UNITED STATES SECURITIES AND
EXCHANGE COMMISSION
Washington, D.C.
20549
Form 10-K
| |
|
|
|
(Mark One)
|
|
|
|
þ
|
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE
SECURITIES EXCHANGE ACT OF 1934
|
|
|
|
For the fiscal year ended
December 31, 2009
|
|
-OR-
|
|
o
|
|
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE
SECURITIES EXCHANGE ACT OF 1934
|
|
|
|
For the transition period
from to
|
Commission File No. 001-33958
RXi PHARMACEUTICALS
CORPORATION
(Exact name of registrant as
specified in its charter)
| |
|
|
|
Delaware
|
|
20-8099512
|
(State or other jurisdiction
of
incorporation or organization)
|
|
(I.R.S. Employer
Identification Number)
|
|
60 Prescott Street
Worcester, Massachusetts
(Address of principal
executive offices)
|
|
01605
(Zip
Code)
|
Registrant’s telephone number, including area code:
(508) 767-3861
SECURITIES
REGISTERED PURSUANT TO SECTION 12(b) OF THE ACT:
| |
|
|
|
Title of Each Class
|
|
Name of Exchange on Which Registered
|
|
Common Stock, $0.0001 Par Value Per Share
|
|
NASDAQ Capital Market
|
SECURITIES
REGISTERED PURSUANT TO SECTION 12(g) OF THE ACT:
None
Indicate by check mark if the registrant is a well-known
seasoned issuer, as defined in Rule 405 of the Securities
Act. Yes o No þ
Indicate by check mark if the registrant is not required to file
reports pursuant to Section 13 or Section 15(d) of the
Act. Yes o No þ
Indicate by check mark whether the registrant (1) has filed
all reports required to be filed by Section 13 or 15(d) of
the Securities Exchange Act of 1934 during the preceding
12 months (or for such shorter period that the registrant
was required to file such reports), and (2) has been
subject to such filing requirements for the past
90 days. Yes þ No o
Indicate by check mark if disclosure of delinquent filers
pursuant to Item 405 of
Regulation S-K
is not contained herein, and will not be contained, to the best
of registrant’s knowledge, in definitive proxy or
information statements incorporated by reference in
Part III of this
Form 10-K
or any amendment to this
Form 10-K. Yes o No þ
Indicate by check mark whether the registrant has submitted
electronically and posted on it corporate Web site, if any,
every Interactive Data File required to be submitted and posted
pursuant to Rule 405 of
Regulation S-T
(§ 232.405 of this chapter) during the preceding
12 months (or for such shorter time that the registrant was
required to submit and post such
files). Yes o No o
Indicate by check mark whether the registrant is a large
accelerated filer, an accelerated filer, a non-accelerated
filer, or a smaller reporting company. See the definitions of
“large accelerated filer,” “accelerated
filer” and “smaller reporting company” in
Rule 12b-2
of the Exchange Act. (Check one):
| |
|
|
|
|
|
|
|
Large
accelerated filer o
|
|
Accelerated filer o
|
|
Non-accelerated filer o
(Do not check if a smaller reporting company)
|
|
Smaller reporting company þ
|
Indicate by check mark whether the registrant is a shell company
(as defined in
Rule 12b-2
of the
Act). Yes o No þ
The aggregate market value of the voting Common Stock held by
non-affiliates of the registrant, based on the last sale price
of the registrant’s Common Stock at the close of business
on June 30, 2009, was $34,131,897.
As of March 29, 2010, the registrant had
18,281,125 shares of Common Stock, $0.0001 par value
per share, outstanding.
DOCUMENTS INCORPORATED BY REFERENCE:
Portions of the registrant’s definitive proxy statement for
its 2010 annual meeting of stockholders, to be filed pursuant to
Regulation 14A with the Securities and Exchange Commission
not later than 120 days after the registrant’s fiscal
year end of December 31, 2009, are incorporated by
reference into this
Form 10-K.
RXi
PHARMACEUTICALS CORPORATION
FORM 10-K —
FISCAL YEAR ENDED DECEMBER 31, 2009
Table of
Contents
All references in this
Form 10-K
to “RXi,” the “Company,” “we,”
“us,” or “our” mean RXi Pharmaceuticals
Corporation, unless we state otherwise or the context otherwise
requires.
i
PART I
Overview
We were incorporated as Argonaut Pharmaceuticals, Inc. in
Delaware on April 3, 2006, changed our name to RXi
Pharmaceuticals Corporation on November 28, 2006, and began
operations in January 2007. We are a discovery-stage
biopharmaceutical company pursuing proprietary therapeutics
based on RNA interference, or “RNAi”, a naturally
occurring cellular mechanism that has the potential to
effectively and selectively interfere with, or
“silence”, expression of targeted disease-associated
genes. It is widely accepted by the scientific community that
this specific silencing can be used to potentially treat human
diseases by “turning off” genes that lead to disease.
While no therapeutic RNAi products have been approved for
marketing to date, there has been significant growth in the
field of RNAi development and potential therapeutic applications
in this field. This growth is driven by the potential ability to
use RNAi to rapidly develop lead compounds that specifically and
selectively inhibit a target gene, many of which are undruggable
by other modalities.
By utilizing our expertise in RNAi and the comprehensive RNAi
therapeutic platform that we have established, we believe we
will be able to discover and develop lead compounds and progress
them into and through clinical development for potential
commercialization more efficiently than traditional drug
development approaches.
Our proprietary therapeutic platform is comprised of two main
components:
|
|
|
| |
•
|
Novel RNAi Compounds, referred to as
rxRNAtm
compounds, that are distinct from, and we believe convey
significant advantages over classic siRNA
(conventionally-designed “small interfering RNA”
compounds), and offer many of the properties that we believe are
important to the clinical development of RNAi-based drugs. We
have developed a number of unique forms of rxRNA compounds, all
of which have been shown in our testing to be highly potent both
in vitro and in vivo. These RNAi
compounds include
rxRNAoritm,
rxRNAsolotm
and
sd-rxRNAtm,
or “self delivering” RNA. Based on our research, we
believe that these different, novel siRNA configurations have
various advantages for therapeutic use. These advantages include
high potency, increased resistance to nucleases and off-target
effects, and in the case of the sd-rxRNA compounds,
access to cells and tissues with no additional formulation
required.
|
| |
| |
•
|
Advanced Delivery Technologies that may enable the
delivery of our rxRNA compounds to treat a variety of acute and
chronic diseases using both local and systemic approaches,
potentially providing a competitive advantage in the development
of many RNAi therapeutic compounds. Our suite of delivery
technologies is comprised of delivery vehicles, which can be
combined with various rxRNA compounds, as well as
sd-rxRNA compounds, which are chemically modified and
have the unique property of entering cells and tissues to effect
silencing without the need for any additional delivery vehicle.
This suite of delivery technologies has broad potential
applications for multiple therapeutic areas targeting both local
and systemic applications.
|
|
|
|
| |
•
|
Local Applications. An area of application of
the RXi therapeutic platform which uses rxRNA compounds to
target genes expressed in tissues that can be silenced by
direct, local delivery. The numerous diseases common to tissues
accessible by local delivery represent significant unmet medical
needs and large market opportunities. Most of our initial
targets are validated gene targets relevant in important
biological pathways and are implicated in multiple diseases
enabling us to leverage these targets and associated compounds
across a broad array of therapeutic areas.
|
| |
| |
•
|
Systemic Applications. We have active internal
efforts to advance our therapeutic platform to optimize robust
systemic delivery to various tissues and organs of the body. In
some cases, such as in targeting a treatment to the liver, the
optimal route of administration is by systemic delivery. Efforts
to improve the systemic delivery of RNAi compounds are currently
ongoing, and these efforts are supported by internal activities
targeting a gene thought to be responsible for elevated
cholesterol. We have also in-licensed intellectual property
developed by Dr. Michael Czech (one of our scientific
|
1
|
|
|
| |
|
co-founders and scientific advisory board members) on genes that
appear to be important regulators of metabolism, and continue to
develop and validate this approach with these other potential
target genes.
|
We intend to use our RNAi therapeutic platform and our expertise
in RNAi to identify lead compounds against multiple target
genes, and advance them towards pre-clinical and clinical
development in therapeutic areas that address broad unmet
medical needs, in both acute and chronic settings. There are
many well-studied genes that have been associated with numerous
diseases but have been difficult to target with conventional
medicinal chemistry or traditional modalities involving both
large and small molecules. We believe RNAi technology may play
an important role in targeting these genes and potentially
treating the related diseases and disorders. We plan to pursue
select disease areas with the goal of creating multiple clinical
development program opportunities, either independently or with
partners through various collaborations and partnerships with
academic institutions or pharmaceutical or biotechnology
companies.
We believe that we have created and established a strong
intellectual property portfolio. We have secured exclusive and
nonexclusive licenses from both academic institutions and
commercial entities to certain issued and pending patents and
patent applications covering RNAi technologies in the following
three categories: (i) therapeutic targets,
(ii) chemistry and configurations of RNAi compounds and
(iii) formulation and delivery of RNAi compounds. We have
also filed patents based on our internal discoveries in the each
of the areas mentioned above, which should enable us to further
strengthen our intellectual property portfolio.
Our founding scientists recognized that the key to therapeutic
success with RNAi lies in delivering intact RNAi compounds to
the target tissue and the interior of the target cells. To
accomplish this, we are developing a comprehensive platform that
includes local, systemic and oral delivery approaches that give
rise to target silencing after RNAi compound administration. We
work with chemically synthesized RNAi compounds that we believe
are optimized for stability and efficacy. We endow these
compounds with favorable delivery profiles and properties either
by covalent chemical modification or combination with
appropriate formulations to achieve optimal delivery to specific
target tissues.
We have an accomplished business team which includes Noah D.
Beerman, MBA, President & CEO, Konstantinos
Andrikopoulos, Ph.D., J.D., VP, Legal
Counsel & Chief IP Counsel, Ramani Varanasi, MS, MBA,
VP Business Development, Dmitry Samarsky, Ph.D., VP
Technology Development, Amy B. Tata, CPA, Chief Accounting
Officer, Joanne Kamens, Ph.D., Senior Director of Research
Collaborations, and Donna Falcetti, MBA, Director of Operations.
We have an accomplished scientific team which includes Anastasia
Khvorova, Ph.D., Chief Scientific Officer, Pamela
Pavco, Ph.D., VP Pharmaceutical Development, Lyn
Libertine, M.D., Director of Pharmacology, and Kevin
Fettes, Ph.D., Director of Chemistry.
We have an accomplished Scientific Advisory Board
(“SAB”) which includes Craig C. Mello, Ph.D., the
Chairman of the SAB, Gregory Hannon, Ph.D., Michael
Czech, Ph.D., Victor Ambros, Ph.D., Nassim
Usman, Ph.D. and Tod Woolf, Ph.D. (our former Chief
Executive Officer), together known as the SAB members. SAB
members participate in scientific planning meetings which are
typically held every three to six months. During such meetings,
our management team and SAB members review the progress of our
research and licensing efforts and provide technological input,
including suggestions for new experiments, suggestions regarding
the therapeutic relevance of target genes and suggestions
regarding new technologies we may want to consider licensing
and/or
developing internally. Further, certain of our SAB members
periodically assist us in business-related activities, such as
discussions with potential strategic partners and introductions
to potential key consultants and collaborators.
We were formed in 2006 by CytRx Corporation (Nasdaq: CYTR)
and four prominent RNAi researchers, including Dr. Craig
Mello, who was awarded the 2006 Nobel Prize in Medicine for his
co-discovery of RNAi. From 2003 through 2006, CytRx sponsored
therapeutic RNAi research at the University of Massachusetts
Medical School (“UMMS”) and Massachusetts General
Hospital. We commenced operations in January 2007 after CytRx
contributed to us its portfolio of RNAi therapeutic assets in
exchange for approximately 7.04 million shares of our
common stock. These assets consisted primarily of RNAi licenses
and related intellectual property and a nominal amount of
equipment.
2
To date, our principal activities have consisted of conducting
discovery research and pre-clinical development activities
utilizing our RNAi therapeutic platform, acquiring RNAi
technologies and patent rights through exclusive, co-exclusive
and non-exclusive licenses, recruiting an RNAi-focused
management and scientific/clinical advisory team, capital
raising activities and conducting business development
activities aimed at establishing research and development
partnerships with pharmaceutical and biotechnology companies.
We have not generated revenue to date and may not generate
product revenue in the foreseeable future, if ever. We expect to
incur significant operating losses as we advance our product
candidates through the drug development and regulatory process.
In addition to increasing research and development expenses, we
expect general and administrative costs to increase as we add
personnel. We will need to generate significant revenues to
achieve profitability and might never do so. In the absence of
product revenues, our potential sources of operational funding
are expected to be the proceeds from the sale of equity, funded
research and development payments and payments received under
partnership and collaborative agreements.
We had cash and cash equivalents of approximately
$5.7 million as of December 31, 2009 and approximately
$14.4 million as of March 31, 2010.
During 2009 and to date in 2010 we have entered into the
following significant financing transactions:
On January 30, 2009, we entered into the Standby Equity
Distribution Agreement (“SEDA”) with YA Global Master
SPV Ltd. (“YA Global”), pursuant to which we may, at
our option over a two-year period, ending on January 30,
2011, periodically sell to YA Global shares of our common stock,
for a total purchase price of up to $25.0 million. To date
we have not sold any shares under the SEDA.
On August 4, 2009, we closed registered direct financing in
which we sold to certain investors 2,385,715 shares of our
common stock at $3.50 per share and warrants to purchase
954,286 shares of our common stock and at an exercise price
of $4.50 per share (the “2009 Offering”) resulting in
approximately $7.8 million in net proceeds after deducting
the placement agent fee and offering expenses.
On March 26, 2010 we closed a registered direct financing
pursuant to which we sold to certain investors
2,700,000 shares of common stock at $6.00 per share and
warrants to purchase 540,000 shares of common stock with an
exercise price of $6.00 per share (the “2010
Offering”). The financing provided approximately
$15.2 million in net proceeds to the Company after
deducting the placement agent fee and offering expenses.
Pursuant to a stock redemption agreement between us and CytRx
Corporation dated March 22, 2010, we were required to use
25% of the net proceeds from the offering to repurchase from
CytRx a number of shares of our common stock held by CytRx equal
to 25% of the shares sold by us in the offering. We are also
required to use 25% of the proceeds from the exercise of
warrants issued in the offering to repurchase from CytRx a
number of shares of our common stock held by CytRx equal to 25%
of the shares issued upon the exercise of such warrants. As
required by the agreement with CytRx, on March 29, 2010 we
repurchased 675,000 shares of our common stock from CytRx
for an aggregate price of approximately $3.8 million. We
estimate that we will repurchase an additional
135,000 shares of our common stock from CytRx for an
aggregate price of $0.8 million if all of the warrants
issued in the Offering are exercised.
We have not generated any revenues since inception nor are any
revenues expected for the foreseeable future. We expect to incur
significant operating losses for the foreseeable future while we
advance our future product candidates from discovery through
pre-clinical studies and clinical trials and seek regulatory
approval and potential commercialization, even if we are
collaborating with other pharmaceutical and biotechnology
companies. In addition to these increasing research and
development expenses, we expect general and administrative costs
to increase as we recruit additional management and
administrative personnel.
We believe that our existing cash and cash equivalents should be
sufficient to fund our operations through at least the first
half of 2011. In addition, we also have available to us the SEDA
which expires on January 30, 2011. In the future, we will
be dependent on obtaining funding from third parties such as
proceeds from the sale of equity, funded research and
development payments and payments under partnership and
collaborative agreements, in order to maintain our operations
and meet our obligations to licensors. There is no guarantee
that debt, additional equity or other funding will be available
to us on acceptable terms, or at all. If we fail to
3
obtain additional funding when needed, we would be forced to
scale back or terminate our operations or to seek to merge with
or to be acquired by another company.
Our
Competitive Strengths
We believe we are well positioned to compete successfully in the
RNAi-based therapeutics market due to the following competitive
strengths:
|
|
|
| |
•
|
Novel, proprietary technology platform with the potential to
generate multiple RNAi therapeutic product opportunities,
comprised of:
|
|
|
|
| |
•
|
Our rxRNA compound platform that includes multiple distinct
approaches all of which offer novelty and potential high
potency; and
|
| |
| |
•
|
Multiple delivery technologies, including self delivering RNAi
approaches, which do not require a delivery vehicle and can be
administered for various local and systemic applications.
|
|
|
|
| |
•
|
Accomplished scientific and business team with significant
experience in RNAi therapeutics and in building and managing
emerging life sciences companies.
|
| |
| |
•
|
Scientific advisors who are recognized leaders in RNAi research,
including Dr. Craig Mello, recipient of the 2006 Nobel
Prize in Medicine for co-discovering RNAi, and Dr. Victor
Ambros, who was awarded the 2008 Albert Lasker Award for Basic
Medical Research for his work leading to the groundbreaking
discovery of the first microRNA (miRNA).
|
| |
| |
•
|
Strong early intellectual property position covering
|
• Novel approaches to RNAi chemistry and configuration,
|
|
|
| |
•
|
Proprietary, formulation and delivery of active RNAi compounds,
and
|
• Key therapeutic target genes.
|
|
|
| |
•
|
A focus on unmet medical needs and significant market
opportunity.
|
Introduction
to the Field of RNAi Therapeutics
RNAi is a naturally-occurring phenomenon where short
double-stranded RNA molecules interfere with the expression of
targeted genes. RNAi technology takes advantage of this
phenomenon and potentially allows us to effectively interfere
with particular genes within living cells by designing
RNA-derived molecules targeting those genes. RNAi is regarded as
a significant advancement in the scientific community, as
evidenced by the journal Science’s selection of RNAi
as the “Breakthrough of the Year” in 2002, and by the
awarding of the 2006 Nobel Prize in Medicine to the
co-discoverers of RNAi, including Dr. Craig Mello, an RXi
founder and our SAB Chairman.
RNAi offers a novel approach to the drug development process
because, as described below under “The RNAi
Mechanism,” RNAi compounds can potentially be designed to
target any one of the thousands of human genes, many of which
are undruggable by other modalities. In contrast, an article
published in the December 2005 edition of Drug Discovery
Today by Andreas P. Russ and Stefan Lampel has demonstrated
that only a subset of the proteins encoded in the human genetic
code (human genome) are able to be targeted efficiently by
traditional medicinal chemistry or antibody-based approaches.
The specificity of RNA interference is achieved by an intrinsic
well-understood biological mechanism based on designing the
sequence of an RNAi compound to match the sequence of the
targeted gene. The specificity of RNAi may be sufficient to
permit therapeutic targeting of only a single gene and,
importantly, may even selectively reduce or eliminate expression
from a single abnormal copy of a gene while preserving
expression from a normal copy (“allele-specific”
targeting). This is critical in diseases such as cancer and
neurodegenerative disorders that are often caused by abnormal
copies of genes.
4
The RNAi
Mechanism
The genome is made of a double-strand of DNA (the double helix)
that acts as an instruction manual for the production of the
roughly 30,000 to 50,000 human proteins. Proteins are important
molecules that allow cells and organisms to live and function.
With rare exceptions, each cell in the human body has the entire
complement of genes. However, only a subset of these genes
directs the production of proteins in any particular cell type.
For example, a muscle cell produces muscle-specific protein,
whereas a skin cell does not.
In order for a gene to guide the production of a protein, it
must first be copied into a single-stranded chemical messenger
(messenger RNA or mRNA), which is then translated into protein.
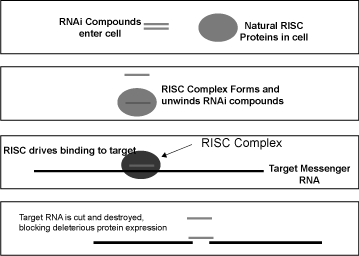
RNA interference is a naturally occurring process by which a
particular messenger RNA can be destroyed before it is
translated into protein. The process of RNAi can be artificially
induced by introducing a small double-stranded fragment of RNA
corresponding to a particular messenger RNA into a cell. A
protein complex within the cell called RISC (RNA-Induced
Silencing Complex) recognizes this double-stranded RNA fragment
and splits the double-strands apart, retaining one strand in the
RISC complex. The RISC then helps this guide strand of RNA bind
to and destroy its corresponding cellular messenger RNA target.
Thus, RNAi provides a method to potentially block the creation
of the proteins that cause disease, as depicted in the following
figure.
Figure
1 — Mechanism of RNA interference within a
cell
Since gene expression controls most cellular processes, the
ability to inhibit gene expression provides a potentially
powerful tool to treat human diseases. Furthermore, since the
human genome has already been decoded, and based on numerous
gene-silencing reports, we believe that RNAi compounds can
readily be designed to interfere with the expression of any
specific gene. Based on our internal research and our review of
certain scientific literature, we also believe that our RNAi
platform may allow us to develop create therapeutics with
significant potential advantages over traditional drug
development methods, including:
|
|
|
| |
•
|
High specificity for targeted genes,
|
| |
| |
•
|
High potency (low doses),
|
| |
| |
•
|
Ability to interfere with the expression of potentially any gene,
|
| |
| |
•
|
Accelerated generation of lead compounds, and
|
| |
| |
•
|
Low toxicity, natural mechanism of action.
|
5
RXi’s
RNAi Therapeutic Platform
RNAi
Compound Design
RNAi compounds are made from a strand or strands of RNA that are
manufactured by a nucleic acid synthesizer. The synthesizer is
programmed to assemble a strand of RNA of a particular sequence
using the four kinds of nucleotide units (Adenine
(“A”), Uracil (“U”), Cytidine
(“C”) and Guanosine (“G”)) that match a
small segment of the targeted gene. The hallmark of an RNAi
compound is that it has a double stranded region. The compounds
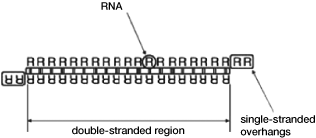
can be of various lengths of nucleotide units (nt). As seen in
Figure 2 below, the two strands can have overhangs (as shown on
the far left), or they can have blunt ends (as shown in the
middle and right). A single strand can form an RNAi compound by
forming a structure referred to as a hairpin.
Figure
2 — Types of RNAi Compounds
The length and shape of the compound can affect the activity and
hence the potency of the RNAi in cells. The first design of RNAi
compounds to be pursued for development as a human therapeutic
was a short double-stranded RNA that included at least one
overhanging single-stranded region, known as small interfering
RNA, or siRNA which we also refer to as classic siRNA and can be
seen in Figure 2 above.
Figure
3 — First generation of RNAi pursued for human
therapeutics: classic siRNA
In the case of classic siRNA, double-stranded RNA with
single-stranded overhangs is used. The two strands comprising
the RNA have bases that are complementary to each other in order
to create double-stranded regions; that is, an “A” on
one strand is paired with a “U” on the other, and a
“C” on one strand is paired with a “G” on
the other, creating double-stranded regions. The pairing holds
the two strands together creating double-stranded RNA. The
overhangs that are at the ends of the double-stranded RNA do not
have a matching partner and thus these single-stranded bases in
the overhang area are exposed to nucleases in the environment
which can degrade the molecule. Classic siRNA therapeutics are
about 19 to 23 base pairs long.
We believe that classic siRNAs have drawbacks that may limit the
usefulness of those agents as human therapeutics, and that we
may be able to utilize the technologies we have licensed and
developed internally to optimize RNAi compounds for use as human
therapeutic agents. It is the combination of the length, the
nucleotide sequence, and the configuration of chemical
modifications that are important for effective RNAi
therapeutics. For example, the RNA can be chemically modified in
a manner that reduces its sensitivity to nucleases, which are
enzymes that attack and degrade RNA. Likewise, it is our
expectation that removing the single-stranded overhang regions
will be a way of reducing the rate of spurious degradation of
the RNAi, as single-stranded RNA is more susceptible to
degradation than double-stranded RNA. The length range of 19 to
23 nucleotides can also be varied to yield more potent RNAi
compounds. Introducing “mismatches” in the
6
double-stranded region, that is, discrete internal portions of
the duplex region that do not form good base pairs between the
two strands, also may be a useful way of improving the potency
of the resulting RNA.
Depending on the delivery method selected, stabilizing RNAi
compounds by chemical modification may be critical for RNAi
activity in animal models and in humans. The stabilization may
be necessary to protect the RNAi compounds from being degraded
by enzymes that exist in bodily fluids. Many of our employees
and SAB Members are accomplished in the field of chemically
modified RNAi design. For example, Dr. Craig Mello, an
expert in the field, is an inventor of the Fire-Mello seminal
family of patents (“Fire-Mello”), which we have a
license to, Dr. Tod Woolf, our former President and CEO,
was a co-inventor of the
Stealthtm
RNAi brand of chemically modified RNAi compounds and
Dr. Anastasia Khvorova, our Chief Scientific Officer, was a
co-inventor of OnTarget
Plustm,
siStable®
and
Accelltm
brands of chemically modified RNAi compounds. We will continue
to employ their collective expertise to design chemically
modified RNAi compounds. We have in-licensed technology on
chemically stabilized RNAi compounds that will serve as a
foundation for our chemical modification strategy.
Our internal research leads us to believe that next generation
rxRNA compounds offer significant advantages over classic siRNA
used by other companies developing RNAi therapeutics,
highlighted by the following characteristics:
|
|
|
| |
•
|
Up to 100 times more active than classic siRNA,
|
| |
| |
•
|
More resistant to nuclease degradation,
|
| |
| |
•
|
Readily manufactured,
|
| |
| |
•
|
Potentially more specific for the target gene,
|
| |
| |
•
|
More reliable at blocking immune side effects than classic
siRNA, and
|
| |
| |
•
|
In the case of
sd-rxRNAtm,
the unique ability to be ‘self delivering’, without
the need for any additional delivery vehicle.
|
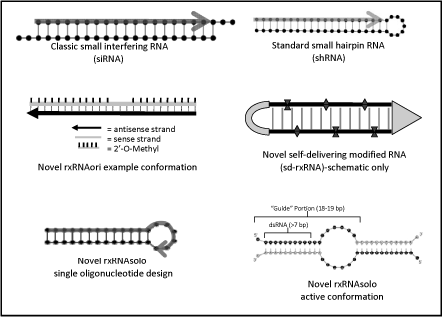
Based on our own research we have developed a variety of novel
siRNA configurations with potential advantages for therapeutic
use. The first of these has been termed rxRNAori. This
configuration has some similarities to classic siRNA in that it
is composed of two, short RNA strands. We have found that by
using a somewhat longer length (25 bp), removing the
overhangs and using proprietary chemical modification patterns,
we achieve a higher hit rate of very potent (picomolar potency)
compounds in a given target sequence. These rxRNAori
compounds are modified to increase resistance to nucleases
and to prevent off-target effects including induction of an
immune response. These novel RNAi compounds are distinct from
the siRNA compounds used by many other companies developing RNAi
therapeutics in that they are designed specifically for
therapeutic use and offer many of the properties that we believe
are important to the clinical development of RNAi-based drugs.
The second novel configuration has been termed rxRNAsolo
to indicate the fact that it is composed of a single RNA
oligonucleotide strand. This configuration also makes use of
carefully placed and selected chemical modifications to
introduce properties for therapeutic use as described above.
Conventional shRNA (short hairpin RNA) and siRNA compounds with
duplex regions of
19-27
nucleotides are efficient substrates for RNAi machinery in
mammalian cells. Efficacy of single-stranded oligonucleotides is
substantially lower (by 3-4 orders of magnitude). We have
developed a single-oligo (27 nt long) construct with silencing
potency equal to conventional RNAi triggers. These molecules are
designed to have at least 16 bases complementary to the target
mRNA and are then extended at the 3' end to allow efficient
self annealing (dimerization). This new class of RNAi compounds
has the potential to further reduce off-target effects and
manufacturing costs and may thus offer additional advantages for
use in research and therapeutic applications.
A third novel configuration has been called
“sd-rxRNA” to indicate its novel
“self-delivering” properties which do not require
additional delivery vehicles for efficient cellular uptake and
RISC-mediated silencing. A combination of at least three
characteristics is required for activity: 1) specific,
proprietary chemical modifications, 2) a precise number of
chemical modifications and 3) reduction in oligonucleotide
content.
7
Kineteic analyses of fluorescently-labeled compounds demonstrate
that efficient cellular internalization is observed within
minutes of exposure. These molecules are taken up efficiently
and cause target gene silencing in diverse cell types (cell
lines and primary cells). This novel class of RNAi compounds may
afford a broad opportunity for therapeutic development.
Figure
4 — Classic and Novel RNAi Compound
Configurations
8
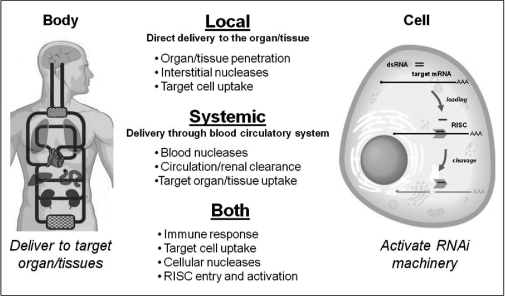
Delivery
Figure
5 — Principles and Challenges of RNAi in vivo
Delivery
We believe both chemical modification and formulation of RNAi
compounds may be utilized to develop RNA drugs suitable for
therapeutic use. A series of delivery hurdles must be overcome
to achieve in vivo efficacy: (1) delivery to the target
tissue or organ (2) tissue penetration and distribution
(3) crossing of the plasma membrane (4) intracellular
trafficking to the RISC (RNAi machinery complex)
(5) incorporation into and activation of the RISC.
Different cell types and tissues may each require unique
approaches. Three categories of tools currently exist: variation
in administration route, selection of delivery vehicle, and
chemical modification of the RNA compound. A combination of some
or all of these is likely to be required for successful delivery.
The route by which an RNAi therapeutic is brought into contact
with the body depends on the intended organ or tissue to be
treated. Delivery routes can be simplified into two major
categories: local (when a drug is delivered directly to the
tissue of interest) or systemic (when a drug accesses the tissue
of interest through the circulatory system). Local delivery may
avoid some hurdles associated with systemic approaches such as
circulation clearance and tissue extravasation (crossing the
endothelial barrier from the blood stream). However, this
approach can only be applied to a limited number of organs or
tissues (e.g. skin, eye, lung, and potentially, the central
nervous system).
RNAi delivery vehicles, a large and diverse group of particles
(e.g. polymer-based particles, lipoplexes, other), can
contribute in additional ways to successful delivery.
Formulation can help prevent nuclease degradation, improve
nucleotide retention in circulation and alter tissue and cell.
In some cases, a formulation can result in more efficient
cellular uptake and intracellular release.
RNA chemical modifications of the sd-rxRNA compounds, as
described above, can include base, backbone and sugar
modifications, as well as covalently bound moieties such as
cholesterol, antibody fragments or peptides. Some of these
modifications can be utilized to enhance stability, reduce
immunogenicity, improve circulation properties (presumably
through binding to blood transporter proteins), increase tissue
access and improve uptake to cells and the RNAi machinery. A
combination of chemical modifications, delivery vehicles or both
might be dictated by the target organs/tissues and specific
requirements for the therapeutic application, including the
intended administration route.
Our founding scientists recognized that the key to therapeutic
success with RNAi lies in delivering intact RNAi compounds to
the target tissue and the interior of the target cells. To
accomplish this, we have
9
developed a comprehensive platform that includes local, systemic
and oral delivery approaches. We work with chemically
synthesized RNAi compounds that are optimized for stability and
efficacy, and combine delivery at the site of action and
formulation with delivery agents to achieve optimal delivery to
specific target tissues.
Local
Delivery
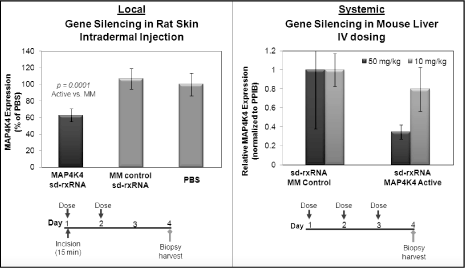
sd-rxRNA molecules have unique properties which improve
tissue and cell uptake. Delivery of sd-rxRNA by a local
route of administration may avoid hurdles associated with
systemic approaches such as rapid clearance from the bloodstream
and inefficient extravasation (e.g. crossing the endothelial
barrier from the blood stream). We have studied sd-rxRNA
molecules in a rat model of dermal delivery. Direct application
of sd-rxRNA with no additional delivery vehicle to the
skin (incision introduced) demonstrates that target gene
silencing can be measured after topical delivery. Figure 6
illustrates that direct injection of sd-rxRNA into the
dermis layers of the skin with no additional delivery vehicle
resulted in efficient uptake and significant target gene
silencing. The dose levels required for these direct injection
methods are small and suitable for clinical development
suggesting that local delivery indications will be very
accessible with the sd-rxRNA technology platform. Target
tissues that are potentially accessible for local delivery using
sd-rxRNA compounds include lung, eye, skin, CNS, mucosal
tissues, sites of inflammation, and tumors (direct
administration).
Systemic
Delivery
Systemic delivery occurs when a drug accesses the tissue of
interest through the circulatory system. In some cases, such as
in targeting a treatment to the liver, the optimal route of
delivery may be by a systemic route. We have developed a
portfolio of systemic delivery solutions utilizing our RNAi
therapeutic platforms. One novel approach involves the use of
sd-rxRNA compounds. The self-delivering technology
introduces properties required for in vivo efficacy such
as cell and tissue penetration and improved blood clearance and
distribution properties. Systemic delivery of these compounds to
mice has resulted in gene specific inhibition with no additional
delivery vehicle required as shown in Figure 6 below . In
addition, we have developed novel nanotransporter formulations
to aid in transport of RNAi compounds to both liver and various
other target tissues in the body. These nanotransporters are
chemically synthesized molecules that form nanometer-sized
particles when mixed with RNAi compounds and alter the
clearance, distribution and tissue penetration properties of the
RNAi compounds. Delivery of RNAi compounds to the liver might be
critical for the treatment of many diseases and using rxRNA in
conjunction with such delivery vehicles has enabled us to
demonstrate gene specific inhibition at low doses in a mouse
model after intravenous, systemic delivery. Target tissues that
are potentially accessible using rxRNA compounds by systemic
delivery include liver, lung, adipocytes, cardiomyocytes, bone
marrow, sites of inflammation, tumors, vascular endothelium, and
kidney.
10
Figure 6:
Data demonstrating in vivo gene silencing with sd-rxRNA
in local and systemic settings
Oral
Delivery
Most RNAi therapeutic products being developed today require
recurring intravenous injections or other forms of
administration which are not patient friendly. To address the
desire for RNAi therapeutics with improved modes of
administration, we are testing a novel formulation technology,
Glucan Encapsulated RNAi Particles (GeRPs), that may allow our
rxRNA compounds to be incorporated into orally administered
pills. Early data to date suggest that the GeRP delivery system
appears to be more potent than previous methods used for
systemic delivery of RNAi therapeutics by intravenous injection.
Additional studies will need to be conducted to clearly
establish the flexibility of the GeRP system and to determine
whether they can either be used to administer a single RNAi
compound, multiple RNAi compounds, or could potentially allow
co-delivery of RNAi, DNA, protein and small molecule
combinations.
Therapeutic
Programs and Markets
By utilizing our expertise in RNAi compound design and delivery,
we intend to identify lead compounds for both tractable and
intractable targets implicated in diseases that address broad
unmet medical needs in both acute and chronic settings. The
broad applicability of our RNAi therapeutic platform has the
potential to enable delivery to various tissues in both a local
and systemic setting. Target tissues that are potentially
accessible using our rxRNA compounds in the context of a local
delivery approach include lung, eye, skin, CNS, mucosal tissues,
sites of inflammation, and tumors (locally). Similarly, target
tissues that are potentially accessible using our rxRNA
compounds in the context of a systemic delivery approach include
liver, lung, adipocytes, cardiomyocytes, bone marrow, sites of
inflammation, tumors, vascular endothelium, and kidney. We will
continue to focus our efforts selecting targets to pursue
internally, and as we identify relevant compounds, we intend to
begin preclinical development in specific areas as appropriate.
Advancing
RXi’s Therapeutic Pipeline
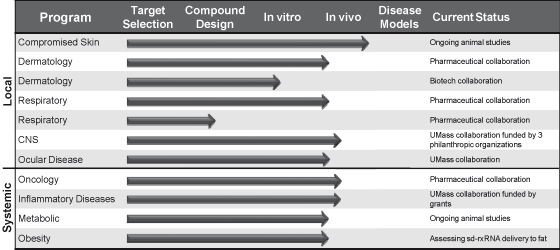
Table 1 below outlines our current therapeutic pipeline. Our
local and systemic programs include a combination of internal
development and collaborative efforts. As we focus our internal
efforts on a manageable number of diseases areas, we intend to
partner with other pharmaceutical and biotechnology companies in
certain disease areas.
11
Table
1: Advancing RXi’s Therapeutic
Pipeline:
Alliance
Partners in Therapeutic Areas
We are actively seeking to leverage our technology platform by
seeking to work with pharmaceutical and biotechnology partners
in the partners’ fields of interest. Our team has
experience targeting genes in virtually every major therapeutic
area, and based on this experience, we believe we can discover
more drug candidates by working with partners than we can
develop with our own resources. We are seeking to work with
partners in the discovery and development of rxRNA based drugs
in a number of therapeutic areas.
Business
Strategy
We intend to use our RNAi technology platform and expertise in
RNAi to discover, develop and potentially commercialize RNA
targeting therapeutics. The key elements of our business
strategy are as follows:
|
|
|
| |
•
|
We are focused on the discovery, development and
commercialization of a pipeline of RNA based therapeutics to
address various diseases using our proprietary rxRNA compounds
and delivery technologies. We have promising preliminary data
with compounds in skin models and certain other disease areas
and continue to pursue various research and development efforts
to further validate and develop both the local and systemic
delivery approaches using multiple in vivo model systems both
internally as well as with academic, biotechnology and
pharmaceutical partners.
|
| |
| |
•
|
We intend to fund the initial development of a limited number of
RNAi drug candidates with our own capital resources. We intend
to develop drugs in these areas internally to establish
significant value, at which point we may seek to partner them.
|
| |
| |
•
|
We are seeking partnerships with pharmaceutical and
biotechnology companies to leverage our intellectual property
and expand our development pipeline. Such partnerships may
include traditionally structured drug development and
commercialization licenses, discovery and development
collaborations, research and technology collaborations all of
which could be focused on specific therapeutic areas, diseases
and/or
targets of interest to the partner, and intellectual property
licenses.
|
| |
| |
•
|
We intend to continue to develop and enhance our RNAi technology
platform by expanding our intellectual property position in RNAi
compound chemistry, delivery and target sequences through
in-licensing
and/or
acquiring novel technologies through scientific collaborations
as well as through internal innovation.
|
| |
| |
•
|
We intend to develop future RNAi technology improvements through
a combination of internal efforts as well as external technology
focused collaborations and partnerships and believe we are well
|
12
|
|
|
| |
|
positioned to do so. Our management and advisors have developed
much of the core technology in the field of oligonucleotide
therapeutics, and RNAi therapeutics more specifically.
|
|
|
|
| |
•
|
As we make progress on compounds in our internal pipeline, we
may consider seeking product oriented partnerships, to help
advance our pre-clinical efforts and to leverage the expertise
of pharmaceutical partners, who may have the development,
manufacturing and commercial capabilities that will be important
for further product advancement.
|
| |
| |
•
|
We may also seek to collaborate with government and charitable
institutions through grants and funded research for our
development programs such as our collaboration with
Dr. Czech and his colleagues at UMMS, which is funded in
part by a grant from the Mass Life Sciences Center.
|
Intellectual
Property and Proprietary Rights
We actively seek protection for our proprietary information by
means of United States and foreign patents, trademarks, and
copyrights, as appropriate. In addition, we rely upon trade
secret protection and contractual arrangements to protect
certain of our proprietary information and products. We have
pending patent applications that relate to potential drug
targets, compounds we are developing to modulate those targets
(described throughout herein as rxRNA), methods of making or
using those compounds and proprietary elements of our drug
discovery platform.
Much of our technology and many of our processes depend upon the
knowledge, experience and skills of key scientific and technical
personnel. To protect our rights to our proprietary know-how and
technology, we require all employees, as well as our consultants
and advisors when feasible, to enter into confidentiality
agreements that require disclosure and assignment to us of
ideas, developments, discoveries and inventions made by these
employees, consultants and advisors in the course of their
service to us.
We have also obtained rights to various patents and patent
applications under licenses with third parties, which provide
for the payment of milestones and royalties by us. The ultimate
degree of patent protection that will be afforded to
biotechnology products and processes, including ours, in the
United States and in other important markets remains uncertain
and is dependent upon the scope of protection decided upon by
the patent offices, courts and lawmakers in these countries.
There is no certainty that our existing patents or others, if
obtained, will afford us substantial protection or commercial
benefit. Similarly, there is no assurance that our pending
patent applications or patent applications licensed from third
parties will ultimately be granted as patents or that those
patents that have been issued or are issued in the future will
stand if they are challenged in court. We assess our license
agreements on an ongoing basis, and may from time to time
terminate licenses to technology that we do not intend to employ
in our RNAi technology platform, or in our product discovery or
development activities.
rxRNA
Platform
We have 12 pending patent applications, 10 of which were filed
as PCTs to date, encompassing what we believe to be important
new compounds and their use as therapeutics in RNAi, chemical
modifications of these and existing RNAi compounds that improve
the compounds’ suitability for therapeutic uses (including
delivery), and compounds directed to specific targets (that
address specific disease states). Any patents that may issue
from these pending patent applications will be set to expire
between 2028 and 2031, not including any patent term extensions
that may be afforded to the patents that encompass products (or
processes for making or using the same) that are human drug
products subject to regulation under the Federal Food, Drug and
Cosmetic Act (and the equivalent provisions in foreign
jurisdictions) for any delays incurred during the regulatory
approval process of the drug product.
In-Licensed
Technologies and License Agreements
We have also secured exclusive and non-exclusive rights to
develop RNAi therapeutics by licensing key RNAi technologies and
patent rights from third parties. These rights relate to
chemistry and configurations of RNAi compounds, delivery
technologies of RNAi compounds to cells, and therapeutic targets.
13
Chemistry
and Configurations of RNAi
We have a non-exclusive license to the Fire-Mello patent (US
6,506,559, set to expire in 2018) and related applications
covering the use of double stranded RNA to induce gene silencing
that describes RNAi products, compositions and therapeutic RNAi
methods.
In addition, we have secured exclusive and co-exclusive rights
to technologies, patents and pending patent applications
directed to producing and delivering in vivo stable and
potent RNAi therapeutics including those from TriLink
Biotechnologies, Inc., Invitrogen IP Holdings, Inc. (now known
as Life Technologies, Inc), and UMMS. See “License
Agreements” below.
Delivery
of RNAi Compounds to Cells
We have exclusive and non-exclusive licenses to technologies for
the efficient delivery of RNAi therapeutics to cells in cell
culture
and/or in
the intact organism. These technologies include but are not
limited to:
|
|
|
| |
•
|
methods and compositions for the oral delivery of RNAi compounds
that enable efficient therapeutic gene silencing in cells and
animals, which is licensed for all therapeutic areas utilizing
GeRPs; and
|
| |
| |
•
|
methods and compositions using nanotransporters, for RNAi
compound delivery which enable therapeutic gene silencing in
cells and animals.
|
Therapeutic
Targets
In addition to our internally-developed proprietary compounds
against a broad range of targets that can be utilized in
multiple therapeutic areas, we have exclusive, co-exclusive and
non exclusive licenses from UMMS covering therapeutic targets in
the area of inflammatory diseases, amyotrophic lateral sclerosis
or ALS diabetes and obesity from UMMS against which we may seek
to develop therapeutics for in the future.
License
Agreements
University
of Massachusetts Medical School
Pursuant to the Contribution Agreement dated January 8,
2007, CytRx assigned to us its rights under four exclusive
license agreements, one co-exclusive license agreement and one
non-exclusive license agreement entered into between CytRx and
UMMS which cover potential therapeutic applications for
proprietary RNAi technology in the treatment of specified
diseases. Additionally, CytRx assigned to us its rights under
the Collaboration and Invention Disclosure Agreement entered
into between CytRx and UMMS.
As consideration for the licenses and Collaboration and
Invention Disclosure Agreement assigned to us by CytRx, we
agreed to assume and be responsible for all of the liabilities
and obligations to the extent that such liabilities and
obligations relate to the assigned licenses and agreement,
including all of CytRx’s payment, performance and other
obligations under the assigned licenses and collaboration
agreements.
In connection with the licenses entered into with UMMS that were
assigned to us by CytRx, we have assumed the obligation to pay
to UMMS annual license maintenance fees and certain additional
amounts upon the attainment of certain specified product
development milestones. These licenses will expire upon the
expiration of all patents licensed under the licenses or ten
years after the effective date of such license if no patents
have been issued within that ten year period and are terminable
by either party upon an uncured breach by the other party. We
are generally required to indemnify UMMS for losses incurred by
UMMS based on the exercise of the licensed patents by us.
On January 10, 2007, we entered into certain licenses with
UMMS pursuant to which UMMS granted to us rights under certain
UMMS patent applications to make, use and sell products related
to applications of RNAi technologies in particular fields,
including HCMV and retinitis, ALS, diabetes and obesity.
Under these licenses, UMMS granted to us exclusive, worldwide
licenses, with the right to
sub-license,
to three different patent families and a non-exclusive,
worldwide license to a fourth patent family. As
14
consideration for these licenses, we paid UMMS an up-front fee,
reimbursed UMMS for previously incurred patent expenses, agreed
to undertake to raise working capital by a specified date, to
expend a specified amount on the development of royalty-bearing
products, and agreed to meet a defined timeline relating to the
clinical development of royalty-bearing products. Our obligation
to raise working capital was satisfied when CytRx invested
$17.0 million in us (before a $2.0 million
reimbursement for expenses by us to CytRx) on April 30,
2007. Upon the completion of the $17.0 million financing
from CytRx, we became obligated to pay UMMS additional licenses
fees in an aggregate amount of $175,000, issued to UMMS
approximately 308,075 shares of our common stock at $5.00
per share, for a total value of $1,540,375 and thereafter to pay
UMMS annual maintenance fees, commencing on January 1,
2008, and certain additional amounts upon the attainment of
certain specified product development milestones. We also will
be required to pay to UMMS a percentage of income received from
any sublicensees under these licenses and to pay expenses
incurred by UMMS in prosecuting and maintaining the licensed
patents.
On October 3, 2008, we acquired co-exclusive worldwide
rights to technology for the oral delivery of RNAi therapeutics
from UMMS. This Agreement was amended on July 1, 2009,
allowing us to extend the periods for which certain milestone
Payments are due to UMMS.
These licenses will expire upon the expiration of all patents
licensed under the licenses, are terminable by either party upon
an uncured breach by the other party, and may be terminated by
us for any reason following a specified notice period. We are
generally required to indemnify UMMS for losses incurred by UMMS
based on the exercise of the licensed patents by us.
In connection with all of our licenses with UMMS, including
those assigned to us by CytRx as well as those entered into
directly between us and UMMS, we are obligated to pay specified
royalties on net sales of products covered by the licensed
patents, subject to minimum annual royalties.
We recently terminated a number of these UMMS licenses and still
hold licenses to patents and patent applications that belong to
six distinct families of patent applications from the original
thirteen.
We also recently terminated the Collaboration and Invention
Disclosure Agreement which CytRx had assigned to us.
Other
License Agreements and Acquisitions of Intellectual
Property
Consistent with our overall business strategy, we have enhanced
our RNAi technology platform by entering into additional
licenses for various aspects of RNAi technology, including:
|
|
|
| |
•
|
In August 2007, we entered into a license agreement with TriLink
Biotechnologies, Inc. for three RNAi chemistry technologies for
all therapeutic RNAi applications, for which we paid an up-front
fee and agreed to pay yearly maintenance fees, as well as future
clinical milestone payments and royalty payments based on sales
of therapeutic products developed from the licensed technologies.
|
| |
| |
•
|
In October 2007, we entered into a license agreement with
Dharmacon, Inc. (now part of Thermo Fisher Scientific Inc.),
pursuant to which we obtained an exclusive license to certain
RNAi sequences to a number of target genes for the development
of our rxRNA compounds. Further, we have obtained the right to
license additional RNAi sequences, under the same terms,
disclosed by Thermo Fisher Scientific Inc. in its pending patent
applications against target genes and have received an option
for exclusivity for other siRNA configurations. As consideration
for this license, we paid an up-front fee of $150,000 and agreed
to pay future clinical milestone payments and royalty payments
based on sales of siRNA compositions developed in connection
with the licensed technology
|
| |
| |
•
|
In November 2007, we entered into a license agreement with Life
Technologies, Inc., pursuant to which we were granted rights
under four patents relating to RNA target sequences, RNA
chemical modifications, RNA configurations
and/or RNA
delivery to cells. As consideration for this license, we paid an
up-front fee of $250,000 and agreed to pay yearly maintenance
fees of the same amount beginning in 2008. Further, we are
obligated to pay a fee for each additional gene target added to
the license as well as a fee on the first and second
anniversaries of the date we were granted consent to add the
gene target
|
15
|
|
|
| |
|
to the list of those covered by the license. We have also been
granted, for each gene target, an option to secure pre-clinical
rights
and/or the
clinical rights, for which we would be required to pay
additional fees. Further, we are required to make future
clinical milestone payments and royalty payments based on sales
of therapeutic products developed from the licensed technologies.
|
|
|
|
| |
•
|
In September 2009, we obtained an assignment and direct
ownership of technology for which we had previously exercised
our option to exclusively license from Advirna, LLC
(“Advirna”). The acquired technology complements our
internally developed sd-rxRNA technology platform and
further strengthens our IP position in this promising field for
the delivery of RNAi therapeutics.
|
| |
| |
•
|
We recently terminated the License agreement with Imperial
College Innovations Limited and Imperial College of Science and
Technology that was assigned to us by CytRx in connection with
the Contribution Agreement dated January 8, 2007.
|
As we continue to develop our own proprietary compounds, we
continue to evaluate both our in-licensed portfolio as well as
the field for new technologies that could be in-licensed to
further enhance our intellectual property portfolio and unique
position in the RNAi space.
Competition
There are a number of competitors in the RNAi therapeutics
field, and other approaches to gene silencing, such as
antisense. These competitors include large and small
pharmaceutical, chemical and biotechnology companies, as well as
universities, government agencies, and other private and public
research organizations that are focusing their efforts in the
RNAi field.
A number of medical institutions and pharmaceutical companies
are seeking to develop therapeutic products using RNAi
technologies. Companies working in this area include: Alnylam
Pharmaceuticals, MDRNA, Cequent Pharmaceuticals, Tacere
Therapeutics, Benitec, OPKO Health, Silence Therapeutics, Quark
Pharmaceuticals, Rosetta Genomics, Lorus Therapeutics, Tekmira
Pharmaceuticals Corporation, Calando Pharmaceuticals, Regulus
Therapeutics, and Santaris Pharmaceuticals, as well as a number
of large pharmaceutical companies. A number of the large
pharmaceutical companies also either have in-house RNAi
development programs or are collaborating with smaller
biopharmaceutical companies. This competition will manifest
itself in the discovery and development of RNAi technology, in
recruiting and retaining key scientific and management
personnel, in securing strategic alliances, and in obtaining
rights to key intellectual property.
Our RNAi-focused competitors as well as major pharmaceutical
companies may be targeting the same diseases we intend to
target. Competitors both in and outside of the RNAi field have
financial resources, research and development staffs, and
facilities that are, in most cases, substantially greater than
ours or potentially those of our strategic partners or licensees
and are engaged in the research and development of
pharmaceutical products that could compete with our potential
products. The industry is characterized by rapid technological
advances and competitors may develop products more rapidly and
such products may be more effective than those currently under
development or that may be developed in the future by our
strategic partners or licensees.
Government
Regulation
The United States and other developed countries extensively
regulate the pre-clinical and clinical testing, manufacturing,
labeling, storage, record-keeping, advertising, promotion,
export, marketing and distribution of drugs and biologic
products. The United States Food and Drug Administration
(“FDA”) regulates pharmaceutical and biologic products
under the Federal Food, Drug, and Cosmetic Act, the Public
Health Service Act and other federal statutes and regulations.
To obtain approval of our future product candidates from the
FDA, we must, among other requirements, submit data supporting
safety and efficacy for the intended indication as well as
detailed information on the manufacture and composition of the
product candidate. In most cases, this will require extensive
laboratory tests and pre-clinical and clinical trials. The
collection of these data, as well as the preparation of
applications for review by the FDA involve significant time and
expense. The FDA also may require post-marketing testing
16
to monitor the safety and efficacy of approved products or place
conditions on any approvals that could restrict the therapeutic
claims and commercial applications of these products. Regulatory
authorities may withdraw product approvals if we fail to comply
with regulatory standards or if we encounter problems at any
time following initial marketing of our products.
The first stage of the FDA approval process for a new biologic
or drug involves completion of pre-clinical studies and the
submission of the results of these studies to the FDA. This
data, together with proposed clinical protocols, manufacturing
information, analytical data and other information submitted to
the FDA, in an investigational new drug application, or IND,
must become effective before human clinical trials may commence.
Pre-clinical studies generally involve FDA regulated laboratory
evaluation of product characteristics and animal studies to
assess the efficacy and safety of the product candidate.
After the IND becomes effective, a company may commence human
clinical trials. These are typically conducted in three
sequential phases, but the phases may overlap. Phase I trials
consist of testing the product candidate in a small number of
patients or healthy volunteers, primarily for safety at one or
more doses. Phase II trials, in addition to safety,
evaluate the efficacy of the product candidate in a patient
population somewhat larger than Phase I trials. Phase III
trials typically involve additional testing for safety and
clinical efficacy in an expanded population at multiple test
sites. A company must submit to the FDA a clinical protocol,
accompanied by the approval of the Institutional Review Boards
at the institutions participating in the trials, prior to
commencement of each clinical trial.
To obtain FDA marketing authorization, a company must submit to
the FDA the results of the pre-clinical and clinical testing,
together with, among other things, detailed information on the
manufacture and composition of the product candidate, in the
form of a new drug application, or NDA, or, in the case of a
biologic, a biologics license application, or BLA.
The amount of time taken by the FDA for approval of an NDA or
BLA will depend upon a number of factors, including whether the
product candidate has received priority review, the quality of
the submission and studies presented, the potential contribution
that the compound will make in improving the treatment of the
disease in question, and the workload at the FDA.
The FDA may, in some cases, confer upon an investigational
product the status of a fast track product. A fast track product
is defined as a new drug or biologic intended for the treatment
of a serious or life threatening condition that demonstrates the
potential to address unmet medical needs for this condition. The
FDA can base approval of an NDA or BLA for a fast track product
on an effect on a surrogate endpoint, or on another endpoint
that is reasonably likely to predict clinical benefit. If a
preliminary review of clinical data suggests that a fast track
product may be effective, the FDA may initiate review of entire
sections of a marketing application for a fast track product
before the sponsor completes the application.
We anticipate that our products will be manufactured by our
strategic partners, licensees or other third parties. Before
approving an NDA or BLA, the FDA will inspect the facilities at
which the product is manufactured and will not approve the
product unless the manufacturing facilities are in compliance
with the FDA’s current good manufacturing practice
(“cGMP”), which are regulations that govern the
manufacture, holding and distribution of a product.
Manufacturers of biologics also must comply with the FDA’s
general biological product standards. Our manufacturers also
will be subject to regulation under the Occupational Safety and
Health Act, the Environmental Protection Act, the Nuclear Energy
and Radiation Control Act, the Toxic Substance Control Act and
the Resource Conservation and Recovery Act. Following approval,
the FDA periodically inspects drug and biologic manufacturing
facilities to ensure continued compliance with the good
manufacturing practices regulations. Our manufacturers will have
to continue to comply with those requirements. Failure to comply
with these requirements subjects the manufacturer to possible
legal or regulatory action, such as suspension of manufacturing
or recall or seizure of product. Adverse patient experiences
with the product must be reported to the FDA and could result in
the imposition of marketing restrictions through labeling
changes or market removal. Product approvals may be withdrawn if
compliance with regulatory requirements is not maintained or if
problems concerning safety or efficacy of the product occur
following approval.
17
The labeling, advertising, promotion, marketing and distribution
of a drug or biologic product also must be in compliance with
FDA and Federal Trade Commission requirements which include,
among others, standards and regulations for off-label promotion,
industry sponsored scientific and educational activities,
promotional activities involving the internet, and
direct-to-consumer
advertising. We also will be subject to a variety of federal,
state and local regulations relating to the use, handling,
storage and disposal of hazardous materials, including chemicals
and radioactive and biological materials. In addition, we will
be subject to various laws and regulations governing laboratory
practices and the experimental use of animals. In each of these
areas, as above, the FDA has broad regulatory and enforcement
powers, including the ability to levy fines and civil penalties,
suspend or delay issuance of product approvals, seize or recall
products, and deny or withdraw approvals.
We will also be subject to a variety of regulations governing
clinical trials and sales of our products outside the United
States. Whether or not FDA approval has been obtained, approval
of a product candidate by the comparable regulatory authorities
of foreign countries and regions must be obtained prior to the
commencement of marketing the product in those countries. The
approval process varies from one regulatory authority to another
and the time may be longer or shorter than that required for FDA
approval. In the European Union, Canada and Australia,
regulatory requirements and approval processes are similar, in
principle, to those in the United States.
Environmental
Compliance
Our research and development activities involve the controlled
use of potentially harmful biological materials as well as
hazardous materials, chemicals and various radioactive
compounds. We are subject to federal, state and local laws and
regulations governing the use, storage, handling and disposal of
these materials and specific waste products. We are also subject
to numerous environmental, health and workplace safety laws and
regulations, including those governing laboratory procedures,
exposure to blood-borne pathogens and the handling of
bio-hazardous materials. The cost of compliance with these laws
and regulations could be significant and may adversely affect
capital expenditures to the extent we are required to procure
expensive capital equipment to meet regulatory requirements.
Human
Resources
As of March 29, 2010, we had 28 employees, 27 of whom
were full-time employees and one of whom was a part-time
employee. 19 of our employees are engaged in research and
development and 9 of our employees are engaged in management,
administration and finance. None of our employees are
represented by a labor union or covered by a collective
bargaining agreement, nor have we experienced any work stoppages.
Insurance
The Company currently purchases insurance policies for property
and liability risks arising out of current operations.
Our business is subject to numerous risks. We caution you
that the following important factors, among others, could cause
our actual results to differ materially from those expressed in
forward-looking statements made by us or on our behalf in
filings with the SEC, press releases, communications with
investors and oral statements. Any or all of our forward-looking
statements in this annual report on
Form 10-K
and in any other public statements we make may turn out to be
wrong. They can be affected by inaccurate assumptions or by
known or unknown risks and uncertainties. Many factors mentioned
in the discussion below will be important in determining future
results. Consequently, no forward-looking statement can be
guaranteed. Actual future results may vary materially from those
anticipated in forward-looking statements. When used in this
Form 10-K,
the words “believe,” “anticipate,”
“expect,” “estimate,” “intend,”
“plan,” “project,” “will be,”
“will continue,” “will result,”
“seek,” “could,” “may,”
“might” and similar expressions are intended to
identify forward-looking statements. We explicitly disclaim any
obligation to update any forward-looking statements to
18
reflect events or circumstances that arise after the date
hereof. You are advised, however, to consult any further
disclosure we make in our reports filed with the SEC.
Risks
Relating to RXi’s Business and Industry
The
approach we are taking to discover and develop novel
therapeutics using RNAi is unproven and may never lead to
marketable products.
The scientific discoveries that form the basis for our efforts
to discover and develop new drugs are relatively new. The RNAi
technologies that we have licensed or have created internally
and that we intend to develop have not yet been clinically
tested by us, nor are we aware of any clinical trials for
efficacy having been completed by third parties involving these
technologies. To date, neither we nor any other company has
received regulatory approval to market therapeutics utilizing
RNAi and a number of clinical trials of third parties’ RNAi
technology has been unsuccessful. The scientific evidence to
support the feasibility of developing drugs based on these
discoveries is both preliminary and limited. Successful
development of RNAi-based products by us will require solving a
number of issues, including providing suitable methods of
stabilizing the RNAi material and delivering it into target
cells in the human body. We may spend large amounts of money
trying to solve these issues and never succeed in doing so. In
addition, any compounds that we develop may not demonstrate in
patients the chemical and pharmacological properties ascribed to
them in laboratory studies, and they may interact with human
biological systems in unforeseen, ineffective or even harmful
ways.
Further, our exclusive focus on RNAi technology for developing
products as opposed to multiple, more proven technologies for
drug development increases the risk associated with our
business. If we are not successful in developing a product
candidate using RNAi technology, we may not be able to identify
and successfully implement an alternative product development
strategy.
We
will be subject to competition and may not be able to compete
successfully.
A number of medical institutions and pharmaceutical companies
are seeking to develop therapeutic products using RNAi
technologies, including for at least some of the disease
indications we have been focusing our efforts on to date.
Companies working in the RNAi area include: Alnylam
Pharmaceuticals, MDRNA, Cequent Pharmaceuticals, Tacere
Therapeutics, Benitec, OPKO Health, Silence Therapeutics, Quark
Pharmaceuticals, Rosetta Genomics, Lorus Therapeutics, Tekmira
Pharmaceuticals Corporation, Calando Pharmaceuticals, Regulus
Therapeutics, and Santaris Pharmaceuticals, as well as a number
of the large pharmaceutical companies. In addition, a number of
companies are developing therapeutics for the same diseases we
are targeting using technologies other than RNA interference,
and, for some of these diseases, there are existing therapeutics
being marketed currently. Most of these competitors have
substantially greater research and development capabilities and
financial, scientific, technical, manufacturing, marketing,
distribution, and other resources than us, and we may not be
able to successfully compete with them. In addition, even if we
are successful in developing our product candidates, in order to
compete successfully we may need to be first to market or to
demonstrate that our RNAi based products are superior to
therapies based on different technologies. A number of our
competitors have already commenced clinical testing of RNAi
product candidates and may be more advanced than are we in the
process of developing products. If we are not first to market or
are unable to demonstrate such superiority, any products for
which we are able to obtain approval may not be successful.
We may
not be able to establish or maintain the third party
relationships that are necessary to develop or potentially
commercialize some or all of our product
candidates.
We expect to depend on collaborators, partners, licensees,
clinical research organizations and other third parties to
support our discovery efforts, to formulate product candidates,
and to conduct clinical trials for some or all of our product
candidates. We cannot guarantee that we will be able to
successfully negotiate agreements for or maintain relationships
with collaborators, partners, licensees, clinical investigators
and other third parties on favorable terms, if at all. If we are
unable to obtain or maintain these agreements, we may not be
able to
19
clinically develop, formulate, manufacture, obtain regulatory
approvals for or commercialize our product candidates. Under
certain license agreements that we have already entered into, we
have minimum dollar amounts per year that we are obligated to
spend on the development of the technology we have licensed from
our contract partners. If we fail to meet this requirement under
any of our licenses that contain such requirements or any other
obligations under these licenses, we may be in breach of our
obligations under such agreement, which may result in the loss
of the technology licensed. We cannot necessarily control the
amount or timing of resources that our contract partners will
devote to our research and development programs, product
candidates or potential product candidates, and we cannot
guarantee that these parties will fulfill their obligations to
us under these arrangements in a timely fashion. We may not be
able to readily terminate any such agreements with contract
partners even if such contract partners do not fulfill their
obligations to us.
We are
dependent on technologies we license, and if we lose the right
to license such technologies or we fail to license new
technologies in the future, our ability to develop new products
would be harmed.
We currently are dependent on licenses from third parties for
our key technologies relating to fundamental RNAi technologies.
Our current licenses impose, and any future licenses we enter
into are likely to impose, various development, funding,
royalty, diligence, sublicensing, insurance and other
obligations on us. If our license with respect to any of these
technologies is terminated for any reason, the development of
the products contemplated by the licenses would be delayed, or
suspended altogether, while we seek to license similar
technology or develop new non-infringing technology. The costs
of obtaining new licenses are high, and many patents in the RNAi
field have already been exclusively licensed to third parties,
including our competitors. If any of our existing licenses are
terminated, the development of the products contemplated by the
licenses could be delayed or terminated and we may not be able
to negotiate additional licenses on acceptable terms, if at all,
which would have a material adverse effect on our business.
We
will rely upon third parties for the manufacture of our clinical
product candidates.
We do not have the facilities or expertise to manufacture
supplies of any of our potential product candidates.
Accordingly, we will be dependent upon contract manufacturers
for these supplies. We currently manufacture limited quantities
of our product candidates for our research activities at our
facility, and we have no manufacturing supply arrangements for
any of our product candidates. There can be no assurance that we
will be able to secure needed supply arrangements on attractive
terms, or at all. Our failure to secure these arrangements as
needed could have a materially adverse effect on our ability to
complete the development of our product candidates or, if we
obtain regulatory approval for our product candidates, to
commercialize them.
Our current plans call for the manufacture of our rxRNA
compounds and, as necessary, any delivery vehicles that may be
used to deliver our rxRNA compounds by contract manufacturers
offering research grade, Good Laboratory grade and Good
Manufacturing Practices grade materials for preclinical studies
(e.g. toxicology studies) and for clinical use. We anticipate
the chemistry, manufacturing and controls for each RNAi active
pharmaceutical ingredient will be addressed by our clinical
development team in close collaboration with a contract
manufacturer with extensive experience in RNA drug synthesis.
RNA is a complex molecule requiring many synthesis steps, which
may lead to challenges with purification and
scale-up.
These challenges could result in increased costs and delays in
manufacturing. Additionally, although we are not currently aware
of any such litigation or threatened litigation or challenge, if
we were involved in litigation or faced threatened litigation
regarding or challenge to the composition or intellectual
property covering such composition of our products candidates in
the future, manufacturers may refuse to manufacture such
compounds.
Any
drug candidates we develop may fail in development or be delayed
or may not be commercially viable.
Before obtaining regulatory approval for the sale of any drug
candidate, we must conduct, at our own expense, extensive
pre-clinical tests and clinical trials to demonstrate the safety
and efficacy in humans of our drug candidates. However, we are
required to do extensive testing in animal models with our
product candidates before we can be approved by the FDA to
initiate clinical trials in humans. Furthermore, we cannot
20
be sure that our product candidates will be safely tolerated by
humans or be efficacious. All of our products in development
must be approved by the FDA or similar foreign governmental
agencies before they can be marketed. The process for obtaining
FDA approval is both time-consuming and costly, with no
certainty of a successful outcome. This process typically
includes the conduct of extensive preclinical and clinical
testing, which may take longer or cost more than we anticipate,
and may prove unsuccessful due to numerous factors. A failure of
one or more of our pre-clinical studies or clinical trials can
occur at any stage of testing. Product candidates that may
appear to be promising at early stages of development may not
successfully reach the market for a number of reasons. The
results of pre-clinical and initial clinical testing of these
products may not necessarily indicate the results that will be
obtained from later or more extensive testing. Companies in the
pharmaceutical and biotechnology industries have suffered
significant setbacks in advanced clinical trials, even after
obtaining promising results in earlier trials.
We, the FDA or other applicable regulatory authorities, or an
institutional review board (“IRB”), an independent
committee under the oversight of the United States Department of
Health and Human Services (“HHS”), which has been
formally registered with HHS and functions to approve, monitor
and review biomedical and behavioral research involving humans,
may suspend clinical trials of a drug candidate at any time for
various reasons, including if we or they believe the subjects or
patients participating in such trials are being exposed to
unacceptable health risks. Among other reasons, adverse side
effects of a drug candidate on subjects or patients in a
clinical trial could result in the FDA or other regulatory
authorities suspending or terminating the trial and refusing to
approve a particular drug candidate for any or all indications
of use.
Clinical trials of a new drug candidate require the enrollment
of a sufficient number of patients, including patients who are
suffering from the disease the drug candidate is intended to
treat and who meet other eligibility criteria. Rates of patient
enrollment are affected by many factors, and delays in patient
enrollment can result in increased costs and longer development
times.
Clinical trials also require the review and oversight of IRBs,
which approve and continually review clinical investigations and
protect the rights and welfare of human subjects. An inability
or delay in obtaining IRB approval could prevent or delay the
initiation and completion of clinical trials, and the FDA may
decide not to consider any data or information derived from a
clinical investigation not subject to initial and continuing IRB
review and approval.
Numerous factors could affect the timing, cost or outcome of our
drug development efforts, including the following:
|
|
|
| |
•
|
Delays in filing initial drug applications,
|
| |
| |
•
|
Difficulty in securing centers to conduct trials,
|
| |
| |
•
|
Conditions imposed on us by the FDA or comparable foreign
authorities regarding the scope or design of our clinical trials,
|
| |
| |
•
|
Problems in engaging IRBs to oversee trials or problems in
obtaining or maintaining IRB approval of studies,
|
| |
| |
•
|
Difficulty in enrolling patients in conformity with required
protocols or projected timelines,
|
| |
| |
•
|
Third party contractors failing to comply with regulatory
requirements or meet their contractual obligations to us in a
timely manner,
|
| |
| |
•
|
Our drug candidates having very different chemical and
pharmacological properties in humans than in laboratory testing
and interacting with human biological systems in unforeseen,
ineffective or harmful ways,
|
| |
| |
•
|
The need to suspend or terminate our clinical trials if the
participants are being exposed to unacceptable health risks,
|
| |
| |
•
|
Insufficient or inadequate supply or quality of our drug
candidates or other necessary materials necessary to conduct our
clinical trials,
|
21
|
|
|
| |
•
|
Effects of our drug candidates not being the desired effects or
including undesirable side effects or the drug candidates having
other unexpected characteristics.
|
| |
| |
•
|
The cost of our clinical trials may be greater than we
anticipate,
|
| |
| |
•
|
Negative or inconclusive results from our clinical trials or the
clinical trials of others for drug candidates similar to our own
or inability to generate statistically significant data
confirming the efficacy of the product being tested,
|
| |
| |
•
|
Changes in the FDA’s requirements for our testing during
the course of that testing,
|
| |
| |
•
|
Modification of the drug during testing,
|
| |
| |
•
|
Reallocation of our limited financial and other resources to
other clinical programs, and
|
| |
| |
•
|
Adverse results obtained by other companies developing RNAi
drugs.
|
The substances we are intending to develop may represent a new
class of drug, and the FDA has not yet established any
definitive policies, practices or guidelines in relation to
these drugs. While we expect any product candidates that we
develop will be regulated as a new drug under the Federal Food,
Drug, and Cosmetic Act, the FDA could decide to regulate them or
other products we may develop as biologics under the Public
Health Service Act. The lack of policies, practices or
guidelines may hinder or slow review by the FDA of any
regulatory filings that we may submit. Moreover, the FDA may
respond to these submissions by defining requirements that we
may not have anticipated.
It is possible that none of the product candidates that we
develop will obtain the appropriate regulatory approvals
necessary for us to begin selling them or that any regulatory
approval to market a product may be subject to limitations on
the indicated uses for which we may market the product. The time
required to obtain FDA and other approvals is unpredictable but
often can take years following the commencement of clinical
trials, depending upon the complexity of the drug candidate. Any
analysis we perform of data from clinical activities is subject
to confirmation and interpretation by regulatory authorities,
which could delay, limit or prevent regulatory approval. Any
delay or failure in obtaining required approvals could have a
material adverse effect on our ability to generate revenue from
the particular drug candidate.
We are also subject to numerous foreign regulatory requirements
governing the conduct of clinical trials, manufacturing and
marketing authorization, pricing and third-party reimbursement.
The foreign regulatory approval process includes all of the
risks associated with FDA approval described above as well as
risks attributable to the satisfaction of local regulations in
foreign jurisdictions. Approval by the FDA does not assure
approval by regulatory authorities outside of the United States.
The
FDA approval process may be delayed for any drugs we develop
that require the use of specialized drug delivery devices or
vehicles.
Some drug candidates that we develop may need to be administered
using specialized vehicles that deliver RNAi therapeutics
directly to diseased parts of the body. For example, we may use
an implantable pump to deliver certain potential drug candidates
to the nervous system. The drug delivery vehicles that we expect
to deliver our drug candidates have not been approved by the FDA
or other regulatory agencies. In addition, the FDA may regulate
the product as a combination product of a drug and a device or
require additional approvals or clearances for the modified
delivery.
Further, to the extent the specialized delivery vehicle is owned
by another company, we would need that company’s
cooperation to implement the necessary changes to the vehicle,
or its labeling, and to obtain any additional approvals or
clearances. Any delays in finding suitable drug delivery
vehicles to administer RNAi therapeutics directly to diseased
parts of the body could negatively affect our ability to
successfully develop our RNAi therapeutics.
22
If we
are not successful in developing pre-clinical product
candidates, we will not be able to commence clinical trials in
humans or obtain approval for our product
candidates.
We are in the new drug discovery phase and we have not yet
identified any lead compounds for therapeutic development. RNA
interference is a relatively new scientific field, and the
technologies are still in the early stage of development. We
have no compounds in pre-clinical toxicology studies, and we may
not be able to advance any product candidate through the
pre-clinical stage into clinical trials. Additionally, our
development efforts may never result in the identification of a
pre-clinical candidate which we are able to successfully develop
into a drug. Even if we are able to designate a lead candidate,
we may not be able to identify data that would support entering
such a candidate into clinical trials. Furthermore, even if we
successfully enter into clinical studies, the results from
pre-clinical testing of a drug candidate may not predict the
results that will be obtained on human clinical trials.
Even
if we obtain regulatory approvals, our marketed drugs will be
subject to ongoing regulatory review. If we fail to comply with
continuing U.S. and foreign regulations, we could lose our
approvals to market drugs and our business would be materially
adversely affected.
Following any initial regulatory approval of any drugs we may
develop, we will also be subject to continuing regulatory
review, including the review of adverse drug experiences and
clinical results that are reported after our drug products are
made available to patients. This would include results from any
post marketing tests or vigilance required as a condition of
approval. The manufacturer and manufacturing facilities we use
to make any of our drug candidates will also be subject to
periodic review and inspection by the FDA. The discovery of any
new or previously unknown problems with the product,
manufacturer or facility may result in restrictions on the drug
or manufacturer or facility, including withdrawal of the drug
from the market. Our product promotion and advertising also will
be subject to regulatory requirements and continuing regulatory
review. If we fail to comply with applicable continuing
regulatory requirements, we may be subject to fines, suspension
or withdrawal of regulatory approval, product recalls and
seizures, operating restrictions and other adverse consequences.
Even
if we receive regulatory approval to market our product
candidates, our product candidates may not be accepted
commercially, which may prevent us from becoming
profitable.
The product candidates that we are developing are based on new
technologies and therapeutic approaches. RNAi products may be
more expensive to manufacture than traditional small molecule
drugs, which may make them more costly than competing small
molecule drugs. Additionally, for various applications, RNAi
products are likely to require injection or implantation, and do
not readily cross the so-called blood brain barrier, which will
make them less convenient to administer than drugs administered
orally. Key participants in the pharmaceutical marketplace, such
as physicians, medical professionals working in large reference
laboratories, public health laboratories and hospitals,
third-party payors and consumers, may not accept products
intended to improve therapeutic results based on RNAi
technology. As a result, it may be more difficult for us to
convince the medical community and third-party payors to accept
and use our product, or to provide favorable reimbursement. And
if medical professionals working with large reference
laboratories, public health laboratories and hospitals choose
not to adopt and use our RNAi technology, our products may not
achieve broader market acceptance.
Other factors that we believe will materially affect market
acceptance of our product candidates include:
|
|
|
| |
•
|
The timing of our receipt of any marketing approvals, the terms
of any approvals and the countries in which approvals are
obtained,
|
| |
| |
•
|
The safety, efficacy and ease of administration of our product
candidates,
|
| |
| |
•
|
The advantages of our product candidates over those of our
competitors,
|
| |
| |
•
|
The willingness of patients to accept relatively new therapies,
|
| |
| |
•
|
The success of our physician education programs,
|
23
|
|
|
| |
•
|
The availability of government and third-party payor
reimbursement,
|
| |
| |
•
|
The pricing of our products, particularly as compared to
alternative treatments, and
|
| |
| |
•
|
The availability of effective alternative treatments and the
relative risks
and/or
benefits of the treatments.
|
We may
be unable to protect our intellectual property rights licensed
from others parties, our intellectual property rights may be
inadequate to prevent third parties from using our technologies
or developing competing products, and we may need to license
additional intellectual property from others.
We have a non-exclusive license to the Fire-Mello patent owned
by UMMS and the Carnegie Institution of Washington, which claims
various aspects of RNAi or genetic inhibition by double stranded
RNA. This license continues to be available to third parties,
and as such it does not provide us with the ability to exclude
others from its use or protect us from competition. Therapeutic
applications of gene silencing technologies, delivery methods,
and other technologies that we license from third parties are
claimed in a number of pending patent applications, but there
can be no assurance that these applications will result in any
issued patents or that those patents would withstand possible
legal challenges or protect our technologies from competition.
United States Patent and Trademark Office and patent granting
authorities in other countries have upheld stringent standards
for the RNAi patents that have been prosecuted so far.
Consequently, pending patents that we have licensed and those
that we own may continue to experience long and difficult
prosecution challenges and may ultimately issue with much
narrower claims than those in the pending applications. We are
aware of a number of issued patents covering various particular
forms and compositions of RNAi-mediating molecules and
therapeutic methods. Third parties may hold or seek to obtain
additional patents that could make it more difficult or
impossible for us to develop products based on RNAi technology
without obtaining a license to such patents, which licenses may
not be available on attractive terms or at all.
In addition, others may challenge the patents or patent
applications that we currently license or may license in the
future or that we own and, as a result, these patents could be
narrowed, invalidated or rendered unenforceable, which would
negatively affect our ability to exclude others from use of RNAi
technologies described in these patents. There can be no
assurance that these patent or other pending applications or
issued patents we license or that we own will withstand possible
legal challenges. Moreover, the laws of some foreign countries
may not protect our proprietary rights to the same extent as do
the laws of the United States. Any patents issued to us or our
licensors may not provide us with any competitive advantages,
and there can be no assurance that the patents of others will
not have an adverse effect on our ability to do business or to
continue to use our technologies freely. Our efforts to enforce
and maintain our intellectual property rights may not be
successful and may result in substantial costs and diversion of
management time. Even if our rights are valid, enforceable and
broad in scope, competitors may develop products based on
technology that is not covered by our licenses or patents or
patent application that we own.
We may need to license additional intellectual property rights
from third parties in order to be able to complete the
development or enhance the efficacy of our product candidates or
avoid possible infringement of the rights of others.
Additionally, many of our UMMS licenses are limited to ALS,
obesity, diabetes and cancer, and in order to pursue other
diseases against proprietary gene targets, we may need licenses
from other third parties that may be unavailable. There is no
assurance that we will be able to acquire any additional
intellectual property rights on satisfactory terms, or at all.
To the extent that we are required and are able to obtain
multiple licenses from third parties to develop or commercialize
a product candidate, the aggregate licensing fees and milestones
and royalty payments made to these parties may materially reduce
our economic returns or even cause us to abandon development or
commercialization of a product candidate.
In addition to our licenses, we also rely on copyright and
trademark protection, trade secrets, know-how, continuing
technological innovation and licensing opportunities. In an
effort to maintain the confidentiality and ownership of our
trade secrets and proprietary information, we require our
employees, consultants, advisors and others to whom we disclose
confidential information to execute confidentiality and
proprietary information agreements. However, it is possible that
these agreements may be breached, invalidated or rendered
unenforceable, and if so, there may not be an adequate
corrective remedy available. Furthermore, like many companies
24
in our industry, we may from time to time hire scientific
personnel formerly employed by other companies involved in one
or more areas similar to the activities we conduct. In some
situations, our confidentiality and proprietary information
agreements may conflict with, or be subject to, the rights of
third parties with whom our employees, consultants or advisors
have prior employment or consulting relationships. Although we
require our employees and consultants to maintain the
confidentiality of all confidential information of previous
employers, we or these individuals may be subject to allegations
of trade secret misappropriation or other similar claims as a
result of their prior affiliations. Finally, others may
independently develop substantially equivalent proprietary
information and techniques, or otherwise gain access to our
trade secrets. Our failure to protect our proprietary
information and techniques may inhibit or limit our ability to
exclude certain competitors from the market and execute our
business strategies.
Our
success depends upon our ability to obtain and maintain
intellectual property protection for our products and
technologies.
Our success will depend on our ability to obtain and maintain
adequate protection of our intellectual property covering our
product candidates and technologies. The ultimate degree of
patent protection that will be afforded to biotechnology
products and processes, including ours, in the United States and
in other important markets remains uncertain and is dependent
upon the scope of protection decided upon by the patent offices,
courts and lawmakers in these countries. There is no certainty
that our existing patents, or patent applications if obtained,
will afford us substantial protection or commercial benefit.
Similarly, there is no assurance that our pending patent
applications or patent applications licensed from third parties
will ultimately be granted as patents or that those patents that
have been issued or are issued in the future will stand if they
are challenged in court. These applications claim many different
methods, compositions and processes relating to the discovery,
development, delivery and commercialization of RNAi
therapeutics. Because the field is so new, very few of these
patent applications have been fully processed by government
patent offices around the world, and there is a great deal of
uncertainty about which patents will issue, when, to whom, and
with what claims. While we are not aware of any litigation,
threatened litigation or challenge to our intellectual property
rights, it is likely that there will be significant litigation
and other proceedings, such as interference and opposition
proceedings in various patent offices, relating to patent rights
in the RNAi field.
There may be patent or other intellectual property rights
belonging to others that require us to alter our products, pay
licensing fees or cease certain activities. If our products
infringe patent or other intellectual property rights of others,
the owners of those rights could bring legal actions against us
claiming damages and seeking to enjoin manufacturing and
marketing of the affected products. If these legal actions are
successful, in addition to any potential liability for damages,
we could be required to obtain a license in order to continue to
manufacture or market the affected products. We may not prevail
in any action brought against us, and any license required under
any rights that we infringe may not be available on acceptable
terms or at all. Others may attempt to invalidate our
intellectual property rights or those of our licensors. Even if
our rights, or those of our licensors, are not directly
challenged, disputes among third parties could lead to the
weakening or invalidation of our intellectual property rights.
Any attempt by third parties to circumvent or invalidate our
intellectual property rights could be costly to defend, require
significant time and attention of our management and have a
material adverse effect on our business.
We are
subject to potential liabilities from clinical testing and
future product liability claims.
If any of our future products are alleged to be defective, they
may expose us to claims for personal injury by patients in
clinical trials of our products or by patients using our
commercially marketed products. Even if the marketing of one or
more of our products is approved by the FDA, users may claim
that such products caused unintended adverse effects. We will
seek to obtain clinical trial insurance for clinical trials that
we conduct, as well as liability insurance for any products that
we market. There can be no assurance that we will be able to
obtain insurance in the amounts we seek, or at all. We
anticipate that licensees who develop our products will carry
liability insurance covering the clinical testing and marketing
of those products. There is no assurance, however, that any
insurance maintained by us or our licensees will prove adequate
in the event
25
of a claim against us. Even if claims asserted against us are
unsuccessful, they may divert management’s attention from
our operations and we may have to incur substantial costs to
defend such claims.
Any
drugs we develop may become subject to unfavorable pricing
regulations, third-party reimbursement practices or healthcare
reform initiatives, which could have a material adverse effect
on our business.
We intend to sell our products primarily to hospitals which
receive reimbursement for the health care services they provide
to their patients from third-party payors, such as Medicare,
Medicaid and other domestic and international government
programs, private insurance plans and managed care programs.
Most third-party payors may deny reimbursement if they determine
that a medical product was not used in accordance with
cost-effective treatment methods, as determined by the
third-party payor, or was used for an unapproved indication.
Third-party payors also may refuse to reimburse for experimental
procedures and devices. Furthermore, because our programs are in
the early stages of development, we are unable at this time to
determine their cost-effectiveness and the level or method of
reimbursement for them. Increasingly, the third-party payors who
reimburse patients are requiring that drug companies provide
them with predetermined discounts from list prices, and are
challenging the prices charged for medical products. If the
price we are able to charge for any products we develop is
inadequate in light of our development and other costs, our
profitability could be adversely effected.
We currently expect that any drugs we develop may need to be
administered under the supervision of a physician. Under
currently applicable law, drugs that are not usually
self-administered may be eligible for coverage by the Medicare
program if:
|
|
|
| |
•
|
They are “incidental” to a physician’s services,
|
| |
| |
•
|
They are “reasonable and necessary” for the diagnosis
or treatment of the illness or injury for which they are
administered according to accepted standard of medical practice,
|
| |
| |
•
|
They are not excluded as immunizations, and
|
| |
| |
•
|
They have been approved by the FDA.
|
There may be significant delays in obtaining insurance coverage
for newly-approved drugs, and insurance coverage may be more
limited than the purpose for which the drug is approved by the
FDA. Moreover, eligibility for insurance coverage does not imply
that any drug will be reimbursed in all cases or at a rate that
covers our costs, including research, development, manufacture,
sale and distribution. Interim payments for new drugs, if
applicable, may also not be sufficient to cover our costs and
may not be made permanent. Reimbursement may be based on
payments for other services and may reflect budgetary
constraints or imperfections in Medicare data. Net prices for
drugs may be reduced by mandatory discounts or rebates required
by government health care programs or private payors and by any
future relaxation of laws that presently restrict imports of
drugs from countries where they may be sold at lower prices than
in the United States. Third-party payors often rely upon
Medicare coverage policy and payment limitations in setting
their own reimbursement rates. Our inability to promptly obtain
coverage and profitable reimbursement rates from both
government-funded and private payors for new drugs that we
develop could have a material adverse effect on our operating
results, our ability to raise capital needed to develop
products, and our overall financial condition.
Additionally, third-party payors are increasingly attempting to
contain health care costs by limiting both coverage and the
level of reimbursement for medical products and services. Levels
of reimbursement may decrease in the future, and future
legislation, regulation or reimbursement policies of third-party
payors may adversely affect the demand for and price levels of
our products. If our customers are not reimbursed for our
products, they may reduce or discontinue purchases of our
products, which could have a material adverse effect on our
business, financial condition and results of operations.
Comprehensive health care reform legislation, which was recently
adopted by Congress and was subsequently signed into law, could
adversely affect our business and financial condition. Among
other provisions, the legislation provides that a
“biosimilar” product may be approved by the FDA on the
basis of
26
analytical tests and certain clinical studies demonstrating that
such product is highly similar to an existing, approved product
and that switching between an existing product and the
biosimilar product will not result in diminished safety or
efficacy. This abbreviated regulatory approval process may
result in increased competition if we are able to bring a
product to market. The legislation also includes more stringent
compliance programs for companies in various sectors of the life
sciences industry with which we may need to comply and enhanced
penalties for non-compliance with the new health care
regulations. Complying with new regulations may divert
management resources, and inadvertent failure to comply with new
regulations may result in penalties being imposed on us.
Some states and localities have established drug importation
programs for their citizens, and federal drug import legislation
has been introduced in Congress. The Medicare Prescription Drug
Plan legislation, which became law in December 2003, required
the Secretary of Health and Human Services to promulgate
regulations for drug reimportation from Canada into the United
States under some circumstances, including when the drugs are
sold at a lower price than in the United States. The Secretary,
however, retained the discretion not to implement a drug
reimportation plan if he finds that the benefits do not outweigh
the costs, and has so far declined to approve a reimportation
plan. Proponents of drug reimportation may attempt to pass
legislation that would directly allow reimportation under
certain circumstances. Legislation or regulations allowing the
reimportation of drugs, if enacted, could decrease the price we
receive for any products that we may develop and adversely
affect our future revenues and prospects for profitability.
If we
fail to attract, hire and retain qualified personnel, we may not
be able to design, develop, market or sell our products or
successfully manage our business.
We are highly dependent on our named executive officers and
Scientific Advisory Board (“SAB”) members. The
continued service of our named executive officers and SAB
members is critical to our success. We have entered into
employment agreements with our named executive officers, all of
which can be terminated by such persons on shortnotice. The loss
of any of our named executive officers or SAB members, or our
inability to identify, attract, retain and integrate additional
qualified key personnel, could make it difficult for us to
manage our business successfully and achieve our business
objectives.
Competition for skilled research, product development,
regulatory and technical personnel also is intense, and we may
not be able to recruit and retain the personnel we need. The
loss of the services of any key research, product development,
regulatory, and technical personnel, or our inability to hire
new personnel with the requisite skills, could restrict our
ability to develop our product candidates.
We use
biological and hazardous materials and if we do not comply with
laws regulating the protection of the environment and health and
human safety, our business could be adversely
affected.
Our research and development activities involve the controlled
use of potentially harmful biological materials as well as
hazardous materials, chemicals and various radioactive
compounds. We cannot completely eliminate the risk of accidental
contamination or injury; we could be held liable for any damages
that result, and any liability could exceed our resources. We
are subject to federal, state and local laws and regulations
governing the use, storage, handling and disposal of these
materials and specific waste products. We are also subject to
numerous environmental, health and workplace safety laws and
regulations, including those governing laboratory procedures,
exposure to blood-borne pathogens and the handling of
biohazardous materials. The cost of compliance with these laws
and regulations could be significant and may adversely affect
capital expenditures to the extent we are required to procure
expensive capital equipment to meet regulatory requirements.
We are also subject to numerous environmental, health and
workplace safety laws and regulations, including those governing
laboratory procedures, exposure to blood-borne pathogens and the
handling of biohazardous materials. We maintain workers’
compensation insurance to cover us for costs and expenses we may
incur due to injuries to our employees resulting from the use of
these materials. The limits of our workers’ compensation
insurance are mandated by state law, and our workers’
compensation liability is capped at these state-mandated limits.
We do not maintain insurance for environmental liability or
toxic tort claims
27
that may be asserted against us in connection with our storage
or disposal of biological, hazardous or radioactive materials.
Additional federal, state and local laws and regulations
affecting our operations may be adopted in the future. We may
incur substantial costs to comply with, and substantial fines or
penalties if we violate any of these laws or regulations.
Risks
Relating Our Financial Position and Capital
Requirements
We may
not be able to obtain sufficient financing, and may not be able
to develop our product candidates.
We believe that our existing cash and cash equivalent should be
sufficient to fund our operations through at least the first
half of 2011. In the future, we will be dependent on obtaining
further financing from third parties in order to maintain our
operations and to meet our financial obligations. In addition,
until its expiration on January 30, 2011, we also have
available to us the SEDA that we entered into on
January 30, 2009. However, before we are able to access
additional capital from the SEDA, we must satisfy certain
conditions, including the requirement that shares of our stock
to be sold to YA Global be registered with the
U.S. Securities and Exchange Commission (“SEC”),
and there is risk of delays in our satisfying these conditions.
We cannot assure that additional debt or equity or other funding
to maintain our operations and to meet our obligations to our
licensors will be available to us in the future on acceptable
terms, or at all. If we fail to obtain additional funding when
needed, we would be forced to scale back, or terminate, our
operations, or to seek to merge with or to be acquired by
another company.
We anticipate that we will need to raise substantial amounts of
money to fund a variety of future activities integral to the
development of our business, which may include but are not
limited to the following:
|
|
|
| |
•
|
To conduct research and development to successfully develop our
RNAi technologies,
|
| |
| |
•
|
To obtain regulatory approval for our product candidates,
|
| |
| |
•
|
To file and prosecute patent applications and to defend and
assess patents to protect our technologies,
|
| |
| |
•
|
To retain qualified employees, particularly in light of intense
competition for qualified scientists,
|
| |
| |
•
|
To manufacture products ourselves or through third parties,
|
| |
| |
•
|
To market our products, either through building our own sales
and distribution capabilities or relying on third parties, and
|
| |
| |
•
|
To acquire new technologies, licenses, products or companies.
|
We cannot assure you that any financing needed for the
development of our business will be available to us on
acceptable terms or at all. If we cannot obtain additional
financing in the future, our operations may be restricted and we
may ultimately be unable to continue to develop and potentially
commercialize our product candidates.
We
expect to continue to incur significant research and development
expenses, which may make it difficult for us to attain
profitability, and may lead to uncertainty about or as to our
ability to continue as a going concern.
Substantial funds were expended to develop our RNAi
technologies, and additional substantial funds will be required
for further research and development, including pre-clinical
testing and clinical trials of any product candidates, and to
manufacture and market any products that are approved for
commercial sale. Because the successful development of our
products is uncertain, we are unable to precisely estimate the
actual funds we will require to develop and potentially
commercialize them. In addition, we may not be able to generate
enough revenue, even if we are able to commercialize any of our
product candidates, to become profitable.
In the event that we are unable to achieve or sustain
profitability or to secure additional financing, we may not be
able to meet our obligations as they come due, raising
substantial doubts as to our ability to
28
continue as a going concern. Any such inability to continue as a
going concern may result in our common stock holders losing
their entire investment. There is no guaranty that we will
become profitable or secure additional financing. Our financial
statements contemplate that we will continue as a going concern
and do not contain any adjustments that might result if we were
unable to continue as a going concern. Changes in our operating
plans, our existing and anticipated working capital needs, the
acceleration or modification of our expansion plans, increased
expenses, potential acquisitions or other events will all affect
our ability to continue as a going concern.
Future
financing may be obtained through, and future development
efforts may be paid for by, the issuance of debt or equity,
which may have an adverse effect on our stockholders or may
otherwise adversely affect our business.
If we raise funds through the issuance of debt or equity, any
debt securities or preferred stock issued will have rights,
preferences and privileges senior to those of holders of our
common stock in the event of a liquidation. In such event, there
is a possibility that once all senior claims are settled, there
may be no assets remaining to pay out to the holders of common
stock. In addition, if we raise funds through the issuance of
additional equity, whether through private placements or
additional public offerings, such an issuance would dilute your
ownership in us.
The terms of debt securities may also impose restrictions on our
operations, which may include limiting our ability to incur
additional indebtedness, to pay dividends on or repurchase our
capital stock, or to make certain acquisitions or investments.
In addition, we may be subject to covenants requiring us to
satisfy certain financial tests and ratios, and our ability to
satisfy such covenants may be affected by events outside of our
control.
You
may have difficulty evaluating our business, because we have
limited history and our historical financial information may not
be representative of our future results.
The historical financial information included in this annual
report on
Form 10-K
for the year ended December 31, 2009 does not necessarily
reflect the financial condition, results of operations or cash
flows that we would have achieved as a separate company during
the periods presented or those that we will achieve in the
future. Prior to the contribution of our RNAi assets from CytRx,
our RNAi research and development activities were conducted by
CytRx as part of its broader operations, rather than as an
independent division or subsidiary, and were primarily conducted
through sponsored research arrangements rather than through
internal activities. CytRx also performed various corporate
functions relating to our business, as discussed above. Our
historical financial information reflects allocations of
indirect expenses from CytRx for these and similar functions. We
believe that these allocations are comparable to the expenses we
would have incurred had we operated as a separate company,
although we may incur higher expenses as a separate company.
We
have limited operating experience and may not be able to
effectively operate.
We are a discovery-stage company with limited operating history.
We will focus solely on developing and, if we obtain regulatory
approval for our product candidates, commercializing therapeutic
products based upon RNAi technologies, and there is no assurance
that we will be able to successfully implement our business
plan. While our management collectively possesses substantial
business experience, there is no assurance that we will be able
to manage our business effectively, or that we will be able to
identify, hire and retain any needed additional management or
scientific personnel to develop and implement our product
development plans, obtain third-party contracts or any needed
financing, or achieve the other components of our business plan.
The
obligations associated with being an independent public company
require significant resources and management
attention.
As a publicly traded company, we are subject to the reporting
requirements of the U.S. Securities Exchange Act of 1934,
as amended (the “Exchange Act”), and the
Sarbanes-Oxley Act of 2002. In addition,
29
the Exchange Act requires that we file annual, quarterly and
current reports. Our failure to prepare and disclose this
information in a timely manner could subject us to penalties
under federal securities laws, expose us to lawsuits and
restrict our ability to access financing. The Sarbanes-Oxley Act
requires that we, among other things, establish and maintain
effective internal controls and procedures for financial
reporting. From time to time we evaluate our existing internal
controls in light of the standards adopted by the Public Company
Accounting Oversight Board. It is possible that we or our
independent registered public accounting firm may identify
significant deficiencies or material weaknesses in our internal
control over financial reporting in the future. Any failure or
difficulties in implementing and maintaining these controls
could cause us to fail to meet the periodic reporting
obligations or result in material misstatements in our financial
statements.
Section 404 of the Sarbanes-Oxley Act requires annual
management assessments of the effectiveness of our internal
control over financial reporting. Our failure to satisfy the
requirements of Section 404 on a timely basis could result
in the loss of investor confidence in the reliability of our
financial statements, which in turn could have a material
adverse effect on our business and our common stock.
Risks
Related to Ownership of Our Common Stock
The
market price and trading volume of our common stock may be
volatile
The market price of our common stock could fluctuate
significantly for many reasons, including the following factors:
|
|
|
| |
•
|
Announcements of regulatory developments or technological
innovations by us or our competitors,
|
| |
| |
•
|
Changes in our relationship with our licensors and other
strategic partners,
|
| |
| |
•
|
Changes in our ownership or other relationships with CytRx,
|
| |
| |
•
|
Our quarterly operating results,
|
| |
| |
•
|
Developments in patent or other technology ownership rights,
|
| |
| |
•
|
Public concern regarding the safety of our products,
|
| |
| |
•
|
Additional funds may not be available on terms that are
favorable to us and, in the case of equity financings, may
result in dilution to our stock holders,
|
| |
| |
•
|
Government regulation of drug pricing, and
|
| |
| |
•
|
General changes in the economy, the financial markets or the
pharmaceutical or biotechnology industries.
|
In addition, factors beyond our control may also have an impact
on the price of our stock. For example, to the extent that other
large companies within our industry experience declines in their
stock price, our stock price may decline as well. In addition,
when the market price of a company’s common stock drops
significantly, stockholders often institute securities class
action lawsuits against the company. A lawsuit against us could
cause us to incur substantial costs and could divert the time
and attention of our management and other resources.
Sales
of our shares by CytRx could adversely affect our stock
price.
As of March 30, 2010, CytRx owned 5,093,881 shares of
our common stock, or approximately 28% of our outstanding
shares. The availability of our shares held by CytRx for public
resale on the open market, as well as any actual sales of these
shares, could adversely affect the market price of our shares.
We
have granted CytRx preemptive rights to acquire shares that we
may sell in the future, which may impair our ability to raise
funds.
Under an agreement between us, CytRx and our founding
stockholders, with some exceptions, CytRx has preemptive rights
to acquire a portion of any new securities sold or issued by us
so as to maintain its
30
percentage ownership of us at the time of any such sale and
issuance, which is currently approximately 28% of our
outstanding shares. The exercise by CytRx of its preemptive
rights may impair our ability to raise funds, or adversely
affect the terms on which we are able to raise funds, as we may
not be able to offer to new investors the quantity of our stock
that they may desire to purchase.
CytRx’s
ownership of our common stock could delay or prevent a change in
corporate control.
CytRx owns approximately 28% of our common stock, and has
preemptive rights, as described above, to maintain its
percentage ownership. CytRx has agreed with UMMS, us and our
other founding stockholders to vote its shares of our common
stock so that a majority of the members of our board of
directors are not affiliated with CytRx. However, by virtue of
its stock ownership, CytRx may be able to significantly
influence the outcome of matters required to be submitted to a
vote of our stockholders, including any proposed amendments to
our certificate of incorporation and approval of mergers and
other significant corporate transactions. This concentration of
ownership may adversely affect the market price of our common
stock by:
|
|
|
| |
•
|
Delaying, deferring or preventing a change in control of our
company,
|
| |
| |
•
|
Impeding a merger, consolidation, takeover or other business
combination involving our company, or
|
| |
| |
•
|
Discouraging a potential acquirer from making a tender offer or
otherwise attempting to obtain control of our company.
|
CytRx
could unilaterally effect a change of control of our company by
selling or disposing of our shares owned by it.
If CytRx were to sell or otherwise dispose of all or a
significant portion of our shares owned by it to a single buyer
or group of affiliated buyers, it could effect a potential
change of control of our company without the advice or
participation by our board of directors or other stockholders,
since transferees of the shares owned by CytRx will not be bound
by CytRx’s agreements with UMMS, us and our other founding
stockholders with respect to voting such shares.
Anti-takeover
provisions of our certificate of incorporation and by-laws and
provisions of Delaware law could delay or prevent a change of
control that you may favor.
Anti-takeover provisions of our certificate of incorporation and
by-laws and provisions of Delaware law may discourage, delay or
prevent a merger or other change of control that stockholders
may consider favorable, or may impede the ability of the holders
of our common stock to change our management. These provisions
of our certificate of incorporation and by-laws, among other
things:
|
|
|
| |
•
|
Divide our board of directors into three classes, with members
of each class to be elected for staggered three-year terms,
|
| |
| |
•
|
Limit the right of stockholders to remove directors,
|
| |
| |
•
|
Regulate how stockholders may present proposals or nominate
directors for election at annual meetings of
stockholders, and
|
| |
| |
•
|
Authorize our board of directors to issue preferred stock in one
or more series, without stockholder approval.
|
In addition, Section 203 of the Delaware General
Corporation Law provides that, subject to limited exceptions,
persons that acquire, or are affiliated with a person that
acquires, more than 15% of the outstanding voting stock of a
Delaware corporation such as our company shall not engage in any
business combination with that corporation, including by merger,
consolidation or acquisitions of additional shares for a
three-year period following the date on which that person or its
affiliate crosses the 15% stock ownership threshold.
Section 203 could operate to delay or prevent a change of
control of our company.
31
We may
acquire other businesses or form joint ventures that may be
unsuccessful and could adversely dilute your ownership of our
company.
As part of our business strategy, we may pursue future
acquisitions of other complementary businesses and technology
licensing arrangements. We also may pursue strategic alliances.
We have no experience with respect to acquiring other companies
and limited experience with respect to the formation of
collaborations, strategic alliances and joint ventures. If we
were to make any acquisitions, we may not be able to integrate
these acquisitions successfully into our existing business and
we could assume unknown or contingent liabilities. We also could
experience adverse effects on our reported results of operations
from acquisition related charges, amortization of acquired
technology and other intangibles and impairment charges relating
to write-offs of goodwill and other intangible assets from time
to time following the acquisition. Integration of an acquired
company also may require management resources that otherwise
would be available for ongoing development of our existing
business. We may not identify or complete these transactions in
a timely manner, on a cost-effective basis, or at all, and we
may not realize the anticipated benefits of any acquisition,
technology license or strategic alliance.
To finance acquisitions, we may choose to issue shares of our
common stock as consideration, which would dilute your ownership
interest in us. Alternatively, it may be necessary for us to
raise additional funds through public or private financings.
Additional funds may not be available on terms that are
favorable to us and, in the case of equity financings, may
result in dilution to our stockholders. Any future acquisitions
by us also could result in large and immediate write-offs, the
incurrence of contingent liabilities or amortization of expenses
related to acquired intangible assets, any of which could harm
our operating results.
|
|
|
ITEM 1B.
|
UNRESOLVED
STAFF COMMENTS
|
None.
On September 25, 2007, we entered into a lease agreement
with Newgate Properties, LLC (an affiliate of Worcester
Polytechnic Institute), for our facility located at 60 Prescott
Street, Worcester, Massachusetts. The lease has a term of
20 months. The facility is approximately 6,800 square
feet, of which 5,600 is laboratory space used for research and
development and the additional 1,200 square feet is used
for general and administrative offices. On January 23,
2009, we extended our lease for an additional two years through
July 31, 2011. The monthly rental fee is approximately
$19,000. We believe that the space is suitable for our current
needs.
|
|
|
ITEM 3.
|
LEGAL
PROCEEDINGS
|
Although we are not currently involved in any legal proceedings,
from time to time, we may become a party to various legal
actions and complaints arising in the ordinary course of
business.
PART II
|
|
|
ITEM 5.
|
MARKET
FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS
AND ISSUER PURCHASES OF EQUITY SECURITIES
|
Market
Information
Our common stock has been listed on the Nasdaq Capital Market
under the symbol “RXII” since March 12, 2008.
Prior to that, there was no established public market for our
common stock. The following
32
table sets forth for the periods indicated the high and low
sales prices of our common stock on the Nasdaq Capital Market:
| |
|
|
|
|
|
|
|
|
|
Year Ended December 31, 2008
|
|
High
|
|
|
Low
|
|
|
|
|
First Quarter
|
|
$
|
23.95
|
|
|
$
|
6.01
|
|
|
Second Quarter
|
|
$
|
10.12
|
|
|
$
|
5.22
|
|
|
Third Quarter
|
|
$
|
9.05
|
|
|
$
|
6.42
|
|
|
Fourth Quarter
|
|
$
|
12.25
|
|
|
$
|
5.41
|
|
| |
|
|
|
|
|
|
|
|
|
Year Ended December 31, 2009
|
|
High
|
|
|
Low
|
|
|
|
|
First Quarter
|
|
$
|
7.19
|
|
|
$
|
2.71
|
|
|
Second Quarter
|
|
$
|
7.57
|
|
|
$
|
4.00
|
|
|
Third Quarter
|
|
$
|
4.93
|
|
|
$
|
2.44
|
|
|
Fourth Quarter
|
|
$
|
4.84
|
|
|
$
|
1.51
|
|
Holders
As of March 29, 2010, there were approximately
631 holders of record of our common stock. Because many of
our shares are held by brokers and other institutions on behalf
of stockholders, we are unable to estimate the total number of
individual stockholders represented by these records.
Dividends
We have never paid any dividends and do not anticipate paying
any dividends on our common stock in the foreseeable future. We
expect to retain future earnings, if any, for use in our
development activities and the operation of our business. The
payment of any future dividends will be subject to the
discretion of our board of directors and will depend, among
other things, upon our results of operations, financial
condition, cash requirements, prospects and other factors that
our board of directors may deem relevant. Additionally, our
ability to pay future dividends may be restricted by the terms
of any debt financing.
Securities
authorized for issuance under equity compensation
plans
Information relating to our equity compensation plans will be
included in our proxy statement in connection with our 2010
Annual Meeting of Stockholders, under the caption “Equity
Compensation Plan Information”. The relevant portion of our
proxy statement is incorporated herein by reference.
Performance
Graph
Because we are a smaller reporting company, we are not required
to provide this information.
Recent
Sales of Unregistered Securities
Set forth below is information regarding shares of common stock
and preferred stock issued, and options and warrants granted, by
us during the period covered by this report. Also included is
the consideration, if any, received by us for such shares,
options and warrants and information relating to the section of
the Securities Act, or rule of the SEC under which exemption
from registration was claimed.
Preferred
Stock
There were no unregistered shares of preferred stock issued by
us during the period covered by this report.
Common
Stock
There were no unregistered shares of common stock issued by us
during the period covered by this report.
33
Options
There were no unregistered options granted by us during the
period covered by this report.
Common
Stock Warrants
There were no unregistered common stock warrants issued by us
during the period covered by this report, except which have
previously been disclosed in a Current Report on Form 8-K.
|
|
|
ITEM 6.
|
SELECTED
FINANCIAL DATA
|
Because we are a smaller reporting company, we are not required
to provide the information required by this Item.
|
|
|
ITEM 7.
|
MANAGEMENT’S
DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF
OPERATIONS
|
You should read the following discussion in conjunction with the
RXi financial statements and the notes to financial statements
included elsewhere in this annual report. This
“Management’s Discussion and Analysis of Financial
Condition and Results of Operations” section contains
forward-looking statements within the meaning of the Private
Securities Litigation Reform Act of 1995. For a discussion of
indicators of forward-looking statements and specific important
factors that could cause actual results to differ materially
from those contained in forward-looking statements, see
“Risk Factors” under Part I —
Item 1A of this Annual Report on
Form 10-K
for the fiscal year ended December 31, 2009. This
Management’s Discussion and Analysis of Financial Condition
and Results of Operations should be read and interpreted in
light of such factors.
Our actual results and the timing of certain events could differ
materially from those anticipated in these forward-looking
statements as a result of certain factors, including those
discussed below and elsewhere in this annual report.
Overview
We are a discovery-stage biopharmaceutical company pursuing
proprietary therapeutics based on RNA interference, or
“RNAi,” a naturally occurring cellular mechanism that
has the potential to effectively and selectively interfere with,
or “silence,” expression of targeted
disease-associated genes. It is believed that this specific
silencing can be used to potentially treat human diseases by
“turning off” genes that lead to disease.
We intend to use our RNAi therapeutic platform and our expertise
in RNAi to identify lead compounds against multiple target
genes, and advance them towards pre-clinical and clinical
development in therapeutic areas that address broad unmet
medical needs, in both acute and chronic settings. By utilizing
our expertise in RNAi and the comprehensive RNAi therapeutic
platform that we have established, we believe we will be able to
discover and develop lead compounds and progress them into and
through clinical development for potential commercialization
more efficiently than traditional drug development approaches.
We were formed in 2006 by CytRx Corporation and four prominent
RNAi researchers, including Dr. Craig Mello, who was
awarded the 2006 Nobel Prize in Medicine for his co-discovery of
RNAi. From 2003 through 2006, CytRx sponsored therapeutic RNAi
research at the University of Massachusetts Medical School, or
“UMMS,” and Massachusetts General Hospital. We
commenced operations in January 2007 after CytRx contributed to
us its portfolio of RNAi therapeutic assets in exchange for
approximately 7.04 million shares of our common stock.
These assets consisted primarily of RNAi licenses and related
intellectual property and a nominal amount of equipment. The
cost of the licenses had previously been expensed by CytRx as
in-process research and development and was recorded in the
predecessor financial statements at cost.
To date, our principal activities have consisted of conducting
discovery research and pre-clinical development activities
utilizing our RNAi therapeutic platform, acquiring RNAi
technologies and patent rights through exclusive, co-exclusive
and non-exclusive licenses, recruiting an RNAi-focused
management and scientific/clinical advisory team, capital
raising activities and conducting business development
activities aimed
34
at establishing research and development partnerships with
pharmaceutical and larger biotechnology companies.
The
Founding and Funding of RXi
On April 30, 2007, we issued approximately 3,273,000
additional shares of our common stock to CytRx at $5.19 per
share, based in part, upon the advice of a third party valuation
advisor and assuming the issuance of 462,112 shares to UMMS
pursuant to our license agreements with them, in exchange for
CytRx’s additional investment of $17.0 million. On
September 25, 2007, we issued an additional
188,387 shares of common stock to CytRx at $5.19 per share
to satisfy in full certain reimbursement amounts owed to CytRx
by us. CytRx currently owns approximately 28% of our outstanding
shares of common stock. In the event that we propose to sell or
issue shares of RXi common stock in the future, CytRx will have
the right to purchase a portion of such shares sufficient to
maintain its percentage ownership at the time of such sale or
issuance. This right will terminate on the earlier of
January 8, 2012 or the first date at which CytRx owns less
than 10% of our outstanding shares.
On June 24, 2008, we sold 1,073,299 shares of our
common stock to institutional investors at $8.12 per share,
resulting in aggregate gross proceeds of approximately
$8.7 million.
On August 4, 2009, we sold 2,385,715 shares of our
common stock to institutional investors at $3.50 per share,
resulting in aggregate gross proceeds of approximately
$7.8 million.
On March 26, 2010, we sold 2,700,000 shares of our
common stock to institutional investors at $6.00 per share,
resulting in aggregate gross proceeds of approximately
$16.2 million.
Research
and Development
Our research programs focus on identifying product candidates
and optimizing the delivery method and technology necessary to
make RNAi compounds available by local, systemic or oral
administration, as appropriate for diseases for which we intend
to develop an RNAi therapeutic.
Since we commenced operations, research and development has
comprised a significant proportion of our total operating
expenses and is expected to comprise the majority of our
spending for the foreseeable future.
There are risks in any new field of drug discovery that preclude
certainty regarding the successful development of a product. We
cannot reasonably estimate or know the nature, timing and costs
of the efforts necessary to complete the development of, or the
period in which material net cash inflows are expected to
commence from, any product candidate. Our inability to make
these estimates results from the uncertainty of numerous
factors, including but not limited to:
|
|
|
| |
•
|
Our ability to advance product candidates into pre-clinical
research and clinical trials;
|
| |
| |
•
|
The scope and rate of progress of our pre-clinical program and
other research and development activities;
|
| |
| |
•
|
The scope, rate of progress and cost of any clinical trials we
commence;
|
| |
| |
•
|
The cost of filing, prosecuting, defending and enforcing patent
claims and other intellectual property rights;
|
| |
| |
•
|
Clinical trial results;
|
| |
| |
•
|
The terms and timing of any collaborative, licensing and other
arrangements that we may establish;
|
| |
| |
•
|
The cost and timing of regulatory approvals;
|
| |
| |
•
|
The cost of establishing clinical and commercial supplies of our
product candidates and any products that we may develop;
|
| |
| |
•
|
The cost and timing of establishing sales, marketing and
distribution capabilities;
|
35
|
|
|
| |
•
|
The effect of competing technological and market
developments; and
|
| |
| |
•
|
The effect of government regulation and insurance industry
efforts to control healthcare costs through reimbursement policy
and other cost management strategies.
|
Failure to complete any stage of the development of our product
candidates in a timely manner could have a material adverse
effect on our operations, financial position and liquidity.
License
Agreements
We have entered into licensing relationships with academic
institutions, research foundations and commercial entities, and
may seek to enter into additional licenses with pharmaceutical
and biotechnology companies. We also may enter into strategic
alliances to expand our RNAi intellectual property portfolio and
to potentially accelerate our development programs by gaining
access to technology and funding, including equity sales,
license fees and other revenues. For each product that we
develop that is covered by the patents licensed to us including
the material licenses discussed below, we are obligated to make
additional payments upon the attainment of certain specified
product development milestones.
University
of Massachusetts Medical School
As part of the Contribution Agreement dated January 8,
2007, CytRx assigned to us their rights under four exclusive
license agreements, one co-exclusive license agreement and one
non-exclusive license agreement entered into between CytRx and
UMMS, which cover potential therapeutic applications for
proprietary RNAi technology in the treatment of specified
diseases. Additionally, CytRx assigned to us their rights under
the Collaboration and Invention Disclosure Agreement, dated
January 10, 2007 between CytRx and UMMS. Under these
licenses, UMMS granted to us exclusive, worldwide licenses, with
the right to
sub-license,
to three different patent families and a non-exclusive,
worldwide license to a fourth patent family. As consideration
for these licenses, we paid UMMS an up-front fee, reimbursed
UMMS for previously incurred patent expenses and agreed to
undertake to raise working capital by a specified date, agreed
to expend a specified amount on the development of
royalty-bearing products, and to meet a defined timeline
relating to the clinical development of royalty-bearing
products. Our obligation to raise working capital was satisfied
when CytRx invested $17.0 million in us (before a
$2.0 million reimbursement for expenses by us to CytRx) on
April 30, 2007. Upon the completion of the
$17.0 million financing from CytRx, we became obligated to
pay UMMS additional licenses fees in an aggregate amount of
$175,000, issued to UMMS approximately 308,075 shares of
our common stock valued at $5.00 per share, for a total value of
$1,540,375 and thereafter to pay UMMS annual maintenance fees,
commencing on January 1, 2008, and certain additional
amounts upon the attainment of certain specified product
development milestones. We also will be required to pay to UMMS
a percentage of income received from any sub-licensees under
these licenses and to pay expenses incurred by UMMS in
prosecuting and maintaining the licensed patents.
Further, on January 10, 2007, we entered into three
exclusive licenses and one non-exclusive license with UMMS
pursuant to which UMMS granted to us rights under certain UMMS
patent applications to make, use and sell products related to
applications of RNAi technologies in particular fields,
including HCMV, retinitis, ALS, diabetes and obesity.
In connection with all of our licenses with UMMS, including
those assigned to us by CytRx as well as those entered into
directly between us and UMMS, we are obligated to pay specified
royalties on net sales of products covered by the licensed
patents, subject to minimum annual royalties.
We recently terminated a number of these UMMS licenses and still
hold licenses to patents and patent applications that belong to
six distinct families of patent applications from the original
thirteen.
We also recently terminated the Collaboration and Invention
Disclosure Agreement we had entered into in January 10,
2007, with UMMS.
36
We recently terminated the License agreement with Imperial
College Innovations Limited and Imperial College of Science and
Technology that was assigned to us by CytRx in connection with
the Contribution Agreement dated January 8, 2007.
Financial
Information
The financial information of RXi as of December 31, 2009
and 2008 and the cumulative financial information for the period
from January 1, 2003 (date of inception) to
December 31, 2009 have been audited by our independent
registered public accounting firm, BDO Seidman, LLP.
Critical
Accounting Policies and Estimates
Use of
Estimates
Management’s discussion and analysis of our financial
condition and results of operations include the financial
statements as of and for the years ended December 31, 2009
and 2008. The preparation of these financial statements requires
management to make estimates, allocations and judgments that
affect the reported amounts of assets, liabilities, revenues and
expenses, and related disclosure of contingent assets and
liabilities. On an ongoing basis, management evaluates its
estimates, including those related to impairment of long-lived
assets, accrued liabilities and certain expenses. We base our
estimates about the carrying values of assets and liabilities
that are not readily apparent from other sources on historical
experience and on other assumptions believed to be reasonable
under the circumstances. Actual results may differ materially
from these estimates under different assumptions or conditions.
Additionally, the financial information included here may not
necessarily reflect the financial position, operating results,
changes in our invested equity and cash flows in the future or
what they would have been had we been a separate, stand-alone
entity during the periods presented.
Our significant accounting policies are summarized in the
footnotes to our financial statements. We believe the following
critical accounting policies involve significant judgments and
estimates used in the preparation of our financial statements.
Research
and Development Expenses
Research and development costs are expensed as incurred.
Included in research and development costs are wages, benefits
and other operating costs, facilities, supplies, external
services and overhead directly related to our research and
development departments as well as costs to acquire technology
licenses.
Stock-Based
Compensation
We follow the provisions of the Financial Accounting Standards
Board (“FASB”) Accounting Standards Codification
(“ASC”) Topic 718, “Compensation —
Stock Compensation” (“ASC 718”), which
requires the measurement and recognition of compensation expense
for all share-based payment awards made to employees,
non-employee directors, and consultants, including employee
stock options. Stock compensation expense based on the grant
date fair value estimated in accordance with the provisions of
ASC 718 is recognized as an expense over the requisite service
period.
For stock options granted as consideration for services rendered
by non-employees, we recognize compensation expense in
accordance with the requirements of FASB ASC Topic
505-50
(“ASC
505-50”)
“Equity Based Payments to Non-Employees.”
Non-employee option grants that do not vest immediately upon
grant are recorded as an expense over the vesting period of the
underlying stock options. At the end of each financial reporting
period prior to vesting, the value of these options, as
calculated using the Black-Scholes option-pricing model, will be
re-measured using the fair value of our common stock and the
non-cash compensation recognized during the period will be
adjusted accordingly. Since the fair market value of options
granted to non-employees is subject to change in the future, the
amount of the future compensation expense will include fair
value re-measurements until the stock options are fully vested.
37
The fair value of each option grant is estimated using the
Black-Scholes option-pricing model, with the following weighted
average assumptions:
| |
|
|
|
|
|
|
|
2009
|
|
2008
|
|
|
|
Weighted average risk free interest rate
|
|
1.55% - 3.91%
|
|
1.55% - 3.99%
|
|
Weighted average volatility
|
|
116.72% - 122.93%
|
|
101.79% - 116.74%
|
|
Expected lives (years)
|
|
6 - 10
|
|
6 - 10
|
|
Expected dividend yield
|
|
0%
|
|
0%
|
Our expected common stock price volatility assumption is based
upon the volatility of a basket of comparable companies. The
expected life assumptions for employee grants were based upon
the simplified method provided for under ASC 718, which averages
the contractual term of our options of ten years with the
average vesting term of four years for an average of six years.
The expected life assumptions for non-employees were based upon
the contractual term of the option. The dividend yield
assumption of zero is based upon the fact that we have never
paid cash dividends and presently have no intention of paying
cash dividends in the future. The risk-free interest rate used
for each grant was also based upon prevailing short-term
interest rates.
We have estimated an annualized forfeiture rate of 4.0% for
options granted to our employees, 2.1% for options granted to
senior management and no forfeiture rate for the directors. We
record additional expense if the actual forfeitures are lower
than estimated and will record a recovery of prior expense if
the actual forfeiture rates are higher than estimated.
Valuation
of Common Stock
Our common stock was registered and began trading publicly on
March 12, 2008. As a result, the actual value of a common
share may be materially different than the fair value per share
prior to that date.
Derivative
Financial Instruments
During the normal course of business, from time to time, we
issue warrants and options to vendors as consideration to
perform services. We may also issue warrants as part of a debt
or equity financing. We do not enter into any derivative
contracts for speculative purposes.
We recognize all derivatives as assets or liabilities measured
at fair value with changes in fair value of derivatives
reflected as current period income or loss unless the
derivatives qualify for hedge accounting and are accounted for
as such. During the year ended December 31, 2009, we issued
warrants to purchase 978,142 shares of common stock in
connection with an equity transaction. In accordance with ASC
Topic 815-40,
“Derivatives and Hedging — Contracts in
Entity’s Own Stock” (“ASC
815-40”),
the value of these warrants is required to be recorded as a
liability, as the holders have an option to put the warrants
back to us in certain events, as defined.
Results
of Operations
For the year ended December 31, 2009, our net loss was
approximately $18,387,000, compared with a net loss of
$14,373,000 for the year ended December 31, 2008. The loss
increased by $4,014,000 or approximately 28%. Reasons for the
variations in the losses between the years are discussed below.
Revenue
Since we are a development-stage biopharmaceutical company, we
have not generated any revenues since inception through
December 31, 2009.
38
Research
and Development Expense
| |
|
|
|
|
|
|
|
|
|
|
|
For the Years Ended December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
Research and development expense
|
|
$
|
6,728
|
|
|
$
|
5,105
|
|
|
Research and development employee stock-based compensation
expense
|
|
|
867
|
|
|
|
336
|
|
|
Research and development non-employee stock-based compensation
expense
|
|
|
1,297
|
|
|
|
1,613
|
|
|
|
|
|
|
|
|
|
|
|
|
Total research and development expense
|
|
$
|
8,892
|
|
|
$
|
7,054
|
|
|
|
|
|
|
|
|
|
|
|
During 2009, research and development expense consists primarily
of compensation-related costs for our employees dedicated to
research and development activities and for our Scientific
Advisory Board (“SAB”) members as well as licensing
fees, patent prosecution costs, and the cost of lab supplies
used in our research and development programs. We expect to
continue to devote a substantial portion of our resources to
research and development expenses and we expect research and
development expenses to increase as we expand our discovery and
development activities for RNAi therapeutics.
Total research and development expenses for the year ended
December 31, 2009 were approximately $8,892,000 or 51% of
our total operating expenses incurred. For the year ended
December 31, 2008, total research and development expenses
were approximately $7,054,000, or 48%, of our total operating
expenses incurred, an increase of approximately $1,838,000, or
26%. Research and development expenses increased $1,623,000, or
32%, from $5,105,000 in the year ended December 31, 2008 to
$6,728,000 in the year ended December 31, 2009. This
increase was primarily due to increases in costs associated with
employee compensation, resulting from an increase in headcount
as well as an increase in patent costs related to patent
applications on internal discoveries.
Research
and development employee stock-based compensation
expense
Research and development employee stock-based compensation
expense increased $531,000, or 158%, in the year ended
December 31, 2009, compared with the year ended
December 31, 2008. This increase was due to an increase in
the number of outstanding common stock options during the year,
which include new common stock options issued to new employees
and to certain existing employees.
Research
and Development Non-Employee Stock-Based Compensation
Expense
We issued options to purchase shares of our common stock as
compensation to SAB members and consultants. For financial
statement purposes, we valued these shares at their fair value.
Fluctuations in non-employee stock-based compensation expense
results from variations in the number of common stock options
issued, vesting schedules and the Black-Scholes fair values of
common stock options granted to SAB members.
Research and development non-employee stock based compensation
expenses decreased $316,000, or 20%, from $1,613,000 for the
year ended December 31, 2008 to $1,297,000 for the year
ended December 31, 2009. The decrease was due to a decrease
in the Black-Scholes value of the founding SAB members’
2009 annual grant compared with the value of their 2008 annual
grant offset by the re-measurement of
Black-Scholes
fair value of the founding SAB members’ original grants
that vest on April 2012.
39
General
and Administrative Expense
| |
|
|
|
|
|
|
|
|
|
|
|
For the Years Ended
|
|
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
General and administrative expenses
|
|
$
|
5,483
|
|
|
$
|
4,874
|
|
|
Common stock warrants issued for general and administrative
expense
|
|
|
826
|
|
|
|
750
|
|
|
Fair value of common stock issued in exchange for general and
administrative expenses
|
|
|
281
|
|
|
|
—
|
|
|
Common stock and stock options issued for general and
administrative expense
|
|
|
2,038
|
|
|
|
1,875
|
|
|
|
|
|
|
|
|
|
|
|
|
Total general and administrative expense
|
|
$
|
8,628
|
|
|
$
|
7,499
|
|
|
|
|
|
|
|
|
|
|
|
General and administrative expenses include compensation-related
costs for our employees dedicated to general and administrative
activities, legal fees, audit and tax fees, consultants and
professional services, business development and corporate
partnership opportunities and general corporate expenses.
General and administrative expenses were $8,628,000 for the year
ended December 31, 2009 compared with $7,499,000 for the
year ended December 31, 2008. The increase of $1,129,000,
or 15%, was due to higher staff-related costs, including
$2,038,000 in non-cash share-based compensation expense, and
$826,000 in warrant expense related to a warrant issued for
business advisory services and $281,000 in common stock issued
as a commitment fee under the SEDA.
General and administrative expense as a percentage of total
operating expense for the years ended December 31, 2009 and
2008 was 49% and 52%, respectively. Although, we expect general
and administrative expense to increase for the foreseeable
future as we add personnel, the percentage of general and
administrative expense to total operating expense is expected to
decrease as our discovery and development activities for RNAi
therapeutics progress and expand.
From time to time, we expect to issue shares of our common stock
or warrants or options to purchase shares of our common stock to
consultants and other service providers in exchange for
services. For financial statement purposes, we will value these
shares of common stock, common stock options, and warrants at
their fair value, or at the value of the services received,
whichever is more reliably measurable.
Interest
Income
Interest income was negligible for the year ended
December 31, 2009, compared with approximately $180,000 for
the year ended December 31, 2008. This decrease was
primarily due to current interest rates available to us on our
cash and cash equivalents during the twelve months ended
December 31, 2009, as compared with the twelve months ended
December 31, 2008. The key objectives of our investment
policy are to preserve principal and ensure sufficient
liquidity, so our invested cash may not earn as high a level of
income as longer-term or higher risk securities, which generally
have less liquidity and more volatility. The interest rates
available on lower risk, shorter-term investments in
today’s market are lower than rates available in the prior
period. We expect to have interest income in future periods
based on our current account balances and potentially from
additional capital we may receive in the future.
Other
Expense
Other expenses were $862,000 for the year ended
December 31, 2009. Approximately $858,000 of this expense
relates to the change in fair value of warrants issued in
connection with the 2009 Offering.
Income
Taxes
There was no income tax expense for the years ended
December 31, 2009 and 2008 due to the fact that we have
incurred significant tax losses since we began operations. A tax
benefit would have been recorded for losses however, due to the
uncertainty of realizing these assets, a valuation allowance was
recognized which fully offset the deferred income tax assets.
40
Liquidity
and Capital Resources
We had cash and cash equivalents of approximately
$5.7 million as of December 31, 2009 and approximately
$14.4 million as of March 31, 2010.
During 2009 and to date in 2010, we have entered into the
following significant financing transactions:
On January 30, 2009, we entered into the SEDA, pursuant to
which we may, at our option over a two-year period, ending on
January 30, 2011, periodically sell to YA Global shares of
our common stock, for a total purchase price of up to
$25.0 million. To date we have not sold any shares under
the SEDA.
On August 4, 2009, we closed the 2009 Offering in which we
sold 2,385,715 shares of our common stock and warrants to
purchase 954,286 shares of our common stock and a at an
exercise price of $4.50 per share resulting in approximately
$7.8 million in net proceeds after deducting the placement
agent fee and offering expenses.
On March 26, 2010 we closed the 2010 Offering pursuant to
which we sold to certain investors 2,700,000 shares of
common stock at $6.00 per share and warrants to purchase
540,000 shares of common stock with an exercise price of
$6.00 per share. The financing provided approximately
$15.2 million in net proceeds to the Company after
deducting the placement agent fee and offering expenses.
Pursuant to a stock redemption agreement between us and CytRx
Corporation dated March 22, 2010, we were required to use
25% of the net proceeds from the offering to repurchase from
CytRx a number of shares of our common stock held by CytRx equal
to 25% of the shares sold by us in the offering. We are also
required to use 25% of the proceeds from the exercise of
warrants issued in the offering to repurchase from CytRx a
number of shares of our common stock held by CytRx equal to 25%
of the shares issued upon the exercise of such warrants. As
required by the agreement with CytRx, on March 29, 2010 we
repurchased 675,000 shares of our common stock from CytRx
for an aggregate price of approximately $3.8 million. We
estimate that we will repurchase an additional
135,000 shares of our common stock from CytRx for an
aggregate price of $0.8 million if all of the warrants
issued in the offering are exercised.
We have not had not generated any revenues since inception nor
are any revenues expected for the foreseeable future. We expect
to incur significant operating losses for the foreseeable future
while we advance our future product candidates from discovery
through pre-clinical studies and clinical trials and seek
regulatory approval and potential commercialization, even if we
are collaborating with pharmaceutical and larger biotechnology
companies. In addition to these increasing research and
development expenses, we expect general and administrative costs
to increase as we recruit additional management and
administrative personnel.
We believe that our existing cash and cash equivalents should be
sufficient to fund our operations through at least the first
half of 2011. In addition, we also have available to us the SEDA
which expires on January 30, 2011. In the future, we will
be dependent on obtaining funding from third parties such as
proceeds from the sale of equity, funded research and
development payments and payments under partnership and
collaborative agreements, in order to maintain our operations
and meet our obligations to licensors. There is no guarantee
that debt, additional equity or other funding will be available
to us on acceptable terms, or at all. If we fail to obtain
additional funding when needed, we would be forced to scale
back, or terminate, our operations or to seek to merge with or
to be acquired by another company.
Net
Cash Flow from Operating Activities
Net cash used in operating activities was approximately
$11,769,000 for the year ended December 31, 2009 compared
with $9,429,000 net cash used in operating activities for
the year ended December 31, 2008. The increase of
approximately $2,340,000 resulted primarily from a net loss of
$18,387,000, less the add back of non-cash items of $6,333,000,
of which $4,202,000 related to stock-based compensation,
$826,000 related to stock warrant expense in exchange for
services, $218,000 issued as a commitment fee in connection with
the SEDA, $162,000 related to depreciation and $285,000 related
to changes in current assets and liabilities and $858,000 that
reflects the fair value of warrants issued with the 2009
Offering.
41
Net
Cash Flow from Investing Activities
Net cash used in investing activities was approximately $83,000
for the year ended December 31, 2009, compared with net
cash provided by investing activities of $9,600,000 for the year
ended December 31, 2008. The decrease of approximately
$9,683,000 in cash provided by investing activities was
primarily due to net redemption of short-term investments
in 2008.
Net
Cash Flow from Financing Activities
Net cash provided by financing activities was $7,680,000 for the
year ended December 31, 2009, compared with $7,922,000 for
the year ended December 31, 2008. This decrease was
primarily due to net proceeds from the issuance of common stock
in the amount of $7,918,000 to institutional investors in the
second quarter of 2008 compared with net proceeds from the
issuance of common stock in the amount of $7,714,000 to
institutional investors in the third quarter of 2009.
Recently
Issued Accounting Standards
In June 2009, the FASB issued Accounting Standard Update
(“ASU”)
No. 2009-01,
Topic 105 — “Generally Accepted Accounting
Principles, amendments based on Statement of Financial
Accounting Standards (“SFAS”)
No. 168 — The FASB Accounting Standards
Codification and the Hierarchy of Generally Accepted Accounting
Principles” and establishes only two levels of
U.S. GAAP, authoritative and non-authoritative. The
amendments established the FASB Accounting Standards
Codification (the “Codification”) as the source of
authoritative accounting principles recognized by the FASB to be
applied by nongovernmental entities in the preparation of
financial statements in conformity with U.S. GAAP. Rules
and interpretive releases of the SEC under authority of federal
securities laws are also sources of authoritative U.S. GAAP
for SEC registrants. On the effective date of this statement,
the Codification superseded all then-existing non-SEC accounting
and reporting standards. All other non-grandfathered non-SEC
accounting literature not included in the Codification has
become non-authoritative. This statement is effective for
financial statements for interim or annual reporting periods
ending after September 15, 2009. We adopted this update
during the three-month period ended September 30, 2009. As
the Codification was not intended to change or alter existing
U.S. GAAP, the adoption of this update did not have any
impact on our financial position, results of operations or
cash flows.
In June 2009, the FASB ASC Topic 810 “Amendments to
FASB Interpretation No. 46(R)”,
(“ASC 810”), which amends the consolidation
guidance applicable to variable interest entities and is
effective as of January 1, 2010. We are currently in the
process of evaluating the impact of this pronouncement.
In June 2009, the FASB issued ASC Topic 860,
“Accounting for Transfers of Financial Assets”
(“ASC 860”), which eliminates the concept of a
qualifying special-purpose entity, changes the requirements for
derecognizing financial assets, and requires additional
disclosures in order to enhance information reported to users of
financial statements by providing greater transparency about
transfers of financial assets, including securitization
transactions, and an entity’s continuing involvement in and
exposure to the risks related to transferred financial assets.
This statement is effective for fiscal years beginning after
November 15, 2009. We are currently in the process of
evaluating the impact of ASC-860.
In May 2009, FASB ASC Topic 855, “Subsequent
Events” (“ASC 855”) was issued. This
statement sets forth (i) the period after the balance sheet
date during which management of a reporting entity should
evaluate events or transactions that may occur for potential
recognition or disclosure in the financial statements,
(ii) the circumstances under which an entity should
recognize events or transactions occurring after the balance
sheet date in its financial statements, and (iii) the
disclosures that an entity should make about events or
transactions that occurred after the balance sheet date. ASC 855
is effective for interim and annual periods ending after
June 15, 2009. We adopted ASC 855 in the quarter ending
June 30, 2009. The adoption of ASC 855 did not have any
impact on our financial position, results of operations or cash
flows. We have evaluated all events or transactions that
occurred after the period covered by this report, up through the
time of filing our annual
42
report on
Form 10-K
with the SEC. During this period that we did not have any
material recognizable subsequent events other than what is
disclosed in footnote 17.
In April 2009, the FASB issued the following: (i) FASB ASC
Topic
820-10-65,
“Determining Fair Value When the Volume and Level of
Activity for the Asset or Liability have Significantly Decreased
and Identifying Transactions That Are Not Orderly”
(“ASC
820-10-65”),
(ii) FASB ASC Topic
320-10-65,
“Recognition and Presentation of
Other-Than-Temporary
Impairment” (“ASC
320-10-65”),
and (iii) FASB ASC Topic
825-10-65,
“Interim Disclosures about Fair Value of Financial
Instruments” (“ASC
825-10-65”),
which was effective for interim and annual periods ending after
June 15, 2009. ASC
820-10-65
provides guidance on how to determine the fair value of assets
and liabilities under ASC
820-10 in
the current economic environment and reemphasizes that the
objective of a fair value measurement remains an exit price. If
we were to conclude that there has been a significant decrease
in the volume and level of activity of the asset or liability in
relation to normal market activities, quoted market values may
not be representative of fair value and we may conclude that a
change in valuation technique or the use of multiple valuation
techniques may be appropriate. ASC
820-10-65
modifies the requirements for recognizing
other-than-temporarily
impaired debt securities and revise the existing impairment
model for such securities, by modifying the current intent and
ability indicator in determining whether a debt security is
other-than-temporarily
impaired. ASC
825-10-65
enhances the disclosure of instruments under the scope of ASC
820 for both interim and annual periods. We are currently
evaluating these staff positions and the impact, if any, that
adoption will have on our financial position and results of
operation.
Effective January 1, 2009, we adopted FASB ASC Topic
815-40-15,
“Derivatives and Hedging Evaluating Whether an
Instrument is Indexed to an Entity’s Own Stock”
(“ASC
815-40-15”),
which addresses the accounting for certain instruments as
derivatives. Under ASC
815-40-15
specific guidance is provided regarding requirements for an
entity to consider embedded features as indexed to the
entity’s own stock. See footnote 6 for the impact the
adoption of ASC
815-40-15
had on our financial position, results of operations and cash
flows.
In December 2007, the FASB ratified the consensus reached by the
EITF on ASC Topic 808, “Accounting for
Collaborative Agreements” (“ASC 808-10-2”),
which provides the definition of a collaborative agreement and
ASC 808-10-15 provides guidelines to assist an entity in
determining whether or not it is a party in a collaborative
agreement. ASC 808-10-45 states that costs incurred and
revenues generated from transactions with third parties shall be
reported in accordance with ASC Topic 605-45, “Reporting
Revenue Gross as a Principal versus Net as an Agent,”
(“ASC 808-10-50”) also provides minimum
disclosure requirements for an entity’s collaboration
agreements and transition guidance. The adoption of ASC
Topic 808 did not have a material impact on our financial
position, results of operations and cash flows.
In June 2007, the Emerging Issues Task Force issued ASC Topic
730-20
“Research and Development Arrangements”
(“ASC
730-20”).
ASC 730-20
addresses the diversity which exists with respect to the
accounting for the non-refundable portion of a payment made by a
research and development entity for future research and
development activities. Under ASC
730-20, an
entity would defer and capitalize non-refundable advance
payments made for research and development activities until the
related goods are delivered or the related services are
performed subject to an assessment of recoverability. ASC
730-20 was
effective for fiscal years beginning after December 15,
2007 and interim periods within those years. The adoption of ASC
730-20 did
not have a material impact on our financial statements.
Off-Balance
Sheet Arrangements
In connection with certain license agreement, we are required to
indemnify the licensor for certain damages arising in connection
with the intellectual property rights licensed under the
agreement. In addition, we are a party to a number of agreements
entered into in the ordinary course of business that contain
typical provisions that obligate us to indemnify the other
parties to such agreements upon the occurrence of certain
events. These indemnification obligations are considered
off-balance sheet arrangements in accordance with ASC Topic 460
(“ASC 460”), “Guarantor’s Accounting and
Disclosure Requirements for Guarantees, Including Indirect
Guarantees of Indebtedness of Others.” To date, we have
not encountered material costs as
43
a result of such obligations and have not accrued any
liabilities related to such obligations and have not accrued any
liabilities related to such obligations in our financial
statements. See Note 9 to our financial statements included
in this annual report on
Form 10-K
for further discussion of these indemnification agreements.
On January 30, 2009, we entered into the SEDA with
YA Global pursuant to which we may, at our sole and
exclusive option, periodically sell to YA Global shares of
our common stock at a price based on it then current market
price for a total purchase price of up to $25.0 million.
Advance notices may be given to YA Global once every five
trading days, and advances shall not be more than $500,000. The
purchase price for shares of common stock shall be 95% of the
lowest volume weighted average price of the common stock during
the five consecutive trading days after the advance notice date.
YA Global is not obligated to fund any advance from us
until such time as a registration statement which registers the
resale of the shares of our common stock to be issued to
YA Global is declared effective by the SEC, which has not
yet occurred. The term of the SEDA is two years.
We issued YA Global an aggregate of 58,398 shares of our
common stock as a commitment fee in connection with the
transaction. RXi has also paid to Yorkville Advisors, LLC,
YA Global’s general partner, a due diligence and
structuring fee of $25,000.
|
|
|
ITEM 7A.
|
QUANTITATIVE
AND QUALITATIVE DISCLOSURE ABOUT MARKET RISK
|
Because we are a smaller reporting company, we are not required
to provide the information required by this Item.
44
|
|
|
ITEM 8.
|
FINANCIAL
STATEMENTS AND SUPPLEMENTARY DATA
|
RXi’s financial information as of December 31, 2008
and 2009 and for the years then ended and for the cumulative
financial information for the period from January 1, 2003
(date of inception) to December 31, 2009 have been audited
by our independent registered public accounting firm, BDO
Seidman, LLP.
| |
|
|
|
|
|
|
|
Page No.
|
|
|
|
Index to Financial Statements
|
|
|
|
|
|
|
|
|
46
|
|
|
|
|
|
47
|
|
|
|
|
|
48
|
|
|
|
|
|
49
|
|
|
|
|
|
51
|
|
|
|
|
|
53
|
|
45
REPORT OF
INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Board of Directors and Stockholders
RXi Pharmaceuticals Corporation
Worcester, Massachusetts
We have audited the accompanying balance sheets of RXi
Pharmaceuticals Corporation (a development stage Company) as of
December 31, 2009 and 2008 and the related statements of
expenses, stockholders’ equity and cash flows for the years
then ended and for the period from January 1, 2003 (date of
inception) to December 31, 2009. These financial statements
are the responsibility of the Company’s management. Our
responsibility is to express an opinion on these financial
statements based on our audits.
We conducted our audits in accordance with the standards of the
Public Company Accounting Oversight Board (United States). Those
standards require that we plan and perform the audit to obtain
reasonable assurance about whether the financial statements are
free of material misstatement. The Company is not required to
have, nor were we engaged to perform, an audit of its internal
control over financial reporting. Our audits included
consideration of internal control over financial reporting as a
basis for designing audit procedures that are appropriate in the
circumstances, but not for the purpose of expressing an opinion
on the effectiveness of the Company’s internal control over
financial reporting. Accordingly, we express no such opinion. An
audit also includes examining, on a test basis, evidence
supporting the amounts and disclosures in the financial
statements, assessing the accounting principles used and
significant estimates made by management, as well as evaluating
the overall presentation of the financial statements. We believe
that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above
present fairly, in all material respects, the financial position
of RXi Pharmaceuticals Corporation as of December 31, 2009
and 2008 and the results of its operations and its cash flows
for the years then ended and for the period from January 1,
2003 (date of inception) to December 31, 2009, in
conformity with accounting principles generally accepted in the
United States of America.
Boston, Massachusetts
March 31, 2010
46
RXi
PHARMACEUTICALS CORPORATION
(A Development Stage Company)
BALANCE SHEETS
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(Amounts in thousands, except share data)
|
|
|
|
|
ASSETS
|
|
Current assets:
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents
|
|
$
|
5,684
|
|
|
$
|
9,856
|
|
|
Prepaid expenses
|
|
|
120
|
|
|
|
73
|
|
|
|
|
|
|
|
|
|
|
|
|
Total current assets
|
|
|
5,804
|
|
|
|
9,929
|
|
|
|
|
|
|
|
|
|
|
|
|
Equipment and furnishings, net of accumulated depreciation and
amortization of $320 and $158 in 2009 and 2008, respectively
|
|
|
432
|
|
|
|
414
|
|
|
Deposits
|
|
|
16
|
|
|
|
16
|
|
|
|
|
|
|
|
|
|
|
|
|
Total assets
|
|
$
|
6,252
|
|
|
$
|
10,359
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
LIABILITIES AND STOCKHOLDERS’ EQUITY
|
|
Current liabilities:
|
|
|
|
|
|
|
|
|
|
Accounts payable
|
|
$
|
625
|
|
|
$
|
394
|
|
|
Accrued expense and other current liabilities
|
|
|
1,077
|
|
|
|
976
|
|
|
Current maturities of capital lease obligations
|
|
|
52
|
|
|
|
17
|
|
|
Warrants potentially settleable in cash
|
|
|
3,721
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
Total current liabilities
|
|
|
5,475
|
|
|
|
1,387
|
|
|
Capital lease obligations, net of current maturities
|
|
|
36
|
|
|
|
4
|
|
|
|
|
|
|
|
|
|
|
|
|
Total liabilities
|
|
|
5,511
|
|
|
|
1,391
|
|
|
|
|
|
|
|
|
|
|
|
|
Commitments and contingencies (Notes 6, 8 & 14)
|
|
|
|
|
|
|
|
|
|
Stockholders’ equity:
|
|
|
|
|
|
|
|
|
|
Preferred stock, $0.0001 par value; 5,000,000 shares
authorized; no shares issued and outstanding
|
|
|
—
|
|
|
|
—
|
|
|
Common stock, $0.0001 par value; 50,000,000 shares
authorized; 16,207,625 and 13,763,231 shares issued and
outstanding in 2009 and 2008, respectively
|
|
|
2
|
|
|
|
1
|
|
|
Additional paid-in capital
|
|
|
44,489
|
|
|
|
34,330
|
|
|
Deficit accumulated during the developmental stage
|
|
|
(43,750
|
)
|
|
|
(25,363
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Total stockholders’ equity
|
|
|
741
|
|
|
|
8,968
|
|
|
|
|
|
|
|
|
|
|
|
|
Total liabilities and stockholders’ equity
|
|
$
|
6,252
|
|
|
$
|
10,359
|
|
|
|
|
|
|
|
|
|
|
|
See accompanying notes to financial statements.
47
RXi
PHARMACEUTICALS CORPORATION
(A Development Stage Company)
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from
|
|
|
|
|
|
|
|
|
|
|
January 1,
|
|
|
|
|
|
|
|
|
|
|
2003 (Date of
|
|
|
|
|
Year Ended
|
|
|
Year Ended
|
|
|
Inception) to
|
|
|
|
|
December 31,
|
|
|
December 31,
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2009
|
|
|
|
|
(Amounts in thousands, except share and per share data)
|
|
|
|
|
Expenses:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development expense
|
|
$
|
6,728
|
|
|
$
|
5,105
|
|
|
$
|
20,637
|
|
|
Research and development employee stock-based compensation
expense
|
|
|
867
|
|
|
|
336
|
|
|
|
1,323
|
|
|
Research and development non-employee stock-based compensation
expense
|
|
|
1,297
|
|
|
|
1,613
|
|
|
|
5,320
|
|
|
Fair value of common stock issued in exchange for licensing
rights
|
|
|
—
|
|
|
|
—
|
|
|
|
3,954
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total research and development expense
|
|
|
8,892
|
|
|
|
7,054
|
|
|
|
31,234
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
General and administrative
|
|
|
5,483
|
|
|
|
4,874
|
|
|
|
15,617
|
|
|
Fair value of common stock warrants issued for general and
administrative expenses
|
|
|
826
|
|
|
|
750
|
|
|
|
1,576
|
|
|
Fair value of common stock issued in exchange for general and
administrative expenses
|
|
|
281
|
|
|
|
—
|
|
|
|
281
|
|
|
General and administrative employee stock-based compensation
|
|
|
2,038
|
|
|
|
1,875
|
|
|
|
4,844
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total general and administrative expense
|
|
|
8,628
|
|
|
|
7,499
|
|
|
|
22,318
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses
|
|
|
(17,520
|
)
|
|
|
(14,553
|
)
|
|
|
(53,552
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest income (expense)
|
|
|
(5
|
)
|
|
|
180
|
|
|
|
623
|
|
|
Other income (expense)
|
|
|
(862
|
)
|
|
|
—
|
|
|
|
(862
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss before provision for income taxes
|
|
|
(18,387
|
)
|
|
|
(14,373
|
)
|
|
|
(53,791
|
)
|
|
Provision for income taxes
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
$
|
(18,387
|
)
|
|
$
|
(14,373
|
)
|
|
$
|
(53,791
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss per common share:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted loss per share
|
|
$
|
(1.24
|
)
|
|
$
|
(1.09
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average common shares outstanding: basic and diluted
|
|
|
14,796,541
|
|
|
|
13,239,942
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
See accompanying notes to financial statements.
48
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Deficit
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additional
|
|
|
During
|
|
|
Parent
|
|
|
|
|
|
|
|
Common Stock
|
|
|
Paid-In
|
|
|
Development
|
|
|
Company’s
|
|
|
|
|
|
|
|
Shares Issued
|
|
|
Amount
|
|
|
Capital
|
|
|
Stage
|
|
|
Net Deficit
|
|
|
Total
|
|
|
|
|
(Amounts in thousands, except share data)
|
|
|
|
|
Predecessor
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2003
|
|
|
—
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
$
|
(89
|
)
|
|
$
|
(89
|
)
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
(3,272
|
)
|
|
|
(3,272
|
)
|
|
Net transactions with Parent
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
2,393
|
|
|
|
2,393
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2004
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
(968
|
)
|
|
|
(968
|
)
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
(2,209
|
)
|
|
|
(2,209
|
)
|
|
Net transactions with Parent
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
2,727
|
|
|
|
2,727
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2005
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
(450
|
)
|
|
|
(450
|
)
|
|
Net Loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
(2,405
|
)
|
|
|
(2,405
|
)
|
|
Net transactions with Parent
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
2,587
|
|
|
|
2,587
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2006
|
|
|
—
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
$
|
(268
|
)
|
|
$
|
(268
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Successor
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at April 3, 2006
|
|
|
—
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
$
|
—
|
|
|
Issuance of common stock
|
|
|
1,624,278
|
|
|
|
—
|
|
|
|
2
|
|
|
|
—
|
|
|
|
|
|
|
|
2
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2006
|
|
|
1,624,278
|
|
|
|
—
|
|
|
|
2
|
|
|
|
—
|
|
|
|
|
|
|
|
2
|
|
|
Common stock issued to CytRx for contribution of RXi and other
assets
|
|
|
7,040,318
|
|
|
|
1
|
|
|
|
47
|
|
|
|
—
|
|
|
|
|
|
|
|
48
|
|
|
Common stock issued for cash
|
|
|
3,273,292
|
|
|
|
—
|
|
|
|
15,348
|
|
|
|
—
|
|
|
|
|
|
|
|
15,348
|
|
|
Common stock issued to CytRx for reimbursement of expenses
|
|
|
188,387
|
|
|
|
—
|
|
|
|
978
|
|
|
|
—
|
|
|
|
|
|
|
|
978
|
|
|
Expenses incurred by CytRx for RXi
|
|
|
—
|
|
|
|
—
|
|
|
|
831
|
|
|
|
—
|
|
|
|
|
|
|
|
831
|
|
|
Common stock issued to UMMS for additional intellectual
properties
|
|
|
462,112
|
|
|
|
—
|
|
|
|
2,311
|
|
|
|
—
|
|
|
|
|
|
|
|
2,311
|
|
|
Common stock issued to directors
|
|
|
30,000
|
|
|
|
—
|
|
|
|
150
|
|
|
|
—
|
|
|
|
|
|
|
|
150
|
|
|
Common stock issued upon exercise of stock options
|
|
|
66,045
|
|
|
|
—
|
|
|
|
331
|
|
|
|
—
|
|
|
|
|
|
|
|
331
|
|
|
Stock based compensation for directors and employees
|
|
|
—
|
|
|
|
—
|
|
|
|
1,048
|
|
|
|
—
|
|
|
|
|
|
|
|
1,048
|
|
|
Stock based compensation expense for services
|
|
|
—
|
|
|
|
—
|
|
|
|
766
|
|
|
|
—
|
|
|
|
|
|
|
|
766
|
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(10,990
|
)
|
|
|
|
|
|
|
(10,990
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2007
|
|
|
12,684,432
|
|
|
|
1
|
|
|
|
21,812
|
|
|
|
(10,990
|
)
|
|
|
|
|
|
|
10,823
|
|
49
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Deficit
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additional
|
|
|
During
|
|
|
Parent
|
|
|
|
|
|
|
|
Common Stock
|
|
|
Paid-In
|
|
|
Development
|
|
|
Company’s
|
|
|
|
|
|
|
|
Shares Issued
|
|
|
Amount
|
|
|
Capital
|
|
|
Stage
|
|
|
Net Deficit
|
|
|
Total
|
|
|
|
|
(Amounts in thousands, except share data)
|
|
|
|
|
Issuance of common stock, net of offering costs of $796
|
|
|
1,073,299
|
|
|
|
—
|
|
|
|
7,918
|
|
|
|
—
|
|
|
|
|
|
|
|
7,918
|
|
|
Common stock issued upon exercise of stock options
|
|
|
5,500
|
|
|
|
—
|
|
|
|
26
|
|
|
|
—
|
|
|
|
|
|
|
|
26
|
|
|
Stock based compensation for directors and employees
|
|
|
—
|
|
|
|
—
|
|
|
|
2,211
|
|
|
|
—
|
|
|
|
|
|
|
|
2,211
|
|
|
Stock based compensation expense for services
|
|
|
—
|
|
|
|
—
|
|
|
|
1,613
|
|
|
|
—
|
|
|
|
|
|
|
|
1,613
|
|
|
Common stock warrant expense in exchange for services
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
750
|
|
|
|
|
|
|
|
750
|
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(14,373
|
)
|
|
|
|
|
|
|
(14,373
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2008
|
|
|
13,763,231
|
|
|
|
1
|
|
|
|
34,330
|
|
|
|
(25,363
|
)
|
|
|
|
|
|
|
8,968
|
|
|
Issuance of common stock, net of offering costs of $636
|
|
|
2,385,715
|
|
|
|
1
|
|
|
|
7,713
|
|
|
|
—
|
|
|
|
|
|
|
|
7,714
|
|
|
Common stock warrants issued in connection with the 2009 Offering
|
|
|
—
|
|
|
|
—
|
|
|
|
(2,863
|
)
|
|
|
—
|
|
|
|
|
|
|
|
(2,863
|
)
|
|
Common stock issued upon exercise of stock options
|
|
|
281
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
—
|
|
|
Common stock issued as commitment fee in connection with SEDA
|
|
|
58,398
|
|
|
|
—
|
|
|
|
281
|
|
|
|
—
|
|
|
|
|
|
|
|
281
|
|
|
Stock based compensation for directors and employees
|
|
|
—
|
|
|
|
—
|
|
|
|
2,906
|
|
|
|
—
|
|
|
|
|
|
|
|
2,906
|
|
|
Stock based compensation expense for services
|
|
|
—
|
|
|
|
—
|
|
|
|
1,296
|
|
|
|
—
|
|
|
|
|
|
|
|
1,296
|
|
|
Common stock warrant expense in exchange for services
|
|
|
|
|
|
|
—
|
|
|
|
826
|
|
|
|
—
|
|
|
|
|
|
|
|
826
|
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(18,387
|
)
|
|
|
|
|
|
|
(18,387
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2009
|
|
|
16,207,625
|
|
|
|
2
|
|
|
$
|
44,489
|
|
|
$
|
(43,750
|
)
|
|
|
|
|
|
$
|
741
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
See accompanying notes to financial statements.
50
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from
|
|
|
|
|
|
|
|
|
|
|
January 1,
|
|
|
|
|
|
|
|
|
|
|
2003 (Date of
|
|
|
|
|
|
|
|
|
|
|
Inception)
|
|
|
|
|
Year Ended
|
|
|
Year Ended
|
|
|
through
|
|
|
|
|
December 31,
|
|
|
December 31,
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2009
|
|
|
|
|
(Amounts in thousands)
|
|
|
|
|
Cash flows from operating activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
$
|
(18,387
|
)
|
|
$
|
(14,373
|
)
|
|
$
|
(53,791
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Adjustment to reconcile net loss to net cash used in operating
activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Depreciation and amortization expense
|
|
|
162
|
|
|
|
131
|
|
|
|
329
|
|
|
Loss on disposal of equipment
|
|
|
4
|
|
|
|
8
|
|
|
|
12
|
|
|
Non-cash rent expense
|
|
|
—
|
|
|
|
29
|
|
|
|
29
|
|
|
Accretion and receipt of bond discount
|
|
|
—
|
|
|
|
207
|
|
|
|
35
|
|
|
Non-cash share based compensation
|
|
|
4,202
|
|
|
|
3,824
|
|
|
|
11,489
|
|
|
Fair value of common stock warrants issued in exchange for
services
|
|
|
826
|
|
|
|
750
|
|
|
|
1,576
|
|
|
Fair value of common stock issued as commitment fee in
connection with SEDA
|
|
|
281
|
|
|
|
—
|
|
|
|
281
|
|
|
Change in fair value of common stock warrants issued in
connection with the 2009 offering
|
|
|
858
|
|
|
|
—
|
|
|
|
858
|
|
|
Fair value of common stock issued in exchange for licensing
rights
|
|
|
—
|
|
|
|
—
|
|
|
|
3,954
|
|
|
Changes in assets and liabilities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Prepaid expenses
|
|
|
(47
|
)
|
|
|
(51
|
)
|
|
|
(120
|
)
|
|
Accounts payable
|
|
|
231
|
|
|
|
339
|
|
|
|
625
|
|
|
Due to former parent
|
|
|
—
|
|
|
|
(207
|
)
|
|
|
(207
|
)
|
|
Accrued expenses and other current liabilities
|
|
|
101
|
|
|
|
(86
|
)
|
|
|
1,077
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash used in operating activities
|
|
|
(11,769
|
)
|
|
|
(9,429
|
)
|
|
|
(33,853
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash flows from investing activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Purchase of short-term investments
|
|
|
—
|
|
|
|
(19,785
|
)
|
|
|
(31,542
|
)
|
|
Maturities of short-term investments
|
|
|
—
|
|
|
|
29,530
|
|
|
|
31,507
|
|
|
Cash paid for purchase of equipment and furnishings
|
|
|
(82
|
)
|
|
|
(166
|
)
|
|
|
(580
|
)
|
|
Disposal of equipment and furnishings
|
|
|
(1
|
)
|
|
|
—
|
|
|
|
(1
|
)
|
|
Cash refunded (paid) for lease deposit
|
|
|
—
|
|
|
|
21
|
|
|
|
(45
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash provided by (used in) investing activities
|
|
|
(83
|
)
|
|
|
9,600
|
|
|
|
(661
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash flows from financing activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net proceeds from issuance of common stock
|
|
|
7,714
|
|
|
|
7,918
|
|
|
|
31,132
|
|
|
Net proceeds from exercise of common stock options
|
|
|
—
|
|
|
|
26
|
|
|
|
356
|
|
|
Repayments of capital lease obligations
|
|
|
(34
|
)
|
|
|
(22
|
)
|
|
|
(56
|
)
|
|
Cash advances from Parent, net
|
|
|
|
|
|
|
|
|
|
|
8,766
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash provided by financing activities
|
|
|
7,680
|
|
|
|
7,922
|
|
|
|
40,198
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net increase in cash and cash equivalents
|
|
|
(4,172
|
)
|
|
|
8,093
|
|
|
|
5,684
|
|
|
Cash and cash equivalents at the beginning of period
|
|
|
9,856
|
|
|
|
1,763
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents at end of period
|
|
$
|
5,684
|
|
|
$
|
9,856
|
|
|
$
|
5,684
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Supplemental disclosure of cash flow information:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash received during the periods for interest
|
|
|
1
|
|
|
$
|
449
|
|
|
$
|
724
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash paid during the periods for interest
|
|
$
|
—
|
|
|
$
|
7
|
|
|
$
|
7
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
51
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from
|
|
|
|
|
|
|
|
|
|
|
January 1,
|
|
|
|
|
|
|
|
|
|
|
2003 (Date of
|
|
|
|
|
|
|
|
|
|
|
Inception)
|
|
|
|
|
Year Ended
|
|
|
Year Ended
|
|
|
through
|
|
|
|
|
December 31,
|
|
|
December 31,
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2009
|
|
|
|
|
(Amounts in thousands)
|
|
|
|
|
Supplemental disclosure of non-cash investing and financing
activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Settlement of corporate formation expenses in exchange for
common stock
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
978
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Fair value of warrants issued in connection with common stock
recorded as cost of equity
|
|
$
|
2,863
|
|
|
$
|
—
|
|
|
$
|
2,863
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Allocation of management expenses
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
551
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Equipment and furnishings exchanged for common stock
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
48
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Acquisition of equipment and furnishings through accrued
liabilities
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Equipment and furnishings acquired through capital lease
|
|
$
|
101
|
|
|
$
|
43
|
|
|
$
|
144
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Non-cash lease deposit
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
50
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
See accompanying notes to financial statements.
52
RXi Pharmaceuticals Corporation (“RXi” or the
“Company”) was formed by CytRx Corporation
(“CytRx” or the “Former Parent”) and four
prominent RNAi researchers, including Craig C.
Mello, Ph.D., who was awarded the 2006 Nobel Prize in
Medicine for his co-discovery of RNAi. The purpose of forming
RXi was to pursue the development of proprietary therapeutics
based on RNAi for the treatment of human diseases. By utilizing
the Company’s expertise in RNAi and the comprehensive RNAi
technology platform it has established, the Company believes it
will be able to discover and develop lead compounds and progress
them into and through clinical development for potential
commercialization more efficiently than traditional drug
development approaches, primarily in partnerships with
pharmaceutical and biotechnology companies.
RXi was incorporated as Argonaut Pharmaceuticals, Inc., in
Delaware, on April 3, 2006. The Company changed its name to
RXi Pharmaceuticals Corporation on November 28, 2006. From
April 3, 2006 (date of incorporation) until January 8,
2007, no business was conducted at the RXi level. On
January 8, 2007, RXi entered into a contribution agreement
with CytRx under which CytRx assigned and contributed to RXi
substantially all of its RNAi-related technologies and assets
and commenced operations; these contributed assets were recorded
by RXi at the historical cost basis of $48,000.
Because the RNAi activities prior to 2007 were conducted by
CytRx, the financial statements of RXi for the periods through
December 31, 2006 have been disaggregated, or
“carved-out,” of the financial statements of CytRx.
These carved-out financial statements form what are referred to
herein as the financial statements of the
“Predecessor,” and include both direct and indirect
expenses. The historical direct expenses consist primarily of
the various costs for technology license agreements, sponsored
research agreements and fees paid to scientific advisors.
Indirect expenses during this period represent expenses incurred
by CytRx on behalf of RXi, including salary, benefits, rent,
accounting and other general and administrative expenses that
have been allocated to RXi based upon estimates of the
percentage of time spent by individual CytRx employees working
on RXi matters. Management believes the assumptions underlying
the allocations of indirect expenses in the carve-out financial
information are reasonable; however, RXi’s financial
position, results of operations and cash flows may have been
materially different if it was operated as a stand-alone entity
as of and for the periods ended December 31, 2007.
RXi’s financial information from January 8, 2007 is
referred to in these financial statements as the financial
information of the “Successor” and includes expenses
incurred by RXi in its RNAi therapeutic programs, as well as an
allocation of indirect expenses relating to corporate services
provided by CytRx through December 31, 2007. In addition,
the net intercompany activities between Predecessor and CytRx
have been accumulated into a single caption entitled
“Parent Company’s Net Deficit.”
To date, RXi’s principal activities have consisted of
conducting discovery research and pre-clinical development
activities utilizing the Company’s RNAi therapeutic
platform, acquiring RNAi technologies and patent rights through
exclusive, co-exclusive and non-exclusive licenses, recruiting
an RNAi-focused management and scientific/clinical advisory
team, capital raising activities and conducting business
development activities aimed at establishing research and
development partnerships with pharmaceutical and larger
biotechnology companies.
As the Company has not generated any revenues from inception
through December 31, 2009, the Company is considered a
development-stage company for accounting purposes.
The Company had cash and cash equivalents of approximately
$5.7 million as of December 31, 2009 and approximately
$14.4 million as of March 31, 2010.
53
RXi
PHARMACEUTICALS CORPORATION
NOTES TO
FINANCIAL STATEMENTS — (Continued)
During 2009 and to date in 2010, the Company entered into the
following significant financing transactions:
On January 30, 2009, the Company entered into the SEDA,
pursuant to which the Company may, at its option over a two-year
period, ending on January 30, 2011, periodically sell to YA
Global shares of the Company’s common stock, for a total
purchase price of up to $25.0 million. To date no shares
have been sold under the SEDA.
On August 4, 2009, the Company closed the 2009 Offering in
which it sold 2,385,715 shares of its common stock and
warrants to purchase 954,286 shares of its common stock and
a at an exercise price of $4.50 per share resulting in
approximately $7.8 million in net proceeds after deducting
the placement agent fee and offering expenses.
On March 26, 2010, the Company closed a registered direct
financing (the “2010 Offering”) pursuant to which it
sold to certain investors 2,700,000 shares of common stock
at $6.00 per share and warrants to purchase 540,000 shares
of common stock with an exercise price of $6.00 per share. The
financing provided approximately $15.2 million in net
proceeds to the Company after deducting the placement agent fee
and offering expenses. Pursuant to a stock redemption agreement
between us and CytRx Corporation dated March 22, 2010, the
Company is required to use 25% of the net proceeds from the
offering to repurchase from CytRx a number of shares of its
common stock held by CytRx equal to 25% of the shares sold by th
e Company in the offering. The Company is also required to use
25% of the proceeds from the exercise of warrants issued in the
offering to repurchase from CytRx a number of shares of its
common stock held by CytRx equal to 25% of the shares issued
upon the exercise of such warrants. As required by the agreement
with CytRx, on March 31, 2010 the Company repurchased
675,000 shares of its common stock from CytRx for an
aggregate price of approximately $3.8 million. The Company
estimates that it will repurchase an additional
135,000 shares of its common stock from CytRx for an
aggregate price of $0.8 million if all of the warrants
issued in the offering are exercised.
We have not had not generated any revenues since inception nor
are any revenues expected for the foreseeable future. We expect
to incur significant operating losses for the foreseeable future
while we advance our future product candidates from discovery
through pre-clinical studies and clinical trials and seek
regulatory approval and potential commercialization, even if we
are collaborating with pharmaceutical and larger biotechnology
companies. In addition to these increasing research and
development expenses, we expect general and administrative costs
to increase as we recruit additional management and
administrative personnel.
We believe that our existing cash and cash equivalents should be
sufficient to fund our operations through at least the first
half of 2011. In addition, we also have available to us the SEDA
which expires on January 30, 2011. In the future, we will
be dependent on obtaining funding from third parties such as
proceeds from the sale of equity, funded research and
development payments and payments under partnership and
collaborative agreements, in order to maintain our operations
and meet our obligations to licensors. There is no guarantee
that debt, additional equity or other funding will be available
to us on acceptable terms, or at all. If we fail to obtain
additional funding when needed, we would be forced to scale
back, or terminate, our operations or to seek to merge with or
to be acquired by another company.
The Company believes that its existing cash, is sufficient to
fund operations through at least the first half of 2011. In the
future, the Company will be dependent on obtaining funding from
third parties such as proceeds from the sale of equity, funded
research and development payments and payments under partnership
and collaborative agreements, in order to maintain its
operations. There is no guarantee that debt, additional equity
or other funding will be available to the Company on acceptable
terms, or at all. If the Company fails to obtain additional
funding when needed, it would be forced to scale back, or
terminate, its operations or to seek to merge with or to be
acquired by another company.
54
RXi
PHARMACEUTICALS CORPORATION
NOTES TO
FINANCIAL STATEMENTS — (Continued)
The Company expects to incur significant operating losses for
the foreseeable future while it advances its future product
candidates from discovery through pre-clinical studies and
clinical trials and seek regulatory approval and potential
commercialization, even if the Company is collaborating with
pharmaceutical and larger biotechnology companies. In addition
to these increasing research and development expenses, the
Company expects general and administrative costs to increase as
it recruits additional management and administrative personnel.
The Company will need to generate significant revenues to
achieve profitability and may never do so.
|
|
|
2.
|
Summary
of Significant Accounting Policies
|
Uses of estimates in preparation of financial
statements — The preparation of financial
statements in accordance with accounting principles generally
accepted in the United States of America requires management to
make estimates and assumptions that affect the reported amounts
of assets and liabilities and disclosure of contingent assets
and liabilities at the date of the financial statements and the
reported amounts of revenues and expenses during the reporting
period. Actual results could differ materially from these
estimates.
Cash and Cash Equivalents — The Company
considers all highly-liquid debt instruments with an original
maturity of 90 days or less to be cash equivalents. Cash
equivalents consist primarily of amounts invested in money
market accounts.
Fair Value of Financial Instruments — The
carrying amounts reported in the balance sheet for cash
equivalents, accounts payable and accrued liabilities
approximate their fair values due to their short-term nature.
Equipment and Furnishings — Equipment and
furnishings are stated at cost and depreciated using the
straight-line method based on the estimated useful lives
(generally three to five years for equipment and furniture) of
the related assets.
Depreciation and amortization expense for the year ended
December 31, 2009 and 2008 was approximately $162,000 and
$131,000, respectively.
Impairment of Long-Lived Assets — The Company
reviews long-lived assets, including finite lived intangible
assets, for impairment on an annual basis, as of
December 31, or on an interim basis if an event occurs that
might reduce the fair value of such assets below their carrying
values. An impairment loss would be recognized based on the
difference between the carrying value of the asset and its
estimated fair value, which would be determined based on either
discounted future cash flows or other appropriate fair value
methods. The Company believes no impairment existed as of
December 31, 2009.
Patents and Patent Application Costs — Although
the Company believes that its patents and underlying technology
have continuing value, the amount of future benefits to be
derived from the patents is uncertain. Patent costs are,
therefore, expensed as incurred.
Share-based Compensation — The Company follows
the provisions of the Financial Accounting Standards Board
(“FASB”) Accounting Standards Codification
(“ASC”) Topic 718, “Compensation —
Stock Compensation” (“ASC 718”), which
requires the measurement and recognition of compensation expense
for all share-based payment awards made to employees,
non-employee directors, and consultants, including employee
stock options. Stock compensation expense based on the grant
date fair value estimated in accordance with the provisions of
ASC 718 is recognized as an expense over the requisite service
period.
For stock options granted as consideration for services rendered
by non-employees, the Company recognizes compensation expense in
accordance with the requirements of FASB ASC Topic
505-50
(“ASC 505-50”)
“Equity Based Payments to Non-Employees”.
Non-employee option grants that do not vest immediately upon
grant are recorded as an expense over the vesting period of the
underlying stock options. At
55
RXi
PHARMACEUTICALS CORPORATION
NOTES TO
FINANCIAL STATEMENTS — (Continued)
the end of each financial reporting period prior to vesting, the
value of these options, as calculated using the Black-Scholes
option-pricing model, will be re-measured using the fair value
of the Company’s common stock and the non-cash compensation
recognized during the period will be adjusted accordingly. Since
the fair market value of options granted to non-employees is
subject to change in the future, the amount of the future
compensation expense will include fair value re-measurements
until the stock options are fully vested.
The Company recognized $1.3 million and $1.6 million
of stock based compensation expense related to non-employee
stock options for the years ended December 31, 2009 and
2008, respectively.
Derivative Financial Instruments — During the
normal course of business, from time to time, the Company issues
warrants and options to vendors as consideration to perform
services. It may also issue warrants as part of a debt or equity
financing. The Company does not enter into any derivative
contracts for speculative purposes.
The Company recognizes all derivatives as assets or liabilities
measured at fair value with changes in fair value of derivatives
reflected as current period income or loss unless the
derivatives qualify for hedge accounting and are accounted for
as such. During the year ended December 31, 2009, the
Company issued warrants to purchase 978,142 shares of
common stock in connection with an equity transaction. In
accordance with FASB ASC Topic
815-40,
“Derivatives and Hedging — Contracts in
Entity’s Own Stock”
(“ASC 815-40”),
the value of these warrants is required to be recorded as a
liability, as the holders have an option to put the warrants
back to the Company in certain events, as defined.
As of December 31, 2009, the value of these warrants is
approximately $3.7 million and is recorded as a current
liability on the accompanying balance sheet.
Research and Development Expenses — Research
and development costs are expensed as incurred. Included in
research and development costs are wages, benefits and other
operating costs, facilities, supplies, external services and
overhead directly related to the Company’s research and
development departments as well as costs to acquire technology
licenses.
Income Taxes — The Company recognizes
liabilities or assets for the deferred tax consequences of
temporary differences between the tax basis of assets or
liabilities and their reported amounts in the financial
statements in accordance with ASC
740-10,
“Accounting for Income Taxes” (“ASC
740-10”).
These temporary differences will result in taxable or deductible
amounts in future years when the reported amounts of the assets
or liabilities are recovered or settled. ASC
740-10
requires that a valuation allowance be established when
management determines that it is more likely than not that all
or a portion of a deferred asset will not be realized. RXi
evaluates the realizability of its net deferred income tax
assets and valuation allowances as necessary, at least on an
annual basis. During this evaluation, the Company reviews its
forecasts of income in conjunction with other positive and
negative evidence surrounding the realizability of its deferred
income tax assets to determine if a valuation allowance is
required. Adjustments to the valuation allowance will increase
or decrease the Company’s income tax provision or benefit.
The recognition and measurement of benefits related to the
Company’s tax positions requires significant judgment, as
uncertainties often exist with respect to new laws, new
interpretations of existing laws, and rulings by taxing
authorities. Differences between actual results and RXi’s
assumptions or changes in the Company’s assumptions in
future periods are recorded in the period they become known
Concentrations of Credit Risk — Financial
instruments that potentially subject the Company to significant
concentrations of credit risk consist principally of cash and
cash equivalents. The Company maintains cash balances in several
accounts with one bank, which at times are in excess of
federally insured limits. As of December 31, 2009, the
Company’s cash equivalents were invested in money market
mutual funds. The Company’s investment policy disallows
investment in any debt securities rated less than
“investment grade” by national ratings services. The
Company has not experienced any losses on its deposits of cash
and cash equivalents or its short-term investments.
56
RXi
PHARMACEUTICALS CORPORATION
NOTES TO
FINANCIAL STATEMENTS — (Continued)
Comprehensive Loss — The Company’s
comprehensive loss is equal to its net loss for all periods
presented.
Parent Company’s Net Deficit — The Parent
Company’s Net Deficit of the Predecessor consists of
CytRx’s initial investment in RXi and subsequent changes in
RXi’s net investment resulting from RXi being an integrated
part of CytRx. All disbursements for the Predecessor were made
by CytRx.
|
|
|
3.
|
Recent
Accounting Pronouncements
|
In June 2009, the FASB issued ASC Topic 105, “Generally
Accepted Accounting Principles, amendments based on Statement of
Financial Accounting Standards (“SFAS”)
No. 168 — The FASB Accounting Standards
Codification and the Hierarchy of Generally Accepted Accounting
Principles”, (“ASC 105”) which establishes
only two levels of U.S. GAAP, authoritative and
non-authoritative. The amendments established the FASB
Accounting Standards Codification (the “Codification”)
as the source of authoritative accounting principles recognized
by the FASB to be applied by nongovernmental entities in the
preparation of financial statements in conformity with
U.S. GAAP. Rules and interpretive releases of the SEC under
authority of federal securities laws are also sources of
authoritative U.S. GAAP for SEC registrants. On the
effective date of this statement, the Codification superseded
all then-existing non-SEC accounting and reporting standards.
All other non-grandfathered non-SEC accounting literature not
included in the Codification has become non-authoritative. This
statement is effective for financial statements for interim or
annual reporting periods ending after September 15, 2009.
The Company adopted this update during the three-month period
ended September 30, 2009. As the Codification was not
intended to change or alter existing U.S. GAAP, the
adoption of ASC 105 did not have any impact on the
Company’s financial position, results of operations or cash
flows.
In June 2009, the FASB ASC Topic 810 “Amendments to FASB
Interpretation No. 46R”, (“ASC 810”),
which amends the consolidation guidance applicable to variable
interest entities and is effective as of January 1, 2010.
The Company is currently in the process of evaluating the impact
of this pronouncement.
In June 2009, the FASB issued ASC Topic 860, “Accounting
for Transfers of Financial Assets”
(“ASC 860”), which eliminates the concept of
a qualifying special-purpose entity, changes the requirements
for derecognizing financial assets, and requires additional
disclosures in order to enhance information reported to users of
financial statements by providing greater transparency about
transfers of financial assets, including securitization
transactions, and an entity’s continuing involvement in and
exposure to the risks related to transferred financial assets.
This statement is effective for fiscal years beginning after
November 15, 2009. The Company is currently in the process
of evaluating the impact of ASC-860.
In May 2009, FASB ASC Topic 855, “Subsequent
Events” (“ASC 855”) which sets forth
(i) the period after the balance sheet date during which
management of a reporting entity should evaluate events or
transactions that may occur for potential recognition or
disclosure in the financial statements, (ii) the
circumstances under which an entity should recognize events or
transactions occurring after the balance sheet date in its
financial statements and (iii) the disclosures that an
entity should make about events or transactions that occurred
after the balance sheet date. ASC 855 is effective for interim
and annual periods ending after June 15, 2009. The Company
adopted ASC 855 in the quarter ending June 30, 2009. The
adoption of ASC 855 did not have any impact on the
Company’s financial position, results of operations or cash
flows. The Company evaluated all events or transactions that
occurred after the period covered by this report, up through the
time of filing this annual report on
Form 10-K
with the SEC. During this period the Company did not have any
material recognizable subsequent events other than what is
disclosed in footnote 17.
In April 2009, the FASB issued the following: (i) FASB
ASC Topic
820-10-65,
“Determining Fair Value When the Volume and Level of
Activity for the Asset or Liability have Significantly Decreased
and Identifying Transactions That Are Not Orderly”
(“ASC
820-10-65”),
(ii) FASB ASC Topic
320-10-65,
“Recognition and Presentation of
Other-Than-Temporary
Impairment” (“ASC
320-10-65”),
and (iii) FASB ASC
Topic 825-10-65,
57
RXi
PHARMACEUTICALS CORPORATION
NOTES TO
FINANCIAL STATEMENTS — (Continued)
“Interim Disclosures about Fair Value of Financial
Instruments” (“ASC
825-10-65”),
which was effective for interim and annual periods ending after
June 15, 2009. ASC
820-10-65
provides guidance on how to determine the fair value of assets
and liabilities under ASC
820-10 in
the current economic environment and reemphasizes that the
objective of a fair value measurement remains an exit price. If
RXI were to conclude that there has been a significant decrease
in the volume and level of activity of the asset or liability in
relation to normal market activities, quoted market values may
not be representative of fair value and the Company may conclude
that a change in valuation technique or the use of multiple
valuation techniques may be appropriate. ASC
820-10-65
modifies the requirements for recognizing
other-than-temporarily
impaired debt securities and revises the existing impairment
model for such securities, by modifying the current intent and
ability indicator in determining whether a debt security is
other-than-temporarily
impaired.
ASC 825-10-65
enhances the disclosure of instruments under the scope of ASC
820 for both interim and annual periods. The Company is
currently evaluating these staff positions and the impact, if
any, that adoption will have on its financial position and
results of operation.
Effective January 1, 2009, the Company adopted FASB ASC
Topic
815-40-15,
“Derivatives and Hedging Evaluating Whether an
Instrument is Indexed to an Entity’s Own Stock”
(“ASC
815-40-15”),
which addresses the accounting for certain instruments as
derivatives. Under ASC
815-40-15
specific guidance is provided regarding requirements for an
entity to consider embedded features as indexed to the
entity’s own stock. See footnote 6 for the impact the
adoption of ASC
815-40-15
had on the Company’s financial position, results of
operations and cash flows.
In December 2007, the FASB ratified the consensus reached by the
EITF on ASC Topic 808, “Accounting for Collaborative
Agreements” (“ASC 808-10-2”), which provides
the definition of a collaborative agreement and ASC 808-10-15
provides guidelines to assist an entity in determining whether
or not it is a party in a collaborative agreement. ASC 808-10-45
states that costs incurred and revenues generated from
transactions with third parties shall be reported in accordance
with ASC Topic 605-45, “Reporting Revenue Gross as a
Principal versus Net as an Agent,” (“ASC
808-10-50”) also provides minimum disclosure requirements
for an entity’s collaboration agreements and transition
guidance. The adoption of ASC Topic 808 did not have a material
impact on the Company’s financial position, results of
operations and cash flows.
In June 2007, the Emerging Issues Task Force issued ASC Topic
730-20
“Research and Development Arrangements”
(“ASC
730-20”).
ASC 730-20
addresses the diversity which exists with respect to the
accounting for the non-refundable portion of a payment made by a
research and development entity for future research and
development activities. Under ASC
730-20, an
entity would defer and capitalize non-refundable advance
payments made for research and development activities until the
related goods are delivered or the related services are
performed subject to an assessment of recoverability. ASC
730-20 was
effective for fiscal years beginning after December 15,
2007 and interim periods within those years. The adoption of ASC
730-20 did
not have a material impact on the Company’s financial
statements
|
|
|
4.
|
Fair
Value Measurements
|
Effective January 1, 2008, the Company implemented FASB ASC
Topic 820, “Fair Value Measurements and
Disclosures” (“ASC 820”) for the
Company’s financial assets and liabilities that are
re-measured and reported at fair value at each reporting period,
and are re-measured and reported at fair value at least annually
using a fair value hierarchy that is broken down into three
levels. Level inputs are as defined as follows:
Level 1 — quoted prices in active markets for
identical assets or liabilities.
Level 2 — other significant observable inputs for
the assets or liabilities through corroboration with market data
at the measurement date.
58
RXi
PHARMACEUTICALS CORPORATION
NOTES TO
FINANCIAL STATEMENTS — (Continued)
Level 3 — significant unobservable inputs that
reflect management’s best estimate of what market
participants would use to price the assets or liabilities at the
measurement date.
The Company categorized its cash equivalents as a Level 1
hierarchy. The valuation for Level 1 was determined based
on a “market approach” using quoted prices in active
markets for identical assets. Valuations of these assets do not
require a significant degree of judgment. The Company
categorized its warrants potentially settled in cash as a
Level 2 hierarchy. The warrants are measured at market
value on a recurring basis and are being marked to market each
quarter-end until they are completely settled. The warrants are
valued using the Black-Scholes method, using assumptions
consistent with our application of ASC 718. See footnote 9.
In accordance with the provisions of ASC 820 the Company has
elected to defer implementation of ASC 820, as it relates
to its financial assets and liabilities that are recognized and
disclosed at fair value in the financial statements on a
nonrecurring basis until January 1, 2010. The Company is
evaluating the impact, if any, this standard will have on its
financial assets and liabilities. The adoption of ASC 820, as it
relates to the Company’s financial assets and liabilities
that are re-measured and reported at fair value at least
annually did not have an impact on the Company’s financial
results.
At December 31, 2009 and 2008, the Company had $16,000 on
deposit with its landlords related to leased facilities, all of
which are classified as deposits.
|
|
|
6.
|
Capital
Lease Obligations
|
The Company acquires equipment under capital lease obligations.
Accordingly, the Company capitalized approximately $100,000 and
$43,000 of equipment during the accompanying years ended
December 31, 2009 and 2008, respectively, and this is
included in equipment and furnishings on the balance sheet.
Amortization of capitalized leased equipment for the years ended
December 31, 2009 and 2008 was approximately $10,000 and
$7,000, respectively. Accumulated amortization of capitalized
lease equipment was approximately $17,000 and $7,000 at
December 31, 2009 and 2008, respectively. Future minimum
lease payments under the capital lease are $52,000, $24,000 and
$2,000 for the years ending December 31, 2010, 2011 and
2012, respectively.
|
|
|
7.
|
Accrued
Expenses and Other Current Liabilities
|
Accrued expenses and other current liabilities consist of the
following (in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
For the Year Ended
|
|
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
Professional fees
|
|
$
|
390
|
|
|
$
|
398
|
|
|
Research and development costs
|
|
|
28
|
|
|
|
47
|
|
|
Payroll related costs
|
|
|
659
|
|
|
|
531
|
|
|
|
|
|
|
|
|
|
|
|
|
Total accrued expenses and other current liabilities
|
|
$
|
1,077
|
|
|
$
|
976
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
8.
|
Commitments
and Contingencies
|
The Company acquires assets still in development and enters into
research and development arrangements with third parties that
often require milestone and royalty payments based on the
progress of the asset through development stages. Milestone
payments may be required, for example, upon approval of the
product for marketing by a regulatory agency. In certain
agreements, RXi is required to make royalty payments based
59
RXi
PHARMACEUTICALS CORPORATION
NOTES TO
FINANCIAL STATEMENTS — (Continued)
upon a percentage of the sales. Because of the contingent nature
of these payments, they are not included in the table of
contractual obligations shown below. See footnote 15.
These arrangements may be material individually, and in the
unlikely event that milestones for multiple products covered by
these arrangements were reached in the same period, the
aggregate charge to expense could be material to the results of
operations. In addition, these arrangements often give RXi the
discretion to unilaterally terminate development of the product,
which would allow RXi to avoid making the contingent payments;
however, RXi is unlikely to cease development if the compound
successfully achieves clinical testing objectives. The
Company’s contractual obligations that will require future
cash payments as of December 31, 2009 are as follows (in
thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Non-Cancelable
|
|
|
|
|
|
Cancelable
|
|
|
|
|
|
|
|
Operating
|
|
|
Employment
|
|
|
|
|
|
License
|
|
|
|
|
|
Years Ending December 31,
|
|
Leases(1)
|
|
|
Agreements(2)
|
|
|
Subtotal
|
|
|
Agreements(3)
|
|
|
Total
|
|
|
|
|
2010
|
|
$
|
239
|
|
|
$
|
1,389
|
|
|
$
|
1,628
|
|
|
$
|
2,068
|
|
|
$
|
3,696
|
|
|
2011
|
|
|
22
|
|
|
|
198
|
|
|
|
220
|
|
|
|
875
|
|
|
|
1,095
|
|
|
2012
|
|
|
|
|
|
|
73
|
|
|
|
73
|
|
|
|
930
|
|
|
|
1,003
|
|
|
2013
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
1,400
|
|
|
|
1,400
|
|
|
2014
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
735
|
|
|
|
735
|
|
|
thereafter
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
13,481
|
|
|
|
13,481
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
261
|
|
|
$
|
1,660
|
|
|
$
|
1,921
|
|
|
$
|
19,489
|
|
|
$
|
21,410
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) |
|
Operating leases are primarily facility and equipment related
obligations with third party vendors. Operating lease expenses
during the years ended December 31, 2009 and 2008 were
approximately $260,000 and $216,000, respectively. |
| |
|
(2) |
|
Employment agreement obligations include management contracts,
as well as scientific advisory board member compensation
agreements. Certain agreements, which have been revised from
time to time, provide for minimum salary levels, adjusted
annually at the discretion of the Compensation Committee, as
well as for minimum bonuses that are payable. |
| |
|
(3) |
|
License agreements generally relate to the Company’s
obligations with UMMS associated with RNAi and, for future
periods, represent minimum annual royalty and milestone payment
obligations, of the total amount due $2,250,000 can be paid in
equity, provided that the securities are registered for resale
at the time of such payment. The Company continually assesses
the progress of its licensed technology and the progress of its
research and development efforts as it relates to its licensed
technology and may terminate with notice to the licensor at any
time. In the event these licenses are terminated, no amounts
will be due. |
The Company applies the disclosure provisions ASC Topic 460
(“ASC 460”), Guarantor’s Accounting and
Disclosure Requirements for Guarantees, Including Indirect
Guarantees of Indebtedness of Others”, to its
agreements that contain guarantee or indemnification clauses.
The Company provides (i) indemnifications of varying scope
and size to certain investors and other parties for certain
losses suffered or incurred by the indemnified party in
connection with various types of third-party claims and
(ii) indemnifications of varying scope and size to officers
and directors against third party claims arising from the
services they provide to us. These indemnifications give rise
only to the disclosure provisions of ASC 460. To date, the
Company has not incurred costs as a result of these obligations
and does not expect to incur material costs in the future.
Accordingly, the Company has not accrued any liabilities in its
financial statements related to these indemnifications.
Preferred Stock — The Company has authorized up
to 5,000,000 shares of preferred stock, $0.00001 par
value per share, for issuance. The preferred stock will have
such rights, preferences, privileges and restrictions,
60
RXi
PHARMACEUTICALS CORPORATION
NOTES TO
FINANCIAL STATEMENTS — (Continued)
including voting rights, dividend rights, conversion rights,
redemption privileges and liquidation preferences, as shall be
determined by the Company’s board of directors upon its
issuance. At December 31, 2009, there were no shares of
preferred stock outstanding.
Common Stock Warrants — On August 7, 2008,
the Company issued 190,000 warrants to an investment bank as
consideration for investment and business advisory services. The
warrants have an exercise price of $7.036 per share and expire
5 years from the date of issuance, on August 7, 2013.
The warrant vested as to 94,000 shares upon issuance, and
vested at a rate of 32,000 shares per month starting on the
90 day anniversary of issuance, and is exercisable for a
period of five years. The Company also agreed to give the holder
of the warrant unlimited “piggy back” registration
rights with respect to the shares of the Company’s common
stock underlying the warrant in any registration statement the
Company files in connection with an underwritten offering of its
common stock. The fair value of these warrants has been
estimated based on the Black-Scholes options pricing model and
changes in the fair value are recorded in the condensed
statement of expenses in accordance with the requirements of ASC
718 and ASC
505-50.
Total expense related to these warrants was approximately
$318,000 and $750,000 during the years ended December 31,
2009 and 2008, respectively
On October 3, 2008, the Company acquired the rights to
license exclusive worldwide technology for the oral delivery of
RNAi therapeutics. As consideration for this license, the
Company agreed to pay a total license fee of $2.5 million
over a 12 month period, which can be paid in cash, in
equity or a combination thereof, provided that a specified
amount of the license fee must be made in cash. During 2009,
this 12 month period was extended to a date after
January 1, 2010 to be agreed upon by the parties. Payments
made in equity may only be made if, at the time of such payment,
the shares of common stock issuable upon conversion of the
warrant have been registered for resale under the Securities Act
of 1933. No warrants have been issued under this agreement
through the date of this report. The Company continually
assesses the progress of its research and development efforts as
it relates to its licensed technology and may terminate with
notice to the Licensor at any time. Accordingly, the amounts are
being expensed, as payments are made. During the year ended
December 31, 2009 and 2008, the Company paid and expensed
$250,000 each year under this agreement.
On January 29, 2009, the Company issued 142,500 warrants to
an investment bank as consideration for investment and business
advisory services. The warrants have an exercise price of $4.273
per share and expire five years from the date of issuance on
January 29, 2014. The warrants vested as to
71,250 shares upon issuance, and vested at a rate of
23,750 shares per month starting on the 90 day
anniversary of issuance, and are exercisable for a period of
five years. The Company has also agreed to give the holder of
the warrants unlimited “piggy back” registration
rights with respect to the shares of Common Stock underlying the
warrants in any registration statement the Company files in
connection with an underwritten offering of the common stock.
The fair value of these warrants has been estimated based on the
Black-Scholes options pricing model and changes in the fair
value are recorded in the condensed statement of expenses in
accordance with the requirements of ASC Topic 718 and ASC Topic
505-50.
Total expense related to these warrants was approximately
$509,000 during the year ended December 31, 2009.
In connection with the 2009 Offering, the Company issued
warrants to purchase 978,142 shares of the Company’s
common stock. Details of the transaction can be found under the
heading 2009 Registered Direct Offering below.
Private Investment in Public Equity — On
June 24, 2008, the Company entered into a Securities
Purchase Agreement pursuant to which RXi issued and sold to
certain investors an aggregate of 1,073,299 shares of
common stock in a private placement at a price of $8.12 per
share. Net proceeds to the Company were approximately
$7.9 million. The Company agreed to file a registration
statement covering the resale of all shares issued in the
private placement, with all expenses incurred in connection with
such registration to be borne by the Company. The registration
statement went effective on August 6, 2008.
61
RXi
PHARMACEUTICALS CORPORATION
NOTES TO
FINANCIAL STATEMENTS — (Continued)
2009 Registered Direct Offering — On
March 17, 2009, the Company entered into a placement agency
agreement, which was subsequently amended on May 26, 2009
and July 22, 2009, with Rodman & Renshaw, LLC
(“Rodman”) as the exclusive placement agent, relating
to a proposed offering by the Company of new securities to
potential investors. On July 30, 2009, the Company entered
into definitive agreements for the sale and issuance by the
Company to certain investors of 2,385,715 units, with each
unit consisting of one share of the Company’s common stock
and a warrant to purchase 0.40 of a share of common stock, at a
purchase price of $3.50 per unit (the “2009
Offering”). The 2009 Offering closed on August 4,
2009. The warrants have an exercise price of $4.50 per share and
are exercisable for a period beginning on February 3, 2010
until their expiration on August 3, 2014. The Company
raised gross proceeds of approximately $8,350,000 in the 2009
Offering and net cash proceeds, after deducting the placement
agents’ fees and other offering expenses payable by the
Company, of approximately $7.7 million. Total warrants
issued in connection with the transaction were 954,285.
As part of the placement agency agreement, the Company issued a
warrant to purchase 23,857 shares of the Company’s
common stock to Rodman. The warrant has an exercise price of
$4.38 per share. The warrant is immediately vested and is
exercisable until its expiration on August 3, 2014.
The Company follows the guidance of ASC Topic
815-40, as
certain warrants issued in connection with the stock offering on
August 4, 2009 were determined not to be indexed to the
Company’s common stock as they are potentially settleable
in cash. The fair value of the warrants at the dates of issuance
totaling $2,862,640 was recorded as a liability and a cost of
equity and was determined by the Black-Scholes option pricing
model. Due to the fact that the Company has limited trading
history, the Company’s expected stock volatility assumption
is based on a combination of implied volatilities of similar
entities whose share or option prices are publically traded. The
Company used a weighted average expected stock volatility of
122.69%. The expected life assumption is based on the contract
term of five years. The dividend yield of zero is based on the
fact that RXi has no present intention to pay cash dividends in
the future. The risk free rate of 1.72% used for the warrant is
equal to the zero coupon rate in effect at the time of the
grant. The increase in the fair value of the warrants from the
date of issuance to December 31, 2009 of $858,000 has been
included as an offset to other expense in the accompanying
condensed statements of expenses for the respective period. The
fair value of the warrants at December 31, 2009 of
$3,721,000 is included as a current liability in the
accompanying balance sheet as of that date and was determined by
the Black-Scholes option pricing model. The following
assumptions were used to determine the fair value as of
December 31, 2009: weighted average expected stock
volatility of 119.10%; an expected life of five years based on
the contractual terms; a dividend yield of zero; and a risk free
rate of 2.69%.
62
RXi
PHARMACEUTICALS CORPORATION
NOTES TO
FINANCIAL STATEMENTS — (Continued)
|
|
|
10.
|
Development
Stage Supplemental Equity Disclosure
|
Summarized below are the Company’s equity (common stock and
common stock options) transactions since the Company’s
inception through December 31, 2009 (in thousands except
per share data).
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Price per
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Share or
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Dollar
|
|
|
Exercise
|
|
|
|
|
|
|
|
|
|
|
|
|
Shares of
|
|
|
Amount of
|
|
|
Price per
|
|
|
Counter
|
|
Nature of
|
|
|
|
|
|
|
|
Common
|
|
|
Consideration
|
|
|
Share
|
|
|
Party to
|
|
Non-Cash
|
|
Basis of
|
|
Type of Security
|
|
Date of Issuance
|
|
Stock
|
|
|
($)
|
|
|
($)
|
|
|
Transaction
|
|
Consideration
|
|
Assigning Cost
|
|
|
|
Common Stock
|
|
April 3, 2006
|
|
|
1,624,278
|
|
|
|
2
|
|
|
|
0.002
|
|
|
Founders
|
|
NA
|
|
Cash
|
|
Common Stock
|
|
January 8, 2007
|
|
|
7.040,318
|
|
|
|
48
|
(A)
|
|
|
0.007
|
|
|
CytRx
|
|
Contributed
Assets
|
|
Predecessor
Cost
|
|
Common Stock
|
|
April 30, 2007
|
|
|
3,273,292
|
|
|
|
15,348
|
(B)
|
|
|
5.19
|
|
|
CytRx
|
|
NA
|
|
Cash
|
|
Common Stock
|
|
April 30, 2007
|
|
|
462,112
|
|
|
|
2,311
|
|
|
|
5.00
|
|
|
UMMS
|
|
Intellectual
Properties
|
|
Independent
Third Party
Valuation
|
|
Common Stock
|
|
August 18, 2007
|
|
|
30,000
|
|
|
|
150
|
|
|
|
5.00
|
|
|
Directors
|
|
—
|
|
Cash
|
|
Common Stock
|
|
September 28, 2007
|
|
|
188,387
|
|
|
|
978
|
|
|
|
5.19
|
|
|
CytRx
|
|
NA
|
|
Independent
Third Party
Valuation
|
|
Common Stock
|
|
November 21, 2007
|
|
|
66,045
|
|
|
|
331
|
|
|
|
5.00
|
|
|
Exercise of
Stock Options
|
|
NA
|
|
Cash
|
|
Common Stock
|
|
June 26, 2008
|
|
|
1,073,299
|
|
|
|
7,918
|
|
|
|
8.12
|
|
|
PIPE
|
|
NA
|
|
Net Cash
|
|
Common Stock
|
|
October 6, 2008 and
November 16, 2008
|
|
|
5,500
|
|
|
|
26
|
|
|
|
5.00
|
|
|
Exercise of
Stock Options
|
|
NA
|
|
Cash
|
|
Common Stock
|
|
January 30, 2009
|
|
|
58,398
|
|
|
|
NA
|
|
|
|
NA
|
|
|
|
|
NA
|
|
Market Value
|
|
Common Stock
|
|
May 1, 2009
|
|
|
281
|
|
|
|
NA
|
|
|
|
4.19
|
|
|
Exercise of
Stock Options
|
|
NA
|
|
Cash
|
|
Common Stock
|
|
August 3, 2009 and
August 4, 2009
|
|
|
2,385,715
|
|
|
|
7,714
|
|
|
|
3.50
|
|
|
Register Direct
|
|
NA
|
|
Net Cash
|
|
|
|
|
(A) |
|
Transactions between related parties are accounted for at the
historical cost of CytRx, with the intellectual property which
was previously expensed on CytRx’s books being recorded at
zero cost and equipment and furnishings being recorded at
$48,000. |
| |
|
(B) |
|
RXi received gross proceeds of $17.0 million for the
issuance of the 3,273,292 shares of common stock which
equals $5.19 per share. The gross proceeds were reduced by a
reimbursement to CytRx of (1) $1.3 million for
RXi’s pro rata share of offering costs related to the
April 17, 2007 private placement conducted by CytRx to fund
its capital contribution to the Company and (2) $363,000 of
expenses incurred on behalf of RXi for the year ended
December 31, 2006. Net proceeds to RXi after these charges
were $15.3 million or $4.69 a share. |
|
|
|
11.
|
Stock
Based Compensation
|
Options to Purchase Shares of Common Stock —
The Company follows the provisions ASC 718 which requires the
measurement and recognition of compensation expense for all
share-based payment awards made to employees, non-employee
directors, and consultants, including employee stock options.
Stock compensation expense based on the grant date fair value
estimated in accordance with the provisions of ASC 718 is
recognized as an expense over the requisite service period.
For stock options granted as consideration for services rendered
by non-employees, the Company recognizes compensation expense in
accordance with the requirements of FASB ASC Topic
505-50
(“ASC 505-50”)
“Equity Based Payments to Non- Employees.”
Non-employee option grants that do not vest
63
RXi
PHARMACEUTICALS CORPORATION
NOTES TO
FINANCIAL STATEMENTS — (Continued)
immediately upon grant are recorded as an expense over the
vesting period of the underlying stock options. At the end of
each financial reporting period prior to vesting, the value of
these options, as calculated using the Black-Scholes
option-pricing model, will be re-measured using the fair value
of the Company’s common stock and the non-cash compensation
recognized during the period will be adjusted accordingly. Since
the fair market value of options granted to non-employees is
subject to change in the future, the amount of the future
compensation expense will include fair value re-measurements
until the stock options are fully vested.
The Company is currently using the Black-Scholes option-pricing
model to determine the fair value of all its option grants. For
options grants issued for the year ended December 31, 2009
and 2008, the following assumptions were used:
| |
|
|
|
|
|
|
|
2009
|
|
2008
|
|
|
|
Weighted average risk free interest rate
|
|
1.55% - 3.91%
|
|
1.55% - 3.99%
|
|
Weighted average volatility
|
|
116.72% - 122.93%
|
|
101.79% - 116.74%
|
|
Expected lives (years)
|
|
6 - 10
|
|
6 - 10
|
|
Expected dividend yield
|
|
0%
|
|
0%
|
The weighted average fair value of options granted during the
years ended December 31, 2009 and 2008 was $4.11 and $6.37
per share, respectively.
RXi’s expected common stock price volatility assumption is
based upon the volatility of a basket of comparable companies.
The expected life assumptions for employee grants were based
upon the simplified method provided for under ASC 718, which
averages the contractual term of RXi’s options of ten years
with the average vesting term of four years for an average of
six years. The expected life assumptions for non-employees were
based upon the contractual term of the option. The dividend
yield assumption of zero is based upon the fact that RXi has
never paid cash dividends and presently has no intention of
paying cash dividends in the future. The risk-free interest rate
used for each grant was also based upon prevailing short-term
interest rates. RXi has estimated an annualized forfeiture rate
of 4.0% for options granted to its employees, 2.1% for options
granted to senior management and no forfeiture rate for the
directors. RXi will record additional expense if the actual
forfeitures are lower than estimated and will record a recovery
of prior expense if the actual forfeiture rates are higher than
estimated.
RXi recorded approximately $4,202,000 and $3,824,000 of
stock-based compensation for the years ended December 31,
2009 and 2008, respectively. As of December 31, 2009, there
was $4.7 million of unrecognized compensation cost related
to outstanding options that is expected to be recognized as a
component of RXi’s operating expenses through 2013.
On November 4, 2009, as part of a plan succession in
leadership, Tod Woolf, Ph.D., resigned as our President,
Chief Executive Officer and a member of our Board of Directors.
According to the Separation Agreement between Dr. Woolf and
the Company, Dr. Woolf received in one lump sum payment his
full severance equivalent to a six (6) month salary
($187,500), six (6) months acceleration of vesting of all
of his outstanding unvested Stock Options as of November 4,
2009, and an offer to join the Company’s Scientific
Advisory Board (SAB) for 3 years (the “New
Position”). In addition, and as part of the Separation
Agreement, the Company agreed to extend the exercise period for
all of Dr. Woolf’s vested Stock Options as of
November 4, 2009, to the later of: (i) a period of two
(2) years from his resignation (until November 4,
2011), or (ii) ninety (90) days following the end of
the term of the SAB Agreement (February 4,
2013) or such earlier date as the SAB Agreement may be
terminated pursuant to the terms of the SAB Agreement
provided Dr. Woolf has not violated the non-competition
provisions of the SAB Agreement prior to the date of
exercise (whether or not the SAB Agreement is still in
effect at that time). Notwithstanding any provision of the
Company’s 2007 Incentive Plan, the Company also agreed that
Dr. Woolf’s previously awarded Stock Options shall
continue to vest during his continuing role in the Company in
the New Position. The option modification resulted in an
incremental value of the options of approximately $153,000 of
which $37,000 was expensed during 2009. The remaining $411,000
will be expensed over the remaining vesting period of
approximately 3 years.
64
RXi
PHARMACEUTICALS CORPORATION
NOTES TO
FINANCIAL STATEMENTS — (Continued)
As of December 31, 2009, an aggregate of
4,750,000 shares of common stock were reserved for issuance
under the RXi Pharmaceuticals Corporation 2007 Incentive Plan,
including 3,630,839 shares subject to outstanding common
stock options and restricted stock units granted under this plan
and 1,047,335 shares available for future grants. The
administrator of the plan determines the times when an option
may become exercisable. Vesting periods of options granted to
date include vesting upon grant to vesting at the end of a four
year period. The options will expire, unless previously
exercised, no later than ten years from the grant date.
The following table summarizes the activity of the
Company’s stock option plan:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted
|
|
|
|
|
Stock
|
|
|
Average
|
|
|
|
|
Options
|
|
|
Exercise Price
|
|
|
|
|
Outstanding — January 1, 2008
|
|
|
1,335,184
|
|
|
$
|
5.00
|
|
|
Granted
|
|
|
899,609
|
|
|
|
7.76
|
|
|
Exercised
|
|
|
(5,500
|
)
|
|
|
5.00
|
|
|
Forfeited
|
|
|
(5,841
|
)
|
|
|
6.03
|
|
|
|
|
|
|
|
|
|
|
|
|
Outstanding — December 31, 2008
|
|
|
2,223,452
|
|
|
|
6.11
|
|
|
Granted
|
|
|
1,622,546
|
|
|
|
3.84
|
|
|
Exercised
|
|
|
(281
|
)
|
|
|
4.19
|
|
|
Forfeited
|
|
|
(263,378
|
)
|
|
|
5.05
|
|
|
Outstanding — December 31, 2009
|
|
|
3,582,339
|
|
|
|
5.16
|
|
|
|
|
|
|
|
|
|
|
|
|
Exercisable — December 31, 2008
|
|
|
1,238,187
|
|
|
|
5.65
|
|
|
|
|
|
|
|
|
|
|
|
|
Exercisable — December 31, 2009
|
|
|
2,131,298
|
|
|
|
5.42
|
|
|
|
|
|
|
|
|
|
|
|
The following table summarizes the activity for non-vested stock
options:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted Average
|
|
|
|
|
Stock
|
|
|
Grant Date Fair
|
|
|
|
|
Options
|
|
|
Value per Share
|
|
|
|
|
Non-vested at January 1, 2008
|
|
|
839,361
|
|
|
$
|
3.54
|
|
|
Granted
|
|
|
899,609
|
|
|
|
6.37
|
|
|
Vested
|
|
|
(740,364
|
)
|
|
|
4.94
|
|
|
Exercised
|
|
|
(5,500
|
)
|
|
|
3.93
|
|
|
Pre-vested forfeitures
|
|
|
(5,841
|
)
|
|
|
4.76
|
|
|
|
|
|
|
|
|
|
|
|
|
Non-vested at December 31, 2008
|
|
|
987,265
|
|
|
|
5.15
|
|
|
Granted
|
|
|
1,622,546
|
|
|
|
3.44
|
|
|
Vested
|
|
|
(895,111
|
)
|
|
|
4.27
|
|
|
Exercised
|
|
|
(281
|
)
|
|
|
3.71
|
|
|
Pre-vested forfeitures
|
|
|
(263,378
|
)
|
|
|
4.07
|
|
|
|
|
|
|
|
|
|
|
|
|
Non-vested at December 31, 2009
|
|
|
1,451,041
|
|
|
|
3.94
|
|
|
|
|
|
|
|
|
|
|
|
The weighted average remaining contractual life of options
outstanding and exercisable at December 31, 2009 was
8.47 years and 8.14 years, respectively. The weighted
average remaining contractual life of options outstanding and
exercisable at December 31, 2008 was 8.832 years and
8.733 years, respectively.
The aggregate intrinsic value of outstanding options as of
December 31, 2009 and 2008 is $1,262,270 and $653,974,
respectively. The aggregate intrinsic value of exercisable
options as of December 31, 2009 and 2008 is $138,881 and
$654,000, respectively. The aggregate intrinsic value is
calculated based on the positive difference between the closing
fair market value of RXi’s common stock and the exercise
price of the underlying options.
65
RXi
PHARMACEUTICALS CORPORATION
NOTES TO
FINANCIAL STATEMENTS — (Continued)
The aggregate intrinsic value of options exercised during 2009
and 2008 was approximately $1,000 and $18,000, respectively.
Restricted Stock Units — In addition to options
to purchase shares of common stock, the Company may grant
restricted stock units (“RSU”) as part of its
compensation package. Each RSU is granted at the fair market
value based on the date of grant. Vesting is determined on a
grant by grant basis. As of December 31, 2009, the Company
had granted a total of 48,500 RSUs with an aggregate intrinsic
value of $222,000 and recognized total expense of $105,000. The
RSUs vest in full on January 2, 2010.
The Company accounts for and discloses net loss per common share
in accordance with FASB ASC Topic 260 “Earnings per
Share.” Basic net loss per common share is computed by
dividing net loss attributable to common stockholders by the
weighted average number of common shares outstanding. Diluted
net loss per common share is computed by dividing net loss
attributable to common stockholders by the weighted average
number of common shares that would have been outstanding during
the period assuming the issuance of common shares for all
potential dilutive common shares outstanding. Potential common
shares consist of shares issuable upon the exercise of stock
options and warrants. Because the inclusion of potential common
shares would be anti-dilutive for all periods presented, diluted
net loss per common share is the same as basic net loss per
common share.
The following table sets forth the potential common shares
excluded from the calculation of net loss per common share
because their inclusion would be anti-dilutive:
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
Options to purchase common stock
|
|
|
3,582,339
|
|
|
|
2,223,452
|
|
|
Restricted stock units
|
|
|
48,500
|
|
|
|
—
|
|
|
Warrants to purchase common stock
|
|
|
1,310,642
|
|
|
|
190,000
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
|
4,941,481
|
|
|
|
2,413,452
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The components of federal and state income tax expense are as
follows (in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
As of December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
Current
|
|
|
|
|
|
|
|
|
|
Federal
|
|
$
|
—
|
|
|
$
|
—
|
|
|
State
|
|
|
—
|
|
|
|
—
|
|
|
Deferred
|
|
|
|
|
|
|
|
|
|
Federal
|
|
|
(5,533
|
)
|
|
|
(4,466
|
)
|
|
State
|
|
|
(2,257
|
)
|
|
|
(1,513
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Total deferred
|
|
|
(7,789
|
)
|
|
|
(5,979
|
)
|
|
Valuation allowance
|
|
|
7,789
|
|
|
|
5,979
|
|
|
|
|
|
|
|
|
|
|
|
|
Total income tax expense
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
66
RXi
PHARMACEUTICALS CORPORATION
NOTES TO
FINANCIAL STATEMENTS — (Continued)
The components of net deferred tax assets are as follows (in
thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
As of December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
Net operating loss carryforwards
|
|
$
|
10,348
|
|
|
$
|
6,710
|
|
|
Tax credit carryforwards
|
|
|
753
|
|
|
|
948
|
|
|
Non-qualified stock based compensation
|
|
|
4,222
|
|
|
|
2,471
|
|
|
Other
|
|
|
74
|
|
|
|
28
|
|
|
Licensing deduction deferral
|
|
|
2,089
|
|
|
|
1,225
|
|
|
|
|
|
|
|
|
|
|
|
|
Gross deferred tax assets
|
|
|
17,486
|
|
|
|
11,382
|
|
|
Valuation allowance
|
|
|
(17,486
|
)
|
|
|
(11,382
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Net deferred tax asset
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
The provision for income taxes differs from the provision
computed by applying the federal statutory rate to net loss
before income taxes as follows (in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
As of December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
Expected federal income tax benefit
|
|
$
|
(6,252
|
)
|
|
$
|
(4,887
|
)
|
|
Non-qualified stock compensation
|
|
|
621
|
|
|
|
186
|
|
|
Effect of change in valuation allowance
|
|
|
6,103
|
|
|
|
6,707
|
|
|
State income tax credits
|
|
|
821
|
|
|
|
(426
|
)
|
|
State income taxes after credits
|
|
|
(324
|
)
|
|
|
(867
|
)
|
|
Other
|
|
|
(969
|
)
|
|
|
(713
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
The Company has incurred net operating losses from inception. At
December 31, 2009, the Company had domestic federal and
state net operating loss carryforwards of approximately
$25.7 million available to reduce future taxable income,
which expire at various dates beginning in 2012 through 2029.
The Company also had federal and state research and development
tax credit carryforwards of approximately $500,000 and $400,000,
respectively, available to reduce future tax liabilities and
which expire at various dates beginning in 2022 through 2029.
Under the provisions of the Internal Revenue Code, certain
substantial changes in the Company’s ownership may result
in a limitation on the amount of net operating loss
carryforwards and research and development credit carryforwards
which could be utilized annually to offset future taxable income
and taxes payable.
Based on an assessment of all available evidence including, but
not limited to the fact the RXi operating results have been
included in CytRx consolidated U.S. federal and state
income tax return for the year ended December 31, 2007, the
Company’s limited operating history in its core business
and lack of profitability, uncertainties of the commercial
viability of its technology, the impact of government regulation
and healthcare reform initiatives, and other risks normally
associated with biotechnology companies, the Company has
concluded that it is more likely than not that these net
operating loss carryforwards and credits will not be realized
and, as a result, a 100% deferred income tax valuation allowance
has been recorded against these assets.
The Company adopted certain provisions of the ASC 740,
effective January 1, 2007 which clarifies the
accounting for uncertainty in income taxes recognized in
financial statements and requires the impact of a tax
67
RXi
PHARMACEUTICALS CORPORATION
NOTES TO
FINANCIAL STATEMENTS — (Continued)
position to be recognized in the financial statements if that
position is more likely than not of being sustained by the
taxing authority. The adoption of ASC
740-10
did not have any effect on the Company’s financial
position or results of operations.
The Company files income tax returns in the U.S. federal
and Massachusetts jurisdictions. The Company is subject to tax
examinations through the 2009 tax year. The Company does not
believe there will be any material changes in its unrecognized
tax positions over the next 12 months. The Company has not
incurred any interest or penalties. In the event that the
Company is assessed interest or penalties at some point in the
future, they will be classified in the financial statements as
general and administrative expense.
During the year ended December 31, 2007, RXi entered into a
license agreement with Cold Spring Harbor Laboratory
(“CSHL”) for small hairpin RNA, or “shRNA”,
for which the Company paid $50,000 and agreed to make future
milestone and royalty payments upon successful development and
commercialization of products. The Company also entered into
four exclusive license agreements and an invention disclosure
agreement with UMMS for which the Company paid cash of $453,000
and issued 462,112 shares of its common stock valued at
$2.3 million, or $5.00 per share. For each RNAi product
developed in connection with the license granted by CSHL, the
possible aggregate milestone payments equal $2,650,000. The
invention disclosure agreement has an initial term of three
years and provides the option to negotiate licenses to certain
RNAi technologies discovered at UMMS.
On August 29, 2007, RXi entered into a license agreement
with TriLink Biotechnologies, Inc. for three RNAi chemistry
technologies for all therapeutic RNAi applications, for which
the Company paid $100,000 and agreed to pay yearly maintenance
fees of $30,000, as well as future clinical milestone payments
and royalty payments based on sales of therapeutic products
developed from the licensed technologies. The Company expensed
$30,000 for each of the years ended December 31, 2009 and
2008, respectively.
In October 2007, RXi entered into a license agreement with
Dharmacon, Inc. (now part of Thermo Fisher Scientific Inc.),
pursuant to which the Company obtained an exclusive license to
certain RNAi sequences to a number of target genes for the
development of the Company’s rxRNA compounds. Further, the
Company has obtained the right to license additional RNAi
sequences, under the same terms, disclosed by Thermo Fisher
Scientific Inc. in its pending patent applications against
target genes and has received an option for exclusivity for
other siRNA configurations. As consideration for this license,
the Company paid an up-front fee of $150,000 and agreed to pay
future clinical milestone payments and royalty payments based on
sales of siRNA compositions developed in connection with the
licensed technology. No amounts were expensed in 2008 and 2009
related to this license.
In November 2007, RXi entered into a license agreement with Life
Technologies, Inc., pursuant to which the Company was granted
rights under four patents relating to RNA target sequences, RNA
chemical modifications, RNA configurations
and/or RNA
delivery to cells. As consideration for this license, RXi paid
an up-front fee of $250,000 and agreed to pay yearly maintenance
fees of the same amount beginning in 2008. Further, the Company
is obligated to pay a fee for each additional gene target added
to the license as well as a fee on the first and second
anniversaries on the date of which consent to add the gene
target to the list of those covered by the license was granted.
The Company has also been granted, for each gene target, an
option to secure pre-clinical rights
and/or the
clinical rights, for which RXi would be required to pay
additional fees. Further, the Company is required to make future
clinical milestone payments and royalty payments based on sales
of therapeutic products developed from the licensed
technologies. The Company expensed $250,000 for each of the
years ended December 31, 2009 and 2008 related to this
license.
On October 3, 2008, the Company acquired co-exclusive
rights to technology for the oral delivery of RNAi therapeutics
from UMMS. As consideration for this license, the Company agreed
to pay a total license
68
RXi
PHARMACEUTICALS CORPORATION
NOTES TO
FINANCIAL STATEMENTS — (Continued)
fee of $2,500,000 over a 12 month period, which can be paid
in cash, in equity or a combination thereof, provided that a
specified amount of the license fee must be made in cash. This
Agreement was amended on July 1, 2009, allowing the Company
to extend the periods for which certain milestone payments are
due to UMMS. Payments made in equity may only be made if, at the
time of such payment, the shares of common stock issuable upon
conversion of the warrant have been registered for resale under
the Securities Act of 1933. No warrants have been issued under
this agreement through the date of this report. The Company
continually assesses the progress of its research and
development efforts as it relates to its licensed technology and
may terminate with notice to the licensor at any time.
Accordingly, the amounts are being expensed, as payments are
made. The Company expensed $250,000 for each of the years ended
December 31, 2009 and 2008 related to this license
In September, 2009, the Company entered into a Patent and
Technology Assignment Agreement with Advirna, LLC
(“Advirna”), a Colorado limited liability company
co-founded by RXi’s Chief Scientific Officer. Pursuant to
the terms of the agreement, Advirna assigned to the Company
certain patent and technology rights related to chemically
modified polynucleotides (the “Rights”) and the
Company granted to Advirna a fully
paid-up
license to the Rights in a specified field. Under the terms of
the agreement, the Company will pay to Advirna an annual
maintenance fee beginning on January 1, 2011, certain
payments upon the achievement of regulatory milestones and
royalty payments on the sales of certain products. In addition,
the Company may terminate the agreement upon 90 days’
prior written notice to Advirna and Advirna may terminate upon
90 days’ prior written notice to the Company in the
event the Company ceases to use reasonable efforts to research,
develop, license or otherwise commercialize the Rights. If the
agreement expires in accordance with its terms or is terminated
by a party in the absence of a material breach or for cause in
the event that the Company fails to pay Advirna certain fees,
the Company will assign the Rights back to Advirna. During the
years ended December 31, 2009 and 2008, the Company paid
and expensed $5,000 and $75,000, respectively, under this
agreement.
|
|
|
15.
|
Related
Party Transactions
|
On February 15, 2007, the Company entered into a letter
agreement with CytRx and certain current RXi stockholders. Under
the stockholders agreement, the Company agreed to grant to CytRx
preemptive rights to acquire any new securities, as defined
therein, that RXi proposes to sell or issue so that CytRx may
maintain its percentage ownership of the Company. The preemptive
rights are effective so long as CytRx owns less than 50% of the
Company’s outstanding shares of common stock, and will
expire on January 8, 2012 or such earlier time at which
CytRx owns less than 10% of RXi’s outstanding common stock.
Under this letter agreement, CytRx also undertakes to vote its
shares of the Company’s stock in the election of its
directors and dispose of its shares of RXi stock in accordance
with the terms of its letter agreement with UMMS described
above. CytRx has further agreed in this letter agreement to
approve of actions that may be adopted and recommended by
RXi’s board of directors to facilitate any future financing.
On April 30, 2007, the Company entered into a Registration
Rights Agreement with CytRx. Under the Registration Rights
Agreement, RXi agreed, with certain exceptions, that at any time
after its common stock is registered under the Exchange Act, to
use its best efforts to cause all of RXi’s shares issued to
CytRx pursuant to the Contribution Agreement to be registered
under the Securities Act if CytRx shall so request. All expenses
incurred in connection with any such registration will be borne
by the Company.
One of the members of RXi’s board of directors is the
President, Chief Executive Officer and a member of the board of
directors for CytRx.
The Company’s current Chief Scientific Officer or CSO,
prior to her employment by the Company, was a consultant to RXi
from January 2008 until the date of her employment. This
consulting contract resulted in payments to the CSO’s
consulting firm of approximately $13,400 which was recorded in
the year ended December 31, 2008, in consulting fees and
$5,000 recorded as license expense as discussed below. As the
69
RXi
PHARMACEUTICALS CORPORATION
NOTES TO
FINANCIAL STATEMENTS — (Continued)
CSO is not the sole owner of the consulting firm, the
approximate dollar value of her interest in this consulting
contract is approximately $9,250.
In addition, RXi and the CSO’s 50% owned Company, Advirna,
are parties to an option agreement whereby the Company paid
$5,000 for consideration to be granted the exclusive worldwide
rights to license certain technology by paying an initial
maintenance fee of $75,000 before June 12, 2009.
The Company’s current Chief Intellectual Property Counsel
and Vice President of Legal Counsel, prior to his employment by
the Company, was a consultant to RXi from September 2008 until
the date of his employment. This consulting contract resulted in
payments to him of approximately $5,000 which was recorded in
the year ended December 31, 2008 in patent and legal fees.
The approximate dollar value of his interest in this consulting
contract is also approximately $5,000.
On February 26, 2007, the Company entered into Scientific
Advisory Board Agreements (the “SAB Agreements”),
with four of its founders. At the time of the execution of the
SAB Agreements, each of the founders were beneficial owners
of more than five percent of the Company’s outstanding
stock. Pursuant to the SAB Agreements, on May 23,
2007, the Company granted to each of the founders a stock option
under the 2007 Plan to purchase 52,832 shares of its common
stock. In addition, under the SAB Agreements, the Company
will grant each of the founders a stock option under the 2007
Plan to purchase 52,832 shares of its common stock on
February 26, 2008, June 5, 2009 and June 4, 2010
with a per share exercise price equal to the closing price of
such stock on the public market on the date of grant unless a
founder terminates a SAB Agreement without good reason (as
defined) or the Company terminates a SAB Agreement with
cause (as defined therein) in which case no further option
grants will be made to the founder. If the Company’s common
stock is not publicly available on the dates specified above,
its Board of Directors will grant the stock options to the
founders at the first scheduled board meeting after such date
and the per share exercise price of the options will be
determined in good faith by the Company’s board of
directors. All options granted pursuant to the
SAB Agreements are fully vested on the date of grant and
have a term of ten years. The fair value of stock options
granted during 2009 and 2008 under the SAB Agreement for
each founder is approximately $245,000 and $338,000 which was
estimated using the Black-Scholes option-pricing model as more
fully discussed above under significant accounting policies and
the stock based compensation footnote. Included in the
accompanying financial statements for the years ended
December 31, 2009 and 2008 is approximately $978,000 and
$1,350,000, respectively, of expense related to the granting of
these stock options.
Additionally, pursuant to a letter agreement between the Company
and each founder dated as of April 30, 2007, the
“SAB Letters”, in further consideration of the
services to be rendered by the founders under the
SAB Agreements, the Company granted additional stock
options on May 23, 2007 under the 2007 Plan to each of the
founders to purchase 26,416 shares of its common stock.
Unless a founder terminates a SAB Agreement without good
reason (as defined) or the Company terminates a
SAB Agreement with cause (as defined therein), the options
granted pursuant to the SAB Letters will fully vest from
and after April 29, 2012 and will have a term of ten years
from the date of grant. At December 31, 2009, the fair
market value of stock options under the SAB Agreement for
each founder is approximately $60,000 which was estimated using
the Black-Scholes option-pricing model as more fully discussed
above under the summary of significant accounting policies and
the stock based compensation footnote. Included in the
accompanying financial statements for the years ended
December 31, 2009 and 2008 is approximately $73,000 and
$156,000, respectively, of expense related to these stock
options.
|
|
|
16.
|
Employee
Benefit Plan
|
RXi sponsors a 401(k) retirement savings plan (the
“Plan”). Participation in the Plan is available to
full-time employees who meet eligibility requirements. Eligible
employees may defer a portion of their salary as defined by
Internal Revenue Service regulations. The Company may make
matching contributions on behalf of all participants in the
401(k) Plan in an amount determined by the Company’s board
of directors. The
70
RXi
PHARMACEUTICALS CORPORATION
NOTES TO
FINANCIAL STATEMENTS — (Continued)
Company may also make additional discretionary profit sharing
contributions in amounts as determined by the board of
directors, subject t to statutory limitations. Matching and
profit-sharing contributions, if any, are subject to a vesting
schedule; all other contributions are at all times fully vested.
The Company intends the 401(k) Plan, and the accompanying trust,
to qualify under Sections 401(k) and 501 of the Internal
Revenue Code so that contributions by employees to the 401(k)
Plan, and income earned (if any) on plan contributions, are not
taxable to employees until withdrawn from the 401(k) Plan, and
so that the Company will be able to deduct its contributions, if
any, when made. The trustee under the 401(k) Plan, at the
direction of each participant, invests the assets of the 401(k)
Plan in any of a number of investment options. To date, the
Company has not made any matching contributions.
On January 14, 2010, the Company granted options to
purchase 572,440 shares of common stock to employees and
members of the Board of Directors. These options had an exercise
price of $5.66 per share, which represented the Company’s
closing stock price on that date. These options vest quarterly
over a one to four year period and expire no later than
10 years from the grant date.
On January 20, 2010, the Company granted 43,541 RSUs with a
contingent right to receive one share of Company common stock
for each restricted stock unit to certain employees. The RSUs
vested on February 9, 2010.
On March 2, 2010, the Company granted an option to purchase
50,000 shares of common stock to a member of the Board of
Directors. This options had an exercise price of $5.28 per
share, which represented the Company’s closing stock price
on that date. This option vested immediately and expires no
later than 10 years from the grant date.
On January 29, 2010, the Company granted warrants to
purchase 250,000 shares of common stock at an exercise
price of $5.66 per share in exchange for business advisory
services to the Company for a period of up to twelve months. The
warrants vested as to 71,250 shares upon issuance, and then
will vest at a rate of 23,750 shares per month starting on
the 90 day anniversary of issuance. The Company has also
agreed to give “piggy back” registration rights with
respect to the shares of common stock underlying the warrants in
any registration statement filed by the company in connection
with an underwritten offering of the common stock.
On March 22, 2010, the Company entered into a placement
agency agreement, with Rodman as the exclusive placement agent,
relating to a proposed offering by the Company of new securities
to potential investors. On March 23, 2010, the Company
entered into definitive agreements for the sale and issuance by
the Company to certain investors of 2,700,000 units, with
each unit consisting of one share of the Company’s common
stock and a warrant to purchase 0.20 of a share of common stock,
at a purchase price of $6.00 per unit (the “2010
Offering”). The 2010 Offering closed on March 26,
2010. The Company issued 540,000 warrants with an exercise price
of $6.00 per share and that are exercisable for a period
beginning on September 26, 2010 until their expiration on
March 26, 2016. The Company raised gross proceeds of
approximately $16.2 million in the 2010 Offering and net
cash proceeds, after deducting the placement agent fees and
other offering expenses payable by the Company, of approximately
$15.2 million.
As part of the 2010 Offering, the Company is required to use 25%
of the net proceeds from the 2010 Offering to repurchase from
CytRx 675,000 shares of the Company’s common stock
held by CytRx (“CytRx shares”). The Company is also
required to use 25% of the proceeds from the exercise of
warrants issued in the 2010 Offering to repurchase from CytRx a
number of CytRx Shares equal to 25% of shares issued upon the
exercise of such warrants. Subject to the satisfaction of
certain closing conditions, the Company is required to
repurchase the CytRx Shares on March 29, 2010 and if any
warrant issued in this 2010 Offering is exercised, on the first
business day after the exercise of such warrant.
The Company intends to use the net proceeds remaining from the
2010 Offering, after the repurchase of the CytRx Shares for
general corporate purposes.
71
|
|
|
ITEM 9.
|
CHANGES
IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND
FINANCIAL DISCLOSURES.
|
None
|
|
|
ITEM 9A(T).
|
CONTROLS
AND PROCEDURES
|
Evaluation
of Disclosure Controls and Procedures
Rule 13a-15(e)
under the Securities and Exchange Act of 1934, as amended (the
“Exchange Act”), defines the term “disclosure
controls and procedures” as those controls and procedures
designed to ensure that information required to be disclosed by
a company in the reports that it files or submits under the
Exchange Act is recorded, processed, summarized and reported
within the time periods specified in the Securities and Exchange
Commission rules and forms and that such information is
accumulated and communicated to the company’s management,
including its principal executive and principal financial
officers, or persons performing similar functions, as
appropriate to allow timely decisions regarding required
disclosure.
Our Chief Executive Officer and Principal Accounting Officer
have evaluated the effectiveness of the design and operation of
our disclosure controls and procedures (as defined under
Rules 13a-15(e)
and
15d-15(e)
promulgated under the Securities Exchange Act of 1934, as
amended) as of the end of the period covered by this report.
Based upon that evaluation, our Chief Executive Officer and
Principal Accounting Officer have concluded that our disclosure
controls and procedures are effective.
There have been no changes in our internal controls over
financial reporting during the fourth quarter of the year ended
December 31, 2009 that have materially affected, or are
reasonably likely to materially affect, our internal controls
over financial reporting.
Evaluation
of Disclosure Controls and Procedure Management’s report on
Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining
adequate internal control over financial reporting, as such term
is defined in Exchange Act
Rule 13a-15(f).
Under the supervision and with the participation of our
management, including our Chief Executive Officer and Principal
Accounting Officer, we conducted evaluations of the
effectiveness of our internal control over financial reporting
based on the framework in Internal Control-Integrated Framework
issued by the Committee of Sponsoring Organizations of the
Treadway Commission (“COSO”). Based on our evaluations
under the framework in Internal Control-Integrated Framework
issued by the COSO, our management concluded that our internal
control over financial reporting was effective as of
December 31, 2009.
Because of its inherent limitations, internal control over
financial reporting may not prevent or detect misstatements.
Also, projections of any evaluation of effectiveness to future
periods are subject to the risk that controls may become
inadequate because of changes in conditions, or that the degree
of compliance with the policies or procedures may deteriorate.
This annual report does not include an attestation report of the
company’s registered public accounting firm regarding
internal control over financial reporting. Management’s
report was not subject to attestation by the company’s
registered public accounting firm pursuant to temporary rules of
the Securities and Exchange Commission that permit the company
to provide only management’s report in this annual report.
|
|
|
ITEM 9B.
|
OTHER
INFORMATION
|
None
72
PART III
|
|
|
ITEM 10.
|
DIRECTORS
AND EXECUTIVE OFFICERS OF THE REGISTRANT
|
We will file with the SEC a definitive Proxy Statement, which we
refer to herein as the Proxy Statement, not later than
120 days after the close of the fiscal year ended
December 31, 2009. The information required by this item is
incorporated herein by reference to the information contained in
the Proxy Statement.
|
|
|
ITEM 11.
|
EXECUTIVE
COMPENSATION
|
The information required by this item is incorporated herein by
reference to the information contained in the Proxy Statement.
|
|
|
ITEM 12.
|
SECURITY
OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND
RELATED STOCKHOLDER MATTERS.
|
The information required by this item is incorporated herein by
reference to the information contained in the Proxy Statement.
|
|
|
ITEM 13.
|
CERTAIN
RELATIONSHIPS, RELATED TRANSACTIONS AND DIRECTOR
INDEPENDENCE
|
The information required by this item is incorporated herein by
reference to the information contained in the Proxy Statement.
|
|
|
ITEM 14.
|
PRINCIPAL
ACCOUNTANT FEES AND SERVICES
|
The information required by this item is incorporated herein by
reference to the information contained in the Proxy Statement.
PART IV
|
|
|
ITEM 15.
|
EXHIBITS AND
FINANCIAL STATEMENT SCHEDULES
|
(a) (1) Financial Statements
See Item 8 in Part II of this annual report on
Form 10-K,
“Financial Statements and Supplementary Data”, for an
index to the financial statements filed in this annual report.
(2) Financial Statement Schedules
Certain schedules are omitted because they are not applicable,
or not required by smaller reporting companies.
(3) Exhibits
The Exhibits listed in the Exhibit Index immediately
preceding the Exhibits are filed as a part of this annual report
on
Form 10-K.
73
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the
Securities Exchange Act of 1934, the Registrant has duly caused
this report to be signed on its behalf by the undersigned,
thereunto duly authorized.
RXi PHARMACEUTICALS CORPORATION
Noah D. Beerman
President, Chief Executive Officer and Director
(Principal Executive Officer)
Dated: March 31, 2010
Pursuant to the requirements of the Securities Exchange Act of
1934, this report has been signed below by the following persons
on behalf of the Registrant and in the capacities and on the
dates indicated.
| |
|
|
|
|
|
|
|
Signatures
|
|
Title
|
|
Date
|
|
|
|
|
|
|
|
|
|
/s/ Noah
D. Beerman
Noah
D. Beerman
|
|
President, Chief Executive Officer and Director (Principal
Executive Officer)
|
|
March 31, 2010
|
|
|
|
|
|
|
|
/s/ Amy
Tata
Amy
Tata
|
|
Principal Accounting Officer
(Principal Financial Officer and Accounting Officer)
|
|
March 31, 2010
|
|
|
|
|
|
|
|
/s/ Sanford
J. Hillsberg
Sanford
J. Hillsberg
|
|
Director
|
|
March 31, 2010
|
|
|
|
|
|
|
|
/s/ Mark
J. Ahn
Mark
J. Ahn
|
|
Director
|
|
March 31, 2010
|
|
|
|
|
|
|
|
/s/ Richard
Chin
Richard
Chin
|
|
Director
|
|
March 31, 2010
|
|
|
|
|
|
|
|
/s/ Stephen
S. Galliker
Stephen
S. Galliker
|
|
Director
|
|
March 31, 2010
|
|
|
|
|
|
|
|
/s/ Steven
A. Kriegsman
Steven
A. Kriegsman
|
|
Director
|
|
March 31, 2010
|
|
|
|
|
|
|
|
/s/ Rudolph
Nisi
Rudolph
Nisi
|
|
Director
|
|
March 31, 2010
|
74
EXHIBIT INDEX
| |
|
|
|
|
Exhibit
|
|
|
|
Number
|
|
Description
|
|
|
|
|
2
|
.1
|
|
Contribution Agreement between CytRx Corporation and RXi
Pharmaceuticals Corporation, dated January 8, 2007(1)
|
|
|
2
|
.2
|
|
Contribution Agreement between CytRx Corporation and RXi
Pharmaceuticals Corporation, dated April 30, 2007(1)
|
|
|
2
|
.3
|
|
Reimbursement Agreement between CytRx Corporation and RXi
Pharmaceuticals Corporation, dated January 8, 2007(1)
|
|
|
3
|
.1
|
|
Form of Amended and Restated Certificate of Incorporation of RXi
Pharmaceuticals Corporation(1)
|
|
|
3
|
.2
|
|
Form of Amended and Restated By-laws of RXi Pharmaceuticals
Corporation(1)
|
|
|
4
|
.1
|
|
Specimen common stock certificate(3)
|
|
|
4
|
.2
|
|
Stockholders Agreement between CytRx Corporation, RXi
Pharmaceuticals Corporation, the other Stockholders and the
Scientific Advisory Board Members, dated February 23,
2007(1)
|
|
|
4
|
.3
|
|
Exhibit A to Contribution Agreement —
Registration Rights Terms between CytRx Corporation and RXi
Pharmaceuticals Corporation, dated April 30, 2007(1)
|
|
|
4
|
.4
|
|
Annex I to form of Subscription Agreement —
Registration Rights Terms between RXi Pharmaceuticals
Corporation and Stephen Galliker, Mark Ahn and Sanford
Hillsberg(1)
|
|
|
4
|
.5
|
|
Form of Securities Purchase Agreement between RXi
Pharmaceuticals Corporation and various investors, dated
June 24, 2008(5)
|
|
|
4
|
.6
|
|
Amendment to Stockholders Agreement between CytRx Corporation,
RXi Pharmaceuticals Corporation, the Stockholders and the
Scientific Advisory Board Members, dated July 28, 2008(7)
|
|
|
4
|
.7
|
|
Amendment to Exhibit A to Contribution
Agreement — Registration Rights Terms between CytRx
Corporation and RXi Pharmaceuticals Corporation, dated
July 28, 2008(7)
|
|
|
4
|
.8
|
|
Form of Warrant Certificate issued to certain purchasers of the
RXI Pharmaceuticals Corporation’s common stock in August
2009(14)
|
|
|
10
|
.1
|
|
Voting Agreement between CytRx Corporation and the University of
Massachusetts Medical School, dated January 10, 2007(1)
|
|
|
10
|
.2
|
|
Invention Disclosure Agreement between the University of
Massachusetts Medical School and RXi Pharmaceuticals
Corporation, dated January 10, 2007(2)
|
|
|
10
|
.3
|
|
Placement Agency Agreement between RXi Pharmaceuticals
Corporation, Jeffries & Company, Inc., Natixis
Bleichroeder Inc., Broadpoint Securities Group, Inc. and Griffin
Securities, Inc., dated June 24, 2008(5)
|
|
|
10
|
.4
|
|
RXI Pharmaceuticals Corporation’s Amended 2007 Incentive
Plan, dated June 5, 2009(13)
|
|
|
10
|
.5
|
|
Warrant
No. A-1
in favor of J.P. Turner Partners, dated August 7, 2008(8)
|
|
|
10
|
.6
|
|
Standby Equity Distribution Agreement between RXi
Pharmaceuticals Corporation and YA Global Master SPV Ltd. dated
January 30, 2009(10)
|
|
|
10
|
.7
|
|
Registration Rights Agreement between RXi Pharmaceuticals
Corporation and YA Global Master SPV Ltd. dated January 30,
2009(10)
|
|
|
10
|
.8
|
|
Form of Securities Purchase Agreement between RXi
Pharmaceuticals Corporation and various investors, dated
July 31, 2009(14)
|
|
|
10
|
.9
|
|
Form of Common Stock Purchase Warrant dated July 31, 2009
|
|
|
10
|
.10
|
|
Exclusive License Agreement (No.: UMMC
06-21-01)
between the University of Massachusetts and RXi Pharmaceuticals
Corporation, dated January 10, 2007+(2)
|
|
|
10
|
.11
|
|
Exclusive License Agreement (No.: UMMC
03-68-02)
between the University of Massachusetts and RXi Pharmaceuticals
Corporation, dated January 10, 2007+(2)
|
|
|
10
|
.12
|
|
Exclusive License Agreement (No.: UMMC
03-75-01)
between the University of Massachusetts and RXi Pharmaceuticals
Corporation, dated January 10, 2007+(2)
|
|
|
10
|
.13
|
|
Non-Exclusive License Agreement (No.: UMMC
06-08-03)
between the University of Massachusetts and RXi Pharmaceuticals
Corporation, dated January 10, 2007+(2)
|
75
| |
|
|
|
|
Exhibit
|
|
|
|
Number
|
|
Description
|
|
|
|
|
10
|
.14
|
|
Non-Exclusive License Agreement, between CytRx Corporation and
the University of Massachusetts Medical School related to UMMS
disclosure number
01-36, dated
April 15, 2003, as amended February 1, 2004+(2)
|
|
|
10
|
.15
|
|
Exclusive License Agreement between CytRx Corporation and the
University of Massachusetts Medical School related to UMMS
disclosure number
02-01, dated
April 15, 2003, as amended September 10, 2004+(2)
|
|
|
10
|
.16
|
|
Amended and Restated Exclusive License Agreement between CytRx
Corporation and the University of Massachusetts Medical School
related to UMMS disclosure number
03-05,
00-37,
01-31,
03-134,
93-09 and
02-38, dated
September 15, 2003, as amended September 17, 2003 and
February 1, 2004+(2)
|
|
|
10
|
.17
|
|
Exclusive License Agreement between CytRx Corporation and the
University of Massachusetts Medical School related to UMMS
disclosure number
03-17, dated
April 15, 2003, as amended January 7, 2004 and
February 1, 2004+(2)
|
|
|
10
|
.18
|
|
Exclusive License Agreement between CytRx Corporation and the
University of Massachusetts Medical School related to UMMS
disclosure number
03-60, dated
April 15, 2003 as amended February 1, 2004+(2)
|
|
|
10
|
.19
|
|
Co-Exclusive License Agreement between CytRx Corporation and the
University of Massachusetts Medical School related to UMMS
disclosure number
03-33, and
all amendments thereto, dated May 18, 2006+(2)
|
|
|
10
|
.20
|
|
License Agreement between CytRx Corporation, Imperial College
Innovations Limited and Imperial College of Science and
Technology, dated May 19, 2004+(2)
|
|
|
10
|
.21
|
|
Employment Agreement between RXi Pharmaceuticals Corporation and
Pamela Pavco, dated March 7, 2007*(1)
|
|
|
10
|
.22
|
|
Employment Agreement between RXi Pharmaceuticals Corporation and
James Warren, dated May 23, 2007*(1)
|
|
|
10
|
.23
|
|
Employment Agreement between RXi Pharmaceuticals Corporation and
Dmitry Samarsky, dated June 25, 2007*(1)
|
|
|
10
|
.24
|
|
Employment Agreement between RXi Pharmaceuticals Corporation and
Stephen J. DiPalma, dated August 28, 2007*(1)
|
|
|
10
|
.25
|
|
Employment Agreement between RXi Pharmaceuticals Corporation and
Anastasia Khvorova, dated October 17, 2008*(11)
|
|
|
10
|
.26
|
|
Employment Agreement between RXi Pharmaceuticals Corporation and
Konstantinos Andrikopoulos, dated November 10, 2008*(11)
|
|
|
10
|
.27
|
|
Employment Agreement between RXi Pharmaceuticals Corporation and
Noah D. Beerman, dated November 5, 2009*(16)
|
|
|
10
|
.28
|
|
Separation Agreement and General Release between RXi
Pharmaceuticals Corporation and Tod Woolf, dated
November 5, 2009*(16)
|
|
|
10
|
.29
|
|
RXi Pharmaceuticals Corporation’s 2007 Incentive Plan*(1)
|
|
|
10
|
.30
|
|
Form of Incentive Stock Option*(1)
|
|
|
10
|
.31
|
|
Form of Non-qualified Stock Option*(2)
|
|
|
10
|
.32
|
|
Lease between RXi Pharmaceuticals Corporation and Newgate
Properties, LLC for One Gateway Place, Worcester, Massachusetts,
01605, dated September 25, 2007(3)
|
|
|
10
|
.33
|
|
Amendment to Lease between Xi Pharmaceuticals Corporation and
Newgate Properties, LLC for One Gateway Place, Worcester,
Massachusetts, 01605, dated January 23, 2009(9)
|
|
|
10
|
.34
|
|
Amendment to Lease between Xi Pharmaceuticals Corporation and
Newgate Properties, LLC for One Gateway Place, Worcester,
Massachusetts, 01605, dated March 5, 2009(12)
|
|
|
10
|
.35
|
|
Form of Subscription Agreement between RXi Pharmaceuticals
Corporation and each of Mark K. Ahn, Ph.D., Stephen S.
Galliker and Sanford J. Hillsberg(3)
|
|
|
10
|
.36
|
|
Scientific Advisory Board Agreement between RXi Pharmaceuticals
Corporation and Tariq Rana, Ph.D., dated February 26,
2007 and corresponding Letter Agreement, dated April 30,
2007(3)
|
76
| |
|
|
|
|
Exhibit
|
|
|
|
Number
|
|
Description
|
|
|
|
|
10
|
.37
|
|
Scientific Advisory Board Agreement between RXi Pharmaceuticals
Corporation and Gregory Hannon, Ph.D., dated
February 26, 2007 and corresponding Letter Agreement dated
April 30, 2007(3)
|
|
|
10
|
.38
|
|
Scientific Advisory Board Agreement between RXi Pharmaceuticals
Corporation and Michael Czech, Ph.D., dated
February 26, 2007 and corresponding Letter Agreement dated
April 30, 2007(3)
|
|
|
10
|
.39
|
|
Scientific Advisory Board Agreement between RXi Pharmaceuticals
Corporation and Craig C. Mello, Ph.D., dated
February 26, 2007 and corresponding Letter Agreement dated
April 30, 2007(3)
|
|
|
10
|
.40
|
|
Letter Agreement between CytRx Corporation and RXi
Pharmaceuticals Corporation, dated December 27, 2007(3)
|
|
|
10
|
.41
|
|
Patent License Agreement between RXi Pharmaceuticals Corporation
and Invitrogen IP Holdings, Inc. dated November 1, 2007(4)
|
|
|
10
|
.42
|
|
Patent and Technology Assignment Agreement between RXi
Pharmaceuticals Corporation and Advirna, LLC dated
September 21, 2009+(15)
|
|
|
14
|
.1
|
|
Code of Conduct(5)
|
|
|
23
|
.1
|
|
Consent of BDO Seidman, LLP, Independent Registered Public
Accounting Firm(16)
|
|
|
31
|
.1
|
|
Sarbanes-Oxley Act Section 302 Certification of Noah D.
Beerman(16)
|
|
|
31
|
.2
|
|
Sarbanes-Oxley Act Section 302 Certification of Amy Tata(16)
|
|
|
32
|
.1
|
|
Sarbanes-Oxley Act Section 906 Certification of Noah D.
Beerman and Amy Tata(16)
|
|
|
|
|
(1) |
|
Previously filed as an Exhibit to the Company’s
Registration Statement on
Form S-1
filed on October 30, 2007 (File
No. 333-147009)
and incorporated by reference herein |
| |
|
(2) |
|
Previously filed as an Exhibit to Amendment No. 1 to the
Company’s Registration Statement on
Form S-1
filed on November 19, 2007(File
No. 333-147009)
and incorporated by reference herein. |
| |
|
(3) |
|
Previously filed as an Exhibit to Amendment No. 2 to the
Company’s Registration Statement on
Form S-1
filed on January 20, 2008 (File
No. 333-147009)
and incorporated by reference herein. |
| |
|
(4) |
|
Previously filed as an Exhibit to Amendment No. 3 to the
Company’s Registration Statement on
Form S-1
filed on February 1, 2008 (File
No. 333-147009)
and incorporated by reference herein. |
| |
|
(5) |
|
Previously filed as an Exhibit to the Company’s
Form 8-K
filed on June 26, 2008 (File No.
001-33958)
and incorporated by reference herein. |
| |
|
(6) |
|
Previously filed as an Exhibit to the Company’s
Form 8-K
filed on July 24, 2008 (File No.
001-33958)
and incorporated by reference herein. |
| |
|
(7) |
|
Previously filed as an Exhibit to Amendment No. 1 to the
Company’s Registration Statement on
Form S-1
filed on August 4, 2008 (File
No. 333-152555)
and incorporated by reference herein. |
| |
|
(8) |
|
Previously filed as an Exhibit to the Company’s
Form 10-Q
filed on November 14, 2008
(File No. 001-33958)
and incorporated by reference herein. |
| |
|
(9) |
|
Previously filed as an Exhibit to the Company’s
Form 8-K
filed on January 23, 2009
(File No. 001-33958)
and incorporated by reference herein. |
| |
|
(10) |
|
Previously filed as an Exhibit to the Company’s
Form 8-K
filed on February 5, 2009
(File No. 001-33958)
and incorporated by reference herein. |
| |
|
(11) |
|
Previously filed as an Exhibit to the Company’s
Form 10-K
filed on March 18, 2009
(File No. 001-33958)
and incorporated by reference herein. |
| |
|
(12) |
|
Previously filed as an Exhibit to the Company’s
Form 10-Q
filed on May 15, 2009
(File No. 001-33958)
and incorporated by reference herein. |
| |
|
(13) |
|
Previously filed as an Exhibit to the Company’s
Form 8-K
filed on June 10, 2009 (File No.
001-33958)
and incorporated by reference herein. |
| |
|
(14) |
|
Previously filed as an Exhibit to the Company’s
Form 8-K
filed on July 31, 2009 (File No.
001-33958)
and incorporated by reference herein. |
77
|
|
|
|
(15) |
|
Previously filed as an Exhibit to the Company’s
Form 10-Q
filed on November 16, 2009 (File
No. 001-33958)
and incorporated by reference herein. |
| |
|
(16) |
|
Filed herewith. |
| |
|
* |
|
Indicates a management contract or compensatory plan or
arrangement. |
| |
|
+ |
|
This exhibit was filed separately with the Commission pursuant
to an application for confidential treatment. The confidential
portions of the exhibit have been omitted and have been marked
by an asterisk. |
78