Attached files
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
| þ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2009
Commission file number: 1-33818
ENTEROMEDICS INC.
(Exact name of registrant as specified in its charter)
| Delaware | 48-1293684 | |
| (State or other jurisdiction of incorporation) | (IRS Employer Identification No.) |
2800 Patton Road, St. Paul, Minnesota 55113
(Address of principal executive offices, including zip code)
(651) 634-3003
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of Class |
Name of Exchange on Which Registered | |
| Common stock, $0.01 par value per share | The NASDAQ Capital Market |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No þ
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes ¨ No þ
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes þ No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ¨ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer ¨ |
Accelerated filer ¨ | |
| Non-accelerated filer þ (Do not check if a smaller reporting company) |
Smaller Reporting Company ¨ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No þ
At June 30, 2009, the last business day of the registrant’s most recently completed second fiscal quarter, the aggregate market value of the registrant’s common stock held by non-affiliates of the registrant, based upon the closing price of a share of the registrant’s common stock as reported by the NASDAQ Global Market on that date was $50,816,406.
As of February 26, 2010, 44,856,657 shares of the registrant’s Common Stock were outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Specified portions of the registrant’s Definitive Proxy Statement, which will be filed with the Commission pursuant to Regulation 14A in connection with the registrant’s 2010 Annual Meeting of Stockholders, to be held May 6, 2010 (the Proxy Statement), are incorporated by reference into Part III of this report. Except with respect to information specifically incorporated by reference in this report, the Proxy Statement is not deemed to be filed as a part hereof.
ENTEROMEDICS INC.
FORM 10-K
TABLE OF CONTENTS
| PART I | ||||
| Item 1. |
1 | |||
| Item 1A. |
22 | |||
| Item 1B. |
39 | |||
| Item 2. |
39 | |||
| Item 3. |
39 | |||
| Item 4. |
39 | |||
| PART II | ||||
| Item 5. |
40 | |||
| Item 6. |
43 | |||
| Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
44 | ||
| Item 7A. |
55 | |||
| Item 8. |
56 | |||
| Item 9. |
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
93 | ||
| Item 9A(T). |
93 | |||
| Item 9B. |
94 | |||
| PART III | ||||
| Item 10. |
95 | |||
| Item 11. |
95 | |||
| Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
95 | ||
| Item 13. |
Certain Relationships and Related Transactions, and Director Independence |
96 | ||
| Item 14. |
96 | |||
| PART IV | ||||
| Item 15. |
Exhibits, Financial Statements and Financial Statement Schedules |
97 | ||
| 98 | ||||
| EXHIBITS |
||||
Registered Trademarks and Trademark Applications: In the United States we have registered trademarks for VBLOC, ENTEROMEDICS and MAESTRO each registered with the United States Patent and Trademark Office, and have received a Notice of Allowance and third extension of time to file a Statement of Use on our application to register the mark EMPOWER. In addition, the marks VBLOC, MAESTRO and ENTEROMEDICS are the subject of either a trademark registration or application for registration in Australia, Brazil, China, Mexico, the European Community, Saudi Arabia, the United Arab Emirates and Switzerland. This Form 10-K contains other trade names and trademarks and service marks of EnteroMedics and of other companies.
PART I.
| ITEM 1. | BUSINESS |
This Annual Report on Form 10-K contains forward-looking statements. These forward-looking statements are based on our current expectations about our business and industry. In some cases, these statements may be identified by terminology such as “may,” “will,” “should,” “expects,” “could,” “intends,” “might,” “plans,” “anticipates,” “believes,” “estimates,” “predicts,” “potential,” or “continue,” or the negative of such terms and other comparable terminology. These statements involve known and unknown risks and uncertainties that may cause our results, levels of activity, performance or achievements to be materially different from those expressed or implied by the forward-looking statements. Factors that may cause or contribute to such differences include, among others, those discussed in this report in Item 1A “Risk Factors.” Except as may be required by law, we undertake no obligation to update any forward-looking statement to reflect events after the date of this report.
Overview
We are a development stage medical device company focused on the design and development of devices that use neuroblocking technology to treat obesity, its associated co-morbidities, and other gastrointestinal disorders. Our proprietary neuroblocking technology, which we refer to as VBLOC therapy, is designed to intermittently block the vagus nerve using high-frequency, low-energy, electrical impulses. The vagus nerve controls much of the activity of the stomach, intestines and pancreas and plays a role in food processing. Our initial product under development is the Maestro System, which uses VBLOC therapy to limit the expansion of the stomach, help control hunger sensations between meals, reduce the frequency and intensity of stomach contractions and produce a feeling of early and prolonged fullness. Based on our understanding of vagal nerve function and nerve blocking from our preclinical studies and the results of our initial clinical trials, we believe the Maestro System may offer obese patients a minimally-invasive treatment alternative that has the potential to result in significant and sustained weight loss. In addition, data from sub-group analyses demonstrate that VBLOC therapy may hold promise in improving the obesity-related co-morbidities of diabetes and hypertension, independent of, and prior to, substantial weight loss. We are conducting, or plan to conduct, feasibility studies in each of these co-morbidities to assess VBLOC therapy’s potential in addressing multiple indications.
We are currently evaluating the Maestro System in human clinical trials conducted in the United States, Australia, Mexico, Norway and Switzerland. To date, we have not observed any mortality or any unanticipated adverse device effects in these clinical trials. We have also not observed any long-term problematic clinical side effects in any patients, including in those patients who have been using the Maestro System for more than one year.
On October 2, 2009, we announced preliminary results from our pivotal clinical study, the EMPOWER trial; indicating that based on an initial analysis, the study did not meet its primary and secondary efficacy endpoints. We also announced that there were no therapy-related serious adverse events reported during the study. The EMPOWER trial is a multi-center, randomized, double-blind, prospective, placebo-controlled pivotal study being conducted in the United States and selected international centers. We further announced on November 12, 2009, the ongoing detailed review suggests that vagal blocking therapy may promote safe and effective weight loss as an adjunct to behavioral support, diet and exercise in morbidly obese patients. The review further suggests that these effects were evident in both the treatment and control arms. We are continuing a comprehensive analysis of all clinical, statistical, and engineering data to understand this finding. Based on the analysis to date, the control arm of the trial, which was intended to be inactive, apparently provided a low-intensity blocking signal that introduced VBLOC therapy in human subjects.
In January 2010, we met with the U.S. Food and Drug Administration (FDA) to discuss the EMPOWER trial results and the regulatory process going forward. Based on this discussion, we recently submitted an Investigational Device Exemption (IDE) application for a clinical trial using the next-generation Maestro RC
1
System in the treatment of morbid obesity. Assuming that we obtain an approved IDE, successfully enroll and implant the trial and achieve favorable results, we plan to use data from that trial to support a premarket approval (PMA) application for the Maestro System, which we expect to submit no earlier than the second half of 2012. If the FDA grants us approval, we anticipate we will be able to commercialize the Maestro System in the United States no earlier than the second half of 2013. In the event that the Maestro System receives FDA approval, we expect to recruit and retain personnel responsible for commercial operations, sales and marketing, customer service, reimbursement and technical service in order to support the commercial launch of our product. We will also need to increase production volumes of our products in connection with commercialization. We rely primarily on third-party manufacturers and suppliers to produce our products and will continue to select qualified suppliers and contract manufacturers that can supply products on a commercial scale according to our proprietary specifications.
Background—The Obesity Epidemic
Obesity has been identified by the U.S. Surgeon General as the fastest growing cause of disease and death in the United States. In 1980, approximately 15% of the adult population in the United States was obese according to National Health and Nutrition Examination Survey. By 2005, the incidence of obesity had more than doubled to 33%. Currently, the Centers for Disease Control and Prevention (CDC) estimates that there are more than 72 million obese adults in the United States, having a Body Mass Index (BMI) of 30 or higher. BMI is calculated by dividing a person’s weight in kilograms by the square of their height in meters. It is estimated that by 2015, over 40% of American adults could be obese. Obesity is also a significant health problem outside of the United States, with as many as 400 million people worldwide estimated to be obese and 1.6 billion adults estimated to be overweight, according to the World Health Organization. The World Health Organization estimates that by 2015, approximately 2.3 billion adults will be overweight and more than 700 million people worldwide will be obese.
The CDC has identified obesity as a leading public health threat in the United States and has estimated that there are approximately 112,000 obesity-related deaths each year in the United States. The World Health Organization has estimated that about 2.5 million deaths worldwide are attributed to people being overweight or obese. According to data from the U.S. Department of Health and Human Services, almost 80% of adults with a BMI above 30 have an obesity-related disease or disorder, also called a co-morbidity, and almost 40% have two or more of these co-morbidities. According to the North American Association for the Study of Obesity and the CDC, obesity is associated with many significant weight-related co-morbidities including Type 2 diabetes, high blood-pressure, sleep apnea, certain cancers, high cholesterol, coronary artery disease, osteoarthritis and stroke. In addition, a number of disorders involving the central nervous system may also be complicated by obesity, such as anxiety, bipolar disorder, agoraphobia, depression and insomnia. As of 2000, the Department of Health and Human Services estimated the overall economic costs of obesity in the United States to be $117 billion per year. In an abstract sponsored and co-authored by the CDC, it was noted that in 2008 these costs could have risen to $147 billion per year.
We believe that the obesity epidemic will continue to grow worldwide given dietary trends in developed nations that favor highly processed sugars, larger meals and fattier foods, as well as increasingly sedentary lifestyles. Despite the growing obesity rate, increasing public interest in the obesity epidemic and significant medical repercussions and economic costs associated with obesity, there continues to be a significant unmet need for more effective treatments. We believe existing options for the treatment of obesity have seen limited adoption to date due to a range of efficacy and potential side effects including morbidity. The principal treatment alternatives available today for obesity include:
| • | Behavioral modification. Behavioral modification, which includes diet and exercise, is an important component in the treatment of obesity; however, most obese patients find it difficult to achieve and maintain significant weight loss with a regimen of diet and exercise alone. |
| • | Pharmaceutical therapy. Pharmaceutical therapies often represent a first option in the treatment of obese patients within lower BMI ranges but carry significant safety risks and may present troublesome side effects. |
2
| • | Bariatric surgery. In more severe cases of obesity, patients may pursue more aggressive surgical treatment options such as gastric bypass, sleeve gastrectomy and gastric banding. These procedures promote weight loss by surgically restricting the stomach’s capacity and outlet size. While largely effective, they may present substantial side effects and carry short- and long-term safety risks that have limited their adoption. |
Given the limitations of behavioral modification, pharmaceutical therapy and bariatric surgical approaches, we believe there is a substantial need for a safer and more effective solution that:
| • | preserves normal anatomy; |
| • | is “non-punitive” in that it supports continued ingestion and digestion of foods and micronutrients such as vitamins and minerals found in a typical, healthy diet while allowing the user to modify his or her eating behavior appropriately without inducing punitive physical restrictions that physically force a limitation of food intake; |
| • | enables non-invasive adjustability while reducing the need for frequent clinic visits; |
| • | minimizes unpleasant side-effects such as persistent vomiting; |
| • | minimizes the risks of re-operations, malnutrition and mortality; and |
| • | reduces the natural hunger drive of patients. |
EnteroMedics’ Solution
We are designing our Maestro System to address many of the unmet needs of physicians and patients for an effective long-term obesity treatment that minimizes the complications presented by existing alternatives. The Maestro System delivers VBLOC therapy, which we believe is the first therapy of its kind for the treatment of obesity using neuroblocking. VBLOC therapy interrupts nerve signals along the vagus nerve to selectively block the gastrointestinal effects of the vagus nerve, unlike neurostimulation, which attempts to increase neural activity through stimulation to impact the digestive system.
The Vagus Nerve and the Digestive System
Beginning in the brain, the vagus nerve travels down alongside the esophagus to the stomach and other gastrointestinal organs and is primarily responsible for autonomic regulation involved in heart, lung and gastrointestinal function. The vagus nerve controls much of the activity of the stomach, intestine and pancreas and plays a role in food processing, including:
| • | expansion of the stomach as food enters; |
| • | contractions of the stomach to break food into smaller particles; |
| • | release of gastric acid required for food processing; |
| • | emptying of the stomach contents into the small intestine; |
| • | secretion of digestive pancreatic enzymes that enable absorption of calories; and |
| • | controlling sensations of hunger, satisfaction and fullness. |
VBLOC Therapy
Several studies of the vagus nerve and its effect on the digestive system have focused on the effects of surgical vagotomy, the permanent severing of the vagus nerve at the level of the junction between the esophagus and the stomach. Given the role of the vagus nerve in regulating the release of gastric acid, early researchers originally used vagotomy as a treatment for peptic ulcers. They discovered that their patients often experienced
3
weight loss or, at a minimum, failure to gain weight following vagotomy. However, weight loss after vagotomy alone has been disappointing, particularly over the long-term and likely dissipates as the body compensates for the anatomical disruption by partial restoration of nervous system function.
VBLOC therapy is designed to block the gastrointestinal effects of the vagus nerve by using high-frequency, low-energy electrical impulses to intermittently interrupt naturally occurring neural impulses on the vagus nerve between the brain and the digestive system. Our therapy is designed to control hunger sensations between meals, limit the expansion of the stomach and to reduce the frequency and intensity of stomach contractions. In addition, we believe VBLOC therapy also reduces the absorption of calories by decreasing the secretion of digestive enzymes. The resulting physiologic effects of VBLOC therapy are intended to produce a feeling of early and prolonged fullness following smaller meal portions. By intermittently blocking the vagus nerve and allowing it to return to full function between therapeutic episodes, we believe we have limited the body’s natural tendency to circumvent the therapy, which can result in long-term weight loss.
We have designed our Maestro System to address a significant market opportunity that we believe exists for a safe, effective and less-invasive therapy that is intended to address the underlying causes of hunger and obesity. Our Maestro System is designed to offer each of the following benefits, which we believe could lead to the adoption of VBLOC as the therapy of choice for obesity:
| • | Preserves Normal Anatomy. The Maestro neuroblocking pulse generator is designed to deliver therapy that blocks the neural signals that influence a patient’s hunger and sense of fullness without altering digestive system anatomy. Accordingly, patients should experience fewer and less severe side effects compared to treatments that incorporate anatomical alterations. |
| • | Allows Continued Ingestion and Digestion of Foods Found in a Typical, Healthy Diet. Because our therapy leaves the digestive anatomy unaltered, we believe that patients will be able to maintain a more consistent nutritional balance compared to existing surgical approaches, thus allowing them to effect positive changes in their eating behavior in a non-forced and potentially more consistent way. |
| • | May be Implanted on an Outpatient Basis and Adjusted Non-Invasively. The Maestro System is designed to be laparoscopically implanted in approximately one hour, allowing patients to leave the hospital or clinic on the same day. The implantable system is designed to be turned off and left in place for patients who reach their target weight. When desired, the follow-up physician can simply and non-invasively turn the therapy back on. Alternatively, the implantable system can be removed in a laparoscopic procedure. |
| • | Offers Favorable Safety Profile. We have designed our EMPOWER clinical trial to demonstrate the safety of the Maestro System. In our clinical trials to date, including the EMPOWER trial, we have not observed any mortality or any medically serious device related adverse events that have required surgical attention in the patients we have implanted with the Maestro System. We have also not observed any long-term problematic clinical side effects in any patients, including in those patients who have been using the Maestro System for more than one year. |
| • | Targets Multiple Factors that Contribute to Hunger and Obesity. We designed VBLOC therapy to target the multiple digestive functions of the vagus nerve and to affect the perception of hunger and fullness, which together contribute to obesity. |
VBLOC therapy, delivered via our Maestro System, is intended to offer patients what we believe could be an effective, safe, outpatient solution that minimizes complications. We believe that if approved it could enable patients to lose weight and maintain long-term weight loss while enjoying a normal, healthy diet. We also believe that the Maestro System, if approved, will appeal to physicians based on the inherent physiological approach of VBLOC therapy and its anticipated favorable safety profile.
4
Our Strategy
Our goal is to establish VBLOC therapy, delivered via our Maestro System pulse generator, as the leading obesity management solution. The key business strategies by which we intend to achieve these objectives include:
Achieve Regulatory Approval for VBLOC Therapy Using Our Maestro System. We received an IDE from the FDA for use of the Maestro System in the United States in our EMPOWER trial, and announced on October 2, 2009 that based on an initial analysis, the study did not meet its primary and secondary efficacy endpoints. We further announced on November 12, 2009, the ongoing detailed review suggests that vagal blocking therapy may promote safe and effective weight loss as an adjunct to behavioral support, diet and exercise in morbidly obese patients. The review further suggests that these effects were evident in both the treatment and control arms. We are continuing a comprehensive analysis of all clinical, statistical, and engineering data to understand this finding. Based on the analysis to date, the control arm of the trial, which was intended to be inactive, apparently provided a low-intensity blocking signal that introduced VBLOC therapy in human subjects. After meeting with the FDA in January 2010 to discuss the EMPOWER trial results and the regulatory process going forward, we recently submitted an IDE application for a clinical trial using the next-generation Maestro RC System in the treatment of morbid obesity. Assuming that we obtain an approved IDE, successfully enroll and implant the trial and achieve favorable results, we plan to use data from that trial to pursue a PMA from the FDA to allow us to commence sales in the United States. We have also received the European CE Mark for our Maestro RF System to enable the eventual commercialization of our systems in the European Economic Area. We also plan to use our CE Mark certification to make other international regulatory filings to permit sales in those jurisdictions.
Drive the Adoption and Endorsement of VBLOC Therapy Through Obesity Therapy Experts. Our clinical development strategy is to collaborate closely with regulatory bodies, obesity therapy experts and scientific experts. We have established credible and open relationships with obesity therapy experts and scientific experts and we believe these obesity therapy experts and scientific experts will be important in promoting patient awareness and gaining widespread adoption if the Maestro System is approved and commercialized.
Commercialize Our Products using a Direct Sales and Marketing Effort. We plan to build a sales force to call directly on key opinion leaders and bariatric surgeons, primarily within bariatric Centers of Excellence. We believe this currently represents approximately 378 facilities within the United States, which we believe will enable us to target them effectively with a small sales force. We expect that our direct sales force will promote the Maestro System to physicians and patients who have concerns with current bariatric surgical procedures. We also plan to call on physicians, weight-management specialists and nurses who influence patient adoption.
Identify Appropriate Coding, Obtain Coverage and Payment for the Maestro System. While payors are not our direct customers, their coverage and reimbursement policies influence patient and physician selection of obesity treatment. We plan to employ a focused campaign to obtain payor support for VBLOC therapy. We plan to seek specific and appropriate coding, coverage and payment for our Maestro System from the Centers for Medicare and Medicaid Services (CMS) and from private insurers.
Expand and Protect Our Intellectual Property Position. We believe that our issued patents and our patent applications encompass a broad platform of neuromodulation therapies, including vagal blocking and combination therapy focused on obesity and other gastrointestinal disorders. We intend to continue to pursue further intellectual property protection through U.S. and foreign patent applications.
Leverage our VBLOC Technology for Other Disease States. We intend to continue to conduct research and development for other potential applications for our VBLOC therapy and believe we have a broad technology platform that will support the development of additional clinical applications and therapies for other gastrointestinal disorders in addition to obesity.
5
The Maestro System, Implantation Procedure and Usage
The Maestro System. Our Maestro System pulse generator delivers VBLOC therapy via two small electrodes that are laparoscopically implanted and placed in contact with the trunks of the vagus nerve just above the junction between the esophagus and the stomach, near the diaphragm. We are developing the Maestro System in two different energy configurations, the Maestro RF System and the Maestro RC System. The Maestro RF System is the device currently being used in our EMPOWER trial, which we announced on October 2, 2009 did not meet its primary and secondary efficacy endpoints.
The major components of the Maestro System include:
| • | Neuroregulator. The neuroregulator, sometimes referred to as a neuroblocking pulse generator, is an implanted device that controls the delivery of VBLOC therapy to the vagus nerve. It is surgically implanted just below, and parallel to, the skin, typically on the side of the body over the ribs. The neuroregulator emits short, charge-balanced electrical pulses at a high pulse rate that travel down the leads to the electrodes and intermittently block natural nerve signals on the vagus nerve. |
| • | Lead system. Our neuroblocking leads are powered by the neuroregulator and deliver electrical pulses to the vagus nerve via the electrodes. The leads and electrodes are similar to those used in traditional cardiac rhythm management and some neurostimulation products, are intended to be implanted and may be removed laparoscopically. |
| • | Controller/Mobile charger. Our controller regulates the rate and intensity of the electrical pulses delivered by the neuroregulator and maintains a log of device and treatment changes. In the Maestro RF System, the controller is an external unit. In the Maestro RC System, the external controller is replaced by an external mobile charger and the control logic is contained within the implanted neuroregulator. |
| • | Transmit coil. The transmit coil is positioned over the implanted neuroregulator and delivers radiofrequency energy and therapy control information across the skin into the device. The coil is held in position over the neuroregulator using either an adhesive or an adjustable elastic belt worn around the torso. |
| • | Clinician programmer. The clinician programmer connects to the controller to enable clinicians to customize therapy settings as necessary and download reports stored in system components. The reports include patient use and system performance information used to manage therapy. The clinician programmer incorporates our proprietary software and is operated with a commercially available laptop computer. |
The Maestro RF System and the Maestro RC System differ in the following ways:
| • | The neuroblocking pulse generator, or neuroregulator, within the Maestro RF System is powered by a battery in the externally-worn controller, which is connected to the external transmit coil. The transmit coil needs to be properly positioned over the approximately 20 cubic centimeter neuroregulator and worn daily during the patient’s waking hours to deliver therapy. The controller is recharged nightly using AC wall power. |
| • | The neuroregulator in the Maestro RC System is powered by an internal rechargeable battery. The RC neuroregulator is approximately 80 cubic centimeters in volume to accommodate its internal battery. An external mobile charger is connected to the external transmit coil to recharge the battery. The mobile charger is recharged using AC wall power. |
We intend to evaluate each system as part of our clinical trial plan.
Implantation Procedure. The Maestro System is designed to be implanted by a bariatric surgeon in approximately one hour during an outpatient procedure that will be typically performed using a short-acting general anesthetic. During the procedure, the surgeon laparoscopically implants the electrodes in contact with the
6
vagal nerve trunks and then connects the lead wires to the neuroregulator. After the electrodes have been attached adjacent to the vagal trunks and connected to the neuroregulator, the surgeon confirms final system operation by sending electrical pulses to the leads by the neuroregulator. Once system operation has been confirmed, the surgeon implants the neuroregulator under the skin and closes all incisions. We believe that patients who are implanted with the Maestro System will be able to return home from the hospital or clinic on the same day. The implantation procedure and usage of the Maestro System carry some risks, such as the risks generally associated with laparoscopic procedure as well as the possibility of device malfunction. In addition, in rare circumstances during implantation, the vagus nerve or esophagus may be damaged causing problems such as difficulty in swallowing, vomiting, heartburn, belching, abdominal fullness or discomfort, diarrhea, or decreased appetite. We expect that any of these problems would be temporary without lasting effects, although there is the risk of permanent injury to the vagus nerve. Some post-operative effects that may occur after implantation of our Maestro System include movement of the leads or neuroregulator from their original positions, erosion or wire breakage and potential allergic reaction with internal or external device contacts.
Usage of the Maestro System. The physician activates the Maestro System after an approximate two-week healing period following implantation. VBLOC therapy is then delivered intermittently each day during the patient’s waking hours through the neuroregulator. The scheduled delivery of the intermittent electrical pulses blocking the vagus nerve is customized for each patient by the physician using the clinician programmer and when necessary, therapy can also be easily and non-invasively modified by the physician. The physician determines the duration of the therapy in consultation with the patient based on the patient’s weight loss and overall treatment objectives. Patients using the Maestro RF System can elect to suspend or circumvent therapy at any time by simply not carrying the controller. Without the controller, the RF neuroregulator receives no power and cannot provide therapy. Patients using the Maestro RC System are more limited in their ability to suspend or circumvent therapy because the control logic is embedded in the implanted neuroregulator.
The physician is able to download reports to monitor patient use and system performance information. This information is particularly useful to physicians to ensure that patients are properly using the system. Although usage of our Maestro System generally proceeds without complications, as part of the therapy or intentional weight loss, subjects in our clinical trials have observed side-effects such as heartburn, bloating, diarrhea, sweating, nausea, constipation, greasy bowel movements, tiredness and excessive feelings of fullness, especially after meals. In addition, patient noncompliance with wearing the external components of the Maestro RF System may render VBLOC therapy less effective in achieving long-term weight loss.
Clinical Development
We are developing our Maestro System to deliver VBLOC therapy for the long-term treatment of obesity. Based on our preliminary preclinical and clinical findings, we believe that our Maestro System has the potential to offer a compelling combination of efficacy and safety. We are continuing to evaluate the Maestro System in human clinical studies conducted in the United States and internationally. We announced on October 2, 2009 that based on an initial analysis, our EMPOWER trial did not meet its primary and secondary efficacy endpoints. We also announced that there were no therapy-related serious adverse events reported during the study. After meeting with the FDA in January 2010 to discuss the EMPOWER trial results and the regulatory process going forward, we recently submitted an IDE application for a clinical trial using the next-generation Maestro RC System in the treatment of morbid obesity.
Preclinical Experience
We have completed several preclinical animal studies, primarily in pigs and rats, to evaluate the safety of our Maestro System and to refine our implantation procedure. These studies have also shown that VBLOC therapy could completely block activated nerve signals, with the nerve regaining normal function within minutes after each intermittent application of therapy. Over a 12-week period of VBLOC therapy, over 91% of all nerve axons showed normal histology and the animals demonstrated unimpaired heart rate, respiration, blood pressure
7
and glucose regulation. Additionally, we observed that VBLOC therapy resulted in a greater than 80% reduction in pancreatic exocrine secretions, which are composed of digestive enzymes, water and bicarbonate that facilitate food digestion and caloric intake.
As a result of the findings of our preclinical studies, we were able to refine the implant technique, demonstrate the biocompatibility of our Maestro System in animals and collect the data necessary to begin human clinical trials. Several publications resulting from these preclinical studies were peer-reviewed and accepted for podium presentation at the Digestive Disease Week meeting in 2006, the American Society for Bariatric Surgery meeting in 2006 and the International Federation for Surgery of Obesity meeting in 2006.
Clinical Experience
We began evaluating VBLOC therapy with our initial Maestro System, the RF1 system, in a clinical trial in February 2006. The next generation RF2 system is distinguished from the RF1 system by an improved user interface, improvements in the energy management within the neuroregulator and a more robust transmission link for delivering energy from the coil to the neuroregulator in the RF2 system. Our early clinical experience has shown that VBLOC therapy using the Maestro System offers physicians a programmable method to selectively and reversibly block the vagus nerve and results in clinically and statistically significant excess weight loss. Excess weight represents the difference between a subject’s actual weight and the subject’s weight assuming a BMI of 25, which is considered healthy. Excess weight loss (EWL) is reported as the percentage of excess weight that is lost by the subject.
We have not observed any mortality or any medically serious device related adverse events in any of our completed or ongoing studies. Reported events include those associated with laparoscopic surgery or any implantable electronic device. The effects of VBLOC therapy include changes in appetite, and, in some subjects, effects that may be expected with decreased intra-abdominal vagus nerve activity, such as temporary abdominal discomfort and short episodes of belching, bloating, cramping or nausea.
Findings from our clinical feasibility trials have resulted in more than 20 publications peer-reviewed and accepted for presentation between 2006 and 2009 at the following meetings: Digestive Disease Week, American Society for Metabolic and Bariatric Surgery, International Federation for Surgery of Obesity, Obesity Surgery Society of Australia & New Zealand and Obesity Society (formerly the North American Association for the Study of Obesity).
We used our clinical studies data in a submission to our Notified Body and obtained European CE Mark approval for our Maestro RF System on March 4, 2009.
Below is a summary of our planned and ongoing clinical studies.
VBLOC-RF2 Trial
Enrollment of 38 subjects in the VBLOC-RF2 trial began in November 2006 and is designed to evaluate the safety and efficacy of the Maestro RF2 System in treating patients with obesity over a period of 60 months. The trial is an international, open-label, prospective, multi-center study. We are implementing weight management programs and plan to evaluate the efficacy of VBLOC therapy by measuring average percentage EWL at one month, three, six and 12 months and possibly longer. We are using results from this trial to further optimize selection of VBLOC therapy parameters. Preliminary data indicate that the RF2 system improvements have resulted in improved therapy delivery and improved weight loss. To date, no deaths or medically serious device related adverse events have been reported during the VBLOC-RF2 trial. As of January 12, 2009, the most recent follow-up of nine RF2 patients, among the earliest patients implanted in the VBLOC-RF2 trial, showed an EWL of 37.6% at 18 months of VBLOC therapy. At that time, the most recent results for the prior follow-up periods demonstrated an EWL of 28.1% in 17 RF2 patients at 12 months and an EWL of 17.9% in 35 RF2 patients at six months of VBLOC therapy.
8
VBLOC-RC Trial
We initiated the VBLOC-RC trial in November 2007. The trial is an international feasibility study designed to demonstrate that the clinical performance of the Maestro RC System in five subjects is similar to that of the RF2 system. It is also intended to demonstrate that the subject can effectively recharge the implanted RC device and the physician and staff can perform device programming and operation. We are implementing weight management programs such as diet, behavior modification or exercise programs and plan to evaluate system performance and efficacy by measuring average percentage EWL at one, three and six months. To date, no deaths or medically serious device related adverse events have been reported during the VBLOC-RC trial.
VBLOC-DM2 ENABLE Trial
Enrollment of the VBLOC-DM2 ENABLE trial began in the second quarter of 2008 and is designed to evaluate the effects of VBLOC therapy on glucose regulation and blood pressure using the Maestro RC2 System in approximately 30 subjects. The trial is an international, open-label, prospective, multi-center study. We plan to evaluate the efficacy of VBLOC therapy by measuring average percentage EWL, HbA1c and FPG and blood pressure at one week, one month, three, six and 12 months and possibly longer. To date, no deaths or medically serious device related adverse events have been reported during the VBLOC-DM2 ENABLE trial and the safety profile is similar to that seen in the other VBLOC trials. As of January 14, 2010 the most recent follow-up of patients shows an average device usage of approximately 14 hours per day and the below data.
Change in HbA1c (Baseline 7.7%)
| Visit (post-device activation) |
HbA1c Change |
Percent HbA1c |
n | p (compared to baseline) | ||||
| Week 1 |
-0.3 | 7.4 | 19 | 0.0206 | ||||
| Week 4 |
-0.7 | 7.0 | 19 | 0.0002 | ||||
| Week 12 |
-0.9 | 6.8 | 18 | 0.0019 | ||||
| 6 Months |
-0.8 | 6.9 | 19 | 0.0062 |
Percent EWL (BMI Method from Implant)
| Visit (post-device activation) |
EWL | n | p (compared to baseline) | ||||
| Week 12 |
23.6 | % | 18 | <0.0001 | |||
| 6 Months |
26.4 | % | 19 | <0.0001 |
Change in Mean Arterial Pressure (MAP) in Hypertensive Subjects in mmHg (Baseline 99.5 mmHg, average)
| Visit (post-device activation) |
MAP Change |
n | p (compared to baseline) | |||
| Week 1 |
-7.8 | 10 | 0.0102 | |||
| Week 4 |
-12.3 | 10 | 0.0046 | |||
| Week 12 |
-9.9 | 9 | 0.0007 | |||
| 6 Months |
-12.9 | 10 | 0.0018 |
EMPOWER Trial
On October 2, 2009, we announced preliminary results from our pivotal clinical study, the EMPOWER trial; indicating that based on an initial analysis, the study did not meet its primary and secondary efficacy endpoints. We also announced that there were no therapy-related serious adverse events reported during the study. The
9
EMPOWER trial is a multi-center, randomized, double-blind, prospective, placebo-controlled pivotal study including a maximum of 300 subjects at up to 15 U.S. and international sites. We completed enrollment and implantation of 294 subjects in the EMPOWER trial in 2008.
We further announced on November 12, 2009, the ongoing detailed review suggests that vagal blocking therapy may promote safe and effective weight loss as an adjunct to behavioral support, diet and exercise in morbidly obese patients. The review further suggests that these effects were evident in both the treatment and control arms with overall study results showing that for all patients (n=253), the average EWL at 12 months was 16.6% EWL (BMI) from implant (12.1% from initiation, MetLife) for the treatment arm and 16.4% EWL (BMI) from implant (12.0% from initiation, MetLife) for the control arm. We are continuing a comprehensive analysis of all clinical, statistical, and engineering data to understand this finding. Based on the analysis to date, the control arm of the trial, which was intended to be inactive, apparently provided a low-intensity blocking signal that introduced VBLOC therapy in human subjects.
It is our belief after continuing to analyze the EMPOWER trial data that there is a direct correlation between weight loss and hours of daily device usage. On January 14, 2010 we announced the below observations and additional data from our ongoing detailed review of the EMPOWER trial.
Weight loss corresponded directly to hours of use for patients in the treatment arm. At 12 months, results were as follows:
| <6 Hours /Day |
Greater-than or Equal to 6 and <9 Hours/Day |
Greater-than or Equal to 9 and <12 Hours/Day |
Greater-than or Equal to 12 Hours/Day |
|||||||||
| Percent EWL (BMI Method) |
4.7 | % | 12.9 | % | 21.5 | % | 29.5 | % |
Weight loss corresponded directly to hours of use when both the treatment and control arms are combined. At 12 months, results were as follows:
| 12 Months from Implant (BMI Method) |
Greater-than or Equal to 9 Hours/Day (n=128) |
<9 Hours/Day (n=125) |
p | |||||
| Subjects Achieving Greater-than or Equal to 25% EWL |
39.1 | % | 12.0 | % | <0.0001 | |||
| Average Daily Use in Subjects |
11.2 hrs | 7.7 hrs | <0.0001 |
The purpose of the EMPOWER trial was to measure the safety and efficacy of our Maestro System in obese subjects after 12 months of VBLOC therapy. After all subjects completed 12 months of follow up, the trial was unblinded and all subjects, including those in the control group, had the option to receive ongoing VBLOC therapy. Subjects will continue to be followed out to 60 months as part of the trial and we will continue to monitor average percentage EWL and safety during this extended period.
Next Generation Maestro RC System Trial
In January 2010, we met with the FDA to discuss the EMPOWER trial results and the regulatory process going forward. Based on this discussion, we recently submitted an IDE application for a clinical trial using the next-generation Maestro RC System in the treatment of morbid obesity. Assuming that we obtain an approved IDE, successfully enroll and implant the trial and achieve favorable results, we plan to use data from that trial to support a PMA application for the Maestro System, which we expect to submit no earlier than the second half of 2012. If the FDA grants us approval, we anticipate we will be able to commercialize the Maestro System in the United States no earlier than the second half of 2013.
10
Research and Development
We have an experienced research and development team, including clinical, regulatory affairs and quality, comprised of scientists, electrical engineers, software engineers and mechanical engineers with significant clinical knowledge and expertise. Our research and development efforts are focused in the following major areas:
| • | identifying the effect of vagal blocking on nerve and organ function; |
| • | developing the Maestro System; and |
| • | investigating the Maestro platform for gastrointestinal disorders in addition to obesity. |
We have spent a significant portion of our capital resources on research and development. Our research and development expenses were $15.6 million in 2009, $27.7 million in 2008 and $21.1 million in 2007. Our research and development expenditures in 2010 and beyond will largely depend on our regulatory path forward. If the FDA grants us approval on an IDE application for a clinical trial using the next-generation Maestro RC System in the treatment of morbid obesity we would expect research and development expenditures to increase in support of a new clinical trial in addition to the continued follow-up on existing trials, such as VBLOC-DM2 ENABLE and EMPOWER.
Other Diseases and Disorders
We believe that our VBLOC therapy may have the potential, if validated through appropriate clinical studies, to treat a number of additional gastrointestinal disorders or co-morbidities frequently associated with obesity, including the following:
| • | Type 2 Diabetes. Metabolic syndrome refers to a group of risk factors for cardiovascular disease and Type 2 diabetes mellitus and affects an estimated 50 million people in the United States. We believe that VBLOC therapy has significant potential in treating metabolic syndrome. We have launched an international feasibility trial, VBLOC-DM2 ENABLE, to further explore the efficacy of VBLOC therapy in this patient population and have reported preliminary findings in the “Clinical Development” section above. |
| • | Hypertension. Blood pressure normally rises and falls throughout the day. When it consistently stays too high for too long, it is called hypertension. It is estimated that one out of three American adults has high blood pressure or hypertension. We believe that VBLOC therapy may improve mean systolic and diastolic blood pressure in hypertensive patients. We have included an evaluation of the blood pressure effects of VBLOC therapy in our international feasibility trial, VBLOC-DM2 ENABLE, to further explore the efficacy of VBLOC therapy in this patient population and have reported preliminary findings in the “Clinical Development” section above. |
| • | Pancreatitis. Primary and recurrent cases of acute pancreatitis are estimated to number from 150,000 to 200,000 annually, resulting in approximately 80,000 hospital admissions each year in the United States. In animal and human studies, we have shown that VBLOC therapy suppresses pancreatic exocrine secretion, suggesting its potential efficacy in treating pancreatitis. |
| • | Other Gastrointestinal Disorders. We believe that VBLOC therapy may have potential in a number of other gastrointestinal disorders, including irritable bowel syndrome and inflammatory bowel disease. |
Mayo Clinic Relationship
Our research and development team works with clinicians from Mayo Clinic Rochester, Minnesota pursuant to exclusive know-how, license, and consulting agreements. Mayo clinicians with multiple specialties such as bariatric surgery, gastroenterology and laparoscopic surgery consult with our research and development team on an exclusive basis to advise us as we develop our devices for vagal blocking therapy to treat obesity. Specifically, Mayo clinicians, along with other of our consultants, have offered their expertise to advise us with regard to our clinical trials and surgical techniques for our implantation procedure and participate on our medical advisory board and therapeutic algorithm panel. The agreements with Mayo Clinic also include a similar collaboration for
11
the development of products to address a wide variety of disorders susceptible to treatment by electrically blocking neural impulses on the vagus nerve. We retain the exclusive rights to obesity-related device inventions developed through this collaboration. We have also licensed-in two obesity-related patent applications from Mayo Clinic. These patent applications cover a number of medical device concepts for treating obesity, all of which are unrelated to our VBLOC technology. The five-year agreement entered into with the Mayo Clinic in 2005 was extended for two additional years effective February 3, 2010.
Medical Advisors
In addition to our collaboration with Mayo Clinic, we also have medical advisors who provide strategic guidance to our development programs, consult with us on clinical investigational plans and individual study protocols, and advise on clinical investigational site selection. Members of our medical advisory group also:
| • | serve on our Data Safety Monitoring Board and Clinical Events Committee; |
| • | meet with governmental regulatory authorities; |
| • | provide consultation on professional meeting presentations and journal manuscript submissions; and |
| • | develop and participate in clinical site training programs, including study surgical technique training and study subject follow-up training. |
Sales and Marketing
We currently do not have a sales organization and have no experience as a company in the marketing, sale or distribution of our proposed products. In the event that the Maestro System receives FDA approval, we expect to recruit and retain personnel responsible for commercial operations, sales and marketing, customer service, reimbursement and technical service in order to support the commercial launch of our product.
Finally, we expect that account management and patient registration processes used during the clinical trial will be transitioned to commercial registration structure. Centers responsible for implanting our product will be expanded, and trained to perform the patient selection, implant and manage appropriate follow-up procedures.
Initially, we anticipate that our sales representatives will exclusively target selected bariatric surgery Centers of Excellence and nationally recognized bariatric surgery centers. To be approved as a bariatric surgery Center of Excellence, a surgery center needs to perform a minimum of 125 bariatric surgical procedures per year. As of December 31, 2009, there were approximately 378 bariatric surgery Centers of Excellence approved by the Surgical Review Corporation and 75 Level I Centers of Excellence approved by the American College of Surgeons. In addition we expect to market our products to a small number of nationally-recognized hospitals that do not intend to pursue the Center of Excellence certification.
We plan to support our sales representatives with field clinical experts who will be responsible for training and support at various implant centers. We also expect that our sales representatives will spend time implementing joint consumer marketing programs with surgical centers and implanting surgeons. We also intend to market to potential referral source clinicians such as general practitioners, internists, endocrinologists and nurses.
The primary focus of our sales efforts will be in the United States. Outside of the United States, we may sell and support our products either through direct sales or medical device distributors. We plan to target countries with reasonable regulatory and reimbursement barriers and a population interested in managing their obesity. Each country we target will require specific regulatory approval from the local government or agency. In some situations, we may be able to rely on FDA approval, European CE Mark or ISO quality certificates to satisfy local regulatory requirements.
12
To achieve commercial success for any product that receives regulatory approval, we must either develop a sales organization or enter into arrangements with others to sell our products. Developing a direct sales force can be expensive and time consuming and can delay the success of any product launch. Any sales force we develop will likely be competing against the experienced and well-funded sales and marketing operations of our competitors.
Competition
We compete primarily in the market for obesity treatment with surgical obesity procedures and various devices used to implement neurostimulation and gastric stimulation systems. We also compete with pharmaceutical therapies. The market for obesity treatments is intensely competitive, subject to rapid technological change and significantly affected by new product development. Although we expect to compete in the market for gastric stimulation systems and other neurotechnology devices that treat obesity, there are currently no FDA-approved neuromodulation or neuroblocking therapies for the treatment of obesity. We believe we are the first and only company currently pursuing neuroblocking therapy for the treatment of obesity.
We also compete against the manufacturers of pharmaceuticals that are directed at treating obesity. We are aware of two drugs that are approved for long-term treatment of obesity in the United States: Sibutramine, marketed by Abbott Labs as Meridia, and Orlistat, marketed by Roche as Xenical and GlaxoSmithKline as alli. In addition, numerous pharmaceutical companies are working on additional drug therapies that may prove effective in addressing obesity.
We compete with several private early-stage companies developing neurostimulation devices for application to the gastric region and related nerves for the treatment of obesity. These companies may prove to be significant competitors, particularly through collaborative arrangements with large and established companies. They also compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and subject registration for clinical trials, as well as in acquiring technologies and technology licenses complementary to our programs or advantageous to our business.
In addition, there are many larger potential competitors experimenting in the field of neurostimulation to treat various diseases and disorders. For example, Medtronic, which develops deep brain stimulators and spinal cord stimulators, acquired TransNeuronix, which sought to treat obesity by stimulating the smooth muscle of the stomach wall and nearby tissue. St. Jude Medical, through its acquisition of Advanced Neuromodulation Systems, is developing spinal cord stimulators. Cyberonics is developing vagus nerve stimulators to modulate epileptic seizures and other neurological disorders. Boston Scientific, through its Advanced Bionics division, is developing neurostimulation devices such as spinal cord stimulators and cochlear implants. Ethicon-Endo Surgery acquired Cyberonics’ patents and patent applications pertaining to vagus nerve stimulation for the treatment of obesity and two related co-morbidities, diabetes and hypertension, in overweight patients.
In addition to competition from developers of neurostimulation and gastric modulation systems, we expect our Maestro System will also compete with surgical obesity procedures, including gastric bypass, gastric banding, vertical-banded gastroplasty and biliopancreatic diversion. The leader in the field of gastric banding is Allergan, whose Lap-Band received FDA approval for marketing in 2001. Allergan also recently acquired EndoArt, a European band company that has developed the EasyBand, which uses RF telemetry to adjust the gastric band. Additionally, we are aware that Johnson & Johnson received approval on September 28, 2007 of their gastric band product known as the Realize Adjustable Gastric Band.
We believe that the principal competitive factors in our market include:
| • | acceptance by healthcare professionals, patients and payors; |
| • | published rates of safety and efficacy; |
| • | reliability and high quality performance; |
13
| • | effectiveness at controlling co-morbidities such as diabetes and hypertension; |
| • | invasiveness and the inherent reversibility of the procedure or device; |
| • | cost and average selling price of products and relative rates of reimbursement; |
| • | effective marketing, education, sales and distribution; |
| • | regulatory and reimbursement expertise; |
| • | technological leadership and superiority; and |
| • | speed of product innovation and time to market. |
Many of our competitors are either publicly-traded or are divisions of publicly-traded companies, and they enjoy several competitive advantages over us, including:
| • | significantly greater name recognition; |
| • | established relations with healthcare professionals, customers and third-party payors; |
| • | established distribution networks; |
| • | greater experience in research and development, manufacturing, preclinical testing, clinical trials, obtaining regulatory approvals, obtaining reimbursement and marketing approved products; and |
| • | greater financial and human resources. |
As a result, we cannot assure you that we will be able to compete effectively against these companies or their products.
Third-party Coverage and Reimbursement
We plan to set a market price for the Maestro System in the United States that is comparable to other high-end, active implantable devices such as implantable cardioverter defibrillators (ICDs), neurostimulation devices for chronic pain, and cochlear implant systems. We expect that the procedure will be performed in the outpatient setting.
We believe that establishing appropriate third-party coverage for the therapy should be achievable as important structural elements are already in place. Physician claims for payment use Current Procedural Terminology, Fourth Edition (CPT) billing codes to describe procedures and services performed. Currently, there are established CPT codes for the implantation of cranial nerve pulse generators and related leads, and we expect providers may seek payment for our therapy based on these codes. With respect to possible usage of our product in the hospital inpatient setting, hospital inpatient billing is referenced by International Classifications of Diseases, 9th Revision, Clinical Modification (ICD-9-CM) procedure codes. There is an existing ICD-9-CM diagnosis code for morbid obesity and our studies are intended to provide the necessary outcomes data to link appropriate billing codes with the ICD-9 diagnosis code for morbid obesity. Our clinical trial data substantiating VBLOC therapy will also be used to seek coverage of VBLOC therapy for patients with morbid obesity and appropriate reimbursement for surgeons and hospitals under the codes already in place.
CMS, the federal agency that administers the Medicare program, has issued a national coverage determination for several specific types of bariatric surgery, which we view as positive, potential precedent and guidance to factors that CMS might use in deciding to cover our therapy. The policy indicates that Medicare will cover these bariatric surgical procedures when they are performed in an approved Bariatric Center of Excellence by a bariatric surgeon who also meets established requirements. Subjects with a BMI greater than or equal to 35, at least one obesity-related disease or disorder and who were previously unsuccessful with medical treatment for obesity are considered eligible. However, the policy reiterates that treatments for obesity alone are not covered,
14
because such treatments are not considered reasonable and necessary. Although Medicare policies are often emulated or adopted by other third-party payors, other governmental and private insurance coverage currently varies by carrier and geographic location. We intend to actively work with major insurance carriers as well as CMS to obtain coverage for procedures using our product.
Other manufacturers of neurostimulator devices for a variety of indications have been successful in securing third-party coverage and reimbursement for use of their devices after early commercialization. We will actively pursue all similar opportunities to secure appropriate payment for our device.
Intellectual Property
Our success will depend in part on our ability to obtain and defend patent protection for our products and processes, to preserve our trade secrets and to operate without infringing or violating the proprietary rights of third parties. To date, we have nine issued U.S. patents, seven of which pertain to treating gastrointestinal disorders and we believe provide us with broad intellectual property protection covering electrically-induced vagal blocking and for treating obesity. Material among these are our U.S. Patent No. 7,167,750, U.S. Patent No. 7,489,969, US Patent No. 7,613,515, US Patent No. 7,444,183 and US Patent No. 7,672,727. Assuming timely payment of maintenance fees as they become due, these patents will expire in 2023. We also have 22 U.S. patent applications, four pending international patent applications (PCT) and fourteen national stage patent applications (including seven European applications) in foreign jurisdictions. These applications primarily pertain to our vagal blocking technology and its application to obesity as well as other gastrointestinal disorders. In addition to our patents and applications, we have a license agreement with Mayo Foundation for Medical Education and Research for two pending U.S. patent applications on medical device obesity treatments, which are unrelated to our VBLOC therapy.
We also register the trademarks and trade names through which we conduct our business. To date, in the United States we have registered trademarks for VBLOC, ENTEROMEDICS and MAESTRO each registered with the United States Patent and Trademark Office, and have received a Notice of Allowance and third extension of time to file a Statement of Use on our application to register the mark EMPOWER. In addition, the marks VBLOC, MAESTRO and ENTEROMEDICS are the subject of either a trademark registration or application for registration in Australia, Brazil, China, Mexico, the European Community, Saudi Arabia, the United Arab Emirates and Switzerland. This Form 10-K contains other trade names and trademarks and service marks of EnteroMedics and of other companies. We may file additional trademark applications from time to time as deemed appropriate by management.
We are dedicated to continuing our patent activity to ensure that our patent portfolio remains reflective of our intellectual property development. New developments and modifications of prior developments are periodically reviewed to identify necessary additions and modifications to our patent portfolio.
In addition to our patents, we rely on confidentiality and proprietary information agreements to protect our trade secrets and proprietary knowledge. These confidentiality and proprietary information agreements generally provide that all confidential information developed or made known to individuals by us during the course of their relationship with us is to be kept confidential and not disclosed to third parties, except in specific circumstances. The agreements also provide for ownership of inventions conceived during the course of such agreements. If our proprietary information is shared or our confidentiality agreements are breached, we may not have adequate remedies, or our trade secrets may otherwise become known to or independently developed by competitors.
Manufacturers and Suppliers
We have designed and developed all of the elements of our Maestro System, except for the clinician programmer hardware, which uses a commercially available laptop computer. To date, all of the materials and components of the system used in our clinical trials are procured from qualified suppliers and contract
15
manufacturers in accordance with our proprietary specifications. We use third parties to manufacture our Maestro System to minimize our capital investment, help control costs and take advantage of the expertise these third parties have in the large-scale production of medical devices. We do not currently plan to manufacture our Maestro System ourselves. All of our key manufacturers and suppliers have experience working with commercial implantable device systems, are ISO certified and are regularly audited by us. Our key manufacturers and suppliers have a demonstrated record of compliance with international regulatory requirements.
In the event that the Maestro System receives FDA approval, we expect to increase our production volume by a significant amount. Given that we rely primarily on third-party manufacturers and suppliers for the production of our products, our ability to increase production will depend upon the experience, certification levels and large scale production capabilities of our suppliers and manufacturers. Qualified suppliers and contract manufacturers have been and will continue to be selected to supply products on a commercial scale according to our proprietary specifications. We also intend to increase our inventory levels to support commercial forecasts as we expand our implanting centers. Our FDA approval process requires us to name and obtain approval for the suppliers of key components of our Maestro System.
Many of our parts are custom designed and in certain instances, are obtained through long-term supply arrangements that are exclusive. Due to these factors, we may not be able to quickly qualify and establish additional or replacement suppliers for the components of our Maestro System. A delay in the approval process with the FDA for our Maestro System as a result of the need to qualify or obtain alternate vendors for any of our components would delay our ability to sell and market the Maestro System and could have a material adverse effect on our business.
We believe that our current manufacturing and supply arrangements will be adequate to continue our ongoing and planned clinical trials. In order to produce the Maestro System in the quantities we anticipate to meet future market demand, we will need our manufacturers and suppliers to increase, or scale up, manufacturing production and supply arrangements by a significant factor over the current level of production. There are technical challenges to scaling up manufacturing capacity and developing commercial-scale manufacturing facilities that may require the investment of substantial additional funds by our manufacturers and suppliers and hiring and retaining additional management and technical personnel who have the necessary experience. If our manufacturers or suppliers are unable to do so, we may not be able to meet the requirements for the launch of the product or to meet future demand, if at all. We may also represent only a small portion of our suppliers’ or manufacturers’ business and if they become capacity constrained they may choose to allocate their available resources to other customers that represent a larger portion of their business. We currently anticipate that we will continue to rely on third-party manufacturers and suppliers for the production of the Maestro System following commercialization. If we develop and obtain regulatory approval for our product and are unable to obtain a sufficient supply of our product, our revenue, business and financial prospects would be adversely affected.
Government Regulations
United States
Our Maestro System is regulated by the FDA as a medical device under the Federal Food, Drug, and Cosmetic Act (FFDCA) and the regulations promulgated under the FFDCA. Pursuant to the FFDCA, the FDA regulates the research, design, testing, manufacture, safety, labeling, storage, record keeping, advertising, sales and distribution, post-market adverse event reporting, production and advertising and promotion of medical devices in the United States. Noncompliance with applicable requirements can result in warning letters, fines, injunctions, civil penalties, recall or seizure of products, total or partial suspension of production, failure of the government to grant premarket approval for devices and criminal prosecution.
Medical devices are classified into one of three classes, Class I, II or III, on the basis of the amount of risk and the controls deemed by the FDA to be necessary to reasonably ensure their safety and effectiveness. Class I, low risk, devices are subject to general controls (e.g., labeling and adherence to good manufacturing practices
16
(GMPs)). Class II, intermediate risk, devices are subject to general controls and to special controls (e.g., performance standards, and premarket notification). Generally, Class III devices are those which must receive premarket approval by the FDA to ensure their safety and effectiveness (e.g., life-sustaining, life-supporting and implantable devices, or new devices which have not been found substantially equivalent to legally marketed devices), and require clinical testing to ensure safety and effectiveness and FDA approval prior to marketing and distribution. The FDA also has the authority to require clinical testing of Class II devices. In both the United States and certain international markets, there have been a number of legislative and regulatory initiatives and changes, such as the Modernization Act, which could and have altered the healthcare system in ways that could impact our ability to sell our medical devices profitably. Recent, widely-publicized events concerning the safety of certain drug, food and medical device products have raised concerns among members of Congress, medical professionals, and the public regarding the FDA’s handling of these events and its perceived lack of oversight over regulated products. The increased attention to safety and oversight issues could result in a more cautious approach by the FDA to device clearances and approvals, as well as post- market compliance, which could prevent, delay clearance or approval of our new products or product modifications, or require us to expend additional resources on post-market studies and controls.
The FFCDA provides two basic review procedures for medical devices. Certain products may qualify for a submission authorized by Section 510(k) of the FFCDA, where the manufacturer submits to the FDA a premarket notification of the manufacturer’s intention to commence marketing the product. The manufacturer must, among other things, establish that the product to be marketed is substantially equivalent to another legally marketed product. Marketing may commence when the FDA issues a letter finding substantial equivalence. If a medical device does not qualify for the 510(k) procedure, the manufacturer must file a premarket approval (PMA) application with the FDA. This procedure requires more extensive pre-filing clinical and preclinical testing than the 510(k) procedure and involves a significantly longer FDA review process.
Premarket Approval
Our product will require prior premarket approval from the FDA. Because our Maestro System is an implanted device, it is deemed to pose a significant risk. To market the Maestro System in the United States, the FDA must approve the device after submission of a PMA. The FDA can also impose restrictions on the sale, distribution or use of devices at the time of their clearance or approval, or subsequent to marketing. The process of obtaining premarket approval is costly, lengthy and uncertain. A PMA must be supported by extensive data including, but not limited to, technical, pre-clinical and clinical trials to demonstrate to the FDA’s satisfaction the safety and effectiveness of the device. Among other information, the PMA must also contain a full description of the device and its components, a full description of the methods, facilities and controls used for manufacturing, and proposed device labeling.
If the FDA determines that a PMA is complete, the FDA accepts the application and begins an in-depth review of the submitted information. The FDA, by statute and regulation, has 180 days to review an accepted PMA application, although the review and response activities generally occur over a significantly longer period of time, typically one year, and can take up to several years. During this review period, the FDA may request additional information or clarification of information already provided. Also during the review period, an advisory panel of experts from outside the FDA may be convened to review and evaluate the application and provide recommendations to the FDA as to the approvability of the device. In addition, the FDA will conduct a pre-approval inspection of our, and our manufacturers’, facilities to evaluate compliance with the quality system regulation. Under the Medical Device User Fee and Modernization Act of 2002, the fee to submit a PMA can be up to $259,600 per PMA, however, we have qualified for a small business exemption. If the FDA’s evaluation of the PMA is favorable, the PMA is approved, and the device may be marketed in the United States. The FDA may approve the PMA with post-approval conditions intended to ensure the safety and effectiveness of the device. Failure to comply with the conditions of approval can result in material adverse enforcement action, including the loss or withdrawal of the approval. Even after approval of a PMA, new PMAs or supplemental PMAs are required for significant modifications to the manufacturing process, labeling, use and design of a device that is
17
approved through the premarket approval process. Premarket approval supplements often require submission of the same type of information as a PMA except that the supplement is limited to information needed to support any changes from the device covered by the original PMA.
Clinical Trials
A clinical trial is almost always required to support a PMA. Clinical trials for a “significant risk” device such as ours require submission of an application for an IDE, to the FDA. The IDE application must be supported by appropriate data, such as animal and laboratory testing results, showing that it is safe to test the device in humans and that the testing protocol is scientifically sound. Clinical trials for a significant risk device may begin once the IDE application is allowed to proceed by the FDA and the institutional review boards overseeing the clinical trial at the various investigational sites.
Clinical trials require extensive recordkeeping and detailed reporting requirements. Our clinical trials must be conducted under the oversight of an institutional review board at the relevant clinical trial site and in accordance with applicable regulations and policies including, but not limited to, the FDA’s good clinical practice (GCP) requirements. We, the trial data safety monitoring board, the FDA or the institutional review board at each site at which a clinical trial is being performed may suspend a clinical trial at any time for various reasons, including a belief that the risks to study subjects outweigh the anticipated benefits.
Pervasive and Continuing FDA Regulation
Both before and after FDA approval, numerous regulatory requirements apply. These include:
| • | quality system regulation, which requires manufacturers to follow design, testing, control, documentation, complaint handling and other quality assurance procedures during the design and manufacturing processes; |
| • | regulations which govern product labels and labeling, prohibit the promotion of products for unapproved or “off-label” uses and impose other restrictions on labeling and promotional activities; |
| • | medical device reporting regulations, which require that manufacturers report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur; and |
| • | notices of correction or removal and recall regulations. |
Advertising and promotion of medical devices are also regulated by the Federal Trade Commission and by state regulatory and enforcement authorities. Recently, some promotional activities for FDA-regulated products have resulted in enforcement actions brought under healthcare reimbursement laws and consumer protection statutes. In addition, under the federal Lanham Act, competitors and others can initiate litigation relating to advertising claims.
Compliance with regulatory requirements is enforced through periodic, unannounced facility inspections by the FDA. Failure to comply with applicable regulatory requirements can result in enforcement action by the FDA, which may include any of the following sanctions:
| • | warning letters or untitled letters; |
| • | fines, injunction and civil penalties; |
| • | recall or seizure of our products; |
| • | customer notification, or orders for repair, replacement or refund; |
| • | operating restrictions, partial suspension or total shutdown of production or clinical trials; |
18
| • | refusing our request for premarket approval of new products; |
| • | withdrawing premarket approvals that are already granted; and |
| • | criminal prosecution. |
International
International sales of medical devices are subject to foreign government regulations, which vary substantially from country to country. The time required to obtain approval by a foreign country may be longer or shorter than that required for FDA approval, and the requirements may differ. The primary regulatory environment in Europe is that of the European Economic Community (EEC), which consists of 25 countries encompassing nearly all the major countries in Europe. Other countries that are not part of the EEC, such as Switzerland, have voluntarily adopted laws and regulations that mirror those of the EEC with respect to medical devices. The EEC has adopted Directive 90/385/EEC for active implantable medical devices and numerous standards that govern and harmonize the national laws and standards regulating the design, manufacture, clinical trials, labeling and adverse event reporting for medical devices that are marketed in member states. Medical devices that comply with the requirements of the national law of the member state in which they are first marketed will be entitled to bear CE marking, indicating that the device conforms to applicable regulatory requirements, and, accordingly, can be commercially marketed within EEC states and other countries that recognize this mark for regulatory purposes.
We obtained European CE Mark approval for our Maestro RF System on March 4, 2009. The method of assessing conformity with applicable regulatory requirements varies depending on the class of the device, but for our Maestro System (which falls into Class III), the method involved a combination of self-assessment by the manufacturer of the safety and performance of the device, and a third-party assessment by a Notified Body, usually of the design of the device and of the manufacturer’s quality system. A Notified Body is a private commercial entity that is designated by the national government of a member state as being competent to make independent judgments about whether a product complies with applicable regulatory requirements. The manufacturer’s assessment included a clinical evaluation of the conformity of the device with applicable regulatory requirements. We used KEMA in the Netherlands as the Notified Body for our CE marking approval process.
Employees
As of December 31, 2009, we had a total of 34 employees. All of these employees are located in the United States.
From time to time we also employ independent contractors, consultants and temporary employees to support our operations. None of our employees are subject to collective bargaining agreements. We have never experienced a work stoppage and believe that our relations with our employees are good.
Executive Officers
Our executive officers are as follows:
| Name |
Age | Position | ||
| Mark B. Knudson, Ph.D. |
61 | President, Chief Executive Officer, Chairman and Director | ||
| Greg S. Lea |
57 | Senior Vice President and Chief Financial Officer | ||
| Adrianus (Jos) Donders |
56 | Senior Vice President of Operations | ||
| Daniel L. Cohen |
51 | Senior Vice President of Government Relations and Health Policy | ||
| Katherine S. Tweden, Ph.D. |
49 | Vice President of Research and Clinical |
19
Mark B. Knudson, Ph.D. has served as our President, Chief Executive Officer and Chairman of the board since December 2002. Dr. Knudson also served as President and Chief Executive Officer of Venturi Group, LLC and Venturi Development, Inc., positions he held from 1999 and 2001 until their dissolutions in 2008 and 2009, respectively. Dr. Knudson served as Chairman of the board of Restore Medical, Inc., a publicly-held medical device company focused on the treatment of sleep disordered breathing, from 1999 through July 2008 when it was acquired by Medtronic, Inc. Dr. Knudson was also a member of the audit committee of Restore Medical. Dr. Knudson received a Bachelor of Science in biology from Pacific Lutheran University and a Ph.D. in physiology from Washington State University.
Greg S. Lea has served as our Senior Vice President and Chief Financial Officer since May 21, 2007. Prior to joining us, Mr. Lea served as Chief Financial Officer of Pemstar Inc. from July 2002 through January 2007 when it was acquired by Benchmark Electronics, Inc. Mr. Lea also served as a director of Pemstar from April 2001 through January 2007 and held the position of Corporate Controller from April 2002 through July 2002. From 1993 to April 2002, Mr. Lea served as a corporate Vice President for Jostens Corporation, a commemorative and affiliation products manufacturer, serving most recently as corporate Vice President-Business Ventures. Prior to that, Mr. Lea held several financial management and administrative positions at IBM Corporation from 1974 to 1993 and was President and a director of the Ability Building Center, Inc. from 1981 to 1993. Mr. Lea holds a B.S. in Accounting/Business Management from Minnesota State University, Mankato.
Adrianus (Jos) Donders has served as our Senior Vice President of Operations since April 2005. From September 2003 to April 2005, Mr. Donders was Director Communication Systems Engineering for Medtronic USA. From June 2000 to August 2003, Mr. Donders served as Director Clinical Study Management and Research and Development Europe for Medtronic Europe. Mr. Donders received a degree equivalent to a Masters of Electrical Engineering from the Institute of Technology Eindhoven Netherlands.
Daniel L. Cohen has served as our Senior Vice President of Government Relations and Health Policy since September 2009. Mr. Cohen worked as a consultant for the company from March 2009 to September 2009. Prior to joining EnteroMedics, Mr. Cohen served as Senior Vice President for Government Relations and Public Policy with US Oncology. He also served as a consultant with Inamed Corporation, a division of the Allergan Corporation, from 2001 to 2003, when he joined the company as Vice President Global, Corporate and Government Affairs, and again after the Allergan acquisition in 2006 through 2008. Mr. Cohen has experience as a Principal in a full-service government affairs firm, has served on the American Israel Public Affairs Committee (AIPAC), held staff positions with Members of the U.S. House of Representatives and managed political campaigns of all levels. Mr. Cohen holds a Master of Arts in Liberal Studies/International Affairs from Georgetown University and a Bachelor of Science Degree from Willamette University.
Katherine S. Tweden, Ph.D. has served as our Vice President of Research since January 2003 and Vice President of Clinical since September 2008. From November 2002 to January 2003, Dr. Tweden was a consultant to Venturi Group, a medical device incubator company. From January 2003 through August 2004, Dr. Tweden worked for Venturi Development Inc. as a consultant to us. From July 1997 to October 2002, Dr. Tweden held positions including Director of Research and Vice President of Research for HeartStent Corporation. From September 1990 to June 1997, Dr. Tweden held the positions of Sr. Research Scientist and Principal Research Scientist at St Jude Medical, Inc. Dr. Tweden received a Bachelor of Arts in chemistry from Gustavus Adolphus College and a Masters degree and Ph.D. in biomedical engineering from Iowa State University.
Our Corporate Information
We were incorporated in Minnesota in December 2002 under the name Beta Medical, Inc. In 2003, we changed our name to EnteroMedics Inc. and in 2004 we reincorporated in Delaware. We file reports and other information with the Securities and Exchange Commission (SEC) including annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and proxy or information statements. Those reports and statements as well as all amendments to those documents filed or furnished pursuant to Section 13(a) or
20
15(d) of the Securities Exchange Act of 1934, as amended (1) are available at the SEC’s Public Reference Room at 100 F Street, N.E., Washington, DC 20549, (2) may be obtained by sending an electronic message to the SEC at publicinfo@sec.gov or by sending a fax to the SEC at 1-202-777-1027, (3) are available at the SEC’s internet site (http://www.sec.gov), which contains reports, proxy and information statements and other information regarding issuers that file electronically with the SEC and (4) are available free of charge through our website as soon as reasonably practicable after electronic filing with, or furnishing to, the SEC. You may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330.
Our principal executive offices are located at 2800 Patton Road, St. Paul, Minnesota 55113, and our telephone number is (651) 634-3003. Our website address is www.enteromedics.com. The information on, or that may be accessed through, our website is not incorporated by reference into this Annual Report on Form 10-K and should not be considered a part of this Annual Report on Form 10-K.
21
| ITEM 1A. | RISK FACTORS |
Risks Related to our Business and Industry
We are a development stage company with a limited history of operations and no approved products, and we cannot assure you that we will ever have a commercialized product.
We are a development stage company with a limited operating history upon which you can evaluate our business. We currently do not have any products cleared or approved for commercialization or any other source of revenue, and we do not expect to have a commercialized product earlier than the second half of 2013. We have been engaged in research and development since our inception in 2002 and have invested substantially all of our time and resources in developing our VBLOC therapy, which we intend to commercialize initially in the form of our Maestro System. The success of our business will depend on our ability to obtain regulatory approval to market our Maestro System and any products we may develop in the future and our ability to create product sales, successfully introduce new products, establish our sales force and control costs, all of which we may be unable to do. If we are unable to successfully develop, receive regulatory approval for and commercialize our Maestro System for its indicated use, we may never generate revenue or be profitable and we may have to cease operations. Our lack of a significant operating history also limits your ability to make a comparative evaluation of us, our products and our prospects.
We have incurred losses since inception and we anticipate that we will continue to incur increasing losses for the foreseeable future. If we are unable to raise additional capital in the second half of 2010, we may be unable to continue as a going concern.
We have incurred losses in each year since our formation in 2002. As of December 31, 2009, we had a deficit accumulated during the development stage of $133.2 million. Our net losses applicable to common stockholders for the fiscal years ended December 31, 2009, 2008 and 2007 were $31.9 million, $37.9 million and $28.6 million, respectively. We have funded our operations to date principally from the sale of our securities and through the issuance of indebtedness. Development of a new medical device, including conducting clinical trials and seeking regulatory approvals, is a long, expensive and uncertain process. If our Maestro System is approved for marketing by the U.S. Food and Drug Administration (FDA) we expect to incur significant sales and marketing expenses prior to recording sufficient revenue to offset these expenses. We expect our general and administrative expenses to increase as we continue to add the infrastructure necessary to support operating as a public company and develop our intellectual property portfolio. For these reasons, we expect to continue to incur significant and increasing operating losses for the next several years. These losses, among other things, have had and will continue to have an adverse effect on our stockholders’ equity and working capital. Because of the numerous risks and uncertainties associated with developing new medical devices, we are unable to predict the extent of any future losses or when we will become profitable, if ever.
Without additional capital, we may run out of cash in the second half of 2010, which has raised a substantial doubt about our ability to continue as a going concern. We have prepared our consolidated financial statements for the year ended December 31, 2009 on a going concern basis, which contemplates the realization of assets and satisfaction of liabilities and other commitments in the normal course of business. The funding of our operations beyond the second half of 2010 will require additional investments in our company in the form of equity or debt financing or through collaboration, licensing or other similar arrangements. There is no assurance that we will be able to raise sufficient capital to continue as a going concern.
If we are unable to comply with the continued listing requirements of the NASDAQ Capital Market, our common stock could be delisted, which could affect its market price and liquidity and reduce our ability to raise capital.
We are required to meet certain qualitative and financial tests (including a minimum closing bid price for our common stock of $1.00 per share) to maintain the listing of our common stock on the NASDAQ Capital Market. On November 13, 2009, we received a notice from the NASDAQ Stock Market advising that for the
22
prior 30 consecutive business days, the minimum closing bid price of our listed securities had been below the minimum $1.00 per share requirement for continued listing on the NASDAQ Global Market pursuant to NASDAQ Listing Rule 5450(a)(1). In anticipation of not regaining compliance with two other continued listing requirements of the NASDAQ Global Market prior to the expiration of their grace periods, we requested and were approved to transfer to the NASDAQ Capital Market, effective January 22, 2010. In connection with this transfer, we have been afforded the balance of our 180 calendar day grace period, until May 12, 2010, to regain compliance with the minimum closing bid price rule by having our stock price close at or above $1.00 per share for a minimum of ten consecutive business days. If we are unable to regain compliance with the minimum closing bid price rule or are unable to maintain compliance with the other continued listing requirements of the NASDAQ Capital Market within specified periods and subject to permitted extensions, our common stock may be recommended for delisting (subject to any appeal we would file). If our common stock were delisted, it could be more difficult to buy or sell our common stock and to obtain accurate quotations, and the price of our stock could suffer a material decline. Delisting would also impair our ability to raise capital.
We have not received, and may never receive, approval from the FDA or the regulatory body in any other country to market our Maestro System for the treatment of obesity.
We do not have the necessary regulatory approvals to market our Maestro System in the United States or in any foreign market other than the European Community for which we received CE Mark approval for our Maestro RF System on March 4, 2009. We plan initially to launch our product, if approved, in the United States, but ultimately will also seek to commercialize our Maestro System in countries outside the United States.
We cannot market our product in the United States unless it has been approved by the FDA. The FDA approval process involves, among other things, successfully completing clinical trials and obtaining a premarket approval (PMA). The PMA process requires us to prove the safety and efficacy of our Maestro System to the FDA’s satisfaction. This process can be expensive and uncertain, requires detailed and comprehensive scientific and human clinical data, generally takes one to three years after a PMA application is filed, and notwithstanding the effort and expense incurred, may never result in the FDA granting a PMA. Because VBLOC therapy represents a novel way to effect weight loss in the treatment of obesity, and because there is a large population of obese patients who might be eligible for treatment, it is possible that the FDA and other regulatory bodies will review an application for approval of our Maestro System with greater scrutiny, which could cause that process to be lengthier and more involved than that for products without such characteristics. The FDA can delay, limit or deny approval of a PMA application for many reasons, including:
| • | our inability to demonstrate safety or effectiveness to the FDA’s satisfaction; |
| • | the data from our preclinical studies and clinical trials may be insufficient to support approval; |
| • | the facilities of our third-party manufacturers or suppliers may not meet applicable requirements; |
| • | our compliance with preclinical, clinical or other regulations; |
| • | our inability to meet the FDA’s statistical requirements or changes in statistical tests or significance levels the FDA requires for approval of a medical device, including ours; and |
| • | changes in the FDA approval policies, expectations with regard to the type or amount of scientific data required or adoption of new regulations may require additional data or additional clinical studies. |
In order to market our Maestro System outside of the United States, we will need to establish and comply with the numerous and varying regulatory requirements of other countries regarding safety and efficacy. Approval procedures vary among countries and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries may differ from that required to obtain FDA approval. The regulatory approval process in other countries may also include all of the risks detailed above regarding FDA approval in the United States. Regulatory approval in one country does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country may
23
negatively impact the regulatory process in others. While we have received the European CE Mark for our Maestro RF System, we cannot assure you when, or if, we will be able to commence sales in the European Economic Area or obtain approval to market our Maestro System in other countries outside the United States.
We may not obtain the necessary regulatory approvals to market our Maestro System in the United States or anywhere else. Even if we obtain approval, the FDA or other regulatory authorities may require expensive or burdensome post-market testing or controls. Any delay in, failure to receive or maintain, or significant limitation on approval for our Maestro System could prevent us from generating revenue or achieving profitability and we may be forced to cease operations.
The preliminary results of the blinded segment of our EMPOWER trial may not be sufficient to support approval of a PMA application, and this will likely prevent or delay regulatory approval of our Maestro System and impair our financial position.
In September 2009, we completed the blinded segment of our EMPOWER pivotal trial, a randomized, prospective, placebo-controlled multi-center trial of our Maestro System in the United States. Based on our initial analysis, the EMPOWER trial did not meet its primary and secondary efficacy endpoints; however, we are currently conducting a thorough analysis of the EMPOWER study data and have met with the FDA to discuss the EMPOWER trial results and the regulatory process going forward. Based on this discussion, we recently submitted an Investigational Device Exemption (IDE) application for a clinical trial using the next-generation Maestro RC System in the treatment of morbid obesity. Even if we are able to obtain FDA approval of an IDE for a clinical trial using the next-generation Maestro RC System in the treatment of morbid obesity, the inability to achieve our primary and secondary efficacy endpoints in the EMPOWER trial means that it will take us longer to ultimately commercialize a product and generate revenue, our financial projections may be impaired, and we may never be able to produce sufficient data to support a PMA application with the FDA or commercialize a product.
We may be unable to receive approval for or complete a pivotal trial using our next-generation Maestro RC System or other trials, or we may experience significant delays in completing our clinical trials, which could prevent or delay regulatory approval of our Maestro System and impair our financial position.
We recently submitted an IDE application for a clinical trial using the next-generation Maestro RC System in the treatment of morbid obesity. Assuming that we obtain an approved IDE, successfully enroll and implant the trial and achieve favorable results, we plan to use data from that trial to support a PMA application for the Maestro System. We expect to commence the trial upon receipt of an IDE approval from the FDA and upon receipt of approval from the relevant institutional review boards at the various sites at which we would be conducting the trial. Conducting a clinical trial of this size, which involves screening, assessing, testing, treating and monitoring patients at several sites across the country and possibly internationally, and coordinating with patients and clinical institutions, is a complex and uncertain process.
The commencement of our trial could be delayed for a variety of reasons, including:
| • | obtaining an IDE approval from the FDA with acceptable terms; |
| • | reaching agreement on acceptable terms with prospective clinical trial sites; |
| • | manufacturing sufficient quantities of our Maestro System; |
| • | obtaining institutional review board approval to conduct the trial at a prospective site; and |
| • | obtaining sufficient patient enrollment, which is a function of many factors, including the size of the patient population, the nature of the protocol, the proximity of patients to clinical sites and the eligibility criteria for the trial. |
24
Once the trial has begun, the completion of the trial, and our other ongoing clinical trials, could be delayed, suspended or terminated for several reasons, including:
| • | ongoing discussions with regulatory authorities regarding the scope or design of our preclinical results or clinical trial or requests for supplemental information with respect to our preclinical results or clinical trial results; |
| • | our failure or inability to conduct the clinical trials in accordance with regulatory requirements; |
| • | sites currently participating in the trial may drop out of the trial, which may require us to engage new sites or petition the FDA for an expansion of the number of sites that are permitted to be involved in the trial; |
| • | patients may not remain in or complete, clinical trials at the rates we expect; |
| • | patients may experience serious adverse events or side effects during the trial, which, whether or not related to our product, could cause the FDA or other regulatory authorities to place the clinical trial on hold; |
| • | clinical investigators may not perform our clinical trials on our anticipated schedule or consistent with the clinical trial protocol and good clinical practices; and |
| • | we may be unable to obtain a sufficient supply of our Maestro System necessary for the timely conduct of the clinical trials. |
If our clinical trials are delayed it will take us longer to ultimately commercialize a product and generate revenue or the delay could result in our being unable to do so. Moreover, our development costs will increase if we have material delays in our clinical trials or if we need to perform more or larger clinical trials than planned.
Even if we obtain the necessary regulatory approvals, our efforts to commercialize our Maestro System may not succeed or may encounter delays which could significantly harm our ability to generate revenue.
If we obtain regulatory approval to market our Maestro System, our ability to generate revenue will depend upon the successful commercialization of this product. Our efforts to commercialize our Maestro System may not succeed for a number of reasons, including:
| • | our Maestro System may not be accepted in the marketplace by physicians, patients and third-party payors; |
| • | the price of our Maestro System, associated costs of the surgical procedure and treatment and the availability of sufficient third-party reimbursement for the procedure and therapy implantation and follow-up procedures; |
| • | appropriate reimbursement coding options may not exist to enable billing for the system implantation and follow-up procedures; |
| • | we may not be able to sell our Maestro System at a price that allows us to meet the revenue targets necessary to generate revenue for profitability; |
| • | the frequency and severity of any side effects of our VBLOC therapy; |
| • | physicians and potential patients may not be aware of the perceived effectiveness and sustainability of the results of VBLOC therapy provided by our Maestro System; |
| • | we, or the investigators of our product, may not be able to have information on the outcome of the trials published in medical journals; |
| • | the availability and perceived advantages and disadvantages of alternative treatments; |
| • | any rapid technological change may make our product obsolete; |
25
| • | we may not be able to have our Maestro System manufactured in commercial quantities or at an acceptable cost; |
| • | we may not have adequate financial or other resources to complete the development and commercialization of our Maestro System; and |
| • | we may be sued for infringement of intellectual property rights and could be enjoined from manufacturing or selling our products. |
Besides requiring physician adoption, market acceptance of our Maestro System will depend on successfully communicating the benefits of our VBLOC therapy to three additional constituencies involved in deciding whether to treat a particular patient using such therapy: (1) the potential patients themselves; (2) institutions such as hospitals, where the procedure would be performed and opinion leaders in these institutions; and (3) third-party payors, such as private healthcare insurers and Medicare, which would ultimately bear most of the costs of the various providers and equipment involved in our VBLOC therapy. Marketing to each of these constituencies requires a different marketing approach, and we must convince each of these groups of the efficacy and utility of our VBLOC therapy to be successful.
If our VBLOC therapy, or any other neuroblocking therapy for other gastrointestinal diseases and disorders that we may develop, does not achieve an adequate level of acceptance by the relevant constituencies, we may not generate significant product revenue and may not become profitable. The earliest we expect to be able to commercialize our Maestro System is in the second half of 2013, if at all. If we are not successful in the commercialization of our Maestro System for the treatment of obesity we may never generate any revenue and may be forced to cease operations.
We depend on clinical investigators and clinical sites to enroll patients in our clinical trials, and on other third parties to manage the trials and to perform related data collection and analysis, and, as a result, we may face costs and delays that are outside of our control.
We rely on clinical investigators and clinical sites to enroll patients in our clinical trials, including a potentially new clinical trial using our next generation Maestro RC System if approved by the FDA, and other third parties to manage the trials and to perform related data collection and analysis. However, we may not be able to control the amount and timing of resources that clinical sites may devote to our clinical trials. If these clinical investigators and clinical sites fail to enroll a sufficient number of patients in our clinical trials, to ensure compliance by patients with clinical protocols or comply with regulatory requirements, we will be unable to complete these trials, which could prevent us from obtaining regulatory approvals for our product. Our agreements with clinical investigators and clinical trial sites for clinical testing place substantial responsibilities on these parties and, if these parties fail to perform as expected, our trials could be delayed or terminated. If these clinical investigators, clinical sites or other third parties do not carry out their contractual duties or obligations or fail to meet expected deadlines, or if the quality or accuracy of the clinical data they obtain is compromised due to their failure to adhere to our clinical protocols, regulatory requirements or for other reasons, our clinical trials may be extended, delayed or terminated, or the clinical data may be rejected by the FDA, and we may be unable to obtain regulatory approval for, or successfully commercialize, our product.
Assuming we receive regulatory approval for the Maestro System, modifications to the Maestro System may require additional approval from the FDA, which may not be obtained or may delay our commercialization efforts.
The FDA requires medical device companies to initially make and document a determination of whether or not a modification requires a new approval, supplement or clearance; however, the FDA can review a company’s decision. Any modifications to an FDA-approved device that could significantly affect its safety or efficacy, or that would constitute a major change in its intended use would require a supplemental IDE and possibly additional clinical studies and a separate PMA application. Product changes or revisions will require all the regulatory steps and associated risks discussed above including testing, an IDE supplement and clinical study.
26
We may not be able to obtain approval of supplemental IDEs or PMAs for product modifications, new indications for our product or new products. Delays in obtaining future clearances would adversely affect our ability to introduce new or enhanced products in a timely manner, which in turn would harm our commercialization efforts and future growth.
Physicians may not widely adopt our Maestro System and VBLOC therapy unless they determine, based on experience, long-term clinical data and published peer reviewed journal articles, that VBLOC therapy provides a safe and effective alternative to other existing treatments for obesity.
Physicians tend to be slow to change their medical treatment practices because of the time and skill required to learn a new procedure, the perceived liability risks arising from the use of new products and procedures, and the uncertainty of third-party coverage and reimbursement. Physicians may not widely adopt our Maestro System and VBLOC therapy unless they determine, based on experience, long-term clinical data and published peer reviewed journal articles, that the use of our VBLOC therapy provides a safe and effective alternative to other existing treatments for obesity, including pharmaceutical solutions and bariatric surgical procedures.
We cannot provide any assurance that the data collected from our current and planned clinical trials will be sufficient to demonstrate that our VBLOC therapy is an attractive alternative to other obesity treatment procedures. We rely on experienced and highly trained surgeons to perform the procedures in our clinical trials and both short- and long-term results reported in our clinical trials may be significantly more favorable than typical results of practicing physicians, which could negatively impact rates of adoption of our Maestro System and VBLOC therapy. We believe that published peer-reviewed journal articles and recommendations and support by influential physicians regarding our Maestro System and VBLOC therapy will be important for market acceptance and adoption, and we cannot assure you that we will receive these recommendations and support, or that supportive articles will be published.
If we fail to obtain adequate coding, coverage or payment levels for our product by governmental healthcare programs and other third-party payors, there may be no commercially viable markets for our Maestro System or other products we may develop or our target markets may be much smaller than expected.
Healthcare providers generally rely on third-party payors, including governmental payors, such as Medicare and Medicaid, and private healthcare insurers, to adequately cover and reimburse the cost of medical devices. Importantly, third-party payors are increasingly challenging the price of medical products and services and instituting cost containment measures to control or significantly influence the purchase of medical products and services. We expect that third-party payors will continue to attempt to contain or reduce the costs of healthcare by challenging the prices charged for healthcare products and services. If reimbursement for our Maestro System and the related surgery and facility costs is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, market acceptance of our Maestro System will be impaired and our future revenue, if any, would be adversely affected. As such, even if we obtain FDA clearance or approval for our Maestro System and begin to market it, the availability and level of third-party coverage and reimbursement could substantially affect our ability to commercialize our Maestro System and other products we may develop.
The efficacy, safety, ease of use and cost-effectiveness of our Maestro System and of any competing products will, in part, determine the availability and level of coverage and payment. In particular, we expect that securing coding, coverage and payment for our Maestro System will be more difficult if our clinical trials do not demonstrate a percentage of excess weight loss from a pre-implementation baseline that healthcare providers and obese individuals consider clinically meaningful, whether or not regulatory agencies consider the improvement of patients treated in clinical trials to have been clinically meaningful.
In some international markets, pricing of medical devices is subject to government control. In the United States and international markets, we expect that both government and third-party payors will continue to attempt to contain or reduce the costs of healthcare by challenging the prices charged for healthcare products and
27
services. If payment for our Maestro System and the related surgery and facility costs is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, market acceptance of our Maestro System will be impaired and our future revenue, if any, would be adversely affected.
We cannot predict the likelihood or pace of any significant regulatory or legislative action in any of these areas, nor can we predict whether or in what form healthcare legislation being formulated by various governments will be passed. We also cannot predict with precision what effect such governmental measures would have if they were ultimately enacted into law. However, in general, we believe that such legislative activity will likely continue. If adopted, such measures can be expected to have an impact on our business.
Even if our Maestro System is approved by regulatory authorities, if we or our suppliers fail to comply with ongoing regulatory requirements, or if we experience unanticipated product problems, our Maestro System could be subject to restrictions or withdrawal from the market.
Completion of our clinical trials and commercialization of our Maestro System will require access to manufacturing facilities that meet applicable regulatory standards to manufacture a sufficient supply of our product. We rely solely on third parties to manufacture and assemble our Maestro System, and do not currently plan to manufacture or assemble our Maestro System ourselves in the future.
Any product for which we obtain marketing approval, along with the manufacturing processes, post-approval clinical data and promotional activities for such product, will be subject to continual review and periodic inspections by the FDA and other regulatory bodies. In particular we and our manufacturers and suppliers are required to comply with Good Manufacturing Practices (GMP), which for medical devices is called the Quality System Regulation (QSR), and other regulations which cover the methods and documentation of the design, testing, production, control, quality assurance, labeling, packaging, storage and shipping of any product for which we obtain marketing approval. The FDA enforces the QSR through unannounced inspections. We and our third-party manufacturers and suppliers have not yet been inspected by the FDA and will have to successfully complete such inspections before we receive regulatory approvals for our Maestro System. Failure by us or one of our manufacturers or suppliers to comply with statutes and regulations administered by the FDA and other regulatory bodies, or failure to adequately respond to any observations, could result in enforcement actions against us or our manufacturers or suppliers, including, restrictions on our product or manufacturing processes, withdrawal of the product from the market, voluntary or mandatory recall, fines, suspension of regulatory approvals, product seizures, injunctions or the imposition of civil or criminal penalties.
If any of these actions were to occur it would harm our reputation and cause our product sales to suffer. Furthermore, our key component suppliers may not currently be or may not continue to be in compliance with applicable regulatory requirements. If the FDA or any other regulatory body finds their compliance status to be unsatisfactory, our commercialization efforts could be delayed, which would harm our business and our results of operations.
Even if regulatory approval of a product is granted, the approval may be subject to limitations on the indicated uses for which the product may be marketed. If the FDA determines that our promotional materials, training or other activities constitute promotion of an unapproved use, we could be subject to significant liability, the FDA could request that we cease, correct or modify our training or promotional materials or subject us to regulatory enforcement actions. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider our training or other promotional materials to constitute promotion of an unapproved use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false claims for reimbursement.
We are subject to medical device reporting (MDR) regulations that require us to report to the FDA or governmental authorities in other countries if our products cause or contribute to a death or serious injury or malfunction in a way that would be reasonably likely to contribute to death or serious injury if the malfunction were to recur. The FDA and similar governmental authorities in other countries have the authority to require the
28
recall of our products in the event of material deficiencies or defects in design or manufacturing. A government mandated, or voluntary, recall by us could occur as a result of component failures, manufacturing errors or design defects, including defects in labeling. Any recall would divert managerial and financial resources and could harm our reputation with customers. There can be no assurance that there will not be product recalls in the future or that such recalls would not have a material adverse effect on our business. Furthermore, we may later discover previously unknown problems with our products, including medically serious device related events. For example, we do not have long-term data on the safety of the Maestro System. Thus, there is a risk that long-term use of our Maestro System could cause injuries or harm, including possible damage to the vagus nerve. Any discovery of previously unknown problems with our product, including medically serious device related events, may result in restrictions on such products, withdrawal of the products from the market, voluntary or mandatory recalls, fines, suspension of regulatory approvals, product seizures, injunctions or the imposition of civil or criminal penalties.
We depend on a limited number of manufacturers and suppliers of various critical components for our Maestro System. The loss of any of these manufacturer or supplier relationships could delay our clinical trials or prevent or delay commercialization of our Maestro System.
We rely entirely on third parties to manufacture our Maestro System and to supply us with all of the critical components of our Maestro System, including our leads, implantable batteries, neuroregulators and controllers. If any of our existing suppliers were unable or unwilling to meet our demand for product components, or if the components or finished products that they supply do not meet quality and other specifications, clinical trials or commercialization of our product could be delayed. Alternatively, if we have to switch to a replacement manufacturer or replacement supplier for any of our product components, we may face additional regulatory delays, and the manufacture and delivery of our Maestro System could be interrupted for an extended period of time, which could delay completion of our clinical trials or commercialization of our Maestro System. In addition, we may be required to obtain regulatory approval from the FDA to use different suppliers or components.
If our device manufacturers or our suppliers are unable to provide an adequate supply of our product following the start of commercialization, our growth could be limited and our business could be harmed.
In order to produce our Maestro System in the quantities that we anticipate will be required to meet anticipated market demand, we will need our manufacturers to increase, or scale-up, the production process by a significant factor over our current level of production. There are technical challenges to scaling-up manufacturing capacity and developing commercial-scale manufacturing facilities that may require the investment of substantial additional funds by our manufacturers and hiring and retaining additional management and technical personnel who have the necessary manufacturing experience. If our manufacturers are unable to do so, we may not be able to meet the requirements for the launch of the product or to meet future demand, if at all. We may also represent only a small portion of our supplier’s or manufacturer’s business and if they become capacity constrained they may choose to allocate their available resources to other customers that represent a larger portion of their business. We currently anticipate that we will continue to rely on third-party manufacturers and suppliers for the production of the Maestro System following commercialization. If we develop and obtain regulatory approval for our product and are unable to obtain a sufficient supply of our product, our revenue, business and financial prospects would be adversely affected.
If we are unable to establish sales and marketing capabilities or enter into and maintain arrangements with third parties to market and sell our Maestro System, our business may be harmed.
We do not have a sales organization and have no experience as a company in sales, marketing and distribution of our product. To generate sales we will need to develop a sales and marketing infrastructure or contract with third parties to perform that function. Developing a sales force is expensive and time consuming and could delay or limit the success of any product launch. Even if we obtain approval from the FDA to market our Maestro System, we may be unable to develop an effective sales and marketing organization on a timely basis, if at all. If we develop our own sales and marketing capabilities, our sales force will be competing with the
29
experienced and well-funded marketing and sales organizations of our more established competitors. If we are unable to establish our own sales and marketing capabilities, we will need to contract with third parties to market and sell our product. In this event, our profit margins would likely be lower than if we performed these functions ourselves. In addition, we would necessarily be relying on the skills and efforts of others for the successful marketing of our product. If we are unable to establish and maintain effective sales and marketing capabilities, independently or with others, we may not be able to generate product revenue and may not become profitable.
We will need substantial additional funding and may be unable to raise capital when needed, which would force us to delay, reduce or eliminate our product development programs or liquidate some or all of our assets.
Our operations have consumed substantial amounts of cash since inception. We expect to continue to spend substantial amounts on research and development, including conducting current and future clinical trials for our Maestro System. Even before we receive regulatory approval to market our Maestro System, we expect to spend significant funds commercializing the product, including development of a direct sales force. In 2009, our cash used in operations was $24.7 million. Our cash used in operations in 2010 and beyond will largely depend on our regulatory path forward. If the FDA grants us approval on an IDE application for a clinical trial using the next-generation Maestro RC System in the treatment of morbid obesity we would expect research and development expenditures to increase in support of a new clinical trial in addition to the continued follow-up on existing trials, such as VBLOC-DM2 ENABLE and EMPOWER. In 2010 and the years following, we expect that our cash used in operations will be significant, and we will need to raise substantial additional capital to continue our research and development programs, commercialize our Maestro System, if approved by the FDA, and fund our on going operations.
Our future funding requirements will depend on many factors, including:
| • | the scope, rate of progress, results and cost of our clinical trials and other research and development activities; |
| • | the cost and timing of regulatory approvals; |
| • | the cost and timing of establishing sales, marketing and distribution capabilities; |
| • | the cost of establishing clinical and commercial supplies of our Maestro System and any products that we may develop; |
| • | the rate of market acceptance of our Maestro System and VBLOC therapy and any other product candidates; |
| • | the cost of filing and prosecuting patent applications and defending and enforcing our patent and other intellectual property rights; |
| • | the cost of defending, in litigation or otherwise, any claims that we infringe third-party patent or other intellectual property rights; |
| • | the effect of competing products and market developments; |
| • | the cost of explanting clinical devices; |
| • | the terms and timing of any collaborative, licensing or other arrangements that we may establish; |
| • | any revenue generated by sales of our future products; and |
| • | the extent to which we acquire or invest in businesses, products and technologies, although we currently have no commitments or agreements relating to any of these types of transactions. |
Until the time, if ever, when we can generate a sufficient amount of product revenue, we expect to finance our future cash needs through public or private equity offerings, debt financings or corporate collaboration, licensing arrangements and grants, as well as through interest income earned on cash balances.
30
Additional capital may not be available on terms favorable to us, or at all. If we raise additional funds by issuing equity securities, our stockholders may experience dilution. Debt financing, if available, may involve restrictive covenants or additional security interests in our assets. Any additional debt or equity financing that we complete may contain terms that are not favorable to us or our stockholders. Issuing public equity or debt securities may also be more costly or time-consuming for us because the aggregate market value of our common stock held by non-affiliates (public float) is less than $75.0 million (calculated in accordance with the SEC rules and regulations), which limits the size of offerings we may make using a Form S-3 registration statement to 1/3 of our public float for any twelve month period. If we raise additional funds through collaboration and licensing arrangements with third parties, it may be necessary to relinquish some rights to our technologies or products, or grant licenses on terms that are not favorable to us. If we are unable to raise adequate funds, we may have to delay, reduce the scope of, or eliminate some or all of, our development programs or liquidate some or all of our assets.
We may be unable to attract and retain management and other personnel we need to succeed.
Our success depends on the services of our senior management and other key research and development employees. The loss of the services of one or more of our officers or key research and development employees could delay or prevent the successful completion of our clinical trials and the commercialization of our Maestro System. Upon receiving regulatory approval for our product, we expect to rapidly expand our operations and grow our research and development, product development and administrative operations. Our growth will require hiring a significant number of qualified clinical, scientific, commercial and administrative personnel. Accordingly, recruiting and retaining such personnel in the future will be critical to our success. There is intense competition from other companies and research and academic institutions for qualified personnel in the areas of our activities. If we fail to identify, attract, retain and motivate these highly skilled personnel, we may be unable to continue our development and commercialization activities.
We may be unable to manage our growth effectively.
Our business strategy entails significant future growth. For example, we will have to expand existing operations in order to conduct additional clinical trials, increase our contract manufacturing capabilities, hire and train new personnel to handle the marketing and sales of our product, assist patients in obtaining reimbursement for the use of our product and create and develop new applications for our technology. This growth may place significant strain on our management and financial and operational resources. Successful growth is also dependent upon our ability to implement appropriate financial and management controls, systems and procedures. Our ability to effectively manage growth depends on our success in attracting and retaining highly qualified personnel, for which the competition may be intense. If we fail to manage these challenges effectively, our business could be harmed.
We face the risk of product liability claims that could be expensive, divert management’s attention and harm our reputation and business. We may not be able to obtain adequate product liability insurance.
Our business exposes us to a risk of product liability claims that is inherent in the testing, manufacturing and marketing of medical devices. The medical device industry has historically been subject to extensive litigation over product liability claims. We may be subject to product liability claims if our Maestro System, or any other products we may sell, causes, or appears to have caused, an injury. Claims may be made by consumers, healthcare providers, third-party strategic collaborators or others selling our products.
We have $5 million of product liability insurance, which covers the use of our Maestro System and VBLOC therapy in our clinical trials, which amount we believe is appropriate. Our current product liability insurance may not continue to be available to us on acceptable terms, if at all, and, if available, the coverage may not be adequate to protect us against any future product liability claims. If we are unable to obtain insurance at an acceptable cost and on acceptable terms for an adequate coverage amount, or otherwise to protect against
31
potential product liability claims, we could be exposed to significant liabilities, which may harm our business. A product liability claim, recall or other claim with respect to uninsured liabilities or for amounts in excess of insured liabilities could have a material adverse effect on our business, financial condition and results of operations. These liabilities could prevent or interfere with our product commercialization efforts. Defending a suit, regardless of merit, could be costly, could divert management attention and might result in adverse publicity, which could result in the withdrawal of, or inability to recruit, clinical trial volunteers or result in reduced acceptance of our Maestro System and VBLOC therapy in the market.
We may be subject to product liability claims even if it appears that the claimed injury is due to the actions of others. For example, we rely on the expertise of surgeons and other associated medical personnel to perform the medical procedure to implant and remove our Maestro System and to perform the related VBLOC therapy. If these medical personnel are not properly trained or are negligent, the therapeutic effect of our Maestro System and VBLOC therapy may be diminished or the patient may suffer critical injury, which may subject us to liability. In addition, an injury that is caused by the negligence of one of our suppliers in supplying us with a defective component that injures a patient could be the basis for a claim against us. A product liability claim, regardless of its merit or eventual outcome, could result in decreased demand for our products; injury to our reputation; diversion of management’s attention; withdrawal of clinical trial participants; significant costs of related litigation; substantial monetary awards to patients; product recalls or market withdrawals; loss of revenue; and the inability to commercialize our products under development.
We may be subject, directly or indirectly, to federal and state healthcare fraud and abuse and false claims laws and regulations. Prosecutions under such laws have increased in recent years and we may become subject to such litigation. If we are unable to, or have not fully complied with such laws, we could face substantial penalties.
If we are successful in achieving regulatory approval to market our Maestro System, our operations will be directly, or indirectly through our customers, subject to various state and federal fraud and abuse laws, including, without limitation, the federal Anti-Kickback Statute and federal False Claims Act. These laws may impact, among other things, our proposed sales, marketing and education programs.
The federal Anti-Kickback Statute prohibits persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual, or the furnishing or arranging for a good or service, for which payment may be made under a federal healthcare program such as the Medicare and Medicaid programs. Several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered business, the statute has been violated. The Anti-Kickback Statute is broad and, despite a series of narrow safe harbors, prohibits many arrangements and practices that are lawful in businesses outside of the healthcare industry. Penalties for violations of the federal Anti-Kickback Statute include criminal penalties and civil sanctions such as fines, imprisonment and possible exclusion from Medicare, Medicaid and other federal healthcare programs. Many states have also adopted laws similar to the federal Anti-Kickback Statute, some of which apply to the referral of patients for healthcare items or services reimbursed by any source, not only the Medicare and Medicaid programs.
The federal False Claims Act prohibits persons from knowingly filing, or causing to be filed, a false claim to, or the knowing use of false statements to obtain payment from the federal government. Suits filed under the False Claims Act, known as “qui tam” actions, can be brought by any individual on behalf of the government and such individuals, commonly known as “whistleblowers,” may share in any amounts paid by the entity to the government in fines or settlement. The frequency of filing qui tam actions has increased significantly in recent years, causing greater numbers of medical device, pharmaceutical and healthcare companies to have to defend a False Claim Act action. When an entity is determined to have violated the federal False Claims Act, it may be required to pay up to three times the actual damages sustained by the government, plus civil penalties for each separate false claim. Various states have also enacted laws modeled after the federal False Claims Act.
32
We are unable to predict whether we could be subject to actions under any of these laws, or the impact of such actions. If we are found to be in violation of any of the laws described above and other applicable state and federal fraud and abuse laws, we may be subject to penalties, including civil and criminal penalties, damages, fines, exclusion from government healthcare reimbursement programs and the curtailment or restructuring of our operations.
We incur significant costs as a result of operating as a public company, and our management is required to devote substantial time to new compliance initiatives.
As a public company, we incur significant legal, accounting and other expenses. In addition, the Sarbanes-Oxley Act of 2002, as well as rules subsequently implemented by the SEC and NASDAQ have imposed various requirements on public companies, including establishment and maintenance of effective disclosure and financial controls and changes in corporate governance practices. Our management and other personnel devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations result in increased legal and financial compliance costs and will make some activities more time-consuming and costly.
The Sarbanes-Oxley Act of 2002 requires, among other things, that we maintain effective internal controls for financial reporting and disclosure. In particular, we are required to perform system and process evaluation and testing of our internal controls over financial reporting to allow management to report on the effectiveness of our internal controls over financial reporting, as required by Section 404 of the Sarbanes-Oxley Act. Our testing may reveal deficiencies in our internal controls over financial reporting that are deemed to be material weaknesses. We have incurred and continue to expect to incur significant expense and devote substantial management effort toward ensuring compliance with Section 404. We currently do not have an internal audit function, and we may need to hire additional accounting and financial staff with appropriate public company experience and technical accounting knowledge. Moreover, if we do not comply with the requirements of Section 404, or if we identify deficiencies in our internal controls that are deemed to be material weaknesses, the market price of our stock could decline and we could be subject to sanctions or investigations by NASDAQ, the SEC or other regulatory authorities, which would entail expenditure of additional financial and management resources.
We operate in a highly competitive industry that is subject to rapid change. If our competitors are able to develop and market products that are safer or more effective than our products, our commercial opportunities will be reduced or eliminated.
The health care industry is highly competitive, subject to rapid change and significantly affected by new product introductions and other market activities of industry participants. The obesity treatment market in which we operate has grown significantly in recent years and is expected to continue to expand as technology continues to evolve and awareness of the need to treat the obesity epidemic grows. Although we are not aware of any competitors in the neuroblocking market, we face potential competition from pharmaceutical and surgical obesity treatments. Many of our competitors in the obesity treatment field have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, clinical trials, obtaining regulatory approvals and marketing approved products than we do. Smaller or early-stage companies may also prove to be significant competitors, particularly if they pursue competing solutions through collaborative arrangements with large and established companies, such as Allergan, Cyberonics, Johnson & Johnson, Medtronic or St. Jude Medical. Our competitors may develop and patent processes or products earlier than us, obtain regulatory approvals for competing products more rapidly than we are able to and develop more effective, safer and less expensive products or technologies that would render our products non-competitive or obsolete.
We may not be successful in our efforts to utilize our VBLOC therapy to treat co-morbidities associated with obesity and other gastrointestinal diseases and disorders.
As part of our long-term business strategy, we plan to research the application of our VBLOC therapy to treat co-morbidities associated with obesity and other gastrointestinal diseases and disorders. Research to identify new target applications requires substantial technical, financial and human resources, whether or not any new
33
applications for our VBLOC therapy are ultimately identified. We may be unable to identify or pursue other applications of our technology. Even if we identify potential new applications for our VBLOC therapy, investigating the safety and efficacy of our therapy requires extensive clinical testing, which is expensive and time-consuming. If we terminate a clinical trial in which we have invested significant resources, our prospects will suffer, as we will have expended resources on a program that will not provide a return on our investment and missed the opportunity to allocate those resources to potentially more productive uses. We will also need to obtain regulatory approval for these new applications, as well as achieve market acceptance and an acceptable level of reimbursement.
Risks Related to Intellectual Property
If we are unable to obtain or maintain intellectual property rights relating to our technology and neuroblocking therapy, the commercial value of our technology and any future products will be adversely affected and our competitive position will be harmed.
Our commercial success depends in part on our ability to obtain protection in the United States and other countries for our Maestro System and VBLOC therapy by establishing and maintaining intellectual property rights relating to or incorporated into our technology and products. To date, we have nine issued U.S. patents, seven of which pertain to treating gastrointestinal disorders, 22 U.S. patent applications, four pending international patent applications (PCT) and fourteen national stage patent applications, including seven European applications, in foreign jurisdictions. In addition, we are the exclusive licensee to two U.S. patent applications owned by Mayo Foundation for Medical Education and Research, which are unrelated to our VBLOC therapy. Our pending and future patent applications may not issue as patents or, if issued, may not issue in a form that will provide us any competitive advantage. We expect to incur substantial costs in obtaining patents and, if necessary, defending our proprietary rights. The patent positions of medical device companies, including ours, can be highly uncertain and involve complex and evolving legal and factual questions. We do not know whether we will obtain the patent protection we seek, or that the protection we do obtain will be found valid and enforceable if challenged. If we fail to obtain adequate protection of our intellectual property, or if any protection we obtain is reduced or eliminated, others could use our intellectual property without compensating us, resulting in harm to our business. We may also determine that it is in our best interests to voluntarily challenge a third party’s products or patents in litigation or administrative proceedings, including patent interferences or re-examinations. In the event that we seek to enforce any of our owned or exclusively licensed patents against an infringing party, it is likely that the party defending the claim will seek to invalidate the patents we assert, which, if successful could result in the loss of the entire patent or the relevant portion of our patent, which would not be limited to any particular party. Any litigation to enforce or defend our patent rights, even if we were to prevail, could be costly and time-consuming and could divert the attention of our management and key personnel from our business operations. Even if we were to prevail in any litigation, we cannot assure you that we can obtain an injunction that prevents our competitors from practicing our patented technology. Our competitors may independently develop similar or alternative technologies or products without infringing any of our patent or other intellectual property rights, or may design around our proprietary technologies.
We cannot assure you that we will obtain any patent protection that we seek, that any protection we do obtain will be found valid and enforceable if challenged or that it will confer any significant commercial advantage. U.S. patents and patent applications may also be subject to interference proceedings and U.S. patents may be subject to re-examination proceedings in the U.S. Patent and Trademark Office (USPTO) and foreign patents may be subject to opposition or comparable proceedings in the corresponding foreign patent offices, which proceedings could result in either loss of the patent or denial of the patent application, or loss or reduction in the scope of one or more of the claims of, the patent or patent application. In addition, such interference, re-examination and opposition proceedings may be costly. Moreover, the U.S. patent laws may change, possibly making it easier to challenge patents. Some of our technology was, and continues to be, developed in conjunction with third parties, and thus there is a risk that such third parties may claim rights in our intellectual property. Thus, any patents that we own or license from others may provide limited or no protection against competitors. Our pending patent applications, those we may file in the future, or those we may license from third parties, may
34
not result in patents being issued. If issued, they may not provide us with proprietary protection or competitive advantages against competitors with similar technology.
Non-payment or delay in payment of patent fees or annuities, whether intentional or unintentional, may result in loss of patents or patent rights important to our business. Many countries, including certain countries in Europe, have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In addition, many countries limit the enforceability of patents against third parties, including government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of the patent. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as do the laws of the United States, particularly in the field of medical products and procedures.
Many of our competitors have significant resources and incentives to apply for and obtain intellectual property rights that could limit or prevent our ability to commercialize our current or future products in the United States or abroad.
Many of our competitors who have significant resources and have made substantial investments in competing technologies may seek to apply for and obtain patents that will prevent, limit or interfere with our ability to make, use or sell our products either in the United States or in international markets. Our current or future U.S. or foreign patents may be challenged, circumvented by competitors or others or may be found to be invalid, unenforceable or insufficient. Since patent applications are confidential until patents are issued in the United States, or in most cases, until after 18 months from filing of the application, or corresponding applications are published in other countries, and since publication of discoveries in the scientific or patent literature often lags behind actual discoveries, we cannot be certain that we were the first to make the inventions covered by each of our pending patent applications, or that we were the first to file patent applications for such inventions.
If we are unable to protect the confidentiality of our proprietary information and know-how, the value of our technology and products could be adversely affected.
In addition to patented technology, we rely on our unpatented proprietary technology, trade secrets, processes and know-how. We generally seek to protect this information by confidentiality agreements with our employees, consultants, scientific advisors and third parties. These agreements may be breached, and we may not have adequate remedies for any such breach. In addition, our trade secrets may otherwise become known or be independently developed by competitors. To the extent that our employees, consultants or contractors use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions.
Intellectual property litigation is a common tactic in the medical device industry to gain competitive advantage. If we become subject to a lawsuit, we may be required to expend significant financial and other resources and our management’s attention may be diverted from our business.
There has been a history of frequent and extensive litigation regarding patent and other intellectual property rights in the medical device industry, and companies in the medical device industry have employed intellectual property litigation to gain a competitive advantage. Accordingly, we may become subject to patent infringement claims or litigation in a court of law, or interference proceedings declared by the USPTO to determine the priority of inventions or an opposition to a patent grant in a foreign jurisdiction. We may also become subject to claims or litigation seeking payment of royalties based on sales of our product in connection with licensing or similar joint development arrangements with third parties or in connection with claims of patent infringement. The defense and prosecution of intellectual property suits, USPTO interference or opposition proceedings and related legal and administrative proceedings, are both costly and time consuming and could result in substantial uncertainty to us. Litigation or regulatory proceedings may also be necessary to enforce patent or other intellectual property rights of ours or to determine the scope and validity of other parties’ proprietary rights. Any
35
litigation, opposition or interference proceedings, with or without merit, may result in substantial expense to us, cause significant strain on our financial resources, divert the attention of our technical and management personnel and harm our reputation. We may not have the financial resources to defend our patents from infringement or claims of invalidity. An adverse determination in any litigation could subject us to significant liabilities to third parties, require us to seek licenses from or pay royalties to third parties or prevent us from manufacturing, selling or using our proposed products, any of which could have a material adverse effect on our business and prospects. We are not currently a party to any patent or other litigation.
Our VBLOC therapy or Maestro System may infringe or be claimed to infringe patents that we do not own or license, including patents that may issue in the future based on patent applications of which we are currently aware, as well as applications of which we are unaware. For example, we are aware of other companies that are investigating neurostimulation, including neuroblocking, and of patents and published patent applications held by companies in those fields. While we believe that none of such patents and patent applications are applicable to our products and technologies under development, third parties who own or control these patents and patent applications in the United States and abroad could bring claims against us that would cause us to incur substantial expenses and, if such claims are successfully asserted against us, they could cause us to pay substantial damages, could result in an injunction preventing us from selling, manufacturing or using our proposed products and would divert management’s attention. Because patent applications in many countries such as the United States are maintained under conditions of confidentiality and can take many years to issue, there may be applications now pending of which we are unaware and which may later result in issued patents that our products infringe. If a patent infringement suit were brought against us, we could be forced to stop our ongoing or planned clinical trials, or delay or abandon commercialization of the product that is subject of the suit.
As a result of patent infringement claims, or to avoid potential claims, we may choose or be required to seek a license from a third party and be required to pay license fees or royalties, or both. A license may not be available at all or on commercially reasonable terms, and we may not be able to redesign our products to avoid infringement. Modification of our products or development of new products could require us to conduct additional clinical trials and to revise our filings with the FDA and other regulatory bodies, which would be time-consuming and expensive. Even if we were able to obtain a license, the rights may be nonexclusive, which could result in our competitors gaining access to the same intellectual property. Ultimately, we could be forced to cease some aspect of our business operations if, as a result of actual or threatened patent infringement claims, we are unable to enter into licenses on acceptable terms. This could harm our business significantly.
Risks Related to Ownership of our Common Stock
The trading price of our common stock has been volatile and is likely to be volatile in the future.
The trading price of our common stock has been highly volatile. Further, our common stock has a limited trading history. Since our public offering in November 2007 through February 26, 2010 our stock price has fluctuated from a low of $0.40 to a high of $10.77. The market price for our common stock will be affected by a number of factors, including:
| • | the denial or delay of regulatory clearances or approvals of our product or receipt of regulatory approval of competing products; |
| • | our ability to accomplish clinical, regulatory and other product development milestones and to do so in accordance with the timing estimates we have publicly announced; |
| • | changes in policies affecting third-party coverage and reimbursement in the United States and other countries; |
| • | changes in government regulations and standards affecting the medical device industry and our product; |
| • | ability of our product, if it receives regulatory approval, to achieve market success; |
| • | the performance of third-party contract manufacturers and component suppliers; |
36
| • | our ability to develop sales and marketing capabilities; |
| • | actual or anticipated variations in our results of operations or those of our competitors; |
| • | announcements of new products, technological innovations or product advancements by us or our competitors; |
| • | developments with respect to patents and other intellectual property rights; |
| • | sales of common stock or other securities by us or our stockholders in the future; |
| • | additions or departures of key scientific or management personnel; |
| • | disputes or other developments relating to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our technologies; |
| • | trading volume of our common stock; |
| • | changes in earnings estimates or recommendations by securities analysts, failure to obtain analyst coverage of our common stock or our failure to achieve analyst earnings estimates; |
| • | public statements by analysts or clinicians regarding their perceptions of our clinical results or the effectiveness of our products; |
| • | decreases in market valuations of medical device companies; and |
| • | general market conditions and other factors unrelated to our operating performance or the operating performance of our competitors. |
The stock prices of many companies in the medical device industry have experienced wide fluctuations that have often been unrelated to the operating performance of these companies. Following periods of volatility in the market price of a company’s securities, securities class action litigation often has been initiated against a company. If class action litigation is initiated against us, we may incur substantial costs and our management’s attention may be diverted from our operations, which could significantly harm our business.
Our directors and executive officers hold substantial control over us and could limit your ability to influence the outcome of key transactions, including changes of control.
Our executive officers and directors and entities affiliated with them beneficially own, in the aggregate, approximately 39.1% of our outstanding common stock as of February 26, 2010. Our executive officers, directors and affiliated entities, if acting together, would be able to influence significantly all matters requiring approval by our stockholders, including the election of directors and the approval of mergers or other significant corporate transactions. The concentration of ownership of our common stock may have the effect of delaying, preventing or deterring a change of control of our company, could deprive our stockholders of an opportunity to receive a premium for their common stock as part of a sale of our company and may affect the market price of our common stock. This significant concentration of stock ownership may adversely affect the trading price of our common stock due to investors’ perception that conflicts of interest may exist or arise.
Sales of a substantial number of shares of our common stock in the public market by existing stockholders, or the perception that they may occur, could cause our stock price to decline.
Sales of substantial amounts of our common stock by us or by our stockholders, announcements of the proposed sales of substantial amounts of our common stock or the perception that substantial sales may be made, could cause the market price of our common stock to decline. We may issue additional shares of our common stock in follow-on offerings to raise additional capital or in connection with acquisitions or corporate alliances and we plan to issue additional shares to our employees, directors or consultants in connection with their services to us. All of the currently outstanding shares of our common stock are freely tradable under federal and state securities laws, except for shares held by our directors, officers and certain greater than five percent stockholders,
37
which may be subject to volume limitations, and shares issued in connection with our recent private placement offering. Due to these factors, sales of a substantial number of shares of our common stock in the public market could occur at any time and could reduce the market price of our common stock.
In addition, certain holders of our common stock and warrants to purchase our common stock have rights, subject to some conditions, to require us to file registration statements covering their shares or to include their shares in registration statements that we may file for ourselves or other stockholders. If we were to include in a company-initiated registration statement shares held by those holders pursuant to the exercise of their registration rights, the sale of those shares could impair our ability to raise needed capital by depressing the price at which we could sell our common stock.
Our organizational documents and Delaware law make a takeover of our company more difficult, which may prevent certain changes in control and limit the market price of our common stock.
Our certificate of incorporation and bylaws and Section 203 of the Delaware General Corporation Law contain provisions that may have the effect of deterring or delaying attempts by our stockholders to remove or replace management, engage in proxy contests and effect changes in control. These provisions include:
| • | our board of directors will be authorized, without prior stockholder approval, to create and issue preferred stock which could be used to implement anti-takeover devices; |
| • | advance notice will be required for director nominations or for proposals that can be acted upon at stockholder meetings; |
| • | our board of directors will be classified such that not all members of our board are elected at one time, which may make it more difficult for a person who acquires control of a majority of our outstanding voting stock to replace all or a majority of our directors; |
| • | stockholder action by written consent will be prohibited; |
| • | special meetings of the stockholders will be permitted to be called only by the chairman of our board of directors or by a majority of our board of directors; and |
| • | stockholders will not be permitted to accumulate their votes for the election of directors; and stockholders will be permitted to amend our bylaws only upon receiving a majority of the votes entitled to be cast by holders of all outstanding shares then entitled to vote generally in the election of directors, voting together as a single class. |
In addition, as a Delaware corporation, we are subject to Delaware law, including Section 203 of the Delaware General Corporation Law. In general, Section 203 prohibits a Delaware corporation from engaging in any business combination with any interested stockholder for a period of three years following the date that the stockholder became an interested stockholder unless certain specific requirements are met as set forth in Section 203. These provisions, alone or together, could have the effect of deterring or delaying changes in incumbent management, proxy contests or changes in control.
These provisions also could discourage proxy contests and make it more difficult for you and other stockholders to elect directors and take other corporate actions. The existence of these provisions could limit the price that investors might be willing to pay in the future for shares of our common stock. Some provisions in our certificate of incorporation and bylaws may deter third parties from acquiring us, which may limit the market price of our common stock.
We have not paid dividends in the past and do not expect to pay dividends in the future, and any return on investment may be limited to the value of our common stock.
We have never paid dividends on our common stock and do not anticipate paying dividends on our common stock in the foreseeable future. The payment of dividends on our common stock will depend on our earnings,
38
financial condition and other business and economic factors affecting us at such time as our board of directors may consider relevant. If we do not pay dividends, our common stock may be less valuable because a return on your investment will only occur if our stock price appreciates.
| ITEM 1B. | UNRESOLVED STAFF COMMENTS |
Not applicable.
| ITEM 2. | PROPERTIES |
We lease approximately 28,388 square feet of lab and office space in St. Paul, Minnesota. The lease agreement began October 1, 2008 and ends September 30, 2015.
| ITEM 3. | LEGAL PROCEEDINGS |
We are not currently a party to any litigation and we are not aware of any pending or threatened litigation against us that could have a material adverse effect on our business, operating results or financial condition. The medical device industry in which we operate is characterized by frequent claims and litigation, including claims regarding patent and other intellectual property rights as well as improper hiring practices. As a result, we may be involved in various legal proceedings from time to time.
| ITEM 4. | RESERVED |
39
PART II.
| ITEM 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Market For Our Common Stock
Our common stock has been traded on the NASDAQ Stock Market under the symbol “ETRM” since our initial public offering (IPO) on November 15, 2007. Prior to that date, there was no public market for our common stock. Our stock was traded on the NASDAQ Global Market from its initial listing at the time of our IPO until January 21, 2010. Subsequently, in anticipation of not curing our deficiencies with the continued listing requirements of the NASDAQ Global Market, we requested and were approved to transfer to the NASDAQ Capital Market, effective January 22, 2010.
As of February 26, 2010, there were approximately 60 holders of record of our common stock and 44,856,657 shares of common stock outstanding. No dividends have been paid on our common stock to date, and we do not anticipate paying any dividends in the foreseeable future.
The following table sets forth the high and low sales prices of our common stock as quoted on the NASDAQ Global Market for the periods indicated.
Price Range of Common Stock
| Price Range | ||||||
| High | Low | |||||
| Fiscal 2008 |
||||||
| First Quarter |
$ | 10.26 | $ | 3.45 | ||
| Second Quarter |
$ | 5.75 | $ | 3.75 | ||
| Third Quarter |
$ | 5.24 | $ | 3.02 | ||
| Fourth Quarter |
$ | 3.24 | $ | 0.83 | ||
| Fiscal 2009 |
||||||
| First Quarter |
$ | 5.38 | $ | 1.10 | ||
| Second Quarter |
$ | 4.37 | $ | 1.25 | ||
| Third Quarter |
$ | 5.58 | $ | 2.68 | ||
| Fourth Quarter |
$ | 4.90 | $ | 0.40 | ||
The closing price for our common stock as reported by the NASDAQ Capital Market on February 26, 2010 was $0.53 per share.
Securities Authorized for Issuance Under Equity Compensation Plans
The information required by this Item regarding equity compensation plans is incorporated by reference to the information set forth in PART III, Item 12 of this Annual Report on Form 10-K.
Unregistered Sales of Equity Securities
From January 1, 2009 through December 31, 2009, we sold and issued the following unregistered securities. Also included is the consideration, if any, received by us for such shares, warrants, promissory notes and options and information relating to the section of the Securities Act of 1933, as amended (the Securities Act), or rules of the SEC, under which exemption from registration was claimed.
As previously described in our Current Report on Form 8-K filed November 24, 2008, on November 18, 2008, we entered into a new loan and security agreement with Silicon Valley Bank (SVB), Venture Lending & Leasing V, Inc. (a private equity fund under the management of Western Technology Investment (WTI)) and
40
Compass Horizon Funding Company LLC (Horizon and, collectively with SVB and WTI, the Lenders), pursuant to which the Lenders agreed to make term loans (each, a Term Loan) to the Company in an aggregate principal amount of up to $20.0 million, on the terms and conditions set forth in the loan agreement. On April 28, 2009, Horizon funded a Term Loan in the aggregate principal amount of $5.0 million. In conjunction with the funding, Horizon received a warrant to purchase an aggregate number of shares equal to $495,000 divided by the per share exercise price of the warrant, or 296,763 common stock warrants with an exercise price of $1.668 per share and a ten year life. The warrants give Horizon the option to purchase either (i) shares of our common stock with a per share exercise price equal to $1.668, or (ii) shares of our stock (including common stock) issued in an equity financing that occurs after the warrant issue date and on or before May 18, 2010 at the per share price of the stock sold in the financing. The warrants that we issued are in the form attached as Exhibit 10.20 to our December 31, 2008 Annual Report on Form 10-K filed March 12, 2009. Pursuant to the terms of the warrant agreement, the number of shares issued to Horizon and the exercise price of the warrant were adjusted in connection with the closing of our private placement in February 2009, our registered direct offering in October 2009 and our registered direct offering in January 2010. See Note 7 to our consolidated financial statements included in Item 8 of this Annual Report on Form 10-K.
As previously described in our Current Report on Form 8-K filed February 25, 2009, on February 19, 2009, we entered into several securities purchase agreements for the sale of 13,110,393 shares of our common stock, together with warrants to purchase an aggregate of 6,555,197 shares of our common stock, in a private placement transaction with several accredited investors (the Private Placement). The purchase price per share was $1.15, which equaled the consolidated closing bid price of our common stock as reported by the NASDAQ Global Market on February 19, 2009. The warrants will be exercisable at any time and from time to time beginning on the date that is six months and one day after the closing of the Private Placement and ending four years after the closing of the Private Placement. The warrants have an exercise price of $1.38 per share, which equals 120% of the consolidated closing bid price of our common stock as reported by the NASDAQ Global Market on February 19, 2009. On February 24, 2009, we completed the final closing of the Private Placement receiving gross proceeds of $15.9 million, less a placement agent fee and certain other expenses. In addition, the placement agent received a warrant to purchase 218,242 shares of common stock in the same form as that issued to participants in the Private Placement.
The sales and issuances of securities described in the paragraphs above were deemed to be exempt from registration under the Securities Act by virtue of Section 4(2) of the Securities Act, as transactions by an issuer not involving any public offering.
Uses of Proceeds from Sale of Registered Securities
None.
Dividend Policy
We have never paid cash dividends on our common stock. The board of directors presently intends to retain all earnings for use in our business and does not anticipate paying cash dividends in the foreseeable future. We do not have a dividend reinvestment plan or a direct stock purchase plan.
Issuer Purchases of Equity Securities
None.
41
Stock Performance Graph
The following performance graph and related information shall not be deemed “soliciting material” or to be “filed” with the SEC, nor shall such information be incorporated by reference into any future filing under the Securities Act or Securities Exchange Act of 1934, each as amended, except to the extent that we specifically incorporate it by reference into such filing.
The following graph shows a comparison of cumulative total return for our common stock, the NASDAQ Composite Index, and the NASDAQ Medical Equipment Index. Such returns are based on historical results and are not intended to suggest future performance. The graph assumes $100 was invested in our common stock and in each of the indexes on November 15, 2007 (the date our common stock commenced trading on the NASDAQ Stock Market).
Data for the NASDAQ Composite Index and the NASDAQ Medical Equipment Index assume reinvestment of dividends. The Company has never paid dividends on its common stock and has no present plans to do so.
The stockholder return shown on the graph below is not necessarily indicative of future performance, and we do not make or endorse any predictions as to future stockholder returns.
COMPARISON OF 25 MONTH CUMULATIVE TOTAL RETURN*
Among EnteroMedics Inc., the NASDAQ Composite Index
and the NASDAQ Medical Equipment Index
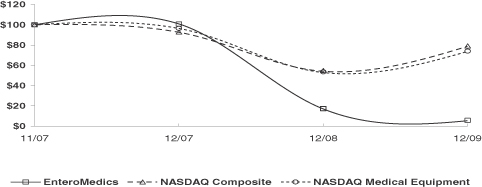
* $100 invested on 11/15/07 in stock or 10/31/07 in index, including reinvestment of dividends. No dividends have been declared or paid on our common stock. Stock performance shown in the above chart for the common stock is historical and should not be considered indicative of future price performance. This graph was prepared by Research Data Group, Inc.
| November 15, 2007 |
December 31, 2007 |
December 31, 2008 |
December 31, 2009 | |||||||||
| EnteroMedics Inc. |
$ | 100.00 | $ | 100.63 | $ | 18.25 | $ | 7.00 | ||||
| NASDAQ Composite |
100.00 | 92.70 | 54.79 | 79.14 | ||||||||
| NASDAQ Medical Equipment |
100.00 | 96.43 | 53.56 | 74.06 | ||||||||
42
| ITEM 6. | SELECTED FINANCIAL DATA |
The following table sets forth certain financial data with respect to our business. The information set forth below is not necessarily indicative of results of future operations and should be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in Item 7 and the consolidated financial statements and related notes thereto in Item 8 of this Annual Report on Form 10-K.
| Fiscal Years | ||||||||||||||||||||
| 2009(1) | 2008(1) | 2007(1)(2) | 2006(1) | 2005 | ||||||||||||||||
| (In thousands, except per share data) | ||||||||||||||||||||
| Operations: |
||||||||||||||||||||
| Loss from operations |
$ | (24,212 | ) | $ | (36,270 | ) | $ | (28,026 | ) | $ | (18,122 | ) | $ | (11,152 | ) | |||||
| Net loss |
(31,929 | ) | (37,874 | ) | (28,575 | ) | (17,690 | ) | (11,215 | ) | ||||||||||
| Basic and diluted net loss per share |
(1.07 | ) | (2.25 | ) | (11.69 | ) | (34.19 | ) | (28.82 | ) | ||||||||||
| Shares used to compute basic and diluted net loss per share |
29,846 | 16,836 | 2,445 | 517 | 389 | |||||||||||||||
| Financial Position: |
||||||||||||||||||||
| Cash, cash equivalents and short-term investments |
14,618 | 26,295 | 57,031 | 34,732 | 10,719 | |||||||||||||||
| Working capital (current assets less current liabilities) |
8,821 | 20,916 | 49,802 | 29,921 | 8,640 | |||||||||||||||
| Total assets |
16,214 | 28,279 | 59,051 | 36,064 | 11,561 | |||||||||||||||
| Long-term debt, net of current portion and discounts |
3,881 | 10,996 | 6,018 | 1,727 | 7,344 | |||||||||||||||
| Convertible preferred stock |
— | — | — | 103 | 46 | |||||||||||||||
| Deficit accumulated during development stage |
(133,368 | ) | (101,307 | ) | (63,433 | ) | (34,858 | ) | (17,168 | ) | ||||||||||
| Total stockholders’ equity |
5,581 | 11,405 | 45,282 | 28,574 | 1,975 | |||||||||||||||
| (1) | Loss from operations, net loss and basic and diluted net loss per share for 2009, 2008, 2007 and 2006 include the impact of fair value accounting for stock-based compensation charges, which were not presented in prior years. Refer to Notes 2 and 11 of our consolidated financial statements included in Item 8 of this Annual Report on Form 10-K. |
| (2) | Basic and diluted net loss per share and shares used to compute basic and diluted net loss per share include the impact of converting 10,488,178 shares of convertible preferred stock into common stock immediately prior to the closing of our initial public offering on November 20, 2007. |
43
| ITEM 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
Except for the historical information contained herein, the matters discussed in this “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” and elsewhere in this Form 10-K are forward-looking statements that involve risks and uncertainties. The factors listed in Item 1A “Risk Factors,” as well as any cautionary language in this Form 10-K, provide examples of risks, uncertainties and events that may cause our actual results to differ materially from those projected. Except as may be required by law, we undertake no obligation to update any forward-looking statement to reflect events after the date of this report.
Overview
We are a development stage medical device company focused on the design and development of devices that use neuroblocking technology to treat obesity, its associated co-morbidities, and other gastrointestinal disorders. Our proprietary neuroblocking technology, which we refer to as VBLOC therapy, is designed to intermittently block the vagus nerve using high frequency, low energy, electrical impulses. We have a limited operating history and we currently have no products approved for sale. Our initial product under development is the Maestro System, which uses VBLOC therapy to limit the expansion of the stomach, help control hunger sensations between meals, reduce the frequency and intensity of stomach contractions and produce a feeling of early and prolonged fullness. We were formerly known as Beta Medical, Inc. and were incorporated in Minnesota on December 19, 2002. We later reincorporated in Delaware on July 22, 2004. Since inception, we have devoted substantially all of our resources to the development and commercialization of our Maestro System.
Based on our understanding of vagal nerve function and nerve blocking from our preclinical studies and the results of our initial clinical trials, we believe the Maestro System may offer obese patients a minimally-invasive treatment alternative that has the potential to result in significant and sustained weight loss. We believe that our Maestro System will allow bariatric surgeons to help obese patients who are concerned about the risks and complications associated with gastric banding and gastric bypass surgery. In addition, data from sub-group analyses demonstrate that VBLOC therapy may hold promise in improving the obesity-related co-morbidities of diabetes and hypertension, independent of, and prior to, substantial weight loss. We are conducting, or plan to conduct, feasibility studies in each of these co-morbidities to assess VBLOC therapy’s potential in addressing multiple indications.
We are currently evaluating the Maestro System in human clinical trials conducted in the United States, Australia, Mexico, Norway and Switzerland. To date, we have not observed any mortality or any unanticipated adverse device effects in these clinical trials. We have also not observed any long-term problematic clinical side effects in any patients, including in those patients who have been using the Maestro System for more than one year.
On October 2, 2009, we announced preliminary results from our pivotal clinical study, the EMPOWER trial; indicating that based on an initial analysis, the study did not meet its primary and secondary efficacy endpoints. We also announced that there were no therapy-related serious adverse events reported during the study. The EMPOWER trial is a multi-center, randomized, double-blind, prospective, placebo-controlled pivotal study being conducted in the United States and selected international centers. We further announced on November 12, 2009, the ongoing detailed review suggests that vagal blocking therapy may promote safe and effective weight loss as an adjunct to behavioral support, diet and exercise in morbidly obese patients. The review further suggests that these effects were evident in both the treatment and control arms. We are continuing a comprehensive analysis of all clinical, statistical, and engineering data to understand this finding. Based on the analysis to date, the control arm of the trial, which was intended to be inactive, apparently provided a low-intensity blocking signal that introduced VBLOC therapy in human subjects.
In January 2010, we met with the U.S. Food and Drug Administration (FDA) to discuss the EMPOWER trial results and the regulatory process going forward. Based on this discussion, we recently submitted an Investigational Device Exemption (IDE) application for a clinical trial using the next-generation Maestro RC
44
System in the treatment of morbid obesity. Assuming that we obtain an approved IDE, successfully enroll and implant the trial and achieve favorable results, we plan to use data from that trial to support a premarket approval (PMA) application for the Maestro System, which we expect to submit no earlier than the second half of 2012. If the FDA grants us approval, we anticipate we will be able to commercialize the Maestro System in the United States no earlier than the second half of 2013.
If and when we obtain FDA approval of our Maestro System we intend to market our products in the United States through a direct sales force supported by field technical and marketing managers who provide training, technical and other support services to our customers. Outside the United States we intend to use direct, dealer and distributor sales models as the targeted geography best dictates. To date, we have relied on third-party manufacturers and suppliers for the production of our Maestro System. We currently anticipate that we will continue to rely on third-party manufacturers and suppliers for the production of the Maestro System. We obtained European CE Mark approval for our Maestro RF System on March 4, 2009. The method of assessing conformity with applicable regulatory requirements varies depending on the class of the device, but for our Maestro System (which falls into Class III), the method involved a combination of self-assessment by the manufacturer of the safety and performance of the device, and a third-party assessment by a Notified Body, usually of the design of the device and of the manufacturer’s quality system. We used KEMA in the Netherlands as the Notified Body for our CE marking approval process.
To date, we have generated no revenue from the sale of products, and we have incurred net losses in each year since our inception. As of December 31, 2009, we had a deficit accumulated during the development stage of $133.2 million. We expect our losses to continue and to increase as we continue our development activities. We have financed our operations primarily through public and private placement of our equity securities and issuance of debt.
Critical Accounting Policies and Significant Judgments and Estimates
Our management’s discussion and analysis of our financial condition and results of operations are based on our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements as well as the reported expenses during the reporting periods. We evaluate our estimates and judgments on an ongoing basis. Actual results may differ materially from these estimates under different assumptions or conditions.
While our significant accounting policies are more fully described in Note 2 to our consolidated financial statements included elsewhere in this Annual Report on Form 10-K, we believe that the following accounting policies and estimates are most critical to a full understanding and evaluation of our reported financial results.
Stock-Based Compensation
Prior to January 1, 2006, we accounted for stock-based employee compensation arrangements using the intrinsic value method and recognizing the expense over the option vesting period. The intrinsic value method is calculated as the difference, if any, between the fair value of our common stock and the exercise price on the date of the grant. We also followed the minimum value disclosure provisions. Using the intrinsic value method, we were not required to recognize stock-based compensation expense for employee stock options granted from inception through 2005 as the exercise prices, for financial reporting purposes, were determined to be at or above the deemed fair value of the underlying common stock on the date of grant. The fair value of our common stock was assessed and approved by our board of directors, the members of which have extensive experience in the life sciences industry and all but one of whom are nonemployee directors. In determining the appropriateness of the fair value of our common stock, the board of directors considered several factors, such as our life cycle, results of research and development, recent financings and financial projections.
45
Effective January 1, 2006, we adopted the fair value method of accounting for share-based payments, which superseded the previous accounting method, and requires compensation expense to be recognized using a fair-value-based method for costs related to all share-based payments including stock options. Companies are required to estimate the fair value of share-based payment awards on the date of grant using an option-pricing model. We adopted the new provisions using the prospective transition method. Under this method, compensation cost is recognized for all share-based payments granted or modified subsequent to December 31, 2005. All option grants valued after January 1, 2006 are expensed on a straight-line basis over the vesting period.
Calculating stock-based compensation expense requires the input of highly subjective assumptions, which represent our best estimates and involve inherent uncertainties and the application of management’s judgment. Estimates of stock-based compensation expenses are significant to our consolidated financial statements, but these expenses are based on the Black-Scholes pricing model and will never result in the payment of cash by us.
The application of share-based payment principles may be subject to further interpretation and refinement over time. There are significant differences among option valuation models, and this may result in a lack of comparability with other companies that use different models, methods and assumptions. If factors change and we employ different assumptions in the application of share-based payment accounting in future periods, or if we decide to use a different valuation model, the compensation expense that we record in the future may differ significantly from what we have recorded in the current period and could materially affect our operating loss, net loss and net loss per share.
The fair value method is applied to all share-based payment awards issued to employees and where appropriate, nonemployees, unless another source of literature applies. When determining the measurement date of a nonemployee’s share-based payment award, the Company measures the stock options at fair value and remeasures such stock options to the current fair value until the performance date has been reached. For stock options granted to nonemployees, the fair value of the stock options is estimated using the Black-Scholes valuation model. This model utilizes the estimated fair value of common stock and requires that, at the date of grant and each subsequent reporting period until the services are completed or a significant disincentive for nonperformance occurs, we make assumptions with respect to the expected term of the option, the volatility of the fair value of our common stock, risk free interest rates and expected dividend yields of our common stock. Different estimates of volatility and expected life of the option could materially change the value of an option and the resulting expense.
Common Stock Warrant Liability
Effective January 1, 2009, we adopted new authoritative accounting guidance regarding the financial reporting for outstanding equity-linked financial instruments. This adoption required certain warrants issued by us to be recorded as a liability and recorded at fair value. Calculating the fair value of the warrant liability requires the input of highly subjective assumptions, which requires our best estimates, and involves inherent uncertainties and the application of management’s judgment. The common stock warrant liability and related changes in fair value are significant to our consolidated financial statements and is based on a weighted-average Black-Scholes valuation model, however the warrant liability will never result in the payment of cash by us.
Net Operating Losses and Tax Credit Carryforwards
At December 31, 2009, we had federal and state net operating loss carryforwards of approximately $51.7 million and $50.7 million, respectively. These net operating loss carryforwards will expire in varying amounts from 2022 through 2029, if not utilized. Under the provisions of Sections 382 and 383 of the Internal Revenue Code, substantial changes in our ownership may limit the amount of net operating loss carryforwards and certain tax credits that can be utilized annually in the future to offset taxable income. A valuation allowance has been established to reserve the potential benefits of these carryforwards and tax credits in our consolidated financial statements to reflect the uncertainty of future taxable income required to utilize available tax loss
46
carryforwards and other deferred tax assets. If a change in our ownership is deemed to have occurred or occurs in the future, our ability to use our net operating loss carryforwards and tax credits in any fiscal year may be significantly limited.
Financial Overview
Revenue
To date, we have not commercialized any products and we have not generated any revenue. On October 2, 2009 we announced that our EMPOWER trial did not meet its primary and secondary efficacy endpoints. As such, we do not expect to generate revenue earlier than the second half of 2013 and then, only if we receive an approved IDE for a clinical trial using the next-generation Maestro RC System, successfully enroll and implant the clinical trial, achieve favorable results and receive FDA approval of our Maestro System. Any revenue from initial sales of a new product is difficult to predict and in any event will only modestly reduce our continued and increasing losses resulting from our research and development and other activities.
Research and Development Expenses
Our research and development expenses primarily consist of engineering, product development and clinical and regulatory expenses, incurred in the development of our Maestro System. Research and development expenses also include employee compensation, including stock-based compensation, consulting services, outside services, materials, supplies, depreciation and travel. We expense research and development costs as they are incurred. From inception through December 31, 2009, we have incurred a total of $91.6 million in research and development expenses. Our research and development expenditures in 2010 and beyond will largely depend on our regulatory path forward. If the FDA grants us approval on an IDE application for a clinical trial using the next-generation Maestro RC System in the treatment of morbid obesity we would expect research and development expenditures to increase in support of a new clinical trial in addition to the continued follow-up on existing trials, such as VBLOC-DM2 ENABLE and EMPOWER.
Selling, General and Administrative Expenses
Our selling, general and administrative expenses consist primarily of compensation for executive, finance, market development and administrative personnel, including stock-based compensation. Other significant expenses include costs associated with attending medical conferences, professional fees for legal, including legal services associated with our efforts to obtain and maintain broad protection for the intellectual property related to our products, and accounting services, cash management fees, consulting fees and travel expenses. From inception through December 31, 2009, we have incurred $31.9 million in selling, general and administrative expenses.
Results of Operations
Comparison of the Years Ended December 31, 2009 and 2008
Research and Development Expenses. Research and development expenses were $15.6 million for the year ended December 31, 2009, compared to $27.7 million for the year ended December 31, 2008. The decrease of $12.1 million, or 43.7%, is primarily due to decreases of $7.7 million, $2.2 million and $1.5 million in professional services, device costs and compensation and benefits expense, respectively. Professional services and device cost decreases are driven by the completion of enrollment and implants in our EMPOWER trial during 2008. We are currently incurring costs related to follow-up visits, which are less expensive than the cost of the implantation procedure, and do not require us to incur new device costs. The reduction in compensation and benefits expense is the result of a reduction-in-force completed December 1, 2008 and a 40% reduction-in-force completed October 27, 2009.
47
Selling, General and Administrative Expenses. Selling, general and administrative expenses were $8.6 million for the years ended December 31, 2009 and 2008. Although total annual selling, general and administrative expenses were consistent, various components fluctuated year-over-year. There were increases of $259,000 and $219,000 in compensation and benefits expense and professional services, respectively. The increases were driven by limited activities in support of commercializing the Maestro System in the event our EMPOWER trial met its primary and secondary efficacy endpoints and the FDA granted us approval to market our Maestro System. These limited activities were put on hold when we determined that our EMPOWER trial failed to meet the primary and secondary efficacy endpoints. The increases were offset by decreases of $331,000 and $104,000 in employee stock-based compensation expense and reduced travel expense, respectively. The decrease in employee stock-based compensation expense is primarily the result of the cancellation and related forfeiture of several stock options granted in 2008 with milestone features that were not achieved in 2009.
Interest Income. Interest income was $79,000 for the year ended December 31, 2009, compared to $1.1 million for the year ended December 31, 2008. The decrease of $1.0 million, or 92.8%, is primarily due to a decrease in short-term interest rates and a reduction in total cash available to invest. The average cash, cash equivalents and short-term investments balance was $30.1 million and $39.2 million for the years ended December 31, 2009 and 2008, respectively. The decreased average cash, cash equivalents and short-term investments balance is the result of $50.5 million in net cash used in operating and investing activities from January 1, 2008 through December 31, 2009 together with $23.5 million in debt principal payments during the same time period, offset by $15.0 million of debt funding received in November 2008, $15.1 million of net private placement proceeds received February 24, 2009, $5.0 million of additional debt funding received in April 2009 and $4.8 million of net registered direct proceeds received October 7, 2009.
Interest Expense. Interest expense was $4.1 million for the year ended December 31, 2009, compared to $2.7 million for the year ended December 31, 2008. The increase of $1.4 million, or 52.9%, was primarily the result of entering into a $20.0 million debt facility, of which $15.0 million was funded in November 2008 that resulted in net proceeds of $7.1 million after transaction expenses, facility charges and existing debt pay off and the funding of the remaining $5.0 million in April 2009. The effective rates on the $15.0 million and $5.0 million debt fundings are approximately 19% and 22%, respectively, compared to the old debt facility containing several outstanding loans with effective interest rates primarily ranging from approximately 15% to 17%. On December 1, 2009 we voluntarily prepaid two of the outstanding term loans in full, or approximately 50% of the outstanding principal balance. The prepayment of the term loans resulted in a final payment fee of $500,000 and the acceleration of $602,000 of unamortized discount on notes payable, both recorded as interest expense in 2009.
Change in Value of Warrant Liability. The change in value of warrant liability was $3.6 million for the year ended December 31, 2009, compared to zero for the year ended December 31, 2008. This is the result of a change in accounting principle effective January 1, 2009, which resulted in warrants issued November 2008 with a recorded value of $1.4 million on December 31, 2008 being reclassified from equity to a liability. On September 29, 2009, Silicon Valley Bank completed a cashless exercise of 956,522 common stock warrants with an exercise price of $1.15 per share. The related warrant liability was marked-to-market on the date of exercise and reclassified to equity. The change in fair value of these warrants from January 1, 2009 to the date of exercise was $3.8 million. On October 2, 2009, Western Technology Investment completed a cashless exercise of 478,261 common stock warrants with an exercise price of $1.15 per share. The related warrant liability was marked-to-market on the date of exercise and reclassified to equity. The change in fair value of these warrants from January 1, 2009 to the date of exercise was $1,000. The fair market value of the remaining 687,500 warrants, with a weighted-average exercise price of $0.80, was $472,000 as of December 31, 2009. The fair market value for these remaining awards was calculated using the Black-Scholes valuation model, which resulted in a $120,000 decrease for the year ended December 31, 2009. The decrease was primarily the result of our stock price decreasing from a closing price of $1.46 on January 1, 2009 to $0.56 on December 31, 2009.
48
Comparison of the Years Ended December 31, 2008 and 2007
Research and Development Expenses. Research and development expenses were $27.7 million for the year ended December 31, 2008, compared to $21.1 million for the year ended December 31, 2007. The increase of $6.6 million, or 31.4%, is primarily due to a $1.3 million increase in compensation expenses associated with increased headcount to support the EMPOWER trial and a $6.4 million increase in professional services primarily due to EMPOWER patient recruiting and payments to the EMPOWER clinical study sites, partially offset by product development and research reductions for contractor separations upon completion of the Maestro RC System, contractor conversions and pre-clinical animal study reductions. Included in research and development expenses during 2008 were $869,000 of stock-based compensation charges compared to $2.3 million in 2007. The $1.5 million decrease in stock-based compensation is primarily the result of a $1.7 million one-time expense for the issuance of 206,044 shares of common stock to the Mayo Foundation for Medical Education and Research upon the completion of our initial public offering (IPO) in November 2007, partially offset by increases from additional employee options granted during 2008 with a higher weighted average option price and fair value.
Selling, General and Administrative Expenses. Selling, general and administrative expenses were $8.6 million for the year ended December 31, 2008, compared to $7.0 million for the year ended December 31, 2007. The increase of $1.6 million, or 23.3%, is primarily due to increases of $581,000 in compensation expense from increased headcount and $620,000 in professional services, including legal expenses, travel and insurance, all driven by the new requirements of being a publicly traded company. Included in selling, general and administrative expenses during 2008 were $1.7 million of stock-based compensation charges compared to $1.5 million in 2007. Stock-based compensation expense increased $141,000 due to a $1.2 million increase resulting from additional employee options granted during 2008 with a higher weighted average option price and fair value offset by a $1.0 million reduction due to an increased number of nonemployee options becoming fully vested during 2008.
Interest Income. Interest income was $1.1 million for the year ended December 31, 2008, compared to $1.6 million for the year ended December 31, 2007. The decrease of $455,000, or 29.2%, is primarily due to a decrease in the short-term interest rate environment despite an increase in the average cash, cash equivalents and short-term investment balance from $32.1 million during 2007 to $39.2 million during 2008. The increased average cash, cash equivalents and short-term investments balance is the result of the net $39.1 million raised in our IPO in November 2007, $10.0 million of debt funding received in 2007 and $15.0 million of debt funding received in November 2008, of which we received net proceeds of $7.1 million after transaction expenses, facility charges and existing debt pay off.
Interest Expense. Interest expense was $2.7 million for the year ended December 31, 2008, compared to $1.6 million for the year ended December 31, 2007. The increase of $1.0 million was primarily the result of entering into a $20.0 million debt facility, of which $15.0 million was funded in November 2008 that resulted in net proceeds of $7.1 million after transaction expenses, facility charges and existing debt pay off. The existing debt pay off resulted in a one-time interest payment of $763,000 and the acceleration of $255,000 in unamortized discounts on notes payable.
Change in Value of Warrant Liability. Change in value of the convertible preferred stock warrant liability was zero for the year ended December 31, 2008, compared to $362,000 for the year ended December 31, 2007. The preferred stock warrant liability was recorded on December 11, 2006 when we sold an additional 123,569 shares of Series C preferred stock. Upon the closing of the sale, we had insufficient authorized and unissued shares of Series C preferred stock available to share settle outstanding warrants to purchase Series C preferred stock, resulting in the warrants being reclassified as a liability at the estimated fair value of $735,000 on December 11, 2006. On May 14, 2007 we filed an amended certificate of incorporation to increase the number of authorized shares of Series C preferred stock to 6,043,957. As a result of the amendment, we had sufficient authorized and unissued shares of Series C preferred stock available to share settle the warrants. The fair market value of the warrants on May 14, 2007 was determined to be $1.1 million. The $362,000 change in fair value
49
from December 31, 2006 to the amendment date was recorded as expense and the convertible preferred stock liability was reclassified to additional paid-in capital.
Liquidity and Capital Resources
We have incurred losses since our inception in December 2002 and, as of December 31, 2009 we had a deficit accumulated during the development stage of $133.2 million. We have financed our operations to date principally through the sale of capital stock, debt financing and interest earned on investments. Prior to our IPO in November 2007, we had received net proceeds of $63.2 million from the sale of common stock and preferred stock and $30.8 million in debt financing, $746,000 to finance equipment purchases and $30.0 million to finance working capital. Through our initial public offering we received net proceeds of $39.1 million after expenses and underwriters’ discounts and commissions and including the exercise of the underwriters’ over-allotment option. In November 2008, we entered into a $20.0 million working capital debt facility, replacing the existing debt financing. We received net proceeds of $7.1 million from the first draw of $15.0 million after transaction expenses, facility charges and existing debt pay off. The debt facility provided that the additional $5.0 million draw was to be available and automatically fund under the terms of the loan agreement if and when the trading price of our common stock on the NASDAQ Global Market met or exceeded a target amount on or before June 30, 2009. The Company’s trading price achieved this target and therefore, on April 28, 2009, the automatic funding of the additional $5.0 million was made to the Company under the debt facility. On February 24, 2009, we completed the sale of 13,110,393 shares of our common stock, together with warrants to purchase an aggregate of 6,555,197 shares of our common stock, in a private placement transaction with several accredited investors. We received gross proceeds of $15.9 million less a placement agent fee of $617,000 and certain other expenses. On October 7, 2009, we completed the sale of 6,161,068 shares of our common stock in a registered direct offering, at a purchase price of $0.80 per share. We received gross proceeds of $4.9 million before deducting estimated offering expenses.
As of December 31, 2009, we had $14.6 million in cash, cash equivalents and short-term investments. Of this amount $13.8 million was invested in short-term money market funds that are not considered to be bank deposits and are not insured or guaranteed by the federal deposit insurance company or other government agency. These money market funds seek to preserve the value of the investment at $1.00 per share; however, it is possible to lose money investing in these funds. Cash in excess of immediate requirements is invested in accordance with our investment policy, primarily with a view to liquidity and capital preservation. At times, such deposits may be in excess of insured limits. We have not experienced any losses on our deposits of cash and cash equivalents. On January 20, 2010, we completed the sale of 7,438,299 shares of our common stock in a registered direct offering, at a purchase price of $0.65 per share. We received gross proceeds of $4.8 million before deducting estimated offering expenses. We believe that the funds received January 20, 2010, together with our pre-existing cash, cash equivalents and short-term investment balances and interest income we earn on these balances, will be sufficient to meet our anticipated cash requirements into the second half of 2010 assuming we do not receive any additional funds. The potential lack of liquidity through 2010 has raised a substantial doubt about our ability to continue as a going concern and is discussed further in “Operating Capital and Capital Expenditure Requirements” below and in Note 3 to our consolidated financial statements included in Item 8 of this Annual Report on Form 10-K. In order to fund on-going operating cash requirements beyond that point or to further accelerate and execute our business plan, including a potentially new clinical trial using the next-generation Maestro RC System if approved by the FDA, we will need to raise significant additional funds. In view of these matters, our ability to continue as a going concern is dependent upon our ability to secure additional financing sufficient to support our research and development activities, approval of developed products for sale by regulatory authorities, including the FDA, and ultimately to generate revenues sufficient to cover all costs. See further discussion in the below section titled “Operating Capital and Capital Expenditure Requirements.”
As of December 31, 2009, we had repaid the outstanding principal amount due to Venture Lending & Leasing V, Inc. (a private equity fund under the management of Western Technology Investment) and Compass Horizon Funding Company pursuant to the Loan and Security Agreement, effective as of November 18, 2008
50
(the Loan Agreement or Prior Loan Agreement). The remaining unpaid balance of $8.2 million in debt financing as of December 31, 2009 owed to Silicon Valley Bank (SVB) pursuant to the Loan Agreement is collateralized by a first security priority lien on all of our assets, excluding intellectual property. We have entered into account control agreements in order to perfect the lender’s first security interest in our cash and investment accounts. In the event we have less than five remaining months of liquidity, we are required to grant a temporary lien on our intellectual property. The number of remaining months of liquidity is calculated by dividing cash and cash equivalents as of the end of any particular month by the sum of our total operating expenses for each of the immediately preceding five months. The debt financing agreement also requires us to (1) maintain a cash and cash equivalents balance that exceeds our aggregate operating expenses for the most recent five calendar month period ending prior to the determination date and (2) secure aggregate net proceeds of at least $20.0 million by January 9, 2010 from new capital transactions, of which $10.0 million was required by June 30, 2009. The second covenant requiring $20.0 million from new capital transactions was achieved with the completion of our private placement transaction on February 24, 2009 together with the completion of our registered direct transaction on October 7, 2009. There are no additional liquidity covenants that we are required to maintain under the terms of our debt financing agreements.
On February 8, 2010 we entered into a First Amendment (the Amendment) with SVB to the Loan Agreement. The Amendment provides that SVB’s term loan shall be repaid with a payment of $383,532 on February 1, 2010 followed by consecutive equal monthly payments of $380,421 each, commencing on March 1, 2010 and ending on December 1, 2011. It also amends the interest rate due on the remaining principal amount of the term loan from 11.0% to a fixed annual rate of 10.0%, payable monthly. Pursuant to the Amendment, the conditions pursuant to which the Excluded Collateral (as defined in the Prior Loan Agreement) will be deemed to be included as Collateral (as defined in the Prior Loan Agreement) are changed from the failure to have five months of remaining liquidity to the occurrence of an Event of Default (as defined in the Prior Loan Agreement) after the date of the Amendment or the lender’s awareness after such date of an Event of Default that occurred on or before such date with written notice of such event delivered to the Company. In addition, the Amendment revises the financial covenants in the Prior Loan Agreement to delete the covenant relating to five months of remaining liquidity and to change the liquidity ratio covenant to equal a ratio of (i) the sum of our unrestricted cash and cash equivalents held with SVB and SVB’s affiliates, divided by (ii) the outstanding principal amount of the term loan, which is not permitted to be less than 1.50:1.00. Finally, the Amendment adds a new covenant, the breach of which would constitute an Event of Default. The new covenant requires that we receive aggregate net proceeds of at least $4.0 million from new capital transactions after January 1, 2010 and before March 31, 2010 and to keep the proceeds of such transactions at SVB until used. We satisfied this new covenant with the closing, on January 20, 2010, of our sale of 7,438,299 shares of common stock to certain institutional investors in a registered direct offering for gross proceeds of approximately $4.8 million, before deducting estimated offering expenses and placement agent fees.
Net Cash Used in Operating Activities
Net cash used in operating activities was $24.7 million, $33.7 million and $23.4 million for the years ended December 31, 2009, 2008 and 2007, respectively. Net cash used in operating activities primarily reflects the net loss for those periods, which was partially offset by depreciation and amortization, change in the carrying value of warrant liability, stock-based compensation and changes in operating assets and liabilities.
Net Cash Provided by Investing Activities
Net cash provided by investing activities was $5.0 million, $2.8 million and $8.2 million for the years ended December 31, 2009, 2008 and 2007, respectively. Net cash provided by investing activities was primarily related to the proceeds from the maturity of short-term investments partially offset by the purchase of short-term investments and, to a lesser extent, the purchase of property and equipment.
51
Net Cash Provided by Financing Activities
Net cash provided by financing activities was $13.2 million, $3.2 million and $46.3 million for the years ended December 31, 2009, 2008 and 2007, respectively. Net cash provided by financing activities was attributable to proceeds from debt financing in each of the three years ended December 31, 2009, 2008 and 2007, offset by repayments on long-term debt. We also received net proceeds of $39.1 million from the issuance of common shares in our IPO in the year ended December 31, 2007. During 2009 net cash was provided by the completion of a private placement transaction that resulted in gross proceeds of $15.9 million for the issuance of common stock and common stock warrants, offset by $806,000 in financing costs, and the completion of a registered direct transaction that resulted in gross proceeds of $4.9 million for the issuance of common stock, offset by $92,000 in financing costs. On January 20, 2010, we completed the sale of 7,438,299 shares of our common stock in a registered direct offering, at a purchase price of $0.65 per share. We received gross proceeds of $4.8 million before deducting estimated offering expenses.
Operating Capital and Capital Expenditure Requirements
To date, we have not commercialized any products and we have not earned any operating revenues. On October 2, 2009, we announced preliminary results from our pivotal clinical study, the EMPOWER trial; indicating that based on an initial analysis, the study did not meet its primary and secondary efficacy endpoints, while meeting its safety endpoint. Following this announcement, management has taken steps to preserve capital by halting all commercialization and development activities and focusing on a comprehensive analysis of all clinical, statistical, and engineering data to understand the trial outcome. This resulted in a 40% reduction in force on October 27, 2009. On November 12, 2009 we announced that the ongoing detailed review suggests that vagal blocking therapy may promote safe and effective weight loss as an adjunct to behavioral support, diet and exercise in morbidly obese patients. The review further suggests that these effects were evident in both the treatment and control arms and that based on the analysis to date, the control arm of the trial, which was intended to be inactive, apparently provided a low-intensity blocking signal that introduced VBLOC therapy in human subjects. In January 2010, we met with the FDA to discuss the EMPOWER trial results and the regulatory process going forward. Based on this discussion, we recently submitted an IDE application for a clinical trial using the next-generation Maestro RC System in the treatment of morbid obesity. Assuming we are able to obtain an approved IDE, successfully enroll and implant the new clinical trial and achieve favorable results, and obtain FDA approval for our Maestro System, we do not expect to generate any product revenue earlier than the second half of 2013. As a result, we anticipate that we will continue to incur substantial net losses for the next several years.
We believe the net proceeds from our registered direct offering closed on January 20, 2010 of $4.8 million before deducting estimated offering expenses, together with our pre-existing cash, cash equivalents and short-term investments balance of $14.6 million as of December 31, 2009 and interest income we earn on these balances will be sufficient to meet our anticipated cash requirements into the second half of 2010 assuming we do not receive any additional funds, which has raised a substantial doubt about our ability to continue as a going concern. In order to fund on-going operating cash requirements beyond that point or to further accelerate and execute our business plan, including a potentially new clinical trial using the next-generation Maestro RC System if approved by the FDA, we will need to raise significant additional funds. In view of these matters, the ability for us to continue as a going concern is dependent upon our ability to secure additional financing sufficient to support our research and development activities, approval of developed products for sale by regulatory authorities, including the FDA, and ultimately to generate revenues sufficient to cover all costs. We may seek to raise funds through the sale of additional equity or debt securities, or by entering into an additional credit facility or through collaboration, licensing or other similar arrangements. The sale of additional equity and debt securities may result in dilution to our stockholders. If we raise additional funds through the issuance of debt securities, these securities could have rights senior to those of our common stock and could contain covenants that would restrict our operations. We may be required to raise additional capital on more than one occasion and beyond our currently forecasted amounts. Any such required additional capital may not be available on reasonable terms, if at all. If we are unable to obtain additional financing or the FDA does not approve our IDE
52
application for a clinical trial using the next-generation Maestro RC System, we may be required to reduce the scope of, delay, or eliminate some or all of, our planned research, development and commercialization activities, which could materially harm our business. In addition, if we raise additional funds through collaboration, licensing or other similar arrangements, it may be necessary to relinquish valuable rights to products or proprietary technologies, or grant licenses on terms that are not favorable.
Our forecast of the period of time through which our financial resources will be adequate to support our operations, the costs to complete development of products and the cost to commercialize our products are forward-looking statements and involve risks and uncertainties, and actual results could vary materially and negatively as a result of a number of factors, including the factors discussed in Item 1A “Risk Factors” of this Annual Report on Form 10-K. We have based these estimates on assumptions that may prove to be wrong, and we could utilize our available capital resources sooner than we currently expect.
Because of the numerous risks and uncertainties associated with the development of medical devices, such as our Maestro System, we are unable to estimate the exact amounts of capital outlays and operating expenditures necessary to complete the development of the products and successfully deliver a commercial product to the market. Our future capital requirements will depend on many factors, including but not limited to the following:
| • | the scope, rate of progress, results and cost of our clinical trials and other research and development activities; |
| • | the cost and timing of regulatory approvals; |
| • | the cost and timing of establishing sales, marketing and distribution capabilities; |
| • | the cost of establishing clinical and commercial supplies of our Maestro System and any products that we may develop; |
| • | the rate of market acceptance of our Maestro System and VBLOC therapy and any other product candidates; |
| • | the cost of filing and prosecuting patent applications and defending and enforcing our patent and other intellectual property rights; |
| • | the cost of defending, in litigation or otherwise, any claims that we infringe third-party patent or other intellectual property rights; |
| • | the effect of competing products and market developments; |
| • | the cost of explanting clinical devices; |
| • | the terms and timing of any collaborative, licensing or other arrangements that we may establish; |
| • | any revenue generated by sales of our future products; and |
| • | the extent to which we acquire or invest in businesses, products and technologies, although we currently have no commitments or agreements relating to any of these types of transactions. |
Contractual Obligations
The following table summarizes our contractual obligations as of December 31, 2009 and the effect those obligations are expected to have on our financial condition and liquidity position in future periods:
| Payments Due By Period | |||||||||||||||
| Contractual Obligations |
Total | Less Than 1 Year |
1-3 Years | 3-5 Years | More than 5 Years | ||||||||||
| Operating lease |
$ | 1,601,384 | $ | 247,951 | $ | 554,619 | $ | 577,025 | $ | 221,789 | |||||
| Long-term debt, including interest |
9,704,775 | 4,602,387 | 5,102,388 | — | — | ||||||||||
| Total contractual cash obligations |
$ | 11,306,159 | $ | 4,850,338 | $ | 5,657,007 | $ | 577,025 | $ | 221,789 | |||||
53
The table above reflects only payment obligations that are fixed and determinable. Our operating lease commitments relate to our corporate headquarters in St. Paul, Minnesota.
On February 8, 2010 we entered into an amended loan agreement reducing our annual interest rate from 11.0% to 10.0%, which in turn reduces our monthly principal and interest payments from $383,532 to $380,421, commencing on March 1, 2010 and ending on December 1, 2011. See Note 17 to our consolidated financial statements included in Item 8 of this Annual Report on Form 10-K for a more detailed description of the amendment.
The table above excludes a recent two-year extension of our five-year license agreement with the Mayo Foundation for Medical Education and Research (Mayo Foundation) entered into in 2005. Under the terms of the extension, entered into on March 11, 2010 and effective February 3, 2010, the Mayo Foundation will receive an annual retainer of $100,000 in 2010 and 2011. We may also be obligated to pay the Mayo Foundation, contingent upon the occurrence of certain future events, earned royalty payments, including a minimum annual royalty as defined by the agreement, for the commercial sale of products developed and patented by the Mayo Foundation, jointly patented by the Company and the Mayo Foundation, or a product where the Mayo Foundation provided know-how as defined by the agreement. If no products are patented, the minimum royalty is not due.
Off-balance-sheet Arrangements
Since our inception, we have not engaged in any off-balance-sheet arrangements, including the use of structured finance, special purpose entities or variable interest entities as defined by rules enacted by the Securities and Exchange Commission and Financial Accounting Standards Board, and accordingly, no such arrangements are likely to have a current or future effect on our financial position, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources.
Recent Accounting Pronouncements
In June 2009, the Financial Accounting Standards Board (FASB) approved the FASB Accounting Standards Codification (ASC or the Codification) as the single source of authoritative, nongovernmental accounting principles generally accepted in the United States of America (GAAP), excluding the guidance issued by the Securities and Exchange Commission (SEC). FASB approved an Exposure Draft that replaced Statement of Financial Accounting Standards No. 162, The Hierarchy of Generally Accepted Accounting Principles, and modified GAAP by establishing only two levels of GAAP, authoritative and nonauthoritative. This was accomplished by authorizing the Codification to become the single source of authoritative U.S. accounting and reporting standards, except for rules and interpretive releases of the SEC, which are sources of authoritative GAAP for SEC registrants. All other nongrandfathered, non-SEC accounting literature not included in the Codification has become nonauthoritative. The Codification was effective for us beginning with the quarter ended September 30, 2009. The adoption of the Codification did not have a material impact on our consolidated financial statements.
Effective January 1, 2009, we adopted new authoritative accounting guidance regarding the financial reporting for outstanding equity-linked financial instruments. See Note 2 to our consolidated financial statements included in Item 8 of this Annual Report on Form 10-K for more details on this new authoritative accounting guidance.
There have been no other significant changes in recent accounting pronouncements during the fiscal year ended December 31, 2009.
54
| ITEM 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURE ABOUT MARKET RISK |
Our exposure to market risk is confined to our cash, cash equivalents and short-term investments. As of December 31, 2009, we had $14.6 million in cash, cash equivalents and short-term investments. The goals of our investment policy are preservation of capital, fulfillment of liquidity needs and fiduciary control of cash and investments. We also seek to maximize income from our investments without assuming significant risk. To achieve our goals, we may maintain a portfolio of cash equivalents and investments in a variety of securities of high credit quality. The securities in our investment portfolio, if any, are not leveraged, are classified as either available for sale or held-to-maturity and are, due to their very short-term nature, subject to minimal interest rate risk. We currently do not hedge interest rate exposure. Because of the short-term maturities of our cash equivalents and investments, we do not believe that an increase in market rates would have any material negative impact on the value of our investment portfolio. We have no investments denominated in foreign currencies and therefore our investments are not subject to foreign currency exchange risk.
55
| ITEM 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
Index to Financial Statements
| Page | ||
| 57 | ||
| Financial Statements |
||
| 58 | ||
| 59 | ||
| 60 | ||
| 67 | ||
| 68 |
56
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of
EnteroMedics Inc.
St. Paul, Minnesota
We have audited the accompanying consolidated balance sheets of EnteroMedics Inc. and subsidiary (a development stage company) (the “Company”) as of December 31, 2009 and 2008, and the related consolidated statements of operations, stockholders’ equity (deficit), and cash flows for each of the three years in the period ended December 31, 2009, and for the period from December 19, 2002 (date of inception) to December 31, 2009. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, such consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2009 and 2008, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2009, and for the period from December 19, 2002 (date of inception) to December 31, 2009, in conformity with accounting principles generally accepted in the United States of America.
As discussed in Note 2 to the consolidated financial statements, effective January 1, 2009, the Company adopted new authoritative accounting guidance regarding the financial reporting for outstanding equity-linked financial instruments.
These accompanying financial statements have been prepared assuming the Company will continue as a going concern. As discussed in Note 3 to the consolidated financial statements, the Company’s recurring losses from operations and stockholder’s capital deficiency raise substantial doubt about its ability to continue as a going concern. Management’s plans concerning these matters are also discussed in Note 3 to the consolidated financial statements. These financial statements do not include any adjustments that might result from the outcome of this uncertainty.
| /s/ DELOITTE & TOUCHE LLP |
| Minneapolis, MN March 29, 2010 |
57
(A development stage company)
Consolidated Balance Sheets
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| ASSETS | ||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 14,617,594 | $ | 21,055,108 | ||||
| Short-term investments available for sale |
— | 5,239,892 | ||||||
| Interest receivable |
— | 57,965 | ||||||
| Other receivables |
10,007 | 19,308 | ||||||
| Prepaid expenses and other current assets |
474,336 | 421,817 | ||||||
| Total current assets |
15,101,937 | 26,794,090 | ||||||
| Property and equipment, net |
965,829 | 1,263,903 | ||||||
| Other assets |
146,234 | 220,907 | ||||||
| Total assets |
$ | 16,214,000 | $ | 28,278,900 | ||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| Current liabilities: |
||||||||
| Current portion of notes payable |
$ | 3,880,656 | $ | 2,674,597 | ||||
| Accounts payable |
33,618 | 163,377 | ||||||
| Accrued expenses |
2,077,916 | 2,862,102 | ||||||
| Accrued interest payable |
288,305 | 177,869 | ||||||
| Total current liabilities |
6,280,495 | 5,877,945 | ||||||
| Notes payable, less current portion (net discounts of $455,469 and $1,329,592 at December 31, 2009 and 2008, respectively) |
3,880,810 | 10,995,811 | ||||||
| Common stock warrant liability |
471,585 | — | ||||||
| Total liabilities |
10,632,890 | 16,873,756 | ||||||
| Stockholders’ equity: |
||||||||
| Common stock, $0.01 par value 85,000,000 and 50,000,000 shares authorized at December 31, 2009 and 2008, respectively; 37,378,387 and 16,899,935 shares issued and outstanding at December 31, 2009 and 2008, respectively |
373,784 | 168,999 | ||||||
| Additional paid-in capital |
138,576,593 | 112,552,256 | ||||||
| Deferred compensation |
(1,667 | ) | (21,667 | ) | ||||
| Accumulated other comprehensive income |
— | 12,988 | ||||||
| Deficit accumulated during development stage |
(133,367,600 | ) | (101,307,432 | ) | ||||
| Total stockholders’ equity |
5,581,110 | 11,405,144 | ||||||
| Total liabilities and stockholders’ equity |
$ | 16,214,000 | $ | 28,278,900 | ||||
See accompanying notes to consolidated financial statements.
58
(A development stage company)
Consolidated Statements of Operations
| Years ended December 31, | Period from December 19, 2002 (inception) to December 31, 2009 |
|||||||||||||||
| 2009 | 2008 | 2007 | ||||||||||||||
| Operating expenses: |
||||||||||||||||
| Research and development |
$ | 15,580,746 | $ | 27,673,725 | $ | 21,053,395 | $ | 91,609,479 | ||||||||
| Selling, general and administrative |
8,631,597 | 8,596,703 | 6,972,803 | 31,907,364 | ||||||||||||
| Total operating expenses |
24,212,343 | 36,270,428 | 28,026,198 | 123,516,843 | ||||||||||||
| Other income (expense): |
||||||||||||||||
| Interest income |
79,355 | 1,101,923 | 1,556,551 | 4,018,425 | ||||||||||||
| Interest expense |
(4,104,300 | ) | (2,683,658 | ) | (1,648,818 | ) | (9,581,505 | ) | ||||||||
| Change in value of warrant liability |
(3,644,549 | ) | — | (361,504 | ) | (3,999,456 | ) | |||||||||
| Other, net |
(47,363 | ) | (21,865 | ) | (95,379 | ) | (157,253 | ) | ||||||||
| Net loss |
$ | (31,929,200 | ) | $ | (37,874,028 | ) | $ | (28,575,348 | ) | $ | (133,236,632 | ) | ||||
| Net loss per share—basic and diluted |
$ | (1.07 | ) | $ | (2.25 | ) | $ | (11.69 | ) | |||||||
| Shares used to compute basic and diluted net loss per share |
29,845,954 | 16,835,661 | 2,445,001 | |||||||||||||
See accompanying notes to consolidated financial statements.
59
(A development stage company)
Consolidated Statements of Stockholders’ Equity (Deficit)
Period from December 19, 2002 (inception) to December 31, 2009
| Series C Convertible Preferred Stock |
Series B Convertible Preferred Stock |
Series A Convertible Preferred Stock |
Common Stock | Additional Paid-in Capital |
Deferred Compensation |
Accumulated Other Comprehensive Income |
Deficit Accumulated During the Development Stage |
Total Stockholders’ Equity (Deficit) |
||||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | Shares | Amount | |||||||||||||||||||||||||||||||||||
| Common stock issued at inception of Alpha Medical, Inc. on December 19, 2002 at $0.09 per share for cash |
— | $ | — | — | $ | — | — | $ | — | 109,890 | $ | 1,099 | $ | 8,901 | $ | — | $ | — | $ | — | $ | 10,000 | ||||||||||||||||||||
| Common stock issued at inception of Beta Medical, Inc. on December 19, 2002 at $0.09 per share for cash |
— | — | — | — | — | — | 109,890 | 1,099 | 8,901 | — | — | — | 10,000 | |||||||||||||||||||||||||||||
| Alpha Medical, Inc. Series A convertible preferred stock issued on December 31, 2002 at $9.10 per share for cash |
— | — | — | — | 33,150 | 332 | — | — | 301,342 | — | — | — | 301,674 | |||||||||||||||||||||||||||||
| Beta Medical, Inc. Series A convertible preferred stock issued on December 31, 2002 at $9.10 per share for cash |
— | — | — | — | 33,150 | 332 | — | — | 301,342 | — | — | — | 301,674 | |||||||||||||||||||||||||||||
| Net loss for the period ended December 31, 2002 |
— | — | — | — | — | — | — | — | — | — | — | (603,348 | ) | (603,348 | ) | |||||||||||||||||||||||||||
| Balance, December 31, 2002 |
— | $ | — | — | $ | — | 66,300 | $ | 664 | 219,780 | $ | 2,198 | $ | 620,486 | $ | — | $ | — | $ | (603,348 | ) | $ | 20,000 | |||||||||||||||||||
| Alpha Medical, Inc. Series A convertible preferred stock issued on October 1, 2003 at $9.10 per share for cash |
— | — | — | — | 38,461 | 384 | — | — | 349,616 | — | — | — | 350,000 | |||||||||||||||||||||||||||||
| Beta Medical, Inc. Series A convertible preferred stock issued on October 1, 2003 at $9.10 per share for cash |
— | — | — | — | 93,406 | 934 | — | — | 849,066 | — | — | — | 850,000 | |||||||||||||||||||||||||||||
| Cancellation of Alpha Medical, Inc. Series A convertible preferred stock and common stock upon merger with Beta Medical, Inc. effective October 1, 2003 |
— | — | — | — | (71,613 | ) | (716 | ) | (109,890 | ) | (1,099 | ) | (659,859 | ) | — | — | — | (661,674 | ) | |||||||||||||||||||||||
| Issuance of Series A convertible preferred stock upon merger of Alpha Medical, Inc. and Beta Medical, Inc. effective October 1, 2003 |
— | — | — | — | 65,934 | 659 | — | — | 661,015 | — | — | — | 661,674 | |||||||||||||||||||||||||||||
| Common stock issued in October 2003 at $0.09 per share for cash |
— | — | — | — | — | — | 115,376 | 1,154 | 9,346 | — | — | — | 10,500 | |||||||||||||||||||||||||||||
| Warrants issued for the purchase of 23,516 shares of Series B convertible preferred stock for cash at $0.00455 per share in connection with the November 13, 2003 convertible bridge notes |
— | — | — | — | — | — | — | — | 107 | — | — | — | 107 | |||||||||||||||||||||||||||||
| Net loss for the year ended December 31, 2003 |
— | — | — | — | — | — | — | — | — | — | — | (1,900,288 | ) | (1,900,288 | ) | |||||||||||||||||||||||||||
| Balance, December 31, 2003 |
— | $ | — | — | $ | — | 192,488 | $ | 1,925 | 225,266 | $ | 2,253 | $ | 1,829,777 | $ | — | $ | — | $ | (2,503,636 | ) | $ | (669,681 | ) | ||||||||||||||||||
See accompanying notes to consolidated financial statements.
60
ENTEROMEDICS INC.
(A development stage company)
Consolidated Statements of Stockholders’ Equity (Deficit) (Continued)
Period from December 19, 2002 (inception) to December 31, 2009
| Series C Convertible Preferred Stock |
Series B Convertible Preferred Stock |
Series A Convertible Preferred Stock |
Common Stock | Additional Paid-in Capital |
Deferred Compensation |
Accumulated Other Comprehensive Income |
Deficit Accumulated During the Development Stage |
Total Stockholders’ Equity (Deficit) |
||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | Shares | Amount | |||||||||||||||||||||||||||||||
| Balance, December 31, 2003 |
— | $ | — | — | $ | — | 192,488 | $ | 1,925 | 225,266 | $ | 2,253 | $ | 1,829,777 | $ | — | $ | — | $ | (2,503,636 | ) | $ | (669,681 | ) | ||||||||||||||
| Warrants issued for the purchase of 6,484 shares of Series B convertible preferred stock for cash at $0.00455 per share in connection with the April 23, 2004 convertible bridge notes |
— | — | — | — | — | — | — | — | 30 | — | — | — | 30 | |||||||||||||||||||||||||
| Exercise of 125,778 Series A convertible preferred stock warrants on April 23, 2004 for cash at $1.4919359 per share |
— | — | — | — | 125,778 | 1,258 | — | — | 186,394 | — | — | — | 187,652 | |||||||||||||||||||||||||
| Warrants issued for the purchase of 4,396 shares of Series B convertible preferred stock for cash at $0.00455 per share in connection with the June 30, 2004 convertible bridge notes |
— | — | — | — | — | — | — | — | 20 | — | — | — | 20 | |||||||||||||||||||||||||
| Fair value of warrants related to convertible bridge notes |
— | — | — | — | — | — | — | — | 153,722 | — | — | — | 153,722 | |||||||||||||||||||||||||
| Series B convertible preferred stock issued upon conversion of $1,564,843 of convertible bridge notes and $34,809 of accrued interest payable on July 30, 2004 at $3.9430 per share |
— | — | 405,690 | 4,057 | — | — | — | — | 1,595,595 | — | — | — | 1,599,652 | |||||||||||||||||||||||||
| Series B convertible preferred stock issued on July 30, 2004 for cash at $3.9430 per share, net of financing costs of $94,776 |
— | — | 1,914,767 | 19,148 | — | — | — | — | 7,436,077 | — | — | — | 7,455,225 | |||||||||||||||||||||||||
| Warrants issued for the purchase of 45,333 shares of Series B convertible preferred stock on December 1, 2004 valued at $1.0747 per warrant for debt commitment |
— | — | — | — | — | — | — | — | 48,720 | — | — | — | 48,720 | |||||||||||||||||||||||||
| Issuance of 22,912 common stock options to nonemployees in 2004 valued at $0.1574 per option |
— | — | — | — | — | — | — | — | 3,610 | (3,610 | ) | — | — | — | ||||||||||||||||||||||||
| Amortization of deferred compensation |
— | — | — | — | — | — | — | — | — | 830 | — | — | 830 | |||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | — | — | — | (3,448,752 | ) | (3,448,752 | ) | |||||||||||||||||||||||
| Balance, December 31, 2004 |
— | $ | — | 2,320,457 | $ | 23,205 | 318,266 | $ | 3,183 | 225,266 | $ | 2,253 | $ | 11,253,945 | $ | (2,780 | ) | $ | — | $ | (5,952,388 | ) | $ | 5,327,418 | ||||||||||||||
See accompanying notes to consolidated financial statements.
61
ENTEROMEDICS INC.
(A development stage company)
Consolidated Statements of Stockholders’ Equity (Deficit) (Continued)
Period from December 19, 2002 (inception) to December 31, 2009
| Series C Convertible Preferred Stock |
Series B Convertible Preferred Stock |
Series A Convertible Preferred Stock |
Common Stock | Additional Paid-in Capital |
Deferred Compensation |
Accumulated Other Comprehensive Income |
Deficit Accumulated During the Development Stage |
Total Stockholders’ Equity (Deficit) |
||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | Shares | Amount | |||||||||||||||||||||||||||||||
| Balance, December 31, 2004 |
— | $ | — | 2,320,457 | $ | 23,205 | 318,266 | $ | 3,183 | 225,266 | $ | 2,253 | $ | 11,253,945 | $ | (2,780 | ) | $ | — | $ | (5,952,388 | ) | $ | 5,327,418 | ||||||||||||||
| Series B convertible preferred stock issued on June 17, 2005 for cash at $3.9430 per share, net of financing costs of $5,218 |
— | — | 760,834 | 7,608 | — | — | — | — | 2,987,174 | — | — | — | 2,994,782 | |||||||||||||||||||||||||
| Warrants issued for the purchase of 69,744 shares of Series B convertible preferred stock in September 2005 valued at $1.0702 per warrant for debt commitment and funding |
— | — | — | — | — | — | — | — | 74,636 | — | — | — | 74,636 | |||||||||||||||||||||||||
| Warrants issued for the purchase of 170,336 shares of common stock on December 12, 2005 for cash at $0.09 per warrant |
— | — | — | — | — | — | — | — | 15,500 | — | — | — | 15,500 | |||||||||||||||||||||||||
| Series B convertible preferred stock issued on December 12, 2005 at $3.9430 per share, net of financing costs of $11,085 |
— | — | 1,204,655 | 12,046 | — | — | — | — | 4,726,870 | — | — | — | 4,738,916 | |||||||||||||||||||||||||
| Common stock issued to nonemployees in 2005 valued at $0.46 per share |
— | — | — | — | — | — | 225,274 | 2,252 | 100,248 | (102,500 | ) | — | — | — | ||||||||||||||||||||||||
| Issuance of 46,543 common stock options to nonemployees in 2005 valued at $0.1565 per option |
— | — | — | — | — | — | — | — | 7,288 | (7,288 | ) | — | — | — | ||||||||||||||||||||||||
| Exercise of 29,561 common stock options in 2005 for cash at $0.46 per share |
— | — | — | — | — | — | 29,561 | 296 | 13,154 | — | — | — | 13,450 | |||||||||||||||||||||||||
| Amortization of deferred compensation |
— | — | — | — | — | — | — | — | — | 25,041 | — | — | 25,041 | |||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | — | — | — | (11,215,191 | ) | (11,215,191 | ) | |||||||||||||||||||||||
| Balance, December 31, 2005 |
— | $ | — | 4,285,946 | $ | 42,859 | 318,266 | $ | 3,183 | 480,101 | $ | 4,801 | $ | 19,178,815 | $ | (87,527 | ) | $ | — | $ | (17,167,579 | ) | $ | 1,974,552 | ||||||||||||||
See accompanying notes to consolidated financial statements.
62
ENTEROMEDICS INC.
(A development stage company)
Consolidated Statements of Stockholders’ Equity (Deficit) (Continued)
Period from December 19, 2002 (inception) to December 31, 2009
| Series C Convertible Preferred Stock |
Series B Convertible Preferred Stock |
Series A Convertible Preferred Stock |
Common Stock | Additional Paid-in Capital |
Deferred Compensation |
Accumulated Other Comprehensive Income |
Deficit Accumulated During the Development Stage |
Total Stockholders’ Equity (Deficit) |
|||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | Shares | Amount | ||||||||||||||||||||||||||||||||
| Balance, December 31, 2005 |
— | $ | — | 4,285,946 | $ | 42,859 | 318,266 | $ | 3,183 | 480,101 | $ | 4,801 | $ | 19,178,815 | $ | (87,527 | ) | $ | — | $ | (17,167,579 | ) | $ | 1,974,552 | |||||||||||||||
| Warrants issued for the purchase of 34,872 shares of Series B convertible preferred stock in March 2006 valued at $2.9257 per warrant for debt funding |
— | — | — | — | — | — | — | — | 102,022 | — | — | — | 102,022 | ||||||||||||||||||||||||||
| Series C convertible preferred stock issued upon conversion of $5,250,003 of convertible bridge notes and $131,013 of accrued interest payable on July 6, 2006 at $8.0926 per share |
664,919 | 6,649 | — | — | — | — | — | — | 5,374,367 | — | — | — | 5,381,016 | ||||||||||||||||||||||||||
| Series C convertible preferred stock issued on July 6, 2006 for cash at $8.0926 per share, net of financing costs of $2,222,342 |
4,921,142 | 49,211 | — | — | — | — | — | — | 37,553,450 | — | — | — | 37,602,661 | ||||||||||||||||||||||||||
| Warrants issued for the purchase of 147,635 shares of Series C convertible preferred stock on July 6, 2006 valued at $4.9813 per warrant for equity financing |
— | — | — | — | — | — | — | — | 735,438 | — | — | — | 735,438 | ||||||||||||||||||||||||||
| Series C convertible preferred stock issued on December 11, 2006 for cash at $8.0926 per share |
123,569 | 1,236 | — | — | — | — | — | — | 998,764 | — | — | — | 1,000,000 | ||||||||||||||||||||||||||
| Series C convertible preferred stock warrants reclassified to convertible preferred stock warrant liability on December 11, 2006 |
— | — | — | — | — | — | — | — | (735,438 | ) | — | — | — | (735,438 | ) | ||||||||||||||||||||||||
| Common stock issued to nonemployees in 2006 valued at $0.46 per share |
— | — | — | — | — | — | 9,889 | 100 | 4,400 | (4,500 | ) | — | — | — | |||||||||||||||||||||||||
| Common stock issued to nonemployees in 2006 valued at $1.91 per share |
— | — | — | — | — | — | 2,747 | 27 | 5,223 | (5,250 | ) | — | — | — | |||||||||||||||||||||||||
| Employee stock-based compensation expense |
— | — | — | — | — | — | — | — | 47,479 | — | — | — | 47,479 | ||||||||||||||||||||||||||
| Nonemployee stock-based compensation expense |
— | — | — | — | — | — | — | — | 86,125 | — | — | — | 86,125 | ||||||||||||||||||||||||||
| Exercise of 87,022 common stock options in 2006 for cash at $0.46 per share |
— | — | — | — | — | — | 87,022 | 870 | 38,726 | — | — | — | 39,596 | ||||||||||||||||||||||||||
| Amortization of deferred compensation |
— | — | — | — | — | — | — | — | — | 30,798 | — | — | 30,798 | ||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | — | — | — | (17,690,477 | ) | (17,690,477 | ) | ||||||||||||||||||||||||
| Balance, December 31, 2006 |
5,709,630 | $ | 57,096 | 4,285,946 | $ | 42,859 | 318,266 | $ | 3,183 | 579,759 | $ | 5,798 | $ | 63,389,371 | $ | (66,479 | ) | $ | — | $ | (34,858,056 | ) | $ | 28,573,772 | |||||||||||||||
See accompanying notes to consolidated financial statements.
63
ENTEROMEDICS INC.
(A development stage company)
Consolidated Statements of Stockholders’ Equity (Deficit) (Continued)
Period from December 19, 2002 (inception) to December 31, 2009
| Series C Convertible Preferred Stock |
Series B Convertible Preferred Stock |
Series A Convertible Preferred Stock |
Common Stock | Additional Paid-in Capital |
Deferred Compensation |
Accumulated Other Comprehensive Income |
Deficit Accumulated During the Development Stage |
Total Stockholders’ Equity (Deficit) |
|||||||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | Shares | Amount | ||||||||||||||||||||||||||||||||||||||
| Balance, December 31, 2006 |
5,709,630 | $ | 57,096 | 4,285,946 | $ | 42,859 | 318,266 | $ | 3,183 | 579,759 | $ | 5,798 | $ | 63,389,371 | $ | (66,479 | ) | $ | — | $ | (34,858,056 | ) | $ | 28,573,772 | |||||||||||||||||||||
| Employee stock-based compensation expense |
— | — | — | — | — | — | — | — | 883,310 | — | — | — | 883,310 | ||||||||||||||||||||||||||||||||
| Nonemployee stock-based compensation expense |
— | — | — | — | — | — | — | — | 1,289,349 | — | — | — | 1,289,349 | ||||||||||||||||||||||||||||||||
| Warrants issued for the purchase of 67,963 shares of Series C convertible preferred stock in May 2007 valued at $8.0926 per warrant for debt facility commitment |
— | — | — | — | — | — | — | — | 550,212 | — | — | — | 550,212 | ||||||||||||||||||||||||||||||||
| Warrants issued for the purchase of 33,982 shares of Series C convertible preferred stock in May 2007 valued at $8.2783 per warrant for debt funding |
— | — | — | — | — | — | — | — | 281,321 | — | — | — | 281,321 | ||||||||||||||||||||||||||||||||
| Warrants issued for the purchase of 16,991 shares of Series C convertible preferred stock in August 2007 valued at $11.6377 per warrant for debt funding |
— | — | — | — | — | — | — | — | 197,731 | — | — | — | 197,731 | ||||||||||||||||||||||||||||||||
| Warrants issued for the purchase of 16,991 shares of Series C convertible preferred stock in October 2007 valued at $11.4599 per warrant for debt funding |
— | — | — | — | — | — | — | — | 194,716 | — | — | — | 194,716 | ||||||||||||||||||||||||||||||||
| Series C convertible preferred stock warrants reclassified from convertible preferred stock warrant liability |
— | — | — | — | — | — | — | — | 1,090,345 | — | — | — | 1,090,345 | ||||||||||||||||||||||||||||||||
| Issuance of common stock in initial public offering (“IPO”) in November 2007 for cash at $8.00 per share, net of financing costs of $4,552,663 |
— | — | — | — | — | — | 5,000,000 | 50,000 | 35,397,337 | — | — | — | 35,447,337 | ||||||||||||||||||||||||||||||||
| Conversion of preferred stock to common stock in November 2007 in connection with the IPO |
(5,709,630 | ) | (57,096 | ) | (4,285,946 | ) | (42,859 | ) | (318,266 | ) | (3,183 | ) | 10,488,178 | 104,882 | (1,744 | ) | — | — | — | — | |||||||||||||||||||||||||
| Reclassification of amounts due to shareholders for fractional shares upon reverse stock split |
— | — | — | — | — | — | — | — | (355 | ) | — | — | — | (355 | ) | ||||||||||||||||||||||||||||||
| Common stock issued to Mayo Foundation upon closing the IPO in November 2007 with a fair value of $8.05 per share |
— | — | — | — | — | — | 206,044 | 2,060 | 1,656,594 | — | — | — | 1,658,654 | ||||||||||||||||||||||||||||||||
| Exercise of over-allotment option by underwriters in December 2007 in connection with the IPO for cash at $8.00 per share, net of financing costs of $274,315 |
— | — | — | — | — | — | 489,849 | 4,899 | 3,639,578 | — | — | — | 3,644,477 | ||||||||||||||||||||||||||||||||
| Exercise of 35,132 common stock options in 2007 for cash at $0.60 per share |
— | — | — | — | — | — | 35,132 | 351 | 20,836 | — | — | — | 21,187 | ||||||||||||||||||||||||||||||||
| Amortization of deferred compensation |
— | — | — | — | — | — | — | — | — | 24,812 | — | — | 24,812 | ||||||||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | — | — | — | (28,575,348 | ) | (28,575,348 | ) | ||||||||||||||||||||||||||||||
| Balance, December 31, 2007 |
— | $ | — | — | $ | — | — | $ | — | 16,798,962 | $ | 167,990 | $ | 108,588,601 | $ | (41,667 | ) | $ | — | $ | (63,433,404 | ) | $ | 45,281,520 | |||||||||||||||||||||
See accompanying notes to consolidated financial statements.
64
ENTEROMEDICS INC.
(A development stage company)
Consolidated Statements of Stockholders’ Equity (Deficit) (Continued)
Period from December 19, 2002 (inception) to December 31, 2009
| Series C Convertible Preferred Stock |
Series B Convertible Preferred Stock |
Series A Convertible Preferred Stock |
Common Stock | Additional Paid-in Capital |
Deferred Compensation |
Accumulated Other Comprehensive Income |
Deficit Accumulated During the Development Stage |
Total Stockholders’ Equity (Deficit) |
|||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | Shares | Amount | ||||||||||||||||||||||||||||||||
| Balance, December 31, 2007 |
— | $ | — | — | $ | — | — | $ | — | 16,798,962 | $ | 167,990 | $ | 108,588,601 | $ | (41,667 | ) | $ | — | $ | (63,433,404 | ) | $ | 45,281,520 | |||||||||||||||
| Comprehensive loss: |
|||||||||||||||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | — | — | — | (37,874,028 | ) | (37,874,028 | ) | ||||||||||||||||||||||||
| Unrealized gains on available for sale investments |
— | — | — | — | — | — | — | — | — | — | 12,988 | — | 12,988 | ||||||||||||||||||||||||||
| Total comprehensive loss |
(37,861,040 | ) | |||||||||||||||||||||||||||||||||||||
| Employee stock-based compensation expense |
— | — | — | — | — | — | — | — | 2,648,410 | — | — | — | 2,648,410 | ||||||||||||||||||||||||||
| Nonemployee stock-based compensation expense |
— | — | — | — | — | — | — | — | (147,855 | ) | — | — | — | (147,855 | ) | ||||||||||||||||||||||||
| Warrants issued for the purchase of 1,398,702 shares of common stock in November 2008 valued at $1.30 per warrant for debt funding |
— | — | — | — | — | — | — | — | 1,398,702 | — | — | — | 1,398,702 | ||||||||||||||||||||||||||
| Exercise of 100,973 common stock options in 2008 for cash from $0.46 to $1.91 per share |
— | — | — | — | — | — | 100,973 | 1,009 | 64,398 | — | — | — | 65,407 | ||||||||||||||||||||||||||
| Amortization of deferred compensation |
— | — | — | — | — | — | — | — | — | 20,000 | — | — | 20,000 | ||||||||||||||||||||||||||
| Balance, December 31, 2008 |
— | $ | — | — | $ | — | — | $ | — | 16,899,935 | $ | 168,999 | $ | 112,552,256 | $ | (21,667 | ) | $ | 12,988 | $ | (101,307,432 | ) | $ | 11,405,144 | |||||||||||||||
See accompanying notes to consolidated financial statements.
65
ENTEROMEDICS INC.
(A development stage company)
Consolidated Statements of Stockholders’ Equity (Deficit) (Continued)
Period from December 19, 2002 (inception) to December 31, 2009
| Series C Convertible Preferred Stock |
Series B Convertible Preferred Stock |
Series A Convertible Preferred Stock |
Common Stock | Additional Paid-in Capital |
Deferred Compensation |
Accumulated Other Comprehensive Income |
Deficit Accumulated During the Development Stage |
Total Stockholders’ Equity (Deficit) |
||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | Shares | Amount | |||||||||||||||||||||||||||||||||
| Balance, December 31, 2008 |
— | $ | — | — | $ | — | — | $ | — | 16,899,935 | $ | 168,999 | $ | 112,552,256 | $ | (21,667 | ) | $ | 12,988 | $ | (101,307,432 | ) | $ | 11,405,144 | ||||||||||||||||
| Comprehensive loss: |
||||||||||||||||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | — | — | — | (31,929,200 | ) | (31,929,200 | ) | |||||||||||||||||||||||||
| Change in unrealized gain (loss) on available for sale investments |
— | — | — | — | — | — | — | — | — | — | (12,988 | ) | — | (12,988 | ) | |||||||||||||||||||||||||
| Total comprehensive loss |
(31,942,188 | ) | ||||||||||||||||||||||||||||||||||||||
| Employee stock-based compensation expense |
— | — | — | — | — | — | — | — | 2,209,216 | — | — | — | 2,209,216 | |||||||||||||||||||||||||||
| Nonemployee stock-based compensation expense |
— | — | — | — | — | — | — | — | 210,075 | — | — | — | 210,075 | |||||||||||||||||||||||||||
| Issuance of common stock in private investment public equity offering in February 2009 for cash at $1.15 per share, net of financing costs of $806,499 |
— | — | — | — | — | — | 13,110,393 | 131,104 | 14,139,349 | — | — | — | 14,270,453 | |||||||||||||||||||||||||||
| Warrants issued for the purchase of 6,555,197 shares of common stock in February 2009 for cash at $0.12 per warrant |
— | — | — | — | — | — | — | — | 819,400 | — | — | — | 819,400 | |||||||||||||||||||||||||||
| Issuance of common stock in registered direct offering in October 2009 for cash at $0.80 per share, net of financing costs of $92,470 |
— | — | — | — | — | — | 6,161,068 | 61,611 | 4,774,774 | — | — | — | 4,836,385 | |||||||||||||||||||||||||||
| Common stock warrants reclassified to common stock warrant liability on January 1, 2009 |
— | — | — | — | — | — | — | — | (1,398,702 | ) | — | — | (130,968 | ) | (1,529,670 | ) | ||||||||||||||||||||||||
| Cashless exercise of 956,522 warrants with an exercise price of $1.15 per share in exchange for 752,818 shares of common stock in September 2009 |
— | — | — | — | — | — | 752,818 | 7,528 | 4,742,598 | — | — | — | 4,750,126 | |||||||||||||||||||||||||||
| Cashless exercise of 628,210 warrants with exercise prices ranging from $1.15 to $3.94 per share in exchange for 373,467 shares of common stock in October 2009 |
— | — | — | — | — | — | 373,467 | 3,735 | 490,917 | — | — | — | 494,652 | |||||||||||||||||||||||||||
| Exercise of 80,706 common stock options in 2009 for cash from $0.46 to $3.70 per share |
— | — | — | — | — | — | 80,706 | 807 | 36,710 | — | — | — | 37,517 | |||||||||||||||||||||||||||
| Amortization of deferred compensation |
— | — | — | — | — | — | — | — | — | 20,000 | — | — | 20,000 | |||||||||||||||||||||||||||
| Balance, December 31, 2009 |
— | $ | — | — | $ | — | — | $ | — | 37,378,387 | $ | 373,784 | $ | 138,576,593 | $ | (1,667 | ) | $ | — | $ | (133,367,600 | ) | $ | 5,581,110 | ||||||||||||||||
See accompanying notes to consolidated financial statements.
66
(A development stage company)
Consolidated Statements of Cash Flows
| Years ended December 31, | Period from December 19, 2002 (inception) to December 31, 2009 |
|||||||||||||||
| 2009 | 2008 | 2007 | ||||||||||||||
| Cash flows from operating activities: |
||||||||||||||||
| Net loss |
$ | (31,929,200 | ) | $ | (37,874,028 | ) | $ | (28,575,348 | ) | $ | (133,236,632 | ) | ||||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||||||
| Depreciation |
424,354 | 513,060 | 403,041 | 1,580,310 | ||||||||||||
| Loss on sale of equipment |
4,651 | 5,313 | 7,911 | 20,236 | ||||||||||||
| Employee stock-based compensation |
2,209,216 | 2,648,410 | 883,310 | 5,788,415 | ||||||||||||
| Nonemployee stock-based compensation |
230,075 | (127,855 | ) | 2,972,815 | 3,217,829 | |||||||||||
| Amortization of commitment fees, debt issuance costs and original issue discount |
1,490,940 | 654,333 | 738,166 | 3,264,022 | ||||||||||||
| Amortization of short-term investment discount |
904 | (3,679 | ) | (148,834 | ) | (308,051 | ) | |||||||||
| Change in value of warrant liability |
3,644,549 | — | 361,504 | 3,999,456 | ||||||||||||
| Change in operating assets and liabilities: |
||||||||||||||||
| Interest receivable |
57,965 | (4,788 | ) | 56,224 | — | |||||||||||
| Other receivables |
9,301 | 23,827 | 2,985 | (10,007 | ) | |||||||||||
| Prepaid expenses and other current assets |
(52,519 | ) | 4,901 | (359,072 | ) | (474,336 | ) | |||||||||
| Other assets |
— | 5,000 | 1,395 | — | ||||||||||||
| Accounts payable |
(73,709 | ) | (183,034 | ) | (378,420 | ) | 43,599 | |||||||||
| Accrued expenses |
(784,186 | ) | 492,058 | 665,813 | 2,077,916 | |||||||||||
| Accrued interest payable |
110,436 | 177,869 | — | 454,127 | ||||||||||||
| Net cash used in operating activities |
(24,657,223 | ) | (33,668,613 | ) | (23,368,510 | ) | (113,583,116 | ) | ||||||||
| Cash flows from investing activities: |
||||||||||||||||
| Purchases of short-term investments available for sale |
— | (9,127,233 | ) | — | (14,882,233 | ) | ||||||||||
| Maturities of short-term investments available for sale |
5,226,000 | 8,938,414 | 690,000 | 14,854,414 | ||||||||||||
| Purchases of short-term investments held to maturity |
— | (1,185,838 | ) | (6,944,194 | ) | (22,414,130 | ) | |||||||||
| Maturities of short-term investments held to maturity |
— | 4,450,000 | 15,300,000 | 22,750,000 | ||||||||||||
| Purchases of property and equipment |
(186,981 | ) | (244,439 | ) | (800,393 | ) | (2,576,356 | ) | ||||||||
| Net cash provided by (used in) investing activities |
5,039,019 | 2,830,904 | 8,245,413 | (2,268,305 | ) | |||||||||||
| Cash flows from financing activities: |
||||||||||||||||
| Proceeds from stock options exercised |
37,517 | 65,407 | 21,187 | 177,157 | ||||||||||||
| Proceeds from warrants issued |
819,400 | — | — | 835,057 | ||||||||||||
| Proceeds from warrants exercised |
— | — | — | 187,652 | ||||||||||||
| Proceeds from sale of common stock, net of underwriting fees of $3,074,315 |
— | — | 40,844,477 | 40,874,977 | ||||||||||||
| Proceeds from sale of common stock in private placement and registered direct offerings |
20,005,807 | — | — | 20,005,807 | ||||||||||||
| Common stock financing costs |
(898,969 | ) | — | (1,752,663 | ) | (2,651,632 | ) | |||||||||
| Payment to shareholders for fractional shares upon reverse stock split |
— | — | (355 | ) | (355 | ) | ||||||||||
| Proceeds from sale of Series A, B and C convertible preferred stock |
— | — | — | 57,928,353 | ||||||||||||
| Series B and C convertible preferred stock financing costs |
— | — | — | (1,597,983 | ) | |||||||||||
| Proceeds from convertible notes payable |
— | — | — | 6,814,846 | ||||||||||||
| Proceeds from notes payable |
5,000,000 | 15,000,000 | 10,000,000 | 35,831,121 | ||||||||||||
| Repayments on notes payable |
(11,783,065 | ) | (11,674,658 | ) | (2,793,712 | ) | (27,614,186 | ) | ||||||||
| Debt issuance costs |
— | (230,241 | ) | — | (321,799 | ) | ||||||||||
| Net cash provided by financing activities |
13,180,690 | 3,160,508 | 46,318,934 | 130,469,015 | ||||||||||||
| Net (decrease) increase in cash and cash equivalents |
(6,437,514 | ) | (27,677,201 | ) | 31,195,837 | 14,617,594 | ||||||||||
| Cash and cash equivalents: |
||||||||||||||||
| Beginning of period |
21,055,108 | 48,732,309 | 17,536,472 | — | ||||||||||||
| End of period |
$ | 14,617,594 | $ | 21,055,108 | $ | 48,732,309 | $ | 14,617,594 | ||||||||
| Supplemental disclosure: |
||||||||||||||||
| Interest paid |
$ | 2,502,924 | $ | 1,826,674 | $ | 935,433 | $ | 5,863,355 | ||||||||
| Noncash investing and financing activities: |
||||||||||||||||
| Cancellation of Alpha Medical, Inc. Series A convertible preferred stock and common stock |
$ | — | $ | — | $ | — | $ | (661,674 | ) | |||||||
| Issuance of Beta Medical, Inc. Series A convertible preferred stock in exchange for Alpha Medical, Inc. Series A convertible preferred stock and common stock |
— | — | — | 661,674 | ||||||||||||
| Value of warrants issued with debt |
542,144 | 1,398,702 | 673,768 | 2,907,676 | ||||||||||||
| Value of warrants issued for debt commitment |
— | — | 550,212 | 636,250 | ||||||||||||
| Value of warrants issued with Series C financing |
— | — | — | 735,438 | ||||||||||||
| Value of warrants issued with private investment public equity financing |
154,525 | — | — | 154,525 | ||||||||||||
| Cashless exercise of warrants |
5,244,778 | — | — | 5,244,778 | ||||||||||||
| Conversion of notes and interest payable to Series B and C convertible preferred shares |
— | — | — | 6,980,668 | ||||||||||||
| Options issued for deferred compensation |
— | — | — | 10,898 | ||||||||||||
| Common stock issued to Mayo Foundation and for deferred compensation |
— | — | 1,658,654 | 1,770,904 | ||||||||||||
| Reclassification of warrant liability |
1,529,670 | — | 1,090,345 | 2,620,015 | ||||||||||||
| Conversion of convertible preferred stock to common stock |
— | — | 103,138 | 103,138 | ||||||||||||
See accompanying notes to consolidated financial statements.
67
(A development stage company)
Notes to Consolidated Financial Statements
| (1) | Formation and Business of the Company |
EnteroMedics Inc. (EnteroMedics or the Company) is developing implantable systems to treat obesity and other gastrointestinal disorders. The Company was incorporated in the state of Minnesota on December 19, 2002, originally as two separate legal entities, Alpha Medical, Inc. and Beta Medical, Inc., both of which were owned 100% by a common stockholder. Effective October 1, 2003, the two entities were combined and the combined entity changed its name to EnteroMedics Inc. The Company reincorporated in Delaware on July 22, 2004. The Company is in the development stage and since inception has devoted substantially all of its resources to recruiting personnel, developing its product technology, obtaining patents to protect its intellectual property and raising capital, and has not derived revenues from its primary business activity. Accordingly, the Company is in the development stage, as defined by the Accounting Standards Codification. The Company is headquartered in St. Paul, Minnesota.
EnteroMedics Europe Sárl (EnteroMedics Europe), a wholly owned subsidiary of the Company, was formed in January 2006. EnteroMedics Europe is a Swiss entity established as a means to conduct clinical trials in Switzerland. Upon establishment there were 20 shares of EnteroMedics Europe issued and outstanding with a par value of 1,000 Swiss Francs. EnteroMedics purchased 100% of the shares and then issued one share to a fiduciary agent. The one share is the property of EnteroMedics and is held by the fiduciary in a fiduciary capacity under terms of the Fiduciary Agreement. The functional currency of EnteroMedics Europe has been determined to be the U.S. Dollar.
In November 2007, the Company effected a 1-for-9.1 reverse split of its common stock and convertible preferred stock which has been retroactively applied to these consolidated financial statements. Also, in November 2007, the Company completed its initial public offering of common stock (IPO), issuing a total of 5,489,849 shares for net proceeds of approximately $39.1 million after expenses and underwriters’ discounts and commissions, and including the exercise of the underwriters’ over-allotment option.
Since inception, EnteroMedics has incurred losses through December 31, 2009 totaling approximately $133.2 million and has not generated positive cash flows from operations. The Company expects such losses to continue into the foreseeable future as it continues to develop and commercialize its technologies. The Company may need to obtain additional financing and there can be no assurance that the Company will be successful in obtaining additional financing on favorable terms, or at all. If adequate funds are not available, the Company may have to delay development or commercialization of products or license to third parties the rights to commercialize products or technologies that the Company would otherwise seek to commercialize.
| (2) | Summary of Significant Accounting Policies |
Basis of Presentation
The Company has prepared the accompanying consolidated financial statements in conformity with accounting principles generally accepted in the United States of America. The Company’s fiscal year ends on December 31.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Actual results could differ from those estimates.
68
EnteroMedics Inc.
(A development stage company)
Notes to Consolidated Financial Statements (Continued)
Principles of Consolidation
The consolidated financial statements include the accounts of the Company and its wholly owned subsidiary. All intercompany transactions and accounts have been eliminated in consolidation.
Concentration of Credit Risk and Other Risks and Uncertainties
Financial instruments that potentially subject the Company to significant concentrations of credit risk consist primarily of cash and cash equivalents and short-term investments. Cash and cash equivalents are primarily deposited in demand and money market accounts. At times, such deposits may be in excess of insured limits. Investments in short-term money market funds are not considered to be bank deposits and are not insured or guaranteed by the federal deposit insurance company or other government agency. These money market funds seek to preserve the value of the investment at $1.00 per share; however, it is possible to lose money investing in these funds. The Company has not experienced any losses on its deposits of cash and cash equivalents.
Most of the products developed by the Company will require approval from the U.S. Food and Drug Administration (FDA) or corresponding foreign regulatory agencies prior to commercial sales. There can be no assurance the Company’s products will receive the necessary approvals. If the Company is denied approval or approval is delayed, it will have a material adverse impact on the Company.
The medical device industry is characterized by frequent and extensive litigation and administrative proceedings over patent and other intellectual property rights. Whether a product infringes a patent involves complex legal and factual issues, the determination of which is often difficult to predict, and the outcome may be uncertain until the court has entered final judgment and all appeals are exhausted. The Company’s competitors may assert that its products or the use of the Company’s products are covered by U.S. or foreign patents held by them.
Fair Value of Financial Instruments
Carrying amounts of certain of the Company’s financial instruments, including cash and cash equivalents, prepaid expenses and other current assets, accounts payable and accrued liabilities approximate fair value due to their short maturities. The fair values of investments in debt and equity securities are disclosed in Note 4. The fair value of the Company’s long-term debt is approximately $8.3 million as of December 31, 2009 based on the present value of estimated future cash flows using a discount rate commensurate with borrowing rates available to the Company.
Cash and Cash Equivalents
The Company considers highly liquid investments with maturities of 90 days or less when purchased to be cash equivalents. Cash equivalents are stated at cost, which approximates market value. The Company’s cash equivalents are primarily in money market funds and commercial paper. The Company deposits its cash and cash equivalents in high-quality credit institutions. Under terms of the Company’s notes payable agreements (see Note 7), in the event of default, the lender has the right to enforce account control agreements and restrict the Company’s access to their cash and investment accounts.
Short-Term Investments
The Company considers all investments with maturities greater than three months and less than one year at the time of purchase as short-term investments and classifies them as either available for sale or held to maturity.
69
EnteroMedics Inc.
(A development stage company)
Notes to Consolidated Financial Statements (Continued)
The Company also considers certain investments with maturities greater than one year but which are also held for liquidity purposes and are available for sale as short-term investments.
Available-for-sale securities are carried at fair value based on quoted market prices, with the unrealized gains and losses included in other comprehensive income within stockholders’ equity (deficit) in the consolidated balance sheets. Realized gains and losses and declines in value judged to be other than temporary on available-for-sale securities are included in interest and other income. Interest and dividends on securities classified as available for sale are included in interest income. The cost of securities sold is based on the specific identification method.
Short-term investments in debt securities which the Company has the positive intent and ability to hold to maturity are reported at cost, adjusted for premiums and discounts that are recognized in interest income, using the interest method, over the period to maturity. Unrealized losses on held-to-maturity securities reflecting a decline in value determined to be other than temporary are charged to income.
Property and Equipment, Net
Property and equipment are stated at cost less accumulated depreciation and amortization. Depreciation of property and equipment is computed using the straight-line method over their estimated useful lives of three to seven years. Leasehold improvements are amortized on a straight-line basis over the lesser of their useful life or the term of the lease. Upon retirement or sale, the cost and related accumulated depreciation are removed from the consolidated balance sheets and the resulting gain or loss is reflected in the consolidated statements of operations. Repairs and maintenance are expensed as incurred.
Impairment of Long-Lived Assets
The Company evaluates its long-lived assets for impairment by comparison of the carrying amounts to future net undiscounted cash flows expected to be generated by such assets when events or changes in circumstances indicate the carrying amount of an asset may not be recoverable. Should an impairment exist, the impairment loss would be measured based on the excess carrying value of the asset over the asset’s fair value or estimates of future discounted cash flows. The Company has not identified any such impairment losses to date.
Income Taxes
Income taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carry-forwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. A valuation allowance for deferred income tax assets is recorded when it is more likely than not that some portion or all of the deferred income tax assets will not be realized. The Company has provided a full valuation allowance against the gross deferred tax assets as of December 31, 2009 and 2008 (see Note 10). The Company’s policy is to classify interest and penalties related to income taxes as income tax expense in the consolidated statements of operations.
Comprehensive Loss
Comprehensive loss is defined as the change in equity of a company during a period from transactions and other events and circumstances excluding transactions resulting from investment owners and distributions to
70
EnteroMedics Inc.
(A development stage company)
Notes to Consolidated Financial Statements (Continued)
owners. The difference from reported net loss for the year ended December 31, 2009 related entirely to the maturity of short-term investments and the realization of net unrealized gains on those short-term investments. The difference from reported net loss for the year ended December 31, 2008 related entirely to net unrealized gains on short-term investments. There was no difference from reported net loss for the year ended December 31, 2007.
Research and Development Expenses
Research and development expenses are charged to expense as incurred. Research and development expenses include, but are not limited to, product development, clinical and regulatory expenses, payroll and other personnel expenses, materials, supplies, and consulting costs.
Patent Costs
Costs associated with the submission of a patent application are expensed as incurred given the uncertainty of the patents resulting in probable future economic benefits to the Company. Patent-related legal expenses included in general and administrative costs were $231,291, $346,119 and $378,362 for the years ended December 31, 2009, 2008 and 2007, respectively, and $1,586,996 for the period from December 19, 2002 (inception) to December 31, 2009.
Derivative Instruments
The Company accounts for outstanding warrants that are not indexed to the Company’s stock or warrants issued when the Company has insufficient authorized and unissued stock available to share settle the outstanding warrants as derivative instruments, which require that the warrants be classified as a liability and measured at fair value with changes in fair value recognized currently in earnings and recorded separately in the consolidated statements of operations.
On July 6, 2006, the Company issued 147,635 Series C warrants upon closing the Series C convertible preferred stock financing with a fair value of $735,438 calculated using a Black-Scholes valuation model and the following assumptions: volatility of 55%, dividend rate of 0%, risk-free interest rate of 5.18%, and the maximum seven-year warrant life. In December 2006, the Company had insufficient authorized and unissued Series C stock available to share settle the outstanding Series C warrants which required the warrants to be classified as a liability. The fair market value of the warrants as of December 31, 2006 was $728,841. On May 14, 2007 the Company filed an amended certificate of incorporation to increase the number of authorized shares of Series C stock, resulting in sufficient authorized and unissued shares of Series C stock available to share settle the Series C warrants. The fair market value of the warrants on May 14, 2007 was determined to be $1,090,345. The change in fair value from December 31, 2006 to the amendment date of $361,504 was recorded as expense and the warrant liability was reclassified to additional paid-in capital.
Effective January 1, 2009, as a result of a change in accounting guidance, the Company assessed any outstanding equity-linked financial instruments and concluded that warrants issued in November 2008 with a recorded value of $1.4 million on December 31, 2008 were to be reclassified from equity to a liability. The cumulative effect of the change in accounting principle on January 1, 2009 was a $130,968 increase to the deficit accumulated during development stage. See Note 7 for details regarding the change in fair value of the warrant liability during the year ended December 31, 2009.
71
EnteroMedics Inc.
(A development stage company)
Notes to Consolidated Financial Statements (Continued)
Stock-Based Compensation
Prior to January 1, 2006, the Company accounted for stock-based employee compensation using the intrinsic value method and recognizing the expense over the option vesting period. The intrinsic value method is calculated as the difference, if any, between the fair value of the Company’s stock and the exercise price on the date of the grant. The Company also followed the minimum value disclosure provisions.
Effective January 1, 2006, the Company adopted the fair value method of accounting for share-based payments, which superseded the previous accounting method, and requires compensation expense to be recognized using a fair-value-based method for costs related to all share-based payments including stock options. Companies are required to estimate the fair value of share-based payment awards on the date of grant using an option-pricing model. The Company adopted the new provisions using the prospective transition method, which requires that for nonpublic entities that used the minimum value method for either pro forma or financial statement recognition purposes, to apply the new accounting provisions only to option grants or modifications to existing options that occur after the required effective date. For options granted prior to January 1, 2006, the Company has continued to apply the intrinsic value provisions on any remaining unvested awards. All option grants valued after January 1, 2006 are expensed on a straight-line basis over the vesting period.
The fair value method is applied to all share-based payment awards issued to employees and where appropriate, nonemployees, unless another source of literature applies. When determining the measurement date of a nonemployee’s share-based payment award, the Company measures the stock options at fair value and remeasures such stock options to the current fair value until the performance date has been reached.
Net Loss Per Share
Basic net loss per share is computed by dividing net loss by the weighted-average number of common shares outstanding during the period. Diluted net loss per share is based on the weighted-average common shares outstanding during the period plus dilutive potential common shares calculated using the treasury stock method. Such potentially dilutive shares are excluded when the effect would be to reduce a net loss per share. The Company’s potential dilutive shares, which include outstanding common stock options, unvested common shares subject to repurchase and warrants, have not been included in the computation of diluted net loss per share for all periods as the result would be anti-dilutive.
The following table sets forth the computation of basic and diluted net loss per share for the years ended December 31, 2009, 2008 and 2007:
| Year ended December 31, |
||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| Numerator: |
||||||||||||
| Net loss |
$ | (31,929,200 | ) | $ | (37,874,028 | ) | $ | (28,575,348 | ) | |||
| Denominator for historical and basic and diluted net loss per share: |
||||||||||||
| Weighted-average common shares outstanding |
29,845,954 | 16,835,661 | 2,447,515 | |||||||||
| Weighted-average unvested common shares subject to repurchase |
— | — | (2,514 | ) | ||||||||
| Denominator for net loss per common share—basic and diluted |
29,845,954 | 16,835,661 | 2,445,001 | |||||||||
| Net loss per share—basic and diluted |
$ | (1.07 | ) | $ | (2.25 | ) | $ | (11.69 | ) | |||
72
EnteroMedics Inc.
(A development stage company)
Notes to Consolidated Financial Statements (Continued)
The following table sets forth the potential shares of common stock that are not included in the calculation of diluted net loss per share because to do so would be anti-dilutive as of the end of each period presented:
| December 31, | ||||||
| 2009 | 2008 | 2007 | ||||
| Stock options outstanding |
5,974,173 | 2,797,178 | 2,101,926 | |||
| Warrants to purchase common stock |
7,994,225 | 1,759,216 | 683,235 | |||
Recently Issued Accounting Standards
In June 2009, the Financial Accounting Standards Board (FASB) approved the FASB Accounting Standards Codification (ASC or the Codification) as the single source of authoritative, nongovernmental accounting principles generally accepted in the United States of America (GAAP), excluding the guidance issued by the Securities and Exchange Commission (SEC). FASB approved an Exposure Draft that replaced Statement of Financial Accounting Standards No. 162, The Hierarchy of Generally Accepted Accounting Principles, and modified GAAP by establishing only two levels of GAAP, authoritative and nonauthoritative. This was accomplished by authorizing the Codification to become the single source of authoritative U.S. accounting and reporting standards, except for rules and interpretive releases of the SEC, which are sources of authoritative GAAP for SEC registrants. All other nongrandfathered, non-SEC accounting literature not included in the Codification has become nonauthoritative. The Codification was effective for the Company beginning with the quarter ended September 30, 2009. The adoption of the Codification did not have a material impact on the consolidated financial statements.
Effective January 1, 2009, the Company adopted new authoritative accounting guidance regarding the financial reporting for outstanding equity-linked financial instruments. See the discussion above in this Note 2, under the heading “Derivative Instruments,” for more details on the adoption of this new authoritative accounting guidance.
There have been no other significant changes in recent accounting pronouncements during the fiscal year ended December 31, 2009.
| (3) | Liquidity and Management’s Plans |
The accompanying consolidated financial statements have been prepared assuming the Company will continue as a going concern. For the period from inception (December 19, 2002) through December 31, 2009 the Company experienced net losses of $133.2 million and cash used in operations of $113.6 million. As of December 31, 2009, the Company has not emerged from the development stage and had approximately $14.6 million of cash and cash equivalents. On January 14, 2010, the Company entered into a securities purchase agreement with certain institutional investors for the sale of 7,438,299 shares of its common stock in a registered direct offering, at a purchase price of $0.65 per share. On January 20, 2010 the Offering closed and the Company received gross proceeds of $4.8 million before deducting estimated offering expenses (see Note 17). Assuming the Company does not receive any additional funds, it estimates that it has sufficient funds to operate into the second half of 2010, which has raised a substantial doubt about the Company’s ability to continue as a going concern. In order to fund on-going operating cash requirements beyond that point or to further accelerate and execute its business plan, including a potentially new clinical trial using the next-generation Maestro RC System, if approved by the FDA, the Company will need to and plans to raise significant additional funds. In view of these matters, the ability of the Company to continue as a going concern is dependent upon the Company’s ability to secure additional financing sufficient to support its research and development activities, approval of developed products for sale by regulatory authorities, including the FDA, and ultimately to generate revenues sufficient to cover all costs.
73
EnteroMedics Inc.
(A development stage company)
Notes to Consolidated Financial Statements (Continued)
Since inception, the Company has financed its activities principally from the sale of equity securities. While the Company has been successful in the past in obtaining the necessary capital to support its operations, and has similar future plans, there is no assurance that the Company will be able to obtain additional equity capital or other financing under commercially reasonable terms and conditions, or at all. Furthermore, if the Company issues equity or debt securities to raise additional funds, existing shareholders may experience dilution and the new equity or debt securities it issues may have rights, preferences and privileges senior to those of existing stockholders. In addition, if the Company raises additional funds through collaboration, licensing or other similar arrangements, it may be necessary to relinquish valuable rights to products or proprietary technologies, or grant licenses on terms that are not favorable. If the Company cannot execute its plan to raise funds on acceptable terms, the Company will not be able to continue as a going concern, develop or enhance products, obtain the required regulatory clearances or approvals, execute the Company’s business plan, take advantage of future opportunities, or respond to competitive pressure or unanticipated customer requirements. If the Company is unable to obtain additional financing or the FDA does not approve the IDE application for a clinical trial using the next-generation Maestro RC System, the Company may be required to reduce the scope of, delay, or eliminate some or all of, our planned research, development and commercialization activities, which could materially harm our business. Any of these events would adversely affect the Company’s ability to achieve the Company’s development and commercialization goals, which could have a material adverse effect on the Company’s business, results of operations and financial condition. The Company’s financial statements do not include any adjustments relating to the recoverability or classification of assets or the amounts of liabilities that might result from the outcome of these uncertainties.
| (4) | Short-term Investments and Fair Value Measurements |
Effective January 1, 2008, the Company adopted fair value measurement and disclosure provisions for its financial assets and liabilities as described below.
Fair value of financial assets and liabilities is defined as the price that would be received to sell an asset or transfer a liability in an orderly transaction between market participants at the measurement date. A fair value hierarchy has been established that prioritizes the inputs to valuation techniques used to measure fair value. The hierarchy gives the highest priority to unadjusted quoted prices in active markets for identical assets or liabilities (Level 1 measurements) and the lowest priority to unobservable inputs (Level 3 measurements). The three levels of the fair value hierarchy are described below:
| • | Level 1—Unadjusted quoted prices in active markets that are accessible at the measurement date for identical, unrestricted assets or liabilities. |
| • | Level 2—Quoted prices for similar assets and liabilities, quoted prices for identical or similar assets or liabilities in markets that are not active or model-derived valuations for which all significant inputs are observable, either directly or indirectly. |
| • | Level 3—Prices or valuation techniques that require inputs that are both significant to the fair value measurement and unobservable. |
The Company’s assets that are measured at fair value on a recurring basis are classified within Level 1 or Level 2 of the fair value hierarchy. The Company does not hold any assets that are measured at fair value using Level 3 inputs. The types of instruments the Company invests in that are valued based on quoted market prices in active markets include U.S. treasury securities. Such instruments are classified by the Company within Level 1 of the fair value hierarchy. U.S. treasuries are valued using unadjusted quoted prices for identical assets in active markets that the Company can access.
74
EnteroMedics Inc.
(A development stage company)
Notes to Consolidated Financial Statements (Continued)
The types of instruments the Company invests in that are valued based on quoted prices in less active markets, broker or dealer quotations, or alternative pricing sources with reasonable levels of price transparency include the Company’s U.S. agency securities, commercial paper, U.S. corporate bonds and municipal obligations. Such instruments are classified by the Company within Level 2 of the fair value hierarchy. The Company values these types of assets using consensus pricing or a weighted average price, which is based on multiple pricing sources received from a variety of industry standard data providers (e.g. Bloomberg), security master files from large financial institutions, and other third-party sources. The multiple prices obtained are then used as inputs in to a distribution-curve-based algorithm to determine the daily market price.
In addition, the Company recorded a financial liability in 2009 related to warrants outstanding, which is fair valued using Level 3 inputs (see Note 7).
The Company did not hold any short-term investments classified as available for sale as of December 31, 2009 and did not hold any short-term investments classified as held to maturity as of December 31, 2009 and 2008. The following table sets forth by level, within the fair value hierarchy, the Company’s financial assets accounted for at fair value as of December 31, 2008. Assets and liabilities are classified in their entirety based on the lowest level of input that is significant to the fair value measurement.
All short-term investments at December 31, 2008 are classified as Level 2 and are as follows:
| Significant Other Observable Inputs Level 2 | |||
| U.S. agency securities |
$ | 2,692,482 | |
| U.S. corporate bonds |
1,400,829 | ||
| Commercial paper |
797,686 | ||
| Asset-backed securities |
348,895 | ||
| Total |
$ | 5,239,892 | |
The amortized cost and fair value of short-term investments available for sale, and the related gross unrealized gains and losses, were as follows:
As of December 31, 2008
| Gross Unrealized | |||||||||||||
| Cost | Gains | Losses | Fair value | ||||||||||
| U.S. agency securities |
$ | 2,676,917 | $ | 15,565 | $ | — | $ | 2,692,482 | |||||
| U.S. corporate bonds |
1,402,670 | — | (1,841 | ) | 1,400,829 | ||||||||
| Commercial paper |
797,520 | 166 | — | 797,686 | |||||||||
| Asset-backed securities |
349,797 | — | (902 | ) | 348,895 | ||||||||
| Total investment securities available for sale |
$ | 5,226,904 | $ | 15,731 | $ | (2,743 | ) | $ | 5,239,892 | ||||
With the exception of the asset-backed security position, the short-term investments available for sale at December 31, 2008 had contractual maturities of less than one year. At December 31, 2008, the asset-back security had a contractual maturity between two and three years with an effective maturity of less than one year. In February 2009 the asset-backed security was called at 100% of the original face amount.
75
EnteroMedics Inc.
(A development stage company)
Notes to Consolidated Financial Statements (Continued)
| (5) | Property and Equipment |
Property and equipment consist of the following as of:
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| Furniture and equipment |
$ | 1,713,162 | $ | 1,643,344 | ||||
| Computer hardware and software |
468,289 | 457,403 | ||||||
| Leasehold improvements |
32,258 | — | ||||||
| 2,213,709 | 2,100,747 | |||||||
| Less accumulated depreciation and amortization |
(1,247,880 | ) | (836,844 | ) | ||||
| Property and equipment, net |
$ | 965,829 | $ | 1,263,903 | ||||
Depreciation expense included in the consolidated statements of operations was $424,354, $513,060 and $403,041 for the years ended December 31, 2009, 2008 and 2007, respectively, and $1,580,310 for the period from December 19, 2002 (inception) to December 31, 2009.
| (6) | Accrued expenses |
Accrued expenses consist of the following as of:
| December 31, | ||||||
| 2009 | 2008 | |||||
| Professional service related expenses |
$ | 1,419,576 | $ | 1,150,223 | ||
| Payroll related expenses |
423,377 | 1,176,255 | ||||
| Other expenses |
234,963 | 535,624 | ||||
| Accrued expenses |
$ | 2,077,916 | $ | 2,862,102 | ||
| (7) | Notes payable |
Notes payable consists of the following as of:
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| Growth capital loan dated November 18, 2008 (net discount of $455,469 and $1,329,592 at December 31, 2009 and 2008, respectively) |
$ | 7,761,466 | $ | 13,670,408 | ||||
| Less current portion |
(3,880,656 | ) | (2,674,597 | ) | ||||
| Total long-term debt |
$ | 3,880,810 | $ | 10,995,811 | ||||
On May 17, 2007 the Company entered into a $15.0 million debt facility with Western Technology Investment (WTI) of which $5.0 million was drawn on May 22, 2007, $2.5 millions was drawn on August 31, 2007 and $2.5 million was drawn on October 31, 2007. Each loan consisted of interest only payments for the first six months, except for the October 31, 2007 draw which was for the first four months, at an annual percentage rate of 12.48%, followed by 30 equal principal and interest installments at an annual percentage rate of 10.25%. Each loan included a final payment of 3.1% of the amount funded.
76
EnteroMedics Inc.
(A development stage company)
Notes to Consolidated Financial Statements (Continued)
Upon the closing of the initial commitment, the Company issued 67,963 Series C stock warrants with an exercise price of $8.0926 per share and a seven year life. The fair value of the warrants at the time of issuance was determined to be $550,212 and is recorded as interest expense in 2007. The fair value of the warrants was calculated using a Black-Scholes valuation model and the following assumptions: volatility of 55.5%, dividend rate of 0%, risk-free interest rate of 4.76% and a seven year life. In accordance with the agreement, upon the closing of the IPO, the warrants were converted into warrants to purchase common stock, with all other terms unchanged.
In conjunction with the three growth capital loan draws between May and October 2007, the Company issued a total of 67,964 Series C stock warrants at an exercise price of $8.0926 per share and a seven year life. The combined fair value of the warrants issued was determined to be $673,768 and is recorded as interest expense over the term of the loan, with $286,319 and $132,217 recorded as interest expense in 2008 and 2007, respectively. This fair value was calculated using a Black-Scholes valuation model and the following assumptions: volatility between 50.3% and 55.5%, dividend rate of 0%, risk-free interest rate between 4.47% and 4.83% and a seven year life. In accordance with the agreement, upon the closing of the IPO, the warrants were converted into warrants to purchase common stock, with all other terms unchanged.
On November 21, 2008, the Company had outstanding $7,702,641 (principal of $6,939,610 and interest of $763,031) in total payments and $255,232 of unamortized discount on notes payable with WTI. Proceeds from a new debt agreement, discussed below, were used to repay all outstanding indebtedness under the previous WTI loan agreements, resulting in the one-time interest payment of $763,031 and the acceleration of $255,232 of unamortized discount on notes payable, both recorded as interest expense in 2008.
On November 18, 2008 the Company entered into a new Loan and Security Agreement (the Loan Agreement) with Silicon Valley Bank (SVB), Venture Lending & Leasing V, Inc. (a private equity fund under the management of WTI) and Compass Horizon Funding Company LLC (Horizon and, collectively with SVB and WTI, the Lenders), in an aggregate principal amount of up to $20.0 million. On November 21, 2008, SVB and WTI each funded a Term Loan in the aggregate principal amount of $10.0 million and $5.0 million, respectively. The additional $5.0 million Term Loan was automatically funded by Horizon on April 28, 2009 when the trading price of the Company’s common stock on the NASDAQ Global Market exceeded a target amount specified in the Loan Agreement.
Interest-only payments are required on the Term Loans during a period beginning on the Term Loan funding date and continuing through June 30, 2009, followed thereafter by equal monthly payments of principal and interest over the remaining term of the Term Loan. Amounts borrowed under the Loan Agreement bear interest per annum at a rate equal to 12.0% during the period of interest-only payments, and thereafter, at a rate of 11.0% per annum for the remainder of the term. The Loan Agreement will terminate and all outstanding Term Loans must be repaid no later than December 1, 2011 (the Maturity Date). On the Maturity Date, the Company will also make a final payment in an aggregate amount equal to 5.0% of the Term Loans funded by the Lenders (the Final Payment Fee). The Company may voluntarily prepay the Term Loans in full, but not in part and any voluntary or mandatory prepayment is subject to applicable prepayment premiums. The Company will also be required to pay the Final Payment Fee in connection with any voluntary or mandatory prepayment.
On December 1, 2009 the Company voluntarily prepaid both the WTI and Horizon Term Loans in full, or $9,100,468 (principal of $8,522,346, interest accrued and not yet paid of $78,122 and the 5.0% Final Payment Fee of $500,000). The prepayment of the Term Loans also resulted in the acceleration of $601,505 of unamortized discount on notes payable, recorded as interest expense in 2009. Both WTI and Horizon released their right to future interest when the Term Loans were paid in full.
77
EnteroMedics Inc.
(A development stage company)
Notes to Consolidated Financial Statements (Continued)
The Loan Agreement requires the issuance of warrants to the Lenders with an aggregate exercise price equal to 11.0% of the loan commitment. The warrants give the Lenders the option to purchase either (i) shares of the Company’s common stock with a per share exercise price equal to $1.5846, or (ii) shares of the Company’s stock (including common stock) issued in an equity financing that occurs within 18 months after November 18, 2008 at the per share price of the stock sold in the financing. On November 18, 2008 (i) SVB was issued a warrant to purchase an aggregate number of shares equal to $1,100,000 divided by the per share exercise price of the warrant, (ii) WTI was issued a warrant to purchase an aggregate number of shares equal to $550,000 divided by the per share exercise price of the warrant, and (iii) Horizon received a warrant to purchase an aggregate number of shares equal to $55,000 divided by the per share exercise price of the warrant. On April 28, 2009 Horizon was issued an additional warrant to purchase an aggregate number of shares equal to $495,000 divided by the per share exercise price of the warrant in connection with the additional $5.0 million Term Loan that was automatically funded by Horizon pursuant to the Loan Agreement.
On November 18, 2008, the Company issued a total of 1,075,981 common stock warrants with an exercise price of $1.5846 per share and a ten year life to the Lenders, or a calculated fair value of $1.4 million. This fair value was calculated using a Black-Scholes valuation model and the following assumptions: volatility of 78.9%, dividend rate of 0%, risk-free interest rate of 3.54% and a ten year life. The exercise price of the common stock warrants issued on November 18, 2008 was adjusted to $1.15, the price per share sold in an equity financing that closed on February 24, 2009 (see Note 8), resulting in an additional 406,628 common stock warrants for the Lenders. On April 28, 2009, the Company issued a total of 296,763 common stock warrants with an exercise price of $1.668 per share and a ten year life to Horizon, or a calculated fair value of $542,144. This fair value was calculated using a Black-Scholes valuation model and the following assumptions: volatility of 99.1%, dividend rate of 0%, risk-free interest rate of 3.00% and a ten year life. The exercise price of the common stock warrants issued to Horizon on both November 18, 2008 and April 28, 2009 was further adjusted to $0.80, the price per share sold in an equity financing that closed October 7, 2009 (see Note 8), resulting in an additional 342,911 common stock warrants for Horizon. On January 20, 2010, the Company closed an equity financing transaction at $0.65 per share resulting in an additional adjustment to Horizon’s outstanding warrants (see Note 17).
As discussed in Note 2, effective January 1, 2009, as a result of a change in accounting guidance, the Company revalued the warrants issued in November 2008 and reclassified them from equity to a liability. The fair value of the warrant liability on January 1, 2009 was $1.5 million and the change in fair value was recorded as an increase to the deficit accumulated during development stage. This fair value was calculated using a weighted-average Black-Scholes valuation model and the following assumptions: volatility of 79.6%, dividend rate of 0%, risk-free interest rate of 2.24% and a remaining life of 9.88 years.
On September 29, 2009, SVB completed a cashless exercise of the warrants issued to them as part of the Loan Agreement. SVB held a total of 956,522 common stock warrants with an exercise price of $1.15 per share. The cashless exercise of the warrants resulted in the Company issuing 752,818 shares of its common stock. The fair value of the warrant liability on the date of exercise was $4.8 million. This fair value was calculated using a weighted-average Black-Scholes valuation model and the following assumptions: volatility of 108.0%, dividend rate of 0%, risk-free interest rate of 3.29% and a remaining life of 9.14 years. As a result of the warrants being exercised, the warrant liability was reclassified to equity with $3.8 million being recorded as a change in value of the warrant liability for the year ended December 31, 2009.
On October 2, 2009, WTI completed a cashless exercise of the warrants issued to them as part of the Loan Agreement entered into on November 18, 2008. WTI held a total of 478,261 common stock warrants with an exercise price of $1.15 per share. The cashless exercise of the warrants resulted in the Company issuing 355,493 shares of its common stock. The fair value of the warrant liability on the date of exercise was $494,652. This fair
78
EnteroMedics Inc.
(A development stage company)
Notes to Consolidated Financial Statements (Continued)
value was calculated using a weighted-average Black-Scholes valuation model and the following assumptions: volatility of 108.0%, dividend rate of 0%, risk-free interest rate of 3.22% and a remaining life of 9.13 years. As a result of the warrants being exercised, the warrant liability was reclassified to equity with $1,210 being recorded as a change in value of the warrant liability for the year ended December 31, 2009. WTI also completed a cashless exercise of an additional 149,949 common stock warrants with an exercise price of $3.94 per share. The cashless exercise of the warrants resulted in the Company issuing 17,974 shares of its common stock. These warrants were not included as part of the warrant liability.
As of December 31, 2009, Horizon had outstanding 687,500 common stock warrants with an exercise price of $0.80 per share. The fair value of the warrant liability associated with these warrants is $471,585 as of December 31, 2009. This fair value was calculated using a weighted-average Black-Scholes valuation model and the following assumptions: volatility between 103.9% and 104.8%, dividend rate of 0%, risk-free interest rate of 3.84% and a remaining life between 8.89 and 9.33 years. The Company recorded a decrease of $119,904 in the change in value of the warrant liability for the year ended December 31, 2009 for this portion of the warrant liability.
The debt financing is collateralized by a first security priority lien on all of the Company’s assets, excluding intellectual property. The Company has entered into account control agreements in order to perfect the Lenders first security interest in the Company’s cash and investment accounts. In the event the Company has less than five remaining months of liquidity, the Company is required to grant a temporary lien on its intellectual property. The number of remaining months of liquidity is calculated by dividing cash and cash equivalents as of the end of any particular month by the sum of the Company’s total operating expenses for each of the immediately preceding five months. The debt financing agreement also requires the Company to both maintain a cash and cash equivalents balance that exceeds the outstanding principal balance and secure aggregate net proceeds of at least $20.0 million by January 9, 2010 from new capital transactions, of which $10.0 million was required by June 30, 2009. The financial and capital covenants may change upon the achievement of certain milestones defined in the debt financing agreement.
The Company was in compliance with all covenants related to the notes payable at December 31, 2009, and has not incurred any events of default as described in the terms of the notes payable agreements.
Subsequent to year end on February 8, 2010, the Company entered into an amendment to the Loan Agreement with SVB, the only remaining lender, amending both the terms and covenants (see Note 17).
Scheduled debt principal payments are as follows as of December 31, 2009:
| Years Ending December 31: |
||||
| 2010 |
$ | 3,880,656 | ||
| 2011 |
4,336,279 | |||
| 8,216,935 | ||||
| Less: Original issue discount |
(455,469 | ) | ||
| Notes payable, net |
$ | 7,761,466 | ||
79
EnteroMedics Inc.
(A development stage company)
Notes to Consolidated Financial Statements (Continued)
| (8) | Stock Purchases |
On February 19, 2009, the Company entered into several securities purchase agreements for the sale of 13,110,393 shares of its common stock, together with warrants to purchase an aggregate of 6,555,197 shares of its common stock, in a private placement transaction with several accredited investors (the Private Placement) including certain directors and officers of the Company (see Note 13). The purchase price per share was $1.15, which equaled the consolidated closing bid price of the Company’s common stock as reported by the NASDAQ Global Market on February 19, 2009. The warrants will be exercisable at any time and from time to time beginning on the date that is six months and one day after the closing of the Private Placement and ending four years after the closing of the Private Placement. The warrants have an exercise price of $1.38 per share, which equals 120% of the consolidated closing bid price of the Company’s common stock as reported by the NASDAQ Global Market on February 19, 2009. On February 24, 2009, the Company completed the final closing of the Private Placement receiving gross proceeds of $15.9 million, less a placement agent fee and certain other expenses. In addition, the placement agent received a warrant to purchase 218,242 shares of common stock in the same form as that issued to participants in the Private Placement.
On October 2, 2009, the Company entered into a securities purchase agreement with certain institutional investors for the sale of 6,161,068 shares of its common stock in a registered direct offering (the Offering), at a purchase price of $0.80 per share. On October 7, 2009, the Offering closed and the Company received gross proceeds of $4.9 million before deducting estimated offering expenses. No warrants were issued with the Offering. On January 20, 2010, the Company closed an additional equity financing transaction (see Note 17).
| (9) | Convertible Preferred Stock |
The Company’s Amended and Restated Certificate of Incorporation, currently authorizes 5,000,000 shares of $0.01 par value convertible preferred stock. As of December 31, 2009 and 2008, there were no shares of convertible preferred stock issued or outstanding as all shares of Series A, Series B and Series C convertible preferred stock converted into shares of common stock upon completion of the Company’s IPO utilizing the quotient obtained by dividing the original purchase price per share of $6.5593, $3.9430 and $8.0926 by $4.2379, $3.9430 and $8.0926 per share, respectively.
| (10) | Income Taxes |
The Company has incurred net operating losses (NOLs) since inception. The Company has not reflected any benefit of such net operating loss carryforwards in the accompanying consolidated financial statements.
The income tax expense benefit differed from the amount computed by applying the U.S. federal income tax rate of 34% to income before income taxes as a result of the following:
| 2009 | 2008 | 2007 | |||||||
| Computed ‘expected’ tax benefit |
34.0 | % | 34.0 | % | 34.0 | % | |||
| Other permanent adjustments |
-6.3 | % | -0.8 | % | -2.7 | % | |||
| Research and development credit |
1.2 | % | 2.4 | % | 2.2 | % | |||
| Effect of foreign operations |
0.0 | % | 0.0 | % | 0.0 | % | |||
| Federal valuation allowance |
-28.9 | % | -35.6 | % | -33.5 | % | |||
| 0.0 | % | 0.0 | % | 0.0 | % | ||||
80
EnteroMedics Inc.
(A development stage company)
Notes to Consolidated Financial Statements (Continued)
The tax effect of temporary differences that give rise to significant portions of the deferred tax assets as of December 31 is presented below:
| 2009 | 2008 | |||||||
| Deferred tax assets (liabilities): |
||||||||
| Start-up costs |
$ | 10,729,000 | $ | 7,727,000 | ||||
| Capitalized research and development costs |
16,376,000 | — | ||||||
| Reserves and accruals |
1,436,000 | 1,380,000 | ||||||
| Property and equipment |
499,000 | 59,000 | ||||||
| Research and development credit |
2,089,000 | 2,599,000 | ||||||
| Net operating loss carryforwards |
20,773,000 | 30,138,000 | ||||||
| Total gross deferred tax assets |
51,902,000 | 41,903,000 | ||||||
| Valuation allowance |
(51,902,000 | ) | (41,903,000 | ) | ||||
| Net deferred tax assets |
$ | — | $ | — | ||||
In assessing the realization of deferred tax assets, management considers whether it is more likely than not that some portion or all of the deferred tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during periods in which those temporary differences become deductible. In addition, certain limitations imposed under the Internal Revenue Code could further limit the Company’s realization of these deferred tax assets in the event of a change in ownership of the Company (as described below).
Based on the level of historical taxable losses and projections of future taxable income (losses) over the periods in which the deferred tax assets can be realized, management currently believes that it is more likely than not that the Company will not realize the benefits of these deductible differences. Accordingly, the Company has provided a valuation allowance against the gross deferred tax assets as of December 31, 2009 and 2008.
As of December 31, 2009, the Company has U.S. federal net operating loss carryforwards of approximately $51,704,000. Of the total federal net operating loss, $258,000 would result in tax benefits recorded to additional paid-in capital. The federal net operating loss carryforwards expire in the years 2022 through 2029. However, the taxing authorities do have the ability to adjust our net operating loss calculations upon utilization.
Federal tax laws impose significant restrictions on the utilization of net operating loss carryforwards and research and development credits in the event of a change in ownership of the Company, as defined by the Internal Revenue Code Sections 382 and 383. The Company’s net operating loss carryforwards and research and development credits may be subject to the above limitations.
As of December 31, 2009 and 2008, there were no unrecognized tax benefits. Accordingly, a tabular reconciliation from beginning to ending periods is not provided. The Company will classify any future interest and penalties as a component of income tax expense if incurred. To date, there have been no interest or penalties charged or accrued in relation to unrecognized tax benefits.
The Company is subject to federal and state examinations for the years 2006 forward. There are no tax examinations currently in progress.
81
EnteroMedics Inc.
(A development stage company)
Notes to Consolidated Financial Statements (Continued)
| (11) | Stock Options |
The Company has adopted the EnteroMedics Inc. 2003 Stock Incentive Plan (the Plan) that includes both incentive stock options and nonqualified stock options to be granted to employees, officers, consultants, independent contractors, directors and affiliates of the Company. At December 31, 2009 and 2008, according to the Plan, 6,901,103 and 3,901,103 shares, respectively, have been authorized and reserved. The board of directors establishes the terms and conditions of all stock option grants, subject to the Plan and applicable provisions of the Internal Revenue Code. Incentive stock options must be granted at an exercise price not less than the fair market value of the common stock on the grant date. The options granted to participants owning more than 10% of the Company’s outstanding voting stock must be granted at an exercise price not less than 110% of fair market value of the common stock on the grant date. The options expire on the date determined by the board of directors, but may not extend more than 10 years from the grant date, while incentive stock options granted to participants owning more than 10% of the Company’s outstanding voting stock expire five years from the grant date. The vesting period for employees is generally over four years. The vesting period for nonemployees is determined based on the services being provided.
82
EnteroMedics Inc.
(A development stage company)
Notes to Consolidated Financial Statements (Continued)
Stock option activity is as follows:
| Shares Available For Grant |
Outstanding Options | Aggregate Intrinsic Value | ||||||||||
| Number of Shares |
Weighted-Average Exercise Price |
|||||||||||
| Shares reserved at Plan inception |
42,858 | — | $ | — | ||||||||
| Balance, December 31, 2003 |
42,858 | — | — | |||||||||
| Shares reserved |
439,561 | — | — | |||||||||
| Options granted |
(344,796 | ) | 344,796 | 0.46 | ||||||||
| Options exercised |
— | — | — | |||||||||
| Options cancelled |
3,847 | (3,847 | ) | 0.46 | ||||||||
| Balance, December 31, 2004 |
141,470 | 340,949 | 0.46 | |||||||||
| Shares reserved |
678,891 | — | — | |||||||||
| Options granted |
(504,285 | ) | 504,285 | 0.46 | ||||||||
| Options exercised |
— | (29,561 | ) | 0.46 | ||||||||
| Options cancelled |
43,255 | (43,255 | ) | 0.46 | ||||||||
| Balance, December 31, 2005 |
359,331 | 772,418 | 0.46 | |||||||||
| Shares reserved |
566,697 | — | — | |||||||||
| Options granted |
(679,911 | ) | 679,911 | 1.15 | ||||||||
| Options exercised |
— | (87,022 | ) | 0.46 | ||||||||
| Options cancelled |
103,436 | (103,436 | ) | 0.46 | ||||||||
| Balance, December 31, 2006 |
349,553 | 1,261,871 | 0.83 | |||||||||
| Shares reserved |
2,173,096 | — | — | |||||||||
| Options granted |
(912,805 | ) | 912,805 | 6.50 | ||||||||
| Options exercised |
— | (35,132 | ) | 0.60 | ||||||||
| Options cancelled |
37,618 | (37,618 | ) | 3.88 | ||||||||
| Balance, December 31, 2007 |
1,647,462 | 2,101,926 | 3.24 | |||||||||
| Shares reserved |
— | — | — | |||||||||
| Options granted |
(1,331,308 | ) | 1,331,308 | 7.59 | ||||||||
| Options exercised |
— | (100,973 | ) | 0.64 | ||||||||
| Options cancelled |
535,082 | (535,083 | ) | 6.44 | ||||||||
| Balance, December 31, 2008 |
851,236 | 2,797,178 | 4.80 | $ | 766,256 | |||||||
| Shares reserved |
3,000,000 | — | — | |||||||||
| Options granted |
(4,156,200 | ) | 4,156,200 | 2.45 | ||||||||
| Options exercised |
— | (80,706 | ) | 0.46 | ||||||||
| Options cancelled |
898,499 | (898,499 | ) | 5.20 | ||||||||
| Balance, December 31, 2009 |
593,535 | 5,974,173 | $ | 3.16 | $ | 68,243 | ||||||
83
EnteroMedics Inc.
(A development stage company)
Notes to Consolidated Financial Statements (Continued)
The options outstanding, vested and currently exercisable by exercise price at December 31, 2009:
| Outstanding Options and Expected to Vest |
Options Exercisable and Vested | ||||||||||||||
| Exercise |
Number of Shares Outstanding |
Weighted-Average Remaining Contractual Life (Years) |
Aggregate Intrinsic Value |
Number of Options |
Weighted-Average Exercise Price |
Aggregate Intrinsic Value | |||||||||
| $0.46 |
682,434 | 5.5 | $ | 68,243 | 670,441 | $ | 0.46 | $ | 67,044 | ||||||
| $0.63 |
1,270,000 | 9.9 | — | 317,500 | $ | 0.63 | — | ||||||||
| $1.10 |
517,792 | 9.1 | — | 106,683 | $ | 1.10 | — | ||||||||
| $1.40 |
25,000 | 9.1 | — | 11,979 | $ | 1.40 | — | ||||||||
| $1.77 |
5,250 | 8.9 | — | 2,583 | $ | 1.77 | — | ||||||||
| $1.91 |
252,683 | 6.8 | — | 205,840 | $ | 1.91 | — | ||||||||
| $1.93 |
80,000 | 9.3 | — | 31,664 | $ | 1.93 | — | ||||||||
| $2.30 |
175,000 | 9.4 | — | 34,028 | $ | 2.30 | — | ||||||||
| $3.33 |
6,250 | 9.5 | — | 6,250 | $ | 3.33 | — | ||||||||
| $3.56 |
19,969 | 8.6 | — | 9,636 | $ | 3.56 | — | ||||||||
| $3.70 |
1,262,130 | 9.5 | — | 212,188 | $ | 3.70 | — | ||||||||
| $4.40 |
81,656 | 8.4 | — | 42,698 | $ | 4.40 | — | ||||||||
| $5.06 |
278,000 | 9.7 | — | 20,000 | $ | 5.06 | — | ||||||||
| $5.19 |
407,190 | 7.1 | — | 300,166 | $ | 5.19 | — | ||||||||
| $7.46 |
63,738 | 7.3 | — | 56,229 | $ | 7.46 | — | ||||||||
| $8.00 |
137,500 | 8.2 | — | 98,440 | $ | 8.00 | — | ||||||||
| $8.16 |
19,095 | 7.8 | — | 13,354 | $ | 8.16 | — | ||||||||
| $8.27 |
553,123 | 8.1 | — | 212,819 | $ | 8.27 | — | ||||||||
| $8.46 |
137,363 | 7.4 | — | 98,443 | $ | 8.46 | — | ||||||||
| 5,974,173 | $ | 68,243 | 2,450,941 | $ | 3.17 | $ | 67,044 | ||||||||
Stock-Based Compensation for Nonemployees
Stock-based compensation expenses related to stock options granted to nonemployees is recognized as the stock options are earned. The Company believes that the fair value of the stock options is more reliably measurable than the fair value of the services received. The fair value of the stock options granted is calculated at each reporting date, using the Black-Scholes option-pricing model, until the award vests or there is a substantial disincentive for the nonemployee not to perform the required services. The fair value for the years ended December 31, 2009, 2008 and 2007 was calculated using the following assumptions:
| Years Ended December 31, | ||||||
| 2009 | 2008 | 2007 | ||||
| Risk-free interest rates |
2.68%–3.71% | 2.24%–3.98% | 4.03%–5.03% | |||
| Expected life |
6 years–9.98 years | 10 years | 10 years | |||
| Expected dividends |
0% | 0% | 0% | |||
| Expected volatility |
99.70%–108.10% | 72.88%–75.25% | 60.50%–63.25% | |||
Stock-based compensation expense charged to operations on options granted to nonemployees for the years ended December 31, 2009, 2008 and 2007 was $210,075, $(147,855) and $1,289,349, respectively, and $1,448,593 for the period from December 19, 2002 (inception) to December 31, 2009.
84
EnteroMedics Inc.
(A development stage company)
Notes to Consolidated Financial Statements (Continued)
Employee Stock-Based Awards Granted on or Subsequent to January 1, 2006
On January 1, 2006, the Company adopted the fair value method of accounting for the issuance of stock-based payments, using the prospective transition method. Under this transition method, beginning January 1, 2006, compensation cost recognized includes: (a) compensation cost for all stock-based awards granted prior to, but not yet vested as of December 31, 2005, based on the intrinsic value method, and (b) compensation cost for all stock-based payments granted or modified subsequent to December 31, 2005, based on the estimated grant-date fair value.
Compensation cost for employee stock-based awards is based on the estimated grant-date fair value and is recognized over the vesting period of the applicable award on a straight-line basis. The weighted average estimated fair value of the employee stock options granted for the years ended December 31, 2009, 2008 and 2007 was $1.94, $4.62 and $4.51 per share, respectively.
The Company uses the Black-Scholes pricing model to determine the fair value of stock options. The determination of the fair value of stock-based payment awards on the date of grant is affected by the Company’s stock price as well as assumptions regarding a number of complex and subjective variables. These variables include the Company’s expected stock price volatility over the term of the awards, actual and projected employee stock option exercise behaviors, risk-free interest rates and expected dividends. The estimated grant-date fair values of the employee stock options were calculated using the Black-Scholes valuation model, based on the following assumptions for the years ended December 31, 2009, 2008 and 2007:
| Years Ended December 31, | ||||||
| 2009 | 2008 | 2007 | ||||
| Risk-free interest rates |
1.90%–2.99% | 2.57%–4.01% | 4.15%–4.79% | |||
| Expected life |
5.25 years–6.25 years | 5 years–6.25 years | 6 years–6.5 years | |||
| Expected dividends |
0% | 0% | 0% | |||
| Expected volatility |
88.10%–103.20% | 67.63%–74.50% | 55.13%–58.63% | |||
Expected Life. The expected life is based on the “simplified” method described in the SEC Staff Accounting Bulletin, Topic 14: Share-Based Payment.
Volatility. Since the Company was a private entity for most of 2007 and a limited amount of historical data regarding the volatility of its common stock is available, the expected volatility used for 2009, 2008 and 2007 is based on both the volatility of similar entities, referred to as “guideline” companies, and the Company’s historical volatility. In evaluating similarity, the Company considered factors such as industry, stage of life cycle and size.
Risk-Free Interest Rate. The risk-free rate is based on the daily yield curve rate from the U.S. Treasury with remaining terms similar to the expected term on the options.
Dividend Yield. The Company has never declared or paid any cash dividends and does not plan to pay cash dividends in the foreseeable future, and, therefore, used an expected dividend yield of zero in the valuation model.
Forfeitures. The Company is required to estimate forfeitures at the time of grant, and revise those estimates in subsequent periods if actual forfeitures differ from those estimates. The Company uses historical data to estimate pre-vesting option forfeitures and record stock-based compensation expense only for those
85
EnteroMedics Inc.
(A development stage company)
Notes to Consolidated Financial Statements (Continued)
awards that are expected to vest. All stock-based payment awards are amortized on a straight-line basis over the requisite service periods of the awards, which are generally the vesting periods. If the Company’s actual forfeiture rate is materially different from its estimate, the stock-based compensation expense could be significantly different from what the Company has recorded in the current period.
As of December 31, 2009 there was $6,965,951 of total unrecognized compensation costs related to non-vested stock option awards granted after January 1, 2006, which are expected to be recognized over a weighted-average period of 2.69 years.
The aggregate intrinsic value of stock options (the amount by which the market price of the stock on the date of exercise exceeded the exercise price of the option) exercised during the years ended December 31, 2009, 2008 and 2007, was $92,929, $284,437 and $184,970, respectively.
86
EnteroMedics Inc.
(A development stage company)
Notes to Consolidated Financial Statements (Continued)
| (12) | Warrants |
Stock warrant activity is as follows:
| Common Shares |
Price(1) | Series A Preferred Shares |
Price(1) | Series B Preferred Shares |
Price(1) | Series C Preferred Shares |
Price(1) | |||||||||||||||||
| Balance as of: |
||||||||||||||||||||||||
| December 31, 2002 |
— | — | — | — | ||||||||||||||||||||
| Granted |
— | 125,778 | $ | 0.91 | 23,516 | $ | 3.9430 | — | ||||||||||||||||
| Exercised |
— | — | — | — | ||||||||||||||||||||
| Cancelled |
— | — | — | — | ||||||||||||||||||||
| December 31, 2003 |
— | 125,778 | $ | 0.91 | 23,516 | $ | 3.9430 | — | ||||||||||||||||
| Granted |
— | — | 101,205 | $ | 3.9430 | — | ||||||||||||||||||
| Exercised |
— | (125,778 | ) | $ | 1.46 | — | — | |||||||||||||||||
| Cancelled |
— | — | — | — | ||||||||||||||||||||
| December 31, 2004 |
— | — | 124,721 | $ | 3.9430 | — | ||||||||||||||||||
| Granted |
170,336 | $ | 0.46 | — | 69,744 | $ | 3.9430 | — | ||||||||||||||||
| Exercised |
— | — | — | — | ||||||||||||||||||||
| Cancelled |
— | — | — | — | ||||||||||||||||||||
| December 31, 2005 |
170,336 | $ | 0.46 | — | 194,465 | $ | 3.9430 | — | ||||||||||||||||
| Granted |
— | — | 34,872 | $ | 3.9430 | 147,635 | $ | 8.0926 | ||||||||||||||||
| Exercised |
— | — | — | — | ||||||||||||||||||||
| Cancelled |
— | — | — | — | ||||||||||||||||||||
| December 31, 2006 |
170,336 | $ | 0.46 | — | 229,337 | $ | 3.9430 | 147,635 | $ | 8.0926 | ||||||||||||||
| Granted(2) |
— | — | — | 135,927 | $ | 8.0926 | ||||||||||||||||||
| Exercised |
— | — | — | — | ||||||||||||||||||||
| Cancelled |
— | — | — | — | ||||||||||||||||||||
| Converted upon close of IPO |
512,899 | $ | 6.24 | — | (229,337 | ) | $ | 3.9430 | (283,562 | ) | $ | 8.0926 | ||||||||||||
| December 31, 2007 |
683,235 | $ | 4.80 | — | — | — | ||||||||||||||||||
| Granted(2) |
1,075,981 | $ | 1.58 | — | — | — | ||||||||||||||||||
| Exercised |
— | — | — | — | ||||||||||||||||||||
| Cancelled |
— | — | — | — | ||||||||||||||||||||
| December 31, 2008 |
1,759,216 | $ | 2.83 | — | — | — | ||||||||||||||||||
| Granted(2)(3) |
9,240,311 | $ | 1.31 | — | — | — | ||||||||||||||||||
| Exercised(3) |
(1,584,732 | ) | $ | 1.41 | — | — | — | |||||||||||||||||
| Cancelled(3) |
(1,420,570 | ) | $ | 1.59 | — | — | — | |||||||||||||||||
| December 31, 2009 |
7,994,225 | $ | 1.57 | — | — | — | ||||||||||||||||||
| (1) | Represents weighted-average exercise price per share. |
| (2) | See Notes 7 and 8 for discussions relating to the issuance of warrants in 2009, 2008 and 2007. |
| (3) | See Note 7 for discussions relating to both the cashless exercises of warrants in 2009 and the cancellation and reissuance of warrants following down round equity financings. |
At December 31, 2009 and 2008, the weighted-average remaining contractual life of outstanding warrants was 3.65 and 7.49 years, respectively. All of the warrants outstanding are currently exercisable.
87
EnteroMedics Inc.
(A development stage company)
Notes to Consolidated Financial Statements (Continued)
The aggregate number of common shares that could be issued if all warrants were exercised and converted to common stock at the option of the holder would be 7,994,225.
| (13) | Related Party Transactions |
The Company shared space with Restore Medical, Inc. (Restore) until Restore was acquired by Medtronic, Inc. in July 2008, a related party who had directors and stockholders that are officers of the Company. The Company reimbursed Restore for various facility expenses, including property taxes, common area maintenance charges, payroll for the use of personnel, and shipping charges. Beginning in 2005 the Company also reimbursed Restore for rent expense related to the sublease agreement discussed in Note 14. Total expenses recorded were approximately $294,000 and $432,000 for the years ended December 31, 2008 and 2007, respectively, and approximately $1,125,000 for the period from December 19, 2002 (inception) to December 31, 2009. The majority of expenses are included in general and administrative costs on the consolidated statements of operations. On December 31, 2009 and 2008 the Company had no outstanding payable balance to Restore.
Prior to 2009, the Company obtained consulting services from Venturi Development, Inc. (VDI), whose stockholders and officers are investors in the Company. The consultants received cash compensation for services provided. Total expenses recorded, including consulting expenses, were approximately $1,000 and $29,000 for the years ended December 31, 2008 and 2007, respectively, and approximately $2,680,000 for the period from December 19, 2002 (inception) to December 31, 2009. On December 31, 2009 and 2008 the Company had no outstanding payable balance to VDI.
Private Placement
As discussed in Note 8, on February 19, 2009, the Company entered into several securities purchase agreements for the sale of 13,110,393 shares of its common stock, together with warrants to purchase an aggregate of 6,555,197 shares of its common stock, in a private placement transaction with several accredited investors (the Private Placement). The following officers, directors and principal stockholders, each purchased shares of common stock and warrants in the Private Placement at a price of $1.15 per share and $0.125 per share, respectively. The shares purchased, together with the proceeds, before expenses to the Company, are shown in the table below:
| Beneficial Owner |
Shares Purchased |
Warrants Purchased |
Net Proceeds, before expenses, to the Company | ||||
| MPM Capital |
1,765,499 | 882,750 | $ | 2,140,669 | |||
| Bay City Capital |
1,649,485 | 824,742 | $ | 2,000,000 | |||
| Aberdare Ventures |
1,237,113 | 618,557 | $ | 1,500,000 | |||
| InterWest Partners |
678,402 | 339,201 | $ | 822,563 | |||
| Bobby I. Griffin |
206,186 | 103,093 | $ | 250,000 | |||
| Mark B. Knudson, Ph.D. |
65,979 | 32,990 | $ | 80,000 | |||
| Greg S. Lea |
16,495 | 8,247 | $ | 20,000 | |||
Luke Evnin, Ph.D. is a director of the Company and is a member of MPM BioVentures III LLC and a manager of MPM Asset Management Investors 2002 BVIII LLC. Carl Goldfischer, M.D. is a director of the Company and is a managing director of Bay City Capital LLC. Paul H. Klingenstein is a director of the Company and is a managing partner of the Aberdare Funds. Ellen Koskinas served as a director of the Company until her resignation, effective April 3, 2009, and was a venture member of InterWest Management Partners IX, LLC.
88
EnteroMedics Inc.
(A development stage company)
Notes to Consolidated Financial Statements (Continued)
Bobby I. Griffin is a director of the Company. Mark B. Knudson, Ph.D. is the Company’s President, Chief Executive Officer and Chairman of the Board. Greg S. Lea is the Company’s Senior Vice President and Chief Financial Officer.
Consulting Agreement—Nicholas L. Teti, Jr.
On May 28, 2009 the Company entered into a one-year consulting agreement effective June 1, 2009 with Nicholas L. Teti, Jr., who is a member of the board of directors. Pursuant to the agreement, in exchange for consulting services provided, Mr. Teti was entitled to receive a consulting fee of $275,000 per year and the reimbursement of reasonable expenses. Mr. Teti also received an option to purchase 150,000 shares of common stock at $2.30 per share that vests in 36 equal monthly installments following the date of grant. The full grant date fair value of the option grant was approximately $314,000. Total stock-based compensation expense recorded was approximately $41,000 for the year ended December 31, 2009. In addition to the option grant, the Company paid Mr. Teti approximately $268,000 in fees and expenses for consulting services rendered during the year ended December 31, 2009.
On February 10, 2010, the Company entered into a new agreement with Mr. Teti, which was effective as of February 1, 2010. In connection with entering into the new agreement, Mr. Teti and the Company agreed to terminate Mr. Teti’s prior consulting agreement. However, the options that Mr. Teti received in connection with the prior agreement will continue to vest in accordance with their terms. Pursuant to the new agreement, in exchange for consulting services provided, Mr. Teti is entitled to receive a consulting fee of $15,416.67 per month and one-third of Mr. Teti’s administrative assistant expenses. Mr. Teti also received an option to purchase 75,000 shares of common stock at $0.54 per share that vests such that one-third of the options vested immediately with the remainder vesting in 36 equal monthly installments following the date of grant.
Consulting Agreement—Bobby I. Griffin
Effective September 21, 2006, the Company entered into a consulting agreement with Bobby I. Griffin, who is a member of the board of directors. The consulting agreement provided for the consultant to receive compensation in the form of an option to purchase common stock for services provided. Pursuant to this consulting agreement, Mr. Griffin received a one-time option grant to purchase 54,946 shares of common stock at $1.91 per share that vested 25% on the first anniversary of the date the consulting agreement was entered into and 1/36th per month each month thereafter for 36 months. The consulting agreement terminated after one year and does not provide for the forfeiture of any vested or unvested options if after one year Mr. Griffin stops performing services as a consultant. The Company recorded the options at their fair value on the measurement date. The Company remeasured the fair value of the options granted at each reporting period until performance under the consulting agreement was completed and the measurement date was reached. The Company expensed the fair value of the options granted over the requisite service period which was the term of the consulting agreement, or one year. Total expense recorded was approximately $778,000 for the year ended December 31, 2007, and approximately $783,000 for the period from December 19, 2002 (inception) to December 31, 2009. All of the expenses were included in general and administrative costs on the consolidated statements of operations.
| (14) | Commitments and Contingencies |
In September 2005, the Company entered into a three-year non-cancelable operating sublease agreement for office/warehouse space with Restore that expired on September 30, 2008. Effective October 1, 2008 the Company entered into a seven-year non-cancelable operating lease agreement for office/warehouse space. The
89
EnteroMedics Inc.
(A development stage company)
Notes to Consolidated Financial Statements (Continued)
lease expires on September 30, 2015 with monthly base rent ranging from $19,570 to $24,643. Total rent expense recognized for the years ended December 31, 2009, 2008 and 2007 was $270,872, $157,910 and $120,256, respectively, and $811,312 for the period from December 19, 2002 (inception) to December 31, 2009. Facility related expenses are included as general and administrative costs on the consolidated statements of operations.
The following is a schedule of total future minimum lease payments due as of December 31, 2009:
| Years ending December 31: |
|||
| 2010 |
$ | 247,951 | |
| 2011 |
274,564 | ||
| 2012 |
280,055 | ||
| 2013 |
285,656 | ||
| 2014 |
291,369 | ||
| 2015 |
221,789 | ||
| $ | 1,601,384 | ||
The Company is exposed to product liability claims that are inherent in the testing, production, marketing and sale of medical devices. Management believes any losses that may occur from these matters are adequately covered by insurance, and the ultimate outcome of these matters will not have a material effect on the Company’s financial position or results of operations. The Company is not currently a party to any litigation and is not aware of any pending or threatened litigation that could have a material adverse effect on the Company’s business, operating results or financial condition.
In 2005, EnteroMedics entered into an exclusive collaborative obesity device research and development agreement with the Mayo Foundation for Medical Education and Research (Mayo Foundation), Rochester, Minnesota. Through this agreement, EnteroMedics will collaborate with a group of physicians and researchers at Mayo Clinic in the field of obesity. Under the terms of this five-year agreement, EnteroMedics and this group of Mayo specialists will collectively work toward the development of new and innovative medical devices for the treatment of obesity. The agreement also includes a similar collaboration for the development of products to address a wide variety of disorders susceptible to treatment by electrically blocking neural impulses on the vagus nerve.
Under this agreement, the Company issued 219,780 shares of common stock to the Mayo Foundation in 2005 and recorded $100,000 as deferred compensation, which is being amortized over the term of the five-year agreement. Unamortized deferred compensation related to the agreement was $1,667 and $21,667 at December 31, 2009 and 2008, respectively. In accordance with the agreement, upon the closing of the IPO in November 2007, the Company was also obligated to issue 206,044 shares of common stock as consideration to the Mayo Foundation and recorded a one-time stock-based compensation expense of $1.7 million. The stock-based compensation expense is recorded on the consolidated statements of operations as research and development expense.
The Company may also be obligated to pay the Mayo Foundation, contingent upon the occurrence of certain future events, earned royalty payments, including a minimum annual royalty as defined by the agreement, for the commercial sale of products developed and patented by the Mayo Foundation, jointly patented by the Company and the Mayo Foundation, or a product where the Mayo Foundation provided know-how as defined by the agreement. If no products are patented, the minimum royalty is not due. The Mayo Foundation receives an annual $250,000 retainer fee which commenced in 2005 and continued through January 2009. The annual retainer fee paid to the Mayo Foundation is recorded on the consolidated statements of operations as research and
90
EnteroMedics Inc.
(A development stage company)
Notes to Consolidated Financial Statements (Continued)
development expense. This agreement with the Mayo Foundation was extended for two additional years effective February 3, 2010 (see Note 17).
In December 2007, EnteroMedics entered into a second research and development agreement with the Mayo Foundation. In accordance with the three year agreement, the Mayo Foundation receives an annual $50,000 retainer fee. The annual retainer fee paid to the Mayo Foundation is recorded on the consolidated statements of operations as research and development expense. The Company may also be obligated to pay the Mayo Foundation, contingent upon the occurrence of certain future events as defined by the agreement, consideration with respect to licensed know-how regarding product development and testing of products and rights to licensed patents, where the Mayo Foundation provided know-how as defined by the agreement.
| (15) | Retirement Plan |
The Company has a 401(k) profit-sharing plan that provides retirement benefits to employees. Eligible employees may contribute a percentage of their annual compensation, subject to Internal Revenue Service limitations. The Company’s matching is at the discretion of the Company’s board of directors. For the years ended December 31, 2009, 2008 and 2007 and for the period from December 19, 2002 (inception) to December 31, 2009, the Company did not provide any matching of employees’ contributions.
| (16) | Quarterly Data (unaudited) |
The following table represents certain unaudited quarterly information for each of the eight quarters in the period ended December 31, 2009. In management’s opinion, this information has been prepared on the same basis as the audited financial statements and includes all the adjustments necessary to fairly state the unaudited quarterly results of operations (in thousands, except per share data).
| First Quarter |
Second Quarter |
Third Quarter |
Fourth Quarter |
|||||||||||||
| 2009: |
||||||||||||||||
| Net loss |
$ | (6,669 | ) | $ | (10,362 | ) | $ | (12,008 | ) | $ | (2,890 | ) | ||||
| Basic and diluted net loss per share |
$ | (0.30 | ) | $ | (0.34 | ) | $ | (0.40 | ) | $ | (0.08 | ) | ||||
| 2008: |
||||||||||||||||
| Net loss |
$ | (8,498 | ) | $ | (11,354 | ) | $ | (10,200 | ) | $ | (7,822 | ) | ||||
| Basic and diluted net loss per share |
$ | (0.51 | ) | $ | (0.68 | ) | $ | (0.61 | ) | $ | (0.46 | ) | ||||
| (17) | Subsequent Events |
On January 14, 2010, the Company entered into a securities purchase agreement with certain institutional investors for the sale of 7,438,299 shares of its common stock in a registered direct offering (the Offering), at a purchase price of $0.65 per share. On January 20, 2010, the Offering closed and the Company received gross proceeds of $4.8 million before deducting estimated offering expenses. No warrants were issued with the Offering.
As a result of the Offering completed on January 20, 2010, the exercise price of 687,500 common stock warrants held by Horizon, was adjusted from $0.80 to $0.65 per share, resulting in the issuance of 158,653 additional common stock warrants to Horizon. See Note 7 for a description of the warrants issued to Horizon under terms of the loan agreement effective as of November 18, 2009.
91
EnteroMedics Inc.
(A development stage company)
Notes to Consolidated Financial Statements (Continued)
On February 8, 2010 the Company and Silicon Valley Bank (SVB) entered into a First Amendment (the Amendment) to the Loan and Security Agreement, effective as of November 18, 2008, by and among the Company and SVB, Venture Lending & Leasing V, Inc. and Compass Horizon Funding Company LLC, as lenders (the Prior Loan Agreement), reducing our annual interest rate from 11.0% to a fixed annual rate of 10.0%, payable monthly. This has the effect of reducing the monthly payment obligation from $383,532 to $380,421 commencing on March 1, 2010 and ending on December 1, 2011. On December 1, 2009, the Company repaid the outstanding principal amount due to Venture Lending & Leasing V, Inc. and Compass Horizon Funding Company pursuant to the Prior Loan Agreement. The purpose of the Amendment is to establish the terms by which the Company will repay the remaining outstanding Term Loan to SVB.
Pursuant to the Amendment, the conditions pursuant to which the Excluded Collateral (as defined in the Prior Loan Agreement) will be deemed to be included as Collateral (as defined in the Prior Loan Agreement) are changed from the failure to have five months of remaining liquidity to the occurrence of an Event of Default (as defined in the Prior Loan Agreement) after the date of the Amendment or the lender’s awareness after such date of an Event of Default that occurred on or before such date with written notice of such event delivered to the Company. In addition, the Amendment revises the financial covenants in the Prior Loan Agreement to delete the covenant relating to five months of remaining liquidity and to change the liquidity ratio covenant to equal a ratio of (i) the sum of the Company’s unrestricted cash and cash equivalents held with SVB and SVB’s affiliates, divided by (ii) the outstanding principal amount of the Term Loan, which is not permitted to be less than 1.50:1.00. Finally, the Amendment adds a new covenant, the breach of which would constitute an Event of Default. The new covenant requires that the Company receive aggregate net proceeds of at least $4.0 million from new capital transactions after January 1, 2010 and before March 31, 2010 and to keep the proceeds of such transactions at SVB until used. The Company satisfied this new covenant with the closing, on January 20, 2010, of its sale of 7,438,299 shares of its common stock to certain institutional investors in a registered direct offering for gross proceeds of approximately $4.8 million, before deducting estimated offering expenses and placement agent fees.
On March 11, 2010, the Company entered into Amendment No. 1 (the Amendment) to the License Agreement by and between Mayo Foundation for Medical Education and Research (the Mayo Foundation), Rochester, Minnesota, and the Company, effective as of February 3, 2005 (as amended, the License Agreement). The Amendment is effective as of February 3, 2010 (the Effective Date). The Amendment extends the Company’s collaboration with the Mayo Foundation pursuant to the License Agreement for a period of two years from the Effective Date. Pursuant to the Amendment, the Mayo Foundation granted the Company certain royalty-bearing, worldwide exclusive and non-exclusive licenses and committed to the joint collaboration between the Company and a designated group of physicians and researchers at the Mayo Clinic for the development and testing of products for the treatment of obesity, including devices that use electrical signaling to block the vagal nerve, and for the treatment of other gastrointestinal diseases, solely using devices that use electrical signaling to block the vagal nerve. Pursuant to the Amendment, the Mayo Foundation will receive an annual retainer of $100,000 in 2010 and 2011. The Company may also be obligated to pay the Mayo Foundation, contingent upon the occurrence of certain future events, earned royalty payments, including a minimum annual royalty as defined in the License Agreement, for the commercial sale of products developed and patented by the Mayo Foundation, jointly patented by the Company and the Mayo Foundation, or a product where the Mayo Foundation provided know-how as defined by the License Agreement. If no products are patented, the minimum royalty is not due.
92
| ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
None.
| ITEM 9A(T). | CONTROLS AND PROCEDURES |
Evaluation of Disclosure Controls and Procedures
As of the end of the period covered by this report (the Evaluation Date), we carried out an evaluation, under the supervision and with the participation of management, including the Chief Executive Officer and the Chief Financial Officer, of the effectiveness of the design and operation of our disclosure controls and procedures (as defined in Rule 13a-15(e) of the Securities Exchange Act of 1934, as amended (the Exchange Act)). Based upon that evaluation, the Chief Executive Officer and the Chief Financial Officer concluded that, as of the Evaluation Date, our disclosure controls and procedures were effective to ensure that information required to be disclosed in the reports that we file or submit under the Exchange Act is (i) recorded, processed, summarized and reported within the time periods specified in applicable rules and forms, and (ii) accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, to allow timely decisions regarding required disclosure.
Changes in Internal Control Over Financial Reporting
There was no change in our internal control over financial reporting (as defined in Rule 13a-15(f) of the Exchange Act) that occurred during our fourth quarter that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Management’s Report on Internal Control Over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting for the Company as defined in Rules 13a-15(c) and 15d-15(c) of the Securities Exchange Act of 1934. Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the United States of America.
The Company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the Company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the Company are being made only in accordance with authorizations of management and directors of the Company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the Company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Therefore, even those systems of internal control determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation.
Management has evaluated the design and operating effectiveness of our internal control over financial reporting as of December 31, 2009 in accordance with Section 404, Management Assessment of Internal Controls, of the Sarbanes-Oxley Act of 2002 utilizing the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control—Integrated Framework. Based upon the evaluation, management has concluded that our internal control over financial reporting was effective as of December 31, 2009.
93
This annual report does not include an attestation report of the Company’s registered public accounting firm regarding internal control over financial reporting. Management’s report was not subject to attestation by the Company’s registered public accounting firm pursuant to temporary rules of the Securities and Exchange Commission that permit the Company to provide only management’s report in this annual report.
| ITEM 9B. | OTHER INFORMATION |
None.
94
PART III.
Certain information required by Part III is omitted from this report, and is incorporated by reference to our Definitive Proxy Statement to be filed with the Securities and Exchange Commission pursuant to Regulation 14A (the Proxy Statement) in connection with our 2010 Annual Meeting of Stockholders.
| ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE |
The information required by this Item concerning our directors and executive officers is hereby incorporated by reference to the sections of our Proxy Statement under the headings “Nominees,” “Executive Officers,” “Section 16(a) Beneficial Ownership Reporting Compliance” and “Board Meetings and Committees—Audit Committee.”
We have adopted a code of business conduct and ethics, which applies to all directors and employees, including executive officers, including, without limitation, our principal executive officer, principal financial officer, principal accounting officer and persons performing similar functions. A copy of this code of business conduct and ethics is available on our website at www.enteromedics.com (under “Investors”, “Corporate Governance”) and we intend to satisfy the disclosure requirement under Item 5.05 of Form 8-K regarding any waivers from or amendments to any provision of the code of business conduct and ethics by disclosing such information on the same website.
In addition, we intend to promptly disclose (1) the nature of any amendment to our code of business conduct and ethics that applies to our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions and (2) the nature of any waiver, including an implicit waiver, from a provision of our code of business conduct and ethics that is granted to one of these specified officers, the name of such person who is granted the waiver and the date of the waiver on our website in the future.
| ITEM 11. | EXECUTIVE COMPENSATION |
The information required by this Item is hereby incorporated by reference to the sections of our Proxy Statement entitled “Executive Compensation,” “Compensation Committee Interlocks and Insider Participation” and “Compensation Committee Report.”
| ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
(a) Equity Compensation Plans
The following table sets forth information as of December 31, 2009, with respect to our equity compensation plans:
| Plan Category |
Number of Securities to be Issued Upon Exercise of Outstanding Options, Warrants and Rights |
Weighted- Average Exercise Price of Outstanding Options, Warrants and Rights |
Number of Securities Remaining Available for Future Issuance Under Equity Compensation Plans (Excluding Securities Reflected in Second Column) |
||||||
| Equity compensation plans approved by security holders |
13,968,398 | (1) | $ | 2.25 | 593,535 | (2) | |||
| Equity compensation plans not approved by security holders |
— | — | — | ||||||
| Total |
13,968,398 | $ | 2.25 | 593,535 | |||||
| (1) | Consists of options awarded under the 2003 Stock Incentive Plan and outstanding warrants to purchase common stock. |
| (2) | Represents the maximum number of shares of common stock available to be awarded as of December 31, 2009. |
95
(b) Security Ownership
The information required by this Item is hereby incorporated by reference to the section of our Proxy Statement entitled “Security Ownership of Certain Beneficial Owners and Management.”
| ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
The information required by this Item is hereby incorporated by reference to the section of our Proxy Statement entitled “Certain Relationships and Related Transactions, and Director Independence.”
| ITEM 14. | PRINCIPAL ACCOUNTANT FEES AND SERVICES |
The information required by this Item is hereby incorporated by reference to the section of our Proxy Statement entitled “Principal Accountant Fees and Services” and “Administration of Engagement of Independent Auditor.”
96
PART IV.
| ITEM 15. | EXHIBITS, FINANCIAL STATEMENTS AND FINANCIAL STATEMENT SCHEDULES |
(a) Financial Statements and Schedules: Consolidated Financial Statements for the three years ended December 31, 2009 are included in Part II, Item 8. All schedules are omitted because they are not applicable or the required information is shown in the consolidated financial statements or notes thereto.
(b) Exhibits: The list of exhibits on the Exhibit Index on page 99 of this report is incorporated herein by reference.
97
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| ENTEROMEDICS INC. | ||
| By: |
/S/ MARK B. KNUDSON, PH.D. | |
| Mark B. Knudson, Ph.D. | ||
| President and Chief Executive Officer | ||
Dated: March 29, 2010
POWERS OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Mark B. Knudson and Greg S. Lea, and each of them, as his true and lawful attorney-in-fact and agent, with full power of substitution and resubstitution, for him and in his name, place and stead, in any and all capacities, to sign any and all amendments to this report, and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, or any of them or their substitutes may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
| Signature |
Title |
Date | ||
| /S/ MARK B. KNUDSON, PH.D. Mark B. Knudson, Ph.D. |
President, Chief Executive Officer, Chairman and Director (principal executive officer) | March 29, 2010 | ||
| /S/ GREG S. LEA Greg S. Lea |
Senior Vice President and Chief Financial Officer (principal financial and accounting officer) |
March 29, 2010 | ||
| /S/ LUKE EVNIN, PH.D. Luke Evnin, Ph.D. |
Director | March 29, 2010 | ||
| /S/ CATHERINE FRIEDMAN Catherine Friedman |
Director | March 29, 2010 | ||
| /S/ CARL GOLDFISCHER, M.D. Carl Goldfischer, M.D. |
Director | March 29, 2010 | ||
| /S/ BOBBY I. GRIFFIN Bobby I. Griffin |
Director | March 29, 2010 | ||
| /S/ DONALD C. HARRISON M.D. Donald C. Harrison M.D. |
Director | March 29, 2010 | ||
| /S/ PAUL H. KLINGENSTEIN Paul H. Klingenstein |
Director | March 29, 2010 | ||
| /S/ NICHOLAS L. TETI, JR. Nicholas L. Teti, Jr. |
Director | March 29, 2010 | ||
| /S/ JON T. TREMMEL Jon T. Tremmel |
Director | March 29, 2010 | ||
98
EXHIBIT INDEX
| Exhibit |
Description of Document | |
| 3.1 | Fifth Amended and Restated Certificate of Incorporation of the Company. (Incorporated herein by reference to Exhibit 3.2 to Amendment No. 6 to the Company’s Registration Statement on Form S-1 filed on November 9, 2007 (File No. 333-143265)). | |
| 3.2 | Certificate of Amendment to the Fifth Amended and Restated Certificate of Incorporation of the Company. (Incorporated herein by reference to Exhibit 3.2 to the Company’s Quarterly Report on Form 10-Q filed on August 7, 2009 (File No. 1-33818)). | |
| 3.3 | Amended and Restated Bylaws of the Company, as currently in effect. (Incorporated herein by reference to Exhibit 3.4 to Amendment No. 1 to the Company’s Registration Statement on Form S-1 filed on July 6, 2007 (File No. 333-143265)). | |
| 4.1 | Amended and Restated Investors’ Rights Agreement, dated as of July 6, 2006, by and between the Company and the parties named therein. (Incorporated herein by reference to Exhibit 4.2 to the Company’s Registration Statement on Form S-1 filed on May 25, 2007 (File No. 333-143265)). | |
| 10.1 | Licensing Agreement, by and between Mayo Foundation for Medical Education and Research and the Company, dated February 3, 2005. (Incorporated herein by reference to Exhibit 10.1 to Amendment No. 2 to the Company’s Registration Statement on Form S-1 filed on August 14, 2007 (File No. 333-143265)). | |
| 10.2 | Supply Agreement, by and between Atrotech OY and the Company, dated September 11, 2006. (Incorporated herein by reference to Exhibit 10.2 to the Company’s Registration Statement on Form S-1 filed on May 25, 2007 (File No. 333-143265)). | |
| 10.3 | Loan and Security Agreement, dated December 1, 2004, between the Company and Venture Lending and Leasing IV, Inc. (Incorporated herein by reference to Exhibit 10.3 to the Company’s Registration Statement on Form S-1 filed on May 25, 2007 (File No. 333-143265)). | |
| 10.4 | Supplement to the Loan and Security Agreement, dated December 1, 2004, between the Company and Venture Lending and Leasing IV, Inc. (Incorporated herein by reference to Exhibit 10.4 to the Company’s Registration Statement on Form S-1 filed on May 25, 2007 (File No. 333-143265)). | |
| 10.5 | Amendment No. 1, dated as of September 29, 2005, to Supplement to the Loan and Security Agreement, dated December 1, 2004, between the Company and Venture Lending and Leasing IV, Inc. (Incorporated herein by reference to Exhibit 10.5 to the Company’s Registration Statement on Form S-1 filed on May 25, 2007 (File No. 333-143265)). | |
| 10.6 | Loan and Security Agreement, dated as of May 17, 2007, between the Company, Venture Lending and Leasing IV, Inc. and Venture Lending and Leasing V, Inc. (Incorporated herein by reference to Exhibit 10.6 to the Company’s Registration Statement on Form S-1 filed on May 25, 2007 (File No. 333-143265)). | |
| 10.7 | Supplement to the Loan and Security Agreement, dated as of May 17, 2007, between the Company, Venture Lending and Leasing IV, Inc. and Venture Lending and Leasing V, Inc. (Incorporated herein by reference to Exhibit 10.7 to the Company’s Registration Statement on Form S-1 filed on May 25, 2007 (File No. 333-143265)). | |
| 10.7A | Amendment No. 1 to Supplement to Loan and Security Agreement dated August 28, 2007 between the Company, Venture Lending and Leasing IV, Inc. and Venture Lending and Leasing V, Inc. (Incorporated herein by reference to Exhibit 10.7A to Amendment No. 3 to the Company’s Registration Statement on Form S-1 filed on September 11, 2007 (File No. 333-143265)). | |
| 10.8† | Executive Employment Agreement, dated June 22, 2005, by and between the Company and Mark B. Knudson. (Incorporated herein by reference to Exhibit 10.8 to the Company’s Registration Statement on Form S-1 filed on May 25, 2007 (File No. 333-143265)). | |
99
| Exhibit |
Description of Document | |
| 10.9† | Amended and Restated Executive Employment Agreement, dated May 4, 2009, by and between the Company and Mark B. Knudson. (Incorporated herein by reference to Exhibit 10.3 to the Company’s Quarterly Report on Form 10-Q filed on May 7, 2009 (File No. 1-33818)). | |
| 10.10† | Executive Employment, dated May 21, 2007, by and between the Company and Greg Lea. (Incorporated herein by reference to Exhibit 10.9 to the Company’s Registration Statement on Form S-1 filed on May 25, 2007 (File No. 333-143265)). | |
| 10.11† | Executive Employment Agreement, dated February 9, 2007, by and between the Company and Adrianus Donders. (Incorporated herein by reference to Exhibit 10.10 to the Company’s Registration Statement on Form S-1 filed on May 25, 2007 (File No. 333-143265)). | |
| 10.12† | Executive Employment Agreement, dated August 5, 2008, by and between the Company and Katherine S. Tweden. (Incorporated herein by reference to Exhibit 10.4 to the Company’s Quarterly Report on Form 10-Q filed on May 7, 2009 (File No. 1-33818)). | |
| 10.13† | 2003 Stock Incentive Plan, as amended. (Incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on May 11, 2009 (File No. 1-33818)). | |
| 10.14† | Standard form of Incentive Stock Option Agreement pursuant to the 2003 Stock Incentive Plan. (Incorporated herein by reference to Exhibit 10.13 to the Company’s Registration Statement on Form S-1 filed on May 25, 2007 (File No. 333-143265)). | |
| 10.15† | Standard form of Non-Incentive Stock Option Agreement pursuant to the 2003 Stock Incentive Plan. (Incorporated herein by reference to Exhibit 10.14 to the Company’s Registration Statement on Form S-1 filed on May 25, 2007 (File No. 333-143265)). | |
| 10.16† | Standard form of Restricted Stock Agreement. (Incorporated herein by reference to Exhibit 10.15 to the Company’s Registration Statement on Form S-1 filed on May 25, 2007 (File No. 333-143265)). | |
| 10.17† | Management Incentive Plan, (Incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on February 12, 2008 (File No. 1-33818)). | |
| 10.18 | Form of Indemnification Agreement entered into by and between the Company and each of its executive officers and directors. (Incorporated herein by reference to Exhibit 10.17 to Amendment No. 1 to the Company’s Registration Statement on Form S-1 filed on July 6, 2007 (File No. 333-143265)). | |
| 10.19 | Consulting Agreement, dated September 21, 2006, by and between the Company and Bobby I. Griffin. (Incorporated herein by reference to Exhibit 10.19 to Amendment No. 1 to the Company’s Registration Statement on Form S-1 filed on July 6, 2007 (File No. 333-143265)). | |
| 10.20 | Loan and Security Agreement, dated November 18, 2008, between the Company and Silicon Valley Bank, Compass Horizon Funding Company LLC, and Venture Lending & Leasing V, Inc. (Incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on November 24, 2008 (File No. 1-33818)). | |
| 10.21 | Form of Warrant to purchase stock under Loan and Security Agreement, dated November 18, 2008, between the Company and Silicon Valley Bank, Compass Horizon Funding Company LLC, and Venture Lending & Leasing V, Inc. (Incorporated herein by reference to Exhibit 10.20 to the Company’s Annual Report on Form 10-K filed on March 12, 2009 (File No. 1-33818)). | |
| 10.22 | Form of Securities Purchase Agreement, dated February 19, 2009, by and between the Company and several accredited investors. (Incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on February 25, 2009 (File No. 1-33818)). | |
100
| Exhibit |
Description of Document | |
| 10.23 | Form of Warrant, dated February 24, 2009, by and between the Company and several accredited investors. (Incorporated herein by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K filed on February 25, 2009 (File No. 1-33818)). | |
| 10.24 | Lease Agreement, effective October 1, 2008, by and between the Company and Roseville Properties Management Company. (Incorporated herein by reference to Exhibit 10.23 to the Company’s Annual Report on Form 10-K filed on March 12, 2009 (File No. 1-33818)). | |
| 10.25 | Consulting Agreement, dated June 1, 2009, by and between the Company and Nicholas L. Teti, Jr. (Incorporated herein by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q filed on August 7, 2009 (File No. 1-33818)). | |
| 10.26* | Consulting Agreement, dated as of February 1, 2010, by and between the Company and Nicholas L. Teti, Jr. | |
| 10.27 | Securities Purchase Agreement, dated as of October 2, 2009. (Incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on October 5, 2009 (File No. 1-33818)). | |
| 10.28 | First Amendment to Loan and Security Agreement, dated as of February 8, 2010, by and between Silicon Valley Bank and the Company. (Incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on February 12, 2010 (File No. 1-33818)). | |
| 10.29 | Amendment No. 1, effective as of February 3, 2010, to License Agreement between Mayo Foundation for Medical Education and Research and the Company. (Incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on March 17, 2010 (File No. 1-33818)). | |
| 14.1 | Code of Conduct and Ethics of the Company. (Incorporated herein by reference to Exhibit 14.1 to the Company’s Registration Statement on Form S-1 filed on May 25, 2007 (File No. 333-143265)). | |
| 23.1* | Consent of Deloitte & Touche LLP, Independent Registered Public Accounting Firm. | |
| 24.1* | Power of Attorney (included on signature page to this Form 10-K). | |
| 31.1* | Certification of Chief Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 31.2* | Certification of Chief Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 32.1* | Certification of Chief Executive Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| 32.2* | Certification of Chief Financial Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| * | Filed herewith. |
| † | Indicates management contract or compensation plan or agreement. |
101