UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark one)
| |
|
|
| þ |
|
ANNUAL REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2009
| |
|
|
| o |
|
TRANSITION REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to .
Commission file number 001-31812
BIOSANTE PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
| |
|
|
| Delaware
|
|
58-2301143 |
| (State or other jurisdiction of incorporation or organization)
|
|
(I.R.S. Employer Identification No.) |
| |
|
|
| 111 Barclay Boulevard |
|
|
| Lincolnshire, Illinois
|
|
60069 |
| (Address of principal executive offices)
|
|
(Zip Code) |
(847) 478-0500
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| |
|
|
| Title of each class
|
|
Name of each exchange on which registered |
| Common Stock, par value $0.0001 per share
|
|
The NASDAQ Stock Market |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of
the Securities Act. YES o NO þ
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or
Section 15(d) of the Act. YES o NO þ
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by
Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for
such shorter period that the registrant was required to file such reports), and (2) has been
subject to such filing requirements for the past 90 days. YES þ NO o
Indicate by check mark whether the registrant has submitted electronically and posted on its
corporate Web site, if any, every Interactive Data File required to be submitted and posted
pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period
that the registrant was required to submit and post such files). YES o NO o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K
(§229.405 of this chapter) is not contained herein, and will not be contained, to the best of
registrant’s knowledge, in definitive proxy or information statements incorporated by reference in
Part III of this Form 10-K or any amendment to this Form 10-K. þ
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a
non-accelerated filer, or a smaller reporting company. See definitions of “large accelerated
filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
(Check one):
| |
|
|
|
|
|
|
| Large accelerated filer o
|
|
Accelerated filer o
|
|
Non-accelerated filer o
(Do not check if a smaller reporting company)
|
|
Smaller reporting company þ |
Indicate by check mark whether registrant is a shell company (as defined in Rule 12b-2 of the Act).
YES o NO þ
The aggregate market value of the registrant’s common stock, excluding shares beneficially owned by
affiliates, computed by reference to the closing sale price at which the common stock was last sold
as of June 30, 2009 (the last business day of the registrant’s second fiscal quarter) as reported
by The NASDAQ Global Market on that date was approximately $48.8 million.
As of
March 15, 2010, 63,667,194 shares of common stock of the registrant were outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Part III of this annual report on Form 10-K incorporates by reference information (to the extent
specific sections are referred to herein) from the registrant’s Proxy Statement for its 2010 Annual
Meeting of Stockholders to be held in June 2010.
This annual report on Form 10-K contains or incorporates by reference forward-looking statements.
For this purpose, any statements contained in this Form 10-K that are not statements of historical
fact may be deemed to be forward-looking statements. You can identify forward-looking statements
by those that are not historical in nature, particularly those that use terminology such as
“believe,” “may,” “could,” “would,” “might,” “possible,” “potential,” “project,” “will,” “should,”
“expect,” “intend,” “plan,” “predict,” “anticipate,” “estimate,” “approximate,” “contemplate” or
“continue”, the negative of these words, other words and terms of similar meaning or the use of
future dates. In evaluating these forward-looking statements, you should consider various factors,
including those listed below under the headings “Part I. Item I. Business — Forward-Looking
Statement” and “Part I. Item 1A. Risk Factors.” These factors may cause our actual results to
differ materially from any forward-looking statement.
As used in this report, references to “BioSante,” the “company,” “we,” “our” or “us,” unless the
context otherwise requires, refer to BioSante Pharmaceuticals, Inc.
We own or have the rights to use various trademarks, trade names or service marks, including
BioSante®, LibiGel®, Elestrin™, Bio-T-Gel™, The Pill-Plus™, BioLook™,
BioVant™, BioOral™, BioAir™ and GVAX™. This report also contains trademarks, trade names and
service marks that are owned by other persons or entities.
ii
PART I
Item 1. BUSINESS
Company Overview
We are a specialty pharmaceutical company focused on developing products for female sexual health,
menopause, contraception and male hypogonadism. In addition, we are evaluating and seeking
opportunities for our GVAX cancer immunotherapies, 2A/Furin and other technologies we acquired in
our merger with Cell Genesys, Inc. in October 2009. We also are developing our calcium phosphate
technology (CaP) for aesthetic medicine (BioLook), as a vaccine adjuvant, including for an H1N1
(swine flu) vaccine, and drug delivery.
Our products for female sexual health, menopause, contraception and male hypogonadism include:
| • |
|
LibiGel — once daily transdermal testosterone gel in Phase III clinical development under
a Special Protocol Assessment (SPA) for the treatment of female sexual dysfunction (FSD). |
| • |
|
Elestrin — once daily transdermal estradiol (estrogen) gel approved by the U.S. Food and
Drug Administration (FDA) indicated for the treatment of moderate-to-severe vasomotor symptoms
(hot flashes) associated with menopause and marketed in the U.S. |
| • |
|
The Pill-Plus (triple component contraceptive) — once daily use of various combinations of
estrogens, progestogens and androgens in development for the treatment of FSD in women using
oral or transdermal contraceptives. |
| • |
|
Bio-T-Gel — once daily transdermal testosterone gel in development for the treatment of
hypogonadism, or testosterone deficiency, in men. |
We believe LibiGel remains the lead pharmaceutical product in the U.S. in active development for
the treatment of hypoactive sexual desire disorder (HSDD) in menopausal women, and that it has the
potential to be the first product approved by the FDA for this common and unmet medical need. We
believe based on agreements with the FDA, including an SPA, that two Phase III safety and efficacy
trials and one year of LibiGel exposure in a Phase III cardiovascular and breast cancer safety
study with a four-year follow-up post-NDA filing and potentially post-FDA approval are the
essential requirements for submission and, if successful, approval by the FDA of a new drug
application (NDA) for LibiGel for the treatment of FSD, specifically, HSDD in menopausal women. We
have three SPAs in place concerning LibiGel. The first SPA agreement covers the pivotal Phase III
safety and efficacy trials of LibiGel in the treatment of FSD for “surgically”
menopausal women. The second SPA covers our LibiGel program in the treatment of FSD, specifically
HSDD, in “naturally” menopausal women. The third SPA agreement covers the LibiGel stability, or
shelf life, studies for the intended commercialization of LibiGel product.
1
Currently, three LibiGel Phase III studies are underway and enrolling women: two LibiGel Phase III
safety and efficacy clinical trials and one Phase III cardiovascular and breast cancer safety
study. Both Phase III safety and efficacy trials are double-blind, placebo-controlled trials that
will enroll up to approximately 500 surgically menopausal women each for a six-month clinical
trial. The Phase III safety study is a randomized, double-blind, placebo-controlled, multi-center,
cardiovascular events driven study of between 2,400 and 3,100 women exposed to LibiGel or placebo
for 12 months after which time we intend to submit an NDA to the FDA. In February 2010, we
announced that based upon the second
review of study conduct and unblinded data from the LibiGel Phase III cardiovascular and breast
cancer safety study, the independent data monitoring committee (DMC) unanimously recommended
continuing the study as described in the FDA-agreed study protocol, with no modifications. The DMC
reviewed all unblinded adverse events in the safety study including “serious adverse events” and
all “adverse cardiovascular and breast cancer events” in almost 1,200 women-years of exposure. As
of such date, there had been no deaths, only six adjudicated cardiovascular events and four breast
cancers reported. In view of the DMC recommendation, we will continue the LibiGel Phase III
development program as planned. We continue to target submission to the FDA of an NDA by mid-2011.
Following NDA submission and potential FDA approval, we will continue to follow the subjects in
the safety study for an additional four years.
Elestrin is our first FDA approved product. Azur Pharma International II Limited is marketing
Elestrin in the U.S. using its women’s health sales force that targets estrogen prescribing
physicians in the U.S. comprised mostly of gynecologists. In December 2009, we entered into an
amendment to our original licensing agreement with Azur which permanently reduced the royalty
percentage due to us related to Azur’s sales of Elestrin. Upon signing the amended agreement, Azur
made a $1.0 million nonrefundable payment in December 2009 in exchange for a permanent reduction in
future royalty rates, and received options to make a total of $2.16 million in additional
non-refundable payments (which were exercised during the first quarter of 2010) in exchange for the
elimination of substantially all remaining future royalties and milestone payments due us under the
terms of the original license. The $1.0 million nonrefundable payment was recorded as revenue during
the fourth quarter of 2009 as we had no remaining performance obligations with respect to this
amount. We maintain the right to receive up to $140 million in sales-based milestone payments from
Azur if Elestrin reaches certain predefined sales per calendar year.
As a result of our October 2009 merger with Cell Genesys, we acquired a portfolio of products,
including GVAX cancer immunotherapies. GVAX cancer immunotherapies are cancer vaccines designed to
stimulate the patient’s immune system to effectively fight cancer. Multiple Phase II trials of
these technologies which had been ongoing at the date of the acquisition are continuing at minimal
cost to us at the Johns Hopkins Sidney Kimmel Comprehensive Cancer Center in various cancer
programs, including pancreatic cancer, leukemia and breast cancer.
Our CaP technology is based on the use of extremely small, solid, uniform particles, which we call
“nanoparticles.” We are pursuing the development of three potential initial applications for our
CaP technology. First, CaP technology is being tested in the area of aesthetic medicine. Second,
our CaP technology is being tested for its “adjuvant” activity that enhances the ability of a
vaccine to stimulate an immune response. The same nanoparticles allow for delivery of the vaccine
via alternative routes of administration including non-injectable routes of administration. Third,
we are pursuing the creation of oral, buccal, intranasal, inhaled and longer acting delivery of
drugs that currently must be given by injection (e.g., insulin).
The following is a list of our CaP products in development:
| • |
|
BioLook — facial line filler in development using proprietary CaP technology in the area
of aesthetic medicine, e.g., a facial line filler. |
| • |
|
BioVant — proprietary CaP adjuvant and delivery technology in development for improved
vaccines against viral and bacterial infections and autoimmune diseases, among others.
BioVant also serves as a delivery system for non-injected delivery of vaccines. |
2
| • |
|
BioOral — a delivery system using CaP technology for oral/buccal/intranasal administration
of proteins and other therapies that currently must be injected. |
| • |
|
BioAir — a delivery system using CaP technology for inhalable versions of proteins and
other therapies that currently must be injected. |
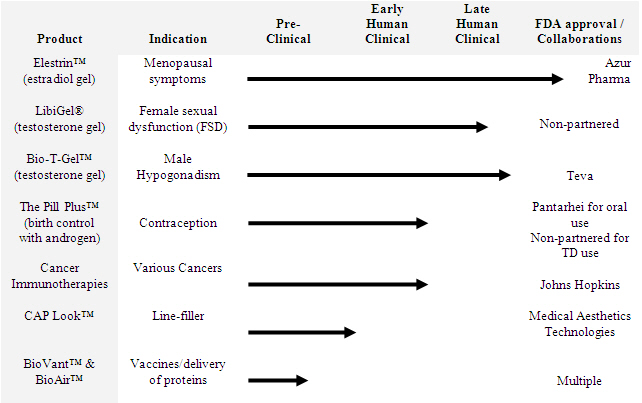
BioSante’s Product Portfolio
One of our strategic goals is to continue to seek and implement strategic alternatives with respect
to our products and our company, including licenses, business collaborations and other business
combinations or transactions with other pharmaceutical and biotechnology companies. Therefore, as
a matter of course, we may engage in discussions with third parties regarding the licensure, sale
or acquisition of our products and technologies or a merger or sale of our company. As part of
this process, we merged with Cell Genesys in October 2009. As a result of this transaction,
although we will continue to focus primarily on our LibiGel clinical development program, we also
will seek future development opportunities for our GVAX cancer immunotherapies, including potential
combination with BioVant, our vaccine adjuvant, as well as possible external collaborations, and
also will seek to outlicense or sell other technologies acquired from Cell Genesys. In addition,
as a result of our merger with Cell Genesys, we acquired an investment equivalent to approximately
16 percent of the total equity of Ceregene, Inc., a privately held biotechnology company focused on
the treatment of major neurodegenerative disorders using the delivery of nervous system growth
factors.
3
Description of Our Female Sexual Health, Menopause, Contraception and Male Hypogonadism Products
Overview. Our products for female sexual health, menopause, contraception and male hypogonadism
include our gel formulations of estradiol, testosterone and a combination of estradiol and
testosterone: Elestrin, Bio-T-Gel and LibiGel, and our triple component contraceptive that uses
various combinations
of estrogens, progestogens and androgens in development for the treatment of FSD in women using
oral or transdermal contraceptives, The Pill-Plus.
Our gel products are designed to be quickly absorbed through the skin after application on the
upper arm for the women’s products, delivering the hormone to the bloodstream evenly and in a
non-invasive, painless manner. The gels are formulated to be applied once per day and to be
absorbed into the skin without a trace of residue and to dry in under one to two minutes. We
believe our gel products have a number of benefits over competitive products, including the
following:
| • |
|
our transdermal gels can be spread over areas of skin where they dry rapidly and decrease
the chance for skin irritation versus transdermal patches; |
| • |
|
our transdermal gels may have fewer side effects than many pills which have been known to
cause gallstones, blood clots and complications related to metabolism; |
| • |
|
our transdermal gels have been shown to be well absorbed, thus allowing clinical hormone
levels to reach the systemic circulation; |
| • |
|
transdermal gels may allow for better dose adjustment than either transdermal patches or
oral tablets or capsules; and |
| • |
|
transdermal gels may be more appealing to patients since they are less conspicuous than
transdermal patches, which may be aesthetically unattractive. |
LibiGel. We believe LibiGel, if approved by the FDA, could be a very successful product. LibiGel
is a once daily transdermal testosterone gel designed to treat FSD, specifically HSDD in menopausal
women. The majority of women with FSD are postmenopausal, experiencing FSD due to hormonal changes
due to aging or following surgical menopause. LibiGel successfully has completed a Phase II
clinical trial, and three Phase III safety and efficacy clinical studies are currently underway and
enrolling women.
We believe LibiGel remains the lead pharmaceutical product in the U.S. in active development for
the treatment of HSDD in menopausal women, and that it has the potential to be the first product
approved by the FDA for this common and unmet medical need. We believe based on agreements with
the FDA, including an SPA, that two Phase III safety and efficacy trials and one year of LibiGel
exposure in a Phase III cardiovascular and breast cancer safety study with a four-year follow-up
post-NDA filing and potentially post-FDA approval are the essential requirements for submission
and, if successful, approval by the FDA of an NDA for LibiGel for the treatment of FSD,
specifically HSDD in menopausal women. The SPA process and agreement affirms that the FDA agrees
that the LibiGel Phase III safety and efficacy clinical trial design, clinical endpoints, sample
size, planned conduct and statistical analyses are acceptable to support regulatory approval.
Further, it indicates that these agreed measures will serve as the basis for regulatory review and
any decision by the FDA to approve an NDA for LibiGel. These SPA trials use our validated
instruments to measure the clinical endpoints. We have three SPA agreements in place concerning
LibiGel. The first SPA agreement covers the pivotal Phase III safety and efficacy trials of
LibiGel in the treatment of FSD for “surgically” menopausal women. The second SPA covers LibiGel
for the treatment of FSD, specifically, HSDD in “naturally” menopausal women. The third SPA
agreement covers the LibiGel stability, or shelf life, studies for the intended commercialization
of LibiGel product.
4
Currently, three LibiGel Phase III studies are underway and enrolling women: two LibiGel Phase III
safety and efficacy clinical trials and one Phase III cardiovascular and breast cancer safety
study. Both Phase III safety and efficacy trials are double-blind, placebo-controlled trials that
will enroll up to
approximately 500 surgically menopausal women each for a six-month clinical trial. The Phase III
safety study is a randomized, double-blind, placebo-controlled, multi-center, cardiovascular events
driven study of between 2,400 and 3,100 women exposed to LibiGel or placebo for 12 months after
which time we intend to submit an NDA to the FDA. In February 2010, we announced that based upon
the second review of study conduct and unblinded data from the LibiGel Phase III cardiovascular and
breast cancer safety study, the independent data monitoring committee unanimously recommended
continuing the study as described in the FDA-agreed study protocol, with no modifications. The DMC
reviewed all unblinded adverse events in the safety study including “serious adverse events” and
all “adverse cardiovascular and breast cancer events” in almost 1,200 women-years of exposure. As
of such date, there had been no deaths, only six adjudicated cardiovascular events and only four
breast cancers reported. In view of DMC recommendation, we will continue the LibiGel Phase III
development program as planned. We continue to target submission to the FDA of an NDA by mid-2011.
Following NDA submission and potential FDA approval, we will continue to follow the subjects in
the safety study for an additional four years.
There is no pharmaceutical product currently approved in the United States for FSD, specifically
HSDD. While several therapies have been tested to treat FSD, thus far testosterone therapy appears
to be the only treatment that results in a consistent significant increase in the number of
satisfying sexual events in women, which represents one of the two key efficacy endpoints chosen by
the FDA for pivotal clinical trials of FSD therapies. We are not aware of another testosterone
therapy product for the treatment of FSD in active clinical development in the U.S. other than
LibiGel.
Although generally characterized as limited to men, testosterone also is present in women and its
deficiency has been found to cause low libido or sex drive. Studies have shown that testosterone
therapy in women can boost sexual desire, sexual activity and pleasure, increase bone density,
raise energy levels and improve mood. According to a study published in the Journal of the
American Medical Association, 43 percent of American women between the ages of 18-59, or about 40
million women, experience some degree of impaired sexual function. Among the more than 1,400 women
surveyed, 32 percent lacked interest in sex (low sexual desire) and 26 percent could not experience
orgasm. Furthermore, according to a study published in the New England Journal of Medicine, 43
percent of American women between the ages of 57-85 experience low sexual desire. Importantly,
according to IMS data, two million testosterone prescriptions were written off-label for women by
U.S. physicians in 2007. Female sexual dysfunction is defined as a lack of sexual desire, arousal
or pleasure. The majority of women with FSD are postmenopausal, experiencing symptoms due to
hormonal changes that occur with aging or following surgical menopause.
Treatment with LibiGel in our Phase II clinical trial significantly increased satisfying
sexual events in surgically menopausal women suffering from FSD. The Phase II trial results showed
LibiGel significantly increased the number of satisfying sexual events by 238 percent versus
baseline; this increase also was significant versus placebo. In this study, the effective dose of
LibiGel produced testosterone blood levels within the normal range for pre-menopausal women and had
a safety profile similar to that observed in the placebo group. In addition, no serious adverse
events and no discontinuations due to adverse events occurred in any subject receiving LibiGel.
The Phase II clinical trial was a double-blind, placebo-controlled trial, conducted in the United
States, in surgically menopausal women distressed by their low sexual desire and activity.
5
Elestrin. Elestrin is our first FDA approved product. Elestrin is a once daily transdermal gel
that delivers estrogen without the skin irritation associated with, and the physical presence of,
transdermal patches, and to avoid the effects of oral estrogen. Elestrin contains estradiol versus
conjugated equine estrogen contained in the most commonly prescribed oral estrogen.
In December 2006, we received FDA approval for the marketing of Elestrin in the United States.
Elestrin is indicated for the treatment of moderate-to-severe vasomotor symptoms (hot flashes)
associated with menopause. Elestrin is administered using a metered dose applicator that delivers
0.87 grams of gel per actuation, thereby allowing for precise titration from dose to dose. Two
doses of Elestrin, 0.87 grams per day and 1.7 grams per day, were approved by the FDA. The 0.87
gram dose of Elestrin, which delivers 12.5 mcg of estradiol per day, is one of the lowest daily
doses of estradiol approved by the FDA for the treatment of hot flashes and is 67 percent lower
than the lowest dose, FDA-approved estrogen patch for hot flashes on the market. The Elestrin FDA
approval was a non-conditional and full approval. In addition, we received three years of
marketing exclusivity for Elestrin.
In December 2008, we entered into a license agreement and an asset purchase agreement with Azur for
the marketing of Elestrin and the sale of certain assets related to Elestrin pursuant to which we
received approximately $3.3 million. Under the license agreement, Azur was to pay us royalties on
sales of Elestrin ranging from 10 percent to 20 percent depending primarily upon the annual sales
levels and additional sales-based milestone payments. In April 2009, we announced the initiation
of sales and marketing activity of Elestrin by Azur. In December 2009, we entered into an
amendment to our original licensing agreement with Azur which permanently reduced the royalty
percentage due to us related to Azur’s sales of Elestrin. Upon signing the amended agreement, Azur
made a $1.0 million nonrefundable payment in December 2009 in exchange for a permanent reduction in
future royalty rates, and received options to make a total of $2.16 million in additional
non-refundable payments (which were exercised during the first quarter of 2010) in exchange for the
elimination of substantially all remaining future royalties and milestone payments due us under the
terms of the original license. The $1.0 million nonrefundable payment was recorded as revenue during
the fourth quarter of 2009 as we had no remaining performance obligations with respect to this
amount. We maintain the right to receive up to $140 million in sales-based milestone payments from
Azur if Elestrin reaches certain predefined sales per calendar year.
In December 2008, we signed an exclusive agreement with PharmaSwiss SA for the marketing of
Elestrin in Israel. PharmaSwiss is responsible for regulatory and marketing activities in Israel.
In June 2009, PharmaSwiss submitted a new drug application to the Israeli authorities based on our
approved U.S. NDA and manufacturing information.
According to The North American Menopause Society, there are more than 40 million postmenopausal
women in the U.S., and this group is expected to grow 25 percent by 2010. Menopause begins when
the ovaries cease to produce estrogen, or when both ovaries are surgically removed prior to natural
menopause. The average age at which women experience natural menopause is 51 years. The average
age of surgical menopause is 41 years. The most common physical symptoms of natural or surgical
menopause and the resultant estrogen deficiency are hot flashes, vaginal atrophy and osteoporosis.
According to the North American Menopause Society, studies show that hot flashes occur in
approximately two-thirds of menopausal women. Hormone therapy in women decreases the chance that
women will experience the symptoms of menopause due to estrogen deficiency. According to industry
estimates, approximately six million women in the U.S. currently are receiving some form of
estrogen or combined estrogen hormone therapy. According to IMS Health, the current market in the
U.S. for single-entity estrogen products was approximately $1.5 billion in 2009, of which the
transdermal segment, mostly patches, is reported at about $316 million. As the “baby boomer”
generation ages, the number of women reaching menopause, a large percentage of whom may need
estrogen or combined estrogen therapy, is between 5,000 and 6,000 women per day in the U.S.
6
There are several treatment options for women experiencing menopausal symptoms, which vary
according to which symptoms a woman experiences and whether or not she has had a hysterectomy.
Estrogen is most commonly given orally in pill or tablet form. There are several potential side
effects,
however, with the use of oral estrogen, including insufficient absorption by the circulatory
system, upset stomach, gallstones, blood clots as well as an increase in C-reactive protein, a
possible marker for cardiovascular inflammation. Reports suggest that oral estrogen causes an
increase in strokes and blood clots. Although transdermal, or skin, patches have been shown to
avoid some of these problems or effects, transdermal patches have a physical presence, can fall
off, and can result in skin irritation. However, transdermal delivery of estrogen via patches or
gels may reduce the risks associated with oral estrogen, including having no effect on C-reactive
protein and potentially reduce the risk of breast cancer and cardiovascular disease.
Bio-T-Gel. Bio-T-Gel is our once daily transdermal testosterone gel in development for the
treatment of hypogonadism, or testosterone deficiency, in men. Unlike LibiGel and Elestrin,
Bio-T-Gel is owned by us with no royalty or milestone obligations to any other party.
In December 2002, we entered into a development and license agreement, which was subsequently
amended, with Teva Pharmaceuticals USA, Inc., a wholly-owned subsidiary of Teva Pharmaceutical
Industries Ltd., pursuant to which Teva USA agreed to develop and market Bio-T-Gel for the U.S.
market. The financial terms of the development and license agreement included a $1.5 million
upfront payment by Teva USA, certain milestones and royalties on sales of the product, if and when
approved and marketed, in exchange for rights to develop and market the product. Teva USA also is
responsible under the terms of the agreement for continued development, regulatory filings and all
manufacturing and marketing associated with the product.
Testosterone deficiency in men is known as hypogonadism. Low levels of testosterone may result in
lethargy, depression, decreased sex drive, impotence, low sperm count and increased irritability.
Men with severe and prolonged reduction of testosterone also may experience loss of body hair,
reduced muscle mass, osteoporosis and bone fractures due to osteoporosis. Approximately five
million men in the United States, primarily over age 40, have lower than normal levels of
testosterone. Testosterone therapy has been shown to restore levels of testosterone with minimal
side effects.
There are currently several products on the market for the treatment of low testosterone levels in
men. As opposed to estrogen therapy products, oral administration of testosterone is currently not
possible as the hormone is, for the most part, rendered inactive in the liver making it difficult
to achieve adequate levels of the compound in the bloodstream. Current methods of administration
include testosterone injections, patches and gels. Testosterone injections require large needles,
are often painful and not effective for maintaining adequate testosterone blood levels throughout
the day. Delivery of testosterone through transdermal patches was developed primarily to promote
the therapeutic effects of testosterone therapy without the often painful side effects associated
with testosterone injections. Transdermal patches, however, similar to estrogen patches, have a
physical presence, can fall off, and can result in skin irritation. Testosterone formulated gel
products for men are designed to deliver testosterone without the pain of injections and the
physical presence, skin irritation and discomfort associated with transdermal patches. We are
aware of two gel testosterone products for men currently on the market in the United States.
According to IMS Health, the U.S. market for transdermal testosterone therapies grew approximately
28 percent in 2009 to $968 million from $755 million in 2008.
7
The Pill-Plus. The Pill-Plus is based on three issued U.S. patents claiming triple component
therapy via any route of administration (the combination use of estrogen plus progestogen plus
androgen, e.g. testosterone) and three issued U.S. patents pertaining to triple component
contraception. The Pill-Plus adds a third component, an androgen, to the normal two component
(estrogen and progestogen) oral contraceptive to prevent androgen deficiency which often leads to a
decrease in sexual desire, sexual activity and mood changes. In a completed Phase II double-blind
randomized clinical trial, the addition of an oral androgen resulted in restoration of testosterone
levels to the normal and physiological range for
healthy women. Paradoxically, many women who use oral contraceptives have reduced sexual desire,
arousabilty and activity due to the estrogen and progestogen in normal oral contraceptives. The
Pill-Plus is designed to improve female sexual dysfunction in oral contraceptive users, among other
potential benefits.
We have an exclusive license from Wake Forest University Health Sciences (formerly known as Wake
Forest University) and Cedars-Sinai Medical Center for the three issued U.S. patents for triple
component contraception. The financial terms of the license include an upfront payment, regulatory
milestone payments, maintenance payments and royalty payments by us if a product incorporating the
licensed technology gets approved and subsequently is marketed.
In May 2007, we announced that we sublicensed U.S. rights to The Pill-Plus to Pantarhei Bioscience
B.V. (Pantarhei), a Netherlands-based pharmaceutical company. Pantarhei is responsible under the
agreement for all expenses to develop and market the product. We may receive certain development
and regulatory milestones for the first product developed under the license. In addition, we will
receive royalty payments on any sales of the product in the U.S., if and when approved and
marketed. If the product is sublicensed by Pantarhei to another company, we will receive a
percentage of any and all payments received by Pantarhei for the sublicense from a third party. We
have retained all rights under our licensed patents to the transdermal delivery of triple component
contraceptives.
Other Products. In September 2000, we sublicensed the marketing rights to our gel products in
Canada to Paladin Labs Inc. In exchange for the sublicense, Paladin agreed to make an initial
investment in our company, make future milestone payments and pay royalties on sales of the
products in Canada. The milestone payments are required to be in the form of a series of equity
investments by Paladin in our common stock at a 10 percent premium to the market price of our stock
at the time the equity investment is made. If and when we receive FDA approval for any of our gel
products in the United States, Paladin may be able to use this information to obtain the
appropriate regulatory approvals of such products in Canada.
In August 2001, we entered into a sublicense agreement with Solvay Pharmaceuticals, B.V. (which was
purchased by Abbott Laboratories in February 2010) covering the U.S. and Canadian rights to the
estrogen/progestogen combination therapy gel product licensed from Antares. Under the terms of the
agreement, Solvay sublicenses our estrogen/progestogen combination therapy gel product for an
initial payment of $2.5 million, future milestone payments (of which $950,000 has been received to
date) and sales-based royalties. Solvay has been responsible for all costs of development to date.
We believe that the product licensed to Solvay is not in active development by Solvay, and we do
not expect its active development to occur at any time in the near future.
8
Description of GVAX Cancer Immunotherapies and Other Technologies Acquired in the Cell Genesys
Merger
GVAX Cancer Immunotherapy Technology. GVAX cancer immunotherapies are cancer vaccines designed to
stimulate the patient’s immune system to effectively fight cancer. GVAX cancer immunotherapies are
comprised of tumor cells that are genetically modified to secrete an immune-stimulating cytokine
known as granulocyte-macrophage colony-stimulating factor, or GM-CSF, and are then irradiated for
safety. Since GVAX cancer immunotherapies consist of whole tumor cells, the cancer patient’s
immune system can be activated against multiple tumor cell components, or antigens, potentially
resulting in greater clinical benefit than if the immunotherapy consisted of only a single tumor
cell component. Additionally, the secretion of GM-CSF by the modified tumor cells can enhance
greatly the immune response by recruiting and activating dendritic cells at the injection site, a
critical step in the optimal response by the immune system to any immunotherapy product. The
antitumor immune response
which occurs throughout the body following administration of a GVAX immunotherapy potentially can
result in the destruction of tumor cells that persist or recur following surgery, radiation therapy
or chemotherapy treatment.
GVAX cancer immunotherapies can be administered conveniently in an outpatient setting as an
injection into the skin, a site where immune cells, including in particular dendritic cells, can be
optimally accessed and activated. GVAX cancer immunotherapies are being tested as
patient-specific, or autologous, products and as non patient-specific, or allogeneic, products.
Multiple Phase II trials of these technologies which had been ongoing at the date of the
acquisition are continuing at minimal cost to us at the Johns Hopkins Sidney Kimmel Comprehensive
Cancer Center in various cancer programs, including pancreatic cancer, leukemia and breast cancer.
2A/Furin Protein Expression Technology. The 2A/furin technology is a novel expression system for
producing high levels of multimeric proteins. The 2A/furin technology allows for continuous,
equimolar expression of at least two proteins at high concentrations from a single expression
vector making it particularly useful for recombinant antibody expression. The technology
expression technology has been used successfully to express antibodies from several species,
including murine, rat and human, as well as a variety of antibody isotypes. The 2A/furin
expression technology has several potential applications including preclinical lead and target
validation, gene therapy, production of stable, high producer antibody cell lines, and commercial
production of antibodies and other proteins. The 2A/furin technology can increase the efficiency of
antibody production by cutting the cost and reducing the time to manufacture antibodies. According
to the Global Monoclonal Antibodies Review, the antibody market in the U.S. is estimated to be more
than $30 billion per year.
Oncolytic VirusTechnology. Our oncolytic virus technology uses replication-competent adenoviruses
derived from Adenovirus type 5, a common “cold” virus that replicate in and selectively kill tumor
cells. The replication of the virus is controlled by replacing the promoter of a gene required for
replication with a promoter that is preferentially expressed only in tumor cells. Furthermore, the
virus may optionally include a gene encoding a cytokine, which enhances immune stimulation to the
tumor, thereby providing a dual mechanism of action for killing targeted cancer cells by direct
cell lysis as well as via cellular and humoral immune responses to the tumor.
Description of Our CaP Technology and Products in Development
We believe our CaP technology can serve as a facial line filler in the area of aesthetic medicine
and as an effective vehicle for delivering drugs and vaccines and enhancing the effects of
vaccines. Our CaP nanoparticles successfully have passed the first stage of toxicity studies for
administration orally, into muscles, under the skin, and into the lungs by inhalation. We
successfully have completed a Phase I human clinical safety trial of CaP. We have entered into
several subcontract or development agreements with various corporate partners and governmental
entities concerning our CaP technology.
Overview of CaP Technology. Research and development involving our CaP technology originated under
an agreement dated April 6, 1989 between the University of California and one of our predecessor
companies, relating to viral protein surface absorption studies. The discovery research was funded
at UCLA School of Medicine and was based, in essence, on the use of extremely small, solid, uniform
particles as components that could increase the stability of drugs and act as systems to deliver
drugs into the body. Research in these areas at UCLA or our laboratory has resulted in the
issuance of a number of patents, which we either license from the University of California or own.
9
These ultra fine particles are made from inert, biologically acceptable materials, such as
ceramics, pure crystalline carbon or biodegradable calcium phosphate-like particles. The size of
the particles is in the
nanometer range. A nanometer is one millionth of a millimeter and typically particles measure
approximately 300 nanometers (nm). Because the size of these particles is measured in nanometers,
we use the term “nanoparticles” to describe them.
We use the nanoparticles as the basis of a delivery system. The critical property of these
nanoparticles is that biologically active molecules, proteins, peptides or pharmacological agents,
for example, vaccine components like bacterial or viral antigens or proteins like insulin, attached
to them, retain their activity and can be protected from natural alterations to their molecular
structure by adverse environmental conditions. It has been shown in studies conducted by us and
confirmed by others that when these combinations are injected into animals, the attachment can
enhance the biological activity as compared to injection of the molecule alone.
We believe our CaP technology has a number of benefits, including the following:
| • |
|
it is biodegradable (capable of being decomposed by natural biological processes) and
non-toxic and therefore potentially safe to use and introduce into the human body; |
| • |
|
it is fast, easy and inexpensive to manufacture, which should keep costs down and
potentially lead to higher profit margins compared to other delivery systems; |
| • |
|
the nanometer (one-millionth of a millimeter) size range makes it ideal for delivering
drugs through aerosol sprays, inhalation or intranasally, instead of using often painful and
inconvenient injections; and |
| • |
|
it has excellent “loading” capacity — the amount of molecules that can bond with the
nanoparticles — thereby potentially decreasing the dose needed to be taken by patients while
enhancing the release capabilities. |
Potential Commercial Applications for CaP. We plan to develop commercial applications of our CaP
technology and any proprietary technology developed as a result of our ongoing research and
development efforts. Initially, we plan to pursue primarily the development of:
| • |
|
a facial line filler using CaP technology in the area of aesthetic medicine; |
| • |
|
injected and non-injected vaccines using CaP as a delivery system and vaccine adjuvant; and |
| • |
|
drug delivery systems, including a method of delivering proteins (e.g., insulin) orally or
buccally, or through intranasal and subcutaneous routes of administration. |
Our pre-clinical research team in our laboratory in Doylestown, Pennsylvania currently is pursuing
the development of our CaP technology in these areas as well as exploring other areas.
10
CaP Products in Development. The following is a list of our CaP products in development:
| • |
|
BioLook — a facial line filler in development using proprietary CaP technology in the area
of aesthetic medicine. |
| • |
|
BioVant — proprietary CaP adjuvant and delivery technology in development for improved
versions of current vaccines and new vaccines against viral and bacterial infections and
autoimmune diseases, among others. BioVant also serves as a delivery system for non-injected
delivery of vaccines. |
| • |
|
BioOral — a delivery system using CaP technology for oral/buccal/intranasal administration
of proteins and other therapies that currently must be injected. |
| • |
|
BioAir — a delivery system using CaP technology for inhalable versions of proteins and
other therapies that currently must be injected. |
Aesthetic Medicine. In November 2007, we signed a license agreement with Medical Aesthetics
Technology Corporation (MATC) covering the use of our CaP as a facial line filler in aesthetic
medicine (BioLook). Under the license agreement, MATC is responsible for continued development of
BioLook, including required clinical trials, regulatory filings and all manufacturing and marketing
associated with the product. In exchange for the license, we received an ownership position in
MATC of approximately five percent of the common stock of MATC. In addition to the ownership
position, we may receive certain milestone payments and royalties as well as share in certain
payments if MATC sublicenses the technology.
Pre-clinical work to date by MATC indicates that our BioLook nanotechnology performs well as a
facial line filler and may be at least as long lasting and safe as other injectable fillers.
Preliminary results indicate long lasting effects with no adverse events. BioLook should be
extremely user friendly with minimal risk of side effects and may improve both facial wrinkles and
fulfill larger facial volume needs. Human clinical testing of BioLook for this use is being
planned and is expected to be initiated by MATC in 2010.
Vaccine Adjuvant and Delivery System. We believe that our CaP nanoparticles may offer a means of
preparing new vaccines that are equal or better in their safety and immunogenicity, that is, in
their capacity to elicit an immune response, compared to alum-formulated (the vaccine adjuvant used
in the vast majority of adjuvanted vaccines in the United States) and non-adjuvanted vaccines but
may be injected in lower concentrations and less often which could result in certain benefits,
including cost savings and improved patient compliance. Further, we believe that CaP will allow
for vaccines to be delivered by alternate routes of administration such as intranasally rather than
by injection.
Our nanoparticles when combined with vaccine antigens have been shown in animal studies conducted
by us and others to possess an ability to elicit a higher immune response than non-adjuvanted
vaccines and an immune response of the same magnitude as alum-formulated vaccines. These
preclinical studies also have shown that our CaP nanoparticles also may sustain higher antibody
levels over a longer time period than both alum-formulated vaccines and non-adjuvanted vaccines.
Because our CaP nanoparticles are made of calcium phosphate-like material, which has a chemical
nature similar to normal bone material and therefore is natural to the human body, as opposed to
aluminum hydroxide, or alum, which is not natural to the human body, we believe that our
nanoparticles may be safer to use than alum especially for intranasal delivery. In our animal
studies, we observed no material adverse reactions when our CaP nanoparticles were administered at
effective levels.
11
We filed an investigational new drug, or IND, application with the FDA and have conducted a Phase I
human clinical trial of CaP as a vaccine adjuvant and delivery system, which we call BioVant. As
discussed in more detail under the heading “Government Regulation,” the purpose of a Phase I trial
is to evaluate the metabolism and safety of the experimental product in humans, the side effects
associated with increasing doses, and, if possible, to gain early evidence of possible
effectiveness. The Phase I trial of our CaP specifically looked at safety parameters, including
local irritation and blood chemistry changes. The Phase I trial was a double blind, placebo
controlled trial, in 18 subjects to determine the safety of CaP as a vaccine adjuvant. The trial
results showed that there was no apparent difference in side-effect profile between CaP and
placebo. Phase I and or Phase II clinical trials will need to be repeated for each CaP/vaccine and
CaP/protein drug developed.
Drug Delivery Systems. The third field of use in which we are exploring applying our CaP
technology involves creating novel and improved forms of delivery of drugs, especially proteins
(e.g., insulin). The attachment of drugs to CaP may enhance their effects in the body or enable
the addition of further protective coatings to permit oral, delayed-release and mucosal (through
mucous membranes) applications. Currently, insulin is given by frequent, inconvenient and often
painful injections. However, several companies are in the process of developing and testing
products that will deliver insulin orally or through inhalation. We have shown pre-clinical
efficacy in the oral delivery of insulin in normal and diabetic mouse models.
We were awarded a $150,000 Small Business Innovation Research (SBIR) grant from the National
Institutes of Health (NIH) to support our development of formulations for the pulmonary delivery of
interferon alpha (IFN-a) using our CaP technology. The grant was used to fund product development
for IFN-a formulated with CaP particles for administration via inhalation. The desired outcome is
safe and effective treatment of hepatitis B and C. An inhaled product may allow for convenient
self treatment which would be an improvement over the current injectable IFN-a. We are in the
process of applying for additional government grants to help fund our CaP drug delivery
development.
License and Development Activities. In addition to continuing our own research and development in
the potential commercial applications of our CaP technology, we have sought and continue to seek
opportunities to enter into business collaborations or joint ventures with vaccine companies and
others interested in development and marketing arrangements with respect to our CaP technology. We
believe these collaborations may enable us to accelerate the development of potential improved
vaccines and the delivery of injectable drugs by other routes of administration, such as orally,
buccally, intranasally or through needle-free administration. Our out-licensing activities with
respect to our CaP vaccine adjuvant and delivery system for use in other companies’ vaccines, have
to date included meeting with target sub-licensees and, in some cases, agreeing that the target
sub-licensee will test our CaP adjuvant or delivery system in their animal models. Thereafter, the
target sub-licensee may send to us its vaccine antigen or DNA that we will then formulate with our
nanoparticles and return for use in the target sub-licensee’s animal models. Once this is
completed, if the results are positive, we would seek to negotiate an out-license agreement with
the target sub-licensee.
It is important to point out that vaccine development is an expensive and long-term process. We
have used our strategy of utilizing primarily outside resources to fund CaP’s development in order
to leverage the expertise of other companies and the United States government and to minimize our
spending on this expensive and long-term development work. Our strategic plan is to focus on our
nearer-term products and to seek collaborations and funding for our CaP technology.
Sales and Marketing
We currently have no sales and marketing personnel to sell any of our products on a commercial
basis. Under our license agreements, our licensees have agreed to market the products covered by
the agreements in certain countries. For example, under our license agreement with Azur, Azur has
agreed to use commercially reasonable efforts to manufacture, market, sell and distribute Elestrin
for commercial sale and distribution throughout the United States.
12
If and when we are ready to launch commercially a product not covered by our license agreements, we
will either contract with or hire qualified sales and marketing personnel or seek a joint marketing
partner or licensee to assist us with this function.
Research and Product Development
We spend a significant amount of our financial resources on product development activities, with
the largest portion being spent on clinical trials of our products, including in particular
LibiGel. We spent approximately $13.7 million in 2009, $15.8 million in 2008 and $4.8 million in
2007 on research and development activities. We spent an average of approximately $1.1 million per
month on our research and development activities during 2009. The decrease in 2009 research and
development expenses compared to 2008 was primarily the result of our decision in April 2009 to
delay screening new subjects for our LibiGel Phase III safety study to conserve cash. We
reinitiated screening and enrollment in our safety study in August 2009. We expect our monthly
research and development expenses to increase significantly in 2010 compared to 2009. The amount
of our actual research and development expenditures, however, may fluctuate from quarter-to-quarter
and year-to-year depending upon: (1) the amount of resources, including cash and cash equivalents,
available; (2) our development schedule, including the timing of our clinical trials; (3) results
of studies, clinical trials and regulatory decisions; (4) whether we or our licensees are funding
the development of our products; and (5) competitive developments.
Manufacturing
We currently do not have any facilities suitable for manufacturing on a commercial scale basis any
of our products nor do we have any experience in volume manufacturing. We currently use
third-party current Good Manufacturing Practices, or cGMP, manufacturers to manufacture our
products in accordance with FDA and other appropriate regulations. LibiGel for clinical studies
and Elestrin for commercial supplies are currently manufactured by an approved U.S.-based
manufacturer under FDA-approved, cGMP conditions.
Patents, Licenses and Proprietary Rights
Our success depends and will continue to depend in part upon our ability to maintain our exclusive
licenses, to obtain and maintain patent protection for our products and processes, to preserve our
proprietary information, trademarks and trade secrets and to operate without infringing the
proprietary rights of third parties. Our policy is to attempt to protect our technology by, among
other things, filing patent applications or obtaining license rights for technology that we
consider important to the development of our business.
Gel Products. We licensed the technology underlying LibGel, Elestrin and certain of our other gel
products, other than Bio-T-Gel, from Antares Pharma, Inc. Under the agreement, Antares granted us
an exclusive license to certain patents and patent applications covering these gel products,
including rights to sublicense, in order to develop and market the products in certain territories.
We are the exclusive licensee in certain territories for issued U.S. patents for these products
and additional patent applications have been filed for this licensed technology in the U.S. and
several foreign jurisdictions. Under the agreement, we are required to pay Antares certain
development and regulatory milestone payments and royalties based on net sales of any products we
or our sub-licensees sell incorporating the in-licensed technology. The patents covering the
formulations used in these gel products are expected to expire in 2022. Bio-T-Gel was developed
and is fully-owned by us and not covered under the Antares license.
13
The Pill Plus. We licensed the technology underlying our triple component contraceptives, or The
Pill Plus, from Wake Forest University Health Sciences and Cedars-Sinai Medical Center. The
financial terms of this license include regulatory milestone payments, maintenance payments and
royalty payments by us if a product incorporating the licensed technology gets approved and
subsequently is marketed. The patents covering the technology underlying The Pill Plus are
expected to expire in 2016.
GVAX Cancer Immunotherapy Technology. We own development and commercialization rights to our GVAX
cancer immunotherapy technology as a result of our merger with Cell Genesys in October 2009. The
original core patent applications covering our GVAX cancer immunotherapy technology were licensed
exclusively to Cell Genesys from Johns Hopkins University and The Whitehead Institute for
Biomedical Research in 1992. Rights to additional patents and patent applications were licensed
from Johns Hopkins University in 2001. In addition, we own several patents and patent applications
that build upon our in-licensed technology, and provide for significant additional patent term.
Our GVAX patent estate broadly covers our GVAX cancer immunotherapy products and pipeline. The
GVAX patent estate includes 17 patent families, comprising over 60 issued US and foreign patents,
directed to various aspects of the GVAX cancer immunotherapy technology. The patents are expected
to expire between 2012 and 2026.
Under the various agreements, we are required to pay Johns Hopkins University and The Whitehead
Institute for Biomedical Research certain development and regulatory milestone payments and
royalties based on net sales of any products we or our sub-licensees sell incorporating the
in-licensed technology.
2A/Furin Protein Expression Technology. We own development and commercialization rights to our
2A/furin protein expression technology as a result of our merger with Cell Genesys in October 2009.
Our 2A/furin patent estate includes five patent families, including four issued US patents and
additional patent applications, directed to various aspects of the 2A/furin technology, including
compositions and methods for producing recombinant antibodies. The patents are expected to expire
between 2023 and 2026.
Oncolytic Virus Technology. We also own development and commercialization rights to our oncolytic
virus technology as a result of our merger with Cell Genesys in October 2009. The oncolytic virus
patent estate includes eight patent families, comprising over 24 issued US and foreign patents,
directed to various aspects of the oncolytic virus technology, including CG0070 which has completed
a Phase 1 trial for non-muscle, invasive transitional cell bladder cancer. The patents are
expected to expire between 2018 and 2023.
CaP Technology. In June 1997, we entered into a licensing agreement with the Regents of the
University of California, which subsequently has been amended, pursuant to which the University
granted us an exclusive license to certain United States patents owned by the University, including
rights to sublicense such patents, in fields of use pertaining to vaccine adjuvants and drug
delivery systems. The last of the expiration dates for these patents is 2014. Importantly, we own
several of our own additional patents and patent applications covering the CaP technology expiring
beginning in 2021. The University of California also has filed patent applications for this
licensed technology in several foreign jurisdictions, including Canada, Europe and Japan. The
license agreement requires us to pay royalties to the University based on a percentage of the net
sales of any products we sell or a licensee sells incorporating the licensed technology until
expiration of the licensed patents.
14
As described earlier in this report, we have entered into agreements with respect to our CaP
technology, including a license agreement covering the use of our CaP as a facial line filler
(BioLook) in aesthetic medicine.
Other License Agreements. As described earlier in this report, we have entered into several other
license agreements pursuant to which we have sublicensed to third parties certain rights with
respect to our products, none of which we view as material to our business. The financial terms of
these agreements generally include an upfront license fee and subsequent milestone and royalty
payments to us if a product incorporating the licensed technology gets approved and subsequently is
marketed and a portion of any payments received from subsequent successful out-licensing efforts.
Trademarks and Trademark Applications/Registrations. We own trademark registrations in the U.S.
and/or in certain foreign jurisdictions for the marks BIOSANTE®, LIBIGEL®,
BIO-E-GEL®, BIOAIR™ and GVAX™. In addition, we have filed trademark applications for
several other marks including ELESTRIN™ (pursuant to our license of Elestrin to Azur in the U.S.,
we transferred the Elestrin trademark in the U.S. to Azur), BIO-T-GEL™, BIOVANT™ and covering goods
that include or are closely related to products, vaccines and vaccine adjuvants and drug delivery
platforms. In addition, we own common law rights to several trademarks, including
BIOSANTE®, LIBIGEL®, ELESTRIN™, BIO-E-GEL®, BIO-T-GEL™, THE
PILL-PLUS™, LIBIGEL-E/T™, BIO-E/P-GEL™, BIOLOOK™, CAP-ORAL™, BIOVANT™, BIOAIR™ and GVAX™. For
those trademarks for which registration has been sought, registrations have issued for some of
those trademarks in certain jurisdictions and others currently are in the application/prosecution
phase.
Confidentiality and Assignment of Inventions Agreements. We require our employees, consultants and
advisors having access to our confidential information to execute confidentiality agreements upon
commencement of their employment or consulting relationships with us. These agreements generally
provide that all confidential information we develop or make known to the individual during the
course of the individual’s employment or consulting relationship with us must be kept confidential
by the individual and not disclosed to any third parties. We also require all of our employees and
consultants who perform research and development for us to execute agreements that generally
provide that all inventions and works-for-hire conceived by these individuals during their
employment by us will be our property.
Competition
There is intense competition in the biopharmaceutical industry, the market for prevention and/or
treatment of the same infectious diseases we target and in the acquisition or licensing of new
products. Potential competitors in the United States are numerous and include major pharmaceutical
and specialized biotechnology companies, universities and other institutions. In general,
competition in the pharmaceutical industry can be divided into four categories: (1) corporations
with large research and developmental departments that develop and market products in many
therapeutic areas; (2) companies that have moderate research and development capabilities and focus
their product strategy on a small number of therapeutic areas; (3) small companies with limited
development capabilities and only a few product offerings; and (4) university and other research
institutions. Many of our competitors have longer operating histories, greater name recognition,
substantially greater financial resources and larger research and development staffs than we do, as
well as substantially greater experience than us in developing products, obtaining regulatory
approvals, and manufacturing and marketing pharmaceutical products. A significant amount of
research is carried out at academic and government institutions. These institutions are aware of
the commercial value of their findings and are becoming more aggressive in pursuing patent
protection and negotiating licensing arrangements to collect royalties for use of technology that
they have developed.
15
There are several firms currently marketing or developing products that may be competitive with
ours; they include Upsher-Smith Laboratories, Inc., Noven Pharmaceuticals, Inc. (a subsidiary of
Hisamitsu Pharmaceutical Co., Inc.), Pfizer Inc., Auxilium Pharmaceuticals, Inc., Ascend
Therapeutics, Inc., Watson Pharmaceuticals, Inc., KV Pharmaceutical Co., and Abbott Laboratories.
Competitor products include oral tablets, transdermal patches, a spray and gels. We expect our
FDA-approved product, Elestrin, and our other products, if and when approved for sale, to compete
primarily on the basis of product efficacy, safety, patient convenience, reliability and patent
position. In addition, the first product to reach the market in a therapeutic or preventative area
is often at a significant competitive advantage relative to later entrants in the market and may
result in certain marketing exclusivity as per federal legislation.
Acceptance by physicians and other health care providers, including managed care groups, also is
critical to the success of a product versus competitor products.
With regard to our CaP technology, the international vaccine industry is dominated by three
companies: GlaxoSmithKline plc, Sanofi-aventis (through its subsidiaries, including Institut
Merieux International S.A., Pasteur Merieux Serums et Vaccins, S.A., Connaught Laboratories Limited
and Connaught Laboratories, Inc.) and Merck & Co., Inc. The larger, better known pharmaceutical
companies generally have focused on a traditional synthetic drug approach, although some have
substantial expertise in biotechnology. During the last decade, however, significant research
activity in the biotechnology industry has been completed by smaller research and development
companies, like us, formed to pursue new technologies.
With regard to our GVAX cancer immunotherapy technology and other recently acquired technologies,
we face substantial competition in the development of products for cancer and other diseases. This
competition from other manufacturers is expected to continue in both U.S. and international
markets. Cancer immunotherapies and oncolytic virus therapies are evolving areas in the
biotechnology industry and are expected to undergo many changes in the coming years as a result of
technological advances. We currently are aware of a number of groups that are developing cancer
immunotherapies and oncolytic virus therapies including early-stage and established biotechnology
companies, pharmaceutical companies, academic institutions, government agencies and research
institutions. Examples in the cancer immunotherapy area include Dendreon Corporation, which has
completed a Phase III trial for its product in prostate cancer and has filed a Biologics License
Application (BLA) with the FDA, and Onyvax Ltd., which has commenced Phase II trials in prostate
cancer. Antigenics, Inc., Oncothyreon Inc., Warner Chilcott plc and Boerhinger Ingelheim USA
Corporation also are developing immunotherapy products for other types of cancers.
Governmental Regulation
Pharmaceutical companies are subject to extensive regulation by national, state and local agencies
in countries in which they do business. Pharmaceutical products intended for therapeutic use in
humans are governed by extensive FDA regulations in the United States and by comparable regulations
in foreign countries. Any products developed by us will require FDA approvals in the United States
and comparable approvals in foreign markets before they can be marketed. The process of seeking
and obtaining FDA approval for a previously unapproved new human pharmaceutical product generally
requires a number of years and involves the expenditure of substantial resources.
16
Following drug discovery, the steps required before a drug product may be marketed in the United
States include:
| • |
|
completion of preclinical laboratory and animal testing; |
| • |
|
the submission to the FDA of an investigational new drug application, commonly known as an
IND application, which must be evaluated and found acceptable by the FDA before human clinical
trials may commence; |
| • |
|
the completion of clinical and other studies to assess safety and parameters of use; |
| • |
|
the completion of multiple adequate and well-controlled human clinical trials to establish
the safety and effectiveness of the drug product for its intended use; |
| • |
|
the submission to the FDA of a new drug application, commonly known as an NDA, or an
abbreviated NDA, commonly known as an ANDA; |
| • |
|
satisfactory completion of an FDA pre-approval inspection of manufacturing facilities at
which the drug product is produced, and potentially other involved facilities as well, to
assess compliance with current good manufacturing practice, or cGMP, regulations and other
applicable regulations; and |
| • |
|
FDA approval of the NDA or ANDA prior to any commercial sale or shipment of the product. |
Pre-Clinical Studies and Clinical Trials. Typically, preclinical studies are conducted in the
laboratory and in animals to gain preliminary information on a product’s uses and physiological
effects and harmful effects, if any, and to identify any potential safety problems that would
preclude testing in humans. The results of these studies, together with the general investigative
plan, protocols for specific human studies and other information, are submitted to the FDA as part
of the IND application. The FDA regulations do not, by their terms, require FDA approval of an
IND. Rather, they allow a clinical investigation to commence if the FDA does not notify the sponsor
to the contrary within 30 days of receipt of the IND. As a practical matter, however, FDA approval
is often sought before a company commences clinical investigations. That approval may come within
30 days of IND receipt but may involve substantial delays if the FDA requests additional
information.
Our submission of an IND, or those of our collaboration partners, may not result in FDA
authorization to commence a clinical trial. A separate submission to an existing IND also must be
made for each successive clinical trial conducted during product development. Depending on its
significance, the FDA also must approve changes to an existing IND. Further, an independent
institutional review board, or IRB, for each medical center proposing to conduct the clinical trial
must review and approve the plan for any clinical trial before it commences at that center and it
must monitor the study until completed. Alternatively, a central IRB may be used instead of
individual IRBs. The FDA, the IRB or the sponsor may suspend a clinical trial at any time on
various grounds, including a finding that the subjects or patients are being exposed to an
unacceptable health risk. Clinical testing also must satisfy extensive Good Clinical Practice
requirements and regulations for informed consent.
The sponsor of a drug product typically conducts human clinical trials in three sequential phases,
but the phases may overlap or not all phases may be necessary. The initial phase of clinical
testing, which is known as Phase I, is conducted to evaluate the metabolism, uses and physiological
effects of the experimental product in humans, the side effects associated with increasing doses,
and, if possible, to gain early evidence of possible effectiveness. Phase I studies can also
evaluate various routes, dosages and schedules of product administration. These studies generally
involve a small number of healthy volunteer subjects, but may be conducted in people with the
disease the product is intended to treat. The total number of subjects is generally in the range
of 20 to 80. A demonstration of therapeutic benefit is not required in order to complete Phase I
trials successfully. If acceptable product safety is demonstrated, Phase II trials may be
initiated.
17
Phase II trials are designed to evaluate the effectiveness of the product in the treatment of a
given disease and involve people with the disease under study. These trials often are well
controlled, closely monitored studies involving a relatively small number of subjects, usually no
more than several hundred. The optimal routes, dosages and schedules of administration are
determined in these studies. If Phase II trials are completed successfully, Phase III trials are
often commenced, although Phase III trials are not always required.
Phase III trials are expanded, controlled trials that are performed after preliminary evidence of
the effectiveness of the experimental product has been obtained. These trials are intended to
gather the
additional information about safety and effectiveness that is needed to evaluate the overall
risk/benefit relationship of the experimental product and provide the substantial evidence of
effectiveness and the evidence of safety necessary for product approval. Phase III trials are
usually conducted with several hundred to several thousand subjects.
A clinical trial may combine the elements of more than one phase and typically two or more Phase
III studies are required. A company’s designation of a clinical trial as being of a particular
phase is not necessarily indicative that the trial will be sufficient to satisfy the FDA
requirements of that phase because this determination cannot be made until the protocol and data
have been submitted to and reviewed by the FDA. In addition, a clinical trial may contain elements
of more than one phase notwithstanding the designation of the trial as being of a particular phase.
The FDA closely monitors the progress of the phases of clinical testing and may, at its
discretion, re-evaluate, alter, suspend or terminate the testing based on the data accumulated and
its assessment of the risk/benefit ratio to patients. It is not possible to estimate with any
certainty the time required to complete Phase I, II and III studies with respect to a given
product.
New Drug Applications. Upon the successful completion of clinical testing, an NDA is submitted to
the FDA for approval. This application requires detailed data on the results of preclinical
testing, clinical testing and the composition of the product, specimen labeling to be used with the
drug, information on manufacturing methods and samples of the product. The FDA reviews all NDAs
submitted before it accepts them for filing and may request additional information rather than
accepting an NDA for filing. Once the submission is accepted for filing, the FDA begins an
in-depth review of the NDA. Under the policies agreed to by the FDA under the Prescription Drug
User Fee Act, or PDUFA, the FDA has 10 months in which to complete its initial review of a standard
NDA and respond to the applicant. The review process and the PDUFA goal date may be extended by
three months if the FDA requests or the NDA sponsor otherwise provides additional information or
clarification regarding information already provided in the submission within the last three months
of the PDUFA goal date. The FDA typically takes from 10 to 18 months to review an NDA after it has
been accepted for filing. Following its review of an NDA, the FDA invariably raises questions or
requests additional information. The NDA approval process can, accordingly, be very lengthy.
Further, there is no assurance that the FDA will ultimately approve an NDA.
During its review of an NDA, the FDA may refer the application to an advisory committee for review,
evaluation and recommendation as to whether the application should be approved. The FDA may refuse
to approve an NDA and issue a not approvable letter if the applicable regulatory criteria are not
satisfied, or it may require additional clinical or other data, including one or more additional
pivotal Phase III clinical trials. Even if such data are submitted, the FDA may ultimately decide
that the NDA does not satisfy the criteria for approval. Data from clinical trials are not always
conclusive and the FDA may interpret data differently than we or our collaboration partners
interpret data. If the FDA’s evaluations of the NDA and the clinical and manufacturing procedures
and facilities are favorable, the FDA may issue either an approval letter or an approvable letter,
which contains the conditions that must be met in order to secure final approval of the NDA. If
and when those conditions have been met to the FDA’s satisfaction, the FDA will issue an approval
letter, authorizing commercial marketing of the drug for certain indications. The FDA may withdraw
drug approval if ongoing regulatory requirements are not met or if safety problems occur after the
drug reaches the market. In addition, the FDA may require testing, including Phase IV clinical
trials, and surveillance programs to monitor the effect of approved products that have been
commercialized, and the FDA has the power to prevent or limit further marketing of a drug based on
the results of these post-marketing programs. Drugs may be marketed only for the FDA-approved
indications and in accordance with the FDA-approved label. Further, if there are any modifications
to the drug, including changes in indications, other labeling changes, or manufacturing processes
or facilities, we may be required to submit and obtain FDA approval of a new NDA or NDA
supplement, which may require us to develop additional data or conduct additional preclinical
studies and clinical trials.
18
Special Protocol Assessments. The special protocol assessment, or SPA, process generally involves
FDA evaluation of a proposed Phase III clinical trial protocol and a commitment from the FDA that
the design and analysis of the trial are adequate to support approval of an NDA, if the trial is
performed according to the SPA and meets its endpoints. The FDA’s guidance on the SPA process
indicates that SPAs are designed to evaluate individual clinical trial protocols primarily in
response to specific questions posed by the sponsors. In practice, the sponsor of a product
candidate may request an SPA for proposed Phase III trial objectives, designs, clinical endpoints
and analyses. A request for an SPA is submitted in the form of a separate amendment to an IND, and
the FDA’s evaluation generally will be completed within a 45-day review period under applicable
PDUFA goals, provided that the trials have been the subject of discussion at an end-of-Phase II and
pre-Phase III meeting with the FDA, or in other limited cases.
If an agreement is reached, the FDA will reduce the agreement to writing and make it part of the
administrative record. While the FDA’s guidance on SPAs states that documented SPAs should be
considered binding on the review division, the FDA has the latitude to change its assessment if
certain exceptions apply. Exceptions include identification of a substantial scientific issue
essential to safety or efficacy testing that later comes to light, a sponsor’s failure to follow
the protocol agreed upon, or the FDA’s reliance on data, assumptions or information that are
determined to be wrong.
The Hatch-Waxman Act. Under the Hatch-Waxman Act, newly-approved drugs and new conditions of use
may benefit from a statutory period of non-patent marketing exclusivity. The Hatch-Waxman Act
provides three years of marketing exclusivity for the approval of new and supplemental NDAs for,
among other things, new indications, dosages or strengths of an existing drug, if new clinical
investigations that were conducted or sponsored by the applicant are essential to the approval of
the application. It is under this provision that we received three years marketing exclusivity for
Elestrin and expect to receive three years of marketing exclusivity for LibiGel.
Other Regulatory Requirements. Regulations continue to apply to pharmaceutical products after FDA
approval occurs. Post-marketing safety surveillance is required in order to continue to market an
approved product. The FDA also may, in its discretion, require post-marketing testing and
surveillance to monitor the effects of approved products or place conditions on any approvals that
could restrict the commercial applications of these products.
All facilities and manufacturing techniques used to manufacture products for clinical use or sale
in the United States must be operated in conformity with “current good manufacturing practice”
regulations, commonly referred to as “cGMP” regulations, which govern the production of
pharmaceutical products. We currently do not have any manufacturing capability. In the event we
undertake any manufacturing activities or contract with a third-party manufacturer to perform our
manufacturing activities, we intend to establish a quality control and quality assurance program to
ensure that our products are manufactured in accordance with the cGMP regulations and any other
applicable regulations.
19
Foreign Regulation. Products marketed outside of the United States are subject to regulatory
approval requirements similar to those in the United States, although the requirements governing
the conduct of clinical trials, product licensing, pricing and reimbursement vary widely from
country to country. No action can be taken to market any product in a country until an appropriate
application has been approved by the regulatory authorities in that country. The current approval
process varies from country to country, and the time spent in gaining approval varies from that
required for FDA approval. In certain European countries, the sales price of a product must also
be approved. The pricing review period often begins after
market approval is granted. We intend to seek and utilize foreign partners to apply for foreign
approvals of our products.
Employees
We had 25 employees as of December 31, 2009, including 20 in product development and five in
management or administrative positions. None of our employees is covered by a collective
bargaining agreement. We also engage independent contractors from time to time on an as needed
basis.
Forward-Looking Statements
This annual report on Form 10-K contains or incorporates by reference not only historical
information, but also forward-looking statements within the meaning of Section 27A of the
Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as
amended, and are subject to the safe harbor created by those sections. In addition, we or others
on our behalf may make forward-looking statements from time to time in oral presentations,
including telephone conferences and/or web casts open to the public, in press releases or reports,
on our Internet web site or otherwise. All statements other than statements of historical facts
included in this report that address activities, events or developments that we expect, believe or
anticipate will or may occur in the future are forward-looking statements including, in particular,
the statements about our plans, objectives, strategies and prospects regarding, among other things,
our financial condition, results of operations and business. We have identified some of these
forward-looking statements with words like “believe,” “may,” “could,” “would,” “might,” “possible,”
“potential,” “project,” “will,” “should,” “expect,” “intend,” “plan,” “predict,” “anticipate,”
“estimate,” “approximate,” “contemplate” or “continue”, the negative of these words, other words
and terms of similar meaning or the use of future dates. These forward-looking statements may be
contained in the notes to our financial statements and elsewhere in this report, including under
the heading “Part II. Item 7. Management’s Discussion and Analysis of Financial Condition and
Results of Operations.” Our forward-looking statements generally relate to:
| • |
|
the timing of the commencement, enrollment and successful completion of our clinical
studies, the submission of new drug applications and other regulatory status of our products
in development; |
| • |
|
approval by the FDA of our products that are currently in clinical development; |
| • |
|
our spending capital on research and development programs, pre-clinical studies and
clinical studies, regulatory processes and licensure or acquisition of new products; |
| • |
|
our efforts to continue to evaluate various strategic alternatives with respect to our
products and our company; |
| • |
|
the future market size and market acceptance of our products; |
| • |
|
the effect of new accounting pronouncements and future health care, tax and other
legislation; |
20
| • |
|
whether and how long our existing cash will be sufficient to fund our operations; |
| • |
|
our need, ability and expected timing of any actions to raise additional capital through
future equity and other financings; and |
| • |
|
our substantial and continuing losses. |
Forward-looking statements involve risks and uncertainties. These uncertainties include factors
that affect all businesses as well as matters specific to us. Some of the factors known to us that
could cause our actual results to differ materially from what we have anticipated in our
forward-looking statements are described under the heading “Part I. Item 1A. Risk Factors” below.
We wish to caution readers not to place undue reliance on any forward-looking statement that speaks
only as of the date made and to recognize that forward-looking statements are predictions of future
results, which may not occur as anticipated. Actual results could differ materially from those
anticipated in the forward-looking statements and from historical results, due to the risks and
uncertainties described under the heading “Part I. Item 1A. Risk Factors” below, as well as others
that we may consider immaterial or do not anticipate at this time. Although we believe that the
expectations reflected in our forward-looking statements are reasonable, we do not know whether our
expectations will prove correct. Our expectations reflected in our forward-looking statements can
be affected by inaccurate assumptions we might make or by known or unknown risks and uncertainties,
including those described below under the heading “Part I. Item 1A. Risk Factors.” The risks and
uncertainties described under the heading “Item 1A. Risk Factors” below are not exclusive and
further information concerning us and our business, including factors that potentially could
materially affect our financial results or condition, may emerge from time to time. We assume no
obligation to update forward-looking statements to reflect actual results or changes in factors or
assumptions affecting such forward-looking statements. We advise you, however, to consult any
further disclosures we make on related subjects in our quarterly reports on Form 10-Q and current
reports on Form 8-K we file with or furnish to the Securities and Exchange Commission.
Available Information
We are a Delaware corporation that was initially formed as a corporation organized under the laws
of the Province of Ontario in 1996. In October 2009, we acquired Cell Genesys, Inc. through a
direct merger. Our principal executive offices are located at 111 Barclay Boulevard, Lincolnshire,
Illinois 60069. Our telephone number is (847) 478-0500, and our Internet web site address is
www.biosantepharma.com. The information contained on our web site or connected to our web site is
not incorporated by reference into and should not be considered part of this annual report on Form
10-K.
We make available, free of charge and through our Internet web site, our annual reports on Form
10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and any amendments to any such
reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of
1934, as amended, as soon as reasonably practicable after we electronically file such material
with, or furnish it to, the SEC. We also make available, free of charge and through our Internet
web site, to any stockholder who requests, our corporate governance guidelines, the charters of our
board committees and our Code of Conduct and Ethics. Requests for copies can be directed to
Investor Relations at (847) 478-0500, extension 120.
21
The following are significant risk factors known to us that could have a material adverse effect on
our business, financial condition or operating results.
Risks Related to Our Financial Condition and Future Capital Requirements
We have a history of operating losses, expect continuing losses and may never become profitable.
We incurred a net loss of $47.5 million for the year ended December 31, 2009 and as of December 31,
2009, our accumulated deficit was $119.4 million. Substantially all of our revenue to date has
been derived from upfront and milestone payments earned on licensing transactions, revenue earned
from subcontracts and royalty revenue. We expect to continue to incur substantial and continuing
losses as our own product development programs continue and various preclinical and clinical trials
commence or continue, including in particular our Phase III clinical study program for LibiGel.
In order to generate new and significant revenues, we must develop successfully our own products or
enter into collaborative agreements with others who can commercialize them successfully. Even if
our products are introduced commercially, they may never achieve market acceptance and we may not
generate additional revenues or ever achieve profitability.
We may need to continue to raise substantial additional capital to fund our operations and we may
be unable to raise such funds when needed and on acceptable terms.
We currently do not have sufficient resources to obtain regulatory approval of LibiGel or any of
our other products or to complete the commercialization of any of our products. We expect the
Phase III clinical study program of LibiGel to continue to require significant resources. Our
future capital requirements will depend upon numerous factors, including:
| • |
|
the progress, timing, cost and results of our preclinical and clinical development
programs, including in particular our Phase III clinical study program for LibiGel, and our
other product development efforts; |
| • |
|
subject recruitment and enrollment in our current and future clinical studies, including in
particular our Phase III clinical study program for LibiGel; |
| • |
|
our ability to license LibiGel or our other products for development and commercialization; |
| • |
|
the success, progress, timing and costs of our business development efforts to implement
business collaborations, licenses and other business combinations or transactions, including
our efforts, to continue to evaluate various strategic alternatives available with respect to
our GVAX cancer immunotherapics and other technologies that we acquired as a result of our
merger with Cell Genesys, our products and our company; |
| • |
|
the cost, timing and outcome of regulatory reviews of our products; |
| • |
|
the rate of technological advances; |
| • |
|
the commercial success of our products; |
| • |
|
our general and administrative expenses; |
22
| • |
|
the timing and cost of obtaining third party reimbursement for our products; and |
| • |
|
the activities of our competitors. |
Therefore, we may need to continue to raise substantial additional capital to fund our operations.
Although we believe that our cash and cash equivalents of $29.9 million at December 31, 2009 and
the additional $17.5 million in net proceeds we received from our March 2010 registered direct
offering will be sufficient to meet our liquidity requirements through at least the next 12 months,
this estimate may prove incorrect or we may decide to raise additional financing earlier.
We have on file an effective shelf registration statement that allows us to raise up to $75.0
million from the sale of common stock, preferred stock, warrants or units comprised of the
foregoing. However, as of March 15, 2010, we had used approximately $46.8 million of this amount
and under applicable SEC rules, if we have a public float of less than $75.0 million, we can only
offer to sell under the registration statement up to one-third of our public float during any 12
month period. We can provide no assurance that additional financing, if needed, will be available
on terms favorable to us, or at all. If adequate funds are not available or are not available on
acceptable terms when we need them, we may need to delay our Phase III clinical study program for
LibiGel or otherwise make changes to our operations to cut costs. As an alternative to raising
additional financing, we may choose to license LibiGel, Elestrin (outside the territories already
licensed) or another product, e.g., our GVAX immunotherapies, to a third party who may finance a
portion or all of the continued development and, if approved, commercialization of that licensed
product, sell certain assets or rights we have under our existing license agreements or enter into
other business collaborations or combinations, including the possible sale of our company.
Raising additional funds by issuing equity securities may cause dilution to existing stockholders,
raising additional funds by issuing additional debt financing may restrict our operations and
raising additional funds by through licensing arrangements may require us to relinquish proprietary
rights.
If we raise additional funds through the issuance of equity or convertible debt securities, the
percentage ownership of our stockholders could be diluted significantly, and these newly issued
securities may have rights, preferences or privileges senior to those of our existing stockholders.
If we incur additional debt financing, the payment of principal and interest on such indebtedness
may limit funds available for our business activities, and we could be subject to covenants that
restrict our ability to operate our business and make distributions to our stockholders. These
restrictive covenants may include limitations on additional borrowing and specific restrictions on
the use of our assets, as well as prohibitions on the ability of us to create liens, pay dividends,
redeem our stock or make investments. As an alternative to raising additional financing by issuing
equity or debt securities, we may choose to license LibiGel, Elestrin (outside the territories
already licensed) or another product to a third party, e.g., our GVAX immunotherapies, who may
finance a portion or all of the continued development and, if approved, commercialization of that
licensed product, sell certain assets or rights we have under our existing license agreements or
enter into other business collaborations or combinations, including the possible sale of our
company. If we raise additional funds through licensing arrangements, we may be required to
relinquish greater or all rights to our products at an earlier stage of development or on less
favorable terms than we otherwise would choose.
23
Our committed equity financing facility with Kingsbridge Capital Limited may not be available to us
if we elect to make a draw down.
We have a committed equity financing facility with Kingsbridge that expires in December 2010. The
committed equity financing facility entitles us to sell and obligates Kingsbridge to purchase, from
time to time through the expiration date, up to the lesser of (i) an aggregate of $25 million in or
(ii) 5,405,840
shares of our common stock for cash consideration, subject to certain conditions and restrictions.
Kingsbridge will not be obligated to purchase shares under the facility unless certain conditions
are met, which include a minimum price for our common stock of $1.15 per share; the accuracy of
representations and warranties made to Kingsbridge; compliance with laws; continued effectiveness
of the registration statement registering the resale of shares of our common stock issued or
issuable to Kingsbridge; and the continued listing of our stock on the NASDAQ Global Market. In
addition, Kingsbridge is permitted to terminate the facility if it determines that a material and
adverse event has occurred affecting our business, operations, properties or financial condition
and if such condition continues for a period of 10 trading days from the date Kingsbridge provides
us notice of such material and adverse event. If we are unable to access funds through the
committed equity financing facility, or if the facility is terminated by Kingsbridge, we may be
unable to access capital on favorable terms or at all. As of the date of this report, we had not
sold any shares to Kingsbridge under the committed equity financing facility and we did not have an
effective registration statement registering the resale of shares of our common stock issued or
issuable to Kingsbridge under the facility.
As a result of our merger with Cell Genesys, we have substantial indebtedness, which we may not be
able to pay when it becomes due and payable.
As a result of our merger with Cell Genesys, we assumed $22.0 million aggregate principal amount of
outstanding convertible notes, $1.2 million of which will be due in November 2011 and $20.8 million
of which will be due in May 2013. The annual interest payment on these notes is approximately $0.7
million. We do not have any significant source of revenues and thus although we intend to continue
to seek additional financing to support our operations, it is possible that we may not have
sufficient funds to pay the principal on our convertible notes when it becomes due, especially if
an event of default were to occur under the indentures governing the convertible notes.
The indentures governing our convertible notes contain covenants, which if not complied with, could
result in an event of default and the acceleration of all amounts due under the notes.
The indentures governing our assumed convertible notes contain covenants, such as the requirement
to pay accrued interest on May 1 and November 1 of each year, the requirement to repurchase the
notes upon a “fundamental change,” as defined in the indenture, if a note holder so elects and the
requirement to file periodic reports electronically with the SEC. If we do not comply with the
covenants in the indentures, an event of default could occur and all amounts due under the notes
could become immediately due and payable. Upon the occurrence of an event of default under the
indentures, the trustee has available a range of remedies customary in these circumstances,
including declaring all such indebtedness, together with accrued and unpaid interest thereon, to be
due and payable. Although it is possible we could negotiate a waiver with the trustee and the
holders of the notes, such a waiver likely would involve significant costs. It also is possible
that we could refinance our obligations under the notes; however, such a refinancing also would
involve significant costs and likely result in increased interest rates.
24
As a result of our merger with Cell Genesys, we possess not only all of the assets but also all of
the liabilities of Cell Genesys. Discovery of previously undisclosed liabilities could have an
adverse effect on our business, operating results and financial condition.
Acquisitions often involve known and unknown risks, including inaccurate assessment of undisclosed,
contingent or other liabilities or problems. In October 2008, in view of the termination of both
its VITAL-1 and VITAL-2 Phase III clinical trials, Cell Genesys discontinued further development of
GVAX immunotherapy for prostate cancer. Cell Genesys subsequently implemented a substantial
restructuring plan to wind down its business operations and seek strategic alternatives. Under the
restructuring plan, Cell Genesys terminated approximately 280 employees, closed two facilities and
terminated two leases. As a result our merger with Cell Genesys, we possess not only all of the
assets, but also all of the potential liabilities of Cell Genesys. Although we conducted a due
diligence investigation of Cell Genesys and its known and potential liabilities and obligations, it
is possible that undisclosed, contingent or other liabilities or problems may arise, which could
have an adverse effect on our business, operating results and financial condition.
Risks Related to Our Business
Most of our products are in the development stages and likely will not be introduced commercially
for several years, if at all.
Our products are in the development stages and will require further development, preclinical and
clinical testing and investment prior to commercialization in the United States and abroad. Other
than Elestrin, none of our products has been introduced commercially nor do we expect them to be
for several years. Some of our products are not in active development. For example, at this time
and as previously disclosed, we believe that our estrogen/progestogen combination transdermal gel
product licensed to Solvay is not in active development by Solvay (now owned by Abbott
Laboratories), and we do not expect its active development to occur at any time in the near future.
We cannot assure you that any of our products will:
| • |
|
be developed successfully; |
| • |
|
prove to be safe and effective in clinical studies; |
| • |
|
meet applicable regulatory standards or obtain required regulatory approvals; |
| • |
|
demonstrate substantial protective or therapeutic benefits in the prevention or treatment
of any disease; |
| • |
|
be capable of being produced in commercial quantities at reasonable costs; |
| • |
|
obtain coverage and favorable reimbursement rates from insurers and other third-party
payors; or |
| • |
|
be successfully marketed or achieve market acceptance by physicians and patients. |
If we fail to obtain regulatory approval to manufacture commercially or sell any of our future
products, or if approval is delayed or withdrawn, we will be unable to generate revenue from the
sale of our products.
We must obtain regulatory approval to sell any of our products in the United States and abroad. In
the United States, we must obtain the approval of the FDA for each product or drug that we intend
to commercialize. The FDA approval process typically is lengthy and expensive, and approval never
is certain. Products to be commercialized abroad are subject to similar foreign government
regulation.
25
Generally, only a very small percentage of newly discovered pharmaceutical products that enter
preclinical development eventually are approved for sale. Because of the risks and uncertainties
in biopharmaceutical development, our products could take a significantly longer time to gain
regulatory approval than we expect or may never gain approval. If regulatory approval is delayed
or never obtained, the credibility of our management, the value of our company and our operating
results and liquidity would be affected adversely. Even if a product gains regulatory approval,
the product and the
manufacturer of the product may be subject to continuing regulatory review. In addition, even
after obtaining regulatory approval, we may be restricted or prohibited from marketing or
manufacturing a product if previously unknown problems with the product or our manufacture
subsequently are discovered. The FDA also may require us to commit to perform lengthy
post-approval studies, for which we would have to expend significant additional resources, which
could have an adverse effect on our operating results and financial condition.
To obtain regulatory approval to market many of our products, costly and lengthy pre-clinical
studies and human clinical trials are required, and the results of the studies and trials are
highly uncertain. As part of the FDA approval process, we must conduct, at our own expense or the
expense of current or potential licensees, clinical trials in human subjects on each of our
products. Pre-clinical studies on animals must be conducted on some of our products. We expect
the number of pre-clinical studies and human clinical trials that the FDA will require will vary
depending on the product, the disease or condition the product is being developed to address and
regulations applicable to the particular product. We may need to perform multiple pre-clinical
studies using various doses and formulations before we can begin human clinical trials, which could
result in delays in our ability to market any of our products. Furthermore, even if we obtain
favorable results in pre-clinical studies on animals, the results in humans may be different.
After we have conducted pre-clinical studies in animals, we must demonstrate that our products are
safe and effective for use on the target human patients in order to receive regulatory approval for
commercial sale. The data obtained from pre-clinical and human clinical testing are subject to
varying interpretations that could delay, limit or prevent regulatory approval. We face the risk
that the results of our clinical trials in later phases of clinical trials may be inconsistent with
those obtained in earlier phases. A number of companies in the biopharmaceutical industry have
suffered significant setbacks in advanced clinical trials, even after experiencing promising
results in early animal or human testing. Adverse or inconclusive human clinical results would
prevent us from filing for regulatory approval of our products. Additional factors that can cause
delay or termination of our human clinical trials include:
| • |
|
slow subject enrollment; |
| • |
|
timely completion of clinical site protocol approval and obtaining informed consent from
subjects; |
| • |
|
longer treatment time required to demonstrate efficacy or safety; |
| • |
|
adverse medical events or side effects in treated subjects; |
| • |
|
lack of effectiveness of the product being tested; and |
Delays in our clinical trials could allow our competitors additional time to develop or market
competing products and thus can be extremely costly in terms of lost sales opportunities and
increased clinical trial costs.
26
Although we successfully have completed and reached agreement with the FDA under the Special
Protocol Assessment process for our Phase III safety and efficacy clinical trial program for
LibiGel, we still may not obtain FDA approval of LibiGel within a reasonable period of time or
ever, which would harm our business and likely decrease our stock price.
LibiGel has not been approved for marketing by the FDA and is still subject to risks associated
with its clinical development and obtaining regulatory approval. We believe based on agreements
with the FDA,
including a Special Protocol Assessment received in January 2008, that two Phase III safety and
efficacy trials and one year of LibiGel exposure in a Phase III cardiovascular and breast cancer
safety study with a four-year follow-up post-NDA filing and potentially post-FDA approval are the
essential requirements for submission and, if successful, approval by the FDA of an NDA for LibiGel
for the treatment of FSD, specifically, HSDD in menopausal women. The SPA process and agreement
affirms that the FDA agrees that the LibiGel Phase III safety and efficacy clinical trial design,
clinical endpoints, sample size, planned conduct and statistical analyses are acceptable to support
regulatory approval. Further, it provides assurance that these agreed measures will serve as the
basis for regulatory review and the decision by the FDA to approve an NDA for LibiGel. These SPA
trials use our validated instruments to measure the clinical endpoints. The January 2008 SPA
agreement covers the pivotal Phase III safety and efficacy trials of LibiGel in the treatment of
FSD for “surgically” menopausal women. In July 2008, we received another SPA for our LibiGel
program in the treatment of FSD, specifically, HSDD in “naturally” menopausal women. We have an
additional SPA agreement which covers the LibiGel stability, or shelf life studies for the intended
commercialization of LibiGel product. The SPA agreements, however, are not guarantees of LibiGel
approval by the FDA or approval of any permissible claims about LibiGel. In particular, SPA
agreements are not binding on the FDA if previously unrecognized public health concerns later comes
to light, other new scientific concerns regarding product safety or effectiveness arise, we fail to
comply with the protocol agreed upon, or the FDA’s reliance on data, assumptions or information are
determined to be wrong. Even after an SPA agreement is finalized, the SPA agreement may be changed
by us or the FDA on written agreement of both parties, and the FDA retains significant latitude and
discretion in interpreting the terms of the SPA agreement and the data and results from any study
that is the subject of the SPA agreement. In addition, the data obtained from clinical trials are
susceptible to varying interpretations, which could delay, limit or prevent FDA regulatory
approval.
Delays in the completion of these clinical trials, which can result from unforeseen issues, FDA
interventions, problems with enrolling subjects and other reasons, could delay significantly
commercial launch and affect our product development costs. Moreover, results from these clinical
studies may not be as favorable as the results we obtained in prior, completed studies. We cannot
ensure that, even after extensive clinical trials, regulatory approval will ever be obtained for
LibiGel.
Uncertainties associated with the impact of published studies regarding the adverse health effects
of certain forms of hormone therapy could adversely affect the market for our hormone therapy
products and the trading price of our common stock.
The market for hormone therapy products has been affected negatively by the Women’s Health
Initiative (WHI) study and other studies that have found that the overall health risks from the use
of certain hormone therapy products may exceed the benefits from the use of those products among
postmenopausal women. In July 2002, the NIH released data from its WHI study on the risks and
benefits associated with long-term use of oral hormone therapy by women. The NIH announced that it
was discontinuing the arm of the study investigating the use of oral estrogen/progestin combination
hormone therapy products after an average follow-up period of 5.2 years because the product used in
the study was shown to cause an increase in the risk of invasive breast cancer. The study also
found an increased risk of stroke, heart attacks and blood clots and concluded that overall health
risks exceeded benefits from use of combined estrogen plus progestin for an average of 5.2 year
follow-up among postmenopausal women. Also, in July 2002, results of an observational study
sponsored by the National Cancer Institute on the effects of estrogen therapy were announced. The
main finding of the study was that postmenopausal women who used estrogen therapy for 10 or more
years had a higher risk of developing ovarian cancer than women who never used hormone therapy. In
October 2002, a significant hormone therapy study being conducted in the United Kingdom also was
halted. Our products differ from the products used in the WHI study and the primary products
observed in the National Cancer Institute and United Kingdom studies. In March 2004, the NIH
announced that the estrogen-alone study was discontinued after nearly seven years because
the NIH concluded that estrogen alone does not affect (either increase or decrease) heart disease,
the major question being evaluated in the study. The findings indicated a slightly increased risk
of stroke as well as a decreased risk of hip fracture and breast cancer. Preliminary data from the
memory portion of the WHI study suggested that estrogen alone may possibly be associated with a
slight increase in the risk of dementia or mild cognitive impairment.
27
Researchers continue to analyze data from both arms of the WHI study and other studies. Recent
reports indicate that the safety of estrogen products may be affected by the age of the woman at
initiation of therapy. There currently are no studies published comparing the safety of our
products against other hormone therapies. The markets for female hormone therapies for menopausal
symptoms declined as a result of these published studies, although the market now seems to have
stabilized. The release of any follow-up or other studies that show adverse affects from hormone
therapy, including in particular, hormone therapies similar to our products, also could affect
adversely our business and likely decrease our stock price.
If clinical trials for our products are prolonged or delayed, we may be unable to commercialize our
products on a timely basis, which would require us to incur additional costs and delay our receipt
of any revenue from potential product sales or licenses.
We may encounter problems with our completed, ongoing or planned clinical trials for our products
that may cause us or a regulatory authority to delay or suspend those clinical trials or delay the
analysis of data derived from them. A number of events, including any of the following, could delay
the completion of, or terminate, our ongoing and planned clinical trials for our products and
negatively impact our ability to obtain regulatory approval or enter into collaborations for, or
market or sell, a particular product:
| • |
|
conditions imposed on us by the FDA or any foreign regulatory authority regarding the scope
or design of our clinical trials; |
| • |
|
delay in developing, or our inability to obtain, a clinical dosage form, insufficient
supply or deficient quality of our products or other materials necessary to conduct our
clinical trials; |
| • |
|
negative or inconclusive results from clinical trials, or results that are inconsistent
with earlier results, that necessitate additional clinical study or termination of a clinical
program; |
| • |
|
serious and/or unexpected product-related side effects experienced by subjects in clinical
trials; or |
| • |
|
failure of our third-party contractors or our investigators to comply with regulatory
requirements or otherwise meet their contractual obligations to us in a timely manner. |
Regulatory authorities, clinical investigators, institutional review boards, data safety monitoring
boards and the sites at which our clinical trials are conducted all have the power to stop our
clinical trials prior to completion. Our clinical trials for our products may not begin as
planned, may need to be restructured, and may not be completed on schedule, if at all. This is
particularly true if we no longer have the financial resources to dedicate to our clinical trial
program.
28
We entered into an exclusive license agreement with Azur Pharma International II, Limited for the
marketing of Elestrin in the United States. Our ability to obtain sales-based milestones of up to
$140 million from Azur is dependent upon Azur’s ability to market and sell Elestrin.
Elestrin is our first FDA approved product. Azur Pharma International II Limited is marketing
Elestrin in the U.S. using its women’s health sales force that targets estrogen prescribing
physicians in the U.S.
comprised mostly of gynecologists. Azur launched sales and marketing activities related to
Elestrin in April 2009. We recognized royalty and other revenue from sales of Elestrin of $1.1
million for the year ended December 31, 2009. In December 2009, we entered into an amendment to
our original licensing agreement with Azur which permanently reduced the royalty percentage due to
us related to Azur’s sales of Elestrin. Upon signing the amended agreement, Azur made a $1.0
million nonrefundable payment in December 2009 in exchange for a permanent reduction in future
royalty rates, and received options to make a total of $2.16 million in additional non-refundable
payments (which were exercised during the first quarter of 2010) in exchange for the elimination of
substantially all remaining future royalties and milestone payments due us under the terms of the
original license. The $1.0 million nonrefundable payment was recorded as revenue during the fourth
quarter of 2009 as we had no remaining performance obligations with respect to this amount. We
maintain the right to receive up to $140 million in sales-based milestone payments from Azur if
Elestrin reaches certain predefined sales per calendar year. We cannot assure you that Azur will
be successful in marketing Elestrin or that Azur will remain focused on the commercialization of
Elestrin, especially if Azur does not experience significant Elestrin sales. Based on sales of
Elestrin to date, we believe it is unlikely that we will receive any sales-based milestone payments
from Azur.
Our other products, if they receive FDA approval and are introduced commercially may not achieve
expected levels of market acceptance, which could harm our business, financial position and
operating results and could cause the market value of our common stock to decline.
The commercial success of our products, if they receive the required FDA or other regulatory
approvals, is dependent upon market acceptance by physicians and patients. Levels of market
acceptance for our products could be affected by several factors, including:
| • |
|
the availability of alternative products from competitors; |
| • |
|
the price of our products relative to that of our competitors; |
| • |
|
the timing of market entry; and |
| • |
|
the ability to market our products effectively. |
Some of these factors are not within our control, especially if we have transferred all of the
marketing rights associated with the product, as we have with the U.S. marketing rights to Elestrin
to Azur and the U.S. marketing rights to The Pill Plus to Pantarhei Science. Our products may not
achieve expected levels of market acceptance. Additionally, continuing studies of the proper
utilization, safety and efficacy of pharmaceutical products are being conducted by the industry,
government agencies and others. Such studies, which increasingly employ sophisticated methods and
techniques, can call into question the utilization, safety and efficacy of previously marketed
products. In some cases, these studies have resulted, and may in the future result, in the
discontinuance of product marketing. These situations, should they occur, could harm our business,
financial position and results of operations, and the market value of our common stock could
decline.
29
We and our licensees depend on third-party manufacturers to produce our products and if these third
parties do not manufacture successfully these products our business would be harmed.
We have no manufacturing experience or manufacturing capabilities for the production of our
products for clinical trials or commercial sale. In order to continue to develop products, apply
for regulatory approvals and commercialize our products following approval, we or our licensees
must be able to manufacture or contract with third parties to manufacture our products in clinical
and commercial
quantities, in compliance with regulatory requirements, at acceptable costs and in a timely manner.
The manufacture of our products may be complex, difficult to accomplish and difficult to scale-up
when large-scale production is required. Manufacture may be subject to delays, inefficiencies and
poor or low yields of quality products. The cost of manufacturing our products may make them
prohibitively expensive. If supplies of any of our products become unavailable on a timely basis
or at all or are contaminated or otherwise lost, our clinical trials could be seriously delayed.
To the extent that we or our licensees seek to enter into manufacturing arrangements with third
parties, we and such licensees will depend upon these third parties to perform our obligations in a
timely and effective manner and in accordance with government regulations. Contract manufacturers
may breach their manufacturing agreements because of factors beyond our control or may terminate or
fail to renew a manufacturing agreement based on their own business priorities at a time that is
costly or inconvenient for us. If third-party manufacturers fail to perform their obligations, our
competitive position and ability to generate revenue may be adversely affected in a number of ways,
including:
| • |
|
we and our collaborators may not be able to initiate or continue clinical trials of product
candidates that are under development; |
| • |
|
we and our collaborators may be delayed in submitting applications for regulatory approvals
for our product candidates; and |
| • |
|
we and our collaborators may not be able to meet commercial demands for any approved
products. |
We have very limited staffing and will continue to be dependent upon key employees.
Our success is dependent upon the efforts of a relatively small management team and staff. We have
employment arrangements in place with both of our two executive officers, but neither of our
executive officers is bound legally to remain employed for any specific term. We do not have key
man life insurance policies covering our executive officers or any of our other employees. If key
individuals leave our company, our business could be affected adversely if suitable replacement
personnel are not recruited quickly.
There is competition for qualified personnel in all functional areas, which makes it difficult to
attract and retain the qualified personnel necessary for the development and growth of our
business. Our future success depends upon our ability to continue to attract and retain qualified
personnel.
30
Risks Related to Our Industry
Because our industry is very competitive, we may not succeed in bringing certain of our products to
market and any products we introduce commercially may not be successful.
Competition in the pharmaceutical industry is intense. Potential competitors in the United States
and abroad are numerous and include pharmaceutical and biotechnology companies, most of which have
substantially greater capital resources and more experience in research and development,
manufacturing and marketing than us. Academic institutions, hospitals, governmental agencies and
other public and private research organizations also are conducting research and seeking patent
protection and may develop and commercially introduce competing products or technologies on their
own or through joint ventures. We cannot assure you that our potential competitors, some of whom
are our development collaborators, will not succeed in developing similar technologies and products
more rapidly than we do, commercially introducing such technologies and products to the marketplace
prior to us, or that these
competing technologies and products will not be more effective or successful than any of those that
we currently are developing or will develop.
Because the pharmaceutical industry is heavily regulated, we face significant costs and
uncertainties associated with our efforts to comply with applicable regulations. Should we fail to
comply, we could experience material adverse effects on our business, financial position and
results of operations, and the market value of our common stock could decline.
The pharmaceutical industry is subject to regulation by various federal and state governmental
authorities. For example, we must comply with FDA requirements with respect to the development of
our products and our clinical trials, and if any of our products are approved, the manufacture,
labeling, sale, distribution, marketing, advertising and promotion of our products. Failure to
comply with FDA and other governmental regulations can result in fines, disgorgement, unanticipated
compliance expenditures, recall or seizure of products, total or partial suspension of production
and/or distribution, suspension of the FDA’s review of NDAs, enforcement actions, injunctions and
criminal prosecution. Under certain circumstances, the FDA also has the authority to revoke
previously granted drug approvals. Despite our efforts at compliance, there is no guarantee that we
may not be deemed to be deficient in some manner in the future. If we were deemed to be deficient
in any significant way, our business, financial position and results of operations could be
materially affected and the market value of our common stock could decline.
Risks Related to Our Intellectual Property
We license rights to the technology underlying LibiGel and many of our other products and
technologies from third parties. The loss of these rights, including in particular, our rights
underlying LibiGel, could have an adverse effect on our business and future prospects and could
cause the market value of our common stock to decline.
We license rights to certain of the technology underlying our gel products, including LibiGel, from
Antares Pharma, Inc., our GVAX cancer immunotherapies from Johns Hopkins University and The
Whitehead Institute for Biomedical Research, a portion of our CaP technology from the University of
California and The Pill Plus from Wake Forest University. We may lose our rights to these
technologies if we breach our obligations under the license agreements. Although we intend to use
commercially reasonable efforts to meet these obligations, if we violate or fail to perform any
term or covenant of the license agreements, the other party to these agreements under certain
circumstances may terminate these agreements or certain projects contained in these agreements.
The termination of these agreements, however, will not relieve us of our obligation to pay any
royalty or license fees owed at the time of termination. For example, if we were to enter into an
license agreement with a third party under which we agree to license rights to our gel technology,
GVAX cancer immunotherapies or CaP technology for a license fee, the termination of the main
license agreement with Antares Pharma, Inc., Johns Hopkins University and The Whitehead Institute
for BioMedical Research, the University of California or Wake Forest University could either,
depending upon the terms of the license agreement, cause us to breach our obligations under the
license agreement or give the other party a right to terminate that agreement, thereby causing us
to lose future revenue generated by the license fees. Our failure to retain the right to these
technologies could harm our business and future prospects. This is true in particular with respect
to LibiGel, which we believe if approved by the FDA could be a very successful product.
31
We have licensed some of our products to third parties and any breach by these parties of their
obligations under these license agreements or a termination of these license agreements by these
parties could adversely affect the development and marketing of our licensed products. In
addition, these third parties also may compete with us with respect to some of our products.
We have licensed our CaP technology for use as a facial line filler to MATC and some of our gel
products to third parties, including Azur, Solvay Pharmaceuticals, B.V. (now owned by Abbott
Laboratories), Teva Pharmaceuticals USA, Inc., Pantarhei Bioscience B.V. and PharmaSwiss SA. All
of these parties, except for Azur, have agreed to be responsible for continued development,
regulatory filings and all have agreed to manufacturing and marketing associated with the products.
In addition, in the future we may enter into additional similar license agreements. Our products
that we have licensed to others thus are subject to not only customary and inevitable uncertainties
associated with the drug development process, regulatory approvals and market acceptance of
products, but also depend on the respective licensees for timely development, obtaining required
regulatory approvals, commercialization and otherwise continued commitment to the products. Our
current and future licensees may have different and, sometimes, competing priorities. We cannot
assure you that our partners or any future third party to whom we may license our products will
remain focused on the development and commercialization of our partnered products or will not
otherwise breach the terms of our agreements with them, especially since these third parties also
may compete with us with respect to some of our products. For example, at this time and as
previously disclosed, we believe that our estrogen/progestogen combination transdermal gel product
licensed to Abbott Laboratories is not in active development by Abbott, and we do not expect its
active development to occur at any time in the near future. As an additional example, in 2005, we
were notified that Teva USA had discontinued development of our male testosterone gel, Bio-T-Gel,
product. Although in June 2007, we signed an amendment to the agreement under which we and Teva
reinitiated our collaboration on the development of Bio-T-Gel for the U.S. market, no assurance can
be provided that Teva will continue such development. Any future breach of this agreement by Teva
or any other breach by our partners or any other third party of their obligations under these
agreements or a termination of these agreements by these parties could harm development of the
products in these agreements if we are unable to license the products to another party on
substantially the same or better terms or continue the development and future commercialization of
the products ourselves.
If we are unable to protect our proprietary technology, we may not be able to compete as
effectively.
The pharmaceutical industry places considerable importance on obtaining patent and trade secret
protection for new technologies, products and processes. Our success will depend, in part, upon
our ability to obtain, enjoy and enforce protection for any products we develop or acquire under
United States and foreign patent laws and other intellectual property laws, preserve the
confidentiality of our trade secrets and operate without infringing the proprietary rights of third
parties. We rely on patent protection, as well as a combination of copyright and trademark laws
and nondisclosure, confidentiality and other contractual arrangements to protect our proprietary
technology. These legal means, however, afford only limited protection and may not adequately
protect our rights or permit us to gain or keep any competitive advantage.
Where appropriate, we seek patent protection for certain aspects of our technology. Our owned and
licensed patents and patent applications, however, may not ensure the protection of our
intellectual property for a number of other reasons:
| • |
|
We do not know whether our licensor’s patent applications will result in issued patents. |
| • |
|
Competitors may interfere with our patents and patent process in a variety of ways. Our
issued patents and those that may be issued in the future may be challenged, invalidated or
circumvented, which could limit our ability to stop competitors from marketing related
products. Competitors also may have our patents reexamined by demonstrating to the patent
examiner that the invention was not original or novel or was obvious. |
32
| • |
|
We are engaged in the process of developing products. Even if we receive a patent, it may
not provide much practical protection. There is no assurance that third parties will not be
able to design around our patents. If we receive a patent with a narrow scope, then it will be
easier for competitors to design products that do not infringe on our patent. Even if the
development of our products is successful and approval for sale is obtained, there can be no
assurance that applicable patent coverage, if any, will not have expired or will not expire
shortly after this approval. Though patent term extension may be possible for particular
products, any expiration of the applicable patent could have a material adverse effect on the
sales and profitability of our products. |
| • |
|
Litigation also may be necessary to enforce patent rights we hold or to protect trade
secrets or techniques we own. Intellectual property litigation is costly and may adversely
affect our operating results. Such litigation also may require significant time by our
management. In litigation, a competitor could claim that our issued patents are not valid for
a number of reasons. If the court agrees, we would lose protection on products covered by
those patents. |
| • |
|
We also may support and collaborate in research conducted by government organizations or
universities. We cannot guarantee that we will be able to acquire any exclusive rights to
technology or products derived from these collaborations. If we do not obtain required
licenses or rights, we could encounter delays in product development while we attempt to
design around other patents or we may be prohibited from developing, manufacturing or selling
products requiring these licenses. There is also a risk that disputes may arise as to the
rights to technology or products developed in collaboration with other parties. |
We also rely on unpatented proprietary technology. It is unclear whether efforts to secure our
trade secrets will provide useful protection. We rely on the use of registered trademarks with
respect to the brand names of some of our products. We also rely on common law trademark
protection for some brand names, which are not protected to the same extent as our rights in the
use of our registered trademarks. We cannot assure you that we will be able to meaningfully protect
all of our rights in our unpatented proprietary technology or that others will not independently
develop and obtain patent protection substantially equivalent proprietary products or processes or
otherwise gain access to our unpatented proprietary technology. We seek to protect our know-how
and other unpatented proprietary technology, in part with confidentiality agreements and
intellectual property assignment agreements with our employees and consultants. Such agreements,
however, may not be enforceable or may not provide meaningful protection for our proprietary
information in the event of unauthorized use or disclosure or other breaches of the agreements or
in the event that our competitors discover or independently develop similar or identical designs or
other proprietary information. Enforcing a claim that someone else illegally obtained and is using
our trade secrets, like patent litigation, is expensive and time consuming, and the outcome is
unpredictable. In addition, courts outside the United States are sometimes less willing to protect
trade secrets.
Claims by others that our products infringe their patents or other intellectual property rights
could adversely affect our financial condition.
The pharmaceutical industry has been characterized by frequent litigation regarding patent and
other intellectual property rights. Patent applications are maintained in secrecy in the United
States and also are maintained in secrecy outside the United States until the application is
published. Accordingly, we cannot determine whether our technology would infringe on patents
arising from these unpublished patent applications of others. Any claims of patent infringement
asserted by third parties would be time-consuming and could likely:
| • |
|
result in costly litigation; |
33
| • |
|
divert the time and attention of our technical personnel and management; |
| • |
|
cause product development delays; |
| • |
|
require us to develop non-infringing technology; or |
| • |
|
require us to enter into royalty or licensing agreements. |
Although patent and intellectual property disputes in the pharmaceutical industry often have been
settled through licensing or similar arrangements, costs associated with these arrangements may be
substantial and often require the payment of ongoing royalties, which could hurt our potential
gross margins. In addition, we cannot be sure that the necessary licenses would be available to us
on satisfactory terms, or that we could redesign our products or processes to avoid infringement,
if necessary. Accordingly, an adverse determination in a judicial or administrative proceeding, or
the failure to obtain necessary licenses, could prevent us from developing, manufacturing and
selling some of our products, which could harm our business, financial condition and operating
results.
Risks Related to Our Common Stock
The price of our common stock has been volatile. As a result, we could become subject to class
action litigation, which even if without merit, could be costly to defend and could divert the time
and attention of our management, which could harm our business and financial condition.
Since January 1, 2009, the sale price of our common stock has ranged from a low of $1.03 to a high
of $2.70. It is likely that the price of our common stock will continue to fluctuate in the
future. The securities of small capitalization, biopharmaceutical companies, including our
company, from time to time experience significant price fluctuations, often unrelated to the
operating performance of these companies. In particular, the market price of our common stock may
fluctuate significantly due to a variety of factors, including:
| • |
|
general stock, market and general economic conditions in the United States and abroad, not
directly related to our company or our business. |
| • |
|
our ability to obtain needed financing; |
| • |
|
governmental agency actions, including in particular decisions or actions by the FDA or FDA
advisory committee panels with respect to our products or our competitors’ products; |
| • |
|
the results of our clinical trials or those of our competitors; |
| • |
|
announcements of technological innovations or new products by us or our competitors; |
| • |
|
announcements by licensors or licensees of our technology; |
| • |
|
public concern as to the safety or efficacy of or market acceptance of products developed
by us or our competitors; |
| • |
|
developments or disputes concerning patents or other proprietary rights; |
| • |
|
period-to-period fluctuations in our financial results, including our cash and cash
equivalents, operating expenses, cash burn rate or revenues; |
34
| • |
|
loss of key management; |
| • |
|
common stock sales in the public market by one or more of our larger stockholders, officers
or directors; and |
| • |
|
other potentially negative financial announcements, including delisting of our common stock
from the NASDAQ Global Market, review of any of our filings by the SEC, changes in accounting
treatment or restatement of previously reported financial results, delays in our filings with
the SEC or our failure to maintain effective internal control over financial reporting. |
In addition, the occurrence of any of the risks described in this report or otherwise in reports we
file with or submit to the SEC from time to time could have a material and adverse impact on the
market price of our common stock. Securities class action litigation is sometimes brought against
a company following periods of volatility in the market price of its securities or for other
reasons. We may become the target of similar litigation. Securities litigation, whether with or
without merit, could result in substantial costs and divert management’s attention and resources,
which could harm our business and financial condition, as well as the market price of our common
stock.
If we fail to meet continued listing standards of the Nasdaq Global Market, our common stock may be
delisted which could have a material adverse effect on the liquidity of our common stock.
In order for our securities to be eligible for continued listing on the NASDAQ Global Market, we
must remain in compliance with certain listing standards, including a $1.00 minimum closing bid
price per share requirement, a $50 million market capitalization and a $15 million public float
requirement or a $12 million minimum stockholders’ equity requirement, and certain corporate
governance standards. If our common stock were to be delisted from the NASDAQ Global Market, we
could apply to list our common stock on the NASDAQ Capital Market or our common stock could be
traded in the over-the-counter market on an electronic bulletin board established for unlisted
securities, such as the Pink Sheets or the OTC Bulletin Board. Any delisting could adversely
affect the market price of, and liquidity of the trading market for, our common stock, our ability
to obtain financing for the continuation of our operations and could result in the loss of
confidence by investors.
Provisions in our charter documents and Delaware law could discourage or prevent a takeover, even
if an acquisition would be beneficial to our stockholders.
Provisions of our certificate of incorporation and bylaws, as well as provisions of Delaware law,
could make it more difficult for a third party to acquire us, even if doing so would be beneficial
to our stockholders. These provisions include:
| • |
|
authorizing the issuance of “blank check” preferred shares that could be issued by our
Board of Directors to increase the number of outstanding shares and thwart a takeover attempt; |
| • |
|
prohibiting cumulative voting in the election of directors, which would otherwise allow
less than a majority of stockholders to elect director candidates; and |
| • |
|
advance notice provisions in connection with stockholder proposals that may prevent or
hinder any attempt by our stockholders to bring business to be considered by our stockholders
at a meeting or replace our board of directors. |
35
We may issue additional equity securities which would dilute your share ownership.
We may issue additional equity securities to raise capital and through the exercise of options and
warrants that are outstanding or may be outstanding. These additional issuances would dilute your
share ownership.
We do not intend to pay any cash dividends in the foreseeable future and, therefore, any return on
an investment in our common stock must come from increases in the fair market value and trading
price of our common stock.
We do not intend to pay any cash dividends in the foreseeable future and, therefore, any return on
an investment in our common stock must come from increases in the fair market value and trading
price of our common stock.
|
|
|
| Item 1B. |
|
UNRESOLVED STAFF COMMENTS |
This Item 1B is not applicable to BioSante as a smaller reporting company.
Our principal executive office is located in a leased facility in Lincolnshire, Illinois, where we
lease approximately 12,000 square feet of office space for approximately $23,000 per month. Our
lease for this space expires in April 2012. Our CaP development operations are located within the
Bucks County Biotech Park in Doylestown, Pennsylvania where we lease approximately 2,000 square
feet of laboratory space for approximately $3,900 per month. This lease is renewable in one year
increments each July and expires in July 2010. Management of our company considers our leased
properties suitable and adequate for our current and foreseeable needs.
36
|
|
|
| Item 3. |
|
LEGAL PROCEEDINGS |
We are presently involved in one legal action and from time to time may be subject to various
pending or threatened legal actions and proceedings, including those that arise in the ordinary
course of our business. Such matters are subject to many uncertainties and to outcomes that are
not predictable with assurance and that may not be known for extended periods of time.
On July 1, 2009, a putative shareholder class action lawsuit concerning our then proposed merger
with Cell Genesys, Inc. was filed in California Superior Court in San Mateo County naming Cell
Genesys, its officers and directors, and our company as defendants. On July 6, 2009, a second
putative shareholder class action lawsuit naming the same parties and containing essentially
identical allegations was filed in California Superior Court in San Mateo County. On July 8, 2009,
a third putative shareholder class action lawsuit was filed in California Superior Court in San
Mateo County, which also named the same parties and contained essentially identical allegations as
the two prior lawsuits. On July 15, 2009, the Court consolidated these three lawsuits into one
action and appointed interim lead counsel. On August 13, 2009, plaintiffs filed a consolidated
class action complaint alleging that defendants breached their fiduciary duties and/or aided and
abetted the breach of fiduciary duties owed to Cell Genesys stockholders in connection with the
then proposed merger, including by failing to engage in a fair sales process, failing to obtain a
fair price for the sale of Cell Genesys, and failing to provide Cell Genesys stockholders with
material information regarding the merger. Plaintiffs sought an order certifying the lawsuit as a
class action, injunctive relief to enjoin the merger or, in the event the then pending merger was
completed, a rescission of the merger or rescissory damages. Plaintiffs further sought an
accounting for all damages and an award of attorneys’ fees and costs.
Solely to avoid the costs, risks and uncertainties inherent in litigation, on September 18, 2009,
we and Cell Genesys entered into a memorandum of understanding with plaintiffs’ counsel in the San
Mateo County action pursuant to which we, Cell Genesys, the other named defendants and the
plaintiffs agreed to settle the lawsuits subject to court approval. If the Court approves the
settlement, the lawsuits will be dismissed with prejudice. Pursuant to the memorandum of
understanding, Cell Genesys agreed to pay to plaintiffs’ counsel an amount not more than $240,000
as is approved by Court order for plaintiffs’ attorneys’ fees, costs and expenses in the San Mateo
County action and to make additional disclosures in a current report on Form 8-K, without admitting
in any way that the certain disclosures are material or otherwise required by law. Cell Genesys
filed the Form 8-K on September 21, 2009. Pursuant to the memorandum of understanding, plaintiffs’
counsel conducted confirmatory discovery to confirm the fairness and adequacy of the settlement.
The parties filed a stipulation of settlement with the Court and moved the Court for preliminary
approval and issuance of a notice of settlement to the potential class members, after which the
parties intend to seek final settlement approval and dismissal of the action with prejudice. As a
result of our merger with Cell Genesys, we assumed Cell Genesys’s rights and obligations relative
to this lawsuit. We have recorded a liability of $240,000 for the potential settlement and believe
that the resolution of this matter will not have a material impact on our financial position, cash
flows or results of operations.
37
|
|
|
| Item 4A. |
|
EXECUTIVE OFFICERS OF THE REGISTRANT |
Our executive officers, their ages and the offices held, as of March 15, 2010, are as follows:
| |
|
|
|
|
|
|
| Name |
|
Age |
|
Title |
|
|
|
|
|
|
|
Stephen M. Simes
|
|
|
58 |
|
|
Vice Chairman, President and Chief Executive Officer |
| |
Phillip B. Donenberg
|
|
|
49 |
|
|
Chief Financial Officer, Treasurer and Secretary |
Each of our executive officers serves at the discretion of our Board of Directors and holds office
until his successor is elected and qualified or until his earlier resignation or removal. There
are no family relationships among any of our directors or executive officers.
Information regarding the business experience of our executive officers is set forth below.
Stephen M. Simes has served as our Vice Chairman, President and a director of our company since
January 1998 and Chief Executive Officer since March 1998. From October 1994 to January 1997, Mr.
Simes was President, Chief Executive Officer and a Director of Unimed Pharmaceuticals, Inc.,
(currently a wholly owned subsidiary of Abbott Laboratories) a company with a product focus on
infectious diseases, AIDS, endocrinology and oncology. From 1989 to 1993, Mr. Simes was Chairman,
President and Chief Executive Officer of Gynex Pharmaceuticals, Inc., a company which concentrated
on the AIDS, endocrinology, urology and growth disorders markets. In 1993, Gynex was acquired by
Savient Pharmaceuticals Inc. (formerly Bio-Technology General Corp.), and from 1993 to 1994, Mr.
Simes served as Senior Vice President and director of Savient Pharmaceuticals Inc. Mr. Simes’s
career in the pharmaceutical industry started in 1974 with G.D. Searle & Co. (now a part of Pfizer
Inc.). Mr. Simes currently serves as our designee on the board of directors of Ceregene, Inc., a
privately-held biotechnology company focused on the treatment of major neurodegenerative disorders
using the delivery of nervous system growth factors. As a result of our merger with Cell Genesys,
we acquired an investment equivalent to approximately 16 percent of the total equity of Ceregene,
and by virtue of such ownership, we have the right to designate one member of Ceregene’s board of
directors.
Phillip B. Donenberg, CPA, has served as our Chief Financial Officer, Treasurer and Secretary since
July 1998. Before joining our company, Mr. Donenberg was Controller of Unimed Pharmaceuticals,
Inc. (currently a wholly owned subsidiary of Abbott Laboratories) from January 1995 to July 1998.
Prior to Unimed Pharmaceuticals, Inc., Mr. Donenberg held similar positions with other
pharmaceutical companies, including Gynex Pharmaceuticals, Inc. (currently Savient Pharmaceuticals,
Inc.), Applied NeuroSolutions, Inc. (formerly Molecular Geriatrics Corporation) and Xtramedics,
Inc.
38
PART II
|
|
|
| Item 5. |
|
MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF
EQUITY SECURITIES |
Market Price
Our common stock is listed for trading on the NASDAQ Global Market, under the symbol “BPAX.” The
following table sets forth the high and low daily sale prices for our common stock, as reported by
the NASDAQ Global Market, for each calendar quarter during 2009 and 2008.
| |
|
|
|
|
|
|
|
|
| 2009 |
|
High |
|
|
Low |
|
|
|
|
|
|
|
|
|
|
First Quarter |
|
$ |
2.33 |
|
|
$ |
1.03 |
|
Second Quarter |
|
$ |
2.67 |
|
|
$ |
1.30 |
|
Third Quarter |
|
$ |
2.70 |
|
|
$ |
1.45 |
|
Fourth Quarter |
|
$ |
2.15 |
|
|
$ |
1.33 |
|
| |
|
|
|
|
|
|
|
|
| 2008 |
|
High |
|
|
Low |
|
|
|
|
|
|
|
|
|
|
First Quarter |
|
$ |
5.05 |
|
|
$ |
2.05 |
|
Second Quarter |
|
$ |
5.85 |
|
|
$ |
3.50 |
|
Third Quarter |
|
$ |
5.79 |
|
|
$ |
3.26 |
|
Fourth Quarter |
|
$ |
4.85 |
|
|
$ |
0.81 |
|
Number of Record Holders; Dividends
As of March 15, 2010, there were 847 record holders of our common stock and six record holders of
our class C stock. To date, we have not declared or paid any cash dividends on our common stock
and our class C stock is not eligible to receive dividends.
Recent Sales of Unregistered Equity Securities
During the fourth quarter ended December 31, 2009, we did not issue or sell any equity securities
of ours without registration under the Securities Act of 1933, as amended.
Issuer Purchases of Equity Securities
We did not purchase any shares of our common stock or other equity securities of ours during the
fourth quarter ended December 31, 2009. Our Board of Directors has not authorized any repurchase
plan or program for the purchase of our shares of common stock or other securities on the open
market or otherwise, other than in connection with the cashless exercise of outstanding warrants
and stock options.
39
|
|
|
| Item 6. |
|
SELECTED FINANCIAL DATA |
The following selected financial information has been derived from our audited financial
statements. The information below is not necessarily indicative of results of future operations,
and should be read together with “Part II. Item 7. Management’s Discussion and Analysis of
Financial Condition and Results of Operations” and the financial statements and related notes
included in “Part II. Item 8. Financial Statements and Supplementary Data” of this report in order
to fully understand factors that may affect the comparability of the information presented below:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Year Ended December 31, |
|
| |
|
2009 |
|
|
2008 |
|
|
2007 |
|
|
2006 |
|
|
2005 |
|
| |
|
(in thousands, except per share data) |
|
Statement of Operations Data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenue |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Licensing revenue |
|
$ |
— |
|
|
$ |
3,384 |
|
|
$ |
199 |
|
|
$ |
14,136 |
|
|
$ |
45 |
|
Grant revenue |
|
|
116 |
|
|
|
65 |
|
|
|
59 |
|
|
|
247 |
|
|
|
181 |
|
Royalty revenue |
|
|
1,142 |
|
|
|
34 |
|
|
|
69 |
|
|
|
— |
|
|
|
— |
|
Other revenue |
|
|
— |
|
|
|
298 |
|
|
|
166 |
|
|
|
55 |
|
|
|
32 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total revenue |
|
|
1,258 |
|
|
|
3,781 |
|
|
|
493 |
|
|
|
14,438 |
|
|
|
258 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest income |
|
|
12 |
|
|
|
588 |
|
|
|
1,095 |
|
|
|
429 |
|
|
|
401 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Expenses |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development |
|
|
13,681 |
|
|
|
15,790 |
|
|
|
4,751 |
|
|
|
3,908 |
|
|
|
6,409 |
|
General and administration |
|
|
5,374 |
|
|
|
5,125 |
|
|
|
4,331 |
|
|
|
4,550 |
|
|
|
3,801 |
|
Acquired in-process research and development |
|
|
9,000 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Excess consideration paid over fair value |
|
|
20,192 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Licensing expense |
|
|
300 |
|
|
|
836 |
|
|
|
— |
|
|
|
3,500 |
|
|
|
— |
|
Depreciation and amortization |
|
|
137 |
|
|
|
43 |
|
|
|
90 |
|
|
|
118 |
|
|
|
101 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total expenses |
|
|
48,684 |
|
|
|
21,794 |
|
|
|
9,172 |
|
|
|
12,076 |
|
|
|
10,311 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Other income — Fair value adjustment |
|
|
33 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Other expense — Interest expense |
|
|
147 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net (loss) income |
|
$ |
(47,528 |
) |
|
$ |
(17,425 |
) |
|
$ |
(7,584 |
) |
|
$ |
2,791 |
|
|
$ |
(9,651 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted net (loss) income per share |
|
$ |
(1.40 |
) |
|
$ |
(0.64 |
) |
|
$ |
(0.30 |
) |
|
$ |
0.13 |
|
|
$ |
(0.50 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average number of shares outstanding |
|
|
33,952 |
|
|
|
27,307 |
|
|
|
25,486 |
|
|
|
21,484 |
|
|
|
19,392 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
As of December 31, |
|
| |
|
2009 |
|
|
2008 |
|
|
2007 |
|
|
2006 |
|
|
2005 |
|
| |
|
(in thousands) |
|
Balance Sheet Data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash, cash equivalents and short-term investments |
|
$ |
29,858 |
|
|
$ |
14,787 |
|
|
$ |
30,655 |
|
|
$ |
11,450 |
|
|
$ |
9,102 |
|
Total assets |
|
|
36,437 |
|
|
|
17,679 |
|
|
|
31,241 |
|
|
|
22,371 |
|
|
|
9,575 |
|
Total current liabilities |
|
|
3,930 |
|
|
|
3,853 |
|
|
|
1,516 |
|
|
|
4,300 |
|
|
|
2,666 |
|
Convertible senior notes |
|
|
16,676 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Stockholders’ equity |
|
|
15,830 |
|
|
|
13,826 |
|
|
|
29,725 |
|
|
|
18,071 |
|
|
|
6,819 |
|
40
|
|
|
| Item 7. |
|
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
This Management’s Discussion and Analysis provides material historical and prospective disclosures
intended to enable investors and other users to assess our financial condition and results of
operations. Statements that are not historical are forward-looking and involve risks and
uncertainties discussed under the headings “Part I. Item 1. Business—Forward-Looking Statements”
and “Part I. Item 1A. Risk Factors” of this report. The following discussion of our results of
operations and financial condition should be read in conjunction with our financial statements and
the related notes thereto included elsewhere in this report. This Management’s Discussion and
Analysis is organized in the following major sections:
| • |
|
Business Overview. This section provides a brief overview description of our
business, focusing in particular on developments during the most recent fiscal year. |
| |
| • |
|
Summary of 2009 Financial Results and Outlook for 2010. This section provides a
brief summary of our financial results and financial condition for 2009 and our outlook for
2010. |
| |
| • |
|
Critical Accounting Policies and Estimates. This section discusses the
accounting estimates that are considered important to our financial condition and results of
operations and require us to exercise subjective or complex judgments in their application.
All of our significant accounting policies, including our critical accounting estimates, are
summarized in Note 2 to our financial statements. |
| |
| • |
|
Results of Operations. This section provides our analysis of the significant
line items in our statements of operations. |
| |
| • |
|
Liquidity and Capital Resources. This section provides an analysis of our
liquidity and cash flows and a discussion of our outstanding indebtedness and commitments. |
| |
| • |
|
Recent Accounting Pronouncements. This section discusses recently issued
accounting pronouncements that have had or may affect our results of operations and financial
condition. |
Business Overview
We are a specialty pharmaceutical company focused on developing products for female sexual health,
menopause, contraception and male hypogonadism. In addition, we are evaluating and seeking
opportunities for our GVAX cancer immunotherapies, 2A/Furin and other technologies we acquired in
our merger with Cell Genesys, Inc. We also are developing our calcium phosphate technology (CaP)
for aesthetic medicine (BioLook), as a vaccine adjuvant, including for an H1N1 (swine flu) vaccine,
and drug delivery.
Our products for female sexual health, menopause, contraception and male hypogonadism include:
| • |
|
LibiGel — once daily transdermal testosterone gel in Phase III clinical
development under a Special Protocol Assessment (SPA) for the treatment of female sexual
dysfunction (FSD). |
| |
| • |
|
Elestrin — once daily transdermal estradiol (estrogen) gel approved by the U.S.
Food and Drug Administration (FDA) indicated for the treatment of moderate-to-severe vasomotor
symptoms (hot flashes) associated with menopause and marketed in the U.S. |
41
| • |
|
The Pill-Plus (triple component contraceptive) — once daily use of various
combinations of estrogens, progestogens and androgens in development for the treatment of FSD
in women using oral or transdermal contraceptives. |
| |
| • |
|
Bio-T-Gel — once daily transdermal testosterone gel in development for the
treatment of hypogonadism, or testosterone deficiency, in men. |
We believe LibiGel remains the lead pharmaceutical product in the U.S. in active development for
the treatment of hypoactive sexual desire disorder (HSDD) in menopausal women, and that it has the
potential to be the first product approved by the FDA for this common and unmet medical need. We
believe based on agreements with the FDA, including an SPA, that two Phase III safety and efficacy
trials and one year of LibiGel exposure in a Phase III cardiovascular and breast cancer safety
study with a four-year follow-up post-NDA filing and potentially post-FDA approval are the
essential requirements for submission and, if successful, approval by the FDA of a new drug
application (NDA) for LibiGel for the treatment of FSD, specifically HSDD in menopausal women.
Currently, three LibiGel Phase III studies are underway and enrolling women: two LibiGel Phase III
safety and efficacy clinical trials and one Phase III cardiovascular and breast cancer safety
study. Both Phase III safety and efficacy trials are double-blind, placebo-controlled trials that
will enroll up to approximately 500 surgically menopausal women each for a six-month clinical
trial. The Phase III safety study is a randomized, double-blind, placebo-controlled, multi-center,
cardiovascular events driven study of between 2,400 and 3,100 women exposed to LibiGel or placebo
for 12 months after which time we intend to submit an NDA to the FDA. In February 2010, we
announced that based upon the second review of study conduct and unblinded data from the LibiGel
Phase III cardiovascular and breast cancer safety study, the independent data monitoring committee
(DMC) unanimously recommended continuing the study as described in the FDA-agreed study protocol,
with no modifications. The DMC reviewed all unblinded adverse events in the safety study including
“serious adverse events” and all “adverse cardiovascular and breast cancer events” in almost 1,200
women-years of exposure. As of such date, there had been no deaths, only six adjudicated
cardiovascular events and only four breast cancers reported. In view of DMC recommendation, we
will continue the LibiGel Phase III development program as planned. We continue to target
submission to the FDA of an NDA by mid-2011. Following NDA submission and potential FDA approval,
we will continue to follow the subjects in the safety study for an additional four years.
Elestrin is our first FDA approved product. Azur Pharma International II Limited is marketing
Elestrin in the U.S. using its women’s health sales force that targets estrogen prescribing
physicians in the U.S. comprised mostly of gynecologists. In December 2009, we entered into an
amendment to our original licensing agreement with Azur which permanently reduced the royalty
percentage due to us related to Azur’s sales of Elestrin. Upon signing the amended agreement, Azur
made a $1.0 million nonrefundable payment in December 2009 in exchange for a permanent reduction in
future royalty rates, and received options to make a total of $2.16 million in additional
non-refundable payments (which were exercised during the first quarter of 2010) in exchange for the
elimination of substantially all remaining future royalties and milestone payments due us under the
terms of the original license. The $1.0 million nonrefundable payment was recorded as revenue during
the fourth quarter of 2009 as we had no remaining performance obligations with respect to this
amount. We maintain the right to receive up to $140 million in sales-based milestone payments from
Azur if Elestrin reaches certain predefined sales per calendar year.
42
We license the technology underlying certain of our gel products, including LibiGel and Elestrin,
from Antares Pharma, Inc. Our license agreement with Antares requires us to pay Antares certain
development and regulatory milestone payments and royalties based on net sales of any products we
or our licensees
sell incorporating the licensed technology. Specifically, we are obligated to pay Antares 25
percent of all licensing-related proceeds and 4.5 percent of any associated royalties that we may
receive. Bio-T-Gel was developed and is fully-owned by us. We license the technology underlying
The Pill Plus from Wake Forest University Health Sciences and Cedars-Sinai Medical Center. The
financial terms of this license include regulatory milestone payments, maintenance payments and
royalty payments by us if a product incorporating the licensed technology gets approved and is
subsequently marketed.
As a result of our October 2009 merger with Cell Genesys, we acquired a portfolio of products,
including GVAX cancer immunotherapies. GVAX cancer immunotherapies are cancer vaccines designed to
stimulate the patient’s immune system to effectively fight cancer. Multiple Phase II trials of
these technologies which had been ongoing at the date of the acquisition are continuing at minimal
cost to us at the Johns Hopkins Sidney Kimmel Comprehensive Cancer Center in various cancer
programs, including pancreatic cancer, leukemia and breast cancer. We license our GVAX cancer
immunotherapy technology from Johns Hopkins University. Under various agreements, we are required
to pay Johns Hopkins University and The Whitehead Institute for Biomedical Research certain
development and regulatory milestone payments and royalties based on net sales of any products we
or our licensees sell incorporating the in-licensed technology.
Our strategy with respect to our CaP technology is to continue development of our nanoparticle
technology and actively seek collaborators and licensees to fund and accelerate the development and
commercialization of products incorporating the technology. In addition to continuing our own
product development in the potential commercial applications of our CaP technology, we have sought
and continue to seek opportunities to enter into business collaborations or joint ventures with
vaccine companies and others interested in development and marketing arrangements with respect to
our CaP technology. For example, we have entered into a license agreement with Medical Aesthetics
Technology Corporation (MATC) covering the use of our CaP as a facial line filler in aesthetic
medicine (BioLook). Under the license agreement, MATC is responsible for continued development of
BioLook, including required clinical trials, regulatory filings and all manufacturing and marketing
associated with the product. In exchange for the license, we received an ownership position in
MATC of approximately five percent of the common stock of MATC. In addition to the ownership
position, we may receive certain milestone payments and royalties as well as share in certain
payments if MATC sublicenses the technology.
One of our strategic goals is to continue to seek and implement strategic alternatives with respect
to our products and our company, including licenses, business collaborations and other business
combinations or transactions with other pharmaceutical and biotechnology companies. Therefore, as
a matter of course, we may engage in discussions with third parties regarding the licensure, sale
or acquisition of our products and technologies or a merger or sale of our company.
Summary of 2009 Financial Results and Outlook for 2010
Substantially all of our revenue to date has been derived from upfront, milestone and royalty
payments earned on licensing and sublicensing transactions and from subcontracts. To date, we have
used primarily equity financings, and to a lesser extent, licensing income, interest income and the
cash received from our merger with Cell Genesys, to fund our ongoing business operations and
short-term liquidity needs, and we expect to continue this practice for the foreseeable future.
43
We have not introduced commercially any products. Azur, our marketing licensee for Elestrin,
commercially launched Elestrin in April 2009. As a result, we received royalties on net sales of
Elestrin from Azur. We recognized $1.1 million in royalty revenue from sales of Elestrin during
the year ended December 31, 2009, which includes $141,665 in royalty payments pursuant to our
original agreement
with Azur, and $1.0 million of additional royalty income resulting from payments received as a
result of the amendment of the agreement in December 2009. This royalty revenue amount represents
the gross royalty revenue we received from Elestrin through December 31, 2009 and not our
corresponding obligation to pay Antares royalties. Our corresponding obligation to pay Antares a
portion of the royalties received, which equaled $63,749 for the year ended December 31, 2009, is
recorded within general and administrative expenses in our statements of operations. In December
2009, we entered into an amendment to our original licensing agreement with Azur which permanently
reduced the royalty percentage due to us related to Azur’s sales of Elestrin. Upon signing the
amended agreement, Azur made a $1.0 million nonrefundable payment in December 2009 in exchange for
a permanent reduction in future royalty rates, and received options to make a total of $2.16
million in additional non-refundable payments (which were exercised during the first quarter of
2010) in exchange for the elimination of substantially all remaining future royalties and milestone
payments due us under the terms of the original license. The $1.0 million nonrefundable payment was
recorded as revenue during the fourth quarter of 2009 as we had no remaining performance
obligations with respect to this amount. Upon receiving the final payment in February 2010, our
future Elestrin royalty stream was reduced to zero. We expect to receive a portion of our Elestrin
royalties from Azur for January 2010 and a portion of February 2010. Pursuant to a separate
agreement with Antares and related to the Azur royalty stream and milestone buydown, we paid
Antares an aggregate of $268,750 in February 2010.
Our business operations to date have consisted mostly of licensing and research and development
activities and we expect this to continue for the immediate future. If and when our products for
which we have not entered into marketing relationships receive FDA approval, we may begin to incur
other expenses, including sales and marketing related expenses if we choose to market the products
ourselves. We currently do not have sufficient resources on a long-term basis to complete the
commercialization of any of our products for which we have not entered into marketing
relationships. We believe that our cash and cash equivalents of $29.9 million at December 31, 2009
and the additional $17.5 million in net proceeds from our March 2010 registered direct offering
will be sufficient to meet our liquidity requirements through at least the next 12 months.
We incurred expenses of approximately $1.1 million per month on research and development activities
during the year ended December 31, 2009. Our research and development expenses decreased 13
percent to $13.7 million for the year ended December 31, 2009 compared to the year ended December
31, 2008, primarily as a result of our decision in April 2009 to delay screening new subjects for
our LibiGel Phase III safety study to conserve cash. We have reinitiated screening and enrollment
in our safety study, and we expect our monthly research and development expenses to increase
significantly in 2010 compared to 2009. The amount of our actual research and development
expenditures, however, may fluctuate from quarter-to-quarter and year-to-year depending upon: (1)
the amount of resources, including cash and cash equivalents, available; (2) our development
schedule, including the timing of our clinical trials; (3) results of studies, clinical trials and
regulatory decisions; (4) whether we or our licensees are funding the development of our products;
and (5) competitive developments.
Our general and administrative expenses for the year ended December 31, 2009 increased 5 percent
compared to the year ended December 31, 2008. This increase was due primarily to a 9 percent or
$111,878 increase in our non-cash, stock option and warrant expense for the year ended December 31,
2009, compared to the year ended December 31, 2008. The primary reason for this increase was the
grant of options and warrants to purchase an aggregate of 1,170,929 and 180,000 shares of our
common stock, respectively, to new and certain existing employees in 2009 and an investor and
public relations firm in the third quarter 2009. Our general and administrative expenses may
fluctuate from year-to-year and quarter-to-quarter depending upon the amount of non-cash,
stock-based compensation expense, legal, public and investor relations, business development,
accounting and corporate governance and other fees and expenses incurred.
44
We recognized a net loss for the year ended December 31, 2009 of approximately $47.5 million
compared to a net loss of approximately $17.4 million for the year ended December 31, 2008. This
increase was primarily due to the impact of transaction costs incurred in connection with the
merger with Cell Genesys, and associated expenses related to our acquisition of in-process research
and development and the excess of the purchase price over the fair value of the assets and
liabilities acquired. We expect to continue to incur substantial and continuing losses for the
foreseeable future. This is true especially as our own product development programs expand and
various clinical trials continue, including in particular the Phase III clinical study program for
LibiGel.
Critical Accounting Policies and Estimates
Our significant accounting policies are described in Note 2 to our financial statements included
under the heading “Part II. Item 8. Financial Statements and Supplementary Data” of this report.
The discussion and analysis of our financial statements and results of operations are based upon
our financial statements, which have been prepared in accordance with accounting principles
generally accepted in the United States of America. The preparation of these financial statements
requires management to make estimates and judgments that affect the reported amount of assets,
liabilities, revenues and expenses, and related disclosure of contingent assets and liabilities.
The Securities and Exchange Commission has defined a company’s most critical accounting policies as
those that are most important to the portrayal of its financial condition and results of
operations, and which requires the company to make its most difficult and subjective judgments,
often as a result of the need to make estimates of matters that are inherently uncertain. Based on
this definition, we have identified the critical accounting policies described below. Although we
believe that our estimates and assumptions are reasonable, they are based upon information
available when they are made. Actual results may differ significantly from these estimates under
different assumptions or conditions.
Revenue Recognition
We have entered and may enter into various licensing agreements that generate license revenue or
other upfront fees and which also may involve subsequent milestone payments earned upon our
completion of development milestones or upon the occurrence of certain regulatory actions, such as
the filing of a regulatory application or the receipt of a regulatory approval. We recognize
non-refundable license fees as revenue when we have a contractual right to receive such payment,
the contract price is fixed or determinable, the collection of the resulting receivable is
reasonably assured and we have no further performance obligations under the license agreement.
Non-refundable license fees that meet these criteria and are due to us upon execution of an
agreement are recognized as revenue immediately. Milestones, in the form of additional license
fees, typically represent non-refundable payments to be received in conjunction with the
achievement of a specific event identified in the contract, such as completion of specified
clinical development activities and/or regulatory submissions and/or approvals. We recognize
revenues from milestone payments that meet the criteria above when the milestone is achieved. We
record royalty revenue based upon sales of products under a license when such royalties are earned,
which is generally in the quarter when the related products are sold.
Deferred revenue arises from payments received in advance of the culmination of the earnings
process. We classify as a current liability any deferred revenue that is expected to be recognized
within the next 12 months. If applicable, we will recognize deferred revenue in future periods
when the applicable revenue recognition criteria have been met.
45
Research and Development Costs
Research and development costs are charged to expense as incurred. Government grants are recorded
as an offset to the related research and development costs when we have complied with the
conditions attached to the grant and there is reasonable assurance that the funds will be received.
Non-refundable advance payments for goods and services to be used in future research and
development activities are recorded as assets and the payments are expensed when the research and
development activities are performed.
During 2009, in connection with the merger with Cell Genesys, we acquired the rights to in-process
research and development of Cell Genesys, as well as associated patents and technology. The
estimated fair value of the in-process research and development was charged to expense as it was
deemed to have no alternative future use.
Accounting Treatment Related to Acquisition of Assets and Liabilities of Cell Genesys
On October 14, 2009, we
completed our legal merger with Cell Genesys, as a result of which we
acquired all of the assets and liabilities of Cell Genesys. Concurrently with the merger, the
common stock of Cell Genesys was converted into common stock of BioSante, and Cell Genesys ceased
to exist. The primary reason we merged with Cell Genesys was our need
for additional funding to continue our Phase III clinical studies for
LibiGel and the lack of other available acceptable alternatives for
us to access capital prior to and at the time the merger agreement
was entered into by both of us in June 2009, especially in light of
the then state of the markets for equity offerings,
which historically had been our primary method for raising additional
financing. We have accounted for our transaction with Cell Genesys under U.S. generally accepted
accounting principles as an acquisition of the net assets of Cell Genesys, whereby we have recorded
the individual assets and liabilities of Cell Genesys as of the completion of the merger based on
their estimated fair values. As Cell Genesys had ceased operations, the acquisition was not
considered to be a business combination, and the allocation of the purchase price did not result in
recognition of goodwill. As a result of this treatment, we recognized as an expense approximately
$20.2 million representing the excess of the consideration and costs of the transaction over the
fair value of assets and liabilities received.
Following the completion of the merger, our future net income (loss) reflects charges resulting
from the purchase price allocation related to the merger, which includes adjustments to carrying
values of the acquired net assets based on the fair value of consideration measured as of the
completion of the merger.
Accounting for Convertible Notes Assumed in Connection with the Cell Genesys Acquisition
We assumed $22.0 million
principal of convertible notes in connection with the Cell Genesys
acquisition. We elected to apply the fair value option to the debt at the time of the acquisition,
with recognition of subsequent changes in the fair value of the convertible notes recognized in our statements
of operations immediately. As a result of this election, we must periodically estimate the fair
value of our convertible notes, which requires us to make certain judgments and estimates about
appropriate discount rates, our creditworthiness, and assumptions regarding potential conversion of
the notes. We believe that our estimates and assumptions are reasonable; however changes in these
estimates and assumptions could result in significant differences in the carrying value of the
convertible notes.
Results of Operations
The following table sets forth, for the periods indicated, our results of operations.
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Year Ended December 31, |
|
| |
|
2009 |
|
|
2008 |
|
|
2007 |
|
Revenue |
|
$ |
1,258,054 |
|
|
$ |
3,780,829 |
|
|
$ |
493,054 |
|
Expenses |
|
|
48,683,608 |
|
|
|
21,794,471 |
|
|
|
9,172,498 |
|
Research and development |
|
|
13,680,573 |
|
|
|
15,789,980 |
|
|
|
4,751,313 |
|
General and administrative |
|
|
5,373,945 |
|
|
|
5,124,934 |
|
|
|
4,331,361 |
|
Acquired in-process research and development |
|
|
9,000,000 |
|
|
|
— |
|
|
|
— |
|
Excess consideration paid over fair value |
|
|
20,192,194 |
|
|
|
— |
|
|
|
— |
|
Licensing expense |
|
|
299,616 |
|
|
|
836,420 |
|
|
|
— |
|
Interest income |
|
|
11,648 |
|
|
|
588,464 |
|
|
|
1,095,009 |
|
Other income — Fair value adjustment |
|
|
33,163 |
|
|
|
— |
|
|
|
— |
|
Other expense — Interest expense |
|
|
147,025 |
|
|
|
— |
|
|
|
— |
|
Net loss |
|
$ |
(47,527,768 |
) |
|
$ |
(17,425,178 |
) |
|
$ |
(7,584,435 |
) |
Net loss per share (basic and diluted) |
|
$ |
(1.40 |
) |
|
$ |
(0.64 |
) |
|
$ |
(0.30 |
) |
Weighted average number of shares outstanding |
|
|
33,951,652 |
|
|
|
27,307,494 |
|
|
|
25,485,513 |
|
46
Year Ended December 31, 2009 Compared to Year Ended December 31, 2008
Revenue for the year ended December 31, 2009 decreased 67 percent compared to revenue for 2008
primarily as a result of our receipt of $3.3 million from the Azur license of Elestrin in 2008
compared to our receipt of approximately $1.1 million from Azur in 2009 which was primarily for a
$1.0 million nonrefundable payment received in exchange for a
permanent reduction in future royalty rates.
Research and development expenses for the year ended December 31, 2009 decreased 13 percent
compared to research and development expenses for 2008 primarily as a result of our decision in
April 2009 to delay screening new subjects for our LibiGel Phase III safety study to conserve cash.
Screening has been re-initiated.
Our general and administrative expenses for the year ended December 31, 2009 increased 5 percent
compared to general and administrative expenses for 2008 due primarily to a 9 percent or $111,878
increase in our non-cash, stock option and warrant expense for the year ended December 31, 2009,
compared to the year ended December 31, 2008. This increase was due to an increase in the number of
stock options and warrants granted and the number of stock options and warrants outstanding during
the year ended December 31, 2009 compared to 2008.
We recognized $299,616 in licensing expense for the year ended December 31, 2009 compared to
$836,420 in licensing expense for 2008 due to expenses associated with both the Nycomed termination
agreement and Azur licensing agreement.
Interest income for the year ended December 31, 2009 decreased 98 percent compared to interest
income for 2008 primarily as a result of our decision to keep cash and cash equivalents in a 100%
FDIC-insured non-interest bearing checking account for the majority of 2009, in order to ensure
maximum safety of principal.
We recognized total additional expenses of $29.2 million related to our merger with Cell Genesys,
consisting of $9.0 million related to the write-off of acquired in-process research and
development, and $20.2 million related to transaction related expenses and additional charges
related to the excess of merger consideration over fair values of the net assets acquired.
Year Ended December 31, 2008 Compared to Year Ended December 31, 2007
Revenue for the year ended December 31, 2008 increased 667 percent compared to revenue for 2007
primarily as a result of our license of Elestrin to Azur and an increase in royalty and other
revenue from Elestrin sales.
Research and development expenses for the year ended December 31, 2008 increased 232 percent
compared to research and development expenses for 2007 primarily as a result of increased spending
on our Phase III LibiGel clinical study program.
47
Our general and administrative expenses for the year ended December 31, 2008 increased 18 percent
compared to general and administrative expenses for 2007 primarily as a result of an increase in
investor and public relations expenses and business development and other personnel-related costs.
Our non-cash, stock option and warrant expense for the year ended December 31, 2008 increased
$462,563, or 62 percent, compared to non-cash, stock option and warrant expense for the year ended
December 31, 2007 due to an increase in the number of stock options granted and the number of stock
options and warrants outstanding during the year ended December 31, 2008 compared to 2007.
We recognized $836,420 in licensing expense for the year ended December 31, 2008 compared to no
licensing expense for 2007 due to expenses associated with both the Nycomed termination agreement
and Azur licensing agreement.
Interest income for the year ended December 31, 2008 decreased 46 percent compared to interest
income during 2007 primarily as a result of a lower average invested cash balances and lower
average interest rates on our invested funds.
Liquidity and Capital Resources
Working Capital
Substantially all of our revenue to date has been derived from upfront, milestone and royalty
payments earned on licensing transactions and from subcontracts. Our business operations to date
have consisted mostly of licensing and research and development activities and we expect this to
continue for the immediate future. If and when our other products for which we have not entered
into marketing relationships receive FDA approval, we may begin to incur other expenses, including
sales and marketing and other expenses if we choose to market the products ourselves. We currently
do not have sufficient resources to establish our own sales and marketing function or complete the
commercialization of any of our products that are not licensed to others for development and
marketing. We expect the ongoing Phase III clinical study program of LibiGel to continue to
require significant resources.
To date, we have used primarily equity financings, and to a lesser extent, licensing income,
interest income and the cash received from our merger with Cell Genesys, to fund our ongoing
business operations and short-term liquidity needs, and we expect to continue this practice for the
foreseeable future. As of December 31, 2009, we had $29.9 million of cash and cash equivalents
compared to $14.7 million as of December 31, 2008.
Subsequent to December 31, 2009, we completed a registered
direct offering of 10.4 million shares of our common stock and warrants to purchase an aggregate of
5.2 million shares of our common stock at a purchase price of $1.73 per share. The offering
resulted in net proceeds of approximately $17.5 million, after deduction of placement agent fees
and offering expenses.
We completed our merger with Cell Genesys in October 2009. One of the primary benefits of our
merger with Cell Genesys was the cash acquired in the merger which we have used to continue our
Phase III clinical studies for LibiGel. As of the completion of the merger, Cell Genesys had
approximately $23.3 million in net cash and cash equivalents after deducting merger-related and
other expenses. As of such date, Cell Genesys also had, and we assumed by virtue of the merger,
$1.2 million in principal amount of 3.125% convertible senior notes due in November 2011 and $20.8
million in principal amount of 3.125% convertible senior notes due in May 2013. For a more
complete description of these notes, see “—Convertible Senior Notes Due November 2011 and May
2013.”
48
We expect our cash and cash equivalents balance to decrease as we continue to use cash to fund our
operations, including in particular our LibiGel clinical study program. Our future capital
requirements will depend upon numerous factors, including:
| • |
|
the progress, timing, cost and results of our preclinical and clinical development
programs, including in particular our Phase III clinical study program for LibiGel, and our
other product development efforts; |
| |
| • |
|
subject recruitment and enrollment in our current and future clinical studies, including in
particular our Phase III clinical study program for LibiGel; |
| |
| • |
|
our ability to license LibiGel or our other products for development and commercialization; |
| |
| • |
|
the cost, timing and outcome of regulatory reviews of our products; |
| |
| • |
|
the rate of technological advances; |
| |
| • |
|
the commercial success of our products; |
| |
| • |
|
our general and administrative expenses; and |
| |
| • |
|
the success, progress, timing and costs of our business development efforts to implement
business collaborations, licenses and other business combinations or transactions, including
our efforts to obtain value for any acquired GVAX cancer immunotherapies and other
technologies as a result of our merger with Cell Genesys and our efforts to continue to
evaluate various strategic alternatives available with respect to our products and our
company. |
We expect that our current cash resources will provide us sufficient capital to maintain our
projected business operations through at least the next 12 months, including continued Phase III
clinical development of LibiGel. Although we believe we have sufficient cash resources for the
next 12 months, our estimate may prove incorrect or we, nonetheless, may choose to raise additional
financing earlier.
As of December 31, 2009, we did not have any existing credit facilities under which we could borrow
funds, other than our committed equity financing facility described below. If we are unable to
raise additional financing when needed or secure another funding source for our clinical study
program, we may need to delay our Phase III clinical study program for LibiGel or otherwise make
changes to our operations to cut costs. As an alternative to raising additional financing, we may
choose to license LibiGel, Elestrin (outside the territories already sublicensed) or another
product to a third party who may finance a portion or all of the continued development and, if
approved, commercialization of that licensed product, sell certain assets or rights under our
existing license agreements or enter into other business collaborations or combinations, including
the possible sale of our company.
Committed Equity Financing Facility with Kingsbridge Capital Limited
In December 2008, we entered into a committed equity financing facility with Kingsbridge Capital
Limited in which Kingsbridge has committed to purchase, subject to certain conditions and at our
sole discretion, up to the lesser of $25.0 million or 5,405,840 shares of our common stock through
the end of December 2010. Under the terms of the facility, we are not obligated to utilize any of
the $25.0 million available under the facility and there are no minimum commitments or minimum use
penalties. We have access, at our discretion, to the funds through the sale of newly-issued shares
of our common stock. The funds that can be raised under the facility over the two-year term set to
expire in December 2010, will depend on the then-current price for our common stock and the number
of shares actually sold, which may not exceed an aggregate of 5,405,840 shares. We may access
capital under the facility by providing Kingsbridge with common stock at discounts ranging from
eight to 14 percent,
49
depending on the average market price of our common stock during the
applicable pricing period. Kingsbridge will not be
obligated to purchase shares under the facility unless certain conditions are met, which include a
minimum price for our common stock of $1.15 per share; the accuracy of representations and
warranties made to Kingsbridge; compliance with laws; continued effectiveness of the registration
statement registering the resale of shares of common stock issued or issuable to Kingsbridge; and
the continued listing of our common stock on the NASDAQ Global Market. In addition, Kingsbridge is
permitted to terminate the facility if it determines that a material and adverse event has occurred
affecting our business, operations, properties or financial condition and if such condition
continues for a period of 10 trading days from the date Kingsbridge provides us notice of such
material and adverse event. In connection with the committed equity financing facility, we issued
a warrant to Kingsbridge to purchase 300,000 shares of our common stock at an exercise price of
$4.00. The warrant became exercisable on June 15, 2009 and will remain exercisable, subject to
certain exceptions, for a period of five years thereafter. Other than attorneys’ fees and other
direct costs related to the registration of these shares, we did not make any other payments to
secure the facility. The facility does not impose any material restrictions on our operating or
financial activities. During the term of the facility, Kingsbridge is prohibited from engaging in
any short selling or derivative transactions related to our common stock. As of December 31, 2009,
we had not sold any shares to Kingsbridge under the committed equity financing facility. As of the
date of this report, we did not have an effective registration statement registering the resale of
shares of our common stock issue or issuable to Kingsbridge under the facility.
Convertible Senior Notes Due November 2011 and May 2013
As a result of our merger with Cell Genesys, we assumed $1.2 million in principal amount of 3.125%
convertible senior notes due in November 2011 and $20.8 million in principal amount of 3.125%
convertible senior notes due in May 2013 issued by Cell Genesys. Contractual interest payments on
the convertible senior notes are due on May 1 and November 1 of each year through maturity. Annual
interest on the notes is approximately $0.7 million. As a result of the merger and in accordance
with the terms of the indentures governing such notes as supplemented by supplemental indentures
entered into between us and the trustees thereunder, the November 2011 convertible notes are
convertible into an aggregate of 24,789 shares of our common stock at a conversion price of $49.78
per share and the May 2013 convertible notes are convertible into an aggregate of 5,586,559 shares
of our common stock at a conversion price of $3.72 per share, in each case subject to adjustments
for stock dividends, stock splits and other similar events. The convertible notes are our general,
unsecured obligations, ranking equally with all of our existing and future unsubordinated,
unsecured indebtedness and senior in right of payment to any subordinated indebtedness, but are
effectively subordinated to all of our existing and future secured indebtedness to the extent of
the value of the related security, and structurally subordinated to all existing and future
liabilities and other indebtedness of our subsidiaries. The convertible notes are subject to
repurchase by us at each holder’s option, if a fundamental change (as defined in the indentures),
occurs, at a repurchase price equal to 100% of the principal amount of the convertible notes, plus
accrued and unpaid interest (and additional amounts, if any) to, but not including, the repurchase
date and are subject to redemption for cash by us at any time in the case of the convertible notes
due in 2011 and at any time on or after May 1, 2011, in the case of the convertible notes due in
2013, in whole or in part, at a redemption price equal to 100% of the principal amount of such
notes if the closing price of our common stock has exceeded 150% of the conversion price then in
effect with respect to such notes for at least 20 trading days in any period of 30 consecutive
trading days ending on the trading day prior to the mailing of the notice of redemption. The
indentures governing the convertible notes, as supplemented by the supplemental indentures, do not
contain any financial covenants and do not restrict us from paying dividends, incurring additional
debt or issuing or repurchasing our other securities. In addition, the indentures, as supplemented
by the supplemental indentures, do not protect the note holders in the event of a highly leveraged
transaction or a fundamental change of our company except in certain circumstances specified in the
indentures.
50
Uses of Cash and Cash Flow
Net cash used in operating activities was $18.4 million for the year ended December 31, 2009
compared to net cash used in operating activities of $15.5 million for the year ended December 31,
2008 and net cash provided by operating activities of $739,991 for the year ended December 31,
2007. Net cash used in operating activities for 2009 was primarily the result of the net loss for
that period. Technology and transaction related expenses and charges of $29.2 million were
incurred as a result of our merger with Cell Genesys in
October 2009 but did not result in an operating cash payment by
us as we issued shares as consideration for the transaction and cash
payments for transaction costs were classified as a financing
activity based on the nature of the transaction. Net cash used in operating
activities for 2008 was primarily the result of the net loss for that period, and to a lesser
extent, an increase in prepaid expenses and other assets related to an increase in our prepaid
clinical study related costs, partially offset by an increase in accounts payable and accrued
liabilities. Net cash provided by operating activities of $739,991 for 2007 was due primarily to
the receipt of approximately $10.5 million from Nycomed, 25 percent of which was due to our
licensor, partially offset by our net loss of $7.6 million for 2007.
Net cash provided by investing activities was $2.9 million for the year ended December 31, 2009
compared to net cash provided by investing activities of $11.3 million for the year ended December
31, 2008 and net cash used in investing activities of $11.2 million for the year ended December 31,
2007. Net cash provided by investing activities for 2009 was primarily due to the redemption of
short-term investments. Net cash provided by investing activities for 2008 was due to the
redemption of approximately $11.0 million in short-term investments, partially offset by purchases
of capital assets associated with clinical trial software and manufacturing equipment due to the
conduct of our LibiGel clinical trial program. Net cash used in investing activities for 2007
consisted primarily of purchases and sales, respectively, of short-term investments.
Net cash provided by financing activities was $33.7 million for the year ended December 31, 2009
compared to $319,377 for the year ended December 31, 2008 and $18.5 million for the year ended
December 31, 2007. Net cash provided by investing activities for 2009 resulted from a combination
of recognizing $24.7 million in cash acquired as a result of our merger with Cell Genesys and $11.4
million in net proceeds to us, after deducting placement agent fees and offering expenses, from the
completion of our August 2009 registered direct offering, partially offset by $2.4 million in cash
paid for Cell Genesys acquisition-related costs. Net cash provided by investing activities for 2008
resulted from warrant exercises. Net cash provided by financing activities for 2007 resulted
primarily from the completion of a private placement resulting in net proceeds to us of
approximately $17.3 million, after deduction of transaction expenses, and to a lesser extent,
warrant and stock option exercises.
Commitments and Contractual Obligations
We did not have any material commitments for capital expenditures as of December 31, 2009. We
have, however, several financial commitments, including our convertible senior notes, product
development milestone payments to the licensors of certain of our products, payments under our
license agreement with Wake Forest University Health Sciences, as well as minimum annual lease
payments.
51
The following table summarizes the timing of these future contractual obligations and commitments
as of December 31, 2009:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Payments Due by Period |
|
| |
|
|
|
|
|
Less than |
|
|
|
|
|
|
|
|
|
|
More than |
|
| |
|
Total |
|
|
1 Year |
|
|
1-3 Years |
|
|
3-5 Years |
|
|
5 Years |
|
Convertible Senior Notes |
|
$ |
22,016,000 |
|
|
$ |
— |
|
|
$ |
1,234,000 |
|
|
$ |
20,782,000 |
|
|
$ |
— |
|
Interest payment obligations
related to Convertible Senior
Notes |
|
|
2,235,491 |
|
|
|
688,000 |
|
|
|
1,331,011 |
|
|
|
216,480 |
|
|
|
|
|
Operating Leases |
|
|
667,618 |
|
|
|
299,618 |
|
|
|
368,000 |
|
|
|
— |
|
|
|
— |
|
Settlement of shareholder lawsuit |
|
|
240,000 |
|
|
|
240,000 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Obligation under License
Agreement with Antares |
|
|
18,033 |
|
|
|
18,033 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Commitments Under License
Agreements with Johns Hopkins
University |
|
|
550,000 |
|
|
|
95,000 |
|
|
|
235,000 |
|
|
|
80,000 |
|
|
|
140,000 |
|
Commitments Under License
Agreement with Massachusetts
Institute of Technology |
|
|
250,000 |
|
|
|
50,000 |
|
|
|
150,000 |
|
|
|
50,000 |
|
|
|
— |
|
Commitments Under License
Agreement with University of
California |
|
|
360,000 |
|
|
|
20,000 |
|
|
|
60,000 |
|
|
|
40,000 |
|
|
|
240,000 |
|
Commitments Under License
Agreement with Wake Forest |
|
|
720,000 |
|
|
|
200,000 |
|
|
|
160,000 |
|
|
|
160,000 |
|
|
|
200,000 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total Contractual Cash Obligations |
|
$ |
27,057,142 |
|
|
$ |
1,610,651 |
|
|
$ |
3,538,011 |
|
|
$ |
21,328,480 |
|
|
$ |
580,000 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements that have or reasonably are likely to have a
material effect on our financial condition, changes in financial condition, revenues or expenses,
results of operations, liquidity, capital expenditures or capital resources. As a result, we are
not exposed materially to any financing, liquidity, market or credit risk that could arise if we
had engaged in these arrangements.
Recent Accounting Pronouncements
Effective July 1, 2009, the Financial Accounting Standards Board (FASB) issued Statement of
Financial Accounting Standards (SFAS) No. 168, “The FASB Accounting Standards Codification and the
Hierarchy of Generally Accepted Accounting Principles—a replacement of FASB Statement No. 162”
(SFAS No. 168). SFAS No. 168 reduces the U.S. GAAP hierarchy to two levels, one that is
authoritative and one that is not. We began to use the new guidance and reflect the new accounting
guidance references when referring to GAAP for the quarterly period ended September 30, 2009, and
all subsequent periods. As the guidance was not intended to change or alter existing GAAP, adoption
of this pronouncement did not have an effect on our consolidated financial statements.
52
In October 2009, the FASB codified and issued Accounting Standards Updated (ASU) No. 2009-13,
Revenue Recognition (Topic 605), “Multiple-Deliverable Revenue Arrangements” (ASU No. 2009-13). ASU
No. 2009-13 amends the guidance that in the absence of vendor-specific objective and third-party
evidence for deliverables in multiple-deliverable arrangements, companies will be required to
develop a best estimate of the selling price to separate deliverables and allocate arrangements
consideration using the relative selling price method. ASU No. 2009-13 expands the disclosure
requirements for multiple-deliverable revenue arrangements. The guidance will be effective for
financial statements issued for fiscal years beginning after June 15, 2010. Early adoption is
permitted. Adoption of ASU No. 2009-13 is not expected to have a material impact on our financial
position or operations.
|
|
|
| Item 7A. |
|
QUANTITATIVE AND QUALITATIVE DISCLOSURE ABOUT MARKET RISK |
This Item 7A is not applicable to BioSante as a smaller reporting company and has been omitted
pursuant to Item 305(e) of SEC Regulation S-K.
53
|
|
|
| Item 8. |
|
FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
| |
|
|
|
|
| Description |
|
Page |
|
|
|
|
|
|
|
|
|
55 |
|
|
|
|
|
|
|
|
|
56 |
|
|
|
|
|
|
|
|
|
57 |
|
|
|
|
|
|
|
|
|
58 |
|
|
|
|
|
|
|
|
|
59 |
|
|
|
|
|
|
|
|
|
60 |
|
|
|
|
|
|
|
|
|
61-87 |
|
|
|
|
|
|
54
MANAGEMENT’S REPORT ON INTERNAL CONTROL OVER FINANCIAL REPORTING
As management of BioSante Pharmaceuticals, Inc., we are responsible for establishing and
maintaining an adequate system of internal control over financial reporting as defined in Rules
13a-15(f) and 15d-15(f) under the Securities Exchange Act of 1934, as amended, for BioSante
Pharmaceuticals, Inc. This system is designed to provide reasonable assurance regarding the
reliability of financial reporting and the preparation of financial statements for external
purposes in accordance with U.S. generally accepted accounting principles.
BioSante’s internal control over financial reporting includes those policies and procedures that
(i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect
the transactions and dispositions of the assets of BioSante; (ii) provide reasonable assurance that
transactions are recorded as necessary to permit preparation of financial statements in accordance
with generally accepted accounting principles, and that receipts and expenditures of BioSante are
being made only in accordance with authorizations of management and directors of BioSante; and
(iii) provide reasonable assurance regarding prevention or timely detection of unauthorized
acquisition, use, or disposition of BioSante’s assets that could have a material effect on the
financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or
detect misstatements and even when determined to be effective, can only provide reasonable
assurance with respect to financial statement preparation and presentation. Also, projection of
any evaluation of the effectiveness of internal control over financial reporting to future periods
is subject to the risk that controls may become inadequate because of changes in conditions, or
that the degree or compliance with the policies or procedures may deteriorate.
With our participation, management evaluated the effectiveness of BioSante’s internal control over
financial reporting as of December 31, 2009. In making this evaluation, management used the
criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO)
in Internal Control—Integrated Framework. Based on this assessment, management concluded that
BioSante’s internal control over financial reporting was effective as of December 31, 2009.
| |
|
|
|
|
|
|
/s/ Stephen M. Simes
Stephen M. Simes
|
|
|
|
/s/ Phillip B. Donenberg
Phillip B. Donenberg
|
|
|
Vice Chairman, President and Chief
Executive Officer
|
|
|
|
Chief Financial Officer, Treasurer and
Secretary |
|
|
March 29, 2010
Further discussion of our internal controls and procedures is included under the heading “Part II.
Item 9A. Controls and Procedures” of this report.
55
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
BioSante Pharmaceuticals, Inc.
Lincolnshire, Illinois
We have audited the accompanying balance sheets of BioSante Pharmaceuticals, Inc. (the “Company”)
as of December 31, 2009 and 2008, and the related statements of operations, stockholders’ equity,
and cash flows for each of the three years in the period ended December 31, 2009. These financial
statements are the responsibility of the Company’s management. Our responsibility is to express an
opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight
Board (United States). Those standards require that we plan and perform the audit to obtain
reasonable assurance about whether the financial statements are free of material misstatement. An
audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in
the financial statements, assessing the accounting principles used and significant estimates made
by management, as well as evaluating the overall financial statement presentation. We believe that
our audits provide a reasonable basis for our opinion.
In our opinion, such financial statements present fairly, in all material respects, the financial
position of BioSante Pharmaceuticals, Inc. as of December 31, 2009 and 2008, and the results of
their operations and their cash flows for each of the three years in the period ended December 31,
2009, in conformity with accounting principles generally accepted in the United States of America.
/s/ DELOITTE & TOUCHE LLP
Deloitte & Touche LLP
Chicago, Illinois
March 29, 2010
56
BIOSANTE PHARMACEUTICALS, INC.
Balance Sheets
December 31, 2009 and 2008
| |
|
|
|
|
|
|
|
|
| |
|
December 31, |
|
|
December 31, |
|
| |
|
2009 |
|
|
2008 |
|
ASSETS |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CURRENT ASSETS |
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
29,858,465 |
|
|
$ |
11,760,920 |
|
Short-term investments |
|
|
— |
|
|
|
3,026,334 |
|
Accounts receivable |
|
|
64,645 |
|
|
|
229,775 |
|
Prepaid expenses and other assets |
|
|
1,487,160 |
|
|
|
1,070,051 |
|
|
|
|
|
|
|
|
|
|
|
31,410,270 |
|
|
|
16,087,080 |
|
|
|
|
|
|
|
|
|
|
PROPERTY AND EQUIPMENT, NET |
|
|
747,979 |
|
|
|
814,894 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
OTHER ASSETS |
|
|
|
|
|
|
|
|
Investments |
|
|
3,626,000 |
|
|
|
140,000 |
|
Deposits |
|
|
652,679 |
|
|
|
637,397 |
|
|
|
|
|
|
|
|
|
|
$ |
36,436,928 |
|
|
$ |
17,679,371 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
LIABILITIES AND STOCKHOLDERS’ EQUITY |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CURRENT LIABILITIES |
|
|
|
|
|
|
|
|
Accounts payable |
|
$ |
2,440,096 |
|
|
$ |
3,182,089 |
|
Due to licensor — Antares |
|
|
18,033 |
|
|
|
5,393 |
|
Accrued compensation |
|
|
529,066 |
|
|
|
290,583 |
|
Other accrued expenses |
|
|
942,922 |
|
|
|
374,887 |
|
|
|
|
|
|
|
|
|
|
|
3,930,117 |
|
|
|
3,852,952 |
|
|
|
|
|
|
|
|
|
|
Convertible senior notes due 2011 and 2013 |
|
|
16,676,417 |
|
|
|
— |
|
|
|
|
|
|
|
|
TOTAL LIABILITIES |
|
|
20,606,534 |
|
|
|
3,852,952 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
STOCKHOLDERS’ EQUITY |
|
|
|
|
|
|
|
|
Capital stock |
|
|
|
|
|
|
|
|
Issued and outstanding |
|
|
|
|
|
|
|
|
2009 — 391,286; 2008 — 391,286 Class C special stock |
|
|
391 |
|
|
|
391 |
|
2009 — 53,262,568; 2008 — 27,042,764 Common stock |
|
|
135,264,431 |
|
|
|
85,732,688 |
|
|
|
|
|
|
|
|
|
|
|
135,264,822 |
|
|
|
85,733,079 |
|
|
|
|
|
|
|
|
|
|
Accumulated deficit |
|
|
(119,434,428 |
) |
|
|
(71,906,660 |
) |
|
|
|
|
|
|
|
|
|
|
15,830,394 |
|
|
|
13,826,419 |
|
|
|
|
|
|
|
|
|
|
$ |
36,436,928 |
|
|
$ |
17,679,371 |
|
|
|
|
|
|
|
|
See accompanying notes to the financial statements.
57
BIOSANTE PHARMACEUTICALS, INC.
Statements of Operations
Years ended December 31, 2009, 2008 and 2007
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Year Ended December 31, |
|
| |
|
2009 |
|
|
2008 |
|
|
2007 |
|
| |
REVENUE |
|
|
|
|
|
|
|
|
|
|
|
|
Licensing revenue |
|
$ |
— |
|
|
$ |
3,384,091 |
|
|
$ |
199,091 |
|
Grant revenue |
|
|
116,389 |
|
|
|
65,051 |
|
|
|
59,060 |
|
Royalty revenue |
|
|
1,141,665 |
|
|
|
34,200 |
|
|
|
69,353 |
|
Other revenue |
|
|
— |
|
|
|
297,487 |
|
|
|
165,550 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1,258,054 |
|
|
|
3,780,829 |
|
|
|
493,054 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
EXPENSES |
|
|
|
|
|
|
|
|
|
|
|
|
Research and development |
|
|
13,680,573 |
|
|
|
15,789,980 |
|
|
|
4,751,313 |
|
General and administration |
|
|
5,373,945 |
|
|
|
5,124,934 |
|
|
|
4,331,361 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Acquired in-process research and development |
|
|
9,000,000 |
|
|
|
— |
|
|
|
— |
|
Excess consideration paid over fair value |
|
|
20,192,194 |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Licensing expense |
|
|
299,616 |
|
|
|
836,420 |
|
|
|
— |
|
Depreciation and amortization |
|
|
137,280 |
|
|
|
43,137 |
|
|
|
89,824 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
48,683,608 |
|
|
|
21,794,471 |
|
|
|
9,172,498 |
|
|
|
|
|
|
|
|
|
|
|
OTHER |
|
|
|
|
|
|
|
|
|
|
|
|
Fair value adjustment |
|
|
33,163 |
|
|
|
— |
|
|
|
— |
|
Interest expense |
|
|
(147,025 |
) |
|
|
— |
|
|
|
— |
|
Interest income |
|
|
11,648 |
|
|
|
588,464 |
|
|
|
1,095,009 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NET LOSS |
|
$ |
(47,527,768 |
) |
|
$ |
(17,425,178 |
) |
|
$ |
(7,584,435 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss per common share: |
|
|
|
|
|
|
|
|
|
|
|
|
Basic |
|
$ |
(1.40 |
) |
|
$ |
(0.64 |
) |
|
$ |
(0.30 |
) |
Diluted |
|
$ |
(1.40 |
) |
|
$ |
(0.64 |
) |
|
$ |
(0.30 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average number of common and
common equivalent shares outstanding: |
|
|
|
|
|
|
|
|
|
|
|
|
Basic |
|
|
33,951,652 |
|
|
|
27,307,494 |
|
|
|
25,485,513 |
|
Diluted |
|
|
33,951,652 |
|
|
|
27,307,494 |
|
|
|
25,485,513 |
|
See accompanying notes to the financial statements.
58
BIOSANTE PHARMACEUTICALS, INC.
Statements of Stockholders’ Equity
Years ended December 31, 2009, 2008 and 2007
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Class C |
|
|
|
|
|
|
|
|
|
|
| |
|
Special Shares |
|
|
Common Stock |
|
|
Accumulated |
|
|
|
|
| |
|
Shares |
|
|
Amount |
|
|
Shares |
|
|
Amount |
|
|
Deficit |
|
|
Total |
|
Balance, December 31, 2006 |
|
|
391,286 |
|
|
$ |
391 |
|
|
|
22,975,040 |
|
|
$ |
64,967,887 |
|
|
$ |
(46,897,047 |
) |
|
$ |
18,071,231 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Issuance of common shares |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Option exercises — various |
|
|
— |
|
|
|
— |
|
|
|
53,081 |
|
|
|
192,371 |
|
|
|
— |
|
|
|
192,371 |
|
Warrant exercises — various |
|
|
— |
|
|
|
— |
|
|
|
711,487 |
|
|
|
1,019,225 |
|
|
|
— |
|
|
|
1,019,225 |
|
Stock option expense |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
711,259 |
|
|
|
— |
|
|
|
711,259 |
|
Private placement of common shares, net |
|
|
— |
|
|
|
— |
|
|
|
3,054,999 |
|
|
|
17,282,935 |
|
|
|
— |
|
|
|
17,282,935 |
|
Stock warrant expense |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
32,906 |
|
|
|
— |
|
|
|
32,906 |
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(7,584,435 |
) |
|
|
(7,584,435 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance, December 31, 2007 |
|
|
391,286 |
|
|
$ |
391 |
|
|
|
26,794,607 |
|
|
$ |
84,206,583 |
|
|
$ |
(54,481,482 |
) |
|
$ |
29,725,492 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Issuance of common shares |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Warrant exercises — various |
|
|
— |
|
|
|
— |
|
|
|
248,157 |
|
|
|
379,720 |
|
|
|
— |
|
|
|
379,720 |
|
Stock option expense |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
1,102,444 |
|
|
|
— |
|
|
|
1,102,444 |
|
Stock warrant expense |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
104,284 |
|
|
|
— |
|
|
|
104,284 |
|
Credit equity financing facility |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(60,343 |
) |
|
|
— |
|
|
|
(60,343 |
) |
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(17,425,178 |
) |
|
|
(17,425,178 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance, December 31, 2008 |
|
|
391,286 |
|
|
$ |
391 |
|
|
|
27,042,764 |
|
|
$ |
85,732,688 |
|
|
$ |
(71,906,660 |
) |
|
$ |
13,826,419 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stock option expense |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
1,254,503 |
|
|
|
— |
|
|
|
1,254,503 |
|
Stock warrant expense |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
64,103 |
|
|
|
— |
|
|
|
64,103 |
|
Registered direct offering of common shares, net |
|
|
— |
|
|
|
— |
|
|
|
6,000,000 |
|
|
|
11,352,751 |
|
|
|
— |
|
|
|
11,352,751 |
|
Issuance of common shares pursuant to Cell Genesys, Inc. transaction |
|
|
— |
|
|
|
— |
|
|
|
20,219,804 |
|
|
|
36,800,043 |
|
|
|
— |
|
|
|
36,800,043 |
|
Credit equity financing facility |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
60,343 |
|
|
|
— |
|
|
|
60,343 |
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(47,527,768 |
) |
|
|
(47,527,768 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance, December 31, 2009 |
|
|
391,286 |
|
|
$ |
391 |
|
|
|
53,262,568 |
|
|
$ |
135,264,431 |
|
|
$ |
(119,434,428 |
) |
|
$ |
15,830,394 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
See accompanying notes to the financial statements.
59
BIOSANTE PHARMACEUTICALS, INC.
Statements of Cash Flows
Years ended December 31, 2009, 2008 and 2007
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Year Ended December 31, |
|
| |
|
2009 |
|
|
2008 |
|
|
2007 |
|
CASH FLOWS (USED IN) PROVIDED BY OPERATING ACTIVITIES |
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(47,527,768 |
) |
|
$ |
(17,425,178 |
) |
|
$ |
(7,584,435 |
) |
Adjustments to reconcile net loss to net cash
(used in) provided by operating activities |
|
|
|
|
|
|
|
|
|
|
|
|
Acquired in-process research and development |
|
|
9,000,000 |
|
|
|
— |
|
|
|
— |
|
Excess consideration paid over fair value |
|
|
20,192,194 |
|
|
|
— |
|
|
|
— |
|
Depreciation and amortization |
|
|
137,280 |
|
|
|
43,137 |
|
|
|
89,824 |
|
Employee and director stock-based compensation |
|
|
1,254,503 |
|
|
|
1,102,444 |
|
|
|
711,259 |
|
Stock warrant expense — noncash |
|
|
64,103 |
|
|
|
104,284 |
|
|
|
32,906 |
|
Loss on disposal of equipment |
|
|
— |
|
|
|
— |
|
|
|
21,748 |
|
Other non-cash charges |
|
|
60,739 |
|
|
|
— |
|
|
|
— |
|
Fair value adjustment |
|
|
(33,163 |
) |
|
|
— |
|
|
|
— |
|
MATC license revenue — noncash |
|
|
— |
|
|
|
— |
|
|
|
(140,000 |
) |
Changes in assets and liabilities
affecting cash flows from operations |
|
|
|
|
|
|
|
|
|
|
|
|
Prepaid expenses and other assets |
|
|
(316,101 |
) |
|
|
(1,330,491 |
) |
|
|
(103,514 |
) |
Accounts receivable |
|
|
285,838 |
|
|
|
(215,209 |
) |
|
|
10,495,963 |
|
Accounts payable and accrued liabilities |
|
|
(1,561,175 |
) |
|
|
2,189,843 |
|
|
|
449,856 |
|
Provision for contingencies |
|
|
— |
|
|
|
— |
|
|
|
(550,588 |
) |
Due to licensor — Antares |
|
|
12,640 |
|
|
|
4,330 |
|
|
|
(2,623,937 |
) |
Deferred revenue |
|
|
— |
|
|
|
(9,091 |
) |
|
|
(59,091 |
) |
|
|
|
|
|
|
|
|
|
|
Net cash (used in) provided by operating activities |
|
|
(18,430,910 |
) |
|
|
(15,535,931 |
) |
|
|
739,991 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CASH FLOWS PROVIDED BY (USED IN) INVESTING ACTIVITIES |
|
|
|
|
|
|
|
|
|
|
|
|
Redemption of short term investments |
|
|
3,026,334 |
|
|
|
11,979,642 |
|
|
|
981 |
|
Purchase of short term investments |
|
|
— |
|
|
|
— |
|
|
|
(11,210,979 |
) |
Purchase of capital assets |
|
|
(165,724 |
) |
|
|
(651,116 |
) |
|
|
(29,428 |
) |
|
|
|
|
|
|
|
|
|
|
Net cash provided by (used in) investing activities |
|
|
2,860,610 |
|
|
|
11,328,526 |
|
|
|
(11,239,426 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CASH FLOWS PROVIDED BY FINANCING ACTIVITIES |
|
|
|
|
|
|
|
|
|
|
|
|
Cash paid for transaction related costs |
|
|
(2,431,252 |
) |
|
|
— |
|
|
|
— |
|
Cash received in transaction |
|
|
24,746,346 |
|
|
|
— |
|
|
|
— |
|
Proceeds from sale or conversion of shares, net |
|
|
11,352,751 |
|
|
|
319,377 |
|
|
|
18,494,531 |
|
|
|
|
|
|
|
|
|
|
|
Net cash provided by financing activities |
|
|
33,667,845 |
|
|
|
319,377 |
|
|
|
18,494,531 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NET INCREASE (DECREASE) IN CASH AND CASH EQUIVALENTS |
|
|
18,097,545 |
|
|
|
(3,888,028 |
) |
|
|
7,995,096 |
|
CASH AND CASH EQUIVALENTS
AT BEGINNING OF PERIOD |
|
|
11,760,920 |
|
|
|
15,648,948 |
|
|
|
7,653,852 |
|
|
|
|
|
|
|
|
|
|
|
CASH AND CASH EQUIVALENTS AT END OF PERIOD |
|
$ |
29,858,465 |
|
|
$ |
11,760,920 |
|
|
$ |
15,648,948 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
SUPPLEMENTAL SCHEDULE OF
CASH FLOW INFORMATION |
|
|
|
|
|
|
|
|
|
|
|
|
Interest paid, including acquired accrued interest |
|
$ |
248,388 |
|
|
$ |
— |
|
|
$ |
— |
|
Noncash Investing and Financing Activities: |
|
|
|
|
|
|
|
|
|
|
|
|
Liabilities acquired through Cell Genesys transaction |
|
$ |
18,487,298 |
|
|
$ |
— |
|
|
$ |
— |
|
Shares issued for Cell Genesys transaction |
|
$ |
36,800,043 |
|
|
$ |
— |
|
|
$ |
— |
|
Investment aquired through Cell Genesys transaction |
|
$ |
3,486,000 |
|
|
$ |
— |
|
|
$ |
— |
|
Other assets acquired in Cell Genesys transaction |
|
$ |
293,658 |
|
|
$ |
— |
|
|
$ |
— |
|
Purchase of capital assets on account, non-cash investing activity |
|
$ |
— |
|
|
$ |
152,019 |
|
|
$ |
— |
|
Investment in MATC — noncash |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
140,000 |
|
See accompanying notes to the financial statements.
60
BIOSANTE PHARMACEUTICALS, INC.
Notes to the Financial Statements
December 31, 2009
| 1. |
|
DESCRIPTION OF BUSINESS |
| |
|
BioSante Pharmaceuticals, Inc. (the Company) is a specialty pharmaceutical company focused
on developing products for female sexual health, menopause, contraception and male
hypogonadism. The Company’s products for female sexual health, menopause, contraception and
male hypogonadism include: (1) LibiGel, a once daily transdermal testosterone gel in Phase
III clinical development under a Special Protocol Assessment (SPA) for the treatment of
female sexual dysfunction (FSD); (2) Elestrin, a once daily transdermal estradiol (estrogen)
gel approved by the U.S. Food and Drug Administration (FDA) indicated for the treatment of
moderate-to-severe vasomotor symptoms associated with menopause and marketed in the U.S.;
(3) The Pill-Plus (triple component contraceptive), a once daily use of various combinations
of estrogens, progestogens and androgens in development for the treatment of FSD in women
using oral or transdermal contraceptives; and (4) Bio-T-Gel, a once daily transdermal
testosterone gel in development for the treatment of hypogonadism, or testosterone
deficiency, in men. The Company also is developing its calcium phosphate nanotechnology, or
CaP, primarily for aesthetic medicine, as a vaccine adjuvant and for drug delivery. |
| |
|
On October 14, 2009, the Company completed a transaction with Cell Genesys, Inc. (Cell
Genesys) resulting in the acquisition of the assets and liabilities of Cell Genesys. See
Note 4 entitled “Acquisition of Net Assets of Cell Genesys.” |
| 2. |
|
SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES |
| |
|
These financial statements are expressed in U.S. dollars. The Company is organized into one
operating and one reporting segment. |
| |
|
The financial statements have been prepared in accordance with accounting principles
generally accepted in the United States of America (generally accepted accounting
principles). The preparation of financial statements in conformity with generally accepted
accounting principles requires management to make estimates and assumptions that affect the
reported amounts of assets and liabilities, the disclosure of contingent assets and
liabilities at the date of the financial statements, and the reported amounts of revenues
and expenses during the reporting period. Actual results could differ from those estimates. |
| |
| |
|
Cash and Cash Equivalents |
| |
|
The Company generally considers all instruments with original maturities of three months or
less to be cash equivalents. Certain investments that could meet the definition of a cash
equivalent are classified as investments due to the nature of the account in which the
investment is held and the Company’s intended use of the investment. Interest income on
invested cash balances is recognized on the accrual basis as earned. |
61
BIOSANTE PHARMACEUTICALS, INC.
Notes to the Financial Statements
December 31, 2009
| 2. |
|
SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued) |
| |
| |
|
Short-Term Investments |
| |
|
Short-term investments are classified as “available for sale” under the provisions of ASC
320 (formerly FAS No. 115). Accordingly, short-term investments are reported at fair value,
with any related unrealized gains and losses included as a separate component of
stockholders’ equity, net of applicable taxes. Realized gains and losses and interest and
dividends are included in interest income. Realized gains and losses are recorded based
upon the specific identification method. |
| |
|
As of December 31, 2009 and December 31, 2008, the Company had $0 and $3.0 million of
short-term investments, respectively. As of December 31, 2009, all of the Company’s cash
and cash equivalents resided in a 100% FDIC-insured non-interest bearing checking account in
order to ensure maximum safety of principal. The Company had auction rate securities
investments of $3.0 million and money market fund investments of approximately $26,000 as of
December 31, 2008. There were no gains or losses recorded in accumulated other
comprehensive income as of December 31, 2009 or December 31, 2008, and there were no
realized gains or losses included in earnings as the result of sales of available for sale
securities for the years ended December 31, 2009, December 31, 2008 or December 31, 2007. |
| |
|
As of December 31, 2008, the Company’s auction rate securities with a $3.0 million par value
were held in an account with UBS Financial Services, Inc. (UBS). In August 2008, UBS and
its affiliates reached agreements with the U.S. Securities and Exchange Commission (SEC),
the New York Attorney General, the Massachusetts Securities Division, the Texas State
Securities Board and other state regulatory agencies represented by the North American
Securities Administrators Association to restore liquidity to UBS clients who held auction
rate securities. Pursuant to these agreements, in October 2008, the Company received rights
from UBS entitling the Company to sell to UBS or its affiliates and requiring UBS or its
affiliates to purchase the Company’s $3.0 million in auction rate securities for their face
(or par) value plus any accrued and unpaid interest. On January 8, 2009, pursuant to those
rights, the Company received $3.0 million principal plus accrued and unpaid interest from
UBS. |
| |
| |
|
Property and Equipment |
| |
|
Property and equipment that currently is being used in the Company’s operations is stated at
cost less accumulated depreciation and amortization. Depreciation is computed primarily on a
straight line basis over the estimated useful lives of the respective assets, typically five
years for software and 10 years for laboratory equipment. |
| |
| |
|
Long-Lived Assets |
| |
|
Long-lived assets are reviewed for possible impairment whenever events indicate that the
carrying amount of such assets may not be recoverable. If such a review indicates an
impairment, the carrying amount of such assets is reduced to estimated recoverable value. |
62
BIOSANTE PHARMACEUTICALS, INC.
Notes to the Financial Statements
December 31, 2009
| 2. |
|
SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued) |
| |
| |
|
Convertible Senior Notes |
| |
|
The Company assumed two series of convertible senior note obligations with an aggregated
principal balance of $22,016,000, which contain certain redemption, repurchase and
conversion adjustment features as a result of its transaction with Cell Genesys. The
Company has made an irrevocable election to account for these debt instruments at fair value
commencing from the date of the merger, resulting in recognition of a single liability for
each of the two series of convertible senior notes which will be reported at fair value at
each reporting date. Subsequent changes in the carrying value of the notes are reflected in
fair value adjustment in the accompanying statements of operations. See Note 7 entitled
“Convertible Senior Notes” for a description of these financial liabilities. |
| |
| |
|
Research and Development |
| |
|
Research and development costs are charged to expense as incurred. Direct government grants
are recorded as an offset to the related research and development costs when the Company has
complied with the conditions attached to the grant and there is reasonable assurance that
the funds will be received. |
| |
| |
|
Legal Costs |
| |
|
For ongoing matters, legal costs are charged to expense as incurred. |
| |
| |
|
Basic and Diluted Net Loss Per Share |
| |
|
The basic and diluted net loss per share is computed based on the weighted average number of
the aggregate of common stock and class C special stock outstanding, all being considered as
equivalent of one another. Basic loss per share is computed by dividing loss available to
common stockholders by the weighted average number of shares outstanding for the reporting
period. Diluted loss per share reflects the potential dilution that could occur if
securities or other contracts to issue common stock were exercised or converted into common
stock. The computation of diluted loss per share does not include the Company’s stock
options, warrants, or convertible debt when there is an antidilutive effect on loss per
share. |
| |
| |
|
Stock-Based Compensation |
| |
|
The Company recognizes stock-based compensation expense granted to employees generally on a
straight-line basis over the estimated service period of the award, or when certain
performance-based vesting provisions occur, for awards that contain these features. The
Company has granted options to non-employees in exchange for services. Expense related to
such grants is recognized within the Company’s statements of operations in accordance with
the nature of the service received by the Company. |
63
BIOSANTE PHARMACEUTICALS, INC.
Notes to the Financial Statements
December 31, 2009
| 2. |
|
SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued) |
| |
|
Warrants issued to non-employees as compensation for services rendered are valued at their
fair value on the date of issue. Warrants of this nature to purchase an aggregate of
180,000 and 80,000 shares of the Company’s common stock were issued in 2009 and 2008,
respectively. |
| |
| |
|
Revenue Recognition |
| |
|
The Company has entered into various licensing agreements that generate license revenue or
other upfront fees and which also may involve subsequent milestone payments earned upon
completion of development milestones by the Company or upon the occurrence of certain
regulatory actions, such as the filing of a regulatory application or the receipt of a
regulatory approval. Non-refundable license fees are recognized as revenue when the Company
has a contractual right to receive such payment, the contract price is fixed or
determinable, the collection of the resulting receivable is reasonably assured and the
Company has no further performance obligations under the license agreement. Non-refundable
license fees that meet these criteria and are due to the Company upon execution of an
agreement are recognized as revenue immediately. |
| |
|
Milestones, in the form of additional license fees, typically represent non-refundable
payments to be received in conjunction with the achievement of a specific event identified
in the contract, such as completion of specified clinical development activities and/or
regulatory submissions and/or approvals, or as sales-based milestone payments. Revenues
from milestone payments that meet the criteria in the preceding paragraph are recognized
when the milestone is achieved. |
| |
|
Additionally, royalty revenue based upon sales of products under license is recorded when
such royalties are earned and are deemed collectible, which is generally in the quarter when
the related products are sold. |
| |
|
Deferred revenue arises from payments received in advance of the culmination of the earnings
process. Deferred revenue is recognized as revenue in future periods when the applicable
revenue recognition criteria have been met. |
| |
| |
|
Income Taxes |
| |
|
Deferred tax assets or liabilities are determined based on the difference between the
financial statement and tax basis of assets and liabilities, as measured by enacted tax
rates. A valuation allowance is provided against deferred income tax assets in circumstances
where management believes the recoverability of a portion of the assets is not more likely
than not. The Company has provided a full valuation allowance against its net deferred tax
assets as of December 31, 2009 and 2008. |
64
BIOSANTE PHARMACEUTICALS, INC.
Notes to the Financial Statements
December 31, 2009
| 2. |
|
SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued) |
| |
| |
|
Investments |
| |
|
As a result of the Company’s merger with Cell Genesys, the Company acquired an investment
equivalent to approximately 16% of the total equity of Ceregene, Inc., a privately held
biotechnology company. The Company has recorded its investment using the cost method, as no
active market exists for this investment, and the Company does not possess significant
influence over operating and financial policies of Ceregene, although the Company by virtue
of its stock ownership of Ceregene has the right to designate one member on the Ceregene
board of directors. The valuation of investments accounted for under the cost method is
based on all available financial information related to the investee, including valuations
based on recent third party equity investments in the investee. If an unrealized loss on
any investment is considered to be other-than-temporary, the loss is recognized in the
period the determination is made. All investments are reviewed for changes in circumstances
or occurrence of events that suggest the investment may not be recoverable. |
| |
| |
|
Recent Accounting Pronouncements |
| |
|
Effective July 1, 2009, the Financial Accounting Standards Board (FASB) issued Statement of
Financial Accounting Standards (SFAS) No. 168, “The FASB Accounting Standards
Codification and the Hierarchy of Generally Accepted Accounting Principles—a
replacement of FASB Statement No. 162” (SFAS No. 168). SFAS No. 168 reduces the
U.S. GAAP hierarchy to two levels, one that is authoritative and one that is not. The
Company began to use the new guidance and reflect the new accounting guidance
references when referring to GAAP for the quarterly period ended September 30, 2009,
and all subsequent periods. As the guidance was not intended to change or alter
existing GAAP, adoption of this pronouncement did not have an effect on the Company’s
consolidated financial statements. |
| |
|
In October 2009, the FASB codified and issued Accounting Standards Updated (ASU)
No. 2009-13, Revenue Recognition (Topic 605), “Multiple-Deliverable Revenue Arrangements”
(ASU No. 2009-13). ASU No. 2009-13 amends the guidance that in the absence of
vendor-specific objective and third-party evidence for deliverables in multiple-deliverable
arrangements, companies will be required to develop a best estimate of the selling price to
separate deliverables and allocate arrangements consideration using the relative selling
price method. ASU No. 2009-13 expands the disclosure requirements for multiple-deliverable
revenue arrangements. The guidance will be effective for financial statements issued for
fiscal years beginning after June 15, 2010. Early adoption is permitted. Adoption of ASU
No. 2009-13 is not expected to have a material impact on the Company’s financial position or
operations. |
65
BIOSANTE PHARMACEUTICALS, INC.
Notes to the Financial Statements
December 31, 2009
| 3. |
|
LIQUIDITY AND CAPITAL RESOURCES |
| |
|
Substantially all of the Company’s revenue to date has been derived from upfront, milestone
and royalty payments earned on licensing transactions and from subcontracts. The
Company’s business operations to date have consisted mostly of licensing and research
and development activities and the Company expects this to continue for the immediate
future. The Company has not introduced commercially any products. If and when the
Company’s products for which it has not entered into marketing relationships receive
FDA approval, the Company may begin to incur other expenses, including sales and
marketing related expenses if it chooses to market the products itself. The Company
currently does not have sufficient resources to complete the commercialization of any
of its products for which the Company has not entered into marketing relationships. |
| |
|
To date, the Company has used primarily equity financings, and to a lesser extent,
licensing income, interest income and the cash received from its merger with Cell
Genesys, to fund its ongoing business operations and short-term liquidity needs, and
the Company expects to continue this practice for the foreseeable future. In addition,
as a direct result of the transaction with Cell Genesys, the Company acquired $23.3
million in net cash and cash equivalents after deducting merger-related and other
expenses. As a result of the merger, the Company assumed $1.2 million in principal
amount of 3.125% convertible senior notes due in November 2011 and $20.8 million in
principal amount of 3.125% convertible senior notes due in May 2013. Contractual
interest payments on the convertible senior notes are due on May 1 and November 1 of
each year through maturity. For additional discussion regarding the merger with Cell
Genesys and the assets and liabilities acquired, see Note 4 entitled “Acquisition of
Net Assets of Cell Genesys.” For additional discussion regarding the convertible
senior notes, see Note 7 entitled “Convertible Senior Notes.” |
| |
|
In August 2009, the Company completed an offering of an aggregate of 6,000,000
shares of the Company’s common stock and warrants to purchase an aggregate of 2,400,000
additional shares of its common stock, resulting in net proceeds to the Company of
approximately $11.4 million, after deducting placement agent fees and other offering
expenses. For additional discussion regarding the registered direct offering, see Note
9 entitled “Stockholders’ Equity.” |
| |
|
As of December 31, 2009, the Company had $29.9 million of cash and cash
equivalents. On March 8, 2010, the Company completed an additional offering of an
aggregate of 10,404,626 shares of the Company’s common stock and warrants to purchase
an aggregate of 5,202,313 additional shares of its common stock, resulting in net
proceeds to the Company of approximately $17.5 million, after deducting placement agent
fees and other offering expenses. For additional discussion regarding the March 2010
registered direct offering, see Note 18 entitled “Subsequent Events.” |
66
BIOSANTE PHARMACEUTICALS, INC.
Notes to the Financial Statements
December 31, 2009
| 3. |
|
LIQUIDITY AND CAPITAL RESOURCES (continued) |
| |
|
The Company expects its cash and cash equivalents balance to decrease as the
Company continues to use cash to fund its operations, including in particular its
LibiGel clinical study program. The Company expects that its current cash resources
will provide it sufficient capital to maintain its projected business operations
through at least the next 12 months, including continued Phase III clinical development
of LibiGel. Although the Company believes it has sufficient cash resources for the
next 12 months, this estimate may prove incorrect or the Company, nonetheless, may
choose to raise additional financing earlier. The Company does not have sufficient
resources to obtain regulatory approval of LibiGel or any of its other products or to
complete the commercialization of any of its products. |
| |
|
As of December 31, 2009, the Company did not have any existing credit facilities
under which it could borrow funds, other than the committed equity financing facility
described below. If the Company is unable to raise additional financing when needed or
secure another funding source for its clinical study program, the Company may need to
delay its Phase III clinical study program for LibiGel or otherwise make changes to its
operations to cut costs As an alternative to raising additional financing, the Company
may choose to license LibiGel, Elestrin (outside the territories already sublicensed)
or another product to a third party who may finance a portion or all of the continued
development and, if approved, commercialization of that licensed product, sell certain
assets or rights under its existing license agreements or enter into other business
collaborations or combinations, including the possible sale of the Company. |
| |
|
In December 2008, the Company entered into a Committed Equity Financing Facility (CEFF) with
Kingsbridge Capital Limited in which Kingsbridge has committed to purchase, subject to
certain conditions and at the Company’s sole discretion, up to the lesser of $25.0 million
or 5,405,840 shares of the Company’s common stock through the end of December 2010. The
Company may access capital under the CEFF by providing Kingsbridge with common stock at
discounts ranging from eight to 14 percent, depending on the average market price of the
Company’s common stock during the applicable pricing period. Kingsbridge will not be
obligated to purchase shares under the CEFF unless certain conditions are met, which include
a minimum price for the Company’s common stock of $1.15 per share; the accuracy of
representations and warranties made to Kingsbridge; compliance with laws; continued
effectiveness of the registration statement registering the resale of shares of common stock
issued or issuable to Kingsbridge; and the continued listing of the Company’s common stock
on the NASDAQ Global Market. In addition, Kingsbridge is permitted to terminate the CEFF if
it determines that a material and adverse event has occurred affecting the Company’s
business, operations, properties or financial condition and if such condition continues for
a period of 10 trading days from the date Kingsbridge provides the Company notice of such
material and adverse event. As of December 31, 2009, the Company had not sold any shares to
Kingsbridge under the CEFF. |
67
BIOSANTE PHARMACEUTICALS, INC.
Notes to the Financial Statements
December 31, 2009
| 4. |
|
ACQUISITION OF NET ASSETS OF CELL GENESYS |
| |
|
On October 14, 2009, the Company acquired 100% of the common stock of Cell Genesys in a
direct merger transaction. The primary reason the Company merged with Cell Genesys was
the Company’s need for additional funding to continue its Phase III clinical studies
for LibiGel and the lack of other available acceptable alternatives for the Company
to access capital prior to and at the time the merger agreement was entered into by
the parties in June 2009, especially in light of the then state of the markets for
equity offerings, which historically had been the Company’s primary method for
raising additional financing. The merger was accounted for under U.S. generally accepted
accounting principles as an acquisition of the net assets of Cell Genesys, whereby the
individual assets and liabilities of Cell Genesys were recorded by the Company as of the
completion of the merger based on their estimated fair values. As Cell Genesys had ceased
substantially its operations prior to the date of the transaction, the merger was not
considered to be a business combination, and the allocation of the purchase price did not
result in recognition of goodwill. Effective October 14, 2009, the balance sheet and net
loss of the Company reflect the purchase price allocation and charges resulting from the
purchase price allocation related to the merger, which included adjustments to carrying
values of the acquired net assets based on their estimated fair values as of that date. |
| |
|
The total purchase price is allocated to the acquired assets and assumed liabilities of Cell
Genesys based on their estimated relative fair values as of the merger closing date. The
table below displays the purchase price of the merger. |
| |
|
|
|
|
Fair value of BioSante common stock issued (20,219,804 shares) |
|
$ |
36,800,043 |
|
Transaction costs of BioSante |
|
|
2,431,252 |
|
|
|
|
|
Total purchase price |
|
$ |
39,231,295 |
|
|
|
|
|
| |
|
The total purchase price was allocated as follows: |
| |
|
|
|
|
Cash |
|
$ |
24,746,346 |
|
Investment in Ceregene |
|
|
3,486,000 |
|
In process research and development |
|
|
9,000,000 |
|
Receivables, equipment and other assets |
|
|
293,658 |
|
Accounts payable and accrued liabilities |
|
|
1,777,323 |
|
Convertible senior notes |
|
|
16,709,580 |
|
|
|
|
|
Total net assets acquired |
|
$ |
19,039,101 |
|
|
|
|
|
| |
|
In addition to the $24.7 million in cash acquired, the Company obtained, as a result of the
merger, the rights to all in-process research and development of Cell Genesys, which
included a portfolio of GVAX cancer immunotherapies and other technologies. The $9.0
million value attributed to this portfolio was expensed as of the date of the acquisition as
acquired in-process technology, as it was considered to have had no alternative future use.
The $20.2 million representing the premium of the total value of consideration in excess of
fair values of the net assets acquired was also expensed as of the date of the acquisition. |
68
BIOSANTE PHARMACEUTICALS, INC.
Notes to the Financial Statements
December 31, 2009
| 4. |
|
ACQUISITION OF NET ASSETS OF CELL GENESYS (continued) |
| |
|
In addition, as a result of the merger, the Company assumed $1.2 million in principal amount
of outstanding 3.125% convertible senior notes due in November 2011 and $20.8 million in
principal amount of 3.125% convertible senior notes due in May 2013 issued by Cell Genesys.
Contractual interest payments on the convertible senior notes are payable on May 1 and
November 1 of each year through maturity. As a result of the merger and in accordance with
the terms of the indentures governing such notes as supplemented by supplemental indentures
entered into between the Company and the trustees thereunder, the November 2011 convertible
notes became convertible into an aggregate of 24,789 shares of the Company’s common stock at
an initial conversion price of $49.78 per share and the May 2013 convertible notes became
convertible into an aggregate of 5,586,559 shares of the Company’s common stock at a
conversion price of $3.72 per share, in each case subject to adjustments for stock
dividends, stock splits, and other similar events. For more details see Note 7, entitled
“Convertible Senior Notes.” |
| |
|
The Company licensed the technology underlying LibiGel, Elestrin and certain of its other
gel products, other than Bio-T-Gel, from Antares Pharma, Inc. (Antares). Under the
agreement, Antares granted the Company an exclusive license to certain patents and patent
applications covering these gel products, including rights to sublicense, in order to
develop and market the products in certain territories. Under the agreement, the Company is
required to pay Antares certain development and regulatory milestone payments and royalties
based on net sales of any products the Company or any sub-licensee sells incorporating the
in-licensed technology and as such, the Company owed Antares $18,033 as of December 31, 2009
and $5,393 as of December 31, 2008 pursuant to this agreement. The patents covering the
formulations used in these gel products are expected to expire in 2022. Bio-T-Gel was
developed and is fully-owned by the Company and is not covered under the Antares license. |
| |
| |
|
The Pill Plus |
| |
|
The Company licensed the technology underlying its triple component contraceptive, or The
Pill Plus, from Wake Forest University Health Sciences and Cedars-Sinai Medical Center. The
financial terms of this license include regulatory milestone payments, maintenance payments
and royalty payments by the Company if a product incorporating the licensed technology gets
approved and subsequently is marketed. The patents covering the technology underlying The
Pill Plus are expected to expire in 2016. |
69
BIOSANTE PHARMACEUTICALS, INC.
Notes to the Financial Statements
December 31, 2009
| 5. |
|
LICENSE AGREEMENTS (continued) |
| |
| |
|
CaP Technology |
| |
|
In June 1997, the Company entered into a licensing agreement with the Regents of the
University of California (the University), which agreement subsequently has been amended,
pursuant to which the University has granted the Company an exclusive license to seven
United States patents owned by the University, including rights to sublicense such patents,
in fields of use pertaining to vaccine adjuvants and drug delivery systems. The last of the
expiration dates for these patents is 2014. The University of California also has filed
patent applications for this licensed technology in several foreign jurisdictions, including
Canada, Europe and Japan. The license agreement requires the Company to pay royalties to
the University based on a percentage of the net sales of any products the Company sells or a
licensee sells incorporating the licensed technology until expiration of the licensed
patents. |
| |
| |
|
GVAX Cancer Immunotherapy Technology |
| |
|
The Company owns development and commercialization rights to its GVAX cancer immunotherapy
technology as a result of its transaction with Cell Genesys. The original core patent
applications covering the GVAX cancer immunotherapy technology were licensed exclusively to
Cell Genesys from Johns Hopkins University and The Whitehead Institute for Biomedical
Research in 1992. Rights to additional patents and patent applications were licensed from
Johns Hopkins University in 2001. The patents are expected to expire between 2012 and 2026.
Under the various agreements, the Company is required to pay Johns Hopkins University and
The Whitehead Institute for Biomedical Research certain development and regulatory milestone
payments and royalties based on net sales of any products the Company or any sub-licensee
sells incorporating the in-licensed technology. |
| |
| |
|
Other License Agreements |
| |
|
The Company has entered into several other license agreements in which the Company has
out-licensed certain of the rights and technologies the Company has licensed. Under these
agreements, the Company typically is entitled to receive royalty payments on any sales of
the products and, in some cases, may be entitled to receive certain development and
regulatory milestones. |
70
BIOSANTE PHARMACEUTICALS, INC.
Notes to the Financial Statements
December 31, 2009
| 6. |
|
PROPERTY AND EQUIPMENT |
| |
|
Property and equipment, net of accumulated depreciation at December 31, 2009 and 2008
consist of the following: |
| |
|
|
|
|
|
|
|
|
| |
|
2009 |
|
|
2008 |
|
Computer equipment |
|
$ |
432,625 |
|
|
$ |
375,311 |
|
Office equipment |
|
|
143,548 |
|
|
|
131,239 |
|
Laboratory and equipment |
|
|
518,775 |
|
|
|
518,034 |
|
|
|
|
|
|
|
|
|
|
|
1,094,948 |
|
|
|
1,024,584 |
|
Accumulated depreciation and amortization |
|
|
(346,969 |
) |
|
|
(209,690 |
) |
|
|
|
|
|
|
|
|
|
$ |
747,979 |
|
|
$ |
814,894 |
|
|
|
|
|
|
|
|
| |
|
There was no construction in progress as of December 31, 2009. As of December 31,
2008, $243,556 of computer equipment, and $486,084 of laboratory and equipment, related to
construction in progress that had not been placed into service. |
| 7. |
|
CONVERTIBLE SENIOR NOTES |
| |
|
As a result of the Company’s merger with Cell Genesys, the Company assumed liabilities
related to two series of convertible senior notes of Cell Genesys. The conversion features
of the convertible senior notes have been adjusted for the exchange ratio used in the
merger, as described in Note 4, entitled “Acquisition of Net Assets of Cell Genesys.” The
terms of the convertible senior notes are as follows: |
| |
• |
|
$20,782,000 principal amount of 3.125% Convertible Senior Notes due May 1, 2013
(the “2013 Notes”), exchangeable at the option of the holder or upon certain
specified events into 0.1828 shares of the Company’s common stock at a conversion
price of $3.72 per share. The Company has the right to redeem the 2013
Notes for cash as a whole or in part after May 1, 2011. The Company may be
obligated to redeem the 2013 Notes if there is an occurrence of a fundamental
event, as described in the indentures. |
| |
• |
|
$1,234,000 principal amount of 3.125% Convertible Senior Notes due November 1,
2011 (the “2011 Notes”), exchangeable at the option of the holder (or upon certain
specified events) into 0.1828 shares of the Company’s common stock at a conversion
price of $49.78 per share. The Company has the right to redeem the 2011 Notes for
cash as a whole or in part after November 1, 2009. The Company may be obligated to
redeem the 2011 Notes if there is an occurrence of a fundamental event, as
described in the indentures. |
| |
|
Interest on both series of Notes is payable on May 1 and November 1 each year through
maturity. Under certain circumstances, the Company may redeem some or all of the Notes on or
after specified dates at a redemption price equal to 100% of the principal amount of the
notes plus accrued and unpaid interest. Holders of the notes may require the Company to
purchase some or all of their notes if certain changes in control occur at a repurchase
price equal to 100% of the principal amount of the notes plus accrued and unpaid interest. |
71
BIOSANTE PHARMACEUTICALS, INC.
Notes to the Financial Statements
December 31, 2009
| 7. |
|
CONVERTIBLE SENIOR NOTES (continued) |
| |
|
The Company has made an election to record the value of the 2011 Notes and the 2013 Notes at
fair value in accordance with ASC 825 (Formerly SFAS 159, The Fair Value Option for
Financial Assets and Financial Liabilities) in order to simplify the accounting for the
convertible debt, inclusive of the redemption, repurchase and conversion adjustment features
which would otherwise require specialized valuation, bifurcation, and recognition.
Accordingly, the Company has adjusted the carrying value of the convertible senior notes to
their fair value as of December 31, 2009, with changes in the fair value of the notes
occurring since October 14, 2009, the date these liabilities were assumed by the Company,
reflected in fair value adjustment in the statements of operations. |
| |
|
The Company recorded fair value adjustments of $33,163 related to the convertible senior
notes for the year ended December 31, 2009. |
| |
|
The Company establishes the value the convertible senior notes based upon contractual terms
of the notes, as well as certain key assumptions. |
| |
|
The assumptions as of October 14, 2009 were: |
| |
|
|
|
|
|
|
|
|
| |
|
2013 Notes |
|
|
2011 Notes |
|
Average risk free rate |
|
|
1.9 |
% |
|
|
1.0 |
% |
Volatility of BioSante common stock |
|
|
86.8 |
% |
|
|
100.7 |
% |
Discount rate for principal payments in cash |
|
|
19.8 |
% |
|
|
19.8 |
% |
The assumptions as of December 31, 2009 were:
| |
|
|
|
|
|
|
|
|
| |
|
2013 Notes |
|
|
2011 Notes |
|
Average risk free rate |
|
|
1.7 |
% |
|
|
1.1 |
% |
Volatility of BioSante common stock |
|
|
81.4 |
% |
|
|
89.8 |
% |
Discount rate for principal payments in cash |
|
|
17.6 |
% |
|
|
17.6 |
% |
| |
|
The discount rate is based on observed yields as of the measurement date for debt securities
of entities having a C and Ca rating for long-term corporate obligations as assigned by
Moody’s Investors Service. |
| |
|
The following table represents the scheduled maturities of required principal payments by
year related to the convertible senior notes at December 31, 2009: |
| |
|
|
|
|
2010 |
|
$ |
— |
|
2011 |
|
|
1,234,000 |
|
2012 |
|
|
— |
|
2013 |
|
|
20,782,000 |
|
|
|
|
|
Total |
|
$ |
22,016,000 |
|
|
|
|
|
72
BIOSANTE PHARMACEUTICALS, INC.
Notes to the Financial Statements
December 31, 2009
The Company has analyzed its filing positions in all significant federal and state
jurisdictions where it is required to file income tax returns, as well as open tax years in
these jurisdictions. The only periods subject to examination by the major tax jurisdictions
where the Company does business are the 2006 through 2009 tax years. The Company determined
there are no uncertain tax positions existing as of December 31, 2009 or December 31, 2008.
The components of the Company’s net deferred tax asset at December 31, 2009 and 2008 were as
follows:
| |
|
|
|
|
|
|
|
|
| |
|
2009 |
|
|
2008 |
|
Net operating loss carryforwards |
|
$ |
29,856,745 |
|
|
$ |
23,609,594 |
|
Tax basis in intangible assets |
|
|
3,618,489 |
|
|
|
403,498 |
|
Convertible
senior notes |
|
|
6,295,348 |
|
|
|
— |
|
Deferred
financing costs for tax |
|
|
7,622,553 |
|
|
|
— |
|
Research & development credits |
|
|
4,242,829 |
|
|
|
3,415,143 |
|
Stock option expense |
|
|
1,935,640 |
|
|
|
1,462,065 |
|
Other |
|
|
109,356 |
|
|
|
56,063 |
|
|
|
|
|
|
|
|
|
|
|
53,680,960 |
|
|
|
28,946,363 |
|
Valuation allowance |
|
|
(53,680,960 |
) |
|
|
(28,946,363 |
) |
|
|
|
|
|
|
|
|
|
$ |
— |
|
|
$ |
— |
|
|
|
|
|
|
|
|
The Company has no current tax provision due to its accumulated losses, which result in
net operating loss carryforwards. At December 31, 2009, the Company had approximately
$79,091,000 of net operating loss carryforwards that are available to reduce future taxable
income for a period of up to 20 years. The net operating loss carryforwards expire in the
years 2018-2029. The net operating loss carryforwards as well as amortization of various
intangibles, principally acquired in-process research and development, generate deferred tax
benefits, which have been recorded as deferred tax assets and are entirely offset by a tax
valuation allowance. The valuation allowance has been provided at 100% to reduce the
deferred tax assets to zero, the amount management believes is more likely than not to be
realized. Additionally, the Company has provided a full valuation allowance against
$4,242,829 of research and development credits, which are available to reduce future income
taxes, if any, through the year 2029.
The valuation
allowance as of December 31, 2009, includes $18,439,249 of additional amounts
resulting from current year tax losses which are not expected to be realized in the
foreseeable future, and $6,295,348 of additional valuation allowances arising from acquisition of the certain net assets of Cell Genesys with acquired tax basis differences.
73
BIOSANTE PHARMACEUTICALS, INC.
Notes to the Financial Statements
December 31, 2009
| 8. |
|
INCOME TAXES (continued) |
The provision for income taxes differs from the amount computed by applying the statutory
federal income tax rate of 34.5% to pre-tax income as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
2009 |
|
|
2008 |
|
|
2007 |
|
Tax at U.S. federal statutory rate |
|
$ |
(16,397,080 |
) |
|
$ |
(6,030,952 |
) |
|
$ |
(2,616,630 |
) |
State taxes, net of federal benefit |
|
|
(1,544,652 |
) |
|
|
(568,133 |
) |
|
|
(246,494 |
) |
Research and development credits |
|
|
(515,235 |
) |
|
|
(526,196 |
) |
|
|
(162,675 |
) |
Other, net |
|
|
17,718 |
|
|
|
(2,998 |
) |
|
|
(132,577 |
) |
Change in valuation allowance |
|
|
18,439,249 |
|
|
|
7,128,279 |
|
|
|
3,158,376 |
|
|
|
|
|
|
|
|
|
|
|
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
|
|
|
|
|
|
|
|
|
Authorized and Outstanding Capital Stock
The Company is authorized to issue 200,000,000 shares of common stock, $0.0001 par value per
share, 4,687,684 shares of class C special stock, $0.0001 par value per share, and
10,000,000 shares of undesignated preferred stock, $0.0001 par value per share.
No shares of preferred stock were outstanding as of December 31, 2009 and 2008.
There were 391,286 shares of class C special stock issued and outstanding as of December 31,
2009 and 2008. Each share of class C special stock entitles its holder to one vote per
share. Each share of class C special stock is exchangeable, at the option of the holder, for
one share of the Company’s common stock, at an exchange price of $2.50 per share, subject to
adjustment upon certain capitalization events. Holders of class C special stock are not
entitled to receive dividends or to participate in the distribution of the Company’s assets
upon any liquidation, dissolution or winding-up of the Company. The holders of class C
special stock have no cumulative voting, preemptive, subscription, redemption or sinking
fund rights.
There were 53,262,568 and 27,042,764 shares of common stock issued and outstanding as of
December 31, 2009 and 2008, respectively. The Company has presented the par values of its
common stock and the related additional paid in capital on a combined basis for all periods
presented.
Registered Direct Offerings
In August 2009, the Company completed a registered direct offering of an aggregate of
6,000,000 shares of its common stock and warrants to purchase an aggregate of 2,400,000
shares of its common stock, at a purchase price of $2.00 per share to three institutional
investors resulting in net proceeds to the Company of approximately $11.4 million, after
deducting placement agent fees and other offering expenses. The warrants were exercisable
immediately, have an exercise price of $2.50 per share and will expire on August 12, 2014.
In connection with the offering, the Company issued the placement agent warrants to purchase
an aggregate of 240,000 shares of the Company’s common stock at an exercise price of $2.50, which warrants were exercisable on
February 13, 2010 and will expire on June 9, 2014.
74
BIOSANTE PHARMACEUTICALS, INC.
Notes to the Financial Statements
December 31, 2009
| 9. |
|
STOCKHOLDERS’ EQUITY (continued) |
In addition, on March 8, 2010, the Company completed a registered direct offering of an
aggregate of 10,404,626 shares of its common stock and warrants to an aggregate of
5,202,313 shares of its common stock, at a purchase price of $1.73 per share to funds
affiliated with two institutional investors resulting in net proceeds to the Company of
approximately $17.5 million, after deducting placement agent fees and other offering
expenses. The warrants are exercisable beginning on September 9, 2010, have an exercise
price of $2.08 per share and will expire on September 8, 2015. In connection with the
offering, the Company issued the placement agent warrants to purchase an aggregate of
208,093 shares of the Company’s common stock at an exercise price of $2.16, which warrants
are exercisable beginning on September 8, 2010 and will expire on June 9, 2014. See Note
18 entitled “Subsequent Events.”
Acquisition of Net Assets of Cell Genesys
In October 2009, the Company acquired Cell Genesys in a direct merger. As a result of the
merger, each share of common stock of Cell Genesys issued and outstanding immediately prior
to the effective time of the merger was converted into the right to receive 0.1828 of a
share of the Company’s common stock. In the aggregate, the Company issued approximately
20.2 million shares of its common stock to former Cell Genesys stockholders in connection
with the merger. All options to purchase shares of Cell Genesys common stock, other than
certain designated options held by certain of Cell Genesys’s former officers (Assumed
Options), became fully vested and exercisable until immediately prior to the effective time
of the merger. At the effective time of the merger, such unexercised options other than
the Assumed Options terminated. The Assumed Options were assumed by the Company and will
remain outstanding following the merger, but converted into and became options to purchase shares of the Company’s common stock on terms substantially identical to those in effect
prior to the merger, except for adjustments to the underlying number of shares and the
exercise price based on the 0.1828 exchange ratio. As a result of the merger, the Assumed
Options converted into options to purchase an aggregate of 234,429 shares of the Company’s
common stock at a weighted average exercise price of $19.73 per share. All warrants to
purchase shares of Cell Genesys common stock which by their terms survived the merger
(Assumed Warrants) were assumed by the Company, but were converted into and became warrants
to purchase shares of the Company’s common stock on terms substantially identical to those
in effect prior to the merger, except for adjustments to the underlying number of shares
and the exercise price based on the 0.1828 exchange ratio. As a result of the merger,
these Assumed Warrants converted into warrants to purchase an aggregate of 395,246 shares
of the Company’s common stock at a weighted average exercise price of $39.27 per share.
75
BIOSANTE PHARMACEUTICALS, INC.
Notes to the Financial Statements
December 31, 2009
| 9. |
|
STOCKHOLDERS’ EQUITY (continued) |
In addition, as a result of the merger, the Company assumed two series of 3.125%
convertible senior notes issued by Cell Genesys due in November 2011 and May 2013.
Contractual interest payments on the convertible senior notes are payable on May 1 and
November 1 of each year through maturity. As a result of the merger and in accordance with
the terms of the indentures governing such notes as supplemented by supplemental indentures
entered into between the
Company and the trustees thereunder, the November 2011 convertible notes became convertible
into an aggregate of 24,789 shares of the Company’s common stock at an initial conversion
price of $49.78 per share and the May 2013 convertible notes became convertible into an
aggregate of 5,586,559 shares of the Company’s common stock at a conversion price of $3.72
per share, in each case subject to adjustments for stock dividends, stock splits, and other
similar events.
For additional discussion regarding the merger with Cell Genesys and the assets and
liabilities acquired, see Note 4 entitled “Acquisition of Net Assets of Cell Genesys.” For
additional discussion regarding the convertible senior notes, see Note 7 entitled
“Convertible Senior Notes.”
Committed Equity Financing Facility
In December 2008, the Company entered into a Committed Equity Financing Facility
arrangement, or CEFF, with Kingsbridge Capital Limited (Kingsbridge) in which Kingsbridge
has committed to purchase, subject to certain conditions and at the Company’s sole
discretion, up to the lesser of $25.0 million or 5,405,840 shares of the Company’s common
stock through the end of December 2010. Under the terms of the CEFF, the Company is not
obligated to utilize any of the $25.0 million available under the CEFF and there are no
minimum commitments or minimum use penalties. The Company has access, at its discretion, to
the funds through the sale of newly-issued shares of the Company’s common stock. The funds
that can be raised under the CEFF over the two-year term will depend on the then-current
price for the Company’s common stock and the number of shares actually sold, which may not
exceed an aggregate of 5,405,840 shares. The Company may access capital under the CEFF by
providing Kingsbridge with common stock at discounts ranging from eight to 14 percent,
depending on the average market price of the Company’s common stock during the applicable
pricing period. Kingsbridge will not be obligated to purchase shares under the CEFF unless
certain conditions are met, including a minimum price for the Company’s common stock of
$1.15 per share. In connection with the CEFF, the Company issued a warrant to Kingsbridge to
purchase 300,000 shares of the
Company’s common stock at an exercise price of $4.00. The warrant became exercisable on
June 15, 2009, the six-month anniversary of the date of the Purchase Agreement, and will
remain exercisable, subject to certain exceptions, for a period of five years thereafter.
Pursuant to the CEFF, the Company filed a registration statement with respect to the resale
of shares issued pursuant to the CEFF and underlying the warrant. As of December 31, 2009,
the Company had not sold any shares to Kingsbridge under the CEFF.
Convertible Senior Notes
See the “Acquisition of Net Assets of Cell Genesys” section of this Note 9 and see Note 7,
entitled “Convertible Senior Notes” for information regarding the convertible senior notes
assumed in the Cell Genesys merger.
76
BIOSANTE PHARMACEUTICALS, INC.
Notes to the Financial Statements
December 31, 2009
| 9. |
|
STOCKHOLDERS’ EQUITY (continued) |
Warrants
As of December 31, 2009, warrants to purchase 5,378,955 shares of the Company’s common
stock were outstanding, as follows:
| |
|
|
|
|
|
|
|
|
| Amount |
|
Exercise Price |
|
|
Expiration |
|
853,292 |
|
$ |
2.75 |
|
|
October 21, 2011 |
763,750 |
|
$ |
8.00 |
|
|
December 14, 2010 |
180,000 |
|
$ |
8.00 |
|
|
July 18, 2010 |
66,667 |
|
$ |
4.78 |
|
|
May 14, 2011 |
300,000 |
|
$ |
4.00 |
|
|
June 14, 2014 |
180,000 |
|
$ |
2.00 |
|
|
July 20, 2012 |
2,640,000 |
|
$ |
2.50 |
|
|
August 12, 2014 |
395,246 |
|
$ |
39.27 |
|
|
April 1, 2012 |
Pursuant to the Company’s private placement financing in July 2006, warrants to
purchase an aggregate of 1,334,542 shares of common stock were issued at an exercise price
of $2.75 per share with a term of four years and nine months, beginning January 22, 2007.
Of these warrants initially granted, warrants to purchase an aggregate of 853,292 shares of
common stock remained outstanding as of December 31, 2009.
Pursuant to the Company’s private placement financing in June 2007, warrants to purchase an
aggregate of 763,750 shares of common stock were issued at an exercise price of $8.00 per
share with a term of two years and six months, beginning June 14, 2007. All of these
warrants to purchase shares of common stock remained outstanding as of December 31, 2009.
In July 2007, the Company issued warrants to purchase 180,000 shares of common stock to an
investor relations firm in return for various investor relations services. The warrants are
exercisable at an exercise price equal to $8.00 per share with 50 percent of the warrants
exercisable on July 19, 2008 and the remainder exercisable on July 19, 2009. The warrants
are exercisable through and including July 18, 2010. In July 2009, the Company issued a
warrant to purchase 180,000 shares of common stock to an investor relations firm in
consideration for various investor relations services. The warrant has an exercise price
equal to $2.00 per share, vests in two equal installments on the six-month and one-year
anniversary of the date of grant and will remain exercisable through July 20, 2012. The
Company uses the Black-Scholes pricing model to value these warrants and remeasures the
award each quarter until the measurement date is established. In the year ended December
31, 2009 and 2008, the Company recorded $56,620 and $43,988, respectively, in non-cash
general and administrative expense pertaining to these consultant warrants.
77
BIOSANTE PHARMACEUTICALS, INC.
Notes to the Financial Statements
December 31, 2009
| 9. |
|
STOCKHOLDERS’ EQUITY (continued) |
In May 2008, the Company issued warrants to purchase an aggregate of 80,000 shares of common
stock to two individuals, the sole principal and a key executive officer, of an investor and
public relations firm in return for various investor and public relations services. These
warrants are exercisable at an exercise price equal to $4.78 per share with 1/12 of the
warrants becoming exercisable on June 15, 2008 and the remainder becoming exercisable on a
monthly basis thereafter through May 15, 2009 so long as the investor and public relations
firm continues to provide services to the Company. The Company terminated its relationship
with the firm effective March 31, 2009, at which time 66,667 of the warrants were
exercisable. The warrants that were exercisable as of March 31, 2009 will remain
exercisable through and including May 14, 2011. The Company uses the Black-Scholes pricing
model to value this warrant consideration and remeasures the award each quarter until the
measurement date is established. In the year ended December 31, 2009 and 2008, the Company
recorded $7,483 and $60,296, respectively, in non-cash general and administrative expense
pertaining to these warrants.
During 2009, no warrants were exercised to purchase the Company’s common stock and 534,996
warrants associated with the Company’s 2004 private placement of its common stock,
exercisable at an exercise price equal to $7.00 per share, expired unexercised on August 10,
2009.
During 2008, warrants to purchase an aggregate of 176,614 shares of common stock were
exercised for total cash proceeds of $379,720. Warrants to purchase an aggregate of 71,543
shares of common stock were exercised on a cashless basis, for which 74,957 additional
warrants were cancelled by the Company in payment of the exercise price for the exercised
warrants. Warrants to purchase an aggregate of 500 shares of common stock expired without
being exercised. All of the exercised warrants were granted pursuant to the Company’s
private placement financing in August 2003 and none were outstanding as of December 31,
2009.
Pursuant to the Company’s registered direct offering in August 2009, the Company issued
warrants to purchase an aggregate of 2,400,000 shares of common stock were issued at an
exercise price of $2.50 per share, immediately exercisable with a term of five years. In
connection with the offering, the Company issued the placement agent warrants to purchase an
aggregate of 240,000 shares of the Company’s common stock at an exercise price of $2.50,
which warrants were exercisable on February 13, 2010 and will expire on June 9, 2014.
Warrants to purchase an aggregate of 2,640,000 shares of common stock remained outstanding
as of December 31, 2009.
As described above, in connection with the Company’s merger with Cell Genesys, all warrants
to purchase shares of Cell Genesys common stock which by their terms survived the merger
were assumed by the Company, but were converted into and became warrants to purchase shares
of the Company’s common stock on terms substantially identical to those in effect prior to
the merger, except for adjustments to the underlying number of shares and the exercise price
based on the 0.1828 exchange ratio. As a result of the merger, these Assumed Warrants
converted into warrants to purchase an aggregate of 395,246 shares of the Company’s common
stock at a weighted average exercise price of $39.27 per share.
78
BIOSANTE PHARMACEUTICALS, INC.
Notes to the Financial Statements
December 31, 2009
| 9. |
|
STOCKHOLDERS’ EQUITY (continued) |
Stock Options
During 2009 and 2008, no options to purchase shares of the Company’s common stock were
exercised. See Note 10 entitled “Stock Based Compensation” for more detail regarding the
number of stock options granted during 2009 and 2008.
See the “Acquisition of Net Assets of Cell Genesys” section of this Note 9 for information
regarding the options and warrants issued related to the Cell Genesys merger.
| 10. |
|
STOCK-BASED COMPENSATION |
As of December 31, 2009, the Company has two stockholder-approved equity-based compensation
plans under which stock options have been granted — the BioSante Pharmaceuticals, Inc.
Amended and Restated 1998 Stock Plan (1998 Plan) and the BioSante Pharmaceuticals, Inc. 2008
Stock Incentive Plan (2008 Plan) (collectively, the Plans). The 2008 Plan replaced the 1998
Plan as pertains to future option grants, which was terminated with respect to future grants
upon the effectiveness of the 2008 Plan. As of December 31, 2009, there were 2,000,000
shares of the Company’s common stock authorized for issuance under the 2008 Plan, subject to
adjustment as provided in the 2008 Plan. Of the 2,000,000 authorized shares, none had been
issued and 1,170,929 shares were subject to outstanding stock options as of December 31,
2009. Outstanding employee stock options generally vest over a period of three years and
have 10-year contractual terms. From time to time, the Company grants employee stock
options that have performance condition-based vesting provisions which result in expense
when such performance conditions are probable of being achieved. None of these options were
outstanding as of December 31, 2009. The non-cash, stock-based compensation cost that was
incurred by the Company in connection with the 1998 and 2008 Plans was $1,254,503,
$1,102,444 and $711,259 for the years ended December 31, 2009, 2008 and 2007, respectively.
No income tax benefit was recognized in the Company’s statements of operations for
stock-based compensation arrangements due to the Company’s net loss position.
The weighted average fair value of the options at the date of grant for options granted
during 2009, 2008 and 2007 was $1.04, $2.41, and $2.37, respectively. The fair value of
each option grant is estimated on the date of grant using the Black-Scholes option-pricing
model with the following weighted average assumptions:
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
2009 |
|
|
2008 |
|
|
2007 |
|
Expected option life (years) |
|
|
6.00 |
|
|
|
6.00 |
|
|
|
9.83 |
|
Risk free interest rate |
|
|
2.74 |
% |
|
|
3.45 |
% |
|
|
4.74 |
% |
Expected stock price volatility |
|
|
76.75 |
% |
|
|
67.63 |
% |
|
|
69.31 |
% |
Dividend yield |
|
|
— |
|
|
|
— |
|
|
|
— |
|
79
BIOSANTE PHARMACEUTICALS, INC.
Notes to the Financial Statements
December 31, 2009
| 10. |
|
STOCK-BASED COMPENSATION (continued) |
The Company uses a volatility rate calculation based on the closing price for its common
stock at the end of each calendar month as reported by the NASDAQ Global Market (or The
American Stock Exchange prior to November 5, 2007). Since the Company has a limited history
with option exercises, the expected life was set to the entire life of the option grant
through the fourth quarter 2007. Beginning with options granted during the fourth quarter
2007, the Company began estimating the expected life of its options in a manner consistent
with Staff Accounting Bulletin (SAB) 107, and SAB 110 beginning January 1, 2008, which
allows companies to use a simplified method to estimate the life of options meeting certain
criteria. The Company believes that the use of the simplified method provides a reasonable
term for purposes of determining compensation costs for these grants, and expects to use the
simplified method to estimate the expected life of future options for eligible grants. The
discount rate used is the yield on a United States Treasury note as of the grant date with a
maturity equal to the estimated life of the option. The Company has not in the past issued
a cash dividend nor does it have any current plans to do so in the future and therefore, an
expected dividend yield of zero was used.
The following table summarizes the stock option compensation expense for employees and
non-employees recognized in the Company’s statements of operations for each period:
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
2009 |
|
|
2008 |
|
|
2007 |
|
Research and development |
|
$ |
361,773 |
|
|
$ |
356,287 |
|
|
$ |
202,335 |
|
General and administrative |
|
|
892,730 |
|
|
|
746,157 |
|
|
|
508,924 |
|
|
|
|
|
|
|
|
|
|
|
Total stock-based
compensation expense |
|
$ |
1,254,503 |
|
|
$ |
1,102,444 |
|
|
$ |
711,259 |
|
|
|
|
|
|
|
|
|
|
|
A summary of activity under the Plans during the year ended December 31, 2009 is
presented below:
| |
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
Weighted Average |
|
| Options |
|
Option Shares |
|
|
Exercise Price |
|
Outstanding December 31, 2008 |
|
|
2,038,191 |
|
|
$ |
3.66 |
|
|
|
|
|
|
|
|
Granted |
|
|
1,117,929 |
|
|
|
5.34 |
|
Exercised |
|
|
— |
|
|
|
— |
|
Forfeited or expired |
|
|
150,000 |
|
|
|
4.73 |
|
|
|
|
|
|
|
|
|
Outstanding December 31, 2009 |
|
|
3,006,120 |
|
|
|
4.24 |
|
|
|
|
|
|
|
|
(weighted average contractual term) |
|
|
7.34 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Exercisable at December 31, 2009 |
|
|
1,640,284 |
|
|
|
5.80 |
|
|
|
|
|
|
|
|
(weighted average contractual term) |
|
|
5.76 |
|
|
|
|
|
There was no aggregate intrinsic value of the Company’s outstanding or exercisable
options as of December 31, 2009.
80
BIOSANTE PHARMACEUTICALS, INC.
Notes to the Financial Statements
December 31, 2009
| 10. |
|
STOCK-BASED COMPENSATION (continued) |
A summary of the Plans’ non-vested options at December 31, 2009 and activity under the Plans
during the year ended December 31, 2009 is presented below:
| |
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
Weighted Average |
|
| |
|
|
|
|
|
Grant Date Fair- |
|
| Options |
|
Option Shares |
|
|
Value |
|
Outstanding December 31, 2008 |
|
|
1,015,165 |
|
|
$ |
3.74 |
|
|
|
|
|
|
|
|
|
Granted |
|
|
1,117,929 |
|
|
|
5.34 |
|
Vested |
|
|
650,593 |
|
|
|
9.45 |
|
Forfeited |
|
|
116,665 |
|
|
|
4.42 |
|
|
|
|
|
|
|
|
|
Non-Vested at December 31, 2009 |
|
|
1,365,836 |
|
|
|
2.37 |
|
|
|
|
|
|
|
|
|
As of December 31, 2009, there was $998,029 of total unrecognized compensation cost
related to non-vested stock-based compensation arrangements granted under the Plans. The
cost is expected to be recognized over a weighted-average period of 1.56 years.
No stock options were exercised during 2009 and 2008. Cash received from option exercises
under the Plans for the year ended December 31, 2007 was $192,371. The intrinsic value of
options exercised during the year ended December 31, 2007 was $136,020. The Company did not
receive a tax benefit related to the exercise of these options because of its net operating
loss position. The total fair value of shares vested during the years ended December 31,
2009, 2008 and 2007 was $788,461, $659,898 and $326,254, respectively.
Options and warrants to purchase an aggregate of 8,385,075 and 4,750,229 shares,
respectively, were excluded from the earnings per share calculation for the years ended
December 31, 2009 and December 31, 2008, respectively, since including these options and
warrants would have had an anti-dilutive effect under the treasury stock method due to the
Company’s net loss position.
The Company offers a discretionary 401(k) Plan to all of its employees. Under the 401(k)
Plan, employees may defer income on a tax-exempt basis, subject to IRS limitation. Under
the 401(k) Plan, the Company can make discretionary matching contributions. Company
contributions expensed in 2009, 2008 and 2007 totaled $117,969, $108,019 and $59,683,
respectively.
The Company has entered into lease commitments for rental of its office space which expires
in 2012 and its laboratory facility which expires in 2010. The future minimum lease
payments during 2010, 2011 and 2012 are $299,618, $276,000 and $92,000, respectively.
Rent expense amounted to $325,093, $277,370 and $259,971 for the years ended December 31,
2009, 2008 and 2007, respectively.
81
BIOSANTE PHARMACEUTICALS, INC.
Notes to the Financial Statements
December 31, 2009
| 13. |
|
RELATED PARTY TRANSACTIONS |
Included in current liabilities on the balance sheet are $60,632 and $15,638, which
represent amounts due to current directors and officers of the Company for reimbursement of
business expenses and payment for director meeting fees as of December 31, 2009 and 2008,
respectively.
Antares Pharma, Inc. License
The Company’s license agreement with Antares Pharma, Inc. requires the Company to fund the
development of the licensed products, make milestone payments and pay royalties on the sales
of products related to this license. In 2008, the Company paid $462,500 to Antares as a
result of the Azur license of Elestrin and paid or accrued $21,830 to Antares as a result of
royalties received by the Company. In 2009, the Company paid or accrued $63,749 to Antares
as a result of royalties received by the Company from Azur.
Wake Forest License
In April 2002, the Company exclusively in-licensed from Wake Forest University and
Cedars-Sinai Medical Center three issued U.S. patents claiming triple component therapy (the
combination use of estrogen plus progestogen plus androgen, e.g. testosterone) and obtained
an option to license the patents for triple component contraception. The financial terms of
the license include an upfront payment by the Company in exchange for exclusive rights to
the license and regulatory milestone payments, maintenance payments and royalty payments by
the Company if a product incorporating the licensed technology gets approved and
subsequently marketed. In July 2005, the Company exercised the option for an exclusive
license for the three U.S. patents for triple component contraception. The financial terms
of this license include an upfront payment, regulatory milestone payments, maintenance
payments and royalty payments by the Company if a product incorporating the licensed
technology gets approved and subsequently marketed.
Future minimum maintenance payments due under this agreement are as follows:
| |
|
|
|
|
| Year |
|
Minimum Amount Due |
|
2010 |
|
|
70,000 |
|
2011 |
|
|
80,000 |
|
2012 |
|
|
80,000 |
|
2013 |
|
|
80,000 |
|
2014 |
|
|
80,000 |
|
2015 |
|
|
80,000 |
|
Thereafter |
|
|
120,000 |
|
82
BIOSANTE PHARMACEUTICALS, INC.
Notes to the Financial Statements
December 31, 2009
| 14. |
|
COMMITMENTS (continued) |
Under the terms of the license agreement with the Wake Forest University and Cedars-Sinai
Medical Center, the Company has the right to terminate the license at any time.
The Company has agreed to indemnify, hold harmless and defend Wake Forest University and
Cedars-Sinai Medical Center against any and all claims, suits, losses, damages, costs, fees
and expenses resulting from or arising out of exercise of the license agreement, including
but not limited to, any product liability claims. The Company has not recorded any
liability in connection with this obligation as no events occurred that would require
indemnification.
| 15. |
|
FAIR VALUE MEASUREMENTS |
The Company has adopted the fair value methods required under ASC 820 (formerly FAS
157) to value its financial assets and liabilities. As defined in ASC 820, fair value
is based on the price that would be received to sell an asset or paid to transfer a
liability in an orderly transaction between market participants at the measurement
date. In order to increase consistency and comparability in fair value measurements,
ASC 820 establishes a fair value hierarchy that prioritizes observable and unobservable
inputs used to measure fair value into three broad levels, which are described below:
Level 1: Quoted prices (unadjusted) in active markets that are accessible at the
measurement date for assets or liabilities. The fair value hierarchy gives the highest
priority to Level 1 inputs.
Level 2: Observable prices that are based on inputs not quoted on active markets,
but corroborated by market data.
Level 3: Unobservable inputs are used when little or no market data is available.
The fair value hierarchy gives the lowest priority to Level 3 inputs.
In determining fair value, the Company utilizes valuation techniques that maximize
the use of observable inputs and minimize the use of unobservable inputs to the extent
possible as well as considers counterparty credit risk.
Financial assets and liabilities recorded at fair value as of December 31, 2009 and December
31, 2008 are classified in the table below in one of the three categories described
above:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
Quoted Prices in |
|
|
Significant |
|
|
|
|
| |
|
|
|
|
|
Active Markets |
|
|
Other |
|
|
Significant |
|
| |
|
December 31, |
|
|
for Identical |
|
|
Observable |
|
|
Unobservable |
|
| Description |
|
2009 Balance |
|
|
Assets (Level 1) |
|
|
Inputs (Level 2) |
|
|
Inputs (Level 3) |
|
2011 Senior Notes |
|
$ |
974,579 |
|
|
|
|
|
|
$ |
974,579 |
|
|
|
|
|
2013 Senior Notes |
|
|
15,701,838 |
|
|
|
|
|
|
|
15,701,838 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$ |
16,676,417 |
|
|
|
|
|
|
$ |
16,676,417 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
83
BIOSANTE PHARMACEUTICALS, INC.
Notes to the Financial Statements
December 31, 2009
| 15. |
|
FAIR VALUE MEASUREMENTS (continued) |
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
Quoted Prices in |
|
|
Significant |
|
|
|
|
| |
|
|
|
|
|
Active Markets |
|
|
Other |
|
|
Significant |
|
| |
|
December 31, |
|
|
for Identical |
|
|
Observable |
|
|
Unobservable |
|
| Description |
|
2008 Balance |
|
|
Assets (Level 1) |
|
|
Inputs (Level 2) |
|
|
Inputs (Level 3) |
|
Available for Sale
Securities |
|
$ |
2,534,820 |
|
|
|
— |
|
|
|
— |
|
|
$ |
2,534,820 |
|
Put Asset on
Available
for Sale Securities |
|
|
465,180 |
|
|
|
— |
|
|
|
— |
|
|
|
465,180 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total |
|
$ |
3,000,000 |
|
|
|
— |
|
|
|
— |
|
|
$ |
3,000,000 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The Company made an election to record the values of the November 2011 and May 2013
convertible senior notes at fair value with gains and losses related to fluctuations in
the value of these financial liabilities recorded in earning immediately pursuant to
ASC 825 (formerly FAS 159). The fair values of the 2011 and 2013 Senior Notes are
estimated based on the risk-free borrowing rate, the volatility of the Company’s stock,
and the current borrowing rates for similar companies. See Note 7 entitled
“Convertible Senior Notes” for more information and disclosures regarding key
assumptions used in this fair value determination.
At January 1, 2008, the value of the Company’s auction rate securities were based
on observable prices in active markets and as such would have been considered based on
Level 1 inputs. At December 31, 2008, due to the lack of currently observable market
quotes, generally those obtained or corroborated through the auction process. The
Company determines the fair value using unobservable inputs based on expected cash
flows and collateral values, including assessments of counterparty credit quality,
default risk underlying the security, overall capital market liquidity, and
expectations of early redemption of the securities. Factors that may impact the
Company’s valuation include changes to credit ratings of the securities as well as to
the underlying assets supporting those securities, rates of default of the underlying
assets, underlying collateral value, counterparty risk and ongoing strength and quality
of market credit and liquidity. As a result of these declines in fair value of the
Company’s auction rate securities, which the Company attributed to liquidity issues
affecting the credit markets associated with the securities rather than counterparty
credit issues, the Company recorded an other-than-temporary impairment loss of $465,180
on its remaining auction rate securities investment, which was offset by a $465,180
gain on the right to sell the auction rate securities back to UBS at par value, both of
which are recorded in other income.
The Company made an election to record the asset related to its right to sell its
remaining auction rate securities to UBS at fair value with gains and losses related to
this instrument recorded in earnings immediately pursuant to ASC 825 (formerly FAS
159), so the right to sell the auction rate securities back to UBS would offset the
change in value of the underlying auction rate securities investments during the
period. As a result, a gain of $232,480 related to the change in value of the
put/right from October 14, 2008 (the date that the company entered into the settlement
agreement) to December 31, 2008 has been recorded in other income. If the company had
not elected to record this instrument at fair value, its carrying value would have been
$232,700 at December 31, 2008, which had been recorded in other income upon creation of the
asset.
84
BIOSANTE PHARMACEUTICALS, INC.
Notes to the Financial Statements
December 31, 2009
| 15. |
|
FAIR VALUE MEASUREMENTS (continued) |
As of December 31, 2008, the Company’s remaining auction rate securities with a $3.0 million
par value were held in an account with UBS Financial Services, Inc. (UBS). In August 2008,
UBS and its affiliates reached agreements with the SEC, the New York Attorney General, the
Massachusetts Securities Division, the Texas State Securities Board and other state
regulatory agencies represented by the North American Securities Administrators Association
to restore liquidity to UBS clients who held auction rate securities. Pursuant to these
agreements, in October 2008, the Company received rights from UBS entitling the Company to
sell to UBS or its affiliates and requiring UBS or its affiliates to purchase the Company’s
$3.0 million in remaining auction rate securities for their face (or par) value plus any
accrued and unpaid interest. On January 8, 2009, pursuant to those rights, the Company
received $3.0 million principal plus accrued and unpaid interest from UBS. Therefore, as of
December 31, 2009, the Company’s auction rate securities balance was $0. No net realized
gains or losses were included in the Company’s statement of operations for the year ended
December 31, 2009 and 2008, respectively.
The table below presents a reconciliation of the level 3 fair value measurements,
which are based on significant unobservable inputs, at December 31, 2009 and December
31, 2008. Both of the assets that existed as of December 31, 2008 are recorded in
short-term investments. The remaining short-term investment balance of $26,334 as of
December 31, 2008 was invested in a money market fund.
| |
|
|
|
|
|
|
|
|
| |
|
Fair Value |
|
|
Fair Value |
|
| |
|
Measurements Using |
|
|
Measurements Using |
|
| |
|
Significant |
|
|
Significant |
|
| |
|
Unobservable Inputs |
|
|
Unobservable Inputs |
|
| |
|
|
|
|
|
Put Asset Related to |
|
| |
|
Auction Rate Securities |
|
|
Auction Rate Securities |
|
January 1, 2008 |
|
$ |
— |
|
|
$ |
— |
|
Transfers into Level 3 |
|
|
14,000,000 |
|
|
|
232,700 |
|
Purchases, redemptions, issuances of settlements |
|
|
(11,000,000 |
) |
|
|
— |
|
Total gains or (losses) (realized/unrealized)
included in net loss |
|
|
(465,180 |
) |
|
|
232,480 |
|
|
|
|
|
|
|
|
December 31, 2008 |
|
$ |
2,534,820 |
|
|
$ |
465,180 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
January 1, 2009 |
|
$ |
2,534,820 |
|
|
$ |
465,180 |
|
Transfers into Level 3 |
|
|
— |
|
|
|
— |
|
Purchases, redemptions, issuances or settlements |
|
|
(2,534,820 |
) |
|
|
(465,180 |
) |
Total gains or (losses) (realized/unrealized)
included in net loss |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
December 31, 2009 |
|
$ |
— |
|
|
$ |
— |
|
|
|
|
|
|
|
|
85
BIOSANTE PHARMACEUTICALS, INC.
Notes to the Financial Statements
December 31, 2009
The Company is presently involved in the following legal action and from time to time may be
subject to various pending or threatened legal actions and proceedings, including those that
arise in the ordinary course of its business. Such matters are subject to many
uncertainties and to outcomes that are not predictable with assurance and that may not be
known for extended periods of time. The Company records a liability in its financial
statements for costs related to claims, including future legal costs, settlements and
judgments, where the Company has assessed that a loss is probable and an amount can be
reasonably estimated.
On July 1, 2009, a putative shareholder class action lawsuit concerning the Company’s then
proposed merger with Cell Genesys was filed in California Superior Court in San Mateo County
naming Cell Genesys, its officers and directors, and the Company as defendants. On July 6,
2009, a second putative shareholder class action lawsuit naming the same parties and
containing essentially identical allegations was filed in California Superior Court in San
Mateo County. On July 8, 2009, a third putative shareholder class action lawsuit was filed
in California Superior Court in San Mateo County, which also named the same parties and
contained essentially
identical allegations as the two prior lawsuits. On July 15, 2009, the Court consolidated
these three lawsuits into one action and appointed interim lead counsel. On August 13,
2009, plaintiffs filed a consolidated class action complaint alleging that defendants
breached their fiduciary duties and/or aided and abetted the breach of fiduciary duties owed
to Cell Genesys stockholders in connection with the then proposed merger, including by
failing to engage in a fair sales process, failing to obtain a fair price for the sale of
Cell Genesys, and failing to provide Cell Genesys
stockholders with material information regarding the merger. Plaintiffs sought an order
certifying the lawsuit as a class action, injunctive relief to enjoin the merger or, in the
event the then pending merger was completed, a rescission of the merger or rescissory
damages. Plaintiffs further sought an accounting for all damages and an award of attorneys’
fees and costs.
Solely to avoid the costs, risks and uncertainties inherent in litigation, on September 18,
2009, the Company and Cell Genesys entered into a memorandum of understanding with
plaintiffs’ counsel in the San Mateo County action pursuant to which the Company, Cell
Genesys, the other named defendants and the plaintiffs agreed to settle the lawsuits subject
to court approval. If the Court approves the settlement, the lawsuits will be dismissed with
prejudice. Pursuant to the memorandum of understanding, Cell Genesys agreed to pay to
plaintiffs’ counsel an amount not more than $240,000 as is approved by Court order for
plaintiffs’ attorneys’ fees, costs and expenses in the San Mateo County action and to make
additional disclosures in a current report on Form 8-K, without admitting in any way that
the certain disclosures are material or otherwise required by law. Cell Genesys filed the
Form 8-K on September 21, 2009. Pursuant to the memorandum of understanding, plaintiffs’
counsel conducted confirmatory discovery to confirm the fairness and adequacy of the
settlement. The parties filed a stipulation of settlement with the Court and moved the
Court for preliminary approval and issuance of a notice of settlement to the potential class
members, after which the parties intend to seek final settlement approval and dismissal of
the action with prejudice. The Company assumed Cell Genesys’s rights and obligations
relative to this lawsuit as a result of its merger with Cell Genesys, and accordingly has
recorded a liability of $240,000 for the potential settlement. The Company believes that
the resolution of this matter will not have a material impact on its financial position, cash
flows or results of operations.
86
BIOSANTE PHARMACEUTICALS, INC.
Notes to the Financial Statements
December 31, 2009
| 17. |
|
SELECTED QUARTERLY FINANCIAL DATA (UNAUDITED) |
Selected quarterly data for 2009 and 2008 is as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
2009 |
|
| |
|
First |
|
|
Second |
|
|
Third |
|
|
Fourth |
|
Revenue |
|
$ |
68,428 |
|
|
$ |
115,163 |
|
|
$ |
10,492 |
|
|
$ |
1,063,971 |
|
Research and development expenses |
|
|
3,072,240 |
|
|
|
3,493,576 |
|
|
|
3,371,217 |
|
|
|
3,743,540 |
|
General and administrative expenses |
|
|
1,029,202 |
|
|
|
1,208,956 |
|
|
|
1,506,056 |
|
|
|
1,629,731 |
|
Acquisition related charges |
|
|
— |
|
|
|
— |
|
|
|
1,470,467 |
|
|
|
27,721,727 |
|
Licensing expense |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
299,616 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating loss |
|
|
(4,062,260 |
) |
|
|
(4,620,701 |
) |
|
|
(6,370,556 |
) |
|
|
(33,630,091 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
|
(4,050,612 |
) |
|
|
(4,620,701 |
) |
|
|
(6,370,556 |
) |
|
|
(32,485,899 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss per share: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted |
|
$ |
(0.15 |
) |
|
$ |
(0.17 |
) |
|
$ |
(0.21 |
) |
|
$ |
(0.84 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
2008 |
|
| |
|
First |
|
|
Second |
|
|
Third |
|
|
Fourth |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenue |
|
$ |
62,997 |
|
|
$ |
25,869 |
|
|
$ |
82,212 |
|
|
$ |
3,609,751 |
|
Research and development expenses |
|
|
2,677,946 |
|
|
|
3,934,118 |
|
|
|
5,322,472 |
|
|
|
3,855,444 |
|
General and administrative expenses |
|
|
1,325,493 |
|
|
|
1,593,156 |
|
|
|
1,438,816 |
|
|
|
767,469 |
|
Licensing expense |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
836,420 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating loss |
|
|
(3,950,215 |
) |
|
|
(5,513,714 |
) |
|
|
(6,690,835 |
) |
|
|
(1,858,878 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
|
(3,626,638 |
) |
|
|
(6,048,067 |
) |
|
|
(6,585,084 |
) |
|
|
(1,165,389 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss per share: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted |
|
$ |
(0.13 |
) |
|
$ |
(0.22 |
) |
|
$ |
(0.24 |
) |
|
$ |
(0.05 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
In December 2009, the Company entered into an amendment to its original licensing agreement with
Azur which permanently reduced the royalty percentage due to us related to Azur’s sales of
Elestrin. Upon signing the amended agreement, Azur made a $1.0 million nonrefundable payment
in December 2009 in exchange for a permanent reduction in future royalty rates, and received
options to make a total of $2.16 million in additional non-refundable payments (which were
exercised during the first quarter of 2010) in exchange for the elimination of substantially
all remaining future royalties and milestone payments due to the Company under the terms of the original
license. The $1.0 million nonrefundable payment was recorded as revenue during the fourth
quarter of 2009 as we had no remaining performance obligations with respect to this amount.
Pursuant to a separate agreement with Antares and related to the Azur royalty stream
buydown, the Company paid Antares an aggregate $268,750 in February 2010.
On March 8, 2010, the Company completed an offering of 10,404,626 shares of its common
stock and warrants to purchase an aggregate of 5,202,313 shares of its common stock at a
purchase price of $1.73 per share to funds affiliated with two institutional investors for
gross proceeds of $18.0 million. The offering resulted in net proceeds to the Company of
approximately $17.5 million, after deducting placement agent fees and offering expenses.
The warrants are exercisable beginning on September 9, 2010 and continuing for a period of
five years, at an exercise price of $2.08 per share. In connection with the offering, the
Company issued the placement agent warrants to purchase an aggregate of 208,093 shares of
the Company’s common stock at an exercise price of $2.16, which warrants are exercisable
beginning on September 8, 2010 and will expire on June 9, 2014. The number of shares
issuable upon exercise of the warrants and the exercise price of the warrants are adjustable
in the event of stock splits, combinations and reclassifications, but not in the event of
the issuance of additional securities.
87
|
|
|
| Item 9. |
|
CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL
DISCLOSURE |
None.
|
|
|
| Item 9A. |
|
CONTROLS AND PROCEDURES |
Evaluation of Disclosure Controls and Procedures
We maintain disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under
the Securities Exchange Act of 1934, as amended) that are designed to provide reasonable assurance
that the information required to be disclosed by us in the reports we file or submit under the
Securities Exchange Act of 1934, as amended, is recorded, processed, summarized and reported within
the time periods specified in the Securities and Exchange Commission’s rules and forms and that
such information is accumulated and communicated to our management, including our principal
executive officer and principal financial officer, or persons performing similar functions, as
appropriate to allow timely decisions regarding required disclosure. In designing and evaluating
our disclosure controls and procedures, we recognize that any controls and procedures, no matter
how well designed and operated, can provide only reasonable assurance of achieving the desired
control objectives, and we are required to apply our judgment in evaluating the cost-benefit
relationship of possible internal controls. Our management evaluated, with the participation of
our Chief Executive Officer and Chief Financial Officer, the effectiveness of the design and
operation of our disclosure controls and procedures as of the end of the period covered in this
annual report on Form 10-K. Based on that evaluation, our Chief Executive Officer and Chief
Financial Officer concluded that our disclosure controls and procedures were effective as of the
end of such period to provide reasonable assurance that information required to be disclosed in our
Exchange Act reports is recorded, processed, summarized, and reported within the time periods
specified in the SEC’s rules and forms, and that material information relating to our company is
made known to management, including our Chief Executive Officer and Chief Financial Officer,
particularly during the period when our periodic reports are being prepared.
Management’s Report on Internal Control Over Financial Reporting
Our management report on internal control over financial reporting is included in this report in
Part II. Item 8, under the heading “Management’s Report on Internal Control over Financial
Reporting.” This annual report does not include an attestation report of the Company’s registered
public accounting firm regarding internal control over financial reporting. Management’s report
was not subject to attestation by the company’s registered public accounting firm pursuant to
temporary rules of the Securities and Exchange Commission that permit the company to provide only
management’s report in this annual report.
Change in Internal Control Over Financial Reporting
There was no change in our internal control over financial reporting that occurred during our
fourth quarter ended December 31, 2009 that has materially affected, or is reasonably likely to
materially affect, our internal control over financial reporting.
|
|
|
| Item 9B. |
|
OTHER INFORMATION |
Not applicable.
88
PART III
|
|
|
| Item 10. |
|
DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE |
Directors
The information in the “Proposal One — Election of Directors” section of our definitive proxy
statement to be filed with the SEC with respect to our next annual meeting of stockholders is
incorporated in this report by reference, or, if such proxy statement is not filed with the SEC
within 120 days after the end of the fiscal year covered by this report, such information will be
filed as part of an amendment to this report not later than the end of the 120-day period.
Executive Officers
The information concerning our executive officers is included in this report under Item 4a,
“Executive Officers of the Registrant” and is incorporated in this report by reference.
Section 16(a) Beneficial Ownership Reporting Compliance
The information in the “Stock Ownership—Section 16(a) Beneficial Ownership Reporting Compliance”
section of our definitive proxy statement to be filed with the SEC with respect to our next annual
meeting of stockholders is incorporated in this report by reference, or, if such proxy statement is
not filed with the SEC within 120 days after the end of the fiscal year covered by this report,
such information will be filed as part of an amendment to this report not later than the end of the
120-day period.
Code of Conduct and Ethics
Our Code of Conduct and Ethics applies to all of our employees, officers and directors, including
our principal executive officer and principal financial officer, and meets the requirements of the
Securities and Exchange Commission. A copy of our Code of Conduct and Ethics is filed as an
exhibit to this report. We intend to satisfy the disclosure requirements of Item 5.05 of Form 8-K
regarding amendments to or waivers from any provision of our Code of Conduct and Ethics by posting
such information on our corporate website located at www.biosantepharma.com.
Changes to Nomination Procedures
During the fourth quarter of 2009, we made no material changes to the procedures by which
stockholders may recommend nominees to the Board of Directors, as described in our most recent
proxy statement.
Audit Committee Matters
The information in the “Corporate Governance — Audit Committee” section of our definitive proxy
statement to be filed with the SEC with respect to our next annual meeting of stockholders is
incorporated in this report by reference, or, if such proxy statement is not filed with the SEC
within 120 days after the end of the fiscal year covered by this report, such information will be
filed as part of an amendment to this report not later than the end of the 120-day period.
89
|
|
|
| Item 11. |
|
EXECUTIVE COMPENSATION |
The information in the “Executive Compensation” and the “Director Compensation” sections of our
definitive proxy statement to be filed with the SEC with respect to our next annual meeting of
stockholders is incorporated in this report by reference, or, if such proxy statement is not filed
with the SEC within 120 days after the end of the fiscal year covered by this report, such
information will be filed as part of an amendment to this report not later than the end of the
120-day period.
|
|
|
| Item 12. |
|
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
Stock Ownership
The information in the “Stock Ownership” section of our definitive proxy statement to be filed with
the SEC with respect to our next annual meeting of stockholders and is incorporated in this report
by reference, or, if such proxy statement is not filed with the SEC within 120 days after the end
of the fiscal year covered by this report, such information will be filed as part of an amendment
to this report not later than the end of the 120-day period.
Securities Authorized for Issuance Under Equity Compensation Plans
The following table and notes provide information about shares of our common stock that may be
issued under all of our equity compensation plans as of December 31, 2009. Except otherwise stated
below, options granted in the future under the BioSante Pharmaceuticals, Inc. 2008 Stock Incentive
Plan are within the discretion of the Compensation Committee of our Board of Directors and our
Board of Directors therefore cannot be ascertained at this time.
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
(c) |
|
| |
|
|
|
|
|
(b) |
|
|
Number of Securities |
|
| |
|
(a) |
|
|
Weighted-Average |
|
|
Remaining Available for |
|
| |
|
Number of Securities to be |
|
|
Exercise |
|
|
Future Issuance Under |
|
| |
|
Issued Upon Exercise of |
|
|
Price of Outstanding |
|
|
Equity Compensation Plans |
|
| |
|
Outstanding Options, |
|
|
Options, Warrants |
|
|
(excluding securities |
|
| Plan Category |
|
Warrants and Rights |
|
|
and Rights |
|
|
reflected in column (a)) |
|
Equity
compensation plans
approved by
security holders |
|
|
2,771,691 |
(1)(2) |
|
$ |
2.88 |
|
|
|
829,071 |
(3) |
|
|
|
|
|
|
|
|
|
|
|
|
|
Equity compensation
plans not approved
by security holders |
|
|
234,429 |
|
|
$ |
19.73 |
|
|
|
0 |
|
|
|
|
|
|
|
|
|
|
|
Total |
|
|
3,006,120 |
|
|
$ |
4.20 |
|
|
|
829,071 |
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
| (1) |
|
Amount includes shares of our common stock issuable upon the exercise of stock options
outstanding as of December 31, 2009 under the BioSante Pharmaceuticals, Inc. 2008 Stock
Incentive Plan and the BioSante Pharmaceuticals, Inc. Amended and Restated 1998 Stock Plan. |
| |
| (2) |
|
Excludes options assumed by us in connection with our merger with Cell Genesys, Inc.
As of December 31, 2009, a total of 234,429 shares of our common stock were issuable upon
exercise of the assumed options. The weighted average exercise price of the outstanding
assumed options as of such date was $19.73 per share and they have an average weighted
life remaining of 6.7 years. All of the options assumed and outstanding in connection with
our merger with Cell Genesys were exercisable as of December 31, 2009. No additional
options, restricted stock units or other equity incentive awards may be granted under the
assumed Cell Genesys, Inc. plans. |
| |
| (3) |
|
As of December 31, 2009, these shares remain available for future issuance under the
BioSante Pharmaceuticals, Inc. 2008 Stock Incentive Plan. No shares remain available for
grant under the BioSante Pharmaceuticals, Inc. Amended and Restated 1998 Stock Plan since
such plan expired with respect to future grants in 2008. |
90
|
|
|
| Item 13. |
|
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
The information in the “Related Party Relationships and Transactions” and “Corporate Governance-
Director Independence” sections of our definitive proxy statement to be filed with the SEC with
respect to our next annual meeting of stockholders is incorporated in this report by reference, or,
if such proxy statement is not filed with the SEC within 120 days after the end of the fiscal year
covered by this report, such information will be filed as part of an amendment to this report not
later than the end of the 120-day period.
|
|
|
| Item 14. |
|
PRINCIPAL ACCOUNTING FEES AND SERVICES |
The information in the “Proposal Three — Ratification of Selection of Independent Registered Public
Accounting Firm — Audit, Audit-Related, Tax and Other Fees” and “Proposal Three — Ratification of
Selection of Independent Registered Public Accounting Firm — Pre-Approval Policy and Procedures” of
our definitive proxy statement to be filed with the SEC with respect to our next annual meeting of
stockholders is incorporated herein by reference, or, if such proxy statement is not filed with the
SEC within 120 days after the end of the fiscal year covered by this report, such information will
be filed as part of an amendment to this report not later than the end of the 120-day period.
91
PART IV
|
|
|
| Item 15. |
|
EXHIBITS, FINANCIAL STATEMENT SCHEDULES |
Our financial statements are included in Item 8 of Part II of this report.
The exhibits to this report are listed on the Exhibit Index to this report. A copy of any of the
exhibits listed will be furnished at a reasonable cost, upon receipt from any person of a written
request for any such exhibit. Such request should be sent to BioSante Pharmaceuticals, Inc., 111
Barclay Boulevard, Lincolnshire, Illinois 60069, Attn: Stockholder Information.
The following is a list of each management contract or compensatory plan or arrangement required to
be filed as an exhibit to this annual report on Form 10-K pursuant to Item 15(a):
| A. |
|
Amended and Restated Employment Letter Agreement dated July 16, 2008 between BioSante
Pharmaceuticals, Inc. and Stephen M. Simes (incorporated by reference to Exhibit 10.1 to
BioSante’s Current Report on Form 8-K as filed with the SEC on July 18, 2008 (File No.
001-31812)). |
| |
| B. |
|
Amended and Restated Employment Letter Agreement dated July 16, 2008 between BioSante
Pharmaceuticals, Inc. and Phillip B. Donenberg (incorporated by reference to Exhibit 10.2 to
BioSante’s Current Report on Form 8-K as filed with the SEC on July 18, 2008 (File No.
001-31812)). |
| |
| C. |
|
BioSante Pharmaceuticals, Inc. 2008 Stock Incentive Plan (incorporated
by reference to Exhibit 10.1 contained in BioSante’s 8-K as filed with
the Securities and Exchange Commission on June 13, 2008 (File No.
001-31812)). |
| |
| D. |
|
Form of Incentive Stock Option Agreement between BioSante
Pharmaceuticals, Inc. and its Executive Officers Under the BioSante
Pharmaceuticals, Inc. 2008 Stock Incentive Plan (incorporated by
reference to Exhibit 10.2 contained in BioSante’s 8-K as filed with
the Securities and Exchange Commission on June 13, 2008 (File No.
001-31812)). |
| |
| E. |
|
Form of Non-Statutory Stock Option Agreement between BioSante
Pharmaceuticals, Inc. and its Executive Officers Under the BioSante
Pharmaceuticals, Inc. 2008 Stock Incentive Plan (incorporated by
reference to Exhibit 10.3 contained in BioSante’s 8-K as filed with
the Securities and Exchange Commission on June 13, 2008 (File No.
001-31812)). |
| |
| F. |
|
Form of Non-Statutory Stock Option Agreement between BioSante
Pharmaceuticals, Inc. and its Directors Under the BioSante
Pharmaceuticals, Inc. 2008 Stock Incentive Plan (incorporated by
reference to Exhibit 10.4 contained in BioSante’s 8-K as filed with
the Securities and Exchange Commission on June 13, 2008 (File No.
001-31812)). |
| |
| G. |
|
BioSante Pharmaceuticals, Inc. Amended and Restated 1998 Stock Plan
(incorporated by reference to Exhibit 10.1 contained in BioSante’s 8-K
as filed with the Securities and Exchange Commission on June 12, 2006
(File No. 001-31812)). |
92
| H. |
|
Form of Stock Option Agreement between BioSante Pharmaceuticals, Inc.
and each of BioSante’s Executive Officers Under the BioSante
Pharmaceuticals, Inc. Amended and Restated
1998 Stock Plan (incorporated by reference to Exhibit 10.5 to BioSante’s Annual
Report on Form 10-KSB for the fiscal year ended December 31, 2001 (File No.
000-28637)). |
| |
| I. |
|
Form of Stock Option Agreement between BioSante
Pharmaceuticals, Inc. and each of BioSante’s
Executive Officers Under the BioSante
Pharmaceuticals, Inc. Amended and Restated 1998 Stock
Plan (incorporated by reference to Exhibit 10.30 to
BioSante’s Annual Report on Form 10-KSB for the
fiscal year ended December 31, 2003 (File No.
001-31812)). |
| |
| J. |
|
Form of Stock Option Agreement between BioSante
Pharmaceuticals, Inc. and each of BioSante’s
Directors Under the BioSante Pharmaceuticals, Inc.
Amended and Restated 1998 Stock Plan (incorporated by
reference to Exhibit 10.31 to BioSante’s Annual
Report on Form 10-KSB for the fiscal year ended
December 31, 2003 (File No. 001-31812)). |
| |
| K. |
|
Form of Indemnification Agreement between BioSante Pharmaceuticals, Inc. and each of
BioSante’s Directors and Executive Officers (incorporated by reference to Exhibit 10.30 to
BioSante’s Annual Report on Form 10-K for the fiscal year ended December 31, 2007 (File No.
001-31812)). |
| |
| L. |
|
Description of Non-Employee Director Compensation Arrangements (filed herewith). |
| |
| M. |
|
Cell Genesys, Inc. 2005 Equity Incentive Plan, as amended (incorporated by reference to
Exhibit 10.3 contained in Cell Genesys’s Quarterly Report on Form 10-Q for the quarter ended
June 30, 2007 (File No. 000-19986)). |
| |
| N. |
|
Cell Genesys, Inc. Amended and Restated 1998 Incentive Stock Plan (incorporated by reference
to Exhibit 10.2 contained in Cell Genesys’s Quarterly Report on Form 10-Q for the quarter
ended June 30, 2003 (File No. 000-19986)). |
93
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the
registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto
duly authorized.
| |
|
|
|
|
| Dated: March 30, 2010 |
BIOSANTE PHARMACEUTICALS, INC.
|
|
| |
By: |
/s/ Stephen M. Simes
|
|
| |
|
Stephen M. Simes |
|
| |
|
Vice Chairman, President and Chief Executive Officer
(Principal Executive Officer) |
|
| |
|
|
| |
By: |
/s/ Phillip B. Donenberg
|
|
| |
|
Phillip B. Donenberg |
|
| |
|
Chief Financial Officer, Treasurer and Secretary
(Principal Financial and Accounting Officer) |
|
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed
below by the following persons on behalf of the registrant and in the capacities and on the dates
indicated.
| |
|
|
|
|
| Name and Signature |
|
Title |
|
Date |
|
|
|
|
|
/s/ Stephen M. Simes
Stephen M. Simes
|
|
Vice Chairman,
President and
Chief
Executive Officer
|
|
March 30, 2010 |
|
|
|
|
|
/s/ Louis W. Sullivan, M.D.
Louis W. Sullivan, M.D.
|
|
Chairman of the Board
|
|
March 30, 2010 |
|
|
|
|
|
/s/ Fred Holubow
Fred Holubow
|
|
Director
|
|
March 30, 2010 |
|
|
|
|
|
/s/ Peter Kjaer
Peter Kjaer
|
|
Director
|
|
March 30, 2010 |
|
|
|
|
|
/s/ Ross Mangano
Ross Mangano
|
|
Director
|
|
March 30, 2010 |
|
|
|
|
|
/s/ John T. Potts, Jr., M.D.
John T. Potts, Jr., M.D.
|
|
Director
|
|
March 30, 2010 |
|
|
|
|
|
/s/ Edward C. Rosenow, III, M.D.
Edward C. Rosenow, III, M.D.
|
|
Director
|
|
March 30, 2010 |
|
|
|
|
|
/s/ Stephen A. Sherwin, M.D.
Stephen A. Sherwin, M.D.
|
|
Director
|
|
March 30, 2010 |
94
BIOSANTE PHARMACEUTICALS, INC.
EXHIBIT INDEX TO ANNUAL REPORT ON FORM 10-K
FOR THE YEAR ENDED DECEMBER 31, 2009
| |
|
|
|
|
|
|
| Exhibit No. |
|
Exhibit |
|
Method of Filing |
| |
| |
1.1 |
|
|
Placement Agent Agreement dated as of
August 13, 2009 between BioSante
Pharmaceuticals, Inc. and Rodman &
Renshaw, LLC
|
|
Incorporated by
reference to
Exhibit 1.1 to
BioSante’s Current
Report on Form 8-K
as filed with the
Securities and
Exchange Commission
on August 14, 2009
(File No.
001-31812) |
| |
|
|
|
|
|
|
| |
1.2 |
|
|
Placement Agent Agreement dated as of
March 4, 2010 between BioSante
Pharmaceuticals, Inc. and Rodman &
Renshaw, LLC
|
|
Incorporated by
reference to
Exhibit 1.1 to
BioSante’s Current
Report on Form 8-K
as filed with the
Securities and
Exchange Commission
on March 5, 2010
(File No.
001-31812) |
| |
|
|
|
|
|
|
| |
2.1 |
|
|
Agreement and Plan of Merger dated as
of June 29, 2009 by and between
BioSante Pharmaceuticals, Inc. and Cell
Genesys, Inc.(1)
|
|
Incorporated by
reference to
Exhibit 2.1 to
BioSante’s Current
Report on Form 8-K
as filed with the
Securities and
Exchange Commission
on June 30, 2009
(File No.
001-31812) |
| |
|
|
|
|
|
|
| |
3.1 |
|
|
Restated Certificate of Incorporation
of BioSante Pharmaceuticals, Inc.
|
|
Incorporated by
reference to
Exhibit 3.1 to
BioSante’s Current
Report on Form 8-K
as filed with the
Securities and
Exchange Commission
on October 14, 2009
(File No.
001-31812) |
| |
|
|
|
|
|
|
| |
3.2 |
|
|
Bylaws of BioSante Pharmaceuticals, Inc.
|
|
Incorporated by
reference to
Exhibit 3.2 to
BioSante’s
Registration
Statement on Form
SB-2, as amended
(Reg. No.
333-64218) |
| |
|
|
|
|
|
|
| |
4.1 |
|
|
Indenture, dated as of October 20,
2004, between Cell Genesys, Inc. and
U.S. Bank National Association, as
trustee
|
|
Incorporated by
reference to
Exhibit 4.1 to Cell
Genesys’s
Registration
Statement on Form
S-3 as filed with
the Securities and
Exchange Commission
on December 29,
2004 (File No.
000-19986) |
95
| |
|
|
|
|
|
|
| Exhibit No. |
|
Exhibit |
|
Method of Filing |
| |
| |
4.2 |
|
|
Supplemental Indenture dated as of
October 14, 2009 to Indenture dated as
of October 20, 2004, by and between
BioSante Pharmaceuticals, Inc. and U.S.
Bank National Association, Relating to
Cell Genesys, Inc. 3.125% Convertible
Senior Subordinated Notes due 2011
|
|
Incorporated by
reference to
Exhibit 4.1 to
BioSante’s Current
Report on Form 8-K
as filed with the
Securities and
Exchange Commission
on October 14, 2009
(File No.
001-31812) |
| |
|
|
|
|
|
|
| |
4.3 |
|
|
Indenture, dated as of June 24, 2009,
between Cell Genesys, Inc. and U.S.
Bank National Association, as trustee
|
|
Incorporated by
reference to
Exhibit 4.1 to Cell
Genesys’s Current
Report on Form 8-K
as filed with the
Securities and
Exchange Commission
on June 29, 2009
(File No.
000-19986) |
| |
|
|
|
|
|
|
| |
4.4 |
|
|
Supplemental Indenture dated as of
October 14, 2009 to Indenture dated as
of June 24, 2009, by and between
BioSante Pharmaceuticals, Inc. and U.S.
Bank National Association, Relating to
Cell Genesys, Inc. 3.125% Convertible
Senior Subordinated Notes due 2013
|
|
Incorporated by
reference to
Exhibit 4.2 to
BioSante’s Current
Report on Form 8-K
as filed with the
Securities and
Exchange Commission
on October 14, 2009
(File No.
001-31812) |
| |
|
|
|
|
|
|
| |
4.5 |
|
|
Form of Warrant dated as of July 21,
2006 issued by BioSante
Pharmaceuticals, Inc. to each of the
Subscribers Party to the Subscription
Agreements dated July 7, 2006
|
|
Incorporated by
reference to
Exhibit 10.2 to
BioSante’s Form 8-K
as filed with the
Securities and
Exchange Commission
on July 24, 2006
(File No.
001-31812) |
| |
|
|
|
|
|
|
| |
4.6 |
|
|
Form of Warrant dated as of June 13,
2007 issued by BioSante
Pharmaceuticals, Inc. to each of the
Subscribers Party to the Subscription
Agreements dated May 25, 2007
|
|
Incorporated by
reference to
Exhibit 10.2 to
BioSante’s Form 8-K
as filed with the
Securities and
Exchange Commission
on June 14, 2007
(File No.
001-31812) |
| |
|
|
|
|
|
|
| |
4.7 |
|
|
Warrant dated December 15, 2008 issued
by BioSante Pharmaceuticals, Inc. to
Kingsbridge Capital Limited
|
|
Incorporated by
reference to
Exhibit 4.1 to
BioSante’s Form 8-K
as filed with the
Securities and
Exchange Commission
on December 18,
2008 (File No.
001-31812) |
| |
|
|
|
|
|
|
| |
4.8 |
|
|
Form of Common Stock Purchase Warrant
issued by BioSante Pharmaceuticals,
Inc. to Investors and the Placements
Agent in the August 2009 Registered
Direct Offering
|
|
Incorporated by
reference to
Exhibit 4.1 to
BioSante’s Form 8-K
as filed with the
Securities and
Exchange Commission
on August 14, 2009
(File No.
001-31812) |
96
| |
|
|
|
|
|
|
| Exhibit No. |
|
Exhibit |
|
Method of Filing |
| |
| |
4.9 |
|
|
Form of Replacement Warrant issued to
Investors in Cell Genesys, Inc.’s April
2007 Registered Direct Offering
|
|
Filed herewith |
| |
|
|
|
|
|
|
| |
4.10 |
|
|
Form of Common Stock Purchase Warrant
issued by BioSante Pharmaceuticals,
Inc. to Investors and the Placements
Agent in the March 2010 Registered
Direct Offering
|
|
Incorporated by
reference to
Exhibit 4.1 to
BioSante’s Form 8-K
as filed with the
Securities and
Exchange Commission
on March 5, 2010
(File No.
001-31812) |
| |
|
|
|
|
|
|
| |
10.1 |
|
|
Amended and Restated Employment Letter
Agreement dated July 16, 2008 between
BioSante Pharmaceuticals, Inc. and
Stephen M. Simes
|
|
Incorporated by
reference to
Exhibit 10.1 to
BioSante’s Current
Report on Form 8-K
as filed with the
SEC on July 18,
2008 (File No.
001-31812) |
| |
|
|
|
|
|
|
| |
10.2 |
|
|
Amended and Restated Employment Letter
Agreement dated July 16, 2008 between
BioSante Pharmaceuticals, Inc. and
Phillip B. Donenberg
|
|
Incorporated by
reference to
Exhibit 10.2 to
BioSante’s Current
Report on Form 8-K
as filed with the
SEC on July 18,
2008 (File No.
001-31812) |
| |
|
|
|
|
|
|
| |
10.3 |
|
|
BioSante Pharmaceuticals, Inc. 2008
Stock Incentive Plan
|
|
Incorporated by
reference to
Exhibit 10.1 to
BioSante’s Current
Report on Form 8-K
as filed with the
SEC on June 13,
2008 (File No.
001-31812) |
| |
|
|
|
|
|
|
| |
10.4 |
|
|
Form of Incentive Stock Option
Agreement between BioSante
Pharmaceuticals, Inc. and its Executive
Officers Under the BioSante
Pharmaceuticals, Inc. 2008 Stock
Incentive Plan
|
|
Incorporated by
reference to
Exhibit 10.2 to
BioSante’s Current
Report on Form 8-K
as filed with the
SEC on June 13,
2008 (File No.
001-31812) |
| |
|
|
|
|
|
|
| |
10.5 |
|
|
Form of Non-Statutory Stock Option
Agreement between BioSante
Pharmaceuticals, Inc. and its Executive
Officers Under the BioSante
Pharmaceuticals, Inc. 2008 Stock
Incentive Plan
|
|
Incorporated by
reference to
Exhibit 10.3 to
BioSante’s Current
Report on Form 8-K
as filed with the
SEC on June 13,
2008 (File No.
001-31812) |
| |
|
|
|
|
|
|
| |
10.6 |
|
|
Form of Non-Statutory Stock Option
Agreement between BioSante
Pharmaceuticals, Inc. and its Directors
Under the BioSante Pharmaceuticals,
Inc. 2008 Stock Incentive Plan
|
|
Incorporated by
reference to
Exhibit 10.4 to
BioSante’s Current
Report on Form 8-K
as filed with the
SEC on June 13,
2008 (File No.
001-31812) |
97
| |
|
|
|
|
|
|
| Exhibit No. |
|
Exhibit |
|
Method of Filing |
| |
|
|
|
|
|
|
| |
10.7 |
|
|
BioSante Pharmaceuticals, Inc. Amended
and Restated 1998 Stock Plan
|
|
Incorporated by
reference to
Exhibit 10.1 to
BioSante’s 8-K as
filed with the
Securities and
Exchange Commission
on June 12, 2006
(File No.
001-31812) |
| |
|
|
|
|
|
|
| |
10.8 |
|
|
Form of Stock Option Agreement between
BioSante Pharmaceuticals, Inc. and each
of BioSante’s Executive Officers Under
the BioSante Pharmaceuticals, Inc.
Amended and Restated 1998 Stock Plan
|
|
Incorporated by
reference to
Exhibit 10.5 to
BioSante’s Annual
Report on Form
10-KSB for the
fiscal year ended
December 31, 2001
(File No. 0-28637) |
| |
|
|
|
|
|
|
| |
10.9 |
|
|
Form of Stock Option Agreement between
BioSante Pharmaceuticals, Inc. and each
of BioSante’s Executive Officers Under
the BioSante Pharmaceuticals, Inc.
Amended and Restated 1998 Stock Plan
|
|
Incorporated by
reference to
Exhibit 10.30 to
BioSante’s Form
10-KSB for the
fiscal year ended
December 31, 2003
(File No.
001-31812) |
| |
|
|
|
|
|
|
| |
10.10 |
|
|
Form of Stock Option Agreement between
BioSante Pharmaceuticals, Inc. and each
of BioSante’s Directors Under the
BioSante Pharmaceuticals, Inc. Amended
and Restated 1998 Stock Plan
|
|
Incorporated by
reference to
Exhibit 10.31 to
BioSante’s Form
10-KSB for the
fiscal year ended
December 31, 2003
(File No.
001-31812) |
| |
|
|
|
|
|
|
| |
10.11 |
|
|
Form of Indemnification Agreement
between BioSante Pharmaceuticals, Inc.
and each of its Directors and Executive
Officers
|
|
Incorporated by
reference to
Exhibit 10.30 to
BioSante’s Form
10-K for the fiscal
year ended December
31, 2007 (File No.
001-31812) |
| |
|
|
|
|
|
|
| |
10.12 |
|
|
Description of Non-Employee Director
Compensation Arrangements
|
|
Filed herewith |
| |
|
|
|
|
|
|
| |
10.13 |
|
|
Cell Genesys, Inc. 2005 Equity
Incentive Plan, as amended
|
|
Incorporated by
reference to
Exhibit 10.3 to
Cell Genesys’s
Quarterly Report on
Form 10-Q for the
quarter ended June
30, 2007 (File No.
000-19986) |
| |
|
|
|
|
|
|
| |
10.14 |
|
|
Cell Genesys, Inc. Amended and Restated
1998 Incentive Stock Plan
|
|
Incorporated by
reference to
Exhibit 10.2 to
Cell Genesys’s
Quarterly Report on
Form 10-Q for the
quarter ended June
30, 2003 (File No.
000-19986) |
98
| |
|
|
|
|
|
|
| Exhibit No. |
|
Exhibit |
|
Method of Filing |
| |
|
|
|
|
|
|
| |
10.15 |
|
|
Office Lease, dated December 19, 2003,
between BioSante and LaSalle National
Bank Association, as successor trustee
to American National Bank and Trust
Company of Chicago
|
|
Incorporated by
reference to
Exhibit 10.29 to
BioSante’s Form
10-KSB for the
fiscal year ended
December 31, 2003
(File No.
001-31812) |
| |
|
|
|
|
|
|
| |
10.16 |
|
|
First Amendment to Lease, dated
February 26, 2004, between BioSante and
LaSalle National Bank Association, as
successor trustee to American National
Bank and Trust Company of Chicago
|
|
Incorporated by
reference to
Exhibit 10.1 to
BioSante’s Form
10-QSB for the
fiscal quarter
ended March 31,
2004 (File No.
001-31812) |
| |
|
|
|
|
|
|
| |
10.17 |
|
|
Second Amendment to Lease dated as of
January 4, 2005, by and between
BioSante Pharmaceuticals, Inc. and
LaSalle Bank National Association, as
successor trustee to American National
Bank and Trust Company of Chicago
|
|
Incorporated by
reference to
Exhibit 10.1 to
BioSante’s Current
Report on Form 8-K
as filed with the
Securities and
Exchange Commission
on January 6, 2005
(File No.
001-31812) |
| |
|
|
|
|
|
|
| |
10.18 |
|
|
Third Amendment to Lease dated as of
January 27, 2006 by and between
BioSante Pharmaceuticals, Inc. and
LaSalle Bank National Association, as
successor trustee to American National
Bank and Trust Company of Chicago
|
|
Incorporated by
reference to
Exhibit 10.1 to
BioSante’s Current
Report on Form 8-K
as filed with the
Securities and
Exchange Commission
on January 27, 2006
(File No.
001-31812) |
| |
|
|
|
|
|
|
| |
10.19 |
|
|
Fourth Amendment to Lease dated as of
March 7, 2007 by and between BioSante
Pharmaceuticals, Inc. and LaSalle Bank
National Association, as successor
trustee to American National Bank and
Trust Company of Chicago
|
|
Incorporated by
reference to
Exhibit 10.1 to
BioSante’s Current
Report on Form 8-K
as filed with the
Securities and
Exchange Commission
on March 7, 2007
(File No.
001-31812) |
| |
|
|
|
|
|
|
| |
10.20 |
|
|
Fifth Amendment to Lease dated as of
November 2, 2007 by and between
BioSante Pharmaceuticals, Inc. and
LaSalle Bank National Association, as
successor trustee to American National
Bank and Trust Company of Chicago.
|
|
Incorporated by
reference to
Exhibit 10.1 to
BioSante’s Current
Report on Form 8-K
as filed with the
Securities and
Exchange Commission
on November 6, 2007
(File No.
001-31812) |
| |
|
|
|
|
|
|
| |
10.21 |
|
|
Sixth Amendment to Lease dated as of
April 18, 2008 by and between BioSante
Pharmaceuticals, Inc. and LaSalle Bank
National Association, as successor
trustee to American National Bank and
Trust Company of Chicago
|
|
Incorporated by
reference to
Exhibit 10.1 to
BioSante’s Current
Report on Form 8-K
as filed with the
SEC on April 21,
2008 (File No.
001-31812) |
99
| |
|
|
|
|
|
|
| Exhibit No. |
|
Exhibit |
|
Method of Filing |
| |
|
|
|
|
|
|
| |
10.22 |
|
|
Seventh Amendment to Lease dated as of
November 17, 2008 by and between
BioSante Pharmaceuticals, Inc. and
LaSalle Bank National Association, as
successor trustee to American National
Bank and Trust Company of Chicago
|
|
Filed herewith |
| |
|
|
|
|
|
|
| |
10.23 |
|
|
Eighth Amendment to Lease dated as of
September 8, 2009 by and between
BioSante Pharmaceuticals, Inc. and
LaSalle Bank National Association, as
successor trustee to American National
Bank and Trust Company of Chicago
|
|
Filed herewith |
| |
|
|
|
|
|
|
| |
10.24 |
|
|
License Agreement, dated June 18, 1997,
between BioSante Pharmaceuticals, Inc.
and The Regents of the University of
California (2)
|
|
Incorporated by
reference to
Exhibit 10.1 to
BioSante’s
Registration
Statement on Form
10-SB, as amended
(File No. 0-28637) |
| |
|
|
|
|
|
|
| |
10.25 |
|
|
Amendment to License Agreement, dated
October 26, 1999, between BioSante
Pharmaceuticals, Inc. and the Regents
of the University of California (2)
|
|
Incorporated by
reference to
Exhibit 10.2 to
BioSante’s
Registration
Statement on Form
10-SB, as amended
(File No. 0-28637) |
| |
|
|
|
|
|
|
| |
10.26 |
|
|
Amendment No. 2 to the License
Agreement, dated May 7, 2001, between
BioSante Pharmaceuticals, Inc. and The
Regents of the University of California
(2)
|
|
Incorporated by
reference to
Exhibit 10.23 to
BioSante’s Annual
Report on Form
10-KSB for the
fiscal year ended
December 31, 2001
(File No. 0-28637) |
| |
|
|
|
|
|
|
| |
10.27 |
|
|
Third Amendment to the License
Agreement dated June 30, 2004, between
BioSante and The Regents of the
University of California (2)
|
|
Incorporated by
reference to
Exhibit 10.3 to
BioSante’s Form
10-QSB for the
fiscal quarter
ended June 30, 2004
(File No.
001-31812) |
| |
|
|
|
|
|
|
| |
10.28 |
|
|
Fourth Amendment to Exclusive License
Agreement for Selected Applications of
Coated Nanocrystalline Particles
between The Regents of the University
of California and BioSante
Pharmaceuticals, Inc. dated as of
August 11, 2006 (2)
|
|
Incorporated by
reference to
Exhibit 10.1 to
BioSante’s Form
10-Q for the fiscal
quarter ended
September 30, 2006
(File No.
001-31812) |
| |
|
|
|
|
|
|
| |
10.29 |
|
|
License Agreement, dated June 13, 2000,
between Permatec Technologie, AG (now
known as Antares Pharma, Inc.) and
BioSante Pharmaceuticals, Inc. (2)
|
|
Incorporated by
reference to
Exhibit 10.1 to
BioSante’s Current
Report on Form 8-K
as filed with the
Securities and
Exchange Commission
on July 11, 2000
(File No. 0-28637) |
100
| |
|
|
|
|
|
|
| Exhibit No. |
|
Exhibit |
|
Method of Filing |
| |
|
|
|
|
|
|
| |
10.30 |
|
|
Amendment No. 1 to the License
Agreement, dated May 20, 2001, between
Antares Pharma IPL AG and BioSante
Pharmaceuticals, Inc. (2)
|
|
Incorporated by
reference to
Exhibit 10.18 to
BioSante’s Annual
Report on Form
10-KSB for the
fiscal year ended
December 31, 2001
(File No. 0-28637) |
| |
|
|
|
|
|
|
| |
10.31 |
|
|
Amendment No. 2 to the License
Agreement, dated July 5, 2001, between
Antares Pharma IPL AG and BioSante
Pharmaceuticals, Inc. (2)
|
|
Incorporated by
reference to
Exhibit 10.19 to
BioSante’s Annual
Report on Form
10-KSB for the
fiscal year ended
December 31, 2001
(File No. 0-28637) |
| |
|
|
|
|
|
|
| |
10.32 |
|
|
Amendment No. 3 to the License
Agreement, dated August 30, 2001,
between Antares Pharma IPL AG and
BioSante Pharmaceuticals, Inc. (2)
|
|
Incorporated by
reference to
Exhibit 10.20 to
BioSante’s Annual
Report on Form
10-KSB for the
fiscal year ended
December 31, 2001
(File No. 0-28637) |
| |
|
|
|
|
|
|
| |
10.33 |
|
|
Amendment No. 4 to the License
Agreement, dated August 8, 2002,
between Antares Pharma IPL AG and
BioSante Pharmaceuticals, Inc. (2)
|
|
Incorporated by
reference to
Exhibit 10.20 to
BioSante’s
Registration
Statement on Form
SB-2, as amended
(File No.
333-87542) |
| |
|
|
|
|
|
|
| |
10.34 |
|
|
Amendment No. 5 to the License
Agreement, dated December 30, 2002
between Antares Pharma IPL AG and
BioSante Pharmaceuticals, Inc. (2)
|
|
Incorporated by
reference to
Exhibit 10.25 to
BioSante’s Annual
Report on Form
10-KSB for the
fiscal year ended
December 31, 2002
(File No. 0-28637) |
| |
|
|
|
|
|
|
| |
10.35 |
|
|
Amendment No. 6 to the License
Agreement, dated October 20, 2006
between Antares Pharma IPL AG and
BioSante Pharmaceuticals, Inc. (2)
|
|
Incorporated by
reference to
Exhibit 10.27 to
BioSante’s Form
10-K for the fiscal
year ended December
31, 2006 (File No.
001-31812) |
| |
|
|
|
|
|
|
| |
10.36 |
|
|
Form of Subscription Agreement dated as
of July 7, 2006 by and between BioSante
Pharmaceuticals, Inc. and each of the
subscribers party to the Subscription
Agreement
|
|
Incorporated by
reference to
Exhibit 10.1 to
BioSante’s Current
Report on Form 8-K
as filed with the
Securities and
Exchange Commission
on July 10, 2006
(File No.
001-31812) |
101
| |
|
|
|
|
|
|
| Exhibit No. |
|
Exhibit |
|
Method of Filing |
| |
|
|
|
|
|
|
| |
10.37 |
|
|
Form of Subscription Agreement dated as
of May 25, 2007 by and between BioSante
Pharmaceuticals, Inc. and each of the
subscribers party to the Subscription
Agreement
|
|
Incorporated by
reference to
Exhibit 10.1 to
BioSante’s Current
Report on Form 8-K
as filed with the
Securities and
Exchange Commission
on May 25, 2007
(File No.
001-31812) |
| |
|
|
|
|
|
|
| |
10.38 |
|
|
Common Stock Purchase Agreement dated
as of December 15, 2008 between
BioSante Pharmaceuticals, Inc. and
Kingsbridge Capital Limited
|
|
Incorporated by
reference to
Exhibit 10.1 to
BioSante’s Current
Report on Form 8-K
as filed with the
Securities and
Exchange Commission
on December 18,
2008 (File No.
001-31812) |
| |
|
|
|
|
|
|
| |
10.39 |
|
|
Amendment No. 1 to Common Stock
Purchase Agreement dated as of
March 24, 2010 between BioSante
Pharmaceuticals, Inc. and Kingsbridge
Capital Limited
|
|
Filed herewith |
| |
|
|
|
|
|
|
| |
10.40 |
|
|
Registration Rights Agreement dated as
of December 15, 2008 between BioSante
Pharmaceuticals, Inc. and Kingsbridge
Capital Limited
|
|
Incorporated by
reference to
Exhibit 10.2 to
BioSante’s Current
Report on Form 8-K
as filed with the
Securities and
Exchange Commission
on December 18,
2008 (File No.
001-31812) |
| |
|
|
|
|
|
|
| |
10.41 |
|
|
Amendment dated as of June 26, 2009
Registration Rights Agreement dated as
of December 15, 2008 between BioSante
Pharmaceuticals, Inc. and Kingsbridge
Capital Limited
|
|
Incorporated by
reference to
Exhibit 10.3 to
BioSante’s
Quarterly Report on
Form 10-Q for the
fiscal quarter
ended June 30, 2009
(File No.
001-31812) |
| |
|
|
|
|
|
|
| |
10.42 |
|
|
Form of Securities Purchase Agreement,
dated August 13, 2009, by and between
BioSante Pharmaceuticals, Inc. and each
of the investors in the offering
|
|
Incorporated by
reference to
Exhibit 10.1 to
BioSante’s Current
Report on Form 8-K
as filed with the
Securities and
Exchange Commission
on August 14, 2009
(File No.
001-31812) |
| |
|
|
|
|
|
|
| |
10.43 |
|
|
Form of Securities Purchase Agreement,
dated March 4, 2010, by and between
BioSante Pharmaceuticals, Inc. and each
of the investors in the offering
|
|
Incorporated by
reference to
Exhibit 10.1 to
BioSante’s Current
Report on Form 8-K
as filed with the
Securities and
Exchange Commission
on March 5, 2010
(File No.
001-31812) |
102
| |
|
|
|
|
|
|
| Exhibit No. |
|
Exhibit |
|
Method of Filing |
| |
|
|
|
|
|
|
| |
14.1 |
|
|
Code of Conduct and Ethics
|
|
Incorporated by
reference to
Exhibit 14.1 to
BioSante’s Form
10-KSB for the
fiscal year ended
December 31, 2003
(File No.
001-31812) |
| |
|
|
|
|
|
|
| |
23.1 |
|
|
Consent of Deloitte & Touche LLP
|
|
Filed herewith |
| |
|
|
|
|
|
|
| |
31.1 |
|
|
Certification of Chief Executive
Officer Pursuant to SEC Rule 13a-14
|
|
Filed herewith |
| |
|
|
|
|
|
|
| |
31.2 |
|
|
Certification of Chief Financial
Officer Pursuant to SEC Rule 13a-14
|
|
Filed herewith |
| |
|
|
|
|
|
|
| |
32.1 |
|
|
Certification of Chief Executive
Officer Pursuant to Rule 18 U.S.C.
Section 1350, as adopted pursuant to
Section 906 of the Sarbanes-Oxley Act
of 2002
|
|
Furnished herewith |
| |
|
|
|
|
|
|
| |
32.2 |
|
|
Certification of Chief Financial
Officer Pursuant to Rule 18 U.S.C.
Section 1350, as adopted pursuant to
Section 906 of the Sarbanes-Oxley Act
of 2002
|
|
Furnished herewith |
| |
|
|
| (1) |
|
All exhibits and schedules to this exhibit have been omitted pursuant to Item 601(b)(2) of
Regulation S-K. BioSante will furnish the omitted exhibits and schedules to the Securities and
Exchange Commission upon request by the Securities and Exchange Commission. |
| |
| (2) |
|
Confidential treatment under Rule 24b-2 of the Securities Exchange Act of 1934, as amended, has
been granted with respect to designated portions of this document. |
103