As filed with the Securities and Exchange Commission on
April 8, 2010
Registration
No. 333-
UNITED STATES SECURITIES AND
EXCHANGE COMMISSION
Washington, D.C.
20549
Form S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
KIPS BAY MEDICAL,
INC.
(Exact name of registrant as
specified in its charter)
| |
|
|
|
|
Delaware
(State or other jurisdiction
of
incorporation or organization)
|
|
3841
(Primary Standard
Industrial
Classification Code Number)
|
|
20-8947689
(I.R.S. Employer
Identification No.)
|
3405 Annapolis Lane North,
Suite 200
Minneapolis, Minnesota 55447
(763) 235-3540
(Address, including
zip code, and telephone number, including area code, of
registrant’s principal executive offices)
Manny Villafaña
Chairman and Chief Executive Officer
Kips Bay Medical, Inc.
3405 Annapolis Lane North, Suite 200
Minneapolis, Minnesota 55447
(763) 235-3540
(Name, address,
including zip code, and telephone number, including area code,
of agent for service)
Copies to:
| |
|
|
Robert K. Ranum, Esq.
Thomas Steichen, Esq.
Alexander Rosenstein, Esq.
Fredrikson & Byron, P.A.
200 South Sixth Street, Suite 4000
Minneapolis, Minnesota 55402
(612) 492-7000
|
|
Frank F. Rahmani, Esq.
John T. McKenna, Esq.
Cooley Godward Kronish, LLP
3175 Hanover Street
Palo Alto, California 94304
(650) 843-5000
|
Approximate date of commencement of proposed sale to the
public: As soon as practicable after the
effective date of this registration statement.
If any of the securities being registered on this Form are to be
offered on a delayed or continuous basis pursuant to
Rule 415 under the Securities Act of 1933, as amended,
check the following
box. o
If this Form is filed to register additional securities for an
offering pursuant to Rule 462(b) under the Securities Act,
check the following box and list the Securities Act registration
statement number of the earlier effective registration statement
for the same
offering. o
If this Form is a post-effective amendment filed pursuant to
Rule 462(c) under the Securities Act, check the following
box and list the Securities Act registration statement number of
the earlier effective registration statement for the same
offering. o
If this Form is a post-effective amendment filed pursuant to
Rule 462(d) under the Securities Act, check the following
box and list the Securities Act registration statement number of
the earlier effective registration statement for the same
offering. o
Indicate by check mark whether the registrant is a large
accelerated filer, an accelerated filer, a non-accelerated
filer, or a smaller reporting company. See the definitions of
“large accelerated filer,” “accelerated
filer” and “smaller reporting company” in
Rule 12b-2
of the Exchange Act. (Check one):
| |
|
|
|
|
|
|
|
Large accelerated
filer o
|
|
Accelerated
filer o
|
|
Non-accelerated
filer o
(Do not check if a smaller reporting company)
|
|
Smaller reporting
company þ
|
CALCULATION
OF REGISTRATION FEE
| |
|
|
|
|
|
|
|
|
|
|
Proposed Maximum
|
|
|
|
Title of Each Class of
|
|
|
Aggregate
|
|
|
Amount of
|
|
Securities to be Registered
|
|
|
Offering Price(1)(2)
|
|
|
Registration Fee
|
|
Common stock, par value $0.01 per share
|
|
|
$57,500,000
|
|
|
$4,100
|
|
|
|
|
|
|
|
|
|
|
|
|
(1)
|
|
In accordance with
Rule 457(o) under the Securities Act of 1933, the number of
shares being registered and the proposed maximum offering price
per share are not included in this table. Includes the offering
price of additional shares that the underwriters have the option
to purchase. |
| |
|
(2)
|
|
Estimated solely for the purpose
of computing the registration fee pursuant to Rule 457(o) under
the Securities Act. |
The Registrant hereby amends this Registration Statement on
such date or dates as may be necessary to delay its effective
date until the registrant shall file a further amendment which
specifically states that this Registration Statement shall
thereafter become effective in accordance with Section 8(a)
of the Securities Act of 1933, as amended, or until the
Registration Statement shall become effective on such date as
the Commission, acting pursuant to said Section 8(a), may
determine.
The
information in this preliminary prospectus is not complete and
may be changed. We may not sell these securities until the
registration statement filed with Securities and Exchange
Commission is effective. This preliminary prospectus is not an
offer to sell these securities and is not soliciting an offer to
buy these securities in any state where the offer or sale is not
permitted.
|
SUBJECT
TO COMPLETION, DATED APRIL 8, 2010
Preliminary
Prospectus
Shares
Kips
Bay Medical, Inc.
Common
Stock
We are
offering shares
of our common stock. This is our initial public offering, and no
public market currently exists for our common stock. We expect
that the initial public offering price will be between
$ and
$ per common share. We have
applied for listing of our common stock on the NASDAQ Global
Market under the symbol “KIPS.”
Investing in our common stock involves a high degree of risk.
Please read “Risk Factors” beginning on
page 6.
Neither the Securities and Exchange Commission nor any state
securities commission has approved or disapproved of these
securities or determined if this prospectus is truthful or
complete. Any representation to the contrary is a criminal
offense.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
PER SHARE
|
|
TOTAL
|
|
|
|
Public Offering Price
|
|
$
|
|
|
|
$
|
|
|
|
Underwriting Discounts and Commissions
|
|
$
|
|
|
|
$
|
|
|
|
Proceeds to Kips Bay Medical, Inc. (Before Expenses)
|
|
$
|
|
|
|
$
|
|
|
|
|
Delivery of the shares of common stock is expected to be made on
or
about ,
2010. We have granted the underwriters an option for a period of
30 days to purchase, on the same terms and conditions set
forth above, up to an
additional shares
of our common stock to cover overallotments. If the underwriters
exercise the option in full, the total underwriting discounts
and commissions payable by us will be
$ and the total proceeds to us,
before expenses, will be $ .
Sole Book-Running
Manager
Jefferies &
Company
Prospectus
dated ,
2010
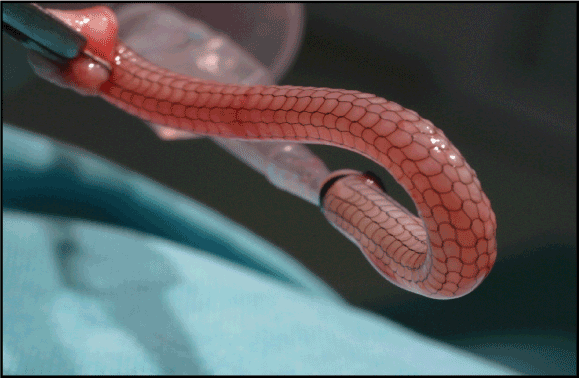
Our External Saphenous Vein Support Technology, or eSVS MESH, is a nitinol mesh sleeve that
is placed over the saphenous vein graft during
coronary artery bypass grafting, or
CABG surgery. We believe the use of our eSVS MESH with saphenous vein grafts in CABG surgery could improve
the long-term outcome of CABG procedures, including improved openness and improved
blood flow through the saphenous vein graft, resulting in a reduced need for costly
and potentially complicated reoperations or revascularization procedures.
Caution: Investigational device. Limited by law to investigational use and not approved for
commercial sale by any regulatory agency.
Table of
Contents
Until ,
2010 (25 days after the date of this prospectus), all
dealers that buy, sell, or trade the common shares, whether or
not participating in this offering, may be required to deliver a
prospectus. This is in addition to the dealers’ obligation
to deliver a prospectus when acting as underwriters and with
respect to their unsold allotments or subscriptions.
We have not authorized anyone to give any information or to make
any representations other than those contained in this
prospectus. Do not rely upon any information or representations
made outside of this prospectus. This prospectus is not an offer
to sell, and it is not soliciting an offer to buy, (1) any
securities other than our common shares or (2) our common
shares in any circumstances in which our offer or solicitation
is unlawful. The information contained in this prospectus may
change after the date of this prospectus. Do not assume after
the date of this prospectus that the information contained in
this prospectus is still correct.
Prospectus
Summary
This summary highlights certain information about us, this
offering and selected information contained in the prospectus.
This summary is not complete and does not contain all of the
information that you should consider before deciding whether to
invest in our common stock. For a more complete understanding of
our company and this offering, we encourage you to read and
consider the more detailed information in the prospectus,
including “Risk Factors” and the financial statements
and related notes. Unless we specify otherwise, all references
in this prospectus to “Kips Bay,” “we,”
“our,” “us” and “our company”
refer to Kips Bay Medical, Inc.
Overview
We are a medical device company focused on developing,
manufacturing and commercializing our innovative external
saphenous vein support technology, or eSVS MESH, for use in
coronary artery bypass grafting, or CABG, surgery. Our eSVS MESH
is a nitinol mesh sleeve that, when placed over a saphenous vein
graft during CABG surgery, is designed to improve the structural
characteristics and long-term performance of the vein graft.
Based on the data collected in a 90 patient multi-center
clinical trial conducted outside the United States, we submitted
our CE Mark application in February 2010. We expect to receive
CE Mark approval and begin marketing our eSVS MESH in select
European Union markets in the second half of 2010. The United
States Food and Drug Administration, or FDA, is reviewing our
application for an investigational device exemption, or IDE,
which, if granted, will allow us to begin clinical trials of our
eSVS MESH in the United States. We anticipate beginning
enrollment in a United States IDE trial in the second half of
2010. Currently, we have no products available for commercial
sale and to date we have not generated any revenue from the sale
of products.
Industry
Background
According to the American Heart Association, approximately
17.6 million people in the United States have coronary
artery disease, and approximately 587,000 people in the
United States die each year as a result of the disease. In
addition, according to a 2007 World Health Organization report,
approximately 7.2 million people worldwide died of coronary
heart disease in 2002. Physicians and patients may select among
a variety of treatments to address coronary artery disease,
including pharmaceutical therapy, balloon angioplasty, stenting
with bare metal or drug-eluting stents, and CABG procedures,
with the selection often depending upon the stage of the disease
and the age of the patient.
CABG is one of the most commonly performed surgeries in the
United States, with the American Heart Association estimating
that 448,000 were performed in the United States in 2006. In
addition, the Millennium Research Group, an independent market
research firm, estimates that there will be 165,000 CABG
procedures in Europe per year by 2013. An independent study
comparing CABG and implantation of drug-eluting stents shows
that CABG is the more effective long-term treatment for coronary
artery disease, achieving the best long-term patient outcomes as
measured by survival rate and need for re-intervention
12 months after surgery. Moreover, patients with severe and
multi-vessel coronary artery disease often cannot be effectively
treated with methods other than CABG. According to the
Millennium Research Group, moderate growth in CABG procedures is
expected in the United States through 2012 and in Europe through
2013, largely due to the increase in procedure volumes caused by
rising rates of coronary disease and the need for repeat
revascularizations.
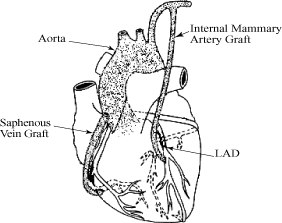
In CABG procedures, surgeons harvest blood vessels, including
the internal mammary artery from the chest and the saphenous
vein from the leg, and attach the harvested vessels to bypass,
or provide blood flow around, blocked coronary arteries. The
effectiveness of the procedure, however, is often limited by the
failure rate of saphenous vein grafts, which has been shown in
various studies to range from 6% to 30% one year after surgery
and 60% ten years after surgery. Failure of these grafts,
typically evidenced by partial or complete blockage and reduced
or stopped blood flow, can lead to the need for further coronary
interventions up to and including additional CABG procedures. We
believe the use of our eSVS MESH with saphenous vein grafts in
CABG surgery could improve the long-term outcome of CABG
procedures, including improved openness, or patency, and
improved blood flow through the saphenous vein graft, resulting
in a reduced need for costly and potentially complicated
reoperations or revascularization procedures.
1
According to results published in the European Journal of
Cardio-Thoracic Surgery in 2009, each CABG procedure involves an
average of 3.3 bypass grafts, typically consisting of the left
internal mammary artery, or LIMA, for one graft and the
saphenous vein for the remaining 2.3 grafts per procedure. Some
of the main advantages of using the saphenous vein include its
ease of accessibility, its ease of handling, and the number of
grafts, typically three, that can be constructed from a single
vein. Despite these advantages and the widespread use of
saphenous veins in CABG surgery, saphenous vein grafts fail more
frequently than LIMA grafts due to differences in structure and
size of saphenous vein grafts as compared to LIMA grafts. Unlike
the LIMA, which is a thick-walled artery intended to handle the
high pressure blood flow from the heart, saphenous veins are
thin-walled vessels that are intended for a low-pressure venous
environment. Saphenous veins are also typically larger than the
coronary arteries to which they are attached and this difference
in size slows blood flow, adding stress to the vessel wall and
increasing the risk of thrombosis, or blood clotting. When the
vein grafts used to bypass a blocked artery are exposed to the
high pressure of arterial flow, there is significant stress on
the thin wall of the veins. The vein responds to this injury by
causing its walls to thicken in a manner that often leads to
failure of the bypass graft.
Our
Solution
Our eSVS MESH is designed to address these limitations by
providing the vein graft with physiological attributes similar
to those of an artery by constricting the vein and preventing
expansion of the vein graft and resulting injury due to
increased pressure.
We believe the key benefits of our eSVS MESH technology include:
|
|
|
| |
•
|
Structural support designed to inhibit vessel expansion and
resulting damage to the vessel, which can prevent a thickening
of the vessel wall over time, or hyperplasia, and resulting
graft failure.
|
| |
| |
•
|
Radial constriction designed to cause the diameter of the graft,
or lumen, to be consistent in size and more closely match the
diameter of the target coronary artery to which it is attached,
thereby increasing blood flow velocities, reducing the potential
for clot formation, and inhibiting vein wall thickening.
|
| |
| |
•
|
Compatibility with current CABG procedures, including on-pump or
off-pump procedures, and open or endoscopic vein harvest
methods. Except for the placement of our eSVS MESH on the
saphenous vein graft, the surgical steps to use a saphenous vein
graft with our eSVS MESH are the same as would be performed for
any coronary artery bypass procedure utilizing unsupported
saphenous vein grafts. We do not expect, nor have we seen, a
significant increase in CABG procedure time due to eSVS MESH use.
|
We are also pursuing additional applications for our eSVS MESH,
including applications for use in peripheral artery bypass
surgery, for use with coronary allografts, and for use in
arteriovenous, or AV, fistula dialysis applications.
Clinical
Development of Our eSVS MESH
We have completed enrollment in a 90 patient multi-center
clinical trial conducted outside the United States. The primary
effectiveness endpoint of this trial is statistical
non-inferiority of the patency of eSVS MESH vessels as compared
to control vessels at nine-months post-implant. Effectiveness
data, which is based on angiographic patency, is being collected
at this time. Preliminary safety data has indicated that our
eSVS MESH and implant procedure do not result in an increase in
patient complications during or after surgery. We completed
enrollment in this trial in July 2009, and as of March 1,
2010, all 90 patients have been implanted for six months or
more, 83 patients have been implanted for nine months or
more, and 50 patients have been implanted for 12 months or
more. This trial formed the basis for our CE Mark application
submitted in February 2010.
We expect to receive CE Mark approval and to begin marketing our
eSVS MESH in select European Union markets in the second half of
2010. The FDA is reviewing our application for an IDE, which, if
granted, will allow us to begin clinical trials of our eSVS MESH
in the United States. We anticipate beginning enrollment in a
United States IDE trial in the second half of 2010. We could be
delayed by adverse clinical results or regulatory complications,
and we may never receive marketing approval.
2
Our
Strategy
Our objective is to achieve significant market adoption of our
eSVS MESH technology in CABG and other vascular applications.
Key elements of our strategy to achieve this objective include
the following:
|
|
|
| |
•
|
Work with respected medical centers and key thought leaders to
demonstrate and communicate the potential benefits of our eSVS
MESH.
|
| |
| |
•
|
Commercialize our eSVS MESH in select European markets.
|
| |
| |
•
|
Obtain regulatory approval and commercialize our eSVS MESH in
the United States.
|
| |
| |
•
|
Conduct trials to expand indications for our eSVS MESH.
|
Risks Associated
with Our Business
Our business is subject to a number of risks discussed under the
heading “Risk Factors” and elsewhere in this
prospectus, including, but not limited to, the following:
|
|
|
| |
•
|
We have a limited operating history, expect future losses, and
may be unable to achieve or maintain profitability.
|
| |
| |
•
|
We may be unable to successfully complete our clinical trials
and commercialize our eSVS MESH, or we may experience
significant delays in doing so.
|
| |
| |
•
|
We may be unable to obtain market acceptance of our eSVS MESH.
|
| |
| |
•
|
Third-party payors may not provide sufficient coverage or
reimbursement to healthcare providers for the use of our eSVS
MESH.
|
| |
| |
•
|
We may be unable to protect our intellectual property rights,
and claims of infringement or misappropriation of the
intellectual property rights of others could prohibit us from
commercializing our eSVS MESH.
|
| |
| |
•
|
We will be subject to U.S. and foreign government
regulation.
|
You should carefully consider these factors, as well as all of
the other information set forth in this prospectus, before
making an investment decision.
Company
Information
We were incorporated in Delaware in May 2007. Our principal
executive offices are located at 3405 Annapolis Lane North,
Suite 200, Minneapolis, MN 55447. Our telephone number is
(763) 235-3540,
and our website is www.kipsbaymedical.com. The information
contained in or connected to our website is not incorporated by
reference into, and should not be considered part of, this
prospectus. Kips Bay
Medical®,
eSVS®,
the Kips Bay Medical logo, and other trademarks or service marks
of Kips Bay Medical, Inc. appearing in this prospectus are our
property. Trade names, trademarks, and service marks of other
companies appearing in this prospectus are the property of the
respective holders.
3
The
Offering
|
|
|
|
Common stock offered by us |
|
shares |
| |
|
Common stock to be outstanding immediately after this offering |
|
shares |
Use of
Proceeds
We expect the net proceeds to us from this offering will be
approximately $ million,
after deducting the estimated underwriting discounts and
commissions and estimated offering expenses. We intend to use
the net proceeds from this offering to seek regulatory approval
to market our eSVS MESH in the United States and abroad,
including human clinical trials in the United States; develop
and test additional applications of our eSVS MESH; make certain
milestone payments for our acquired intellectual property; and
for working capital and general corporate purposes. See
“Use of Proceeds” on page 22 of this prospectus.
NASDAQ Global
Market Listing
We intend to apply for the listing of our common stock on the
Nasdaq Global Market under the symbol “KIPS.”
Risk
Factors
Investing in our common stock involves a high degree of risk.
See “Risk Factors” on page 6 of this prospectus.
Outstanding
Shares
The number of shares of our common stock that will be
outstanding immediately after this offering is based on
13,581,791 shares outstanding as of March 1, 2010 and
excludes:
|
|
|
| |
•
|
783,000 shares of common stock issuable upon the exercise
of outstanding stock options as of March 1, 2010 at a
weighted average exercise price of $4.01 per share; and
|
| |
| |
•
|
1,216,000 additional shares of common stock reserved and
available for future issuances under our 2007 Long-Term
Incentive Plan.
|
Except as otherwise noted, all information in this prospectus
assumes no exercise of the underwriters’ option to purchase
additional shares.
4
Summary Financial
Data
The following table summarizes our financial data. We have
derived the statements of operations data for the period from
May 1, 2007 (inception) through December 31, 2007, the
years ended December 31, 2008 and 2009, and the period from
May 1, 2007 (inception) through December 31, 2009, and
the balance sheet data as of December 31, 2009 from our
audited financial statements appearing elsewhere in this
prospectus. Our historical results are not necessarily
indicative of the results that may be experienced in the future.
You should read this data in conjunction with “Selected
Financial Data,” “Management’s Discussion and
Analysis of Financial Condition and Results of Operations,”
and our financial statements and related notes, all included
elsewhere in this prospectus.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from May 1,
|
|
|
|
|
|
|
|
|
Period from May 1,
|
|
|
|
|
2007 (Date of
|
|
|
|
|
|
|
|
|
2007 (Date of
|
|
|
|
|
Inception) to
|
|
|
|
|
|
|
|
|
Inception) to
|
|
|
|
|
December 31,
|
|
|
Year Ended December 31,
|
|
|
December 31,
|
|
|
|
|
2007
|
|
|
2008
|
|
|
2009
|
|
|
2009
|
|
|
|
|
Statements of Operations Data:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating expenses:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development
|
|
$
|
196
|
|
|
$
|
2,635
|
|
|
$
|
3,004
|
|
|
$
|
5,835
|
|
|
Selling, general and administrative
|
|
|
381
|
|
|
|
754
|
|
|
|
779
|
|
|
|
1,914
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating loss
|
|
|
(577
|
)
|
|
|
(3,389
|
)
|
|
|
(3,783
|
)
|
|
|
(7,749
|
)
|
|
Interest income
|
|
|
65
|
|
|
|
52
|
|
|
|
17
|
|
|
|
134
|
|
|
Interest expense
|
|
|
(164
|
)
|
|
|
(390
|
)
|
|
|
(181
|
)
|
|
|
(735
|
)
|
|
Impairment of available for sale securities
|
|
|
—
|
|
|
|
(85
|
)
|
|
|
—
|
|
|
|
(85
|
)
|
|
Change in fair value of investor stock purchase option
|
|
|
—
|
|
|
|
—
|
|
|
|
610
|
|
|
|
610
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
$
|
(676
|
)
|
|
$
|
(3,812
|
)
|
|
$
|
(3,337
|
)
|
|
$
|
(7,825
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted net loss per share
|
|
$
|
(0.16
|
)
|
|
$
|
(0.62
|
)
|
|
$
|
(0.30
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average common shares
|
|
|
4,106,557
|
|
|
|
6,100,767
|
|
|
|
11,069,342
|
|
|
|
|
|
|
outstanding—basic and diluted
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(In thousands, except share and per
share amounts)
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
As of December 31, 2009
|
|
|
|
|
Actual
|
|
|
As Adjusted(1)
|
|
|
|
|
Balance Sheet Data:
|
|
|
|
|
|
|
|
|
|
Cash, cash equivalents and short term investments
|
|
$
|
3,417
|
|
|
$
|
|
|
|
Working capital
|
|
|
2,226
|
|
|
|
|
|
|
Total assets
|
|
|
3,740
|
|
|
|
|
|
|
Long-term debt, net
|
|
|
—
|
|
|
|
|
|
|
Total stockholders’ equity
|
|
|
2,512
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(In thousands
|
)
|
|
|
|
|
(1) |
|
As adjusted to reflect the sale
of shares
of our common stock in this offering at an assumed initial
public offering price of $ per
share, the midpoint of the range on the front cover of this
prospectus, after deducting the estimated underwriting discounts
and commissions and estimated offering expenses payable by us. A
$1.00 increase or decrease in the assumed initial public
offering price of $ per share
would increase or decrease cash and cash equivalents, working
capital, total assets and total stockholders’ equity by
$ million, assuming the
number of shares offered by us, as set forth on the front cover
of this prospectus, remains the same, and after deducting the
estimated underwriting discounts and commissions and estimated
offering expenses payable by us. |
5
Risk
Factors
You should carefully consider the following information about
risks, together with the other information contained in this
prospectus, before making an investment in our common stock. If
any of the circumstances or events described below actually
arises or occurs, our business, results of operations, cash
flows and financial condition could be harmed. In any such case,
the market price of our common stock could decline, and you may
lose all or part of your investment.
Risks Related to
Our Business and Strategy
We have a
limited operating history, expect future losses, and may be
unable to achieve or maintain profitability.
We were founded on May 1, 2007 and to date we have engaged
primarily in development of and initial clinical trials of our
external saphenous vein support system, or eSVS MESH.
Accordingly, we have limited operating history on which to base
an evaluation of our business and prospects. As of
December 31, 2009, we had an accumulated deficit of
$9.2 million. We have incurred net losses in each year
since our inception, and we expect to continue to incur
operating losses for the foreseeable future. These losses, among
other things, have had and will continue to have an adverse
effect on our stockholders’ equity and working capital.
Because of the numerous risks and uncertainties associated with
developing medical devices, we are unable to predict the extent
of any future losses or when we will become profitable, if at
all. To date, we have not generated any product revenues and we
have financed our operations and internal growth primarily
through private placements of equity securities and convertible
promissory notes. Our prospects must be considered in light of
the significant risks, expenses, and difficulties frequently
encountered by medical device companies in their early stage of
development. We may not be successful in addressing the risks we
will encounter, and our failure to do so would likely harm our
business and our ability to continue to operate.
If we are
unable to successfully complete our clinical trials and
commercialize our eSVS MESH in Europe, the United States, or
other major markets or experience significant delays in doing
so, our ability to generate revenue will be significantly
delayed and our business will be harmed.
Our time and financial resources since our inception have
largely been devoted to the development of our eSVS MESH. We
have conducted only one human clinical trial of our eSVS MESH
that was conducted outside the United States, and as of the date
of this prospectus, our eSVS MESH has been implanted in only
90 patients. Our ability to generate revenues depends on
the receipt of regulatory approvals and the successful
development and commercialization of our eSVS MESH in select
markets in Europe, in the United States, and in other major
markets. If we are unable to prove the safety and effectiveness
of our eSVS MESH through clinical trials, we will not receive
marketing approvals from the FDA or foreign health regulatory
authorities, and we will be unable to sell our eSVS MESH. We
have no other products ready for clinical testing or
commercialization; therefore, our ability to remain in business
would be doubtful if our eSVS MESH is not proven to be safe and
effective. Even if our eSVS MESH receives CE Mark approval in
the European Union, if the data from our clinical trials is not
satisfactory, we may not proceed with our planned filing of
applications for regulatory approvals in the United States or
other major markets, or we may be forced to delay the filings.
Even if we file an application for approval with satisfactory
clinical data, the FDA or foreign regulatory authorities may not
accept our filing, or may request additional information,
including data from additional clinical trials. Delays in
collecting or analyzing our clinical trial data could result in
delays in filing regulatory applications with the FDA or other
regulatory authorities. The FDA or foreign regulatory
authorities may also approve our eSVS MESH for very limited
purposes with many restrictions on its use or in limited sizes,
may delay approvals, or ultimately may not grant marketing
approval for our eSVS MESH. Even if we do receive CE Mark, FDA,
or other foreign regulatory approval, we may be unable to
successfully commercialize our eSVS MESH in the European Union,
the United States, or other major markets, and our ability to
generate revenue will be significantly impaired.
Our
success depends on the coronary bypass graft market and the
superior outcomes of coronary bypass surgery over competitive
procedures, and such superior outcomes may not
continue.
Physicians treat coronary artery disease with methods other than
CABG procedures, including interventional
6
techniques such as balloon angioplasty with or without the use
of stents, pharmaceuticals, atherectomy catheters, and lasers.
Several of these alternative treatments are widely accepted in
the medical community and have a long history of use. In
addition, technological advances may result in improvements in
these alternative treatments or new therapies that produce
superior treatment outcomes as compared to CABG surgery. The
medical device industry is highly competitive and subject to
rapid and profound technological change. Our success depends, in
part, upon physicians continuing to perform a significant number
of CABG procedures and our ability to achieve and maintain a
competitive position in the development of technologies and
products in the coronary artery bypass field. If physicians,
patients, or hospitals opt to use our competitors’
products, our commercial opportunity will be reduced and our
potential revenues will suffer.
The
market acceptance of new medical technologies is uncertain, and
we may be unable to obtain market acceptance of our eSVS
MESH.
Even if our clinical trials demonstrate that the use of our eSVS
MESH provides equivalent or more effective results as compared
to coronary bypass operations using only the unsupported
saphenous vein grafts and if all regulatory approvals are
obtained, the success of our eSVS MESH will depend upon the
acceptance by cardiovascular and cardiothoracic surgeons of our
eSVS MESH as equivalent or better than the current saphenous
vein procedure and other available treatments. We believe that
physicians’ recommendations will be essential for the
development and successful marketing of our eSVS MESH, and
physicians will not begin to use our eSVS MESH unless they
determine that it is a safe and effective alternative to current
treatment methods. The degree of physician and market acceptance
of our eSVS MESH will depend on a number of factors, including:
|
|
|
| |
•
|
the perceived effectiveness of our eSVS MESH relative to its
cost;
|
| |
| |
•
|
the prevalence and severity of any side effects;
|
| |
| |
•
|
potential advantages over alternative treatments;
|
| |
| |
•
|
effectiveness of our sales and marketing efforts;
|
| |
| |
•
|
publication in peer-reviewed medical journals of data regarding
the successful use and longer term clinical benefits of our eSVS
MESH;
|
| |
| |
•
|
development of new products and technologies by our competitors
or new alternative treatments;
|
| |
| |
•
|
regulatory developments related to manufacturing, marketing and
selling our eSVS MESH both within and outside the United States;
|
| |
| |
•
|
perceived liability risks arising out of the use of new products;
|
| |
| |
•
|
the willingness of physicians to adopt new technologies and the
ability of physicians to acquire the skills necessary to use our
eSVS MESH;
|
| |
| |
•
|
the strength of our sales and marketing support; and
|
| |
| |
•
|
the adequacy of third-party coverage or reimbursement.
|
If our eSVS MESH does not achieve an adequate level of
acceptance by physicians, healthcare payors, and patients, we
may not generate meaningful revenue and we may not become
profitable. In addition, we have not yet determined pricing for
our eSVS MESH and our pricing policies could adversely impact
market acceptance of our eSVS MESH as compared to competing
products and treatments. Any of the foregoing factors, or other
factors, could limit or detract from market acceptance of our
eSVS MESH. If our eSVS MESH is not accepted by the market, our
business would be harmed.
We will
be subject to intense competition and the risk of obsolescence
if our competitors develop products superior to our eSVS
MESH.
We face competition from established medical technology,
pharmaceutical and biotechnology companies, as well as from
academic institutions, government agencies, and private and
public research institutions in the United States and abroad.
The industry in which we operate has undergone, and is expected
to continue to undergo, rapid and significant technological
change, and we expect competition to intensify as technical
advances are made. Our competitors may develop and commercialize
medical device or pharmaceutical products that are safer or more
7
effective, have fewer side effects or are less expensive than
coronary artery bypass surgery. For example, we are aware of
companies that are developing various other less-invasive
technologies for treating cardiovascular disease, which could
make our technology obsolete. We also compete in recruiting and
retaining qualified scientific and management personnel,
establishing clinical trial sites and patient registration for
clinical trials, as well as in acquiring technologies and
technology licenses complementary to our programs or
advantageous to our business.
Furthermore, companies with significantly greater financial
resources and expertise in research and development,
manufacturing, pre-clinical testing, conducting clinical trials,
obtaining regulatory approvals and marketing approved products
than we have may be working on products similar to our eSVS
MESH. Our eSVS MESH may not replace current surgical techniques
and other products or techniques may render our eSVS MESH
obsolete. In addition, our distributors will also face
competition from established companies with significantly
greater financial and marketing resources. Our competitors may
produce more advanced products than ours or demonstrate superior
safety of their products. Our ability to effectively compete
depends on our ability to innovate successfully. There are few
barriers that would prevent new or existing competitors from
developing products that compete directly with ours. Demand for
our eSVS MESH could be diminished by equivalent or superior
products and technologies offered by competitors.
Smaller or early-stage companies may also prove to be
significant competitors, particularly through collaborative
arrangements with, or mergers with or acquisitions by, large and
established companies or through the development of novel
products and technologies.
Our competitive position also depends on:
|
|
|
| |
•
|
obtaining any necessary United States or foreign marketing
approvals;
|
| |
| |
•
|
widespread awareness, acceptance and adoption by the
cardiovascular and cardiothoracic markets of our eSVS MESH;
|
| |
| |
•
|
product coverage and reimbursement from third-party payors,
insurance companies and others;
|
| |
| |
•
|
published studies supporting the effectiveness and safety and
long-term clinical benefit of our eSVS MESH;
|
| |
| |
•
|
properly identifying customer needs and delivering new products
or product enhancements to address those needs;
|
| |
| |
•
|
limiting the time required from proof of feasibility to routine
production;
|
| |
| |
•
|
limiting the timing and cost of regulatory approvals;
|
| |
| |
•
|
our ability to attract and retain qualified personnel;
|
| |
| |
•
|
the extent of our patent protection or our ability to otherwise
develop proprietary products and processes;
|
| |
| |
•
|
our ability to maintain adequate manufacturing capacity and to
source the materials and equipment required to manufacture our
eSVS MESH; and
|
| |
| |
•
|
securing sufficient capital resources to expand our research and
development, sales and marketing efforts, and manufacturing
capacity.
|
If our eSVS MESH is not competitive based on these or other
factors, our business would be harmed.
We have
limited manufacturing resources and experience, and if our
manufacturing facilities are unable to provide an adequate
supply of our eSVS MESH, our growth could be limited and our
business could be harmed.
We have limited experience in manufacturing our eSVS MESH and
rely on outside vendors for several materials and processes. We
currently manufacture our eSVS MESH for our clinical trials and
for research and development purposes at our manufacturing
facility in Minnesota. If our existing manufacturing facility
experiences a disruption, we would have no other means of
manufacturing our eSVS MESH until we are able to restore the
manufacturing capability at our current facility or develop
alternative manufacturing facilities.
If we are unable to produce sufficient quantities of our eSVS
MESH for use in our current and planned clinical
8
trials or for commercialization, or if our manufacturing process
yields a substandard product, our regulatory, development and
commercialization efforts would be delayed.
In order to produce our eSVS MESH in the quantities that would
be required for commercialization, we would have to increase, or
“scale up,” the production process over the current
level of production. Manufacturers often encounter difficulties
in scaling up production, including problems involving yields,
controlling and anticipating costs, quality control and
assurance, supply and shortages of qualified personnel. If the
scaled-up
production process is not efficient or produces a product that
does not meet quality and other standards, we may be unable to
meet market demand and our revenues, business and financial
prospects would be adversely affected. The contract vendors with
which we are and will be developing relationships may not have
the ability to produce the quantities of the materials needed
for human clinical trials or commercial sales or may not do so
at prices that allow our eSVS MESH to compete successfully in
the market.
Additionally, any damage to or destruction of our facilities or
our equipment, prolonged power outage or contamination at our
facilities would significantly impair our ability to produce our
eSVS MESH.
We depend
upon third-party suppliers, making us vulnerable to supply
problems and price fluctuations.
We rely on third-party suppliers to provide us certain
components of our eSVS MESH. We depend on these suppliers to
provide us with materials in a timely manner that meet our
quality, quantity and cost requirements. These suppliers may
encounter problems during manufacturing for a variety of
reasons, including unanticipated demand from larger customers,
failure to follow specific protocols and procedures, failure to
comply with applicable regulations, equipment malfunction,
quality or yield problems, and environmental factors, any of
which could delay or impede their ability to meet our demand.
Our reliance on these outside suppliers also subjects us to
other risks that could harm our business, including:
|
|
|
| |
•
|
interruption of supply resulting from modifications to, or
discontinuation of, a supplier’s operations;
|
| |
| |
•
|
delays in product shipments resulting from defects, reliability
issues or changes in components from suppliers;
|
| |
| |
•
|
price fluctuations due to a lack of long-term supply
arrangements for key components with our suppliers;
|
| |
| |
•
|
errors in manufacturing components, which could negatively
impact the effectiveness or safety of our eSVS MESH or cause
delays in shipment of our eSVS MESH;
|
| |
| |
•
|
discontinued production of components, which could significantly
delay our production and sales and impair operating margins;
|
| |
| |
•
|
inability to obtain adequate supplies in a timely manner or on
commercially acceptable terms;
|
| |
| |
•
|
difficulty locating and qualifying alternative suppliers for our
sole-source supplies;
|
| |
| |
•
|
delays in production and sales caused by switching components,
which may require product redesign and new regulatory
submissions;
|
| |
| |
•
|
delays due to evaluation and testing of products from
alternative suppliers and corresponding regulatory
qualifications;
|
| |
| |
•
|
non-timely delivery of components due to our suppliers
manufacturing products for a range of customers; and
|
| |
| |
•
|
inability of suppliers to fulfill orders and meet requirements
because of supplier financial hardships.
|
Other than existing, unfulfilled purchase orders, our suppliers
have no contractual obligations to supply us with, and we are
not contractually obligated to purchase from them, any of our
supplies. Any supply interruption from our suppliers or failure
to obtain additional suppliers for any of the components used in
our eSVS MESH would limit our ability to manufacture our eSVS
MESH and could have a material adverse effect on our business,
financial condition and results of operations. We have no reason
to believe that any of our current suppliers could not be
replaced if they were unable to deliver components to us in a
timely manner or at an acceptable price and level of quality.
However, if we lost one of these suppliers and were unable to
obtain an alternate source on a timely basis or on terms
acceptable to us, our production schedules could be delayed, our
margins could be
9
negatively impacted, and we could fail to meet our
customers’ demand. Our customers will rely upon our ability
to meet committed delivery dates and any disruption in the
supply of key components would adversely affect our ability to
meet these dates and could result in legal action by our
customers, cause us to lose customers or harm our ability to
attract new customers, any of which could decrease our revenue
and negatively impact our growth. In addition, to the extent
that our suppliers use technology or manufacturing processes
that are proprietary, we may be unable to obtain comparable
materials or components from alternative sources.
Manufacturing operations are often faced with a supplier’s
decision to discontinue manufacturing a component, which may
force us to make last time purchases, qualify a substitute part,
or make a design change which may divert engineering time away
from the development of new products.
Quality
issues in our manufacturing processes could delay our clinical
trials and our commercialization.
Even if we are able to contract with manufacturers for key
materials or supplies, we may experience future manufacturing
difficulties. Any difficulties in locating and hiring material
manufacturers or in the ability of manufacturers to supply
materials at the times and in the quantities we need, and at
prices that allow us to compete, could have a material adverse
effect on our business.
The production of our eSVS MESH must occur in a highly
controlled, clean environment to minimize particles and other
yield- and quality-limiting contaminants. In spite of stringent
quality controls, weaknesses in process control or minute
impurities in materials may cause a substantial percentage of
defective products in a lot. In addition, we must meet certain
lot release specifications before our eSVS MESH can be shipped
to our clinical trial sites. If a particular lot fails to meet
lot release specifications, we will not be able to ship that lot
to our clinical trial sites. If we are not able to maintain
stringent quality controls, if contamination problems arise or
if we are not able to meet our lot release specifications, our
clinical trials could be delayed, which would harm our business
and our results of operations.
Our
business is subject to risks relating to operating
internationally.
As part of our product development and regulatory strategy, we
intend to market our eSVS MESH internationally following CE Mark
approval. There are a number of risks associated with conducting
business internationally, including:
|
|
|
| |
•
|
potential differences in treatment protocols and methods across
the markets in which we expect to market our eSVS MESH;
|
| |
| |
•
|
potential differences in reimbursement levels and the
requirements necessary to obtain such reimbursement;
|
| |
| |
•
|
general economic and political conditions in the markets in
which we operate;
|
| |
| |
•
|
potential international conflicts, including terrorist acts;
|
| |
| |
•
|
potential increased costs associated with overlapping tax
structures;
|
| |
| |
•
|
potential trade restrictions, exchange controls and legal
restrictions on the repatriation of funds into the United States;
|
| |
| |
•
|
difficulties and costs associated with staffing and managing
foreign operations, including risks of violations of local laws
or the U.S. Foreign Corrupt Practices Act by employees
overseas or the OECD Convention on Combating Bribery of Foreign
Public Officials in International Business Transactions;
|
| |
| |
•
|
unexpected changes in regulatory requirements;
|
| |
| |
•
|
the difficulties of compliance with a wide variety of foreign
laws and regulations;
|
| |
| |
•
|
unfavorable regulations in foreign jurisdictions regarding
distributors;
|
| |
| |
•
|
the deferral of revenue recognition;
|
| |
| |
•
|
longer accounts receivable cycles in certain foreign countries;
and
|
| |
| |
•
|
import and export licensing requirements.
|
10
Any of these risks could adversely affect our international
operations or financial results, which would harm our business.
We could
become subject to product liability claims, product recalls,
other field actions and warranty claims that could be expensive,
divert management’s attention, and harm our
business.
We face an inherent risk of exposure to product liability claims
in the event that the use of our eSVS MESH results or is alleged
to have resulted in adverse effects to a patient. In many
jurisdictions, producers of medical products are strictly liable
for personal injuries caused by medical devices. A product
liability claim against us, even if we are ultimately successful
in defending it, could have a material adverse effect on our
business, results of operations and reputation.
We may be held liable if our eSVS MESH causes injury or death or
is found otherwise unsuitable during usage. Because our eSVS
MESH is designed to be used in complex surgical procedures,
defects could result in a number of complications, including
serious injury or death. It is also possible that defects in the
design, manufacture or labeling of our eSVS MESH might
necessitate a product recall or other field corrective action,
which may result in warranty claims beyond our expectations and
may harm our reputation. A product liability claim, regardless
of its merit or eventual outcome, could result in significant
legal defense costs. The coverage limits of our insurance
policies may not be adequate to cover future claims. We may be
unable to maintain product liability insurance in the future at
satisfactory rates or with adequate amounts. A product liability
claim, any product recalls or other field actions or excessive
warranty claims, whether arising from defects in design or
manufacture or otherwise, could divert management’s
attention from our core business, be expensive to defend and
result in sizable damage awards against us, any of which could
harm our reputation and business.
If
third-party payors do not provide sufficient coverage or
reimbursement to healthcare providers for the use of our eSVS
MESH, our acceptance in the marketplace would be
harmed.
The availability of insurance coverage and reimbursement for
newly approved medical devices and procedures is uncertain. Our
success depends upon the use of our eSVS MESH and whether
third-party insurance coverage and reimbursement for the use of
this product is available.
Our success in international markets depends upon the
eligibility of reimbursement for our eSVS MESH through
government-sponsored healthcare payment systems and third-party
payors. Reimbursement and healthcare payment systems in
international markets vary significantly by country and, within
some countries, by region. In many international markets,
payment systems may control reimbursement for procedures
performed using new products as well as procurement of these
products. In addition, as economies of emerging markets develop,
these countries may implement changes in their healthcare
delivery and payment systems. Furthermore, healthcare cost
containment efforts similar to those underway in the United
States are prevalent in many of the other countries in which we
intend to sell our eSVS MESH and these efforts are expected to
continue. Market acceptance of our eSVS MESH in a particular
country may depend on the availability and level of
reimbursement in that country. In the event that our customers
are unable to obtain adequate reimbursement for our eSVS MESH in
international markets in which we are seeking to sell our eSVS
MESH, market acceptance of our eSVS MESH would be adversely
affected.
In the United States, our eSVS MESH would be purchased primarily
by medical institutions, which would then bill various
third-party payors, such as the Centers for Medicare &
Medicaid Services, or CMS, which administer the Medicare
program, and other government programs and private insurance
plans, for the healthcare services provided to their patients.
The process involved in applying for coverage and incremental
reimbursement from CMS is lengthy and expensive. Even if our
eSVS MESH receives FDA and other regulatory approval, it may not
be granted coverage and reimbursement in the foreseeable future,
if at all. Moreover, many private payors look to CMS in setting
their reimbursement policies and amounts. If CMS or other
agencies limit coverage or decrease or limit reimbursement
payments for doctors and hospitals, this may affect coverage and
reimbursement determinations by many private payors.
CMS may not provide coverage and reimbursement for our eSVS
MESH. If a medical device does not receive incremental
reimbursement from CMS, then a medical institution would have to
absorb the cost of our eSVS
11
MESH as part of the cost of the procedure in which the products
are used. Acute care hospitals are now generally reimbursed by
CMS for inpatient operating costs under a Medicare hospital
inpatient prospective payment system. Under the Medicare
hospital inpatient prospective payment system, acute care
hospitals receive a fixed payment amount for each covered
hospitalized patient based upon the Diagnosis-Related Group, or
DRG, to which the inpatient stay is assigned, regardless of the
actual cost of the services provided. At this time, we do not
know the extent to which medical institutions would consider
insurers’ payment levels adequate to cover the cost of our
eSVS MESH. Failure by hospitals and physicians to receive an
amount that they consider to be adequate reimbursement for
procedures in which our eSVS MESH is used could deter them from
purchasing our eSVS MESH and limit our revenue growth. In
addition, pre-determined DRG payments may decline over time,
which could deter medical institutions from purchasing our eSVS
MESH. If medical institutions are unable to justify the costs of
our eSVS MESH, they may refuse to purchase it, which would
significantly harm our business.
We may
not be able to attract and retain the technical, regulatory, and
sales personnel necessary for our success, which may divert
management’s attention and negatively impact our
operations.
We are highly dependent on our senior management, specifically
Manny Villafaña, our Chairman and Chief Executive Officer,
and Michael Winegar, our Chief Operating Officer and Vice
President of Regulatory Affairs. The loss of services of either
of these individuals would impair our ability to commercialize
our eSVS MESH and develop new products and would harm our
business. Our success will depend on our ability to retain our
senior management and to attract and retain qualified personnel
in the future. Competition for senior management personnel, as
well as clinical and regulatory specialists, engineers and sales
personnel, is intense and we may not be able to retain our
personnel. The loss of a member of our senior management or our
professional staff would require the remaining senior executive
officers to divert immediate and substantial attention to
seeking a replacement. Each of our senior officers may terminate
his employment at any time without notice and without cause or
good reason. We do not carry key person life insurance on any of
our employees. If we lose the services of any key personnel, our
business, financial condition and results of operations may
suffer.
We will
need to increase the size of our organization and we may
experience difficulties managing growth. If we are unable to
manage the anticipated growth of our business, our future
revenue and operating results may be adversely
affected.
We expect to significantly expand our manufacturing operations,
sales support and marketing staff, and administrative and
financial resources to meet anticipated growth in demand for our
eSVS MESH. We may face difficulties in recruiting, training,
managing and retaining an adequate number of qualified personnel
to support this growth. Rapid expansion in personnel may mean
that less experienced people could be manufacturing and
providing clinical and sales and marketing support for our eSVS
MESH, and managing our administrative and financial functions,
which could result in unanticipated costs and disruptions to our
operations. If we cannot scale and manage our business
appropriately, our anticipated growth may be impaired and our
financial results will suffer.
Becoming
a public company will cause us to incur increased costs and
demands on our management and divert management’s attention
from our core business.
The obligations of being a public company, including substantial
public reporting and auditing obligations, will require
significant additional expenditures, place additional demands on
our management and divert management’s time and attention
away from our core business. These additional obligations will
require us to hire additional personnel in order to ensure
compliance with the regulatory requirements of the Securities
and Exchange Commission and the NASDAQ Global Market. We will be
required to report on our internal controls, as required by
Section 404 of the Sarbanes-Oxley Act, beginning with our
annual report for the year ending December 31, 2011. We
cannot be certain as to the timing of completion of our
evaluation, testing and remediation actions relating to our
internal controls or the impact of the same on our operations.
Our management may not be able to effectively and timely
implement controls and procedures that adequately respond to the
increased regulatory compliance and reporting requirements that
will be applicable to us as a public company. If we fail to
staff our accounting and finance function adequately or maintain
internal controls adequate to meet the demands that will be
placed upon us as a public company, including the requirements
of the Sarbanes-Oxley Act, we may be unable
12
to report our financial results accurately or in a timely manner
and our business and stock price may suffer. The costs of being
a public company, as well as diversion of management’s time
and attention, may harm our business, financial condition and
results of operations.
Risks Related to
Our Intellectual Property
If we are
unable to protect our intellectual property rights, our ability
to compete will be harmed.
We rely upon patents, trade secret laws and confidentiality
agreements to protect our technology. We have six patent
applications pending in the United States and nine patent
applications pending in countries outside the United States on
our eSVS MESH. We also have one international patent application
pending, which gives us the opportunity to file in more
individual countries. Some of our pending patent applications
have been examined and currently stand rejected. We will
continue to pursue obtaining patents from these applications,
but it is possible that our pending patent applications may not
issue as patents or, if issued, may not issue in a form that
will be advantageous to us. We expect to launch our eSVS MESH in
Europe before any of our pending European patent applications
issue as patents. Any patents we obtain in the future might be
invalidated or circumvented by third parties. If any challenges
are successful, competitors might be able to market products and
use manufacturing processes that are substantially similar to
ours. We may not be able to prevent the unauthorized disclosure
or use of our technical knowledge or other trade secrets by
consultants, vendors or former or current employees, despite the
existence generally of confidentiality agreements and other
contractual restrictions. Monitoring unauthorized use and
disclosure of our intellectual property is difficult, and we do
not know whether the steps we have taken to protect our
intellectual property will be adequate. In addition, the laws of
many foreign countries may not protect our intellectual property
rights to the same extent as the laws of the United States. To
the extent that our intellectual property protection is
inadequate, we are exposed to a greater risk of direct
competition. In addition, competitors could purchase our eSVS
MESH and attempt to replicate some or all of the competitive
advantages we derive from our development efforts or attempt to
design around our protected technology. If our intellectual
property is not adequately protected against competitors’
products and methods, our competitive position could be
adversely affected, as could our business.
The laws of some foreign countries may not protect our
intellectual property rights to the same extent as do the laws
of the United States. In the event a competitor infringes upon
our patent or other intellectual property rights, enforcing
those rights may be difficult and time consuming. Even if
successful, litigation to enforce our intellectual property
rights or to defend our patents against challenge could be
expensive and time consuming and could divert our
management’s attention. We may not have sufficient
resources to enforce our intellectual property rights or to
defend our patents against a challenge.
We also rely upon trade secrets, technical know-how and
continuing technological innovation to develop and maintain our
competitive position. We require our employees to execute
appropriate confidentiality and
assignment-of-inventions
agreements with us. These agreements typically provide that all
materials and confidential information developed or made known
to the individual during the course of the individual’s
relationship with us be kept confidential and not disclosed to
third parties except in specific circumstances and that all
inventions arising out of the individual’s relationship
with us shall be our exclusive property. Additionally, we seek
to have our consultants and advisors execute similar
confidentiality and
assignment-of-inventions
agreements with us, but in some instances these agreements have
not included
assignment-of-invention
provisions. These agreements may be breached, and in some
instances, we may not have an appropriate remedy available for
breach of the agreements. Furthermore, our competitors may
independently develop substantially equivalent proprietary
information and techniques, reverse engineer our information and
techniques, or otherwise gain access to our proprietary
technology.
Claims of
infringement or misappropriation of the intellectual property
rights of others could prohibit us from commercializing our eSVS
MESH and harm our business.
The medical device industry has been characterized by extensive
litigation regarding patents and other intellectual property
rights, and companies in the industry have used intellectual
property litigation to gain a competitive advantage. We may
become a party to patent infringement claims and litigation or
interference proceedings
13
declared by the U.S. Patent and Trademark Office to
determine the priority of inventions. The defense and
prosecution of these matters are both costly and time consuming.
Additionally, we may need to commence proceedings against others
to enforce our patents, to protect our trade secrets or know-how
or to determine the enforceability, scope and validity of the
proprietary rights of others. These proceedings would result in
substantial expense to us and significant diversion of effort by
our technical and management personnel.
We are aware of patents issued to third parties that contain
subject matter related to our technology. These or other third
parties may assert that our eSVS MESH infringes the claims in
their patents or seek to expand their patent claims to cover
aspects of our eSVS MESH. An adverse determination in litigation
or interference proceedings to which we may become a party could
subject us to significant liabilities or require us to seek
licenses. In addition, if we are found to willfully infringe
third-party patents, we could be required to pay treble damages
in addition to other penalties. Although patent and intellectual
property disputes in the medical device area have often been
settled through licensing or similar arrangements, costs
associated with these arrangements may be substantial and could
include ongoing royalties. We may be unable to obtain necessary
licenses on satisfactory terms, if at all. If we do not obtain
necessary licenses, we may be required to redesign our eSVS MESH
to avoid infringement, and it may not be possible to do so
effectively. Adverse determinations in a judicial or
administrative proceeding or failure to obtain necessary
licenses could cause us to incur significant costs, place
significant strain on our resources, divert management’s
attention from our business and harm our reputation and prevent
us from commercializing our eSVS MESH or any other product we
may develop, which would have a significant adverse impact on
our business. Some of our competitors may be able to sustain the
costs of complex patent litigation more effectively than we can
because they have substantially greater resources.
Risks Related to
Regulatory Approval and Other Government Regulations
We will
be subject to government regulation, and we may not receive
approval for our eSVS MESH on a timely basis, if at
all.
Our eSVS MESH, product development activities and manufacturing
processes are, and will continue to be, subject to extensive and
rigorous scrutiny and regulation by the FDA and by comparable
agencies in foreign countries. In the United States, the FDA
regulates the introduction of medical devices, as well as
manufacturing, labeling and record keeping procedures for such
products. The process of obtaining marketing clearance or
approval for new medical products from the FDA is costly and
time consuming, and there can be no assurance that such approval
will be granted for our eSVS MESH on a timely basis, if at all,
or that the FDA review will not involve delays that would
adversely affect our ability to commercialize our eSVS MESH.
Even if regulatory clearance or approval to market a product is
obtained from the FDA, this clearance or approval may entail
limitations on the indicated uses of the product. Marketing
clearance or approval can also be withdrawn by the FDA due to
failure to comply with regulatory standards or the occurrence of
unforeseen problems following initial clearance.
The FDA will require us to file a Pre-Market Approval, or PMA,
application with regard to our eSVS MESH, and there is no
assurance whatsoever that approval will be obtained. Even if
approval is obtained, the process of obtaining a PMA is
expensive, uncertain and lengthy, frequently requiring several
years from the date of submission. Changing FDA policies and
requirements for PMA products may add additional uncertainty.
Significant delay or failure to obtain FDA approval to market
our eSVS MESH would harm our business.
The FDA
may not approve our investigational device exemption application
for our eSVS MESH, which would prevent us from conducting our
clinical trials in the United States, and even if the FDA does
grant such approval, our clinical trials may be more costly and
burdensome than we currently anticipate, which would limit or
delay our ability to complete clinical trials and ultimately
market our eSVS MESH in the United States.
The FDA is reviewing our application for an investigational
device exemption, or IDE, which, if granted, will allow us to
begin clinical trials of our eSVS MESH in the United States. The
FDA has not yet approved our application for an IDE and may
never grant such approval.
14
If the FDA approves our IDE application, the clinical trials we
conduct may have unanticipated complications and delays and may
be more costly than we currently anticipate. The FDA may approve
our IDE application with conditions relating to the scope or
design of our clinical trials for which we have not planned.
These conditions may require us to collect additional data,
enroll more patients, spend more time and expend more resources
than we currently anticipate, and these conditions may make a
clinical trial in the United States more costly and time
consuming than we currently plan. Any unanticipated costs and
length of U.S. clinical trials would delay our ability to
market our eSVS MESH in the United States, which would harm our
business.
If the FDA does not approve our IDE application, we would be
unable to conduct clinical trials of our eSVS MESH in the United
States. If our IDE application is not approved and we are unable
to conduct U.S. clinical trials, we would not be able to
submit a PMA application and we would be unable to market our
eSVS MESH in the United States, which would have an adverse
effect on our business.
Even if
our eSVS MESH is approved by regulatory authorities, if we fail
to comply with ongoing regulatory requirements, or if we
experience unanticipated problems with our eSVS MESH, it could
be subject to restrictions or withdrawal from the
market.
Any product for which we obtain marketing approval, along with
the manufacturing processes, post-approval clinical data and
promotional activities for such product, will be subject to
continual review and periodic inspections by the FDA and other
regulatory bodies. Even if regulatory approval of our eSVS MESH
is granted, the approval may be subject to limitations on the
indicated uses for which the product may be marketed or contain
requirements for costly post-marketing testing and surveillance
to monitor the safety or effectiveness of the product. Later
discovery of previously unknown problems with our eSVS MESH,
including unanticipated adverse events or adverse events of
unanticipated severity or frequency, manufacturer or
manufacturing processes, or failure to comply with regulatory
requirements, may result in restrictions on such products or
manufacturing processes, withdrawal of the products from the
market, voluntary or mandatory recall, fines, suspension of
regulatory approvals, product seizures, injunctions or the
imposition of civil or criminal penalties.
We will
be highly dependent on third-party institutions to conduct our
clinical testing, and the results of such testing may delay or
prevent regulatory approval of our eSVS MESH.
We rely on clinical investigators and clinical trial sites to
enroll patients in our clinical trials and other third parties
to manage our trials and to perform related data collection and
analysis. However, we are not able to control the amount and
timing of resources that clinical trial sites devote to our
clinical trials. If these clinical investigators and clinical
trial sites fail to enroll a sufficient number of patients in
our clinical trials or fail to ensure compliance by patients
with clinical protocols, we will be unable to complete our
planned trials, which could prevent us from obtaining regulatory
approvals for our eSVS MESH. Our agreements with clinical
investigators and clinical trial sites for clinical testing
place substantial responsibilities on these parties and, if
these parties fail to perform as expected, our planned trials
could be delayed or terminated. If these clinical investigators,
clinical trial sites, or other third parties do not carry out
their contractual duties or obligations or fail to meet expected
deadlines, or if the quality or accuracy of the clinical data
they obtain is compromised due to their failure to adhere to our
clinical protocols, the FDA’s good clinical practice
regulations or for other reasons, our clinical trials may be
extended, delayed or terminated, and we may be unable to obtain
regulatory approval for, or successfully commercialize, our eSVS
MESH.
In addition, the data obtained from human clinical testing is
subject to varying interpretations that could delay, limit or
prevent regulatory approval, and delays or rejection may be
encountered based upon changes in FDA policy for device approval
during the period of development.
Our
facilities will be subject to inspection by the FDA and
international authorities, and we could face penalties if we are
found to be non-compliant with the regulations of the FDA or
international authorities.
The FDA and various other authorities will inspect our
facilities from time to time to determine whether we are in
compliance with regulations relating to medical device
manufacturing, including regulations concerning design,
manufacturing, testing, quality control, product labeling,
distribution, promotion, and record keeping practices. A
15
determination that we are in material violation of such
regulations could lead to the imposition of civil penalties,
including fines, product recalls, product seizures or, in
extreme cases, criminal sanctions. Even if regulatory approvals
to market a product are obtained from the FDA, such approvals
may contain limitations on the indicated uses of our eSVS MESH.
The FDA could also limit or prevent the manufacture or
distribution of our eSVS MESH and has the power to require the
recall of products. FDA regulations depend heavily on
administrative interpretation, and there can be no assurance
that the future interpretations made by the FDA or other
regulatory bodies with possible retroactive effect will not
adversely affect us.
Our
promotional and marketing activities will be subject to
regulation by the FDA and international authorities, and we
could face severe penalties if we are found to be promoting our
eSVS MESH for an unapproved use.
If the FDA or international authorities determine that our
promotional materials or activities constitute promotion of our
eSVS MESH for an unapproved use, it could demand that we cease
the use of or modify our promotional materials or subject us to
regulatory enforcement actions, including the issuance of a
warning letter, injunction, civil fine and criminal penalties.
It is also possible that other federal, state or foreign
enforcement authorities might take action if they consider
promotional or other materials to constitute promotion of our
eSVS MESH for an unapproved use, which could result in
significant fines or penalties under other statutory
authorities, such as laws prohibiting false claims for
reimbursement.
Our
success will also be dependent on complying with foreign
regulatory requirements, and our inability to do so could result
in sales of our eSVS MESH being restricted
internationally.
Our revenues will initially be dependent upon sales of our eSVS
MESH outside the United States. Foreign regulatory bodies have
established varying regulations governing product standards,
packaging requirements, labeling requirements, import
restrictions, tariff regulations, duties and tax requirements.
We will rely heavily upon independent foreign distributors to
comply with such foreign regulatory requirements. Our inability
or failure or the inability or failure of such foreign
distributors to comply with varying foreign regulation or the
imposition of new regulations could restrict the sale of our
eSVS MESH internationally and thereby harm our business.
Proposed
legislation may negatively affect coverage and reimbursement
levels for our eSVS MESH.
Even if third-party payors provide adequate coverage and
reimbursement for our eSVS MESH, adverse changes in third-party
payors’ general policies toward reimbursement could
preclude market acceptance for our eSVS MESH and harm our
potential sales and revenue growth, which in turn would harm our
business. Recently, healthcare reform legislation was signed
into law in the United States and we expect that there will
continue to be legislative proposals for governmental controls
over healthcare in the United States and other countries. Some
third-party payors also require pre-approval of coverage or
companies to demonstrate the superiority of their product before
they will reimburse healthcare providers who use such devices or
procedures.
The trend toward managed healthcare in the United States and
other countries and legislation intended to reduce the cost of
government insurance programs could significantly influence the
purchase of healthcare services and products, and may result in
necessary price reductions for our eSVS MESH or the exclusion of
our eSVS MESH from reimbursement programs. It is uncertain
whether our eSVS MESH will be viewed as sufficiently
cost-effective to warrant adequate coverage and reimbursement
levels.
Our
operations involve hazardous materials, and we must comply with
environmental laws and regulations, which can be expensive, time
consuming, and may impair our operations.
We are subject to a variety of federal, state and local
regulations relating to the use, handling, storage, disposal and
human exposure to hazardous and toxic materials. We could incur
costs, fines and civil and criminal sanctions, third-party
property damage or personal injury claims, or could be required
to incur substantial investigation or remediation costs, if we
were to violate or become liable under environmental laws. We do
not have insurance for environmental liabilities and liability
under environmental laws can be joint and several and without
regard to comparative fault. Environmental laws could become
more stringent over time, imposing greater compliance costs
16
and increasing risks and penalties associated with violations,
which could harm our business. Compliance with current or future
environmental and safety laws and regulations could restrict our
ability to expand our facilities, impair our research,
development or production efforts, or require us to incur other
significant expenses. There can be no assurance that violations
of environmental laws or regulations will not occur in the
future as a result of the inability to obtain permits, human
error, accident, equipment failure or other causes.
Risks Related to
Our Common Stock and this Offering
Because
there has not been a public market for our common stock and our
stock price may be volatile, you may not be able to resell your
shares at or above the initial public offering price.
Prior to this initial public offering there has not been a
public market for our common stock. We cannot predict the extent
to which investors’ interests will lead to an active
trading market for our common stock or whether the market price
of our common stock will be volatile following this offering. If
an active trading market does not develop or is not sustained,
you may have difficulty selling any of our common stock that you
buy. The initial public offering price for our common stock will
be determined by negotiations between representatives of the
underwriters and us and may not be indicative of prices that
will prevail in the open market following this offering.
Consequently, you may not be able to sell our common stock at
prices equal to or greater than the price you paid in this
offering. In addition to the risk factors discussed elsewhere in
this section, the following factors, most of which are outside
of our control, could cause the market price of our common stock
to decrease significantly from the price you pay in this
offering:
|
|
|
| |
•
|
inconclusive or failed clinical trial outcomes of our eSVS MESH;
|
| |
| |
•
|
failure to achieve market acceptance for our eSVS MESH;
|
| |
| |
•
|
inability to manufacture our eSVS MESH in adequate quantities or
to commercial standards;
|
| |
| |
•
|
departure of key personnel;
|
| |
| |
•
|
inability to hire, train and retain qualified personnel to
support our growth;
|
| |
| |
•
|
variations in our quarterly operating results or those of
companies that are perceived to be similar to us;
|
| |
| |
•
|
announcements by our competitors of significant technological
developments;
|
| |
| |
•
|
changes in governmental regulations and standards;
|
| |
| |
•
|
litigation related to patent infringement and product liability
claims;
|
| |
| |
•
|
changes to financial estimates by equity research analysts;
|
| |
| |
•
|
sales of common stock or other securities by us in the future;
|
| |
| |
•
|
decreases in market valuations of similar companies; and
|
| |
| |
•
|
fluctuations in stock market prices and volumes.
|
Each of these factors could cause the market price of our stock
to decline, and you may lose some or all of your investment.
In addition, the stock markets have been extremely volatile.
Securities class action litigation is often initiated against a
company following a period of volatility in the market price of
the company’s securities. If class action litigation is
initiated against us, we would incur substantial costs and our
management’s attention would be diverted from our
operations.
If equity
research analysts do not publish research or reports about our
business or if they issue unfavorable research or downgrade our
common stock, the price of our common stock could
decline.
The trading market for our common stock will rely in part on the
research and reports that equity research analysts publish about
us and our business. We do not control these analysts. Equity
research analysts may elect not to provide research coverage of
our common stock, which may adversely affect the market price of
our common stock. If equity research analysts do provide
research coverage of our common stock, the price of our common
stock could decline if one or more of these analysts downgrade
our common stock or if they issue other
17
unfavorable commentary about us or our business. If one or more
of these analysts ceases coverage of our company, we could lose
visibility in the market, which in turn could cause our stock
price to decline.
Future
sales of our common stock by our existing stockholders could
cause our stock price to decline and cause you to lose part or
all of your investment.
If our stockholders sell substantial amounts of our common stock
in the public market, the market price of our common stock could
decrease significantly. The perception in the public market that
our stockholders might sell shares of our common stock could
also depress the market price of our common stock. Substantially
all of our existing stockholders prior to this offering are
subject to
lock-up
agreements that restrict their ability to transfer their shares
of our common stock for at least 180 days after the date of
this prospectus, subject to certain exceptions. Upon expiration
of the
lock-up
period, shares of our common stock will be eligible for sale in
the public market. In addition, we intend to file registration
statements with the SEC covering (a) any shares of our
common stock acquired upon option exercises prior to the closing
of this offering, (b) all of the shares subject to options
outstanding, but not exercised, as of the closing of this
offering and (c) all of the shares available for future
issuance under our stock incentive plan upon the closing of this
offering. The market price of shares of our common stock may
decrease significantly when the restrictions on resale by our
existing stockholders lapse and our stockholders, warrant
holders and option holders are able to sell shares of our common
stock into the market. A decline in the price of shares of our
common stock might impede our ability to raise capital through
the issuance of additional shares of our common stock or other
equity securities, and may cause you to lose part or all of your
investment in our shares of common stock.
We have
broad discretion in the use of the proceeds of this offering and
may apply the proceeds in ways with which you do not agree or
which cause our stock price to decline.
A significant portion of our net proceeds from this offering
will be used, as determined by management in its sole
discretion, for working capital and general corporate purposes.
Our management will have broad discretion over the use and
investment of these net proceeds, and, accordingly, you will
have to rely upon the judgment of our management with respect to
our use of these net proceeds, with only limited information
concerning management’s specific intentions. You will not
have the opportunity, as part of your investment decision, to
assess whether we use the net proceeds from this offering
appropriately. We may place the net proceeds in investments that
do not produce income or that lose value, which may cause our
stock price to decline.
Our
directors, executive officers and significant stockholders will
continue to have substantial control over us after this offering
and could limit your ability to influence the outcome of key
transactions, including changes of control.
We anticipate that our directors and executive officers and
their affiliated entities will, in the aggregate, beneficially
own % of our outstanding common
stock following the completion of this offering, assuming the
underwriters do not exercise their option to purchase additional
shares. In addition, Kips Bay Investments, LLC will beneficially
own % and Mr. Villafaña,
our Chairman and Chief Executive Officer, will beneficially
own % of our outstanding common
stock following the completion of this offering, and together
will be able to control or influence significantly all matters
requiring approval by our stockholders. Our directors, executive
officers, significant stockholders and affiliated entities, if
acting together, would be able to control or influence
significantly all matters requiring approval by our
stockholders, including the election of directors and the
approval of mergers or other significant corporate transactions.
These stockholders may have interests that differ from yours,
and they may vote in a way with which you disagree and that may
be adverse to your interests. The concentration of ownership of
our common stock may have the effect of delaying, preventing or
deterring a change of control of our company, could deprive our
stockholders of an opportunity to receive a premium for their
common stock as part of a sale of our company, and may affect
the market price of our common stock. This concentration of
ownership of our common stock may also have the effect of
influencing the completion of a change in control that may not
necessarily be in the best interests of all of our stockholders.
18
Our
charter documents and Delaware law may inhibit a takeover that
stockholders consider favorable.
Upon the closing of this offering, provisions of our certificate
of incorporation and amended and restated bylaws and applicable
provisions of Delaware law may make it more difficult for or
prevent a third party from acquiring control of us without the
approval of our board of directors. These provisions:
|
|
|
| |
•
|
set limitations on the removal of directors;
|
| |
| |
•
|
limit who may call a special meeting of stockholders;
|
| |
| |
•
|
establish advance notice requirements for nominations for
election to our board of directors or for proposing matters that
can be acted upon at stockholder meetings;
|
| |
| |
•
|
do not permit cumulative voting in the election of our
directors, which would otherwise permit less than a majority of
stockholders to elect directors;
|
| |
| |
•
|
prohibit stockholder action by written consent unless unanimous,
thereby requiring all stockholder actions to be taken at a
meeting of our stockholders; and
|
| |
| |
•
|
provide our board of directors the ability to designate the
terms of and issue a new series of preferred stock without
stockholder approval.
|
In addition, Section 203 of the Delaware General
Corporation Law generally limits our ability to engage in any
business combination with certain persons who own 15% or more of
our outstanding voting stock or any of our associates or
affiliates who at any time in the past three years have owned
15% or more of our outstanding voting stock. These provisions
may have the effect of entrenching our management team and may
deprive you of the opportunity to sell your shares to potential
acquirers at a premium over prevailing prices. This potential
inability to obtain a control premium could reduce the price of
our common stock.
You will
experience immediate and substantial dilution in the net
tangible book value of the common stock you purchase in this
offering.
If you purchase common stock in this offering, you will incur
immediate dilution of $ in as
adjusted net tangible book value per share of common stock,
based on an assumed initial public offering price of $ per
share, the midpoint of the range on the front cover of this
prospectus, because the price that you pay will be substantially
greater than the as adjusted net tangible book value per share
of common stock that you purchase. This dilution is due in large
part to the fact that our earlier investors paid substantially
less than the price of the shares being sold in this offering
when they purchased their shares of our common stock. In
addition, if outstanding options to purchase our common stock
are exercised, you will experience additional dilution.
We do not
intend to declare dividends on our common stock after this
offering, and you should not expect to receive dividends on your
common stock for the foreseeable future.
We currently intend to retain all future earnings for the
operation and expansion of our business and, therefore, do not
anticipate declaring or paying cash dividends on our common
stock in the foreseeable future. Any payment of cash dividends
on our common stock will be at the discretion of our board of
directors and will depend upon our results of operations,
earnings, capital requirements, financial condition, future
prospects, contractual restrictions and other factors deemed
relevant by our board of directors. Therefore, you should not
expect to receive dividend income from shares of our common
stock.
We
anticipate future losses and may require additional financing,
and our failure to obtain additional financing when needed could
force us to delay, reduce or eliminate our product development
programs or commercialization efforts.
We expect to incur losses for the foreseeable future, and we may
require financing in addition to the proceeds of this offering
in order to satisfy our capital requirements. In particular, we
may require additional capital in order to continue to conduct
the research and development and obtain regulatory clearances
and approvals necessary to bring any future products to market
and to establish effective marketing and sales capabilities for
existing and future products. Additional funds may not be
available when we need them on terms that are acceptable to us,
or
19
at all. If adequate funds are not available on a timely basis,
we may terminate or delay the development of our eSVS MESH, or
delay establishment of sales and marketing capabilities or other
activities necessary to commercialize our eSVS MESH.
Our future capital requirements will depend on many factors,
including:
|
|
|
| |
•
|
the costs of expanding our distribution network and our
manufacturing operations;
|
| |
| |
•
|
the degree of success we experience in commercializing our eSVS
MESH;
|
| |
| |
•
|
the number and types of future products we develop and
commercialize;
|
| |
| |
•
|
the costs, timing and outcomes of regulatory reviews associated
with our future product candidates;
|
| |
| |
•
|
the costs of preparing, filing and prosecuting patent
applications and maintaining, enforcing and defending
intellectual property-related claims; and
|
| |
| |
•
|
the extent and scope of our general and administrative expenses.
|
Raising
additional capital may cause dilution to our stockholders or
restrict our operations.
To the extent that we raise additional capital through the sale
of equity or convertible debt securities, your ownership
interest will be diluted, and the terms may include liquidation
or other preferences that adversely affect your rights as a
stockholder. Debt financing, if available, may involve
agreements that include covenants limiting or restricting our
ability to take specific actions such as incurring additional
debt, making capital expenditures or declaring dividends. Any of
these events could adversely affect our ability to achieve our
product development and commercialization goals and harm our
business.
20
Special Note
Regarding Forward-Looking Statements
This prospectus, including the sections entitled
“Prospectus Summary,” “Risk Factors,”
“Management’s Discussion and Analysis of Financial
Condition and Results of Operations” and
“Business,” contains forward-looking statements that
involve risks and uncertainties. In some cases, you can identify
forward-looking statements by the following words:
“anticipate,” “believe,”
“continue,” “could,” “estimate,”
“expect,” “intend,” “may,”
“ongoing,” “plan,” “potential,”
“predict,” “project,” “should,”
“will,” “would,” or the negative of these
terms or other comparable terminology, although not all
forward-looking statements contain these words. These statements
involve known and unknown risks, uncertainties and other factors
that may cause our or our industry’s results, levels of
activity, performance or achievements to be materially different
from the information expressed or implied by these
forward-looking statements. Although we believe that we have a
reasonable basis for each forward-looking statement contained in
this prospectus, we caution you that these statements are based
on a combination of facts and factors currently known by us and
our projections of the future, about which we cannot be certain.
Many important factors affect our ability to achieve our
objectives, including:
|
|
|
| |
•
|
our ability to commercialize our eSVS MESH technology;
|
| |
| |
•
|
our ability to obtain and maintain foreign and domestic
regulatory approval of our eSVS MESH technology;
|
| |
| |
•
|
our ability to obtain coverage and reimbursement from
third-party payors for our eSVS MESH technology and the extent
of such coverage;
|
| |
| |
•
|
the successful development of our distribution and marketing
capabilities;
|
| |
| |
•
|
our ability to attract and retain scientific, regulatory, and
sales and marketing support personnel;
|
| |
| |
•
|
our ability to obtain and maintain intellectual property
protection for our eSVS MESH technology;
|
| |
| |
•
|
any future litigation regarding our business, including product
liability claims;
|
| |
| |
•
|
changes in governmental laws and regulations relating to
healthcare;
|
| |
| |
•
|
the availability and cost of third-party products and the
ability of our suppliers to timely meet our demands;
|
| |
| |
•
|
changes affecting the medical device industry;
|
| |
| |
•
|
general and economic business conditions; and
|
| |
| |
| |
•
|
the other risks described under “Risk Factors” in this
prospectus.
|
These factors could cause actual results to differ materially
from the results anticipated by these forward-looking
statements. You should read these risk factors and the other
cautionary statements made in this prospectus as being
applicable to all related forward-looking statements wherever
they appear in this prospectus. We cannot assure you that the
forward-looking statements in this prospectus will prove to be
accurate. Furthermore, if our forward-looking statements prove
to be inaccurate, the inaccuracy may be material. In light of
the significant uncertainties in these forward-looking
statements, you should not regard these statements as a
representation or warranty by us or any other person that we
will achieve our objectives and plans in any specified time
frame, if at all.
You should read this prospectus completely. Other than as
required by law, we undertake no obligation to update these
forward-looking statements, even though our situation may change
in the future. We qualify all the forward-looking statements
contained in this prospectus by the foregoing cautionary
statements.
Information and management estimates contained in this
prospectus concerning the medical device industry and the
coronary artery bypass graft market, including our general
expectations and market position, market opportunity and market
share, are based on publicly available information, such as
clinical studies, academic research reports and other research
reports. The management estimates are also derived from our
internal research, using assumptions made by us that we believe
to be reasonable and our knowledge of the industry and markets
in which we operate and expect to compete. None of the sources
cited in this prospectus has consented to the inclusion of any
data from its reports, nor have we sought their consent. Our
internal research has not been verified by any independent
source, and we have not independently verified any third-party
information. In addition, while we believe the market position,
market opportunity and market share information included in this
prospectus is generally reliable, such information is inherently
imprecise. Such data involves risks and uncertainties and are
subject to change based on various factors, including those
discussed under the heading “Risk Factors.”
21
Use of
Proceeds
Assuming an initial public offering price of
$ per share, the midpoint of the
range on the front cover of this prospectus, we estimate our net
proceeds from the sale
of shares
of our common stock in this offering will be
$ million, after deducting
the estimated underwriting discounts and commissions and our
estimated offering expenses payable by us.
If the underwriters exercise their option to purchase additional
shares in full, we estimate that our net proceeds from this
offering will be $ million,
after deducting the estimated underwriting discounts and
estimated offering expenses payable by us. A $1.00 increase or
decrease in the assumed initial public offering price of
$ per share would increase or
decrease the net proceeds to us from this offering by
$ million, assuming the
number of shares offered by us, as set forth on the front cover
of this prospectus, remains the same and after deducting the
estimated underwriting discounts and commissions and estimated
offering expenses payable by us.
We intend to use the net proceeds from this offering primarily
for the following purposes:
|
|
|
| |
•
|
approximately $15.0 million to fund the process of seeking
regulatory approval to market our eSVS MESH in the United States
and abroad, including human clinical trials in the United States;
|
| |
| |
•
|
approximately $ million to
fund the development and testing of additional applications of
our eSVS MESH;
|
| |
| |
•
|
from $5.0 million to $10.0 million to fund certain
milestone payments payable to Medtronic, Inc. for the assignment
of certain intellectual property rights to our eSVS
MESH; and
|
| |
| |
•
|
the remainder for working capital and general corporate
purposes, including commercialization activities for our eSVS
MESH in select European and other international markets and for
the purchase of capital equipment and expansion of facilities.
|
The amounts we actually expend in these areas may vary
significantly from our expectations and will depend on a number
of factors, including operating costs and capital expenditures.
Accordingly, management will retain broad discretion in the
allocation of the net proceeds of this offering. You will not
have the opportunity to evaluate the economic, financial or
other information on which we base our decisions on how to use
the proceeds. A portion of the net proceeds may also be used to
acquire or invest in complementary businesses, technologies,
services or products. We have no current plans, agreements or
commitments with respect to any such acquisition or investment,
and we are not currently engaged in any negotiations with
respect to any such transaction.
Dividend
Policy
We have never declared or paid cash dividends on our capital
stock. Following the completion of this offering, we intend to
retain our future earnings, if any, to finance the further
development and expansion of our business and do not expect to
pay cash dividends in the foreseeable future. Payment of future
cash dividends, if any, will be at the discretion of our board
of directors after taking into account various factors,
including our financial condition, operating results, current
and anticipated cash needs, outstanding indebtedness and plans
for expansion and restrictions imposed by lenders, if any.
22
Capitalization
The following table sets forth our capitalization as of
December 31, 2009 on:
|
|
|
| |
•
|
an actual basis; and
|
| |
| |
•
|
an as adjusted basis to reflect the receipt of the net proceeds
from the sale
of shares
of common stock in this offering at an assumed initial public
offering price of $ per share, the
midpoint of the range on the front cover of this prospectus,
after deducting the estimated underwriting discounts and
commissions and estimated offering expenses payable by us.
|
You should read this capitalization table together with our
financial statements and the related notes appearing elsewhere
in this prospectus, as well as “Management’s
Discussion and Analysis of Financial Condition and Results of
Operations” and other financial information included in
this prospectus.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
As of December 31,
|
|
|
|
|
2009
|
|
|
|
|
Actual
|
|
|
As Adjusted(1)
|
|
|
|
|
Stockholders’ equity:
|
|
|
|
|
|
|
|
|
|
Common stock, $0.01 par value, 40,000,000 common shares and
10,000,000 undesignated shares authorized, 12,398,919 common
shares issued and outstanding,
actual; shares
authorized, issued
and outstanding, as adjusted
|
|
$
|
124
|
|
|
$
|
|
|
|
Additional paid-in capital
|
|
|
11,556
|
|
|
|
|
|
|
Accumulated other comprehensive gain
|
|
|
33
|
|
|
|
|
|
|
Deficit accumulated during development stage
|
|
|
(9,201
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total stockholders’ equity
|
|
|
2,512
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total capitalization
|
|
$
|
2,512
|
|
|
$
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(In thousands, except share and per
share data)
|
|
|
| (1) |
A $1.00 increase or decrease in the assumed initial public
offering price would result in an approximately
$ million increase or
decrease in as adjusted additional paid-in capital, as adjusted
total stockholders’ equity and as adjusted total
capitalization, assuming the number of shares offered by us, as
set forth on the front cover of this prospectus, remains the
same and after deducting the estimated underwriting discounts
and commissions and estimated offering expenses payable by us.
|
The outstanding share information in the table above is based on
the number of shares outstanding as of December 31, 2009,
and excludes:
|
|
|
| |
•
|
598,000 shares of common stock issuable upon the exercise
of outstanding stock options as of December 31, 2009 at a
weighted average exercise price of $3.08 per share; and
|
| |
| |
•
|
1,401,000 additional shares of common stock reserved and
available for future issuances under our 2007 Long-Term
Incentive Plan.
|
23
Dilution
If you invest in our common stock, your ownership interest will
be diluted to the extent of the difference between the initial
public offering price per share of our common stock and the as
adjusted net tangible book value per share of our common stock
immediately after completion of this offering.
As of December 31, 2009, we had a net tangible book value
of $2.5 million, or $0.20 per share of common stock. Net
tangible book value per share is equal to our total tangible
assets (total assets less intangible assets) less our total
liabilities divided by the number of shares of common stock
outstanding.
After giving effect to our sale of shares of common stock at an
assumed initial public offering price of
$ per share, the midpoint of the
range on the front cover of this prospectus, deducting the
estimated underwriting discounts and commissions and offering
expenses, the as adjusted net tangible book value of our common
stock, as of December 31, 2009, would have been
$ million, or
$ per share. This amount
represents an immediate increase in net tangible book value to
our existing stockholders of $ per
share and an immediate dilution to new investors of
$ per share.
The following table illustrates this dilution on a per share
basis:
| |
|
|
|
|
|
|
|
|
|
Assumed initial public offering price per share
|
|
|
|
|
|
$
|
|
|
|
Historical net tangible book value per share as of
December 31, 2009
|
|
$
|
0.20
|
|
|
|
|
|
|
Increase in net tangible book value per share attributable to
new investors purchasing shares in this offering
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
As adjusted net tangible book value per share after this offering
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Dilution per share to new investors in this offering
|
|
|
|
|
|
$
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
A $1.00 increase or decrease in the assumed initial public
offering price of $ per share
would increase or decrease, respectively, our as adjusted net
tangible book value by
$ million, the as adjusted
net tangible book value per share by
$ per share and the dilution in
the net tangible book value to investors in this offering by
$ per share, assuming the number
of shares offered by us, as set forth on the cover page of this
prospectus, remains the same and after deducting the estimated
underwriting discounts and commissions and estimated offering
expenses payable by us.
The following table summarizes, as of December 31, 2009, on
an as adjusted basis, the number of shares of common stock
purchased from us, the total consideration paid to us and the
average price per share paid by our existing stockholders and by
new investors, based upon an assumed initial public offering
price of $ per share, the midpoint
of the range on the front cover of this prospectus, and before
deducting estimated underwriting discounts and commissions and
offering expenses payable by us.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted
|
|
|
|
|
Shares Purchased
|
|
|
Total Consideration
|
|
|
Average Price
|
|
|
|
|
Number
|
|
|
Percent
|
|
|
Amount
|
|
|
Percent
|
|
|
per Share
|
|
|
|
|
Existing stockholders
|
|
|
|
|
|
|
|
%
|
|
$
|
|
|
|
|
|
%
|
|
$
|
|
|
|
New investors
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
|
|
|
|
|
100
|
%
|
|
$
|
|
|
|
|
100
|
%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
A $1.00 increase or decrease in the assumed initial public
offering price of $ per share
would increase or decrease, respectively, total consideration
paid by new investors and total consideration paid by all
stockholders by approximately
$ million, assuming that the
number of shares offered by us, as set forth on the front cover
of this prospectus, remains the same.
In the preceding tables, the shares of common stock outstanding
exclude, as of December 31, 2009:
|
|
|
| |
•
|
598,000 shares of common stock issuable upon exercise of
stock options outstanding as of December 31, 2009 at a
weighted average exercise price of $3.08 per share;
|
24
|
|
|
| |
•
|
1,401,000 additional shares of common stock reserved and
available for future issuances under our 2007 Long-Term
Incentive Plan; and
|
| |
| |
•
|
shares of our common stock that may be purchased by the
underwriters to cover over-allotments, if any.
|
If the underwriters exercise their option to purchase additional
shares in full:
|
|
|
| |
•
|
the number of shares of our common stock held by existing
stockholders would decrease to % of
the total number of shares of our common stock outstanding after
this offering;
|
| |
| |
•
|
the number of shares of our common stock held by new investors
would increase to % of the total
number of shares of our common stock outstanding after this
offering; and
|
| |
| |
•
|
our as adjusted net tangible book value at December 31,
2009 would have been
$ million, or
$ per share of common stock,
representing an immediate increase in as adjusted net tangible
book value of $ per share of
common stock to our existing stockholders and an immediate
dilution of $ per share to
investors purchasing shares in this offering.
|
Because we expect the exercise prices of the outstanding options
to be significantly below the assumed initial public offering
price of $ per share, the midpoint
of the range on the front cover of this prospectus, investors
purchasing common stock in this offering will suffer additional
dilution when and if these options are exercised. If the options
exercisable for 598,000 shares of common stock were
exercised prior to this offering, but assuming no exercise of
the underwriters’ option to purchase additional shares, our
existing stockholders would, after this offering,
own % of the total number of
outstanding shares of our common stock while
contributing % of the total
consideration for all shares, and our new investors would
own % of the total number of
outstanding shares of our common stock while
contributing % of the total
consideration for all shares.
25
Selected
Financial Data
The following table summarizes our selected financial data for
the periods and as of the dates indicated. The selected
financial data should be read in conjunction with, and are
qualified by reference to, our financial statements and related
notes and “Management’s Discussion and Analysis of
Financial Condition and Results of Operations” appearing
elsewhere in this prospectus. The statements of operations data
for the period from May 1, 2007 (inception) through
December 31, 2007, the years ended December 31, 2008
and 2009, and the period from May 1, 2007 (inception)
through December 31, 2009, and the balance sheet data at
December 31, 2008 and 2009, are derived from the audited
financial statements included elsewhere in this prospectus. The
balance sheet data at December 31, 2007 is derived from
audited financial statements not included in this prospectus.
The historical results are not necessarily indicative of results
to be expected in any future period.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from
|
|
|
|
|
|
|
|
|
Period from
|
|
|
|
|
May 1, 2007 (Date of
|
|
|
|
|
|
|
|
|
May 1, 2007 (Date
|
|
|
|
|
Inception) to
|
|
|
|
|
|
|
|
|
of Inception) to
|
|
|
|
|
December 31,
|
|
|
Year Ended December 31,
|
|
|
December 31,
|
|
|
|
|
2007
|
|
|
2008
|
|
|
2009
|
|
|
2009
|
|
|
|
|
Statements of Operations Data:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating expenses:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development
|
|
$
|
196
|
|
|
$
|
2,635
|
|
|
$
|
3,004
|
|
|
$
|
5,835
|
|
|
Selling, general and administrative
|
|
|
381
|
|
|
|
754
|
|
|
|
779
|
|
|
|
1,914
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating loss
|
|
|
(577
|
)
|
|
|
(3,389
|
)
|
|
|
(3,783
|
)
|
|
|
(7,749
|
)
|
|
Interest income
|
|
|
65
|
|
|
|
52
|
|
|
|
17
|
|
|
|
134
|
|
|
Interest expense
|
|
|
(164
|
)
|
|
|
(390
|
)
|
|
|
(181
|
)
|
|
|
(735
|
)
|
|
Impairment of available for sale securities
|
|
|
—
|
|
|
|
(85
|
)
|
|
|
—
|
|
|
|
(85
|
)
|
|
Change in fair value of investor stock purchase option
|
|
|
—
|
|
|
|
—
|
|
|
|
610
|
|
|
|
610
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss attributable to common stockholders
|
|
$
|
(676
|
)
|
|
$
|
(3,812
|
)
|
|
$
|
(3,337
|
)
|
|
$
|
(7,825
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted net loss per share
|
|
$
|
(0.16
|
)
|
|
$
|
(0.62
|
)
|
|
$
|
(0.30
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average common shares outstanding—basic and diluted
|
|
|
4,106,557
|
|
|
|
6,100,767
|
|
|
|
11,069,342
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(In thousands, except share and per
share amounts)
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
As of December 31,
|
|
|
|
|
2007
|
|
|
2008
|
|
|
2009
|
|
|
|
|
Balance Sheet Data:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash, cash equivalents, and short-term investments
|
|
$
|
2,370
|
|
|
$
|
1,124
|
|
|
$
|
3,417
|
|
|
Working capital
|
|
|
2,262
|
|
|
|
607
|
|
|
|
2,226
|
|
|
Total assets
|
|
|
2,637
|
|
|
|
1,452
|
|
|
|
3,740
|
|
|
Long-term debt, net
|
|
|
2,770
|
|
|
|
2,862
|
|
|
|
—
|
|
|
Total stockholders’ equity (deficit)
|
|
$
|
(305
|
)
|
|
$
|
(2,016
|
)
|
|
$
|
2,512
|
|
|
|
|
(In thousands)
|
26
Management’s
Discussion and Analysis of
Financial Condition and Results of Operations
The following discussion of our financial condition and
results of operations should be read in conjunction with the
“Selected Financial Data” and our financial statements
and the related notes included elsewhere in this prospectus.
This discussion contains forward-looking statements, which are
based on our assumptions about the future of our business. Our
actual results will likely differ materially from those
contained in the forward-looking statements. Please read
“Special Note Regarding Forward-Looking Statements”
included elsewhere in this prospectus for additional information
regarding forward-looking statements used in this prospectus.
Overview
We are a medical device company focused on developing,
manufacturing and commercializing our innovative external
saphenous vein support technology, or eSVS MESH, for use in
coronary artery bypass grafting, or CABG, surgery. Our eSVS MESH
is a nitinol mesh sleeve that, when placed over a saphenous vein
graft during CABG surgery, is designed to improve the structural
characteristics and long-term performance of the vein graft.
CABG surgery is one of the most commonly performed surgeries in
the United States, with the American Heart Association
estimating that 448,000 CABG procedures were performed in the
United States in 2006. In addition, the Millennium Research
Group estimates that there will be 165,000 CABG procedures per
year in Europe by 2013. In CABG procedures, surgeons harvest
blood vessels, including the internal mammary artery from the
chest and the saphenous vein from the leg, and attach the
harvested vessels to bypass, or provide blood flow around,
blocked coronary arteries. We believe the use of our eSVS MESH
with saphenous vein grafts in CABG surgery will improve the
long-term outcome of CABG procedures, including improved
openness, or patency, and improved blood flow characteristics
through the saphenous vein graft, resulting in a reduced need
for costly and potentially complicated reoperations or
revascularization procedures.
We have completed enrollment in a 90 patient multi-center
clinical trial conducted outside the United States. The primary
effectiveness endpoint of this trial is statistical
non-inferiority of the patency of eSVS MESH vessels as compared
to control vessels at nine-months post-implant. Effectiveness
data, which is based on angiographic patency, is being collected
at this time. Preliminary safety data has indicated that our
eSVS MESH and implant procedure do not result in an increase in
patient complications during or after surgery. We completed
enrollment in this trial in July 2009, and as of March 1,
2010, all 90 patients have been implanted for six months or
more, 83 patients have been implanted for nine months or
more, and 50 patients have been implanted for
12 months or more. This trial formed the basis for our CE
Mark application submitted in February 2010.
We expect to receive CE Mark approval and to begin marketing our
eSVS MESH in select European Union markets in the second half of
2010. The U.S. Food and Drug Administration, or FDA, is
reviewing our application for an investigational device
exemption, or IDE, which, if granted, will allow us to begin
clinical trials of our eSVS MESH in the United States. We are
currently amending our IDE application and anticipate obtaining
IDE approval in the first half of 2010, and if approved, we
expect to commence enrollment in our IDE trial in the second
half of 2010. We anticipate beginning enrollment in a United
States IDE trial in the second half of 2010. We could be delayed
by adverse clinical results or regulatory complications, and we
may never receive marketing approval.
We were incorporated and commenced operations in May 2007. Since
our inception, we have generated losses. From inception to
December 31, 2009, we had an accumulated deficit of
$9.2 million. We have not generated any revenue from
operations to date and we will not generate any revenue from
operations until we receive our CE Mark and begin selling our
eSVS MESH in select European Union markets. We expect to incur
losses for the foreseeable future as we pursue the development
and commercialization of our eSVS MESH. Our activities since
inception have consisted principally of acquiring product and
technology rights, raising capital, performing research and
development and conducting preclinical and clinical trials.
Accordingly, we are considered to be a development stage company
as of December 31, 2009. Successful completion of our
development programs and, ultimately, the attainment of
profitable operations are dependent on future events, including,
among other things, our ability to obtain regulatory approval
and achieve commercial adoption of our eSVS MESH.
27
Key Components of
Our Results of Operations
Research and
Development Expenses
Since our inception, we have focused our activities on the
development of our eSVS MESH. We expense both internal and
external research and development costs as incurred. Research
and development costs include the costs to design, develop,
test, seek approval for, and enhance our eSVS MESH and
production processes. Expenses related to research and
development consist primarily of personnel costs, including
salaries, benefits and stock-based compensation; product
development; preclinical and clinical trials; professional
service fees; materials and supplies; and facilities-related
costs. We expense amounts paid to obtain patents or acquire
licenses, as the ultimate recoverability of the amounts paid is
uncertain.
While our research and development expenses to date have been
focused on product development and evaluating the feasibility of
our eSVS MESH, we expect that a large percentage of our research
and development expenses in the future will be incurred in
support of our current and future clinical trials. These
expenditures are subject to numerous uncertainties in timing and
costs to complete. As we obtain results from clinical trials, we
may elect to discontinue or delay clinical trials for certain
product applications or programs in order to focus our resources
on more promising product applications. Completion of clinical
trials may take several years or more, but the length of time
generally varies according to the type, complexity, novelty and
intended use of a product. The cost of clinical trials may vary
significantly over the life of the trial as a result of
differences arising during the clinical trial, including:
|
|
|
| |
•
|
the number of sites included in the clinical trials;
|
| |
| |
•
|
the length of time required to enroll suitable patient subjects;
|
| |
| |
•
|
the number of patients that participate in the clinical
trials; and
|
| |
| |
•
|
the duration of patient
follow-up.
|
Our expenses related to clinical trials are based on estimates
of the services received and efforts expended pursuant to
contracts with multiple clinical trial sites and contract
research organizations, or CROs, that administer clinical trials
on our behalf. The financial terms of these agreements are
subject to negotiation and vary from contract to contract and
may result in uneven payment flows. Generally, these agreements
set forth the scope of work to be performed at a fixed fee or
unit price. Payments under the contracts depend on factors such
as the successful enrollment of patients and the completion of
clinical trial milestones. Expenses related to clinical trials
generally are accrued based on contracted amounts and the
achievement of milestones, such as number of patients enrolled.
If timelines or contracts are modified based upon changes to the
clinical trial design or scope of work to be performed, we
modify our estimates of accrued expenses accordingly.
We anticipate the cost of completing our U.S. clinical
trial will be approximately $15.0 million, based upon our
current expectation for the trial design. Because of the
numerous risks and uncertainties associated with developing
medical devices, we are unable to determine the duration and
completion costs of our development projects or when and to what
extent sales of our eSVS MESH will commence and become
significant.
Selling,
General and Administrative Expenses
Our selling, general and administrative expenses consist
primarily of salaries and benefits and other costs, including
stock-based compensation, for our executive and administrative
personnel; legal and other professional fees; business
development; insurance and other corporate costs. After
completion of this offering, we anticipate incurring a
significant increase in general and administrative expenses as
we operate as a public company. These increases will likely
include increased costs for insurance, costs related to the
hiring of additional personnel and payments to outside
consultants, lawyers and accountants. We also expect to incur
significant costs to comply with the corporate governance,
internal controls and similar requirements applicable to public
companies. While our selling, general and administrative
expenses to date have been primarily comprised of general and
administrative costs, we expect that we will incur significant
additional sales and marketing expenses as we prepare for and
commence commercialization of our eSVS MESH.
28
Interest
Income
Interest income consists of interest earned on investments in
certificates of deposits and money market accounts.
Interest
Expense
Interest expense results from interest associated with secured
convertible notes in the aggregate principal amount of
$3.0 million, or the Notes. Our reported interest expense
includes interest payable in cash based upon the stated rate in
the Notes and the amortization of discount recorded on the Notes
created by the allocation of a portion of the Note proceeds to
the fair value of the stock purchase options granted in
conjunction with the issuance of the Notes and due to the
beneficial conversion feature specified in the Notes. Beneficial
conversion feature accounting rules require the recognition of
the intrinsic value of the conversion feature at the time of the
Notes’ issuance, which is then amortized as additional
interest expense over the life of the Notes.
Critical
Accounting Policies and Estimates
Our management’s discussion and analysis of financial
condition and results of operations is based on our financial
statements, which have been prepared in accordance with
accounting principles generally accepted in the United States,
or GAAP. The preparation of these financial statements requires
us to make estimates and judgments that affect the reported
amounts of assets, liabilities and expenses. On an ongoing
basis, we evaluate these estimates and judgments, including
those described below. We base our estimates on our historical
experience and on various other assumptions that we believe to
be reasonable under the circumstances. These estimates and
assumptions form the basis for making judgments about the
carrying values of assets and liabilities that are not readily
apparent from other sources. Actual results and experiences may
differ materially from these estimates.
While our significant accounting policies are more fully
described in Note 2 to our financial statements included at
the end of this prospectus, we believe that the following
accounting policies are the most critical to aid you in fully
understanding and evaluating our reported financial results and
affect the more significant judgments and estimates that we use
in the preparation of our financial statements.
Research and
Development Expenses
We expense research and development costs, including clinical
trial costs, when incurred, consistent with the guidance of FASB
ASC 730, Research and Development. All of our clinical
trials are performed at clinical trial sites and are
administered by CROs. We accrue costs for clinical trials
performed by CROs based on estimates of work performed under the
contracts. Costs of setting up clinical trial sites are accrued
immediately. Expenses related to clinical trials generally are
accrued based on contracted amounts and the achievement of
milestones, such as number of patients enrolled.
All material clinical trial and CRO contracts are terminable by
us upon written notice and we are generally only liable for
actual effort expended by the CROs and certain non-cancelable
expenses incurred at any point of termination.
Stock-Based
Compensation
Stock-based incentive awards are accounted for under the
provisions of FASB ASC 718, Compensation—Stock
Compensation, which requires companies to measure and
recognize the cost of employee and non-employee services
received in exchange for awards of equity instruments based on
the grant date fair value of those awards. Compensation cost is
recognized ratably using the straight-line attribution method
over the expected vesting period, which is considered to be the
requisite service period. In addition, we are required to
estimate the amount of expected forfeitures when calculating the
compensation costs, instead of accounting for forfeitures as
incurred. All of our options previously awarded were classified
as equity instruments and continue to maintain their equity
classification.
The fair value of options is estimated at the date of grant
using the Black-Scholes option pricing model with the
assumptions described in the following sentences. Risk free
interest rates are based upon U.S. Treasury rates
appropriate for the expected term. Expected volatility and
forfeiture rates are based on the volatility rates of a set
29
of guideline companies, which consist of public and recently
public medical technology companies. The assumed dividend yield
is zero, as we do not expect to declare any dividends in the
foreseeable future. The expected term is determined using the
simplified method allowed by SEC Staff Accounting
Bulletin No. 110. The fair market value of the common
stock underlying the stock options has been determined by our
board of directors at each award grant date based upon a variety
of factors, as discussed below. If we had made different
assumptions and estimates, the amount of our recognized and to
be recognized stock-based compensation expense could have been
materially different. We believe that we have used reasonable
methodologies, approaches and assumptions in determining the
fair market value of our common stock.
Significant
Factors, Assumptions and Methodologies Used in Determining Fair
Market Value of Common Stock
The following table summarizes by ranges of grant date, the
number of shares subject to options granted in 2007, 2008 and
2009 and the associated per share exercise price, estimated fair
market value and ASC Topic 718 Black-Scholes Value. The exercise
prices were set by our board of directors at prices believed to
equal the fair market value of our common stock at each of the
grant dates, taking into account all information available at
those times. We did not obtain any third party contemporaneous
valuations.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ASC Topic 718 Black-
|
|
|
Date Range of Option Grants
|
|
Number of Shares
|
|
|
Exercise Price
|
|
|
Fair Market Value
|
|
|
Scholes Value
|
|
|
|
|
September 2007 through February 2008
|
|
|
349,000
|
|
|
$
|
1.00
|
|
|
$
|
1.00
|
|
|
$
|
0.52 - $0.55
|
|
|
March 2008
|
|
|
10,000
|
|
|
|
2.00
|
|
|
|
2.00
|
|
|
|
1.07
|
|
|
June 2008 through January 2009
|
|
|
219,000
|
|
|
|
5.83
|
|
|
|
5.83
|
|
|
|
2.81 - 3.20
|
|
|
September 2009
|
|
|
40,000
|
|
|
|
6.00
|
|
|
|
6.00
|
|
|
|
2.99
|
|
|
|
A brief narrative of the factors considered in estimating the
fair market value of our common stock as of the date of each
grant and the option exercise price is set forth below. The
factors generally included, but were not limited to, the most
recent purchase prices of our common stock issued to third
parties in arms-length transactions, the lack of marketability
of our common stock, the progress of our product development,
the progress of our preclinical and clinical testing, and the
risks associated with the completion of our business plan.
September
2007 through February 2008
The estimated fair market value of our common stock as
determined by our board of directors was $1.00 from September
2007 through February 2008. The estimated fair value of $1.00
primarily reflects the issuance of the Notes in July 2007, which
were convertible into common stock at $0.625 per share, and
continued progress towards the product development of our eSVS
MESH, including the first successful manufacturing of our eSVS
MESH and the commencement of negotiations for our Assignment and
License Agreement with Medtronic.
March
2008
The estimated fair market value of our common stock as
determined by our board of directors was $2.00 per share in
March 2008. The increase in the estimated fair market value
primarily reflects continued progress toward the product
development of our eSVS MESH, including the first successful
implants of our eSVS MESH in a preclinical trial in February
2008.
June 2008
through January 2009
The estimated fair market value of our common stock as
determined by our board of directors was $5.83 per share from
June 2008 through January 2009. The increase in the estimated
fair market value reflects the exercise in May 2008 of a stock
purchase option by a third party, pursuant to which we issued
600,000 shares of common stock at a purchase price of $5.83
per share. We issued this stock purchase option in July 2007,
and it became exercisable by its terms following our
determination that our eSVS MESH was suitable for human
implantation. Please see
30
“Certain Relationships and Related Party
Transactions—Investment Agreement with Kips Bay
Investments, LLC” for a description of the stock purchase
option.
September
2009
The estimated fair market value of our common stock as
determined by our board of directors was $6.00 per share in
September 2009. The increase in estimated fair market value
reflects the sale of 516,241 shares of common stock to
third party investors in August 2009 at $6.00 per share.
Results
of Operations
Comparison of
the Year Ended December 31, 2008 with the Year Ended
December 31, 2009
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
Percent
|
|
|
|
|
2008
|
|
|
2009
|
|
|
Change
|
|
|
|
|
Operating expenses:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development
|
|
$
|
2,635
|
|
|
$
|
3,004
|
|
|
|
14.0
|
%
|
|
Selling, general and administrative
|
|
|
754
|
|
|
|
779
|
|
|
|
3.3
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses
|
|
|
3,389
|
|
|
|
3,783
|
|
|
|
11.6
|
|
|
Other income (expense):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest income
|
|
|
52
|
|
|
|
17
|
|
|
|
(67.3
|
)
|
|
Interest expense
|
|
|
(390
|
)
|
|
|
(181
|
)
|
|
|
(53.6
|
)
|
|
Impairment of available for sale securities
|
|
|
(85
|
)
|
|
|
—
|
|
|
|
|
|
|
Gain from change in market value of investor option agreement
|
|
|
—
|
|
|
|
610
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
$
|
(3,812
|
)
|
|
$
|
(3,337
|
)
|
|
|
(12.5
|
)%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(In thousands)
|
Research and development and selling, general and administrative
expenses include non-cash compensation expense as a result of
our issuance of stock options. We expense the fair value of
stock options over their vesting periods. The terms and vesting
schedules for share-based awards vary by type of grant and the
employment status of the grantee. The awards granted through
December 31, 2009 vest upon time-based conditions. We
expect to record additional non-cash compensation expense in the
future, which may be significant. The following table summarizes
the stock-based compensation expense in our statement of
operations for 2008 and 2009:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2008
|
|
|
2009
|
|
|
|
|
Research and development
|
|
$
|
298
|
|
|
$
|
390
|
|
|
Selling, general and administrative
|
|
|
40
|
|
|
|
47
|
|
|
|
|
|
|
|
|
|
|
|
|
Total stock-based compensation
|
|
$
|
338
|
|
|
$
|
437
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(In thousands)
|
Research
and Development
Our research and development expenses increased 14.0% from
$2.6 million in 2008 to $3.0 million in 2009. This
increase was caused primarily by an increase of approximately
$350,000 from 2008 to 2009 in the expenses associated with our
international feasibility trial for our eSVS MESH, which began
enrolling patients in August 2008. These expenses include per
patient and per procedure fees payable to the clinical trial
sites, clinical trial administration costs payable to a CRO, and
costs related to the
set-up of
clinical trial sites and training of medical staff.
Selling,
General and Administrative
Selling, general and administrative expenses increased 3.3% from
$754,000 in 2008 to $779,000 in 2009. This
31
increase was the result of an increase in salary expense related
to the temporary addition of an individual performing marketing
related work, partially offset by reduced travel expenses and
professional fees.
Interest
Income
Interest income declined from $52,000 in 2008 to $17,000 in
2009. This decline resulted primarily from decline in short-term
interest rates from 2008 to 2009.
Interest
Expense
Interest expense declined from $390,000 in 2008 to $181,000 in
2009. The Notes were converted into shares of common stock in
February 2009, resulting in only two months of interest expense
in 2009, as compared with a full year of interest in 2008. This
decline was offset by the write-off of the $138,000 balance in
unamortized discount on the Notes at the time of the conversion.
Comparison of
the Period from May 1, 2007 (Date of Inception) to
December 31, 2007 with the Year Ended December 31,
2008
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from
|
|
|
|
|
|
|
|
|
|
|
May 1, 2007 (Date of
|
|
|
|
|
|
|
|
|
|
|
Inception) to
|
|
|
Year Ended
|
|
|
|
|
|
|
|
December 31,
|
|
|
December 31,
|
|
|
Percent
|
|
|
|
|
2007
|
|
|
2008
|
|
|
Change
|
|
|
|
|
Operating expenses:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development
|
|
$
|
196
|
|
|
$
|
2,635
|
|
|
|
1,244.4
|
%
|
|
Selling, general and administrative
|
|
|
381
|
|
|
|
754
|
|
|
|
97.9
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses
|
|
|
577
|
|
|
|
3,389
|
|
|
|
487.3
|
|
|
Other income (expense):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest income
|
|
|
65
|
|
|
|
52
|
|
|
|
(20.0
|
)
|
|
Interest expense
|
|
|
(164
|
)
|
|
|
(390
|
)
|
|
|
137.8
|
|
|
Impairment of available for sale securities
|
|
|
—
|
|
|
|
(85
|
)
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
$
|
(676
|
)
|
|
$
|
(3,812
|
)
|
|
|
463.9
|
%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(In thousands)
|
The following table summarizes the stock-based compensation
expense in our statement of operations for 2007 and 2008:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2007
|
|
|
2008
|
|
|
|
|
Research and development
|
|
$
|
14
|
|
|
$
|
298
|
|
|
Selling, general and administrative
|
|
|
2
|
|
|
|
40
|
|
|
|
|
|
|
|
|
|
|
|
|
Total stock-based compensation
|
|
$
|
16
|
|
|
$
|
338
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(In thousands)
|
Research
and Development
Our research and development expenses increased from $196,000 in
2007 to $2.6 million in 2008. Research and development
expenses in 2007 were primarily comprised of personnel expenses
of $150,000. In 2008, we operated for a full year and also
expanded our research and development staff, which resulted in
our personnel expenses, including stock-based compensation,
increasing by approximately $1.2 million. The balance of
the increase related to expenses for the development of our eSVS
MESH and the initiation of our international clinical trial.
These expenses included product development, preclinical trials,
clinical trial site selection, training and operation of our
international clinical trial and intellectual property related
costs.
32
Selling,
General and Administrative
Selling, general and administrative expenses increased 97.9%
from $381,000 in 2007 to $754,000 in 2009. This increase was
primarily a function of having 12 months of operations in
2008, compared with seven months in 2007. In addition, we
incurred approximately $80,000 in fees for an outside consultant
working on certain marketing matters.
Interest
Income
Interest income declined 20.0% from $65,000 in 2007 to $52,000
in 2008. This decline resulted from a decline in available cash
and equivalents and short-term investments, which more than
offset our having a full 12 months during which interest
was earned in 2008.
Interest
Expense
Interest expense increased 137.8% from $164,000 in 2008 to
$390,000 in 2009. This increase resulted from the Notes being
outstanding for the entire year in 2008, as compared with
slightly more than five months in 2007.
Impairment of
Available for Sale Investments
In conjunction with a simplified employee retirement plan that
we maintain for the benefit of our employees, we invested
$250,000 in a mutual fund under an arrangement that resulted in
reduced maintenance and transaction costs for our employees. Due
primarily to the significant decline in the U.S. stock
market and the accompanying declines in the U.S. economy
during 2008, we concluded as of December 31, 2008 that the
value of this mutual fund investment was impaired and that this
decline in value was other than temporary. We recorded a charge
of $85,000, which resulted in a new adjusted cost basis for this
investment. We had no other realized gains or losses in 2007 or
2008.
Income
Taxes
Since inception, we have incurred operating losses and,
accordingly, have not recorded a provision for income taxes for
any of the periods presented. As of December 31, 2009, we
had net operating loss carryforwards for federal and state
income tax purposes of approximately $6.5 million. We also
had federal research and development tax credit carryforwards of
approximately $400,000. If not utilized, the federal net
operating loss and tax credit carryforwards will expire
beginning in 2027. Utilization of net operating loss and credit
carryforwards may be subject to a substantial annual limitation
due to limitations provided by the Internal Revenue Code of
1986, as amended, that are applicable if we experience an
“ownership change” that may occur, for example, as a
result of this offering aggregated with certain other sales of
our stock before or after this offering. If not utilized, the
state net operating loss carryforward will expire beginning in
2022. The annual limitation may result in the expiration of our
net operating loss and tax credit carryforwards before they can
be used.
Liquidity and
Capital Resources
The following table summarizes our liquidity and capital
resources as of and for each of the last two fiscal years, and
is intended to supplement the more detailed discussion that
follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
Liquidity and Capital Resources
|
|
2008
|
|
|
2009
|
|
|
|
|
Cash and cash equivalents
|
|
$
|
943
|
|
|
$
|
2,469
|
|
|
Short-term investments
|
|
|
181
|
|
|
|
948
|
|
|
Working capital
|
|
|
607
|
|
|
|
2,226
|
|
|
|
|
(In thousands)
|
33
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from
|
|
|
|
|
|
|
|
|
Period from
|
|
|
|
|
May 1, 2007
|
|
|
|
|
|
|
|
|
May 1, 2007
|
|
|
|
|
(Date of
|
|
|
|
|
|
|
|
|
(Date of
|
|
|
|
|
Inception)
|
|
|
Year Ended
|
|
|
Inception)
|
|
|
|
|
to December 31,
|
|
|
December 31,
|
|
|
to December 31,
|
|
|
Cash Flow Data
|
|
2007
|
|
|
2008
|
|
|
2009
|
|
|
2009
|
|
|
|
|
Cash provided by (used in):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating activities
|
|
$
|
(505
|
)
|
|
$
|
(2,838
|
)
|
|
$
|
(3,382
|
)
|
|
$
|
(6,725
|
)
|
|
Investment activities
|
|
|
(760
|
)
|
|
|
196
|
|
|
|
(840
|
)
|
|
|
(1,404
|
)
|
|
Financing activities
|
|
|
3,100
|
|
|
|
1,750
|
|
|
|
5,748
|
|
|
|
10,598
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net increase (decrease) in cash and cash equivalents
|
|
$
|
1,835
|
|
|
$
|
(892
|
)
|
|
$
|
1,526
|
|
|
$
|
2,469
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(In thousands)
|
Cash and Cash
Equivalents
Our total cash resources, excluding short-term investments, as
of December 31, 2009 were $2.5 million, compared to
$943,000 as of December 31, 2008. As of December 31,
2009, we had approximately $1.2 million in current
liabilities and $2.2 million in net working capital. We
incurred a net loss of $3.3 million and had negative cash
flow from operating activities of $3.4 million for the year
ended December 31, 2009. Since May 1, 2007 (date of
inception) through December 31, 2009, we had an accumulated
deficit of $9.2 million, while negative cash flow from
operating activities has amounted to $6.7 million. The
difference between our accumulated deficit and negative cash
flow from operations results primarily from the adoption of FASB
ASC 815-40
on January 1, 2009. Under the provisions of FASB ASC
815-40,
certain instruments previously reported as equity are now
accounted for as derivative instruments. These provisions were
initially applied by recording a non-cash, cumulative effect
adjustment of $1.4 million to retained earnings. Our
accumulated deficit also includes non-cash charges for stock
based compensation and depreciation of $791,000 and $118,000,
respectively, and approximately $217,000 of accrued interest
expense which was settled through conversion into our common
stock.
As we continue to pursue regulatory approvals, prepare for
commercialization in international markets, develop our
manufacturing capabilities and develop additional applications
for our eSVS MESH, we expect to continue to incur substantial
and increasing losses, which will continue to generate negative
net cash flows from operating activities.
To date, we have funded our operations primarily through private
sales of common stock and convertible debt. As of
December 31, 2009, we had received net proceeds of
approximately $7.6 million from the sale of equity
securities, and net proceeds of approximately $3.0 million
from the issuance of the Notes, all of which have been converted
into common stock. We have not generated any revenue from
operations to date and we will not generate any revenue from
operations until we receive approval from the FDA, or an
equivalent foreign regulatory body, to begin selling our eSVS
MESH. We intend to use the net proceeds of this offering to seek
regulatory approval to market our technology in the United
States and abroad, including human clinical trials in the United
States; to develop and test additional applications of our eSVS
MESH; to make certain milestone payments for our acquired
intellectual property; and for working capital and general
corporate purposes. We may seek to raise additional funds
through various sources, such as equity and debt financings, or
through strategic collaborations and license agreements. We can
give no assurances that we will be able to secure such
additional sources of funds to support our operations, or if
such funds are available to us, that such additional financing
will be sufficient to meet our needs.
Net Cash Used
in Operating Activities
Net cash used in operating activities was $505,000 in 2007,
$2.8 million in 2008, and $3.4 million in 2009. From
May 1, 2007 (date of inception) to December 31, 2009,
net cash used in operating activities was $6.7 million. The
net cash used in each of these periods primarily reflects the
net loss for those periods, offset in part by depreciation,
non-cash stock-based compensation and the effects of changes in
operating assets and liabilities.
34
Net Cash
Provided by (Used in) Investment Activities
Net cash provided by (used in) investment activities was
$(760,000) in 2007, $196,000 in 2008, and $(840,000) in 2009.
From May 1, 2007 (date of inception) to December 31,
2009, net cash used in investment activities was
$1.4 million. Cash used in investment activities is related
to purchases and sales of short-term investments and purchases
of property and equipment.
Net Cash
Provided by Financing Activities
Net cash provided by financing activities was $3.1 million
in 2007, $1.8 million in 2008, $5.7 million in 2009.
From May 1, 2007 (date of inception) to December 31,
2009, net cash provided by financing activities was
$10.6 million. Net cash provided by financing activities
was primarily attributable to proceeds from issuances of the
Notes and from private sales of our common stock.
Capital
Requirements
We expect to incur substantial expenses and generate significant
operating losses as we continue to execute our business strategy
including:
|
|
|
| |
•
|
collecting effectiveness data from our 90 patient,
multi-center clinical trial conducted outside the United States
and seeking an IDE approval from the FDA to begin clinical
trials in the United States;
|
| |
| |
•
|
obtaining CE Mark approval;
|
| |
| |
•
|
commercializing our eSVS MESH in select European markets;
|
| |
| |
•
|
obtaining regulatory approval and commercializing our eSVS MESH
in the United States;
|
| |
| |
•
|
conducting clinical trials to expand indications for our eSVS
MESH;
|
| |
| |
•
|
hiring additional personnel for managerial, research and
development, operations and other functions;
|
| |
| |
•
|
expanding our facilities to increase our manufacturing and
development capabilities; and
|
| |
| |
•
|
implementing new operational, financial and management systems
to comply with SEC requirements.
|
Our future capital uses and requirements depend on numerous
forward-looking factors. These factors include the following:
|
|
|
| |
•
|
the timing of our receipt of approval to sell in select European
markets, if received at all;
|
| |
| |
•
|
our ability to demonstrate safety and effectiveness of our eSVS
MESH;
|
| |
| |
•
|
the selling price of our eSVS MESH to distributors and the price
that distributors charge hospitals;
|
| |
| |
•
|
the rate of progress in establishing reimbursement arrangements
with third-party payors;
|
| |
| |
•
|
the effect of competing technological and market developments;
|
| |
| |
•
|
the cost and delays in product development that may result from
changes in regulatory oversight applicable to our eSVS MESH;
|
| |
| |
•
|
the costs involved in filing and prosecuting patent applications
and enforcing or defending patent claims;
|
| |
| |
•
|
the cost of expanding our commercial operations, including our
selling and marketing efforts;
|
| |
| |
•
|
our ability to establish and maintain effective relationships
with independent distributors;
|
| |
| |
•
|
the rate at which physicians adopt our eSVS MESH for use in CABG
surgery; and
|
| |
| |
•
|
the progress of preclinical and clinical trials required to
support our applications for regulatory approvals, including our
human clinical trials in the United States.
|
We have not generated any revenue from operations to date. We do
not expect to generate revenue unless or until we obtain
regulatory approval of and commercialize our eSVS MESH. While
there can be no guarantee, we anticipate commencing sales in
select international markets by the end of the current fiscal
year. However, we do not expect to generate meaningful sales in
the current fiscal year and we expect our operating losses and
negative
35
cash flows from operations to continue for the foreseeable
future. Our future capital requirements will depend upon a
number of factors as indicated above.
We expect the proceeds of this offering, together with our
existing resources as of the date of this prospectus, to be
sufficient to fund our planned operations for at least the next
12 months. However, we may require significant additional
funds earlier than we currently expect in order to conduct
additional clinical trials to obtain regulatory approvals of our
eSVS MESH. To the extent that we raise additional capital
through the sale of equity or convertible debt securities,
stockholders’ interest will be diluted, and the terms may
include liquidation or other preferences that adversely affect
the rights of our stockholders. Debt financing, if available,
may involve agreements that include covenants limiting or
restricting our ability to take specific actions such as
incurring additional debt, making capital expenditures or
declaring dividends. Any of these events could adversely affect
our ability to achieve our product development and
commercialization goals and harm our business.
If adequate funds are not available, we may be required to
terminate, significantly modify or delay our development
programs, reduce our planned commercialization efforts, or
obtain funds through collaborators that may require us to
relinquish rights to our technologies or product candidates that
we might otherwise seek to develop or commercialize
independently. We may elect to raise additional funds even
before we need them if the conditions for raising capital are
favorable.
Contractual
Obligations, Commitments and Contingencies
To date, we have not entered into long-term minimum purchase
commitments with suppliers. Our principal commitments consist of
obligations relating to our international clinical trial and
obligations under the lease for our facility in Minneapolis,
Minnesota, and certain office equipment.
The following table summarizes our outstanding contractual
obligations as of December 31, 2009:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Payments Due by Period
|
|
|
|
|
|
|
|
Less than
|
|
|
|
|
|
|
|
|
After
|
|
|
|
|
Total
|
|
|
1 Year
|
|
|
1-3 Years
|
|
|
4-5 Years
|
|
|
5 Years
|
|
|
|
|
International clinical trial obligations(1)
|
|
$
|
241
|
|
|
$
|
241
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
Operating lease obligations(2)
|
|
|
52
|
|
|
|
46
|
|
|
|
6
|
|
|
|
—
|
|
|
|
—
|
|
|
Key supplier purchase commitment(3)
|
|
|
43
|
|
|
|
43
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
336
|
|
|
$
|
330
|
|
|
$
|
6
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(In thousands)
|
|
|
| (1)
|
The outstanding obligations for our international clinical trial
include our estimated costs to complete patient monitoring and
follow-up
and trial data collection and analysis for our 90 patient
multi-center clinical trial conducted outside the United States.
Patient enrollment in this trial was completed in July 2009.
|
| |
| (2)
|
Operating lease obligations represent future minimum lease
payments under non-cancelable operating leases for our facility
and certain office equipment. The operating lease obligation for
our corporate facility ends September 30, 2010. We are
currently in negotiations with our landlord to renew this lease
and secure additional space adjacent to our current offices. As
of the date of this prospectus, we have not reached such an
agreement with our landlord.
|
| |
| (3)
|
The amount presented reflects the undelivered portion of an
outstanding purchase order with one of our key suppliers. To
date we have not entered into any long term supply agreements
with any of our vendors. As of December 31, 2009, remaining
outstanding purchase orders were not significant.
|
Royalty
Payments
The core intellectual property relating to our eSVS MESH,
including five patent applications pending in the United States
and nine patent applications pending in countries outside the
United States, was acquired from Medtronic, Inc. pursuant to an
Assignment and License Agreement dated October 9, 2007.
Pursuant to the Assignment and License Agreement, Medtronic has
also assigned to us certain patents relating to a brushed ePTFE
36
vascular graft, or the Brushed Graft Product, and licensed to us
certain technology to produce platelet-poor plasma for use in
connection with the Brushed Graft Product.
As consideration for the assignment of intellectual property
relating to the eSVS MESH and the Brushed Graft Product, we have
agreed to pay Medtronic an aggregate of $20.0 million upon
the achievement of certain sales milestones relating to the eSVS
MESH and the Brushed Graft Product and a royalty of 4% on sales
of our eSVS MESH and the Brushed Graft Product. The royalty will
terminate upon the earlier of the expiration of all of the
patents and patent applications, or when the aggregate royalties
paid reach $100.0 million.
In the recitals to the Assignment and License Agreement, we
stated that we would use our reasonable best efforts to develop
and commercialize both the Brushed Graft Product and the eSVS
MESH. While we have undertaken activities to develop and
commercialize the Brushed Graft Product, we are currently
primarily focused on the development of our eSVS MESH for use in
CABG surgery and additional applications of our eSVS MESH. Any
or all licenses granted to us pursuant to our agreement with
Medtronic may be terminated and potentially all of the core
intellectual property and patent rights related to our eSVS MESH
shall revert to Medtronic, upon notice by Medtronic, (i) if
we become insolvent, make an assignment for the benefit of
creditors, go into liquidation or receivership or otherwise lose
legal control of our business; and (ii) if we determine to
cease commercializing either the Brushed Graft Product or our
eSVS MESH, then Medtronic may terminate our license to patent
rights related to the Brushed Graft Product or the patent rights
related to our eSVS MESH, as applicable. If we cease
commercialization of the Brushed Graft Product and not the eSVS
MESH, we believe that Medtronic is entitled to terminate only
the license to patent rights related to the Brushed Graft
Product and not the patent rights related to the eSVS MESH.
Similarly, if we cease commercialization of the eSVS MESH but
not the Brushed Graft Product, we believe that Medtronic is
entitled to cause the reversion of the patent rights related to
the eSVS MESH but not terminate our license to patent rights
related to the Brushed Graft Product. We recognize, however,
that the terms of our agreement with Medtronic are susceptible
to differing interpretations.
Off-Balance Sheet
Arrangements
Since inception, we have not engaged in any off-balance sheet
activities as defined in
Regulation S-K
Item 303(a)(4).
Recent Accounting
Pronouncements
In June 2009, the FASB issued FASB ASC 105, Generally
Accepted Accounting Principles, which establishes the FASB
Accounting Standards Codification as the sole source of
authoritative generally accepted accounting principles. Pursuant
to the provisions of FASB ASC 105, we have updated references to
GAAP in our financial statements issued for the period ended
December 31, 2009. The adoption of FASB ASC 105 did not
impact our financial position or results of operations.
In June 2008, the FASB issued FASB ASC
815-40,
Derivatives and Hedging, which provides guidance on how
to determine if certain instruments (or embedded features) are
considered indexed to a company’s own stock, including
instruments similar to warrants to purchase the company’s
stock. FASB ASC
815-40
clarifies the determination of whether equity-linked instruments
(or embedded features), such as our convertible notes or options
to purchase our common stock, are considered indexed to our own
stock, which would qualify as a scope exception and therefore be
exempt from the application of FASB ASC 815. FASB ASC
815-40
became effective January 1, 2009. Any outstanding
instrument at the date of adoption requires a retrospective
application of the accounting through a cumulative effect
adjustment to retained earnings upon adoption. Our adoption of
this guidance had a material impact on our financial position
and results of operations, as described in Note 6 to our
financial statements.
37
Business
Overview
We are a medical device company focused on developing,
manufacturing and commercializing our innovative external
saphenous vein support technology, or eSVS MESH, for use in
coronary artery bypass grafting, or CABG, surgery. Our eSVS MESH
is a nitinol mesh sleeve that, when placed over a saphenous vein
graft during CABG surgery, is designed to improve the structural
characteristics and long-term performance of the vein graft.
CABG is one of the most commonly performed surgeries in the
United States, with the American Heart Association estimating
that 448,000 CABG procedures were performed in the United States
in 2006. In addition, the Millennium Research Group, an
independent market research firm, estimates that there will be
165,000 CABG procedures per year in Europe by 2013. In CABG
procedures, surgeons harvest blood vessels, including the
internal mammary artery from the chest and the saphenous vein
from the leg, and attach the harvested vessels to bypass, or
provide blood flow around, blocked coronary arteries. The
effectiveness of the procedure, however, is often limited by the
failure rate of saphenous vein grafts, which has been shown in
various studies to range from 6% to 30% one year after surgery
and 60% ten years after surgery. Failure of these grafts,
typically evidenced by partial or complete blockage and reduced
or stopped blood flow, can lead to the need for further coronary
interventions up to and including additional CABG procedures. We
believe the use of our eSVS MESH with saphenous vein grafts in
CABG surgery could improve the long-term outcome of CABG
procedures, including improved openness, or patency, and
improved blood flow through the saphenous vein graft, resulting
in a reduced need for costly and potentially complicated
reoperations or revascularization procedures.
According to the American Heart Association, approximately
17.6 million people in the United States have coronary
artery disease, and approximately 587,000 people in the
United States die each year as a result of the disease. In
addition, according to a 2007 World Health Organization report,
approximately 7.2 million people worldwide died of coronary
heart disease in 2002. The direct and indirect cost of coronary
artery disease to the U.S. economy is estimated to be over
$177 billion in 2010. Physicians and patients may select
among a variety of treatments to address coronary artery
disease, including pharmaceutical therapy, balloon angioplasty,
stenting with bare metal or drug-eluting stents, and CABG
procedures, with the selection often depending upon the stage of
the disease and the age of the patient. An independent study
comparing CABG and implantation of drug-eluting stents shows
that CABG is the more effective long-term treatment for coronary
artery disease, achieving the best long-term patient outcomes as
measured by survival rate and need for re-intervention
12 months after surgery. Moreover, patients with severe and
multi-vessel coronary artery disease often cannot be effectively
treated with methods other than CABG. The prevalence of coronary
artery disease and the success rates for CABG procedures versus
other treatments for coronary artery disease has made CABG
surgery one of the most commonly performed surgeries in the
United States. Based on a report published by the Millennium
Research Group, moderate growth in coronary artery bypass
procedures is expected in the United States through 2012 and in
Europe through 2013, largely due to the increase in procedure
volumes caused by rising rates of coronary artery disease and
the need for repeat revascularizations.
According to results published in the European Journal of
Cardio-Thoracic Surgery in 2006, each CABG procedure involves an
average of 3.3 bypass grafts, typically consisting of the left
internal mammary artery, or LIMA, for one graft and the
saphenous vein for the remaining 2.3 grafts per procedure.
Saphenous vein grafts fail more frequently than LIMA grafts due
to differences in structure and size of saphenous vein grafts as
compared to LIMA grafts. Unlike the LIMA, which is a
thick-walled artery intended to handle the high pressure blood
flow from the heart, saphenous veins are thin-walled vessels
that are intended for a low-pressure venous environment.
Saphenous veins are also typically larger than the coronary
arteries to which they are attached and this difference in size
slows blood flow, adding stress to the vessel wall and
increasing the risk of thrombosis, or blood clotting. When the
vein grafts used to bypass a blocked artery are exposed to the
high pressure of arterial flow, there is significant stress on
the thin wall of the veins. The vein responds to this injury by
causing its walls to thicken in a manner that often leads to
failure of the bypass graft.
Our eSVS MESH is a nitinol mesh sleeve that is placed over the
saphenous vein graft during CABG surgery and is designed to
constrict the vein and prevent expansion of the vein graft and
resulting injury due to increased
38
pressure. The constriction of the vein graft also causes the
diameter of the graft, or lumen, to more closely match the
diameter of the target coronary artery to which it is attached,
thereby reducing blood flow disruption. Our eSVS MESH is
designed to be applied quickly and is compatible with most
current CABG surgery protocols. In addition, nitinol is commonly
used in many other implantable medical devices.
We have completed enrollment in a 90 patient multi-center
clinical trial conducted outside the United States. The primary
effectiveness endpoint of this trial is statistical
non-inferiority of the patency of eSVS MESH vessels as compared
to control vessels at nine-months post-implant. Effectiveness
data, which is based on angiographic patency, is being collected
at this time. Preliminary safety data has indicated that our
eSVS MESH and implant procedure do not result in an increase in
patient complications during or after surgery. We completed
enrollment in this trial in July 2009, and as of March 1,
2010, all 90 patients have been implanted for six months or
more, 83 patients have been implanted for nine months or
more, and 50 patients have been implanted for
12 months or more. This trial formed the basis for our CE
Mark application submitted in February 2010.
We expect to receive CE Mark approval and to begin marketing our
eSVS MESH in select European Union markets in the second half of
2010. The U.S. Food and Drug Administration, or FDA, is
reviewing our application for an investigational device
exemption, or IDE, which, if granted, will allow us to begin
clinical trials of our eSVS MESH in the United States. We
anticipate beginning enrollment in a United States IDE trial in
the second half of 2010. We could be delayed by adverse clinical
results or regulatory complications, and we may never receive
marketing approval.
We are pursuing additional applications for our eSVS MESH,
including applications for use in peripheral artery bypass
surgery, for use with coronary allografts, and for use in
arteriovenous, or AV, fistula dialysis applications. In
peripheral artery bypass surgery, saphenous vein grafts are used
to bypass obstructed arterial vessels in the legs. Coronary
allografts are saphenous veins obtained from cadavers that are
used in CABG procedures for patients who do not have appropriate
arterial or venous conduits. An AV fistula is a surgically
created connection, or fistula, between an artery and a vein
used to provide access to the circulatory system of patients
with kidney disease for chronic dialysis treatment. We believe
that these applications could have significant commercial
potential.
Our
Strategy
Our objective is to achieve significant market adoption of our
eSVS MESH technology in CABG and other vascular applications.
Key elements of our strategy to achieve this objective include
the following:
|
|
|
| |
•
|
Work with respected medical centers and key thought leaders
to demonstrate and communicate the potential benefits of our
eSVS MESH. We are in the process of collecting effectiveness
data from our 90 patient, multi-center trial conducted
outside the United States and are currently seeking an IDE from
the FDA to begin clinical trials in the United States. We
believe that it will be important to increase the awareness of
our eSVS MESH by collaborating with key opinion leaders at
leading academic and medical institutions and supporting
post-approval marketing studies and publication of peer-reviewed
articles. We have formed clinical relationships with key
surgeons at several leading cardiovascular surgery and CABG
centers.
|
| |
| |
•
|
Commercialize our eSVS MESH in select European markets.
We expect to receive CE Mark approval and to begin marketing our
eSVS MESH in select European markets in the second half of 2010.
We plan to engage independent distributors experienced in their
respective European markets to promote and sell our eSVS MESH.
Concurrent with this effort, we intend to commence activities to
seek regulatory approval to begin marketing in other
international markets.
|
| |
| |
•
|
Obtain regulatory approval and commercialize our eSVS MESH in
the United States. The FDA is reviewing our application for
an IDE, and we expect to receive approval to commence our IDE
trial in the first half of 2010. We are in discussions with 12
clinical trial sites to participate in our IDE trial, six of
which have been engaged as of April 1, 2010. We expect to
commence enrollment in our IDE trial in the second half of 2010.
Over the longer term, we will assemble data from our United
States human clinical trial in support of an application for PMA
approval from the FDA. If we receive the necessary regulatory
approval, we plan to commercially introduce our eSVS MESH in the
United States through independent distributors with access to
key CABG centers and key physicians.
|
39
|
|
|
| |
•
|
Conduct trials to expand indications for our eSVS MESH.
We plan to begin preclinical trials in 2010 designed to provide
proof of concept for the use of our eSVS MESH in peripheral
artery bypass surgery, for use with coronary allografts, and for
use with AV fistulas in dialysis patients.
|
Treatment of
Coronary Artery Disease
According to the American Heart Association, approximately
17.6 million people in the United States have coronary
artery disease, and approximately 587,000 people in the
United States die each year as a result of the disease. In
addition, according to a 2007 World Health Organization report,
approximately 7.2 million people worldwide died of coronary
heart disease in 2002. The direct and indirect cost of coronary
artery disease to the U.S. economy is estimated to be over
$177 billion in 2010. Primary treatment options for
coronary artery disease are pharmaceutical therapy, balloon
angioplasty, intravascular stents, and CABG surgery. A
description of each of these options is provided below:
Pharmaceutical
Therapy
In patients with less severe disease, pharmaceuticals remain the
primary treatment approach and include drugs such as platelet
adhesion inhibitors or drugs that reduce the blood cholesterol
or triglyceride levels. For more serious disease, however,
pharmacological therapy alone is often inadequate.
Balloon
Angioplasty
Percutaneous transluminal coronary angioplasty, commonly
referred to as balloon angioplasty, is a surgical procedure that
involves the dilation of the obstructed artery with a balloon
catheter. Angioplasty is generally successful in increasing
immediate blood flow and, relative to current surgical
procedures, offers the benefits of shorter periods of
hospitalization, quicker recovery times, reduced patient
discomfort and lower cost. However, according to a trial
published in the journal Circulation in 2006, over 40% of
vessels treated with balloon angioplasty return to their
pre-treatment, narrowed size, a process known as restenosis,
within six months following the procedure.
Intravascular
Stents
High rates of restenosis following treatment by balloon
angioplasty led to the introduction of stents, mesh-like
metallic tubes that are placed within the narrowed portion of
the coronary vessel to hold the vessel open after the
angioplasty balloon has been removed. Although clinical outcomes
for procedures using stents reflect an improvement over balloon
angioplasty alone, the effectiveness of stents is still limited
by restenosis, which for bare metal stents occurs in about 20%
of cases within six months of the procedure.
Drug eluting stents are coated with specially formulated,
slow-release drugs designed to prevent restenosis. According to
the FDA in 2008, drug eluting stents were shown in clinical
trials to reduce the rate of restenosis within one year after
placement to 10%. Drug eluting stents are widely used, with a
current market share relative to total stent usage in the range
of 70%. However, some studies have been presented that associate
drug eluting stents with late stage thrombosis, or clotting,
which can be an adverse event.
Despite the advancements and market success of drug-eluting
stents and angioplasty therapies, these interventional
procedures may be less effective than surgical procedures in
addressing diffuse progressive coronary artery disease. In this
advanced stage of coronary artery disease, intervention is
required for multiple vessels, many of which are less than two
millimeters in internal diameter, a diameter currently
unsuitable for angioplasty and stenting. In addition, stents
have been shown to be difficult to place in patients with
coronary lesions in sections with vessel branches and in
patients with narrowings in the left main coronary artery. In a
study published in the New England Journal of Medicine in
January 2008 that compared drug-eluting stents with CABG in
multivessel coronary disease, death rates and revascularization
rates were higher in patients receiving drug-eluting stents than
in patients receiving CABG, even though the cohort of patients
receiving CABG was older and had more severe coronary disease.
40
CABG
Surgery
Coronary Artery Bypass Grafting involves the construction of an
alternative path to bypass a narrowed or occluded coronary
artery and restore blood flow from the aorta to an area past the
occlusion. This procedure is normally accomplished using
saphenous veins from the leg and the LIMA from the chest as
bypass grafts. Most commonly, the LIMA is utilized for bypassing
the left anterior descending artery of the heart, or LAD, while
saphenous veins are utilized for bypassing other coronary
arteries.
For vein grafts, one end of the harvested vessel is then
generally attached to the aorta for blood inflow, and the
opposite end is attached to the target coronary vessel. If a
mammary artery is used as the bypass graft, it must be dissected
from the chest wall, leaving one end in place on the aorta,
while the opposite end is attached to the target vessel,
providing uninterrupted blood flow from the arterial
circulation. Once in place, these grafts provide sufficient
blood flow to bypass the narrowed or occluded portion of the
coronary artery. The following diagram illustrates the use of
the internal mammary artery graft and saphenous vein graft in
CABG surgery:
Current
Disadvantages of Saphenous Vein Grafts
Since its first successful use in the 1960’s, the saphenous
vein graft has been one of the most commonly used conduits in
CABG surgery. Some of the main advantages of using the saphenous
vein include its ease of accessibility, its ease of handling,
and the number of grafts, typically three, that can be
constructed from a single vein. Despite these advantages and the
widespread use of saphenous veins in CABG surgery, several
issues have been identified, such as:
|
|
|
| |
•
|
Pressure normally exerted on veins is much lower than the
pressure on arteries. Arterial pressure is normally
80-120 mm Hg
while central venous pressure is normally about 3-7 mm Hg.
|
| |
| |
•
|
Veins do not have the strong muscular wall seen in arteries.
Therefore, when placed under higher arterial pressures, the
veins typically dilate, or expand.
|
| |
| |
•
|
Veins have large lumens as compared to arteries, resulting in a
mismatch of lumen diameters when a saphenous vein graft is
connected to a coronary artery. This size mismatch results in
slow, sluggish blood flow in the vein graft with more stress
placed on the wall of the vein due to blood volume.
|
The higher pressure of arterial blood flow and the size mismatch
that results when a saphenous vein is used as a graft in CABG
surgery often cause the vein to expand, damaging the lining of
the vein. The vein responds to this damage by causing its walls
to thicken in a manner that often leads to failure of the bypass
graft. Smooth muscle cells proliferate in the middle layer of
the vein wall and migrate to the inner surface of the vein in a
process known as neointimal hyperplasia. The resulting
accumulation of activated smooth muscle cells secrete
inflammatory and growth factors leading to a stenotic
build-up and
graft failure over time. The failure rates of saphenous vein
grafts
41
in CABG procedures is well documented in the scientific
literature. A sampling of data from some of the larger benchmark
studies is provided below:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Saphenous Vein Graft Failure Rates
|
|
|
|
|
|
1 Year
|
|
5 Year*
|
|
10 Year*
|
|
|
|
|
|
Number of
|
|
Failure
|
|
Number of
|
|
Failure
|
|
Number of
|
|
Failure
|
|
Year
|
|
Author
|
|
Patients
|
|
Rate
|
|
Patients
|
|
Rate
|
|
Patients
|
|
Rate
|
|
|
|
1984
|
|
Barner, et al.
|
|
|
248
|
|
|
|
7
|
%
|
|
|
112
|
|
|
|
26
|
%
|
|
|
—
|
|
|
|
—
|
|
|
1996
|
|
Fitzgibbon, et al.
|
|
|
3993
|
|
|
|
19
|
%
|
|
|
1978
|
|
|
|
25
|
%
|
|
|
—
|
|
|
|
—
|
|
|
2004
|
|
Goldman, et al.
|
|
|
660
|
|
|
|
8
|
%
|
|
|
336
|
|
|
|
25
|
%
|
|
|
368
|
|
|
|
61
|
%
|
|
2005
|
|
Alexander
|
|
|
2000
|
|
|
|
30
|
%
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
2009
|
|
Serruys, et al.
|
|
|
870
|
|
|
|
6
|
%
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
2009
|
|
Puskas, et al.
|
|
|
183
|
|
|
|
18
|
%
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
* Five and ten year data is not available for those studies
for which data is not presented in these columns.
Failure of these grafts, typically evidenced by partial or
complete blockage and reduced or stopped blood flow, can lead to
chest pain, congestive heart failure, irregular heart beat,
myocardial infarction, or MI, revascularization or death. A
repeat of a CABG procedure to repair a failing or failed graft
is a technically more difficult procedure with mortality rates
three to five times higher than the original CABG procedure.
eSVS
MESH—Our Solution
Our eSVS MESH is designed to improve the long-term outcome of
CABG procedures by addressing limitations of unsupported
saphenous veins. Our eSVS MESH is a highly flexible,
semi-compliant, kink-resistant extravascular tubular prosthesis
made of knitted nickel/titanium, or nitinol, wire mesh. Our eSVS
MESH is designed to be fitted like a sleeve over vein grafts,
thereby providing the vein graft with physiological attributes
similar to those of an artery.
An artery has a thick muscular wall to handle higher pressures,
and a relatively small lumen that produces higher blood
velocities, offering less chance for blood to pool and clot. In
contrast, a vein has a thinner, less muscular wall due to the
lower pressures normally found in veins and a larger lumen
designed to maintain these lower pressures. We believe that
larger, thinner-walled veins will have greater potential benefit
from our eSVS MESH. Although we are pursuing commercialization
of a 3.0 mm diameter eSVS MESH in select European markets, in
the United States we will initially only be pursuing eSVS MESH
diameters of 3.5, 4.0 and 4.5 mm.
Our eSVS MESH is designed to provide the vein graft with
physiological attributes similar to those of an artery by
reducing the lumen diameter and strengthening the vessel wall.
We believe the key benefits of our eSVS MESH technology include:
|
|
|
| |
•
|
Structural support designed to inhibit vessel expansion and
resulting damage to the vessel, which can prevent a thickening
of the vessel wall over time, or hyperplasia, and resulting
graft failure.
|
| |
| |
•
|
Radial constriction designed to cause the diameter of the graft,
or lumen, to be consistent in size and more closely match the
diameter of the target coronary artery to which it is attached,
thereby increasing blood flow velocities, reducing the potential
for clot formation, and inhibiting hyperplasia.
|
| |
| |
•
|
Compatibility with current CABG procedures, including on-pump or
off-pump procedures, and open or endoscopic vein harvest
methods. Except for the placement of our eSVS MESH on the
saphenous vein graft, the surgical steps to use a saphenous vein
graft with our eSVS MESH are the same as would be performed for
any coronary artery bypass procedure utilizing unsupported
saphenous vein grafts. We do not expect, nor have we seen, a
significant increase in CABG procedure time due to eSVS MESH use.
|
Our eSVS MESH technology consists of the following:
|
|
|
| |
•
|
eSVS MESH (25 cm length, and either 3.0, 3.5, 4.0, or 4.5 mm in
diameter);
|
42
|
|
|
| |
•
|
INTRODUCER for use in placing our eSVS MESH on the saphenous
vein (one for each diameter of our eSVS MESH);
|
|
|
|
| |
•
|
SUTURE SNARE for use in loading our eSVS MESH onto the saphenous
vein; and
|
|
|
|
| |
•
|
SIZING TOOL for use in choosing the correct device size based on
saphenous vein diameter.
|
Clinical
Development of our eSVS MESH
International
Human Clinical Trial
The first human clinical trial of our eSVS MESH is a
prospective, randomized, controlled non-inferiority trial, where
each patient is randomized to receive a saphenous vein graft
with our eSVS MESH to bypass either the right coronary artery,
or RCA, or the circumflex artery, or Cx, two arteries commonly
bypassed during CABG. The RCA or Cx saphenous vein graft not
chosen to receive our eSVS MESH serves as the control artery. To
ensure Good Clinical Practices compliance, outside resources are
utilized for data collection and analysis, including a contract
research organization for data entry and verification, a
physician clinical events committee for the review and
evaluation of adverse events, and an angiographic core lab for
assessment of saphenous vein graft patency.
Seven international centers enrolled 90 patients in this
trial. Enrollment in this trial closed on July 21, 2009. As
of March 1, 2010, all 90 patients have been implanted
for six months or more, 83 patients have been implanted for
nine months or more, and 50 patients have been implanted
for 12 months or more.
The international sites involved in this trial, and the number
of patients enrolled at each site, is provided below:
| |
|
|
|
|
|
|
|
|
|
|
Number of Patients
|
|
|
Center Name
|
|
Enrolled
|
|
|
|
|
Schleswig-Holstein University Hospital, Kiel, Germany
|
|
|
25
|
|
|
National University Hospital, Singapore
|
|
|
21
|
|
|
University Of Cape Town, Cape Town, South Africa
|
|
|
20
|
|
|
Hospital Regional De Sion, Sion, Switzerland
|
|
|
9
|
|
|
Auckland City Hospital, Auckland, New Zealand
|
|
|
8
|
|
|
Hospital Universitario 12 de Octubre, Madrid, Spain
|
|
|
5
|
|
|
Prince Charles Hospital, Brisbane, Australia
|
|
|
2
|
|
|
|
|
|
|
|
|
Total
|
|
|
90
|
|
|
|
|
|
|
|
|
|
43
The primary safety endpoint of this trial is statistical
non-inferiority based on the total rate of major adverse cardiac
and cerebral events, or MACCE, at
30-days
post-implant as compared to published literature. MACCE is a
composite of MI, stroke, revascularization, including surgery or
stenting, and death. In summary, there were four adverse events
that met the protocol definition of MACCE, which compared
favorably to the protocol objective performance criteria, or
OPC. OPC are performance criteria based on broad sets of data
from historical databases, such as literature or registries,
that are generally recognized as acceptable values. For this
trial, the OPC utilized was a compilation of published
literature that presented
30-day
post-implant MACCE rates for CABG surgery patients, separating
the MACCE category into the composite factors listed above. In
addition, none of the adverse events were determined to be
“Definitely Device Related” or “Probably Device
Related.”
A table summarizing these results is shown below:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Objective
|
|
|
|
|
Trial Data
|
|
|
Performance Criteria
|
|
|
|
|
MI
|
|
|
2(2.2
|
%)
|
|
|
2.8
|
%
|
|
Stroke
|
|
|
2(2.2
|
%)
|
|
|
1.8
|
%
|
|
Revascularization
|
|
|
0
|
|
|
|
0
|
%
|
|
Death*
|
|
|
0
|
|
|
|
4.8
|
%
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
|
4(4.4
|
%)
|
|
|
9.4
|
%
|
|
|
|
|
|
|
|
|
|
|
|
|
* One patient death eight months after surgery due to
non-cardiac causes
The primary effectiveness endpoint of this trial is statistical
non-inferiority of angiographic stenosis, or patency, of eSVS
MESH vessels as compared to control vessels at nine-months
post-implant. A vessel is considered to be patent if there is
less than 50% stenosis. Collection of this data is ongoing.
We have made the following observations during this trial that
have led to improvements in the device and implant procedure:
|
|
|
| |
•
|
One center had implant methods incompatible with our eSVS MESH.
Specifically, surgeons at this site did not make an adequate
lateral slit on the side of the saphenous vein graft that lies
on the heart, or heel slit, in grafts that had our eSVS MESH,
causing a higher than anticipated number of graft closures at
the aortic anastomotic site, the location where the saphenous
vein graft is sewn onto the aorta. By design, a saphenous vein
graft with an eSVS MESH in place will not dilate. Without this
heel slit in place, a saphenous vein graft with our eSVS MESH in
place does not permit adequate blood flow into the vein graft at
the aortic anastomotic site, potentially resulting in failure of
the graft. We have modified our instructions for use to provide
clear direction to surgeons on how to make the heel slit when
using our eSVS MESH.
|
| |
| |
•
|
The amount of reduction in the diameter of the saphenous vein
grafts, or downsizing, prescribed in our instructions for use
and sizing tool was too aggressive, resulting in a higher than
anticipated closure rate in saphenous vein grafts utilizing the
smaller eSVS MESH diameters. This resulted in lumen diameters
that were very small and did not remain patent. We have modified
our instructions for use and sizing tool to decrease the amount
of downsizing applied to saphenous vein grafts by our eSVS MESH.
|
United States
IDE Trial
The FDA is reviewing our application for an IDE which, if
granted, will allow us to begin clinical trials of our eSVS MESH
in the United States. We are currently amending our IDE
application and anticipate obtaining IDE approval in the first
half of 2010, and if approved, we expect to commence enrollment
in our IDE trial in the second half of 2010. The FDA has not
approved our IDE submission and the summary below of the trial
design is based upon our current expectations of the IDE
protocol. The ultimate trial design, if and when approved by the
FDA, may be materially different than our expectations set forth
below.
We expect that the FDA will require a feasibility phase as part
of our IDE trial. We are proposing that the first
60 patients enrolled in the trial will have a
90-day
post-implant assessment of device patency, in addition to normal
follow-ups
prescribed in the clinical trial protocol. We are also proposing
that success criteria for this
44
feasibility analysis be non-inferiority. Once the
90-day
follow-up is
completed and the data is reported to the FDA, subject to FDA
consent, we expect to continue enrollment at all sites and these
feasibility phase patients will be included in the IDE trial
data set. The feasibility phase will delay completion of our IDE
trial by at least five to six months compared to a trial that
does not incorporate a feasibility phase.
The primary safety endpoint of the IDE trial is expected to be
statistical non-inferiority based on the total rate of major
adverse cardiac events, or MACE, at
30-days
post-implant as compared to published literature. The primary
effectiveness endpoint of this trial is expected to be
statistical superiority of the patency of eSVS MESH vessels as
compared to control vessels at nine-months post-implant. This
effectiveness endpoint is more rigorous than the effectiveness
endpoint of our international trial, which was a non-inferiority
comparison. The IDE protocol reflects our prior observations
from the international trial. For example, the IDE protocol will
only include eSVS MESH sizes of 3.5 mm or greater in diameter
and will include detailed instruction regarding preparation of
anastomotic sites. We expect the IDE trial to require enrollment
of 366 patients and include up to 20 clinical trial sites.
Of these 366 patients, we expect that we will be required
to perform nine-month post-implant angiograph procedures on at
least 293 patients. We would be dependent upon our clinical
sites and enrolled subjects for compliance in returning and
agreeing to the nine-month angiograms and expect that a certain
percent of subjects will either not return or will refuse the
nine-month angiogram. We are in discussions with 12 clinical
trial sites to participate in our IDE trial, six of which have
been engaged as of April 1, 2010. We expect to commence
enrollment in the IDE trial, in the second half of 2010.
Enrollment in the IDE trial is expected to take approximately
18 months, and
follow-up is
expected to take up to an additional year from the completion of
enrollment. Prior to commercializing our eSVS MESH in the United
States, we will be required to submit a Pre-Market Approval, or
PMA, application to the FDA. Approval of a PMA by the FDA
generally takes approximately one year after the application. We
could be delayed by adverse clinical results or regulatory
complications, and we may never receive marketing approval.
Preclinical
Testing
Preclinical trials of our eSVS MESH technology have been
presented in peer-reviewed journals, including The Journal of
Thoracic and Cardiovascular Surgery in February 2008 and the
Journal of Vascular Surgery in June 2009. Between 2002 and 2007,
Medtronic, Inc. sponsored multiphase trials with the
Cardiovascular Research Unit of the Christiaan Barnard
Department of Cardiothoracic Surgery at the University of Cape
Town in South Africa, or UCT, to evaluate the effects of various
designs of external nitinol mesh sleeves on the vascular
architecture of vein grafts used in CABG and peripheral bypass
procedures. This multiphase research concluded that the use of
our eSVS MESH showed a statistically significant decrease in
intimal hyperplasia after six months of implantation. In
addition to these trials, Medtronic, Inc. and UCT collaborated
on stress, fatigue, durability, and finite element analysis of
knitted eSVS MESH designs.
In October 2007, we acquired ownership of the core intellectual
property relating to our eSVS MESH from Medtronic, Inc. and
initiated additional work on the technology. This work included
developing additional sizes of our eSVS MESH, completing
required preclinical and biological testing of the product and
accessories, developing packaging and labeling for our eSVS
MESH, and creating product documentation intended to comply with
relevant FDA and international standards.
In addition, we initiated and completed a series of animal
trials utilizing sheep to confirm that our eSVS MESH, as
manufactured by us, performed as expected, and produced the
expected results. These animal trials showed a statistically
significant inhibition of the formation of intimal hyperplasia
when our eSVS MESH was used with a saphenous vein graft in CABG
procedures. However, sheep arterial pressures and vasculature
differ from humans, and human clinical studies may not be
consistent with animal trial results.
45
Additional eSVS
MESH Applications
Additional development projects based on our eSVS MESH
technology that we are exploring and may advance include:
Peripheral
Grafts
In this clinical application, saphenous vein grafts are used to
bypass obstructed arterial vessels in the legs. We have
conducted initial preclinical trials for this application,
utilizing saphenous vein grafts with our eSVS MESH in place. We
plan to conduct further preclinical trials of this application
in 2010, in support of future potential regulatory submissions.
Coronary
Allografts
In this clinical application, cadaver, or allograft, saphenous
vein grafts are used in CABG procedures for patients who do not
have appropriate arterial or venous conduits. We have had
discussions with suppliers of this allograft material to
determine usage patterns. We plan to conduct initial preclinical
trials of this application in 2010.
Arteriovenous
Fistula
In this clinical application, a fistula, or connection, is made
between an artery and a vein, normally in the non-dominant arm,
for circulatory system access in patients requiring chronic
dialysis. We plan to conduct initial preclinical trials of this
application in 2010.
Sales and
Marketing
Europe and
Other International Markets
We have submitted an application to obtain the CE Mark for our
eSVS MESH. If approved, the CE Mark will allow us to sell our
eSVS MESH for use in CABG procedures in 32 countries within the
European Union, the European Economic Area, and the European
Free Trade Association. Provided we receive CE Mark approval, we
plan to begin sales in select European nations beginning in the
second half of 2010. Our plan is to utilize independent
distributors to commercialize our technology in Europe, and we
have identified independent distributors that may be contracted
to conduct sales in these markets. These distributors will be
supported by our
U.S.-based
staff with regard to training and promotional materials. We
intend to work with our distributors with respect to product
reimbursement and have also identified other third parties that
may be contracted to assist in obtaining country-specific
product reimbursement.
As the European cardiac surgery market is characterized by
centralized, high-volume cardiac surgery centers, we believe
this market can be effectively addressed through a small,
highly-focused independent distributor network.
We will be an active participant in post-market clinical trials
aimed at validating the long-term outcomes of patients who
receive our eSVS MESH. These studies will be designed to show
that eSVS MESH patients require less revascularization
procedures than standard CABG patients, thereby also reducing
the costs associated with revascularization procedures for eSVS
MESH patients. We envision that the results of these studies
will be presented at scientific sessions and presented in
peer-reviewed journals, thereby increasing the visibility and
adoption of our eSVS MESH. These studies will also be used to
support applications for public hospital reimbursement in those
countries that require outcomes data for such reimbursement.
We believe that the CE Mark will also be a gateway approval that
will allow us to begin regulatory submissions to obtain
marketing approval in other select markets, including South
Africa, Canada, Australia, New Zealand and Argentina.
United
States
We are required to conduct a PMA IDE trial in the United States.
Enrollment in this trial,
follow-up of
trial patients, and subsequent PMA approval are anticipated to
take approximately three years. If the U.S. IDE trial is
46
successful and our resulting PMA is approved, we expect to
launch our eSVS MESH in the United States no sooner than 2013.
Based upon the 2009 Society of Thoracic Surgeons Adult Cardiac
Surgery Database, we believe CABG surgeries were performed in
approximately 1,150 U.S. hospitals in 2009.
According to an article published in the journal Health Affairs
in 2007, the volume of CABG surgeries performed per
U.S. hospital in 2003 was:
| |
|
|
|
|
|
|
|
|
|
|
|
|
CABG Volume per Year
|
|
Percent of Hospitals in this Category
|
|
|
Number of Hospitals in this Category
|
|
|
|
|
<130 cases
|
|
|
29
|
%
|
|
|
310
|
|
|
130-199 cases
|
|
|
20
|
%
|
|
|
214
|
|
|
200-314 cases
|
|
|
22
|
%
|
|
|
235
|
|
|
315-484 cases
|
|
|
15
|
%
|
|
|
160
|
|
|
>484 cases
|
|
|
14
|
%
|
|
|
150
|
|
|
|
Based upon this information, approximately 545 hospitals in the
United States perform at least 200 CABG surgeries each year. Of
these, approximately 310 hospitals perform more than 315
surgeries each year. Our initial marketing focus will be on
these 300 to 500 hospitals.
Our plan is to utilize independent distributors to commercialize
our eSVS MESH in the U.S., and we have identified independent
distributors that may be contracted to conduct sales. These
distributors will be supported by Kips Bay staff with regard to
training and promotional materials. We have contracted outside
reimbursement experts to assist in obtaining Centers for
Medicare & Medicaid Services, or CMS, product
reimbursement.
Intellectual
Property
As of December 31, 2009, we had six patent applications
pending in the United States and nine patent applications
pending in countries outside the United States covering various
aspects of our eSVS MESH. We also have one international patent
application pending, which gives us the opportunity to file in
more individual countries. Some of our pending patent
applications have been examined, and currently stand rejected.
We will continue to pursue obtaining patents from these
applications, but it is possible that our pending patent
applications may not issue as patents or, if issued, may not
issue in a form that will be advantageous to us. We expect to
launch our eSVS MESH in Europe before any of our pending
European patent applications issue as patents.
The core intellectual property relating to our eSVS MESH,
including five patent applications pending in the United States
and nine patent applications pending in countries outside the
United States, was acquired from Medtronic, Inc. pursuant to an
Assignment and License Agreement dated October 9, 2007.
Pursuant to the Assignment and License Agreement, Medtronic has
also assigned to us certain patent rights relating to a brushed
ePTFE vascular graft, or the Brushed Graft Product, and licensed
to us certain technology to produce platelet-poor plasma for use
in connection with the Brushed Graft Product.
As consideration for the assignment of intellectual property
relating to the eSVS MESH and the Brushed Graft Product, we have
agreed to pay Medtronic an aggregate of $20.0 million upon
the achievement of certain sales milestones relating to the eSVS
MESH and the Brushed Graft Product and a royalty of 4% on sales
of our eSVS MESH and the Brushed Graft Product. The royalty will
terminate upon the earlier of the expiration of all of the
patents and patent applications, or when the aggregate royalties
paid reach $100.0 million. There is no assurance that any
of these patent applications will issue as patents or that any
such patent(s) will be effective to prevent a competitor from
developing and using substantially equivalent technology.
In the recitals to the Assignment and License Agreement, we
stated that we would use our reasonable best efforts to develop
and commercialize both the Brushed Graft Product and the eSVS
MESH. While we have undertaken activities to develop and
commercialize the Brushed Graft Product, we are currently
primarily focused on the development of our eSVS MESH for use in
CABG surgery and additional applications of our eSVS MESH. Any
or all licenses granted to us pursuant to our agreement with
Medtronic may be terminated and potentially all of
47
the core intellectual property and patent rights related to our
eSVS MESH shall revert to Medtronic, upon notice by Medtronic,
(i) if we become insolvent, make an assignment for the
benefit of creditors, go into liquidation or receivership or
otherwise lose legal control of our business; and (ii) if
we determine to cease commercializing either the Brushed Graft
Product or our eSVS MESH, then Medtronic may terminate our
license to patent rights related to the Brushed Graft Product or
the patent rights related to our eSVS MESH, as applicable. If we
cease commercialization of the Brushed Graft Product and not the
eSVS MESH, we believe that Medtronic is entitled to terminate
only the license to patent rights related to the Brushed Graft
Product and not the patent rights related to the eSVS MESH.
Similarly, if we cease commercialization of the eSVS MESH but
not the Brushed Graft Product, we believe that Medtronic is
entitled to cause the reversion of the patent rights related to
the eSVS MESH but not terminate our license to patent rights
related to the Brushed Graft Product. We recognize, however,
that the terms of our agreement with Medtronic are susceptible
to differing interpretations.
Competition
The development and commercialization of medical devices to
treat cardiovascular disease is a highly competitive industry.
If approved, our eSVS MESH will compete with current forms of
treatment, including pharmaceutical treatment, balloon
angioplasty, coronary stents and conventional CABG surgery.
The key competitive factors affecting the success of our eSVS
MESH are likely to be the effectiveness, safety profile and
price of our eSVS MESH, as compared to existing methods for the
prevention or treatment of cardiovascular disease. The
commercial success of our eSVS MESH will depend upon the results
of clinical trials of the technology and experience with the
technology in the commercial marketplace.
If the commercialization of our eSVS MESH technology is
successful, it can be expected that other medical device
companies, many of whom are larger and have greater financial
resources than us, will seek to enter into this market by
introducing competing technologies.
Manufacturing and
Suppliers
We fabricate our eSVS MESH both at our facility and at a
contract manufacturer. We conduct final assembly and packaging
inside a controlled environment area within our facility that
satisfies the requirements of a Class 10,000 level clean
room. We have implemented systems to ensure that our
manufacturing operations comply with relevant United States and
International Good Manufacturing Practices requirements.
We have vendors for all of our key components and outsourced
processes. We have no sole source suppliers and have identified
alternate suppliers for each key component and outsourced
process; however, in some cases, components are provided by
single source suppliers at this time due to quality
considerations, costs, or regulatory requirements. We have
established redundancy for custom equipment used in the
manufacture of our eSVS MESH. A third-party supplier performs
sterilization services for our eSVS MESH. We currently use two
knitting machines that knit the mesh sleeve of our eSVS MESH,
with one located at our facility and the other located off-site.
We intend to order two additional knitting machines in the first
half of 2010. We believe that these four machines will produce
sufficient quantities of our eSVS MESH to meet our expected
needs for the foreseeable future. In the event that one or all
of our knitting machines were to become unavailable, we believe
that we can obtain one or more replacement knitting machines,
although the custom work required to enable the machines to
produce our eSVS MESH would likely result in some delays in our
production process.
Research and
Development
During 2007, 2008 and 2009, we incurred approximately $196,000,
$2.6 million and $3.0 million, respectively, of
research and development expenses. Research and development
costs include the costs to design, develop, test, seek approval
for, and enhance our eSVS MESH and production process. Expenses
related to research and development consist primarily of
personnel costs, including salaries, benefits and stock-based
compensation, product development, pre-clinical and clinical
trials, materials and supplies, and facilities-related costs.
While our research and development expenses to date have been
focused on product development and evaluating the feasibility of
our eSVS MESH, we expect that a large percentage of our research
and development expenses in the future will be incurred in
support of our current and future clinical trials. As we develop
further applications for
48
our eSVS MESH, we intend to utilize internal resources, outside
contract resources and facilities, and our Scientific Advisory
Board.
Employees
As of March 1, 2010, we had ten employees. We plan to
continue to expand our research and development and
commercialization activities. To support this growth, we will
need to expand managerial, research and development, operations
and other functions. None of our employees is represented by a
labor union, and we consider our relationship with our employees
to be good.
Facilities
We lease approximately 5,000 square feet of office,
laboratory, manufacturing and warehouse space at 3405 Annapolis
Lane North, Suite 200, Minneapolis, Minnesota. The term of
our lease expires on August 31, 2010. Negotiations are
underway to extend this lease and to secure additional space.
Our corporate offices, research and development facilities,
prototype development, manufacturing, warehousing, and shipping
facilities are located at this facility.
Government
Regulation
United States
Medical Device Regulation
The Federal Food, Drug, and Cosmetic Act, or FDCA, and the
FDA’s implementing regulations, govern medical device
design and development, preclinical and clinical testing,
premarket clearance or approval, registration and listing,
manufacturing, labeling, storage, advertising and promotion,
sales and distribution, and post-market surveillance. Medical
devices and their manufacturers are also subject to inspection
by the FDA. The FDCA, supplemented by other federal and state
laws, also provides civil and criminal penalties for violations
of its provisions. We intend to manufacture and market a medical
device that is regulated by the FDA, comparable state agencies
and regulatory bodies in other countries.
Our eSVS MESH will require marketing authorization from the FDA
prior to commercial distribution in the United States. The two
primary types of FDA marketing authorization are premarket
notification (also called 510(k) clearance) and premarket
approval (also called PMA approval). The type of marketing
authorization applicable to a device—510(k) clearance or
PMA approval—is generally linked to classification of the
device. The PMA approval process is generally more stringent,
time-consuming and expensive than the 510(k) clearance process.
The FDA classifies medical devices into one of three classes
(Class I, II or III) based on the degree of risk
FDA determines to be associated with a device and the extent of
control deemed necessary to ensure the device’s safety and
effectiveness. Devices requiring fewer controls because they are
deemed to pose lower risk are placed in Class I or II.
Class I devices are deemed to pose the least risk and are
subject only to general controls applicable to all devices, such
as requirements for device labeling, premarket notification, and
adherence to the FDA’s current good manufacturing practice
requirements, as reflected in its Quality System Regulation, or
QSR. Class II devices are intermediate risk devices that
are subject to general controls and may also be subject to
special controls such as performance standards, product-specific
guidance documents, special labeling requirements, patient
registries or postmarket surveillance. Class III devices
are those for which insufficient information exists to assure
safety and effectiveness solely through general or special
controls, and include life-sustaining, life-supporting, or
implantable devices, and devices not “substantially
equivalent” to a device that is already legally marketed.
Most Class I devices and some Class II devices are
exempted by regulation from the 510(k) clearance requirement and
can be marketed without prior authorization from FDA.
Class I and Class II devices that have not been so
exempted are eligible for marketing through the 510(k) clearance
pathway. By contrast, devices placed in Class III generally
require PMA approval prior to commercial marketing. To obtain
510(k) clearance for a medical device, an applicant must submit
a premarket notification to the FDA demonstrating that the
device is “substantially equivalent” to a predicate
device legally marketed in the United States. A device is
substantially equivalent if, with respect to the predicate
device, it has the same intended use and (1) the same
technological characteristics, or (2) has different
technological characteristics and the information submitted
demonstrates that the device is as safe
49
and effective as a legally marketed device and does not raise
different questions of safety or effectiveness. A showing of
substantial equivalence sometimes, but not always, requires
clinical data. Generally, the 510(k) clearance process can
exceed 90 days and may extend to a year or more.
After a device has received 510(k) clearance for a specific
intended use, any modification that could significantly affect
its safety or effectiveness, such as a significant change in the
design, materials, method of manufacture or intended use, will
require a new 510(k) clearance or (if the device as modified is
not substantially equivalent to a legally marketed predicate
device) PMA approval. While the determination as to whether new
authorization is needed is initially left to the manufacturer,
the FDA may review this determination and evaluate the
regulatory status of the modified product at any time and may
require the manufacturer to cease marketing and recall the
modified device until 510(k) clearance or PMA approval is
obtained. The manufacturer may also be subject to significant
regulatory fines or penalties.
Our coronary eSVS MESH has been designated a Class III
product by the FDA and will be required to go through the PMA
process. Other indications of our eSVS MESH, including
peripheral and arteriovenous fistula applications, have not been
classified at this time.
The FDA will require us to file a PMA application with respect
to our eSVS MESH and there is no assurance that PMA approval
will be granted. A PMA application requires the payment of
significant User Fees, and must be supported by valid scientific
evidence, which typically requires extensive data, including
technical, preclinical, clinical and manufacturing data, to
demonstrate to the FDA’s satisfaction the safety and
effectiveness of the device. A PMA application also must include
a complete description of the device and its components, a
detailed description of the methods, facilities and controls
used to manufacture the device, and proposed labeling. After a
PMA application is submitted and found to be sufficiently
complete, the FDA begins an in-depth review of the submitted
information. During this review period, the FDA may request
additional information or clarification of information already
provided. Also during the review period, an advisory panel of
experts from outside the FDA may be convened to review and
evaluate the application and provide recommendations to the FDA
as to the approvability of the device. In addition, the FDA will
conduct a pre-approval inspection of the manufacturing facility
to ensure compliance with the QSR, which requires manufacturers
to follow design, testing, control, documentation and other
quality assurance procedures.
FDA review of a PMA application is required by statute to take
no longer than 180 days, although the process typically
takes significantly longer, and may require several years to
complete. The FDA can delay, limit or deny approval of a PMA
application for many reasons, including:
|
|
|
| |
•
|
the device may not be safe or effective to the FDA’s
satisfaction;
|
| |
| |
•
|
the data from our preclinical trials and clinical trials may be
insufficient to support approval;
|
| |
| |
•
|
the manufacturing process or facilities we use may not meet
applicable requirements; and
|
| |
| |
•
|
changes in FDA approval policies or adoption of new regulations
may require additional data.
|
If the FDA evaluations of both the PMA application and the
manufacturing facilities are favorable, the FDA will either
issue an approval letter, or an approvable letter, which usually
contains a number of conditions that must be met in order to
secure final approval of the PMA. When and if those conditions
have been fulfilled to the satisfaction of the FDA, the agency
will issue a PMA approval letter authorizing commercial
marketing of the device for certain indications. If the
FDA’s evaluation of the PMA or manufacturing facilities is
not favorable, the FDA will deny approval of the PMA or issue a
not approvable letter. The FDA may also determine that
additional clinical trials are necessary, in which case the PMA
approval may be delayed for several months or years while the
trials are conducted and then the data submitted in an amendment
to the PMA. The PMA process can be expensive, uncertain and
lengthy and a number of devices for which FDA approval has been
sought by other companies have never been approved for
marketing. Even if a PMA application is approved, the FDA may
approve the device with an indication that is narrower or more
limited than originally sought. The agency can also impose
restrictions on the sale, distribution, or use of the device as
a condition of approval, or impose post approval requirements
such as continuing evaluation and periodic reporting on the
safety, effectiveness and reliability of the device for its
intended use.
50
New PMA applications or PMA supplements may be required for
modifications to the manufacturing process, labeling, device
specifications, materials or design of a device that is approved
through the PMA process. PMA approval supplements often require
submission of the same type of information as an initial PMA
application, except that the supplement is limited to
information needed to support any changes from the device
covered by the original PMA application and may not require as
extensive clinical data or the convening of an advisory panel.
Clinical trials are almost always required to support a PMA
application and are sometimes required for a 510(k) clearance.
These trials generally require submission of an application for
an investigational device exemption, or IDE, to the FDA. The IDE
application must be supported by appropriate data, such as
animal and laboratory testing results, showing that it is safe
to test the device in humans and that the testing protocol is
scientifically sound. The IDE application must be approved in
advance by the FDA for a specified number of patients, unless
the product is deemed a non-significant risk device and eligible
for more abbreviated IDE requirements. Generally, clinical
trials for a significant risk device may begin once the IDE
application is approved by the FDA and the trial protocol and
informed consent are approved by appropriate institutional
review boards at the clinical trial sites.
FDA approval of an IDE allows clinical testing to go forward,
but does not bind the FDA to accept the results of the trial as
sufficient to prove the product’s safety and effectiveness,
even if the trial meets its intended success criteria. With
certain exceptions, changes made to an investigational plan
after an IDE is approved must be submitted in an IDE supplement
and approved by the FDA (and by governing institutional review
boards when appropriate) prior to implementation.
All clinical trials must be conducted in accordance with
regulations and requirements collectively known as Good Clinical
Practice, or GCP. GCPs include the FDA’s IDE regulations,
which describe the conduct of clinical trials with medical
devices, including the recordkeeping, reporting and monitoring
responsibilities of sponsors and investigators, and labeling of
investigation devices. They also prohibit promotion, test
marketing, or commercialization of an investigational device,
and any representation that such a device is safe or effective
for the purposes being investigated. GCPs also include
FDA’s regulations for institutional review board approval
and for protection of human subjects (informed consent), as well
as disclosure of financial interests by clinical investigators.
Required records and reports are subject to inspection by the
FDA. The results of clinical testing may be unfavorable or, even
if the intended safety and effectiveness success criteria are
achieved, may not be considered sufficient for the FDA to grant
approval or clearance of a product. The commencement or
completion of any of our clinical trials may be delayed or
halted, or be inadequate to support approval of a PMA
application or clearance of a premarket notification for
numerous reasons, including, but not limited to, the following:
|
|
|
| |
•
|
the FDA or other regulatory authorities do not approve a
clinical trial protocol or a clinical trial (or a change to a
previously approved protocol or trial that requires approval),
or place a clinical trial on hold;
|
| |
| |
•
|
patients do not enroll in clinical trials or follow up at the
rate expected;
|
| |
| |
•
|
institutional review boards and third-party clinical
investigators may delay or reject our trial protocol or changes
to our trial protocol;
|
| |
| |
•
|
third-party clinical investigators decline to participate in a
trial or do not perform a trial on our anticipated schedule or
consistent with the clinical trial protocol, investigator
agreements, good clinical practices or other FDA requirements;
|
| |
| |
•
|
third-party organizations do not perform data collection and
analysis in a timely or accurate manner;
|
| |
| |
•
|
regulatory inspections of our clinical trials or manufacturing
facilities, which may, among other things, require us to
undertake corrective action or suspend or terminate our clinical
trials;
|
| |
| |
•
|
changes in governmental regulations or administrative actions;
|
| |
| |
•
|
the interim or final results of the clinical trial are
inconclusive or unfavorable as to safety or
effectiveness; and
|
| |
| |
•
|
the FDA concludes that our trial design is inadequate to
demonstrate safety and effectiveness.
|
51
After a device is approved and placed in commercial
distribution, numerous regulatory requirements apply. These
include:
|
|
|
| |
•
|
establishment registration and device listing;
|
| |
| |
•
|
the QSR, which requires manufacturers to follow design, testing,
control, documentation and other quality assurance procedures;
|
| |
| |
•
|
labeling regulations, which prohibit the promotion of products
for unapproved or “off-label” uses and impose other
restrictions on labeling;
|
| |
| |
•
|
medical device reporting regulations, which require that
manufacturers report to the FDA if a device may have caused or
contributed to a death or serious injury or malfunctioned in a
way that would likely cause or contribute to a death or serious
injury if malfunctions were to recur; and
|
| |
| |
•
|
corrections and removal reporting regulations, which require
that manufacturers report to the FDA field corrections and
product recalls or removals if undertaken to reduce a risk to
health posed by the device or to remedy a violation of the FDCA
caused by the device that may present a risk to health.
|
Also, the FDA may require us to conduct postmarket surveillance
studies or order us to establish and maintain a system for
tracking our eSVS MESH through the chain of distribution to the
patient level. The FDA enforces regulatory requirements by
conducting periodic, announced and unannounced inspections and
market surveillance. Inspections may include the manufacturing
facilities of our subcontractors.
Failure to comply with applicable regulatory requirements,
including those applicable to the conduct of our clinical
trials, can result in enforcement action by the FDA, which may
lead to any of the following sanctions:
|
|
|
| |
•
|
warning letters or untitled letters;
|
| |
| |
•
|
fines and civil penalties;
|
| |
| |
•
|
unanticipated expenditures;
|
| |
| |
•
|
delays in clearing or approving or refusal to clear or approve
products;
|
| |
| |
•
|
withdrawal or suspension of FDA approval;
|
| |
| |
•
|
product recall or seizure;
|
| |
| |
•
|
orders for physician notification or device repair, replacement,
or refund;
|
| |
| |
•
|
production interruptions;
|
| |
| |
•
|
operating restrictions;
|
| |
| |
•
|
injunctions; and
|
| |
| |
•
|
criminal prosecution.
|
We and our contract manufacturers, specification developers and
suppliers are also required to manufacture our eSVS MESH in
compliance with current Good Manufacturing Practice requirements
set forth in the QSR. The QSR requires a quality system for the
design, manufacture, packaging, labeling, storage, installation
and servicing of marketed devices, and includes extensive
requirements with respect to quality management and
organization, device design, buildings, equipment, purchase and
handling of components, production and process controls,
packaging and labeling controls, device evaluation,
distribution, installation, complaint handling, servicing and
record keeping. The FDA enforces the QSR through periodic
announced and unannounced inspections that may include the
manufacturing facilities of our subcontractors. If the FDA
believes we or any of our contract manufacturers or regulated
suppliers is not in compliance with these requirements, it can
shut down our manufacturing operations, require recall of our
eSVS MESH, refuse to clear or approve new marketing
applications, institute legal proceedings to detain or seize
products, enjoin future violations, or assess civil and criminal
penalties against us or our officers or other employees. Any
such action by the FDA would have a material adverse effect on
our business.
Fraud and
Abuse
Our operations will be directly, or indirectly through our
customers, subject to various state and federal fraud and
52
abuse laws, including, without limitation, the FDCA, federal
Anti-Kickback Statute and False Claims Act. These laws may
impact, among other things, our proposed sales, marketing and
education programs. In addition, these laws require us to screen
individuals and other companies, suppliers and vendors in order
to ensure that they are not “debarred” by the federal
government and therefore prohibited from doing business in the
healthcare industry. The association or conduct of business with
a “debarred” entity could be detrimental to our
operations and result in a negative impact on our business.
The federal Anti-Kickback Statute prohibits persons from
knowingly and willfully soliciting, offering, receiving or
providing remuneration, directly or indirectly, in exchange for
or to induce either the referral of an individual, or the
furnishing or arranging for a good or service, for which payment
may be made under a federal healthcare program such as the
Medicare and Medicaid programs. Several courts have interpreted
the statute’s intent requirement to mean that if any one
purpose of an arrangement involving remuneration is to induce
referrals of federal healthcare covered business, the statute
has been violated. The Anti-Kickback Statute is broad and
prohibits many arrangements and practices that are lawful in
businesses outside of the healthcare industry. Many states have
also adopted laws similar to the federal Anti-Kickback Statute,
some of which apply to the referral of patients for healthcare
items or services reimbursed by any source, not only the
Medicare and Medicaid programs.
The federal False Claims Act prohibits persons from knowingly
filing or causing to be filed a false claim to, or the knowing
use of false statements to obtain payment from, the federal
government. Various states have also enacted laws modeled after
the federal False Claims Act.
In addition to the laws described above, the Health Insurance
Portability and Accountability Act of 1996 created two new
federal crimes: healthcare fraud and false statements relating
to healthcare matters. The healthcare fraud statute prohibits
knowingly and willfully executing a scheme to defraud any
healthcare benefit program, including private payors. The false
statements statute prohibits knowingly and willfully falsifying,
concealing or covering up a material fact or making any
materially false, fictitious or fraudulent statement in
connection with the delivery of or payment for healthcare
benefits, items or services.
Voluntary industry codes, federal guidance documents and a
variety of state laws address the tracking and reporting of
marketing practices relative to gifts given and other
expenditures made to doctors and other healthcare professionals.
In addition to impacting our marketing and educational programs,
internal business processes will be affected by the numerous
legal requirements and regulatory guidance at the state, federal
and industry levels.
If our operations are found to be in violation of any of the
laws described above or other applicable state and federal fraud
and abuse laws, we, as well as our employees, may be subject to
penalties, including civil and criminal penalties, damages,
fines, exclusion from government healthcare programs, and the
curtailment or restructuring of our operations. Individual
employees may need to defend such suits on behalf of us or
themselves, which could lead to significant disruption in our
present and future operations. We cannot assure you that we will
be able to comply with the above laws and regulations.
European
Medical Device Regulation
The European Union has adopted directives and numerous standards
that govern and harmonize the national laws and standards
regulating the design, manufacture, clinical trials, labeling,
adverse event reporting and post-market surveillance activities
for medical devices that are marketed in member states.
Compliance with voluntary harmonized standards including ISO
13845 issued by the International Organization for Standards
establishes the presumption of conformity with the essential
requirements for a CE Mark. The International Organization for
Standardization, or ISO, is a worldwide federation of national
standards bodies from some 130 countries, established in 1947.
The mission of the ISO is to promote the development of
standardization and related activities in the world with a view
to facilitating the international exchange of goods and
services. ISO certification is commonly a pre-requisite to use
of the CE Mark and indicates that a quality system complies with
standards applicable to activities ranging from initial product
design and development through production and distribution.
Devices that comply with the requirements of a relevant
directive will be entitled to bear the CE Mark and, accordingly,
can be commercially distributed throughout the member states of
the European Union, and other
53
countries that comply with or have adopted these directives. The
method of assessing conformity varies depending on the type and
class of the product, but typically involves a combination of
self-assessment by the manufacturer and a third-party assessment
by a “Notified Body,” an independent and neutral
institution appointed to conduct conformity assessment. This
third-party assessment consists of an audit of the
manufacturer’s quality system and technical review of the
manufacturer’s product. An assessment by a Notified Body
residing within the European Union is required in order for a
manufacturer to commercially distribute the product throughout
the European Union. The manufacturer’s assessment will
include a clinical evaluation of the conformity of the device
with applicable regulatory requirements, which for our eSVS MESH
will include clinical study results. The clinical data presented
by us must provide evidence that the products meet the
performance specifications claimed by us, provide sufficient
evidence of adequate assessment of unwanted side effects and
demonstrate that the benefits to the patient outweigh the risks
associated with the device. We are subject to continued
surveillance by the Notified Body and are required to report any
serious adverse incidents to the appropriate authorities of the
European Union member states.
The Medical Devices Directive, or MDD, covers the regulatory
requirements of the European Union for medical devices.
Compliance with the requirements of the MDD is declared by
placing the CE Mark on the product and supplying the device with
a declaration of conformity, in which the manufacturer certifies
that its product complies with the MDD.
Products intended for sale must bear the CE mark to show
compliance with the MDD. If a Notified Body is involved in the
approval, the number of the Notified Body must also appear
adjacent to the CE Mark.
The routes to compliance under the MDD depend on the
classification of the product:
Class I devices are low risk, such as stethoscopes,
hospital beds and wheelchairs. The manufacturer must produce a
technical file, including product test results compared to
relevant standards. In addition, manufacturers of sterile
products and devices with a measuring function must apply to a
Notified Body for certification of the aspects of manufacture
relating to sterility or measurement.
Class IIa devices are low to medium risk, such as
hearing aids, electrocardiographs and ultrasonic diagnostic
equipment. As with Class I devices, the manufacturer
produces a technical file, but a conformity assessment must be
carried out by a Notified Body, according to one of the
following routes, at the manufacturer’s option:
|
|
|
| |
•
|
examination and testing of each product or homogenous batch of
products;
|
| |
| |
•
|
audit of the full quality assurance system;
|
| |
| |
•
|
audit of the production quality assurance system; or
|
| |
| |
•
|
audit of final inspection and testing.
|
Class IIb devices are medium-high risk devices, such
as surgical lasers, infusion pumps, ventilators, intensive care
monitoring equipment and many implantable devices. Routes to
compliance are the same as for Class IIa devices, with the
addition of required examination and testing of the product by
the Notified Body; however, the full quality assurance route
does not require type examination and testing.
Class III devices are high risk, such as balloon
catheters and prosthetic heart valves. Our eSVS MESH is
classified as a Class III device. Routes to compliance are:
|
|
|
| |
•
|
audit of the full quality assurance system and examination of a
design dossier by the Notified Body. A design dossier is a
submission similar to a PMA application with the FDA; or
|
| |
| |
•
|
examination and testing of the product, together with audit of
the production quality assurance system.
|
We are seeking approval to apply the CE Mark to our eSVS MESH in
order to market our eSVS MESH in the European Union and other
countries that accept the CE Mark.
Third
Party Reimbursement
The availability of insurance coverage and reimbursement for
newly approved medical devices is variable. The commercial
success of our eSVS MESH in both domestic and international
markets will be substantially dependent
54
on whether third-party coverage and reimbursement is available
for patients receiving bypass grafts with our eSVS MESH.
Medicare, Medicaid, health maintenance organizations and other
third-party payors are increasingly attempting to contain
healthcare costs by limiting both coverage and the level of
reimbursement of new medical devices, and, as a result, they may
not cover or provide additional payment for our eSVS MESH. In
order to position our device for coverage by third-party payors,
we may have to agree to a lower net sales price than we might
otherwise charge. The continuing efforts of governmental and
commercial third-party payors to contain or reduce the costs of
healthcare may limit our revenue.
In many countries including the United States, third-party
payors consist of both government funded insurance programs and
private insurance programs who cover a significant portion of a
patient’s medical expenses. The trends toward managed
healthcare in the U.S., and proposed legislation intended to
reduce the cost of government insurance programs, could
significantly influence the purchase of healthcare services and
products, and may result in lower prices for our eSVS MESH or
the exclusion of our eSVS MESH from reimbursement programs. Even
before reimbursement may be obtained for our eSVS MESH in the
United States, FDA approval will be required.
In the United States, CMS is the government entity responsible
for oversight of the Medicare program. Medicare establishes
coverage and reimbursement policies at a federal and local level
for medical products and procedures, and such policies are
periodically reviewed and updated. While private payors vary in
their coverage and payment policies, the Medicare program is
viewed as a benchmark.
There are established codes for CABG procedures and products
that are payable for both Medicare and commercial payors. There
are no assurances that our eSVS MESH technology would fall under
existing policies or reimbursement codes. There are also no
assurances that existing payment rates for such reimbursement
codes will continue to hold at the current levels, such as if
regulatory changes are implemented regarding the methodology for
calculating hospital payments for current inpatient procedures.
Medicare payment rates have decreased approximately 10% to 14%
for those procedures using drug eluting stents. The reductions
are being transitioned over a three-year period that began in
fiscal year 2007. In 2007, CMS also implemented a revised
payment methodology that more accurately reflects the severity
of the patient’s condition.
In the United States, governmental and private sector payors
have instituted initiatives to limit the growth of healthcare
costs, using, for example, price regulation or controls and
competitive pricing programs. Some third-party payors require
pre-approval of coverage for new or innovative devices or
therapies before they will reimburse healthcare providers who
use such devices or therapies.
Providers have also sought ways to manage costs, such as through
the use of group purchasing organizations. It is our belief that
the planned economic benefits provided by our eSVS MESH to
physicians and hospitals through lower revascularization costs
(PCI and/or
CABG) will be viewed by providers and third-party payors as
cost-effective. However, there remains uncertainty whether our
eSVS MESH will be viewed positively in a cost-avoidance model so
as to warrant adequate coverage and reimbursement levels.
Medicare reimburses hospital inpatient stays under the Medicare
Severity Diagnosis-Related Group (MS-DRG) system. The MS-DRG
system assigns individual cases to an MS-DRG according to the
patient’s diagnoses, the procedures performed, and the
severity of a patient’s condition as identified by the
presence or absence of complications and comorbidities, or CCs,
or major CCs, or MCCs. MS-DRGs provide a single bundled payment
which serves as reimbursement for all items and services
provided to the Medicare beneficiary during a single
hospitalization.
Additionally, a relative weight is calculated for each
individual MS-DRG, which represents the average resources
required to care for cases within a particular MS-DRG relative
to the average resources required to treat cases in all MS-DRGs.
Generally, MS-DRG relative weights are adjusted annually to
reflect changes in medical practice in a budget neutral manner.
CMS has made no decisions with respect to MS-DRG assignment for
patients who undergo CABG procedures in which our eSVS MESH
would be used, and there can be no assurance that the MS-DRG to
which such patients will be assigned will result in Medicare
payment levels that are considered by hospitals to be adequate
to further support purchase of our eSVS MESH.
55
Under current CMS reimbursement policies, the agency offers a
process to obtain add-on payment for a new medical technology
when the existing MS-DRG prospective payment rate is inadequate.
To obtain add-on payment, a technology must be considered
“new,” demonstrate substantial improvement above the
current standard of care and exceed certain payment thresholds.
Add-on payments are made for no less than two years and no more
than three years. We must demonstrate the safety and
effectiveness of our eSVS MESH to the FDA in addition to the CMS
requirements listed above before add-on payments will be
approved.
For reporting of physician services, the American Medical
Association, or AMA, has developed a coding system known as
Current Procedural Terminology, or CPT. CPT codes are
established by the AMA and statutorily adopted by all government
and commercial payors to describe and develop payment amounts
for physician services. Physician services are reimbursed by
Medicare based on a physician fee schedule whereby payment is
based generally on the number of “relative value
units” assigned by the AMA to each CPT code. No decision
has been made concerning whether existing CPT codes would be
appropriate for use in coding CABG procedures when our eSVS MESH
is used or if separate, new CPT codes are required. We cannot
assure you that codes used for submitting claims for CABG
procedures using our eSVS MESH will result in incremental
payment to physicians. Failure by physicians to receive what
they consider to be adequate reimbursement for CABG procedures
in which our eSVS MESH is used could have a material adverse
effect on our business, financial condition and results of
operations.
Outside of the United States, there are many reimbursement
programs through private payors as well as government programs.
In some countries, government reimbursement is the predominant
program available to patients and hospitals. While the majority
of countries have existing reimbursement for CABG procedures and
products, a number of countries may require us to gather
additional clinical data before recognizing coverage and
reimbursement for our eSVS MESH. It is our intent to complete
the requisite clinical trials and obtain coverage and
reimbursement approval in countries where it makes economic
sense to do so.
Post-FDA approval in the United States, we intend to pursue an
application for a hospital inpatient New Technology Add-On
Payment. This will allow facilities to be paid an additional
amount to account for the cost of our eSVS MESH device on top of
the MS-DRG for the CABG procedure. Should the clinical trial
results continue to prove a lowering of revascularization rates,
we believe there is a reasonable chance that CMS will grant our
request.
Legal
Proceedings
We are not a party to any pending or threatened litigation.
Scientific
Advisory Board
Our Scientific Advisory Board is currently comprised of the
following five practicing cardiac surgeons and one practicing
cardiologist, who provide feedback on disease states, product
concepts, product requirements, and preclinical/clinical trial
designs.
William Cohn, M.D.: Dr. Cohn is the Director of
Minimally Invasive Surgical technology at the Texas Heart
Institute. His specialties include adult cardiac surgery,
minimally invasive cardiac surgery, off-pump coronary artery
bypass surgery, and minimally invasive valve surgery. He has
been involved with the development of numerous products,
including many products for minimally invasive cardiac surgery.
Robert Emery, M.D.: Dr. Emery has been a
cardiovascular and thoracic surgeon for more than 25 years
and is presently a senior partner of Cardiac Surgical
Associates, P.A. with practices at St. Josephs Hospital in St.
Paul, Minnesota, where he is Medical Director of Cardiovascular
Surgery. Dr. Emery has contributed more than 175 articles
and 140 scientific abstracts to medical literature and has
lectured in many parts of the United States and around the
world. He is active in several professional societies, including
the American College of Chest Physicians, The Society of
Thoracic Surgeons, and is the past president of the
International Society for Minimally Invasive Cardiac Surgery.
Richard Gray, M.D.: Dr. Gray has been a
cardiologist since 1975. He is currently a Medical Director with
Tyler Heart Institute at Community Hospital of Monterey
Peninsula, California. His specialties include valvular heart
disease, artificial heart valves, coronary artery disease, and
preventive cardiology. Dr. Gray has been a cardiologist
56
with California Pacific Medical Center in San Francisco,
California, Director of Cardiovascular Services for
HealthPartners Medical Group, Minneapolis, Minnesota, Chief of
Cardiology for Regions Hospital, St. Paul, Minnesota, Chairman
of the University of North Dakota School of Medicine Department
of Medicine, Director, Surgical Cardiology, Division of
Cardiology for Cedars-Sinai Medical Center, Los Angeles,
California, and Medical Director, Heart Transplant Program at
Cedars-Sinai Medical Center, Los Angeles, California. He has
also acted as principal investigator on several cardiac research
programs over his career.
Stuart Jamieson, M.D.: Dr. Jamieson is the
Endowed Chair, Distinguished Professor of Surgery and Chief of
the Division of Cardiothoracic Surgery for the University of
California, San Diego School of Medicine. He is also
Director of the California Heart and Lung Institute. He is known
throughout the world for his pioneering research and innovative
surgical therapy. Internationally recognized for his expertise
in pulmonary thromboendarterectomy, or PTE, he has performed
more of these specialized operations than all other surgeons in
the world combined. He is also well known for his skill in both
adult and pediatric cardiac surgery. The longest surviving
heart-lung and double lung transplanted patients in the world
are patients of Dr. Jamieson.
Uwe Klima, M.D.: Dr. Klima is Chief of
Cardiothoracic Surgery at the American Hospital in Dubai, UAE.
Prior to his current position, he was Professor of Surgery for
Singapore National University Hospital’s Department of
Cardiac, Thoracic and Vascular Surgery and Associate Professor
of Surgery at Hanover Medical School. He started his training in
cardiothoracic surgery in Austria. He completed residencies at
Harvard Medical School and Vienna General Hospital. His
specialties include adult cardiac surgery, general thoracic
surgery, minimally invasive surgery, beating heart surgery, and
peripheral vascular surgery. Dr. Klima is also the author
or co-author of over 300 publications.
Theo Kofidis, MD, Ph.D, FAHA: Dr. Kofidis is the
Associate Professor of Surgery for National University Hospital
in Singapore. He is also Director of the Robotic Surgery Program
for National University Hospital. His specialties include adult
cardiac surgery, transplantation, heart failure surgery, cardiac
assist devices, minimally invasive cardiac surgery, and
arrhythmia surgery. He has been recipient of many professional
honors, holds several patents, and has been author/co-author on
nearly 100 papers and books in the field of cardiothoracic
surgery.
Our Scientific Advisory Board members have each received
nonqualified stock options to purchase 50,000 shares of our
common stock, which generally vest 25% at grant and 25% each
year over the subsequent three years on the anniversary of the
grant date.
57
Management
The name, age and position of each of our directors and
executive officers as of March 1, 2010, and the names of
certain persons who have agreed to serve as directors upon the
closing of the offering are as follows:
Executive
Officers and Directors
| |
|
|
|
|
|
|
|
|
|
Name
|
|
Age
|
|
Position
|
|
|
|
Manny Villafaña
|
|
|
69
|
|
|
Chairman of the Board and Chief Executive Officer
|
|
Michael P. Winegar
|
|
|
49
|
|
|
Chief Operating Officer, Vice President of Regulatory Affairs,
and Prospective Director
|
|
Scott Kellen
|
|
|
44
|
|
|
Chief Financial Officer, Vice President of Finance, and Secretary
|
|
Arch C. Smith(1)(2)(3)
|
|
|
55
|
|
|
Prospective Director
|
|
Robert E. Munzenrider(1)(2)(3)
|
|
|
65
|
|
|
Prospective Director
|
|
Robert J. Sheehy(1)(2)(3)
|
|
|
52
|
|
|
Prospective Director
|
|
|
|
|
| (1)
|
Prospective member of our compensation committee.
Mr. Sheehy has agreed to serve as the chairman of the
committee upon his appointment to the board.
|
| |
| (2)
|
Prospective member of our nominating and governance committee.
Mr. Munzenrider has agreed to serve as the chairman of the
committee upon his appointment to the board.
|
| |
| (3)
|
Prospective member of our audit committee. Mr. Munzenrider
has agreed to serve as the chairman of the committee upon his
appointment to the board.
|
Manny Villafaña is our founder, and has been our
Chairman of the Board and Chief Executive Officer since our
inception in 2007. Prior to founding us and since 1999,
Mr. Villafaña founded and served as Chairman of the
Board and Chief Executive Officer of CABG Medical, Inc., formed
to develop an artificial coronary graft for use in bypass
surgery. From 1987 to 2004, Mr. Villafaña founded and
served as Chairman of the Board and Chief Executive Officer of
ATS Medical, Inc., which developed the latest generation of
open-pivot mechanical heart valves. From 1976 to 1982,
Mr. Villafaña founded and served as President and
Chairman of the Board of St. Jude Medical, Inc., leading the
company’s development of a novel heart valve that still
dominates the mechanical valve replacement market. From 1972 to
1976, Mr. Villafaña founded and served as President
and Chairman of the Board of Cardiac Pacemakers, Inc., or CPI, a
cardiac rhythm management company that introduced a long life
lithium iodine pacemaker that became and continues to be the
standard of care in the pacemaker market. CPI was ultimately
acquired by Eli Lilly and Company, which spun out CPI as Guidant
Corporation. Guidant was, in turn, purchased by Boston
Scientific Corporation.
Mr. Villafaña has received numerous awards and honors,
including the “Living Legend of Medicine” award from
the International Society of Cardio Thoracic Surgeons, the Ellis
Island Medal of Honor, the Grand Prize
Recipient—Mediterranean Institute of Cardiology, the
Ernst & Young LLP National Master Entrepreneur of the
Year, the Top 100 Hispanics in the USA, the Boys and Girls Club
of America Hall of Fame, and induction into the Minnesota
Business Hall of Fame. We believe that
Mr. Villafaña’s nearly 40 years of
experience in healthcare, his proven and respected leadership,
and his deep commitment to us as our founder will be valuable in
helping to guide us in the years ahead.
Michael P. Winegar joined us as Chief Operating Officer
and Vice President of Regulatory Affairs in September 2007. From
2006 to September 2007, Mr. Winegar was the Vice President
of Regulatory and Quality at Enpath Medical, Inc., up to
and through the company’s acquisition by Greatbatch, Inc.
While at Enpath, Mr. Winegar oversaw the regulatory and
quality functions of various Class II and III devices
and coordinated relevant functions for facility consolidations.
From 2001 to 2005, Mr. Winegar was a founding employee of
ev3 Inc., holding various management positions in the Regulatory
Affairs, Clinical Research, and Quality Assurance departments.
From 2000 to 2001, Mr. Winegar was the Vice President of
Regulatory Affairs, Clinical Research, and Quality Assurance for
Myocor, Inc., where he oversaw the first chronic implants of
Myocor heart failure therapy technologies.
58
Mr. Winegar began his career with positions at Medical
Incorporated, Medtronic, Inc., SciMed Life Systems Inc., and
Boston Scientific Corporation. Mr. Winegar has also acted
as a medical device industry consultant in the areas of
regulatory affairs, quality assurance, and clinical research.
Mr. Winegar has agreed to join our board of directors upon
the closing of this offering. We believe that
Mr. Winegar’s experience in the regulatory affairs and
clinical research fields, coupled with his knowledge of our eSVS
MESH technology, will bring valuable insight to our board.
Scott Kellen joined us as Chief Financial Officer, Vice
President of Finance, and Secretary in February 2010. From 2007
to 2009, Mr. Kellen served as Director of Finance for
Transoma Medical, Inc., including during the preparation of its
proposed initial public offering, which was withdrawn in
February 2008 due to deteriorated market conditions. From 2005
to 2007, Mr. Kellen served as the Corporate Controller for
ev3 Inc. during the company’s initial public offering and
during additional follow-on offerings. From 2003 to 2005,
Mr. Kellen served as Senior Audit Manager of
Deloitte & Touche, LLP (now Deloitte LLP), providing
auditing and consulting services to mid-size public companies
after the passage of the Sarbanes-Oxley Act. Altogether,
Mr. Kellen has spent more than 12 years in the medical
device industry, serving early stage and growth companies that
produced Class II and III devices. Mr. Kellen
began his career with Deloitte & Touche in 1987.
Arch C. Smith is currently, and has been since April
2005, a Venture Partner at Sight Line Partners, a venture
capital firm focused on investments in later stage private
medical device companies. From 1984 to 2003, Mr. Smith
worked for Piper Jaffray, a Minneapolis-based investment bank.
Mr. Smith contributed in roles of increasing responsibility
and most recently as a senior healthcare analyst and Managing
Director for equity research, specializing in medical technology
companies. Mr. Smith initially covered large capitalization
stocks in the cardiovascular device arena, but later shifted the
focus of his practice to small capitalization medical technology
companies where he was responsible for identifying many of the
new and emerging technologies that have changed the way
healthcare is delivered today. Mr. Smith served on the
board of directors for CABG Medical, Inc. from 2004 to 2006.
Mr. Smith serves on the board of the Minneapolis Heart
Institute Foundation. Mr. Smith has agreed to join our
board of directors upon the closing of this offering. We believe
that, as a successful venture capitalist, Mr. Smith will
bring important strategic insight to our board, as well as a
wealth of experience working with the investment community.
Robert E. Munzenrider is a retired financial and
operating executive. From 2000 to 2002, Mr. Munzenrider was
President of Harmon AutoGlass, a subsidiary of Apogee
Enterprises, Inc. In 1999, he served as Vice President and Chief
Financial Officer of the Glass Services Segment of Apogee
Enterprises. He also served as Executive Vice President and
Chief Financial Officer of Eliance Corp., an
e-commerce
service provider, during part of 1999. From 1998 to 1999,
Mr. Munzenrider served as Vice President and Chief
Financial Officer of St. Jude Medical, Inc. Mr. Munzenrider
has served on the board of directors for ATS Medical, Inc. since
2003 and on the board of directors for Viad Corp since 2004.
Mr. Munzenrider also served on the board of directors for
Criticare Systems, Inc. from 2007 to 2008 and the board of
directors for CABG Medical, Inc. from 2004 to 2006. He is also a
Trustee Emeritus on the University of Montana Foundation.
Mr. Munzenrider has agreed to join our board of directors
upon the closing of this offering. We believe that
Mr. Munzenrider’s significant leadership experience in
consumer-focused industries will add valuable expertise and
insight to our board.
Robert J. Sheehy is a retired health insurance industry
executive. From 2007 to 2008, Mr. Sheehy served as Senior
Vice President for UnitedHealth Group, Inc. From 2000 to 2007,
Mr. Sheehy served as Chief Executive Officer of
UnitedHealthcare, Inc., a division of UnitedHealth Group. From
April 1998 to December 2000, Mr. Sheehy was President of
UnitedHealthcare. Prior to April 1998, Mr. Sheehy served in
various capacities with UnitedHealth Group. Mr. Sheehy has
agreed to join our board of directors upon the closing of this
offering. We believe that Mr. Sheehy will bring strategic
insight and leadership and a wealth of experience in healthcare
to our board, as well as knowledge of regulations and issues
facing healthcare providers and medical device companies.
Director
Independence
Our board of directors has reviewed the materiality of any
relationship that each of our directors and prospective
directors has with us, either directly or indirectly. Based on
this review, our board has determined that the following
prospective directors will be “independent directors”
as defined by Rule 5605(a)(2) of the Marketplace
59
Rules of The NASDAQ Stock Market, or NASDAQ, at the time they
become directors upon the closing of the offering:
Messrs. Smith, Munzenrider, and Sheehy.
Committees of the
Board of Directors
Our board of directors has provided for the establishment of an
audit committee, a compensation committee and a nominating and
governance committee effective upon the closing of this
offering. The composition and function of each of these
committees is described below.
Audit
Committee
Our audit committee will be comprised of Mr. Munzenrider
(chairman), Mr. Smith and Mr. Sheehy. Our board of
directors has determined that Mr. Munzenrider is an audit
committee financial expert, as defined by the rules of the
Securities and Exchange Commission. Our audit committee will be
authorized to:
|
|
|
| |
•
|
approve and retain the independent registered public accounting
firm to conduct the annual audit of our financial statements;
|
| |
| |
•
|
review the proposed scope and results of the audit;
|
| |
| |
•
|
review and pre-approve audit and non-audit fees and services;
|
| |
| |
•
|
review accounting and financial controls with the independent
auditors and our financial and accounting staff;
|
| |
| |
•
|
review and approve transactions between us and our directors,
officers and affiliates;
|
| |
| |
•
|
recognize and prevent prohibited non-audit services; and
|
| |
| |
•
|
establish procedures for complaints received by us regarding
accounting matters; oversee internal audit functions, if any.
|
We believe that the composition of our audit committee will meet
the independence requirements of the applicable rules of the
Securities and Exchange Commission and NASDAQ upon completion of
this offering.
Compensation
Committee
Upon the closing of the offering, our compensation committee
will be comprised of Mr. Sheehy (chairman),
Mr. Munzenrider and Mr. Smith. All members of the
compensation committee will qualify as independent under the
current definition promulgated by NASDAQ. Our compensation
committee will be authorized to:
|
|
|
| |
•
|
review and recommend the compensation arrangements for
management;
|
| |
| |
•
|
establish and review general compensation policies with the
objective to attract and retain superior talent, to reward
individual performance and to achieve our financial goals;
|
| |
| |
•
|
administer our stock incentive and purchase plans; and
|
| |
| |
•
|
oversee the evaluation of the board of directors and management.
|
Nominating and
Governance Committee
Upon the closing of the offering, our nominating and governance
committee will be comprised of Mr. Munzenrider (chairman),
Mr. Smith and Mr. Sheehy. All members of the
nominating and governance committee will qualify as independent
directors under the current definition promulgated by NASDAQ.
Our nominating and governance committee will be authorized to:
|
|
|
| |
•
|
identify and nominate candidates for election to the board of
directors; and
|
| |
| |
•
|
develop and recommend to the board of directors a set of
corporate governance principles applicable to our company.
|
60
Compensation
Committee Interlocks and Insider Participation
No prospective member of our compensation committee has at any
time been an employee of ours. None of our executive officers
serves as a member of the board of directors or compensation
committee of any other entity that has one or more executive
officers serving as a member of our board of directors or
compensation committee.
Code of Business
Conduct and Ethics
We have adopted a code of business conduct and ethics that
applies to all of our employees, officers and directors,
including those officers responsible for financial reporting.
The code of business conduct and ethics is available on our
website at www.kipsbaymedical.com. We expect that any amendments
to the code, or any waivers of its requirements, will be
disclosed on our website.
Limitation of
Directors’ and Officers’ Liability and
Indemnification
The Delaware General Corporation Law authorizes corporations to
limit or eliminate, subject to specified conditions, the
personal liability of directors to corporations and their
stockholders for monetary damages for breach of their fiduciary
duties. Our certificate of incorporation and amended and
restated bylaws limit the liability of our directors to the
fullest extent permitted by Delaware law.
We have obtained director and officer liability insurance to
cover liabilities our directors and officers may incur in
connection with their services to us. Our certificate of
incorporation and amended and restated bylaws also provide that
we will indemnify and advance expenses to any of our directors
and officers who, by reason of the fact that he or she is one of
our officers or directors, is involved in a legal proceeding of
any nature. We will repay certain expenses incurred by a
director or officer in connection with any civil, criminal,
administrative or investigative action or proceeding, including
actions by us or in our name. Such indemnifiable expenses
include, to the maximum extent permitted by law, attorney’s
fees, judgments, fines, settlement amounts and other expenses
reasonably incurred in connection with legal proceedings. A
director or officer will not receive indemnification if he or
she is found not to have acted in good faith and in a manner he
or she reasonably believed to be in, or not opposed to, our best
interest.
We have entered into agreements to indemnify our directors and
officers and intend to enter into these agreements in the
future. These agreements provide that we will, among other
things, indemnify and advance expenses to our directors and
officers for certain expenses, including attorneys’ fees,
judgments, fines and settlement amounts incurred by any such
person in any action or proceeding, including any action by us
arising out of such person’s services as our director or
officer, or any other company or enterprise to which the person
provides services at our request. We believe that these
provisions and agreements are necessary to attract and retain
qualified persons as directors and officers.
Such limitation of liability and indemnification does not affect
the availability of equitable remedies. In addition, we have
been advised that in the opinion of the Securities and Exchange
Commission, indemnification for liabilities arising under the
Securities Act of 1933, as amended, is against public policy as
expressed in the Securities Act and is therefore unenforceable.
There is no pending litigation or proceeding involving any of
our directors, officers, employees or agents in which
indemnification will be required or permitted. We are not aware
of any threatened litigation or proceeding that may result in a
claim for such indemnification.
61
Director
Compensation
Until the closing of this offering, Manny Villafaña will be
our sole director and the chairman of our board of directors.
Mr. Villafaña is not compensated for his services as a
director. Messrs. Smith, Munzenrider, Sheehy and Winegar
have agreed to join our board of directors immediately following
this offering.
Upon the closing of the offering, Messrs. Smith,
Munzenrider, and Sheehy, as our non-employee directors, will
each be paid:
|
|
|
| |
•
|
an annual retainer of $18,000;
|
| |
| |
•
|
a meeting attendance fee of $1,250 per meeting;
|
| |
| |
•
|
a committee meeting attendance fee of $1,500 and $1,000 per
meeting for chairs and members, respectively; and
|
| |
| |
•
|
stipends of $3,000 and $5,000 for the chairmen of the
compensation and audit committees, respectively.
|
Upon joining the board, each non-employee director will receive
30,000 shares of common stock, which will vest in four
annual increments, beginning with the one-year anniversary of
the director’s appointment to the board. At this time, we
do not have a policy regarding annual grants of common stock to
our non-employee directors.
62
Executive
Compensation
In light of our limited operating history, small management
team, and single-member board structure, we have elected not to
provide a compensation discussion and analysis in this
prospectus as allowed under the smaller reporting company
disclosure rules of the SEC applicable to us.
Overview
In this section, we describe our compensation programs and
policies and the material elements of compensation for the year
ended December 31, 2009 for our Chairman and Chief
Executive Officer, who also served as our Chief Financial
Officer in fiscal year 2009, and our Chief Operating Officer and
Vice President of Regulatory Affairs. We refer to these persons
as our “named executive officers” elsewhere in this
prospectus. We did not have any other executive officers in
fiscal year 2009, and, except for Scott Kellen, who joined us as
our Chief Financial Officer, Vice President of Finance, and
Secretary in February 2010, we have not hired any additional
executive officers to date.
Decisions on the components of our compensation programs are,
until the closing of this offering, the responsibility of our
Chief Executive Officer and Chairman. Upon the closing of this
offering, our compensation committee will be responsible for
reviewing and evaluating these components, including employee
base salaries and benefit plans. The compensation committee will
provide advice and recommendations to the board of directors on
such matters. See “Committees of the Board of
Directors—Compensation Committee” for further details
on the role of the compensation committee.
Employment
Agreements
Manny
Villafaña
We entered into an employment agreement with
Mr. Villafaña on July 19, 2007. This agreement
provided for an initial base salary of $275,000, which was
increased to $304,500 in 2009. Mr. Villafaña may be
awarded discretionary bonuses as determined by our board of
directors and is entitled to participate in any employee benefit
plans we sponsor.
Pursuant to the employment agreement, if we terminate
Mr. Villafaña’s employment without cause, he is
entitled to his base salary for the entire term of the
employment agreement, which expires on July 1, 2012. For
benefits payable upon a change in control, see “Severance
Benefits and Change in Control Arrangements.”
The employment agreement also contains provisions relating to
confidential information, requiring Mr. Villafaña to
refrain from disclosing any of our proprietary information, and
to assignment of inventions, obligating Mr. Villafaña
to assign to us any inventions which directly concern our eSVS
MESH or future products, research, or development, or which
result from work he performs for us or using our facilities.
Further, Mr. Villafaña’s employment agreement
contains certain provisions concerning his post-employment
activities. Pursuant to the agreement, he has agreed not to
compete with us for a period of two years after the termination
of his employment, provided that we make a monthly payment to
Mr. Villafaña equal to his base salary rate at the
time of termination, adjusted based upon changes in the consumer
price index, beginning with the first month after termination of
employment and continuing until the non-competition provision
expires. Such two-year non-competition period will automatically
be extended by one year increments, up to a total of five years,
unless terminated by us, provided we continue making the monthly
payments set forth above. Mr. Villafaña will also be
entitled to continue his participation in our medical benefits
plan for the term of the non-competition provision, provided he
continues to pay the employee portion of the premium. Following
the termination of his employment with us,
Mr. Villafaña has also agreed to consult on
non-confidential matters at the request of our board of
directors.
Michael P.
Winegar and Scott Kellen
We entered into employment agreements with Mr. Winegar on
September 1, 2007 and Mr. Kellen on February 8,
63
2010. Mr. Winegar’s employment agreement provided for
an initial base salary of $175,000, which was increased to
$210,000 in 2009. Mr. Kellen’s employment agreement
provided for an annual base salary of $185,000. As explained
further in “Overview” above, these compensation
amounts were determined by our Chief Executive Officer and
Chairman.
Mr. Winegar and Mr. Kellen are both at-will employees.
Therefore, their employment agreements do not have defined
terms, and may be terminated by the executive or by us for any
reason or no reason with ten days prior notice.
Although we may, in our discretion, provide Mr. Winegar or
Mr. Kellen with severance benefits upon termination of his
employment, neither executive is entitled to severance benefits.
For benefits payable upon a change in control, see
“Severance Benefits and Change in Control
Arrangements.”
Pursuant to non-competition and non-solicitation provisions of
their employment agreements, both Mr. Winegar and
Mr. Kellen have agreed not to compete with us for a period
of one year following termination of their employment. The
employment agreements also contain provisions relating to
confidential information and assignment of inventions, which
require Mr. Winegar and Mr. Kellen to refrain from
disclosing any of our proprietary information and to assign to
us any inventions which directly concern our eSVS MESH or future
products, research, or development, or which result from work
they perform for us or using our facilities.
Base
Salary
Base salaries for our named executive officers are established
based on the executive’s level of responsibility and years
of experience, taking into account competitive trends. Base
salaries of all employees, including executive officers, are
reviewed annually and may be increased for merit reasons or due
to overall company performance.
Equity
Awards
Equity awards to our named executive officers generally consist
of incentive stock options. For a description of the terms and
conditions of our stock option plan, see “Employee Benefit
Plans—2007 Long-Term Incentive Plan.”
Upon commencement of his employment with us, Mr. Winegar
received an incentive stock option to purchase
50,000 shares of our common stock at an exercise price of
$1.00 per share. One-quarter of the total number of shares
subject to the option vested on each of September 1, 2008
and 2009, and an additional one-quarter of the total number of
shares will vest on each of September 1, 2010 and 2011. In
June 2008, January 2009, September 2009, and January 2010,
Mr. Winegar received additional incentive stock options to
purchase 10,000, 30,000, 10,000, and 25,000 shares,
respectively, of our common stock at exercise prices per share
of $5.83, $5.83, $6.00, and $7.00, respectively. Each of these
option grants vests, in the aggregate, as to one-fourth of the
shares on the first anniversary of the grant date and annually
thereafter until the fourth anniversary of the grant date.
There were no options exercised by any of our named executive
officers during 2009.
Non-Equity
Incentive Compensation
We did not award any non-equity incentive compensation to any of
our named executive officers in 2009.
Retirement Plan
and Other Benefits
We offer a SIMPLE IRA plan and health, disability, and life
insurance to our full-time employees, including our named
executive officers. For a description of the terms and
conditions of our SIMPLE IRA plan, see “Employee Benefit
Plans—Retirement Plan and Other Benefits.”
Nonqualified
Deferred Compensation
None of our named executive officers participate in or have
account balances in nonqualified defined contribution plans or
other deferred compensation plans maintained by us.
64
Perquisites and
Other Personal Benefits
We provide Mr. Winegar with a car allowance of $500 per
month.
Severance
Benefits and Change in Control Arrangements
We have agreed to provide the severance benefits and change in
control arrangements described below to our named executive
officers.
Manny
Villafaña
Pursuant to his employment agreement, if we terminate
Mr. Villafaña’s employment without cause, he is
entitled to his base salary for the entire term of the
agreement. The agreement will expire on July 1, 2012.
Pursuant to the agreement, Mr. Villafaña has agreed
not to compete with us for a period of two years after the
termination of his employment, subject to extension by us for
three additional years, provided that we make a monthly payment
to Mr. Villafaña equal to his base salary rate at the
time of termination, adjusted based upon changes in the consumer
price index, beginning with the first month after termination of
employment and continuing until the non-competition provision
expires. Mr. Villafaña will also be entitled to
continue his participation in our medical benefits plan,
provided he continues to pay the employee portion of the
premium. Such benefits will continue until the expiration of the
non-competition provision, which, as discussed in
“Employment Agreements” above, will be a period of not
less than two years and not more than five years.
We also entered into a change in control agreement with
Mr. Villafaña on September 12, 2008. Under the
terms of this agreement, if, within 24 months of a change
in control, Mr. Villafaña’s employment is
terminated by us other than for cause, or if he resigns for good
reason, Mr. Villafaña will be entitled to a prorated
portion of any annual incentive bonus for the fiscal year in
which the termination occurs and a severance benefit equal to
three years of his base salary. The change in control agreement
will expire on September 12, 2011, but will be
automatically extended by one-year increments thereafter unless
either party provides written notice to the other of the intent
not to extend the agreement.
Michael P.
Winegar and Scott Kellen
Under the terms of their employment agreements, neither
Mr. Winegar nor Mr. Kellen is entitled to any
severance benefits upon termination of employment. However, we
may, in our sole discretion, provide them with severance
benefits.
We entered into change in control agreements with
Mr. Winegar, effective September 12, 2008, and
Mr. Kellen, effective February 8, 2010. Under the
terms of these agreements, if, within 24 months of a change
in control, either executive is terminated by us for a reason
other than cause or resigns for good reason, he will be entitled
to a prorated portion of any annual incentive bonus for the
fiscal year in which the termination occurs and a severance
benefit equal to two years of his base salary. The change in
control agreements expire three years from their effective
dates, but will be automatically extended by one-year increments
unless either party provides written notice to the other of the
intent not to extend the agreement.
65
Summary
Compensation Table for 2009
The following table provides information regarding the
compensation earned during the year ended December 31, 2009
by our Chairman and Chief Executive Officer, who also served as
our Chief Financial Officer in fiscal year 2009, and our Chief
Operating Officer and Vice President of Regulatory Affairs. We
refer to these persons as our “named executive
officers” elsewhere in this prospectus. We did not have any
other executive officers in 2009, and, except for Scott Kellen,
who joined us as our Chief Financial Officer, Vice President of
Finance, and Secretary in February 2010, we have not hired any
additional executive officers to date.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Option
|
|
All Other
|
|
|
|
|
|
|
|
Salary
|
|
Awards
|
|
Compensation
|
|
Total
|
|
Name and Principal Position
|
|
Year
|
|
($)
|
|
($)(1)
|
|
($)(2)
|
|
($)
|
|
|
|
Manny Villafaña
|
|
|
2009
|
|
|
$
|
304,500
|
|
|
$
|
—
|
|
|
$
|
8,901
|
|
|
$
|
313,401
|
|
|
Chairman and Chief
Executive Officer(3)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Michael Winegar
|
|
|
2009
|
|
|
|
210,000
|
|
|
|
114,131
|
|
|
|
12,128
|
|
|
|
336,259
|
|
|
Chief Operating Officer
and Vice President of Regulatory Affairs
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) |
|
The value of each of the option awards was computed in
accordance with FASB ASC Topic 718 without consideration of
forfeitures. Valuation assumptions are described in the notes to
financial statements appearing elsewhere in this prospectus. See
the table entitled “Outstanding Equity Awards at Fiscal
Year End” and our discussion of stock-based compensation
under “Equity Awards.” Each stock option is an
incentive stock option with a ten-year term, and vests, in the
aggregate, as to one-fourth of the shares on the first
anniversary of the grant date and annually thereafter until the
fourth anniversary of the grant date. |
| |
|
(2) |
|
Represents our match of Mr. Villafaña’s and
Mr. Winegar’s 2009 contributions to their SIMPLE IRA
accounts. See our discussion of our SIMPLE IRA plan under
“Employee Benefit Plans—Retirement Plan and Other
Benefits. The amount provided for Mr. Winegar also
represents the $6,000 car allowance discussed under
“Perquisites and Other Benefits.” |
| |
|
(3) |
|
In addition to his roles as Chairman and Chief Executive
Officer, Mr. Villafaña also served as Chief Financial
Officer in fiscal year 2009. |
Outstanding
Equity Awards at December 31, 2009
The following table sets forth certain information regarding
outstanding equity awards granted to our named executive
officers as of December 31, 2009. Since our inception,
Mr. Winegar is the only named executive officer who has
been granted an option award. Each award was granted pursuant to
our 2007 Long-Term Incentive Plan.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Option Awards
|
|
|
|
|
|
Number of
|
|
Number of
|
|
|
|
|
|
|
|
|
|
Securities
|
|
Securities
|
|
|
|
|
|
|
|
|
|
Underlying
|
|
Underlying
|
|
|
|
|
|
|
|
|
|
Unexercised
|
|
Unexercised
|
|
Option
|
|
Option
|
|
|
|
|
|
Options
|
|
Options
|
|
Exercise
|
|
Expiration
|
|
Name
|
|
Grant Date
|
|
Exercisable
|
|
Unexercisable
|
|
Price
|
|
Date
|
|
|
|
Michael Winegar
|
|
|
9/1/2007
|
|
|
|
25,000
|
(1)
|
|
|
25,000
|
(1)
|
|
$
|
1.00
|
|
|
|
9/1/2017
|
|
|
|
|
|
6/1/2008
|
|
|
|
2,500
|
(2)
|
|
|
7,500
|
(2)
|
|
|
5.83
|
|
|
|
6/1/2018
|
|
|
|
|
|
1/1/2009
|
|
|
|
7,500
|
(3)
|
|
|
22,500
|
(3)
|
|
|
5.83
|
|
|
|
1/1/2019
|
|
|
|
|
|
9/4/2009
|
|
|
|
—
|
|
|
|
10,000
|
(4)
|
|
|
6.00
|
|
|
|
9/4/2019
|
|
|
|
|
|
|
|
(1) |
|
Represents shares granted pursuant to an incentive stock option
agreement for an aggregate of 50,000 shares of our common
stock. This option vests in four annual installments beginning
on September 1, 2008. |
| |
|
(2) |
|
Represents shares granted pursuant to an incentive stock option
agreement for an aggregate of 10,000 shares of our common
stock. This option vests in four annual installments beginning
on June 1, 2009. |
66
|
|
|
|
(3) |
|
Represents shares granted pursuant to an incentive stock option
agreement for an aggregate of 30,000 shares of our common
stock. This option vests in four annual installments beginning
on January 1, 2010. |
| |
|
(4) |
|
Represents shares granted pursuant to an incentive stock option
agreement for an aggregate of 10,000 shares. This option
vests in four annual installments beginning on September 4,
2010. |
Employee Benefit
Plans
2007 Long-Term
Incentive Plan
Our 2007 Long-Term Incentive Plan, or the 2007 Plan, was adopted
by our board of directors and approved by our stockholders on
July 27, 2007. The 2007 Plan will expire in July 2017,
unless sooner terminated by our board of directors.
Stock
Awards
Under the 2007 Plan, we may grant incentive stock options,
nonqualified stock options, restricted stock awards, stock
appreciation rights, and other performance awards to our
officers, directors, employees, consultants, and advisors.
Administration
The 2007 Plan is administered by our board of directors and our
compensation committee, collectively referred to as the plan
administrator, which has the authority to grant awards,
determine award recipients, dates of grant, the numbers and
types of stock awards to be granted and to set the terms of
these awards, including the exercise price, vesting periods and
the period of exercisability.
Share
Reserve
As of December 31, 2009, there were 1,999,000 shares
of our common stock reserved, but only 1,401,000 shares of
our common stock available for issuance under the 2007 Plan. As
of December 31, 2009, there were outstanding options to
purchase 598,000 shares of our common stock under the 2007
Plan. Options for an additional 185,000 shares of our
common stock were granted in 2010 and were outstanding as of
March 1, 2010. Accordingly, as of March 1, 2010, there
were outstanding options to purchase an aggregate of
783,000 shares of our common stock under the 2007 Plan and
1,216,000 shares of our common stock available for future
grant. The maximum number of shares that may be issued pursuant
to the exercise of incentive stock options under the 2007 Plan
is equal to 2,000,000 shares.
If a stock award granted under the 2007 Plan expires or
otherwise terminates without being exercised in full, or is
settled in cash, the shares of common stock not acquired
pursuant to the stock award again become available for
subsequent issuance under our 2007 Plan. In addition, the
following types of shares under the 2007 Plan may become
available for the grant of new stock awards under the 2007 Plan:
(a) shares that are forfeited prior to becoming fully
vested; (b) shares withheld to satisfy income or employment
withholding taxes; (c) shares used to pay the exercise
price of an option in a net share settlement; and
(d) shares tendered to us to pay the exercise price of an
option. Shares issued under the 2007 Plan may be previously
unissued shares or reacquired shares bought on the open market.
Stock
Options
Incentive and nonqualified stock options are granted pursuant to
incentive and nonqualified stock option agreements adopted by
the plan administrator. The plan administrator determines the
exercise price for a stock option, within the terms and
conditions of the 2007 Plan, provided that the exercise price of
an incentive stock option cannot be less than 100% of the fair
market value of our common stock on the date of grant. Options
granted under our 2007 Plan vest at the rate specified by the
plan administrator.
The plan administrator determines the term of stock options
granted under our 2007 Plan. Incentive stock options may be
granted for terms up to a maximum of 10 years, except in
the case of certain incentive stock options, as described below.
Stock options generally are not transferable except by will, or
the laws of descent and distribution unless, in the case of
nonqualified stock options, permitted by the plan administrator.
67
Acceptable consideration for the purchase of common stock issued
upon the exercise of a stock option will be determined by the
plan administrator and may include (a) cash, personal check
or certified check or (b) the tender of common stock
previously owned by the optionholder.
Limitations on
Incentive Stock Options
No incentive stock option may be granted to any person who, at
the time of the grant, owns or is deemed to own stock possessing
more than 10% of our total combined voting power or that of any
of our affiliates unless (a) the option exercise price is
at least 110% of the fair market value of the stock subject to
the option on the date of grant and (b) the term of the
incentive stock option does not exceed five years from the date
of grant.
Restricted Stock
Awards
Restricted stock awards are granted pursuant to restricted stock
agreements adopted by the plan administrator. Shares of common
stock acquired under a restricted stock award may, but need not,
be subject to a risk of forfeiture in our favor in accordance
with a vesting schedule to be determined by the plan
administrator. No restricted stock award may be transferred,
other than by will or the laws of descent and distribution,
prior to the date any risks of forfeiture described in the
restricted stock agreement have lapsed.
Stock
Appreciation Rights
Stock appreciation rights are granted pursuant to stock
appreciation right agreements adopted by the plan administrator.
The plan administrator determines the strike price for a stock
appreciation right which, unless otherwise determined by the
plan administrator, cannot be less than 100% of the fair market
value of our common stock on the date of grant. Upon the
exercise of a stock appreciation right, we will pay the
participant an amount equal to the product of (a) the
excess of the per share fair market value of our common stock on
the date of exercise over the strike price, multiplied by
(b) the number of shares of common stock with respect to
which the stock appreciation right is exercised. A stock
appreciation right granted under the 2007 Plan vests at the rate
specified in the stock appreciation right agreement as
determined by the plan administrator.
Performance
Awards
The 2007 Plan permits the grant of performance unit awards that
shall consist of monetary awards that may be earned if the
company or award recipient achieves certain performance
objectives established by the plan administrator over a
specified performance periods and performance share awards shat
consist of shares of our common stock that may be earned or
become vested in whole or in part if the Company or award
recipient achieves certain performance objectives established by
the plan administrator over a specified performance period.
Changes to
Capital Structure
In the event that there is a specified type of change in our
capital structure, such as a stock split, appropriate
adjustments may be made to (a) the number of shares
reserved under the 2007 Plan and (b) the number of shares
and exercise price or strike price, if applicable, of all
outstanding stock awards.
Effect on Stock
Awards of Certain Corporate Transactions
In the event of the acquisition of the Company through the sale
of substantially all of the Company’s assets and the
consequent discontinuance of its business or through a merger,
consolidation, exchange, reorganization, reclassification,
extraordinary dividend, divestiture, liquidation,
recapitalization, stock split, stock dividend or otherwise,
certain significant corporate transactions, our board of
directors has the discretion to:
|
|
|
| |
•
|
accelerate the vesting of a stock or performance award;
|
| |
| |
•
|
arrange for the lapse of any reacquisition or repurchase rights
held by us with respect to the stock award;
|
| |
| |
•
|
cancel or arrange for the cancellation of the stock award, to
the extent not vested or exercised prior to the effective time
of the corporate transaction;
|
68
|
|
|
| |
•
|
provide for the surrender of a stock award in exchange for a
payment equal to the excess of (a) the value of the
property that the award holder would have received upon the
exercise of the stock award over (b) the exercise price
otherwise payable in connection with the stock award; or
|
| |
| |
•
|
arrange for the assumption, continuation or substitution of a
stock award by a surviving or acquiring entity or parent company.
|
Retirement
Plan and Other Benefits
We sponsor a SIMPLE IRA retirement plan, which covers
substantially all qualified full-time employees. This plan
provides that each employee may elect to contribute to an
individual retirement plan through salary reduction
contributions. We currently match each employee’s
contribution to the plan up to 3% of the employee’s base
annual wage. We also offer health, disability, and life
insurance to our full-time employees.
69
Certain
Relationships and Related Party Transactions
The following is a summary of transactions since our inception
to which we have been a party in which the amount involved
exceeded $25,960, which is equal to 1% of the average of our
total assets at December 31, 2008 and 2009, and in which
any of our directors, executive officers or beneficial holders
of more than 5% of our capital stock had or will have a direct
or indirect material interest, other than compensation
arrangements that are described under the section of this
prospectus entitled “Compensation Discussion and
Analysis.”
Equity Issuances
to Directors, Executive Officers and 5% Stockholders
The following table shows all issuances of common stock during
the past two fiscal years to each of our directors, executive
officers and holders of more than 5% of our capital stock.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Aggregate
|
|
|
|
|
Date of
|
|
|
Number of
|
|
|
Purchase
|
|
|
|
|
Issuance
|
|
|
Shares
|
|
|
Price
|
|
|
|
|
Directors and Executive Officers
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Manny Villafaña
|
|
|
7/18/2007
|
|
|
|
5,400,000
|
|
|
$
|
90,000
|
|
|
Michael Winegar
|
|
|
7/18/2007
|
|
|
|
300,000
|
|
|
|
5,000
|
|
|
5% Stockholders
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Kips Bay Investments, LLC
|
|
|
5/21/2008
|
|
|
|
60,000
|
|
|
|
350,000
|
|
|
|
|
|
7/21/2008
|
|
|
|
60,000
|
|
|
|
350,000
|
|
|
|
|
|
9/3/2008
|
|
|
|
60,000
|
|
|
|
350,000
|
|
|
|
|
|
10/15/2008
|
|
|
|
60,000
|
|
|
|
350,000
|
|
|
|
|
|
12/1/2008
|
|
|
|
60,000
|
|
|
|
350,000
|
|
|
|
|
|
1/12/2009
|
|
|
|
60,000
|
|
|
|
350,000
|
|
|
|
|
|
3/2/2009
|
|
|
|
4,800,000
|
|
|
|
3,000,000
|
|
|
|
|
|
3/2/2009
|
|
|
|
347,389
|
|
|
|
217,188
|
|
|
|
|
|
3/5/2009
|
|
|
|
60,000
|
|
|
|
350,000
|
|
|
|
|
|
4/17/2009
|
|
|
|
60,000
|
|
|
|
350,000
|
|
|
|
|
|
6/18/2009
|
|
|
|
120,000
|
|
|
|
700,000
|
|
|
|
|
|
6/30/2009
|
|
|
|
41,667
|
|
|
|
250,000
|
|
|
|
|
|
2/16/2010
|
|
|
|
600,000
|
|
|
|
3,500,000
|
|
|
|
|
|
2/16/2010
|
|
|
|
400,000
|
|
|
|
250,000
|
|
|
|
Agreements with
Directors and Executive Officers
Please see “Executive Compensation” for information
regarding the employment agreements with, and compensation of,
our executive officers.
We have entered into indemnification agreements with our
directors and executive officers. See
“Management—Limitation of Directors’ and
Officers’ Liability and Indemnification.”
Agreements with
5% Stockholders
Investment
Agreement with Kips Bay Investments, LLC
On July 19, 2007, we executed an Investment Agreement and
two secured convertible notes, each subject to a Loan and
Security Agreement securing the repayment of such notes with all
of our assets, with Kips Bay Investments, LLC, or KBI. Pursuant
to the Investment Agreement, which was executed prior to KBI
becoming a 5% or more beneficial holder of our common stock,
KBI agreed to sell
us all right and title to certain intellectual
70
property assets for $100,000 in the form of a first secured
convertible note, and to loan us $2.9 million in exchange
for a second secured convertible note, or collectively the
Notes. The Notes accrued interest at a rate of nine percent per
annum, and were convertible into an aggregate of
4,800,000 shares of our common stock for repayment of
principal, representing a conversion price of $0.625 per share,
and conversion of accrued interest also at $0.625 per share. The
Investment Agreement also granted KBI two separate stock
purchase options, each for the right to purchase up to
600,000 shares of common stock for $3.5 million when
we achieved defined milestones. The Investment Agreement also
provides certain registration rights to KBI. See
“Description of Capital Stock—Registration
Rights.”
In April 2008, we achieved our first defined milestone and KBI
subsequently exercised its first stock purchase option under the
Investment Agreement, purchasing 600,000 shares of our
common stock for a purchase price of $3.5 million. On
February 25, 2009, KBI elected to convert the entire amount
of principal and $217,188 of the $467,188 in accrued interest on
the Notes into 5,147,389 shares of our common stock which
conversion occurred in March 2009. The remaining accrued
interest of $250,000 was paid in cash concurrent with KBI’s
conversion election. In February 2010, KBI exercised its
second stock purchase option under the Investment Agreement, the
second milestone having previously been achieved by us,
purchasing 600,000 shares of our common stock for a
purchase price of $3.5 million. At that time we also agreed
to convert the $250,000 of interest on the Notes previously paid
in cash to common stock, and pursuant to this conversion, KBI
repaid the $250,000 to us and we issued 400,000 shares of
our common stock to KBI. The exercise of stock purchase options
were made pursuant to a Stock Purchase Agreement between KBI and
us, whereby each party agreed to indemnify the other for any
breach of the agreement or violation of securities law, and
whereby KBI made certain standard investment representations and
warranties.
In connection with the execution of the Notes, we entered into a
Loan and Security Agreement with KBI, whereby we granted a
security interest in all of our existing and to-be-acquired
property and proceeds therefrom, including all intellectual
property assets transferred by KBI to us pursuant to the first
secured convertible note. The Loan and Security Agreement
terminated upon conversion of the Notes.
Due to the conversion of the Notes, exercise of the stock
purchase options, and other purchases of our common stock, as of
March 1, 2010, KBI beneficially owns 49.9% of our common
stock.
Private
Placements of Our Common Stock
In March 2009, we commenced a private offering of a minimum of
500,000 shares of our common stock and up to a maximum of
1,666,667 shares of our common stock to certain accredited
investors at an offering price of $6.00 per share. We sold an
aggregate of 516,241 shares of common stock in the private
offering, which was completed in August 2009. KBI purchased
41,667 shares of our common stock in this private offering.
Promoters and
Certain Control Persons
We were incorporated in the State of Delaware in May 2007 and
5,400,000 shares of common stock were issued to Manny
Villafaña on July 17, 2007 for consideration of
$90,000.
Mr. Villafaña may be deemed a promoter as defined in
Rule 405 under the Securities Act of 1933, as amended.
71
Principal
Stockholders
The following table sets forth information regarding the
beneficial ownership of our common stock as of March 1,
2010 and as adjusted to reflect the sale of the common stock in
this offering for:
|
|
|
| |
•
|
each person, or group of affiliated persons, known by us to
beneficially own more than 5% of our common stock;
|
| |
| |
•
|
each of our named executive officers;
|
| |
| |
•
|
each of our directors; and
|
| |
| |
•
|
all of our directors and executive officers as a group.
|
The percentage ownership information shown in the table is based
upon 13,581,791 shares of common stock outstanding as of
March 1, 2010, and the issuance
of shares
of common stock in this offering. The percentage ownership
information assumes no exercise of the underwriter’s option
to purchase additional shares.
Information with respect to beneficial ownership has been
furnished by each director, officer or beneficial owner of more
than 5% of our common stock. We have determined beneficial
ownership in accordance with the rules of the SEC. These rules
generally attribute beneficial ownership of securities to
persons who possess sole or shared voting power or investment
power with respect to those securities. In addition, the rules
include shares of common stock issuable pursuant to the exercise
of stock options that are either immediately exercisable or
exercisable on or before April 30, 2010, which is
60 days after March 1, 2010. These shares are deemed
to be outstanding and beneficially owned by the person holding
those options for the purpose of computing the percentage
ownership of that person. Unless otherwise indicated, the
persons or entities identified in this table have sole voting
and investment power with respect to all shares shown as
beneficially owned by them, subject to applicable community
property laws.
Unless otherwise noted below, the address for each person or
entity listed in the table is
c/o Kips
Bay Medical, Inc., 3405 Annapolis Lane North, Suite 200,
Minneapolis, Minnesota 55447.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Beneficial Ownership
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Options
|
|
|
Percentage of Shares
|
|
|
|
|
Number of Shares
|
|
|
Exercisable
|
|
|
Beneficially Owned
|
|
|
|
|
Held Before the
|
|
|
within 60
|
|
|
Before this
|
|
|
After this
|
|
|
Beneficial Owner
|
|
Offering
|
|
|
Days
|
|
|
Offering
|
|
|
Offering
|
|
|
|
|
5% Stockholders
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Kips Bay Investments, LLC(1)
|
|
|
6,789,056
|
|
|
|
—
|
|
|
|
49.9
|
%
|
|
|
|
|
|
7803 Glenroy Road, Suite 300
Bloomington, Minnesota 55438
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Directors and Named Executive Officers
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Manny Villafaña
|
|
|
5,400,000
|
|
|
|
—
|
|
|
|
39.7
|
|
|
|
|
|
|
Michael Winegar
|
|
|
300,000
|
|
|
|
35,000
|
|
|
|
2.5
|
|
|
|
|
|
|
All directors and executive officers as a group (3 persons)
|
|
|
5,700,000
|
|
|
|
35,000
|
|
|
|
42.2
|
|
|
|
|
|
|
|
|
|
|
|
(1) |
|
Voting and dispositive authority for the shares held of record
by Kips Bay Investments, LLC are held by Nasser Kazeminy,
members of his family, and certain trusts for the benefit of his
children and grandchildren. |
72
Description of
Capital Stock
Upon the closing of this offering, our authorized capital stock
will consist of 40,000,000 shares of common stock, par
value $0.01 per share, and 10,000,000 shares of
undesignated stock, par value $0.01 per share.
The following summarizes important provisions of our common
stock and describes certain material provisions of our
certificate of incorporation and amended and restated bylaws.
This summary is qualified by our certificate of incorporation
and amended and restated bylaws, copies of which have been filed
as exhibits to the registration statement of which this
prospectus is a part, and by the provisions of applicable law.
Common
Stock
Outstanding
Shares
As of March 1, 2010, there were 13,581,791 shares of
common stock outstanding held of record by 64 stockholders. All
of our issued and outstanding shares of common stock are duly
authorized, validly issued, fully paid and non-assessable. After
giving effect to the sale of common stock offered in this
offering, there will
be shares
of common stock outstanding.
Dividend
Rights
The holders of our outstanding shares of common stock are
entitled to receive dividends, if any, as may be declared out of
legally available funds at the times and the amounts as our
board of directors may from time to time determine.
Voting
Rights
Each holder of common stock is entitled to one vote for each
share of common stock held on all matters submitted to a vote of
the stockholders, including the election of directors. Our
certificate of incorporation and amended and restated bylaws do
not provide for cumulative voting rights. Because of this, the
holders of a majority of the shares of common stock entitled to
vote in any election of directors can elect all of the directors
standing for election, if they should so choose.
No Preemptive
or Similar Rights
The common stock is not entitled to preemptive rights and is not
subject to conversion or redemption.
Right to
Receive Liquidation Distributions
In the event of our liquidation, dissolution or winding up,
holders of common stock will be entitled to share ratably in the
net assets legally available for distribution to stockholders
after the payment of all of our debts and other liabilities.
Undesignated
Stock
Our board of directors has the authority, without first
obtaining approval of our stockholders, to establish from the
undesignated shares, one or more series of preferred stock and
to fix the powers, preferences, rights and limitations of such
class or series, including dividend rights, voting rights, and
the right to receive liquidation distributions.
Options
As of March 1, 2010, we had outstanding options to purchase
an aggregate of 783,000 shares of our common stock at a
weighted average exercise price of $4.01 per share under our
2007 Long-Term Incentive Plan. All outstanding options provide
for adjustments in the event of a merger, consolidation,
reorganization, recapitalization, stock dividend, stock split or
other similar change in our corporate structure. As of
March 1, 2010, 1,216,000 additional shares are
reserved and available for issuance under our 2007 Long-Term
Incentive Plan.
73
Registration
Rights
The Investment Agreement, dated July 19, 2007, between KBI
and us provides that we will file a registration statement under
the Securities Act of 1933, as amended, covering the re-sale of
1,000,000 or more shares of our common stock within 90 days
of a request by KBI. We are not obligated to take any
registration-related actions during the following periods:
|
|
|
| |
•
|
prior to the date that is six months following the effective
date of our first registered public offering pursuant to a firm
commitment underwritten offering;
|
| |
| |
•
|
during the period starting with the date that is 90 days
prior to our good faith estimated date of filing of, and ending
on the date three months immediately following the effective
date of, any registration statement pertaining to our
securities, provided certain conditions are met;
|
| |
| |
•
|
after we have effected two registrations of our securities for
KBI, excluding registrations effected on
Form S-3
or any successor form, and such registrations have been declared
effective; and
|
| |
| |
•
|
if we furnish to KBI a certificate stating that in the good
faith judgment of the board of directors that it would be
seriously detrimental to us or our stockholders for a
registration statement to be filed in the near future, which
certificate may only be used once in any
12-month
period, and which certificate may only defer our registration
obligation for a maximum of 120 days.
|
Anti-Takeover
Effects of Provisions of the Certificate of Incorporation and
Amended and Restated Bylaws
Provisions of our certificate of incorporation and amended and
restated bylaws may delay or discourage transactions involving
an actual or potential change in our control or change in our
management, including transactions in which stockholders might
otherwise receive a premium for their shares, or transactions
that our stockholders might otherwise deem to be in their best
interests. Therefore, these provisions could adversely affect
the price of our common stock. Among other things, our
certificate of incorporation and amended and restated bylaws:
|
|
|
| |
•
|
provide that all vacancies, including newly created
directorships, may, except as otherwise required by law, be
filled by the affirmative vote of a majority of directors then
in office, even if less than a quorum;
|
| |
| |
•
|
require that any action to be taken by our stockholders must be
effected at a duly called annual or special meeting of
stockholders and not be taken by written consent;
|
| |
| |
•
|
provide that stockholders seeking to present proposals before a
meeting of stockholders or to nominate candidates for election
as directors at a meeting of stockholders must provide notice in
writing in a timely manner, and also specify requirements as to
the form and content of a stockholder’s notice;
|
| |
| |
•
|
do not provide for cumulative voting rights (therefore allowing
the holders of a majority of the shares of common stock entitled
to vote in any election of directors to elect all of the
directors standing for election, if they should so choose);
|
| |
| |
•
|
provide that special meetings of our stockholders may be called
only by the Chairman of the Board, our Chief Executive Officer
or by the board of directors pursuant to a resolution adopted by
a majority of the total number of authorized directors; and
|
| |
| |
•
|
provide that stockholders will be permitted to amend our amended
and restated bylaws only upon receiving at least
662/3%
of the votes entitled to be cast by holders of all outstanding
shares then entitled to vote generally in the election of
directors, voting together as a single class.
|
The amendment of any of these provisions requires approval by
the holders of at least
662/3%
of our then outstanding common stock, voting as a single class.
Section 203
of the General Corporation Law of the State of
Delaware
We are subject to Section 203 of the Delaware General
Corporation Law. Section 203 generally prohibits a public
74
Delaware corporation from engaging in a “business
combination” with an “interested stockholder” for
a period of three years after the date of the transaction in
which the person became an interested stockholder, unless:
|
|
|
| |
•
|
prior to the date of the transaction, the board of directors of
the corporation approved either the business combination or the
transaction that resulted in the stockholder becoming an
interested stockholder;
|
| |
| |
•
|
the interested stockholder owned at least 85% of the voting
stock of the corporation outstanding at the time the transaction
commenced, excluding for purposes of determining the number of
shares outstanding (a) shares owned by persons who are
directors and also officers and (b) shares owned by
employee stock plans in which employee participants do not have
the right to determine confidentially whether shares held
subject to the plan will be tendered in a tender or exchange
offer; or
|
| |
| |
•
|
on or subsequent to the date of the transaction, the business
combination is approved by the board and authorized at an annual
or special meeting of stockholders, and not by written consent,
by the affirmative vote of at least
662/3%
of the outstanding voting stock which is not owned by the
interested stockholder.
|
Section 203 defines a business combination to include:
|
|
|
| |
•
|
any merger or consolidation involving the corporation and the
interested stockholder;
|
| |
| |
•
|
any sale, transfer, pledge or other disposition involving the
interested stockholder of 10% or more of the assets of the
corporation;
|
| |
| |
•
|
subject to exceptions, any transaction that results in the
issuance or transfer by the corporation of any stock of the
corporation to the interested stockholder; and
|
| |
| |
•
|
the receipt by the interested stockholder of the benefit of any
loans, advances, guarantees, pledges or other financial benefits
provided by or through the corporation.
|
In general, Section 203 defines an interested stockholder
as any entity or person beneficially owning 15% or more of the
outstanding voting stock of the corporation and any entity or
person affiliated with or controlling or controlled by the
entity or person.
Limitation on
Liability of Directors and Indemnification
Our certificate of incorporation limits the liability of our
directors to the fullest extent permitted by Delaware law.
Delaware law provides that directors of a corporation will not
be personally liable for monetary damages for breach of their
fiduciary duties as directors, except for liability for any:
|
|
|
| |
•
|
breach of their duty of loyalty to us or our stockholders;
|
| |
| |
•
|
act or omission not in good faith or that involves intentional
misconduct or a knowing violation of law;
|
| |
| |
•
|
unlawful payment of dividends or redemption of shares as
provided in Section 174 of the Delaware General Corporation
Law; or
|
| |
| |
•
|
transaction from which the directors derived an improper
personal benefit.
|
These limitations of liability do not apply to liabilities
arising under federal securities laws and do not affect the
availability of equitable remedies such as injunctive relief or
rescission.
Our amended and restated bylaws provide that we will indemnify
our directors and executive officers, and may indemnify other
officers, employees and other agents, to the fullest extent
permitted by law. Our amended and restated bylaws also permit us
to secure insurance on behalf of any officer, director, employee
or other agent for any liability arising out of his or her
actions in connection with their services to us, regardless of
whether our amended and restated bylaws permit indemnification.
We have obtained a directors’ and officers’ liability
insurance policy.
We have entered, and intend to continue to enter, into separate
indemnification agreements with our directors and executive
officers, in addition to the indemnification provided for in our
certificate of incorporation and amended and restated bylaws.
These agreements, among other things, require us to indemnify
our directors and executive officers for certain expenses,
including attorneys’ fees, judgments, fines and settlement
amounts incurred by a director or executive officer in any
action or proceeding arising out of their services as one of our
directors or
75
executive officers, or any of our subsidiaries or any other
company or enterprise to which the person provides services at
our request.
At present, there is no pending litigation or proceeding
involving any of our directors or executive officers as to which
indemnification is required or permitted, and we are not aware
of any threatened litigation or proceeding that may result in a
claim for indemnification.
Insofar as indemnification for liabilities arising under the
Securities Act of 1933, as amended, may be permitted to
directors, officers and controlling persons of the registrant
pursuant to the foregoing provisions, or otherwise, the
registrant has been advised that in the opinion of the
Securities and Exchange Commission such indemnification is
against public policy as expressed in the Securities Act and is,
therefore, unenforceable. In the event that a claim for
indemnification against such liabilities (other than the payment
by the registrant of expenses incurred or paid by a director,
officer or controlling person of the registrant in the
successful defense of any action, suit or proceeding) is
asserted by such director, officer or controlling person in
connection with the securities being registered, the registrant
will, unless in the opinion of its counsel the matter has been
settled by controlling precedent, submit to a court of
appropriate jurisdiction the question whether such
indemnification by it is against public policy as expressed in
the Securities Act and will be governed by the final
adjudication of such issue.
Stock
Exchange
We intend to apply for the listing of our common stock on the
NASDAQ Global Market under the symbol “KIPS.”
Transfer Agent
and Registrar
The transfer agent and registrar for our common stock is Wells
Fargo Shareowner Services.
76
Shares Eligible
For Future Sale
Prior to this offering, no public market existed for our common
stock. Market sales of shares of our common stock after this
offering and from time to time, and the availability of shares
for future sale, may reduce the market price of our common
stock. Sales of substantial amounts of our common stock, or the
perception that these sales could occur, could adversely affect
prevailing market prices for our common stock and could impair
our future ability to obtain capital, especially through an
offering of equity securities.
Based on the number of shares of common stock outstanding as
of ,
2010, upon completion of this
offering, shares
of common stock will be outstanding, assuming no exercise of the
underwriter’s option to purchase additional shares and no
exercise of options prior to the completion of this offering.
All of the shares sold in this offering will be freely tradable
without restrictions or further registration under the
Securities Act of 1933, as amended, unless held by our
affiliates as that term is defined under Rule 144 under the
Securities Act.
The
remaining shares
of common stock outstanding upon the closing of this offering
are restricted securities as defined under Rule 144 of the
Securities Act. Restricted securities may be sold in the
U.S. public market only if registered or if they qualify
for an exemption from registration, including by reason of
Rule 144 or 701 under the Securities Act, which rules are
summarized below. These remaining shares will generally become
available for sale in the public market as follows:
|
|
|
| |
•
|
approximately
restricted shares will be eligible for sale in the public market
upon completion of this offering under Rule 144;
|
| |
| |
•
|
approximately
restricted shares will be eligible for sale in the public market
90 days after the date of this prospectus, subject to the
volume, manner of sale and other limitations under Rule 144
and Rule 701; and
|
| |
| |
•
|
approximately
restricted shares will be eligible for sale in the public market
upon expiration of
lock-up
agreements 180 days after the date of this prospectus,
which date may be extended in specified circumstances, subject
in certain circumstances to the volume, manner of sale and other
limitations under Rule 144 and Rule 701.
|
Additionally, of the 783,000 shares of common stock
issuable upon exercise of options outstanding as of
March 1, 2010,
approximately shares
will be vested and eligible for sale 180 days after the
date of this prospectus.
Rule 144
In general, under Rule 144 under the Securities Act, as in
effect on the date of this prospectus, beginning 90 days
after the date of this prospectus, a person who is not one of
our affiliates at any time during the three months preceding a
sale, and who has beneficially owned shares of our common stock
to be sold for at least six months, would be entitled to sell an
unlimited number of shares of our common stock, provided current
public information about us is available. In addition, under
Rule 144, a person who is not one of our affiliates at any
time during the three months preceding a sale, and who has
beneficially owned the shares of our common stock to be sold for
at least one year, would be entitled to sell an unlimited number
of shares immediately upon the closing of this offering without
regard to whether current public information about us is
available. Beginning 90 days after the date of this
prospectus, our affiliates who have beneficially owned shares of
our common stock for at least six months are entitled to sell
within any three-month period a number of shares that does not
exceed the greater of:
|
|
|
| |
•
|
one percent of the number of shares of our common stock then
outstanding, which will equal
approximately
shares immediately after this offering; and
|
| |
| |
•
|
the average weekly trading volume of our common stock on The
NASDAQ Global Market during the four calendar weeks preceding
the filing of a notice on Form 144 with respect to the
sale, or if no such notice is required, the date of receipt of
the order to execute the sale.
|
Sales of restricted shares under Rule 144 by our affiliates
are also subject to requirements regarding the manner of sale,
notice and the availability of current public information about
us. Rule 144 also provides that affiliates relying
77
on Rule 144 to sell shares of our common stock that are not
restricted shares must nonetheless comply with the same
restrictions applicable to restricted shares, other than the
holding period requirement.
Notwithstanding the availability of Rule 144, holders
of
of our restricted shares have entered into
lock-up
agreements as described below under “Underwriting” and
their restricted shares will become eligible for sale at the
expiration of the restrictions set forth in those agreements,
subject to any exceptions set forth therein or waivers by the
underwriters.
Rule 701
Rule 701 under the Securities Act, as in effect on the date
of this prospectus, permits resales of shares in reliance upon
Rule 144 but without compliance with some of the
restrictions of Rule 144, including the holding period
requirement. Most of our employees, executive officers,
directors or consultants who purchased shares under a written
compensatory plan or contract, such as the shares issued under
our 2007 Long-Term Incentive Plan, may be entitled to rely on
the resale provisions of Rule 701, but all holders of
Rule 701 shares are required to wait until
90 days after the date of this prospectus before selling
their shares under Rule 701. However, all of the
Rule 701 shares are subject to
lock-up
agreements as described below and under “Underwriting”
and will become eligible for sale at the expiration of the
restrictions set forth in those agreements.
Lock-up
Agreements
We, along with our directors, executive officers and the holders
of substantially all of our outstanding common stock and stock
options, have agreed with the underwriters that for a period of
180 days, subject to a possible extension under certain
circumstances, following the date of this prospectus, we or they
will not offer, sell, assign, transfer, pledge, contract to sell
or otherwise dispose of or hedge any shares of our common stock
or any securities convertible into or exchangeable for shares of
common stock, subject to specified exceptions, without the prior
written consent of Jefferies & Company, Inc. After the
180-day
lock-up
period, these shares may be sold, subject to applicable
securities laws.
Registration
Rights
KBI received certain registration rights in connection with the
execution of the Investment Agreement dated July 19, 2007.
See “Description of Capital Stock—Registration
Rights.”
Equity Incentive
Plans
We intend to file registration statements under the Securities
Act as promptly as possible after the effective date of this
offering to register shares to be issued pursuant to our
employee benefit plans. As a result, any options or rights
exercised under our 2007 Long-Term Incentive Plan or any other
benefit plan after the effectiveness of the registration
statements will also be freely tradable in the public market,
subject to the
lock-up
agreements discussed above. However, such shares held by
affiliates will still be subject to the volume limitation,
manner of sale, notice and public information requirements of
Rule 144 and the
180-day
lock-up
arrangement described above, if applicable.
78
Underwriting
Subject to the terms and conditions set forth in the
underwriting agreement to be dated on or
about ,
2010, between us and Jefferies & Company, Inc., as
underwriter, we have agreed to sell to the underwriter and the
underwriter has agreed to purchase from us, the entire number of
shares of common stock offered by this prospectus.
The underwriting agreement provides that the obligations of
Jefferies & Company, Inc. are subject to certain
conditions precedent such as the receipt by
Jefferies & Company, Inc. of officers’
certificates and legal opinions and approval of certain legal
matters by their counsel. The underwriting agreement provides
that Jefferies & Company, Inc. will purchase all of
the shares if any of them are purchased. We have agreed to
indemnify Jefferies & Company, Inc. and certain of its
controlling persons against certain liabilities, including
liabilities under the Securities Act, and to contribute to
payments that Jefferies & Company, Inc. may be
required to make in respect of those liabilities.
Jefferies & Company, Inc. has advised us that it
currently intends to make a market in the common stock. However,
Jefferies & Company, Inc. is not obligated to do so
and may discontinue any market-making activities at any time
without notice. No assurance can be given as to the liquidity of
the trading market for the common stock.
Jefferies & Company, Inc. is offering the common stock
subject to its acceptance of the shares from us and subject to
prior sale. Jefferies & Company, Inc. reserves the
right to withdraw, cancel or modify offers to the public and to
reject orders in whole or in part. In addition,
Jefferies & Company, Inc. has advised us that it does
not expect sales to accounts over which they have discretionary
authority to exceed 5% of the common stock being offered.
Commission and
Expenses
Jefferies & Company, Inc. has advised us that it
proposes to offer the common stock to the public at the initial
public offering price set forth on the cover page of this
prospectus and to certain dealers at that price less a
concession not in excess of $ per
share of common stock. Jefferies & Company, Inc. may
allow, and certain dealers may reallow, a discount from the
concession not in excess of $ per
share of common stock to certain brokers and dealers. After the
offering, the initial public offering price, concession and
reallowance to dealers may be reduced by Jefferies &
Company, Inc. No such reduction will change the amount of
proceeds to be received by us as set forth on the cover page of
this prospectus.
The following table shows the public offering price, the
underwriting discounts and commissions that we are to pay
Jefferies & Company, Inc. and the proceeds, before
expenses, to us in connection with this offering. Such amounts
are shown assuming both no exercise and full exercise of
Jefferies & Company, Inc.’s option to purchase
additional shares.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Per Share
|
|
|
Total
|
|
|
|
|
Without
|
|
|
With
|
|
|
Without
|
|
|
With
|
|
|
|
|
Option to Purchase
|
|
|
Option to Purchase
|
|
|
Option to Purchase
|
|
|
Option to Purchase
|
|
|
|
|
Additional Shares
|
|
|
Additional Shares
|
|
|
Additional Shares
|
|
|
Additional Shares
|
|
|
|
|
Public offering price
|
|
$
|
|
|
|
$
|
|
|
|
$
|
|
|
|
$
|
|
|
|
Underwriting discounts and commissions paid by us
|
|
$
|
|
|
|
$
|
|
|
|
$
|
|
|
|
$
|
|
|
|
Proceeds to us, before expenses
|
|
$
|
|
|
|
$
|
|
|
|
$
|
|
|
|
$
|
|
|
|
|
We estimate expenses payable by us in connection with this
offering, other than the underwriting discounts and commissions
referred to above, will be approximately
$ .
Determination of
Offering Price
Prior to the offering, there has not been a public market for
our common stock. Consequently, the initial public
79
offering price for our common stock will be determined by
negotiations between us and Jefferies & Company, Inc.
Among the factors to be considered in these negotiations will be
prevailing market conditions, our financial information, market
valuations of other companies that we and Jefferies &
Company, Inc. believe to be comparable to us, estimates of our
business potential, the present state of our development and
other factors deemed relevant.
We offer no assurances that the initial public offering price
will correspond to the price at which the common stock will
trade in the public market subsequent to the offering or that an
active trading market for the common stock will develop and
continue after the offering.
Listing
We intend to apply to have our common stock approved for listing
on the Nasdaq Global Market under the trading symbol
“KIPS.”
Option to
Purchase Additional Shares
We have granted to Jefferies & Company, Inc. an
option, exercisable for 30 days from the date of this
prospectus, to purchase up to an aggregate
of
additional shares of common stock at the public offering price
set forth on the cover page of this prospectus, less
underwriting discounts and commissions. This option may be
exercised only if Jefferies & Company, Inc. sells more
shares than the total number set forth on the cover page of this
prospectus.
No Sales of
Similar Securities
We, our officers, directors and holders of all or substantially
all our outstanding capital shares and other securities have
agreed, subject to specified exceptions, not to directly or
indirectly:
|
|
|
| |
•
|
sell, offer, contract or grant any option to sell (including any
short sale), pledge, transfer, establish an open “put
equivalent position” within the meaning of
Rule 16a-l(h)
under the Securities Exchange Act of 1934, as amended, or
|
| |
| |
•
|
otherwise dispose of any common shares, options or warrants to
acquire common shares, or securities exchangeable or exercisable
for or convertible into common shares currently or hereafter
owned either of record or beneficially, or
|
| |
| |
•
|
publicly announce an intention to do any of the foregoing for a
period of 180 days after the date of this prospectus
without the prior written consent of the underwriter.
|
This restriction terminates after the close of trading of the
common shares on and including the 180 days after the date
of this prospectus. However, subject to certain exceptions, in
the event that either:
|
|
|
| |
•
|
during the last 17 days of the
180-day
restricted period, we issue an earnings release or material news
or a material event relating to us occurs, or
|
| |
| |
•
|
prior to the expiration of the
180-day
restricted period, we announce that we will release earnings
results during the
16-day
period beginning on the last day of the
180-day
restricted period,
|
then in either case the expiration of the
180-day
restricted period will be extended until the expiration of the
18-day
period beginning on the date of the issuance of an earnings
release or the occurrence of the material news or event, as
applicable, unless Jefferies & Company, Inc. waives,
in writing, such an extension.
Jefferies & Company, Inc. may, in its sole discretion
and at any time or from time to time before the termination of
the 180-day
period, without public notice, release all or any portion of the
securities subject to
lock-up
agreements. There are no existing agreements between
Jefferies & Company, Inc. and any of our shareholders
who will execute a
lock-up
agreement, providing consent to the sale of shares prior to the
expiration of the
lock-up
period.
80
Stabilization
The underwriter has advised us that, pursuant to
Regulation M under the Securities Exchange Act of 1934, as
amended, certain persons participating in the offering may
engage in transactions, including overallotment, stabilizing
bids, syndicate covering transactions or the imposition of
penalty bids, which may have the effect of stabilizing or
maintaining the market price of the common stock at a level
above that which might otherwise prevail in the open market.
Overallotment involves syndicate sales in excess of the offering
size, which creates a syndicate short position.
“Covered” short sales are sales made in an amount not
greater than the underwriter’s option to purchase
additional common shares in this offering. The underwriter may
close out any covered short position by either exercising their
option to purchase additional common shares or purchasing common
shares in the open market. In determining the source of shares
to close out the covered short position, the underwriter will
consider, among other things, the price of shares available for
purchase in the open market as compared to the price at which
they may purchase shares through the option to purchase
additional shares. “Naked” short sales are sales in
excess of the option to purchase additional common shares. The
underwriter must close out any naked short position by
purchasing shares in the open market. A naked short position is
more likely to be created if the underwriter is concerned that
there may be downward pressure on the price of the common shares
in the open market after pricing that could adversely affect
investors who purchase in this offering. A stabilizing bid is a
bid for the purchase of common stock on behalf of the
underwriter for the purpose of fixing or maintaining the price
of the common stock. A syndicate covering transaction is the bid
for or the purchase of common stock on behalf of the underwriter
to reduce a short position incurred by the underwriter in
connection with the offering. A penalty bid is an arrangement
permitting the underwriter to reclaim the selling concession
otherwise accruing to a syndicate member in connection with the
offering if the notes originally sold by such syndicate member
are purchased in a syndicate covering transaction and therefore
have not been effectively placed by such syndicate member.
Neither we nor the underwriter makes any representation or
prediction as to the direction or magnitude of any effect that
the transactions described above may have on the price of our
common stock. The underwriter is not obligated to engage in
these activities and, if commenced, any of the activities may be
discontinued at any time.
Electronic
Distribution
A prospectus in electronic format may be made available by
e-mail or on
the web sites or through online services maintained by
Jefferies & Company, Inc. or its affiliates. In those
cases, prospective investors may view offering terms online and
may be allowed to place orders online. Jefferies &
Company, Inc. may agree with us to allocate a specific number of
common stock for sale to online brokerage account holders. Any
such allocation for online distributions will be made by
Jefferies & Company on the same basis as other
allocations. Other than the prospectus in electronic format, the
information on Jefferies & Company, Inc.’s web
site and any information contained in any other web site
maintained by Jefferies & Company, Inc. is not part of
this prospectus, has not been approved
and/or
endorsed by us or Jefferies & Company, Inc. and should
not be relied upon by investors.
Affiliations
Jefferies & Company, Inc. or its affiliates from time
to time may in the future provide investment banking, commercial
lending and financial advisory services to us and our affiliates
in the ordinary course of business. Jefferies &
Company, Inc. and its affiliates, as applicable, will receive
customary compensation and reimbursement of expenses in
connection with such services.
European Economic
Area
In relation to each Member State of the European Economic Area
which has implemented the Prospectus Directive (as defined
below) (each, a Relevant Member State), with effect from and
including the date on which the Prospectus Directive is
implemented in that Relevant Member State, or the Relevant
Implementation Date, an offer of our common stock to the public
may not be made in that Relevant Member State prior to the
publication of a prospectus in relation to our common stock
which has been approved by the competent authority in that
Relevant Member State or, where appropriate, approved in another
Relevant Member State and notified to the competent authority in
that Relevant Member State, all in accordance with the
Prospectus Directive, except that an
81
offer to the public in that Relevant Member State of any shares
of our common stock may be made at any time under the following
exemptions under the Prospectus Directive if they have been
implemented in the Relevant Member State:
(a) to legal entities which are authorized or regulated to
operate in the financial markets or, if not so authorized or
regulated, whose corporate purpose is solely to invest in
securities;
(b) to any legal entity which has two or more of
(1) an average of at least 250 employees during the
last financial year; (2) a total balance sheet of more than
€43,000,000 and (3) an annual net turnover of more
than €50,000,000, as shown in its last annual or
consolidated accounts;
(c) to fewer than 100 natural or legal persons per Relevant
Member State (other than qualified investors as defined in the
Prospectus Directive); or
(d) in any other circumstances falling within
Article 3(2) of the Prospectus Directive,
provided that no such offer of our common stock shall result in
a requirement for the publication by us or any underwriter of a
prospectus pursuant to Article 3 of the Prospectus
Directive.
For the purposes of this provision, the expression an
“offer of our common stock to the public” in relation
to any shares of our common stock in any Relevant Member State
means the communication in any form and by any means of
sufficient information on the terms of the offer and our common
stock to be offered so as to enable an investor to decide to
purchase or subscribe our common stock, as the same may be
varied in that Member State by any measure implementing the
Prospectus Directive in that Member State and the expression
“Prospectus Directive” means Directive 2003/71/EC and
includes any relevant implementing measure in each Relevant
Member State.
United
Kingdom
Shares of our common stock may not be offered or sold and will
not be offered or sold to any persons in the United Kingdom
other than to persons whose ordinary activities involve them in
acquiring, holding, managing or disposing of investments (as
principal or as agent) for the purposes of their businesses or
otherwise in circumstances which have not resulted or will not
result in an offer to the public in the United Kingdom within
the meaning of the Financial Services and Markets Act 2000, or
the FSMA.
In addition, any invitation or inducement to engage in
investment activity (within the meaning of section 21 of
the FSMA) in connection with the issue or sale of shares of our
common stock may only be communicated or caused to be
communicated in circumstances in which Section 21(1) of the
FSMA does not apply to us. Without limitation to the other
restrictions referred to herein, this prospectus is directed
only at (1) persons outside the United Kingdom or
(2) persons who:
(a) are qualified investors as defined in
section 86(7) of FSMA, being persons falling within the
meaning of article 2.1(e)(i), (ii) or (iii) of
the Prospectus Directive; and
(b) are either persons who fall within article 19(1)
of the Financial Services and Markets Act 2000 (Financial
Promotion) Order 2005, as amended, or Order, or are persons who
fall within article 49(2)(a) to (d) (“high net worth
companies, unincorporated associations, etc.”) of the
Order; or
(c) to whom it may otherwise lawfully be communicated in
circumstances in which Section 21(1) of the FSMA does not
apply.
Without limitation to the other restrictions referred to herein,
any investment or investment activity to which this offering
circular relates is available only to, and will be engaged in
only with, such persons, and persons within the United Kingdom
who receive this communication (other than persons who fall
within (2) above) should not rely or act upon this
communication.
Germany
Any offer or solicitation of securities within Germany must be
in full compliance with the German Securities
82
Prospectus Act (Wertpapierprospektgesetz—WpPG). The offer
and solicitation of securities to the public in Germany requires
the publication of a prospectus that has to be filed with and
approved by the German Federal Financial Services Supervisory
Authority (Bundesanstalt für
Finanzdienstleistungsaufsicht—BaFin). This prospectus has
not been and will not be submitted for filing and approval to
the BaFin and, consequently, will not be published. Therefore,
this prospectus does not constitute a public offer under the
German Securities Prospectus Act (Wertpapierprospektgesetz).
This prospectus and any other document relating to our common
stock, as well as any information contained therein, must
therefore not be supplied to the public in Germany or used in
connection with any offer for subscription of our common stock
to the public in Germany, any public marketing of our common
stock or any public solicitation for offers to subscribe for or
otherwise acquire our common stock. This prospectus and other
offering materials relating to the offer of our common stock are
strictly confidential and may not be distributed to any person
or entity other than the designated recipients hereof.
France
This prospectus has not been prepared in the context of a public
offering of financial securities in France within the meaning of
Article L.411-1
of the French Code Monétaire et Financier and Title I
of Book II of the Règlement Général of the
Autorité des marchés financiers (the “AMF”)
and therefore has not been and will not be filed with the AMF
for prior approval or submitted for clearance to the AMF.
Consequently, the shares of our common stock may not be,
directly or indirectly, offered or sold to the public in France
and offers and sales of the shares of our common stock may only
be made in France to qualified investors (investisseurs
qualifiés) acting for their own, as defined in and in
accordance with
Articles L.411-2
and D.411-1 to D.411-4, D.734-1, D.744-1, D.754-1 and D.764-1 of
the French Code Monétaire et Financier. Neither this
prospectus nor any other offering material may be released,
issued or distributed to the public in France or used in
connection with any offer for subscription on sale of the shares
of our common stock to the public in France. The subsequent
direct or indirect retransfer of the shares of our common stock
to the public in France may only be made in compliance with
Articles L.411-1,
L.411-2, L.412-1 and L.621-8 through L.621-8-3 of the French
Code Monétaire et Financier.
Sweden
This is not a prospectus under, and has not been prepared in
accordance with the prospectus requirements provided for in, the
Swedish Financial Instruments Trading Act [lagen (1991:980) om
handel med finasiella instrument] nor any other Swedish
enactment. Neither the Swedish Financial Supervisory Authority
nor any other Swedish public body has examined, approved, or
registered this document.
83
Material United
States Federal Tax Considerations
for Non-U.S.
Holders of Common Stock
This section summarizes certain material U.S. federal
income and estate tax considerations relating to the ownership
and disposition of our common stock. This summary does not
provide a complete analysis of all potential tax considerations.
The information provided below is based on existing authorities.
These authorities may change, possibly with retroactive effect,
or the Internal Revenue Service, or IRS, might interpret the
existing authorities differently. In either case, the tax
considerations of owning or disposing of our common stock could
differ from those described below. For purposes of this summary,
a
“non-U.S. holder”
is any holder that is not, for U.S. federal income tax
purposes, any of the following:
|
|
|
| |
•
|
an individual citizen or resident of the United States;
|
| |
| |
•
|
a corporation organized under the laws of the United States or
any state;
|
| |
| |
•
|
a trust that is (i) subject to the primary supervision of a
U.S. court and the control of one or more U.S. persons
or (ii) has a valid election in effect under applicable
U.S. Treasury regulations to be treated as a
U.S. person; or
|
| |
| |
•
|
an estate, the income of which is subject to U.S. federal
income taxation regardless of source.
|
If a partnership or other flow-through entity is the owner of
our common stock, the tax treatment of a partner in the
partnership or an owner of the entity will depend upon the
status of the partner or other owner and the activities of the
partnership or other entity. Accordingly, partnerships and
flow-through entities that hold our common stock and partners or
owners of such partnerships or entities, as applicable, should
consult their own tax advisors.
This summary is based upon provisions of the Internal Revenue
Code of 1986, as amended, or the Code, and regulations, rulings
and judicial decisions as of the date of this prospectus. Those
authorities may be changed, perhaps retroactively, so as to
result in U.S. federal income and estate tax consequences
different from those summarized below. In addition, the summary
does not represent a description of the U.S. federal income
and estate tax consequences applicable to you if you are subject
to special treatment under the U.S. federal income tax laws
(including if you are a U.S. expatriate, “controlled
foreign corporation,” “passive foreign investment
company,” bank, insurance company or other financial
institution, dealer or trader in securities, a person who holds
our common stock as a position in a hedging transaction,
straddle or conversion transaction, or other person subject to
special tax treatment). We cannot assure you that a change in
law will not alter significantly the tax considerations that we
describe in this summary. Finally, this summary does not
describe the effects of any applicable foreign, state, or local
laws.
INVESTORS CONSIDERING THE PURCHASE OF OUR COMMON STOCK SHOULD
CONSULT THEIR OWN TAX ADVISORS REGARDING THE APPLICATION OF THE
U.S. FEDERAL INCOME AND ESTATE TAX LAWS TO THEIR PARTICULAR
SITUATION AND THE CONSEQUENCES OF FOREIGN, STATE OR LOCAL LAWS
AND TAX TREATIES.
Dividends
Any dividend paid to a
non-U.S. holder
in respect of our common stock generally will be subject to
U.S. withholding tax at a 30% rate. The withholding tax
might apply at a reduced rate under the terms of an applicable
income tax treaty between the United States and the
non-U.S. holder’s
country of residence. To obtain a reduced rate of withholding
under a treaty, a
non-U.S. holder
must certify its entitlement to treaty benefits by providing a
properly completed
Form W-8BEN
or other applicable form to us or our paying agent. If the
non-U.S. holder
holds the stock through a financial institution or other agent
acting on the holder’s behalf, the holder will be required
to provide appropriate documentation to the agent. The
holder’s agent will then be required to provide
certification to us or our paying agent, either directly or
through other intermediaries. For payments made to a foreign
partnership or other flow-through entity, the certification
requirements generally apply to the partners or other owners
rather than to the partnership or other entity, and the
partnership or other entity must provide the partners’ or
other owners’ documentation to us or our paying agent.
Special rules, described
84
below, apply if a dividend is effectively connected with a
U.S. trade or business conducted by the
non-U.S. holder.
A
non-U.S. holder
eligible for a reduced rate of U.S. withholding tax
pursuant to an income tax treaty generally may obtain a refund
of any excess amounts withheld from the Internal Revenue Service
by filing an appropriate claim for refund with the Internal
Revenue Service.
Sale of Common
Stock
Non-U.S. holders
generally will not be subject to U.S. federal income tax on
any gains realized on the sale, exchange, or other disposition
of our common stock. This general rule, however, is subject to
several exceptions. For example, the gain would be subject to
U.S. federal income tax if:
|
|
|
| |
•
|
the gain is effectively connected with the conduct by the
non-U.S. holder
of a U.S. trade or business or, if a treaty applies, is
attributable to a permanent establishment of the
non-U.S. holder
in the United States, in which case the special rules described
below apply;
|
| |
| |
•
|
the
non-U.S. holder
is an individual who holds our common stock as a capital asset
and who is present in the United States for 183 days or
more in the taxable year of the sale, exchange, or other
disposition, and certain other requirements are met; or
|
| |
| |
•
|
the rules of the Foreign Investment in Real Property Tax Act, or
FIRPTA (described below), treat the gain as effectively
connected with a U.S. trade or business.
|
A
non-U.S. holder
described in the first bullet point immediately above will be
subject to tax on the net gain derived from the sale under
regular graduated U.S. federal income tax rates, and if
such
non-U.S. holder
is a corporation, it may also be subject to the branch profits
tax generally equal to 30% of its effectively connected earnings
and profits or at such lower rate as may be specified by an
applicable income tax treaty. An individual
non-U.S. holder
described in the second bullet point immediately above will be
subject to a flat 30% tax on the gain derived from the sale,
which may be offset by United States source capital losses, even
though the individual is not considered a resident of the United
States.
The FIRPTA rules may apply to a sale, exchange or other
disposition of our common stock if we are, or were within five
years before the transaction, a “U.S. real property
holding corporation,” or USRPHC. In general, we would be a
USRPHC if interests in U.S. real property comprised most of
our assets. We do not believe that we are a USRPHC or that we
will become one in the future. Even if we become a USRPHC, if
our common stock is regularly traded on an established
securities market, such common stock will be treated as United
States real property interests only if the
non-U.S. holder
actually or constructively held more than five percent of such
regularly traded common stock.
Dividends or Gain
Effectively Connected With a U.S. Trade or Business
If any dividend on our common stock, or gain from the sale,
exchange or other disposition of our common stock, is
effectively connected with a U.S. trade or business
conducted by the
non-U.S. holder,
then the dividend or gain will be subject to U.S. federal
income tax at the regular graduated rates. If the
non-U.S. holder
is eligible for the benefits of a tax treaty between the United
States and the holder’s country of residence, any
effectively connected dividend or gain would generally be
subject to U.S. federal income tax only if it is also
attributable to a permanent establishment or fixed base
maintained by the holder in the United States. Payments of
dividends that are effectively connected with a U.S. trade
or business will not be subject to the 30% withholding tax if
the holder claims exemption from withholding by providing a
properly completed
Form W-8ECI.
If the
non-U.S. holder
is a corporation, that portion of its earnings and profits that
is effectively connected with its U.S. trade or business
would generally be subject to a “branch profits tax.”
The branch profits tax rate is generally 30%, although an
applicable income tax treaty might provide for a lower rate.
U.S. Federal
Estate Tax
The estates of nonresident alien individuals generally are
subject to U.S. federal estate tax on property with a
U.S. situs. Because we are a U.S. corporation, our
common stock will be U.S. situs property and therefore will
be included for U.S. estate tax purposes in the taxable
estate of a nonresident alien decedent. The U.S. federal
estate
85
tax liability of the estate of a nonresident alien may be
affected by a tax treaty between the United States and the
decedent’s country of residence. The test for whether an
individual is a resident of the United States for federal estate
tax purposes differs from the test used for federal income tax
purposes.
Backup
Withholding and Information Reporting
The Code and the Treasury regulations require those who make
specified payments to report the payments to the IRS. Among the
specified payments are dividends and proceeds paid by brokers to
their customers. The required information returns enable the IRS
to determine whether the recipient properly included the
payments in income. This reporting regime is reinforced by
“backup withholding” rules. These rules require the
payors to withhold tax from payments subject to information
reporting if the recipient fails to provide his taxpayer
identification number to the payer, furnishes an incorrect
identification number, or repeatedly fails to report interest or
dividends on his returns. The withholding tax rate is currently
28%. The backup withholding rules do not apply to payments to
corporations, whether domestic or foreign.
Payments to
non-U.S. holders
of dividends on our common stock will generally not be subject
to backup withholding, and payments of proceeds made to
non-U.S. holders
by a broker upon a sale of our common stock will not be subject
to backup withholding, in each case so long as the
non-U.S. holder
certifies its nonresident status and the payer does not have
actual knowledge or reason to know that such holder is a
U.S. person as defined under the Code or such holder
otherwise establishes an exemption. We must report annually to
the IRS any dividends paid to each
non-U.S. holder
and the tax withheld, if any, with respect to such dividends.
Copies of these reports may be made available to tax authorities
in the country where the
non-U.S. holder
resides.
Any amounts withheld from a payment to a holder of our common
stock under the backup withholding rules generally may be
credited against any U.S. federal income tax liability of
the holder.
New Legislation
Relating to Foreign Accounts
Newly enacted legislation may impose withholding taxes on
certain types of payments made to “foreign financial
institutions” and certain other
non-U.S. entities.
Under this legislation, the failure to comply with additional
certification, information reporting and other specified
requirements could result in withholding tax being imposed on
payments of dividends and sales proceeds to foreign
intermediaries and certain
non-U.S. holders.
The legislation imposes a 30% withholding tax on dividends on,
or gross proceeds from the sale or other disposition of, our
common stock paid to a foreign financial institution or to a
foreign non-financial entity, unless (i) the foreign
financial institution undertakes certain diligence and reporting
obligations or (ii) the foreign non-financial entity either
certifies it does not have any substantial U.S. owners or
furnishes identifying information regarding each substantial
U.S. owner. If the payee is a foreign financial
institution, it must enter into an agreement with the
U.S. Treasury requiring, among other things, that it
undertake to identify accounts held by certain U.S. persons
or
U.S.-owned
foreign entities, annually report certain information about such
accounts and withhold 30% on payments to account holders whose
actions prevent it from complying with these reporting and other
requirements. The legislation applies to payments made after
December 31, 2012. Prospective investors should consult
their tax advisors regarding this legislation.
THE PRECEDING DISCUSSION OF U.S. FEDERAL INCOME TAX
CONSIDERATIONS IS FOR GENERAL INFORMATION ONLY. IT IS NOT TAX
ADVICE. EACH PROSPECTIVE INVESTOR SHOULD CONSULT ITS OWN TAX
ADVISOR REGARDING THE PARTICULAR U.S. FEDERAL, STATE, LOCAL
AND FOREIGN TAX CONSEQUENCES OF PURCHASING, HOLDING, AND
DISPOSING OF OUR COMMON STOCK, INCLUDING THE CONSEQUENCES OF ANY
PROPOSED CHANGE IN APPLICABLE LAWS.
86
Legal
Matters
The validity of the shares of common stock offered hereby and
certain other legal matters will be passed upon for us by
Fredrikson & Byron, P.A., Minneapolis, Minnesota. The
underwriters have been represented in connection with this
offering by Cooley Godward Kronish LLP, Palo Alto, California.
Experts
Ernst & Young LLP, independent registered public
accounting firm, has audited our financial statements at
December 31, 2008 and 2009, and for the period from
May 1, 2007 (date of inception) to December 31, 2007,
the years ended December 31, 2008 and 2009, and for the
period from May 1, 2007 (date of inception) to
December 31, 2009, as set forth in their report. We have
included our financial statements in the prospectus and
elsewhere in the registration statement in reliance on
Ernst & Young LLP’s report, given on their
authority as experts in accounting and auditing.
Where You Can
Find More Information
We have filed with the SEC a registration statement on
Form S-1
under the Securities Act with respect to the shares of common
stock offered by this prospectus. This prospectus does not
contain all of the information included in the registration
statement, portions of which are omitted as permitted by the
rules and regulations of the SEC. For further information
pertaining to us and the common stock to be sold in this
offering, you should refer to the registration statement and its
exhibits. Whenever we make reference in this prospectus to any
of our contracts, agreements or other documents, the references
are not necessarily complete, and you should refer to the
exhibits attached to the registration statement for copies of
the actual contract, agreement or other document filed as an
exhibit to the registration statement or such other document,
each such statement being qualified in all respects by such
reference. On the closing of this offering, we will be subject
to the informational requirements of the Securities Exchange Act
and will be required to file annual, quarterly and current
reports, proxy statements and other information with the SEC. We
anticipate making these documents publicly available, free of
charge, on our website at www.kipsbaymedical.com as soon as
reasonably practicable after filing such documents with the SEC.
The information contained in, or that can be accessed through,
our website is not part of this prospectus.
You can read the registration statement and our future filings
with the SEC over the Internet at the SEC’s website at
www.sec.gov. You may request copies of the filing, at no cost,
by telephone at
(763) 235-3540
or by mail at Kips Bay Medical, Inc., 3405 Annapolis Lane North,
Suite 200, Minneapolis, MN 55447. You may also read and
copy any document we file with the SEC at its public reference
facility at 100 F Street, N.E., Washington, D.C.
20549. Copies of the registration statement may be obtained from
the SEC at prescribed rates from the public reference room at
such address. You may obtain information regarding the operation
of the public reference room by calling
1-800-SEC-0330.
87
Report of
Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders
Kips Bay Medical, Inc.
We have audited the accompanying balance sheets of Kips Bay
Medical, Inc. (a development stage company) as of
December 31, 2008 and 2009, and the related statements of
operations, stockholders’ equity (deficit), and cash flows
for each of the three years in the period ended
December 31, 2009, and for the period from May 1, 2007
(inception) to December 31, 2009. These financial
statements are the responsibility of the company’s
management. Our responsibility is to express an opinion on these
financial statements based on our audits.
We conducted our audits in accordance with the standards of the
Public Company Accounting Oversight Board (United States). Those
standards require that we plan and perform the audit to obtain
reasonable assurance about whether the financial statements are
free of material misstatement. We were not engaged to perform an
audit of the Company’s internal control over financial
reporting. Our audits included consideration of internal control
over financial reporting as a basis for designing audit
procedures that are appropriate in the circumstances, but not
for the purpose of expressing an opinion on the effectiveness of
the Company’s internal control over financial reporting.
Accordingly, we express no such opinion. An audit also includes
examining, on a test basis, evidence supporting the amounts and
disclosures in the financial statements, assessing the
accounting principles used and significant estimates made by
management and evaluating the overall financial statement
presentation. We believe that our audits provide a reasonable
basis for our opinion.
In our opinion, the financial statements referred to above
present fairly, in all material respects, the financial position
of Kips Bay Medical, Inc. at December 31, 2008 and 2009,
and the results of its operations and its cash flows for each of
the three years in the period ended December 31, 2009 and
for the period from May 1, 2007 (inception) to
December 31, 2009, in conformity with U.S. generally
accepted accounting principles.
As discussed in Note 6 to the financial statements,
effective January 1, 2009, the Company adopted the
provisions of the Financial Accounting Standards Board’s
Accounting Standards Codification Topic
815-40 and
changed its method of accounting for certain instruments indexed
to the Company’s own stock.
Minneapolis, Minnesota
April 8, 2010
F-2
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from
|
|
|
|
|
|
|
|
|
Period from
|
|
|
|
|
May 1, 2007
|
|
|
|
|
|
|
|
|
May 1, 2007
|
|
|
|
|
(Date of
|
|
|
|
|
|
|
|
|
(Date of
|
|
|
|
|
Inception) to
|
|
|
|
|
|
|
|
|
Inception) to
|
|
|
|
|
December 31,
|
|
|
Year Ended December 31,
|
|
|
December 31,
|
|
|
|
|
2007
|
|
|
2008
|
|
|
2009
|
|
|
2009
|
|
|
|
|
Operating expenses:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development
|
|
$
|
196
|
|
|
$
|
2,635
|
|
|
$
|
3,004
|
|
|
$
|
5,835
|
|
|
Selling, general and administrative
|
|
|
381
|
|
|
|
754
|
|
|
|
779
|
|
|
|
1,914
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating loss
|
|
|
(577
|
)
|
|
|
(3,389
|
)
|
|
|
(3,783
|
)
|
|
|
(7,749
|
)
|
|
Interest income
|
|
|
65
|
|
|
|
52
|
|
|
|
17
|
|
|
|
134
|
|
|
Interest expense
|
|
|
(164
|
)
|
|
|
(390
|
)
|
|
|
(181
|
)
|
|
|
(735
|
)
|
|
Impairment of available for sale securities
|
|
|
—
|
|
|
|
(85
|
)
|
|
|
—
|
|
|
|
(85
|
)
|
|
Change in fair value of investor stock purchase option
|
|
|
—
|
|
|
|
—
|
|
|
|
610
|
|
|
|
610
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
$
|
(676
|
)
|
|
$
|
(3,812
|
)
|
|
$
|
(3,337
|
)
|
|
$
|
(7,825
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted net loss per share
|
|
$
|
(0.16
|
)
|
|
$
|
(0.62
|
)
|
|
$
|
(0.30
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average shares outstanding—basic and diluted
|
|
|
4,106,557
|
|
|
|
6,100,767
|
|
|
|
11,069,342
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(In thousands, except share and per
share amounts)
|
See accompanying notes to financial statements.
F-3
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2008
|
|
|
2009
|
|
|
|
|
ASSETS
|
|
Current assets:
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents
|
|
$
|
943
|
|
|
$
|
2,469
|
|
|
Short-term investments
|
|
|
181
|
|
|
|
948
|
|
|
Prepaid expenses and other current assets
|
|
|
89
|
|
|
|
37
|
|
|
|
|
|
|
|
|
|
|
|
|
Total current assets
|
|
|
1,213
|
|
|
|
3,454
|
|
|
Property and equipment, net
|
|
|
239
|
|
|
|
286
|
|
|
|
|
|
|
|
|
|
|
|
|
Total assets
|
|
$
|
1,452
|
|
|
$
|
3,740
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
LIABILITIES AND STOCKHOLDERS’ EQUITY
|
|
Current liabilities:
|
|
|
|
|
|
|
|
|
|
Accounts payable
|
|
$
|
23
|
|
|
$
|
84
|
|
|
Accrued liabilities
|
|
|
159
|
|
|
|
184
|
|
|
Accrued interest payable
|
|
|
424
|
|
|
|
—
|
|
|
Investor stock purchase option liability
|
|
|
—
|
|
|
|
960
|
|
|
|
|
|
|
|
|
|
|
|
|
Total current liabilities
|
|
|
606
|
|
|
|
1,228
|
|
|
Long-term debt, net
|
|
|
2,862
|
|
|
|
—
|
|
|
Commitments and contingencies (note 5)
|
|
|
|
|
|
|
|
|
|
Stockholders’ equity:
|
|
|
|
|
|
|
|
|
|
Undesignated stock, $0.01 par value, 10,000,000 shares
authorized, no shares issued and outstanding as of
December 31, 2008 and 2009, respectively
|
|
|
—
|
|
|
|
—
|
|
|
Common stock, $0.01 par value, 40,000,000 shares
authorized, 6,300,000 and 12,398,919 issued and outstanding as
of December 31, 2008 and 2009, respectively
|
|
|
63
|
|
|
|
124
|
|
|
Additional paid-in capital
|
|
|
2,409
|
|
|
|
11,556
|
|
|
Accumulated other comprehensive income
|
|
|
—
|
|
|
|
33
|
|
|
Deficit accumulated during development stage
|
|
|
(4,488
|
)
|
|
|
(9,201
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Total stockholders’ equity (deficit)
|
|
|
(2,016
|
)
|
|
|
2,512
|
|
|
|
|
|
|
|
|
|
|
|
|
Total liabilities and stockholders’ equity
|
|
$
|
1,452
|
|
|
$
|
3,740
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(In thousands except share and per
share amounts)
|
See accompanying notes to financial statements.
F-4
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Deficit
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated
|
|
|
Accumulated
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additional
|
|
|
During the
|
|
|
Other
|
|
|
Total
|
|
|
|
|
Common Stock
|
|
|
Paid-In
|
|
|
Development
|
|
|
Comprehensive
|
|
|
Stockholders’
|
|
|
|
|
Shares
|
|
|
Amount
|
|
|
Capital
|
|
|
Stage
|
|
|
Income
|
|
|
Equity
|
|
|
|
|
Balance at May 1, 2007 (date of inception)
|
|
|
—
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
Comprehensive loss
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(676
|
)
|
|
|
—
|
|
|
|
(676
|
)
|
|
Unrealized loss on investments, net
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(13
|
)
|
|
|
(13
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Comprehensive loss
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(689
|
)
|
|
Common stock issued to founders at incorporation, $0.167 per
share
|
|
|
6,000,000
|
|
|
|
60
|
|
|
|
40
|
|
|
|
—
|
|
|
|
—
|
|
|
|
100
|
|
|
Issued stock purchase options in conjunction with issuance of
secured convertible notes
|
|
|
|
|
|
|
|
|
|
|
194
|
|
|
|
|
|
|
|
|
|
|
|
194
|
|
|
Effect of beneficial conversion feature on secured convertible
notes
|
|
|
|
|
|
|
|
|
|
|
74
|
|
|
|
|
|
|
|
|
|
|
|
74
|
|
|
Stock-based compensation expense
|
|
|
—
|
|
|
|
—
|
|
|
|
16
|
|
|
|
—
|
|
|
|
—
|
|
|
|
16
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2007
|
|
|
6,000,000
|
|
|
|
60
|
|
|
|
324
|
|
|
|
(676
|
)
|
|
|
(13
|
)
|
|
|
(305
|
)
|
|
Comprehensive loss
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(3,812
|
)
|
|
|
—
|
|
|
|
(3,812
|
)
|
|
Unrealized gain on investments, net
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
13
|
|
|
|
13
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Comprehensive loss
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(3,799
|
)
|
|
Common stock issued at $5.83 per share
|
|
|
300,000
|
|
|
|
3
|
|
|
|
1,747
|
|
|
|
—
|
|
|
|
—
|
|
|
|
1,750
|
|
|
Stock-based compensation expense
|
|
|
—
|
|
|
|
—
|
|
|
|
338
|
|
|
|
—
|
|
|
|
—
|
|
|
|
338
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2008
|
|
|
6,300,000
|
|
|
|
63
|
|
|
|
2,409
|
|
|
|
(4,488
|
)
|
|
|
—
|
|
|
|
(2,016
|
)
|
|
Comprehensive loss
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(3,337
|
)
|
|
|
—
|
|
|
|
(3,337
|
)
|
|
Unrealized gain on investments, net
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
33
|
|
|
|
33
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Comprehensive loss
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(3,304
|
)
|
|
Cumulative effect adjustment for adoption of FASB ASC
815-40
|
|
|
|
|
|
|
|
|
|
|
(194
|
)
|
|
|
(1,376
|
)
|
|
|
|
|
|
|
(1,570
|
)
|
|
Common stock issued at $5.83 per share
|
|
|
300,000
|
|
|
|
3
|
|
|
|
1,747
|
|
|
|
—
|
|
|
|
—
|
|
|
|
1,750
|
|
|
Common stock issued upon conversion of note payable, conversion
price of $0.625
|
|
|
4,800,000
|
|
|
|
48
|
|
|
|
2,952
|
|
|
|
—
|
|
|
|
—
|
|
|
|
3,000
|
|
|
Common stock issued upon conversion of accumulated interest on
note payable, conversion price of $0.625
|
|
|
347,389
|
|
|
|
4
|
|
|
|
213
|
|
|
|
—
|
|
|
|
—
|
|
|
|
217
|
|
|
Common stock issued, $1.00 per share, employee exercise of
incentive stock option
|
|
|
1,000
|
|
|
|
—
|
|
|
|
1
|
|
|
|
—
|
|
|
|
—
|
|
|
|
1
|
|
|
Common stock issued, $6.00 per share, under private placement
offering (net of issuance costs of $29)
|
|
|
516,241
|
|
|
|
5
|
|
|
|
3,063
|
|
|
|
—
|
|
|
|
—
|
|
|
|
3,068
|
|
|
Common stock issued, $7.00 per share, under private placement
offering (net of issuance costs of $11)
|
|
|
134,289
|
|
|
|
1
|
|
|
|
928
|
|
|
|
—
|
|
|
|
—
|
|
|
|
929
|
|
|
Stock-based compensation expense
|
|
|
—
|
|
|
|
—
|
|
|
|
437
|
|
|
|
—
|
|
|
|
—
|
|
|
|
437
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2009
|
|
|
12,398,919
|
|
|
$
|
124
|
|
|
$
|
11,556
|
|
|
$
|
(9,201
|
)
|
|
$
|
33
|
|
|
$
|
2,512
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(In thousands except share amounts)
|
See accompanying notes to financial statements.
F-5
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from
|
|
|
|
|
|
Period from
|
|
|
|
|
May 1, 2007
|
|
|
|
|
|
May 1, 2007
|
|
|
|
|
(Date of
|
|
|
|
|
|
(Date of
|
|
|
|
|
Inception)
|
|
|
|
|
|
Inception)
|
|
|
|
|
to December 31,
|
|
|
Year Ended December 31,
|
|
|
to December 31,
|
|
|
|
|
2007
|
|
|
2008
|
|
|
2009
|
|
|
2009
|
|
|
|
|
Cash flows from operating activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
$
|
(676
|
)
|
|
$
|
(3,812
|
)
|
|
$
|
(3,337
|
)
|
|
$
|
(7,825
|
)
|
|
Adjustments to reconcile net loss to net cash used in operating
activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Depreciation expense
|
|
|
9
|
|
|
|
49
|
|
|
|
60
|
|
|
|
118
|
|
|
Stock-based compensation
|
|
|
16
|
|
|
|
338
|
|
|
|
437
|
|
|
|
791
|
|
|
Amortization of discount on secured convertible notes
|
|
|
38
|
|
|
|
92
|
|
|
|
138
|
|
|
|
268
|
|
|
Impairment of available for sale securities
|
|
|
—
|
|
|
|
85
|
|
|
|
—
|
|
|
|
85
|
|
|
Change in fair value of investor stock purchase option
|
|
|
—
|
|
|
|
—
|
|
|
|
(610
|
)
|
|
|
(610
|
)
|
|
Changes in operating assets and liabilities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Prepaid expenses and other current assets
|
|
|
(65
|
)
|
|
|
(24
|
)
|
|
|
52
|
|
|
|
(37
|
)
|
|
Accounts payable
|
|
|
17
|
|
|
|
7
|
|
|
|
60
|
|
|
|
84
|
|
|
Accrued liabilities
|
|
|
30
|
|
|
|
129
|
|
|
|
25
|
|
|
|
184
|
|
|
Accrued interest payable
|
|
|
126
|
|
|
|
298
|
|
|
|
(207
|
)
|
|
|
217
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash used in operating activities
|
|
|
(505
|
)
|
|
|
(2,838
|
)
|
|
|
(3,382
|
)
|
|
|
(6,725
|
)
|
|
Cash flows from investing activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net change in short-term investments
|
|
|
(548
|
)
|
|
|
282
|
|
|
|
(734
|
)
|
|
|
(1,000
|
)
|
|
Purchase of property and equipment
|
|
|
(212
|
)
|
|
|
(86
|
)
|
|
|
(106
|
)
|
|
|
(404
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash provided by (used in) investing activities
|
|
|
(760
|
)
|
|
|
196
|
|
|
|
(840
|
)
|
|
|
(1,404
|
)
|
|
Cash flows from financing activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Proceeds from sale of common stock
|
|
|
100
|
|
|
|
—
|
|
|
|
—
|
|
|
|
100
|
|
|
Proceeds from issuances of notes payable
|
|
|
3,000
|
|
|
|
—
|
|
|
|
—
|
|
|
|
3,000
|
|
|
Proceeds from exercise of investor option to purchase common
stock
|
|
|
—
|
|
|
|
1,750
|
|
|
|
1,750
|
|
|
|
3,500
|
|
|
Proceeds from sale of common stock under private placement
offerings, net of issuance costs
|
|
|
—
|
|
|
|
—
|
|
|
|
3,997
|
|
|
|
3,997
|
|
|
Proceeds from exercise of employee stock options
|
|
|
—
|
|
|
|
—
|
|
|
|
1
|
|
|
|
1
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash provided by financing activities
|
|
|
3,100
|
|
|
|
1,750
|
|
|
|
5,748
|
|
|
|
10,598
|
|
|
Net increase (decrease) in cash and cash equivalents
|
|
|
1,835
|
|
|
|
(892
|
)
|
|
|
1,526
|
|
|
|
2,469
|
|
|
Cash and cash equivalents at beginning of period
|
|
|
—
|
|
|
|
1,835
|
|
|
|
943
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents at end of period
|
|
$
|
1,835
|
|
|
$
|
943
|
|
|
$
|
2,469
|
|
|
$
|
2,469
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Supplemental disclosures:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest paid in cash
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
250
|
|
|
$
|
250
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Supplemental non-cash disclosures:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Conversion of note payable into common stock
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
3,000
|
|
|
$
|
3,000
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accrued interest paid by conversion into common stock
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
217
|
|
|
$
|
217
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(In thousands)
|
See accompanying notes to financial statements.
F-6
|
|
|
1.
|
Organization and
Business
|
Kips Bay Medical, Inc. was incorporated in the state of Delaware
on May 1, 2007. We are a medical device company focused on
developing, manufacturing and commercializing our innovative
external saphenous vein support technology, or eSVS MESH, for
use in coronary artery bypass grafting, or CABG, surgery. Our
eSVS MESH is a nitinol mesh sleeve that, when placed over a
saphenous vein graft during CABG surgery, is designed to improve
the structural characteristics and long-term performance of the
vein graft. In CABG procedures, surgeons harvest blood vessels,
including the internal mammary artery from the chest and the
saphenous vein from the leg, and attach the harvested vessels to
bypass, or provide blood flow around, blocked coronary arteries.
Our activities since inception have consisted principally of
acquiring product and technology rights, raising capital,
performing research and development and conducting preclinical
and clinical trials. Accordingly, we are considered to be in the
development stage as of December 31, 2009, as defined by
the Financial Accounting Standard Board, or FASB, Accounting
Standard Codification, or ASC, 915. At December 31, 2009,
we had an accumulated deficit of $9.2 million and we expect
to incur losses for the foreseeable future. To date, we have
been funded by private equity and debt financings. Although we
believe that we will be able to successfully fund our
operations, there can be no assurance that we will be able to do
so or that we will ever operate profitably.
|
|
|
2.
|
Summary of
Significant Accounting Policies
|
Use of
Estimates
The preparation of financial statements in conformity with
accounting principles generally accepted in the United States of
America requires management to make estimates and assumptions
that affect the reported amounts and disclosures in the combined
consolidated financial statements and accompanying notes. Actual
results could differ from those estimates.
Cash and Cash
Equivalents
Cash and cash equivalents consist of cash and money market funds
with original maturities of three months or less. The carrying
value of these instruments approximates fair value. We have not
experienced any losses on our cash and cash equivalents.
Short-term
Investments
Short-term investments consist of certificates of deposits and
mutual fund investments with a maturity of greater than three
months and less than one year. Short-term investments have been
classified and accounted for as
available-for-sale
securities and are reported on the balance sheet at fair value
with unrealized gains or losses reported as a component of other
comprehensive income. We continually evaluate our investments in
marketable securities for impairment due to declines in market
value considered to be
other-than-temporary.
That evaluation includes persistent declining stock prices and
general economic and company-specific evaluations.
Property and
Equipment
Property and equipment is stated at cost less accumulated
depreciation. Depreciation is computed based upon the estimated
useful lives of the respective assets, or the lesser of the
estimated useful life or the remaining life of the underlying
facility lease for leasehold improvements, ranging from three to
seven years, and is recorded using the straight-line method.
Repairs and maintenance costs are expensed as incurred.
F-7
Kips Bay Medical,
Inc.
(A Development Stage Company)
Notes To Financial Statements—(Continued)
Impairment of
Long-Lived Assets
In accordance with FASB ASC 360, Property, Plant and
Equipment, long-lived assets, such as property and
equipment, are reviewed for impairment whenever events or
changes in circumstances indicate that the carrying amount of an
asset may not be recoverable. Recoverability of assets to be
held and used is measured by a comparison of the carrying amount
of an asset to estimated undiscounted future cash flows expected
to be generated by the asset. If the carrying amount of an asset
exceeds its estimated future cash flows, then an impairment
charge is recognized by the amount by which the carrying amount
of the asset exceeds the fair value of the asset. As of
December 31, 2008 and 2009, management believes that no
modification of the remaining useful lives or write-down of
long-lived assets is required.
Fair Value of
Financial Instruments
We adopted the provisions of FASB ASC 820, Fair Value
Measurements and Disclosures, effective January 1,
2008. FASB ASC 820 defines fair value, establishes a framework
for measuring fair value under generally accepted accounting
principles, or GAAP, and enhances disclosures about fair value
measurements.
Fair value is defined as the price that would be received to
sell an asset or paid to transfer a liability in an orderly
transaction between market participants at the measurement date.
Valuation techniques used to measure fair value, as required by
FASB ASC 820, must maximize the use of observable inputs and
minimize the use of unobservable inputs.
The standard describes a fair value hierarchy based on three
levels of inputs, of which the first two are considered
observable and the last unobservable, that may be used to
measure fair value. Our assessment of the significance of a
particular input to the fair value measurements requires
judgment, and may affect the valuation of the assets and
liabilities being measured and their placement within the fair
value hierarchy. The three levels of input are:
Level 1—Quoted prices in active markets for identical
assets or liabilities.
Level 2—Inputs other than Level 1 that are
observable, either directly or indirectly, such as quoted prices
for similar assets or liabilities; quoted prices in markets that
are not active; or other inputs that are observable or can be
corroborated by observable market data for substantially the
full term of the assets or liabilities.
Level 3—Unobservable inputs that are supported by
little or no market activity and that are significant to the
fair value of the assets or liabilities.
The adoption of this statement did not have a material impact on
our results of operations and financial condition.
Following is a description of our valuation methodologies for
assets and liabilities measured at fair value.
Where quoted prices are available in an active market, fair
value is based upon quoted market prices and is classified in
level 1 of the fair value hierarchy. If quoted market
prices are not available, fair value is based upon observable
inputs such as quoted prices for similar assets or liabilities,
quoted prices in markets that are not active, or other inputs
that are observable or can be corroborated by observable market
data, and the assets or liabilities are classified in
level 2 of the valuation hierarchy. When quoted prices and
observable inputs are unavailable, fair values are based on
internally developed cash flow models and are classified in
level 3 of the valuation hierarchy. The internally
developed cash flow models primarily use, as inputs, estimates
for interest rates and discount rates, including yields of
comparable traded instruments adjusted for illiquidity and other
risk factors, amount of cash flows and expected holding periods
of the assets. These inputs reflect our own assumptions about
the assumptions market participants would use in pricing the
assets, including assumptions about risk developed based on the
best information available in the circumstances.
F-8
Kips Bay Medical,
Inc.
(A Development Stage Company)
Notes To Financial Statements—(Continued)
Other financial instruments, including accounts payable and
accrued liabilities, are carried at cost, which we believe
approximates fair value because of the short-term maturity of
these instruments.
Research and
Development Expenses
Research and development costs are expensed as incurred and
include the costs to design, develop, test, deploy and enhance
our eSVS MESH. It also includes costs related to the execution
of clinical trials and to obtain regulatory approval for our
eSVS MESH. We expense amounts paid to obtain patents or acquire
licenses as the ultimate recoverability of the amounts paid is
uncertain.
We charge research and development costs, including clinical
trial costs, to expense when incurred, consistent with the
guidance of FASB ASC 730, Research and Development.
Clinical trial costs are a significant component of research and
development expenses. All of our clinical trials are performed
at clinical trial sites and are administered by contract
research organizations, or CROs. Costs of setting up clinical
trial sites are accrued immediately. Expenses related to
clinical trials generally are accrued based on contracted
amounts and the achievement of milestones, such as number of
patients enrolled. We monitor levels of performance under each
significant contract, including the extent of patient enrollment
and other activities through communications with the CROs, and
adjust the estimates, if required, on a quarterly basis so that
clinical expenses reflect the actual effort expended by each CRO.
All material CRO contracts are terminable by us upon written
notice and we are generally only liable for actual effort
expended by the CROs and certain non-cancelable expenses
incurred at any point of termination.
Comprehensive
Income/Loss
Comprehensive income/loss consists of other comprehensive income
or losses and net loss. Other comprehensive income or losses
include certain changes in equity that are excluded from net
loss. Specifically, we include unrealized gains and losses on
available-for-sale
securities in other comprehensive income/loss. Comprehensive
loss for each period presented is set forth in the Statements of
Stockholders’ Equity (Deficit).
Income
Taxes
Income taxes are accounted for under the asset and liability
method. Deferred tax assets and liabilities are recognized for
the future tax consequences attributable to differences between
the financial statement carrying amounts of existing assets and
liabilities and their respective tax bases and operating loss
and tax credit carry-forwards. Deferred tax assets and
liabilities are measured using enacted tax rates expected to
apply to taxable income in the years in which those temporary
differences are expected to be recovered or settled. The effect
on deferred tax assets and liabilities of a change in tax rates
is recognized in income in the period that includes the
enactment date. In light of our cumulative losses, we believe
that it is more likely than not that our net deferred tax asset
will not be realized. Accordingly, a full valuation allowance
has been recorded against our net deferred tax assets.
Stock-Based
Compensation
Stock-based incentive awards are accounted for under the
provisions of FASB ASC 718, Compensation — Stock
Compensation, which requires companies to measure and
recognize the cost of employee and non-employee services
received in exchange for awards of equity instruments based on
the grant date fair value of those awards. Compensation cost is
recognized ratably using the straight-line attribution method
over the expected vesting period, which is considered to be the
requisite service period. In addition, we are required to
estimate the amount of expected forfeitures when calculating the
compensation costs, instead of accounting for forfeitures as
incurred.
F-9
Kips Bay Medical,
Inc.
(A Development Stage Company)
Notes To Financial Statements—(Continued)
All of our options previously awarded were classified as equity
instruments and continue to maintain their equity classification.
The fair value of options is estimated at the date of grant
using the Black-Scholes option pricing model with the
assumptions listed in note 7. Risk free interest rates are
based upon U.S. Treasury rates appropriate for the expected
term. Expected volatility and forfeiture rates are based on the
volatility rates of a set of guideline companies, which consist
of public and recently public medical technology companies. The
assumed dividend yield is zero, as we do not expect to declare
any dividends in the foreseeable future. The expected term is
determined using the simplified method allowed by SEC Staff
Accounting Bulletin No. 110. The fair value of the
common stock underlying the stock options has been determined by
our Board of Directors at each award grant date based upon a
variety of factors, primarily the most recent purchase prices of
our common stock issued to third parties in arms-length
transactions, but also the progress of our product development,
the progress of our preclinical and clinical testing, and the
risks associated with our business plan.
Stock-based compensation expense in our statements of operations
is summarized as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from May 1,
|
|
|
|
|
|
|
|
|
Period from May 1,
|
|
|
|
|
2007 (Date of
|
|
|
|
|
|
|
|
|
2007 (Date of
|
|
|
|
|
Inception) to
|
|
|
December 31,
|
|
|
Inception) to
|
|
|
|
|
December 31, 2007
|
|
|
2008
|
|
|
2009
|
|
|
December 31, 2009
|
|
|
|
|
Research and development
|
|
$
|
14
|
|
|
$
|
298
|
|
|
$
|
390
|
|
|
$
|
702
|
|
|
Sales, general and administrative
|
|
|
2
|
|
|
|
40
|
|
|
|
47
|
|
|
|
89
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total stock-based compensation
|
|
$
|
16
|
|
|
$
|
338
|
|
|
$
|
437
|
|
|
$
|
791
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(In thousands)
|
Net Loss Per
Share
We compute net loss per share in accordance with FASB ASC 260,
Earnings Per Share, under which basic net loss
attributable to common stockholders, on a per share basis, is
computed by dividing income available to common stockholders
(the numerator) by the weighted-average number of common shares
outstanding (the denominator) during the period. Shares issued
during the period and shares reacquired during the period are
weighted for the portion of the period that they were
outstanding. The computation of diluted earnings per share, or
EPS, is similar to the computation of basic EPS except that the
denominator is increased to include the number of additional
common shares that would have been outstanding if the dilutive
potential common shares had been issued. In addition, in
computing the dilutive effect of convertible securities, the
numerator is adjusted to add back the after-tax amount of
interest recognized in the period associated with any
convertible debt. Diluted EPS is the same as basic EPS due to
common equivalent shares being excluded from the calculation, as
their effect is anti-dilutive.
The following table summarizes our calculation of net loss per
common share:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from May 1,
|
|
|
|
|
|
|
|
|
|
|
2007 (Date of
|
|
|
|
|
|
|
|
|
|
|
Inception) to
|
|
|
December 31,
|
|
|
|
|
December 31, 2007
|
|
|
2008
|
|
|
2009
|
|
|
|
|
Net loss
|
|
$
|
(676
|
)
|
|
$
|
(3,812
|
)
|
|
$
|
(3,337
|
)
|
|
Weighted average shares outstanding—basic and diluted
|
|
|
4,106,557
|
|
|
|
6,100,767
|
|
|
|
11,069,342
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss per share—basic and diluted
|
|
$
|
(0.16
|
)
|
|
$
|
(0.62
|
)
|
|
$
|
(0.30
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(In thousands, except per share
amounts)
|
F-10
Kips Bay Medical,
Inc.
(A Development Stage Company)
Notes To Financial Statements—(Continued)
Outstanding potential common shares not included in diluted net
loss per share attributable to common stockholders calculations
included:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from May 1,
|
|
|
|
|
|
|
|
2007 (Date of
|
|
|
|
|
|
|
|
Inception) to
|
|
December 31,
|
|
|
|
December 31, 2007
|
|
2008
|
|
2009
|
|
|
|
Employee and non-employee stock options
|
|
|
295,000
|
|
|
|
531,750
|
|
|
|
598,000
|
|
|
Common shares issuable for conversion of debt (See note 6)
|
|
|
4,800,000
|
|
|
|
4,800,000
|
|
|
|
—
|
|
|
Common shares issuable under investor option purchase agreements
(See note 6)
|
|
|
1,200,000
|
|
|
|
900,000
|
|
|
|
600,000
|
|
|
|
Fiscal
Year
We operate on a manufacturing calendar with our fiscal year
always ending on December 31. Each quarter is
13 weeks, consisting of one five-week and two four-week
periods.
New Accounting
Pronouncements
In June 2009, the FASB issued FASB ASC 105, Generally
Accepted Accounting Principles, which establishes the FASB
Accounting Standards Codification as the sole source of
authoritative generally accepted accounting principles. Pursuant
to the provisions of FASB ASC 105, we have reflected references
to GAAP in our financial statements issued for the period ended
December 31, 2009. The adoption of FASB ASC 105 did not
impact our financial position or results of operations.
In June 2008, the FASB issued FASB ASC
815-40,
Derivatives and Hedging, which provides guidance on how
to determine if certain instruments (or embedded features) are
considered indexed to a company’s own stock, including
instruments similar to warrants to purchase the company’s
stock. FASB ASC
815-40
clarifies the determination of whether equity-linked instruments
(or embedded features), such as our convertible notes or options
to purchase our common stock, are considered indexed to our own
stock, which would qualify as a scope exception and therefore be
exempt from the application of FASB ASC 815. As a result of the
adoption of the provisions of FASB ASC
815-40 on
January 1, 2009, certain instruments previously reported as
equity are now accounted for as derivative instruments. ASC
815-40 was
initially applied by recording a cumulative effect adjustment to
retained earnings upon adoption. The impact of adopting this
guidance is described in note 6.
|
|
|
3.
|
Short-Term
Investments and Impairment
|
Short-term investments include a mutual fund investment and
certificates of deposit, which are generally measured using
quoted prices in active markets and are classified as
Level 1.
Due to the short maturities of our investments in certificates
of deposits, their amortized cost approximates fair value.
As of December 31, 2008 we concluded that the value of our
mutual fund investment was impaired and that this impairment was
other than temporary. We recorded an other than temporary
impairment charge of $85,000, which resulted in a new adjusted
cost basis for this investment. We had no other unrealized gains
or losses as of December 31, 2008. For the year ended
December 31, 2009, unrealized gains of $33,000 were
reported in other comprehensive income as the market value
exceeded the cost of our mutual fund investment.
F-11
Kips Bay Medical,
Inc.
(A Development Stage Company)
Notes To Financial Statements—(Continued)
|
|
|
4.
|
Property and
Equipment
|
At December 31, 2008 and 2009, property and equipment
consist of the following:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2008
|
|
|
2009
|
|
|
|
|
Furniture and fixtures
|
|
$
|
33
|
|
|
$
|
36
|
|
|
Machinery, equipment and tooling
|
|
|
154
|
|
|
|
254
|
|
|
Computers and software
|
|
|
62
|
|
|
|
66
|
|
|
Leasehold improvements
|
|
|
48
|
|
|
|
48
|
|
|
Accumulated depreciation
|
|
|
(58
|
)
|
|
|
(118
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Property and equipment, net
|
|
$
|
239
|
|
|
$
|
286
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(In thousands)
|
Depreciation expense for the years ended December 31, 2007,
2008 and 2009 was $9,000, $49,000 and $60,000, respectively.
|
|
|
5.
|
Commitments and
Contingencies
|
Leases
We entered into an operating lease agreement for our facility.
The term of this lease runs from October 1, 2007 through
September 30, 2010. The monthly base rent amount is fixed
over the entire term of the lease. Terms of this lease
arrangement include market rate renewal options, payment of
executory costs such as real estate taxes, insurance and common
area maintenance. We are currently in negotiations with our
landlord to renew this lease. We also lease certain other office
equipment under non-cancelable operating lease arrangements
which are not recognized on our balance sheets.
Annual future minimum lease obligations under our operating
lease agreements as of December 31, 2009 are as follows:
| |
|
|
|
|
|
|
|
2010
|
|
$
|
46
|
|
|
2011
|
|
|
3
|
|
|
2012
|
|
|
3
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
52
|
|
|
|
|
|
|
|
|
|
|
(In thousands)
|
Rent expense was $14,000, $57,000 and $57,000 for the years
ended December 31, 2007, 2008 and 2009, respectively, and
$128,000 for the period from May 1, 2007 (date of
inception) to December 31, 2009.
Royalty
Payments
The core intellectual property relating to our eSVS MESH,
including five patent applications pending in the United States
and nine patent applications pending in countries outside the
United States, was acquired from Medtronic, Inc. pursuant to an
Assignment and License Agreement dated October 9, 2007. As
consideration for the assignment of such intellectual property,
we have agreed to pay Medtronic an aggregate of up to
$15.0 million upon the achievement of certain sales
milestones and a royalty of 4% on sales of our eSVS MESH. The
royalty will terminate upon the earlier of the expiration of all
of the patents and patent applications, or when the aggregate
royalties paid reaches $100.0 million.
F-12
Kips Bay Medical,
Inc.
(A Development Stage Company)
Notes To Financial Statements—(Continued)
Legal
Proceedings
We are not currently engaged in any litigation.
Employment
Agreements
We entered into an employment agreement with Manny
Villafaña, our founder, Chief Executive Officer and sole
director, on July 19, 2007, which provides for a base
salary and discretionary bonuses as determined by our Board of
Directors. Mr. Villafaña is also entitled to
participate in any employee benefit plans we sponsor. If we
terminate Mr. Villafaña’s employment without
cause, he is entitled to his base salary for the entire term of
the employment agreement, which expires on July 1, 2012.
Mr. Villafaña’s employment agreement contains
usual and customary provisions relating to confidential
information and assignment of inventions to us. In addition, the
agreement contains certain provisions concerning
Mr. Villafaña’s post-employment activities.
Pursuant to the agreement, he has agreed not to compete with us
for a period of two years after the termination of his
employment. Such two-year period will automatically be extended
by one year increments, up to a total of five years, unless
terminated by us. As consideration for this non-competition
provision, we will make a monthly payment to
Mr. Villafaña equal to one twelfth of his base salary
at the time of termination, adjusted to the Consumer Price
Index, or CPI, beginning with the first month after termination
of employment and continuing until the non-competition provision
expires. Mr. Villafaña will also be entitled to
continue his participation in our medical benefits plan for the
term of the non-competition provision, provided he continues to
pay the employee portion of the premium. Following the
termination of his employment with us, Mr. Villafaña
has also agreed to consult on non-confidential matters at the
request of our Board of Directors.
We also entered into a change in control agreement with
Mr. Villafaña on September 12, 2008. Under the
terms of this agreement, if, within 24 months of a change
in control, Mr. Villafaña’s employment is
terminated by us other than for cause, or if he resigns for good
reason, Mr. Villafaña will be entitled to a prorated
portion of any annual incentive bonus for the fiscal year in
which the termination occurs and a severance benefit equal to
three years of his base salary. The change in control agreement
will expire on September 12, 2011, but will be
automatically extended by one-year increments thereafter unless
either party provides written notice to the other of the intent
not to extend the agreement.
We have entered into employment agreements with certain key
employees providing for an annual salary and such benefits in
the future as may be approved by the Board of Directors. We have
also entered into change of control agreements with employees
which provide that if the employee is terminated for a reason
other than cause, or resigns for good reason, upon a merger,
acquisition, sale of substantially all of our assets, or
liquidation, the employee will receive severance payments equal
to his or her monthly salary for 12 to 36 months.
Indemnification
Agreements
The Company, as permitted under Delaware law and in accordance
with its Bylaws, indemnifies its officers and directors for
certain events or occurrences, subject to certain limits, while
the officer or director is or was serving at the Company’s
request in such capacity. The term of the indemnification period
is for the officer’s or director’s lifetime. The
Company may terminate the indemnification agreements with its
officers and directors upon 90 days written notice, but
termination will not affect claims for indemnification relating
to events occurring prior to the effective date of termination.
The maximum amount of potential future indemnification is
unlimited; however, the Company has a director and officer
insurance policy that limits its exposure and may enable it to
recover a portion of any future amounts paid. The Company
believes the fair value of these indemnification agreements is
minimal. Accordingly, the Company has not recorded any
liabilities for these agreements as of December 31, 2009.
F-13
Kips Bay Medical,
Inc.
(A Development Stage Company)
Notes To Financial Statements—(Continued)
|
|
|
6.
|
Convertible
Promissory Notes and Equity Financing
|
Investment
Agreement with Kips Bay Investments, LLC
On July 19, 2007, we executed an Investment Agreement under
which we issued two secured convertible notes to Kips Bay
Investments, LLC, or KBI. Pursuant to the Investment Agreement,
which was executed prior to KBI becoming a 5% or more beneficial
holder of our common stock, KBI agreed to sell us all right and
title to certain intellectual property assets for the sum of
$100,000 in the form of a first secured convertible note, and to
loan us the sum of $2.9 million in exchange for a second
secured convertible note, or collectively, the Notes. The Notes
accrued interest at a rate of nine percent per annum, and were
convertible into an aggregate of 4,800,000 shares of our
common stock for repayment of principal, representing a
conversion price of $0.625 per share, and conversion of accrued
interest also at $0.625 per share. Pursuant to the Investment
Agreement, we also granted KBI two separate options to purchase
common stock, each for the right to purchase up to
600,000 shares of our common stock for the sum of
$3.5 million at such time as we have achieved certain
defined milestones. The Investment Agreement also provides
certain registration rights to KBI. The relative fair value of
the stock purchase options and the beneficial conversion feature
on the Notes were recorded as discounts on the Notes and were
amortized over the term of the Notes using the effective
interest method.
In connection with the execution of the Notes, we entered into a
Loan and Security Agreement with KBI, whereby we granted a
security interest in all of our existing and to-be-acquired
property and proceeds therefrom, including all intellectual
property assets transferred by KBI to us pursuant to the first
secured convertible note. The Loan and Security Agreement
terminated upon conversion of the Notes.
In April 2008, we achieved the first defined milestone and KBI
subsequently exercised its first stock purchase option under the
Investment Agreement, purchasing 600,000 shares of our
common stock for a purchase price of $3.5 million.
In February 2009, KBI elected to convert the entire amount of
principal and $217,000 of $467,000 of accrued interest on the
Notes into our common stock, and we subsequently issued
5,147,389 shares of our common stock to KBI. The remaining
accrued interest of $250,000 was paid in cash concurrent with
KBI’s conversion election.
We account for derivative instruments in accordance with FASB
ASC 815, Derivatives and Hedging, which provides
accounting and reporting standards for derivative instruments,
including certain derivative instruments embedded in other
contracts. We do not use derivative instruments for hedging of
market risks or for trading or speculative purposes. Effective
January 1, 2009, we were required to adopt FASB ASC
815-40,
(formerly EITF Issue
No. 07-5,
Determining Whether an Instrument (or Embedded Feature) is
Indexed to an Entity’s Own Stock), which clarified the
determination of whether equity-linked instruments (or embedded
features), such as the options to purchase our common stock
granted to KBI, are considered indexed to our own stock, which
would qualify as a scope exception under FASB ASC 815. As a
result of adopting FASB ASC
815-40, the
second option to purchase our common stock granted to KBI is
considered a derivative liability and must be measured at fair
value. As noted above, the first option was exercised in 2008
and was not outstanding at the effective date for FASB ASC
815-40.
The estimated fair value of our investor stock purchase option
liability is recorded as a current liability on our balance
sheets. Changes in the estimated fair values of this liability
are recorded in our statements of operations.
On January 1, 2009, the date of adoption, we estimated the
fair value of the second option to be $1.6 million and this
amount was recorded as a cumulative effect adjustment on
January 1, 2009, which increased our deficit accumulated
during development stage by $1.4 million. The effect of
marking this liability to “market” at
December 31, 2009 resulted in a net decrease in the
estimated fair values of this liability of $610,000, resulting
in an estimated fair value of this liability of $960,000.
F-14
Kips Bay Medical,
Inc.
(A Development Stage Company)
Notes To Financial Statements—(Continued)
In February 2010, KBI exercised its second stock purchase option
under the Investment Agreement, the second milestone having
previously been achieved by us, purchasing 600,000 shares
of our common stock for a purchase price of $3.5 million.
At that time we also agreed to convert the $250,000 of interest
on the Notes previously paid in cash to common stock, and
pursuant to such conversion, KBI repaid the $250,000 to us and
we subsequently issued 400,000 shares of our common stock
to KBI. The exercise of this stock purchase option was made
pursuant to a Stock Purchase Agreement between KBI and us.
Common Stock
Offerings
In March 2009, we commenced a private offering of a minimum of
500,000 shares of our common stock and up to a maximum of
1,666,667 shares of our common stock to certain accredited
investors at an offering price of $6.00 per share. We sold an
aggregate of 516,241 shares of common stock in the private
offering, which was completed in August 2009. KBI purchased
41,667 shares of our common stock in this private offering.
In October 2009, we commenced a private offering of
714,286 shares of our common stock to certain accredited
investors at an offering price of $7.00 per share. As of
December 31, 2009, we had sold an aggregate of
134,289 shares of common stock in the private offering.
Subsequent to December 31, 2009, we sold an additional
182,872 shares of common stock for an aggregate total of
317,161 shares. This offering was completed in
February 2010.
|
|
|
7.
|
Stock-Based
Compensation
|
2007 Long-Term
Incentive Plan
Our 2007 Long-Term Incentive Plan, or the Plan, was adopted by
the Board of Directors in July 2007. The Plan permits the
granting of incentive and non-statutory stock options,
restricted stock, stock appreciation rights, performance units,
performance shares and other stock awards to eligible employee,
directors and consultants. We grant options to purchase shares
of common stock under the Plan at no less than the fair market
value of the underlying common stock as of the date of grant.
Options granted under the Plan have a maximum term of ten years
and generally vest over four years at the rate of 25% of total
shares underlying the option. Under the Plan, a total of
2,000,000 shares of common stock were initially reserved
for issuance.
A summary of option activity is as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted
|
|
|
|
|
|
|
|
Shares Under
|
|
|
Average
|
|
|
Available
|
|
|
|
|
Option
|
|
|
Exercise Price
|
|
|
For Grant
|
|
|
|
|
Options outstanding at May 1, 2007 (date of inception)
|
|
|
—
|
|
|
$
|
—
|
|
|
|
2,000,000
|
|
|
Granted
|
|
|
295,000
|
|
|
|
1.00
|
|
|
|
(295,000
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Options outstanding at December 31, 2007
|
|
|
295,000
|
|
|
|
1.00
|
|
|
|
1,705,000
|
|
|
Granted
|
|
|
253,000
|
|
|
|
4.65
|
|
|
|
(253,000
|
)
|
|
Exercised
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Cancelled
|
|
|
(16,250
|
)
|
|
|
2.21
|
|
|
|
16,250
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Options outstanding at December 31, 2008
|
|
|
531,750
|
|
|
|
1.72
|
|
|
|
1,468,250
|
|
|
Granted
|
|
|
70,000
|
|
|
|
5.93
|
|
|
|
(70,000
|
)
|
|
Exercised
|
|
|
(1,000
|
)
|
|
|
1.00
|
|
|
|
—
|
|
|
Cancelled
|
|
|
(2,750
|
)
|
|
|
1.00
|
|
|
|
2,750
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Options outstanding at December 31, 2009
|
|
|
598,000
|
|
|
|
3.08
|
|
|
|
1,401,000
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
F-15
Kips Bay Medical,
Inc.
(A Development Stage Company)
Notes To Financial Statements—(Continued)
The grant date total fair value of employee options vested
during the years ended December 31, 2007, 2008 and 2009 was
$0, $27,000 and $67,000, respectively. The total intrinsic value
of options exercised during the years ended December 31,
2007, 2008 and 2009 was $0, $0 and $5,000, respectively. Total
proceeds received for options exercised during years ended
December 31, 2008 and 2009 was $0 and $1,000, respectively.
Information about stock options outstanding, vested and expected
to vest as of December 31, 2009, is as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Outstanding, Vested and Expected to Vest
|
|
|
Options Vested
|
|
|
|
|
|
|
|
|
Weighted
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Average
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Remaining
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Contractual
|
|
|
Weighted
|
|
|
|
|
Per Share
|
|
|
|
|
|
Life
|
|
|
Average
|
|
|
Options
|
|
|
Exercise Price
|
|
|
Shares
|
|
|
(Years)
|
|
|
Exercise Price
|
|
|
Exercisable
|
|
|
|
|
$
|
1.00
|
|
|
|
334,000
|
|
|
|
7.80
|
|
|
$
|
1.00
|
|
|
|
166,000
|
|
|
|
2.00
|
|
|
|
10,000
|
|
|
|
8.19
|
|
|
|
2.00
|
|
|
|
2,500
|
|
|
|
5.83
|
|
|
|
214,000
|
|
|
|
8.81
|
|
|
|
5.83
|
|
|
|
84,750
|
|
|
|
6.00
|
|
|
|
40,000
|
|
|
|
9.68
|
|
|
|
6.00
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
598,000
|
|
|
|
8.30
|
|
|
|
2.63
|
|
|
|
253,250
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stock-Based
Compensation Expense
Total employee stock-based compensation expense recognized under
FASB ASC 718 was as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from May 1,
|
|
|
|
|
|
|
|
|
Period from May 1,
|
|
|
|
|
2007 (Date of
|
|
|
|
|
|
|
|
|
2007 (Date of
|
|
|
|
|
Inception) to
|
|
|
December 31,
|
|
|
Inception) to
|
|
|
|
|
December 31, 2007
|
|
|
2008
|
|
|
2009
|
|
|
December 31, 2009
|
|
|
|
|
Research and development
|
|
$
|
5
|
|
|
$
|
37
|
|
|
$
|
83
|
|
|
$
|
125
|
|
|
Selling, general and administrative
|
|
|
2
|
|
|
|
9
|
|
|
|
12
|
|
|
|
23
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total stock-based compensation
|
|
$
|
7
|
|
|
$
|
46
|
|
|
$
|
95
|
|
|
$
|
148
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(In thousands)
|
As of December 31, 2007, 2008 and 2009, total compensation
expense related to unvested employee stock options not yet
recognized was $86,000, $199,000 and $297,000, respectively,
which is expected to be allocated to expenses over a
weighted-average period of 3.68, 2.88 and 2.22 years,
respectively.
The assumptions used in the Black-Scholes option-pricing model
for the years ended December 31, 2007, 2008 and 2009, and
for the period from May 1, 2007 (Date of Inception) to
December 31, 2009, are as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from May 1,
|
|
|
|
|
|
Period from May 1,
|
|
|
|
2007
|
|
|
|
|
|
2007
|
|
|
|
(Date of Inception) to
|
|
|
|
|
|
(Date of Inception) to
|
|
|
|
December 31,
|
|
December 31,
|
|
December 31,
|
|
|
|
2007
|
|
2008
|
|
2009
|
|
2009
|
|
|
|
Risk free interest rate
|
|
|
3.39-4.32
|
%
|
|
|
2.66-3.58
|
%
|
|
|
1.75-2.80
|
%
|
|
|
1.75-4.32
|
%
|
|
Dividend yield
|
|
|
0
|
%
|
|
|
0
|
%
|
|
|
0
|
%
|
|
|
0
|
%
|
|
Expected volatility
|
|
|
52
|
%
|
|
|
53
|
%
|
|
|
48
|
%
|
|
|
48-53
|
%
|
|
Expected term
|
|
|
5.75-6.25 years
|
|
|
|
5.75-6.25 years
|
|
|
|
6.25 years
|
|
|
|
5.75-6.25 years
|
|
|
Weighted average grant date fair value
|
|
$
|
0.55
|
|
|
$
|
2.42
|
|
|
$
|
2.91
|
|
|
$
|
1.58
|
|
|
|
F-16
Kips Bay Medical,
Inc.
(A Development Stage Company)
Notes To Financial Statements—(Continued)
Nonemployee
Stock-Based Compensation
We account for stock options granted to nonemployees as required
by FASB ASC 718. In connection with stock options granted to
nonemployees we recorded $8,000, $292,000 and $342,000 for
nonemployee stock-based compensation during the years ended
December 31, 2007, 2008 and 2009, respectively, and
$642,000 for nonemployee stock-based compensation for the period
from May 1, 2007 (Date of Inception) to December 31,
2009. These amounts were based upon the fair value of the vested
portion of the grants.
Amounts expensed during the remaining vesting period will be
determined based on the fair value at the time of vesting.
We maintain a simplified employee retirement plan, or SEP, which
commenced on January 1, 2008. The SEP is a defined
contribution plan; employee contributions are voluntary and are
determined on an individual basis, limited by the maximum
amounts allowable under federal tax regulations. We contribute
up to 3% of each individual’s base salary as required under
the safe-harbor provisions of Internal Revenue Service rules
governing SEP plans. Our contributions vest immediately and are
expensed when paid. We have recorded contributions of $33,000
and $38,000 for the years ended December 31, 2008 and 2009,
respectively, and $71,000 for the period from May 1, 2007
(Date of Inception) to December 31, 2009.
We have incurred net operating losses since inception. We have
not reflected any benefit of such net operating loss
carryforwards in the accompanying financial statements and have
established a full valuation allowance against our deferred tax
assets.
Deferred income taxes reflect the net tax effects of
(a) temporary differences between the carrying amounts of
assets and liabilities for financial reporting purposes and the
amounts used for income tax purposes, and (b) operating
losses and tax credit carryforwards.
F-17
Kips Bay Medical,
Inc.
(A Development Stage Company)
Notes To Financial Statements—(Continued)
The significant components of our deferred tax assets for the
years ended December 31, 2008 and 2009 are as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2008
|
|
|
2009
|
|
|
|
|
Deferred tax assets:
|
|
|
|
|
|
|
|
|
|
Net operating loss carryforwards
|
|
$
|
1,438
|
|
|
$
|
2,838
|
|
|
Intangible assets—license agreement
|
|
|
62
|
|
|
|
89
|
|
|
Stock-based compensation
|
|
|
134
|
|
|
|
283
|
|
|
Short term investment impairment
|
|
|
37
|
|
|
|
37
|
|
|
Research credit carryforwards
|
|
|
200
|
|
|
|
408
|
|
|
Other
|
|
|
61
|
|
|
|
51
|
|
|
|
|
|
|
|
|
|
|
|
|
Total deferred tax assets
|
|
|
1,932
|
|
|
|
3,706
|
|
|
Deferred tax liabilities:
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
Total deferred taxes
|
|
|
1,932
|
|
|
|
3,706
|
|
|
Valuation allowance
|
|
|
(1,932
|
)
|
|
|
(3,706
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Net deferred tax asset
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(In thousands)
|
A reconciliation of the statutory tax rates and the effective
tax rates for the years ended December 31, 2007, 2008 and
2009 is as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from
|
|
|
|
|
|
|
|
|
Period from
|
|
|
|
|
May 1, 2007
|
|
|
|
|
|
|
|
|
May 1, 2007
|
|
|
|
|
(Date of Inception)
|
|
|
|
|
|
|
|
|
(Date of Inception)
|
|
|
|
|
to December 31,
|
|
|
Year Ended December 31,
|
|
|
to December 31,
|
|
|
|
|
2007
|
|
|
2008
|
|
|
2009
|
|
|
2009
|
|
|
|
|
Statutory rate
|
|
|
34.0
|
%
|
|
|
34.0
|
%
|
|
|
34.0
|
%
|
|
|
34.0
|
%
|
|
Permanent differences
|
|
|
(7.2
|
)
|
|
|
(3.3
|
)
|
|
|
(1.4
|
)
|
|
|
(2.7
|
)
|
|
State and local income taxes
|
|
|
7.7
|
|
|
|
8.9
|
|
|
|
9.4
|
|
|
|
9.0
|
|
|
Credits and other
|
|
|
1.8
|
|
|
|
5.0
|
|
|
|
5.5
|
|
|
|
5.0
|
|
|
Valuation allowance
|
|
|
(36.3
|
)
|
|
|
(44.6
|
)
|
|
|
(47.5
|
)
|
|
|
(45.3
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
0.0
|
%
|
|
|
0.0
|
%
|
|
|
0.0
|
%
|
|
|
0.0
|
%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Realization of the future tax benefits is dependent on our
ability to generate sufficient taxable income within the
carryforward period. Because of our history of operating losses,
management believes that the deferred tax assets arising from
the above-mentioned future tax benefits are currently not likely
to be realized and, accordingly, we have provided a full
valuation allowance. The net valuation allowance increased by
$1.7 million and $1.8 million for the years ended
December 31, 2008 and 2009, respectively, and
$3.7 million for the period from May 1, 2007 (Date of
Inception) to December 31, 2009.
Net operating losses and tax credit carryforwards as of
December 31, 2009, are as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
Amount
|
|
Expiration Years
|
|
|
|
Net operating losses—federal
|
|
$
|
6,481
|
|
|
Beginning 2027
|
|
Tax credits—federal
|
|
$
|
396
|
|
|
Beginning 2027
|
|
|
|
(In thousands)
|
F-18
Kips Bay Medical,
Inc.
(A Development Stage Company)
Notes To Financial Statements—(Continued)
Utilization of the net operating loss carryforwards and credits
may be subject to a substantial annual limitation due to the
ownership change limitations provided by the Internal Revenue
Code of 1986, as amended, or the IRC, and similar state
provisions. We have not performed a detailed analysis to
determine whether an ownership change under Section 382 of
the IRC has occurred. The effect of an ownership change would be
the imposition of an annual limitation on the use of net
operating loss carryforwards attributable to periods before the
change.
We would classify interest and penalties related to uncertain
tax positions in income tax expense, if applicable. There was no
interest expense or penalties related to unrecognized tax
benefits recorded through December 31, 2009. The tax years
2007 through 2009 remain open to examination by federal and
state tax authorities.
As discussed in note 6 above, in October 2009, we commenced
a private offering of 714,286 shares of our common stock to
certain accredited investors at an offering price of $7.00 per
share. Subsequent to December 31, 2009, we sold an
additional 182,872 shares of common stock, raising gross
proceeds of $1.3 million. This offering was completed in
February 2010.
Also as discussed in note 6 above, in February 2010, KBI
exercised its second stock purchase option under the Investment
Agreement. At that time we also agreed to convert the interest
on the Notes previously paid in cash to common stock. In
conjunction with this transaction we issued
1,000,000 shares of our common stock in exchange for gross
proceeds of $3.8 million.
F-19
Shares
Kips
Bay Medical, Inc.
Common
Stock
Prospectus
Jefferies &
Company
,
2010
Part II
Information Not
Required In Prospectus
|
|
|
Item 13.
|
Other Expenses
of Issuance and Distribution.
|
The following table sets forth the costs and expenses, other
than the underwriting discounts and commissions, payable by us
in connection with the sale of common stock being registered.
All amounts shown are estimates, except the SEC registration
fee, the Financial Industry Regulatory Authority, or FINRA,
filing fee and the NASDAQ Global Market listing fee.
| |
|
|
|
|
|
|
|
|
|
|
Amount
|
|
|
|
|
SEC registration fee
|
|
$
|
4,100
|
|
|
FINRA filing fee
|
|
|
6,250
|
|
|
NASDAQ Global Market listing fee
|
|
|
*
|
|
|
Blue sky fees and expenses
|
|
|
*
|
|
|
Legal fees and expenses
|
|
|
*
|
|
|
Accounting fees and expenses
|
|
|
*
|
|
|
Printing expenses
|
|
|
*
|
|
|
Transfer agent and registrar fees and expenses
|
|
|
*
|
|
|
Miscellaneous
|
|
|
*
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
*
|
|
|
|
|
|
|
|
|
|
|
|
|
|
* |
|
To be filed by amendment. |
|
|
|
Item 14.
|
Indemnification
of Directors and Officers.
|
Section 145 of the Delaware General Corporation Law
provides that a corporation may indemnify any person made a
party to an action by reason of the fact that he or she was a
director, executive officer, employee or agent of the
corporation or is or was serving at the request of the
corporation against expenses (including attorneys’ fees),
judgments, fines and amounts paid in settlement actually and
reasonably incurred by him or her in connection with such action
if he or she acted in good faith and in a manner he or she
reasonably believed to be in, or not opposed to, the best
interests of the corporation and, with respect to any criminal
action or proceeding, had no reasonable cause to believe his or
her conduct was unlawful, except that, in the case of an action
by or in right of the corporation, no indemnification may
generally be made in respect of any claim as to which such
person is adjudged to be liable to the corporation.
Our certificate of incorporation and amended and restated bylaws
limit the liability of our directors to the fullest extent
permitted by Delaware law. Delaware law provides that directors
of a corporation will not be personally liable for monetary
damages for breach of their fiduciary duties as directors,
except for liability for any:
|
|
|
| |
•
|
breach of their duty of loyalty to us or our stockholders;
|
| |
| |
•
|
act or omission not in good faith or that involves intentional
misconduct or a knowing violation of law;
|
| |
| |
•
|
unlawful payment of dividends or redemption of shares as
provided in Section 174 of the Delaware General Corporation Law;
or
|
| |
| |
•
|
transaction from which the directors derived an improper
personal benefit.
|
These limitations of liability do not apply to liabilities
arising under federal securities laws and do not affect the
availability of equitable remedies such as injunctive relief or
rescission. Our amended and restated bylaws, which will become
effective upon the closing of this offering, provide that we
will indemnify our directors and executive officers, and may
indemnify other officers, employees and other agents, to the
fullest extent permitted by law.
As permitted by the Delaware General Corporation Law, we have
entered into indemnification agreements with each of our
directors and executive officers that require us to indemnify
such persons against expenses, judgments,
II-1
penalties, fines, settlements and other amounts actually and
reasonably incurred, including expenses of a derivative action,
in connection with an actual or threatened proceeding if any of
them may be made a party because he or she is or was one of our
directors. We will be obligated to pay these amounts only if the
director acted in good faith and in a manner that he or she
reasonably believed to be in or not opposed to our best
interests. With respect to any criminal proceeding, we will be
obligated to pay these amounts only if the director had no
reasonable cause to believe his or her conduct was unlawful. The
indemnification agreements also set forth certain procedures
that will apply in the event of a claim for indemnification.
The underwriting agreement (Exhibit 1.1 hereto) provides
for indemnification by the underwriters of us and our executive
officers and directors, and by us of the underwriters for
certain liabilities, including liabilities arising under the
Securities Act, in connection with matters specifically provided
in writing by the underwriters for inclusion in the registration
statement.
Section 145(g) of the Delaware General Corporation Law
permits a corporation to purchase and maintain insurance on
behalf of any person who is or was a director, officer,
employee, or agent of the corporation arising out of his or her
actions in connection with their services to us, regardless of
whether our amended and restated bylaws permit indemnification.
We have purchased and intend to maintain insurance on behalf of
any person who is or was a director or officer against any loss
arising from any claim asserted against him or her and incurred
by him or her in any such capacity, subject to certain
exclusions.
|
|
|
Item 15.
|
Recent Sales
of Unregistered Securities.
|
Since our inception in May 2007, we have issued and sold the
following unregistered securities:
1. In July 2007, we issued an aggregate of
5,700,000 shares of our common stock to our officers and
director at a per share price of $0.0167 per share for aggregate
consideration of $95,000.
2. We have granted stock options to purchase an aggregate
of 803,000 shares of our common stock at exercise prices
ranging from $1.00 to $7.00 per share through a total of 43
grants to executive officers, employees, director and
consultants pursuant to our 2007 Long-Term Incentive Plan.
3. We have issued and sold an aggregate of
1,000 shares of our common stock to an employee, at a price
of $1.00 per share pursuant to exercises of options granted
under our 2007 Long Term Incentive Plan.
4. In April 2008, KBI exercised its option to purchase
shares in connection with the Investment Agreement and related
convertible promissory notes entered into with KBI in July 2007.
Between May 2008 and June 2009, we issued an aggregate
of 600,000 shares of our common stock to KBI for aggregate
consideration of $3.5 million in satisfaction of this
exercise.
5. In March 2009, we issued an aggregate of
5,147,389 shares of our common stock to KBI for aggregate
consideration of $3.2 million in connection with the
Investment Agreement and related convertible promissory notes
entered into with KBI in July 2007.
6. In February 2010, we issued 1,000,000 shares of our
common stock to KBI for aggregate consideration of
$3.8 million.
7. During March through August 2009, we issued and sold in
a series of closings, an aggregate of 516,241 shares of our
common stock at a per share price of $6.00, for aggregate
consideration of $3.1 million in a private placement
offering.
8. During October 2009 through February 2010, we issued and
sold an aggregate of 317,161 shares of our common stock in
at a per share price of $7.00, for aggregate consideration of
$2.2 million in a private placement offering.
The offers, sales and issuances of the securities described in
1-3 above were deemed to be exempt from registration under the
Securities Act under either (i) Rule 701 promulgated
under the Securities Act as offers and sale of securities
pursuant to certain compensatory benefit plans and contracts
relating to compensation in compliance with Rule 701 or
(ii) Section 4(2) of the Securities Act as
transactions by an issuer not involving any public offering. The
recipients of securities in each of these transactions
represented their intention to acquire the
II-2
securities for investment only and not with view to or for sale
in connection with any distribution thereof and appropriate
legends were affixed to the share certificates and instruments
issued in such transactions. All recipients had adequate access,
through their relationships with us, to information about us.
The offers, sales, and issuances of the securities described in
4-8 above were deemed to be exempt from registration under the
Securities Act in reliance on Section 4(2) of the
Securities Act and Regulation D promulgated thereunder as
transactions by an issuer not involving a public offering. The
recipients of securities in each of these transactions acquired
the securities for investment only and not with a view to or for
sale in connection with any distribution thereof and appropriate
legends were affixed to the securities issued in these
transactions. Each of the recipients of securities in these
transactions was an accredited or sophisticated person and had
adequate access, through employment, business or other
relationships, to information about us.
|
|
|
Item 16.
|
Exhibits and
Financial Statement Schedules.
|
(a) Exhibits.
Exhibit Index
| |
|
|
|
|
Exhibit
|
|
|
|
Number
|
|
Description of Exhibit
|
|
|
|
|
1
|
.1*
|
|
Form of Underwriting Agreement.
|
|
|
3
|
.1
|
|
Certificate of Incorporation of the Registrant.
|
|
|
3
|
.2
|
|
Amended and Restated Bylaws of the Registrant.
|
|
|
4
|
.1*
|
|
Form of Common Stock Certificate.
|
|
|
5
|
.1*
|
|
Opinion of Fredrikson & Byron, P.A., counsel to the
Registrant, with respect to the legality of securities being
registered.
|
|
|
|
|
|
Lease Agreements
|
|
|
10
|
.1
|
|
Lease Agreement by and between the Registrant and St. Paul
Properties, Inc., as assigned to St. Paul Fire and Marine
Insurance Company, dated as of July 26, 2007.
|
|
|
|
|
|
Loan and Financing Agreements
|
|
|
10
|
.2
|
|
Investment Agreement by and between the Registrant and Kips Bay
Investments, LLC, dated as of July 19, 2007.
|
|
|
10
|
.3
|
|
Loan and Security Agreement by and between the Registrant and
Kips Bay Investments, LLC, dated as of June 19, 2007.
|
|
|
10
|
.4
|
|
First Secured Convertible Promissory Note executed by the
Registrant in favor of Kips Bay Investments, LLC, dated as of
June 19, 2007.
|
|
|
10
|
.5
|
|
Second Secured Convertible Promissory Note executed by the
Registrant in favor of Kips Bay Investments, LLC, dated as of
June 19, 2007.
|
|
|
10
|
.6
|
|
Agreement by and between the Registrant and Kips Bay
Investments, LLC, dated as of February 12, 2010.
|
|
|
10
|
.7
|
|
Debt Conversion Agreement by and between the Registrant and Kips
Bay Investments, LLC, dated as of February 12, 2010.
|
|
|
|
|
|
Agreements with Respect to Collaborations, Licenses, Research
and Development
|
|
|
10
|
.8+
|
|
Assignment and License Agreement by and between the Registrant
and Medtronic, Inc., dated as of October 9, 2007.
|
|
|
10
|
.9
|
|
Assignment by Medtronic, Inc. to the Registrant, dated as of
August 26, 2008.
|
|
|
10
|
.10
|
|
Trademark Transfer Agreement by Medtronic, Inc. to the
Registrant, dated as of October 10, 2007.
|
|
|
|
|
|
Agreements with Executive Officers
|
|
|
10
|
.11@
|
|
Employment Agreement by and between the Registrant and Manuel A.
Villafaña, dated as of July 19, 2007.
|
|
|
10
|
.12@
|
|
Employment Agreement by and between the Registrant and Michael
Winegar, dated as of September 1, 2007.
|
|
|
10
|
.13@
|
|
Employment Agreement by and between the Registrant and Scott
Kellen, dated as of February 8, 2010.
|
II-3
| |
|
|
|
|
Exhibit
|
|
|
|
Number
|
|
Description of Exhibit
|
|
|
|
|
10
|
.14@*
|
|
Form of Indemnification Agreement between the Registrant and its
Directors and Executive Officers.
|
|
|
10
|
.15@
|
|
Change in Control Agreement by and between the Registrant and
Manuel A. Villafaña, dated as of September 12, 2008.
|
|
|
10
|
.16@
|
|
Change in Control Agreement by and between the Registrant and
Michael Winegar, dated as of September 12, 2008.
|
|
|
10
|
.17@
|
|
Change in Control Agreement by and between the Registrant and
Scott Kellen, dated as of February 8, 2010.
|
|
|
|
|
|
Equity Compensation Plans
|
|
|
10
|
.18@
|
|
2007 Long-Term Incentive Plan.
|
|
|
10
|
.19@
|
|
Form of Incentive Stock Option Agreement under the 2007
Long-Term Incentive Plan.
|
|
|
10
|
.20@
|
|
Form of Non-Qualified Stock Option Agreement under the 2007
Long-Term Incentive Plan.
|
|
|
10
|
.21@
|
|
Form of Restricted Stock Agreement under the 2007 Long-Term
Incentive Plan.
|
|
|
21
|
|
|
Subsidiaries of the Registrant - None.
|
|
|
23
|
.1
|
|
Consent of Ernst & Young LLP.
|
|
|
23
|
.2*
|
|
Consent of Fredrikson & Byron, P.A. (see Exhibit 5.1).
|
|
|
24
|
.1
|
|
Powers of Attorney (see signature page to initial filing).
|
|
|
99
|
.1
|
|
Consent of Michael Winegar
|
|
|
99
|
.2
|
|
Consent of Arch C. Smith
|
|
|
99
|
.3
|
|
Consent of Robert E. Munzenrider
|
|
|
99
|
.4
|
|
Consent of Robert J. Sheehy
|
|
|
|
|
* |
|
To be filed by amendment. |
| |
|
+ |
|
Confidential treatment will be requested for portions of this
exhibit. |
| |
|
@ |
|
Denotes management compensation plan or contract. |
(b) Financial Statement Schedules.
All schedules are omitted as the required information is
inapplicable or the information is presented in the financial
statements or related notes.
Schedule II. Valuation and Qualifying Accounts
All other schedules are omitted as the required information is
inapplicable or the information is presented in the financial
statements or related notes.
The undersigned registrant hereby undertakes to provide to the
underwriters at the closing specified in the underwriting
agreement, certificates in such denominations and registered in
such names as required by the underwriters to permit prompt
delivery to each purchaser.
Insofar as indemnification for liabilities arising under the
Securities Act may be permitted to directors, officers and
controlling persons of the registrant pursuant to the foregoing
provisions, or otherwise, the registrant has been advised that
in the opinion of the Securities and Exchange Commission such
indemnification is against public policy as expressed in the
Securities Act and is unenforceable. In the event that a claim
for indemnification against such liabilities (other than the
payment by the registrant of expenses incurred or paid by a
director, officer or controlling person of the registrant in the
successful defense of any action, suit or proceeding) is
asserted by such director, officer or controlling person in
connection with the securities being registered, the registrant
will, unless in the opinion of its counsel the matter has been
settled by controlling precedent, submit to a court of
appropriate jurisdiction the question whether such
indemnification by it is against public policy as expressed in
the Securities Act and will be governed by the final
adjudication of such issue.
II-4
The undersigned registrant hereby undertakes that:
(1) For purposes of determining any liability under the
Securities Act, the information omitted from the form of
prospectus filed as part of this registration statement in
reliance upon Rule 430A and contained in a form of
prospectus filed by the registrant pursuant to
Rule 424(b)(1) or (4) or 497(h) under the Securities
Act shall be deemed to be part of this registration statement as
of the time it was declared effective.
(2) For the purpose of determining any liability under the
Securities Act, each post-effective amendment that contains a
form of prospectus shall be deemed to be a new registration
statement relating to the securities offered, and the offering
of these securities at that time shall be deemed to be the
initial bona fide offering.
II-5
Signatures
Pursuant to the requirements of the Securities Act of 1933, as
amended, the registrant has duly caused this registration
statement on
Form S-1
to be signed on its behalf by the undersigned, thereunto duly
authorized, in the City of Minneapolis, State of Minnesota on
this 8th day of April, 2010.
Kips Bay Medical, Inc.
Manny Villafaña
Chairman of the Board and Chief
Executive Officer
Power Of
Attorney
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose
signature appears below hereby constitutes and appoints Manny
Villafaña and Scott Kellen, and each of them acting
individually, as his true and lawful attorneys-in-fact and
agents, with full power of each to act alone, with full powers
of substitution and resubstitution, for him and in his name,
place and stead, in any and all capacities, to sign the
Registration Statement filed herewith and any and all amendments
to said Registration Statement (including post-effective
amendments and any related registration statements thereto filed
pursuant to Rule 462 and otherwise), and file the same,
with all exhibits thereto, and other documents in connection
therewith, with the Securities and Exchange Commission, granting
unto said attorneys-in-fact and agents, with full power of each
to act alone, full power and authority to do and perform each
and every act and thing requisite and necessary to be done in
connection therewith, as fully for all intents and purposes as
he might or could do in person, hereby ratifying and confirming
all that said attorneys-in-fact and agents, or his or their
substitutes, may lawfully do or cause to be done by virtue
hereof.
Pursuant to the requirements of the Securities Act of 1933, this
registration statement has been signed by the following persons
in the capacities and on the dates indicated.
| |
|
|
|
|
|
|
|
Signature
|
|
Title
|
|
Date
|
|
|
|
|
|
|
|
|
|
/s/ Manny
Villafaña
Manny
Villafaña
|
|
Chairman of the Board, Chief Executive Officer, and Director
(principal executive officer)
|
|
April 8, 2010
|
|
|
|
|
|
|
|
/s/ Scott
Kellen
Scott
Kellen
|
|
Chief Financial Officer and
Vice President of Finance
(principal financial officer and principal accounting officer)
|
|
April 8, 2010
|
II-6
Kips Bay Medical,
Inc.
Registration Statement On
Form S-1
Exhibit Index
| |
|
|
|
|
Exhibit
|
|
|
|
Number
|
|
Description of Exhibit
|
|
|
|
|
1
|
.1*
|
|
Form of Underwriting Agreement.
|
|
|
3
|
.1
|
|
Certificate of Incorporation of the Registrant.
|
|
|
3
|
.2
|
|
Amended and Restated Bylaws of the Registrant.
|
|
|
4
|
.1*
|
|
Form of Common Stock Certificate.
|
|
|
5
|
.1*
|
|
Opinion of Fredrikson & Byron, P.A., counsel to the
Registrant, with respect to the legality of securities being
registered.
|
|
|
|
|
|
Lease Agreements
|
|
|
10
|
.1
|
|
Lease Agreement by and between the Registrant and St. Paul
Properties, Inc., as assigned to St. Paul Fire and Marine
Insurance Company, dated as of July 26, 2007.
|
|
|
|
|
|
Loan and Financing Agreements
|
|
|
10
|
.2
|
|
Investment Agreement by and between the Registrant and Kips Bay
Investments, LLC, dated as of July 19, 2007.
|
|
|
10
|
.3
|
|
Loan and Security Agreement by and between the Registrant and
Kips Bay Investments, LLC, dated as of June 19, 2007.
|
|
|
10
|
.4
|
|
First Secured Convertible Promissory Note executed by the
Registrant in favor of Kips Bay Investments, LLC, dated as of
June 19, 2007.
|
|
|
10
|
.5
|
|
Second Secured Convertible Promissory Note executed by the
Registrant in favor of Kips Bay Investments, LLC, dated as of
June 19, 2007.
|
|
|
10
|
.6
|
|
Agreement by and between the Registrant and Kips Bay
Investments, LLC, dated as of February 12, 2010.
|
|
|
10
|
.7
|
|
Debt Conversion Agreement by and between the Registrant and Kips
Bay Investments, LLC, dated as of February 12, 2010.
|
|
|
|
|
|
Agreements with Respect to Collaborations, Licenses, Research
and Development
|
|
|
10
|
.8+
|
|
Assignment and License Agreement by and between the Registrant
and Medtronic, Inc., dated as of October 9, 2007.
|
|
|
10
|
.9
|
|
Assignment by Medtronic, Inc. to the Registrant, dated as of
August 26, 2008.
|
|
|
10
|
.10
|
|
Trademark Transfer Agreement by Medtronic, Inc. to the
Registrant, dated as of October 10, 2007.
|
|
|
|
|
|
Agreements with Executive Officers
|
|
|
10
|
.11@
|
|
Employment Agreement by and between the Registrant and Manuel A.
Villafaña, dated as of July 19, 2007.
|
|
|
10
|
.12@
|
|
Employment Agreement by and between the Registrant and Michael
Winegar, dated as of September 1, 2007.
|
|
|
10
|
.13@
|
|
Employment Agreement by and between the Registrant and Scott
Kellen, dated as of February 8, 2010.
|
|
|
10
|
.14@*
|
|
Form of Indemnification Agreement between the Registrant and its
Directors and Executive Officers.
|
|
|
10
|
.15@
|
|
Change in Control Agreement by and between the Registrant and
Manuel A. Villafaña, dated as of September 12, 2008.
|
|
|
10
|
.16@
|
|
Change in Control Agreement by and between the Registrant and
Michael Winegar, dated as of September 12, 2008.
|
|
|
10
|
.17@
|
|
Change in Control Agreement by and between the Registrant and
Scott Kellen, dated as of February 8, 2010.
|
|
|
|
|
|
Equity Compensation Plans
|
|
|
10
|
.18@
|
|
2007 Long-Term Incentive Plan.
|
|
|
10
|
.19@
|
|
Form of Incentive Stock Option Agreement under the 2007
Long-Term Incentive Plan.
|
|
|
10
|
.20@
|
|
Form of Non-Qualified Stock Option Agreement under the 2007
Long-Term Incentive Plan.
|
| |
|
|
|
|
Exhibit
|
|
|
|
Number
|
|
Description of Exhibit
|
|
|
|
|
10
|
.21@
|
|
Form of Restricted Stock Agreement under the 2007 Long-Term
Incentive Plan.
|
|
|
21
|
|
|
Subsidiaries of the Registrant - None.
|
|
|
23
|
.1
|
|
Consent of Ernst & Young LLP.
|
|
|
23
|
.2*
|
|
Consent of Fredrikson & Byron, P.A. (see Exhibit 5.1).
|
|
|
24
|
.1
|
|
Powers of Attorney (see signature page to initial filing).
|
|
|
99
|
.1
|
|
Consent of Michael Winegar
|
|
|
99
|
.2
|
|
Consent of Arch C. Smith
|
|
|
99
|
.3
|
|
Consent of Robert E. Munzenrider
|
|
|
99
|
.4
|
|
Consent of Robert J. Sheehy
|
|
|
|
|
* |
|
To be filed by amendment. |
| |
|
+ |
|
Confidential treatment will be requested for portions of this
exhibit. |
| |
|
@ |
|
Denotes management compensation plan or contract. |