Attached files
| file | filename |
|---|---|
| EX-32.1 - CERTIFICATION - SENORX INC | seno_10k-ex3201.htm |
| EX-31.2 - CERTIFICATION - SENORX INC | seno_10k-ex3102.htm |
| EX-23.1 - CONSENT - SENORX INC | seno_10k-ex2301.htm |
| EX-31.1 - CERTIFICATION - SENORX INC | seno_10k-ex3101.htm |
UNITED
STATES
SECURITIES
AND EXCHANGE COMMISSION
Washington,
D.C. 20549
FORM 10-K
(Mark
One)
x ANNUAL REPORT PURSUANT TO
SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF
1934
For the
fiscal year ended December 31, 2009
Or
¨ TRANSITION REPORT PURSUANT TO
SECTION 13 Or 15(d) OF THE SECURITIES EXCHANGE ACT OF
1934
Commission
file number: 001-33382
SENORX,
INC.
(Exact
name of registrant as specified in its charter)
|
Delaware
|
33-0787406
|
|
(State
of Incorporation)
|
(I.R.S.
Employer
Identification
Number)
|
|
3
Morgan
Irvine,
California
|
92618
|
|
(Address
of principal executive offices)
|
(Zip
Code)
|
Registrant’s
telephone number, including area code: (949) 362-4800
Securities
registered pursuant to Section 12(b) of the Act:
|
Title
of each class:
|
Name
of each exchange on which registered:
|
|
Common
Stock, par value $0.001
|
The
NASDAQ Stock Market, LLC
(NASDAQ
Global Market)
|
Securities
registered pursuant to Section 12(b) of the Act:
None
Indicate
by check mark if the Registrant is a well-known seasoned issuer, as defined in
Rule 405 of the Securities Act. Yes ¨ No x
Indicate
by check mark if the Registrant is not required to file reports pursuant to
Section 13 or Section 15(d) of the
Act. Yes ¨ No x
Indicate
by check mark whether the Registrant (1) has filed all reports required to
be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934
during the preceding 12 months (or for such shorter period that the Registrant
was required to file such reports), and (2) has been subject to such
requirements for the past 90 days. Yes x No ¨
Indicate
by check mark whether the registrant has submitted electronically and posted on
is corporate Web site, if any, every Interactive Data File required to be
submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this
chapter) during the preceding 12 months (or for such shorter period that the
registrant was required to submit and post such
files). Yes ¨ No o
Indicate
by check mark if disclosure of delinquent filers pursuant to Item 405 of
Regulation S-K is not contained herein, and will not be contained, to the
best of the Registrant’s knowledge, in definitive proxy or information
statements incorporated by reference in Part III of this Form 10-K or any
amendment to this Form 10-K. ¨
Indicate
by check mark whether the Registrant is a large accelerated filer, an
accelerated filer, a non-accelerated filer, or a smaller reporting company. See
the definitions of “large accelerated filer,” “accelerated filer” and “smaller
reporting company” in Rule 12b-2 of the Exchange Act (check
one):
Large
accelerated filer ¨ Accelerated
filer ¨ Non-accelerated
filer x Smaller
reporting company ¨
(Do not
check if a smaller reporting company)
Indicate
by check mark whether the Registrant is a shell company (as defined in
Rule 12b-2 of the Exchange
Act). Yes ¨ No x
The
aggregate market value of the registrant’s common stock, held by non-affiliates
of the registrant as of June 30, 2009 (which is the last business day of
registrant’s most recently completed second fiscal quarter) based upon the
closing price of such stock on the NASDAQ Global Market on that date, was
$34,391,947. For purposes of this disclosure, shares of common stock held by
entities and individuals who own 5% or more of the outstanding common stock and
shares of common stock held by each officer and director have been excluded in
that such persons may be deemed to be “affiliates” as that term is defined under
the Rules and Regulations of the Securities Exchange Act of 1934. This
determination of affiliate status is not necessarily conclusive.
At
February 28, 2010, the Registrant had 17,540,128 shares of Common Stock
outstanding.
DOCUMENTS
INCORPORATED BY REFERENCE
Items 9,
10, 11, 12 and 13 of Part III of this Form 10-K incorporate
information by reference from the registrant’s definitive proxy statement to be
filed with the Securities and Exchange Commission within 120 days after the
close of the fiscal year covered by this annual report.
SENORX,
INC.
FISCAL
YEAR 2009 FORM 10-K ANNUAL REPORT
TABLE
OF CONTENTS
|
PART
I
|
||
|
Item
1.
|
Business
|
1
|
|
Item
1A.
|
Risk
Factors
|
20
|
|
Item
1B.
|
Unresolved
Staff Comments
|
31
|
|
Item
2.
|
Properties
|
31
|
|
Item
3.
|
Legal
Proceedings
|
31
|
|
Item
4.
|
[Reserved]
|
32
|
|
PART
II
|
||
|
Item
5.
|
Market
for the Registrant’s Common Equity, Related Stockholder Matters and Issuer
Purchases of Equity Securities
|
32
|
|
Item
6.
|
Selected
Financial Data
|
34
|
|
Item
7.
|
Management’s
Discussion and Analysis of Financial Condition and Results of
Operations
|
35
|
|
Item
7a.
|
Quantitative
and Qualitative Disclosures about Market Risk
|
46
|
|
Item
8.
|
Financial
Statements and Supplementary Data
|
46
|
|
Item
9.
|
Changes
in and Disagreements with Accountants on Accounting and Financial
Disclosure
|
47
|
|
Item
9A.
|
Controls
and Procedures
|
47
|
|
Item
9B.
|
Other
Information
|
47
|
|
PART
III
|
||
|
Item
10.
|
Directors,
Executive Officers and Corporate Governance
|
49
|
|
Item
11.
|
Executive
Compensation
|
49
|
|
Item
12.
|
Security
Ownership of Certain Beneficial Owners and Management and Related
Stockholder Matters
|
49
|
|
Item
13.
|
Certain
Relationships and Related Transactions and Director
Independence
|
49
|
|
Item
14.
|
Principal
Accountant Fees and Services
|
49
|
|
PART
IV
|
||
|
Item
15.
|
Exhibits
and Financial Statement Schedules
|
49
|
|
Signatures
|
73
|
|
-i-
PART
I
This
Annual Report on Form 10-K contains forward-looking statements within the
meaning of the federal securities laws. These statements include, but are not
limited to, those concerning the following: regarding future events, our future
financial performance, business strategy, product introductions and plans and
objectives of management for future operations, regulatory approvals, and
clinical timelines. Forward-looking statements are subject to risks and
uncertainties that could cause actual results and events to differ materially.
For a detailed discussion of these risks and uncertainties, see the
“Management’s Discussion and Analysis of Financial Condition and Results of
Operations” section of this Form 10-K. We undertake no obligation to update
forward-looking statements to reflect events or circumstances occurring after
the date of this Form 10-K.
|
ITEM 1.
|
BUSINESS
|
Overview
We
develop, manufacture and sell minimally-invasive medical devices that are used
in the diagnosis and treatment of breast cancer. Our initial product focus has
been biopsy systems and breast tissue markers and in January 2008 we launched a
radiation balloon catheter for localized partial breast irradiation therapy.
With the emergence of clinicians coordinating multi-disciplinary patient care
through integrated breast centers, we believe that our ability to provide a
broad array of products will enhance our competitive positioning. Since we
launched our first breast tissue marker products in 2002, we have established
over 1,000 customer accounts. In 2009, we generated net revenues of $55.6
million. The sale of disposable products, including our breast biopsy probes,
radiation therapy products and tissue markers, accounted for 90.0% of our
revenues in 2009.
The EnCor
system, our flagship product for use in breast biopsy procedures, is a
minimally-invasive vacuum-assisted breast biopsy system. EnCor allows users to
obtain multiple biopsy samples with a quick, single probe insertion. In contrast
to existing competitive systems, EnCor is the only commercialized “open/closed”
tissue collection system compatible in all three
imaging modalities, providing the operator
with a clear view of tissue samples through a proprietary transparent collection
chamber, and the ability to either open the chamber to examine and remove one or
more samples or to continue uninterrupted collection of multiple samples. EnCor
also incorporates novel programmability, which allows the user to select
automated cutting patterns, tissue density, number of samples, and to deliver
anesthetic. The EnCor system’s handpieces and disposable probes are compatible
with the most commonly used imaging modalities, including x-ray, ultrasound,
magnetic resonance imaging, or MRI, and positron emission tomography, or PET.
With its simplicity and versatility, we believe that EnCor can play an important
role in the paradigm shift from invasive open surgical to minimally-invasive
biopsy procedures. We launched the EnCor system on a limited basis and conducted
marketing preference testing in late 2004 and subsequently progressed with a
full commercial launch in November 2005. In 2007, we further enhanced the
versatility of the EnCor system with the FDA clearance in the United States of
our SenoSonix system, a combination of EnCor with state-of-the art ultrasound
imaging technology, and with the commercial launch of VisiLoc, an MRI visible
obturator that helps to facilitate accurate probe placement under MRI guidance.
As of December 31, 2009, we had an installed base of 991EnCor systems and we had
sold more than 310,000 EnCor disposable probes.
Our
Contura Multi-Lumen Radiation Balloon Catheter, or Contura MLB, our flagship
radiation therapy product, for which we received FDA 510(k) clearance in May
2007, launched in January 2008 and received its CE Mark in Europe in March 2009,
is designed to be a novel localized partial breast irradiation therapy device
that uses vacuum to remove excess seroma and air to enhance conformance of often
irregularly shaped lumpectomy cavity walls to the balloon’s surface in order to
deliver precise irradiation dosing through multiple radiation source lumens. We
believe that Contura MLB can play an important role in the paradigm shift from
traditional whole breast radiation therapy to localized partial breast radiation
therapy. In 2008 and 2009, our Contura MLB program was the subject matter of a
law suit filed by Hologic and its affiliates that generally alleged patent
infringement of certain Hologic brachytherapy patent claims. On December 17,
2009, the jury in the case returned a verdict ruling in our favor on all counts
that were then remaining in the matter; however, Hologic has subsequently filed
an appeal in February of this year.
Our
headquarters is in Irvine, California and we were incorporated in Delaware in
January 1998.
-1-
Industry
Overview
Breast
Cancer
One in
eight women in the United States will develop breast cancer during her lifetime,
a risk that was one in fourteen in 1960. It is estimated that in the United
States, approximately 192,000 new cases of breast cancer were diagnosed in 2008.
Breast cancer is the second-leading cause of cancer-related death in U.S. women
overall, and the leading cause of cancer-related death for women of ages 20 to
59. Over 70% of breast cancers occur in women who have no
identifiable risk factor other than age. The older a woman is, the greater her
chance of getting breast cancer. One of the major challenges in the treatment of
breast cancer is that, while the disease typically does not show symptoms in
early stages, survival is dramatically impacted by the stage at which the
disease is diagnosed and treated. If breast cancer is detected at an early
“localized” stage and treated, the 5-year survival rate is 98%. If the cancer
has spread to nearby lymph nodes, the 5-year survival rate decreases to 81%. If
the cancer has spread, or metastasized, to organs such as the lungs, bone
marrow, liver or brain, the 5-year survival rate falls to 26%. These statistics
underscore the need for early diagnosis and treatment of breast cancer.
Currently, 62% of breast cancers are discovered at an early, localized stage.
The number of breast biopsies performed annually has increased significantly
since 1997 when the American Cancer Society updated its guidelines for breast
cancer screening, recommending that women should begin annual screening at age
40 rather than the previously recommended age 50. However, some studies conclude
that annual breast cancer screening by mammography for women under age 50 may be
more harmful, due to increased radiation exposure, than beneficial and as a
result of this and other factors, the trend towards earlier and broader
screening programs may not continue or mammography may be supplemented by other
screening modalities in the future. Recently, an independent panel of
experts appointed by the federal Department of Health and Human Services
recommended a return to screening starting at age 50. In response to this
recommendation, the American Cancer Society has stated that it does not plan to
revise its guidelines. In addition, outside of the United States, mammography
screening programs are in the early stage of adoption, which has been
accelerating in the last several years.
Breast
Cancer Screening, Diagnosis and Treatment
The
principal means of breast cancer screening are physical examination and
mammography. In a physical examination, the patient’s breast is examined to
search for palpable lesions or any other abnormalities. However, physical
examination cannot detect small, early stage lesions that may be cancerous. As a
result, mammography, a low-dose x-ray imaging technique, is recognized as the
best screening method for detecting breast cancer in its earliest stages, when
the disease is most successfully treated and there are more treatment options.
Historically, mammograms are believed to find 85% to 90% of breast cancers in
women over 50, and can discover a lesion one to four years before a lump can be
felt. Newer technologies, such as digital mammography and tomosynthesis, are
being introduced into the market and may help increase the lesion detection
rate. Nevertheless, for patients with dense breast tissue, breast
implants or patients who are breastfeeding, the images produced by a mammogram
can be difficult for a radiologist to interpret. Consequently, physicians will
often order a secondary screening using ultrasound, or, in some cases,
MRI.
If breast
cancer screening detects a lesion, a physician will typically recommend that the
patient undergo a breast biopsy, a diagnostic procedure in which breast tissue
samples are extracted to determine whether a lesion is benign or malignant. The
breast biopsy procedure is performed by either a radiologist or surgeon. As a
final step in the biopsy procedure, the physician usually places a tissue marker
at the location from which the sample was removed as a point of reference. If
the sample is found to be cancerous and more tissue must be removed from the
breast, the marker will help the physician identify the specific area from which
tissue should be removed. This can minimize the amount of healthy tissue removed
from the breast during surgery. If surgery is not required, the marker will be
visible on future screenings to enable the physician to identify the site of the
previous biopsy.
If a
breast biopsy indicates that a patient’s lesion is malignant, the patient is
often scheduled for surgery to remove the tumor and to sample nearby lymph nodes
to determine if the cancer has spread. Surgical procedures include a breast
conserving therapy, known as lumpectomy, in which the cancerous lesion and a
margin of surrounding normal tissue is removed, and mastectomy, in which the
entire breast is removed. It is estimated that at least 50% of women with breast
cancer, typically women whose breast cancer was detected at an early stage, are
good candidates for lumpectomies. In most cases, a course of radiation therapy
after lumpectomy is part of the treatment, as a means of destroying any cancer
cells that may remain. Additionally, 75% of women who have mastectomies go on to
have surgical reconstruction of one or both breasts, either using artificial
implants or their own body tissue to rebuild the breast. Some women who have
lumpectomies also choose breast reconstruction for cosmetic
improvement.
-2-
Evolution
of Breast Biopsy Procedures
According
to the Millennium Research Group, there were approximately 1.9 million breast
biopsies performed in the United States in 2009. This number has increased
significantly since 1997, at which time approximately 750,000 biopsies were
performed, and is expected to continue to increase year-over-year. In 1997, the
American Cancer Society updated its guidelines for breast cancer screening,
recommending that women should begin annual screening at age 40. The previous
guideline had recommended annual mammography for women beginning at age 50. This
updated guidance, along with increased public awareness and technological
improvements in screening and diagnostic equipment, likely has contributed to
the increase in breast biopsies over the past decade. Biopsy methods include
surgical biopsy, needle biopsy, and vacuum-assisted biopsy, all of which,
according to the American Cancer Society, have similar accuracy
rates.
Surgical Biopsy.
Traditionally, most breast biopsies were performed as open surgical procedures,
and such procedures remain common today, often being preferred for large
lesions. The procedure has several drawbacks. Surgery is highly invasive,
requires at least one full day of recovery and can leave a visible scar and
depression at the site of the removed tissue. It can also lead to scar tissue
formation within the breast, which can complicate the interpretation of
follow-up mammograms.
Needle Biopsy. Needle biopsy
emerged as the first minimally-invasive biopsy technique, enabling extraction of
tissue samples without surgery, but rather through insertion of a needle to
remove tissue. The typical procedure involves repeated needle insertions to
acquire multiple samples. If the breast lesion is large enough to feel, the
physician can do a needle biopsy by directly guiding the needle into the lesion.
If the lesion is too small or too deep within the breast to be felt, a needle
biopsy is done using breast imaging methods to guide the needle into the lesion.
There are two types of needle biopsy:
|
|
·
|
Fine
needle aspiration uses a fine-gauge, hollow needle and a syringe to sample
clusters of cells from a lesion. While fine needle aspiration is the
fastest and simplest procedure among all biopsy methods, it is typically
used only on lesions that are large enough to be felt. An experienced
breast cytopathologist is required to determine if cells are cancerous,
but this method cannot distinguish between cancer that remains confined to
particular cells, known as in situ cancer, from invasive cancer that has
spread to surrounding tissue, two types of cancer that are generally
treated differently.
|
|
|
·
|
Core
needle biopsy uses an inner notched needle and a larger outer hollow
needle, which are sequentially advanced, to cut and collect single tissue
samples. Compared to fine needle aspiration, core needle biopsy allows for
a more accurate assessment of a breast lesion because the larger core
needle usually removes enough tissue for the pathologist to evaluate
abnormal cells in relation to the surrounding small sample of breast
tissue taken in the specimen. However, this method is not well-suited to
characterizing small lesions or calcifications that may indicate early
cancers. False negatives may occur if the needle misses the lesion and
instead takes a sample of normal tissue, which may lead to the undiagnosed
cancer going untreated. Also, it is sometimes difficult to penetrate dense
breast tissue with core needles.
|
Vacuum-Assisted Biopsy. In a
single sample, vacuum-assisted biopsy is able to remove approximately six to ten
times as much breast tissue as core needle biopsy. Vacuum-assisted biopsy
incorporates a special probe that can capture tissue samples from a single
insertion into the breast through a small nick in the skin, promoting minimal
patient discomfort and a relatively short procedure time. Vacuum-assisted biopsy
is typically performed with imaging technology, either using a specially
designed stereotactic x-ray table to pinpoint the site of the lesion, or with
real-time ultrasound guidance. During the procedure, vacuum is used to draw
tissue from the lesion into an opening located on or at the end of the biopsy
probe, and a cutter is then used to sever this tissue sample. Among the
minimally-invasive biopsy options, vacuum-assisted biopsy is the only method
that can obtain multiple contiguous tissue samples with a single insertion,
which makes the procedure an attractive alternative for most lesions, including
those that may be indicative of early stage cancer.
A
vacuum-assisted biopsy device can either be an “open” or “closed” system. An
open system requires each sample to be individually cut, extracted and removed
from the device, a relatively time consuming process that requires the
assistance of a technician. The first open system vacuum-assisted biopsy device,
the Mammotome, was introduced in 1995. A closed system automatically transports
samples to a sealed tissue collection chamber, allowing multiple samples to be
collected without having to interrupt collection by removing tissue from the
device. In 2002, a closed system vacuum-assisted biopsy device called the ATEC
was introduced. Both open and closed systems have been widely adopted. While
closed systems may result in faster procedure times and minimize fluid loss,
open systems allow visualization of the sample, which may be preferred by a
technician to ensure that the proper area of the breast is being targeted.
Today, in many cases customers prefer a vacuum-assisted biopsy system that is
compatible with all three major imaging modalities, stereotactic, ultrasound,
and MRI.
-3-
According
to the Millennium Research Group, vacuum-assisted biopsies became the
predominant biopsy method, surpassing open surgical breast biopsy procedures in
2007. Vacuum-assisted biopsies are already the predominant minimally-invasive
method, accounting for 681,700 of 1,839,500 such procedures performed in the
United States in 2008. Millennium projects that vacuum-assisted
biopsies will account for 1,075,900 of 2,342,200 such procedures performed in
the United States in 2013.
Evolution
from Whole to Partial Breast Irradiation Therapy
Following
a lumpectomy to remove a cancerous breast tumor, many patients are subsequently
treated with breast radiation therapy to destroy any cancer cells that may
remain. Similar to the evolution in breast biopsy toward minimally-invasive
procedures, radiation therapy is beginning to transition from whole breast
radiation, which is currently used in the vast majority of cases, to more
localized radiation therapy.
Whole Breast Radiation.
Following a lumpectomy, the current standard of care is to treat patients with
external beam radiation that is widely directed at the whole breast. Although
the use of radiation has improved long-term survival rates, this treatment is
inconvenient for patients, often requiring daily outpatient radiation treatments
for six to eight weeks, and can expose healthy tissue and organs to damaging
effects from the radiation.
Accelerated Partial Breast
Irradiation, or APBI. APBI delivers localized radiation to a targeted
surgical site. For appropriate patients, this method offers a number of
advantages including greatly-diminished treatment time, concentrated radiation
exposure and the reduction of skin irritation and burning. There are currently
three approaches to APBI in the market.
|
|
·
|
Radiation
Balloon Brachytherapy. This approach involves a catheter attached to a
fluid expandable balloon that is inserted into the lumpectomy cavity. A
mixture of saline and contrast media is injected to expand the balloon to
contact the walls of the lumpectomy cavity, a radioactive source is then
inserted into the balloon. Radiation is administered for five to ten
minutes, allowing a therapeutic radiation dose to penetrate tissue
approximately one centimeter from the exterior of the balloon. Typically,
this procedure is repeated twice a day for five days on an outpatient
basis. While the technical challenges are fewer than with other partial
breast radiation treatment options, delivering a uniform radiation dose
remains an obstacle due to the difficulties in conforming the shape of the
balloon to the walls of the often irregularly-shaped cavity, the site of
the cavity, or accumulation of fluid from the body around the balloon.
Radiation balloon brachytherapy is in the early stage of adoption by the
market.
|
|
|
·
|
Conformal
Radiotherapy. Like whole breast radiation, this approach uses a radiation
source outside the body. Rather than targeting the entire breast, however,
this approach typically uses a CT scan to pinpoint the tumor site in three
dimensions and a computer program to aim radiation beams that “conform”
closely to the shape of the tumor and thereby helping to minimize damage
to healthy tissue. This approach is well-established in the treatment of
prostate cancer, but is in the early stages of adoption and clinical study
for breast cancer. Conformal radiotherapy may play a greater role in
brachytherapy in the future.
|
|
|
·
|
Multi-Catheter
Interstitial Brachytherapy. This approach involves placing 20 to 30 small
catheters completely through the breast at carefully selected locations
around the lumpectomy site. A radioactive source is inserted into the
catheters twice a day for about 20 minutes to deliver radiation, typically
over a one-week period, during which time the catheters remain in the
breast. The multiple catheter placements may cause infection, as well as
potential cosmetic damage. Additionally, the procedure requires a high
level of technical expertise for appropriate catheter placement and
radiation dose administration.
|
-4-
Breast
Care Market Trends
The
breast care market has undergone a significant evolution over recent years,
driven by advancements in imaging technologies, which has led a paradigm shift
to less invasive devices and procedures for screening and for diagnosis, and to
comprehensive patient care through integrated breast centers.
Advances in Imaging Technology for
Screening. Digital mammography is being rapidly adopted as clinical
studies suggest advantages over traditional film mammography. When the patient
has dense breast tissue, breast implants or is breastfeeding, the images
produced by either traditional or digital mammography can be difficult for a
radiologist to interpret. Consequently, physicians will often order a secondary
screening using ultrasound. MRI may also be used as a secondary screening method
for these patients, and is being recommended as a primary screening technique
for high-risk women as young as 30. Breast tomosynthesis, a newer
form of digital mammography which is an emerging technology that is currently in
the experimental development stage. Tomosynthesis is a
process in which multiple 2D x-rays of the breast are combined using computer
software to produce a more focused 3D image.
Advances in Imaging Technology for
Biopsy. Technological advances in imaging have allowed for more effective
and less invasive diagnosis and treatment of breast cancer. While stereotactic
x-ray imaging and ultrasound guidance are used most frequently in conjunction
with biopsy procedures, MRI may also become an important alternate imaging
modality for both diagnosis and treatment.
|
|
·
|
Stereotactic
x-ray imaging uses x-ray to capture images of breast tissue. With the
patient lying on a specialized treatment table known as a stereotactic
table, x-ray images are taken from two angles which permits integrated
computerized equipment to map the exact location of the target lesion,
thereby enabling the physician to fire a biopsy needle or probe into the
lesion. Stereotactic imaging is used in the vast majority of
vacuum-assisted biopsies today.
|
|
|
·
|
Ultrasound
imaging bounces low-power, high-frequency sound waves off internal tissue
to provide real-time images of the interior of the breast to guide the
physician’s manual placement of a biopsy probe at the site of the lesion.
Ultrasound is widely available and relatively inexpensive, and does not
expose the patient to radiation. As a screening tool, ultrasound is used
as a primary breast imaging application for biopsy of women who have dense
breast tissue, typically under the age of 40, women with breast implants
or women who are breastfeeding.
|
|
|
·
|
Magnetic
resonance imaging (MRI) uses a magnet, radio waves and a computer to make
a series of detailed images of the inside of the breast. Technological
advances have made MRI an emerging alternative for image-guided biopsy.
Several academic institutions and leading breast centers have begun
performing biopsies under MRI guidance. Although there are several
hurdles, including the relatively high cost of and significant time
required for this procedure, the clinical benefits of this approach are
gaining acceptance.
|
Paradigm Shift to Less Invasive
Procedures. Advancements in imaging technology have allowed abnormal
breast tissue to be identified at an early stage and have helped facilitate the
emergence of novel, less invasive diagnostic and therapeutic devices for
accessing and removing this tissue. For example, open surgical biopsies and
needle biopsies are giving way to minimally-invasive vacuum-assisted procedures.
Similarly, excision of tumors is shifting from invasive mastectomies to less
invasive lumpectomies. In addition, radiation treatment is shifting from whole
breast radiation to partial breast radiation.
Emergence of Integrated Breast
Centers. Effective screening, diagnosis and treatment of breast cancer
require interaction among specialists in multiple departments of a healthcare
system. These include, but are not limited to, surgery, oncology, radiology,
pathology and plastic surgery. While many of these services exist in any given
healthcare system, the concept of a breast center is to organize these services
into a coordinated, multidisciplinary approach where the patient’s care is
integrated. This coordinated approach allows for higher-quality and more
patient-focused care than she might receive from the same specialists working in
isolation. Breast centers can be at one physical location, with all services
available at one site, or they can be virtual, organizing the interaction of
diverse services found at different locations. A factor in the accelerating
establishment of integrated breast centers is the increasing public awareness of
the importance of quality breast care. Breast centers are also actively
educating the general public as it relates to the latest clinical and
technological advances available in minimally-invasive diagnosis and
treatment.
-5-
We
believe that there is significant opportunity for a company that offers breast
centers a full range of minimally-invasive diagnostic and therapeutic devices
that are both compatible with multiple imaging modalities and flexible enough to
be tailored to the diverse needs of the physicians on the breast care
team.
Our
Solution
We have
commercialized, and are continuing to develop, a broad product line of
minimally-invasive breast care devices to be used by breast care specialists. By
focusing on the continuum of care from diagnostic to excision and therapeutic
procedures, we believe that we will be an attractive and convenient supplier for
integrated breast centers.
Our
Breast Care Management Product Continuum

Our
current commercial products and products under development include:
|
|
·
|
Diagnostic.
Breast biopsy systems, location and lymph node gamma ray detection
devices.
|
|
|
·
|
Marking.
Tissue markers which identify the biopsy site, under the major imaging
modalities, for future diagnostic and surgical reference, and are
compatible with most imaging technologies and biopsy
devices.
|
|
|
·
|
Lesion
Location. Devices used to facilitate lesion excision and
treatment.
|
|
|
·
|
Treatment.
Radiation balloons and other devices for localized partial breast
irradiation therapy.
|
|
|
·
|
Excising.
Tissue cutting devices designed to facilitate the contoured removal of
lesions and facilitate the use of balloons in radiation
therapy.
|
The EnCor
system, our flagship product for use in breast biopsy procedures, is a
vacuum-assisted system which allows users to obtain multiple tissue samples with
a quick, single insertion. EnCor can be used with multiple imaging modalities,
including stereotactic x-ray, ultrasound and MRI. EnCor is the only
“open/closed” tissue collection system compatible in all three imaging
modalities, providing the operator with a clear view of tissue samples through a
proprietary transparent collection chamber, and the ability to either open the
chamber to examine and remove one or more samples or to continue uninterrupted
collection of multiple samples. Our EnCor system also incorporates proprietary
programmability and automation features which provide a competitive advantage to
other marketed biopsy systems. In 2007, we further enhanced the versatility of
the EnCor system with the FDA clearance in the United States of our SenoSonix
system, a combination of EnCor with state-of-the art ultrasound imaging
technology, and with the commercial launch of VisiLoc, an MRI visible obturator
that helps to facilitate accurate probe placement under MRI
guidance.
The
Contura Multi-Lumen Radiation Balloon Catheter, or Contura MLB, our flagship
radiation therapy product, for which we received FDA 510(k) clearance in May
2007, launched in January 2008 and received its CE Mark in Europe in March 2009,
is designed to be a novel localized partial breast irradiation therapy device
that uses vacuum to remove excess seroma and air to enhance conformance of often
irregularly shaped lumpectomy cavity walls to the balloon’s surface in order to
deliver precise radiation dosing through multiple radiation source lumens. In
addition, we began marketing Contura Shape Select MLB in 2009, a new larger
version of Contura MLB with a novel proprietary design that provides clinicians
with enhanced flexibility.
-6-
Our
Strategy
Our goal
is to become the leader in providing minimally-invasive solutions across the
continuum of care in the breast care market. The key elements of our business
strategy to achieve this goal are to:
|
|
·
|
Provide
Differentiated, Tailored Solutions in the Breast Biopsy Market. We believe
that our EnCor breast biopsy system represents a significant advancement
in the breast biopsy market. We seek to leverage this recent product
introduction to establish a leadership position in the minimally-invasive
breast biopsy market. We believe that by making the EnCor system modular
and upgradeable, which enables the addition of features over time,
customers will view it as an attractive platform product that can be
tailored to the needs of their practice. The EnCor system allows for
significant flexibility across multiple imaging modalities,
programmability, automation and the ability to shift between open and
closed tissue collection. In addition to EnCor, we believe that EnCor 360
and SenoSonix with EnCor is a complementary product that will continue to
address a need in the ultrasound guided vacuum-assisted biopsy market. We
intend to continue to develop and commercialize advanced products in the
minimally-invasive breast biopsy market, such as VisiLoc, which helps to
facilitate accurate biopsy probe placement under MRI. Additionally, we
believe that the EnCor hardware architecture and the ease of software
upgrades allows us to continuously and easily bring technological
improvements to market and to easily interface with imaging equipment sold
by a broad spectrum of
manufacturers.
|
|
|
·
|
Focus
on leveraging existing R&D Competencies to Develop Products Across the
Continuum of Interventional Breast Care. While our initial product focus
has involved devices used in diagnosis of breast cancer, such as biopsy
systems and breast tissue markers, we launched Contura MLB in January 2008
and are also developing a series of additional lesion location, excision
and therapeutic products that we are seeking to commercialize. With the
emergence of integrated breast centers designed to provide comprehensive
and specialized patient care, we believe that our ability to provide novel
solutions to a broad set of needs in breast cancer management will enhance
our competitive positioning in the market. We are also working on a next
generation EnCor product.
|
|
|
·
|
Leverage
Clinical Education with Key Opinion Leaders in Breast Care Centers. We
believe that integrated breast centers are emerging as the focal point for
breast care, with teams of surgeons, radiologists, oncologists and
technicians providing coordinated care. Our products, spanning the
continuum of breast care from diagnostics to therapeutics, positions us to
meet many clinical needs of breast centers. As a key element of our
strategy, we focus on educating and training clinicians on our products
through frequent hands-on classes and industry events. We have worked with
key opinion leaders in training several thousand clinicians in the
effective use of our products.
|
|
|
·
|
Capitalize
on Product Cross-Selling Opportunities within Our Existing Customer Base.
We believe that we have a significant opportunity to grow our revenues by
selling additional products to existing customers. We have established a
strong base of customers who have a history of placing repeat orders for
our products. We believe this customer base is an attractive target for
early adoption of our complementary products as they are commercially
launched. For example, our EnCor and EnCor 360 biopsy hardware,
disposables and tissue markers are sold to the same surgeon customers that
are purchasing Contura MLB.
|
|
|
·
|
Pursue
Strategic Acquisitions and Partnerships. In addition to adding to our
product portfolio through internal development efforts, we intend to
explore the acquisition of other product lines, technologies or companies
that may leverage our sales force or be complementary to our strategic
objectives. We may also evaluate distribution agreements, licensing
transactions and other strategic partnerships, which may include expansion
of our selling and marketing efforts beyond the United States. We are also
collaborating with a number of large and smaller corporations who sell
imaging products to offer our customers bundled packages of products and
joint clinical education
programs.
|
-7-
Our
Products and Products under Development
We are
focused on developing and offering a broad portfolio of products that address
needs across the continuum of breast care, from the diagnosis to the treatment
of breast cancer. The sale of disposable products, including our breast biopsy
probes, tissue markers and radiation therapy products, accounted for 90.0% of
our revenues in 2009, 87.3% in 2008 and 86% in 2007. The following table
provides information concerning our primary products and products under
development.
|
Product
Category/Name
|
Primary
Component(s)
|
Year
(or
Expected Year)
of
Full
Commercial
Launch
|
||
|
DIAGNOSTIC
PRODUCTS
|
||||
|
Breast
Biopsy
|
||||
|
SenoRx
Breast Biopsy Console
|
Console
for EnCor and EnCor 360 Biopsy Devices
|
2002 | ||
|
EnCor
|
Reusable
Handpiece and Disposable Probe
|
2005(1)
|
||
|
EnCor
360(3)
|
Reusable
Handpiece and Disposable Probe
|
2003(1)
|
||
|
VisiLoc
|
Obturator
Compatible with EnCor MRI
|
2008
|
||
|
SenoSonix
|
EnCor
Hardware Integration with ultrasound imaging
|
2008
|
||
|
SenoSonix
Hand-Carry
|
EnCor
hardware with hand-carry ultrasound component
|
2009
|
||
|
EnCor
II
|
Next-generation
EnCor with touch-screen
|
2010
|
||
|
Tissue
Markers and Location Devices
|
||||
|
Gel
Mark
|
Applicator
and Combination Metal/Bioresorbable Markers
|
2002
|
||
|
Gel
Mark Ultra
|
Applicator
and Combination Metal/Bioresorbable Markers
|
2004
|
||
|
Gel
Mark UltraCor
|
Applicator
and Combination Metal/Bioresorbable Markers
|
2004
|
||
|
SenoMark
|
Applicator
and Combination Metal/Bioresorbable Markers
|
2006
|
||
|
Gel
Mark UltraCor MRI
|
Applicator
and Combination Metal/Bioresorbable Markers
|
2006(1)
|
||
|
Starch
Mark
|
Metal/Bioresorbable
Markers
|
2009
|
||
|
RFID
tag location device
|
Radio
frequency identification lesion location device and accompanying handheld
reader
|
2010
|
||
|
Tissue
Marker Line Extension
|
Applicator
and Combination Metal/Bioresorbable Markers
|
2011
|
||
|
Gamma
Ray Detection
|
||||
|
Gamma
Finder
|
Reusable
Probe and Disposable Sleeve
|
2003
|
||
|
THERAPEUTIC/EXCISION
PRODUCTS
|
||||
|
Radiation
Therapy
|
||||
|
Contura
MLB
|
Radiation
Balloon
|
2008
|
||
|
Contura
Shape Select MLB
|
Contura
MLB that expands to multiple shapes
|
2009
|
||
|
CED
Device
|
Radiation
Balloon Introducer
|
2010
|
||
|
CavityMark
|
Next-Generation
Radiation Therapy Product
|
2011
|
||
|
Excision
and Reconstruction
|
||||
|
SenoPulse
RF Generator
|
Console
for Excision/Reconstruction Devices
|
2011(2)
|
||
|
Single
Step
|
Reusable
Handpiece and Disposable Probe
|
2011(2)
|
________________
|
(1)
|
FDA
510(k) clearance received and product available prior to full commercial
launch.
|
|
(2)
|
FDA
510(k) clearance received prior to full commercial launch. Initial
preference testing may occur one year
earlier.
|
|
(3)
|
Previously
referred to by us and marketed as
SenoCor 360.
|
-8-
Breast
Biopsy Systems
Components
of Our Breast Biopsy Systems
Our
breast biopsy systems primarily consist of two components—reusable handpieces
and disposable probes—and are used in conjunction with our SenoRx Breast Biopsy
Console.
|
|
·
|
SenoRx
Breast Biopsy Console. The SenoRx Breast Biopsy Console is compatible with
both our EnCor and EnCor 360 reusable handpieces and disposable probes.
This modular console is a portable hardware system which may be
conveniently transported to various areas of the healthcare facility. The
primary modules of our console
include:
|
|
|
·
|
a
control module, which facilitates convenient user interface with
proprietary software, a visual display screen, and controls that allows
the user to customize the various parameters of the diagnostic procedure;
and
|
|
|
·
|
a
vacuum system, which pulls tissue into the probe for excision and
subsequent delivery of tissue to the sample collection
chamber.
|
|
|
·
|
Reusable
Handpieces. Handpieces are instruments which facilitate placement or
insertion of the biopsy probe. The handpieces primarily consist of motors,
circuitry, sensors and proprietary software incorporated into a housing.
We commercialize three different biopsy handpieces: EnCor
Stereotactic/Ultrasound, EnCor MRI, and EnCor
360.
|
|
|
·
|
Disposable
Probes and Accessories. Our probes are sterile, single-use,
vacuum-assisted disposables, which are used with our EnCor and EnCor 360
handpieces. They consist of a sharp stainless steel tissue cutter and
needle, coupled with EnCor, a tissue sample collection chamber. The probe
accessories also include additional tubing and a vacuum
canister.
|
We offer
probes in a variety of sizes, ranging from 7-gauge to 10-gauge, depending on
user preference. The probe cutters and tips incorporate one or more of our
proprietary tissue cutting technologies.
|
|
·
|
Tri-Concave
Tip. A three-edged tip used on our EnCor probes, EnCor 360 probes and MRI
insertion device, designed especially for facilitating easy placement into
dense breast tissue.
|
|
|
·
|
360°
Tissue Cutter. A hollow, cylindrical cutting edge used on our EnCor 360
probes, which automatically rotates and advances to generate large 360°
contiguous samples.
|
|
|
·
|
Oscillating
Cutters. Cone-shaped cutters used on our EnCor probes, which shear tissue
in a manner similar to a scissors
cut.
|
We also
received clearance for and launched additional products that are used together
or in conjunction with our EnCor system.
|
|
·
|
VisiLoc
MRI Visible Obturator (2007). An MRI visible obturator that is used during
an MRI-guided EnCor procedure, designed to facilitate biopsy probe
placement under MRI guidance.
|
|
|
·
|
SenoSonix
with EnCor (2007). An integration of our EnCor system with a
state-of-the-art third-party ultrasound imaging
systems.
|
|
|
·
|
SenoSonix
with EnCor Handcarry (2009). SenoSonix with a portable hand-carry laptop
ultrasound component.
|
-9-
EnCor
Breast Biopsy System
Our
flagship product for use in breast biopsy procedures, the EnCor system, is a
vacuum-assisted breast biopsy system that facilitates adoption of
minimally-invasive biopsy procedures over open surgical biopsy. The EnCor system
is comprised of a reusable handpiece and disposable probes that are used in
conjunction with our SenoRx Breast Biopsy Console. We believe the EnCor system
offers a comprehensive set of features which make it an attractive solution for
meeting the diverse demands of breast care providers. Key features of the EnCor
system include:
|
|
·
|
“Open/Closed”
System. Functions either as an open or closed system, providing the
operator with a clear view of tissue samples through a proprietary
transparent collection chamber, and the ability to either open the chamber
to examine and remove one or more samples or to continue uninterrupted
collection and automatic transfer of multiple samples from inside the
breast to the collection chamber.
|
|
|
·
|
Highly-Automated
and Programmable. Automated and programmable tissue collection and
automated anesthetic delivery. Aligns and rotates automatically through a
variety of optional programmed cutting patterns. Provides multiple
programmed options for users to choose their own approach to the array of
lesions they may encounter.
|
|
|
·
|
Multi-Modality
System. Compatible with each of the three major imaging modalities used in
the market—stereotactic, ultrasound and MRI—and transportable, eliminating
the need for multiple systems.
|
|
|
·
|
Modular
Hardware and Upgradeable Design. Customers are able to purchase all or
part of the system according to their needs, thus minimizing up-front
costs. In addition, the modular system facilitates easy repair and
replacement of components. Software-based design allows us to continuously
innovate by adding new features which may extend the useful life of the
device. The user may update the system by upgrading software rather than
purchasing new hardware.
|
|
|
·
|
Precise
Probe Placement. Tools to facilitate accurate placement of EnCor probe
under MRI imaging.
|
The EnCor
system also incorporates a number of additional features, including
compatibility with our tissue markers, multiple gauge sizes, automated sample
rinsing, lighting, an ergonomically-designed handpiece, noise reduction and
novel MRI probe insertion accessories and proprietary ultrasound options. We
received FDA 510(k) clearance and conducted marketing preference testing of the
EnCor system in 2004, with full commercial launch in 2005.
EnCor
360 Breast Biopsy System
Our EnCor
360 system utilizes a vacuum to provide the physician with a contiguous 360°
breast biopsy sample. The EnCor 360 system incorporates our mechanical
Tri-Concave Tip to penetrate virtually any lesion, regardless of size, location
or density. EnCor 360 secures tissue through the end of the probe, providing a
large, high-quality sample. Since the EnCor 360 is interchangeable and
compatible with the SenoRx Breast Biopsy console, users may select EnCor 360 or
EnCor depending upon their clinical and economic objectives.
We
received FDA 510(k) clearance in 2002 and launched EnCor 360 in 2003, as our
initial product in the vacuum-assisted breast biopsy segment. We intend to
continue to offer the EnCor 360 as a low-cost, ultrasound-guided breast biopsy
device. We believe that our EnCor 360 and future product enhancements will
continue to appeal to clinicians doing ultrasound biopsies in their offices,
which is a more price-sensitive segment of the biopsy market.
SenoSonix
with EnCor
In
October 2007 we received 510(k) clearance from the FDA for SenoSonix with EnCor,
an integration of our EnCor system with a state-of-the-art ultrasound imaging
system developed by third-party. We received the right to affix CE Mark in the
European Union for SenoSonix with EnCor in April of 2008 and patents are
currently pending. We are marketing SenoSonix with EnCor primarily for physician
in-office procedures, particularly in Europe, where we believe a greater
percentage of biopsy procedures are done under ultrasound imaging guidance.
SenoSonix with EnCor may be used with either our EnCor or EnCor 360
probes. In April 2009, we introduced a new version of SenoSonix that
incorporates “hand-carry” laptop ultrasound technology from a second third party
ultrasound OEM source. This version of SenoSonix has increased ease of
portability and is well suited for the target physician in-office
market.
-10-
EnCor
II
We are
currently developing a next Generation EnCor System that contains all the
functional capabilities of
the
current EnCor, but is integrated into a new ergonomic, modern design package
that incorporates a PC-based, 15 inch Touch Screen Display. The display provides
a new, large intuitive user’s interface for easy and flexible control of all
features of the EnCor, EnCor360 and EnCor MRI platforms. The display feature
also enables:
|
|
·
|
Interactive
Help and Troubleshooting
|
|
|
·
|
Upgrades
through USB Memory Sticks or Internet File Types: pdf, jpg, avi,
wma
|
|
|
·
|
Integrated
Operator IFU and (password protected) Hardware
Service
|
|
|
·
|
MRI
Quick Reference Guide and Grid
Sheets
|
|
|
·
|
Training
Videos including for stereotactic and MRI
use.
|
|
|
·
|
Ease-of-integration
with multiple PC based ultrasound imaging
systems
|
We expect to commercially launch this
product in the second half of 2010.
VisiLoc
MRI Visible Obturator
We
launched the VisiLoc MRI Visible Obturator in the United States in November
2007. VisiLoc is an MRI visible obturator that is used during an MRI-guided
EnCor procedure and is designed to help facilitate biopsy probe placement under
MRI guidance.
Gel
Mark Ultra, SenoMark, StarchMark, and RFID Tag (Biopsy Site Tissue
Markers)
Biopsy
site tissue markers are placed at a biopsy site to provide a visible landmark
for future surgical reference. If cancer is found and more tissue must be
removed from the breast, the marker will help the physician identify the
specific area from which tissue should be removed. If surgery is not necessary,
the marker will be visible on future mammograms to enable the breast care
specialist to identify the site of the biopsy.
We offer
a full portfolio of tissue markers that are compatible not only with our EnCor
and EnCor 360 biopsy product lines, but also with competing biopsy systems. Our
products consist of markers that come in a variety of materials, including
gelatin and synthetic materials, titanium and stainless steel, and associated
delivery applicators. The markers are designed to facilitate easy placement and
optimize visibility under different imaging modalities.
We were
first to commercialize markers visible not only under x-ray, but also ultrasound
imaging. Gel Mark Ultra is designed to provide pellet-shaped,
ultrasound-visible, bioresorbable tissue marker alternatives. Gel Mark UltraCor
provides core needle users with an ultrasound-visible tissue marking
alternative. SenoMark provides those users who prefer a pad-shape with an
ultrasound-visible, bioresorbable tissue marker alternative. StarchMark provides
polysaccharide (starch) pellets to help manage and control bleeding. Our
UltraCor MRI may be an attractive alternative for clinicians interested in
marking lesions under MRI guidance. We received FDA 510(k) clearance and began
commercializing Gel Mark in 2002, Gel Mark Ultra and Gel Mark UltraCor in 2004,
and SenoMark in 2006. We received FDA 510(k) clearance and conducted marketing
preference testing of the Gel Mark UltraCor MRI in 2004, with full commercial
launch occurring in the second half of 2006. We received FDA 510(k) clearance
and conducted marketing preference testing of StarchMark in 2008, and in April
2009 commercially launched the product.
We
submitted an application for FDA 510(k) clearance in the fourth quarter 2009 for
a new radio frequency identification (RFID) localization system that allows
placement of an RFID tag into a breast lesion preoperatively, typically by a
radiologist, for subsequent intraoperative lesion localization and removal by a
surgeon. We have not received clearance from the FDA on this product and cannot
predict when such approval will ultimately be obtained, if at all. The system
employs a needle/syringe-type applicator to percutaneously place the tag in
conjunction with a RFID reader. This novel system eliminates the need for
preoperative localization wire placement and eliminates the need for close
schedule coordination between the radiologist and the surgeon as surgery can be
scheduled up to 7 days post tag implantation instead of the same day. The system
consists of an applicator, RFID tag, RFID reader, and sterile probe covers. The
disposable applicator is used to insert the RFID tag into the breast at the site
of a lesion to mark its location. Placement is conducted under ultrasound or
x-ray guidance. The reader displays the distance between the tag and the reader
and the tag location is also indicated by an audible sound whose pitch and
volume is proportional to the “reader to tag” distance. The RFID tag is designed
to prevent migration within the tissue after
implantation
by incorporating a wire winding with two projecting ends. The RFID reader is a
reusable handheld device that has two probes with different detecting ranges; a
loop probe that has a detecting range of 0-6cm, and a pencil probe attachment
that has a detecting range of 0-3cm, to allow for closer localization in the
surgical field. Two different covers are provided to maintain reader sterility
during explantation. We are currently conducting market research on the suite of
products and intend to commercially launch later in 2010.
-11-
Gamma
Finder (Gamma Ray Detection Device)
Immediately
prior to removal of a malignant lesion in the breast, a patient may be injected
with gamma ray emitting isotopes near the site of the lesion to determine if the
cancer has spread. Our Gamma Finder is currently the only cordless handpiece
probe to detect the emission of gamma rays, and, consequently, whether breast
cancer has spread to the lymph nodes. The Gamma Finder detects and gives a
numerical indication and an acoustic signal when close to a gamma ray emitting
source. Our Gamma Finder has all the features of traditional, larger, corded
gamma ray detection devices, with the convenience of a portable and compact
device. The Gamma Finder consists of a reusable probe and a disposable sterility
sleeve. We began commercializing the Gamma Finder in 2003 upon receipt of FDA
510(k) clearance, and, in 2005, added automatic ten second count and binary
pitch mode features.
Contura
MLB Radiation Balloon
Current
radiation therapy includes less invasive alternatives to whole breast radiation
therapy, known as partial breast irradiation therapy, consisting of balloon
brachytherapy, conformal radiotherapy and multi-catheter interstitial
brachytherapy. We believe that balloon brachytherapy will be widely adopted over
time due to its ease of use, low-cost and clinical effectiveness. In January
2008 we launched our Contura Multi-Lumen Radiation Balloon Catheter, or Contura
MLB.
Our
Contura MLB design consists of a multilumen catheter with several access ports
on one end and an inflatable balloon on the other. The balloon is positioned
into the cavity formed in the breast following a lumpectomy and subsequently
inflated with saline through one of the ports. Small openings in the catheter
allow for the suction of excess seroma and air from the lumpectomy cavity
through a second access port. We believe that this suction feature will help
conform the walls of the lumpectomy cavity to the exterior of the balloon.
Multiple lumens are designed to provide for precise placement of radioactive
seeds, which we believe will allow clinicians to expand their use of radiation
therapy to a greater number of patients. Such patients may include those with
smaller anatomies and with tumors closer to the skin, which were not previously
able to receive treatment with other minimally-invasive radiation balloon
products. The overall design and the use of special balloon materials is
intended to control the distance between the radiation source and the tissue in
contact with the balloon and to result in controlled radiation
dosing.
We
received FDA 510(k) clearance for Contura MLB in May 2007, launched in January
2008 and received CE Mark in Europe in March 2009. We began selling Contura MLB
in several select countries outside the United States in 2009 and plan to
further expand distribution in 2010. Also in 2009, we began marketing Contura
Shape Select MLB, a new larger version of Contura MLB with a novel proprietary
design that provides clinicians with enhanced flexibility in appropriately
fitting the lumpectomy cavity with one balloon catheter, which may be adjusted
into different sizes. We anticipate developing and commercializing additional
line extensions for this product over the next several years. Clinical
investigators for Contura MLB have either presented, published or had accepted
33 articles or abstracts related to Contura MLB.
CavityMark
The
cavity left after tumor resection post lumpectomy often makes 3D conformal
radiation targeting of the margins difficult because it may be irregularly
shaped, change with body movement and position and be radiographically
indistinct. Also, cosmetically undesirable dimpling of the skin after healing
commonly results from the surgery. The CavityMark is designed to address these
issues by helping form the post lumpectomy cavity into a spherical shape and
radiographically delineate a target for delivery of 3D conformal radiation
therapy. Secondarily, the device helps to provide a scaffold for tissue growth
and thereby minimize post lumpectomy skin dimpling. This product is currently in
development and we have not yet filed for any regulatory clearances to market
it.
-12-
SenoPulse
RF Generator (Excision/Therapeutic Console)
The
SenoPulse RF Generator will be used to power our radiofrequency cutting
technologies in order to provide advanced breast tissue cutting capabilities.
The SenoPulse RF Generator offers high-frequency and impedance-matching
circuitry to enable cutting into a wide variety of tissue types, high start
voltage and sustained power for continuous cutting ability, and low heat
generation to minimize thermal damage. The SenoPulse RF Generator directs
modulated, monopolar radiofrequency energy and will be used in the Single Step
cutting and excision devices. We received FDA 510(k) clearance for the SenoPulse
RF Generator in 2005.
Tissue
Cutting Devices for Excision and Reconstruction
Single
Step is an alternative excision device to a scalpel or a straight-bladed
electrosurgical scalpel commonly known as a Bovie. The Single Step system is an
automated surgical excision device that uses a long-wire RF disposable probe and
reusable handpiece to cut and remove a large, intact volume of tissue through a
small surgical incision. The Single Step system is powered by the SenoPulse RF
Generator. The Single Step probe is inserted into the breast, where the surgeon
anchors the device and excises tissue of a predetermined size. The surgeon
controls the amount and shape of the tissue removed by selecting the appropriate
option on the SenoPulse RF Generator. The Single Step is designed to produce a
smooth and optimally-shaped cavity to facilitate lesion removal and subsequent
use of balloon brachytherapy. We intend to build upon data obtained with one of
our previous products by evaluating the clinical benefit of anchoring or
stabilizing lesions in conjunction with the automatic lesion cutting ability of
our Single Step system. We received FDA 510(k) clearance for the Single Step and
anticipate making several design enhancements and conducting clinical testing
prior to fully commercializing the product in 2011. We have also received FDA
510(k) clearance for a second, long wire RF cutting device that may be useful in
lesion excision and tissue reconstruction.
Sales
and Marketing
We focus
our sales and marketing efforts on increasing awareness of our products among
breast care specialists, including radiologists, surgeons and oncologists. We
market and sell our products through a direct sales force in the United States.
As of December 31, 2009, we employed a president and chief operating
officer, a vice president of global marketing, and support staff of 69 direct
sales employees, including 19 clinical specialists, nine brachytherapy
specialists, six regional sales managers and 35 sales
representatives.
In our
selling process, we use clinical studies, cost-benefit data and case studies. To
date, we have 39 clinical studies that have either been published or presented
as abstracts at major medical meetings. Peer-to-peer selling is also a critical
element of our strategy. We have developed popular training seminars, including
a Continuing Medical Education-accredited course led by our internal personnel
and by nationally-known breast cancer specialists Drs.
Nathalie Duchesne, Mark Gittleman, Phillip Israel, Terese Kaske, Frank Vicini,
Doug Arthur, Dorin Todor, Phillipe
Sebag, Rakesh Patel and Robin Wilson who present our clinical data and/or
products from time to time. We hosted 73 seminars in 2009, educating and
providing hands-on training to over 1,500 clinicians about our
products. We initiated a Contura MLB long-term physician registry study with
more than 80 patients enrolled at the end of 2009.
An
additional element of our educational efforts is our relationships with several
manufacturers of ultrasound imaging systems. With these companies, we co-sponsor
several breast practice seminars across the country to educate clinicians on the
changes that are driving the specialization of breast care and the emergence of
integrated breast centers. We also contribute to organizations designed to
increase awareness of breast cancer, including our sponsorship of the newsletter
and website of the American Society of Breast Surgeons.
International
sales have not historically accounted for a significant portion of total sales,
but have been increasing incrementally in recent years. We do not have a direct
sales force outside of the United States but rather contract with distributors
for sales coverage in a number of countries worldwide. We have the authorization
to affix the CE Mark to Gel Mark Ultra, Gel Mark UltraCor, and our EnCor system
and to commercialize these devices in the European Economic Community, Hong
Kong, Singapore, Taiwan, Mexico, South Korea, and Australia. In 2007, we
partnered with local distributors who have breast imaging and/or interventional
radiology franchises in Austria, Belgium, the United Kingdom, Hong Kong,
Ireland, Luxembourg, The Netherlands, Singapore, Switzerland, and Taiwan to sell
EnCor and our Gel Mark Ultra products in these ten countries. In 2008 we added
distributors in Denmark, Finland, France, Germany, Iceland, Italy, Mexico,
Norway, Portugal, Russia, South Korea, Spain and Sweden. In 2009 we added
distributors in China, Egypt, Greece, Poland, Saudi Arabia, Thailand, Turkey and
the United Arab Emirates. We are currently in negotiations with distributors for
a number of additional countries in which our products are already approved and
intend to further expand beyond these countries in 2010. Our International
Distributors are managed by two sales managers who report to our President, and
Chief Operating Officer.
-13-
Competition
We
compete primarily on the basis of our ability to provide minimally-invasive
products to diagnose and treat breast cancer safely and effectively, with ease
and predictability of product use, brand name recognition and cost. We believe
that we compete favorably with respect to these factors, although we cannot
assure you that we will be able to continue to do so in the future or that new
products that perform better than those we offer will not be
introduced.
The
markets in which our products compete are highly competitive, subject to change
and significantly affected by new product introductions and other activities of
industry participants. We face different competitors within different product
lines. To our knowledge, we do not have one competitor that produces products
that compete with all our products. Several of our competitors have significant
financial and human capital resources and have established reputations with our
target customers, as well as worldwide distribution channels that are more
effective than ours. We are aware that several companies are developing products
that, if successfully commercialized, would compete with our current and future
products.
Our
vacuum assisted breast biopsy and tissue marker products compete with, among
others, products sold by Johnson & Johnson, C.R. Bard and
Hologic. Contura MLB competes against well-established whole breast
external beam radiation devices, as well as current and potential future
manufacturers of balloon brachytherapy devices and image-guided targeted
radiation beam therapy. We compete directly with the current industry leader,
Hologic, as well as other companies that have minimally-invasive therapeutic
devices in various stages of development. Short-term brachytherapy products from
Cianna Medical and Xoft are also currently sold in the market. Our commercial
success will depend on a general market shift from whole to partial breast
irradiation and our ability to overcome Hologic’s current market leadership with
its current balloon product. Furthermore, we compete against Hologic with their
product bundling programs for digital mammography and stereotactic table
platforms. Our excision products will compete with manufacturers of handheld
surgical excision instrumentation and standard RF cutting devices.
Our
competitors dedicate, and we believe they will continue to dedicate, significant
resources to promote their products aggressively. The breast cancer market is
also characterized by extensive research efforts and technological progress. As
a result, new products are likely to be developed and introduced into the market
that could compete with our products more effectively.
Manufacturing
We
assemble and package a significant portion of our finished products at our
current corporate headquarters in Irvine, California, where we moved to in
August 2008. Our Gamma Finder is licensed and produced exclusively for us by
World of Medicine, a German medical device company. We manufacture in-house
several components used in our products, and we rely on several outside vendors
to produce many components, and in some cases completed products that we quality
check, sterilize, and package at our corporate headquarters. We also have
established a production engineering department to focus on integrating product
changes into the manufacturing process and to continually improve upon product
quality and cost.
We
manufacture our proprietary products in a controlled environment and have
implemented quality control systems as part of our manufacturing processes. We
believe our manufacturing facility and control systems comply with the FDA’s
Quality System Regulations, or QSRs. We are certified to ISO 13485:2003, the
medical device manufacturing standard, and applicable medical device directives
promulgated by the European Economic Community, which facilitates entry of our
products into the European Economic Community. We have received our CMDCAS
Certificate of Registration permitting importation of our devices into
Canada.
Since
2005, we have continued to transfer portions of our manufacturing operations to
Infus Medical, a contract manufacturer with facilities in Thailand, which
currently provides us with certain tissue marker production, biopsy probe
production and assembly and packaging services. We anticipate that over time we
will continue to transfer additional responsibility to this manufacturer related
to production, assembly and packaging. We believe that transferring production
of our more established products abroad in a stepwise manner, along with
increased sales volume, will result in cost savings and will allow us to focus
our domestic efforts on developing, modifying and promoting our newer products.
We will continue to produce certain of our products at our facility in Irvine,
California for the foreseeable future.
-14-
We have
one product and several components of other products that we obtain from
sole-source suppliers. We rely on one vendor, World of Medicine, for our Gamma
Finder product, one vendor, Faulhaber, for our biopsy handpiece motors, one
vendor, NuSil Technology, for a coating used in our biopsy probes, one vendor,
Advance Polymers, for material used in our Contura MLB and three vendors,
UltraSonix, Teratech and Esaote, for the ultrasound technology used in SenoSonix
with EnCor. We do not believe that we could replace these suppliers without
significant effort and delay in production. Other products and components come
from single suppliers, but alternate suppliers are easier to identify, though in
many cases we have not yet qualified alternate suppliers. We do not carry a
significant inventory of most components used in our products, with the
exception of the gamma finder product. Most of our suppliers have no contractual
obligations to supply us with, and we are not contractually obligated to
purchase from them, the components used in our devices.
On March
5, 2008, we entered into a Lease Agreement with The Irvine Company LLC for the
lease of approximately 41,402 square feet space at 3 Morgan, Irvine, California.
The term of the lease commenced on November 1, 2008, although we moved into the
property in August pursuant to a period of rent-free early occupancy and will
expire on January 31, 2014.
Government
Regulation
Our
products are medical devices subject to extensive and rigorous regulation by the
FDA, as well as other federal and state regulatory bodies in the United States
and comparable authorities in other countries. The FDA regulations govern, among
other things, the following activities that we perform, or that are performed on
our behalf, to ensure that medical products distributed domestically or exported
internationally are safe and effective for their intended uses:
|
|
·
|
product
design, development and
manufacture;
|
|
|
·
|
product
safety, testing, labeling and
storage;
|
|
|
·
|
premarketing
clearance or approval;
|
|
|
·
|
recordkeeping
procedures;
|
|
|
·
|
product
marketing, sales and distribution;
and
|
|
|
·
|
post-marketing
surveillance, reporting of deaths or serious injuries and medical device
reporting.
|
The FDA’s Premarket Clearance and
Approval Requirements. Unless an exemption applies, each medical device
we wish to distribute commercially in the United States will require either
prior 510(k) clearance or a premarket approval, or a PMA, from the FDA. Medical
devices are classified into one of three classes—Class I, Class II, or
Class III—depending on the degree or risk associated with each medical
device and the extent of control needed to ensure safety and effectiveness.
Devices deemed to pose lower risks are placed in either Class I or II,
which requires the manufacturer to submit to the FDA a premarket notification
requesting permission to commercially distribute the device. This process is
generally known as 510(k) clearance. Some low-risk devices are exempted from
this requirement. Devices deemed by the FDA to pose the greatest risk, such as
life-sustaining, life-supporting or implantable devices, or devices deemed not
substantially equivalent to a previously cleared 510(k) device, are placed in
Class III, requiring premarket approval. Our minimally-invasive breast care
products are Class I and II devices.
510(k) Clearance Pathway.
When a 510(k) clearance is required, we must submit a premarket notification to
the FDA demonstrating that our proposed device is substantially equivalent to a
previously cleared and legally marketed 510(k) device or a device that was in
commercial distribution before May 28, 1976 for which the FDA has not yet
required the submission of a PMA application. By statute, the FDA is required to
clear or deny a 510(k) premarket notification within 90 days of submission of
the application. As a practical matter, clearance often takes significantly
longer. The FDA may require further information, including clinical data, to
make a determination regarding substantial equivalence. If the FDA determines
that the device, or its intended use, is not substantially equivalent to a
previously-cleared device or use, the FDA will place the device, or the
particular use, into Class III.
-15-
Premarket Approval Pathway. A
PMA application must be submitted to the FDA if the device cannot be cleared
through the 510(k) process. The PMA application process is much more demanding
than the 510(k) premarket notification process. A PMA application must be
supported by extensive data, including but not limited to technical,
preclinical, clinical trials, manufacturing and labeling to demonstrate to the
FDA’s satisfaction the safety and effectiveness of the device.
After a
PMA application is submitted and the FDA determines that the application is
sufficiently complete to permit a substantive review, the FDA will accept the
application for review. The FDA has 180 days to review an “accepted” PMA
application, although the review of an application generally occurs over a
significantly longer period of time and can take up to several years. During
this review period, the FDA may request additional information or clarification
of the information already provided. Also, an advisory panel of experts from
outside the FDA may be convened to review and evaluate the application and
provide recommendations to the FDA as to the approvability of the device. In
addition, the FDA will conduct a preapproval inspection of the manufacturing
facility to ensure compliance with the QSRs. New PMA applications or PMA
application supplements are required for significant modification to the
manufacturing process, labeling and design of a device that is approved through
the premarket approval process. Premarket approval supplements often require
submission of the same type of information as a premarket approval application,
except that the supplement is limited to information needed to support any
changes from the device covered by the original premarket approval application
and may not require as extensive clinical data or the convening of an advisory
panel. We do not anticipate that any of our products in development will require
the submission and approval of a PMA.
Clinical Trials. Clinical
trials are almost always required to support an FDA premarket application and
are sometimes required for 510(k) clearance. These trials generally require
submission of an application for an Investigational Device Exemption, or IDE, to
the FDA. The IDE application must be supported by appropriate data, such as
animal and laboratory testing results, showing that it is safe to test the
device in humans and that the testing protocol is scientifically sound. The IDE
must be approved in advance by the FDA for a specific number of patients unless
the product is deemed a non-significant risk device eligible for more
abbreviated IDE requirements. Clinical trials for significant risk devices may
not begin until the IDE application is approved by the FDA and the appropriate
institutional review boards, or IRBs, at the clinical trial sites. Our clinical
trials must be conducted under the oversight of an IRB at the relevant clinical
trial sites and in accordance with the FDA’s regulations, including but not
limited to those relating to good clinical practices. We are also required to
obtain patients’ informed consent that complies with both the FDA’s requirements
and state and federal privacy regulations. We, the FDA or the IRB at each site
at which a clinical trial is being performed may suspend a clinical trial at any
time for various reasons, including a belief that the risks to study subjects
outweigh the benefits. Even if a trial is completed, the results of clinical
testing may not demonstrate the safety and efficacy of the device, may be
equivocal or may otherwise not be sufficient to obtain approval or clearance of
the product.
Pervasive and Continuing
Regulation. After a device is placed on the market, numerous regulatory
requirements continue to apply. These include:
|
|
·
|
the
FDA’s Quality System Regulations, which require manufacturers, including
third-party manufacturers, to follow stringent design, testing, control,
documentation and other quality assurance procedures during all aspects of
the manufacturing process;
|
|
|
·
|
labeling
regulations and FDA prohibitions against the promotion of products for
uncleared, unapproved or off-label
uses;
|
|
|
·
|
clearance
or approval of product modifications that could significantly affect
safety or efficacy or that would constitute a major change in intended
use;
|
|
|
·
|
medical
device reporting, or MDR, regulations, which require that manufacturers
report to the FDA if their device may have caused or contributed to a
death or serious injury or malfunctioned in a way that would likely cause
or contribute to a death or serious injury if the malfunction were to
recur; and
|
|
|
·
|
post-market
surveillance regulations, which apply when necessary to protect the public
health or to provide additional safety and effectiveness data for the
device.
|
-16-
After a
device receives 510(k) clearance or a PMA, any modification that could
significantly affect its safety or effectiveness, or that would constitute a
major change in its intended use, will require a new clearance or approval. The
FDA requires each manufacturer to make this determination initially, but the FDA
can review any such decision and can disagree with a manufacturer’s
determination. We have modified various aspects of some of our marketed products
since receiving regulatory clearance, but we believe that new 510(k) clearances
are not required for these modifications. If the FDA disagrees with our
determination not to seek a new 510(k) clearance or PMA, the FDA may
retroactively require us to seek 510(k) clearance or premarket approval. The FDA
could also require us to cease marketing and distribution and/or recall the
modified device until 510(k) clearance or premarket approval is obtained. Also,
in these circumstances, we may be subject to significant regulatory fines,
penalties and Warning Letters.
The MDR
regulations require that we report to the FDA any incident in which our product
may have caused or contributed to a death or serious injury or in which our
product malfunctioned and, if the malfunction were to recur, would likely cause
or contribute to death or serious injury.
We have
registered with the FDA as a medical device manufacturer and have obtained a
manufacturing license from the California Department of Health Services, or
CDHS. The FDA has broad post-market and regulatory enforcement powers. We are
subject to unannounced inspections by the FDA and the Food and Drug Branch of
CDHS, or FDB, to determine our compliance with the QSRs and other regulations,
and these inspections may include the manufacturing facilities of our suppliers.
We underwent an inspection of our facilities by the FDA in April 2005, which
resulted in the issuance in July 2005 of a Warning Letter from the FDA related
to, among other things, our failure to adequately validate manufacturing changes
we undertook to prevent the tip of the Gel Mark Ultra biopsy site marker
shearing off in the patient’s breast during surgery, which we had experienced.
The letter required us to take prompt action to strengthen our Quality System
and product engineering area. We responded to the FDA with a comprehensive
corrective action plan in August 2005. In 2008, no incidents of tip sheer were
reported to SenoRx. In addition, we recently underwent an inspection
of our manufacturing facilities by the FDA, which resulted in the issuance on
September 30, 2009 of an FDA Form 483, Notice of Inspectional Observations, from
the FDA related to our failure to properly implement and maintain adequate
methods and documentation of the design, testing, production, control, quality
assurance, storage, shipping and post-market surveillance for some of our
products, including Contura MLB and Gel Mark product line. The Notice
of Inspectional Observations will require us to take prompt action to strengthen
our Quality System and result in increased expenses. We have
completed responding to the observations and are currently implementing a
comprehensive corrective action plan to update our facilities and systems and
intend to be in full compliance with QSRs. If, upon reinspection, the
FDA determines we have not properly addressed their concerns or they identify
new violations, we can be subject to any of the following
sanctions:
|
|
·
|
Warning
Letters, fines, injunctions, consent decrees and civil
penalties;
|
|
|
·
|
repair,
replacement, refunds, recall or seizure of our
products;
|
|
|
·
|
operating
restrictions, partial suspension or total shutdown of
production;
|
|
|
·
|
refusal
of our requests for 510(k) clearance or premarket approval of new
products, new intended uses or modifications to existing
products;
|
|
|
·
|
withdrawal
of 510(k) clearance or premarket approvals that have already been granted;
and
|
|
|
·
|
criminal
prosecution.
|
Fraud and Abuse. We may
directly or indirectly be subject to various federal and state laws pertaining
to healthcare fraud and abuse, including anti-kickback laws. In particular, the
federal Anti-Kickback Statute prohibits persons from knowingly and willfully
soliciting, offering, receiving or providing remuneration, directly or
indirectly, in exchange for or to induce either the referral of an individual,
or the furnishing, arranging for or recommending a good or service, for which
payment may be made in whole or part under federal healthcare programs, such as
the Medicare and Medicaid programs. Penalties for violations include criminal
penalties and civil sanctions such as fines, imprisonment and possible exclusion
from Medicare, Medicaid and other federal healthcare programs. The Anti-Kickback
Statute is broad and prohibits many arrangements and practices that are lawful
in businesses outside of the healthcare industry. In implementing the statute,
the Office of Inspector General, or OIG, has issued a series of regulations,
known as the “safe harbors.” These safe harbors set forth provisions that, if
all their applicable requirements are met, will assure healthcare providers and
other parties that they will not be prosecuted under the Anti-Kickback Statute.
The failure of a transaction or arrangement to fit precisely within one or more
safe harbors does not necessarily mean that it is illegal or that prosecution
will be pursued. However, conduct and business arrangements that do not fully
satisfy each applicable element of a safe harbor may result in increased
scrutiny by government enforcement authorities, such as the
OIG.
-17-
International. International
sales of medical devices are subject to foreign governmental regulations, which
vary substantially from country to country. The time required to obtain
clearance or approval by a foreign country may be longer or shorter than that
required for FDA clearance or approval, and the requirements may be
different.
The
primary regulatory environment in Europe is that of the European Economic
Community, which has adopted numerous directives and has promulgated voluntary
standards regulating the design, manufacture, clinical trials, labeling and
adverse event reporting for medical devices. Devices that comply with the
requirements of a relevant directive will be entitled to bear CE conformity
marking, indicating that the device conforms with the essential requirements of
the applicable directives and, accordingly, can be commercially distributed
throughout the member states of the European Economic Community, and other
countries that comply with or mirror these directives. The method of assessing
conformity varies depending on the type and class of the product, but normally
involves a combination of self-assessment by the manufacturer and a third-party
assessment by our designated notified body, an independent and neutral
institution appointed by a country to conduct the conformity assessment. This
third-party assessment may consist of an audit of the manufacturer’s quality
system and specific testing of the manufacturer’s device. Such an assessment is
required in order for a manufacturer to commercially distribute the product
throughout these countries. ISO 9001 and ISO 13845 certifications are voluntary
harmonized standards. Compliance establishes the presumption of conformity with
the essential requirements for a CE Marking. We have the authorization to affix
the CE Mark to Gel Mark Ultra, Gel Mark UltraCor, EnCor and Contura MLB and to
commercialize these devices in the European Economic Community, Australia, Hong
Kong, Mexico, Singapore, South Korea, Taiwan, and several other
countries. We have an active program to comply with the “WEE”
directive should it be adopted in Europe and Asia in the next five
years.
In 2007
we began partnering with local distributors who have breast imaging and/or
interventional radiology franchises in countries outside North
America. Through these distributors, we are selling our EnCor and
GelMark Ultra products in Austria, Belgium, Denmark, Finland, France, Germany,
Hong Kong, Iceland, Ireland, Italy, Luxembourg,
Mexico, The Netherlands, Norway, Portugal, Russia, Singapore, South Korea,
Spain, Sweden, Switzerland,
Taiwan and the United Kingdom. In 2009 we added distributors in
China, Egypt, Greece, Poland, Saudi Arabia, Thailand, Turkey and the United Arab
Emirates.
Third-Party
Reimbursement
Payment
for patient care in the United States is generally made by third-party payors,
including private insurers and government insurance programs, such as Medicare
and Medicaid. The Medicare program, the largest single payor in the United
States, is a federal governmental health insurance program administered by the
Centers for Medicare and Medicaid Services, or CMS. Reimbursement for procedures
related to breast cancer has been favorable as a result of the growing awareness
of the impact of the disease as well as the recognition that proactive diagnosis
and treatment is critical for effective care. The costs associated with the
purchase of our products are reimbursed through Medicare, Medicaid and other
third-party payors. International market acceptance of our products may depend,
in part, upon the availability of reimbursement within the prevailing healthcare
payment systems. Reimbursement and healthcare payment systems in international
markets vary significantly by country, and include both government-sponsored
healthcare and private insurance.
Research
and Development
As of
December 31, 2009, we had 19 employees, as well as several key on-going
consultants, in our research and development department, which is overseen by
our chief technical officer. Historically, we developed and successfully brought
to market diagnostic products, including our breast biopsy systems and our
tissue markers and therapeutic products, such as Contura MLB. We continue to
focus on developing additional diagnostic, therapeutic and excision products to
further support our focus on serving the continuum of care in the breast care
market. We are currently developing next generations of our current products as
well as various radio-frequency based excision and reconstructive cutting
devices, markers and new therapeutic devices.
-18-
Research
and development expenses for 2009, 2008 and 2007 were $7.4 million, $6.1million
and $6.4 million, respectively. We expect research and development efforts and
expenses to increase in absolute dollar terms but decrease as a percentage of
net revenues.
Patents
and Proprietary Technology
We plan
to pursue and maintain intellectual property protection in the United States,
Europe, Japan, Canada and other countries such as China and Australia. As of
December 31, 2009, we have 65 issued United States patents primarily
covering devices relating to breast biopsy, including biopsy site marking
devices, excision devices and balloon products, the earliest of which will
expire in 2018 and the last of which will expire in 2026, 2 granted European
patents which are validated in 14 countries, 3 Canadian patents, and 1 Japanese
patent.
In
addition, we have 95 pending United States patent applications, 8 pending PCT
(international) patent applications, 29 pending European regional patent
applications, 27 pending Canadian patent applications, 1 pending Japanese patent
applications, 10 pending Australian patent applications, as well as pending
patent applications in Brazil, China, Mexico, Russia, South Korea and Singapore.
We believe we have a strong intellectual portfolio that has permitted us to make
modifications to our products in response to competition without significant
disruption to our operations.
We have
several issued United States patents related either to the design or use of
Contura MLB and additional United States patent applications and continuations
are pending. In December 2008, we were granted a patent relating to Contura
MLB’s asymmetrical radiation of a breast cavity with a balloon. During the
development of Contura MLB, we appropriately considered the intellectual
property landscape, including citing as appropriate in our own patent filings
the Hologic/Proxima patents that are the subject matter of our litigation with
Hologic.
Together,
our patents and patent applications protect aspects of our technologies. Key
areas of our issued and pending patent coverage include:
|
|
·
|
biopsy
systems, covering current embodiments and variations to the design of the
EnCor and EnCor 360 probes, handpieces and control
module;
|
|
|
·
|
mechanical
cutters, covering the Tri-Concave penetrating tip and the EnCor and EnCor
360 tissue cutting mechanisms;
|
|
|
·
|
radiofrequency
technologies, covering the SenoPulse RF Generator, devices powered by the
generator, including the Single Step and long wire scalpels, EnCor 360
probe and lesion location devices;
|
|
|
·
|
bioresorbable
biopsy site markers, covering marker materials, methods for imparting
ultrasound visibility, and marker delivery systems;
and
|
|
|
·
|
Contura
MLB, covering the use of vacuum to help conform tissue surrounding the
lumpectomy cavity to the walls of the radiation delivery balloon, our
proprietary balloon manufacturing process and delivery of radiation
through multiple lumens in a balloon
catheter.
|
We also
rely on copyrights, trade secrets, technical know-how and continuing innovation
to develop and maintain our competitive position. We seek to protect our
proprietary information and other intellectual property by generally requiring
our employees, consultants, contractors, outside scientific collaborators and
other advisors to execute non-disclosure agreements on commencement of their
employment or engagement.
Employees
As of
December 31, 2009, we had 161 employees, including 81 employees in sales
and marketing, 19 employees in research and development, 35 employees in
manufacturing, 15 employees in clinical, regulatory and quality assurance and 11
employees in general and administrative. We believe that our future success will
depend on our continued ability to attract, hire and retain qualified personnel.
None of our employees are represented by a labor union or are parties to a
collective bargaining agreement, and we believe our employee relations are
good.
Available
Information
We are
subject to the reporting requirements under the Securities Exchange Act of 1934.
Consequently, we are required to file reports and information with the
Securities and Exchange Commission (SEC), including reports on the following
forms: annual report on Form 10-K, quarterly reports on Form 10-Q,
current reports on Form 8-K, and amendments to those reports filed or
furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act
of 1934. These reports and other information concerning the company may be
accessed through the SEC’s website at http://www.sec.gov. You may also read and
copy any materials we file with the SEC at the SEC’s Public Reference Room at
100 F Street, NE, Washington, D.C. 20549. Information on the operation of the
Public Reference Room can be obtained by calling
1-800-SEC-0330.
-19-
You may
also find on our website at http://www.senorx.com/ electronic copies of our
annual report on Form 10-K, quarterly reports on Form 10-Q, current
reports on Form 8-K, and amendments to those reports filed or furnished
pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934.
Such filings are placed on our website as soon as reasonably practicable after
they are filed with the SEC. Our charter for our Nominating and Corporate
Governance, Audit and Compensation Committees and our Code of Ethics are
available on our website. In the event that we grant a waiver under our Code of
Ethics, to any of our officers and directors, we will publish it on our
website.
|
ITEM 1A.
|
RISK
FACTORS
|
RISKS
RELATED TO OUR BUSINESS
We
have a limited history of operations and a history of net losses, therefore, we
may not be able to generate significant revenues or profitability.
We have a
limited history of operations upon which you can evaluate our
business. We began selling our first products in 2002, fully launched
our flagship product for use in breast biopsy procedures, the EnCor system, in
November 2005, and launched our flagship radiation therapy product, the Contura
MLB, in January 2008. We incurred net losses of $2.9 million in 2009,
$8.7 million in 2008, $9.9 million in 2007 and, as of December 31, 2009, had an
accumulated deficit of approximately $87.1 million. In order for us
to become increasingly profitable, we believe that our EnCor system and Contura
MLB must be widely adopted. We cannot assure you that we will be able to achieve
or sustain profitability even if we are able to generate significant revenues.
Our failure to sustain profitability would negatively impact the market price of
our common stock and require us to obtain additional funding. If our future
funding requirements increase beyond currently expected levels, as a result of
our failure to sustain profitability relating to sales, litigation expenses or
otherwise, we cannot make any assurance that additional funding will be
available on a timely basis on terms acceptable to us, or at all, particularly
in the short-term due to the current credit and equity market funding
environments.
Our
success depends upon market adoption of our EnCor system and Contura MLB,
without which our results of operations will suffer.
Until
2007, we derived our revenues primarily from our tissue marker products.
However, our EnCor system and Contura MLB now account for a majority of our
revenue growth, and we expect this to continue for the foreseeable future. Our
ability to meet this expectation is based upon a number of assumptions,
including:
|
|
·
|
we
have limited experience selling to radiological oncologists, the primary
market for Contura MLB;
|
|
|
·
|
attracting
and retaining qualified sales professionals to sell
it;
|
|
|
·
|
differentiating
Contura MLB from competing products and obtaining a significant share of
this market;
|
|
|
·
|
protecting
our products with intellectual property
rights;
|
|
|
·
|
sustaining
adequate third-party reimbursement
;
|
|
|
·
|
producing
compelling clinical data on safety and
effectiveness;
|
|
|
·
|
partnering,
as necessary, with suppliers; and
|
|
|
·
|
manufacturing
it consistently within our specifications and in accordance with the FDA’s
Quality System Regulations.
|
Even if
we are able to present potential customers with compelling clinical data,
technological advancements or influential user experiences, they may be
reluctant to switch from a competing device, which they have grown accustomed
to. We may not be successful in our near-term strategy of marketing EnCor and
Contura MLB to our existing customer base of tissue marker users, and users of
our earlier vacuum-assisted breast biopsy system. Our commercial success also
depends on the continued general market shift to less invasive biopsy
procedures.
-20-
We
may be subject to costly claims of infringement or misappropriation of the
intellectual property rights of others, which could impact our business and harm
our operations.
Our
industry has been characterized by frequent demands for licenses and litigation.
Our competitors, potential competitors or other patent holders may, in the
future, assert that our products and the methods we employ are covered by their
patents or misappropriate their intellectual property. In addition, we do not
know whether our competitors will apply for and obtain patents that will
prevent, limit or interfere with our ability to make, use, sell or import our
products. Because patent applications may take years to issue, there may be
applications now pending of which we are unaware that may later result in issued
patents that our products infringe. There also could be existing patents that
one or more components of our systems may inadvertently infringe. Although we
may seek to settle any future claims, we may not be able to do so on reasonable
terms, or at all. If we lose a claim against us, we may be ordered to pay
substantial damages, including compensatory damages, which may be trebled in
certain circumstances, plus prejudgment interest. We also could be enjoined,
temporarily, preliminarily or permanently, from making, using, selling, offering
to sell or importing our products or technologies essential to our products,
which could significantly harm our business and operating
performance. On February
26, 2010, Hologic filed a notice of its appeal in the matter and the outcome of any such appeal process is
unknown. If we lose this appeal, we may be completely prevented from
selling Contura MLB and as a result, our future prospects will be significantly
harmed. Moreover, any such appeal would be costly to defend and also be a
significant distraction to management.
We may
become involved in litigation not only as a result of alleged infringement of a
third party’s intellectual property rights but also to protect our own
intellectual property. Enforcing our patent rights against infringers, even when
such litigation is resolved in our favor, could involve substantial costs and
divert management’s attention from our core business and harm our
reputation.
Our
future success will depend in part upon our ability to continue to successfully
commercialize our Contura
MLB.
Contura
MLB, which we received FDA 510(k) clearance in May 2007, has become a
significant contributor to our revenues. The Contura MLB commercialization
effort is fully engaged, but there remain significant challenges that must be
overcome before we can obtain significant revenues from this product,
including:
|
|
·
|
we
have limited experience selling to radiological oncologists and
physicists, the primary market for this
product;
|
|
|
·
|
attracting
and retaining qualified sales professionals to sell
it;
|
|
|
·
|
differentiating
Contura MLB from competing products and obtaining a significant share of
this market;
|
|
|
·
|
protecting
it with intellectual property
rights;
|
|
|
·
|
obtaining
adequate third-party reimbursement;
|
|
|
·
|
producing
compelling clinical data on safety and
effectiveness;
|
|
|
·
|
partnering,
as necessary, with suppliers; and
|
|
|
·
|
manufacturing
it consistently within our specifications and in accordance with the FDA’s
Quality System Regulations.
|
If we are
able to overcome these challenges, we may nevertheless be unable to convince
potential customers that the Contura MLB represents a compelling alternative to
competing products. Short-term brachytherapy products from Cianna Medical and
Xoft began to be commercialized in 2008 and we have recently seen competitors
discount their brachytherapy products in the market, which may cause our
products to be less competitive or require us to also reduce pricing in the
future. Our commercial success will also depend on a general market shift from
whole to partial breast irradiation. If we are unable to obtain a significant
share of the brachytherapy market for the reasons listed above, or that
competing products are more compelling and achieve better acceptance by the
market, our long-term commercialization experience with the Contura MLB could be
significantly below expectations or not achieved at all, which would have a
material adverse effect on our future financial performance. Additionally, the
adoption of conformal radiotherapy may grow at a faster rate than the overall
market for partial breast irradiation therapies, and as a result, could impact
the speed of adoption of balloon brachytherapy devices, including Contura
MLB.
-21-
We
have limited clinical data regarding the safety and efficacy of our products. If
future data or clinical experience is negative, we may lose significant market
share.
Our
success depends on the acceptance of our products by the medical community as
safe and effective. Physicians that may be interested in using our products may
hesitate to do so without long-term data on safety and efficacy. The limited
clinical studies on some of our products that have been published or presented
as abstracts at major medical meetings typically have been based on the work of
a small number of physicians examining small patient populations over relatively
short periods. Accordingly, the results of these clinical studies do not
necessarily predict long-term clinical results, or even short-term clinical
results from the broader physician community. If future safety or efficacy data
or clinical experience is negative, we may lose significant market
share.
We
compete against companies that have more established products and greater
resources, which may prevent us from achieving significant market penetration or
improved operating results.
Many of
our products compete, and our future products may compete, against products that
are more established and accepted within our target markets. With fewer
resources and operating history than many of our competitors and potential
future competitors, and a less-established reputation, it may be difficult for
our products to gain significant market penetration. We may be unable to
convince physicians to switch their practice away from competing devices.
Competing effectively will require us to distinguish our company and our
products from our competitors and their products, and turns on factors such
as:
|
|
·
|
ease
of use and performance;
|
|
|
·
|
price;
|
|
|
·
|
quality
and scale of our sales and marketing
efforts;
|
|
|
·
|
our
ability to offer a broad portfolio of products across the continuum of
breast care;
|
|
|
·
|
establishing
a strong reputation through compelling clinical study publications and
endorsements from influential physicians;
and
|
|
|
·
|
brand
and name recognition.
|
Competition
could result in price-cutting, reduced profit margins and loss of market share,
any of which could have a material adverse effect on our results of operations.
In addition, our competitors with greater financial resources could acquire
other companies that would enhance their name recognition and market share, and
allow them to compete more effectively by bundling together related products.
For example one competitor provides incentives for the purchase of its biopsy
capital equipment and disposables when purchased with its digital mammography
and stereotactic tables. Certain potential customers may view this value
proposition as attractive, which could result in their decision not to purchase
our products. We also anticipate that new products and improvements to existing
products could be introduced that would compete with our current and future
products. If we are unable to compete effectively, we will not be able to
generate expected sales and our future financial performance will
suffer.
Changes
in coverage and reimbursement for procedures using our products could affect the
adoption of our products and our future revenues.
Breast
biopsy procedures and markers are typically reimbursed by third-party payors,
including Medicare, Medicaid and private healthcare insurance companies. These
payors may adversely change their coverage amounts and reimbursement policies.
Reimbursement may be impacted by the general national health care reform
initiatives or otherwise by various branches of the federal government. In
addition, the Federal Deficit Reduction Act of 2006 may in the future affect
future reimbursement rates for our vacuum- assisted biopsy products and Contura
MLB products. We cannot assure you that the current scope of coverage or levels
of reimbursement will continue to be available or that coverage of, or
reimbursement for, our products will be available at all. If physicians,
hospitals and other providers are unable to obtain adequate reimbursement for
our current products or future products, or for the procedures in which such
products are used, they may be less likely to purchase the products, which could
have a material adverse impact on our market share. For example, in 2009, there
was an increase in reimbursement rates to non hospital based Radiation Oncology
centers for multiple-dwell radiation balloon catheter procedures, which includes
Contura MLB, relative to single catheter procedures, but a decrease for non
hospital based Radiology Oncology centers in rates relative to whole breast
radiation therapy.
-22-
Our
ability to compete depends upon our ability to innovate, develop and
commercialize new products and product enhancements.
The
markets in which we compete involve rapid and substantial technological
development and product innovations. There are few barriers to prevent new
entrants or existing competitors from developing or acquiring products or
technological improvements that compete effectively against our products or
technology. If we are unable to innovate successfully to anticipate or respond
to competitive threats, obtain regulatory approvals, or protect such innovation
with defensible intellectual property, our revenues could fail to grow or could
decline. Our business strategy is in part based upon our expectation that we
will continue to make frequent new product introductions and improvements to
existing products that will be demanded by our target customers. If we are
unable to continue to develop new products and technologies as anticipated, our
ability to grow and our future financial performance could be materially
harmed.
Our
success will depend on our ability to attract and retain key personnel,
particularly members of management and scientific staff.
We
believe our future success will depend upon our ability to attract and retain
employees, including members of management, engineers and other highly skilled
personnel. Our employees may terminate their employment with us at any time.
Hiring qualified personnel may be difficult due to the limited number of
qualified professionals and the fact that competition for these types of
employees is intense. If we fail to attract and retain key personnel, we may not
be able to execute our business plan. For
example, we recently appointed Mr. John Buhler as our Chief Executive Officer.
With his new responsibilities, Mr. Buhler will have less time to devote to
managing the sales organization and as a result, we will need to hire a new Vice
President of Sales or similar position. We may need to internally promote
candidates or otherwise recruit and hire from the outside, which may take
significant time and become a distraction for the sales team and its sales
effort and could have a material adverse impact on our results of
operations.
Our
business strategy is heavily focused on integrated breast centers and other
large institutions.
We are
focusing our sales efforts on becoming a preferred provider to integrated breast
centers and other large customer accounts. We cannot assure you that we will be
able to secure or maintain these accounts or that this strategy will maximize
our revenue growth. These targeted customers often have a rigorous and lengthy
qualification process for approving new vendors and products. Additionally,
breast centers are in many cases not located at one physical location, but
instead involve the coordinated efforts of various geographically dispersed
offices and physicians, which may complicate the qualification process and may
strain our sales and support organizations. Further, these customers have not
entered, and we do not expect them in the future to enter, long-term contracts
to purchase our products. Therefore, obtaining approval from these potential
customers to sell them our products may not result in significant or long-term
sales of our products to them. Our strategy of focusing on large institutions
may result in relatively few customers contributing a significant amount to our
revenues. For example, Kaiser Permanente is our largest customer, in
the years ended December 31, 2009 and 2008, represented approximately 4.7% and
4.9%, respectively, of our total revenues. We cannot assure you that
Kaiser or other large customer accounts will continue to purchase our
products. The loss of any of these customers could have a material
adverse impact on our results of operations.
Our
strategy of providing a broad array of products to the breast care market may be
difficult to achieve, given our size and limited resources.
We aim to
be an attractive and convenient supplier for integrated breast centers by
offering a broad product line of minimally-invasive devices for breast care
specialists. Commercializing several product lines simultaneously may be
difficult because we are a relatively small company. Additionally, offering a
broad product line will require us to manufacture, sell and support some
products that are not as profitable or in as high demand as some of our other
products, which could have a material adverse effect on our overall results of
operations. To succeed in our approach, we will need to grow our organization
considerably and enhance our relationships with third-party manufacturers and
suppliers. If we fail to make product introductions successfully or in a timely
manner because we lack resources, or if we fail to adequately manufacture, sell
and support our existing products, our reputation may be negatively affected and
our results of operations could be materially harmed. Additionally, managing
such a large line of products may be challenging for an organization of our
size. For example, we recently underwent an inspection of our
manufacturing facilities by the FDA, which resulted in the issuance on September
30, 2009 of an FDA Form 483, Notice of Inspectional Observations, from the FDA
related to our failure to properly implement and maintain adequate methods and
documentation of the design, testing, production, control, quality assurance,
storage, shipping and post-market surveillance for some of our products,
including the Contura MLB and Gel Mark product line.
-23-
We
believe that demand for minimally-invasive products for the diagnosis and
treatment of breast cancer must grow in order for our business to grow as
anticipated.
While
there have been trends in recent years that favor increased screening, diagnosis
and treatment of breast cancer, these trends may not continue. The incidence of
breast cancer in the United States appears to have fallen from its highest level
over the last few years. Additionally, while the number of breast biopsies
performed annually has increased significantly since 1997 when the American
Cancer Society updated its guidelines for breast cancer screening, recommending
that women should begin annual screening at age 40 rather than the previously
recommended age 50, new guidance could be published that could support a
reversal of this trend. For example, recently an independent panel of experts
appointed by the federal Department of Health and Human Services recommended a
return to screening starting at age 50. The American Cancer Society has stated
that it does not currently plan to revise its guidelines, but this does not
preclude the possibility of future revisions to the guideline. In
addition, some studies conclude that annual breast cancer screening by
mammography for women under age 50 may be more harmful, due to increased
radiation exposure, than beneficial. These factors, in addition to possible
future innovations in screening technologies or in breast cancer treatment
options, could result in a decline in breast biopsy procedures and radiation
therapy, which could reduce our overall market.
We
have limited sales and marketing experience and failure to build and manage our
sales force or to market and distribute our products effectively could have a
material adverse effect on our results of operations.
We rely
on a direct sales force to sell our products. In order to meet our anticipated
sales objectives, we expect to grow our sales organization significantly over
the next several years. There are significant risks involved in building and
managing our sales organization, including our ability to:
|
|
·
|
hire
and successfully integrate qualified individuals as
needed;
|
|
|
·
|
provide
adequate training for the effective sale of our
products;
|
|
|
·
|
retain
and motivate our sales employees;
and
|
|
|
·
|
integrate
our new brachytherapy sales professionals and successfully sell into the
radiation oncology market.
|
We expect
that our Contura MLB will continue to be a key driver of future growth. However,
our sales force has historically sold diagnostic products and therefore has
limited experience selling a therapeutic device. Our Contura MLB competes with
products that are well-established and with new entrants to the market.
Accordingly, it is difficult for us to predict how well our sales force will
perform. Our failure to adequately address these risks could have a
material adverse effect on our ability to sell our products, causing our
revenues to be lower than expected and harming our results of
operations.
If
we are unable to obtain and maintain intellectual property protection covering
our products, others may be able to make, use or sell our products, which could
have a material adverse effect on our business and results of
operations.
We rely
on patent, copyright, trade secret and trademark laws and confidentiality
agreements to protect our technology, products and our competitive position in
the market. Additionally, our patent applications, including those covering our
EnCor system, may not result in patents being issued to us or, if they are
issued, may not be in a form that is advantageous to us. Any patents we obtain
may be challenged or invalidated by third parties. Competitors also may design
around our protected technology or develop their own technologies that fall
outside our intellectual property rights. In addition, we may not be able to
prevent the unauthorized disclosure or use of our technical knowledge or other
trade secrets by consultants, vendors, former employees or current employees,
despite the existence of confidentiality agreements and other contractual
restrictions. Monitoring unauthorized uses and disclosures of our intellectual
property is difficult, and we cannot be certain that the steps we have taken to
protect our intellectual property will be effective or that any remedies we may
have in these circumstances would be adequate. Moreover, the laws of foreign
countries may not protect our intellectual property rights to the same extent as
the laws of the United States.
-24-
We may
not have adequate intellectual property protection for some of our products and
products under development and consequently may need to obtain licenses from
third parties. If any such licenses are required, we may be unable to negotiate
terms acceptable to us and such failure could have a material adverse effect on
our future results of operations.
We
may be unsuccessful in our long-term goal of expanding our product offerings
outside the United States and Canada.
For the
year ended December 31, 2009, we derived approximately 90.2% of our net revenues
from sales within the United States and Canada. We have entered into
distribution agreements with third parties outside the United States and Canada,
but do not anticipate sales of our products through these distributors becoming
a significant portion of our revenues in the foreseeable future. If we do begin
to offer our products more broadly outside the United States and Canada, we
expect that we will remain dependent on third-party distribution relationships
and will need to attract additional distributors to increase the number of
territories in which we sell our products. Distributors may not commit the
necessary resources to market and sell our products to the level of our
expectations. If current or future distributors do not perform adequately, or we
are unable to locate distributors in particular geographic areas, our ability to
realize long-term international revenue growth could be materially adversely
affected.
Although
some of our products have regulatory clearances and approvals from jurisdictions
outside the United States and Canada, others do not. These products may not be
sold in these jurisdictions until the required clearances and approvals are
obtained. We cannot assure you that we will be able to obtain these clearances
or approvals on a timely basis, or at all.
We are dependent on sole-source and single-source suppliers for certain of our products and components, thereby exposing us to supply interruptions that could have a material adverse effect on our business.
We have
one product and several components of other products that we obtain from sole
suppliers. We rely on one vendor for our Gamma Finder product, one vendor for
our biopsy handpiece motors, one vendor for a coating used in our biopsy probes,
one vendor for material used in our Contura MLB and three vendors for the
ultrasound technology used in SenoSonix with EnCor. Other products and
components come from single suppliers, but alternate suppliers are easier to
identify. However, in many of these cases we have not yet qualified alternate
suppliers and rely upon purchase orders, rather than longer-term supply
agreements. We also do not carry a significant inventory of most components used
in our products and generally could not replace our suppliers without
significant effort and delay in production. In addition, switching components
may require product redesign and new regulatory clearances by the FDA, either of
which could significantly delay or prevent production and involve substantial
costs.
Reliance
on third-party vendors may lead to unanticipated interruptions in supply or
failure to meet demand on a timely basis. Any supply interruption from our
vendors or failure to obtain additional vendors for any of the components could
limit our ability to manufacture our products and fulfill customer orders on a
timely basis, which could harm our reputation and revenues.
We
have limited experience manufacturing certain components of our products in
significant quantities, which could adversely impact the rate at which we
grow.
We may
encounter difficulties in manufacturing relating to our products and products
under development for the following reasons:
|
|
·
|
our
limited experience in manufacturing such products in significant
quantities and in compliance with the FDA’s Quality System
Regulation;
|
|
|
·
|
to
increase our manufacturing output significantly, we will have to attract
and retain qualified employees, who are in short supply, for the
manufacturing, assembly and testing operations;
and
|
|
|
·
|
some
of the components and materials that we use in our manufacturing
operations are currently provided by sole and single sources of
supply.
|
-25-
Our
limited manufacturing experience has in the past resulted in unexpected and
costly delays. For example, in 2006, as a part of our settlement of litigation
with Suros Surgical Systems, a wholly-owned subsidiary of Hologic, we
implemented a redesign to the EnCor system cutter. This effort resulted in a
short-term decrease in yields and a delay in implementing certain cost
improvements, which had an adverse effect on our costs of goods sold. In
addition, although we believe that our current manufacturing capabilities will
be adequate to support our commercial manufacturing activities for the
foreseeable future, we may be required to expand our manufacturing facilities if
we experience faster-than-expected growth. If we are unable to provide customers
with high-quality products in a timely manner, we may not be able to achieve
wide market adoption for our EnCor system or other products and products under
development. Our inability to successfully manufacture or commercialize our
devices could have a material adverse effect on our product sales.
We
rely on third-party manufacturers for certain components, and the loss of any of
these manufacturers, or their inability to provide us with an adequate supply of
high-quality components, could have a material adverse effect on our
business.
Although
we manufacture certain components and assemble some of our products at our
corporate headquarters in Irvine, California, we rely on third parties to
manufacture most of the components of our products. Since the end of 2005, we
have transferred, and continue to transfer, a significant portion of our
manufacturing and product assembly operations to a third party contract
manufacturer in Thailand. Because of the distance between California and
Thailand, we may have difficulty adequately supervising and supporting its
operations. There are several risks inherent in relying on third-party
manufacturers, including:
|
|
·
|
failure
to meet our requirements on a timely basis as demand grows for our
products;
|
|
|
·
|
errors
in manufacturing components that could negatively affect the performance
of our products, cause delays in shipment of our products, or lead to
malfunctions or returns;
|
|
|
·
|
inability
to manufacture products to our quality specifications and strictly
enforced regulatory requirements;
|
|
|
·
|
inability
to implement design modifications that we develop in the
future;
|
|
|
·
|
unwillingness
to negotiate a long-term supply contract that meets our needs or to supply
components on a short-term basis on commercially reasonable
terms;
|
|
|
·
|
prioritization
of other customers orders over
ours;
|
|
|
·
|
inability
to fulfill our orders due to unforeseen events, including foreign
political events, that result in a disruption of their operations;
and
|
|
|
·
|
continued
fluctuations in the value of the U.S. dollar could impact our future
third-party manufacturing
costs.
|
If a
manufacturer fails to meet our needs with high-quality products on a timely
basis, we may be unable to meet customer demand, which could have a material
adverse effect on our reputation and customer relationships.
Any
acquisitions that we make could disrupt our business and have an adverse effect
on our financial
condition.
We expect
that in the future we may identify and evaluate opportunities for strategic
acquisitions of complementary product lines, technologies or companies. We may
also consider joint ventures and other collaborative projects. However, we may
not be able to identify appropriate acquisition candidates or strategic
partners, or successfully negotiate, finance or integrate any businesses,
products or technologies that we acquire. Furthermore, the integration of any
acquisition and the management of any collaborative project may divert
management’s time and resources from our core business and disrupt our
operations. We do not have any experience with acquiring other product lines,
technologies or companies. We may spend time and money on projects that do not
increase our revenues. Any cash acquisition we pursue would diminish the funds
available to us for other uses, and any stock acquisition would be dilutive to
our stockholders.
-26-
Our
financial controls and procedures may not be sufficient to ensure timely and
reliable reporting of financial information, which, as a public company, could
materially harm our stock price and NASDAQ listing.
As a
public company, we will require greater financial resources than we have had as
a private company. We will need to hire additional employees for our finance
department. We cannot provide you with assurance that our finance department has
or will maintain adequate resources to ensure that we will not have any future
material weakness in our system of internal controls. The effectiveness of our
controls and procedures may in the future be limited by a variety of factors
including:
|
·
|
faulty
human judgment and simple errors, omissions or
mistakes;
|
|
·
|
fraudulent
action of an individual or collusion of two or more
people;
|
|
·
|
inappropriate
management override of procedures;
and
|
|
·
|
the
possibility that any enhancements to controls and procedures may still not
be adequate to assure timely and accurate financial
information.
|
If we
fail to have effective controls and procedures for financial reporting in place,
we could be unable to provide timely and accurate financial information and be
subject to NASDAQ delisting, SEC investigation, and civil or criminal
sanctions.
Product
liability claims may lead to expensive and time-consuming litigation,
substantial damages, increased insurance rates, and may have a material adverse
effect on our financial condition.
Our
business exposes us to potential product liability claims that are inherent in
the manufacturing, marketing and sale of medical devices. For example, in the
past we experienced, and in the future could experience, an issue related to the
tip of our Gel Mark Ultra Biopsy Site Marker shearing off in the patient’s
breast during the biopsy procedure, which could lead to a claim of damages,
though none has previously been made. We may be unable to avoid product
liability claims, including those based on manufacturing defects or claims that
the use, misuse or failure of our products resulted in a misdiagnosis or harm to
a patient. Although we believe that our liability coverage is adequate for our
current needs, and while we intend to expand our product liability insurance
coverage to any products we intend to commercialize, insurance may be
unavailable, prohibitively expensive or may not fully cover our potential
liabilities. If we are unable to maintain sufficient insurance coverage on
reasonable terms or to otherwise protect against potential product liability
claims, we may be unable to continue to market our products and to develop new
products. Defending a product liability lawsuit could be costly and have a
material adverse effect on our financial condition, as well as significantly
divert management’s attention from conducting our business. In addition, product
liability claims, even if they are unsubstantiated, may damage our reputation by
raising questions about our products’ safety and efficacy, which could
materially adversely affect our results of operations, interfere with our
efforts to market our products and make it more difficult to obtain commercial
relationships necessary to maintain our business.
We
may be adversely affected by the impact of environmental and safety
regulations.
We are
subject to federal, state, local and foreign laws and regulations governing the
protection of the environment and occupational health and safety, including laws
regulating the disposal of hazardous wastes and the health and safety of our
employees. We may be required to obtain permits from governmental authorities
for certain operations. If we violate or fail to comply with these laws and
regulations, we could incur fines, penalties or other sanctions, which could
adversely affect our business and our financial condition and cause our stock
price to decline. We also may incur material expenses in the future relating to
compliance with future environmental laws. In addition, we could be held
responsible for substantial costs and damages arising from any contamination at
our present facilities or third-party waste disposal sites. We cannot completely
eliminate the risk of contamination or injury resulting from hazardous
materials, and we may incur material liability as a result of any contamination
or injury.
Our
ability to use net operating loss carryforwards may be limited.
Section
382 of the Internal Revenue Code generally imposes an annual limitation on the
amount of net operating loss carryforwards that may be used to offset taxable
income when a corporation has undergone significant changes in its stock
ownership. We have internally reviewed the applicability of the annual
limitations imposed by Section 382 caused by previous changes in our stock
ownership and believe such limitations should not be significant. Future
ownership changes, including changes resulting from or affected by our IPO, may
adversely affect our ability to use our remaining net operating loss
carryforwards. If our ability to use net operating loss carryforwards is
limited, we may be subject to tax on our income earlier than we would otherwise
be had we been able to fully utilize our net operating loss
carryforwards.
-27-
RISKS
RELATED TO REGULATORY MATTERS
The
FDA may find that we do not comply with regulatory requirements and take action
against us.
We
recently underwent an inspection of our manufacturing facilities by the FDA,
which resulted in the issuance on September 30, 2009 of an FDA Form 483, Notice
of Inspectional Observations, from the FDA related to our failure to properly
implement and maintain adequate methods and documentation of the design,
testing, production, control, quality assurance, storage, shipping and
post-market surveillance for some of our products, including the Contura MLB and
Gel Mark product line. The Notice of Inspectional Observations will
require us to take prompt action to strengthen our Quality System. We
have completed a comprehensive corrective action plan that has been presented to
the FDA outlining the steps and timing for coming into compliance with Quality
System regulations. FDA may determine we have failed to adequately or
timely implement or complete the corrective action plan. Such a
determination could lead to the FDA promptly commencing an enforcement action
against us without warning, which may include the following
sanctions:
|
|
·
|
injunctions,
fines, other civil penalties or issuance of a Warning
Letter;
|
|
|
·
|
the
refusal of, or delay by, the FDA in granting further 510(k) clearances or
approving further premarket approval
applications;
|
|
|
·
|
suspension
or withdrawal of our FDA clearances or
approvals;
|
|
|
·
|
operating
restrictions, including total or partial suspension of production,
distribution, sales and marketing of our products;
or
|
|
|
·
|
product
recalls, product seizures or criminal prosecution of our company, our
officers or our employees.
|
Any of
these could have a material adverse effect on our reputation, results of
operation and financial condition.
If
we fail to obtain or maintain necessary FDA clearances or approvals for
products, or if clearances or approvals are delayed, we will be unable to
commercially distribute and market our products in the United
States.
Our
products are medical devices, and as such are subject to extensive regulation in
the United States and in the foreign countries where we do business. Unless an
exemption applies, each medical device that we wish to market in the United
States must first receive 510(k) clearance or premarket approval from the FDA.
Either process can be lengthy and expensive. The FDA’s 510(k) clearance process
usually takes from three to twelve months from the date the application is
complete, but it may take longer. The premarket approval process is much more
costly, lengthy and uncertain. It generally takes from one to three years from
the date the application is completed or even longer. Achieving a completed
application is a process that may require numerous clinical trials and the
filing of amendments over time. We expect that our products in the foreseeable
future will be subject to 510(k) procedures and not premarket approval, or PMA,
applications. We may not be able to obtain additional FDA clearances or
approvals in a timely fashion, or at all. Delays in obtaining clearances or
approvals could adversely affect our revenues and profitability.
Modifications
to our devices may require new 510(k) clearances, which may not be
obtained.
The FDA
requires device manufacturers to initially make and document a determination of
whether or not a modification requires a new clearance; however, the FDA can
review a manufacturer’s decision. Any modifications to an FDA-cleared device
that could significantly affect its safety or effectiveness, or that would
constitute a major change in its intended use would require a 510(k) clearance
or possibly a premarket approval.
-28-
We have
modified aspects of some of our products since receiving FDA clearance, but we
believe that new 510(k) clearances are not required. We may make additional
modifications, and in appropriate circumstances, determine that new clearance or
approval is unnecessary. The FDA may not agree with our decisions not to seek
new clearances or approvals. If the FDA requires us to seek 510(k) clearances or
approval for any modifications to a previously cleared product, we may be
required to cease marketing or recall the modified device until we obtain
clearance or approval. Also, in these circumstances we may be subject to adverse
publicity, regulatory Warning Letters and significant fines and
penalties.
Government
regulation imposes significant restrictions and costs on the development and
commercialization of our products.
Any
products cleared or approved by the FDA are subject to on-going regulation. Any
discovery of previously unknown or unrecognized problems with the product or a
failure of the product to comply with any applicable regulatory requirements can
result in, among other things:
|
|
·
|
Warning
Letters, injunctions, fines or other civil
penalties;
|
|
|
·
|
the
refusal of, or delay by, the FDA in granting further 510(k) clearances or
approving further premarket approval
applications;
|
|
|
·
|
suspension
or withdrawal of our FDA clearances or
approvals;
|
|
|
·
|
operating
restrictions, including total or partial suspension of production,
distribution, sales and marketing of our products;
or
|
|
|
·
|
product
recalls, product seizures or criminal prosecution of our company, our
officers or our employees.
|
Any of
these could have a material adverse effect on our reputation and results of
operations.
RISKS
RELATED TO THE SECURITIES MARKETS AND OWNERSHIP OF OUR COMMON STOCK
Our
common stock has been publicly traded for a short time and an active trading
market may not be
sustained.
Prior to
March 2007, there had been no public market for our common stock. An active
trading market may not be sustained. The lack of an active market may impair the
value of your shares and your ability to sell your shares at the time you wish
to sell them. An inactive market may also impair our ability to raise capital by
selling shares and may impair our ability to acquire other companies, products
or technologies by using our shares as consideration.
If
our public guidance or our future operating performance does not meet investor
expectations, our stock price could decline.
As a
public company, we provide guidance to the investing community regarding our
anticipated future operating performance. Our business typically has a short
sales cycle, so that we do not have significant backlog of orders at the start
of a quarter, and our ability to sell our products successfully is subject to
many uncertainties. In light of these factors, it is difficult for us to
estimate with accuracy our future results. Our expectations regarding these
results will be subject to numerous risks and uncertainties that could make
actual results differ materially from those anticipated. If our actual results
do not meet our public guidance or our guidance or actual results do not meet
the expectations of third-party financial analysts, our stock price could
decline significantly.
We
expect that the price of our common stock will fluctuate
substantially.
The
market price of our common stock is likely to be highly volatile and may
fluctuate substantially due to many factors, including:
|
|
·
|
volume
and timing of sales of our
products;
|
-29-
|
|
·
|
the
introduction of new products or product enhancements by us or our
competitors;
|
|
|
·
|
disputes
or other developments with respect to our intellectual property rights or
the intellectual property rights of
others;
|
|
|
·
|
our
ability to develop, obtain regulatory clearance or approval for, and
market, new and enhanced products on a timely
basis;
|
|
|
·
|
product
liability claims or other
litigation;
|
|
|
·
|
quarterly
variations in our or our competitors’ results of
operations;
|
|
|
·
|
sales
of large blocks of our common stock, including sales by our executive
officers and directors;
|
|
|
·
|
announcements
of technological or medical innovations for the diagnosis and treatment of
breast cancer;
|
|
|
·
|
changes
in governmental regulations or in the status of our regulatory approvals
or applications;
|
|
|
·
|
changes
in the availability of third-party reimbursement in the United States or
other countries;
|
|
|
·
|
changes
in earnings estimates or recommendations by securities
analysts;
|
|
|
·
|
general
market conditions and other factors, including factors unrelated to our
operating performance or the operating performance of our competitors;
and
|
|
|
·
|
limited
liquidity and low trading volumes for our common
stock.
|
These and
other factors may make the price of our stock volatile and subject to unexpected
fluctuation.
Our
directors, executive officers and principal stockholders have significant voting
power and may take actions that may not be in the best interests of our other
stockholders.
Our
officers, directors and principal stockholders that currently hold more than 5%
of our common stock together control nearly a majority of our outstanding common
stock. As a result, these stockholders, if they act together, will be able to
exercise significant influence over the management and affairs of our company
and all matters requiring stockholder approval, including the election of
directors and approval of significant corporate transactions. This concentration
of ownership may have the effect of delaying or preventing a change in control,
might have a material adverse effect on the market price of our common stock and
may not be in the best interest of our other stockholders.
A
sale of a substantial number of shares of our common stock may cause the price
of our common stock
to
decline.
All
shares of our common stock that were outstanding before the IPO are eligible for
resale, subject to compliance with Rule 144 under the Securities Act. If our
stockholders sell substantial amounts of our common stock, the market price of
our common stock could decline.
Our
Amended and Restated Certificate of Incorporation and Bylaws, and Delaware law,
contain provisions that could discourage a takeover.
Our
Amended and Restated Certificate of Incorporation and Bylaws, and Delaware law,
contain provisions that might enable our management to resist a takeover, and
might make it more difficult for an investor to acquire a substantial block of
our common stock. These provisions include:
|
|
·
|
a
classified board of directors;
|
|
|
·
|
advance
notice requirements to stockholders for matters to be brought at
stockholder meetings;
|
|
|
·
|
a
supermajority stockholder vote requirement for amending certain provisions
of our Amended and Restated Certificate of Incorporation and
Bylaws;
|
|
|
·
|
limitations
on stockholder actions by written consent;
and
|
|
|
·
|
the
right to issue preferred stock without stockholder approval, which could
be used to dilute the stock ownership of a potential hostile
acquirer.
|
-30-
These
provisions might discourage, delay or prevent a change in control of our company
or a change in our management. The existence of these provisions could adversely
affect the voting power of holders of our common stock and limit the price that
investors might be willing to pay in the future for shares of the common
stock.
We
do not intend to pay cash dividends.
We have
never declared or paid cash dividends on our capital stock. We currently intend
to retain all available funds and any future earnings for use in the operation
and expansion of our business and do not anticipate paying any cash dividends in
the foreseeable future. In addition, the terms of any future debt or credit
facility may preclude us from paying any dividends. As a result, we anticipate
that capital appreciation of our common stock, if any, will be your sole source
of potential gain for the foreseeable future.
|
ITEM 1B.
|
UNRESOLVED
STAFF COMMENTS
|
None.
|
ITEM 2.
|
PROPERTIES
|
On
March 5, 2008, we entered into a Lease Agreement with The Irvine Company
LLC for the lease of approximately 41,402 square feet space at 3 Morgan,
Irvine, California. The term of the lease commenced on November 1, 2008 and
will expire on January 31, 2014. The lease provided for a period of
rent-free early occupancy before the commencement date. We believe the facility
is adequate
for our current and future needs for at least the next 12 months and has the
capacity to accommodate additional headcount and manufacturing that we may need
over this period of time.
|
ITEM 3.
|
LEGAL
PROCEEDINGS
|
On
January 8, 2008, Hologic and its wholly-owned subsidiaries, including Cytyc
Corporation and Cytyc LP, filed a lawsuit against us in the United States
District Court, Northern District of California, San Jose Division. The
complaint generally alleges patent infringement of certain Hologic brachytherapy
patent claims, seeking unspecified monetary damages and an injunction against us
for infringement of those claims. On February 6, 2008, Hologic filed a motion
seeking a preliminary injunction in the case and requested that the Court stop
the sale of Contura MLB. On March 7, 2008, Hologic filed an amended complaint
restating its allegations regarding patent infringement, and adding new claims
related to unfair competition under the Lanham Act and California state unfair
competition and false advertising statutes. On April 25, 2008, the court denied
Hologic's request for a preliminary injunction and ordered the parties to
schedule a trial within 60 to 90 days of such date. On May 22, 2008, the Court
issued an order scheduling the Markman claims construction hearing on the patent
counts for June 25, 2008, and the trial in the case to start July 14, 2008.
Pursuant to an agreement of the parties, the order also dismissed Hologic's
unfair competition and false advertising claims under the Lanham Act and
California state law, without prejudice. On June 24, 2008, the Court granted our
joint request with Hologic to stay all proceedings, including the previously
scheduled Markman claims construction hearing and the trial, until at least
August 22, 2008 in order to provide the parties time to discuss possible
resolution of the matter. On August 22, 2008, we jointly requested
with Hologic that the Court resume proceedings in the pending
lawsuit. On October 15, 2008, a Markman claims construction hearing
and a hearing on our motion for summary judgment of invalidity of certain claims
was held and a ruling was issued on February 18, 2009. In light of the Court's
Markman ruling, on May 20, 2009, the parties separately filed motions seeking
partial summary judgment: Hologic filed a motion seeking partial
summary judgment of infringement for certain claims, and we filed motions
seeking partial summary judgment that certain claims were not infringed and that
certain claims were invalid. Briefing on the summary judgment motions
was completed, and argument was held on August 21, 2009. The Court
issued an order on October 30, 2009 ruling that one of the asserted claims of
U.S. Patent No. 6,482,142 was invalid, and that physicians using the Contura MLB
infringed the other asserted claim of the '142 patent. The Court
further ruled that one asserted claim of U.S. Patent No. 6,413,204 and all
asserted claims of U.S. Patent No. 5,913,813 were not infringed. The
remaining claims in the case were tried by a jury between December 2, 2009 and
December 16, 2009. On December 17, 2009, the jury returned a verdict
ruling in our favor on all counts remaining in the case. In
particular, the jury found that we did not infringe the '204 patent, and that
the remaining claims of the '204 patent and '142 patent were invalid for reasons
of anticipation and obviousness. The
Court issued a final judgment in the case on February 24, 2010.
Accordingly, following the result of the jury trial, the Contura MLB does
not infringe any valid claim of any of the Hologic patents that were
the subject matter of the litigation. On February 26, 2010, Hologic filed a
notice of its appeal of the Court's final judgment and other adverse orders,
opinions, and rulings relating to the final judgment, claim construction and the
summary judgment order. No further dates have been scheduled at this
juncture. We believe that the probability of incurring any loss related to
this litigation is not determinable, nor is the amount of loss quantifiable at
this time. Accordingly, we have not accrued a loss related to this litigation as
of December 31, 2009. We intend to continue to vigorously defend
ourselves in this matter.
-31-
|
ITEM
4.
|
[RESERVED]
|
PART
II
|
ITEM
5.
|
MARKET
FOR THE REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER
PURCHASES OF EQUITY SECURITIES
|
Stock
Exchange Listing
Our
common stock has traded on the NASDAQ Global Market under the symbol “SENO”
since our initial public offering on March 29, 2007. Prior to that time, there
was no public market for our stock. On February 26, 2010, the closing sale price
of our common stock was $7.19 per share.
Common
Stockholders
As of
February 28, 2010 there were approximately 80 stockholders of record of our
common stock.
Stock
Prices
The
following table sets forth quarterly high and low closing sales prices of our
common stock for the indicated periods.
|
2009:
|
High
|
Low
|
|
|
Fourth
Quarter
|
$8.10
|
$3.85
|
|
|
Third
Quarter
|
$5.40
|
$3.28
|
|
|
Second
Quarter
|
$3.71
|
$2.90
|
|
|
First
Quarter
|
$3.50
|
$2.40
|
|
|
2008:
|
|||
|
Fourth
Quarter
|
$5.00
|
$2.14
|
|
|
Third
Quarter
|
$7.80
|
$4.66
|
|
|
Second
Quarter
|
$7.75
|
$5.45
|
|
|
First
Quarter
|
$9.06
|
$6.08
|
Dividend
Policy
We have
never paid a cash dividend and have no present intention to pay cash dividends
in the foreseeable future. The Board of Directors currently intends to retain
any future earnings for use in our business.
Sale
of Unregistered Securities
We did
not sell any unregistered securities during the period covered by this Annual
Report on Form 10-K.
Purchase
of Equity Securities
We did
not purchase any equity securities during the period covered by this Annual
Report on Form 10-K.
Stock
Performance Graph
The
following graph compares the cumulative total stockholder return on our common
stock with the cumulative total return of the NASDAQ Composite Index and the
NASDAQ Medical Equipment Index for the period beginning on March 29, 2007,
our first day of trading after our initial public offering, and ending on
December 31, 2009.
-32-
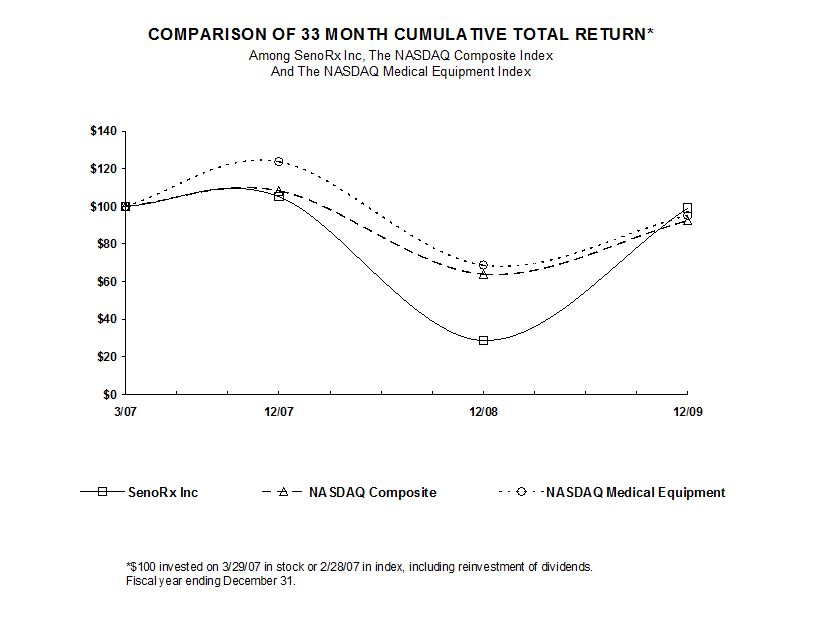
_____________________________
The graph
assumes that $100 was invested on March 29, 2007 in our common stock, the
NASDAQ Composite Index, and the NASDAQ Medical Equipment Index, and that all
dividends were reinvested. No dividends have been declared or paid on our common
stock. Stock performance shown in the above chart for the common stock is
historical and should not be considered indicative of future price performance.
This graph was prepared by Research Data Group, Inc.
Annual Percentage
Return
|
March
29,
2007
|
December
31,
2007
|
December
31,
2008
|
December
31,
2009
|
|
|
SenoRx,
Inc
|
100.00
|
105.13
|
28.61
|
98.97
|
|
NASDAQ
Composite
|
100.00
|
108.27
|
63.99
|
92.43
|
|
NASDAQ
Medical Equipment
|
100.00
|
123.75
|
68.74
|
95.04
|
-33-
|
ITEM 6.
|
SELECTED
FINANCIAL DATA
|
The
following table presents selected historical financial data. We derived the
selected statements of operations data for the years ended December 31,
2009, 2008, and 2007and balance sheet data as of December 31, 2009 and 2008
from our audited financial statements and notes thereto that are included
elsewhere in this annual report. We derived the selected statements of
operations data for the years ended December 31, 2006 and 2005 and the balance
sheet data as of December 31, 2007, 2006 and 2005 from our audited financial
statements that do not appear in this annual report.
You
should read the following financial information together with the information
under “Management’s Discussion and Analysis of Financial Condition and Results
of Operations” and our financial statements and related notes included elsewhere
in this annual report. The information set forth below is not
necessarily indicative of our future financial condition or results of
operations.
|
Years Ended
December 31,
|
||||||||||||||||||||
|
2009
|
2008
|
2007
|
2006
|
2005
|
||||||||||||||||
|
(in
thousands, except per share data)
|
||||||||||||||||||||
|
Statement
of Operations Data:
|
||||||||||||||||||||
|
Net
revenues
|
$ | 55,577 | $ | 46,685 | $ | 35,036 | $ | 25,508 | $ | 19,253 | ||||||||||
|
Cost
of goods sold (1)
|
16,366 | 16,503 | 15,124 | 13,506 | 10,105 | |||||||||||||||
|
Gross
profit
|
39,211 | 30,181 | 19,912 | 12,002 | 9,148 | |||||||||||||||
|
Operating
expenses:
|
||||||||||||||||||||
|
Selling
and marketing (1)
|
23,880 | 23,117 | 19,023 | 15,041 | 10,148 | |||||||||||||||
|
Research
and development (1)
|
7,361 | 6,111 | 6,354 | 5,323 | 4,903 | |||||||||||||||
|
General
and administrative (1)(3)
|
10,687 | 10,094 | 4,187 | 2,050 | 2,116 | |||||||||||||||
|
Total
operating expenses
|
41,928 | 39,322 | 29,564 | 22,414 | 17,167 | |||||||||||||||
|
Loss
from operations
|
(2,717 | ) | (9,141 | ) | (9,652 | ) | (10,412 | ) | (8,019 | ) | ||||||||||
|
Interest
(income) expense, net
|
171 | (435 | ) | 7 | 850 | 594 | ||||||||||||||
|
Loss
on debt extinguishment
|
— | — | 1,265 | 197 | — | |||||||||||||||
|
Change
in fair value of convertible promissory notes
|
— | — | (991 | ) | 3,960 | — | ||||||||||||||
|
Loss
before provision for income taxes
|
(2,888 | ) | (8,706 | ) | (9,933 | ) | (15,419 | ) | (8,613 | ) | ||||||||||
|
Provision
for income taxes
|
— | — | — | — | 10 | |||||||||||||||
|
Net
loss
|
$ | (2,888 | ) | $ | (8,706 | ) | $ | (9,933 | ) | $ | (15,419 | ) | $ | (8,613 | ) | |||||
|
Net
loss per share—basic and diluted
|
$ | (0.17 | ) | $ | (0.50 | ) | $ | (0.75 | ) | $ | (6.61 | ) | $ | (4.19 | ) | |||||
|
Weighted-average
shares outstanding basic and diluted (2)
|
17,381 | 17,250 | 13,309 | 2,332 | 2,060 | |||||||||||||||
|
(1)
|
||||||||||||||||||||
|
Includes
all non-cash stock-based compensation expense as follows:
|
||||||||||||||||||||
|
Cost
of goods sold
|
$ | 139 | $ | 95 | $ | 110 | $ | 52 | $ | 34 | ||||||||||
|
Selling
and marketing
|
785 | 863 | 589 | 409 | 438 | |||||||||||||||
|
Research
and development
|
542 | 449 | 509 | 395 | 286 | |||||||||||||||
|
General
and administrative
|
832 | 865 | 883 | 220 | 563 | |||||||||||||||
|
Total
|
$ | 2,298 | $ | 2,272 | $ | 2,091 | $ | 1,076 | $ | 1,321 | ||||||||||
|
(2)
|
See
Note 1 of the notes to our audited financial statements included
elsewhere in this annual report for an explanation of the determination of
the number of shares used in computing per share
data.
|
|
(3)
|
Includes
patent litigation expense of $5.5 million and $4.9 million in 2009 and
2008, respectively.
|
-34-
|
As of
December 31,
|
||||||||||||||||||||
|
2009
|
2008
|
2007
|
2006
|
2005
|
||||||||||||||||
|
(in
thousands)
|
||||||||||||||||||||
|
Balance Sheet
Data:
|
||||||||||||||||||||
|
Cash
and cash equivalents
|
$ | 18,297 | $ | 15,323 | $ | 17,185 | $ | 7,413 | $ | 482 | ||||||||||
|
Working
capital
|
27,006 | 28,008 | 32,894 | 7,386 | 2,308 | |||||||||||||||
|
Total
assets
|
37,237 | 35,417 | 42,062 | 19,981 | 8,163 | |||||||||||||||
|
Long
term obligations, less current portion
|
1,125 | 1,632 | 27 | 12,125 | 2,741 | |||||||||||||||
|
Convertible
promissory notes (at fair value)
|
— | — | — | 11,960 | — | |||||||||||||||
|
Convertible
preferred stock
|
— | — | — | 46,817 | 46,817 | |||||||||||||||
|
Total
stockholders’ equity (deficit)
|
27,957 | 28,299 | 34,363 | (13,582 | ) | 658 | ||||||||||||||
|
ITEM 7.
|
MANAGEMENT’S
DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF
OPERATIONS
|
This
Annual Report on Form 10-K contains forward-looking statements within the
meaning of the federal securities laws. These statements are subject to risks
and uncertainties that could cause actual results and events to differ
materially from those expressed or implied by such forward-looking statements.
For a detailed discussion of these risks and uncertainties, see the “Risk
Factors” section in Item 1A of Part I of this Form 10-K. We
caution the reader not to place undue reliance on these forward-looking
statements, which reflect management’s analysis only as of the date of this
Form 10-K. We undertake no obligation to update forward-looking statements
to reflect events or circumstances occurring after the date of this
Form 10-K.
Overview
We
develop, manufacture and sell minimally-invasive medical devices that are used
in the diagnosis and treatment of breast cancer. We were incorporated in 1998.
From our inception until 2002, our principal activity was the development and
regulatory clearance of our initial products, primarily our biopsy tissue
markers and our first breast biopsy system, the EnCor 360. We launched our first
biopsy tissue markers in 2002 and our EnCor 360 in 2003. The EnCor 360 hardware
subsequently served as a platform to facilitate the later launch of the EnCor
probes, handpieces and other probes, which are compatible with the major imaging
modalities.
In 2004,
we received 510(k) clearance from the FDA to market our EnCor breast biopsy
system, our flagship product for use in breast biopsy procedures, conducting
market preference testing commencing in the fourth quarter of 2004. Over the
subsequent period ending in October 2005, we began selling the product on a
limited basis while we focused on enhancing certain components of the product to
optimize its performance, and we subsequently progressed with a full commercial
launch of our EnCor system in November 2005.
We have
and are continuing to develop minimally-invasive products for surgical excision
of lesions and for breast cancer treatment. We received 510(k) clearance for our
Contura Multi-Lumen Radiation Balloon Catheter, or Contura MLB, in May 2007 and
launched in January 2008. Contura MLB is one of a new class of devices designed
to reduce radiation treatment time to five days from six to eight weeks in
patients eligible for the treatment. We also believe that Contura MLB may
present radiation oncologists with opportunities to optimize dosing for certain
patients. We are also developing next generation tissue marker products,
additional EnCor line extensions, line extensions of Contura MLB, devices to
assist in lesion location and certain other excision and reconstructive tissue
cutting devices.
Before
2007, we derived our revenues primarily from our tissue marker products.
However, our EnCor system accounted for a majority of our revenue growth in
2007and 2008. Our ability to continue to grow revenues is based upon a number of
assumptions, which may not ultimately occur, including retention of our sales
force, growth in the market for minimally-invasive breast biopsy procedures and
rapid adoption of the product by physicians who specialize in breast care. We
expect our Contura MLB to increasingly contribute to our revenues and we are
marketing this device as a compelling alternative to competing
devices.
For the
year ended December 31, 2009, we generated net revenues of $55.6 million
and a net loss of $2.9 million. As of December 31, 2009, our accumulated
deficit was $87.1 million. We have not been profitable since inception. We
expect our operating expenses to increase as we expand our business to meet
anticipated increased demand for our EnCor system, expand sales of our Contura
MLB and devote resources to our sales and marketing and research and development
activities.
-35-
Net
Revenues
We derive our revenues primarily from
the sales of our breast biopsy systems, breast biopsy capital equipment, our
tissue markers, Contura MLB and other products for breast care. Our largest
market for these products is in the United States and Canada, where we employ a
direct sales force. Our breast biopsy systems, the EnCor and EnCor 360, consist
of two primary components: reusable handpieces and disposable probes, and are
used in conjunction with our SenoRx Breast Biopsy Console. The disposable probes
form the basis of a recurring revenue stream and also contribute to the sales of
tissue markers. Diagnostic adjunct revenue consists primarily of tissue marker
sales, both used with our breast biopsy systems and with competitor’s biopsy
products. Our breast biopsy capital equipment includes a reusable handpiece, a
control module and vacuum source used in conjunction with our disposable biopsy
probe. We expect that the sales of biopsy disposable products will continue to
grow in 2010. Sales of biopsy capital equipment and sales of our
adjunct products, such as our tissue markers and Gamma Finder, have begun to
show signs of recovery as a result of the improvement in the general economic
environment and we expect additional growth in the coming quarters. We have
experienced significant revenue growth for Contura MLB since its commercial
launch in January 2008. We anticipate that
Contura MLB will continue to provide revenue increases quarter over quarter in
the coming quarters, however, at a lower percentage growth rate than in 2009. We
believe that overall placements of EnCor systems will continue to increase in
2010.
Cost
of Goods Sold
Our cost
of goods sold consists of the cost to manufacture and assemble our products,
primarily including materials, components and labor. We assemble and package all
of our finished products with the exception of our Gamma Finder product. We
expect that our cost of goods sold as a percentage of revenues will decrease,
and, correspondingly, gross profits will increase, as a percentage of net
revenues with increased sales volume, product enhancements and outsourced
manufacturing efficiencies. At the end of 2005, we entered into an agreement
with a contract manufacturer in Thailand and began to transfer a portion of our
manufacturing for certain components of our products to this site, and we
anticipate that we will transfer additional manufacturing to this site in order
to increase gross margins. We anticipate that our gross margin will continue to
increase, though at a slower rate, in 2010 due to design and production process
improvements, changes in product mix, the manufacturing efficiencies that we
expect to see with increased production and the continued successful transfer of
manufacturing of certain products and product components to our Thailand
contract manufacturer.
Operating
Expenses
Our
operating expenses consist of selling and marketing, research and development,
and general and administrative expenses. Stock-based compensation, a non-cash
item, is primarily included in these expenses.
Our
selling and marketing expenses consist of salaries and related expenses of our
direct sales team and sales management, travel, clinical education and training
expenses, marketing and promotional expenses, and costs associated with
tradeshows. We expect selling and marketing expenses to increase in absolute
terms as we expand our sales organization and promotional activities, although
at a rate less than our revenue growth rate.
Our
research and development expenses consist of salaries and related expenses of
our research and development personnel and consultants and costs of product
development, which include patent filing and maintenance costs, production
engineering, clinical and regulatory support and post-clearance clinical product
enhancements. We expense all our research and development costs as they are
incurred. We expect research and development expenses to increase in absolute
terms and as a percent of revenues in 2010 as we continue to develop, enhance,
obtain clinical results and commercialize existing and new
products.
In
addition, we recently underwent an inspection of our manufacturing facilities by
the FDA, which resulted in the issuance on September 30, 2009 of an FDA Form
483, Notice of Inspectional Observations, from the FDA related to our failure to
properly implement and maintain adequate methods and documentation of the
design, testing, production, control, quality assurance, storage, shipping and
post-market surveillance for some of our products, including Contura MLB and Gel
Mark product line. The Notice of Inspectional Observations will
require us to take prompt action to strengthen our Quality System and will
result in increased expenses.
-36-
Our
general and administrative expenses consist of the cost of corporate operations,
litigation and professional services. We expect general and administrative
expenses to decrease in absolute dollars as a result of the completion of the
trial relating to the Hologic patent infringement lawsuit. In 2009
and 2008, we incurred $5.5 million and $4.9 million, respectively, in patent
litigation expenses related to alleged patent infringement by
Hologic. On December 18, 2009 a jury delivered a verdict in our
favor. On February 26, 2010, Hologic filed a notice of its appeal in the matter
As a result, we believe that the
probability of incurring any loss related to this litigation is not
determinable, nor is the amount of loss quantifiable at this time. Accordingly,
we have not accrued a loss related to this litigation as of December 31, 2009.
We expect to incur stock-based compensation expense for option grants and
RSU’s. We expect these expenses to remain essentially flat with 2009
levels. We also expect to incur stock-based compensation expense related to the
issuance of common stock under our employee stock purchase plan.
Interest
Interest
income represents income generated from our cash and cash equivalents and
short-term investments that are invested generally in liquid money-market funds
and commercial paper. During 2009, we had a term loan with Silicon Valley Bank
or SVB, resulting in interest expense. Interest expense also includes the fair
value for any equity interests, such as warrants, granted in conjunction with
the debt obligations. The fair value of the equity interests were amortized to
interest expense over the term of the related debt obligations. Interest expense
has decreased due to the retirement of certain debt obligations in 2007 and
early 2008 and lower available interest rates.
Income
Tax Expense
Due to
uncertainty surrounding the realization of deferred tax assets through future
taxable income, we have provided a full valuation allowance and no benefit has
been recognized for our net operating loss and other deferred tax
assets.
Results
of Operations
The
following table sets forth our results of operations expressed as percentages of
revenues for the years ended December 31, 2009, 2008 and 2007:
|
For
the Years Ended
December 31,
|
||||||||||||
|
2009
|
2008
|
2007
|
||||||||||
|
Net
revenues
|
100.0 | % | 100.0 | % | 100.0 | % | ||||||
|
Cost
of goods sold
|
29.4 | 35.4 | 43.2 | |||||||||
|
Gross
profit
|
70.6 | 64.6 | 56.8 | |||||||||
|
Operating
expenses:
|
||||||||||||
|
Selling
and marketing
|
43.0 | 49.5 | 54.3 | |||||||||
|
Research
and development
|
13.2 | 13.1 | 18.1 | |||||||||
|
General
and administrative
|
19.2 | 21.6 | 12.0 | |||||||||
|
Total
operating expenses
|
75.4 | 84.2 | 84.4 | |||||||||
|
Loss
from operations
|
4.9 | 19.6 | 27.5 | |||||||||
|
Interest
expense
|
0.4 | 0.2 | 4.7 | |||||||||
|
Loss
on debt extinguishment
|
— | — | 3.6 | |||||||||
|
Change
in fair value of convertible promissory notes and warrant
liability
|
— | — | (2.8 | ) | ||||||||
|
Interest
income
|
— | (1.1 | ) | (4.7 | ) | |||||||
|
Provision
for income taxes
|
— | — | — | |||||||||
|
Net
loss
|
(5.2 | )% | (18.6 | )% | (28.4 | )% | ||||||
-37-
Year
ended December 31, 2009 Compared to year Ended December 31,
2008
Net
Revenues. Net revenues increased $8.9 million, or 19.0%, to $55.6 million
in 2009 from $46.7 million in 2008. Therapeutic disposable revenues increased
$6.4 million, or 123.0%, resulting from increased adoption of our Contura
MLB. We began sales of the Contura MLB to a limited number of
clinical sites in June 2007 following the May 2007 FDA 510(k) clearance, with
full commercialization in January 2008. Biopsy disposable revenues
increased $2.9 million or 13.9% from 2008 due to a larger installed base of
EnCor systems. Total disposable product sales, which includes Contura
MLB and EnCor probes, increased 22.7% to $50.0 million or 90.0% of net revenues
in 2009 from $40.7 million or 87.3% of net revenues for the same period a year
ago. These increases were off set by a decrease in biopsy capital
revenues of $287,000, or 6.1%, and a decrease of diagnostic adjunct revenues of
$118,000, or 0.7%. We believe that the decreases in biopsy capital and Gamma
Finder sales were a result of the global recession, tight credit markets and
currency fluctuations negatively impacting our global capital equipment
sales.
Cost of Goods
Sold and Gross Profit. Cost of goods sold decreased $138,000, or 0.8%, to
$16.4 million in 2009 from $16.5 million in 2008. The decrease in total cost of
goods sold primarily consisted of a decrease in direct labor, manufacturing
overhead and material costs associated with our increase in product sales, which
was partially offset by an increase of $925,000 in the provision for inventory
obsolescence. Gross profit increased $9.0 million, or 29.9% to $39.2 million for
2009 from $30.2 million or 64.6% for 2008. Gross margins continue to benefit
from improved efficiencies in the production of our disposable biopsy probe and
leverage from the allocation of fixed manufacturing overhead over greater
product revenues and inventory unit production.
Selling and
Marketing Expenses. Selling and marketing expenses increased $763,000, or
3.3%, to $23.9 million in 2009 from $23.1 million in 2008. The increase
primarily consisted of $602,000 in salaries and related employee costs due to
the modest increases to our sales organization, a $349,000 increase in selling
and promotional related expenses and a $64,000 increase in professional
services, which were partially offset by a $174,000 decrease in departmental
related costs and a $78,000 decrease in equity based compensation charges
including deferred compensation and the discount associated with shares
purchased by employees under our Employee Stock Purchase Plan.
Research and
Development Expenses. Research and development expenses increased $1.2
million, or 20.4%, to $7.4 in 2009 from $6.1 million in 2008. The increase in
these expenses consisted primarily of $401,000 in salaries and the related
employee costs, an increase of $362,000 for new product development and support
of existing product lines, an increase of $197,000 in departmental related
costs, an increase of $195,000 for patent related professional fees
and an increase of $94,000 in equity based compensation charges including
deferred compensation and the discount associated with shares purchased by
employees under our Employee Stock Purchase Plan.
General and
Administrative Expenses. General and administrative expenses increased
$594,000, or 5.9%, to $10.7 million in 2009 from $10.1 million in
2008. The increase primarily consisted of an increase of $593,000 to
$5.5million in attorney and related litigation costs incurred responding to the
allegations by Hologic of patent infringement relating to our Contura MLB, an
increase of $130,000 in salaries and the related employee costs, an increase of
$101,000 for state franchise taxes and an increase of $36,000 for departmental
costs. These increases were partially offset by a decrease of $143,000 for
professional services, including general legal fees and a decrease of $92,000
for public company related costs, including legal, SOX and financial reporting
expenses and a decrease of $33,000 in equity based compensation charges
including deferred compensation and the discount associated with shares
purchased by employees under our Employee Stock Purchase Plan.
Interest Expense.
Interest expense increased $113,000 to $198,000 in 2009 from $85,000 in
2008. The increase is primarily due to the $2.0 million advance in November 2008
from our term loan with SVB.
Interest Income.
Interest income decreased $493,000 to $27,000 in 2009 from $520,000 in
2008 primarily due to lower interest rates and lower cash balances resulting
from working capital needs for operations and spending on patent
litigation.
Year
ended December 31, 2008 Compared to year Ended December 31,
2007
Net
Revenues. Net revenues increased $11.6 million, or 33.2%, to $46.7
million in 2008 from $35.0 million in 2007. The increase primarily consisted of
$4.8 million in biopsy disposable revenues, an increase of 29.8% from 2007 due
to a larger installed base of EnCor systems. Biopsy capital equipment revenues
increased $1.4 million, or 42.1% in 2008 due to a greater number of customers
purchasing our breast biopsy systems as compared to those customers acquiring
the capital through a “product supply agreement” and increased equipment sales
to international distributors. Diagnostic adjunct revenues increased $798,000,
or 5.3%, primarily due to an increase in marker sales resulting from increased
EnCor disposable biopsy sales and sales of markers used with competitive biopsy
disposables and increased Gamma Finder sales. Therapeutic revenues increased
$4.6 million as we began sales of our Contura MLB to a limited number of
clinical sites in June 2007 following the May 2007 FDA 510(k) clearance, with
full commercialization in January 2008.
-38-
Cost of Goods
Sold and Gross Profit. Cost of goods sold increased $1.4 million, or
9.1%, to $16.5 million in 2008 from $15.1 million in 2007. The increase in total
cost of goods sold primarily consisted of an increase in direct labor,
manufacturing overhead and material costs associated with our increase in unit
sales. Gross profit increased $10.3 million or 51.6% to $30.2 million in 2008
from $19.9 in 2007. Gross profit as a percentage of net revenues increased by
7.8% to 64.6% in 2008 from 56.8% in 2007. The increase in gross profit as a
percentage of net revenues was primarily attributable to improved efficiencies
in the production of our disposable biopsy probe and gross margin contribution
from the sales of our Contura MLB. Additionally, gross margins continue to
benefit from the allocation of manufacturing overhead over greater product
revenues and inventory unit production.
Selling and
Marketing Expenses. Selling and marketing expenses increased $4.1
million, or 21.5%, to $23.1 million in 2008 from $19.0 million in 2007. The
increase primarily consisted of $3.5 million in salaries and related employee
costs due to the expansion of our sales organization, a $133,000 increase in
departmental related expenses, a $147,000 increase in selling and promotional
related expenses and a $274,000 increase in equity based
compensation.
Research and
Development Expenses. Research and development expenses decreased
$242,000, or 3.8%, to $6.1 in 2008 from $6.4 million in 2006. The decrease in
these expenses consisted primarily of project costs, predominately for the
Contura MLB, which decreased $502,000 and a decrease of $60,000 in equity based
compensation. This decrease was partially offset by an increase of $62,000 for
salaries and the related employee costs, a $116,000 increase for patent related
expenses and an increase of $142,000 in departmental related
expenses.
General and
Administrative Expenses. General and administrative expenses increased
$5.9 million, or 141.1%, to $10.1 million in 2008 from $4.2 million in
2007. The increase primarily consisted of $4.9 million in attorney
and related litigation costs incurred in connection with responding to the
allegations by Hologic of patent infringement relating to our Contura MLB,
$265,000 for public company related costs, including legal and reporting
expenses, which includes first year Sarbanes Oxley implementation costs, $275,000 for salaries and
the related employee costs, $287,000 for departmental costs and $173,000 for
increased professional fees.
Interest Expense.
Interest expense decreased $1.6 million to $85,000 in 2008 from $1.6
million in 2007. The decrease was primarily due to the repayment of the December
2006 subordinated note.
Loss on Debt
Extinguishment. In November 2007, we incurred a $1.3 million expense on
the retirement of a December 2006 Subordinate Note facility that had an original
principal amount of $10.0 million outstanding under it, representing the
unamortized debt issuance and debt discounts which would have been otherwise
charged to interest expense over the term of the facility. In 2008,
we did not incur any expense related to this item.
Change in Fair
Value of Convertible Promissory Notes and Warrant Valuation. In 2007, we
recorded income of $160,000 for the change in fair value of our May 2006
convertible promissory notes that had an original aggregate principal amount of
$8.0 million outstanding, in accordance with FAS No. 155, and income of $831,000
for the reduction in the fair value of a related warrant liability. In 2008, we
did not incur any expense related to this item.
Interest Income.
Interest income decreased $1.1 million to $520,000 in 2008 from $1.6
million in 2007 primarily due to lower cash and short-term investment balances
resulting from the repayment of $10.3 million for the retirement of the December
2006 Subordinated Note, $2.0 million to repay a February 2003 convertible
subordinated note and 2002 note obligations owing to Century Medical, as well as
working capital needs.
-39-
Liquidity
and Capital Resources
General
We have
incurred losses since our inception in January 1998 and, as of December 31,
2009, we had an accumulated deficit of $87.1 million. From inception
through December 31, 2009, we generated cumulative gross profit from the sale of
our product offerings of $126.1 million. To date, our operations have
been funded primarily with proceeds from the issuance of our preferred stock,
debt issuances and our IPO that closed in April 2007. Cumulative net proceeds
from the issuance of preferred stock totaled $46.8 million. Proceeds from the
issuance of promissory notes totaled $8.0 million. Net proceeds from our IPO,
including the sale of shares pursuant to the subsequent underwriters’
overallotment and after deducting total expenses, was $44.8 million. All of our
preferred stock converted into common stock upon the closing of the IPO. In
November 2007 we used $10.3 million to retire a December 2006 Subordinated Note
and in February 2008 we used $2.0 million to repay a February 2003 convertible
subordinated note and 2002 note obligations owing to Century Medical. In
September 2008 we amended our existing loan agreement with SVB to, among other
items, increase the total maximum amount available for borrowing from $4.0
million to $12.0 million. As of December 31, 2009, $2.0 million has been drawn
down under this facility, of which $1.6 million is outstanding, and $8.1 million
was available based on the borrowing formula for the facility.
We
believe that our cash and cash equivalents will be sufficient to meet our
projected operating requirements for at least the next 12 months.
Net
Cash Provided By Operating Activities—Year Ended 2009
Net cash
provided by operating activities was $3.8 million, for the year ended December
31, 2009, which was primarily a function of an increase in accounts payable and
accrued expenses of $2.3 million, a decrease in inventory of $944,000, and an
increase in deferred revenue of $282,000. These sources of cash were
partially offset by an increase in accounts receivable of $1.6 million and an
increase in other assets of $110,000. The $2.3 million increase in
accounts payable and accrued expenses was primarily due to the timing of payment
obligations, the $944,000 decrease in inventory was due to a focus on cost
containment measures and the $282,000 increase in deferred revenue resulted from
a higher number of equipment service contracts. The increase in
accounts receivable was primarily due to an increase in net sales and an
increase in sales outside the United States, which include extended payment
terms for initial stocking orders. While we expect that the amount of accounts
receivable will fluctuate based on the timing of sales and collections, we
expect our ratio of overall investment in accounts receivable as compared to
revenues will continue to modestly increase.
Net
Cash Used in Investing Activities—Year Ended 2009
Net cash
used in investing activities amounted to $616,000 during the year ended December
31, 2009, was primarily attributable to the purchase of new manufacturing molds
and demonstration units.
Net
Cash Used in Financing Activities—Year Ended 2009
Net cash
used in financing activities was $199,000 during the year ended December 31,
2009, which was primarily attributable to the scheduled repayments of $375,000
on our term loan with SVB, $50,000 cash payment for the annual renewal of the
SVB working capital facility, $57,000 payment of payroll taxes for the net share
settlement of equity awards, $11,000 repayment of capital leases and the
repayment of a $10,000 equipment loan. These uses of cash were offset by
$305,000 related to the proceeds from the issuance of common stock from option
exercises and purchases of stock pursuant to our Employee Stock Purchase
Plan.
Net
Cash Used in Operating Activities—Year Ended 2008
Net cash
used in operating activities was $11.9 million, for the year ended December 31,
2008, which was primarily a function of an increase in inventory of $3.9
million, an increase in accounts receivable of $2.9 million, an increase in
other assets of $225,000, and a decrease in accounts payable and accrued
expenses of $560,000. These uses of cash were partially offset by a decrease in
prepaid expenses of $158,000 and an increase in deferred revenue of $68,000. The
aggregate increased investment in inventory of $3.9 million resulted primarily
from two major factors, including (i) our decision to build shelf stock for our
higher volume products in order to better service our customers as well as
accommodate our move to the new manufacturing facility in late August 2008, and
(ii) the need to purchase longer-term quantities of certain parts due to
long lead times. We expect inventory will modestly decrease in 2009. The
increase in accounts receivable was primarily due to an increase in net sales
and an increase in sales outside the United States, which include extended
payment terms for initial stocking orders. While we expect that the amount of
accounts receivable will fluctuate based on the timing of sales and collections,
we expect our ratio of overall investment in accounts receivable as compared to
revenues will continue to modestly increase. The $560,000 decrease in accounts
payable and accrued expenses resulted from the use of funds, including funds
borrowed under our bank loan facility with SVB that allowed us to reduce
outstanding accounts payable with certain vendors.
-40-
Net
Cash Provided by Investing Activities—Year Ended 2008
Net cash
provided by investing activities amounted to $9.8 million during the year ended
December 31, 2008, was primarily attributable to the maturities of short-term
investments which was partially offset by the $970,000 addition of new
manufacturing molds and equipment, business intelligence software, leasehold
improvements to our new Irvine facility and demonstration units.
Net
Cash Provided by Financing Activities—Year Ended 2008
Net cash
provided by financing activities was $242,000 during the year ended December 31,
2008, primarily attributable to $2.0 million advance on our term loan and
$370,000 related to the proceeds from the issuance of common stock from option
exercises and ESPP stock purchases. These proceeds were partially offset by the
repayment of $2.0 million for the Century Medical notes and $30,000 cash paid
for the annual renewal of the SVB working capital facility
Net
Cash Used in Operating Activities—Year Ended 2007
Net cash
used in operating activities was $10.3 million, for the year ended
December 31, 2007, which was a function of an increase in inventory of $2.7
million, an increase in accounts receivable of $1.2 million, a decrease in
accounts payable and accrued expenses of $748,000, and an increase in prepaid
expenses of $324,000. These uses of cash were partially offset by a decrease in
other assets of $384,000 and an increase in deferred revenue of $58,000. The
aggregate increased investment in inventory of $2.7 million resulted primarily
from two major factors, including our decision to build shelf stock of our
higher volume products in order to better service our customers, and the need to
purchase longer-term quantities of certain parts due to long lead times. We
expect inventory will continue to increase in 2008. The increase in accounts
receivable was primarily due to an increase in net sales. While we expect that
the amount of accounts receivable will fluctuate based on the timing of sales
and collections, we expect our ratio of overall investment in accounts
receivable as compared to revenues will remain constant as compared to 2007. The
$748,000 decrease in accounts payable and accrued expenses resulted from the use
of proceeds from our April 2007 IPO, which allowed us to reduce outstanding
accounts payable with certain vendors.
Net
Cash Used in Investing Activities—Year Ended 2007
Net cash
used in investing activities amounted to $11.3 million during the year ended
December 31, 2007, primarily attributable to the purchase of short-term
investments and the addition of demonstration units and new manufacturing
molds.
Net
Cash Provided by Financing Activities—Year Ended 2007
Net cash
provided by financing activities was $31.4 million during the year ended
December 31, 2007, primarily attributable to proceeds of $47.1 million from
our April 2007 IPO and underwriters’ overallotment, a $2.8 million advance on
our working capital facility and $61,000 related to the proceeds from the
issuance of common stock from option exercises. These proceeds were partially
offset by an aggregate of $17.8 million in repayments under our various debt
facilities and $941,000 in cash paid for deferred offering costs including
legal, accounting and printing fees, which were offset against offering proceeds
at the completion of our IPO in April 2007.
Accounts
Receivable
Our
accounts receivable days outstanding were 52 days at December 31, 2009, 51
days at December 31, 2008 and 47 days at December 31, 2007. Our
products are typically sold for terms net 30 days from shipment. We review our
accounts receivable balances and customers regularly to establish and maintain
an appropriate allowance for doubtful accounts. Our account analysis includes
reviewing the customer’s historical payment history, the amount and number of
days an account is outside of payment terms, the magnitude of the account
balance, historical order patterns and any specific knowledge about the
customer’s financial condition. Our allowance for doubtful accounts as a
percentage of gross receivables was 2.4%, 2.7% and 2.0% at December 31,
2009, 2008 and 2007, respectively. Our reserve requirements are based on our
review of every account and we place particular emphasis on each customer
account with an account receivable balance more than 90 days old and on that
customer’s specific payment history and other financial information. As our
revenues increase, we anticipate our days sales outstanding will fluctuate
moderately.
-41-
Contractual
Obligations
The
following summarizes our long-term contractual obligations at December 31,
2009:
|
Payments due
by period
|
||||||||||||||||||||
|
Total
|
Less than
1 year
|
1-3 years
|
3-5 years
|
More than
5 years
|
||||||||||||||||
|
Long-term
debt obligations
|
$ | 1,625,000 | $ | 500,000 | $ | 1,000,000 | $ | 125,000 | $ | — | ||||||||||
|
Capital
lease obligations
|
1,054 | 1,054 | — | — | — | |||||||||||||||
|
Operating
lease obligations
|
2,419,529 | 555,612 | 1,185,746 | 678,171 | — | |||||||||||||||
|
Total
|
$ | 4,045,583 | $ | 1,056,666 | $ | 2,185,746 | $ | 803,171 | $ | — | ||||||||||
The
operating leases shown above reflect payments related to our real estate lease
in Irvine, California, which expires in January 2014 and will amount to $3.0
million over the 63 month duration of the lease, which commenced in November
2008.
Working Capital
Facility with Silicon Valley Bank. We have a working capital facility
with SVB that, as a result of amendments to this facility in February 2007 and
September 2008, has an aggregate limit of $12.0 million. In
connection with the September 2008 amendment, we also entered into an
Export-Import Bank of the United States Working Capital Guarantee Program
Borrower Agreement, which helped increase the total maximum amount available for
borrowing under the facility, which is now $12.0 million. Previously, our credit
facility with SVB provided for a maximum borrowing amount of $4.0
million.
Revolving Line. The SVB
facility provides for a domestic receivables-based revolving line of credit in
an aggregate amount of up to $10.0 million, or, if less, the sum of 80% of
eligible domestic accounts receivable plus 25% of eligible domestic inventory,
coupled with a foreign receivables-based revolving line of credit, guaranteed by
Export-Import Bank of the United States, in an aggregate amount of up to $2.5
million, or if less, the sum of 90% of eligible foreign accounts receivable plus
50% of eligible export-related inventory. No more than $10.0 million, in the
aggregate, will be available for combined draw-downs under these two revolving
lines of credit, a maturity date of September 2010 and bears interest at an
annual rate equal to the prime rate plus 0.25% (4.25% at December 31, 2009),
provided that the annual rate will increase to prime rate plus 1.00% if a
financial ratio relating to our liquidity falls below a specified level.
Interest is due monthly, with the balance due at the maturity date. The domestic
portion of revolving line is subject to a $1.0 million sublimit available for
cash management services provided by SVB, including the issuance of short-term
letters of credit and foreign exchange contracts. At December 31, 2009, there
was $8.1 million available to borrow on the revolving line.
Term Loan. The SVB facility
also provides for a term loan in an aggregate amount of up to $2.0 million, with
a maturity date of March 2013, provided that if the revolving line is not
renewed at its maturity date, all amounts outstanding under the term loan will
become due at such time. The term loan bears interest at an annual rate equal to
the prime rate plus 0.75% (4.75% at December 31, 2009), provided that the annual
rate applicable to the term loan increases to an annual rate equal to the prime
rate plus 1.50% if a financial ratio relating to our liquidity falls below a
specified level. A single advance is permitted and was drawn down in November
2008. Monthly payments of interest only on the amount outstanding
were due through March 2009, followed by forty-eight (48) consecutive monthly
installments of amortized principal and interest through the maturity date. A
principal amount of $1.6 million was outstanding at December 31,
2009.
-42-
Loan Agreement Obligations.
The obligations under the SVB facility are secured by security interest
on substantially all of our assets, excluding intellectual property for which we
gave a negative pledge against encumbering. The SVB facility contains certain
restrictive loan covenants, including, among others, financial covenants
requiring a minimum tangible net worth and a minimum liquidity ratio, and
covenants limiting our ability to dispose of assets, make acquisitions, be
acquired, incur indebtedness, grant liens or enter into negative pledge
agreements, make investments, make distributions in respect of our capital stock
(including repurchases of such capital stock) or enter into transactions with
affiliates. The SVB facility also contains events of default that include, among
others, failure to make payments when due, inaccuracy of representations and
warranties, violation of covenants, events constituting a material adverse
change, bankruptcy and insolvency events, material judgments, and cross defaults
to certain other agreements. The occurrence of an event of default could result
in the acceleration of our obligations and an increase to the applicable
interest rate, and would permit SVB to exercise remedies with respect to the
collateral.
Off-Balance
Sheet Arrangements
Since
inception, we have not engaged in material off-balance sheet activities,
including the use of structured finance, special purpose entities or variable
interest entities.
Income
Taxes
Realization
of our deferred tax assets is dependent upon the timing and amount of our future
earnings, if any. Accordingly, we have established full deferred tax asset
valuation allowances as of December 31, 2009, 2008 and 2007 to reflect
these uncertainties.
As of
December 31, 2009, we had federal and state net operating loss
carryforwards of approximately $65.2 million and $45.9 million, respectively,
and $1.8 million in federal tax credit carryforwards and $2.2 million in state
tax credit carryforwards. The federal net operating loss carryforwards and tax
credit carryforwards will begin to expire in 2018. The state net operating loss
carryforwards began to expire in 2009. The state tax credit
carryforwards do not expire. The utilization of the net operating loss and tax
credit carryforwards may be subject to a substantial annual limitation due to
ownership change limitations provided by the Internal Revenue Code. This annual
limitation may result in the expiration of net operating loss and tax credit
carryforwards before we are able to utilize them. As of December 31, 2009 the
Company conducted an analysis with regard to its ownership and concluded an
ownership change had not occurred.
Critical
Accounting Policies
We
prepare our financial statements in accordance with accounting principles
generally accepted in the United States. In doing so, we have to make estimates
and assumptions that affect our reported amounts of assets and liabilities and
disclosure of contingent assets and liabilities at the date of the financial
statements and the reported amounts of revenues and expenses during the
reporting period. We regularly evaluate our estimates and assumptions based upon
historical experience and various other factors that we believe to be reasonable
under the circumstances, the results of which form the basis for making
judgments about the carrying values of assets and liabilities that are not
readily apparent from other sources. To the extent actual results differ from
those estimates, our future results of operations may be affected.
While our
significant accounting policies are more fully described in Note 1 of the notes
to our audited financial statements, we believe that the following accounting
policies and estimates are most critical to a full understanding and evaluation
of our reported financial results.
Revenue
Recognition
Revenue
is recognized when (a) persuasive evidence of an arrangement exists;
(b) title has transferred; (c) the fee is fixed or determinable; and
(d) collectability is reasonably assured. Our recognition policy is
significant because our revenue is a key component of our operations and the
timing of revenue recognition determines the timing of certain expenses, such as
sales commissions. Revenue results are difficult to predict, and any shortfall
in revenues could cause our operating results to vary significantly from period
to period.
-43-
For those
sales that include multiple deliverables, we allocate revenue based on the
relative fair values of the individual components as determined in accordance
with ASC No. 605-25 (formerly, EITF Issue No. 00-21), “Revenue Arrangements
with Multiple Deliverables.” When more than one element, such as hardware and
disposables, are contained in a single arrangement, revenues are allocated
between the elements based on each element’s relative fair value, provided that
each element meets the criteria for treatment as a separate unit of accounting.
An item is considered a separate unit of accounting if it has value to the
customer on a standalone basis and there is objective and reliable evidence of
the fair value of the undelivered items. Fair value is generally determined
based upon the price charged when the element is sold separately. In the absence
of fair value for a delivered element, we allocate revenue first to the fair
value of the undelivered elements and allocate the residual revenue to the
delivered elements. In the absence of fair value for an undelivered element, the
arrangement is accounted for as a single unit of accounting, resulting in a
deferral of revenue recognition for the delivered elements until all undelivered
elements have been fulfilled.
We place
certain equipment with customers in return for the customer purchasing a minimum
number of disposable devices during a specified contract period. Title to the
equipment passes to the customer at the end of the contract period if the
minimum purchase requirements are met. The cost of the equipment, which is
included in other long-term assets in the accompanying balance sheets, is
amortized to cost of goods sold based on the monthly disposable unit shipments
compared to the total purchase commitment of disposables. In the event the
customer does not fulfill the minimum purchase requirements, collection efforts
may be undertaken and we will attempt to recover the equipment. If the
collection efforts or recovery of the equipment is not successful, the
unamortized equipment cost would be expensed to cost of goods sold.
Deferred
Revenue
We also
account for a customer’s advance payment on product purchases and service
contracts as deferred revenue. As product is purchased or the service contract
period matures, the applicable sales value is recognized as
revenue.
Stock-Based
Compensation
Effective
January 1, 2006, we adopted ASC 718 (formerly SFAS No. 123R), which
requires that all stock-based compensation to employees, including grants of
employee stock options, be expensed in our financial statements based on their
respective grant date fair values. Under ASC 718, we estimate the fair value of
each stock-based payment award using the Black-Scholes option pricing
model.
The
determination of the fair value of stock-based payment awards using the
Black-Scholes model is affected by our stock price and a number of assumptions,
including expected volatility, expected life, risk-free interest rate and
expected dividends. We did not have a history of market prices of our common
stock as we were not a public company until our April 2007 initial public
offering, and as such, we estimate volatility in accordance with ASC 718,
(formerly SAB No. 107) using historical volatilities of other publicly
traded companies in our industry. The expected life of the awards is based on
the simplified method as defined in ASC 718. The risk-free interest rate
assumption is based on observed interest rates appropriate for the terms of our
awards. The dividend yield assumption is based on our history and expectation of
not paying any dividends. Forfeitures are estimated at the time of grant and
revised, if necessary, in subsequent periods if actual forfeitures differ from
those estimates. We recognized stock-based compensation expense in our financial
statements based on awards that are ultimately expected to vest.
A summary of significant assumptions
used in determining the fair value of the options is as follows:
|
Year Ended
December 31,
|
|||||
|
2009
|
2008
|
2007
|
|||
|
Expected
life (years)
|
4.5 - 4.75
|
4.5 - 4.75
|
4.75 - 6.25
|
||
|
Risk-free
interest rate
|
1.97% - 2.30%
|
1.62% - 3.26%
|
3.49% - 4.69%
|
||
|
Volatility
|
46% - 47%
|
45% - 48%
|
42% - 48%
|
||
|
Dividend
yield
|
0%
|
0%
|
0%
|
||
|
Forfeiture
rate
|
2.5%
|
2.5%
|
2.5%
|
||
If
factors change and we employ different assumptions, stock-based compensation
expense may differ significantly form what we recorded in the past. If there are
any modifications or cancellations of the underlying unvested securities, we may
be required to accelerate, increase or cancel any remaining unearned stock-based
compensation expense. Future stock-based compensation expense and unearned
stock-based compensation will increase to the extent that we grant addition
equity awards to employees or we assume unvested equity awards in connection
with acquisitions.
-44-
Inventories
We assess
the recoverability of our inventories at least quarterly through a review of
inventory levels in relation to foreseeable demand, generally over twelve
months. Foreseeable demand is based upon all available information, including
sales backlog and forecasts, product marketing plans and product life-cycle
information. When the inventory on hand exceeds the foreseeable demand, we write
down the value of those inventories which, at the time of our review, we expect
to be unable to sell. The amount of the inventory write-down is the excess of
historical cost over estimated realizable value. Once established, these
write-downs are considered permanent adjustments to the cost basis of the excess
inventory. Demand for our products may fluctuate significantly over time, and
actual demand and market conditions may be more or less favorable than those
projected by management. In the event that actual demand or product pricing is
lower than originally projected, additional inventory write-downs may be
required. Further, on a quarterly basis, we assess the net realizable value of
our inventories. When the estimated average selling price, plus costs to sell
our inventory, falls below our inventory cost, we adjust our inventory to its
current estimated market value.
Allowance
for Doubtful Accounts
We
maintain allowances for doubtful accounts for estimated losses resulting from
the inability of our customers to make required payments. We use a specific
identification method for some items, and a percentage of aged receivables for
others. The percentages are determined based on our past experience. If the
financial condition of our customers were to deteriorate, our actual losses
might exceed our estimates, and additional allowances would be
required.
Software
Development
Certain
of our products incorporate software which is incidental to the product as a
whole. Software development costs incurred prior to the establishment of
technological feasibility are expensed as research and development costs. We
define the establishment of technological feasibility as the completion of a
final working model that has been incorporated into a product that has been
cleared by the FDA, at which time the product can be sold to third parties. As a
result, we have expensed all software development costs.
Impairment
of Long-lived Assets
Long-lived
assets, including fixed assets, are continually monitored and are reviewed for
impairment whenever events or changes in circumstances indicate that the
carrying amount of any such asset may not be recoverable. The determination of
recoverability is based on an estimate of undiscounted cash flows expected to
result from the use of an asset and its eventual disposition. The estimate of
cash flows is based upon, among other things, certain assumptions about expected
future operating performance, growth rates and other factors. Our estimates of
undiscounted cash flows may differ from actual cash flows due to, among other
things, technological changes, economic conditions, changes to our business
model or changes in our operating performance. If the sum of the undiscounted
cash flows (excluding interest) is less than the carrying value, we recognize an
impairment loss, measured as the amount by which the carrying value exceeds the
fair value of the asset. We determine fair value by using available market data,
comparable asset quotes and/or discounted cash flow models.
Deferred
Income Taxes
We
evaluate the realizability of our deferred tax assets and assess the need for a
valuation allowance quarterly. We record a valuation allowance to reduce our
deferred tax assets to the net amount that is more likely than not to be
realized. Our assessment of the need for a valuation allowance is based upon our
history of operating results, expectations of future taxable income and the
ongoing prudent and feasible tax planning strategies available to us. In the
event that we determine that we will not be able to realize all or part of our
deferred tax assets in the future, an adjustment to the deferred tax assets
would be charged against income in the period such determination is made.
Likewise, in the event we were to determine that we will be able to realize our
deferred tax assets in the future in excess of the net recorded amount, an
adjustment to the deferred tax assets would increase income in the period such
determination is made.
-45-
Fair
Value of Financial Instruments
At each
reporting date, we were required to estimate the fair value of our May 2006
convertible promissory notes in its entirety with changes in fair value
recognized in the statement of operations. Our estimate of fair value was based
upon a valuation which encompasses the probability weighted scenarios of the
conversion features, as well as the timing and method of payment of interest
associated with our May 2006 notes.
At each
reporting date, we were also required to estimate the fair value of the warrant
issued in conjunction with our 2006 Subordinated Note. Our estimate of fair
value was determined using the Black-Scholes option pricing model, which
requires inputs for risk-free interest rate, dividend yield, volatility, the
life of the warrant and the fair value of the underlying security.
Recent
Accounting Pronouncements
In
January 2010, the FASB issued ASU No. 2010-06, Improving Disclosures about Fair
Value Measurements, which amends the guidance in ASC 820, Fair Value Measurements and
Disclosures. ASU No. 2010-06 requires companies to disclose
transfers between Level 1 and Level 2 within the fair value hierarchy and
describe the reasons for the transfers. In addition, in the reconciliation
of assets and liabilities in Level 3 of the fair value hierarchy, companies are
required to present sales, purchases, issuances and settlements on a gross
rather than net basis. ASU No. 2010-06 also clarifies that companies
should provide fair value measurement disclosures for each class of assets and
liabilities and that companies should disclose the inputs and valuation
techniques used to measure assets and liabilities that fall either in Level 2 or
Level 3. ASU No. 2010-06 will be effective for interim and annual periods
beginning after December 15, 2009, except for the new Level 3 reconciliation
disclosures, which will be effective for interim and annual periods beginning
after December 15, 2010. This update is not expected to have a material
impact on our financial position and results of operations.
In
October 2009, the FASB issued ASU No. 2009-13, Multiple-Deliverable Revenue
Arrangements a consensus of the FASB Emerging Issues Task Force, which amends
the guidance in ASC 605, Revenue Recognition. ASU 2009-13 eliminates the
residual method of accounting for revenue on undelivered products and instead,
requires companies to allocate revenue to each of the deliverable products based
on their relative selling price. In addition, this ASU expands the
disclosure requirements surrounding multiple-deliverable arrangements.
This update is effective beginning January 1, 2011 and can be applied
prospectively or retrospectively. Adoption is not expected to materially impact
our financial position, results of operations or cash flows directly when it
becomes effective, as we will not elect retrospective adoption. However, this
update may impact how we reflect multiple-element arrangements entered into
subsequent to January 1, 2011, in the financial statements.
|
ITEM 7A.
|
QUANTITATIVE
AND QUALITATIVE DISCLOSURES ABOUT MARKET
RISK
|
The
primary objective of our investment activities is to preserve our capital for
the purpose of funding operations while at the same time maximizing the income
we receive from our investments without significantly increasing risk. To
achieve these objectives, our investment policy allows us to maintain a
portfolio of cash equivalents and investments in a variety of marketable
securities, including commercial paper, money market funds and corporate debt
securities and U.S. government securities. Our cash and cash equivalents as of
December 31, 2009, included liquid money market accounts. Due to the liquid
nature of our cash and cash equivalents, we believe we have no material exposure
to interest rate risk. Additionally, since the majority of our debt carries
interest at fixed rates, we also believe changes in interest rates will not
cause significant changes in our interest expense. Our revenues are denominated
in U.S. dollars. Accordingly, we have not had exposure to foreign currency rate
fluctuations. We expect to continue to realize our revenues in U.S.
dollars.
|
ITEM 8.
|
FINANCIAL
STATEMENTS AND SUPPLEMENTARY DATA
|
Our
Financial Statements as of December 31, 2009 and 2008 and for each of the
three years in the period ended December 31, 2009, together with the
reports of our independent registered public accounting firm, are included under
Part IV, Item 14 of this report.
-46-
|
ITEM 9.
|
CHANGES
IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL
DISCLOSURE
|
None.
|
ITEM 9A.
|
CONTROLS
AND PROCEDURES
|
Evaluation of Disclosure Controls and
Procedures. Our management evaluated, with the participation of our Chief
Executive Officer and our Chief Financial Officer, the effectiveness of our
disclosure controls and procedures (as defined in Rule 13a-15(e) of the
Exchange Act of 1934, as amended) as of the end of the period covered by this
Annual Report on Form 10-K. Based on this evaluation, our Chief Executive
Officer and our Chief Financial Officer have concluded that our disclosure
controls and procedures are effective to ensure that information we are required
to disclose in reports that we file or submit under the Securities Exchange Act
of 1934 is recorded, processed, summarized and reported within the time periods
specified in Securities and Exchange Commission rules and forms, and that such
information is accumulated and communicated to management as appropriate to
allow for timely decisions regarding required disclosure.
Management’s Report on Internal
Control Over Financial Reporting. Our management is responsible for
establishing and maintaining adequate internal control over financial reporting
as defined in Rule 13a-15(f) and 15d-15(f) under the Securities Exchange
Act of 1934, as amended. Our internal control over financial reporting is
designed to provide reasonable assurance regarding the reliability of financial
reporting and the preparation of financial statements for external purposes in
accordance with generally accepted accounting principles. Because of its
inherent limitations, internal control over financial reporting may not prevent
or detect misstatements. Also, projections of any evaluation of effectiveness to
future periods are subject to the risk that controls may become inadequate
because of changes in conditions, or that the degree of compliance with policies
or procedures my deteriorate.
To
evaluate the effectiveness of internal control over financial reporting,
management used the criteria set forth in Internal Control-Integrated
Framework, issue by the Committee of Sponsoring Organizations of the
Treadway Commission. Based on its assessment using those criteria, management
has concluded that we maintained effective internal control over financial
reporting as of December 31, 2009.
The
effectiveness of the Company’s internal control over financial reporting as of
December 31, 2009 has been audited by Deloitte & Touche LLP, an
independent registered public accounting firm, as stated in their report which
is included herein.
Changes in Internal Control Over
Financial Reporting. There was no change in our internal control over
financial reporting (as defined in Rule 13a-15(f) and 15d-15(f) of the
Exchange Act of 1934, as amended) that occurred during the fourth quarter ended
December 31, 2009 that has materially affected, or is reasonably likely to
materially affect, our internal control over financial reporting.
ITEM 9B. OTHER
INFORMATION
None.
-47-
REPORT
OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the
Board of Directors and Shareholders of
SenoRx,
Inc.
Irvine,
California
We have
audited the internal control over financial reporting of SenoRx, Inc.
(the “Company”) as of December 31, 2009, based on criteria established
in Internal Control—Integrated
Framework issued by the Committee of Sponsoring Organizations of the
Treadway Commission. The Company’s management is responsible for maintaining
effective internal control over financial reporting and for its assessment of
the effectiveness of internal control over financial reporting, included in the
accompanying Management’s
Report on Internal Control over Financial Reporting. Our responsibility
is to express an opinion on the Company’s internal control over financial
reporting based on our audit.
We
conducted our audit in accordance with the standards of the Public Company
Accounting Oversight Board (United States). Those standards require that we plan
and perform the audit to obtain reasonable assurance about whether effective
internal control over financial reporting was maintained in all material
respects. Our audit included obtaining an understanding of internal control over
financial reporting, assessing the risk that a material weakness exists, testing
and evaluating the design and operating effectiveness of internal control based
on the assessed risk, and performing such other procedures as we considered
necessary in the circumstances. We believe that our audit provides a reasonable
basis for our opinion.
A
company’s internal control over financial reporting is a process designed by, or
under the supervision of, the company’s principal executive and principal
financial officers, or persons performing similar functions, and effected by the
company’s board of directors, management, and other personnel, to provide
reasonable assurance regarding the reliability of financial reporting and the
preparation of financial statements for external purposes in accordance with
generally accepted accounting principles. A company’s internal control over
financial reporting includes those policies and procedures that (1) pertain
to the maintenance of records that, in reasonable detail, accurately and fairly
reflect the transactions and dispositions of the assets of the company,
(2) provide reasonable assurance that transactions are recorded as
necessary to permit preparation of financial statements in accordance with
generally accepted accounting principles, and that receipts and expenditures of
the company are being made only in accordance with authorizations of management
and directors of the company, and (3) provide reasonable assurance
regarding prevention or timely detection of unauthorized acquisition, use, or
disposition of the company’s assets that could have a material effect on the
financial statements.
Because
of the inherent limitations of internal control over financial reporting,
including the possibility of collusion or improper management override of
controls, material misstatements due to error or fraud may not be prevented or
detected on a timely basis. Also, projections of any evaluation of the
effectiveness of the internal control over financial reporting to future periods
are subject to the risk that the controls may become inadequate because of
changes in conditions, or that the degree of compliance with the policies or
procedures may deteriorate.
In our
opinion, the Company maintained, in all material respects, effective internal
control over financial reporting as of December 31, 2009, based on the
criteria established in Internal Control—Integrated
Framework issued by the Committee of Sponsoring Organizations of the
Treadway Commission.
We have
also audited, in accordance with the standards of the Public Company Accounting
Oversight Board (United States), the financial statements as of and for the year
ended December 31, 2009, of the Company and our report dated March 10,
2010, expressed an unqualified opinion on those financial
statements.
/s/
DELOITTE & TOUCHE LLP
Costa
Mesa, California
March 10,
2010
-48-
PART
III
|
ITEM 10.
|
DIRECTORS,
EXECUTIVE OFFICERS AND CORPORATE
GOVERNANCE
|
The
information required by this Item is incorporated by reference to the definitive
proxy statement for our 2009 Annual Meeting of Stockholders to be filed with the
Securities and Exchange Commission within 120 days after the end of our 2009
fiscal year (the “2010 Proxy Statement”).
|
ITEM 11.
|
EXECUTIVE
COMPENSATION
|
The
information required by this Item is incorporated by reference to the 2010 Proxy
Statement.
|
ITEM 12.
|
SECURITY
OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED
STOCKHOLDER MATTERS
|
The
information required by this Item is incorporated by reference to the 2010 Proxy
Statement.
|
ITEM 13.
|
CERTAIN
RELATIONSHIPS AND RELATED TRANSACTIONS AND DIRECTOR
INDEPENDENCE
|
The
information required by this Item is incorporated by reference to the 2010 Proxy
Statement.
|
ITEM 14.
|
PRINCIPAL
ACCOUNTANT FEES AND SERVICES
|
The
information required by this Item is incorporated by reference to the 2010 Proxy
Statement.
PART
IV
|
ITEM
15.
|
EXHIBITS
AND FINANCIAL STATEMENT SCHEDULES
|
|
(1)
|
The
financial statements listed in the “Index to Financial Statements” at
page 50 are filed as a part of this
report.
|
|
(2)
|
All
schedules are omitted because they are not applicable. All the required
information is shown in the financial statements or notes
thereto.
|
|
(3)
|
Exhibits
included or incorporated herein. See Exhibit
Index.
|
-49-
SENORX,
INC.
INDEX
TO FINANCIAL STATEMENTS
|
Page
|
|
|
Financial
Statements:
|
|
|
Report
of Independent Registered Public Accounting Firm
|
51
|
|
Balance
Sheets as of December 31, 2009 and 2008
|
52
|
|
Statements
of Operations for the Years Ended December 31, 2009, 2008 and
2007
|
53
|
|
Statements
of Stockholders’ Equity (Deficit) for the Years Ended December 31,
2009, 2008 and 2007
|
54
|
|
Statements
of Cash Flows for the Years Ended December 31, 2009, 2008 and
2007
|
55
|
|
Notes
to Financial Statements
|
57
|
-50-
REPORT
OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the
Board of Directors and Stockholders of
SenoRx,
Inc.
Irvine,
California
We have
audited the accompanying balance sheets of SenoRx, Inc. (the “Company”) as of
December 31, 2009 and 2008, and the related statements of operations,
stockholders’ equity (deficit), and cash flows for each of the three years in
the period ended December 31, 2009. These financial statements are the
responsibility of the Company’s management. Our responsibility is to
express an opinion on the financial statements based on our audits.
We
conducted our audits in accordance with the standards of the Public Company
Accounting Oversight Board (United States). Those standards require that we
plan and perform the audit to obtain reasonable assurance about whether the
financial statements are free of material misstatement. An audit includes
examining, on a test basis, evidence supporting the amounts and disclosures in
the financial statements. An audit also includes assessing the
accounting principles used and significant estimates made by management, as well
as evaluating the overall financial statement presentation. We believe that
our audits provide a reasonable basis for our opinion.
In our
opinion, such financial statements present fairly, in all material respects, the
financial position of SenoRx, Inc. as of December 31, 2009 and 2008, and
the results of its operations and its cash flows for each of the three years in
the period ended December 31, 2009, in conformity with accounting
principles generally accepted in the United States of America.
We have
also audited, in accordance with the standards of the Public Company Accounting
Oversight Board (United States), the effectiveness of the Company’s internal
control over financial reporting as of December 31, 2009, based on the
criteria established in Internal Control—Integrated
Framework issued by the Committee of Sponsoring Organizations of the
Treadway Commission and our report dated March 10, 2010, expressed an
unqualified opinion on the effectiveness of the Company’s internal control over
financial reporting.
|
/S/
DELOITTE & TOUCHE LLP
|
|
Costa
Mesa, California
March
10, 2010
|
-51-
SENORX,
INC.
BALANCE
SHEETS
|
December 31,
|
||||||||
|
2009
|
2008
|
|||||||
|
ASSETS
|
||||||||
|
Current
assets:
|
||||||||
|
Cash
and cash equivalents
|
$ | 18,297,413 | $ | 15,323,143 | ||||
|
Accounts
receivable, net of allowance for doubtful accounts of $241,443, and
$225,793, respectively
|
9,761,488 | 8,179,099 | ||||||
|
Inventory
|
6,315,988 | 9,433,184 | ||||||
|
Prepaid
expenses and deposits
|
513,883 | 386,594 | ||||||
|
Total
current assets
|
34,888,772 | 33,322,020 | ||||||
|
Property
and equipment, net
|
1,100,691 | 1,554,201 | ||||||
|
Other
assets, net of accumulated depreciation of $327,548, and $259,469,
respectively
|
1,247,049 | 540,344 | ||||||
|
Total
assets
|
$ | 37,236,512 | $ | 35,416,565 | ||||
|
LIABILITIES
AND STOCKHOLDERS’ EQUITY
|
||||||||
|
Current
liabilities:
|
||||||||
|
Accounts
payable
|
$ | 3,229,858 | $ | 2,039,280 | ||||
|
Accrued
expenses, including accrued employee compensation of $1,872,096 and
$1,598,338, respectively
|
3,585,090 | 2,498,911 | ||||||
|
Deferred
revenue
|
566,839 | 385,600 | ||||||
|
Current
portion of long-term debt
|
501,180 | 390,246 | ||||||
|
Total
current liabilities
|
7,882,967 | 5,314,037 | ||||||
|
Long-term
debt—less current portion
|
1,125,000 | 1,632,410 | ||||||
|
Deferred
revenue—less current portion
|
272,027 | 171,465 | ||||||
|
Total long-term liabilities
|
1,397,027 | 1,803,875 | ||||||
|
Commitments
and Contingencies (Note 10)
|
||||||||
|
Stockholders’
Equity:
|
||||||||
|
Common
stock, $0.001 par value—100,000,000 shares authorized; 17,504,436
(2009) and 17,327,191 (2008) issued and
outstanding
|
17,504 | 17,327 | ||||||
|
Additional
paid-in capital
|
115,002,745 | 112,456,924 | ||||||
|
Accumulated
deficit
|
(87,063,731 | ) | (84,175,598 | ) | ||||
|
Total
stockholders’ equity
|
27,956,518 | 28,298,653 | ||||||
|
TOTAL
|
$ | 37,236,512 | $ | 35,416,565 | ||||
See
accompanying notes to financial statements.
-52-
SENORX,
INC.
STATEMENTS
OF OPERATIONS
|
Year Ended
December 31,
|
||||||||||||
|
2009
|
2008
|
2007
|
||||||||||
|
Net
revenues
|
$ | 55,577,299 | $ | 46,684,588 | $ | 35,035,836 | ||||||
|
Cost
of goods sold
|
16,365,970 | 16,503,327 | 15,123,897 | |||||||||
|
Gross
profit
|
39,211,329 | 30,181,261 | 19,911,939 | |||||||||
|
Operating
expenses:
|
||||||||||||
|
Selling
and marketing
|
23,880,060 | 23,117,137 | 19,022,994 | |||||||||
|
Research
and development
|
7,360,859 | 6,111,225 | 6,353,430 | |||||||||
|
General
and administrative
|
10,687,429 | 10,093,882 | 4,187,133 | |||||||||
|
Total
operating expenses
|
41,928,348 | 39,322,244 | 29,563,557 | |||||||||
|
Loss
from operations
|
(2,717,019 | ) | (9,140,983 | (9,651,618 | ) | |||||||
|
Interest
expense
|
198,096 | 85,196 | 1,646,670 | |||||||||
|
Loss
on debt extinguishment
|
— | — | 1,264,777 | |||||||||
|
Change
in fair value of convertible promissory notes and warrant
liability
|
— | — | (990,875 | ) | ||||||||
|
Interest
income
|
(26,982 | ) | (520,053 | ) | (1,639,194 | ) | ||||||
|
Loss
before provisions for income taxes
|
(2,888,133 | ) | (8,706,126 | (9,932,996 | ) | |||||||
|
Provisions
for income taxes
|
— | — | — | |||||||||
|
Net
loss
|
$ | (2,888,133 | ) | $ | (8,706,126 | ) | $ | (9,932,996 | ) | |||
|
Net
loss per share-basic and diluted
|
$ | (0.17 | ) | $ | (0.50 | ) | $ | (0.75 | ) | |||
|
Weighted
average shares outstanding-basic and diluted
|
17,381,106 | 17,249,569 | 13,308,790 | |||||||||
See
accompanying notes to financial statements.
-53-
SENORX,
INC.
STATEMENTS
OF STOCKHOLDERS’ EQUITY (DEFICIT)
|
Series A
Convertible
Preferred
Stock
|
Series B
Convertible
Preferred
Stock
|
Series C
Convertible
Preferred
Stock
|
Common
Stock
|
Additional
Paid-in
|
Deferred
|
Accumulated
|
Total
Stockholders’
Equity
|
|||||||||||||||||||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
Shares
|
Amount
|
Shares
|
Amount
|
Capital
|
Compensation
|
Deficit
|
(Deficit)
|
|||||||||||||||||||||||||||||||||||||
|
Balance
as of December 31, 2006
|
3,000,000 | 3,000,000 | 3,523,040 | 8,807,600 | 17,861,899 | 35,009,323 | 2,371,002 | 2,371 | 5,262,394 | (126,658 | ) | (65,536,476 | ) | (13,581,446 | ) | |||||||||||||||||||||||||||||||||
|
Proceeds
from initial public offering, net of offering costs of
$2,266,158
|
6,325,000 | 6,325 | 44,785,517 | 44,791,842 | ||||||||||||||||||||||||||||||||||||||||||||
|
Proceeds
from exercise of common stock options and warrants
|
112,116 | 112 | 758,986 | 759,098 | ||||||||||||||||||||||||||||||||||||||||||||
|
Proceeds
from purchase of shares under the employee stock purchase
plan
|
34,708 | 34 | 235,980 | 236,014 | ||||||||||||||||||||||||||||||||||||||||||||
|
Amortization
of deferred compensation
|
126,658 | 126,658 | ||||||||||||||||||||||||||||||||||||||||||||||
|
Stock-based
compensation
|
1,818,206 | 1,818,206 | ||||||||||||||||||||||||||||||||||||||||||||||
|
Employee
stock purchase plan compensation
|
145,966 | 145,966 | ||||||||||||||||||||||||||||||||||||||||||||||
|
Conversion
of promissory note
|
9,998,750 | 10,000,000 | ||||||||||||||||||||||||||||||||||||||||||||||
|
Conversion
of preferred stock
|
(3,000,000 | ) | (3,000,000 | ) | (3,523,040 | ) | (8,807,600 | ) | (17,861,899 | ) | (35,009,323 | ) | 7,109,570 | 7,110 | 46,809,813 | — | ||||||||||||||||||||||||||||||||
|
Net
loss
|
(9,932,996 | ) | (9,932,996 | ) | ||||||||||||||||||||||||||||||||||||||||||||
|
Balance
as of December 31, 2007
|
— | — | — | — | — | — | 17,202,395 | 17,202 | 109,815,612 | — | (75,469,472 | ) | 34,363,342 | |||||||||||||||||||||||||||||||||||
|
Proceeds
from exercise of common stock options
|
35,969 | 36 | 49,964 | 50,000 | ||||||||||||||||||||||||||||||||||||||||||||
|
Proceeds
from purchase of shares under the employee stock purchase
plan
|
88,827 | 89 | 319,758 | 319,847 | ||||||||||||||||||||||||||||||||||||||||||||
|
Stock-based
compensation
|
2,135,938 | 2,135,938 | ||||||||||||||||||||||||||||||||||||||||||||||
|
Employee
stock purchase plan compensation
|
135,652 | 135,652 | ||||||||||||||||||||||||||||||||||||||||||||||
|
Net
loss
|
(8,706,126 | ) | (8,706,126 | ) | ||||||||||||||||||||||||||||||||||||||||||||
|
Balance
as of December 31, 2008
|
— | $ | — | — | $ | — | — | $ | — | 17,327,191 | $ | 17,327 | $ | 112,456,924 | $ | — | $ | (84,175,598 | ) | $ | 28,298,653 | |||||||||||||||||||||||||||
|
Proceeds
from exercise of common stock options
|
64,170 | 64 | 44,876 | 44,940 | ||||||||||||||||||||||||||||||||||||||||||||
|
Proceeds
from purchase of shares under the employee stock purchase
plan
|
113,075 | 113 | 259,822 | 259,935 | ||||||||||||||||||||||||||||||||||||||||||||
|
Stock-based
compensation
|
2,189,295 | 2,189,295 | ||||||||||||||||||||||||||||||||||||||||||||||
|
Net
share settlement of equity awards
|
(57,224 | ) | (57,224 | ) | ||||||||||||||||||||||||||||||||||||||||||||
|
Employee
stock purchase plan compensation
|
109,052 | 109,052 | ||||||||||||||||||||||||||||||||||||||||||||||
|
Net loss
|
(2,888,133 | ) | (2,888,133 | ) | ||||||||||||||||||||||||||||||||||||||||||||
|
Balance
as of December 31, 2009
|
— | $ | — | — | $ | — | — | $ | — | 17,504,436 | $ | 17,504 | $ | 115,002,745 | $ | — | $ | (87,063,731 | ) | $ | 27,956,518 | |||||||||||||||||||||||||||
See
accompanying notes to financial statements.
-54-
SENORX,
INC.
STATEMENT
OF CASH FLOWS
|
Year Ended
December 31,
|
||||||||||||
|
2009
|
2008
|
2007
|
||||||||||
|
Cash
Flows From Operating Activities:
|
||||||||||||
|
Net
loss
|
$ | (2,888,133 | ) | $ | (8,706,126 | ) | $ | (9,932,996 | ) | |||
|
Adjustments
to reconcile net loss to net cash used in operating
activities:
|
||||||||||||
|
Depreciation
and amortization
|
1,539,404 | 1,564,258 | 1,400,195 | |||||||||
|
Stock-based
compensation
|
2,298,347 | 2,271,590 | 2,090,831 | |||||||||
|
Loss
on fixed asset abandonment
|
12,273 | 32,692 | 37,430 | |||||||||
|
Provision
for doubtful accounts
|
51,254 | 124,387 | — | |||||||||
|
Provision
for inventory obsolescence
|
925,272 | 193,992 | 45,205 | |||||||||
|
Amortization
of debt discounts
|
— | — | 361,695 | |||||||||
|
Loss
on debt extinguishment
|
— | — | 1,264,777 | |||||||||
|
Change
in fair value of convertible promissory notes and warrant
liability
|
— | — | (990,875 | ) | ||||||||
|
Changes
in operating assets and liabilities:
|
||||||||||||
|
Accounts
receivable
|
(1,633,643 | ) | (2,882,302 | ) | (1,179,877 | ) | ||||||
|
Inventory
|
943,625 | (3,937,211 | ) | (2,731,525 | ) | |||||||
|
Prepaid
expenses and deposits
|
(127,289 | ) | 157,682 | (323,617 | ) | |||||||
|
Other
assets
|
109,780 | (225,213 | ) | 384,196 | ||||||||
|
Accounts
payable
|
1,190,578 | (549,899 | ) | (1,545,224 | ) | |||||||
|
Accrued
expenses
|
1,086,179 | (10,542 | ) | 797,377 | ||||||||
|
Deferred
revenue
|
281,801 | 68,027 | 57,838 | |||||||||
|
Net
cash provided by (used in) operating activities
|
3,789,448 | (11,898,665 | ) | (10,264,570 | ) | |||||||
|
Cash
Flows From Investing Activities:
|
||||||||||||
|
Purchases
of short-term investments
|
— | — | (24,071,842 | ) | ||||||||
|
Maturities
of short-term investments
|
— | 10,764,490 | 13,307,352 | |||||||||
|
Acquisition
of property and equipment
|
(616,353 | ) | (970,278 | ) | (550,205 | ) | ||||||
|
Net
cash (used in) provided by investing activities
|
(616,353 | ) | 9,794,212 | (11,314,695 | ) | |||||||
|
Cash
Flows From Financing Activities:
|
||||||||||||
|
Proceeds
from issuance of common stock from stock option and warrant
exercises
|
44,940 | 50,000 | 60,724 | |||||||||
|
Proceeds
from initial public offering
|
— | — | 47,058,000 | |||||||||
|
Proceeds
from issuance of common stock under the ESPP plan
|
259,935 | 319,847 | 236,014 | |||||||||
|
Payment
of payroll taxes for net share settlement of equity awards
|
(57,224 | ) | — | — | ||||||||
|
Payment
of debt issuance costs
|
(50,000 | ) | (30,000 | ) | (49,497 | ) | ||||||
|
Payment
of initial public offering costs
|
— | — | (941,027 | ) | ||||||||
|
Proceeds
from borrowings
|
— | 2,000,000 | 2,750,000 | |||||||||
|
Repayment
of borrowings
|
(385,058 | ) | (2,088,158 | ) | (17,755,523 | ) | ||||||
|
Repayment
of capital leases
|
(11,418 | ) | (9,352 | ) | (7,153 | ) | ||||||
|
Net
cash (used in) provided by financing activities
|
(198,825 | ) | 242,337 | 31,351,538 | ||||||||
|
Net
increase (decrease) in cash and cash equivalents
|
2,974,270 | (1,862,116 | ) | 9,772,273 | ||||||||
|
Cash
and cash equivalents—beginning of year
|
15,323,143 | 17,185,259 | 7,412,986 | |||||||||
|
Cash
and cash equivalents—end of year
|
$ | 18,297,413 | $ | 15,323,143 | $ | 17,185,259 | ||||||
See
accompanying notes to financial statements.
-55-
SENORX,
INC.
STATEMENT
OF CASH FLOWS (Continued)
|
Year Ended
December 31,
|
||||||||||||
|
2009
|
2008
|
2007
|
||||||||||
|
Supplemental
Disclosure of Cash Flow Information:
|
||||||||||||
|
Cash
paid for income taxes
|
$ | — | $ | — | $ | — | ||||||
|
Cash
paid for interest
|
$ | 136,193 | $ | 67,060 | $ | 1,241,449 | ||||||
|
Deferred
offering costs offset against offering proceeds of initial public
offering
|
$ | — | $ | — | $ | 1,325,131 | ||||||
|
Promissory
note converted to common stock
|
$ | — | $ | — | $ | 10,000,000 | ||||||
|
Preferred
stock converted to common stock
|
$ | — | $ | — | $ | 46,809,813 | ||||||
|
Warrant
liability transferred to equity and subsequently exercised
|
$ | — | $ | — | $ | 698,374 | ||||||
|
Net
other assets transferred to fixed assets
|
$ | — | $ | 90,949 | $ | — | ||||||
|
Net
fixed assets transferred to other assets
|
$ | — | $ | — | $ | 26,512 | ||||||
|
Property
and equipment acquired included in accounts payable
|
$ | — | $ | 8,930 | $ | 2,996 | ||||||
|
Capital
leases
|
$ | — | $ | — | $ | 14,477 | ||||||
|
Inventory
transferred to fixed assets and other assets
|
$ | 1,248,299 | $ | 960,990 | $ | 1,024,060 | ||||||
See
accompanying notes to financial statements.
-56-
SENORX,
INC.
NOTES
TO FINANCIAL STATEMENTS
|
1.
|
GENERAL
AND SIGNIFICANT ACCOUNTING POLICIES
|
Business—SenoRx,
Inc. (the “Company”) is a medical device company focused on developing,
manufacturing and selling minimally-invasive medical devices for the diagnosis
and treatment of breast cancer. The Company was incorporated on January 21,
1998 as BiopSolation Medical, Inc. and subsequently changed its name to SenoRx,
Inc.
Basis of
Presentation—The accompanying financial statements have been prepared in
accordance with accounting principles generally accepted in the United States of
America.
Cash and Cash
Equivalents—All highly liquid investments purchased with a maturity, at
date of purchase, of three months or less are considered to be cash equivalents.
Cash equivalents, in the accompanying financial statements, include money market
funds and commercial paper.
Short-term
Investments—The Company’s short-term investments consist of commercial
paper issued by major U.S. financial institutions with a credit rating of A1/P1
and maturities of three to six months. The Company accounts for its investments
in marketable securities under ASC 320, (formerly FASB No. 115 “Accounting
for Certain Investments in Debt and Equity Securities”). Investments are
recorded at amortized cost, which approximates fair value, and are classified as
held to maturity based on the Company’s intent and ability to hold such
investments until maturity.
Concentration of
Credit Risks—The Company’s cash and cash equivalents at December 31,
2009 are invested in money market accounts that can be withdrawn without penalty
at any time. The Company maintains cash balances in excess of federally insured
limits in a reputable financial institution. As such, there is nominal credit
risk with respect to cash and cash equivalents.
The
Company has one product and several component parts for other products which are
obtained from single-source suppliers. The Company has managed the risk
associated with single-source suppliers by closely monitoring existing supply
levels as compared to customer purchase orders. The Company is exposed to loss
of revenue from the sale of these products if the supplier cannot fulfill
demand.
The
Company’s customer base is diverse and consists of hospitals and physicians. No
single customer represents greater than 10% of net revenues during the years
ended December 31, 2009, 2008 and 2007. The Company is exposed to risks
associated with extending credit to its customers related to the sale of
products. Management believes that credit risks on trade accounts receivable are
mitigated by the diversity of its customers. The Company performs credit
evaluations on its customers’ financial condition, and to date, credit losses
have been within management’s expectations.
Following
is a summary of activity in the allowance for doubtful accounts:
|
Year Ended
December 31,
|
||||||||||||
|
2009
|
2008
|
2007
|
||||||||||
|
Allowance
for doubtful accounts, beginning
|
$ | 225,793 | $ | 107,728 | $ | 120,000 | ||||||
|
Provision
for doubtful accounts
|
51,254 | 124,387 | — | |||||||||
|
Write-offs,
net of recoveries
|
(35,604 | ) | (6,322 | ) | (12,272 | ) | ||||||
|
Allowance
for doubtful accounts, ending
|
$ | 241,443 | $ | 225,793 | $ | 107,728 | ||||||
Inventory—Inventory
consists principally of raw materials, work-in-process and finished goods, and
is carried at the lower of standard cost or market. Standard costs are
determined using the first-in, first-out method and are updated at regular
intervals such that standard costs approximate actual costs. Provisions for slow
moving or obsolete inventory are charged to cost of sales and are permanent
reductions to the carrying value of inventory.
Property and
Equipment—Property and equipment are recorded at cost less accumulated
depreciation. Maintenance and repairs are expensed as incurred. Upon sale or
disposition of assets, any gain or loss is included in the statements of
operations.
-57-
SENORX,
INC.
NOTES
TO FINANCIAL STATEMENTS (Continued)
The cost
of property, plant and equipment is depreciated using the straight-line method
over the following estimated useful lives of the respective assets. Leasehold
improvements are depreciated over the lesser of the estimated useful lives of
the respective assets or the related lease terms.
|
Computers
and software
|
3
years
|
|
Manufacturing
molds
|
3
years
|
|
Machinery
and equipment
|
3
years
|
|
Demonstration
and evaluation equipment
|
1
year
|
|
Office
furniture and equipment
|
3
years
|
|
Trade
show booth and equipment
|
1
to 3 years
|
Long-Lived
Assets—The Company’s long-lived assets include property and equipment. In
accordance with ASC 360, (formerly, Statement of Financial Accounting Standards
(“SFAS”) No. 144, “Accounting for the Impairment or Disposal of Long-Lived
Assets”), the Company estimates the future undiscounted cash flows derived from
an asset to assess whether or not a potential impairment exists when events or
circumstances indicate the carrying value of a long-lived asset may be impaired.
An impairment loss is recognized when the undiscounted future cash flows are
less than its carrying amount and is equal to the amount the carrying value
exceeds the fair value of the asset or asset group. The Company periodically
reviews the carrying value of long-lived assets to determine whether or not an
impairment to such value has occurred and, based on its most recent assessment
at December 31, 2009, has determined that there was no material impairment
at December 31, 2009.
Fair Value of
Financial Instruments—ASC 820-10-65-4 (formerly SFAS No. 107,
“Disclosures about Fair Value of Financial Instruments”), requires management to
disclose the estimated fair value of certain assets and liabilities defined by
ASC 820 as financial instruments. Financial instruments are generally defined as
cash, evidence of ownership interest in an entity, or a contractual obligation
that both conveys to one entity a right to receive cash or other financial
instruments from another entity and imposes on the other entity the obligation
to deliver cash or other financial instruments to the first entity. At
December 31, 2009, management believes that the carrying value of cash and
cash equivalents, receivables and payables approximate fair value because of the
short maturity of these financial instruments. At December 31, 2009,
management believes that the fair value of the Company’s debt approximated its
carrying value based on interest rates available to the Company at the
time
Net Loss Per
Share—Basic loss per share is based on the weighted-average number of
shares of common stock outstanding during the period. Diluted loss per share
also includes the effect of stock options, warrants and other common stock
equivalents outstanding during the period. In periods of a net loss position,
basic and diluted weighted average shares are the same.
The
following table sets forth the computation of denominator used in the
computation of net loss per share:
|
Year Ended
December 31,
|
||||||||||||
|
2009
|
2008
|
2007
|
||||||||||
|
Weighted-average
common stock outstanding
|
17,381,404 | 17,252,550 | 13,325,186 | |||||||||
|
Less:
Unvested common shares subject to repurchase
|
(298 | ) | (2,981 | ) | (16,396 | ) | ||||||
|
Total
weighted-average number of shares used in computing net loss per
share-basic and diluted
|
17,381,106 | 17,249,569 | 13,308,790 | |||||||||
Revenue
Recognition and Deferred Revenue—The Company recognizes revenues in
accordance with ASC 605, (formerly Staff Accounting Bulletin, or SAB,
No. 104, “Revenue Recognition”). ASC 605 requires that four basic criteria
must be met before revenue can be recognized: (1) persuasive evidence of an
arrangement exists; (2) title has transferred; (3) the fee is fixed or
determinable; and (4) collectability is reasonably assured. The Company’s
terms of sales specify that title transfers at the time of shipment by the
Company. The Company generally uses contracts and purchase orders to determine
the existence of an arrangement. The Company assesses whether the fee is fixed
or determinable based upon the terms of the agreement associated with the
transaction. To determine whether collection is reasonably assured, the Company
assesses a number of factors, including past transaction history with the
customer and creditworthiness of the customer. The Company generally does not
provide any rights of return by the customer other than returns for product
warranty related issues. In addition to these product warranty related returns,
the Company occasionally accepts other returns at its discretion. Such returns
have historically been insignificant, and reserves for these returns are
established at the time of sale.
-58-
SENORX,
INC.
NOTES
TO FINANCIAL STATEMENTS (Continued)
For those
sales that include multiple deliverables, the Company allocates revenue based on
the relative fair values of the individual components as determined in
accordance with ASC 605-25-30 (formerly known as EITF Issue No. 00-21,
“Revenue Arrangements with Multiple Deliverables”). When more than
one element, such as hardware and disposables, are contained in a single
arrangement, revenues are allocated between the elements based on each element’s
relative fair value, provided that each element meets the criteria for treatment
as a separate unit of accounting. An item is considered a separate unit of
accounting if it has value to the customer on a standalone basis and there is
objective and reliable evidence of the fair value of the undelivered items. Fair
value is generally determined based upon the price charged when the element is
sold separately. In the absence of fair value for a delivered element, revenue
is allocated first to the fair value of the undelivered elements and the
residual revenue is allocated to the delivered elements. In the absence of fair
value for an undelivered element, the arrangement is accounted for as a single
unit of accounting, resulting in a deferral of revenue recognition for the
delivered elements until all undelivered elements have been
fulfilled.
The
Company also places certain equipment with customers in return for the customer
purchasing a minimum number of disposable procedure devices during a specified
contract period. Title to the equipment passes to the customer at the end of the
contract period provided the minimum purchase requirement is met. The cost of
the equipment, which is included in other assets in the accompanying balance
sheets is amortized to cost of goods sold based on the monthly disposable unit
shipments compared to the total purchase commitment of disposables. In the event
the customer does not fulfill the minimum purchase requirements, collection
efforts may be undertaken and the Company will attempt to recover the equipment.
If the collection efforts or recovery of the equipment is not successful, the
unamortized equipment cost would be expensed to cost of goods sold.
Income
Taxes—Income taxes are accounted for in accordance with ASC 740,
(formerly SFAS No. 109, “Accounting for Income Taxes”). This statement
requires the recognition of deferred tax assets and liabilities to reflect the
future tax consequences of events that have been recognized in the Company’s
financial statements or tax returns. Measurement of the deferred items is based
on enacted tax laws. In the event the future consequences of differences between
financial reporting bases and tax bases of the Company’s assets and liabilities
result in a deferred tax asset, ASC 740 requires an evaluation of the
probability of being able to realize the future benefits indicated by such
assets. A valuation allowance related to a deferred tax asset is recorded when
it is more likely than not that some portion or all of the deferred tax asset
will not be realized.
Research and
Development—Research and development costs are charged to operating
expense in the year incurred. Research and development expense consists
principally of expenditures for equipment, parts, tooling costs and outside
third-party consultants, which are used in testing and the development of the
Company’s devices under development, and compensation to specific Company
personnel. The Company also expenses the costs of internally developed patents
since recoverability is uncertain. The cost of equipment used in research and
development activities which has alternative uses is capitalized as equipment.
Such equipment is depreciated over estimated useful lives of three
years.
Software
Development Costs—Certain of the Company’s products incorporate software
which is incidental to the product as a whole. Software development costs
incurred prior to the establishment of technological feasibility are expensed as
research and development costs. The Company defines the establishment of
technological feasibility as the completion of a final working model that has
been incorporated into a product that has been cleared by the U.S. Food and Drug
Administration, at which time the product can be sold to third parties. As a
result, all software development costs incurred by the Company to date have been
expensed as incurred given the short duration between technological feasibility
and sales to third parties.
Shipping and
Handling—In accordance with ASC 605-45-45 (formerly known as Emerging
Issues Task Force (“EITF”) No. 00-10, “Accounting for Shipping and Handling Fees
and Costs”), the Company includes shipping and handling fees billed to customers
in net revenues. Amounts incurred by the Company for freight are included in
cost of goods sold.
Advertising—Advertising
costs are expensed as incurred and are included in selling and marketing
expense. Advertising costs have not been material for any period
presented.
-59-
SENORX,
INC.
NOTES
TO FINANCIAL STATEMENTS (Continued)
Stock-Based
Compensation—Effective January 1, 2006, we adopted ASC 718,
(formerly SFAS 123R), which requires that all stock-based compensation to
employees, including grants of employee stock options, be expensed in the
financial statements based on their respective grant date fair values. The
Company does not have a history of market prices of its common stock as it did
not become a public company until April 2007. Therefore, volatility was
estimated in accordance with ASC 718, (formerly SAB No. 107) using
historical volatilities of other publicly traded companies within the same
industry. The expected life of the awards is based on the simplified method as
defined in ASC 718. The risk-free rate assumption is based on observed interest
rates appropriate for the terms of the awards. The dividend yield assumption is
based on history and the expectation of not paying any dividends. Forfeitures
are estimated at the time of grant and revised, if necessary, in subsequent
periods if actual forfeitures differ from those estimates. Stock-based
compensation expense is recognized in the financial statements based on awards
that are ultimately expected to vest. A summary of significant assumptions used
in determining the fair value of the options is as follows:
|
Year Ended
December 31,
|
|||||
|
2009
|
2008
|
2007
|
|||
|
Expected
life (years)
|
4.5 - 4.75
|
4.5 - 4.75
|
4.75 - 6.25
|
||
|
Risk-free
interest rate
|
1.97% - 2.30%
|
1.62% - 3.26%
|
3.49% - 4.69%
|
||
|
Volatility
|
46% - 47%
|
45% - 48%
|
42% - 48%
|
||
|
Dividend
yield
|
0%
|
0%
|
0%
|
||
|
Forfeiture
rate
|
2.5%
|
2.5%
|
2.5%
|
||
The
Company had a choice of two attribution methods for allocating compensation
costs under ASC 718: the “straight-line” method, which allocates expense on a
straight-line basis over the requisite service period of the last separately
vesting portion of an award, or the “graded vesting attribution method”, which
allocates expense on a straight-line basis over the requisite service period for
each separately vesting portion of the award as if the award was, in-substance,
multiple awards. The Company chose the latter method (i.e. graded vesting). The
Company amortizes the fair value of each option over each option’s vesting
period (requisite service period).
The
Company has not recognized any income tax benefit for the stock-based
compensation arrangements due to the fact that the Company does not believe it
is more likely than not it will recognize any deferred tax assets from such
compensation cost recognized in the current period.
Stock-based
compensation relating to options included in the Company’s statement of
operations under ASC 718 for the years ended December 31, 2009, 2008 and
2007 was:
|
Year Ended
December 31,
|
||||||||||||
|
2009
|
2008
|
2007
|
||||||||||
|
Cost
of goods sold
|
$ | 135,630 | $ | 87,257 | $ | 95,522 | ||||||
|
Research
and development expense
|
704,602 | 766,954 | 436,211 | |||||||||
|
Selling
and marketing expense
|
524,596 | 429,618 | 428,443 | |||||||||
|
General
and administrative expense
|
824,467 | 852,109 | 858,030 | |||||||||
| $ | 2,189,295 | $ | 2,135,938 | $ | 1,818,206 | |||||||
Comprehensive
Loss—For the years ended December 31, 2009, 2008 and 2007, there was
no difference between the Company’s net loss and comprehensive
loss.
Use of
Estimates—The preparation of financial statements in conformity with
accounting principles generally accepted in the United States of America
necessarily requires management to make estimates and assumptions that affect
the reported amounts of assets and liabilities and disclosure of contingent
assets and liabilities at the date of the financial statements and the reported
amounts of expenses and revenues during reporting periods. Actual results could
differ from these estimates.
Segment
Reporting—ASC 280 (formerly SFAS No. 131, “Disclosures about
Segments of an Enterprise and Related Information”), established standards for
reporting information about operating segments in financial statements.
Operating segments are defined as components of an enterprise about which
separate financial information is available that is evaluated regularly by the
chief decision maker in deciding how to allocate resources and in assessing
performance. The Company’s chief decision maker is the chief executive officer.
The Company’s chief decision maker reviews the results of operations based on
one industry segment: the production and sale of breast care
products.
-60-
SENORX,
INC.
NOTES
TO FINANCIAL STATEMENTS (Continued)
The
Company reclassified $395,150 of warranty contracts at December 31, 2008 from
accrued expenses to deferred revenue to conform to the current presentation in
the December 31, 2009 balance sheet.
Recent Accounting
Pronouncements— In January 2010, the FASB issued ASU No. 2010-06,
Improving Disclosures about
Fair Value Measurements, which amends the guidance in ASC 820, Fair Value Measurements and
Disclosures. ASU No. 2010-06 requires companies to disclose
transfers between Level 1 and Level 2 within the fair value hierarchy and
describe the reasons for the transfers. In addition, in the reconciliation
of assets and liabilities in Level 3 of the fair value hierarchy, companies are
required to present sales, purchases, issuances and settlements on a gross
rather than net basis. ASU No. 2010-06 also clarifies that companies
should provide fair value measurement disclosures for each class of assets and
liabilities and that companies should disclose the inputs and valuation
techniques used to measure assets and liabilities that fall either in Level 2 or
Level 3. ASU No. 2010-06 will be effective for interim and annual periods
beginning after December 15, 2009, except for the new Level 3 reconciliation
disclosures, which will be effective for interim and annual periods beginning
after December 15, 2010. This update is not expected to have a material
impact on the Company’s financial position and results of
operations.
In October 2009, the FASB issued
Accounting Standards Update (ASU) No. 2009-13, Multiple-Deliverable Revenue
Arrangements a consensus of the FASB Emerging Issues Task Force, which amends
the guidance in ASC 605, Revenue Recognition. ASU 2009-13 eliminates the
residual method of accounting for revenue on undelivered products and instead,
requires companies to allocate revenue to each of the deliverable products based
on their relative selling price. In addition, this ASU expands the
disclosure requirements surrounding multiple-deliverable arrangements.
This update is effective beginning January 1, 2011 and can be applied
prospectively or retrospectively. Adoption is not expected to materially impact
the company’s financial position, results of operations or cash flows directly
when it becomes effective, as the company will not elect retrospective adoption.
However, this update may impact how the company reflects multiple-element
arrangements entered into subsequent to January 1, 2011, in the financial
statements.
|
2.
|
INITIAL
PUBLIC OFFERING
|
The
Company registered the initial public offering of its common stock, par value
$.001 per share, in a Registration Statement on Form S-1 (Registration
No. 333-134466), which was declared effective on March 28, 2007. The
Company completed its initial public offering (“IPO”) and sold 5,500,000 shares
at $8.00 per share on April 3, 2007. Additionally, on April 20, 2007,
the underwriters the IPO exercised their overallotment option to purchase an
additional 825,000 shares at $8.00. Total expenses from the offering were
approximately $5.8 million, which included underwriting discounts and
commissions of $3.5 million, and approximately $2.2 million in other
offering-related expense. Net offering proceeds, including the sale of shares
pursuant to the subsequent underwriters’ overallotment and after deducting total
expenses was $44.8 million. Upon the closing of the IPO, all of the outstanding
shares of the Company’s convertible preferred stock converted to 7,109,570
shares of the Company’s common stock. In addition, the May 2006 Notes were
converted into 1,249,999 shares of common stock.
-61-
SENORX,
INC.
NOTES
TO FINANCIAL STATEMENTS (Continued)
|
3.
|
PROPERTY
AND EQUIPMENT
|
Property
and equipment consist of the following:
|
Year Ended
December 31,
|
||||||||
|
2009
|
2008
|
|||||||
|
Computers
and software
|
$ | 663,609 | $ | 627,330 | ||||
|
Manufacturing
molds
|
1,950,762 | 1,641,464 | ||||||
|
Machinery
and equipment
|
708,943 | 616,415 | ||||||
|
Demonstration
and evaluation equipment
|
1,733,417 | 1,785,243 | ||||||
|
Office
furniture and equipment
|
143,857 | 130,173 | ||||||
|
Trade
show booth and equipment
|
195,709 | 157,196 | ||||||
|
Leasehold
improvements
|
159,199 | 183,482 | ||||||
| 5,555,497 | 5,141,303 | |||||||
|
Less
accumulated depreciation
|
(4,454,805 | ) | (3,587,102 | ) | ||||
| $ | 1,100,691 | $ | 1,554,201 | |||||
|
4.
|
INVENTORY
|
Inventories
consist of the following:
|
Year Ended
December 31,
|
||||||||
|
2009
|
2008
|
|||||||
|
Raw
materials
|
$ | 2,927,151 | $ | 4,261,809 | ||||
|
Work-in-process
|
743,578 | 382,188 | ||||||
|
Finished
goods
|
2,645,259 | 4,789,187 | ||||||
| $ | 6,315,988 | $ | 9,433,184 | |||||
Long-term
inventory of $429,606 is included in other assets in the accompanying balance
sheet as of December 31, 2009.
|
5.
|
INCOME
TAXES
|
The
components of the federal and state income tax expense are as
follows:
|
Year Ended
December 31,
|
||||||||||||
|
2009
|
2008
|
2007
|
||||||||||
|
Current:
|
||||||||||||
|
Federal
|
$ | — | $ | — | $ | — | ||||||
|
State
|
— | — | — | |||||||||
| — | — | — | ||||||||||
|
Deferred:
|
||||||||||||
|
Federal
|
(554,501 | ) | (1,999,514 | ) | (2,748,189 | ) | ||||||
|
State
|
(459,825 | ) | (736,813 | ) | (949,164 | ) | ||||||
| (1,014,326 | ) | (2,736,326 | ) | (3,697,353 | ) | |||||||
|
Valuation
allowance
|
1,014,326 | 2,736,326 | 3,697,353 | |||||||||
|
Total
|
$ | — | $ | — | $ | — | ||||||
-62-
SENORX,
INC.
NOTES
TO FINANCIAL STATEMENTS (Continued)
Taxes on
income vary from the statutory federal income tax rate applied to earnings
before taxes on income as follows:
|
Year Ended
December 31,
|
||||||||||||
|
2009
|
2008
|
2007
|
||||||||||
|
Statutory
federal income tax rate applied to earnings before income
taxes
|
$ | (991,232 | ) | $ | (3,047,144 | ) | $ | (3,476,549 | ) | |||
|
State
income taxes—net of federal benefit
|
(252,500 | ) | (486,296 | ) | (626,448 | ) | ||||||
|
Meals
and entertainment
|
63,160 | 96,568 | 82,274 | |||||||||
|
Research
and development credit
|
(343,383 | ) | (158,645 | ) | (147,853 | ) | ||||||
|
Equity
based compensation
|
544,524 | 772,116 | 708,738 | |||||||||
|
Change
in fair value of convertible promissory note
|
— | — | (336,898 | ) | ||||||||
|
Other
|
(34,895 | ) | 87,075 | 99,383 | ||||||||
|
Change
in valuation allowance
|
1,014,326 | 2,736,326 | 3,697,353 | |||||||||
| $ | — | $ | — | $ | — | |||||||
Deferred
income tax assets and liabilities arising from differences between accounting
for financial statement purposes and tax purposes, less valuation allowances,
are as follows:
|
December 31,
|
||||||||
|
2009
|
2008
|
|||||||
|
Deferred
tax assets:
|
||||||||
|
Net
operating loss
|
$ | 25,459,504 | $ | 26,468,355 | ||||
|
Property
and equipment
|
168,585 | 69,350 | ||||||
|
Capitalized
assets
|
2,032,234 | 1,804,320 | ||||||
|
Tax
credits
|
4,047,016 | 3,405,543 | ||||||
|
Accrued
expenses
|
2,181,972 | 1,056,065 | ||||||
|
Total
deferred tax assets
|
33,889,311 | 32,803,633 | ||||||
|
Deferred
tax liabilities—state taxes
|
(2,230,931 | ) | (2,157,047 | ) | ||||
|
Net
deferred tax assets
|
31,658,380 | 30,646,586 | ||||||
|
Valuation
allowance
|
(31,658,380 | ) | (30,646,586 | ) | ||||
|
Net
|
$ | — | $ | — | ||||
At
December 31, 2009, the Company has federal and state net operating loss
(“NOL”) carryforwards available to offset future taxable income of approximately
$65.3 million and $46.0 million, respectively. These carryforwards will begin to
expire in 2018 and 2010, respectively.
As
of December 31, 2009, the Company's net operating loss carryforwards include
approximately $857,000 of potential tax deductions related to stock option
transactions that will be credited directly to additional paid in capital, if
realized.
The
Company has federal and state research credit carryforwards of approximately
$1,813,000 and $2,203,000 respectively. The federal research credit
carryforwards will begin to expire in the year ending December 31, 2018 and the
state research credit will carry forward until exhausted.
Pursuant
to Section 382 of the Internal Revenue Code, use of the Company's NOL’s and
credit carryforwards may be limited if the Company experiences a cumulative
change in ownership of greater than 50% in a moving three-year
period. Ownership changes could impact the Company's ability to
utilize the NOL’s and credit carryforwards remaining at the ownership change
date. As of December 31, 2009 the Company conducted an analysis with
regard to its ownership and concluded a cumulative change in ownership of
greater than 50% in a moving three-year period had not occurred.
-63-
SENORX,
INC.
NOTES
TO FINANCIAL STATEMENTS (Continued)
As of
January 1, 2007, December 31, 2007, December 31, 2008 and December 31, 2009, the
Company had unrecognized tax benefits of $410,000. During each period from
January 1, 2007 to December 31, 2009, there was no change in the unrecognized
tax benefit as there was no change in judgment, lapse in statute of limitations,
or examination by taxing authorities. The Company estimates that the
unrecognized tax benefit will not change significantly within the next twelve
months. Future changes in the unrecognized tax benefit will have no
impact on the effective tax rate due to the existence of the valuation
allowance. The Company will continue to classify income tax penalties and
interest as part of interest expense in its Statements of Operations. Accrued
interest on uncertain tax positions is not material as of December 31, 2009.
There are no penalties accrued as of December 31, 2009.
The
following table summarizes the open tax years for each major
jurisdiction:
|
Jurisdiction
|
Open Tax Years
|
|
Federal
|
1998
- 2008
|
|
California
|
2000
- 2008
|
6. LONG-TERM
DEBT
A summary
of long-term debt follows:
|
December 31,
|
||||||||
|
2009
|
2008
|
|||||||
|
Equipment
facility
|
$ | — | $ | 10,058 | ||||
|
Term
loan
|
1,625,000 | 2,000,000 | ||||||
|
Capital
leases
|
1,180 | 12,598 | ||||||
| 1,626,180 | 2,022,656 | |||||||
|
Current
portion of long-term debt
|
(501,180 | ) | (390,246 | ) | ||||
|
Long-term
debt
|
$ | 1,125,000 | $ | 1,632,410 | ||||
Silicon
Valley Bank and Working Capital Facility—On September 30, 2008, the
Company and Silicon Valley Bank (“SVB”) entered into an amendment to the Amended
and Restated Loan and Security Agreement dated February 20, 2007 (as so
amended, the “Domestic Loan Agreement”), a Loan and Security Agreement (Ex-Im
Loan Facility) (the “Export-Import Loan Agreement”), and an Export-Import Bank
of the United States Working Capital Guarantee Program Borrower Agreement (the
“Borrower Agreement,” and together with the Domestic Loan Agreement and the
Export-Import Loan Agreement, the “Loan Agreement”). The total maximum amount
available for borrowing under the Loan Agreement is $12.0 million.
Revolving
Line
The Loan
Agreement provides for a domestic receivables-based revolving line of credit in
an aggregate amount of up to $10.0 million, or, if less, the sum of 80% of
eligible domestic accounts receivable plus 25% of eligible domestic inventory
(the “Domestic Revolving Line”), coupled with a foreign receivables-based
revolving line of credit in an aggregate amount of up to $2.5 million, or if
less, the sum of 90% of eligible foreign accounts receivable plus 50% of
eligible export-related inventory (the “Export-Import Revolving Line,” and
together with the Domestic Revolving Line, the “Revolving Line”). No more than
$10.0 million, in the aggregate, will be available for combined draw-downs under
the Domestic Revolving Line and the Export-Import Revolving Line. The Domestic
Revolving Line is subject to a $1.0 million sublimit available for cash
management services provided by SVB, including the issuance of short-term
letters of credit and foreign exchange contracts. The Export-Import Revolving
Line is guaranteed by the U.S. Export-Import Bank. The Revolving Line maturity
date is September 30, 2010.
The
Revolving Line bears interest at an annual rate equal to the prime rate plus
0.25% (4.25% at December 31, 2009). However, the annual rate
applicable to the Revolving Line increases to the prime rate plus 1.00% if a
financial ratio relating to the Company’s liquidity falls below a specified
level. Interest on the Revolving Line is due monthly, with the principal due at
the maturity date. At December 31, 2009, there was $8.1 million
available to borrow on the revolving line. No amounts have been
borrowed on the Revolving Line at December 31, 2009.
-64-
SENORX,
INC.
NOTES
TO FINANCIAL STATEMENTS (Continued)
Term
Loan
The Loan
Agreement provides for a term loan in an aggregate amount of up to $2.0 million
(the “Term Loan”), with a maturity date of March 30, 2013, unless the Revolving
Line is not renewed at its maturity date in which case, all amounts outstanding
under the Term Loan will become due at such time. The Term Loan bears interest
at an annual rate equal to the prime rate plus 0.75% (4.75% at December 31,
2009). However, the annual rate applicable to the Term Loan increases to the
prime rate plus 1.50% if a financial ratio relating to the Company’s liquidity
falls below a specified level. The Loan Agreement allows for a single advance
under the Term Loan which was requested in November 2008. Monthly payments of
interest only on the amount outstanding on the Term Loan are due through March
31, 2009, followed by forty-eight (48) consecutive monthly installments of
amortized principal and interest through the maturity date of the Term Loan.
There was $1.6 million in principal outstanding at December 31,
2009.
Aggregate
annual maturities of long-term debt at December 31, 2009 are as
follows:
|
Years Ending December 31
|
||||
|
2010
|
$ | 500,000 | ||
|
2011
|
500,000 | |||
|
2012
|
500,000 | |||
|
2013
|
125,000 | |||
| $ | 1,625,000 | |||
Loan
Agreement Obligations
The
obligations under the Loan Agreement are secured by a security interest on
substantially all assets of the Company, excluding intellectual property. The
Loan Agreement contains certain restrictive loan covenants, including, among
others, financial covenants requiring a minimum tangible net worth and a minimum
liquidity ratio, and covenants limiting the Company’s ability to dispose of
assets, make acquisitions, be acquired, incur indebtedness, grant liens or enter
into negative pledge agreements, make investments, make distributions in respect
of the Company’s capital stock (including repurchases of such capital stock) or
enter into transactions with affiliates. At December 31, 2009, the Company was
in compliance with all covenants.
The Loan
Agreement contains events of default that include, among others, failure to make
payments when due, inaccuracy of representations and warranties, violation of
covenants, events constituting a material adverse change, bankruptcy and
insolvency events, material judgments, and cross defaults to certain other
agreements. The occurrence of an event of default could result in the
acceleration of the Company’s obligations under the Loan Agreement and an
increase to the applicable interest rate, and would permit SVB to exercise
remedies with respect to the collateral under the Loan Agreement.
|
7.
|
STOCKHOLDERS’
EQUITY
|
Capital
Stock—In March 2007, the Company amended its certificate of incorporation
to reflect a 1-for-3.5 reverse stock split of common stock. All share and per
share amounts relating to common stock and stock options included in the
accompanying financial statements and footnotes have been restated to reflect
the reverse stock split.
Preferred
Stock—At December 31, 2006, the Company had 3,000,000 shares of
Series A convertible preferred stock outstanding, 3,523,040 shares of Series B
convertible stock outstanding and 17,861,899 shares of Series C Convertible
preferred stock outstanding. Upon the closing of the IPO in April 2007, all the
outstanding shares of preferred stock converted to 7,109,570 shares of the
Company’s common stock.
Restricted Common
Stock—Certain options to purchase common stock have been exercised,
subject to restricted stock purchase agreements. The restricted shares are
subject to the risk of forfeiture, certain restrictions on transferability and
to the Company’s repurchase rights. The restrictions and repurchase options
generally lapse 25% per year over a four-year vesting period. The Company
has a repurchase option, exercisable upon discontinuance of the purchaser’s
service with the Company, to repurchase the unvested shares at the original
price paid by the purchaser. Vested shares are not subject to the Company’s
repurchase rights. However, before any vested shares may be sold or otherwise
transferred, the Company has a right of first refusal to purchase the shares at
the price offered by the proposed transferee or fair value. Holders of
restricted stock have the voting rights of a common
stockholder.
-65-
SENORX,
INC.
NOTES
TO FINANCIAL STATEMENTS (Continued)
The
following summarizes information about aggregate number of chares of restricted
common stock issued pursuant to restricted stock purchase
agreements:
|
Year Ended
December 31,
|
||||||||||||
|
2009
|
2008
|
2007
|
||||||||||
|
Unvested
shares outstanding—beginning
|
2,981 | 16,396 | 25,318 | |||||||||
|
Restricted
shares issued upon exercise of stock options
|
— | — | — | |||||||||
|
Shares
vested
|
(2,683 | ) | (13,415 | ) | (8,922 | ) | ||||||
|
Unvested
common shares—ending
|
298 | 2,981 | 16,396 | |||||||||
|
8.
|
STOCK
OPTIONS, WARRANTS AND RESTRICTED STOCK
UNITS
|
The
Company’s 2006 Stock Option Plan (the “2006 Plan”), which was adopted by the
Company’s board of directors in May 2006 and approved by the Company’s
shareholders in June 2006 is designed to enable the Company to offer an
incentive-based compensation system to employees, officers and directors of the
Company and to consultants who do business with the Company. The 2006 Plan
provides for the grant of incentive stock options, nonqualified stock options
and restricted stock units to purchase up to an aggregate of 5,009,224 shares of
common stock. The 2006 plan will terminate in 2016 and also provides for annual
increases in the number of shares available for issuance thereunder on the first
day of each fiscal year, beginning with the 2007 fiscal year, equal to the
lesser of:
|
|
·
|
3.5%
of the outstanding shares of the Company’s common stock on the first day
of the fiscal year;
|
|
|
·
|
630,000
shares; or
|
|
|
·
|
Such
other amount as the board of directors may
determine.
|
Stock Options— As of
December 31, 2009, options to purchase a total of 2,433,120 shares of
common stock were outstanding under the 2006 Plan and options to purchase a
total of 1,216,799 shares were available for issuance under the 2006
Plan.
The
Company also has a 1998 Stock Option Plan (“1998 Plan”) which plan was approved
by the Company’s board of directors and shareholders in 1998. As of
December 31, 2009, options to purchase a total of 614,638 shares of common
stock were outstanding under the 1998 Plan and no options to purchase shares
were available for issuance under the 1998 Plan.
The 2006
Plan and the 1998 Plan are administered by a committee appointed by the board of
directors that determines the recipients and the terms of the options granted.
Options may be granted to eligible employees, directors and consultants to
purchase shares of the Company’s common stock at a price that is at least equal
to the fair market value of the common stock on the date of grant for incentive
stock options (or 110% of the fair market value in the case of an optionee who
holds more than 10% of the voting power of the Company on the date of grant).
Subject to termination of employment, options may expire up to ten years from
the date of grant.
The
exercise price, term and other conditions applicable to each option granted
under the 2006 and 1998 Plans are generally determined by the Administrator at
the time of grant of each option and may vary with each option granted. The
stock options granted generally vest 25% per year over a four-year period
and expire after seven to 10 years. The options are exercisable according to the
vesting schedule. The vesting of certain outstanding options will accelerate in
the event of a change in control. Alternatively, the options may be
exercised in whole or in part at any time into restricted, unvested common
shares which are subject to the risk of forfeiture, and to the Company’s
repurchase rights. The restrictions and repurchase options on currently
outstanding restricted stock grants issued pursuant to the 1998 Plan generally
lapse 25% per year over a four-year period (consistent with the vesting
period for the original stock option grants).
-66-
SENORX,
INC.
NOTES
TO FINANCIAL STATEMENTS (Continued)
The 1998
Plan also permits for the issuance of restricted stock pursuant to grants of
stock purchase rights. As of December 31, 2009, there were no outstanding
stock purchase rights.
During
the years ended December 31, 2009, 2008 and 2007, the Company recorded
stock-based compensation expense of $2,189,295, $2,135,938 and $1,818,206,
respectively, associated with employee performance-based and non-employee stock
option and RSU awards.
A summary
of stock option activity is as follows:
|
Number
of
Shares
|
Weighted-
Average
Exercise
Price
|
Weighted-
Average
Remaining
Contractual
Term (Years)
|
Aggregate
Intrinsic
Value
|
|||||||||||||
|
Outstanding—December
31, 2006
|
600,168 | 3.05 | ||||||||||||||
|
Granted
(weighted-average fair value of $4.64 per share)
|
764,086 | 9.67 |
|
|||||||||||||
|
Exercised
|
(47,583 | ) | 1.14 | |||||||||||||
|
Forfeited
|
(63,890 | ) | 7.07 | |||||||||||||
|
Outstanding—December
31, 2007
|
1,252,781 | 6.95 | ||||||||||||||
|
Granted
(weighted-average fair value of $1.97 per share)
|
1,141,441 | 4.60 | ||||||||||||||
|
Exercised
|
(35,969 | ) | 1.39 | |||||||||||||
|
Forfeited
|
(68,924 | ) | 8.61 | |||||||||||||
|
Outstanding—December
31, 2008
|
2,289,329 | $ | 5.82 | |||||||||||||
|
Granted
(weighted-average fair value of $1.28 per share)
|
580,373 | 3.11 | ||||||||||||||
|
Exercised
|
(46,809 | ) | 0.96 | |||||||||||||
|
Forfeited
|
(61,939 | ) | 7.51 | |||||||||||||
|
Outstanding—December
31, 2009
|
2,760,954 | $ | 5.29 | 5.9 | $ | 7,752,517 | ||||||||||
|
Vested
(exercisable)—December 31, 2009
|
1,340,119 | $ | 5.89 | 5.8 | $ | 2,958,650 | ||||||||||
|
Unvested
(unexercisable)—December 31, 2009
|
1,420,835 | $ | 4.73 | 6.0 | $ | 4,793,867 | ||||||||||
The
following table summarizes information with respect to stock options outstanding
and exercisable at December 31, 2009:
|
Exercise
Price
|
Number
Outstanding
|
Weighted-
Average
Remaining
Contractual
Life
(Years)
|
Weighted-
Average
Exercise
Price
|
Number
Exercisable
|
Weighted-
Average
Exercise
Price
|
|||||||||||||||||
| $0.60 – 1.75 | 216,896 | 3.5 | $ | 1.22 | 216,896 | $ | 1.22 | |||||||||||||||
| $2.28 – 2.63 | 657,635 | 5.9 | $ | 2.30 | 199,959 | $ | 2.33 | |||||||||||||||
| $3.00 – 5.97 | 705,849 | 6.2 | $ | 3.32 | 222,414 | $ | 3.46 | |||||||||||||||
| $6.98 – 8.89 | 853,176 | 5.9 | $ | 7.94 | 465,786 | $ | 8.01 | |||||||||||||||
| $9.55 – 12.01 | 327,398 | 7.2 | $ | 11.35 | 235,064 | $ | 11.32 | |||||||||||||||
| 2,760,954 | 1,340,119 | |||||||||||||||||||||
As of
December 31, 2009, there was unrecognized compensation expense of $1.2
million related to unvested stock options, which the Company expects to
recognize over a weighted average period of 1.5 years. The aggregate intrinsic
value of the options outstanding and options exercisable as of December 31,
2009 was $7.8 million and $3.0 million, respectively. The weighted average grant
date fair value of stock options granted during the year ended December 31,
2009 was $1.28.
-67-
SENORX,
INC.
NOTES
TO FINANCIAL STATEMENTS (Continued)
As of
December 31, 2009, the total number of outstanding options vested or
expected to vest (considering anticipated forfeitures) was 2,622,906, which had
a weighted average exercise price of $5.29. The average remaining life of these
options was 5.9 years and the aggregate intrinsic value was $7.8 million at
December 31, 2009.
Warrants — Upon the
closing of the IPO in April 2007, the outstanding preferred stock warrants
converted into warrants to purchase 462,046 shares of common stock. A summary of
the activity of common stock purchase warrants is as follows:
|
Warrant
Price
|
||||||||||||
|
Number
of
Shares
|
Per
Share
|
Total
|
||||||||||
|
Balance
outstanding, December 31, 2006
|
462,046 | 6.86 | 3,169,635 | |||||||||
|
Warrants
issued
|
— | — | — | |||||||||
|
Warrants
exercised
|
(243,446 | ) | 6.86 | (1,670,039 | ) | |||||||
|
Balance
outstanding, December 31, 2007
|
218,600 | 6.86 | 1,499,596 | |||||||||
|
Warrants
issued
|
— | — | — | |||||||||
|
Warrants
exercised
|
— | — | — | |||||||||
|
Balance
outstanding, December 31, 2008
|
218,600 | $ | 6.86 | $ | 1,499,596 | |||||||
|
Warrants
issued
|
— | — | — | |||||||||
|
Warrants
exercised
|
— | — | — | |||||||||
|
Balance
outstanding, December 31, 2009
|
218,600 | $ | 6.86 | $ | 1,499,596 | |||||||
Restricted Stock Units —
The 2006 Plan provides for awards of restricted stock units. Restricted stock
units have time-based vesting and are subject to forfeiture if employment
terminates prior to the end of the service period. Restricted stock units are
valued at the grant date based upon the market price of the Company’s common
stock and the fair value of each award is charged to expense over the service
period.
In 2009
and 2008, respectfully, the Company granted a total of 268,179 and 51,750 shares
of restricted stock units to employees. The restricted stock units vest 50% per
year over a two year period.
Restricted
stock unit activity for the year ended December 31, 2009 is as
follows:
|
Number
of Shares of
Restricted
Stock Units
|
Weighted-
Average
Grant-Date
Fair
Value
|
|||||||
|
Outstanding—December
31, 2007
|
— | |||||||
|
Granted
|
51,750 | $ | 2.38 | |||||
|
Vested
|
— | |||||||
|
Canceled
or forfeited
|
— | |||||||
|
Outstanding—December
31, 2008
|
51,750 | |||||||
|
Granted
|
268,179 | $ | 3.76 | |||||
|
Vested
|
(27,875 | ) | ||||||
|
Canceled
or forfeited
|
(5,250 | ) | ||||||
|
Outstanding—December
31, 2009
|
286,804 | |||||||
As of
December 31, 2009, there was unrecognized compensation expense of $540,000
related to all unvested restricted stock units, which the Company expects to
recognize on a “graded vesting attribution method”, which allocates expense on a
straight-line basis over the requisite service period for each separately
vesting portion of the award as if the award was, in-substance, multiple awards
over a weighted average period of approximately
1.2 years.
-68-
SENORX,
INC.
NOTES
TO FINANCIAL STATEMENTS (Continued)
|
9.
|
EMPLOYEE
STOCK PURCHASE PLAN
|
Effective
April 3, 2007, the closing date of the IPO, the Employee Stock Purchase
Plan (“Purchase Plan”) was established. The Company’s Purchase Plan was adopted
by the Company’s board of directors effective as of May 2006 and approved by the
Company’s stockholders in June 2006. The Purchase Plan provides eligible
employees of the Company with an incentive by providing a method whereby they
may voluntarily purchase common stock of the Company upon terms described in the
Purchase Plan. The Purchase Plan is designed to be operated on the basis of six
consecutive month offering periods commencing May 15 and November 15
of each year. The 2006 Purchase Plan terminates in 2016. The Purchase Plan
provides that eligible employees may authorize payroll deductions of up to 10%
of their salary to purchase shares of the Company’s common stock at 85% of the
fair market value of common stock on the first or last day of the applicable
purchase period. During 2009, 2008 and 2007, the Company recorded $109,052,
$135,652 and $145,966, respectively, for compensation related to the discounted
purchase price and look-back feature of the Purchase Plan. The 2009 fair value
of these discounts were estimated using a Black-Scholes pricing method with the
following assumptions: $2.51 and $3.74 strike price; $2.95 and $4.40 share
price; 184 days until expiration; 48% and 47.0% volatility and 0.29% and 0.17%
interest rate on May 16, 2009 and November 16, 2009,
respectively. The 2008 fair value of these discounts were estimated
using a Black-Scholes pricing method with the following assumptions: $6.16 and
$2.08 strike price; $7.25 and $2.44 share price; 184 days until expiration; 48%
and 46.5% volatility and 1.89% and 0.87% interest rate on May 16, 2008 and
November 17, 2008, respectively. The 2007 fair value of these
discounts were estimated using a Black-Scholes pricing method with the following
assumptions: $7.60 and $6.80 strike price; $8.94 and $8.00 share price; 228 and
182 days until expiration; 44% and 43% volatility and 4.08% and 5.05% interest
rate on October 1, 2007 and April 3, 2007, respectively. As of
December 31, 2009 and 2008 Purchase Plan participant contributions of
$78,249 and $84,925, respectively, are included in other current liabilities in
the accompanying balance sheets. A total of 550,000 shares of common stock are
authorized for issuance under the Purchase Plan, and as of December 31,
2009, 236,609 shares have been issued under the Purchase Plan.
|
10.
|
COMMITMENTS
AND CONTINGENCIES
|
Leases—In
March 2008, the Company entered into an agreement to lease a new corporate and
manufacturing facility in Irvine, California consisting of 41,402 square feet of
space. The lease has an initial term of 63 months commencing November 1,
2008, with the Company’s right of earlier occupancy, and one five-year option to
extend. The monthly base rent is initially approximately $44,300 and increases
at a rate of 4% per annum. The lease agreement contains certain scheduled
rent increases which are accounted for on a straight-line basis.
Future
minimum lease payments are as follows:
|
Years
Ending December 31,
|
|
Operating
Leases
|
|||
|
2010
|
$ | 555,612 | |||
|
2011
|
580,452 | ||||
|
2012
|
605,294 | ||||
|
2013
|
626,004 | ||||
|
2014
|
52,167 | ||||
|
Total
minimum lease payments
|
$ | 2,419,529 | |||
Rent
expense was $721,818, $610,087 and $447,741, for the years ended
December 31, 2009, 2008 and 2007, respectively.
Indemnities and
Guarantees—During its normal course of business, the Company has made
certain indemnities, commitments and guarantees under which it may be required
to make payments in relation to certain transactions. These indemnities include
those given to various lessors in connection with facility leases for certain
claims arising from such facility or lease and indemnities to directors and
officers of the Company to the maximum extent permitted under the laws of the
State of California. The duration of these indemnities, commitments and
guarantees varies. Some of these indemnities, commitments and guarantees do not
provide for any limitation of the maximum potential future payments the Company
could be obligated to make. The Company has not recorded any liability for these
indemnities, commitments and guarantees in the accompanying balance
sheets.
-69-
SENORX,
INC.
NOTES
TO FINANCIAL STATEMENTS (Continued)
The
Company provides a limited warranty against manufacturer’s defects on its
products. Product warranty costs have not been significant.
FDA
Inspection—The Company recently underwent an inspection of the Irvine
manufacturing facility by the FDA, which resulted in the issuance on September
30, 2009 of an FDA Form 483, Notice of Inspectional Observations, from the FDA
related to the Company’s failure to properly implement and maintain adequate
methods and documentation of the design, testing, production, control, quality
assurance, storage, shipping and post-market surveillance for some products,
including Contura MLB and Gel Mark product line. The Notice of
Inspectional Observations will require the Company to take prompt action to
strengthen its quality system. The Company completed a comprehensive corrective
action plan that has been presented to the FDA outlining the steps and timing
for coming into compliance with Quality Systems regulations. The
Company’s failure to adequately or timely implement or complete a corrective
action plan could have a material adverse effect on the Company’s results of
operations, cash flows and financial position. The Company believes
that the probability of incurring any loss related to this inspection is not
determinable nor is the loss quantifiable at this time. Accordingly,
the Company has not accrued a loss related to matter as of December 31,
2009.
Litigation—The
Company may be subject to legal proceedings, claims and litigation arising in
the ordinary course of business. While the amounts claimed may be substantial,
the ultimate liability cannot presently be determined because of considerable
uncertainties that exist. Therefore, it is possible the outcome of such legal
proceedings, claims and litigation could have a material effect on quarterly or
annual operating results or cash flows when resolved in a future
period.
On
January 8, 2008, Hologic and its wholly-owned subsidiaries, including Cytyc
Corporation and Cytyc LP, filed a lawsuit against the Company in the United
States District Court, Northern District of California, San Jose Division. The
complaint generally alleges patent infringement of certain Hologic brachytherapy
patent claims, seeking unspecified monetary damages and an injunction against
the Company for infringement of those claims. On February 6, 2008, Hologic filed
a motion seeking a preliminary injunction in the case and requested that the
Court stop the sale of Contura MLB. On March 7, 2008, Hologic filed an amended
complaint restating its allegations regarding patent infringement, and adding
new claims related to unfair competition under the Lanham Act and California
state unfair competition and false advertising statutes. On April 25, 2008, the
court denied Hologic's request for a preliminary injunction and ordered the
parties to schedule a trial within 60 to 90 days of such date. On May 22, 2008,
the Court issued an order scheduling the Markman claims construction hearing on
the patent counts for June 25, 2008, and the trial in the case to start July 14,
2008. Pursuant to an agreement of the parties, the order also dismissed
Hologic's unfair competition and false advertising claims under the Lanham Act
and California state law, without prejudice. On June 24, 2008, the Court granted
the Company and Hologic’s joint request to stay all proceedings, including the
previously scheduled Markman claims construction hearing and the trial, until at
least August 22, 2008 in order to provide the parties time to discuss possible
resolution of the matter. On August 22, 2008, the Company jointly
requested with Hologic that the Court resume proceedings in the pending
lawsuit. On October 15, 2008, a Markman claims construction hearing
and a hearing on the Company’s motion for summary judgment of invalidity of
certain claims was held and a ruling was issued on February 18, 2009. In light
of the Court's Markman ruling, on May 20, 2009, the parties separately filed
motions seeking partial summary judgment: Hologic filed a motion
seeking partial summary judgment of infringement for certain claims, and the
Company filed motions seeking partial summary judgment that certain claims were
not infringed and that certain claims were invalid. Briefing on the
summary judgment motions was completed, and argument was held on August 21,
2009. The Court issued an order on October 30, 2009 ruling that one
of the asserted claims of U.S. Patent No. 6,482,142 patent was invalid, and that
physicians using the Contura MLB infringed the other asserted claim of the '142
patent. The Court further ruled that one asserted claim of U.S.
Patent No. 6,413,204 and all asserted claims of U.S. Patent No. 5,913,813 were
not infringed. The remaining claims in the case were tried by a jury
between December 2, 2009 and December 16, 2009. On December 17, 2009,
the jury returned a verdict ruling in the Company’s favor on all counts
remaining in the case. In particular, the jury found that the Company
did not infringe the '204 patent, and that the remaining claims of the '204
patent and '142 patent were invalid for reasons of anticipation and
obviousness. The court issued a final judgment in the case on
February 24, 2010. Accordingly, following the result of the jury
trial, the Contura MLB does not infringe any valid claim of any of the Hologic
patents that were the subject matter of the litigation. On February
26, 2010 Hologic filed a notice of its appeal of the Court's final judgment and
other adverse orders opinions and rulings relating to the final judgment, claim
construction and the summary judgment order. No further dates
have been scheduled at this juncture. The Company believes that the
probability of incurring any loss related to this litigation is not
determinable, nor is the amount of loss quantifiable at this
time. Accordingly, the Company has not accrued a loss related to this
litigation as of December 31, 2009. The Company intends to continue
to vigorously defend itself in this matter.
-70-
SENORX,
INC.
NOTES
TO FINANCIAL STATEMENTS (Continued)
|
11.
|
SEGMENT
INFORMATION
|
Net
revenues by geographic area are presented based upon the country of destination.
No foreign country represented 10% or more of net revenues for any period
presented. Net revenues by geographic area were as follows:
|
2009
|
2008
|
2007
|
||||||||||
|
United
States
|
$ | 48,300,930 | $ | 41,209,560 | $ | 32,321,888 | ||||||
|
Canada
|
1,826,367 | 1,200,218 | 1,052,543 | |||||||||
|
Rest
of world
|
5,450,002 | 4,274,810 | 1,661,405 | |||||||||
|
Total
|
$ | 55,577,299 | $ | 46,684,588 | $ | 35,035,836 | ||||||
No
customer accounted for 10% or more of net revenues for any period
presented.
At
December 31, 2009, the Company has four product classes. Biopsy disposable
products include the Company’s EnCor products. Biopsy capital equipment products
include the consoles and other pieces (non-disposable) of the EnCor products.
Diagnostic adjunct products include the Marker product and the Gamma Finder
product. Therapeutic disposable products include the Company’s Contura
Multi-Lumen Balloon (MLB) Catheter, which received FDA 510(k) clearance in May
2007.
Net
revenues by product class are as follows:
|
Year Ended
December 31,
|
||||||||||||
|
2009
|
2008
|
2007
|
||||||||||
|
Biopsy
disposable products
|
$ | 23,971,962 | $ | 21,041,449 | $ | 16,215,740 | ||||||
|
Biopsy
capital equipment products
|
4,405,793 | 4,692,778 | 3,301,908 | |||||||||
|
Diagnostic
adjunct products
|
15,656,933 | 15,774,788 | 14,976,567 | |||||||||
|
Therapeutic
disposable products
|
11,542,611 | 5,175,573 | 541,621 | |||||||||
|
Total
|
$ | 55,577,299 | $ | 46,684,588 | $ | 35,035,836 | ||||||
Substantially
all of the Company’s assets are in the United States.
|
12.
|
EMPLOYEE
BENEFIT PLANS
|
In
January 2003, the Company adopted a 401(k) retirement and savings plan which
provides for voluntary employee participation subject to a waiting period. The
plan provides for discretionary matching subject to the approval by the
Company’s Board of Directors. The Company did not make contributions to this
plan in the years ended December 31, 2009, 2008 and 2007.
-71-
SENORX,
INC.
NOTES
TO FINANCIAL STATEMENTS (Continued)
|
13.
|
QUARTERLY
FINANCIAL DATA (Unaudited)
|
A summary
of quarterly financial data (unaudited) is as follows:
|
Quarter
Ended
|
||||||||||||||||
|
March
31
|
June
30
|
September
30
|
December
31
|
|||||||||||||
|
Year
ended December 31, 2009
|
||||||||||||||||
|
Total
revenues
|
$ | 12,876,712 | $ | 13,502,201 | $ | 13,718,264 | $ | 15,480,122 | ||||||||
|
Gross
profit
|
9,057,903 | 9,538,400 | 9,732,193 | 10,882,833 | ||||||||||||
|
Operating
loss
|
(878,974 | ) | (678,212 | ) | (198,694 | ) | (961,139 | ) | ||||||||
|
Net
loss
|
(928,372 | ) | (709,708 | ) | (235,443 | ) | (1,014,610 | ) | ||||||||
|
Net
loss per share, basic and diluted
|
$ | (0.05 | ) | $ | (0.04 | ) | $ | (0.01 | ) | $ | (0.06 | ) | ||||
|
Year
ended December 31, 2008
|
||||||||||||||||
|
Total
revenues
|
$ | 10,682,775 | $ | 11,186,118 | $ | 11,264,471 | $ | 13,551,224 | ||||||||
|
Gross
profit
|
6,629,325 | 6,859,816 | 7,321,549 | 9,370,571 | ||||||||||||
|
Operating
income (loss)
|
(2,229,534 | ) | (5,265,563 | ) | (1,753,930 | ) | 108,044 | |||||||||
|
Net
income (loss)
|
(1,994,537 | ) | (5,131,592 | ) | (1,677,021 | ) | 97,024 | |||||||||
|
Net
income (loss) per share, basic and diluted
|
$ | (0.12 | ) | $ | (0.30 | ) | $ | (0.10 | ) | $ | 0.01 | |||||
Earnings
per basic and diluted shares are computed independently for each of the quarters
presented based on basic and diluted shares outstanding per quarter and,
therefore, may not sum to the totals for the year.
-72-
SIGNATURES
Pursuant
to the requirements of Section 13(a) or 15(d) of the Securities Exchange
Act of 1934, the Registrant has duly caused this Annual Report on Form 10-K
to be signed on our behalf by the undersigned, thereunto duly
authorized.
|
Date:
March 16,
2010
|
SenoRx,
Inc.
|
|||
|
By:
|
/s/
John T. Buhler
|
|||
|
John
T. Buhler
|
||||
|
President,
Chief Executive Officer and Chief Operating
Officer
(Principal
Executive Officer)
|
||||
KNOW ALL
MEN AND WOMEN BY THESE PRESENTS, that each person whose signature appears below
constitutes and appoints each of John T. Buhler and Kevin J. Cousins, his
or her attorney-in-fact, with the power of substitution, for him or her in any
and all capacities, to sign any amendments to this annual report on
Form 10-K, and to file the same, with exhibits thereto and other documents
in connection therewith, with the U.S. Securities and Exchange Commission,
hereby ratifying and confirming all that said attorney-in-fact, or his
substitute, may do or cause to be done by virtue thereof.
Pursuant
to the requirements of the Securities Exchange Act of 1934, this Annual Report
on Form 10-K has been signed below by the following persons on behalf of
the Registrant in the capacities and on the dates indicated:
|
Signature
|
Title
|
Date
|
|
|
/S/
JOHN T. BUHLER
John
T. Buhler
|
Director,
President, Chief Executive Officer and Chief Operating
Officer (Principal Executive Officer)
|
March
16, 2010
|
|
|
/S/ KEVIN J. COUSINS
Kevin
J. Cousins
|
Chief
Financial Officer (Principal Accounting Officer)
|
March
16, 2010
|
|
|
/S/ VICKIE L. CAPPS
Vickie
L. Capps
|
Director
|
March
16, 2010
|
|
|
/S/ KIM D. BLICKENSTAFF
Kim
D. Blickenstaff
|
Director
|
March
16, 2010
|
|
|
/S/ FREDERICK J. DOTZLER
Frederick
J. Dotzler
|
Director
|
March
16, 2010
|
|
|
/S/ JOHN L. ERB
John
L. Erb
|
Director
|
March
16, 2010
|
|
|
/S/ A. Thomas
Bender
A.
Thomas Bender
|
Director
|
March
16, 2010
|
|
|
/S/ GREGORY D. WALLER
Gregory
D. Waller
|
Director
|
March
16, 2010
|
|
-73-
EXHIBIT INDEX
|
Exhibit
Number
|
Description
|
|
|
3.2(1)
|
Amended
and Restated Certificate of Incorporation.
|
|
|
3.4(1)
|
Bylaws.
|
|
|
4.1(1)
|
Specimen
Common Stock certificate of the Registrant.
|
|
|
4.2(1)
|
Fourth
Amended and Restated Investors’ Rights Agreement, dated May 3, 2006, by
and among the Registrant and certain stockholders.
|
|
|
10.1(1)
|
Form
of Indemnification Agreement for directors and executive
officers.
|
|
|
10.2(1)
|
1998
Stock Plan.
|
|
|
10.3(5)
|
2006
Equity Incentive Plan.
|
|
|
10.4(1)
|
Employee
Stock Purchase Plan.
|
|
|
10.5(1)
|
Fourth
Amended and Restated Investors’ Rights Agreement, dated May 3, 2006, by
and among the Registrant and certain stockholders.
|
|
|
10.6(1)
|
Standard
Industrial/Commercial Multi-Tenant Lease, dated September 15, 1999, as
amended on March 28, 2003 and November 1, 2003, by and between the
Registrant and Columbia Investors, LLC.
|
|
|
10.7(1)
|
Loan
and Security Agreement, dated March 15, 2002, and various amendments
thereto, by and between the Registrant and Silicon Valley
Bank.
|
|
|
10.7.1(1)
|
Amended
and Restated Loan and Security Agreement, dated February 20, 2007, by and
between the Registrant and Silicon Valley Bank.
|
|
|
10.8(1)
|
Convertible
Subordinated Note Agreement, dated May 9, 2002, by and between the
Registrant and Century Medical, Inc.
|
|
|
10.9(1)
|
$2,500,000
Loan and Security Agreement, dated December 27, 2004, by and between the
Registrant and Venture Lending & Leasing IV, Inc.
|
|
|
10.10(1)
|
Note
Purchase Agreement and Form of Subordinated Convertible Promissory Note,
each dated May 4, 2006, by and between the Registrant and certain
stockholders.
|
|
|
10.11(1)†
|
Agreement
for Vacuum Assisted Breast Biopsy Needle, System, and Accessory Products,
effective April 1, 2005 by and between the Registrant and KP
Select.
|
|
|
10.12(1)
|
Executive
Employment Agreement, dated May 1, 1999, by and between the Registrant and
Lloyd Malchow.
|
|
|
10.13(1)†
|
Distribution
Agreement, dated June 11, 2003, and various amendments thereto, by and
among the Registrant, W.O.M. World of Medicine USA, Inc. and W.O.M. World
of Medicine AG.
|
|
|
10.14(1)
|
Settlement
Agreement, effective as of May 22, 2006, by and between the Registrant and
Suros Surgical Systems, Inc.
|
|
|
10.15(1)
|
Loan
and Security Agreement, dated December 8, 2006, by and between the
Registrant and Escalate Capital I, L.P.
|
|
|
10.16(2)
|
Lease
Agreement between The Irvine Company LLC and Registrant dated March 5,
2008.
|
|
|
10.17(3)
|
Change
in Control Agreement between Registrant and Lloyd
Malchow.
|
|
|
10.18(3)
|
Change
in Control Agreement between Registrant and Paul
Lubock.
|
|
|
10.19(3)
|
Change
in Control Agreement between Registrant and Kevin
Cousins.
|
|
|
10.20(3)
|
Change
in Control Agreement between Registrant and William
Gearhart.
|
|
|
10.21(3)
|
Change
in Control Agreement between Registrant and Eben
Gordon.
|
-74-
|
10.22(4)
|
Amendment
dated September 30, 2008, to the Amended and Restated Loan and Security
Agreement dated February 27, 2007, by and between the Registrant and
Silicon Valley Bank.
|
|
|
10.23(4)
|
Loan
and Security Agreement (Ex-Im Loan Facility) dated September 30, 2008, by
and between the Registrant and Silicon Valley Bank.
|
|
|
10.24(4)
|
Export-Import
Bank of the United States Working Capital Guarantee Program Borrower
Agreement dated September 30, 2008, by and between the Registrant and
Silicon Valley Bank.
|
|
|
23.1
|
Consent
of Deloitte & Touche LLP, Independent Registered Public Accounting
Firm.
|
|
|
31.1
|
Certification
of Chief Executive Officer pursuant to Section 302 of the
Sarbanes-Oxley Act of 2002.
|
|
|
31.2
|
Certification
of Chief Financial Officer pursuant to Section 302 of the
Sarbanes-Oxley Act of 2002.
|
|
|
32.1
|
Certification
of Chief Executive Officer and Chief Financial Officer pursuant to 18
U.S.C. Section 1350, as adopted pursuant to Section 906 of the
Sarbanes-Oxley Act of 2002.
|
______________________
|
(1)
|
Incorporated
by reference from our Registration Statement on Form S-1 (Registration No.
333-134466), which was declared effective on March 28,
2007.
|
|
(2)
|
Incorporated
by reference from our Current Report on Form 8-K filed on March 10,
2008.
|
|
(3)
|
Incorporated
by reference from our Current Report on Form 8-K filed on August 29,
2008.
|
|
(4)
|
Incorporated
by reference from our Current Report on Form 8-K filed on October 2,
2008.
|
|
(5)
|
Incorporated
by reference from our Annual Report on Form 10-K filed on March 16,
2009.
|
|
†
|
Portions
of the exhibit have been omitted pursuant to a request for confidential
treatment. The confidential portions have been filed with the
SEC.
|
-75-