Attached files
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, DC 20549
Form 10-K
(Mark One)
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
| For the fiscal year ended December 31, 2009 |
or
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
| For the transition period from to |
Commission file number: 001-33185
MEDICINOVA, INC.
(Exact Name of Registrant as Specified in its Charter)
| Delaware | 33-0927979 | |
| (State or Other Jurisdiction of Incorporation or Organization) |
(I.R.S. Employer Identification No.) | |
| 4350 La Jolla Village Drive, Suite 950, San Diego, CA | 92122 | |
| (Address of Principal Executive Offices) | (Zip Code) | |
(858) 373-1500
(Registrant’s Telephone Number, Including Area Code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of Each Class |
Name of Each Exchange on Which Registered | |
| Common Stock, par value $0.001 per share | The NASDAQ Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act:
Series A Participating Preferred Stock Purchase Rights
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes [ ] No [X]
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes [ ] No [X]
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes [X] No [ ]
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). [ ] Yes [ ] No
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. [X]
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See definitions of “large accelerated filer”, “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer [ ] Accelerated filer [ ] Non-accelerated filer [ ] Smaller reporting company [X]
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Securities Exchange Act of 1934). Yes [ ] No [X]
The aggregate market value of the registrant’s common stock held by non-affiliates of the registrant was approximately $43,192,907 based on the closing price of the registrant’s common stock on the Nasdaq Global Market of $4.25 per share on June 30, 2009. Shares of common stock held by each executive officer and director and each person who beneficially owns 10% or more of the outstanding common stock have been excluded from this calculation. This determination of affiliate status may not be conclusive for other purposes.
The number of outstanding shares of the registrant’s common stock, par value $0.001 per share, as of March 22, 2010 was 12,371,508.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s proxy statement to be filed with the Securities and Exchange Commission pursuant to Regulation 14A in connection with the registrant’s 2009 Annual Meeting of Stockholders, which will be filed subsequent to the date hereof, are incorporated by reference into Part III of this Form 10-K. Such proxy statement will be filed with the Securities and Exchange Commission not later than 120 days following the end of the registrant’s fiscal year ended December 31, 2009.
Table of Contents
FORM 10-K—ANNUAL REPORT
For the Fiscal Year Ended December 31, 2009
Table of Contents
| Page | ||||
| PART I |
||||
| Item 1 |
1 | |||
| Item 1A |
32 | |||
| Item 1B |
56 | |||
| Item 2 |
56 | |||
| Item 3 |
57 | |||
| Item 4 |
57 | |||
| PART II |
||||
| Item 5 |
Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities | 58 | ||
| Item 6 |
61 | |||
| Item 7 |
Management’s Discussion and Analysis of Financial Condition and Results of Operations | 63 | ||
| Item 7A |
77 | |||
| Item 8 |
78 | |||
| Item 9 |
Changes in and Disagreements With Accountants on Accounting and Financial Disclosure | 114 | ||
| Item 9A(T) |
114 | |||
| Item 9B |
114 | |||
| PART III |
||||
| Item 10 |
115 | |||
| Item 11 |
115 | |||
| Item 12 |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | 115 | ||
| Item 13 |
Certain Relationships and Related Transactions, and Director Independence |
116 | ||
| Item 14 |
116 | |||
| PART IV |
||||
| Item 15 |
117 | |||
| 121 | ||||
The MediciNova logo is a registered trademark of MediciNova, Inc. All other product and company names are registered trademarks or trademarks of their respective companies.
i
Table of Contents
PART I
Forward-Looking Statements
This Annual Report on Form 10-K includes forward-looking statements that involve a number of risks and uncertainties, many of which are beyond our control. Our actual results may differ from those anticipated or expressed in these forward-looking statements as a result of various factors, including those set forth below under the caption “Item 1A. Risk Factors,” and the differences may be material. Forward-looking statements discuss matters that are not historical facts. Forward-looking statements include discussions regarding our operating strategy, growth strategy, licensing and acquisition strategy, cost savings initiatives, industry and economic conditions, market factors, financial condition, liquidity and capital resources, results of operations, expected progress of the development of our product candidates, potential licensing, collaboration and partnering plans, anticipated trends and challenges in our business and the markets in which we operate, competitive position, intellectual property protection, critical accounting policies and the impact of recent accounting pronouncements. In this report, for example, we make forward-looking statements regarding the potential for our product candidates to receive regulatory approval for one or more indications on a timely basis or at all; the progress and results of pending clinical trials for certain of our product candidates, including any delays in commencing or completing enrollment for our ongoing or planned clinical trials; plans for future clinical trials and regulatory submissions; unexpected adverse side effects or inadequate therapeutic efficacy of certain of our product candidates that could delay or prevent regulatory approval or commercialization or that could result in product liability claims; other difficulties or delays in development, testing, manufacturing and marketing of and obtaining regulatory approval for our product candidates; the scope and validity of patent protection for our product candidates; the market potential for our target markets and our ability to compete; the potential to attract one or more strategic partners and terms of any related transactions; intense competition if any of our product candidates are ever commercialized; our ability to realize the anticipated strategic and financial benefits of our acquisition of Avigen, Inc., or Avigen; our ability to integrate Avigen’s ibudalist development program with ours; the potential impact of uncertainties in the credit and capital markets or a future deterioration of these markets on our investment portfolio; and our ability to raise sufficient capital or debt financing when needed, or at all. Such forward-looking statements include statements preceded by, followed by or that otherwise include the words “may,” “might,” “will,” “intend,” “should,” “could,” “can,” “would,” “expect,” “believe,” “estimate,” “anticipate,” “predict,” “potential,” “plan” or similar words. For all forward-looking statements, we claim the protection of the safe harbor for forward-looking statements contained in the Private Securities Litigation Reform Act of 1995. You should not rely unduly on these forward-looking statements, which speak only as of the date on which they are made. We undertake no obligation to revise or update publicly any forward-looking statements, whether as a result of new information, future events or otherwise, unless required by law.
Overview
We are a biopharmaceutical company focused on acquiring and developing novel, small molecule therapeutics for the treatment of diseases with unmet medical need with a specific focus on the U.S. market. Through strategic alliances, primarily with Japanese pharmaceutical companies, we hold rights to a diversified portfolio of clinical and preclinical product candidates, each of which we believe has a well-characterized and differentiated therapeutic profile, attractive commercial potential and patent assets having claims of commercially adequate scope. We were incorporated in Delaware in September 2000. In December 2009, we completed our acquisition of Avigen, a biopharmaceutical company that had focused on identifying and developing differentiated products to treat patients with serious disorders.
We believe that our ability to gain access to and acquire potentially high-value product candidates from Japanese and European pharmaceutical companies is largely attributable to the established relationships and broad industry experience of our management team. In particular, we believe our relationships with Japanese
1
Table of Contents
pharmaceutical companies and their executives provide us with a competitive advantage in opportunistically sourcing product candidates from Japanese pharmaceutical companies at attractive terms. Since our inception, we have established relationships with a number of pharmaceutical companies, including Kissei Pharmaceutical Co., Ltd., or Kissei Pharmaceutical, Kyorin Pharmaceutical Co., Ltd., or Kyorin Pharmaceutical, Mitsubishi Tanabe Pharma Corporation and Meiji Seika Kaisha, Ltd., or Meiji Seika Kaisha, in Japan and Angiogene Pharmaceuticals, Ltd., or Angiogene Pharmaceuticals, in the United Kingdom, pursuant to which we have obtained rights to develop and commercialize our current product candidates.
Since our inception, we have acquired licenses to eight compounds for the development of ten product candidates in what we believe are large and underserved markets. Our development pipeline consists of eight product development programs which have been in clinical development for the treatment of asthma, acute exacerbations of asthma, multiple sclerosis, or MS, other central nervous system, or CNS, disorders, interstitial cystitis, or IC, solid tumor cancers, Generalized Anxiety Disorder/insomnia, preterm labor and urinary incontinence. Our two earlier stage product development programs have been in preclinical development for the treatment of thrombotic disorders. In addition, we have expanded the development program for one of our prioritized product candidates, MN-221, to evaluate MN-221 for the treatment of Chronic Obstructive Pulmonary Disease, or COPD exacerbations.
Our current strategy is to focus our resources on the development of two prioritized product development programs:
| Product Candidate |
Disease/Indication |
Phase of Development |
Licensor |
Licensed Territory | ||||
| MN-221 | Acute exacerbations of asthma and COPD exacerbations | Phase II clinical trial in emergency rooms to evaluate MN-221 at planned escalating doses in patients with severe, acute exacerbations of asthma completed in Q2, 2009
Phase II clinical trial in emergency rooms to evaluate safety and efficacy in patients with severe, acute exacerbations of asthma initiated in Q1, 2009
Phase Ib clinical trial to evaluate the safety and efficacy of MN-221 in patients with stable, moderate to severe COPD initiated in Q4, 2009 and completed in Q1, 2010 |
Kissei Pharmaceutical |
Worldwide, except Japan | ||||
| MN-166/ AV411* |
Multiple sclerosis and other CNS disorders** | Phase II clinical trial of MN-166 completed in Q2, 2008.
Prototype once-per-day oral formulation developed for future clinical trials. |
Kyorin Pharmaceutical (MN-166) |
Worldwide, except Japan, China, Taiwan and South Korea (MN-166) | ||||
2
Table of Contents
| * | MN-166 and AV411 are both ibudalist, an orally available, small molecule therapeutic. With the acquisition of AV411, we intend to integrate the two ibudalist-based product development programs and pursue discussions with potential partners to secure a strategic collaboration to advance clinical development of the combined development programs. |
AV411 has advanced through multiple Phase 1 and 2a clinical trials in healthy volunteers and patients with neuropathic pain and is currently in a Phase 1b/2a opioid withdrawal clinical trial funded by the National Institute on Drug Abuse, or NIDA. AV411 is also in collaborative studies with NIDA for methamphetamine addiction.
| ** | Other CNS disorders encompass neuropathic pain, opioid withdrawal and methamphetamine addiction. |
Upon completion of proof-of-concept Phase II clinical trials, we will either continue to pursue clinical development independently in the United States, as we presently intend with MN-221, or establish a strategic collaboration to support further clinical development, as we presently intend with MN-166/AV411.
We intend to limit development activities for the balance of our product candidates. For each of these remaining product candidates, we plan to conduct development activities only to the extent deemed necessary to maintain our license rights or maximize our value while pursuing a variety of initiatives to monetize such product candidate on appropriate terms. We cannot assure you that we will be successful in monetizing these product candidates on attractive terms, or at all. See “Risk Factors.”
These eight product development programs consist of:
| Product Candidate |
Disease/Indication |
Phase of Development*** |
Licensor |
Licensed Territory | ||||
| MN-001 | Bronchial asthma | Phase III clinical trial initiated in Q4, 2006 and terminated in Q2, 2007; Once-per-day oral dosing formulation prototypes developed | Kyorin Pharmaceutical |
Worldwide, except Japan, China, Taiwan and South Korea | ||||
| MN-001 | Interstitial cystitis | Phase II/III clinical trial completed in Q1, 2007† | Kyorin Pharmaceutical |
Worldwide, except Japan, China, Taiwan and South Korea | ||||
| MN-029 | Solid tumors | Phase I clinical trial completed in Q2, 2006; Second Phase I clinical trial completed in Q4, 2007 | Angiogene Pharmaceuticals |
Worldwide | ||||
| MN-305 | Generalized Anxiety Disorder/ Insomnia | Phase II/III clinical trial completed in Generalized Anxiety Disorder in Q2, 2006†; Phase II clinical trial in insomnia completed in Q4, 2007†† | Mitsubishi Tanabe Pharma Corporation | Worldwide, except Japan and certain countries in Asia | ||||
| MN-221 | Preterm labor | Phase I clinical trial completed in Q2, 2007 | Kissei Pharmaceutical | Worldwide, except Japan | ||||
| MN-246 | Urinary incontinence | Phase I clinical trial completed in Q4, 2006; Phase I food effects study completed in Q1, 2007 | Mitsubishi Tanabe Pharma Corporation | Worldwide, except Japan and certain countries in Asia | ||||
3
Table of Contents
| MN-447 | Thrombotic disorders | Preclinical | Meiji Seika Kaisha | Worldwide, except Japan and certain countries in Asia | ||||
| MN-462 | Thrombotic disorders | Preclinical | Meiji Seika Kaisha | Worldwide, except Japan and certain countries in Asia | ||||
| *** | We define a product candidate to be in Phase II/III when the clinical trial design is such that, if the primary endpoint is met, the results may provide confirmatory evidence of efficacy if we choose to submit the clinical trial as a pivotal trial and the U.S. Food and Drug Administration, or FDA, chooses to review the clinical trial as a pivotal trial. However, in regulatory filings with the FDA, we have nominally described these clinical trials as Phase II clinical trials. |
| † | Although positive signs of efficacy were obtained in the clinical trials conducted on MN-001 in interstitial cystitis and MN-305 in Generalized Anxiety Disorder, the predefined primary statistical endpoints of the clinical trials were not achieved; therefore, we would not anticipate submitting either clinical trial as a pivotal trial supporting a New Drug Application, or NDA, to the FDA. |
| †† | In the Phase II clinical trial conducted on MN-305 in insomnia, the predefined statistical endpoint of the clinical trial was not achieved; therefore, we terminated any further development of MN-305 for the treatment of insomnia. |
Our Strategy
Our goal is to build a sustainable biopharmaceutical business through the successful development and commercialization of differentiated products for the treatment of diseases with unmet medical need in high-value therapeutic areas. Key elements of our strategy are as follows:
| • | Concentrate our resources on our two prioritized product development programs, MN-221 and MN-166/AV411. We may either pursue the development and commercialization of these product candidates ourselves or enter into strategic alliances with larger pharmaceutical companies to do the same. We intend to pursue further development of MN-221 for the treatment of acute exacerbations of asthma and COPD exacerbations independently in the United States; however, following completion of the Phase II clinical trial of MN-166 for the treatment of MS in the second quarter of 2008 and the acquisition of AV411 in December 2009, we have not undertaken, nor do we plan to undertake, any further significant clinical development of MN-166/AV411 until such time that we secure a strategic collaboration to advance the combined MN-166/AV411 ibudalist-based development program. We intend to actively pursue strategic collaborations for these product development programs to draw on the development, regulatory and commercialization expertise and financial resources of larger biotechnology and pharmaceutical partners. We may also decide to pursue potential partners and potential acquirers of license rights to our programs in markets outside the United States, with the goal of retaining significant commercial participation in these product opportunities. |
| • | Pursue additional indications and commercial opportunities for our prioritized product candidates. We will seek to maximize the value of MN-221 and MN-166/AV411 by pursuing other potential indications and commercial opportunities for such product candidates. For example, we have rights to develop and commercialize MN-221 for any disease or indication. In addition to the ongoing evaluation of MN-221 for the treatment of acute exacerbations of asthma, we have recently expanded our development program for MN-221 to evaluate MN-221 for the treatment of COPD exacerbations utilizing our existing Investigational New Drug Application, or IND, for MN-221. |
| • | Maximize the value of the remainder of our diversified pipeline of existing product candidates. We will conduct development activities strategically on the remainder of our existing product candidates, to the extent that we deem any further activities necessary to maintain our license rights or maximize their value, while aggressively pursuing a variety of initiatives to monetize these product candidates on appropriate terms. |
4
Table of Contents
| • | Opportunistically in-license additional product candidates through our global industry relationships. Over the long term, we intend to expand our pipeline of in-licensed product candidates by continuing to cultivate and strengthen our business relationships with pharmaceutical companies in Japan and other markets. We believe our ability to take advantage of industry relationships to acquire product candidates with high potential and existing preclinical or early clinical data from Japanese pharmaceutical companies provides us with a competitive advantage over other drug development companies in the U.S. market. We believe that additional diversification and expansion of our pipeline of product candidates will help maximize the commercial opportunity and mitigate the risks inherent in drug discovery and development. |
| • | Selectively add commercial capabilities as our product development programs mature. To ensure our ability to build a sustainable business, we plan to selectively add commercial capabilities to our management team to support our evolution into a commercial entity as our product development programs mature. We may develop our own marketing and sales organization to promote certain of our product candidates. |
Product Development Programs
Our product development programs address diseases that we believe are not well served by currently available therapies and represent significant commercial opportunities. We believe that our product candidates offer innovative therapeutic approaches that may provide significant advantages relative to current therapies.
Our product acquisitions have focused primarily on product candidates with significant preclinical and early clinical testing data that have been developed by the licensors outside of the United States. We utilize the existing data in preparing INDs or foreign equivalents and designing additional clinical trials to advance the regulatory approval process in the United States or abroad. Following are details of our product development programs:
Prioritized Product Candidates
The current state of the development program for each of our two prioritized product candidates is described below.
MN-221 for Acute Exacerbations of Asthma
Indication Overview and Market Opportunity. An acute exacerbation of asthma is a long-lasting and severe asthma episode in which asthma symptoms are not responsive to initial bronchodilator or corticosteroid therapy. Acute exacerbations of asthma are an emergency situation that can lead to emergency department treatment and, in some cases, hospital admission or death. Beta-agonist agents are the mainstays of acute treatment for these types of asthma attacks and are included in the recommended standard of care according to the National Guideline Clearinghouse from the Department of Health and Human Services for patients suffering from acute exacerbations of asthma.
Data from the National Center for Health Statistics show that visits to emergency departments for asthma increased from approximately 1.5 million in 1992 to approximately 1.7 million in 2006. Despite significant improvements in the treatment for asthma over the past 20 years, there has not been a corresponding decrease in either hospitalizations or deaths due to asthma according to the National Center for Health Statistics. Data from the National Center for Health Statistics show that approximately 444,000 hospital discharges were attributed to asthma in 2006. In addition, there were approximately 2,563 deaths due to asthma during 2006. According to the National Heart, Lung and Blood Institute, the direct costs associated with hospital care due to asthma were $4.7 billion in 2007. We believe that there remains an unmet medical need for a safe and effective treatment for acute exacerbations of asthma that could prevent some of these hospitalizations.
5
Table of Contents
Overview of MN-221 in Acute Exacerbations of Asthma. MN-221 is a novel, highly selective ß2-adrenergic receptor agonist being developed for the treatment of acute exacerbations of asthma. We licensed MN-221 from Kissei Pharmaceutical in February 2004. Preclinical studies conducted in vitro and in vivo showed MN-221 to be highly selective for the ß 2-adrenergic receptor. In these studies, the ß1-adrenergic receptor stimulating activity of MN-221 was less than that of other ß2-adrenergic receptor agonists in isolated rat atrium and in vivo cardiac function tests in rats, dogs and sheep, thereby suggesting that the stimulating action of older, less selective ß2-adrenergic receptor agonists on the heart via ß1-adrenergic receptors may be reduced with MN-221 due to its greater ß2-adrenergic receptor selectivity. In vitro studies also suggested that MN-221 may act as only a partial ß1-adrenergic receptor agonist in cardiac tissue, while acting as a full ß2-adrenergic receptor in lung tissue. In addition, a preclinical drug interaction study in dogs completed during 2008 demonstrated that, while each of albuterol and MN-221 induced an increase in heart rate independently, the addition of MN-221 by intravenous administration in combination with inhaled albuterol did not add to the heart rate increase associated with inhaled albuterol alone, which further suggests that MN-221 acts as a partial agonist at ß1-adrenergic receptors. We believe that this improved receptor binding and functional selectivity may result in fewer cardiovascular side effects than are commonly observed with other ß2 -adrenergic receptor agonists used to treat this condition. We have developed and studied an intravenous formulation of MN-221 appropriate for hospital use.
Clinical Results. We initiated a randomized, double-blind, placebo-controlled, dose escalation, multi-center Phase II clinical trial of MN-221 in January 2007 to evaluate the safety and efficacy of MN-221. We completed this Phase II clinical trial, which involved 23 stable mild-to-moderate asthmatics, in August 2007. At each dose level in the escalation, patients were randomized to receive either a 15-minute intravenous infusion of MN-221 or placebo. This clinical trial achieved statistical significance in its primary endpoint of mean change in forced expiratory volume in one second, or FEV1, from baseline to measurement at 15 minutes (the end of the infusion) at doses of 10, 16, 30 and 60 micrograms per minute of MN-221 (p-value less than or equal to 0.0006) compared to placebo. MN-221 produced a significant linear, dose-related increase in mean change in post-infusion FEV1 from baseline (p-value less than or equal to 0.0001) following a 15-minute intravenous infusion of MN-221. Significant improvements in mean change in post-infusion (15 minute) FEV1 from baseline were observed at doses of 10, 16, 30 and 60 micrograms per minute (p-value less than or equal to 0.0006) and at the dose of 3.5 micrograms per minute (p-value=0.0106) compared to placebo. In the protocol correct population for this clinical trial, which consisted of 21 patients, the dose-related increases in FEV1 were maintained for four hours (p-value=0.0393) and at eight hours (p-value=0.0424) following the 15-minute infusion of MN-221. MN-221 was well tolerated in this Phase II clinical trial, with only the expected ß2-adrenergic receptor pharmacology noted in some patients (e.g., fall in serum potassium, elevation in plasma glucose, mild headache and mild tremors). There were no clinically significant cardiovascular, electrocardiogram, or ECG, or vital sign changes observed at any dose tested. In addition, no serious adverse effects were observed in this clinical trial.
We initiated a randomized, open-label, placebo-controlled Phase II clinical trial in June 2008 to evaluate the safety and efficacy of MN-221 in patients with moderate to severe, but stable asthma. We completed this clinical trial, which involved 17 patients in two dose cohorts, in September 2008. In one dosing cohort, each patient received MN-221 at a dose of 1,125 micrograms or placebo over one hour by a continuous intravenous infusion. In the other dosing cohort, each patient received MN-221 at a dose of 1,080 micrograms or placebo over two hours by a continuous intravenous infusion. Both infusion rates of MN-221 produced a marked and clinically significant improvement in FEV1. FEV1 results were expressed as “percent predicted” based on standard reference equations accounting for an individual’s race, gender, age and height. At the end of the one-hour infusion, FEV1 increased by 17.5 percent predicted for MN-221 compared to an increase of three percent predicted for placebo. At the end of the two-hour infusion, FEV1 increased by an average of 12.1 percent predicted for MN-221 compared to an increase of 1.4 percent predicted for placebo. In accordance with the study protocol, no inferential statistical testing was performed. MN-221 was well tolerated by the patients who received either infusion rate of MN-221. There were no clinically significant safety concerns noted among adverse events, ECG data, vital sign data or laboratory assessments collected throughout this clinical trial.
6
Table of Contents
We initiated a randomized, modified single-blind, placebo-controlled, dose escalation Phase II clinical trial in March 2008 to evaluate MN-221 in patients with severe, acute exacerbations of asthma in emergency departments by holding an investigator’s meeting. We completed this Phase II clinical trial, which included 29 patients (13 treated with standard care only and 16 treated with MN-221 plus standard care) at planned escalating doses of 240 to 1,080 micrograms, in April 2009. All patients received standardized care consisting of inhaled albuterol, ipratropium and oral steroid treatment. No safety concerns with adding MN-221 to standardized care were identified following review of ECG laboratory and adverse experience data. The hospitalization rate among patients treated with standardized care only was 46 percent (six of 13), which was the anticipated rate, compared to a hospitalization rate of 25 percent (four of 16) among patients receiving MN-221 plus standardized care. Improvement in FEV1 values generally appeared to be greater for patients receiving MN-221 in addition to standardized treatment. As specified in the protocol for this clinical trial, no inferential statistics (e.g., p-values) were calculated for this study.
Development Plans. In January 2009, we initiated a randomized, double-blind, placebo-controlled Phase II clinical trial designed to evaluate the safety and efficacy of MN-221 in patients with severe, acute exacerbations of asthma in emergency departments by holding an investigator’s meeting. We plan to employ clinical sites in North America, Australia and New Zealand (including a majority of the clinical sites that participated in the smaller Phase II clinical trial concluded in April 2009) to enroll approximately 200 patients in this clinical trial, which is designed to compare standardized care to standardized care plus MN-221 at a dose of 1,200 micrograms administered over one hour. Once a patient has received the initial standardized care treatment regimen, the patient will be assessed for response to that treatment. If the patient’s FEV1 is less than or equal to 50 percent of predicted and the patient meets all other study entry criteria, the patient will be randomized to receive either MN-221 or placebo. Patients enrolled in the clinical trial will continue to receive standardized care as needed. The primary efficacy endpoint will be improvement in FEV1.
If we are successful in completing our planned Phase II clinical trials in a timely manner, we would anticipate conducting an End-of-Phase II meeting with the FDA and subsequently initiating our planned Phase III program. If we are successful in completing our planned Phase III trials in a timely manner, we would anticipate filing an NDA with the FDA to seek regulatory approval for MN-221 in the United States.
MN-221 for Chronic Obstructive Pulmonary Disease Exacerbations
Indication Overview and Market Opportunity. A COPD exacerbation is a sustained worsening of the patient’s condition, from the stable state and beyond normal day-to-day variations, that is acute in onset and necessitates a change in regular medication in a patient with underlying COPD. Exacerbations are associated with a significant increase in mortality, hospitalization and healthcare utilization. According to data from the Centers for Disease Control and Prevention, an estimated ten million adults had a diagnosis of COPD in the United States in the year 2000. In addition, according to the Centers for Disease Control and Prevention, in the year 2000, there were 119,000 deaths, 726,000 hospitalizations, and 1.5 million hospital emergency department visits due to COPD in the United States. According to a more recent report on respiratory diseases from the Centers for Disease Control and Prevention and National Institutes of Health, the prevalence and age-adjusted death rate for COPD increased more than 30 percent since 1980. The same report also indicated that the direct costs of health care services and indirect costs through loss of productivity related to COPD amounted to approximately $26 billion in 1998. In 2002, according to the Global Initiative for Chronic Obstructive Lung Disease (GOLD) direct costs for COPD were approximately $18.0 billion and indirect costs were approximately $14.1 billion in the United States. In 2007, according to the American Lung Association, the direct costs for COPD were approximately $26.7 billion and indirect costs were approximately $15.9 billion in the United States. We believe there remains an unmet medical need for a safe and effective treatment for COPD exacerbations that could prevent some of these hospitalizations.
Overview of MN-221 in COPD Exacerbations. In July 2009, we announced our plan to evaluate MN-221 for the treatment of COPD exacerbations. Inhaled ß2-adrenergic receptor agonists, which are the current standard of care, are often inadequate to control the symptoms of COPD exacerbations. We believe that MN-221 may offer
7
Table of Contents
an immediate intravenous delivery for this life-threatening condition for patients who cannot get the full benefit from treatment with inhaled ß2-adrenergic receptor agonists due to severe bronchoconstriction. In addition, we believe that MN-221 may offer the potential for fewer cardiovascular side effects than older ß2-adrenergic receptor agonists due to its greater selectivity for the ß2-adrenergic receptor. This could be very significant due to the relative older age population seen in COPD patients who tend to have more underlying heart disease.
Clinical Results. We initiated in the fourth quarter of 2009, a randomized, double-blind, placebo-controlled Phase Ib study involving 48 moderate-to-severe COPD patients who received a one (1) hour intravenous infusion of MN-221 at three different escalating dose levels (300 µg, 600 µg, or 1200 µg) or placebo. We completed this Phase 1b in the first quarter of 2010. On March 17, 2010, based on preliminary findings, we announced that all doses of MN-221 produced a clinically significant improvement in FEV1(L) as compared to the baseline and placebo. At the end of the one hour infusion, FEV1(L) increased as compared to baseline by an average of 21.5% (p=0.0025) for the 1200 µg dose, 16.2% (p=0.020) for the 600 µg dose, and 9.2% (p=NS) for the 300 µg dose compared to a decrease of 4.0% for the placebo. MN-221 at doses of 600 µg and 1200 µg appeared to have an effect for at least six (6) hours as compared to placebo. MN-221 was well tolerated by all patients who received infusions of MN-221.
Development Plans. Utilizing our existing IND for MN-221, we successfully completed a Phase Ib clinical trial to evaluate the safety and efficacy of MN-221 at planned escalating doses in patients with stable, moderate to severe COPD. We are now considering the next steps for the COPD development program.
MN-166 for Multiple Sclerosis
Indication Overview and Market Opportunity. MS is an inflammatory disease of the CNS in which the body’s immune system attacks the protective sheath surrounding nerve fibers. According to the National Multiple Sclerosis Society, MS affects approximately 400,000 people in the United States and approximately 2.5 million people worldwide. In addition, according to the National Multiple Sclerosis Society, approximately 200 people are diagnosed with MS in the United States each week. The most obvious effect of MS is its destruction of nerve fibers leading to the loss of muscle control. However, MS also affects multiple CNS functions. Currently, there is no known cure for the disease. According to a Cognos study published by Decision Resources, Inc., relapsing-remitting MS, or RRMS, is the most common type of the disease, accounting for approximately 65 percent of MS patients, and most patients with RRMS eventually progress to the secondary progressive form of the disease. According to sales data included in the most recent annual reports of the leading MS drug companies, including Biogen Idec Inc., Merck Serono S.A., Teva Pharmaceuticals Industries Ltd. and Bayer Schering Pharma AG, worldwide sales of drugs to treat MS exceeded $8.0 billion in 2008.
The aim of treatment is to relieve symptoms of acute attacks by reducing the frequency of relapses and limiting the disabling effects of relapses and to minimize disability caused by disease progression. Steroids are used in treating MS to decrease the severity and shorten the duration of the attacks, but they do not change the course of the disease. Corticosteroid use is normally limited to the short-term treatment of MS, perhaps over a period of one to three weeks, as it generally is believed that the side effects and safety risks of long-term corticosteroid therapy outweigh clinical benefits in extended MS treatment. More recently, immunosuppressive agents and techniques have been introduced for the treatment of MS. However, these treatments are only partially effective and certain side effects may preclude their widespread use. These treatments may slow the course of disease progression and mitigate its effects temporarily, but additional drugs are often required to address the various CNS dysfunctions caused by the disease. In addition, many patients continue to experience relapses and progression of the disease despite taking these immunomodulators, as they are generally successful in only reducing the relapse rate by approximately one-third. Currently, the most widely used treatments for MS are beta-interferons; however, beta-interferons require injection, which may result in inflammation at the injection site. Severe flu symptoms also may occur with the beta-interferons. We believe drugs for the treatment of MS that can be taken with less discomfort, particularly those that can be taken orally, with efficacy equal or better than the available treatments for MS would have widespread appeal.
8
Table of Contents
Overview of MN-166. We licensed MN-166 from Kyorin Pharmaceutical in October 2004. MN-166 has been marketed in Japan and Korea since 1989 to treat cerebrovascular disorders and bronchial asthma. In preclinical in vivo and in vitro studies, MN-166 inhibited leukotriene activity, phosphodiesterases and nitric oxide synthase, all of which are inflammatory mechanisms known to be involved in MS. These studies also suggested that MN-166 may suppress the production of pro-inflammatory cytokines (IL-1ß, TNF-~) and enhance the production of the anti-inflammatory cytokines (IL-4, IL-10). Based on the potential mechanisms of action of MN-166, its clinical safety history in Japan, the results of pilot studies conducted by Kyorin Pharmaceutical in MS patients and the issuance of a U.S. patent covering the method of using MN-166 to treat the disease, we decided to pursue development of MN-166 as a novel, oral agent for the treatment of MS.
Clinical Results. Based on its anti-inflammatory activity and safety profile, MN-166 was evaluated for potential activity in MS in two pilot clinical trials sponsored by academic investigators in Japan. In one open-label pilot clinical trial, the investigators studied the effects of MN-166 on relapse rates in six MS patients who had a mean of four relapses per year. Following 12 to 20 months of treatment with MN-166, the average relapse rate was reduced. Over this time frame, there was no significant change in the mean Expanded Disability Status Score, or EDSS, a measure of MS drug efficacy and disease progression. No side effects of MN-166 were reported in this clinical trial. In a second pilot trial involving 12 MS patients receiving MN-166 for four weeks, MN-166 tended to normalize the levels of several chemical mediators of inflammation, including tumor necrosis factor alpha and interferon gamma. These two pilot clinical trials in MS were not performed and analyzed in accordance with standards that will allow us to use them to support a marketing application to the FDA.
We initiated a two-year Phase II multi-center, randomized, double-blind, placebo-controlled clinical trial of MN-166 for the treatment of patients with relapsing MS in August 2005. This clinical trial involved 297 patients with relapsing MS in several countries in Eastern Europe. Patients received either 30 mg of MN-166 per day, 60 mg of MN-166 per day or a placebo. In March 2007, we announced one-year results from this clinical trial. The one-year results, which included a number of efficacy endpoints for this clinical trial, showed a significant increase in the proportion of patients who remained relapse-free over the first 12 months of treatment with 60 mg per day of MN-166 compared to placebo (p-value=0.03). The time to first relapse was also significantly increased in patients treated with 60 mg of MN-166 per day compared to placebo (p-value=0.04). Positive trends were also observed in the annualized relapse rate (p-value=0.08) and number of relapses (p-value=0.10) among patients who completed the first 12 months of treatment with 60 mg of MN-166 per day compared to those patients completing the first 12 months of treatment on placebo. A significant reduction in brain volume loss (p-value=0.04), as measured by cranial magnetic resonance imaging, or MRI, scans, was observed in patients treated with 60 mg per day of MN-166 compared to placebo. Loss of brain volume on MRI scans has been shown to correlate with clinical progression and disability in MS patients. Positive trends were also observed in several other radiological outcome measures, including the volume of gadolinium-enhancing (T1) lesions (p-value=0.09), in patients treated with 60 mg of MN-166 per day compared with placebo. However, no reduction in the cumulative number of active (gadolinium-enhancing (T1) and non-enhancing new/enlarging (T2)) lesions on cranial MRI scans over 12 months of treatment was observed in patients treated with MN-166 compared to placebo, which was the protocol-defined primary endpoint of this clinical trial. No clinical or radiological benefit was observed in patients treated with 30 mg per day of MN-166. MN-166 was well tolerated at all doses in this clinical trial. Eighty-nine percent of patients completed the first 12 months of this clinical trial with only mild gastrointestinal side effects observed with MN-166 compared to placebo (3-6 percent vs. 1-3 percent, respectively). In October 2007, these one-year results were presented at the 23rd Congress of the European Committee for Treatment and Research of Multiple Sclerosis, or ECTRIMS, and the 12th Conference of Rehabilitation in Multiple Sclerosis, or RIMS.
In February 2008, we announced additional data from a double-blind analysis of the first year of treatment from the two-year Phase II clinical trial of MN-166 for the treatment of MS. Following the recommendation of our Scientific Advisory Board, we performed this analysis using MRI data collected from 292 patients with relapsing forms of MS who were randomly assigned to receive daily oral treatment with placebo or 30 or 60 mg per day during year one of this two-year clinical trial. The analysis showed that MN-166 decreased the formation
9
Table of Contents
of black holes, which are permanent brain lesions believed to indicate the death of nerves in the brain, on MRI scans in patients participating in this clinical trial, thereby adding support to our belief that MN-166 may provide neuroprotection in relapsing MS. The data demonstrated that a 60 mg per day dosing regimen of MN-166 significantly reduced the proportion of new T1 gadolinium-enhancing or new T2 lesions identified at month two of the clinical trial that evolved into persistent black holes at month ten compared to placebo (RR=0.63, p-value=0.011). Treatment with a 30 mg per day dosing regimen of MN-166 showed a trend toward reduced risk of new lesion evolution to persistent black holes compared to placebo (RR=0.735, p-value=0.074). In June 2008, additional data from an analysis of the first year of treatment was presented at the 18th Meeting of the European Neurological Society.
In April 2008, we announced the results of the completed two-year Phase II clinical trial. In the second year of the study, all patients received active drug. Patients who received 30 or 60 mg of MN-166 per day during the first year of the study remained on the assigned dose for the second 12 months of the study; patients who received placebo during the first 12 months of the study were randomized to receive either 30 or 60 mg of MN-166 per day (double-blind maintained) during the second 12 months of the study. Clinical and radiological outcomes were evaluated. MN-166 treatment resulted in positive findings on three independent measures indicative of a potential disease-progression modifying effect. First, sustained disability progression was significantly less likely (by approximately 50 percent) in those patients receiving MN-166 at either 30 or 60 mg per day for 24 months than in those patients receiving the drug for 12 months (p=0.026). Sustained disability progression was measured as a greater than or equal to 1.0 point increase from baseline in the EDSS score for four consecutive months. Second, the significant reduction in brain volume loss (p=0.035), as measured by cranial MRI scans, observed after 12 months in patients treated with 60 mg per day of MN-166 compared to placebo was again demonstrated in year two of the study. Brain volume loss was significantly less (p=0.030) in patients receiving 60 mg per day of MN-166 for 24 months compared to the other treatment groups. Third, MN-166 treatment at 60 mg per day significantly reduced the relative risk for conversion of new inflammatory lesions identified at month two to Persistent Black Holes, or PBHs, an MRI indicator of neuronal loss, eight months later at month ten by 37 percent (p=0.011); such lesions that remain unchanged for eight months are considered PBHs as compared to transient inflammatory lesions that are more closely associated with relapses. MN-166 treatment at 30 mg per day resulted in a trend toward reducing evolution to PBH (p=0.074). MN-166 was well tolerated at all doses over the two years of this clinical trial, with the most common adverse events possibly related to MN-166 involving mild, transient gastrointestinal disturbances and depression. Of the 297 patients enrolled in this clinical trial, 245 patients completed the full two years of treatment. In September 2008, data from this completed two-year clinical trial was presented at the World Congress for Treatment and Research in MS.
Development Plans. At present, with the acquisition of AV411 in December 2009, we are not planning to undertake any further significant clinical development of MN-166 until such time that we are successful in entering into a strategic collaboration to support further clinical development of our combined MN-166/AV411 ibudalist-based programs. We are actively pursuing potential partners for such purpose.
AV411 for Other Central Nervous System Disorders
The AV411 portfolio, which includes the phase 2-staged lead drug compound and proprietary analogs, represents novel, first-in-class, non-opioid drugs for the treatment of several large pain and drug addiction indications. AV411 is a first-in-class, orally bioavailable small molecule, a glial attenuator that suppresses pro-inflammatory cytokines IL-1 beta, TNF alpha, and IL-6, and may upregulate the anti-inflammatory cytokine IL-10. It has additionally been shown to be a toll-like receptor 4 (TLR4) functional antagonist that may contribute to its attenuation of neuroinflammation. While considered a New Molecular Entity, or NME in the United States and Europe, it involves redirection of an approved drug, ibudilast, which was first approved in Japan more than 20 years ago. Ibudilast has been prescribed to over one million patients for a different indication and has a good post-marketing safety profile as reported in nearly 15,000 patients studied at the prescribed doses.
10
Table of Contents
Based on our research, we have filed for patents protecting multiple uses of AV411 in neurological conditions, as well as for patents on AV411 analogs which we believe have the potential to be effective second generation molecules. As NMEs, AV411 and its analogs are additionally entitled to five years of marketing exclusivity from launch in the U.S. under the Drug Price Competition and Patent Term Restoration Act of 1984, as amended, or the Hatch-Waxman Act, provisions and up to 10 years of exclusivity in the European Union.
Neuropathic pain: Glial activation in the brain and spinal cord contribute to the establishment and amplification of the chronic pain state. As part of Avigen’s program investigating glial attenuation as a novel approach to the treatment of neuropathic pain, Avigen conceived and demonstrated that AV411 (ibudilast) was efficacious in preclinical models of neuropathic pain and may be effective in a wide range of neuropathic pain syndromes including neuropathy, post-herpetic neuralgia, HIV neuropathy, radiculopathy, spinal cord injury and chemotherapy-induced neuropathy. While ibudilast was initially developed as a non-selective phosphodiesterase (PDE) inhibitor for the treatment of bronchial asthma, its efficacy in some neuropathic pain models appears to be independent of this activity and yet still linked to glial attenuation.
AV411 has advanced through multiple Phase 1 and 2a clinical trials in both healthy volunteers and patients for neuropathic pain and the program, under current U.S. Food and Drug Administration standards, is able to enter Phase 2 development for neuropathic pain in the United States based on completed Avigen preclinical and clinical development.
Opioid withdrawal: AV411 is currently in a Phase 1b/2a clinical trial funded by NIDA and conducted at Columbia University by leading specialists in the study and treatment of substance abuse. AV411 and analogs have been shown in preclinical models of opioid (morphine or oxycodone) withdrawal to significantly reduce withdrawal symptoms. Moreover, AV411 attenuates both behavioral and neurochemical markers of opioid reward. AV411 and analogs are differentiated from other drug candidates in clinical trials that may demonstrate similar effects, in that AV411 and analogs are not narcotics and do not, themselves, provide reward or “reinforcement” in behavioral models of dependence. Thus, while current therapies involve substitution of one opioid for another (e.g. methadone for heroin), AV411 represents a novel, non-opioid, approach for the treatment of opioid withdrawal and dependence.
Methamphetamine addiction: In collaborative studies with NIDA, AV411 has demonstrated utility in methamphetamine relapse in animals which may be translated to a NIDA-funded exploratory clinical trial with investigators at the University of California – Los Angeles.
Development Plans. We are not planning to undertake any further significant clinical development of AV411 until such time that we are successful in entering into a strategic collaboration to support further clinical development of our combined MN-166/AV411 ibudalist-based programs. We are actively pursuing potential partners for such purpose.
Other Product Candidates
We intend to limit development activities on the balance of our ten product candidates. For each of these product candidates, we plan to conduct development activities only to the extent that we deem any further activities necessary to maintain our license rights or maximize its value, while pursuing a variety of initiatives to monetize such product candidate on appropriate terms. The status of the development program for each of these non-prioritized product candidates is described below.
MN-001 for Asthma
Indication Overview and Market Opportunity. Asthma is a chronic inflammatory disease of the airways in which symptom control is the key to effective disease management. Alleviation of acute asthmatic symptoms and blocking of late phase inflammation are both important to asthma therapy. According to the American Lung Association and the Global Initiative for Asthma, there are approximately 22.9 million asthma patients in the United States and over 300 million asthma patients worldwide.
11
Table of Contents
According to the most recent annual reports of the leading asthma drug companies, GlaxoSmithKline plc, Merck & Co., Inc., AstraZeneca plc and Roche Holding Ltd., worldwide sales of asthma therapeutics increased to over $17.0 billion in 2008. Leading treatments currently include inhaled corticosteroids, bronchodilators and leukotriene antagonists. Worldwide sales of the Flovent® and Pulmicort® inhaled corticosteroids were over $2.6 billion in 2008 according to the annual reports of GlaxoSmithKline plc and AstraZeneca plc. Combination products, consisting of inhaled corticosteroids plus long-acting beta agonists, added an additional $8.6 billion in sales in 2008. Inhaled steroids, such as Flovent® (fluticasone) and Vanceril® (beclomethasone), are more broadly effective in blocking late phase inflammation, but their general side effects require careful monitoring. Leukotriene antagonists, such as Singulair® (montelukast) or Accolate® (zafirlukast), became available as a new asthma therapy in the late 1990s. These drugs block the actions of leukotrienes, which are pro-inflammatory chemical mediators, and the subsequent inflammation caused by eosinophil migration to the lungs. According to Merck & Co., Inc.’s 2008 Annual Report, worldwide sales of Singulair®, a leading leukotriene antagonist, were $4.7 billion in 2009.
Overview of MN-001 in Asthma. MN-001 is a novel, orally bioavailable compound being developed for the treatment of bronchial asthma. We licensed MN-001 from Kyorin Pharmaceutical in March 2002. In in vivo preclinical studies conducted by Kyorin Pharmaceutical and us, MN-001 combined the positive attributes of the leukotriene antagonists and inhaled steroids, while maintaining an acceptable safety profile.
In preclinical pharmacology studies, MN-001 inhibited airway hyper-reactivity through a reduction of airway inflammation. In vitro studies and animal studies also suggested that MN-001 may affect many of the downstream mechanisms activated by mast cell degranulation, which is the release of chemicals that cause inflammation. MN-001 also demonstrated that it is a potent inhibitor of pro-inflammatory enzymes in vitro (e.g., 5-lipoxygenase and phosphodiesterase 4), as it prevented migration of inflammatory cells to the lungs of rodents in these studies. In addition, in guinea pig asthma models, MN-001 was more selective than steroids in affecting cells involved in the inflammatory process and not those involved in cellular immunity.
Clinical Results. MN-001 has proven to be well tolerated in early clinical testing. Treatment-related adverse effects, primarily consisting of gastrointestinal discomfort such as diarrhea, loose stools, nausea and upper abdominal pain, were mild, transient and reversible. These adverse effects were consistent with findings in preclinical studies.
We conducted a randomized, double-blind, placebo-controlled, multi-center Phase II clinical trial in patients with mild-to-moderate asthma, which was completed in the fourth quarter of 2005. In this clinical trial, 147 patients were randomly assigned to receive placebo or MN-001 tablets in one of three oral dosing regimens for four weeks. The primary endpoint of the trial was achieved with a statistically significant improvement in FEV1 after four weeks of treatment with 500 mg of MN-001 at three times daily dosage, or TID, compared to placebo (p-value=0.021; intent-to-treat, observed cases). A similar trend was observed for the 750 mg two times daily dosage, or BID, of MN-001 (p-value=0.058). Positive trends in secondary outcome measures were also observed in the 500 mg TID treatment group, including serial spirometry, morning and evening peak flow rates, and provocative concentration causing a 20 percent fall in FEV1, or PC20, values in a methacholine challenge test, each of which is a common measure of respiratory function. MN-001 was well tolerated in this clinical trial with 89 percent of patients completing four weeks of treatment. There was no apparent difference between placebo and any of the active treatment groups in adverse events leading to discontinuation or in adverse events attributable to treatment.
MN-001 for Interstitial Cystitis
Indication Overview and Market Opportunity. IC is a chronic disease of the bladder characterized by urinary frequency and urgency, nighttime urination and pelvic and bladder pain. It is widely believed that IC is due to an altered or defective bladder lining and an increased number of activated bladder mast cells, which are specialized cells that release biochemicals and cause inflammation. According to the National Kidney and Urologic Diseases
12
Table of Contents
Information Clearinghouse, which is a division of the National Institutes of Health, an estimated 1.3 million patients suffer from IC in the United States, 90 percent of whom are women. The prevalence of IC in Europe is approximately one-third that of the United States. We believe that IC is currently underdiagnosed and that the market for drugs that treat IC will likely expand with the introduction of effective new treatments.
Overview of MN-001 in Interstitial Cystitis. MN-001 is a novel, orally bioavailable, anti-inflammatory compound being developed for the treatment of IC. We licensed MN-001 from Kyorin Pharmaceutical in March 2002. Data that we collected in connection with the development of MN-001 for bronchial asthma and data collected by Kyorin Pharmaceutical provided us with a strong scientific rationale for evaluating MN-001 as an oral treatment for IC. MN-001 has been shown to block a number of the inflammatory mechanisms activated by mast cell degranulation that are important in the pathogenesis of inflammatory disorders, including IC and asthma (e.g., leukotriene receptor antagonism and inhibition of phosphodiesterases III and IV, 5-lipoxygenase, phospholipase C and thromboxane A2). In addition, MN-001 produced anti-inflammatory effects in a variety of rodent models of IC and asthma; in these models, MN-001 reduced bladder hyper-reactivity much in the same way that it reduced airway hyper-reactivity in the lung.
Clinical Results. We conducted a randomized, double-blind, placebo-controlled multi-center Phase II/III clinical trial in patients with moderate-to-severe IC, which was completed in the first quarter of 2007. This clinical trial involved 305 patients at 37 clinical sites in the United States. Results from this clinical trial indicated that, while well-tolerated, MN-001 did not show a statistically significant clinical benefit compared to placebo on the primary endpoint (to be much or very much improved overall on a patient-rated global response assessment) at the doses tested in this clinical trial (500 mg once or twice a day for eight weeks). Results from this clinical trial also indicated that IC patients were more than twice as likely to respond on 500 mg of MN-001 administered twice a day compared to placebo (25 percent compared to 12 percent, p-value=0.04) after four weeks of treatment. This difference, however, was not observed at eight weeks due to continued improvement among placebo-treated patients. The response rate of patients treated with 500 mg of MN-001 once a day did not significantly differ from placebo at either four or eight weeks.
MN-029 for Solid Tumors
Indication Overview and Market Opportunity. The American Cancer Society estimates that more than 1.5 million Americans were diagnosed with cancer in 2009, of which more than 750,000 patients were diagnosed with lung, prostate, colon, rectum or breast solid tumor cancers. The American Cancer Society also estimates that approximately 560,000 patients were ultimately to die from cancer in 2009. According to Med Ad News, the market for solid tumor cancer therapeutics exceeded $26.0 billion in 2007.
Tumor blood vessels are a promising target for cancer therapy. Compounds that act to deprive tumors of their blood supply fall into two classes: angiogenesis inhibitors and vascular disrupting agents, or VDAs. Angiogenesis inhibitors block the formation of new blood vessels formed in response to tumor growth, whereas VDAs disrupt blood flow through existing tumor blood vessels. We believe that VDAs have a potential advantage over angiogenesis inhibitors because VDAs work on existing tumor blood vessels and can kill hundreds of cancer cells that depend on that blood supply with even a brief interruption in blood flow, rather than simply slowing tumor growth by blocking new blood vessel formation.
Overview of MN-029. MN-029 is a novel, small molecule VDA being developed for the treatment of solid tumors. We licensed MN-029 from Angiogene Pharmaceuticals in June 2002. Several preclinical pharmacology studies conducted by Angiogene Pharmaceuticals and us have assessed the mechanism of action and anti-tumor activity of MN-029 in vivo in rodent models of breast adenocarcinoma, colon carcinoma, lung carcinoma and KHT sarcoma. In these studies, MN-029 damaged poorly formed tumor blood vessels by weakening tumor blood vessel walls and causing leakage, clotting and eventual vascular shutdown within the tumor. These studies suggest that MN-029 acts quickly and is rapidly cleared from the body, which may reduce the potential for some adverse effects commonly associated with chemotherapy. Shutdown of tumor blood flow in tumor models was confirmed through the use of dynamic contrast-enhanced magnetic resonance imaging, or DCE-MRI.
13
Table of Contents
Clinical Results. To date, we have conducted two Phase I clinical trials of MN-029 for the treatment of solid tumors. We completed one Phase I clinical trial of MN-029 in the second quarter of 2006 and one Phase I clinical trial in the fourth quarter of 2007.
In the first Phase I clinical trial, MN-029 was administered as an intravenous infusion once every three weeks with a 20-day recovery period between doses (one cycle). Results from this clinical trial showed that MN-029 was well tolerated at doses that reduced tumor blood flow. A maximum tolerated dose of 180 mg/m2 per dose was established in this clinical trial. The most common side effects of MN-029 were characteristic of other VDAs and included nausea, vomiting, fatigue and diarrhea. Nine of 34 patients with advanced solid tumors for whom no standard therapy was available had stable disease after three cycles of treatment. Six patients had prolonged (greater than six months) stable disease. To date, two of these patients remain on therapy with MN-029 under compassionate use Investigator INDs and had stable disease (one with melanoma after 24 months of treatment and one with carcinoid tumors after 33 months of treatment) upon their transition from our clinical trial to compassionate use programs in the fourth quarter of 2007. Following the transition of these patients to compassionate use programs, we have not received, nor will we receive, any further data on these patients unless a serious adverse effect occurs. Although no patients showed objective responses based on Response Evaluation Criteria in Solid Tumors, or RECIST criteria, which is tumor length on computed tomography, or CT, or MRI scans, semi-automated measurements of tumor volumes from CT scans showed a measureable reduction in tumor burden in the subject with the largest reduction in tumor blood flow (Ktrans -40 percent). Tumor blood flow reduction assessed by dynamic contrast-enhanced magnetic resonance imaging, or DCE-MRI, was recorded at doses greater than or equal to 120 mg/m2.
In the second Phase I clinical trial, MN-029 was administered as an intravenous infusion every seven days (days 1, 8, 15) followed by a 13-day recovery period (one cycle). Results from this clinical trial showed that MN-029 was well tolerated. The maximum dose was limited to 180 mg/m2 per dose based on the results of the other Phase I trial that employed a less aggressive dosing schedule. The most common side effects of MN-029 in this clinical trial included nausea, vomiting, arthralgia and headache. Eleven of 20 patients with advanced solid tumors for whom no standard therapy was available had stable disease after two cycles of treatment. Four subjects continued on extended cycles of MN-029 treatment. Based on RECIST criteria, one patient with metastatic pancreatic cancer had an overall partial response with a duration of 74 days. Seven patients had stable disease with a median duration of 83 days.
MN-305 for Generalized Anxiety Disorder/Insomnia
Indication Overview and Market Opportunity. The essential characteristic of Generalized Anxiety Disorder is excessive, uncontrollable worry about everyday events. This constant worry affects daily functioning and can cause severe physical symptoms. Generalized Anxiety Disorder can occur with other anxiety disorders, depressive disorders or substance abuse. Generalized Anxiety Disorder is often difficult to diagnose because it is not triggered by a specific object or situation. The intensity, duration and frequency of the worry are disproportionate to the issue. As a result, Generalized Anxiety Disorder tends to interfere with the patient’s performance of tasks and ability to concentrate. According to the National Institute of Mental Health, anxiety disorders affect approximately 40 million American adults, of whom approximately 6.8 million suffer from Generalized Anxiety Disorder. Anxiety disorders are the most prevalent of neuropsychiatric conditions, yet are generally considered to be under-diagnosed and therefore undertreated. Therefore, we believe that there is a significant opportunity for the introduction of new anxiety reducing drugs.
A variety of pharmacologic agents are used to manage patients with anxiety disorders. Benzodiazepines have been the mainstay of the treatment of acute anxiety since the late 1960s. However, their efficacy as a treatment has been limited by problems faced in chronic use due to their sedative effects. In the late 1980s, buspirone was introduced and widely used even though it takes effect slowly. Buspirone was well tolerated and relatively safe. During the late 1990s, newer anti-depressants, notably the specific serotonin reuptake inhibitors,
14
Table of Contents
or SSRIs, were increasingly used to treat anxiety as well. While effective, the use of SSRIs may result in a variety of undesirable side effects, including agitation and sexual dysfunction. Also, SSRIs may take weeks to exert their beneficial effects.
Overview of MN-305 in Generalized Anxiety Disorder/Insomnia. MN-305 is a serotonin receptor agonist with high affinity and selectivity for the serotonin 5-HT1A receptor subtype. Drugs that act through this mechanism, such as buspirone, have been proven to be clinically effective in treating Generalized Anxiety Disorder. We licensed MN-305 from Mitsubishi Pharma Corporation, now Mitsubishi Tanabe Pharma Corporation, in April 2004. MN-305 has been shown to be more potent than buspirone and to exhibit anti-anxiety efficacy in a wide range of preclinical rodent models. For example, in a social interaction test, MN-305 prolonged the duration of social interaction in rats. Preclinical and clinical studies conducted by Mitsubishi Tanabe Pharma Corporation and us also suggest that MN-305 may have a more rapid onset of action than buspirone.
Clinical Results. Preliminary evidence of anti-anxiety efficacy was provided by a six-week, open-label, fixed-flexible dose Phase II clinical trial conducted by Mitsubishi Tanabe Pharma Corporation in Japan in 61 patients with neurotic disorders. The neurotic disorders included Generalized Anxiety Disorder, panic disorder, agoraphobia, mixed anxiety and depressive disorder and dysthymia. MN-305 was well tolerated, with headaches being the most common side effect in this clinical trial. At the end of the clinical trial, the mean Hamilton Rating Scale for Anxiety score, or HAM-A score, which is a scale used to measure the intensity of anxiety symptoms, was reduced compared to the pre-treatment value. Similarly, a majority of the patients were rated “Moderately Improved” or better following treatment with MN-305. In addition, MN-305 was well tolerated in several clinical trials conducted by Mitsubishi Tanabe Pharma Corporation in healthy volunteers and patients with anxiety disorders and Major Depressive Disorder. These studies did not evaluate the reduction of anxiety symptoms in patients that were not treated with MN-305.
The IND for MN-305 was transferred to us from Mitsubishi Tanabe Pharma Corporation, which enabled us to initiate a Phase II/III randomized, double-blind, placebo-controlled clinical trial in 416 patients with Generalized Anxiety Disorder in the first quarter of 2005. We completed this clinical trial in the second quarter of 2006. The results revealed trends for improvement in all efficacy outcome measures. Statistically significant improvements in the total HAM-A score and in anxious mood, which is item 1 of the HAM-A score and was a secondary endpoint in this clinical trial, were observed through eight weeks of treatment. However, statistical significance on change from baseline of the total HAM-A score after ten weeks of treatment, which was the primary outcome measure of this clinical trial, was not achieved. MN-305 was well tolerated at all doses in this clinical trial, and we believe the findings were sufficiently positive to warrant further clinical evaluation of this product candidate.
We analyzed the results from our Phase II/III clinical trial of MN-305 in Generalized Anxiety Disorder and performed in-depth analyses of subgroups that showed statistically significant improvement in certain aspects of the HAM-A score (e.g., insomnia). Based on these analyses, we initiated a Phase II proof-of-concept clinical trial of MN-305 for the treatment of insomnia in the first quarter of 2007 to assess the effects of three dosages of MN-305 (1 mg, 3 mg and 6 mg) and placebo, all administered orally approximately 60 minutes before bedtime. This clinical trial, which involved 74 subjects at ten study centers in the United States, was completed in the fourth quarter of 2007. This clinical trial failed to achieve statistical significance in its primary endpoint of reducing Wake (time) After Sleep Onset, or WASO. MN-305 was well tolerated in this clinical trial with no clinically significant adverse events observed at any dose tested, and there was no evidence of any decrements in psychomotor performance, as assessed in digit symbol substitution and symbol copying tests, in patients treated with MN-305. Based upon the results of this clinical trial, we decided to terminate the evaluation of MN-305 for the treatment of insomnia.
MN-221 for Preterm Labor
Indication Overview and Market Opportunity. Preterm labor is caused by the onset of uterine contractions before term. According to a November 2002 publication in Obstetrics & Gynecology, preterm labor is the leading cause of neonatal mortality and a substantial portion of all birth-related short and long-term morbidity.
15
Table of Contents
Successful inhibition of premature birth is known to reduce the risk of complications. Despite extensive research into preterm labor during the past several decades, the rate of premature births has not decreased. According to the National Vital Statistics Reports issued by the U.S. Department of Health and Human Services, there were more than four million births in the United States in 2005, almost 13 percent of which were considered premature births. The U.S. Department of Health and Human Services estimates that the cost of intensive care unit, or ICU, services for premature infants is over $15.0 billion annually. In addition, according to a September 2004 publication in British Medical Journal, approximately six percent to seven percent of all births in Europe occur before term.
Currently, therapy for preterm labor remains targeted at uterine contractions. ß2-adrenergic receptor agonists are generally used as first-line treatments for premature labor. The only FDA-approved treatment for preterm labor is ritodrine, a ß2 agonist. However, ritodrine has not been available for sale in the U.S. market since 1999. The more widely used treatment for preterm labor is another ß2 agonist, terbutaline; however, this drug is not approved by the FDA for preterm labor. Atosiban, an oxytocin antagonist, is available in Europe, but was denied regulatory approval in the United States. The usefulness of these ß2-adrenergic receptor agonists is often limited by the adverse reactions they produce, which include cardiovascular side effects such as heart palpitations. As a result, we believe that there is a need for treatments with better safety and tolerability profiles that are effective in reducing the premature birth rate and/or providing for longer gestation.
Overview of MN-221 in Preterm Labor. MN-221 is highly-selective ß2-adrenergic receptor agonist being developed for the treatment of preterm labor. We licensed MN-221 from Kissei Pharmaceutical in February 2004. Preclinical testing in vitro and in vivo showed MN-221 to be more selective for the ß2-adrenergic receptor than other ß2-adrenergic receptor agonists currently used to treat preterm labor. Moreover, in vitro studies also suggested that MN-221 may act as only a partial ß1-adrenergic receptor agonist in cardiac tissue, while acting as a full ß2-adrenergic receptor in the uterus. This improved receptor binding and functional selectivity may result in fewer cardiovascular side effects than are commonly observed with other ß2-adrenergic receptor agonists used to treat this condition. In preclinical pharmacology studies in pregnant rats and sheep conducted by Kissei Pharmaceutical, MN-221 reduced the number of spontaneous or drug-induced uterine contractions. Furthermore, in these studies, MN-221 delayed both normal and preterm labor in rats and caused a marked increase in the bodyweight of rat pups as a result of prevention of premature birth. In rat and sheep studies which compared MN-221 to ritodrine and/or terbutaline, the potency of MN-221 was greater than those ß2-adrenergic receptor agonists.
Clinical Results. To date, pharmacokinetic and safety data has been generated from human experience with MN-221 through Phase I clinical studies in healthy male and non-pregnant female volunteers conducted by Kissei Pharmaceutical in Japan and the U.K. and a Phase I clinical trial in the United States conducted by us. A total of 244 healthy subjects received intravenous infusions of either MN-221 or a placebo. MN-221 was generally well tolerated. A pilot double-blind, placebo-controlled Phase II clinical trial of MN-221 was completed in 2004 by Kissei Pharmaceutical in seven women in preterm labor in the U.K. A trend towards a reduction in the number of uterine contractions was observed in MN-221-treated women and, as a result, only limited conclusions could be drawn from this clinical trial. No serious adverse events related to MN-221 were observed in this clinical trial.
We initiated a Phase I clinical trial in healthy pregnant women in the third quarter of 2006. Ten healthy, pregnant volunteers who were not in labor participated in this clinical trial, which was completed in the second quarter of 2007. The volunteers received a single-dose intravenous infusion regimen of MN-221, consisting of two consecutive rounds of a 15-minute priming and a 105-minute maintenance infusion to deliver 294 micrograms of MN-221 over four hours. The primary objectives of this clinical trial were to determine the pharmacokinetics, safety and tolerability of this infusion regimen of MN-221 in pregnant women. No significant safety concerns with MN-221 were identified in this clinical trial.
MN-246 for Urinary Incontinence
Indication Overview and Market Opportunity. Urinary incontinence occurs when normal regulation of bladder function is lost. According to the National Kidney and Urologic Disease Information Clearinghouse, there are over 13 million persons in the United States suffering from urinary incontinence.
16
Table of Contents
The market for drugs to treat urinary incontinence is expected to grow substantially as more patients seek treatment and as newer drugs are introduced to the market. According to Datamonitor, the global market for urinary incontinence is projected to grow to $4.0 billion in 2010. The current marketplace is dominated by anti-cholinergic drugs that are modestly effective and produce treatment-limiting side effects such as dry mouth. According to Pfizer Inc.’s 2008 annual report, sales of its market leading drug, Detrol®, were approximately $1.2 billion in 2008.
Overview of MN-246 in Urinary Incontinence. MN-246 is a novel ß3-adrenergic receptor agonist being developed for the treatment of urinary incontinence. We licensed MN-246 from Mitsubishi Pharma Corporation, now Mitsubishi Tanabe Pharma Corporation, in December 2004. We believe that MN-246 represents a new approach to treating urinary incontinence and may have advantages over existing therapies, including potential improvements in efficacy through increases in bladder volume with decreases in involuntary bladder contractions and the absence of anti-cholinergic side effects, such as dry mouth. In preclinical studies in rats conducted by Mitsubishi Tanabe Pharma Corporation, MN-246 was more potent and active than oxybutynin and propiverine in increasing bladder volume. In addition, the studies showed that MN-246 produced little or no increase in residual urine volume and no anti-cholinergic side effects in rats. MN-246 also increased bladder volume in preclinical studies conducted on dogs and monkeys.
Clinical Results. We initiated a double-blind, randomized, placebo-controlled, single escalating dose Phase I clinical trial of MN-246 for the treatment of urinary incontinence in the first quarter of 2006. This clinical trial, which involved healthy volunteers to evaluate the safety, tolerability and pharmacokinetics of MN-246, was completed in the fourth quarter of 2006. We also conducted a Phase I food effects study in healthy volunteers, which was completed in the first quarter of 2007. MN-246 was tolerated in both clinical trials.
MN-447 and MN-462 for Thrombotic Disorders
Indication Overview and Market Opportunity. Despite advances in the treatment of cardiovascular disease, or CVD, more than 910,000 Americans still die of heart disease annually according to the American Heart Association, constituting 37 percent of all deaths. In addition, there are over 70 million individuals in the United States that currently live with some form of heart disease, which can include high blood pressure, CVD, stroke, angina (chest pain), myocardial infarction (heart attack) and congenital heart defects. According to Datamonitor, worldwide sales of antithrombotic drugs are forecasted to reach approximately $14.8 billion in 2011. We believe that there remains an unmet medical need for safe and effective treatments for thrombotic conditions, including acute coronary syndrome, myocardial infarction, peripheral arterial disease and percutaneous coronary interventions.
According to the Centers for Disease Control, one out of every three Americans has CVD, and heart disease and stroke account for almost six million hospitalizations each year and cause disability for almost ten million Americans over the age of 65. According to the Centers for Disease Control, CVD remains the leading cause of death in the United States for both men and women among all racial and ethnic groups. In addition, heart disease is the leading cause of death for all Americans and causes more deaths than cancer and accidents combined based on data from the National Center for Health Statistics, the National Center for Health Promotion and the Centers for Disease Control and Prevention. Given the high mortality and morbidity rates associated with CVD. We believe there is an urgent need for more targeted therapies that can intervene in known molecular pathways and minimize damage to the heart and related tissues.
Overview of MN-447 and MN-462 in Thrombotic Disorders. MN-447 and MN-462 are novel, small molecule antithrombic agents being developed for the treatment of various thrombotic disorders. We licensed MN-447 and MN-462 from Meiji Seika Kaisha in November 2006.
MN-447 is a cardioprotective, anti-platelet agent that acts as a dual antagonist of glycoprotein, or GP, IIbIIIa and integrin alpha-v-beta-3, or avß3, receptors that play key roles in blood clot formation and various cell behaviors and functions such as leukocyte adhesion. Preclinical studies have demonstrated that MN-447 acts
17
Table of Contents
downstream by inhibiting the final common pathway of platelet aggregation - the cross-linking of platelets via fibrinogen bridges to GP IIbIIIa receptors. Inhibition of integrin avß3 receptors has been linked to an inhibition of leukocyte adhesion to endothelium (the layer of cells lining blood vessels), reduction of hyperplasia (abnormal cellular proliferation) and lumen stenosis (blood vessel constriction) in response to vascular injury. In animal models of myocardial infarction and unstable angina, the dual inhibitory activity of MN-447 produced superior cardioprotective efficacy, such as reduction in infarct size after reperfusion (restoration of blood flow) compared to inhibition of the GP IIbIIIa receptor alone, and showed a low risk of bleeding.
MN-462 is a selective inhibitor of a key enzyme in the intrinsic antifibrinolytic mechanism, plasma carboxypeptidase B, or CPB, and also called activated thrombin-activatable fibrinolysis inhibitor, or TAFIa, which inhibits physiological fibrinolysis, or the lysis or dissolving of blood clots. By enhancing intrinsic fibrinolysis through plasma CPB inhibition, MN-462 has the potential to reduce and prevent thrombus or blood clot formation, as well as dissolve formed thrombus. In preclinical studies, MN-462 demonstrated significant fibrinolytic-enhancing and anti-thrombotic activities as monotherapy in several thrombosis models, as well as activities when used as an adjunct to fibrinolytics such as tissue plasminogen activator, or t-PA. The effect of MN-462 in enhancing the intrinsic fibrinolytic process was also observed to result in a low risk of bleeding.
Sales and Marketing
We currently have no marketing and sales capabilities. Within the United States, we may develop, at the appropriate time, a focused product-driven marketing and sales organization to promote a product development program. For example, we may develop a commercial organization in the United States to focus on promoting MN-221 for the treatment of acute exacerbations of asthma and COPD exacerbations to physicians, nurses and pharmacy directors in the emergency room or hospital setting. We believe that we can achieve our strategic goals for MN-221 by deploying an experienced sales organization supported by an internal marketing infrastructure to target hospitals and institutions with emergency room departments. The size and other features of our sales and marketing organization, if any, will be influenced by the timing of regulatory approvals for our product candidates, the willingness of our partners to agree to co-promotion, if applicable, and the investment involved.
Manufacturing
We rely on third parties to manufacture bulk active pharmaceutical ingredients, or API, and finished investigational products for research, development, preclinical and clinical trials. We expect to continue to rely on third-party manufacturers for the manufacture of the API and finished products for our clinical and any future commercial production requirements. We believe that there are several manufacturing sources available at commercially reasonable terms to meet our clinical requirements and any future commercial production requirements for the API of our products and the finished drug products.
Pursuant to the terms of our license agreement with Kissei Pharmaceutical for MN-221, we are currently negotiating with Kissei Pharmaceutical for the commercial supply of the API for MN-221. If we enter into a supply agreement with Kissei Pharmaceutical, we will purchase from Kissei Pharmaceutical all API that we require for the commercial supply of MN-221, if such product candidate is approved for commercial sale by the FDA or other regulatory authorities.
In March 2009, we entered into an agreement with Hospira Worldwide, Inc., or Hospira, for the completion of pre-commercialization manufacturing development activities and the manufacture of commercial supplies of finished product for MN-221 utilizing Hospira’s proprietary ADD-Vantage drug delivery system, if such product candidate is approved for commercial sale by the FDA or other regulatory authorities. Pursuant to the terms of the agreement with Hospira, Hospira will receive development fees from us upon completion of specified development activities, which we will expense as the costs are incurred. We are also obligated under the agreement to purchase a minimum number of units each year following regulatory approval, which number is based on our forecasts submitted to Hospira on a rolling basis. In addition to the agreement with Hospira, we anticipate entering into a commercial supply agreement with a contract manufacturer for finished product of
18
Table of Contents
MN-221 in standard vials. However, at present, we do not have any agreements established regarding the commercial supply of MN-221 in standard vials or for the API or finished product of any of our product candidates.
Intellectual Property and License Agreements
Since our inception in September 2000, we have entered into eight license agreements which cover our current product candidates. In general, we seek to procure patent protection for our anticipated products, or obtain such protection from the relevant patents owned by our licensors. To date, we have obtained licensed rights under 18 issued U.S. patents and fifteen pending U.S. patent applications. We also have obtained licensed rights to over 165 issued and pending foreign patents and applications corresponding to these U.S. patents and patent applications. In addition to these licensed rights, we hold five issued U.S. patents and two U.S. patent applications relating to MN-001 and its metabolite, MN-002. These patents and pending patent applications contain claims directed to compounds, compositions, methods of use and/or methods of manufacture. We are not aware of any third-party infringement of the patents owned or licensed by us and are not party to any material claims by third parties of infringement by us of such third parties’ intellectual property rights. The following is a description of our existing license agreements and intellectual property rights for each of our product candidates:
MN-221
On February 25, 2004, we entered into an exclusive license agreement with Kissei Pharmaceutical for the development and commercialization of MN-221. Kissei Pharmaceutical is a fully integrated Japanese pharmaceutical company and is listed on the First Section of the Tokyo Stock Exchange. We obtained an exclusive, worldwide (excluding Japan), sublicensable license to various patent rights and know-how related to MN-221 and other compounds disclosed or included in, or covered by, these patent rights, for all indications, including preterm labor. This license includes an exclusive license under one U.S. patent and one U.S. patent application and certain corresponding patents and patent applications in foreign countries and is sublicensable upon receipt of the written consent of Kissei Pharmaceutical. The U.S. patent for MN-221 has composition of matter and method of use claims.
The U.S. composition of matter patent underlying the license issued on October 17, 2000 and is set to expire no earlier than February 18, 2017. Corresponding composition of matter patents in various other countries are set to expire no earlier than February 18, 2017. Under the terms of the agreement, we granted to Kissei Pharmaceutical a royalty-free, non-exclusive right and license to use our know-how and patents relating to MN-221 to develop products incorporating the MN-221 compound outside of our territory. Kissei Pharmaceutical also has the right to co-promote licensed products in our territory on terms to be agreed upon by the parties and the exclusive right to manufacture and supply us with the API that we require for clinical development of MN-221 and commercial sale of any approved product.
The license agreement may be terminated by either party following an uncured breach of any material provision in the agreement by the other party, and we may terminate the agreement for scientific or commercial reasons upon 100 days’ prior written notice to Kissei Pharmaceutical during the development phase and 180 days’ prior written notice to Kissei Pharmaceutical during the commercialization phase.
The term of the agreement is determined on a country-by-country basis and extends until the expiration of the last Kissei Pharmaceutical patent (or equivalent) under license to expire or in the event that a valid claim does not exist or, if a valid claim expired more than ten years from the date of first commercial sale, ten years from the date of first commercial sale. In either case, the term of the agreement would not extend for any particular country past the date on which generic competition exists in such country.
Under the license agreement, we have paid Kissei Pharmaceutical $1.0 million to date, and we are obligated to make payments of up to $17.0 million based on the achievement of certain clinical and regulatory milestones. We are also obligated to pay a royalty on net sales of the licensed products.
19
Table of Contents
MN-166
On October 22, 2004, we entered into an exclusive license agreement with Kyorin Pharmaceutical for the development and commercialization of MN-166. Kyorin Pharmaceutical is a fully integrated Japanese pharmaceutical company and is listed on the First Section of the Tokyo Stock Exchange. We obtained an exclusive, worldwide (excluding Japan, China, South Korea and Taiwan), sublicensable license to the patent rights and know-how related to MN-166 for the treatment of MS, except for ophthalmic solution formulations. MN-166 is not covered by a composition of matter patent. The U.S. method of use patent for MN-166 underlying the license is set to expire on August 10, 2018. Corresponding method of use patents in several other countries are set to expire no earlier than August 10, 2018. We also have rights under one pending U.S. application directed to a method of treating MS using a combination of MN-166 and Interferon-ß. Under the terms of the agreement, we granted to Kyorin Pharmaceutical an exclusive, royalty-free, sublicensable license to use the preclinical, clinical and regulatory databases to develop opthmalmic products incorporating the MN-166 compound anywhere in the world and non-opthalmic products incorporating the MN-166 compound outside of our territory.
The license agreement may be terminated by either party following an uncured breach of any material provision in the agreement by the other party. We may terminate the agreement for any reason with 90 days’ written notice to Kyorin Pharmaceutical or, in the event that a third party claims that the licensed patent rights or know-how infringe upon such third party’s intellectual property rights, with 30 days’ written notice.
The term of this agreement is determined on a country-by-country basis and extends until the later of the expiration of the obligation to make payments under the agreement or the last date on which the manufacture, use or sale of the product would infringe a valid patent claim held by Kyorin Pharmaceutical but for the license granted by the agreement or the last date of the applicable market exclusivity period. In the absence of a valid patent claim and generic competition in a particular country, the agreement will expire on the earlier of five years from the date of the first commercial sale of the product by us or the end of the second consecutive calendar quarter in which generic competition exists in such country.
Under the license agreement, we have paid Kyorin Pharmaceutical $700,000 to date, and we are obligated to make payments of up to $5.0 million based on the achievement of certain clinical and regulatory milestones. We are also obligated to pay a royalty on net sales of the licensed products.
We have also filed a patent application directed to the use of MN-166 for the treatment of progressive neurodegenerative diseases in the United States and are pursuing counterparts of this patent application in certain foreign jurisdictions.
AV411
With the acquisition of Avigen, we own, co-own or hold licenses to three issued U.S. patents and 11 pending U.S. patent applications, as well as corresponding pending non-U.S. patent applications. The three patents were issued in 2009 in the United States (7,534,806—Use of Ibudilast for the Treatment of Neuropathic Pain Syndromes; 7,585,875—Substituted pyrazolo-pyridine compounds and their methods of use; and 7,622,256- Method for selecting compounds that modulate MIF-induced expression of ICAM-1 and/or VCAM-1) and will expire in 2025, 2027, and 2027, respectively. The patent applications are primarily related to Avigen’s development portfolio of small molecule-based products and are currently directed to methods of treating various indications using AV411 and analogs.
MN-001
On March 14, 2002, we entered into an exclusive license agreement with Kyorin Pharmaceutical for the development and commercialization of MN-001. We obtained an exclusive, worldwide (excluding Japan, China, South Korea and Taiwan) sublicenseable license to the patent rights and know-how related to MN-001 and its
20
Table of Contents
active metabolite, MN-002, disclosed and included in, or covered by, these patents, in all indications, except for ophthalmic solution formulations. This license includes an exclusive, sublicensable license under one U.S. patent and certain corresponding patents and patent applications in foreign countries. The U.S. composition of matter patent for MN-001 underlying the license expired on February 23, 2009, and the U.S. composition of matter patent for MN-002 underlying the license is set to expire on December 30, 2011. Certain annuities were not paid in a timely manner with respect to certain foreign patents licensed under MN-002, resulting in the lapse of patents in certain countries. In such jurisdictions, we intend to rely upon the applicable period of post-approval exclusivity, in addition to any patents that may issue from our own patent applications. Under the terms of the agreement, we granted to Kyorin Pharmaceutical an exclusive, royalty-free, sublicenseable license to use the preclinical, clinical and regulatory databases to develop opthmalmic products incorporating the MN-001 compound anywhere in the world and non-opthalmic products incorporating the MN-001 compound outside of our territory.
The license agreement may be terminated by either party following an uncured breach of any material provision in the agreement by the other party, and we may terminate the agreement for any reason with 90 days’ written notice to Kyorin Pharmaceutical or, in the event that a third party claims that the licensed patent rights or know-how infringe upon such third party’s intellectual property rights, with 30 days’ written notice.
The term of this agreement is determined on a country-by-country basis and extends until the later of the expiration of the obligation to make payments under the agreement or the last date on which the manufacture, use or sale of the product would infringe a valid patent claim held by Kyorin Pharmaceutical but for the license granted by the agreement or the last date of the applicable market exclusivity period. In the absence of a valid patent claim and generic competition in a particular country, the agreement will expire on the earlier of five years from the date of the first commercial sale of the product by us or the end of the second consecutive calendar quarter in which generic competition exists in such country.
Under the license agreement, we have paid Kyorin Pharmaceutical $4.0 million to date, and we are obligated to make payments of up to $5.0 million based on the achievement of certain clinical and regulatory milestones. We are also obligated to pay a royalty on net sales of the licensed products.
We filed, and the U.S. Patent and Trademark Office issued, five U.S. patents covering certain compositions, uses and manufacturing processes associated with MN-001, four of which are set to expire on June 24, 2023 and one of which is set to expire on April 27, 2025. Patent applications corresponding to these U.S. patents were filed in certain foreign countries. We also filed one U.S. continuation application and one U.S. divisional application from these patents.
MN-029
On June 19, 2002, we entered into an exclusive license agreement with Angiogene Pharmaceuticals for the development and commercialization of the ANG-600 series of compounds. Angiogene Pharmaceuticals is a privately held, British drug discovery company. We obtained an exclusive, worldwide, sublicenseable license to the patent rights and know-how related to the ANG-600 series of compounds disclosed in and included or covered by these patents for all indications. MN-029 is one of the ANG-600 series compounds covered by this license. This license includes an exclusive, sublicensable license under three U.S. patents, two U.S. patent applications and certain corresponding patents and patent applications in foreign countries. The U.S. composition of matter patent for MN-029, which issued on November 11, 2003, is set to expire on January 14, 2020. Patent applications corresponding to this U.S. patent were filed in certain foreign countries. The U.S. patent covering methods of treating solid cancer tumors by administering MN-029, which issued on July 25, 2006, is set to expire on January 14, 2020.
The license agreement may be terminated by either party following an uncured breach of any material provision in the agreement by the other party, and we may terminate the agreement at any time by giving 30 days’ advance written notice to Angiogene Pharmaceuticals.
21
Table of Contents
The term of this agreement is determined on a country-by-country basis and extends until the earlier of the expiration of the last Angiogene Pharmaceuticals patent (or equivalent) under license which has a valid claim to expire or 15 years from the date of first commercial sale.
Under the license agreement, we have paid Angiogene Pharmaceuticals $1.4 million to date and are obligated to make payments of up to $16.5 million based on the achievement of certain clinical and regulatory milestones. We are also obligated to pay a royalty on net sales of the licensed products.
MN-305
On April 27, 2004, we entered into an exclusive license agreement with Mitsubishi Pharma Corporation, the predecessor to Mitsubishi Tanabe Pharma Corporation, for the development and commercialization of MN-305. Mitsubishi Tanabe Pharma Corporation is a fully integrated Japanese pharmaceutical company and is listed on the First Section of the Tokyo Stock Exchange. We obtained an exclusive, worldwide (excluding Japan, Singapore, Brunei, Thailand, Malaysia, Indonesia, the Philippines, Vietnam, Bangladesh, Pakistan, South Korea, China and Taiwan), sublicenseable license to the patent rights and know-how related to MN-305 and its active metabolite disclosed and included or covered by these patents for all indications except for ophthalmic solution formulations. The license is sublicensable upon receipt of the written consent of Mitsubishi Tanabe Pharma Corporation. This license includes an exclusive, sublicensable license under five U.S. patents and certain corresponding patents and patent applications in foreign countries. The U.S. composition of matter patent for MN-305, which issued on December 1, 1992, is set to expire on March 14, 2011. Patent applications corresponding to this U.S. patent were filed in certain foreign countries, and these foreign counterparts are set to expire no earlier than between March 12, 2011 and March 14, 2011. The U.S. patent covering the use of MN-305 to treat anxiety, which issued on August 10, 1993, is set to expire on March 14, 2011.
Under the terms of the agreement, we granted to Mitsubishi Tanabe Pharma Corporation a license to use our know-how and patents relating to MN-305 to develop products incorporating the MN-305 compound outside of our territory. Mitsubishi Tanabe Pharma Corporation also has the right to co-promote licensed products in our territory on terms to be agreed upon by the parties.
The license agreement may be terminated by either party following an uncured breach of any material provision in the agreement by the other party. We may terminate the agreement if, in our reasonable opinion, the safety, patient tolerability, efficacy, profile or commercial viability of MN-305 does not justify continued development with 90 days’ written notice to Mitsubishi Tanabe Pharma Corporation or, in the event that a third party claims that the licensed intellectual property related to MN-305 infringes such third party’s intellectual property rights, with 30 days’ written notice.
The term of this agreement is determined on a country-by-country basis and extends until the later of ten years from the date of first commercial sale in a specific country or the expiration of a valid patent claim in such country. In the event that we enter into a sublicense with a third party, the term of the agreement will extend for as long as we receive royalty payments from such third party.
Under the license agreement, we have paid Mitsubishi Tanabe Pharma Corporation $1.0 million to date, and we are obligated to make payments of up to $18.8 million based on the achievement of certain clinical, regulatory and sales milestones. We are also obligated to pay a royalty on net sales of the licensed products.
MN-246
On December 8, 2004, we entered into an exclusive license agreement with Mitsubishi Pharma Corporation, the predecessor to Mitsubishi Tanabe Pharma Corporation, for the development and commercialization of MN-246. We obtained an exclusive, worldwide (excluding Japan, Singapore, Brunei, Thailand, Malaysia, Indonesia, the Philippines, Vietnam, Bangladesh, Pakistan, South Korea, China and Taiwan), sublicenseable
22
Table of Contents
license to the intellectual property surrounding MN-246, its derivatives and any other compounds disclosed or claimed in the licensed Mitsubishi Tanabe Pharma Corporation patent assets. The license is sublicensable upon receipt of the written consent of Mitsubishi Tanabe Pharma Corporation and includes an exclusive license under one U.S. patent and certain corresponding patents and patent applications in foreign countries. The U.S. patent covering MN-246 and methods of making and using MN-246, which issued on May 30, 2000, is set to expire on October 24, 2016. Patent applications corresponding to this U.S. patent were filed in certain foreign countries, and these foreign counterparts are set to expire no earlier than October 24, 2016. In addition, we filed a U.S. patent application and corresponding patent applications in Thailand and Taiwan for a new method of use for MN-246.
The issued U.S. patent covers generic phenylethanolamines encompassed by a given chemical formula, including MN-246, processes for the production of such phenylethanolamines, a pharmaceutical composition of such phenylethanolamines and methods of use for such phenylethanolamines for the treatment of a variety of human or animal ailments, including accelerated or spasmodic gastrointestinal motility, dysuria, pollakisuria, urinary incontinence, obesity and diabetes. This U.S. patent is set to expire on October 24, 2016. Foreign counterparts have been filed or patented in other countries and are also set to expire no earlier than October 24, 2016. Under the terms of the agreement, we granted to Mitsubishi Tanabe Pharma Corporation a license to use our know-how and patents relating to MN-246 to develop products incorporating the MN-246 compound outside of our territory. Mitsubishi Tanabe Pharma Corporation also has the right to co-promote licensed products in our territory on terms to be agreed upon by the parties.
The license agreement may be terminated by either party following an uncured breach of any material provision in the agreement by the other party. We may terminate the agreement if, in our reasonable opinion, the safety, patient tolerability, efficacy, profile or commercial viability of MN-246 does not justify continued development with 90 days’ written notice to Mitsubishi Tanabe Pharma Corporation or in the event that a third party claims that the licensed intellectual property related to MN-246 infringes such third party’s intellectual property rights with 30 days’ written notice.
The term of this agreement is determined on a country-by-country basis and extends until the later of ten years from the date of first commercial sale in a specific country or the expiration of a valid patent claim in such country. In the event that we enter into a sublicense with a third party, the term of the agreement will extend for as long as we receive royalty payments from such third party.
Under the license agreement, we have paid Mitsubishi Tanabe Pharma Corporation $750,000 to date, and we are obligated to make payments of up to $14.5 million based on the achievement of certain clinical, regulatory and sales milestones. We are also obligated to pay a royalty on net sales of the licensed products.
MN-447
On November 1, 2006, we entered into an exclusive license agreement with Meiji Seika Kaisha for the development and commercialization of MN-447. Meiji Seika Kaisha is a fully integrated Japanese pharmaceutical company and is listed on the First Section of the Tokyo Stock Exchange. We obtained an exclusive, worldwide (excluding Japan, Bangladesh, Brunei, Cambodia, People’s Republic of China, Indonesia, Laos, Malaysia, Myanmar, the Philippines, Singapore, South Korea, Taiwan, Thailand and Vietnam), sublicensable license from Meiji Seika Kaisha for MN-447 (and any other compound claimed or covered by U.S. patent 6,420,558) for any human use. This license includes an exclusive sublicensable license under one U.S. patent and certain corresponding patents and patent applications in foreign countries. The U.S. patent covering MN-447 and methods of treating an integrin avß3-mediated disease, platelet thrombosis, aggregation and related disorders, which issued on July 16, 2002, is set to expire on April 9, 2019. Patent applications corresponding to this U.S. patent were filed in certain foreign countries. Under the terms of the license, we granted a license to Meiji Seika Kaisha to use our know-how and patents relating to MN-447 to develop products incorporating the MN-447 compound outside of our territory.
23
Table of Contents
This license agreement may be terminated by either party following an uncured breach of any material provision of the agreement by the other party upon 90 days’ written notice or the inability or delay in performing under the agreement due to a force majeure event which lasts longer than 12 months. We also have the right to terminate the agreement in the event of third party intellectual property claims which are not timely remedied by us and Meiji Seika Kaisha or if, in our reasonable opinion, the safety, patient tolerability, efficacy, profile or commercial viability of MN-447 does not justify continued development. Meiji Seika Kaisha also has the right to terminate the agreement in the event that we cease development of MN-447 for a period of one year or longer.
The term of the agreement is determined on a country-by-country basis and extends until the expiration of the last Meiji Seika Kaisha patent (or equivalent) under license to expire or in the event that a valid claim does not exist or, if a valid claim expired more than 15 years from the date of first commercial sale, 15 years from the date of first commercial sale.
Under the license agreement, we have paid Meiji Seika Kaisha $400,000 to date, and we are obligated to make payments of up to $8.7 million based on the achievement of certain clinical and regulatory milestones. We are also obligated to pay a royalty on net sales of the licensed products.
MN-462
On November 1, 2006, we entered into an exclusive license agreement with Meiji Seika Kaisha for the development and commercialization of MN-462. We obtained an exclusive, worldwide (excluding Japan, Bangladesh, Brunei, Cambodia, People’s Republic of China, Indonesia, Laos, Malaysia, Myanmar, the Philippines, Singapore, South Korea, Taiwan, Thailand and Vietnam), sublicenseable license from Meiji Seika Kaisha for MN-462 (and any other compound claimed or covered by U.S. patent 6,576,627) for any human use. This license includes an exclusive sublicensable license under two U.S. patents and certain corresponding patents and patent applications in foreign countries. The U.S. patent covering MN-462 medicament compositions containing MN-462, and methods of therapeutic treatment or preventive treatment of thrombotic disease, which issued on June 10, 2003, is set to expire on September 13, 2020. Patent applications corresponding to this U.S. patent were filed in certain foreign countries. Under the terms of the license, we granted a license to Meiji Seika Kaisha to use our know-how and patents relating to MN-462 to develop products incorporating the MN-462 compound outside of our territory.
This license agreement may be terminated by either party following an uncured breach of any material provision of the agreement by the other party upon 90 days’ written notice or the inability or delay in performing under the agreement due to a force majeure event which lasts longer than 12 months. We also have the right to terminate the agreement in the event of third party intellectual property claims which are not timely remedied by us and Meiji Seika Kaisha or if, in our reasonable opinion, the safety, patient tolerability, efficacy, profile or commercial viability of MN-462 does not justify continued development. Meiji Seika Kaisha also has the right to terminate the agreement in the event that we cease development of MN-462 for a period of one year or longer.
The term of the agreement is determined on a country-by-country basis and extends until the expiration of the last Meiji Seika Kaisha patent (or equivalent) under license to expire or in the event that a valid claim does not exist or, if a valid claim expired more than 15 years from the date of first commercial sale, 15 years from the date of first commercial sale.
Under this license agreement, we have paid Meiji Seika Kaisha $400,000 to date, and we are obligated to make payments of up to $8.7 million based on the achievement of certain clinical and regulatory milestones. We are also obligated to pay a royalty on net sales of the licensed products.
General
Our proposed commercial activities may conflict with patents which have been or may be granted to competitors, universities and/or others. Third parties could bring legal action against us, our licensors or our sublicensees claiming patent infringement and could seek damages or enjoin manufacturing and marketing of the
24
Table of Contents
affected product or its use or the use of a process for the manufacturing of such products. If any such actions were to be successful, in addition to any potential liability for indemnification, damages and attorneys’ fees in certain cases, we could be required to obtain a license, which may not be available on commercially reasonable terms or at all, in order to continue to manufacture, use or market the affected product. We also rely upon unpatented proprietary technology because, in some cases, our interests would be better served by reliance on trade secrets or confidentiality agreements than by patents. However, others may independently develop substantially equivalent proprietary information and techniques or gain access to or disclose such proprietary technology. We may not be able to meaningfully protect our rights in such unpatented proprietary technology. We may also conduct research on other pharmaceutical compounds or technologies, the rights to which may be held by, or be subject to patent rights of, third parties. Accordingly, if products based on such research are commercialized, such commercial activities may infringe patents or other rights, which may require us to obtain a license to such patents or other rights. We are not aware of any third-party infringements of patents we hold or licenses and have not received any material claims by third parties of infringement by us of such parties’ intellectual property rights.
There can be no assurance that patent applications filed by us or others, in which we have an interest as assignee, licensee or prospective licensee, will result in patents being issued or that, if issued, any of such patents will afford protection against competitors with similar technology or products or could not be circumvented or challenged. For example, we have U.S. patents covering the method of using MN-166 to treat MS and the method of using AV411 to treat neuropathic pain, but we do not have any unqualified composition of matter patent claims for MN-166 or AV411. As a result, unrelated third parties may develop products with the same API as MN-166 so long as such parties do not infringe our method of use patent, other patents we have exclusive rights to through our licensor or any patents we may obtain for MN-166 or AV411.
In addition, if we develop certain products that are not covered by any patents, we will be dependent on obtaining market exclusivity under the data exclusivity provisions of Hatch-Waxman Act for such products. If we are unable to obtain strong proprietary rights protection for our products after obtaining regulatory approval, competitors may be able to market competing generic products by taking advantage of an abbreviated procedure for obtaining regulatory clearance, including the ability to demonstrate bioequivalency to our product(s) without being required to conduct lengthy clinical trials. Certain of our license agreements provide for reduced royalties or, in some cases, foregone royalties in the event of generic competition.
Competition
The development and commercialization of new drugs is extremely competitive and characterized by extensive research efforts and rapid technological progress. Competition in our industry occurs on a variety of fronts, including developing and bringing new products to market before others, developing new products to provide the same benefits as existing products at lower cost and developing new products to provide benefits superior to those of existing products. We face competition from pharmaceutical and biotechnology companies, as well as numerous academic and research institutions and governmental agencies, in the United States and abroad. Some of these competitors have products or are pursuing the development of drugs that target the same diseases and conditions that are the focus of our product development programs. Many of our competitors have products that have been approved or are in advanced development and may succeed in developing drugs that are more effective, safer and more affordable or more easily administered than ours or that achieve patent protection or commercialization sooner than our products. Our competitors may also develop alternative therapies that could further limit the market for any products that we are able to obtain approval for, if at all.
In many of our target disease areas, potential competitors are working to develop new compounds with different mechanisms of action and attractive efficacy and safety profiles. Many of our competitors have substantially greater financial, research and development resources (including personnel and technology), clinical trial experience, manufacturing, sales and marketing capabilities and production facilities than we do. Smaller companies also may prove to be significant competitors, particularly through proprietary research discoveries and collaboration arrangements with large pharmaceutical and established biotechnology companies.
25
Table of Contents
MN-221 for Acute Exacerbations of Asthma
Our MN-221 product candidate is being developed for the treatment of acute exacerbations of asthma in the emergency room setting. The current standard of care for acute exacerbations of asthma is inhaled albuterol (a ß2-adrenergic receptor agonist), inhaled ipratropium (an anticholinergic) and oral or injected corticosteroids. In addition, subcutaneously administered terbutaline (a ß2-adrenergic receptor agonist) is sometimes used to treat this condition, particularly in pediatric patients. Certain oral anti-inflammatory asthma drugs are being investigated in an intravenous form for the treatment of acute exacerbations of asthma.
MN-221 for Chronic Obstructive Pulmonary Disease Exacerbations
Our MN-221 product candidate is also being developed for the treatment of COPD exacerbations. The standard of care for COPD exacerbations is similar to that of acute exacerbations of asthma in that inhaled bronchodilators and anticholinergics are administered; however, antibiotics are also administered and parenteral terbutaline is excluded because of the exclusively adult patient population. A greater percentage of patients diagnosed with COPD exacerbations are hospitalized than patients diagnosed with asthma exacerbations, and such patients continue the same treatment paradigm as in the emergency department.
MN-166 for Multiple Sclerosis
Our MN-166 product candidate has been in development for the treatment of MS. Current treatments for MS include the beta interferons, such as Biogen Idec Inc.’s Avonex® (beta interferon), Teva Pharmaceutical Industries Ltd.’s and Sanofi-Aventis’ Copaxone® (glatiramer acetate), Merck Serono’s and Pfizer Inc.’s Rebif® (beta interferon), Bayer Schering Pharma AG’s Betaseron/Betaferon® and Biogen Idec Inc.’s Tysabri® (natalizumab), all of which are administered by injection. Of the many new agents in development for MS, only a few, such as Sanofi-Aventis’ teriflunomide, Novartis AG’s fingolimod/FTY720, Teva Pharmaceutical Industries Ltd.’s laquinimod and Biogen Idec Inc.’s BG-12, are intended for oral administration like MN-166.
AV411 for Other Central Nervous System Disorders
Our AV411 product candidate has been in development for treatment of neuropathic pain and opioid withdrawal and methamphetamine addiction. Current treatments for neuropathic pain include anti-epileptics such as Pfizer Inc.’s Neurontin® (gabapentin) and Lyrica® (pregabalin), and antidepressants, including Eli Lilly & Co.’s Cymbalta® (duloxetine). We are aware of additional compounds for chronic neuropathic pain that are currently in development at numerous companies including Bayer Schering Pharma AG , GlaxoSmithKline plc, Merck & Co., Inc., Novartis AG, Pfizer Inc., Cognetix, Inc., GW Pharmaceuticals plc, Indevus Pharmaceuticals, Inc., Nastech Pharmaceutical Company Inc., Avanir Pharmaceuticals, Solace Pharmaceuticals, Pain Therapeutics, Inc., and XenoPort, Inc.
Current treatments for withdrawal symptoms include narcotics such as generic methadone and Reckitt Benckiser Pharmaceuticals, Inc.’s Suboxone® (buprenorphine) and Subutex® (buprenorphine + the narcotic antagonist naloxone). Limited non-narcotic drug candidates for withdrawal symptoms exist. Britannia Pharmaceuticals Limited’s BritLofex® (Lofexidine), licensed for development in U.S. clinical trials to US WorldMeds LLC, is an alpha adrenoceptor agonist like clonidine which may have somewhat less orthostatic hypotension limitations.
MN-001 for Bronchial Asthma
Our MN-001 product candidate has been in development for the treatment of bronchial asthma. There are two currently marketed leukotriene inhibitors, Merck & Co. Inc.’s Singulair® (montelukast) and AstraZeneca PLC’s Accolate® (zafirlukast). There are also several products in clinical development to treat bronchial asthma, including Mitsubishi Tanabe Pharma Corporation’s MCC 847 (masilukast), which is another leukotriene inhibitor currently in Phase III clinical testing in Japan.
26
Table of Contents
MN-001 for Interstitial Cystitis
Our MN-001 product candidate has been in development for the treatment of IC. There are two currently marketed products, Teva Pharmaceuticals Industries Ltd.’s Elmiron® and Bioniche Pharma Group Limited’s RIMSO-50®. There is also a product in clinical development to treat IC, Taiho Pharmaceutical Co., Ltd.’s IPD-1151 (suplatast tosilate), which is currently in Phase III clinical testing in Japan. In addition, Urigen Pharmaceuticals, Inc.’s URG-101 for the treatment of painful bladder syndrome/interstitial cystitis is in Phase II clinical testing.
MN-029 for Solid Tumors
Our MN-029 product candidate has been in development for the treatment of solid tumors. There are a number of compounds in clinical development with a mechanism similar to MN-029, including Oxigene Inc.’s ZBRESTAT™ (fosbretabulin) and Sanofi-Aventis’ AVE 8062, which are in Phase III clinical testing.
MN-305 for General Anxiety Disorder
Our MN-305 product candidate has been in development for the treatment of General Anxiety Disorder. There are a number of approved products to treat Generalized Anxiety Disorder, including Eli Lilly and Company’s Cymbalta® (duloxetine).
MN-221 for Preterm Labor
Our MN-221 product candidate has been in development for the treatment of preterm labor. There are a number of oxytocin antagonists undergoing clinical evaluation, including GlaxoSmithKline plc’s GSK221149, which is currently in Phase II clinical testing.
MN-246 for Urinary Incontinence
Our MN-246 product candidate has been in development for the treatment of urinary incontinence. There are a number of compounds in various stages of clinical development to treat urinary incontinence. Pfizer Inc.’s Detrol® (tolterodine tartrate) is the market leader, and other marketed drugs include Astellas Pharma Inc.’s VESIcare® (solifenacin succinate) and Novartis AG’s Enablex® (darifenacin) were introduced in the first quarter of 2005, both of which are anti-cholinergic agents. Ono Pharmaceutical Co., Ltd. and Kyorin Pharmaceutical have received approval for Staybla® (muscarinic antagonist). Schwarz Pharma AG’s Toviaz® (fesoterodine fumarate), another anti-cholinergic, has also recently been approved. Kissei Pharmaceutical, Astellas Pharma Inc. and GlaxoSmithKline plc also have ß3-adrenergic receptor agonists for the treatment of this indication.
MN-447 and MN-462 for Thrombotic Disorders
Our MN-447 and MN-462 product candidates have been in development for the treatment of thrombotic disorders. Both product candidates are currently in preclinical development; therefore, we have not identified the particular thrombotic disorders that we intend to target upon reaching the clinical development stage for these product candidates. Consequently, we cannot accurately evaluate the competition we will face. Currently, the market leaders for anti-thrombotic drugs are Bristol-Myers Squibb Company’s and Sanofi-Aventis’ Plavix® (clopidogrel) and Sanofi-Aventis’ Lovenox® (enoxaparin).
Government Regulation
Government authorities in the United States and other countries extensively regulate the research, development, testing, manufacture, labeling, promotion, advertising, distribution, sampling, marketing, and import and export of pharmaceutical products and biologics such as those we are developing. In the United
27
Table of Contents
States, the FDA, under the Federal Food, Drug and Cosmetic Act, as amended, and other federal statutes and regulations, subjects pharmaceutical products to extensive and rigorous review. Any failure to comply with applicable requirements, both before and after approval, may subject us, our third-party manufacturers, contractors, suppliers and partners to administrative and judicial sanctions, such as a delay in approving or refusal to approve pending applications, fines, warning letters, product recalls, product seizures, total or partial suspension of manufacturing or marketing, injunctions and/or criminal prosecution.
U.S. Regulatory Approval
Overview. In the United States, drugs and drug testing are regulated by the FDA under the Federal Food, Drug and Cosmetic Act, as well as state and local government authorities. All of in development will require regulatory approval by government agencies prior to commercializing the product. To obtain approval of a new product from the FDA, we must, among other requirements, submit data supporting safety and efficacy, as well as detailed information on the manufacture and composition of the product and proposed labeling. Our product candidates are in the early stages of testing and none has been approved. The steps required before a drug can be approved generally involve the following:
| • | completion of preclinical laboratory and animal tests; |
| • | submission of an IND, which must become effective before human clinical trials may begin in the United States; |
| • | completion of adequate and well-controlled human clinical trials to establish the safety and efficacy of the product candidate for each indication for which approval is sought; |
| • | submission to the FDA of an NDA; |
| • | development of manufacturing processes which conform to FDA-mandated commercial good manufacturing practices, or cGMPs, and satisfactory completion of our FDA inspection to assess compliance; and |
| • | FDA review and approval of an NDA, which process may involve input from advisory panels to the FDA and may include post-approval commitments. |
The testing, collection of data, preparation of necessary applications and approval process requires substantial time, effort and financial resources. Additionally, statutes, rules, regulations and policies may change and new regulations may be issued that could delay such approvals. The FDA may not act quickly or favorably in reviewing our applications, and we may encounter significant difficulties and costs in our efforts to obtain FDA approvals that could delay or preclude us from marketing our products.
Preclinical Tests. Preclinical tests include laboratory evaluation of the product candidate, its chemistry, toxicity, formulation and stability, as well as animal studies to assess the potential safety and efficacy of the product candidate. The results of the preclinical tests, together with manufacturing information, analytical data and other available information about the product candidate, are submitted to the FDA as part of an IND. Preclinical tests and studies can take several years to complete and, despite completion of those tests and studies, the FDA may not permit clinical testing to begin.
The IND Process. An IND must be effective to administer an investigational drug to humans. The IND will automatically become effective 30 days after its receipt by the FDA unless the FDA, before that time, raises concerns or questions about the information provided and/or the conduct of the studies as outlined in the IND. At any time thereafter, the FDA may raise concerns or questions about the conduct of the trials as outlined in the IND and even impose a clinical hold if the FDA deems it appropriate. In such case, the IND sponsor and the FDA must resolve any outstanding concerns before clinical trials can begin or continue. The IND application process may become extremely costly and substantially delay development of our products. Moreover, positive results in preclinical tests or prior human studies do not necessarily predict positive results in subsequent clinical trials.
28
Table of Contents
Clinical Trials. Human clinical trials are typically conducted in three sequential phases that may overlap:
| • | Phase I: The drug is initially introduced into a small number of human subjects or patients and tested for safety, dosage tolerance, absorption, distribution, excretion and metabolism. |
| • | Phase II: The drug is introduced into a limited patient population to assess the efficacy of the drug in specific, targeted indications, assess dosage tolerance and optimal dosage, and identify possible adverse effects and safety risks. |
| • | Phase III: The drug is introduced into an expanded patient population at geographically dispersed clinical trial sites to further evaluate clinical efficacy and safety. |
Prior to initiation of each clinical trial, an independent Institutional Review Board, or IRB, for each medical site proposing to conduct the clinical trials must review and approve the study protocol and study subjects must provide informed consent for participation in the study.
We cannot be certain that we will successfully complete Phase I, Phase II or Phase III testing of our drug candidates within any specific time period, if at all. Clinical trials must be conducted in accordance with the FDA’s good clinical practices requirements. The FDA may order the partial, temporary or permanent discontinuation of a clinical trial at any time or impose other sanctions if it believes that the clinical trial is not being conducted in accordance with FDA requirements or presents an unacceptable risk to the clinical trial patients. The IRB generally must approve the clinical trial design and patient informed consent at each clinical site and may also require the clinical trial at that site to be halted, either temporarily or permanently, for failure to comply with the IRB’s requirements, or may impose other conditions.
The NDA Process. If clinical trials are successful, the next step in the drug regulatory approval process is the preparation and submission to the FDA of a NDA. The NDA is the vehicle through which drug sponsors formally propose that the FDA approve a new pharmaceutical product for marketing and sale in the United States. The NDA must contain a description of the manufacturing process and quality control methods, as well as results of preclinical tests, toxicology studies, clinical trials and proposed labeling, among other things. A substantial user fee must also be paid with the submission of the NDA, unless an exemption applies.
Upon submission of the NDA, the FDA will make a threshold determination as to whether the application is sufficiently complete to permit review and, if not, will issue a refuse to file letter. If the submission is accepted for filing, the FDA begins an in-depth review of the NDA and will attempt to review and take action on the application in accordance with performance goals established in connection with the user fee laws. Among the conditions for a NDA approval is the requirement that the prospective manufacturer’s quality control and manufacturing procedures conform on an ongoing basis with cGMPs.
If the FDA’s evaluations of the NDA and the clinical and manufacturing procedures and facilities cGMPs are favorable, the FDA may issue either an approval letter or an approvable letter, which contains the conditions that must be met in order to secure final approval of the NDA. If and when those conditions have been met to the FDA’s satisfaction, the FDA will issue an approval letter, authorizing commercial marketing of the drug for certain indications. The FDA may also grant approval with requirements to complete post-marketing studies, referred to as Phase IV clinical trials, or restrictive product labeling, or may impose other restrictions on marketing or distribution, such as the adoption of a special risk management plan. The FDA may deny or delay approval of applications that do not meet applicable regulatory criteria or if the FDA determines that the clinical data do not adequately establish the safety and efficacy of the drug.
The Hatch-Waxman Act. Under the Hatch-Waxman Act, certain newly approved drugs and indications benefit from a statutory period of non-patent marketing exclusivity. The Hatch-Waxman Act provides five-year marketing exclusivity to the first applicant to gain approval of an NDA for a new chemical entity, meaning that the FDA has not previously approved any other new drug containing the same active moiety. The Hatch-
29
Table of Contents
Waxman Act also provides three years of marketing exclusivity for the approval of new and supplemental NDAs for, among other things, new indications, dosages or strengths of an existing drug, if new clinical investigations that were conducted or sponsored by the applicant are essential to the approval of the application. Pediatric exclusivity of six months may also be available if agreement is reached with the FDA and qualifying studies of product candidates in pediatric populations are conducted.
Manufacturing and Other Regulatory Requirements. Both before and after approval, we and our third-party manufacturers must comply with a number of regulatory requirements. For example, if we seek to make certain changes to an approved product, such as promoting or labeling a product for a new indication, manufacturing changes or additional labeling claims, we will need FDA review and approval. Advertising and other promotional materials must comply with FDA requirements and established requirements applicable to drug samples. In addition, we may not label or promote the product for an indication that has not been approved by the FDA. Securing FDA approval for new indications or product enhancements and, in some cases, for new labeling claims, is generally a time-consuming and expensive process that may require us to conduct clinical trials under the FDA’s IND regulations. Even if such studies are conducted, the FDA may not approve any change in a timely fashion, or at all. In addition, adverse experiences associated with use of the products must be reported to the FDA, and FDA rules govern how we can label, advertise or otherwise commercialize our products.
The NDA holders and manufacturers of approved products will be subject to continual review and periodic inspections by the FDA and other authorities, where applicable, and must comply with ongoing requirements, including the FDA’s cGMP requirements. Manufacturers must provide certain safety and efficacy information and make certain other required reports. To comply with cGMP requirements, manufacturers must continue to spend time, money and effort to meet requirements relating to personnel, facilities, equipment, production and process, labeling and packaging, quality control, record-keeping and other requirements. The FDA periodically inspects drug manufacturing facilities to evaluate compliance with cGMP requirements. Product approvals may be withdrawn if compliance with regulatory requirements is not maintained or if problems concerning safety or efficacy of the product occur following approval. Because we intend to contract with third parties for manufacturing of our products, our ability to control third-party compliance with FDA requirements will be limited to contractual remedies and rights of inspection.
In addition to FDA restrictions on marketing of pharmaceutical products, several other state and federal laws have been applied to restrict certain sales and marketing practices in the pharmaceutical industry in recent years. These laws include licensing requirements, compliance program requirements, annual certificates and disclosures, anti-kickback statutes and false claims statutes. The Medicare and Medicaid Patient Protection Act of 1987, as amended, or the Anti-Kickback Statute, prohibits, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any health care item or service reimbursable under Medicare, Medicaid or other federally financed health care programs. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers and formulary managers on the other. Violations of the Anti-Kickback Statute are punishable by imprisonment, criminal fines, civil monetary penalties and exclusion from participation in federal health care programs. Although there are a number of statutory exemptions and regulatory safe harbors protecting certain common activities from prosecution or other regulatory sanctions, the exemptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exemption or safe harbor.
Federal false claims laws prohibit any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making, or causing to be made, a false statement to have a false claim paid. Recently, several pharmaceutical and other health care companies have been prosecuted under these laws for allegedly inflating drug prices they report to pricing services, which in turn were used by the government to set Medicare and Medicaid reimbursement rates, and for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. In addition,
30
Table of Contents
certain marketing practices, including off-label promotion, may also violate false claims laws. The majority of states also have statutes or regulations similar to the Anti-Kickback Statue and false claims laws, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor.
We are also subject to various laws and regulations regarding laboratory practices, the experimental use of animals and the use and disposal of hazardous or potentially hazardous substances in connection with our research.
Foreign Regulatory Approval
We will have to complete approval processes, similar or related to the U.S. approval processes, in virtually every foreign market for our products in order to conduct clinical or preclinical research and to commercialize our drug candidates in those countries. The approval procedures and the time required for approvals vary from country to country and may involve additional testing. In addition, regulatory approval of prices is required in most countries other than the United States. We face the risk that the resulting prices would be insufficient to generate an acceptable return to us or our collaborators.
Similar to the U.S. regulatory framework, the various phases of preclinical and clinical research are subject to significant regulatory controls within the European Union. Variations among national regimes exist. However, most jurisdictions require regulatory and ethics committees approval of interventional clinical trials. Most European regulators also require the submission of adverse event reports during a study and a copy of the final study report.
Under European Union regulatory systems, marketing authorizations may be submitted either under a centralized or decentralized procedure. The centralized procedure is currently mandatory for products developed by means of a biotechnological process and optional for new active substances and other “innovative medicinal products with novel characteristics.” It provides for the grant of a single marketing authorization that is valid for all European Union member states. The decentralized procedure provides for mutual recognition of national approval decisions. Under this procedure, the holder of a national marketing authorization may submit applications in other European Union member states, requesting them to mutually recognize the marketing authorization already granted. Within 90 days of receiving the applications and assessment report, each member state must decide whether to recognize the existing approval.
Where possible, we will strive to choose the European regulatory filing route that will most rapidly enable us to obtain the needed regulatory approvals. However, the chosen regulatory strategy may not secure regulatory approvals or approvals of the chosen product indications. In addition, these approvals, if obtained, may take longer than anticipated.
Employees
We have assembled an experienced and cohesive management and support team, with core competencies in general management, clinical development, regulatory affairs and corporate development. As of March 15, 2010, we had 24 full-time employees, one part-time employee and one intern. We believe that our relations with our employees are good, and we have no history of work stoppages.
More Information
We maintain a website at www.medicinova.com. Information contained in or that can be accessed through our website is not a part of this Annual Report on Form 10-K. We make available through our website, free of charge, all public filings with the Securities and Exchange Commission, or SEC, as soon as reasonably practicable after filing.
31
Table of Contents
We operate in a dynamic and rapidly changing environment that involves numerous risks and uncertainties. Certain factors may have a material adverse effect on our business, financial condition and results of operations, and you should carefully consider them. Accordingly, in evaluating our business, we encourage you to consider the following discussion of risk factors, in its entirety, in addition to other information contained in this Annual Report on Form 10-K and our other public filings with the SEC. Other events that we do not currently anticipate or that we currently deem immaterial may also affect our results of operations and financial condition.
Risks Related to Our Business and Industry
We have incurred significant operating losses since our inception and expect that we will incur continued losses for the foreseeable future.
We are a development stage biopharmaceutical company with a limited operating history. We have incurred significant net losses since our inception. For the year ended December 31, 2009, we had a net loss of $20.4 million. At December 31, 2009, our accumulated deficit was approximately $247.4 million. If we are successful in raising additional capital to support the expansion of our business, our annual net losses may increase over the next several years as we expand our infrastructure and incur significant costs related to the development of our product candidates.
We expect our research and development expenses to increase in connection with ongoing and planned clinical trials for our prioritized product candidates, primarily related to MN-221 for the treatment of acute exacerbations of asthma and chronic obstructive pulmonary disease, or COPD, exacerbations, and any other development activities that it may initiate. In addition, our general and administrative expenses may increase in future periods as a result of several factors, including our research and development activities, our business development activities and any expansions in our infrastructure related to such activities. Consequently, we expect to continue to incur significant and increasing operating losses for the foreseeable future. Because of the numerous risks and uncertainties associated with developing drug products, we are unable to predict the extent of any future losses or when we will become profitable, if at all.
We do not have any products that are approved for commercial sale and therefore do not expect to generate any revenues from product sales in the foreseeable future, if ever.
To date, we have funded our operations primarily from sales of our securities. We have not received, and do not expect to receive for at least the next several years, if at all, any revenues from the commercialization of our product candidates. Our only source of revenues since inception has been from development management services rendered to Asahi Kasei Pharma Corporation and Argenes, Inc., both Japanese pharmaceutical companies, in connection with their clinical development of pharmaceutical product candidates. We completed our agreement with Asahi Kasei Pharma Corporation and terminated our agreement with Argenes, Inc.; therefore, we will not generate any further revenues from these agreements. We anticipate that, prior to our commercialization of a product candidate, out-licensing upfront and milestone payments will be our primary source of revenue. To obtain revenues from sales of our product candidates, we must succeed, either alone or with third parties, in developing, obtaining regulatory approval for, manufacturing and marketing drugs with commercial potential. We may never succeed in these activities, and we may not generate sufficient revenues to continue our business operations or achieve profitability.
We are largely dependent on the success of our two prioritized product candidates, MN-221 and MN-166/AV411, and we cannot be certain that either of these product candidates will receive regulatory approval or be successfully commercialized.
We currently have no products for sale, and we cannot guarantee that we will ever have any drug products approved for sale. The research, testing, manufacturing, labeling, approval, selling, marketing and distribution of drug products are subject to extensive regulation by the FDA, and comparable regulatory authorities in other
32
Table of Contents
countries. We are not permitted to market any of our product candidates in the United States until we submit and receive approval of a NDA, for a product candidate from the FDA or its foreign equivalent from a foreign regulatory authority. Obtaining FDA approval is a lengthy, expensive and uncertain process. We currently have two prioritized product candidates, MN-221 for the treatment of acute exacerbations of asthma and COPD exacerbations and MN-166/AV411, a combined ibudalist product development program covering MS and other CNS disorders and the success of our business currently depends on their successful development and commercialization. Neither of these product candidates has completed the clinical development process; therefore, we have not submitted an NDA or foreign equivalent or received marketing approval for either of these two prioritized product candidates. In addition, we are not currently planning to pursue any further significant clinical development of MN-166/AV411 until such time that we are able to secure a strategic collaboration to advance the combined development programs, which may delay or impede the process of completing clinical trials and seeking regulatory approval for this product candidate. We also cannot assure you that we will be able to secure such a strategic collaboration on attractive financial and other terms, or at all.
The clinical development programs for MN-221 and MN-166/AV411 may not lead to commercial products for a number of reasons, including if we fail to obtain necessary approvals from the FDA or similar foreign regulatory authorities because our clinical trials fail to demonstrate to their satisfaction that these product candidates are safe and effective. We may also fail to obtain the necessary approvals if we have inadequate financial or other resources to advance our product candidates through the clinical trial process or are unable to secure a strategic collaboration or partnership with a third party. Any failure or delay in completing clinical trials or obtaining regulatory approval for either MN-221 or MN-166/AV411 in a timely manner would have a material and adverse impact on our business and our stock price.
In order to commercialize a therapeutic drug successfully, a product candidate must receive regulatory approval after the successful completion of clinical trials, which are long, complex and costly, have a high risk of failure and can be delayed or terminated at any time.
Our product candidates are subject to extensive government regulations related to development, clinical trials, manufacturing and commercialization. The process of obtaining FDA and other regulatory approvals is costly, time-consuming, uncertain and subject to unanticipated delays. To receive regulatory approval for the commercial sale of any of our product candidates, we must conduct, at our own expense, adequate and well-controlled clinical trials in human patients to demonstrate the efficacy and safety of the product candidate. Clinical testing is expensive, takes many years and has an uncertain outcome. To date, we have obtained regulatory authorization to conduct clinical trials for eight of our product development programs. INDs, were approved by the FDA and are active for seven of our product candidates. We also have obtained Clinical Trial Authorizations, or CTAs, for the ongoing Phase II clinical trial for MN-221 in Canada, Australia and New Zealand. Furthermore, through the acquisition of Avigen, we have assumed responsibility for AV411 clinical trials including one active IND for neuropathic pain and cross-reference and drug product support of the NIDA-funded opioid withdrawal investigator-initiated IND with Columbia University drug addiction clinical researchers.
It may take years to complete the clinical development necessary to commercialize a drug, and delays or failure can occur at any stage, which may result in our inability to market and sell any products derived from any of our product candidates that are ultimately approved by the FDA or foreign regulatory authorities. Our clinical trials may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical and/or non-clinical testing. For example, in October 2007, we announced that our Phase II clinical trial of MN-305 for the treatment of insomnia failed to achieve statistical significance in its primary endpoint, and, as a result, we terminated development of MN-305 for the treatment of insomnia. Of the large number of drugs in development, only a small percentage result in the submission of an NDA to the FDA and even fewer are approved for commercialization. Interim results of clinical trials do not necessarily predict final results, and success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful. A number of companies in the pharmaceutical industry have suffered significant setbacks in advanced clinical trials even after promising results in earlier clinical trials. In addition, any delays in completing clinical trials or the rejection of data from a clinical trial by a
33
Table of Contents
regulatory authority will result in increased development costs and could have a material adverse effect on the development of the impacted product candidate.
In connection with the conduct of clinical trials for each of our product candidates, we face many risks, including the risks that:
| • | the product candidate may not prove to be effective in treating the targeted indication; |
| • | patients may die or suffer other adverse effects for reasons that may or may not be related to the product candidate being tested; |
| • | the results may not confirm the positive results of earlier trials; |
| • | the FDA or other regulatory authorities may not agree with our proposed development plans or accept the results of completed clinical trials; and |
| • | our planned clinical trials and the data collected from such clinical trials may be deemed by the FDA or other regulatory authorities not to be sufficient, which would require additional development for the product candidate before it can be evaluated in late stage clinical trials or before the FDA will consider an application for marketing approval. |
If we do not complete clinical development of our product candidates successfully, we will be unable to obtain regulatory approval to market products and generate revenues from such product candidates. We may also fail to obtain the necessary regulatory approvals if we have inadequate financial or other resources to advance our product candidates through the clinical trial process. In addition, even if we believe that the preclinical and clinical data are sufficient to support regulatory approval for a product candidate, the FDA and foreign regulatory authorities may not ultimately approve such product candidate for commercial sale in any jurisdiction, which would limit our ability to generate revenues and adversely affect our business. In addition, even if our product candidates receive regulatory approval, they remain subject to ongoing FDA regulations, including obligations to conduct additional clinical trials, changes to the product label, new or revised regulatory requirements for manufacturing practices, written advisements to physicians, and/or a product recall or withdrawal.
Delays in the commencement or completion of clinical trials, or suspension or termination of our clinical trials, could result in increased costs to us and delay or limit our ability to obtain regulatory approval for our product candidates.
If we experience delays in the commencement or completion of our clinical trials, we could incur significantly higher product development costs and our ability to obtain regulatory approvals for our product candidates could be delayed or limited. The commencement and completion of clinical trials requires us to identify and maintain a sufficient number of study sites and enroll a sufficient number of patients at such sites. We do not know whether enrollment in our ongoing and planned clinical trials for our product candidates will be completed on time, or whether our additional planned and ongoing clinical trials for our product candidates will be completed on schedule, if at all. For example, we recently have experienced delays in the enrollment of patients for its ongoing Phase II clinical trial evaluating the safety and efficacy of MN-221 in patients with severe, acute exacerbations of asthma due to changes in the dosing regimen. These delays extended the anticipated date for completion of enrollment by approximately two months.
The commencement and completion of clinical trials can be delayed for a variety of other reasons, including delays in:
| • | obtaining regulatory approval to commence or amend a clinical trial; |
| • | reaching agreements on acceptable terms with prospective clinical research organizations, or CROs, and trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
34
Table of Contents
| • | recruiting and enrolling patients to participate in clinical trials; |
| • | retaining patients who have initiated a clinical trial but who may be prone to withdraw due to the treatment protocol, lack of efficacy, personal issues, or side effects from the therapy or who are lost to further follow-up; |
| • | manufacturing sufficient quantities of a product candidate; and |
| • | IRB approval or approval from foreign counterparts to conduct or amend a clinical trial at a prospective site. |
In addition, a clinical trial may be delayed, suspended or terminated by us, the FDA or other regulatory authorities due to a number of factors, including:
| • | ongoing discussions with regulatory authorities regarding the scope or design of our clinical trials or requests by them for supplemental information with respect to our clinical trial results, which may result in the imposition of a clinical hold on the IND for any clinical trial, as well as the inability to resolve any outstanding concerns with the FDA so that a clinical hold already placed on the IND may be lifted and the clinical trial may begin; |
| • | inspections of our own clinical trial operations, the operations of our CROs or our clinical trial sites by the FDA or other regulatory authorities, which may result in the imposition of a clinical hold or potentially prevent us from using some of the data generated from our clinical trials to support requests for regulatory approval of our product candidates; |
| • | our failure or inability, or the failure or inability of our CROs, clinical trial site staff or other third party service providers involved in the clinical trial, to conduct clinical trials in accordance with regulatory requirements or our clinical protocols; |
| • | lower than anticipated enrollment or retention rates of patients in clinical trials; |
| • | new information suggesting unacceptable risk to subjects or unforeseen safety issues or any determination that a trial presents unacceptable health risks; |
| • | insufficient supply or deficient quality of product candidates or other materials necessary for the conduct of our clinical trials; and |
| • | lack of adequate funding to continue the clinical trial, including the incurrence of unforeseen costs due to enrollment delays, requirements to conduct additional trials and studies and increased expenses associated with the services of our CROs and other third parties. |
If we experience delays in the completion of our clinical trials for a product candidate, the commercial prospects for such product candidate may be harmed, we may incur increased costs for development of such product candidate, and our ability to obtain regulatory approval for such product candidate could be delayed or limited. Many of the factors that cause or lead to delays in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval for a product candidate. In addition, any amendment to a clinical trial protocol may require us to resubmit our clinical trial protocols to IRBs or their foreign counterparts for reexamination, which may delay or otherwise impact the costs, timing or successful completion of a clinical trial.
The loss of any rights to develop and market any of our product candidates could significantly harm our business.
With the exception of AV411, we license the rights to develop and market our product candidates. Currently, we have licensed rights relating to eight compounds for the development of ten product candidates.
We are obligated to develop and commercialize these product candidates in accordance with mutually agreed upon terms and conditions. Our ability to satisfy some or all of the terms and conditions of our license
35
Table of Contents
agreements is dependent on numerous factors, including some factors that are outside of our control. Any of our license agreements may be terminated if we breach our obligations under the agreement materially and fail to cure any such breach within a specified period of time.
If any of our license agreements is terminated, we would have no further rights to develop and commercialize the product candidate that is the subject of the license. The termination of the license agreements related to either of our two prioritized product candidates would significantly and adversely affect our business. The termination of any of the remainder of our license agreements could materially and adversely affect our business.
If our competitors develop and market products that are more effective than our product candidates, they may reduce or eliminate our commercial opportunities.
The biotechnology and pharmaceutical industries are subject to rapid and intense technological change. We face, and will continue to face, competition from pharmaceutical and biotechnology companies, as well as numerous academic and research institutions and governmental agencies, in the United States and abroad. Some of these competitors have products or are pursuing the development of drugs that target the same diseases and conditions that are the focus of our product development programs. We cannot assure you that developments by others will not render our product candidates obsolete or noncompetitive. Many of our competitors have products that have been approved or are in advanced development and may succeed in developing drugs that are more effective, safer and more affordable or more easily administered than ours, or that achieve patent protection or commercialization sooner than our products. Our competitors may also develop alternative therapies that could further limit the market for any products that we are able to obtain approval for, if at all. In addition, new developments, including the development of other drug technologies and methods of preventing the incidence of disease, occur in the pharmaceutical industry at a rapid pace. These developments may render our product candidates obsolete or noncompetitive.
In many of our target disease areas, potential competitors are working to develop new compounds with different mechanisms of action and attractive efficacy and safety profiles. Many of our competitors have substantially greater financial, research and development resources (including personnel and technology), clinical trial experience, manufacturing, sales and marketing capabilities and production facilities than we do. Smaller companies also may prove to be significant competitors, particularly through proprietary research discoveries and collaboration arrangements with large pharmaceutical and established biotechnology companies.
Our competitors may obtain regulatory approval of their products more rapidly than we are able to or may obtain patent protection or other intellectual property rights that limit our ability to develop or commercialize our product candidates. Our competitors may also develop drugs that are more effective and less costly than ours and may also be more successful than us in manufacturing and marketing their products. We also expect to face similar competition in our efforts to identify appropriate collaborators or partners to help develop or commercialize our product candidates.
We will depend on strategic collaborations with third parties to develop and commercialize selected product candidates and will not have control over a number of key elements relating to the development and commercialization of these product candidates if we are able to achieve such third-party arrangements.
A key aspect of our strategy is to seek collaborations with partners, such as large pharmaceutical companies, that are willing to conduct later-stage clinical trials and further develop and commercialize selected product candidates. Following completion of the Phase II clinical trial for MN-166 for the treatment of MS in the second quarter of 2008 and the acquisition of AV411 in December 2009, we have not undertaken, nor do we plan to undertake, any further significant clinical development activities for any of our product candidates other than MN-221 for the treatment of acute exacerbations of asthma and COPD exacerbations, other than those activities deemed necessary to maximize each product candidate’s value, until such time that we are successful in entering
36
Table of Contents
into a partnership or collaboration to further development of such product candidates. To date, we have not entered into any such collaborative arrangements, and we may not be able to enter into any collaborations or otherwise monetize these product candidates on acceptable terms, if at all.
By entering into a strategic collaboration with a partner, we may rely on the partner for financial resources and for development, regulatory and commercialization expertise. Even if we are successful in entering into a strategic collaboration for one of our product candidates, our partner may fail to develop or effectively commercialize the product candidate because such partner:
| • | does not have sufficient resources or decides not to devote the necessary resources due to internal constraints such as limited cash or human resources; |
| • | decides to pursue a competitive potential product developed outside of the collaboration; |
| • | cannot obtain the necessary regulatory approvals; |
| • | determines that the market opportunity is not attractive; or |
| • | cannot manufacture the necessary materials in sufficient quantities from multiple sources or at a reasonable cost. |
We also face competition in our search for partners from other biotechnology and pharmaceutical companies worldwide, many of whom are larger and able to offer more attractive deals in terms of financial commitments, contribution of human resources, or development, manufacturing, regulatory or commercial expertise and support.
If we are not successful in attracting partners and entering into collaborations on acceptable terms for these product candidates or otherwise monetizing these product candidates, we may not be able to complete development of or obtain regulatory approval for such product candidates. In such event, our ability to generate revenues from such products and achieve or sustain profitability would be significantly hindered.
Negative conditions in the global credit markets may impair further the liquidity of our investment portfolio.
All of our investment securities, which consist of auction rate securities, or ARS, are designated as trading securities. These ARS represent 100 percent of our overall investment portfolio. At December 31, 2009, approximately $26.8 million of our ARS and the ARS Put were classified as current assets because they can be readily converted to cash within 12 months. Of the approximately $2.1 million of ARS which continue to be classified as long-term assets as of December 31, 2009, approximately $1.8 million consisted of private placement investment securities. None of the underlying collateral for our ARS consisted of subprime mortgages or collateralized debt obligations.
Due to continued negative conditions in the global credit markets, our ARS have continued to fail at auction with few to no trades in either the primary or the secondary markets. As a result, we have only been unable to liquidate at a loss a portion of ARS that was not subject to the ARS Rights Offer. As such we could be required to hold these securities until such time that they are redeemed by the issuer, successfully sold at auction, sold through a secondary market or ultimately mature. In addition, with the adoption of Accounting Standards Codification, or ASC, 820, authoritative guidance for fair value, measurements and disclosures (formerly Statement of Financial Accounting Standards, or SFAS, 157), we determined the fair value of our ARS portfolio primarily on Level 3 criteria, which resulted in our reliance on a discounted cash flow valuation model with assumptions related to interest rates, maturities and liquidity, determined by us based on the credit quality of the security, the credit quality of the associated insurer, if applicable, the respective prospectus and the credit market outlook. With all of our investment securities designated as trading securities, any additional increase or decrease in the fair value of our investment securities is recorded as either a gain or an impairment charge, respectively, in our consolidated statement of operations. For the year ended December 31, 2009, we recorded a net gain on our
37
Table of Contents
investment securities of approximately $3.5 million to increase the carrying value of our investment securities. In addition, for the year ended December 31, 2009, we recorded an impairment charge of approximately $3.2 million on the ARS Put to decrease its carrying value based on our discounted cash flow model with liquidity discount.
In August 2008, UBS AG and its affiliates, or UBS, the brokerage firm through which we purchased the majority of its ARS investments, entered into a settlement with the SEC, the New York Attorney General and other state agencies. Under the settlement, UBS issued to us Auction Rate Security Rights, which would allow us to sell to UBS our ARS held in accounts with UBS, or the ARS Rights Offer. Pursuant to the ARS Rights Offer, we received the right to sell to UBS the ARS held in accounts with UBS at par value at any time during the period beginning June 30, 2010 and ending July 2, 2012, or the ARS Put. As part of the settlement, UBS also offered to us a no net cost loan program, or ARS Loan, whereby we would be able to borrow up to 75 percent of the market value, as determined by UBS at its sole discretion, of our ARS that have been pledged as collateral at an interest cost that would not exceed the interest being paid on the underlying ARS investments. In January 2009, we were approved for the ARS Loan in the amount of $15.9 million and drew down the entire preapproved amount. In addition, in February 2009, we borrowed an additional $2.2 million under the ARS Loan, bringing the total amount outstanding under the ARS Loan to $18.1 million, following UBS’ decision to increase our availability under the ARS Loan. All cash received under the ARS Loan was invested in money market accounts. At December 31, 2009, our ARS Loan balance was $17.6 million.
UBS may demand full or partial payment of the ARS Loan, at its sole option and without cause, at any time. All ARS Loan advances are subject to collateral maintenance requirements. UBS may also, at any time, in its discretion, terminate and cancel the ARS Loan. If at any time UBS exercises its right to terminate the credit line agreement governing the ARS Loan, then UBS is required to provide, as soon as reasonably possible, alternative financing on substantially the same terms and conditions as those under the credit line agreement and the agreement will remain in full force and effect until such time as such alternative financing has been established. We cannot assure you that we will not default on our obligations under the credit line agreement, which could result in the acceleration of our repayment obligations, or that UBS will not call the amounts outstanding under the ARS Loan, either of which would negatively impact our financial condition and cash flow. In addition, we cannot assure you that UBS will consummate the ARS Rights Offer and repurchase its ARS subject to such offer at par value, or that we will be able to renew this facility at maturity on similar terms, or at all.
If we fail to obtain the capital necessary to fund our operations, we will be unable to develop and commercialize our product candidates.
We have consumed substantial amounts of capital since our inception. From our inception to December 31, 2009, we had an accumulated deficit of $247.4 million. Our cash, cash equivalents, investment securities- current and ARS Put, net of the ARS Loan, totaled approximately $28.4 million at December 31, 2009. We intend to manage our product development programs such that our existing cash, cash equivalents and investment securities as of December 31, 2009 will be sufficient to meet our operating requirements through at least December 31, 2010. We have based this estimate on assumptions that may prove to be wrong, and we could spend our available financial resources faster than we currently anticipate. Our future capital requirements will depend on, and could increase significantly as a result of, many factors including:
| • | progress in, and the costs of, our ongoing and planned clinical trials and other research and development activities; |
| • | the scope, prioritization and number of our product development programs; |
| • | our obligations under our license agreements, pursuant to which we may be required to make future milestone payments upon the achievement of various milestones related to clinical, regulatory or commercial events; |
38
Table of Contents
| • | our ability to establish and maintain strategic collaborations, including licensing and other arrangements, and to complete acquisitions of additional product candidates; |
| • | the time and costs involved in obtaining regulatory approvals; |
| • | the costs of securing manufacturing arrangements for clinical or commercial production of our product candidates; |
| • | the costs associated with expanding our management, personnel, systems and facilities; |
| • | the costs associated with any litigation; |
| • | the costs associated with the operations or wind-down of any business it may acquire; |
| • | the costs involved in filing, prosecuting, enforcing and defending patent claims and other intellectual property rights; and |
| • | the costs of establishing or contracting for sales and marketing capabilities and commercialization activities if we obtain regulatory approval to market our product candidates. |
Until we can generate significant continuing revenues, we expect to satisfy our future cash needs through strategic collaborations, private or public sales of our securities, debt financings or licensing transactions, involving all or a portion of our product candidates, to the extent we are able to do so. We may not be successful in obtaining strategic collaboration agreements or in receiving milestone or royalty payments under such agreements. We cannot assure you that additional sources of capital will be available to us on acceptable terms, or at all. If sources of capital are not available, we may not be in a position to pursue present or future business opportunities that require financial commitments, and we may be required to terminate, delay or reduce the scope of one or more of our product development programs; delay establishing sales and marketing capabilities or other activities to commercialize a product candidate; curtail our efforts to acquire new product candidates; or relinquish some or even all rights to our product candidates.
The terms under which we raise additional capital may harm our business and may significantly dilute stockholders’ ownership interests.
If we raise additional funds through collaborations or licensing arrangements with third parties, we may need to relinquish some rights to our product candidates, including commercialization rights, which may hinder our ability to generate revenues and achieve or sustain profitability. If we raise additional funds by issuing equity securities, stockholders may experience substantial dilution. Debt financing, if available, may involve significant cash payment obligations and restrictive covenants and other financial terms that may impede our ability to operate our business. Any debt financing or additional equity that we raise may contain terms that are not favorable to us or our stockholders.
We are subject to stringent regulation of our product candidates, which could delay the development and commercialization of our product candidates.
We, our third-party manufacturers, service providers, suppliers and partners, and our product candidates are subject to stringent regulation by the FDA and other regulatory agencies in the United States and by comparable authorities in other countries. None of our product candidates can be marketed in the United States until it has been approved by the FDA. None of our product candidates has been approved by the FDA to date, and we may never receive FDA approval for any of our product candidates. Obtaining FDA approval for a product takes many years of clinical development and requires substantial resources. Additionally, changes in regulatory requirements and guidance may occur or new information regarding the product candidate or the target indication may emerge, and we may need to perform additional, unanticipated non-clinical or clinical testing of our product candidates or amend clinical trial protocols to reflect these changes. Any additional unanticipated testing would add costs and could delay or result in the denial of regulatory approval for a product candidate. These regulatory
39
Table of Contents
requirements may limit the size of the market for the product or result in the incurrence of additional costs. Any delay or failure in obtaining required approvals could substantially reduce or negate our ability to generate revenues from the particular product candidate.
In addition, both before and after regulatory approval, we, our partners and our product candidates are subject to numerous FDA requirements, including requirements related to testing, manufacturing, quality control, labeling, advertising, promotion, distribution and export. The FDA’s requirements may change and additional government regulations may be promulgated that could affect us, our partners and our product candidates. Given the number of recent high profile adverse safety events with certain drug products, the FDA may require, as a condition of approval, costly risk management programs, which may include safety surveillance, restricted distribution and use, patient education, enhanced labeling, special packaging or labeling, expedited reporting of certain adverse events, preapproval of promotional materials and restrictions on direct-to-consumer advertising. Furthermore, heightened Congressional scrutiny on the adequacy of the FDA’s drug approval process and the agency’s efforts to assure the safety of marketed drugs has resulted in the enactment of new legislation addressing drug safety issues, the Food and Drug Administration Amendments Act of 2007. This legislation provides the FDA with expanded authority over drug products after approval and the FDA’s exercise of this authority could result in delays or increased costs during the period of product development, clinical trials and regulatory review and approval, and increased costs to assure compliance with new post-approval regulatory requirements. Furthermore, we cannot predict the likelihood, nature or extent of government regulation that may arise from this or future legislation or administrative action, either in the United States or abroad.
In order to market any of our products outside of the United States, we and our strategic partners and licensees must establish and comply with numerous and varying regulatory requirements of other countries regarding safety and efficacy. Approval procedures vary among countries and can involve additional product testing and additional administrative review periods beyond the requirements of the FDA and the time required to obtain approval in other countries might differ from that required to obtain FDA approval. The regulatory approval process in other countries may include all of the risks detailed above regarding FDA approval in the United States. Regulatory approval in one country, including FDA approval in the United States, does not ensure regulatory approval in another. In addition, a failure or delay in obtaining regulatory approval in one country may negatively impact the regulatory process in others. A product candidate may not be approved for all indications that we request, which would limit the uses of our product and adversely impact our potential royalties and product sales, and any approval that we receive may be subject to limitations on the indicated uses for which the product may be marketed or require costly, post-marketing follow-up studies.
If we fail to comply with applicable regulatory requirements in the United States or other countries, we may be subject to regulatory and other consequences, including fines and other civil penalties, delays in approving or failure to approve a product, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions, interruption of manufacturing or clinical trials, injunctions and criminal prosecution, any of which would harm our business.
We rely on third parties to conduct our clinical trials, and we may incur additional development costs, experience delays in the commencement and completion of clinical trials, and be unable to obtain regulatory approval for or commercialize our product candidates on our anticipated timeline if these third parties do not successfully carry out their contractual duties or meet expected deadlines.
We rely extensively on CROs, medical institutions, clinical investigators, contract laboratories and other service providers to perform important functions related to the conduct of its clinical trials, the collection and analysis of data and the preparation of regulatory submissions. Although we design and manage our current clinical trials to ensure that each clinical trial is conducted in accordance with its investigational plan and protocol, we do not have the ability to conduct all aspects of our clinical trials directly for our product candidates.
The FDA requires us and our CROs to comply with regulations and standards, commonly referred to as good clinical practices, or GCPs, for conducting, monitoring, recording and reporting the results of clinical trials
40
Table of Contents
to ensure that the data and results are scientifically credible and accurate and that the trial subjects are adequately informed of the potential risks of participating in clinical trials. Our reliance on CROs does not relieve us of these responsibilities and requirements. The CROs, medical institutions, clinical investigators, contract laboratories and other service providers that we employ in the conduct of our clinical trials are not our employees, and we cannot control the amount or timing of resources that they devote to our product development programs. If any of these third parties fails to devote sufficient care, time and resources to our product development programs, if its performance is substandard, or if any third party is inspected by the FDA and found not to be in compliance with GCPs, it will delay the completion of the clinical trial in which they are involved and the progress of the affected development program. The CROs with which we contract for execution of our clinical trials play a significant role in the conduct of the clinical trials and the subsequent collection and analysis of data. Any failure of the CROs to meet their obligations could adversely affect clinical development of our product candidates. Moreover, the CROs, clinical investigators and other service providers may have relationships with other commercial entities, some of which may have competitive products under development or currently marketed, and our competitive position could be harmed if they assist our competitors. If any of these third parties does not successfully carry out their contractual duties or obligations or meet expected deadlines, or if the quality or accuracy of the clinical data is compromised for any reason, our clinical trials may be extended, delayed or terminated, and we may not be able to obtain regulatory approval for our product candidates. In addition, while we believe that there are numerous alternative sources to provide these services, we might not be able to enter into replacement arrangements without delays or additional expenditures if we were to seek such alternative sources.
We rely on third-party manufacturers to produce our product candidates, which may result in delays in our clinical trials and the commercialization of products, as well as increased costs.
We have no manufacturing facilities, and we do not intend to develop facilities for the manufacture of our product candidates for clinical trials or commercial purposes in the foreseeable future. We contract with third-party manufacturers to produce, in collaboration with us, sufficient quantities of our product candidates for clinical trials, and we plan to contract with third-party manufacturers to produce sufficient quantities of any product candidates approved by the FDA or other regulatory authorities for commercial sale. While we believe that there are competitive sources available to manufacture our product candidates, we may not be able to enter into arrangements without delays or additional expenditures. We cannot estimate these delays or costs with certainty.
Reliance on third-party manufacturers limits our ability to control certain aspects of the manufacturing process and therefore exposes us to a variety of significant risks, including risks related to our ability to commercialize any products approved by regulatory authorities or conduct clinical trials, reliance on such third parties for regulatory compliance and quality assurance, and the refusal or inability of a third-party manufacturer to supply our requirements on a long-term basis. In addition, manufacturers of pharmaceutical products often encounter difficulties in production, particularly in scaling up initial production. These problems include difficulties with production costs and yields, quality control, including stability of the product candidate and quality assurance testing, shortages of qualified personnel and compliance with federal, state and foreign regulations. Also, our manufacturers may not perform as agreed. If our manufacturers were to encounter any of these difficulties, our ability to timely produce our product candidates for clinical trials and commercial sale may be interrupted, which could result in delayed clinical trials or receipt of regulatory approval and lost or delayed revenues.
To date, we have entered into an agreement with Hospira for the development and supply of finished product of MN-221 for the treatment of acute exacerbations of asthma utilizing Hospira’s proprietary ADD-Vantage drug delivery system that we intend to use in clinical trials and the commercial market. In addition to Hospira’s proprietary drug delivery system, we anticipate entering into a commercial supply agreement for finished product of MN-221 in standard vials. However, other than Hospira, we do not have agreements established regarding commercial supply of finished product of MN-221 in standard vials or for the API or
41
Table of Contents
finished product for any of our product candidates. In particular, pursuant to our license agreement with Kissei Pharmaceutical, Kissei Pharmaceutical has the exclusive right to manufacture the commercial supply of the API for MN-221. Therefore, we will need to successfully negotiate a commercial supply agreement with Kissei Pharmaceutical on commercially reasonable terms, or another third-party manufacturer in the event that we are unable to reach agreement with Kissei Pharmaceutical, in order to manufacture the API for MN-221 on a commercial scale if MN-221 is approved by the FDA or other regulatory authorities for commercial sale. We will also need to successfully negotiate a supply agreement with a third-party manufacturer on commercially reasonable terms in order to manufacture the finished product of MN-221 in standard vials. We may not be able to establish or maintain any commercial manufacturing and supply arrangements on commercially reasonable terms that we require for purposes of commercializing a product. Any failure by us to secure or maintain any such required commercial supply agreements could result in interruption of supply and lost or delayed revenues, which would adversely affect our business.
Any problems or delays we experience in preparing for commercial-scale manufacturing of a product candidate may result in a delay in FDA or other regulatory approval of the product candidate or may impair our ability to manufacture commercial quantities, which would adversely affect our business. For example, our manufacturers will need to produce specific batches of a product candidate to demonstrate acceptable stability under various conditions and for commercially viable lengths of time. We and our third-party manufacturers will need to demonstrate to the FDA and other regulatory authorities this acceptable stability data for the product candidate, as well as validate methods and manufacturing processes, in order to receive regulatory approval to commercialize such product candidate.
Our manufacturers are obligated to operate in accordance with FDA-mandated current good manufacturing practices, or cGMPs and, in some cases, International Convention on Harmonization, or ICH, standards. A failure of any of our third-party manufacturers to establish and follow cGMPs and/or ICH standards and to document their adherence to such practices may lead to significant delays in our ability to timely conduct and complete clinical trials, obtain regulatory approval of product candidates or launch of our products into the market. In addition, changing third-party manufacturers is difficult. For example, a change in third-party manufacturer for a particular product candidate requires re-validation of the manufacturing processes and procedures in accordance with cGMPs, which may be costly and time-consuming and, in some cases, our manufacturers may not provide us with adequate assistance to transfer the manufacturing processes and procedures for our product candidates to new manufacturers or may possess intellectual property rights covering parts of these processes or procedures for which we may need to obtain a license. Failure by our third-party manufacturers or us to comply with applicable regulations could result in sanctions being imposed on us, including fines, injunctions, civil penalties, delays, suspension or withdrawal of regulatory approvals, seizures or recalls of products, operating restrictions and criminal prosecutions.
We may not be able to manufacture our product candidates in commercial quantities, which would prevent us from commercializing our product candidates.
To date, our product candidates have been manufactured in small quantities for preclinical studies and clinical trials. If any of our product candidates is approved by the FDA or comparable regulatory authorities in other countries for commercial sale, we will need to manufacture such product candidate in larger quantities. We may not be able to increase successfully the manufacturing capacity for any of our product candidates in a timely or economic manner, or at all. Significant scale-up of manufacturing may require additional validation studies, which the FDA must review and approve. If we are unable to increase successfully the manufacturing capacity for a product candidate, the regulatory approval or commercial launch of that product candidate may be delayed or there may be a shortage in supply. Our product candidates require precise, high quality manufacturing. Our failure to achieve and maintain these high manufacturing standards in collaboration with our third-party manufacturers, including the incidence of manufacturing errors, could result in patient injury or death, product recalls or withdrawals, delays or failures in product testing or delivery, cost overruns or other problems that could harm our business, financial condition and results of operations.
42
Table of Contents
Materials necessary to manufacture our product candidates may not be available on commercially reasonable terms, or at all, which may delay the development and commercialization of our product candidates.
We rely on the third-party manufacturers of our product candidates to purchase from third-party suppliers the materials necessary to produce the API and product candidates for our clinical trials, and we will rely on such manufacturers to purchase such materials to produce the API and finished product for any commercial distribution of our products if we obtain marketing approval. Suppliers may not sell these materials to our manufacturers at the time they need them in order to meet our required delivery schedule or on commercially reasonable terms, if at all. We do not have any control over the process or timing of the acquisition of these materials by our manufacturers. Moreover, we currently do not have any agreements for the production of these materials. If our manufacturers are unable to obtain these materials for our clinical trials, testing of the affected product candidate would be delayed, which may significantly impact our ability to develop the product candidate. If we or our manufacturers are unable to purchase these materials after regulatory approval has been obtained for one of our products, the commercial launch of such product would be delayed or there would be a shortage in supply of such product, which would harm our ability to generate revenues from such product and achieve or sustain profitability.
Even if our product candidates receive regulatory approval, they may still face future development and regulatory difficulties.
Even if U.S. regulatory approval is obtained, the FDA may still impose significant restrictions on a product’s indicated uses or marketing or impose ongoing requirements for potentially costly post-approval studies, including additional research and development and clinical trials. Any of these restrictions or requirements could adversely affect our potential product revenues. For example, the label ultimately approved for MN-221 or MN-166/AV411, our other product candidates or any other product candidates that we may in-license or acquire, if any, may include a restriction on the terms of its use, or it may not include one or more of our intended indications.
Our product candidates will also be subject to ongoing FDA requirements for the labeling, packaging, storage, advertising, promotion, record-keeping and submission of safety and other post-market information on the drug. In addition, approved products, manufacturers and manufacturers’ facilities are subject to continual review and periodic inspections. If a regulatory agency discovers previously unknown problems with a product, such as adverse events of unanticipated severity or frequency or problems with the facility where the product is manufactured, a regulatory agency may impose restrictions on that product or us, including requiring withdrawal of the product from the market. If our product candidates fail to comply with applicable regulatory requirements, such as cGMPs, a regulatory agency may:
| • | issue warning letters or untitled letters; |
| • | require us to enter into a consent decree, which can include imposition of various fines, reimbursements for inspection costs, required due dates for specific actions and penalties for noncompliance; |
| • | impose other civil or criminal penalties; |
| • | suspend regulatory approval; |
| • | suspend any ongoing clinical trials; |
| • | refuse to approve pending applications or supplements to approved applications filed by us; |
| • | impose restrictions on operations, including costly new manufacturing requirements; or |
| • | seize or detain products or require a product recall. |
43
Table of Contents
Our product candidates, if approved for sale, may not gain acceptance among physicians, patients and the medical community, thereby limiting our potential to generate revenues.
If one of our product candidates is approved for commercial sale by the FDA or other regulatory authorities, the degree of market acceptance of any approved product by physicians, healthcare professionals and third-party payors and our profitability and growth will depend on a number of factors, including:
| • | demonstration of efficacy; |
| • | changes in the standard of care for the targeted indication; |
| • | relative convenience and ease of administration; |
| • | the prevalence and severity of any adverse side effects; |
| • | availability, cost and potential advantages of alternative treatments, including less expensive generic drugs; |
| • | pricing and cost effectiveness, which may be subject to regulatory control; |
| • | effectiveness of our or any of our partners’ sales and marketing strategies; |
| • | the product labeling or product insert required by the FDA or regulatory authority in other countries; and |
| • | the availability of adequate third-party insurance coverage or reimbursement. |
If any product candidate that we develop does not provide a treatment regimen that is as beneficial as, or is perceived as being as beneficial as, the current standard of care or otherwise does not provide patient benefit, that product candidate, if approved for commercial sale by the FDA or other regulatory authorities, likely will not achieve market acceptance. Our ability to effectively promote and sell any approved products will also depend on pricing and cost-effectiveness, including our ability to produce a product at a competitive price and our ability to obtain sufficient third-party coverage or reimbursement. If any product candidate is approved but does not achieve an adequate level of acceptance by physicians, patients and third-party payors, our ability to generate revenues from that product would be substantially reduced. In addition, our efforts to educate the medical community and third-party payors on the benefits of our product candidates may require significant resources and may never be successful.
If our products are not accepted by the market or if users of our products are unable to obtain adequate coverage of and reimbursement for our products from government and other third-party payors, our revenues and profitability will suffer.
Our ability to commercialize our products successfully will depend in significant part on pricing and cost effectiveness, including our ability to produce a product at a competitive price and our ability to obtain appropriate coverage of and reimbursement for our products and related treatments are obtained from governmental authorities, private health insurers and other organizations, such as health maintenance organizations, or HMOs. Third-party payors are increasingly challenging the prices charged for medical products and services. We cannot provide any assurances that third-party payors will consider our products cost-effective or provide coverage of and reimbursement for our products, in whole or in part.
Uncertainty exists as to the coverage and reimbursement status of newly approved medical products and services and newly approved indications for existing products. Third-party payors may conclude that our products are less safe, less clinically effective or less cost-effective than existing products, and third-party payors may not approve our products for coverage and reimbursement. If we are unable to obtain adequate coverage of and reimbursement for our products from third-party payors, physicians may limit how much or under what circumstances they will prescribe or administer them. Such reduction or limitation in the use of our products
44
Table of Contents
could cause our sales to suffer. Even if third-party payors make reimbursement available, payment levels may not be sufficient to make the sale of our products profitable.
Also, the trend towards managed health care in the United States and the concurrent growth of organizations such as HMOs, which could control or significantly influence the purchase of medical services and products, may result in inadequate coverage of and reimbursement for our products. Many third-party payors, including HMOs, are pursuing various ways to reduce pharmaceutical costs, including the use of formularies. The market for our products depends on access to such formularies, which are lists of medications for which third-party payors provide reimbursement. These formularies are increasingly restricted, and pharmaceutical companies face significant competition in their efforts to place their products on formularies of HMOs and other third-party payors. This increased competition has led to a downward pricing pressure in the industry. The cost containment measures that third-party payors are instituting could have a material adverse effect on our ability to operate profitably.
If we fail to identify and license or acquire other product candidates, we will not be able to expand our business over the long term.
Because we do not have internal discovery capabilities, our business over the long term is substantially dependent on our ability to license or acquire product candidates and further develop them for commercialization. The success of this strategy depends upon our ability to identify, select and acquire the right product candidates. We have limited experience identifying, negotiating and implementing economically viable product candidate acquisitions or licenses, which is a lengthy and complex process. Also, the market for licensing and acquiring product candidates is intensely competitive, and many of our competitors have greater resources than we do. We may not have the requisite capital resources to consummate product candidate acquisitions or licenses that we identify to fulfill our strategy.
Moreover, product candidate acquisitions that we do complete involve numerous risks, including:
| • | difficulties in integrating the development program for the acquired product candidate into our existing operations; |
| • | diversion of financial and management resources from existing operations; |
| • | risks of entering new markets or technologies and of receiving regulatory approval; |
| • | inability to generate sufficient revenues to offset acquisition costs; and |
| • | delays that may result from our having to perform unanticipated preclinical trials or other tests on the product candidate. |
If we are not successful in identifying and licensing or acquiring other product candidates over the long term, we will not be able to grow our revenues with sales from new products beyond those revenues, if any, from any approved products derived from our existing product candidates, and we may fail to achieve or sustain profitability.
We are dependent on our management team, particularly Yuichi Iwaki, M.D., Ph.D., and if we are unable to attract, retain and motivate Dr. Iwaki and other key management and scientific staff, our product development programs may be delayed and we may be unable to develop successfully or commercialize our product candidates.
We are dependent upon the continued services of our executive officers and other key personnel, particularly Yuichi Iwaki, M.D., Ph.D., a founder of the company and our President and Chief Executive Officer, who has been instrumental in our ability to in-license product candidates from Japanese pharmaceutical companies and secure financing from Japanese institutions. The relationships that certain of our key managers
45
Table of Contents
have cultivated with pharmaceutical companies from whom we license product candidates and to whom we expect to out-license product candidates make us particularly dependent upon their continued employment with us. We are also substantially dependent on the continued services of our existing clinical development personnel because of the highly technical nature of our product development programs. We are not presently aware of any plans of our executive officers or key personnel to retire or leave employment with the company. Each of our executive officers is party to an employment agreement that continues in effect until the earliest of termination of employment upon (i) consent of the parties, (ii) cause or other material breach of the agreement, (iii) death or permanent disability and (iv) three months’ written notice. Following termination of employment, these individuals may engage in other businesses that may compete with us.
If we acquire or license new product candidates, our success will depend on our ability to attract, retain and motivate highly qualified management and scientific personnel to manage the development of these new product candidates. In particular, our product development programs depend on our ability to attract and retain highly experienced clinical development and regulatory personnel. However, we face competition for experienced scientists and other technical and professional personnel from numerous companies and academic and other research institutions. Competition for qualified personnel is particularly intense in the San Diego, California area, where our corporate headquarters is located. Our short operating history and the uncertainties attendant to being a development-stage biopharmaceutical company could impair our ability to attract and retain personnel and impede the achievement of our development and commercialization objectives. In addition, we have scientific and clinical advisors who assist us in our product development and clinical strategies. These third parties are not our employees and may have commitments to, or contracts with, other entities that may limit their availability to us, or may have arrangements with other companies to assist in the development of products that may compete with our product candidates.
Although we have employment agreements with key members of management, each of our employees, subject to applicable notice requirements, may terminate his or her employment at any time. We do not carry “key person” insurance covering members of senior management. If we lose any of our key management personnel, we may not be able to find suitable replacements, which would adversely affect our business.
If we are unable to establish our sales and distribution capabilities, we will be unable to successfully commercialize our product candidates.
To date, we have not sold, marketed or distributed any pharmaceutical products. If we are successful in obtaining regulatory approvals for any of our product candidates or acquiring other approved products, we will need to establish sales, marketing and distribution capabilities on our own or with partners in order to commercialize an approved product. The acquisition or development of an effective sales and marketing infrastructure will require a significant amount of our financial resources and time and could negatively impact our commercialization efforts, including delay of a product launch. We may be unable to establish and manage a sufficient or effective sales force in a timely or cost-effective manner, if at all, and any sales force we do establish may not be capable of generating demand for our products, therefore hindering our ability to generate revenues and achieve or sustain profitability. In addition, if we are unable to develop internal sales capabilities, we will need to contract with third parties or establish a partnership to market and sell the product. If we are unable to establish adequate sales and marketing capabilities, whether independently or with third parties, we may not be able to generate any product revenues, may generate increased expenses and may never become profitable. In addition, although we intend to establish strategic collaborations to market any products approved for sale by regulatory authorities outside of the United States, we may be required to market our product candidates outside of the United States directly if we are unable to establish such collaborations. In that event, we may need to build a corresponding international sales and marketing capability with technical expertise and with supporting distribution capabilities.
46
Table of Contents
We may need to change our business practices to comply with health care fraud and abuse regulations, and our failure to comply with such laws could adversely affect our business, financial condition and results of operations.
If we market one or more of our product candidates, our operations will be directly, or indirectly through our customers, subject to various state and federal fraud and abuse laws, including the Anti-Kickback Statute and the False Claims Act, as amended. These laws may impact any proposed sales, marketing and education programs as well as other aspects of our operations.
The Anti-Kickback Statute prohibits persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual, or the furnishing or arranging for a good or service, for which payment may be made under a federal healthcare program such as the Medicare and Medicaid programs. Several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered business, the statute has been violated. The Anti-Kickback Statute is broad and prohibits many arrangements and practices that are lawful in businesses outside of the healthcare industry. Recognizing that the Anti-Kickback Statute is broad and may technically prohibit many innocuous or beneficial arrangements, Congress authorized the U.S. Department of Health and Human Services, Office of Inspector General, or OIG, to issue a series of regulations, known as the “safe harbors in certain instances” to shield healthcare providers and other parties from prosecution under the Anti-Kickback Statute in certain instances. However, conduct and business arrangements that do not fully satisfy each applicable safe harbor may result in increased scrutiny by government enforcement authorities such as the OIG. Many states have also adopted laws similar to the federal Anti-Kickback Statute, some of which apply to the referral of patients for healthcare items or services reimbursed by any source, not only the Medicare and Medicaid programs.
The federal False Claims Act prohibits persons from knowingly filing or causing to be filed a false claim to, or the knowing use of false statements to obtain payment from, the federal government. Suits filed under the False Claims Act can be brought by any individual on behalf of the government and such individuals may share in any amounts paid by the entity to the government in fines or settlement. The frequency of filing of such actions has increased significantly in recent years, causing greater numbers of healthcare companies to have to defend a False Claims Act action. Various states have also enacted laws modeled after the federal False Claims Act.
In addition to the laws described above, the Health Insurance Portability and Accountability Act of 1996, as amended, created two new federal crimes: healthcare fraud and false statements relating to healthcare matters. The healthcare fraud statute prohibits knowingly and willfully executing a scheme to defraud any healthcare benefit program, including private payors. A violation of this statute is a felony and may result in fines, imprisonment or exclusion from government-sponsored programs. The false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services.
If our operations are found to be in violation of any of the laws described above and other applicable state and federal fraud and abuse laws, we may be subject to penalties, including civil and criminal penalties, damages, fines, exclusion from government healthcare programs, imprisonment and the curtailment or restructuring of our operations.
Health care reform measures could adversely affect our business.
The business and financial condition of pharmaceutical and biotechnology companies are affected by the efforts of governmental and third-party payers to contain or reduce the costs of health care. In the United States and in foreign jurisdictions, there have been, and we expect that there will continue to be, a number of legislative
47
Table of Contents
and regulatory proposals aimed at changing the health care system. For example, in some countries, pricing of prescription drugs is subject to government control, and we expect proposals to implement similar controls in the United States to continue. Another example of proposed reform that could affect our business is the current discussion of drug reimportation into the United States. In 2000, Congress directed the FDA to adopt regulations allowing the reimportation of approved drugs originally manufactured in the United States back into the United States from other countries where the drugs were sold at lower prices. Although the Secretary of Health and Human Services has refused to implement this directive, the House of Representatives passed a similar bill that does not require the Secretary of Health and Human Services to act in July 2003. The reimportation bills have not yet resulted in any new laws or regulations; however, these and other initiatives could decrease the price we or any potential collaborators receive for our product candidates if and when they are approved for sale, adversely affecting our future revenue growth and potential profitability. Moreover, the pendency or approval of such proposals could result in a decrease in our stock price or our ability to raise capital or to obtain strategic partnerships or licenses.
We may be sued for product liability, which could result in substantial liabilities that exceed our available resources and damage our reputation.
The development and commercialization of drug products entails significant product liability risks. Product liability claims may arise from use of any of our product candidates in clinical trials and the commercial sale of any approved products. If we cannot successfully defend ourselves against these claims, we will incur substantial liabilities. Regardless of merit or eventual outcome, product liability claims may result in:
| • | withdrawal of clinical trial participants; |
| • | termination of clinical trial sites or entire clinical trial programs; |
| • | decreased demand for our product candidates; |
| • | impairment of our business reputation; |
| • | costs of related litigation; |
| • | substantial monetary awards to patients or other claimants; |
| • | loss of revenues; and |
| • | the inability to commercialize our product candidates. |
We currently have insurance that covers our clinical trials. We believe our current insurance coverage is reasonably adequate at this time; however, our insurance coverage may not reimburse us or may not be sufficient to reimburse us for all expenses or losses we may suffer. In addition, we will need to increase and expand this coverage as we commence additional clinical trials, as well as larger scale clinical trials, and in the event that any of our product candidates is approved for commercial sale. This insurance may be prohibitively expensive or may not fully cover our potential liabilities. In addition, our inability to obtain sufficient insurance coverage at an acceptable cost or otherwise to protect against potential product liability claims could prevent or inhibit the regulatory approval or commercialization of products that we or one of our collaborators develop. Successful product liability claims could have a material adverse effect on our business and results of operations. Liability from such claims could exceed our total assets if we do not prevail in any lawsuit brought by a third party alleging that an injury was caused by one of our product candidates.
We may need to increase the size of our organization, and we may encounter difficulties managing our growth, which could adversely affect our results of operations.
As of March 15, 2010, we had 24 full-time employees, one part-time employee and one intern. We may need to continue to expand our managerial, operational, financial and other resources in order to manage and
48
Table of Contents
fund our operations and clinical trials, continue our development activities and commercialize our product candidates. Our management, personnel, systems and facilities currently in place may not be adequate to support this future growth. For example, we may hire additional personnel in clinical development, regulatory affairs and business development to further strengthen our core competencies or choose to develop sales, marketing and distribution capabilities for certain of our product candidates. Our need to effectively manage our operations, growth and product development programs requires that we:
| • | manage our clinical trials effectively; |
| • | manage our internal development efforts effectively while carrying out our contractual obligations to licensors and other third parties; |
| • | ensures that its consultants, CROs and other service providers successfully carry out their contractual obligations, provide high quality results and meet expected deadlines; and |
| • | continue to improve our operational, financial and management controls, reporting systems and procedures. |
We may be unable to successfully implement these tasks on a larger scale, which may impact our ability to timely achieve our development and commercialization goals, if at all.
We expect that our results of operations will fluctuate, which may make it difficult to predict our future performance from period to period.
Our quarterly operating results have fluctuated in the past and are likely to continue to do so in the future. Some of the factors that could cause our operating results to fluctuate from period to period include:
| • | the status of development of our product candidates and, in particular, the advancement or termination of activities related to our product development programs and the timing of any milestone payments payable under our licensing agreements; |
| • | the execution of other collaboration, licensing and similar arrangements and the timing of payments we may make or receive under these arrangements; |
| • | variations in the level of expenses related to our product development programs; |
| • | the unpredictable effects of collaborations during these periods; |
| • | the timing of our satisfaction of applicable regulatory requirements, if at all; |
| • | the rate of expansion of our clinical development and other internal research and development efforts; |
| • | the costs of any litigation; |
| • | the effect of competing technologies and products and market developments; and |
| • | general and industry-specific economic conditions. |
We believe that quarterly or yearly comparisons of our financial results are not necessarily meaningful and should not be relied upon as indications of our future performance.
Our management has broad discretion over the use of our cash, and we may not use our cash effectively, which could adversely affect our results of operations.
Our management has significant flexibility in applying our cash resources and could use these resources for corporate purposes that do not increase our market value or in ways with which our stockholders may not agree. We may use our cash resources for corporate purposes that do not yield a significant return or any return at all for our stockholders, which may cause our stock price to decline.
49
Table of Contents
We will continue to incur significant increased costs as a result of operating as a public company, and our management will be required to devote substantial time to new compliance initiatives.
As a public company, we are required to comply with the Sarbanes-Oxley Act of 2002, as well as rules and regulations implemented by the SEC, The Nasdaq Stock Market, or Nasdaq, and the Osaka Securities Exchange, or OSE, and incur significant legal, accounting and other expenses as a result. These rules impose various requirements on public companies, including requiring the establishment and maintenance of effective disclosure and financial controls and appropriate corporate governance practices. Our management and other personnel have devoted and will continue to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations increase our legal and financial compliance costs and may make it more difficult and expensive for us to renew our director and officer liability insurance, and result in imposition of reduced policy limits and coverage.
The Sarbanes-Oxley Act requires that we maintain effective internal controls for financial reporting and disclosure controls and procedures. As a result, we are required to perform an evaluation of our internal control over financial reporting to allow management to report on the effectiveness of those controls, as required by Section 404 of the Sarbanes-Oxley Act. Our efforts to comply with Section 404 and related regulations have required, and continue to require, the commitment of significant financial and managerial resources. While we anticipate maintaining the integrity of our internal control over financial reporting and all other aspects of Section 404 applicable to us, we cannot be certain that a material weakness will not be identified when we test the effectiveness of our control systems in the future. If a material weakness is identified, we could be subject to sanctions or investigations by Nasdaq, the SEC, the OSE or other regulatory authorities, which would require additional financial and management resources, costly litigation or a loss of public confidence in our internal controls, which could have an adverse effect on the market price of our stock. As a smaller reporting company, our report regarding internal control over financial reporting for the year ended December 31, 2009 was not subject to attestation by our registered public accounting firm pursuant to temporary SEC rules.
Our business and operations would suffer in the event of system failures.
Despite the implementation of security measures, our internal computer systems are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. Any system failure, accident or security breach that causes interruptions in our operations could result in a material disruption of our drug development programs, including delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach results in a loss or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we may incur liability and the further development of our product candidates may be delayed.
We may not realize all of the anticipated benefits of the combined clinical development programs based on ibudalist.
We may not be able to successfully secure a strategic collaboration to advance the combined ibudalist development programs. Following completion of the Phase II clinical trial of MN-166 for the treatment of MS in the second quarter of 2008 and the acquisition of AV411 in December 2009, we have not undertaken, nor do we plan to undertake, any further significant clinical development of MN-166/AV411 until such time that we secure a strategic collaboration to advance the combined clinical development of MN-166/AV411 ibudalist-based development program. We cannot assure you that we will be able to secure such a strategic collaboration or otherwise further advance, or recognize value from, a combined MN-166/AV411 clinical development program.
50
Table of Contents
Risks Related to Our Intellectual Property
Our ability to compete may decline if we do not adequately protect our proprietary rights.
There is the risk that our patents (both those owned by us and those in-licensed) may not provide a competitive advantage, including the risk that our patents expire before we obtain regulatory and marketing approval for one or more of our product candidates, particularly our in-licensed patents. Also, our competitors may develop products similar to ours using methods and technologies that are beyond the scope of our intellectual property rights. Composition of matter patents on APIs may provide protection for pharmaceutical products without regard to formulation, method of use, or other type of limitation. We do not have compound patent protection for the API in our MN-166/AV411 and MN-001 product candidates, although we do have patent protection for a particular crystalline polymorph of MN-001. As a result, competitors that obtain the requisite regulatory approval will be able to offer products with the same API as found in our MN-166/AV411 and MN-001 product candidates so long as such competitors do not infringe any methods of use, methods of manufacture, formulation or, in the case of MN-001, specific polymorph patents that we hold or have exclusive rights to through our licensors. For example, we currently rely on a method of use patent for MN-166, which covers the use of the API found in our MN-166 product candidate for the treatment of MS. We also have a method of use patent for AV411 for the treatment of neuropathic pain syndromes.
It is our policy to consult with our licensors in the maintenance of granted patents we have licensed and in their pursuit of patent applications that we have licensed, but each of our licensors generally remains primarily responsible for or in control of the maintenance of the granted patents and prosecution of the applications. We have limited control, if any, over the amount or timing of resources that each licensor devotes on our behalf, and a licensor may not assign as great a priority to prosecution of these patent applications as we would if we were undertaking such prosecution ourselves. As a result of this lack of control and general uncertainties in the patent prosecution process, we cannot be sure that our licensed patents will be maintained and that any additional patents will ever mature from our licensed applications. Issued U.S. patents require the payment of maintenance fees to continue to be in force. We typically rely on our licensors to do this and their failure to do so could result in the forfeiture of patents not timely maintained. Many foreign patent offices also require the payment of periodic annuities to keep patents and patent applications in good standing. As we generally do not maintain control over the payment of annuities, we cannot be certain that our licensors will timely pay such annuities and that the granted patents and pending patent applications will not become abandoned. For example, certain annuities were not paid in a timely manner with respect to foreign patents licensed under MN-002 (the active metabolite of MN-001). In addition, our licensors may have selected a limited amount of foreign patent protection, and therefore applications have not been filed in, and foreign patents may not have been perfected in, all commercially significant countries.
The patent protection of our product candidates and technology involves complex legal and factual questions. Most of our license agreements give us a right, but not an obligation, to enforce our patent rights. To the extent it is necessary or advantageous for any of our licensors’ cooperation in the enforcement of our patent rights, we cannot control the amount or timing of resources our licensors devote on our behalf or the priority they place on enforcing our patent rights. We may not be able to protect our intellectual property rights against third party infringement, which may be difficult to detect, especially for infringement of patent claims for methods of manufacturing. Additionally, challenges may be made to the ownership of our intellectual property rights, our ability to enforce them or our underlying licenses, which in some cases have been made under foreign laws and may provide different protections than that of U.S. law.
We cannot be certain that any of the patents or patent applications owned by us or our licensors related to our product candidates and technology will provide adequate protection from competing products. Our success will depend, in part, on whether we or our licensors can:
| • | obtain and maintain patents to protect our product candidates; |
51
Table of Contents
| • | obtain and maintain any required or desirable licenses to use certain technologies of third parties, which may be protected by patents; |
| • | protect our trade secrets and know-how; |
| • | operate without infringing the intellectual property and proprietary rights of others; |
| • | enforce the issued patents under which we hold rights; and |
| • | develop additional proprietary technologies that are patentable. |
The degree of future protection for our proprietary rights is uncertain. For example:
| • | we or our licensor might not have been the first to make the inventions covered by each of our pending patent applications or issued patents; |
| • | we or our licensor might not have been the first to file patent applications for these inventions; |
| • | others may independently develop similar or alternative technologies or duplicate any of our technologies; |
| • | it is possible that none of our pending patent applications will result in issued patents; |
| • | any patents under which we hold rights may not provide us with a basis for maintaining market exclusivity for commercially viable products, may not provide us with any competitive advantages or may be challenged by third parties as invalid, not infringed or unenforceable under U.S. or foreign laws; or |
| • | any of the issued patents under which we hold rights may not be valid or enforceable or may be circumvented successfully in light of the continuing evolution of domestic and foreign patent laws. |
Confidentiality agreements with employees and others may not adequately prevent disclosure of our trade secrets and other proprietary information and may not adequately protect our intellectual property, which could limit our ability to compete.
Because we operate in the highly technical field of research and development of small molecule drugs, we rely in part on trade secret protection in order to protect our proprietary trade secrets and unpatented know-how. However, trade secrets are difficult to protect, and we cannot be certain that others will not develop the same or similar technologies on their own. We have taken steps, including entering into confidentiality agreements with our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors, to protect our trade secrets and unpatented know-how. These agreements generally require that the other party keep confidential and not disclose to third parties all confidential information developed by the party or made known to the party by us during the course of the party’s relationship with us. We also typically obtain agreements from these parties which provide that inventions conceived by the party in the course of rendering services to us will be our exclusive property. However, these agreements may not be honored and may not effectively assign intellectual property rights to us. Further, we have limited control, if any, over the protection of trade secrets developed by our licensors. Enforcing a claim that a party illegally obtained and is using our trade secrets or know-how is difficult, expensive and time consuming, and the outcome is unpredictable. In addition, courts outside the United States may be less willing to protect trade secrets or know-how. The failure to obtain or maintain trade secret protection could adversely affect our competitive position.
A dispute concerning the infringement or misappropriation of our proprietary rights or the proprietary rights of others could be time consuming and costly, and an unfavorable outcome could harm our business.
There is significant litigation in our industry regarding patent and other intellectual property rights. While we are not currently subject to any pending intellectual property litigation, and are not aware of any such threatened litigation, we may be exposed to future litigation by third parties based on claims that our product
52
Table of Contents
candidates, their methods of use, manufacturing or other technologies or activities infringe the intellectual property rights of such third parties. There are many patents relating to chemical compounds and methods of use. If our compounds or their methods of use or manufacture are found to infringe any such patents, we may have to pay significant damages or seek licenses under such patents. We have not conducted comprehensive searches for unexpired patents issued to third parties relating to our product candidates. Consequently, no assurance can be given that unexpired, third-party patents containing claims covering our product candidates, their methods of use or manufacture do not exist. Moreover, because some patent applications in the United States may be maintained in secrecy until the patents are issued, and because patent applications in the United States and many foreign jurisdictions are typically not published until 18 months after filing, we cannot be certain that others have not filed patent applications that will mature into issued patents that relate to our current or future product candidates and which could have a material effect in developing and commercializing one or more of our product candidates. The owner of a patent that is arguably infringed can bring a civil action seeking to enjoin an accused infringer from importing, making, marketing, distributing, using or selling an infringing product. We may need to resort to litigation to enforce our intellectual property rights or to seek a declaratory judgment concerning the scope, validity or enforceability of third-party proprietary rights. Similarly, we may be subject to claims that we have inappropriately used or disclosed trade secrets or other proprietary information of third parties. If we become involved in litigation, it could consume a substantial portion of our managerial and financial resources, regardless of whether we win or lose. Some of our competitors may be able to sustain the costs of complex intellectual property litigation more effectively than we can because they have substantially greater resources. We may not be able to afford the costs of litigation. Any legal action against us or our collaborators could lead to:
| • | payment of actual damages, royalties, lost profits, potential enhanced damages and attorneys’ fees, if a case against us is determined by a judge to be exceptional; |
| • | injunctive or other equitable relief that may effectively block our ability to further develop, commercialize and sell our products; |
| • | having to enter into license arrangements that may not be available on reasonable or commercially acceptable terms; or |
| • | significant cost and expense, as well as distraction of our management from our business. |
As a result, we could lose our ability to develop and commercialize current or future product candidates.
We may be subject to claims that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
As is common in the biotechnology and pharmaceutical industry, we employ individuals who were previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that these employees or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
Risks Related to the Securities Markets and Investment in Our Common Stock
Our stock price may be volatile, and you may not be able to resell our shares at a profit or at all.
Despite the listing of our common stock on the Nasdaq Global Market and the Hercules Market of the Osaka Securities Exchange in Japan, trading volume in our securities has been light and an active trading market may not develop for our common stock. In December 2009, our average trading volume was approximately 16,800 shares per day on the Nasdaq and approximately 62,100 shares per day on the Hercules Market of the Osaka Stock Exchange.
53
Table of Contents
The market prices for securities of biopharmaceutical and biotechnology companies, and early-stage drug discovery and development companies like us in particular, have historically been highly volatile and may continue to be highly volatile in the future. For example, since the date of our initial public offering in Japan on February 4, 2005 through December 31, 2009, our common stock has traded as high as approximately $42.00 and as low as approximately $1.40. The following factors, in addition to other risk factors described in this section, may have a significant impact on the market price of our common stock:
| • | the development status of our product candidates, including clinical trial results and determinations by regulatory authorities with respect to our product candidates, and particularly our prioritized product candidates; |
| • | the initiation, termination, or reduction in the scope of any collaboration arrangements or any disputes or developments regarding such collaborations; |
| • | FDA or foreign regulatory actions, including failure to receive regulatory approval for any of our product candidates; |
| • | announcements of technological innovations, new commercial products or other material events by us or our competitors; |
| • | disputes or other developments concerning our intellectual property rights; |
| • | market conditions in the pharmaceutical and biotechnology sectors; |
| • | actual and anticipated fluctuations in our quarterly or annual operating results; |
| • | price and volume fluctuations in the overall stock markets; |
| • | any potential delisting of our securities; |
| • | changes in, or failure to meet, securities analysts’ or investors’ expectations of our financial performance; |
| • | additions or departures of key personnel; |
| • | discussions of our business, management, products, financial performance, prospects or stock price by the financial and scientific press and online investor communities; |
| • | litigation or public concern about the safety of our potential products; |
| • | public concern as to, and legislative action with respect to, the pricing and availability of prescription drugs or the safety of drugs and drug delivery techniques; or |
| • | regulatory developments in the United States and in foreign countries. |
Broad market and industry factors, as well as economic and political factors, also may materially adversely affect the market price of our common stock.
We may become involved in securities class action litigation that could divert management’s attention and harm our business.
The stock markets have from time to time experienced significant price and volume fluctuations that have affected the market prices for the common stock of biotechnology and biopharmaceutical companies. These broad market fluctuations may cause the market price of our common stock to decline. In the past, securities class action litigation has often been brought against a company following a decline in the market price of its securities. This risk is especially relevant for us because biotechnology and biopharmaceutical companies have experienced significant stock price volatility in recent years. We may become involved in this type of litigation in the future. Litigation often is expensive and diverts management’s attention and resources, which could adversely affect our business.
54
Table of Contents
In addition, on August 24, 2009, The Pennsylvania Avenue Funds, an Avigen stockholder, filed a complaint in Alameda County Superior Court alleging that Avigen’s directors breached their fiduciary duties in connection with the proposed transaction with us. On October 15, 2009, The Pennsylvania Avenue Funds filed an amended complaint adding us as a defendant. In the amended complaint, The Pennsylvania funds alleged, among other things, that we aided and abetted the alleged breach of fiduciary duties by the Avigen directors. Avigen and The Pennsylvania Avenue Funds have signed a stipulation of settlement agreement and moved the court for preliminary approval. The Court heard oral argument on the Motion for Preliminary Approval of Settlement and held a case management conference on March 8, 2010, during which the Court raised a few issues regarding the settlement provisions. The parties have addressed those concerns and will appear before the Court on April 6, 2010 for preliminary approval of the settlement and a further case management conference.
Future sales of our common stock may cause our stock price to decline and may make it difficult to sell your shares.
Sales of substantial amounts of our common stock, or the availability of such common stock for sale, could adversely affect the prevailing market prices for our common stock. If this occurs and continues, it could impair our ability to raise additional capital through the sale of securities should we desire to do so. In addition, it may be difficult, or even impossible, to find a buyer for shares of our common stock.
We have also registered all common stock that we may issue under our current employee benefits plans. As a result, these shares can be freely sold in the public market upon issuance, subject to the terms of the underlying agreements governing the grants and the restrictions of the securities laws. In addition, our directors and officers may in the future establish programmed selling plans under Rule 10b5-1 of the Securities Exchange Act of 1934, for the purpose of effecting sales of our common stock. If any of these events cause a large number of our shares to be sold in the public market, the sales could reduce the trading price of our common stock and impede our ability to raise future capital.
Our stockholder rights plan and anti-takeover provisions in our charter documents and under Delaware law may make an acquisition of us more complicated and the removal and replacement of our directors and management more difficult.
Our restated certificate of incorporation and amended and restated bylaws contain provisions that may delay or prevent a change in control, discourage bids at a premium over the market price of our common stock or adversely affect the market price of our common stock and the voting and other rights of the holders of our common stock. These provisions may also make it difficult for stockholders to remove and replace our board of directors and management. These provisions:
| • | establish that members of the board of directors may be removed only for cause upon the affirmative vote of stockholders owning at least a majority of our capital stock; |
| • | authorize the issuance of “blank check” preferred stock that could be issued by our board of directors in a discriminatory fashion designed to increase the number of outstanding shares and prevent or delay a takeover attempt; |
| • | limit who may call a special meeting of stockholders; |
| • | establish advance notice requirements for nominations for election to the board of directors or for proposing matters that can be acted upon at stockholder meetings; |
| • | prohibit our stockholders from making certain changes to our Restated Certificate of Incorporation or Amended and Restated Bylaws except with 66 2/3 percent stockholder approval; and |
| • | provide for a classified board of directors with staggered terms. |
55
Table of Contents
Effective November 24, 2006, our board of directors adopted our stockholder rights plan. On March 30, 2007, our stockholders ratified the plan at our annual meeting of stockholders. Under the plan, we declared a dividend distribution of one “right” for each outstanding share of our common stock to stockholders of record at the close of business on December 11, 2006. Since that time, we have issued one right with each newly issued share of common stock. Each right, when exercisable, entitles the holder to purchase from us one one-thousandth (1/1,000) of a share of our Series A Preferred Stock at a purchase price of $77.00, subject to adjustment. In general, under the plan, if a person or affiliated group acquires beneficial ownership of 20 percent or more of our shares of common stock, then each right (other than those held by such acquiring person or affiliated group) will entitle the holder to receive, upon exercise, shares of common stock (or, under certain circumstances, a combination of securities or other assets) having a value of twice the underlying purchase price of the right. In addition, if following the announcement of the existence of an acquiring person or affiliated group we are involved in a business combination or sale of 50 percent or more of our assets or earning power, each right (other than those held by the acquiring person or affiliated group) will entitle the holder to receive, upon exercise, shares of common stock of the acquiring entity having a value of twice the underlying purchase price of the right. The board of directors also has the right, after an acquiring person or affiliated group is identified, to cause each right to be exchanged for common stock or substitute consideration. We may redeem the rights at a price of $0.001 per right prior to the identification of an acquiring person or affiliated group. The rights expire on November 23, 2016. If our stockholders do not re-ratify our stockholder rights plan at our 2010 annual meeting, our rights will expire on the date of such meeting.
We also may be subject to provisions of the Delaware corporation law that, in general, prohibit any business combination with a beneficial owner of 15 percent or more of our common stock for three years unless the holder’s acquisition of our stock was approved in advance by our board of directors. Although we believe these provisions collectively provide for an opportunity to receive higher bids by requiring potential acquirers to negotiate with our board of directors, they would apply even if the offer may be considered beneficial by some stockholders. In any event, these provisions may delay or prevent a third party from acquiring us. Any such delay or prevention could cause the market price of our common stock to decline.
We have never paid dividends on our capital stock, and we do not anticipate paying any cash dividends in the foreseeable future.
We have paid no cash dividends on any of our classes of capital stock to date, and we currently intend to retain our future earnings, if any, to fund the development and growth of our business. We do not anticipate paying any cash dividends on our common stock in the foreseeable future. As a result, capital appreciation, if any, of our common stock will be your sole source of gain for the foreseeable future.
Item 1B. Unresolved Staff Comments
Not applicable.
We lease approximately 12,699 square feet of office space at our headquarters in San Diego, California under a lease that expires in August 2011. We have no laboratory, research or manufacturing facilities, and we currently do not plan to purchase or lease any such facilities, as such services are provided to us by third-party service providers. We believe that our current facilities are adequate for our needs for the immediate future and that, should it be needed, suitable additional space will be available to accommodate expansion of our operations on commercially reasonable terms. In addition to our headquarters, we also lease approximately 1,726 square feet of office space in Tokyo, Japan under a lease that expires in May 2011. Furthermore, pursuant to our acquisition of Avigen, we acquired a month-to-month lease for 4,000 square feet of office space in Alameda, California. We vacated the Alameda premises on March 8, 2010 and, accordingly, we were released from our month-to-month lease by the landlord.
56
Table of Contents
On August 24, 2009, The Pennsylvania Avenue Funds, an Avigen stockholder, filed a complaint in Alameda County Superior Court alleging that Avigen’s directors breached their fiduciary duties in connection with the proposed transaction with us. On October 15, 2009, The Pennsylvania Avenue Funds filed an amended complaint adding us as a defendant. In the amended complaint, The Pennsylvania Avenue funds alleged, among other things, that we aided and abetted the alleged breach of fiduciary duties by the Avigen directors. Avigen and Pennsylvania Avenue Funds have signed a stipulation of settlement agreement and moved the court for preliminary approval. The Court heard oral argument on the Motion for Preliminary Approval of Settlement and held a case management conference on March 8, 2010, during which the Court raised a few issues regarding the settlement provisions. The parties have addressed those concerns and will appear before the Court on April 6, 2010 for preliminary approval of the settlement and a further case management conference.
In addition, we may be a party to lawsuits in the normal course of business. Litigation and governmental investigations can be expensive and disruptive to normal business operations. Moreover, the results of legal proceedings are difficult to predict. Significant judgments or settlements in connection with legal proceedings may have a material adverse effect on our business, financial condition and results of operations. We are not a party to any material legal proceedings.
57
Table of Contents
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market Information
Our common stock is traded on the Hercules Market of the Osaka Securities Exchange under the symbol “4875” and on the Nasdaq Global Market under the symbol “MNOV.” Our stock has been traded on the Hercules Market since February 8, 2005 and on the Nasdaq Global Market since December 7, 2006.
The following table sets forth the high and low sale prices per share of our common stock as reported on the Nasdaq Global Market.
| Common Stock Price | ||||||
| High | Low | |||||
| Fiscal year ended December 31, 2008 |
||||||
| First quarter |
$ | 4.78 | $ | 3.30 | ||
| Second quarter |
$ | 4.96 | $ | 3.31 | ||
| Third quarter |
$ | 4.76 | $ | 2.21 | ||
| Fourth quarter |
$ | 2.63 | $ | 1.50 | ||
| Fiscal year ended December 31, 2009 |
||||||
| First quarter |
$ | 3.20 | $ | 1.43 | ||
| Second quarter |
$ | 4.25 | $ | 1.93 | ||
| Third quarter |
$ | 7.46 | $ | 4.00 | ||
| Fourth quarter |
$ | 8.44 | $ | 5.60 | ||
Holders of Common Stock
As of March 22, 2010, there were approximately 6,000 holders of record of our common stock.
Dividend Policy
We have never declared or paid any cash dividends on our capital stock, and we do not anticipate paying any cash dividends in the foreseeable future. We expect to retain our future earnings, if any, to fund the growth and development of our business.
Securities Authorized for Issuance Under Equity Compensation Plans
Information regarding the securities authorized for issuance under our equity compensation plans can be found under Item 12 of this Annual Report on Form 10-K.
58
Table of Contents
Performance Graph
The following graph illustrates a comparison of the total cumulative stockholder return on our common stock since February 8, 2005, which is the date our common stock first began trading on the Hercules Market of the Osaka Securities Exchange, to two indices: the Hercules Total Index and the Hercules Standard Index. The graph assumes an initial investment of $100 on February 8, 2005, and that all dividends were reinvested.
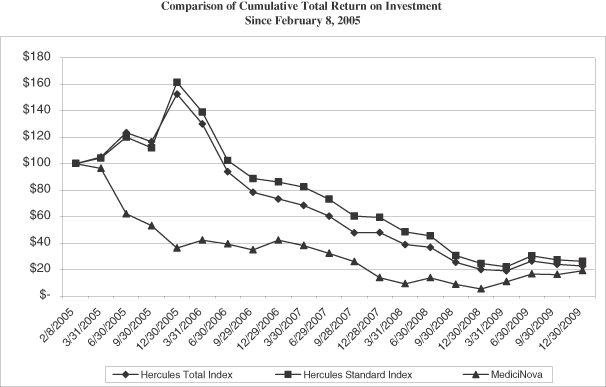
| 2/8/2005 | 12/30/2005 | 12/29/2006 | 12/28/2007 | 12/30/2008 | 12/30/2009 | |||||||||||||
| MediciNova, Inc |
$ | 100 | $ | 36 | $ | 42 | $ | 14 | $ | 5 | $ | 19 | ||||||
| Hercules Total Index |
$ | 100 | $ | 153 | $ | 73 | $ | 48 | $ | 20 | $ | 23 | ||||||
| Hercules Standard Index |
$ | 100 | $ | 162 | $ | 86 | $ | 59 | $ | 25 | $ | 26 | ||||||
| * | No cash dividends have been declared or paid on our common stock. Stockholder returns over the indicated period should not be considered indicative of future stockholder returns. |
59
Table of Contents
The following graph illustrates a comparison of the total cumulative stockholder return on our common stock since December 7, 2006 which is the date our common stock first began trading on the Nasdaq Global Market, to two indices: the NASDAQ Composite Index and the NASDAQ Biotechnology Index. The graph assumes an initial investment of $100 on December 7, 2006, and that all dividends were reinvested.
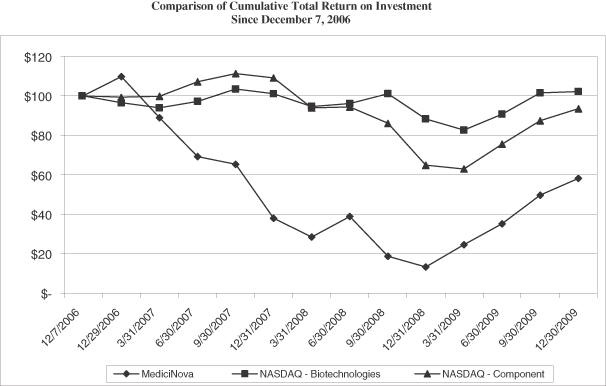
| * | No cash dividends have been declared or paid on our common stock. Stockholder returns over the indicated period should not be considered indicative of future stockholder returns. |
| 12/7/2006 | 12/31/2007 | 12/31/2008 | 12/31/2009 | |||||||||
| MediciNova, Inc |
$ | 100 | $ | 38 | $ | 13 | $ | 58 | ||||
| NASDAQ Biotechnologies Index |
$ | 100 | $ | 101 | $ | 88 | $ | 102 | ||||
| NASDAQ Composite Index |
$ | 100 | $ | 109 | $ | 65 | $ | 93 | ||||
60
Table of Contents
Item 6. Selected Financial Data.
The selected financial data set forth below is derived from our audited consolidated financial statements and may not be indicative of future operating results. The following selected financial data should be read in conjunction with the Consolidated Financial Statements and notes thereto and Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included elsewhere in this Annual Report on Form 10-K. Amounts are in thousands, except per share amounts.
| Years ended December 31, | September 26, 2000 (inception) to December 31, 2009 |
|||||||||||||||||||||||
| 2009 | 2008 | 2007 | 2006 | 2005 | ||||||||||||||||||||
| Statements of Operations Data: |
||||||||||||||||||||||||
| Revenues |
$ | — | $ | — | $ | — | $ | 264 | $ | 804 | $ | 1,558 | ||||||||||||
| Operating expenses: |
||||||||||||||||||||||||
| Cost of revenues |
— | — | — | 147 | 674 | 1,258 | ||||||||||||||||||
| Research and development |
10,873 | 13,828 | 42,121 | 32,171 | 22,738 | 144,546 | ||||||||||||||||||
| General and administrative |
10,366 | 8,773 | 11,373 | 9,624 | 7,479 | 89,027 | ||||||||||||||||||
| Total operating expenses |
21,239 | 22,601 | 53,494 | 41,942 | 30,891 | 234,831 | ||||||||||||||||||
| Operating loss |
(21,239 | ) | (22,601 | ) | (53,494 | ) | (41,678 | ) | (30,088 | ) | (233,273 | ) | ||||||||||||
| Gain/(impairment charge), net on investment securities and ARS put |
310 | (1,260 | ) | — | — | — | (950 | ) | ||||||||||||||||
| Foreign exchange loss |
(14 | ) | (88 | ) | — | — | — | (102 | ) | |||||||||||||||
| Other income, net |
581 | 2,038 | 4,611 | 5,988 | 4,396 | 18,377 | ||||||||||||||||||
| Income Taxes |
(7 | ) | (14 | ) | (20 | ) | — | — | (40 | ) | ||||||||||||||
| Net loss |
(20,369 | ) | (21,925 | ) | (48,903 | ) | (35,690 | ) | (25,692 | ) | (215,988 | ) | ||||||||||||
| Accretion to redemption value of redeemable convertible preferred stock |
— | — | — | — | (20 | ) | (99 | ) | ||||||||||||||||
| Deemed dividend resulting from beneficial conversion on Series C redeemable convertible preferred stock |
— | — | — | — | — | (31,264 | ) | |||||||||||||||||
| Net loss applicable to common stockholders |
$ | (20,369 | ) | $ | (21,925 | ) | $ | (48,903 | ) | $ | (35,690 | ) | $ | (25,712 | ) | $ | (247,351 | ) | ||||||
| Basic and diluted net loss per share |
$ | (1.68 | ) | $ | (1.82 | ) | $ | (4.16 | ) | $ | (3.52 | ) | $ | (2.88 | ) | |||||||||
| Shares used to compute basic and diluted net loss per share |
12,105,835 | 12,072,027 | 11,752,139 | 10,130,920 | 8,928,533 | |||||||||||||||||||
61
Table of Contents
| As of December 31, | ||||||||||||||||||||
| 2009 | 2008 | 2007 | 2006 | 2005 | ||||||||||||||||
| Balance Sheet Data: |
||||||||||||||||||||
| Cash, cash equivalents and current investment securities |
$ | 43,497 | $ | 19,297 | $ | 70,635 | $ | 104,051 | $ | 138,701 | ||||||||||
| ARS put—current |
2,557 | — | — | — | — | |||||||||||||||
| Working capital |
24,500 | 17,836 | 65,938 | 100,102 | 134,633 | |||||||||||||||
| Restricted cash, investment and letter of credit |
31,223 | — | — | — | — | |||||||||||||||
| Long-term investments |
2,085 | 24,047 | — | — | — | |||||||||||||||
| ARS put—long-term |
— | 5,793 | — | — | — | |||||||||||||||
| Total assets |
94,327 | 50,224 | 73,752 | 111,591 | 142,394 | |||||||||||||||
| ARS loan payable |
17,605 | — | — | — | — | |||||||||||||||
| Convertible notes |
29,258 | — | — | — | — | |||||||||||||||
| Deficit accumulated during the development stage |
(247,351 | ) | (226,982 | ) | (205,057 | ) | (156,154 | ) | (120,465 | ) | ||||||||||
| Total stockholders’ equity |
40,013 | 48,045 | 66,608 | 100,981 | 135,708 | |||||||||||||||
62
Table of Contents
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
You should read the following discussion and analysis together with “Item 5. Selected Financial Data” and the consolidated financial statements and related notes included elsewhere in this Annual Report on Form 10-K. The following discussion contains forward-looking statements that involve risks and uncertainties. Our actual results could differ materially from those expressed or implied in any forward-looking statements as a result of various factors, including those set forth under the caption “Item 1A. Risk Factors.”
Overview
Background
We are a biopharmaceutical company focused on acquiring and developing novel, small molecule therapeutics for the treatment of diseases with unmet medical need with a specific focus on the U.S. market. Through strategic alliances primarily with Japanese pharmaceutical companies, we hold rights to a diversified portfolio of clinical and preclinical product candidates, each of which we believe has a well-characterized and differentiated therapeutic profile, attractive commercial potential and patent assets having claims of commercially adequate scope.
We are a development stage company. We have incurred significant net losses since our inception. At December 31, 2009, from inception, our accumulated deficit was approximately $247.4 million, including $46.3 million of non-cash stock-based compensation charges related to employee stock-based compensation and founders’ warrants. We expect to incur substantial net losses for the next several years as we continue to develop certain of our existing product development programs, primarily MN-221 for the treatment of acute exacerbations of asthma and COPD exacerbations, and over the long-term if we are successful in expanding our research and development programs and acquiring or in-licensing products, technologies or businesses that are complementary to our own.
We have acquired licenses to eight compounds for the development of ten product candidates. Our development pipeline consists of eight product development programs which have been in clinical development for the treatment of acute exacerbations of asthma, MS, bronchial asthma, IC, solid tumor cancers, Generalized Anxiety Disorders/insomnia, preterm labor and urinary incontinence, and two product development programs which have been in preclinical development for the treatment of thrombotic disorders. In addition, we have expanded our development program for MN-221 for the treatment of COPD exacerbations.
In December 2009 we acquired Avigen, a biopharmaceutical company that has focused on identifying and developing differentiated products to treat patients with serious disorders, whose potential product candidate is AV411, a glial attenuator, for CNS disorders, such as neuropathic pain, and opioid withdrawal and methamphetamine addiction. AV411 and MN-166 are both ibudalist, an orally available, small molecule therapeutic. With the acquisition of AV411, we intend to integrate the two ibudalist-based product development programs and pursue discussions with potential partners to secure a strategic collaboration to advance clinical development of the combined development programs.
Avigen Transaction
On December 18, 2009, Absolute Merger, Inc., a wholly-owned subsidiary of ours, merged with and into Avigen with Avigen continuing as the surviving entity and wholly-owned subsidiary of ours. We refer to this transaction as “the Merger”. At the effective time of the Merger, each share of Avigen’s common stock (and the associated preferred stock purchase right) was cancelled and extinguished and automatically converted into the right to receive either cash, convertible notes or a combination of both in an amount equal to the First Payment Consideration (as defined below) and the Second Payment Consideration (as defined below) and one Contingent Payment Right, or CPR, granting the holder thereof the rights as described under the section entitled “Contingent Payment Rights” below.
63
Table of Contents
The First Payment Consideration, which was approximately $1.19 per share of Avigen’s common stock, was equal to $35,461,000 divided by 29,852,115, the number of shares of Avigen’s common stock outstanding immediately prior to the effective time of the Merger. The Second Payment Consideration is equal to the amount remaining in the escrow account described below following satisfaction of the Demand Amount (as defined below), as adjusted by the Selected Amount (as defined below).
Escrow Agreement
Under the terms of the escrow agreement, or Escrow Agreement, entered into at the closing of the Merger, we and Avigen funded $1,500,000 in an escrow account. After closing, we also deposited into the escrow account certain payments, including certain cash amounts that exceed specified amounts agreed upon by the parties. We (Avigen and us) identified certain additional liabilities of approximately $400,000 prior to closing of the Merger. At the closing of the Merger, Andrew A. Sauter, Avigen’s former Chief Executive Officer and Chief Financial Officer, as the stockholder representative appointed in accordance with the procedures set forth in the Escrow Agreement, authorized the release of $400,000 from the Escrow Agreement in satisfaction of these additional liabilities. As a result, the Second Payment Consideration is estimated to be no more than approximately $1.1 million, or $0.04 per share.
On or prior to June 30, 2010, we will be entitled to submit one demand certificate to claim all or a portion of the funds in the escrow account, or Demand Amount, with respect to certain additional liabilities, including liabilities in excess of specified amounts agreed upon by the parties. Upon delivery of the demand certificate, amounts in the escrow account that are not being demanded in satisfaction of additional liabilities will be released to Avigen’s former stockholders on a pro rata basis. The stockholder representative will be entitled to dispute the Demand Amount, and provision has been made in the Escrow Agreement for an independent accounting firm to resolve any unresolved dispute between us and the stockholder representative with respect to the Demand Amount. Amounts disputed will not be distributed until the dispute is resolved, and the timing of the full distribution of the Second Payment Consideration is therefore subject to delay.
Following resolution of the dispute regarding the Demand Amount, which requires the independent accounting firm to select either the amount demanded by us or the amount of such demand as adjusted by the amounts contested by the stockholder representative as the numerical amount it believes is the accurate amount of additional liabilities, or Selected Amount, we will receive an amount reflecting any adjustments resulting from the Selected Amount. Any remaining amounts in the escrow account then will be released to Avigen’s former stockholders on a pro rata basis.
Indenture
At the closing of the Merger, we and American Stock Transfer & Trust Company, LLC, as trustee, entered into an indenture. Under the terms of a separate trust agreement, or Trust Agreement, $29.4 million, which represents the First Payment Consideration less $6.0 million paid out to Avigen shareholders who elected cash payment and the initial principal amount of the convertible notes, or Convertible Notes, was deposited with a trust agent for the benefit of the holders and us (the amount of such deposit together with interest accrued and capitalized thereon, the Property). Provided no event of default has occurred and is continuing, we will be able to direct the investment and reinvestment of the Property in certain approved investment options, including certain money market funds. At the maturity of the Convertible Notes on June 18, 2011, the 18-month anniversary of the closing of the Merger, we will use the Property to pay the principal amount of, and accrued interest on, the Convertible Notes.
The Convertible Notes are our secured obligations, and the Indenture does not limit other indebtedness of ours, secured or unsecured. The indenture contains limited covenants, including a requirement that we deliver to holders of the Convertible Notes quarterly statements setting forth the principal amount of the Convertible Notes at the close of the fiscal quarter as well as information regarding the amount of interest capitalized to such Convertible Notes during the fiscal quarter.
64
Table of Contents
Holders of the Convertible Notes may submit conversion notices, which are irrevocable, instructing the trustee to convert such Convertible Notes into shares of our common stock at an initial conversion price of $6.80 per share. Following each conversion date, which date generally is the final business day of each calendar month, we will issue the number of whole shares of common stock issuable upon conversion as promptly as practicable (and in any event within 10 business days). Any fractional shares (after aggregating all Convertible Notes being converted by a holder on such date) will be rounded down and we will deliver cash out of the separate trust for the current market value of the fractional share. The Indenture includes customary anti-dilution adjustments and events of default.
Contingent Payment Rights
At the closing of the Merger, we, Avigen and American Stock Transfer & Trust Company, LLC, as rights agent, entered into a Contingent Payment Rights Agreement, or CPR Agreement. The CPR Agreement sets forth the rights that former Avigen stockholders will have with respect to each CPR held after the closing of the Merger. The CPR Agreement provides for the payment of the following amounts on a pro rata basis:
| • | if the first milestone payment under Avigen’s agreement with Genzyme, or the Genzyme Agreement, is received before August 18, 2011, $6,000,000 or such lesser cash amount paid by Genzyme; |
| • | if the first milestone payment has not occurred and the Parkinson’s Product, as defined in the Genzyme Agreement, is sold or otherwise disposed of by us before August 18, 2011, 50 percent of the net proceeds of such sale or disposition received before August 18, 2011; and |
| • | if the trust established pursuant to Avigen’s Management Transition Plan is terminated, the amount remaining in such trust upon termination (less any payments required to be made under Avigen’s Management Transition Plan Trust Agreement), such amount currently estimated at $550,000. |
All payments will be made on a pro rata basis. In each case, the payments will be net of any related taxes and out-of-pocket costs, damages, fines, penalties and expenses incurred by us. The CPRs are not transferable, except in limited circumstances.
Revenues and Cost of Revenues
We recognized no revenues for each of the years in the three-year period ended December 31, 2009.
Research and Development
Our research and development expenses consist primarily of the license fees related to our product candidates, salaries and related employee benefits, costs associated with the preclinical and clinical development of our product candidates, costs associated with non-clinical activities, such as regulatory expenses, and pre-commercialization manufacturing development activities. We use external service providers to manufacture our product candidates to be used in clinical trials and for the majority of the services performed in connection with the preclinical and clinical development of our product candidates; therefore, these research and development expenses consist substantially of external costs, such as fees paid to consultants, contract research organizations, contract manufacturers and other external service providers, including professional fees and costs associated with legal services, patents and patent applications for our intellectual property. Internal research and development expenses consist of costs of compensation and other expenses for research and development personnel, supplies, materials, facility costs and depreciation. Research and development costs are expensed as incurred or accrued based on certain contractual factors such as for estimates of work performed, milestones achieved, patient enrollment and experience with similar contracts. As actual costs become known, accruals are adjusted. To date, our estimates have not differed significantly from the actual costs incurred.
65
Table of Contents
The following table summarizes our research and development expenses for the periods indicated for each of our product candidates. To the extent that costs, including personnel costs, are not tracked to a specific product development program, such costs are included in the “Unallocated” category (in thousands):
| Product |
Disease/Indication |
Year ended December 31, | |||||||||||
| 2009 | 2008 | 2007 | |||||||||||
| MN-221 |
Acute exacerbations of asthma and COPD |
$ | 8,419 | $ | 6,542 | $ | 4,188 | ||||||
| MN-166/AV411 |
Multiple sclerosis/other CNS disorders |
635 | 3,363 | 9,512 | |||||||||
| MN-001 |
Bronchial asthma |
64 | 73 | 14,436 | |||||||||
| MN-001 |
Interstitial cystitis |
27 | 11 | 377 | |||||||||
| MN-029 |
Solid tumors |
86 | 796 | 2,591 | |||||||||
| MN-305 |
Generalized Anxiety Disorder/Insomnia |
(1 | ) | 18 | 5,309 | ||||||||
| MN-221 |
Preterm labor |
1 | 99 | 873 | |||||||||
| MN-246 |
Urinary incontinence |
15 | (17 | ) | 1,771 | ||||||||
| MN-447 |
Thrombotic disorders |
— | 123 | 416 | |||||||||
| MN-462 |
Thrombotic disorders |
— | 5 | 297 | |||||||||
| Unallocated |
1,627 | 2,815 | 2,351 | ||||||||||
| Total research and development |
$ | 10,873 | $ | 13,828 | $ | 42,121 | |||||||
As of the end of the second quarter of 2007, we determined to focus our resources on the development of our two prioritized product candidates, MN-221 for the treatment of acute exacerbations of asthma and MN-166 for the treatment of MS. However, following completion of the Phase II clinical trial of MN-166 ibudalist for the treatment of MS in the second quarter of 2008 and the December 2009 acquisition of AV411 ibudalist for the treatment of other CNS disorders, we plan to combine the two ibudalist-based development programs and pursue discussions with potential partners to secure a strategic collaboration. As such, we have not undertaken, nor do we plan to undertake, any further significant clinical development of MN-166/AV411 until such time that we secure a strategic collaboration to advance the combined ibudalist-based development program. In addition, as of the third quarter of 2009, we determined to expand the product development program for MN-221 to evaluate MN-221 for the treatment of COPD exacerbations. We anticipate that our research and development expenses will increase with respect to MN-221 in future periods as we continue development and launch clinical trials in support of potential commercialization of this product candidate for the treatment of acute exacerbations of asthma and COPD exacerbations and decrease with respect to MN-166/AV411 in future periods as we will limit expenditures on this product candidate to those development activities deemed necessary, if any, to maximize its value for purposes of securing a partner for clinical development. However, at this time, due to the risks inherent in the clinical development process and given the early stage of our MN-221 product development programs, we are unable to estimate with any certainty the costs that we will incur in the continued development of such product candidate for potential commercialization.
We intend to limit our expenditures on the remainder of our existing product candidates to only those activities deemed necessary to maintain our license rights or maximize the value of such product candidates, if any, while pursuing a variety of initiatives to monetize such product candidates on appropriate terms. As a result, we expect that research and development expenses will decrease or otherwise remain low for the remainder of our existing product candidates in future periods. These eight non-prioritized product development programs consist of the following:
| • | MN-001 for the treatment of bronchial asthma, for which we initiated a Phase III clinical program in the fourth quarter of 2006 that we subsequently terminated in the second quarter of 2007 and for which we developed prototypes of once-per-day oral dosing formulations; |
| • | MN-001 for the treatment of IC, for which we completed a Phase II/III clinical trial in the first quarter of 2007; |
66
Table of Contents
| • | MN-029 for the treatment of solid tumors, for which we completed one Phase I clinical trial in the second quarter of 2006 and one Phase I clinical trial in the fourth quarter of 2007; |
| • | MN-305 for the treatment of Generalized Anxiety Disorder/insomnia, for which we completed a Phase II/III clinical trial for the treatment of Generalized Anxiety Disorder in the second quarter of 2006 and a Phase II clinical trial for the treatment of insomnia in the fourth quarter of 2007; |
| • | MN-221 for the treatment of preterm labor, for which we completed a Phase I clinical trial to investigate the pharmacokinetic profile of MN-221 in healthy pregnant women not in labor in the second quarter of 2007; |
| • | MN-246 for the treatment of urinary incontinence, for which we completed a Phase I clinical trial in the fourth quarter of 2006 and a Phase I food effects study in the first quarter of 2007; |
| • | MN-447 for the treatment of thrombotic disorders, which remains in preclinical development; and |
| • | MN-462 for the treatment of thrombotic disorders, which remains in preclinical development. |
General and Administrative
Our general and administrative expenses primarily consist of salaries, benefits and consulting and professional fees related to our administrative, finance, human resources, business development, legal and information systems support functions. In addition, general and administrative expenses include facilities and insurance costs. General and administrative costs are expensed as incurred or accrued based on monitoring the status of the specified project, contractual factors such as milestones or retainer fees, services provided and invoices received. As actual costs become known to us, we adjust our accruals. To date, general and administrative accruals have not differed significantly from the actual costs incurred.
We anticipate that our general and administrative expenses may increase in future periods if we are required to expand our infrastructure based on the success of our current prioritized product development programs and in raising capital to support those and other development programs or otherwise in connection with increased business development activities related to partnering, out-licensing or disposition of our product candidates.
Investment Securities and ARS Put
Our investment securities consist of ARS, all of which had AAA ratings at the time of original purchase. ARS are generally long-term debt instruments that historically have provided liquidity through a “Dutch” auction process that resets the applicable interest rate at predetermined calendar intervals, typically seven, 28, 35 or 49 days. All of our ARS principally represent interests in municipal bonds, government-guaranteed student loans, insurance notes and portfolios of securities (primarily commercial paper). When our ARS were originally purchased, there was an active market for purchasing and selling ARS; therefore, we considered these investment securities to be available-for-sale.
Due to continued negative conditions in the global credit markets, our ARS have continued to fail at auction with few to no trades in either the primary or the secondary markets. As such, with the adoption of Accounting Standards Codification, or ASC, 820, authoritative guidance for fair value measurements and disclosures (formerly Statement of Financial Accounting Standards, or SFAS, No. 157), we determine the fair value of our ARS portfolio primarily on Level 3 criteria, which results in our reliance on a discounted cash flow valuation model with assumptions related to interest rates, maturities and liquidity determined by us based on the credit quality of the security, the credit quality of the associated insurer, if applicable, the respective prospectus and the credit market outlook. Given the lack of a primary and secondary market for our ARS investment securities, we designated all of our ARS investment securities as trading securities at December 31, 2008. As a result, any additional increase or decrease in the fair value of our ARS investment securities is recorded as either a gain or an impairment charge, respectively, in our consolidated statement of operations. For the year ended
67
Table of Contents
December 31, 2009, we recorded a net gain on our investment securities of $3.5 million to increase the overall carrying value of our investment securities. We have classified our investment securities covered by the ARS Rights Offer (as described below) as current assets given that they can be converted into cash within twelve months from December 31, 2009. Our remaining investment securities are considered long-term assets, as they cannot be readily converted to cash within 12 months from December 31, 2009.
In August 2008, UBS, the brokerage firm through which we purchased the majority of our ARS, entered into a settlement with the SEC, the New York Attorney General and other state agencies. Under the settlement, UBS issued to us Auction Rate Security Rights, which would allow us to sell to UBS our ARS held in accounts with UBS, or the ARS Rights Offer. Pursuant to the ARS Rights Offer, we received the right to sell to UBS the ARS held in accounts with UBS at par value at any time during the period beginning June 30, 2010 and ending July 2, 2012, or the ARS Put. As part of the settlement, UBS also offered to us a no net cost loan program, or ARS Loan, whereby we would be able to borrow up to 75 percent of the market value, as determined by UBS at its sole discretion, of our ARS that have been pledged as collateral at an interest cost that would not exceed the interest being paid on the underlying ARS investments. Under the terms of the ARS Loan, UBS may demand full or partial payment of the ARS Loan, at its sole option and without cause, at any time. If at any time UBS exercises its right to terminate the credit line agreement governing the ARS Loan, then UBS is required to provide, as soon as reasonably possible, alternative financing on substantially the same terms and conditions as those under the credit line agreement and the agreement will remain in full force and effect until such time as such alternative financing has been established. In January 2009, we were approved for the ARS Loan in the amount of $15.9 million and drew down the entire preapproved amount. In addition, in February 2009, we borrowed an additional $2.2 million under the ARS Loan, bringing the total amount outstanding under the ARS Loan to $18.1 million, following UBS’ decision to increase our availability under the ARS Loan. All cash received under the ARS Loan was invested in money market accounts. At December 31, 2009, the outstanding balance of the ARS Loan was $17.6 million.
Although we have the right to sell to UBS the ARS subject to the ARS Put at par beginning June 30, 2010, we determined the fair market value of the ARS without consideration of the ARS Put because they are deemed separate contractual agreements under ASC 820.
We elected to measure the ARS Put under the fair value option of ASC 825, authoritative guidance on financial instruments (formerly SFAS No. 159), to mitigate the volatility in reported earnings due to the linkage of certain of our ARS and the ARS Put. Under ASC 825, any subsequent increase or decrease in the fair value of the ARS Put would be recorded as either a gain or an impairment charge, respectively, in our consolidated statement of operations. The fair value of the ARS Put was also determined by a discounted cash flow valuation model with assumptions being made related to interest rate, maturity and liquidity. For the year ended December 31, 2009, based on our discounted cash flow valuation, we recorded an impairment charge of $3.2 million in our consolidated statement of operations due to a decrease in the carrying value of the ARS Put to $2.6 million.
The net gain on our investment securities and ARS Put was $0.3 million for the year ended December 31, 2009, which we recorded in our consolidated statement of operations.
Foreign Exchange Loss
To date, we have conducted most of our clinical trials in the United States. However, the Phase II clinical trial for MN-166 for the treatment of MS was conducted in Eastern Europe. When we entered into the euro-denominated contract with the CRO managing this clinical trial on our behalf, the U.S. dollar to euro conversion rate had remained fairly constant; therefore, we did not enter into a hedging program to mitigate our foreign exchange exposure at such time. We completed this clinical trial in the second quarter of 2008. Our foreign exchange loss in 2009 is primarily attributable to the decline in the value of the U.S. dollar against the euro on the accrued payable for this foreign currency denominated contract. At December 31, 2009, the accrued payable had been settled.
68
Table of Contents
Critical Accounting Policies and Estimates
Our management’s discussion and analysis of our financial condition and results of operations is based on our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of the consolidated financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses and the related disclosure of contingent liabilities at the date of the consolidated financial statements, as well as the revenues and expenses during the reporting periods. We evaluate our estimates and judgments on an ongoing basis, including those related to our significant accruals. We base our estimates on historical experience and on various other assumptions that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
Our significant accounting policies are more fully described in Note 1 to our consolidated financial statements included elsewhere in this Annual Report on Form 10-K. Our most critical accounting estimates include our recognition of research and development expenses, which impacts operating expenses and accrued liabilities, and stock-based compensation, which impacts operating expenses. We review our estimates, judgments and assumptions periodically and reflect the effects of revisions in the period in which they are deemed to be necessary. We believe that the following accounting policies are critical to the judgments and estimates used in preparation of our consolidated financial statements.
Research and Development Expenses
Research and development expenses consist of costs incurred to further our research and development activities and include salaries and related employee benefits, costs associated with clinical trials, costs associated with non-clinical activities such as toxicology testing, regulatory activities, research-related overhead expenses and fees paid to external service providers who conduct certain research and development activities on our behalf. We use external service providers and vendors to conduct clinical trials, to manufacture product candidates to be used in clinical trials and to provide various other products and services related to our product development programs. Research and development expenses also include fees for licensed technology for which technological feasibility has not been established and there are no alternative uses. Research and development costs are expensed as incurred or accrued based on certain contractual factors such as for estimates of work performed, milestones achieved, patient enrollment and experience with similar contracts. As actual costs become known, accruals are adjusted. To date, our estimates have not differed significantly from the actual costs incurred.
Stock-Based Compensation
We grant options to purchase our common stock to our employees and directors under our Amended and Restated 2004 Stock Incentive Plan. Additionally, we have outstanding stock options that were granted under our 2000 General Stock Incentive Plan. The benefits provided under both of these plans requires stock-based compensation for an award of equity instruments, including stock options and employee stock purchase rights, issued to employees to be recognized as a cost in the consolidated financial statements. The cost of these awards is measured according to the grant date fair value of the stock award and is recognized over the period during which an employee is required to provide service in exchange for the award, which is usually the vesting period. In the absence of an observable market price for the stock award, the grant date fair value of the award would be based upon a valuation methodology that takes into consideration various factors, including the exercise price of the award, the expected term of the award, the current price of the underlying shares, the expected volatility of the underlying share price, the expected dividends on the underlying shares and the risk-free interest rate.
Valuation of our stock option grants require us to estimate certain variables, such as estimated volatility and expected life. If any of our estimations change, such changes could have a significant impact on the stock-based compensation amount we recognize.
69
Table of Contents
Stock option compensation expense is recognized on a straight-line basis over the vesting period of the underlying option, generally four years.
Business Combinations
Our consolidated financial statements include an acquired business’ operations after the completion of the acquisition. We account for acquired businesses using the acquisition method of accounting. The acquisition method of accounting for acquired businesses requires, among other things, that most assets acquired and liabilities assumed be recognized at their fair values as of the acquisition date and that the fair value of acquired in-process research and development (IPR&D) be recorded on the balance sheet. Also, transaction costs are expensed as incurred. Any excess of the purchase price over the assigned values of the net assets acquired is recorded as goodwill. See Notes to Consolidated Financial Statements—Note 2. Avigen Transaction for further information on IPR&D and goodwill.
Fair Value Measurements
We are required to measure certain assets and liabilities at fair value, either upon initial measurement or for subsequent accounting or reporting. We use fair value extensively in the initial measurement of net assets acquired in a business combination and when accounting for and reporting on investment securities and certain financial instruments or assets. We estimate fair value using an exit price approach, which requires, among other things, that we determine the price that would be received to sell an asset or paid to transfer a liability in an orderly market of market participants, considering the highest and best use of assets and, for liabilities, assuming the risk of non-performance will be the same before and after the transfer. Many, but not all, of our financial instruments are carried at fair value. In addition, as required under accounting rules for business combinations, the assets acquired and liabilities assumed from Avigen on December 18, 2009 have been recorded at their estimated fair values as of the acquisition date. For additional information on the valuation approach to determine fair value, including a description of the inputs used, see Long Lived Assets below and Notes to Consolidated Financial Statements—Note 2. Avigen Transaction. Also, for information on fair value for our financial instruments, see Notes to Consolidated Financial Statements—Note 3. Fair Value Measurements—Other Than Intangibles and Goodwill.
Long-Lived Assets and Impairment of Long-Lived Assets
We review long-lived assets, including property and equipment, and other intangible assets, for impairment whenever events or changes in business circumstances indicate that the carrying amount of the assets may not be fully recoverable and we perform impairment testing for goodwill and IPR&D annually. When it is determined that impairment has occurred, a charge to operations will be recorded. Impairment on property and equipment or other intangible assets, if any, is assessed using discounted cash flows. Impairment on goodwill is assessed on our overall market capitalization, as we operate under one reporting segment. Impairment on IPR&D is assessed on a fair value cost approach.
New Accounting Standards Not Yet Adopted
In October 2009, the FASB ratified Accounting Standards Update, or ASU, 2010-13, which eliminates the residual method of allocation and the requirement to use the relative selling price method when allocating revenue in a multiple deliverable arrangement. When applying the relative selling price method, the selling price for each deliverable shall be determined using vendor specific objective evidence of selling price, if it exists, otherwise third-party evidence of selling price. If neither vendor specific objective evidence nor third-party evidence of selling price exists for a deliverable, companies shall use its best estimate of the selling price for that deliverable when applying the relative selling price method. ASU 2010-13 shall be effective in fiscal years
70
Table of Contents
beginning on or after June 15, 2010, with earlier application permitted. Companies may elect to adopt this guidance prospectively for all revenue arrangements entered into or materially modified after the date of adoption, or retrospectively for all periods presented. We do not believe the adoption of this accounting standard will have a material effect on our consolidated results of operations or financial condition.
In March 2010, the FASB issued ASU No. 2010-11, “Derivatives and Hedging (Topic 815): Scope Exception Related to Embedded Credit Derivatives”. The FASB believes this ASU clarifies the type of embedded credit derivative that is exempt from embedded derivative bifurcation requirements. Specifically, only one form of embedded credit derivative qualifies for the exemption—one that is related only to the subordination of one financial instrument to another. As a result, entities that have contracts containing an embedded credit derivative feature in a form other than such subordination may need to separately account for the embedded credit derivative feature. The amendments in the ASU are effective for each reporting entity at the beginning of its first fiscal quarter beginning after June 15, 2010. Early adoption is permitted at the beginning of each entity’s first fiscal quarter beginning after March 5, 2010. We do not believe the adoption of this accounting standard will have a material effect on our consolidated results of operations or financial condition.
Results of Operations
Comparison of the Years ended December 31, 2009 and 2008
Revenues
There were no revenues for the year ended December 31, 2009 or December 31, 2008.
Research and Development
Research and development expenses for the year ended December 31, 2009 were $10.9 million, a decrease of $2.9 million when compared to $13.8 million for the year ended December 31, 2008. This decrease in research and development expenses primarily resulted from the following:
| • | a decrease of $2.7 million due to the completion of the two year Phase II clinical trial for MN-166 for the treatment of MS; |
| • | a decrease of $0.9 million related primarily to the completion of clinical trials for MN-029 for the treatment of solid tumors and other non-prioritized assets; and |
| • | a decrease of $1.2 million in research and development personnel costs not tracked to a specific development program, |
which decrease was offset primarily by a net increase of $1.9 million related to the conduct of Phase II clinical trials for MN-221 for the treatment of acute exacerbations of asthma and COPD.
General and Administrative
General and administrative expenses were $10.4 million for the year ended December 31, 2009, an increase of $1.6 million when compared to $8.8 million for the year ended December 31, 2008. The $1.6 million increase was primarily related to expenses in connection with the Avigen transaction, including expenses related to legal fees to review and draft the merger agreement and related registration statement, accounting fees related to review of and consent for the related registration statement, the cost of the fairness opinion, and printing and mailing costs related to the special shareholders’ meeting needed to approve the Avigen transaction.
71
Table of Contents
Gain/Impairment Charge on Investment Securities and ARS Put
For the year-ended December 31, 2009, we recorded a net gain of $0.3 million on our investment securities and ARS Put, as compared to a net impairment charge of $1.3 million for the year-ended December 31, 2008. The net gain in 2009 on our investment securities and ARS Put is primarily due to a change in assumed maturity in our discounted cash flow valuation analysis. In 2009 we utilized a five year assumed maturity on our ARS subject to UBS settlement, as opposed to a seven year assumed maturity in 2008. The change in assumed maturity was based on the outlook for the ARS market.
Foreign Exchange Loss
Foreign exchange loss was $14,000 for the year ended December 31, 2009, a decrease of $74,000 when compared $88,000 for the year ended December 31, 2008. The decrease in foreign exchange loss was due to less weakening of the U.S. dollar against the euro and the settlement of the foreign currency denominated contract.
Other Income, net
Other income, net consisted of interest income earned on our cash and investment balances and totaled $581,000 for the year ended December 31, 2009, a decrease of $1.4 million when compared to $2.0 million for the year ended December 31, 2008. The decrease was primarily due to a decrease in interest earned on most of our cash and investment balances due to lower interest rates as a result of the continued economic downturn. In addition, during the year ended December 31, 2009, $235,000 of interest expense was recorded on the ARS Loan.
Comparison of the Years ended December 31, 2008 and 2007
Revenues
There were no revenues for the year ended December 31, 2008 or December 31, 2007.
Research and Development
Research and development expenses for the year ended December 31, 2008 were $13.8 million, a decrease of $28.3 million when compared to $42.1 million for the year ended December 31, 2007. The decrease in research and development expenses primarily resulted from our business decision to focus on the development of our two prioritized assets, MN-221 for the treatment of acute exacerbations of asthma and MN-166 for the treatment of MS. This decrease in research and development expenses primarily resulted from the following:
| • | a decrease of $14.4 million related to the termination of a Phase III clinical trial for MN-001 for the treatment of bronchial asthma; |
| • | a decrease of $5.3 million related to the completion of the Phase II clinical trial for insomnia and the ceased further clinical development of MN-305 for the treatment of Generalized Anxiety Disorder/insomnia; and |
| • | a decrease of $6.1 million due to the completion of the two year Phase II clinical trial for MN-166 for the treatment of MS; and |
| • | a decrease of $4.9 million related primarily to the completion of clinical trials for MN-029 for the treatment of solid tumors, MN-221 for the treatment of preterm labor and MN-246 for the treatment of urinary incontinence; |
which decrease was offset primarily by a net increase of $2.4 million related to the conduct of Phase II clinical trials for MN-221 for the treatment of acute exacerbations of asthma.
72
Table of Contents
General and Administrative
General and administrative expenses were $8.8 million for the year ended December 31, 2008, a decrease of $2.6 million when compared to $11.4 million for the year ended December 31, 2007. The decrease was primarily due to a $1.2 million decrease in stock-based compensation and a $1.4 million decrease related to reduced administrative headcount and fees paid to third-party consultants.
Impairment Charge, Net on Long-Term Investments and ARS Put
For the year-ended December 31, 2008, we recorded a $7.1 million other-than-temporary write-down of the carrying value of our ARS based upon a discounted cash flow valuation analysis of our entire ARS portfolio conducted on a security-by-security basis, the outlook of the ARS market and our expectation as to when we may be required to liquidate our ARS for operating purposes, which was offset by a gain of $5.8 million recognized on the ARS Put which is linked to certain of our ARS.
Foreign Exchange Loss
At December 31, 2007, the conversion rate was approximately $1.30 U.S. dollars for each euro, which approximated the conversion rate at the time we entered into the contract with the CRO managing our Phase II clinical trial for MN-166 for the treatment of MS which was completed in the second quarter of 2008. At December 31, 2008, the conversion rate was approximately $1.41 U.S. dollars for each euro, and we reduced the accrued liability related to this clinical research contract based on reconciliations performed through year end. This resulted in a $0.1 million foreign exchange loss related to the revaluation of our euro-denominated liability for the year ended December 31, 2008.
Other Income
Other income primarily consisted of interest income earned on our cash and investment balances and totaled $2.0 million for the year ended December 31, 2008, a decrease of $2.6 million when compared to $4.6 million for the year ended December 31, 2007. The decrease was due to a decrease in our investment balances and overall lower yields on our investments due to the economic recession.
Liquidity and Capital Resources
At December 31, 2009, we had $28.4 million in cash, cash equivalents, investment securities- current and an ARS Put, net of ARS loan, as compared to $49.1 million of cash, cash equivalents, investment securities and a long-term asset consisting of the ARS Put as of December 31, 2008, which decrease of $20.7 million was primarily a result of our $3.0 million payment to acquire Avigen and our operating loss of $20.4 million, offset by noncash expenses. At December 31, 2008, we had $49.1 million in cash, cash equivalents, investment securities and a long-term asset consisting of the ARS Put as compared to $70.6 million of cash, cash equivalents and marketable securities available-for-sale at December 31, 2007, which decrease of $21.5 million was primarily a result of our operating loss of $21.9 million. Restricted cash and letter of credit of $30.5 million would be included in our liquidity analysis and capital resources upon the conversion of the associated convertible notes into our stock. Through March 2010 we issued a total of 249,291 shares of our stock as a result of notes conversions and, accordingly, a total of approximately $1.7 million was transferred from restricted cash to cash and cash equivalents.
Net cash used in operating activities amounted to $17.0 million for the year ended December 31, 2009, primarily due to the net loss incurred during the year ended December 31, 2009 of $20.4 million. In addition, $1.8 million of cash used in operating activities was directly related to the Avigen transaction. Net cash used in investing activities of $1.1 million for the year ended December 31, 2009 consisted of the net cash used to
73
Table of Contents
acquire Avigen, offset by the net maturities/sales of investment securities. Net cash provided by financing activities amounted to $18.1 million for the year ended December 31, 2009, primarily due to the net proceeds received from our ARS Loan of $17.6 million.
Our future capital uses and requirements will depend on, and could increase significantly as a result of, many forward-looking factors, including the following:
| • | progress of our clinical trials and other research and development activities, including expenses to support the clinical development of MN-221 for the treatment of acute exacerbations of asthma and milestone payments that may become payable to Kissei Pharmaceutical based on the progress of such product development program; |
| • | our ability to establish and maintain strategic collaborations, including licensing and other arrangements; |
| • | the scope, prioritization and number of our product development programs; |
| • | the time and costs involved in obtaining regulatory approvals; |
| • | the costs involved in filing, prosecuting, enforcing and defending patent claims and other intellectual property rights; |
| • | the costs of securing manufacturing arrangements for clinical and commercial production of our product candidates; |
| • | the costs of establishing sales and marketing capabilities and commercialization activities if we obtain regulatory clearances to market our product candidates; and |
| • | the extent to which we may in-license, acquire or invest in other indications, products, technologies and businesses. |
Until we can generate significant continuing revenues, we expect to satisfy our future cash needs through strategic collaborations, private or public sales of our securities, debt financings or licensing transactions, involving all or a portion of our product candidates, to the extent we are able to do so. We cannot be certain that additional sources of capital will be available to us on acceptable terms, or at all. If sources of capital are not available, we may not be in a position to pursue present or future business opportunities that require financial commitments, and we may be required to delay, reduce the scope of or terminate one or more of our product development programs or our commercialization efforts, curtail our efforts to acquire new product candidates or relinquish rights to our technologies or product candidates.
Sources of Liquidity
Since our inception, our operations have been financed primarily through the private placement of our equity securities and through the public sale of our common stock, net of treasury stock repurchases. Over the three years ended December 31, 2009, we have completed the following sales of equity securities:
| • | on February 1, 2007, we completed a public offering of 1,000,000 shares of common stock for aggregate proceeds of $10.6 million, net of underwriting discounts and commissions and certain other costs associated with the offering. |
In January 2007, a founder exercised warrants to purchase 359,248 shares of our common stock at $1.00 per share in a cashless exercise that resulted in the issuance of 332,196 shares of common stock. In September 2007, a founder exercised warrants to purchase 367,828 shares of our common stock at $1.00 per share in a cashless exercise that resulted in the issuance of 317,851 shares of common stock. At December 31, 2007, no underlying shares of common stock remained subject to purchase under the terms of the founders’ warrants.
74
Table of Contents
Auction Rate Securities. At December 31, 2009, our investment securities (both current and long-term) totaled $30.0 million (at par value) of ARS. With the continued negative conditions in the global credit markets, these ARS have experienced multiple failed auctions, as the amount of securities submitted for sale has exceeded the amount of purchase orders. As a result of the failed auctions, these securities are currently not liquid in the primary market. The majority of our ARS are secured by parts of government-guaranteed student loans, and all of our ARS continue to pay interest according to their stated terms (generally 120 basis points over the 91-day U.S. Treasury bill rate or 200 basis points over LIBOR) with interest rates resetting every seven to 63 days. While it is not our intent to hold our ARS until their ultimate stated maturities, these securities are scheduled to mature between 2022 and 2024.
Because an active primary market for ARS does not exist, we utilized a discounted cash flow valuation model utilizing liquidity discounts ranging from two percent to 25 percent to determine the estimated fair value of our ARS investments on a security-by-security basis. We also took into consideration the brokerage firm’s pricing model, if applicable, the tax status (taxable vs. tax exempt) of the security, credit quality of the issuer, assumed maturity (primarily five years), insurance wraps and the portfolio composition. We also made assumptions regarding future cash flows and the likelihood of the ARS being redeemed or refinanced. In 2009 we modified the assumed maturity of student loan backed ARS to five years from seven years due to the outlook of the credit markets. With the change is assumed maturity, for the year ended December 31, 2009 we have recognized a net gain of $3.5 million in our consolidated statement of operations as the investment securities are designated as trading securities. The carrying value of our investment securities at December 31, 2009 was $26.3 million and the carrying value of our ARS Put (as described below) was $2.6 million.
ARS Rights Offer, ARS Put and ARS Loan. In August 2008, UBS, the brokerage firm through which we purchased the majority of our ARS investments, entered into a settlement with the SEC, the New York Attorney General and other state agencies. Under the settlement, UBS issued to us the ARS Rights Offer. Pursuant to the ARS Rights Offer, we received the ARS Put. As part of the settlement, UBS also offered to us the ARS Loan, whereby we would be able to borrow up to 75 percent of the market value, as determined by UBS at its sole discretion, of our ARS that have been pledged as collateral at an interest cost that would not exceed the interest being paid on the underlying ARS investments. Under the ARS Loan program, UBS may demand full or partial payment of the ARS Loan, at its sole option and without cause, at any time. In November 2008, we accepted the ARS Rights Offer. In January 2009, we were approved for the ARS Loan in the amount of $15.9 million and drew down the entire preapproved amount. In addition, in February 2009, we borrowed an additional $2.2 million under the ARS Loan, bringing the total amount outstanding under the ARS Loan to $18.1 million, following UBS’ decision to increase our availability under the ARS Loan. All cash received under the ARS Loan was invested in money market accounts.
We elected to measure the ARS Put under the fair value option of ASC 825 to mitigate the volatility in reported earnings due to the linkage of certain of our ARS and the ARS Put. The fair value of the ARS Put was also determined by a discounted cash flow valuation model effectively using a liquidity discount of approximately seven percent and an interest rate of approximately five percent, which took into consideration the brokerage firm’s weighted average cost of capital. Based on our discounted cash flow valuation, we recorded a realized loss of $3.2 million in our consolidated statement of operations.
The fair value of our ARS and the ARS Put are based in part on management’s estimates and assumptions. In the event of actual market exchanges, if any, these assumptions may prove materially different from those assumed in our valuation models and amounts may be materially different than our estimates. For example, a reduction of the expected term to redemption by two years for our ARS portfolio yielded in our models a net increase in valuation of our ARS of $3.4 million and an increase in expected term to redemption by two years for our ARS portfolio yielded in our model a decrease in valuation of our ARS of $1.1 million. Other factors that may impact the valuation of our ARS and the ARS Put include changes to the credit quality of the underlying assets, discount rates, counterparty risk and the condition of the overall credit market.
75
Table of Contents
Convertible Notes. Upon conversion of the Convertible Notes at each monthly conversion date, the Indenture and Trust Agreement permit the release of the principal and interest represented by such Convertible Notes to us. As no Convertible Notes were converted into shares of our common stock as of December 31, 2009, no amounts were released to us in 2009. Following the January and February 2010 conversion dates, an aggregate of $1.7 million was released to us in accordance with the Indenture and Trust Agreement, and any subsequent conversions will enhance our liquidity position.
Capital Resources
We have consumed substantial amounts of capital since our inception. Our current cash and cash equivalent balances are our principal sources of liquidity. We believe that our existing cash and cash equivalents as of December 31, 2009 will be sufficient to fund our anticipated operating requirements through at least December 31, 2010. Although we believe that our existing capital resources will be sufficient to fund our operating requirements through at least December 31, 2010, including all of our planned research and development activities, we anticipate that we may require significant additional financing in the future to fund our operations and intended research and development activities.
Other Significant Cash and Contractual Obligations
The following summarizes our scheduled long-term contractual obligations that may affect our future liquidity as of December 31, 2009 (in thousands):
| Payment Due By Period | |||||||||||||||
| Contractual Obligations |
Total | Less than 1 Year |
1-3 Years |
3-5 Years |
More than 5 Years | ||||||||||
| Operating leases |
$ | 1,165 | $ | 687 | $ | 478 | $ | — | $ | — | |||||
| License obligations(1) |
— | — | — | — | — | ||||||||||
| Convertible Notes due 2011 (2) |
$ | 29,258 | — | $ | 29,258 | — | — | ||||||||
| Escrow Agreement (2) |
$ | 1,094 | $ | 1,094 | — | — | — | ||||||||
| Total(3) |
$ | 31,517 | $ | 1,781 | $ | 29,736 | $ | — | $ | — | |||||
| (1) | Under the license agreements for our product candidates, we may be required to make future payments based upon the occurrence of certain milestones related to clinical development, regulatory or commercial events. We will also be required to pay royalties on any net sales of the licensed products, if any are approved by the FDA or foreign regulatory authorities for commercial sale. These milestone payments and royalty payments under our license agreements are not included in the table above because we cannot determine when, or if, the related milestones will be achieved or the events triggering the commencement of payment obligations will occur at present. |
| (2) | These are recorded at fair value which is less than face value due to a lack of marketability discount employed in the binomial option pricing model we used to value these contractual obligations. |
| (3) | We also enter into agreements with third parties to conduct our clinical trials, manufacture our product candidates, perform data collection and analysis and other services in connection with our product development programs. Our payment obligations under these agreements depend upon the progress of our product development programs. Therefore, we are unable at this time to estimate with certainty the future costs we will incur under these agreements. |
76
Table of Contents
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
Our primary exposure to market risk due to changes in interest rates relates primarily to the increase or decrease in the amount of interest income we can earn on our investment portfolio. The primary objective of our investment activities is to preserve principal while at the same time maximizing the income we receive without significantly increasing risk. Our risk associated with fluctuating interest rates is limited to our investments in interest rate sensitive financial instruments. Under our current policies, we do not use interest rate derivative instruments to manage exposure to interest rate changes. We mitigate default risk by investing in investment grade securities. A hypothetical 100 basis point adverse move in interest rates along the entire interest rate yield curve would not materially affect the fair value of our interest sensitive financial instruments due to their relatively short term nature. Declines in interest rates over time will, however, reduce our interest income, while increases in interest rates over time will increase our interest income.
Our investment securities are trading securities and consist of ARS, which are debt instruments with long-term maturities in which the interest rates reset in short intervals through “Dutch” auctions by matching buyers and sellers. All of our ARS had AAA ratings at the time of purchase and principally represent interests in government-guaranteed student loans, insurance notes and portfolios of securities (primarily commercial paper). None of the underlying collateral for our ARS consisted of subprime mortgages or collateralized debt obligations. At December 31, 2009, $24.6 million of our ARS consisted primarily of government-guaranteed student loan securities and $1.8 million of our ARS consisted of private placement securities.
The continued negative conditions in the global credit markets have prevented most investors, including ourselves, from liquidating certain holdings of ARS because the amount of securities submitted for sale has exceeded the amount of purchase orders for the securities. If there is insufficient demand for the securities at the time of the “Dutch” auction, the auction may not be completed and the interest rates may be reset to the maximum interest rate applicable to the specific securities being auctioned as per the official statement issued at the initial bond sale. When auctions for these securities fail, as they did in 2009, the investments may not be readily convertible to cash until a future auction of these investments is successful or they are redeemed or repurchased, sold through a secondary market or mature. For the year ended December 31, 2009, we liquidated $1.3 million of ARS, which we reinvested in cash equivalents.
In the fourth quarter of 2008, we received and accepted the ARS Rights Offer from UBS. Pursuant to the ARS Rights Offer, we received the ARS Put. In January 2009, we were approved by UBS for the ARS Loan in the amount of $15.9 million and drew down the entire preapproved amount. In addition, in February 2009, we borrowed an additional $2.2 million under the ARS Loan, bringing the total amount outstanding under the ARS Loan to $18.1 million, following UBS’ decision to increase our availability under the ARS Loan. Under the ARS Loan program, UBS may demand full or partial payment of the ARS Loan, at its sole option and without cause, at any time. All cash received under the UBS Loan was invested in money market accounts. At December 31, 2009, our outstanding ARS Loan balance was $17.6 million. Because the interest that we pay on the ARS Loan will not exceed the interest that we receive on the ARS pledged as security for the ARS Loan and which are held in the collateral account, we do not believe that this arrangement subjects us to additional interest rate risk.
77
Table of Contents
Item 8. Financial Statements and Supplementary Data
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders
MediciNova, Inc.:
We have audited the accompanying consolidated balance sheet of MediciNova, Inc. and subsidiaries (a development stage company) (the Company) as of December 31, 2009, and the related consolidated statements of operations, stockholders’ equity and cash flows for the year ended December 31, 2009 and for the period from September 26, 2000 (inception) through December 31, 2009. These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audit. The cumulative statements of operations and cash flows for the period from September 26, 2000 (inception) through December 31, 2008 and the statements of stockholders’ equity for the period from September 26, 2000 (inception) to December 31, 2000 and for each of the years in the eight-year period ended December 31, 2008, were audited by other auditors whose report has been furnished to us, and our opinion insofar as it relates to the amounts included for the period September 26, 2000 through December 31, 2008 is based solely on the report of the other auditors.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audit provides a reasonable basis for our opinion.
In our opinion, based on our audit and the report of other auditors, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of MediciNova, Inc. and subsidiaries (a development stage company) as of December 31, 2009, and the results of their operations and their cash flows for the year ended December 31, 2009 and for the period from September 26, 2000 (inception) to December 31, 2009, in conformity with U.S. generally accepted accounting principles.
/s/ KPMG LLP
San Diego, California
March 24, 2010
78
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders
MediciNova, Inc.
We have audited the accompanying consolidated balance sheet of MediciNova, Inc. (a development stage company) as of December 31, 2008, and the related consolidated statements of operations, and cash flows for the years ended December 31, 2008 and 2007 and for the period from September 26, 2000 (inception) through December 31, 2008 (not included herein), and the statements of stockholders’ equity for the period from September 26, 2000 (inception to December 31, 2000) and for each of the eight years in the period ended December 31, 2008. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. We were not engaged to perform an audit of the Company’s internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the consolidated financial position of MediciNova, Inc. (a development stage company) at December 31, 2008, and the results of its consolidated operations and its cash flows for the years ended December 31, 2008 and 2007, and the period from September 26, 2000 (inception) through December 31, 2008 (not included herein), and the consolidated statements of stockholders’ equity for the period from September 26, 2000 (inception) to December 31, 2000 and each of the eight years in the period ended December 31, 2008, in conformity with U.S. generally accepted accounting principles.
/s/ Ernst & Young LLP
San Diego, California
March 27, 2009
79
Table of Contents
(a development stage company)
CONSOLIDATED BALANCE SHEETS
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| Assets |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 19,241,581 | $ | 19,297,284 | ||||
| Investment securities-current (Note 3) |
24,254,987 | — | ||||||
| ARS put—current (Note 3) |
2,557,007 | — | ||||||
| Prepaid expenses and other current assets |
869,649 | 718,317 | ||||||
| Total current assets |
46,923,224 | 20,015,601 | ||||||
| Restricted cash (Notes 1 and 2) |
30,045,965 | — | ||||||
| In-process research and development (Notes 1 and 2) |
4,800,000 | — | ||||||
| Restricted investment (Notes 1 and 2) |
676,499 | — | ||||||
| Restricted letter of credit (Notes 1 and 2) |
500,042 | — | ||||||
| Goodwill (Notes 1 and 2) |
9,142,205 | — | ||||||
| Property and equipment, net |
153,547 | 368,299 | ||||||
| Long-term investments (Note 3) |
2,085,425 | 24,047,314 | ||||||
| ARS put—long-term (Note 3) |
— | 5,792,701 | ||||||
| Total assets |
$ | 94,326,907 | $ | 50,223,915 | ||||
| Liabilities and Stockholders’ Equity |
||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 1,300,271 | $ | 392,572 | ||||
| ARS loan payable |
17,605,485 | — | ||||||
| Escrow holdback (Notes 1 and 2) |
1,094,045 | — | ||||||
| Accrued expenses |
1,276,036 | 1,011,916 | ||||||
| Income taxes payable |
— | 9,748 | ||||||
| Accrued compensation and related expenses |
1,146,960 | 765,147 | ||||||
| Total current liabilities |
22,422,797 | 2,179,383 | ||||||
| Management transition plan liability (Note 2) |
676,499 | — | ||||||
| Deferred tax liability (Note 8) |
1,956,000 | — | ||||||
| Convertible notes (Notes 1 and 2) |
29,258,137 | — | ||||||
| Total liabilities |
54,313,433 | 2,179,383 | ||||||
| Commitments and contingencies (Note 6) |
||||||||
| Stockholders’ equity: |
||||||||
| Preferred stock, $0.01 par value; 500,000 shares authorized at December 31, 2009 and December 31, 2008; no shares outstanding at December 31, 2009 and December 31, 2008 |
— | — | ||||||
| Common stock, $0.001 par value; 30,000,000 shares authorized at December 31, 2009 and December 31, 2008; 12,172,510 and 12,072,027 shares issued at December 31, 2009 and December 31, 2008, respectively, and 12,122,217 and 11,984,713 shares outstanding at December 31, 2009 and December 31, 2008, respectively |
12,170 | 12,072 | ||||||
| Additional paid-in capital |
288,652,712 | 276,361,775 | ||||||
| Accumulated other comprehensive loss |
(64,914 | ) | (29,744 | ) | ||||
| Treasury stock, at cost; 50,293 shares at December 31, 2009 and 87,314 shares at December 31, 2008 |
(1,235,395 | ) | (1,317,362 | ) | ||||
| Deficit accumulated during the development stage |
(247,351,099 | ) | (226,982,209 | ) | ||||
| Total stockholders’ equity |
40,013,474 | 48,044,532 | ||||||
| Total liabilities and stockholders’ equity |
$ | 94,326,907 | $ | 50,223,915 | ||||
See accompanying notes to consolidated financial statements.
80
Table of Contents
(a development stage company)
CONSOLIDATED STATEMENTS OF OPERATIONS
| Years ended December 31, | Period from September 26, 2000 (inception) to December 31, 2009 |
|||||||||||||||
| 2009 | 2008 | 2007 | ||||||||||||||
| Revenues |
$ | — | $ | — | $ | — | $ | 1,558,227 | ||||||||
| Operating expenses: |
||||||||||||||||
| Cost of revenues |
— | — | — | 1,258,421 | ||||||||||||
| Research and development |
10,873,169 | 13,827,651 | 42,121,095 | 144,545,867 | ||||||||||||
| General and administrative |
10,366,291 | 8,773,695 | 11,372,873 | 89,026,998 | ||||||||||||
| Total operating expenses |
21,239,460 | 22,601,346 | 53,493,968 | 234,831,286 | ||||||||||||
| Operating loss |
(21,239,460 | ) | (22,601,346 | ) | (53,493,968 | ) | (233,273,059 | ) | ||||||||
| Gain/(impairment charge), net on investment securities and ARS put |
310,250 | (1,259,984 | ) | — | (949,734 | ) | ||||||||||
| Foreign exchange loss |
(13,622 | ) | (88,159 | ) | — | (101,781 | ) | |||||||||
| Other income, net |
580,949 | 2,038,219 | 4,610,724 | 18,377,163 | ||||||||||||
| Income taxes |
(7,007 | ) | (13,559 | ) | (20,000 | ) | (40,566 | ) | ||||||||
| Net loss |
(20,368,890 | ) | (21,924,829 | ) | (48,903,244 | ) | (215,987,977 | ) | ||||||||
| Accretion to redemption value of redeemable convertible preferred stock |
— | — | — | (98,445 | ) | |||||||||||
| Deemed dividend resulting from beneficial conversion feature on Series C redeemable convertible preferred stock |
— | — | — | (31,264,677 | ) | |||||||||||
| Net loss applicable to common stockholders |
$ | (20,368,890 | ) | $ | (21,924,829 | ) | $ | (48,903,244 | ) | $ | (247,351,099 | ) | ||||
| Basic and diluted net loss per common share |
$ | (1.68 | ) | $ | (1.82 | ) | $ | (4.16 | ) | |||||||
| Shares used to compute basic and diluted net loss per share |
12,105,835 | 12,072,027 | 11,752,139 | |||||||||||||
See accompanying notes to consolidated financial statements.
81
Table of Contents
(a development stage company)
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
| Convertible preferred stock |
Common stock | Additional paid-in capital |
Deferred Compensation |
Accumulated other comprehensive loss |
Treasury stock |
Deficit accumulated during the development stage |
Total stockholders’ equity |
||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | ||||||||||||||||||||||||||||
| Issuance of common stock for cash to founders at $1.00 per share in September |
— | $ | — | 50,000 | $ | 50 | $ | 49,950 | $ | — | $ | — | $ | — | $ | — | $ | 50,000 | |||||||||||||
| Issuance of Series A convertible preferred stock at $10 per share in October |
500,000 | 5,000 | — | — | 4,995,000 | — | — | — | — | 5,000,000 | |||||||||||||||||||||
| Net loss and comprehensive loss |
— | — | — | — | — | — | — | — | (201,325 | ) | (201,325 | ) | |||||||||||||||||||
| Balance at December 31, 2000 |
500,000 | 5,000 | 50,000 | 50 | 5,044,950 | — | — | — | (201,325 | ) | 4,848,675 | ||||||||||||||||||||
| Issuance of Series A convertible preferred stock at $10 per share in August |
500,000 | 5,000 | — | — | 4,995,000 | — | — | — | — | 5,000,000 | |||||||||||||||||||||
| Net loss and comprehensive loss |
— | — | — | — | — | — | — | — | (1,794,734 | ) | (1,794,734 | ) | |||||||||||||||||||
| Balance at December 31, 2001 |
1,000,000 | 10,000 | 50,000 | 50 | 10,039,950 | — | — | — | (1,996,059 | ) | 8,053,941 | ||||||||||||||||||||
| Net loss and comprehensive loss |
— | — | — | — | — | — | — | — | (6,931,476 | ) | (6,931,476 | ) | |||||||||||||||||||
| Balance at December 31, 2002 |
1,000,000 | 10,000 | 50,000 | 50 | 10,039,950 | — | — | — | (8,927,535 | ) | 1,122,465 | ||||||||||||||||||||
| Issuance of Series B convertible preferred stock at $100 per share, net of issuance costs of $1,093,453, in March, April, May and December |
107,500 | 1,075 | — | — | 9,655,472 | — | — | — | — | 9,656,547 | |||||||||||||||||||||
| Net loss and comprehensive loss |
— | — | — | — | — | — | — | — | (6,209,130 | ) | (6,209,130 | ) | |||||||||||||||||||
| Balance at December 31, 2003 |
1,107,500 | 11,075 | 50,000 | 50 | 19,695,422 | — | — | — | (15,136,665 | ) | 4,569,882 | ||||||||||||||||||||
| Issuance of Series B convertible preferred stock at $100 per share, net of issuance costs of $1,208,896, in January, February, March, April and May |
183,650 | 1,837 | — | — | 17,154,267 | — | — | — | — | 17,156,104 | |||||||||||||||||||||
| Stock-based compensation related to founders’ warrants |
— | — | — | — | 34,069,916 | — | — | — | — | 34,069,916 | |||||||||||||||||||||
| Deferred employee stock-based compensation |
— | — | — | — | 1,419,300 | (1,419,300 | ) | — | — | — | — | ||||||||||||||||||||
| Amortization of deferred employee stock-based |
— | — | — | — | — | 224,579 | — | — | — | 224,579 | |||||||||||||||||||||
| Deemed dividend resulting from beneficial conversion feature on Series C redeemable convertible preferred stock |
— | — | — | — | 31,264,677 | — | — | — | (31,264,677 | ) | — | ||||||||||||||||||||
| Accretion to redemption value of redeemable convertible preferred stock |
— | — | — | — | — | — | — | — | (78,756 | ) | (78,756 | ) | |||||||||||||||||||
| Net loss and comprehensive loss |
— | — | — | — | — | — | — | — | (48,272,603 | ) | (48,272,603 | ) | |||||||||||||||||||
| Balance at December 31, 2004 |
1,291,150 | 12,912 | 50,000 | 50 | 103,603,582 | (1,194,721 | ) | — | — | (94,752,701 | ) | 7,669,122 | |||||||||||||||||||
82
Table of Contents
MEDICINOVA, INC.
(a development stage company)
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY—(Continued)
| Convertible preferred stock |
Common stock | Additional paid-in capital |
Deferred Compensation |
Accumulated other comprehensive loss |
Treasury stock |
Deficit accumulated during the development stage |
Total stockholders’ equity |
|||||||||||||||||||||
| Shares | Amount | Shares | Amount | |||||||||||||||||||||||||
| Issuance of common stock in initial public offering at $38.80 per share in February |
— | — | 3,000,000 | 3,000 | 104,483,895 | — | — | — | — | 104,486,895 | ||||||||||||||||||
| Issuance of common stock upon partial exercise of over-allotment option at $38.80 per share in March |
— | — | 157,300 | 157 | 5,557,616 | — | — | — | — | 5,557,773 | ||||||||||||||||||
| Issuance costs for registration statement filed on behalf of restricted stockholders |
— | — | — | — | (165,476 | ) | — | — | — | — | (165,476 | ) | ||||||||||||||||
| Conversion of redeemable convertible preferred stock into common stock in February |
— | — | 2,766,785 | 2,767 | 43,499,998 | — | — | — | — | 43,502,765 | ||||||||||||||||||
| Conversion of convertible preferred stock into common stock in February |
(1,291,150 | ) | (12,912 | ) | 3,911,500 | 3,911 | 9,001 | — | — | — | — | — | ||||||||||||||||
| Stock-based compensation related to acceleration of option vesting upon employee termination and subsequent reissuance of a fully vested option |
— | — | — | — | 127,875 | — | — | — | — | 127,875 | ||||||||||||||||||
| Amortization of deferred employee stock-based compensation, net of cancelations |
— | — | — | — | — | 311,282 | — | — | — | 311,282 | ||||||||||||||||||
| Cancelation of stock options issued to employees and related deferred compensation |
— | — | — | — | (84,000 | ) | 84,000 | — | — | — | — | |||||||||||||||||
| Accretion to redemption value of redeemable convertible preferred stock |
— | — | — | — | — | — | — | — | (19,689 | ) | (19,689 | ) | ||||||||||||||||
| Purchase of treasury stock at $11.10 per share in December |
— | — | — | — | — | — | — | (55,445 | ) | — | (55,445 | ) | ||||||||||||||||
| Comprehensive loss: |
||||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | (25,692,135 | ) | (25,692,135 | ) | ||||||||||||||||
| Accumulated other comprehensive loss |
— | — | — | — | — | — | (15,188 | ) | — | — | (15,188 | ) | ||||||||||||||||
| Total comprehensive loss |
— | — | — | — | — | — | — | — | — | (25,707,323 | ) | |||||||||||||||||
| Balance at December 31, 2005 |
— | — | 9,885,585 | 9,885 | 257,032,491 | (799,439 | ) | (15,188 | ) | (55,445 | ) | (120,464,525 | ) | 135,707,779 | ||||||||||||||
83
Table of Contents
MEDICINOVA, INC.
(a development stage company)
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY—(Continued)
| Convertible preferred stock |
Common stock | Additional paid-in capital |
Deferred Compensation |
Accumulated other comprehensive loss |
Treasury stock |
Deficit accumulated during the development stage |
Total stockholders’ equity |
|||||||||||||||||||
| Shares | Amount | Shares | Amount | |||||||||||||||||||||||
| Cashless warrant exercises of 260,000 in February, April and August |
— | — | 260,000 | 260 | (260 | ) | — | — | — | — | — | |||||||||||||||
| Warrant exercises of 275,000 shares at $1.00 per share in March and August |
— | — | 275,000 | 275 | 274,725 | — | — | — | — | 275,000 | ||||||||||||||||
| Write off balance of deferred employee stock-based compensation as of 12/31/05 |
— | — | — | — | (799,439 | ) | 799,439 | — | — | — | — | |||||||||||||||
| Option exercises of 1,400 shares at $10.00 per share in May and August |
— | — | 1,400 | 2 | 13,998 | — | — | — | — | 14,000 | ||||||||||||||||
| Amortization of deferred employee stock-based compensation |
— | — | — | — | 2,090,182 | — | — | — | — | 2,090,182 | ||||||||||||||||
| Purchase of treasury stock from $10.30—$13.10 per share in February, March, May, June, July, September and October |
— | — | — | — | — | — | — | (1,382,425 | ) | — | (1,382,425 | ) | ||||||||||||||
| Comprehensive loss: |
||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | (35,689,611 | ) | (35,689,611 | ) | ||||||||||||||
| Accumulated other comprehensive loss |
— | — | — | — | — | — | (34,017 | ) | — | — | (34,017 | ) | ||||||||||||||
| Total Comprehensive loss |
— | — | — | — | — | — | — | — | — | (35,723,628 | ) | |||||||||||||||
| Balance at December 31, 2006 |
— | — | 10,421,985 | 10,422 | 258,611,697 | — | (49,205 | ) | (1,437,870 | ) | (156,154,136 | ) | 100,980,908 | |||||||||||||
| Cashless warrant exercises of 650,047 in January and September |
— | — | 650,047 | 650 | (650 | ) | — | — | — | — | — | |||||||||||||||
| Issuance of common stock in a public offering at $12.00 per share in February |
— | — | 1,000,000 | 1,000 | 10,638,600 | — | — | — | — | 10,639,600 | ||||||||||||||||
| Employee stock-based compensation |
— | — | — | 3,939,416 | — | — | — | — | 3,939,416 | |||||||||||||||||
| Issuance of shares under an employee stock purchase plan at $6.72 |
— | — | — | — | — | — | — | 33,782 | — | 33,782 | ||||||||||||||||
| Comprehensive loss: |
||||||||||||||||||||||||||
| Net loss |
— | — | (5 | ) | — | — | — | — | — | (48,903,244 | ) | (48,903,244 | ) | |||||||||||||
| Accumulated other comprehensive loss |
— | — | — | — | — | — | (82,261 | ) | — | — | (82,261 | ) | ||||||||||||||
| Total comprehensive loss |
— | — | — | — | — | — | — | — | — | (48,985,505 | ) | |||||||||||||||
| Balance at December 31, 2007 |
— | — | 12,072,027 | 12,072 | 273,189,063 | — | (131,466 | ) | (1,404,088 | ) | (205,057,380 | ) | 66,608,201 | |||||||||||||
84
Table of Contents
MEDICINOVA, INC.
(a development stage company)
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY—(Continued)
| Convertible preferred stock |
Common stock | Additional paid-in capital |
Deferred Compensation |
Accumulated other comprehensive loss |
Treasury stock |
Deficit accumulated during the development stage |
Total stockholders’ equity |
|||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | |||||||||||||||||||||||||||||
| Employee stock-based compensation |
— | — | — | — | 3,172,712 | — | — | — | — | 3,172,712 | ||||||||||||||||||||||
| Issuance of shares under an employee stock purchase plan at $2.33 average |
— | — | — | — | — | — | — | 86,726 | — | 86,726 | ||||||||||||||||||||||
| Comprehensive loss: |
||||||||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | (21,924,829 | ) | (21,924,829 | ) | ||||||||||||||||||||
| Accumulated other comprehensive loss |
— | — | — | — | — | — | 101,722 | — | — | 101,722 | ||||||||||||||||||||||
| Total comprehensive loss |
— | — | — | — | — | — | — | — | — | (21,823,107 | ) | |||||||||||||||||||||
| Balance at December 31, 2008 |
— | — | 12,072,027 | 12,072 | 276,361,775 | — | (29,744 | ) | (1,317,362 | ) | (226,982,209 | ) | 48,044,532 | |||||||||||||||||||
| Employee stock-based compensation |
— | — | — | — | 2,371,636 | — | — | — | — | 2,371,636 | ||||||||||||||||||||||
| Option exercises |
— | — | 100,483 | 98 | 406,259 | — | — | — | — | 406,357 | ||||||||||||||||||||||
| Fair value of redemption feature of Avigen purchase (Note 2) |
— | — | — | — | 9,513,042 | — | — | — | — | 9,513,042 | ||||||||||||||||||||||
| Issuance of shares under an employee stock purchase plan at $2.21 average |
— | — | — | — | — | — | — | 81,967 | — | 81,967 | ||||||||||||||||||||||
| Comprehensive loss: |
||||||||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | (20,368,890 | ) | (20,368,890 | ) | ||||||||||||||||||||
| Accumulated other comprehensive loss |
— | — | — | — | — | — | (35,170 | ) | — | — | (35,170 | ) | ||||||||||||||||||||
| Total comprehensive loss |
— | — | — | — | — | — | — | — | — | (20,404,060 | ) | |||||||||||||||||||||
| Balance at December 31, 2009 |
— | $ | — | 12,172,510 | $ | 12,170 | $ | 288,652,712 | $ | — | $ | (64,914 | ) | $ | (1,235,395 | ) | $ | (247,351,099 | ) | $ | 40,013,474 | |||||||||||
See accompanying notes to consolidated financial statements.
85
Table of Contents
(a development stage company)
CONSOLIDATED STATEMENTS OF CASH FLOWS
| Years ended December 31, | Period from September 26, 2000 (inception) to December 31, 2009 |
|||||||||||||||
| 2009 | 2008 | 2007 | ||||||||||||||
| Operating activities: |
||||||||||||||||
| Net loss |
$ | (20,368,890 | ) | $ | (21,924,829 | ) | $ | (48,903,244 | ) | $ | (215,987,977 | ) | ||||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||||||
| Non-cash stock-based compensation |
2,371,636 | 3,172,712 | 3,939,416 | 46,307,598 | ||||||||||||
| Depreciation and amortization |
219,202 | 305,018 | 516,013 | 1,795,298 | ||||||||||||
| Amortization of premium/discount on marketable securities |
— | (691,706 | ) | (170,576 | ) | (2,476,420 | ) | |||||||||
| (Gain)/impairment charge, net on investment securities and ARS Put |
(310,250 | ) | 1,259,984 | — | 949,734 | |||||||||||
| Loss on disposal of assets |
11,997 | — | — | 11,997 | ||||||||||||
| Impairment of sublease |
— | — | — | 35,259 | ||||||||||||
| Changes in operating assets and liabilities: |
||||||||||||||||
| Prepaid expenses and other assets |
(114,383 | ) | 1,725,295 | 4,225,382 | (832,700 | ) | ||||||||||
| Accounts payable, income tax payable, accrued expenses and deferred rent |
890,854 | (5,109,397 | ) | (3,678,280 | ) | 2,305,090 | ||||||||||
| Accrued compensation and related expenses |
285,672 | 144,543 | 212,600 | 1,050,819 | ||||||||||||
| Net cash used in operating activities |
(17,014,162 | ) | (21,118,380 | ) | (43,858,689 | ) | (166,841,302 | ) | ||||||||
| Investing activities: |
||||||||||||||||
| Cash paid for acquired business, net of acquired cash |
(2,371,749 | ) | — | — | (2,371,749 | ) | ||||||||||
| Purchases of investment securities |
— | (2,000,000 | ) | (41,712,645 | ) | (377,205,766 | ) | |||||||||
| Maturities or sales of investment securities |
1,252,846 | 23,550,000 | 85,662,087 | 349,806,297 | ||||||||||||
| Acquisition of property and equipment |
(16,447 | ) | — | (380,709 | ) | (2,252,946 | ) | |||||||||
| Proceeds from sales of property and equipment |
— | — | 62,024 | 256,845 | ||||||||||||
| Net cash provided by / (used in) investing activities |
(1,135,350 | ) | 21,550,000 | 43,630,757 | (31,767,319 | ) | ||||||||||
| Financing activities: |
||||||||||||||||
| Net proceeds from the sale of common stock |
406,357 | — | 10,672,374 | 121,296,923 | ||||||||||||
| Sale of preferred stock, net of issuance costs |
— | — | — | 80,216,971 | ||||||||||||
| Proceeds from ARS loan, net |
17,605,485 | — | — | 17,605,485 | ||||||||||||
| Purchase of treasury stock, net of employee stock purchases |
81,967 | 86,726 | — | (1,269,177 | ) | |||||||||||
| Net cash provided by financing activities |
18,093,809 | 86,726 | 10,672,374 | 217,850,202 | ||||||||||||
| Net increase / (decrease) in cash and cash equivalents |
(55,703 | ) | 518,346 | 10,444,442 | 19,241,581 | |||||||||||
| Cash and cash equivalents, beginning of period |
19,297,284 | 18,778,938 | 8,334,496 | — | ||||||||||||
| Cash and cash equivalents, end of period |
$ | 19,241,581 | $ | 19,297,284 | $ | 18,778,938 | $ | 19,241,581 | ||||||||
| Supplemental disclosure of non-cash investing and financing activities: |
||||||||||||||||
| Conversion of convertible preferred stock into common stock upon initial public offering |
$ | — | $ | — | $ | — | $ | 43,515,677 | ||||||||
| Unrealized loss on marketable securities available-for-sale |
$ | — | $ | — | $ | (39,813 | ) | $ | (89,018 | ) | ||||||
| Supplemental disclosure of non-cash operating and investing activities: |
||||||||||||||||
| Reclassification of current marketable securities available-for-sale to long-term investments |
$ | — | $ | (24,047,314 | ) | $ | — | $ | (24,047,314 | ) | ||||||
| Supplemental disclosures of cash flow information: |
||||||||||||||||
| Income taxes paid |
$ | 9,434 | $ | 24,528 | $ | — | $ | 33,962 | ||||||||
| Interest paid |
$ | 235,364 | $ | — | $ | — | $ | 235,364 | ||||||||
| Supplemental disclosure of investing activities related to business acquisition: |
||||||||||||||||
| Fair value of assets acquired |
$ | 36,687,706 | $ | — | $ | — | $ | 36,687,706 | ||||||||
| Liabilities assumed |
$ | (1,008,687 | ) | $ | — | $ | — | $ | (1,008,687 | ) | ||||||
| Deferred tax liability |
$ | (1,956,000 | ) | $ | — | $ | — | $ | (1,956,000 | ) | ||||||
| Fair value of total merger consideration (Note 2) |
$ | (42,865,224 | ) | $ | — | $ | — | $ | (42,865,224 | ) | ||||||
See accompanying notes to consolidated financial statements.
86
Table of Contents
(a development stage company)
Notes to Consolidated Financial Statements
1. The Company, Basis of Presentation and Summary of Significant Accounting Policies
The Company
We were incorporated in the state of Delaware in September 2000. We are a development stage biopharmaceutical company focused on acquiring and developing novel, small molecule therapeutics for the treatment of diseases with unmet medical need with a specific focus on the U.S. market. Through strategic alliances primarily with Japanese pharmaceutical companies, we hold rights to a diversified portfolio of clinical and preclinical product candidates, each of which we believe has a well-characterized and differentiated therapeutic profile, attractive commercial potential and patent assets having claims of commercially adequate scope.
Basis of Presentation
Our primary activities since incorporation have been organizational activities, including recruiting personnel, establishing office facilities, conducting research and development, performing business and financial planning and raising capital. Accordingly, in connection with preparation of the consolidated financial statements we operate under one reporting segment and are considered to be in the development stage, under the authoritative guidance for development stage entities, Accounting Standards Codification (“ASC”) 915 (formerly Statement of Financial Accounting Standards (“SFAS”) No. 7.)
During the first quarter of 2005, we completed our initial public offering (“IPO”) of 3,000,000 shares of common stock in Japan for proceeds of $104.5 million, net of underwriting discounts and commissions and offering costs. In December 2006, we were listed on the Nasdaq Global Market. Accordingly, we are a public company in both the United States and Japan, as our stock is traded on both the Nasdaq Global Market and the Hercules Market of the Osaka Securities Exchange.
Avigen Transaction. On December 18, 2009, Absolute Merger, Inc., a wholly-owned subsidiary of ours, merged with and into Avigen, Inc., or Avigen, with Avigen continuing as the surviving entity and wholly-owned subsidiary of ours, or the Merger. Under the terms of the merger agreement, Avigen shareholders, at their election, received an amount per share either in cash, convertible notes issued by us or a combination thereof, upon closing. Of the 29,852,115 shares of Avigen common stock outstanding, approximately 17% of Avigen shareholders elected to receive cash at closing in the amount of approximately $1.19 per share with an additional $0.04 per share expected to be paid in two increments on June 30, 2010 and after November 30, 2010, while the remaining 83% elected to receive convertible notes issued by us. See Notes to Consolidated Financial Statements – Note 2, Avigen Transaction, for additional information on the merger.
We have sustained operating losses since inception and expect such losses to continue over the next several years. Management plans to continue financing the operations with equity issuances, debt arrangements or a combination thereof. We expect current working capital to be sufficient to fund our operations through December 31, 2009. If adequate future funds are not available, we may be required to delay, reduce the scope of or eliminate one or more of our research or development programs, or cease operations.
Principles of Consolidation
The consolidated financial statements include the accounts of MediciNova, Inc. and its wholly-owned subsidiaries. MediciNova, Inc. and its subsidiaries are collectively referred to herein as “we,” “our” or “us.” We do not have any interests in any variable interest entities.
87
Table of Contents
MEDICINOVA, INC.
(a development stage company)
Notes to Consolidated Financial Statements
On December 13, 2006, MediciNova (Europe) Limited, a wholly-owned subsidiary of MediciNova, Inc., was incorporated under the laws of England and Wales and established for the purpose of facilitating the clinical development of our compounds for the European marketplace. MediciNova (Europe) Limited’s functional currency is the U.S. dollar, the reporting currency of its parent.
On January 4, 2007, MediciNova Japan, Inc., a wholly-owned subsidiary of MediciNova, Inc., was incorporated under the laws of Japan and established to strengthen business development and investor and public relations activities in Japan and other Asian countries. MediciNova Japan, Inc.’s functional currency is the Japanese yen.
On August 17, 2009, Absolute Merger, Inc., a wholly-owned subsidiary of MediciNova, Inc. was incorporated under the General Corporation Law of the State of Delaware for the purpose of facilitating the Merger (the “Merger”) with Avigen, Inc. (“Avigen”). See Notes to Consolidated Financial Statements- Note 2. Avigen Transaction, for more information regarding the merger.
All intercompany transactions and investments in our subsidiaries have been eliminated in consolidation.
Use of Estimates
The preparation of consolidated financial statements in conformity with U.S. generally accepted accounting principles requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Actual results may differ from these estimates under different assumptions or conditions.
Cash and Cash Equivalents
Cash and cash equivalents consist of cash and other highly liquid investments with original maturities of three months or less from the date of purchase. Cash equivalents at December 31, 2009 consisted of money market funds.
Investment Securities and ARS Put
Investments with maturity of more than three months on the date acquired are considered short-term investments and have been classified by us as marketable securities available-for-sale. Such investments are carried at fair value, with unrealized gains and losses, if any, included as a separate component of stockholders’ equity. The cost of marketable securities available-for-sale is based on the specific identification method. At December 31, 2009, there were no marketable securities available for sale recorded on our consolidated balance sheets.
Our investment securities, are trading securities, and consist of auction rate securities (“ARS”), all of which had AAA ratings at the time of purchase, that principally represent interests in government-guaranteed student loans, insurance notes and portfolios of securities (primarily commercial paper), and these securities have been designate as trading securities. ARS are generally long-term debt instruments that historically have provided liquidity through a “Dutch” auction process that resets the applicable interest rate at predetermined calendar intervals, typically seven, 28, 35 or 49 days. Due to continued negative conditions in the global credit markets, our ARS have continued to fail at auction with few trades in either the primary or the secondary markets. As such, as required by ASC 820, authoritative guidance for fair value measurement and disclosures, we
88
Table of Contents
MEDICINOVA, INC.
(a development stage company)
Notes to Consolidated Financial Statements
determined the fair value of our ARS portfolio primarily on Level 3 criteria as prescribed by the accounting standard, which resulted in our reliance on a discounted cash flow valuation model with assumptions related to interest rates, maturities and liquidity determined by us based on the credit quality of the security, the credit quality of the associated insurer, if applicable, the respective prospectuses, and the credit market outlook. At December 31, 2009, $24.6 million of our ARS consisted primarily of government-guaranteed student loan securities and $1.8 million of our ARS consisted of private placement securities. None of the underlying collateral for our ARS consisted of subprime mortgages or collateralized debt obligations. At December 31, 2009, $24.3 million of ARS subject to the UBS settlement (described below) have been classified as current assets given the estimated time frame in which we can readily convert these securities into cash. The remaining $2.1 million of ARS have been classified as long-term assets given the estimated time frame in which we can readily convert these securities into cash.
In August 2008, UBS and its affiliates (“UBS”), the brokerage firm through which we purchased the majority of our ARS investments, entered into a settlement with the SEC, the New York Attorney General and other state agencies. Under the settlement, UBS issued to us the Auction Rate Security Rights, which would allow us to sell to UBS our ARS held in accounts with UBS (“ARS Rights Offer”). Pursuant to the ARS Rights Offer, we received the right to sell to UBS the ARS held in accounts with UBS at par value at any time during the period beginning June 30, 2010 and ending July 2, 2012 (“ARS Put”). As part of the settlement, UBS also offered to us a no net cost loan program (“ARS Loan”), whereby we would be able to borrow up to 75% of the market value, as determined by UBS at its sole discretion, of our ARS that have been pledged as collateral at an interest cost that would not exceed the interest being paid on the underlying ARS investments. Under the ARS Loan program, UBS may demand full or partial payment of the ARS Loan, at its sole option and without cause, at any time. In November 2008, we accepted the ARS Rights Offer. In January 2009, we were approved for the ARS Loan in the amount of $15.9 million and drew down the entire preapproved amount. In addition, in February 2009, we borrowed an additional $2.2 million under the ARS Loan, bringing the total amount outstanding under the ARS Loan to $18.1 million, following UBS’ decision to increase our availability under the ARS Loan. All cash received under the ARS Loan was invested in money market accounts. Our ARS Loan balance at December 31, 2009 was $17.6 million, with an effective average interest rate of 1.29 percent charged, or approximately $235,000 of interest charged, on the no net cost loan.
We elected to measure the ARS Put under the fair value option of ASC 825, authoritative guidance on financial instruments, to mitigate the volatility in reported earnings due to the linkage of certain of our ARS and the ARS Put. The fair value of the ARS Put was also determined by a discounted cash flow valuation model effectively using a liquidity discount of approximately 5% and an interest rate of approximately 5%, which took into consideration the brokerage firm’s weighted average cost of capital. Based on our discounted cash flow valuation, we recorded a loss of $3.2 million in our consolidated statement of operations. In addition, we recorded the ARS Put as a current asset in our consolidated balance sheet as the ARS Put is exercisable beginning June 2010.
Restricted Cash
Restricted cash consists of cash held in a separate trust account, managed by a third-party, in connection with the $32.4 million of cash funded by Avigen and the $3.0 million of cash paid by us, or the First Payment Consideration, less the $6.0 million paid out to Avigen shareholders who elected a cash payout at the merger closing date—see Notes to Consolidated Financial Statements – Note 2. Avigen Transaction for further information.
89
Table of Contents
MEDICINOVA, INC.
(a development stage company)
Notes to Consolidated Financial Statements
Restricted Investment
Restricted investment consists of cash held in an irrevocable grantor trust, or rabbi trust, which is intended to fund benefit obligations under the Avigen, Inc. Management Transition Plan, or MTP. These funds represent reserves for benefits eligible to terminated employees as defined by the MTP. The cash equivalents in the rabbi trust is reported at fair value and classified as restricted investment in current assets. Upon termination of the trust, the merger agreement provides that these funds be paid to the former Avigen stockholders on a pro rata basis—see Notes to Consolidated Financial Statements – Note 2. Avigen Transaction for further information.
Restricted Letter of Credit
Restricted letter of credit consists of cash provided as a credit guarantee and security for an irrevocable letter of credit related to the original lease of office space which expires November 30, 2010. Any funds remaining after the letter of credit expires will revert to the escrow holdback account described below.
Convertible Notes
At the closing of the Merger, we and American Stock Transfer & Trust Company, LLC, as trustee, entered into an indenture. Under the terms of a separate trust agreement, $29.4 million, which represents the initial principal amount of the convertible notes, or Convertible Notes, or 83% of the First Payment Consideration, was deposited with a trust agent for the benefit of the holders and us (the amount of such deposit together with interest accrued and capitalized thereon, the Property). Provided no event of default has occurred and is continuing, we will be able to direct the investment and reinvestment of the Property in certain approved investment options, including certain money market funds. At the maturity of the Convertible Notes on June 18, 2011, the 18-month anniversary of the closing of the Merger, we will use the Property to pay the principal amount of, and accrued interest on, the remaining Convertible Notes. At acquisition date, we recorded the Convertible Notes in our consolidated balance sheet at fair value—see Notes to Consolidated Financial Statements – Note 2. Avigen Transaction for further information on the valuation of the Convertible Notes.
Escrow Holdback
At the closing of the merger, we and Avigen funded in cash and letter of credit $1,500,000 in a separate escrow account, or Second Payment Consideration, pursuant to an escrow agreement. The Second Payment Consideration is considered the “Escrow Holdback”. We (Avigen and us) identified certain additional liabilities of approximately $400,000 prior to closing of the Merger. As such, in accordance with the procedures set forth in the escrow agreement, $400,000 was released from the escrow account in satisfaction of these additional liabilities. As a result, the Second Payment Consideration is estimated to be no more than approximately $1.1 million, or $0.04 per share. At acquisition date, we recorded the Escrow Holdback in our consolidated balance sheet at fair value—see Notes to Consolidated Financial Statements – Note 2. Avigen Transaction for further information on the valuation of the Escrow Holdback.
Concentration of Credit Risk
Financial instruments that potentially subject us to a significant concentration of credit risk consist primarily of cash, cash equivalents and investment securities. We maintain deposits in federally insured financial institutions in excess of federally insured limits. However, management believes we are not exposed to significant credit risk due to the financial position of the depository institutions in which those deposits are held.
90
Table of Contents
MEDICINOVA, INC.
(a development stage company)
Notes to Consolidated Financial Statements
Additionally, we have established guidelines regarding diversification of our investments and their maturities, which are designed to maintain safety and liquidity.
Business Combinations
Our consolidated financial statements include an acquired business’s operations after the completion of the acquisition. We account for acquired businesses using the acquisition method of accounting. The acquisition method of accounting requires, among other things, that assets acquired and liabilities assumed be recognized at their fair values as of the acquisition date and that the fair value of acquired in-process research and development (IPR&D) be recorded on the balance sheet. Also, transaction costs are expensed as incurred. Any excess of the purchase price over the assigned values of the net assets acquired is recorded as goodwill. In connection with the Avigen transaction we recorded, at fair value, IPR&D and goodwill—See Notes to Consolidated Financial Statements – Note 2. Avigen Transaction for a more detailed discussion on IPR&D and goodwill.
Fair Value
Financial instruments, including cash and cash equivalents, accounts payable and accrued liabilities, are carried at cost, which we believe approximates fair value given their short-term nature. The carrying amount of our ARS Loan also approximates its fair value due to the loan’s short-term nature. We are required to measure certain assets and liabilities at fair value, either upon initial measurement or for subsequent accounting or reporting. We use fair value in the initial measurement of net assets acquired in a business combination and when accounting for and reporting on investment securities and certain financial instruments or assets. We estimate fair value using an exit price approach, which requires, among other things, that we determine the price that would be received to sell an asset or paid to transfer a liability in an orderly market of market participants, considering the highest and best use of assets and, for liabilities, assuming the risk of non-performance will be the same before and after the transfer. Many, but not all, of our financial instruments are carried at fair value. In addition, as required under accounting rules for business combinations, most of the assets acquired and liabilities assumed from Avigen on December 18, 2009 have been recorded at their estimated fair values as of the acquisition date. For additional information on the valuation approach to determine fair value, including a description of the inputs used, see Long Lived Assets below and Notes to Consolidated Financial Statements – Note 2. Avigen Transaction. Also, for information on fair value for our financial instruments, see Notes to Consolidated Financial Statements – Note 3. Fair Value Measurements – Other Than Intangibles and Goodwill.
91
Table of Contents
MEDICINOVA, INC.
(a development stage company)
Notes to Consolidated Financial Statements
The following table presents our financial instruments measured at fair value on a recurring basis classified by the fair value measurements and disclosures valuation hierarchy (in thousands):
| As of December 31, 2009 | ||||||||||||
| Fair Value Measurements Using | ||||||||||||
| Total | Level 1 | Level 2 | Level 3 | |||||||||
| Cash and cash equivalents |
$ | 19,242 | $ | 19,242 | $ | — | $ | — | ||||
| Current assets: |
||||||||||||
| Investment securities (ARS) |
$ | 24,255 | $ | — | $ | — | $ | 24,255 | ||||
| ARS Put |
2,557 | — | — | 2,557 | ||||||||
| Total current assets |
$ | 26,812 | $ | — | $ | — | $ | 26,812 | ||||
| Long-term investments: |
||||||||||||
| Investment securities (ARS) |
$ | 2,085 | $ | — | $ | — | $ | 2,085 | ||||
| Total long-term investments |
$ | 2,085 | $ | — | $ | — | $ | 2,085 | ||||
| As of December 31, 2008 | ||||||||||||
| Fair Value Measurements | ||||||||||||
| Total | Level 1 | Level 2 | Level 3 | |||||||||
| Cash and cash equivalents |
$ | 19,297 | $ | 19,297 | $ | — | $ | — | ||||
| Long-term investments: |
||||||||||||
| Investment securities (ARS) |
$ | 24,047 | $ | — | $ | — | $ | 24,047 | ||||
| ARS Put |
5,793 | — | — | 5,793 | ||||||||
| Total long-term investments |
$ | 29,840 | $ | — | $ | — | $ | 29,840 | ||||
The carrying amount of our ARS Loan as of December 31, 2009 and 2008 approximates its fair value due to its short term nature.
The following table presents our financial instruments measured at fair value on a non-recurring basis classified by the fair value measurements and disclosures valuation hierarchy (in thousands):
| As of December 31, 2009 | ||||||||||||
| Fair Value Measurements Using | ||||||||||||
| Total | Level 1 | Level 2 | Level 3 | |||||||||
| Current liabilities: |
||||||||||||
| Escrow holdback |
$ | 1,094 | $ | — | $ | — | $ | 1,094 | ||||
| Total current liability |
$ | 1,094 | $ | — | $ | — | $ | 1,094 | ||||
| Non-current liability: |
||||||||||||
| Convertible notes |
$ | 29,258 | $ | — | $ | — | $ | 29,258 | ||||
| Total non-current liability |
$ | 29,258 | $ | — | $ | — | $ | 29,258 | ||||
There were no financial instruments measured at fair value on a non-recurring basis at December 31, 2008.
92
Table of Contents
MEDICINOVA, INC.
(a development stage company)
Notes to Consolidated Financial Statements
The judgments made in determining an estimate of fair value can materially impact our results of operations.
Property and Equipment
Property and equipment, net, which consists of leasehold improvements, furniture and equipment and software, is stated at cost. Leasehold improvements, furniture and equipment, and software are depreciated using the straight-line method over the estimated useful lives of the related assets. The useful life for furniture, equipment (other than computers) and software is five years, computers is three years and leasehold improvements are amortized over the lesser of the useful life or the term of the lease. Our current lease expires in August 2011. We also lease office space in Tokyo, Japan under a lease that expires in May 2011. Furthermore, pursuant to our acquisition of Avigen we acquired a month-to-month lease for 4,000 square feet of office space in Alameda, California. We vacated the Alameda premises on March 8, 2010 and we were released from our month-to-month lease by the landlord.
Long-Lived Assets and Impairment of Long-Lived Assets
We review long-lived assets, including property and equipment, and other intangible assets, for impairment whenever events or changes in business circumstances indicate that the carrying amount of the assets may not be fully recoverable and we perform impairment testing for goodwill and IPR&D at least annually. When it is determined that impairment has occurred, a charge to operations will be recorded. Impairment on property and equipment or other intangible assets, if any, is assessed using discounted cash flows. Impairment on goodwill is assessed on our overall market capitalization, as we operate as one reporting segment. Impairment on IPR&D is assessed on a fair value cost approach.
The fair value of intangible assets is determined on a level 3 basis in which significant unobservable inputs were utilized primarily using the “income approach,” which starts with a forecast of all the expected future net cash flows, some of which are more certain than others. Some of the more significant estimates and assumptions inherent in the intangible asset impairment estimation process include: the amount and timing of projected future cash flows; the discount rate selected to measure the risks inherent in the future cash flows; and the assessment of the asset’s life cycle and the competitive trends impacting the asset, including consideration of any technical, legal, regulatory or economic barriers to entry, as well as expected changes in standards of practice for indications addressed by the asset.
Revenue Recognition
We recognized no revenues for each of the years in the three-year period ended December 31, 2009.
Research and Development
Research and development expenses consist of costs incurred to further our research and development activities and include salaries and related employee benefits, costs associated with clinical trials, costs associated with non-clinical activities such as toxicology testing, regulatory activities, research-related overhead expenses, and fees paid to external service providers who conduct certain research and development activities on our behalf. We use external service providers and vendors to conduct clinical trials, to manufacture product candidates to be used in clinical trials and to provide various other products and services related to our product development programs. Research and development expenses also include fees for licensed technology for which technological feasibility has not been established and there are no alternative uses. Research and development costs are expensed as incurred or accrued based on certain contractual factors such as for estimates of work performed, milestones achieved, patient enrollment and experience with similar contracts. As actual costs become known, accruals are adjusted. To date, our estimates have not differed significantly from the actual costs incurred.
93
Table of Contents
MEDICINOVA, INC.
(a development stage company)
Notes to Consolidated Financial Statements
Income Taxes
In accordance with the authoritative guidance for income taxes under ASC 740 (formerly SFAS No. 109), a deferred tax asset or liability is determined based on the difference between the financial statement and the tax basis of assets and liabilities as measured by the enacted tax rates, which will be in effect when these differences reverse. We provide a valuation allowance against net deferred tax assets unless, based upon the available evidence, it is more likely than not that the deferred tax assets will be realized.
Effective January 1, 2007, we adopted the authoritative guidance on accounting for uncertainty in income taxes, which prescribes a comprehensive model for how we should recognize, measure, present and disclose in our financial statements for uncertain tax positions that we have taken or expect to take on a tax return. The cumulative effect of adopting the guidance on accounting for uncertainty in income taxes resulted in no adjustment to retained earnings as of January 1, 2007.
Our practice is to recognize interest and/or penalties related to income tax matters in income tax expense. We had no accrued interest or penalties since implementation of guidance on accounting for uncertainty in income taxes.
We are subject to taxation in the United States, California and foreign jurisdictions, of which currently no years are under examination. Our tax years for 2000 and forward are subject to examination by the U.S. and state tax authorities due to the carryforward of unutilized net operating losses and research and development credits. At December 31, 2009, income taxes relate to service income earned by our Japanese subsidiary, MediciNova Japan, Inc.
Stock-Based Compensation
We grant stock options to our employees, directors and consultants under the MediciNova, Inc. Amended and Restated 2004 Stock Incentive Plan (the “2004 Plan”), the successor to the MediciNova, Inc. 2000 General Stock Incentive Plan (the “2000 Plan”). No additional stock options have been or will be issued under the 2000 Plan subsequent to our IPO. Stock options issued to non-employees were recorded at their fair value as determined in accordance with the authoritative guidance for equity under ASC 505 (formerly EITF Issue No. 96-18.)
The exercise price of stock options granted during the years ended December 31, 2009, 2008 and 2007 were equal to market value on the date of grant. During the years ended December 31, 2009, 2008 and 2007, options to purchase 521,373, 615,540 and 151,000 shares of common stock, respectively, were granted and stock-based compensation expense for such stock options is reflected in operating results during fiscal years 2009, 2008 and 2007. The estimated fair value of each stock option award was determined on the date of grant using the Black-Scholes option valuation model with the following weighted-average assumptions for stock option grants:
| Year Ended December 31, |
||||||
| 2009 | 2008 | |||||
| Risk-free interest rate |
1.79 | % | 3.00 | % | ||
| Expected volatility of common stock |
70.00 | % | 69.00 | % | ||
| Dividend yield |
0.00 | % | 0.00 | % | ||
| Expected option term (in years) |
4.13 | 4.00 | ||||
The risk-free interest rate assumption is based upon observed interest rates appropriate for the expected term of our employee stock options. The expected volatility is based on the weighted average volatility of our stock price, the volatility of stock prices of certain peers within our industry sector and management’s judgment. We
94
Table of Contents
MEDICINOVA, INC.
(a development stage company)
Notes to Consolidated Financial Statements
have not paid any dividends on common stock since our inception and do not anticipate paying dividends on our common stock in the foreseeable future. The expected term of employee stock options is based on the simplified method for “plain vanilla options” as provided by the authoritative guidance on stock compensation, as we concluded that our historical stock option exercise experience does not provide a reasonable basis for us to estimate the expected term.
As stock-based compensation expense recognized in the accompanying consolidated statement of operations for the years ended December 31, 2009, 2008 and 2007 were based on awards ultimately expected to vest, such expense should be reduced for estimated forfeitures. The authoritative guidance for compensation under ASC 718 (formerly SFAS No. 123R ) requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. We have very few employees and our stock options vest monthly; therefore, we did not estimate any forfeitures in 2009, and we will adjust our stock-based compensation expense should any forfeitures occur. Our determination of fair value is affected by our stock price, as well as a number of assumptions that require judgment. The weighted-average fair value of each stock option granted during the years ended December 31, 2009, 2008 and 2007, estimated as of the grant date using the Black-Scholes option valuation model, was $1.53 per option, $2.37 per option and $5.27 per option, respectively.
For the years ended December 31, 2009, 2008 and 2007, stock-based compensation expense related to stock options was $2.4 million, $3.2 million and $3.9 million, respectively, and was recorded as a component of general and administrative expense ($1.9 million, $1.8 million and $3.0 million, respectively) and research and development expense ($0.5 million, $1.4 million and $0.9 million, respectively). There were 100,483 stock options exercised during the year ended December 31, 2009, from which proceeds of $0.4 million were received. No stock options were exercised during the years ended December 31, 2008 and 2007.
As of December 31, 2009, there was $2.3 million of unamortized compensation cost related to unvested stock option awards, which is expected to be recognized over a remaining weighted-average vesting period of 1.4 years, on a straight-line basis.
Comprehensive Income (Loss)
The authoritative guidance for comprehensive income under ASC 220 (formerly SFAS No. 130) requires that all components of comprehensive income (loss), including net income (loss), be reported in the financial statements in the period in which they are recognized. Comprehensive income (loss) is defined as the change in equity (net assets) during a period from transactions and other events and circumstances from non-owner sources. Net income (loss) and other comprehensive income (loss), including foreign currency translation adjustments and unrealized gains and losses on investments, are reported, net of their related tax effect, to arrive at comprehensive income (loss). Our comprehensive loss includes unrealized losses on marketable securities and currency translation. The table below sets forth the components of our accumulated other comprehensive loss at:
| December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| Beginning Balance |
$ | (29,744 | ) | $ | (131,466 | ) | $ | (49,205 | ) | |||
| Currency translation |
(35,170 | ) | 101,722 | 6,757 | ||||||||
| Unrealized loss on marketable securities |
— | — | (89,018 | ) | ||||||||
| Ending Balance |
$ | (64,914 | ) | $ | (29,744 | ) | $ | (131,466 | ) | |||
95
Table of Contents
MEDICINOVA, INC.
(a development stage company)
Notes to Consolidated Financial Statements
As of December 31, 2009, 2008 and 2007, our comprehensive loss was $20,404,060, $21,823,107 and $48,985,505, respectively.
Net Loss Per Share
Net loss per share is presented as basic and diluted net loss per share. Basic net loss per share is calculated by dividing the net loss by the weighted average number of common shares outstanding for the period, without consideration for common stock equivalents. Diluted net loss per share is computed by dividing the net loss attributable to common stockholders by the weighted average number of common share equivalents outstanding for the period determined using the treasury-stock method. For purposes of this calculation, convertible preferred stock, stock options and warrants are considered to be common stock equivalents and are only included in the calculation of diluted net loss per share when their effect is dilutive. For the year ended December 31, 2009, there were 4,821,330 potentially dilutive securities excluded from determining diluted earnings per share because of their anti-dilutive effect, of which 4,330,300 potentially dilutive securities were based on the assumption that all of the convertible notes issued pursuant to the Avigen merger were converted at the closing date. There were no potentially dilutive securities for the years ended December 31, 2008 and 2007.
New Accounting Standards Not Yet Adopted
In October 2009, the FASB ratified Accounting Standards Update, or ASU, 2010-13, which eliminates the residual method of allocation and the requirement to use the relative selling price method when allocating revenue in a multiple deliverable arrangement. When applying the relative selling price method, the selling price for each deliverable shall be determined using vendor specific objective evidence of selling price, if it exists, otherwise third-party evidence of selling price. If neither vendor specific objective evidence nor third-party evidence of selling price exists for a deliverable, companies shall use its best estimate of the selling price for that deliverable when applying the relative selling price method. ASU 2010-13 shall be effective in fiscal years beginning on or after June 15, 2010, with earlier application permitted. Companies may elect to adopt this guidance prospectively for all revenue arrangements entered into or materially modified after the date of adoption, or retrospectively for all periods presented. We do not believe the adoption of this accounting standard will have a material effect on our consolidated results of operations or financial condition.
In March 2010, the FASB issued ASU No. 2010-11, “Derivatives and Hedging (Topic 815): Scope Exception Related to Embedded Credit Derivatives”. The FASB believes this ASU clarifies the type of embedded credit derivative that is exempt from embedded derivative bifurcation requirements. Specifically, only one form of embedded credit derivative qualifies for the exemption—one that is related only to the subordination of one financial instrument to another. As a result, entities that have contracts containing an embedded credit derivative feature in a form other than such subordination may need to separately account for the embedded credit derivative feature. The amendments in the ASU are effective for each reporting entity at the beginning of its first fiscal quarter beginning after June 15, 2010. Early adoption is permitted at the beginning of each entity’s first fiscal quarter beginning after March 5, 2010. We do not believe the adoption of this accounting standard will have a material effect on our consolidated results of operations or financial condition.
2. Avigen Transaction
On December 18, 2009 we acquired 100% of the outstanding shares of Avigen, a biopharmaceutical company that had focused on identifying and developing differentiated products to treat patients with serious disorders, whose potential product candidate is AV411, a glial attenuator and ibudalist small molecule
96
Table of Contents
MEDICINOVA, INC.
(a development stage company)
Notes to Consolidated Financial Statements
therapeutic, for CNS disorders. The primary reasons for the Avigen acquisition were to combine the ibudalist development programs each company was respectively pursuing, to utilize the preclinical and clinical data for AV411 as support for the development pathway of MN-166 resulting in cost savings for us, and to capture a potential financing opportunity given Avigen’s cash balance prior to the Merger.
The aggregate Merger consideration consisted of a First Payment Consideration of $35.4 million of which $3.0 million was funded in cash by us and $32.4 million was funded in cash by Avigen, and a reduced Second Payment Consideration of $1.1 million of which $0.6 million was funded in cash by Avigen and $0.5 million is to be funded upon the release of the restricted letter of credit, which is recorded as such in our consolidated balance sheet, by the letter of credit’s beneficiary. The cash payments were deposited in a separate trust account and are considered restricted cash by us. Of the 29,852,115 shares of Avigen common stock outstanding at the closing date, approximately 17% of Avigen shareholders elected to receive cash. Thereby, the First Payment Consideration was reduced by the number of shareholders who elected to receive cash, or $6.0 million, resulting in $29.4 million of Convertible Notes at face value to be issued by us. The $1.1 million Second Payment Consideration acts as an escrow holdback and is neither paid out in cash to the 17% of Avigen shareholders who elected cash nor issued as Convertible Notes by us until the respective holdback period lapses on June 30, 2010 and November 30, 2010 for the restricted letter of credit. The Convertible Notes can be converted into shares of our common stock at a conversion price of $6.80 per share. At the date of closing, our closing stock price was $7.99, resulting in a beneficial conversion feature on the Convertible Notes issued pursuant to the First Payment Consideration and the Convertible Notes to be issued pursuant to the Second Payment Consideration. In addition to the First and Second Payment Considerations, the Merger agreement includes a Contingent Payment Rights Agreement, or CPR Agreement, between us, Avigen and American Stock Transfer & Trust Company, LLC, as rights agent. The CPR Agreement sets forth the rights that former Avigen stockholders will have with respect to each CPR held after the closing of the Merger. The CPR Agreement provides for the payment of the following amounts on a pro rata basis:
| • | if the first milestone payment under Avigen’s agreement with Genzyme, or the Genzyme Agreement, is received before August 18, 2011, $6,000,000 or such lesser cash amount paid by Genzyme; |
| • | if the first milestone payment has not occurred and the Parkinson’s Product, as defined in the Genzyme Agreement, is sold or otherwise disposed of by us before August 18, 2011, 50 percent of the net proceeds of such sale or disposition received before August 18, 2011; and |
| • | if the trust established pursuant to Avigen’s Management Transition Plan, or Avigen’s MTP, is terminated, the amount remaining in such trust upon termination (less any payments required to be made under Avigen’s Management Transition Plan Trust Agreement), such amount currently estimated at $550,000. |
With respect to the first two contingent payment rights described above, we have not ascribed any value to them as we have deemed them not probable and we cannot determine when, or if, the related milestones will be achieved or the events triggering the commencement of payment obligations will occur. With respect to the contingent payment rights related to Avigen’s MTP, as none of the assets will revert to us, we have recorded a restricted investment and a corresponding liability in our consolidated balance sheet.
We have included Avigen’s business operations in our consolidated financial statements since the acquisition date and we have accounted for the Merger under the acquisition method of accounting. Included in our consolidated statement of operations is approximately $4,000 of operating expenses since the acquisition date of December 18, 2009. Acquisition method of accounting requires that assets acquired and liabilities assumed are
97
Table of Contents
MEDICINOVA, INC.
(a development stage company)
Notes to Consolidated Financial Statements
recognized at their fair values as of the acquisition date, that the fair value of acquired in-process research and development (IPR&D) is recorded on the balance sheet, all transaction costs are expensed as incurred and any excess of the purchase price over the assigned values of net assets acquired is recorded as goodwill. In addition, Avigen’s historical stockholder’s equity accounts were eliminated.
For the year ended December 31, 2009, we expensed $1.8 million of transaction costs as they were incurred. The estimated fair value of the aggregate Merger consideration (“Purchase Price”) was as follows (table in thousands):
| First Payment Consideration (Convertible Notes issued by us) |
$ | 29,258 | |
| Second Payment Consideration (Escrow Holdback) |
1,094 | ||
| Cash paid by us |
3,000 | ||
| Conversion Feature related to First Payment Convertible Notes |
9,227 | ||
| Conversion Feature related to Second Payment Convertible Notes |
286 | ||
| Total Purchase Price |
$ | 42,865 | |
The fair value of the First Payment Consideration and Second Payment Consideration and the related fair value of their respective beneficial conversion feature, was based on a binomial option pricing model (“BOPM”). Assumptions used in the BOPM included the maturity date of the Convertible Notes, time between nodes, volatility, face value of the Convertible Notes at the closing date and the risk-free rate. The maturity date utilized was 1.5 years based on the maturity of the notes in June 2011. As our projected period was 1.5 years, we used the average of the one and two year U.S. Treasury bonds as of the closing date and we based volatility on the historical volatility of publicly-traded comparable companies to Avigen and our stock price volatility. To calculate the fair value of the Convertible Notes and their respective beneficial conversion feature under the BOPM we first had to generate a price tree, which is produced by working forward from the date of closing to the Convertible Notes maturity date. At each step it is assumed that the Convertible Notes will move up or down by a specific factor of volatility. In the second step of the BOPM we had to determine the option value at each final node, which is the intrinsic or exercise value. The intrinsic value is calculated by subtracting the conversion price, or $6.80 per share, from the expected stock price as determine in the aforementioned step. The third step of the BOPM was to calculate option value at each node, starting at the end node, working back to the first node of the price tree, where the result would be the value of the option, discounted by the risk-free rate. In the last step of the BOPM we determined the fair value of the Convertible Notes without the conversion feature. To calculate the value of the Convertible Notes without the conversion feature, we multiplied the expected payments from the Convertible Notes by a discount factor, that discount factor being one divided by one plus the discount rate raised to the power of time. We then applied to the result a lack of marketability discount for the conversion feature using a protective put model to account for the lower degree of liquidity which would detract from the face value of the Convertible Notes.
The First Payment Consideration was recorded on our consolidated balance sheet as Convertible Notes at its fair value of $29.3 million. The $0.2 million difference between fair value and face value will be accreted to interest expense through the Convertible Note period. At acquisition-date, following ASC 805, the fair value of the conversion feature was accounted for within equity and will not be re-measured during interim periods and subsequent settlements (conversions to our stock) will be accounted for in equity.
The Second Payment Consideration was recorded on our consolidated balance sheet as an Escrow Holdback at its fair value of $1.1 million. At acquisition-date, although this contingent consideration was recorded as a liability following ASC 805, the fair value of the conversion feature was accounted for within equity and will not
98
Table of Contents
MEDICINOVA, INC.
(a development stage company)
Notes to Consolidated Financial Statements
be re-measured during interim periods and subsequent settlements for those who elected Convertible Notes (conversions to our stock) will be accounted for in equity.
Based on a third-party valuation, as of the date of closing, amounts of estimated fair value of assets acquired and liabilities assumed at the acquisition date were as follows (table in thousands):
| Cash and cash equivalents |
$ | 628 | ||
| Restricted cash |
30,046 | |||
| Restricted investment |
676 | |||
| Restricted letter of credit |
500 | |||
| Identifiable intangible assets |
4,800 | |||
| Accrued interest |
2 | |||
| Prepaid expenses |
35 | |||
| Deferred tax liability |
(1,956 | ) | ||
| MTP liability |
(676 | ) | ||
| Accounts payable |
(236 | ) | ||
| Accrued compensation |
(96 | ) | ||
| Identifiable net assets acquired and liabilities assumed |
33,723 | |||
| Goodwill |
9,142 | |||
| Total purchase price |
$ | 42,865 | ||
The carrying value of all assets acquired, except for identifiable intangible assets discussed below, and all liabilities assumed approximates fair value.
Identifiable intangible assets. Identifiable intangible assets acquired have been attributed as follows: (table in thousands):
| IPR&D |
$ | 4,800 | |
| Genzyme Agreement |
— | ||
| Total |
$ | 4,800 | |
IPR&D. The fair value attributed to IPR&D represents an estimate of fair value of in-process technology related to Avigen’s AV411 program, which at the Merger closing date, had not received U.S. Food and Drug Administration (“FDA”) approval for any indication. As such, pursuant to ASC 805, amortization of the IPR&D will not occur until it reaches market feasibility. Although we plan to integrate the two ibudalist-based development programs (our MN-166 and the acquired AV411) and pursue discussions with potential partners to secure a strategic collaboration to advance clinical development of the combined development programs, the fair value for the AV411 IPR&D was determined using the income approach, although the cost and market approaches were also reviewed. Under the income approach we used a multi-period excess earning method in which the forecast of all expected future cash flows was predicated on a collaboration partner structure in which revenue streams were generated in the short-term by milestone payments and royalty payments in the long-term. As several significant milestones need to be achieved prior to expected commercialization, a probability adjustment was applied to the forecasted revenue to account for the risk associated with being able to successfully commercialize. We also applied a discount rate on the overall valuation based on the industry composite weighted average cost of capital to account for the perceived risk of the technology with respect to
99
Table of Contents
MEDICINOVA, INC.
(a development stage company)
Notes to Consolidated Financial Statements
successful commercialization, market acceptance and growth and profitability. To validate the reasonableness of the IPR&D fair value under the income approach, we also valued the technology under the cost and market approaches. Under the cost approach we estimated the cost to re-create the technology’s preclinical and clinical data package, which this cost was considered a savings benefit by us and was part of our rationale for doing the Merger. Under the market approach we considered the formal and informal bids that Avigen received while it marketed its AV411 program for sale. After reviewing the results derived from all three approaches, we concluded that the income approach was a reasonable basis to fair value IPR&D.
Genzyme Agreement. In the event the first milestone is not reached and we can dispose of the respective Parkinson’s product or FDA approval is received on the respective Parkison’s product, then, the Genzyme Agreement could potentially have value. At the date of closing, however, we are unable to estimate the likelihood that we will be able to sell or dispose of our rights under the Genzyme Agreement and we are unable to estimate the likelihood of the respective Parkinson’s product receiving FDA approval. Because we cannot determine the probability of selling or disposing of the Parkinson’s product and we are unable to determine the probability that the Parkinson’s product will receive FDA approval, we have not ascribed any value to this contingent asset at the acquisition date as its fair value cannot be reasonably estimated.
Goodwill. The authoritative guidance for business combinations requires that contingent consideration be recognized at acquisition-date fair value as part of the consideration transferred. As such, as stated above, we included in the purchase price the fair value of the aggregate Merger consideration, which included both the Convertible Notes associated with the First and Second Payment Considerations, the cash paid by us and the beneficial conversion feature on the Convertible Notes. The goodwill is primarily a direct result of the fair value of the beneficial conversion feature of the Convertible Notes. We were willing to set the conversion price of the Convertible Notes issued and to be issued at $6.80 per share, which at acquisition-date was less than our closing stock price, as we viewed the Merger as a financing opportunity given the cash balance held by Avigen prior to the Merger. We also believe that the cost for a development stage company to raise $30 million in today’s economic environment exceeds the goodwill recorded on our books. To-date, we have raised approximately $1.7 million as a result of the conversions that have taken place in January and February 2010.
We tested goodwill for impairment at December 31, 2009, utilizing a market based approach in which our total market capitalization was significantly higher than our goodwill carrying value; thus, noting, no impairment. We also tested IPR&D for impairment at December 31, 2009, utilizing a cost approach in which the total cost to re-create the technologies preclinical and clinical data package was significantly higher than our IPR&D carrying value; thus, noting no impairment.
The accompanying consolidated statement of operations for the year ended December 31, 2009, includes the operations of Avigen from the date of acquisition. Assuming the acquisition of Avigen had occurred January 1, 2009 and 2008, the pro forma unaudited condensed results of operations would have been as follows (in thousands, except per share amounts):
| Year Ending December 31, |
||||||||
| 2009 | 2008 | |||||||
| Revenues |
$ | 144 | $ | 7,100 | ||||
| Operating Expenses |
$ | (31,917 | ) | $ | (50,191 | ) | ||
| Net Loss |
$ | (29,978 | ) | $ | (47,024 | ) | ||
| Basic and diluted net loss per common share. |
$ | (1.82 | ) | $ | (2.86 | ) | ||
100
Table of Contents
MEDICINOVA, INC.
(a development stage company)
Notes to Consolidated Financial Statements
The above proforma unaudited results of operations do not include proforma adjustments relating to costs of integration or post-integration cost reductions that may be incurred or realized by us in excess of actual amounts incurred or realized through December 31, 2009.
3. Fair Value Measurements – Other Than Intangibles and Goodwill
As defined in the authoritative guidance for fair value measurements and disclosures under ASC 820 (formerly SFAS No. 157), fair value is based on the price that would be received to sell an asset or would be paid to transfer a liability in an orderly transaction between market participants at the measurement date. To increase the comparability and consistency of fair value measurements, ASC 820 prescribes a fair value hierarchy that prioritizes observable and unobservable inputs used to measure fair value into three broad levels which are described below:
| Level 1: | Inputs are quoted prices (unadjusted) in active markets for identical assets or liabilities at the measurement date. | |
| Level 2: | Inputs are quoted prices for similar items in active markets or inputs are quoted prices for identical or similar items in markets that are not active. | |
| Level 3: | Inputs are unobservable due to little or no market data availability and inputs are usually developed by management or a third-party which reflect those inputs that a market participant would use. The fair value hierarchy gives the lowest priority to Level 3 inputs. | |
At December 31, 2009, cash and cash equivalents (instruments with maturities of three months or less at the date of purchase) were $19.2 million and primarily invested in money market accounts. At December 31, 2009, restricted cash and restricted investments were $30.7 million and primarily invested in money market funds. We measure our cash equivalents, restricted cash and restricted investments on a recurring basis. The fair value of our cash equivalents, which are current assets, is based on Level 1 criteria in which their carrying amount is a reasonable estimate of their fair value based on daily quoted market prices.
At December 31, 2009, we held investment securities-current of $24.3 million consisting of Auction Rate Securities (“ARS”), all of which had AAA ratings at the time of purchase, that principally represent interests in government-guaranteed student loans and we held an ARS Put (as defined below) in the amount of $2.6 million. In August 2008, UBS AG and its affiliates (“UBS”), the brokerage firm through which we purchased the majority of our ARS, entered into a settlement with the Securities and Exchange Commission (“SEC”), the New York Attorney General and other state agencies. Pursuant to the settlement, UBS issued to us Auction Rate Security Rights, which would allow us to sell to UBS our ARS held in accounts with UBS (“ARS Rights Offer”). As part of the ARS Rights Offer, we received the right to sell to UBS our ARS held in accounts with UBS at par value any time during the period beginning June 30, 2010 and ending July 2, 2012 (“ARS Put”). As part of the settlement, UBS also offered to us a no net cost loan program, whereby we would be able to borrow up to 75 percent of the market value, as determined by UBS at its sole discretion, of our ARS that have been pledged as collateral at an interest cost that would not exceed the interest being paid on the underlying ARS investments (“ARS Loan”). Under the terms of the ARS Loan, UBS may demand full or partial payment of the ARS Loan, at its sole option and without cause, at any time. If at any time UBS exercises its right to terminate the credit line agreement governing the ARS Loan, then UBS is required to provide, as soon as reasonably possible, alternative financing on substantially the same terms and conditions as those under the credit line agreement and the agreement will remain in full force and effect until such time as such alternative financing has been established. In January 2009, we were approved for the ARS Loan in the amount of $15.9 million and drew down the entire preapproved amount. In addition, in February 2009, we
101
Table of Contents
MEDICINOVA, INC.
(a development stage company)
Notes to Consolidated Financial Statements
borrowed an additional $2.2 million under the ARS Loan, bringing the total amount outstanding under the ARS Loan to $18.1 million, following UBS’ decision to increase our availability under the ARS Loan. All cash received under the ARS Loan was invested in money market accounts. At June 30, 2009, the ARS associated with the ARS Rights Offer and the ARS Put were reclassified out of long-term assets to current assets due to the time frame in which they can be readily converted to cash.
At December 31, 2009, the carrying cost of the ARS Loan, which approximates its fair value due to its short-term nature, was $17.6 million. For the three months and year ended December 31, 2009, $50,000 and $350,000, respectively, of our current investment securities were redeemed at par value, with the proceeds being used to pay down the outstanding balance of the ARS Loan.
At December 31, 2009, we held long-term investments of $2.1 million which consisted of ARS that principally represent interests of government-guaranteed student loan securities, insurance notes and portfolios of securities (primarily commercial paper).
At December 31, 2009, our total ARS portfolio (both current and long-term) totaled $26.3 million at fair value ($29.6 million at par value), of which $1.8 million at fair value ($2.2 million at par value) consisted of private placement securities. None of the underlying collateral of our ARS portfolio consisted of subprime mortgages or collateralized debt obligations. Our ARS were designated as trading investment securities at December 31, 2008. We measure all of our ARS and the ARS Put on a recurring basis based on Level 3 criteria because neither an active primary nor active secondary market exists for these securities. The table below reconciles fair value of our ARS trading investment securities and the ARS Put at December 31, 2008 with fair value at December 31, 2009, as determined by Level 3 (unobservable) inputs:
| Fair Value at 12/31/08 |
Transfers in/ (out) of Level 3 1/1/09-12/31/09 |
Transfers in/(out) of Long-term to Current 1/1/09-12/31/09 |
Sales/ Redemptions 1/1/09- 12/31/09 |
Impairment Charge at 12/31/09 |
Gain at 12/31/09 |
Fair Value at 12/31/09 | ||||||||||||||||||
| Investment securities(1) |
$ | 21,055,569 | $ | — | $ | (21,055,569 | ) | $ | — | $ | — | $ | — | $ | — | |||||||||
| Investment rate securities(2) |
2,991,745 | — | — | (902,846 | ) | (3,474 | ) | — | 2,085,425 | |||||||||||||||
| Total long-term investments |
$ | 24,047,314 | $ | — | $ | (21,055,569 | ) | $ | (902,846 | ) | $ | (3,474 | ) | $ | — | $ | 2,085,425 | |||||||
| Long-term asset, ARS Put(3) |
$ | 5,792,701 | $ | — | $ | (5,792,701 | ) | $ | — | $ | — | $ | — | — | ||||||||||
| Investment securities-current(1) |
$ | — | $ | — | $ | 21,055,569 | $ | (350,000 | ) | $ | — | $ | 3,549,418 | $ | 24,254,987 | |||||||||
| ARS Put-current(3) |
$ | — | $ | — | $ | 5,792,701 | $ | — | $ | (3,235,694 | ) | $ | — | $ | 2,557,007 | |||||||||
| (1) | Aggregated fair value reported at December 31, 2009 reflects fair value as determined by our discounted cash flow model with liquidity discounts, pursuant to which we took into consideration the brokerage firm’s pricing model, the tax status (taxable vs. tax exempt) of the security, credit quality of the issuer, assumed maturity (five years), insurance wraps and the portfolio composition. We also made assumptions regarding future cash flows and the likelihood of the ARS being redeemed or refinanced. In addition, we performed a sensitivity analysis by calculating fair value with a maturity of one year through ten years. The annual coupon rate utilized was set at the U.S. Treasury Department published average of the bond equivalent rates |
102
Table of Contents
MEDICINOVA, INC.
(a development stage company)
Notes to Consolidated Financial Statements
| of the 91-day Treasury bills auctioned during the quarter ending December 31, 2009 (which was the Federal Family Education Loan Program special allowance rate for the quarter ending December 31, 2009) plus 120 basis points. We believe that using this interest rate is reasonable given that a majority of our ARS portfolio is collateralized by student loans guaranteed by the U.S. government under the Federal Family Education Loan Program. Using our discounted cash flow model with liquidity discounts ranging from 2% to 23%, we calculated aggregate fair value for these securities, which ranged between $25.8 million with a two-year maturity, $23.3 million with a seven-year maturity and $21.8 million with a ten-year maturity. As of December 31, 2009, these ARS continued to pay interest according to their stated interest terms, and we received a partial redemption at par value of $350,000 on one of the securities in this portfolio. In addition, as these investment securities are trading securities, the increase of approximately $3.5 million in the overall fair value of the ARS was a recorded as a gain, of which approximately $2.7 million of the gain was recorded in the fourth quarter of 2009, in our consolidated statement of operations and was primarily due to the change in the assumed maturity from seven years to five years. We believe the change in maturity from seven years to five years to be reasonable after discussing with certain financial advisors the outlook of the ARS market. Pursuant to the ARS Rights Offer, the earliest date that we can redeem these investment securities at par is June 30, 2010; therefore, at June 30, 2009, we reclassified these investment securities out of long-term assets and into current assets in our consolidated balance sheets. |
| (2) | Aggregated fair value reported at December 31, 2009 reflects fair value as determined by our discounted cash flow model, which employed liquidity discounts ranging from 3% to 25% depending on the security type and included assumptions regarding future cash flows and the likelihood of the redemption or refinancing of such ARS. For the student loan ARS we changed assumed maturity from seven years to five years and for the private placement ARS assumed a maturity remained at seven years. We believe the assumed maturities we utilized to be reasonable after discussing with certain financial advisors the outlook of the ARS market. We also performed a sensitivity analysis by calculating fair value with a maturity of one year through ten years. The interest rate utilized in the model was either the London Interbank Offered Rate (“LIBOR”) plus the spread, as indicated in the respective security prospectus which was generally 200 basis points, or the U.S. Treasury Department published average of the bond equivalent rates of the 91-day Treasury bills auctioned during the quarter ending December 31, 2009 (which was the Federal Family Education Loan Program special allowance rate for the quarter ending December 31, 2009) plus 120 basis points for the ARS collateralized by student loans. The LIBOR rate was per bankrate.com, which we deemed as a reasonable source given it is a widely utilized third-party rate source. We believe that utilizing the Federal Family Education Loan Program special allowance rate for the student loan ARS is reasonable given the collateral of the ARS is student loans. Using this methodology, we calculated aggregate fair value for these securities, which ranged between $2.5 million with a two-year maturity for all securities, $2.3 million with a five-year maturity for all securities and $2.0 million with a ten-year maturity. As of December 31, 2009, the ARS continue to pay interest according to their stated interest terms. Because these investment securities are trading securities, the approximately $3,000 decrease in fair value was recorded as a loss in our consolidated statement of operations. In addition, because of our expectation as to when we may be required to liquidate these ARS for operating purposes, these securities are classified as long-term investments in our consolidated balance sheets. |
| (3) | We elected to measure the ARS Put under the fair value option of ASC 825, authoritative guidance on financial instruments (formerly SFAS No. 159), to mitigate the volatility in reported earnings due to the linkage of certain of our ARS and the ARS Put. Fair value of the ARS Put, which equaled $2.6 million at December 31, 2009, was also determined through the use of a discounted cash flow valuation model with assumptions being made related to interest rate, maturity and liquidity. We effectively used a liquidity discount of approximately 5%, an interest rate of approximately 5% which took into consideration the brokerage firm’s weighted average cost of capital and a maturity of 12 months. Based on our discounted |
103
Table of Contents
MEDICINOVA, INC.
(a development stage company)
Notes to Consolidated Financial Statements
| cash flow valuation, at December 31, 2009, we recorded an impairment charge of approximately $3.2 million in our consolidated statement of operations, of which approximately $2.8 million was recorded in the fourth quarter of 2009, which minimized the gain we recognized on the linked ARS. In addition, at June 30, 2009, we reclassified the ARS Put out of long-term assets to current assets because it can be exercised within 12 months. |
The portion of trading gains and losses for the year ended December 31, 2009 related to our investment securities classified as trading securities which were still held at December 31, 2009 is as follows (in thousands):
| Net gain recognized on trading securities |
$ | 3,546 | |
| Net loss recognized on trading securities sold |
44 | ||
| Net unrealized gain on trading securities still held |
$3,590 | ||
4. Balance Sheet Details
Property and Equipment
Property and equipment, net, consist of the following:
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| Leasehold improvements |
$ | 498,581 | $ | 498,581 | ||||
| Furniture and equipment |
867,083 | 880,337 | ||||||
| Software |
367,146 | 380,245 | ||||||
| 1,732,810 | 1,759,163 | |||||||
| Less accumulated depreciation and amortization |
(1,579,263 | ) | (1,390,864 | ) | ||||
| $ | 153,547 | $ | 368,299 | |||||
| Depreciation and amortization expense |
$ | 219,202 | $ | 305,018 | ||||
Accrued Expenses
A substantial portion of our ongoing research and development activities are performed under agreements we enter into with external service providers, including clinical research organizations, which conduct many of our research and development activities. A portion of our ongoing general and administrative activities relate to legal, accounting and consulting services. We accrue for costs incurred as the services are being provided by monitoring the status of clinical trials or specific projects or services provided, contractual factors such as milestones or retainer fees and the invoices received from our external service providers. Accrued expenses consist of the following:
| December 31, | ||||||
| 2009 | 2008 | |||||
| Research and development costs |
$ | 864,471 | $ | 740,207 | ||
| Professional services fees |
187,367 | 176,236 | ||||
| Other |
224,198 | 95,473 | ||||
| $ | 1,276,036 | $ | 1,011,916 | |||
104
Table of Contents
MEDICINOVA, INC.
(a development stage company)
Notes to Consolidated Financial Statements
5. Related Party Transactions
There were no related party transactions during the three years ending December 31, 2009.
On May 4, 2007, our board of directors approved the modification of certain stock option grants received by Dr. Iwaki while serving in his consulting capacity as President and Chief Executive Officer as a result of the change in Dr. Iwaki’s status from consultant to employee. Two nonqualified stock option (“NSO”) grants received by Dr. Iwaki for 40,000 shares of common stock and 333,503 shares of common stock, which were granted on January 4, 2006 and November 12, 2006, respectively, were modified such that the NSO grants were cancelled and new grants of incentive stock options equal in number to the prior NSO grants were granted at the prior exercise prices and with the original vesting schedules approved for the cancelled NSO grants. Pursuant to ASC 718, there is no impact to our consolidated financial results related to the modification from nonqualified stock options to incentive stock options as there is no incremental value attributed to the modified awards.
6. Commitments and Contingencies
Facility Lease
In January 2004, we leased 16,609 square feet of space for our corporate headquarters under a non-cancelable operating lease that was set to expire in February 2008. In January 2008, we entered into a third amendment to lease for our corporate headquarters at the same location in which we reduced the amount of space under lease to 12,699 square feet of office space through August 2011. In June 2005, we leased 1,726 square feet of office space in Tokyo, Japan under a non-cancelable operating lease that expires in May 2011. Furthermore, pursuant to our acquisition of Avigen we acquired a month-to-month lease for 4,000 square feet of office space in Alameda, California. We vacated the Alameda premises on March 8, 2010 and, accordingly, we were released from our month-to-month lease by the landlord. Rent expense for the years ended December 31, 2009 and 2008 was $578,493 and $551,234, respectively, and rent expense, net of sub-lease income for the period from September 26, 2000 (inception) to December 31, 2009 was $3,596,442.
Future minimum payments are as follows:
| Years ending December 31: |
|||
| 2010 |
$ | 613,490 | |
| 2011 |
$ | 385,311 | |
| Thereafter |
$ | — | |
| Total minimum payments |
$ | 998,801 | |
License Agreements
We have entered into numerous license agreements to acquire the rights to develop and commercialize a variety of product candidates. Pursuant to these agreements, we have obtained exclusive licenses to the patent rights and know-how for all indications under the agreements within our licensed territories. We generally make an upfront payment and are required to make additional payments upon the achievement of specific development and regulatory approval milestones. We are also obligated to pay royalties under the agreements until the later of the expiration of the applicable patent or the applicable last date of market exclusivity after the first commercial sale, on a country-by-country basis.
105
Table of Contents
MEDICINOVA, INC.
(a development stage company)
Notes to Consolidated Financial Statements
The amounts expended under these agreements and charged to research and development expense during the years ended December 31, 2009, 2008, 2007, and the period from September 26, 2000 (inception) to December 31, 2009 were $0, $100,000, $3,000,000 and $9,850,000, respectively. As of December 31, 2009, future potential milestone payments totaled approximately $94.1 million, and there are no minimum royalties required under any of the license agreements. We are unable at this time to estimate with certainty the timing on when these milestone payments will occur as these payments are dependent upon the progress of our product development programs. From June 19, 2002 (the date of our first license agreement) through December 31, 2009, we have entered into nine license agreements with Japanese and British pharmaceutical companies and a non-profit research institute.
Termination of Phase III Trial for MN-001, Bronchial Asthma
On June 26, 2007, we announced a strategic initiative to focus our resources on the development and commercialization of two prioritized assets in our development pipeline, MN-221 for the treatment of acute exacerbations of asthma and MN-166 for the treatment of multiple sclerosis. As part of this strategy, we terminated the Phase III clinical trial of MN-001 for the treatment of bronchial asthma. At December 31, 2007, the termination of the Phase III clinical trial was completed and our financial results for the year then ended reflect additional research and development expense of $2.1 million (or $0.18 loss per share) to complete the wind-down of this clinical trial.
Legal Proceedings
On August 24, 2009, The Pennsylvania Avenue Funds, an Avigen stockholder, filed a complaint in Alameda County Superior Court alleging that Avigen’s directors breached their fiduciary duties in connection with the proposed transaction with us. On October 15, 2009, The Pennsylvania Avenue Funds filed an amended complaint adding us as a defendant. In the amended complaint, The Pennsylvania Avenue funds alleged, among other things, that we aided and abetted the alleged breach of fiduciary duties by the Avigen directors. Avigen and Pennsylvania Avenue Funds have signed a stipulation of settlement agreement and moved the court for preliminary approval. The Court heard oral argument on the Motion for Preliminary Approval of Settlement and held a case management conference on March 8, 2010, during which the Court raised a few issues regarding the settlement provisions. The parties have addressed those concerns and will appear before the Court on April 6, 2010 for preliminary approval of the settlement and a further case management conference.
On April 30, 2007, a participant in one of our clinical trials filed a lawsuit against us, the clinical investigatory site where the individual participated in the clinical trial and the chief investigator at such clinical investigatory site. The complaint alleged that the plaintiff’s daughter suffered permanent injuries in utero as a result of the plaintiff’s participation in our clinical trial. Our insurance carrier assumed defense of this lawsuit, which was settled on September 27, 2007 with no admission of liability. On October 29, 2007, the court entered an order of dismissal of the claims asserted against us and all other defendants and subsequently entered a final judgment approving the settlement. Settlement of the lawsuit did not have a material adverse effect on our business, financial condition or operating results.
We may become involved in various disputes and legal proceedings which arise in the ordinary course of business. While it is not possible to accurately predict or determine the outcome of these matters, an adverse result in any of these matters may occur which could harm our business. We are currently not a party to any legal proceedings.
106
Table of Contents
MEDICINOVA, INC.
(a development stage company)
Notes to Consolidated Financial Statements
7. Redeemable Convertible Preferred Stock, Convertible Notes and Stockholders’ Equity
Initial Public Offering in Japan
On February 4, 2005, we completed an IPO of 3,000,000 shares of common stock in Japan and received aggregate proceeds of $104,486,895, net of underwriting discounts and commissions and offering expenses. In addition, on March 8, 2005, we closed the sale of an additional 157,300 shares of our common stock pursuant to the partial exercise by our underwriters of an over-allotment option which resulted in aggregate proceeds to us of $5,557,773, net of underwriting discounts and commissions. In connection with our IPO, redeemable convertible and convertible preferred stock outstanding as of February 4, 2005 was automatically converted into 6,678,285 shares of common stock.
Public Offering in the United States
On February 1, 2007, we completed a public offering of 1,000,000 shares of common stock in the United States at a purchase price of $12.00 per share and received aggregate net proceeds of approximately $10,639,600 million, net of underwriting discounts and commissions and offering expenses.
Redeemable Convertible Preferred Stock
On September 2, 2004, we sold 27,667,856 shares of Series C redeemable convertible preferred stock at a purchase price of $1.62 per share for total net proceeds of $43,404,320, net of issuance costs. The Series C preferred stock was sold at a price per share below our IPO price. Accordingly, pursuant to the authoritative guidance for debt under ASC 470 (formerly EITF Issue No. 98-5), we recorded a deemed dividend on the Series C preferred stock of $31,264,677, which is equal to the number of shares of Series C preferred stock sold multiplied by the difference between the estimated fair value of the underlying common stock and the Series C preferred stock conversion price per share. The deemed dividend increased the net loss applicable to common stockholders in the calculation of basic and diluted net loss per common share and was reported as a charge to accumulated deficit and a credit to additional paid-in capital, with no net impact on total stockholders’ equity.
Founders’ Common Stock and Warrants
At inception, we issued a total of 50,000 shares of our common stock to two of our founders who became officers and directors, for proceeds of $50,000. We also granted the two individuals warrants to purchase 50,000 shares of our common stock at an exercise price of $1.00 per share. The warrants contained an anti-dilution clause providing the founders with the right to purchase additional shares of common stock any time there was a dilution event so that they could maintain their original ownership percentage. At December 31, 2003, as a result of the Series A and Series B preferred stock sales, the warrants were adjusted to allow the holders to purchase up to 365,000 shares of common stock. At December 31, 2007, no underlying shares of common stock remained subject to purchase under the terms of these warrants.
From January through May 2004, in conjunction with the sale of Series B preferred stock, the shares of common stock issuable upon exercise of the warrants were adjusted up to 732,300 shares. Based on subsequent financing activities and the price of our IPO, we believe that the estimated fair value of the 732,300 shares exceeded the $1.00 exercise price of the warrants and, as a result, we recorded stock-based compensation in general and administrative expense in the amount of $19,405,950.
107
Table of Contents
MEDICINOVA, INC.
(a development stage company)
Notes to Consolidated Financial Statements
On September 2, 2004, in conjunction with the sale of Series C preferred stock, we and our two founders amended the terms of our warrant agreements. In exchange for relinquishing any future anti-dilution rights, the number of underlying common shares that could be purchased under the terms of the warrants was increased and fixed at 1,285,657, up from 732,300. Since all of the warrants were previously variable, we recorded additional stock-based compensation in general and administrative expense of $14,663,966 based on the estimated value of the underlying common stock on September 2, 2004 for a total of $34,069,916. Since the number of warrants became fixed at September 2, 2004, no additional compensation has been recorded.
Other Warrants
In May 2004, as compensation for fundraising efforts related to the sale of Series B preferred stock, we issued to BioVen Advisory, Inc. a warrant to purchase 50,000 shares of common stock with an exercise price of $10.00 per share and these warrants expired May 2009. The warrant was valued at the $250,000 cash value of the services performed. The warrant issuance had no net impact on the consolidated financial statements because the transaction resulted in both a charge and a credit to additional paid-in capital.
Stock Options
We grant options to our employees, directors and consultants under the 2004 Plan, the successor to the 2000 Plan.
2000 General Stock Incentive Plan
In September 2000, we adopted the 2000 Plan under which incentive stock options could be granted to our employees and nonstatutory stock options and other stock-based awards could be granted to employees, directors and consultants. Stock options have been granted with an exercise price of $10.00 per share and vest 25% after the first year of service from the grant date, with the remaining shares vesting in equal monthly installments over the subsequent 36 months of service. An employee may exercise stock options prior to vesting in which case we have the right to repurchase the unvested shares at the original exercise price if the employee is terminated before vesting in all shares occurs.
Following the vesting period, options are exercisable until the earlier of 90 days after the employee’s termination with us or the ten-year anniversary of the initial grant, subject to adjustment under certain conditions. We have the right to purchase all of those shares that the employees have or will acquire under these stock options. The purchase price for any vested shares repurchased will be the greater of the fair market value of such shares on the date of purchase or the aggregate exercise price for such shares.
At December 31, 2009, stock options to purchase a total of 37,500 shares of common stock were outstanding under the 2000 Plan at a weighted average exercise price of $10.00 per share. No additional stock options have been or will be issued under the 2000 Plan subsequent to our IPO. However, stock options previously granted under the 2000 Plan will remain outstanding until the earlier of expiration or exercise.
2004 Stock Incentive Plan
In connection with our IPO, we adopted the 2004 Plan, which serves as the successor program to the 2000 Plan. The 2004 Plan became effective upon the completion of our IPO in February 2005 and was amended and restated in February 2007.
108
Table of Contents
MEDICINOVA, INC.
(a development stage company)
Notes to Consolidated Financial Statements
The 2004 Plan is administered by the compensation committee of our board of directors and provides for the grant of (i) options to purchase shares of common stock; (ii) restricted stock; (iii) stock appreciation rights; and (iv) stock units. Incentive stock options may only be granted to employees. Nonstatutory stock options and other stock-based awards may be granted to employees, non-employee directors and consultants.
The number of shares reserved for issuance under the 2004 Plan will be increased on the first day of each of our fiscal years from 2006 through 2014, with the first such increase occurring on January 1, 2006, by the lesser of: (i) 100,000 shares; (ii) 3% of our outstanding common stock on the last day of the immediately preceding fiscal year; or (iii) the number of shares determined by our board of directors. In addition, in February 2007 and June 2008, the total number of shares available for grant under the 2004 Plan was increased by 300,000 and 1,000,000, respectively.
Options granted to optionees other than non-employee directors will generally vest monthly over a four-year period, beginning on the vesting commencement date. The exercise price of an incentive stock option shall not be less than 100% of the fair market value at the time of grant and the exercise price of a nonstatutory stock option shall not be less than 85% of the fair market value at the time of grant.
Fully vested automatic grants of nonstatutory stock options will be made to non-employee directors in an initial amount of 1,000 shares upon first becoming a member of our board of directors. Immediately after each of our regularly scheduled annual meetings of stockholders, each non-employee director will be automatically granted a nonstatutory option to purchase 1,000 shares of our common stock, at 100% of the fair market value at the time of grant, provided that the director has served on our board for at least six months. Each annual option will be fully vested and exercisable on the date which is six months after the date of grant.
The 2004 Plan terminates ten years after its initial adoption by the board of directors, unless terminated earlier by the board of directors. The board of directors may amend or terminate the plan at any time, subject to stockholder approval where required by applicable law.
A summary of our stock option activity and related information as of December 31, 2009 is as follows:
| Number of Option Shares |
Weighted Average Exercise Price | |||||
| Outstanding at January 1, 2009 |
2,579,511 | $ | 10.59 | |||
| Granted |
521,373 | $ | 2.77 | |||
| Exercised |
(100,483 | ) | $ | 4.05 | ||
| Cancelled |
(944,825 | ) | $ | 11.42 | ||
| Outstanding at December 31, 2009 |
2,055,576 | $ | 8.63 | |||
| Exercisable at December 31, 2009 |
1,319,391 | $ | 10.32 | |||
The weighted average contractual life of options outstanding at December 31, 2009 was 7.4 years and the weighted average contractual life of exercisable options at December 31, 2009 was 6.9 years. The intrinsic value of stock options exercised, outstanding and exerciseable during the year ended December 31, 2009 was $0.3 million, $2.7 million and $0.8 million, respectively, based on the Nasdaq Global Market on such date.
109
Table of Contents
MEDICINOVA, INC.
(a development stage company)
Notes to Consolidated Financial Statements
Common Stock Reserved for Future Issuance
The following table summarizes common stock reserved for future issuance at December 31, 2009:
| Common Stock under the employee stock purchase program |
250,685 | |
| Common stock reserved for issuance upon conversion of convertible notes |
5,500,000 | |
| Common stock options outstanding (under the 2000 Plan and 2004 Plan) |
2,055,576 | |
| Common stock options authorized for future grant (under the 2004 Plan) |
1,968,941 | |
| 9,775,202 | ||
Convertible Notes
At the closing of the Merger, we and American Stock Transfer & Trust Company, LLC, as trustee, entered into the Indenture. Under the terms of a separate trust agreement (the “Trust Agreement”), $29.4 million, which represents the initial principal amount of the Convertible Notes, was deposited with a trust agent for the benefit of the holders and us (the amount of such deposit together with interest accrued and capitalized thereon, the “Property”). Provided no event of default has occurred and is continuing, we are able to direct the investment and reinvestment of the Property in certain approved investment options, including certain money market funds. At the maturity of the Convertible Notes on June 18, 2011, the 18-month anniversary of the closing of the Merger, we will use the Property to pay the principal amount of, and accrued interest on, the Convertible Notes.
The Convertible Notes are our secured obligation, and the Indenture does not limit our other indebtedness, secured or unsecured. The Indenture contains limited covenants, including a requirement that we deliver to holders of the Convertible Notes quarterly statements setting forth the principal amount of the Convertible Notes at the close of the fiscal quarter as well as information regarding the amount of interest capitalized to such Convertible Notes during the fiscal quarter. At December 31, 2009, $137 was the amount of dividends capitalized on the Convertible Notes. The interest rate on the Convertible Notes is equal to the interest earned on the money market funds in the trust account, which was less than half of a percentage point. The $0.2 million in discount will be accreted to interest expense over the conversion period of the Convertible Notes.
Holders of the Convertible Notes may submit conversion notices, which are irrevocable, instructing the trustee to convert such Convertible Notes into shares of our common stock at an initial conversion price of $6.80 per share. Following each conversion date, which date generally is the final business day of each calendar month, we will issue the number of whole shares of common stock issuable upon conversion as promptly as practicable (and in any event within 10 business days). The trustee will in turn release to us the respective amount of restricted cash to cover the stock issuance. We will then invest the unrestricted cash into either a money market fund or a money market account. Any fractional shares (after aggregating all Convertible Notes being converted by a holder on such date) will be rounded down and we will deliver cash for the current market value of the fractional share. The Indenture includes customary anti-dilution adjustments and events of default.
As of December 31, 2009, none of the Convertible Notes were converted into our common stock.
110
Table of Contents
MEDICINOVA, INC.
(a development stage company)
Notes to Consolidated Financial Statements
8. Income Taxes
The significant components of our deferred income taxes at December 31, 2009 and 2008 are as follows:
| December 31, | ||||||
| 2009 | 2008 | |||||
| Deferred Tax Assets: |
||||||
| Net operating loss carry forwards |
64,627,000 | 51,884,000 | ||||
| Capitalized licenses |
2,559,000 | 2,805,000 | ||||
| Research tax credits |
6,037,000 | 5,380,000 | ||||
| Stock Options |
420,000 | 1,093,000 | ||||
| Unrealized loss on marketable securities |
387,000 | 513,000 | ||||
| Other, net |
305,000 | 257,000 | ||||
| Total Deferred Tax Assets |
74,335,000 | 61,932,000 | ||||
| Deferred Tax Liabilities |
||||||
| IPR&D |
(1,956,000 | ) | — | |||
| Total Deferred Tax Liabilities |
(1,956,000 | ) | — | |||
| Net deferred tax assets |
72,379,000 | 61,932,000 | ||||
| Valuation Allowance |
(74,335,000 | ) | (61,932,000 | ) | ||
| Net Deferred Tax Liability |
(1,956,000 | ) | — | |||
We have established a deferred tax liability for the book to tax basis difference related to IPR&D acquired through the acquisition of Avigen.
We have established a valuation allowance against our deferred tax assets due to the uncertainty that such assets will be realized. We periodically evaluate the recoverability of the deferred tax assets. At such time as it is determined that it is more likely than not that deferred tax assets will be realizable, the valuation allowance will be reduced.
At December 31, 2009, we had federal and California net operating loss carryforwards of approximately $158.8 million and $157.9 million, respectively. Included in these amounts are federal and California tax benefits of approximately $22,000 attributable to stock option deductions which will be credited to equity when realized. The federal net operating loss carryforwards begin to expire in 2020, and the California net operating loss carryforwards begin to expire in 2013. At December 31, 2009, we also had federal and California research tax credit carryforwards of approximately $5.4 million and $1 million, respectively. The federal research tax credit carryforwards begin to expire in 2024, and the California research tax credit carryforward does not expire and can be carried forward indefinitely until utilized.
Additionally, utilization of the net operating losses, or NOL, and tax credit carryforwards will be subject to a substantial annual limitation under Section 382 and 383 of the Internal Revenue Code of 1986, and similar state provisions due to ownership change limitations that have occurred. These ownership changes will limit the amount of NOL and tax credit carryforwards that can be utilized to offset future taxable income and tax, respectively. In general, an ownership change, as defined by Section 382 and 383, results from transactions increasing ownership of certain stockholders or public groups in the stock of the corporation by more than 50 percentage points over a three-year period. We are in the process of updating our formal Section 382 analysis to determine whether such an ownership change occurred during the period September 26, 2000–December 31, 2009. We believe an ownership change may have occurred during this period as a result of various equity financings. If so the amount of NOL and tax credit carryforwards available for utilization would be subject to an annual limitation. Due to the existence of the valuation allowance, limitations created by future ownership changes, if any, related to our operations in the U.S. will not impact our effective tax rate.
111
Table of Contents
MEDICINOVA, INC.
(a development stage company)
Notes to Consolidated Financial Statements
In July 2006, the FASB issued ASC 740, which clarifies the accounting for uncertainty in income taxes recognized in a company’s financial statements. ASC 740 prescribes a recognition threshold and measurement process for recording in the financial statements uncertain tax positions taken or expected to be taken in a tax return. Additionally, ASC 740 provides guidance on the de-recognition, classification, interest and penalties, accounting in interim periods, and disclosure requirements for uncertain tax positions. We adopted the provisions of ASC 740 beginning January 1, 2007. The adoption of ASC 740 did not materially impact our financial condition, results of operations or cash flows. As of December 31, 2009, we have not recorded any uncertain tax benefits.
We file income tax returns in the United States, California and foreign jurisdictions. Due to our losses incurred, we are essentially subject to income tax examination by tax authorities from our inception to date. Our policy is to recognize interest expense and penalties related to income tax matters as tax expense. At December 31, 2009, we do not have any significant accruals for interest related to unrecognized tax benefits or tax penalties.
9. Employee Savings Plan and Employee Stock Purchase Plan
We have an employee savings plan available to substantially all employees. Under the plan, an employee may elect salary reductions which are contributed to the plan. The plan provides for discretionary contributions by us, which totaled $149,994, $151,488, $155,598 and $862,126 for the years ended December 31, 2009, 2008, 2007 and the period from September 26, 2000 (inception) to December 31, 2009, respectively.
Under the MediciNova, Inc. 2007 Employee Stock Purchase Plan (“ESPP”), 300,000 shares of our common stock have been reserved for issuance. In addition, the shares reserved will automatically increase by a number equal to the lesser of: (i) 15,000 shares, (ii) 1% of the outstanding shares of our common stock on the last day of the immediately preceding fiscal year or (iii) such lesser amount as determined by the Board. The ESPP permits full-time employees to purchase our common stock through payroll deductions (which cannot exceed 15% of each employee’s compensation) at the lower of 85% of fair market value at the beginning of the offering period or the end of each six-month offering period. For the year ended December 31, 2009, 37,021 shares were issued under the ESPP, leaving 250,685 shares available for future issuance.
112
Table of Contents
MEDICINOVA, INC.
(a development stage company)
Notes to Consolidated Financial Statements
10. Quarterly Financial Data (Unaudited)
The following financial information reflects all normal recurring adjustments, which are, in the opinion of management, necessary for a fair statement of the results of the interim periods. Summarized quarterly data for fiscal 2009 and 2008 are as follows (in thousands, except per share data):
| Year Ended December 31, 2009 | ||||||||||||||||
| 1st Quarter |
2nd Quarter |
3rd Quarter |
4th Quarter |
|||||||||||||
| Selected quarterly financial data: |
||||||||||||||||
| Revenue |
$ | — | $ | — | $ | — | $ | — | ||||||||
| Total operating expenses |
5,265 | 4,945 | 4,943 | 6,086 | ||||||||||||
| Net loss |
(4,993 | ) | (4,665 | ) | (4,795 | ) | (5,916 | ) | ||||||||
| Net loss applicable to common stockholders |
(4,993 | ) | (4,665 | ) | (4,795 | ) | (5,916 | ) | ||||||||
| Basic and diluted net loss per common share(1) |
(0.41 | ) | (0.39 | ) | (0.40 | ) | (0.49 | ) | ||||||||
| Year Ended December 31, 2008 | ||||||||||||||||
| 1st Quarter |
2nd Quarter |
3rd Quarter |
4th Quarter |
|||||||||||||
| Selected quarterly financial data: |
||||||||||||||||
| Revenue |
$ | — | $ | — | $ | — | $ | — | ||||||||
| Total operating expenses |
8,660 | 4,460 | 5,697 | 3,785 | ||||||||||||
| Net loss |
(10,803 | ) | (4,892 | ) | (4,815 | ) | (1,415 | ) | ||||||||
| Net loss applicable to common stockholders |
(10,803 | ) | (4,892 | ) | (4,815 | ) | (1,415 | ) | ||||||||
| Basic and diluted net loss per common share(1) |
(0.89 | ) | (0.40 | ) | (0.40 | ) | (0.12 | ) | ||||||||
| (1) | Loss per share is computed independently for each of the quarters presented. Therefore, the sum of the quarterly net loss per share will not necessarily equal the total for the year. |
113
Table of Contents
Item 9. Changes in and Disagreements With Accountants on Accounting and Financial Disclosure
None
Item 9A(T). Controls and Procedures
Evaluation of Disclosure Controls and Procedures
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in our periodic and current reports that we file with the SEC is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure. In designing and evaluating the disclosure controls and procedures, management recognized that any controls and procedures, no matter how well designed and operated, can provide only reasonable and not absolute assurance of achieving the desired control objectives. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, or misstatements due to error, if any, have been detected. While we believe that our disclosure controls and procedures and internal control over financial reporting are and have been effective, we intend to continue to examine and refine our disclosure controls and procedures and internal control over financial reporting and to monitor ongoing developments in these areas.
As of December 31, 2009, management conducted an evaluation, under the supervision and with the participation of our Chief Executive Officer and Chief Financial Officer, of the effectiveness of the design and operation of our disclosure controls and procedures, as defined under Rule 13a-15(e) promulgated under the Securities Exchange Act of 1934, as amended, or the Exchange Act. Based on this evaluation, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures were effective as of the end of the period covered by this Annual Report on Form 10-K.
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f) and 15d-15(f). Under the supervision and with the participation of our Chief Executive Officer and Chief Financial Officer, management conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 2009 based on the framework in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on our evaluation under the framework in Internal Control—Integrated Framework, our management concluded that our internal control over financial reporting was effective as of December 31, 2009.
Based on our market capitalization as of June 30, 2009, we qualify as a smaller reporting company under the Securities Act of 1933, as amended, and the Exchange Act for the fiscal year ended December 31, 2009. As a result of qualifying as a smaller reporting company, this annual report does not include an attestation report of our registered independent public accounting firm regarding internal control over financial reporting. Management’s report was not subject to attestation by our registered independent public accounting firm pursuant to temporary rules of the SEC that permit us to provide only management’s report in this annual report.
Changes in Internal Control Over Financial Reporting
There has been no change in our internal control over financial reporting in our most recent fiscal quarter that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Not applicable.
114
Table of Contents
PART III
Item 10. Directors, Executive Officers and Corporate Governance
The information required by this item will be contained in our definitive proxy statement to be filed with the SEC in connection with the Annual Meeting of Stockholders within 120 days after the conclusion of our fiscal year ended December 31, 2009, or the Proxy Statement, and is incorporated in this Annual Report on Form 10-K by reference.
We have adopted a Code of Ethics for Senior Officers, or Code of Ethics, that applies to our Chief Executive Officer, President, Chief Financial Officer and key management employees (including other senior financial officers) who have been identified by our Board of Directors. We have also adopted a Code of Business Conduct that applies to all of our officers, directors, employees, consultants and representatives. Each of the Code of Ethics and Code of Business Conduct are available on our website at www.medicinova.com under the Corporate Governance section of our Investor Relations page. We will promptly post on our website (i) any waiver, if and when granted, to any provision of the Code of Ethics or Code of Business Conduct (for executive officers or directors) and (ii) any amendment to the Code of Ethics or Code of Business Conduct.
Item 11. Executive Compensation
The information required by this item will be contained in the Proxy Statement and is incorporated in this Annual Report on Form 10-K by reference.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
Certain of the information required by this item will be contained in the Proxy Statement and is incorporated in this Annual Report on Form 10-K by reference.
The following table provides information as of December 31, 2009 with respect to the shares of our common stock that may be issued under our existing equity compensation plans.
| Plan Category |
Number of Securities to be Issued Upon Exercise of Outstanding Options and Rights |
Weighted Average Exercise Price of Outstanding Options and Rights |
Number of Securities Remaining Available for Future Issuance Under Equity Compensation Plans | |||||
| Equity Compensation Plans Approved by Stockholders(1) |
2,018,076 | $ | 8.61 | 1,968,941 | ||||
| Equity Compensation Plans Approved by Stockholders(2) |
3,568 | $ | 5.15 | (2) | 250,685 | |||
| Equity Compensation Plans Not Approved by Stockholders(3) |
37,500 | $ | 10.00 | — | ||||
| Total |
2,059,144 | $ | 8.54 | 2,219,626 | ||||
| (1) | Consists of the MediciNova, Inc. Amended and Restated 2004 Stock Incentive Plan , or 2004 Plan. Awards under the 2004 Plan shall not exceed 3,330,000 shares, plus an annual increase on the first day of each fiscal year, with the first increase occurring on January 1, 2006, in an amount equal to the lesser of (i) 100,000 shares, (ii) 3% of the outstanding shares on the last day of the immediately preceding year, or (iii) an amount determined by the Board. Stock options under the 2004 Plan have an exercise price equal to the fair market value of the underlying common stock at the date of grant, generally vest over a period of four years and have a ten-year life. |
115
Table of Contents
| (2) | Consists of the MediciNova, Inc. 2007 Employee Stock Purchase Plan, or ESPP. Under the ESPP, 300,000 shares of our common stock have been reserved for issuance. In addition, the shares reserved will automatically increase by a number equal to the lesser of: (i) 15,000 shares, (ii) 1% of the outstanding shares of our common stock on the last day of the immediately preceding fiscal year or (iii) such lesser amount as determined by the Board. The ESPP permits full-time employees to purchase our common stock through payroll deductions (which cannot exceed 15% of each employee’s compensation) at the lower of 85% of fair market value at the beginning of the offering period or the end of each six-month offering period. |
| (3) | Consists solely of the MediciNova, Inc. 2000 General Stock Incentive Plan, or 2000 Plan, which was terminated upon the completion of our initial public offering on February 4, 2005. The material terms of the 2000 Plan are described in Note 7 to our consolidated financial statements contained in this Annual Report on Form 10-K. |
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information required by this item will be contained in the Proxy Statement and is incorporated in this Annual Report on Form 10-K by reference.
Item 14. Principal Accounting Fees and Services
The information required by this item will be contained in the Proxy Statement and is incorporated in this Annual Report on Form 10-K by reference.
116
Table of Contents
PART IV
Item 15. Exhibits, Financial Statement Schedules
(a) Documents filed as part of this report.
1. Financial Statements. The following financial statements of MediciNova, Inc. and Reports of KPMG LLP and Ernst & Young LLP, each an independent registered public accounting firm, are included in this Annual Report on Form 10-K:
| Page | ||
| 78 | ||
| 80 | ||
| 81 | ||
| 82 | ||
| 86 | ||
| 87 |
2. Financial Statement Schedules. None.
3. Exhibits.
| Exhibit Number |
Description | |
| 2.1(23) | Agreement and Plan of Merger dated as of August 20, 2009 by and among Registrant, Absolute Merger, Inc. and Avigen, Inc. (attached as Annex A to the joint proxy statement/prospectus). | |
| 3.1(19) | Restated Certificate of Incorporation of the Registrant, as amended. | |
| 3.2(1) | Amended and Restated Bylaws of the Registrant. | |
| 4.1(11) | Specimen of Common Stock Certificate. | |
| 4.2(1) | Amended and Restated Registration Rights Agreement by and among the Registrant, its founders and the investors named therein, dated September 2, 2004. | |
| 4.3(12) | Rights Agreement between the Registrant and American Stock Transfer & Trust Company, which includes the form of Rights Certificate as Exhibit B and the Summary of Rights as Exhibit C, dated November 24, 2006. | |
| 4.4(23) | Form of Indenture by and between Registrant and American Stock Transfer and Trust Company, LLC (attached as Annex C to the joint proxy statement/prospectus). | |
| 4.5(23) | Form of Convertible Note (included in Exhibit 4.4). | |
| 10.1(1) | 2000 General Stock Incentive Plan of the Registrant. | |
| 10.2(2) | 2004 Stock Incentive Plan of the Registrant. | |
| 10.3(5) | Form of Indemnification Agreement between the Registrant and its officers and directors. | |
| 10.4(2)† | License Agreement between the Registrant and Kyorin Pharmaceutical Co., Ltd., dated March 14, 2002. | |
| 10.5(2)† | License Agreement between the Registrant and Angiogene Pharmaceuticals, Ltd., dated June 19, 2002. | |
| 10.6(2)† | Exclusive License Agreement between the Registrant and Kissei Pharmaceutical Co., Ltd., dated February 25, 2004. | |
| 10.7(2)† | License Agreement between the Registrant and Mitsubishi Tanabe Pharma Corporation, dated April 27, 2004. | |
117
Table of Contents
| Exhibit Number |
Description | |
| 10.8(3)† | License Agreement between the Registrant and Kyorin Pharmaceutical Co., Ltd., dated October 22, 2004. | |
| 10.9(2) | Office Lease Agreement between the Registrant and CA-LA Jolla II Limited Partnership, dated January 28, 2004 and the First Amendment thereto, dated August 10, 2004. | |
| 10.10(3)† | License Agreement between the Registrant and Mitsubishi Tanabe Pharma Corporation, dated December 8, 2004. | |
| 10.11(4) | Second Amendment to Office Lease Agreement between the Registrant and CA-La Jolla II Limited Partnership, dated March 21, 2005. | |
| 10.12(5) | Executive Employment Agreement between the Registrant and Shintaro Asako, CPA, dated July 18, 2005. | |
| 10.13(11) | Executive Employment Agreement between the Registrant and Masatsune Okajima, dated September 1, 2006. | |
| 10.14(7)† | License Agreement, dated October 31, 2006 by and between the Registrant and Meiji Seika Kaisha, Ltd. | |
| 10.15(7)† | License Agreement, dated October 31, 2006 by and between the Registrant and Meiji Seika Kaisha, Ltd. | |
| 10.16(8) | Executive Employment Agreement between the Registrant and Yuichi Iwaki, M.D., Ph.D., dated April 1, 2007. | |
| 10.17(13) | 2007 Employee Stock Purchase Plan of the Registrant. | |
| 10.18(9) | Form of Severance Protection Agreement between the Registrant and certain of its executive officers, dated September 12, 2007. | |
| 10.19(10) | Third Amendment to Office Lease Agreement between the Registrant and 4350 La Jolla Village LLC, dated January 31, 2008. | |
| 10.20(14) | Severance Agreement and Release by and between the Registrant and Kenneth W. Locke, dated April 30, 2008. | |
| 10.21(14) | Consulting Agreement by and between the Registrant and Kenneth W. Locke, dated May 1, 2008. | |
| 10.22(15) | Amendment to the Amended and Restated 2004 Stock Incentive Plan of the Registrant, dated June 6, 2008. | |
| 10.23(16) | Fourth Amendment to Lease Agreement between the Registrant and 4350 La Jolla Village LLC, dated October 3, 2008. | |
| 10.24(17) | Credit Line Account Application and Agreement for Organizations and Businesses, executed by the Registrant on January 8, 2009, by and between the Registrant and UBS Bank USA. | |
| 10.25(17) | Addendum to Credit Line Account Application and Agreement, executed by the Registrant on January 8, 2009, by and between the Registrant, UBS Bank USA and UBS Financial Services Inc. | |
| 10.26(17) | Addendum to Credit Line Agreement, executed by the Registrant on January 8, 2009, by and between the Registrant and UBS Bank USA. | |
| 10.27(17) | Important Notice on Interest Rates and Payments, executed by the Registrant on January 8, 2009, by and between the Registrant and UBS Bank USA. | |
| 10.28(18)* | Development and Supply Agreement between the Registrant and Hospira Worldwide, Inc., dated as of March 26, 2009. | |
| 10.29(20) | Consulting Agreement dated June 12, 2009, by and between Registrant and Danerius, LLC. | |
118
Table of Contents
| Exhibit Number |
Description | |
| 10.30(21) | Separation Agreement and Release dated June 26, 2009, by and between Registrant and Richard E. Gammans, Ph.D. | |
| 10.31(22) | Separation Agreement and Release dated July 12, 2009, by and between Registrant and Michael E. Kalafer, M.D. | |
| 10.33(23) | Form of Contingent Payment Rights Agreement by and among Registrant, Avigen, Inc. and American Stock Transfer and Trust Company, LLC (attached as Annex B to the joint proxy statement/prospectus). | |
| 10.34(23) | Form of Trust Agreement by and between Registrant and American Stock Transfer and Trust Company, LLC (attached as Annex D to the joint proxy statement/prospectus). | |
| 10.32(23) | Form of Escrow Agreement by and between Registrant and American Stock Transfer and Trust Company, LLC (attached as Annex E to the joint proxy statement/prospectus). | |
| 10.35(24) | First Amendment to Consulting Agreement dated as of September 23, 2009 by and between Registrant and Danerius, LLC. | |
| 10.36(25)† | Assignment Agreement, dated December 19, 2005, by and between Genzyme Corporation and Avigen, Inc. | |
| 10.38(26) | Avigen, Inc. Management Transition Plan effective July 15, 1998, last amended October 30, 2008 | |
| 10.38(27)† | Asset Purchase Agreement, dated December 17, 2008, by and between Baxter Healthcare Corporation, Baxter International Inc., and Baxter Healthcare S.A. and Avigen, Inc. | |
| 10.40(28) | Amendment to Management Transition Plan, effective as of August 20, 2009. | |
| 10.41(29) | Amendment to Management Transition Plan, effective as of November 9, 2009. | |
| 14.1(11) | Code of Ethics of the Registrant. | |
| 21.1 | List of subsidiaries of the Registrant. | |
| 23.1 | Consent of Independent Registered Public Accounting Firm. | |
| 23.2 | Consent of Independent Registered Public Accounting Firm. | |
| 24.1 | Powers of Attorney (included in Signature page). | |
| 31.1 | Certification of the Chief Executive Officer pursuant to Rules 13a-14 and 15d-14 promulgated under the Securities Act of 1933. | |
| 31.2 | Certification of the Chief Financial Officer pursuant to Rules 13a-14 and 15d-14 promulgated under the Securities Act of 1933. | |
| 32.1 | Certification of the Chief Executive Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| 32.2 | Certification of the Chief Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| (1) | Filed with the Registrant’s Registration Statement on Form S-1 filed October 1, 2004 and incorporated herein by reference. |
| (2) | Filed with the Registrant’s Amendment to Registration Statement on Form S-1/A filed November 24, 2004 and incorporated herein by reference. |
| (3) | Filed with the Registrant’s Amendment to Registration Statement on Form S-1/A filed January 6, 2005 and incorporated herein by reference. |
| (4) | Filed with the Registrant’s Quarterly Report on Form 10-Q filed May 12, 2005 and incorporated herein by reference. |
| (5) | Filed with the Registrant’s Registration Statement on Form S-1 filed on September 1, 2005 and incorporated herein by reference. |
119
Table of Contents
| (6) | Filed with the Registrant’s Registration Statement on Form S-3 filed November 14, 2006 and incorporated herein by reference. |
| (7) | Filed with the Registrant’s Current Report on Form 8-K filed November 2, 2006 and incorporated herein by reference. |
| (8) | Filed with the Registrant’s Current Report on Form 8-K filed April 4, 2007 and incorporated herein by reference. |
| (9) | Filed with the Registrant’s Current Report on Form 8-K filed September 14, 2007 and incorporated herein by reference. |
| (10) | Filed with the Registrant’s Current Report on Form 8-K filed February 4, 2008 and incorporated herein by reference. |
| (11) | Filed with the Registrant’s Annual Report on Form 10-K filed February 15, 2007 and incorporated herein by reference. |
| (12) | Filed with the Registrant’s Registration Statement on Form 8-A filed November 29, 2006 and incorporated herein by reference. |
| (13) | Filed with the Registrant’s Definitive Proxy Statement on Schedule 14A filed March 13, 2007 and incorporated herein by reference. |
| (14) | Filed with the Registrant’s Current Report on Form 8-K filed May 1, 2008 and incorporated herein by reference. |
| (15) | Filed with the Registrant’s Current Report on Form 8-K filed June 10, 2008 and incorporated herein by reference. |
| (16) | Filed with the Registrant’s Current Report on Form 8-K filed October 8, 2008 and incorporated herein by reference. |
| (17) | Filed with the Registrant’s Current Report on Form 8-K filed January 21, 2009 and incorporated herein by reference. |
| (18) | Filed with the Registrant’s Current Report on Form 8-K filed March 30, 2009 and incorporated herein by reference. |
| (19) | Filed with the Registrant’s Current Report on Form 8-K filed May 29, 2009 and incorporated herein by reference. |
| (20) | Filed with the Registrant’s Current Report on Form 8-K filed June 16, 2009 and incorporated herein by reference. |
| (21) | Filed with the Registrant’s Current Report on Form 8-K filed July 2, 2009 and incorporated herein by reference. |
| (22) | Filed with the Registrant’s Current Report on Form 8-K filed July 16, 2009 and incorporated herein by reference. |
| (23) | Filed with the Registrant’s Registration Statement on Form S-4 initially filed September 17, 2009 and incorporated herein by reference. |
| (24) | Filed with the Registrant’s Current Report on Form 8-K filed September 25, 2009 and incorporated herein by reference. |
| (25) | Filed with Avigen, Inc.’s Annual Report on Form 10-K filed March 16, 2006 and incorporated herein by reference. |
| (26) | Filed with Avigen, Inc.’s Solicitation/Recommendation Statement on Schedule 14D-9 filed February 6, 2009 and incorporated herein by reference. |
| (27) | Filed with Avigen, Inc.’s Annual Report on Form 10-K filed with the SEC on March 16, 2009. |
| (28) | Filed with Avigen, Inc.’s Current Report on Form 8-K filed August 25, 2009 and incorporated herein by reference. |
| (29) | Filed with Avigen, Inc.’s Quarterly Report on Form 10-Q filed November 9, 2009 and incorporated herein by reference. |
| † | Portions of this Exhibit have been omitted pursuant to a grant of confidential treatment by the SEC. * Portions of this Exhibit have been omitted pursuant to a Confidential Treatment Request submitted to the SEC. Omitted information has been filed separately with the SEC. |
120
Table of Contents
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this Annual Report on Form 10-K to be signed on its behalf by the undersigned, thereunto duly authorized.
| MEDICINOVA, INC. | ||||
| A Delaware Corporation | ||||
| Date: March 24, 2010 |
By: |
/s/ Yuichi Iwaki | ||
| Yuichi Iwaki, M.D., Ph.D. President & Chief Executive Officer | ||||
POWER OF ATTORNEY
KNOW ALL MEN BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Yuichi Iwaki and Shintaro Asako and each of them, his true and lawful attorneys-in-fact, each with full power of substitution, for him in any and all capacities, to sign any amendments to this Annual Report on Form 10-K and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that each of said attorneys-in-fact or their substitute or substitutes may do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this Annual Report on Form 10-K has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature |
Title |
Date | ||
| /s/ Yuichi Iwaki Yuichi Iwaki, M.D., Ph.D. |
Director, President & Chief Executive Officer (Principal Executive Officer) | March 24, 2010 | ||
| /s/ Shintaro Asako Shintaro Asako, CPA |
Vice President and Chief Financial Officer (Principal Financial Officer and Principal Accounting Officer) | March 24, 2010 | ||
| /s/ Alan Dunton Alan Dunton, M.D. |
Director | March 24, 2010 | ||
| /s/ Jeff Himawan Jeff Himawan, Ph.D. |
Director | March 24, 2010 | ||
| /s/ Arlene Morris Arlene Morris |
Director | March 24, 2010 | ||
| /s/ John K.A. Prendergast John K.A. Prendergast, Ph.D. |
Director | March 24, 2010 | ||
| /s/ Hiroaki Shigeta Hiroaki Shigeta |
Director | March 24, 2010 | ||
121