UNITED STATES SECURITIES AND
EXCHANGE COMMISSION
Washington, D.C.
20549
Form 10-K
| |
|
|
|
(Mark One)
|
|
|
|
þ
|
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE
SECURITIES EXCHANGE ACT OF 1934
|
|
|
|
For the fiscal year ended
December 31,
2009
|
|
or
|
|
o
|
|
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE
SECURITIES EXCHANGE ACT OF 1934
|
|
|
|
For the transition period
from to
|
Commission File Number
001-33054
Trubion Pharmaceuticals,
Inc.
(Exact name of registrant as
specified in its charter)
| |
|
|
DELAWARE
(State or other jurisdiction
of
incorporation or organization)
|
|
52-2385898
(IRS Employer
Identification No.)
|
|
2401 FOURTH AVENUE, SUITE 1050
SEATTLE, WASHINGTON
(Address of
registrant’s principal executive offices)
|
|
98121
(Zip
Code)
|
(206) 838-0500
(Telephone number, including area code)
Securities registered pursuant to Section 12(b) of the
Act:
| |
|
|
|
Title of Each Class
|
|
Name of Each Exchange on Which Registered
|
|
|
|
COMMON STOCK, $0.001 PAR VALUE
|
|
NASDAQ GLOBAL MARKET
|
Securities registered pursuant to Section 12(g) of the
Act:
None
Indicate by check mark whether the registrant is a well-known
seasoned issuer, as defined in Rule 405 of the Securities
Act. Yes o No þ
Indicate by check mark whether the registrant is not required to
file reports pursuant to Section 13 or Section 15(d)
of the
Act. Yes o No þ
Indicate by check mark whether the registrant (1) has filed
all reports required to be filed by Section 13 or 15(d) of
the Securities Exchange Act of 1934 during the preceding
12 months (or for such shorter period that the registrant
was required to file such reports), and (2) has been
subject to such filing requirements for the past
90 days. Yes þ No o
Indicate by check mark if disclosure of delinquent filers
pursuant to Item 405 of
Regulation S-K
is not contained herein, and will not be contained, to the best
of the registrant’s knowledge, in definitive proxy or
information statements incorporated by reference in
Part III of this
Form 10-K
or any amendment to this
Form 10-K. o
Indicate by check mark whether the registrant is a large
accelerated filer, an accelerated filer, a non-accelerated
filer, or a smaller reporting company. See the definitions of
“large accelerated filer,” “accelerated
filer” and “smaller reporting company” in Rule
12b-2 of the
Exchange Act. (Check one):
|
|
|
|
| Large
accelerated
filer o
|
Accelerated
filer o
|
Non-accelerated
filer þ
|
Smaller
reporting
company o
|
(Do not check if a smaller reporting company)
Indicate by check mark whether the registrant is a shell company
(as defined in
Rule 12b-2
of the
Act). Yes o No þ
The aggregate market value of the common stock held by
non-affiliates of the registrant based on the closing sale price
of the registrant’s common stock on June 30, 2009, as
reported on the National Association of Securities Dealers
Automated Market, was $47,268,191.
As of February 26, 2010, 20,386,800 shares of the
registrant’s common stock were outstanding.
DOCUMENTS
INCORPORATED BY REFERENCE
Specified portions of the registrant’s definitive proxy
statement with respect to the 2009 Annual Meeting of
Stockholders to be held May 26, 2010, which are to be filed
pursuant to Regulation 14A within 120 days after the
end of the registrant’s fiscal year ended December 31,
2009, are incorporated by reference into Part III of this
annual report.
TRUBION
PHARMACEUTICALS, INC.
2009
Form 10-K
Annual Report
Table of
Contents
PART I
Forward-Looking
Statements
This annual report on
Form 10-K
and the documents incorporated herein by reference contain
“forward-looking statements” within the meaning of
Section 21E of the Securities Exchange Act of 1934, as
amended, or the Exchange Act, and Section 27A of the
Securities Act of 1933, as amended, or the Securities Act. These
forward-looking statements are based on current expectations,
estimates and projections about our industry, management’s
beliefs, and certain assumptions made by management. Words such
as “may,” “anticipate,” “expect,”
“could,” “intend,” “plan,”
“believe,” “seek” and “estimate”
or similar expressions are intended to identify forward-looking
statements. These statements are not guarantees of future
performance and actual actions or results may differ materially.
These statements are subject to certain risks, uncertainties,
and assumptions that are difficult to predict, including those
identified in the sections of Item 1 entitled
“Strategic Collaborations,” “Competition,”
“Intellectual Property,” “Manufacturing,”
“Government Regulation” and “Reimbursement,”
and in the sections of Item 1A entitled “Risk
Factors.” We undertake no duty to update any
forward-looking statement to conform the statement to actual
results or changes in our expectations. Readers should, however,
carefully review the risk factors included in other reports or
documents filed by us from time to time with the Securities and
Exchange Commission, or SEC, particularly our quarterly reports
on
Form 10-Q
and any current reports on
Form 8-K.
Overview
We are a biopharmaceutical company creating a pipeline of novel
protein therapeutic product candidates to treat autoimmune and
inflammatory diseases and cancer. Our mission is to develop a
variety of
first-in-class
and
best-in-class
product candidates customized in an effort to optimize safety,
efficacy, and convenience that we believe may offer improved
patient experiences. Our current product development efforts are
focused on three proprietary technologies that comprise the
expanded foundation for Trubion product development —
Small Modular Immunopharmaceutical, or
SMIPtm,
protein therapeutics,
SCORPIONtm
protein therapeutics, and
TRU-ADhanCetm
potency enhancing technology for immunopharmaceuticals. Our
current clinical-stage therapeutics target specific antigens on
B cells, CD20 and CD37, and are designed using our custom drug
assembly technology. In order to fund ongoing development
activities and commercialize our products, we will, in some
cases, enter into collaboration agreements that would likely
include licenses to our technology and arrangements to provide
research and development services for others.
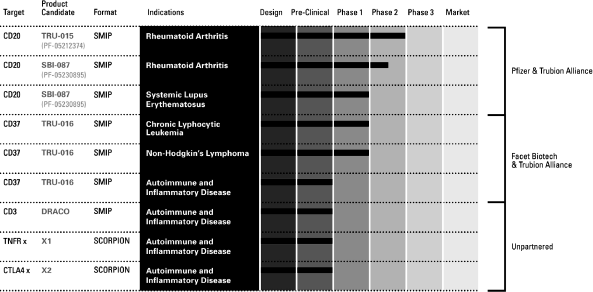
Our lead product candidate, TRU-015, which we are developing
with our partner, Pfizer Inc., or Pfizer, has completed a Phase
2b clinical trial for the treatment of rheumatoid arthritis, or
RA. A second Phase 2b (study 2203) clinical trial for RA is
under way and enrollment was completed in September 2009. The
randomized, parallel, double-blind, placebo-controlled,
dose-regimen finding study is evaluating the safety and efficacy
of two dosing regimens of TRU-015 administered to patients with
active seropositive rheumatoid factor, or RF, on a background of
methotrexate. This study was designed in a way that we believe
could be supportive of a registration package with the Food and
Drug Administration, or FDA.
In October 2009, we announced positive data from the second
course of re-treatment in the first Phase 2b clinical trial for
RA demonstrating that administration of TRU-015 every
24 weeks produces well-tolerated results that are
comparable with the more than four and a half years of
re-treatment data compiled to date.
In collaboration with us, Pfizer is also developing SBI-087, our
next generation CD20-directed product candidate. SBI-087 for RA
builds on our and Pfizer’s clinical experience with TRU-015
and is based on our SMIP technology. Enrollment was completed in
a Phase 1 study of SBI-087 for RA and patient dosing commenced
in a Phase 2 study in December 2009. In addition, patient
recruitment is under way in an additional Phase 1 study of
SBI-087 for RA in Japan. Finally, Pfizer is conducting a Phase 1
clinical trial of SBI-087 in systemic lupus erythematosus, or
SLE, in which patient dosing has commenced and recruitment is
ongoing.
2
TRU-016, which we are developing with our partner Facet Biotech
Corporation, or Facet, is a novel CD37-directed SMIP protein
therapeutic. A TRU-016 Phase 1 clinical trial for patients with
chronic lymphocytic leukemia, or CLL, is currently under way and
has recently been expanded to treat patients with non-Hodgkins
lymphoma, or NHL. TRU-016 uses a different mechanism of action
than CD20-directed therapies. As a result, we believe its novel
design may provide patients with improved therapeutic options
and enhance efficacy when used alone or in combination with
chemotherapy
and/or
CD20-directed therapeutics.
In June and December 2009, we announced positive results
following each of two preliminary analyses from the Phase 1
clinical trial of TRU-016 for the treatment of CLL. The
objectives of the Phase 1 TRU-016 CLL study are to define safety
and tolerability, identify a maximum tolerated dose, or MTD,
evaluate pharmacology and pharmacodynamics, and assess
preliminary clinical activity. As of March 2010, we have not
reached a MTD.
Our product candidates are as follows:
|
|
|
| |
•
|
TRU-015 for the Treatment of Rheumatoid
Arthritis. According to Datamonitor, RA is
estimated to affect approximately 5.2 million people in the
United States, Japan, and the five major European markets. In
2009, total reported worldwide therapeutic sales for the
treatment of RA were over $10 billion. Unfortunately, a
significant segment of the RA population are refractory to
current biological disease modifying anti-rheumatic drugs, or
DMARDs, and clinical benefit (as measured by America College of
Rheumatology, or ACR, 50 score) is estimated at less than 50% at
six months post therapy.
|
We and our partner Pfizer have completed a Phase 2b clinical
trial of TRU-015 for the treatment of RA (study 15002). A second
Phase 2b clinical trial (study 2203) is ongoing and
enrollment was completed in September 2009. The interim analysis
of the second Phase 2b clinical trial of TRU-015 in RA (study
2203) met the pre-defined primary endpoint demonstrating
superiority of TRU-015 over placebo. The study’s
pre-defined primary endpoint is ACR50 response at week 24. We
expect to receive final data from study 2203 in mid 2010. Pfizer
will determine whether to commence a Phase 3 study of TRU-015
for the treatment of RA after reviewing data from the ongoing
Phase 2 SBI-087 RA study, in addition to final data from the
ongoing Phase 2b TRU-015 (study 2203) RA study. Patient
dosing in the Phase 2 SBI-087 RA study commenced in December
2009 and an interim data review is planned that we believe could
occur in late 2010 or early 2011 and final data is anticipated
at the end of 2011.
|
|
|
| |
•
|
SBI-087 for the Treatment of Rheumatoid
Arthritis. In collaboration with us, our partner
Pfizer has commenced two clinical studies of SBI-087 for the
treatment of RA. The first is a Phase 2 randomized,
placebo-controlled, double-blind, parallel-group, outpatient
dose regimen-finding study in which patient dosing commenced in
December 2009, with interim data review anticipated to occur
late 2010 or early 2011 and final data is anticipated at the end
of 2011. The second is a Phase 1 dose escalation clinical trial
designed to evaluate the safety, tolerability, pharmacokinetics,
or PK, and pharmacodynamics, or PD, of a single dose of SBI-087
in patients with RA, in which patient enrollment is complete. In
addition, patient recruitment is also under way in an additional
Phase 1 study of SBI-087 for RA in Japan.
|
| |
| |
•
|
SBI-087 for the Treatment of Systemic Lupus
Erythematosus. According to Datamonitor, SLE is
estimated to affect approximately 434,000 people in the
United States, Japan, and the five major European markets. The
prevalence of SLE varies significantly on a
country-by-country
basis and could be up to five times greater with expanding
disease definitions and increasing diagnosis. No new
pharmaceutical or biologic treatments have been approved for SLE
in over 40 years. Our partner Pfizer is conducting a Phase
1 clinical trial of SBI-087 in SLE in which patient dosing has
commenced and recruitment is ongoing. Currently, no protein
therapeutics have been approved specifically for the treatment
of SLE.
|
| |
| |
•
|
TRU-016 for the Treatment of B-cell
Malignancies. According to the National Cancer
Institute, CLL is estimated to affect approximately
70,000 people in the United States. Approximately 12,000
new cases of CLL are diagnosed each year according to
Datamonitor. In addition, NHL, another B-cell malignancy, is one
of the most common types of cancer accounting for approximately
4% of all cancers. About 66,000 people are expected to be
diagnosed with NHL in 2009 according to the American Cancer
society. Total reported worldwide sales of one of the most
commonly used biologics
|
3
|
|
|
| |
|
in NHL,
Rituxan®,
surpassed $3 billion in 2009. Our TRU-016 product candidate
targets CD37 for the treatment of B-cell malignancies such as
CLL and NHL. TRU-016 uses a different mechanism of action than
CD20-directed therapies. As a result, we believe its novel
design may provide patients with improved therapeutic options
and enhance efficacy when used alone or in combination with
chemotherapy
and/or other
CD20-directed therapeutics. We are currently conducting a Phase
1 clinical trial of TRU-016 in CLL and have filed an amendment
to include treatment of patients with NHL.
|
In addition to our current product candidates, we are also
developing additional alliance and proprietary product
candidates that build on our existing product experience. To
date, none of our product candidates has been approved for
marketing and sale to patients nor have we received any product
revenue.
In August 2009, we entered into a collaboration agreement with
Facet for the joint worldwide development and commercialization
of TRU-016, a product candidate in Phase 1 clinical development
for CLL. TRU-016 is a CD37-directed SMIP protein therapeutic.
The collaboration agreement includes TRU-016 in all indications
and all other CD37-directed protein therapeutics. Under the
terms of the collaboration agreement, the parties will not
develop or commercialize protein therapeutics directed to CD37
outside of the collaboration agreement.
In December 2005, we entered into a collaboration agreement with
Wyeth, now a wholly-owned subsidiary of Pfizer, for the
development and worldwide commercialization of TRU-015 and other
therapeutics directed to CD20, an antigen that is a validated
clinical target present on B cells. Pursuant to the agreement,
we are also collaborating with Pfizer on the development and
worldwide commercialization of certain other product candidates
directed to a small number of targets other than CD20. During
the period in which we will be providing research services for
Pfizer, Pfizer has the right, subject to our reasonable consent,
to replace a limited number of these targets. In addition, we
also have the option to co-promote with Pfizer, on customary
terms to be agreed, CD20-directed therapies in the United States
for niche indications. We retain the right to develop and
commercialize, on our own or with others, product candidates
directed to all targets not included within the agreement,
including CD37. Unless it is terminated earlier, our agreement
with Pfizer will remain in effect on a
product-by-product
basis and on a
country-by-country
basis until the later of the date that any such product shall no
longer be covered by a valid claim of a United States or foreign
patent or application and, generally, ten years after the first
commercial sale of any product licensed under the agreement.
Product
Technologies
Our current product development efforts are focused on three
proprietary technologies that comprise the expanded foundation
for Trubion product development —
SMIPtm
protein therapeutics,
SCORPIONtm
protein therapeutics, and
TRU-ADhanCetm
potency enhancing technology for immunopharmaceuticals. We
believe our product candidates offer the potential for safer and
more effective therapies than existing or other potential
products. Additionally, we believe that these technologies will
provide the basis for the long-term development of additional
first-in-class
and
best-in-class
product candidates.
SMIPtm
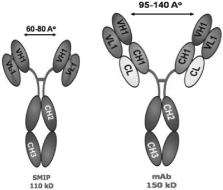
Protein Therapeutics vs. mAbs
Our custom SMIP drug assembly technology was designed to
specifically address the limitations of monoclonal antibodies,
or mAbs. SMIP therapeutics are single chain polypeptides
comprising a binding domain, a hinge domain and an effector
domain designed in an effort to meet predetermined therapeutic
criteria for specific diseases. SMIP proteins are mono-specific
therapeutics — a drug that recognizes and attaches to
a single antigen target and initiates biological activity. SMIP
therapeutics have engineered structural design characteristics
and are significantly smaller than whole antibodies, which we
believe allows for better in vivo penetration.
SMIP technology enables us to design and develop differentiated
product candidates for a range of targets and biological
activities that have the following advantages:
|
|
|
| |
•
|
Engineered Structural Characteristics: When
engaging cell surface targets, SMIP proteins are capable of
bringing together cell surface molecules in ways not always
possible with mAbs. The binding
|
4
|
|
|
| |
|
domains of SMIP product candidates have a different geometry
than the binding domains of mAbs — that is, the
binding domains are closer together. The structural format of
SMIP proteins permits engineering a range of distances between
the binding domains. SMIP proteins are also capable of binding
and neutralizing soluble molecules.
|
|
|
|
| |
•
|
Differentiated Product Candidates: SMIP
product candidates can be engineered to deliver desired cellular
signaling responses. These properties can be used to generate
biological responses not observed with mAbs. In addition, SMIP
proteins can be engineered to balance target signal induction,
complement-dependent cytotoxicity, or CDC and antibody-dependent
cellular cytotoxicity, or ADCC, mediated activity. The ability
to customize this balance of biological activities could result
in safer and more effective immunopharmaceuticals.
|
|
|
|
| |
•
|
Improved Biodistribution: SMIP product
candidates have a particle size that is approximately one-half
the size of mAbs. Smaller molecules have been demonstrated to
penetrate tissues more readily, a feature we believe will
provide increased therapeutic benefits.
|
| |
| |
•
|
Reliable Manufacturing: SMIP product
candidates can be produced at large scale in mammalian cell
expression systems from readily available materials.
|
| |
| |
•
|
Broad Therapeutic Application: SMIP product
candidates have potential application in diabetes, solid organ
transplant, oncology, and other high unmet need areas.
|
SCORPIONtm
Multispecific Protein Therapeutics
SCORPION therapeutics are a novel platform for the development
of multi-specific protein therapeutics. SCORPION therapeutics
are single chain proteins comprised of one binding domain, an
effector domain, and another binding domain. This proprietary
molecular class leverages Trubion’s
SMIPtm
product format by combining single-chain binding and effector
domain libraries, and adding additional binding domains. We
utilize human protein sequences selected for stability,
manufacturability, geometry of the binding domains, and low
immunogenicity.
5
SCORPION therapeutics offer several potential advantages:
|
|
|
| |
•
|
Dual Targeting: Scorpion constructs enable
simultaneous multi-valent engagement of two or more different
soluble or cell-surface targets, providing the capability for
differentiated signaling events.
|
| |
| |
•
|
Desirable Pharmacodynamic Properties: Scorpion
constructs retain immunoglobulin effector functions such as long
in vivo half-life and Fc-dependent cellular cytotoxicity
(FcDCC) activity, if desired.
|
| |
| |
•
|
Development of Multiple Product
Candidates: Scorpion technology provides for a
multitude of product candidates by utilizing binding domains in
a variety of target combinations .
|
| |
| |
•
|
Reliable Manufacturing: Scorpion constructs
can be produced as stable, homogeneous products with a standard
manufacturing profile.
|
| |
| |
•
|
Broad Therapeutic Application: Scorpion
therapeutics have applications in autoimmune and inflammatory
diseases, or AIID, transplant, oncology, and other high unmet
need areas.
|
TRU-ADhanCetm
Technology: Greater ADCC Potency
Our
TRU-ADhanCetm
technology enhances the antibody-dependent cellular
cytotoxicity, or ADCC, potency of immunopharmaceutical product
candidates. In contrast to existing approaches that impose
challenges on product development, Trubion has created a
proprietary manufacturing methodology with the following
advantages:
|
|
|
| |
•
|
TRU-ADhanCe requires no change to the amino acid sequence of a
product;
|
| |
| |
•
|
TRU-ADhanCe requires no change to a manufacturing cell
line; and
|
| |
| |
•
|
TRU-ADhanCe can be applied late in product development.
|
Our TRU-ADhanCe technology is capable of yielding a well defined
glycovariant product with ADCC via the addition of the
glycosidase inhibitor castanospermine during the cell-culture
stage of production. Although many other ADCC technologies
require genetic modifications to the producing cell line,
TRU-ADhanCe technology instead interferes with carbohydrate
maturation in production cell lines, yielding products with
enhanced ADCC.
Our
Product Candidates
Our current clinical product candidates target B cells. B cells
are important to the basic functioning of the body’s immune
system. In addition to producing antibodies that attack and kill
bacteria and viruses circulating within the body, they also help
recruit and coordinate other types of immune system cells to
perform specialized functions in the body’s fight against
disease and infection. When B cells fail to appropriately
distinguish the body’s own cells, tissues or organs from
foreign pathogens or proteins, the mistaken identification can
result in the B cells initiating an immune response against
healthy cells, which results in an autoimmune disease that can
lead to progressive disability. Autoimmune diseases include RA,
SLE, multiple sclerosis, type 1 diabetes, and Graves’
disease. As a group, autoimmune diseases are among the most
prevalent illnesses in the United States, affecting up to 5-8%
of the population, or up to 24 million people. In addition,
when B cells become malignant or otherwise multiply
uncontrollably, they can result in cancers known as lymphomas,
leukemias and myelomas.
6
The following table sets forth the development stages of our
product candidates:
TRU-015
We designed TRU-015 for a desired therapeutic label surrounding
B-cell depletion in multiple indications, including autoimmune
and inflammatory diseases and different types of cancer. TRU-015
binds to its target, CD20, and is engineered to promote specific
biological activity designed for optimized safety and efficacy.
Specifically, general systemic complement activation is thought
to initiate or exacerbate symptoms in RA patients. There is
evidence that CDC may be associated with certain side effects,
particularly intravenous infusion reactions observed in
currently marketed protein immunopharmaceuticals. We have
designed TRU-015 for reduced CDC activity, while preserving
potent ADCC activity and apoptotic signaling. In addition,
TRU-015’s smaller size may provide improved therapeutic
options for patients through more rapid diffusion to disease
sites.
Rheumatoid
Arthritis
Background. RA is an autoimmune disease
characterized by inflammation of the joint lining, called the
synovium. In RA, a person’s immune system attacks the
synovium, resulting in the thickening of the normally thin
membrane and degradation of the cartilage and bone at the joint.
Though the primary symptoms of RA are pain, stiffness and
swelling of joints, additional symptoms may include fatigue,
weakness, muscle pain, and lumps of tissue under the skin.
Tissue damage from the inflammation ultimately results in
deformity and disability.
Potential Market. According to Datamonitor, RA
is estimated to affect approximately 5.2 million people in
the United States, Japan and and the five major European
markets. In 2009 total reported worldwide sales of therapeutics
used for the treatment of RA were greater than $10 billion.
Notwithstanding the administration of currently available
treatments, approximately two-thirds of the RA patient
population experiences pain, stiffness and fatigue on a daily
basis. As a result, we believe there is a large unmet medical
need in the RA patient population for an effective drug therapy.
Current Treatments. Initially, a patient
presenting symptoms of RA is typically prescribed non-steroidal
anti-inflammatory drugs, or NSAIDS. As the disease progresses,
the RA patient may be prescribed a regimen of disease modifying
anti-rheumatic drugs, or DMARDS, an anti-tumor necrosis factor,
or anti-TNF, or other biologics. It is estimated that 20% of the
RA patient population takes a combination of therapies that
include biologics. Most biologics currently on the market for RA
attempt to block the activity of immune system cytokines, which
are chemical messengers thought to be associated with the
autoimmune reactions, joint
7
inflammation and bone damage characteristic of RA. These
biologics include anti-TNF drugs such as
Remicade®,
Enbrel®,
Humira,®
and
Cimzia®.
Biologics are typically administered to patients with moderate
to severe RA who need therapy in addition to NSAIDS or DMARDS.
In addition to biologics that target immune system cytokines
such as
Kineret®,
Orencia®,
a drug that targets co-receptors on T cells,
Actemra®,
which targets IL-6 receptors and
Rituxan®
that, like TRU-015, is targeted to the CD20 antigen, have been
approved for RA.
TRU-015 for RA Clinical Trial Results. A
second Phase 2b clinical trial (study 2203) is ongoing and
enrollment was completed in September 2009. The interim analysis
of the second Phase 2b clinical trial of TRU-015 in RA (study
2203) met the pre-defined primary endpoint demonstrating
superiority of TRU-015 over placebo. The study’s
pre-defined primary endpoint is ACR50 response at week 24. We
expect to receive final data from study 2203 in mid 2010.
In October 2009 we announced positive data from the second
course of re-treatment in the first Phase 2b clinical trial
(study 15002) for RA demonstrating that administration of
TRU-015 every 24 weeks produces well-tolerated results that
are comparable with the more than four and a half years of
re-treatment data compiled to date. Clinical disease activity
parameters such as tender and swollen joint counts, patient and
physician global assessments, patient assessment of pain and
disability, and laboratory measures of inflammation may be
combined to form composite measures of clinical response derived
from the American College of Rheumatology that are known as
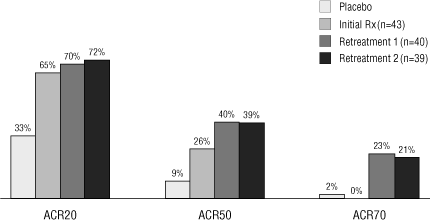
ACR20, ACR50, and ACR70. In these measures of clinical response,
ACR70 indicates a greater response from a baseline measure than
ACR20, which is defined as an improvement of at least 20% from
baseline in counts of both tender and swollen joints, as well as
in at least three of five other disease activity parameters. At
24 weeks after the second re-treatment course, subjects in
the group that had received 800 mg of TRU-015 in the
initial treatment and the first and second re-treatments
achieved ACR 20, 50 and 70 response rates of 72%, 39% and 21%,
respectively.
In November 2007 we presented initial results from our first
Phase 2b clinical trial of TRU-015 for the treatment of RA at
the ACR Annual Meeting. Data from the Phase 2b trial
demonstrated that TRU-015 provided statistically significant
efficacy after a single infusion of 800 mg or 1600 mg.
Our clinical program is designed to optimize dose and schedule
for maximal efficacy and patient convenience without resulting
in long term B-cell depletion.
American College of Rhuematology (ACR) Criteria for
Improvement
FDA Approved CD20-Directed Therapies in
RA. Rituxan®
is a mAb that is targeted to the CD20 antigen and was previously
approved for the treatment of NHL. In February 2006, it was
approved for marketing in the United States by the FDA, for the
treatment of patients with moderate to severe RA who have failed
one or more anti-TNF therapies. The recommended dose and
schedule for Rituxan in RA is two intravenous infusions of 1 gm
each separated by two weeks, in combination with continued
methotrexate (10 to 25 mg weekly). Patients given this
regimen show B-cell depletion for at least six months with some
showing B- cell depletion for over three years. There is no
recommended treatment for patients with symptomatic RA and
8
concomitant B-cell depletion. We believe that the dose-dependent
B-cell depletion shown by TRU-015 may allow us to choose a dose
and schedule that offers similar or greater efficacy while
improving safety as a result of a shorter period of B-cell
depletion. Additionally, the Rituxan -product label contains
warnings related to infusion reactions, including fatal infusion
reactions. We believe that the attenuated CDC activity of
TRU-015 relative to Rituxan may allow for safer infusion
protocols. TRU-015 is a smaller molecule than Rituxan and may
diffuse more rapidly to disease sites. We believe this
characteristic of TRU-015 may allow it to show greater efficacy
or more rapid onset of action in future studies.
TRU-015 for RA Ongoing Clinical
Development. Our partner, Pfizer, has fully
enrolled the second Phase 2b (study 2203) clinical trial
for TRU-015. The randomized, parallel, double-blind,
placebo-controlled, dose-regimen finding study will evaluate the
safety and efficacy of two dosing regimens administered to
patients with active seropositive rheumatoid factor, or RF, on a
background of methotrexate. This study has been designed in a
way that we believe could be supportive of a registration
package with the FDA. The interim analysis of the second Phase
2b clinical trial of TRU-015 in RA (study 2203) met the
pre-defined primary endpoint demonstrating superiority of
TRU-015 over placebo. The study’s pre-defined primary
endpoint is ACR50 response at week 24. We expect to receive
final data from study 2203 in mid 2010. Pfizer will determine
whether to commence a Phase 3 study of TRU-015 for the treatment
of RA after reviewing data from the ongoing Phase 2 SBI-087 RA
study, in addition to final data from the ongoing Phase 2b
TRU-015 (2203) RA study. Patient dosing in the Phase 2
SBI-087 RA study commenced in December 2009 and an interim data
review is planned that we believe could occur in late 2010 or
early 2011 and final data is anticipated at the end of 2011.
SBI-087
SBI-087 is our next generation, humanized, CD20-directed product
candidate for the treatment of RA, SLE, and other autoimmune and
inflammatory diseases. Pfizer is evaluating both intravenous and
subcutaneous formulations of SBI-087 in multiple clinical
studies. Preclinical studies conducted by Pfizer evaluated the
PK and PD of SBI-087 following a single intravenous dose.
Administration of SBI-087 in preclinical studies resulted in
dose-dependent B-lymphocyte depletion in peripheral blood and
lymphoid tissues that was more profound and sustained in
SBI-087-treated groups compared with rituximab. In collaboration
with us, our partner Pfizer has commenced two clinical studies
of SBI-087 for the treatment of RA including a Phase 2
randomized, placebo-controlled, double-blind, parallel-group,
outpatient dose regimen-finding study in which patient dosing
commenced in December 2009, with interim data review anticipated
to occur in late 2010/early 2011.
Rheumatoid
Arthritis
Background. As discussed with TRU-015, RA is
an autoimmune disease characterized by inflammation of the joint
lining, called the synovium. In RA, a person’s immune
system attacks the synovium, resulting in the thickening of the
normally thin membrane and degradation of the cartilage and bone
at the joint. Though the primary symptoms of RA are pain,
stiffness and swelling of joints, additional symptoms may
include fatigue, weakness, muscle pain, and lumps of tissue
under the skin. Tissue damage from the inflammation ultimately
results in deformity and disability.
SBI-087 for RA Ongoing Clinical
Development. Our partner Pfizer has completed a
Phase 1 study of SBI-087 for RA and a Phase 2 study of SBI-087
for RA has commenced and is ongoing. In addition, patient
recruitment is under way in an additional Phase 1 study of
SBI-087 for RA in Japan.
Systemic
Lupus Erythematosus
Background. SLE is a debilitating, chronic,
inflammatory autoimmune disease characterized by the presence of
auto-reactive antibodies. It can cause disease in the skin,
internal organs, and the nervous system. Some of the most common
symptoms include extreme fatigue, painful or swollen joints,
fever, skin rashes, and kidney problems.
9
SLE is a chronic condition with episodic periods of disease
activity, known as flares, and periods of remission. Currently,
there is no cure for SLE, and symptomatic treatment is used in
an effort to prevent flares or treat them when they occur. We
believe that B- cell-depletion therapy is a promising approach
toward a targeted therapy in SLE.
Potential Market. According to Datamonitor,
SLE is estimated to affect 236,000 people in the United
States. The prevalence of SLE varies significantly on a
country-by-country
basis and could be up to five times greater with expanding
disease definitions and increasing diagnosis. No new
pharmaceutical or biologic treatments have been approved for SLE
in over 40 years. We believe there is a large, unmet
medical need in the SLE patient population as SLE patients have
a death rate three times higher than that of the general
population despite the fact that most patients are young and
middle-aged individuals.
Current Treatment. No protein therapeutics
have been approved specifically for use in the treatment of SLE.
Current drug therapies are predominantly palliative in nature
and are targeted to the patient’s specific symptoms.
Different medications are used to treat specific manifestations
of SLE. Treatments include acetaminophen
and/or
NSAIDs, immunosuppressants such as methotrexate and
cylcophosphamide, corticosteroids such as methylprednisolone,
and antimalarials such as hydroxychloroquine.
SBI-087 for SLE Ongoing Clinical
Development. Our partner, Pfizer, is conducting a
Phase 1 clinical trial of SBI-087 in systemic lupus
erythematosus, or SLE, in which patient dosing has commenced and
recruitment is ongoing.
Commercialization
Rights
Our collaboration agreement with Pfizer includes a worldwide
licensing and commercialization agreement for the development of
TRU-015 and other therapies. We retain an option to co-promote
with Pfizer, on customary terms to be agreed, CD20-directed
therapies in the United States for niche indications. See
“Business — Our Strategic Collaboration with
Pfizer.”
TRU-016
Our TRU-016 program, in collaboration with Facet, is focused on
the development of a novel CD37-directed therapy for B-cell
malignancies, such as CLL and NHL. CD37 is a clinically
validated target for the treatment of B-cell malignancies and
our TRU-016 product candidate has been designed for a desired
therapeutic label surrounding B-cell depletion in these B-cell
malignancies. CD37 is found at high levels on B cells and at
lower levels on a subpopulation of T cells and myeloid cells.
Experiments suggest that CD37 plays an important role in B-cell
regulation. In addition, CD37 is known to be overexpressed in
patients with CLL. TRU-016 uses a different mechanism of action
than CD20-directed therapies. As a result, we believe its novel
design may provide patients with improved therapeutic options
and enhance efficacy when used alone or in combination with
chemotherapy
and/or other
CD20-directed therapeutics. We are currently conducting a Phase
1 clinical trial of TRU-016 in CLL and have filed an amendment
to include treatment of patients with NHL. Expansion of the
development program to other indications in oncology and AIID is
planned.
B-cell
Malignancies: Chronic Lymphocytic Leukemia and
Non-Hodgkin’s Lymphoma
Background. B cells and T cells are the two
major types of lymphocytes responsible for defending the body
against infection. Lymphocytic malignancies arise when these
cells multiply uncontrollably. CLL is a type of cancer affecting
the blood and bone marrow. It is a slowly progressing disease
and in most patients the abnormal proliferating lymphocytes are
clonal B cells arrested in the differentiation pathway between
pre B cells and mature B cells. NHL is a diverse group of
lymphocytic malignancies, approximately 85% of which are B-cell
malignancies.
Preclinical data has demonstrated that TRU-016 induces potent
ADCC against primary B-CLL cells, demonstrates significant in
vivo therapeutic efficacy, and induces potent apoptosis in
primary CLL cells. In addition, as shown below, combination
therapy with a CD37-directed
SMIPtm
product candidate and CD20-directed therapy with
Rituxan®
has shown greater preclinical efficacy than either therapy alone.
10
Potential Market. According to the National
Cancer Institute, CLL is estimated to affect approximately
70,000 people in the United States. Approximately 12,000
new cases of CLL are diagnosed each year in the United States
according to Datamonitor. In addition, NHL, another B-cell
malignancy, is one of the most common types of cancer accounting
for about 4% of all cancers. About 66,000 people in the
United States are expected to be diagnosed with NHL in 2009
according to the American Cancer Society. Total reported
worldwide sales of one of the most common used biologics in NHL,
Rituxan®
surpassed $3 billion in 2009.
Current Treatments. While available CLL and
NHL therapies include chemotherapy, radiation therapy, surgery
and bone and stem cell transplantation, biologics have become
the standard of care to treat these cancers. Biologic therapies
for NHL include interferon and mAbs such as
Rituxan®/Mabthera,
Bexxar®,
Zevalin®
and
Arzerra®.
These mAbs all target CD20 on B cells, and Bexxar and Zevalin
are radiolabeled. In addition,
Campath®
is a CD52-targeted mAb indicated for CLL, and
Treanda®,
a cytotoxic, is also indicated for CLL. FCR, a combination of
fludaraline, cyclophosphamide and rituximab is currently the
most effective combination for the treatment of CLL.
TRU-016 for CLL Ongoing Clinical
Development. A TRU-016 Phase 1 clinical trial for
patients with CLL is currently under way. The open label
clinical trial is composed of two parts- a dose escalation study
designed to evaluate the safety, tolerability and
pharmacokinetics of TRU-016; and an expansion cohort designed to
further evaluate safety and to estimate clinical activity of
TRU-016 in patients with previously treated CLL or small
lymphocytic leukemia.
TRU-016 for CLL Clinical Trial Results. In
December 2009, we announced positive data from a phase 1 study
of TRU-016 in patients with relapsed and refractory CLL.
Evidence of TRU-016 biological activity was seen beginning with
patients dosed at the 0.3 mg/kg dose level, including in
high-risk patients. Partial response was observed in five
patients, including one patient with the 17p deletion
cytogenetic abnormality. Partial response was determined
following investigator assessment and the two-month confirmation
of these responses is pending. Two patients with leukemia cutis
experienced clearing, one complete and one partial. At the
10 mg/kg dose, four of five patients with elevated
peripheral lymphocyte counts were reduced to normal levels. A
total of 16 serious adverse events have been reported. The MTD
has not yet been reached.
TRU-016
Phase 1 Clinical Results: Dose Response in Evaluable
Patients — Reduction in Peripheral
Lymphocytes
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
Normalized
|
|
Median
|
|
Best
|
|
Dose Cohort
|
|
N
|
|
Lymph Count
|
|
Reduction
|
|
Response
|
|
|
|
1 mg/kg
|
|
3
|
|
0/3 (0%)
|
|
67%
|
|
0
|
|
3 mg/kg
|
|
4
|
|
0/4 (0%)
|
|
78%
|
|
0
|
|
6 mg/kg
|
|
4
|
|
1/4 (25%)
|
|
85%
|
|
1 PR
|
|
10 mg/kg
|
|
5
|
|
4/5 (80%)
|
|
95%
|
|
2 PR
|
|
3 mg/kg TIW
|
|
4
|
|
1/4 (25%)
|
|
49%
|
|
1 PR
|
|
6 mg/kg TIW
|
|
4
|
|
1/1 (100%)
|
|
77%
|
|
1 PR
|
Strategic
Collaborations
Facet
In August 2009, we entered into a collaboration agreement with
Facet for the joint worldwide development and commercialization
of TRU-016, a product candidate in Phase 1 clinical development
for CLL. TRU-016 is a CD37-directed SMIP protein therapeutic.
The collaboration agreement includes TRU-016 in all indications
and all other CD37-directed protein therapeutics. Under the
terms of the collaboration agreement, the parties will not
develop or commercialize protein therapeutics directed to CD37
outside of the collaboration agreement.
We received an up-front payment of $20 million in cash in
September 2009 and may receive up to $176.5 million in
additional contingent payments upon the achievement of specified
development, regulatory
11
and sales milestones. We and Facet share equally the costs of
all development, commercialization and promotional activities
and all global operating profits. In connection with the
collaboration agreement, we and Facet also entered into a stock
purchase agreement, pursuant to which Facet purchased
2,243,649 shares of our common stock for an aggregate
purchase price of $10 million, or $4.46 per share. The per
share price of $4.46 represents a 35% equity premium over the
sixty-day
trading average of our common stock on NASDAQ for the trading
period ending immediately prior to the execution of the stock
purchase agreement.
With respect to control over decisions and responsibilities, the
collaboration agreement provides for a joint steering committee,
or JSC, consisting of representatives of Trubion and Facet,
which makes decisions by consensus. If the JSC is unable to
reach a consensus, then the matter will be referred to
Trubion’s and Facet’s chief executive officers for
resolution. If the chief executive officers are unable to
resolve the matter, then it will be resolved by arbitration.
Both Trubion and Facet, at their sole discretion, may
discontinue participation on the JSC with 90 days written
notice to the other party.
At predefined times, the parties have the right to opt-out of
the collaboration entirely or on a
product-by-product
basis. Upon a change of control of a party, the other party will
have the right to opt-out of the collaboration entirely and if
the successor party is conducting a program that competes with
the programs under the collaboration agreement, then the
successor party must either (i) opt-out of the
collaboration entirely or (ii) divest the competing program
to a third party. If a party exercises its opt-out right with
respect to a product, then the parties will no longer share
development and commercialization costs for such product and
such opting-out party will receive certain royalty payments upon
the sale of such product, rather than half of the profits
derived from such product. Even if Facet exercises its opt-out
right, its obligation to make milestone payments to us
continues. In addition, if the party that opts-out is the lead
manufacturing party for the opt-out product, then such party
must continue to supply the product to the continuing party for
up to eighteen months following the opt-out.
On March 9, 2010, Abbott Laboratories, or Abbott, announced
a definitive agreement to purchase Facet. Abbott further
announced that it expects the transaction to close in the second
quarter of 2010, subject to certain conditions. We intend to
continue to pursue the objectives in the approved development
plan.
Facet can terminate the collaboration agreement at any time, in
which event all rights to TRU-016 and other CD37-directed
protein therapeutics under the collaboration agreement would
revert to us. If Facet terminates the collaboration agreement in
the first 18 months, then Facet must pay us a
$10 million termination fee.
If there is a material breach of the collaboration agreement,
then the non-breaching party may either terminate the
collaboration agreement or continue the collaboration agreement
and force the breaching party to opt-out and accept royalties at
a reduced rate.
Either party may assign its interest in the collaboration
agreement to a third party, provided that the non-assigning
party maintains a right of first negotiation over any proposed
assignment. In addition, either we or Facet can freely assign
the collaboration agreement without the consent of the other
party in connection with certain specified change of control
transactions, such as an acquisition.
Pfizer
In December 2005 we entered into a collaboration agreement with
Wyeth, now a wholly-owned subsidiary of Pfizer, for the
development and worldwide commercialization of TRU-015 and other
CD20-directed therapeutics. Pursuant to the agreement, we are
also collaborating with Pfizer on the development and worldwide
commercialization of certain other product candidates directed
to a small number of targets other than CD20. During the period
in which we will be providing research services for Pfizer,
Pfizer has the right, subject to our reasonable consent, to
replace a limited number of these targets. In addition, we have
the option to co-promote with Pfizer, on customary terms to be
agreed, CD20-directed therapies in the United States for niche
indications. We retain the right to develop and commercialize,
on our own or with others, product candidates directed to all
targets not included within the agreement. Unless it is
terminated earlier, our agreement with Pfizer will remain in
effect on a
product-by-product
basis and on a
country-by-country
basis until the later of the date that any such product shall no
longer be covered by a valid claim of a U.S. or foreign
patent or application and, generally, ten years after the first
commercial sale of any product licensed under the agreement.
Pfizer may terminate the agreement without cause at any time
upon 90 days’ prior written notice.
12
In connection with the agreement, Wyeth paid us a
$40 million non-refundable, non-creditable, up-front fee in
January 2006 and purchased directly from us in a private
placement, concurrent with our initial public offering,
800,000 shares of our common stock at the initial public
offering price of $13.00 per share, resulting in net proceeds to
us of $10.4 million. Under the agreement, we provided
research services for an initial three-year period ended
December 22, 2008 with the option for Wyeth to extend the
service period for two additional one-year periods. Wyeth’s
financial obligations during the initial research service term
included collaborative research funding commitments of
$9.0 million in exchange for such committed research
services. This $9.0 million was subject to an increase if
the service period was extended beyond three years as well as
annual increases pursuant to percentage changes in the CPI. In
June 2008, Wyeth exercised the first option under the terms of
the agreement to extend the research period for an additional
one-year period through December 22, 2009. In June 2009,
Wyeth exercised the second option under the terms of the
agreement to extend the research period for an additional
one-year period through December 22, 2010. Due to the
research period extension in 2009, the collaboration research
funding commitments to us initially from Wyeth and now from
Pfizer, increased to approximately $3.3 million per year in
exchange for committed research services from us through
December 22, 2010.
Pfizer’s financial obligations include additional amounts
for reimbursement of
agreed-upon
external research and development costs and patent costs.
Pursuant to the agreement, Pfizer is also obligated to make
payments to us of up to $250 million based on the
achievement of specified regulatory and sales milestones for
CD20-directed therapies and payments to us of up to
$535 million based on the specified achievement of
regulatory and sales milestones for therapies directed to the
small number of targets other than CD20. In addition, we will
receive royalty payments in the event of future licensed product
sales. Pfizer may terminate the agreement without cause at any
time upon 90 days’ prior written notice.
In October 2009, Pfizer completed its acquisition of Wyeth. Our
collaboration agreement remains in effect with Pfizer and in
response to our request, Pfizer has provided further written
assurances reaffirming its commitment to comply with the terms
and conditions of the agreement.
If during the 12 month period following Pfizer’s
acquisition of Wyeth, Pfizer is required or voluntarily decides
to divest itself of one or more of the products under the
collaboration agreement, then subject to any governmental
limitations, Pfizer must offer us an exclusive opportunity to
negotiate the acquisition or license of all of Pfizer’s
rights to that product on commercially reasonable terms. If we
do not conclude an agreement with Pfizer covering the product,
Pfizer can divest itself of the product but the terms of that
divestiture cannot be more favorable than those that were last
offered to us unless we are given the opportunity to accept
those more favorable terms.
Upon a change of control of Trubion, the agreement would remain
in effect, subject to the right of Pfizer to terminate specified
provisions of the agreement.
Assuming TRU-015 and other product candidates under the
collaboration with Pfizer continue to progress in development,
expenses for future clinical trials may be higher than those
incurred in prior clinical trials. These expenses will, however,
likely be incurred by Pfizer and expenses incurred by us, if
any, will be substantially offset by reimbursement revenue from
Pfizer. In addition, Pfizer is responsible for a substantial
portion of costs related to patent prosecution and patent
litigation for products directed to targets selected by Pfizer
pursuant to the collaboration agreement.
Competition
The pharmaceutical and biotechnology industries are intensely
competitive, and any product candidate developed by us would
likely compete with other drugs and therapies. There are many
pharmaceutical companies, biotechnology companies, public and
private universities, government agencies, and research
organizations actively engaged in research and development of
products targeting the same markets as our product candidates.
Many of these organizations have substantially greater
financial, technical, manufacturing, marketing and personnel
resources than we have. Several of them have developed or are
developing therapies that could be used for treatment of the
same diseases that we are targeting. In addition, many of these
13
competitors have significantly greater commercial
infrastructures than we have. Our ability to compete
successfully will depend largely on our ability to:
|
|
|
| |
•
|
design and develop products that are superior to other products
in the market;
|
| |
| |
•
|
successfully collaborate with others in the design, development
and commercialization of new products;
|
| |
| |
•
|
attract and retain qualified scientific, medical, product
development, commercial and sales and marketing personnel;
|
| |
| |
•
|
obtain patent
and/or other
proprietary protection for our processes, product candidates and
technologies;
|
| |
| |
•
|
operate without infringing the patents and proprietary rights of
third parties; and
|
| |
| |
•
|
obtain required regulatory approvals.
|
We expect to compete on, among other things, product efficacy,
safety, convenience, time to market and price. In order to
compete successfully we will need to identify, secure the rights
to and develop products and exploit these products commercially
before others are able to develop competitive products. In
addition, our ability to compete may be affected if insurers and
other third-party payors seek to encourage the use of generic
products, making branded products less attractive to buyers from
a cost perspective.
We believe our product development programs will be subject to
significant competition from companies utilizing alternative
technologies. In addition, as the principles of our
SMIPtm
product candidates become more widely known and appreciated
based on patent and scientific publications and regulatory
filings, we expect the field to become highly competitive.
Pharmaceutical companies, biotechnology companies, and academic
and research institutions may succeed in developing products
based upon the principles underlying our proprietary
technologies earlier than us, obtaining approvals for such
products from the FDA more rapidly than us or developing
products that are safer, more effective,
and/or more
cost effective than those under development or proposed to be
developed by us.
Product Candidates for Autoimmune and Inflammatory
Diseases. If approved for the treatment of RA, we
anticipate that our product candidates would compete with other
marketed protein therapeutics for the treatment of RA in this
$10 billion market including:
Rituxan®
(Genentech, Roche and Biogen Idec),
Enbrel®
(Amgen and Pfizer),
Remicade®
(JNJ and Schering-Plough),
Humira®
(Abbott),
Orencia®
(BMS),
Cimzia®
(UCB),
Simponi®
(JNJ and Schering-Plough) and
Actemra®
(Roche and Chugai).
If approved for the treatment of SLE, we anticipate that our
product candidates would have to compete with other B-cell
depleting therapies, including CD20-directed therapeutics.
Product Candidates for B-cell Malignancies. If
approved for the treatment of CLL, NHL, or other B-cell
malignancies, we anticipate that our product candidates would
compete with other B-cell depleting therapies in these billion
dollar markets. Although we are not aware of any CD37-directed
therapeutics in development or on the market, for the treatment
of CLL, NHL, or other B-cell malignancies, other biologic
therapies are marketed for the treatment of NHL or CLL or both,
such as
Rituxan®
(Genentech),
Zevalin®
(Spectrum Pharmaceuticals, Inc. and Bayer Schering AG),
Bexxar®
(GSK),
Campath®
(Genzyme and Bayer Schering AG) and
Arzerra®
(GSK and Genmab).
Intellectual
Property
Because of the length of time and expense associated with
bringing new products through development and the governmental
approval process, pharmaceutical and biotechnology companies
have traditionally placed considerable importance on obtaining
and maintaining patent protection for significant new
technologies, products and processes.
We intend to seek patent protection for appropriate proprietary
technologies by filing patent applications when possible in the
United States and selected other jurisdictions. Our policy is to
seek patent protection for the inventions that we consider
important to the development of our business. We intend to
continue using our scientific expertise to pursue and file
patent applications on new developments with respect to uses,
methods,
14
and compositions to enhance our intellectual property position
in the areas that are important to the development of our
business. We have applied, and are applying for, patents
directed to our
SMIPtm
technology and product candidates, our
SCORPIONtm
technology and our
TRU-ADhanCetm
technology as well as other aspects of our technology both in
the United States and, when appropriate, in other jurisdictions.
Even if we are granted patents by government authorities or
obtain the right to utilize them through licensing, our patents
may not provide significant protection, competitive advantage or
commercial benefit. The validity and enforceability of patents
issued to pharmaceutical and biotechnology companies has proven
highly uncertain. For example, legal considerations surrounding
the validity of patents in the fields of pharmaceuticals and
biotechnology are in transition, and we cannot assure you that
the historical legal standards surrounding questions of validity
will continue to be applied or that current defenses relating to
issued patents in these fields will be sufficient in the future.
In addition, we cannot assure you as to the degree and range of
protections any of our patents, if issued, may afford us or
whether patents will be issued. For example, patents that may
issue to us may be subjected to further governmental review that
may ultimately result in the reduction of their scope of
protection, and pending patent applications may have their
requested breadth of protection significantly limited before
being issued, if issued at all. Further, since publication of
discoveries in scientific or patent literature often lags behind
actual discoveries, we cannot assure you that we were the first
creator of inventions covered by our pending patent
applications, or that we were the first to file patent
applications for these inventions.
Many pharmaceutical and biotechnology companies and university
and research institutions have filed patent applications or have
received patents in our areas of product development. Many of
these entities’ applications, patents and other
intellectual property rights could prevent us from obtaining
patents or could call into question the validity of any of our
patents, if issued, or could otherwise adversely affect the
ability to develop, manufacture or commercialize product
candidates. If use of technology incorporated into or used to
produce our product candidates is challenged, or if a
conflicting patent issued to others is upheld in the courts or
if a conflicting patent application filed by others is issued as
a patent and is upheld, we may be unable to market one or more
of our product candidates, or we may be required to obtain a
license to market those product candidates. To contend with
these possibilities, we may have to enter into license
agreements in the future with third parties for technologies
that may be useful or necessary for the manufacture or
commercialization of some of our product candidates. In
addition, we are routinely in discussions with academic and
commercial entities that hold patents on technology or processes
that we may find necessary in order to engage in some of our
activities. We cannot, however, assure you that these licenses,
or any others that we may be required to obtain to market our
product candidates, will be available on commercially reasonable
terms, if at all, or that we will be able to develop alternative
technologies if we cannot obtain required licenses.
To protect our rights to any of our patents, if issued, and
proprietary information, we may need to litigate against
infringing third parties, or otherwise avail ourselves of the
courts or participate in administrative proceedings to determine
the scope and validity of those patents or other proprietary
rights. These types of proceedings are often costly and could be
very time-consuming to us, and we cannot assure you that the
deciding authorities will rule in our favor. An unfavorable
decision could allow third parties to use our technology without
being required to pay us licensing fees or may compel us to
license needed technologies to avoid infringing third-party
patent and proprietary rights. Although we believe we would have
valid defenses to allegations that our current product
candidates, production methods and other activities infringe the
valid and enforceable intellectual property rights of any third
parties, we cannot be certain that a third party will not
challenge our position in the future. Even if some of these
activities were found to infringe a third party’s patent
rights, we may be found to be exempt from infringement under
35 U.S.C. § 271(e) to the extent that these are
found to be pre-commercialization activities related to our
seeking regulatory approval for a product candidate. The scope
of protection under 35 U.S.C. § 271(e), however,
is uncertain and we cannot assure you that any defense under
35 U.S.C. § 271(e) would be successful. Further,
the defense under 35 U.S.C. § 271(e) is only
available for pre-commercialization activities, and could not be
used as a defense for sale and marketing of any of our product
candidates. There has been, and we believe that there will
continue to be,
15
significant litigation in the biopharmaceutical and
pharmaceutical industries regarding patent and other
intellectual property rights.
Third parties could bring legal actions against us claiming we
infringe their patents or proprietary rights, and seek monetary
damages
and/or
enjoin clinical testing, manufacturing and marketing of the
affected product or products. If we become involved in any
litigation, it could consume a substantial portion of our
resources, and cause a significant diversion of effort by our
technical and management personnel regardless of the outcome of
the litigation. If any of these actions were successful, in
addition to any potential liability for damages, we could be
required to obtain a license to continue to manufacture or
market the affected product, in which case we may be required to
pay substantial royalties or grant cross-licenses to our
patents. We cannot, however, assure you that any such license
will be available on acceptable terms, if at all. Ultimately, we
could be prevented from commercializing a product, or forced to
cease some aspect of our business operations as a result of
claims of patent infringement or violation of other intellectual
property rights, which could have a material and adverse effect
on our business, financial condition, and results of operations.
Further, the outcome of intellectual property litigation is
subject to uncertainties that cannot be adequately quantified in
advance, including the demeanor and credibility of witnesses and
the identity of the adverse party. This is especially true in
intellectual property cases that may turn on the testimony of
experts as to technical facts upon which experts may reasonably
disagree.
While we pursue patent protection and enforcement of our product
candidates and aspects of our technologies when appropriate, we
also rely on trade secrets, know-how and continuing
technological advancement to develop and maintain our
competitive position. To protect this competitive position, we
regularly enter into confidentiality and proprietary information
agreements with third parties, including employees, suppliers
and collaborators. Our employment policy requires each new
employee to enter into an agreement that contains provisions
generally prohibiting the disclosure of confidential information
to anyone outside of Trubion and providing that any invention
conceived by an employee within the scope of his or her
employment duties is our exclusive property. Furthermore, our
know-how that is accessed by third parties through
collaborations and research and development contracts and
through our relationships with scientific consultants is
generally protected through confidentiality agreements with the
appropriate parties. We cannot, however, assure you these
protective arrangements will be honored by third parties,
including employees, suppliers, and collaborators, or that these
arrangements will effectively protect our rights relating to
unpatented proprietary information, trade secrets and know-how.
In addition, we cannot assure you that other parties will not
independently develop substantially equivalent proprietary
information and techniques or otherwise gain access to our
proprietary information and technologies.
We are aware of intellectual property, including European patent
No. EP-B-1176981,
in which Genentech has an ownership interest with claims
directed to the second medical use of an anti-CD20 antibody for
treatment of RA. On August 8, 2006, we filed an opposition
to this patent raising objections as to its validity. Subsequent
to the submission of our opposition, other parties filed
oppositions to the Genentech patent prior to August 30,
2006, including MedImmune, Inc., Genmab A/S, Centocor, Inc.,
Glaxo Group Limited, Serono S.A, and Pfizer. On
September 11, 2008 we announced that the Opposition
Division, or OD, of the EPO had revoked the European patent in
its entirety. On February 19, 2009, Genentech and Biogen
Idec appealed the decision. Final resolution of the opposition
proceedings will likely take a number of years. In addition to
its opposition, Glaxo Group Limited filed an action with the
United Kingdom High Court to revoke the U.K. counterpart of
EP-B-1176981. Pfizer also initiated a revocation action. On
May 19, 2008 the U.K. counterpart was revoked by Court
order.
The Genentech European patent claims the benefit of priority to
two U.S. provisional patent applications that are
unpublished and the status of which will remain confidential
unless a U.S. patent or patent application claiming
priority to the provisional patent applications publishes. In
the event any such corresponding U.S. patent issues, and if
our activities are determined to be covered by such a patent, we
cannot assure you that Genentech would be willing to grant us or
Pfizer a license on terms we or they would consider commercially
reasonable, if at all, which could prevent us from manufacturing
and marketing TRU-015 for the treatment of RA in the United
States and have a material adverse effect on our business,
financial condition,
16
operating results and our collaboration with Pfizer. The
Genentech patent has also been applied for in other countries
including Japan, where the application is pending.
Manufacturing
We do not currently own or operate manufacturing facilities for
the production of clinical or commercial quantities of our
product candidates. We currently rely on a small number of
third-party manufacturers to produce our compounds and expect to
continue to do so to meet the clinical requirements of our
product candidates and for all of our commercial needs. Our
product candidates are currently manufactured in mammalian cell
expression systems from readily available starting materials. To
the extent that TRU-015 and TRU-016 advance through clinical
trials, and to the extent we bring our future product candidates
into clinical trials and partner the development and
commercialization of any of the product candidates, we and our
existing and prospective partners will be required to assess the
manufacturing needs of the product candidates for clinical
requirements as well as for commercial production. We may need
to obtain one or more licenses to intellectual property rights
held by third parties in order to manufacture each of our
product candidates. While such licenses may be available, they
may not be available on terms that are commercially acceptable
to our existing or prospective partners or us. Should such
licenses prove unavailable, we or our existing or prospective
partners may choose to modify our manufacturing processes to use
alternative manufacturing methods. Such modifications may result
in greater expenditures of capital by us or our partners, delay
commercialization, or prevent us or our partners from
successfully commercializing our product candidates.
We have multiple potential sources for manufacturing our product
candidates. Pfizer manufactures TRU-015 and has significant
process development capabilities and extensive commercial-scale
production capabilities at numerous facilities worldwide.
Pfizer’s manufacturing commitment is contingent upon the
effectiveness of our collaboration agreement which they may
terminate without cause at any time upon 90 days’
prior written notice. However, in the event we or Pfizer
terminate our collaboration agreement for certain reasons
specified in the collaboration agreement, Pfizer would have
limited manufacturing obligations to us. In addition to Pfizer,
we have entered into agreements with Lonza Biologics and related
entities for certain license rights related to Lonza’s
manufacturing technology, research and development services, and
for the manufacture of TRU-015 as well as other product
candidates. As of December 31, 2009, we had committed to
purchase $2.1 million of manufacturing services for TRU-016
from Lonza in 2010. Under our manufacturing agreement with
Lonza, we could incur cancellation fees if we cancel the
production run with insufficient advance notice. Our commitments
with Lonza for manufacturing expire in the second quarter of
2010 and we currently do not have any future manufacturing
agreements at this time. We plan on negotiating additional
manufacturing capacity with Lonza or other third party
manufacturers during 2010.
We rely and expect to continue to rely on a number of contract
manufacturers to produce sufficient quantities of our product
candidates in accordance with current good manufacturing
practices, or cGMP, for use in clinical trials. We will
ultimately depend on contract manufacturers for the manufacture
of our products for commercial sale. Contract manufacturers are
subject to extensive government regulation.
Government
Regulation
Government authorities in the United States at the federal,
state and local level, and other countries, extensively
regulate, among other things, the research, development,
testing, manufacture, labeling, promotion, advertising,
distribution, marketing, and export and import of
immunopharmaceutical products such as those we are developing.
United
States Government Regulation
In the United States the information that must be submitted to
the FDA in order to obtain approval to market a new drug varies
depending on whether the drug is a new product whose safety and
effectiveness has not previously been demonstrated in humans or
a drug whose active ingredient(s) and certain other properties
are the same as those of a previously approved drug. A new
biologic will follow the Biologics License Application, or BLA,
route for approval, a new drug will follow the New Drug
Application, or NDA, route for
17
approval, and a drug that claims to be the same as an already
approved drug may be able to follow the Abbreviated New Drug
Application route for approval.
BLA
and NDA Approval Process
In the United States, the FDA regulates drugs and biologics
under the Federal Food, Drug and Cosmetic Act, and, in the case
of biologics, also under the Public Health Service Act, and the
FDA’s implementing regulations. If we fail to comply with
the applicable U.S. requirements at any time during the
product development process, approval process or after approval,
we may become subject to administrative or judicial sanctions.
These sanctions could include the FDA’s refusal to approve
pending applications, license suspension or revocation,
withdrawal of an approval, a clinical hold, warning letters,
product recalls, product seizures, total or partial suspension
of production or distribution, injunctions, fines, civil
penalties or criminal prosecution. Any agency or judicial
enforcement action could have a material adverse effect on us.
The major steps required before a biologic drug may be marketed
in the United States include:
|
|
|
| |
•
|
completion of laboratory tests and animal studies under the
FDA’s good laboratory practices regulations;
|
| |
| |
•
|
submission to the FDA of an IND for human clinical testing,
which must become effective before human clinical trials may
begin;
|
| |
| |
•
|
performance of adequate and well-controlled clinical trials to
establish the safety and efficacy of the product for each
indication;
|
| |
| |
•
|
submission to the FDA of a BLA or NDA, which includes the
results of all required preclinical animal studies, laboratory
tests, clinical trials, and data relating to the product’s
pharmacology, chemistry, manufacture, and control;
|
| |
| |
•
|
satisfactory completion of an FDA inspection of the
manufacturing facility or facilities at which the product is
produced to assess compliance with cGMP; and
|
| |
| |
•
|
FDA review and approval of the BLA or NDA.
|
Preclinical tests include laboratory evaluations of product
chemistry, toxicity and formulation, as well as animal studies.
An IND sponsor must submit the results of the preclinical tests,
together with manufacturing information and analytical data, to
the FDA as part of the IND. Long term preclinical tests, such as
animal tests for reproductive toxicity and carcinogenicity, may
continue after the IND is submitted. The IND must become
effective before human clinical trials may begin. An IND will
automatically become effective 30 days after receipt by the
FDA, unless before that time the FDA raises concerns or
questions about issues such as the conduct of the trials as
outlined in the IND. In that case, the IND sponsor and the FDA
must resolve any outstanding FDA concerns or questions before
clinical trials can proceed. Submission of an IND does not
guarantee that the FDA will allow clinical trials to commence.
The FDA may order the temporary or permanent discontinuation of
a clinical trial at any time or impose other sanctions if it
believes that the clinical trial is not being conducted in
accordance with FDA requirements or presents an unacceptable
risk to the study subjects.
Clinical trials involve the administration of the
investigational product to human subjects under the supervision
of qualified investigators. Clinical trials must be conducted in
compliance with federal regulations, good clinical practices, or
GCPs, and under protocols detailing, among other things, the
objectives of the study, the parameters to be used in monitoring
safety, and the effectiveness criteria to be evaluated. Each
clinical protocol must be submitted to the FDA as part of the
IND.
Clinical trials typically are conducted in three sequential
phases, but the phases may overlap or be combined. Each trial
must be reviewed and approved by an independent institutional
review board, or IRB, before it can begin at that site. An IRB
may require the clinical trial be halted, either temporarily or
permanently, for failure to comply with the IRB’s
requirements, or may impose other conditions.
18
Phase 1 clinical trials usually involve the initial introduction
of the investigational drug into humans to evaluate the
product’s safety, dosage tolerance and pharmacodynamics
and, if possible, to gain an early indication of its efficacy.
Phase 2 clinical trials usually involve controlled trials in a
limited patient population to:
|
|
|
| |
•
|
evaluate dosage tolerance and appropriate dosage;
|
| |
| |
•
|
identify possible adverse effects and safety risks; and
|
| |
| |
•
|
evaluate preliminarily the efficacy of the drug for specific
indications.
|
Phase 3 clinical trials usually further evaluate clinical
efficacy and further test for safety in an expanded patient
population. Phase 1, Phase 2 and Phase 3 trials may not be
completed successfully within any specified period, if at all.
The FDA or we, or our partners may suspend or terminate clinical
trials at any time on various grounds, including a finding that
the subjects or patients are being exposed to an unacceptable
health risk. Prior to conducting Phase 3 trials, an applicant
may seek a special protocol assessment which is an agreement
between an applicant and the FDA on the design and size of
clinical trial(s) that is/are intended to form the basis of a
BLA or NDA.
Assuming successful completion of the required clinical trials,
the results of the preclinical studies and of the clinical
studies, together with other detailed information, including
information on the chemistry, manufacture, and control criteria
of the product, are submitted to the FDA in the form of a BLA or
NDA requesting approval to market the product for one or more
indications. The FDA reviews a BLA or NDA to determine, among
other things, whether the product is safe, pure, and potent and
whether the facility in which it is manufactured, processed,
packed, or held meets standards designed to assure the
product’s continued safety, purity and potency. The FDA
also reviews a BLA or NDA to determine whether a product is safe
and effective for its intended use.
Before approving an application, the FDA will inspect the
facility or the facilities at which the product is manufactured.
The FDA will not approve the product unless cGMP compliance is
satisfactory. If the FDA determines the application,
manufacturing process, or manufacturing facilities are not
acceptable, it will outline the deficiencies in the submission
and often will request additional testing or information.
Notwithstanding the submission of any requested additional
information, the FDA ultimately may decide that the application
does not satisfy the regulatory criteria for approval. Before
approving a BLA or NDA, the FDA will also typically inspect one
or more clinical sites to assure compliance with GCP.
The testing and approval process requires substantial time,
effort and financial resources, and each may take several years
to complete. The FDA may not grant approval on a timely basis,
if at all. We may encounter difficulties or unanticipated costs
in our efforts to secure necessary governmental approvals, which
could delay or preclude us from marketing our product
candidates. The FDA may limit the indications for use or place
other conditions on any approvals that could restrict the
commercial application of our product candidates. After
approval, some types of changes to the approved product, such as
adding new indications, manufacturing changes and additional
labeling claims, are subject to further FDA review and approval.
Priority
Review
The FDA has established priority and standard review
classifications for original BLAs and NDAs and efficacy
supplements. The classification of an application indicates the
anticipated time frame for FDA review of completed marketing
applications. The classification system, which does not preclude
the FDA from doing work on other projects, provides a way of
prioritizing certain BLAs and NDAs upon receipt and throughout
the FDA application review process.
Under FDA policies, a biologic or drug candidate is eligible for
priority review, or review within a six-month time frame from
the time a complete BLA or NDA, as applicable, is accepted for
filing, if the drug candidate provides a significant improvement
compared to marketed drugs in the treatment, diagnosis or
prevention of a disease. Even if a BLA or NDA is initially
classified as a priority application, this status can change
during the FDA review process, such as in the situation where
another product is approved for the
19
same disease for which previously there was no available
therapy. In addition, priority review does not guarantee that a
product candidate will receive regulatory approval.
Post-Approval
Requirements
After regulatory approval of a product is obtained, we would be
required to comply with a number of post-approval requirements.
For example, as a condition of approval of a BLA or NDA, the FDA
may require post-marketing clinical studies and surveillance to
monitor the product’s safety or efficacy.
In addition, holders of an approved BLA or NDA are required to
report certain adverse reactions and production problems to the
FDA, to provide updated safety and efficacy information and to
comply with requirements concerning advertising and promotional
labeling for their products. Drugs may be marketed only for the
approved indications and in accordance with the provisions of
the approved labeling. Changes to some of the conditions
established in an approved application, including changes in
indications, labeling, or manufacturing processes or facilities,
require submission and FDA approval of a new BLA/NDA or BLA/NDA
supplement before the change can be implemented. A BLA/NDA
supplement for a new indication typically requires clinical data
similar to that in the original application, and the FDA uses
the same procedures and actions in reviewing BLA/NDA supplements
as it does in reviewing BLAs/NDAs.
Also, quality control and manufacturing procedures must continue
to conform to cGMP after approval. The FDA periodically inspects
manufacturing facilities to assess compliance with cGMP, which
imposes certain procedural, substantive and recordkeeping
requirements. Accordingly, biologics and drug companies and
their manufacturers must continue to expend time, money, and
effort in the area of production and quality control to maintain
compliance with cGMP and other aspects of regulatory compliance.
Foreign
Regulation
In addition to regulations in the United States, we will be
subject to a variety of foreign regulations governing clinical
trials and commercial sales and distribution of our product
candidates. Whether or not we obtain FDA approval for a product
candidate, we must obtain approval by the comparable regulatory
authorities of foreign countries before we can commence clinical
trials or marketing of the product candidate in those countries.
The approval process varies from country to country, and the
time may be longer or shorter than that required for FDA
approval. The requirements governing the conduct of clinical
trials, product licensing, pricing and reimbursement vary
greatly from country to country.
Under European Union regulatory systems, a marketing
authorization for a medical product derived from biotechnology
processes must be submitted under a centralized procedure. The
centralized procedure provides for the grant of a single
marketing authorization that is valid for all European Union
member states
Reimbursement
Sales of biopharmaceutical products depend in significant part
on the availability of third-party reimbursement. Each
third-party payor may have its own policy regarding what
products it will cover, the conditions under which it will cover
such products, and how much it will pay for such products. It
will be time consuming and expensive for us to seek
reimbursement from third-party payors. Reimbursement may not be
available or sufficient to allow us to sell our products on a
competitive and profitable basis.
The passage of the Medicare Prescription Drug and Modernization
Act of 2003, or the MMA, sets forth the requirements for the
distribution and pricing of prescription drugs for Medicare
beneficiaries, which may affect the marketing of our products.
The MMA also introduced a new reimbursement methodology.
Moreover, while the MMA applies only to drug benefits for
Medicare beneficiaries, private payors often follow Medicare
coverage policy and payment limitations in setting their own
payment rates. Any reduction in payment that results from the
MMA may result in a similar reduction in payments from
non-governmental payors.
In addition, in some foreign countries, the proposed pricing for
a drug must be approved before it may be lawfully marketed. The
requirements governing drug pricing vary widely from country to
country. For example, the European Union provides options for
its member states to restrict the range of medicinal products
20
for which their national health insurance systems provide
reimbursement and to control the prices of medicinal products
for human use. A member state may approve a specific price for
the medicinal product or it may instead adopt a system of direct
or indirect controls on the profitability of the company placing
the medicinal product on the market.
We expect there will continue to be a number of federal and
state proposals to implement governmental pricing controls.
While we cannot predict whether such legislative or regulatory
proposals will be adopted, the adoption of such proposals could
have a material adverse effect on our business, financial
condition, and profitability.
Employees
As of December 31, 2009, we had 73 full-time
employees, 18 of whom held Ph.D. or M.D. degrees and 57 of whom
were engaged in full-time research and development activities.
None of our employees is represented by a labor union and we
consider our employee relations to be good.
Available
Information
Our corporate website address is www.trubion.com. We make
available free of charge on our website our annual, quarterly
and current reports as soon as reasonably practicable after we
electronically file such material with, or furnish it to, the
SEC. These SEC reports can be accessed through the
“Investors” section of our website. We also make
available on our website our corporate governance guidelines,
the charters for our audit committee, compensation committee,
and nominating and corporate governance committee, our
whistleblower and corporate communications policies and our code
of business conduct and ethics, and such information is
available in print to any stockholder of Trubion who requests
it. In addition, we intend to disclose on our website any
amendments to, or waivers from, our code of business conduct and
ethics that are required to be publicly disclosed pursuant to
rules of the SEC and The Nasdaq Global Market. The information
found on our corporate website is not, however, part of this or
any other report.
We were founded as a limited liability company in the state of
Washington in March 1999, and operated as a development-stage
company. We converted into a corporation and redomiciled in the
state of Delaware in October 2002.
Investment in our common stock involves a high degree of risk
and uncertainty. You should carefully consider the risks
described below together with all of the other information
included in this annual report on
Form 10-K
as well as our quarterly reports on
Form 10-Q
and current reports on
Form 8-K.
The risks and uncertainties described below are not the only
ones facing us. If any of the following risks actually occurs,
our business, financial condition, or operating results could be
harmed. In such case, the trading price of our common stock
could decline, and investors in our common stock could lose all
or part of their investment.
Risks
Related to Our Business
Our
success depends on the success of our clinical product
candidates, TRU-015, TRU-016, and SBI-087, and we cannot be
certain that they will be safe or effective, complete clinical
trials, receive regulatory approval or be successfully
commercialized.
Although our lead product candidate, TRU-015, has completed a
Phase 2b clinical trial for the treatment of RA, additional
clinical trials would be required before we are able to submit a
Biologic License Application, or BLA, to the FDA for approval.
In addition, our Facet collaboration clinical candidate,
TRU-016, and our Pfizer collaboration clinical candidate,
SBI-087, commenced initial clinical testing in 2008 and even if
we and Facet, in the case of TRU-016, or Pfizer, in the case of
SBI-087, determine to proceed with further clinical testing, a
number of additional clinical trials will be required before a
BLA can be submitted to the FDA for product approval.
21
The regulatory approval process can take many years and require
the expenditure of substantial resources. We are a party to a
collaboration agreement with Pfizer pursuant to which Pfizer is
responsible for regulatory approval of, and any subsequent
commercialization of TRU-015 and SBI-087. Ultimate
decision-making authority as to most matters within the
collaboration, including development plans and timeline, is
vested with Pfizer. Pfizer may not advance the development and
commercialization of TRU-015 and SBI-087, or either of these
product candidates as quickly as we would like, if at all. For
example, Pfizer will determine whether to commence a Phase 3
study of TRU-015 for the treatment of RA after reviewing data
from the ongoing Phase 2 SBI-087 RA study, in addition to final
data from the ongoing Phase 2b TRU-015 (2203) RA study.
Patient dosing in the Phase 2 SBI-087 RA study commenced in
December 2009 and an interim data review is planned that we
believe could occur in late 2010 or early 2011 and final data is
anticipated at the end of 2011.
In addition, prior to its acquisition by Pfizer, Wyeth had
determined not to pursue TRU-015 for any oncology indications
and discontinued the TRU-015 Phase
1/2
clinical trial for the treatment of non-Hodgkins’ lymphoma,
or NHL, that it had initiated in December 2007. We are also
party to a collaboration agreement with Facet pursuant to which
we and Facet must jointly agree to all development and
commercialization plans and timelines for TRU-016. Acting
jointly, we and Facet may be unable to advance the development
and commercialization of TRU-016 as quickly as we would, acting
alone.
Clinical trials required for FDA approval of TRU-015 for RA,
SBI-087 for RA or SLE or TRU-016 for CLL and NHL may not be
successfully completed. If required clinical trials are not
completed or their results do not meet safety and efficacy
thresholds required by the FDA, our product candidates will
likely not receive regulatory approval. Even if any of these
product candidates receive regulatory approval, the approved
product candidate may never be successfully commercialized. If
our product candidates do not receive regulatory approval or are
not successfully commercialized, we may not be able to generate
revenue, or become profitable, which would negatively affect our
ability to continue operations.
If we
fail to obtain the capital necessary to fund our operations, we
may be unable to develop our product candidates and we could be
forced to share our rights to these product candidates with
third parties on terms that may not be favorable to
us.
We need large amounts of capital to support our research and
development efforts. We may seek to raise funds through
additional strategic partnerships, by selling additional equity
or debt securities, or both, or by incurring other indebtedness.
If we are unable to raise additional capital in sufficient
amounts or on terms acceptable to us, we will be prevented from
pursuing research and development efforts or we may instead
elect to enter into collaborations that could require us to
share rights to our product candidates to a greater extent than
we currently intend, which could harm our business prospects and
financial condition. The sale of additional equity or
equity-linked securities could result in the issuance of
additional shares of our capital stock and could result in
dilution to our stockholders. The incurrence of indebtedness
would result in increased fixed payment obligations and could
also result in certain restrictive covenants, such as
limitations on our ability to incur additional debt, limitations
on our ability to acquire or license intellectual property
rights, and other operating restrictions that could adversely
impact our ability to conduct our business. The capital markets
have been experiencing disruption for more than 16 months.
The scope and extent of this disruption in the capital markets
could make it difficult or impossible to raise additional
capital in public or private capital markets until conditions
stabilize and appetite for additional investment grows. Whether
or not the capital markets stabilize, future financing may not
be available in sufficient amounts or on terms acceptable to us,
if at all.
We
have incurred operating losses in each year since our inception
and expect to continue to incur substantial and increasing
losses for the foreseeable future.
We have been engaged in designing and developing compounds and
product candidates since 1999 and have not generated any product
revenue to date. Our net losses were $29.2 million,
$25.6 million and $23.3 million in the years ended
December 31, 2009, 2008 and 2007, respectively. As of
December 31, 2009, we had an accumulated deficit of
$121.7 million. We expect our research and development
expenses to increase in the future due to increased
manufacturing and clinical development costs primarily related
to TRU-
22
016, our Facet collaboration clinical candidate, as well as the
advancement of our preclinical programs, and to product
candidate manufacturing costs. As a result, we expect to
continue to incur substantial and increasing losses for the
foreseeable future. We are uncertain when or if we will be able
to achieve or sustain profitability. Failure to become and
remain profitable would adversely affect the price of our common
stock and our ability to raise capital and continue operations.
Continued operating losses and depletion of our cash balance may
also result in non-compliance with our existing debt covenants
and may require us to dedicate a substantial portion of our cash
to repay our debt. As of December 31, 2009, our outstanding
indebtedness under agreements with financial debt covenants that
could be affected by continued operating losses or our cash
position totaled $8.3 million. In addition, our net
operating loss carry forwards and credits were substantially
exhausted as a result of the payments we received from Wyeth in
January 2006 pursuant to our Pfizer collaboration agreement, and
any remaining net operating loss carry forwards and credits may
be subject to an annual limitation due to the “change in
ownership” provisions of the Internal Revenue Code of 1986,
as amended, and similar state law provisions, which would have
an adverse effect on our ability to reduce future tax expenses.
We
depend on our collaborative relationship with Pfizer to develop,
manufacture, and commercialize TRU-015, SBI-087, and other
selected product candidates.
In October 2009, Pfizer completed its acquisition of Wyeth and
Pfizer is now our collaboration partner for TRU-015 and SBI-087.
We have no prior relationship with Pfizer and, as a result, we
cannot predict how or whether Pfizer will proceed with the
collaboration or the development of any of the collaboration
product candidates. In addition to our collaboration agreement
with Pfizer for the development and worldwide commercialization
of TRU-015, SBI-087 and other therapeutics directed to CD20, we
are also collaborating with Pfizer on the development and
worldwide commercialization of certain other product candidates
directed to a small number of targets other than CD20 that have
been established pursuant to the agreement. Our ability to
receive any significant revenue from our product candidates
covered by the collaboration agreement depends on the efforts of
Pfizer and on our ability to collaborate effectively. Any future
payments, including royalties to us, will depend on the extent
to which we and Pfizer advance product candidates through
development and commercialization. Pfizer may terminate the
collaboration relationship, in whole or in part, without cause,
by giving 90 days’ written notice to us. Pfizer also
has the right to terminate the agreement, on a
target-by-target
basis, upon 60 days’ written notice, if any safety or
regulatory issue arises that would have a material adverse
effect on Pfizer’s ability to develop, manufacture, or
commercialize one or more product candidates.
With respect to control over decisions and responsibilities, the
collaboration agreement provides for a research committee and a
CD20-directed therapy development committee consisting of
representatives of Pfizer and us. Ultimate decision-making
authority as to most matters within the collaboration, including
development plans and timelines, however, is vested in Pfizer.
Pfizer has the right to develop multiple product candidates
against the targets licensed to it under our collaboration.
Pfizer has begun clinical development of SBI-087, another
CD20-directed therapy, for RA and is currently enrolling
patients in a phase 1 trial for the evaluation of SBI-087 for
SLE. Pfizer could decide to focus its resources on only one
CD-20-directed therapy and, as a result, discontinue efforts to
further develop TRU-015. For example, Pfizer will determine
whether to commence a Phase 3 study of TRU-015 for the treatment
of RA after reviewing data from the ongoing Phase 2 SBI-087 RA
study, in addition to final data from the ongoing Phase 2b
TRU-015 (2203) RA study. SBI-087 is at an earlier stage in
clinical development than TRU-015 and a decision by Pfizer to
develop SBI-087 instead of TRU-015 would therefore likely delay
the potential commercialization of any product under our
collaboration with Pfizer, which could adversely affect our
business and cause the price of our common stock to decline.
Pfizer may not develop and commercialize our product candidates
as quickly as we would like, if at all. If Pfizer terminates the
agreement or fails to fulfill its obligations under the
agreement, we would need to obtain the capital necessary to fund
the development and commercialization of our product candidates
or enter into alternative arrangements with a third party. We
could also become involved in disputes with Pfizer, which could
lead to delays in or termination of our development and
commercialization programs and time-consuming and expensive
litigation or arbitration. If Pfizer terminates or breaches its
agreement with us, or
23
otherwise fails to complete its obligations in a timely manner,
our collaboration product development programs would be
substantially delayed and the chances of successfully developing
or commercializing our collaboration product candidates would be
materially and adversely affected.
We
depend on our collaborative relationship with Facet to develop,
manufacture, and commercialize TRU-016 and other CD37-directed
protein therapeutics.
In August 2009, we entered into a collaboration agreement with
Facet for the joint worldwide development and commercialization
of TRU-016, our product candidate in Phase 1 clinical
development for chronic lymphocytic leukemia, or CLL, and other
CD37-directed protein therapeutics. Under the terms of the
collaboration agreement, neither we nor Facet have the right to
develop or commercialize protein therapeutics directed to CD37
outside of the collaboration.
Our ability to receive funding for TRU-016 under the
collaboration depends on our ability to collaborate effectively
with Facet. Any future payments, including milestones payable to
us, will depend on the extent to which we and Facet advance
TRU-016 through development and commercialization. Facet may
terminate the collaboration agreement without cause, and would
not be obligated to pay us a termination fee if such a
termination was more than 18 months after the beginning of
the collaboration. Facet also has the right upon
90 days’ written notice to terminate the agreement for
any uncured material breach by us.
With respect to control over decisions and responsibilities, the
collaboration agreement provides for a joint steering committee,
or JSC, that must make decisions by consensus. The failure of
the JSC to reach consensus on material aspects of the
development or commercialization of TRU-016 would lead to
dispute resolution by our and Facet’s chief executive
officers, and potentially arbitration, any of which may delay
the development of TRU-016, which may harm our business.
Under certain circumstances, the parties have the right to
opt-out of the collaboration or may be deemed to have opted-out
of the collaboration. If Facet opts-out of the collaboration
with respect to a product, then we would become responsible for
all development and commercialization costs for that product and
be obligated to pay Facet certain royalty payments upon the sale
of that product. We are currently the lead manufacturing party
for TRU-016 and if we opt-out of the collaboration and are the
lead TRU-016 manufacturing party at that time, we would be
obligated to continue to supply TRU-016 to Facet for up to
18 months.
On March 9, 2010, Abbott Laboratories, or Abbott, announced
a definitive agreement to purchase Facet. Abbott further
announced that it expects the transaction to close in the second
quarter of 2010, subject to certain conditions. We intend to
continue to pursue the objectives in the approved development
plan.
If Facet opts-out of or terminates the agreement or fails to
fulfill its obligations under the agreement, we would need to
obtain the capital necessary to fully fund the development and
commercialization of TRU-016 or enter into alternative
arrangements with a third party. We could also become involved
in disputes with Facet, which could lead to delays in or
termination of our development and commercialization programs
and time-consuming and expensive litigation or arbitration. If
Facet terminates or breaches its agreement with us, or otherwise
fails to complete its obligations in a timely manner, our
collaboration product development programs would be
substantially delayed and the chances of successfully developing
or commercializing our collaboration product candidates would be
materially and adversely affected.
We
currently rely on third-party manufacturers to supply our
product candidates for clinical trials and will rely on
third-party manufacturers to manufacture our product candidates
in commercial quantities, which could delay, prevent or increase
the costs associated with the clinical development and future
commercialization of our product candidates.
We currently depend on Pfizer for the supply of TRU-015 and
SBI-087. We also currently depend on contract manufacturers for
certain biopharmaceutical development and manufacturing services
for TRU-016, our Facet collaboration clinical candidate. Any
disruption in production, inability of these third-party
manufacturers to produce adequate quantities to meet our needs,
or other impediments with respect to development or
manufacturing could adversely affect our ability to successfully
complete clinical trials, delay submissions of our regulatory
applications, increase our costs or otherwise adversely affect
our ability to
24
commercialize our product candidates in a timely manner, if at
all. For example, our commitments with Lonza for manufacturing
TRU-016 expire in the second quarter of 2010 and we currently do
not have any future manufacturing agreements at this time. We
plan on negotiating for additional manufacturing capacity with
Lonza during 2010. We may be unable to do so in a timely manner
or on terms that are consistent with our existing agreements. If
we are unable to negotiate for additional manufacturing capacity
with Lonza, we will need to contract with other third-party
manufacturers, which will result in additional costs and may
cause delays in the future supply of TRU-016 and the clinical
development of TRU-016.
Our product candidates have not yet been manufactured for
commercial use. If any of our product candidates becomes a
product approved for commercial sale, in order to supply our or
our collaborators’ commercial requirements for such an
approved product, the third-party manufacturer may need to
increase its manufacturing capacity, which may require the
manufacturer to fund capital improvements to support the
scale-up of
manufacturing and related activities. The third-party
manufacturer may not be able to successfully increase its
manufacturing capacity for such an approved product in a timely
or economic manner, if at all. If any manufacturer is unable to
provide commercial quantities of such an approved product, we
will have to successfully transfer manufacturing technology to a
new manufacturer. Engaging a new manufacturer for such an
approved product could require us to conduct comparative studies
or utilize other means to determine bioequivalence of the new
and prior manufacturers’ products, which could delay or
prevent our ability to commercialize such an approved product.
If any of these manufacturers is unable or unwilling to increase
its manufacturing capacity or if we are unable to establish
alternative arrangements on a timely basis or on acceptable
terms, the development and commercialization of such an approved
product may be delayed or there may be a shortage in supply. Any
inability to manufacture our products in sufficient quantities
when needed would seriously harm our business.
Any manufacturer of our product candidates and approved
products, if any, must comply with cGMP requirements enforced by
the FDA through its facilities inspection program. These
requirements include quality control, quality assurance, and the
maintenance of records and documentation. Manufacturers of our
product candidates and approved products, if any, may be unable
to comply with these cGMP requirements and with other FDA,
state, and foreign regulatory requirements. We have little
control over our manufacturers’ compliance with these
regulations and standards. A failure to comply with these
requirements may result in fines and civil penalties, suspension
of production, suspension or delay in product approval, product
seizure or recall, or withdrawal of product approval. If the
safety of any quantities supplied is compromised due to our
manufacturers’ failure to adhere to applicable laws or for
other reasons, we may not be able to obtain regulatory approval
for or successfully commercialize our products, which would
seriously harm our business.
Our
success depends on the proper management of our current and
future business operations, and the expenses associated with
them.
Our business strategy requires us to manage our operations to
provide for the continued development and potential
commercialization of our product candidates and to manage our
expenses generated by these activities. In an effort to reduce
costs, we announced in February 2009 a workforce reduction of
approximately 25%, which included the elimination of certain
existing positions across our research and administrative
functions. As a result of this reduction in force, we recorded a
restructuring charge of $0.8 million in the first quarter
of 2009. We continue to believe that strict cost containment in
the near term is essential if our current funds are to be
sufficient to allow us to continue our currently planned
operations.
If we are unable to effectively manage our current operations,
we may not be able to implement our business strategy and our
financial condition and results of operations may be adversely
affected. If we are unable to effectively manage our expenses,
we may find it necessary to reduce our expenses through another
reduction in our workforce, which could adversely affect our
operations.
We
rely on highly skilled personnel, and if we are unable to retain
or motivate key personnel or hire qualified personnel, we may
not be able to maintain our operations.
Our operations and our ability to execute our business strategy
are highly dependent on the efforts of our executive management
team. In November 2009, our Chief Executive Officer, or CEO, and
Chairman of the Board retired after serving since February 2003.
Following his departure, our Board of Directors appointed our
25
prior Lead Director to serve as Executive Chairman and Acting
President until a qualified replacement is found. We have
retained a recruiting firm and are currently conducting a
nationwide search for a new CEO. The departure of our CEO and
Chairman could have a disruptive effect on our ability to
attract and retain qualified team members and execute our
strategic plan. Though we intend to hire a qualified candidate
for CEO in the near term, no assurances can be given that we
will be able to attract and retain a suitable CEO. An extended
period of time without a permanent CEO could materially and
adversely affect our business, financial condition or results of
operations. Furthermore, in recruiting a new CEO we will incur
expenses related to recruiting, relocation, training and
possibly experience operational inefficiencies. In the event we
are unable to effect a smooth transition from our Interim
Executive Chairman and Acting President to a new CEO, or if a
new CEO should unexpectedly prove to be unsuitable, the
resulting disruption could negatively impact our operations and
impede our ability to execute our strategic plan. In addition,
although the members of our senior management team have
employment agreements with us, these agreements may not provide
sufficient incentives for these officers to continue employment
with us. The loss of one or more of the members of our senior
management team could adversely affect our operations.
In February 2009, as part of our efforts to reduce our operating
expenses through prioritization of our development portfolio and
streamlining our infrastructure, we announced a reduction of
approximately 25% of our workforce, across our research and
administrative functions. This reduction in workforce may impair
our ability to recruit and retain qualified employees and to
effectively complete research and administrative functions. As a
result, our ability to respond to unexpected challenges may be
impaired and we may be unable to take advantage of new
opportunities. If we need to rehire terminated individuals or
hire individuals with similar skills, we may be unable to do so
or we may be required to pay above market rates. Our future
success depends on our continuing ability to develop, motivate,
and retain qualified management, clinical, and scientific
personnel for all areas of our organization. If we do not
succeed in retaining and motivating our remaining personnel, our
existing operations may suffer and we may be unable to
effectively engage in planned operations.
We
cannot assure you any of our product candidates will be safe or
effective, or receive regulatory approval.
The clinical trials and the manufacturing of our product
candidates are, and marketing of our products will be, subject
to extensive and rigorous review and regulation by numerous
government authorities in the United States and in other
countries where we intend to test and market our product
candidates. Before obtaining regulatory approvals for the
commercial sale of any product candidate, we must demonstrate
through preclinical testing and clinical trials that the product
candidate is safe and effective for use in each target
indication. This process can take many years and require the
expenditure of substantial resources, and may include
post-marketing studies and surveillance. To date, we have not
successfully demonstrated in clinical trials safety or efficacy
sufficient for regulatory approval. Although our lead product
candidate, TRU-015, has completed a Phase 2b clinical trial for
the treatment of RA, additional clinical trials would be
required before we are able to submit a BLA to the FDA for
approval and Pfizer will determine whether to commence a Phase 3
study of TRU-015 for the treatment of RA after reviewing data
from the ongoing Phase 2 SBI-087 RA study, in addition to final
data from the ongoing Phase 2b TRU-015 (2203) RA study. In
addition, our Facet collaboration clinical candidate TRU-016 and
our Pfizer collaboration clinical candidate SBI-087 commenced
initial clinical testing in 2008 and as a result we only have
limited clinical trial results regarding the safety or efficacy
of either of these product candidates. Even if, based on the
results of the initial clinical trials for TRU-016 and SBI-087,
we and Facet, in the case of TRU-016, or Pfizer, in the case of
SBI-087, determine to proceed with further clinical testing, a
number of additional clinical trials will be required before a
BLA can be submitted to the FDA for product approval. The
results from preclinical testing and clinical trials that we
have completed may not be predictive of results in future
preclinical tests and clinical trials, and we cannot assure you
we will demonstrate sufficient safety and efficacy to seek or
obtain the requisite regulatory approvals. A number of companies
in the biotechnology and pharmaceutical industries have suffered
significant setbacks in advanced clinical trials, even after
promising results in earlier trials. All of our other product
candidates remain in the discovery and pre-clinical testing
stages. We may also encounter delays or rejections due to
additional government regulation from future legislation,
administrative action, or changes in FDA policy. We cannot
assure you that regulatory approval will be obtained for any of
our product candidates,
26
and even if the FDA approves a product, the approval will be
limited to those indications covered in the approval. If our
current product candidates are not shown to be safe and
effective in clinical trials, the resulting delays in developing
other product candidates and conducting related preclinical
testing and clinical trials, as well as the potential need for
additional financing, would have a material adverse effect on
our business, financial condition, and operating results. If we
are unable to discover or successfully develop drugs that are
effective and safe in humans and receive regulatory approval, we
will not have a viable business. We do not expect any of our
current product candidates to be commercially available in major
markets before 2014, if at all.
Any
failure or delay in commencing or completing clinical trials for
product candidates could severely harm our
business.
Each of our product candidates must undergo extensive
preclinical studies and clinical trials as a condition to
regulatory approval. Preclinical studies and clinical trials are
expensive and take many years to complete. To date we have not
initiated any Phase 3 clinical trials of any product candidate.
The commencement and completion of clinical trials for our
product candidates may be delayed by many factors, including:
|
|
|
| |
•
|
having the capital resources available to fund additional
clinical trials;
|
| |
| |
•
|
our or our collaborators’ ability to obtain regulatory
approval to commence a clinical trial;
|
| |
| |
•
|
our or our collaborators’ ability to manufacture or obtain
from third parties materials sufficient for use in preclinical
studies and clinical trials;
|
| |
| |
•
|
delays in patient enrollment and variability in the number and
types of patients available for clinical trials;
|
| |
| |
•
|
poor effectiveness of product candidates during clinical trials;
|
| |
| |
•
|
unforeseen safety issues or side effects;
|
| |
| |
•
|
governmental or regulatory delays related to clinical trials,
including trial design, results, and materials supply;
|
| |
| |
•
|
changes in regulatory requirements, policy, and
guidelines; and
|
| |
| |
•
|
varying interpretation of data by us, any or all of our
collaborators, the FDA, and similar foreign regulatory agencies.
|
It is possible that none of our product candidates will complete
the required clinical trials in any of the markets in which we
or our collaborators intend to commercialize those product
candidates. Accordingly, we or our collaborators may not seek or
receive the regulatory approvals necessary to market our product
candidates. Any failure or delay in commencing or completing
clinical trials or obtaining regulatory approvals for product
candidates would prevent or delay their commercialization and
severely harm our business and financial condition.
We
rely on third parties to conduct our clinical trials. If these
third parties do not perform as contractually required or
otherwise expected, we may not be able to obtain regulatory
approval for or commercialize our product
candidates.
We do not currently have the ability to conduct clinical trials
and we must rely on third parties, such as contract research
organizations, medical institutions, clinical investigators, and
contract laboratories, to conduct our clinical trials. We have,
in the ordinary course of business, entered into agreements with
these third parties. Nonetheless, we are responsible for
confirming that each of our clinical trials is conducted in
accordance with its general investigational plan and protocol.
Moreover, the FDA requires us to comply with regulations and
standards, commonly referred to as good clinical practices, for
conducting, recording, and reporting the results of clinical
trials to ensure that data and reported results are credible and
accurate and that the trial participants are adequately
protected. Our reliance on third parties does not relieve us of
these responsibilities and requirements. If these third parties
do not successfully carry out their contractual duties or
regulatory
27
obligations or meet expected deadlines, if the third parties
need to be replaced or if the quality or accuracy of the data
they obtain is compromised due to the failure to adhere to our
clinical protocols or regulatory requirements or for other
reasons, our clinical trials may be extended, delayed,
suspended, or terminated, and we may not be able to obtain
regulatory approval for our product candidates.
If we
enter into additional strategic partnerships we may be required
to relinquish important rights to and control over the
development of our product candidates or otherwise be subject to
terms unfavorable to us.
If we enter into any strategic partnerships, we will be subject
to a number of risks, including:
|
|
|
| |
•
|
we may not be able to control the amount and timing of resources
that our strategic partners devote to the development or
commercialization of product candidates;
|
| |
| |
•
|
strategic partners may delay clinical trials, design clinical
trials in a manner with which we do not agree, provide
insufficient funding, terminate a clinical trial or abandon a
product candidate, repeat or conduct new clinical trials, or
require a new version of a product candidate for clinical
testing;
|
| |
| |
•
|
strategic partners may not pursue further development and
commercialization of products resulting from the strategic
partnering arrangement or may elect to discontinue research and
development programs;
|
| |
| |
•
|
strategic partners may not commit adequate resources to the
marketing and distribution of any future products, limiting our
potential revenues from these products;
|
| |
| |
•
|
disputes may arise between us and our strategic partners that
result in the delay or termination of the research, development,
or commercialization of our product candidates or that result in
costly litigation or arbitration that diverts management’s
attention and consumes resources;
|
| |
| |
•
|
strategic partners may experience financial difficulties;
|
| |
| |
•
|
strategic partners may not properly maintain or defend our
intellectual property rights or may use our proprietary
information in a manner that could jeopardize or invalidate our
proprietary information or expose us to potential litigation;
|
| |
| |
•
|
business combinations or significant changes in a strategic
partner’s business strategy may also adversely affect a
strategic partner’s willingness or ability to complete its
obligations under any arrangement;
|
| |
| |
•
|
strategic partners could independently move forward with a
competing product candidate developed either independently or in
collaboration with others, including our competitors; and
|
| |
| |
•
|
strategic partners could terminate the arrangement or allow it
to expire, which would delay the development and may increase
the cost of developing our product candidates.
|
The occurrence of any of these risks could negatively impact the
development of our product candidates which would have an
adverse effect on our business prospects.
If our
technology or our product candidates conflict with the rights of
others we may not be able to manufacture or market our product
candidates, which could have a material adverse effect on
us.
Our commercial success will depend in part on not infringing the
patents or violating the proprietary rights of third parties.
Issued patents held by others may limit our ability to develop
commercial products. All issued U.S. patents are entitled
to a presumption of validity under U.S. law. If we need
licenses to such patents to permit us to manufacture, develop,
or market our product candidates we may be required to pay
significant fees or royalties, and we cannot be certain that we
would be able to obtain such licenses. Competitors or third
parties may obtain patents that may cover subject matter we use
in developing the technology required to bring our products to
market, producing our products, or treating patients with our
products. We know that others have filed patent applications in
various jurisdictions that relate to several areas in which we
are developing products. Some of these patent applications have
already resulted in patents and some are still
28
pending. We may be required to alter our processes or product
candidates, pay licensing fees, or cease activities. For
example, certain parts of our
SMIPtm
product technology, including the current expression system
responsible for the production of the recombinant proteins used
in our product candidates and certain nucleic acids, originated
from third-party sources. These third-party sources include
academic, government, and other research laboratories, as well
as the public domain. If use of technology incorporated into or
used to produce our product candidates is challenged, or if our
processes or product candidates conflict with patent rights of
others, third parties could bring legal actions against us in
Europe, the United States, and elsewhere claiming damages and
seeking to enjoin manufacturing and marketing of the affected
products. Additionally, it is not possible to predict with
certainty what patent claims may issue from pending
applications. In the United States, for example, patent
prosecution can proceed in secret prior to issuance of a patent.
As a result, third parties may be able to obtain patents with
claims relating to our product candidates which they could
attempt to assert against us. Further, as we develop our
products, third parties may assert that we infringe the patents
currently held or licensed by them and we cannot predict the
outcome of any such action.
We are aware of previously filed U.S. provisional patent
applications owned by Genentech and Biogen Idec, which patent
applications are related to a revoked European patent that was
generally directed to the use of an anti-CD20 antibody for the
treatment of RA. A U.S. patent or patent application
claiming the benefit of priority to the provisional patent
applications, if it exists, has not yet published. In the event
any such U.S. patent issues, and if our activities are
determined to be covered by such a patent, we cannot assure you
Genentech would be willing to grant us or Pfizer a license on
terms we or they would consider commercially reasonable, if at
all, which could prevent us from manufacturing and marketing
TRU-015 or SBI-087 for the treatment of RA in the United States,
and have a material adverse effect on our business, financial
condition, operating results, and our collaboration with Pfizer.
With regard to the related European patent, we announced on
September 11, 2008 that the Opposition Division, or OD, of
the European Patent Office, or EPO, had revoked the European
patent in its entirety. On February 19, 2009, Genentech and
Biogen Idec appealed the decision to the Board of Appeals of the
EPO. If Genentech and Biogen Idec succeed in their appeal of the
OD’s decision the opposition proceeding will be reopened
before the OD. If upon these further proceedings the European
patent is held to be valid, either as amended prior to the OD
hearing or with a more limited scope, and if our activities are
determined to be covered by that patent, we cannot assure you
that Genentech would be willing to grant us or Pfizer a license
on terms we or they would consider commercially reasonable, if
at all. As a consequence, we and Pfizer could be prevented from
manufacturing and marketing TRU-015 or SBI-087 for the treatment
of RA in the designated and extended states of the European
Patent Convention where the patent is validated, which could
have a material adverse effect on our business, financial
condition, and operating results. The revoked Genentech European
patent claimed the benefit of priority to two
U.S. provisional patent applications and a U.S. patent
or patent application claiming priority to the provisional
patent applications that have not been published. In the event
any such U.S. patent issues, and if our activities are
determined to be covered by such a patent, we cannot assure you
Genentech would be willing to grant us or Pfizer a license on
terms we or they would consider commercially reasonable, if at
all, which could prevent us from manufacturing and marketing
TRU-015 or SBI-087 for the treatment of RA in the United States,
and have a material adverse effect on our business, financial
condition and operating results, and our collaboration with
Pfizer.
If we
are unable to obtain, maintain, and enforce our proprietary
rights, we may not be able to compete effectively or operate
profitably.
Our success depends in part on obtaining, maintaining, and
enforcing our patents and other proprietary rights, and will
depend in large part on our ability to:
|
|
|
| |
•
|
obtain and maintain patent and other proprietary protection for
our technology, processes, and product candidates;
|
| |
| |
•
|
enforce patents once issued and defend those patents if their
enforceability is challenged;
|
| |
| |
•
|
preserve trade secrets; and
|
| |
| |
•
|
operate without infringing the patents and proprietary rights of
third parties.
|
29
The degree of future protection for our proprietary rights is
uncertain. For example:
|
|
|
| |
•
|
we might not have been the first to make the inventions claimed
in our patents, if issued, or disclosed in our pending patent
applications;
|
| |
| |
•
|
we might not have been the first to file patent applications for
these inventions;
|
| |
| |
•
|
others may independently develop similar or alternative
technologies or duplicate any of our technologies;
|
| |
| |
•
|
it is possible that none of our pending patent applications will
result in issued patents or, if issued, these patents may not be
sufficient to protect our technology or provide us with a basis
for commercially viable products, and may not provide us with
any competitive advantages;
|
| |
| |
•
|
if our pending applications issue as patents, they may be
challenged by third parties as not infringed, invalid, or
unenforceable under U.S. or foreign laws;
|
| |
| |
•
|
if issued, the patents under which we hold rights may not be
valid or enforceable; or
|
| |
| |
•
|
we may develop additional proprietary technologies that are not
patentable and that may not be adequately protected through
trade secrets, if, for example, a competitor were to
independently develop duplicative, similar, or alternative
technologies.
|
The patent position of biotechnology and pharmaceutical firms is
highly uncertain and involves many complex legal and technical
issues. There is no clear policy involving the breadth of claims
allowed in patents or the degree of protection afforded under
patents. Although we believe our potential rights under patent
applications provide a competitive advantage, we cannot assure
you that patent applications owned by or licensed to us will
result in patents being issued or that, if issued, the patents
will give us an advantage over competitors with similar
technology, nor can we assure you that we can obtain, maintain,
and enforce all ownership and other proprietary rights necessary
to develop and commercialize our product candidates.
Even if any or all of our patent applications issue as patents,
others may challenge the validity, inventorship, ownership,
enforceability, or scope of our patents or other technology used
in or otherwise necessary for the development and
commercialization of our product candidates. Further, we cannot
assure you that any such challenge would not be successful.
Moreover, the cost of litigation to uphold the validity of
patents to prevent infringement or to otherwise protect our
proprietary rights can be substantial. If the outcome of
litigation is adverse to us, third parties may be able to use
the challenged technologies without payment to us. We cannot
assure you that our patents, if issued, will not be infringed or
successfully avoided through design innovation. Intellectual
property lawsuits are expensive and would consume time and other
resources, even if the outcome were successful. In addition,
there is a risk that a court would decide that our patents, if
issued, are not valid and that we do not have the right to stop
the other party from using the inventions. There is also the
risk that, even if the validity of a patent were upheld, a court
would refuse to stop the other party from using the inventions,
including on the ground that its activities do not infringe that
patent. If any of these events were to occur, our business,
financial condition, and operating results would be materially
adversely affected.
We also will rely on current and future trademarks to establish
and maintain recognized brands. If we fail to acquire and
protect such trademarks, our ability to market and sell our
products, and therefore our business, financial condition and
operating results, would be materially adversely affected. For
example, in November 2005, Merck KGaA filed a proceeding with
the Office for Harmonisation in the Internal Market opposing our
European registration of the trademark TRUBION and seeking to
place certain restrictions on the identification of goods,
services, and channels of trade description in our European
trademark registration. Merck claims rights resulting from its
prior trademark registration of TRIBION HARMONIS. Our action
with the Court of First Instance of the European Community to
annul the Board decision has been denied. Merck also filed a
similar opposition in Brazil in February 2009. We intend to
vigorously pursue registration of the mark TRUBION for products
in the European Union and Brazil and to challenge Merck’s
claimed rights as necessary to obtain such registration;
however, if we are unable to effectively defend against the
opposition,
30
we may be prohibited from using the TRUBION trademark in certain
European Union jurisdictions, which could have an adverse effect
on our ability to promote the Trubion brand in those
jurisdictions.
In addition to the intellectual property and other rights
described above, we also rely on unpatented technology, trade
secrets, and confidential information, particularly when we do
not believe that patent or trademark protection is appropriate
or available. Trade secrets are difficult to protect and we
cannot assure you that others will not independently develop
substantially equivalent information and techniques or otherwise
gain access to or disclose our unpatented technology, trade
secrets, and confidential information. In addition, we cannot
assure you that the steps we take with employees, consultants,
and advisors will provide effective protection of our
confidential information or, in the event of unauthorized use of
our intellectual property or the intellectual property of third
parties, provide adequate or effective remedies or protection.
We may
incur substantial costs as a result of litigation or other
proceedings relating to patent and other intellectual property
rights.
There has been significant litigation in the biotechnology
industry over patents and other proprietary rights, and if we
become involved in any litigation it could consume a substantial
portion of our resources, regardless of the outcome of the
litigation. Some of our competitors may be better able to
sustain the costs of complex patent litigation because they have
substantially greater resources. If these legal actions are
successful, in addition to any potential liability for damages,
we could be required to obtain a license, grant cross-licenses,
and pay substantial royalties in order to continue to
manufacture or market the affected products. We cannot assure
you we would prevail in any legal action or that any license
required under a third-party patent would be made available on
acceptable terms, if at all. In addition, uncertainties
resulting from the initiation and continuation of any litigation
could have a material adverse effect on our ability to continue
our operations. Ultimately we could be prevented from
commercializing a product or be forced to cease some aspect of
our business operations as a result of claims of patent
infringement or violation of other intellectual property rights,
which could have a material adverse effect on our business,
financial condition, and operating results. Should third parties
file patent applications, or be issued patents claiming
technology also claimed by us in pending applications, we may be
required to participate in interference proceedings in the
United States Patent and Trademark Office to determine priority
of invention, which could result in substantial costs to us and
an adverse decision as to the priority of our inventions. An
unfavorable outcome in an interference proceeding could require
us to cease using the technology or to license rights from
prevailing third parties. We cannot assure you that any
prevailing party would offer us a license or that we could
acquire any license made available to us on commercially
acceptable terms.
We
face substantial competition, which may result in others
discovering, developing, or commercializing products before, or
more successfully than, we do.
Our future success depends on our ability to demonstrate and
maintain a competitive advantage with respect to the design,
development, and commercialization of our product candidates. We
expect any product candidate that we commercialize with our
collaborative partners, or on our own, will compete with other
products.
Product Candidates for Autoimmune and Inflammatory
Diseases. If approved for the treatment of RA, we
anticipate that our product candidates would compete with other
marketed protein therapeutics for the treatment of RA,
including:
Rituxan®
(Genentech, Roche and Biogen Idec),
Enbrel®
(Amgen and Pfizer),
Remicade®
(JNJ and Schering-Plough),
Humira®
(Abbott),
Orencia®
(BMS),
Cimzia®
(UCB),
Simponi®
(JNJ and Schering-Plough), and
Actemra®
(Roche and Chugai). If approved for the treatment of SLE, our
product candidates will compete with other therapies.
Product Candidates for B-cell Malignancies. If
approved for the treatment of CLL, NHL, or other B-cell
malignancies, we anticipate that our product candidates would
compete with other B-cell depleting therapies. While we are not
aware of any CD37-directed therapeutics in development or on the
market, other biologic therapies are marketed for the treatment
of NHL or CLL or both, such as
Rituxan/Mabthera®
31
(Genentech, Roche and Biogen Idec),
Zevalin®
(Spectrum Pharmaceuticals, Inc. and Bayer Schering AG),
Bexxar®
(GSK), ,
Campath®
(Genzyme and Bayer Schering AG) and
Arzerra®
(GSK and Genmab).
Many of our potential competitors have substantially greater
financial, technical, manufacturing, marketing and personnel
resources than we have. In addition, many of these competitors
have significantly greater commercial infrastructures than we
have. Our ability to compete successfully will depend largely on
our ability to:
|
|
|
| |
•
|
design and develop products that are superior to other products
in the market;
|
| |
| |
•
|
attract and retain qualified scientific, medical, product
development, commercial, and sales and marketing personnel;
|
| |
| |
•
|
obtain patent
and/or other
proprietary protection for our processes, product candidates,
and technologies;
|
| |
| |
•
|
operate without infringing the patents and proprietary rights of
third parties;
|
| |
| |
•
|
obtain required regulatory approvals; and
|
| |
| |
•
|
successfully collaborate with others in the design, development,
and commercialization of new products.
|
Established competitors may invest heavily to quickly discover
and develop novel compounds that could make our product
candidates obsolete. In addition, any new product that competes
with a generic market-leading product must demonstrate
compelling advantages in efficacy, convenience, tolerability,
and safety in order to overcome severe price competition and to
be commercially successful. If we are not able to compete
effectively against our current and future competitors, our
business will not grow, and our financial condition and
operating results will suffer.
We may
fail to select or capitalize on the most scientifically,
clinically, or commercially promising or profitable product
candidates.
We have limited technical, managerial, and financial resources
to determine which of our product candidates should proceed to
initial clinical trials, later-stage clinical development, and
potential commercialization and, further, we may make incorrect
determinations as a result of our limited resources or
information available to us at the time of our determination.
Our decisions to allocate our research and development,
management, and financial resources toward particular product
candidates or therapeutic areas may not lead to the development
of viable commercial products and may divert resources from
better opportunities. Similarly, our decisions to delay or
terminate drug development programs may also be incorrect and
could cause us to miss valuable opportunities.
Even
if our product candidates receive regulatory approval, they
could be subject to restrictions or withdrawal from the market
and we may be subject to penalties if we fail to comply with
regulatory requirements or if we experience unanticipated
problems with our products.
Any product candidate for which we receive regulatory approval,
together with the manufacturing processes, post-approval
clinical data, and advertising and promotional activities for
such product, will be subject to continued review and regulation
by the FDA and other regulatory agencies. Even if regulatory
approval of a product candidate is granted, the approval may be
subject to limitations on the indicated uses for which the
product candidate may be marketed or on the conditions of
approval, or contain requirements for costly post-marketing
testing and surveillance to monitor the safety or efficacy of
the product candidate. Later discovery of previously unknown
problems with our products or their manufacture, or failure to
comply with regulatory requirements, may result in, among other
things:
|
|
|
| |
•
|
restrictions on the products or manufacturing processes;
|
| |
| |
•
|
withdrawal of the products from the market;
|
32
|
|
|
| |
•
|
voluntary or mandatory recalls;
|
| |
| |
•
|
fines;
|
| |
| |
•
|
suspension of regulatory approvals;
|
| |
| |
•
|
product seizures; or
|
| |
| |
•
|
injunctions or the imposition of civil or criminal penalties.
|
If we are slow or otherwise unable to adapt to changes in
existing regulatory requirements, we may lose marketing approval
for any products that may be approved in the future.
Failure
to obtain regulatory approval in foreign jurisdictions would
prevent us from marketing our products
internationally.
We intend to have our product candidates marketed outside the
United States. In order to market our products in the European
Union and many other
non-U.S. jurisdictions,
we must obtain separate regulatory approvals and comply with
numerous and varying regulatory requirements. To date, we have
not filed for marketing approval of any of our product
candidates and may not receive the approvals necessary to
commercialize our product candidates in any market. The approval
procedure varies among countries and can involve additional
testing and data review. The time required to obtain foreign
regulatory approval may differ from that required to obtain FDA
approval. The foreign regulatory approval process may include
all of the risks associated with obtaining FDA approval, or may
include different or additional risks. We may not obtain foreign
regulatory approvals on a timely basis, if at all. Approval by
the FDA does not ensure approval by regulatory agencies in other
countries, and approval by one foreign regulatory authority does
not ensure approval by regulatory agencies in other foreign
countries or by the FDA. A failure or delay in obtaining
regulatory approval in one jurisdiction may have a negative
effect on the regulatory approval process in other
jurisdictions, including approval by the FDA. The failure to
obtain regulatory approval in foreign jurisdictions could
seriously harm our business.
Our
product candidates may never achieve market acceptance even if
we obtain regulatory approvals.
Even if we obtain regulatory approvals for the commercial sale
of our product candidates, the commercial success of these
product candidates will depend on, among other things, their
acceptance by physicians, patients, third-party payors, and
other members of the medical community as a therapeutic and
cost-effective alternative to competing products and treatments.
If our product candidates fail to gain market acceptance, we may
be unable to earn sufficient revenue to continue our business.
Market acceptance of, and demand for, any product that we may
develop and commercialize will depend on many factors, including:
|
|
|
| |
•
|
our ability to provide acceptable evidence of safety and
efficacy;
|
| |
| |
•
|
the prevalence and severity of adverse side effects;
|
| |
| |
•
|
availability, relative cost, and relative efficacy of
alternative and competing treatments;
|
| |
| |
•
|
the effectiveness of our marketing and distribution strategy;
|
| |
| |
•
|
publicity concerning our products or competing products and
treatments; and
|
| |
| |
•
|
our ability to obtain sufficient third-party insurance coverage
or reimbursement.
|
If our product candidates do not become widely accepted by
physicians, patients, third-party payors, and other members of
the medical community, our business, financial condition, and
operating results would be materially adversely affected.
33
If we
are unable to establish a sales and marketing infrastructure or
enter into collaborations with partners to perform these
functions, we will not be able to commercialize our product
candidates.
We currently do not have any internal sales, marketing, or
distribution capabilities. In order to commercialize any of our
product candidates that are approved for commercial sale, we
must either acquire or internally develop a sales, marketing,
and distribution infrastructure or enter into collaborations
with partners able to perform these services for us. In December
2005, we entered into a collaboration agreement with Wyeth, now
Pfizer, to develop and commercialize therapeutics directed to
the CD20 protein and other targets. In August 2009 we entered
into a collaboration agreement with Facet to develop and
commercialize TRU-016. If we do not enter into collaborations
with respect to product candidates not covered by the Pfizer or
Facet collaborations, or if any of our product candidates are
the subject of collaborations with partners that are not able to
commercialize such product candidates, we will need to acquire
or internally develop a sales, marketing, and distribution
infrastructure. For example, neither we nor Facet currently have
a sales, marketing or distribution infrastructure. Factors that
may inhibit our efforts to commercialize our product candidates
without partners that are able to commercialize the product
candidates include:
|
|
|
| |
•
|
our inability to recruit and retain adequate numbers of
effective sales and marketing personnel;
|
| |
| |
•
|
the inability of sales personnel to obtain access to or persuade
adequate numbers of physicians to prescribe our products;
|
| |
| |
•
|
the lack of complementary products to be offered by sales
personnel, which may put us at a competitive disadvantage
relative to companies with more extensive product lines; and
|
| |
| |
•
|
unforeseen costs and expenses associated with creating a sales
and marketing organization.
|
If we are not able to partner with a third party able to
commercialize our product candidates, or are not successful in
recruiting sales and marketing personnel or in building a sales,
marketing, and distribution infrastructure, we will have
difficulty commercializing our product candidates, which would
adversely affect our business and financial condition.
If any
products we develop become subject to unfavorable pricing
regulations, third-party reimbursement practices, or healthcare
reform initiatives, our business could be harmed.
Our ability to commercialize any product candidate profitably
will depend in part on the extent to which reimbursement for
such product candidate and related treatments will be available
from government health administration authorities, private
health insurers, or private payors, and other organizations in
the United States and internationally. Even if we succeed in
bringing one or more product candidates to market, these
products may not be considered cost-effective, and the amount
reimbursed for any product may be insufficient to allow us to
sell it profitably. Because our product candidates are in the
early stages of development, we are unable at this time to
determine their cost-effectiveness and the level or method of
reimbursement. There may be significant delays in obtaining
coverage for newly approved products, and coverage may be more
limited than the purposes for which the product candidate is
approved by the FDA or foreign regulatory agencies. Moreover,
eligibility for coverage does not mean that any product will be
reimbursed in all cases or at a rate that covers our costs,
including research, development, manufacture, sale, and
distribution. Increasingly, the third-party payors who reimburse
patients, such as government and private payors, are requiring
that companies provide them with predetermined discounts from
list prices and are challenging the prices charged for medical
products. If the reimbursement we are able to obtain for any
product we develop is inadequate in light of our development and
other costs, our business could be harmed.
We
face potential product liability exposure, and if successful
claims are brought against us, we may incur substantial
liability for a product candidate and may have to limit its
commercialization.
The use of our product candidates in clinical trials and the
sale of any products for which we obtain marketing approval
expose us to the risk of product liability claims. Product
liability claims might be brought against us by consumers,
health-care providers, pharmaceutical companies, or others
selling our products. If
34
we cannot successfully defend ourselves against these claims, we
will incur substantial liabilities. Regardless of merit or
eventual outcome, product liability claims may result in:
|
|
|
| |
•
|
decreased demand for our product candidates;
|
| |
| |
•
|
impairment of our business reputation;
|
| |
| |
•
|
withdrawal of clinical trial participants;
|
| |
| |
•
|
costs of related litigation;
|
| |
| |
•
|
substantial monetary awards to patients or other claimants;
|
| |
| |
•
|
loss of revenues; and
|
| |
| |
•
|
the inability to commercialize our product candidates.
|
Although we currently have product liability insurance coverage
for our clinical trials for expenses or losses, our insurance
coverage may not reimburse us or may not be sufficient to
reimburse us for any or all expenses or losses we may suffer.
Moreover, insurance coverage is becoming increasingly expensive
and, in the future, we may not be able to maintain insurance
coverage at a reasonable cost or in sufficient amounts to
protect us against losses due to liability. We intend to expand
our insurance coverage to include the sale of commercial
products if we obtain marketing approval for our product
candidates in development, but we may be unable to obtain
commercially reasonable product liability insurance for any
products approved for marketing. On occasion, large judgments
have been awarded in class action lawsuits based on products
that had unanticipated side effects. A successful product
liability claim or series of claims brought against us could
cause our stock price to fall and, if judgments exceed our
insurance coverage, could decrease our cash and adversely affect
our business.
If we
use biological and hazardous materials in a manner that causes
contamination or injury or violates laws, we may be liable for
damages.
Our research and development activities involve the use of
potentially harmful biological materials, as well as hazardous
materials, chemicals, and various radioactive compounds. We
cannot completely eliminate the risk of accidental contamination
or injury from the use, storage, handling, or disposal of these
materials. In the event of contamination or injury, we could be
held liable for damages that result, and any liability could
exceed our resources. We do not maintain liability insurance
coverage for our handling of biological or hazardous materials.
We, the third parties that conduct clinical trials on our
behalf, and the third parties that manufacture our product
candidates are subject to federal, state, and local laws and
regulations governing the use, storage, handling, and disposal
of these materials and waste products. The cost of compliance
with these laws and regulations could be significant. The
failure to comply with any of these laws and regulations could
result in significant fines and work stoppages and may harm our
business.
Risks
Related to Our Common Stock
The
trading price of our common stock may be subject to significant
fluctuations and volatility, and our stockholders may be unable
to resell their shares at a profit.
The trading prices of many smaller publicly traded companies are
highly volatile, particularly companies such as ours that have
limited operating histories. Accordingly, the trading price of
our common stock has been subject to significant fluctuations
and may continue to fluctuate or decline. Since our initial
public offering, which was completed in October 2006, the price
of our common stock has ranged from an
intra-day
low of $1.00 to an
intra-day
high of $22.50. Factors that could cause fluctuations in the
trading price of our common stock include the following:
|
|
|
| |
•
|
low trading volumes;
|
| |
| |
•
|
our ability to develop and market new and enhanced product
candidates on a timely basis;
|
35
|
|
|
| |
•
|
announcements by us or our collaborators or competitors of new
commercial products, clinical progress or the lack thereof,
changes in or terminations of relationships, significant
contracts, commercial relationships, or capital commitments;
|
| |
| |
•
|
commencement of, or our involvement in, litigation;
|
| |
| |
•
|
changes in earnings estimates or recommendations by securities
analysts;
|
| |
| |
•
|
changes in governmental regulations or in the status of our
regulatory approvals;
|
| |
| |
•
|
any major change in our board or management;
|
| |
| |
•
|
quarterly variations in our operating results or those of our
collaborators or competitors;
|
| |
| |
•
|
general economic conditions and slow or negative growth of our
markets; and
|
| |
| |
•
|
political instability, natural disasters, war,
and/or
events of terrorism.
|
In addition, the U.S. stock market in the last
16 months has experienced extreme price and volume
fluctuations that have often been unrelated or disproportionate
to the operating performance of trading companies. Broad market
and industry factors may seriously affect the market price of
companies’ stock, including ours, regardless of actual
operating performance. In addition, in the past, following
periods of volatility in the overall market and the market price
of a particular company’s securities, securities class
action litigation has often been instituted against these
companies. This litigation, if instituted against us, could
result in substantial costs and a diversion of our
management’s attention and resources.
If
securities analysts do not publish research or reports about our
business, or if they downgrade our stock, the price of our stock
could decline.
The trading market for our common stock will rely in part on the
availability of research and reports that third-party industry
or financial analysts publish about us. There are many large,
publicly traded companies active in the biopharmaceutical
industry, which may mean it will be less likely that we receive
widespread analyst coverage. Furthermore, if one or more of the
analysts who do cover us downgrade our stock, our stock price
would likely decline. If one or more of these analysts cease
coverage of us, we could lose visibility in the market, which in
turn could cause our stock price to decline.
The
concentration of our capital stock ownership with insiders will
likely limit your ability to influence corporate
matters.
As of December 31, 2009, our executive officers, directors,
current five percent or greater stockholders, and affiliated
entities together beneficially owned approximately 81% of our
outstanding common stock. As a result, these stockholders,
acting together, have control over most matters that require
approval by our stockholders, including the election of
directors and approval of significant corporate transactions.
Corporate action might be taken even if other stockholders
oppose them. This concentration of ownership might also have the
effect of delaying or preventing a change of control of us that
other stockholders may view as beneficial.
Anti-takeover
provisions in our charter documents and under Delaware law could
make an acquisition of us, which may be beneficial to our
stockholders, more difficult and may prevent attempts by our
stockholders to replace or remove our current
management.
Provisions in our certificate of incorporation and bylaws may
delay or prevent an acquisition of us or a change in our
management. These provisions include a classified board of
directors, a prohibition on actions by written consent of our
stockholders and the ability of our board of directors to issue
preferred stock without stockholder approval. In addition,
because we are incorporated in Delaware, we are governed by the
provisions of Section 203 of the Delaware General
Corporation Law, which prohibits stockholders owning in excess
of 15% of our outstanding voting stock from merging or combining
with us. Although we believe these provisions collectively
provide for an opportunity to receive higher bids by requiring
potential acquirers to negotiate with our board of directors,
they would apply even if the offer may be considered beneficial
by some
36
stockholders. In addition, these provisions may frustrate or
prevent any attempts by our stockholders to replace or remove
our current management by making it more difficult for
stockholders to replace members of our board of directors, which
is responsible for appointing the members of our management.
We are
exposed to potential risks from legislation requiring companies
to evaluate controls under Section 404 of the
Sarbanes-Oxley Act.
The Sarbanes-Oxley Act requires that we maintain effective
internal controls over financial reporting and disclosure
controls and procedures. Among other things, we must perform
system and process evaluation and testing of our internal
controls over financial reporting to allow management to report
on, and our independent registered public accounting firm to
attest to, our internal controls over financial reporting, as
required by Section 404 of the Sarbanes-Oxley Act.
Compliance with Section 404 requires substantial accounting
expense and significant management efforts. Our testing, or the
subsequent review by our independent registered public
accounting firm, may reveal deficiencies in our internal
controls that would require us to remediate in a timely manner
so as to be able to comply with the requirements of
Section 404 each year. If we are not able to comply with
the requirements of Section 404 in a timely manner each
year, we could be subject to sanctions or investigations by the
SEC, NASDAQ or other regulatory authorities that would require
additional financial and management resources and could
adversely affect the market price of our common stock.
|
|
|
ITEM 1B.
|
UNRESOLVED
STAFF COMMENTS
|
None.
In June 2003 we entered into a lease agreement for
31,507 square feet of office and laboratory facilities in
Seattle, Washington. On February 10, 2006, we amended the
lease agreement to add an additional 15,892 square feet in
the same building. The lease expires on April 30, 2013,
subject to our option to extend the term for up to
10 years. On February 2, 2007, we leased an additional
3,067 square feet in the same building, through
April 30, 2013. The annual lease payments for these
facilities are approximately $1.5 million in the aggregate.
We believe that the facilities we currently lease are sufficient
for our anticipated near-term needs.
|
|
|
ITEM 3.
|
LEGAL
PROCEEDINGS
|
In November 2005, Merck KGaA filed a proceeding with the Office
of Harmonisation for the Internal Market, or the Office,
opposing our European application for registration of the
trademark TRUBION for certain products and services. The Office
has held in our favor with respect to the services, and in
Merck’s favor with respect to the products. Our action with
the Court of First Instance (CFI) of the European Community to
annul the decision at the Office has been denied. In February
2009, Merck filed a similar opposition in Brazil. We intend to
vigorously pursue registration of the mark TRUBION for products
in the European Union and Brazil, and to challenge Merck’s
claimed rights as necessary to obtain such registration.
On August 8, 2006, we filed with the European Patent
Office, or EPO, an opposition to European patent
No. EP-B-1176981,
in which Genentech has an ownership interest with claims
directed to the second medical use of an anti-CD20 antibody for
the treatment of RA, raising objections as to its validity.
Subsequent to the submission of our opposition, other parties
filed oppositions to the Genentech patent prior to
August 30, 2006, including MedImmune, Inc., Genmab A/S,
Centocor, Inc., Glaxo Group Limited, Serono S.A., and Pfizer. On
September 11, 2008, we announced that the Opposition
Division, or OD, of the EPO had revoked the European patent in
its entirety. On February 19, 2009, Genentech and Biogen
Idec appealed the decision. Final resolution of the opposition
proceeding may take a number of years.
|
|
|
ITEM 4.
|
SUBMISSION
OF MATTERS TO A VOTE OF SECURITY HOLDERS
|
No matters were submitted for a vote of security holders,
through the solicitation of proxies or otherwise, during the
quarter ended December 31, 2009.
37
PART II
|
|
|
ITEM 5.
|
MARKET
FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS
AND ISSUER PURCHASES OF EQUITY SECURITIES
|
Market
Information for Common Stock
Our common stock trades on The Nasdaq Global Market under the
symbol “TRBN.”
The following table sets forth, for the periods indicated, the
range of high and low quarterly closing sales prices of the
common stock as quoted on The Nasdaq Global Market:
| |
|
|
|
|
|
|
|
|
|
|
|
High
|
|
|
Low
|
|
|
|
|
Year ended December 31, 2009
|
|
|
|
|
|
|
|
|
|
First Quarter
|
|
$
|
1.76
|
|
|
$
|
1.16
|
|
|
Second Quarter
|
|
$
|
2.97
|
|
|
$
|
1.30
|
|
|
Third Quarter
|
|
$
|
6.25
|
|
|
$
|
2.36
|
|
|
Fourth Quarter
|
|
$
|
5.11
|
|
|
$
|
3.65
|
|
|
Year ended December 31, 2008
|
|
|
|
|
|
|
|
|
|
First Quarter
|
|
$
|
12.55
|
|
|
$
|
5.99
|
|
|
Second Quarter
|
|
$
|
8.80
|
|
|
$
|
4.39
|
|
|
Third Quarter
|
|
$
|
5.40
|
|
|
$
|
3.32
|
|
|
Fourth Quarter
|
|
$
|
3.67
|
|
|
$
|
1.01
|
|
Stockholders
As of February 26, 2010, there were approximately 35
holders of record of our common stock.
Dividend
Policy
No cash dividends have been paid on the common stock. We
currently intend to retain all future income to fund the
development and growth of our business and do not anticipate
paying any cash dividends in the foreseeable future. In 2008, we
entered into a loan and security agreement that may restrict our
ability to pay cash dividends.
38
Stock
Performance Graph
The graph set forth below compares the cumulative total
stockholder return on our common stock between October 18,
2006 (the date of our initial public offering) and
December 31, 2009, with the cumulative total return of
(i) the Nasdaq Biotechnology Index and (ii) the Nasdaq
Stock Market Index, over the same period. This graph assumes the
investment of $100 on October 18, 2006 in our common stock,
the Nasdaq Biotechnology Index and the Nasdaq Stock Market
Index, and assumes the reinvestment of dividends, if any. The
graph assumes the initial value of our common stock on
October 18, 2006 was the closing sales price of $13.09 per
share.
The comparisons shown in the graph below are based on historical
data. We caution that the stock price performance shown in the
graph below is not necessarily indicative of, nor is it intended
to forecast, the potential future performance of our common
stock. Information used in the graph was obtained from the
Nasdaq website, a source believed to be reliable, but we are not
responsible for any errors or omissions in such information.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10/18/2006
|
|
|
12/29/2006
|
|
|
12/31/2007
|
|
|
12/31/2008
|
|
|
12/31/2009
|
|
Trubion Pharmaceuticals, Inc.
|
|
|
$
|
100.00
|
|
|
|
$
|
137.59
|
|
|
|
$
|
76.39
|
|
|
|
$
|
9.78
|
|
|
|
$
|
29.41
|
|
|
Nasdaq Stock Market Index
|
|
|
$
|
100.00
|
|
|
|
$
|
87.97
|
|
|
|
$
|
141.40
|
|
|
|
$
|
70.60
|
|
|
|
$
|
56.63
|
|
|
Nasdaq Biotechnology Index
|
|
|
$
|
100.00
|
|
|
|
$
|
99.47
|
|
|
|
$
|
104.03
|
|
|
|
$
|
90.89
|
|
|
|
$
|
105.10
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Recent
Sales of Unregistered Securities
In connection with the collaboration agreement, we and Facet
entered into a stock purchase agreement, pursuant to which Facet
purchased 2,243,649 shares of our common stock for an
aggregate purchase price of $10 million, or $4.46 per
share. The per share price of $4.46 represents a 35% equity
premium over the
sixty-day
trading average of our common stock on NASDAQ for the trading
period ending immediately prior to the execution of the stock
purchase agreement.
39
|
|
|
ITEM 6.
|
SELECTED
FINANCIAL DATA
|
The following selected financial data should be read in
conjunction with Item 7, “Management’s Discussion
and Analysis of Financial Condition and Results of
Operations” and our audited financial statements and the
related notes thereto included in this annual report.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
2006
|
|
|
2005
|
|
|
|
|
(In thousands, except per share data)
|
|
|
|
|
Statements of Operations Data:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenue:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Collaboration revenue
|
|
$
|
18,003
|
|
|
$
|
16,467
|
|
|
$
|
20,148
|
|
|
$
|
36,530
|
|
|
$
|
222
|
|
|
Grant revenue
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
127
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total revenue
|
|
|
18,003
|
|
|
|
16,467
|
|
|
|
20,148
|
|
|
|
36,530
|
|
|
|
349
|
|
|
Operating expenses:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development
|
|
|
34,396
|
|
|
|
31,608
|
|
|
|
36,466
|
|
|
|
33,309
|
|
|
|
15,212
|
|
|
General and administrative
|
|
|
12,429
|
|
|
|
11,374
|
|
|
|
10,833
|
|
|
|
9,473
|
|
|
|
4,146
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses
|
|
|
46,825
|
|
|
|
42,982
|
|
|
|
47,299
|
|
|
|
42,782
|
|
|
|
19,358
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss from operations
|
|
|
(28,822
|
)
|
|
|
(26,515
|
)
|
|
|
(27,151
|
)
|
|
|
(6,252
|
)
|
|
|
(19,009
|
)
|
|
Net interest income (expense)
|
|
|
(361
|
)
|
|
|
956
|
|
|
|
3,837
|
|
|
|
2,222
|
|
|
|
278
|
|
|
Other income (expense)
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
101
|
|
|
|
(134
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss before cumulative effect of change in accounting principle
|
|
|
(29,183
|
)
|
|
|
(25,559
|
)
|
|
|
(23,314
|
)
|
|
|
(3,929
|
)
|
|
|
(18,865
|
)
|
|
Cumulative effect of change in accounting principle
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(62
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss(1)
|
|
$
|
(29,183
|
)
|
|
$
|
(25,559
|
)
|
|
$
|
(23,314
|
)
|
|
$
|
(3,929
|
)
|
|
$
|
(18,927
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted net loss per share
|
|
$
|
(1.55
|
)
|
|
$
|
(1.43
|
)
|
|
$
|
(1.32
|
)
|
|
$
|
(0.83
|
)
|
|
$
|
(23.30
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Shares used in computation of basic and diluted net loss per
share
|
|
|
18,797
|
|
|
|
17,856
|
|
|
|
17,688
|
|
|
|
4,744
|
|
|
|
812
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) |
|
Effective January 1, 2006, we began recognizing
compensation expenses for all future share-based payments made
to employees and directors be based on estimated fair values.
For the years ended December 31, 2009, 2008 and 2007, we
recorded non-cash stock-based employee compensation expense of
$3.5 million, $3.2 million and $2.9 million,
respectively. See Note 10 of the Notes to Financial
Statements for further discussion. |
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
At December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
2006
|
|
|
2005
|
|
|
|
|
(In thousands)
|
|
|
|
|
Balance Sheet Data:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash, cash equivalents and investments
|
|
$
|
54,846
|
|
|
$
|
52,897
|
|
|
$
|
78,515
|
|
|
$
|
105,801
|
|
|
$
|
9,792
|
|
|
Receivable from collaborations
|
|
|
3,428
|
|
|
|
3,084
|
|
|
|
4,237
|
|
|
|
4,354
|
|
|
|
40,000
|
|
|
Working capital
|
|
|
40,530
|
|
|
|
45,287
|
|
|
|
69,132
|
|
|
|
93,188
|
|
|
|
37,881
|
|
|
Total assets
|
|
|
65,380
|
|
|
|
67,290
|
|
|
|
95,174
|
|
|
|
121,394
|
|
|
|
54,009
|
|
|
Deferred revenue
|
|
|
35,262
|
|
|
|
19,493
|
|
|
|
24,854
|
|
|
|
31,778
|
|
|
|
39,778
|
|
|
Non-current portion of notes payable
|
|
|
6,975
|
|
|
|
8,261
|
|
|
|
7,567
|
|
|
|
6,708
|
|
|
|
1,276
|
|
|
Preferred stock warrant liability
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
282
|
|
|
Convertible preferred stock
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
45,753
|
|
|
Total stockholders’ equity (deficit)
|
|
|
15,094
|
|
|
|
31,468
|
|
|
|
53,313
|
|
|
|
72,654
|
|
|
|
(37,902
|
)
|
40
|
|
|
ITEM 7.
|
MANAGEMENT’S
DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF
OPERATIONS
|
The following discussion and analysis should be read in
conjunction with our audited financial statements and the
related notes thereto that appear elsewhere in this annual
report. This discussion contains forward-looking statements
reflecting our current expectations that involve risks and
uncertainties. Actual results may differ materially from those
discussed in these forward-looking statements due to a number of
factors, including those set forth in the section entitled
“Risk Factors” and elsewhere in this annual report.
Overview
We are a biopharmaceutical company creating a pipeline of novel
protein therapeutic product candidates to treat autoimmune and
inflammatory diseases and cancer. Our mission is to develop a
variety of
first-in-class
and
best-in-class
product candidates customized in an effort to optimize safety,
efficacy, and convenience that we believe may offer improved
patient experiences. Our current product development efforts are
focused on three proprietary technologies that comprise the
expanded foundation for Trubion product development —
SMIPtm
protein therapeutics,
SCORPIONtm
protein therapeutics, and
TRU-ADhanCetm
potency enhancing technology for immunopharmaceuticals. Our
current clinical-stage therapeutics target specific antigens on
B cells, CD20 and CD37, and are designed using our custom drug
assembly technology. In order to fund ongoing development
activities and commercialize our products, we will, in some
cases, enter into collaboration agreements that would likely
include licenses to our technology and arrangements to provide
research and development services for others.
We were founded as a limited liability company in the state of
Washington in March 1999. We converted into a corporation and
redomiciled in the state of Delaware in October 2002. To date,
we have funded our operations primarily through the sale of
equity securities, strategic alliances, equipment financings and
interest earned on investments.
Product
Candidates and Recent Developments
Our lead product candidate, TRU-015, which we are developing
with our partner Pfizer Inc., or Pfizer, has completed a Phase
2b clinical trial for the treatment of rheumatoid arthritis, or
RA. A second Phase 2b (study 2203) clinical trial for RA is
under way and enrollment was completed in September 2009. The
randomized, parallel, double-blind, placebo-controlled,
dose-regimen finding study is evaluating the safety and efficacy
of two dosing regimens of TRU-015 administered to patients with
active seropositive rheumatoid factor on a background of
methotrexate. This study was designed in a way that we believe
could be supportive of a registration package with the Food and
Drug Administration, or FDA. Pfizer will determine whether to
commence a Phase 3 study of TRU-015 for the treatment of RA
after reviewing data from the ongoing Phase 2 SBI-087 RA study,
in addition to final data from the ongoing Phase 2b TRU-015
(2203) RA study. Patient dosing in the Phase 2 SBI-087 RA
study commenced in December 2009 and an interim data review is
planned that we believe could occur in late 2010 or early 2011
and final data is anticipated at the end of 2011.
In October 2009, we announced positive data from the second
course of re-treatment in the first Phase 2b clinical trial for
RA demonstrating that administration of TRU-015 every
24 weeks produces well-tolerated results that are
comparable with the more than four and a half years of
re-treatment data compiled to date.
In collaboration with us, Pfizer is also developing SBI-087, our
next generation CD20-directed product candidate. SBI-087 for RA
builds on our and Pfizer’s clinical experience with TRU-015
and is based on our SMIP technology. Enrollment was completed in
a Phase 1 study of SBI-087 for RA. Patient dosing has commenced
and recruitment is ongoing in a Phase 2 study of SBI-087 for RA.
In addition, patient recruitment is under way in an additional
Phase 1 study of SBI-087 for RA in Japan. Finally, Pfizer is
conducting a Phase 1 clinical trial of SBI-087 in systemic lupus
erythematosus, or SLE, in which patient dosing has commenced and
recruitment is ongoing.
TRU-016, which we are developing with our partner Facet Biotech
Corporation, or Facet, is a novel CD37-directed SMIP protein
therapeutic. A TRU-016 Phase 1 clinical trial for patients with
chronic
41
lymphocytic leukemia, or CLL, is currently under way. TRU-016
uses a different mechanism of action than CD20-directed
therapies. As a result, we believe its novel design may provide
patients with improved therapeutic options and enhance efficacy
when used alone or in combination with chemotherapy
and/or
CD20-directed therapeutics.
In June and December 2009, we announced positive results
following each of two preliminary analysis from the Phase 1
clinical trial of TRU-016 for the treatment of CLL. The
objectives of the Phase 1 TRU-016 CLL study were to define
safety and tolerability, identify a maximum tolerated dose,
evaluate pharmacology and pharmacodynamics, and assess
preliminary clinical activity. As of February 2010, we have not
reached a maximum tolerated dose and have filed an amendment to
include treatment of patients with non-Hodgkins lymphoma, or NHL.
Collaborations
Facet
In August 2009, we entered into a collaboration agreement with
Facet for the joint worldwide development and commercialization
of TRU-016, a product candidate in Phase 1 clinical development
for chronic lymphocytic leukemia, or CLL. TRU-016 is a
CD37-directed SMIP protein therapeutic. The collaboration
agreement includes TRU-016 in all indications and all other
CD37-directed protein therapeutics. Under the terms of the
collaboration agreement, the parties will not develop or
commercialize protein therapeutics directed to CD37 outside of
the collaboration agreement.
We received an up-front payment of $20 million in cash in
September 2009 and may receive up to $176.5 million in
additional contingent payments upon the achievement of specified
development, regulatory and sales milestones. We and Facet share
equally the costs of all development, commercialization and
promotional activities and all global operating profits. In
connection with the collaboration agreement, we and Facet also
entered into a stock purchase agreement, pursuant to which Facet
purchased 2,243,649 shares of our common stock for an
aggregate purchase price of $10 million, or $4.46 per
share. The per share price of $4.46 represents a 35% equity
premium over the
sixty-day
trading average of our common stock on NASDAQ for the trading
period ending immediately prior to the execution of the stock
purchase agreement.
With respect to control over decisions and responsibilities, the
collaboration agreement provides for a joint steering committee,
or JSC, consisting of representatives of Trubion and Facet,
which makes decisions by consensus. If the JSC is unable to
reach a consensus, then the matter will be referred to
Trubion’s and Facet’s Chief Executive Officers for
resolution. If the Chief Executive Officers are unable to
resolve the matter, then it will be resolved by arbitration.
Both Trubion and Facet, at their sole discretion, may
discontinue participation on the JSC with 90 days written
notice to the other party.
At predefined times, the parties have the right to opt-out of
the collaboration entirely or on a
product-by-product
basis. Upon a change of control of a party, the other party will
have the right to opt-out of the collaboration entirely and if
the successor party is conducting a program that competes with
the programs under the collaboration agreement, then the
successor party must either (i) opt-out of the
collaboration entirely or (ii) divest the competing program
to a third party. If a party exercises its opt-out right with
respect to a product, then the parties will no longer share
development and commercialization costs for such product and
such opting-out party will receive certain royalty payments upon
the sale of such product, rather than half of the profits
derived from such product. Even if Facet exercises its opt-out
right, its obligation to make milestone payments to us
continues. In addition, if the party that opts-out is the lead
manufacturing party for the opt-out product, then such party
must continue to supply the product to the continuing party for
up to eighteen months following the opt-out.
Facet can terminate the collaboration agreement at any time, in
which event all rights to TRU-016 and other CD37-directed
protein therapeutics under the collaboration agreement would
revert to us. If Facet terminates the collaboration agreement in
the first 18 months, then Facet must pay us a
$10 million termination fee.
42
If there is a material breach of the collaboration agreement,
then the non-breaching party may either terminate the
collaboration agreement or continue the collaboration agreement
and force the breaching party to opt-out and accept royalties at
a reduced rate.
Either party may assign its interest in the collaboration
agreement to a third party, provided that the non-assigning
party maintains a right of first negotiation over any proposed
assignment. In addition, either we or Facet can freely assign
the collaboration agreement without the consent of the other
party in connection with certain specified change of control
transactions, such as an acquisition.
Pfizer
In December 2005 we entered into a collaboration agreement with
Wyeth, now a wholly-owned subsidiary of Pfizer, for the
development and worldwide commercialization of TRU-015 and other
CD20-directed therapeutics. Pursuant to the agreement, we are
also collaborating with Pfizer on the development and worldwide
commercialization of certain other product candidates directed
to a small number of targets other than CD20. During the period
in which we will be providing research services for Pfizer,
Pfizer has the right, subject to our reasonable consent, to
replace a limited number of these targets. In addition, we have
the option to co-promote with Pfizer, on customary terms to be
agreed, CD20-directed therapies in the United States for niche
indications. We retain the right to develop and commercialize,
on our own or with others, product candidates directed to all
targets not included within the agreement. Unless it is
terminated earlier, our agreement with Pfizer will remain in
effect on a
product-by-product
basis and on a
country-by-country
basis until the later of the date that any such product shall no
longer be covered by a valid claim of a U.S. or foreign
patent or application and, generally, ten years after the first
commercial sale of any product licensed under the agreement.
Pfizer may terminate the agreement without cause at any time
upon 90 days’ prior written notice.
In connection with the agreement, Wyeth paid us a
$40 million non-refundable, non-creditable, up-front fee in
January 2006 and purchased directly from us in a private
placement, concurrent with our initial public offering,
800,000 shares of our common stock at the initial public
offering price of $13.00 per share, resulting in net proceeds to
us of $10.4 million. Under the agreement, we provided
research services for an initial three-year period ended
December 22, 2008 with the option for Wyeth to extend the
service period for two additional one-year periods. Wyeth’s
financial obligations during the initial research service term
included collaborative research funding commitments of
$9.0 million in exchange for such committed research
services. This $9.0 million was subject to an increase if
the service period was extended beyond three years as well as
annual increases pursuant to percentage changes in the CPI. In
June 2008, Wyeth exercised the first option under the terms of
the agreement to extend the research period for an additional
one-year period through December 22, 2009. In June 2009,
Wyeth exercised the second option under the terms of the
agreement to extend the research period for an additional
one-year period through December 22, 2010. Due to the
research period extension in 2009, the collaboration research
funding commitments to us initially from Wyeth and now from
Pfizer, increased to approximately $3.3 million per year in
exchange for committed research services from us through
December 22, 2010.
Pfizer’s financial obligations include additional amounts
for reimbursement of
agreed-upon
external research and development costs and patent costs.
Pursuant to the agreement, Pfizer is also obligated to make
payments to us of up to $250 million based on the
achievement of specified regulatory and sales milestones for
CD20-directed therapies and payments to us of up to
$535 million based on the specified achievement of
regulatory and sales milestones for therapies directed to the
small number of targets other than CD20. In addition, we will
receive royalty payments in the event of future licensed product
sales. Pfizer may terminate the agreement without cause at any
time upon 90 days’ prior written notice.
In October 2009, Pfizer completed its acquisition of Wyeth. Our
collaboration agreement remains in effect with Pfizer and, as
allowed under the collaboration agreement, we requested and
Pfizer has provided further written assurances reaffirming
Pfizer’s commitment to comply with the terms and conditions
of the agreement.
43
If Pfizer has ongoing development
and/or
commercialization activities that would violate the mutual
exclusivity provisions of the collaboration agreement, we have
the right to require Pfizer to engage in good faith discussions
regarding the terms and conditions on which Pfizer would pay
reasonable financial consideration to us with respect to those
development and commercialization activities. If we and Pfizer
do not agree to terms, we have the right to require Pfizer to
enter into an agreement to divest such development and
commercialization activities, or to divest the relevant
collaboration agreement products to a third party. If Pfizer
does not divest such development and commercialization
activities or such collaboration agreement products, we have the
right to terminate all licenses related to CD20
and/or the
additional specified target, as applicable.
If during the 12 month period following Pfizer’s
acquisition of Wyeth, Pfizer is required or voluntarily decides
to divest itself of one or more of the products under the
collaboration agreement, then subject to any governmental
limitations, Pfizer must offer us an exclusive opportunity to
negotiate the acquisition or license of all of Pfizer’s
rights to that product on commercially reasonable terms. If we
do not conclude an agreement with Pfizer covering the product,
Pfizer can divest itself of the product but the terms of that
divestiture cannot be more favorable than those that were last
offered to us unless we are given the opportunity to accept
those more favorable terms.
Upon a change of control of Trubion, the agreement would remain
in effect, subject to the right of Pfizer to terminate specified
provisions of the agreement.
Assuming TRU-015 and other product candidates under the
collaboration with Pfizer continue to progress in development,
expenses for future clinical trials may be higher than those
incurred in prior clinical trials. These expenses will, however,
likely be incurred by Pfizer and expenses incurred by us, if
any, will be substantially offset by reimbursement revenue from
Pfizer. In addition, Pfizer is responsible for a substantial
portion of costs related to patent prosecution and patent
litigation for products directed to targets selected by Pfizer
pursuant to the collaboration agreement.
Outlook
The continued research and development of our product candidates
will require significant additional expenditures, including
preclinical studies, clinical trials, manufacturing costs, and
the expenses of seeking regulatory approval. We rely on third
parties to conduct a portion of our preclinical studies, all of
our clinical trials and all of the manufacturing of current Good
Manufacturing Process, or cGMP material. We expect expenditures
associated with these activities to increase in future years as
we continue developing our product candidates. Expenses
associated with our product candidates included in the Pfizer
collaboration are substantially offset by reimbursement revenue
from Pfizer. Expenses associated with our product candidates
included in the Facet collaboration are shared equally.
We have incurred significant losses since our inception. As of
December 31, 2009, our accumulated deficit was
$121.7 million and total stockholders’ equity was
$15.1 million. During the years ended December 31,
2009, 2008 and 2007, we recognized net losses of
$29.2 million, $25.6 million and $23.3 million,
respectively. We expect our net losses to increase in the future
as we continue our existing and anticipated preclinical studies,
manufacturing, and clinical trials. Our revenues and research
and development expenses under the Facet collaboration may
fluctuate depending on which party in the collaboration is
incurring the majority of the development costs in any
particular quarterly period. However, we expect revenue to
increase in the future as a result of the additional
collaboration revenue from our Facet collaboration. This
increase in revenue from the Facet collaboration will be
partially offset by decreases in revenue from Pfizer due to the
transfer to Pfizer of the responsibility for the majority of the
clinical development efforts and related costs for product
candidates covered by our collaboration agreement with Pfizer.
Critical
Accounting Policies and Significant Judgments and
Estimates
Our management’s discussion and analysis of our financial
condition and results of operations are based on our unaudited
financial statements, which have been prepared in accordance
with GAAP. The preparation of these financial statements
requires us to make estimates and judgments that affect the
reported amounts of
44
assets and liabilities and the disclosure of contingent assets
and liabilities at the date of the financial statements, as well
as reported revenues and expenses during the reporting periods.
We base our estimates on historical experience and on various
other factors that we believe are reasonable under the
circumstances. An accounting policy is considered to be critical
if it is important to a company’s financial condition and
results of operations, and if it requires the exercise of
significant judgment and the use of estimates on the part of
management in its application. We have discussed the selection
and development of the critical accounting policies with the
audit committee of our board of directors, and the audit
committee has reviewed our related disclosures in this annual
report on
Form 10-K.
Although we believe our judgments and estimates are appropriate,
actual results may differ from those estimates.
Our significant accounting policies are described in Note 1
to our audited financial statements for the year ended
December 31, 2009 in this annual report on
Form 10-K.
Of our significant accounting policies, we believe that the
following accounting policies relating to revenue recognition,
preclinical study, clinical trial and manufacturing accruals,
stock-based compensation and valuation of investments are the
most critical to understanding and evaluating our reported
financial results.
Revenue
Recognition
We recognize revenue from our collaboration agreements with
Pfizer and Facet, which consists of non-refundable,
non-creditable up-front fees and license fees, collaborative
research funding, regulatory and sales milestones future product
royalties and future product sales. Revenue related to our
collaboration agreements is recognized as follows:
Up-Front Fees and License Fee. Non-refundable,
non-creditable up-front fees and license fees received in
connection with collaborative research and development
agreements are deferred and recognized on a straight-line basis
over the estimated term of the research and development service
period. The estimated term of the research and development
service period is reviewed and adjusted based on the status of
the project against the estimated timeline as additional
information becomes available. We also consider the time frame
of our substantive contractual obligations related to research
and development agreements when estimating the term of the
research and development period. For each collaboration
agreement, we review our ongoing performance obligations on a
regular basis and make adjustments to the estimated term as
additional information becomes available. During the third
quarter of 2008, the estimated term of the research and
development service period related to the Pfizer agreement was
adjusted from six years and three months to seven years, or
through December 2012, due to an extension of the estimated
service period of our obligations to conduct clinical activities
under our agreement with Pfizer. The adjustment during the third
quarter of 2008 was the second adjustment to the estimated
research and development service period since the inception of
the collaboration agreement with Pfizer. Adjustments to the
research and development service period are made prospectively.
We have made adjustments to the research and development service
periods in the past and we expect to revise our estimate of the
development term in future periods due to the inherently
uncertain nature of development terms. As a result, revenue may
fluctuate materially in the future due to adjustments to the
estimated term of the research and development service periods
and our substantive contractual obligations under our
collaborations.
Collaborative Research Funding. Certain
internal and external research and development costs and patent
costs are reimbursed in connection with our collaboration
agreements. Reimbursed costs under the Pfizer collaboration are
recognized as revenue in the same period the costs are incurred.
With respect to the reimbursement of development costs under the
Facet collaboration, each quarter, we and Facet reconcile what
each party has incurred for development costs, and we record
either a net receivable or a net payable in our financial
statements. For each quarterly period, if we have a net
receivable from Facet, we recognize revenues by such amount, and
if we have a net payable to Facet, we recognize additional
research and development expenses by such amount. As a result,
our revenues and research and development expenses may fluctuate
depending on which party in the collaboration is incurring the
majority of the development costs in any particular quarterly
period. Reimbursed costs are subject to the estimation processes
described in the preclinical study, clinical trial and
manufacturing accruals processes
45
described below and are subject to change in future periods when
actual activity is known. To date we have not made any material
adjustments to these estimates.
Milestones. Payments for milestones that are
based on the achievement of substantive and at-risk performance
criteria will be recognized in full at such time as the
specified milestone has been achieved according to the terms of
the agreement. When payments are not for substantive or at-risk
milestones, revenue will be recognized on a straight-line basis
over the remaining estimated term of the research and
development service period. The estimated term of the research
and development service period is reviewed and adjusted based on
the status of the project against the estimated timeline as
additional information becomes available.
Preclinical
Study, Clinical Trial and Manufacturing Accruals
We estimate our preclinical study, clinical trial and
manufacturing accrued expenses based on our estimates of the
services received pursuant to contracts with multiple research
organizations and contract manufacturers that conduct, manage,
and provide materials for preclinical studies and clinical
trials on our behalf. The financial terms of these agreements
vary from contract to contract and may result in uneven payment
flows. Research and development costs are expensed as the
related goods are delivered or the related services are
performed. Our preclinical study, clinical trial and
manufacturing expenses include fees paid to the following:
|
|
|
| |
•
|
contract research organizations in connection with preclinical
studies;
|
| |
| |
•
|
clinical research organizations and other clinical sites in
connection with clinical trials; and
|
| |
| |
•
|
contract manufacturers in connection with the production of
components and drug materials for preclinical studies and
clinical trials.
|
We record accruals for these preclinical studies, clinical trial
and manufacturing expenses based on the estimated amount of work
completed. All such costs are included in research and
development expenses based on these estimates. Costs of setting
up a preclinical study or clinical trial are expensed as the
related services are performed. Costs related to patient
enrollment in clinical trials are accrued as patients are
enrolled in the trial. We monitor patient enrollment levels and
related activities to the extent possible through internal
reviews, correspondence and discussions with research
organizations. If we have incomplete or inaccurate information,
we may, however, underestimate or overestimate activity levels
associated with various preclinical studies and clinical trials
at a given point in time. In the event we underestimate, we
could record significant research and development expenses in
future periods when the actual activity level becomes known. To
the extent any of these expenses are reimbursable under our
collaboration agreements with Pfizer or Facet, we could also
record significant adjustments to revenue when the actual
activity becomes known. To date, we have not made any material
adjustments to our estimates of preclinical study, clinical
trial and manufacturing expenses. We make good-faith estimates
that we believe to be accurate, but the actual costs and timing
of preclinical studies, clinical trials and manufacturing runs
are highly uncertain, subject to risks, and may change depending
on a number of factors, including our clinical development plan.
If any of our product candidates enter Phase 3 clinical trials,
the process of estimating clinical trial costs will become more
difficult because the trials will involve larger numbers of
patients and clinical sites.
Stock-Based
Compensation
We account for stock-based compensation for employees and
directors based on estimated fair values. Employee stock-based
compensation expense recognized in the years ended
December 31, 2009, 2008 and 2007 was calculated based on
awards ultimately expected to vest, and has been reduced for
estimated forfeitures. Forfeitures are estimated at the time of
grant and revised, if necessary, in subsequent periods if actual
forfeitures differ from those estimates. The forfeiture estimate
is based on historical employee turnover rates and could differ
from actual forfeitures. Compensation costs for employee stock
options granted prior to January 1, 2006 were accounted for
using the option’s intrinsic value or the difference, if
any, between the fair market value of our common stock and the
exercise price of the option.
46
The fair value of each employee option grant in the years ended
December 31, 2009, 2008 and 2007, respectively, was
estimated on the date of grant using the Black-Scholes option
pricing model with the following assumptions:
| |
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
2009
|
|
2008
|
|
2007
|
|
|
|
Risk-free interest rate
|
|
2.13%-2.72%
|
|
2.80%-3.40%
|
|
3.78%-4.78%
|
|
Weighted-average expected life (in years)
|
|
5.92
|
|
6.04
|
|
6.14
|
|
Expected dividend yield
|
|
0%
|
|
0%
|
|
0%
|
|
Expected volatility rate
|
|
88%-105%
|
|
70%-74%
|
|
65%-75%
|
|
Weighted-average estimated fair value of employee options
|
|
$1.85
|
|
$5.29
|
|
$12.87
|
For stock options granted to non-employees, the fair value of
the stock options is estimated using the Black-Scholes valuation
model. The Black-Scholes model utilizes the estimated fair value
of common stock and requires that, at the date of grant, we make
assumptions with respect to the expected life of the option, the
volatility of the fair value of the underlying common stock,
risk-free interest rates and expected dividend yields of our
common stock. We have assumed that non-employee stock options
have an expected life of one to ten years and assumed common
stock volatility between 65% and 105%.
Stock-based compensation expense is recognized over the period
of expected service by the non-employee. As the service is
performed, we are required to update our valuation assumptions,
remeasure unvested options and record the stock-based
compensation using the valuation as of the vesting date. These
adjustments may result in higher or lower stock-based
compensation expense in the statement of operations than
originally estimated. Changes in the market price of our stock
could materially change the value of an option and the resulting
stock-based compensation expense. We expect stock-based
compensation expense associated with non-employee options to
fluctuate in the future based on the volatility of our future
stock price.
Valuation
of Investments
We classify our investment portfolio as
available-for-sale.
The cost of securities sold is based on the specific
identification method. We carry our investments in debt
securities at fair value, estimated as the amount at which an
asset or liability could be bought or sold in a current
transaction between willing parties. In accordance with our
investment policy, we diversify our credit risk and invest in
debt securities with high credit quality. The majority of our
investments held as of December 31, 2009 are in active
markets and our estimate of fair value is based upon quoted
market prices. The remainder of our investments held as of
December 31, 2009 are valued using observable inputs. We
regularly evaluate the performance of our investments
individually for impairment, taking into consideration the
investment, volatility and current returns. If a determination
is made that a decline in fair value is
other-than-temporary,
the related investment is written down to its estimated fair
value. To date, the carrying values of our investments have not
been written down due to declines in value because such declines
are judged to be temporary. Declines in the fair value of our
investments judged to be other than temporary could adversely
affect our future operating results. We continue to monitor our
credit risks and evaluate the potential need for impairment
charges related to credit risks in future periods.
Recent
Accounting Pronouncements
In October 2009, the FASB issued new guidance for
multiple-deliverable revenue arrangements. The new guidance
addresses the accounting for multiple-deliverable arrangements
to enable vendors to account for products or services
(deliverables) separately rather than as a combined unit. This
guidance establishes a selling price hierarchy for determining
the selling price of a deliverable, which is based on:
(a) vendor-specific objective evidence;
(b) third-party evidence; or (c) estimates. This
guidance also eliminates the residual method of allocation and
requires that arrangement consideration be allocated at the
inception of the arrangement to all deliverables using the
relative selling price method. In addition, this guidance
significantly
47
expands required disclosures related to a vendor’s
multiple-deliverable revenue arrangements. We expect to adopt
this guidance on January 1, 2011 and it will be applied
prospectively for revenue arrangements entered into or
materially modified after the date of adoption. We are
evaluating the impact this guidance will have on our financial
position, results of operations, cash flows and disclosures.
Results
of Operations for the Years Ended December 31, 2009, 2008
and 2007
Revenue
Revenue recognized under our collaboration agreements for the
years ended December 31, 2009, 2008 and 2007 was as follows
(in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
|
|
Pfizer
|
|
$
|
15,855
|
|
|
$
|
16,467
|
|
|
$
|
20,148
|
|
|
Facet
|
|
|
2,148
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total revenue
|
|
$
|
18,003
|
|
|
$
|
16,467
|
|
|
$
|
20,148
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenue increased to $18.0 million in 2009 from
$16.5 million in 2008. Revenue was $20.1 million in
2007. The increase in 2009 compared to 2008 was due to revenue
recognized from our Facet collaboration of $2.1 million.
The $2.1 million is comprised of $0.8 million for
recognition of the $20 million up-front fee and
$1.4 million equity premium and $1.3 million for
collaborative research funding. The increase in revenue related
to our Facet collaboration was partially offset by lower revenue
recognized from our Pfizer collaboration due to an extension of
the recognition period of the up-front fee and lower
reimbursement revenue due to lower costs related to the Phase 2b
clinical trial for TRU-015 in the treatment of RA. Revenue from
our Pfizer collaboration for the year ended December 31,
2009 was comprised of $11.0 million for collaborative
research funding and $4.9 million for recognition of the
$40 million up-front fee.
The decrease in 2008 compared to 2007 was due to a decrease in
reimbursement revenue from our Pfizer collaboration related to
the Phase 2b clinical trial for TRU-015 in the treatment of RA,
an extension of the recognition of the up-front fee and a
decline in reimbursable legal costs. Revenue for the year ended
December 31, 2008 was comprised of $11.1 million for
Pfizer collaborative research funding and $5.4 million for
recognition of the $40 million Pfizer up-front fee.
The Pfizer and Facet up-front fees are being deferred and
recognized on a straight-line basis over the estimated term of
the research and development service periods. The Pfizer
estimated service period is through 2012 and the Facet estimated
service period is through 2018. Reimbursement revenue is
expected to fluctuate in the future due to the timing of
reimbursed development and legal costs, and the recognition of
the associated collaborative research revenue under our
collaboration agreements. However, we expect revenue to increase
in the future as a result of the additional collaboration
revenue from our Facet collaboration. This increase in revenue
from the Facet collaboration will be partially offset by
decreases in revenue from Pfizer due to the transfer to Pfizer
of the responsibility for the majority of the clinical
development efforts and related costs for product candidates
covered by our collaboration agreement with Pfizer. Our actual
revenue, however, could differ materially from anticipated
revenue.
Research
and Development Expenses
Research and development expenses increased to
$34.4 million from $31.6 million in 2008. Research and
development expenses were $36.5 million in 2007. The
increase in 2009 compared to 2008 was primarily due to higher
outside manufacturing and clinical development costs related to
our TRU-016 product candidate, partially offset by decreased lab
expense and personnel costs. In connection with the
restructuring in February 2009, we incurred a $0.8 million
charge in the first quarter of 2009 related to employee
severance, benefits and outplacement services, $0.6 million
of which was classified as research and development expense.
The decrease in 2008 compared to 2007 was primarily due to
decreased outside manufacturing costs related to our TRU-016
product candidate, decreased clinical costs related to our Phase
2b clinical trial for TRU-015 and decreased costs for lab
expenses for TRU-016. We expect research and development
expenses to
48
increase in the future due to additional clinical trials and
manufacturing expenses associated with TRU-016 development and
increases in preclinical research. These costs may fluctuate
depending on which party in the Facet collaboration is incurring
the majority of the development costs in any particular period.
Our actual research and development expenses could differ
materially from those anticipated.
At any time, we have many ongoing research projects. Our
internal resources, employees and infrastructure are not
directly tied to any individual research project and are
typically deployed across multiple projects. Through our
clinical development programs, we are developing each of our
product candidates in parallel for multiple disease indications,
and through our basic research activities, we are seeking to
design potential drug candidates for multiple new disease
indications. Due to the number of ongoing projects and our
ability to utilize resources across several projects, we do not
record or maintain information regarding the costs incurred for
our research and development programs on a program-specific
basis. In addition, we believe that allocating costs on the
basis of time incurred by our employees does not accurately
reflect the actual costs of a project.
Our research and development activities can be divided into
research and preclinical programs and clinical development
programs. The costs associated with research and preclinical
programs and clinical development programs approximate the
following (in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
|
|
Research and preclinical programs
|
|
$
|
17,954
|
|
|
$
|
20,257
|
|
|
$
|
21,344
|
|
|
Clinical development programs:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
TRU-015
|
|
|
7,168
|
|
|
|
7,423
|
|
|
|
9,296
|
|
|
TRU-016
|
|
|
7,659
|
|
|
|
2,009
|
|
|
|
4,587
|
|
|
Indirect
|
|
|
1,615
|
|
|
|
1,919
|
|
|
|
1,239
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total clinical development programs
|
|
|
16,442
|
|
|
|
11,351
|
|
|
|
15,122
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total research and development
|
|
$
|
34,396
|
|
|
$
|
31,608
|
|
|
$
|
36,466
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and preclinical program costs consist of costs
associated with our product development efforts, conducting
preclinical studies, personnel costs, lab expenses and indirect
costs such as rent, utilities and depreciation. Research and
preclinical program costs decreased in 2009 compared to 2008 due
to decreased lab expense and personnel costs as a result of the
restructuring in February 2009. Research and preclinical program
costs decreased in 2008 compared to 2007 due to decreased lab
expenses for TRU-016 as this program moved from a preclinical
program to a clinical program.
Clinical development costs consist of direct expenses such as
clinical manufacturing costs, clinical trial site and
investigator fees. Indirect costs include items such as
personnel costs, rent, utilities and depreciation. Costs for
TRU-015 decreased in 2009 compared to 2008 and 2007 due to lower
costs related to the Phase 2b clinical trial for TRU-015 in the
treatment of RA. Costs for TRU-016 increased in 2009 compared to
2008 due to increased manufacturing costs and clinical
development costs. Costs for TRU-016 decreased in 2008 compared
to 2007 due to decreased outside manufacturing costs and
decreased lab supplies. Costs for SBI-087 are incurred by our
partner, Pfizer, and as such are not included in the table above.
The majority of our research and development programs are at an
early stage and may not result in any approved products. Product
candidates that may appear promising at early stages of
development may not reach the market for a variety of reasons.
Product candidates may be found to be ineffective or to cause
harmful side effects during clinical trials, may take longer to
pass through clinical trials than had been anticipated, may fail
to receive necessary regulatory approvals and may prove
impracticable to manufacture in commercial quantities at
reasonable cost and with acceptable quality. As part of our
business strategy, we may enter into collaborative arrangements
with third parties to complete the development and
commercialization of our product candidates and it is uncertain
which of our product candidates may be subject to future
collaborative arrangements. The participation of a collaborative
partner may accelerate the time to completion and reduce the
cost to us of a product candidate or it may delay the time to
completion and increase the cost to us due to the alteration of
our existing strategy.
49
As a result of the uncertainties discussed above, the
uncertainty associated with clinical trial enrollments, and the
risks inherent in the development process, we are unable to
determine the duration and completion costs of the current or
future clinical stages of our product candidates or when, or to
what extent, we will generate revenue from the commercialization
and sale of any of our product candidates. Development
timelines, probability of success and development costs vary
widely. Under our collaboration with Pfizer, we are responsible
for completing the Phase 2a and 2b clinical retreatment trials
of TRU-015 for RA. In addition, we are responsible for
conducting clinical studies for TRU-015 niche indications. Under
our collaboration agreement with Facet, we are the lead party
responsible for the ongoing clinical trial for patients with
CLL, manufacturing activities, and regulatory activities. While
we are currently focused on developing TRU-015, SBI-087 and
other preclinical product candidates with Pfizer and our TRU-016
product candidate with Facet, together with other
SMIPtm
product candidates that are outside of our collaborations, we
will make determinations as to which programs to pursue and how
much funding to direct to each program on an ongoing basis in
response to the scientific and clinical success of each product
candidate, as well as an ongoing assessment as to the product
candidate’s commercial potential and value to potential
partners. In addition, due to the limited availability of
capital we may not be able to fund programs adequately, or at
all. We anticipate developing additional product candidates,
which will also increase our research and development expenses
in future periods. We do not expect any of our current product
candidates to be commercially available in major markets before
2014, if at all.
General
and Administrative Expenses
General and administrative expenses increased to
$12.4 million in 2009 from $11.4 million in 2008.
General and administrative expenses were $10.8 million in
2007. The increase in 2009 compared to 2008 was primarily due to
charges associated with the resignation of our former CEO,
partially offset by lower personnel-related costs as a result of
the restructuring in February 2009. In connection with the
restructuring in February 2009, we incurred a $0.8 million
charge in the first quarter of 2009 related to employee
severance, benefits and outplacement services, $0.2 million
of which was classified as general and administrative expense.
The increase in 2008 compared to 2007 was primarily due to
increased consulting and outside service fees and increased
non-cash stock-based compensation expense partially offset by
decreased fees related to filings for the protection of our
intellectual property. We expect our general and administrative
expenses to remain relatively stable in the future. Our actual
general and administrative expenses could differ materially from
those anticipated.
Net
Interest Income (Expense)
Net interest income (expense) decreased to an expense of
$0.4 million in 2009 from a gain of $1.0 million in
2008 and a gain of $3.8 million in 2007. The decreases were
the result of a decline in interest rates and a decrease in our
average cash and investment balance We expect net interest
expense to increase in the near future as a result of lower
interest income from a declining cash balance and low interest
rates that are not great enough to exceed interest expense on
our debt.
Income
Taxes
Since inception, we have incurred operating losses and,
accordingly, have not recorded a provision for income taxes for
any of the periods presented. As of December 31, 2009, we
had net operating loss carry forwards for federal income tax
purposes of $64.2 million. We also had federal research and
development tax credit carry forwards of $2.7 million. If
not utilized, the net operating loss and tax credit carry
forwards will expire between 2021 and 2029.
Liquidity
and Capital Resources
As of December 31, 2009, we had $54.8 million in cash,
cash equivalents and investments. We have received the majority
of our funding from the issuance of common stock, proceeds from
our collaboration agreements, asset-based lease financings and
interest earned on investments. Our cash and investment balances
are held in a variety of interest bearing instruments, including
obligations of United States government
50
agencies, high credit rating corporate borrowers, and money
market accounts. We do not hold auction rate securities within
our investment portfolio and as of December 31, 2009 we did
not hold any corporate securities. Cash in excess of immediate
requirements is invested with regard to liquidity and capital
preservation.
Operating Activities. Net cash used in
operating activities was $5.1 million, $23.9 million
and $26.4 million for the years ended December 31,
2009, 2008 and 2007, respectively. Net cash used in operations
during 2009 was due to personnel-related costs, clinical trial
costs, manufacturing costs, legal and professional fees,
facilities costs, lab supplies to support our research
activities and administrative costs, partially offset by
up-front fees received under the Facet collaboration. Net cash
used in operations during 2008 was primarily used for
personnel-related costs, clinical trial costs, legal and
professional fees, lab supplies to support our research
activities, facilities costs and administrative costs. We expect
net cash used in operations to increase in the future as we
continue to expand our clinical activities.
Investing Activities. Net cash used in
investing activities was $9.9 million in the year ended
December 31, 2009. Net cash provided by investing
activities was $12.4 million and $9.1 million for the
years ended December 31, 2008 and 2007. Investing
activities consist primarily of purchases, sales and maturities
of marketable securities and capital purchases. Our purchases of
securities increased during 2009 as a result of the
$30.0 million in cash provided to us as part of the
collaboration agreement with Facet. Additionally, we had lower
maturities in 2009 as a result of a lower average cash and
investment balance. Purchases of property and equipment were
$85,000, $1.3 million and $3.8 million in the years
ended December 31, 2009, 2008 and 2007, respectively.
Financing Activities. Net cash provided by
financing activities was $7.4 million in the year ended
December 31, 2009 compared to net cash used in financing
activities of $373,000 in the year ended December 31, 2008.
Net cash provided by financing activities was $2.7 million
in the year ended December 31, 2007. In 2009 financing
activities consisted primarily of a private placement of common
stock to Facet of $8.6 million and payments on an equipment
financing arrangement of $1.3 million. In 2008, financing
activities consisted primarily of $10.0 million in proceeds
under a new debt facility, offset by $9.5 million in
payments against pre-existing equipment financing arrangements
and $900,000 in other debt payments. In 2007, financing
activities consisted primarily of net proceeds from an equipment
financing arrangement of $2.2 million.
We entered into a loan and security agreement with Silicon
Valley Bank, or SVB, effective July 25, 2008, the terms of
which provide for a $10.0 million debt facility secured by
a security interest in our assets, other than intellectual
property, and used $8.5 million of the proceeds from this
debt facility to fully extinguish our obligations with Comerica
Bank, or Comerica, under our existing debt facility. In
conjunction with extinguishing our obligations under the
Comerica debt facility, we also terminated the Comerica loan and
security agreement and related interest rate swap agreement. We
incurred a fee of $165,000 in connection with the termination of
the interest rate swap agreement, which is included in interest
expense in the statements of operations for the year ended
December 31, 2008. The full $10.0 million available
under the SVB facility was drawn at closing and is payable in
fixed equal payments of principal plus accrued interest at a
fixed rate of 5.75% based on an
84-month
amortization schedule with all principal and interest due
July 25, 2013. As of December 31, 2009,
$8.3 million was outstanding under the SVB loan and
security agreement.
The loan and security agreement with SVB contains
representations and warranties and affirmative and negative
covenants that are customary for credit facilities of this type.
The debt covenants in place by the loan and security agreement
with SVB require us to maintain a liquidity coverage of at least
1.5:1 and remaining months liquidity of at least 3:1. We were in
compliance with all covenants under the loan and security
agreement as of December 31, 2009. The loan and security
agreement could restrict our ability to, among other things,
sell certain assets, engage in a merger or change in control
transaction, incur debt, pay cash dividends, and make
investments. The loan and security agreement also contains
events of default that are customary for credit facilities of
this type, including payment defaults, covenant defaults,
insolvency type defaults, and events of default relating to
liens, judgments, material misrepresentations, and the
occurrence of certain material adverse events. In addition, the
loan and security agreement with SVB contains a material
51
adverse change clause which may accelerate the maturity of the
loan upon the occurrence of certain events. We have no
indication that we are in default of the material adverse change
clause and no scheduled loan payments have accelerated as a
result of this provision.
Based on our current operating plans, we believe that our
existing capital resources, together with interest thereon, will
be sufficient to meet our financial obligations for at least the
next 18 months. The key assumption underlying this estimate
is that expenditures related to continued preclinical,
manufacturing, and clinical development of our product
candidates during this period will be within budgeted levels.
Our forecast of the period of time that our financial resources
will be adequate to support operations is a forward-looking
statement and involves risks and uncertainties, and actual
results could vary as a result of a number of factors, including
the factors discussed in the section of Item 1A entitled
“Risk Factors.” In light of the numerous risks and
uncertainties associated with the development and
commercialization of our product candidates and the extent to
which we enter into collaborations with third parties to
participate in their development and commercialization, we are
unable to estimate the amounts of increased capital outlays and
operating expenditures associated with product development. Our
future funding requirements will depend on many factors,
including:
|
|
|
| |
•
|
the ability to raise capital through strategic partnerships or
in the debt/equity markets;
|
| |
| |
•
|
the terms and timing of any additional collaborative or
licensing agreements that we may establish;
|
| |
| |
•
|
milestone payments projected to be received under the Pfizer and
Facet collaboration agreements;
|
| |
| |
•
|
the determination by any of our current collaboration partners
to cease developing any product candidate that is the subject of
that collaboration;
|
| |
| |
•
|
the scope, rate of progress, results and costs of our
preclinical testing, clinical trials, and other research and
development activities;
|
| |
| |
•
|
the number of programs we pursue;
|
| |
| |
•
|
the cost of establishing clinical and commercial supplies of our
product candidates;
|
| |
| |
•
|
the cost of preparing, filing, prosecuting, defending, and
enforcing any patent claims and other intellectual property
rights;
|
| |
| |
•
|
the cost, timing, and outcomes of regulatory approvals; and
|
| |
| |
•
|
the extent to which we acquire or invest in businesses,
products, or technologies.
|
We will need to raise additional funds to support our
operations, and such funding may not be available to us on
acceptable terms, if at all. The capital markets have been
experiencing extreme volatility and disruption. The scope and
extent of this disruption in the capital markets could make it
difficult or impossible to raise additional capital in public or
private capital markets until conditions stabilize. If we are
unable to raise additional funds when needed, we may not be able
to continue development of our product candidates or we could be
required to delay, scale back, or eliminate some or all of our
development programs and other operations. We may seek to raise
additional funds through public or private financing, strategic
partnerships, or other arrangements. Any additional equity
financing may be dilutive to stockholders and debt financing, if
available, may involve restrictive covenants. If we raise funds
through collaborative or licensing arrangements we may be
required to relinquish, on terms that are not favorable to us,
rights to some of our technologies or product candidates that we
would otherwise seek to develop or commercialize ourselves. Our
failure to raise capital when needed may harm our business and
operating results.
52
Our future contractual obligations as of December 31, 2009
were as follows (in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Payments Due by Period
|
|
|
|
|
Total
|
|
|
1 Year
|
|
|
2-3 Years
|
|
|
4-5 Years
|
|
|
Thereafter
|
|
|
|
|
Notes payable (including interest)
|
|
$
|
9,531
|
|
|
$
|
1,744
|
|
|
$
|
3,486
|
|
|
$
|
4,301
|
|
|
$
|
—
|
|
|
Manufacturing commitment
|
|
|
2,100
|
|
|
|
2,100
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Operating lease obligations
|
|
|
4,934
|
|
|
|
1,490
|
|
|
|
2,952
|
|
|
|
492
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
16,565
|
|
|
$
|
5,334
|
|
|
$
|
6,438
|
|
|
$
|
4,793
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Off-Balance
Sheet Arrangements
Since inception, we have not engaged in any off-balance sheet
arrangements, including the use of structured finance, special
purpose entities or variable interest entities.
|
|
|
ITEM 7A.
|
QUANTITATIVE
AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
|
Our exposure to market risk is primarily confined to our
investment securities. The primary objective of our investment
activities is to preserve our capital to fund operations. We
also seek to maximize income from our investments without
assuming significant risk. To achieve our objectives, we
maintain a portfolio of investments in a variety of securities
of high credit quality. As of December 31, 2009 our
portfolio of investments consisted of money market accounts and
United States treasury securities. We have no exposure to
auction rate securities within our investment portfolio. The
securities in our investment portfolio are not leveraged, are
classified as available for sale and, due to their short-term
nature, are subject to minimal interest rate risk. We currently
do not hedge interest rate exposure on our investment
securities. We actively monitor changes in interest rates.
We are exposed to potential loss due to changes in interest
rates. Our principal interest rate exposure is to changes in
U.S. interest rates related to our investment securities.
To estimate the potential loss due to changes in interest rates,
we performed a sensitivity analysis using the instantaneous
adverse change in interest rates of 100 basis points across
the yield curve. On this basis, we estimate the potential loss
in fair value that would result from a hypothetical 1%
(100 basis points) increase in interest rates to be $86,000
and $47,000 as of December 31, 2009 and 2008, respectively.
|
|
|
ITEM 8.
|
FINANCIAL
STATEMENTS AND SUPPLEMENTARY DATA
|
Our financial statements, together with related notes, are
listed in Item 15(a) and included herein beginning on
page 57.
|
|
|
ITEM 9.
|
CHANGES
IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND
FINANCIAL DISCLOSURE
|
None.
|
|
|
ITEM 9A.
|
CONTROLS
AND PROCEDURES
|
Evaluation of Disclosure Controls and
Procedures. As of the end of the period covered
by this annual report, we carried out an evaluation under the
supervision and with the participation of our management,
including our Executive Chairman and Acting President and Chief
Financial Officer, of the effectiveness of our disclosure
controls and procedures pursuant to Exchange Act
Rules 13a — 15 (e) and 15d — 15
(e). Based on this evaluation, our Executive Chairman and Acting
President and Chief Financial Officer have concluded that, as of
the end of the period covered by this annual report, the
disclosure controls and procedures were effective.
Internal Control Over Financial
Reporting. Management is responsible for
establishing and maintaining adequate internal control over
financial reporting, as such term is defined in Exchange Act
Rules 13a-15(f)
and
15d-15(f).
Internal control over financial reporting is a process designed
to provide reasonable assurance
53
regarding the reliability of financial reporting and the
preparation of financial statements for external purposes in
accordance with generally accepted accounting principles.
Internal control over financial reporting cannot provide
absolute assurance of achieving financial reporting objectives
because of its inherent limitations. Internal control over
financial reporting is a process that involves human diligence
and compliance and is subject to lapses in judgment and
breakdowns resulting from human failures. Internal control over
financial reporting also can be circumvented by collusion or
improper management override. Because of such limitations, there
is a risk that material misstatements may not be prevented or
detected on a timely basis by internal control over financial
reporting. However, these inherent limitations are known
features of the financial reporting process. Therefore, it is
possible to design into the process safeguards to reduce, though
not eliminate, this risk.
Management conducted an assessment of the effectiveness of our
internal control over financial reporting as of
December 31, 2009. In making this assessment, management
used the criteria set forth by the Committee of Sponsoring
Organizations of the Treadway Commission in Internal
Control — Integrated Framework. Based on the
results of this assessment and on those criteria, management
concluded that our internal control over financial reporting was
effective as of December 31, 2009.
The effectiveness of the our internal control over financial
reporting as of December 31, 2009 has been audited by
Ernst & Young LLP, an independent registered public
accounting firm, as stated in their report that appears herein.
There was no change in our internal control over financial
reporting during our last fiscal quarter that materially
affected, or is reasonably likely to materially affect, our
internal control over financial reporting.
54
Report of
Independent Registered Public Accounting Firm on Internal
Control Over Financial Reporting
The Board of Directors and Stockholders
Trubion Pharmaceuticals, Inc.
We have audited Trubion Pharmaceuticals, Inc.’s internal
control over financial reporting as of December 31, 2009,
based on criteria established in Internal Control —
Integrated Framework issued by the Committee of Sponsoring
Organizations of the Treadway Commission (the COSO criteria).
Trubion Pharmaceuticals Inc.’s management is responsible
for maintaining effective internal control over financial
reporting, and for its assessment of the effectiveness of
internal control over financial reporting included in the
accompanying Management’s Report on Internal Control over
Financial Reporting. Our responsibility is to express an opinion
on the Company’s internal control over financial reporting
based on our audit.
We conducted our audit in accordance with the standards of the
Public Company Accounting Oversight Board (United States). Those
standards require that we plan and perform the audit to obtain
reasonable assurance about whether effective internal control
over financial reporting was maintained in all material
respects. Our audit included obtaining an understanding of
internal control over financial reporting, assessing the risk
that a material weakness exists, testing and evaluating the
design and operating effectiveness of internal control based on
the assessed risk, and performing such other procedures as we
considered necessary in the circumstances. We believe that our
audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a
process designed to provide reasonable assurance regarding the
reliability of financial reporting and the preparation of
financial statements for external purposes in accordance with
generally accepted accounting principles. A company’s
internal control over financial reporting includes those
policies and procedures that (1) pertain to the maintenance
of records that, in reasonable detail, accurately and fairly
reflect the transactions and dispositions of the assets of the
company; (2) provide reasonable assurance that transactions
are recorded as necessary to permit preparation of financial
statements in accordance with generally accepted accounting
principles, and that receipts and expenditures of the company
are being made only in accordance with authorizations of
management and directors of the company; and (3) provide
reasonable assurance regarding prevention or timely detection of
unauthorized acquisition, use, or disposition of the
company’s assets that could have a material effect on the
financial statements.
Because of its inherent limitations, internal control over
financial reporting may not prevent or detect misstatements.
Also, projections of any evaluation of effectiveness to future
periods are subject to the risk that controls may become
inadequate because of changes in conditions, or that the degree
of compliance with the policies or procedures may deteriorate.
In our opinion, Trubion Pharmaceuticals, Inc. maintained, in all
material respects, effective internal control over financial
reporting as of December 31, 2009, based on the COSO
criteria.
We also have audited, in accordance with the standards of the
Public Company Accounting Oversight Board (United States), the
2009 financial statements of Trubion Pharmaceuticals, Inc. and
our report dated March 15, 2010 expressed an unqualified
opinion thereon.
Seattle, Washington
March 15, 2010
55
|
|
|
ITEM 9B.
|
OTHER
INFORMATION
|
None.
PART III
|
|
|
ITEM 10.
|
DIRECTORS,
EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
|
The information required by this Item is contained in part in
the sections captioned “Board of Directors,”
“Summary of Trubion’s Corporate Governance
Guidelines,” “Committees of the Board of
Directors,” “Executive Officers” and
“Section 16(a) Beneficial Ownership Reporting
Compliance” in the proxy statement for Trubion’s
Annual Meeting of Stockholders scheduled to be held on or around
May 26, 2010, and such information is incorporated herein
by reference.
|
|
|
ITEM 11.
|
EXECUTIVE
COMPENSATION
|
The information required by this Item is contained in the
section captioned “Executive Compensation,” “2009
Director Compensation” and “Committee of the Board of
Directors — Compensation Committee Matters —
Compensation Committee Interlocks and Insider
Participation” of the proxy statement for Trubion’s
Annual Meeting of Stockholders scheduled to be held on or around
May 26, 2010, and such information is incorporated herein
by reference.
|
|
|
ITEM 12.
|
SECURITY
OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND
RELATED STOCKHOLDER MATTERS
|
The information required by this Item is contained in part in
the sections captioned “Voting Securities and Principal
Holders” and “Other Matters — Securities
Authorized for Issuance under Equity Compensation Plans” in
the proxy statement for Trubion’s Annual Meeting of
Stockholders scheduled to be held on or around May 26,
2010, and such information is incorporated herein by reference.
|
|
|
ITEM 13.
|
CERTAIN
RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR
INDEPENDENCE
|
The information required by this Item is contained in the
sections captioned “Transactions with Related Persons”
and “Summary of Trubion’s Corporate Governance
Guidelines” of the proxy statement for Trubion’s
Annual Meeting of Stockholders scheduled to be held on or around
May 26, 2010, and such information is incorporated herein
by reference.
|
|
|
ITEM 14.
|
PRINCIPAL
ACCOUNTANT FEES AND SERVICES
|
The information required by this Item is contained in the
section captioned “Audit Committee Matters —
Independent Auditor Fees” of the proxy statement for
Trubion’s Annual Meeting of Stockholders scheduled to be
held on or around May 26, 2010, and such information is
incorporated herein by reference.
56
PART IV
|
|
|
ITEM 15.
|
EXHIBITS AND
FINANCIAL STATEMENT SCHEDULES
|
(a) 1. Financial Statements
The financial statements required by this item are included
herein:
| |
|
|
|
|
|
|
|
Page
|
|
|
|
No.
|
|
|
|
|
|
|
61
|
|
|
|
|
|
62
|
|
|
|
|
|
63
|
|
|
|
|
|
64
|
|
|
|
|
|
67
|
|
|
|
|
|
68
|
|
(a) 2. Financial Statement Schedules
None.
(a) 3. Exhibits
| |
|
|
|
|
Exhibit
|
|
|
|
Number
|
|
Description
|
|
|
|
|
3
|
.1(1)
|
|
Amended and Restated Certificate of Incorporation
(exhibit 3.1)
|
|
|
3
|
.2(11)
|
|
Amended and Restated Bylaws (exhibit 3.1)
|
|
|
4
|
.1(2)
|
|
Form of common stock certificate (exhibit 4.1)
|
|
|
4
|
.2(1)
|
|
Amended and Restated Investor Rights Agreement, dated
July 13, 2004 (exhibit 4.2)
|
|
|
4
|
.3(1)
|
|
Amendment No. 1 to Amended and Restated Investor Rights
Agreement, dated December 19, 2005 (exhibit 4.3)
|
|
|
10
|
.1(1)+
|
|
Form of Indemnification Agreement entered into between the
registrant and its officers and its non-fund designated
directors (exhibit 10.1)
|
|
|
10
|
.2(13)+
|
|
Form of Indemnification Agreement entered into between the
registrant and its fund-designated directors (exhibit 10.1)
|
|
|
10
|
.3(1)+
|
|
2002 Stock Plan (exhibit 10.2)
|
|
|
10
|
.4(1)+
|
|
Form of Stock Option Agreement under the 2002 Stock Plan
(exhibit 10.3)
|
|
|
10
|
.5(1)+
|
|
2002 Equity Incentive Plan (exhibit 10.4)
|
|
|
10
|
.6(1)+
|
|
Form of Stock Option Agreement under the 2002 Equity Incentive
Plan (exhibit 10.5)
|
|
|
10
|
.7(2)+
|
|
2006 Equity Incentive Plan (exhibit 10.6)
|
|
|
10
|
.8(2)+
|
|
Form of Stock Option Agreement under the 2006 Equity Incentive
Plan (exhibit 10.7)
|
|
|
10
|
.9(1)
|
|
Lease Agreement between the registrant and Selig Real Estate
Holdings Eight, dated April 28, 2003 (exhibit 10.8)
|
|
|
10
|
.10(1)
|
|
Amendment to Lease Agreement between the registrant and Selig
Real Estate Holdings Eight, dated December 8, 2004
(exhibit 10.9)
|
|
|
10
|
.11(1)
|
|
Amendment to Lease Agreement between the registrant and Selig
Real Estate Holdings Eight, dated February 1, 2006
(exhibit 10.10)
|
|
|
10
|
.12(10)
|
|
Amendment to Lease Agreement between the registrant and Selig
Real Estate Holdings Eight, L.L.C, dated February 2, 2007
(exhibit 10.1)
|
|
|
10
|
.13(3)
|
|
Collaboration and License Agreement between the registrant and
Wyeth, acting through Wyeth Pharmaceuticals Division, dated
December 19, 2005 (exhibit 10.11)
|
57
| |
|
|
|
|
Exhibit
|
|
|
|
Number
|
|
Description
|
|
|
|
|
10
|
.14(5)†
|
|
Amendment No. 1 to the Collaboration and License Agreement
between the registrant and Wyeth, acting through Wyeth
Pharmaceuticals Division, dated November 30, 2006
(exhibit 10.12)
|
|
|
10
|
.15(1)
|
|
Common Stock Purchase Agreement between the registrant and
Wyeth, dated December 19, 2005 (exhibit 10.12)
|
|
|
10
|
.16(14)†
|
|
Collaboration and License Agreement between the registrant and
Facet Biotech Corporation, dated August 27, 2009
(exhibit 10.1)
|
|
|
10
|
.17(14)†
|
|
Manufacturing Services Agreement between the registrant and
Lonza Biologics dated November 21, 2005 (exhibit 10.2)
|
|
|
10
|
.18(10)
|
|
Novation Agreement effective as of January 1, 2007, among
the registrant, Lonza Biologics, Inc. and Lonza Sales AG,
effective January 1, 2007 (exhibit 10.3)
|
|
|
10
|
.19(14)†
|
|
Amendment, dated December 5, 2008, to the Manufacturing
Services Agreement dated November 21, 2005 between the
registrant and Lonza Sales AG (exhibit 10.4)
|
|
|
10
|
.20(14)†
|
|
Quality Agreement for TRU-016 between the registrant and Lonza
Sales AG dated December 5, 2008 (exhibit 10.5)
|
|
|
10
|
.21(14)†
|
|
Second Amendment, dated April 3, 2009, to the Manufacturing
Services Agreement dated November 21, 2005 between the
registrant and Lonza Sales AG (exhibit 10.6)
|
|
|
10
|
.23(7)+
|
|
Employment Agreement between the registrant and Peter A.
Thompson, M.D., FACP dated March 21, 2008
(exhibit 10.1)
|
|
|
10
|
.24(7)+
|
|
Employment Agreement between the registrant and Michelle G.
Burris dated March 21, 2008 (exhibit 10.2)
|
|
|
10
|
.26(7)+
|
|
Employment Agreement between the registrant and Kathleen M.
Deeley dated March 21, 2008 (exhibit 10.4)
|
|
|
10
|
.27(7)+
|
|
Employment Agreement between the registrant and Kendall M.
Mohler, Ph.D. dated March 21, 2008 (exhibit 10.5)
|
|
|
10
|
.28(8)+
|
|
Form of Amendment No. 1 to Employment Agreement between the
registrant and each of Michelle G. Burris, Kathleen M. Deeley
and Kendall M. Mohler (exhibit 10.1)
|
|
|
10
|
.29(12)+
|
|
Employment Agreement between the registrant and Scott C.
Stromatt, M.D. dated March 11, 2009 (exhibit 10.1)
|
|
|
10
|
.30*+
|
|
Employment Agreement between the registrant and John A. Bencich
dated November 16, 2009
|
|
|
10
|
.31(1)
|
|
Consulting Agreement with Lee R. Brettman, M.D., FACP,
dated January 1, 2003 (exhibit 10.31)
|
|
|
10
|
.32(1)
|
|
Letter from Oxford Finance Corporation, dated April 2, 2003
(exhibit 10.33)
|
|
|
10
|
.33(1)
|
|
Letter from Oxford Finance Corporation, dated November 3,
2004 (exhibit 10.34)
|
|
|
10
|
.34(1)
|
|
Master Security Agreement with Oxford Finance Corporation, dated
June 18, 2003 (exhibit 10.35)
|
|
|
10
|
.35(1)
|
|
Form of Oxford Finance Corporation Promissory Note
(exhibit 10.36)
|
|
|
10
|
.36(1)†
|
|
Technology & Investment Agreement by and among the
registrant, Jeffrey A. Ledbetter, Ph.D., Martha
Hayden-Ledbetter, Ph.D., and the Pacific Northwest Research
Institute, dated December 31, 2001 (exhibit 10.39)
|
|
|
10
|
.37(4)
|
|
Independent Contractor Agreement between the registrant and
Martha Hayden-Ledbetter, Ph.D., dated May 1, 2004
(exhibit 10.40)
|
|
|
10
|
.38(9)
|
|
Loan and Security Agreement between the registrant and Silicon
Valley Bank, dated July 25, 2008 (exhibit 99.1)
|
|
|
10
|
.39(6)
|
|
First Amendment to Loan and Security Agreement between the
registrant and Comerica Bank, entered into as of July 24,
2007 (exhibit 10.1)
|
|
|
23
|
.1*
|
|
Consent of Independent Registered Public Accounting Firm
|
|
|
24
|
.1*
|
|
Power of Attorney (included on signature page)
|
58
| |
|
|
|
|
Exhibit
|
|
|
|
Number
|
|
Description
|
|
|
|
|
31
|
.1*
|
|
Certification of Chief Executive Officer of Trubion
Pharmaceuticals, Inc., Pursuant to Exchange Act
Rules 13a-14(a)
and 15d-14(a), as Adopted Pursuant to Section 302 of the
Sarbanes-Oxley Act of 2002
|
|
|
31
|
.2*
|
|
Certification of Chief Financial Officer of Trubion
Pharmaceuticals, Inc., Pursuant to Exchange Act
Rules 13a-14(a)
and 15d-14(a), as Adopted Pursuant to Section 302 of the
Sarbanes-Oxley Act of 2002
|
|
|
32*
|
|
|
Certification of Chief Executive Officer and Chief Financial
Officer of Trubion Pharmaceuticals, Inc., Pursuant to
18 U.S.C. Section 1350, as Adopted Pursuant to
Section 906 of the Sarbanes-Oxley Act of 2002
|
|
|
|
|
* |
|
Filed herewith |
| |
|
(1) |
|
Incorporated by reference to the designated exhibit to the
registrant’s Registration Statement on
Form S-1
filed with the SEC on June 2, 2006 (File
No. 333-134709). |
| |
|
(2) |
|
Incorporated by reference to the designated exhibit to the
registrant’s Registration Statement on
Form S-1
filed with the SEC on October 2, 2006 (File
No. 333-134709). |
| |
|
(3) |
|
Incorporated by reference to the designated exhibit to the
registrant’s Registration Statement on
Form S-1
filed with the SEC on October 5, 2006 (File
No. 333-134709). |
| |
|
(4) |
|
Incorporated by reference to the designated exhibit to the
registrant’s Registration Statement on
Form S-1
filed with the SEC on July 18, 2006 (File
No. 333-134709). |
| |
|
(5) |
|
Incorporated by reference to the designated exhibit to the
registrant’s Annual Report on
Form 10-K
filed with the SEC on March 26, 2007 (File
No. 001-33054). |
| |
|
(6) |
|
Incorporated by reference to the designated exhibit to the
registrant’s Current Report on
Form 8-K
filed with the SEC on July 26, 2007 (File
No. 001-33054). |
| |
|
(7) |
|
Incorporated by reference to the designated exhibit to the
registrant’s Current Report on
Form 8-K/A
filed with the SEC on April 8, 2008 (File
No. 001-33054). |
| |
|
(8) |
|
Incorporated by reference to the designated exhibit to the
registrant’s Current Report on
Form 8-K/A
filed with the SEC on April 8, 2008 (File
No. 001-33054). |
| |
|
(9) |
|
Incorporated by reference to the designated exhibit to the
registrant’s Current Report on
Form 8-K
filed with the SEC on July 29, 2008 (File
No. 001-33054). |
| |
|
(10) |
|
Incorporated by reference to the designated exhibit to the
registrant’s Quarterly Report on
Form 10-Q
filed with the SEC on August 7, 2008 (File
No. 001-33054). |
| |
|
(11) |
|
Incorporated by reference to the designated exhibit to the
registrant’s Current Report on
Form 8-K
filed with the SEC on November 26, 2008 (File
No. 001-33054). |
| |
|
(12) |
|
Incorporated by reference to the designated exhibit to the
registrant’s Quarterly Report on
Form 10-Q
filed with the SEC on May 7, 2009 (File
No. 001-33054). |
| |
|
(13) |
|
Incorporated by reference to the designated exhibit to the
registrant’s Current Report on
Form 8-K
filed with the SEC on August 13, 2009 (File
No. 001-33054). |
| |
|
(14) |
|
Incorporated by reference to the designated exhibit to the
registrant’s Quarterly Report on
Form 10-Q
filed with the SEC on November 5, 2009 (File
No. 001-33054). |
| |
|
† |
|
Portions of the agreement are subject to confidential treatment |
| |
|
+ |
|
Executive Compensation Plan or Agreement |
59
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the
Securities Exchange Act of 1934, as amended, the registrant has
duly caused this report to be signed on its behalf by the
undersigned, thereunto duly authorized in the city of Seattle,
state of Washington, on March 15, 2010.
TRUBION PHARMACEUTICALS, INC.
Steven Gillis, Ph.D.
Executive Chairman and
Acting President
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose
signature appears below constitutes and appoints Michelle Burris
and Kathleen Deeley, and each of them, with full power of
substitution and resubstitution and full power to act without
the other, as his or her true and lawful attorney-in-fact and
agent to act in his or her name, place, and stead and to execute
in the name and on behalf of each person, individually and in
each capacity stated below, and to file, any and all amendments
to this report, and to file the same, with all exhibits thereto,
and other documents in connection therewith, with the Securities
and Exchange Commission, granting unto said attorneys-in-fact
and agents, and each of them, full power and authority to do and
perform each and every act and thing, ratifying and confirming
all that said attorneys-in-fact and agents or any of them or
their and her substitute or substitutes, may lawfully do or
cause to be done by virtue thereof.
Pursuant to the requirements of the Securities Exchange Act of
1934, this report has been signed below by the following persons
on behalf of the registrant and in the capacities indicated and
on the dates indicated.
| |
|
|
|
|
|
|
|
Signature
|
|
Title
|
|
Date
|
|
|
|
|
|
|
|
|
|
/s/ Steven
Gillis
Steven
Gillis, Ph.D.
|
|
Executive Chairman and Acting President (Principal Executive
Officer)
|
|
March 15, 2010
|
|
|
|
|
|
|
|
/s/ John
A. Bencich
John
A. Bencich
|
|
Vice President and Chief Financial Officer (Principal Accounting
and Financial Officer)
|
|
March 15, 2010
|
|
|
|
|
|
|
|
/s/ Lee
R. Brettman
Lee
R. Brettman, M.D., FACP
|
|
Director
|
|
March 15, 2010
|
|
|
|
|
|
|
|
/s/ Patrick
J. Heron
Patrick
J. Heron
|
|
Director
|
|
March 15, 2010
|
|
|
|
|
|
|
|
/s/ Anders
D. Hove
Anders
D. Hove, M.D.
|
|
Director
|
|
March 15, 2010
|
|
|
|
|
|
|
|
/s/ David
A. Mann
David
A. Mann
|
|
Director
|
|
March 15, 2010
|
|
|
|
|
|
|
|
/s/ Samuel
R. Saks
Samuel
R. Saks, M.D.
|
|
Director
|
|
March 15, 2010
|
|
|
|
|
|
|
|
/s/ David
Schnell
David
Schnell, M.D.
|
|
Director
|
|
March 15, 2010
|
60
Report of
Independent Registered Public Accounting Firm
The Board of Directors and Stockholders
Trubion Pharmaceuticals, Inc.
We have audited the accompanying balance sheets of Trubion
Pharmaceuticals, Inc. as of December 31, 2009 and 2008, and
the related statements of operations, stockholders’ equity
(deficit), and cash flows for each of the three years in the
period ended December 31, 2009. These financial statements
are the responsibility of the Company’s management. Our
responsibility is to express an opinion on these financial
statements based on our audits.
We conducted our audits in accordance with the standards of the
Public Company Accounting Oversight Board (United States). Those
standards require that we plan and perform the audit to obtain
reasonable assurance about whether the financial statements are
free of material misstatement. An audit also includes examining,
on a test basis, evidence supporting the amounts and disclosures
in the financial statements, assessing the accounting principles
used and significant estimates made by management, and
evaluating the overall financial statement presentation. We
believe that our audits provide a reasonable basis for our
opinion.
In our opinion, the financial statements referred to above
present fairly, in all material respects, the financial position
of Trubion Pharmaceuticals, Inc. at December 31, 2009 and
2008, and the results of its operations and its cash flows for
each of the three years in the period ended December 31,
2009, in conformity with U.S. generally accepted accounting
principles.
As discussed in Note 1 to the financial statements, the
Company adopted the guidance related to the accounting for
nonrefundable advance payments for goods or services received
for use in future research and development activities as of
January 1, 2008.
We have also audited, in accordance with the standards of the
Public Company Accounting Oversight Board (United States), the
Company’s internal control over financial reporting as of
December 31, 2009, based on the criteria established in
Internal Control — Integrated Framework issued
by the Committee of Sponsoring Organizations of the Treadway
Commission and our report dated March 15, 2010 expressed an
unqualified opinion on the Company’s internal control over
financial reporting.
Seattle, Washington
March 15, 2010
61
TRUBION
PHARMACEUTICALS, INC.
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands, except share and par value)
|
|
|
|
|
ASSETS
|
|
Current assets:
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents
|
|
$
|
22,304
|
|
|
$
|
29,969
|
|
|
Investments
|
|
|
29,037
|
|
|
|
22,928
|
|
|
Receivable from collaborations
|
|
|
3,428
|
|
|
|
3,084
|
|
|
Prepaid expenses
|
|
|
977
|
|
|
|
2,112
|
|
|
|
|
|
|
|
|
|
|
|
|
Total current assets
|
|
|
55,746
|
|
|
|
58,093
|
|
|
Property and equipment, net
|
|
|
6,129
|
|
|
|
9,190
|
|
|
Long-term investments
|
|
|
3,505
|
|
|
|
—
|
|
|
Other assets
|
|
|
—
|
|
|
|
7
|
|
|
|
|
|
|
|
|
|
|
|
|
Total assets
|
|
$
|
65,380
|
|
|
$
|
67,290
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
LIABILITIES AND STOCKHOLDERS’ EQUITY
|
|
Current liabilities:
|
|
|
|
|
|
|
|
|
|
Accounts payable
|
|
$
|
379
|
|
|
$
|
301
|
|
|
Accrued liabilities
|
|
|
4,143
|
|
|
|
4,981
|
|
|
Accrued compensation
|
|
|
2,106
|
|
|
|
1,169
|
|
|
Current portion of notes payable
|
|
|
1,286
|
|
|
|
1,302
|
|
|
Current portion of deferred rent
|
|
|
135
|
|
|
|
180
|
|
|
Current portion of deferred revenue
|
|
|
7,167
|
|
|
|
4,873
|
|
|
|
|
|
|
|
|
|
|
|
|
Total current liabilities
|
|
|
15,216
|
|
|
|
12,806
|
|
|
Non-current portion of notes payable
|
|
|
6,975
|
|
|
|
8,261
|
|
|
Non-current portion of deferred rent
|
|
|
—
|
|
|
|
135
|
|
|
Non-current portion of deferred revenue
|
|
|
28,095
|
|
|
|
14,620
|
|
|
Commitments and contingencies
|
|
|
|
|
|
|
|
|
|
Stockholders’ equity:
|
|
|
|
|
|
|
|
|
|
Preferred stock, $0.001 par value per share; shares
authorized — 5,000,000 at December 31, 2009 and
2008; issued and outstanding — none at
December 31, 2009 and 2008
|
|
|
—
|
|
|
|
—
|
|
|
Common stock, $0.001 par value per share; shares
authorized — 150,000,000; outstanding —
20,381,561 and 17,882,307 at December 31, 2009 and 2008,
respectively
|
|
|
20
|
|
|
|
18
|
|
|
Additional paid-in capital
|
|
|
136,732
|
|
|
|
123,846
|
|
|
Deferred stock-based compensation
|
|
|
—
|
|
|
|
(30
|
)
|
|
Accumulated other comprehensive income (loss)
|
|
|
(6
|
)
|
|
|
103
|
|
|
Accumulated deficit
|
|
|
(121,652
|
)
|
|
|
(92,469
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Total stockholders’ equity
|
|
|
15,094
|
|
|
|
31,468
|
|
|
|
|
|
|
|
|
|
|
|
|
Total liabilities and stockholders’ equity
|
|
$
|
65,380
|
|
|
$
|
67,290
|
|
|
|
|
|
|
|
|
|
|
|
See accompanying notes
62
TRUBION
PHARMACEUTICALS, INC.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
|
|
(In thousands, except per share data)
|
|
|
|
|
Revenue:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Collaboration revenue
|
|
$
|
18,003
|
|
|
$
|
16,467
|
|
|
$
|
20,148
|
|
|
Operating expenses:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development
|
|
|
34,396
|
|
|
|
31,608
|
|
|
|
36,466
|
|
|
General and administrative
|
|
|
12,429
|
|
|
|
11,374
|
|
|
|
10,833
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses
|
|
|
46,825
|
|
|
|
42,982
|
|
|
|
47,299
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss from operations
|
|
|
(28,822
|
)
|
|
|
(26,515
|
)
|
|
|
(27,151
|
)
|
|
Interest income
|
|
|
173
|
|
|
|
1,781
|
|
|
|
4,607
|
|
|
Interest expense
|
|
|
(534
|
)
|
|
|
(825
|
)
|
|
|
(770
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
$
|
(29,183
|
)
|
|
$
|
(25,559
|
)
|
|
$
|
(23,314
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted net loss per share
|
|
$
|
(1.55
|
)
|
|
$
|
(1.43
|
)
|
|
$
|
(1.32
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Shares used in computation of basic and diluted net loss per
share
|
|
|
18,797
|
|
|
|
17,856
|
|
|
|
17,688
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
See accompanying notes
63
TRUBION
PHARMACEUTICALS, INC.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Other
|
|
|
|
|
|
Total
|
|
|
|
|
|
|
|
|
|
|
Additional
|
|
|
Deferred
|
|
|
Comprehensive
|
|
|
|
|
|
Stockholders’
|
|
|
|
|
Common Stock
|
|
|
Paid-in
|
|
|
Stock-Based
|
|
|
Income
|
|
|
Accumulated
|
|
|
Equity
|
|
|
|
|
Shares
|
|
|
Amount
|
|
|
Capital
|
|
|
Compensation
|
|
|
(Loss)
|
|
|
Deficit
|
|
|
(Deficit)
|
|
|
|
|
(In thousands, except share data)
|
|
|
|
|
Balance at January 1, 2007
|
|
|
17,554,318
|
|
|
$
|
18
|
|
|
$
|
117,061
|
|
|
$
|
(850
|
)
|
|
$
|
21
|
|
|
$
|
(43,596
|
)
|
|
$
|
72,654
|
|
|
Issuance of common stock upon exercise of stock options
|
|
|
237,852
|
|
|
|
—
|
|
|
|
468
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
468
|
|
|
Stock-based compensation to non-employees at fair value
|
|
|
—
|
|
|
|
—
|
|
|
|
91
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
91
|
|
|
Stock-based compensation expense
|
|
|
—
|
|
|
|
—
|
|
|
|
2,881
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
2,881
|
|
|
Amortization of deferred stock-based compensation
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
526
|
|
|
|
—
|
|
|
|
—
|
|
|
|
526
|
|
|
Reversal of deferred stock-based compensation due to employee
terminations
|
|
|
—
|
|
|
|
—
|
|
|
|
(30
|
)
|
|
|
30
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Comprehensive loss
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Change in valuation of interest rate swap liability for the
twelve months ended December 31, 2007
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(129
|
)
|
|
|
—
|
|
|
|
(129
|
)
|
|
Unrealized holding gain on
available-for-sale
securities
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
136
|
|
|
|
—
|
|
|
|
136
|
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(23,314
|
)
|
|
|
(23,314
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Comprehensive loss
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(23,307
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2007 (carried forward)
|
|
|
17,792,170
|
|
|
$
|
18
|
|
|
$
|
120,471
|
|
|
$
|
(294
|
)
|
|
$
|
28
|
|
|
$
|
(66,910
|
)
|
|
$
|
53,313
|
|
See accompanying notes
64
TRUBION
PHARMACEUTICALS, INC.
STATEMENT
OF STOCKHOLDERS’ EQUITY
(DEFICIT) — (Continued)
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Other
|
|
|
|
|
|
Total
|
|
|
|
|
|
|
|
|
|
|
Additional
|
|
|
Deferred
|
|
|
Comprehensive
|
|
|
|
|
|
Stockholders’
|
|
|
|
|
Common Stock
|
|
|
Paid-in
|
|
|
Stock-Based
|
|
|
Income
|
|
|
Accumulated
|
|
|
Equity
|
|
|
|
|
Shares
|
|
|
Amount
|
|
|
Capital
|
|
|
Compensation
|
|
|
(Loss)
|
|
|
Deficit
|
|
|
(Deficit)
|
|
|
|
|
(In thousands, except share data)
|
|
|
|
|
Balance at December 31, 2007 (brought forward)
|
|
|
17,792,170
|
|
|
$
|
18
|
|
|
$
|
120,471
|
|
|
$
|
(294
|
)
|
|
$
|
28
|
|
|
$
|
(66,910
|
)
|
|
$
|
53,313
|
|
|
Issuance of common stock upon exercise of stock options
|
|
|
90,137
|
|
|
|
—
|
|
|
|
91
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
91
|
|
|
Stock-based compensation to non-employees at fair value
|
|
|
—
|
|
|
|
—
|
|
|
|
106
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
106
|
|
|
Stock-based compensation expense
|
|
|
—
|
|
|
|
—
|
|
|
|
3,216
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
3,216
|
|
|
Amortization of deferred stock-based compensation
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
226
|
|
|
|
—
|
|
|
|
—
|
|
|
|
226
|
|
|
Reversal of deferred stock-based compensation due to employee
terminations
|
|
|
—
|
|
|
|
—
|
|
|
|
(38
|
)
|
|
|
38
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Comprehensive loss
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Realized loss on interest rate swap liability
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
129
|
|
|
|
—
|
|
|
|
129
|
|
|
Unrealized holding loss on
available-for-sale
securities
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(54
|
)
|
|
|
—
|
|
|
|
(54
|
)
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(25,559
|
)
|
|
|
(25,559
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Comprehensive loss
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(25,484
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2008 (carried forward)
|
|
|
17,882,307
|
|
|
$
|
18
|
|
|
$
|
123,846
|
|
|
$
|
(30
|
)
|
|
$
|
103
|
|
|
$
|
(92,469
|
)
|
|
$
|
31,468
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
See accompanying notes
65
TRUBION
PHARMACEUTICALS, INC.
STATEMENT
OF STOCKHOLDERS’ EQUITY
(DEFICIT) — (Continued)
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Other
|
|
|
|
|
|
Total
|
|
|
|
|
|
|
|
|
|
|
Additional
|
|
|
Deferred
|
|
|
Comprehensive
|
|
|
|
|
|
Stockholders’
|
|
|
|
|
Common Stock
|
|
|
Paid-in
|
|
|
Stock-Based
|
|
|
Income
|
|
|
Accumulated
|
|
|
Equity
|
|
|
|
|
Shares
|
|
|
Amount
|
|
|
Capital
|
|
|
Compensation
|
|
|
(Loss)
|
|
|
Deficit
|
|
|
(Deficit)
|
|
|
|
|
(In thousands, except share data)
|
|
|
|
|
Balance at December 31, 2008 (brought forward)
|
|
|
17,882,307
|
|
|
$
|
18
|
|
|
$
|
123,846
|
|
|
$
|
(30
|
)
|
|
$
|
103
|
|
|
$
|
(92,469
|
)
|
|
$
|
31,468
|
|
|
Issuance of common stock upon exercise of stock options
|
|
|
255,605
|
|
|
|
—
|
|
|
|
90
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
90
|
|
|
Stock-based compensation to non-employees at fair value
|
|
|
—
|
|
|
|
—
|
|
|
|
661
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
661
|
|
|
Stock-based compensation expense
|
|
|
—
|
|
|
|
—
|
|
|
|
3,544
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
3,544
|
|
|
Amortization of deferred stock-based compensation
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
30
|
|
|
|
—
|
|
|
|
—
|
|
|
|
30
|
|
|
Issuance of common stock for cash in private placement offering
|
|
|
2,243,649
|
|
|
|
2
|
|
|
|
8,591
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
8,593
|
|
|
Comprehensive loss
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Unrealized holding loss on
available-for-sale
securities
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(109
|
)
|
|
|
—
|
|
|
|
(109
|
)
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(29,183
|
)
|
|
|
(29,183
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Comprehensive loss
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(29,292
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2009
|
|
|
20,381,561
|
|
|
$
|
20
|
|
|
$
|
136,732
|
|
|
$
|
—
|
|
|
$
|
(6
|
)
|
|
$
|
(121,652
|
)
|
|
$
|
15,094
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
See accompanying notes
66
TRUBION
PHARMACEUTICALS, INC.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
|
|
(In thousands)
|
|
|
|
|
Operating activities
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
$
|
(29,183
|
)
|
|
$
|
(25,559
|
)
|
|
$
|
(23,314
|
)
|
|
Adjustments to reconcile net loss to net cash used in operating
activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Non-cash stock-based compensation expense
|
|
|
4,235
|
|
|
|
3,548
|
|
|
|
3,498
|
|
|
Depreciation and amortization
|
|
|
3,146
|
|
|
|
3,230
|
|
|
|
2,936
|
|
|
Net amortization of premium (discount) on investments
|
|
|
132
|
|
|
|
81
|
|
|
|
(7
|
)
|
|
Amortization of debt discount
|
|
|
11
|
|
|
|
34
|
|
|
|
21
|
|
|
Changes in operating assets and liabilities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Receivable from collaborations
|
|
|
(344
|
)
|
|
|
1,153
|
|
|
|
117
|
|
|
Prepaid expenses and other assets
|
|
|
1,142
|
|
|
|
(860
|
)
|
|
|
(354
|
)
|
|
Accounts payable
|
|
|
78
|
|
|
|
(730
|
)
|
|
|
(506
|
)
|
|
Accrued liabilities and compensation
|
|
|
99
|
|
|
|
791
|
|
|
|
(1,658
|
)
|
|
Deferred revenue
|
|
|
15,769
|
|
|
|
(5,361
|
)
|
|
|
(6,924
|
)
|
|
Deferred rent
|
|
|
(180
|
)
|
|
|
(180
|
)
|
|
|
(180
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash used in operating activities
|
|
|
(5,095
|
)
|
|
|
(23,853
|
)
|
|
|
(26,371
|
)
|
|
Investing activities
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Purchases of property and equipment
|
|
|
(85
|
)
|
|
|
(1,257
|
)
|
|
|
(3,765
|
)
|
|
Purchases of investments
|
|
|
(51,980
|
)
|
|
|
(64,385
|
)
|
|
|
(81,240
|
)
|
|
Sales of investments
|
|
|
649
|
|
|
|
20,255
|
|
|
|
4,458
|
|
|
Maturities of investments
|
|
|
41,476
|
|
|
|
57,755
|
|
|
|
89,624
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash provided by (used in) investing activities
|
|
|
(9,940
|
)
|
|
|
12,368
|
|
|
|
9,077
|
|
|
Financing Activities
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Proceeds from issuance of notes payable
|
|
|
—
|
|
|
|
10,000
|
|
|
|
3,516
|
|
|
Payments on notes payable
|
|
|
(1,313
|
)
|
|
|
(10,464
|
)
|
|
|
(1,277
|
)
|
|
Proceeds from private placement of common stock
|
|
|
8,593
|
|
|
|
—
|
|
|
|
—
|
|
|
Proceeds from issuance of common stock and exercise of stock
options
|
|
|
90
|
|
|
|
91
|
|
|
|
468
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash provided by (used in) financing activities
|
|
|
7,370
|
|
|
|
(373
|
)
|
|
|
2,707
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net decrease in cash and cash equivalents
|
|
|
(7,665
|
)
|
|
|
(11,858
|
)
|
|
|
(14,587
|
)
|
|
Cash and cash equivalents at beginning of year
|
|
|
29,969
|
|
|
|
41,827
|
|
|
|
56,414
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents at end of year
|
|
$
|
22,304
|
|
|
$
|
29,969
|
|
|
$
|
41,827
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Supplemental disclosure information:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash paid for interest
|
|
$
|
525
|
|
|
$
|
807
|
|
|
$
|
713
|
|
See accompanying notes
67
TRUBION
PHARMACEUTICALS, INC.
|
|
|
1.
|
Organization
and Summary of Significant Accounting Policies
|
Organization
We are a biopharmaceutical company creating a pipeline of novel
protein therapeutic product candidates to treat autoimmune and
inflammatory diseases and cancer. Our mission is to develop a
variety of
first-in-class
and
best-in-class
product candidates customized in an effort to optimize safety,
efficacy, and convenience that we believe may offer improved
patient experiences. Our current product development efforts are
focused on three proprietary technologies that comprise the
expanded foundation for Trubion product development —
SMIPtm
protein therapeutics,
SCORPIONtm
protein therapeutics, and
TRU-ADhanCetm
potency enhancing technology for immunopharmaceuticals. Our
current clinical-stage therapeutics target specific antigens on
B cells, CD20 and CD37, and are designed using our custom drug
assembly technology. In order to fund ongoing development
activities and commercialize our products, we will, in some
cases, enter into collaboration agreements that would likely
include licenses to our technology and arrangements to provide
research and development services for others
We were founded as a limited liability company in the state of
Washington in March 1999. We converted into a corporation and
redomiciled in the state of Delaware in October 2002.
In December 2005 we entered into a collaboration agreement with
Wyeth, now a wholly-owned subsidiary of Pfizer Inc., or Pfizer,
for the development and worldwide commercialization of certain
therapeutics, including our lead product candidate, TRU-015. In
August 2009 we entered into a collaboration agreement with Facet
Biotech Corporation, or Facet, for the joint worldwide
development and commercialization of TRU-016, a product
candidate in Phase 1 clinical development for chronic
lymphocytic leukemia, or CLL and non-Hodgkins lymphoma, or NHL.
To date, none of our product candidates has been approved for
marketing and sale and we have not received any product revenue.
We operate in a single reporting segment, which is the
development of pharmaceutical products on our own behalf, or in
collaboration with others.
Use of
Estimates
Our financial statements have been prepared in accordance with
U.S. generally accepted accounting principles. The
preparation of these financial statements requires us to make
estimates and judgments in certain circumstances that affect the
reported amounts of assets, liabilities, revenues and expenses,
and related disclosure of contingent assets and liabilities. In
preparing these financial statements, management has made its
best estimates and judgments of certain amounts included in the
financial statements, giving due consideration to materiality.
On an ongoing basis, we evaluate our estimates, including those
related to revenue recognition, valuation of investments, fair
values of assets, income taxes, clinical trial and manufacturing
accruals, and other contingencies. Management bases its
estimates on historical experience and on various other
assumptions that it believes to be reasonable under the
circumstances. Actual results could differ from these estimates.
Fair
Value of Financial Instruments
We carry cash, cash equivalents, and investments
available-for-sale
at fair value. Our other financial instruments, including
accounts receivable, accounts payable, accrued liabilities, and
notes payable, are carried at cost, which approximates fair
value given their short-term nature.
Cash,
Cash Equivalents and Investments
We consider all highly liquid investments with original
maturities of 90 days or less from date of purchase to be
cash equivalents. Cash equivalents consist of interest-bearing
instruments, including obligations of U.S. government
agencies, high credit rating corporate borrowers, and money
market funds, which are carried at market value.
68
TRUBION
PHARMACEUTICALS, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
We classify our investment portfolio as
available-for-sale.
Available-for sale securities are carried at estimated fair
value, with the unrealized gains and losses, if any, reported in
stockholders’ equity and included in accumulated other
comprehensive income (loss). We regularly evaluate the
performance of our investments individually for impairment,
taking into consideration the investment, volatility and current
returns. If a determination is made that a decline in fair value
is
other-than-temporary,
the related investment is written down to its estimated fair
value. We consider an investment with a remaining maturity
greater than one year as long-term and a remaining maturity less
than one year as short-term at the balance sheet date. The cost
of securities in this category is adjusted for amortization of
premiums and accretion of discounts from the date of purchase to
maturity. Such amortization is included in interest income.
Realized gains and losses and declines in value judged to be
other than temporary on
available-for-sale
securities are also included in interest income. The cost of
securities sold is based on the specific identification method.
Property
and Equipment
Property and equipment, including leasehold improvements, are
stated at cost, less accumulated depreciation and amortization.
Property and equipment is depreciated using the straight-line
method over the estimated useful lives of the assets, which
range from three to five years. Leasehold improvements are
depreciated over the shorter of their estimated useful lives or
the related lease term ranging from four to seven years.
Impairment
of Long-Lived Assets
We record losses from impairment of long-lived assets used in
operations when indicators of impairment are present and the
undiscounted cash flows estimated to be generated by those
assets are less than the assets’ carrying amount. We
periodically evaluate the carrying value of long-lived assets to
be held and used when events and circumstances indicate that the
carrying amount of an asset may not be recovered.
Deferred
Rent
Lease incentives, including rent holidays and tenant improvement
allowances provided by lessors, and rent escalation provisions
are accrued as deferred rent. We recognize rent expense on a
straight-line basis over the term of the lease. The related
benefits are included in research and development expense or
general and administrative expense based on the nature of the
related expense.
Revenue
Recognition
Revenue is recognized when there is persuasive evidence that an
arrangement exists, delivery has occurred, the price is fixed or
determinable, and collection is reasonably assured. Revenue
arrangements with multiple elements are divided into separate
units of accounting if certain criteria are met, including
whether the delivered element has stand-alone value to the
customer and whether there is objective and reliable evidence of
the fair value of the undelivered items. Multiple contracts with
a single collaborative partner are combined and accounted for as
one arrangement. The consideration received is allocated among
the separate units of accounting based on their respective fair
values when there is reliable evidence of fair value for the
undelivered elements of the arrangement. If separable, the
applicable revenue recognition criteria are then applied to each
of the separate units. For combined units of accounting, the
revenue is generally recognized in the same manner as the final
deliverable. Generally, revenue related to licensing activity
and our research and development services under collaboration
agreements is recognized ratably over the estimated term of the
research and development service period. Payments received in
advance of work performed are recorded as deferred revenue and
recognized when earned.
We recognize revenue from our collaboration agreements with
Pfizer and Facet, which consists of non-refundable,
non-creditable up-front fees and license fees, collaborative
research funding, regulatory and sales
69
TRUBION
PHARMACEUTICALS, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
milestones, future product royalties and future product sales.
Revenue related to our collaboration agreements is recognized as
follows:
Up-Front Fees and License
Fees. Non-refundable, non-creditable up-front
fees and license fees received in connection with collaborative
research and development agreements are deferred and recognized
on a straight-line basis over the estimated term of the research
and development service period. The estimated term of the
research and development service period is reviewed and adjusted
based on the status of the project against the estimated
timeline as additional information becomes available. We also
consider the time frame of our substantive contractual
obligations related to research and development agreements when
estimating the term of the research and development period. For
each collaboration agreement, we review our ongoing performance
obligations on a regular basis and make adjustments to the
estimated term as additional information becomes available.
Adjustments to the research and development service period are
made prospectively. We have made adjustments to the research and
development service periods in the past and we expect to revise
our estimate of the development term in future periods due to
the inherently uncertain nature of development terms. As a
result, revenue may fluctuate materially in the future due to
adjustments to the estimated term of the research and
development service periods and our substantive contractual
obligations under our collaborations.
Collaborative Research Funding. Certain
internal and external research and development costs and patent
costs are reimbursed in connection with our collaboration
agreements. Reimbursed costs under the Pfizer collaboration are
recognized as revenue in the same period the costs are incurred.
With respect to the reimbursement of development costs under the
Facet collaboration, each quarter, we and Facet reconcile what
each party has incurred for development costs, and we record
either a net receivable or a net payable in our financial
statements. For each quarterly period, if we have a net
receivable from Facet, we recognize revenues by such amount, and
if we have a net payable to Facet, we recognize additional
research and development expenses by such amount. As a result,
our revenues and research and development expenses may fluctuate
depending on which party in the collaboration is incurring the
majority of the development costs in any particular quarterly
period. Reimbursed costs are subject to the estimation processes
within our preclinical study, clinical trial and manufacturing
accruals processes and are subject to change in future periods
when actual activity is known. To date we have not made any
material adjustments to these estimates.
Milestones. Payments for milestones that are
based on the achievement of substantive and at-risk performance
criteria are recognized in full at such time as the specified
milestone has been achieved according to the terms of the
agreement. When payments are not for substantive or at-risk
milestones, revenue will be recognized on a straight-line basis
over the remaining estimated term of the research and
development service period. The estimated term of the research
and development service period is reviewed and adjusted based on
the status of the project against the estimated timeline as
additional information becomes available.
Research
and Development
Research and development costs are expensed as the related goods
are delivered or the related services are performed. Effective
January 1, 2008 we adopted the guidance related to the
accounting for nonrefundable advance payments for goods or
services received for use in future research and development
activities. Research and development costs include, but are not
limited to, salaries and benefits, lab supplies, preclinical
fees, clinical trial and related clinical manufacturing costs,
allocated overhead costs, and professional service fees.
70
TRUBION
PHARMACEUTICALS, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
Income
Taxes
We use the liability method of accounting for deferred income
taxes. The provision for income taxes includes income taxes
deferred as a result of temporary differences between financial
reporting and tax basis of assets and liabilities. Deferred
taxes are measured using enacted tax rates expected to be in
effect in a year in which the basis difference is expected to
reverse. We continue to record a valuation allowance for the
full amount of deferred assets, which would otherwise be
recorded for tax benefits relating to operating loss and tax
credit carry forwards, as realization of such deferred tax
assets cannot be determined to be more likely than not.
During the twelve months ended December 31, 2009 and 2008,
we had no unrecognized tax benefits and expect no significant
changes in unrecognized tax benefits in the next 12 months.
We classify any interest and penalties as a component of tax
expense. To date there have been no interest or penalties
charged to us in relation to the underpayment of income taxes.
We file our tax returns as prescribed by the tax laws of the
jurisdictions in which we operate. We are subject to audit by
the Internal Revenue Service for all years since inception.
Comprehensive
Income (Loss)
Comprehensive income (loss) is comprised of net loss and
unrealized gains (losses) on marketable securities and
derivatives. Total comprehensive income (loss) for all other
periods presented has been disclosed in the statements of
stockholders’ equity.
Accumulated comprehensive income (loss), net of taxes at
December 31, 2009 and 2008 was ($6,000) and $103,000,
respectively, which was comprised of net unrealized gains and
losses on investments
available-for-sale.
Stock-Based
Compensation
We account for stock-based compensation for employees and
directors based on estimated fair values. Employee stock-based
compensation expense recognized in the years ended
December 31, 2009, 2008 and 2007 was calculated based on
awards ultimately expected to vest, and has been reduced for
estimated forfeitures. Forfeitures are estimated at the time of
grant and revised, if necessary, in subsequent periods if actual
forfeitures differ from those estimates. The forfeiture estimate
is based on historical employee turnover rates and could differ
from actual forfeitures. Compensation costs for employee stock
options granted prior to January 1, 2006 were accounted for
using the option’s intrinsic value or the difference, if
any, between the fair market value of our common stock and the
exercise price of the option.
For stock options granted to non-employees, the fair value of
the stock options is estimated using the Black-Scholes valuation
model. The Black-Scholes model utilizes the estimated fair value
of common stock and requires that, at the date of grant, we make
assumptions with respect to the expected life of the option, the
volatility of the fair value of the underlying common stock,
risk-free interest rates and expected dividend yields of our
common stock. Stock-based compensation expense is recognized
over the period of expected service by the non-employee. As the
service is performed, we are required to update our valuation
assumptions, remeasure unvested options and record the
stock-based compensation using the valuation as of the vesting
date.
Concentration
of Credit Risk
Financial instruments that subject us to potential credit risk
consist of cash, cash equivalents and investments. Our cash,
cash equivalents and investments are placed with high
credit-quality financial institutions and issuers. We believe
that our established guidelines for investment of excess cash
maintain safety and liquidity through policies on
diversification and investment maturity.
71
TRUBION
PHARMACEUTICALS, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
Major
Customers
We define our customers as our collaborative partners and our
licensees from whom we have received and may receive
reimbursement for research and development services, license
fees, royalties and milestone payments.
Revenue recognized under our collaboration agreements for the
years ended December 31, 2009, 2008 and 2007 was as follows
(in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
|
|
Pfizer
|
|
$
|
15,855
|
|
|
$
|
16,467
|
|
|
$
|
20,148
|
|
|
Facet
|
|
|
2,148
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total revenue
|
|
$
|
18,003
|
|
|
$
|
16,467
|
|
|
$
|
20,148
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash received from collaborative partners for the years ended
December 31, 2009, 2008 and 2007 was as follows (in
thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
|
|
Pfizer
|
|
$
|
11,570
|
|
|
$
|
12,259
|
|
|
$
|
13,342
|
|
|
Facet
|
|
|
30,452
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total cash received
|
|
$
|
42,022
|
|
|
$
|
12,259
|
|
|
$
|
13,342
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Included in cash received from collaborative partners is
$10 million received from Facet pursuant to a stock
purchase agreement entered into during 2009 for which Facet
purchased 2,243,649 shares of our common stock for an
aggregate purchase price of $10 million.
Accounts receivable from collaborative partners as of
December 31, 2009 and 2008 were as follows (in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
Pfizer
|
|
$
|
2,496
|
|
|
$
|
3,084
|
|
|
Facet
|
|
|
932
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
Total accounts receivable
|
|
$
|
3,428
|
|
|
$
|
3,084
|
|
|
|
|
|
|
|
|
|
|
|
Recent
Accounting Pronouncements
In October 2009, the FASB issued new guidance for
multiple-deliverable revenue arrangements. The new guidance
addresses the accounting for multiple-deliverable arrangements
to enable vendors to account for products or services
(deliverables) separately rather than as a combined unit. This
guidance establishes a selling price hierarchy for determining
the selling price of a deliverable, which is based on:
(a) vendor-specific objective evidence;
(b) third-party evidence; or (c) estimates. This
guidance also eliminates the residual method of allocation and
requires that arrangement consideration be allocated at the
inception of the arrangement to all deliverables using the
relative selling price method. In addition, this guidance
significantly expands required disclosures related to a
vendor’s multiple-deliverable revenue arrangements. We
expect to adopt this guidance on January 1, 2011 and it
will be applied prospectively for revenue arrangements entered
into or materially modified after the date of adoption. We are
evaluating the impact this guidance will have on our financial
position, results of operations, cash flows and disclosures.
72
TRUBION
PHARMACEUTICALS, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
|
|
|
2.
|
Fair
Value Measurements
|
We measure and record cash equivalents and investment securities
considered
available-for-sale
at fair value in the accompanying financial statements. Fair
value is defined as the exchange price that would be received
for an asset or paid to transfer a liability (an exit price) in
the principal or most advantageous market for the asset or
liability in an orderly transaction between market participants
on the measurement date. Valuation techniques used to measure
fair value must maximize the use of observable inputs and
minimize the use of unobservable inputs. The three-tier fair
value hierarchy, which prioritizes the inputs used in measuring
fair value include:
Level 1 — Observable inputs for identical assets
or liabilities such as quoted prices in active markets;
Level 2 — Inputs other than quoted prices in
active markets that are either directly or indirectly
observable; and
Level 3 — Unobservable inputs in which little or
no market data exists, which are therefore developed by us using
estimates and assumptions that reflect those that a market
participant would use.
The following table represents our fair value hierarchy for our
financial assets measured at fair value on a recurring basis as
of December 31, 2009 and 2008 (in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31, 2009
|
|
Level 1
|
|
|
Level 2
|
|
|
Level 3
|
|
|
Total
|
|
|
|
|
Money market funds
|
|
$
|
22,259
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
22,259
|
|
|
Government and agency debt securities
|
|
|
—
|
|
|
|
32,542
|
|
|
|
—
|
|
|
|
32,542
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
22,259
|
|
|
$
|
32,542
|
|
|
$
|
—
|
|
|
$
|
54,801
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31, 2008
|
|
Level 1
|
|
|
Level 2
|
|
|
Level 3
|
|
|
Total
|
|
|
|
|
Money market funds
|
|
$
|
27,444
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
27,444
|
|
|
Government and agency debt securities
|
|
|
—
|
|
|
|
12,424
|
|
|
|
—
|
|
|
|
12,424
|
|
|
Corporate debt securities
|
|
|
—
|
|
|
|
13,003
|
|
|
|
—
|
|
|
|
13,003
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
27,444
|
|
|
$
|
25,427
|
|
|
$
|
—
|
|
|
$
|
52,871
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash of $45,000 and $26,000 is not included in our fair value
hierarchy disclosure as of December 31, 2009 and 2008.
Separate disclosure is required of assets and liabilities
measured at fair value on a recurring basis, as documented
above, from those measured at fair value on a nonrecurring
basis. As of December 31, 2009 and 2008, no assets or
liabilities were measured at fair value on a nonrecurring basis.
73
TRUBION
PHARMACEUTICALS, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
We invest in a variety of highly liquid investment-grade
securities. The following is a summary of our
available-for-sale
securities at December 31, 2009 and 2008 (in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Gross
|
|
|
Gross
|
|
|
|
|
|
|
|
Amortized
|
|
|
Unrealized
|
|
|
Unrealized
|
|
|
Estimated Fair
|
|
|
December 31, 2009
|
|
Cost
|
|
|
Gains
|
|
|
Losses
|
|
|
Market Value
|
|
|
|
|
Money market funds
|
|
$
|
22,259
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
22,259
|
|
|
Government and agency debt securities
|
|
|
32,549
|
|
|
|
6
|
|
|
|
(13
|
)
|
|
|
32,542
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
|
54,808
|
|
|
|
6
|
|
|
|
(13
|
)
|
|
|
54,801
|
|
|
Less: cash equivalents
|
|
|
(22,259
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
(22,259
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Amounts classified as investments
|
|
$
|
32,549
|
|
|
$
|
6
|
|
|
$
|
(13
|
)
|
|
$
|
32,542
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Gross
|
|
|
Gross
|
|
|
|
|
|
|
|
Amortized
|
|
|
Unrealized
|
|
|
Unrealized
|
|
|
Estimated Fair
|
|
|
December 31, 2008
|
|
Cost
|
|
|
Gains
|
|
|
Losses
|
|
|
Market Value
|
|
|
|
|
Money market funds
|
|
$
|
27,444
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
27,444
|
|
|
Government and agency debt securities
|
|
|
12,384
|
|
|
|
40
|
|
|
|
—
|
|
|
|
12,424
|
|
|
Corporate debt securities
|
|
|
12,940
|
|
|
|
63
|
|
|
|
—
|
|
|
|
13,003
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
|
52,768
|
|
|
|
103
|
|
|
|
—
|
|
|
|
52,871
|
|
|
Less: cash equivalents
|
|
|
(29,935
|
)
|
|
|
(8
|
)
|
|
|
—
|
|
|
|
(29,943
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Amounts classified as investments
|
|
$
|
22,833
|
|
|
$
|
95
|
|
|
$
|
—
|
|
|
$
|
22,928
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The following table summarizes the fair value and gross
unrealized losses related to
available-for-sale
securities, aggregated by investment category and length of time
that individual securities have been in a continuous unrealized
loss position at December 31, 2009:
| |
|
|
|
|
|
|
|
|
|
|
|
Less Than 12 Months
|
|
|
|
|
|
|
|
Gross
|
|
|
|
|
Fair
|
|
|
Unrealized
|
|
|
|
|
Value
|
|
|
Losses
|
|
|
|
|
Government and agency debt securities
|
|
$
|
13,804
|
|
|
$
|
13
|
|
The estimated fair market value amounts have been determined
using available market information. The declines in value of
these investments are not related to credit quality and are
primarily related to changes in interest rates and are
considered to be temporary in nature. Because it is more likely
than not that we will hold these investments until a forecasted
recovery of fair value, which may be the maturity or call date,
we do not consider these investments to be
other-than-temporarily
impaired as of December 31, 2009. Unrealized gains and
losses on cash equivalents and available for sale securities are
included in accumulated other comprehensive income (loss) in the
accompanying balance sheets. Gross realized gains and losses on
cash equivalents or investments were not material for 2009, 2008
or 2007.
Basic net loss per share is calculated by dividing net loss by
the weighted-average number of common shares outstanding.
Because we report a net loss, diluted net loss per share is the
same as basic net loss per share. We have excluded all
outstanding stock options and unvested restricted stock from the
calculation of
74
TRUBION
PHARMACEUTICALS, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
diluted net loss per common share because all such securities
are antidilutive to the computation of net loss per share.
Potentially dilutive securities include the following:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
As of December 31,
|
|
|
|
2009
|
|
2008
|
|
2007
|
|
|
|
Stock options
|
|
|
2,654,035
|
|
|
|
2,093,940
|
|
|
|
1,551,968
|
|
|
|
|
5.
|
Collaboration
Agreements
|
Facet
In August 2009, we entered into a collaboration agreement with
Facet Biotech Corporation, or Facet, for the joint worldwide
development and commercialization of TRU-016, a product
candidate in Phase 1 clinical development for chronic
lymphocytic leukemia, or CLL. TRU-016 is a CD37-directed Small
Modular Immunopharmaceutical, or SMIP protein therapeutic. The
collaboration agreement includes TRU-016 in all indications and
all other CD37-directed protein therapeutics. Under the terms of
the collaboration agreement, the parties will not develop or
commercialize protein therapeutics directed to CD37 outside of
the collaboration agreement.
We received an up-front payment of $20 million in cash in
September 2009 and may receive up to $176.5 million in
additional contingent payments upon the achievement of specified
development, regulatory and sales milestones. We and Facet share
equally the costs of all development, commercialization and
promotional activities and all global operating profits. In
connection with the collaboration agreement, we and Facet also
entered into a stock purchase agreement, pursuant to which Facet
purchased 2,243,649 shares of our common stock for an
aggregate purchase price of $10 million, or $4.46 per
share. The per share price of $4.46 represents a 35% equity
premium over the
sixty-day
trading average of our common stock on NASDAQ for the trading
period ending immediately prior to the execution of the stock
purchase agreement. As a result of the ownership of shares Facet
is considered to be a related party. The $20 million
up-front payment and $1.4 million of equity premium
representing the difference between the purchase price and the
closing price of our common stock on the date the stock was
purchased by Facet have been deferred and are being recognized
ratably over the estimated term of our substantive contractual
obligations under the collaboration. Our current obligations
under the collaboration include the performance of non-clinical,
clinical, manufacturing and regulatory activities. We currently
estimate these obligations to extend through 2018. The estimated
term of the research and development service period will be
reviewed on a regular basis and adjusted as necessary.
With respect to control over decisions and responsibilities, the
collaboration agreement provides for a joint steering committee,
or JSC, consisting of representatives of Trubion and Facet,
which makes decisions by consensus. If the JSC is unable to
reach a consensus, then the matter will be referred to
Trubion’s and Facet’s Chief Executive Officers for
resolution. If the Chief Executive Officers are unable to
resolve the matter, then it will be resolved by arbitration.
Both Trubion and Facet, at their sole discretion, may
discontinue participation on the JSC with 90 days written
notice to the other party.
At predefined times, the parties have the right to opt-out of
the collaboration entirely or on a
product-by-product
basis. Upon a change of control of a party, the other party will
have the right to opt-out of the collaboration entirely and if
the successor party is conducting a program that competes with
the programs under the collaboration agreement, then the
successor party must either (i) opt-out of the
collaboration entirely or (ii) divest the competing program
to a third party. If a party exercises its opt-out right with
respect to a product, then the parties will no longer share
development and commercialization costs for such product and
such opting-out party will receive certain royalty payments upon
the sale of such product, rather than half of the profits
derived from such product. Even if Facet exercises its opt-out
right, its obligation to make milestone payments to us
continues. In addition, if the party that opts-out is the lead
manufacturing party for
75
TRUBION
PHARMACEUTICALS, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
the opt-out product, then that party must continue to supply the
product to the continuing party for up to eighteen months
following the opt-out.
On March 9, 2010, Abbott Laboratories, or Abbott, announced
a definitive agreement to purchase Facet. Abbott further
announced that it expects the transaction to close in the second
quarter of 2010, subject to certain conditions. We intend to
continue to pursue the objectives in the approved development
plan.
Facet can terminate the collaboration agreement at any time, in
which event all rights to TRU-016 and other CD37-directed
protein therapeutics under the collaboration agreement would
revert to us. If Facet terminates the collaboration agreement in
the first 18 months, then Facet must pay us a
$10 million termination fee.
If there is a material breach of the collaboration agreement,
then the non-breaching party may either terminate the
collaboration agreement or continue the collaboration agreement
and force the breaching party to opt-out and accept royalties at
a reduced rate.
Either party may assign its interest in the collaboration
agreement to a third party, provided that the non-assigning
party maintains a right of first negotiation over any proposed
assignment. In addition, either we or Facet can freely assign
the collaboration agreement without the consent of the other
party in connection with certain specified change of control
transactions, such as an acquisition.
We deferred the recognition of the up-front payment of
$20 million and $1.4 million equity premium. These
payments are being recognized as revenue over the period of our
substantive contractual obligations, which we estimate to be
through 2018. During the year ended December 31, 2009 we
recognized as revenue $2.1 million for research and
development services pursuant to our Facet collaboration. The
$2.1 million recognized in the year ended December 31,
2009 is comprised of $0.8 million for recognition of the
$20 million up-front fee received from Facet and the
$1.4 million equity premium, and $1.3 million for
collaborative research funding from the Facet collaboration.
Pfizer
In December 2005, we entered into a collaboration agreement with
Wyeth, now a wholly-owned subsidiary of Pfizer, for the
development and worldwide commercialization of our lead product
candidate, TRU-015, and other CD20-directed therapeutics.
Pursuant to the agreement, we are also collaborating with Pfizer
on the development and worldwide commercialization of certain
other product candidates directed to a small number of targets
other than CD20. During the period in which we will be providing
research and development services for Pfizer, Pfizer has the
right, subject to our reasonable consent, to replace a limited
number of these targets. In addition, we have the option to
co-promote with Pfizer, on customary terms to be agreed,
CD20-directed therapies in the United States for niche
indications. We retain the right to develop and commercialize,
on our own or with others, product candidates directed to all
targets not included within the agreement. Unless it is
terminated earlier, the agreement will remain in effect on a
product-by-product
basis and on a
country-by-country
basis until the later of the date that any such product shall no
longer be covered by a valid claim of a U.S. or foreign
patent or application and, generally, ten years after the first
commercial sale of any product licensed under the agreement.
Pfizer may terminate the agreement without cause at any time
upon 90 days’ prior written notice.
In connection with the agreement, Wyeth paid us a
$40 million non-refundable, non-creditable, up-front fee in
January 2006 and purchased directly from us in a private
placement, concurrent with our initial public offering,
800,000 shares of our common stock at the initial public
offering price of $13.00 per share, resulting in net proceeds to
us of $10.4 million. As a result of the ownership of shares
Pfizer is considered to be a related party. Under the agreement,
we provided research services for an initial three-year period
ended December 22, 2008 with the option for Wyeth to extend
the service period for two additional one-year periods.
Wyeth’s financial obligations during the initial research
service term included collaborative research funding commitments
of $9.0 million in exchange for such committed research
services. This $9.0 million was subject to an increase if
the service period was extended beyond three years, as well as
annual increases
76
TRUBION
PHARMACEUTICALS, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
pursuant to percentage changes in the CPI. In June 2008, Wyeth
exercised the first option under the terms of the agreement to
extend the research period for an additional one-year period
through December 22, 2009. In June 2009, Wyeth exercised
the second option under the terms of the agreement to extend the
research period for an additional one-year period through
December 22, 2010. Due to the research period extension in
2009, the collaboration research funding commitments to us
initially from Wyeth and now from Pfizer, increased to
approximately $3.3 million per year in exchange for
committed research services from us through December 22,
2010.
Pfizer’s financial obligations include additional amounts
for reimbursement of
agreed-upon
external research and development costs and patent costs.
Pursuant to the agreement, Pfizer’s financial obligations
also include payments to us of up to $250 million based on
the achievement of specified regulatory and sales milestones for
CD20-directed therapies and payments to us of up to
$535 million based on the achievement of specified
regulatory and sales milestones for therapies directed to the
small number of targets other than CD20. In addition, we will
receive royalty payments in the event of future licensed product
sales. The $40 million up-front fee is being recognized
ratably over the estimated term of our substantive contractual
obligations under the agreement and the related research and
development service period. Currently, our clinical development
obligations under the agreement are limited to conducting
ongoing re-treatment studies for TRU-015. The ongoing second
Phase 2b (study 2203) study and future studies will be
conducted by Pfizer. The estimated term of the research and
development service period is reviewed and adjusted as
additional information becomes available. During the third
quarter of 2008, the estimated term of the research and
development service period was adjusted from six years and three
months to seven years, or through December 2012. The change in
the estimated research and development service period was
primarily due to an extension of our obligations to conduct
clinical activities under our agreement with Pfizer. The change
in estimate reduced the recognition of the up-front fee during
2008 by $487,000. During the third quarter of 2007, the
estimated term of the research and development service period
was increased 15 months resulting in reduced recognition of
the up-front fee during 2007 of $1.1 million.
During the years ended December 31, 2009, 2008 and 2007, we
recognized revenue of $15.9 million, $16.5 million and
$20.1 million, respectively, for research and development
services pursuant to our Pfizer collaboration. The
$15.9 million recognized in the year ended
December 31, 2009 is comprised of $4.9 million for
recognition of the $40 million up-front fee received from
Wyeth and $11.0 million for collaborative research funding
from the Pfizer collaboration. The $16.5 million recognized
in the year ended December 31, 2008 is comprised of
$5.4 million for recognition of the $40 million
up-front fee received from Wyeth and $11.1 million for
collaborative research funding from the Pfizer collaboration.
The $20.1 million recognized in the year ended
December 31, 2007 is comprised of $6.9 million for
recognition of the $40 million up-front fee received from
Wyeth and $13.2 million for collaborative research funding
from the Pfizer collaboration.
In an effort to reduce costs, we announced in February 2009 a
workforce reduction of approximately 25%, which included the
elimination of certain existing positions across our research
and administrative functions. We incurred a $0.8 million
restructuring charge in the first quarter of 2009 related to
employee severance, benefits and outplacement services. Of the
total restructuring charges, approximately $0.6 million and
$0.2 million were recorded as research and development
expense and general and administrative expense, respectively, in
the first quarter of 2009. We paid cash of $0.8 million
related to the restructuring charge during the twelve months
ended December 31, 2009. No restructuring obligations
remain as of December 31, 2009.
Effective November 16, 2009, our Chief Executive Officer
and Chairman of the Board resigned from his positions with the
Company. As a result of this resignation we incurred a
$1.3 million one-time charge in the fourth quarter of 2009,
$733,000 of which was related to severance, benefits and
consulting services and the remaining $584,000 was related to
the accelerated vesting of stock options and extended period to
exercise
77
TRUBION
PHARMACEUTICALS, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
vested stock options. The $1.3 million charge was recorded
as general and administrative expense. We paid cash of $39,000
to Dr. Thompson in 2009 related to this one-time charge.
The remaining amount payable as of December 31, 2009 was
approximately $694,000 and will be paid during 2010.
|
|
|
7.
|
Property
and Equipment
|
Property and equipment consisted of the following (in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
Lab equipment
|
|
$
|
10,534
|
|
|
$
|
10,495
|
|
|
Leasehold improvements
|
|
|
6,673
|
|
|
|
6,611
|
|
|
Computer equipment and software
|
|
|
1,163
|
|
|
|
1,141
|
|
|
Furniture and fixtures
|
|
|
449
|
|
|
|
447
|
|
|
Construction in progress
|
|
|
—
|
|
|
|
40
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
18,819
|
|
|
|
18,734
|
|
|
Accumulated depreciation and amortization
|
|
|
(12,690
|
)
|
|
|
(9,544
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
6,129
|
|
|
$
|
9,190
|
|
|
|
|
|
|
|
|
|
|
|
Property and equipment included equipment acquired under
equipment financing agreements of $14.1 million at
December 31, 2009 and 2008. Accumulated depreciation
related to assets purchased under the equipment financing
agreements was $9.8 million and $7.6 million at
December 31, 2009 and 2008, respectively. Amortization of
property and equipment under equipment financing agreements is
included in depreciation and amortization expense in the
statement of cash flows.
Accrued liabilities consisted of the following (in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
Accrued clinical trials
|
|
$
|
3,078
|
|
|
$
|
2,979
|
|
|
Accrued professional fees
|
|
|
421
|
|
|
|
556
|
|
|
Accrued manufacturing
|
|
|
65
|
|
|
|
933
|
|
|
Other
|
|
|
579
|
|
|
|
513
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
4,143
|
|
|
$
|
4,981
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
9.
|
Notes
Payable — Equipment Financing Arrangements
|
We entered into a loan and security agreement with Silicon
Valley Bank, or SVB, effective July 25, 2008, the terms of
which provide for a $10.0 million debt facility secured by
a security interest in our assets, other than intellectual
property, and used $8.5 million of the proceeds from this
debt facility to fully extinguish our obligations with Comerica
under our previous debt facility. In conjunction with
extinguishing our obligations under the Comerica debt facility,
we also terminated the Comerica loan and security agreement and
related interest rate swap agreement. We incurred a breakage fee
of $165,000 in connection with the termination of the interest
rate swap agreement, which is included in interest expense in
the statements of operations for the year ended
December 31, 2008. As of December 31, 2009, the full
$10.0 million available under the SVB facility was drawn
and is payable in fixed equal payments of principal plus
interest at a fixed rate of 5.75% based on an
84-month
amortization schedule with all principal and accrued interest
due July 25, 2013. The
78
TRUBION
PHARMACEUTICALS, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
loan and security agreement contains representations and
warranties and affirmative and negative covenants that are
customary for credit facilities of this type. In addition, the
loan and security agreement with SVB contains a material adverse
change clause which may accelerate the maturity of the loan upon
the occurrence of certain events. We have no indication that we
are in default of the material adverse change clause and no
scheduled loan payments have accelerated as a result of this
provision. As of December 31, 2009, approximately
$8.3 million was outstanding under the loan and security
agreement.
We have previously entered into various equipment financing
arrangements with a lender, each of which is secured by the
underlying equipment financed through the arrangement. The
credit facilities bore interest at annual rates between 8.83%
and 9.67% and were payable in monthly installments ranging from
36 to 42 months, and as of December 31, 2009 no
obligations were outstanding under the credit facilities.
As of December 31, 2009 and 2008, we financed
$14.1 million of equipment purchased under the lender
credit facilities. As of December 31, 2009 we had no credit
facilities available to us.
The future principal payments due under the equipment financing
arrangements were as follows as of December 31, 2009 (in
thousands):
| |
|
|
|
|
|
|
|
Notes
|
|
|
|
|
Payable
|
|
|
|
|
Year ending December 31, 2010
|
|
$
|
1,294
|
|
|
2011
|
|
|
1,372
|
|
|
2012
|
|
|
1,453
|
|
|
2013
|
|
|
4,172
|
|
|
|
|
|
|
|
|
Total payments
|
|
$
|
8,291
|
|
|
|
|
|
|
|
|
|
|
10.
|
Commitments
and Contingencies
|
Operating
Lease Commitments
We lease office and laboratory space under two operating lease
agreements, which expire on April 30, 2013. Under the
lease, we have two options to extend the term of the lease, each
for an additional term of five years at the then fair market
value of the leased premises. On February 2, 2007 we
entered into a lease to add an additional 3,067 square feet
of space in the same building it currently leases space
effective February 1, 2007 and expiring April 30,
2013. Rent expense was $1.3 million for each of the years
ended December 31, 2009, 2008 and 2007. We also entered
into operating lease obligations through August 2010 for certain
office equipment.
Future minimum lease payments under these leases as of
December 31, 2009, were as follows (in thousands):
| |
|
|
|
|
|
|
|
Operating
|
|
|
|
|
Leases
|
|
|
|
|
Year ending December 31, 2010
|
|
$
|
1,490
|
|
|
2011
|
|
|
1,476
|
|
|
2012
|
|
|
1,476
|
|
|
2013
|
|
|
492
|
|
|
|
|
|
|
|
|
Total minimum lease payments
|
|
$
|
4,934
|
|
|
|
|
|
|
|
79
TRUBION
PHARMACEUTICALS, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
Manufacturing
Commitments
We have entered into agreements with Lonza Biologics, or Lonza,
and related entities for certain license rights related to
Lonza’s manufacturing technology, and research and
development services. We have reserved future manufacturing
capacity from Lonza under pre-specified terms and conditions. As
of December 31, 2009, we had committed to purchase
$2.1 million of manufacturing services for TRU-016 from
Lonza in 2010.
Guarantees
and Indemnifications
We, as permitted under Delaware law and in accordance with its
bylaws, indemnify our officers and directors for certain events
or occurrences, subject to certain limits, while the officer or
director is or was serving at the Company’s request in such
capacity. The term of the indemnification period is equal to the
officer’s or director’s lifetime.
The maximum amount of potential future indemnification is
unlimited; however, we have obtained director and officer
insurance that limits our exposure and may enable us to recover
a portion of any future amounts paid. We believe that the fair
value of these indemnification obligations is minimal.
Accordingly, we have not recognized any liabilities relating to
these obligations as of December 31, 2009.
We have certain agreements with certain research organizations
with which we do business that contain indemnification
provisions pursuant to which we typically agree to indemnify the
party against certain types of third-party claims. We accrue for
known indemnification issues when a loss is probable and can be
reasonably estimated. We also accrue for estimated incurred but
unidentified indemnification issues based on historical
activity. There were no accruals for or expenses related to
indemnification issues for any period presented.
|
|
|
11.
|
Convertible
Preferred Stock and Stockholders’ Equity
(Deficit)
|
Preferred
Stock
As of December 31, 2009 and 2008 we had
5,000,000 shares, $0.001 par value, of authorized
preferred stock. Our board of directors has the authority,
without further action by the stockholders, to issue from time
to time preferred stock in one or more series, to fix the number
of shares of any such series and the designation thereof and to
fix the rights, preferences, privileges and restrictions granted
to or imposed upon such preferred stock, including dividend
rights, dividend rate, conversion rights, voting rights, rights
and terms of redemption, redemption prices, liquidation
preference and sinking fund terms. No preferred stock was issued
or outstanding as of December 31, 2009 and 2008.
Common
Stock
As of December 31, 2009 and 2008, we had
150,000,000 shares of authorized common stock. As of
December 31, 2009 and 2008, respectively, we had 20,381,561
and 17,882,307 shares of common stock outstanding.
In August 2009, we and Facet entered into a stock purchase
agreement, pursuant to which Facet purchased
2,243,649 shares of our common stock for an aggregate
purchase price of $10 million, or $4.46 per share. The per
share price of $4.46 represents a 35% equity premium over the
sixty-day
trading average of our common stock on NASDAQ for the trading
period ending immediately prior to the execution of the stock
purchase agreement. The stock purchase was recorded at fair
value and the equity premium of $1.4 million was recorded
as deferred revenue.
80
TRUBION
PHARMACEUTICALS, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
Equity
Incentive Plans
In September 2006 our Board of Directors adopted the 2006 Equity
Incentive Plan, or the 2006 Plan. The 2006 Plan is intended to
serve as the successor equity incentive program to our 2002
Stock Plan and 2002 Equity Incentive Plan. The 2006 Plan
provides for the grant of incentive stock options, nonstatutory
stock options, restricted stock, restricted stock units, stock
appreciation rights, performance units and performance shares.
The 2006 Plan became effective upon the completion of our
initial public offering, at which time options could no longer
be granted under the 2002 Stock Plan and the 2002 Equity
Incentive Plan. A total of 437,500 shares of common stock
have been authorized for issuance pursuant to the 2006 Plan,
plus the number of shares of common stock available for issuance
under the 2002 Stock Plan and the 2002 Equity Incentive Plan.
Also, any shares returned to the 2002 Stock Plan and the 2002
Equity Incentive Plans as a result of termination of options or
repurchase of shares will be included in the 2006 Plan. In
addition, on the first day of each fiscal year beginning in
2007, the number of shares available for issuance may be
increased by an amount equal to the lesser of:
(i) 1,500,000 shares; (ii) 5% of the outstanding
shares of our common stock on the first day of each fiscal year;
or (iii) such other amount as our board of directors may
determine. On January 1, 2010 the number of shares
available for issuance under the 2006 Plan increased by
1,019,078 shares.
The following summarizes information about employee, consultant
and director options outstanding, including aggregate intrinsic
values based on the estimated fair value at December 31,
2009 of $3.85 per share (aggregate intrinsic value in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted-
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted-
|
|
|
Average
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Average
|
|
|
Remaining
|
|
|
|
|
|
|
|
Shares
|
|
|
|
|
|
Exercise
|
|
|
Contractual
|
|
|
Aggregate
|
|
|
|
|
Available
|
|
|
|
|
|
Price per
|
|
|
Life
|
|
|
Intrinsic
|
|
|
|
|
for Grant
|
|
|
Options Granted
|
|
|
Share
|
|
|
(In years)
|
|
|
Value
|
|
|
|
|
Balance at January 1, 2007
|
|
|
490,522
|
|
|
|
1,587,626
|
|
|
$
|
3.90
|
|
|
|
8.34
|
|
|
$
|
22,449
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Authorized increase in Plan
|
|
|
877,716
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
Granted at fair value
|
|
|
(237,000
|
)
|
|
|
237,000
|
|
|
|
18.55
|
|
|
|
|
|
|
|
|
|
|
Exercised
|
|
|
—
|
|
|
|
(237,852
|
)
|
|
|
1.97
|
|
|
|
|
|
|
|
|
|
|
Cancelled
|
|
|
34,806
|
|
|
|
(34,806
|
)
|
|
|
5.79
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2007
|
|
|
1,166,044
|
|
|
|
1,551,968
|
|
|
$
|
6.39
|
|
|
|
7.44
|
|
|
$
|
8,106
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Authorized increase in Plan
|
|
|
889,609
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
Granted at fair value
|
|
|
(770,375
|
)
|
|
|
770,375
|
|
|
|
8.21
|
|
|
|
|
|
|
|
|
|
|
Exercised
|
|
|
—
|
|
|
|
(90,137
|
)
|
|
|
1.02
|
|
|
|
|
|
|
|
|
|
|
Cancelled
|
|
|
138,266
|
|
|
|
(138,266
|
)
|
|
|
9.76
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2008
|
|
|
1,423,544
|
|
|
|
2,093,940
|
|
|
$
|
7.07
|
|
|
|
7.51
|
|
|
$
|
513
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Authorized increase in Plan
|
|
|
894,115
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
Granted at fair value
|
|
|
(1,223,042
|
)
|
|
|
1,223,042
|
|
|
|
2.47
|
|
|
|
|
|
|
|
|
|
|
Exercised
|
|
|
—
|
|
|
|
(255,605
|
)
|
|
|
0.35
|
|
|
|
|
|
|
|
|
|
|
Cancelled
|
|
|
407,342
|
|
|
|
(407,342
|
)
|
|
|
6.05
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2009
|
|
|
1,501,959
|
|
|
|
2,654,035
|
|
|
$
|
5.75
|
|
|
|
7.69
|
|
|
$
|
2,861
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Vested and expected to vest at December 31, 2009
|
|
|
—
|
|
|
|
2,333,704
|
|
|
$
|
5.75
|
|
|
|
7.69
|
|
|
$
|
2,462
|
|
|
Exercisable at December 31, 2009
|
|
|
—
|
|
|
|
1,462,544
|
|
|
$
|
6.64
|
|
|
|
6.68
|
|
|
$
|
1,443
|
|
81
TRUBION
PHARMACEUTICALS, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
During the years ended December 31, 2009, 2008 and 2007,
the total intrinsic value of stock options exercised was
$479,000, $569,000 and $3.8 million, respectively. The
total fair value of shares vested during 2009, 2008 and 2007 was
approximately $2.8 million, $2.8 million and
$2.0 million respectively.
The following summarizes information about employee, consultant
and director options outstanding, including aggregate intrinsic
values based on the fair value at December 31, 2009 of
$3.85 per share (aggregate intrinsic value in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Options Outstanding
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted-Average
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Remaining
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Contractual
|
|
|
|
|
|
Options Exercisable
|
|
|
|
|
Number of
|
|
|
Life
|
|
|
Aggregate
|
|
|
Number of
|
|
|
Aggregate
|
|
|
Exercise Price per Share
|
|
Shares
|
|
|
(In Years)
|
|
|
Intrinsic Value
|
|
|
Shares
|
|
|
Intrinsic Value
|
|
|
|
|
$0.07 — $ 0.32
|
|
|
257,328
|
|
|
|
3.90
|
|
|
$
|
932
|
|
|
|
255,734
|
|
|
$
|
927
|
|
|
$1.33 — $ 1.33
|
|
|
704,334
|
|
|
|
9.08
|
|
|
|
1,775
|
|
|
|
168,331
|
|
|
|
424
|
|
|
$1.37 — $ 3.85
|
|
|
191,175
|
|
|
|
7.73
|
|
|
|
154
|
|
|
|
97,136
|
|
|
|
92
|
|
|
$3.86 — $ 8.98
|
|
|
1,213,267
|
|
|
|
7.80
|
|
|
|
—
|
|
|
|
721,054
|
|
|
|
—
|
|
|
$9.35 — $21.43
|
|
|
287,931
|
|
|
|
7.22
|
|
|
|
—
|
|
|
|
220,289
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$0.07 — $21.43
|
|
|
2,654,035
|
|
|
|
7.69
|
|
|
$
|
2,861
|
|
|
|
1,462,544
|
|
|
$
|
1,443
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Employee
Stock-Based Compensation
The components of the stock-based compensation recognized in
general and administrative expense (G&A) and research and
development expense (R&D) on our statements of operations
are as follows (in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31, 2009
|
|
|
|
|
G&A
|
|
|
R&D
|
|
|
Total
|
|
|
|
|
Employee stock options granted prior to January 1, 2006
|
|
$
|
30
|
|
|
$
|
—
|
|
|
$
|
30
|
|
|
Employee stock options granted on or subsequent to
January 1, 2006
|
|
|
2,112
|
|
|
|
1,432
|
|
|
|
3,544
|
|
|
Non-employee stock options(1)
|
|
|
584
|
|
|
|
77
|
|
|
|
661
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
2,726
|
|
|
$
|
1,509
|
|
|
$
|
4,235
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31, 2008
|
|
|
|
|
G&A
|
|
|
R&D
|
|
|
Total
|
|
|
|
|
Employee stock options granted prior to January 1, 2006
|
|
$
|
144
|
|
|
$
|
82
|
|
|
$
|
226
|
|
|
Employee stock options granted on or subsequent to
January 1, 2006
|
|
|
2,008
|
|
|
|
1,208
|
|
|
|
3,216
|
|
|
Non-employee stock options
|
|
|
70
|
|
|
|
36
|
|
|
|
106
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
2,222
|
|
|
$
|
1,326
|
|
|
$
|
3,548
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31, 2007
|
|
|
|
|
G&A
|
|
|
R&D
|
|
|
Total
|
|
|
|
|
Employee stock options granted prior to January 1, 2006
|
|
$
|
265
|
|
|
$
|
261
|
|
|
$
|
526
|
|
|
Employee stock options granted on or subsequent to
January 1, 2006
|
|
|
1,748
|
|
|
|
1,133
|
|
|
|
2,881
|
|
|
Non-employee stock options
|
|
|
9
|
|
|
|
82
|
|
|
|
91
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
2,022
|
|
|
$
|
1,476
|
|
|
$
|
3,498
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) |
|
Includes $584,000 related to the accelerated vesting of options
in the fourth quarter of 2009 and extended period to exercise
vested stock options. |
82
TRUBION
PHARMACEUTICALS, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
Employee
Stock Options Granted Prior to January 1,
2006
Compensation cost for employee stock options granted prior to
January 1, 2006, were accounted for using the option’s
intrinsic value or the difference, if any, between the fair
market value of our common stock and the exercise price of the
option. We recorded the total value of these options as a
component of stockholders’ equity, which has been amortized
over the vesting period of the applicable option on a straight
line basis. As of December 31, 2009 all expense related to
employee options granted prior to January 1, 2006 was fully
amortized.
Employee
Stock Options Granted On or Subsequent to January 1,
2006
Compensation cost for employee stock options granted on or
subsequent to January 1, 2006 is based on the estimated
grant-date fair value and will be recognized over the vesting
period of the applicable option on a straight-line basis.
Compensation costs recognized during the years ended
December 31, 2009, 2008 and 2007 includes:
(a) compensation cost for all share-based payment awards
granted prior to, but not yet vested as of January 1, 2006,
based on the intrinsic value method; and (b) compensation
cost for all share-based payment awards granted subsequent to
January 1, 2006, based on the estimated grant-date fair
value.
As stock-based compensation expense recognized in the statement
of operations for the years ended December 31, 2009, 2008
and 2007 is based on options ultimately expected to vest, it has
been reduced for estimated forfeitures. The fair value of
options is estimated utilizing the Black-Scholes model as our
chosen option-pricing model.
In regards to the calculation of expected term, we chose to
utilize the “simplified” method for “plain
vanilla” options. Under this approach, the expected term is
presumed to be the average of the vesting term and the
contractual term of the option. We have utilized the simplified
method for estimating the expected term due to our limited
historical exercise activity. For the calculation of expected
volatility, we based our estimate of expected volatility on the
estimated volatility of similar entities whose share prices are
publicly available and the historical volatility of our stock.
We used the following factors to identify similar public
entities: industry, stage of life cycle and the existence of at
least one significant partnership.
The fair value of each employee option grant was estimated on
the date of grant using the Black-Scholes option pricing model
with the following assumptions:
| |
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
2009
|
|
2008
|
|
2007
|
|
|
|
Risk-free interest rate
|
|
2.13%-2.72%
|
|
2.80%-3.40%
|
|
3.78%-4.78%
|
|
Weighted-average expected life (in years)
|
|
5.92
|
|
6.04
|
|
6.14
|
|
Expected dividend yield
|
|
0%
|
|
0%
|
|
0%
|
|
Expected volatility rate
|
|
88%-105%
|
|
70%-74%
|
|
65%-75%
|
|
Weighted-average estimated fair value of employee options
|
|
$1.85
|
|
$5.29
|
|
$12.87
|
As of December 31, 2009 total compensation related to
nonvested employee options not yet recognized in the financial
statements was approximately $2.5 million, and the
weighted-average period over which it is expected to be
recognized is approximately 2.3 years. We recorded no tax
benefit related to options during any of the years presented
since we currently maintain a full valuation allowance on all
deferred tax assets.
Non-employee
Stock-Based Compensation
For stock options granted to non-employees, we measure fair
value of the equity instruments utilizing the Black-Scholes
valuation model. Stock-based compensation expense is recognized
over the period of expected service by the non-employee. As the
service is performed, we are required to update our valuation
83
TRUBION
PHARMACEUTICALS, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
assumptions, remeasure unvested options and record the
stock-based compensation using the valuation as of the vesting
date. These adjustments may result in higher or lower
stock-based compensation expense in the statement of operations
than originally estimated. Changes in the market price of our
stock could materially change the value of an option and the
resulting stock-based compensation expense.
We valued the non-employee stock options granted during 2009,
2008 and 2007 using the Black-Scholes valuation model, using a
volatility rate between 65% and 105%, an expected life of one to
ten years, an expected dividend yield of 0% and a risk-free
interest rate ranging from 0.15% to 5.03%. Stock-based
compensation expense associated with these non-employee options
was $661,000, $106,000 and $67,000 for the years ended
December 31, 2009, 2008 and 2007, respectively. The
$661,000 recorded in 2009 includes a charge of $584,000 related
to the accelerated vesting of options in the fourth quarter of
2009 and extended period to exercise vested stock options.
Stock-based compensation expense related to restricted stock
awards granted to members of our Scientific Advisory Board was
$24,000 for the year ended December 31, 2007. Compensation
expense was recorded using straight-line amortization. There was
no restricted stock awards outstanding subsequent to
December 31, 2007.
We sponsor a 401(k) Plan that stipulates that eligible employees
can elect to contribute to the 401(k) Plan, subject to certain
limitations, up to 100% of eligible compensation on a pretax
basis. Pursuant to the 401(k) Plan, we do not match any employee
contributions.
At December 31, 2009, we had a net operating loss and
research and development, or R&D, tax credit carry forwards
of approximately $64.2 million and $2.7 million,
respectively. If not utilized, the net operating loss and
R&D tax credit carry forwards expire between 2021 and 2029.
Deferred income taxes reflect the net tax effects of temporary
differences between the carrying amounts of assets and
liabilities for financial reporting purposes and the amounts
used for income tax purposes. We have recognized a valuation
allowance equal to its deferred tax assets due to the
uncertainty of realizing the benefits of the assets. The
increase in the valuation allowance on the deferred tax asset
was approximately $10.1 million and $8.8 million for
2009 and 2008, respectively.
The effects of temporary differences and carry forwards that
give rise to deferred tax assets and liabilities are as follows
(in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
Deferred tax assets
|
|
|
|
|
|
|
|
|
|
Net operating loss carry forwards
|
|
$
|
22,500
|
|
|
$
|
19,035
|
|
|
Deferred revenue
|
|
|
12,341
|
|
|
|
6,823
|
|
|
Stock compensation
|
|
|
2,047
|
|
|
|
1,377
|
|
|
R&D tax credit carry forwards
|
|
|
2,677
|
|
|
|
1,978
|
|
|
Other current assets and liabilities (net)
|
|
|
224
|
|
|
|
272
|
|
|
Other non-current assets and liabilities (net)
|
|
|
1,287
|
|
|
|
1,442
|
|
|
Less: Valuation allowance
|
|
|
(41,076
|
)
|
|
|
(30,927
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Net deferred tax asset (liability)
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
84
TRUBION
PHARMACEUTICALS, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
|
|
|
16.
|
Quarterly
Information (Unaudited)
|
The following table summarizes the unaudited statements of
operations for each quarter of 2009 and 2008 (in thousands,
except per share amounts):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
March 31,(1)
|
|
|
June 30,
|
|
|
September 30,
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenue
|
|
$
|
4,212
|
|
|
$
|
4,119
|
|
|
$
|
4,452
|
|
|
$
|
5,220
|
|
|
Total operating expenses
|
|
|
15,189
|
|
|
|
10,719
|
|
|
|
10,556
|
|
|
|
10,361
|
|
|
Loss from operations
|
|
|
(10,977
|
)
|
|
|
(6,600
|
)
|
|
|
(6,104
|
)
|
|
|
(5,141
|
)
|
|
Net loss
|
|
|
(10,999
|
)
|
|
|
(6,702
|
)
|
|
|
(6,227
|
)
|
|
|
(5,255
|
)
|
|
Basic and diluted net loss per share
|
|
|
(0.61
|
)
|
|
|
(0.37
|
)
|
|
|
(0.33
|
)
|
|
|
(0.29
|
)
|
|
2008
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenue
|
|
$
|
3,963
|
|
|
$
|
4,468
|
|
|
$
|
3,766
|
|
|
$
|
4,270
|
|
|
Total operating expenses
|
|
|
10,488
|
|
|
|
11,415
|
|
|
|
10,384
|
|
|
|
10,695
|
|
|
Loss from operations
|
|
|
(6,525
|
)
|
|
|
(6,947
|
)
|
|
|
(6,618
|
)
|
|
|
(6,425
|
)
|
|
Net loss
|
|
|
(5,968
|
)
|
|
|
(6,632
|
)
|
|
|
(6,582
|
)
|
|
|
(6,377
|
)
|
|
Basic and diluted net loss per share
|
|
|
(0.33
|
)
|
|
|
(0.37
|
)
|
|
|
(0.37
|
)
|
|
|
(0.36
|
)
|
|
|
|
|
(1) |
|
The quarterly period ending March 31, 2009 included
$3.6 million for outside manufacturing costs for TRU-016. |
85