Attached files
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2009
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission file number 001-33657
Abraxis BioScience, Inc.
(Exact name of registrant as specified in its charter)
| Delaware |
30-0431735 | |
| (State of incorporation) |
(I.R.S. Employer Identification No.) | |
| 11755 Wilshire Boulevard, Suite 2000 Los Angeles, CA 90025 |
(310) 883-1300 | |
| (Address of principal executive offices, including zip code) | (Registrant’s telephone number, including area code) | |
Securities registered pursuant to Section 12(b) of the Act:
| Common Stock, par value $0.001 per share |
The NASDAQ Stock Market LLC | |
| (Title of Class) | (Name of each exchange on which registered) |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ¨ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained to the best of the registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| ¨ Large accelerated filer | x Accelerated filer | ¨ Non-accelerated filer | ¨ Smaller reporting company | |||
| (Do not check if a smaller reporting company) |
Indicate by check mark whether the registrant is a shell company (as determined by rule 12b-2 of the Exchange Act). Yes ¨ No x
The approximate aggregate market value of voting and non-voting stock held by non-affiliates of the registrant was $247 million as of June 30, 2009. All executive officers and directors of the registrant have been deemed, solely for the purpose of the foregoing calculation, to be “affiliates” of the registrant.
The number of shares of common stock outstanding as of February 26, 2010 was 40,127,810
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the Proxy Statement for the registrant’s 2010 Annual Meeting of Stockholders are incorporated by reference into Part III of this Annual Report on Form 10-K.
Table of Contents
Abraxis BioScience, Inc.
FORM 10-K
For the Year Ended December 31, 2009
2
Table of Contents
Unless the context otherwise requires, references to “New Abraxis,” “Abraxis BioScience,” “Abraxis,” “we,” “us” and “our” refer to Abraxis BioScience, Inc. (formerly New Abraxis, Inc.) and its subsidiaries, including our operating subsidiary Abraxis BioScience, LLC; references to “Old Abraxis” refer to Abraxis BioScience, Inc. (formerly American Pharmaceuticals, Inc.) prior to the 2007 separation; references to “APP” refer to APP Pharmaceuticals, Inc. and its subsidiaries, including its operating subsidiary APP Pharmaceuticals Partners, LLC (which we sometimes refer to as APP LLC); and references to the “distribution,” “separation,” “2007 separation” or “spin-off” refer to the transactions in which we separated from Old Abraxis and became an independent publicly-traded company in November 2007.
Note Regarding Forward-Looking Statements
This Annual Report on Form 10-K and other materials filed or to be filed by us with the Securities and Exchange Commission, or the SEC, as well as information included in oral statements or other written statements made or to be made by us, contain forward-looking statements within the meaning of federal securities laws. We intend these forward-looking statements to be covered by the safe harbor provisions for forward-looking statements in the federal securities laws. These forward-looking statements are not historical facts but rather are based on current expectations, estimates and projections about our industry, our beliefs and assumptions. These risks and uncertainties include those described in “Risk Factors” and elsewhere in this Annual Report on Form 10-K. Forward-looking statements, whether express or implied, are not guarantees of future performance and are subject to risks and uncertainties, which can cause actual results to differ materially from those currently anticipated, due to a number of factors, which include, but are not limited to:
| • | the amount and timing of costs associated with the continuing launch of Abraxane® in Europe and abroad |
| • | our ability to maintain and/or improve sales and earnings performance; |
| • | the actual results achieved in further clinical trials of Abraxane® may or may not be consistent with the results achieved to date; |
| • | the market adoption of any new pharmaceutical products; |
| • | the difficulty in predicting the timing or outcome of product development efforts and regulatory approvals; |
| • | our ability and that of our suppliers to comply with laws, regulations and standards, and the application and interpretation of those laws, regulations and standards, that govern or affect the pharmaceutical industry, the non-compliance with which may delay or prevent the sale of their products; |
| • | the availability and price of acceptable raw materials and components from third-party suppliers; |
| • | any adverse outcome in litigation; |
| • | general economic, political and business conditions that adversely affect our company or our suppliers, distributors or customers; |
| • | changes in costs, including changes in labor costs, raw material prices or advertising and marketing expenses; |
| • | inventory reductions or fluctuations in buying patterns by wholesalers or distributors; |
| • | the impact on our products and revenues of patents and other proprietary rights licensed or owned by us, our competitors and other third parties; |
| • | the ability to successfully manufacture our products in an efficient, time-sensitive and cost effective manner, including the impact of product recalls and other manufacturing issues; |
3
Table of Contents
| • | the impact of recent legislative changes to the governmental reimbursement system; and |
| • | risks inherent in acquisitions, divestitures and spin-offs, including the capital resources required for acquisitions, business risks, legal risks and risks associated with the tax and accounting treatment of such transactions. |
You should read this Annual Report on Form 10-K with the understanding that actual future results may be materially different from expectations. Readers should carefully review the factors described in “Item 1A: Risk Factors” below and other documents we file from time to time with the SEC for a more detailed description of these risks and other factors that may affect the forward-looking statements. All forward-looking statements made in this Annual Report on Form 10-K are qualified by these cautionary statements. These forward-looking statements are made only as of the date of this Annual Report on Form 10-K, and we do not undertake any obligation (and we expressly disclaim any such obligation), other than as may be required by law, to update or revise any forward-looking statements to reflect changes in assumptions, the occurrence of unanticipated events or changes in future operating results over time or otherwise. We use words such as “should,” “anticipate,” “expect,” “intend,” “plan,” “believe,” “seek,” “estimate” and variations of these words and similar expressions to identify forward-looking statements, but these are not the exclusive means of identifying forward-looking statements.
Available Information
Our Internet address is www.abraxisbio.com. Our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, proxy statements and other information are available free of charge on our website as soon as reasonably practical after they are electronically filed or furnished to the SEC. The information found on our website shall not be deemed incorporated by reference by any general statement into any filing under the Securities Act of 1933 or under the Securities Exchange Act of 1934, except to the extent we specifically incorporate the information found on our website by reference, and shall not be deemed filed under such Acts.
The SEC also maintains an Internet site that contains reports, proxy and information statements, and other information about issuers that file reports electronically with the SEC. The address of that site is www.sec.gov.
2007 Separation
On November 13, 2007, Abraxis BioScience, Inc. (formerly “American Pharmaceutical Partners, Inc.”) (“Old Abraxis”) was separated into two independent publicly-traded companies: one holding the former Abraxis Pharmaceutical Products business (which is referred to as the “hospital-based business”); and the other holding the former Abraxis Oncology and Abraxis Research businesses (which is referred to as the “proprietary business”). Following the separation, the proprietary business changed its name from New Abraxis, Inc. to Abraxis BioScience, Inc. and the hospital-based business was operated under the name APP Pharmaceuticals, Inc.
In connection with the separation, stockholders of Old Abraxis as of November 13, 2007 received one share of our company for every four shares of Old Abraxis held as of that date. In addition, in connection with the separation, we entered into a separation and distribution agreement that provides for, among other things, the principal corporate transactions required to effect the separation and other specified terms governing our relationship with APP after the spin-off. We also entered into various agreements with APP, including (i) a transition services agreement pursuant to which we and APP agreed to continue to provide each other with various services on an interim, transitional basis, for periods up to 24 months depending on the particular service; (ii) a manufacturing agreement whereby we and APP agreed to manufacture Abraxane® and certain other products and to provide other manufacturing-related services for a period of four or five years; (iii) an employees matters agreement providing for each company’s respective obligations to employees and former employees who
4
Table of Contents
are or were associated with their respective businesses, and for other employment and employee benefit matters; (iv) various real estate leases; and (v) a tax allocation agreement. Also, in connection with the separation, APP contributed $700 million in cash to us.
Proposed 2010 Spin-Off
In January 2009, we announced that our board of directors approved a plan to spin-off a newly-formed subsidiary Abraxis Health, Inc. as a new independent, stand-alone company holding our drug discovery, pilot manufacturing and development business. We currently anticipate the spin-off will be completed in 2010. If the spin-off occurs, our stockholders would own (i) shares of Abraxis Health and (ii) shares of our common stock, and we would continue to operate our existing business, excluding the drug discovery, pilot manufacturing and development business to be held by Abraxis Health. In connection with the proposed spin-off, Abraxis Health would enter into several agreements with us related to, among other things, manufacturing, transition services, tax allocations and a number of ongoing commercial relationships.
The proposed spin-off is subject to, among other things, final approval by our board of directors and the effectiveness of the registration statement registering the common stock of Abraxis Health to be distributed to our stockholders in connection with the spin-off. Our board of directors, in its sole discretion and for any reason or no reason at all, may amend, modify or abandon the proposed spin-off without liability at any time prior to the time the spin-off is completed. Approval by our stockholders is not required as a condition to the consummation of the proposed spin-off. In connection with the proposed spin-off, Abraxis Health filed a registration statement on Form 10 with the SEC on August 4, 2009, that was withdrawn on August 12, 2009. Abraxis Health plans to file a new Form 10 registration statement, and stockholders are urged to read it, including any amendments or supplements thereto, carefully when available because it will contain important information about the proposed spin-off.
Overview
We are a Delaware corporation that was formed in June 2007. Our business was conducted as part of Old Abraxis prior to the separation. References in this Annual Report on Form 10-K to the historical assets, liabilities, products, businesses or activities of our company are generally intended to refer to the historical assets, liabilities, products, businesses or activities of the business as it was conducted as part of Old Abraxis prior to the separation.
We are one of the few fully integrated biotechnology companies, with a breakthrough marketed product (Abraxane®), global ownership of our proprietary technology platform and clinical pipeline, and dedicated nanoparticle manufacturing capabilities for worldwide supply integrated with seasoned in-house capabilities, including discovery, clinical drug development, regulatory and sales and marketing.
We are dedicated to the discovery, development and delivery of next-generation therapeutics and core technologies that offer patients safer and more effective treatments for cancer and other critical illnesses. Our portfolio includes the world’s first and only protein-based nanoparticle chemotherapeutic compound (Abraxane®), which is based on our proprietary tumor targeting technology known as the nab® technology platform. This nab® technology platform is the first to exploit the tumor’s biology against itself, taking advantage of an albumin-specific, receptor-mediated transport system and allowing the delivery of a drug from the vascular space across the blood vessel wall to the underlying tumor tissue. Abraxane® is the first clinical and commercial validation of our nab® technology platform. From the discovery and research phase to development and commercialization, we are committed to rapidly enhancing our product pipeline and accelerating the delivery of breakthrough therapies that will transform the lives of patients who need them.
We own the worldwide rights to Abraxane®, a next-generation, nanometer-sized, solvent-free taxane that was approved by the U.S. Food and Drug Administration, or the FDA, in January 2005 for its initial indication in
5
Table of Contents
the treatment of metastatic breast cancer and launched in February 2005. We believe the successful launch of Abraxane® validates our nab® tumor targeting technology, a novel biologically interactive (receptor-mediated) system to deliver chemotherapeutic agents. Abraxane® revenue for the year ended December 31, 2009, 2008 and 2007 was $314.5 million, $335.6 million and $324.7 million, respectively, which in 2008 and 2007 included the recognition of deferred revenue related to our co-promotion agreement with AstraZeneca UK Limited. In January 2009, we re-acquired from AstraZeneca the exclusive rights to market Abraxane® in the United States, thereby ending the co-promotion agreement.
Our research and development approach is based on the integration of our nab® tumor targeting technology, our natural product drug discovery platform, our multi-functional chemistry capabilities with our in-house clinical trial and regulatory strengths, combined with our unique nanoparticle manufacturing capabilities. Our product pipeline includes over five clinical oncology and cardiovascular product candidates in various stages of testing and development, including Abraxane® and Coroxane™ for various indications. We also have several discovery product candidates and novel chemical entities for various diseases, including cancer, multiple sclerosis and Alzheimer’s. We believe the application of our nab® technology will serve as the platform for the development of numerous drugs for the treatment of cancer and other critical illnesses. To leverage in-house manufacturing, clinical trial and regulatory expertise, we will continue to supplement our discovery efforts through technology acquisitions and external collaborations with academia and start-up biotechnology companies.
Abraxane®, A Proprietary Injectable Oncology Product
Overview
Abraxane® is a next-generation, nanometer-sized, solvent-free taxane that was approved by the FDA in January 2005 for its initial indication in the treatment of metastatic breast cancer and launched in February 2005. As of December 2009, Abraxane was approved for marketing in 39 countries. Taxanes are one of the most widely used chemotherapy agents. We believe the successful launch of Abraxane® validates our nab® tumor targeting technology described below. Effective January 1, 2006, we received a unique reimbursement “J” code for Abraxane® in the U.S., which facilitates reimbursement from Medicare and Medicaid as well as private payors.
First clinical and commercial validation of our nab® tumor targeting technology
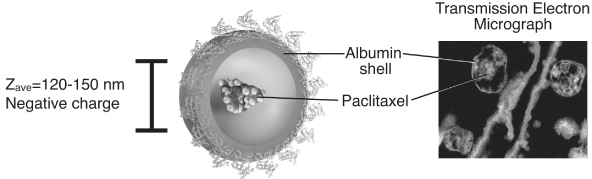
Abraxane® represents the first in a new class of protein-bound drug particles that takes advantage of albumin, a natural carrier of water insoluble molecules (e.g., various nutrients, vitamins and hormones) found in humans. Because tumors have an increased need for nutrients to support rapid malignant growth, albumin complexes accumulate preferentially in tumors. Recent molecular analyses have identified receptor-mediated pathways (gp60) facilitating active transport of albumin complexes from the blood into tumor tissue and also preferential accumulation in tumors by tumor-secreted proteins such as Secreted Protein Acidic and Rich in Cysteine (SPARC), which can attract albumin. Abraxane® consists only of paclitaxel nanometer-sized albumin-bound particles and, in a pivotal 460 patient Phase III clinical trial, demonstrated an almost doubling of the response rate, a longer time to tumor progression in metastatic breast cancer as well as an increased survival in patients previously treated for metastatic disease when compared to Taxol®. We believe that our nab® tumor targeting technology and these albumin pathways allow more rapid delivery of paclitaxel to the cancer cells, with preferential accumulation while at the same time allowing less drug indiscriminately into normal, healthy cells. This is supported by the clinical findings demonstrated by Abraxane®.
Overcoming the obstacles and toxicity of solvents
Many oncology drugs are water insoluble and thus require solvents to formulate the drugs for injection. Taxol® and its generic solvent-based equivalents contain the active ingredient paclitaxel dissolved in the solvent Cremophor. The toxicity of Cremophor limits the dose of Taxol® that can be administered, potentially limiting
6
Table of Contents
the efficacy of the drug. Furthermore, patients receiving Taxol® require pre-medication with steroids and antihistamines to prevent the toxic side effects associated with Cremophor and, in some cases, require a growth factor such as G-CSF to overcome low white blood cell levels resulting from chemotherapy.
Abraxane® utilizes our nab® tumor targeting technology to encapsulate an amorphous form of paclitaxel in albumin, a human protein found in blood and avoids the need for Cremophor. Because Abraxane® contains no Cremophor solvent, this next-generation taxane product enables the administration of 50% more chemotherapy with a well-tolerated safety profile, requires no routine premedication to prevent hypersensitivity reactions, can be given over a shorter infusion time using standard IV tubing and reduces the need for G-CSF support.
Abraxane® clinical development, label expansion and global commercialization plans
We will continue to pursue an aggressive and comprehensive clinical development plan to maximize the commercial potential and clinical knowledge of Abraxane®. As of December 31, 2009, approximately 24 company-sponsored Abraxane® clinical studies and approximately 61 investigator-initiated Abraxane® clinical studies were planned or underway for various indications, of which more than 44 had patient participation. The investigator-initiated studies generally are intended to advance the clinical knowledge of Abraxane® and complement the knowledge gained through our clinical development program.
Results from five company-supported studies of Abraxane® were presented at the 32nd Annual San Antonio Breast Cancer Symposium in December 2009. A pilot neoadjuvant trial evaluated the efficacy and safety of lapatinib combined with nab-paclitaxel in HER2-positive breast cancer patients. Response data was available for 29 patients: 82.8 percent of patients (n=24) achieved an overall response rate; 13.8 percent (n=4) experienced a complete response; 69 percent (n=20) experienced a partial response. Additionally, 17.2 percent (n=5) of patients experienced stable disease, and 5 out of 27 patients achieved a pathological complete response (pCR). The most common toxicities were rash, neuropathy, fatigue and myalgias. No cardiac toxicity was observed. Another study assessed the predictive value of Mammaprint scores (poor vs. good), as well as basal vs. luminal vs. HER2 genotype profiling, for response to treatment on three different neoadjuvant chemotherapy (NCT) regimens. Gene profiling indicated 28 percent of the tumors were HER2-like, 26 percent were basal-like, 42% luminal-like, and 4 percent borderline luminal-like. Results indicated that HER2-like and basal-like genotype breast cancer is more likely to respond to NCT and is associated with poor-risk characteristics as determined by the 70-gene assay. We also presented pre-clinical data evaluating the effectiveness of different combination regimens for nab-rapamycin against human breast tumor xenografts. Another pre-clinical study further explored the role of SPARC characterizing the identity of its albumin-binding and angiogenic domain. Health economic data was presented as well. The study was conducted to determine if differences exist in the total cost of care in patients receiving taxane-based chemotherapy for metastatic breast cancer (MBC). Additionally, a comparison between the taxanes - solvent-based paclitaxel (Taxol® and others); docetaxel (Taxotere®); and nab-paclitaxel (Abraxane®) - was performed on the use of ancillary medications (for neutropenia, anemia, and nausea and vomiting) and their associated costs. Costs per utilizing patient with MBC per month were calculated and compared. For patients with MBC, differences in total medical costs between the three taxanes were modest. Total medical costs were lowest for patients receiving solvent-based paclitaxel and comparable between docetaxel and nab-paclitaxel. Patients on nab-paclitaxel received more doses than the other taxanes. Nab-paclitaxel was associated with lower utilization and costs for colony stimulating factors compared with solvent-based paclitaxel and docetaxel. Additionally in November 2009, at the AACR-NCI-EORTC Molecular Targets and Cancer Therapeutics conference in Boston, preclinical data was presented detailing the stromal targeting of the Abraxane-gemcitabine combination in pancreatic cancer.
We will continue to study the use of Abraxane® in a variety of oncology settings and intend to focus our Phase III trials in first-line non-small cell lung cancer (NSCLC), melanoma and pancreatic cancer, all of which are superiority trials utilizing weekly dosing schedules of Abraxane®. The Phase III NSCLC pivotal trial is a randomized, open-label trial comparing weekly 100 mg/m2 Abraxane® (days 1, 8 and 15 of each cycle) and 200 mg/m2 Taxol® (paclitaxel) injection every three weeks. Carboplatin is administered at AUC=6 on day 1 of each
7
Table of Contents
cycle repeated every three weeks in both treatment arms. The study has enrolled over 1,000 patients with Stage IIIb and IV non-small cell lung cancer. The primary endpoint of the study is overall response rate, while also being powered for statistically significant differences in progression-free survival (PFS) as a secondary endpoint. The Data Monitoring Committee (DMC) performed a pre-specified interim analysis on the first 400 patients and recommended that the trial proceed to completion with no changes to the protocol, treatment arms, or sample size. The Phase III melanoma trial comparing Abraxane to dacarbazine (DTIC) in patients with metastatic disease began enrolling patients in April 2009, and has a planned enrollment of 514 patients. Based on encouraging Phase I/II clinical trial data of Abraxane® in combination with gemcitabine in the treatment of metastatic pancreatic cancer, a Phase III registration study comparing Abraxane® plus gemcitabine versus gemcitabine monotherapy in metastatic pancreatic cancer was successfully filed with the FDA and began enrolling patients in May 2009. A total of 630 patients are expected to be enrolled in the study. Interim data for the Phase I/II study of Abraxane® plus gemcitabine in the treatment of metastatic pancreatic cancer was presented at the American Society of Clinical Oncology Annual Meeting in May 2009. We continue to expand our clinical experience with Abraxane® and its potential in treating multiple tumor types as a single agent and in combination.
With regard to the global commercialization, Abraxane® is presently approved in 39 countries worldwide. Subsequent to the FDA approval of Abraxane® in the US in January 2005, Abraxis received regulatory approval from the Therapeutic Products Directorate of Health Canada to market Abraxane® for the treatment of metastatic breast cancer in Canada in June 2006. In October 2007, Abraxis received regulatory approval from India’s Drug Controller General to market Abraxane® for the treatment of breast cancer after failure of combination chemotherapy for metastatic disease or relapse within 6 months of adjuvant chemotherapy. In January 2008, the European Commission granted marketing approval in 30 European countries (via the centralized procedure) for Abraxane® in patients who have failed first-line treatment for metastatic disease and for whom standard, anthracycline containing therapy is not indicated. In March 2008, we received regulatory approval with the Korean FDA to market Abraxane® in South Korea for the treatment of breast cancer after failure of combination chemotherapy for metastatic disease. In June 2008, we received regulatory approval with the State Food and Drug Administration of the People’s Republic of China to market Abraxane® for the treatment of breast cancer after failure of combination chemotherapy for metastatic disease or relapse within 6 months of adjuvant chemotherapy. Prior therapy should have included an anthracycline unless clinically contraindicated. In September 2008, we received regulatory approval from the Therapeutic Goods Administration, Australia to market Abraxane® powder for injection (suspension) for the treatment of metastatic carcinoma of the breast after failure of anthracycline therapy. We received approval to market Abraxane in Bhutan in April 2009 and in the United Arab Emirates in July 2009. We launched Abraxane® in India (through our partner Biocon) in July 2008, in the United Arab Emirates with partner Neobiocon in October 2008, in the United Kingdom in December 2008, in Germany in February 2009 and in Australia in collaboration with Specialised Therapeutics Australia Pty Ltd. in February 2009. Additionally, we are working towards launching Abraxane® in South Korea with partner Green Cross Corporation.
Marketing authorization applications have been submitted to several countries including Russia in June 2006, Japan in February 2008, and via the centralized procedure, in the Gulf Corporation Council countries (Saudi Arabia, Bahrain, Sultanate of Oman and Kuwait) in December 2008, in Sri Lanka and in Nepal in January 2009, and in New Zealand in June 2009.
nab® Tumor Targeting Technology Platform
We have identified a biological pathway specific to tumors through which tumors preferentially accumulate albumin-bound complexes. This is accomplished through our nab® tumor targeting technology, which encapsulates chemotherapy agents in nanometer-sized particles of albumin that are approximately 1/100th the size of a single red blood cell. Millions of these particles can be injected in a single dose of medication. This albumin-bound medication enters the bloodstream where it is transported across the blood vessel wall through a specific albumin receptor (gp60) on the blood vessel wall and results in higher accumulation of the medication to
8
Table of Contents
the tumor. This effect may be further enhanced due to potential binding of the transported albumin to the tumor-secreted protein SPARC. A body of research has suggested that the secretion of SPARC occurs in most solid tumors that are difficult to treat, including in breast, lung, pancreatic, ovarian, head and neck, melanoma, gastric, esophageal, glioma and cervical tumors. Exploiting these tumors’ attraction for albumin, we have developed a novel mechanism to deliver chemotherapy agents in high concentrations.
The following diagram depicts our nab® tumor targeting technology and the proposed mechanism by which high concentrations of chemotherapy agents are delivered preferentially to tumors through the biological gp60 receptor pathway and through albumin binding to SPARC.
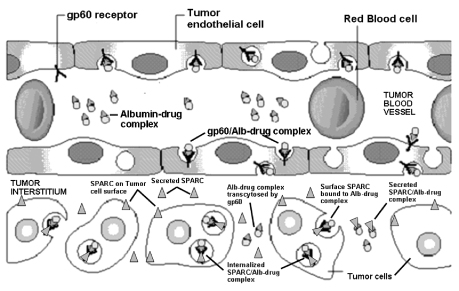
In April 2007, at the 98th Annual Meeting of the American Association for Cancer Research (AACR), we announced results from various pre-clinical studies that support the role of SPARC and our nab® tumor targeting technology. The results from one pre-clinical study demonstrated that SPARC is an albumin-binding protein and that the level of the SPARC expression could be correlated with the response of tumors to Abraxane®. The pre-clinical study results also provided evidence for chemotherapy induced angiogenesis and a rationale for combining Abraxane® with VEGF inhibitor drugs like Avastin®. Several ongoing clinical studies are investigating the role of SPARC in Abraxane® based treatment regimens. Preliminary data released in September 2009 at the European Society for Medical Oncology (ESMO) congress suggests that it is possible to identify subgroups of patients treated with Abraxane® based regimens that are at high or low risk of progression using SPARC as a biomarker. We continue to explore the role of our nab® technology for targeting different biological pathways, as well as for the development of new therapeutic candidates, including our clinical product candidates described below.
We believe we can apply our nab® tumor targeting technology to numerous chemotherapy agents. By exploiting the abnormal vascular growth (angiogenesis) and the overexpression of albumin-binding proteins (gp60 and SPARC) in advanced tumor cells and by overcoming water insolubility of many active chemotherapy agents, we believe that our technology may revolutionize the delivery of chemotherapy agents to cancer patients. As shown below, Abraxane® represents the first clinical and commercial validation of our nab® technology that
9
Table of Contents
takes advantage of albumin, a natural carrier of water insoluble molecules (e.g., various nutrients, vitamins and hormones) found in humans. Because tumors have an increased need for nutrients to support rapid malignant growth, albumin complexes accumulate preferentially in tumors.
We believe that our nab® tumor targeting technology will serve as the platform for the development of numerous other drugs for the treatment of cancer and other critical illnesses. We are committed to maximizing the potential application of our technology. We are also actively exploring partnerships with pharmaceutical companies that have an interest in using the nab® technology for their drug candidates.
Select Clinical Product Candidates
We intend to continue to leverage our nab® tumor targeting technology and our internal clinical development and regulatory expertise to develop numerous drug candidates for the treatment of cancer and other critical illnesses. The following table is a selection of our clinical product candidates and their status.
| Compound |
Critical Illness |
Status | ||
| Abraxane® |
First-Line Non-Small Cell Lung Cancer | Phase III began in the fourth quarter of 2007. Patient enrollment fully accrued. | ||
| Abraxane® |
Malignant Melanoma | Phase III enrollment began in April 2009. | ||
| Abraxane® |
Pancreatic Cancer | Phase III enrollment began in May 2009. | ||
| ABI-008 (nab®-docetaxel) |
Solid Tumors | Phase I completed in hormone-refractory prostate cancer (HRPC). Phase II started in the second half of 2008. | ||
| ABI-009 (nab®-rapamycin) |
Solid Tumors | Phase I in progress. | ||
| ABI-010 (nab®-17AAG) |
Solid Tumors | IND filed in the first quarter of 2008. Phase I study in planning. | ||
| ABI-011 (nab®-thiocolchicine dimer) |
Solid Tumors |
IND filed in the third quarter of 2009. | ||
| Coroxane™ |
Coronary Artery Restenosis | Phase II. Seeking strategic partner. | ||
| Coroxane™ |
Peripheral Artery Restenosis | Phase II. Seeking strategic partner. | ||
10
Table of Contents
ABI-008 (nab®-docetaxel). ABI-008 is a solvent-free, Tween®-free nanometer-sized form of docetaxel. Docetaxel is the active ingredient in the currently largest selling taxane, Taxotere®. An investigational new drug application, or IND, was filed for ABI-008 in the fourth quarter of 2006. ABI-008 is currently in development for the first-line treatment of hormone-refractory prostate cancer (HRPC). A Phase I study has been completed and a Phase II study started in the second half of 2008.
ABI-009 (nab®-rapamycin). Rapamycin, a protein kinase inhibitor, inhibits downstream signals from mTOR, a pathway that promotes tumor growth. Currently, it is marketed as an orally bioavailable immunosuppresive drug and no intravenous form is available because of the water insolubility of this molecule. The anti-cancer effects of rapamycin are limited by the immunosuppression associated with chronic oral administration. ABI-009 is a nanometer-sized, albumin-bound form of rapamycin able to be administered intravenously. At the 98th Annual Meeting of the AACR in April 2007, first-time data were presented from a pre-clinical study evaluating the toxicity and anti-tumor effect of ABI-009. These results demonstrated that ABI-009 was well-tolerated in animals, showed linear pharmacokinetics, and was highly effective against a number of tumor models in vivo. The Phase I study is currently ongoing with ABI-009 in patients with solid tumors.
ABI-010 (nab®-17AAG). 17AAG is a polyketide inhibitor of Hsp90 (heat shock protein 90) and interrupts several biological processes implicated in cancer cell growth and survival. Hsp90 is a protein chaperone that binds to several sets of signaling proteins, known as “client proteins.” 17AAG binds to Hsp90 and causes its dissociation from, and consequent degradation of, the client proteins. Because the Hsp90 client proteins are so important in signal transduction and in transcription (processes critical to the growth and survival of cancer cells), 17AAG may serve as a chemotherapy agent in the treatment of multiple cancers. Current formulations of 17AAG require solvents such as Cremophor and DMSO. ABI-010 is a solvent-free albumin-bound form of this drug. The IND for nab®-17AAG and the clinical development plan was approved by the FDA in May 2008.
ABI-011 (nab®-thiocolchicine dimer). Thiocolchicine dimers are novel thiocolchicines with dual mechanisms of action showing both microtubule destabilization and the disruption of topoisomerase-1 activity. nab®-thiocolchicine dimer is a nanoparticle, albumin-bound form of the water insoluble dimer. ABI-011 was selected for its cytotoxic activity in nanomole range concentrations on several human tumor cell lines and, in particular, on a tumor cell line resistant to cisplatin and topotecan. This particular behavior was related to a dual mechanism of action. ABI-011 has the additional oncotherapeutic property of inhibiting topoisomerase-1 nuclear enzymes in a different manner than the classic topo-1 inhibitors, camptothecins. Pre-clinical studies of ABI-011 have shown that it also possesses anti-angiogenic and vascular disrupting activity, which will be exploited in its clinical development. The IND for nab®-thiocolchicine dimer was filed in the third quarter of 2009.
Coroxane™ (nanometer-sized paclitaxel, Abraxane®, under the trade name Coroxane™). Coroxane™ is currently closing its Phase II clinical studies for coronary restenosis as well as peripheral artery (superficial femoral artery) restenosis. The SNAPIST series of studies examines the use of Coroxane™ in the treatment of coronary artery restenosis, including the use of Coroxane™ in patients receiving bare metal stents. Coroxane™ administered with bare metal stents may address the issue of incomplete re-endothelialization and acute thrombosis associated with drug-eluting stents. Coroxane™ administered following balloon angioplasty in the superficial femoral artery may help reduce the incidence of restenosis in these patients. We currently intend to seek a strategic partner for the further development and marketing of Coraxane™.
Research and Development
Our research and development efforts are based on the tight integration and rapid translation of discovery from the bench to the clinic among its chemistry, biology, pharmaceutical, regulatory and clinical development groups. We have recruited seasoned discovery scientists, medicinal chemists, formulation scientists and integrated these talents with a team of clinical development and regulatory experts. Our approach is based on the integration of various capabilities and resources, including:
| • | our nab® tumor targeting technology; |
11
Table of Contents
| • | our proprietary natural product drug discovery platform; |
| • | our multi-functional chemistry capabilities; and |
| • | internal clinical trial and regulatory competencies. |
Natural Product Drug Discovery Platform
Almost half of today’s best-selling drugs originate from natural products or their derivatives. A key feature of natural products is their enormous structural and chemical diversity that is not represented in combinatorial libraries of synthetic compounds. This makes natural products a fertile resource for finding novel compounds that interact with new targets for drug discovery. In addition, we believe our nab® tumor targeting technology platform is ideally suited to the formulation and delivery of natural products, which tend to be more water insoluble, larger and more complex than typical synthetic drugs.
We have established a proprietary natural product library of large chemical diversity for drug discovery. The library currently represents more than 100,000 semi-purified screening samples derived from microorganisms (over 65,000 strains of actinomycetes, fungi and bacteria) retrieved from over 38,000 soil samples representing geographic, habitat and genetic diversity from all over the world. New strains of microorganisms are continuously being added from our soil sample collection, and we believe the millions of microorganisms in each soil sample provide us with an almost limitless resource for continuing to create new and targeted libraries of natural product chemical diversity for drug discovery. The natural product library continues to provide chemical diversity for our ongoing high throughput screening activities. Natural products are an important component of our drug discovery strategy and, with the capability to overcoming water insolubility through the use of our nab® technology, we believe we have the unique opportunity to translate water insoluble compounds discovered from our natural product libraries into clinical applications.
Multi-functional Chemistry Capabilities. We possess a full range of chemistry capabilities, including organic and medicinal chemistry, analytical chemistry, formulation, process development and natural product isolation chemistry. Our approach, which involves chemistry-driven discovery combined with biology-driven validation and integration with our nab® technology, has been applied successfully on many drug discovery programs.
Internal Clinical Trial and Regulatory Competencies. We have established key internal clinical development and regulatory competencies to execute rapid clinical translation of our technology in the most cost-efficient manner. Clinical Operations, Safety Reporting, Data Management and Biostatistics are located in our Durham, North Carolina office. Clinical Operations manages all of our company-sponsored clinical trials by overseeing the study conduct at each site and verifying data integrity. Data Management enters all clinical trial data into an in-house database, which is then analyzed by our biostatistics team who produce reports for FDA submissions. Safety reporting and post-marketing safety surveillance are carried out using a Drug Safety Reporting System. The regulatory operations team is responsible for making submissions to the FDA and global regulatory authorities. This seasoned team accomplished the first electronic submission of a New Drug Application (NDA) with the FDA of an oncology product, resulting in FDA approval of Abraxane®. An important strategy underway is the development of informatics and electronic systems to streamline our clinical trial management processes, including establishing alliances to enhance patient accrual and provide efficient methods to capture data.
Drug Discovery Strategies
Our drug discovery strategies are based upon the following:
| • | our nab® tumor targeting technology; |
| • | our natural product drug discovery platform; |
| • | SPARC protein; |
12
Table of Contents
| • | biomarkers and molecular profiling; |
| • | cyclodextrin-based delivery technology; |
| • | nucleic acid complementation technology; |
| • | pluripotent adult stem cells; |
| • | our cancer drug discovery targeting the tumor suppressor p53; |
| • | immunotherapeutics and related assay systems; |
| • | Abraxis translational molecular bioscience at California NanoSystems Institute; |
| • | biosimilars; and |
| • | central nervous system therapies. |
SPARC
SPARC (Secreted Protein Acidic Rich in Cysteine) appears to be the intratumoral target of our nab® technology platform due to its secretion by a variety of tumors and the affinity of albumin for SPARC. In addition, it has been discovered that the SPARC protein, when administered systemically in combination with chemotherapy, can greatly sensitize a chemotherapy resistant tumor in xenograft tumor models. Our scientists, in collaboration with university scientists, are exploring the therapeutic potential of SPARC as a potential target in the treatment of chemo-resistant tumors. We will continue to explore the role of our nab® technology for targeting SPARC and other biological pathways, as well as for the development of new therapeutic candidates. Additionally, SPARC as a biomarker is being investigated in several ongoing Abraxane® related clinical trials.
Biomarkers and Molecular Profiling
In May 2007, we entered into a license agreement with the University of Southern California (USC) under which we licensed the exclusive worldwide development and commercialization rights for an intellectual property portfolio of diagnostic protein biomarkers for therapy response, therapy toxicity and disease recurrence in colorectal cancers. The intellectual property licensed is based on USC research by Associate Professor of Medicine Heinz-Joseph Lenz and colleagues. We believe these licensed biomarkers are consistent with our strategy to identify compounds with specific activity linked to specific biological markers related to particular disease states and may be useful predictors of response to treatments (evaluated through clinical response, toxicity and time to disease progression) and prognostic markers, which are important in determining the aggressiveness of cancers as well as other diseases.
Cyclodextrin-Based Delivery Technology
Through our wholly-owned subsidiary Shimoda Biotech (Pty) Limited (Shimoda), we are developing cyclodextrin-based formulations of existing therapeutics to improve their bioavailability characteristics. Shimoda has used cyclodextrin formulations to improve the dissolution rate and bioavailability of orally-deliverable drugs, such as diclofenac sodium, to increase aqueous solubility of poorly water-soluble drugs, to increase the chemical and physical stability of injectable drugs and to mask the unpleasant taste of oral drugs. Cyclodextrin is a water-soluble complex oligosaccharide that structurally forms a unique nanoparticle capable of delivering hydrophobic compounds via multiple routes, including oral ingestion and intravenous injection.
Shimoda’s first cyclodextrin-based product, Dyloject® (diclofenac sodium solution for injection), is an injectable painkiller for the treatment of post-surgical pain. Dyloject® is the world’s first solubilized intravenous formulation of diclofenac. Diclofenac is a non-steroidal anti-inflammatory drug (NSAID). Dyloject® was launched in December 2007 in the United Kingdom by Javelin Pharmaceuticals under an exclusive worldwide license agreement pursuant to which Shimoda will receive milestone payments and royalties. In January 2009,
13
Table of Contents
Javelin announced that it had entered into an exclusive European marketing partnership for Dyloject® with Therabel Pharma N.V. In December 2009, Javelin announced that it had entered into a definitive agreement with Myriad Pharmaceuticals to acquire Javelin Pharmaceuticals. In February 2010, Javelin announced that its New Drug Application (NDA) submitted on December 2009 to the US Food and Drug Administration (FDA) for Dyloject® had been accepted for formal review.
Nucleic Acid Complementation Technology
We are pursuing the development of an engineered split-toxin platform that leads to the functional reconstitution of a cytotoxin from multiple parts under very specific conditions (for example, in cancer cells arising from known genetic anomalies or in virus-infected cells). This strategy does not require traditional high-throughput drug screening methodologies and has the added capability of being further modified to serve as a diagnostic tool for monitoring patients’ responses to therapy. This research is being pursued through DiThera, Inc. (DiThera). In December 2008, we purchased preferred stock of DiThera representing 50% of DiThera’s outstanding capital stock. DiThera is consolidated in our financial statements for the years ended December 31, 2009 and 2008.
Pluripotent Adult Stem Cells
In September 2006, we entered into a license with the University of Maryland under which we exclusively licensed the worldwide intellectual property rights to the CD34+ non-adherent adult bone marrow stem cell technology. This technology may present an opportunity to reverse the effects of neurodegenerative diseases such as macular degeneration, Alzheimer’s and Parkinson’s diseases, and multiple sclerosis, as well as to rehabilitate non-CNS diseased organs such as the pancreas for autologous production of insulin in diabetic patients.
Cancer Drug Discovery Targeting the Tumor Suppressor p53
In July 2007, we entered into a license agreement with the Buck Institute for Age Research under which we exclusively licensed the worldwide intellectual property rights for technologies designed to generate novel therapeutics and identify new drug discovery targets. There are no up-front or milestone payments required by us under the license agreement. If products are successfully developed incorporating any of the licensed technologies, we will pay the Buck Institute royalties based on a percentage of net sales. The term of the agreement will continue until the last patent claiming a licensed product under the agreement expires. However, the license agreement may be terminated earlier by either party upon the other party’s failure to timely cure a material breach under the agreement or upon the other party’s bankruptcy or insolvency.
Through this license agreement, we own the rights to a proprietary discovery platform designed to discover new chemical entities that remediate the signaling activities of the tumor suppressor p53 in p53-dysfunctional cancer cells. Loss of p53 activity is associated with one-half of all human tumors, often rendering these cancer cells resistant to conventional therapies. The licensed technologies have made the discovery of reactivators of appropriate p53 signaling behavior possible. Inherent in the design of the technologies is a strategy to develop therapeutics that selectively stimulate programmed cell death in p53-dysfunctional cancer cells and that would leave healthy cells expressing normal p53 unaffected.
The therapies developed in this program will target a specific population of aberrant (tumor) cells and forge a novel class of chemotherapeutics that have the potential to be much more potent than general inhibitors of cell proliferation or inducers of cell death. In the era of personalized medicine, and in combination with our emerging diagnostic methodologies, we believe this program will generate a novel pipeline of drugs that promise cancer patients greater proficiency with fewer side effects.
Immunotherapeutics and Related Assay Systems
The technologies licensed from the Buck Institute also included a novel immunotherapeutic/anti-cancer compound (T9) and highly sensitive cell-based assay systems for the discovery of additional immune-modulating
14
Table of Contents
drugs. Immune-modulating drugs represent an emerging class of therapies with broad clinical application in the treatment of cancer, allergies, inflammation, autoimmunity and tissue transplantation. T9 is a highly potent bi-functional molecule with the ability to kill cancer cells and to activate the immune response to recognize cancer cells in a manner analogous to childhood vaccination.
T9 was originally discovered using robust ultra-sensitive cell-based assay systems that respond to minute amounts of potential immune-modulating drugs. Over time, these systems were further modified to allow for their use in high throughput screens for the identification of compounds that can control the magnitude and quality of the immune response. The immune-modulating high throughput screening systems (IMHTSS) technologies are important tools for the discovery of novel agents that modulate the immune response through controlling the type and degree of inflammation. “Hits” resulting from the screening of synthetic and natural product libraries using the IMHTSS technologies are anticipated to be further developed to act as immune adjuvants in improving existing vaccination platforms or to inhibit the immune response in the context of allergies (such as asthma) or autoimmune diseases (such as type I diabetes, multiple sclerosis and lupus erythematosus).
Translational Molecular Bioscience at CNSI
The California NanoSystems Institute (CNSI) is a multidisciplinary research center at UCLA whose mission is to encourage university-industry collaboration and to enable the rapid commercialization of discoveries in nanosystems. CNSI members include some of the world’s preeminent scientists, and the work conducted at the institute represents world-class expertise in five targeted areas of nanosystems-related research: renewable energy, environmental nanotechnology and nanotoxicology, nanobiotechnology and biomaterials, nanomechanical and nanofluidic systems, and nanoelectronics, photonics and architectonics.
In July 2007, we entered into a research collaboration agreement with CNSI under which the parties agreed to collaborate on early research in nanobiotechnology for the advancement of new technologies in medicine. Under the agreement, we committed to fund up to $10 million over ten years in two tranches of $5 million for research projects selected by a committee comprised equally of our and CNSI representatives. Any intellectual property resulting from the projects will be owned by CNSI. However, we have first right to negotiate an option or license to all of such intellectual property on commercially reasonable terms, including royalties. In addition, if the parties are unable to successfully negotiate a license, CNSI may not license the intellectual property to another party at a lower rate than offered to us for a specified period of time. The term of the agreement is ten years, but either party can terminate on 30 days notice and the funding commitment for the second $5 million is subject to mutual agreement within 30 days of the end of the fifth year of the agreement.
This partnership provides CNSI and our researchers the opportunity to jointly pursue innovative approaches to the diagnosis and treatment of life-threatening diseases, leveraging the complementary resources and skills of both organizations. Working side by side with CNSI researchers, our scientists will focus on rapidly and seamlessly translating early scientific discovery into practical application. The Abraxis/CNSI Research Collaboration Lab has been designed to integrate and support multidisciplinary science, including cellular and molecular biology (including high-throughput discovery), nanodetection methodologies and tools for diagnostic discoveries, medicinal and synthetic chemistry, computational structural biology (including rational approaches to drug discovery) and bioengineering of nanodevices and nanomaterials.
Biosimilars
In June 2007, we entered into a license agreement with Biocon Limited under which we licensed the right to develop and commercialize a biosimilar version of G-CSF (granulocyte-colony stimulating factor) in North America and the European Union. G-CSF is an haematopoietic growth factor that works by encouraging the bone marrow to produce more white blood cells. Therapeutic G-CSF is primarily used for the treatment of neutropenia, the lowering of the white blood cells that fight infections. Biocon has received regulatory approval
15
Table of Contents
of its G-CSF from the Drugs Controller General of India (DCGI) for the treatment of neutropenia in cancer patients. The biological activity of Biocon’s G-CSF used in clinical trials was evaluated by the National Institute of Biological Standards and Control (NIBSC), UK, which provides independent testing of biological medicines. The NIBSC found that the potency of Biocon’s drug met the necessary requirements of a biosimilar G-CSF.
In November 2007, we entered into a license agreement with Green Cross Corporation under which we licensed the right to develop and commercialize five biosimilars for the United States and Canada on an exclusive basis: Enbrel® (etanercept), pegylated G-CSF, recombinant Factor VIII, Interferon-Alpha and Erythropoietin. Etanercept is a chimeric protein designed to neutralize tumor necrosis factor (TNF), an important regulator of local inflammatory response. Pegylated G-CSF, like G-CSF, is primarily used for the treatment of neutropenia, but its half-life is greatly increased compared to G-CSF because of the polyethylene glycol treatment (a process also commonly referred to as “pegylating”), thereby reducing the frequency and dose of the injections required to sustain a patient’s neutrophil levels. Recombinant Factor VIII is a clotting factor that is essential for reestablishing hemostasis in hemophiliac patients. Interferon-Alpha is a powerful, multifunctional protein produced by innate cells of the immune system to combat viral infections. Erythropoietin is a protein produced mainly by cells of the kidneys to maintain red blood cell levels by inhibiting their ability to commit cellular “suicide” and stimulate their production from precursors and is approved for patients experiencing anemia due to a variety of mechanisms including chronic renal failure and certain anti-cancer chemotherapies.
Central Nervous System Therapies
In April 2007, we formed a joint venture with Cenomed, Inc. (Cenomed) to create Cenomed BioSciences, LLC. This venture is designed to further the research and development of novel drugs that interact with the central nervous system focused on psychiatric and neurological diseases. Substantially all of the assets of Cenomed were contributed to this joint venture. The previous focus of the research by Cenomed included the development of drugs for the treatment of schizophrenia, neuroprotection, mild cognitive impairment and memory and attention impairments associated with aging, attention deficit hyperactivity disorder and pain. We hold a 70% membership interest in this joint venture, which is consolidated in our financial statements.
Sales and Marketing
We have dedicated sales and marketing groups in North America, Europe and China. In each of these regions, our team targets key segments of the oncology market: specifically, leading oncologists, cancer centers and the oncology distribution channel. In North America, our team consists of approximately 189 sales, marketing and medical affairs professionals. We continue to maintain our European infrastructure with an initial focus on five key European markets (United Kingdom, France, Spain, Italy and Germany). The sales and commercial services functions are provided by Quintiles Transnational Corp., which is the leading global commercial solutions provider to the pharmaceutical, biotech and medical device industries. We have medical representatives trained and deployed in the United Kingdom, Germany, Spain, Italy and France. In the second quarter of 2009, we launched Abraxane® in China, which has a team of approximately 51 employees including 46 medical representatives and managers.
Strategic Relationships
Taiho Pharmaceutical Co., Ltd.
In May 2005, we entered into a license agreement with Taiho Pharmaceutical Co., Ltd. under which we granted to Taiho the exclusive rights to market and sell Abraxane® in Japan. In March 2008, Taiho filed a Japanese New Drug Application (J-NDA) with the Ministry of Health, Labour and Welfare to market Abraxane® for the treatment of breast cancer in Japan. We established a joint steering committee with Taiho to oversee the development of Abraxane® in Japan for the treatment of breast, lung and gastric cancer and other solid tumors. Under this license agreement, Taiho paid us a non-refundable, upfront payment and will make additional
16
Table of Contents
payments to us upon achievement of various clinical, regulatory and sales milestones, with total potential payments in excess of $50 million. In addition, we will receive royalties from Taiho based on net sales under the license agreement. The agreement will remain in effect until terminated by one of the parties in accordance with its terms. The agreement may be terminated: (a) at any time by mutual agreement of the parties; (b) by either party if the other party fails to timely cure a material breach under the agreement or the other party is bankrupt or insolvent; (c) by Taiho upon nine months notice if Taiho determines, for safety, legal or other reasons, not to continue pursuing the development and commercialization of Abraxane® in Japan; (d) by Taiho if we fail to timely remedy certain manufacturing deficiencies; and (e) at any time by us if Taiho sells (i) Abraxane® while concurrently selling Abraxane® in a generic form or (ii) an injectable formulation of a taxane compound, including paclitaxel and docetaxel.
Biocon Limited
In June 2007, we entered into a license agreement with Biocon Limited under which we granted Biocon the right to market and sell Abraxane® in India, Pakistan, Bangladesh, Sri Lanka, United Arab Emirates, Saudi Arabia, Kuwait and certain other South Asian and Persian Gulf countries. Biocon’s rights are exclusive with respect to India, Pakistan, Bangladesh, Nepal, Bhutan, Sri Lanka and the Maldive Islands and are co-exclusive with us and our licensees with respect to all other countries. If Biocon fails to achieve a specified target market share by a specified time in any exclusive country, then we have the right to make Biocon’s license in that country non-exclusive unless Biocon makes a payment to us equal approximately to the royalty that would have been payable had Biocon achieved the target market share. If Biocon fails to achieve a minimum market share by a specified time in any exclusive country for two consecutive calendar years, then we also have the right to terminate Biocon’s rights with respect to such country. The agreement will remain in effect until terminated by one of the parties in accordance its terms. We may terminate the agreement if Biocon (a) fails to timely cure a material breach; (b) becomes bankrupt; or (c) sells Abraxane® while concurrently selling a generic form of Abraxane®. Biocon may terminate the agreement if we fail to timely cure a material breach. We will receive payments from Biocon based on the higher of a percentage net sales or a specified profit split under the license agreement. In October 2007, we received regulatory approval from India’s Drug Controller General to market Abraxane® for the treatment of metastatic breast cancer in India. In July 2008, we launched Abraxane in India for the treatment of metastatic breast cancer.
Green Cross Corporation
In November 2007, we entered into an agreement with Green Cross Corporation whereby we granted an exclusive license to Green Cross to market and sell Abraxane® in South Korea. Under the agreement, Green Cross will pay a royalty on net sales of Abraxane® as well as upfront and milestone payments. Additionally, we licensed from Green Cross an exclusive license to develop and commercialize the following biosimilars in the United States and Canada: Erythropoetin, pegylated G-CSF (granulocyte-colony stimulating factor), Interferon-Alpha, recombinant Factor VIII and Enbrel® (etanercept). We will pay to Green Cross a milestone on each product once approval has been received and a royalty on net sales.
Specialised Therapeutics of Australia, Pty Ltd.
In February 2008, we entered into an exclusive license agreement with Specialised Therapeutics of Australia, Pty Ltd. under which we granted Specialised Therapeutics the right to market and sell Abraxane® in Australia and New Zealand. In October 2008, the Therapeutic Goods Administration (TGA) in Australia approved Abraxane® for the treatment of metastatic carcinoma of the breast after failure of anthracycline therapy. In February 2009, we launched Abraxane® in Australia through Specialised Therapeutics.
ProMetic Life Sciences Inc.
In September 2008, we entered into agreements with ProMetic Life Sciences Inc. (ProMetic) to develop and commercialize four biopharmaceutical products targeting underserved medical conditions. We entered into the
17
Table of Contents
following agreements: (i) a securities purchase agreement, (ii) a license agreement, (iii) a supply and license agreement (iv) an exclusive manufacturing agreement and (v) a services agreement. The transaction included an initial investment in ProMetic of $7 million and optional future investment rights of up to $25 million. Of the $7 million initial investment, $5.2 million was allocated to the purchase of ProMetic’s common stock and $1.8 million was allocated to the future investment rights option. We will have access to ProMetic’s proprietary protein technologies to commercialize the biopharmaceuticals and will fund all development costs to regulatory approval. In consideration, we will pay potential milestone and royalty payments to ProMetic and royalties on the net sales of the four products. Additionally, ProMetic will perform product development activities on behalf of Abraxis under the service agreement.
In December 2009, we entered into a collaboration agreement with ProMetic to develop and commercialize various applications deriving from ProMetic’s prion capture technology platform. As part of the agreement, we will share equally with ProMetic the costs for the development of such new applications and the revenues arising from their commercialization.
Competition
Competition among biotechnology, pharmaceutical and other companies that research, develop, manufacture or market proprietary pharmaceuticals is intense and is expected to increase. We compete with these entities in all areas of business, including competing to attract and retain qualified scientific and technical personnel.
Our products’ and product candidates’ competitive position among other pharmaceutical products may be based on, among other things, patent position, product efficacy, safety, reliability, availability, patient convenience/delivery devices and price, as well as, the development and marketing of new competitive products. Certain of our products and product candidates may face substantial competition from products marketed by large pharmaceutical companies, many of which have greater clinical, research, regulatory, manufacturing, marketing, financial and human resources, and experience than we do. In addition, the introduction of new products or the development of new processes by competitors or new information about existing products may result in product replacements or price reductions, even for products protected by patents.
Some of our competitors are actively engaged in research and development in areas where we are developing product candidates. The competitive marketplace for our product candidates is significantly dependent upon the timing of entry into the market. Early entry may have important advantages in gaining product acceptance and market share and, as a result, may contribute significantly to the product’s eventual success and profitability. Accordingly, in some cases, the relative speed with which we can develop products, complete the testing, receive approval, and supply commercial quantities of the product to the market is expected to be important to our competitive position.
In addition, we compete with large pharmaceutical and biotechnology companies when entering into cooperative arrangements with smaller companies and research organizations in the biotechnology industry for the development and commercialization of products and product candidates. Small companies, academic institutions, governmental agencies and other public and private research organizations conduct a significant amount of research and development in the biotechnology industry. These entities may seek to enter into licensing arrangements to collect royalties for use of technology or for the sale of products they have discovered or developed. We may face competition in licensing or acquisition activities from pharmaceutical companies and large biotechnology companies that also seek to acquire technologies or product candidates from these entities.
We believe that Abraxane® competes, directly or indirectly, with the primary taxanes in the market place, including Bristol-Myers Squibb’s Taxol® and its generic equivalents, Sanofi-Aventis’ Taxotere® and other cancer therapies. Many pharmaceutical companies have developed and are marketing, or are developing, alternative formulations of paclitaxel and other cancer therapies that may compete directly or indirectly with Abraxane®. In addition, Abraxane® currently competes with other cytotoxic agents outside of the taxane class such as capecitabine, gemcitabine, ixabepilone and navelbine.
18
Table of Contents
Regulatory Considerations
Proprietary pharmaceutical products are subject to extensive pre- and post-market regulation by the FDA, including regulations that govern the testing, manufacturing, safety, efficacy, labeling, storage, record keeping, advertising and promotion of the products under the Federal Food Drug and Cosmetic Act and the Public Health Services Act, and by comparable agencies in foreign countries. FDA approval is required before any dosage form of any drug can be marketed in the United States. All applications for FDA approval of drugs must include information relating to the pharmaceutical formulation, stability, manufacturing, processing, packaging, labeling, quality control, safety and efficacy and other information as appropriate.
The process required by the FDA before a new drug may be marketed in the United States generally involves:
| • | completion of pre-clinical laboratory and animal testing; |
| • | submission of an investigational new drug application, or IND, which must become effective before trials may begin; |
| • | performance of adequate and well-controlled human clinical trials to establish the safety and efficacy of the proposed drug product’s intended use; and |
| • | submission to and approval by the FDA of a new drug application, or NDA. |
Clinical trials are typically conducted in three sequential phases that may overlap. These phases generally include:
| • | Phase I during which the drug is introduced into healthy human subjects or, on occasion, patients, and generally is tested for safety, stability, dose tolerance and metabolism; |
| • | Phase II during which the drug is introduced into a limited patient population to determine the efficacy of the product in specific targeted diseases, to determine dosage tolerance and optimal dosage and to identify possible adverse effects and safety risks; and |
| • | Phase III during which the clinical trial is expanded to a more diverse patient group in geographically dispersed trial sites to further evaluate clinical efficacy, optimal dosage and safety. |
The drug sponsor, the FDA or the Institutional Review Board at each institution at which a clinical trial is being performed may suspend a clinical trial at any time for various reasons, including a belief that the subjects are being exposed to an unacceptable health risk.
The results of product development, preclinical animal studies and human studies are submitted to the FDA as part of the NDA. The NDA also must contain extensive manufacturing information. The FDA may approve on the basis of the submission made or disapprove the NDA if applicable FDA regulatory criteria are not satisfied. The FDA may also require additional clinical data. Under certain circumstances, drug sponsors may obtain approval pursuant to Section 505(b)(2) of the Federal Food, Drug & Cosmetic Act based in part upon literature or an FDA finding and/or effectiveness for another approved product, even where the products are not duplicates in terms of formulation and bioequivalence. Once approved, the FDA may withdraw the product approval if compliance with pre- and post-market regulatory standards is not maintained or if problems occur after the product reaches the marketplace. In addition, the FDA may require post-marketing studies to monitor the effect of approved products and may limit further marketing of the product based on the results of these post-marketing studies. The FDA has broad post-market regulatory and enforcement powers, including the ability to levy fines and civil penalties, suspend or delay issuance of approvals, seize or recall products, and withdraw approvals.
Satisfaction of FDA pre-market approval requirements typically takes several years and the actual time required may vary substantially based upon the type, complexity and novelty of the product or disease. Government regulation may delay or prevent marketing of potential products for a considerable period of time
19
Table of Contents
and impose costly procedures upon a manufacturer’s activities. Success in early stage clinical trials does not assure success in later stage clinical trials. Data obtained from clinical activities is not always conclusive and may be susceptible to varying interpretations that could delay, limit or prevent regulatory approval. Even if a product receives regulatory approval, later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market.
In addition to regulating and auditing human clinical trials, the FDA regulates and inspects equipment, facilities, laboratories and processes used in the manufacturing and testing of such products prior to providing approval to market a product. If after receiving clearance from the FDA, a material change is made in manufacturing equipment, location or process, additional regulatory review may be required. We and our suppliers also must adhere to current good manufacturing practice and product-specific regulations enforced by the FDA through its facilities inspection program. The FDA also conducts regular, periodic visits to re-inspect equipment, facilities, laboratories and processes following the initial approval. If, as a result of these inspections, the FDA determines that the equipment, facilities, laboratories or processes do not comply with applicable FDA regulations and conditions of product approval, the FDA may seek civil, criminal or administrative sanctions and/or remedies against our manufacturers, including the suspension of manufacturing operations. In addition, our discovery pipeline includes product candidates in the follow-on biologics area, also known as biosimilars. Although, like all pharmaceutical products, these product candidates must receive appropriate regulatory clearance before they can be marketed and sold in a particular country, including the United States, there does not currently exist a defined abbreviated regulatory approval process for obtaining this approval in the United States. As a result, considerable uncertainty exists regarding the pathway, timing and likelihood of approval for these types of product candidates.
While the above process describes the regulatory process in the United States, similar processes for drug development and approval to market generally apply internationally.
We are also subject to various federal and state laws pertaining to health care “fraud and abuse,” including anti-kickback laws and false claims laws. Anti-kickback laws make it illegal to solicit, offer, receive or pay any remuneration in exchange for, or to induce, the referral of business, including the purchase or prescription of a particular drug. The federal government has published regulations that identify “safe harbors” or exemptions for certain arrangements that do not violate the anti-kickback statutes. Due to the breadth of the statutory provisions and the absence of guidance in the form of regulations or court decisions addressing some practices, it is possible that our practices might be challenged under anti-kickback or similar laws. False claims laws prohibit anyone from knowingly and willingly presenting, or causing to be presented for payment to third-party payers (including Medicare and Medicaid), claims for reimbursed drugs or services that are false or fraudulent, claims for items or services not provided as claimed or claims for medically unnecessary items or services. The activities of our strategic partners relating to the sale and marketing of our products may be subject to scrutiny under these laws. Violations of fraud and abuse laws may be punishable by criminal and/or civil sanctions, including fines and civil monetary penalties, as well as the possibility of exclusion from federal health care programs (including Medicare and Medicaid). If the government were to allege against or convict us of violating these laws, there could be a material adverse effect on us. Our activities could be subject to challenge for the reasons discussed above and due to the broad scope of these laws and the increasing attention being given to them by law enforcement authorities.
We are also subject to regulation under the Occupational Safety and Health Act, the Toxic Substances Control Act, the Resource Conservation and Recovery Act, and other current and potential future federal, state, or local laws, rules and/or regulations. Our research and development activities involve the controlled use of chemicals, biological materials and other hazardous materials. We believe that our procedures comply with the standards prescribed by federal, state and local laws, rules and/or regulations; however, the risk of injury or accidental contamination cannot be completely eliminated.
20
Table of Contents
Manufacturing
We have manufacturing facilities in Melrose Park, Illinois, and Phoenix, Arizona. For the length of the applicable lease, our manufacturing facility in Melrose Park has been leased to APP and APP has agreed to provide certain contract manufacturing services to us in accordance with the terms of the manufacturing agreement. In addition, we lease from APP a portion of APP’s Grand Island, New York manufacturing facility to enable us to perform our responsibilities under the manufacturing agreement with APP for its term. The initial term of the manufacturing agreement will expire on December 31, 2011, but the term of the manufacturing agreement will be automatically extended for one year if either APP exercises its option to extend the lease for our Melrose Park manufacturing facility for an additional year or we exercise our option to extend the lease for APP’s Grand Island manufacturing facility for an additional year.
In addition, as part of the separation agreement, APP’s Puerto Rico facility was divided into two separate facilities and we took ownership of one part of the facility. The portion of the Puerto Rico facility that is owned by us following the separation is the 90,000 square foot active pharmaceutical ingredients manufacturing plant that was leased to Pfizer for a term expiring in February 2012. Pfizer vacated the premises in December 2009 and did not incur any penalty for the early termination of the lease.
We are required to comply with the applicable FDA manufacturing requirements contained in the FDA’s current Good Manufacturing Practice, or cGMP, regulations. cGMP regulations require quality control and quality assurance as well as the corresponding maintenance of records and documentation. Our manufacturing facilities must meet cGMP requirements to permit us to manufacture our products. We are subject to the periodic inspection of our facilities, procedures and operations and/or the testing of our products by the FDA, the Drug Enforcement Administration and other authorities to assess our compliance with applicable regulations.
Failure to comply with the statutory and regulatory requirements subjects the manufacturer to possible legal or regulatory action, including the seizure or recall of products, injunctions, consent decrees placing significant restrictions on or suspending manufacturing operations, and civil and criminal penalties. In September and October 2009, we voluntarily initiated a global recall of a total of twenty-five lots of Abraxane® manufactured at the Grand Island, New York facility as a result of particulate matter observed in a small number of vials from the recalled lots. Adverse experiences with a product must be reported to the FDA and could result in the imposition of market restriction through labeling changes or in product removal. Product approvals may be withdrawn if compliance with regulatory requirements is not maintained or if problems concerning safety or efficacy of the product occur following approval.
Raw Materials
We are developing a raw material supply business, including active pharmaceutical ingredients, for biological and biosimilar applications. Ultimately, these raw materials may be used in our product candidates and/or sold to third parties. We acquired a manufacturing facility in Oelwein, Iowa in part to support these raw material activities. At various times, we may pursue revenue opportunities from sales of our raw materials, but we cannot assure any such opportunities will materialize to any meaningful degree or at all.
The manufacture of our products requires raw materials and other components that must meet stringent FDA requirements. Some of these raw materials and other components currently are available only from a limited number of sources. Additionally, regulatory approvals for a particular product denote the raw materials and components, and the suppliers for such materials that may be used for that product. Even when more than one supplier exists, we may elect to list, and in some cases have only listed, one supplier in our applications with the FDA. Any change in or addition of a supplier not previously approved must then be submitted through a formal approval process with the FDA. From time to time, it is necessary to maintain increased levels of certain raw materials due to the anticipation of raw material shortages or in response to market opportunities.
21
Table of Contents
Intellectual Property
We rely on trade secrets, unpatented proprietary know-how, continuing technological innovation and patent protection to preserve our competitive position. We have over 100 issued U.S. and foreign patents, including patents relating to Abraxane® and the technology surrounding Abraxane®. The issued patents covering Abraxane®, and methods of use and preparation of Abraxane®, currently expire through 2018, but we have additional pending U.S. and foreign patent applications covering Abraxane® that could extend the expiration dates.
Our success depends on our ability to operate without infringing the patents and proprietary rights of third parties. We cannot determine with certainty whether patents or patent applications of other parties will materially affect our ability to make, use, offer to sell or sell any products. A number of pharmaceutical companies, biotechnology companies, universities and research institutions may have filed patent applications or may have been granted patents that cover aspects of our or our licensors’ products, product candidates or other technologies.
Intellectual property protection is highly uncertain and involves complex legal and factual questions. Our patents and those for which we have or will have licensed rights may be challenged, invalidated, infringed or circumvented, and the rights granted in those patents may not provide proprietary protection or competitive advantages to us. We and our licensors may not be able to develop patentable products. Even if patent claims are allowed, the claims may not issue, or in the event of issuance, may not be sufficient to protect the technology owned by or licensed to us.
Third-party patent applications and patents could reduce the coverage of the patents licensed, or that may be licensed to or owned by us. If patents containing competitive or conflicting claims are issued to third parties, we may be enjoined from commercialization of products or be required to obtain licenses to these patents or to develop or obtain alternative technology. In addition, other parties may duplicate, design around or independently develop similar or alternative technologies to our licensors or our technologies.
Litigation may be necessary to enforce patents issued or licensed to us or to determine the scope or validity of another party’s proprietary rights. U.S. Patent and Trademark Office interference proceedings may be necessary if we and another party both claim to have invented the same subject matter. Similarly, our patents and patent applications, or those of our licensors, could face other challenges, such as opposition proceedings and reexamination proceedings. Any such challenge, if successful, could result in the invalidation of, or a narrowing of scope of, any such patents and patent applications. We could incur substantial costs and our management’s attention would be diverted if:
| • | patent litigation is brought by third parties; |
| • | we are a party to or participate in patent suits brought against or initiated by us or our licensors; |
| • | we are a party to or participate in an interference proceeding; or |
| • | we are a party to an opposition or reexamination proceeding. |
In addition, we may not prevail in any of these actions or proceedings.
We are currently involved in a patent infringement lawsuit with Élan Pharmaceutical Int’l Ltd. See “Item 3—Legal Proceedings” below.
Employees
As of December 31, 2009, we had a total of approximately 885 full-time employees, of which approximately 349 were dedicated to research and development activities, including clinical trials, approximately 150 were in manufacturing, approximately 239 were in sales and marketing and approximately 147 were in
22
Table of Contents
administration and finance. None of our employees are represented by a labor union or subject to a collective bargaining agreement. We have not experienced any work stoppage and consider our relations with our employees to be good.
Environment
We believe that our operations comply in all material respects with applicable laws and regulations concerning the environment. While it is impossible to predict accurately the future costs associated with environmental compliance and potential remediation activities, compliance with environmental laws is not expected to require significant capital expenditures and has not had, and is not expected to have, a material adverse effect on our financial condition or competitive position.
You should carefully consider the risks described below before investing in our publicly traded securities. The risks described below are not the only ones facing us. Our business is also subject to the risks that affect many other companies, such as competition and, general economic conditions. Additional risks not currently known to us or that we currently believe are immaterial also may impair our business operations and our liquidity.
Risks Related to our Business and Industry
We are highly dependent upon the strong market acceptance of Abraxane®.
Our future profitability is highly dependent on the strong market acceptance of Abraxane®. For the years ended December 31, 2009, 2008 and 2007, total Abraxane® revenue was $314.5 million, $335.6 million and $324.7 million, respectively. Included in Abraxane revenue in each year for the years ended December 31, 2008 and 2007 was $36.4 million of recognized deferred revenue relating to the co-promotion agreement with AstraZeneca. The co-promotion agreement ended in January 2009. Because Abraxane® is our only approved product, Abraxane® revenue represented approximately 87.6%, 97.2% and 97.3% of our revenues for the year ended December 31, 2009, 2008 and 2007, respectively. We anticipate that sales of Abraxane® will remain a substantial portion of our total revenue over the next several years. However, a number of pharmaceutical companies are working to develop alternative formulations of paclitaxel and other cancer drugs and therapies, any of which may compete directly or indirectly with Abraxane® and which might adversely affect the commercial success of Abraxane®.
Abraxane® could lose market share or revenue due to numerous factors, many of which are beyond our control, including:
| • | lower prices offered on similar products by other manufacturers, including generic forms of Taxol or other taxanes; |
| • | substitute or alternative products or therapies; |
| • | development by others of new pharmaceutical products or treatments that are more effective than Abraxane®; |
| • | introduction of other generic equivalents or products that may be therapeutically interchanged with Abraxane®; |
| • | interruptions in manufacturing or supply; |
| • | changes in the prescribing practices of physicians; |
| • | changes in third-party reimbursement practices; and |
| • | migration of key customers to other manufacturers or sellers. |
In addition, we will continue to make a significant investment in Abraxane®, including costs associated with conducting clinical trials and obtaining necessary regulatory approvals for the use of Abraxane® in other
23
Table of Contents
indications and settings and in other jurisdictions, expansion of our marketing, sales and manufacturing staff, the acquisition of paclitaxel raw material, expansion of manufacturing facilities and the manufacture of finished product. The success of Abraxane® in the Phase III trial for metastatic breast cancer and other clinical trials may not be representative of future clinical trial results for Abraxane® with respect to other clinical indications. The results from clinical, pre-clinical studies and early clinical trials conducted to date may not be predictive of results to be obtained in later clinical trials, including those ongoing at present. Further, the commencement and completion of clinical trials may be delayed by many factors that are beyond our control, including:
| • | slower than anticipated patient enrollment; |
| • | difficulty in finding and retaining patients fitting the trial profile or protocols; and |
| • | adverse events occurring during the clinical trials. |
Proprietary product development efforts may not result in commercial products.
We intend to maintain an aggressive research and development program for proprietary pharmaceutical products. Successful product development in the biotechnology industry is highly uncertain, and statistically very few research and development projects produce a commercial product. Product candidates that appear promising in the early phases of development, such as in early stage human clinical trials, may fail to reach the market for a number of reasons, including:
| • | the product candidate did not demonstrate acceptable clinical trial results even though it demonstrated positive pre-clinical trial results; |
| • | the product candidate was not effective in treating a specified condition or illness; |
| • | the product candidate had harmful side effects in humans or animals; |
| • | the necessary regulatory bodies, such as the FDA, did not approve a product candidate for an intended use; |
| • | the product candidate was not economical to manufacture and commercialize; |
| • | other companies or people have or may have proprietary rights to a product candidate, such as patent rights, and will not let the product candidate be sold on reasonable terms, or at all; or |
| • | the product candidate is not cost effective in light of existing therapeutics. |
In addition, our discovery pipeline includes product candidates in the follow-on biologics area, also known as biosimilars. Although, like all pharmaceutical products, these product candidates must receive appropriate regulatory clearance before they can be marketed and sold in a particular country, including the United States, there does not currently exist a defined abbreviated regulatory approval process for obtaining this approval in the United States. As a result, considerable uncertainty exists regarding the pathway, timing and likelihood of approval for these types of product candidates in the United States. Because of the absence of a defined abbreviated regulatory approval process for biosimilars in the United States, it could take a longer amount of time to obtain regulatory approval for our biosimilar product candidates than for our other product candidates in our discovery pipeline, if at all. Any delay in, or the inability to receive, regulatory approval for our biosimilar product candidates could materially adversely affect our business plan and growth strategy.
We may be required to perform additional clinical trials or change the labeling of our products if side effects or manufacturing problems are identified after our products are on the market.
If side effects are identified after any of our products are on the market, or if manufacturing problems occur, regulatory approval may be withdrawn and product recalls, reformulation of products, additional clinical trials, changes in labeling of products, and changes to or re-approvals of our manufacturing facilities may be required, any of which could have a material adverse effect on sales of the affected products and on our business and results of operations.
24
Table of Contents
After products are approved for commercial use, we or regulatory bodies could decide that changes to the product labeling are required. Label changes may be necessary for a number of reasons, including the identification of actual or theoretical safety or efficacy concerns by regulatory agencies or the discovery of significant problems with a similar product that implicates an entire class of products. Any significant concerns raised about the safety or efficacy of our products could also result in the need to recall products, reformulate those products, to conduct additional clinical trials, to make changes to the manufacturing processes, or to seek re-approval of the manufacturing facilities. Significant concerns about the safety and effectiveness of a product could ultimately lead to the revocation of our marketing approval. The revision of product labeling or the regulatory actions described above could be required even if there is no clearly established connection between the product and the safety or efficacy concerns that have been raised. The revision of product labeling or the regulatory actions described above could have a material adverse effect on sales of the affected products and on our business and results of operations.
If we or our suppliers are unable to comply with ongoing and changing regulatory standards, sales of our products could be delayed or prevented.
Virtually all aspects of our business, including the development, testing, manufacturing, processing, quality, safety, efficacy, packaging, labeling, record-keeping, distribution, storage and advertising and promotion of our products and disposal of waste products arising from these activities, are subject to extensive regulation by federal, state and local governmental authorities in the United States, including the FDA and the Department of Health and Human Services Office of Inspector General (HHS OIG). Our business is also subject to similar and/or additional regulation in other countries. Compliance with these regulations is costly and time-consuming.
Our manufacturing facilities and procedures and those of our suppliers are subject to ongoing regulation, including periodic inspection by the FDA and other regulatory agencies. For example, manufacturers of pharmaceutical products must comply with detailed regulations governing current good manufacturing practices, including requirements relating to quality control and quality assurance. We must spend funds, time and effort in the areas of production, safety, quality control and quality assurance to ensure compliance with these regulations. We cannot assure that our manufacturing facilities or those of our suppliers will not be subject to regulatory action in the future.
Our products generally must receive appropriate regulatory clearance before they can be sold in a particular country, including the United States. We may encounter delays in the introduction of a product as a result of, among other things, insufficient or incomplete submissions to a regulatory agency for approval of a product, objections by another company with respect to our submissions for approval, new patents by other companies, patent challenges by other companies that result in a 180-day exclusivity period, and changes in regulatory policy during the period of product development or during the regulatory approval process. Regulatory agencies have the authority to revoke drug approvals previously granted and remove from the market previously approved products for various reasons, including issues related to current good manufacturing practices for that particular product or in general. We may be subject from time to time to product recalls initiated by us or by regulatory authorities. In September and October 2009, we voluntarily initiated a global recall of a total of twenty-five lots of Abraxane® manufactured at the Grand Island, New York facility as a result of particulate matter observed in a small number of vials from the recalled lots. Delays in obtaining regulatory approvals, the revocation of a prior approval, or product recalls could impose significant costs on us and adversely affect our ability to generate revenue.
Our inability or the inability of our suppliers to comply with applicable regulatory requirements can result in, among other things, warning letters, fines, consent decrees restricting or suspending our manufacturing operations, delay of approvals for new products, injunctions, civil penalties, recall or seizure of products, total or partial suspension of sales and criminal prosecution. Any of these or other regulatory actions could materially adversely affect our business and financial condition.
25
Table of Contents
We depend on third parties to supply raw materials and other components and may not be able to obtain sufficient quantities of these materials, which will limit our ability to manufacture our products on a timely basis and harm our operating results.
The manufacture of Abraxane® requires, and our product candidates will require, raw materials and other components that must meet stringent regulatory requirements. Some of these raw materials and other components are available only from a limited number of sources. We have not historically experienced any paclitaxel supply shortages. Additionally, we maintain a safety stock supply of paclitaxel to mitigate any supply disruption. Our regulatory approvals for each particular product denote the raw materials and components, and the suppliers for such materials, we may use for that product. Obtaining approval to change, substitute or add a raw material or component, or the supplier of a raw material or component, can be time consuming and expensive, as testing and regulatory approval are necessary. If our suppliers are unable to deliver sufficient quantities of these materials on a timely basis or we encounter difficulties in our relationships with these suppliers, the manufacture and sale of our products may be disrupted, and our business, operating results and reputation could be adversely affected.
The injectable pharmaceutical products markets are highly competitive, and if we are unable to compete successfully, our revenue will decline and our business will be harmed.
The markets for injectable pharmaceutical products are highly competitive, rapidly changing and undergoing consolidation. Abraxane® competes, directly or indirectly, with the primary taxanes in the market place, including Bristol-Myers Squibb’s Taxol® and its generic equivalents and Sanofi-Aventis’ Taxotere® and other cancer therapies. Many pharmaceutical companies have developed and are marketing, or are developing, alternative formulations of paclitaxel and other cancer therapies that may compete directly or indirectly with Abraxane®. In addition, Abraxane currently competes with other cytotoxic agents outside of the taxane class such as capecitabine, gemcitabine, ixabepilone and navelbine. Many of our competitors have significantly greater research and development, financial, sales and marketing, manufacturing, regulatory and other resources than we do. As a result, they may be able to devote greater resources to the development, manufacture, marketing or sale of their products, receive greater resources and support for their products, initiate or withstand substantial price competition, more readily take advantage of acquisition or other opportunities, or otherwise more successfully market their products.
Any reduction in demand for our products could lead to a decrease in prices, fewer customer orders, reduced revenues, reduced margins, reduced levels of profitability, or loss of market share. These competitive pressures could adversely affect our business and operating results.
Other companies may claim that we infringe on their intellectual property or proprietary rights, which could cause us to incur significant expenses or prevent us from selling our products.
Our success depends in part on our ability to operate without infringing the patents and proprietary rights of third parties. The manufacture, use and sale of new products with conflicting patent rights have been subject to substantial litigation in the pharmaceutical industry. These lawsuits relate to the validity and infringement of patents or proprietary rights of third parties. A number of pharmaceutical companies, biotechnology companies, universities and research institutions may have filed patent applications or may have been granted patents that cover aspects of our products or our licensors’ products, product candidates or other technologies.
Future or existing patents issued to third parties may contain claims that conflict with our products. We may be subject to infringement claims from time to time in the ordinary course of our business, and third parties could assert infringement claims against us in the future with respect to Abraxane®, products that we may develop or products that we may license. We are in the process of appealing the jury ruling in the patent infringement lawsuit with Élan Pharmaceutical Int’l Ltd. See “Item 3—Legal Proceedings”. Litigation or interference proceedings could force us to:
| • | stop or delay selling, manufacturing or using products that incorporate or are made using the challenged intellectual property; |
26
Table of Contents
| • | pay damages; or |
| • | enter into licensing or royalty agreements that may not be available on acceptable terms, if at all. |
Any litigation or interference proceedings, regardless of their outcome, would likely delay the regulatory approval process, be costly and require significant time and attention of key management and technical personnel.
Our inability to protect our intellectual property rights in the United States and foreign countries could limit our ability to manufacture or sell our products.
We rely on trade secrets, unpatented proprietary know-how, continuing technological innovation and patent protection to preserve our competitive position. We have over 100 issued U.S. and foreign patents, including patents relating to Abraxane® and the technology surrounding Abraxane®. The issued patents covering Abraxane®, and methods of use and preparation of Abraxane®, currently expire through 2018, but we have additional pending U.S. and foreign patent applications pending that could extend the expiration dates. Our patents and those for which we have licensed or will license rights, including for Abraxane®, may be challenged, invalidated, infringed or circumvented, and the rights granted in those patents may not provide proprietary protection or competitive advantages to us. We and our licensors may not be able to develop patentable products. Even if patent claims are allowed, the claims may not issue, or in the event of issuance, may not be sufficient to protect the technology owned by or licensed to us. Third-party patents could reduce the coverage of the patents licensed, or that may be licensed to or owned by us. If patents containing competitive or conflicting claims are issued to third parties, we may be prevented from commercializing the products covered by such patents, or may be required to obtain or develop alternate technology. In addition, other parties may duplicate, design around or independently develop similar or alternative technologies.
We may not be able to prevent third parties from infringing or using our intellectual property. We generally control and limit access to, and the distribution of, our product documentation and other proprietary information. Despite our efforts to protect this proprietary information, however, unauthorized parties may obtain and use information that we regard as proprietary. Other parties may independently develop similar know-how or may even obtain access to these technologies.
The laws of some foreign countries do not protect proprietary information to the same extent as the laws of the United States, and many companies have encountered significant problems and costs in protecting their proprietary information in these foreign countries.
The U.S. Patent and Trademark Office and the courts have not established a consistent policy regarding the breadth of claims allowed in pharmaceutical patents. The allowance of broader claims may increase the incidence and cost of patent interference proceedings and the risk of infringement litigation. On the other hand, the allowance of narrower claims may limit the value of our proprietary rights.
The strategy to license rights to or acquire and commercialize proprietary, biological injectable or other specialty injectable products may not be successful and we may never receive any return on our investment in these products.
We may license rights to or acquire products or technologies from third parties. Other companies, including those with substantially greater financial and sales and marketing resources, will compete with us to license rights to or acquire these products and technologies. We may not be able to license rights to or acquire these proprietary or other products or technologies on acceptable terms, if at all. Even if we obtain rights to a pharmaceutical product and commit to payment terms, including, in some cases, significant up-front license payments, we may not be able to generate product sales sufficient to create a profit or otherwise avoid a loss.
27
Table of Contents
A product candidate may fail to result in a commercially successful drug for other reasons, including the possibility that the product candidate may:
| • | be found during clinical trials to be unsafe or ineffective; |
| • | fail to receive necessary regulatory approvals; |
| • | be difficult or uneconomical to produce in commercial quantities; |
| • | be precluded from commercialization by proprietary rights of third parties; or |
| • | fail to achieve market acceptance. |
The marketing strategy, distribution channels and levels of competition with respect to any licensed or acquired product may be different from those of Abraxane® and we may not be able to compete favorably in any new product category.
We may become subject to federal or state false claims or other similar litigation brought by private individuals and the government.
The Federal False Claims Act (and equivalent state statutes) allows persons meeting specified requirements to bring suit alleging false or fraudulent Medicare or Medicaid claims and to share in any amounts paid to the government in fines or settlement. These suits, known as qui tam actions, have increased significantly in recent years and have increased the risk that a health care company will have to defend a false claim action, pay fines and/or be excluded from Medicare and Medicaid programs. False claims litigation can lead to civil monetary penalties, criminal fines and imprisonment and/or exclusion from participation in Medicare, Medicaid and other federally funded health programs. Other alternate theories of liability may also be available to private parties seeking redress for such claims. A number of parties have brought claims against numerous pharmaceutical manufacturers, and we cannot be certain that such claims will not be brought against us, or if they are brought, that such claims might not be successful.
We may need to change our business practices to comply with changes to, or may be subject to charges under, the fraud and abuse laws.
We are subject to various federal and state laws pertaining to health care fraud and abuse, including the federal Anti-Kickback Statute and its various state analogues, the federal and state False Claims Act and additional marketing and pricing laws. Violations of these laws are punishable by criminal and/or civil sanctions, including, in some instances, imprisonment and exclusion from participation in federal and state health care programs such as Medicare and Medicaid. We may have to change our advertising and promotional business practices, or our existing business practices could be challenged as unlawful due to changes in laws, regulations or rules or due to administrative or judicial findings, which could materially adversely affect our business.
We face uncertainty related to pricing and reimbursement and health care reform.
In both domestic and foreign markets, sales of our products will depend in part on the availability of reimbursement from third-party payors such as government health administration authorities, private health insurers, health maintenance organizations and other health care-related organizations. Effective January 1, 2006 in the U.S., we received a unique reimbursement “J” code for Abraxane® from the Centers for Medicare and Medicaid Services. That code allows providers to bill Medicare for the use of Abraxane®. We believe that most major insurers reimburse providers for Abraxane® use consistent with the FDA-approved indication, which we believe is consistent with the reimbursement practices of major insurers for other drugs containing paclitaxel for the same indication. However, reimbursement by such payors is presently undergoing reform, and there is significant uncertainty at this time how this will affect sales of certain pharmaceutical products. There is possible U.S. legislation or regulatory action affecting, among other things, pharmaceutical pricing and reimbursement, including under Medicaid and Medicare, the importation of prescription drugs that are marketed outside the U.S. and sold at prices that are regulated by governments of various foreign countries.
28
Table of Contents
Medicare, Medicaid and other governmental reimbursement legislation or programs govern drug coverage and reimbursement levels in the United States. Federal law requires all pharmaceutical manufacturers to rebate a percentage of their revenue arising from Medicaid-reimbursed drug sales to individual states. For Abraxane® and any other proprietary products that are marketed and sold under new drug applications, manufacturers are required to rebate the greater of (a) 15.1% of the average manufacturer price or (b) the difference between the average manufacturer price and the lowest manufacturer price for products sold during a specified period.
Both the federal and state governments in the United States and foreign governments continue to propose and pass new legislation, rules and regulations designed to contain or reduce the cost of health care. Existing regulations that affect the price of pharmaceutical and other medical products may also change before any of our products are approved for marketing. Cost control initiatives could decrease the price that we receive for any product we develop in the future. In addition, third-party payers are increasingly challenging the price and cost-effectiveness of medical products and services and litigation has been filed against a number of pharmaceutical companies in relation to these issues. Additionally, significant uncertainty exists as to the reimbursement status of newly approved injectable pharmaceutical products. Our products may not be considered cost effective or adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an adequate return on our investment.
State pharmaceutical marketing compliance and reporting requirements may expose us to regulatory and legal action by state governments or other government authorities.
In recent years, several states, including California, Vermont, Maine, Minnesota, Nevada and West Virginia, in addition to the District of Columbia, have enacted legislation requiring pharmaceutical companies to establish marketing compliance programs and file periodic reports on sales, marketing, pricing and other activities. Similar legislation is being considered in other states. Many of these requirements are new and uncertain, and available guidance is limited. We are continuing to assess our compliance with these state laws. A failure to act in full compliance with these laws, could result in enforcement action and fines and other penalties and adverse publicity, all of which could harm our business.
We may be required to defend lawsuits or pay damages for product liability claims.
Product liability is a major risk in testing and marketing biotechnology and pharmaceutical products. We may face substantial product liability exposure in human clinical trials and for products that we sell after regulatory approval. We maintain insurance coverage for product liability claims in the aggregate amount of $100 million, including primary and excess coverages, which we believe is reasonably adequate coverage. Product liability claims, regardless of their merits, could exceed policy limits, divert management’s attention and adversely affect our reputation and the demand for these products.
We depend heavily on the principal members of our management and research and development teams, the loss of whom could harm our business.
We depend heavily on the principal members of our management and research and development teams, including Dr. Patrick Soon-Shiong, Executive Chairman, and Bruce J. Wendel, Chief Executive Officer. Each of the members of the executive management team is employed “at will.” The loss of the services of any member of the executive management team may significantly delay or prevent the achievement of product development or business objectives.
To be successful, we must attract, retain and motivate key employees, and the inability to do so could seriously harm our business and operations.
To be successful, we must attract, retain and motivate executives and other key employees. We face competition for qualified scientific, technical and other personnel, which may adversely affect our ability to attract and retain key personnel. We also must continue to attract and motivate employees and keep them focused on our strategies and goals.
29
Table of Contents
If we are unable to integrate successfully potential future acquisitions, our business may be harmed.
As part of our business strategy and growth plan, we may acquire businesses, technologies or products that we believe will complement our business. The process of integrating an acquired business, technology or product may result in unforeseen operating difficulties and expenditures and may require significant management attention that would otherwise be available for ongoing development of our existing business. In addition, we may not be able to maintain the levels of operating efficiency that any acquired company achieved or might have achieved separately. Successful integration of the companies that we may acquire will depend upon our ability to, among other things, eliminate redundancies and excess costs. As a result of difficulties associated with combining operations, we may not be able to achieve cost savings and other benefits that the company might hope to achieve with acquisitions. Future acquisitions could result in potentially dilutive issuances of equity securities, the incurrence of debt and contingent liabilities or have an undesirable impact on our financial statements.
We are subject to risks associated with international operations, which could harm both our domestic and international operations.
As part of our business strategy and growth plan, we continue to evaluate international expansion opportunities as we obtain regulatory approvals to market our Abraxane® and other product candidates in foreign countries, including countries in the European Union and Asia. Expansion of our international operations could impose substantial burdens on our resources, divert management’s attention from domestic operations and otherwise harm our business. In addition, international operations are subject to risks, including:
| • | regulatory requirements of differing nations; |
| • | inadequate protection of intellectual property; |
| • | difficulties and costs associated with complying with a wide variety of complex domestic and foreign laws and treaties; |
| • | legal uncertainties regarding, and timing delays associated with, tariffs, export licenses and other trade barriers; and |
| • | currency fluctuations. |
Any of these or other factors could adversely affect our ability to compete in international markets and our operating results.
We may be required to indemnify APP and may not be able to collect on indemnification rights from APP.
Under the terms of the separation and distribution agreement, we have agreed to indemnify APP from and after the distribution with respect to all liabilities of Old Abraxis not related to its hospital-based products business and the use by APP of any trademarks or other source identifiers owned by us. Similarly, APP has agreed to indemnify us from and after the distribution with respect to all liabilities of Old Abraxis related to its hospital-based products business and the use by us of any trademarks or other source identifiers owned by APP.
Under the terms of the tax allocation agreement, we agreed to indemnify APP against all tax liabilities to the extent they relate to the proprietary products business, and APP agreed to indemnify us against all tax liabilities to the extent they relate to the hospital-based products business. The tax allocation agreement also generally allocates between us and APP any liability for taxes that may arise in connection with the distribution. In September 2008, APP entered into a merger agreement with Fresenius Kabi, a subsidiary of Fresenius SE. Pursuant to the merger agreement, Fresenius acquired all of the outstanding common stock of APP. Pursuant to the tax allocation agreement and the merger agreement, APP received a tax opinion, in form and substance reasonably acceptable to us, that the acquisition should not affect the qualification of the distribution under Section 355 and Section 368(a)(1)(D) of the Internal Revenue Code and the nonrecognition of gain to APP in the
30
Table of Contents
distribution. Under the terms of the tax allocation agreement, we are generally liable for, and are required to indemnify APP against, any tax liability arising as a result of the distribution failing to qualify for tax-free treatment unless, notwithstanding such tax opinion, such tax liability is imposed as a result of an acquisition of APP, including the acquisition of APP by Fresenius, or certain other specified acts of APP.
Under the terms of the manufacturing agreement, we have agreed to indemnify APP from any damages resulting from a third-party claim caused by or alleged to be caused by (i) our failure to perform our obligations under the manufacturing agreement; (ii) any product liability claim arising from the negligence, fraud or intentional misconduct of us or any of our affiliates or any product liability claim arising from our manufacturing obligations (or any failure or deficiency in our manufacturing obligations) under the manufacturing agreement; (iii) any claim that the manufacture, use or sale of Abraxane® or our pipeline products infringes a patent or any other proprietary right of a third party; or (iv) any recall, product liability claim or other third-party claim not arising from the gross negligence or bad faith of, or intentional misconduct or intentional breach of the manufacturing agreement by APP, by reason of the $100 million limitation of liability described below. We have also agreed to indemnify APP for liabilities that it becomes subject to as a result of its activities under the manufacturing agreement and for which it is not responsible under the terms of the manufacturing agreement. APP has agreed to indemnify us from any damages resulting from a third-party claim caused by or alleged to be caused by (i) APP’s gross negligence, bad faith, intentional misconduct or intentional failure to perform its obligations under the manufacturing agreement; or (ii) any product liability claim arising from the gross negligence or bad faith of, or intentional misconduct or intentional breach of the manufacturing agreement by APP. APP generally will not have any liability for monetary damages to us or third parties in connection with the manufacturing agreement for damages in excess of $100 million in the aggregate.
There are no time limits on when an indemnification claim must be brought and no other monetary limits on the amount of indemnification that may be provided. These indemnification obligations could be significant. Our ability to satisfy any of these indemnification obligations will depend upon the future financial strength of our company. We cannot determine whether we will have to indemnify APP for any substantial obligations. We also cannot assure you that, if APP becomes obligated to indemnify us for any substantial obligations, APP will have the ability to satisfy those obligations. Any indemnification payment by us, or any failure by APP to satisfy its indemnification obligations, could have a material adverse effect on our business.
We will be dependent upon APP to manufacture Abraxane® for a remaining term of two or three years, and the manufacture of pharmaceutical products is highly regulated.
In connection with the separation and distribution agreement, we entered into a manufacturing agreement with APP for the manufacture of Abraxane® and our pipeline products whereby APP agreed to undertake certain of the tasks necessary to manufacture Abraxane® and our pipeline products until December 31, 2011, with this agreement automatically extended by one year if either APP elects to exercise its option to extend the lease on our Melrose Park manufacturing facility or we elect to exercise our option to extend the lease on APP’s Grand Island manufacturing facility. Accordingly, we will be dependent upon APP to manufacture our products. The amount and timing of resources that APP devotes to the manufacture of our products is not within our direct control. Further, in the event of capacity constraints at the manufacturing facilities, the manufacturing agreement provides that the available capacity will be prorated between us and APP according to the parties’ then current use of manufacturing capacity at the relevant facilities. Any loss in manufacturing capacity pursuant to these proration provisions could be detrimental to our business and operating results. While the manufacturing agreement allows us to override these proration provisions, we may only do so by paying APP additional fees under the manufacturing agreement. If we are forced to pay APP additional fees to retain our capacity rights under the manufacturing agreement, it could be detrimental to our operating results.
The manufacture of pharmaceutical products is highly exacting and complex, due in part to strict regulatory requirements and standards that govern both the manufacture of a particular product and the manufacture of these types of products in general. Problems may arise during their manufacture due to a variety of reasons, including
31
Table of Contents
equipment malfunction, failure to follow specific protocols and procedures and environmental factors. If problems arise during the production of a batch of product, that batch of product may have to be discarded. This could, among other things, lead to loss of the cost of raw materials and components used and lost revenue. If such problems are not discovered before the product is released to the market, recall costs may also be incurred. Under the terms of the manufacturing agreement, we have the final responsibility for release of the products manufactured pursuant to the manufacturing agreement and will bear all expenses in connection with any recall of products, unless the recall is a result of APP’s gross negligence, bad faith, intentional misconduct or intentional breach, in which case APP would bear all costs and expenses related to such product recall, subject to the $100 million limit on liability under the manufacturing agreement. To the extent APP encounters difficulties or problems with respect to the manufacture of our pharmaceutical products, including Abraxane®, this may be detrimental to our business, operating results and reputation.
Risks Related To An Investment In Our Common Stock
Our executive chairman and entities affiliated with him collectively own a significant percentage of our common stock and could exercise significant influence over matters requiring stockholder approval, regardless of the wishes of other stockholders.
Our executive chairman and entities affiliated with him collectively own approximately 82% of our outstanding common stock. Accordingly, they have the ability to significantly influence all matters requiring stockholder approval, including the election and removal of directors and approval of significant corporate transactions such as mergers, consolidations and sales of assets. This concentration of ownership could have the effect of delaying, deferring or preventing a change in control or impeding a merger or consolidation, takeover or other business combination, which could cause the market price of our common stock to fall or prevent our stockholders from receiving a premium in such a transaction. This significant concentration of stock ownership may adversely affect the market for and trading price of our common stock if investors perceive that conflicts of interest may exist or arise.
Substantial sales of our common stock could depress the market price of our common stock.
All of the shares of our common stock issued in connection with the separation, other than shares issued to our affiliates, are eligible for immediate resale in the public market. Although shares issued to our affiliates are not immediately freely tradable, we have granted registration rights to the former ABI shareholders, including our executive chairman. Under the registration rights agreement, the former ABI shareholders will have the right to require us to register all or a portion of the shares of our common stock they received in connection with the separation. In addition, the former ABI shareholders may require us to include their shares in future registration statements that we file and our executive chairman may require us to register shares for resale on a Form S-3 registration statement. 2,000,000 shares of our common stock held by our executive chairman have been registered on the registration statement on Form S-3 that was declared effective by the SEC on July 2, 2009. Upon registration, the registered shares generally will be freely tradeable in the public market without restriction. However, in connection with any underwritten offering, the holders of registrable securities will agree to lock up any other shares for up to 90 days and will agree to a limit on the maximum number of shares that can be registered for the account of the holders of registrable securities under so-called “shelf” registration statements. Substantial sales of our shares, or the perception that such sales might occur, could depress the market price for our shares. Our executive chairman and entities affiliated with him beneficially own over 82% of our outstanding common stock.
Our stock price may be volatile in response to market and other factors.
The market price for our common stock may be volatile and subject to price and volume fluctuations in response to market and other factors, including the following, some of which are beyond our control:
| • | variations in our quarterly operating results from the expectations of securities analysts or investors; |
32
Table of Contents
| • | revisions in securities analysts’ estimates; |
| • | announcements of technological innovation or new products by us or our competitors; |
| • | announcements by us or our competitors of significant acquisition, strategic partnerships, joint ventures or capital commitments; |
| • | general technological, market or economic trends; |
| • | investor perception of our industry or our prospects; |
| • | insider selling or buying; |
| • | investors entering into short sale contracts; |
| • | regulatory developments affecting our industry; and |
| • | additions or departures of key personnel. |
We are not subject to the provisions of Section 203 of the Delaware General Corporation Law, which could negatively affect your investment.
We elected in our certificate of incorporation to not be subject to the provisions of Section 203 of the Delaware General Corporation Law. In general, Section 203 prohibits a publicly held Delaware corporation from engaging in a “business combination” with an “interested stockholder” for a period of three years after the date of the transaction in which the person became an interested stockholder, unless the business combination is approved in a prescribed manner. A “business combination” includes a merger, asset sale or other transaction resulting in a financial benefit to the interested stockholder. An “interested stockholder” is a person who, together with affiliates and associates, owns (or, in certain cases, within three years prior, did own) 15% or more of the corporation’s voting stock. Our decision not to be subject to Section 203 will allow, for example, our executive chairman (who with members of his immediate family and entities affiliated with him beneficially own approximately 82% of our common stock) to transfer shares in excess of 15% of our voting stock to a third-party free of the restrictions imposed by Section 203. This may make us more vulnerable to takeovers that are completed without the approval of our board of directors and/or without giving us the ability to prohibit or delay such takeovers as effectively. These provisions could also limit the price that investors would be willing to pay in the future for shares of our common stock.
Item 1B. UNRESOLVED STAFF COMMENTS
None.
33
Table of Contents
We operate various facilities in the United States, Puerto Rico and Canada, which have an aggregate size of approximately 1.2 million square feet. We believe that our existing facilities are sufficient to meet our expected needs. The location and uses of our principal facilities as of December 31, 2009 were as follows:
| Location |
Use |
Own/ Lease |
Lease Expiration | Approximate Square Feet | ||||
| Los Angeles, CA |
Principal executive office | Lease | November 2017 | 60,900 | ||||
| Marina Del Rey, CA |
Research & regulatory affairs | Lease | August 2011 | 50,700 | ||||
| Bridgewater, NJ |
Administration | Lease | September 2011 | 33,000 | ||||
| Elk Grove Village, Illinois |
Warehouse | Own | — | 90,000 | ||||
| Melrose Park, Illinois(1) |
Manufacturing | Own | — | 122,000 | ||||
| Melrose Park, Illinois(2) |
Research and development and warehouse | Own | — | 147,000 | ||||
| Grand Island, New York(3) |
Manufacturing | Lease | December 2011 | 5,700 | ||||
| Phoenix, Arizona |
Manufacturing | Own | — | 200,000 | ||||
| Barceloneta, Puerto Rico(4) |
Manufacturing | Own | — | 90,000 | ||||
| Durham, North Carolina |
Clinical trial management | Lease | April 2016 | 36,000 | ||||
| Auburn, California |
Research and development | Lease | April 2010 | 12,800 | ||||
| Ontario, Canada |
Administration | Lease | October 2013 | 13,600 | ||||
| George, South Africa(5) |
Research and development | Lease | November 2011 | 6,500 | ||||
| Oelwein, Iowa |
Manufacturing | Own | — | 48,500 | ||||
| Costa Mesa, California |
Research and development | Own | — | 180,000 | ||||
| Elk Grove Village, Illinois |
Manufacturing | Own | — | 60,100 | ||||
| 1,156,800 | ||||||||
| (1) | For the length of the applicable lease, the manufacturing facility in Melrose Park has been leased to APP, and APP has agreed to provide certain contract manufacturing services to us in accordance with the terms of the manufacturing agreement. |
| (2) | We have leased approximately 71,000 square feet of our warehouse space located at this Melrose Park facility to APP for a term initially expiring in December 2011. We have also leased approximately 48,000 square feet of our research and development space located at this Melrose Park facility to APP for a term expiring in December 2010. |
| (3) | We have leased from APP a portion of APP’s Grand Island, New York manufacturing facility to enable us to perform our responsibilities under the manufacturing agreement for its term. The initial term of the manufacturing agreement will expire on December 31, 2011, but the term of the manufacturing agreement will be automatically extended for one year if either APP exercises its option to extend the lease for our Melrose Park manufacturing facility for an additional year or we elect to exercise our option to extend the lease for APP’s Grand Island manufacturing facility for an additional year. |
| (4) | APP’s Puerto Rico facility was divided into two separate facilities, and we took ownership of one part of the facility. The portion of the Puerto Rico facility owned by us following the separation is the 90,000 square foot active pharmaceutical ingredients manufacturing plant currently leased to Pfizer for a term expiring in February 2012. Pfizer vacated the premises in December 2009 and did not incur any penalty for the early termination of the lease. |
| (5) | Through our wholly-owned subsidiary, Shimoda Biotech (Pty) Limited, we also lease approximately 250 square feet of office space in Port Elizabeth, South Africa under a lease expiring in July 2010. |
34
Table of Contents
On July 19, 2006, Élan Pharmaceutical Int’l Ltd. filed a lawsuit against Old Abraxis in the U.S. District Court for the District of Delaware alleging that Old Abraxis willfully infringed upon two of their patents by asserting that Abraxane® uses technology protected by Élan-owned patents. Élan sought unspecified damages and an injunction. In August 2006, Old Abraxis filed a response to Élan’s complaint contending that it did not infringe on the Élan patents and that the Élan patents are invalid. On June 13, 2008, the jury ruled that we infringed upon one of Élan’s patents, which runs until 2011, and awarded Élan $55.2 million in damages for sales of Abraxane® through the judgment date. We have filed various post-trial motions and are appealing the jury ruling. We assumed any liability of Old Abraxis relating to this litigation in connection with the separation.
We are from time to time subject to claims and litigation arising in the ordinary course of business. We intend to defend vigorously any such litigation that may arise under all defenses that would be available to us. In the opinion of management, the ultimate outcome of proceedings of which management is aware, even if adverse to us, would not have a material adverse effect on our consolidated financial position or results of operations.
Item 4. SUBMISSION OF MATTERS TO A VOTE OF SECURITY HOLDERS
Our 2009 annual meeting of stockholders was held on December 10, 2009.
| (a) | Our stockholders voted as follows with respect to elect six directors to serve until our next annual meeting of stockholders or until their successors are duly elected and qualified: |
| DIRECTORS |
FOR | Authority Withheld | ||
| Patrick Soon-Shiong, M.D. |
35,502,994 | 3,872,305 | ||
| Leon O. Moulder, Jr. (1) . |
39,162,384 | 212,915 | ||
| Kirk K. Calhoun |
35,887,674 | 3,487,625 | ||
| Stephen D. Nimer, M.D. |
35,522,029 | 3,853,270 | ||
| Leonard Shapiro |
35,887,524 | 3,487,775 | ||
| David S. Chen, Ph.D. |
35,522,833 | 3,852,466 |
| (1) | Leon Moulder resigned on January 27, 2010. |
| (b) | Our stockholders voted as follows with respect to a proposal to ratify the appointment of Ernst & Young LLP as our independent registered public accounting firm for the fiscal year ended December 31, 2009. |
| FOR |
AGAINST |
ABSTENTIONS |
BROKER NON-VOTES | |||
| 39,369,502 |
5,070 | 727 | -0- |
35
Table of Contents
Item 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market for Common Stock
Our common stock is listed and traded on The NASDAQ Global Market under the symbol “ABII.” Regular-way trading of our common stock commenced on November 14, 2007, the day after our separation from Old Abraxis. The following table sets forth the prices for our common stock as reported by NASDAQ for fiscal years 2009 and 2008:
| 2009 Price Per Share |
2008 Price Per Share | |||||||||||
| For the quarter ended: | High | Low | High | Low | ||||||||
| March 31 |
$ | 73.98 | $ | 42.40 | $ | 69.00 | $ | 53.00 | ||||
| June 30 |
$ | 57.60 | $ | 35.25 | $ | 69.91 | $ | 58.33 | ||||
| September 30 |
$ | 39.90 | $ | 24.52 | $ | 78.95 | $ | 59.03 | ||||
| December 31 |
$ | 43.00 | $ | 31.20 | $ | 74.50 | $ | 46.28 | ||||
As of February 26, 2010, there were approximately 68 stockholders of record of our common stock and the closing price of our stock as reported by NASDAQ on that date was $32.35.
Dividend Policy
We have never declared or paid a cash dividend and we have no current intention of paying cash dividends. We currently anticipate that we will retain future earnings to support our growth strategy. We do not anticipate paying regular cash dividends on our common stock in the foreseeable future
Issuer Purchases of Equity Securities
We did not repurchase any of our equity securities during the fourth quarter of 2009.
Securities Authorized for Issuance under Equity Compensation Plans
Please see Part III, Item 12 of this Annual Report on Form 10-K for disclosure relating to our equity compensation plans. Such information is incorporated by reference from our Proxy Statement for our 2010 Annual Meeting of Stockholders.
36
Table of Contents
Performance Graph Notwithstanding anything to the contrary set forth in any of our previous filings under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, that might incorporate future filings, including this Annual Report of Form 10-K, in whole or in part, the following graph shall not be deemed to be incorporated by reference into any such filings.
The following graph compares the percentage change in the cumulative total stockholder return on our common stock as compared with the NASDAQ Pharmaceutical Index and the NASDAQ stock market (U.S. Index). The comparison assumes an investment of $100 on November 14, 2007 (the date on which our common stock first began to be publicly traded) in our common stock and in each of the foregoing indices, and assumes reinvestment of dividends, of which we paid none. The stock performance shown on the graph below is not necessarily indicative of future price performance.
Abraxis BioScience, Inc.
NASDAQ Pharmaceutical Index
NASDAQ Stock Market (U.S. Index)
37
Table of Contents
Item 6. SELECTED FINANCIAL DATA
We derived the following selected consolidated and combined financial data for the five years ended December 31, 2009 from our audited consolidated and combined financial statements. You should read the data in conjunction with “Item 7: Management’s Discussion and Analysis of Financial Condition and Results of Operations,” our consolidated and combined financial statements, related notes and other financial information included herein. Historical results of operations and financial position are not necessarily indications of the results that may be expected in future periods.
| Year ended December 31, | ||||||||||||||||||||
| 2009 | 2008 | 2007 | 2006 | 2005 | ||||||||||||||||
| (in thousands, except per share data) | ||||||||||||||||||||
| Consolidated and Combined Statement of Operations Data: |
||||||||||||||||||||
| Abraxane revenue |
$ | 314,545 | $ | 335,631 | $ | 324,692 | $ | 174,906 | $ | 133,731 | ||||||||||
| Other revenue |
44,505 | 9,678 | 8,994 | 7,381 | 1,944 | |||||||||||||||
| Net revenue |
359,050 | 345,309 | 333,686 | 182,287 | 135,675 | |||||||||||||||
| Cost of sales |
63,665 | 39,068 | 34,450 | 21,183 | 24,066 | |||||||||||||||
| Gross profit |
295,385 | 306,241 | 299,236 | 161,104 | 111,609 | |||||||||||||||
| Operating expenses: |
||||||||||||||||||||
| Research and development |
154,615 | 98,976 | 85,424 | 63,073 | 50,121 | |||||||||||||||
| Selling, general and administrative |
200,734 | 221,418 | 233,324 | 119,462 | 69,239 | |||||||||||||||
| Reacquisition costs |
— | 158,909 | — | — | — | |||||||||||||||
| Litigation costs |
— | 57,635 | — | — | — | |||||||||||||||
| Acquired in-process research and development |
— | 13,900 | — | 83,447 | — | |||||||||||||||
| Impairment charge |
13,999 | 9,214 | — | — | — | |||||||||||||||
| Amortization of intangible assets |
39,782 | 39,429 | 38,615 | 27,349 | — | |||||||||||||||
| Merger related costs |
— | — | — | 16,722 | — | |||||||||||||||
| Total operating expenses |
409,130 | 599,481 | 357,363 | 310,053 | 119,360 | |||||||||||||||
| Loss from operations |
(113,745 | ) | (293,240 | ) | (58,127 | ) | (148,949 | ) | (7,751 | ) | ||||||||||
| Equity in net income of unconsolidated entities |
2,090 | 908 | 3,771 | 2,776 | 1,843 | |||||||||||||||
| Interest income |
3,052 | 18,809 | 4,990 | 399 | 287 | |||||||||||||||
| Other income (expense) |
1,255 | (5,186 | ) | (190 | ) | (4,741 | ) | (6,563 | ) | |||||||||||
| Loss before income taxes |
(107,348 | ) | (278,709 | ) | (49,556 | ) | (150,515 | ) | (12,184 | ) | ||||||||||
| (Benefit) provision for income taxes |
(2,580 | ) | (1,938 | ) | (7,952 | ) | (25,964 | ) | 478 | |||||||||||
| Net loss |
$ | (104,768 | ) | $ | (276,771 | ) | $ | (41,604 | ) | $ | (124,551 | ) | $ | (12,662 | ) | |||||
| Net loss attributable to noncontrolling interest |
$ | (1,652 | ) | $ | — | $ | — | $ | — | $ | — | |||||||||
| Net loss attributable to common shareholders |
$ | (103,116 | ) | $ | (276,771 | ) | $ | (41,604 | ) | $ | (124,551 | ) | $ | (12,662 | ) | |||||
| Basic and diluted net loss per common share |
$ | (2.57 | ) | $ | (6.91 | ) | $ | (1.04 | ) | $ | (3.11 | ) | $ | (0.32 | ) | |||||
| Weighted-average common shares outstanding(1): |
||||||||||||||||||||
| Basic |
40,100 | 40,032 | 39,991 | 39,990 | 39,990 | |||||||||||||||
| Diluted |
40,100 | 40,032 | 39,991 | 39,990 | 39,990 | |||||||||||||||
| Consolidated balance sheet data: |
||||||||||||||||||||
| Working capital |
$ | 192,747 | $ | 386,148 | $ | 735,181 | $ | 25,093 | $ | 55,232 | ||||||||||
| Total assets |
1,068,380 | 1,438,584 | 1,502,255 | 764,783 | 179,080 | |||||||||||||||
| Total debt |
— | — | — | — | 190,000 | |||||||||||||||
| Total equity (deficit) |
846,265 | 929,472 | 1,197,387 | 459,021 | (65,644 | ) | ||||||||||||||
| Other data: |
||||||||||||||||||||
| Cash flow (used in) provided by operating activities |
$ | 4,488 | $ | (315,468 | ) | $ | (2,893 | ) | $ | 170,870 | $ | (22,272 | ) | |||||||
| Purchases of property plant and equipment |
(94,473 | ) | (43,729 | ) | (40,581 | ) | (64,431 | ) | (17,212 | ) | ||||||||||
| Cash flow provided by (used in) financing activities |
(561 | ) | 2,360 | 752,082 | (94,398 | ) | 40,728 | |||||||||||||
| (1) | As of the completion of the separation on November 13, 2007, we had 40.0 million common shares outstanding. The same number of shares is being used for both diluted earnings per share and basic earnings per share for all periods prior to the separation date. All potentially dilutive employee stock awards were excluded from the computation of diluted loss per common share for all periods as the effect on net loss per share was anti-dilutive. |
38
Table of Contents
Item 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
OVERVIEW
The following management’s discussion and analysis of financial condition and results of operations, or MD&A is intended to assist the reader in understanding our company. The MD&A is provided as a supplement to, and should be read in conjunction with the other sections of this Annual Report on Form 10-K, including “Item 1: Business”; “Item 1A: Risk Factors”, “Item 6: Selected Financial Data”; and “Item 8: Financial Statements and Supplemental Data.”
Background
We are one of the few fully integrated global biotechnology companies, with a breakthrough marketed product (Abraxane®), global ownership of our proprietary technology platform and clinical pipeline, and dedicated nanoparticle manufacturing capabilities for worldwide supply integrated with seasoned in-house capabilities, including discovery, clinical drug development, regulatory and sales and marketing.
We are dedicated to the discovery, development and delivery of next-generation therapeutics and core technologies that offer patients safer and more effective treatments for cancer and other critical illnesses. Our portfolio includes the world’s first and only protein-based nanoparticle chemotherapeutic compound (Abraxane®), which is based on our proprietary tumor targeting technology known as the nab® technology platform. This nab® technology platform is the first to exploit the tumor’s biology against itself, taking advantage of an albumin-specific, receptor-mediated transport system and allowing the delivery of a drug from the vascular space across the blood vessel wall to the underlying tumor tissue. Abraxane® is the first clinical and commercial validation of our nab® technology platform. From the discovery and research phase to development and commercialization, we are committed to rapidly enhancing our product pipeline and accelerating the delivery of breakthrough therapies that will transform the lives of patients who need them.
We own the worldwide rights to Abraxane®, a next generation, nanometer-sized, solvent-free taxane that was approved by the U.S. Food and Drug Administration, or the FDA, in January 2005 for its initial indication in the treatment of metastatic breast cancer and launched in February 2005. We believe the successful launch of Abraxane® validates our nab® tumor targeting technology, a novel biologically interactive (receptor-mediated) system to deliver chemotherapeutic agents.
Proposed 2010 Spin-Off
In January 2009, we announced that our board of directors approved a plan to spin-off a newly-formed subsidiary Abraxis Health, Inc. as a new independent, stand-alone company holding our drug discovery, manufacturing and development business. We currently anticipate the spin-off will be completed in 2010. If the spin-off occurs, our stockholders would own (i) shares of Abraxis Health and (ii) shares of our common stock, and we would continue to operate our existing business, excluding the drug discovery, manufacturing and development business to be held by Abraxis Health. In connection with the proposed spin-off, Abraxis Health would enter into several agreements with us related to, among other things, manufacturing, transition services, licensing, tax allocations and a number of ongoing commercial relationships.
The proposed spin-off is subject to, among other things, final approval by our board of directors and the effectiveness of the registration statement registering the common stock of Abraxis Health to be distributed to our stockholders in connection with the spin-off. Our board of directors, in its sole discretion and for any reason or no reason at all, may amend, modify or abandon the proposed spin-off without liability at any time prior to the time the spin-off is completed. Approval by our stockholders is not required as a condition to the consummation of the proposed spin-off. In connection with the proposed spin-off, Abraxis Health filed a registration statement on Form 10 with the SEC on August 4, 2009, that was withdrawn on August 12, 2009. Abraxis Health plans to
39
Table of Contents
file a new Form 10 registration statement, and stockholders are urged to read it, including any amendments or supplements thereto, carefully when available because it will contain important information about the proposed spin-off.
2007 Separation
On November 13, 2007, Old Abraxis was separated into two independent publicly-traded companies: one holding the former Abraxis Pharmaceutical Products business, or the hospital-based business; and our company, which holds the former Abraxis Oncology and Abraxis Research businesses, or the proprietary business, which we refer to as the “2007 Separation”. In connection with the separation, we changed our name from New Abraxis, Inc. to Abraxis BioScience, Inc. (and the hospital-based business is operated under the name APP Pharmaceuticals).
In connection with the separation, stockholders of Old Abraxis as of November 13, 2007 received one share of our company for every four shares of Old Abraxis held as of that date. In addition, in connection with the separation, we entered into a separation and distribution agreement that provides for, among other things, the principal corporate transactions required to effect the separation and other specified terms governing our relationship with APP after the spin-off. We also entered into various agreements with APP, including: (i) a transition services agreement pursuant to which we and APP agreed to continue to provide each other with various services on an interim, transitional basis, for periods up to 24 months depending on the particular service; (ii) a manufacturing agreement whereby we and APP agreed to manufacture Abraxane® and certain other products and to provide other manufacturing-related services for a period of four or five years; (iii) an employees matters agreement providing for each company’s respective obligations to employees and former employees who are or were associated with their respective businesses, and for other employment and employee benefit matters; (iv) various real estate leases; and (v) a tax allocation agreement. Also, in connection with the separation, APP contributed $700 million in cash to us.
40
Table of Contents
RESULTS OF OPERATIONS
Overview
The following table sets forth the results of our operations for each of the three years ended December 31, 2009, and forms the basis for the following discussion of our operating activities:
| Change Favorable (Unfavorable) | ||||||||||||||||||||||||||
| Year Ended December 31, | 2009 vs. 2008 | 2008 vs. 2007 | ||||||||||||||||||||||||
| 2009 | 2008 | 2007 | $ | % | $ | % | ||||||||||||||||||||
| (in thousands, except per share data) | ||||||||||||||||||||||||||
| Consolidated and combined statements of operations data: |
||||||||||||||||||||||||||
| Revenue: |
||||||||||||||||||||||||||
| Abraxane |
$ | 314,545 | $ | 335,631 | $ | 324,692 | $ | (21,086 | ) | (6 | )% | $ | 10,939 | 3 | % | |||||||||||
| Other |
44,505 | 9,678 | 8,994 | 34,827 | 360 | % | 684 | 8 | % | |||||||||||||||||
| Net revenue |
359,050 | 345,309 | 333,686 | 13,741 | 4 | % | 11,623 | 3 | % | |||||||||||||||||
| Cost of sales |
63,665 | 39,068 | 34,450 | (24,597 | ) | (63 | )% | (4,618 | ) | (13 | )% | |||||||||||||||
| Gross profit |
295,385 | 306,241 | 299,236 | (10,856 | ) | (4 | )% | 7,005 | 2 | % | ||||||||||||||||
| Operating expenses: |
||||||||||||||||||||||||||
| Research and development |
154,615 | 98,976 | 85,424 | (55,639 | ) | (56 | )% | (13,552 | ) | (16 | )% | |||||||||||||||
| Selling, general and administrative |
200,734 | 221,418 | 233,324 | 20,684 | 9 | % | 11,906 | 5 | % | |||||||||||||||||
| Reacquisition costs |
— | 158,909 | — | 158,909 | 100 | % | (158,909 | ) | — | |||||||||||||||||
| Litigation costs |
— | 57,635 | — | 57,635 | 100 | % | (57,635 | ) | — | |||||||||||||||||
| Acquired in-process research and development |
— | 13,900 | — | 13,900 | 100 | % | (13,900 | ) | — | |||||||||||||||||
| Impairment charge |
13,999 | 9,214 | — | (4,785 | ) | (52 | )% | (9,214 | ) | — | ||||||||||||||||
| Amortization of intangible assets |
39,782 | 39,429 | 38,615 | (353 | ) | (1 | )% | (814 | ) | (2 | )% | |||||||||||||||
| Total operating expense |
409,130 | 599,481 | 357,363 | 190,351 | 32 | % | (242,118 | ) | (68 | )% | ||||||||||||||||
| Loss from operations |
(113,745 | ) | (293,240 | ) | (58,127 | ) | 179,495 | 61 | % | (235,113 | ) | (404 | )% | |||||||||||||
| Equity in net income of unconsolidated entities |
2,090 | 908 | 3,771 | 1,182 | 130 | % | (2,863 | ) | (76 | )% | ||||||||||||||||
| Interest income |
3,052 | 18,809 | 4,990 | (15,757 | ) | (84 | )% | 13,819 | 277 | % | ||||||||||||||||
| Other income (expense) |
1,255 | (5,186 | ) | (190 | ) | 6,441 | 124 | % | (4,996 | ) | (2629 | )% | ||||||||||||||
| Loss before income taxes |
(107,348 | ) | (278,709 | ) | (49,556 | ) | 171,361 | 61 | % | (229,153 | ) | (462 | )% | |||||||||||||
| Benefit for income taxes |
(2,580 | ) | (1,938 | ) | (7,952 | ) | 642 | 33 | % | (6,014 | ) | (76 | )% | |||||||||||||
| Net loss |
$ | (104,768 | ) | $ | (276,771 | ) | $ | (41,604 | ) | $ | 172,003 | 62 | % | $ | (235,167 | ) | (565 | )% | ||||||||
| Net loss attributable to noncontrolling interest |
$ | (1,652 | ) | $ | — | $ | — | $ | 1,652 | — | $ | — | — | |||||||||||||
| Net loss attributable to common shareholders |
$ | (103,116 | ) | $ | (276,771 | ) | $ | (41,604 | ) | $ | 173,655 | 63 | % | $ | (235,167 | ) | (565 | )% | ||||||||
| Basic and diluted loss per common share |
$ | (2.57 | ) | $ | (6.91 | ) | $ | (1.04 | ) | |||||||||||||||||
| Weighted-average common shares outstanding: |
||||||||||||||||||||||||||
| Basic |
40,100 | 40,032 | 39,991 | |||||||||||||||||||||||
| Diluted |
40,100 | 40,032 | 39,991 | |||||||||||||||||||||||
41
Table of Contents
Operating Results
Net Revenue
Net revenue for the year ended December 31, 2009 increased $13.7 million, or 4.0%, to $359.0 million as compared to $345.3 million in 2008. Included in net revenue for the year ended December 2008 was $36.4 million of recognized deferred revenue associated with the co-promotion agreement with AstraZeneca (“Co-Promotion Agreement”), which was terminated effective January 2009.
Abraxane® revenue for the year ended December 31, 2009 decreased $21.1 million, or 6.3%, to $314.5 million as compared to $335.6 million in 2008, primarily due to the elimination of the recognized deferred revenue related to the Co-Promotion Agreement. Excluding the recognition of the deferred revenue associated with the Co-Promotion Agreement, Abraxane® revenue for the year ended December 31, 2009 increased $15.2 million to $314.5 million compared to $299.3 million for the same period in 2008. The increase in Abraxane® revenue was driven by incremental revenue from the global expansion into China, Australia and the European Union and higher average net selling price from sales within the United States. Abraxane® revenue represented 87.6% of net revenue for the year ended December 31, 2009 as compared to 97.2% in 2008.
Other revenue increased $34.8 million to $44.5 million as compared to $9.7 million for the same period in 2008 due primarily to raw material product sales of $32.5 million, which began in 2009. Revenue from contract manufacturing contributed an additional increase of $2.3 million.
Net revenue for the year ended December 31, 2008 increased $11.6 million, or 3.5%, to $345.3 million as compared to $333.7 million in 2007. Included in net revenue for the year ended December 31, 2008 was $39.5 million of recognized deferred revenue relating to the Co-Promotion Agreement and the license agreements with Taiho and Green Cross as compared to $39.4 million for the year ended December 31, 2007.
Abraxane® revenue for the year ended December 31, 2008 increased $10.9 million to $335.6 million as compared to $324.7 million in 2007. The increase was primarily the result of a price increase and increased unit sales of Abraxane®. Excluding the recognition of deferred revenue relating to the Co-Promotion Agreement, total Abraxane® revenue for the year ended December 31, 2008 increased $10.9 million, or 3.8%, to $299.3 million compared to $288.3 from the prior year. Abraxane® revenue represented 97.2% of net revenue for the year ended December 31, 2008 as compared to 97.3% in 2007.
Other revenue increased to $9.7 million in 2008 from $9.0 million in 2007, primarily due to higher management fees revenue from our Melrose Park manufacturing and R&D facilities, and service revenue from our Phoenix plant, partially offset by lower licensing revenues due to a milestone payment received in 2007.
Gross Profit
Gross profit for the year ended December 31, 2009 was $295.4 million, or 82.3% of net revenue, as compared to $306.2 million, or 88.7% of net revenue, for the same period in 2008. The decrease in gross margin as a percentage of net revenue was due primarily to the elimination of deferred revenue recognized from the Co-Promotion Agreement, higher volume of sales of raw material products, a voluntary recall initiated on certain lots of Abraxane® and increased sales of Abraxane® outside the United States.
Gross profit for the year ended December 31, 2008 was $306.2 million as compared to $299.2 million in 2007. Included in gross profit was the recognition of $36.4 million in each of 2008 and 2007 of deferred revenue under the Co-Promotion Agreement. Excluding the recognized deferred revenue from the Co-Promotion Agreement, gross profit as a percentage of net revenue for the year ended December 31, 2008 was 87.3% versus 88.4% in 2007. The decrease in gross margin percentage over the same period of 2007 was primarily due to a milestone payment received in 2007, increased sales of lower margin products associated with our contract manufacturing in Phoenix, offset partially by a pricing increase of Abraxane® in 2008.
42
Table of Contents
Research and Development
Research and development expense for the year ended December 31, 2009 increased $55.6 million, or 56.2%, to $154.6 million as compared to $99.0 million in 2008. Spending on Phase III clinical trials for non-small cell lung cancer, pancreatic cancer and melanoma accounted for a majority of the increase. The remainder of the increase was attributable to investments in early stage discovery and other research and development projects.
Research and development expense for the year ended December 31, 2008 increased $13.6 million, or 15.9%, to $99.0 million as compared to $85.4 million in 2007. The increase was primarily due to increased investments in research and development companies, and increased spending in clinical and investigator sponsored studies, offset partially by reductions in stock compensation expense.
Our research and development expenses are comprised primarily of costs related to our drug discovery efforts, drug development efforts, clinical trials, and other research and development activities. We do not track total research and development expenses separately for each of our product development programs. Drug discovery and drug development expenses mostly include personnel expense, lab supplies, non-refundable upfront payments, consulting fees, occupancy costs and other third-party costs.
The scope and magnitude of our future research and development expenses are difficult to predict at this time given the number of studies that will need to be conducted for any of our potential product candidates. In general, biotechnology product development involves a series of steps. The process begins with discovery and preclinical research leading up to the submission of an Investigational New Drug (IND) application to the U.S. Food and Drug Administration (FDA), which allows for the initiation of the clinical evaluation of a potential drug candidate in humans. Clinical trials are typically comprised of three phases of study: Phase 1, Phase 2 and Phase 3. Generally, the majority of a drug candidate’s total development costs are incurred during Phase 3, which consists of trials that are typically both the longest and largest conducted during the drug development process. The length of time to complete clinical trials may take as many as seven to ten years. However, the length of time may vary substantially according to factors relating to the particular clinical trial, such as the type and intended use of the drug candidate, the clinical trial design and the ability to enroll suitable patients.
The estimation of completion dates or costs to complete our research and development projects would be highly speculative and subjective due to the numerous risks and uncertainties associated with developing biotechnology products. These risks could include (i) significant changing government regulation, (ii) the uncertainty of future preclinical and clinical study results, (iii) uncertainties associated with developing biotechnology products and (iv) uncertainties associated with process development and manufacturing. Our research and development expenses can vary from period to period given the rate at which clinical trial materials are acquired and utilized and the rate at which we are successful in enrolling suitable patients. The following table summarizes our research and development expenses for the years ended December 31, 2009, 2008 and 2007:
| Year Ended December 31, | |||||||||
| 2009 | 2008 | 2007 | |||||||
| (in thousands) | |||||||||
| Discovery |
$ | 20,216 | $ | 14,675 | $ | 11,918 | |||
| Drug development |
38,567 | 29,808 | 24,044 | ||||||
| Clinical trials |
65,291 | 28,795 | 24,093 | ||||||
| Other research and development |
30,541 | 25,698 | 25,369 | ||||||
| $ | 154,615 | $ | 98,976 | $ | 85,424 | ||||
43
Table of Contents
Selling, General and Administrative
Selling, general and administrative expense for the year ended December 31, 2009 decreased $20.7 million to $200.7 million, or 55.9% of net revenue, from $221.4 million, or 64.1% of net revenue in 2008. The reacquisition of Abraxane® marketing rights in the United States yielded savings due to the elimination of costs associated with the Co-Promotion Agreement. The decrease was partially offset by increased investments in the global expansion of Abraxane® primarily in China and the European Union, and increased spending on U.S. sales and marketing.
Selling, general and administrative expense for year ended December 31, 2008 decreased $11.9 million to $221.4 million, or 64.1% of net revenue, from $233.3 million, or 69.9% of net revenue in 2007. The decrease was primarily the result of a legal charge taken in 2007, and overall lower domestic expenditures of marketing expenses including costs associated with the sharing provisions of the Co-Promotion Agreement. Partially offsetting the decrease in selling, general and administrative expense was increased international spending with the launch of Abraxane®, additional administration costs associated with the 2007 Separation from APP Pharmaceutical, and expenses related to the preparation for the spin-off of Abraxis Health.
Reacquisition costs
In November 2008, we entered into an agreement under which we would, subject to the terms and conditions of the agreement, reacquire exclusive rights to market Abraxane® in the United States. In accordance with this agreement, the Co-Promotion Agreement ended effective January 2009 and we agreed to pay AstraZeneca a $268.0 million fee by March 31, 2009. For the year ended December 31, 2008, we recorded a charge of $158.9 million representing the net amount of previously deferred revenue associated with the Co-Promotion Agreement included in our balance sheet and the $268.0 million termination payment.
Litigation Costs
On June 13, 2008, a jury ruled that we infringed upon one of Élan’s patents, which runs until 2011, and awarded Élan $55.2 million in damages for sales of Abraxane® through the judgment date. We have filed various post-trial motions and are appealing the jury ruling (See Note 13 – Contingencies to the consolidated financial statements for further discussion). For the year ended December 31, 2008, we expensed $57.6 million for this matter including interest expense.
Acquired in-Process Research and Development
In connection with the purchase of Shimoda Biotech and Platco Technologies in April 2008, we acquired certain research and development projects that were required to be expensed in accordance with generally accepted accounting principles. Approximately $13.9 million of the purchase price was expensed as in-process research and development for projects that, as of the acquisition date, had not yet reached technological or regulatory feasibility and had no alternative future uses in their current states.
Impairment Charge
We assess for potential impairment of our long-lived assets whenever events or changes in circumstances indicate that the carrying value of the assets might not be recoverable. A long-lived asset is impaired when the carrying value of the asset is determined not to be recoverable and the carrying amount exceeds the asset’s fair value. For the year ended December 31, 2009, we recorded an impairment charge of $14.0 million on property, plant and equipment for which the intended use had changed. For the year ended December 31, 2008, we recognized an impairment charge $9.2 million as a result of an anticipated sale of certain property, plant and equipment.
44
Table of Contents
Amortization of Intangible Assets
Amortization of intangibles assets totaled $39.8 million, $39.4 million and $38.6 million for the years ended December 31, 2009, 2008 and 2007, respectively, and included $31.5 million in each of the years for amortization associated with the U.S. rights to Abraxane®. The increase in 2008 amortization of intangible assets from 2007 relates to incremental amortization associated with the acquisition of Shimoda Biotech and Platco Technologies in April 2008.
Equity in Net Income of Unconsolidated Entities
Income from equity investments accounted for under the equity method for the years ended December 31, 2009, 2008 and 2007 was $2.1 million, $0.9 million and $3.8 million, respectively. For the year ended December 31, 2009, our income from equity method investments consisted of $2.3 million from Drug Source Company, LLC offset by losses from other investments acquired during the fourth quarter of 2009. Drug Source Company was our only equity method investment in 2008 and 2007.
Other Non-Operating Items
Interest income in 2009 consisted primarily of interest earned on invested cash and notes receivables. Interest income decreased $15.8 million compared to 2008 primarily due to lower investment balances and lower interest rates. Interest income in 2008 was $18.8 million compared to $5.0 million in 2007. The increase in interest income in 2008 of $13.8 million was primarily due to a full year’s interest income in 2008 versus only one and one-half months of interest income in 2007.
Other expense was higher in 2008 primarily due to other than temporary losses of $5.1 million recognized on marketable securities whose values were determined to be impaired.
Provision for Income Taxes
At December 31, 2009, we were in a net deferred tax liability position. Management believes it is likely that a portion of the deferred tax assets will not be realized. Therefore, we recorded a valuation allowance of $131.4 million against ending deferred assets. For 2009, we reported an income tax benefit of $2.6 million on a pre-tax loss from continuing operations of $107.3 million.
At December 31, 2008, we were in a net deferred tax asset position. Management believes it is likely that a portion of the deferred tax assets will not be realized. Therefore, we recorded a valuation allowance of $103.6 million against ending deferred assets. For 2008, we reported an income tax benefit of $1.9 million on a pre-tax loss from continuing operations of $278.7 million.
At December 31, 2007, we were in a net deferred tax asset position. In the judgment of management, it is not likely that a portion of these assets will be realized. Therefore, we recorded a valuation allowance of $2.3 million against our deferred tax assets at December 31, 2007.
45
Table of Contents
LIQUIDITY AND CAPITAL RESOURCES
Overview
The following tables summarize key elements of our financial position and sources and (uses) of cash and cash equivalents as of:
| December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| (in thousands) | ||||||||||||
| Summary Financial Position |
||||||||||||
| Cash and cash equivalents |
$ | 203,312 | $ | 306,390 | $ | 705,125 | ||||||
| Cash collateral for reacquisition of agreement |
— | 300,631 | — | |||||||||
| Working capital |
192,747 | 386,148 | 735,181 | |||||||||
| Total assets |
1,068,380 | 1,438,584 | 1,502,255 | |||||||||
| Total equity |
846,265 | 929,472 | 1,197,387 | |||||||||
| Year Ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| (in thousands) | ||||||||||||
| Summary of (Uses) and Sources of Cash and Cash Equivalents: |
||||||||||||
| Operating activities |
$ | 4,488 | $ | (315,468 | ) | $ | (2,893 | ) | ||||
| Purchases of property, plant and equipment |
(94,473 | ) | (43,729 | ) | (40,581 | ) | ||||||
| Purchases of debt securities |
(4,740 | ) | (9,147 | ) | — | |||||||
| Purchase of other equity investments |
(10,691 | ) | (15,097 | ) | (150 | ) | ||||||
| Purchases of other assets |
(125 | ) | — | (4,174 | ) | |||||||
| Purchase of warrants and investment rights |
(260 | ) | (2,591 | ) | — | |||||||
| Proceeds from sale of subsidiary |
2,046 | — | — | |||||||||
| Proceeds from sale of marketable securities |
3,677 | — | — | |||||||||
| Cash paid for acquisition, net of cash acquired |
(2,640 | ) | (14,998 | ) | — | |||||||
| Financing activities |
(561 | ) | 2,360 | 752,082 | ||||||||
(Uses) and Sources of Cash
Operating Activities
Net cash provided by operating activities was $4.5 million for year ended December 31, 2009 compared to net cash used in operating activities of $315.5 million for the same period in 2008, resulting in a $320.0 million increase in cash provided by operations. This increase was primarily due to $300.6 million in cash used to pay for letters of credits in 2008 to secure future payments under the agreement to reacquire the exclusive rights to market Abraxane® in the United States. An increase in accounts payable in 2009 also caused an increase in cash provided by operating activities. These increases were partially offset by an increase in accounts receivable in 2009.
Net cash used in operating activities was $315.5 million for the year ended December 31, 2008 compared to cash used in operating activities of $2.9 million in 2007. The increase in cash used in operations of $312.6 million was primarily due to cash collateral set aside for the $268.0 million payment to reacquire the exclusive rights to market Abraxane® in the United States and estimated remaining payments under the Co-Promotion Agreement of $18 million. Cash outflow for prepaid expenses, accounts payable and accrued expenses accounted for additional increases in cash used for operating activities. The uses of cash were partially offset by cash provided from decreases in accounts receivable, inventory and higher interest income earned on our cash balance in 2008.
46
Table of Contents
Investing Activities
Our investing activities have included capital expenditures necessary to expand and maintain our manufacturing capabilities and infrastructure and to acquire various intellectual property rights and to make other investments.
Net cash used for the acquisition of property, plant and equipment for the year ended December 31, 2009, 2008 and 2007 totaled $94.5, $43.7 million and $40.6 million, respectively. The expenditures in 2009 related primarily to the purchase of our Costa Mesa research facility and the modernization of our Melrose Park, Illinois and Phoenix, Arizona manufacturing facilities. Net cash paid for acquisition of $2.6 million related to net milestone payments for Shimoda Biotech (Pty) Ltd and Platco Technologies (Pty) Ltd. Additionally, we made investments in various entities for $10.7 million and purchased notes receivable of $4.7 million. In 2009, we also had proceeds from the sale of marketable securities of $3.7 million and proceeds of $2.0 million from the sale of our subsidiary in Barbengo, Switzerland.
In 2008, we purchased $19.7 million in marketable securities, which included $5.2 million for our investments in ProMetic Life Sciences, Inc., $5.4 million in other equity marketable securities and $9.1 million in debt marketable securities. Additionally, in 2008, we purchased $2.6 million in warrants and investment rights and $4.5 million in other investments. We acquired 100% of the equity of both Shimoda Biotech (Pty) Ltd and Platco Technologies (Pty) Ltd. for $15.1 million.
Net cash used for the acquisition of product license rights and other non-current assets totaled $4.1 million for the year ended December 31, 2007.
Financing Activities
Net cash used by financing activities for the year ended December 31, 2009 included the purchase of treasury stock and cash received upon the exercise of stock options.
Net cash provided by financing activities for the year ended December 31, 2008 represented cash received upon the exercise of stock options.
Sources of Financing and Capital Requirements
Our primary source of liquidity has been cash from operations and the $700 million cash contribution we received in connection with our separation from APP Pharmaceuticals, Inc. in November 2007. In connection with the proposed spin-off of Abraxis Health, we plan to contribute a portion of our cash balance to Abraxis Health as well as provide a secured credit facility to Abraxis Health. We believe our cash and cash equivalents on hand, together with any cash generated from operations, will be sufficient to finance our operations and our obligations under the proposed secured credit facility for at least the next twelve months. In the event we engage in future acquisitions or significant capital projects or significantly reduce our available cash resources in connection with the proposed spin-off of Abraxis Health, we may have to raise additional capital through additional borrowings or the issuance of debt or equity securities.
On June 18, 2008, we filed a registration statement on Form S-3 with the Securities and Exchange Commission (SEC). On July 2, 2009, the SEC declared this registration statement effective. Under this registration statement, we may, from time to time, offer shares of our common stock and preferred stock, various series of debt securities or warrants or rights to purchase any such securities, either individually or in units, in one or more offerings, in amounts we will determine from time to time, up to a total of $400 million. In addition, certain stockholders may, from time to time, sell in one or more offerings up to a total of 2,000,000 shares of our common stock.
47
Table of Contents
Capital Requirements
Our future capital requirements will depend on numerous factors, including:
| • | expansion of product sales into international markets; |
| • | working capital requirements and production, sales, marketing and development costs required to support Abraxane®; |
| • | research and development, including clinical trials, spending to develop further product candidates and ongoing studies; |
| • | the need for manufacturing expansion and improvement; |
| • | the allocation of cash resources between us and Abraxis Health if the proposed spin-off occurs; |
| • | the requirements of any potential future acquisitions, asset purchases or equity investments; and |
| • | the amount of cash generated by operations, including potential milestone and license revenue. |
On April 20, 2009, we announced that our board of directors approved a program to repurchase up to $100 million of the company’s common stock. Share repurchases, if any, will be funded by internal cash resources and will be made through open market purchases. The timing, volume and nature of share repurchases are subject to market prices and conditions, applicable securities laws and other factors, and are at the discretion of management. Share repurchases may be commenced, suspended or discontinued at any time without prior notice.
Contractual Obligations
Contractual obligations represent future cash commitments and liabilities under agreements with third parties, and exclude contingent liabilities for which we cannot reasonably predict future payment. Accordingly, the table below excludes contractual obligations relating to milestones and royalty payments due to third parties contingent upon certain future events. Such events could include, but are not limited to, development milestones, regulatory approvals, and product sales. The following information summarizes our contractual obligations and other commitments as of December 31, 2009:
| Payments due by period | |||||||||||||||
| Contractual Obligations |
Total | Less than 1 year |
1-3 years | 3-5 years | More than 5 years | ||||||||||
| (in thousands) | |||||||||||||||
| Unconditional purchase obligations |
$ | 269,565 | $ | 52,536 | $ | 85,151 | $ | 65,016 | $ | 66,862 | |||||
| Operating lease obligations |
26,790 | 10,333 | 9,087 | 5,288 | 2,082 | ||||||||||
| Other long term obligations |
12,054 | 8,306 | 3,748 | — | — | ||||||||||
| Total contractual cash obligations |
$ | 308,409 | $ | 71,175 | $ | 97,986 | $ | 70,304 | $ | 68,944 | |||||
Collaborative Agreements and Other Licensing Obligations
We have entered into various collaborative agreements under which we have agreed to provide funding to academic and clinical trial organizations of up to $53.8 million for research and other projects over a period of time of up to five years. Although we cannot predict the timing, we generally can control the timing and amount of spending as the projects require our approval in advance of funding.
Clinical Trial and Other Commitments
We have entered into various clinical trial agreements with third parties for the management, planning and execution of clinical trials. These agreements generally include milestone payments based on the number of patients enrolled in the clinical study. If all milestones in these agreements were achieved, related milestone commitments to be paid by us in 2010 through 2014 would approximate $83.0 million. In addition, we own a
48
Table of Contents
library of natural drug discovery soil samples and related strains acquired from around the world, which is intended for use in the discovery of new chemical entities. We are committed to paying up to $4.2 million upon the achievement of certain milestones related to this library.
Off-Balance Sheet Arrangements
We provide irrevocable standby letters of credits to secure our obligations under certain lease agreements and other obligations. These standby letters of credit are generally for a term of one-year or less and may be extended to match the term of our obligation. As of December 31, 2009 and 2008, we had outstanding letters of credits of $3.1 million and $3.8 million, respectively. In 2008, in connection with the reacquisition of the exclusive rights to market Abraxane® in the U.S. (see Item 8—Financial Statements and Supplementary Data, Note 4—Acquisitions and Other Transactions for further details), we issued $286.0 million in irrevocable standby letters of credit to secure the future payments under the termination agreement. These letters of credits were collateralized by $300.6 million of our cash balance, which was included in “Cash collateral for reacquisition of agreement” in the balance sheet as of December 31, 2008. In March 2009, we made the final payments under the agreement and were released from our obligation to maintain the cash collateral.
CRITICAL ACCOUNTING POLICIES AND ESTIMATES
The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenue and expense during the reporting period. The most significant estimates in our combined financial statements are discussed below. Actual results could vary from those estimates.
Revenue Recognition
Abraxane® Revenue Recognition
We recognize revenue from the sale of a product when title and risk of loss have transferred to the customer, collection is reasonably assured and we have no further performance obligation. This is typically when the product is received by the customer. At the time of sale, as further described below, we reduce sales and provide for estimated chargebacks, customer or Medicaid rebates, product returns and customer credits and cash discounts. Our methodology used to estimate and provide for these sales provisions was consistent across all periods presented. Accruals for sales provisions are presented in our consolidated financial statements as a reduction of net revenue and accounts receivable and, for customer or Medicaid rebates, an increase in accrued liabilities. Our sales provisions totaled $41.0 million, $32.8 million and $30.5 milllion for the years ended December 31, 2009, 2008 and 2007, respectively, and related reserves totaled $13.8 million and $9.2 million at December 31, 2009 and 2008, respectively. The increase in reserves is primarily the result of an increase in performance related rebates. We regularly review information related to these estimates and adjust our reserves accordingly if, and when, actual experience differs from estimates.
We have internal historical information on chargebacks, rebates and customer returns and credits, which we use as the primary factor in determining the related reserve requirements. Due to the nature of our product and its primary use in hospital and clinical settings with generally consistent demand, we believe that this internal historical data, in conjunction with periodic review of available third-party data and updated for any applicable changes in available information, provides a reliable basis for such estimates.
Research Revenue Recognition
Research revenue generally consists of amounts earned from the development and research to advance the commercial application of pharmaceutical compounds for third parties. Such research revenue may include
49
Table of Contents
upfront license fees, milestone payments and reimbursement of development and other costs, and royalties. Non-refundable upfront license fees under which we have continuing involvement in the form of development, manufacturing or commercialization are recognized as revenue ratably over either the development period, if the development risk is significant, or the estimated product useful life, if development risk has been substantially eliminated. Achievement-based milestone payments are recognized as revenue when the milestone objective is attained because the earnings process relating to such payments is complete upon attainment and because the payments are non-refundable and are not dependent upon future activities or the achievement of future objectives. These milestone payments are at risk and require substantive activity to achieve. Reimbursement of development and other costs are recognized as revenue as the related costs are incurred. Royalties are recognized as earned in accordance with the contract terms when royalties from licensees can be reliably measured and collectibility is reasonably assured. Royalty estimates are made in advance of amounts collected using historical and forecasted trends.
Chargebacks
Following industry practice, we typically sell our product to independent pharmaceutical wholesalers at wholesale list price. The wholesaler in turn sells our product to an end user, normally a hospital or alternative healthcare facility, at a lower contractual price previously established between us and the end user via a group purchasing organization, or GPO. GPOs enter into collective purchasing contracts with pharmaceutical suppliers to secure more favorable product pricing on behalf of their end-user members.
Our initial sale to the wholesaler, and the resulting receivable, are recorded at our wholesale list price. However, as most of these selling prices will be reduced to a lower end-user contract price, at the time of sale revenue is reduced by, and a provision recorded for, the difference between the list price and estimated end-user contract price. When the wholesaler ultimately sells the product to the end user at the end-user contract price, the wholesaler charges us, a chargeback, for the difference between the list price and the end-user contract price and such chargeback is offset against our initial estimated contra asset. The most significant estimates inherent in the initial chargeback provision relate to wholesale units pending chargeback and the ultimate end-user contract selling price. We base our estimation for these factors primarily on internal sales and chargeback processing experience, estimated wholesaler inventory stocking levels, current contract pricing and our expectation for future contract pricing changes.
Our net chargeback reserve totaled $1.4 million and $1.3 million at December 31, 2009 and 2008, respectively. Due to information constraints in the distribution channel, it has not been practical, and has not been necessary, for us to capture and quantify the impact of current versus prior year activity on the chargeback provision. Information constraints within the distribution channel primarily relate to our inability to track product through the channel on a unit or specific lot basis. In addition, for the most part, we do not receive information from our customers with respect to what level of their sales are subject to chargeback. The lack of information on a specific lot basis precludes us from tracking actual chargeback activity to the period of our initial sale. As a result, we rely on internal data, external IMS data and management estimates in order to estimate the amount of inventory in the channel subject to future chargeback. The amount of inventory in the channel is comprised of physical inventory at the distributor and that the distributor has yet to report as end user sales. Physical inventory in the channel is determined by the difference between our sales less end user sales as reported by IMS. As IMS data is reported approximately one month after end user sales, the last month of end user sales is determined by a mathematical trend incorporating IMS data for prior months. We estimate yet to be reported end user sales based on a historical average number of days to process charge back activities from the date of the end user sale. We also review current year chargeback activity to determine whether material changes in the provision relate to prior period sales; such changes have not been material to our statements of operations. As a proprietary product, Abraxane® does not require significant chargeback reserves.
50
Table of Contents
Rebates, Returns and Credits
Rebates or administrative fees are offered to certain wholesale customers, GPOs and end-user customers, consistent with pharmaceutical industry practices. Settlement of rebates and fees may generally occur from one to 15 months from date of sale. We provide a provision for rebates at the time of sale based on the contracted rates and historical redemption rates. Upon receipt of chargeback, due to the availability of product and customer specific information on these programs, we then establish a specific provision for fees or rebates based on the specific terms of each agreement. Our rebate reserve totaled $9.5 million and $5.5 million at December 31, 2009 and 2008, respectively. Rebates are reflected in the consolidated financial statements as a reduction of net revenue and as a current accrued liability.
Consistent with industry practice, our return policy permits our customers to return product within a window of time before and after the expiration of product dating. We provide for product returns and other customer credits at the time of sale by applying historical experience factors generally based on our historic data on credits issued by credit category or product, relative to related sales and we provide specifically for known outstanding returns and credits. Our reserve for customer credits and product returns totaled $1.0 million and $0.7 million at December 31, 2009 and 2008, respectively.
Inventories
Inventories consist of products currently approved for marketing and may include certain products pending regulatory approval. From time to time, we capitalize inventory costs associated with products prior to regulatory approval based on our judgment of probable future commercial success and realizable value. Such judgment incorporates our knowledge and best judgment of where the product is in the regulatory review process, our required investment in the product, market conditions, competing products and our economic expectations for the product post-approval relative to the risk of manufacturing the product prior to approval. In evaluating the market value of inventory pending regulatory approval as compared to its cost, we considered the market, pricing and demand for competing products, its anticipated selling price for the product and the position of the product in the regulatory review process. If final regulatory approval for such products is denied or delayed, we may need to provide for and expense such inventory. No inventory held at December 31, 2009 and 2008 was pending regulatory approval.
We routinely review our inventory and establish reserves when the cost of the inventory is not expected to be recovered or our product cost exceeds realizable market value. In instances where inventory is at or approaching expiry, is not expected to be saleable based on our quality and control standards or for which the selling price has fallen below cost a reserve is established. We reserve for any inventory impairment based on the specific facts and circumstances. In evaluating the market value of inventory pending regulatory approval as compared to its cost, we consider the market, pricing and demand for competing products, our anticipated selling price for the product and the position of the product in the regulatory review process. Provisions for inventory reserves are reflected in the consolidated and combined financial statements as an element of cost of sales with inventories presented net of related reserves.
Allocated Expenses
Up to the date of the separation and related transactions on November 13, 2007, our product development and general and administrative functions were fully integrated with Old Abraxis, including product development, accounting, treasury, payroll, internal audit, information technology, corporate income tax, legal services and investor relations. To the extent that an asset, liability, revenue, or expense is directly associated with us, it is reflected in the accompanying consolidated and combined financial statements. In addition, Old Abraxis provided manufacturing services to us at cost. After the distribution, certain of these arrangements have continued on a temporary basis for a period generally not to exceed 24 months (or four or five years with respect to the manufacturing arrangement and real estate leases). The accompanying consolidated and combined financial statements reflect the application of certain estimates and allocations for periods before November 13,
51
Table of Contents
2007 and management believes the methods used to allocate these operating expenses are reasonable. The allocation methods include relative head count, sales, square footage and management knowledge. These allocated operating expenses totaled $33.3 million and $25.8 million for the years ended December 31, 2007 and 2006, respectively. The financial information in these consolidated and combined financial statements does not include all the expenses that would have been incurred had we been a separate, stand-alone entity for the periods prior to November 13, 2007. As such, the consolidated and combined financial statements for periods prior to and including November 13, 2007 may not be indicative of our future performance and do not necessarily reflect what our consolidated and combined results of operations, financial position, and cash flows would have been had we operated as an independent, publicly-traded company during the periods presented, including changes in our capitalization as a result of the separation and related transactions.
Stock-Based Compensation
We account for stock based compensation in accordance with FASB ASC 718, Compensation – Stock Compensation, which requires the recognition of compensation cost for all share-based payments (including employee stock options) at fair value. We use the straight-line attribution method to recognize share-based compensation expenses over the applicable vesting period of the award. Options currently granted generally expire ten years from the grant date and vest ratably over a four-year period. Restricted Stock Units (RSU) granted under the 2001 and 2007 Stock Incentive Plans generally vest ratably over a four-year period, while awards under the RSU II plan generally vested with respect to one half of the units on April 18, 2008 (the second anniversary of the closing of the merger), and the remaining shares generally will vest on April 18, 2010.
To determine stock-based compensation, we use the Black-Scholes option pricing model to estimate the fair value of options granted under equity incentive plans and rights to acquire stock granted under our stock participation plan. Awards under RSU Plan I and RSU Plan II entitle the holders on each vesting date to convert the vested portion of their awards into that number of shares of our common stock equal to the value of the vested portion of the award divided by the lower of (i) the average closing price of our common stock over the three consecutive trading days ending on and including the second full trading day preceding the vesting date and (ii) $66.63 (which is the adjusted price following the separation). Accordingly, compensation expense related to these RSU plans is based on the lower of the market price or $66.63 and is expensed under the liability method in accordance with ASC 718 on a straight-line basis over the applicable vesting period. Compensation expense associated with RSU awards under the 2001 and 2007 Stock Incentive Plans is accounted for under the equity method in accordance with ASC 718.
Acquisitions
We account for acquired businesses using the purchase method of accounting, which requires that the assets acquired and liabilities assumed be recorded at the date of acquisition at their respective fair values. Any excess of the purchase price over the estimated fair values of the net assets acquired is recorded as goodwill. When we acquire net assets that do not constitute a business under Generally Accepted Accounting Principles in the U.S. (GAAP), no goodwill is recognized.
The judgments made in determining the estimated fair value assigned to each class of assets acquired and liabilities assumed, as well as asset lives, can materially impact our results of operations. Accordingly, for significant items, we typically obtain assistance from third-party valuation specialists. The valuations are based on information available near the acquisition date and are based on expectations and assumptions that have been deemed reasonable by management.
Determining the useful life of an intangible asset also requires judgment, as different types of intangible assets will have different useful lives and certain assets may even be considered to have indefinite useful lives. For example, the useful life of the right associated with a pharmaceutical product’s exclusive patent will be finite
52
Table of Contents
and will result in amortization expense being recorded in our results of operations over a determinable period. However, the useful life associated with a brand that has no patent protection but that retains, and is expected to retain, a distinct market identity could be considered to be indefinite and the asset would not be amortized.
Impairment of long-lived assets
We review all of our long-lived assets, including goodwill and other intangible assets, for impairment indicators at least annually and we perform detailed impairment testing for goodwill annually and for all other long-lived assets whenever impairment indicators are present. Examples of those events or circumstances that may be indicative of impairment include:
| • | A significant adverse change in legal factors or in the business climate that could affect the value of the asset, including, by way of example, a successful challenge of our patent rights that could result in generic competition earlier than expected. |
| • | A significant adverse change in the extent or manner in which an asset is used, including, by way of example, restrictions imposed by the FDA or other regulatory authorities that could affect our ability to manufacture or sell a product. |
| • | A projection or forecast that demonstrates losses associated with an asset, including, by way of example, a change in a government reimbursement program that could result in an inability to sustain projected product revenues or profitability or the introduction of a competitor’s product that could result in a significant loss of market share. |
The value of intangible assets is determined primarily using the “income method,” which starts with a forecast of all expected future net cash flows. Accordingly, the potential for impairment for intangible assets may exist if actual revenues are significantly less than those initially forecasted or actual expenses are significantly more than those initially forecasted. Some of the more significant estimates and assumptions inherent in the intangible asset impairment estimation process include: the amount and timing of projected future cash flows; the discount rate selected to measure the risks inherent in the future cash flows; and the assessment of the asset’s life cycle and the competitive trends impacting the asset, including consideration of any technical, legal, regulatory, or economic barriers to entry as well as expected changes in standards of practice for indications addressed by the asset.
RECENT ACCOUNTING PRONOUNCEMENTS
Effective January 1, 2009, we adopted new guidance under FASB ASC 810, Consolidation, relating to the accounting and reporting of noncontrolling interest in a subsidiary. The noncontrolling interest is the portion of equity in a subsidiary not attributable directly or indirectly to a parent. The standard requires the noncontrolling interest to be presented separately from the parent’s equity in the consolidated statements of financial position and statements of shareholders’ equity. Additionally, the consolidated net income attributable to the parent and to the noncontrolling interest is presented separately on the consolidated statements of income. Adoption of this standard did not have a material impact on our consolidated financial statements.
Effective January 1, 2009, we adopted new guidance under ASC 808-10, Collaborative Arrangement, which defines collaborative arrangements and establishes the reporting requirements for transactions between participants in a collaborative arrangement, and between participants in the arrangement and third parties. A collaborative arrangement is defined as one in which the participants are actively involved and are exposed to significant risks and rewards that depend on the ultimate commercial success of the endeavor. For revenues and costs incurred with third parties in connection with collaborative arrangements, the participant that is deemed to be the principal participant in the transaction shall record that transaction on a gross basis on its financial statements. Payments to or from collaborators are evaluated and presented based on the nature of the
53
Table of Contents
arrangement and its terms, the nature of the entity’s business, and whether those payments are within the scope of other accounting literature. The nature and purpose of collaborative arrangements are disclosed along with the accounting policies and the classification of significant financial statement amounts related to the arrangements. Activities in arrangements conducted in a separate legal entity are accounted for under other accounting literature, however, required disclosure applies to the entire collaborative agreement. Adoption of this standard did not have a material impact on our consolidated financial statements.
In June 2009, the Financial Accounting Standards Board (FASB) issued a new accounting standard that amends guidance regarding consolidation of variable interest entities to address the elimination of the concept of a qualifying special purpose entity (the “QSPE”). This standard also replaces the quantitative-based risks and rewards calculation for determining which enterprise has a controlling financial interest in a variable interest entity with an approach focused on identifying which enterprise has the power to direct the activities of the variable interest entity, and the obligation to absorb losses of the entity or the right to receive benefits from the entity. Additionally, the standard requires any enterprise that holds a variable interest in a variable interest entity to provide enhanced disclosures that will provide users of financial statements with more transparent information about an enterprise’s involvement in a variable interest entity. This standard is effective for us for interim and annual reporting periods beginning on or after January 1, 2010. We are assessing the impact of this adoption on our consolidated financial statements.
In August 2009, the FASB issued additional guidance on the fair value measurement of liabilities. The amendments to ASC 820, Measuring Liabilities at Fair Value, provides clarification that in circumstances in which a quoted price in an active market for the identical liability is not available, a reporting entity is required to measure fair value using one or more of the specified techniques. These amendments are effective for us beginning October 1, 2009. Adoption of the standard did not have a material impact on our consolidated financial statements.
In October 2009, the FASB issued a new accounting standard which amends guidance on accounting for revenue arrangements involving the delivery of more than one element of goods and/or services. The standard amends the criteria for separating consideration in multiple-deliverable arrangements and establishes a selling price hierarchy for determining the selling price of a deliverable. The amendments will eliminate the residual method of allocation and require that arrangement consideration be allocated at the inception of the arrangement to all deliverables using the relative selling price method. The standard also significantly expands the disclosures related to a vendor’s multiple-deliverable arrangement. The standard is effective on a prospective basis for revenue arrangements entered into or materially modified in fiscal years beginning on or after June 15, 2010. Alternatively, adoption may be on a retrospective basis, and early application is permitted. Adoption of this standard is not expected to have a material impact on our consolidated financial statements.
Item 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
We are exposed to market risks associated with changes in interest rates and foreign currency exchange rates. Interest rate changes affect primarily our investments in marketable securities and our debt obligations. Changes in foreign currency exchange rates can affect our operations outside of the United States.
Foreign Currency Risk: We have operations in Canada, the European Union, China and other parts of the world; however, both revenue and expenses of those operations are typically denominated in the currency of the country of operations, providing a partial hedge. Nonetheless, these foreign operations are presented in our consolidated and combined financial statement in U.S. dollars and can be impacted by foreign currency exchange fluctuations through both (i) translation risk, which is the risk that the financial statements for a particular period or as of a certain date depend on the prevailing exchange rates of the various currencies against the U.S. dollar, and (ii) transaction risk, which is the risk that the currency impact of transactions denominated in currencies other than the subsidiaries functional currency may vary according to currency fluctuations.
54
Table of Contents
With respect to translation risk, even though there may be fluctuations of currencies against the U.S. dollar, which may impact comparisons with prior periods, the translation impact is included in accumulated other comprehensive income, a component of stockholders’ equity, and does not affect the underlying results of operations. Gains and losses related to transactions denominated in a currency other than the functional currency of the countries in which we operate are included in the consolidated statements of operations. We do not hedge our investments in our foreign subsidiaries.
Investment Risk: The primary objectives of our investment program are the safety and preservation of principal, maintaining liquidity to meet operating and projected cash flow requirements, and maximizing return on invested funds, while diversifying risk. Some of the securities that we invest in may have interest rate risk. This means that a change in prevailing interest rates may cause the fair value of the principal amount of the investment to fluctuate. For example, if we hold a security that was issued with a fixed interest rate at the prevailing rate and the prevailing rate later rises, the fair value of the principal amount of our investment will probably decline.
To minimize this risk, we maintain an investment portfolio of cash equivalents and short-term investments consisting of high credit quality securities, including money market funds, commercial paper, government and non-government debt securities. We do not use derivative financial instruments.
We are also exposed to equity price risks on marketable equity securities included in our portfolio of investments entered into for the promotion of business and strategic objectives. These investments are generally in small capitalization stocks in the biotechnology industry sector. We regularly review the market prices of these investments for impairment purposes. For the years ended December 31, 2009 and 2008, we realized other than temporary losses of $3.4 million and $4.7 million, respectively, on available for sale securities whose values, based on market quotation, had declined below their carrying value. No losses were recognized in 2007. As of December 31, 2009 and 2008 our investment in marketable equity securities totaled $6.9 million and $6.2 million, respectively.
Interest Rate Risk: As of December 31, 2009, we had no debt obligations outstanding. Consequently, we have minimal current exposure to changes in interest rates on borrowings.
55
Table of Contents
Item 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders of Abraxis BioScience, Inc.
We have audited the accompanying consolidated balance sheets of Abraxis BioScience, Inc. (formerly known as New Abraxis, Inc.) as of December 31, 2009 and 2008, and the related consolidated and combined statements of operations, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2009. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Abraxis BioScience, Inc. at December 31, 2009 and 2008, and the consolidated and combined results of its operations and its cash flows for each of the three years in the period ended December 31, 2009, in conformity with U.S. generally accepted accounting principles.
As discussed in Note 2 to the consolidated financial statements, the Company changed its method of accounting for noncontrolling interest with the adoption of the guidance originally issued in FASB Statement No. 160, Noncontrolling Interests in Consolidated Financial Statements (codified in FASB ASC Topic 810, Consolidation) effective January 1, 2009.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Abraxis BioScience, Inc.’s internal control over financial reporting as of December 31, 2009, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated March 12, 2010 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Los Angeles, California
March 12, 2010
56
Table of Contents
Abraxis BioScience, Inc.
Consolidated Balance Sheets
(in thousands, except share data)
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| Assets |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 203,312 | $ | 306,390 | ||||
| Cash collateral for reacquisition of agreement |
— | 300,631 | ||||||
| Accounts receivable, net of chargebacks of $1,445 in 2009 and $1,258 in 2008 and credit returns of $1,026 in 2009 and $695 in 2008 |
47,220 | 37,011 | ||||||
| Related party receivable |
51 | 1,915 | ||||||
| Inventories |
54,209 | 63,506 | ||||||
| Prepaid expenses and other current assets |
40,994 | 33,795 | ||||||
| Deferred income taxes |
25,510 | 26,758 | ||||||
| Total current assets |
371,296 | 770,006 | ||||||
| Property, plant and equipment, net |
236,798 | 166,720 | ||||||
| Investments in unconsolidated entities |
22,774 | 10,183 | ||||||
| Intangible assets, net of accumulated amortization of $144,908 in 2009 and $105,113 in 2008 |
144,633 | 175,291 | ||||||
| Goodwill |
241,361 | 241,361 | ||||||
| Other non-current assets |
51,518 | 36,196 | ||||||
| Total assets |
$ | 1,068,380 | $ | 1,399,757 | ||||
| Liabilities and equity |
||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 28,577 | $ | 39,142 | ||||
| Accrued liabilities |
88,723 | 53,020 | ||||||
| Accrued litigation costs |
57,635 | 57,635 | ||||||
| Reacquisition payable |
— | 268,000 | ||||||
| Related party payable |
334 | — | ||||||
| Income taxes payable |
142 | 679 | ||||||
| Deferred revenue |
3,138 | 4,209 | ||||||
| Total current liabilities |
178,549 | 422,685 | ||||||
| Deferred income taxes, non-current |
29,507 | 23,858 | ||||||
| Long-term portion of deferred revenue |
4,867 | 8,223 | ||||||
| Other non-current liabilities |
9,192 | 15,519 | ||||||
| Total liabilities |
222,115 | 470,285 | ||||||
| Equity |
||||||||
| Stockholders’ equity: |
||||||||
| Common stock—$0.001 par value; 100,000,000 shares authorized; issued and outstanding—40,107,334 shares in 2009 and 40,066,451 shares in 2008 |
40 | 40 | ||||||
| Additional paid-in capital |
1,213,707 | 1,203,092 | ||||||
| Accumulated deficit |
(375,805 | ) | (272,689 | ) | ||||
| Accumulated other comprehensive income (loss) |
5,011 | (971 | ) | |||||
| Total stockholders’ equity |
842,953 | 929,472 | ||||||
| Noncontrolling interest |
3,312 | — | ||||||
| Total equity |
846,265 | 929,472 | ||||||
| Total liabilities and equity |
$ | 1,068,380 | $ | 1,399,757 | ||||
See accompanying notes to consolidated and combined financial statements
57
Table of Contents
Abraxis BioScience, Inc.
Consolidated and Combined Statements of Operations
(in thousands, except per share data)
| Year ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| Abraxane revenue |
$ | 314,545 | $ | 335,631 | $ | 324,692 | ||||||
| Other revenue |
44,505 | 9,678 | 8,994 | |||||||||
| Net revenue |
359,050 | 345,309 | 333,686 | |||||||||
| Cost of sales |
63,665 | 39,068 | 34,450 | |||||||||
| Gross profit |
295,385 | 306,241 | 299,236 | |||||||||
| Operating expenses: |
||||||||||||
| Research and development |
154,615 | 98,976 | 85,424 | |||||||||
| Selling, general and administrative |
200,734 | 221,418 | 233,324 | |||||||||
| Reacquisition costs |
— | 158,909 | — | |||||||||
| Litigation costs |
— | 57,635 | — | |||||||||
| Acquired in-process research and development |
— | 13,900 | — | |||||||||
| Impairment charge |
13,999 | 9,214 | — | |||||||||
| Amortization of intangible assets |
39,782 | 39,429 | 38,615 | |||||||||
| Total operating expenses |
409,130 | 599,481 | 357,363 | |||||||||
| Loss from operations |
(113,745 | ) | (293,240 | ) | (58,127 | ) | ||||||
| Equity in net income of unconsolidated entities |
2,090 | 908 | 3,771 | |||||||||
| Interest income |
3,052 | 18,809 | 4,990 | |||||||||
| Other income (expense) |
1,255 | (5,186 | ) | (190 | ) | |||||||
| Loss before income taxes |
(107,348 | ) | (278,709 | ) | (49,556 | ) | ||||||
| Benefit for income taxes |
(2,580 | ) | (1,938 | ) | (7,952 | ) | ||||||
| Net loss |
$ | (104,768 | ) | $ | (276,771 | ) | $ | (41,604 | ) | |||
| Net loss attributable to noncontrolling interest |
$ | (1,652 | ) | $ | — | $ | — | |||||
| Net loss attributable to common shareholders |
$ | (103,116 | ) | $ | (276,771 | ) | $ | (41,604 | ) | |||
| Basic and diluted net loss per common share |
$ | (2.57 | ) | $ | (6.91 | ) | $ | (1.04 | ) | |||
| The composition of stock-based compensation included above is as follows: |
||||||||||||
| Cost of sales |
$ | 231 | $ | 252 | $ | 1,068 | ||||||
| Research and development |
4,550 | 3,997 | 10,453 | |||||||||
| Selling, general and administrative |
10,294 | 9,025 | 10,772 | |||||||||
| $ | 15,075 | $ | 13,274 | $ | 22,293 | |||||||
See accompanying notes to consolidated and combined financial statements
58
Table of Contents
Abraxis BioScience, Inc.
Consolidated and Combined Statements of Equity
Year Ended December 31, 2009, 2008 and 2007
(in thousands)
| Stockholders’ Equity | Non controlling Interest |
Total Equity |
||||||||||||||||||||||||||||
| Common Stock | Additional Paid-in Capital |
(Accumulated Deficit) Retained Earnings |
Accumulated Other Comprehensive (Loss) Income |
Parent Company Investment |
||||||||||||||||||||||||||
| Shares | Amount | |||||||||||||||||||||||||||||
| Balance at January 1, 2007 |
— | $ | — | $ | — | $ | — | $ | 856 | $ | 458,165 | $ | — | $ | 459,021 | |||||||||||||||
| Net transactions with parent company. |
— | — | — | — | — | 779,187 | — | 779,187 | ||||||||||||||||||||||
| Issuance of common stock in connection with separation and related transactions |
39,990 | 40 | 1,191,626 | — | — | (1,191,666 | ) | — | — | |||||||||||||||||||||
| Exercise of stock options |
3 | — | 59 | — | — | — | — | 59 | ||||||||||||||||||||||
| Repurchase of common stock relating to the vesting of restricted units, net |
1 | — | (9 | ) | — | — | — | — | (9 | ) | ||||||||||||||||||||
| Equity-based compensation |
785 | — | — | — | — | 785 | ||||||||||||||||||||||||
| Comprehensive (loss) income: |
||||||||||||||||||||||||||||||
| Net loss |
4,082 | — | (45,686 | ) | — | (41,604 | ) | |||||||||||||||||||||||
| Unrealized loss on available-for-sale securities, net of tax of $47 |
— | (368 | ) | — | — | (368 | ) | |||||||||||||||||||||||
| Foreign currency translation adjustments |
— | 316 | — | — | 316 | |||||||||||||||||||||||||
| Comprehensive loss |
(41,656 | ) | ||||||||||||||||||||||||||||
| Balance at December 31, 2007 |
39,994 | $ | 40 | $ | 1,192,461 | $ | 4,082 | $ | 804 | $ | — | $ | — | $ | 1,197,387 | |||||||||||||||
| Exercise of stock options |
46 | — | 2,360 | — | — | — | — | 2,360 | ||||||||||||||||||||||
| Net settlement of vested restricted stocks |
26 | — | (946 | ) | — | — | — | — | (946 | ) | ||||||||||||||||||||
| Other |
(354 | ) | — | — | — | — | (354 | ) | ||||||||||||||||||||||
| Equity-based compensation |
9,571 | — | — | — | — | 9,571 | ||||||||||||||||||||||||
| Comprehensive (loss) income: |
||||||||||||||||||||||||||||||
| Net loss |
(276,771 | ) | — | — | — | (276,771 | ) | |||||||||||||||||||||||
| Unrealized loss on available-for-sale securities, net of tax of $43 |
— | (2,883 | ) | — | — | (2,883 | ) | |||||||||||||||||||||||
| Foreign currency translation adjustments |
— | 1,108 | — | — | 1,108 | |||||||||||||||||||||||||
| Comprehensive loss |
(278,546 | ) | ||||||||||||||||||||||||||||
| Balance at December 31, 2008 |
40,066 | $ | 40 | $ | 1,203,092 | $ | (272,689 | ) | $ | (971 | ) | $ | — | $ | — | $ | 929,472 | |||||||||||||
| Exercise of stock options |
8 | 313 | 313 | |||||||||||||||||||||||||||
| Net settlement of vested restricted stocks |
54 | (1,815 | ) | (1,815 | ) | |||||||||||||||||||||||||
| Equity-based compensation |
11,921 | 11,921 | ||||||||||||||||||||||||||||
| Repurchase and retirement of common stock |
(21 | ) | (874 | ) | (874 | ) | ||||||||||||||||||||||||
| Noncontrolling interest related to business combination |
4,964 | 4,964 | ||||||||||||||||||||||||||||
| Other |
1,070 | 1,070 | ||||||||||||||||||||||||||||
| Comprehensive (loss) income: |
||||||||||||||||||||||||||||||
| Net loss |
(103,116 | ) | (1,652 | ) | (104,768 | ) | ||||||||||||||||||||||||
| Unrealized gain on available for sale securities, net of tax of $2,804 |
6,937 | 6,937 | ||||||||||||||||||||||||||||
| Foreign currency translation adjustments |
(955 | ) | (955 | ) | ||||||||||||||||||||||||||
| Comprehensive loss |
(98,786 | ) | ||||||||||||||||||||||||||||
| Balance at December 31, 2009 |
40,107 | $ | 40 | $ | 1,213,707 | $ | (375,805 | ) | $ | 5,011 | $ | — | $ | 3,312 | $ | 846,265 | ||||||||||||||
See accompanying notes to consolidated and combined financial statements
59
Table of Contents
Abraxis BioScience, Inc.
Consolidated and Combined Statements of Cash Flows
(in thousands)
| Year Ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| Cash flows from operating activities: |
||||||||||||
| Net loss |
$ | (104,768 | ) | $ | (276,771 | ) | $ | (41,604 | ) | |||
| Adjustments to reconcile net loss to net cash provided by (used in) operating activities: |
||||||||||||
| Depreciation |
17,037 | 13,571 | 10,443 | |||||||||
| Amortization of intangible assets |
39,782 | 39,429 | 38,702 | |||||||||
| Impairment charges |
13,999 | 9,214 | — | |||||||||
| Acquired in-process research and development charge |
— | 13,900 | — | |||||||||
| Other than temporary loss on marketable securities |
3,406 | 4,658 | — | |||||||||
| (Gain) loss on derivatives |
(3,347 | ) | 1,664 | — | ||||||||
| Stock-based compensation |
15,075 | 13,274 | 22,293 | |||||||||
| Deferred income taxes |
2,917 | (3,457 | ) | (12,981 | ) | |||||||
| Non-cash legal expense |
— | — | 14,763 | |||||||||
| Equity in net income of unconsolidated entities |
(2,090 | ) | (908 | ) | (3,771 | ) | ||||||
| Gain on sale of marketable securities |
(792 | ) | — | — | ||||||||
| Gain on sale of subsidiary |
(2,667 | ) | — | — | ||||||||
| (Gain) loss on disposal of property, plant and equipment |
(771 | ) | — | 481 | ||||||||
| Changes in operating assets and liabilities: |
||||||||||||
| Cash collateral for reacquisition of agreement |
300,631 | (300,631 | ) | — | ||||||||
| Accounts receivable, net |
(9,844 | ) | 6,861 | (16,345 | ) | |||||||
| Related party, net |
2,198 | (6,943 | ) | 5,251 | ||||||||
| Inventories |
9,387 | 10,101 | 6,444 | |||||||||
| Prepaid expenses and other current assets |
(12,396 | ) | (5,126 | ) | (11,430 | ) | ||||||
| Accounts payable and accrued expenses |
16,984 | (8,894 | ) | 18,210 | ||||||||
| Accrued litigation costs |
— | 57,635 | — | |||||||||
| Reacquisition payable |
(268,000 | ) | 268,000 | — | ||||||||
| Deferred revenue |
(4,427 | ) | (149,996 | ) | (34,933 | ) | ||||||
| Other non-current assets and liabilities |
(7,826 | ) | (1,049 | ) | 1,584 | |||||||
| Net cash provided by (used in) operating activities |
4,488 | (315,468 | ) | (2,893 | ) | |||||||
| Cash flows from investing activities: |
||||||||||||
| Cash paid for acquisition, net of cash acquired |
(2,640 | ) | (14,998 | ) | — | |||||||
| Purchases of property, plant and equipment |
(94,473 | ) | (43,729 | ) | (40,581 | ) | ||||||
| Purchase of debt securities |
(4,740 | ) | (9,147 | ) | — | |||||||
| Purchase of other equity investments |
(10,691 | ) | (15,097 | ) | (150 | ) | ||||||
| Purchase of other assets |
(125 | ) | — | (4,174 | ) | |||||||
| Purchase of warrants and investment rights |
(260 | ) | (2,591 | ) | — | |||||||
| Proceeds from sale of subsidiary |
2,046 | — | — | |||||||||
| Proceeds from sale of marketable securities |
3,677 | — | — | |||||||||
| Net cash used in investing activities |
(107,206 | ) | (85,562 | ) | (44,905 | ) | ||||||
| Cash flows from financing activities: |
||||||||||||
| Proceeds from the exercise of stock options |
313 | 2,360 | 59 | |||||||||
| Issuance of restricted stocks |
— | — | (9 | ) | ||||||||
| Repurchase of common stock |
(874 | ) | — | — | ||||||||
| Net transactions with parent company |
— | — | 752,032 | |||||||||
| Net cash (used in) provided by financing activities |
(561 | ) | 2,360 | 752,082 | ||||||||
| Effect of exchange rates on cash and cash equivalents |
201 | (65 | ) | 316 | ||||||||
| Net (decrease) increase in cash and cash equivalents |
(103,078 | ) | (398,735 | ) | 704,600 | |||||||
| Cash and cash equivalents, beginning of year |
306,390 | 705,125 | 525 | |||||||||
| Cash and cash equivalents, end of year |
$ | 203,312 | $ | 306,390 | $ | 705,125 | ||||||
See accompanying notes to consolidated and combined financial statements
60
Table of Contents
Abraxis BioScience, Inc.
Notes to Consolidated and Combined Financial Statements
December 31, 2009
1. Description of Business
Abraxis BioScience, Inc. (formerly known as New Abraxis, Inc.) is a Delaware corporation that was formed in June 2007. Unless the context requires otherwise, references to “we,” “us” or “our” refer to Abraxis BioScience, Inc. and its subsidiaries, including its operating subsidiary Abraxis BioScience, LLC; references to “APP” refer to APP Pharmaceuticals, Inc. and its subsidiaries, including its operating subsidiary APP Pharmaceuticals, LLC (which we sometimes refer to as “APP LLC”); and references to “Old Abraxis” refer to Abraxis BioScience, Inc. (formerly known as American Pharmaceutical Partners, Inc.) prior to the 2007 separation. We are a biotechnology company, with a marketed product (Abraxane®), global ownership of our proprietary technology platform and clinical pipeline, dedicated nanoparticle manufacturing capabilities for worldwide supply integrated with in-house capabilities, including discovery, clinical drug development, regulatory and sales and marketing.
We own the worldwide rights to Abraxane®, a next-generation, nanometer-sized, solvent-free taxane that was approved by the U.S. Food and Drug Administration, or the FDA, in January 2005 for its initial indication in the treatment of metastatic breast cancer and launched in February 2005.
2007 Separation
On November 13, 2007, Abraxis BioScience, Inc. (“Old Abraxis”) was separated into two independent publicly-traded companies: one holding the former Abraxis Pharmaceutical Products business (which is referred to as the “hospital-based business”); and the other holding the former Abraxis Oncology and Abraxis Research businesses (which is referred to as the “proprietary business”). Following the separation, the proprietary business changed its name from New Abraxis, Inc. to Abraxis BioScience, Inc. and the hospital-based business is operated under the name APP Pharmaceuticals, Inc. References to the “separation,” “spin-off” or “2007 separation” refer to the transactions in which the proprietary business and hospital-based business of Old Abraxis were separated into two independent public companies. References to the historical assets, liabilities, products, businesses or activities of our company are generally intended to refer to the historical assets, liabilities, products, businesses or activities of the business as it was conducted as part of Old Abraxis prior to the separation.
In connection with the separation, stockholders of Old Abraxis as of November 13, 2007 received one share of our company for every four shares of Old Abraxis held as of that date. In addition, in connection with the separation, we entered into a separation and distribution agreement that provided for, among other things, the principal corporate transactions required to effect the separation and other specified terms governing our relationship with APP after the spin-off. We also entered into various agreements with APP, including (i) a transition services agreement pursuant to which we and APP agreed to continue to provide each other with various services on an interim, transitional basis, for periods up to 24 months depending on the particular service; (ii) a manufacturing agreement whereby we and APP agreed to manufacture Abraxane® and certain other products and to provide other manufacturing-related services for a period of four or five years; (iii) an employee matters agreement providing for each company’s respective obligations to employees and former employees who are or were associated with their respective businesses, and for other employment and employee benefit matters; (iv) various real estate leases; (v) a tax allocation agreement, and (vi) a dual officer agreement.
2. Summary of Significant Accounting Policies
Basis of Consolidation and Combination
The accompanying consolidated and combined financial statements reflect the consolidated operations of Abraxis BioScience Inc. and its subsidiaries as an independent, publicly-traded company as of and subsequent to
61
Table of Contents
November 13, 2007 and a combined reporting entity comprising the assets and liabilities that constituted the proprietary business of Old Abraxis for periods prior to November 13, 2007. The consolidated and combined financial statements include the assets, liabilities and results of operations of our wholly-owned and majority-owned operating subsidiaries and variable interest entities in which we are the primary beneficiary. Equity investments in which we have the ability to exercise significant influence over the entities but do not control are accounted for using the equity method. All material intercompany balances and transactions were eliminated in consolidation and combination.
For variable interest entities, we assess the terms of our interest in the entity to determine if we are the primary beneficiary. The primary beneficiary of a variable interest entity is the party that absorbs a majority of the entity’s expected losses, receives a majority of its expected residual returns, or both, as a result of holding a variable interest. Variable interests are ownership, contractual, or other pecuniary interests in an entity that change with changes in the fair value of the entity’s net assets excluding variable interests. We have an interest in one variable interest entity, DiThera, Inc., and because we are the primary beneficiary, the variable interest entity is consolidated in our financial statements.
The consolidated and combined financial statements for periods prior to and including November 13, 2007 do not necessarily reflect what our consolidated and combined results of operations, financial position, and cash flows would have been had we operated as an independent, publicly-traded company during the periods presented, including changes in our capitalization as a result of the separation and related transactions. To the extent that an asset, liability, revenue, or expense is directly associated with us, it is reflected in the accompanying consolidated and combined financial statements. Certain general corporate overhead and other expenses for periods prior to the separation have been allocated to us. Management believes such allocations were reasonable; however, they may not be indicative of our actual results had we been operating as an independent, publicly traded company for the periods presented prior to the separation. See Note 6—Related Party Transactions for further information regarding allocated expenses.
Reclassifications
Certain prior year amounts have been reclassified to conform to current year presentations. The reclassifications did not impact net loss or total stockholders’ equity.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the reported amount of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the consolidated financial statements. Estimates may also affect the reported amounts of revenues and expenses during the reporting period. Actual results could differ from these estimates.
Cash and Cash Equivalents
It is our policy to include cash and investments having maturity of three months or less at the time of acquisition in cash and cash equivalents.
Accounts Receivable and Concentration of Credit Risk
We typically have multi-year contractual agreements with specialty and traditional group purchasing organizations (GPOs) and comprehensive academic cancer centers to supply our product to end-users. As is traditional in the pharmaceutical industry, a significant amount of our branded pharmaceutical products are sold to end users under GPO contracts through a relatively small number of drug wholesalers and specialty distributors, which comprise the primary pharmaceutical distribution chain in the United States. Four of the
62
Table of Contents
wholesalers represented revenue in excess of 10% of total revenue. These wholesalers collectively represented approximately 76%, 79%, and 76% of our revenue in 2009, 2008, and 2007, respectively. A single wholesaler accounted for 34%, 35%, and 50% of our revenue in 2009, 2008, and 2007, respectively, with the next largest wholesaler accounting for 17%, 17%, and 13% in 2009, 2008, and 2007, respectively. These four wholesalers also represented 59% and 82% of accounts receivable at December 31, 2009 and 2008, respectively. We have a policy to routinely monitor the creditworthiness of customers, review outstanding customer balances and record allowances for bad debts as necessary. Historical credit losses have been insignificant.
Inventories
Inventories are valued at the lower of cost or market as determined under the first-in, first-out, or FIFO, method, as follows:
| December 31, | ||||||
| 2009 | 2008 | |||||
| (in thousands) | ||||||
| Finished goods |
$ | 3,908 | $ | 14,477 | ||
| Work in process |
18,235 | 9,615 | ||||
| Raw materials |
32,066 | 39,414 | ||||
| $ | 54,209 | $ | 63,506 | |||
Inventories consist of products currently approved for marketing and may from time to time, include certain products pending regulatory approval. Occasionally, we capitalize inventory costs associated with products prior to regulatory approval based on our judgment of probable future commercial success and realizable value. Such judgment incorporates our knowledge and best judgment of where the product is in the regulatory review process, our required investment in the product, market conditions, competing products and our economic expectations for the product post-approval relative to the risk of manufacturing the product prior to approval. In evaluating the market value of inventory pending regulatory approval as compared to its cost, we consider the market, pricing and demand for competing products, its anticipated selling price for the product and the position of the product in the regulatory review process. If final regulatory approval for such products is denied or delayed, we may need to provide for and expense such inventory. No inventory held at December 31, 2009 and 2008 was pending regulatory approval.
We routinely review our inventory and establish reserves when the cost of the inventory is not expected to be recovered or its product cost exceeds realizable market value. In instances where inventory is at or approaching expiry, is not expected to be saleable based on quality and control standards or for which the selling price has fallen below cost, we reserve for any inventory impairment based on the specific facts and circumstances. Provisions for inventory reserves are reflected in the financial statements as an element of cost of sales with inventories presented net of related reserves.
Property, Plant and Equipment
Property, plant and equipment is stated on the basis of cost or allocated fair value relative to our acquisitions. Provisions for depreciation are computed for financial reporting purposes using the straight-line method over the estimated useful life of the related asset and for leasehold improvements the lesser of the estimated useful life of the related asset or the term of the related lease as follows:
| Buildings and improvements |
10-30 years | |
| Corporate aircraft |
15 years | |
| Machinery and equipment |
3-10 years | |
| Furniture and fixtures |
5-7 years |
63
Table of Contents
Depreciation expense was $17.0 million, $13.6 million and $10.4 million for the years ended December 31, 2009, 2008 and 2007, respectively. Old Abraxis utilized certain assets owned by us and accordingly, the related depreciation expense of $2.2 million for the year ended December 31, 2007, was not allocated to our operating results.
Property, plant, and equipment consist of the following:
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| (in thousands) | ||||||||
| Land |
$ | 8,064 | $ | 10,594 | ||||
| Building and improvements |
88,976 | 83,421 | ||||||
| Machinery and equipment |
34,989 | 30,242 | ||||||
| Corporate aircraft |
46,552 | 46,552 | ||||||
| Furniture and fixtures |
22,317 | 11,234 | ||||||
| Construction in progress |
95,921 | 42,352 | ||||||
| 296,819 | 224,395 | |||||||
| Less accumulated depreciation |
(60,021 | ) | (57,675 | ) | ||||
| $ | 236,798 | $ | 166,720 | |||||
Impairment Charge
We review our property, plant and equipment for impairment whenever events or changes in circumstances indicate that the carrying value of the assets might not be recoverable. For the year ended December 31, 2009, based on market valuations, we recorded an impairment charge of $14.0 million on assets for which the intended use had changed. For the year ended December 31, 2008, we recognized an impairment charge of $9.2 million as a result of an anticipated sale of certain property, plant and equipment.
Investments in Unconsolidated Entities
Equity investments in which we have the ability to exercise significant influence over the operating and financial policies of the entities but do not control are accounted for using the equity method. The investments are recorded initially at cost and adjusted quarterly to recognize our proportionate share of the investees’ net income or loss. As of December 31, 2009, we had five equity method investments with an aggregate carrying value of $22.8 million. At December 31, 2008, we had one equity method investment totaling $10.2 million.
Goodwill and Intangible Assets
We perform impairment tests related to goodwill annually and whenever events or changes in circumstances suggest that it is more likely than not that the fair value of the reported units are below their carrying values. No impairment charges were required on our $241.4 million of goodwill as of December 31, 2009 and 2008.
Intangible assets are recorded at acquisition cost or allocated fair value relative to our acquisitions. Most of our intangible assets have finite lives and are amortized on a straight-line basis over the period estimated revenues are expected to be derived from such assets. The weighted average amortization period is approximately five years. We review our intangible assets for impairment whenever events or changes in circumstance indicate that the carrying amount of an intangible asset may not recoverable. No impairment charges were required for our intangible assets as of December 31, 2009 and 2008.
Marketable Securities
We determine the appropriate classification of our investments in marketable securities at the time of purchase and re-evaluate such determinations at each balance sheet date. We consider our marketable securities
64
Table of Contents
to be available-for-sale as defined in FASB ASC 320, Investments - Debt and Equity Securities. Accordingly, these investments are recorded at fair value and the unrealized gains and losses are included in accumulated other comprehensive loss in the statement of stockholders’ equity. As of December 31, 2009, we had $23.9 million of marketable securities that were classified as available-for-sale and included in “Other non-current assets”. As of December 31, 2008, total available-for-sale securities was $15.3 million, of which, $3.7 million is included in “Prepaid expenses and other current assets” and $11.6 million is included in “Other non-current assets”. Available for sale securities consisted of the following:
| December 31, 2009 | December 31, 2008 | ||||||||||||||||||
| Adjusted Cost |
Gross Unrealized Gain |
Estimated Fair Market Value |
Adjusted Cost |
Gross Unrealized Loss |
Estimated Fair Market Value | ||||||||||||||
| (in thousands) | |||||||||||||||||||
| Marketable equity securities |
$ | 2,404 | $ | 4,498 | $ | 6,902 | $ | 8,613 | $ | (2,434 | ) | $ | 6,179 | ||||||
| Marketable debt securities |
14,031 | 2,946 | 16,977 | 9,131 | — | 9,131 | |||||||||||||
| Total available for sale securities |
$ | 16,435 | $ | 7,444 | $ | 23,879 | $ | 17,744 | $ | (2,434 | ) | $ | 15,310 | ||||||
In May 2008, we purchased notes having an aggregate principal amount of $5.0 million from a privately held company. The notes are senior secured convertible notes due 2018 and earn interest at nine percent per annum. The notes are convertible into an ownership interest of up to 49% of the privately held company. In addition, at the option of the privately held company, subject to certain restrictions, we may be required to purchase additional notes having an aggregate principal amount of up to $5.0 million within six months from the closing date. This option was subsequently extended to May 2009 during the third quarter of 2008 and to December 31, 2009 (or such earlier date after June 30, 2009 as we designate on 30 days notice) in the first quarter of 2009. The privately held company did not exercise that option. The $5.0 million convertible notes are designated as available-for-sale securities and are measured at fair value each reporting period. For the year ended December 31, 2009, we recorded unrealized gains of $3.9 million based on valuation of the convertible notes.
In November 2008, we purchased notes having an aggregate principal amount of $5.0 million from a publicly-held company. The convertible notes are due in 2015 and bear interest at nine percent per annum. The notes are convertible into common shares at a conversion price of $1.25 per share. The notes have a further provision for warrants representing the right to purchase additional common stock of the company in two tranches. The first tranche is exercisable in whole or in part at $2 per share, which could result in our having a 40% ownership interest in the company. The second tranche is exercisable in whole or in part at $3 per share which could result in an increase of our ownership interest from 40% to 50%, assuming full conversion of the notes. The warrants are marked to market and are included in our earnings each period. The $5.0 million convertible notes are designated as available-for-sale securities and are measured at fair value each reporting period. For the year ended December 31, 2009, we recorded unrealized losses of $0.9 million based on valuation of the convertible notes.
In September 2009, we acquired convertible notes having an aggregate principal balance of $5 million from a privately held company. The convertible notes mature in two parts: 50% of the notes are due in January 2011 and the remaining 50% are due in 2012. The convertible notes bear interest at ten percent per annum. The $5.0 million convertible notes are designated as available-for-sale securities and are measured at fair value each reporting period.
We review quarterly, our available-for-sale securities for other than temporary declines in fair value and whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. This review includes an analysis of the facts and circumstances of each individual investment such as the severity of loss, the length of time the fair value has been below cost, the expectation for that security’s performance and the creditworthiness of the issuer. For the year ended December 31, 2009 and 2008, we recorded other than temporary losses of $3.4 and $4.7 million, respectively, on available-for-sale securities
65
Table of Contents
whose values, based on market quotation, had declined significantly below their carrying value and we assessed the decline as other-than-temporary. The loss is included in “Other expense” in the consolidated and combined statements of operations. We did not have any other-than-temporary loss in 2007.
Fair Value
FASB ASC 820, Fair Value Measurement and Disclosures, establishes a hierarchy for inputs used in measuring fair value that maximizes the use of observable inputs and minimizes the use of unobservable inputs by requiring that observable inputs be used when available. Observable inputs are inputs that market participants would use in pricing the asset or liability based on market data obtained from sources independent of us. Unobservable inputs are inputs that reflect our assumptions about the inputs that market participants would use in pricing the asset or liability and are developed based on the best information available in the circumstances. The fair value hierarchy is broken down into three levels based on the source of inputs as follows:
| Level 1 – |
Valuations based on unadjusted quoted prices in active markets that are accessible at the measurement date for identical, unrestricted assets or liabilities; | |
| Level 2 – |
Valuations based on quoted prices in markets that are not active, or financial instruments for which all significant inputs are observable; either directly or indirectly; and | |
| Level 3 – |
Valuations based on prices or valuation techniques that require inputs that are both significant to the fair value measurement and unobservable; thus, reflecting assumptions about the market participants. | |
The following fair value hierarchy tables present information about each major category of our financial assets measured at fair value on a recurring basis as of December 31, 2009 and 2008. All of the investments below reflect strategic investments.
| Basis of fair value measurements | ||||||||||||
| Quoted prices in active markets for identical assets (Level 1) |
Significant other observable inputs (Level 2) |
Significant unobservable inputs (Level 3) |
Balance as of December 31, 2009 | |||||||||
| (in thousands) | ||||||||||||
| Marketable equity securities |
$ | 6,902 | $ | — | $ | — | $ | 6,902 | ||||
| Marketable debt securities |
— | — | 16,977 | 16,977 | ||||||||
| Warrants and options |
— | 4,289 | — | 4,289 | ||||||||
| Total |
$ | 6,902 | $ | 4,289 | $ | 16,977 | $ | 28,168 | ||||
| Basis of fair value measurements | ||||||||||||
| Quoted prices in active markets for identical assets (Level 1) |
Significant other observable inputs (Level 2) |
Significant unobservable inputs (Level 3) |
Balance as of December 31, 2008 | |||||||||
| (in thousands) | ||||||||||||
| Marketable equity securities |
$ | 6,179 | $ | — | $ | — | $ | 6,179 | ||||
| Marketable debt securities |
— | — | 9,131 | 9,131 | ||||||||
| Warrants and options |
— | 1,143 | — | 1,143 | ||||||||
| Total |
$ | 6,179 | $ | 1,143 | $ | 9,131 | $ | 16,453 | ||||
In 2008, we acquired warrants and options related to our strategic investments. The warrants and options are measured at fair value at each reporting period using market prices and the change in market value is recognized as realized gains or losses and recorded in other income (expense). We recorded realized gains of $3.3 million and realized losses of $1.7 million for the years ended December 31, 2009 and 2008, respectively.
66
Table of Contents
Level 3 assets include the fair value of convertible notes. There is limited market activity for these convertible notes such that the determination of fair value requires significant judgment or estimation. The convertible notes are initially measured at acquisition cost and subsequently valued by considering, among other items, assumptions that market participants would use in their estimates of fair value, such as the collateral underlying the security, the inability to sell the investment in an active market, the creditworthiness of the issuer, and, the timing of expected future cash flows.
Fair value of other financial instruments
Other financial instruments consist mainly of cash and cash equivalents, accounts receivable and accounts payable. Cash equivalents include investments with maturities of three months or less at the time of acquisition. At December 31, 2009 and 2008, the carrying amounts of items comprising current assets and current liabilities approximate fair value due to the short-term nature of these financial instruments.
Assets and liabilities measured at fair value on a nonrecurring basis
In accordance with the provisions of FASB ASC 360, Property, Plant and Equipment, we recognized asset impairment charges of $14.0 million and $9.2 million on long-lived assets held and used for the years ended December 31, 2009 and 2008, respectively. The assets were written down to their fair values based on significant other observable inputs (level 3) and the impairment charges were included in earnings for the respective periods.
Income Taxes
In accordance with the provisions under FASB ASC 740, Income Taxes, deferred tax assets and liabilities are recognized for future tax consequences attributable to differences between the consolidated and combined financial statement carrying amounts of existing assets and liabilities and their respective tax bases as well as net operating loss and capital loss carry forwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in the financial statements in the period that includes the legislative enactment date.
During 2009 we allocated an income tax benefit to continuing operations. In accordance with FASB ASC 740, all items should be considered for purposes of determining the amount of tax benefit that results from a loss from continuing operations and that should be allocated to continued operations. FASB ASC 740 is applied by tax jurisdiction and, in periods in which there is a pre-tax loss from continuing operations and pre-tax income in another category, such as other comprehensive income, tax expense is first allocated to the other sources of income, with a related benefit recorded in continuing operations. As a result, we have allocated an income tax benefit to the loss from continuing operations based on the tax rate applicable to other comprehensive income.
We were not a taxable entity prior to the separation on November 13, 2007; however, income taxes are provided in the accompanying consolidated and combined financial statements as if we were filing a separate return. Old Abraxis will indemnify us for all tax liabilities to the extent they relate to the hospital-based generic injectable products business arising prior to the separation date.
Treasury Stock
We account for treasury stock purchases under the cost method whereby the entire cost of the acquired stock is recorded as treasury stock. Gains and losses on the subsequent reissuance of shares are credited or charged to paid in capital in excess of par value using the average-cost method.
Stock-Based Compensation
We account for stock-based compensation in accordance with FASB ASC 718, Compensation – Stock Compensation, which requires the recognition of compensation cost for all share-based payments (including
67
Table of Contents
employee stock options) at fair value. We use the straight-line attribution method to recognize share-based compensation expense over the vesting period of the award. Options currently granted generally expire ten years from the grant date and vest ratably over a four-year period, while restricted stock units currently granted generally vest ratably over a four-year period. Awards currently granted under the RSU Plan II generally vest with respect to one half of the units on the second anniversary of the merger of American BioScience, Inc. and American Pharmaceutical Partners, Inc. in 2006 and with respect to the remaining one half of the units on the fourth anniversary of the merger.
Stock-based compensation recognized under ASC 718, as well as expense associated with the retrospective application, uses the Black-Scholes option pricing model to estimate the fair value of options granted under equity incentive plans and rights to acquire stock granted under a stock participation plan. Compensation expense related to equity awards of restricted stock units is based upon the market price on the date of grant. Additionally, expense related to the RSU Plan II awards is based on the lower of the market price or $66.63 and is expensed under the liability method in accordance with ASC 718 over the applicable vesting period. Pre-tax stock-based compensation costs for the years ended December 31, 2009, 2008, and 2007, was $15.1 million, $13.3 million, and $22.3 million, respectively. See Note 15— Stock-Based Compensation for a more detailed discussion.
Foreign Currency Translation
The financial statements of our consolidated foreign subsidiaries are prepared using the subsidiaries currency as the functional currency. Balance sheet amounts are translated at the end of period exchange rate. Income statement and cash flow amounts are translated at the average exchange rate for the period. Adjustments from translating these financial statements into U.S. dollars are recognized in the equity section of the balance sheet under the caption, “Accumulated other comprehensive (loss) income.”
Abraxane® Revenue
We recognize revenue from the sale of Abraxane® when title and risk of loss have transferred to the customer, collection is reasonably assured and we have no further performance obligation. This is typically when the wholesalers receive the product. At the time of sale, as further described below, we reduce sales and provide for estimated chargebacks, bad debts, customer or Medicaid rebates, product returns and customer credits. Our methodology used to estimate and provide for these sales provisions was consistent across all periods presented. Accruals for sales allowance are presented in our financial statements as a reduction of sales and accounts receivable and, for customer or Medicaid rebates, in accrued liabilities. We regularly review information related to these estimates and adjust reserves accordingly if, and when, actual experience differs from estimates.
We have internal historical information on chargebacks, rebates and customer returns and credits, which we use as the primary factor in determining the related reserve requirements. We believe that this internal historical data, in conjunction with periodic review of available third-party data and updated for any applicable changes in available information, provides a reliable basis for such estimates.
Our sales provisions totaled $41.0 million, $32.8 million, and $30.5 million in 2009, 2008, and 2007 respectively. Related reserves totaled $13.8 million and $9.2 million at December 31, 2009 and 2008, respectively.
Co-Promotion Agreement: In April 2006, we entered into a Co-Promotion and Strategic Marketing Services Agreement with AstraZeneca UK Limited, a wholly owned subsidiary of AstraZeneca PLC. In November 2008, we entered into an agreement to end the Co-Promotion Agreement, with the agreement effectively ending in January 2009. We recorded revenue of $36.4 million in each of the years ended December 31, 2008 and 2007 relating to deferred revenue associated with an up-front cash payment from AstraZeneca of $200 million in 2006 under the Co-Promotion Agreement.
68
Table of Contents
Other Revenue
Other revenue generally consists of revenue earned from the sales of raw material products, contract manufacturing, and amounts earned from development and research to advance the commercial application of pharmaceutical compounds for third parties. Such research revenue may include up-front license fees, milestone payments and reimbursement of development and other costs, and royalties. Non-refundable up-front license fees under which we have continuing involvement in the form of development, manufacturing or commercialization are recognized as revenue ratably over either: the development period if the development risk is significant; or the estimated product useful life if the development risk has been substantially eliminated. Achievement-based milestone payments are recognized as revenue when the milestone objective is attained because the earnings process relating to such payments is complete upon attainment and because the payments are non-refundable and are not dependent upon future activities or the achievement of future objectives. These milestone payments are at risk and require substantive activity to achieve. Reimbursement of development and other costs are recognized as revenue as the related costs are incurred. Royalties are recognized as earned in accordance with the contract terms when royalties from licensees can be reliably measured and collectability is reasonably assured. Royalty estimates are made in advance of amounts collected using historical and forecasted trends.
On May 27, 2005, American BioScience, Inc. (“ABI”), the former parent of Old Abraxis, licensed the rights to Abraxane® in Japan to Taiho Pharmaceutical Co., Ltd, (“Taiho”), a subsidiary of Otsuka Pharmaceutical Ltd. Under the agreement, we and Taiho established a steering committee, comprised of three representatives from each of us and Taiho, to oversee the preclinical and clinical development of Abraxane® in Japan for the treatment of breast, lung, gastric, and other solid tumors. The steering committee is required to meet at least once annually. In addition, under the agreement, the steering committee established a development working group, comprised of an equal number of representatives from both us and Taiho, who is required to meet at least once quarterly until regulatory approval of Abraxane® is first obtained in Japan. The license provides for a non-refundable $20.0 million up-front payment, $38.5 million in potential milestone payments contingent upon the achievement of various clinical, regulatory and sales objectives, royalties based on net sales under the agreement; and an agreement under which we will supply Taiho with Abraxane®. Due to our continuing involvement under the agreement through the steering committee and the development working group, the $20.0 million non-refundable up-front payment has been deferred. At the time the up-front payment was made, we estimated that it would take approximately five years to receive regulatory approval for the first indication of Abraxane® in Japan (following which approval, the steering committee’s responsibilities would be substantially diminished) and that Abraxane® could begin to face competition in Japan as early as seven years after entering into the agreement. We believe these estimates remain accurate. Accordingly, the up-front payment is being recognized as research revenue ratably over seven years.
We recognized deferred revenue of $3.2 million, $3.1 million and $2.9 million in 2009, 2008 and 2007, respectively, for the amortization of the $22.0 million in deferred up-front payments, of which $20.0 million related to the Taiho up-front payment. Additionally, in 2007 we recorded revenue of $3.0 million relating to milestone payments received from Taiho. We did not receive a milestone payment from Taiho in 2008 and 2009. At December 31, 2009 and 2008, we had deferred revenue of $8.0 million and $12.4 million, respectively, recorded in our balance sheet.
Chargebacks
Following industry practice, we typically sell our product to independent pharmaceutical wholesalers at wholesale list price. The wholesaler in turn sells our product to an end-user, normally a hospital or alternative healthcare facility, at a lower contractual price previously established between us and the end-user via a group purchasing organization, or GPO. GPOs enter into collective purchasing contracts with pharmaceutical suppliers to secure more favorable product pricing on behalf of their end-user members.
Our initial sale to the wholesaler, and the resulting receivable, are recorded at our wholesale list price. As most of these list selling prices will be reduced to a lower end-user contract price, we then reduce reported sales and receivables by the difference between the list price and estimated end-user contract price. Then, when the
69
Table of Contents
wholesaler ultimately sells the product to the end-user at the end-user contract price, the wholesaler charges us a chargeback for the difference between the list price and the end-user contract price and such chargeback is offset against our initial estimated provision. On a monthly basis, we compare the unit and price components of our recorded chargeback provision to estimated ending wholesale and end user units in the distribution channel pending chargeback under end-user contracts and estimated end-user contract prices and adjust the chargeback provision as necessary.
The provision for chargebacks is presented in our financial statements as a reduction of sales. Our provision for chargebacks during each of the three years ended December 31, 2009 was as follows:
| Year Ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| (in thousands) | ||||||||||||
| Balance at beginning of year |
$ | 1,258 | $ | 1,172 | $ | 1,491 | ||||||
| Provision for chargebacks |
12,955 | 10,871 | 9,578 | |||||||||
| Credit or checks issued to third parties |
(12,768 | ) | (10,785 | ) | (9,897 | ) | ||||||
| Balance at end of year |
$ | 1,445 | $ | 1,258 | $ | 1,172 | ||||||
Rebates
Rebates or administrative fees are offered to certain wholesale customers, GPOs and end-user customers, consistent with pharmaceutical industry practices. Settlement of rebates and fees may generally occur from one to 15 months from date of sale. We accrue for customer rebates at the time of sale based on the contracted rates and historical redemption rates. Upon receipt of chargeback, due to the availability of product and customer specific information on these programs, we establish a provision for customer rebates based on the specific terms of each agreement. Rebates are reflected in the financial statements as accrued liabilities. Our provision for customer rebates during each of the three years ended was as follows:
| Year Ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| (in thousands) | ||||||||||||
| Balance at beginning of year |
$ | 5,552 | $ | 6,206 | $ | 3,296 | ||||||
| Provision for customer rebates |
23,868 | 19,096 | 17,721 | |||||||||
| Credit issued to third parties |
(19,907 | ) | (19,750 | ) | (14,811 | ) | ||||||
| Balance at end of year |
$ | 9,513 | $ | 5,552 | $ | 6,206 | ||||||
Returns and Credits, Cash Discounts and Bad Debts
Consistent with industry practice, our return policy permits our customers to return product within a window of time before and after the expiration of product dating. We provide for product returns and other customer credits at the time of sale by applying historical experience factors, generally based on our historic data on credits issued by credit category or product, relative to related sales and we provide specifically for known outstanding returns and credits. The accrual for customer credits and product returns is presented in the financial statements as a reduction of revenue and accounts receivable. Our provision for customer credits and product returns during each of the three years ended was as follows:
| Year Ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| (in thousands) | ||||||||||||
| Balance at beginning of year |
$ | 695 | $ | 414 | $ | 465 | ||||||
| Provision for customer credits and product returns |
1,494 | 569 | 129 | |||||||||
| Credit issued to third parties |
(1,163 | ) | (288 | ) | (180 | ) | ||||||
| Balance at end of year |
$ | 1,026 | $ | 695 | $ | 414 | ||||||
70
Table of Contents
We establish a reserve for bad debts based on general and identified customer credit exposure. Our bad debts expense for the year ended December 31, 2009 was $0.4 million and less than $0.2 million in the prior two years.
Shipping and Handling Fees and Costs
Shipping and handling fees billed to customers are recognized in net revenue. Shipping and handling costs are included in cost of sales.
Research and Development Costs
Research and development costs are expensed as incurred or consumed and consist of pre-production manufacturing scale-up and related salaries and other personnel-related expenses, depreciation, facilities, clinical material and production costs and professional services.
Acquired In-Process Research and Development
For acquisitions prior to January 1, 2009, the estimated fair value of acquired in-process research and development projects, which had not reached technological feasibility at the date of acquisition and which did not have an alternative future use, were immediately expensed. Beginning January 1, 2009, newly issued accounting standards require that the fair value of acquired in-process research and development projects acquired in a business combination be capitalized at the acquisition date.
Clinical Trial Agreements
We have entered into various clinical trial agreements with third parties for the planning, management and execution of clinical trials. We record accruals based on patient enrollment for estimated clinical study costs related to work performed by participating hospitals and clinics. We also account for other costs associated with the clinical trials as incurred. These costs include activities provided by contract research organizations, incremental patient medical costs and costs associated with laboratory services.
Subsequent Events
We have evaluated subsequent events through the date of issuance of our financial statements in this Form 10-K.
Recent Accounting Pronouncements
Effective January 1, 2009, we adopted new guidance under FASB ASC 810, Consolidation, relating to the accounting and reporting of noncontrolling interest in a subsidiary. The noncontrolling interest is the portion of equity in a subsidiary not attributable directly or indirectly to a parent. The standard requires the noncontrolling interest to be presented separately from the parent’s equity in the consolidated statements of financial position and statements of shareholders’ equity. Additionally, the consolidated net income attributable to the parent and to the noncontrolling interest is presented separately on the consolidated statements of income. Adoption of this standard did not have a material impact on our consolidated financial statements.
Effective January 1, 2009, we adopted new guidance under ASC 808-10, Collaborative Arrangement, which defines collaborative arrangements and establishes the reporting requirements for transactions between participants in a collaborative arrangement, and between participants in the arrangement and third parties. A collaborative arrangement is defined as one in which the participants are actively involved and are exposed to significant risks and rewards that depend on the ultimate commercial success of the endeavor. For revenues and costs incurred with third parties in connection with collaborative arrangements, the participant that is deemed to be the principal participant in the transaction shall record that transaction on a gross basis on its financial statements. Payments to or from collaborators are evaluated and presented based on the nature of the arrangement and its terms, the nature of the entity’s business, and whether those payments are within the scope of other accounting literature. The nature and purpose of collaborative arrangements are disclosed along with the accounting policies and the classification of significant financial statement amounts related to the arrangements.
71
Table of Contents
Activities in arrangements conducted in a separate legal entity are accounted for under other accounting literature, however, required disclosure applies to the entire collaborative agreement. Adoption of this standard did not have a material impact on our consolidated financial statements.
In June 2009, the FASB issued a new accounting standard that amends guidance regarding consolidation of variable interest entities to address the elimination of the concept of a qualifying special purpose entity (the “QSPE”). This standard also replaces the quantitative-based risks and rewards calculation for determining which enterprise has a controlling financial interest in a variable interest entity with an approach focused on identifying which enterprise has the power to direct the activities of the variable interest entity, and the obligation to absorb losses of the entity or the right to receive benefits from the entity. Additionally, the standard requires any enterprise that holds a variable interest in a variable interest entity to provide enhanced disclosures that will provide users of financial statements with more transparent information about an enterprise’s involvement in a variable interest entity. This standard is effective for us for interim and annual reporting periods beginning on or after January 1, 2010. We are assessing the impact of this adoption on our consolidated financial statements.
In August 2009, the FASB issued additional guidance on the fair value measurement of liabilities. The amendments to ASC 820, Measuring Liabilities at Fair Value, provides clarification that in circumstances in which a quoted price in an active market for the identical liability is not available, a reporting entity is required to measure fair value using one or more of the specified techniques. These amendments are effective for us beginning October 1, 2009. Adoption of this standard did not have a material impact on our consolidated financial statements.
In October 2009, the FASB issued a new accounting standard which amends guidance on accounting for revenue arrangements involving the delivery of more than one element of goods and/or services. The standard amends the criteria for separating consideration in multiple-deliverable arrangements and establishes a selling price hierarchy for determining the selling price of a deliverable. The amendments will eliminate the residual method of allocation and require that arrangement consideration be allocated at the inception of the arrangement to all deliverables using the relative selling price method. The standard also significantly expands the disclosures related to a vendor’s multiple-deliverable arrangement. The standard is effective on a prospective basis for revenue arrangements entered into or materially modified in fiscal years beginning on or after June 15, 2010. Alternatively, adoption may be on a retrospective basis, and early application is permitted. Adoption of this standard is not expected to have a material impact on our consolidated financial statements.
3. Proposed Spin-Off
In January 2009, we announced that our board of directors approved a plan to spin-off its newly-formed subsidiary, Abraxis Health, Inc., as a new independent, stand-alone company holding a significant portion our drug discovery, manufacturing and development business. We currently anticipate the spin-off will be completed in 2010. If the spin-off occurs, our stockholders would own (i) shares of Abraxis Health and (ii) shares of our common stock, and we would continue to operate our existing business, excluding the drug discovery, manufacturing and development business to be held by Abraxis Health.
The proposed spin-off is subject to, among other things, final approval by our board of directors and the effectiveness of the registration statement registering the common stock of Abraxis Health to be distributed to our stockholders in connection with the spin-off. Our board of directors, in its sole discretion and for any reason or no reason at all, may amend, modify or abandon the proposed spin-off without liability at any time prior to the time the spin-off is completed. Approval by our stockholders is not required as a condition to the consummation of the proposed spin-off.
72
Table of Contents
4. Acquisitions, Collaborative Agreements and Other Transactions
Investments in Expression Pathology
In April 2009, we acquired preferred stock in Expression Pathology, Inc. (EPI), providing us with a 61% ownership interest, for an aggregate purchase price of $6.7 million. EPI is a developer of technology for tissue protein analysis and is developing proprietary, personalized medicine clinical assays that relate measurement of protein biomarkers to specific patient treatment decisions. EPI’s proprietary technologies include the Liquid Tissue® technology for the extraction of proteins from formalin-fixed tissue and the Director™ technology for the laser microdissection of tissue. We accounted for EPI as a business combination and the assets, liabilities and results of operation of EPI are consolidated in our financial statements from the date of acquisition.
The following table summarizes the estimated fair values of the assets acquired and liabilities recorded after our investment at the acquisition date on April 1, 2009 (in thousands):
| Current assets (including cash of $6.6 million) |
$ | 6,675 | |
| Property, plant and equipment |
72 | ||
| Intangible and other long-term assets |
8,861 | ||
| Total assets |
$ | 15,608 | |
| Current liabilities |
$ | 621 | |
| Deferred tax liability |
2,899 | ||
| Other long-term liabilities |
88 | ||
| Total liabilities |
3,608 | ||
| Non-controlling interest |
5,275 | ||
| Abraxis’ investment in Expression Pathology |
6,725 | ||
| Total liabilities and equity |
$ | 15,608 | |
Acquired intangible assets of $8.9 million represent technologies used by EPI to develop new assays for use in testing drug efficacy of certain drugs with certain cancer types. The intangible assets are all definite-lived intangibles and have weighted average lives of approximately 16 years.
The fair value of the 48% noncontrolling interest in EPI was estimated to be $5.3 million at the time of the acquisition and was determined using a cash flow model. As EPI is a private company, the fair value is based on significant inputs that are not observable in the market and thus represents a Level 3 measurement as defined in FASB ASC 820, Fair Value Measurements and Disclosures.
Acquisition of Shimoda Biotech and Platco Technologies
In April 2008, we acquired Shimoda Biotech (Pty) Ltd and its subsidiary, Platco Technologies (Pty) Ltd, located in Plettenberg Bay, South Africa. Shimoda Biotech focuses on the development of new pharmaceutical products by combining successful off-patent molecules with a novel cyclodextrin drug delivery platform, seeking to exploit the faster onset and improved bioavailability characteristics of that platform. Platco Technologies focuses on the development of novel platinum-based anti-cancer drugs. Shimoda’s first cyclodextrin-based product, Dyloject® (diclofenac sodium solution for injection), is an injectable painkiller for the treatment of post-surgical pain. Dyloject® is a solubilized intravenous formulation of diclofenac. Diclofenac is a non-steroidal anti-inflammatory drug (NSAID). Dyloject® was launched in December 2007 in the United Kingdom by Javelin Pharmaceuticals under an exclusive worldwide license agreement pursuant to which Shimoda Biotech will receive milestone payments and royalties. In January 2009, Javelin announced that it had entered into an exclusive European marketing partnership for Dyloject® with Therabel Pharma N.V. In December 2009, Javelin announced that it had entered into a definitive agreement with Myriad Pharmaceuticals to acquire Javelin
73
Table of Contents
Pharmaceuticals. In February 2010, Javelin announced that its New Drug Application (NDA) submitted on December 2009 to the US Food and Drug Administration (FDA) for Dyloject® had been accepted for formal review.
Under the terms of the agreement, we acquired 100% of the equity of both Shimoda Biotech and Platco Technologies for an initial upfront payment at closing of $15.1 million, plus potential additional payments of up to $14.6 million upon the achievement of specified milestones. We recorded milestone payments of $4.6 million in 2009. We accounted for the purchase as a business combination and the purchase price paid, including transaction costs of $0.3 million, was allocated to the net assets and liabilities acquired based on their respective fair values at the acquisition date, which included $8.0 million of intangible assets associated with Dyloject® and $6.9 million in liabilities relating to net milestones payments to be paid in the future. The $8.0 million of identifiable intangible assets represents existing technology with an estimated amortizable life of seventeen years. Additionally, $13.9 million of the purchase price was expensed as in-process research and development for projects that, as of the acquisition date, had not yet reached technological or regulatory feasibility and had no alternative future uses in their current states. The results of Shimoda Biotech and Platco Technologies have been included in the condensed consolidated financial statements as of the acquisition date.
Investment and License Agreements with ProMetic Life Sciences Inc.
In September 2008, we entered into agreements with ProMetic Life Sciences Inc. (ProMetic) to develop and commercialize four biopharmaceutical products targeting underserved medical conditions. We entered into the following agreements: (i) a securities purchase agreement, (ii) a license agreement, (iii) a supply and license agreement, (iv) an exclusive manufacturing agreement, and (v) a services agreement. The transaction included an initial investment in ProMetic of $7 million and optional future investment rights of up to $25 million. Of the $7 million initial investment, $5.2 million was allocated to the purchase of ProMetic’s common stock and $1.8 million was allocated to the future investment rights option. We will have access to ProMetic’s proprietary protein technologies to commercialize the biopharmaceuticals and will fund all development costs to regulatory approval. For the years ended December 31, 2009 and 2008, we incurred $3.6 and $1.1 million, respectively, in development costs under the license agreements. We may also make potential payments upon achievement of specified milestones and make royalty payments to ProMetic based on the net sales of the four potential new products. Additionally, ProMetic will perform product development activities on behalf of Abraxis under the service agreement.
In December 2009, we entered into a collaboration agreement with ProMetic to develop and commercialize various applications deriving from ProMetic’s prion capture technology platform. As part of the agreement, we will share equally with ProMetic the costs for the development of such new applications and the revenues arising from their commercialization.
AstraZeneca UK Limited
In November 2008, we entered into an agreement with AstraZeneca UK Limited under which we would re-acquire the exclusive rights to market Abraxane® in the United States. In accordance with this agreement, the co-promotion agreement was terminated effective January 2009, and we agreed to pay AstraZeneca a $268 million fee by March 31, 2009. The $268 million fee was accrued in our consolidated balance sheet as of December 31, 2008 and the remainder of the previously deferred revenue of $109.1 million was charged to income and netted against the $268 million fee charge reflected in our 2008 consolidated statement of operations as the negotiations were substantively concluded in December 2008 and standby letters of credit were obtained to cover the obligations. We provided $286 million ($268 million for termination payment and $18 million for estimated final payments due under the Co-Promotion Agreement) in irrevocable standby letters of credit to secure the future payments under the agreement. The letters of credits were collateralized by $300.6 million of cash, which was included in “Cash collateral for reacquisition of agreement” in the balance sheet as of December 31, 2008. In March 2009, we made the final payments under the agreement and were released from our obligation to maintain the cash collateral.
74
Table of Contents
Other Agreements
In December 2008, we entered into an agreement to provide a grant to a non-profit US-based incubator for biomedical start-ups. The transaction included an initial grant by us of $4 million, which was charged to research and development expense and additional funding of $0.6 million per quarter for a total of $14 million over 5 years subject to our approval. Additionally, an optional future payment of $3.5 million is possible upon approval of an additional project by us. Our executive chairman, Patrick Soon-Shiong, M.D., serves on the board of this non-profit entity.
In December 2008, we purchased preferred stock of DiThera, Inc. representing 50% of DiThera’s outstanding capital stock for $5 million. We have determined that DiThera is a variable interest entity and we are the primary beneficiary. Therefore, we have consolidated DiThera in our financial statements.
5. Sale of Assets
In March 2009, we sold the shares of our wholly owned subsidiary, Abraxis BioScience Switzerland GmbH, which held the plant, property and equipment located in Barbengo, Switzerland. We received $2.0 million in proceeds from the sale and recorded a gain of approximately $2.7 million, of which $0.7 million were cumulative foreign currency gains. The gain is included in selling, general and administrative expenses in the consolidated and combined statements of operations. We initially recorded an impairment charge of $9.2 million for these assets in the third quarter of 2008. We did not have any financial ties with the transacting party or any continuing involvement in the subsidiary subsequent to the sale.
6. Related Party Transactions
We had payables of $0.3 million and receivables of $1.9 million from related parties at December 31, 2009 and 2008, respectively. Receivables from related parties at December 31, 2009 included $0.1 million due from Drug Source Company and $0.4 million payables to APP. At December 31, 2008, we had receivables of $1.6 million from Drug Source Company and $0.3 million from APP.
Until May 2008, our then chief executive officer and chairman of our board of directors, Dr. Soon-Shiong, and our former chief financial officer, were also the chief executive officer and chief financial officer, respectively of APP. Dr. Soon-Shiong also owned approximately 80% of the outstanding capital stock of APP and served as its chairman of the board until September 2008.
In September 2008, APP was acquired by Fresenius Kabi Pharmaceutical Holding, Inc., a subsidiary of Fresenius SE. With the purchase, Dr. Soon-Shiong ceased being a shareholder of and having a controlling interest in APP. However, he became a non-controlling member of the Fresenius Kabi Board of Directors. APP continues to be a party to the shared services, the manufacturing and other agreements and leases entered into in connection with the separation of us and APP.
75
Table of Contents
Transactions with APP Pharmaceuticals, Inc.
In connection with the separation on November 13, 2007, we entered into a number of agreements that governs the relationship between APP and us for a period of time after the separation. The agreements were entered into while we were still a wholly owned subsidiary of Old Abraxis. These agreements include: (i) a tax allocation agreement, (ii) a dual officer agreement, (iii) an employee matters agreement, (iv) a transition services agreement, (v) a manufacturing agreement, and (vi) various real estate leases. Transactions relating to these agreements recorded in our consolidated and combined statement of operations are summarized in the following table:
| Year Ended December 31, | ||||||||||
| 2009 | 2008 | 2007 | ||||||||
| (in thousands) | ||||||||||
| Other revenue: |
||||||||||
| Net rental income |
$ | 2,710 | $ | 2,588 | $ | 216 | ||||
| Cost of sales: |
||||||||||
| Manufacturing and distribution |
1,398 | 1,676 | 33 | |||||||
| Facility management fees |
2,750 | 3,000 | 250 | |||||||
| Selling, general and administrative expenses: |
||||||||||
| Net general and administrative costs |
1,204 | 549 | (111 | ) | ||||||
Tax Allocation Agreement
The tax allocation agreement allocates the liability for taxes. Under the tax allocation agreement, APP is responsible for and has agreed to indemnify us against all tax liabilities to the extent they relate to APP’s hospital-based business, and we are responsible for and have agreed to indemnify APP against all tax liabilities to the extent they relate to our proprietary products business. The tax allocation agreement also provides the extent to which, and the circumstances under which, the parties would be liable if the distribution were not to constitute a tax-free distribution under Section 355 and Section 368(a)(1)(D) of the Internal Revenue Code. In general, we are required to indemnify APP for any taxes resulting from a failure of the distribution to so qualify, unless such failures results solely from APP’s specified acts.
Dual Officer Agreement
We entered into an agreement with APP under which we and APP acknowledged and agreed that the chief executive officer and chief financial officer of our company may serve as an officer of both companies and receive compensation from either or both companies. APP also acknowledged and agreed in this agreement that neither the chief executive officer nor the chief financial officer of our company will have any obligation to present to APP any business or corporate opportunity that may come to his or her attention, other than certain business opportunities. This agreement was effectively terminated when our former chief executive officer and former chief financial officer resigned from APP.
Employee Matters Agreement
We entered into an employee matters agreement with APP, providing our respective obligations to employees and former employees who are or were associated with our respective businesses, and for other employment and employee benefit matters. Under the terms of the employee matters agreement, APP generally assumed all liabilities and obligations related to employee benefits for current and former employees who are or were associated with the hospital-based business, and we generally assumed all liabilities and obligations related to employee benefits for current and former employees who are or were associated with the proprietary products business. Under the employee matters agreement, the parties have agreed to reimburse one another for all indemnifiable losses that each may incur on behalf of the other as a result of any of the benefit plans or any of the termination or severance obligations.
76
Table of Contents
Transition Services Agreement
Pursuant to the transition services agreement we and APP agreed to continue to provide to one another various services on an interim, transitional basis, for periods up to 24 months depending on the particular service. Services that we provide to APP include legal services (e.g., assistance with SEC filings, labor and employment matters, and litigation support), financial services and corporate development. Services that APP provide to us include information technology support, tax services, legal services, accounts payable services, internal audit services, accounts receivable services, general accounting related assistance, corporate insurance and franchise tax services, human resources, customer operations, sales and marketing support, corporate purchasing and facility services and regulatory support (including assistance with state and federal license renewals). Payments made under the transition services agreement are based on the providing party’s actual costs of providing a particular service.
Manufacturing Agreement
We entered into a manufacturing agreement with APP for the manufacture of Abraxane® and certain of our pipeline products by APP for us at the Melrose Park and Grand Island manufacturing facilities. Under the terms of the manufacturing agreement, we would perform certain manufacturing activities with respect to Abraxane® or other pipeline products, principally related to the formulation and compounding of the active pharmaceutical ingredients in such products, and APP will undertake the remainder of the manufacturing processes. As part of the manufacturing services, APP would provide us with training related to proper manufacturing practices and other related training, assistance with quality assurance and control and information technology support related to manufacturing operations. With regard to the Melrose Park and Grand Island facilities, APP is responsible for obtaining and maintaining necessary approvals to manufacture Abraxane® or other pipeline products in compliance with the regulatory requirements applicable as to the jurisdictions in which such products are sold, subject to the right to receive reimbursement from us for the costs of such approvals in certain circumstances. We are responsible for obtaining and maintaining the remainder of the regulatory approvals required to sell and distribute Abraxane® both in the United States and in other jurisdictions and we are responsible for the final release of the product for sale at the completion of the manufacturing process. In the manufacturing agreement, APP agreed with us to cap the manufacturing growth over the term of the agreement of Abraxane® and other pipeline products that APP is required to manufacture under the agreement. Further, in the event of capacity constraints at the Melrose Park or Grand Island facilities, the manufacturing agreement provides that the available capacity will be prorated between us and APP according to the parties’ then current use of APP’s manufacturing capacity. While the manufacturing agreement allows us to override these proration provisions, we may only do so by paying APP additional fees under the manufacturing agreement. The fee we would be required to pay APP to override the proration provisions of the manufacturing agreement is equal to the profit lost by APP as a result of our election to override the proration provisions.
We would pay APP a customary margin on its manufacturing costs. In addition, during each of the first three years of the manufacturing agreement, we will pay APP a facility management fee equal to $3 million. The amount of this fee may be offset to the extent APP employees are transferred to us based upon the amount of compensation paid to such transferred employees. The term of the manufacturing agreement will end on December 31, 2011, which will be automatically extended for one year if either APP exercises its right to extend the lease on our Melrose Park manufacturing facility for an additional year or we exercise our right to extend the lease for APP’s Grand Island manufacturing facility for an additional year. (See Real Estate Leases below).
The manufacturing agreement includes customary confidentiality provisions pursuant to which we and APP will keep confidential all information of the other party, subject to certain exceptions.
In addition, the manufacturing agreement contains the following indemnification provisions. We will indemnify APP, its affiliates and its officers, directors and employees and agents (which are referred to as the “APP indemnified parties”) from any damages incurred by or assessed against them resulting from a third-party claim caused by or alleged to be caused by (i) our failure to perform our obligations under the manufacturing
77
Table of Contents
agreement (ii) any product liability claim arising from the negligence, fraud or intentional misconduct of our or any of our affiliates or any product liability claim arising from our manufacturing obligations (or any failure or deficiency in our manufacturing obligations) under the manufacturing agreement; (iii) any claim that the manufacture, use or sale of Abraxane® or our pipeline products infringes a patent or any other proprietary right of a third party; or (iv) any recall, product liability claim or other third-party claim not arising from the gross negligence or bad faith of, or intentional misconduct or intentional breach of the manufacturing agreement by, the APP indemnified parties by reason of the $100 million limitation of liability described below. We will also indemnify the APP indemnified parties for liabilities that they become subject to as a result of their activities under the manufacturing agreement and for which they are not responsible under the terms of the manufacturing agreement. APP will indemnify us, and our affiliates, and their respective officers, directors, employees and agents from any damages incurred or assessed against them resulting from a third-party claim caused by or alleged to be caused by (i) APP’s gross negligence, bad faith, intentional misconduct or intentional failure to perform its obligations under the manufacturing agreement; and (ii) any product liability claim arising from the gross negligence or bad faith of, or intentional misconduct or intentional breach of the manufacturing agreement by, APP or its affiliates. The APP indemnified parties will not have any liability for monetary damages to our affiliates or third parties in connection with the manufacturing agreement for damages in excess of $100 million in the aggregate, except to the extent that the damages are the result of (i) one of APP’s or our executive officers in bad faith affirmatively instructing their employees to intentionally breach APP’s obligation to manufacture Abraxane® or pipeline products under the manufacturing agreement or (ii) any intentional failure by APP, in bad faith, to cure any material breach of its obligation to manufacture Abraxane® or pipeline products under the manufacturing agreement following notice of this breach.
Real Estate Leases
Following the 2007 separation, we own the manufacturing facility located on Ruby Street, Melrose Park, Illinois, and the research and development and the warehouse facility, both located in the same building on N. Cornell Avenue, Melrose Park, Illinois. APP owns the manufacturing facility located in Grand Island, New York. In connection with the separation, the parties entered into a series of lease agreements to facilitate continued production of their respective pharmaceutical products while a manufacturing transition plan is being implemented. Under the terms of the lease agreements, we lease the Ruby Street facility, consisting of office, warehouse and pharmaceutical manufacturing space, to APP. The initial term of the lease expires on December 31, 2011 and may be renewed at the option of APP for one additional year if APP is manufacturing a certain level of its products at the Ruby Street facility in the period prior to the expiration of the lease. In order to provide sufficient warehouse space to APP during the term of the Ruby Street lease, we also leased the Cornell Warehouse facility to APP. The initial term of the lease will be until December 31, 2011, and may be renewed at APP’s option for one additional year if the lease for the Ruby Street manufacturing facility is extended. We also leased the Cornell research and development facility to APP. The initial term of the lease will be until December 31, 2010 and may be terminated upon twelve months written notice from and after January 1, 2009. This lease has no option to extend the term of the lease.
We leased a portion of APP’s Grand Island facility, consisting of pharmaceutical manufacturing space, to allow us to perform our obligations under the manufacturing agreement. The initial term of the lease expires on December 31, 2011, and may be renewed at our option for one additional year if we have not received regulatory approvals from the EMEA to manufacture Abraxane® in at least two facilities (not including the Grand Island facility) by June 30, 2011.
Allocated Expenses
Prior to the separation, our product development and general and administrative functions were fully integrated with Old Abraxis, including product development, accounting, treasury, payroll, internal audit, information technology, corporate income tax, legal services and investor relations. In addition, Old Abraxis provided manufacturing services to us at cost. After the 2007 separation, certain of these arrangements (see
78
Table of Contents
above) continued on a temporary basis for a period generally not to exceed 24 months, or four or five years in the case of manufacturing-related activities and lease arrangements. For periods prior to the 2007 separation, the consolidated and combined financial statements reflect the application of certain estimates and allocations and management believes the methods used to allocate these operating expenses are reasonable. The allocation methods include relative head count, sales, square footage and management knowledge. These allocated operating expenses totaled $33.3 million for the year ended December 31, 2007.
7. Intangible Assets
As of December 31, 2009 and 2008, our goodwill had a carrying value of $241.4 million resulting from the merger of American BioScience, Inc. and American Pharmaceutical Partners, Inc. and the carrying value has not changed since that date. Goodwill is not deductible for income tax purposes.
The following table reflects the components of intangible assets, which have finite lives:
| Weighted- average Remaining Life |
December 31, 2009 | December 31, 2008 | ||||||||||||||||||
| Gross Carrying Amount |
Accumulated Amortization |
Net Book Value |
Gross Carrying Amount |
Accumulated Amortization |
Net Book Value | |||||||||||||||
| (in thousands) | ||||||||||||||||||||
| Core technology |
3.3 yrs | $ | 220,202 | $ | 116,653 | $ | 103,549 | $ | 220,202 | $ | 85,197 | $ | 135,005 | |||||||
| Customer relationships |
3.3 yrs | 23,588 | 12,497 | 11,091 | 23,588 | 9,127 | 14,461 | |||||||||||||
| Tradename |
3.3 yrs | 26,478 | 14,028 | 12,450 | 26,478 | 10,245 | 16,233 | |||||||||||||
| Other |
15.3 yrs | 19,273 | 1,730 | 17,543 | 10,136 | 544 | 9,592 | |||||||||||||
| Total |
$ | 289,541 | $ | 144,908 | $ | 144,633 | $ | 280,404 | $ | 105,113 | $ | 175,291 | ||||||||
Amortization expense on intangible assets was $39.8 million, $39.4 million, and $38.7 million for the years ended December 31, 2009, 2008 and 2007, respectively. Estimated amortization expense for each of the three succeeding years (2010 – 2012) is $39.8 million, and for the two years following (2013 and 2014) is $12.4 million and $1.1 million, respectively. At December 31, 2009, the weighted average expected life of intangible assets was approximately 4.7 years.
8. Accrued Liabilities
Accrued liabilities consisted of the following at:
| December 31, | ||||||
| 2009 | 2008 | |||||
| (in thousands) | ||||||
| Sales and marketing |
$ | 5,226 | $ | 2,631 | ||
| Marketing rebates |
9,513 | 5,552 | ||||
| Payroll and employee benefits |
23,660 | 14,797 | ||||
| Legal |
30,491 | 17,632 | ||||
| Liability award plan |
7,986 | — | ||||
| Milestone |
— | 3,950 | ||||
| Other |
11,847 | 8,458 | ||||
| $ | 88,723 | $ | 53,020 | |||
79
Table of Contents
9. Income Taxes
Pretax loss summarized by region is as follows:
| Year Ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| (in thousands) | ||||||||||||
| Domestic |
$ | (94,444 | ) | $ | (252,260 | ) | $ | (38,312 | ) | |||
| Foreign |
(12,904 | ) | (26,449 | ) | (11,244 | ) | ||||||
| Total pretax loss from continuing operations |
$ | (107,348 | ) | $ | (278,709 | ) | $ | (49,556 | ) | |||
The (benefit ) provision for income taxes consists of the following:
| Year Ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| (in thousands) | ||||||||||||
| Current: |
||||||||||||
| Federal |
$ | (1,931 | ) | $ | 678 | $ | 4,168 | |||||
| State |
285 | 841 | 861 | |||||||||
| Foreign |
57 | — | — | |||||||||
| Total current |
(1,589 | ) | 1,519 | 5,029 | ||||||||
| Deferred: |
||||||||||||
| Federal |
(21,457 | ) | (91,078 | ) | (11,766 | ) | ||||||
| State |
1,684 | (9,863 | ) | (3,511 | ) | |||||||
| Foreign |
(8,988 | ) | (2,565 | ) | — | |||||||
| Valuation allowance |
27,770 | 100,049 | 2,296 | |||||||||
| Total deferred |
(991 | ) | (3,457 | ) | (12,981 | ) | ||||||
| Total benefit for income taxes |
$ | (2,580 | ) | $ | (1,938 | ) | $ | (7,952 | ) | |||
Pursuant to FASB ASC 740-30, Income Taxes, a provision should be made for the tax effect of the earnings of the foreign subsidiaries that are not permanently invested outside the United States. It is our intention that earnings of our foreign subsidiaries are permanently invested outside the U.S.; therefore no deferred tax asset was set up against the cumulative earnings of our foreign subsidiaries.
A reconciliation of the federal statutory rate to our effective tax rate is as follows:
| Year Ended December 31, | |||||||||
| 2009 | 2008 | 2007 | |||||||
| Tax provision at statutory federal rate |
(35.0 | )% | (35.0 | )% | (35.0 | )% | |||
| State income taxes |
(2.5 | ) | (3.5 | ) | (4.2 | ) | |||
| Permanent items |
2.0 | 1.0 | — | ||||||
| In-process research and development |
— | 1.8 | — | ||||||
| Research and development credits |
— | (0.4 | ) | (4.6 | ) | ||||
| Stock based compensation |
1.1 | 0.4 | 2.4 | ||||||
| Net operating losses utilized by APP Pharmaceuticals, Inc. |
— | — | 17.9 | ||||||
| Change in state tax rate |
4.5 | — | — | ||||||
| OCI adjusted |
(2.6 | ) | — | — | |||||
| FIN 48 reserve |
1.0 | — | — | ||||||
| Other |
3.2 | (0.9 | ) | 2.9 | |||||
| Change in valuation allowance |
25.9 | 35.9 | 4.6 | ||||||
| Effective tax rate |
(2.4 | )% | (0.7 | )% | (16.0 | )% | |||
80
Table of Contents
During 2009, we allocated an income tax benefit to continuing operations. In accordance with FASB ASC 740, all items should be considered for purposes of determining the amount of tax benefit that results from a loss from continuing operations and that should be allocated to continued operations. FASB ASC 740 is applied by tax jurisdiction and, in periods in which there is a pre-tax loss from continuing operations and pre-tax income in another category, such as other comprehensive income, tax expense is first allocated to the other sources of income, with a related benefit recorded in continuing operations. As a result, we have allocated an income tax benefit to the loss from continuing operations based on the tax rate applicable to other comprehensive income.
During 2008, we acquired Shimoda Biotech, Ltd, a South African corporation. As part of our purchase price allocation, Shimoda’s existing intangibles were grossed up to fair market value. Because the purchase price allocation to intangibles is not recognized under South African tax rules, a deferred tax liability was established.
We were not a taxable entity prior to the separation on November 13, 2007; however, income taxes are provided in the accompanying combined and consolidated financial statements as if we were filing a separate return. Old Abraxis will indemnify us for all tax liabilities to the extent they relate to the hospital-based generic injectable products business arising prior to the separation date.
Current and noncurrent deferred tax assets/(liabilities) consist of the following:
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| (in thousands) | ||||||||
| Current deferred tax assets: |
||||||||
| Inventory |
$ | 5,525 | $ | 5,022 | ||||
| Other accruals and reserves |
5,810 | 3,250 | ||||||
| Deferred revenue |
1,016 | 1,062 | ||||||
| Accrued legal |
32,854 | 28,034 | ||||||
| Reacquisition payable |
40,377 | 31,204 | ||||||
| Total current deferred tax assets |
85,582 | 68,572 | ||||||
| Valuation allowance |
(60,072 | ) | (41,814 | ) | ||||
| Net current deferred tax assets |
$ | 25,510 | $ | 26,758 | ||||
| Noncurrent deferred tax liabilities: |
||||||||
| Net operating loss and credit carryforwards |
$ | 42,953 | $ | 3,637 | ||||
| Stock based compensation |
14,684 | 12,315 | ||||||
| Deferred revenue |
1,589 | 2,733 | ||||||
| Activity from partnerships |
(94 | ) | 2,189 | |||||
| Unrealized losses on marketable securities |
1,610 | 1,660 | ||||||
| Impairments of investments and other assets |
6,943 | 5,296 | ||||||
| Reacquisition payable |
30,210 | 72,809 | ||||||
| Intangible amortization |
(39,472 | ) | (55,374 | ) | ||||
| Purchase accounting intangible |
(4,996 | ) | (2,235 | ) | ||||
| Depreciation |
(11,609 | ) | (5,075 | ) | ||||
| Total noncurrent deferred tax liabilities |
41,818 | 37,955 | ||||||
| Valuation allowance |
(71,325 | ) | (61,813 | ) | ||||
| Net noncurrent deferred tax liabilities |
$ | (29,507 | ) | $ | (23,858 | ) | ||
| Net deferred tax (liabilities) assets |
$ | (3,997 | ) | $ | 2,900 | |||
Deferred tax assets are reduced by a valuation allowance if, based on the weight of available evidence, it is more likely than not that some portion or all of the deferred tax assets will not be realized. Both positive and
81
Table of Contents
negative evidence are considered in forming our judgment as to whether a valuation allowance is appropriate, and more weight is given to evidence that can be objectively verified. Valuation allowances are reassessed whenever there are changes in circumstances that may cause a change in judgment.
As of December 31, 2009, management believed it is more likely than not that a portion of the deferred tax assets will not be realized. The net change in the valuation allowance balance from 2008 to 2009 was $27.7 million. The net increase in the valuation allowance resulted from the increases to the deferred tax asset balances relating to net operating losses and amortization.
At December 31, 2009, we had a net deferred tax liability of $4.0 million compared to a net deferred tax asset of $2.9 million in 2008. The $4.0 million deferred tax liability consists of a net deferred tax liability of $1.8 million from Shimoda Biotech, Ltd. and a net tax deferred liability of $2.2 million from Expression Pathology Inc.
For 2008, the company reclassified the allocation of the valuation allowance between the net current deferred tax assets and net noncurrent deferred tax assets. The reclassification was to allocate the valuation allowance pro-rata based on the gross current and noncurrent deferred tax assets. The reclassification did not change the net deferred tax asset in 2008.
We adopted new accounting guidance under ASC 740-10 beginning January 1, 2007. This interpretation clarifies what criteria must be met prior to the recognition of the financial statement benefit of a position taken in a tax return. Following is a reconciliation of our total gross unrecognized tax benefits from January 1, 2008 to December 31, 2009 (in thousands).
| Balance, December 31, 2007 |
$ | 5,170 | ||
| Additions for tax positions related to current year |
— | |||
| Additions for tax positions related to prior years |
354 | |||
| Reductions for tax periods of prior years |
— | |||
| Balance, December 31, 2008 |
$ | 5,524 | ||
| Additions for tax positions related to current year |
196 | |||
| Additions for tax positions related to prior years |
496 | |||
| Reductions for tax periods of prior years |
(1,070 | ) | ||
| Balance, December 31, 2009 |
5,146 | |||
The majority of this $5.1 million of gross unrecognized tax benefits, if and when recognized, would affect our effective tax rate. We file income tax returns in the U.S. federal jurisdiction and various state and foreign jurisdictions. In general, we are no longer subject to U.S. federal, state and foreign tax examinations for the years prior to 2004. We do not anticipate that any of the total tax benefits of $5.1 million will reverse in the next twelve months.
We recognize interest and penalties related to unrecognized tax benefits in our income tax expense. At December 31, 2009, we had $0.7 million of accrued interest.
10. Supplemental Disclosure of Cash Flow Information
During 2009, we received income tax refunds of $3.8 million. In 2008, we made income tax payments of $5.9 million. In 2007 we were not a separate entity subject to income taxes as we were a subsidiary of Old Abraxis.
82
Table of Contents
11. Per Share Information
Basic loss per common share is computed by dividing net loss by the weighted-average number of common shares outstanding during the reporting period. Diluted loss per common share is computed by dividing net loss by the weighted-average number of common shares used for the basic calculations plus any potentially dilutive shares for the portion of the year that the shares were outstanding, unless the impact is anti-dilutive. For the periods prior to the separation on November 13, 2007, basic and diluted loss per shares were computed using the number of shares of our common stock outstanding on November 13, 2007. All potentially dilutive employee stock awards were excluded from the computation of diluted loss per common share for all periods, as the effect on net loss per share was anti-dilutive. Calculations of basic and diluted loss per common share information are based on the following:
| Year Ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| (in thousands, except per share data) | ||||||||||||
| Basic and dilutive numerator: |
||||||||||||
| Net loss attributable to common shareholders |
$ | (103,116 | ) | $ | (276,771 | ) | $ | (41,604 | ) | |||
| Denominator: |
||||||||||||
| Weighted average common shares outstanding—basic and diluted |
40,100 | 40,032 | 39,991 | |||||||||
| Net loss per common share—basic and diluted |
$ | (2.57 | ) | $ | (6.91 | ) | $ | (1.04 | ) | |||
| Potentially dilutive shares not included |
199 | 243 | — | |||||||||
12. Leases and Commitments
Leases
We have entered into various operating lease agreements for warehouses, office space, automobiles, communications, information technology equipment and software and office equipment, including real estate leases as described in Note 6—Related Party Transactions. Rental expense amounted to $11.4 million, $10.1 million, and $5.6 million for the years ended December 31, 2009, 2008 and 2007, respectively. Related party rent expense totaled $2.8 million, and $3.3 million for the years ended December 31, 2009 and 2008, respectively.
As of December 31, 2009, future annual minimum lease payments related to non-cancelable operating leases, including amounts pertaining to related party leases are as follows:
| Year |
Total Amount |
Related Party | ||||
| (in thousands) | ||||||
| 2010 |
$ | 10,333 | $ | 3,039 | ||
| 2011 |
5,527 | 39 | ||||
| 2012 |
3,560 | 39 | ||||
| 2013 |
2,895 | — | ||||
| 2014 |
2,393 | |||||
| Thereafter |
2,082 | — | ||||
| $ | 26,790 | $ | 3,117 | |||
Purchase Obligations
As of December 31, 2009, we purchased all of our approved paclitaxel supply from one supplier. We believe our supply risk with respect to this supplier is minimal given that the supplier is well-regarded within the industry and we have not historically experienced supply shortages with this supplier. Additionally, we maintain
83
Table of Contents
an adequate safety stock supply of paclitaxel to mitigate any supply disruption. As of December 31, 2009 and 2008, our inventory included $21.0 million and $34.4 million, respectively, of paclitaxel. As of December 31, 2009, we have no commitment agreement to purchase any paclitaxel in 2010.
We also have other material supply agreements that require us to purchase approximately $32.3 million of material supply in each of the next five years.
Clinical Trial and Other Commitments
We have entered into various clinical trial agreements with third parties for the management, planning and execution of clinical trials. These agreements generally include milestone payments based on the number of patients enrolled in the clinical study. If all milestones in these agreements were achieved, related milestone commitments to be paid by us in 2010 through 2014 would approximate $83.0 million. Based on our current estimates of patient enrollment, approximately $53.4 million would be payable in 2010, approximately $24.1 million would be payable in 2011, approximately $2.9 million would be payable in 2012, approximately $1.5 million would be payable in 2013 and approximately $1.1 million would be payable in 2014.
We own a library of natural drug discovery soil samples and related strains acquired from around the world, which is intended for use in the discovery of new chemical entities. We are committed to paying up to $4.2 million upon the achievement of certain milestones related to these libraries, which we expect to occur within the next five years.
We have entered into various collaborative agreements under which we have agreed to provide funding to academic and clinical trial organizations of up to $53.8 million for research and other projects over the next five years. Even though we cannot predict the timing, we generally can control the timing and amount of spending as the projects require our approval in advance of funding.
13. Contingencies
In September and October 2009, we voluntarily recalled certain lots of Abraxane® manufactured at the Grand Island, New York facility. We accrued for all unsold inventory relating to the recall, which are included in cost of goods sold, and for product returns, which are included as an offset to Abraxane revenue. While we do not expect to incur additional costs relating to the recall, these accruals were based on facts known at the time of estimation and our assessment of the final outcome. As new information becomes available in future periods, we may change our accrual estimates.
On July 19, 2006, Élan Pharmaceutical Int’l Ltd. filed a lawsuit against Old Abraxis in the U.S. District Court for the District of Delaware alleging that Old Abraxis willfully infringed upon two of their patents by asserting that Abraxane® uses technology protected by Élan-owned patents. Élan sought unspecified damages and an injunction. In August 2006, Old Abraxis filed a response to Élan’s complaint contending that it did not infringe on the Élan patents and that the Élan patents are invalid. On June 13, 2008, the jury ruled that we infringed upon one of Élan’s patents, which runs until 2011, and awarded Élan $55.2 million in damages for sales of Abraxane® through the judgment date. We have filed various post-trial motions and are appealing the jury ruling. We assumed any liability of Old Abraxis relating to this litigation in connection with the separation. For the year ended December 31, 2008, we recorded litigation costs of $57.6 million for this matter, which includes $2.4 million of interest.
We are from time to time subject to claims and litigation arising in the ordinary course of business. We intend to defend vigorously any such litigation that may arise under all defenses that would be available to us. In the opinion of management, the ultimate outcome of proceedings of which management is aware, even if adverse to us, would not have a material adverse effect on our consolidated financial position or results of operations.
84
Table of Contents
We record accruals for contingencies to the extent that we conclude their occurrence is probable and the related damages are estimable. These assessments involve complex judgments about future events and rely on estimates and assumptions. Although we believe we have substantial defenses in these matters, litigation is inherently unpredictable and we could in the future incur judgments or enter into settlements that could have a material adverse effect on our results of operations.
14. Stockholders’ Equity
For all periods prior to November 13, 2007, Old Abraxis’ investment in our proprietary products businesses is shown as parent company investment in the consolidated and combined financial statements. Parent company investment represents the historical investment of capital in us, our accumulated net earnings after taxes, and the net effect of transactions with and allocations from Old Abraxis. See Note 6—Related Party Transactions for additional information regarding the allocation to us of various expenses incurred by Old Abraxis.
On November 13, 2007, Old Abraxis completed a distribution of one share of our common stock (par value of $0.001) for every four shares of Old Abraxis common stock. Following the separation, we had 40.0 million shares of common stock outstanding. After the separation adjustments were recorded on November 13, 2007, the remaining parent company investment balance, which includes all earnings prior to the separation, was transferred to additional paid in capital. Net loss subsequent to the separation is included in retained earnings.
Registration Rights
Pursuant to a registration rights agreement, Old Abraxis granted registration rights to the former American BioScience, Inc. stockholders (including our executive chairman and entities affiliated with him) with respect to all 86,096,523 shares of Old Abraxis common stock they received in the merger. Under the terms of the registration rights agreement, any securities issued or issuable in respect of Old Abraxis common stock (including our common stock) would have the benefits of the registration rights agreement. Accordingly, in connection with the separation, we entered into a new registration rights agreement with the former American BioScience, Inc. shareholders with respect to 21,524,130 shares of our common stock containing provisions substantially identical to the registration rights agreement entered into in connection with the merger of American BioScience, Inc. and APP in 2006.
These stockholders have the right to require us to register all or a portion of the shares of our common stock they receive in the distribution. In addition, these stockholders may require us to include their shares in future registration statements that we file and may require us to register their shares for resale on a Form S-3 registration statement. Upon registration, the registered shares generally will be freely tradeable in the public market without restriction. However, in connection with any underwritten offering, the stockholders will agree to lock up any other shares for up to 90 days and will agree to a limit on the maximum number of shares that can be registered for the account of these holders under so-called “shelf” registration statements.
Except for underwriters discounts and commissions and certain marketing expenses, we will be obligated to pay all expenses for the first eight registration statements filed upon the request of a holder of registrable securities, and any other or additional expenses of registration are to be borne by the holders of registrable securities on a pro rata basis. These registration rights are subject to some conditions and limitations, among them the right of the underwriters of an offering to limit the number of shares included in registration. We will be obligated to indemnify the holders of these registration rights, and each selling holder will be obligated to indemnify us, against specified liabilities under the Securities Act of 1933, the Securities Exchange Act of 1934 and other applicable federal and state laws.
Stock Repurchase Program
On April 20, 2009, we announced that our board of directors approved a program to repurchase up to $100 million of our common stock. Share repurchases may be commenced, suspended or discontinued at any time
85
Table of Contents
without prior notice. During 2009, we repurchased 20,500 shares of our common stock for an aggregate amount of $0.9 million pursuant to the stock repurchase program, which were subsequently retired in 2009.
15. Stock-Based Compensation
2007 Stock Incentive Plan
We adopted the 2007 Stock Incentive Plan, which was approved by the sole stockholder subsequent to the 2007 separation. We have reserved 6,000,000 shares of our common stock for issuance under our 2007 Stock Incentive Plan, subject to adjustments for stock splits, stock dividends or other similar changes in our common stock or our capital structure. The number of shares initially reserved for issuance or transfer pursuant to awards under the 2007 Stock Incentive Plan will be increased by the number of shares represented by awards that are forfeited or cancelled, or that expire or terminate, including any shares that are forfeited by the award recipient or repurchased by us pursuant to the terms of the relevant award agreement.
Our 2007 Stock Incentive Plan provides for the grant of (a) incentive stock options to our employees, including officers and employee directors, (b) non-qualified stock options to our employees, directors and consultants and (c) other types of awards, collectively referred to as “awards.”
No more than 1,000,000 shares under options can be granted to an individual optionee in any given fiscal year. We may grant up to an additional 750,000 shares, which will not count against the limit set forth in the previous sentence, in connection with an optionee’s initial commencement of service with our company.
Our board of directors or a committee designated by our board of directors administers our 2007 Stock Incentive Plan and has authority to determine the terms and conditions of awards, including the types of awards, the number of shares subject to each award, the vesting schedule of the awards and the selection of grantees.
The exercise price of all options granted under our 2007 Stock Incentive Plan will be determined by our board of directors or a committee designated by our board of directors, but in no event may this price be less than the fair market value of our common stock on the date of grant, unless otherwise determined by our board of directors with respect to non-qualified stock options. If, however, incentive stock options are granted to a person who owns stock possessing more than 10% of the voting power of all our classes of stock, the exercise price of such option must equal at least 110% of the fair market value our common stock on the grant date and the maximum term of these incentive stock options must not exceed five years. The maximum term of an incentive stock option granted to any person who does not own stock possessing more than 10% of the voting power of all our classes of stock must not exceed ten years. Our board of directors or a committee designated by our board of directors determines the term of all other awards granted under our 2007 Stock Incentive Plan. The consideration for the option may consist of cash, check, shares of our common stock, the assignment of part of the proceeds from the sale of shares acquired upon exercise of the option, through a same-day sale and exercise procedure facilitated by a brokerage firm, loan or any combination of these forms of consideration.
In the event of one of the following, options under our 2007 Stock Incentive Plan will terminate and be cancelled, unless the successor corporation assumes or substitutes the options:
| • | a dissolution, liquidation or sale of all or substantially all of our assets, |
| • | a merger or consolidation in which we are not the surviving entity, |
| • | a corporate transaction in which our stockholders give up a majority of their equity interest in our company, or |
| • | an acquisition of 50% or more of our stock by any individual or entity, including by tender offer or a reverse merger. |
However, in lieu of the termination of options upon occurrence of the above events, the terms of some individual award agreements may otherwise provide for full acceleration of an optionee’s outstanding options
86
Table of Contents
immediately upon occurrence of the above events or full acceleration of any outstanding options that are not assumed or substituted by the surviving corporation, and full acceleration of any otherwise assumed or substituted options if optionee’s employment is terminated for cause or by optionee for good reason within twelve months of the occurrence of such events.
Unless terminated sooner, our 2007 Stock Incentive Plan will automatically terminate in 2017. Our board of directors has authority to amend or terminate our 2007 Stock Incentive Plan, subject to stockholder approval of some amendments. However, no action may be taken that will affect any shares of common stock previously issued and sold or any option previously granted under our 2007 Stock Incentive Plan, without the grantee’s consent.
Converted Stock-Based Compensation Plans
Following the 2007 separation, and pursuant to the provisions of the respective plan documents, holders of Old Abraxis restricted stock units (RSUs) and stock options who are current and former directors or employees of Old Abraxis whose employment related to our proprietary products business had their Old Abraxis BioScience RSUs and stock options converted into newly-issued RSUs and stock options of our company pursuant to a conversion formula that was intended to preserve the intrinsic value and maintained their pre-distribution RSUs and stock options. Except for adjustments made in connection with the conversion, the RSUs and stock options so issued had substantially the same terms, including expiration date and vesting schedule, as the converted Old Abraxis RSUs and stock options. Additionally, we assumed the American BioScience Restricted Stock Unit Plan I and American BioScience Restricted Stock Unit Plan II. We also assumed the restricted units previously granted under these plans. In connection with the separation, we adopted the 2007 Stock Incentive Plan.
For the periods prior to the 2007 separation on November 13, 2007, our employees held stock compensation awards in the following plans sponsored by Old Abraxis:
1997 Stock Option Plan
Under the 1997 Stock Option Plan, or the 1997 Plan, options to purchase shares of Old Abraxis’ common stock were granted to certain employees and the directors with an exercise price equal to the estimated fair market value of Old Abraxis’ common stock on the date of grant. The 1997 Plan stock options vested upon completion of Old Abraxis’ initial public offering in December 2001.
2001 Stock Incentive Plan
The 2001 Stock Incentive Plan, or the 2001 Plan, provided for the grant of incentive stock options and restricted stock to employees, including officers and employee directors, non-qualified stock options to employees, directors and consultants and other types of awards.
The compensation committee of Old Abraxis’ board of directors administered the 2001 Plan and had the authority to determine the terms and conditions of awards, including the types of awards, the number of shares subject to each award, the vesting schedule of the awards and the selection of the grantees. The exercise price of all options granted under the 2001 Plan were determined by the compensation committee of Old Abraxis’ board of directors, but in no event was this price less than the fair market value of Old Abraxis’ common stock on the date of grant.
2001 Non-Employee Director Stock Option Program
The 2001 Non-Employee Director Stock Option Program, or the 2001 Program, was adopted as part, and is subject to the terms and conditions, of the 2001 Plan. The 2001 Program established a program for the automatic grant of awards to non-employee directors at the time that they initially join the board and at each annual meeting of the stockholders at which they are elected.
87
Table of Contents
Restricted Unit Plan
In connection with the 2007 separation, we assumed the American BioScience Restricted Unit Plan I and the American BioScience Restricted Unit Plan II (the “Plans”). We also assumed the restricted units previously granted under these plans to our employees.
Shares of our common stock will be issuable upon the vesting of these units. The units granted under American BioScience Restricted Unit Plan I vested upon the completion of the merger of American BioScience, Inc. and American Pharmaceutical Partners in 2006. The units issued under the American BioScience Restricted Unit Plan II generally vested one-half on April 18, 2008 (which was the second anniversary of the closing of the merger). The balance of the shares generally will vest on April 18, 2010. The units entitle their holders to receive a number of our shares of common stock determined on each vesting date based on the notional price that vests on such date divided by our average trading price over the three days prior to vesting; except that if the average trading price of our stock price is less than $66.63, then the notional price is divided by $66.63. The maximum number of our shares that may be issuable under this restricted unit plan is 367,100 shares.
We also assumed an agreement between Old Abraxis and RSU Plan LLC (“RSU LLC”). Under the terms of this agreement, RSU LLC has agreed that prior to the date on which restricted stock units issued pursuant to American BioScience Restricted Unit Plan II become vested, RSU LLC will deliver, or cause to be delivered, to us the number of our shares of common stock or cash (or a combination thereof) in an amount sufficient to satisfy the obligations to participants under the American BioScience Restricted Unit Plan II of the vested restricted units. We are required to satisfy our obligations under the American BioScience Restricted Unit Plan II by paying to the participants in the American BioScience Restricted Unit Plan II cash and/or shares of our common stock in the same proportion as was delivered by the RSU LLC. The intention of this agreement is to have RSU LLC satisfy our obligations under American BioScience Restricted Unit Plan II so that there would not be any further dilution to our stockholders as a result of our assumption of the American BioScience Restricted Unit Plan II.
As of December 31, 2009, we recorded a current liability of $8.0 million for awards relating to the Plans. We had a long-term liability of $5.8 million at December 31, 2008.
Employee Stock Purchase Plan
Certain of our employees participated in the Old Abraxis’ 2001 Employee Stock Purchase Plan, or the ESPP. Eligible employees contributed up to 10% of their base earnings toward the semi-annual purchase and issuance of Old Abraxis common stock. The employees’ purchase price was the lesser of 85% of the fair market value of the stock on the first business day of the offer period or 85% of the fair market value of the stock on the last business day of the semi-annual purchase period. Employees could purchase no more than 625 shares of Abraxis BioScience common stock within any given purchase period. The Compensation Committee of Old Abraxis indefinitely suspended the ESPP from and after the purchase period that ended on July 31, 2007.
Equity Compensation
Restricted Stock Units
At December 31, 2009, 256,705 restricted stock units were outstanding under our 2007 and 2001 Stock Incentive Plan with a weighted average per share market value of $55.29 and with vesting dates ranging from January 2010 to July 2013. Upon vesting, each restricted stock unit entitles the holder to one share of our common stock. Compensation expense related to restricted stock unit grants is based upon the market price on the date of grant and is charged to earnings on a graded amortization basis over the applicable vesting period. Restricted stock units currently granted generally vest ratably over a four year period.
Stock-based compensation costs for all RSU awards (including liability awards) for the years ended December 31, 2009, 2008 and 2007 were $9.7 million, $8.1 million, and $17.2 million, respectively. As of
88
Table of Contents
December 31, 2009, there was $7.1 million of total unrecognized compensation expense related to restricted stocks granted under our stock-based compensation plans, which is expected to be recognized over a weighted average period of 1.6 years.
Following is a reconciliation of unvested restricted stock unit awards as of December 31, 2009:
| Number of Awards |
Weighted- Average Grant Date Fair Value(1) |
Weighted- Average Remaining Amortization Period (Years) | ||||||
| Outstanding unvested awards at December 31, 2008 |
248,631 | $ | 60.85 | |||||
| Granted |
137,278 | 50.80 | ||||||
| Vested |
(90,954 | ) | 60.50 | |||||
| Forfeited |
(38,250 | ) | 62.97 | |||||
| Outstanding unvested awards at December 31, 2009 |
256,705 | $ | 55.29 | 1.77 | ||||
| (1) | Grant date fair values are based on our stock price on the grant date. |
Stock Options
We account for stock-based compensation in accordance with FASB ASC 718, Compensation – Stock Compensation, which requires the recognition of compensation cost for all share-based payments, including employee stock options, at fair value. We use the straight-line and graded attribution methods depending on the vesting provision to recognize share-based compensation expense over the vesting period of the award. Options currently granted generally expire ten years from the grant date and vest ratably over a four-year period.
We adjust stock option compensation on a quarterly basis for revisions to the estimated rate of equity award forfeitures based on actual forfeiture experience. During the fourth quarter of 2009, we revised the forfeiture rate with respect to stock options. The effect of the change in the forfeiture rate resulted in an increase in stock compensation expense of $0.3 million for 2009. No changes in estimated rate of forfeitures were made during 2008. During the fourth quarter of 2007, we revised the forfeiture rate with respect to stock options. The effect of the change in the forfeiture rate resulted in a reduction in stock compensation expense of $1.0 million for 2007. For the years ended December 31, 2009, 2008 and 2007, we recorded stock option compensation expense of $5.3 million, $5.2 million and $4.5 million, respectively.
As of December 31, 2009, we had $8.1 million of total unrecognized compensation expense related to stock options granted under the equity incentive plans, which is expected to be recognized over a weighted average period of 2.0 years.
89
Table of Contents
The fair values of options granted during the years ended December 31, 2009, 2008 and 2007 were determined using the Black-Scholes option pricing model, which incorporates various assumptions. The risk-free rate of interest for the average contractual life of the option is based on the U.S. Treasury yield curve in effect at the time of grant. The dividend yield is zero as we do not and Old Abraxis did not anticipate paying any dividends. The expected term of awards granted is based upon the historical exercise patterns of the participants in the plans and expected volatility is based on the historical volatility of the stock price over the expected term of the award. The weighted average estimated values of employee stock option grants and rights granted under Old Abraxis’ employee stock purchase plan as well as the weighted average assumptions that were used in calculating such values during the last three years were based on estimates at the date of grant as follows:
| Year Ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| (dollars in thousands, except per share data) | ||||||||||||
| Stock Options |
||||||||||||
| Risk-free rate |
2.1 | % | 2.5 | % | 3.8 | % | ||||||
| Dividend yield |
— | — | — | |||||||||
| Expected life in years |
5 | 5 | 5 | |||||||||
| Volatility |
66 | % | 45 | % | 52 | % | ||||||
| Weighted average grant date fair value of options granted |
$ | 24.15 | $ | 27.55 | $ | 26.44 | ||||||
Our stock options outstanding that have vested and are expected to vest as of December 31, 2009 were as follows:
| Number of Shares |
Weighted Average Exercise Price |
Weighted Average Remaining Contractual Term |
Aggregate Intrinsic Value(1) | |||||||
| Vested |
632,090 | $ | 52.82 | 4.55 | $ | 6,178,759 | ||||
| Expected to vest |
509,886 | 50.42 | 8.94 | 734,600 | ||||||
| Total |
1,141,976 | $ | 51.75 | 6.51 | $ | 6,913,359 | ||||
| (1) | These amounts represent the difference between the exercise price and $40.55, the closing price of our common stock on December 31, 2009 for in the money options. |
The above options outstanding that are expected to vest are net of estimated future option forfeitures in accordance with the provisions of ASC 718. Options with aggregate grant date fair values of $3.8 million and $5.9 million and with weighted average grant date fair values of $30.75 and $33.19 vested during the years ended December 31, 2009 and 2008, respectively. Options with aggregate fair values of $0.2 million and $1.3 million, weighted average grant date fair values of $19.54 and $27.46 and aggregate intrinsic values of $0.2 million and $0.9 million were exercised during the years ended December 31, 2009 and 2008, respectively. Non-vested options totaling 0.5 million and 0.4 million shares with weighted average grant date fair values of $25.52 and $29.00 were outstanding at December 31, 2009 and 2008, respectively.
90
Table of Contents
Additional information with respect to our stock option activity is as follows:
| Options Outstanding | Exercisable Options | ||||||||||
| Shares | Weighted- Average Exercise Price |
Shares | Weighted- Average Exercise Price | ||||||||
| Outstanding at January 1, 2007 |
861,172 | $ | 57.98 | 364,366 | $ | 37.50 | |||||
| Granted |
79,287 | 55.02 | |||||||||
| Exercised |
(23,488 | ) | 15.63 | ||||||||
| Forfeited |
(17,518 | ) | 67.55 | ||||||||
| Expired |
(4,509 | ) | 76.54 | ||||||||
| Outstanding at December 31, 2007 |
894,944 | 55.98 | 461,960 | $ | 46.99 | ||||||
| Granted |
179,714 | 65.05 | |||||||||
| Exercised |
(46,713 | ) | 50.51 | ||||||||
| Forfeited |
(99,990 | ) | 67.42 | ||||||||
| Expired |
(11,314 | ) | 97.04 | ||||||||
| Outstanding at December 31, 2008 |
916,641 | 56.28 | 553,864 | $ | 50.48 | ||||||
| Granted |
324,416 | 42.95 | |||||||||
| Exercised |
(8,522 | ) | 36.74 | ||||||||
| Forfeited |
(90,559 | ) | 67.51 | ||||||||
| Expired |
— | — | |||||||||
| Outstanding at December 31, 2009 |
1,141,976 | $ | 51.75 | 632,090 | $ | 52.82 | |||||
In connection with the separation, stock options of our employees were converted to reflect the adjusted value of the options based on our stock price after the 2007 separation. The number of options outstanding on the separation date and the associated exercise prices were converted using a rate of 0.4243, which was based on Old Abraxis’ stock price for the 10 consecutive trading days preceding the separation date and our stock price for the 10 consecutive trading days following the separation. No additional stock compensation resulted from the modifications under the provisions of ASC 718 because it was determined the changes were required by the terms of the awards.
The following table summarizes information about our stock options outstanding at December 31, 2009:
| Options Outstanding | Options Exercisable | |||||||||||
| Exercise Price Ranges |
Number of Shares |
Weighted- Average Remaining Contractual Life |
Weighted- Average Exercise Price |
Number of Shares |
Weighted- Average Exercise Price | |||||||
| $ 0.00 – $ 12.43 |
171,489 | 0.8 | $ | 7.86 | 171,489 | $ | 7.86 | |||||
| $ 12.44 – $ 24.87 |
12,440 | 2.1 | 15.91 | 12,440 | 15.91 | |||||||
| $ 24.88 – $ 37.30 |
95,463 | 7.7 | 31.35 | 25,463 | 30.07 | |||||||
| $ 37.31 – $ 49.74 |
291,931 | 9.1 | 45.90 | 31,510 | 47.62 | |||||||
| $ 49.75 – $ 62.17 |
111,077 | 7.4 | 55.32 | 81,246 | 55.59 | |||||||
| $ 62.18 – $ 74.61 |
343,498 | 7.3 | 67.65 | 194,395 | 68.57 | |||||||
| $ 74.62 – $ 87.04 |
20,133 | 4.8 | 80.51 | 19,602 | 80.56 | |||||||
| $ 87.05 – $ 99.48 |
16,968 | 5.5 | 94.97 | 16,968 | 94.97 | |||||||
| $ 99.49 – $111.91 |
74,668 | 4.8 | 107.43 | 74,668 | 107.43 | |||||||
| $ 111.92 – $124.35 |
4,309 | 5.2 | 120.80 | 4,309 | 120.80 | |||||||
| 1,141,976 | 6.5 | $ | 51.75 | 632,090 | $ | 52.82 | ||||||
91
Table of Contents
16. Employee Benefit Plan
Following the 2007 separation, we adopted and our employees became eligible to participate in a defined contribution plan intended to be tax-qualified. APP cooperated with us to transfer the account balances of our employees to the defined contribution plan adopted by us. We adopted the defined contribution plan in the first quarter of 2008. Prior to the 2007 separation, Old Abraxis sponsored a 401(k) defined-contribution plan, or 401(k) Plan, covering substantially all eligible employees. Employee contributions to the 401(k) Plan are voluntary. Participants’ contributions are limited to their annual tax deferred contribution limit as allowed by the Internal Revenue Service. Our total matching contributions to our 401(k) Plan in 2009 and 2008 and Old Abraxis’ 401(k) Plan in 2007 were $2.2 million, $1.8 million, and $1.4 million for the years ended December 31, 2009, 2008 and 2007, respectively.
17. Segment Information
We operate in one business segment. Therefore, the results of operations are reported on a combined basis for purpose of segment reporting.
18. Subsequent Events
In January 2010, we acquired a 100% ownership interest in a Maryland-based diagnostics company for $5.0 million, plus the assumption of certain liabilities and future contingent payments. The acquisition will be accounted for as a business combination and the acquired company’s results of operations will be consolidated in our financial statements from the date of acquisition.
19. Selected Quarterly Financial Data (Unaudited)
| Year Ended December 31, 2009 | ||||||||||||||||
| First | Second | Third | Fourth | |||||||||||||
| (in thousands, except per share data) | ||||||||||||||||
| Net revenue |
$ | 72,582 | $ | 85,129 | $ | 96,628 | $ | 104,711 | ||||||||
| Gross profit |
63,455 | 70,513 | 76,914 | 84,503 | ||||||||||||
| Net loss |
(23,301 | ) | (24,280 | ) | (38,044 | ) | (19,143 | ) | ||||||||
| Net loss attributable to noncontrolling interest |
(380 | ) | (847 | ) | (330 | ) | (95 | ) | ||||||||
| Net loss attributable to common shareholders |
(22,921 | ) | (23,433 | ) | (37,714 | ) | (19,048 | ) | ||||||||
| Net loss per common share: |
||||||||||||||||
| Basic and diluted |
$ | (0.57 | ) | $ | (0.58 | ) | $ | (0.94 | ) | $ | (0.47 | ) | ||||
| Year Ended December 31, 2008 | ||||||||||||||||
| First | Second | Third | Fourth | |||||||||||||
| (in thousands, except per share data) | ||||||||||||||||
| Net revenue |
$ | 82,142 | $ | 77,622 | $ | 93,381 | $ | 92,164 | ||||||||
| Gross profit |
73,535 | 67,677 | 82,253 | 82,776 | ||||||||||||
| Net income (loss) |
4,309 | (84,143 | ) | (15,059 | ) | (181,878 | ) | |||||||||
| Net income (loss) per common share: |
||||||||||||||||
| Basic and diluted |
$ | 0.11 | $ | (2.10 | ) | $ | (0.38 | ) | $ | (4.54 | ) | |||||
Item 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
92
Table of Contents
Item 9A. CONTROLS AND PROCEDURES
(a) Evaluation of disclosure controls and procedures.
We maintain disclosure controls and procedures, as such term is defined under Exchange Act Rules 13a-15(e) and 15d-15(e), that are designed to ensure that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to management, including our Chief Executive Officer and our Principal Financial Officer, to allow timely decisions regarding required disclosures. In designing and evaluating our disclosure controls and procedures, we recognize that any controls and procedures, no matter how well designed and operated, can only provide reasonable assurance of achieving the desired control objectives and in reaching a reasonable level of assurance we necessarily are required to apply judgment in evaluating the cost-benefit relationship of possible controls and procedures. Our management, with participation of our Chief Executive Officer and Principal Financial Officer evaluated the effectiveness of our disclosure controls and procedures as of the end of the fiscal year covered by this report. Based on their evaluation and subject to the foregoing, management concluded that our disclosure controls and procedures were effective as of December 31, 2009.
(b) Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rules 13a-15(f) under the Securities Exchange Act of 1934. Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the United States. Internal control over financial reporting includes those written policies and procedures that:
| • | pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of our assets; |
| • | provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with accounting principles generally accepted in the United States of America; |
| • | provide reasonable assurance that our receipts and expenditures are being made only in accordance with authorization of our management and directors; and |
| • | provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of assets that could have a material effect on the consolidated financial statements. |
Internal control over financial reporting includes the controls themselves, monitoring and internal auditing practices and actions taken to correct deficiencies as identified.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Management assessed the effectiveness of our internal control over financial reporting as of December 31, 2009. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control—Integrated Framework. Management’s assessment included an evaluation of the design of our internal control over financial reporting and testing of the operational effectiveness of its internal control over financial reporting. Management reviewed the results of its assessment with the Audit Committee of our Board of Directors.
93
Table of Contents
Based on this assessment, management believes that we maintained effective internal control over financial reporting as of December 31, 2009, based on those criteria.
The effectiveness of the Company’s internal control over financial reporting as of December 31, 2009 has been audited by Ernst & Young LLP, an independent registered public accounting firm, as stated in their report below.
(c) Attestation Report of Independent Registered Public Accounting Firm
94
Table of Contents
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders of Abraxis BioScience, Inc.
We have audited Abraxis BioScience, Inc. (formerly known as New Abraxis, Inc.) internal control over financial reporting as of December 31, 2009 based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). Abraxis BioScience, Inc.’s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying “Management’s Report on Internal Control over Financial Reporting”. Our responsibility is to express an opinion on the company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Abraxis BioScience, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2009, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of Abraxis BioScience, Inc. as of December 31, 2009 and 2008, and the related consolidated and combined statements of operations, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2009 of Abraxis BioScience, Inc. and our report dated March 12, 2010, expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Los Angeles, California
March 12, 2010
95
Table of Contents
(d) Changes in internal controls.
Management has determined that, as of December 31, 2009, there were no changes in our internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f) and 15d-15(f), that occurred during our most recent fiscal quarter that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
None.
96
Table of Contents
Item 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required by this item will be contained in the Proxy Statement for our 2010 Annual Meeting of Stockholders and is incorporated herein by reference.
Item 11. EXECUTIVE COMPENSATION
The information required by this item will be contained in the Proxy Statement for our 2010 Annual Meeting of Stockholders and is incorporated herein by reference.
Item 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by this item will be contained in the Proxy Statement for our 2010 Annual Meeting of Stockholders and is incorporated herein by reference.
Item 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required by this item will be contained in the Proxy Statement for our 2010 Annual Meeting of Stockholders and is incorporated herein by reference.
Item 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
The information required by this item will be contained in the Proxy Statement for our 2010 Annual Meeting of Stockholders and is incorporated herein by reference.
97
Table of Contents
Item 15. EXHIBITS, FINANCIAL STATEMENTS AND FINANCIAL STATEMENT SCHEDULE
a. (1) Financial Statements
The following consolidated and combined financial statements of Abraxis BioScience, Inc. are included in Part II, Item 8 of this Report:
Report of Independent Registered Public Accounting Firm
Consolidated Balance Sheets at December 31, 2009 and 2008
Consolidated and Combined Statements of Operations for the Years Ended December 31, 2009, 2008, and 2007
Consolidated and Combined Statements of Stockholders’ Equity for the Years Ended December 31, 2009, 2008, and 2007
Consolidated and Combined Statements of Cash Flows for the Years Ended December 31, 2009, 2008, and 2007
Notes to Consolidated and Combined Financial Statements
a. (2) Financial Statement Schedule
All schedules are omitted because the required information is not present or is not present in amounts sufficient to require submission of the schedule or because the information required is given in the consolidated and combined financial statements or the notes thereto.
a. (3) Exhibits
The exhibits are as set forth in the Exhibit Index.
98
Table of Contents
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Los Angeles, State of California on the 12th day of March, 2010.
| ABRAXIS BIOSCIENCE, INC. | ||
| By: | /s/ BRUCE J. WENDEL | |
| Bruce J. Wendel | ||
| Chief Executive Officer | ||
POWER OF ATTORNEY
Each person whose signature appears below constitutes and appoints Patrick Soon-Shiong, Bruce J. Wendel and Mitchell K. Fogelman, and each of them, as attorneys-in-fact, each with the power of substitution, for him or her in any and all capacities, to sign any amendment to this Annual Report on Form 10-K and to file the same, with exhibits thereto and other documents in connection therewith, with the Commission, granting to said attorneys-in-fact, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact or any of them, or their or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Partnership in the capacities and on the dates indicated.
| Signature |
Title |
Date | ||
| /s/ PATRICK SOON-SHIONG, M.D. Patrick Soon-Shiong, M.D. |
Executive Chairman |
March 12, 2010 | ||
| /s/ BRUCE J. WENDEL Bruce J. Wendel |
Vice Chairman, and Chief Executive Officer |
March 12, 2010 | ||
| /s/ MITCHELL K. FOGELMAN Mitchell K. Fogelman |
Senior Vice President of Finance (Principal Financial and Accounting Officer) |
March 12, 2010 | ||
| /s/ KIRK K. CALHOUN Kirk K. Calhoun |
Director |
March 12, 2010 | ||
| /s/ DAVID S. CHEN, PH.D. David S. Chen Ph.D. |
Director |
March 12, 2010 | ||
| /s/ STEPHEN D. NIMER, M.D. Stephen D. Nimer, M.D. |
Director |
March 12, 2010 | ||
| /s/ LEONARD S. SHAPIRO Leonard S. Shapiro |
Director |
March 12, 2010 | ||
| /s/ MICHAEL SITRICK Michael Sitrick |
Director |
March 12, 2010 | ||
99
Table of Contents
EXHIBIT INDEX
Exhibit Index
| Exhibit Number |
Description | |
| 2.1 | Separation and Distribution Agreement among APP Pharmaceuticals, Inc., Abraxis BioScience, LLC, APP Pharmaceuticals, LLC and the Registrant (incorporated by reference to Exhibit 2.1 to the Registrant’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on December 20, 2007) | |
| 3.1 | Amended and Restated Certificate of Incorporation of the Registrant (incorporated by reference to Exhibit 3.1 to the Registrant’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on December 20, 2007) | |
| 3.2 | Amended and Restated Bylaws of the Registrant (incorporated by reference to Exhibit 3.2 to the Registrant’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on December 20, 2007) | |
| 3.3 | Certificate of Amendment to Amended and Restated Certificate of Incorporation of the Registrant (incorporated by reference to Exhibit 3.3 to the Registrant’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on December 20, 2007) | |
| 4.1 | Reference is made to Exhibits 3.1, 3.2 and 3.3 | |
| 4.2 | Specimen Stock Certificate of the Registrant (incorporated by reference to Exhibit 4.2 to the Registrant’s Amendment #2 to Form 10 Registration Statement with the Securities and Exchange Commission on October 24, 2007) | |
| 4.3 | Registration Rights Agreement by and among the Registrant and certain stockholders of the Registrant as set forth therein (incorporated by reference to Exhibit 4.3 to the Registrant’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on December 20, 2007) | |
| 10.1 | Separation and Distribution Agreement among APP Pharmaceuticals, Inc., Abraxis BioScience, LLC, APP Pharmaceuticals, LLC and the Registrant (Reference is made to Exhibit 2.1 hereto) | |
| 10.2 | Tax Allocation Agreement among APP Pharmaceuticals, Inc., Abraxis BioScience, LLC, APP Pharmaceuticals, LLC and the Registrant (incorporated by reference to Exhibit 10.2 to the Registrant’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on December 20, 2007) | |
| 10.3 | Transition Services Agreement between APP Pharmaceuticals, Inc. and the Registrant (incorporated by reference to Exhibit 10.3 to the Registrant’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on December 20, 2007) | |
| 10.4 | Employee Matters Agreement among APP Pharmaceuticals, Inc., APP Pharmaceuticals, LLC, Abraxis BioScience, LLC and the Registrant (incorporated by reference to Exhibit 10.4 to the Registrant’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on December 20, 2007) | |
| 10.5* | Manufacturing Agreement between APP Pharmaceuticals, LLC and the Registrant (incorporated by reference to Exhibit 10.5 to the Registrant’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on December 20, 2007) | |
| 10.6 | Lease Agreement between APP Pharmaceuticals, LLC and Abraxis BioScience, LLC for the premises located at 2020 Ruby Street, Melrose Park, Illinois (incorporated by reference to Exhibit 10.6 to the Registrant’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on December 20, 2007) | |
100
Table of Contents
| Exhibit Number |
Description | |
| 10.7 | Lease Agreement between APP Pharmaceuticals, LLC and Abraxis BioScience, LLC for the warehouse facilities located at 2045 N. Cornell Avenue, Melrose Park, Illinois (incorporated by reference to Exhibit 10.7 to the Registrant’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on December 20, 2007) | |
| 10.8 | Lease Agreement between APP Pharmaceuticals, LLC and Abraxis BioScience, LLC for the research and development facility located at 2045 N. Cornell Avenue, Melrose Park, Illinois (incorporated by reference to Exhibit 10.8 to the Registrant’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on December 20, 2007) | |
| 10.9 | Lease Agreement between Abraxis BioScience, LLC and APP Pharmaceuticals, LLC for the premises located at 3159 Staley Road, Grand Island, New York (incorporated by reference to Exhibit 10.9 to the Registrant’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on December 20, 2007) | |
| 10.10 | Form of Indemnification Agreement between the Registrant and each of its executive officers and directors (incorporated by reference to Exhibit 10.10 to the Registrant’s Amendment #1 to Form 10 Registration Statement with the Securities and Exchange Commission on October 5, 2007) | |
| 10.11 | Employment Agreement, dated January 25, 2006, between the Registrant and Carlo Montagner (incorporated by reference to Old Abraxis’ Quarterly Report on Form 10-Q/A filed with the Securities and Exchange Commission on August 10, 2006) | |
| 10.12 | Standard Form Office Lease, dated March 24, 2006, between the Registrant and California State Teacher’s Retirement System, as amended on May 26, 2006, for the premises located at 11755 Wilshire Boulevard, Los Angeles, California (incorporated by reference to Old Abraxis’ Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on November 9, 2006) | |
| 10.13 | Co-Promotion Strategic Marketing Services Agreement, dated April 26, 2006, between the Registrant and AstraZeneca UK Limited (incorporated by reference to Old Abraxis’ Quarterly Report on Form 10-Q/A filed with the Securities and Exchange Commission on August 10, 2006) | |
| 10.14 | Abraxis BioScience, Inc. 2007 Stock Incentive Plan, including forms of agreement thereunder (incorporated by reference to Exhibit 10.15 to the Registrant’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on December 20, 2007) | |
| 10.15 | American BioScience, Inc. Restricted Stock Unit Plan I, including a form of agreement thereunder (incorporated by reference to Old Abraxis’ Registration Statement on Form S-8 (File No. 333-133364) filed with the Securities and Exchange Commission on April 18, 2006) | |
| 10.16 | American BioScience, Inc. Restricted Stock Unit Plan II, including a form of agreement thereunder (incorporated by reference to Old Abraxis’ Registration Statement on Form S-8 (File No. 333-133364) filed with the Securities and Exchange Commission on April 18, 2006) | |
| 10.17 | Agreement, dated April 18, 2006, between the Registrant and RSU LLC (incorporated by reference to Old Abraxis’ Quarterly Report on Form 10-Q/A filed with the Securities and Exchange Commission on August 10, 2006) | |
| 10.18* | Aircraft Purchase and Sale Agreement (incorporated by reference to Old Abraxis’ Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on November 9, 2006) | |
| 10.19 | Escrow Agreement, dated April 18, 2006, by and among Abraxis BioScience, our chief executive officer and Fifth Third Bank (incorporated by reference to Old Abraxis’ Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on May 10, 2006). | |
101
Table of Contents
| Exhibit Number |
Description | |
| 10.20* | License Agreement, dated as of May 27, 2005, between the Registrant and Taiho Pharmaceutical Co., Ltd. (incorporated by reference to Exhibit 10.10 to the Registrant’s Amendment #3 to Form 10 Registration Statement with the Securities and Exchange Commission on November 2, 2007) | |
| 10.21 | Agreement between APP Pharmaceuticals, Inc. and the Registrant (incorporated by reference to Exhibit 10.22 to the Registrant’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on December 20, 2007) | |
| 10.22 | Employment Agreement, dated as of May 22, 2008, between the Registrant and David O’Toole (incorporated by reference to Exhibit 10.22 to the Registrant’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on August 14, 2008) | |
| 10.23 | Employment Agreement, dated as of October 6, 2008, between the Registrant and Edward Geehr, M.D. (incorporated by reference to Exhibit 10.23 to the Registrant’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on November 14, 2008) | |
| 10.24 | Termination Agreement dated as of November 19, 2008, between Abraxis BioScience, LLC and AstraZeneca UK Limited (incorporated by reference to Exhibit 10.24 to the Registrant’s Annual Report on Form 10-K filed with the Securities and Exchange Commission on March 6, 2009) | |
| 10.25 | Employment Agreement, dated as of April 29, 2009, between the Registrant and Leon O. Moulder, Jr. (incorporated by reference to Exhibit 10.25 to the Registrant’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on August 7, 2009) | |
| 10.26 | Offer of Employment by Registrant to Richard J. Rodgers (incorporated by reference to Exhibit 10.26 to the Registrant’s Quarterly Report on Form 10-Q filed with Securities and Exchange Commission on November 9, 2009) | |
| 10.27 | Offer of Employment by Registrant to Mary Lynne Hedley (incorporated by reference to Exhibit 10.27 to the Registrant’s Quarterly Report on Form 10-Q filed with Securities and Exchange Commission on November 9, 2009) | |
| 10.28† | Offer of Employment by Registrant to Mitchell K. Fogelman | |
| 14.1 | Code of Business Conduct (incorporated by reference to Exhibit 14.1 to Registrant’s Annual Report on Form 10-K filed with the Securities and Exchange Commission on March 31, 2008) | |
| 21.1† | List of Subsidiaries | |
| 23.1† | Consent of Independent Registered Public Accounting Firm | |
| 24.1 | Power of Attorney (included on signature page) | |
| 31.1† | Certification of Chief Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | |
| 31.2† | Certification of Principal Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | |
| 32.1† | Certification of Chief Executive Officer pursuant to 18 U.S.C. Section 1350, as adopted by Section 906 of the Sarbanes-Oxley Act of 2002 | |
| 32.2† | Certification of Principal Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted by Section 906 of the Sarbanes-Oxley Act of 2002 | |
| * | Certain portions of this exhibit have been omitted pursuant to a request for confidential treatment filed with the Securities and Exchange Commission. |
| † | Filed herewith. |
102