the period ending date. All payments received following the end of the performance period are recognized as revenue when earned.
If we cannot reasonably estimate when our performance obligations either cease or become inconsequential, then revenue is deferred until we can reasonably estimate when the performance obligations become inconsequential and perfunctory. We then recognize revenue over the remaining estimated period of performance.
Significant management judgment is required in determining the level of effort required under an arrangement and the period over which we are expected to complete our performance obligations under an arrangement. In addition, if we are involved in a research or steering committee as part of an arrangement accounted for as a single unit of accounting, we assess whether the nature of our involvement constitutes a performance obligation or a right to participate. If such services are determined to be other than inconsequential, performance obligations are combined with other performance obligations in determining the period over which we expect to complete our aggregate performance obligations.
Reimbursement of Costs. Our business includes providing research and development services on behalf of our collaboration partners to assist in advancing the developmental cycle of the licensed products. We act primarily as a principal in these transactions, and amounts received are classified as a component of revenue to be recognized consistent with the revenue recognition policy summarized above. We record the expenses incurred and reimbursed on a gross basis.
Royalty Income. Royalty income is recognized upon the sale of the related products, provided the royalty amounts are fixed and determinable, collection of the receivable is reasonably assured and we have no remaining performance obligations under the applicable arrangement. If royalties are received while we have remaining performance obligations, such amounts are attributed to the services being performed under the arrangement and are recognized as collaborative research and development revenue over the expected performance period using the cumulative catch-up method.
40
Table of Contents
Deferred Revenue. Amounts received prior to satisfying the above revenue recognition criteria are recorded as deferred revenue in the accompanying consolidated balance sheets. Deferred revenue amounts not expected to be recognized within one year following the balance sheet date are classified as non-current liabilities.
Applying the policies described above, we reported collaborative research and development revenues of $22.6 million for the year ended June 30, 2010 (fiscal 2010), $12.0 million for the year ended June 30, 2009 (fiscal 2009) and $3.3 million for the year ended June 30, 2008 (fiscal 2008). Substantially all of these revenues were attributable to our March 2008 amended and restated collaboration agreement with Alimera. With respect to our Alimera collaboration agreement, management determined that our performance obligations ended as of December 31, 2009. Amounts received after that date are immediately recognized as revenue and for fiscal 2010 included payment in full by Alimera of a $15.0 million conditional note in April 2010.
In connection with our license and collaboration agreement with Pfizer, we have been unable to estimate the period of our performance obligations. Accordingly, we recorded an aggregate of approximately $5.8 million of payments received through June 30, 2010 as non-current deferred revenue on our consolidated balance sheet. The current portion of deferred revenue and the remainder of the non-current deferred revenue at June 30, 2010 is attributable to our Intrinsiq license, which includes license and minimum royalty payments that are being amortized over the life of the associated patents through 2025.
Valuation of Intangible Assets
Our intangible assets include the Durasert and BioSilicon technology systems. We review these intangible assets for impairment whenever events or changes in business circumstances indicate that the asset carrying value may not be fully recoverable or that the useful life of the asset is no longer appropriate. Factors that could trigger an impairment review include the following:
| • | Significant change relative to historical or projected future operating results, |
| • | Significant changes in the use of the assets or the strategy for the overall business, and |
| • | Significant industry or economic trends and developments. |
If an impairment trigger is identified, we determine impairment by comparing projected undiscounted cash flows to be generated by the asset to its carrying value. If an impairment is identified, a loss is recorded equal to the excess of the asset’s carrying value over its fair value, and the carrying value is adjusted. The estimated future cash flows, based on reasonable and supportable assumptions, require management’s judgment. Actual results could vary from these estimates.
We evaluate the recoverability of our intangible assets based on estimated undiscounted cash flows related to existing contractual agreements as well as projected cash flows from potential future research and development collaboration agreements utilizing the underlying technology system. Future adverse changes or other unforeseeable factors could result in an impairment charge with respect to some or all of the $23.9 million of intangible assets that appear on our consolidated balance sheet as of June 30, 2010. Such an impairment charge could materially impact future results of operations and financial position in the reporting period identified.
41
Table of Contents
Results of Operations
Years Ended June 30, 2010 and 2009
| Year ended June 30, | Change | ||||||||||||||
| 2010 | 2009 | Amounts | % | ||||||||||||
| (In thousands except percentages) | |||||||||||||||
| Revenues |
$ | 23,053 | $ | 12,162 | $ | 10,891 | 90 | % | |||||||
| Operating expenses: |
|||||||||||||||
| Research and development |
6,994 | 8,007 | (1,013 | ) | (13 | )% | |||||||||
| General and administrative |
6,968 | 8,791 | (1,823 | ) | (21 | )% | |||||||||
| Total operating expenses |
13,962 | 16,798 | (2,836 | ) | (17 | )% | |||||||||
| Operating income (loss) |
9,091 | (4,636 | ) | 13,727 | 296 | % | |||||||||
| Other (expense) income: |
|||||||||||||||
| Change in fair value of derivatives |
(339 | ) | 959 | (1,298 | ) | (135 | )% | ||||||||
| Interest income |
27 | 162 | (135 | ) | (83 | )% | |||||||||
| Other (expense) income, net |
(3 | ) | 53 | (56 | ) | (106 | )% | ||||||||
| Total other (expense) income |
(315 | ) | 1,174 | (1,489 | ) | (127 | )% | ||||||||
| Income (loss) before income taxes |
8,776 | (3,462 | ) | 12,238 | 353 | % | |||||||||
| Income tax (expense) benefit |
(23 | ) | 951 | (974 | ) | (102 | )% | ||||||||
| Net income (loss) |
$ | 8,753 | $ | (2,511 | ) | $ | 11,264 | 449 | % | ||||||
Revenues
Revenues increased by approximately $10.9 million, or 90%, to approximately $23.1 million for fiscal 2010 from approximately $12.2 million for fiscal 2009. In each fiscal year, revenues were almost entirely attributable to our collaboration agreement with Alimera, consisting of (i) the portion of the upfront license consideration that we recognized in the given fiscal year; and (ii) the aggregate of conditional note payments and reimbursement of our development costs received from Alimera that we recognized in the given fiscal year. For fiscal 2010, the Alimera-related revenues included payment in full by Alimera of the $15.0 million conditional note plus interest.
We are entitled to receive a $25 million milestone payment from Alimera within 30 days following an FDA approval of Iluvien. However, absent an FDA approval of Iluvien during fiscal 2011, we currently expect to record an insignificant amount of collaborative research and development revenue attributable to the Alimera collaboration agreement in fiscal 2011.
Pursuant to a June 2005 side letter to the collaboration agreement with Bausch & Lomb, CDS received $3.0 million from Bausch & Lomb as an advance payment in consideration of $6.25 million of future Retisert royalties that otherwise would have been payable to us. During fiscal 2010, $1.2 million of Retisert royalties otherwise payable was retained by Bausch & Lomb, thereby completing the advance royalty agreement, and $342,000 was recorded as royalty income, which amount was received from Bausch & Lomb in August 2010. The fiscal 2010 total of royalties payable and otherwise payable of approximately $1.5 million compared to approximately $1.6 million of royalties otherwise payable for fiscal 2009, a decrease of 6.1%. For the year ending June 30, 2011 (fiscal 2011), we will record royalty income equal to 100% of the Retisert royalties reported by Bausch & Lomb.
Research and Development
Research and development decreased by approximately $1.0 million, or 13%, to $7.0 million for fiscal 2010 from $8.0 million for fiscal 2009. This decrease was primarily attributable to an approximate $1.1 million
42
Table of Contents
reduction of U.K.-based research and development costs, primarily related to third party costs of our BrachySil clinical program and third party BioSilicon manufacturing development for the period prior to consummation of our Intrinsiq supply agreement. Approximately $82,000 of the total decrease was attributable to the relative strengthening of the U.S. dollar against the Pound Sterling.
General and Administrative
General and administrative costs decreased by approximately $1.8 million, or 21%, to approximately $7.0 million for fiscal 2010 from $8.8 million for fiscal 2009. This net decrease was primarily attributable to the following factors:
| • | the absence of a $1.3 million provision for losses in fiscal 2009 on a note receivable, |
| • | the elimination of approximately $400,000 of legal fees and consulting services incurred in fiscal 2009 directly related to the June 2008 Reincorporation, |
| • | an approximate $500,000 reduction in U.S. salaries and benefits, primarily related to fiscal 2009 salary and severance agreement costs of a former executive officer, |
partially offset by:
| • | an approximate $480,000 increase in stock-based compensation. |
Change in Fair Value of Derivatives
Change in fair value of derivatives represented an expense of $339,000 for fiscal 2010 compared to income of $959,000 for fiscal 2009. Detachable warrants issued in share offerings denominated in A$ were recorded as derivative liabilities, subject to revaluation at subsequent reporting dates. The change in fair value of derivatives, determined using the Black-Scholes valuation model, primarily reflected a net increase in the market price of our shares in fiscal 2010 (resulting in a smaller spread between the market price and the US$-equivalent exercise prices of the warrants) compared to a net decrease in the market price of our shares in fiscal 2009. The change in fair value was also impacted by the 0.7 year weighted average remaining life of the underlying warrants at June 30, 2010.
We are required to re-value these warrants at each subsequent balance sheet date, and changes in their fair values will result in adjustments to our recorded derivative liabilities (approximately $1.3 million at June 30, 2010) and a corresponding income or expense in our statement of operations. Fluctuations in the fair values of these warrants could have a substantial impact on our future quarterly and annual operating results until the last-to-expire of these warrants in July 2012.
Interest Income
Interest income decreased by $135,000, or 83%, to $27,000 for fiscal 2010 from $162,000 for fiscal 2009, primarily due to sharply lower weighted average interest rates earned on money market funds.
Other (Expense) Income
Other expense, net of $3,000 for fiscal 2010 compares to other income of $53,000 for fiscal 2009. This change was primarily attributable to the absence in fiscal 2010 of foreign exchange gains of $69,000 recognized in fiscal 2009 resulting from the reclassification into earnings of foreign currency translation reserve balances in connection with the dissolution of subsidiaries.
Income Tax (Expense) Benefit
Income tax expense of $23,000 in fiscal 2010 compares to $951,000 of income tax benefit for fiscal 2009. The net change was primarily attributable to an approximate $706,000 decrease of foreign research and development tax credits earned by our U.K. subsidiary and an approximate $186,000 increase in U.S. federal alternative minimum tax expense.
43
Table of Contents
Years Ended June 30, 2009 and 2008
| Year ended June 30, | Change | ||||||||||||||
| 2009 | 2008 | Amounts | % | ||||||||||||
| (In thousands except percentages) | |||||||||||||||
| Revenues |
$ | 12,162 | $ | 3,476 | $ | 8,686 | 250 | % | |||||||
| Operating expenses: |
|||||||||||||||
| Research and development |
8,007 | 14,426 | (6,419 | ) | (44 | )% | |||||||||
| General and administrative |
8,791 | 13,951 | (5,160 | ) | (37 | )% | |||||||||
| Impairment of goodwill |
— | 60,106 | (60,106 | ) | na | ||||||||||
| Total operating expenses |
16,798 | 88,483 | (71,685 | ) | (81 | )% | |||||||||
| Operating loss |
(4,636 | ) | (85,007 | ) | 80,371 | (95 | )% | ||||||||
| Other income (expense): |
|||||||||||||||
| Change in fair value of derivatives |
959 | 8,357 | (7,398 | ) | (89 | )% | |||||||||
| Interest income |
162 | 648 | (486 | ) | (75 | )% | |||||||||
| Interest and finance costs |
— | (507 | ) | 507 | (100 | )% | |||||||||
| Other income, net |
53 | 356 | (303 | ) | (85 | )% | |||||||||
| Total other income |
1,174 | 8,854 | (7,680 | ) | (87 | )% | |||||||||
| Loss before income taxes |
(3,462 | ) | (76,153 | ) | 72,691 | (95 | )% | ||||||||
| Income tax benefit |
951 | 483 | 468 | 97 | % | ||||||||||
| Net loss |
$ | (2,511 | ) | $ | (75,670 | ) | $ | 73,159 | (97 | )% | |||||
na = not applicable
Revenues
Revenues increased by approximately $8.7 million, or 250%, to approximately $12.2 million for fiscal 2009 from approximately $3.5 million for fiscal 2008. In each fiscal year, revenues were almost entirely attributable to our collaboration agreement with Alimera, consisting of (i) the portion of the upfront license consideration that we recognized in the given fiscal year; and (ii) the aggregate of conditional note payments and reimbursement of our development costs received from Alimera that we recognized in the given fiscal year.
Impairment of Goodwill
In fiscal 2008, we recorded an impairment charge of $60.1 million equal to the total carrying value of goodwill. See Note 4 to the accompanying consolidated financial statements.
Research and Development
Research and development decreased by approximately $6.4 million, or 44%, to $8.0 million for fiscal 2009 from $14.4 million for fiscal 2008. This decrease was primarily attributable to the following factors:
| • | the absence in fiscal 2009 of $4.7 million of Iluvien co-development costs incurred in fiscal 2008 as a result of the assumption by Alimera of all financial responsibility for the development of licensed products pursuant to the amendment and restatement of our collaboration agreement with Alimera; and |
| • | a decrease in fiscal 2009 of approximately $1.6 million in U.K.-based research and development costs, of which approximately $1.1 million was attributable to the relative strengthening of the U.S. dollar against the Pound Sterling and approximately $470,000 was primarily attributable to reduced levels of personnel, legal, facilities and depreciation expenses. |
44
Table of Contents
General and Administrative
General and administrative costs decreased by approximately $5.2 million, or 37%, to approximately $8.8 million for fiscal 2009 from $14.0 million for fiscal 2008. This net decrease was primarily attributable to the following factors:
| • | the absence of approximately $3.0 million of costs incurred in fiscal 2008 directly attributable to the Reincorporation; |
| • | a decrease of approximately $1.9 million of audit, tax, financial reporting consulting services and legal fees primarily as a result of the Reincorporation and the absence in fiscal 2009 of costs incurred in fiscal 2008 in connection with the amendment and restatement of the collaboration agreement with Alimera; |
| • | the absence of approximately $700,000 of personnel, facility and travel costs associated with the fiscal 2008 closing of our Perth, Australia office; |
partially offset by:
| • | an approximate $1.0 million increase in provision for losses on a note receivable; and |
| • | an approximate $300,000 increase in U.S. personnel and benefit costs related to the March 2009 severance agreement with a former executive officer. |
Change in Fair Value of Derivatives
Change in fair value of derivatives described above decreased by approximately $7.4 million, or 89%, to income of $959,000 for fiscal 2009 from income of $8.4 million for fiscal 2008.
Interest Income
Interest income decreased by $486,000, or 75%, to $162,000 for fiscal 2009 from $648,000 for fiscal 2008, primarily due to (i) decreased interest-bearing cash balances and sharply lower weighted average interest rates; and (ii) the absence in the current year of approximately $100,000 of interest accrued in fiscal 2008 on a $1.5 million note receivable.
Interest Expense
Interest expense of $507,000 was accrued during fiscal 2008 on the portion of shared Iluvien product candidate co-development costs that we elected not to pay under the original Alimera collaboration agreement. In connection with the March 2008 amendment and restatement of that agreement, the total co-development costs, including associated penalties and accrued interest, which we then owed to Alimera were cancelled and, accordingly, no interest expense was incurred during fiscal 2009.
Other Income, net
Other income, net decreased by $303,000, or 85%, to $53,000 for fiscal 2009 from $356,000 for fiscal 2008. This decrease was primarily attributable to the absence in fiscal 2009 of $412,000 of income in fiscal 2008 attributable to a revenue sharing arrangement under the ADR program, which was terminated as a result of the Reincorporation, partially offset by net foreign exchange gains of $69,000 in fiscal 2009 resulting from the reclassification into earnings of foreign currency translation reserve balances in connection with the dissolution of subsidiaries.
45
Table of Contents
Income Tax Benefit
Income tax benefit increased by approximately $468,000, or 97%, to $951,000 for fiscal 2009 from $483,000 for fiscal 2008. The increase was primarily attributable to approximately $840,000 of foreign research and development tax credits earned by our U.K. subsidiary, partially offset by a $427,000 reduction in U.S. deferred tax benefits.
Inflation and Seasonality
Our management believes inflation has not had a material impact on our operations or financial condition and that our operations are not currently subject to seasonal influences.
Recently Adopted Accounting Pronouncements
Effective July 1, 2009, we adopted the provisions of ASC 808-10, “Collaborative Arrangements”, formerly Emerging Issues Task Force (“EITF”) Issue No. 07-01, “Accounting for Collaborative Arrangements”. This standard defines a collaborative arrangement as a contractual arrangement in which the parties are (i) active participants to the arrangement; and (ii) exposed to significant risks and rewards that depend upon the commercial success of the endeavor. It also addresses the appropriate statements of operations presentation for activities and payments between the participants in a collaborative arrangement as well as for costs incurred and revenue generated from transactions with third parties. Adoption of this standard did not have any material impact on our consolidated financial statements.
Recently Issued Accounting Pronouncements
In October 2009, the FASB issued Accounting Standards Update (ASU) No. 2009-13, “Multiple-Deliverable Revenue Arrangements—a consensus of the FASB Emerging Issues Task Force”. ASU 2009-13 updates the existing multiple-element revenue arrangements guidance currently included under ASC 605-25, which originated primarily from the guidance in EITF Issue No. 00-21, “Revenue Arrangements with Multiple Deliverables”. The update provides principles for allocation of consideration among multiple elements of revenue arrangements, allowing more flexibility in identifying and accounting for separate deliverables under an arrangement. ASU 2009-13 introduces an estimated selling price method for valuing the elements of a bundled arrangement if vendor-specific objective evidence or third-party evidence of selling price is not available. In addition, the update also significantly expands related disclosure requirements. ASU 2009-13 is effective on a prospective basis for revenue arrangements entered into or materially modified in fiscal years beginning on or after June 15, 2010. We are evaluating the potential application of this new accounting update to new or materially modified revenue arrangements.
In April 2010, the FASB issued ASU No. 2010-17, Revenue Recognition (Topic 605—Milestone Method of Revenue Recognition: a consensus of the FASB EITF (“ASU 2010-17”). ASU 2010-17 amends ASC 605-28 and established a revenue recognition method for contingent consideration that is payable on achievement of an uncertain future event, referred to as a milestone. The scope of the milestone method is limited to research and development agreements and is applicable to milestones in multiple-deliverable arrangements involving research and development transactions. The guidance does not preclude the application of any other applicable revenue guidance. The guidance will be effective for financial statements issued for fiscal years beginning after June 15, 2010. Early adoption is permitted. We are currently evaluating the potential impact of ASU 2010-17 on our financial statements.
Liquidity and Capital Resources
For fiscal 2008 to 2010, we financed our operations primarily from license fees, research and development funding and contingent cash payments from our collaboration partners and, to a lesser degree, from the July 2007 private placement of our equity securities. At June 30, 2010, our principal sources of liquidity consisted of cash,
46
Table of Contents
cash equivalents and marketable securities totaling approximately $17.6 million. Our cash equivalents are invested in institutional money market funds and our marketable securities are invested in investment-grade corporate debt, government agency securities and commercial paper with maturities at June 30, 2010 ranging from 3 to 11 months.
With the exception of fiscal 2010, we have incurred operating losses since inception, and, at June 30, 2010, we had a total accumulated deficit of $218.3 million. We generally expect negative cash flows from operations on a quarterly basis at least until such time as one or more of our product candidates achieves regulatory approval and achieves sufficient sales. We believe we can fund our operations as currently conducted into at least calendar year 2012. Whether we will require, or desire, to raise additional capital will be influenced by many factors, including, but not limited to:
| • | the continuation of our existing collaborations with Pfizer and Alimera, including their continued funding of our programs and our receipt of milestone, royalty and other payments; |
| • | the timing of FDA regulatory approval and commercialization of Iluvien; |
| • | the amount of quarterly royalty payments from sales of Retisert, which payments resumed in August 2010 following completion of an advance royalty agreement; |
| • | the scope and extent of our internally funded existing operations and programs, any new product candidates and any new business opportunities; |
| • | our ability to establish and maintain strategic arrangements for product candidates for research, development, clinical testing, manufacturing and marketing; |
| • | the success of our products and product candidates, including the timing and costs of regulatory approvals and the commercial success of approved products; |
| • | the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims; and |
| • | changes in our operating plan, including the pursuit of new business opportunities, which may affect our need for capital. |
Absent adequate levels of funding from new and existing collaboration agreements and/or financing transactions, management currently believes that our cash position thereafter will be substantially dependent upon the timing of FDA approval and the initiation and success of commercialization of Iluvien, and the resulting occurrence of certain milestone events under the terms of our collaboration agreement with Alimera. Alimera has agreed to pay us $25.0 million upon FDA approval of Iluvien for DME and a 20% profit share on its sales of Iluvien, subject to offset of 20% of defined pre-profitability commercialization costs incurred by Alimera. In the event Alimera sublicenses commercialization, we would receive 20% of royalties and 33% of non-royalty consideration received by Alimera, less certain permitted deductions. There is no assurance that the FDA will approve Iluvien, or that Iluvien will achieve market acceptance even if it is approved by the FDA.
The downturn in the economy and the disruptions in the financial and credit markets have made it significantly more difficult and more expensive to obtain financing. If we determine that it is desirable or necessary to raise additional capital in the future, we do not know if it will be available when needed or on terms favorable to us or our stockholders. If available, additional equity financing may be dilutive to stockholders, debt financing may involve restrictive covenants or other unfavorable terms and potential dilutive equity, and funding through collaboration agreements may be on unfavorable terms, including requiring us to relinquish rights to certain of our technologies or products. If adequate financing is not available if and when needed, we may be required to delay, reduce the scope of or eliminate one or more of our research or development programs, postpone the pursuit of product candidates and new business opportunities, or otherwise reduce our cash requirements.
47
Table of Contents
Our consolidated statements of historical cash flows are summarized as follows:
| Year Ended June 30, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| (In thousands) | ||||||||||||
| Net income (loss): |
$ | 8,753 | $ | (2,511 | ) | $ | (75,670 | ) | ||||
| Changes in operating assets and liabilities |
(4,015 | ) | (10,452 | ) | 13,455 | |||||||
| Other adjustments to reconcile net income (loss) to cash flows from operating activities |
5,161 | 4,527 | 57,072 | |||||||||
| Cash flows provided by (used in) operating activities |
$ | 9,899 | $ | (8,436 | ) | $ | (5,143 | ) | ||||
| Cash flows used in investing activities |
$ | (2,069 | ) | $ | (195 | ) | $ | (259 | ) | |||
| Cash flows provided by financing activities |
$ | 802 | $ | — | $ | 18,385 | ||||||
Sources and uses of operating cash flows for the years ended June 30, 2010, 2009 and 2008 are summarized as follows:
| Year Ended June 30, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| (In thousands) | ||||||||||||
| Operating cash inflows: |
||||||||||||
| License and collaboration agreements |
$ | 19,123 | $ | 4,315 | $ | 14,428 | ||||||
| Royalty income |
127 | 181 | 372 | |||||||||
| Foreign R&D tax credits |
130 | 588 | — | |||||||||
| 19,380 | 5,084 | 14,800 | ||||||||||
| Operating cash outflows: |
||||||||||||
| Alimera co-development costs |
— | — | (3,862 | ) | ||||||||
| Reincorporation costs |
— | (1,401 | ) | (1,789 | ) | |||||||
| Legal and audit fees |
(1,770 | ) | (2,737 | ) | (4,452 | ) | ||||||
| All other operating cash outflows, net |
(7,711 | ) | (9,382 | ) | (9,840 | ) | ||||||
| (9,481 | ) | (13,520 | ) | (19,943 | ) | |||||||
| Cash flows provided by (used in) operating activities |
$ | 9,899 | $ | (8,436 | ) | $ | (5,143 | ) | ||||
Operating cash inflows for each year consisted primarily of payments received pursuant to license and collaboration agreements, predominantly Alimera and Pfizer. As a percentage of total license and collaboration agreement payments received, amounts attributable to Alimera represented 86.9% in fiscal 2010, 44.5% in fiscal 2009 and 86.2% in fiscal 2008. Funding from Alimera included the payment in full of a $15.0 million conditional note in April 2010 and payment of a $12.0 million up front license fee in March 2008 in connection with an amendment and restatement of the collaboration agreement originally entered into in February 2005.
Operating cash outflows have decreased significantly on a year-over-year basis between fiscal 2008 and fiscal 2010. These decreases were primarily the result of (a) the elimination of approximately $3.9 million of co-development payments made to Alimera during fiscal 2008 until consummation of the March 2008 amended agreement; (b) approximately $3.2 million of legal and other direct costs paid during fiscal 2008 and fiscal 2009 to effect the reincorporation from Australia to the United States; and (c) substantial decreases in other legal and audit fees primarily resulting from the reincorporation. The downward historical trend of all other cash outflows, net was attributable to several factors, primarily including the completion of Phase II clinical studies for an internally financed product candidate and the elimination of personnel, occupancy and related overhead costs attributable to the closing of the Perth, Australia office in connection with the reincorporation.
48
Table of Contents
Cash used in investing activities included purchases of property and equipment totaling $15,000 for fiscal 2010, $195,000 for fiscal 2009 and $272,000 for fiscal 2008. Additionally, fiscal 2010 investing activities included approximately $2.1 million for purchases of marketable securities.
Net cash flows from financing activities totaled $802,000 for fiscal 2010, $0 for fiscal 2009 and $18.4 million for fiscal 2008. Cash flows from financing activities during fiscal 2010 consisted of the exercise of stock options and warrants. Cash flows from financing activities during fiscal 2008 resulted from July 2007 issuances of 4,114,199 units at a price per unit of $5.00, net of $2.2 million of share issue costs.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements that have, or are reasonably likely to have, a current or future effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources that would be material to investors.
Tabular Disclosure Of Contractual Obligations
The following table summarizes our minimum contractual obligations as of June 30, 2010:
| Payments Due by Period | |||||||||||||||
| Contractual Obligations |
Total | Less than 1 year |
1-3 years | 3-5 years | More than 5 years | ||||||||||
| (In thousands) | |||||||||||||||
| Operating Lease Obligations |
$ | 330 | $ | 317 | $ | 13 | $ | — | $ | — | |||||
| Purchase Obligations |
189 | 189 | — | — | — | ||||||||||
| Total |
$ | 519 | $ | 506 | $ | 13 | $ | — | $ | — | |||||
Our purchase obligations primarily consist of purchase orders for pre-clinical costs, supplies and other operating needs.
We also have contractual obligations that are variable in nature and, as such, are not included in the above table. These include the following:
Executive contracts. At June 30, 2010, we had agreements with two executive officers that would require us to make severance payments to them if we terminate their employment without cause or the executives resign for good cause. Such severance agreements would require us to make aggregate payments of up to approximately $980,000. The amounts payable pursuant to severance arrangements change over time depending upon the date of termination and then current salaries.
49
Table of Contents
| ITEM 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
We have exposure to changes in the valuation of derivative liabilities, foreign currency exchange rates and interest rates.
Derivative Liabilities
At June 30, 2010, the balance of our derivative liabilities, which relate to warrants denominated in A$, totaled approximately $1.3 million and was determined using the Black-Scholes valuation model. The change in fair value of derivatives resulted in an expense of $339,000 for fiscal 2010 and income of $959,000 for fiscal 2009 and $8.4 million for fiscal 2008.
Our financial position and results of operations will continue to be sensitive to future revaluations of these derivative liabilities. At June 30, 2010, these warrants had a weighted average remaining contractual life of approximately 9 months and a weighted average exercise price of $8.17 per share compared to the $3.61 NASDAQ closing price of our common shares. The primary factor that impacts the change in fair value of these derivatives is fluctuations in our share price. Reduction of the remaining useful life of the warrants, assuming that share price remains constant, would result in a significant decrease of the derivative liability value during fiscal 2011 based on the short remaining life of the underlying warrants. Changes in risk-free interest rates have a de minimis effect.
The following table summarizes the sensitivity of our consolidated statements of operations for fiscal 2010 to assumed increases or decreases of our share price at June 30, 2010:
| Decrease in Share Price | Current Price |
Increase in Share Price | ||||||||||||||||||||||
| -15% | -10% | -5% | +5% | +10% | +15% | |||||||||||||||||||
| (In thousands) | ||||||||||||||||||||||||
| Change in fair value of derivatives—income (expense) |
$ | 486 | $ | 338 | $ | 176 | $ | — | $ | (190 | ) | $ | (395 | ) | $ | (613 | ) | |||||||
Foreign Currency Exchange Rates
We conduct operations in two principal currencies, the U.S. dollar and the Pound Sterling (£). The U.S. dollar is the functional currency for our U.S. operations, and the Pound Sterling is the functional currency for our U.K. operations. Changes in the foreign exchange rate of the U.S. dollar and Pound Sterling impact the net operating expenses of our U.K. operations. For fiscal 2010, the strengthening of the U.S. dollar compared to fiscal 2009 resulted in a net decrease in research and development expense of approximately $82,000. All cash and cash equivalents, and most other asset and liability balances, are denominated in each entity’s functional currency and, accordingly, we do not consider our statement of operation exposure to realized and unrealized foreign currency gains and losses to be significant.
Changes in the foreign exchange rate of the U.S. dollar and Pound Sterling also impact total stockholders’ equity. During fiscal 2010, the relative strengthening of the U.S. dollar in relation to the Pound Sterling resulted in a net decrease of $1.5 million in stockholders’ equity due to the translation of approximately £9.9 million of net assets of our U.K. operations, predominantly the BioSilicon technology intangible asset, into U.S. dollars. For every incremental 5% strengthening or weakening of the U.S. dollar at June 30, 2010 in relation to the Pound Sterling, our stockholders’ equity at June 30, 2010 would have decreased or increased, respectively, by approximately $750,000.
Interest Rates
Cash and cash equivalent balances are subject to variable interest rates. We do not consider our exposure to interest rates to be significant.
50
Table of Contents
| ITEM 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
The information required by this item may be found on pages F-1 through F-27 of this annual report.
| ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
None.
| ITEM 9A. | CONTROLS AND PROCEDURES |
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our principal executive officer and principal financial officer, evaluated the effectiveness of our disclosure controls and procedures as of June 30, 2010. The term “disclosure controls and procedures”, as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended (the “Exchange Act’), means controls and other procedures of a company that are designed to ensure that information required to be disclosed by us in the reports that we file or submit under the Exchange Act, is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate to allow timely decisions regarding required disclosure. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their desired objectives, and our management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of June 30, 2010, our principal executive officer and principal financial officer concluded that, as of such date, our disclosure controls and procedures were effective at the reasonable assurance level.
| (a) | Management’s Annual Report on Internal Control Over Financial Reporting |
Our management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rule 13a-15(f) or 15d-15(f) of the Exchange Act as a process designed by, or under the supervision of, our principal executive and principal financial officers and effected by our board of directors, management and other personnel to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles in the U.S., and includes those policies and procedures that:
| • | pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets; |
| • | provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures are being made only in accordance with authorizations of management and our directors; and |
| • | provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements. |
All internal control systems, no matter how well designed, have inherent limitations and may not prevent or detect misstatements. Projections of any evaluations of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Our management, with the participation of our principal executive officer and principal financial officer, assessed the effectiveness of our internal control over financial reporting as of June 30, 2010. In making this
51
Table of Contents
assessment, management used the criteria set forth in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission, or COSO. Based on this assessment, our management concluded that, as of June 30, 2010, our internal control over financial reporting is effective based on those criteria.
This annual report does not include an attestation report of the Company’s independent registered public accounting firm regarding internal control over financial reporting. Management’s report was not subject to attestation by the Company’s independent registered public accounting firm pursuant to the rules of the Securities and Exchange Commission that permit the Company, as a smaller reporting company, to provide only management’s report in this annual report.
| (b) | Changes in Internal Control over Financial Reporting |
There has been no change in the Company’s internal control over financial reporting during the last quarter of the fiscal year covered by this Annual Report on Form 10-K that has materially affected, or is reasonably likely to materially affect, the Company’s internal control over financial reporting.
| ITEM 9B. | OTHER INFORMATION |
None.
52
Table of Contents
| ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS, AND CORPORATE GOVERNANCE |
Corporate Governance
We have adopted a written code of ethics that applies to all of our employees, officers and directors. The Code of Conduct is designed to ensure that our business is conducted with integrity, and to comply with SEC regulations and NASDAQ and ASX listing standards. The Code of Conduct covers adherence to laws and regulations as well as professional conduct, including employment policies, conflicts of interest and the protection of confidential information. The Code of Conduct is available on the “Corporate Governance” section of our website at www.psivida.com.
We intend to disclose any future amendments to, or waivers from, the Code of Conduct that affect the directors, senior financial officers or executive officers within four business days of the amendment or waiver by filing with the SEC a Current Report on Form 8-K.
Other Information
The other information required to be disclosed in Item 10 is hereby incorporated by reference to our 2010 Proxy Statement.
| ITEM 11. | EXECUTIVE COMPENSATION |
The information required to be disclosed in Item 11 is hereby incorporated by reference to our 2010 Proxy Statement.
| ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
The information required to be disclosed in Item 12 is hereby incorporated by reference to our 2010 Proxy Statement.
| ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
The information required to be disclosed in Item 13 is hereby incorporated by reference to our 2010 Proxy Statement.
| ITEM 14. | PRINCIPAL ACCOUNTING FEES AND SERVICES |
The information required to be disclosed in Item 14 is hereby incorporated by reference to our 2010 Proxy Statement.
| ITEM 15. | EXHIBITS AND FINANCIAL STATEMENTS |
(a)(1) Financial Statements
The financial statements filed as part of this report are listed on the Index to Consolidated Financial Statements on page F-1.
(a)(2) Financial Statement Schedules
Schedules have been omitted because of the absence of conditions under which they are required or because the required information is included in the consolidated financial statements or notes thereto.
53
Table of Contents
(a)(3) Exhibits.
| Incorporated by Reference to SEC Filing | ||||||||
| Exhibit No. |
Exhibit Description |
Form | SEC Filing Date |
Exhibit No. | ||||
| Articles of Incorporation and By-Laws | ||||||||
| 3.1 | Certificate of Incorporation of pSivida Corp. | 8-K12G3 | 06/19/08 | 3.1 | ||||
| 3.2 | By-Laws of pSivida Corp. | 8-K12G3 | 06/19/08 | 3.2 | ||||
| Instruments Defining the Rights of Security Holders | ||||||||
| 4.1 | Form of Specimen Stock Certificate for Common Stock | 8-K12G3 | 06/19/08 | 4.1 | ||||
| 4.2 + | Form of Warrant, dated as of November 15, 2005 | 6-K | 11/15/05 | 99.3 | ||||
| 4.3 + | Form of Series A Warrant | 6-K/A | 07/31/06 | 99.4 | ||||
| 4.4 | Registration Rights Agreement, dated as of September 26, 2006, by and among pSivida Limited, Australian IT Investments Limited, Absolute Octane Fund and European Catalyst Fund | 6-K | 09/26/06 | 99.5 | ||||
| 4.5 + | Form of pSivida Limited Warrants to Purchase ADRs, dated September 26, 2006 | 6-K | 09/26/06 | 99.4 | ||||
| 4.6 | pSivida Limited Series C Warrants to Purchase ADRs | 6-K | 01/03/07 | 99.2 | ||||
| 4.7 | Series D Warrants | 6-K | 05/16/07 | 99.4 | ||||
| 4.8 | Series E Warrants | 6-K | 05/16/07 | 99.5 | ||||
| 4.9 | Series F Warrants | 6-K | 05/16/07 | 99.6 | ||||
| 4.10 | Series G Warrants | 6-K | 05/16/07 | 99.7 | ||||
| 4.11 | Second Amended and Restated Registration Rights Agreement dated May 15, 2007 by and among pSivida Limited and Castlerigg Master Investments Ltd | 6-K | 05/16/07 | 99.3 | ||||
| 4.12 + | Form of Investor Warrant | 6-K | 07/02/07 | 99.4 | ||||
| 4.13 + | Form of Placement Agents Warrant | 6-K | 07/02/07 | 99.5 | ||||
| 4.14 + | Form of Application for Shares and Options | 8-K | 06/19/08 | 4.16 | ||||
| 4.15 | Securities Purchase Agreement, dated February 16, 2007, by and among pSivida Limited and the investors set forth on the signature pages thereto | 8-K | 06/19/08 | 4.17 | ||||
| Material Contracts—Management Contracts and Compensatory Plans (*) | ||||||||
| 10.1 | Employment Agreement, between pSivida Limited and Paul Ashton, dated January 1, 2006 | 20-F | 12/08/06 | 4.35 | ||||
| 10.2 | Non-Competition Agreement, between pSivida Limited and Paul Ashton, dated October 3, 2005 | 20-F | 01/18/06 | 4.35 | ||||
| 10.3 | Employment Agreement, between pSivida Limited and Lori Freedman, dated as of May 16, 2006 | 6-K | 05/23/06 | 99.3 | ||||
| 10.4 | Rules of the pSivida Corp. Employee Share Option Plan | 8-K | 06/19/08 | 10.40 | ||||
| 10.5 + | Form of Stock Option Certificate for grants to executive officers under the pSivida Corp. 2008 Incentive Plan | 8-K | 09/10/08 | 10.1 | ||||
| 10.6 + | Form of pSivida Corp. Nonstatutory Stock Options granted to Michael J. Soja and Lori Freedman on September 4, 2008 and September 10, 2008 | 10-K | 09/26/08 | 10.36 | ||||
| Material Contracts—Leases | ||||||||
| 10.7 | Commercial Sublease, between Exergen Corporation and Control Delivery Systems, Inc., dated as of April 6, 2005 | 20-F | 01/18/06 | 4.19 | ||||
| 10.8 | Lease Renewal Agreement between pSivida Inc. and Exergen Corporation dated October 18, 2007 | 10-Q | 02/11/08 | 10.1 | ||||
54
Table of Contents
| Incorporated by Reference to SEC Filing | ||||||||
| Exhibit No. |
Exhibit Description |
Form | SEC Filing Date |
Exhibit No. | ||||
| Material Contracts—License and Collaboration Agreements | ||||||||
| 10.9 # | Amended and Restated License Agreement between Control Delivery Systems, Inc. and Bausch & Lomb Incorporated dated December 9, 2003, as amended on June 28, 2005 | 20-F | 01/18/06 | 4.12 | ||||
| 10.10 # | Second Amendment to Amended and Restated License Agreement between pSivda US, Inc. and Bausch & Lomb dated August 1, 2009 | 10-K | 09/25/09 | 10.13 | ||||
| 10.11 # | Collaborative Research and License Agreement, dated as of April 3, 2007, by and among pSivida Limited, pSivida Inc. and Pfizer, Inc. | 6-K | 04/26/07 | 99.1 | ||||
| 10.12 # | Amended and Restated Collaboration Agreement by and between pSivida Inc. and Alimera Sciences, Inc. dated March 14, 2008 | 8-K | 04/26/10 | 9.01 | ||||
| Material Contracts—Other | ||||||||
| 10.13 # | Process Development and Manufacturing Agreement between pSiMedica Limited and AEA Technology QSA GmbH, dated March 4, 2004 | 20-F | 01/18/06 | 4.3 | ||||
| Other Exhibits | ||||||||
| 21.1 (a) | Subsidiaries of pSivida Corp. | |||||||
| 23.1 (a) | Consent of Independent Registered Public Accounting Firm, Deloitte & Touche LLP | |||||||
| 31.1 (a) | Certification of Principal Executive Officer pursuant to Rule 13a-14(a) and Rule 15d-14(a) of the Securities Exchange Act, as amended | |||||||
| 31.2 (a) | Certification of Principal Financial Officer pursuant to Rule 13a-14(a) and Rule 15d-14(a) of the Securities Exchange Act, as amended | |||||||
| 32.1 (a) | Certification of Principal Executive Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | |||||||
| 32.2 (a) | Certification of Principal Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | |||||||
| # | Confidential treatment has been granted for portions of this exhibit |
| + | The final versions of documents denoted as “form of” have been omitted pursuant to Rule 12b-31. Such final versions are substantially identical in all material respects to the filed versions of such documents, provided that the name of the investor, and the investor’s and/or the Company’s signatures are included in the final versions. |
| * | Management contracts and compensatory plans and arrangements required to be filed as exhibits pursuant to Item 15(b) of this annual report. |
| (a) | Filed herewith |
55
Table of Contents
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| PSIVIDA CORP. | ||
| By: | /s/ PAUL ASHTON | |
| Paul Ashton, | ||
| President and Chief Executive Officer | ||
| Date: |
September 27, 2010 | |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant in the capacities and on the dates indicated.
| Name |
Title |
Date | ||
| /s/ DAVID J. MAZZO David J. Mazzo |
Chairman of the Board of Directors |
September 27, 2010 | ||
| /s/ PAUL ASHTON Paul Ashton |
President, Chief Executive Officer and Director (Principal Executive Officer) |
September 27, 2010 | ||
| /s/ LEONARD S. ROSS Leonard S. Ross |
Vice President, Finance |
September 27, 2010 | ||
| /s/ PAUL A. HOPPER Paul Hopper |
Director |
September 27, 2010 | ||
| /s/ MICHAEL ROGERS Michael Rogers |
Director |
September 27, 2010 | ||
| /s/ PETER SAVAS Peter Savas |
Director |
September 27, 2010 | ||
56
Table of Contents
PSIVIDA CORP. AND SUBSIDIARIES
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
| Consolidated Financial Statements: |
||
| F-2 | ||
| F-3 | ||
| F-4 | ||
| F-5 | ||
| F-6 | ||
| F-7 |
F-1
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of pSivida Corp.
Watertown, Massachusetts
We have audited the accompanying consolidated balance sheets of pSivida Corp. and subsidiaries (the “Company”) as of June 30, 2010 and 2009, and the related consolidated statements of operations, stockholders’ equity and cash flows for each of the three years in the period ended June 30, 2010. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, such consolidated financial statements present fairly, in all material respects, the financial position of pSivida Corp. and subsidiaries as of June 30, 2010 and 2009, and the results of their operations and their cash flows for each of the three years in the period ended June 30, 2010, in conformity with accounting principles generally accepted in the United States of America.
/s/ Deloitte & Touche LLP
Boston, Massachusetts
September 27, 2010
F-2
Table of Contents
PSIVIDA CORP. AND SUBSIDIARIES
(In thousands except share amounts)
| June 30, | ||||||||
| 2010 | 2009 | |||||||
| Assets |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 15,514 | $ | 6,899 | ||||
| Marketable securities |
2,051 | — | ||||||
| Accounts and other receivables, net of allowance |
1,111 | 815 | ||||||
| Prepaid expenses and other current assets |
358 | 413 | ||||||
| Total current assets |
19,034 | 8,127 | ||||||
| Property and equipment, net |
43 | 66 | ||||||
| Intangible assets, net |
23,877 | 28,802 | ||||||
| Other assets |
60 | 109 | ||||||
| Total assets |
$ | 43,014 | $ | 37,104 | ||||
| Liabilities and stockholders’ equity |
||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 387 | $ | 284 | ||||
| Accrued expenses |
1,158 | 1,552 | ||||||
| Deferred revenue |
79 | 5,912 | ||||||
| Derivative liabilities |
1,310 | 971 | ||||||
| Total current liabilities |
2,934 | 8,719 | ||||||
| Deferred revenue |
6,817 | 4,622 | ||||||
| Deferred tax liabilities |
222 | 222 | ||||||
| Total liabilities |
9,973 | 13,563 | ||||||
| Commitments and contingencies (Note 14) |
||||||||
| Stockholders’ equity: |
||||||||
| Preferred stock, $.001 par value, 5,000,000 shares authorized, no shares issued and outstanding |
— | — | ||||||
| Common stock, $.001 par value, 60,000,000 shares authorized, 18,531,392 and 18,293,961 shares issued and outstanding at June 30, 2010 and 2009, respectively |
19 | 18 | ||||||
| Additional paid-in capital |
250,796 | 248,500 | ||||||
| Accumulated deficit |
(218,295 | ) | (227,048 | ) | ||||
| Accumulated other comprehensive income |
521 | 2,071 | ||||||
| Total stockholders’ equity |
33,041 | 23,541 | ||||||
| Total liabilities and stockholders’ equity |
$ | 43,014 | $ | 37,104 | ||||
See notes to consolidated financial statements
F-3
Table of Contents
PSIVIDA CORP. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF OPERATIONS
(In thousands except per share data)
| Year Ended June 30, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Revenues: |
||||||||||||
| Collaborative research and development |
$ | 22,570 | $ | 12,002 | $ | 3,328 | ||||||
| Royalty income |
483 | 160 | 148 | |||||||||
| Total revenues |
23,053 | 12,162 | 3,476 | |||||||||
| Operating expenses: |
||||||||||||
| Research and development |
6,994 | 8,007 | 14,426 | |||||||||
| General and administrative |
6,968 | 8,791 | 13,951 | |||||||||
| Impairment of goodwill |
— | — | 60,106 | |||||||||
| Total operating expenses |
13,962 | 16,798 | 88,483 | |||||||||
| Operating income (loss) |
9,091 | (4,636 | ) | (85,007 | ) | |||||||
| Other (expense) income: |
||||||||||||
| Change in fair value of derivatives |
(339 | ) | 959 | 8,357 | ||||||||
| Interest income |
27 | 162 | 648 | |||||||||
| Interest expense |
— | — | (507 | ) | ||||||||
| Other income, net |
(3 | ) | 53 | 356 | ||||||||
| Total other (expense) income |
(315 | ) | 1,174 | 8,854 | ||||||||
| Income (loss) before income taxes |
8,776 | (3,462 | ) | (76,153 | ) | |||||||
| Income tax (expense) benefit |
(23 | ) | 951 | 483 | ||||||||
| Net income (loss) |
$ | 8,753 | $ | (2,511 | ) | $ | (75,670 | ) | ||||
| Net income (loss) per share: |
||||||||||||
| Basic |
$ | 0.48 | $ | (0.14 | ) | $ | (4.17 | ) | ||||
| Diluted |
$ | 0.46 | $ | (0.14 | ) | $ | (4.17 | ) | ||||
| Weighted average common shares outstanding used in net income (loss) per share calculation: |
||||||||||||
| Basic |
18,405 | 18,263 | 18,166 | |||||||||
| Diluted |
18,895 | 18,263 | 18,166 | |||||||||
See notes to consolidated financial statements
F-4
Table of Contents
PSIVIDA CORP. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
(In thousands except share data)
| Common Stock | Additional Paid-In Capital |
Accumulated Deficit |
Accumulated Other Comprehensive Income |
Total Stockholders’ Equity |
||||||||||||||||||
| Number of Shares |
Par Value Amount |
|||||||||||||||||||||
| Balance at July 1, 2007 |
14,140,184 | $ | 14 | $ | 229,913 | $ | (148,867 | ) | $ | 7,205 | $ | 88,265 | ||||||||||
| Comprehensive loss: |
||||||||||||||||||||||
| Net loss |
— | — | — | (75,670 | ) | — | (75,670 | ) | ||||||||||||||
| Foreign currency translation adjustments |
— | — | — | — | (236 | ) | (236 | ) | ||||||||||||||
| Total comprehensive loss |
$ | (75,906 | ) | |||||||||||||||||||
| Stock issued, net of issue costs |
4,114,199 | 4 | 18,383 | — | — | 18,387 | ||||||||||||||||
| Stock-based compensation |
— | — | 756 | — | — | 756 | ||||||||||||||||
| Vesting of nonvested shares |
8,587 | — | — | — | — | — | ||||||||||||||||
| Proceeds allocated to derivative liabilities in connection with warrants issued to investors |
— | — | (1,422 | ) | — | — | (1,422 | ) | ||||||||||||||
| Cash in lieu of fractional shares in connection with reincorporation |
(625 | ) | — | (2 | ) | — | — | (2 | ) | |||||||||||||
| Balance at June 30, 2008 |
18,262,345 | 18 | 247,628 | (224,537 | ) | 6,969 | 30,078 | |||||||||||||||
| Comprehensive loss: |
||||||||||||||||||||||
| Net loss |
— | — | — | (2,511 | ) | — | (2,511 | ) | ||||||||||||||
| Reclassification of foreign currency translation gains to earnings upon dissolution of subsidiaries |
(69 | ) | (69 | ) | ||||||||||||||||||
| Foreign currency translation adjustments |
— | — | — | — | (4,829 | ) | (4,829 | ) | ||||||||||||||
| Total comprehensive loss |
$ | (7,409 | ) | |||||||||||||||||||
| Stock-based compensation |
— | — | 815 | — | — | 815 | ||||||||||||||||
| Issuance of fully vested shares |
31,616 | — | 57 | — | — | 57 | ||||||||||||||||
| Balance at June 30, 2009 |
18,293,961 | 18 | 248,500 | (227,048 | ) | 2,071 | 23,541 | |||||||||||||||
| Comprehensive income: |
||||||||||||||||||||||
| Net income |
— | — | — | 8,753 | — | 8,753 | ||||||||||||||||
| Foreign currency translation adjustments |
— | — | — | — | (1,548 | ) | (1,548 | ) | ||||||||||||||
| Net unrealized loss on marketable securities |
— | — | — | — | (2 | ) | (2 | ) | ||||||||||||||
| Total comprehensive income |
$ | 7,203 | ||||||||||||||||||||
| Stock-based compensation |
— | — | 1,385 | — | — | 1,385 | ||||||||||||||||
| Exercise of warrants |
100,000 | — | 484 | — | — | 484 | ||||||||||||||||
| Exercise of stock options |
110,000 | 1 | 317 | — | — | 318 | ||||||||||||||||
| Issuance of fully vested shares |
27,431 | — | 110 | — | — | 110 | ||||||||||||||||
| Balance at June 30, 2010 |
18,531,392 | $ | 19 | $ | 250,796 | $ | (218,295 | ) | $ | 521 | $ | 33,041 | ||||||||||
See notes to consolidated financial statements
F-5
Table of Contents
PSIVIDA CORP. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
| Year Ended June 30, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Cash flows from operating activities: |
||||||||||||
| Net income (loss) |
$ | 8,753 | $ | (2,511 | ) | $ | (75,670 | ) | ||||
| Adjustments to reconcile net income (loss) to cash flows from operating activities: |
||||||||||||
| Impairment of goodwill |
— | — | 60,106 | |||||||||
| Amortization of intangible assets |
3,289 | 3,336 | 3,886 | |||||||||
| Depreciation of property and equipment |
37 | 102 | 397 | |||||||||
| Change in fair value of derivatives |
339 | (959 | ) | (8,357 | ) | |||||||
| Amortization of bond premium on marketable securities |
1 | — | — | |||||||||
| Non-cash interest expense |
— | — | 507 | |||||||||
| Stock-based compensation |
1,495 | 872 | 756 | |||||||||
| Loss (gain) on sale of equipment |
— | 39 | (13 | ) | ||||||||
| Provision for losses on note receivable |
— | 1,300 | 325 | |||||||||
| Deferred income tax benefit |
— | (94 | ) | (535 | ) | |||||||
| Foreign currency translation gains upon dissolution of subsidiaries |
— | (69 | ) | — | ||||||||
| Changes in operating assets and liabilities: |
||||||||||||
| Accounts and other receivables |
(290 | ) | 124 | (105 | ) | |||||||
| Prepaid expenses and other current assets |
52 | 117 | (97 | ) | ||||||||
| Accounts payable |
110 | (2,156 | ) | 1,400 | ||||||||
| Accrued expenses |
(360 | ) | (649 | ) | (4,676 | ) | ||||||
| Deferred revenue |
(3,527 | ) | (7,888 | ) | 16,933 | |||||||
| Net cash provided by (used in) operating activities |
9,899 | (8,436 | ) | (5,143 | ) | |||||||
| Cash flows from investing activities: |
||||||||||||
| Purchases of marketable securities |
(2,054 | ) | — | — | ||||||||
| Purchases of property and equipment |
(15 | ) | (195 | ) | (272 | ) | ||||||
| Proceeds from sale of property and equipment |
— | — | 13 | |||||||||
| Net cash used in investing activities |
(2,069 | ) | (195 | ) | (259 | ) | ||||||
| Cash flows from financing activities: |
||||||||||||
| Proceeds from issuance of stock |
— | — | 20,622 | |||||||||
| Stock issuance costs |
— | — | (2,237 | ) | ||||||||
| Proceeds from exercise of stock options and warrants |
802 | — | — | |||||||||
| Net cash provided by financing activities |
802 | — | 18,385 | |||||||||
| Effect of foreign exchange rate changes on cash and cash equivalents |
(17 | ) | (79 | ) | (44 | ) | ||||||
| Net increase (decrease) in cash and cash equivalents |
8,615 | (8,710 | ) | 12,939 | ||||||||
| Cash and cash equivalents at beginning of year |
6,899 | 15,609 | 2,670 | |||||||||
| Cash and cash equivalents at end of year |
$ | 15,514 | $ | 6,899 | $ | 15,609 | ||||||
| Supplemental disclosure of cash flow information: |
||||||||||||
| Cash paid for income taxes |
$ | 266 | $ | — | $ | — | ||||||
| Supplemental disclosure of non-cash investing and financing activities: |
||||||||||||
| Purchases of property and equipment |
$ | — | $ | — | $ | 101 | ||||||
See notes to consolidated financial statements
F-6
Table of Contents
PSIVIDA CORP. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(tabular amounts in thousands except share, per share and percentage amounts)
| 1. | Nature of the Business |
pSivida Corp. (together with its subsidiaries, the “Company”), incorporated in Delaware, develops tiny, sustained release, drug delivery products that are administered by implantation, injection or insertion. The Company’s lead product candidate, Iluvien™, delivers fluocinolone acetonide (“FA”) for the treatment of diabetic macular edema (“DME”), a leading cause of vision loss estimated to affect more than one million people in the United States, for which there is currently no FDA-approved drug therapy. Iluvien is licensed to Alimera Sciences, Inc. (“Alimera”), which is completing fully-recruited Phase III clinical trials. Based on 24-month data released in December 2009, Alimera filed a New Drug Application (“NDA”) with the Food and Drug Administration (“FDA”) in June 2010 and was granted Priority Review of the NDA on August 30, 2010. Under Priority Review, Alimera could receive a response to the NDA from the FDA by the end of calendar year 2010. If approved, Alimera has indicated that it expects to commercialize Iluvien as early as the first calendar quarter of 2011. The Company has also licensed certain of its drug delivery technologies to Alimera for the development of certain other ophthalmic products.
The Company has previously developed with partners two of the only three FDA-approved sustained release products to treat chronic back-of-the-eye diseases: Retisert® for the treatment of posterior uveitis and Vitrasert® for the treatment of AIDS-related cytomegalovirus (“CMV”) retinitis. The Company licensed both of these products and the technologies underlying them to Bausch & Lomb Incorporated (“Bausch & Lomb”). The Company also has a worldwide collaborative research and license agreement with Pfizer, Inc. (“Pfizer”) under which Pfizer may develop additional ophthalmic products using certain of the Company’s technologies.
The Company’s technology systems include Durasert™ and BioSilicon™. The Durasert system uses a drug core with one or more surrounding polymer layers through which drug permeates to the target site in the body at controlled rates for predetermined periods of time ranging from days to years. The Company’s back-of-the-eye products and product candidates utilize successive generations of the Durasert technology system. The BioSilicon technology system is a fully-erodible, nanostructured, porous silicon designed to provide sustained delivery of various therapeutics, including small drug molecules, proteins and peptides. Based on results of our preliminary studies, the Company is currently targeting BioSilicon as a second key drug delivery technology.
The Company is subject to risks common to companies in the biotechnology industry, including, but not limited to, risks relating to pre-clinical and clinical trials and the regulatory approval process, reliance on collaboration partners to successfully research, develop and commercialize products based on the Company’s technologies, development by its competitors of new or better technological innovations, ability to protect its proprietary technology, dependence on key personnel, compliance with FDA and other governmental regulations and approval requirements as well as its ability to execute on its business strategies and obtain adequate financing to fund its operations through corporate collaborations, sales of equity or otherwise.
The Company’s future operating results may vary from year to year and quarter to quarter, and such variations could be significant. Future operating results are expected to depend upon the amounts of payments that may be received from, and revenue recognition associated with, its current and any potential future collaboration arrangements and the clinical development costs and outcomes of its current and potential future product candidates. The Company anticipates that existing capital resources of $17.6 million at June 30, 2010 should enable it to maintain its current and planned operations into at least calendar year 2012. The Company’s ability to internally fund its planned operations beyond then may be substantially dependent upon the timing of FDA approval of Iluvien, which would result in a $25 million milestone payment due from Alimera, and the successful commercialization of Iluvien by Alimera.
F-7
Table of Contents
Basis of Presentation
The consolidated financial statements are presented in U.S. dollars in accordance with generally accepted accounting principles in the United States (“U.S. GAAP”) and include the accounts of pSivida Corp. and its wholly-owned subsidiaries. All intercompany accounts and transactions have been eliminated. The Company’s fiscal year ends on June 30 of each year. The years ended June 30, 2010, 2009 and 2008 may be referred to herein as fiscal 2010, fiscal 2009 and fiscal 2008, respectively. Throughout these financial statements, references to “US$” and “$” are to U.S. dollars and references to “A$” are to Australian dollars.
Effective June 19, 2008, the Company reincorporated from Western Australia to the United States. Pursuant to a scheme of arrangement under Australian law, all ordinary shares, including ordinary shares represented by American Depositary Shares (“ADSs”), of pSivida Limited, a company incorporated in Western Australia, were transferred by court order to pSivida Corp., a company incorporated in Delaware, in exchange for shares of pSivida Corp. common stock, including common stock represented by CHESS Depositary Interests (“CDIs”), in a ratio of 40 pSivida Limited ordinary shares to 1 share of pSivida Corp. common stock. All assets and liabilities of pSivida Limited, including outstanding options and warrants to purchase ordinary shares or ADSs of pSivida Limited, were, by court order, transferred to and assumed by pSivida Corp., following which pSivida Limited was deregistered without a winding up. All options and warrants were equitably adjusted to reflect the reincorporation. Each CDI represents one share of common stock. Throughout these financial statements, all share, option and warrant information, including related per share data, have been adjusted to give effect to the reincorporation for all periods presented.
| 2. | Significant Accounting Policies |
Use of Estimates
The preparation of consolidated financial statements in conformity with U.S. GAAP requires management to make certain estimates and assumptions that affect the reported amounts and disclosure of assets and liabilities at the date of the consolidated financial statements and the reported amounts and disclosure of revenues and expenses during the reporting periods. Significant estimates and assumptions made by management include, among others, revenue recognition, recoverability of long-lived assets, valuation of deferred tax assets, valuation of stock option awards and revaluation of derivative liabilities. On an ongoing basis, the Company evaluates its estimates. Actual results may differ from these estimates.
Foreign Currency
The functional currency of each entity is the currency of the primary economic environment in which that entity operates—the U.S. dollar or the Pound Sterling.
Assets and liabilities of the Company’s foreign subsidiary are translated at period end exchange rates. Amounts included in the statements of operations are translated at the average exchange rate for the period. The resulting currency translation adjustments are recorded in accumulated other comprehensive income as a separate component of stockholders’ equity in the consolidated balance sheets. Foreign currency gains or losses arising from transactions denominated in foreign currencies, whether realized or unrealized, are recorded in other income, net in the consolidated statements of operations.
Cash Equivalents
Cash equivalents represent highly liquid investments with an original or remaining maturity of three months or less at the date of purchase, principally consisting of institutional money market funds. Cash equivalents are stated at amortized cost, which approximates fair value.
F-8
Table of Contents
Marketable Securities
Marketable securities consist of investments with an original or remaining maturity of greater than ninety days at the date of purchase. The Company has classified its marketable securities as available-for-sale and, accordingly, records these investments at fair value, with unrealized gains and losses excluded from earnings and reported, net of tax, in accumulated other comprehensive income (loss), which is a component of stockholders’ equity. The fair value of marketable securities is determined based on quoted market prices at the balance sheet dates of the same or similar instruments. The cost of marketable securities sold is determined by the specific identification method. The amortized cost of debt securities is adjusted for amortization of premiums and accretion of discounts through to the earlier of sale or maturity. Such amortization and accretion are included in interest income, net in the accompanying statements of operations.
Concentration of Risk
Financial instruments that potentially subject the Company to concentrations of credit risk consist principally of cash, cash equivalents and marketable securities. At June 30, 2010, substantially all of the Company’s interest-bearing cash equivalent balances, aggregating approximately $15.1 million, were concentrated in two institutional money market funds that have investments consisting primarily of certificates of deposit, commercial paper, time deposits, U.S. government agencies, treasury bills and treasury repurchase agreements. Generally, these deposits may be redeemed upon demand and, therefore, bear minimal risk. Marketable securities at June 30, 2010 consist of high-grade corporate bonds, U.S. Government obligations and commercial paper. With the exception of U.S. Treasury and U.S. Government agency securities, the Company’s investment policy, approved by the Board of Directors, limits the amount that may be invested in a single issuer, thereby reducing credit risk concentrations.
One customer, Alimera, accounted for approximately $22.3 million, or 97%, of total revenues in fiscal 2010, $11.8 million, or 97%, of total revenues in fiscal 2009 and $3.3 million, or 94%, of total revenues in fiscal 2008. See Note 3 for further discussion of the Company’s collaboration agreement with Alimera.
Fair Value of Financial Instruments
The carrying amounts of cash equivalents, accounts receivable, accounts payable and accrued expenses approximate fair value because of their short-term maturity.
Accounts and Other Receivables
Receivables are recorded net of allowance for doubtful accounts and at June 30, 2010 consisted primarily of (i) quarterly royalties earned; (ii) value added tax reimbursements in certain foreign jurisdictions; (iii) U.K. research and development tax credits and (iii) accrued interest on marketable securities.
Debt and Equity Instruments
Debt and equity instruments are classified as either liabilities or equity in accordance with the substance of the contractual arrangement. Warrants issued in connection with share issues that are denominated in a currency (A$) other than the issuer’s functional currency (US$) are treated as derivative liabilities, reflecting the variable amount of functional currency to be received upon potential exercise. After initial recognition, subsequent changes in the fair value of the derivative liabilities are recorded in the consolidated statements of operations in each reporting period. Fair value is determined using a Black-Scholes valuation model.
Property and Equipment
Property and equipment are recorded at cost and depreciated over their estimated useful lives (generally three years) using the straight-line method. Leasehold improvements are amortized over the shorter of the remaining lease term or the useful life of the asset. Repairs and maintenance costs are expensed as incurred.
F-9
Table of Contents
Leases
Leases are classified at their inception as either operating or capital leases based on the economic substance of the agreement. Lease payments made under operating leases are recognized as an expense on a straight-line basis over the lease term. Contingent rentals are recognized as an expense in the financial year in which they are incurred.
Impairment of Intangible Assets
The Company’s finite life intangible assets include its Durasert and BioSilicon patented technologies that are being amortized on a straight-line basis over twelve years. The intangible asset lives were determined based upon the anticipated period that the Company will derive future cash flows from the intangible assets, and considered the effects of legal, regulatory, contractual, competitive and other economic factors. The Company continually monitors whether events or circumstances have occurred that indicate that the remaining estimated useful life of its intangible assets may warrant revision. Recoverability of these assets is assessed when triggering events have occurred that may give rise to an impairment loss and is determined by a comparison of the carrying amount of the asset to the future undiscounted net cash flows expected to be generated by the asset. If an asset is considered to be impaired, the impairment charge to be recognized is measured by the amount by which the carrying value of the asset exceeds its estimated fair value.
Revenue Recognition
The Company recognizes revenues when they are realized or realizable and earned. Revenues are realized or realizable and earned when the Company has persuasive evidence that an arrangement exists, the goods have been delivered or the services have been rendered to the customer, the sales price is fixed or determinable and collectibility is reasonably assured. In addition to this general policy, the following are specific revenue recognition policies:
Collaborative research and development
The Company’s business strategy includes entering into collaborative arrangements with strategic partners for the development and commercialization of product candidates utilizing the Company’s technologies. The terms of these agreements typically include multiple deliverables by the Company (for example, license rights, providing research and development services and manufacturing of clinical materials) in exchange for consideration to the Company of some combination of non-refundable license fees, funding of research and development activities, payments based upon achievement of clinical development milestones and royalties in the form of a designated percentage of product sales or profits.
Non-refundable license fees are recognized as revenue when the Company has a contractual right to receive such payment, the contract price is fixed or determinable, the collection of the resulting receivable is reasonably assured and the Company has no further performance obligations under the license agreement. Multiple element arrangements, such as license and development agreements, are analyzed to determine whether the deliverables can be separated or whether they must be accounted for as a single unit of accounting. The Company recognizes up-front license payments as revenue upon delivery of the license only if the license has stand-alone value and the fair value of the undelivered performance obligations can be determined. If the fair value of the undelivered performance obligations can be determined, such obligations would then be accounted for separately as performed. If the license is considered not to have stand-alone value or if the fair value of any of the undelivered performance obligations cannot be determined, the arrangement would then be accounted for as a single unit of accounting, with total payments recognized as revenue on a straight-line basis over the estimated period of the Company’s performance obligations. The cumulative amount of revenue earned during the performance period is limited to the cumulative amount of payments received as of the period ending date.
F-10
Table of Contents
If the Company cannot reasonably estimate when its performance obligation either ceases or becomes inconsequential, then revenue recognition is deferred until the Company can reasonably estimate when the performance obligation ceases or becomes inconsequential. Revenue is then recognized over the remaining estimated period of performance.
Significant management judgment is required to determine the level of effort required under an arrangement and the period over which the Company is expected to complete its performance obligations under an arrangement.
Reimbursement of Costs
The Company’s business includes providing research and development services on behalf of its collaboration partners to assist in advancing the developmental cycle of licensed products. The Company acts primarily as a principal in these transactions and, accordingly, amounts received are classified as a component of revenue to be recognized consistent with the revenue recognition policy summarized above. The Company records the expenses incurred and reimbursed on a gross basis.
Royalties
Royalty income is recognized upon the sale of the related products, provided that the royalty amounts are fixed and determinable, collection of the related receivable is reasonably assured and the Company has no remaining performance obligations under the arrangement. Such revenues are included as royalty income.
If royalties are received when the Company has remaining performance obligations, the royalty payments would be attributed to the services being provided under the arrangement and therefore would be recognized as such performance obligations are performed. Such revenues are included as collaborative research and development revenues.
Deferred Revenue
Amounts received prior to satisfying the above revenue recognition criteria would be recorded as deferred revenue in the accompanying consolidated balance sheets. Amounts not expected to be recognized within one year following the balance sheet date would be classified as non-current deferred revenue.
Research and Development
Research and development costs are charged to operations as incurred. These costs include all direct costs, including cash compensation, stock-based compensation and benefits for research and development personnel, amortization of intangible assets, supplies and materials, direct external costs including costs of clinical trials, clinical materials, pre-clinical programs, regulatory affairs, external consultants, and other operational costs related to the Company’s research and development of its product candidates.
Stock-Based Compensation
The Company awards stock options and other equity-based instruments to its employees, directors and consultants pursuant to stockholder-approved plans. Compensation cost related to such awards is based on the fair value of the instrument on the grant date and is recognized, net of estimated forfeitures, on a straight-line basis over the requisite service period, which generally equals the vesting period. The Company estimates the fair value of stock option awards using the Black-Scholes option valuation model.
Net Income (Loss) per Share
Basic net income (loss) per share is computed by dividing net income (loss) by the weighted-average number of common shares outstanding during the period. For periods in which the Company reports net income,
F-11
Table of Contents
diluted net income per share is determined by adding to the weighted average number of common shares outstanding the average number of dilutive common equivalent shares using the treasury stock method, unless the effect is anti-dilutive.
The calculation of shares used to compute basic and diluted net income (loss) per share is as follows:
| Year Ended June 30, | ||||||
| 2010 | 2009 | 2008 | ||||
| Number of common shares—basic |
18,404,823 | 18,262,865 | 18,165,867 | |||
| Effect of dilutive securities: |
||||||
| Stock options |
489,783 | — | — | |||
| Number of common shares—diluted |
18,894,606 | 18,262,865 | 18,165,867 | |||
The following potentially dilutive securities outstanding, prior to the application of the treasury stock method, have been excluded from the computation of diluted weighted-average shares outstanding for the years ended June 30, 2010, 2009 and 2008, as they would be anti-dilutive:
| June 30, | ||||||
| 2010 | 2009 | 2008 | ||||
| Options outstanding |
907,219 | 2,078,397 | 473,092 | |||
| Warrants outstanding |
10,997,681 | 11,097,681 | 11,182,181 | |||
| 11,904,900 | 13,176,078 | 11,655,273 | ||||
Comprehensive Income (Loss)
Comprehensive income (loss) is comprised of net income (loss), foreign currency translation adjustments and unrealized gains and losses on available-for-sale marketable securities.
Income Tax
The Company recognizes deferred income tax assets and liabilities for the expected future tax consequences of events that have been included in the Company’s financial statements or tax returns. Deferred tax assets and liabilities are based on the difference between the financial statement and tax bases of assets and liabilities, as well as net operating loss carry forwards, and are measured using tax rates expected to be in effect in the years in which these differences are expected to reverse. A valuation allowance is provided against net deferred tax assets if, based on the available evidence, it is more likely than not that some or all of the net deferred tax assets will not be realized.
The Company determines whether it is more likely than not that a tax position will be sustained upon examination. If it is not more likely than not that a position will be sustained, no amount of the benefit attributable to the position is recognized. The tax benefit to be recognized for any tax position that meets the more likely than not recognition threshold is calculated as the largest amount that is more than 50% likely of being realized upon resolution of the contingency. The Company accounts for interest and penalties related to uncertain tax positions as part of its income tax (expense) benefit.
Recently Adopted Accounting Pronouncements
Effective July 1, 2009, the Company adopted the provisions of ASC 808-10, Collaborative Arrangements, formerly Emerging Issues Task Force (“EITF”) Issue No. 07-01, Accounting for Collaborative Arrangements. This standard defines a collaborative arrangement as a contractual arrangement in which the parties are (i) active
F-12
Table of Contents
participants to the arrangement; and (ii) exposed to significant risks and rewards that depend upon the commercial success of the endeavor. It also addresses the appropriate statements of operations presentation for activities and payments between the participants in a collaborative arrangement as well as for costs incurred and revenue generated from transactions with third parties. Adoption of this standard did not have any material impact on the Company’s consolidated financial statements.
Recently Issued Accounting Pronouncements
In October 2009, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (ASU) No. 2009-13, Multiple-Deliverable Revenue Arrangements—a consensus of the FASB Emerging Issues Task Force. ASU 2009-13 updates the existing multiple-element revenue arrangements guidance currently included under ASC 605-25, which originated primarily from the guidance in EITF Issue No. 00-21, Revenue Arrangements With Multiple Deliverables. The update provides principles for allocation of consideration among multiple elements of revenue arrangements, allowing more flexibility in identifying and accounting for separate deliverables under an arrangement. ASU 2009-13 introduces an estimated selling price method for valuing the elements of a bundled arrangement if vendor-specific objective evidence or third-party evidence of selling price is not available. In addition, the update also significantly expands related disclosure requirements. ASU 2009-13 is effective on a prospective basis for revenue arrangements entered into or materially modified in fiscal years beginning on or after June 15, 2010. The Company is evaluating the potential application of this new accounting update to new or materially modified revenue arrangements.
In April 2010, the FASB issued ASU No. 2010-17, Revenue Recognition (Topic 605—Milestone Method of Revenue Recognition: a consensus of the FASB EITF (“ASU 2010-17”). ASU 2010-17 amends ASC 605-28 and established a revenue recognition method for contingent consideration that is payable on achievement of an uncertain future event, referred to as a milestone. The scope of the milestone method is limited to research and development agreements and is applicable to milestones in multiple-deliverable arrangements involving research and development transactions. The guidance does not preclude the application of any other applicable revenue guidance. The guidance will be effective for financial statements issued for fiscal years beginning after June 15, 2010. Early adoption is permitted. The Company is currently evaluating the potential impact of ASU 2010-17 on its financial statements.
| 3. | License and Collaboration Agreements |
Alimera
Under a collaboration agreement with Alimera, as amended in March 2008 (the “Alimera Agreement”), the Company has licensed Alimera the rights to develop, market and sell certain product candidates, including Iluvien. Alimera is completing fully-recruited Phase III trials for Iluvien in DME. Based on 24-month data released in December 2009, Alimera filed an NDA with the FDA in June 2010. On August 30, 2010, the FDA granted Priority Review status for the NDA. Under Priority Review, Alimera could receive a response to the NDA from the FDA by the end of calendar year 2010.
Upon execution of the Alimera Agreement, the Company received consideration of $12.0 million in cash and Alimera cancelled $5.7 million of accrued development cost liabilities, including related penalties and accrued interest, owed by the Company to Alimera as of March 14, 2008. In addition, the Company received a $15.0 million conditional note providing for aggregate principal and interest payments of up to approximately $21.3 million through September 2012, Alimera agreed to pay a $25.0 million milestone payment upon FDA approval of Iluvien for DME, and Alimera assumed all financial responsibility for the development of licensed products under the Alimera Agreement, which had previously been shared equally, including reimbursement of approved development costs incurred by the Company in support of the ongoing clinical studies of Iluvien and anticipated regulatory submissions. In exchange, the Company decreased its share in any future profits, as defined, on sales of Iluvien by Alimera from 50% to 20%, subject to an offset of 20% of pre-profitability commercialization costs, as defined, incurred by Alimera. In the event Alimera sublicenses commercialization, the Company is entitled to receive 20% of royalties and 33% of non-royalty consideration received by Alimera, less certain permitted deductions.
F-13
Table of Contents
The scheduled payment terms on the $15.0 million conditional note consisted of (i) interest only at an annual rate of 8% payable quarterly through March 2010 and (ii) principal payments of $500,000 per month commencing April 30, 2010 together with interest payable quarterly at an annual rate of 20%. Upon the occurrence of certain defined liquidity events (such as an initial public offering of Alimera, other sales of capital stock of Alimera and/or the sale or other disposition of substantially all of Alimera’s assets) that resulted in aggregate cash and/or noncash proceeds to Alimera in excess of $75 million, the note became immediately due and payable. Failure by Alimera to pay the note upon the occurrence of a defined liquidity event constituted an event of default under the note. If no liquidity event occurred on or before September 30, 2012, the note would automatically be cancelled. Based upon the terms of the note, payment was within the control of Alimera unless there was a liquidity event or an event of default. Through March 31, 2010, the Company received total interest payments of approximately $2.5 million under the terms of the note. On April 27, 2010, following consummation of its initial public offering, Alimera paid the $15.0 million conditional note in full together with $225,000 of accrued and unpaid interest.
The Company considered the Alimera Agreement to be a revenue arrangement with multiple deliverables. The Company’s deliverables under this collaboration included the exclusive license to Iluvien, future “know-how”, a non-exclusive license for certain other products using the same technology, and certain prescribed research and development. The Company assessed each of these elements against the separation criteria for multiple element arrangements and concluded that the licenses did not have stand-alone value to Alimera and the Company did not have objective and reliable evidence of fair value for all undelivered elements of the arrangement. Accordingly, the Company concluded that the deliverables represented a single unit of accounting. The terms of the collaboration agreement specifically defined the end period of any and all of the Company’s performance obligations as (i) December 31, 2009 for Iluvien and (ii) the effective date of the Alimera Agreement for any other licensed product. Accordingly, the services related to Iluvien were provided through the December 31, 2009 performance period and no further obligations existed after this date.
The Company incurred costs related to the Alimera Agreement to provide services, as requested. The Company was the primary obligor under these arrangements and, upon the amendment in March 2008, was no longer sharing in the costs of product development. Accordingly, costs associated with development activities have been recorded as expense as incurred and payments received have been recorded as revenue.
Based upon the above analysis, the initial $18.3 million of deferred revenue, which consisted of the $12.0 million in cash, the $5.7 million cancellation of accrued development cost liabilities and $650,000 of previously received but unamortized milestone payments, was recognized as revenue on a straight-line basis over the 21.5 month performance period from the effective date of the Alimera Agreement through December 31, 2009. Because the $15.0 million note did not represent an unconditional payment obligation of Alimera, it was not recorded as an asset but instead treated by the Company as contingent future revenue consideration. All additional cash consideration received from Alimera during the performance period, which consisted of conditional note payments and development cost reimbursements, was recognized as revenue during the performance period using the cumulative catch-up method. Amounts received from Alimera subsequent to December 31, 2009, including any note, milestone and profit share payments, are recognized as revenue upon receipt or at such earlier date, if applicable, on which any such amount is both fixed and determinable and reasonably assured of collectibility.
Revenue related to the Alimera Agreement totaled approximately $22.3 million, $11.8 million and $3.3 million for the years ended June 30, 2010, 2009 and 2008, respectively. These revenues represented substantially all of the Company’s collaborative research and development revenue for these periods. The balance of deferred revenue at June 30, 2010 and 2009 was $0 and $5.8 million, respectively.
Pfizer
In April 2007, the Company and Pfizer entered into a worldwide collaborative research and license agreement (the “Pfizer Agreement”), which superseded a December 2006 research agreement. Under the Pfizer
F-14
Table of Contents
Agreement, the parties have implemented a joint research program aimed at developing certain ophthalmic products that are not licensed to others using certain of the Company’s technologies. In addition to potential development and sales related milestone payments, Pfizer pays the Company a minimum of $500,000 quarterly in consideration of the Company’s costs in performing the research program. These payments commenced in calendar year 2008 and will continue until the earlier of the commencement of the first Phase III clinical trial for a licensed product candidate or the termination of the Pfizer Agreement.
Following an evaluation of the multiple deliverables, the Company determined that the Pfizer Agreement and the preceding Pfizer research agreement should be combined for accounting purposes as a single unit of accounting. The Company is unable to define the time period of its overall deliverables and other obligations under the Pfizer Agreement and, as a result, all payments received from Pfizer through June 30, 2010 totaling $5.75 million have been classified in non-current deferred revenue.
Intrinsiq
In January 2008, the Company and Intrinsiq Materials Cayman Limited (“Intrinsiq”) entered into an agreement pursuant to which Intrinsiq acquired an exclusive field-of-use license to develop and commercialize nutraceutical and food science applications of BioSilicon, and certain related assets, for $1.2 million. Provided the license agreement remains in effect, Intrinsiq is obligated to pay the Company aggregate minimum royalties of $3.55 million through April 2014, of which the first $450,000 was paid in July 2009.
Under the original agreement, the parties were obligated to enter into a manufacture and supply agreement, which was consummated effective as of February 1, 2009. Pursuant to the supply agreement, the Company leased to Intrinsiq certain equipment for its use in manufacturing BioSilicon material. Subject to its right to terminate the lease, Intrinsiq will acquire title to the equipment upon the remittance of lease payments totaling $122,000 over the 2-year lease term, of which the first three payments of $24,000 each were received through June 2010.
The Company determined that the equipment lease component represented a separate element of this arrangement. Using the relative fair value method prescribed under the authoritative guidance, the Company allocated the arrangement consideration between the lease and license deliverables. The Company determined the performance period of the license arrangement to be 17 years, coinciding with the last to expire of the patents licensed to Intrinsiq, and is recognizing consideration allocated to the license arrangement on a straight-line basis over this period. The Company recognized collaborative research and development revenue of $121,000 in fiscal 2010 and $77,000 in fiscal 2009, and the remaining balance of payments received, including minimum royalties, of approximately $1.1 million was recorded as deferred revenue at June 30, 2010.
Bausch & Lomb
The Company’s Retisert and Vitrasert products were developed and commercialized under a 1992 licensing and development agreement with Bausch & Lomb. Pursuant to a subsequent collaboration agreement, Bausch & Lomb has a worldwide exclusive license to make and sell Vitrasert and our first-generation products (as defined in the agreement, including the Retisert device) in return for royalties based on sales.
In June 2005, the Company received a $3.0 million advance from Bausch & Lomb in consideration of $6.25 million of future Retisert royalties that otherwise would be payable to the Company. During the quarter ended June 30, 2010, Bausch & Lomb retained the final portion of these royalties otherwise payable and the Company recorded $342,000 of royalty income. Subsequent to June 30, 2010, the Company is entitled to receive 100% of the Retisert royalties pursuant to the collaboration agreement.
F-15
Table of Contents
| 4. | Goodwill and Intangible Assets |
Intangible Assets
The reconciliation of intangible assets for the years ended June 30, 2010 and 2009 is as follows:
| June 30, | ||||||||
| 2010 | 2009 | |||||||
| Patented technologies |
||||||||
| Gross carrying amount at beginning of year |
$ | 56,559 | $ | 64,342 | ||||
| Foreign currency translation adjustments |
(3,284 | ) | (7,783 | ) | ||||
| Gross carrying amount at end of year |
53,275 | 56,559 | ||||||
| Accumulated amortization at beginning of year |
(27,757 | ) | (27,540 | ) | ||||
| Amortization expense |
(3,289 | ) | (3,336 | ) | ||||
| Foreign currency translation adjustments |
1,648 | 3,119 | ||||||
| Accumulated amortization at end of year |
(29,398 | ) | (27,757 | ) | ||||
| Net book value at end of year |
$ | 23,877 | $ | 28,802 | ||||
The net book value of the Company’s intangible assets at June 30, 2010 and 2009 is summarized as follows:
| June 30, | Estimated Remaining Useful Life at June 30, 2010 | |||||||
| 2010 | 2009 | (Years) | ||||||
| Patented technologies |
||||||||
| Durasert |
$ | 7,898 | $ | 8,951 | 7.5 | |||
| BioSilicon |
15,979 | 19,851 | 7.5 | |||||
| $ | 23,877 | $ | 28,802 | |||||
The Company amortizes its intangible assets with finite lives on a straight-line basis over their respective estimated useful lives. The aggregate annual amortization expense for intangible assets was $3.3 million, $3.3 million and $3.9 million for the years ended June 30, 2010, 2009 and 2008, respectively. Based upon intangible assets in service as of June 30, 2010, amortization expense for each of the next five years is estimated to be approximately $3.2 million per year.
The ultimate recoupment of the carrying value of intangible assets is dependent on the Company’s successful development and commercial exploitation of its technologies.
Impairment of Goodwill
At June 30, 2008, the Company performed its annual impairment assessment of the carrying value of goodwill. In accordance with the guidance, the Company compared the carrying value of its single reporting unit to its estimated fair value.
The Company assessed fair value as of June 30, 2008 through a combination of a discounted future cash flows analysis (an income approach) and a market approach. As a result of this step one analysis, the Company concluded that the carrying value of its net assets at June 30, 2008, including goodwill, exceeded fair value. The implied fair value of goodwill in the step two analysis was zero, resulting in a fourth quarter impairment charge equal to the then entire $60.1 million carrying value of the Company’s goodwill at June 30, 2008. This impairment charge is not tax deductible because the acquisitions that gave rise to the goodwill were structured as stock transactions.
F-16
Table of Contents
Since the adoption of FASB Statement No. 142, Goodwill and Other Intangible Assets (codified in ASC 350), the Company has recorded a total of $60.1 million of accumulated goodwill impairment losses and at June 30, 2010 and 2009 the carrying value of goodwill was zero.
| 5. | Marketable Securities |
The amortized cost, unrealized loss and fair value of the Company’s available-for-sale marketable securities at June 30, 2010 were as follows:
| Amortized Cost |
Unrealized Loss |
Fair Value | |||||||
| Corporate bonds |
$ | 1,304 | $ | 2 | $ | 1,302 | |||
| U.S. Government obligations |
449 | — | 449 | ||||||
| Commercial Paper |
300 | — | 300 | ||||||
| Total marketable securities |
$ | 2,053 | $ | 2 | $ | 2,051 | |||
The marketable securities have maturity dates ranging between three and eleven months, with a weighted average maturity of 8.2 months. There were no marketable securities at June 30, 2009.
| 6. | Property and Equipment, Net |
| June 30, | ||||||||
| 2010 | 2009 | |||||||
| Property and equipment |
$ | 3,470 | $ | 3,740 | ||||
| Leasehold improvements |
192 | 188 | ||||||
| Gross property and equipment |
3,662 | 3,928 | ||||||
| Accumulated depreciation and amortization |
(3,619 | ) | (3,862 | ) | ||||
| $ | 43 | $ | 66 | |||||
Depreciation expense was $37,000 for fiscal 2010, $102,000 for fiscal 2009 and $397,000 for fiscal 2008.
| 7. | Accrued Expenses |
| June 30, | ||||||
| 2010 | 2009 | |||||
| Personnel costs |
$ | 592 | $ | 911 | ||
| Professional fees |
282 | 264 | ||||
| Clinical |
242 | 182 | ||||
| Other |
42 | 159 | ||||
| Income taxes |
— | 36 | ||||
| $ | 1,158 | $ | 1,552 | |||
| 8. | Derivative Liabilities |
During the years ended June 30, 2008 and 2007, the Company sold units consisting of common shares together with detachable warrants to purchase additional common shares within specified time periods. In several of these transactions, the warrants were denominated in A$, which is different than the Company’s functional currency. Because the potential exercise of such warrants would result in a variable amount of proceeds in the Company’s functional currency, the fair value of the warrants was recorded as a derivative liability, with a
F-17
Table of Contents
corresponding reduction in additional paid-in capital, subject to revaluation of the liability on a recurring basis through the statements of operations. The fair value of the warrants is determined using a Black-Scholes Model. The net change in the fair values of these derivative liabilities resulted in an expense of $339,000 for the year ended June 30, 2010 and income of $959,000 and $8.4 million for the years ended June 30, 2009 and 2008, respectively. The change in the fair value of these derivative liabilities is primarily attributable to the spread between the Company’s share price and the US$-equivalent exercise prices of the underlying warrants, and secondarily to the reduction of the remaining contractual life of the warrants. See Note 9 for additional information related to A$-denominated warrants.
| 9. | Stockholders’ Equity |
Sales of Common Stock and Warrants
In July 2007, the Company completed a sale of 3,600,500 units at a per unit price of $5.00 for gross proceeds of $18.0 million. Each unit consisted of (i) one common share; and (ii) one warrant to purchase 0.40 common share, with a warrant exercise price of $6.60 per share. Of the total offering, 1,300,000 units were purchased by Pfizer in accordance with the terms of the Pfizer Agreement. A total of 72,010 warrants, with a warrant exercise price of $6.60 per share, were issued to the placement agents in connection with the offering. In addition, the Company simultaneously completed a sale of 513,699 units at the equivalent price of A$5.84 per unit for additional gross proceeds of approximately $2.6 million. Aggregate share issue costs for these transactions totaled approximately $2.2 million.
Warrants to Purchase Common Shares
The following table provides a reconciliation of all US$ warrants for the years ended June 30, 2010 and 2009:
| Year Ended June 30, | ||||||||||||
| 2010 | 2009 | |||||||||||
| Number of Warrants |
Weighted Average Exercise Price |
Number of Warrants |
Weighted Average Exercise Price | |||||||||
| Balance at beginning of year |
7,162,248 | $ | 7.50 | 7,195,498 | $ | 7.69 | ||||||
| Exercised |
(100,000 | ) | 4.84 | — | — | |||||||
| Expired |
— | — | (33,250 | ) | 50.00 | |||||||
| Balance and exercisable at end of year |
7,062,248 | $ | 7.53 | 7,162,248 | $ | 7.50 | ||||||
At June 30, 2010, these outstanding warrants had a weighted average remaining life of approximately 1.7 years.
The following table provides a reconciliation of all A$ warrants for the years ended June 30, 2010 and 2009:
| Year Ended June 30, | |||||||||
| 2010 | 2009 | ||||||||
| Number of Warrants |
Weighted Average Exercise Price A$ |
Number of Warrants |
Weighted Average Exercise Price A$ | ||||||
| Balance at beginning of year |
3,935,433 | 9.54 | 3,986,683 | 9.98 | |||||
| Expired |
— | — | (51,250 | ) | 43.60 | ||||
| Balance and exercisable at end of year |
3,935,433 | 9.54 | 3,935,433 | 9.54 | |||||
F-18
Table of Contents
At June 30, 2010 and 2009, the weighted exercise price of these warrants translated to US$ was $8.17 and $7.68, respectively. At June 30, 2010, these outstanding warrants had a weighted average remaining life of 0.7 years.
Registration Rights Agreements
The Company has entered into registration rights agreements with purchasers of certain of its equity and debt securities. These registration rights agreements required the Company to register with the SEC the resale of shares issued or issuable to such persons. The Company’s obligations to register shares in such transactions were subject to various deadlines, and the Company’s failure to maintain the registration of these securities would result in financial penalties against the Company. All required registration statements related to these underlying securities have been filed, declared effective by the SEC and remain in effect as of June 30, 2010.
| 10. | Stock-Based Compensation |
2008 Incentive Plan
The pSivida Corp. 2008 Incentive Plan (the “2008 Plan”), which became operational following the June 2008 reincorporation, provides for the issuance of shares of common stock in satisfaction of stock-based awards to directors, executives, employees and consultants. Awards may include stock options, stock appreciation rights, restricted and unrestricted stock, deferred stock, performance awards, convertible securities and cash grants. At June 30, 2010, the number of shares reserved for issuance under the 2008 Plan was 2,750,000, of which 614,953 shares were available for grant under the 2008 Plan. The 2008 Plan includes an “evergreen provision” that allows for an annual increase in the number of shares of common stock available for issuance under the 2008 Plan. Beginning on July 1, 2010, and on each subsequent anniversary date through July 1, 2017, the number of shares reserved for issuance under the 2008 Plan will be increased by the least of (i) 750,000 shares; (ii) 4% of the then outstanding shares of common stock; and (iii) any such lesser amount of shares of common stock as is determined by the Compensation Committee of the Board of Directors. On July 1, 2010, the number of shares increased by 741,255 shares, representing 4% of the outstanding shares at June 30, 2010.
A total of 450,000 options were granted during fiscal 2010 at exercise prices equal to the closing market price of the Company’s common stock on the NASDAQ Global Market (“NASDAQ”) on the respective option grant dates. Of this total, 315,000 options were issued to the Company’s chief executive officer with ratable annual vesting over 4 years and 135,000 options were issued to non-employee directors with 1-year cliff vesting. All option grants have a 10-year life. During fiscal 2010, the Company awarded 27,431 fully vested shares to the Company’s chief executive officer in lieu of a cash bonus. The fair value of the award was $110,000 and was recognized as stock-based compensation in the consolidated statement of operations.
The Company measures the fair value of options on their grant date using the Black-Scholes option-pricing model. Based upon limited option exercise history, the Company has used the “simplified” method outlined in SEC Staff Accounting Bulletin No. 107 to estimate the expected life of stock option grants. Management believes that the historical volatility of the Company’s stock price on NASDAQ, for which there has been trading history for approximately 5.5 years, best represents the expected volatility over the estimated life of the option. The risk-free interest rate is based upon published U.S. Treasury yield curve rates at the date of grant corresponding to the expected life of the stock option. An assumed dividend yield of zero reflects the fact that the Company has never paid cash dividends and has no intentions to pay dividends in the foreseeable future.
The key assumptions used to apply the option pricing model for options granted under the 2008 Plan during the years ended June 30, 2010 and 2009 were as follows:
| 2010 | 2009 | |||
| Option life (in years) |
5.50 - 6.25 | 5.50 - 6.25 | ||
| Stock volatility |
95% | 80% - 95% | ||
| Risk-free interest rate |
2.36% - 2.62% | 2.36% - 3.10% | ||
| Expected dividends |
0.0% | 0.0% |
F-19
Table of Contents
The Company recognizes compensation expense for only the portion of options that are expected to vest. An estimated annual forfeiture rate of 5% was used to determine awards expected to vest and to calculate stock-based compensation for fiscal 2010 and 2009, except that no forfeiture rate was assumed for option grants to executive officers and directors. Additional expense will be recorded if the actual forfeiture rate is lower than estimated, and a recovery of prior year expense will be recorded if the actual forfeiture rate is higher than estimated. The Company assesses the forfeiture rate at the end of each reporting period.
Estimates of fair value may not represent actual future events or the values to be ultimately realized by persons who receive stock options.
The following table summarizes information about stock options for the years ended June 30, 2010 and 2009:
| 2010 | 2009 | |||||
| Weighted-average grant date fair value, per share |
$ | 3.10 | $ | 1.43 | ||
| Total cash received from exercise of stock options |
318 | — | ||||
| Total intrinsic value of stock options exercised |
78 | — | ||||
At June 30, 2010, there was approximately $1.4 million of unrecognized compensation expense related to unvested share-based payment awards, which is expected to be recognized as expense over a weighted average period of 1.8 years.
The following table provides a reconciliation of stock option activity under the 2008 Plan for fiscal 2010:
| Number of options |
Weighted Average Exercise Price |
Weighted Average Remaining Contractual Life |
Aggregate Intrinsic Value | ||||||||
| (in years) | |||||||||||
| Outstanding at July 1, 2009 |
1,636,000 | $ | 1.94 | ||||||||
| Granted |
450,000 | 4.01 | |||||||||
| Exercised |
(110,000 | ) | 2.89 | ||||||||
| Forfeited |
(8,750 | ) | 2.26 | ||||||||
| Cancelled |
(1,250 | ) | 2.85 | ||||||||
| Outstanding at June 30, 2010 |
1,966,000 | $ | 2.36 | 8.70 | $ | 2,647 | |||||
| Outstanding at June 30, 2010—vested or unvested and expected to vest |
1,904,327 | $ | 2.35 | 8.71 | $ | 2,571 | |||||
| Exercisable at June 30, 2010 |
554,000 | $ | 1.63 | 8.46 | $ | 1,096 | |||||
The total fair value of options vested during fiscal 2010 was $670,000.
Employee Share Option Plan
The Company’s Employee Share Option Plan (the “Plan”) provided for the issuance of non-qualified stock options to eligible employees and directors. The Plan was assumed by pSivida Corp. in the reincorporation. As of June 30, 2008, no further options could be granted under the Plan. Options outstanding under the Plan had vesting periods ranging from immediate vesting to 3-year graded vesting, have a contractual life of five years and are denominated in A$.
The weighted average grant date fair value of stock options granted pursuant to the Plan during the year ended June 30, 2008 was A$2.47 per share.
F-20
Table of Contents
At June 30, 2010, there was less than $10,000 of unrecognized compensation expense related to non-vested share-based payment awards, which is expected to be recognized over a weighted average period of 0.3 years.
The following table provides a reconciliation of stock option activity under the Plan for fiscal 2010:
| Number of options |
Weighted Average Exercise Price |
Weighted Average Remaining Contractual Life |
Aggregate Intrinsic Value | ||||||
| A$ | (in years) | A$ | |||||||
| Outstanding at July 1, 2009 |
424,783 | 29.05 | |||||||
| Granted |
— | — | |||||||
| Forfeited |
— | — | |||||||
| Cancelled |
(239,471 | ) | 39.99 | ||||||
| Outstanding at June 30, 2010 |
185,312 | 14.91 | 1.59 | — | |||||
| Outstanding at June 30, 2010—vested or unvested and expected to vest |
176,093 | 13.76 | 1.66 | — | |||||
| Exercisable at end of year |
138,593 | 16.00 | 1.50 | — | |||||
At June 30, 2010 the weighted average exercise price of outstanding and exercisable options translated into US$ was $12.77 and $13.71, respectively.
The exercise prices of all outstanding options under the Plan at June 30, 2010, converted to US$ at the rate of exchange at that date, were in excess of the market price of the Company’s common shares at that date and, accordingly, the options had no intrinsic value. No options were exercised under the Plan during the year ended June 30, 2010. A total of 45,000 options were vested during fiscal 2010.
Stock-Based Compensation Expense
The Company’s statements of operations included total compensation expense from stock-based payment awards as follows:
| Year ended June 30, | |||||||||
| 2010 | 2009 | 2008 | |||||||
| Compensation expense from: |
|||||||||
| Stock options |
$ | 1,385 | $ | 815 | $ | 540 | |||
| Issuance of fully vested shares |
110 | 57 | — | ||||||
| Restricted stock |
— | — | 216 | ||||||
| $ | 1,495 | $ | 872 | $ | 756 | ||||
| Compensation expense included in: |
|||||||||
| Research and development |
$ | 306 | $ | 216 | $ | 28 | |||
| General and administrative |
1,189 | 656 | 728 | ||||||
| $ | 1,495 | $ | 872 | $ | 756 | ||||
F-21
Table of Contents
Options Issued in Exchange for CDS Options
On December 30, 2005, as part of the consideration for the acquisition of CDS, the Company issued 43,112 fully vested stock options with a fair value of $15.48 per share in consideration of outstanding fully vested CDS options. The following table presents a reconciliation of the activity related to the issuance of these options:
| 2010 | 2009 | ||||||||||
| Number of Options |
Weighted Average Exercise Price |
Number of Options |
Weighted Average Exercise Price | ||||||||
| Outstanding at beginning of year |
17,614 | $ | 11.35 | 17,614 | $ | 11.35 | |||||
| Options cancelled |
(17,614 | ) | 11.35 | — | — | ||||||
| Outstanding and exercisable at end of year |
— | $ | — | 17,614 | $ | 11.35 | |||||
| 11. | Retirement Plans |
The Company operates a defined contribution plan intended to qualify under Section 401(k) of the U.S. Internal Revenue Code. Participating U.S. employees may contribute up to 15% of their pre-tax compensation, as defined, subject to statutory maximums. The Company matches employee contributions up to 5% of eligible compensation, subject to a stated calendar year Internal Revenue Service maximum.
The Company operates a defined contribution pension plan for U.K. employees pursuant to which the Company makes contributions on behalf of employees plus a matching percentage of elective employee contributions.
Under government regulations in Australia, the Company was required to contribute 9% of Australian employees’ gross wages, as defined, to an approved superannuation fund selected by each employee.
The Company contributed a total of $153,000 for fiscal 2010, $155,000 for fiscal 2009 and $210,000 for fiscal 2008 in connection with these retirement plans.
| 12. | Income Taxes |
The components of income tax (expense) benefit are as follows:
| Year Ended June 30, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| U.S. operations: |
||||||||||||
| Current income tax expense (benefit) |
$ | 156 | $ | (19 | ) | $ | 52 | |||||
| Deferred income tax benefit |
— | (94 | ) | (535 | ) | |||||||
| 156 | (113 | ) | (483 | ) | ||||||||
| Non-U.S. operations: |
||||||||||||
| Current income tax benefit |
(133 | ) | (838 | ) | — | |||||||
| Deferred income tax benefit |
— | — | — | |||||||||
| (133 | ) | (838 | ) | — | ||||||||
| Income tax expense (benefit) |
$ | 23 | $ | (951 | ) | $ | (483 | ) | ||||
F-22
Table of Contents
The components of income (loss) before income taxes are as follows:
| Year Ended June 30, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| U.S. operations |
$ | 12,353 | $ | 1,183 | $ | (47,969 | ) | |||||
| Non-U.S. operations |
(3,577 | ) | (4,645 | ) | (28,184 | ) | ||||||
| Income (loss) before income taxes |
$ | 8,776 | $ | (3,462 | ) | $ | (76,153 | ) | ||||
The difference between Company’s expected income tax expense (benefit), as computed by applying the statutory U.S. federal tax rate of 34% to income (loss) before income taxes, and actual tax is reconciled in the following table:
| Year Ended June 30, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Income tax expense (benefit) at statutory rate |
$ | 2,984 | $ | (1,177 | ) | $ | (25,892 | ) | ||||
| State income taxes, net of federal benefit |
953 | 20 | (2,878 | ) | ||||||||
| Non-U.S. income tax rate differential |
180 | 218 | 312 | |||||||||
| Research and development tax credits |
(132 | ) | (838 | ) | — | |||||||
| Goodwill impairment |
— | — | 21,556 | |||||||||
| Changes in valuation allowance, including revisions of prior year estimates |
(4,219 | ) | 771 | 6,339 | ||||||||
| Other, net |
257 | 55 | 80 | |||||||||
| Income tax expense (benefit) |
$ | 23 | $ | (951 | ) | $ | (483 | ) | ||||
The components of deferred income taxes are as follows:
| June 30, | ||||||
| 2010 | 2009 | |||||
| Deferred tax assets: |
||||||
| Net operating loss carryforwards |
$ | 21,652 | $ | 28,073 | ||
| Deferred revenue |
1,300 | 1,007 | ||||
| Stock-based compensation |
842 | 309 | ||||
| Provision for losses on note receivable |
520 | 520 | ||||
| Other |
590 | 719 | ||||
| Total deferred tax assets |
24,904 | 30,628 | ||||
| Deferred tax liabilities: |
||||||
| Intangible assets |
7,581 | 9,086 | ||||
| Deferred tax assets, net |
17,323 | 21,542 | ||||
| Valuation allowance |
17,545 | 21,764 | ||||
| Net deferred tax liability |
$ | 222 | $ | 222 | ||
The valuation allowances generally reflect limitations on the Company’s ability to use the tax attributes and reduce the value of such attributes to the more likely than not realizable amount. The valuation allowance decreased by approximately $4.2 million during fiscal 2010 and $481,000 during fiscal 2009.
The Company has tax loss carry forwards in its individual tax jurisdictions. At June 30, 2010, the Company had U.S. federal net operating loss carry forwards of approximately $39.2 million which expire at various dates between calendar years 2023 and 2028. The utilization of certain of these loss carry forwards may be limited by Section 382 of the Internal Revenue Code as a result of historical or future changes in the Company’s ownership.
F-23
Table of Contents
At June 30, 2010, the Company had state net operating loss carry forwards of approximately $14.8 million which expire in 2012 and 2013. During fiscal 2010, approximately $487,000 of state net operating loss carry forwards expired unutilized. Additionally, at June 30, 2010 the Company had loss carry forwards in the U.K. of £17.6 million (approximately $26.6 million). During fiscal 2010, the Company recognized a current income tax benefit of $132,000 related to foreign research and development tax credits earned by its U.K. subsidiary.
The Company’s U.S. federal income tax returns for calendar years 2002 through 2009 remain subject to examination by the Internal Revenue Service. The Company’s U.K. tax returns for fiscal 2006 to 2009 remain subject to examination. The Australian tax returns for the former parent company for fiscal 2004 through 2008 remain subject to examination.
Through June 30, 2010, the Company had no significant unrecognized tax benefits in its consolidated statements of operations and no material unrecognized tax benefits in its consolidated balance sheets as of June 30, 2010 or 2009.
As of June 30, 2010 and 2009, the Company had no accrued penalties or interest related to uncertain tax positions.
| 13. | Fair Value Measurements |
The Company’s financial assets and liabilities measured at fair value include cash equivalents, marketable securities and derivative liabilities. Fair value measurements may be classified based on the degree of subjectivity associated with the inputs to fair valuation of these assets and liabilities using the following three levels of hierarchy:
| • | Level 1—Inputs are quoted prices in active markets that are accessible at the measurement date for identical assets and liabilities. The fair value hierarchy gives the highest priority to Level 1 inputs. |
| • | Level 2—Inputs are observable prices that are based on inputs not quoted on active markets, but corroborated by market data. |
| • | Level 3—Inputs are unobservable estimates that are supported by little or no market activity and require the Company to develop its own assumptions about how market participants would price the assets or liabilities. The fair value hierarchy gives the lowest priority to Level 3 inputs. |
The Company’s each equivalents and marketable securities are classified within Level 1 or Level 2. This is because the cash equivalents and marketable securities are valued using quoted market price or alternative pricing sources and models utilizing market observable inputs. The Company’s derivative liabilities were classified as Level 3 and valued using the Black-Scholes model.
F-24
Table of Contents
The following table summarizes the Company’s assets and liabilities carried at fair value measured on a recurring basis at June 30, 2010 and 2009 by valuation hierarchy:
| June 30, 2010 | ||||||||||||
| Description |
Total Carrying Value |
Quoted prices in active markets (Level 1) |
Significant other observable inputs (Level 2) |
Significant unobservable inputs (Level 3) | ||||||||
| Assets: |
||||||||||||
| Cash equivalents |
$ | 15,055 | $ | 15,055 | $ | — | $ | — | ||||
| Marketable securities |
2,051 | 1,302 | 749 | — | ||||||||
| $ | 17,106 | $ | 16,357 | $ | 749 | $ | — | |||||
| Liabilities: |
||||||||||||
| Derivative liabilities |
$ | 1,310 | $ | — | $ | — | $ | 1,310 | ||||
| June 30, 2009 | ||||||||||||
| Description |
Total Carrying Value |
Quoted prices in active markets (Level 1) |
Significant other observable inputs (Level 2) |
Significant unobservable inputs (Level 3) | ||||||||
| Assets: |
||||||||||||
| Cash equivalents |
$ | 6,124 | $ | 6,124 | $ | — | $ | — | ||||
| Liabilities: |
||||||||||||
| Derivative liabilities |
$ | 971 | $ | — | $ | — | $ | 971 | ||||
The Company’s derivative liabilities were classified as Level 3 and valued using the Black-Scholes model. At June 30, 2010 and 2009, the fair values were derived by applying the following assumptions:
| June 30, | ||||
| 2010 | 2009 | |||
| Expected term (in years) |
0.50 - 2.04 | 1.50 - 3.04 | ||
| Stock volatility |
95% | 95% | ||
| Risk-free interest rate |
0.22% - 0.63% | 0.84% - 1.66% | ||
| Expected dividends |
0% | 0% | ||
The reconciliation of the Company’s liabilities measured at fair value on a recurring basis using unobservable inputs (Level 3) is as follows:
| June 30, | |||||||
| 2010 | 2009 | ||||||
| Balance at beginning of year |
$ | 971 | $ | 1,930 | |||
| Change in fair value of derivatives—other (expense) income |
(339 | ) | 959 | ||||
| Balance at end of year |
$ | 1,310 | $ | 971 | |||
| 14. | Commitments and Contingencies |
Operating Leases
The Company leases its office and research laboratory space in Watertown, Massachusetts through April 6, 2011, with a three-year option to renew. In addition to base rent, the lease agreement requires the Company to pay for utilities, taxes, insurance, maintenance and other operating expenses. The Company leases laboratory and office space in Malvern, U.K. through June 2012, subject to a 6-month advance notice of cancellation by either party at any time. The Company also leases certain office equipment under operating lease agreements that expire through calendar year 2012.
F-25
Table of Contents
At June 30, 2010, the Company’s total future minimum lease payments under non-cancellable operating leases were as follows:
| Fiscal Year: |
|||
| 2011 |
317 | ||
| 2012 |
7 | ||
| 2013 |
6 | ||
| Thereafter |
— | ||
| $ | 330 | ||
Rent expense related to operating leases charged to operations was approximately $449,000 for fiscal 2010, $463,000 for fiscal 2009 and $529,000 for fiscal 2008.
Litigation
The Company is subject to various routine legal proceedings and claims incidental to its business, which management believes will not have a material adverse effect on the Company’s financial position, results of operations or cash flows.
| 15. | Segment and Geographic Area Information |
| (a) | Business Segment |
The Company operates in only one business segment, being the biotechnology sector. Operating segments are identified as components of an enterprise about which separate discrete financial information is available for evaluation by the chief operating decision maker in making decisions regarding resource allocation and assessing performance. The chief operating decision maker made such decisions and assessed performance at the company level, as one segment.
| (b) | Geographic Area Information |
The following table summarizes the Company’s revenues and long-lived assets by geographic area:
| Revenues | Long-lived assets | |||||||||||||||||
| 2010 | 2009 | 2008 | 2010 | 2009 | 2008 | |||||||||||||
| United States |
$ | 22,932 | $ | 12,085 | $ | 3,476 | $ | 29 | $ | 36 | $ | 68 | ||||||
| United Kingdom |
121 | 77 | — | 14 | 30 | 405 | ||||||||||||
| Consolidated |
$ | 23,053 | $ | 12,162 | $ | 3,476 | $ | 43 | $ | 66 | $ | 473 | ||||||
| 16. | Related Party Transactions |
As of June 30, 2010, Pfizer owns approximately 10.0% of the Company’s outstanding shares, making it the Company’s largest shareholder. The Company received research and development support payments from Pfizer under the Pfizer Agreement of $2.0 million during fiscal 2010, $1.5 million during fiscal 2009 and $1.5 million during fiscal 2008. Cumulative research payments received from Pfizer through June 30, 2010, totaling $5.75 million, have been classified in non-current deferred revenue. (See Notes 3 and 9)
F-26
Table of Contents
| 17. | Quarterly Financial Data (unaudited) |
The following table summarizes the quarterly results of operations for the years ended June 30, 2010 and 2009:
| Fiscal Year 2010 | ||||||||||||||||||
| First Quarter Ended September 30, 2009 |
Second Quarter Ended December 31, 2009 |
Third Quarter Ended March 31, 2010 |
Fourth Quarter Ended June 30, 2010 (1) |
Year Ended June 30, 2010 (1) | ||||||||||||||
| Total revenues |
$ | 3,383 | $ | 3,433 | $ | 515 | $ | 15,722 | $ | 23,053 | ||||||||
| Loss (income) from operations |
(107 | ) | (113 | ) | (2,863 | ) | 12,174 | 9,091 | ||||||||||
| Net (loss) income |
(1,591 | ) | (24 | ) | (2,705 | ) | 13,073 | 8,753 | ||||||||||
| Net (loss) income per share: |
||||||||||||||||||
| Basic |
$ | (0.09 | ) | $ | — | $ | (0.15 | ) | $ | 0.71 | $ | 0.48 | ||||||
| Diluted |
$ | (0.09 | ) | $ | — | $ | (0.15 | ) | $ | 0.68 | $ | 0.46 | ||||||
| Weighted average common shares: |
||||||||||||||||||
| Basic |
18,294 | 18,317 | 18,480 | 18,531 | 18,405 | |||||||||||||
| Diluted |
18,294 | 18,317 | 18,480 | 19,217 | 18,895 | |||||||||||||
| (1) | Results for the fourth quarter of fiscal 2010 included revenue related to the payment in full by Alimera of a $15.0 million conditional note (see Note 3). |
| Fiscal Year 2009 | ||||||||||||||||||||
| First Quarter Ended September 30, 2008 |
Second Quarter Ended December 31, 2008 |
Third Quarter Ended March 31, 2009 |
Fourth Quarter Ended June 30, 2009 |
Year Ended June 30, 2009 |
||||||||||||||||
| Total revenues |
$ | 2,806 | $ | 2,970 | $ | 3,163 | $ | 3,223 | $ | 12,162 | ||||||||||
| Loss from operations |
(2,379 | ) | (1,421 | ) | (781 | ) | (55 | ) | (4,636 | ) | ||||||||||
| Net loss |
(471 | ) | (870 | ) | (636 | ) | (534 | ) | (2,511 | ) | ||||||||||
| Basic and diluted net loss per share |
$ | (0.03 | ) | $ | (0.05 | ) | $ | (0.03 | ) | $ | (0.03 | ) | $ | (0.14 | ) | |||||
| Weighted average common shares: |
||||||||||||||||||||
| Basic and diluted |
18,262 | 18,262 | 18,262 | 18,264 | 18,263 | |||||||||||||||
F-27