UNITED STATES SECURITIES AND
EXCHANGE COMMISSION
Washington, D.C.
20549
Form 10-K
| |
|
|
|
(Mark One)
|
|
|
|
|
|
þ
|
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE
SECURITIES EXCHANGE ACT OF 1934
|
|
|
|
For the fiscal year ended
December 31,
2009
|
|
OR
|
|
o
|
|
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE
SECURITIES EXCHANGE ACT OF 1934
|
|
|
|
For the transition period
from to .
|
Commission file number 1-33895
CPEX Pharmaceuticals,
Inc.
(Exact name of registrant as
specified in its charter)
| |
|
|
|
Delaware
|
|
No. 26-1172076
|
(State or other jurisdiction
of
incorporation or organization)
|
|
(I.R.S. Employer
Identification No.)
|
|
|
|
|
2 Holland Way
Exeter, New Hampshire
(Address of principal
executive offices)
|
|
03833
(Zip
Code)
|
Registrant’s telephone number, including area code:
(603) 658-6100
Securities registered pursuant to Section 12(b) of the
Act:
| |
|
|
|
Title of Each Class
|
|
Name of Exchange on Which Registered
|
|
|
|
Common Stock, $0.01 par value
|
|
The NASDAQ Stock Market LLC
(NASDAQ Capital Market)
|
Securities registered pursuant to Section 12(g) of the
Act:
None
Indicate by check mark if the registrant is a well-known
seasoned issuer, as defined in Rule 405 of the Securities
Act. YES o NO þ
Indicate by check mark if the registrant is not required to file
reports pursuant to Section 13 or Section 15(d) of the
Act. YES o NO þ
Indicate by check mark whether the registrant (1) has filed
all reports required to be filed by Section 13 or 15(d) of
the Securities Exchange Act of 1934 during the preceding
12 months (or for such shorter period that the registrant
was required to file such reports), and (2) has been
subject to such filing requirements for the past
90 days. YES þ NO o
Indicate by check mark whether the registrants have submitted
electronically and posted on their corporate Website, if any,
every Interactive Data File required to be submitted and posted
pursuant to Rule 405 of
Regulation S-T
during the preceding 12 months (or for such shorter period
that the registrants were required to submit and post such
files). YES o NO o
Indicate by check mark if disclosure of delinquent filers
pursuant to Item 405 of
Regulation S-K
is not contained herein, and will not be contained, to the best
of registrant’s knowledge, in definitive proxy or
information statements incorporated by reference in
Part III of this
Form 10-K
or any amendment to this
Form 10-K. o
Indicate by check mark whether the registrant is a large
accelerated filer, an accelerated filer, a non-accelerated
filer, or a smaller reporting company. See the definitions of
“large accelerated filer,” “accelerated
filer” and “smaller reporting company” in Rule
12b-2 of the
Exchange Act. (Check one):
|
|
|
|
| Large
accelerated
filer o
|
Accelerated
filer o
|
Non-accelerated
filer o
|
Smaller reporting
company þ
|
(Do not check if a smaller
reporting company)
Indicate by check mark whether the registrant is a shell company
(as defined in
Rule 12b-2
of the
Act). YES o NO þ
State the aggregate market value of the voting and non-voting
common equity held by non-affiliates computed by reference to
the price at which the common equity was last sold, or the
average bid and asked prices of such common equity, as of the
last business day of the registrant’s most recently
completed second fiscal quarter.
| |
|
|
|
|
|
Title of Class
|
|
Aggregate Market Value
|
|
As of Close of Business on
|
|
|
|
Common Stock, $0.01 par value
|
|
$20,279,080
|
|
June 30, 2009
|
Indicate the number of shares outstanding of each of the
registrant’s classes of common stock, as of the latest
practicable date.
| |
|
|
|
|
|
Title of Class
|
|
Shares Outstanding
|
|
As of Close of Business on
|
|
|
|
Common Stock, $0.01 par value
|
|
2,540,728
|
|
March 24, 2010
|
DOCUMENTS
INCORPORATED BY REFERENCE
Definitive
Proxy Statement for the 2010 Annual Meeting of Stockholders to
be filed with the Securities and Exchange Commission not later
than 120 days after December 31, 2009 —
Incorporated by Reference into Part III of this Annual
Report on
Form 10-K
TABLE OF CONTENTS
Part I
Overview
We are an emerging specialty pharmaceutical company in the
business of research and development of pharmaceutical products
utilizing our validated drug delivery platform technology. We
have U.S. and international patents and other proprietary
rights to technology that facilitates the absorption of drugs.
Our platform drug delivery technology enhances permeation and
absorption of pharmaceutical molecules across the skin, nasal
mucosa and eye through development of proprietary formulations
with molecules such as CPE-215. Our first product is
Testim®,
a gel for testosterone replacement therapy, which is a
formulation of our technology with testosterone. Testim is
licensed to Auxilium Pharmaceuticals, Inc.
(“Auxilium”) which is currently marketing it in the
United States, Europe and other countries. We have an additional
licensed application and are in discussions with other
pharmaceutical and biotechnology companies to form additional
strategic alliances to facilitate the development and
commercialization of other products using our drug delivery
technology.
To date, our research and development programs for our drug
delivery technology have been focused on the development of
Nasulin, an intranasal formulation of insulin being developed to
treat hyperglycemia in patients suffering from Type 1 and Type 2
diabetes. Our Nasulin development program includes 15 completed
clinical trials in approximately 390 subjects. Eight of the
studies were conducted in healthy volunteers, four in patients
with Type 1 diabetes and three in patients with Type 2 diabetes.
Of the 15 studies, 14 were Phase 1 trials and one was a Phase 2
trial. In 2009 we initiated and completed a Phase 1 Nasulin
pharmacokinetic study designed to demonstrate dose
proportionality of a second concentration strength. Also in
2009, we initiated and completed enrollment in a proof of
concept Phase 2a study, designed to assess the glucose control
of Nasulin versus placebo using continuous glucose monitoring in
subjects with Type 2 diabetes. The primary objective of this
study was to demonstrate that subjects receiving Nasulin would
achieve a larger increase from baseline in the mean proportion
of time spent with normal glucose levels (known as euglycemia)
than those receiving placebo, as assessed by continuous glucose
monitoring. In March 2010 the Company reported that preliminary
results of this study showed that although subjects in the
placebo group spent less time in euglycemia at the end of the
study compared to subjects in the Nasulin group, the difference
between the two groups was not statistically significant
(p-value = 0.2). The secondary efficacy measurements of change
from baseline in average glucose as measured by continuous
glucose monitoring and the change from baseline in serum
fructosamine were consistent with the primary endpoint analysis.
We intend to conduct additional analyses on the data from the
Phase 2a study and together with all other Nasulin data will
determine the appropriate next steps for the Nasulin program. In
the interim we have not commenced our planned Phase 2b trial or
other Nasulin development initiatives.
In addition to our Nasulin program, Serenity Pharmaceuticals,
Inc., our licensing and development partner, is recruiting
patients in multiple Phase 3 clinical trials for an undisclosed
urology drug delivered using CPEX’s intranasal technology
for the treatment of nocturia. These randomized, double blind,
placebo controlled studies are being conducted at multiple sites
in the United States.
We expect to incur increased costs from product pipeline
development and testing efforts. Additional studies will be
required should we continue to advance the Nasulin clinical
program. Clinical and preclinical studies are subject to
numerous risks and uncertainties. Many factors can delay or
result in termination of future clinical studies. For example,
patient enrollment delays or unexpected results from clinical
studies could cause us to adjust our current clinical plans. See
“Risk Factors” section, page 20.
We believe, based upon our experience with Testim and Nasulin,
that our technology has the ability to significantly enhance the
permeation of a wide range of therapeutic molecules. To expand
the development and commercialization of products using our
technology, we are pursuing strategic alliances with partners
including large pharmaceutical, specialty pharmaceutical and
biotechnology companies. The alliance opportunities may include
co-development of products, in-licensing of therapeutic
molecules, out-licensing of delivery technology or partnering
late-stage candidates for commercialization.
2
We were incorporated in the State of Delaware in 2007, and our
principal executive offices are located in Exeter, New Hampshire.
Separation
from Bentley Pharmaceuticals, Inc.
Our business was initially the drug delivery business of Bentley
Pharmaceuticals, Inc. We were spun off in June 2008 in
connection with the sale of Bentley’s remaining business.
Shares of our stock were distributed to Bentley stockholders
after the close of business on June 30, 2008 (the
“Separation Date”) by means of a stock dividend, which
was taxable to Bentley and Bentley’s stockholders (the
“Separation”). Each Bentley stockholder of record on
June 20, 2008, the record date, received on the Separation
Date one CPEX share for every ten shares of Bentley common
stock. Bentley has no ownership interest in CPEX subsequent to
the Separation.
Prior to our separation from Bentley, we entered into a
Separation and Distribution Agreement, Tax Sharing Agreement,
Employment Members Agreement and Transition Services Agreement
(together referred to as the “Separation Agreements”),
which have governed our relationship with Bentley since the
Separation Date. The Transition Services Agreement, whereby
Bentley agreed to provide us with certain executive and
administrative services, expired under its terms in December
2008.
Our financial statements for the twelve months ended
December 31, 2009 and the balance sheet as of
December 31, 2008 represent stand-alone financial
information for our Company. The statements of operations,
statements of changes in stockholders’ equity and
statements of cash flows for the year ended December 31,
2008, which includes financial information for the six months
before the Separation Date, and all of 2007 reflect the
historical financial position, results of operations and cash
flows of the business transferred to us from Bentley as part of
the separation and distribution. Our financial statements have
been prepared and are presented as if we had been operating as a
separate entity using the historical cost basis of the assets
and liabilities of Bentley and including the historical
operations of the business transferred to us from Bentley as
part of the separation. Prior to the Separation Date, we were
fully integrated with Bentley and the accompanying financial
statements reflect the application of certain estimates and
allocations. Our statements of operations include all revenues
and costs that are directly attributable to the business of
CPEX. In addition, certain expenses of Bentley have been
allocated to us using various assumptions that, in the opinion
of management, are reasonable. These expenses include an
allocated share of executive compensation, public company costs
and other administrative costs. The allocated costs totaled
$4.4 million and $4.2 million for the years ended
December 31, 2008 and 2007, respectively. These amounts may
differ from the costs associated with operating as an
independent public company. Therefore, the results for the
twelve months ended December 31, 2008 and 2007 are not
indicative of the results that might have occurred if we had
operated as an independent public company during those periods.
There have been no allocations of expenses of Bentley charged to
CPEX since the Separation Date.
Industry
Overview
Specialty
Pharmaceutical Companies Addressing Limitations of Current
Therapies
Our goal is to become a leading specialty pharmaceutical company
by creating a diverse and renewable pipeline of therapeutic
compounds that address a wide variety of unmet medical needs.
Specialty pharmaceutical companies like ours develop products to
address the limitations of current therapies in selected
established markets (endocrinology, urology, dermatology,
neurology, etc.). These specialty markets can usually be
addressed by smaller sales forces, but have the same market
potential. Our pipeline products are designed to enhance safety,
efficacy,
ease-of-use
and patient compliance. Our pipeline products also provide novel
opportunities for pharmaceutical and biotechnology companies to
partner with us to develop new and innovative products and
extend their therapeutic franchises.
According to data obtained from Evaluate Pharma, the global
specialty pharmaceutical market reached an overall value of
approximately $85 billion in 2008 (an increase of 12.7%
from 2007) and is projected to continue to grow for the
foreseeable future.
Developing safer and more efficacious methods of delivering
existing drugs is generally subject to less risk than attempting
to discover new drugs, because of lower development costs. On
average, it takes 10 to
3
15 years for an experimental new drug to progress from the
laboratory to commercialization in the U.S., with an average
cost of approximately $800 million to $900 million.
Typically, only one in 5,000 compounds entering preclinical
testing advances into human testing and only one in five
compounds tested in humans is approved for commercialization. By
contrast, we typically consider drugs for our pipeline that
already have been approved and are commercially available, have
a track record of safety and efficacy and have established
markets for which there is an unmet medical need. In addition,
we may be able to pursue an accelerated 505(b)(2) path to the
market for some of our pipeline products that may translate into
less time spent in the clinic and less money spent on clinical
development. The 505(b)(2) development path in turn translates
into a faster time to market launch and more overall patent life
for the marketed drug.
The vast majority of the drugs currently on the market are
administered orally or by injection. Oral drug delivery methods,
while simple to use, typically subject drugs to degradation
initially by the stomach and secondarily to first-pass
metabolism in the liver before reaching the bloodstream. In
order to achieve efficacy, higher drug dosages are often used,
which can increase the risk of side effects.
Injectable pharmaceuticals, while avoiding first-pass metabolism
in the liver, possess several disadvantages, which can lead to
decreased patient acceptance and compliance with prescribed
therapy. Injectable drugs are more painful for the patient and
often require medical personnel to administer. In addition,
injectable drugs are typically also more expensive due to the
added cost associated with their manufacturing under sterile
conditions and added costs for their administration, including
medical personnel, syringes, needles and other supplies. A
decline in patient acceptance and compliance due to any of these
reasons can delay the initiation of treatment and increase the
risk of medical complications and could lead to higher
healthcare costs.
Alternative
Drug Delivery Technologies
Pharmaceutical and biotechnology companies recognize the
benefits of alternative delivery technologies as a way of
gaining a competitive advantage. It has been reported that while
pharmaceutical sales in the U.S. have risen steadily over
the last several years to more than $720 billion in 2008,
sales growth is expected to slow over the next several years due
to an extraordinary number of patent expirations and to the poor
global economy. At the same time, it is estimated that the
global drug delivery market has tripled in value since 2000 to
approximately $78 billion (Espicom Healthcare Intelligence,
2009). The growth in the drug delivery market has been driven by
the recognition of pharmaceutical and biotechnology companies
that provide alternative delivery technologies that avoid
first-pass liver metabolism, that are less invasive than
injectable options and that enable product line and patent
position extension provide an attractive combination of
advantages to companies and patients. Further, these
pharmaceutical and biotechnology companies often benefit from
specialty pharmaceutical companies that apply their technologies
to off-patent products, formulating their own proprietary
products, which are then typically commercialized by larger
pharmaceutical companies capable of promoting the products.
Permeation
Enhancement Technology — The Path to Improved Drug
Benefits
Permeation enhancement with CPE-215 is the drug delivery
technology of CPEX. It has been proven to enhance the absorption
of drugs through the nasal mucosa, skin and eye. It can be
adapted to products formulated as creams, ointments, gels,
solutions, sprays or patches. CPE-215 also has maintained a
record of safety as a direct and indirect food additive and
fragrance, and is listed on the U.S. Food and Drug
Agency’s inactive ingredient list for approved use in drug
applications.
We believe that potential key benefits of the patented drug
delivery formulation technology of CPEX using CPE-215 may
include the following therapeutic and commercial opportunities
and advantages:
|
|
|
| |
•
|
Improved compliance and convenience to patients requiring
ongoing injection therapies and the potential for earlier
acceptance of prophylactic treatment for patients reluctant to
use injections;
|
| |
| |
•
|
Application to injectable peptides that could be administered
intranasally with CPE-215;
|
| |
| |
•
|
Application to therapeutic molecules that are degraded by
passage through the liver or would benefit from intra-nasal
administration to eliminate first-pass metabolism;
|
4
|
|
|
| |
•
|
Application to a variety of metabolic, neurological and other
serious medical conditions;
|
| |
| |
•
|
Opportunities for life-cycle extension strategies for existing
marketed products;
|
| |
| |
•
|
Opportunities for allowing product differentiation based on
benefits of administration.
|
Licensed
Products
Testim,
Licensed Topical Testosterone Gel
We earn royalty revenues on sales of Testim, a testosterone gel
that incorporates our CPE-215 drug delivery technology. The
product is licensed to Auxilium and was launched in the
U.S. in early 2003 as a testosterone replacement therapy.
Testim has been approved for marketing in Canada and 15
countries in Europe. Royalties received from Testim sales were
$18.6 million, $15.1 million and $11.1 million
for the years ended December 31, 2009, 2008 and 2007,
respectively. Auxilium uses its sales force to market Testim in
the U.S. and has partnered with Paladin Labs Inc. to market
the drug in Canada and with Ferring International S.A. to market
the drug in Europe.
The testosterone replacement market has expanded as more
baby-boomers enter middle age and more attention is focused on
male hormonal deficiency and the benefits of replacement
therapy. Hypogonadism, a condition in men where insufficient
amounts of testosterone are produced, is thought to affect one
out of every five men in the U.S. and Europe aged over 50.
Symptoms associated with low testosterone levels in men include
depression, decreased libido, erectile dysfunction, muscular
atrophy, loss of energy, mood alterations, increased body fat
and reduced bone density. This condition is currently
significantly under-treated. Growing patient awareness together
with education continue to spur demand for testosterone
replacement therapy.
Currently marketed testosterone replacement therapies deliver
hormones through injections, transdermal patches or gels. Gels
provide commercially attractive and efficacious alternatives to
the other current methods of delivery by providing a more steady
state of absorption rather than the bolus surge of injections or
the irritation caused by patches resulting in a less desirable
dosage regimen.
In October 2008, we and Auxilium received notice that
Upsher-Smith Laboratories filed an Abbreviated New Drug
Application, or ANDA, containing a paragraph IV
certification in which it certified that it believes that its
testosterone gel product does not infringe our patent covering
Testim, U.S. Patent No. 7,320,968 (“the ‘968
Patent”). The ‘968 Patent covers a method for
maintaining effective blood serum testosterone levels for
treating a hypogonadal male using Testim and will expire in
January 2025. The ‘968 patent is listed in Approved Drug
Products with Therapeutic Equivalence Evaluations (commonly
known as the Orange Book), published by the U.S. Food and
Drug Administration. The paragraph IV certification sets
forth allegations that the ‘968 Patent will not be
infringed by Upsher-Smith’s manufacture, use or sale of the
product for which the ANDA was submitted. On December 4,
2008, we and Auxilium filed a lawsuit in the United States
District Court for the District of Delaware against Upsher-Smith
under the Hatch Waxman Act for infringement of our patent. In
June 2009, Upsher-Smith amended its answer to the complaint to
include a defense and counterclaim of invalidity of the
‘968 Patent, which we and Auxilium deny. We and Auxilium
filed a reply to the counterclaim in July 2009 denying the
invalidity of the ‘968 Patent. A patent issued by the
Patent and Trademark Office, such as the ‘968 Patent, is
presumed valid. As described more fully under
Item 3 — “Legal Proceedings” in this
report, any U.S. Food and Drug Agency (FDA) approval of
Upsher-Smith’s proposed generic product will be stayed
until the earlier of 30 months beginning on the date of
receipt of the paragraph IV certification (April
2011) or an adverse decision in our patent infringement
lawsuit.
The primary competition for Testim is
AndroGel®,
marketed by Solvay Pharmaceuticals, Inc., and its potential
generic competitors.
|
|
|
| |
•
|
Watson Pharmaceuticals Inc. and Par Pharmaceuticals Inc. have
filed ANDAs against AndroGel but the timing of the entry of
these generics is not expected until 2015.
|
| |
| |
•
|
Perrigo Israel Pharmaceuticals, Ltd. has filed an ANDA against
AndroGel but the timing of the entry of this generic is
uncertain at this time.
|
5
There are several other compounds in various stages of clinical
development which may compete with Testim.
|
|
|
| |
•
|
Endo Pharmaceuticals, Inc. signed an agreement with U.K. based
ProStrakan Group plc for exclusive U.S. rights to
commercialize
FORTESTAtm,
a 2% testosterone gel. Endo filed a New Drug Application
(“NDA”) for
FORTESTAtm
in 2009 and has received a Complete Response Letter from the
FDA. Endo has announced that it expects to respond to the
FDA’s questions in mid-2010.
FORTESTAtm
is approved in Europe and is sold by ProStrakan under the brand
names
Tostrantm,
Tostrextm
and
Itnogentm.
|
| |
| |
•
|
Bayer Schering Pharma discovered and developed
NEBIDOtm
a novel, long-acting injectable testosterone product sold in
Europe. Bayer has licensed U.S. rights for this product to
Endo, which filed an NDA with the FDA in 2009 using the brand
name
AVEEDtm.
Endo has received and is evaluating a Complete Response Letter
from the FDA regarding AVEED. The timing of the entry of
FORTESTAtm
and
AVEEDtm
in the U.S. market is unknown.
|
| |
| |
•
|
Acrux Limited, an Australian company, has developed
AXIRONtm,
a 2% testosterone solution delivered in the armpit. Acrux has
completed a Phase 3 clinical trial and is expected to file an
NDA with the FDA in the near future.
|
| |
| |
•
|
BioSante Pharmaceuticals, Inc. and Teva Pharmaceuticals USA,
Inc. have developed
Bio-T-Geltm,
a once-daily transdermal testosterone gel currently in late
stage human clinical trials.
|
| |
| |
•
|
Clarus Therapeutics is developing an oral form of testosterone
called
OriTextm
which is currently in Phase 2 clinical trials.
|
Other new treatments are being sought for testosterone
replacement therapy; these products are in development and their
future impact on the treatment of testosterone deficiency is
unknown.
Product
in Development
Nasulintm,
Proprietary Intranasal Insulin Product
Nasulin is the patented intranasal insulin spray of CPEX which
incorporates CPE-215 as a permeation enhancer. We are developing
Nasulin to address the need for an insulin product that more
closely resembles the body’s normal physiological response
and provides a more patient-friendly delivery method. These
potential benefits could reshape the insulin market. Based on
market reports and projections, we have estimated the global
meal time injectable insulin market to be approximately
$5.4 billion and the U.S. market to be approximately
$3.1 billion. In general, drugs delivered via the nasal
cavity have the potential to be readily absorbed across the
highly vascularized nasal mucosa and directly into the
circulatory system, thereby avoiding first-pass metabolism in
the liver. When absorbed, the speed of absorption affords a
faster onset of action compared to the most rapid-acting,
injectable insulin formulations. A series of studies have
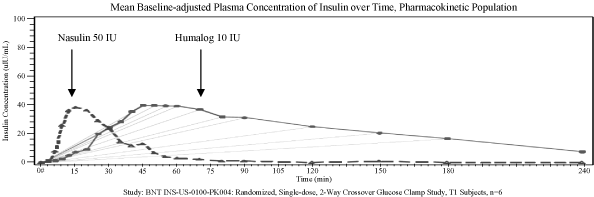
confirmed that Nasulin delivers insulin quickly through the
nasal mucosa, even more rapidly than subcutaneous injection.
6
According to 2007 prevalence data released by the United States
Centers for Disease Control and Prevention, or CDC, the most
recent year for which data is available, approximately
24 million people in the United States, or 8% of the
population, suffer from diabetes. A study published by Diabetes
Care in 2006 projects that the number of diagnosed diabetics in
the U.S. could reach 48.3 million by 2050 due to an
aging population, rising obesity rates and poor health habits.
Prescription trends show a preference for combining rapid-acting
injections during mealtimes with a once daily basal insulin
injection. Due to the time-action profiles of the rapid-acting
mealtime injectable insulins, patients are at increased risk for
hypoglycemic events and for gaining weight. The ultra-rapid
time-action profile of Nasulin has the potential to reduce the
risk of hypoglycemic events and of weight gain. In addition,
because Nasulin delivers needle-free insulin, it has the
potential to improve the acceptability of mealtime insulin among
patients with diabetes and has the potential to improve
adherence to insulin treatment regimens.
We believe an intranasal route of administration will yield an
ultra-rapid time action profile which could provide better
glucose control equivalent to meal time injectable insulins but
with the potential for less hypoglycemia and weight gain. In
addition, based on current data demonstrating that Nasulin does
not enter the deep lung, this needle-free route of delivery
potentially avoids the pulmonary disadvantages of competitive
candidates that use an inhalation route of administration. As
explained above, preliminary findings from our Phase 2a proof of
concept study showed that although subjects in the placebo group
spent less time in euglycemia at the end of the study compared
to subjects in the Nasulin group, the difference between the two
groups was not statistically significant (p-value = 0.2).
Previously our development plan was to finish key efficacy Phase
2 trials in patients with Type 2 diabetes late in 2011 while
simultaneously seeking a pharmaceutical partner to support Phase
3 clinical trials and product commercialization upon regulatory
approval. As a result of the preliminary findings from the Phase
2a study, we intend to conduct additional analyses on the data
together with all other Nasulin data and will determine the
appropriate next steps for the Nasulin program. In the interim
we have not commenced our planned Phase 2b trial or other
Nasulin development initiatives.
Key
Markets and Trends
Testosterone
Substantially all of our revenues are derived through royalty
income from the only commercialized product licensed with our
CPE-215 technology, Testim, which is sold by Auxilium.
Overview
of Testosterone Replacement Market
The testosterone replacement market has expanded as more
baby-boomers enter middle age and more attention is focused on
male hormonal deficiency and the benefits of replacement
therapy. Symptoms associated with low testosterone levels in men
include depression, decreased libido, erectile dysfunction,
muscular atrophy, loss of energy, mood alterations, increased
body fat and reduced bone density. This condition is currently
significantly under-treated and growing patient awareness
together with education continue to spur demand for testosterone
replacement therapy.
Currently marketed testosterone replacement therapies deliver
hormones through injections, transdermal patches or gels. Gels
provide commercially attractive and efficacious alternatives to
the other current methods of delivery by providing a more steady
state of absorption rather than the bolus surge of injections or
the irritation caused by patches, which often results in a less
desirable dosage regimen.
Diabetes
Our most advanced product in development, Nasulin, is an
intranasal formulation of insulin being developed to treat
hyperglycemia in patients suffering from Type 1 and Type 2
diabetes.
7
Overview
of Diabetes
Diabetes is a major disease characterized by the body’s
inability to properly regulate levels of blood glucose, or blood
sugar. The cells of the body use glucose as fuel, which is
consumed 24 hours a day. Between meals, when glucose is not
being supplied from food, the liver releases glucose into the
blood to sustain adequate levels. Insulin is a hormone produced
by the pancreas that regulates the body’s blood glucose
levels. Patients with diabetes develop abnormally high levels of
glucose, a state known as hyperglycemia, either because they
produce insufficient levels of insulin or because they fail to
respond adequately to insulin produced by the body. Over time,
poorly controlled levels of blood glucose can lead to major
complications, including high blood pressure, blindness,
amputations, kidney failure, heart attack, stroke and death.
According to 2007 prevalence data released by the United States
Centers for Disease Control and Prevention, or CDC,
approximately 24 million people in the United States, or 8%
of the population, suffer from diabetes. This represents an
increase of more than 3 million people in approximately two
years. In addition to the 24 million people with diabetes,
approximately 57 million people are estimated to have
pre-diabetes, putting them at increased risk for developing
diabetes. In addition, the incidence of diabetes is also
increasing. A study published by Diabetes Care in 2006 projected
that in 2050 there would be 48.3 million people with
diagnosed diabetes in the United States. Diabetes extracts a
heavy toll from those who suffer from it. The CDC reported that
diabetes was the seventh leading cause of death listed on death
certificates in 2006, but that diabetes was likely to be
underreported as a cause of death. Overall, the CDC found that
the risk of death among people with diabetes is about twice that
of people without diabetes of similar age. The economic costs of
diabetes are high as well. The American Diabetes Association
estimates that in 2007, the total cost of diabetes in the United
States was $174 billion. This amount includes
$116 billion of direct medical costs and $58 billion
for indirect medical costs including disability, work loss and
premature mortality.
There are two major forms of diabetes, Type 1 and Type 2. Type 1
diabetes is an autoimmune disease characterized by a complete
lack of insulin secretion by the pancreas, so insulin must be
supplied from outside the body. In Type 2 diabetes, the pancreas
continues to produce insulin; however, insulin-dependent cells
become resistant to the effects of insulin. Over time, the
pancreas becomes increasingly unable to secrete adequate amounts
of insulin to support metabolism. According to the CDC, Type 2
diabetes is the more prevalent form of the disease, affecting
approximately 90% to 95% of people diagnosed with diabetes.
Challenges
of treating Type 2 Diabetes
Typically, the treatment of Type 2 diabetes starts with
management of diet and exercise and progresses to treatment with
various oral medications and then to treatment with insulin.
Treatment with diet and exercise alone is not an effective
long-term solution for most patients with Type 2 diabetes. Oral
medications — which act predominantly by increasing
the amount of insulin produced by the pancreas, by increasing
the sensitivity of insulin-dependent cells or by reducing the
glucose output of the liver — may have significant
adverse effects and are limited in their ability to manage the
disease effectively.
Insulin therapy usually involves administering several
subcutaneous needle injections of insulin each day. Although
this treatment regimen is accepted as an effective means to
control glucose levels, it has limitations, including:
|
|
|
| |
•
|
the need for injections;
|
| |
| |
•
|
the risk of abnormally low levels of blood glucose, known as
hypoglycemia, that result from excessive insulin administration.
Severe hypoglycemia can result in loss of mental acuity,
confusion, increased heart rate, hunger, sweating and faintness
and, at very low glucose levels, loss of consciousness,
seizures, coma and death;
|
| |
| |
•
|
the need for complex titration of insulin doses in connection
with meals;
|
| |
| |
•
|
inadequate post-meal glucose control;
|
| |
| |
•
|
the need for frequent glucose monitoring with finger
pricks; and
|
8
|
|
|
| |
•
|
the likelihood of weight gain.
|
Particularly because of the dislike of injections and finger
pricks, patients delay the initiation of insulin therapy and may
not adhere adequately to the prescribed treatment regimens once
insulin therapy begins. Moreover, even when properly
administered, current subcutaneous injections of insulin do not
replicate the time-action profile of endogenous insulin. In a
person without diabetes, blood insulin levels rise within
several minutes of the entry into the bloodstream of glucose
from a meal. By contrast, injected insulin enters the
bloodstream more slowly, resulting in peak insulin levels in
about 120 to 180 minutes for regular human insulin or
30-90
minutes for “rapid-acting” insulin analogs. The
bioavailability profile of the currently available rapid-acting
meal-time insulin products can cause patients with diabetes to
have inadequate levels of insulin present at the initiation of a
meal and to be over-insulinized between meals. This lag in
insulin delivery results in hyperglycemia early after meal
onset, followed by a tendency for hypoglycemia to develop during
the period between meals. Physicians who treat patients with
diabetes are concerned about the risks of hypoglycemia and, as a
result, tend to under treat the chronic hyperglycemia that is
associated with the disease. However, the resultant extensive
hyperglycemia significantly contributes to many of the long-term
cardiovascular and other serious complications of diabetes.
There are two components to the hyperglycemia concern. The first
component is related to the duration and magnitude of the
chronic sustained hyperglycemia associated with poorly
controlled diabetes. This component is assessed by measuring
HbA1c levels, which are a measure of the average blood glucose
levels over the preceding three or four months. HbA1c levels are
an indication of overall glucose control, and an important goal
of all diabetes therapies is to lower HbA1c levels. The second
component of hyperglycemia relates to the extent of acute
glucose fluctuations above and below the average level. These
fluctuations occur in response to meals and can be managed by
diabetes medications, including insulin. In a clinical setting,
this component is assessed by determining the mean amplitude of
glucose fluctuations that occur following the ingestion of a
meal.
There is evidence that acute glucose fluctuations may be the
more significant factor contributing to the cardiovascular
complications of diabetes, which are thought to stem from the
activation of a mechanism known as oxidative stress that causes
cellular damage. A recent study of patients with Type 2 diabetes
reported in the Journal of the American Medical Association in
April 2006 found that the urinary levels of a marker for
oxidative stress were significantly correlated with the mean
amplitude of glucose fluctuations during the post-meal period.
This study concluded that acute glucose fluctuations may trigger
oxidative stress, suggesting that doctors and patients should
emphasize the management of acute glucose fluctuations as well
as the goal of lowering HbA1c levels.
The results of the Diabetes Control and Complications Trial
(DCCT), a long-term study conducted by the National Institutes
of Health support the view that controlling acute glucose
fluctuations contributes to a reduced risk for the
cardiovascular complications of diabetes. In the study a group
of patients treated using conventional insulin therapy (one-two
insulin injections per day along with daily urine glucose tests)
was compared to a group treated using intensive insulin therapy
(either an insulin pump or at least three insulin injections and
at least four blood glucose tests per day). In total,
1,441 patients were followed for an average of
6.5 years each. Intensive insulin therapy produced a
significant reduction in HbA1c levels compared to conventional
insulin therapy; the difference between treatment groups
remained evident for the duration of the study. Moreover, the
patients who had been intensively treated also showed
significant decreases in risk for kidney and eye damage compared
to the conventional treatment group. Although when these results
were reported, the DCCT was discontinued, a group of 1,375 of
these subjects (half from each of the original treatment groups)
was subsequently followed in the Epidemiology of Diabetes
Interventions and Complications (EDIC) study. After seven years
in the EDIC study, the HbA1c levels of the former conventional
therapy group did not differ from the HbA1c levels of the former
intensive treatment group. The HbA1c levels of the former
conventional therapy group had improved from the DCCT while
those of the former intensive group had declined. However, the
former conventional therapy group continued to show an elevated
risk of kidney and eye damage compared to the former intensive
therapy group, according to a report published in the Journal of
the American Medical Association in May 2002. These findings led
to the conclusion that intensive insulin therapy, which would be
expected to reduce acute glucose fluctuations, can be beneficial
for patients
9
with diabetes, even years after the therapy has been less
intensified. However, the EDIC also demonstrates how intensive
insulin therapy is difficult for many patients to implement in a
home setting.
Even if intensive insulin therapy is implemented, the available
insulin products are not able to enter the bloodstream fast
enough to replicate the tight coupling between changes in blood
glucose levels and the release of insulin by the pancreas that
is seen in healthy individuals without diabetes. The early
insulin response following glucose ingestion is an important
part of maintaining control over glucose levels during the
post-meal period. It is thought that the early surge of insulin
levels shuts off glucose production by the liver, which
otherwise would continue to release glucose into the bloodstream
at the same time that glucose is being absorbed from the meal.
This avoids hyperglycemia during mealtime and prevents the
pancreas from having to secrete an excessive amount of insulin
during the period between meals. Patients with diabetes,
however, have little or no ability to secrete insulin rapidly in
response to the onset of a meal. Without an adequate means to
deliver insulin to the bloodstream rapidly enough to approximate
the early insulin secretion seen in healthy individuals
following a meal, patients with diabetes end up experiencing an
endless series of glucose fluctuations, triggered by the meals
and the sluggish insulin they take to control meal-time glucose.
This shortcoming is a significant obstacle to the effectiveness
of currently available insulin therapy for the treatment of
diabetes and may be overcome by the rapid entry of Nasulin into
the bloodstream seen consistently in CPEX’s Phase 1
clinical trials to date.
Challenges
of Treating Type 1 Diabetes
Type 1 diabetes is an autoimmune disease characterized by a
complete lack of insulin secretion by the pancreas. In addition
to diet, treatment for patients with Type 1 diabetes usually
includes a regimen of a long-acting nightly dose of insulin
supplemented by either very rapid or regular insulin before each
meal. Type 1 diabetes usually begins early in life and, over
time, these patients become better informed and more disciplined
in their approach to their medical treatment.
Common side effects associated with the treatment of Type 1
diabetes include hypoglycemia and weight gain. Hypoglycemia is
due to an excess of insulin at a time when glucose is either not
being absorbed or produced by the body, usually experienced
between the times when insulin enters the blood stream and just
before the next meal is consumed. This results from the
relatively long duration of action characteristic of most
insulin preparations, including rapid acting insulin. We believe
the rapid onset of Nasulin, peaking at around 15 minutes, and
returning to baseline in less than 2 hours, resembles that
of healthy subjects, which should lessen the risk of
hypoglycemia.
Weight gain after insulin therapy initiation is caused by the
anabolic effect of insulin. Commonly used injections tend to
prolong this anabolic effect rather than allow the body to
experience the alternating anabolic and catabolic activities
found in healthy subjects. We believe the unique pharmacokinetic
profile of Nasulin will allow a patient with Type 1 diabetes to
experience the alternating anabolic and catabolic activity found
in healthy subjects and potentially reduce weight gain.
Additional analyses on the Phase 2a data needs to be completed
before these potential benefits of Nasulin can be evaluated
further.
Insulin
Administration
Proteins and peptides such as insulin are typically delivered by
injection because they cannot be delivered orally without being
degraded in the stomach. Nasal administration of insulin could
present a patient-friendly alternative to the multiple daily
injections required to control diabetes. We believe, although
there can be no assurance, that a rapid-acting insulin delivered
via the nasal route could offer diabetics a new option for
prandial, or meal-time, insulin. An ultra rapid-acting nasal
insulin may have a unique value proposition compared with other
insulin formulations on the market, especially in Type
2 patients who have adequate insulin reserves but a slow
post-meal insulin response. In addition, a nasal dosage form of
insulin would avoid the possible pulmonary side effects
associated with inhalation of insulin while potentially
broadening the applicable patient populations, increasing
patient compliance and improving disease management.
10
Research
and Development
Research and development expenses were $12.3 million,
$9.1 million and $9.6 million for the years ended
December 31, 2009, 2008 and 2007, respectively. These
expenses are attributed to investments in our research and
development programs, primarily for Nasulin, our intranasal
insulin product candidate. We expect our research and
development costs to decline in 2010 due to a reduction in
activities related to Nasulin while we determine the appropriate
next steps for the program.
Sources
and Availability of Raw Materials
Our technology is dependent upon obtaining pharmaceutical grade
CPE-215 from third-party suppliers. We do not manufacture our
own CPE-215. Pharmaceutical grade CPE-215 is available from at
least two major industrial manufacturers. The Company has
obtained a secure supply from an independent manufacturer and
has obtained sufficient inventory to take us through at least
Phase 3 development of Nasulin. Other molecules and compounds
used in our development process are often proprietary to our
development partners and supplied directly by those partners.
Partners/Customers
We enter into research and license agreements with other
companies under which we perform research activities and license
product candidates in exchange for milestone payments and
royalties
and/or a
share of profits derived from product sales.
License
Agreement with Auxilium
We and Auxilium are parties to a License Agreement dated
May 31, 2000 pursuant to which Auxilium obtained a sole and
exclusive, worldwide, royalty-bearing license (including
sub-license
rights) to make, have made on their behalf, use and sell
anywhere in the world any and all pharmaceutical compositions
which contain (A) testosterone as the single active
ingredient; and (B) CPE-215 and which are covered by a
valid CPEX patent, all related patents and technology. Initially
this license was based solely upon the issued U.S. Patent
No. 5,023,252. Since this patent expired in June 2008,
Auxilium’s license is now based upon issued
U.S. Patents, No. 7,320,968 and the recently obtained
six U.S. patents (see “Intellectual Property”).
In addition, Auxilium was granted the exclusive right to enter
into another license agreement to acquire rights in these
patents and technology for the development of combination
products, which right expires upon the termination of the
original License Agreement. The License Agreement continues for
an indefinite term but it is terminable by us if Auxilium fails
to make timely payments and may also be terminated by either
party if (i) the other party becomes insolvent,
(ii) the other party fails to cure a breach within
30 days or (iii) CPEX is dissolved.
Pursuant to the terms of the Agreement, we receive royalties of
12%, of Auxilium’s annual net sales of Testim in the
U.S. and Canada. In the event that we do not have, or do
not maintain an enforceable patent in a country in which
Auxilium products are sold, the royalty rate due to us from
sales in that particular country reduces from the aforementioned
rates to a rate in the low single-digits.
Development
and License Agreement with Serenity Pharmaceuticals
Corporation
On February 4, 2008 we entered into a Development and
License Agreement with Serenity Pharmaceuticals Corporation,
(“Serenity”) to develop a nasal spray delivery product
composition containing a molecule used in the treatment of
nocturia, a condition causing an individual to wake one or more
times at night to urinate. Serenity granted us a non-exclusive
license to its technology and patents rights to conduct initial
formulation activities under the Agreement. We granted Serenity
an exclusive, sublicensable, worldwide license under United
States Patent No. 7,244,703 and foreign equivalents and
CPEX’s proprietary CPE-215 drug delivery technology to
conduct research activities related to the development of the
Serenity product and to make and sell the product. Under the
Agreement, Serenity will pay us certain milestone payments and a
low single-digit royalty on Net Sales of the product. On a
country-by-country
basis, upon the later of ten years after the first commercial
sale of the product or
11
the expiration of United States Patent No. 7,244,703 or its
equivalent, Serenity will have a fully
paid-up,
royalty-free, non-exclusive license under our patent to make
Serenity’s product.
Growth
Strategy
Our objective is to be a leading specialty pharmaceutical
company focused on advanced drug delivery and formulation
technologies to improve the delivery of new as well as existing
pharmaceuticals. Our business strategy to accomplish this
objective includes:
|
|
|
| |
•
|
evaluating the development and commercialization potential of
our most advanced product candidate — Nasulin for
intranasal insulin administration;
|
| |
| |
•
|
identifying and implementing new product candidates for internal
pipeline development that leverage our CPE-215 drug delivery
technology and formulation expertise; and
|
| |
| |
•
|
developing strong alliances for clinical research, product
manufacturing and marketing.
|
Development
and commercialization potential of Nasulin
We believe an intranasal route of administration will yield an
ultra-rapid time action profile which could provide better
glucose control equivalent to meal time injectable insulins but
with the potential for less hypoglycemia and weight gain. In
addition, based on current data demonstrating that Nasulin does
not enter the deep lung, this needle-free route of delivery
potentially avoids the pulmonary disadvantages of competitive
candidates that use an inhalation route of administration. As
explained above, preliminary findings from our Phase 2a proof of
concept study showed that although subjects in the placebo group
spent less time in euglycemia at the end of the study compared
to subjects in the Nasulin group, the difference between the two
groups was not statistically significant (p-value = 0.2).
Previously our development plan was to finish key efficacy Phase
2 trials in patients with Type 2 diabetes late in 2011 while
simultaneously seeking a pharmaceutical partner to support Phase
3 clinical trials and product commercialization upon regulatory
approval. As a result of the preliminary findings from the Phase
2a study, we intend to conduct additional analyses on the data
together with all other Nasulin data to determine the
appropriate next steps for the Nasulin program. In the interim
we have not commenced our planned Phase 2b trial or other
Nasulin development initiatives.
The major issue that could hinder the development and
commercialization potential of Nasulin would be an unacceptable
level of variability in the pharmacokinetic profile of the drug.
This could lead to inconsistent efficacy
and/or a
higher incidence of hypoglycemia in some patients. Other
potential reasons might include (1) a higher maximum dose
may be required to ensure adequate glucose lowering and
developing a higher dose may not be technically and clinically
feasible, and (2) the ultra rapid-on, rapid-off time action
profile of the product may result in insulin levels in the blood
that do not remain high enough for a sustained glucose lowering
effect. Another issue which is being closely followed in
clinical trials is nasal irritation. To date, three month
toxicity studies in rats and dogs at maximum tolerated doses
revealed no evidence of inflammation in the nose. The nature and
pattern of these events will continue to be evaluated in all
trials. While our clinical trials in healthy volunteers and in
patients have reported adverse events associated with the nasal
route of administration, to date only one serious adverse event
has been reported associated with Nasulin. The event was
described as a loss of sense of smell and was determined by the
investigator to be mild in severity.
Our Nasulin development program includes 15 completed clinical
trials in approximately 390 subjects. Eight of the studies
were conducted in healthy volunteers, four in patients with Type
1 diabetes and three in patients with Type 2 diabetes. Of the 15
studies, 14 were Phase 1 trials and one was a Phase 2 trial.
The following is a list of completed studies for which clinical
study reports are being finalized;
|
|
|
| |
•
|
BNT-INS-US-0100-PK007 — A Randomized, Single Site
Open-Label, 5 Way Crossover Meal Challenge Study Comparing Time
Action Profiles of Nasulin vs. Insulin Lispro, both in
Combination with Insulin Glargine in male and Female Subjects
with Type 1 Diabetes Mellitus. As previously reported, this
study was intended to enroll up to 24 patients however due
to slow and intermittent enrollment
|
12
|
|
|
| |
|
rates the company completed an interim analysis and determined
that it was not feasible to continuing to enroll in this trial.
The Company believes the slow enrollment was due to the
stringent inclusion criteria and was not product related.
|
|
|
|
| |
•
|
CPEX INS-US-0100-PK010 — A Single Site, Partially
Randomized, 2 Cohort Study to Determine the Safety and Counter
Regulatory Response of
Nasulintm
(BNT-INS-0100) Compared to Insulin lispro after both Average and
Diminished Sized Meals in Normal Non-Smoking Male and Female
Subjects. All subjects have completed the study.
|
The following is a summary of the recently completed Phase 2a
proof-of-concept
study:
|
|
|
| |
•
|
Nasulin-US-0100-CPEX011 — The effect of Nasulin vs.
Placebo on blood glucose control in patients with moderately
controlled Type 2 diabetes mellitus currently treated with basal
insulin and oral antidiabetic medications, excluding
secretagogues, in a Phase 2a, randomized, parallel,
double-blind, placebo-controlled, multi-center study. This
study, which randomized 94 patients, was designed to assess
the efficacy and safety of Nasulin versus a placebo over a
6-week treatment period and was conducted at multiple centers
across the United States. Preliminary data became available in
March 2010. The primary objective was to demonstrate that
subjects receiving Nasulin would achieve a larger increase from
baseline in the mean proportion of time spent with normal
glucose levels (or in euglycemia) than those receiving placebo,
as assessed by continuous glucose monitoring. Although subjects
in the placebo group spent less time in euglycemia at the end of
the study compared to subjects in the Nasulin group, the
difference between the two groups was not statistically
significant
(p-value =
0.2). The secondary efficacy measurements of change from
baseline in average glucose as measured by continuous glucose
monitoring and the change from baseline in serum fructosamine
were consistent with the primary endpoint analysis. No critical
safety signals were detected with Nasulin in the study. The most
common adverse events were those attributable to administration
site reactions associated with the nasal route of delivery, the
majority of which were mild. The percentage of subjects
reporting hypoglycemia was similar between both the Nasulin and
placebo groups. At the end of the trial, we conducted a meal
challenge which demonstrated significant reductions in the
maximum concentrations of serum glucose in the Nasulin group
versus placebo, as well as significant reductions in the overall
glucose load during 60, 90 and 240 minutes after dosing. These
findings support the pharmacodynamic data demonstrated in
previous Phase 1 clinical studies.
|
Identifying
new product candidates that leverage our CPE-215 technology and
formulation expertise
We intend to apply our CPE-215 drug delivery technology in an
effort to improve the performance of existing pharmaceutical
products and advanced research candidates with respect to their
method of delivery and effectiveness. Candidates will be
prioritized for selection based on compatibility with CPE-215,
clinical need, market size and potential for the associated
intellectual property to be protected through patents.
We are targeting therapeutic areas with high clinical need with
compounds that have established market demand or that face
limited market acceptance as a result of less efficient drug
delivery methods.
Once we bring our products to an advanced stage of development,
we intend to develop collaboration relationships that leverage
the clinical development, marketing and sales capabilities of
strategic partners. We hope to collaborate with partners to
commercialize our internal product candidates by utilizing their
late stage clinical development, regulatory, marketing and sales
capabilities. We believe that this will allow us to license our
products on terms that are more favorable than those that would
be possible earlier in the development cycle. As we succeed with
this strategy, we will identify product candidates that we can
bring to late stage development on our own.
Developing
strong alliances to provide us scale advantages in clinical
research, product manufacturing and marketing
In addition to pursuing our own proprietary compounds, we will
continue to seek strategic collaborations with pharmaceutical
and biotechnology companies to apply our CPE-215 technology to
their branded or
13
generic products. We will assist our collaboration partners in
developing more effective drug delivery methods for their
product candidates that have already completed early stage
clinical trials, or are even currently marketed. We believe
pharmaceutical and biotechnology companies will be motivated to
co-develop products utilizing CPE-215 technology to achieve
these benefits:
|
|
|
| |
•
|
improving efficacy as compared to oral administration, which
subjects the drug to the effects of first-pass metabolism;
|
| |
| |
•
|
improving utilization of costly
and/or
scarce drugs and active ingredients;
|
| |
| |
•
|
expanding the market to patients less suitable for injection,
especially children and the elderly;
|
| |
| |
•
|
improving patient convenience and compliance and lowering costs
relative to a doctor’s office visit for an injection;
|
| |
| |
•
|
potentially extending the period of market exclusivity for a
branded compound based on the grant of a patent that
incorporates new drug delivery methods;
|
| |
| |
•
|
allowing branded and generic drug companies to differentiate
their products from those of competitors; and
|
| |
| |
•
|
reducing the high capital investment needed to introduce and
manufacture injectable drugs.
|
We generally structure our collaborative arrangements to receive
research and development funding and milestone payments during
the development phase and upon commercialization, and
patent-based royalties on future sales of products.
Competition
Competition in the drug industry is intense. There are a number
of competitors who possess capabilities relevant to the drug
delivery field. In particular, we face substantial competition
from companies pursuing the commercialization of products using
nasal drug delivery technology. Established pharmaceutical
companies, such as Archimedes Pharma Ltd, AstraZeneca PLC, Bayer
Consumer Care, GlaxoSmithKline plc, and Pfizer, Inc. also have
in-house nasal drug delivery research and development programs
that have successfully developed non-insulin products that are
being marketed using nasal drug delivery technology. We also
face indirect competition from other companies with expertise in
alternate drug delivery technologies, such as oral, injectable,
patch-based and pulmonary administration. Competitors in these
fields include AstraZeneca PLC and GlaxoSmithKline plc, Alkermes
Inc., Unigene Inc., Generex Biotechnology Corporation, Emisphere
Technologies, Inc. and Coremed Corporation. Many of our
competitors have substantially greater capital resources,
research and development resources and experience, manufacturing
capabilities, regulatory expertise, sales and marketing
resources and established collaborative relationships with
pharmaceutical companies. Our competitors, either alone or with
their collaboration partners, may succeed in developing drug
delivery technologies that are similar or preferable in
effectiveness, safety, cost and ease of commercialization and
our competitors may obtain IP protection or commercialize
competitive products sooner than we do.
Universities and public and private research institutions are
also potential competitors. While these organizations primarily
have educational objectives, they may develop proprietary
technologies related to the drug delivery field or secure
protection that we may need for development of our technologies
and products. We may attempt to license these proprietary
technologies, but these licenses may not be available to us on
acceptable terms, if at all.
Even if we are able to develop products and then obtain the
necessary regulatory approvals, our success will depend to a
significant degree on the commercial success of the products
developed by us and sold or distributed by our collaboration
partners.
Testosterone
Gels
We have described on page 5 the competition for Testim, the
testosterone gel marketed and sold by Auxilium, including the
ANDA filed by Upsher-Smith.
14
Diabetes
Inhaled
and Oral Insulin
If our Nasulin product candidate obtains the necessary
regulatory approvals and becomes commercialized, it will compete
with products already in the market or currently in the
development stage.
Companies known to be working on various versions of inhaled
insulin products, in either liquid or dry form, include MannKind
Corporation and Abbott Laboratories (through its acquisition of
Kos Pharmaceuticals in 2006). MannKind completed Phase 3
clinical studies of their product,
AFREZZAtm
(formerly known as
AFRESA®)
in 2008 and submitted a New Drug Application (NDA) with the FDA
in 2009. Other companies known to be working on similar programs
include Emisphere and Coremed.
There are also several companies, including Generex
Biotechnology Corporation, Biocon Pharmaceuticals, Inc., Diasome
Pharmaceuticals, Oramed Pharmaceuticals, and Emisphere that are
pursuing development of products involving the oral delivery of
insulin.
Oral-lyntm,
Generex’s oral insulin spray product has received
regulatory approval in Ecuador, India, Lebanon and Algeria and
approvals to market the product in Syria and Iraq (North) are
expected in 2010. This product is also in Phase 3 clinical
trials at several sites around the world. Biocon’s product,
IN-105, is a conjugated insulin molecule in tablet form that is
currently in Phase 3 clinical trials. Diasome is developing Oral
HDV-I, a low-dose, short-acting mealtime hepatic insulin in a
pill or gel cap dose form. This product is currently in Phase 2
clinical trials. Oramed’s product, ORMD-0801, is an oral
insulin capsule is also in Phase 2 trials. We believe
Emisphere’s product is currently in early stage clinical
development but the timeline to commercialization has not been
made publicly available.
Non-insulin
medications
We expect that Nasulin will compete with currently available
non-insulin oral medications for Type 2 diabetes. These products
include the following:
|
|
|
| |
•
|
Sulfonylureas (including
Glucotrol®,
Diabeta®,
Glynase®,
Micronase®,
and Amaryl ), also called oral hypoglycemic agents, prompt the
pancreas to secrete insulin. This class of drugs is most
effective in individuals whose pancreas still have some working
beta cells.
|
| |
| |
•
|
Meglitinides (including Prandin and
Starlix®)
are taken with meals and reduce the elevation in blood glucose
that generally follows eating.
|
| |
| |
•
|
Biguanides (including
Glucophage®,
Glucophage XR, and
Fortamet®)
lower blood glucose by improving the sensitivity of cells to
insulin (i.e., by diminishing insulin resistance).
|
| |
| |
•
|
Thiazolidinedione (including Avandia and
Actos®)
improves the uptake of glucose by cells in the body.
|
| |
| |
•
|
Alpha-glucosidase inhibitors (including
Prandase®,
Precose and
Glyset®)
lower the amount of glucose absorbed from the intestines,
thereby reducing the rise in blood glucose that occurs after a
meal.
|
| |
| |
•
|
Incretin mimetics
(Byetta®)
are a new class of drugs that work by several mechanisms
including stimulating the pancreas to secrete insulin when blood
glucose levels are high.
|
| |
| |
•
|
Inhibitors of dipeptidyl peptidase IV
(Januvia®)
are another new class of drugs that work by blocking the
degradation of GLP-1 (glucagon-like peptide-1), which is a
naturally occurring incretin.
|
Injected
insulin
In the subcutaneous insulin market, our competitors have made
considerable efforts in promoting rapid acting injectable
insulin formulations.
Humalog®,
which was developed by Eli Lilly and Company, and
NovoLog®,
which was developed by Novo Nordisk A/S, are the two principal
injectable insulin formulations with which we expect to compete.
There are several companies, including Biodel Inc, and Halozyme
Therapeutics, that are developing new injectable insulin
products. Biodel recently submitted an NDA to the FDA seeking
approval to market
15
VIAject®,
an ultra-rapid-acting injectable human insulin for the treatment
of diabetes. Halozyme’s product, an injectable that
combines their PH20 enzyme with insulin, is currently in Phase 2
clinical trials.
Insulin
pumps
Insulin pumps are predominately used by patients with Type 1
diabetes but it has been reported that people with Type 2
diabetes have started to use them as well. The MiniMed Paradigm
insulin pump, which is sold by Medtronic Inc., is currently the
most prescribed and leading pump in the global market. Patients
well controlled by insulin pumps may be reluctant to change
insulin therapies.
Intellectual
Property
We actively seek to protect our pharmaceutical formulations and
proprietary information by means of U.S. and foreign
patents, trademarks, trade secrets as well as various other
contractual arrangements. We depend on our ability to protect
our intellectual property and proprietary rights, but we may not
be able to maintain the confidentiality, or assure the
protection, of these assets in the United States or elsewhere.
Our success depends, in large part, on our ability to protect
our current and future technologies and products and to defend
our intellectual property rights. If we fail to protect our
intellectual property adequately, competitors may manufacture
and market products similar to ours. Patents covering our
technologies have been issued to us, and we have filed, and
expect to continue to file, patent applications seeking to
protect newly developed technologies and products in various
countries, including the United States. However, patents may not
be issued with respect to any of our patent applications and
existing or future patents issued to or licensed by us may not
provide competitive advantages for our products. Patents that
are issued may be challenged, invalidated or circumvented by our
competitors. Furthermore, our patent rights may not prevent our
competitors from developing, using or commercializing products
that are similar or functionally equivalent to our products.
Where trade secrets are our sole protection, we may not be able
to prevent third parties from marketing generic equivalents to
our products, reducing prices in the marketplace and reducing
our profitability.
We have the following issued patents:
| |
|
|
|
|
|
|
|
Patent/Technology
|
|
Jurisdiction
|
|
Expiration
|
|
|
|
|
Formulations and method of using macrocyclic enhancers,
including CPE-215, with Testosterone,
Testim®
product
|
|
United States
|
|
|
2025
|
(1)
|
|
Pharmaceutical
formulations and methods of use patents relating to the use of
nasal formulations of insulin
(“Nasulintm”)
|
|
United States
|
|
|
2024
|
|
|
Pharmaceutical formulations and methods of use patents relating
to the combination of the macrocyclic enhancer with peptides,
peptidomimetics or proteins
|
|
United States
|
|
|
2022
|
|
|
|
|
|
(1) |
|
Orange Book Listed Patents covering the
Testim®
formulation expire April 21, 2023 and January 18, 2025. |
The use of our technology with various products such as insulin,
testosterone as well as other peptides, is covered by both
U.S. and foreign patents in many major market countries.
The initial patent for use of our technology has expired in the
United States in June 2008, will expire in Canada in 2010 and
has expired in all other markets outside the United States. We
have patent protection for the commercial formulations of
testosterone (Testim) until 2025 and for the nasal insulin
formulation under development (Nasulin) until 2024. We recently
obtained six U.S. patents covering the testosterone
formulation in the United States. These patents were added to
the currently listed Orange Book: Approved Drugs with
Therapeutic Equivalence (“Orange Book”) patent for
Testim, which means that there are a total of seven listed
patents covering the Testim product. We have applied for and
continue to file patent applications covering the use of our
technology worldwide; however, we cannot provide any assurance
that any patents will issue.
16
We also rely on trade secrets, non-patented proprietary
expertise and continuing technological innovation that we seek
to protect, in part, by entering into confidentiality agreements
with licensees, suppliers, employees, consultants and others. To
this end, we require our employees, consultants and advisors to
enter into agreements that prohibit the disclosure of
confidential information and, where applicable, require
disclosure and assignment to us of all ideas, developments,
discoveries and inventions that arise from their activities for
us. Additionally, these confidentiality agreements require that
our employees, consultants and advisors do not bring to our
attention, or use, without proper authorization, any third
party’s proprietary technology. However, these agreements
may be breached and there may not be adequate remedies in the
event of a breach. Disputes may arise concerning the ownership
of intellectual property or the applicability of confidentiality
agreements. Moreover, our trade secrets and proprietary
technology may otherwise become known or be independently
developed by our competitors.
Third parties may claim that we infringe on their proprietary
rights. There has been substantial litigation in the
pharmaceutical industry with respect to the manufacture, use and
sale of new products. These lawsuits relate to the validity and
infringement of patents or proprietary rights of third parties.
We may be required to commence or defend against charges
relating to the infringement of patent or proprietary rights.
Any such litigation could: (i) require us to incur
substantial expenses, even if we are insured or successful in
the litigation; (ii) require us to divert significant time
and effort of our technical and management personnel;
(iii) result in the loss of our rights to develop or make
certain products; (iv) require us to pay substantial
monetary damages or royalties in order to license proprietary
rights from third parties; and (v) prevent us from
launching a developed, tested and approved product.
Assignment
Agreement with Access Pharmaceuticals, Inc.
Under an Assignment Agreement with MacroChem Corporation
(“MacroChem”), dated June 24, 2003, which was
assumed by Access Pharmaceuticals, Inc. (“Access”)
following their acquisition of MacroChem in February 2009, we
own all of Access’s right, title and interest to
U.S. Patent Number 6,495,124 B1 and any and all related
patents and patent applications which are divisions,
continuations,
continuations-in-part,
reissues, renewals, extensions and supplementary protection
certificates (the “Access Patent Rights”). As a result
of the assignment, Access retained no interest whatsoever in the
Access Patent Rights. To date, we have not generated any revenue
from the Access Patent Rights, which expire in 2020.
Government
Regulation
Numerous governmental authorities in the U.S. and other
countries extensively regulate the activities of pharmaceutical
manufacturers. If we fail to comply with the applicable
requirements of governmental authorities, we may be subject to
administrative or judicial sanctions such as refusal of or delay
in the approval of pending marketing applications or supplements
to approved applications, warning letters, total or partial
suspension of production, fines, injunctions, product seizures
or recalls, as well as criminal prosecution.
Prior to marketing most pharmaceutical products in the U.S., the
product must first be approved by the FDA. For new compounds,
the regulatory approval process begins with developing
preclinical laboratory supporting data and animal safety
testing. The approval process generally consists of the
following five principal stages:
|
|
|
| |
•
|
preclinical testing (supporting safety and potential efficacy);
|
| |
| |
•
|
submission and review by the FDA of an Investigational New Drug
Exemption (IND) Application;
|
| |
| |
•
|
clinical trials;
|
| |
| |
•
|
preparation and submission of the New Drug Application
(NDA); and
|
| |
| |
•
|
FDA’s review and approval/disapproval of the NDA.
|
In some cases, further clinical trials may also be required
following approval.
17
The IND is submitted to the FDA when the appropriate preclinical
studies are completed and must be submitted to the FDA
30 days before beginning clinical studies. The IND becomes
effective if the FDA does not put the investigations described
in the IND on clinical hold within 30 days of receiving the
IND for filing.
Human clinical trials typically are conducted in three
sequential phases. Some clinical trials may include aspects of
more than one phase.
|
|
|
| |
•
|
Phase 1 involves the initial introduction of the pharmaceutical
compound into patients or healthy human volunteers; the emphasis
is on testing for dosage tolerance, metabolism, excretion,
clinical pharmacology, safety (adverse effects) and possibly
early evidence of effectiveness.
|
| |
| |
•
|
Phase 2 involves the first controlled clinical trial involving
patients who have the targeted disease or condition and consists
of safety and efficacy studies. The studies may be divided into
early Phase 2 (or 2a), during which studies are performed to
determine initial efficacy and late Phase 2 (or 2b) which may
consist of placebo-controlled trials in a larger number of
patients.
|
| |
| |
•
|
Phase 3 involves large scale, longer-term, well controlled
efficacy and safety studies within an expanded patient
population, frequently at multiple clinical study sites.
|
Throughout the drug development process, the IND must be updated
continually with protocol amendments, information amendments,
IND Safety Reports and Annual Reports. The FDA carefully reviews
all data submitted and holds meetings with the sponsor at key
stages to discuss the preclinical and clinical plans and results.
The Chemistry/Manufacturing/Controls data, clinical studies,
statistical evaluation and all relevant supporting research data
that has been collected over many years of development is
submitted to the FDA in an NDA. Additionally, an NDA will
contain complete information on the proposed manufacturing
process including process, equipment and facilities validation
demonstrating that the applicant is capable of consistently
manufacturing a drug product of appropriate strength, quality
and purity consistent with the product that was studied in the
clinical trials. An NDA is an application requesting FDA
approval to market a new drug for human use in interstate
commerce.
NDAs are allocated varying review priorities based on a number
of factors, including the severity of the disease, the
availability of alternative treatments and the risks and
benefits demonstrated in clinical trials. Additional animal
studies or clinical trials may be requested during the FDA
review process and may delay marketing approval. After FDA
approval for the initial indications, further clinical trials
are necessary to gain approval for the use of the product for
any additional indications. The FDA may also require
post-marketing testing to monitor for adverse effects, and in
some cases to provide additional information on efficacy, which
can involve significant expense. Our products under development
and future products to be developed must go through the approval
process delineated above prior to gaining approval by the FDA
for commercialization.
FDA approval is also required for the marketing of generic
equivalents of an existing drug. An Abbreviated New Drug
Application, or ANDA, is required to be submitted to the FDA for
approval. When processing an ANDA, the FDA, in lieu of the
requirement for conducting complete clinical studies, requires
bioavailability
and/or
bioequivalence studies. Bioavailability indicates the rate and
extent of absorption and levels of concentration of a drug
product in the body. Bioequivalence compares the bioavailability
of one drug product (in this case, the product under review)
with another (usually the innovator product). When
bioequivalence is established, the rate of absorption and levels
of concentration of the drug in the body will closely
approximate those of the previously approved drug. An ANDA may
only be submitted for a drug on the basis that it is the
equivalent to a previously approved drug.
In addition to obtaining FDA approval for each product, each
manufacturer of drugs must register its manufacturing facilities
with the FDA, and must list the drug products it manufactures at
each facility. Domestic manufacturing establishments are subject
to biennial inspections by the FDA and must comply with current
Good Manufacturing Practices or cGMPs for drugs. To supply
products for use in the U.S., foreign manufacturing
establishments must also comply with U.S. cGMPs and are
subject to inspection by the FDA. Such inspections generally
take place upon submission of an NDA or ANDA to the FDA or at
any other time
18
deemed necessary by the FDA and can impact both the approval of
drugs, and a company’s ability to continue manufacturing
following approval.
Employees
We employ 17 people, as of March 24, 2010, all of
which are full-time employees and are based in the United
States. Approximately half of these employees are principally
engaged in research, development, clinical and regulatory
activities. In general, we consider our relations with our
employees to be good.
Executive
Officers
Our Executive Officers are listed below:
| |
|
|
|
|
|
|
|
Name
|
|
Age
|
|
Position(s)
|
|
|
|
John A. Sedor
|
|
|
65
|
|
|
Chief Executive Officer, President and Director
|
|
Nils Bergenhem
|
|
|
51
|
|
|
Chief Scientific Officer and Vice President
|
|
Lance Berman
|
|
|
39
|
|
|
Chief Medical Officer and Senior Vice President
|
|
Robert P. Hebert
|
|
|
37
|
|
|
Chief Financial Officer and Vice President
|
John A. Sedor, CEO and President —
Mr. Sedor has been our Chief Executive Officer and
President since the spin-off from Bentley in 2008.
Mr. Sedor was President of Bentley from 2005 until the
spin-off. From 2001 to May 2005, he was President and CEO of
Sandoz, Inc. (a division of Novartis AG). From
1998-2001
Mr. Sedor was President and Chief Executive Officer at
Verion, Inc., a drug delivery company. Previously,
Mr. Sedor served as President and Chief Executive Officer
at Centeon, LLC, a joint venture between two major multinational
corporations, Rhône-Poulenc Rorer and Hoechst AG.
Previously, Mr. Sedor served as Executive Vice President at
Rhône-Poulenc Rorer, Revlon Health Care and Parke-Davis.
Mr. Sedor holds a Bachelor of Science degree in
Pharmacy/Chemistry from Duquesne University, and has studied
strategic marketing at both Northwestern University’s
Kellogg Graduate School of Management and Harvard Business
School. He has also attended Harvard’s Executive Forum.
Nils Bergenhem, Ph.D., Chief Scientific
Officer — Dr. Berhgenhem joined us as Chief
Scientific Officer in February 2010. From January 2008 until
joining CPEX, Dr. Bergenhem served as Chief Scientific
Officer at Escoublac, Inc., the first biotechnology company in
the Biogen Idec Innovations Incubator. There, he was responsible
for development and execution of the research plan for human
osteocalcin in metabolic disease, Type 2 diabetes and obesity.
From June 2005 until December 2007 he served as CSO at
AdipoGenix where he oversaw internal drug discovery and
development programs for obesity and related co-morbidities, as
well as the AdipoGenix alliance with Johnson and Johnson.
Previously, Dr. Bergenhem served as Vice President for
Research at the Institute for Diabetes Discovery, as Director
for Diabetes Research at OSI Pharmaceuticals Inc. and held
positions of increasing importance at Novo Nordisk in
Copenhagen. Dr. Bergenhem holds a BS degree in Chemistry
from Linkoping University, Sweden, and a Ph.D. in Biochemistry
from Umeå University, Sweden. He has completed postdoctoral
work at University of Michigan in Molecular Biology and Human
Genetics, where he also spent three years at the School of
Medicine as Research Investigator at the Institute of
Gerontology and at the Department of Biological Chemistry.
Lance Berman, M.D., Chief Medical
Officer — Dr. Berman has been our Chief
Medical Officer since February 2009. Previously, Dr. Berman
served at Pfizer from 2003 until 2009. From December 2007 he
served as Senior Medical Director and Global Medical Team Leader
at Pfizer, responsible for the strategic medical development and
evolution of products within the cardiovascular and diabetes
portfolios. Previously, Dr. Berman held roles of increasing
importance at Schering-Plough from 1999 to 2003 and Janssen
Pharmaceuticals (Johnson & Johnson) from 1996 to 1999.
Dr. Berman received his Bachelor of Medicine and Bachelor
of Surgery at University of Cape Town in Cape Town, South
Africa, and holds a Masters Degree in Pharmaceutical Medicine.
Robert P. Hebert, CFO and Vice President —
Mr. Hebert has been in this role since the spin-off from
Bentley in 2008. From June 2006 until the spin-off,
Mr. Hebert was Controller and Principal Accounting Officer
for Bentley. In this role, Mr. Hebert managed all of
Bentley’s accounting and reporting functions. From
19
May 2003 until June 2006, Mr. Hebert was Bentley’s
Director of SEC Reporting & Compliance, Assistant
Secretary and Assistant Treasurer. His responsibilities in this
role included Bentley’s financial reporting and compliance
with the requirements of the Sarbanes-Oxley Act of 2002. Prior
to joining Bentley, Mr. Hebert worked as an auditor for
Deloitte & Touche LLP from 1995 to 2003.
Mr. Hebert received a B.S. in Business Administration with
a concentration in accounting from Merrimack College in 1995.
Available
Information
Copies of reports filed by us pursuant to Section 13(a) or
15(d) of the Securities Exchange Act of 1934, including Annual
Reports on
Form 10-K,
Quarterly Reports on
Form 10-Q,
Current Reports on
Form 8-K
and amendments to those reports may be accessed from our website
at www.cpexpharm.com, free of charge, as soon as reasonably
practicable after we electronically file such reports with, or
furnish such reports to, the Securities and Exchange Commission.
Alternatively, these reports can be accessed through a query at
the website of the Securities and Exchange Commission at
www.sec.gov.
You should carefully consider the following discussion of risks
and uncertainties relating to our business and ownership of our
securities. The risks described below are not the only risks we
face. Additional risks that we do not yet know of or that we
currently think are immaterial may also impair our business
operations. If any of the events or circumstances described
below actually occurs, our business, financial condition, or
results of operations could be materially adversely affected. In
such case, the trading price of our common stock could decline
and you may lose all, or part of your investment.
We
anticipate that we will incur losses for the foreseeable future.
We may never achieve or sustain profitability. If additional
capital is not available, we may have to curtail or cease
operations.
Our business currently is not expected to generate all of the
cash that is necessary to finance our operations in the
short-term. Subject to the success of our development programs,
the continuance of our
Testim®
royalty payments, and potential future licensing transactions,
we will need to raise additional capital to:
|
|
|
| |
•
|
fund our research and development programs;
|
| |
| |
•
|
develop and commercialize our product candidates;
|
| |
| |
•
|
enhance existing services;
|
| |
| |
•
|
respond to competitive pressures; and
|
| |
| |
•
|
acquire complementary businesses or technologies.
|
Our future capital needs depend on many factors, including:
|
|
|
| |
•
|
the scope, duration and expenditures associated with our
research and development programs;
|
| |
| |
•
|
continued scientific progress in these programs;
|
| |
| |
•
|
the amount of royalties or other revenues we are able to earn
and collect;
|
| |
| |
•
|
the outcome of potential licensing transactions, if any;
|
| |
| |
•
|
competing technological developments;
|
| |
| |
•
|
our proprietary patent position, if any, in our
products; and
|
| |
| |
•
|
the regulatory approval process for our products.
|
We may seek to raise necessary funds through public or private
equity offerings, debt financings or additional strategic
alliances and licensing arrangements. We may not be able to
obtain additional financing on terms favorable to us, if at all.
General market conditions may make it very difficult for us to
seek financing
20
from the capital markets. We may be required to relinquish
rights to our technology or drug candidates, or grant licenses
on terms that are not favorable to us, in order to raise
additional funds through an alliance, a joint venture or a
licensing arrangement. If adequate funds are not available, we
may have to delay, reduce or eliminate one or more of our
research or development programs and reduce overall overhead
expenses. These actions would likely reduce the market price of
our common stock.
Substantially
all of our revenues to date have been generated from royalties
on Auxilium’s sales of Testim, which is subject to
projected generic competition. Should the sales of Testim
decline, we may be required to limit, scale back or cease
operations.
Substantially all of our revenues are derived through royalty
income from the only commercialized product utilizing our
CPE-215 technology, Testim, which is sold by Auxilium. Testim
royalties totaled $18.6 million, $15.1 million and
$11.1 million in the years ended December 31, 2009,
2008 and 2007, respectively, and the only expenses regarding
Testim®
that we have incurred during that period have been patent
maintenance costs, which have not been material. Though we
believe that Auxilium intends to continue commercialization of
Testim, sales of this product are subject to the following
risks, among others:
|
|
|
| |
•
|
pressures from existing or new competing products, including
generic products, that may provide therapeutic, convenience or
pricing advantages over Testim or may garner a greater share of
the market;
|
| |
| |
•
|
growth of the overall androgen market where Testim
competes; and
|
| |
| |
•
|
commercialization priorities of Auxilium.
|
In October 2008, we and Auxilium received notice that
Upsher-Smith Laboratories filed an Abbreviated New Drug
Application, or ANDA, containing a paragraph IV
certification in which it certified that it believes that its
testosterone gel product does not infringe our ‘968 patent.
The ‘968 Patent covers a method for maintaining effective
blood serum testosterone levels for treating a hypogonadal male
using Testim and will expire in January 2025. The ‘968
patent is listed in Approved Drug Products with Therapeutic
Equivalence Evaluations (commonly known as the Orange Book),
published by the U.S. Food and Drug Administration. The
paragraph IV certification sets forth allegations that the
‘968 Patent will not be infringed by Upsher-Smith’s
manufacture, use or sale of the product for which the ANDA was
submitted. On December 4, 2008, we and Auxilium filed a
lawsuit in the United States District Court for the District of
Delaware against Upsher-Smith under the Hatch Waxman Act for
infringement of our patent. In June 2009, Upsher-Smith amended
its answer to the complaint to include a defense and
counterclaim of invalidity of the ‘968 Patent, which we and
Auxilium deny. We and Auxilium filed a reply to the counterclaim
in July 2009 denying the invalidity of the ‘968 Patent. A
patent issued by the Patent and Trademark Office, such as the
‘968 Patent, is presumed valid. As described more fully
under Item 3 — “Legal Proceedings” in
this report, any FDA approval of Upsher-Smith’s proposed
generic product will be stayed until the earlier of
30 months beginning on the date of receipt of the
paragraph IV certification (April 2011) or an adverse
decision in our patent infringement lawsuit.
Should Testim sales be adversely impacted by any of the above
risks, our revenues will be reduced, which may force us to delay
our current plans to develop other product candidates.
In addition, our royalty income is dependent upon our ability to
maintain our intellectual property claims for our CPE-215
technology that is used in the Testim product. Should we be
unable to maintain our intellectual property position with
regards to CPE-215, our royalty income would be impaired.
We
cannot give any assurance that Nasulin will advance in clinical
trials or will receive regulatory approval.
We are currently conducting additional analyses of the data from
the Nasulin clinical studies to determine the appropriate next
steps for the Nasulin program. We may decide not to conduct
further trials of Nasulin. If we do conduct trials, the trials
may not be successful and Nasulin may never receive regulatory
approval or be successfully commercialized. Our clinical
development program for Nasulin may not receive regulatory
approval either if we fail to demonstrate that it is safe and
effective in clinical trials and consequently fail to
21
obtain necessary approvals from the FDA, or similar
non-U.S. regulatory
agencies, or if we have inadequate financial or other resources
to advance Nasulin through the clinical trial process.
Our
operations could be adversely affected if we are unable to raise
or obtain needed funding.
Substantial time, financial and other resources will be required
to complete ongoing development and clinical testing of our
proprietary products. Regulatory efforts and collaborative
arrangements also will be necessary for our products that are
currently under development and testing in order for them to be
marketed.
Our revenues from operations and cash may not be sufficient over
the next several years for commercializing all of the products
we are currently developing. Consequently, we may seek strategic
partners for various phases of development, marketing and
commercialization of product candidates employing our
technology. Further, we cannot assure you as to the sufficiency
of our resources or the time required to complete any ongoing
development and clinical testing, since the extent to which we
conduct such testing is dependent on resource allocation
decisions that we make from time to time based on numerous
financial as well as operational conditions.
In addition to development and other costs, we expect to incur
capital expenditures from time to time. These capital
expenditures will be influenced by our regulatory compliance
efforts, our success, if any, at developing collaborative
arrangements with strategic partners, our needs for additional
facilities and capital equipment and the growth, if any, of our
business in general. There can be no assurance that we will
receive additional funding on favorable terms if at all, or that
we will be successful in attracting strategic partners. If we
cannot raise funds or engage strategic partners on acceptable
terms when needed, we may not be able to continue our research
and development activities, develop or enhance our products and
services, take advantage of future opportunities, grow our
business or respond to competitive pressures or unanticipated
requirements.
Our
growth depends on identifying drugs suitable for our drug
delivery technology.
We believe that our growth depends on the identification of
pharmaceutical products that are suitable for delivery using our
proprietary technologies. Our principal drug delivery technology
is our CPE-215 technology. This technology, like certain other
drug delivery technologies, operates to increase the amount and
rate of absorption of certain drugs across biological membranes.
This technology does not operate independently and must be
coupled with suitable pharmaceutical products in order to
provide value. Consequently, our growth will depend to a great
extent on identifying and commercializing these suitable drugs
with respect to which we intend to expend significant resources
and efforts. Identifying suitable products is a lengthy and
complex process that may not succeed. Even if identified,
products may not be available to us or we may otherwise be
unable to enter into licenses or other agreements for their use.
In our efforts to identify suitable products, we compete with
other drug delivery companies with greater research and
development, financial, marketing and sales resources. If we do
not effectively identify drugs to be used with our technologies,
improve the delivery of drugs with our technologies and bring
the improved drugs to commercial success, then we may not be
able to continue our growth and we will be adversely affected.
Products
using our technology are in various stages of development and
may not achieve commercial success.
Independently as well as in conjunction with strategic partners,
we are investigating the use of our technology with respect to a
variety of pharmaceutical compounds and products that are in
various stages of development. We are unable to predict whether
any of these products will receive regulatory approvals or be
successfully developed, manufactured or commercialized. Further,
due to the extended testing and regulatory review process
required before marketing clearance can be obtained, the time
periods before commercialization of any of these products are
long and uncertain. Risks during development include the
possibility that:
|
|
|
| |
•
|
any or all of the proposed products will be found to be
ineffective;
|
22
|
|
|
| |
•
|
the proposed products will have adverse side effects or will
otherwise fail to receive necessary regulatory approvals;
|
| |
| |
•
|
the proposed products may be effective but uneconomical to
market; or
|
| |
| |
•
|
other pharmaceutical companies may market equivalent or superior
products.
|
If
medical doctors do not prescribe our products or the medical
profession does not accept our products, our ability to grow our
revenues will be limited.
Our business is dependent on market acceptance of our products
by physicians, hospitals, pharmacists, patients and the medical
community. Willingness to prescribe our products depends on many
factors, including:
|
|
|
| |
•
|
perceived efficacy of our products;
|
| |
| |
•
|
convenience and ease of administration;
|
| |
| |
•
|
prevalence and severity of adverse side effects in both clinical
trials and commercial use;
|
| |
| |
•
|
availability of alternative treatments;
|
| |
| |
•
|
cost effectiveness;
|
| |
| |
•
|
effectiveness of our marketing strategy and the pricing of our
products;
|
| |
| |
•
|
publicity concerning our products or competing products; and
|
| |
| |
•
|
our ability to obtain third-party coverage or reimbursement.
|
Even though regulatory approval has been received for Testim,
and even if we receive regulatory approval and satisfy the above
criteria for any other product candidates developed by us or
incorporating our drug delivery technology, physicians may not
prescribe these products if we do not promote the products
effectively. Factors that could affect our success in marketing
our products include:
|
|
|
| |
•
|
the effectiveness of our sales force;
|
| |
| |
•
|
the effectiveness of our production, distribution and marketing
capabilities;
|
| |
| |
•
|
the success of competing products; and
|
| |
| |
•
|
the availability and extent of reimbursement from third-party
payors.
|
We
will rely on strategic partners to conduct clinical trials and
commercialize products that use our drug delivery
technology.
In light of our limited development resources and the
significant time, expense, expertise and infrastructure
necessary to bring new drugs and formulations from inception to
market, we are particularly dependent on resources from third
parties to commercialize products incorporating our
technologies. Our strategy involves forming alliances with
others who will develop, manufacture, market and sell our
products in the United States and other countries. We may not be
successful in finding other strategic partners or in otherwise
obtaining financing, in which case the development of our
products would be delayed or curtailed.
We must enter into agreements with strategic partners to conduct
clinical trials, manufacturing, marketing and sales necessary to
commercialize product candidates. In addition, our ability to
apply our drug delivery technologies to any proprietary drugs
will depend on our ability to establish and maintain strategic
partnerships or other collaborative arrangements with the
holders of proprietary rights to such drugs. Arrangements with
strategic partners may be established through a single
comprehensive agreement or may evolve over time through a series
of discrete agreements, such as letters of intent, research
agreements and license agreements. We cannot assure you that we
will be able to establish such strategic partnerships or
collaborative arrangements on favorable terms or at all or that
any agreement entered into with a strategic partner will lead to
further agreements or ultimately result in commercialization of
a product.
23
In collaborative arrangements, we will depend on the efforts of
our strategic partners and will have limited participation in
the development, manufacture, marketing and commercialization of
the products subject to the collaboration. We cannot assure you
that these strategic partnerships or collaborative arrangements
will be successful, nor can we assure you that strategic
partners or collaborators will not pursue alternative
technologies or develop alternative products on their own or
with others, including our competitors. In addition, our
collaborators or contract manufacturers will be subject to
regulatory oversight which could delay or prohibit our
development and commercialization efforts. Moreover, we could
have disputes with our existing or future strategic partners or
collaborators. Any such disagreements could lead to delays in
the research, development or commercialization of potential
products or could result in time-consuming and expensive
litigation or arbitration.
An
interruption in the sourcing and availability of the active
ingredient used in our CPE-215 technology could cause our
product development and commercialization to slow or
stop.
We do not own or operate manufacturing facilities for clinical
or commercial production of our product candidates. We lack the
resources and the capability to manufacture any of our product
candidates on a clinical or commercial scale. We also lack the
resources to manufacture the excipient CPE-215, which is the
major component of our CPE-215 technology. Our technology is
dependent upon obtaining pharmaceutical grade CPE-215 which is
available from at least two major industrial manufacturers. If a
third party supplier is unable to provide us with required
quantities of pharmaceutical grade CPE-215 on commercially
favorable terms, we may be unable to continue our product
development or commercialization activity.
If we
are unable to meet our responsibilities under any of our
agreements, we may lose potential business and be subject to
penalties and other damages.
The Company has a licensing agreement with Auxilium pursuant to
which the Company licenses its
CPE-215 with
Testosterone formulation to Auxilium and receives royalties of
12% from Auxilium based upon Auxilium’s sales of Testim.
This royalty stream is the Company’s major source of
current revenue. If the Company does not maintain adequate
patent protection for Testim, the royalty rate due to the
Company would be reduced to 2%. To date the Company has not
experienced a reduction in the royalty rate due to loss of
patent protection and the Company recently obtained patents that
cover the application of testosterone with CPE-215 in the
U.S. and in foreign countries that continue through 2023.
Disputes may arise with respect to certain of our development
agreements regarding the development and commercialization of
products, which incorporate our intellectual property. These
disputes could lead to delays in commercialization of products
incorporating our technologies or termination of the agreements.
If any
of our product candidates for which we receive regulatory
approval do not achieve broad market acceptance, the revenues
that we generate from their sales will be limited.
The commercial success of our product candidates for which we
obtain marketing approval from the FDA or other regulatory
authorities will depend upon the acceptance of these products by
the medical community and coverage and reimbursement of them by
third-party payors, including government payors. The degree of
market acceptance of any of our approved products will depend on
a number of factors, including:
|
|
|
| |
•
|
limitations or warnings contained in a product’s
FDA-approved labeling;
|
| |
| |
•
|
changes in the standard of care for the targeted indications for
either of our product candidates could reduce the marketing
impact of any superiority claims that we could make following
FDA approval;
|
| |
| |
•
|
limitations inherent in the approved indication for either of
our product candidates compared to more commonly-understood or
addressed conditions; and
|
| |
| |
•
|
potential advantages over, and availability of, alternative
treatments, including, in the case of Nasulin, a number of
products already used to treat diabetes.
|
24
Our ability to effectively promote and sell our product
candidates will also depend on pricing and cost effectiveness,
including our ability to produce a product at a competitive
price and our ability to obtain sufficient third-party coverage
or reimbursement. We will also need to demonstrate acceptable
evidence of safety and efficacy as well as relative convenience
and ease of administration. Market acceptance could be further
limited depending on the prevalence and severity of any expected
or unexpected adverse side effects associated with our product
candidates. If our product candidates are approved but do not
achieve an adequate level of acceptance by physicians, health
care payors and patients, we may not generate sufficient revenue
from these products, and we may not become or remain profitable.
In addition, our efforts to educate the medical community and
third-party payors on the benefits of our product candidates may
require significant resources and may never be successful.
Pharmaceutical
pricing, changes in third-party reimbursement and governmental
mandates are uncertain and may adversely affect
us.
Successful commercialization of many of our products may depend
on the availability of reimbursement for the cost of such
products and related treatment from third-party healthcare
payors, such as the government, private insurance plans and
managed care organizations. Third-party payors are increasingly
challenging the price of medical products and services. Such
reimbursement may not be available for any of our products at
all or for the duration of the recommended treatment with a
drug, which could materially adversely affect our ability to
commercialize that drug. The increasing emphasis on managed care
in the U.S. continues to increase the pressure on
pharmaceutical pricing. Some governmental agencies can compel
companies to continue to produce products that are not
profitable for the company due to insufficient supply. In the
U.S., there have been a number of federal and state proposals to
implement similar government controls. We anticipate that there
will continue to be a number of proposals in the U.S., as has
been the case in many foreign markets. The announcement or
adoption of such proposals could adversely affect us. Further,
our ability to commercialize our products may be adversely
affected to the extent that such proposals materially adversely
affect the business, financial condition and profitability of
companies that are prospective strategic partners.
The cost of healthcare in the U.S. and elsewhere continues
to be a subject of investigation and action by various
governmental agencies. Certain resulting legislative proposals
may adversely affect us. For example, governmental actions to
further reduce or eliminate reimbursement for drugs may directly
diminish our markets. In addition, legislative safety and
efficacy measures may be invoked that lengthen and increase the
costs of drug approval processes. Further, social, economic and
other broad policy legislation may induce unpredictable changes
in the healthcare environment. If any of these measures are
enacted in some form, they may have a material adverse effect on
our results of operations.
If our
clinical trials fail or are delayed for any product candidate,
we will be unable to market it.
Any human pharmaceutical product developed by us would require
clearance by the FDA for sales in the United States and by
comparable regulatory agencies for sales in other countries. The
process of conducting clinical trials and obtaining FDA and
other regulatory approvals is expensive, takes several years and
we cannot be assured of success. In order to obtain FDA approval
of any new product candidates using our technologies, a New Drug
Application (“NDA”) must be submitted to the FDA
demonstrating that the product candidate, based on preclinical
research, animal studies and human clinical trials, is safe for
humans and effective for its intended use. Positive results from
preclinical studies and early clinical trials do not ensure
positive results in more advanced clinical trials designed to
permit application for regulatory approval. We may suffer
significant setbacks in clinical trials, even in cases where
earlier clinical trials show promising results. Any of our new
product candidates may produce undesirable side effects in
humans that could cause us or regulatory authorities to
interrupt, delay or halt clinical trials of a product candidate.
We, the FDA or other regulatory authorities, may suspend our
clinical trials at any time if we or they believe the trial
participants face unacceptable health risks or if they find
deficiencies in any of our regulatory submissions. Other factors
that can cause delay or terminate our clinical trials include:
|
|
|
| |
•
|
slow or insufficient patient enrollment;
|
25
|
|
|
| |
•
|
slow recruitment and completion of necessary institutional
approvals at clinical sites;
|
| |
| |
•
|
longer treatment time required to demonstrate efficacy;
|
| |
| |
•
|
lack of sufficient supplies of the product candidate;
|
| |
| |
•
|
adverse medical reactions or side effects in treated patients;
|
| |
| |
•
|
lack of effectiveness of the product candidate being tested;
|
| |
| |
•
|
regulatory requests for additional clinical trials; and
|
| |
| |
•
|
instability of the pharmaceutical formulations.
|
A delay or termination of any of our clinical trials may have a
material adverse effect on our results of operations.
We
rely on third parties to conduct clinical trials for our product
candidates and plan to rely on third parties to conduct future
clinical trials. If these third parties do not successfully
carry out their contractual duties or meet expected deadlines,
we may be unable to obtain regulatory approval for or
commercialize our current and future product
candidates.
We do not have the ability to conduct clinical trials for any of
our product candidates. We rely on third parties, such as
contract research organizations, medical institutions, clinical
investigators and contract laboratories, to conduct all of our
clinical trials for our product candidates. Although we rely on
these third parties to conduct our clinical trials, we are
responsible for ensuring that each of our clinical trials is
conducted in accordance with its investigational plan and
protocol. Moreover, the FDA and other
non-U.S. regulatory
authorities require us to comply with regulations and standards,
commonly referred to as Good Clinical Practices
(“GCPs”), for conducting, monitoring, recording and
reporting the results of clinical trials to ensure that the data
and results are scientifically credible and accurate and that
the trial subjects are adequately informed of the potential
risks of participating in clinical trials. Our reliance on third
parties does not relieve us of these responsibilities and
requirements. If the third parties do not perform their
contractual duties or obligations, do not meet expected
deadlines or need to be replaced, or if the quality or accuracy
of the clinical data they obtain is compromised due to the
failure to adhere to GCPs or for any other reason, we may need
to enter into new arrangements with alternative third parties
and our clinical trials may be extended, delayed or terminated.
In addition, failure by such third parties to perform their
obligations in compliance with GCPs may cause our clinical
trials to fail to meet regulatory requirements, which may
require us to repeat our clinical trials.
If our
existing patents do not afford adequate protection to us, our
competitors may be able to develop competing
products.
The basic patent disclosing and claiming CPE-215 technology
expired in the U.S. in June 2008 and most foreign markets
in 2006. The patent will also expire in Canada in 2010. The
Company has filed applications in many countries that cover the
application of testosterone with CPE-215. Patents for the
application of testosterone with CPE-215 have been issued to us
in various countries, including the U.S., Canada and Europe that
continue through 2023. As such, we do not anticipate a
significant impact from the expiration of the basic CPE-215
patent on the current Testim royalty rates due to the Company or
on our plan of operation or future business plans. The Company
also has pending applications for other applications involving
CPE-215 technology. If our pending applications covering various
applications involving CPE-215 technology are not issued as
patents or if our patents do not afford adequate protection to
us or our licensees, our competitors may be able to use
information from our expired and soon to expire patents to
develop, manufacture and market products that compete with our
products, as well as other products using CPE-215 that we
otherwise might have developed.
26
Our
patent positions and intended proprietary or similar protections
are uncertain.
We have filed a number of patent applications and have been
granted licenses to, or have acquired, a number of patents. We
cannot assure you, however, that any of our issued or licensed
patents will afford adequate protection to us or our licensees.
Furthermore, enforcing a claim that another person is infringing
one or more of our patents is expensive and time consuming, and
the outcome is unpredictable. We cannot determine the ultimate
scope and validity of patents that are now owned by or may be
granted to third parties, the extent to which we may wish, or be
required, to acquire rights under such patents or the cost or
availability of such rights. In the event that patent protection
for technologies expire, or are not extended, revenues derived
from such technologies may be reduced significantly.
Competitors may interfere with our patent process in a variety
of ways. Competitors may claim that they invented the claimed
invention prior to us. Competitors also may claim that we are
infringing their patents, interfering with or preventing the use
of our technologies. Competitors also may contest our patents by
showing the patent examiner that the invention was not original,
was not novel or was obvious. A competitor could claim that our
issued patents are not valid for a variety of other reasons as
well.
We also rely on trade secrets, unpatented proprietary
technologies and continuing technological innovations in the
development and commercialization of our products. We cannot
assure you that others will not independently develop the same
or similar technologies or obtain access to our proprietary
technologies. It is unclear whether our trade secrets will be
protected under law. While we use reasonable efforts to protect
our trade secrets, our employees or consultants may
unintentionally or willfully disclose our information to
competitors. Our employees and consultants with access to our
proprietary information have entered into or are subject to
confidentiality arrangements with us and have agreed to disclose
and assign to us any ideas, developments, discoveries and
inventions that arise from their activities for us. We cannot
assure you, however, that others may not acquire or
independently develop similar technologies or, if effective
patents in applicable countries are not issued with respect to
our products or technologies, that we will be able to maintain
information pertinent to such research as proprietary
technologies or trade secrets. Enforcing a claim that another
person has illegally obtained and is using our trade secrets,
like patent litigation, is expensive and time consuming, and the
outcome is unpredictable. In addition, we may be subject to the
jurisdiction of courts outside the U.S., some of which may be
less willing to protect trade secrets.
Regulatory
approvals must be obtained and maintained for products
incorporating our technology and, if approvals are delayed or
withdrawn, we will be unable to commercialize these
products.
Government regulations in the United States and other countries
have a significant impact on our business and affect the
research, development and marketing of products incorporating
our technology. In the United States and other countries,
governmental agencies have the authority to regulate the
distribution, manufacture and sale of drugs. Failure to obtain
or experiencing a delay in obtaining regulatory approval for our
products could result in reduction of our expected revenues.
Failure to comply with applicable regulatory requirements can,
among other things, result in fines, suspension or withdrawal of
regulatory approvals, product recalls, operating restrictions
and/or
criminal prosecution. In addition, governmental regulations may
be established that could prevent, delay, modify or rescind
regulatory approval of our products.
If we
cannot keep pace with rapid technological change and meet the
intense competition in our industry, we may not
succeed.
Our success depends, in part, on achieving and maintaining a
competitive position in the development of products and
technologies in a rapidly evolving industry. If we are unable to
continue to develop
and/or
acquire competitive products and technologies, our current and
potential strategic partners may choose to adopt the drug
delivery technologies of our competitors. We also compete
generally with other drug delivery, biotechnology and
pharmaceutical companies engaged in the development of
alternative drug delivery technologies or new drug research and
testing. Many of these competitors have substantially greater
financial, technological, manufacturing, marketing, managerial
and research and development resources and experience than we do
and represent significant competition for us. Our competitors
may succeed in developing
27
competing technologies or obtaining governmental approval for
products before we achieve success, if at all. The products of
our competitors may gain market acceptance more rapidly than our
products. Developments by competitors may render our existing or
proposed products noncompetitive or obsolete.
The competitive position of our drug delivery technologies is
subject to the possible development by others of superior
technologies. Other drug delivery technologies, including oral
and injection methods, have wide acceptance, notwithstanding
certain drawbacks, and are the subject of improvement efforts by
other entities having greater resources. In addition, our drug
delivery technologies are limited by the number and commercial
magnitude of drugs with which they can successfully be combined.
We may
be unable to meet increasing expenses and demands on our
resources from future growth, if any, or to effectively pursue
additional business opportunities.
We have no current agreements or commitments with respect to any
acquisitions or investments. Any future acquisitions or
investments would further challenge our resources. If we do not
properly meet the increasing expenses and demands on our
resources from future growth, we will be adversely affected. To
properly manage our growth, we must, among other things, improve
and implement additional administrative, financial, marketing,
operational and research and development systems, procedures and
controls on a timely basis. While we currently do not have any
plans to hire additional personnel, in the future, we may need
to expand our staff in various areas of the business. We may not
be able to complete the improvements to our systems, procedures
and controls necessary to support our future operations in a
timely manner. We may not be able to hire, train, integrate,
retain, motivate and manage required personnel, successfully
integrate acquisitions or investments, nor successfully
identify, manage and pursue existing and potential market
opportunities. The Company has never been profitable and our
revenues are currently insufficient to generate a profit; if we
fail to generate additional revenue in excess of any increased
operating expenses in any fiscal period, we will continue to
incur losses.
If we
undertake an acquisition, we will incur a variety of costs, and
we may never realize the anticipated benefits of the
acquisition.
One of our strategies for business expansion is the acquisition
of additional technologies, products and product candidates. We
may attempt to acquire these product candidates, or other
potentially beneficial technologies, through the acquisition of
businesses, services or products that we believe are a strategic
fit with our business. Although we currently have no commitments
or agreements with respect to any acquisitions, if we undertake
an acquisition, the process of integrating the acquired
business, technology, service or product may result in
unforeseen operating difficulties and expenditures and may
divert significant management attention from our ongoing
business operations. Moreover, we may fail to realize the
anticipated benefits of any acquisition for a variety of reasons
such as an acquired technology or product candidate proving to
not be safe or effective in later clinical trials. We may fund
any future acquisition by issuing equity or debt securities,
which could dilute your ownership percentage or limit our
financial or operating flexibility as a result of restrictive
covenants related to new debt. Acquisition efforts can consume
significant management attention and require substantial
expenditures, which could detract from our other programs. In
addition, we may devote resources to potential acquisitions that
are never completed.
If we
cannot attract and retain key personnel, we may not be able to
execute our business plan as anticipated.
Our success is dependent on our ability to attract and retain
qualified, experienced personnel. We face significant
competition from other pharmaceutical companies in recruiting
competent personnel. The loss of key personnel, or the inability
to attract and retain additional, competent employees, could
adversely affect our business and financial results.
28
We may
incur substantial liabilities and may be required to limit
commercialization of our products in response to product
liability claims.
The testing and marketing of pharmaceutical products entails an
inherent risk of product liability. We may be held liable to the
extent that there are any adverse reactions from the use of our
products. Our products involve new methods of delivery for
drugs, some of which may require precautions to prevent
unintended use, especially since they are designed for
patients’ self-use rather than being administered by
medical professionals. The FDA may require us to develop a
comprehensive risk management program for our products. The
failure of these measures could result in harmful side effects
or death. As a result, consumers, regulatory agencies,
pharmaceutical companies or others might make claims against us.
If we cannot successfully defend ourselves against product
liability claims, we may incur substantial liabilities, lose
market share or be required to limit commercialization of our
products.
Regardless of merit or eventual outcome, liability claims may
result in:
|
|
|
| |
•
|
withdrawal of clinical trial participants;
|
| |
| |
•
|
termination of clinical trial sites or entire trial programs;
|
| |
| |
•
|
decreased demand for our product candidates;
|
| |
| |
•
|
impairment of our business reputation;
|
| |
| |
•
|
costs of related litigation;
|
| |
| |
•
|
substantial monetary awards to patients or other claimants;
|
| |
| |
•
|
loss of revenues; and
|
| |
| |
•
|
the inability to commercialize our product candidates.
|
Our inability to obtain sufficient product liability insurance
at an acceptable cost to protect against potential product
liability claims could inhibit or prevent the commercialization
of pharmaceutical products we develop alone or with corporate
collaborators. We maintain $5.0 million in product
liability and clinical trials insurance in the U.S. at an
approximate cost of $53,000 per policy year. While management
believes this insurance is reasonable for conducting clinical
trials, we cannot assure you that any of this coverage will be
adequate to protect us in the event of a claim. We, or any
corporate collaborators, may not be able to obtain or maintain
insurance at a reasonable cost, if at all. Even if our
agreements with any future corporate collaborators entitle us to
indemnification against losses, such indemnification may not be
available or adequate if any claim arises.
The
discovery of any new side effects or negative efficacy findings
for our products could significantly harm our
business.
While the safety of our products has been, is being, and will be
extensively studied in clinical trials there can be no assurance
that new or more serious side effects or negative efficacy
findings may not be discovered based on long term safety and
efficacy studies or required reporting of adverse events
regarding any of our products after each such product has been
marketed, any of which could severely harm our business and
result in one or more of the following regulatory events:
|
|
|
| |
•
|
a voluntary or involuntary recall or market withdrawal of the
applicable product;
|
| |
| |
•
|
labeling changes such as restriction on intended uses,
additional contraindications, warnings, precautions, or adverse
reactions that would limit the applicable product’s market
potential;
|
| |
| |
•
|
a “boxed” warning on the label;
|
| |
| |
•
|
imposition of post-marketing surveillance studies or risk
management programs;
|
| |
| |
•
|
distribution restrictions; and
|
| |
| |
•
|
adverse publicity.
|
29
In addition, one or more of the above factors would also have
the potential to negatively impact regulatory registrations for
the applicable product in other countries.
We
have a history of operating losses, expect to continue to have
losses in the future and may never achieve or maintain
profitability.
We have incurred operating losses since the Separation date and
we expect to continue to incur operating losses over the coming
years as we continue to incur significant costs for research and
development, clinical trials, sales and general and
administrative functions. Our ability to achieve profitability
depends upon our ability, alone or with others, to successfully
complete the development of Nasulin, obtain the required
regulatory clearances, and deliver Nasulin to market.
Our revenues, operating results and cash flows may
fluctuate in future periods and we may fail to meet investor
expectations, which may cause the price of our common stock to
decline.
Variations in our quarterly and year-end operating results are
difficult to predict and may fluctuate significantly from period
to period. If our sales or operating results fall below the
expectations of investors or securities analysts, the price of
our common stock could decline substantially. In addition to the
other factors discussed under this “Risk Factors”
section, specific factors that may cause fluctuations in our
operating results include:
|
|
|
| |
•
|
demand and pricing for our products;
|
| |
| |
•
|
government or private healthcare reimbursement policies;
|
| |
| |
•
|
physician, pharmacy and patient acceptance of any of our current
or future products;
|
| |
| |
•
|
patterns or cost structures for our products;
|
| |
| |
•
|
introduction of competing products;
|
| |
| |
•
|
any interruption in the manufacturing or distribution of Testim
or any of our future products;
|
| |
| |
•
|
our operating expenses which fluctuate due to growth of our
business;
|
| |
| |
•
|
timing and size of any new product or technology acquisitions we
may complete; and
|
| |
| |
•
|
variations in our rates of product returns and allowances.
|
We could be negatively affected as a result of a
threatened proxy fight and other actions of activist
shareholders.
We received notice from Arcadia Opportunity Master Fund, Ltd.
and its affiliates, Arcadia Capital Advisors, LLC and Richard
Rofé, collectively referred as Arcadia, of their intention
to nominate Mr. Rofé for election to our Board of
Directors at our 2010 annual stockholders meeting. As of
February 22, 2010, Arcadia owned beneficially 9.98% of our
outstanding common stock. If Arcadia carries through with its
intention and launches a proxy contest, our business could be
adversely affected because:
|
|
|
| |
•
|
responding to proxy contests and other actions by activist
stockholders can be costly and time-consuming, disrupting our
operations and diverting the attention of management and our
employees;
|
| |
| |
•
|
perceived uncertainties as to our future direction may impact
our existing or potential collaborations or strategic
relationships; and
|
| |
| |
•
|
if individuals are elected to our Board of Directors with a
specific agenda, potentially including Mr. Rofé, it
may adversely affect our ability to effectively and timely
implement our strategic plan.
|
Our
historical financial information is not necessarily
representative of the results we would have achieved as a
separate publicly traded company and may not be a reliable
indicator of our future results.
The historical financial information we have included in this
Form 10-K
may not reflect what our results of operations, financial
position and cash flows would have been had we been an
independent publicly traded
30
company during the periods presented or what our results of
operations, financial position and cash flows will be in the
future when we are an independent company. This is primarily
because:
|
|
|
| |
•
|
our historical financial information reflects allocations for
services historically provided to us by Bentley, which
allocations may not reflect the costs we will incur for similar
services in the future as an independent company; and
|
| |
| |
•
|
our historical financial information does not reflect changes
that we expect to incur in the future as a result of our
separation from Bentley, including changes in the cost
structure, personnel needs, financing and operations of the
contributed business as a result of the separation from Bentley
and from reduced economies of scale.
|
Since the Separation date, we have been responsible for the
additional costs associated with being an independent public
company, including costs related to corporate governance and
having listed and registered securities. Therefore, our
financial statements may not be indicative of our future
performance as an independent company. For additional
information about our past financial performance and the basis
of presentation of our financial statements, please see
“Management’s Discussion and Analysis of Financial
Condition and Results of Operations” and our financial
statements and the notes thereto included elsewhere in this
Form 10-K.
Risks
Relating to Our Common Stock
Your
percentage ownership in CPEX common stock may be diluted in the
future.
Your percentage ownership in CPEX may be diluted in the future
because of equity awards that have or we expect will be granted
to our directors, officers and employees and the accelerated
vesting of equity awards. Shareholders of CPEX have approved the
Amended and Restated 2008 Equity Incentive Plan (the “2008
Equity Incentive Plan”), which provides for the grant of
equity based awards, including restricted stock, restricted
stock units, stock options, stock appreciation rights,
unrestricted stock and stock equivalents and other equity-based
awards to our directors, officers and other employees, advisors
and consultants.
Provisions
in our certificate of incorporation and by-laws and of Delaware
law may prevent or delay an acquisition of our Company, which
could decrease the trading price of our common
stock.
Our certificate of incorporation, by-laws and Delaware law
contain provisions that are intended to deter coercive takeover
practices and inadequate takeover bids by making such practices
or bids unacceptably expensive to the raider and to encourage
prospective acquirors to negotiate with our Board of Directors
rather than to attempt a hostile takeover. These provisions
include, among others:
|
|
|
| |
•
|
a Board of Directors that is divided into three classes with
staggered terms;
|
| |
| |
•
|
elimination of the right of our stockholders to act by written
consent;
|
| |
| |
•
|
rules regarding how stockholders may present proposals or
nominate directors for election at stockholder meetings;
|
| |
| |
•
|
the right of our Board to issue preferred stock without
stockholder approval; and
|
| |
| |
•
|
limitations on the right of stockholders to remove directors.
|
Delaware law also imposes some restrictions on mergers and other
business combinations between us and any holder of 15% or more
of our outstanding common stock.
We also maintain a shareholder rights plan which may deter a
potential acquiror from pursuing an offer for our company.
We believe these provisions protect our stockholders from
coercive or otherwise unfair takeover tactics by requiring
potential acquirors to negotiate with our Board and by providing
our Board with more time to assess any acquisition proposal.
These provisions are not intended to make our company immune
from takeovers. However, these provisions apply even if the
offer may be considered beneficial by some stockholders and
31
could delay or prevent an acquisition that our Board determines
is not in the best interests of our company and our stockholders.
We do
not expect to pay any dividends in the short term.
We do not expect to declare dividends in the short term. We
currently intend to retain earnings to support our operations
and to finance the growth and development of our business. There
can be no assurance that we will have sufficient surplus under
Delaware law to be able to pay any dividends. This may result
from extraordinary cash expenses, actual expenses exceeding
contemplated costs funding of capital expenditures, or increases
in reserves. If we do not pay dividends, the price of our common
stock must appreciate for you to receive a gain on your
investment in CPEX. This appreciation may not occur.
|
|
|
Item 1B.
|
Unresolved
Staff Comments
|
Not applicable.
We own a 15,700 square foot commercial building situated on
approximately 14 acres of land in Exeter, New Hampshire
which has sufficient space and fixtures to serve as our
corporate headquarters and research and development laboratory.
It is located approximately 50 miles north of Boston,
Massachusetts.
|
|
|
Item 3.
|
Legal
Proceedings
|
In October 2008, we and Auxilium received notice that
Upsher-Smith Laboratories filed an Abbreviated New Drug
Application, or ANDA, containing a paragraph IV
certification in which it certified that it believes that its
testosterone gel product does not infringe our patent,
U.S. Patent No. 7,320,968 (“the ‘968
Patent”). The ‘968 patent covers a method for
maintaining effective blood serum testosterone levels for
treating a hypogonadal male using Testim and will expire in
January 2025. The ‘968 Patent is listed in Approved Drug
Products with Therapeutic Equivalence Evaluations (commonly
known as the Orange Book), published by the U.S. Food and
Drug Administration. Upsher-Smith Laboratories’
paragraph IV certification sets forth allegations that the
‘968 Patent will not be infringed by Upsher-Smith’s
manufacture, use or sale of the product for which the ANDA was
submitted. On December 4, 2008, we and Auxilium filed a
lawsuit in the United States District Court for the District of
Delaware against Upsher-Smith under the Hatch Waxman Act for
infringement of our patent. In June 2009, Upsher-Smith amended
its answer to the complaint to include a defense and
counterclaim of invalidity of the ‘968 Patent, which CPEX
and Auxilium deny. A patent issued by the U.S. Patent and
Trademark Office, such as the ’968 Patent, is presumed
valid. The lawsuit is currently ongoing. Any U.S. Food and
Drug Administration (FDA) approval of Upsher-Smith’s
proposed generic product will be stayed until the earlier of
30 months from the date of receipt of the paragraph IV
certification (April 2011) or an adverse decision in
our patent infringement lawsuit.
CPEX has filed continuation and divisional applications with the
USPTO relating to the ‘968 patent. Six patents issued from
these applications on October 27, 2009, namely
U.S. Patent Nos. 7,608,605, 7,608,606, 7,608,607,
7,608,608, 7,608,609 and 7,608,610, which may provide us with
further market protection. Each of these six patents has been
listed in listed in Approved Drug Products with Therapeutic
Equivalence Evaluations (commonly known as the Orange Book) with
respect to Testim.
We and Auxilium are committed to protecting our intellectual
property rights and will vigorously pursue this lawsuit.
However, if we are unsuccessful in defending the ‘968
Patent covering Testim, sales of Testim and our royalties
relating to Testim sales will be materially reduced.
|
|
|
Item 4.
|
(Removed
and Reserved)
|
32
Part II
|
|
|
Item 5.
|
Market
for Registrant’s Common Equity, Related Stockholder Matters
and Issuer Purchases of Equity Securities
|
Our common stock has been trading on The NASDAQ Capital Market
since July 1, 2008, under the trading symbol
“CPEX”. The following table sets forth, for the
periods indicated, the range of quarterly high and low sales
prices for our common stock as reported on The NASDAQ Capital
Market since the stock was first traded;
| |
|
|
|
|
|
|
|
|
|
|
|
High
|
|
Low
|
|
|
|
Fiscal Year Ended December 31, 2009
|
|
|
|
|
|
|
|
|
|
First Quarter
|
|
$
|
12.58
|
|
|
$
|
5.90
|
|
|
Second Quarter
|
|
|
11.54
|
|
|
|
6.76
|
|
|
Third Quarter
|
|
|
11.00
|
|
|
|
8.23
|
|
|
Fourth Quarter
|
|
|
12.75
|
|
|
|
8.00
|
|
|
Fiscal Year Ended December 31, 2008
|
|
|
|
|
|
|
|
|
|
Third Quarter
|
|
$
|
19.98
|
|
|
$
|
9.17
|
|
|
Fourth Quarter
|
|
|
19.77
|
|
|
|
7.49
|
|
As of March 24, 2010 the closing price of our Common Stock
was $14.86 and there were 601 holders of record of our common
stock, which does not reflect stockholders whose shares are held
in street name.
Dividends
We did not pay dividends on our common stock during the years
ended December 31, 2009 and 2008 and we do not intend to
pay dividends in the foreseeable future. We intend to retain
future earnings in order to finance the growth and development
of our business.
Securities
Authorized for Issuance Under Equity Compensation
Plans
The following table summarizes the number of securities issuable
under our Amended and Restated 2008 Equity Incentive Plan as of
December 31, 2009.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Number of Securities
|
|
|
|
|
|
|
|
|
|
|
Remaining Available
|
|
|
|
|
|
|
|
|
|
|
for Future Issuance
|
|
|
|
|
|
|
|
|
|
|
Under Equity
|
|
|
|
|
|
|
|
|
|
|
Compensation Plans
|
|
|
|
|
Number of Securities to
|
|
|
Weighted Average
|
|
|
(Excluding
|
|
|
|
|
be Issued Upon Exercise
|
|
|
Exercise Price of
|
|
|
Securities
|
|
|
|
|
of Outstanding Options,
|
|
|
Outstanding Options,
|
|
|
Reflected in the
|
|
|
Plan Category
|
|
Warrants and Rights
|
|
|
Warrants and Rights
|
|
|
First Column)
|
|
|
|
|
Equity compensation plans approved by security holders
|
|
|
509,925
|
|
|
$
|
13.50
|
|
|
|
90,741
|
|
|
Equity compensation plans not approved by security holders
|
|
|
None
|
|
|
|
N/A
|
|
|
|
None
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
|
509,925
|
|
|
$
|
13.50
|
|
|
|
90,741
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Item 6.
|
Selected
Financial Data
|
Not applicable.
|
|
|
Item 7.
|
Management’s
Discussion and Analysis of Financial Condition and Results of
Operations
|
The following discussion and analysis should be read in
conjunction with the Financial Statements and related Notes to
the Condensed Combined and Consolidated Financial Statements, or
Notes, included in Item 15 of this Annual Report. Except
for the historical information contained herein the foregoing
discussion
33
contains forward-looking statements that involve risks and
uncertainties. Our actual results could differ materially from
those projected in the forward-looking statements discussed
herein.
Words such as “expect”, “anticipate”,
“intend”, “believe”, “may”,
“could”, “project”, “estimate” and
similar words are used to identify forward-looking statements
within the meaning of the Private Securities Litigation Reform
Act of 1995. These forward-looking statements, including, but
not limited to, the statements in “Business”,
“Legal Proceedings”, “Management’s
Discussion and Analysis of Financial Condition and Results of
Operations”, “Risk Factors” and other sections in
this Annual Report, are not based on historical facts, but
rather reflect our current expectations concerning future
results and events. The forward-looking statements include
statements about our strategy, the prospects of our technologies
and research and development efforts, our plans to enter into
more collaborative relationships, the prospects for clinical
development of our product candidates, our prospects for revenue
growth, anticipated financial results and the prospects for
growth of our business. Although we believe that the
expectations reflected in the forward-looking statements are
reasonable, such statements involve known and unknown risks,
uncertainties and other factors that may cause our actual
results, performance or achievements to be different from any
future results, performance and achievements expressed or
implied by these statements, including the risks outlined in the
Risk Factors section and elsewhere in this report. You are
cautioned not to place undue reliance on these forward-looking
statements. We undertake no obligation to publicly update or
revise any forward-looking statements, whether as the result of
new information, future events or otherwise, except as may be
required by law.
Overview
We are an emerging specialty pharmaceutical company that employs
17 people as of March 24, 2010, at our principal
executive offices in Exeter, New Hampshire. Our business is the
research, development, licensing and commercialization of
pharmaceutical products utilizing our validated drug delivery
platform technology. We have U.S. and international patents
and other proprietary rights to technology that facilitates the
absorption of drugs. Our platform drug delivery technology
enhances permeation and absorption of pharmaceutical molecules
across the skin, nasal mucosa and eye through formulation
development with proprietary molecules such as CPE-215. Our
first product is
Testim®,
a gel for testosterone replacement therapy, which is a
formulation of CPE-215 with testosterone. Testim is licensed to
Auxilium Pharmaceuticals, Inc. who is currently marketing the
product in the United States, Europe and other countries. Our
second product,
Nasulintm,
currently in Phase 2, is an intranasal spray formulation of
CPE-215 with insulin being developed to treat hyperglycemia in
patients suffering from Type 1 and Type 2 diabetes.
We believe, based upon our experience with Testim and Nasulin,
that our CPE-215 technology is a broad platform technology that
has the ability to significantly enhance the permeation of a
wide range of therapeutic molecules. To expand the development
and commercialization of products using our CPE-215 drug
delivery technology, we are pursuing strategic alliances with
partners including large pharmaceutical, specialty
pharmaceutical and biotechnology companies. The alliance
opportunities may include co-development of products,
in-licensing of therapeutic molecules, out-licensing of delivery
technology or partnering late-stage candidates for
commercialization.
Separation
from Bentley
Our business was initially the drug delivery business of Bentley
Pharmaceuticals, Inc. (referred to as “Bentley”) which
Bentley spun-off in June 2008 in connection with the sale of
Bentley’s remaining businesses. Shares of our stock were
distributed to Bentley stockholders after the close of business
on June 30, 2008 (the “Separation Date”) by means
of a stock dividend, a transaction that was taxable to Bentley
and Bentley’s stockholders (the “Separation”).
Each Bentley stockholder of record on June 20, 2008, the
record date, received on the Separation Date one CPEX share for
every ten shares of Bentley common stock. Bentley has no
ownership interest in CPEX subsequent to the Separation.
Our financial statements for the twelve months ended
December 31, 2009 and the balance sheet as of
December 31, 2008 represent stand-alone financial
information for our Company. The statements of operations,
34
statements of changes in stockholders’ equity and
statements of cash flows for the years ended December 31,
2008, which include six months of financial data prior to the
Separation Date, and all of 2007 reflect the historical
financial position, results of operations and cash flows of the
business transferred to us from Bentley as part of the
Separation. Our financial statements have been prepared and are
presented as if we had been operating as a separate entity using
the historical cost basis of the assets and liabilities of
Bentley and including the historical operations of the business
transferred to us from Bentley as part of the Separation. Prior
to the Separation Date, we were fully integrated with Bentley
and the accompanying financial statements reflect the
application of certain estimates and allocations. Our statements
of operations include all revenues and costs that are directly
attributable to our business. In addition, certain expenses of
Bentley have been allocated to us using various assumptions
that, in the opinion of management, are reasonable. These
expenses include an allocated share of executive compensation,
public company costs and other administrative costs. The
allocated costs totaled $4.4 million and $4.2 million
for the years ended December 31, 2008 and 2007,
respectively. There have been no allocations of expenses charged
to us since the Separation Date. The financial information
included herein may not necessarily reflect our financial
position, results of operations and cash flows in the future or
what our financial position, results of operations and cash
flows would have been had we been a stand-alone company during
the periods presented.
Consolidated
Results of Operations
The following is a discussion of the results of our operations
for the years ended December 31, 2009, 2008 and 2007.
Included in the financial disclosures are direct costs
associated with our business and certain allocated costs from
Bentley related to executive compensation, public company costs
and other administrative costs for periods before June 30,
2008. As these costs only represent an allocation of the costs
incurred by Bentley before the Separation, they are not
necessarily indicative of the costs that would have been
incurred if we were an independent public company in the periods
presented. Inflation and changing prices have not had a material
impact on our revenues or loss from operations in the three
years ended December 31, 2009.
Fiscal
Year Ended December 31, 2009 Compared To Fiscal Year Ended
December 31, 2008
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended
|
|
|
|
|
|
|
|
December 31,
|
|
|
Increase (Decrease)
|
|
|
|
|
2009
|
|
|
2008
|
|
|
$
|
|
|
%
|
|
|
|
|
|
|
|
(In thousands)
|
|
|
|
|
|
|
|
Royalties and other revenue
|
|
$
|
18,658
|
|
|
$
|
15,574
|
|
|
$
|
3,084
|
|
|
|
20
|
%
|
|
Operating expenses:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
General and administrative
|
|
|
8,867
|
|
|
|
6,493
|
|
|
|
2,374
|
|
|
|
37
|
%
|
|
Research and development
|
|
|
12,291
|
|
|
|
9,119
|
|
|
|
3,172
|
|
|
|
35
|
%
|
|
Separation costs
|
|
|
—
|
|
|
|
2,502
|
|
|
|
(2,502
|
)
|
|
|
(100
|
)%
|
|
Depreciation and amortization
|
|
|
699
|
|
|
|
682
|
|
|
|
17
|
|
|
|
2
|
%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses
|
|
|
21,857
|
|
|
|
18,796
|
|
|
|
3,061
|
|
|
|
16
|
%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss from operations
|
|
|
(3,199
|
)
|
|
|
(3,222
|
)
|
|
|
23
|
|
|
|
(1
|
)%
|
|
Other, net
|
|
|
159
|
|
|
|
307
|
|
|
|
(148
|
)
|
|
|
(48
|
)%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
$
|
(3,040
|
)
|
|
$
|
(2,915
|
)
|
|
$
|
(125
|
)
|
|
|
4
|
%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Royalties and other revenue increased 20% to $18.7 million
in 2009 from $15.6 million in 2008 due entirely to
increased royalties earned on sales of Testim. For the year
ended December 31, 2009, Testim prescriptions were reported
to have grown approximately 15% compared to the same period in
2008. The long-term prospects for Testim sales are subject to
resolution of our patent infringement suit against
Upsher-Smith,
which has made an ANDA filing for a generic version of Testim,
as described above in “Legal Proceedings”. Clinical
and other revenue was $16,000 in 2009 compared to $513,000 in
2008 which includes revenue from our development and license
agreement with Serenity Pharmaceuticals, Inc. which we signed in
2008.
35
General and administrative costs increased 37% to
$8.9 million in 2009 compared to $6.5 million in 2008,
primarily due to a $2.8 million increase in legal fees,
mostly relating to the patent infringement suit against
Upsher-Smith Laboratories, which were partially offset by a
$674,000 decrease in share-based compensation expense. Included
in general and administrative expenses for the year ended
December 31, 2008 is a one-time, non-cash charge of
approximately $980,000 resulting from the modification of equity
awards associated with the spin-off from Bentley.
Research and development expenses consist primarily of costs
associated with the development of Nasulin, our lead product
candidate in development. These costs include costs of clinical
trials, manufacturing supplies and other development materials,
compensation and related benefits for research and development
personnel, costs for consultants, and various overhead costs.
Research and development costs are expensed as incurred.
Research and development costs increased to $12.3 million
in 2009 compared to $9.1 million in 2008, mostly due to a
$3.7 million increase in spending on clinical trials, which
was partially offset by a reduction in non-cash share-based
compensation expense of $494,000. The increase in clinical
trials expense is primarily attributable to the Phase 2a study
of Nasulin.
We expect our research and development costs in 2010 to be
between $10 million and $12 million. We completed
enrollment in a Phase 2a trial in the U.S. in 2009 and
preliminary results from this trial were reported in March,
2010. Our expectation was to initiate a Phase 2b trial of
Nasulin in the U.S. in 2010 while continuing the
development of products in our pipeline. However, as explained
above in “Business — Overview”, we
will not commence our planned Phase 2b trial or other Nasulin
development initiatives while we conduct additional analyses on
the data from the Phase 2a study together with all other Nasulin
data to determine the appropriate next steps for the Nasulin
program. Additional expenses for the full development of Nasulin
cannot be estimated at this time. The risks and uncertainties
associated with the planning and execution of a clinical
development program includes, among other things, uncertainties
about results that at any time could require us to abandon or
greatly modify the program. Accordingly, we cannot estimate the
period in which material net cash inflows from Nasulin might
commence, if ever.
Separation costs, consisting of legal, tax and other strategic
consulting costs specifically related to the separation from
Bentley explained above, were $2.5 million in 2008. No
additional separation costs have been incurred since the
Separation Date and we do not expect to incur any additional
separation costs in the future.
Fiscal
Year Ended December 31, 2008 Compared To Fiscal Year Ended
December 31, 2007
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended
|
|
|
|
|
|
|
|
December 31,
|
|
|
Increase (Decrease)
|
|
|
|
|
2008
|
|
|
2007
|
|
|
$
|
|
|
%
|
|
|
|
|
|
|
|
(In thousands)
|
|
|
|
|
|
|
|
Royalties and other revenue
|
|
$
|
15,574
|
|
|
$
|
11,127
|
|
|
$
|
4,447
|
|
|
|
40
|
%
|
|
Operating expenses:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
General and administrative
|
|
|
6,493
|
|
|
|
5,206
|
|
|
|
1,287
|
|
|
|
25
|
%
|
|
Research and development
|
|
|
9,119
|
|
|
|
9,646
|
|
|
|
(527
|
)
|
|
|
(5
|
)%
|
|
Separation costs
|
|
|
2,502
|
|
|
|
1,010
|
|
|
|
1,492
|
|
|
|
148
|
%
|
|
Depreciation and amortization
|
|
|
682
|
|
|
|
752
|
|
|
|
(70
|
)
|
|
|
(9
|
)%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses:
|
|
|
18,796
|
|
|
|
16,614
|
|
|
|
2,182
|
|
|
|
13
|
%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss from operations
|
|
|
(3,222
|
)
|
|
|
(5,487
|
)
|
|
|
2,265
|
|
|
|
(41
|
)%
|
|
Other, net
|
|
|
307
|
|
|
|
559
|
|
|
|
(252
|
)
|
|
|
(45
|
)%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
$
|
(2,915
|
)
|
|
$
|
(4,928
|
)
|
|
$
|
2,013
|
|
|
|
(41
|
)%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Royalties and other revenue increased 40% to $15.6 million
in 2008 from $11.1 million in 2007 due primarily to
increased royalties earned on sales of Testim. For the year
ended December 31, 2008, Testim prescriptions were reported
to have grown approximately 27% compared to the same period in
2007. In addition, it is also reported that Testim’s market
share of the testosterone replacement gel market in December
36
2008 had increased to more than 22% versus approximately 21% in
December 2007. Clinical and other revenue was $513,000 in 2008
which includes revenue from our development and license
agreement with Serenity Pharmaceuticals, Inc. which we signed in
2008 and for which there is no comparable revenue in 2007.
General and administrative costs increased 25% to
$6.5 million in 2008 compared to $5.2 million in 2007,
primarily due to a non-cash charge of approximately $980,000
resulting from the modification of equity awards associated with
the spin-off from Bentley.
Research and development expenses consist primarily of costs
associated with the development of Nasulin, our lead product
candidate. These costs include costs of clinical trials,
manufacturing supplies and other development materials,
compensation and related benefits for research and development
personnel, costs for consultants, and various overhead costs.
Research and development costs are expensed as incurred.
Research and development costs decreased to $9.1 million in
2008 compared to $9.6 million in 2007 due mostly to the
timing of our pre-clinical and clinical activities. Spending on
clinical trials was $1.5 million in 2008 compared to
$2.1 million in 2007.
Separation costs, consisting of legal, tax and other strategic
consulting costs specifically related to the separation from
Bentley explained above, were $2.5 million in 2008 compared
to $1.0 million in 2007.
Liquidity
and Capital Resources
Overview
We had approximately $13.7 million in cash and cash
equivalents at December 31, 2009, which, along with Testim
royalties, we believe will be sufficient to fund our operations
and our cash requirements for at least the next twelve months.
Our cash includes balances maintained in commercial bank
accounts, amounts invested in overnight sweep investments and
cash deposits in money market accounts. Although cost estimates
and timing of our activities are subject to change, we expect
research and development expenses for 2010 to range between
$10 million and $12 million. There can be no assurance
that changes in our research and development plans or other
events affecting our revenues or operating expenses will not
result in the earlier depletion of our funds. However, we will
continue to explore alternative sources for financing our
business activities. In appropriate situations, which will be
strategically determined, we may seek funding from other
sources, including, but not limited to, contribution by others
to joint ventures and other collaborative or licensing
arrangements for the development, testing, manufacturing and
marketing of Nasulin and other products currently under
development or sales of debt or equity securities.
Summary
Cash Flow Information
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
2009
|
|
2008
|
|
2007
|
|
|
|
(In thousands)
|
|
|
|
Summary Financial Position
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents
|
|
$
|
13,695
|
|
|
$
|
15,211
|
|
|
$
|
21,659
|
|
|
Accounts receivable
|
|
|
5,289
|
|
|
|
4,445
|
|
|
|
3,245
|
|
|
Total assets
|
|
|
26,043
|
|
|
|
26,473
|
|
|
|
32,397
|
|
|
Accounts payable and accrued expenses
|
|
|
3,007
|
|
|
|
2,630
|
|
|
|
3,221
|
|
|
Working capital
|
|
|
16,570
|
|
|
|
17,609
|
|
|
|
22,365
|
|
|
Total stockholders’ equity
|
|
|
23,036
|
|
|
|
23,843
|
|
|
|
29,151
|
|
37
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
2009
|
|
2008
|
|
2007
|
|
|
|
Summary of Sources and (Uses) of Cash:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating activities
|
|
$
|
(598
|
)
|
|
$
|
(553
|
)
|
|
$
|
(2,186
|
)
|
|
Investing activities
|
|
|
(922
|
)
|
|
|
(307
|
)
|
|
|
(430
|
)
|
|
Purchases of property, plant and equipment
|
|
|
(398
|
)
|
|
|
(307
|
)
|
|
|
(303
|
)
|
|
Additions to intangible assets
|
|
|
(224
|
)
|
|
|
—
|
|
|
|
(157
|
)
|
|
Financing activities
|
|
|
4
|
|
|
|
(5,588
|
)
|
|
|
13,523
|
|
Sources
and Uses of Cash
Operating
Activities
Net cash used in operating activities was $598,000 for the year
ended December 31, 2009, largely resulting from the net
loss of $3.0 million and an increase in accounts receivable
of $844,000, which were partially offset by non-cash share-based
compensation expense of $1.9 million, increases in accounts
payable and accrued expenses of $670,000 and depreciation and
amortization of $699,000. Net cash used in operating activities
was $553,000 for the year ended December 31, 2008, largely
resulting from the net loss of $2.9 million, an increase in
accounts receivable of $1.2 million and a reduction in
accounts payable and accrued expenses of $591,000, which were
partially offset by non-cash share-based compensation of
$3.2 million and depreciation and amortization of $682,000.
Net cash used in 2007 was $2.2 million resulting from the
net loss of $4.9 million and an increase in accounts
receivable of $1.0 million, which were partially offset by
non-cash share-based compensation expense of $1.5 million,
an increase in accounts payable and accrued expenses of
$1.1 million and depreciation and amortization expenses of
$752,000.
Investing
Activities
Net cash used in investing activities was $922,000 for the year
ended December 31, 2009 which includes $398,000 for the
purchase of manufacturing and laboratory equipment, $300,000 for
the note receivable explained in the notes to our consolidated
and combined financial statements under Commitments,
contingencies and concentrations, and $224,000 for costs
related to obtaining new patents. Net cash used in investing
activities was $307,000 for the year ended December 31,
2008 due to the purchase of necessary equipment to
scale-up our
manufacturing capabilities. Net cash used in the twelve months
ended December 31, 2007 was $430,000, which includes
$303,000 for laboratory expansion and equipment and $157,000 for
costs to acquire intellectual property rights. We expect to
invest approximately $220,000 in capital expenditures in 2010,
primarily for research and development equipment.
Financing
Activities
Net cash provided by financing activities for the year ended
December 31, 2009 includes proceeds from the exercise of
stock options. Net cash used by financing activities was
$5.6 million for the year ended December 31, 2008 due
largely to the change in Bentley’s net investment in our
business of $7.3 million, which was partially offset by
proceeds of $1.7 million from the exercise of stock
options. Financing activities for 2007 reflect the net change in
Bentley’s net investment in our business, consisting
primarily of the funding of our net loss and other operating and
investing activities for CPEX. Additionally, the change in
Bentley’s net investment included a cash transfer of
$5.5 million to us from Bentley.
Off-Balance
Sheet Arrangements
We do not have any material off-balance sheet arrangements, as
defined in Item 303(a)(4)(ii) of SEC
Regulation S-K.
38
Critical
Accounting Policies and Estimates
Certain of our accounting policies are particularly important to
the portrayal of our financial position, results of operations
and cash flows and require the application of significant
judgment by our management. As a result they are subject to an
inherent degree of uncertainty. In applying those policies, our
management uses judgment to determine the appropriate
assumptions to be used in the determination of certain
estimates. Those estimates are based on our historical
experience, terms of existing contracts, our observance of
trends in the industry, information provided by our customers
and information available from other outside sources, as
appropriate. Our critical accounting policies and estimates
include:
Revenue
recognition and accounts receivable
We earn royalty revenues on Auxilium’s sales of Testim,
which incorporates our CPE-215 permeation enhancement
technology. Since 2003, Auxilium has sold Testim to
pharmaceutical wholesalers and chain drug stores. We recognize
revenue upon receiving sales reports from Auxilium which
includes estimates for revenue deductions, including discounts,
rebates and product returns. Estimates related to revenue
deductions are predominately based on historical experience.
Accounts receivable are also recorded upon the receipt of sales
reports from Auxilium at their net realizable value. Receivable
balances are reported net of an estimated allowance for
uncollectible accounts. Estimated uncollectible receivables are
based on the amount and status of past due accounts, contractual
terms with customers, the credit worthiness of customers and the
history of our uncollectible accounts.
Intellectual
property costs
Costs incurred in connection with acquiring licenses, patents
and other proprietary rights are capitalized. Capitalized costs
are amortized on a straight-line basis for periods not exceeding
15 years from the dates of acquisition. Carrying values of
such assets are reviewed at least annually by comparing the
carrying amounts to their estimated undiscounted cash flows and
adjustments are made for any diminution in value.
Research
and development costs
Research and development expenses consist primarily of costs
associated with our clinical trials, manufacturing supplies and
other development materials, compensation and related benefits
for research and development personnel, costs for consultants,
and various overhead costs. Research and development costs are
expensed as incurred consistent with Financial Accounting
Standards Board (the “FASB”) Accounting Standards
Codification (“FASB ASC”)
730-10-25-1
(Prior authoritative literature: SFAS No. 2,
Accounting for Research and Development Costs). Our clinical
trial costs, which are reflected in research and development
expenses, result from obligations under contracts with vendors,
consultants, and clinical site agreements in connection with
conducting clinical trials. The financial terms of these
contracts are subject to negotiations which vary from contract
to contract and may result in cash flows which are not
consistent with the periods in which materials or services are
provided. These costs are capitalized upon payment and expensed
according to the progress of each trial as measured by patient
progression and the timing of various aspects of the trial. The
progress of the trials is obtained through discussions with
internal personnel as well as outside service providers.
Determining the timing and level of services performed often
requires judgment.
Share-based
compensation
Certain former Bentley employees who became our employees
following the separation from Bentley held equity compensation
awards from Bentley. Share-based compensation expense for our
business was allocated based on Bentley’s consolidated
expense related to our employees and certain allocated
share-based compensation expense allocated from Bentley. Such
expense was accounted for in accordance with the fair value
recognition provisions of FASB ASC Topic 718 (Prior
authoritative literature: SFAS No. 123(R), Share-Based
Payment). Under the fair value recognition provisions of
FASB ASC 718, share-based compensation cost is measured at the
grant date based on the fair value of the award and is
recognized as expense over the requisite service period.
Determining the fair value of equity awards at the grant date
requires judgment. We
39
estimate the grant date fair value of stock options using the
Black-Scholes option valuation model. This option valuation
model requires the input of subjective assumptions including:
(1) Expected life — the expected life (estimated
period of time outstanding) of options granted is estimated
based on historical exercise behaviors, including the periods
prior to the separation when we were the drug delivery business
of Bentley; (2) Volatility — the volatility
applied to grants is the average stock volatility of CPEX and a
peer group of comparable life science companies using daily
price observations for each company over a period of time
commensurate with the expected life of the respective award;
(3) Risk-free rate — the risk-free interest rate
is based on the yield curve of U.S. Treasury securities in
effect at the date of the grant, having a duration commensurate
with the estimated life of the award; and
(4) Dividends — as we have not declared
dividends, and we do not expect to declare dividends in the
future, we include an annual dividend rate of 0% when
calculating the grant date fair value of equity awards. Because
share-based compensation expense is based on awards ultimately
expected to vest, it is reduced for estimated forfeitures. FASB
ASC 718 requires forfeitures to be estimated at the time of
grant and revised, if necessary, in subsequent periods if actual
forfeitures differ from those estimates. Forfeitures are
estimated based on historical experience.
Provision
for income taxes
We have provided for current and deferred U.S. federal,
state and foreign income taxes for the current and all prior
periods presented. Current and deferred income taxes have been
provided with respect to jurisdictions where our subsidiary
produces taxable income. We have provided a valuation allowance
with respect to the remainder of our deferred income taxes,
consisting primarily of net operating loss carryforwards in the
U.S. and Ireland, because of uncertainty regarding their
realization. The provision for income taxes for 2007 has been
determined on a pro-forma basis as if CPEX had filed separate
tax returns under its current structure for the periods
presented.
Should we determine that it is more likely than not that we will
realize certain of our net deferred tax assets for which we have
previously provided a valuation allowance, an adjustment would
be required to reduce the existing valuation allowance. In
addition, we operate within multiple taxing jurisdictions and
are subject to audit in those jurisdictions. These audits can
involve complex issues, which may require an extended period of
time for resolution. Although we believe that adequate
consideration has been made for such issues, there is the
possibility that the ultimate resolution of such issues could
have an adverse effect on our financial position, results of
operations or cash flows.
Effective January 1, 2007, we account for uncertain tax
positions in accordance with FASB ASC Topic 740 (Prior
authoritative literature: Financial Accounting Standards Board
Interpretation No. 48, Accounting for Uncertainty in Income
Taxes, an interpretation of FASB Statement No. 109,
Accounting for Income Taxes). The application of income tax
law is inherently complex. As such, we are required to make many
subjective assumptions and judgments regarding our income tax
exposures. Interpretations and guidance surrounding income tax
laws and regulations change frequently. Changes in our
subjective assumptions and judgments could have a material
effect on our financial position, results of operations or cash
flows. In addition, as we operate within multiple taxing
jurisdictions, we are subject to audit in those jurisdictions.
The ultimate resolution of tax audits may require an extended
period of time. Although we believe an adequate provision has
been made for uncertain tax positions, there is the possibility
that the ultimate resolution of such positions could have an
adverse effect on our financial position, results of operations
or cash flows.
Recently
Issued Accounting Pronouncements
In September 2009, the Emerging Issues Task Force
(“EITF”) issued its final consensus for ASU
2009-13
(Prior authoritative literature:
EITF 08-1
Revenue Arrangements with Multiple Deliverables), which will
supersede the guidance in ASC
605-25
(prior authoritative literature:
EITF 00-21,
Revenue Arrangements with Multiple Deliverables). ASU
2009-13
retains the criteria from ASC
605-5 for
when delivered items in a multiple-deliverable arrangement
should be considered separate units of accounting, but modifies
the previous separation criterion under ASC
605-25 that
objective and reliable evidence of fair value of any undelivered
items must exist for the delivered items to be considered a
separate unit or separate units of accounting. ASU
2009-13
introduces a selling price hierarchy for multiple deliverable
arrangements and allows for management selling price estimates
in cases where no vendor specific objective evidence or third
party evidence is
40
available. Additionally, this guidance eliminates the residual
method of allocation. ASU
2009-13 will
be applicable to the Company on January 1, 2011. The
Company has not yet evaluated the impact, if any, that ASU
2009-13 will
have on its financial statements.
In June 2009, the FASB issued ASC Topic 810 (Prior
authoritative literature: SFAS No. 167
Amendments to FASB Interpretation No. 46(R)),which
improves financial reporting by enterprises involved with
variable interest entities. ASC 810 addresses (1) the
effects on certain provisions of FASB Interpretation No. 46
(revised December 2003), Consolidation of Variable Interest
Entities, as a result of the elimination of the qualifying
special-purpose entity concept in SFAS 166 and
(2) concerns about the application of certain key
provisions of FIN 46(R), including those in which the
accounting and disclosures under the Interpretation do not
always provide timely and useful information about an
enterprise’s involvement in a variable interest entity. ASC
810 will be applicable to the Company on January 1, 2010.
Earlier adoption is prohibited. The Company does not expect the
adoption of this topic to have a material impact on its
consolidated and combined financial statements.
In June 2009, the FASB issued ASC Topic 860 (Prior
authoritative literature: SFAS No. 166,
Accounting for Transfers of Financial Assets, an
amendment of FASB Statement No. 140). ASC Topic 860
prescribes the information that a reporting entity must provide
in its financial reports about a transfer of financial assets;
the effects of a transfer on its financial position, financial
performance, and cash flows; and a transferor’s continuing
involvement in transferred financial assets. Specifically, among
other aspects, ASC Topic 860 amends SFAS No. 140,
Accounting for Transfers and Servicing of Financial Assets
and Extinguishments of Liabilities, or
SFAS No. 140, by removing the concept of a qualifying
special-purpose entity from SFAS No. 140 and removes
the exception from applying FASB Interpretation No. 46,
Consolidation of Variable Interest Entities (revised), or
FIN 46(R), to variable interest entities that are
qualifying special-purpose entities. It also modifies the
financial-components approach used in SFAS No. 140.
ASC Topic 860 is effective for transfer of financial assets
occurring on or after January 1, 2010. The Company does not
expect the adoption of this topic to have a material impact on
its consolidated and combined financial statements.
Effective July 1, 2009, the FASB issued ASC Topic 105
(Prior authoritative literature:
SFAS No. 168, The FASB Accounting Standards
Codification and the Hierarchy of Generally Accepted Accounting
Principles). ASC Topic 105 identifies the FASB Accounting
Standards Codification as the authoritative source of Accounting
Principles Generally Accepted in the United States (”
U.S. GAAP”). Rules and interpretive releases of the
SEC under federal securities laws are also sources of
authoritative U.S. GAAP for SEC registrants. ASC Topic 105
became effective for financial statements issued for interim
reporting periods ending after September 15, 2009.
Therefore, beginning with this
Form 10-Q,
all references made to U.S. GAAP in the notes to the
Company’s consolidated financial statements now use the new
ASC numbering system. The ASC does not change or alter existing
U.S. GAAP and, therefore, it does not have an impact on the
Company’s financial position, results of operations or cash
flows.
In May 2009, FASB issued FASB ASC Topic 855 (Prior
authoritative literature: SFAS No. 165,
Subsequent Events). ASC Topic 855 establishes general
standards of accounting for and disclosure of events that occur
after the balance sheet date but before financial statements are
issued or available to be issued. ASC
855-10-25
requires an entity to recognize in the financial statements the
effects of all subsequent events that provide additional
evidence about conditions that existed at the date of the
balance sheet. For unrecognized subsequent events that must be
disclosed to keep the financial statements from being
misleading, an entity will be required to disclose the nature of
the event as well as an estimate of its financial effect, or a
statement that such an estimate cannot be made. ASC 855 is
effective for the interim or annual financial periods ending
after June 15, 2009, and is required to be applied
prospectively. This guidance does not impact the Company’s
current consolidated and combined financial statements.
In February 2008, the FASB issued ASC
820-10-65-1
(Prior authoritative literature: FASB Staff Position
157-2,
“Effective Date of FASB Statement No. 157”),
which delays the effective date of ASC Topic 820 for
nonfinancial assets and nonfinancial liabilities, except for
items that are recognized or disclosed at fair value in the
financial statements on a reoccurring basis. The provisions of
ASC 820 became effective for fiscal years
41
beginning after November 15, 2008. The adoption of this
staff position did not have a material impact on the
Company’s consolidated and combined financial statements.
In April 2009, the FASB issued ASC
820-10-65-4
(Prior authoritative literature: FASB Staff Position
SFAS 157-4,
“Determining Fair Value When the Volume and Level of
Activity for the Asset or Liability Have Significantly Decreased
and Identifying Transactions That Are Not Orderly”).
ASC
820-10-65-4
amends ASC
820-35-15A
to provide additional guidance on estimating fair value when the
volume and level of activity for an asset or liability have
significantly decreased in relation to normal market activity
for the asset or liability. ASC
820-10-65-4
is effective for interim reporting periods ending after
June 15, 2009. The adoption of this staff position did not
have a material impact on the Company’s consolidated and
combined financial statements.
In December 2007, the FASB issued ASC Topic 805 (Prior
authoritative literature: SFAS No. 141(R),
Business Combinations
(“SFAS No. 141(R)”). ASC
805-20-25-2
and 25-3
establish principles and requirements for how an acquirer
recognizes and measures in its financial statements the
identifiable assets acquired, the liabilities assumed, any
non-controlling interest in the acquiree and the goodwill
acquired. ASC Topic 805 also establishes disclosure requirements
which will enable users to evaluate the nature and financial
effects of the business combination. The Company adopted this
topic as of January 1, 2009 and determined that the
adoption did not have a material impact on its consolidated and
combined financial statements.
In December 2007, the FASB ratified the consensus reached by the
EITF on ASC Topic 808 (Prior authoritative literature:
Issue
No. 07-1,
Accounting for Collaborative Agreements”). ASC
808-10-20
provides the definition of a collaborative agreement and ASC
808-10-15
provides guidelines to assist an entity in determining whether
or not it is a party in a collaborative agreement. ASC
808-10-45 states
that costs incurred and revenues generated from transactions
with third parties shall be reported in accordance with ASC
605-45
(Prior authoritative literature: EITF Issue
No. 99-19,
Reporting Revenue Gross as a Principal versus Net as an
Agent). ASC
808-10-50
also provides minimum disclosure requirements for an
entity’s collaboration agreements and transition guidance.
The Company adopted ASC Topic 808 as of January 1, 2009 and
determined that the adoption did not have a material impact on
its consolidated and combined financial statements.
In April 2008, the FASB issued ASC Topic 350 (Prior
authoritative literature: FASB Staff Position
(“FSP”)
142-3,
Determination of the Useful Life of Intangible Assets). ASC
350-30-35
amends the factors that should be considered in developing
renewal or extension assumptions used to determine the useful
life of a recognized intangible asset under Topic ASC 350. The
Company adopted ASC
350-30-35 as
of January 1, 2009 and determined that the adoption did not
have a material impact on its consolidated and combined
financial statements.
In June 2008, the FASB issued ASC Topic 260 (Prior
authoritative literature:
EITF 03-6-1,
Determining Whether Instruments Granted in Share-Based Payment
Transactions Are Participating Securities). ASC
260-10-45
clarified that all outstanding unvested share-based payment
awards that contain rights to non-forfeitable dividends
participate in undistributed earnings with common shareholders.
Awards of this nature are considered participating securities
and the two-class method of computing basic and diluted earnings
per share must be applied. The Company adopted ASC Topic 260 as
of January 1, 2009 and determined that the adoption did not
have a material impact on its consolidated and combined
financial statements.
|
|
|
Item 7A.
|
Quantitative
and Qualitative Disclosures About Market Risk
|
Not applicable.
42
|
|
|
Item 8.
|
Financial
Statements and Supplementary Data
|
The financial statements required by this item are located
beginning on
page F-1
of this report.
|
|
|
Item 9.
|
Changes
in and Disagreements With Accountants on Accounting and
Financial Disclosure
|
Not applicable.
|
|
|
Item 9A.
|
Controls
and Procedures
|
Not applicable.
|
|
|
Item 9A(T).
|
Controls
and Procedures
|
Evaluation
of Disclosure Controls and Procedures.
Our management, with the participation of our Chief Executive
Officer and Chief Financial Officer, has evaluated the
effectiveness of our disclosure controls and procedures (as
defined in
Rules 13a-15(e)
and
15d-15(e)
under the Securities Exchange Act of 1934, as amended (the
“Exchange Act”)) as of the end of the period covered
by this annual report (the “Evaluation Date”). Based
on such evaluation, our Chief Executive Officer and Chief
Financial Officer have concluded that, as of the Evaluation
Date, our disclosure controls and procedures were effective.
Management’s
Report on Internal Control Over Financial
Reporting.
Our management is responsible for establishing and maintaining
adequate internal control over financial reporting, as such term
is defined in
Rule 13a-15(f)
of the Exchange Act. Under the supervision and with the
participation of our Chief Executive Officer and Chief Financial
Officer, we conducted an assessment of the design and
effectiveness of our internal control over financial reporting
as of the Evaluation Date. In making its assessment of internal
control over financial reporting, management used the criteria
set forth by the Committee of Sponsoring Organizations
(“COSO”) of the Treadway Commission in Internal
Control — Integrated Framework. Based on this
assessment, our management concluded that, as of the Evaluation
Date, our internal control over financial reporting was
effective based on the criteria set forth by COSO in Internal
Control — Integrated Framework.
This annual report does not include an attestation report of the
Company’s registered public accounting firm regarding
internal control over financial reporting. Management’s
report was not subject to attestation by the Company’s
registered public accounting firm pursuant to temporary rules of
the Securities and Exchange Commission that permit the Company
to provide only management’s report in this annual report.
Changes
in Internal Control over Financial Reporting.
There have been no changes in our internal control over
financial reporting (as defined in
rules 13a-15(f)
and
15d-15(f)
under the Exchange Act) during the fourth quarter of 2009 that
have materially affected, or are reasonably likely to materially
affect, our internal control over financial reporting.
|
|
|
Item 9B.
|
Other
Information
|
Not applicable.
Part III
|
|
|
Item 10.
|
Directors
and Executive Officers of the Registrant and Corporate
Governance
|
The names, ages, titles and biographies of our executive
officers are provided under “Executive Officers” in
Part I of this
Form 10-K,
and are incorporated herein by reference. Additional information
regarding our directors and executive officers is set forth in
our Proxy Statement for the 2010 Annual Meeting of
43
Stockholders (the “2010 Proxy Statement”) under the
Sections “Election of Directors” and
“Section 16(a) Beneficial Ownership Reporting
Compliance” and is incorporated herein by reference.
|
|
|
Item 11.
|
Executive
Compensation
|
The information regarding executive compensation is set forth
under the Section entitled “Executive Compensation” in
our 2010 Proxy Statement and is incorporated herein by reference.
|
|
|
Item 12.
|
Security
Ownership of Certain Beneficial Owners and Management and
Related Stockholder Matters
|
The information regarding security ownership of certain
beneficial owners, directors and executive officers is set forth
under the Section entitled “Security Ownership of Certain
Beneficial Owners and Management” in our 2010 Proxy
Statement and is incorporated herein by reference.
|
|
|
Item 13.
|
Certain
Relationships and Related Transactions and Director
Independence
|
The information regarding certain relationships and related
transactions is set forth under the Section entitled
“Transactions with Related Persons” in our 2010 Proxy
Statement and is incorporated herein by reference.
|
|
|
Item 14.
|
Principal
Accounting Fees and Services
|
The information regarding auditor fees and services is set forth
under the Section entitled “Independent Registered Public
Accounting Firm” in our 2010 Proxy Statement and is
incorporated herein by reference.
Part IV
|
|
|
Item 15.
|
Exhibits,
Financial Statement Schedules
|
| |
|
|
|
|
|
|
|
|
|
Page Herein
|
|
|
|
(a)
|
|
The following documents are filed as a part of this report:
|
|
|
|
|
|
(1) Financial Statements:
|
|
|
|
|
|
Consolidated Financial
Statements of CPEX Pharmaceuticals, Inc. and Subsidiary
|
|
F-1 to F-25
|
|
|
|
(2) Financial Statement Schedules:
|
|
|
|
|
|
None
|
|
|
|
|
|
(3) Exhibits — See index beginning on page
46
|
|
|
|
|
|
|
|
(b)
|
|
The exhibits filed as a part of this annual report on Form 10-K
are listed on the Exhibit Index immediately preceding the
signature page. The Exhibit Index is incorporated herein by
reference.
|
44
Exhibit Index
to
Form 10-K
for the Year Ended December 31, 2009
| |
|
|
|
|
Exhibit
|
|
|
|
Number
|
|
Description of Exhibit
|
|
|
|
|
2
|
.1
|
|
Form of Separation and Distribution Agreement by and between
CPEX Pharmaceuticals, Inc. and Bentley Pharmaceuticals, Inc.
Filed as Exhibit 2.1 to Amendment No. 2 of the CPEX
Form 10 (File
No. 001-33895)
filed on May 8, 2008 and incorporated herein by this
reference.
|
|
|
3
|
.1
|
|
Amended and Restated Certificate of Incorporation of CPEX
Pharmaceuticals, Inc. filed with the Office of the Secretary of
State of the State of Delaware on September 28, 2007. Filed
as Exhibit 3.1 to Amendment No. 4 of the CPEX
Form 10 (File
No. 001-33895)
filed on May 30, 2008 and incorporated herein by this
reference.
|
|
|
3
|
.2
|
|
Amended and Restated By-laws of CPEX Pharmaceuticals, Inc. Filed
as Exhibit 3.2 to Amendment No. 4 of the CPEX
Form 10 (File
No. 001-33895)
filed on May 30, 2008 and incorporated herein by this
reference.
|
|
|
4
|
.1
|
|
Form of Rights Agreement between CPEX Pharmaceuticals, Inc. and
American Stock Transfer and Trust Company. Filed as
Exhibit 4.1 to Amendment No. 4 of the CPEX
Form 10 (File
No. 001-33895)
filed on May 30, 2008 and incorporated herein by this
reference.
|
|
|
4
|
.2
|
|
Form of Certificate of Designation of Series A Preferred
Stock. Filed as Exhibit A to the Form of Rights Agreement
filed as Exhibit 4.1 to Amendment No. 4 of the CPEX
Form 10 (File
No. 001-33895)
filed on May 30, 2008 and incorporated herein by this
reference.
|
|
|
4
|
.3
|
|
Form of Rights Certificate. Filed as Exhibit B to the Form
of Rights Agreement filed as Exhibit 4.1 to Amendment
No. 4 of the CPEX Form 10 (File
No. 001-33895)
filed on May 30, 2008 and incorporated herein by this
reference.
|
|
|
10
|
.1
|
|
Form of Transition Services Agreement by and between CPEX
Pharmaceuticals, Inc. and Bentley Pharmaceuticals, Inc. Filed as
Exhibit 10.1 to Amendment No. 1 of the CPEX
Form 10 (File
No. 001-33895)
filed on April 11, 2008 and incorporated herein by this
reference.
|
|
|
10
|
.2
|
|
Form of Tax Sharing Agreement by and between CPEX
Pharmaceuticals, Inc. and Bentley Pharmaceuticals, Inc. Filed as
Exhibit 10.2 to Amendment No. 1 of the CPEX
Form 10 (File
No. 001-33895)
filed on April 11, 2008 and incorporated herein by this
reference.
|
|
|
10
|
.3
|
|
Form of Employment Matters Agreement by and between CPEX
Pharmaceuticals, Inc. and Bentley Pharmaceuticals, Inc. Filed as
Exhibit 10.3 to Amendment No. 1 of the CPEX
Form 10 (File
No. 001-33895)
filed on April 11, 2008 and incorporated herein by this
reference.
|
|
|
10
|
.4
|
|
Asset Purchase Agreement between Bentley Pharmaceuticals, Inc.
and Yungtai Hsu dated February 1, 1999, including letter
amendment effective as of December 31, 2007. Filed as
Exhibit 10.4 to Amendment No. 1 of the CPEX
Form 10 (File
No. 001-33895)
filed on April 11, 2008 and incorporated herein by this
reference.
|
|
|
10
|
.5
|
|
License Agreement between Bentley Pharmaceuticals, Inc. and
Auxilium A2, Inc. dated May 31, 2000, including thereto
Amendment No. 1 dated October 2000, Amendment No. 2
dated May 31, 2001, Amendment No. 3 dated
September 6, 2002 and Amendment No. 4 dated
March 25, 2004. Filed as Exhibit 10.5 to the CPEX
Form 10-K
for the fiscal year ended December 31, 2008 and
incorporated herein by this reference.
|
|
|
10
|
.6
|
|
Employment Agreement by and between CPEX Pharmaceuticals, Inc.
and John Sedor dated April 11, 2007. Filed as
Exhibit 10.6 to Amendment No. 1 of the CPEX
Form 10 (File
No. 001-33895)
filed on April 11, 2008 and incorporated herein by this
reference.
|
|
|
10
|
.7
|
|
Employment Agreement by and between CPEX Pharmaceuticals, Inc.
and Robert P. Hebert dated April 11, 2007 Filed as
Exhibit 10.7 to Amendment No. 1 of the CPEX
Form 10 (File
No. 001-33895)
filed on April 11, 2008 and incorporated herein by this
reference.
|
|
|
10
|
.8
|
|
CPEX Pharmaceuticals, Inc. Amended and Restated 2008 Equity
Incentive Plan. Filed as Appendix A to the CPEX definitive
proxy statement filed on May 8, 2009 and incorporated
herein by this reference.
|
|
|
10
|
.9
|
|
Employment Agreement by and between CPEX Pharmaceuticals, Inc.
and Lance Berman dated February 2, 2009. Filed as
Exhibit 10.1 to the CPEX
Form 8-K
filed on February 3, 2009 and incorporated herein by this
reference.
|
45
| |
|
|
|
|
Exhibit
|
|
|
|
Number
|
|
Description of Exhibit
|
|
|
|
|
10
|
.10
|
|
Employment Agreement by and between CPEX Pharmaceuticals, Inc.
and Fred Feldman dated June 30, 2008. Filed as
Exhibit 10.10 to the CPEX
Form 10-K
for the fiscal year ended December 31, 2008 and
incorporated herein by this reference.
|
|
|
10
|
.11*
|
|
Employment Agreement by and between CPEX Pharmaceuticals, Inc.
and Nils Bergenhem dated February 1, 2010.
|
|
|
23
|
.1*
|
|
Consent of BDO Seidman LLP, Independent Registered Public
Accounting Firm.
|
|
|
23
|
.2*
|
|
Consent of Deloitte & Touche LLP, Independent
Registered Public Accounting Firm.
|
|
|
31
|
.1*
|
|
Certification of the Chief Executive Officer pursuant to
Section 302 of the Sarbanes-Oxley Act of 2002.
|
|
|
31
|
.2*
|
|
Certification of the Chief Financial Officer pursuant to
Section 302 of the Sarbanes-Oxley Act of 2002.
|
|
|
32
|
.1*
|
|
Certification of the Chief Executive Officer pursuant to
18 U.S.C. Section 1350 as adopted pursuant to
Section 906 of the Sarbanes-Oxley Act of 2002.
|
|
|
32
|
.2*
|
|
Certification of the Chief Financial Officer pursuant to
18 U.S.C. Section 1350 as adopted pursuant to
Section 906 of the Sarbanes-Oxley Act of 2002.
|
|
|
|
|
* |
|
Filed herewith. |
| |
|
|
|
Exhibits 10.3 and 10.6 through 10.11 above are management
contracts or compensatory plans or arrangements in which
executive officers or directors of CPEX Pharmaceuticals, Inc.
participate. |
46
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the
Securities Exchange Act of 1934, the Registrant has duly caused
this report to be signed on its behalf by the undersigned,
thereunto duly authorized.
CPEX PHARMACEUTICALS, INC.
John A. Sedor
Chief Executive Officer and President
Date: March 29, 2010
Pursuant to the requirements of the Securities Exchange Act of
1934, this report has been signed below by the following persons
on behalf of the Registrant and in the capacities and on the
dates indicated.
| |
|
|
|
|
|
|
|
Signature
|
|
Title
|
|
Date
|
|
|
|
|
|
|
|
|
|
/s/ James
R. Murphy
James
R. Murphy
|
|
Director and Chairman
of the Board
|
|
March 29, 2010
|
|
|
|
|
|
|
|
/s/ Michael
McGovern
Michael
McGovern
|
|
Director
|
|
March 29, 2010
|
|
|
|
|
|
|
|
/s/ Miguel
Fernandez
Miguel
Fernandez
|
|
Director
|
|
March 29, 2010
|
|
|
|
|
|
|
|
/s/ John
W. Spiegel
John
W. Spiegel
|
|
Director
|
|
March 29, 2010
|
|
|
|
|
|
|
|
/s/ John
A. Sedor
John
A. Sedor
|
|
Chief Executive Officer, President and Director (Principal
Executive Officer)
|
|
March 29, 2010
|
|
|
|
|
|
|
|
/s/ Robert
P. Hebert
Robert
P. Hebert
|
|
Chief Financial Officer and Vice President (Principal Financial
and Accounting Officer)
|
|
March 29, 2010
|
47
Index to
Consolidated and Combined Financial Statements of
CPEX Pharmaceuticals, Inc. and Subsidiary
| |
|
|
|
|
|
|
|
Page
|
|
|
|
Report of Independent Registered Public Accounting Firm BDO
Seidman, LLP
|
|
|
F-2
|
|
|
Report of Independent Registered Public Accounting Firm
Deloitte & Touche, LLP
|
|
|
F-3
|
|
|
Consolidated Balance Sheets as of December 31, 2009 and 2008
|
|
|
F-4
|
|
|
Consolidated and Combined Statements of Operations for the years
ended December 31, 2009, 2008 and 2007
|
|
|
F-5
|
|
|
Consolidated and Combined Statements of Changes in
Stockholders’ Equity and Bentley Pharmaceuticals, Inc., net
investment for the years ended December 31, 2009, 2008 and
2007
|
|
|
F-6
|
|
|
Consolidated and Combined Statements of Cash Flows for the years
ended December 31, 2009, 2008 and 2007
|
|
|
F-7
|
|
|
Notes to Consolidated and Combined Financial Statements
|
|
|
F-8
|
|
F-1
Report of
Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of
CPEX Pharmaceuticals, Inc.
Exeter, New Hampshire
We have audited the accompanying consolidated balance sheet of
CPEX Pharmaceuticals, Inc. and its subsidiary (the
“Company”) as of December 31, 2009, and the
related consolidated statements of operations, changes in
stockholders’ equity and cash flows for the year then
ended. These financial statements are the responsibility of the
Company’s management. Our responsibility is to express an
opinion on these financial statements based on our audit.
We conducted our audit in accordance with the standards of the
Public Company Accounting Oversight Board (United States). Those
standards require that we plan and perform the audit to obtain
reasonable assurance about whether the financial statements are
free of material misstatement. The Company is not required to
have nor were we engaged to perform an audit of its internal
control over financial reporting. Our audit included
consideration of internal control over financial reporting as a
basis for designing audit procedures that are appropriate in the
circumstances, but not for the purpose of expressing an opinion
on the effectiveness of the Company’s internal control over
financial reporting. Accordingly, we express no such opinion. An
audit also includes examining, on a test basis, evidence
supporting the amounts and disclosures in the financial
statements, assessing the accounting principles used and
significant estimates made by management, as well as evaluating
the overall financial statement presentation. We believe that
our audit provides a reasonable basis for our opinion.
In our opinion, such consolidated financial statements referred
to above present fairly, in all material respects, the financial
position of CPEX Pharmaceuticals, Inc. and its subsidiary as of
December 31, 2009, and the results of its operations and
their cash flows for the year then ended, in conformity with
accounting principles generally accepted in the United States of
America.
/S/ BDO Seidman, LLP
Boston, Massachusetts
March 29, 2010
F-2
Report of
Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of
CPEX Pharmaceuticals, Inc.
Exeter, New Hampshire
We have audited the accompanying consolidated balance sheet of
CPEX Pharmaceuticals, Inc. and subsidiaries (the
“Company”) as of December 31, 2008, and the
related consolidated and combined statements of operations,
changes in stockholders’ equity and Bentley
Pharmaceuticals, Inc. net investment, and cash flows for each of
the two years in the period ended December 31, 2008. These
financial statements are the responsibility of the
Company’s management. Our responsibility is to express an
opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the
Public Company Accounting Oversight Board (United States). Those
standards require that we plan and perform the audit to obtain
reasonable assurance about whether the financial statements are
free of material misstatement. The Company is not required to
have nor were we engaged to perform an audit of its internal
control over financial reporting. Our audits included
consideration of internal control over financial reporting as a
basis for designing audit procedures that are appropriate in the
circumstances, but not for the purpose of expressing an opinion
on the effectiveness of the Company’s internal control over
financial reporting. Accordingly, we express no such opinion. An
audit also includes examining, on a test basis, evidence
supporting the amounts and disclosures in the financial
statements, assessing the accounting principles used and
significant estimates made by management, as well as evaluating
the overall financial statement presentation. We believe that
our audits provide a reasonable basis for our opinion.
In our opinion, such consolidated and combined financial
statements present fairly, in all material respects, the
financial position of CPEX Pharmaceuticals, Inc. and
subsidiaries as of December 31, 2008, and the results of
their operations and their cash flows for each of the two years
in the period ended December 31, 2008, in conformity with
accounting principles generally accepted in the United States of
America.
As discussed in Note 2 to the consolidated and combined
financial statements, prior to the separation of the Company
from Bentley Pharmaceuticals, Inc. (“Bentley”) on
June 30, 2008, CPEX Pharmaceuticals, Inc. was comprised of
the assets, liabilities and operations of the drug delivery
business of Bentley. The consolidated and combined financial
statements also include allocations from Bentley in those
periods. These allocations may not be reflective of the actual
level of costs which would have been incurred had CPEX
Pharmaceuticals, Inc. operated as a separate entity apart from
Bentley.
/S/ Deloitte &
Touche LLP
Boston, Massachusetts
March 24, 2009
F-3
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands, except per share data)
|
|
|
|
|
ASSETS
|
|
Current assets:
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents
|
|
$
|
13,695
|
|
|
$
|
15,211
|
|
|
Receivables
|
|
|
5,289
|
|
|
|
4,445
|
|
|
Prepaid expenses and other
|
|
|
593
|
|
|
|
583
|
|
|
|
|
|
|
|
|
|
|
|
|
Total current assets
|
|
|
19,577
|
|
|
|
20,239
|
|
|
|
|
|
|
|
|
|
|
|
|
Non-current assets:
|
|
|
|
|
|
|
|
|
|
Fixed assets, net
|
|
|
2,938
|
|
|
|
2,832
|
|
|
Intangible assets, net
|
|
|
2,211
|
|
|
|
2,394
|
|
|
Restricted cash
|
|
|
1,000
|
|
|
|
1,000
|
|
|
Note receivable
|
|
|
300
|
|
|
|
—
|
|
|
Other
|
|
|
17
|
|
|
|
8
|
|
|
|
|
|
|
|
|
|
|
|
|
Total non-current assets
|
|
|
6,466
|
|
|
|
6,234
|
|
|
|
|
|
|
|
|
|
|
|
|
Total assets
|
|
$
|
26,043
|
|
|
$
|
26,473
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
LIABILITIES AND STOCKHOLDERS’ EQUITY
|
|
Current liabilities:
|
|
|
|
|
|
|
|
|
|
Accounts payable
|
|
$
|
1,374
|
|
|
$
|
1,096
|
|
|
Accrued expenses
|
|
|
1,633
|
|
|
|
1,534
|
|
|
|
|
|
|
|
|
|
|
|
|
Total current liabilities
|
|
|
3,007
|
|
|
|
2,630
|
|
|
|
|
|
|
|
|
|
|
|
|
Commitments and contingencies (Note 11)
|
|
|
|
|
|
|
|
|
|
Stockholders’ equity:
|
|
|
|
|
|
|
|
|
|
Series A preferred stock, $1.00 par value, authorized
1,000 shares, issued and outstanding, none
|
|
|
—
|
|
|
|
—
|
|
|
Common stock, $0.01 par value, authorized
35,000 shares, issued and outstanding, 2,537 and
2,484 shares at December 31, 2009 and
December 31, 2008, respectively
|
|
|
25
|
|
|
|
25
|
|
|
Additional paid-in capital
|
|
|
26,765
|
|
|
|
24,532
|
|
|
Accumulated deficit
|
|
|
(3,754
|
)
|
|
|
(714
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Total stockholders’ equity
|
|
|
23,036
|
|
|
|
23,843
|
|
|
|
|
|
|
|
|
|
|
|
|
Total liabilities and stockholders’ equity
|
|
$
|
26,043
|
|
|
$
|
26,473
|
|
|
|
|
|
|
|
|
|
|
|
The accompanying Notes to Consolidated and Combined Financial
Statements are an integral part of these financial statements.
F-4
CPEX
Pharmaceuticals, Inc. and Subsidiary
Consolidated
and Combined Statements of Operations
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For the Year Ended December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
|
|
(In thousands, except per share data)
|
|
|
|
|
Revenues:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Royalties and other revenue
|
|
$
|
18,658
|
|
|
$
|
15,574
|
|
|
$
|
11,127
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating expenses:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
General and administrative
|
|
|
8,867
|
|
|
|
6,493
|
|
|
|
5,206
|
|
|
Research and development
|
|
|
12,291
|
|
|
|
9,119
|
|
|
|
9,646
|
|
|
Separation costs
|
|
|
—
|
|
|
|
2,502
|
|
|
|
1,010
|
|
|
Depreciation and amortization
|
|
|
699
|
|
|
|
682
|
|
|
|
752
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses
|
|
|
21,857
|
|
|
|
18,796
|
|
|
|
16,614
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss from operations
|
|
|
(3,199
|
)
|
|
|
(3,222
|
)
|
|
|
(5,487
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Other income (expenses):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest income
|
|
|
162
|
|
|
|
312
|
|
|
|
591
|
|
|
Interest expense
|
|
|
(3
|
)
|
|
|
(5
|
)
|
|
|
(10
|
)
|
|
Other, net
|
|
|
—
|
|
|
|
—
|
|
|
|
(22
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
$
|
(3,040
|
)
|
|
$
|
(2,915
|
)
|
|
$
|
(4,928
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss per common share:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted
|
|
$
|
(1.21
|
)
|
|
$
|
(1.25
|
)
|
|
$
|
(2.17
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average common shares outstanding:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted
|
|
|
2,511
|
|
|
|
2,338
|
|
|
|
2,274
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The accompanying Notes to Consolidated and Combined Financial
Statements are an integral part of these financial statements.
F-5
CPEX
Pharmaceuticals, Inc. and Subsidiary
Bentley
Pharmaceuticals, Inc. Net Investment
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Bentley
|
|
|
|
|
|
|
|
$0.01 Par Value
|
|
|
Additional
|
|
|
|
|
|
Pharmaceuticals,
|
|
|
|
|
|
|
|
Common Stock
|
|
|
Paid-In
|
|
|
Accumulated
|
|
|
Inc. Net
|
|
|
|
|
|
|
|
Shares
|
|
|
Amount
|
|
|
Capital
|
|
|
Deficit
|
|
|
Investment
|
|
|
Total
|
|
|
|
|
(In thousands)
|
|
|
|
|
Balance at January 1, 2007
|
|
|
—
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
19,052
|
|
|
$
|
19,052
|
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(4,928
|
)
|
|
|
(4,928
|
)
|
|
Net transfers from parent
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
15,027
|
|
|
|
15,027
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2007
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
29,151
|
|
|
|
29,151
|
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(714
|
)
|
|
|
(2,201
|
)
|
|
|
(2,915
|
)
|
|
Net transfers to parent
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(6,295
|
)
|
|
|
(6,295
|
)
|
|
Transfer of parent company investment
|
|
|
—
|
|
|
|
—
|
|
|
|
20,655
|
|
|
|
—
|
|
|
|
(20,655
|
)
|
|
|
—
|
|
|
Issuance of common stock in connection with the spin-off
|
|
|
2,274
|
|
|
|
23
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
23
|
|
|
Exercise of stock options and vesting of restricted stock units
|
|
|
206
|
|
|
|
2
|
|
|
|
1,738
|
|
|
|
—
|
|
|
|
—
|
|
|
|
1,740
|
|
|
Share-based compensation
|
|
|
4
|
|
|
|
—
|
|
|
|
2,139
|
|
|
|
—
|
|
|
|
—
|
|
|
|
2,139
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2008
|
|
|
2,484
|
|
|
|
25
|
|
|
|
24,532
|
|
|
|
(714
|
)
|
|
|
—
|
|
|
|
23,843
|
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(3,040
|
)
|
|
|
—
|
|
|
|
(3,040
|
)
|
|
Share-based compensation
|
|
|
18
|
|
|
|
—
|
|
|
|
1,936
|
|
|
|
—
|
|
|
|
—
|
|
|
|
1,936
|
|
|
Issuance of common stock in lieu of cash compensation
|
|
|
30
|
|
|
|
—
|
|
|
|
293
|
|
|
|
—
|
|
|
|
—
|
|
|
|
293
|
|
|
Exercise of stock options and vesting of restricted stock units
|
|
|
5
|
|
|
|
—
|
|
|
|
4
|
|
|
|
—
|
|
|
|
—
|
|
|
|
4
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2009
|
|
|
2,537
|
|
|
$
|
25
|
|
|
$
|
26,765
|
|
|
$
|
(3,754
|
)
|
|
|
—
|
|
|
$
|
23,036
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The accompanying Notes to Consolidated and Combined Financial
Statements are an integral part of these financial statements.
F-6
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For the Year Ended December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
|
|
(In thousands)
|
|
|
|
|
Cash flows from operating activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
$
|
(3,040
|
)
|
|
$
|
(2,915
|
)
|
|
$
|
(4,928
|
)
|
|
Adjustments to reconcile net loss to net cash used by operating
activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Depreciation and amortization
|
|
|
699
|
|
|
|
682
|
|
|
|
752
|
|
|
Non-cash charge for write-down of intangible assets
|
|
|
—
|
|
|
|
141
|
|
|
|
202
|
|
|
Share-based compensation expense
|
|
|
1,936
|
|
|
|
3,195
|
|
|
|
1,504
|
|
|
Loss on disposal of assets
|
|
|
—
|
|
|
|
—
|
|
|
|
28
|
|
|
Changes in operating assets and liabilities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Receivables
|
|
|
(844
|
)
|
|
|
(1,200
|
)
|
|
|
(983
|
)
|
|
Prepaid expenses and other current assets
|
|
|
(10
|
)
|
|
|
124
|
|
|
|
61
|
|
|
Other assets
|
|
|
(9
|
)
|
|
|
36
|
|
|
|
106
|
|
|
Accounts payable and accrued expenses
|
|
|
670
|
|
|
|
(591
|
)
|
|
|
1,081
|
|
|
Deferred income
|
|
|
—
|
|
|
|
(25
|
)
|
|
|
(9
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash used in operating activities
|
|
|
(598
|
)
|
|
|
(553
|
)
|
|
|
(2,186
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash flows from investing activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additions to fixed assets
|
|
|
(398
|
)
|
|
|
(307
|
)
|
|
|
(303
|
)
|
|
Additions to intangible assets
|
|
|
(224
|
)
|
|
|
—
|
|
|
|
(157
|
)
|
|
Note receivable
|
|
|
(300
|
)
|
|
|
—
|
|
|
|
—
|
|
|
Proceeds from the sale of fixed assets
|
|
|
—
|
|
|
|
—
|
|
|
|
30
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash used in investing activities
|
|
|
(922
|
)
|
|
|
(307
|
)
|
|
|
(430
|
)
|
|
Cash flows from financing activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Proceeds from the exercise of stock options
|
|
|
4
|
|
|
|
1,740
|
|
|
|
—
|
|
|
Net change in investment from Bentley Pharmaceuticals, Inc.
|
|
|
—
|
|
|
|
(7,328
|
)
|
|
|
13,523
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash provided by (used in) financing activities
|
|
|
4
|
|
|
|
(5,588
|
)
|
|
|
13,523
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net (decrease) increase in cash and cash equivalents
|
|
|
(1,516
|
)
|
|
|
(6,448
|
)
|
|
|
10,907
|
|
|
Cash and cash equivalents at beginning of year
|
|
|
15,211
|
|
|
|
21,659
|
|
|
|
10,752
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents at end of year
|
|
$
|
13,695
|
|
|
$
|
15,211
|
|
|
$
|
21,659
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash paid for interest
|
|
$
|
3
|
|
|
$
|
5
|
|
|
$
|
10
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Supplemental Disclosures of Non-Cash Financing and Investing
Activities
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The Company has issued common stock in lieu of cash to its
executive officers and other employees as a portion of their
2008 bonus during the year as follows:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Shares
|
|
|
30
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Amount
|
|
$
|
293
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The Company has issued common stock as share-based compensation
in lieu of cash during the year as follows:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Shares
|
|
|
18
|
|
|
|
4
|
|
|
|
19
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Amount
|
|
|
172
|
|
|
|
52
|
|
|
|
200
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The accompanying Notes to Consolidated and Combined Financial
Statements are an integral part of these financial statements.
F-7
CPEX
Pharmaceuticals, Inc. and Subsidiary
|
|
|
NOTE 1 —
|
DESCRIPTION
OF BUSINESS
|
CPEX Pharmaceuticals, Inc. (which may be referred to as
“CPEX” or the “Company”) was incorporated on
September 28, 2007 in the State of Delaware and has one
wholly-owned subsidiary, CPEX Park, LLC. CPEX is an emerging
specialty pharmaceutical company in the business of research and
development of pharmaceutical products utilizing its validated
drug delivery platform technology. The CPEX platform drug
delivery technology is based upon
CPE-215®,
which enhances permeation and absorption of pharmaceutical
molecules across biological membranes such as the skin, nasal
mucosa and eye.
The first product of CPEX is
Testim®,
a gel for testosterone replacement therapy that is a formulation
of CPE-215 with testosterone. Testim is licensed to Auxilium
Pharmaceuticals, Inc. who is currently marketing the product in
the United States, Europe and other countries.
Nasulintm,
the Company’s lead product candidate, is a patented
intranasal insulin formulation which incorporates CPE-215 as a
permeation facilitator that addresses the need for an improved
delivery method for insulin. The Company has completed a series
of Phase 1 and Phase 2 studies investigating Nasulin. The
Company recently completed a Phase 2a study of Nasulin and is
determining the appropriate next steps for the Nasulin program.
The Company’s business was initially the drug delivery
business of Bentley Pharmaceuticals, Inc. (referred to as
“Bentley”) which was spun-off in June 2008 in
connection with the sale of Bentley’s remaining businesses.
Shares of our stock were distributed to Bentley stockholders
after the close of business on June 30, 2008 (the
“Separation Date”) by means of a stock dividend, a
transaction that was taxable to Bentley and Bentley’s
stockholders (the “Separation”). Each Bentley
stockholder of record on June 20, 2008, the record date,
received on the Separation Date one CPEX share for every ten
shares of Bentley common stock. Bentley has no ownership
interest in CPEX subsequent to the Separation.
In connection with the spin-off, CPEX and Bentley entered into a
series of agreements, including a Separation and Distribution
Agreement, a Transition Services Agreement, an Employee Matters
Agreement and a Tax Sharing Agreement. See Note 6 for
further discussion.
Separation costs, consisting of legal, tax and other strategic
consulting costs specifically related to the separation from
Bentley, were $2.5 million in 2008 and $1.0 million in
2007 and are reported as Separation costs within
operating expenses in the accompanying Consolidated and Combined
Statements of Operations. No additional separation costs have
been incurred since the Separation Date and the Company does not
expect to incur any additional separation costs in the future.
In December 2009, CPEX Pharma, Inc., a wholly-owned subsidiary,
was merged with and into CPEX Pharmaceuticals, Inc. This merger
was administrative in nature and had no impact on the
Company’s results of operations and financial condition.
The Company is subject to a number of risks common to emerging
companies in the life sciences industry. Principal among these
risks are the uncertainties of the drug development process,
technological innovations, development of the same or similar
technological innovations by the Company’s competitors,
protection of proprietary technology, compliance with government
regulations and approval requirements, uncertainty of market
acceptance of products, dependence on key individuals, product
liability, and the need to obtain additional financing necessary
to fund future operations. The Company’s growth and ability
to achieve profitability may be dependent upon the successful
commercialization of new products and partnering arrangements.
F-8
CPEX
Pharmaceuticals, Inc. and Subsidiary
Notes to
Consolidated and Combined Financial
Statements — (Continued)
|
|
|
NOTE 2 —
|
SUMMARY
OF SIGNIFICANT ACCOUNTING POLICIES
|
Basis of
presentation
The consolidated and combined financial statements reflect the
accounts of the Company and its subsidiary. All intercompany
accounts and transactions have been eliminated.
Principles
of consolidation
Prior to the Separation Date, the CPEX combined financial
statements reflect the assets, liabilities and results of
operations of the components of Bentley that constituted the
drug delivery business to be separated into CPEX. The financial
information for periods prior to July 1, 2008 is primarily
comprised of Bentley’s former U.S. drug delivery
business and certain accounts of Bentley’s wholly-owned
subsidiaries, Bentley Pharmaceuticals Ireland Limited and
Bentley Park, LLC. Financial information presented in periods
subsequent to June 30, 2008 reflect the assets and
liabilities of CPEX Pharmaceuticals, Inc. as an independent,
publicly-traded company together with its wholly-owned
subsidiaries, CPEX Pharma, Inc. (which merged with CPEX
Pharmaceuticals, Inc. in December 2009) and CPEX Park, LLC.
All intercompany balances have been eliminated in consolidation
and combination. The drug delivery business of Bentley
Pharmaceuticals Ireland Limited did not have any operations
other than intercompany transactions with CPEX.
Management believes that the assumptions underlying the combined
financial statements are reasonable. The financial information
in the consolidated and combined financial statements for the
years ended December 31, 2008 and 2007 do not include all
the expenses that would have been incurred had CPEX been a
separate, stand-alone entity. As such, the financial information
herein does not reflect the results of operations and cash flows
of what CPEX would have been, had CPEX been a separate,
stand-alone entity during the years ended December 31, 2008
and 2007. Additionally, these historical combined financial
statements include proportional cost allocations of certain
common costs of Bentley and CPEX because specific identification
of these expenses to each entity was not practicable.
Segment
Data
The Company manages its operations on a consolidated, single
segment basis for purposes of assessing performance and making
operating decisions. Accordingly, the Company has only one
reporting segment.
Use of
estimates
The preparation of financial statements in conformity with
accounting principles generally accepted in the United States of
America requires management to make estimates and assumptions
that affect the reported amounts of assets and liabilities and
disclosure of contingent assets and liabilities at the date of
the financial statements and the reported amounts of revenues
and expenses during the reporting period. Actual results could
differ from those estimates.
Cash and
cash equivalents and restricted cash
The Company considers all highly liquid investments with
remaining maturities of three months or less when purchased to
be cash equivalents for purposes of classification in the
Consolidated Balance Sheets and the Consolidated and Combined
Statements of Cash Flows. The cash and cash equivalents of CPEX
include cash balances maintained in commercial bank accounts,
amounts invested in overnight sweep investments and cash
deposits in money market accounts. The Company’s cash
balances exceed the limits of amounts insured by the Federal
Deposit Insurance Corporation; however, because management
believes deposits are maintained at highly rated financial
institutions there is not a significant risk of loss of
uninsured amounts. At December 31, 2009, the Company’s
cash and cash equivalents balance totaled $13.7 million.
F-9
CPEX
Pharmaceuticals, Inc. and Subsidiary
Notes to
Consolidated and Combined Financial
Statements — (Continued)
In connection with intellectual property in-licensed in 2003,
the Company obtained a renewable, irrevocable letter of credit
in the amount of $1.0 million in favor of the assignor to
guarantee future royalty payments by the Company. This letter of
credit will expire on June 30, 2010, and the Company does
not expect to renew it. The $1.0 million used to secure the
letter of credit has been classified as Restricted cash
in the Condensed Consolidated Balance Sheets as of
December 31, 2009 and 2008.
Accounts
receivable and allowances for doubtful accounts
The Company enters into collaboration and research agreements
whereby the Company may receive milestone payments, research
fees and/or
royalties. Accounts receivable from these agreements are
recorded at their net realizable value, generally as services
are performed or as milestones and royalties are earned. When
necessary, receivable balances are reported net of an estimated
allowance for uncollectible accounts. Estimated uncollectible
receivables are based on the amount and status of past due
accounts, contractual terms with customers, the credit
worthiness of customers and the history of uncollectible
accounts. The Company’s accounts receivable and revenues
are primarily royalties due from its licensee, Auxilium for
sales of
Testim®.
Testim royalties represented substantially all of the accounts
receivable as of December 31, 2009 and 2008 and
substantially all of the revenues in the years ended
December 31, 2009, 2008 and 2007. All receivables are
uncollateralized and therefore subject to credit risk.
The Company did not write-off any uncollectible receivables in
the years ended December 31, 2009 and 2008. In addition,
the Company reviewed all receivable balances and concluded that
no allowance for doubtful accounts was necessary as of
December 31, 2009.
Fixed
assets
Fixed assets are stated at cost. Depreciation is computed using
the straight-line method over the following estimated economic
lives of the assets:
| |
|
|
|
|
|
Years
|
|
|
|
Buildings and improvements
|
|
30
|
|
Equipment
|
|
3-7
|
|
Furniture and fixtures
|
|
5-7
|
|
Other
|
|
5
|
Expenditures for replacements and improvements that
significantly add to productive capacity or extend the useful
life of an asset are capitalized, while expenditures for
maintenance and repairs are charged to operations as incurred.
Leasehold improvements are amortized over the lesser of the
useful life of the assets or over the life of the respective
lease. When assets are sold or retired, the cost of the asset
and the related accumulated depreciation are removed from the
accounts and any gain or loss is recognized currently.
Intangible
assets
Costs incurred in connection with acquiring licenses, patents,
and other proprietary rights related to the business of the
Company are capitalized as intangible assets. These assets are
amortized on a straight-line basis for periods not exceeding
fifteen years from the dates of acquisition. Such assets are
reviewed whenever events or changes in circumstances indicate
that the assets may be impaired, by comparing the carrying
amounts to their estimated future undiscounted cash flows, and
adjustments are made for any diminution in value below the
carrying value. During the year ended December 31, 2009 the
Company capitalized approximately $224,000 relating to acquiring
patents and did not record any impairment losses. During the
year ended December 31, 2008 the Company recorded $141,000
related to the write-down of certain intangible assets related
to the Company’s anti-fungal nail lacquer. Impairment
losses related to intangible assets are included in research and
development expenses in the accompanying Consolidated and
Combined Statements
F-10
CPEX
Pharmaceuticals, Inc. and Subsidiary
Notes to
Consolidated and Combined Financial
Statements — (Continued)
of Operations. At December 31, 2009 the Company has also
reassessed the useful lives of its remaining intangible assets
and has determined that the estimated useful lives are
appropriate for determining amortization expense.
Revenue
recognition
The Company recognizes revenue from royalties on Auxilium’s
sales of Testim in accordance with the Financial Standards
Accounting Board (the “FASB”) Accounting Standards
Codification (“FASB ASC”)
605-10-S99-1
(Prior authoritative literature: SAB No. 104,
Revenue Recognition), which requires sales to be recorded
upon shipment, provided that there is evidence of a final
arrangement, there are no uncertainties surrounding acceptance,
title has passed, collectability is reasonably assured and the
price is fixed or determinable. Since 2003, Auxilium has sold
Testim to pharmaceutical wholesalers and chain drug stores,
which have the right to return purchased products prior to the
units being dispensed through patient prescriptions. Based on
historical experience, the Company is able to reasonably
estimate future product returns on sales of Testim and as a
result, the Company did not defer Testim royalties for the years
ended December 31, 2009, 2008 and 2007. Total royalty
revenues recognized for the years ended December 31, 2009,
2008 and 2007 were $18.6 million, $15.1 million and
$11.1 million, respectively.
Fair
value measurements
On January 1, 2007, the Company adopted FASB ASC Topic 820
(Prior authoritative literature: SFAS No. 157,
“Fair Value Measurements”), which provides
guidance for measuring the fair value of assets and liabilities,
and requires expanded disclosures about fair value measurements.
FASB ASC
820-10-35-9
indicates that fair value should be determined based on the
assumptions marketplace participants would use in pricing the
asset or liability, and provides additional guidelines to
consider in determining the market-based measurement. The
adoption of FASB ASC Topic 820 did not have a material impact on
the Company’s consolidated and combined financial
statements. The carrying amounts of cash and cash equivalents,
trade and note receivables, accounts payable and accrued
expenses approximate fair value because of their short-term
nature. FASB ASC
820-10-20
clarifies that the definition of fair value retains the exchange
price notion and focuses on the price that would be received to
sell the asset or paid to transfer the liability (an exit
price), not the price that would be paid to acquire the asset or
received to assume the liability (an entry price). FASB ASC
820-10-35
also emphasizes that fair value is a market-based measurement,
not an entity-specific measurement, therefore a fair value
measurement should be determined based on the assumptions that
market participants would use in pricing the asset or liability
including assumptions about risk, the effect of sale or use
restrictions on an asset and non-performance risk including an
entity’s own credit risk relative to a liability. FASB ASC
820-10-35
also establishes a fair value hierarchy that distinguishes
between (1) market participant assumptions developed based
on market data obtained from sources independent of the
reporting entity (observable inputs) and (2) the reporting
entity’s own assumptions about market participant
assumptions developed based on the best information available in
the circumstances (unobservable inputs). FASB ASC
820-10-35-6
emphasizes that valuation techniques should maximize the use of
observable inputs and minimize the use of unobservable inputs.
The additional disclosure requirements of FASB ASC
820-10-50
focus on the inputs used to measure fair value and for recurring
fair value measurements using significant unobservable inputs
and the effect of the measurement on earnings (or changes in net
assets) for the reporting period. Inputs are categorized by a
fair value hierarchy, Level 1 through Level 3, the
highest priority being given to Level 1 and the lowest
priority to Level 3. Level 1 inputs are quoted prices
(unadjusted) in active markets for identical assets or
liabilities that the reporting entity has the ability to access
at the measurement date. Level 2 inputs are inputs other
than quoted prices included within Level 1 that are
observable for the asset or liability, either directly or
indirectly. Level 3 inputs are unobservable inputs for the
asset or liability. Unobservable inputs shall be used to measure
fair value to the extent that observable inputs are not
available.
F-11
CPEX
Pharmaceuticals, Inc. and Subsidiary
Notes to
Consolidated and Combined Financial
Statements — (Continued)
The following tables present the Company’s assets and
liabilities measured at fair value on a recurring basis as of
December 31, 2009 and the amounts as they correspond to the
respective level within the fair value hierarchy established by
FASB ASC
820-10-50.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Fair Value Measurements at December 31, 2009 Using:
|
|
|
|
|
|
|
|
Quoted Prices in
|
|
|
|
|
|
Significant
|
|
|
|
|
|
|
|
Active Markets for
|
|
|
Significant Other
|
|
|
Unobservable
|
|
|
|
|
Total at
|
|
|
Identical Assets
|
|
|
Observable Inputs
|
|
|
Inputs
|
|
|
|
|
December 31, 2009
|
|
|
(Level 1)
|
|
|
(Level 2)
|
|
|
(Level 3)
|
|
|
|
|
(In thousands)
|
|
|
|
|
Assets:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Money market funds(1)
|
|
$
|
12,770
|
|
|
$
|
12,770
|
|
|
|
—
|
|
|
|
—
|
|
|
Restricted Cash
|
|
|
1,000
|
|
|
|
1,000
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total assets at fair value
|
|
$
|
13,770
|
|
|
$
|
13,770
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) |
|
Included in cash and cash equivalents in the accompanying
consolidated and combined balance sheet. |
Research
and development
Research and development expenses consist primarily of costs
associated with the clinical trials of the Company’s
product candidates, manufacturing supplies and other development
materials, compensation and related benefits for research and
development personnel, costs for consultants, and various
overhead costs. Research and development costs are expensed as
incurred consistent with FASB Topic ASC 730 (Prior
authoritative literature: SFAS No. 2, Accounting for
Research and Development Costs).
Clinical
trial expenses
Clinical trial expenses, which are reflected in research and
development expenses, result from obligations under contracts
with vendors, consultants, and clinical sites in connection with
conducting clinical trials. The financial terms of these
contracts are subject to negotiations which vary from contract
to contract and may result in cash flows which are not
consistent with the periods in which materials or services are
provided. In accordance with FASB ASC
830-20-25-13
(Prior authoritative literature: Emerging Issues Task Force
Issue
No. 07-3,
Accounting for Nonrefundable Advance Payments for Goods or
Services Received for Use in Future Research and Development
Activities), these costs are capitalized upon payment and
recognized as an expense according to the progress of each trial
as measured by patient progression and the timing of various
aspects of the trial. The progress of the trials, including the
level of services performed, is determined based upon
discussions with internal personnel as well as outside service
providers.
Provision
for income taxes
CPEX was not a separate taxable entity prior to its separation
from Bentley. The CPEX operations were historically included in
Bentley’s consolidated U.S. federal and state income
tax returns. On June 12, 2008, CPEX and Bentley entered
into a Tax Sharing Agreement to facilitate CPEX in its
separation from Bentley. Under the Tax Sharing Agreement,
Bentley is responsible for all taxes arising from the CPEX
operations up to the June 30, 2008 Separation Date. CPEX is
responsible for all taxes arising from the CPEX operations
following the Separation Date.
The provision for income taxes in 2009 has been determined based
on the CPEX business operations as a separate, stand-alone
entity. The provisions for income taxes in 2008 have been
determined based on the CPEX business operations following its
separation from Bentley. The provision for income taxes in 2007
has been determined on a pro-forma basis as if CPEX had filed a
separate tax return under its current structure for the period
presented. Accordingly, the effective tax rate of CPEX in future
years could vary from its historical
F-12
CPEX
Pharmaceuticals, Inc. and Subsidiary
Notes to
Consolidated and Combined Financial
Statements — (Continued)
effective tax rates depending on the future legal structure of
CPEX and related tax elections. The historical net operating
losses generated by CPEX through June 30, 2008 remained
with Bentley subsequent to the spin-off transaction.
The Company adopted the provisions of FASB ASC Topic 740
(Prior authoritative literature: Financial Accounting
Standards Board Interpretation No. 48, Accounting for
Uncertainty in Income Taxes, an interpretation of FASB
Statement No. 109, Accounting for Income Taxes)
effective January 1, 2007. The purpose of ASC Topic 740
is to clarify and set forth consistent rules for accounting for
uncertain tax positions by requiring the application of a
“more likely than not” threshold for the recognition
and derecognition of tax positions. The adoption of ASC Topic
740 did not have a material effect on the Company’s
financial statements. The Company recognizes interest and
penalties related to uncertain tax positions as a component of
the provision for income taxes. There were no unrecognized tax
positions relating to the Company at the date of adoption.
Prior to the Separation Date, the Company had agreements with
Bentley Pharmaceuticals, Inc. and its subsidiaries for
allocation of various expenses of each company. Income and
expenses resulting from these agreements were eliminated in
combination; however, the related transactions affected the CPEX
combined income tax provision. As future operating profits can
not be reasonably assured both prior and subsequent to the
Separation Date, no tax benefit has been recorded for the losses
generated by CPEX in the years ended December 31, 2009,
2008 and 2007. Accordingly, CPEX has established valuation
allowances equal to the full amount of its deferred tax assets.
Should CPEX determine that it is more likely than not that it
will realize certain of its net deferred tax assets for which it
has previously provided a valuation allowance, an adjustment
would be required to reduce the existing valuation allowance.
Comprehensive
income (loss)
Comprehensive loss is defined as the change in net assets of the
Company during a period from transactions generated from
non-owner sources. It includes all changes in equity during a
period except those resulting from investments by owners and
distributions to owners. The Company had no components of
comprehensive loss other than its net loss for all periods
presented.
Net loss
per common share
Basic net loss per common share is computed based on the
weighted-average number of common shares outstanding during the
period. Diluted net loss per common share is computed based upon
the weighted-average number of common shares outstanding during
the year, adjusted for the dilutive effect of the Company’s
common stock equivalents, including the exercise of common stock
based upon average market price of the common stock for the
period. Basic and diluted net loss per common share is computed
the same for all periods presented, as the Company had losses
for all periods presented and, consequently, the effect of the
common stock equivalents is anti-dilutive.
Dilutive weighted average shares do not include 509,925 and
443,367 common stock equivalents, which includes stock options
and restricted stock units, for the years ended
December 31, 2009 and 2008, respectively, as their effect
would have been anti-dilutive.
On June 30, 2008, the Company had approximately 2,274,000
common shares outstanding primarily as a result of the
Separation on June 30, 2008, whereby Bentley stockholders
of record on June 20, 2008 received one CPEX common share
for every ten common shares of Bentley. The same number of
shares is being used for the basic and diluted loss per share
computation for all periods presented prior to June 30,
2008 because no CPEX equity awards were outstanding prior to the
Separation Date. In addition, since the Company has been in a
net loss position, any potential common shares would not be used
to compute diluted loss per share because the effect would have
been anti-dilutive.
F-13
CPEX
Pharmaceuticals, Inc. and Subsidiary
Notes to
Consolidated and Combined Financial
Statements — (Continued)
In June 2009, in lieu of cash compensation, the Company awarded
29,624 shares of common stock to its executive officers and
other employees as a portion of their 2008 bonuses. The total
value of the awards, which were expensed in 2008, was
approximately $293,000.
Share-based
compensation
As of December 31, 2009 the Company had one share-based
compensation plan, its Amended and Restated 2008 Equity
Incentive Plan (the “Plan”). The Plan, which is
stockholder approved, permits awards of unrestricted common
stock, restricted stock, restricted stock units, options to
purchase CPEX common stock, stock appreciation rights and stock
equivalents for the Company’s employees, directors and
consultants. Equity awards are generally granted with an
exercise price equal to the high and low trading prices of the
Company’s common stock on the grant date. Equity awards
generally vest ratably over one to four year periods and expire
10 years from the grant date. Shares issued upon exercise
of options or upon vesting of restricted stock units are
generally issued from previously unissued shares of the Company.
Prior to the Separation, all equity awards were granted by
Bentley. In accordance with the Employee Matters Agreement
between the Company and Bentley, upon the Separation,
outstanding Bentley awards held by U.S. employees,
directors and consultants were converted into an adjusted
Bentley award and a CPEX award. The number of common shares
underlying the CPEX awards was calculated as one-tenth of the
number of common shares underlying the original Bentley awards.
The price of the CPEX awards was determined by multiplying the
original exercise price of the Bentley awards by the when-issued
trading price of CPEX common stock on the Separation Date and
then dividing that number by the closing Bentley trading price
on the Separation Date. These CPEX awards were granted under the
Plan.
The Company follows the provisions of Statement of FASB ASC
Topic 718 (Prior authoritative literature:
SFAS No. 123(R), Share-Based Payment).
Compensation expense is recognized, based on the
requirements of ASC Topic 718, for all share-based payments.
The fair value of options granted was calculated using the
Black-Sholes option valuation model. ASC Topic 718 also requires
companies to use an estimated forfeiture rate when calculating
the expense for the period. The Company has applied an estimated
forfeiture rate to remaining unvested awards based on historical
experience in determining the expense recorded in the
Company’s statement of operations. This estimate is
periodically evaluated and adjusted as necessary. Ultimately,
the actual expense recognized over the vesting period will only
be for those shares that vest. See Note 8 for further
discussion.
Recently
issued accounting pronouncements
In September 2009, the Emerging Issues Task Force
(“EITF”) issued its final consensus for ASU
2009-13
(Prior authoritative literature:
EITF 08-1
Revenue Arrangements with Multiple Deliverables), which will
supersede the guidance in ASC
605-25
(prior authoritative literature:
EITF 00-21,
Revenue Arrangements with Multiple Deliverables). ASU
2009-13
retains the criteria from ASC
605-5 for
when delivered items in a multiple-deliverable arrangement
should be considered separate units of accounting, but modifies
the previous separation criterion under ASC
605-25 that
objective and reliable evidence of fair value of any undelivered
items must exist for the delivered items to be considered a
separate unit or separate units of accounting. ASU
2009-13
introduces a selling price hierarchy for multiple deliverable
arrangements and allows for management selling price estimates
in cases where no vendor specific objective evidence or third
party evidence is available. Additionally, this guidance
eliminates the residual method of allocation. ASU
2009-13 will
be applicable to the Company on January 1, 2011. The
Company has not yet evaluated the impact, if any, that ASU
2009-13 will
have on its financial statements.
In June 2009, the FASB issued ASC Topic 810 (Prior
authoritative literature: SFAS No. 167
Amendments to FASB Interpretation No. 46(R)),which
improves financial reporting by enterprises involved with
variable
F-14
CPEX
Pharmaceuticals, Inc. and Subsidiary
Notes to
Consolidated and Combined Financial
Statements — (Continued)
interest entities. ASC 810 addresses (1) the effects on
certain provisions of FASB Interpretation No. 46 (revised
December 2003), Consolidation of Variable Interest Entities, as
a result of the elimination of the qualifying special-purpose
entity concept in SFAS 166 and (2) concerns about the
application of certain key provisions of FIN 46(R),
including those in which the accounting and disclosures under
the Interpretation do not always provide timely and useful
information about an enterprise’s involvement in a variable
interest entity. ASC 810 will be applicable to the Company on
January 1, 2010. Earlier adoption is prohibited. The
Company does not expect the adoption of this topic to have a
material impact on its consolidated and combined financial
statements.
In June 2009, the FASB issued ASC Topic 860 (Prior
authoritative literature: SFAS No. 166,
Accounting for Transfers of Financial Assets, an
amendment of FASB Statement No. 140). ASC Topic 860
prescribes the information that a reporting entity must provide
in its financial reports about a transfer of financial assets;
the effects of a transfer on its financial position, financial
performance, and cash flows; and a transferor’s continuing
involvement in transferred financial assets. Specifically, among
other aspects, ASC Topic 860 amends SFAS No. 140,
Accounting for Transfers and Servicing of Financial Assets
and Extinguishments of Liabilities, or
SFAS No. 140, by removing the concept of a qualifying
special-purpose entity from SFAS No. 140 and removes
the exception from applying FASB Interpretation No. 46,
Consolidation of Variable Interest Entities (revised), or
FIN 46(R), to variable interest entities that are
qualifying special-purpose entities. It also modifies the
financial-components approach used in SFAS No. 140.
ASC Topic 860 is effective for transfer of financial assets
occurring on or after January 1, 2010. The Company does not
expect the adoption of this topic to have a material impact on
its consolidated and combined financial statements.
Effective July 1, 2009, the FASB issued ASC Topic 105
(Prior authoritative literature:
SFAS No. 168, The FASB Accounting Standards
Codification and the Hierarchy of Generally Accepted Accounting
Principles). ASC Topic 105 identifies the FASB Accounting
Standards Codification as the authoritative source of accounting
principles generally accepted in the United States
(“U.S. GAAP”). Rules and interpretive releases of
the SEC under federal securities laws are also sources of
authoritative U.S. GAAP for SEC registrants. ASC Topic 105
became effective for financial statements issued for interim
reporting periods ending after September 15, 2009.
Therefore, beginning with this
Form 10-Q,
all references made to U.S. GAAP in the notes to the
Company’s consolidated financial statements now use the new
ASC numbering system. The ASC does not change or alter existing
U.S. GAAP and, therefore, it does not have an impact on the
Company’s financial position, results of operations or cash
flows.
In May 2009, FASB issued FASB ASC Topic 855 (Prior
authoritative literature: SFAS No. 165,
Subsequent Events). ASC Topic 855 establishes general
standards of accounting for and disclosure of events that occur
after the balance sheet date but before financial statements are
issued or available to be issued. ASC
855-10-25
requires an entity to recognize in the financial statements the
effects of all subsequent events that provide additional
evidence about conditions that existed at the date of the
balance sheet. For unrecognized subsequent events that must be
disclosed to keep the financial statements from being
misleading, an entity will be required to disclose the nature of
the event as well as an estimate of its financial effect, or a
statement that such an estimate cannot be made. ASC 855 is
effective for the interim or annual financial periods ending
after June 15, 2009, and is required to be applied
prospectively. This guidance does not impact the Company’s
current consolidated and combined financial statements.
In February 2008, the FASB issued ASC
820-10-65-1
(Prior authoritative literature: FASB Staff Position
157-2,
“Effective Date of FASB Statement No. 157”),
which delays the effective date of ASC Topic 820 for
nonfinancial assets and nonfinancial liabilities, except for
items that are recognized or disclosed at fair value in the
financial statements on a reoccurring basis. The provisions of
ASC 820 became effective for fiscal years beginning after
November 15, 2008. The adoption of this staff position did
not have a material impact on the Company’s consolidated
and combined financial statements.
F-15
CPEX
Pharmaceuticals, Inc. and Subsidiary
Notes to
Consolidated and Combined Financial
Statements — (Continued)
In April 2009, the FASB issued ASC
820-10-65-4
(Prior authoritative literature: FASB Staff Position
SFAS 157-4,
“Determining Fair Value When the Volume and Level of
Activity for the Asset or Liability Have Significantly Decreased
and Identifying Transactions That Are Not Orderly”).
ASC
820-10-65-4
amends ASC
820-35-15A
to provide additional guidance on estimating fair value when the
volume and level of activity for an asset or liability have
significantly decreased in relation to normal market activity
for the asset or liability. ASC
820-10-65-4
is effective for interim reporting periods ending after
June 15, 2009. The adoption of this staff position did not
have a material impact on the Company’s consolidated and
combined financial statements.
In December 2007, the FASB issued ASC Topic 805 (Prior
authoritative literature: SFAS No. 141(R),
Business Combinations
(“SFAS No. 141(R)”). ASC
805-20-25-2
and 25-3
establish principles and requirements for how an acquirer
recognizes and measures in its financial statements the
identifiable assets acquired, the liabilities assumed, any
non-controlling interest in the acquiree and the goodwill
acquired. ASC Topic 805 also establishes disclosure requirements
which will enable users to evaluate the nature and financial
effects of the business combination. The Company adopted this
topic as of January 1, 2009 and determined that the
adoption did not have a material impact on its consolidated and
combined financial statements.
In December 2007, the FASB ratified the consensus reached by the
EITF on ASC Topic 808 (Prior authoritative literature:
Issue
No. 07-1,
Accounting for Collaborative Agreements”). ASC
808-10-20
provides the definition of a collaborative agreement and ASC
808-10-15
provides guidelines to assist an entity in determining whether
or not it is a party in a collaborative agreement. ASC
808-10-45 states
that costs incurred and revenues generated from transactions
with third parties shall be reported in accordance with ASC
605-45
(Prior authoritative literature: EITF Issue
No. 99-19,
Reporting Revenue Gross as a Principal versus Net as an Agent).
ASC
808-10-50
also provides minimum disclosure requirements for an
entity’s collaboration agreements and transition guidance.
The Company adopted ASC Topic 808 as of January 1, 2009 and
determined that the adoption did not have a material impact on
its consolidated and combined financial statements.
In April 2008, the FASB issued ASC Topic 350 (Prior
authoritative literature: FASB Staff Position
(“FSP”)
142-3,
Determination of the Useful Life of Intangible Assets). ASC
350-30-35
amends the factors that should be considered in developing
renewal or extension assumptions used to determine the useful
life of a recognized intangible asset under Topic ASC 350. The
Company adopted ASC
350-30-35 as
of January 1, 2009 and determined that the adoption did not
have a material impact on its consolidated and combined
financial statements.
In June 2008, the FASB issued ASC Topic 260 (Prior
authoritative literature:
EITF 03-6-1,
Determining Whether Instruments Granted in Share-Based Payment
Transactions Are Participating Securities). ASC
260-10-45
clarified that all outstanding unvested share-based payment
awards that contain rights to non-forfeitable dividends
participate in undistributed earnings with common shareholders.
Awards of this nature are considered participating securities
and the two-class method of computing basic and diluted earnings
per share must be applied. The Company adopted ASC Topic 260 as
of January 1, 2009 and determined that the adoption did not
have a material impact on its consolidated and combined
financial statements.
F-16
CPEX
Pharmaceuticals, Inc. and Subsidiary
Notes to
Consolidated and Combined Financial
Statements — (Continued)
Receivables consist of the following:
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
Royalties receivable
|
|
$
|
5,206
|
|
|
$
|
4,077
|
|
|
Other
|
|
|
83
|
|
|
|
368
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
5,289
|
|
|
$
|
4,445
|
|
|
|
|
|
|
|
|
|
|
|
Fixed assets consist of the following:
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
Land
|
|
$
|
787
|
|
|
$
|
787
|
|
|
Buildings and improvements
|
|
|
1,183
|
|
|
|
1,183
|
|
|
Equipment
|
|
|
2,151
|
|
|
|
1,593
|
|
|
Furniture and fixtures
|
|
|
248
|
|
|
|
248
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4,369
|
|
|
|
3,811
|
|
|
Construction in-progress
|
|
|
—
|
|
|
|
160
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4,369
|
|
|
|
3,971
|
|
|
Less-accumulated depreciation
|
|
|
(1,431
|
)
|
|
|
(1,139
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
2,939
|
|
|
$
|
2,832
|
|
|
|
|
|
|
|
|
|
|
|
Depreciation expense of approximately $292,000, $275,000 and
$243,000 has been charged to operations as a component of
Depreciation and amortization expense in the Consolidated
and Combined Statements of Operations for the years ended
December 31, 2009, 2008 and 2007, respectively.
|
|
|
NOTE 5 —
|
INTANGIBLE
ASSETS
|
Intangible assets consist of the following:
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
Patents and related patent costs
|
|
$
|
5,204
|
|
|
$
|
5,535
|
|
|
Less-accumulated amortization
|
|
|
(2,993
|
)
|
|
|
(3,141
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
2,211
|
|
|
$
|
2,394
|
|
|
|
|
|
|
|
|
|
|
|
Amortization expense for drug licenses and related costs was
approximately $407,000, $407,000 and $509,000 for the years
ended December 31, 2009, 2008 and 2007, respectively, and
has been recorded in Depreciation and amortization expense
in the accompanying Consolidated and Combined Statements of
Operations. During the year ended December 31, 2008 the
Company recorded $141,000 related to the write-down of certain
intangible assets related to the Company’s anti-fungal nail
lacquer. Impairment losses related to intangible assets are
included in research and development expenses in the
accompanying Consolidated and Combined Statements of Operations.
There were no impairment losses for the year ended
December 31, 2009.
F-17
CPEX
Pharmaceuticals, Inc. and Subsidiary
Notes to
Consolidated and Combined Financial
Statements — (Continued)
Amortization expense for existing drug licenses and related
costs for each of the next five years and for all remaining
years thereafter is estimated to be as follows:
| |
|
|
|
|
|
Year Ending December 31,
|
|
Future Amortization Expense
|
|
|
|
(In thousands)
|
|
|
|
2010
|
|
$
|
452
|
|
|
2011
|
|
|
452
|
|
|
2012
|
|
|
452
|
|
|
2013
|
|
|
452
|
|
|
2014
|
|
|
72
|
|
|
2015 and beyond
|
|
|
234
|
|
|
|
|
NOTE 6 —
|
RELATED
PARTY DISCLOSURES
|
Prior to the Separation Date, CPEX operations were fully
integrated with Bentley, including executive services, finance,
treasury, internal audit, corporate income tax, legal services
and investor relations. The accompanying Consolidated and
Combined Financial Statements reflect the application of certain
estimates and allocations of operating expenses including
stock-based compensation. Management believes the methods used
to allocate these operating expenses are reasonable. The
allocation methods include relative time devoted by executive
management on CPEX business and related benefit received by CPEX
for other services, such as public company costs and services.
Allocations of expenses for these services totaled
$4.4 million and $4.2 million for years ended
December 31, 2008 and 2007, respectively, and are reflected
in Total operating expenses in the Consolidated and
Combined Statements of Operations. There have been no
allocations of expenses charged to CPEX since the Separation
Date.
On June 13, 2008, CPEX and Bentley entered into a series of
agreements, — a Separation and Distribution Agreement,
a Tax Sharing Agreement, a Transition Services Agreement and an
Employee Matters Agreement — to facilitate CPEX in its
separation from Bentley. The Transition Services Agreement had
an initial term of six months and had an option to be extended
for an additional six-month term. As a result of the Transition
Services Agreement, CPEX and Bentley were able to provide
services to the other as requested for a fee based upon the
costs incurred in providing such services. As of the Separation
Date, Bentley had prepaid $78,000 to CPEX for services expected
to be provided to Bentley. Through December 31, 2008, CPEX
recognized $78,000 for performance of those services which is
included as an offset to operating expenses in General and
administrative in the accompanying Consolidated and Combined
Statements of Operations. The Transition Services Agreement
expired and was not extended.
|
|
|
NOTE 7 —
|
ACCRUED
EXPENSES
|
Accrued expenses consist of the following:
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
Accrued payroll and related taxes
|
|
$
|
828
|
|
|
$
|
715
|
|
|
Accrued clinical costs
|
|
|
183
|
|
|
|
92
|
|
|
Accrued professional fees
|
|
|
400
|
|
|
|
416
|
|
|
Other accrued expenses
|
|
|
222
|
|
|
|
311
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
1,633
|
|
|
$
|
1,534
|
|
|
|
|
|
|
|
|
|
|
|
F-18
CPEX
Pharmaceuticals, Inc. and Subsidiary
Notes to
Consolidated and Combined Financial
Statements — (Continued)
|
|
|
NOTE 8 —
|
EQUITY
AND SHARE-BASED COMPENSATION
|
As previously stated, as of December 31, 2009 the
Company’s one share-based compensation plan, which is
stockholder approved, permits awards of unrestricted common
stock, restricted stock, restricted stock units, options to
purchase CPEX common stock, stock appreciation rights and stock
equivalents for the Company’s employees, directors and
consultants. On June 18, 2009, the Company’s
stockholders approved, among other things, the addition of
100,000 shares of common stock to the reserve of shares
available for issuance under the Plan. Prior to the Separation,
all equity awards were granted by Bentley. In accordance with
the Employee Matters Agreement between the Company and Bentley,
outstanding Bentley awards held by U.S. employees,
directors and consultants were converted into an adjusted
Bentley award and a CPEX award on the Separation Date. The
number of common shares underlying the CPEX awards was
calculated as one-tenth of the number of common shares
underlying the original Bentley awards. The price of the CPEX
awards was determined by multiplying the original exercise price
of the Bentley awards by the when-issued trading price of CPEX
common stock on the Separation Date and then dividing that
number by the closing Bentley trading price on the Separation
Date. These CPEX awards were granted under the Plan. At
December 31, 2009, approximately 601,000 shares of
common stock were reserved for issuance under the Plan.
Approximately 466,000 of these shares were subject to
outstanding stock options and approximately 44,000 shares
were subject to outstanding restricted stock units. The balance
of approximately 91,000 shares were available for future
issuance of awards under the Plan. The table below presents the
Company’s option activity for the year ended
December 31, 2009:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted
|
|
|
|
|
|
|
|
|
|
|
Weighted
|
|
|
Average
|
|
|
Aggregate
|
|
|
|
|
Number of
|
|
|
Average
|
|
|
Remaining
|
|
|
Intrinsic
|
|
|
|
|
Options
|
|
|
Exercise
|
|
|
Contractual
|
|
|
Value
|
|
|
|
|
(in 000’s)
|
|
|
Price
|
|
|
Term (Years)
|
|
|
(in 000’s)
|
|
|
|
|
Options outstanding, January 1, 2009
|
|
|
430
|
|
|
$
|
15.85
|
|
|
|
|
|
|
|
|
|
|
Granted
|
|
|
61
|
|
|
|
7.81
|
|
|
|
|
|
|
|
|
|
|
Exercised
|
|
|
(2
|
)
|
|
|
2.69
|
|
|
|
|
|
|
|
|
|
|
Forfeited and expired
|
|
|
(23
|
)
|
|
|
17.21
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Options outstanding, December 31, 2009
|
|
|
466
|
|
|
$
|
14.78
|
|
|
|
8.17
|
|
|
$
|
278
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Vested and expected to vest, December 31, 2009
|
|
|
457
|
|
|
$
|
14.78
|
|
|
|
8.16
|
|
|
$
|
272
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Options exercisable, December 31, 2009
|
|
|
194
|
|
|
$
|
14.63
|
|
|
|
7.48
|
|
|
$
|
80
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Options to purchase approximately 2,000 shares of CPEX
common stock were exercised during the year ended
December 31, 2009 for net cash proceeds to the Company of
approximately $4,000, while the total intrinsic value of those
option exercises was approximately $14,000. Since the future
operating profits of CPEX cannot be reasonably assured, no tax
benefit resulting from the settlement of awards has been
recorded.
As of December 31, 2009, unrecognized compensation expense
related to the unvested portion of the Company’s stock
options granted to CPEX employees was approximately
$2.1 million and is expected to be recognized over a
weighted average period of approximately 1.65 years.
F-19
CPEX
Pharmaceuticals, Inc. and Subsidiary
Notes to
Consolidated and Combined Financial
Statements — (Continued)
The fair value of each option award granted to employees is
estimated on the date of grant using the Black-Scholes option
valuation model. Assumptions and the resulting fair value for
option awards granted by CPEX during the years ended
December 31, 2009 and 2008 are provided below:
| |
|
|
|
|
|
|
|
|
|
|
|
For the Year
|
|
|
For the Year
|
|
|
|
|
December 31,
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
Weighted average risk free interest rate
|
|
|
2.45
|
%
|
|
|
3.30
|
%
|
|
Dividend yield
|
|
|
0
|
%
|
|
|
0
|
%
|
|
Expected life (years)
|
|
|
7
|
|
|
|
5 to 7
|
|
|
Volatility
|
|
|
85%-88
|
%
|
|
|
65%-79
|
%
|
|
Weighted average grant-date fair value of options granted
|
|
$
|
5.98
|
|
|
$
|
11.53
|
|
The risk-free interest rate is based on the yield curve of
U.S. Treasury securities in effect at the date of the
grant, having a duration commensurate with the estimated life of
the award. CPEX does not expect to declare dividends in the
future. Therefore, an annual dividend rate of 0% is used when
calculating the grant date fair value of equity awards. The
expected life (estimated period of time outstanding) of options
granted is estimated based on historical exercise behaviors of
Bentley employees. Shares of CPEX common stock began trading on
the NASDAQ Capital Market on July 1, 2008. Since this
period of time is shorter than the expected term of the options
granted, the volatility applied is the average volatility of
CPEX and a peer group of comparable life science companies using
daily price observations for each company over a period of time
commensurate with the expected life of the respective award.
CPEX share-based awards generally vest over three years and the
maximum contractual term of the awards is 10 years.
In addition to the stock options described above, the Company
has granted restricted stock units to its employees. The common
shares subject to the restricted stock units are generally
issued when they vest. The table below presents the
Company’s restricted stock unit activity for the twelve
months ended December 31, 2009:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Number of
|
|
|
|
|
|
Weighted
|
|
|
|
|
|
|
|
Restricted
|
|
|
Weighted
|
|
|
Average
|
|
|
Aggregate
|
|
|
|
|
Stock
|
|
|
Average
|
|
|
Remaining
|
|
|
Intrinsic
|
|
|
|
|
Units
|
|
|
Grant Date
|
|
|
Contractual
|
|
|
Value
|
|
|
|
|
(in 000’s)
|
|
|
Fair Value
|
|
|
Term (Years)
|
|
|
(in 000’s)
|
|
|
|
|
Restricted stock units outstanding, January 1, 2009
|
|
|
14
|
|
|
$
|
12.87
|
|
|
|
|
|
|
|
|
|
|
Granted
|
|
|
35
|
|
|
|
9.03
|
|
|
|
|
|
|
|
|
|
|
Vested(1)
|
|
|
(16
|
)
|
|
|
10.44
|
|
|
|
|
|
|
|
|
|
|
Forfeited
|
|
|
(1
|
)
|
|
|
12.60
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Restricted stock units outstanding, December 31, 2009
|
|
|
32
|
|
|
$
|
9.64
|
|
|
|
1.52
|
|
|
$
|
347
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Vested and expected to vest, December 31, 2009
|
|
|
30
|
|
|
$
|
9.61
|
|
|
|
1.52
|
|
|
$
|
334
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) |
|
Includes approximately 13,000 restricted stock units that have
vested but for which the underlying common shares are not
settled or issued. |
As of December 31, 2009, unrecognized compensation expense
related to the unvested portion of the Company’s restricted
stock units granted to CPEX employees was approximately $204,000
and is expected to be recognized over a weighted average period
of approximately 1.52 years.
Share-based compensation expense relative to grants of stock
options and restricted stock units for the years ended
December 31, 2009, 2008 and 2007 totaled approximately
$1.8 million, $3.0 million and $1.2 million,
respectively. For years ended December 31, 2008 and 2007
the Consolidated and Combined Statements of Operations include
share-based compensation expense recorded for Bentley stock
option awards
F-20
CPEX
Pharmaceuticals, Inc. and Subsidiary
Notes to
Consolidated and Combined Financial
Statements — (Continued)
and Bentley restricted stock unit awards to CPEX employees prior
to the Separation and an allocation of share-based compensation
of executive officers, non-employee directors and consultants of
Bentley. The expenses were recorded in the Condensed
Consolidated and Combined Statements of Operations as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For Year Ended December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
|
|
(In thousands)
|
|
|
|
|
General and administrative expenses
|
|
$
|
1,351
|
|
|
$
|
2,023
|
|
|
$
|
630
|
|
|
Research and development expenses
|
|
|
413
|
|
|
|
968
|
|
|
|
612
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
1,764
|
|
|
$
|
2,991
|
|
|
$
|
1,242
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
No related compensation expense was capitalized as the cost of
an asset and there was no impact on net cash provided by
operating activities or net cash used in financing activities as
a result of these share-based transactions.
The Company sponsors a 401(k) Plan for eligible employees (the
“401k Plan”) and matches eligible contributions with
shares of the Company’s common stock. In July 2008, the
Company’s Board of Directors authorized and reserved
50,000 shares of common stock for the Company’s
contribution to the 401(k) Plan. As of December 31, 2009
approximately 27,000 of these shares were available for
contribution to the 401k Plan.
Share-based compensation expense includes matching contributions
to the 401(k) Plan by the Company. Share-based compensation
expense for periods prior to the Separation Date includes
expense attributable to CPEX employees from Bentley’s
401(k) matching contributions and the related allocated
share-based compensation of executive officers. For the years
ended December 31, 2009, 2008 and 2007 the related expenses
were recorded in the Condensed Consolidated and Combined
Statements of Operations as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For the Year Ended December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
|
|
(In thousands)
|
|
|
|
|
General and administrative expenses
|
|
$
|
76
|
|
|
$
|
99
|
|
|
$
|
91
|
|
|
Research and development expenses
|
|
|
96
|
|
|
|
105
|
|
|
|
171
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
172
|
|
|
$
|
204
|
|
|
$
|
262
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stockholder
Rights Plan
Pursuant to the Rights Agreement that was adopted by the Board
of Directors of the Company on June 12, 2008, the Board of
Directors declared a dividend of one Right for each outstanding
share of common stock payable to stockholders of record at the
close of business on June 23, 2008. Each Right, when
exercisable, entitles the registered holder to purchase one
one-thousandth of a share of Series A preferred stock, par
value $0.01 per share, at a purchase price of $100 per share,
subject to adjustment. The Rights Agreement is designed to
prevent a potential acquirer from gaining control of the Company
without fairly compensating all of the Company’s
stockholders and to protect the Company from coercive takeover
attempts. The Rights will become exercisable only if a person or
group of affiliated persons beneficially acquires 15% or more of
the Company’s common stock (subject to certain exceptions).
In the event that an acquiring person became the beneficial
owner of 15% or more of the then outstanding shares of common
stock (except pursuant to a qualifying offer), each holder of a
Right will thereafter have a right to receive, upon payment of
the purchase price, shares of common stock or, in certain
circumstances, cash, property, or other securities of the
Company having a value (based on a formula set forth in the
Rights Agreement) equal to two times the purchase price of the
Right. The Rights are not exercisable until the distribution
date and will expire at the close of business on June 12,
2018, unless earlier redeemed or exchanged by the Company.
F-21
CPEX
Pharmaceuticals, Inc. and Subsidiary
Notes to
Consolidated and Combined Financial
Statements — (Continued)
|
|
|
NOTE 9 —
|
PROVISION
FOR INCOME TAXES
|
The provisions for income taxes in 2009, 2008 and 2007 have been
determined based on the CPEX business operations as a separate,
stand-alone entity. The provision for income taxes in 2008 has
been determined based on the CPEX business operations following
its separation from Bentley on the Separation Date
(June 30, 2008). The separation was accomplished in a
transaction by which Bentley’s drug delivery business was
contributed to CPEX, a wholly owned subsidiary of Bentley. CPEX
was then spun out from Bentley in a taxable transaction by which
Bentley shareholders received CPEX stock in a pro-rata
distribution. Prior to the Separation Date, the CPEX operations
were fully integrated with Bentley, which operated within
multiple taxing jurisdictions and is subject to audit in those
jurisdictions. These audits can involve complex issues, which
may require an extended period of time for resolution. CPEX and
Bentley entered into a Tax Sharing Agreement under which Bentley
is obligated on all taxes arising from the CPEX operations up to
the Separation Date. CPEX is obligated on all taxes arising from
the CPEX operations following the Separation Date.
Although results from operations were presented for the year
ended December 31, 2008, net operating losses incurred
through the Separation Date will be included in tax returns
filed by Bentley and, accordingly, no tax benefits for these
losses have been recorded in the income tax provision or
deferred tax assets. The provision for income taxes for 2007 has
been determined on a pro-forma basis as if CPEX had filed
separate tax returns under its current structure for the periods
presented.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
|
|
(In thousands)
|
|
|
|
|
Current
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
Deferred:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
State
|
|
|
(158
|
)
|
|
|
(40
|
)
|
|
|
(24
|
)
|
|
Federal
|
|
|
(1,505
|
)
|
|
|
(396
|
)
|
|
|
(4,082
|
)
|
|
Tax effect of operating loss carryforwards:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
State
|
|
|
—
|
|
|
|
—
|
|
|
|
(56
|
)
|
|
Federal
|
|
|
—
|
|
|
|
—
|
|
|
|
1,590
|
|
|
Change in valuation allowance
|
|
|
1,663
|
|
|
|
436
|
|
|
|
2,572
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total provision for income taxes
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
A reconciliation of the income tax provision using the federal
statutory rate to the CPEX effective income tax rate is as
follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
|
|
(In thousands)
|
|
|
|
|
Income tax provision (benefits) at federal statutory rates
|
|
$
|
(1,034
|
)
|
|
$
|
(242
|
)
|
|
$
|
(1,676
|
)
|
|
Foreign income tax rate differential
|
|
|
—
|
|
|
|
—
|
|
|
|
(2,735
|
)
|
|
State income taxes (net of federal benefit)
|
|
|
(158
|
)
|
|
|
(40
|
)
|
|
|
(80
|
)
|
|
Expiration and utilization of operating loss carryforwards
|
|
|
—
|
|
|
|
—
|
|
|
|
2,316
|
|
|
Tax credits
|
|
|
(496
|
)
|
|
|
(156
|
)
|
|
|
(402
|
)
|
|
Other
|
|
|
25
|
|
|
|
2
|
|
|
|
5
|
|
|
Change in valuation allowance
|
|
|
1,663
|
|
|
|
436
|
|
|
|
2,572
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
F-22
CPEX
Pharmaceuticals, Inc. and Subsidiary
Notes to
Consolidated and Combined Financial
Statements — (Continued)
The components of the CPEX deferred taxes follow. Under the
terms of the Separation, the deferred tax assets at
December 31, 2007 and through the Separation Date did not
carryforward to CPEX and are therefore not included in the
December 31, 2009 and 2008 balances.
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
Deferred tax assets:
|
|
|
|
|
|
|
|
|
|
Net operating loss carryforwards
|
|
$
|
656
|
|
|
$
|
214
|
|
|
Book/tax basis difference in assets
|
|
|
309
|
|
|
|
308
|
|
|
Share-based compensation
|
|
|
1,024
|
|
|
|
344
|
|
|
Research and development tax credit carryforwards
|
|
|
652
|
|
|
|
156
|
|
|
Other, net
|
|
|
326
|
|
|
|
282
|
|
|
|
|
|
|
|
|
|
|
|
|
Total deferred tax assets
|
|
|
2,967
|
|
|
|
1,304
|
|
|
Valuation allowance
|
|
|
(2,967
|
)
|
|
|
(1,304
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Deferred tax asset, net
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
As future operating profits cannot be reasonably assured, no tax
benefit has been recorded for net operating losses. Accordingly,
CPEX has established a valuation allowance equal to the full
amount of the deferred tax assets. Should CPEX determine that it
is more likely than not that it will realize certain of its net
deferred tax assets for which it has previously provided a
valuation allowance, an adjustment would be required to reduce
the existing valuation allowance.
The valuation allowance for CPEX increased by $1,663,000 in 2009
and by $436,000 in 2008. The 2009 increase consists of $680,000
attributable to stock options, $496,000 in general business
credits and $442,000 to NOL carryforwards. The valuation
allowance of $1,304,000 in 2008 consists of $344,000
attributable to stock options, 214,000 to NOL carryforwards, and
$156,000 in general business credits. The valuation allowance
increased by approximately $2,572,000 in 2007, which is
primarily attributed to $3,855,000 related to U.S. NOL
carryforwards and $402,000 from general business credits. The
general business credits are attributed to the Company’s
research and development activities.
The Company generated net operating losses (“NOLs”) of
approximately $3,040,000 in 2009 and $714,000 in 2008. As of
December 31, 2009, the CPEX U.S. Federal NOL
carryforwards were approximately $1,677,000. If not offset
against future taxable income, the NOL carryforwards will expire
in tax years 2028 through 2029. All NOLs generated through the
Separation Date related to the CPEX business were retained by
Bentley.
As of December 31, 2009, the CPEX U.S. Federal general
business credit carryforwards were approximately $652,000. If
not offset against future federal taxes, these general business
credit carryforwards will expire in the tax years 2028 and 2029.
All general business credits generated through the Separation
Date related to the CPEX business were retained by Bentley. The
Company did not experience any ownership changes during 2009
under Internal Revenue Code Section 382 that would further
limit their NOLs or general business credit carryforwards.
In June 2006, the FASB issued ASC Topic 740 (Prior
authoritative literature: FIN 48, Accounting for
Uncertainty in Income Taxes), which CPEX adopted effective
January 1, 2007. The adoption of FIN 48 did not have a
material impact on the CPEX combined financial statements. CPEX
recognizes interest and penalties related to uncertain tax
positions as a component of the provision for income taxes.
There were no unrecognized tax positions relating to CPEX at the
date of adoption. As of December 31, 2009, the tax year
F-23
CPEX
Pharmaceuticals, Inc. and Subsidiary
Notes to
Consolidated and Combined Financial
Statements — (Continued)
2008 is the only year that is open to examination due to the
fact that it was the initial year in which CPEX filed as a
taxpayer separate from the Bentley consolidated group.
|
|
|
NOTE 10 —
|
SUBSEQUENT
EVENTS
|
In accordance with ASC Topic 855 (as amended), the Company
evaluated subsequent events through the date the audited
consolidated and combined financial statements were issued.
During this period the Company did not have any material
recognizable subsequent events that require further disclosure
in this filing.
|
|
|
NOTE 11 —
|
COMMITMENTS,
CONTINGENCIES AND CONCENTRATIONS
|
The Company is obligated to pay certain royalty payments upon
commercialization of products using its CPE-215 technology
acquired in 1999 and its intellectual property acquired in 2003.
The royalties are primarily calculated based upon net sales of
certain products generated by the intellectual property. As of
December 31, 2009, no royalties are due under the
agreements.
Legal
Proceedings
In October 2008, we and Auxilium received notice that
Upsher-Smith Laboratories filed an Abbreviated New Drug
Application, or ANDA, containing a paragraph IV
certification in which it certified that it believes that its
testosterone gel product does not infringe our patent,
U.S. Patent No. 7,320,968 (“the ‘968
Patent”). The ‘968 patent covers a method for
maintaining effective blood serum testosterone levels for
treating a hypogonadal male using Testim and will expire in
January 2025. The ‘968 Patent is listed in Approved Drug
Products with Therapeutic Equivalence Evaluations (commonly
known as the Orange Book), published by the U.S. Food and
Drug Administration. Upsher-Smith Laboratories’
paragraph IV certification sets forth allegations that the
‘968 Patent will not be infringed by Upsher-Smith’s
manufacture, use or sale of the product for which the ANDA was
submitted. On December 4, 2008, we and Auxilium filed a
lawsuit in the United States District Court for the District of
Delaware against Upsher-Smith under the Hatch Waxman Act for
infringement of our patent. In June 2009, Upsher-Smith amended
its answer to the complaint to include a defense and
counterclaim of invalidity of the ‘968 Patent, which CPEX
and Auxilium deny. A patent issued by the U.S. Patent and
Trademark Office, such as the ‘968 Patent, is presumed
valid. The lawsuit is currently ongoing. Any U.S. Food and
Drug Administration (FDA) approval of Upsher-Smith’s
proposed generic product will be stayed until the earlier of
30 months from the date of receipt of the paragraph IV
certification (April 2011) or an adverse decision in our
patent infringement lawsuit.
CPEX has filed continuation and divisional applications with the
USPTO relating to the ‘968 patent. Six patents issued from
these applications on October 27, 2009, namely
U.S. Patent Nos. 7,608,605, 7,608,606, 7,608,607,
7,608,608, 7,608,609 and 7,608,610, which may provide us with
further market protection. Each of these six patents has been
listed in listed in Approved Drug Products with Therapeutic
Equivalence Evaluations (commonly known as the Orange Book) with
respect to Testim.
We and Auxilium are committed to protecting our intellectual
property rights and will vigorously pursue this lawsuit.
However, if we are unsuccessful in defending the ‘968
Patent covering Testim, sales of Testim and our royalties
relating to Testim sales will be materially reduced.
Agreement
for a Potential Joint Venture
As previously disclosed, on March 17, 2009, the Company
signed an agreement with Heights Partners, LLC
(“Heights”) to evaluate the desirability for the
Company and Heights to enter into a joint venture arrangement
for the development of specified product candidates of Heights.
Under the agreement, the parties evaluated several product
candidates and on October 15, 2009, the Company advised
Heights that it had selected two products for the collaboration.
Under the terms of the agreement, the parties must execute a
joint
F-24
CPEX
Pharmaceuticals, Inc. and Subsidiary
Notes to
Consolidated and Combined Financial
Statements — (Continued)
venture or other contractual arrangement within 90 days, or
by January 13, 2010, to conduct such collaboration. The
parties agreed to extend this deadline until May 15, 2010.
Under the agreement the Company paid $300,000 as an advance
against its initial contribution to the potential joint venture.
This advance was evidenced by a promissory note payable by
Heights to CPEX and secured by a first priority security
interest in Height’s intellectual property, including,
without limitation, patents, patent applications and know-how,
covering any of the specified product development projects, all
licenses and other agreements with respect to the foregoing and
all proceeds the third party may receive from the sale or
license of the intellectual property or products. Under the
terms of the agreement as amended, the promissory note, which is
included in non-current assets in the accompanying Consolidated
Balance Sheet for the year ended December 31, 2009,
together with interest, was due in October 2009. Upon execution
of a joint venture or other contractual arrangement, Heights
shall contribute the amounts payable under the note to the joint
venture as the Company’s initial investment. If the parties
are unable to execute a joint venture agreement by May 15,
2010, and absent any further extension, all amounts advanced by
the Company will become payable by Heights and either party may
seek to terminate the obligation to form the joint venture.
F-25