Attached files
| file | filename |
|---|---|
| EX-32.1 - CYTRX CORP | ex32-1.htm |
| EX-32.2 - CYTRX CORP | ex32-2.htm |
| EX-23.1 - CONSENT OF BDO SEIDMAN, LLP - CYTRX CORP | ex23-1.htm |
| EX-31.2 - CYTRX CORP | ex31-2.htm |
| EX-31.1 - CYTRX CORP | ex31-1.htm |
| EX-10.37 - BENJAMIN LEVIN EMPLOYMENT AGREEMENT - CYTRX CORP | ex10-37.htm |
| EX-10.39 - JOHN CALOZ EMPLOYMENT AGREEMENT - CYTRX CORP | ex10-39.htm |
| EX-10.38 - SCOTT WIELAND EMPLOYMENT AGREEMENT - CYTRX CORP | ex10-38.htm |
| EX-10.35 - SCOTT GEYER EMPLOYMENT AGREEMENT - CYTRX CORP | ex10-35.htm |
UNITED
STATES SECURITIES AND EXCHANGE COMMISSION
Washington,
D.C. 20549
Form
10-K
|
(Mark One)
|
|
|
R
|
ANNUAL REPORT PURSUANT TO
SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF
1934
|
|
For
the fiscal year ended December 31, 2009
|
|
|
or
|
|
|
£
|
TRANSITION REPORT PURSUANT TO
SECTION 13 OR 15(d) OF SECURITIES EXCHANGE ACT OF
1934
|
|
For
the transition period from __________________ to
__________________
|
|
Commission
file number 0-15327
CytRx
Corporation
(Exact
name of Registrant as specified in its charter)
|
Delaware
|
58-1642740
|
|
(State
or other jurisdiction of
|
(I.R.S.
Employer
|
|
incorporation
or organization)
|
Identification
No.)
|
|
11726
San Vicente Blvd, Suite 650,
|
|
|
Los
Angeles, California
|
90049
|
|
(Address
of principal executive offices)
|
(Zip
Code)
|
Registrant’s telephone number,
including area code: (310) 826-5648
________________
Securities registered pursuant to
Section 12(b) of the Act:
|
Title of each class
|
Name of exchange on which
registered
|
|
|
Common
Stock, $0.001 par value per share
|
The
NASDAQ Capital Market
|
|
|
Series
A Junior Participating Preferred Stock Purchase Rights
|
Securities Registered Pursuant to
Section 12(g) of the Act:
None
Indicate
by check mark if the Registrant is a well-known seasoned issuer (as defined in
Securities Act Rule 405). Yes £ No R
Indicate
by check mark if the Registrant is not required to file reports pursuant to
Section 13 or Section 15(d) of the Securities Exchange Act of 1934.
Yes £ No R
Indicate
by check mark whether the Registrant (1) has filed all reports required to be
filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the
preceding 12 months and (2) has been subject to such filing requirements for the
past 90 days. Yes R No £
Indicate
by check mark whether the registrant has submitted electronically and posted on
its corporate website, if any, every Interactive Data File required to be
submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of
this chapter) during the preceding 12 months (or for such shorter period that
the registrant was required to submit and post such files). Yes £ No £
Indicate
by check mark if disclosure of delinquent filers pursuant to Item 405 of
Regulation S-K is not contained herein, and will not be contained, to the best
of the Registrant’s knowledge, in definitive proxy or information statements
incorporated by reference in Part III of this Form 10-K or any amendment to this
Form 10-K. R
Indicate
by check mark whether the registrant is a large accelerated filer, an
accelerated filer, a non-accelerated filer, or a smaller reporting company. See
the definitions of “large accelerated filer,” “accelerated filer” and “smaller
reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
|
Large
accelerated filer £
|
Accelerated
filer R
|
Non-accelerated
filer £
|
Smaller
reporting company £
|
|
(Do
not check if a smaller reporting company)
|
Indicate
by check mark whether the Registrant is a shell company (as defined in Rule
12b-2 of the Act). Yes £ No R
Based on
the closing price as reported on The Nasdaq Capital Market, the aggregate market
value of the Registrant's common stock held by non-affiliates on June 30, 2009
(the last business day of the Registrant's most recently completed second fiscal
quarter) was approximately $102.0 million. Shares of common stock
held by directors and executive officers and their respective affiliates have
been excluded from this calculation, because such stockholders may be deemed to
be “affiliates” of the Registrant. This is not necessarily
determinative of affiliate status for other purposes. The number of
outstanding shares of the Registrant's common stock as of March 12, 2010
was 108,908,105, exclusive of treasury shares.
CYTRX
CORPORATION
2009
ANNUAL REPORT ON FORM 10-K
TABLE
OF CONTENTS
|
Page
|
||
|
“SAFE
HARBOR” STATEMENT
|
3
|
|
|
PART
I
|
||
|
Item
1. BUSINESS
|
4
|
|
|
Item
1A. RISK FACTORS
|
15
|
|
|
Item
2. PROPERTIES
|
27
|
|
|
Item
3. LEGAL PROCEEDINGS
|
27 | |
|
PART
II
|
||
|
28
|
||
|
Item
6. SELECTED FINANCIAL DATA
|
30
|
|
|
31
|
||
|
42
|
||
|
42
|
||
|
42
|
||
|
Item
9A. CONTROLS AND PROCEDURES
|
43
|
|
|
Item
9B. OTHER INFORMATION
|
43
|
|
|
PART
III
|
||
|
44
|
||
|
Item
11. EXECUTIVE COMPENSATION
|
48
|
|
|
62
|
||
|
63
|
||
|
64
|
||
|
PART
IV
|
||
|
65
|
||
|
68
|
2
“SAFE
HARBOR” STATEMENT
Some of
the information contained in this Annual Report may include forward-looking
statements within the meaning of Section 27A of the Securities Act of 1933, as
amended, or the Securities Act, and Section 21E of the Securities Exchange Act
of 1934, as amended, or the Exchange Act. We base these forward-looking
statements on our current views with respect to our research and development
activities, business strategy, business plan, financial performance and other
matters, both with respect to us, specifically, and the biotechnology sector, in
general. Statements that include the words “expect,” “intend,” “plan,”
“believe,” “project,” “estimate,” “may,” “should,” “anticipate,” “will” and
similar statements of a future or forward-looking nature identify
forward-looking statements for purposes of the federal securities laws or
otherwise, but the absence of these words does not necessarily mean that a
statement is not forward-looking.
All
forward-looking statements involve inherent risks and uncertainties, and there
are or will be important factors that could cause actual results to differ
materially from those indicated in these statements. We believe that these
factors include, but are not limited to, those factors set forth in the sections
entitled “Business,” “Risk Factors,” “Legal Proceedings,” “Management’s
Discussion and Analysis of Financial Condition and Results of Operations,”
“Quantitative and Qualitative Disclosures About Market Risk” and “Controls and
Procedures” in this Annual Report, all of which you should review carefully.
Please consider our forward-looking statements in light of those risks as you
read this Annual Report. We undertake no obligation to publicly update or review
any forward-looking statement, whether as a result of new information, future
developments or otherwise, except as required by law.
If one or
more of these or other risks or uncertainties materializes, or if our underlying
assumptions prove to be incorrect, actual results may vary materially from what
we anticipate. All subsequent written and oral forward-looking statements
attributable to us or individuals acting on our behalf are expressly qualified
in their entirety by the cautionary language above. You should consider
carefully all of the factors set forth or referred to in this Annual Report that
could cause actual results to differ.
3
PART
I
Item
1. BUSINESS
In this
Annual Report, we sometimes refer to CytRx Corporation as “CytRx,” to our former
subsidiary, RXi Pharmaceuticals Corporation, as “RXi,” and to Innovive
Pharmaceuticals, Inc., which we acquired in September 2008, as “Innovive.”
References in this Annual Report to the “company,” “we,” “us” or “our” refer to
CytRx, alone, unless otherwise indicated.
COMPANY
OVERVIEW
We are a
biopharmaceutical research and development company engaged in the development of
high-value human therapeutics, specializing in oncology. Our drug development
pipeline includes clinical development of three product candidates for cancer
indications, including three planned Phase 2 clinical trials for INNO-206 as a
treatment for pancreatic cancer, gastric (stomach) cancer and soft tissue
sarcomas, two Phase 2 proof-of-concept clinical trials with bafetinib in
patients with high-risk B-cell chronic lymphocytic leukemia, or B-CLL, and
patients with glioblastoma multiforme, and a registration study of tamibarotene
for the treatment of acute promyelocytic leukemia, or APL. In addition to our
core oncology programs, we are developing two drug candidates based on our
molecular chaperone regulation technology. Our current business
strategy for our molecular chaperone regulation technology is to seek one or
more strategic partnerships, or a possible spin-out transaction. Apart from our
drug development programs, we currently maintain a 36% equity interest in our
former subsidiary, RXi.
We are a
Delaware corporation, incorporated in 1985. Our corporate offices are
located at 11726 San Vicente Boulevard, Suite 650, Los Angeles, California
90049, and our telephone number is (310) 826-5648.
OUR
PRODUCT CANDIDATE PIPELINE
The
following tables summarize our product candidates and their current or planned
stage of development:
|
Technology
|
Product Candidate
|
Indication(s)
|
Stage of Development
|
|
Doxorubicin
prodrug
|
INNO-2006
|
Pancreatic
cancer
|
Phase
II (1H10)
|
|
Gastric
(stomach) cancer
|
Phase
II (2H10)
|
||
|
Soft
tissue sarcomas
|
Phase
II (2H10)
|
||
|
Tyrosine
kinase inhibitor
|
Bafetinib
|
B-CLL
|
Phase
II (1H10)
|
|
Glioblastoma
Multiforme
|
Phase
II (2H10)
|
||
|
Synthetic
retinoid
|
Tamibarotene
|
APL
(acute promyelocytic leukemia)
|
Pivotal
Phase II
|
|
Molecular
chaperone regulation
|
Arimoclomol
|
ALS
(amyotrophic lateral sclerosis, or Lou Gehrig’s disease)
|
Phase
II
|
|
Molecular
chaperone regulation
|
Iroxanadine
|
Several
potential indications
|
Phase
I
|
OUR
CLINICAL DEVELOPMENT PROGRAMS
Our
current clinical development programs consist of our efforts to develop INNO-206
for gastric (stomach) cancer, pancreatic cancer and soft tissue sarcomas,
bafetinib for B-CLL and glioblastoma multiforme, tamibarotene for APL, and our
molecular chaperone regulation program, which includes a planned Phase II
clinical study of arimoclomol in ALS.
INNO-206. INNO-206
(formerly DOXO-EMCH) is a prodrug of the commonly prescribed chemotherapeutic
agent doxorubicin. Specifically, it is the (6-Maleimidocaproyl) hydrazone of
doxorubicin. Essentially, this chemical is doxorubicin (DOXO) attached to an
acid sensitive linker (EMCH).
INNO-206 for the Treatment of
Cancer. Anthracyclines are a class of drugs that are among the
most commonly used agents in the treatment of cancer. Doxorubicin, the first
anthracycline to gain FDA approval, has demonstrated efficacy in a wide variety
of cancers including breast cancer, lung cancer, sarcomas, and lymphomas.
However, due to the uptake of doxorubicin by various parts of the body, it is
associated with side effects such as cumulative cardiotoxicity, myelosuppression
(decreased production of blood cells by bone marrow), gastrointestinal
disorders, mucositis (inflammation of the mucous membranes lining the digestive
tract, including the mouth), stomatitis (inflammation of the mouth’s soft
tissue), and extravasation (the leakage of intravenous drugs from the vein into
the surrounding tissue).
4
We
believe INNO-206 has attributes that improve on native doxorubicin, including
reduction of adverse events, improvement in efficacy and the ability to reach
the tumor more quickly.
Our
anticipated mechanism of action for INNO-206 is as follows:
|
|
·
|
after
administration, INNO-206 rapidly binds circulating albumin through the
EMCH linker;
|
|
|
·
|
circulating
albumin preferentially accumulates in tumors, bypassing uptake by other
non-tumor sites, including the heart, bone marrow and the gastrointestinal
tract;
|
|
|
·
|
once
albumin-bound INNO-206 reaches the tumor, the acidic environment of the
tumor causes cleavage of the acid sensitive linker;
and
|
|
|
·
|
free
doxorubicin is released at the site of the tumor and is taken up by the
cancer cells.
|
Pre-clinical
data. In a variety of preclinical models, INNO-206 was
superior to doxorubicin in its ability to increase the total doxorubicin dose,
antitumor efficacy, and safety, including a reduction in cardiotoxicity. Animal
studies conducted by INNO-206 inventor Dr. Felix Kratz, Department of Medical
Oncology, Clinical Research, at the Tumor Biology Center in Freiburg, Germany,
demonstrated statistically significant efficacy against breast, ovarian,
pancreatic and small cell lung cancers growing in immunodeficient
mice.
Clinical data. A
Phase I study of INNO-206 that demonstrated safety and objective clinical
responses in a variety of tumor types was completed in 2005 and presented at the
March 2006 Krebskongress meeting in Berlin. In this study, single doses were
administered every 3 weeks at up to six times the standard dosing of doxorubicin
without an increase in side effects over those historically observed with
doxorubicin. Twenty-three of 35 evaluable patients had either an objective
clinical (partial) response or stable disease. Objective
clinical responses were observed in patients with sarcoma, breast, and lung
cancers.
Development
Plan. Based on the objective clinical responses seen in the
Phase I study, and preclinical data, we intend to initiate three Phase 2
clinical trials in 2010 of INNO-206 in patients with pancreatic cancer, gastric
cancer and soft tissue sarcomas.
Bafetinib. Bafetinib
(formerly INNO-406) is a novel drug developed by the Japanese pharmaceutical
company Nippon Shinyaku, to overcome the limitations of Gleevec and second-line
tyrosine kinase inhibitors in resistant chronic myelogenous leukemia, or CML. We
have announced plans to develop bafetinib as a treatment for B-cell chronic
lymphocytic leukemia (B-CLL) and glioblastoma multiforme (GBM), due to the
potent and specific inhibitory properties of bafetinib against Lyn kinase, which
is overexpressed in both B-CLL and GBM.
Bafetinib for the Treatment of
B-CLL. B-CLL is the most common form of leukemia in adults in
Western countries. More than 17,000 new cases of B-CLL are reported in the
United States each year; however up to an estimated 40% of cases may not be
reported due to under-diagnosis and lack of placement in cancer registries.
Virtually all patients are older than 55 years at presentation, with an average
age of 70 years. Patients in the high-risk B-CLL classification have a median
overall survival of one to five years.
Pre-clinical
Data. In mice-leukemia models, bafetinib has been shown to
markedly extend the survival of animals implanted with Gleevec-resistant
leukemic cells. In toxicology studies done in mice, rats, and dogs, bafetinib
appeared to be safe and well-tolerated. A dose described in dogs at
which no side effects were seen was used to calculate the starting dose in
humans for our recently completed clinical trial.
Phase I Study. In
November 2008, we announced that bafetinib demonstrated clinical responses in
patients with CML in a Phase I clinical trial conducted in patients with CML and
other leukemias that have a certain mutation called the Philadelphia Chromosome
(Ph+) and are intolerant of or resistant to Gleevec and, in some cases,
second-line tyrosine kinase inhibitors such as dasatinib (Sprycelâ)
and nilotinib (Tasignaâ)).
The clinical trial was designed to identify the optimal dose for possible future
studies by escalating doses from 30 mg once per day to up to 480 mg twice per
day in a total of 56 patients with Ph+ leukemias. Of the patients, 31 had CML in
chronic phase (CML-CP), nine were in accelerated phase (CML-AP), seven were in
blast phase (CML-BP), and nine had Ph+
acute lymphocytic leukemia. The clinical trial was conducted at seven clinical
sites in the US, Germany, and Israel, with Hagop Kantarjian, M.D., Professor
& Chairman, Department of Leukemia, The University of Texas, M.D. Anderson
Cancer Center, serving as the Principal Investigator. A positive,
dramatic decrease in the number of leukemia cells in the bone marrow was seen in
35% of the patients that were randomly chosen to begin their treatment with the
optimal bafetinib
dose of 240 mg twice per day.
5
The
maximum tolerated
dose was determined to be 240 mg given twice per day, based on evidence of
increasing potential liver toxicity at higher doses. Common adverse events
(observed in greater than 20% of patients in the 240 mg twice per day dose
group) were gastrointestinal toxicity, swelling, and fatigue. There was no
evidence of fluid accumulating around the lungs, or significant changes in a
certain heart rhythm called QTc prolongation, which are serious side effects
known to occur in patients treated with approved drugs for this indication.
Approximately 13% of patients across all dose groups discontinued dosing due to
unacceptable toxicity.
Development
Plan. We recently announced plans to initiate in the first
half of 2010 a Phase 2 proof-of-concept clinical trial to evaluate the efficacy
and safety of bafetinib in patients with B-CLL. In the planned Phase
2 trial, approximately 20 patients who have failed treatment with first-line
agents will be administered daily oral doses of bafetinib. Patients will be
monitored for clinical response, time to disease progression and cancer
progression-free survival. Based on trial results, we plan to conduct a larger
comparative trial to further determine efficacy of this agent. We
also plan to initiate a clinical Phase 2 proof of concept clinical trial with
bafetinib as a treatment for glioblastoma multiforme, a common and aggressive
type of primary brain tumor, in the second half of 2010.
Tamibarotene. Tamibarotene
is an orally available, synthetic retinoid rationally designed to overcome
resistance and avoid toxic side effects of differentiation therapy with
all-trans retinoic acid, or ATRA, a component of the current first-line
treatment for APL.
Tamibarotene for the treatment of
APL. Acute promyelocytic leukemia, or APL, is a specific type
of acute myeloid leukemia characterized by the t(15;17) translocation, which
fuses the promyelocytic leukemia, or PML, gene on chromosome 15 to the retinoic
acid receptor, or RARa gene on chromosome
17. This fusion causes abnormal cell growth.
Differentiation
therapy with ATRA, is the basis for the treatment of APL. Differentiation
therapy causes leukemic promyelocytes to mature and undergo cell death. Patients
typically receive ATRA in combination with chemotherapy as the initial therapy, followed by
anthracyline-based consolidation therapy designed to produce complete remission.
The majority of patients treated this way generally experience a complete
remission of disease. Current National Comprehensive Cancer Network guidelines
recommend that patients then undergo one to two years of maintenance therapy
with ATRA to prevent a recurrence. ATRA therapy is associated with several
toxicities, the most serious of which, retinoic acid syndrome, or
RAS. RAS, which occurs in up to 25% of patients treated with ATRA, is
a serious and potentially fatal complication characterized by fever, dyspnea
(breathing difficulties), weight gain, pulmonary infiltrates (abnormal
accumulation in the lungs), and pleural or pericardial effusions (excess fluid
around the lungs or heart).
Patients
that initially respond to front-line therapy with ATRA plus
chemotherapy sometimes relapse, and some of these patients fail to respond to a
second course of treatment with ATRA. Currently, patients who fail ATRA-based
therapy are treated with arsenic trioxide, a compound administered intravenously
and associated with significant toxicity, including irregular heartbeat. There
currently is no standard of care for patients who do not respond to ATRA and
arsenic trioxide, or who respond but subsequently relapse. In 2007, the FDA
granted Orphan Drug Designation and Fast Track Designation for the use of
tamibarotene in patients with relapsed or refractory APL following treatment
with ATRA and arsenic trioxide.
Tamibarotene
was developed to overcome resistance to ATRA. In vitro, tamibarotene is
approximately ten times more potent than ATRA at causing APL cells to
differentiate and die. In addition, tamibarotene has a lower affinity for
cellular retinoic acid binding protein, or CRABP, which we believe should allow
for sustained plasma levels during administration. This may enhance
tamibarotene’s potential efficacy, because patients may be able to experience
benefits from the drug over a longer period of
time. Tamibarotene does not bind the RAR-g receptor, the major
retinoic acid receptor in the dermal epithelium, which should lessen the
occurrence of skin toxicities.
Pre-clinical
data. In preclinical models, tamibarotene was superior to ATRA
in its ability to cause APL cells to differentiate and die. In the clinical
setting, in vitro response to tamibarotene appeared predictive of clinical
response, including activity in patients who had a poor response to
ATRA.
6
Clinical
data. Tamibarotene is approved in Japan under the brand name
Amnolake for
use in relapsed or refractory APL. The approval was based on data from two
studies in Japanese patients. In the pivotal study, the effectiveness of orally
administered tamibarotene was administered to 42 patients with APL, 39 of whom
were evaluable for response. Patients included individuals who had
never received treatment for APL and patients who had been previously treated
with ATRA. Tamibarotene was administered orally at a dose of 6 mg/m2/day
for eight weeks. The overall complete response rate in these patients was 61.5%.
In patients who had a recurrence of APL following ATRA therapy, the response
rate was 81%. RAS was reported in three patients, or 7.3% of the patient
group.
Development
Plan. There is currently a Special Protocol Assessment (SPA)
in place with the FDA for a Phase 2 registration clinical trial, known as
STAR-1, which is evaluating the efficacy and safety of tamibarotene as a
third-line treatment for APL. The STAR-1 trial is ongoing and currently includes
six clinical sites in the U.S. We recently reported that, of the 11
patients enrolled in the STAR-1 trial to date, three (27%) achieved a
hematologic complete response, and four (36%) a morphologic leukemia-free
state. Depending on the trial’s outcome, this study, in combination
with the data from the two Japanese studies, could form the basis of a new drug
application, or NDA.
In
addition, we have announced plans to develop tamibarotene, in combination with
chemotherapy or arsenic trioxide (ATO), as a first-line treatment for APL. We
have received favorable reviews on tamibarotene as a potential treatment for APL
from key opinion leaders and have been working with notable clinicians in this
field on trial design.
Among
other preparatory activities, in September 2009, we initiated a dose escalation
clinical trial with tamibarotene combined with TRISENOX® (arsenic trioxide)
injection (marketed by Cephalon, Inc.) in patients with relapsed APL. An
objective of this trial is to determine the appropriate combination therapy dose
for evaluation as a potential first-line treatment for APL.
Arimoclomol. Arimoclomol is
an orally-administered small-molecule product candidate that we believe
functions by stimulating a normal cellular protein repair pathway by amplifying
molecular chaperone proteins implicated in neurological disorders.
Arimoclomol for the treatment of
ALS. ALS, or Lou Gehrig’s disease, is a debilitating and ultimately
deadly disease involving the progressive degeneration of motor neurons believed
to be caused by toxic mis-folding of proteins. According to the ALS Association,
approximately 30,000 people in the U.S. are living with ALS and 5,600 new cases
are diagnosed each year. Worldwide, an estimated 120,000 people are living with
ALS. According to the ALS Survival Guide, 50% of ALS patients die within 18
months of diagnosis and 80% die within five years of diagnosis.
The
following is a summary of our clinical development of arimoclomol for treating
ALS:
|
|
·
|
in
July 2006, we completed an 84-patient, multi-center, double-blind,
placebo-controlled, multi-dose Phase IIa clinical trial of safety and
tolerability of arimoclomol in volunteers with ALS, which we refer to as
the Phase IIa trial;
|
|
|
·
|
in
May 2007, we completed an open-label extension of the Phase IIa trial in
approximately 70 ALS patients from the trial who were administered the
highest investigational dose (100 mg three times daily) of arimoclomol for
an additional six months;
|
|
|
·
|
in
June 2007, we completed a multiple ascending-dose clinical trial of safety
and tolerability involving 40 healthy volunteers;
and
|
|
|
·
|
in
November 2007, we completed a 28-day safety clinical trial with 400 mg of
arimoclomol three times daily involving 16 healthy
volunteers.
|
Phase IIa clinical trial.
Participants in the Phase IIa clinical trial of arimoclomol were administered
either a placebo capsule, or one of three dosage levels of arimoclomol capsules,
three times daily for a period of 12 weeks, immediately followed by a one-month
period without the drug. The primary endpoints of the Phase IIa trial were
safety and tolerability. Secondary endpoints included a preliminary evaluation
of efficacy using two widely accepted disease-progression markers. The first
marker, the revised ALS Functional Rating Scale, or ALSFRS-R, is used to
determine patients’ overall functional capacity and independence in 13
activities. The second marker measures vital capacity, an assessment of lung
capacity, which is an important disease indicator since ALS sufferers eventually
lose the ability to breathe on their own. The trial was designed to be able to
detect only extreme responses in these two markers.
7
The
results from our Phase IIa trial and open-label extension clinical trial
indicated that arimoclomol was safe and well tolerated in ALS volunteers, even
at the highest administered dose. Arimoclomol was detected in participants’
cerebral spinal fluid, demonstrating that it passed the so-called blood:brain
barrier, and participants treated with arimoclomol experienced a statistically
significant decrease in adverse events of weakness compared with the placebo
group.
Phase IIb efficacy trial. In
December 2009, the FDA accepted a revised protocol for an ascending dose
efficacy trial to evaluate the safety and efficacy in ALS patients at up to 400
mg administered orally three times daily. The clinical trial protocol accepted
by the FDA is a tiered, placebo-controlled, double-blind ascending dose study.
The study is designed to test progressive groups, each with between 20 and 30
ALS patients over a three-month treatment period. Fifteen patients would receive
a combination of arimoclomol at various dose levels and riluzole at a fixed dose
of 50 mg twice daily, with between five and 15 ALS patients receiving placebo
and riluzole at the same fixed dose. The first group would receive arimoclomol
100 mg capsules three times daily. Every four weeks, another group of ALS
patients would begin three-month testing with six patients receiving arimoclomol
dosing three times daily at a 75 mg per dose increase from the prior group. The
maximum dose in the protocol allows for testing arimoclomol at 400 mg three
times daily. An independent safety monitoring board would review safety results
prior to initiating each consecutive increase in dosage level.
Other Clinical
Development. In February 2009, a Phase II/III adaptive
clinical trial commenced to study arimoclomol in a subset of patients with the
inherited or familial form ALS. Patients with familial ALS (fALS) who harbor
certain mutations in the superoxide dismutase-1 (SOD1) gene suffer from a
rapidly progressing form of the disease. The clinical trial is being financially
supported by grants from the ALS Association and the U.S. Food and Drug
Administration’s (FDA’s) Office of Orphan Products Development (OOPD), and we
are supplying the drug and allowing the sponsor to reference our Investigational
New Drug Application for regulatory purposes.
Arimoclomol for recovery from
stroke. Stroke results from an acute loss of normal blood flow to the
brain caused most often by a blockage in a blood vessel (ischemic) or due to
leaking of blood from a vessel (hemorrhagic). According to the American Heart
Association: stroke is the
third leading cause of death and the number one cause of long-term
disability in the U.S.; between 50% and 70% of stroke survivors regain
functional independence, but between 15% and 30% are permanently disabled and
20% require institutional care within three months after stroke; and the direct
and indirect stroke cost in the U.S. totaled approximately $58 billion in
2006.
After the
normal flow of blood is restored to the brain, post-stroke neurological function
continues to decline. We believe that this continuing decline in neurological
function is the consequence of mis-folded protein aggregates generated as a
result of oxygen deprivation during the original event. Preclinical efficacy
studies completed by us in April 2007 indicated that arimoclomol accelerated the
time to recovery, and improved recovery, in experimental animal models of
stroke. These results were obtained even when arimoclomol was administered as
long as 48 hours after onset.
Arimoclomol for the treatment of
cancer. Through research conducted in our former San Diego laboratory, we
have identified preclinical pipeline compounds that inhibit, rather than
amplify, molecular chaperone proteins. These early stage compounds
are potential oncology therapeutics because cancer cells by their very nature
generate large amounts of toxic, misfolded proteins and are far more dependent
upon the activity of molecular chaperone proteins for survival than are normal
cells. Because several of these compounds have already been shown to selectively
kill cancer cells and spare normal cells in tissue culture, they represent a
novel approach to cancer therapeutics with the potential of having relatively
large therapeutic indices.
Iroxanadine. Iroxanadine also
is an orally-administered small-molecule product candidate. We believe it
functions by stimulating the molecular chaperone protein response in the
endothelium, the thin layer of cells that line the interior surface of human
blood vessels.
Iroxanadine for the treatment of
diabetic ulcers. Type 2 diabetes is a major health problem with
significant secondary complications. The American Diabetes Association estimates
that there are 21 million type 2 diabetes sufferers in the U.S. The World Health
Organization estimates that there are more than 162 million cases of type 2
diabetes worldwide. According to the American Diabetes Association, 15% of all
diabetics will develop a foot ulcer during their lifetime, and over 82,000
non-traumatic lower-limb amputations were performed on diabetics in the U.S. in
2002 due to ulcers and other complications. We believe there are reasonable data
in the scientific literature supporting the concept that diabetic foot ulcers
fail to heal efficiently due to the dysfunction of endothelial cells lining the
blood vessels caused by protein mis-folding.
8
Animal
studies completed by us in May 2007 indicated that iroxanadine significantly
decreased the time it took for wounds to heal in diabetic mice without affecting
healing in healthy mice. Wound healing in the diabetic mice, which normally
required twice the time to heal as healthy mice, was accelerated to the extent
that healing time of diabetic mice treated with iroxanadine was
indistinguishable from that in untreated healthy mice.
In Phase
I clinical trials in healthy volunteers and Phase II clinical trials in patients
with chronic high blood pressure conducted prior to our acquisition of
iroxanadine, the drug candidate was shown to be safe and well-tolerated and
demonstrated significant improvement in the function of endothelial cells in the
brachial artery, a major blood vessel of the upper arm.
Strategic Plans for Molecular
Chaperone Assets. We intend to seek a partner for the development of
arimoclomol and iroxanadine for all indications on a worldwide
basis. We are also exploring the possibility of a spin-out
transaction to accelerate the development of our molecular chaperone
assets.
Other
Technologies
Our other
current technologies are CRL-5861, an intravenous agent for treatment of sickle
cell disease and other acute vaso-occlusive disorders, and TranzFect, a delivery
technology for DNA-based and conventional vaccines and other potential
uses. We currently have no plans for development of these
technologies.
Our
Separation from RXi Pharmaceuticals Corporation
Until
early 2008, we owned approximately 85% of the outstanding shares of common stock
of RXi and our financial statements, including our financial statements as of
and for the year ended December 31, 2007, included the consolidated financial
condition and results of operations of RXi. On February 14, 2008, our board of
directors declared a dividend of one share of RXi common stock for each
approximately 20.05 outstanding shares of our common stock, which was paid on
March 11, 2008 and which reduced our ownership of RXi shares to less than
50%. As a result, our financial statements since March 11, 2008 no longer
consolidate the financial condition and results of operation of RXi, but instead
reflect our ongoing investment in RXi based on the equity method of accounting.
As of March 12, 2010, we owned approximately 36% of the outstanding
shares of RXi common stock.
We are
party to a letter agreement with RXi and some of RXi’s current stockholders
under which we are entitled to preemptive rights to acquire any “new securities”
(as defined) that RXi proposes to sell or issue, so that we may maintain our
percentage ownership in RXi. Our preemptive rights will expire on January 8,
2012 or such earlier time at which we own less than 10% of RXi’s outstanding
common stock.
Under the
letter agreement with RXi, we agreed to vote our RXi shares for the election of
RXi directors and take other actions to ensure that a majority of the board of
directors of RXi are independent of us. We further agreed to approve of actions
that may be adopted and recommended by the RXi board of directors to facilitate
any future financing by RXi.
Manufacturing
We have
no capability to manufacture supplies of any of our products, and rely on
third-party manufacturers to produce materials needed for research and clinical
trials. We have contracted with various contract manufacturing
facilities for supply of our active pharmaceutical ingredient, or API, for our
product candidates. Pursuant to our license with TMRC Co., Ltd., or TMRC,
relating to tamibarotene, TMRC will provide us with tamibarotene at a fixed
price and in a quantity and quality sufficient to meet our clinical and
commercial needs.
To be
commercialized, our products also must be capable of being manufactured in
commercial quantities in compliance with stringent regulatory requirements and
at an acceptable cost. We intend to rely on third-party manufacturers to produce
commercial quantities of any products for which we are able to obtain marketing
approval. We have not commercialized any product, and so we also have not
demonstrated that any of our product candidates can be manufactured in
commercial quantities in accordance with regulatory requirements or at an
acceptable cost.
If our
product candidates cannot be manufactured in suitable quantities and in
accordance with regulatory standards, our clinical trials, regulatory approvals,
and marketing efforts for such products may be delayed. Such delays could
adversely affect our competitive position and our chances of generating
significant recurring revenues. If our products are not able to be manufactured
at an acceptable cost, the commercial success of our products may be adversely
affected.
9
Marketing
Our
tentative plan is to establish our own sales force and marketing capability in
order to commercialize our oncology drug candidates, including INNO-206,
bafetinib and tamibarotene, in the U.S. and to seek a marketing partner for
commercialization in other territories.
Patents
and Proprietary Technology
We
actively seek patent protection for our technologies, processes, uses, and
ongoing improvements and consider our patents and other intellectual property to
be critical to our business. We acquired patents and patent applications, and
have filed several new patent applications, in connection with our molecular
chaperone program.
We
regularly evaluate the patentability of new inventions and improvements
developed by us or our collaborators, and, whenever appropriate, will endeavor
to file U.S. and international patent applications to protect these new
inventions and improvements. We cannot be certain that any of the current
pending patent applications we have filed or licensed, or any new patent
applications we may file or license, will ever be issued in the U.S. or any
other country. There also is no assurance that any issued patents will be
effective to prevent others from using our products or processes. It is also
possible that any patents issued to us, as well as those we have licensed or may
license in the future, may be held invalid or unenforceable by a court, or third
parties could obtain patents that we would need to either license or to design
around, which we may be unable to do. Current and future competitors may have
licensed or filed patent applications or received patents, and may acquire
additional patents and proprietary rights relating to molecular chaperone
amplification and other small molecule technology or other compounds, products
or processes that may be competitive with ours.
In
addition to patent protection, we attempt to protect our proprietary products,
processes and other information by relying on trade secrets and non-disclosure
agreements with our employees, consultants and certain other persons who have
access to such products, processes and information. Under the agreements, all
inventions conceived by employees are our exclusive property, but there is no
assurance that these agreements will afford significant protection against
misappropriation or unauthorized disclosure of our trade secrets and
confidential information.
LICENSE
AGREEMENTS
INNO-206
We
succeeded to Innovive’s agreement with KTB Tumorforschungs GmbH, or KTB, for the
license of patent rights held by KTB for the worldwide development and
commercialization of INNO-206. The license is exclusive and
worldwide, applies to all product that may be subject to the licensed
intellectual property and may be used in all fields of use. We may sublicense
the intellectual property in our sole discretion. The agreement also grants us
an option to include within the license any technology that is claimed or
disclosed in the licensed patents and patent applications for use in the field
of oncology and the right of first refusal on any license that KTB wishes to
make to a third party regarding any technology that is claimed or disclosed in
the licensed patents and patent applications for use in the field of
oncology.
Under the
agreement, we must make payments to KTB in the aggregate of $7.5 million upon
meeting clinical and regulatory milestones up to and including the product’s
second final marketing approval. We also agreed to pay:
|
|
·
|
commercially
reasonable royalties based on a percentage of net sales (as defined in the
agreement);
|
|
|
·
|
a
percentage of non-royalty sub-licensing income (as defined in the
agreement); and
|
|
|
·
|
milestones
of $1 million for each additional final marketing approval that we might
obtain.
|
In the
event that we must pay a third party in order to exercise our rights to the
intellectual property under the agreement, we will deduct a percentage of those
payments from the royalties due KTB, up to an agreed upon cap. This deduction
includes a percentage of any payments that might be required to be made by us to
Bristol-Myers Squibb. Bristol-Myers Squibb holds a patent on technology that
might be considered to block the patents and patent applications that are the
subject of the agreement with KTB.
10
Under the
agreement with KTB, we must use commercially reasonable efforts to conduct the
research and development activities we determine are necessary to obtain
regulatory approval to market the product in those countries that we determine
are commercially feasible. Under the agreement, KTB is to use its commercially
reasonable efforts to provide us with access to suppliers of the API of the
product on the same terms and conditions as may be provided to KTB by those
suppliers.
The
agreement will expire on a product-by-product basis upon the expiration of the
subject patent rights. We have the right to terminate the agreement on 30 days
notice, provided we pay a cash penalty to KTB. KTB may terminate the agreement
if we are in breach and the breach is not cured within a specified cure period
or if we fail to use diligent and commercial efforts to meet specified clinical
milestones.
Bafetinib
We
likewise succeeded to Innovive’s exclusive, worldwide (with the exception of
Japan) royalty-bearing license agreement with Nippon Shinyaku, including the
right to grant sublicenses, for the intellectual property relating to bafetinib
in all fields. The license agreement will expire on a
country-by-country basis upon the expiration of the subject patent rights. The
bafetinib license covers two Patent Cooperation Treaty, or PTC, applications
filed in 2003 and 2004, respectively.
Under the
agreement, we are obliged to pay Nippon Shinyaku an aggregate of $13.35 million
(including $5 million upon the product’s initial final marketing approval) upon
the achievement of clinical and regulatory milestones up to and including
approvals in the U.S. and Europe. We also will be obliged to pay:
|
|
·
|
commercially
reasonable royalties based on a percentage of net sales (as defined in the
Nippon Shinyaku license agreement), dependent on reaching certain revenue
thresholds;
|
|
|
·
|
annual
minimum payments if sales of bafetinib do not meet specified levels;
and
|
|
|
·
|
a
percentage of non-royalty sub-licensing income (as defined in the license
agreement).
|
The
agreement includes covenants that require us to, among other things, file an NDA
by a specific date and use our commercially reasonable efforts to bring a
licensed product to market. In the event that we breach a material
term of the Nippon Shinyaku license agreement, Nippon Shinyaku has the option to
terminate the agreement following the giving of notice and an opportunity to
cure any such breach.
Tamibarotene
We also
succeeded to Innovive’s agreement with TMRC for the license of patent rights
held by TMRC for the North American development and commercialization of
tamibarotene. The license is exclusive, applies to all products that may be
subject to the licensed intellectual property and may be used in the treatment
of APL. We may sublicense the intellectual property in our sole discretion. The
agreement also grants us an option to include within the license the use of the
drug in other fields in oncology including multiple myeloma, myelodysplastic
syndrome, and solid tumors.
Under the
agreement, we must pay TMRC royalties based on net sales and make payments to
TMRC in the aggregate of $4.165 million upon meeting clinical, regulatory, and
sales milestones up to and including the first commercial sale of the product
for the treatment of APL.
Under the
agreement, we must use commercially reasonable efforts to conduct the research
and development activities we determine are necessary to obtain regulatory
approval to market the product in those countries in North America that we
determine are commercially feasible.
The
agreement will expire upon the expiration of the subject patent rights, or 15
years from the date of first commercial sale of product in North America,
whichever is later. The agreement may be terminated if either party is in breach
and the breach is not cured within a required amount of time. We may also
terminate the agreement in the event of a material change in the safety profile
of the technology that makes continued development impossible.
11
Under the
merger agreement by which we acquired Innovive, we agreed to pay the former
Innovive stockholders up to $1.01 per Innovive share of future earnout merger
consideration, subject to our achievement of specified net sales under the
Innovive license agreements. The earnout merger consideration, if
any, will be payable in shares of our common stock, subject to specified
conditions, or, at our election, in cash or by a combination of shares of our
common stock and cash. Our common stock will be valued for purposes
of any future earnout merger consideration based upon the trading price of our
common stock at the time the earnout merger consideration is paid.
Competition
INNO-206
is a prodrug of doxorubicin, a widely used anti-cancer
drug. Doxorubicin is part of the anthracycline class of chemotherapy
agents. Anthracyclines, many of which are generic including
doxorubicin, have been used throughout the world to treat various cancers for
several decades. Due to their track record of broad anti-cancer
activity, new types of anthracyclines and modified or reformulated versions
continue to be developed to overcome toxicities which limit the use of these
drugs.
INNO-206
is a chemically modified version of doxorubicin that incorporates an acid
sensitive linker technology to improve targeting to the tumor. We
believe that the albumin-binding ability of INNO-206 will allow the compound to
overcome many of the side effect issues typically associated with
anthracyclines. We also believe that using albumin as a targeted carrier
will allow for higher dosing and greater efficacy.
Soft
tissue sarcoma patients are typically treated with surgery followed by radiation
therapy. Doxorubicin is the only approved drug for treating soft
tissue sarcoma and is often used in combination with radiation. The
National Comprehensive Cancer Network also includes the use of ifosfamide,
epirubicin, Eli Lilly’s Gemzar, dacarbazine and liposomal doxorubicin marketed
in the US as Doxil by Johnson & Johnson. For patients ineligible
for surgery, radiation and/or chemotherapy is the only option. Based
on both human and animal data, we believe we can give 5-8 times the amount of
doxorubicin with administration of INNO-206. Other approaches to
treating soft tissue sarcoma are in late stage clinical
development. These include ridaforolimus being developed by Ariad
Pharmaceuticals and Merck & Co., GlaxoSmithKline’s pazopanib,
Sanofi-Aventis’ AVE8062, Threshhold Pharmaceuticals’s TH-302, and trabectedin
being co-developed by Johnson and Johnson and PharmaMar.
Advanced
gastric cancer patients are those that have failed both surgery and prior
chemotherapy. They may or may not be suitable candidates for
chemoradiation. These patients are typically treated with a variety
of combination of approved drugs such as cisplatin, docetaxel, 5-FU,
oxaliplatin, iriniotecan and paclitaxel. Epirubicin, an
anthracycline, is part of several gastric cancer treatment
regimens. We believe INNO-206 could be a potentially more effective
anthracycline and potentially more effective in this setting.
Pancreatic
cancer is one of the most lethal types of cancer and current treatment options
have limited benefit due to the rapid progression of this
cancer. Patients are typically treated with surgery, radiation and
chemotherapy. Eli Lilly’s Gemzar is currently approved for the first
line treatment of locally advanced or metastatic pancreatic
cancer. It is also indicated for the use in patients who have
received prior treatment with 5-FU. OSI Pharmaceuticals’ Tarceva was
approved in 2005 for the use in combination with Gemzar. The NCCN
believes the best management for these patients is in a clinical
trial. Because of the tremendous unmet need for these patients, many
companies are developing new drugs to treat pancreatic cancer. Late
stage drugs in clinical trials include Abraxane by Abraxis BioScience, AGS-1C4D4
by Astellas Pharma Inc., TNFerade™ by GenVec, and S-1 by
Sanofi-Aventis.
There are
currently three marketed competitors to bafetinib (formerly INNO-406) in the CML
market, Gleevec®, Sprycel® and Tasigna®. Gleevec is approved for treatment
of newly diagnosed adult patients with Philadelphia chromosome–positive chronic
myeloid leukemia (Ph+ CML) in the chronic phase and patients with Ph+
CML in blast crisis (BC), accelerated phase (AP), or in the chronic phase (CP)
after failure of interferon-alpha therapy. Sprycel® and Tasigna® are approved
for Gleevec-resistant CML. Because of the highly competitive nature
of the CML market including drug candidates in development, CytRx plans to
develop bafetinib initially in cancers other than CML. CytRx has
selected B-cell chronic lymphocytic leukemia (B-CLL) and glioblastoma multiforme
due to the potent and specific inhibitory properties of bafetinib against Lyn
kinase. Lyn kinase is a member of the Src family of kinases which are known to
be involved in cell growth. Lyn kinase is overexpressed in both B-CLL and
glioblastoma multiforme (GBM).
12
We are
unaware of any compounds in development that target Lyn kinase. We
plan to develop bafetinib as a treatment for B-CLL patients that have failed
first-line therapy. There are several drugs approved for the
treatment of CLL. First-line therapy for CLL includes a variety of
combination therapies including fludarabine, cyclophosphamide, Rituxan® and
Campath®. Treatment for relapsed or refractory CLL
includes several chemotherapy regimens including CHOP, CFAR, hyperCFAD and OFAR
in addition to single agents including GlaxoSmithKline’s ArzerraTM. Arzerra
was approved in October 2009 for CLL patients who are refractory to treatment
with fludarabine and Campath.
Current
therapy for glioblastoma multiforme, the most common form of brain cancer, is
surgery followed by radiation therapy and chemotherapy. Merck’s
Temodar® is approved for treating newly diagnosed GBM concomitantly with
radiotherapy and then as a maintenance treatment. Roche’s Avastin was
approved in May 2009 for treatment of recurrent GBM. We believe that
bafetinib’s ability to selectively inhibit Lyn kinase and to penetrate the brain
in an animal model of cancer will be an effective treatment for second-line
therapy in GBM.
To our
knowledge, there are no competitors in clinical development for refractory
APL. Currently, treatment of APL is based on induction and maintenance
therapy with ATRA and chemotherapy (typically idarubicin). ATRA and
idarubicin are both generic compounds. Arsenic trioxide, currently
marketed by Cephalon, is approved for use in patients who have relapsed after
ATRA-based therapy in APL. There are no FDA-approved therapies for
patients who have failed arsenic trioxide. In practice, it appears that
patients who fail arsenic trioxide are retreated with ATRA or receive Mylotarg,
which is marketed by Pfizer Inc.
We are
aware of only one drug, rilutek, developed by Aventis Pharma AG, that has been
approved by the FDA for the treatment of ALS. Many companies are working to
develop pharmaceuticals to treat ALS, including Aeolus Pharmaceuticals,
Mitsubishi Tanabe Pharma Corporation, Ono Pharmaceuticals, Trophos SA, Knopp
Neurosciences Inc., Faust Pharmaceuticals SA, Oxford BioMedica plc, Phytopharm
plc and Teva Pharmaceutical Industries Ltd., as well as RXi. ALS patients often
take over-the-counter supplements, including vitamin E, creatine and coenzyme
Q10, or drugs such as lithium that are approved for other indications. ALS
belongs to a family of neurodegenerative diseases that includes Alzheimer’s,
Parkinson’s and Huntington’s diseases. Due to similarities between these
diseases, a new treatment for one such disease potentially could be useful for
treating others. There are many companies producing and developing drugs used to
treat neurodegenerative diseases other than ALS, including Amgen, Inc., Biogen
Idec, Boehringer Ingelheim, Cephalon, Inc., Ceregene, Inc., Elan
Pharmaceuticals, plc, Forest Laboratories, Inc., H. Lundbeck A/S, Phytopharm
plc, UCB Group and Wyeth.
Many
companies, including large pharmaceutical and biotechnology firms with financial
resources, research and development staffs, and facilities that may be
substantially greater than those of ours or our strategic partners or licensees,
are engaged in the research and development of pharmaceutical products that
could compete with our potential products. To the extent that we seek to
acquire, through license or otherwise, existing or potential new products, we
will be competing with numerous other companies, many of which will have
substantially greater financial resources, large acquisition and research and
development staffs that may give those companies a competitive advantage over us
in identifying and evaluating these drug acquisition opportunities. Any products
that we acquire will be competing with products marketed by companies that in
many cases will have substantially greater marketing resources than we have. The
industry is characterized by rapid technological advances and competitors may
develop their products more rapidly and such products may be more effective than
those currently under development or that may be developed in the future by our
strategic partners or licensees. Competitive products for a number of the
disease indications that we have targeted are currently being marketed by other
parties, and additional competitive products are under development and may also
include products currently under development that we are not aware of or
products that may be developed in the future.
Government
Regulation
The U.S.
and other developed countries extensively regulate the preclinical and clinical
testing, manufacturing, labeling, storage, record-keeping, advertising,
promotion, export, marketing and distribution of drugs and biologic products.
The FDA, under the Federal Food, Drug, and Cosmetic Act, the Public Health
Service Act and other federal statutes and regulations, regulates pharmaceutical
and biologic products.
To obtain
approval of our product candidates from the FDA, we must, among other
requirements, submit data supporting safety and efficacy for the intended
indication as well as detailed information on the manufacture and composition of
the product candidate. In most cases, this will require extensive laboratory
tests and preclinical and clinical trials. The collection of these data, as well
as the preparation of applications for review by the FDA involve significant
time and expense. The FDA also may require post-marketing testing to monitor the
safety and efficacy of approved products or place conditions on any approvals
that could restrict the therapeutic claims and commercial applications of these
products. Regulatory authorities may withdraw product approvals if we fail to
comply with regulatory standards or if we encounter problems at any time
following initial marketing of our products.
13
The first
stage of the FDA approval process for a new biologic or drug involves completion
of preclinical studies and the submission of the results of these studies to the
FDA. These data, together with proposed clinical protocols, manufacturing
information, analytical data and other information submitted to the FDA, in an
investigational new drug application, or IND, must become effective before human
clinical trials may commence. Preclinical studies generally involve FDA
regulated laboratory evaluation of product characteristics and animal studies to
assess the efficacy and safety of the product candidate.
After the
IND becomes effective, a company may commence human clinical trials. These are
typically conducted in three sequential phases, but the phases may overlap.
Phase I trials consist of testing of the product candidate in a small number of
patients or healthy volunteers, primarily for safety at one or more doses. Phase
II trials, in addition to safety, evaluate the efficacy of the product candidate
in a patient population somewhat larger than Phase I trials. Phase III trials
typically involve additional testing for safety and clinical efficacy in an
expanded population at multiple test sites. A company must submit to the FDA a
clinical protocol, accompanied by the approval of the Institutional Review
Boards at the institutions participating in the trials, prior to commencement of
each clinical trial.
To obtain
FDA marketing authorization, a company must submit to the FDA the results of the
preclinical and clinical testing, together with, among other things, detailed
information on the manufacture and composition of the product candidate, in the
form of a new drug application, or NDA.
The
amount of time taken by the FDA for approval of an NDA will depend upon a number
of factors, including whether the product candidate has received priority
review, the quality of the submission and studies presented, the potential
contribution that the compound will make in improving the treatment of the
disease in question, and the workload at the FDA.
The FDA
may, in some cases, confer upon an investigational product the status of a fast
track product. A fast track product is defined as a new drug or biologic
intended for the treatment of a serious or life-threatening condition that
demonstrates the potential to address unmet medical needs for this condition.
The FDA can base approval of an NDA for a fast track product on an effect on a
surrogate endpoint, or on another endpoint that is reasonably likely to predict
clinical benefit. If a preliminary review of clinical data suggests that a fast
track product may be effective, the FDA may initiate review of entire sections
of a marketing application for a fast track product before the sponsor completes
the application. The FDA has granted fast track designation and orphan drug
status to arimoclomol for the treatment of ALS and to tamibarotene for the
treatment of relapsed or refractory APL.
We
anticipate that our products will be manufactured by our strategic partners,
licensees or other third parties. Before approving an NDA, the FDA will inspect
the facilities at which the product is manufactured and will not approve the
product unless the manufacturing facilities are in compliance with the FDA’s
cGMP, which are regulations that govern the manufacture, holding and
distribution of a product. Our manufacturers also will be subject to regulation
under the Occupational Safety and Health Act, the Environmental Protection Act,
the Nuclear Energy and Radiation Control Act, the Toxic Substance Control Act
and the Resource Conservation and Recovery Act. Following approval, the FDA
periodically inspects drug and biologic manufacturing facilities to ensure
continued compliance with the good manufacturing practices regulations. Our
manufacturers will have to continue to comply with those requirements. Failure
to comply with these requirements subjects the manufacturer to possible legal or
regulatory action, such as suspension of manufacturing or recall or seizure of
product. Adverse patient experiences with the product must be reported to the
FDA and could result in the imposition of marketing restrictions through
labeling changes or market removal. Product approvals may be withdrawn if
compliance with regulatory requirements is not maintained or if problems
concerning safety or efficacy of the product occur following
approval.
The
labeling, advertising, promotion, marketing and distribution of a drug or
biologic product also must be in compliance with FDA and Federal Trade
Commission requirements which include, among others, standards and regulations
for off-label promotion, industry sponsored scientific and educational
activities, promotional activities involving the internet, and
direct-to-consumer advertising. We also will be subject to a variety of federal,
state and local regulations relating to the use, handling, storage and disposal
of hazardous materials, including chemicals and radioactive and biological
materials. In addition, we will be subject to various laws and regulations
governing laboratory practices and the experimental use of animals. In each of
these areas, as above, the FDA has broad regulatory and enforcement powers,
including the ability to levy fines and civil penalties, suspend or delay
issuance of product approvals, seize or recall products, and deny or withdraw
approvals.
14
We will
also be subject to a variety of regulations governing clinical trials and sales
of our products outside the U.S. Whether or not FDA approval has been obtained,
approval of a product candidate by the comparable regulatory authorities of
foreign countries and regions must be obtained prior to the commencement of
marketing the product in those countries. The approval process varies from one
regulatory authority to another and the time may be longer or shorter than that
required for FDA approval. In the European Union, Canada and Australia,
regulatory requirements and approval processes are similar, in principle, to
those in the U.S.
Employees
As of
March 12, 2010, we had 15 employees, six of whom were engaged in clinical
development activities and nine of whom were involved in management and
administrative operations.
Available
Information
We
maintain a website at www.cytrx.com and make available there, free of charge,
our periodic reports filed with the Securities and Exchange Commission, or SEC,
as soon as is reasonably practicable after filing. The public may read and copy
any materials we file with the SEC at the SEC’s Public Reference Room at 100 F
Street, NE, Washington, DC 20549. The public may obtain information on the
operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The
SEC maintains a website at http:/www.sec.gov that contains reports, proxy and
information statements, and other information regarding issuers such as us that
file electronically with the SEC. We post on our website our Code of Business
Conduct and Ethics.
Item
1A. RISK FACTORS
An
investment in our shares involves a high degree of risk. Prior to
making a decision about purchasing our shares, you should carefully consider the
risks and uncertainties and all other information contained in this Annual
Report, including the risks and uncertainties discussed below, as well as any
modification, replacement or update to these risks and uncertainties that are
reflected in any subsequent filings we make with the SEC. These risks
and uncertainties are not the only ones facing us. Additional risks
and uncertainties not presently known to us, or that we currently perceive as
immaterial, may also harm our business. If any of these risks or
uncertainties actually occurs, our business, results of operations and financial
condition could be materially and adversely affected. In that case,
the trading price of our common stock could decline, and you could lose all or
part of your investment.
Risks
Associated With Our Business
We
have operated at a loss and will likely continue to operate at a loss for the
foreseeable future.
We have
operated at a loss due to our ongoing expenditures for research and development
of our product candidates and for general and administrative purposes and lack
of significant recurring revenue. We incurred net losses of $4.8
million, $27.0 million, and $21.9 million for the years ended December 31, 2009,
2008 and 2007, respectively. We had an accumulated deficit as of
December 31, 2009 of approximately $197.4 million. We are likely to
continue to incur losses unless and until we are able to commercialize one or
more of our product candidates. These losses, among other things,
have had and will continue to have an adverse effect on our stockholders’ equity
and working capital. Because of the numerous risks and uncertainties associated
with our product development efforts, we are unable to predict when we may
become profitable, if at all. If we do not become profitable or are
unable to maintain future profitability, the market value of our common stock
will be adversely affected.
Because
we have no source of significant recurring revenue, we must depend on financing
to sustain our operations.
Developing
products and conducting clinical trials require substantial amounts of capital.
To date, we have relied primarily upon proceeds from sales of our equity
securities, including securities of RXi, and the exercise of options
and warrants to generate funds needed to finance our business and
operations. We will need to raise additional capital to, among other
things:
|
|
·
|
fund
our clinical trials and pursue regulatory approval of our existing and
possible future product candidates;
|
|
|
·
|
expand
our research and development
activities;
|
|
|
·
|
finance
our general and administrative
expenses;
|
15
|
|
·
|
acquire
or license new technologies;
|
|
|
·
|
prepare,
file, prosecute, maintain, enforce and defend our patent and other
proprietary rights; and
|
|
|
·
|
develop
and implement sales, marketing and distribution capabilities to
successfully commercialize any product for which we obtain marketing
approval and choose to market
ourselves.
|
Our
revenues were $9.5 million, $6.3 million and $7.5 million, respectively, for
years ended December 31, 2009, 2008 and 2007, which included $9.4 million, $6.2
million and $7.2 million, respectively, of deferred revenue recognized from our
sale in August 2006 of a one-percent royalty interest in worldwide sales of
arimoclomol for the treatment of ALS to the privately-funded ALS Charitable
Remainder Trust, or ALSCRT. Pursuant to an amendment signed between
us and the beneficiary of the ALSCRT on August 6, 2009, we were released from
all restrictions on the use of any proceeds previously paid to us in connection
with the arrangement. As a result, we recognized $6.7 million as
service revenue in the third quarter of 2009, which represented the remaining
deferred revenue and previously un-recognized portion of the value received. We
will have no significant recurring revenue unless we are able to commercialize
one or more of our product candidates in development, which may require us to
first enter into license or other strategic arrangements with third
parties.
At
December 31, 2009, we had cash and cash equivalents of approximately $9.9
million, marketable securities of $22.8 million, and held 5,768,881 shares of
restricted common stock of RXi with a market value of $26.4 million
based upon the closing price of the RXi common stock on that date as reported on
The NASDAQ Capital Market. On July 27, 2009, we raised approximately
$18.3 million, net of fees and expenses, in a registered direct offering, and on
September 23, 2009, we raised approximately $1.2 million, net of fees, from the
sale of 500,000 of our shares of common stock of RXi. Management believes that
our current cash on hand, together with our marketable securities and proceeds
from possible future sales of our securities or our shares of RXi common stock,
will be sufficient to fund our operations for the foreseeable
future. The estimate is based, in part, upon our currently projected
expenditures for 2010 of approximately $18.0 million, which includes approximately
$3.2 million for our clinical programs for INNO-206, approximately $1.6 million
for our clinical programs for bafetinib, approximately $2.5 million for our
clinical program for tamibarotene, approximately $1.4 million for our activities
for arimoclomol, approximately $2.2 million for general operation of our
clinical programs, and approximately $7.1 million for other general and
administrative expenses. These projected expenditures are based upon numerous
assumptions and subject to many uncertainties, and our actual expenditures may
be significantly different from these projections.
If we
obtain marketing approval as currently planned and successfully commercialize
our product candidates, we anticipate it will take a minimum of three years, and possibly
longer, for us to generate significant recurring revenue, and we will be
dependent on future financing until such time, if ever, as we can generate
significant recurring revenue. Our ability to raise capital has been materially
and adversely affected by the continuing poor economy. Despite the
recover in the U.S. financial markets in 2009, the market remains severely
depressed for private investment in public equities, or PIPEs, transactions on
which we have relied for raising needed capital. These conditions
also may materially and adversely affect the market for our RXi shares. We have
no commitments from third parties to provide us with any additional financing,
and we may not be able to obtain future financing on favorable terms, or at all.
Failure to obtain adequate financing would adversely affect our ability to
operate as a going concern. If we raise additional funds by issuing equity
securities, dilution to stockholders may result and new investors could have
rights superior to holders of the shares issued in this offering. In addition,
debt financing, if available, may include restrictive covenants. If adequate
funds are not available to us, we may have to liquidate some or all of our
assets or to delay or reduce the scope of or eliminate some portion or all of
our development programs or clinical trials. We also may have to license to
other companies our product candidates or technologies that we would prefer to
develop and commercialize ourselves.
If
we do not achieve our projected development goals in the time frames we
estimate, the commercialization of our products may be delayed and our business
prospects may suffer. Our financial projections also may prove to be
materially inaccurate.
From time
to time, we estimate the timing of the accomplishment of various scientific,
clinical, regulatory and other product development goals, which we sometimes
refer to as milestones. These milestones may include the commencement or
completion of scientific studies and clinical trials and the submission of
regulatory filings. For example, we have stated in our most recent Annual Report
incorporated by reference in this Annual Report supplement the expected timing
of certain milestones relating to our INNO-206, bafetinib, tamibarotene and
arimoclomol clinical development programs.
16
We also
may disclose projected expenditures or other forecasts for future periods such
as the statements above in this Annual Report supplement regarding our current
projected expenditures for fiscal year 2010. These and other financial
projections are based on management’s current expectations and do not contain
any margin of error or cushion for any specific uncertainties, or for the
uncertainties inherent in all financial forecasting.
The
actual timing of milestones and actual expenditures or other financial results
can vary dramatically compared to our estimates, in some cases for reasons
beyond our control. If we do not meet milestones or financial projections as
announced from time to time, the development and commercialization of our
products may be delayed and our business prospects may suffer. The
assumptions management has used to produce these projections may significantly
change or prove to be inaccurate. Accordingly, you should not unduly rely on any
of these financial projections.
If
our products are not successfully developed and approved by the FDA, we may be
forced to reduce or curtail our operations.
All of
our product candidates in development must be approved by the U.S. Food and Drug
Administration, or FDA, or corresponding foreign governmental agencies before
they can be marketed. The process for obtaining FDA and foreign
government approvals is both time-consuming and costly, with no certainty of a
successful outcome. This process typically includes the conduct of extensive
pre-clinical and clinical testing, including post-approval testing, which may
take longer or cost more than we or our licensees, if any, anticipate, and may
prove unsuccessful due to numerous factors. Product candidates that may appear
to be promising at early stages of development may not successfully reach the
market for a number of reasons. The results of preclinical and
initial clinical testing of these product candidates may not necessarily be
predictive of the results that will be obtained from later or more extensive
testing. Companies in the pharmaceutical and biotechnology industries have
suffered significant setbacks in advanced clinical trials, even after obtaining
promising results in earlier trials.
Numerous
factors could affect the timing, cost or outcome of our product development
efforts, including the following:
|
|
·
|
difficulty
in securing centers to conduct
trials;
|
|
|
·
|
difficulty
in enrolling patients in conformity with required protocols or projected
timelines;
|
|
|
·
|
requirements
for clinical trial design imposed by the
FDA;
|
|
|
·
|
unexpected
adverse reactions by patients in
trials;
|
|
|
·
|
difficulty
in obtaining clinical supplies of the
product;
|
|
|
·
|
changes
in or our inability to comply with FDA or foreign governmental product
testing, manufacturing or marketing
requirements;
|
|
|
·
|
regulatory
inspections of clinical trials or manufacturing facilities, which may,
among other things, require us or our manufacturers or licensees to
undertake corrective action or suspend or terminate the affected clinical
trials if investigators find them not to be in compliance with applicable
regulatory requirements;
|
|
|
·
|
inability
to generate statistically significant data confirming the safety and
efficacy of the product being
tested;
|
|
|
·
|
modification
of the product during testing; and
|
|
|
·
|
reallocation
of our limited financial and other resources to other clinical
programs.
|
It is
possible that none of the product candidates we develop will obtain the
regulatory approvals necessary for us to begin selling them. The time
required to obtain FDA and foreign governmental approvals is unpredictable, but
often can take years following the commencement of clinical trials, depending
upon the complexity of the product candidate. Any analysis we perform on data
from clinical activities is subject to confirmation and interpretation by
regulatory authorities, which could delay, limit or prevent regulatory
approval.
17
Furthermore,
even if we obtain regulatory approvals, our products and the manufacturing
facilities used to produce them will be subject to continual review, including
periodic inspections and mandatory post- approval clinical trials by the FDA and
other U.S. and foreign regulatory authorities. Any delay or failure
in obtaining required approvals or to comply with post-approval regulatory
requirements could have a material adverse effect on our ability to generate
revenue from the particular product candidate. The failure to comply
with any post-approval regulatory requirements also could also result in the
rescission of the related regulatory approvals or the suspension of sales of the
offending product.
Our
current and planned clinical trials of our product candidates may fail to show
that these product candidates are clinically safe and effective, or that they
are better than alternative treatments.
INNO-206
was no more toxic than free doxorubicin in a Phase I clinical trial and showed
limited biological responses against certain tumors. However, these
conclusions may not be reproducible in larger clinical trials, including the
planned Phase 2 clinical trials of INNO-206 as a treatment for pancreatic
cancer, gastric cancer and soft tissue sarcomas.
Bafetinib
demonstrated clinical responses in patients with CML in a Phase I clinical trial
conducted in patients with CML and other leukemias that have a certain mutation
called the Philadelphia Chromosome (Ph+) and are intolerant of or resistant to
Gleevec and, in some cases, second-line tyrosine kinase
inhibitors. However, bafetinib has never been tested in human
clinical trials in patients with B-CLL or glioblastoma multiforme, and there are
no assurances that it will be effective in those indications.
Tamibarotene
has been shown to be safe, well tolerated, and efficacious in the Japanese
population. However, it is possible that the response to the drug may
be different in American or European populations. Furthermore, the
efficacy studies that led to approval in Japan occurred prior to the advent of
the use of arsenic trioxide, or ATO, for second line therapy. It is
possible that the current use of ATO could alter the safety or efficacy of
tamibarotene. The FDA might not accept the Japanese studies as a
database for safety in the US. The majority of patients treated with ATRA as a
first-line therapy way generally experience a complete remission of disease. As
a result of the limited population of patients requiring third-line treatment
for APL, there is no assurance that we will be successful in recruiting a
sufficient number of patients into our ongoing clinical trial of tamibarotene as
a third-line treatment for APL in order to demonstrate efficacy. In
part for that reason, we have announced plans to develop tamibarotene, in
combination with chemotherapy or arsenic trioxide (ATO), as a first-line
treatment for APL. There is no assurance, however, that the FDA will accept a
commercially reasonable strategy for first-line development with the FDA, and
any FDA-required changes to our clinical development strategy could delay or
increase the cost of the trial, adversely affect our ability to demonstrate the
efficacy of tamibarotene in the trial or cause us not to pursue clinical
development of tamibarotene for one or more of these
considerations.
Our Phase
IIa clinical trial and open-label extension clinical trial of arimoclomol for
the treatment of ALS indicated that arimoclomol was safe and well-tolerated in
patients, but the results of the open-label extension clinical trial indicated
only a non-statistically significant trend of improvement in the revised ALS
Functional Rating Scale, or ALSFRS-R, in the arimoclomol high-dose group as
compared with reports of previous studies of untreated patients. This trial did
not have a concurrent placebo control group, so we could draw no definitive
conclusions with respect to efficacy. Further development of arimoclomal for
ALS, as well as clinical development of iroxanadine for diabetic foot ulcers,
would require significant additional testing, and it is possible that the
favorable safety data we observed in earlier trials may not be reproduced in any
later trials.
In
December 2009, the FDA accepted our revised protocol for an ascending dose
clinical trial of arimoclomol for the treatment of ALS. Although we
are making preparations related to that clinical trial, while simultaneously
seeking potential strategic partners to advance development and evaluating the
potential for a spin-off transaction for our molecular chaperone assets, it is
possible that we will further amend the clinical trial protocol, or elect not to
proceed with further development of some or all of our molecular chaperone
assets, as a result of future business or market conditions, capital
constraints, an inability to secure a partnership or other factors. The planned
clinical trial includes the administration of arimoclomol at ascending doses,
but there are no assurances that arimoclomol will prove safe at higher
doses.
Later
trials also may not yield statistically significant data indicating that these
product candidates are clinically effective. Accordingly, we, or any development
partners, may ultimately be unable to provide the FDA with satisfactory data on
clinical safety and efficacy sufficient to obtain FDA approval of INNO-206,
tamibarotene, bafetinib, arimoclomol or iroxanadine for these
indications.
18
We
will rely upon third parties for the manufacture of our clinical product
supplies.
We do not
have the facilities or expertise to manufacture supplies of any of our product
candidates. Accordingly, we are dependent upon third-party manufacturers, or
potential future strategic alliance partners, to manufacture these supplies. We
have manufacturing supply arrangements in place with respect to a portion of the
clinical supplies needed for the clinical development programs for INNO-206,
tamibarotene and arimoclomol. However, we have no supply arrangements for the
commercial manufacture of these product candidates or any manufacturing supply
arrangements for any other potential product candidates, and we may not be able
to secure needed supply arrangements on attractive terms, or at all. Our failure
to secure these arrangements as needed could have a materially adverse effect on
our ability to complete the development of our products or to commercialize
them.
If our
product candidates cannot be manufactured in suitable quantities and in
accordance with regulatory standards, our clinical trials, regulatory approvals
and marketing efforts for such products may be delayed. Such delays could
adversely affect our competitive position and our chances of generating
significant recurring revenues. If our products cannot be manufactured at an
acceptable cost, the commercial success of our products may be adversely
affected.
We
may rely upon third parties in connection with the commercialization of our
products.
The
completion of the development of INNO-206, bafetinib and tamibarotene, and our
molecular chaperone product candidates, as well as the marketing of these
products, may require us to enter into strategic alliances, license agreements
or other collaborative arrangements with other pharmaceutical companies under
which those companies will be responsible for one or more aspects of the
commercial development and eventual marketing of our products.
Our
products may not have sufficient potential commercial value to enable us to
secure strategic arrangements with suitable companies on attractive terms, or at
all. If we are unable to enter into such arrangements, we may not have the
financial or other resources to complete the development of any of our products
and may have to sell our rights in them to a third party or abandon their
development altogether.
To the
extent we enter into collaborative arrangements, we will be dependent upon the
timeliness and effectiveness of the development and marketing efforts of our
contractual partners. If these companies do not allocate sufficient personnel
and resources to these efforts or encounter difficulties in complying with
applicable FDA and other regulatory requirements, we may not obtain regulatory
approvals as planned, if at all, and the timing of receipt or the amount of
revenue from these arrangements may be materially and adversely affected. By
entering into these arrangements rather than completing the development and then
marketing these products on our own, the profitability to us of these products
may decline.
We
may be unable to protect our intellectual property rights, which could adversely
affect our ability to compete effectively.
We
believe that obtaining and maintaining patent and other intellectual property
rights for our technologies and potential products is critical to establishing
and maintaining the value of our assets and our business. We will be able to
protect our technologies from unauthorized use by third parties only to the
extent that we have rights to valid and enforceable patents or other proprietary
rights that cover them. Although we own or have rights to patents and patent
applications directed to INNO-206, bafetinib and our molecular chaperone
amplification technologies, these patents and applications may not prevent third
parties from developing or commercializing similar or identical technologies. In
addition, our patents may be held to be invalid if challenged by third parties,
and our patent applications may not result in the issuance of
patents.
The
patent positions of pharmaceutical and biotechnology companies can be highly
uncertain and involve complex legal and factual questions for which important
legal principles remain unresolved. No consistent policy regarding the breadth
of claims allowed in biotechnology patents has emerged to date in the U.S. and
in many foreign countries. The application and enforcement of patent laws and
regulations in foreign countries is even more uncertain. Accordingly, we may not
be able to effectively file, protect or defend our proprietary rights on a
consistent basis. Many of the patents and patent applications on which we rely
were issued or filed by third parties prior to the time we acquired rights to
them. The validity, enforceability and ownership of those patents and patent
applications may be challenged, and if a court decides that our patents are not
valid, we will not have the right to stop others from using our inventions.
There is also the risk that, even if the validity of our patents is upheld, a
court may refuse to stop others on the ground that their activities do not
infringe our patents.
19
Any
litigation brought by us to protect our intellectual property rights could be
costly and have a material adverse effect on our operating results or financial
condition, make it more difficult for us to enter into strategic alliances with
third parties to develop our products, or discourage our existing licensees from
continuing their development work on our potential products. If our patent
coverage is insufficient to prevent third parties from developing or
commercializing similar or identical technologies, the value of our assets is
likely to be materially and adversely affected.
We also
rely on certain proprietary trade secrets and know-how, especially where we
believe patent protection is not appropriate or obtainable. However, trade
secrets and know-how are difficult to protect. Although we have taken measures
to protect our unpatented trade secrets and know-how, including the use of
confidentiality and invention assignment agreements with our employees,
consultants and some of our contractors, it is possible that these persons may
disclose our trade secrets or know-how or that our competitors may independently
develop or otherwise discover our trade secrets and know-how.
If
our product candidates infringe the rights of others, we could be subject to
expensive litigation or be required to obtain licenses from others to develop or
market them.
Our
competitors or others may have patent rights that they choose to assert against
us or our licensees, suppliers, customers or potential collaborators. Moreover,
we may not know about patents or patent applications that our products would
infringe. For example, because patent applications can take many years to issue,
there may be currently pending applications, unknown to us, that may later
result in issued patents that our arimoclomol, iroxanadine or other product
candidates would infringe. In addition, if third parties file patent
applications or obtain patents claiming technology also claimed by us in issued
patents or pending applications, we may have to participate in interference
proceedings in the US Patent and Trademark Office to determine priority of
invention. If third parties file oppositions in foreign countries, we may also
have to participate in opposition proceedings in foreign tribunals to defend the
patentability of our foreign patent applications.
If a
third party claims that we infringe its proprietary rights, any of the following
may occur:
|
|
·
|
we
may become involved in time-consuming and expensive litigation, even if
the claim is without merit;
|
|
|
·
|
we
may become liable for substantial damages for past infringement if a court
decides that our technology infringes a competitor’s
patent;
|
|
|
·
|
a
court may prohibit us from selling or licensing our product without a
license from the patent holder, which may not be available on commercially
acceptable terms, if at all, or which may require us to pay substantial
royalties or grant cross licenses to our patents;
and
|
|
|
·
|
we
may have to redesign our product candidates or technology so that it does
not infringe patent rights of others, which may not be possible or
commercially feasible.
|
If any of
these events occurs, our business and prospects will suffer and the market price
of our common stock will likely decline substantially.
Any
drugs we develop may become subject to unfavorable pricing regulations,
third-party reimbursement practices or healthcare reform initiatives, which
could have a material adverse effect on our business.
We intend
to sell our products primarily to hospitals which receive reimbursement for the
health care services they provide to their patients from third-party payors,
such as Medicare, Medicaid and other domestic and international government
programs, private insurance plans and managed care programs. Most third-party
payors may deny reimbursement if they determine that a medical product was not
used in accordance with cost-effective treatment methods, as determined by the
third-party payor, or was used for an unapproved indication. Third-party payors
also may refuse to reimburse for experimental procedures and devices.
Furthermore, because our programs are in the early stages of development, we are
unable at this time to determine their cost-effectiveness and the level or
method of reimbursement. Increasingly, the third-party payors who reimburse
patients are requiring that drug companies provide them with predetermined
discounts from list prices, and are challenging the prices charged for medical
products. If the price we are able to charge for any products we develop is
inadequate in light of our development and other costs, our profitability could
be adversely affected.
20
We
currently expect that any drugs we develop may need to be administered under the
supervision of a physician. Under currently applicable law, drugs that are not
usually self-administered may be eligible for coverage by the Medicare program
if:
|
|
·
|
they
are “incidental” to a physician’s
services,
|
|
|
·
|
they
are “reasonable and necessary” for the diagnosis or treatment of the
illness or injury for which they are administered according to accepted
standard of medical practice,
|
|
|
·
|
they
are not excluded as immunizations,
and
|
|
|
·
|
they
have been approved by the FDA.
|
We
are subject to intense competition, and we may not compete
successfully.
We and
our strategic partners or licensees may be unable to compete successfully
against our current or future competitors. The pharmaceutical, biopharmaceutical
and biotechnology industries are characterized by intense competition and rapid
and significant technological advancements. Many companies, research
institutions and universities are working in a number of areas similar to our
primary fields of interest to develop new products. There also is intense
competition among companies seeking to acquire products that already are being
marketed. Many of the companies with which we compete have or are likely to have
substantially greater research and product development capabilities and
financial, technical, scientific, manufacturing, marketing, distribution and
other resources than us and at least some of our present or future strategic
partners or licensees.
As a
result, these competitors may:
|
|
·
|
succeed
in developing competitive products sooner than us or our strategic
partners or licensees;
|
|
|
·
|
obtain
FDA or foreign governmental approvals for their products before we can
obtain approval of any of our
products;
|
|
|
·
|
obtain
patents that block or otherwise inhibit the development and
commercialization of our product candidate
candidates;
|
|
|
·
|
develop
products that are safer or more effective than our
products;
|
|
|
·
|
devote
greater resources than us to marketing or selling
products;
|
|
|
·
|
introduce
or adapt more quickly than us to new technologies and other scientific
advances;
|
|
|
·
|
introduce
products that render our products
obsolete;
|
|
|
·
|
withstand
price competition more successfully than us or our strategic partners or
licensees;
|
|
|
·
|
negotiate
third-party strategic alliances or licensing arrangements more effectively
than us; and
|
|
|
·
|
take
better advantage than us of other
opportunities.
|
Companies
that currently sell generic and proprietary compounds for the treatment of
cancer and related diseases include, but are not limited to, Abraxis BioScience,
Amgen, Sanofi-Aventis, Bayer, Bristol-Myers Squibb, Celgene, Cephalon,
Genentech, Eli Lilly, Johnson & Johnson and Novartis. Alternative
technologies are being developed to treat cancer and related diseases by
numerous companies including Bristol-Myers Squibb, Eisai, Merck and Genentech,
several of which are in advanced clinical trials. There also are FDA approved
cancer therapies that are in the late stage of development by larger established
companies for new cancer indications: Alimta (Eli Lilly), Avastin (Genentech),
Eloxatin (Sanofi-Aventis), Erbitux (Bristol-Myers Squibb and Imclone Systems)
and Tarceva (Genentech).
21
Soft
tissue sarcoma patients are typically treated with surgery followed by radiation
therapy. Doxorubicin is the only approved drug for treating soft
tissue sarcoma and is often used in combination with radiation. The
National Comprehensive Cancer Network also includes the use of ifosfamide,
epirubicin, Eli Lilly’s Gemzar, dacarbazine and liposomal doxorubicin marketed
in the US as Doxil by Johnson & Johnson. For patients ineligible
for surgery, radiation and/or chemotherapy is the only option. Other
approaches to treating soft tissue sarcoma are in late stage clinical
development. These include ridaforolimus being developed by Ariad
Pharmaceuticals and Merck & Co., GlaxoSmithKline’s pazopanib,
Sanofi-Aventis’ AVE8062, Threshhold Pharmaceuticals’s TH-302, and trabectedin
being co-developed by Johnson and Johnson and PharmaMar.
Advanced
gastric cancer patients are typically treated with a variety of combination of
approved drugs such as cisplatin, docetaxel, 5-FU, oxaliplatin, iriniotecan and
paclitaxel. Epirubicin, an anthracycline, is part of several gastric
cancer treatment regimens. We believe INNO-206 could be a potentially
more effective anthracycline and potentially more effective in this
setting.
Pancreatic
cancer patients are typically treated with surgery, radiation and
chemotherapy. Eli Lilly’s Gemzar is currently approved for the first
line treatment of locally advanced or metastatic pancreatic
cancer. It is also indicated for the use in patients who have
received prior treatment with 5-FU. OSI Pharmaceuticals’ Tarceva was
approved in 2005 for the use in combination with Gemzar. The NCCN
believes the best management for these patients is in a clinical
trial. Because of the tremendous unmet need for these patients, many
companies are developing new drugs to treat pancreatic cancer. Late
stage drugs in clinical trials include Abraxane by Abraxis BioScience, AGS-1C4D4
by Astellas Pharma Inc., TNFerade™ by GenVec, and S-1 by
Sanofi-Aventis.
To our
knowledge, there are no competitors in clinical development for refractory
APL. Currently, treatment of APL is based on induction and maintenance
therapy with ATRA and chemotherapy (typically idarubicin). ATRA and
idarubicin are both generic compounds. Arsenic trioxide, currently
marketed by Cephalon, is approved for use in patients who have relapsed after
ATRA-based therapy in APL. There are no FDA-approved therapies for
patients who have failed arsenic trioxide. In practice, it appears that
patients who fail arsenic trioxide are retreated with ATRA or receive Mylotarg,
which is marketed by Pfizer Inc.
There are
currently three marketed competitors to bafetinib (formerly INNO-406) in the CML
market, Gleevec®, Sprycel® and Tasigna®. Gleevec is approved for treatment
of newly diagnosed adult patients with Philadelphia chromosome–positive chronic
myeloid leukemia (Ph+ CML) in the chronic phase and patients with Ph+
CML in blast crisis (BC), accelerated phase (AP), or in the chronic phase (CP)
after failure of interferon-alpha therapy. Sprycel® and Tasigna® are approved
for Gleevec-resistant CML. Because of the highly competitive nature
of the CML market including drug candidates in development, CytRx plans to
develop bafetinib initially in cancers other than CML. We
have selected B-cell chronic lymphocytic leukemia (B-CLL) and glioblastoma
multiforme due to the potent and specific inhibitory properties of bafetinib
against Lyn kinase. Lyn kinase is a member of the Src family of kinases which
are known to be involved in cell growth. Lyn kinase is overexpressed in both
B-CLL and glioblastoma multiforme (GBM).
There are
several drugs approved for the treatment of CLL. First-line therapy
for CLL includes a variety of combination therapies including fludarabine,
cyclophosphamide, Rituxan® and Campath®. Treatment
for relapsed or refractory CLL includes several chemotherapy regimens
including CHOP, CFAR, hyperCFAD and OFAR in addition to single agents including
GlaxoSmithKline’s ArzerraTM. Arzerra
was approved in October 2009 for CLL patients who are refractory to treatment
with fludarabine and Campath.
Current
therapy for glioblastoma multiforme, the most common form of brain cancer, is
surgery followed by radiation therapy and chemotherapy. Merck’s
Temodar® is approved for treating newly diagnosed GBM concomitantly with
radiotherapy and then as a maintenance treatment. Roche’s Avastin was
approved in May 2009 for treatment of recurrent GBM. We believe that
bafetinib’s ability to selectively inhibit Lyn kinase and to penetrate the brain
in an animal model of cancer will be an effective treatment for second-line
therapy in GBM.
We are
aware of only one drug, rilutek, developed by Aventis Pharma AG, that has been
approved by the FDA for the treatment of ALS. Many companies are working to
develop pharmaceuticals to treat ALS, including Aeolus Pharmaceuticals,
Mitsubishi Tanabe Pharma Corporation, Ono Pharmaceuticals, Trophos SA, Knopp
Neurosciences Inc., Faust Pharmaceuticals SA, Oxford BioMedica plc, Phytopharm
plc and Teva Pharmaceutical Industries Ltd., as well as RXi. ALS patients often
take over-the-counter supplements, including vitamin E, creatine and coenzyme
Q10, or drugs such as lithium that are approved for other indications. ALS
belongs to a family of neurodegenerative diseases that includes Alzheimer’s,
Parkinson’s and Huntington’s diseases. Due to similarities between these
diseases, a new treatment for one such disease potentially could be useful for
treating others. There are many companies producing and developing drugs used to
treat neurodegenerative diseases other than ALS, including Amgen, Inc., Biogen
Idec, Boehringer Ingelheim, Cephalon, Inc., Ceregene, Inc., Elan
Pharmaceuticals, plc, Forest Laboratories, Inc., H. Lundbeck A/S, Phytopharm
plc, UCB Group and Wyeth.
22
Any of
these competing therapies could prove to be more effective than INNO-206,
bafetinib, tamibarotene, or any future therapy of ours. Most of our
competitors have substantially greater research and product development
capabilities and financial, technical, scientific, manufacturing, marketing,
distribution and other resources than us.
We
will be required to pay substantial milestone and other payments relating to the
commercialization of our products.
The
agreement relating to our worldwide rights to INNO-206 provides for our payment
of an aggregate of $7.5 million upon meeting specified clinical and regulatory
milestones up to and including the product’s second final marketing
approval. We also will be obliged to pay:
|
|
·
|
commercially
reasonable royalties based on a percentage of net sales (as defined in the
agreement);
|
|
|
·
|
a
percentage of non-royalty sub-licensing income (as defined in the
agreement); and
|
|
|
·
|
milestones
of $1,000,000 for each additional final marketing approval that we might
obtain.
|
Our
agreement relating to our worldwide (except Japan) rights to bafetinib provides
for our payment of an aggregate of $13.35 million (including $5 million upon the
product’s initial final marketing approval) upon the achievement of specified
clinical and regulatory milestones up to and including approvals in the U.S. and
Europe. We also will be obliged to pay:
|
|
·
|
commercially
reasonable royalties based on a percentage of net sales (as defined in the
agreement), dependent on reaching certain revenue
thresholds;
|
|
|
·
|
annual
minimum payments if sales of bafetinib do not meet specified levels;
and
|
|
|
·
|
a
percentage of non-royalty sub-licensing income (as defined in the
agreement).
|
The
agreement under which we have North American rights to tamibarotene provides for
our payment of royalties based on net sales of any products, as well as
aggregate payments of $4.4 million upon meeting specified clinical, regulatory
and sales milestones up to and including the first commercial sale of
tamibarotene for the treatment of APL.
If we are
required to pay any third party in order to exercise our rights under the
agreement, we will deduct a percentage of those payments from the royalties due
under the agreement, up to an agreed-upon cap.
Under the
merger agreement by which we acquired Innovive, we agreed to pay the former
Innovive stockholders a total of up to approximately $18.3 million of future
earnout merger consideration, subject to our achievement of specified net sales
under the Innovive license agreements. The earnout merger
consideration, if any, will be payable in shares of our common stock, subject to
specified conditions, or, at our election, in cash or by a combination of shares
of our common stock and cash. Our common stock will be valued for
purposes of any future earnout merger consideration based upon the trading price
of our common stock at the time the earnout merger consideration is
paid.
Our
agreement by which we acquired rights to arimoclomol and our other molecular
chaperone amplification product candidates provides for milestone payments by us
upon the occurrence of specified regulatory filings and approvals related to the
acquired products. In the event that we successfully develop arimoclomol or any
of these other product candidates, these milestone payments could aggregate as
much as $3.7 million, with the most significant payments due upon the first
commercialization of any of these products. In addition, our agreement with the
ALS CRT requires us to pay a one-percent royalty interest on worldwide sales of
arimoclomol for the treatment of ALS. Also, any future license, collaborative or
other agreements we may enter into in connection with our development and
commercialization activities may require us to pay significant milestone,
license and other payments in the future.
We
are subject to potential liabilities from clinical testing and future product
liability claims.
If any of
our products are alleged to be defective, they may expose us to claims for
personal injury by patients in clinical trials of our products or, if we obtain
marketing approval and commercialize our products, by patients using our
commercially marketed products. Even if the if one or more of our products is
approved by the FDA, users may claim that such products caused unintended
adverse
effects. We maintain clinical trial insurance for our clinical trial of
tamibarotene for APL, and we plan to seek to obtain similar insurance for any
other clinical trials that we conduct. We also would seek to obtain product
liability insurance covering the commercial marketing of our product candidates.
We may not be able to obtain additional insurance, however, and any insurance
obtained by us may prove inadequate in the event of a claim against us. Any
claims asserted against us also may divert management’s attention from our
operations, and we may have to incur substantial costs to defend such claims
even if they are unsuccessful.
23
We
may be unable to successfully acquire additional technologies or products. If we
require additional technologies or products, our product development plans may
change and the ownership interests of our shareholders, or our ownership
interest in RXi, could be diluted.
We may
seek to acquire additional technologies by licensing or purchasing such
technologies, or through a merger or acquisition of one or more companies that
own such technologies. We have no current understanding or agreement to acquire
any technologies, however, and we may not be able to identify or successfully
acquire any additional technologies. We also may seek to acquire products from
third parties that already are being marketed or have been approved for
marketing, although we have not currently identified any of these products. We
do not have any prior experience in acquiring or marketing products approved for
marketing and may need to find third parties to market any products that we
might acquire.
Following
our acquisition of Innovive in September 2008, we refocused our product
development efforts on our oncology drug candidates, which we believe has the
greatest near-term revenue potential. If we acquire additional
technologies or product candidates, we may determine to make further changes to
our product development plans and business strategy to capitalize on
opportunities presented by the new technologies and product
candidates.
We may
determine to issue shares of our common stock, or to use shares of RXi common
stock owned by us, or both, to acquire additional technologies or products or in
connection with a merger or acquisition of another company. To the extent we do
so, the ownership interest of our stockholders, or our ownership interest in
RXi, or both, will be diluted accordingly.
Risks
Associated With Our Investment in RXi
We
may sell or dispose of some of our RXi shares, and may not be able to do so on
attractive terms.
As of
December 31, 2009, we held 5,768,881 shares of common stock of RXi, or
approximately 36% of the outstanding shares of RXi common stock. RXi shares are
listed on The NASDAQ Capital Market under the symbol “RXi.” During the 12-month
period ended December 31, 2009, the market prices for RXi shares as reported on
The NASDAQ Capital Market fluctuated from a high of $7.57 per share to a low of
$1.51 per share, and the market price of RXi shares and the value of our RXi
shares may continue to experience significant volatility.
We intend
to look for favorable opportunities to sell or otherwise dispose of our RXi
shares in one or more transactions in order to obtain funds to carry on our
operations or in connection with our acquisition of new technologies or
products. There is no assurance, however, whether, or on what terms,
we might be able to sell or dispose of our RXi shares. In addition,
any sales or other disposition of RXi shares by us, or the possibility of such
sales or disposition, could adversely affect the market price of our remaining
RXi shares.
If
RXi undertakes future financings, our ownership interest in RXi may be
diluted.
Under our
agreement with RXi, with some exceptions, we will have preemptive rights to
acquire a portion of any new securities sold or issued by RXi so as to maintain
our percentage ownership of RXi. Depending upon the terms and provisions of any
proposed sale of new securities by RXi, our financial condition and other
factors, we may be unwilling or unable to exercise our preemptive rights. We
agreed to waive our preemptive rights in connection with a private placement by
RXi in June 2008, which resulted in a reduction in our percentage ownership of
RXi from approximately 49% to approximately 45%. In September, 2009,
we sold 500,000 shares of RXi which reduced our percentage ownership further to
approximately 36%. If RXi undertakes future issuances of equity securities, our
percentage ownership interest in RXi may be diluted further.
24
We
do not control RXi, and the officers, directors and other RXi stockholders may
have interests that are different from ours.
Although
we currently own a significant portion of RXi’s outstanding common stock, we do
not control its management or operations. RXi has its own board of directors and
management, who are responsible for the affairs and policies of RXi and its
development plans. We have entered into letter agreements with RXi and certain
of its stockholders under which we agree to vote our shares of RXi common stock
for the election of directors of RXi and to take other actions to ensure that a
majority of RXi’s board of directors are independent of us. The board of
directors and other stockholders of RXi may have interests that are different
from ours, and RXi may engage in actions in connection with its business and
operations that we believe are not in our best interests.
Risks
Associated with Our Common Stock
Our
anti-takeover measures may make it more difficult to change our management, or
may discourage others from acquiring us, and thereby adversely affect
stockholder value.
We have a
stockholder rights plan and provisions in our bylaws that are intended to
protect our stockholders’ interests by encouraging anyone seeking control of our
company to negotiate with our board of directors. These provisions may
discourage or prevent a person or group from acquiring us without the approval
of our board of directors, even if the acquisition would be beneficial to our
stockholders.
We have a
classified board of directors, which means that at least two stockholder
meetings, instead of one, will be required to effect a change in the majority
control of our board of directors. This applies to every election of directors,
not just an election occurring after a change in control. The classification of
our board increases the amount of time it takes to change majority control of
our board of directors and may cause potential acquirers to lose interest in a
potential purchase of us, regardless of whether our purchase would be beneficial
to us or our stockholders. The additional time and cost to change a majority of
the members of our board of directors makes it more difficult and may discourage
our existing stockholders from seeking to change our existing management in
order to change the strategic direction or operational performance of our
company.
Our
bylaws provide that directors may only be removed for cause by the affirmative
vote of the holders of at least a majority of the outstanding shares of our
capital stock then entitled to vote at an election of directors. This provision
prevents stockholders from removing any incumbent director without cause. Our
bylaws also provide that a stockholder must give us at least 120 days notice of
a proposal or director nomination that such stockholder desires to present at
any annual meeting or special meeting of stockholders. Such provision prevents a
stockholder from making a proposal or director nomination at a stockholder
meeting without us having advance notice of that proposal or director
nomination. This could make a change in control more difficult by providing our
directors with more time to prepare an opposition to a proposed change in
control. By making it more difficult to remove or install new directors, these
bylaw provisions may also make our existing management less responsive to the
views of our stockholders with respect to our operations and other issues such
as management selection and management compensation.
We are
subject to the anti-takeover provisions of Section 203 of the Delaware General
Corporation Law, which may also prevent or delay a takeover of us that may be
beneficial to you.
Our
outstanding options and warrants and the availability for resale of our shares
issued in our private financings may adversely affect the trading price of our
common stock.
As of
December 31, 2009, there were outstanding stock options and warrants to purchase
approximately 24.4 million shares of our common stock at a weighted-average
exercise price of $1.42 per share. Our outstanding options and warrants could
adversely affect our ability to obtain future financing or engage in certain
mergers or other transactions, since the holders of options and warrants can be
expected to exercise them at a time when we may be able to obtain additional
capital through a new offering of securities on terms more favorable to us than
the terms of outstanding options and warrants. For the life of the options and
warrants, the holders have the opportunity to profit from a rise in the market
price of our common stock without assuming the risk of ownership. The issuance
of shares upon the exercise of outstanding options and warrants will also dilute
the ownership interests of our existing stockholders. Many of our outstanding
warrants contain anti-dilution provisions pertaining to dividends with respect
to our common stock. In the event that these anti-dilution provisions are
triggered by us in the future, we would likewise be required to reduce the
exercise price, and increase the number of shares underlying, those warrants,
which would have a dilutive effect on our stockholders.
25
We have
registered with the SEC the resale by the holders of all or substantially all
shares of our common stock issuable upon exercise of our outstanding options and
warrants. The availability of these shares for public resale, as well as actual
resales of these shares, could adversely affect the trading price of our common
stock.
We
may issue preferred stock in the future, and the terms of the preferred stock
may reduce the value of our common stock.
We are
authorized to issue shares of preferred stock in one or more series. Our board
of directors may determine the terms of future preferred stock offerings without
further action by our stockholders. If we issue preferred stock, it could affect
your rights or reduce the value of our outstanding common stock. In particular,
specific rights granted to future holders of preferred stock may include voting
rights, preferences as to dividends and liquidation, conversion and redemption
rights, sinking fund provisions, and restrictions on our ability to merge with
or sell our assets to a third party.
We
may experience volatility in our stock price, which may adversely affect the
trading price of our common stock.
The
market price of our common stock has ranged from $0.23 to $2.98 per share since
January 1, 2008, and it may continue to experience significant volatility from
time to time. Our ability to raise capital has been materially and adversely
affected by the continuing poor economy. Despite the recovery in the
U.S. financial markets in 2009, the market remains depressed for private
investment in public equity, or PIPEs, transactions on which we have relied for
raising needed capital.
Other
factors that may affect the market price of our common stock include the
following:
|
|
·
|
announcements
of regulatory developments or technological innovations by us or our
competitors;
|
|
|
·
|
changes
in our relationship with our licensors and other strategic
partners;
|
|
|
·
|
changes
in our ownership of or other relationships with
RXi;
|
|
|
·
|
our
quarterly operating results;
|
|
|
·
|
litigation
involving or affecting us;
|
|
|
·
|
shortfalls
in our actual financial results compared to our guidance or the forecasts
of stock market analysts;
|
|
|
·
|
developments
in patent or other technology ownership
rights;
|
|
|
·
|
acquisitions
or strategic alliances by us or our
competitors;
|
|
|
·
|
public
concern regarding the safety of our products;
and
|
|
|
·
|
government
regulation of drug pricing.
|
We
do not expect to pay any cash dividends on our common stock.
We have
not declared or paid any cash dividends on our common stock or other securities,
and we currently do not anticipate paying any cash dividends in the foreseeable
future. Because we do not anticipate paying cash dividends for the foreseeable
future, our stockholders will not realize a return on their investment in our
common stock except to the extent of any appreciation in the value of our common
stock. Our common stock may not appreciate in value, or may decline in
value.
26
Item
2. PROPERTIES
We lease
our headquarters in Los Angeles, California. The lease covers approximately
5,700 square feet of office space and expires in March 2015. This lease requires
us to make monthly payments of approximately $23,243, subject to annual
increases. We also lease approximately 10,000 square feet of office and
laboratory space in San Diego, California, for $35,270 per month. The lease
expires in October 2010. In May 2009, we substantially completed the initial
phase of the research and development activities performed at the San Diego
facility, and in November 2009, we signed sublease agreements with two parties
to sublet the facility for the remainder of the term of the
lease. Under those subleases, we are entitled to aggregate annual
rent of approximately $16,900 per month.
We also
acquired a sublease to approximately 5,526 square feet of office space at 555
Madison Avenue, New York, New York, in connection with our acquisition of
Innovive in September 2008. This lease currently requires us to make
annual payments of approximately $210,000, plus certain taxes and operating
expenses, and it expires on August 30, 2012. On December 4, 2008, we
sub-subleased the space through August 29, 2012. Under the sub-sublease, we
are entitled to base annual rent of approximately $350,000, plus certain taxes
and operating expenses.
Item
3. LEGAL PROCEEDINGS
We are
occasionally involved in claims arising in the normal course of business. As of
March 12, 2010, there were no such claims that we expect, individually or in the
aggregate, to have a material adverse affect on us.
27
PART
II
Item
5. MARKET
FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER
PURCHASES OF EQUITY SECURITIES
Market
Information
Our
common stock is traded on The NASDAQ Capital Market under the symbol “CYTR.” The
following table sets forth the high and low sale prices for our common stock for
the periods indicated as reported by The NASDAQ Capital Market:
|
|
High
|
Low
|
|
Fiscal
Year 2009:
|
||
|
Fourth
Quarter
|
$ 1.29
|
$ 0.71
|
|
Third
Quarter
|
$ 1.72
|
$ 0.84
|
|
Second
Quarter
|
$ 1.09
|
$ 0.34
|
|
First
Quarter
|
$ 0.45
|
$ 0.24
|
|
Fiscal
Year 2008:
|
||
|
Fourth
Quarter
|
$ 0.65
|
$ 0.23
|
|
Third
Quarter
|
$ 0.68
|
$ 0.40
|
|
Second
Quarter
|
$ 1.27
|
$ 0.61
|
|
First
Quarter
|
$ 2.98
|
$ 1.00
|
Holders
On March
12, 2010, there were approximately 750 holders of record of our common stock.
The number of record holders does not reflect the number of beneficial owners of
our common stock for whom shares are held by brokerage firms and other
nominees.
Dividends
We have
not paid any cash dividends since our inception and do not contemplate paying
any cash dividends in the foreseeable future. On March 11, 2008, we
distributed to holders of our common stock approximately 36% of the outstanding
shares of RXi on an approximate 1-for-20 basis.
Equity
Compensation Plans
The
following table sets forth certain information as of December 31, 2009,
regarding securities authorized for issuance under our equity compensation
plans:
|
Plan
Category
|
(a)
Number
of Securities
to
be Issued Upon
Exercise
of
Outstanding
Options,
Warrants and Rights
|
(b)
Weighted-Average
Exercise
Price of
Outstanding
Options,
Warrants and Rights
|
Number
of Securities
Remaining
Available
for
Issuance Under
Equity
Compensation
Plans
(Excluding
Securities
Reflected
in Column (a))
|
|||||||||
|
Equity
compensation plans approved by our security holders:
|
||||||||||||
|
2000
Long-Term Incentive Plan
|
7,907,090 | $ | 1.17 | 155,000 | ||||||||
|
2008
Stock Incentive Plan
|
1,100,000 | 0.83 | 8,900,000 | |||||||||
|
Equity
compensation plans not approved by our security holders:
|
||||||||||||
|
Outstanding
warrants (1)
|
15,418,178 | 1.57 | — | |||||||||
|
Total
|
24,425,268 | $ | 1.58 | 9,055,000 | ||||||||
____________
|
(1)
|
The
warrants shown were issued in discreet transactions from time to time as
compensation for services rendered by consultants, advisors or other third
parties, and do not include warrants sold in private placement
transactions. The material terms of such warrants were determined based
upon arm’s-length negotiations with the service providers. The warrant
exercise prices approximated the market price of our common stock at or
about the date of grant, and the warrant terms range from one to ten years
from the grant date. The warrants contain customary anti-dilution
adjustments in the event of a stock split, reverse stock split,
reclassification or combination of our outstanding common stock and
similar events and certain of the warrants contain anti-dilution
adjustments triggered by other corporate events, such as dividends and
sales of equity below market
price.
|
28
Comparison
of Cumulative Total Returns
The
following line graph presentation compares cumulative total stockholder returns
of CytRx with The NASDAQ Stock Market Index and the NASDAQ Pharmaceutical Index
(the “Peer Index”) for the five-year period from December 31, 2004 to December
31, 2009. The graph and table assume that $100 was invested in each of CytRx’s
common stock, the NASDAQ Stock Market Index and the Peer Index on December 31,
2004, and that all dividends were reinvested. This data was furnished by Zacks
Investment Research.
Comparison
of Cumulative Total Returns
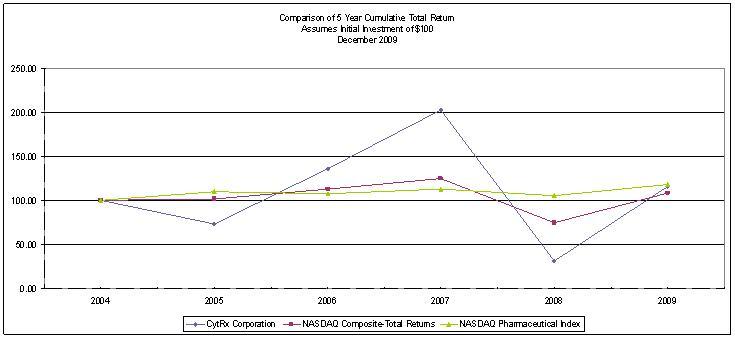
|
December 31,
|
|||||
|
2005
|
2006
|
2007
|
2008
|
2009
|
|
|
CytRx
Corporation
|
73.57
|
136.42
|
202.84
|
31.11
|
116.16
|
|
NASDAQ
Stock Market Index
|
102.12
|
112.73
|
124.73
|
74.87
|
108.83
|
|
NASDAQ
Pharmaceutical Index
|
110.13
|
107.79
|
113.36
|
105.46
|
118.52
|
Recent
Issuances of Unregistered Securities
During
the three-month period ended December 31, 2009, we issued 50,000 shares of our
common stock, and warrants to purchase a total of 800,000 shares of our common
stock at exercise prices ranging from $1.10 to $4.00 per share, in connection
with a consulting arrangement. The issuance of stock and warrants was
exempt from registration under the Securities Act of 1933 pursuant to
Section 4(2) of the Securities Act of 1933 and Regulation D under the
Act.
Repurchase
of Shares
We did
not repurchase any of our shares during the three-month period ended December
31, 2009.
29
Item
6. SELECTED FINANCIAL DATA
General
The
following selected financial data are derived from our audited financial
statements. Our financial statements for 2009, 2008 and 2007 have been audited
by BDO Seidman, LLP, our independent registered public accounting firm. These
historical results do not necessarily indicate future results. When you read
this data, it is important that you also read our financial statements and
related notes, as well as the “Management’s Discussion and Analysis of Financial
Condition and Results of Operations” and “Risk Factors” sections of this Annual
Report. Financial information provided below has been rounded to the nearest
thousand.
|
2009
|
2008
|
2007
|
2006
|
2005
|
||||||||||||||||
|
Statement
of Operations Data:
|
||||||||||||||||||||
|
Revenues
|
||||||||||||||||||||
|
Service
revenue
|
$ | 9,400,000 | $ | 6,166,000 | $ | 7,242,000 | $ | 1,859,000 | $ | 83,000 | ||||||||||
|
Licensing revenue
|
100,000 | 100,000 | 101,000 | 101,000 | 101,000 | |||||||||||||||
|
Grant
revenue
|
— | — | 116,000 | 106,000 | — | |||||||||||||||
|
Total
revenues
|
$ | 9,500,000 | $ | 6,266,000 | $ | 7,459,000 | $ | 2,066,000 | $ | 184,000 | ||||||||||
|
Deemed
dividend for anti-dilution adjustments made to outstanding common stock
warrants
|
— | (757,000 | ) | — | (488,000 | ) | (1,076,000 | ) | ||||||||||||
|
Net
loss applicable to common stockholders
|
$ | (4,800,000 | ) | $ | (27,803,000 | ) | $ | (21,890,000 | ) | $ | (17,240,000 | ) | $ | (16,169,000 | ) | |||||
|
Basic
and diluted loss per share applicable to common stock
|
$ | (0.05 | ) | $ | (0.30 | ) | $ | (0.26 | ) | $ | (0.25 | ) | $ | (0.28 | ) | |||||
|
Balance
Sheet Data:
|
||||||||||||||||||||
|
Cash,
cash equivalents and marketable securities
|
$ | 9,894,000 | $ | 25,042,000 | $ | 60,450,000 | $ | 30,381,000 | $ | 8,299,000 | ||||||||||
|
Total
assets
|
$ | 35,277,000 | $ | 28,324,000 | $ | 64,146,000 | $ | 31,636,000 | $ | 9,939,000 | ||||||||||
|
Total
stockholders’ equity
|
$ | 28,348,000 | $ | 15,698,000 | $ | 40,224,000 | $ | 5,150,000 | $ | 7,208,000 | ||||||||||
Factors Affecting Comparability
On
September 19, 2008, we purchased all of the common stock of Innovive
Pharmaceuticals in a transaction that for accounting purposes is considered an
asset acquisition. The fair value of Innovive’s assets and
liabilities at September 19, 2008, in millions of dollars, are presented
below:
|
In-process
research and development
|
$ | 8.0 | ||
|
Leasehold
interests
|
.1 | |||
|
Prepaid
expenses
|
.3 | |||
|
Accounts
payable
|
(6.1 | ) | ||
|
Net
assets acquired through issuance of common stock
|
$ | 2.3 | ||
As a
result of the March 11, 2008 distribution by us to our stockholders of
approximately 36% of the outstanding shares of RXi, we deconsolidated that
previously majority-owned subsidiary. As part of the transaction, we
deconsolidated $3.7 million of total assets and $4.6 million of total
liabilities of RXi.
In
connection with applicable antidilution adjustments to the price of certain
outstanding warrants in March 2008, we recorded a deemed dividend of
approximately $757,000. The deemed dividend was recorded as a charge to
accumulated deficit and a corresponding credit to additional paid-in
capital.
In July
2009, we completed a $20.0 million registered direct public offering of
approximately 15.3 million shares of our common stock at a price of $1.31 per
share and warrants to purchase an additional approximately 4.7 million shares of
common stock at an exercise price of $1.70 per share. Net of investment banking
commissions, advisory fees, legal, accounting and other fees related to the
transaction, we received proceeds of approximately $18.3 million (without giving
effect to any proceeds that we may receive upon future exercises of the warrants
sold in the offering).
30
In April
2007, we completed a $37.0 million private equity financing in which we issued
8.6 million shares of our common stock at $4.30 per share. Net of investment
banking commissions, legal, accounting and other expenses related to the
transaction, we received approximately $34.2 million of proceeds.
In August
2006, we received approximately $24.5 million in marketable securities (which
were sold by us for approximately $24.3 million) from the privately-funded ALS
Charitable Remainder Trust, or ALSCRT, in exchange for our commitment to
continue research and development of arimoclomol and other potential treatments
for ALS and a one percent royalty from worldwide sales of arimoclomol. We
recorded the value received under the arrangement as deferred service revenue,
which. we recognize using the proportional performance method of revenue
recognition. In August 2009, we were released from all restrictions
on the use of any As a result, we recognized in the third quarter $6.7 million
of service revenue, representing all of the remaining deferred revenue and
previously un-recognized portion of the value received in the arrangement with
ALSCRT. During 2009 and 2008, we recognized approximately $9.4
million and $6.2 million, respectively, of service revenue related to this
transaction, respectively.
Our
Statements of Operations as of and for the years ended December 31, 2009, 2008,
2007 and 2006 reflect the impact of ASC 718 (previously SFAS No. 123(R), Share-Based Payment (“SFAS
123(R)”)). In accordance with the modified prospective transition method, our
results of operations for prior periods have not been restated to reflect the
impact of ASC 718. Share-based compensation expense recognized under ASC
718 for the years ended December 31, 2009, 2008, 2007 and 2006 were $2.6
million, $2.1 million, $2.7 million and $1.2 million, respectively. As of
December 31, 2009, there was $2.5 million of unrecognized compensation cost
related to outstanding options granted to employees that is expected to be
recognized as a component of our operating expenses through 2011. Compensation
costs will be adjusted for future changes in estimated forfeitures.
Item
7. MANAGEMENT’S
DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF
OPERATIONS
The
following discussion and analysis of our financial condition and results of
operations should be read together with the discussion under “Selected Financial
Data” and our consolidated financial statements included in this Annual Report.
This discussion contains forward-looking statements, based on current
expectations and related to future events and our future financial performance,
that involve risks and uncertainties. Our actual results may differ materially
from those anticipated in these forward-looking statements as a result of many
important factors, including those set forth under the caption “Risk Factors”
and elsewhere in this Annual Report.
Overview
CytRx
Corporation
We are a
biopharmaceutical research and development company engaged in the development of
high-value human therapeutics, specializing in oncology. Our drug development
pipeline includes clinical development of three product candidates for cancer
indications, including three planned Phase 2 clinical trials for INNO-206 as a
treatment for pancreatic cancer, gastric (stomach) cancer and soft tissue
sarcomas, two Phase 2 proof-of-concept clinical trials with bafetinib in
patients with high-risk B-cell chronic lymphocytic leukemia, or B-CLL, and
patients with glioblastoma multiforme, and a registration study of tamibarotene
for the treatment of acute promyelocytic leukemia, or APL. In addition to our
core oncology programs, we are developing two drug candidates based on our
molecular chaperone regulation technology, which aim to repair or degrade
mis-folded proteins associated with disease. Apart from our drug development
programs, we currently maintain a 36% equity interest in our former subsidiary,
RXi.
In order
to fund our business and operations, we have relied primarily upon sales of our
equity securities, including proceeds received upon the exercise of options and
warrants, and sales of our shares of RXi common stock. We also have
received limited payments from our strategic partners and
licensees.
31
At
December 31, 2009, we had cash and cash equivalents of approximately $9.9
million, marketable securities of $22.8 million and held 5,768,881 shares of
restricted common stock of RXi with a market value of $26.4 million based upon
the closing price of the RXi common stock on that date. On July 27, 2009, we
raised approximately $18.3 million, net of fees and expenses, in a registered
direct offering, and on September 23, 2009, we raised approximately $1.2
million, net of fees, from the sale of 500,000 shares of our common stock
of RXi. Management believes that our current cash on hand, together with our
marketable securities and proceeds from possible future sales of RXi common
stock, will be sufficient to fund our operations for the foreseeable
future. The estimate is based, in part, upon our currently projected
expenditures for 2010 of approximately $18.0 million (unaudited), which includes approximately
$3.2 million (unaudited) for our clinical programs for INNO-206, approximately
$1.6 million (unaudited) for our clinical programs for bafetinib, approximately
$2.5 million (unaudited) for our clinical program for tamibarotene,
approximately $1.4 million (unaudited) for our activities for arimoclomol,
approximately $2.2 million (unaudited) for general operation of its clinical
programs, and approximately $7.1 million (unaudited) for other general and
administrative expenses. These projected expenditures are also based upon
numerous other assumptions and subject to many uncertainties, and actual
expenditures may be significantly different from these
projections. We will be required to obtain additional funding in
order to execute our long-term business plans, although we do not currently have
commitments from any third parties to provide us with capital. The fair value of
common stock investment in RXi is subject to market fluctuations that could
impact the amount of cash we generate from the sale of RXi shares in the future.
We cannot assure that additional funding will be available on favorable terms,
or at all. If we fail to obtain additional funding when needed, we may not be
able to execute our business plans and our business may suffer, which would have
a material adverse effect on our financial position, results of operations and
cash flows.
Our
Separation from RXi Pharmaceuticals Corporation
RXi
Pharmaceuticals Corporation was founded in April 2006 by us and four researchers
in the field of RNAi, including Dr. Craig Mello, recipient of the 2006 Nobel
Prize for Medicine for his co-discovery of RNAi. RNAi is a naturally occurring
mechanism for the regulation of gene expression that has the potential to
selectively inhibit the activity of any human gene. In January 2007, we
transferred to RXi substantially all of our RNAi-related technologies and
assets, and RXi began operating on a stand-alone basis for the purpose of
accelerating the discovery of RNAi therapeutics previously sponsored by us.
RXi’s initial focus is on developing RNAi-based product candidates for treating
neurological and metabolic disorders and cancer.
Until
early 2008, we owned approximately 85% of the outstanding shares of common stock
of RXi and our financial statements, including our financial statements as of
and for the year ended December 31, 2007, included the consolidated financial
condition and results of operations of RXi. On February 14, 2008, our board of
directors declared a dividend of one share of RXi common stock for each
approximately 20.05 shares of our common stock held by such stockholders, which
was paid on March 11, 2008 and which reduced our ownership of RXi shares to
less than 50%.
For
periods beginning with the first quarter of 2008, we began to account for our
investment in RXi using the equity method, under which we record only our
pro-rata share of the financial results of RXi as “equity in loss of
unconsolidated subsidiary” on our statements of operations. Because a portion of
RXi’s financial results for 2008 and all of RXi’s financial results for 2007
were not recorded by us under the equity method, our results of operations for
the year ended December 31, 2009 are not directly comparable to results of
operations for the same periods in prior years.
Research
and Development
Expenditures
for research and development activities related to continuing operations were
$7.5 million, $10.5 million and $18.8 million for the years ended December 31,
2009, 2008 and 2007, or approximately 44%, 35% and 55%, respectively, of our
total expenses.
Research
and development expenses are further discussed below under “Critical Accounting
Policies and Estimates” and “Results of Operations.”
Our
currently projected expenditures for 2010 include approximately $3.2 million for
our clinical programs for INNO-206, approximately $1.6 million for our clinical
programs for bafetinib, approximately $2.5 million for our clinical program for
tamibarotene, approximately $1.4 million for our activities for arimoclomol, and
approximately $2.2 million for general operation of our clinical
programs. The actual cost of our clinical programs could differ
significantly from our current projections due to any additional requirements or
delays imposed by the FDA in connection with our planned trials, or if actual
costs are higher than current management estimates for other reasons, including
complications with manufacturing. In the event that actual costs of our clinical
program, or any of our other ongoing research activities, are significantly
higher than our current estimates, we may be required to significantly modify
our planned level of operations.
32
There is
a risk that any drug discovery and development program may not produce revenue
because of the risks inherent in drug discovery and development. The successful
development of any product candidate is highly uncertain. We cannot reasonably
estimate or know the nature, timing and costs of the efforts necessary to
complete the development of, or the period in which material net cash inflows
are expected to commence from any product candidate, due to the numerous risks
and uncertainties associated with developing drugs, including the uncertainty
of:
|
|
·
|
our
ability to advance product candidates into pre-clinical and clinical
trials;
|
|
|
·
|
the
scope, rate and progress of our pre-clinical trials and other research and
development activities;
|
|
|
·
|
the
scope, rate of progress and cost of any clinical trials we
commence;
|
|
|
·
|
the
cost of filing, prosecuting, defending and enforcing any patent claims and
other intellectual property rights;
|
|
|
·
|
future
clinical trial results;
|
|
|
·
|
the
terms and timing of any collaborative, licensing and other arrangements
that we may establish;
|
|
|
·
|
the
cost and timing of regulatory
approvals;
|
|
|
·
|
the
cost and timing of establishing sales, marketing and distribution
capabilities;
|
|
|
·
|
the
cost of establishing clinical and commercial supplies of our product
candidates and any products that we may develop;
and
|
|
|
·
|
the
effect of competing technological and market
developments.
|
Any
failure to complete any stage of the development of our products in a timely
manner could have a material adverse effect on our operations, financial
position and liquidity. A discussion of the risks and uncertainties associated
with our business is set forth in the “Risk Factors” section of this Annual
Report.
Critical
Accounting Policies and Estimates
Management’s
discussion and analysis of our financial condition and results of operations are
based on our consolidated financial statements, which have been prepared in
accordance with accounting principles generally accepted in the United States of
America. The preparation of these financial statements requires management to
make estimates and judgments that affect the reported amounts of assets,
liabilities, revenues and expenses, and related disclosure of contingent assets
and liabilities. On an ongoing basis, management evaluates its estimates,
including those related to revenue recognition, stock options, impairment of
long-lived assets, including finite lived intangible assets, accrued liabilities
and certain expenses. We base our estimates on historical experience and on
various other assumptions that are believed to be reasonable under the
circumstances, the results of which form the basis for making judgments about
the carrying values of assets and liabilities that are not readily apparent from
other sources. Actual results may differ materially from these estimates under
different assumptions or conditions.
Our
significant accounting policies are summarized in Note 2 of the Notes to
Financial Statements included in this Annual Report. We believe the following
critical accounting policies are affected by our more significant judgments and
estimates used in the preparation of our consolidated financial
statements:
Revenue
Recognition
Revenue
consists of license fees from strategic alliances with pharmaceutical companies
as well as service and grant revenues. Service revenue consists of contract
research and laboratory consulting. Grant revenues consist of government
and private grants.
33
Monies
received for license fees are deferred and recognized ratably over the
performance period in accordance with Staff Accounting Bulletin (“SAB”) No. 104,
Revenue Recognition.
Milestone payments will be recognized upon achievement of themilestone as long
as the milestone is deemed substantive and we have no other performance
obligations related to the milestone and collectability is reasonably assured,
which is generally upon receipt, or recognized upon termination of the agreement
and all related obligations. Deferred revenue represents amounts received prior
to revenue recognition.
Revenues
from contract research, government grants, and consulting fees are recognized
over the respective contract periods as the services are performed, provided
there is persuasive evidence or an arrangement, the fee is fixed or determinable
and collection of the related receivable is reasonably assured. Once all
conditions of the grant are met and no contingencies remain outstanding, the
revenue is recognized as grant fee revenue and an earned but unbilled revenue
receivable is recorded.
In August
2006, we received approximately $24.3 million in proceeds from the
privately-funded ALS Charitable Remainder Trust (“ALSCRT”) in exchange for the
commitment to continue research and development of arimoclomol and other
potential treatments for ALS and a one percent royalty in the worldwide sales of
arimoclomol. We accounted for the transaction under ASC 730-20
(previously Statement of Financial Accounting Standards No. 68, Research and Development Arrangements).
Accordingly, we recorded the value received under the arrangement as deferred
service revenue and recognize service revenue, using the proportional
performance method of revenue recognition, on a dollar-for-dollar basis for each
dollar of expense incurred for the research and development of arimoclomol and
other potential ALS treatments. In August 2009, we were released from all
restrictions on the use of any proceeds previously paid to us in connection with
the arrangement. As a result, we recognized in the third quarter $6.7
million of service revenue representing the remaining deferred revenue and
previously un-recognized portion of the value received in the transaction with
ALSCRT. For the years ended December 31, 2009 and 2008, we recognized
approximately $9.4 million and $6.2 million, respectively, of service revenue
related to this transaction.
Research
and Development Expenses
Research
and development expenses consist of costs incurred for direct and
overhead-related research expenses and are expensed as incurred. Costs to
acquire technologies, including licenses, that are utilized in research and
development and that have no alternative future use are expensed when incurred.
Technology developed for use in its products is expensed as incurred until
technological feasibility has been established.
Clinical
Trial Expenses
Clinical
trial expenses, which are included in research and development expenses, include
obligations resulting from the Company’s contracts with various clinical
research organizations in connection with conducting clinical trials for its
product candidates. We recognize expenses for these activities based on a
variety of factors, including actual and estimated labor hours, clinical site
initiation activities, patient enrollment rates, estimates of external costs and
other activity-based factors. We believe that this method best approximates the
efforts expended on a clinical trial with the expenses we record. We adjust our
rate of clinical expense recognition if actual results differ from our
estimates. If our estimates are incorrect, clinical trial expenses recorded in
any particular period could vary.
Stock-based
Compensation
Our
stock-based employee compensation plans are described in Note 15 of the Notes to
our Financial Statements. We have adopted the provisions of ASC 718 (previously
SFAS No. 123(R), Share-Based
Payment (“SFAS 123(R)”)), which requires the measurement and recognition
of compensation expense for all stock-based awards made to employees and
non-employees.
For stock
options and stock warrants paid in consideration of services rendered by
non-employees, we recognize compensation expense in accordance with the
requirements of ASC 718 (previously SFAS No. 123(R)), ASC 505-50 (previously
Emerging Issues Task Force Issue No. 96-18 (“EITF 96-18”)), Accounting for Equity
Instruments that are Issued to other than Employees for Acquiring, or in
Conjunction with Selling Goods or Services and ASC 505 (previously EITF
00-18, Accounting Recognition
for Certain Transactions involving Equity Instruments Granted to Other Than
Employees), as amended.
Non-employee
option grants that do not vest immediately upon grant are recorded as an expense
over the vesting period. At the end of each financial reporting period prior to
performance, the value of these options, as calculated using the Black-Scholes
option-pricing model, is determined, and compensation expense recognized or
recovered during the period is adjusted accordingly. Since the fair market value
of options granted to non-employees is subject to change in the future, the
amount of the future compensation expense is subject to adjustment until the
common stock options or warrants are fully vested.
34
Impairment
of Long-Lived Assets
We review
long-lived assets, including finite lived intangible assets, for impairment on
an annual basis, as of December 31, or on an interim basis if an event occurs
that might reduce the fair value of such assets below their carrying values. An
impairment loss would be recognized based on the difference between the carrying
value of the asset and its estimated fair value, which would be determined based
on either discounted future cash flows or other appropriate fair value methods.
If our estimates used in the determination of either discounted future cash
flows or other appropriate fair value methods are not accurate as compared to
actual future results, we may be required to record an impairment
charge. The fixed assets, from our San Diego laboratory and our
molecular library, available for sale were re-allocated from Equipment and
Furnishings to Assets Held for Sale and were written down to their estimated net
realizable value as of September 30, 2009.
Net
Income (Loss) Per Share
Basic net
income (loss) per common share is computed using the weighted-average number of
common shares outstanding. Diluted net income (loss) per common share is
computed using the weighted-average number of common share and common share
equivalents outstanding. Common share equivalents that could
potentially dilute basic earnings per share in the future, and that were
excluded from the computation of diluted loss per share, totaled approximately
24.4 million shares, 15.2 million shares and 17.1 million shares at December 31,
2009, 2008 and 2007, respectively.
As a
result of our March 11, 2008 distribution by our stockholders of approximately
36% of the outstanding shares of RXi, we recorded a deemed dividend of
approximately $757,000. The deemed dividend was reflected as an
adjustment to net loss for the first quarter of 2008 to arrive at net loss
applicable to common stockholders on the consolidated statement of operations
and for purposes of calculating basic and diluted earnings per
shares.
Quarterly
Financial Data
The
following table sets forth unaudited consolidated statements of operations data
for each quarter during our most recent two fiscal years. This quarterly
information has been derived from our unaudited consolidated financial
statements and, in the opinion of management, includes all adjustments,
consisting only of normal recurring adjustments, necessary for a fair
presentation of the information for the periods covered. The quarterly financial
data should be read in conjunction with our consolidated financial statements
and related notes. The operating results for any quarter are not necessarily
indicative of the operating results for any future period.
|
Quarters Ended
|
||||||||||||||||
|
|
March 31
|
June 30
|
September 30
|
December 31
|
||||||||||||
|
(In
thousands, except per share data)
|
||||||||||||||||
|
2009
|
||||||||||||||||
|
Total
revenues
|
$ | 1,483 | $ | 1,000 | $ | 6,954 | $ | 100 | ||||||||
|
Net
income (loss)
|
(3,973 | ) | (2,226 | ) | 3,863 | (2,464 | ) | |||||||||
|
Net
income (loss) applicable to common stockholders
|
$ | (3,973 | ) | $ | (2,226 | ) | $ | 3,863 | $ | (2,464 | ) | |||||
|
Basic
and diluted (income) loss per share applicable to common
stock
|
$ | (0.04 | ) | $ | (0.02 | ) | $ | 0.04 | $ | (0.03 | ) | |||||
|
2008
|
||||||||||||||||
|
Total
revenues
|
$ | 2,181 | $ | 1,740 | $ | 917 | $ | 1,427 | ||||||||
|
Net
loss
|
(5,374 | ) | (5,826 | ) | (12,316 | ) | (3,530 | ) | ||||||||
|
Deemed
dividend for anti-dilution adjustments made to outstanding
common
stock warrants
|
(757 | ) | — | — | — | |||||||||||
|
Net
loss applicable to common stockholders
|
$ | (6,131 | ) | $ | (5,826 | ) | $ | (12,316 | ) | $ | (3,530 | ) | ||||
|
Basic
and diluted loss per share applicable to common stock
|
$ | (0.07 | ) | $ | (0.06 | ) | $ | (0.14 | ) | $ | (0.03 | ) | ||||
Quarterly
and yearly loss per share amounts are computed independently of each other.
Therefore, the sum of the per share amounts for the quarters may not equal the
per share amounts for the year. In 2009 and 2008 we incurred $2.3 million and
$2.1 million, respectively, in employee non-cash compensation
expenses.
In
connection with our adjustment to the exercise terms of certain outstanding
warrants to purchase common stock on March 11, 2008, we recorded deemed
dividends of $757,000. These deemed dividends are reflected as an adjustment to
net loss for the first quarter of 2008 to arrive at net loss applicable to
common stockholders on the consolidated statement of operations and for purposes
of calculating basic and diluted earnings per shares.
35
Liquidity
and Capital Resources
General
In order
to fund our business and operations, we have relied primarily upon sales of our
equity securities, including proceeds received upon the exercise of options and
warrants, and sales of our shares of RXi common stock. We also have
received limited payments from our strategic partners and
licensees.
At
December 31, 2009, we had cash and cash equivalents of approximately $9.9
million, marketable securities of $22.8 million and held 5,768,881 shares of
restricted common stock of RXi with a market value of $26.4 million based upon
the closing price of the RXi common stock on that date. On July 27, 2009, we
raised approximately $18.3 million, net of fees and expenses, in a registered
direct offering, and on September 23, 2009, we raised approximately $1.2
million, net of fees, from the sale of 500,000 shares of our common stock of
RXi. Management believes that our current cash on hand, together with our
marketable securities and proceeds from possible future sales of RXi common
stock, will be sufficient to fund our operations for the foreseeable
future. The estimate is based, in part, upon our currently projected
expenditures for 2010 of approximately $18.0 million (unaudited), which includes approximately
$3.2 million (unaudited) for our clinical programs for INNO-206, approximately
$1.6 million (unaudited) for our clinical programs for bafetinib, approximately
$2.5 million (unaudited) for our clinical program for tamibarotene,
approximately $1.4 million (unaudited) for our activities for arimoclomol,
approximately $2.2 million (unaudited) for general operation of its clinical
programs, and approximately $7.1 million (unaudited) for other general and
administrative expenses. These projected expenditures are also based upon
numerous other assumptions and subject to many uncertainties, and actual
expenditures may be significantly different from these
projections. We will be required to obtain additional funding in
order to execute our long-term business plans, although we do not currently have
commitments from any third parties to provide us with capital. The fair value of
common stock investment in RXi is subject to market fluctuations that could
impact the amount of cash we generate from the sale of RXi shares in the future.
We cannot assure that additional funding will be available on favorable terms,
or at all. If we fail to obtain additional funding when needed, we may not be
able to execute our business plans and our business may suffer, which would have
a material adverse effect on our financial position, results of operations and
cash flows.
If we
obtain marketing approval as currently planned and successfully commercialize
our product candidates, we anticipate it will take a minimum of three years, and possibly
longer, for us to generate significant recurring revenue, and we will be
dependent on future financing until such time, if ever, as we can generate
significant recurring revenue. Our ability to raise capital has been materially
and adversely affected by the continuing poor economy. Despite the
recovery in the U.S. financial markets in 2009, the market remains severely
depressed for private investment in public equities, or PIPEs, transactions on
which we have relied for raising needed capital. These conditions
also may materially and adversely affect the market for our RXi shares. We have
no commitments from third parties to provide us with any additional financing,
and we may not be able to obtain future financing on favorable terms, or at all.
Failure to obtain adequate financing would adversely affect our ability to
operate as a going concern. If we raise additional funds by issuing equity
securities, dilution to stockholders may result and new investors could have
rights superior to holders of the shares issued in this offering. In addition,
debt financing, if available, may include restrictive covenants. If adequate
funds are not available to us, we may have to liquidate some or all of our
assets or to delay or reduce the scope of or eliminate some portion or all of
our development programs or clinical trials. We also may have to license to
other companies our product candidates or technologies that we would prefer to
develop and commercialize ourselves.
Discussion
of Operating, Investing and Financing Activities
Net loss
for the year ended December 31, 2009 was $4.8 million, and cash used for
operating activities for that period was $12.1 million. The net loss for the
year reflects $9.4 million of revenue recognized under the 2006 agreement with
ALSCRT, and $2.8 million for stock option and warrant expense.
Net loss
for the year ended December 31, 2008 was $27.0 million, and cash used for
operating activities for that period was $19.4 million. The net loss for the
year reflects $6.2 million of revenue recognized under the 2006 agreement with
ALSCRT, a expense of $8.0 million related to the acquisition of Innovive’s
in-process research and development, a loss of $3.9 million in our equity in
RXi, and $2.1 million for stock option and warrant expense.
36
Net loss
for the year ended December 31, 2007 was $21.9 million, and cash used for
operating activities for that period was $22.4 million. The net loss for the
year reflects $7.2 million of revenue recognized under the 2006 agreement with
ALSCRT and $3.5 million for stock option and warrant expense.
For the
year ended December 31, 2009, $21.6 million was used in investing
activities. This included $22.8 million used to purchase marketable
securities, which was partially offset by proceeds of $1.2 million from the
sale of 500,000 of our shares of common stock RXi.
For the
year ended December 31, 2008, $7.0 million was used in investing activities
including $10.0 million of RXi funds resulting from converting marketable
securities to cash equivalents that is not available to us due to the
deconsolidation. The total cash outlay to acquire Innovive totaled $5.7 million,
which related primarily to the payment of Innovive’s accounts payable. The other
$0.9 million was used for the purchase of equipment and furnishings, primarily
associated with equipping the San Diego laboratory.
For the
year ended December 31, 2007, $11.0 million was used in investing activities. Of
this amount, $9.8 million was used by RXi for the purchase of marketable
securities. The other $1.3 million was used for the purchase of equipment and
furnishings primarily associated with equipping our San Diego
laboratory.
Cash
provided by financing activities for the year ended December 31, 2009 was $18.6
million. During 2009, we raised $18.3 million in a private placement of our
common stock and an additional $0.3 million from the exercise of previously
outstanding stock options and warrants.
Cash
provided by financing activities for the year ended December 31, 2008 was $1.0
million. During 2008, we received $1.0 million from the exercise of
stock options and warrants.
Cash
provided by financing activities for the year ended December 31, 2007 was $53.5
million. During 2007, we raised $34.2 million in a private placement of our
common stock and an additional $18.8 million from the exercise of previously
outstanding stock options and warrants.
Off-Balance
Sheet Arrangements
We have
no off-balance sheet arrangements that have a material current effect or that
are reasonably likely to have a material future effect on our financial
condition, changes in financial condition, revenues or expenses, results of
operations, liquidity, capital expenditures, or capital resources.
Contractual
Obligations
We
acquire assets still in development and enter into research and development
arrangements with third parties that often require milestone and royalty
payments to the third party contingent upon the occurrence of certain future
events linked to the success of the asset in development. Milestone payments may
be required, contingent upon the successful achievement of an important point in
the development life-cycle of the pharmaceutical product (e.g., approval of the
product for marketing by a regulatory agency). If required by the arrangement,
we may have to make royalty payments based upon a percentage of the sales of the
pharmaceutical product in the event that regulatory approval for marketing is
obtained. Because of the contingent nature of these payments, they are not
included in the table of contractual obligations.
These
arrangements may be material individually, and in the event that milestones for
multiple products covered by these arrangements were reached in the same period,
the aggregate charge to expense could be material to the results of operations
in any one period. In addition, these arrangements often give us the discretion
to unilaterally terminate development of the product, which would allow us to
avoid making the contingent payments; however, we are unlikely to cease
development if the compound successfully achieves clinical testing
objectives.
37
Our
current contractual obligations that will require future cash payments are as
follows (in thousands):
|
Operating
Leases (1)(2)
|
Employment
Agreements (3)
|
Subtotal
|
Research
and Development (4)
|
Total
|
||||||||||||||||
|
2010
|
$ | 873 | $ | 2,210 | $ | 3,083 | $ | 3,018 | $ | 6,101 | ||||||||||
|
2011
|
536 | — | 536 | 49 | 585 | |||||||||||||||
|
2012
|
459 | — | 459 | 149 | 608 | |||||||||||||||
|
2013
|
318 | — | 318 | 149 | 467 | |||||||||||||||
|
2014 and
thereafter
|
372 | — | 372 | 383 | 755 | |||||||||||||||
|
Total
|
$ | 2,558 | $ | 2,210 | $ | 4,768 | $ | 3,748 | $ | 8,516 | ||||||||||
____________
|
(1)
|
Operating
leases are primarily facility lease related obligations, as well as
equipment and software lease obligations with third party
vendors.
|
(2)
We are entitled to receive future rental income under subleases in place
which would be offset against future operating lease obligations as follows:
$519,000 in 2010, $350,000 in 2011 and $235,000 in 2012.
|
(3)
|
Employment
agreements include management contracts, which have been revised from time
to time, provide for minimum salary levels, adjusted annually at the
discretion of our Compensation Committee, as well as for minimum bonuses
that are payable.
|
|
(4)
|
Research
and development obligations relate primarily to clinical trials. Most of
these purchase obligations are
cancelable.
|
We apply
the disclosure provisions of ASC 460 (formerly FASB Interpretation No. (“FIN”)
45, Guarantor’s Accounting and
Disclosure Requirements for Guarantees, Including Indirect Guarantees of
Indebtedness of Other), to our contractual guarantees and
Indemnities. We have provided contractual indemnities to investors
and other parties against possible losses suffered or incurred by the
indemnified parties in connection with various types of third-party claims, as
well as indemnities to our officers and directors against third party
claims arising from the services they provide to us. To date, we have
not incurred material costs as a result of these indemnities, and we do not
expect to incur material costs in the future; further, we maintain insurance to
cover certain losses arising from these indemnities. Accordingly, we
have not accrued any liabilities in our consolidated financial statements
related to these indemnities.
Net
Operating Loss Carryforwards
At
December 31, 2009, we had United States federal and state net operating loss
carryforwards of $91.6 million and $81.9 million, respectively, available to
offset against future taxable income, which expire in 2011 through
2029. Approximately $34.7 million of our federal net operating loss
carryforwards are limited in their availability to $0.3 million annually.
Management currently believes that the remaining $56.9 million in federal net
operating loss carryforwards, and the $57.6 million in state net operating loss
carryforwards as of December 31, 2009, are unrestricted. However,
management is reviewing its recent equity transactions to determine if they may
have resulted in any further restrictions on the Company’s net operating loss
carryforwards. As of December 31, 2009, we also had research and development and
alternative minimum tax credits for federal and state purposes of approximately
$8.9 million and $0, respectively, available for offset against future income
taxes, which expire in 2022 through 2029. Based on an assessment of all
available evidence including, but not limited to, our limited operating history
in our core business and lack of profitability, uncertainties of the commercial
viability of its technology, the impact of government regulation and healthcare
reform initiatives, and other risks normally associated with biotechnology
companies, we have concluded that it is more likely than not that these net
operating loss carryforwards and credits will not be realized and, as a result,
a 100% deferred tax valuation allowance has been recorded against these
assets.
Results
of Operations
We
incurred a net loss of $4.8 million, $27.0 million and $21.9 million for the
years ended December 31, 2009, 2008 and 2007, respectively.
38
During
fiscal 2009, we recognized $9.4 million in service revenues relating to our
$24.3 million sale to the ALSCRT of a one-percent royalty interest in the
worldwide sales of arimoclomol in August 2006. Pursuant to an amendment signed
between us and thebeneficiary of the ALSCRT on August 6, 2009, we were released
from all restrictions on the use of any proceeds previously paid to us in
connection with the arrangement. As a result, we recognized $6.7
million as service revenue in the third quarter of 2009, which represented the
remaining deferred revenue and previously un-recognized portion of the value
received. In the years ended December 31, 2008 and 2007, we
recognized $6.2 million and $7.2 million in service revenues,
respectively.
During
2009, 2008 and 2007, we earned an immaterial amount of license fees and grant
revenue. All future licensing fees under our current licensing agreements are
dependent upon successful development milestones being achieved by the licensor.
During fiscal 2010, we are not anticipating the receipt of any significant
service or licensing fees.
Our net
loss may increase from current levels primarily due to expenses related to our
ongoing and planned clinical trials, research and development programs, possible
technology acquisitions, and other general corporate activities. We
anticipate, therefore, that our operating results will fluctuate for the
foreseeable future and period-to-period comparisons should not be relied upon as
predictive of the results in future periods.
Research
and Development
|
|
Years Ended December 31,
|
|||||||||||
|
|
2009
|
2008
|
2007
|
|||||||||
|
(In
thousands)
|
||||||||||||
|
Research
and development expenses
|
$ | 5,621 | $ | 9,913 | $ | 14,454 | ||||||
|
Non-cash
research and development expenses
|
62 | (224 | ) | 3,778 | ||||||||
|
Impairment
loss on fixed assets
|
1,187 | — | — | |||||||||
|
Employee
stock option expense
|
672 | 777 | 592 | |||||||||
| $ | 7,542 | $ | 10,466 | $ | 18,824 | |||||||
Research
expenses are expenses incurred by us in the discovery of new information that
will assist us in the creation and the development of new drugs or treatments.
Development expenses are expenses incurred by us in our efforts to commercialize
the findings generated through our research efforts.
Research
and development expenses incurred during 2009, 2008 and 2007 relate to our
various development programs. In 2009, we substantially completed the initial
phase of our new-drug discovery research in our laboratory facility in San
Diego, California, which account for the significant decrease in research and
development expenses from 2008. In 2008, only the months of January
and February included RXi-related expenses (totaling approximately $0.6
million), which accounts for the significant decrease in research and
development expenses, and non-cash research and development expenses. In 2009,
our development costs associated included approximately $0.8 million for our
clinical programs for INNO-206, approximately $0.25 million for our clinical
programs for bafetinib, $0.8 million for our clinical program for tamibarotene,
approximately $0.4 million for our activities for arimoclomol, approximately
$0.4 million for general operation of our clinical programs, and approximately
$3.0 million for other general and administrative expenses. None
of our research and development costs have ever been
capitalized.
As
compensation to scientific advisory board (“SAB”) members and consultants, and
in connection with the acquisition of technology, we and RXi sometimes issue
shares of common stock, stock options and warrants to purchase shares of common
stock. For financial statement purposes, we value these shares of common stock,
stock options, and warrants at the fair value of the common stock, stock options
or warrants granted, or the services received, whichever is more reliably
measurable. We recorded charges (recovery) of $0.0 million, $(0.2 million) and
$3.8 million in this regard during 2009, 2008 and 2007, respectively. Included
in the research and development charges for 2007 were $2.3 million of expense
related to RXi’s issuance of 462,112 shares of common stock to UMMS for certain
license agreement rights and a new invention disclosure agreement and $1.0
million for non-qualifying stock options to SAB members of RXi. In 2009, we
recorded $0.7 million of employee stock option expense as compared to $0.8
million in 2008 and $0.6 million in 2007.
We also
incurred an expenditure of $8.0 million in 2008 related to the acquisition of
Innovive’s in-process research and development, which has been reflected as a
separate line item on our Consolidated Statements of Operations.
In 2010,
we expect our research and development expenses to increase as a result of our
clinical programs with INNO-206, bafetinib and tamibarotene.
39
General
and administrative expenses
|
Year Ended December 31,
|
||||||||||||
|
2009
|
2008
|
2007
|
||||||||||
|
(In
thousands)
|
||||||||||||
|
General
and administrative expenses
|
$ | 7,128 | $ | 9,134 | $ | 12,666 | ||||||
|
Stock,
stock option and warrant expenses to non-employees and
consultants
|
421 | 189 | 2 | |||||||||
|
Employee
stock option expense
|
1,579 | 1,610 | 2,154 | |||||||||
| $ | 9,128 | $ | 10,933 | $ | 14,822 | |||||||
General
and administrative expenses include all administrative salaries and general
corporate expenses, including legal expenses associated with the prosecution of
our intellectual property. Our general and administrative expenses, excluding
common stock, stock options and warrants issued, and excluding depreciation
expense, were $7.1 million in 2009, $9.1 million in 2008 and $12.7 million,
respectively, in 2007. General and administrative expenses in 2007 included $4.6
million of RXi-related expenses. In 2008, we recognized RXi-related
expenses for January and February only of $1.3 million. No
RXi-related expenses were recognized in 2009. General and
administrative expenses in 2009 decreased by $2.0 million as compared
to 2008, due primarily to the $1.3 million of RXi-related expenses
recognized in 2008. In 2009, legal and accounting/auditing expenses
decreased by approximately $302,000 and $304,000, respectively, primarily due to
the higher expenses incurred in 2008 in connection with our acquisition of
Innovive, as well as efficiencies in the administrative area.
From time
to time, we issue shares of our common stock or warrants or options to purchase
shares of our common stock to consultants and other service providers in
exchange for services. For financial statement purposes, we value these shares
of common stock, stock options, and warrants at the fair value of the common
stock, stock options or warrants granted, or the services received, whichever we
can measure more reliably.
We
recorded employee stock option expense of $1.6 million in 2009, $1.6 million in
fiscal 2008 and $2.2 million in fiscal 2007. The greater amount in 2007
primarily related to stock options granted by RXi to recruit and retain
directors, officers and additional employees.
Depreciation
and amortization
Depreciation
and amortization expenses for the years ended December 31, 2009, 2008 and 2007
were $475,000, $625,000 and $272,000, respectively. The depreciation expense
reflects the depreciation of our fixed assets and the amortization expenses
related to our molecular library. The decrease in 2009 relates to a
lower depreciation base due to the re-class of fixed assets to
Assets-held-for-sale and the value of the molecular library related to the
closure of the San Diego lab in the third quarter of 2009.
Other
Income
In 2009,
we recognized a gain of $0.7 million on the valuation of our warrant derivative
liability related to warrants issued in July 2009. In July 2009, we
recognized a gain of $1.2 million on the sale of RXi shares. In March 2008, we
recognized a gain of $0.2 million on the transfer of some RXi common stock
to certain employees. In June 2007, we recognized $1.5 million of income arising
from a fee received pursuant to a change-in-control provision included in the
purchase agreement for our 1998 sale of our animal pharmaceutical
unit.
Interest
income
Interest
income was $0.3 million in 2009, $1.2 million in 2008 and $2.7 million in 2007.
The variances between years are attributable primarily to the amount of funds
available for investment each year and, to a lesser extent, changes in
prevailing market rates.
Noncontrolling
Interest in RXi
We offset
$88,000 of losses in noncontrolling interest in RXi against our net loss for the
months of January and February 2008. For the remainder of the year, and for
2009, RXi’s gain and losses were accounted for under the equity method, because
we owned less than 50% of RXi following our March 11, 2008 distribution to
our stockholders of RXi shares. We offset $449,000 of minority interest in
losses of RXi against our net loss for the year ended December 31,
2007.
40
Recent
Accounting Pronouncements
In
December 2007, the FASB issued guidance which is now part of ASC 810-10, Noncontrolling Interests in
Consolidated Financial Statements, an Amendment of Accounting Research Bulletin
No. 51 “ (formerly Statement of Financial Accounting Standards (SFAS)
160, Noncontrolling
Interests in Consolidated Financial Statements—an amendment of ARB No. 51
). This guidance establishes accounting and reporting standards for ownership
interests in subsidiaries held by parties other than the parent, changes in a
parent’s ownership of a noncontrolling interest, calculation and disclosure of
the consolidated net income attributable to the parent and the noncontrolling
interest, changes in a parent’s ownership interest while the parent retains its
controlling financial interest and fair value measurement of any retained
noncontrolling equity investment. The new guidance is effective for financial
statements issued for fiscal years beginning after December 15, 2008, and
interim periods within those fiscal years. Early adoption is prohibited. We
adopted this guidance on January 1, 2009, the beginning of its 2009 fiscal year,
which resulted in certain reclassifications related to the noncontrolling
interest in the consolidated financial statements.
In March
2008, the FASB issued guidance ASC 815-10 (formerly Statement of Financial
Accounting Standards No. 161, Disclosures about Derivative
Instruments and Hedging Activities (“SFAS No. 161”). The new
standard amends Statement of Financial Accounting Standards No. 133, Accounting for Derivative
Instruments and Hedging Activities (“SFAS 133”), and seeks to enhance
disclosure about how and why a company uses derivatives; how derivative
instruments are accounted for under SFAS 133 (and the interpretations of that
standard); and how derivatives affect a company’s financial position, financial
performance and cash flows. SFAS 161 will be effective for financial
statements issued for fiscal years and interim periods beginning after November
15, 2008. Early application of the standard is encouraged, as well as
comparative disclosures for earlier periods at initial adoption. The
adoption of ASC 815-10 did not have a material impact on our consolidated
financial statements.
In April
2008, the FASB issued revised guidance on determining the useful life of
intangible assets. The revised guidance, which is now part of ASC
350-30 General Intangibles Other than Goodwill (previously Staff Position No.
FAS 142-3, Determination of
the Useful Life of Intangible Assets), amends the factors that should be
considered in developing renewal or extension assumptions used to determine the
useful life of a recognized intangible asset. The Position is
effective for fiscal years beginning after December 15, 2008 and applies
prospectively to intangible assets acquired after the effective
date. Early adoption is not permitted. The adoption of SFAS No. ASC
350-30 did not have a material impact on our consolidated financial
statements.
In May
2008, the FASB issued revised guidance on Convertible Debt
Instruments. The revised guidance which is now part of ASC
470-20 (formerly Staff Position No. Accounting Principles Board 14-1, Accounting for Convertible Debt
Instruments That May Be Settled in Cash upon Conversion (Including
Partial Cash Settlement) (“FSP No. APB 14-1”)). ASC 470-20 requires that the
liability and equity components of convertible debt instruments that may be
settled in cash upon conversion (including partial cash settlement) be
separately accounted for in a manner that reflects an issuer’s nonconvertible
debt borrowing rate. ASC 470-20 is effective for us as of January 1,
2009. The adoption of ASCO 470-20 did not have an impact on our consolidated
financial statements.
In June
2008, the FASB ratified guidance which is now part of ASC 815-40, Contracts in Entity’s Own Equity
(formerly EITF (Emerging Issues Task Force) 07-05), Determining Whether an Instrument
(or Embedded Feature) Is Indexed to an Entity’s Own Stock. The objective
of this issue is to provide guidance for determining whether an equity-linked
financial instrument (or embedded feature) is indexed to an entity’s own stock.
This issue applies to any freestanding financial instrument or embedded feature
that has all the characteristics of a derivative instrument or an instrument
which may be potentially settled in an entity’s own stock regardless of whether
the instrument possess derivative characteristics. This issue provides a
two-step approach to assist in making these determinations and is effective for
financial statements issued for fiscal years beginning after December 15, 2008.
The adoption of ASC 815-40 did not have a material impact on our consolidated
financial statements.
In April
2009, the FASB issued guidance which is now part of ASC 825-10 Financial
Instruments (formerly Financial Staff Position SFAS 107-1 and Accounting
Principles Board (APB) Opinion No. 28-1, Interim Disclosures about Fair Value
of Financial Instruments (SFAS 107-1 and APB 28-1). This
statement amends FASB Statement No. 107, Disclosures about Fair Values of
Financial Instruments, to require disclosures about fair value of
financial instruments in interim financial statements as well as in annual
financial statements. The statement also amends APB Opinion No. 28,
“Interim Financial Reporting,” to require those disclosures in all interim
financial statements. This statement is effective for interim periods
ending after June 15, 2009. The adoption of ASC 825-10 did not have an impact on
our financial statements.
41
In May
2009 and February 2010, the FASB issued new guidance for accounting for
subsequent events. The new guidance, which is now part of ASC 855-10, Subsequent Events (formerly,
SFAS No. 165, Subsequent
Events) is consistent with existing auditing standards in defining
subsequent events as events or transactions that occur after the balance sheet
date but before the financialstatements are issued or are available to be
issued. The new guidance defines two types of subsequent events: “recognized
subsequent events” and “non-recognized subsequent events.” Recognized subsequent
events provide additional evidence about conditions that existed at the balance
sheet date and must be reflected in the company’s financial statements.
Non-recognized subsequent events provide evidence about conditions that arose
after the balance sheet date and are not reflected in the financial statements
of a company. Certain non-recognized subsequent events may require
disclosure to prevent the financial statements from being misleading. The
new guidance was effective on a prospective basis for interim or annual periods
ending after June 15, 2009. We adopted the provisions of ASC 855-10
as required.
In June
2009, the FASB amended ASC 860, (formerly SFAS No. 166, Accounting for Transfers of Financial
Assets, an amendment to
SFAS No. 140). ASC 860 eliminates the concept of a “qualifying
special-purpose entity,” changes the requirements for derecognizing financial
assets, and requires additional disclosures in order to enhance information
reported to users of financial statements by providing greater transparency
about transfers of financial assets, including securitization transactions, and
an entity’s continuing involvement in and exposure to the risks related to
transferred financial assets. ASC 860 is effective for fiscal years beginning
after November 15, 2009. The Company will adopt ASC 860 in fiscal
2010. We do not expect that the adoption of ASC 860 will have a
material impact on our financial statements.
In June
2009, the FASB amended ASC 810 (formerly SFAS No.167, Amendments to FASB Interpretation
No. 46). The amendments include: (1) the elimination of the
exemption for qualifying special purpose entities, (2) a new approach for
determining who should consolidate a variable-interest entity, and (3) changes
to when it is necessary to reassess who should consolidate a variable-interest
entity. ASC 810 is effective for the first annual reporting period
beginning after November 15, 2009 and for interim periods within that first
annual reporting period. We will adopt ASC 810 in fiscal 2010. We do not expect
that the adoption of ASC 810 will have a material impact on our financial
statements.
In June
2009, the FASB issued new guidance which is now part of ASC 105-10 (formerly
Statement of Financial Accounting Standards No. 168, The FASB Accounting Standards
Codification and the Hierarchy of Generally Accepted Accounting
Principles ). ASC 105-10 replaces FASB Statement No. 162, “The Hierarchy of Generally Accepted
Accounting Principles”, and establishes the FASB Accounting Standards
Codification as the source of authoritative accounting principles recognized by
the FASB to be applied by nongovernmental entities in the preparation of
financial statements in conformity with generally accepted accounting
principles. ASC 105-10 is effective for interim and annual periods ending after
September 15, 2009. The adoption of ASC 105-10 did not have a material impact on
our financial statements.
In
January, 2010, the FASB issued ASU 2010-06, Improving Disclosures about Fair
Value Measurements. The standard amends ASC Topic 820, Fair Value Measurements
and Disclosures to require additional disclosures related to transfers between
levels in the hierarchy of fair value measurement. The standard does not change
how fair values are measured. The standard is effective for interim and annual
reporting periods beginning after December 15, 2009. As a result, it is
effective for us in the first quarter of fiscal year 2010. We do not believe
that the adoption of ASU 2010-06 will have a material impact on consolidated our
financial statements.
Item
7A. QUANTITATIVE
AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Our
exposure to market risk is limited primarily to interest income sensitivity,
which is affected by changes in the general level of U.S. interest rates,
particularly because a significant portion of our investments are in short-term
debt securities issued by the U.S. government and institutional money market
funds. The primary objective of our investment activities is to preserve
principal. Due to the nature of our marketable securities, we believe that we
are not exposed to any material market risk. We do not have any
derivative financial instruments or foreign currency instruments. If interest
rates had varied by 10% in the year ended December 31, 2009, it would not have
had a material effect on our results of operations or cash flows for that
period.
Item
8. FINANCIAL STATEMENTS AND SUPPLEMENTARY
DATA
Our
consolidated financial statements and supplemental schedule and notes thereto as
of December 31, 2009 and 2008, and for each of the three years in the period
ended December 31, 2009, together with the reports thereon of our independent
registered public accounting firms, are set forth on pages F-1 to F-26 of this
Annual Report.
Item 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND
FINANCIAL DISCLOSURE
None.
42
Item
9A. CONTROLS AND PROCEDURES
Disclosure
Controls and Procedures
We
maintain disclosure controls and procedures that are designed to ensure that the
information disclosed in the reports we file with the Securities and Exchange
Commission under the Exchange Act is recorded, processed, summarized and
reported within the time periods specified in the Securities and Exchange
Commission’s rules and forms, and that such information is accumulated and
communicated to our management, including our Chief Executive Officer and Chief
Financial Officer, as appropriate, to allow timely decisions regarding required
disclosure.
Our
management, with the participation of our Chief Executive Officer and Chief
Financial Officer, performed an evaluation of the effectiveness of the design
and operation of our disclosure controls and procedures (as defined in
Securities Exchange Act Rule 13a-15(e)) as of the end of the period covered by
this Annual Report. Based on that evaluation, our Chief Executive Officer and
Chief Financial Officer have concluded that our disclosure controls and
procedures were effective as of December 31, 2009 to provide reasonable
assurance that information required to be disclosed by us in reports that we
file or submit under the Securities Exchange Act of 1934 is recorded, processed,
summarized and reported within the time periods specified in the Securities and
Exchange Commission’s rules and forms.
There was
no change in our internal control over financial reporting that occurred during
the quarter ended December 31, 2009 that materially affected, or is reasonably
likely to materially affect, our internal control over financial
reporting.
Management’s
Report on Internal Control Over Financial Reporting
Our
management is responsible for establishing and maintaining adequate internal
control over financial reporting, as such term is defined in Exchange Act Rule
13a-15(f).
Under the
supervision and with the participation of our management, including our
principal executive and principal financial officers, we assessed the
effectiveness of our internal control over financial reporting as of December
31, 2009. In making this assessment, management used the criteria set forth by
the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”) in
Internal Control-Integrated
Framework. Based upon management’s assessment using the
criteria contained in COSO, our management has concluded that, as of December
31, 2009, our internal control over financial reporting was
effective.
Our
internal control over financial reporting as of December 31, 2009 has been
audited by BDO Seidman, LLP, an independent registered public accounting firm,
as stated in their report thereon set forth on pages F-25, which is incorporated
herein by reference.
Item
9B. OTHER INFORMATION
None.
43
PART
III
Item
10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE
GOVERNANCE
The
following table sets forth information concerning our directors and executive
officers:
|
Name
|
Age
|
Class
of
Director(1)
|
Position
|
|
Max
Link, Ph.D.
|
69
|
III
|
Director,
Chairman of the Board (2) (3) (4)
|
|
Steven
A. Kriegsman
|
68
|
II
|
Director,
Chief Executive Officer, President
|
|
Marvin
R. Selter
|
82
|
II
|
Director,
Vice Chairman of the Board (2) (3) (4)
|
|
Louis
Ignarro, Ph.D.
|
68
|
I
|
Director
|
|
Joseph
Rubinfeld, Ph.D.
|
77
|
I
|
Director
|
|
Richard
L. Wennekamp
|
67
|
II
|
Director
(2) (3) (4)
|
|
John
Caloz
|
58
|
—
|
Chief
Financial Officer
|
|
Daniel
Levitt, M.D., Ph.D.
|
62
|
—
|
Chief
Medical Officer
|
|
D.
Scott Geyer
|
55
|
—
|
Sr.
Vice President-Manufacturing
|
|
D.
Scott Wieland
|
50
|
—
|
Sr.
Vice President-Drug Development
|
|
Benjamin
S. Levin
|
33
|
—
|
General
Counsel, Vice President — Legal Affairs and Corporate
Secretary
|
|
David
J. Haen
|
31
|
—
|
Vice
President – Business Development
|
____________
|
(1)
|
Our
Class I directors serve until the 2010 annual meeting of stockholders, our
Class II directors serve until the 2011 annual meeting of stockholders,
and our Class III director serves until the 2012 annual meeting of
stockholders.
|
|
(2)
|
Members
of our Audit Committee. Mr. Selter is the Chairman of the
Committee.
|
|
(3)
|
Members
of our Nominating and Corporate Governance Committee. Mr. Wennekamp is
Chairman of the Committee.
|
|
(4)
|
Members
of our Compensation Committee. Mr. Wennekamp is Chairman of the
committee.
|
Max Link, Ph.D has been a
director since 1996. Dr. Link has been retired from business since 2003. From
March 2002 until its acquisition by Zimmer Holdings, Dr. Link served as Chairman
and CEO of Centerpulse, Ltd. From May 1993 to June 1994, Dr. Link
served as the Chief Executive Officer of Corange Ltd. (the holding company for
Boehringer Mannheim Therapeutics, Boehringer Mannheim Diagnostics and DePuy
International). From 1992 to 1993, Dr. Link was Chairman of Sandoz Pharma, Ltd.
From 1987 to 1992, Dr. Link was the Chief Executive Officer of Sandoz Pharma and
a member of the Executive Board of Sandoz, Ltd., Basel. Prior to 1987, Dr. Link
served in various capacities with the United States operations of Sandoz,
including President and Chief Executive Officer. Dr. Link also serves as a
director of Alexion Pharmaceuticals, Inc., Celsion Corporation, Inc. and
Discovery Laboratories, Inc., and in the past five years, also served as a
director of Access Pharmaceuticals, Inc., Cell Therapeutics, Inc., Columbia
Laboratories, Inc., Human Genome Sciences, Inc. and Protein Design
Laboratories, Inc.
Dr. Link
has extensive executive-level experience with a number of large pharmaceutical
companies, including Sandoz Pharma, Ltd. In these positions, he was
responsible for major strategic and other business initiatives, including new
drug development, acquisitions and dispositions of new drug candidates and other
technology, licensing, marketing and distribution agreements and other key
contractual strategic arrangements that affect, or are likely to affect, our
company’s own business efforts. As an executive officer and board
member of these other companies, he has experience with the regulatory schemes
in foreign jurisdictions and also has been exposed to different approaches to
corporate governance matters, potential conflicts of interest, and similar
matters, which enables him to offer importance guidance to our Board of
Directors.
Steven A. Kriegsman has been
CytRx’s President and Chief Executive Officer and a director since July 2002. He
also serves as a director of CytRx’s 36% owned affiliate, RXi Pharmaceuticals
Corporation. He previously served as Director and Chairman of Global Genomics
from June 2000. Mr. Kriegsman is an inactive Chairman and Founder of Kriegsman
Capital Group LLC, a financial advisory firm specializing in the development of
alternative sources of equity capital for emerging growth companies in the
healthcare industry. He has advised such companies as SuperGen Inc., Closure
Medical Corporation, Novoste Corporation, Miravant Medical Technologies, and
Maxim Pharmaceuticals. In the past five years, Mr. Kriegsman has also served on
the Board of Directors of Bradley Pharmaceuticals, Inc. and Hythiam, Inc. Mr.
Kriegsman has a BS degree with honors from New York University in Accounting and
completed the Executive Program in Mergers and Acquisitions at New York
University, The Management Institute. Mr. Kriegsman is a
graduate of the Stanford Law School Directors’ College. Mr. Kriegsman
was formerly a Certified Public Accountant with KPMG in New York City. In
February 2006, Mr. Kriegsman received the Corporate Philanthropist of the Year
Award from the Greater Los Angeles Chapter of the ALS Association and in October
2006, he received the Lou Gehrig Memorial Corporate Award from the Muscular
Dystrophy Association. Mr. Kriegsman has been a guest speaker and lecturer at
various universities including California Institute of Technology (Caltech),
Brown University, and New York University. Mr. Kriegsman has been
active in various charitable organizations including the Biotechnology Industry
Organization, the ALS Association, the Los Angeles Venture Association, the
Southern California Biomedical Council, and the Palisades-Malibu
YMCA.
44
Marvin R. Selter has been a
director since October 2003. He has been President and Chief Executive Officer
of CMS, Inc. since he founded that firm in 1968. CMS, Inc. is a national
management consulting firm. In 1972, Mr. Selter originated the concept of
employee leasing. He served as a member of the Business Tax Advisory
Committee—City of Los Angeles, Small Business Board—State of California and the
Small Business Advisory Commission—State of California. Mr. Selter also serves
on the Valley Economic Development Center as past Chairman and Audit Committee
Chairman, the Board of Valley Industry and Commerce Association as past
Chairman, the Advisory Board of the San Fernando Economic Alliance and the
California State University—Northridge as Past Chairman of the Economic Research
Center and President of the Olive View UCLA Medical Center
Foundation. He has served, and continues to serve, as a member of boards of
directors of various hospitals, universities, private medical companies and
other organizations. Mr. Selter attended Rutgers—The State University, majoring
in Accounting and Business Administration. He was an LPA having served as
Controller, Financial Vice President and Treasurer at distribution,
manufacturing and service firms. He has lectured extensively on finance,
corporate structure and budgeting for the American Management Association and
other professional teaching associations.
Mr.
Selter has founded, operated, and grown his own successful businesses, which
gives him a valuable insight into the financial constraints and operational
challenges facing companies in the development stage and as they
mature. He also has many years of involvement in various governmental
agencies and charitable organizations, which affords him an important
perspective on the business regulatory process and capital-raising
activities. In addition, he has significant education and work
experience in accounting and financial matters that he is able to utilize as the
named financial expert on our Audit Committee.
Louis Ignarro, Ph.D. has been
a director since July 2002. He previously served as a director of Global
Genomics since November 20, 2000. Dr. Ignarro serves as the Jerome J. Belzer,
M.D. Distinguished Professor of Pharmacology in the Department of Molecular and
Medical Pharmacology at the UCLA School of Medicine. Dr. Ignarro has been at the
UCLA School of Medicine since 1985 as a professor, acting chairman and assistant
dean. Dr. Ignarro received the Nobel Prize for Medicine in 1998. Dr. Ignarro
received a B.S. in pharmacy from Columbia University and his Ph.D. in
Pharmacology from the University of Minnesota. Dr. Ignarro is a Nobel Laureate
and an esteemed medical researcher whose experience enables him to offer
importance scientific guidance to our Board of Directors.
Joseph Rubinfeld, Ph.D. has
been a director since July 2002. He co-founded SuperGen, Inc. in 1991 and has
served as its Chief Executive Officer and President and as a director since its
inception until December 31, 2003. He resigned as Chairman Emeritus of SuperGen,
Inc. on February 8, 2005. Dr. Rubinfeld was also Chief Scientific Officer of
SuperGen from 1991 until September 1997. Dr. Rubinfeld is also a founder of JJ
Pharma. Dr. Rubinfeld was one of the four initial founders of Amgen, Inc. in
1980 and served as a Vice President and its Chief of Operations until 1983. From
1987 until 1990, Dr. Rubinfeld was a Senior Director at Cetus Corporation and
from 1968 to 1980, Dr. Rubinfeld was employed at Bristol-Myers Company,
International Division in a variety of positions. Dr. Rubinfeld received a B.S.
degree in chemistry from C.C.N.Y. and an M.A. and Ph.D. in chemistry from
Columbia University.
Dr.
Rubinfeld served as a senior executive of several large pharmaceutical companies
before leaving to co-found and serve as Chief Executive Officer or in other
senior executive capacities with highly successful companies. Dr.
Rubenfeld’s academic training and business experience enhances the breadth and
scope of our Board’s oversight of our company’s management, business, strategic
relationships, and other activities, while his vision adds to the long-range
planning of our Board of Directors and management.
Richard L. Wennekamp has been
a director since October 2003. He retired from Community Bank in June 2008 where
he was the Senior Vice President-Credit Administration since October 2002. From
September 1998 to July 2002, Mr. Wennekamp was an executive officer of Bank of
America Corporation, holding various positions, including Managing
Director-Credit Product Executive for the last four years of his 22-year term
with the bank. From 1977 through 1980, Mr. Wennekamp was a Special Assistant to
former President of the United States, Gerald R. Ford, and the Executive
Director of the Ford Transition Office. Prior thereto, he served as Staff
Assistant to the President of the United States for one year, and as the Special
Assistant to the Assistant Secretary of Commerce of the U.S.
45
Mr.
Wennekamp’s senior executive experience in the banking and financial services
industry sets him apart from our other directors and adds unique capabilities
and a different perspective to the deliberations of our Board of
Directors. As a former Chief Credit Officer at Bank of America and
Community Bank, he understands the credit needs, financing requirements, and
operational constraints of development-stage and mature businesses.
Daniel Levitt, M.D., Ph.D.
joined us in October 2009 as our Chief Medical Officer. Dr. Levitt
brings more than 24 years of senior management experience, having spearheaded
numerous drug development programs to commercialization at leading biotechnology
and pharmaceutical companies. Prior to joining CytRx, Dr. Levitt served
from January 2007 to February 2009 as Executive Vice President, Research and
Development at Cerimon Pharmaceuticals, Inc. Prior to that, from August
2003 to April 2006, he was Chief Medical Officer and Head of Clinical and
Regulatory Affairs at Dynavax Technologies Corporation, managing clinical trials
for four programs and overseeing multi-country regulatory strategies. From
August 2002 to July 2003, Dr. Levitt was Chief Operating Officer and Head of
Research and Development at Affymax, Inc., and prior to that he spent six years
at Protein Design Labs, Inc., completing his tenure as that firm’s President and
Head of Research and Development. Dr. Levitt’s past experience includes a
position as Head of Drug Development at Geron Corporation, and Head of the
Cytokine Development Unit and Global Clinical Oncology at Sandoz Pharmaceuticals
Ltd., and as Director, Clinical Oncology and Immunology at Hoffmann-LaRoche,
Inc. Dr. Levitt graduated Magna Cum Laude and Phi Beta Kappa with a
Bachelor of Arts degree from Brandeis University. He earned both his M.D.
and his Ph.D. in Biology from the University of Chicago Pritzker School of
Medicine. Dr. Levitt has received 10 major research awards and authored or
co-authored nearly 200 papers and abstracts.
John Y. Caloz joined us in
October 2007 as our Chief Accounting Officer. In January of 2009 Mr.
Caloz was named Chief Financial Officer. He has a history of providing senior
financial leadership in the life sciences sector, as Chief Financial Officer of
Occulogix, Inc, a NASDAQ listed, a medical therapy company. Prior to that, Mr.
Caloz served as Chief Financial Officer of IRIS International Inc., a
Chatsworth, CA based medical device manufacturer. He served as Chief Financial
Officer of San Francisco-based Synarc, Inc., a medical imaging company, and from
1993 to 1999 he was Senior Vice President, Finance and Chief Financial Officer
of Phoenix International Life Sciences Inc. of Montreal, Canada, which was
acquired by MDS Inc. in 1999. Mr. Caloz was a partner at Rooney, Greig, Whitrod,
Filion & Associates of Saint Laurent, Quebec, Canada, a firm of Chartered
Accountants specializing in research and development and high tech companies,
from 1983 to 1993. Mr. Caloz, a Chartered Accountant, holds a degree in
Accounting from York University, Toronto, Canada.
Scott Wieland, Ph.D, joined
CytRx in 2005 as the Vice President, Clinical and Regulatory Affairs and was
promoted to the position of Senior Vice President, Drug Development in December
2008. Prior to that, he served in senior level positions in the areas of Drug
Development, Clinical and Regulatory Affairs at various biotech firms. He spent
five years at NeoTherapeutics, Inc. serving as the Director of Product
Development and was later promoted to Vice President of Product Development.
From 1990 to 1997, he served as Director of Regulatory Affairs at CoCensys,
Inc. Dr. Wieland has a Ph.D. in Biopsychology and an M.A. in
Psychology from the University of Arizona. He has an MBA from Webster
University. Dr. Wieland received his B.S. in Physiological Psychology from the
University of California, Santa Barbara.
Scott Geyer joined CytRx in
November 2009 as our Senior Vice President, Manufacturing. Prior to
joining CytRx, he served since May 2009, and also from May 2007 through November
2008, as Vice President, Technical Operations at Cerimon Pharmaceuticals,
Inc. He previously served from December 2008 through April 2009 as Senior
Vice President, Technical Operations & Product Development at TRF Pharma,
Inc., from October 2004 through April 2007 as Vice President, Technical
Operation at Xencor, Inc., and from October 2003 through February 2004 as Vice
President, Manufacturing and Process Development at BioMarin Pharmaceuticals
Inc. Mr. Geyer's past experience includes holding senior positions at Onyx
Pharmaceuticals and Protein Design Labs, Inc., as well as positions at
Ares-Sorono Group and SmithKline Beckman, among others. Mr. Geyer has
co-authored numerous publications in peer-reviewed journals. He holds an
M.S. in veterinary microbiology from Texas A&M University and a B.S. in
microbiology from the University of Southwestern Louisiana
Benjamin S. Levin, has been
our General Counsel, Vice President — Legal Affairs and Corporate Secretary
since July 2004. From November 1999 to June 2004, Mr. Levin was an associate in
the transactions department of the Los Angeles office of O’Melveny & Myers
LLP. Mr. Levin received his S.B. in Economics from the Massachusetts Institute
of Technology, and a J.D. from Stanford Law School.
David J. Haen joined CytRx in
October 2003 as Director of Business Development and was promoted to Vice
President of Business Development in December 2007. From 1999 to 2003, Mr.
Haen worked as an associate for Kriegsman Capital Group LLC, a financial
advisory firm focused on emerging companies in the life sciences field.
Mr. Haen received a B.A. in Communications and Business from Loyola Marymount
University.
46
Diversity
Our board
of directors, acting through the Nomination and Governance Committee, is
responsible for assembling for shareholder consideration a group of
director-nominees that, taken together, have the experience, qualifications,
attributes, and skills appropriate for functioning effectively as a board. The
Nomination and Governance Committee periodically reviews the composition of the
board of directors in light of the company’s changing requirements, its
assessment of the board of directors’ performance, and the input of shareholders
and other key constituencies. The Nomination and Governance Committee
looks for certain characteristics common to all board members, including
integrity, strong professional reputation and record of achievement,
constructive and collegial personal attributes, and the ability and commitment
to devote sufficient time and energy to board service. In addition, the
Nomination and Governance Committee seeks to include on the board of directors a
complementary mix of individuals with diverse backgrounds and skills reflecting
the broad set of challenges that the board of directors confronts. These
individual qualities can include matters such as experience in the company’s
industry, technical experience (i.e., medical or research
expertise), experience gained in situations comparable to the company’s,
leadership experience, and relevant geographical diversity.
Committees
Our
business, property and affairs are managed by or under the direction of the
board of directors. Members of the board are kept informed of our business
through discussion with the chief executive and financial officers and other
officers, by reviewing materials provided to them and by participating at
meetings of the board and its committees.
Our board
of directors currently has three committees. The Audit Committee, Compensation
Committee, and Nomination and Governance Committee consist of Messrs. Selter,
Link, and Wennekamp. Such committees operate under a formal charter, copies of
which are available on our website at www.cytrx.com, that governs their duties
and conduct.
Our board
of directors has determined that Mr. Selter, one of the independent directors
serving on our Audit Committee, is an “audit committee financial expert” as
defined by the SEC’s rules. Our board of directors has determined that Messrs.
Link, Selter and Wennekamp are “independent” under the current independence
standards of both The NASDAQ Capital Market and the SEC.
Section
16(a) Beneficial Ownership Reporting Compliance
Our
executive officers and directors and any person who owns more than 10% of our
outstanding shares of common stock are required under Section 16(a) of the
Securities Exchange Act to file with the SEC initial reports of ownership and
reports of changes in ownership of our common stock and to furnish us with
copies of those reports. Based solely on our review of copies of reports we have
received and written representations from certain reporting persons, we believe
that our directors and executive officers and greater than 10% shareholders for
2009 complied with all applicable Section 16(a) filing
requirements.
Code
of Ethics
We have
adopted a Code of Ethics applicable to all employees, including our principal
executive officer, principal financial officer, and principal accounting officer
or controller, a copy of which is available on our website at www.cytrx.com. We
will furnish, without charge, a copy of our Code of Ethics upon request. Such
requests should be directed to Attention: Corporate Secretary, 11726 San Vicente
Boulevard, Suite 650, Los Angeles, California, or by telephone at
310-826-5648.
Board
Leadership Structure
Our Board
has placed the responsibilities of Chairman with an independent nonexecutive
member of the Board, which we believe provides better accountability between the
Board and our management team. We believe it is beneficial to have an
independent Chairman whose sole responsibility to us is guiding our Board
members as they provide leadership to our executive team. Our
Chairman is responsible for communication among the directors; setting the Board
meeting agendas in consultation with the President and Chief Executive Officer;
and presiding at Board meetings, executive sessions and stockholder
meetings. This delineation of duties allows the President and Chief
Executive Officer to focus his attention on managing the day-to-day business of
the company. We believe this structure provides strong leadership for
our Board, while positioning our President and Chief Executive Officer as the
leader of the company in the eyes of our employees and other
stakeholders.
47
Board
of Directors Role in Risk Oversight
In
connection with its oversight responsibilities, our board of directors,
including the Audit Committee, periodically assesses the significant risks that
we face. These risks include, but are not limited to, financial,
technological, competitive, and operational risks. Our board of directors
administers its risk oversight responsibilities through our Chief Executive
Officer and Chief Financial Officer, who review and assess the operations of our
business as well as operating management’s identification, assessment and
mitigation of the material risks affecting our operations.
Item
11. EXECUTIVE
COMPENSATION
Compensation
Discussion and Analysis
Overview
of Executive Compensation Program
The
Compensation Committee of our board of directors has responsibility for
establishing, implementing and monitoring our executive compensation program
philosophy and practices. The Compensation Committee seeks to ensure that the
total compensation paid to our named executive officers is fair, reasonable and
competitive. Generally, the types of compensation and benefits provided to the
named executive officers are similar to those provided to our other
officers.
Throughout
this Annual Report, the individuals included in the Summary Compensation Table
on page 53 are referred to as the “named executive officers.”
Compensation
Philosophy and Objectives
The
components of our executive compensation consist of salary, annual cash bonuses
awarded based on the Compensation Committee’s subjective assessment of each
individual executive’s job performance, including evaluations of, during the
past year, stock option grants to provide executives with longer-term
incentives, and occasional special compensation awards (either cash, stock or
stock options) to reward extraordinary efforts or results.
The
Compensation Committee believes that an effective executive compensation program
should provide base annual compensation that is reasonable in relation to
individual executive’s job responsibilities and reward the achievement of both
annual and long-term strategic goals of our company. The Compensation Committee
uses annual and other periodic cash bonuses to reward an officer’s achievement
of specific goals, including goals related to the development of the product’s
drug candidates and management of working capital, and employee stock options as
a retention tool and as a means to align the executive’s long-term interests
with those of our stockholders, with the ultimate objective of affording our
executives an appropriate incentive to improve stockholder value. The
Compensation Committee evaluates both performance and compensation to maintain
our company’s ability to attract and retain excellent employees in key positions
and to assure that compensation provided to key employees remains competitive
relative to the compensation paid to similarly situated executives of comparable
companies. To that end, the Compensation Committee believes executive
compensation packages provided by us to our named executive officers should
include both cash compensation and stock options.
Because
of the size of our company, the small number of executive officers in our
company, and our company’s financial priorities, the Compensation Committee has
not implemented any pension benefits, deferred compensation plans, or other
similar plans for our named executive officers.
As a
biopharmaceutical company engaged in developing potential products that, to
date, have not generated significant revenues and are not expected to generate
significant revenues or profits for several years, the Compensation Committee
also takes the company’s financial and working capital condition into account in
its compensation decisions. Accordingly, the Compensation Committee recently has
weighted bonuses more heavily with stock options rather than cash. The
Compensation Committee may periodically reassess the proper weighting of equity
and cash compensation in light of the company’s working capital situation from
time to time.
48
Role
of Executive Officers in Compensation Decisions
The
Compensation Committee makes all compensation decisions for the named executive
officers and approves recommendations made by our President and Chief Executive
Officer regarding equity awards to our other officers. Decisions regarding the
non-equity compensation of our other officers are made by our President and
Chief Executive Officer.
The
Compensation Committee and the President and Chief Executive Officer annually
review the performance of each named executive officer (other than the President
and Chief Executive Officer, whose performance is reviewed only by the
Compensation Committee). The conclusions reached and recommendations based on
these reviews, including with respect to salary adjustments and annual award
amounts, are presented to the Compensation Committee. The Compensation Committee
can exercise its discretion in modifying or declining any recommended
adjustments or awards to executives.
Setting
Executive Compensation
Based on
the foregoing objectives, the Compensation Committee has structured the
company’s annual cash and incentive-based cash and non-cash executive
compensation to seek to motivate our named executives to achieve the company’s
business goals, including goals related to the development of the our drug
candidates and management of working capital, to reward the executives for
achieving such goals, and to retain the executives. In doing so, the
Compensation Committee historically has not employed outside compensation
consultants. However, during 2009, the Compensation Committee obtained two
third-party industry compensation surveys and used them in its compensation
deliberations regarding cash and equity compensation for our executive officers.
The Compensation
Committee utilized this data to set compensation for our executive officers at
levels targeted at or around the third quartile of compensation amounts provided
to executives at comparable companies considering each individual’s individual
experience level related to their position with us. There is no pre-established
policy or target for the allocation between either cash and non-cash incentive
compensation.
2009
Executive Compensation Components
For 2009,
the principal components of compensation for the named executive officers
were:
|
|
·
|
base
salary;
|
|
|
·
|
annual
and special bonuses; and
|
|
|
·
|
equity
incentive compensation.
|
Base
Salary
The
Company provides named executive officers and other employees with base salary
to compensate them for services rendered during the year. Base salary ranges for
the named executive officers are determined for each named executive officer
based on his position and responsibility.
During
its review of base salaries for executives, the Compensation Committee primarily
considers:
|
|
·
|
the
negotiated terms of each executive’s employment agreement, if
any;
|
|
|
·
|
an
internal review of the executive’s compensation, both individually and
relative to other named executive
officers;
|
|
|
·
|
each
executive’s individual performance;
and
|
|
|
·
|
base
salaries paid by comparable
companies.
|
Salary
levels are typically considered annually as part of the company’s performance
review process, as well as upon a change in job responsibility. Merit-based
increases to salaries are based on the company’s available resources and the
Compensation Committee’s assessment of the individual’s performance. Both
assessments are based upon written evaluations of such criteria as job
knowledge, communication, problem solving, initiative, goal-setting, and expense
management. Base salaries for the named executive officers in 2009 were
increased from the base salaries in effect during the prior year by amounts
ranging from 15% for our Vice President – Legal Affairs and our Senior Vice
President – Drug Development, to 18% for our President and Chief Executive
Officer and our Chief Financial Officer.
49
Annual
and Special Bonuses
The
Compensation Committee has not established an incentive compensation program
with fixed performance targets. Because we do not generate significant revenues
and have not commercially released any products, the Compensation Committee
bases its discretionary compensation awards on the achievement of product
development targets and milestones, efforts related to extraordinary
transactions, effective fund-raising efforts, and effective management of
personnel and capital resources, among other criteria. During 2009, the
Compensation Committee granted Mr. Kriegsman an annual cash bonus of $450,000,
and granted cash bonuses to the other named executive officers ranging from $0
to $80,000, principally based on their efforts in helping us advance the
development of our products and raise capital.
Equity
Incentive Compensation
As
indicated above, the Compensation Committee also aims to encourage the company’s
executive officers to focus on long-term company performance by allocating to
them stock options that vest over a period of several years. In 2009, the
Compensation Committee granted to Mr. Kriegsman nonqualified options to purchase
750,000 shares of our common stock at a price of $1.05 per share, which equaled
the closing market price on the date of grant. The option vests monthly over
three years, provided that Mr. Kriegsman continues in our employ through such
monthly vesting periods. In addition, in connection with the annual review of
our other named executive officers, the Compensation Committee also granted
stock options to those named executive officers. All of these other stock
options had an exercise price equal to the closing market price on the date of
grant, and also vest monthly over three years, provided that such executives
remain in our employ through such monthly vesting periods.
Retirement
Plans, Perquisites and Other Personal Benefits
We have
adopted a tax-qualified employee savings and retirement plan, the 401(k) Plan,
for eligible U.S. employees, including our named executive officers. Eligible
employees may elect to defer a percentage of their eligible compensation in the
401(k) Plan, subject to the statutorily prescribed annual limit. We may make
matching contributions on behalf of all participants in the 401(k) Plan in an
amount determined by our board of directors. We did not make any matching
contribution to the 401(k) Plan for 2009. Matching and
profit-sharing contributions, if any, are subject to a vesting schedule; all
other contributions are at all times fully vested. We intend the 401(k) Plan,
and the accompanying trust, to qualify under Sections 401(k) and 501 of the
Internal Revenue Code so that contributions by employees to the 401(k) Plan, and
income earned (if any) on plan contributions, are not taxable to employees until
withdrawn from the 401(k) Plan, and so that we will be able to deduct our
contributions, if any, when made. The trustee under the 401(k) Plan, at the
direction of each participant, may invest the assets of the 401(k) Plan in any
of a number of investment options.
We do not
provide any of our executive officers with any other perquisites or personal
benefits, other than benefits to Mr. Kriegsman provided for in his
employment agreement. As required by his employment agreement, during 2009 we
paid insurance premiums with respect to a life insurance policy for Mr.
Kriegsman which had a face value of approximately $1.4 million as of December
31, 2009 and under which Mr. Kriegsman’s designee is the
beneficiary.
Employment
Agreements and Severance Arrangements
We have
entered into written employment agreements with each of our named executive
officers. The main purpose of these agreements is to protect the company from
business risks such as competition for the executives’ service, loss of
confidentiality or trade secrets, and solicitation of our other employees, and
to define our right to terminate the employment relationship. The employment
agreements also protect the executive from termination without “cause” (as
defined) and, in Mr. Kriegsman’s case, entitles him to resign for “good reason”
(as defined). Each employment agreement was individually negotiated, so there
are some minor variations in the terms among executive officers. Generally
speaking, however, the employment agreements provide for termination and
severance benefits that the Compensation Committee believes are consistent with
industry practices for similarly situated executives. The Compensation Committee
believes that the termination and severance benefits help the company retain the
named executive officers by providing them with a competitive employment
arrangement and protection against unknowns such as termination without “cause”
that go along with the position.
50
In the
event of termination without “cause,” the named executive officers will be
entitled to a lump-sum payment equal to six months of base salary (24 months in
the case of Mr. Kriegsman). Mr. Kriegsman’s employment agreement also provides
for our continuation of Mr. Kriegsman’s life insurance and medical benefits
during his 24-month severance period. If Mr. Kriegsman’s employment is
terminated by us without “cause,” or by Mr. Kriegsman for “good reason,” within
two years following a change of control of CytRx, he also would be entitled
under his employment agreement to receive a “gross-up” payment equal to the sum
of any excise tax on his termination benefits (including any accelerated vesting
of his options under our Plans as described below) plus any penalties and
interest.
Change
of Control Arrangements
The
company’s 2000 Long-Term Incentive Plan and 2008 Stock Incentive Plan provide
generally that, upon a change of control of CytRx, all unvested stock options
and awards under the Plans held by plan participants, including the named
executive officers, will become immediately vested and exercisable immediately
prior to the effective date of the transaction. The Compensation Committee
believes that such “single trigger” change of control policy is consistent with
the objective of aligning the interests of the named executive officer’s and of
the company’s stockholders by allowing the executives to participate equally
with stockholders in the event of a change of control transaction.
The
foregoing severance and change of control arrangements, including the
quantification of the payment and benefits provided under these arrangements,
are described in more detail elsewhere in this Annual Report under the heading
“Executive Compensation – Potential Payments Upon Termination or Change of
Control.”
Ownership
Guidelines
The
Compensation Committee has no requirement that each named executive officer
maintain a minimum ownership interest in our company.
Our
long-term incentive compensation consists solely of periodic grants of stock
options to our named executive officers. The stock option program:
|
|
·
|
links
the creation of stockholder value with executive
compensation;
|
|
|
·
|
provides
increased equity ownership by
executives;
|
|
|
·
|
functions
as a retention tool, because of the vesting features included in all
options granted by the Compensation Committee;
and
|
|
|
·
|
maintains
competitive levels of total
compensation.
|
We
normally grant stock options to new executive officers when they join our
company based upon their position with us and their relevant prior experience.
The options granted by the Compensation Committee generally vest monthly over
the first three years of the ten-year option term. Vesting and exercise rights
cease upon termination of employment (or, in the case of exercise rights, 90
days thereafter), except in the case of death (subject to a one-year
limitation), disability or retirement. Prior to the exercise of an option, the
holder has no rights as a stockholder with respect to the shares subject to such
option, including voting rights and the right to receive dividends or dividend
equivalents. In addition to the initial option grants, our Compensation
Committee may grant additional options to retain our executives and reward, or
provide incentive for, the achievement of corporate goals and strong individual
performance. Our board of directors has granted our President and Chief
Executive Officer discretion to grant up to 100,000 options to employees upon
joining our company, and to make grants from an additional “discretionary pool”
of up to 100,000 options during each annual employee review cycle. Options are
granted based on a combination of individual contributions to our company and on
general corporate achievements, which may include the attainment of product
development milestones (such as commencement and completion of clinical trials)
and attaining other annual corporate goals and objectives. On an annual basis,
the Compensation Committee assesses the appropriate individual and corporate
goals for our executives and provides additional option grants based upon the
achievement by the new executives of both individual and corporate goals. We
expect that we will continue to provide new employees with initial option grants
in the future to provide long-term compensation incentives and will continue to
rely on performance-based and retention grants to provide additional incentives
for current employees. Additionally, in the future, the Compensation Committee
may consider awarding additional or alternative forms of equity incentives, such
as grants of bonus stock, restricted stock and restricted stock
units.
51
It is our
policy to award stock options at an exercise price equal to The NASDAQ Capital
Market’s closing price of our common stock on the date of the grant. In certain
limited circumstances, the Compensation Committee may grant options to an
executive at an exercise price in excess of the closing price of the common
stock on the grant date. The Compensation Committee has never granted options
with an exercise price that is less than the closing price of our common stock
on the grant date, nor has it granted options which are priced on a date other
than the grant date. For purposes of determining the exercise price of stock
options, the grant date is deemed to be the first day of employment for newly
hired employees, or the date on which the Compensation Committee or the Chief
Executive Officer, as applicable, approves the stock option grant to existing
employees.
We have
no program, practice or plan to grant stock options to our executive officers,
including new executive officers, in coordination with the release of material
nonpublic information. We also have not timed the release of material nonpublic
information for the purpose of affecting the value of stock options or other
compensation to our executive officers, and we have no plan to do so. We have no
policy regarding the adjustment or recovery of stock option awards in connection
with the restatement of our financial statements, as our stock option awards
have not been tied to the achievement of specific financial goals.
Tax
and Accounting Implications
Deductibility
of Executive Compensation
As part
of its role, the Compensation Committee reviews and considers the deductibility
of executive compensation under Section 162(m) of the Internal Revenue Code,
which provides that corporations may not deduct compensation of more than
$1,000,000 that is paid to certain individuals. We believe that compensation
paid to our executive officers generally is fully deductible for federal income
tax purposes.
Accounting
for Share-Based Compensation
Beginning
on January 1, 2006, we began accounting for share-based compensation in
accordance with the requirements of FASB Statement 123(R), Share-Based Payment. This
accounting treatment has not significantly affected our compensation decisions.
The Compensation Committee takes into consideration the tax consequences of
compensation to the named executive officers, but tax considerations are not a
significant part of the company’s compensation policy.
Benchmarking
The
Compensation Committee does not attempt to establish or measure executive
compensation against any benchmarks. With certain exceptions, our company’s
compensation policies are not related specifically to our company’s performance,
which is just one of the factors considered by us and our Compensation Committee
in establishing base salaries and awarding discretionary
compensation. We have not established any policy regarding
recoupment, or “clawback,” of any performance-based compensation in the event
our company’s historical performance is subsequently revised or restated in a
way that would have produced a lower compensation amount. We also
have not relied upon wealth accumulation analyses, or “tally sheets,” or
internal pay equity analyses in making executive compensation
decisions.
Compensation
Committee Interlocks and Insider Participation in Compensation
Decisions
There are
no “interlocks,” as defined by the SEC, with respect to any member of the
Compensation Committee. Max Link, Ph.D., Marvin R. Selter and Richard L.
Wennekamp all served as members of the Compensation Committee during
2009.
Compensation
Committee Report
The
Compensation Committee has reviewed and discussed with management the
“Compensation Discussion and Analysis” required by Item 402(b) of Regulation S-K
and, based on such review and discussions, has recommended to our board of
directors that the foregoing “Compensation Discussion and Analysis” be included
in this Annual Report.
|
Richard
L. Wennekamp, Chairman
|
Marvin
R. Selter
|
Dr.
Max Link
|
52
Summary
Compensation Table
The
following table presents summary information concerning all compensation paid or
accrued by us for services rendered in all capacities during 2009, 2008 and 2007
by Steven A. Kriegsman and John Y. Caloz, who are the only individuals who
served as our principal executive and financial officers during the year ended
December 31, 2009, and our three other most highly compensated executive
officers who were serving as executive officers as of December 31,
2009:
Summary
Compensation Table
|
Name and Principal Position
|
Year
|
Salary ($)
|
Bonus
($)(1)
|
Option
Awards
($) (2)
|
All
Other
Compensation ($)(3)
|
Total
($)
|
|||||||||||||||
|
Steven
A. Kriegsman
|
|||||||||||||||||||||
|
President
and Chief Executive Officer
|
2009
|
550,000 | 450,000 | 906,000 | 10,000 | 1,916,000 | |||||||||||||||
|
2008
|
551,000 | 150,000 | 517,800 | 10,000 | 1,228,800 | ||||||||||||||||
|
2007
|
524,767 | 300,000 | 1,328,600 | — | 2,153,367 | ||||||||||||||||
|
John
Y. Caloz
|
|||||||||||||||||||||
|
Chief
Financial Officer and Treasurer
|
2009
|
275,000 | 80,000 | 137,750 | — | 492,750 | |||||||||||||||
|
Daniel
Levitt, M.D., M.D., Ph.D.
|
|||||||||||||||||||||
|
Chief
Medical Officer
|
2009
|
83,894 | — | 405,000 | — | 488,894 | |||||||||||||||
|
Benjamin
S. Levin General Counsel,
|
|||||||||||||||||||||
|
General
Counsel, Vice President — Legal
|
2009
|
276,000 | 75,000 | 119,100 | — | 470,100 | |||||||||||||||
|
Affairs and
Secretary
|
2008
|
276,000 | 55,000 | 125,300 | — | 456,300 | |||||||||||||||
|
2007
|
250,000 | 100,000 | 407,000 | — | 730,000 | ||||||||||||||||
|
Scott
Wieland, Ph.D.
|
|||||||||||||||||||||
|
Senior
Vice President – Drug Development
|
2009
|
275,000 | 75,000 | 105,750 | — | 455,750 | |||||||||||||||
|
2008
|
255,500 | 73,250 | 28,580 | — | 372,480 | ||||||||||||||||
|
2007
|
134,000 | 35,000 | 447,925 | — | 587,750 | ||||||||||||||||
____________
|
(1)
|
Bonuses
to the named executive officers reported above relating to 2009 were paid
in December 2009. Bonuses to the named executive officers reported above
relating to 2008 were paid in December 2008. Bonuses to the named
executive officers reported above relating to 2007 were paid in April
2008.
|
|
(2)
|
The
values shown in this column represent the aggregate grant date fair value
of equity-based awards granted during the fiscal year, in accordance with
ASC 718, “Share
Based-Payment”. At the 2009 Annual Meeting of
Stockholders held on July 1, 2009, the Company’s stockholders approved an
amendment to the Company’s 2000 Long-Term Incentive Plan to allow for a
one-time stock option re-pricing program for employees and
officers. Pursuant to the re-pricing program, 3,265,500
eligible stock options held by ten eligible employees and officers were
amended to reduce the exercise prices of the options to $1.15 per share,
which was the closing sale price of CytRx’s common stock as reported on
The NASDAQ Capital Market on the July 1, 2009 completion date of the
re-pricing program, and to impose a new option vesting
schedule. The values in this column include the
incremental increase in the fair value of the re-priced options
over the fair value of the original award. The fair value of the stock
options at the date of grant was estimated using the Black-Scholes
option-pricing model, based on the assumptions described in Note 15 of the
Notes to Financial Statements included in this Annual
Report.
|
|
(3)
|
This
amount represents life insurance
premiums.
|
53
2009
Grants of Plan-Based Awards
In 2009,
we granted stock options to our named executive officers under our 2000
Long-Term Incentive Plan and 2008 Stock Incentive Plan as follows:
2008
Grants of Plan-Based Awards
|
Name
|
Grant Date
|
All
Other
Option
Awards
(#
of CytRx
Shares)
|
Exercise
Price of
Option
Awards
($/Share)
|
Grant
Date
Fair
Value of
Option
Awards
($)
|
||||||||||
|
Steven
A. Kriegsman
|
12/10/2009
|
750,000 | $ | 1.05 | $ | 609,000 | ||||||||
|
President
and Chief Executive Officer
|
||||||||||||||
|
John
Y. Caloz
|
12/10/2009
|
125,000 | $ | 1.05 | $ | 101,500 | ||||||||
|
Chief
Financial Officer and Treasurer
|
1/2/2009
|
50,000 | 0.30 | 11,750 | ||||||||||
|
Daniel
Levitt, M.D., Ph.D.
|
10/12/2009
|
500,000 | $ | 1.06 | $ | 405,000 | ||||||||
|
Chief
Medical Officer
|
||||||||||||||
|
Benjamin
S. Levin
|
12/10/2009
|
100,000 | $ | 1.05 | $ | 81,200 | ||||||||
|
General
Counsel, Vice President — Legal Affairs and Secretary
|
||||||||||||||
|
Scott
Wieland, Ph.D.
|
12/10/2009
|
100,000 | $ | 1.05 | $ | 81,200 | ||||||||
|
Senior
Vice President – Drug Development
|
||||||||||||||
____________
2000
Long-Term Incentive Plan and 2008 Stock Incentive Plan
The
purpose of our 2000 Long-Term Incentive Plan, or 2000 Plan, and our 2008 Stock
Incentive Plan, or 2008 Plan, is to promote our success and enhance our value by
linking the personal interests of our employees, officers, consultants and
directors to those of our stockholders. The 2000 Plan was originally adopted by
our Board of Directors on August 24, 2000 and by our stockholders on June 7,
2001, with certain amendments to the Plan having been subsequently approved by
our Board of Directors and stockholders. On May 11, 2009, our Board of Directors
approved an amendment to the 2000 Plan to allow for a one-time stock option
re-pricing program for our employees. The 2008 Plan was adopted by
our Board of Directors on November 21, 2008 and by our stockholders on July 1,
2009.
2000
Plan and 2008 Plan Descriptions
The 2000
Plan and the 2008 Plan, or the Plans, are administered by the Compensation
Committee of our Board of Directors. The Compensation Committee has the power,
authority and discretion to:
|
|
·
|
designate
participants;
|
|
|
·
|
determine
the types of awards to grant to each participant and the number, terms and
conditions of any award;
|
|
|
·
|
establish,
adopt or revise any rules and regulations as it may deem necessary or
advisable to administer the Plan;
and
|
|
|
·
|
make
all other decisions and determinations that may be required under, or as
the Compensation Committee deems necessary or advisable to administer, the
Plan.
|
Awards
The
following is summary description of financial instruments that may be granted to
participants by the Compensation Committee of our Board of Directors. The
Compensation Committee to date has only granted stock options to participants in
the Plans.
54
Stock Options. The
Compensation Committee is authorized to grant both incentive stock options and
non-qualified stock options. The terms of any incentive stock option must meet
the requirements of Section 422 of the Internal Revenue Code. The exercise price
of an option may not be less than the fair market value of the underlying stock
on the date of grant, and no option may have a term of more than 10 years from
the grant date.
Stock Appreciation Rights.
The Compensation Committee may grant stock appreciation rights to participants.
Upon the exercise of a stock appreciation right, the participant has the right
to receive the excess, if any, of (1) the fair market value of one share of
common stock on the date of exercise, over (2) the grant price of the stock
appreciation right as determined by the Compensation Committee, which will not
be less than the fair market value of one share of common stock on the date of
grant.
Restricted Stock. The
Compensation Committee may make awards of restricted stock, which will be
subject to such restrictions on transferability and other restrictions as the
Compensation Committee may impose (including limitations on the right to vote
restricted stock or the right to receive dividends, if any, on the restricted
stock).
Performance Units. The
Compensation Committee may grant under the 2000 Plan performance units on such
terms and conditions as may be selected by the Compensation Committee. The
Compensation Committee will have the complete discretion to determine the number
of performance units granted to each participant and to set performance goals
and other terms or conditions to payment of the performance units which,
depending on the extent to which they are met, will determine the number and
value of performance units that will be paid to the participant.
Dividend Equivalents. The
Compensation Committee is authorized to grant under the 2000 Plan dividend
equivalents to participants subject to such terms and conditions as may be
selected by the Compensation Committee. Dividend equivalents entitle the
participant to receive payments equal to dividends with respect to all or a
portion of the number of shares of common stock subject to an option or other
award, as determined by the Compensation Committee. The Compensation Committee
may provide that dividend equivalents be paid or distributed when accrued or be
deemed to have been reinvested in additional shares of common stock, or
otherwise reinvested.
Other Stock-Based Awards. The
Compensation Committee may grant other awards under the 2000 Plan that are
payable in, valued in whole or in part by reference to, or otherwise based on or
related to shares of common stock, as deemed by the Compensation Committee to be
consistent with the purposes of the 2000 Plan. These stock-based awards may
include shares of common stock awarded as a bonus and not subject to any
restrictions or conditions, convertible or exchangeable debt securities, other
rights convertible or exchangeable into shares of common stock, and awards
valued by reference to book value of shares of common stock or the value of
securities of or the performance of our subsidiaries. The Compensation Committee
will determine the terms and conditions of any such awards.
Performance Goals. The
Compensation Committee in its discretion may determine awards under the 2000
Plan based on:
|
|
·
|
the
achievement by CytRx or a parent or subsidiary of a specific financial
target;
|
|
|
·
|
CytRx’s
stock price;
|
|
|
·
|
the
achievement by an individual or a business unit of CytRx or a subsidiary
of a specific financial target;
|
|
|
·
|
the
achievement of specific goals with respect to (i) product development
milestones, (ii) corporate financings, (iii) merger and acquisition
activities, (iv) licensing transactions, (v) development of strategic
partnerships or alliances, or (vi) acquisition or development of new
technologies; and
|
|
|
·
|
any
combination of the goals set forth
above.
|
The
Compensation Committee has the right for any reason to reduce (but not increase)
any award, even if a specific goal has been achieved. If an award is made on the
basis of the achievement of a goal, the Compensation Committee must have
established the goal before the beginning of the period for which the
performance goal relates (or a later date as may be permitted under Internal
Revenue Code Section 162(m)). Any payment of an award for achieving a goal will
be conditioned on the written certification of the Compensation Committee in
each case that the goals and any other material conditions were
satisfied.
55
Limitations on Transfer;
Beneficiaries. Awards under the Plans may not be transferred or assigned
by participants other than by will or the laws of descent and distribution and,
in the case of an incentive stock option, pursuant to a qualified domestic
relations order, provided that the Compensation Committee may (but need not)
permit other transfers where the Compensation Committee concludes that such
transferability (1) does not result in accelerated taxation, (2) does not cause
any option intended to be an incentive stock option to fail to qualify as such,
and (3) is otherwise appropriate and desirable, taking into account any factors
deemed relevant, including any state or federal tax or securities laws or
regulations applicable to transferable awards. A participant may, in the manner
determined by the Compensation Committee, designate a beneficiary to exercise
the participant’s rights and to receive any distribution with respect to any
award upon the participant’s death.
Acceleration Upon Certain
Events. In the event of a “Change in Control” of CytRx, which is a term
defined in the Plans, all outstanding options and other awards in the nature of
rights that may be exercised will become fully vested and exercisable and all
restrictions on all outstanding awards will lapse. The Compensation Committee
may, however, in its sole discretion declare all outstanding options, stock
appreciation rights and other awards in the nature of rights that may be
exercised to become fully vested and exercisable, and all restrictions on all
outstanding awards to lapse, in each case as of such date as the Compensation
Committee may, in its sole discretion, declare. The Compensation Committee may
discriminate among participants or among awards in exercising such
discretion.
Termination
and Amendment
Our Board
of Directors or the Compensation Committee may, at any time and from time to
time, terminate or amend the Plans without stockholder approval; provided,
however, that our board or the Compensation Committee may condition any
amendment on the approval of our stockholders if such approval is necessary or
deemed advisable with respect to tax, securities or other applicable laws,
policies or regulations. No termination or amendment of the Plans may adversely
affect any award previously granted without the written consent of the
participants affected. The Compensation Committee may amend any outstanding
award without the approval of the participants affected, except that no such
amendment may diminish the value of an award determined as if it has been
exercised, vested, cashed in or otherwise settled on the date of such amendment,
and, except as otherwise permitted in the Plan, the exercise price of any option
may not be reduced and the original term of any option may not be
extended.
56
Holdings
of Previously Awarded Equity
Equity
awards held as of December 31, 2009 by each of our named executive officers were
issued under our 2000 Plan, except for the most recent equity award to Mr.
Kriegsman, which was issued under our 2008 Plan. The following table sets forth
outstanding equity awards held by our named executive officers as of December
31, 2009:
2009
Outstanding Equity Awards at Fiscal Year-End
|
|
Option Awards
|
||||||||||||||||
|
|
Number
of Securities
Underlying
Unexercised
Options
(#)
|
Option
Exercis
Price (3)
|
Option
Expiration
|
||||||||||||||
|
Name
|
Exercisable
|
Unexercisable
|
($)
|
Date
|
|||||||||||||
|
Steven
A. Kriegsman
|
— | (1 | ) | 750,000 | 1.05 |
12/10/19
|
|||||||||||
|
President
and Chief Executive Officer
|
99,972 | (1 | ) | 200,028 | 0.37 |
11/21/18
|
|||||||||||
| 250,056 | (1 | ) | 199,944 | 1.15 |
4/07/18
|
||||||||||||
| 252,805 | (1 | ) | 97,195 | 1.15 |
4/18/17
|
||||||||||||
| 200,000 | (1 | ) | — | 1.15 |
6/16/16
|
||||||||||||
| 300,000 | (1 | ) | — | 0.79 |
5/17/15
|
||||||||||||
| 250,000 | (2 | ) | — | 1.15 |
6/19/13
|
||||||||||||
| 750,000 | (2 | ) | — | 1.15 |
6/20/13
|
||||||||||||
|
John
Y. Caloz
|
— | (1 | ) | 125,000 | 1.05 |
12/10/19
|
|||||||||||
|
Chief
Financial Officer and Treasurer
|
15,288 | (1 | ) | 34,713 | 0.30 |
01/02/19
|
|||||||||||
| 8,335 | (2 | ) | 16,665 | 1.15 |
04/07/18
|
||||||||||||
| 8,335 | (2 | ) | 16,665 | 1.15 |
12/06/17
|
||||||||||||
| 25,005 | (2 | ) | 49,995 | 1.15 |
10/26/17
|
||||||||||||
|
Daniel
Levitt, M.D., Ph.D.
|
27,910 | (1 | ) | 472,090 | 1.06 |
11/21/18
|
|||||||||||
|
Chief
Medical Officer
|
|||||||||||||||||
|
Benjamin
S. Levin
|
— | (1 | ) | 100,000 | 1.05 |
12/10/19
|
|||||||||||
|
General
Counsel, Vice President — Legal
|
36,100 | (1 | ) | 63,900 | 0.37 |
11/21/18
|
|||||||||||
|
Affairs
and Secretary
|
30,575 | (1 | ) | 69,425 | 1.15 |
4/07/18
|
|||||||||||
| 72,230 | (1 | ) | 27,770 | 1.15 |
4/18/17
|
||||||||||||
| 90,000 | (1 | ) | — | 1.15 |
6/16/16
|
||||||||||||
| 150,000 | (1 | ) | — | 0.79 |
5/17/15
|
||||||||||||
| 160,000 | (2 | ) | — | 1.15 |
7/15/14
|
||||||||||||
|
Scott
Wieland, Ph.D.
|
— | (1 | ) | 100,000 | 1.05 |
12/10/19
|
|||||||||||
|
Senior
Vice President – Drug Development
|
8,335 | (2 | ) | 16,665 | 1.15 |
11/21/18
|
|||||||||||
| 8,335 | (2 | ) | 16,665 | 1.15 |
12/06/17
|
||||||||||||
| 50,000 | (2 | ) | 25,000 | 1.15 |
4/30/17
|
||||||||||||
____________
|
(1)
|
These
options vest in 36 equal monthly installments, subject to the option
holder’s remaining in our continuous employ through such
dates.
|
|
(2)
|
These
options vest in three annual installments, subject to the option holder’s
remaining in our continuous employ through such
dates.
|
|
|
(3)
The reported options with prices of $1.15 were re-priced to that exercise
price on July 1, 2009 at $1.15.
|
Option
Exercises and Stock Vested
There
were no exercises of stock options by any of our named executive officers during
2009.
57
Employment
Agreements and Potential Payment upon Termination or Change in
Control
Employment
Agreement with Steven A. Kriegsman
Mr.
Kriegsman is employed as our Chief Executive Officer and President pursuant to
an employment agreement that was amended as of May 2009 to continue through
December 31, 2012. The employment agreement will automatically renew in December
2012 for an additional one-year period, unless either Mr. Kriegsman or we elect
not to renew it.
Under his
employment agreement as amended, Mr. Kriegsman is entitled to receive an annual
base salary of $650,000. Our board of directors (or its Compensation Committee)
will review the base salary annually and may increase (but not decrease) it in
its sole discretion. In addition to his annual salary, Mr. Kriegsman is eligible
to receive an annual bonus as determined by our board of directors (or its
Compensation Committee) in its sole discretion, but not to be less than
$150,000. Pursuant to his employment agreement with us, we have agreed that he
shall serve on a full-time basis as our Chief Executive Officer and President
and that he may continue to serve as Chairman of the Kriegsman Group only so
long as necessary to complete certain current assignments.
Mr.
Kriegsman is eligible to receive grants of options to purchase shares of our
common stock. The number and terms of those options, including the vesting
schedule, will be determined by our board of directors (or its Compensation
Committee) in its sole discretion.
Under Mr.
Kriegsman’s employment agreement, we have agreed that, if he is made a party, or
threatened to be made a party, to a suit or proceeding by reason of his service
to us, we will indemnify and hold him harmless from all costs and expenses to
the fullest extent permitted or authorized by our certificate of incorporation
or bylaws, or any resolution of our board of directors, to the extent not
inconsistent with Delaware law. We also have agreed to advance to Mr. Kriegsman
such costs and expenses upon his request if he undertakes to repay such advances
if it ultimately is determined that he is not entitled to indemnification with
respect to the same. These employment agreement provisions are not exclusive of
any other rights to indemnification to which Mr. Kriegsman may be entitled and
are in addition to any rights he may have under any policy of insurance
maintained by us.
In the
event we terminate Mr. Kriegsman’s employment without “cause” (as defined), or
if Mr. Kriegsman terminates his employment with “good reason” (as defined), (i)
we have agreed to pay Mr. Kriegsman a lump-sum equal to his salary and prorated
minimum annual bonus through to his date of termination, plus his salary and
minimum annual bonus for a period of two years after his termination date, or
until the expiration of the amended and restated employment agreement, whichever
is later, (ii) he will be entitled to immediate vesting of all stock options or
other awards based on our equity securities, and (iii) he will also be entitled
to continuation of his life insurance premium payments and continued
participation in any of our health plans through to the later of the expiration
of the amended and restated employment agreement or 24 months following his
termination date. Mr. Kriegsman will have no obligation in such events to seek
new employment or offset the severance payments to him by any compensation
received from any subsequent reemployment by another employer.
Under Mr.
Kriegsman’s employment agreement, he and his affiliated company, The Kriegsman
Group, are to provide us during the term of his employment with the first
opportunity to conduct or take action with respect to any acquisition
opportunity or any other potential transaction identified by them within the
biotech, pharmaceutical or health care industries and that is within the scope
of the business plan adopted by our board of directors. Mr. Kriegsman’s
employment agreement also contains confidentiality provisions relating to our
trade secrets and any other proprietary or confidential information, which
provisions shall remain in effect for five years after the expiration of the
employment agreement with respect to proprietary or confidential information and
for so long as our trade secrets remain trade secrets.
Potential
Payment upon Termination or Change in Control for Steven A.
Kriegsman
Mr.
Kriegsman’s employment agreement contains no provision for payment to him in the
event of a change in control of CytRx. If, however, a change in control (as
defined in our 2000 Long-Term Incentive Plan) occurs during the term of the
employment agreement, and if, during the term and within two years after the
date on which the change in control occurs, Mr. Kriegsman’s employment is
terminated by us without cause or by him for good reason (each as defined in his
employment agreement), then, in addition to the severance benefits described
above, to the extent that any payment or distribution of any type by us to or
for the benefit of Mr. Kriegsman resulting from the termination of his
employment is or will be subject to the excise tax imposed under Section 4999 of
the Internal Revenue Code of 1986, as amended, we have agreed to pay Mr.
Kriegsman, prior to the time the excise tax is payable with respect to any such
payment (through withholding or otherwise), an additional amount that, after the
imposition of all income, employment, excise and other taxes, penalties and
interest thereon, is equal to the sum of (i) the excise tax on such payments
plus (ii) any penalty and interest assessments associated with such excise
tax.
58
Employment
Agreement with Daniel Levitt, M.D., Ph.D.
Daniel
Levitt is employed as our Chief Medical Officer pursuant to an employment
agreement dated as of October 12, 2009 that expires on December 31, 2010. Dr.
Levitt is entitled under his employment agreement to receive an annual base
salary of $375,000 and is eligible to receive an annual bonus as determined by
our board of directors (or our Compensation Committee) in its sole discretion,
but not to be less than 25% of his 2010 base salary. As an incentive to enter
into his employment agreement, we granted Dr. Levitt a ten-year non-qualified
stock option under our 2000 Long-Term Incentive Plan to purchase up to 500,000
shares of our common stock at an exercise price of $1.06 per share, which
equaled the market price of our common stock on October 12, 2009 as reported in
The NASDAQ Capital Market. The option will vest ratably in 36 equal
monthly installments commencing on the first monthly anniversary of the grant
date and continuing on each successive monthly anniversary of the grant date
until the option becomes fully vested, subject to Mr. Geyer remaining in our
continuous employ through such monthly vesting periods.
In the
event we terminate Dr. Levitt’s employment without cause (as defined), we have
agreed to pay him a lump-sum equal to his accrued but unpaid salary and
vacation, plus an amount equal to six months’ salary under his employment
agreement.
Employment
Agreement with John Y. Caloz
John Y.
Caloz is employed as our Chief Financial Officer and Treasurer pursuant to an
employment agreement dated as of January 1, 2010 that expires on December 31,
2010. Mr. Caloz is entitled under his employment agreement to receive an annual
base salary of $325,000 and is eligible to receive an annual bonus as determined
by our board of directors (or our Compensation Committee) in its sole
discretion.
In the
event we terminate Mr. Caloz’s employment without cause (as defined), we have
agreed to pay him a lump-sum equal to his accrued but unpaid salary and
vacation, plus an amount equal to six months’ salary under his employment
agreement.
Employment
Agreement with Scott Wieland, Ph.D.
Scott
Wieland is employed as our Senior Vice President — Drug Development pursuant to
an employment agreement dated as of January 1, 2010 that expires on December 31,
2010. Dr. Wieland is paid an annual base salary of $315,000 and is eligible to
receive an annual bonus as determined by our board of directors (or our
Compensation Committee) in its sole discretion.
In the
event we terminate Dr. Wieland’s employment without “cause” (as defined), we
have agreed to pay him a lump-sum equal to his accrued but unpaid salary and
vacation, plus an amount equal to six months’ base salary.
Employment
Agreement with Benjamin S. Levin
Benjamin
S. Levin is employed as our Vice President — Legal Affairs, General Counsel and
Secretary pursuant to an employment agreement dated as of January 1, 2010 that
expires on December 31, 2010. Mr. Levin is paid an annual base salary of
$315,000 and is eligible to receive an annual bonus as determined by our board
of directors (or our Compensation Committee) in its sole
discretion.
In the
event we terminate Mr. Levin’s employment without “cause” (as defined), we have
agreed to pay him a lump-sum equal to his accrued but unpaid salary and
vacation, plus an amount equal to six months’ base salary.
Employment
Agreement with Scott Geyer
Scott
Geyer is employed as our Senior Vice President — Manufacturing pursuant to an
employment agreement dated as of November 30, 2009 that expires on December 31,
2010. Mr. Geyer is paid an annual base salary of $290,000 and is eligible to
receive an annual bonus as determined by our board of directors (or our
Compensation Committee) in its sole discretion. As an incentive to enter into
his employment agreement, we granted Mr. Geyer a ten-year non-qualified stock
option under our 2000 Long-Term Incentive Plan to purchase up to 150,000 shares
of our common stock at an exercise price of $0.96 per share, which equaled the
market price of our common stock on November 30, 2009 as reported in The NASDAQ
Stock Market. The option will vest ratably in36 equal monthly
installments commencing on the first monthly anniversary of the grant date and
continuing on each successive monthly anniversary of the grant date until the
option becomes fully vested, subject to Mr. Geyer remaining in our continuous
employ through such monthly vesting periods.
59
In the
event we terminate Mr. Geyer’s employment without cause (as defined), we have
agreed to pay him a lump-sum equal to his accrued but unpaid salary and
vacation, plus an amount equal to six months’ salary under his employment
agreement.
Quantification
of Termination Payments and Benefits
The table
below reflects the amount of compensation to each of our named executive
officers in the event of termination of such executive’s employment without
“cause” or his resignation for “good reason,” termination following a change in
control and termination upon the executive’s death of permanent disability. The
named executive officers are not entitled to any payments other than accrued
compensation and benefits in the event of their voluntary resignation. The
amounts shown in the table below assume that such termination was effective as
of December 31, 2009, and thus includes amounts earned through such time, and
are estimates only of the amounts that would be payable to the executives. The
actual amounts to be paid will be determined upon the occurrence of the events
indicated.
Termination
Payments and Benefits
|
|
Termination
w/o Cause or
for Good Reason
|
||||||||||||||||||||
|
Name
|
Benefit
|
Before
Change in
Control ($)
|
After
Change in
Control ($)
|
Death ($)
|
Disability ($)
|
Change
in
Control ($)
|
|||||||||||||||
|
Steven
A. Kriegsman
|
Severance
Payment(4)
|
1,100,000 | 1,100,000 | 1,100,000 | 1,100,000 | — | |||||||||||||||
|
President
and Chief
|
Stock
Options (1)
|
— | — | — | — | — | |||||||||||||||
|
Executive
Officer
|
Health
Insurance (2)
|
87,420 | 87,420 | — | 87,420 | 87,420 | |||||||||||||||
|
Life
Insurance
|
10,000 | 10,000 | — | 10,000 | — | ||||||||||||||||
|
Bonus
|
300,000 | 300,000 | 300,000 | 300,000 | — | ||||||||||||||||
|
Tax
Gross Up (3)
|
— | 0 | — | — | — | ||||||||||||||||
|
John
Y. Caloz
|
Severance
Payment(4)
|
137,500 | 137,500 | — | — | — | |||||||||||||||
|
Chief
Financial Officer
|
Stock
Options (1)
|
— | — | — | — | — | |||||||||||||||
|
Daniel
Levitt, M.D., Ph.D.
|
Severance
Payment(4)
|
187,500 | 187,500 | — | — | — | |||||||||||||||
|
Chief
Medical Officer
|
Stock
Options (1)
|
— | — | — | — | — | |||||||||||||||
|
Benjamin
S. Levin
|
Severance
Payment
|
137,500 | 137,500 | — | — | — | |||||||||||||||
|
General
Counsel, Vice
|
Stock
Options (1)
|
— | — | — | — | — | |||||||||||||||
|
President —
Legal
|
|||||||||||||||||||||
|
Affairs and
Secretary
|
|||||||||||||||||||||
|
Scott
Wieland, Ph.D.
|
Severance
Payment(4)
|
137,500 | 137,500 | — | — | — | |||||||||||||||
|
Senior
Vice President –
|
Stock
Options (1)
|
— | — | — | — | — | |||||||||||||||
|
Drug
Development
|
|||||||||||||||||||||
____________
|
(1)
|
Represents
the aggregate value of stock options that vest and become exercisable
immediately upon each of the triggering events listed as if such events
took place on December 31, 2009, determined by the aggregate difference
between the stock price as of December 31, 2009 and the exercise prices of
the underlying options.
|
|
(2)
|
Represents
the cost as of December 31, 2009 for the family health benefits provided
to Mr. Kriegsman for a period of two
years.
|
|
(3)
|
Mr.
Kriegsman’s employment agreement provides that if a change in control (as
defined in our 2000 Long-Term Incentive Plan) occurs during the term of
the employment agreement, and if, during the term and within two years
after the date on which the change in control occurs, Mr. Kriegsman’s
employment is terminated by us without “cause” or by him for “good reason”
(each as defined in his employment agreement), then, to the extent that
any payment or distribution of any type by us to or for the benefit of Mr.
Kriegsman resulting from the termination of his employment is or will be
subject to the excise tax imposed under Section 4999 of the Internal
Revenue Code of 1986, as amended, we will pay Mr. Kriegsman, prior to the
time the excise tax is payable with respect to any such payment (through
withholding or otherwise), an additional amount that, after the imposition
of all income, employment, excise and other taxes, penalties and interest
thereon, is equal to the sum of (i) the excise tax on such payments plus
(ii) any penalty and interest assessments associated with such excise tax.
Based on Mr. Kriegsman’s past compensation and the estimated payment that
would result from a termination of his employment following a change in
control, we have estimated that a gross-up payment would not be required.
“Good reason” as defined in Mr. Kriegsman’s employment agreement includes
any change in Mr. Kriegsman’s duties or title that are inconsistent with
his position as Chief Executive
Officer.
|
|
(4)
|
Severance
payments are prescribed by our employment agreements with the named
executive officers and represent a factor of their annual base
compensation ranging from six months to two
years.
|
60
Compensation of
Directors
The
following table sets forth the compensation paid to our directors other than our
Chief Executive Officer for 2009:
Director
Compensation Table
|
Name
(1)
|
Fees
Earned or
Paid
in Cash ($) (2)
|
Option
Awards
($) (3)
|
Total
($)
|
|||||||||
|
Max
Link, Ph.D.
|
132,250 | 44,400 | 176,650 | |||||||||
|
Chairman
|
||||||||||||
|
Marvin
R. Selter
|
112,250 | 44,400 | 156,650 | |||||||||
|
Vice
Chairman
|
||||||||||||
|
Louis
Ignarro, Ph.D.
|
37,500 | 44,400 | 81,900 | |||||||||
|
Director
|
||||||||||||
|
Joseph
Rubinfeld, Ph.D.
|
79,000 | 44,400 | 123,400 | |||||||||
|
Director
|
||||||||||||
|
Richard
L. Wennekamp
|
112,250 | 44,400 | 156,650 | |||||||||
|
Director
|
||||||||||||
____________
|
(1)
|
Steven
A. Kriegsman does not receive additional compensation for his role as a
Director. For information relating to Mr. Kriegsman’s compensation as
President and Chief Executive Officer, see the Summary Compensation Table
above.
|
|
(2)
|
The
amounts in this column represent cash payments made to Non-Employee
Directors for attendance at meetings during the
year.
|
|
(3)
|
In
July 2009, we granted stock options to purchase 50,000 shares of our
common stock at an exercise price equal to the current market value of our
common stock to each non-employee director, which had a grant date fair
value of $44,400 calculated in accordance with FASB ASC Topic 718,
excluding the effect of estimated forfeitures related to service-based
vesting conditions. The amount recognized for these awards was calculated
using the Black Scholes option-pricing model, and reflect grants from our
2000 Long-Term Incentive Plan, which is described in Note 15 of the Notes
to Consolidated Financial
Statements.
|
We use a
combination of cash and stock-based compensation to attract and retain qualified
candidates to serve on our board of directors. Directors who also are employees
of our company currently receive no compensation for their service as directors
or as members of board committees. In setting director compensation, we consider
the significant amount of time that directors dedicate to the fulfillment of
their director responsibilities, as well as the competency and skills required
of members of our board. The directors’ current compensation schedule has been
in place since May 2009. The directors’ annual compensation year begins with the
annual election of directors at the annual meeting of stockholders. The annual
retainer year period has been in place for directors since 2003. Periodically,
our board of directors reviews our director compensation policies and, from time
to time, makes changes to such policies based on various criteria the board
deems relevant.
Our
non-employee directors receive a quarterly retainer of $6,000 (plus an
additional $12,500 for the Chairman of the Board, $5,000 for the Chairman of the
Audit Committee, and $1,500 for the Chairmen of the Nomination and Governance
Committee and the Compensation Committee), a fee of $3,000 for each board
meeting attended ($750 for board actions taken by unanimous written consent),
$2,000 for each meeting of the Audit Committee attended, and $1,000 for each
other committee meeting attended. Non-employee directors who serve as the
chairman of a board committee receive an additional $2,000 for each meeting of
the Nomination and Governance Committee or the Compensation Committee attended
and an additional $2,500 for each meeting attended of the audit committee. In
July 2009, we granted stock options to purchase 50,000 shares of our common
stock at an exercise price equal to the current market value of our common stock
to each non-employee director. The options were vested, in full, upon
grant.
61
Joseph
Rubinfeld, Ph.D. Consulting Agreement
On
December 2, 2008, we entered into a written consulting agreement with Joseph
Rubinfeld, Ph.D., under which Dr. Rubinfeld agrees to serve as our Chief
Scientific Advisor. In exchange, we granted to Dr. Rubinfeld under our 2008
Stock Incentive Plan a ten-year stock option to purchase up to 350,000 shares of
our common stock at an exercise price of $0.35 per share, which equaled the
market price of our common stock as of the grant date. The stock option vested
immediately upon grant as to 50,000 of the option shares and will vest as to the
remaining option shares in 36 equal monthly installments, subject in each case
to Dr. Rubinfeld remaining in our service through such dates. We also agree in
the consulting agreement to pay Dr. Rubinfeld a monthly fee of $1,000. The
consulting agreement is terminable at any time by either party upon notice to
the other party.
Item
12. SECURITY
OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED
STOCKHOLDER MATTERS
Based
solely upon information made available to us, the following table sets forth
information with respect to the beneficial ownership of our common stock as of
March 12, 2010 by (1) each person who is known by us to beneficially own more
than five percent of our common stock; (2) each of our directors; (3) the named
executive officers listed in the Summary Compensation Table under Item 11; and
(4) all of our executive officers and directors as a group. Beneficial ownership
is determined in accordance with the SEC rules. Shares of common stock subject
to any warrants or options that are presently exercisable, or exercisable within
60 days of March 12, 2010 (which are indicated by footnote) are deemed
outstanding for the purpose of computing the percentage ownership of the person
holding the warrants or options, but are not treated as outstanding for the
purpose of computing the percentage ownership of any other person. The
percentage ownership reflected in the table is based on 108,908,105 shares of
our common stock outstanding as of March 12, 2010, excluding treasury shares.
Except as otherwise indicated, the holders listed below have sole voting and
investment power with respect to all shares of common stock shown, subject to
applicable community property laws. An asterisk represents beneficial ownership
of less than 1%.
|
|
Shares
of
Common Stock
|
|||||||
|
Name of Beneficial
Owner
|
Number
|
Percent
|
||||||
|
Louis
Ignarro, Ph.D.(1)
|
618,916 | * | ||||||
|
Steven
A. Kriegsman(2)
|
6,305,808 | 5.7 | % | |||||
|
Max
Link, Ph.D.(3)
|
239,519 | * | ||||||
|
Joseph
Rubinfeld, Ph.D.(4)
|
177,000 | * | ||||||
|
Marvin
R. Selter(5)
|
522,451 | * | ||||||
|
Richard
L. Wennekamp(6)
|
170,000 | * | ||||||
|
Dan
Levitt, M.D., Ph.D.(7)
|
83,334 | * | ||||||
|
John
Y. Caloz (8)
|
113,197 | * | ||||||
|
Scott
Wieland, Ph.D.(9)
|
122,502 | * | ||||||
|
Benjamin
S. Levin(10)
|
608,350 | * | ||||||
|
All
executive officers and directors as a group (eleven
persons)(11)
|
8,981,912 | 7.9 | % | |||||
____________
|
(1)
|
Includes
527,000 shares subject to options or
warrants.
|
|
(2)
|
Includes
2,284,708 shares subject to options or warrants. Mr. Kriegsman’s address
is c/o CytRx Corporation, 11726 San Vicente Boulevard, Suite 650, Los
Angeles, CA 90049.
|
|
(3)
|
Includes
184,543 shares subject to options or
warrants.
|
|
(4)
|
Includes
177,000 shares subject to options or
warrants.
|
|
(5)
|
The
shares shown are owned, of record, by the Selter Family Trust or Selter
IRA Rollover.
Includes 165,000 shares subject to options or warrants owned by Mr.
Selter.
|
|
(6)
|
Includes
165,000 shares subject to options or
warrants.
|
|
(7)
|
Includes
83,334 shares subject to options or warrants.
|
(8) Includes 113,197
shares subject to options or warrants.
|
|
|
62
(9)
Includes 122,502 shares subject to options or warrants.
|
(10)
|
Includes
608,350 shares subject to options or
warrants.
|
|
(11)
|
Includes
4,451,469 shares subject to options or
warrants.
|
Item
13. CERTAIN
RELATIONSHIPS AND RELATED TRANSACTIONS
Director
Independence
Our board
of directors has determined that Messrs. Link, Selter, Ignarro and Wennekamp are
“independent” under the current independence standards of both The NASDAQ
Capital Market and the SEC, and have no material relationships with us (either
directly or as a partner, shareholder or officer of any entity) which could be
inconsistent with a finding of their independence as members of our board of
directors or as the members of our Audit Committee. In making these
determinations, our board of directors has broadly considered all relevant facts
and circumstances, recognizing that material relationships can include
commercial, banking, consulting, legal, accounting, and familial relationships,
among others.
Transactions
with Related Persons
General
Our Audit
Committee is responsible for reviewing and approving, as appropriate, all
transactions with related persons, in accordance with its Charter and NASDAQ
Marketplace Rules.
Transactions
between us and one or more related persons may present risks or conflicts of
interest or the appearance of conflicts of interest. Our Code of Ethics requires
all employees, officers and directors to avoid activities or relationships that
conflict, or may be perceived to conflict, with our interests or adversely
affect our reputation. It is understood, however, that certain relationships or
transactions may arise that would be deemed acceptable and appropriate so long
as there is full disclosure of the interest of the related parties in the
transaction and review and approval by disinterested directors to ensure there
is a legitimate business reason for the transaction and that the transaction is
fair to us and our stockholders.
As a
result, the procedures followed by the Audit Committee to evaluate transactions
with related persons require:
|
|
·
|
that
all related person transactions, all material terms of the transactions,
and all the material facts as to the related person’s direct or indirect
interest in, or relationship to, the related person transaction must be
communicated to the Audit Committee;
and
|
|
|
·
|
that
all related person transactions, and any material amendment or
modification to any related person transaction, be reviewed and approved
or ratified by the Audit Committee, as required by NASDAQ Marketplace
Rules.
|
Our Audit
Committee will evaluate related person transactions based on:
|
|
·
|
information
provided by members of our board of directors in connection with the
required annual evaluation of director
independence;
|
|
|
·
|
pertinent
responses to the Directors’ and Officers’ Questionnaires submitted
periodically by our officers and directors and provided to the Audit
Committee by our management;
|
|
|
·
|
background
information on nominees for director provided by the Nominating and
Corporate Governance Committee of our board of directors;
and
|
|
|
·
|
any
other relevant information provided by any of our directors or
officers.
|
63
In
connection with its review and approval or ratification, if appropriate, of any
related person transaction, our Audit Committee is to consider whether the
transaction will compromise standards included in our Code of Ethics. In the
case of any related person transaction involving an outside director or nominee
for director, the Audit Committee also is to consider whether the transaction
will compromise the director’s status as an independent director as prescribed
in the NASDAQ Marketplace Rules.
Exemption
Clause
Item
404(a)(7)(a) of Securities and Exchange Commission Regulation S-K states that:
Disclosure need not be provided if the transaction is one where the rates or
charges involved in the transaction are determined by competitive bid, or the
transaction involves rendering of services as a common or contract carrier, or
public utility, at rates or charges fixed in conformity with law or governmental
authority.
Applicable
Definitions
For
purposes of our Audit Committee’s review:
|
|
·
|
“related
person” has the meaning given to such term in Item 404(a) of Securities
and Exchange Commission Regulation S-K (“Item 404(a)”);
and
|
|
|
·
|
“related
person transaction” means any transaction for which disclosure is required
under the terms of Item 404(a) involving the Company and any related
persons.
|
Item
14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
BDO
Seidman, LLP, or BDO, serves as our independent registered public accounting
firm and audited our financial statements for the years ended December 31, 2009,
2008 and 2007.
Audit
Fees
The fees
for 2009 and 2008 billed to us by BDO for professional services rendered for the
audit of our annual consolidated financial statements and internal controls over
financial reporting were $383,500 and $350,311, respectively.
Audit-Related
Fees
BDO
rendered $25,175 of assurance and other related services in 2009, and
$152,262 of assurance and other related services in 2008, which included
services relating to our shelf registration with the SEC and the Innovive
acquisition.
Tax
Fees
The
aggregate fees billed by BDO for professional services for tax compliance, tax
advice and tax planning were $33,260 and $39,000 for 2009 and 2008,
respectively.
All
Other Fees
No other
services were rendered by BDO for 2009 or 2008.
Pre-Approval
Policies and Procedures
It is the
policy of our Audit Committee that all services to be provided by our
independent registered public accounting firm, including audit services and
permitted audit-related and non-audit services, must be pre-approved by our
Audit Committee. Our Audit Committee pre-approved all services, audit and
non-audit, provided to us by BDO for 2009 and 2008.
64
PART
IV
Item
15. EXHIBITS
AND FINANCIAL STATEMENT SCHEDULES
(a) The
following documents are filed as part of this 10-K:
(1) Financial
Statements
Our
consolidated financial statements and the related report of the independent
registered public accounting firm thereon are set forth on pages F-1 to F-26 of
this Annual Report. These consolidated financial statements are as
follows:
Consolidated
Balance Sheets as of December 31, 2009 and 2008
Consolidated
Statements of Operations for the Years Ended December 31, 2009, 2008 and
2007
Consolidated
Statements of Stockholders’ Equity for the Years Ended December 31, 2009, 2008
and 2007
Consolidated
Statements of Cash Flows for the Years Ended December 31, 2009, 2008 and
2007
Notes to
Consolidated Financial Statements
Reports
of Independent Registered Public Accounting Firms
(2) Financial
Statement Schedules
The
following financial statement schedule is set forth on page F-26 of this Annual
Report.
Schedule
II — Valuation and Qualifying Accounts for the years ended December 31, 2009,
2008 and 2007
All other
schedules are omitted because they are not required, not applicable, or the
information is provided in the financial statements or notes
thereto.
(b) Exhibits
See
Exhibit Index on page 66 of this Annual Report, which is incorporated herein by
reference.
65
CytRx
Corporation
Form
10-K Exhibit Index
|
Exhibit
Number
|
Description
|
Footnote
|
||
|
3.1
|
Amended
and Restated Certificate of Incorporation, as amended
|
(a)
|
||
|
3.2
|
Restated
By-Laws, as amended
|
(a)
|
||
|
4.1
|
Shareholder
Protection Rights Agreement dated April 16, 1997 between CytRx Corporation
and American Stock Transfer &Trust Company as Rights
Agent
|
(b)
|
||
|
4.2
|
Amendment
No. 1 to Shareholder Protection Rights Agreement
|
(e)
|
||
|
4.3
|
Amendment
No. 2 to Shareholder Protection Rights Agreement
|
(r)
|
||
|
4.4
|
Warrant
issued on May 10, 2004 to MBN Consulting, LLC
|
(i)
|
||
|
4.5
|
Form
of Common Stock Purchase Warrant between CytRx Corporation and each of the
investors in the October 4, 2004 private placement
|
(j)
|
||
|
4.6
|
Form
of Common Stock Purchase Warrant between CytRx Corporation and each of the
investors in the January 2005 private placement
|
(k)
|
||
|
4.7
|
Form
of Common Stock Purchase Warrant between CytRx Corporation and each of the
investors in the March 2006 private placement
|
(p)
|
||
|
4.8
|
Securities
Purchase Agreement, dated July 24, 2009, by and among CytRx Corporation
and the purchasers listed on the signature pages thereto
|
(z)
|
||
|
4.9
|
Form
of Common Stock Purchase Warrant to be issued by CytRx Corporation to
purchasers under the Securities Purchase Agreement
|
(z)
|
||
|
10.1*
|
1994
Stock Option Plan, as amended and restated
|
(c)
|
||
|
10.2*
|
1998
Long-Term Incentive Plan
|
(d)
|
||
|
10.3*
|
2000
Long-Term Incentive Plan
|
(e)
|
||
|
10.4*
|
Amendment
No. 1 to 2000 Long-Term Incentive Plan
|
(g)
|
||
|
10.5*
|
Amendment
No. 2 to 2000 Long-Term Incentive Plan
|
(g)
|
||
|
10.6*
|
Amendment
No. 3 to 2000 Long-Term Incentive Plan
|
(h)
|
||
|
10.7*
|
Amendment
No. 4 to 2000 Long-Term Incentive Plan
|
(h)
|
||
|
10.8*
|
2008
Stock Incentive Plan
|
(y)
|
||
|
10.9†
|
License
Agreement dated December 7, 2001 by and between CytRx Corporation and
Vical Incorporated
|
(f)
|
||
|
10.10†
|
Agreement
between CytRx Corporation and Dr. Robert Hunter regarding SynthRx, Inc
dated October 20, 2003
|
(h)
|
||
|
10.11
|
Office
Lease between The Kriegsman Group and Douglas Emmett, dated April 13,
2000
|
(h)
|
||
|
10.12
|
Assignment
to CytRx Corporation effective July 1, 2003 of Office Lease between The
Kriegsman Group and Douglas Emmett, dated April 13, 2000
|
(h)
|
||
|
10.13
|
Asset
Sale and Purchase Agreement dated October 4, 2004, by and among CytRx
Corporation, Biorex Research & Development, RT and BRX Research and
Development Company Ltd
|
(j)
|
||
|
10.14
|
Sublease
dated March 14, 2005 between Innovive Pharmaceuticals, Inc. and Friedman,
Billings, Ramsey Group, Inc.
|
(l)
|
||
|
10.15*
|
Amended
and Restated Employment Agreement dated May 17, 2005 between CytRx
Corporation and Steven A. Kriegsman
|
(m)
|
||
|
10.16
|
First
Amendment to Office Lease dated October 14, 2005, by and between CytRx
Corporation and Douglas Emmett 1993, LLC
|
(n)
|
||
|
10.17†
|
License
Agreement dated December 28, 2005 between Innovive Pharmaceuticals, Inc.
and Nippon Shinyaku Co., Ltd.
|
(l)
|
||
|
10.18†
|
License
Agreement dated April 17, 2006 between Innovive Pharmaceuticals, Inc. and
KTB Tumorforschungs GmbH
|
(o)
|
||
|
10.19
|
Royalty
Agreement dated August 28, 2006 between CytRx Corporation and Kenneth
Council, as Trustee of the ALS Charitable Remainder Trust
|
(q)
|
||
|
10.20†
|
License
Agreement dated December 6, 2006 between Innovive Pharmaceuticals, Inc.
and TMRC Co., Ltd.
|
(s)
|
||
|
10.21
|
Contribution
Agreement, dated as of January 8, 2007, between CytRx Corporation and RXi
Pharmaceuticals Corporation
|
(t)
|
||
|
10.22
|
Voting
agreement, dated as of January 10, 2007, between CytRx Corporation and the
University of Massachusetts
|
(t)
|
||
|
10.23
|
Stockholders
agreement, dated February 23, 2007, among CytRx Corporation, RXi
Pharmaceuticals Corporation, Craig C. Mello, Ph.D., Tariq Rana, Ph.D.,
Gregory J. Hannon, Ph.D., and Michael P. Czech, Ph.D
|
(t)
|
||
|
10.24
|
Form
of Purchase Agreement, dated as of April 17, 2007, by and between CytRx
Corporation and each of the selling stockholders named
therein
|
(u)
|
||
|
10.25
|
Contribution
Agreement, dated as of April 30, 2007, between CytRx Corporation and RXi
Pharmaceuticals Corporation
|
(t)
|
||
|
10.26
|
Lease
dated July 20, 2007, between CytRx Corporation and BMR-3030 Bunker Hill
Street LLC
|
(v)
|
||
|
10.27
|
Agreement
and Plan of Merger, dated as of June 6, 2008, among CytRx
Corporation, CytRx Merger Subsidiary, Inc., Innovive Pharmaceuticals,
Inc., and Steven Kelly
|
(w)
|
||
|
10.28
|
Loan
and Security Agreement, dated as of June 6, 2008, between CytRx
Corporation and Innovive Pharmaceuticals, Inc.
|
(w)
|
||
|
10.29
|
Second
Amendment to Office Lease dated June 30, 2008, by and between CytRx
Corporation and Douglas Emmett 1993, LLC
|
(y)
|
||
|
10.30
|
Amendment
to Contribution Agreement, dated July 28, 2008, between CytRx
Corporation and RXi Pharmaceuticals Corporation
|
(x)
|
||
|
10.31
|
Amendment
to Stockholders Agreement, dated July 28, 2008, among CytRx
Corporation, RXi Pharmaceuticals Corporation, and Michael P. Czech, PhD.,
Gregory J. Hannon, Ph.D., Craig C. Mello, PhD., and Tariq M. Rana,
Ph.D.
|
(x)
|
||
|
10.32
|
Sub-Sublease
dated December 4, 2008, by and between CytRx Oncology Corporation and Red
Pine Advisors LLC
|
(y)
|
||
|
10.33
|
Investment
Banking Agreement, dated January 29, 2009, by and between CytRx
Corporation and Legend Securities, Inc.
|
(y)
|
||
|
10.34*
|
Employment
Agreement dated October 12, 2009 between CytRx Corporation and Daniel
Levitt, M.D., Ph.D.
|
(aa)
|
||
|
10.35*
|
Employment
Agreement dated November 30, 2009, between CytRx Corporation and Scott
Geyer
|
|||
|
10.36
|
Third
Amendment to Office Lease dated December 1, 2009, by and between CytRx
Corporation and Douglas Emmett 1993, LLC
|
(bb)
|
||
|
10.37*
|
Employment
Agreement dated January 1, 2010, between CytRx Corporation and Benjamin S.
Levin
|
|||
|
10.38*
|
Employment
Agreement dated January 1, 2010, between CytRx Corporation and Scott
Wieland
|
|||
|
10.39*
|
Employment
Agreement dated January 1, 2010, between CytRx Corporation and John Y.
Caloz
|
|||
|
23.1
|
Consent
of BDO Seidman, LLP
|
|||
|
31.1
|
Certification
of Chief Executive Officer Pursuant to 15 U.S.C. Section 7241, as adopted
pursuant to Section 302 of the Sarbanes-Oxley Act of 2002
|
|||
|
31.2
|
Certification
of Chief Financial Officer Pursuant to 15 U.S.C. Section 7241, as adopted
pursuant to Section 302 of the Sarbanes-Oxley Act of 2002
|
|||
|
32.1
|
Certification
of Chief Executive Officer Pursuant to 18 U.S.C. Section 1350, as adopted
pursuant to Section 906 of the Sarbanes-Oxley Act of 2002
|
|||
|
32.2
|
Certification
of Chief Financial Officer Pursuant to 18 U.S.C. Section 1350, as adopted
pursuant to Section 906 of the Sarbanes-Oxley Act of 2002
|
____________
|
|
*
Indicates a management contract or compensatory plan or
arrangement.
|
|
|
†
Confidential treatment has been requested or granted for certain portions
which have been blanked out in the copy of the exhibit filed with the
Securities and Exchange Commission. The omitted information has been filed
separately with the Securities and Exchange
Commission.
|
|
(a)
|
Incorporated
by reference to the Registrant’s Form 10-K filed on April 1,
2008
|
|
(b)
|
Incorporated
by reference to the Registrant’s 8-K filed on April 17,
1997
|
|
(c)
|
Incorporated
by reference to the Registrant’s 10-Q filed on August 14,
1997
|
|
(d)
|
Incorporated
by reference to the Registrant’s Proxy Statement filed on April 30,
1998
|
|
(e)
|
Incorporated
by reference to the Registrant’s Form 10-K filed on March 27,
2001
|
|
(f)
|
Incorporated
by reference to the Registrant’s Form 8-K filed on December 21,
2001
|
|
(g)
|
Incorporated
by reference to the Registrant’s Proxy Statement filed June 11,
2002
|
|
(h)
|
Incorporated
by reference to the Registrant’s 10-K filed on May 14,
2004
|
|
(i)
|
Incorporated
by reference to the Registrant’s 10-Q filed on August 16,
2004
|
|
(j)
|
Incorporated
by reference to the Registrant’s 8-K filed on October 5,
2004
|
|
(k)
|
Incorporated
by reference to the Registrant’s 8-K filed on January 21,
2005
|
66
|
(l)
|
Incorporated
by reference to the Innovive Pharmaceuticals Form 10 filed on April 20,
2006
|
|
(m)
|
Incorporated
by reference to the Registrant’s 10-Q filed on August 15,
2005
|
|
(n)
|
Incorporated
by reference to the Registrant’s 8-K filed on October 20,
2005
|
|
(o)
|
Incorporated
by reference to the Innovive Pharmaceuticals 10-Q filed on November 14,
2006
|
|
(p)
|
Incorporated
by reference to the Registrant’s 8-K filed on March 3,
2006
|
|
(q)
|
Incorporated
by reference to the Registrant’s 10-Q filed on November 13,
2006
|
|
(r)
|
Incorporated
by reference to the Registrant’s 10-K filed on April 2,
2007
|
|
(s)
|
Incorporated
by reference to the Innovive Pharmaceuticals 10-K filed on March 21,
2007
|
|
(t)
|
Incorporated
by reference to the Registrant’s 10-Q filed on May 10,
2007
|
|
(u)
|
Incorporated
by reference to the Registrant’s 8-K filed on April 18,
2007
|
|
(v)
|
Incorporated
by reference to the Registrant’s 10-Q filed on August 9,
2007
|
|
(w)
|
Incorporated
by reference to the Registrant’s 8-K filed on June 9,
2008
|
|
(x)
|
Incorporated
by reference to the Registrant’s 10-Q filed on August 11,
2008
|
|
(y)
|
Incorporated
by reference to the Registrant’s 10-K filed on March 13,
2009
|
|
(z)
|
Incorporated
by reference to the Registrant’s 8-K filed on July 27,
2009
|
|
(aa)
|
Incorporated
by reference to the Registrant’s 10-Q filed on November 9,
2009
|
|
(bb
|
Incorporated
by reference to the Registrant’s 8-K filed on December 4,
2009
|
67
Pursuant to
the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934,
the Registrant has duly caused this report to be signed on its behalf by the
undersigned, thereunto duly authorized.
|
CYTRX
CORPORATION
|
|||
|
Date:
March 12, 2010
|
By:
|
/s/ STEVEN
A. KRIEGSMAN
|
|
|
Steven
A. Kriegsman
|
|||
|
President
and Chief Executive Officer
|
|||
68
INDEX
TO FINANCIAL STATEMENTS
AND
FINANCIAL STATEMENT SCHEDULE
|
CytRx
Corporation
|
|
|
Consolidated
Balance Sheets
|
F-2
|
|
Consolidated
Statements of Operations
|
F-3
|
|
Consolidated
Statements of Stockholders’ Equity
|
F-4
|
|
Consolidated
Statements of Cash Flows
|
F-5
|
|
Notes
to Consolidated Financial Statements
|
F-7
|
|
Reports
of Independent Registered Public Accounting Firm
|
F-24
|
|
Financial
Statement Schedule II — Valuation and Qualifying Accounts
|
F-26
|
CYTRX
CORPORATION
CONSOLIDATED
BALANCE SHEETS
|
|
December 31,
|
|||||||
|
|
2009
|
2008
|
||||||
|
ASSETS
|
||||||||
|
Current
assets:
|
||||||||
|
Cash
and cash equivalents
|
$ | 9,893,590 | $ | 25,041,772 | ||||
|
Marketable
securities
|
22,750,000 | — | ||||||
|
Receivable
|
139,680 | 127,280 | ||||||
|
Income
taxes recoverable
|
519,158 | 215,623 | ||||||
|
Interest
receivable
|
130,779 | — | ||||||
|
Assets
held for sale
|
73,634 | — | ||||||
|
Prepaid
expenses and other current assets
|
1,088,074 | 486,609 | ||||||
|
Total
current assets
|
34,594,915 | 25,871,284 | ||||||
|
Equipment
and furnishings, net
|
174,959 | 1,835,052 | ||||||
|
Molecular
library, net
|
— | 103,882 | ||||||
|
Goodwill
|
183,780 | 183,780 | ||||||
|
Other
assets
|
323,235 | 330,032 | ||||||
|
Total
assets
|
$ | 35,276,889 | $ | 28,324,030 | ||||
|
LIABILITIES
AND STOCKHOLDERS’ EQUITY
|
||||||||
|
Current
liabilities:
|
||||||||
|
Accounts
payable
|
$ | 1,066,055 | $ | 668,422 | ||||
|
Accrued
expenses and other current liabilities
|
2,492,450 | 2,556,904 | ||||||
|
Warrant
liability
|
3,370,701 | — | ||||||
|
Deferred
revenue, current portion
|
— | 1,817,600 | ||||||
|
Total
current liabilities
|
6,929,206 | 5,042,926 | ||||||
|
Deferred
revenue, non-current portion
|
— | 7,582,797 | ||||||
|
Total
liabilities
|
6,929,206 | 12,625,723 | ||||||
|
Commitment
and contingencies
|
||||||||
|
Stockholders’
equity:
|
||||||||
|
Preferred
Stock, $.01 par value, 5,000,000 shares authorized, including 15,000
shares of Series A Junior Participating Preferred Stock; no shares issued
and outstanding
|
— | — | ||||||
|
Common
stock, $.001 par value, 175,000,000 shares authorized; 109,538,821 and
93,978,448 shares issued and outstanding at December 31, 2009 and 2008,
respectively
|
109,539 | 93,978 | ||||||
|
Additional
paid-in capital
|
227,441,591 | 210,007,468 | ||||||
|
Treasury
stock, at cost (633,816 shares held, at December 31, 2009 and 2008,
respectively)
|
(2,279,238 | ) | (2,279,238 | ) | ||||
|
Accumulated
deficit
|
(196,924,209 | ) | (192,123,901 | ) | ||||
|
Total
stockholders’ equity
|
28,347,683 | 15,698,307 | ||||||
|
Total
liabilities and stockholders’ equity
|
$ | 35,276,889 | $ | 28,324,030 | ||||
The
accompanying notes are an integral part of these consolidated financial
statements.
F-2
CYTRX
CORPORATION
CONSOLIDATED
STATEMENTS OF OPERATIONS
|
|
Years Ended December 31,
|
|||||||||||
|
|
2009
|
2008
|
2007
|
|||||||||
|
Revenue:
|
||||||||||||
|
Service
revenue
|
$ | 9,400,397 | $ | 6,166,150 | $ | 7,241,920 | ||||||
|
Licensing
revenue
|
100,000 | 100,000 | 101,000 | |||||||||
|
Grant
revenue
|
— | — | 116,118 | |||||||||
| 9,500,397 | 6,266,150 | 7,459,038 | ||||||||||
|
Expenses:
|
||||||||||||
|
Research
and development
|
7,541,998 | 10,465,591 | 18,823,802 | |||||||||
|
General
and administrative
|
9,127,845 | 10,932,522 | 14,822,142 | |||||||||
|
In-process
research and development
|
— | 8,012,154 | — | |||||||||
|
Depreciation
and amortization
|
475,316 | 624,980 | 272,229 | |||||||||
| 17,145,159 | 30,035,247 | 33,918,173 | ||||||||||
|
Loss
before other income
|
(7,644,762 | ) | (23,769,097 | ) | (26,459,135 | ) | ||||||
|
Other
income:
|
||||||||||||
|
Interest
and dividend income
|
349,490 | 1,203,629 | 2,663,542 | |||||||||
|
Other
income, net
|
93,950 | 219,489 | 1,496,979 | |||||||||
|
Gain
on warrant derivative liability
|
656,905 | — | — | |||||||||
|
Gain
on sale of affiliate’s RXi Pharmaceutical shares
|
1,224,951 | — | — | |||||||||
|
Equity
in loss of affiliate – RXi Pharmaceuticals
|
— | (3,915,514 | ) | — | ||||||||
|
Net
loss before provision for income taxes
|
(5,319,466 | ) | (26,261,493 | ) | (22,298,614 | ) | ||||||
|
Provision
for income taxes
|
519,158 | (873,003 | ) | (40,000 | ) | |||||||
|
Net
loss
|
(4,800,308 | ) | (27,134,496 | ) | (22,338,614 | ) | ||||||
|
Deemed
dividend for anti-dilution adjustments made to outstanding common stock
warrants
|
— | (756,954 | ) | — | ||||||||
|
Net
loss
|
(4,800,308 | ) | (27,891,450 | ) | (22,338,614 | ) | ||||||
|
Add:
Loss of noncontrolling interest
|
— | 88,375 | 448,671 | |||||||||
|
Net
loss attributable to CytRx Corporation
|
$ | (4,800,308 | ) | $ | (27,803,075 | ) | $ | (21,889,943 | ) | |||
|
Basic
and diluted loss per share
|
$ | (0.05 | ) | $ | (0.30 | ) | $ | (0.26 | ) | |||
|
Basic
and diluted weighted average shares outstanding
|
99,978,124 | 91,383,934 | 84,006,728 | |||||||||
The
accompanying notes are an integral part of these consolidated financial
statements.
F-3
CYTRX
CORPORATION
CONSOLIDATED
STATEMENTS OF STOCKHOLDERS’ EQUITY
|
Additional
|
||||||||||||||||||||||||||||
|
Common Stock
|
Paid-In
|
Accumulated
|
Treasury
|
Noncontrolling
|
||||||||||||||||||||||||
|
Shares Issued
|
Amount
|
Capital
|
Deficit
|
Stock
|
Interest
|
Total
|
||||||||||||||||||||||
|
Balance
at December 31, 2006
|
70,788,586 | $ | 70,789 | $ | 146,961,657 | $ | (140,139,915 | ) | $ | (2,279,238 | ) | $ | 537,046 | 5,150,339 | ||||||||||||||
|
Common
stock and warrants issued in connection with private
placements
|
8,615,000 | 8,615 | 34,239,442 | — | — | 34,248,057 | ||||||||||||||||||||||
|
Issuance
of stock options/warrants for compensation, services and
licenses
|
2,402,035 | — | — | 2,402,035 | ||||||||||||||||||||||||
|
Options
and warrants exercised
|
10,994,281 | 10,994 | 18,778,180 | — | — | 18,789,174 | ||||||||||||||||||||||
|
Issuance
of stock options by subsidiary
|
— | — | 1,524,377 | — | — | 1,524,377 | ||||||||||||||||||||||
|
Net
loss
|
— | — | — | (21,889,943 | ) | — | (448,671 | ) | (22,338,614 | ) | ||||||||||||||||||
|
Balance
at December 31, 2007
|
90,397,867 | $ | 90,398 | $ | 203,905,691 | $ | (161,492,812 | ) | $ | (2,279,238 | ) | $ | (448,671 | ) | $ | 40,312,414 | ||||||||||||
|
Issuance
of stock options/warrants for compensation, services and
licenses
|
— | — | 2,029,209 | — | — | 2,029,209 | ||||||||||||||||||||||
|
Options
and warrants exercised
|
1,006,402 | 1,006 | 975,782 | — | — | 976,788 | ||||||||||||||||||||||
|
Common
stock issued in connection with the acquisition of
Innovive
|
2,574,179 | 2,574 | 2,339,832 | — | — | 2,342,406 | ||||||||||||||||||||||
|
Deemed
dividend for anti-dilution adjustment
|
— | 756,954 | (756,954 | ) | — | — | ||||||||||||||||||||||
|
Dividend
of RXi stock
|
— | — | — | (2,828,014 | ) | — | (2,828,014 | ) | ||||||||||||||||||||
|
Net
loss
|
— | — | — | (27,046,121 | ) | — | (88,375 | ) | (27,134,496 | ) | ||||||||||||||||||
|
Balance
at December 31, 2008
|
93,978,448 | $ | 93,978 | $ | 210,007,468 | $ | (192,123,901 | ) | $ | (2,279,238 | ) | $ | — | $ | 15,698,307 | |||||||||||||
|
Common
stock and warrants issued in connection with private
placements
|
15,252,040 | 15,253 | 14,230,710 | — | — | 14,245,963 | ||||||||||||||||||||||
|
Issuance
of stock options/warrants for compensation and services
|
— | — | 2, 867,638 | — | — | 2,867,638 | ||||||||||||||||||||||
|
Common
stock issued for services
|
50,000 | 50 | 42,950 | — | — | 43,000 | ||||||||||||||||||||||
|
Options
and warrants exercised
|
258,333 | 258 | 292,825 | — | — | 293,083 | ||||||||||||||||||||||
|
Net
loss
|
— | — | — | (4,800,308 | ) | — | — | (5,319,466 | ) | |||||||||||||||||||
|
Balance
at December 31, 2009
|
109,538,821 | $ | 109,539 | $ | 227,441,591 | $ | (196,924,209 | ) | $ | (2,279,238 | ) | $ | — | $ | 27,828,525 | |||||||||||||
The
accompanying notes are an integral part of these consolidated financial
statements.
F-4
CYTRX
CORPORATION
CONSOLIDATED
STATEMENTS OF CASH FLOWS
|
Years Ended December 31,
|
||||||||||||
|
|
2009
|
2008
|
2007
|
|||||||||
|
Cash
flows from operating activities:
|
||||||||||||
|
Net
loss attributable to CytRx Corporation
|
$ | (4,800,308 | ) | $ | (27,046,121 | ) | $ | (21,889,943 | ) | |||
|
Adjustments
to reconcile net loss to net cash provided by (used in) operating
activities:
|
||||||||||||
|
Depreciation
and amortization
|
475,316 | 624,980 | 272,229 | |||||||||
|
Retirement
of fixed assets
|
103,241 | 262 | — | |||||||||
|
Interest
receivable on marketable securities
|
— | (48,452 | ) | (172,055 | ) | |||||||
|
Loss
of noncontrolling interest
|
— | (88,375 | ) | (448,671 | ) | |||||||
|
Fair
value adjustment of warrant liability
|
(656,905 | ) | — | — | ||||||||
|
Impairment
loss on fixed assets
|
1,187,305 | — | — | |||||||||
|
Equity
in loss of affiliate
|
— | 3,915,515 | — | |||||||||
|
Non-cash
gain on transfer of RXi common stock
|
— | (226,579 | ) | — | ||||||||
|
Gain
on sale of affiliate’s shares
|
(1,224,951 | ) | — | — | ||||||||
|
Non-cash
expense on the acquisition of Innovive’s in-process research &
development
|
— | 8,012,154 | — | |||||||||
|
Stock
option and warrant expense
|
2,734,247 | 2,103,752 | 3,511,541 | |||||||||
|
Common
stock issued for services
|
43,000 | 244,860 | 3,089,639 | |||||||||
|
Changes
in assets and liabilities:
|
||||||||||||
|
Accounts
receivable
|
(12,400 | ) | 238,131 | 4,713 | ||||||||
|
Interest
receivable
|
(130,779 | ) | — | — | ||||||||
|
Income
taxes recoverable
|
(303,535 | ) | (215,623 | ) | — | |||||||
|
Prepaid
expenses and other current assets
|
(461,277 | ) | 478,965 | (1,214,836 | ) | |||||||
|
Accounts
payable
|
397,633 | (1,181,116 | ) | 757,086 | ||||||||
|
Deferred
revenue
|
(9,400,397 | ) | (6,166,151 | ) | (7,241,919 | ) | ||||||
|
Accrued
expenses and other current liabilities
|
(64,454 | ) | (56,146 | ) | 978,388 | |||||||
|
Total
adjustments
|
(7,313,956 | ) | 7,636,177 | (463,885 | ) | |||||||
|
Net
cash used in operating activities
|
(12,114,264 | ) | (19,409,944 | ) | (22,353,828 | ) | ||||||
|
Cash
flows from investing activities:
|
||||||||||||
|
Proceeds
(Purchase) from sale of marketable securities
|
(22,750,000 | ) | 10,000,000 | (9,779,493 | ) | |||||||
|
Proceeds
from sale of fixed assets
|
119,929 | — | — | |||||||||
|
Deconsolidation
of subsidiary
|
— | (10,359,278 | ) | — | ||||||||
|
Proceeds
from sale of unconsolidated sub shares
|
1,224,951 | — | — | |||||||||
|
Cash
outlay in the acquisition of Innovive, relating to its accounts
payable
|
— | (5,669,749 | ) | — | ||||||||
|
Purchases
of equipment and furnishings
|
(195,449 | ) | (994,326 | ) | (1,269,313 | ) | ||||||
|
Net
cash used in investing activities
|
(21,600,569 | ) | (7,023,353 | ) | (11,048,806 | ) | ||||||
|
Cash
flows from financing activities:
|
||||||||||||
|
Net
proceeds from exercise of stock options and warrants
|
293,083 | 976,808 | 18,789,173 | |||||||||
|
Common
stock issued in accordance with financing
|
18,273,568 | — | — | |||||||||
|
Net
proceeds from issuances of common stock
|
— | — | 34,248,058 | |||||||||
|
Capital
contributions from minority interest
|
— | — | 482,271 | |||||||||
|
Net
cash provided by financing activities
|
18,566,651 | 976,808 | 53,519,502 | |||||||||
|
Net
increase (decrease) in cash and cash equivalents
|
(15,148,182 | ) | (25,456,489 | ) | 20,116,868 | |||||||
|
Cash
and cash equivalents at beginning of year
|
25,041,772 | 50,498,261 | 30,381,393 | |||||||||
|
Cash
and cash equivalents at end of year
|
$ | 9,893,590 | $ | 25,041,772 | $ | 50,498,261 | ||||||
|
Supplemental
disclosure of cash flow information:
|
||||||||||||
|
Cash
paid during the years for income taxes
|
$ | — | $ | 1,093,764 | $ | 183,461 | ||||||
|
Supplemental
disclosures of non-cash investing and financing
activities:
|
||||||||||||
|
Acquisition
of property and equipment through accrued liabilities
|
$ | — | $ | 130,955 | $ | 233,974 | ||||||
|
Warrants
issued in connection with financing
|
$ | 4,027,606 | $ | — | $ | — | ||||||
| Warrants issued for prepaid services | $ | 133,391 | $ | — | $ | — | ||||||
The
accompanying notes are an integral part of these consolidated financial
statements. See supplemental information on the following
page.
F-5
Supplemental
schedule of non-cash investing and financing activities:
CytRx
purchased all of the common stock of Innovive Pharmaceuticals, Inc. in a
transaction that for accounting purposes is considered an asset
acquisition. See Note 20 below. The fair values of
Innovive’s assets and liabilities at September 19, 2008, in millions of dollars,
are presented below:
|
In-process
research and development
|
$ | 8.0 | ||
|
Leasehold
interests
|
0.1 | |||
|
Prepaid
expenses
|
0.3 | |||
|
Accounts
payable
|
(6.1 | ) | ||
|
Net
assets acquired through issuance of common stock
|
$ | 2.3 |
As a
result of the March 11, 2008 distribution by CytRx Corporation (the “Company”)
to its stockholders of approximately 36% of the outstanding shares of RXi
Pharmaceuticals Corporation, the Company deconsolidated that previously
majority-owned subsidiary. As part of the transaction, the Company
deconsolidated $3.7 million of total assets and $4.6 million of total
liabilities.
In
connection with applicable antidilution adjustments to the price of certain
outstanding warrants in March 2008, the Company recorded a deemed dividend of
approximately $756,954 in the current year. The deemed dividend was recorded as
a charge to accumulated deficit and a corresponding credit to additional paid-in
capital.
During
2007, the Company allocated $289,254 of additional paid in capital arising from
subsidiary common stock options issued to minority interest.
F-6
CYTRX
CORPORATION
NOTES
TO CONSOLIDATED FINANCIAL STATEMENTS
1.
Nature of Business
CytRx
Corporation (“CytRx” or the “Company”) is a biopharmaceutical research and
development company engaged in the development of high-value human therapeutics,
specializing in oncology. CytRx’s drug development pipeline includes clinical
development of three product candidates for cancer indications, including three
planned Phase 2 clinical trials for INNO-206 as a treatment for pancreatic
cancer, gastric (stomach) cancer and soft tissue sarcomas, two Phase 2
proof-of-concept clinical trials with bafetinib in patients with high-risk
B-cell chronic lymphocytic leukemia, or B-CLL, and patients with glioblastoma
multiforme, and a registration study of tamibarotene for the treatment of acute
promyelocytic leukemia, or APL. In addition to its core oncology programs, CytRx
is developing two drug candidates based on its molecular chaperone regulation
technology, which aim to repair or degrade mis-folded proteins associated with
disease. Apart from its drug development programs, CytRx currently maintain a
36% equity interest in its former subsidiary, RXi. The Company’s
current business strategy for its molecular chaperone regulation technology is
to seek one or more strategic partnerships, or a possible spin-out
transaction.
At
December 31, 2009, the Company had cash and cash equivalents of approximately
$9.9 million, marketable securities of $22.8 million and held 5,768,881 shares
of restricted common stock of RXi Pharmaceuticals Corporation, or RXi, with a
market value of $26.4 million based upon the closing price of the RXi common
stock on that date. On July 27, 2009, the Company raised approximately $18.3
million, net of fees and expenses, in a registered direct offering, and on
September 23, 2009, the Company raised approximately $1.2 million, net of fees,
from the sale of 500,000 shares of its common stock of RXi. Management believes
that the Company’s current cash on hand, together with its marketable securities
and proceeds from possible future sale of RXi common stock, will be sufficient
to fund its operations for the foreseeable future. The estimate is
based, in part, upon the Company’s currently projected expenditures for 2010 of
approximately $18.0 million (unaudited), which includes approximately
$3.2 million (unaudited) for its clinical programs for INNO-206, approximately
$1.6 million (unaudited) for its clinical programs for bafetinib, approximately
$2.5 million (unaudited) for its clinical program for tamibarotene,
approximately $1.4 million (unaudited) for its activities for arimoclomol,
approximately $2.2 million (unaudited) for general operation of its clinical
programs, and approximately $7.1 million (unaudited) for other general and
administrative expenses. These projected expenditures are also based upon
numerous other assumptions and subject to many uncertainties, and actual
expenditures may be significantly different from these
projections. The Company will be required to obtain additional
funding in order to execute its long-term business plans, although it does not
currently have commitments from any third parties to provide it with capital.
The fair value of common stock investment in RXi is subject to market
fluctuations that could impact the amount of cash the Company generates from the
sale of RXi shares in the future. The Company cannot assure that additional
funding will be available on favorable terms, or at all. If the Company fails to
obtain additional funding when needed, it may not be able to execute its
business plans and its business may suffer, which would have a material adverse
effect on its financial position, results of operations and cash
flows.
2.
Summary of Significant Accounting Policies
Basis of Presentation and Principles
of Consolidation — Through February 2008, the Company owned a majority of
the outstanding shares of common stock of RXi, which was founded in April 2006
by the Company and four researchers in the field of RNAi, including Dr. Craig
Mello, recipient of the 2006 Nobel Prize for Medicine for his co-discovery of
RNAi. While RXi was majority-owned, the Company’s consolidated financial
statements reflected 100% of the assets and liabilities and results of
operations of RXi, with the interests of the minority shareholders of RXi
recorded as “noncontrolling interests.” In March 2008, the Company distributed
to its stockholders approximately 36% of RXi’s outstanding shares, which reduced
CytRx’s ownership to less than 50% of RXi. As a result of the reduced ownership,
CytRx began to account for its investment in RXi using the equity method, under
which CytRx records only its pro-rata share of the financial results of RXi as
“equity in loss of unconsolidated subsidiary” on the consolidated statements of
operations (see Note 14 below). Because a portion of RXi’s financial results for
2008 and all of RXi’s financial results for 2007 were not recorded by CytRx
under the equity method, the Company’s results of operations for the year ended
December 31, 2009 are not directly comparable to results of operations for the
same periods in prior years.
Revenue Recognition — Revenue
consists of license fees from strategic alliances with pharmaceutical companies
as well as service and grant revenues. Service revenue consists of contract
research and laboratory consulting. Grant revenues consist of government
and private grants.
F-7
Monies
received for license fees are deferred and recognized ratably over the
performance period in accordance with Staff Accounting Bulletin (“SAB”) No. 104,
Revenue Recognition.
Milestone payments will be recognized upon achievement of the milestone as long
as the milestone is deemed substantive and we have no other performance
obligations related to the milestone and collectability is reasonably assured,
which is generally upon receipt, or recognized upon termination of the agreement
and all related obligations. Deferred revenue represents amounts received prior
to revenue recognition.
Revenues
from contract research, government grants, and consulting fees are recognized
over the respective contract periods as the services are performed, provided
there is persuasive evidence or an arrangement, the fee is fixed or determinable
and collection of the related receivable is reasonably assured. Once all
conditions of the grant are met and no contingencies remain outstanding, the
revenue is recognized as grant fee revenue and an earned but unbilled revenue
receivable is recorded.
In August
2006, the Company received approximately $24.3 million in proceeds from the
privately-funded ALS Charitable Remainder Trust (“ALSCRT”) in exchange for the
commitment to continue research and development of arimoclomol and other
potential treatments for ALS and a one percent royalty in the worldwide sales of
arimoclomol. Under the arrangement, we retain the rights to any products or
intellectual property funded by the arrangement and the proceeds of the
transaction are non-refundable. The ALSCRT has no obligation to provide any
further funding to the Company. The Company has concluded that due to
the research and development components of the transaction that it is properly
accounted for under ASC 730-20 (previously Statement of Financial Accounting
Standards No. 68, Research
and Development
Arrangements). Accordingly, the Company recorded the value received under
the arrangement as deferred service revenue and recognized service revenue using
the proportional performance method of revenue recognition, meaning that service
revenue was recognized on a dollar-for-dollar basis for each dollar of expense
incurred for the research and development of arimoclomol and other potential ALS
treatments. CytRx believes that this method best approximates the efforts
expended related to the services provided. The Company adjusted its estimates of
expense incurred for this research and development on a quarterly
basis.
The
amount of “deferred revenue, current portion” is the amount of deferred revenue
that is expected to be recognized in the next twelve months and is subject to
fluctuation based upon management’s estimates. Management’s estimates include an
evaluation of what pre-clinical and clinical trials are necessary, the timing of
when trials will be performed and the estimated clinical trial expenses. These
estimates are subject to changes and could have a significant effect on the
amount and timing of when the deferred revenues are recognized.
Pursuant
to an amendment signed between the Company and the beneficiary of the ALSCRT on
August 6, 2009, the Company was released from all restrictions on the use of any
proceeds previously received by us in connection with the
arrangement. As a result, the Company recognized $6.7 million as
service revenue in the third quarter of 2009, which represented the remaining
deferred revenue and previously un-recognized portion of the value received. For
the years ended December 31, 2009 and 2008, the Company recognized approximately
$9.4 million and $6.2 million, respectively, of service revenue related to this
transaction.
Other Income — The
Company realized a net gain of $0.1 million in 2009 on the sub-lease of its New
York city rental property inherited on the acquisition of Innovive. In March
2008, the Company recognized a non-cash gain of $0.2 million on the transfer of
some RXi common stock to certain employees. In June 2007, the Company recognized
$1.5 million of income arising from a fee received pursuant to a
change-in-control provision included in the purchase agreement for its 1998 sale
of its animal pharmaceutical unit.
Cash Equivalents — The
Company considers all highly liquid debt instruments with an original maturity
of 90 days or less to be cash equivalents. Cash equivalents consist primarily of
amounts invested in money market accounts.
Marketable securities —
Investment securities held by the Company are classified as available for
sale.
Fair Value of Financial Instruments
— The carrying amounts reported in the balance sheet for cash and cash
equivalents and marketable securities approximate their fair
values.
Equipment and Furnishings —
Equipment and furnishings are stated at cost and depreciated using the
straight-line method based on the estimated useful lives (generally three to
five years for equipment and furniture) of the related assets. Whenever there is
a triggering event that might suggest an impairment, management evaluates the
realizability of recorded long-lived assets to determine whether their carrying
values have been impaired. The Company records impairment losses on long-lived
assets used in operations when events and circumstances indicate that the assets
might be impaired and the non-discounted cash flows estimated to be generated by
those assets are less than the carrying amount of those assets. Any impairment
loss is measured by comparing the fair value of the asset to its carrying
amount.
F-8
Fair Value Measurements — The
Company adopted new guidance which is now part of ASC 820-10 (formerly Financial
Accounting Standards Board Statement of Financial Accounting Standards No. 157),
Fair Value Measurements
(“FAS 157”), effective January 1, 2008. SFAS 157 does not require any
new fair value measurements; instead it defines fair value, establishes a
framework for measuring fair value in accordance with existing generally
accepted accounting principles and expands disclosure about fair value
measurements. The adoption of SFAS 157 for the Company’s financial
assets and liabilities did not have an impact on its financial position or
operating results. Beginning January 1, 2008, assets and liabilities
recorded at fair value in consolidated balance sheets are categorized based upon
the level of judgment associated with the inputs used to measure the fair
value. Level inputs, as defined by SFAS 157, are as
follows:
|
|
·
|
Level
1 – quoted prices in active markets for identical assets or
liabilities.
|
|
|
·
|
Level
2 – other significant observable inputs for the assets or liabilities
through corroboration with market data at the measurement
date.
|
|
|
·
|
Level
3 – significant unobservable inputs that reflect management’s best
estimate of what market participants would use to price the assets or
liabilities at the measurement
date.
|
The
following table summarizes fair value measurements by level at December 31, 2009
for assets and liabilities measured
at fair
value on a recurring basis:
|
(In
thousands)
|
Level
I
|
Level
II
|
Level
III
|
Total
|
||||||||||||
|
Cash
and cash equivalents
|
$
|
9,894
|
$
|
—
|
$
|
—
|
$
|
9,894
|
||||||||
|
Marketable
securities
|
22,750
|
—
|
—
|
22,750
|
||||||||||||
|
Warrant
Liability
|
—
|
3,371
|
—
|
3,371
|
||||||||||||
The
following table summarizes fair value measurements by level at December 31, 2008
for assets and liabilities measured
at fair
value on a recurring basis:
|
(In
thousands)
|
Level
I
|
Level
II
|
Level
III
|
Total
|
||||||||||||
|
Cash
and cash equivalents
|
$
|
25,042
|
$
|
—
|
$
|
—
|
$
|
25,042
|
||||||||
Liabilities
measured at market value on a recurring basis include warrant liabilities
resulting from recent debt and equity financing. In accordance with ASC 815-40
(formerly EITF 00-19,
Accounting for Derivative Financial Instruments Indexed to, and Potentially
Settled in, a Company’s Own Stock), the warrant liabilities are being
marked to market each quarter-end until they are completely settled. The
warrants are valued using the Black-Scholes method, using assumptions consistent
with the Company’s application of ASC 505-50. See Warrant Liabilities
below.
The
Company considers carrying amounts of accounts receivable, accounts payable and
accrued expenses to approximate fair value due to the short-term nature of these
financial instruments.
F-9
The
Company’s non-financial assets, such as assets held for sale are measured at
fair value when there is an indicator of impairment and recorded at fair value
only when an impairment charge is recognized. The following
table summarizes the Company’s financial assets measured at fair value on a
nonrecurring basis as of December 31, 2009:
|
Carrying
Amount as of December 31, 2009
|
Fair Value Measurements | |||||||||||||||||||
| as of December 31, 2009 | ||||||||||||||||||||
|
(in
thousands)
|
Level
1
|
Level
2
|
Level
3
|
Total Losses | ||||||||||||||||
|
Long-lived
assets held for sale:
|
||||||||||||||||||||
|
Plant
and equipment, net
|
$
|
74
|
$
|
—
|
$
|
74
|
$
|
—
|
$ | 1,187 | ||||||||||
| Asssets held for sale |
$
|
74
|
$
|
—
|
$
|
74
|
$
|
—
|
$ | 1,187 | ||||||||||
Impairment of Long-Lived Assets
— The Company reviews long-lived assets, including finite lived
intangible assets, for impairment on an annual basis, as of December 31, or on
an interim basis if an event occurs that might reduce the fair value of such
assets below their carrying values. An impairment loss would be recognized based
on the difference between the carrying value of the asset and its estimated fair
value, which would be determined based on either discounted future cash flows or
other appropriate fair value methods. If the Company’s estimates used in the
determination of either discounted future cash flows or other appropriate fair
value methods are not accurate as compared to actual future results, the Company
may be required to record an impairment charge. The fixed assets,
from the Company’s San Diego laboratory and its molecular library, available for
sale were re-allocated from Equipment and Furnishings to Assets Held for Sale
and were written down to their estimated net realizable value as of September
30, 2009 (see Note7 below).
Patents and Patent Application Costs
— Although the Company believes that its patents and underlying
technology have continuing value, the amount of future benefits to be derived
from the patents is uncertain. Patent costs are therefore expensed as
incurred.
Net Income (Loss) Per Share —
Basic net income (loss) per common share has computed using the weighted-average
number of common shares outstanding. Diluted net income (loss) per common share
computed using the weighted-average number of common share and common share
equivalents outstanding. Common share equivalents which could
potentially dilute basic earnings per share in the future, and which were
excluded from the computation of diluted loss per share, totaled approximately
24.4 million shares, 15.2 million shares and 17.1 million shares at December 31,
2009, 2008 and 2007, respectively.
As a
result of the March 11, 2008 distribution by CytRx to its stockholders of
approximately 36% of the outstanding shares of RXi Pharmaceuticals Corporation,
the Company recorded a deemed dividend of approximately $757,000. The
deemed dividend was reflected as an adjustment to net loss for the first quarter
of 2008 to arrive at net loss applicable to common stockholders on the
consolidated statement of operations and for purposes of calculating basic and
diluted earnings per shares.
Warrant Liabilities
— Liabilities measured at market value on a recurring basis include
warrant liabilities resulting from our recent equity financing. In accordance
with ASC 815-40 (formerly EITF (Emerging Issues Task Force) 00-19, Accounting for Derivative Financial
Instruments Indexed to and Potentially Settled in a Company’s Own Stock,
the warrant liabilities are being marked to market each quarter-end until they
are completely settled. The warrants are valued using the Black-Scholes method.
The gain or loss resulting from the marked to market calculation is shown on the
Consolidated Statements of Operations as Gain on warrant derivative
liability.
Shares Reserved for Future Issuance
— As of December 31, 2009, the Company has reserved approximately 9.1
million of its authorized but unissued shares of common stock for future
issuance pursuant to its employee stock option plans issued to consultants and
investors.
Stock-based Compensation —
The Company’s stock-based employee compensation plans are described in Note 15.
The Company has adopted the provisions of ASC 718 (previously SFAS No. 123(R),
Share-Based Payment
(“SFAS 123(R)”)), which requires the measurement and recognition of compensation
expense for all stock-based awards made to employees and
non-employees.
F-10
For stock
options and stock warrants paid in consideration of services rendered by
non-employees, the Company recognizes compensation expense in accordance with
the requirements of ASC 718 (previously SFAS No. 123(R)), ASC 505-50 (previously
Emerging Issues Task Force Issue No. 96-18 (“EITF 96-18”)), Accounting for Equity
Instruments that are Issued to other than Employees for Acquiring, or in
Conjunction with Selling Goods or Services and ASC 505 (previously EITF
00-18, Accounting Recognition
for Certain Transactions involving Equity Instruments Granted to Other Than
Employees), as amended.
Non-employee
option grants that do not vest immediately upon grant are recorded as an expense
over the vesting period. At the end of each financial reporting period prior to
performance, the value of these options, as calculated using the Black-Scholes
option-pricing model, is determined, and compensation expense recognized or
recovered during the period is adjusted accordingly. Since the fair market value
of options granted to non-employees is subject to change in the future, the
amount of the future compensation expense is subject to adjustment until the
common stock options or warrants are fully vested.
Research and Development Expenses
— Research and development expenses consist of costs incurred for direct
and overhead-related research expenses and are expensed as incurred. Costs to
acquire technologies, including licenses, that are utilized in research and
development and that have no alternative future use are expensed when incurred.
Technology developed for use in its products is expensed as incurred until
technological feasibility has been established.
Clinical Trial Expenses —
Clinical trial expenses, which are included in research and development
expenses, include obligations resulting from the Company’s contracts with
various clinical research organizations in connection with conducting clinical
trials for its product candidates. The Company recognize expenses for these
activities based on a variety of factors, including actual and estimated labor
hours, clinical site initiation activities, patient enrollment rates, estimates
of external costs and other activity-based factors. The Company believe that
this method best approximates the efforts expended on a clinical trial with the
expenses it records. The Company adjusts its rate of clinical expense
recognition if actual results differ from its estimates. If its estimates are
incorrect, clinical trial expenses recorded in any particular period could
vary.
Income Taxes — Income taxes
are accounted for using an asset and liability approach that requires the
recognition of deferred tax assets and liabilities for the expected future tax
consequences of events that have been recognized in the Company’s financial
statements or tax returns. A valuation allowance is established to reduce
deferred tax assets if all, or some portion, of such assets will more than
likely not be realized. Effective January 1, 2007, we adopted the provisions of
ASC 740-10 (formerly FIN 48), which contains a two-step process for recognizing
and measuring uncertain tax positions. The first step is to determine whether or
not a tax benefit should be recognized. A tax benefit will be recognized if the
weight of available evidence indicates that the tax position is more likely than
not to be sustained upon examination by the relevant tax authorities. The
recognition and measurement of benefits related to our tax positions requires
significant judgment, as uncertainties often exist with respect to new laws, new
interpretations of existing laws, and rulings by taxing authorities. Differences
between actual results and our assumptions or changes in our assumptions in
future periods are recorded in the period they become known.
Concentrations of Risks —
Financial instruments that potentially subject the Company to significant
concentrations of credit risk consist principally of cash, cash equivalents and
marketable securities. The Company maintains cash and cash equivalents in large
well-capitalized financial institutions and the Company’s investment policy
disallows investment in any debt securities rated less than “investment-grade”
by national ratings services. The Company has not experienced any losses on its
deposits of cash or cash equivalent or its marketable securities. The Company
also has a 36% investment in RXi, the shares of which can significantly
fluctuate. The sale of these common shares represents a future source of cash
and any decline in the share price can impact the Company.
Use of Estimates — The
preparation of the financial statements in conformity with accounting principles
generally accepted in the United States of America requires management to make
estimates and assumptions that affect the amounts reported in the financial
statements and accompanying notes. Significant estimates include the accrual for
research and development expenses, the basis for the classification of current
deferred revenue, estimated income taxes and the estimate of expense arising
from the common stock options granted to employees and non-employees. Actual
results could materially differ from those estimates.
Other comprehensive income/(loss)
— The Company follows the provisions of ASC 220, (formerly SFAS No. 130,
Reporting Comprehensive
Income), which requires separate representation of certain transactions,
which are recorded directly as components of shareholders’ equity. The Company
has no components of other comprehensive income (loss) and accordingly
comprehensive loss is the same as net loss reported.
F-11
3.
Recent Accounting Pronouncements
In
December 2007, the FASB issued guidance which is now part of ASC 810-10, Noncontrolling Interests in
Consolidated Financial Statements, an Amendment of Accounting Research Bulletin
No. 51 “ (formerly Statement of Financial Accounting Standards (SFAS)
160, Noncontrolling
Interests in Consolidated Financial Statements—an amendment of ARB No. 51
). This guidance establishes accounting and reporting standards for ownership
interests in subsidiaries held by parties other than the parent, changes in a
parent’s ownership of a noncontrolling interest, calculation and disclosure of
the consolidated net income attributable to the parent and the noncontrolling
interest, changes in a parent’s ownership interest while the parent retains its
controlling financial interest and fair value measurement of any retained
noncontrolling equity investment. The new guidance is effective for financial
statements issued for fiscal years beginning after December 15, 2008, and
interim periods within those fiscal years. Early adoption is prohibited. The
Company adopted this guidance on January 1, 2009, the beginning of its 2009
fiscal year, which resulted in certain reclassifications related to the
noncontrolling interest in the consolidated financial statements.
In March
2008, the FASB issued guidance ASC 815-10 (formerly Statement of Financial
Accounting Standards No. 161, Disclosures about Derivative
Instruments and Hedging Activities (“SFAS No. 161”). The new
standard amends Statement of Financial Accounting Standards No. 133, Accounting for Derivative
Instruments and Hedging Activities (“SFAS 133”), and seeks to enhance
disclosure about how and why a company uses derivatives; how derivative
instruments are accounted for under SFAS 133 (and the interpretations of that
standard); and how derivatives affect a company’s financial position, financial
performance and cash flows. SFAS 161 will be effective for financial
statements issued for fiscal years and interim periods beginning after November
15, 2008. Early application of the standard is encouraged, as well as
comparative disclosures for earlier periods at initial adoption. The
adoption of ASC 815-10 did not have a material impact on the Company’s
consolidated financial statements.
In April
2008, the FASB issued revised guidance on determining the useful life of
intangible assets. The revised guidance, which is now part of ASC
350-30 General Intangibles Other than Goodwill (previously Staff Position No.
FAS 142-3, Determination of
the Useful Life of Intangible Assets), amends the factors that should be
considered in developing renewal or extension assumptions used to determine the
useful life of a recognized intangible asset. The Position is
effective for fiscal years beginning after December 15, 2008 and applies
prospectively to intangible assets acquired after the effective
date. Early adoption is not permitted. The adoption of SFAS No. ASC
350-30 did not have a material impact on the Company’s consolidated financial
statements.
In May
2008, the FASB issued revised guidance on Convertible Debt
Instruments. The revised guidance which is now part of ASC
470-20 (formerly Staff Position No. Accounting Principles Board 14-1, Accounting for Convertible Debt
Instruments That May Be Settled in Cash upon Conversion (Including
Partial Cash Settlement) (“FSP No. APB 14-1”)). ASC 470-20 requires that the
liability and equity components of convertible debt instruments that may be
settled in cash upon conversion (including partial cash settlement) be
separately accounted for in a manner that reflects an issuer’s nonconvertible
debt borrowing rate. ASC 470-20 is effective for us as of January 1,
2009. The adoption of ASCO 470-20 did not have an impact on the Company’s
consolidated financial statements.
In June
2008, the FASB ratified guidance which is now part of ASC 815-40, Contracts in Entity’s Own Equity
(formerly EITF (Emerging Issues Task Force) 07-05), Determining Whether an Instrument
(or Embedded Feature) Is Indexed to an Entity’s Own Stock. The objective
of this issue is to provide guidance for determining whether an equity-linked
financial instrument (or embedded feature) is indexed to an entity’s own stock.
This issue applies to any freestanding financial instrument or embedded feature
that has all the characteristics of a derivative instrument or an instrument
which may be potentially settled in an entity’s own stock regardless of whether
the instrument possess derivative characteristics. This issue provides a
two-step approach to assist in making these determinations and is effective for
financial statements issued for fiscal years beginning after December 15, 2008.
The adoption of ASC 815-40 did not have a material impact on the Company’s
consolidated financial statements.
In April
2009, the FASB issued guidance which is now part of ASC 825-10 Financial
Instruments (formerly Financial Staff Position SFAS 107-1 and Accounting
Principles Board (APB) Opinion No. 28-1, Interim Disclosures about Fair Value
of Financial Instruments (SFAS 107-1 and APB 28-1). This
statement amends FASB Statement No. 107, Disclosures about Fair Values of
Financial Instruments, to require disclosures about fair value of
financial instruments in interim financial statements as well as in annual
financial statements. The statement also amends APB Opinion No. 28,
“Interim Financial Reporting,” to require those disclosures in all interim
financial statements. This statement is effective for interim periods
ending after June 15, 2009. The adoption of ASC 825-10 did not have an impact on
the Company’s financial statements.
F-12
In May
2009 and February 2010, the FASB issued new guidance for accounting for
subsequent events. The new guidance, which is now part of ASC 855-10, Subsequent Events (formerly,
SFAS No. 165, Subsequent
Events) is consistent with existing auditing standards in defining
subsequent events as events or transactions that occur after the balance sheet
date but before the financial statements are issued or are available to be
issued. The new guidance defines two types of subsequent events: “recognized
subsequent events” and “non-recognized subsequent events.” Recognized subsequent
events provide additional evidence about conditions that existed at the balance
sheet date and must be reflected in the company’s financial statements.
Non-recognized subsequent events provide evidence about conditions that arose
after the balance sheet date and are not reflected in the financial statements
of a company. Certain non-recognized subsequent events may require
disclosure to prevent the financial statements from being misleading. The
new guidance was effective on a prospective basis for interim or annual periods
ending after June 15, 2009. The Company adopted the provisions of ASC
855-10 as required.
In June
2009, the FASB amended ASC 860, (formerly SFAS No. 166, Accounting for Transfers of Financial
Assets, an amendment to
SFAS No. 140). ASC 860 eliminates the concept of a “qualifying
special-purpose entity,” changes the requirements for derecognizing financial
assets, and requires additional disclosures in order to enhance information
reported to users of financial statements by providing greater transparency
about transfers of financial assets, including securitization transactions, and
an entity’s continuing involvement in and exposure to the risks related to
transferred financial assets. ASC 860 is effective for fiscal years beginning
after November 15, 2009. The Company will adopt ASC 860 in fiscal
2010. The Company does not expect that the adoption of ASC 860 will
have a material impact on its financial statements.
In June
2009, the FASB amended ASC 810 (formerly SFAS No.167, Amendments to FASB Interpretation
No. 46). The amendments include: (1) the elimination of the
exemption for qualifying special purpose entities, (2) a new approach for
determining who should consolidate a variable-interest entity, and (3) changes
to when it is necessary to reassess who should consolidate a variable-interest
entity. ASC 810 is effective for the first annual reporting period
beginning after November 15, 2009 and for interim periods within that first
annual reporting period. The Company will adopt ASC 810 in fiscal 2010. The
Company does not expect that the adoption of ASC 810 will have a material impact
on its financial statements.
In June
2009, the FASB issued new guidance which is now part of ASC 105-10 (formerly
Statement of Financial Accounting Standards No. 168, The FASB Accounting Standards
Codification and the Hierarchy of Generally Accepted Accounting
Principles ). ASC 105-10 replaces FASB Statement No. 162, “The Hierarchy of Generally Accepted
Accounting Principles”, and establishes the FASB Accounting Standards
Codification as the source of authoritative accounting principles recognized by
the FASB to be applied by nongovernmental entities in the preparation of
financial statements in conformity with generally accepted accounting
principles. ASC 105-10 is effective for interim and annual periods ending after
September 15, 2009. The adoption of ASC 105-10 did not have a material impact on
the Company’s financial statements.
In
January, 2010, the FASB issued ASU 2010-06, Improving Disclosures about Fair
Value Measurements. The standard amends ASC Topic 820, Fair Value Measurements
and Disclosures to require additional disclosures related to transfers between
levels in the hierarchy of fair value measurement. The standard does not change
how fair values are measured. The standard is effective for interim and annual
reporting periods beginning after December 15, 2009. As a result, it is
effective for us in the first quarter of fiscal year 2010. We do not believe
that the adoption of ASU 2010-06 will have a material impact on consolidated our
financial statements.
4.
Receivable
At
December 31, 2009 and 2008, the Company had a receivable of $139,680 and
$127,280, respectively, primarily related to annual licensing fees due to the
Company. Due to the certainty of the collectability of the accounts receivable,
no allowance was recorded.
5.
Other Assets
At
December 31, 2009 and 2008, the Company had $323,235 and $330,032, respectively,
of non-current other assets, which consist primarily of security deposits on
contracts for research and development, prepaid insurance and leases for its
facilities.
6.
Marketable securities
The
Company held $22.8 million of marketable securities at December 31,
2009. The Company has classified these investments as available for
sale. These investments are comprised of federally insured
certificates of deposit and these four accounts detailed as follows: $5.0
million with a maturity date of January 28, 2010; $8.8 million with a maturity
date of April 1, 2010; $5 million with a maturity date of July 29, 2010; and $4
million with a maturity date of September 30, 2010.
F-13
7.
Assets Held for Sale
In May
2009, the Company substantially completed the initial phase of the closure of
its drug discovery research at its laboratory facility in San Diego, California.
The Company concluded that it will conduct its research and development
activities through third parties for the foreseeable future. The Company has
sublet the laboratory facility, sold some of the laboratory equipment and is
actively searching for additional buyers. In the third quarter of
2009, the fixed assets related to the San Diego laboratory were re-allocated
from Equipment and Furnishings to Assets Held for Sale and were written down to
their estimated net realizable value as of September 30, 2009, which resulted in
a charge of $1.2 million for that quarter. In November 2009, the Company signed
sublease agreements with two parties to sublet the facility for the remainder of
the term of the lease, which expires in October, 2010. The Company
recognized an onerous lease accrual of $254,000 as of September
2009.
Assets
held for sale consisted of the following:
|
December
31,
|
||||||||||
|
(in
thousands)
|
2009
|
2008
|
||||||||
|
Long-lived
assets held for sale:
|
||||||||||
|
Plant
and equipment, net
|
$
|
74
|
$
|
—
|
||||||
| Asssets held for sale |
$
|
74
|
$
|
—
|
||||||
As of
December 31, 2009, the carrying amount approximates fair value. The costs
associated with disposing of these assets held for sale are
minimal.
8.
Equipment, Furnishings and Molecular Library, net
Equipment,
furnishings and molecular library, net, at December 31, 2009 and 2008 consist of
the following (in thousands):
|
|
2009
|
2008
|
||||||
|
Equipment
and furnishings
|
$ | 334 | $ | 2,606 | ||||
|
Less
— accumulated depreciation
|
(159 | ) | (771 | ) | ||||
|
Equipment
and furnishings, net
|
175 | 1,835 | ||||||
|
Molecular
library
|
$ | 0 | $ | 447 | ||||
|
Less
— accumulated amortization
|
0 | (343 | ) | |||||
|
Molecular
library, net
|
$ | 0 | $ | 104 | ||||
The
molecular library was purchased in 2004 and placed in service by the Company in
March 2005. In the third quarter of 2009, the molecular library was re-allocated
to Assets Held for Sale and was written down to its estimated net realizable
value.
Depreciation
and amortization expense for the years ended December 31, 2009, 2008 and 2007
were approximately $475,000, $625,000 and $272,000, respectively.
9.
Accrued Expenses and Other Current Liabilities
Accrued
expenses and other current liabilities at December 31, 2009 and 2008 are
summarized below (in thousands).
|
|
2009
|
2008
|
||||||
|
Professional
fees
|
$ | 423 | $ | 531 | ||||
|
Research
and development costs
|
1,255 | 1,662 | ||||||
|
Wages,
bonuses and employee benefits
|
208 | 196 | ||||||
|
Income
taxes
|
— | — | ||||||
|
Other
|
606 | 168 | ||||||
|
Total
|
$ | 2,492 | $ | 2,557 | ||||
F-14
10.
Warrant Liabilities
Liabilities
measured at market value on a recurring basis include warrant liabilities
resulting from the Company’s 2009 equity financing. In accordance
with the guidance which is now ASC 815-40 (formerly EITF (Emerging Issues Task
Force) 00-19, Accounting for
Derivative Financial Instruments Indexed and Potentially Settled in a Company’s
Own Stock), the warrant liabilities are being marked to market each
quarter-end until they are completely settled. The warrants are
valued using the Black-Scholes method, using assumptions consistent with our
application of ASC 505-50. The gain or loss resulting from the marked
to market calculation is shown on the Consolidated Statements of Operations as
Gain on warrant derivative liability. The Company recognized a gain
of $657,000 during 2009.
11.
Commitments and Contingencies
The
Company acquires assets still in development and enters into research and
development arrangements with third parties that often require milestone and
royalty payments to the third party contingent upon the occurrence of certain
future events linked to the success of the asset in development. Milestone
payments may be required, contingent upon the successful achievement of an
important point in the development life-cycle of the pharmaceutical product
(e.g., approval of the product for marketing by a regulatory agency). If
required by the arrangement, CytRx may have to make royalty payments based upon
a percentage of the sales of the pharmaceutical product in the event that
regulatory approval for marketing is obtained. Because of the contingent nature
of these payments, they are not included in the table of contractual
obligations.
These
arrangements may be material individually, and in the unlikely event that
milestones for multiple products covered by these arrangements were reached in
the same period, the aggregate charge to expense could be material to the
results of operations in any one period. In addition, these arrangements often
give CytRx the discretion to unilaterally terminate development of the product,
which would allow CytRx to avoid making the contingent payments; however, CytRx
is unlikely to cease development if the compound successfully achieves clinical
testing objectives.
CytRx’s
current contractual obligations that will require future cash payments are as
follows (in thousands):
|
Operating
Leases (1)(2)
|
Employment
Agreements (3)
|
Subtotal
|
Research
and Development (4)
|
Total
|
||||||||||||||||
|
2010
|
$ | 873 | $ | 2,210 | $ | 3,083 | $ | 3,018 | $ | 6,101 | ||||||||||
|
2011
|
536 | — | 536 | 49 | 585 | |||||||||||||||
|
2012
|
459 | — | 459 | 149 | 608 | |||||||||||||||
|
2013
|
318 | — | 318 | 149 | 467 | |||||||||||||||
|
2014 and
thereafter
|
372 | — | 372 | 383 | 755 | |||||||||||||||
|
Total
|
$ | 2,558 | $ | 2,210 | $ | 4,768 | $ | 3,748 | $ | 8,516 | ||||||||||
____________
|
(1)
|
Operating
leases are primarily facility lease related obligations, as well as
equipment and software lease obligations with third party
vendors.
|
(2)
The Company is entitled to receive future rental income under subleases in place
which would be offset against future operating lease obligations as follows:
$519,000 in 2010, $350,000 in 2011 and $235,000 in 2012.
|
(3)
|
Employment
agreements include management contracts, which have been revised from time
to time, provide for minimum salary levels, adjusted annually at the
discretion of the Company's Compensation Committee, as well as for minimum
bonuses that are payable.
|
|
(4)
|
Research
and development obligations relate primarily to clinical trials. Most of
these purchase obligations are
cancelable.
|
The
Company applies the disclosure provisions of ASC 460 (formerly FASB
Interpretation No. 45, Guarantor’s Accounting and
Disclosure Requirements for Guarantees, Including Indirect Guarantees of
Indebtedness of Others), to its agreements that contain guarantee or
indemnification clauses. The Company provides (i) indemnifications of varying
scope and size to certain investors and other parties for certain losses
suffered or incurred by the indemnified party in connection with various types
of third-party claims; and (ii) indemnifications of varying scope and size to
officers and directors against third party claims arising from the services they
provide to us. These indemnifications and guarantees give rise only to the
disclosure provisions of ASC 460. To date, the Company has not incurred material
costs as a result of these obligations and does not expect to incur material
costs in the future; further, the Company maintains insurance to cover certain
losses arising from these indemnifications. Accordingly, the Company has not
accrued any liabilities in its consolidated financial statements related to
these indemnifications or guarantees.
F-15
12.
Equity Transactions
On April
19, 2007, the Company completed a $37.0 million private equity financing in
which it issued 8.6 million shares of its common stock at $4.30 per share. Net
of investment banking commissions, legal, accounting and other expenses related
to the transaction, the Company received approximately $34.2 million of
proceeds.
On March
11, 2008, the Company paid a dividend to its stockholders of approximately 36%
of the outstanding shares of RXi common stock. In connection with that dividend,
the Company adjusted the price of warrants to purchase approximately 10.6
million shares that had been issued in prior equity financings in October 2004,
January 2005 and March 2006. The adjustments were made as a result of
anti-dilution provisions in those warrants that were triggered by the Company’s
distribution of a portion of its assets to its stockholders. The Company
accounted for the anti-dilution adjustments as deemed dividends analogous with
the guidance in ASC 470 (previously Emerging Issues Task Force Issue (“EITF”)
No. 98-5, Accounting for
Convertible Securities with Beneficial Conversion Features or Contingently
Adjustable Conversion Ratio)s, and ASC 470 (previously EITF 00-27, Application of 98-5 to Certain
Convertible Instruments), and recorded an approximate $757,000 charge to
accumulated deficit and a corresponding credit to additional paid-in
capital.
On July
27, 2009, the Company completed a $20.0 million registered direct public
offering in which it issued approximately 15.3 million shares of its common
stock at a price of $1.31 per share, and warrants to purchase an additional
approximately 4.7 million shares of common stock at an exercise price of $1.70
per share. Net of investment banking commissions, advisory fees, legal,
accounting and other fees related to the transaction, the Company received
proceeds of approximately $18.3 million (without giving effect to any proceeds
that may in the future be received by the Company upon exercise of warrants sold
in the offering). Immediately after the sale, the Company had
approximately 109.5 million shares of common stock outstanding, without giving
effect to the possible exercise of the warrants sold in the offering or any of
our other outstanding warrants or stock options.
13.
Noncontrolling Interest in RXi
Through
February 2008, the Company owned approximately 85% of the outstanding shares of
common stock of RXi. While RXi was majority-owned, the Company’s consolidated
financial statements reflected 100% of the assets and liabilities and results of
operations of RXi, with the interests of the minority shareholders of RXi
recorded as “noncontrolling interests.” The Company offset $88,000 of loss in
noncontrolling interest of RXi against its net loss for the months of January
and February 2008.
On March
11, 2008, the Company distributed to its stockholders approximately 4.5 million
shares of RXi common stock, or approximately 36% of RXi’s outstanding shares,
which reduced the Company’s ownership to less than 50% of RXi. As a result, the
Company began to account for its investment in RXi using the equity method,
under which the Company records only its pro-rata share of the financial results
of RXi. Because only a portion of RXi’s financial results for 2008 were recorded
by the Company under the equity method, the Company’s results of operations for
2008 are not directly comparable to results of operations for the same period in
2009. The future results of operations of the Company also will not be directly
comparable to corresponding periods in prior years during which our financial
statements reflected the consolidation of RXi.
14.
Equity Investment in RXi
Management
determined that the distribution of RXi common stock to stockholders of CytRx in
March 2008 represented a partial spin-off of RXi and accounted for the
distribution of the RXi common shares at cost. As a result of its reduced
ownership in RXi, CytRx began to account for its investment in RXi using the
equity method, under which CytRx records only its pro-rata share of the
financial results of RXi. The investment balance in RXi has been reduced to zero
and the Company has not made any advances or guarantees to RXi. Therefore the
Company has stopped recording its share of losses from RXi.
F-16
The
following table presents summarized financial information for RXi for the years
ended December 31, 2009 and 2008:
|
Income Statement Data (in
thousands)
|
Year
Ended
December 31, 2009
|
Year
Ended
December 31, 2008
|
||||||
|
Sales
|
$ | — | $ | — | ||||
|
Gross
profit
|
— | — | ||||||
|
Loss
from continuing operations
|
(18,387 | ) | (14,553 | ) | ||||
|
Net
Loss
|
(18,387 | ) | (14,373 | ) | ||||
|
Balance Sheet Data (in
thousands)
|
December 31, 2009
|
December 31, 2008
|
||||||
|
Current
assets
|
$ | 5,805 | $ | 9,929 | ||||
|
Noncurrent
assets
|
448 | 430 | ||||||
|
Current
liabilities
|
5,475 | 1,387 | ||||||
|
Stockholders’
equity
|
743 | 8,968 | ||||||
At
December 31, 2009, the fair value of CytRx’s 5,768,881 shares of RXi common
stock was $26.4 million based o the closing price of RXi common stock (NASDAQ:
RXII) on that date. As CytRx accounts for its investment in RXi using the equity
method, this value is not reflected 0n the CytRx balance sheet.
15.
Stock Options and Warrants
CytRx
Options
The
Company has a 2000 Long-Term Incentive Plan under which an aggregate of 10
million shares of common stock were originally reserved for
issuance. As of December 31, 2009, there were approximately 7.9
million shares subject to outstanding stock options and approximately 0.2
million shares available for future grant under this plan.
On July
1, 2009, the Company’s stockholders adopted a new 2008 Stock Incentive Plan
under which 10 million shares of common stock were reserved for
issuance. As of December 31, 2009, there were 1.1 million shares
subject to outstanding stock options under the plan and 8.9 million shares
available for future grant under this plan.
The
Company has adopted the provisions of ASC 718 (previously SFAS No. 123(R), Share-Based Payment (“SFAS
123(R)”)), which requires the measurement and recognition of compensation
expense for all stock-based awards made to employees and
non-employees.
For stock
options and stock warrants paid in consideration of services rendered by
non-employees, the Company recognizes compensation expense in accordance with
the requirements of ASC 718 (previously SFAS No. 123(R)), ASC 505-50 (previously
Emerging Issues Task Force Issue No. 96-18 (“EITF 96-18”)), Accounting for Equity
Instruments that are Issued to other than Employees for Acquiring, or in
Conjunction with Selling Goods or Services and ASC 505 (previously EITF
00-18, Accounting Recognition
for Certain Transactions involving Equity Instruments Granted to Other Than
Employees), as amended.
Non-employee
option grants that do not vest immediately upon grant are recorded as an expense
over the vesting period. At the end of each financial reporting period prior to
performance, the value of these options, as calculated using the Black-Scholes
option-pricing model, is determined, and compensation expense recognized or
recovered during the period is adjusted accordingly. Since the fair market value
of options granted to non-employees is subject to change in the future, the
amount of the future compensation expense is subject to adjustment until the
common stock options or warrants are fully vested.
At the
2009 Annual Meeting of Stockholders held on July 1, 2009, the Company’s
stockholders approved an amendment to the Company’s 2000 Long-Term Incentive
Plan to allow for a one-time stock option re-pricing program for employees and
officers. Pursuant to the re-pricing program, 3,265,500 eligible
stock options held by ten eligible employees and officers were amended to reduce
the exercise prices of the options to $1.15 per share, which was the closing
sale price of CytRx’s common stock as reported on the Nasdaq Capital Market on
the July 1, 2009 completion date of the re-pricing program, and to impose a
new option vesting schedule. None of the amended options vested
immediately. To the extent a participating employee’s or officer’s
eligible options were vested on the amendment date, the amended options vested
in full on December 31, 2009, so long as the employee or officer remained
in the Company’s employ through that date. To the extent a participating
employee’s or officer’s eligible options were unvested as of July 1, 2009,
the original scheduled vesting was suspended until December 31, 2009 and
resumed after that date, so long as the employee or officer remained in the
Company’s employ through such date. The incremental cost of the re-pricing
program was approximately $0.4 million.
F-17
ASC 718
(previously SFAS No. 123(R)) requires the re-pricing of equity awards to be
treated as the repurchase of the old award for a new award of equal or greater
value, incurring additional compensation cost for any incremental value. This
incremental difference in value is measured as the excess of the fair value of
the modified award determined in accordance with the provisions of ASC 718 over
the fair value of the original award immediately before its terms are modified,
measured based on the share price and other pertinent factors at that date. ASC
718 provides that this incremental fair value, plus the remaining
unrecognized compensation cost from the original measurement of the fair value
of the old option, must be recognized over the remaining vesting
period.
The fair
value of the stock options at the date of grant was estimated using the
Black-Scholes option-pricing model, based on the following
assumptions:
|
2009
|
2008
|
2007
|
||||||||||
|
Risk-free
interest rate
|
1.95 | % | 2.68 | % | 4.41 | % | ||||||
|
Expected
volatility
|
97 | % | 102 | % | 108 | % | ||||||
|
Expected
lives (years)
|
6 | 6 | 6 | |||||||||
|
Expected
dividend yield
|
0.00 | % | 0.00 | % | 0.00 | % | ||||||
The
Company’s computation of expected volatility is based on the historical daily
volatility of its publicly traded stock. For option grants issued during years
ended December 31, 2009, 2008 and 2007, the Company used a calculated volatility
for each grant. The Company’s computation of expected lives was estimated using
the simplified method provided for under ASC 718 (previously Staff
Accounting Bulletin 107, Share-Based Payment (“SAB
107”)), which averages the contractual term of the Company’s options of ten
years with the average vesting term of three years for an average of six years.
The dividend yield assumption of zero is based upon the fact the Company has
never paid cash dividends and presently has no intention of paying cash
dividends. The risk-free interest rate used for each grant is equal to the U.S.
Treasury rates in effect at the time of the grant for instruments with a similar
expected life. Based on historical experience, for the year ended December 31,
2009, the Company has estimated an annualized forfeiture rate of 14% for options
granted to its employees, 2% for options granted to senior management and 0% for
options granted to directors. For the years ended December 31, 2008 and 2007,
the Company has estimated an annualized forfeiture rate for each period of 10%
for options granted to its employees, 3% for options granted to senior
management and 0% for options granted to directors. Compensation costs will be
adjusted for future changes in estimated forfeitures. The Company will record
additional expense if the actual forfeitures are lower than estimated and will
record a recovery of prior expense if the actual forfeiture rates are higher
than estimated. No amounts relating to employee stock-based compensation have
been capitalized.
At
December 31, 2009, there remained approximately $2.5 million of unrecognized
compensation expense related to unvested stock options granted to current and
former employees and directors, to be recognized as expense over a
weighted-average period of 1.12 years. Presented below is the Company’s stock
option activity for employees and directors:
|
Stock Options
|
Weighted
Average
Exercise Price
|
|||||||||||||||||||||||
|
2009
|
2008
|
2007
|
2009
|
2008
|
2007
|
|||||||||||||||||||
|
Outstanding
— beginning of year
|
6,409,940 | 4,932,273 | 4,500,208 | $ | 1.99 | $ | 2.46 | $ | 1.66 | |||||||||||||||
|
Granted
|
2,351,000 | 2,021,500 | 1,685,500 | 1.00 | 0.79 | 4.02 | ||||||||||||||||||
|
Exercised
|
(8,333 | ) | (54,737 | ) | (1,030,932 | ) | 0.37 | 0.92 | 1.76 | |||||||||||||||
|
Forfeited
|
(713,018 | ) | (473,096 | ) | (222,503 | ) | 2.57 | 2.08 | 1.24 | |||||||||||||||
|
Expired
|
(27,499 | ) | (16,000 | ) | — | 1.07 | 1.00 | — | ||||||||||||||||
|
Outstanding
— end of year
|
8,012,090 | 6,409,940 | 4,932,273 | 1.02 | 1.99 | 2.46 | ||||||||||||||||||
|
Exercisable
at end of year
|
2,261,309 | 4,109,839 | 3,210,320 | $ | 1.16 | $ | 2.07 | $ | 1.93 | |||||||||||||||
|
Weighted
average fair value of stock options granted during the
year:
|
$ | 0.77 | $ | 0.63 | $ | 3.34 | ||||||||||||||||||
A summary
of the activity for unvested employee stock options (excluding re-priced
options) as of December 31, and changes during the year is presented
below:
|
|
Stock Options
|
Weighted
Average
Grant
Date Fair
Value per Share
|
||||||||||||||||||||||
|
|
2009
|
2008
|
2007
|
2009
|
2008
|
2007
|
||||||||||||||||||
|
Nonvested
at January 1,
|
2,300,100 | 1,721,952 | 1,183,214 | $ | 1.52 | $ | 2.92 | $ | 0.99 | |||||||||||||||
|
Granted
|
2,351,000 | 2,021,500 | 1,685,500 | 0.77 | 0.63 | 3.34 | ||||||||||||||||||
|
Vested
|
(924,392 | ) | (970,256 | ) | (924,259 | ) | 2.10 | 2.10 | 1.67 | |||||||||||||||
|
Pre-vested
forfeitures
|
(713,018 | ) | (473,096 | ) | (222,503 | ) | 2.11 | 1.74 | 1.06 | |||||||||||||||
|
Nonvested
at December 31,
|
3,013,690 | 2,300,100 | 1,721,952 | $ | 0.70 | $ | 1.52 | $ | 2.92 | |||||||||||||||
F-18
For stock
options paid in consideration of services rendered by non-employees, the Company
recognizes compensation expense in accordance with the requirements of ASC 718,
ASC 503-50 (formerly Emerging Issues Task Force Issue No. 96-18 (“EITF 96-18”),
Accounting for Equity
Instruments that are Issued to other than Employees for Acquiring, or in
Conjunction with Selling Goods or Services and formerly EITF 00-18, Accounting Recognition for Certain
Transactions involving Equity Instruments Granted to Other Than Employees, as
amended).
Non-employee
option grants that do not vest immediately upon grant are recorded as an expense
over the vesting period. At the end of each financial reporting period prior to
performance, the value of these options, as calculated using the Black-Scholes
option-pricing model, is determined, and compensation expense recognized or
recovered during the period is adjusted accordingly. Since the fair market value
of options granted to non-employees is subject to change in the future, the
amount of the future compensation expense is subject to adjustment until the
common stock options are fully vested.
The
Company recorded approximately $0, ($0.4) million and $0.4 million of non-cash
charges related to the issuance of stock options to certain consultants in
exchange for services during 2009, 2008 and 2007, respectively.
At
December 31, 2009, there remained approximately $0.1 million (subject to change
in the future based on vesting date fair value) of unrecognized compensation
expense related to unvested non-employee stock options to be recognized as
expense over a weighted-average period of 2.0 years. Presented below is the
Company’s non-employee stock option activity:
|
|
Stock Options
|
Weighted
Average
Exercise Price
|
||||||||||||||||||||||
|
|
2009
|
2008
|
2007
|
2009
|
2008
|
2007
|
||||||||||||||||||
|
Outstanding
— beginning of year
|
995,000 | 1,067,000 | 2,358,000 | $ | 0.91 | $ | 1.44 | $ | 1.66 | |||||||||||||||
|
Granted
|
— | 350,000 | — | — | 0.35 | — | ||||||||||||||||||
|
Exercised
|
— | — | (728,500 | ) | — | — | 1.86 | |||||||||||||||||
|
Forfeited
|
— | (402,000 | ) | (562,500 | ) | — | 1.85 | 1.85 | ||||||||||||||||
|
Expired
|
— | (20,000 | ) | — | — | 0.30 | — | |||||||||||||||||
|
Outstanding
— end of year
|
995,000 | 995,000 | 1,067,000 | 0.91 | 0.91 | 1.44 | ||||||||||||||||||
|
Exercisable
at end of year
|
545,080 | 445,000 | 817,000 | $ | 1.00 | $ | 1.18 | $ | 1.54 | |||||||||||||||
|
Weighted
average fair value of stock options granted during the
year:
|
$ | 0 | $ | 0.33 | $ | 0 | ||||||||||||||||||
The fair
value of the stock options at the date of grant was estimated using the
Black-Scholes option-pricing model, based on the following
assumptions:
|
2009
|
2008
|
2007
|
||||||||||
|
Risk-free
interest rate
|
— | 2.68 | % | — | ||||||||
|
Expected
volatility
|
— | 123 | % | — | ||||||||
|
Expected
lives (years)
|
— | 10 | — | |||||||||
|
Expected
dividend yield
|
— | 0 | % | — | ||||||||
A summary
of the activity for nonvested, non-employee stock options as of December 31, and
changes during the years are presented below:
|
|
Stock Options
|
Weighted
Average
Grant
Date Fair
Value per Share
|
||||||||||||||||||||||
|
|
2009
|
2008
|
2007
|
2009
|
2008
|
2007
|
||||||||||||||||||
|
Nonvested
at January 1,
|
550,000 | 250,000 | 916,663 | $ | 0.58 | $ | 1.00 | $ | 1.44 | |||||||||||||||
|
Granted
|
— | 350,000 | — | — | 0.33 | — | ||||||||||||||||||
|
Vested
|
(100,080 | ) | (50,000 | ) | (104,163 | ) | 0.33 | 2.33 | 1.63 | |||||||||||||||
|
Pre-vested
forfeitures
|
— | — | (562,500 | ) | — | — | 1.63 | |||||||||||||||||
|
Nonvested
at December 31,
|
449,920 | 550,000 | 250,000 | $ | 1.38 | $ | 0.58 | $ | 1.00 | |||||||||||||||
F-19
The
following table summarizes significant ranges of outstanding stock options under
the three plans at December 31, 2009:
|
Range
of
Exercise Prices
|
Number of Options
|
Weighted
Average
RemainingContractual Life
(years)
|
Weighted
Average
Exercise Price
|
Number
of
Options
Exercisable
|
Weighted
Average
Contractual Life
|
Weighted
Average
Exercise Price
|
||||||||||||||||||||
| $ | 0.30 — 1.00 | 2,302,607 | 7.89 | $ | 0.57 | 1,290,762 | 7.89 | $ | 0.62 | |||||||||||||||||
| $ | 1.01 — 1.50 | 6,176,483 | 7.20 | 1.13 | 3,723,317 | 7.20 | 1.16 | |||||||||||||||||||
| $ | 2.35 — 3.33 | 528,000 | 4.55 | 2.27 | 528,000 | 4.55 | 2.27 | |||||||||||||||||||
| 9,007,090 | 7.22 | $ | 1.05 | 5,542,079 | 7.22 | $ | 1.16 | |||||||||||||||||||
The
aggregate intrinsic value of outstanding options as of December 31, 2009 was
$1.3 million, which represents options whose exercise price was less than the
closing fair market value of the Company’s common stock on December 31, 2009 of
$1.12.
RXi
Pharmaceuticals
RXi has
its own stock option plan. RXi accounted for stock option expense in the same
manner as CytRx as described above.
As
discussed in Note 2, the Company has accounted for its investment in RXi under
the equity method since March 2008, and accordingly, the following table sets
forth the total stock-based compensation expense for January and February 2008
resulting from RXi stock options that is included in the Company’s consolidated
statements of operations:
|
|
2009
|
2008
|
2007
|
|||||||||
|
Research
and development — employee
|
$ | — | $ | 28,000 | $ | 120,000 | ||||||
|
General
and administrative — employee
|
— | 369,000 | 931,000 | |||||||||
|
Total
employee stock-based compensation
|
$ | — | $ | 397,000 | $ | 1,051000 | ||||||
|
Research
and development — non-employee
|
$ | — | $ | 121,000 | $ | 1,043,000 | ||||||
|
General
and administrative — non-employee
|
— | — | — | |||||||||
|
Total
non-employee stock-based compensation
|
$ | — | $ | 121,000 | $ | 1,043,000 | ||||||
CytRx
Warrants
A summary
of the Company’s warrant activity and related information for the years ended
December 31 are shown below.
|
Warrants
|
Weighted
Average
Exercise Price
|
|||||||||||||||||||||||
|
2009
|
2008
|
2007
|
2009
|
2008
|
2007
|
|||||||||||||||||||
|
Outstanding
— beginning of year
|
10,634,848 | 13,031,515 | 23,360,165 | $ | 1.40 | $ | 1.87 | $ | 1.83 | |||||||||||||||
|
Granted
|
6,328,330 | — | — | 1.61 | — | — | ||||||||||||||||||
|
Exercised
|
(250,000 | ) | (951,665 | ) | (10,233,650 | ) | 1.16 | 0.97 | 1.77 | |||||||||||||||
|
Forfeited
|
— | — | — | — | — | — | ||||||||||||||||||
|
Expired
|
(1,295,000 | ) | (1,445,002 | ) | (95,000 | ) | 1.28 | 2.60 | 2.25 | |||||||||||||||
|
Outstanding
— end of year
|
15,418,178 | 10,634,848 | 13,031,515 | 1.50 | 1.40 | 1.87 | ||||||||||||||||||
|
Exercisable
at end of year
|
15,418,178 | 10,634,848 | 13,031,515 | $ | 1.50 | $ | 1.40 | $ | 1.87 | |||||||||||||||
|
Weighted
average fair value of warrants granted during the year:
|
$ | 1.61 | $ | — | $ | — | ||||||||||||||||||
F-20
The
following table summarizes additional information concerning warrants
outstanding and exercisable at December 31, 2009:
|
Range of Exercise Prices
|
Number
of
Shares
|
Weighted
Average
Remaining
Contractual
Life
(years)
|
Weighted
Average
Exercise Price
|
Warrants
Number
of
Shares
Exercisable
|
Exercisable
Weighted
Average
Exercise Price
|
|||||||||||||||||
| $ | 0.26 — 1.26 | 3,322,767 | 2.22 | $ | 0.94 | 3,322,767 | $ | 0.94 | ||||||||||||||
| $ | 1.51 — 1.62 | 6,861,790 | 0.07 | 1.51 | 6,861,790 | 1.51 | ||||||||||||||||
| $ | 1.70 — 1.84 | 4,735,973 | 4.57 | 1.70 | 4,735,973 | 1.70 | ||||||||||||||||
| $ | 2.25 — 4.00 | 497,648 | 0.90 | 3.07 | 497,648 | 3.07 | ||||||||||||||||
| 15,418,178 | 1.94 | $ | 1.48 | 15,418,178 | $ | 1.50 | ||||||||||||||||
16.
ALSCRT Amendment
Pursuant
to an amendment signed between the Company and the beneficiary of the ALSCRT on
August 6, 2009, the Company was released from all restrictions on the use of any
proceeds previously paid to the Company in connection with the
arrangement. As a result, the Company recognized $6.7 million as
service revenue in the third quarter of 2009, which represented the remaining
deferred revenue and previously un-recognized portion of the value
received.
17.
Stockholder Protection Rights Plan
Effective
April 16, 1997, the Company’s board of directors declared a distribution of one
right (“Rights”) for each outstanding share of the Company’s common stock to
stockholders of record at the close of business on May 15, 1997 and for each
share of common stock issued by the Company thereafter and prior to a Flip-in
Date (as defined below). Each Right entitles the registered holder to purchase
from the Company one-ten thousandth (1/10,000th) of a share of Series A Junior
Participating Preferred Stock, at an exercise price of $30. The Rights are
generally not exercisable until 10 business days after an announcement by the
Company that a person or group of affiliated persons (an “Acquiring Person”) has
acquired beneficial ownership of 15% or more of the Company’s then outstanding
shares of common stock (a “Flip-in Date”).
In the
event the Rights become exercisable as a result of the acquisition of shares,
each Right will enable the owner, other than the Acquiring Person, to purchase
at the Right’s then-current exercise price a number of shares of common stock
with a market value equal to twice the exercise price. In addition, unless the
Acquiring Person owns more than 50% of the outstanding shares of common stock,
the Board of Directors may elect to exchange all outstanding Rights (other than
those owned by such Acquiring Person) at an exchange ratio of one share of
common stock per Right. All Rights that are owned by any person on or after the
date such person becomes an Acquiring Person will be null and void.
The
Rights have been distributed to protect the Company’s stockholders from coercive
or abusive takeover tactics and to give the Board of Directors more negotiating
leverage in dealing with prospective acquirors. In April 2007, the Company
extended the stockholder rights plan through April 2017.
18.
Income Taxes
At
December 31, 2009, the Company had federal and state net operating loss
carryforwards of $91.6 million and $81.9 million, respectively, available to
offset against future taxable income, which expire in 2011 through 2029. As a
result of a change in-control that occurred in the CytRx shareholder base in
July 2002, approximately $34.7 million in federal net operating loss
carryforwards became limited in their availability to $0.3 million annually.
Management currently believes that the remaining $56.9 million in federal net
operating loss carryforwards, and the $57.6 million in state net operating loss
carryforwards, are unrestricted. However, management is reviewing its recent
equity transactions to determine if they may have resulted in any further
restrictions on the Company’s net operating loss carryforwards. As of December
31, 2009, CytRx also had research and development and alternative minimum tax
credits for federal and state purposes of approximately $8.9 million and $0,
respectively, available for offset against future income taxes, which expire in
2022 through 2029. Based on an assessment of all available evidence including,
but not limited to, the Company’s limited operating history in its core business
and lack of profitability, uncertainties of the commercial viability of its
technology, the impact of government regulation and healthcare reform
initiatives, and other risks normally associated with biotechnology companies,
the Company has concluded that it is more likely than not that these net
operating loss carryforwards and credits will not be realized and, as a result,
a 100% deferred tax valuation allowance has been recorded against these
assets.
F-21
Deferred
income taxes reflect the net effect of temporary differences between the
financial reporting carrying amounts of assets and liabilities and income tax
carrying amounts of assets and liabilities. The components of the Company’s
deferred tax assets and liabilities, all of which are long-term, are as follows
(in thousands):
|
|
December 31,
|
|||||||
|
|
2009
|
2008
|
||||||
|
Net
operating loss carryforwards
|
$ | 35,285 | $ | 34,407 | ||||
|
Tax
credit carryforwards
|
8,957 | 6,071 | ||||||
|
Equipment,
furnishings and other
|
8,142 | 4,358 | ||||||
|
Deferred
revenue
|
— | 3,744 | ||||||
|
Total
deferred tax assets
|
52,384 | 48,580 | ||||||
|
Deferred
tax liabilities
|
(560 | ) | (407 | ) | ||||
|
Net
deferred tax assets
|
51,824 | 48,173 | ||||||
|
Valuation
allowance
|
(51,824 | ) | (48,173 | ) | ||||
| $ | — | $ | — | |||||
For all
years presented, the Company did not recognize any deferred tax assets or
liabilities. The net change in valuation allowance for the years ended December
31, 2009 and 2008 was $3,651,000 and ($5,073,000), respectively.
The
provision for income taxes differs from the provision computed by applying the
Federal statutory rate to net loss before income taxes as follows (in
thousands):
|
Years ended December 31,
|
||||||||||||
|
2009
|
2008
|
2007
|
||||||||||
|
Federal
benefit at statutory rate
|
$ | (1,809 | ) | $ | (8,899 | ) | $ | (7,781 | ) | |||
|
State
income taxes, net of Federal taxes
|
(310 | ) | (1,525 | ) | (908 | ) | ||||||
|
Permanent
differences
|
(136 | ) | 16,272 | 65 | ||||||||
|
Provision
related to change in valuation allowance
|
3,651 | (5,073 | ) | 9,416 | ||||||||
|
Net
change in research and development tax credits
|
(2,826 | ) | (2,207 | ) | (1,125 | ) | ||||||
|
Other,
net
|
911 | (2,304 | ) | 373 | ||||||||
| $ | (519 | ) | $ | 872 | $ | 40 | ||||||
As of
January 1, 2007, the Company had no unrecognized tax benefits and there was no
effect on its financial condition or results of operations as a result of
implementing ASC 740. There have been no changes to the Company’s liability for
unrecognized tax benefits during the year ended December 31, 2009.
The
Company and its Subsidiaries file income tax returns in the U.S. Federal
jurisdiction and various state jurisdictions. As of the date of adoption of ASC
740 and the year ended December 31, 2009, the tax returns for 2006 through 2008
remain open to examination by the Internal Revenue Service and various state tax
authorities.
The
Company’s policy is to recognize any interest and penalties related to
unrecognized tax benefits as a component of income tax expense. As of the date
of adoption of ASC 740 and the years ended December 31, 2009 and 2008, the
Company had accrued no interest or penalties related to uncertain tax
positions.
F-22
19.
Quarterly Financial Data (unaudited)
Summarized
quarterly financial data for 2009 and 2008 is as follows (in thousands, except
per share data):
|
|
Quarters Ended
|
|||||||||||||||
|
|
March 31
|
June 30
|
September 30
|
December 31
|
||||||||||||
|
(In
thousands, except per share data)
|
||||||||||||||||
|
2009
|
||||||||||||||||
|
Total
revenues
|
$ | 1,483 | $ | 1,000 | $ | 6,954 | $ | 100 | ||||||||
|
Net
income (loss)
|
(3,973 | ) | (2,226 | ) | 3,863 | (2,464 | ) | |||||||||
|
Net
income (loss) applicable to common stockholders
|
$ | (3,973 | ) | $ | (2,226 | ) | $ | 3,863 | $ | (2,464 | ) | |||||
|
Basic
and diluted loss per share applicable to common stock
|
$ | (0.04 | ) | $ | (0.02 | ) | $ | 0.04 | $ | (0.03 | ) | |||||
|
2008
|
||||||||||||||||
|
Total
revenues
|
$ | 2,181 | $ | 1,740 | $ | 917 | $ | 1,428 | ||||||||
|
Net
loss
|
(5,374 | ) | (5,826 | ) | (12,316 | ) | (3,530 | ) | ||||||||
|
Deemed
dividend for anti-dilution adjustments made to outstanding
common
stock warrants
|
(757 | ) | — | — | — | |||||||||||
|
Net
loss applicable to common stockholders
|
$ | (6,131 | ) | $ | (5,826 | ) | $ | (12,316 | ) | $ | (3,530 | ) | ||||
|
Basic
and diluted income (loss) per share applicable to common
stock
|
$ | (0.07 | ) | $ | (0.06 | ) | $ | (0.14 | ) | $ | (0.03 | ) | ||||
Quarterly
and year-to-date loss per share amounts are computed independently of each
other. Therefore, the sum of the per share amounts for the quarters may not
agree to the per share amounts for the year.
In
connection with the Company’s adjustment to the exercise terms of certain
outstanding warrants to purchase common stock on March 11, 2008, the Company
recorded a deemed dividend of $757,000. That deemed dividend is reflected as an
adjustment to net loss for the first quarter of 2008 to arrive at net loss
applicable to common stockholders on the consolidated statements of operations
and for purposes of calculating basic and diluted earnings per
shares.
20.
Acquisition of Innovive Pharmaceuticals
On
September 19, 2008, the Company completed the merger of Innovive with CytRx
Merger Subsidiary, Inc., the Company’s wholly owned subsidiary, with Innovive
continuing as the surviving corporation. As a result, Innovive became a wholly
owned subsidiary of CytRx and changed its name to CytRx Oncology Corporation.
Because Innovive was a development stage company, under accounting principles
generally accepted in the United States and the SEC regulations,
it was not considered a business. Accordingly, CytRx accounted for
the merger in accordance with ASC 350 (previously Statement of Financial
Accounting Standards No. 142, Goodwill and Other Intangible Assets), for
transactions other than a business combination. The initial merger
consideration, together with direct costs incurred to effect the merger, were
allocated to the individual assets acquired, including identifiable intangible
assets and liabilities assumed based on the relative fair value. No
goodwill was recorded. The Company’s consolidated financial
statements reflect these fair values and were not restated retroactively to
reflect the historical financial position or results of operations of Innovive.
In connection with the merger, the Company recorded a one-time expense for
acquired in-process research and development. Under the merger agreement by
which the Company acquired Innovive, it agreed to pay the former Innovive
stockholders a total of up to approximately $18.3 million of future earnout
merger consideration, subject to the Company’s achievement of specified net
sales under the Innovive license agreements. The earnout merger
consideration, if any, will be payable in shares of CytRx common stock, subject
to specified conditions, or, at the Company’s election, in cash or by a
combination of shares of CytRx common stock and cash. The Company’s
common stock will be valued for purposes of any future earnout merger
consideration based upon the trading price of its common stock at the time the
earnout merger consideration is paid.
F-23
REPORT
OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Board of
Directors and Stockholders
CytRx
Corporation
Los
Angeles, California
We have
audited the accompanying consolidated balance sheets of CytRx Corporation (“the
Company”) as of December 31, 2009 and 2008 and the related consolidated
statements of operations, stockholders’ equity, and cash flows for each of the
three years in the period ended December 31, 2009. We have also
audited the schedule listed in the accompanying index under Item 15a (2). These
financial statements and schedule are the responsibility of the Company’s
management. Our responsibility is to express an opinion on these
financial statements and schedule based on our audits.
We
conducted our audits in accordance with the standards of the Public Company
Accounting Oversight Board (United States). Those standards require
that we plan and perform the audit to obtain reasonable assurance about whether
the financial statements are free of material misstatement. An
audit also includes examining, on a test basis, evidence supporting the amounts
and disclosures in the financial statements, assessing the accounting principles
used and significant estimates made by management, as well as evaluating the
overall financial statement presentation. We believe that our audits
provide a reasonable basis for our opinion.
In our
opinion, the consolidated financial statements referred to above present fairly,
in all material respects, the financial position of CytRx Corporation at
December 31, 2009 and 2008, and the results of its operations and its cash flows
for each of the three years in the period ended December 31, 2009, in conformity with
accounting principles generally accepted in the United States of
America.
As more
fully described in Note 3 to the consolidated financial statements, effective
January 1, 2009, the Company adopted the
Amendments
to the provisions of Accounting Standards Codification 810-10,
Consolidation.
Also, in
our opinion, the financial statement schedule, when considered in relation to
the basic consolidated financial statements taken as a whole, present fairly, in
all material respects, the information set forth therein.
We also
have audited, in accordance with the standards of the Public Company Accounting
Oversight Board (United States), CytRx Corporation's internal control over
financial reporting as of December 31, 2009, based on criteria established in
Internal Control – Integrated
Framework issued by the Committee of Sponsoring Organizations of the
Treadway Commission (COSO) and our report dated March 12, 2010 expressed an
unqualified opinion thereon.
/s/ BDO SEIDMAN, LLP
Los
Angeles, California
March 12,
2010
F-24
REPORT
OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Board of
Directors and Shareholders
CytRx
Corporation
Los
Angeles, California
We have
audited CytRx Corporation’s (“the Company”) internal control over financial
reporting as of December 31, 2009, based on criteria established in Internal Control – Integrated
Framework issued by the Committee of Sponsoring Organizations of the
Treadway Commission (the COSO criteria). CytRx Corporation’s management is
responsible for maintaining effective internal control over financial reporting
and for its assessment of the effectiveness of internal control over financial
reporting, included in the accompanying “Item 9A, Controls and Procedures.” Our
responsibility is to express an opinion on the Company’s internal control over
financial reporting based on our audit.
We
conducted our audit in accordance with the standards of the Public Company
Accounting Oversight Board (United States). Those standards require that we plan
and perform the audit to obtain reasonable assurance about whether effective
internal control over financial reporting was maintained in all material
respects. Our audit included obtaining an understanding of internal control over
financial reporting, assessing the risk that a material weakness exists, and
testing and evaluating the design and operating effectiveness of internal
control based on the assessed risk. Our audit also included performing such
other procedures as we considered necessary in the circumstances. We believe
that our audit provides a reasonable basis for our opinion.
A
company’s internal control over financial reporting is a process designed to
provide reasonable assurance regarding the reliability of financial reporting
and the preparation of financial statements for external purposes in accordance
with generally accepted accounting principles. A company’s internal control over
financial reporting includes those policies and procedures that (1) pertain to
the maintenance of records that, in reasonable detail, accurately and fairly
reflect the transactions and dispositions of the assets of the company; (2)
provide reasonable assurance that transactions are recorded as necessary to
permit preparation of financial statements in accordance with generally accepted
accounting principles, and that receipts and expenditures of the company are
being made only in accordance with authorizations of management and directors of
the company; and (3) provide reasonable assurance regarding prevention or timely
detection of unauthorized acquisition, use, or disposition of the company’s
assets that could have a material effect on the financial
statements.
Because
of its inherent limitations, internal control over financial reporting may not
prevent or detect misstatements. Also, projections of any evaluation of
effectiveness to future periods are subject to the risk that controls may become
inadequate because of changes in conditions, or that the degree of compliance
with the policies or procedures may deteriorate.
In our
opinion, CytRx Corporation maintained, in all material respects, effective
internal control over financial reporting as of December 31, 2009, based on the
COSO criteria.
We also
have audited, in accordance with the standards of the Public Company Accounting
Oversight Board (United States), the balance sheets of the Company as of
December 31, 2009 and 2008, and the related statements of operations,
stockholders’ equity, and cash flows for each of the three years in the period
ended December 31, 2009 and our report dated March 12, 2010 expressed an
unqualified opinion thereon.
/s/ BDO SEIDMAN, LLP
Los
Angeles, California
March 12,
2010
F-25
CYTRX
CORPORATION
SCHEDULE
II — VALUATION AND QUALIFYING ACCOUNTS
For
the Years Ended December 31, 2009, 2008 and 2007
|
Additions
|
|
|
||||||||||||||||||
|
Description
|
Balance at
Beginning
of Year
|
Charged
to Costs
and
Expenses
|
Charged
to
Other
Accounts
|
Deductions
|
Balance at
End of Year
|
|||||||||||||||
|
Reserve
Deducted in the Balance Sheet from the Asset to Which it
Applies:
|
||||||||||||||||||||
|
Allowance
for Deferred Tax Assets
|
||||||||||||||||||||
|
Year
ended December 31, 2009
|
$ | 48,998,000 | $ | — | $ | 2,826,000 | $ | — | $ | 51,824,000 | ||||||||||
|
Year
ended December 31, 2008
|
$ | 53,246,000 | $ | — | $ | — | $ | 4,248,000 | $ | 48,998,000 | ||||||||||
|
Year
ended December 31, 2007
|
$ | 43,830,000 | $ | — | $ | 9,416,000 | $ | — | $ | 53,246,000 | ||||||||||
F-26