Attached files
| file | filename |
|---|---|
| EX-32.1 - EXHIBIT 32.1 - Assertio Therapeutics, Inc | a2197080zex-32_1.htm |
| EX-23.1 - EXHIBIT 23.1 - Assertio Therapeutics, Inc | a2197080zex-23_1.htm |
| EX-32.2 - EXHIBIT 32.2 - Assertio Therapeutics, Inc | a2197080zex-32_2.htm |
| EX-31.2 - EXHIBIT 31.2 - Assertio Therapeutics, Inc | a2197080zex-31_2.htm |
| EX-31.1 - EXHIBIT 31.1 - Assertio Therapeutics, Inc | a2197080zex-31_1.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| (Mark One) | ||
ý |
Annual Report Pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 |
|
For the fiscal year ended December 31, 2009 |
||
OR |
||
o |
Transition Report Pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 |
|
For the transition period from: to |
||
Commission File Number: 001-13111
DEPOMED, INC.
(Exact Name of Registrant as Specified in its Charter)
| California | 94-3229046 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) | |
1360 O'Brien Drive, Menlo Park, California |
94025 |
|
| (Address of principal executive offices) | (Zip Code) |
Registrant's telephone number, including area code: (650) 462-5900
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Name of each exchange on which registered | |
|---|---|---|
| Common Stock, no par value | The NASDAQ Stock Market LLC |
Securities
registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No ý
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes o No ý
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Exchange Act during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes o No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ý
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer or a non-accelerated filer, as defined in Rule 12b-2 of the Exchange Act.
| Large accelerated filer o | Accelerated filer ý | Non-accelerated filer o (Do not check if a smaller reporting company) |
Smaller reporting company o |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes o No ý
The aggregate market value of the voting and non-voting common equity held by non-affiliates of the registrant based upon the closing price of Common Stock on the Nasdaq Stock Market on June 30, 2009 was approximately $143,091,000. Shares of Common Stock held by each officer and director and by each person who owned 10% or more of the outstanding Common Stock as of June 30, 2009 have been excluded in that such persons may be deemed to be affiliates. This determination of affiliate status is not necessarily a conclusive determination for other purposes.
The number of shares outstanding of the registrant's Common Stock, no par value, as of March 5, 2010 was 52,330,443.
Documents Incorporated by Reference
Portions of the registrant's Proxy Statement, which will be filed with the Securities and Exchange Commission pursuant to Regulation 14A in connection with the registrant's 2010 Annual Meeting of Shareholders, expected to be held on or about May 20, 2010, are incorporated by reference in Part III of this Form 10-K.
DEPOMED, INC.
2009 FORM 10-K REPORT
TABLE OF CONTENTS
2
NOTE REGARDING FORWARD-LOOKING STATEMENTS
Statements made in this Annual Report on Form 10-K that are not statements of historical fact are forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended (the Securities Act), and Section 21E of the Securities Exchange Act of 1934, as amended. We have based these forward-looking statements on our current expectations and projections about future events. Our actual results could differ materially from those discussed in, or implied by, these forward-looking statements. Forward-looking statements are identified by words such as "believe," "anticipate," "expect," "intend," "plan," "will," "may" and other similar expressions. In addition, any statements that refer to expectations, projections or other characterizations of future events or circumstances are forward-looking statements. Forward-looking statements include, but are not necessarily limited to, those relating to:
- •
- regulatory filings and approval of DM-1796 for postherpetic neuralgia;
- •
- the commercial success and market acceptance of DM-1796 if it is approved for marketing in the United States,
and the efforts of Abbott Laboratories' subsidiary, Solvay Pharmaceuticals, Inc. (Solvay), with respect to the commercialization of DM-1796;
- •
- results and timing of our clinical trials, including the results of our Serada™ Phase 3 trial for
menopausal hot flashes;
- •
- the commercial success and market acceptance of Serada if we receive approval to market Serada in the United States;
- •
- the commercial success of GLUMETZA® (metformin hydrochloride extended release tablets) in the United States,
and the efforts of Santarus, Inc. (Santarus) with respect to the commercialization of GLUMETZA;
- •
- the results of our ongoing litigation against Lupin Limited (Lupin) related to Lupin's abbreviated New Drug Application
(ANDA) to market generic GLUMETZA in the United States;
- •
- the results of our research and development efforts;
- •
- submission, acceptance and approval of regulatory filings;
- •
- our need for, and ability to raise additional capital;
- •
- our collaborative partners' compliance or non-compliance with their obligations under our agreements with
them; and
- •
- our plans to develop other product candidates.
Factors that could cause actual results or conditions to differ from those anticipated by these and other forward-looking statements include those more fully described in the "ITEM 1A. RISK FACTORS" section and elsewhere in this Annual Report on Form 10-K. We disclaim any intent to update or revise these forward-looking statements to reflect new events or circumstances.
The address of our Internet website is http://www.depomed.com. We make available, free of charge through our website or upon written request, our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and other periodic SEC reports, along with amendments to all of those reports, as soon as reasonably practicable after we file the reports with the SEC.
Unless the context indicates otherwise, "Depomed", "the Company", "we", "our" and "us" refer to Depomed, Inc. Depomed was incorporated in the State of California on August 7, 1995. Our principal executive offices are located at 1360 O'Brien Drive, Menlo Park, California 94025, and our telephone number is (650) 462-5900.
Depomed®, Proquin®, and AcuForm® are registered trademarks of Depomed. SeradaTM is a trademark of Depomed. GLUMETZA® is a registered trademark of Biovail Laboratories, s.r.l. exclusively licensed in the United States to Depomed. All other trademarks and trade names referenced in this Annual Report on Form 10-K are the property of their respective owners.
3
COMPANY OVERVIEW
Depomed is a specialty pharmaceutical company focused on the development and commercialization of differentiated products that address large and growing markets and are based on proprietary oral drug delivery technologies. In 2009, we completed Phase 3 clinical trials for two product candidates. In October 2009, we announced that DM-1796, an extended release formulation of gabapentin for the treatment of postherpetic neuralgia that we have licensed to Abbott Laboratories' subsidiary, Solvay Pharmaceuticals, Inc. (Solvay) met its Phase 3 clinical trial primary endpoint with statistical significance. We anticipate a New Drug Application (NDA) for DM-1796 will be filed with the FDA in March 2010. Also in October 2009, we announced the results of Breeze 1 and Breeze 2, our Phase 3 clinical trials for Serada, our proprietary extended release formulation of gabapentin for the treatment of menopausal hot flashes. The higher dose formulation of Serada evaluated in the studies met five of eight co-primary endpoints across the two studies, while the lower dose formulation evaluated met four of eight co-primary endpoints. In December 2009, we met with the FDA to discuss the results of the Breeze 1 and Breeze 2 studies and any additional clinical development that may be required to obtain approval to market Serada in the United States. Based on guidance from the meeting with the FDA, we expect to initiate one additional Phase 3 trial for Serada by the end of April 2010.
We seek to optimize the use and value of our product candidates and drug delivery technologies in three ways. First, we are seeking to assemble a number of pharmaceutical products that can be highly differentiated from immediate release versions of the compounds upon which they are based and may be promoted together within a specialty pharmaceutical field, such as women's health care providers. Our development of Serada, and our retention of co-promotion rights within the obstetrics/gynecology field in our commercialization arrangements with Covidien, Ltd. (Covidien) and Santarus, Inc. (Santarus), are examples of this aspect of our business strategy. Second, we out-license product candidates after we have increased their value through our formulation and clinical development efforts. Our DM-1796 license and development arrangement with Solvay is an example of this strategy. Third, we enter into collaborative partnerships with other companies where the unique capabilities of our technology can provide superior value to a partner's product candidate, resulting in greater value for Depomed than traditional fee-for-service arrangements. Our license and development arrangement with Covidien and our license agreement with Merck & Co., Inc. (Merck) are examples of this strategy.
We have developed two products which have been approved by the FDA and are currently marketed. GLUMETZA is a once-daily treatment for adults with type 2 diabetes that we commercialize in the United States with Santarus. Proquin® XR (ciprofloxacin hydrochloride) is a once-daily treatment for uncomplicated urinary tract infections.
4
The following table summarizes our product pipeline and marketed products.
| Product Pipeline | ||||
Product |
Indication |
Status |
||
|---|---|---|---|---|
| DM-1796 | Postherpetic neuralgia | Phase 3 study completed. New Drug Application expected to be submitted to the FDA by March 2010. Licensed to Solvay in the United States, Mexico and Canada. |
||
SeradaTM |
Menopausal hot flashes |
Phase 3 studies completed (Breeze 1 and Breeze 2). One additional Phase 3 study (Breeze 3) expected to be initiated in Q2 2010. |
||
DM-3458 |
Gastroesophageal reflux disease |
Proof of concept studies completed. |
||
DM-1992 |
Parkinson's disease |
Phase 1 study completed. Second Phase 1 study expected to be initiated in 2010. |
||
Marketed Products |
||||
GLUMETZA® |
Type 2 diabetes |
Currently sold in the United States and Canada. Co-promoted in the United States with Santarus. Canadian rights held by Biovail. Korean rights held by LG Life Sciences. |
||
Proquin® XR |
Uncomplicated urinary tract infections |
Currently sold in the United States. European rights held by Rottapharm/Madaus. |
||
SIGNIFICANT DEVELOPMENTS DURING 2009
Among the significant developments in our business during 2009 were the following:
- •
- In January 2009, our license agreement with Solvay related to DM-1796 became effective after clearance of the
transaction under the HSR Antitrust Improvements Act of 1976. Pursuant to the agreement, Solvay paid us a $25.0 million upfront fee in February 2009.
- •
- In February 2009 and in March 2009, we completed enrollment of our Breeze 1 and Breeze 2 Phase 3 clinical trials
for Serada for the treatment of menopausal hot flashes.
- •
- In February 2009, we dosed the first patient in a Phase 1 clinical trial for our DM-1992 program in
Parkinson's disease.
- •
- In June 2009, we completed enrollment of our Phase 3 clinical trial for DM-1796 for the treatment of
postherpetic neuralgia.
- •
- In May 2009, Shay Weisbrich was appointed as our Vice President, Marketing.
- •
- In July 2009, we entered into a non-exclusive license agreement with Merck granting Merck a license to certain
patents related to our metformin extended release technology to be used in developing fixed dose combinations of sitagliptin and extended release metformin. Pursuant to the agreement, Merck paid us a
$10.0 million upfront fee in July 2009.
- •
- In August 2009, we announced the results of our Phase 1 clinical trial for our DM-1992 program in Parkinson's disease.
5
- •
- In October 2009, we delivered the first formulation under our agreement with Covidien, which triggered a
$0.5 million milestone payment to us.
- •
- In October 2009, we announced the primary endpoint of reducing pain was met with statistical significance in our
Phase 3 clinical trial for DM-1796 for the treatment of postherpetic neuralgia.
- •
- In October 2009, we announced the results of Breeze 1 and Breeze 2, our Phase 3 clinical trials for Serada for the
treatment of menopausal hot flashes. The higher dose formulation of Serada (1800mg) evaluated in the studies met five of eight co-primary endpoints across the two studies, while the lower
dose formulation (1200mg) evaluated met four of eight co-primary endpoints.
- •
- In November 2009, William Callahan was appointed as our Vice President, Operations.
- •
- In December 2009, we met with the FDA to discuss the results of the Breeze 1 and Breeze 2 studies and any additional
clinical development that may be required to obtain approval to market Serada in the United States. Based on guidance from the FDA meeting, we plan to conduct one additional Phase 3 clinical
trial for the 1800mg formulation of Serada.
- •
- Total revenues for the year ended December 31, 2009 were $57.7 million, as compared to $34.8 million
for the year ended December 31, 2008. Revenue for the year ended December 31, 2009 included recognition of $10.0 million associated with our license agreement with Merck. Revenue
for the year ended December 31, 2008 included recognition of $6.3 million in previously deferred GLUMETZA product sales.
- •
- Operating expenses for the year ended December 31, 2009 were $74.5 million, compared to operating expenses
of $46.2 million for the year ended December 31, 2008. Operating expenses for 2008 included a $7.5 million gain on litigation related to the IVAX settlement, which had the effect
of reducing operating expenses.
- •
- Cash, cash equivalents and marketable securities were $81.8 million as of December 31, 2009, compared to $82.1 million as of December 31, 2008.
RECENT PRODUCT DEVELOPMENTS AND TRANSACTIONS
DM-1796 for Postherpetic Neuralgia
General
DM-1796 is our internally developed, extended release formulation of the compound gabapentin. Gabapentin is marketed by Pfizer Inc. for adjunctive therapy for epileptic seizures and postherpetic pain under the trade name Neurontin. It is also marketed by a number of other companies as a generic, immediate release drug.
In November 2008, we entered into an Exclusive License Agreement with Solvay Pharmaceuticals, Inc. granting Solvay exclusive rights to develop and commercialize DM-1796 in the United States, Canada and Mexico for pain indications.
Postherpetic Neuralgia. Postherpetic neuralgia, or PHN, is a persistent pain condition caused by nerve damage during a shingles, or herpes zoster, viral infection. There are an estimated 600,000 to 1 million cases of shingles each year, according to the Centers for Disease Control and Prevention. The incidence of PHN increases in elderly shingles patients, in whom the incidence of PHN in shingles patients 50 to 69 years old is 50 percent and increases to 75 percent in patients over 70 years old, according to WWMR, Inc., a pharmaceutical market research firm. Pain associated with PHN reportedly can be so severe that patients are unable to resume normal activities for months. Since there is no cure for PHN, treatments are focused on relieving pain.
6
Target Market
Approximately 2.6 million people are estimated to have suffered from moderate to severe neuropathic pain in 2004. The overall neuropathic pain market is expected to reach $5 billion in 2010, according to Datamonitor.
Clinical Program
2008/2009 Postherpetic Neuralgia Study.
Study Design. In March 2008, we initiated dosing of the first patient in a Phase 3 clinical trial for DM-1796 for PHN. The study was a randomized, double-blind, placebo-controlled study of approximately 450 PHN patients. Patients in the study were randomized into two treatment arms: placebo, or 1800mg of DM-1796 dosed once daily. The study was conducted at sites in the United States, Russia and Argentina. In June 2009, the study was fully enrolled.
The primary objective of the study was to assess the efficacy of DM-1796 in reducing the pain associated with PHN, measured from baseline pain scores to the end of a ten-week treatment period on the basis of the Likert pain scale. Secondary objectives were also evaluated, including assessments of sleep interference and quality of life.
In October 2009, we announced that the primary endpoint was met with statistical significance.
Efficacy. DM-1796 demonstrated a statistically significant reduction in pain associated with postherpetic neuralgia versus placebo using the baseline observation carried forward (BOCF) method required by FDA. The primary efficacy outcome observed in the study is set forth in the table below.
Treatment Group
|
Reduction in pain score at 10 weeks | |
|---|---|---|
| 1800mg | -2.1 (p = 0.0125) | |
| placebo | -1.6 |
Safety. DM-1796 was generally well tolerated in the study. The most common side effects observed in the study were somnolence and dizziness. The incidence of those side effects in each of the treatment groups for each study is set forth in the table below.
Treatment Group
|
Somnolence (%) | Dizziness (%) | ||
|---|---|---|---|---|
| 1800mg | 5 | 11 | ||
| placebo | 3 | 2 |
2006/2007 Postherpetic Neuralgia Study. In May 2006, we initiated a Phase 3 clinical trial for DM-1796 for the treatment of PHN. The study was a randomized, double-blind, placebo-controlled study of approximately 400 PHN patients. The study was fully enrolled in early March 2007. Patients in the study were randomized into three treatment arms: placebo, a total daily dose of 1800mg of DM-1796 dosed once daily, and a total daily dose of 1800mg of DM 1796 dosed twice daily.
The primary objective of the study was to assess the efficacy of DM-1796 in reducing the pain associated with PHN, measured from baseline pain scores to the end of a ten-week treatment period on the basis of the Likert pain scale. Secondary objectives include an assessment of changes from baseline in sleep interference, and additional patient and clinician assessments of pain and quality of life.
In July 2007, we announced the primary endpoint was not achieved with statistical significance for either active treatment regimen, as compared to placebo, over the ten-week treatment period. The mean reductions in average daily pain scores from baseline to end of study were 1.83 (once-daily), 1.72 (twice-daily) and 1.43 (placebo). However, statistical significance relative to placebo was achieved in
7
each of the first seven weeks for the once-daily treatment arm and in each of the first four weeks for the twice-daily treatment arm.
The secondary endpoints of sleep interference, Clinical Global Impression of Change (CGIC), a scale used by physicians for overall assessment of patient improvement, and Patient Global Impression of Change (PGIC), a scale used by patients to report their overall assessment of change, were all statistically significant for the once-daily treatment compared to placebo over the ten week study period. Sleep interference scores were reduced by 2.01 points with DM-1796 compared to -1.39 with placebo (p=0.014). Physicians reported that 48.0% of patients taking Gabapentin once-daily were "very much improved" or "much improved" compared to 27.1% of the patients who received placebo (p<0.001), as measured by the CGIC. Similar results were observed for the PGIC in the once-daily and placebo arms (p=0.009).
Phase 2 Postherpetic Neuralgia Study. We conducted a randomized, double-blind, placebo-controlled Phase 2 trial of 158 PHN patients, and reported the results of the study in January 2006. Patients were randomized into three treatment groups for four weeks of treatment: placebo, an 1800mg total daily dose of DM-1796 given once daily, and an 1800mg total daily dose of DM-1796 given twice daily. The primary objective of the study was to assess the relative efficacy of DM-1796 once-daily, twice-daily, and placebo in reducing PHN patients' average daily pain scores from baseline to the end of a four-week treatment period on the basis of the Likert pain scale, and 11-point numerical rating scale used to assess pain intensity. Secondary objectives included assessments of changes from baseline in sleep interference, and additional patient and clinician assessments of pain.
Reductions in average daily pain scores were statistically significant with twice-daily DM-1796 from week two to the end of treatment based on the Likert pain scale. Clinically significant improvements in the score were observed with mean change from baseline to study end of -2.24 compared to -1.29 for placebo (p= 0.014). The secondary endpoint of sleep interference was also statistically significantly different, with sleep interference scores reduced by -2.28 with DM-1796 compared to -1.16 with placebo (p=0.006).
For once-daily DM-1796, there was an improvement in pain that did not reach statistical significance, with a reduction in mean daily pain score of -1.93 with DM-1796 compared to -1.29 with placebo (p= 0.089). Sleep Interference Scores were reduced by -1.94 compared to -1.16 with placebo (p=0.048).
There were no serious adverse events associated with DM-1796. The most common side effects observed were dizziness (22% in the once-daily arm, 11% in the twice-daily arm, and 10% in the placebo arm), and somnolence (9% in the once-daily arm, 8% in the twice-daily arm, and 8% in the placebo arm).
Collaboration and Licensing Arrangements
Solvay Pharmaceuticals, Inc. In November 2008, we entered into an exclusive license agreement with Solvay Pharmaceuticals, Inc. granting Solvay exclusive rights to develop and commercialize DM-1796 in the United States, Canada and Mexico for pain indications. The agreement became effective in January 2009, upon clearance of the transaction under the Hart-Scott-Rodino (HSR) Antitrust Improvements Act of 1976.
Pursuant to the agreement, Solvay Pharmaceuticals paid us a $25 million upfront fee in February 2009. We are also eligible to receive aggregate milestone payments of up to $70 million for acceptance and FDA approval of the New Drug Application for DM-1796 for PHN, and up to $300 million in potential sales milestone payments. Solvay will pay us royalties of 14 to 20 percent of net product sales, depending on the level of net product sales.
8
We were responsible for completion of the Phase 3 clinical trial for DM-1796 in PHN, and are responsible for certain other regulatory support activities through NDA approval. Solvay will be responsible for NDA filing and has the option to develop DM-1796 in further pain indications other than PHN. If Solvay elects to develop DM-1796 in fibromyalgia, we have a right of first negotiation for co-promotion rights in the obstetrics/gynecology field upon fibromyalgia indication regulatory approval.
We are responsible for the manufacture of DM-1796 for up to four years from the effective date of the License Agreement, pursuant to a supply agreement to be entered into by Depomed and Solvay in 2010. The License Agreement will expire with the last to expire of our patents covering DM-1796, subject to early termination in certain circumstances.
Abbott Laboratories (Abbott) acquired the pharmaceutical business of Solvay in a transaction that closed in February 2010. Accordingly, Abbott is now responsible for our DM-1796 license arrangement, through its subsidiary, Solvay Pharmaceuticals, Inc.
Serada™ for Menopausal Hot Flashes
General
We have an exclusive sublicense from PharmaNova, under United States Patent No. 6,310,098, held by the University of Rochester, to develop and commercialize in the United States a product that contains gabapentin as its active pharmaceutical ingredient, and is indicated for the treatment of menopausal hot flashes. We believe that Serada is an excellent non-hormonal product candidate for this indication, because our clinical study and numerous academic studies have demonstrated that gabapentin may be effective in treating hot flashes, and gabapentin has a long history of use in other indications.
Hot Flashes
A hot flash is a sudden flushing and sensation of heat caused by dilation of skin capillaries. Hot flashes are often associated with menopausal endocrine imbalance. The occurrence and frequency of hot flashes are unpredictable. Symptoms often associated with hot flashes include sweating, irritability and frustration.
Hot flashes can begin early in menopause and are most common during the first few years after menopause begins. There are over 40 million postmenopausal women more than 55 years old and about 2 million women enter menopause every year in the United States. Approximately 80% of those women suffer from hot flashes.
Current Treatments; Target Market
Currently, the leading prescription drug product for the treatment of hot flashes associated with menopause is hormone replacement therapy, or HRT, which involves the administration of the hormone estrogen, either alone or in combination with the hormone progestin. In 2001, the HRT market represented more than $2 billion and in excess of 90 million prescriptions. In 2003, the Women's Health Initiative released the results of a clinical study that revealed an increased risk of blood clots, stroke, and breast cancer associated with HRT. Subsequently, in 2003, the HRT market decreased by more than $850 million and 34 million prescriptions relative to 2001. HRT prescriptions have declined to approximately 32 million prescriptions in 2008.
Existing non-hormonal pharmaceutical alternatives to HRT for the treatment of hot flashes include off-label administration of anti-depressants. There is also a considerable market for dietary and herbal supplements for the treatment of hot flashes, although we are not aware of any clinical study demonstrating the safety and efficacy of any such treatment.
9
Clinical Program
Phase 3 Study-Breeze 3 Clinical Trial. In December 2009, the Company met with the FDA regarding Serada for the treatment of menopausal hot flashes. Based on guidance reflected in the meeting minutes, Depomed plans to conduct a single additional pivotal Phase 3 trial evaluating Serada for the treatment of menopausal hot flashes. The company expects to initiate the trial, which will be known as Breeze 3, by the end of April 2010 and expects to complete the trial by the end of the first quarter of 2011. The FDA has agreed to review the proposed Breeze 3 study protocol under the FDA's Special Protocol Assessment (SPA) process.
Study Design. Breeze 3 will be a randomized, double-blind, placebo-controlled study of up to 600 patients. Patients will be randomized into one of two treatment arms, with patients receiving either placebo or a total dose of 1800mg of Serada dosed 600mg in the morning and 1200mg in the evening. The co-primary efficacy endpoints in the study will be reductions in the mean frequency of moderate-to-severe hot flashes, and the average severity of hot flashes, measured after four and 12 weeks of stable treatment. As in the prior Breeze 1 trial, the treatment duration of the study will be 24 weeks, to address the FDA's view that an effective drug should also show statistically significant persistence of efficacy at 24 weeks. The trial will also include a responder analysis to assess the clinical meaningfulness of any reduction in the frequency of hot flashes in the active arm relative to the placebo arm.
Modifications to the design of Breeze 3 relative to Breeze 1 and 2 include: (i) a single active arm rather than two arms, and therefore a required statistical p value of .05 rather than .025 to achieve statistical significance; (ii) up to 65% more patients in the active treatment arm than in Breeze 1 and 2 (iii) a two-week run in period to prior to randomization, rather than one week, which is designed to reduce the regression to the mean observed in Breeze 1 and 2, resulting in a more stable baseline, and thereby potentially reducing the placebo effect; and (iv) an alternative statistical analysis method, known as a non-parametric analysis, that is designed to reduce the influence significant outliers can have on the achievement of efficacy endpoints.
Phase 3 Study-Breeze 1 and 2 Clinical Trials.
Study Design. We recently completed our Breeze 1 and 2 clinical trials evaluating Serada in menopausal hot flashes. Each trial was a Phase 3 randomized, double-blind, placebo-controlled study of approximately 540 patients. In September 2008, we enrolled and dosed the first patient in Breeze 1, and in October 2008, we enrolled and dosed the first patient in Breeze 2. In each study, patients were randomized into three treatment arms: (i) placebo; (ii) 1200mg of Serada dosed once daily; or (iii) a total dose of 1800mg of Serada dosed 600mg in the morning and 1200mg in the evening. We completed enrollment in Breeze 1 in February 2009, and completed enrollment in Breeze 2 in March 2009.
The treatment duration of the Breeze 1 study was six months, with primary efficacy endpoints assessed at 4 and 12 weeks. Persistence of efficacy was assessed at 6 months as one of the secondary endpoints. The treatment duration in the second study, Breeze 2, was three months, with assessment of efficacy at 4 and 12 weeks only.
The primary efficacy endpoints in both studies were reductions in the mean frequency of moderate to severe hot flashes, and the average severity of hot flashes. Various secondary efficacy endpoints were measured as well.
10
Efficacy. As set forth in the table below, in the higher dose treatment arm of the two doses evaluated, the 1800mg dose achieved positive results at 4 weeks. All four co-primary endpoints of the 1800mg dose at 4 weeks demonstrated significant reductions in frequency and severity in both clinical trials. Of the other four co-primary endpoints of the 1800mg dose at 12 weeks, one endpoint was positive (p=0.0026) while the other three endpoints did not achieve statistical significance.
In the lower dose treatment arm, the 1200mg dose at 4 weeks achieved statistical significance in three of the four co-primary endpoints. The Frequency of hot flashes were significantly reduced in both clinical trials (p-values of 0.0024 and 0.0117) at four weeks. Severity was significantly reduced in only one trial (p=0.0016). Of the other four co-primary endpoints of the 1200mg dose at 12 weeks, one endpoint was positive (p=0.0024) while the other three endpoints did not achieve statistical significance.
Both patients' and clinicians' impression of overall improvement in the higher dose treatment arm was highly statistically significant relative to placebo in both studies.
The primary efficacy outcomes observed in the studies are set forth in the tables below.
| Breeze 1 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Frequency (# per day) | Severity (average per incident) | ||||||||||
Treatment Group
|
Baseline | 4 weeks | 12 weeks | Baseline | 4 weeks | 12 weeks | ||||||
| 1800mg | 11.1 | 3.8 (p £ 0.0001) | 3.7 (p = 0.2) | 2.5 | 1.8 (p £ 0.0001) | 1.7 (p = 0.0468) | ||||||
| 1200mg | 11.3 | 4.5 (p = 0.0117) | 3.8 (p = 0.183) | 2.5 | 1.9 (p = 0.0016) | 1.7 (p = 0.0433) | ||||||
| placebo | 11.3 | 5.4 | 4.3 | 2.5 | 2.1 | 1.8 | ||||||
| Breeze 2 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Frequency (# per day) | Severity (average per incident) | ||||||||||
Treatment Group
|
Baseline | 4 weeks | 12 weeks | Baseline | 4 weeks | 12 weeks | ||||||
| 1800mg | 11.2 | 4.1 (p = 0.004) | 3.7 (p = 0.028) | 2.5 | 1.8 (p = 0.0003) | 1.6 (p = 0.0026) | ||||||
| 1200mg | 12.0 | 4.7 (p = 0.0024) | 3.9 (p = 0.0024) | 2.5 | 1.9 (p = 0.06) | 1.7 (p = 0.028) | ||||||
| placebo | 11.2 | 5.7 | 5.0 | 2.5 | 2.0 | 1.9 | ||||||
Safety. Serada was generally well tolerated in the study. The most common side effects observed in the study were headache, somnolence, dizziness and nausea. The incidence of those side effects in each of the treatment groups for each study are set forth in the tables below:
| Breeze 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
Treatment Group
|
Somnolence (%) | Dizziness (%) | Headache (%) | Nausea (%) | ||||
| 1800mg | 19 | 19 | 9 | 9 | ||||
| 1200mg | 13 | 24 | 9 | 7 | ||||
| placebo | 2 | 3 | 6 | 4 | ||||
| Breeze 2 | ||||||||
|---|---|---|---|---|---|---|---|---|
Treatment Group
|
Somnolence (%) | Dizziness (%) | Headache (%) | Nausea (%) | ||||
| 1800mg | 8 | 19 | 7 | 7 | ||||
| 1200mg | 7 | 17 | 5 | 3 | ||||
| placebo | 3 | 3 | 8 | 2 | ||||
11
Withdrawals due to adverse events in each of the treatment groups for each study are set forth in the tables below:
| Breeze 1 | ||||
|---|---|---|---|---|
Treatment Group
|
Somnolence (%) | Dizziness (%) | ||
| 1800mg | 2 | 1 | ||
| 1200mg | 3 | 3 | ||
| placebo | 0 | 0 | ||
| Breeze 2 | ||||
|---|---|---|---|---|
Treatment Group
|
Somnolence (%) | Dizziness (%) | ||
| 1800mg | 2 | 3 | ||
| 1200mg | 0.5 | 3 | ||
| placebo | 0.5 | 0 | ||
Phase 2 Study. In June 2007, we randomized the first patient in a Phase 2 double-blind, placebo-controlled, multi-center trial evaluating Serada for the treatment of women with moderate-to-severe menopausal hot flashes. The 124 patient study was fully enrolled in September 2007. In February 2008, we announced positive results of our Phase 2 trial for Serada for moderate-to-severe menopausal hot flashes.
Study Design. The study included 124 menopausal women (approximately 30 per group) with recurrent, moderate to severe hot flashes and was conducted at eight sites in the United States. The total study treatment duration after screening and baseline was 13 weeks. The primary objective of the study was to investigate the relationship between blood plasma concentrations of gabapentin observed in menopausal women after administration of Serada and the frequency of hot flashes in those women. The plasma concentration data (pharmacokinetics) and the hot flash frequency and severity data (pharmacodynamics) are being used to construct a PK/PD dose response model designed to identify the dosing regimen to utilize in the Phase 3 program.
In order to facilitate the generation of an optimal dose response model, patients in each of the three active treatment arms remained on a stable Serada dose for five weeks at an initial dose, followed by five weeks on a stable, incrementally higher dose, as follows.
| Treatment Group | Weeks 2 – 6 | Weeks 8 – 12 | ||
|---|---|---|---|---|
| A ("1800mg group") | 600mg PM | 600mg AM + 1200mg PM | ||
| B ("2400mg group") | 600mg AM + 600mg PM | 600mg AM + 1800mg PM | ||
| C ("3000mg group") | 1200mg PM | 1200mg AM + 1800mg PM | ||
| D ("placebo group") | placebo | placebo |
Each stable dosing regimen was preceded by a one-week titration period
Efficacy. Serada demonstrated a reduction in the mean frequency of moderate to severe hot flashes, and in the mean total daily severity of hot flashes, in all active treatment groups. Statistical significance relative to placebo from baseline to the end of the study was observed in the 1800mg and 2400mg treatment groups with regard to frequency, and statistical significance was observed in the 1800mg treatment group with regard to severity. The severity of hot flashes is based on a mean daily composite score, where a moderate hot flash is assigned a score of "2" and a severe hot flash is
12
assigned a score of "3". The primary efficacy outcomes observed in the study are set forth in the table below.
| |
Mean Daily Frequency (#) | Mean Total Daily Severity Score | ||||||
|---|---|---|---|---|---|---|---|---|
Treatment Group
|
Baseline | End of treatment | Baseline | End of treatment | ||||
| 1800mg | 10.1 | 2.7 (p = 0.016) | 24.0 | 6.9 (p = 0.044) | ||||
| 2400mg | 11.8 | 3.0 (p = 0.03) | 29.6 | 6.8 (p = 0.041) | ||||
| 3000mg | 11.4 | 3.9 (p = 0.229) | 27.8 | 10.2 (p = 0.426) | ||||
| placebo | 10.6 | 5.1 | 26.7 | 12.2 | ||||
Safety. Serada was generally well tolerated in the study, with one, two, one and three patients, respectively, withdrawing due to adverse events from the placebo, 1800mg, 2400mg and 3000mg groups. The most common side effects observed in the study were headache, somnolence, dizziness and nausea. The incidence of those side effects in each of the treatment groups is set forth in the table below.
Treatment Group
|
Somnolence (%) | Dizziness (%) | Headache (%) | Nausea (%) | ||||
|---|---|---|---|---|---|---|---|---|
| 1800mg | 16 | 10 | 32 | 16 | ||||
| 2400mg | 16 | 39 | 32 | 3 | ||||
| 3000mg | 16 | 9 | 25 | 3 | ||||
| placebo | 3 | 10 | 10 | 7 |
Collaboration and License Arrangements
PharmaNova. In October 2006, we entered into a sublicense agreement with PharmaNova, Inc. Pursuant to the agreement, PharmaNova has granted us an exclusive sublicense, under United States Patent No. 6,310,098, held by the University of Rochester, to develop and commercialize in the United States a product that contains gabapentin as its active pharmaceutical ingredient, and is indicated for the treatment of hot flashes associated with menopause.
We paid PharmaNova an upfront license fee of $0.5 million upon signing of the agreement and paid an additional $0.5 million upon dosing of the first patient in our Phase 3 trials for the product. We are required to pay PharmaNova $1.0 million upon submission to the FDA of a NDA for the product, and $2.0 million upon FDA approval of an NDA. The agreement provides for royalty payments to PharmaNova on net sales of the product, and for milestone payments upon achievement of annual net sales in excess of certain thresholds. We also paid PharmaNova consultancy fees of $0.3 million over approximately ten months beginning in November 2006.
DM-3458 for Gastroesophageal Reflux Disease
General
Gastroesophageal reflux disease, or GERD, is a disorder of the digestive system caused by the failure of the lower esophageal sphincter muscle, or LES, to close properly, which permits stomach contents to leak back into the esophagus. When stomach contents pass through the LES into the esophagus, stomach acid causes the burning sensation in the chest or throat known as heartburn. Heartburn that occurs more than twice a week may be GERD. Other symptoms of GERD can include acid indigestion, bad breath, chest pain, hoarseness in the morning, and trouble swallowing. According to the NIDDK, 20% of the US population suffers from GERD.
GERD Treatments; Target Market
Treatments for GERD include: antacids designed to neutralize stomach acid, such as Alka-Seltzer, Mylanta and Rolaids, among others; foaming agents, such as Gaviscon, that cover stomach contents
13
with foam in order to prevent reflux; H2 blockers, such as cimetidine (Tagamet HB), famotidine (Pepcid AC) and ranitidine (Zantac), among others; proton pump inhibitors, or PPIs, such as omeprazole (Prilosec), lansoprazole (Prevacid), and esomperzole (Nexium), among others; and drugs known as prokinetics that are designed to strengthen the LES and accelerate stomach emptying. GERD treatments are often taken in combination.
The US market for GERD treatments was in excess of $14 billion in 2008, according to IMS Health, Inc., a pharmaceutical market research firm.
Clinical Program
In 2006, we conducted a Phase 1 study designed to provide us with insight into our formulation strategy, and in 2007, we conducted a proof-of-concept study related to our DM-3458 program. No additional DM-3458 clinical studies are currently planned, as we are awaiting the results of our efforts to enter into a development and commercialization partnership for the product candidate.
DM-1992 for Parkinson's Disease
General
Parkinson's disease is a chronic degenerative disorder that affects nearly one million Americans, with significant prevalence growth expected over the next 25 years due to aging population demographics. Nearly 5 million people worldwide are estimated to have Parkinson's. While the average age at onset is 60, disease onset starts by age 40 in an estimated 5 to 10 percent of patients, and people as young as 30 can also be affected.
Parkinson's Treatments; Target Market
Current therapies are effective in addressing only the mild/moderate motor symptoms of the disease and have significant long-term side effects. Levodopa/carbidopa is the common treatment of Parkinson's but currently has limitations with inconsistent efficacy and inconvenient dosing since it absorbed in the upper GI tract. Levodopa/carbidopa is available as a generic (brand name Sinemet) and had $270 million in sales in the United States in 2006.
Clinical Program
In July 2008, The Michael J. Fox Foundation awarded the Company a preclinical development grant to support the DM-1992 program. DM-1992 is our investigative novel gastric retentive extended-release formulation of levodopa/carbidopa.
In January 2009, we initiated a Phase 1 pharmacokinetic study in Parkinson's patients designed to provide us with insight into our formulation strategy for the DM-1992 program. The Phase 1 trial in DM-1992 was a randomized, open-label crossover study that enrolled 18 patients with stable Parkinson's disease at two leading neurology centers in Russia. The objective of the study was to compare the pharmacokinetics of two distinct formulations of DM-1992 and a generic version of Sinemet CR sustained-release levodopa/carbidopa, as well as the safety and tolerability of the formulations. Patients in the trial received a single dose of each of the three treatments being studied. A dose of the first treatment was administered at the beginning of the study, followed by a dose of a second treatment after 7 to 14 days, and a dose of the third treatment after another 7 to 14 days. Blood samples were drawn during the 24 hour period following administration of each treatment. Patients remained on any anti-Parkinson's therapy other than levodopa/carbidopa during the trial.
In August 2009, we completed the Phase 1 study. In the study, DM-1992 extended coverage above levodopa's efficacious threshold and extended the time to peak levodopa concentration relative to currently available sustained release levodopa/carbidopa formulations. One of our formulations tested
14
in the study extended the median time at which levodopa blood levels stayed above the efficacious threshold of 300 ng/mL to approximately nine hours, compared to approximately seven hours for the generic version of Sinemet CR tested in the study. The time to median peak levodopa blood levels in the study was extended to four hours, compared to 2.8 hours for the comparator.
We anticipate commencing a second Phase 1 study in 2010.
OTHER RESEARCH AND DEVELOPMENT AND COLLABORATIVE PROGRAMS
Merck & Co., Inc. In July 2009, we entered into a non-exclusive license agreement with Merck & Co., Inc. granting Merck a license to certain patents related to our metformin extended release technology to be used in developing fixed dose combinations of sitagliptin and extended release metformin.
Under terms of the agreement, Merck received a non-exclusive license as well as other rights to certain of our patents directed to metformin extended release technology. In exchange, we received a $10.0 million upfront fee in the third quarter of 2009. We are also eligible to receive a milestone payment upon filing of the New Drug Application for the therapeutic candidate, as well as modest royalties on any net product sales for an agreed-upon period. Merck will also be granted a right of reference to the New Drug Application covering our GLUMETZA product in Merck's regulatory filings covering fixed dose combinations of sitagliptin and extended release metformin. We have no development obligations under the agreement.
Covidien. In November 2008, we entered into a license agreement with Mallinckrodt, Inc., a subsidiary of Covidien, Ltd. (Covidien) granting Covidien worldwide rights to utilize the Company's Acuform® technology for the exclusive development of four products containing acetaminophen in combination with opiates. In 2008, Covidien paid us a total of $5.5 million in upfront fees, representing a $4.0 million upfront license fee and a $1.5 million upfront payment for formulation work to be performed by Depomed under the agreement. Under the agreement, we may also receive certain developmental milestone payments, if achieved, and is also entitled to receive royalties on sales of the products.
In October 2009, we completed and delivered the first formulation for Covidien, which triggered a $0.5 million milestone payment to us from Covidien in October 2009. In December 2009, Covidien paid us an additional $0.5 million milestone payment, which is related to the second formulation. The second formulation is not complete and we expect to provide formulation work during the first half of 2010 to complete this formulation.
RESEARCH AND DEVELOPMENT EXPENSES
Our research and development expenses were $34.3 million in 2009, $27.3 million in 2008 and $23.3 million in 2007.
MARKETED PRODUCTS
GLUMETZA
General
The 500mg strength of GLUMETZA is our internally developed once-daily metformin product for type 2 diabetes. The FDA approved GLUMETZA for marketing in the United States in June 2005. At that time, a subsidiary of Biovail Corporation (Biovail) held US and Canadian marketing rights to GLUMETZA pursuant to a license agreement we entered into with the Biovail subsidiary in 2002. We reacquired the US rights to GLUMETZA from Biovail in December 2005.
15
In June 2006, we entered into a promotion arrangement with King Pharmaceuticals (King) related to GLUMETZA under which we and King jointly commercialized GLUMETZA in the United States. GLUMETZA was launched in the United States in September 2006. In October 2007, we terminated our promotion agreement with King related to GLUMETZA, and King paid us $29.7 million in termination and other fees. King ended promotion of GLUMETZA in December 2007.
In July 2008, we entered into a promotion agreement with Santarus granting Santarus exclusive rights to promote GLUMETZA in the United States. Santarus began promotion of GLUMETZA in October 2008.
In connection with the restructuring of our GLUMETZA agreements with Biovail in December 2005, we also acquired the exclusive US license to a 1000mg strength of GLUMETZA utilizing proprietary Biovail drug delivery technology. In December 2007, the FDA approved the 1000mg formulation for marketing in the United States, and we began selling the 1000mg GLUMETZA in June 2008.
The 500mg and 1000mg GLUMETZA have also been approved for marketing in Canada, where they are marketed by Biovail.
Diabetes
Diabetes is a disease in which levels of glucose, a type of sugar found in the blood, are above normal. Diabetic patients do not produce insulin, a hormone produced in the pancreas, or do not properly use insulin, making it difficult for the body to convert food into energy. Type 2 diabetes is the most common form of diabetes, accounting for 90 to 95 percent of all diabetes cases, according to the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health, or the NIDDK.
The body breaks down food into glucose, and delivers glucose to cells through the bloodstream. Cells use insulin to help process blood glucose into energy. In the case of type 2 diabetes, cells fail to use insulin properly or the pancreas cannot make as much insulin as the body requires. That causes the amount of glucose in the blood to increase, while starving cells of energy. Over time, high blood glucose levels damage nerves and blood vessels, which can lead to complications such as heart disease, stroke, blindness, kidney disease, nerve problems, gum infections, and amputation.
Target Market
According to the American Diabetes Association (ADA), 23.6 million people in the United States have diabetes. Of those, 17.9 million are diagnosed. The ADA estimates that the number of diagnosed diabetics in the United States is increasing by approximately one million per year. Among adults with diagnosed diabetes, 57 percent take oral medication only, and 12 percent take both insulin and oral medication, according to the 2001-2003 National Health Interview Survey of the Centers for Disease Control and Prevention. In 2008, the metformin market in the United States was approximately $1.4 billion in retail sales.
GLUMETZA Collaboration, Commercialization and Licensing Arrangements
Santarus, Inc. In July 2008, we entered into a promotion agreement with Santarus granting Santarus exclusive rights to promote GLUMETZA in the United States. Santarus paid us a $12.0 million upfront fee, and based on the achievement of specified levels of annual GLUMETZA net product sales, Santarus may be required to pay us additional one-time sales milestones, totaling up to $16.0 million.
Santarus began promotion of GLUMETZA in October 2008. Under the promotion agreement, Santarus is required to meet certain minimum promotion obligations during the term of the agreement,
16
and required to make certain minimum marketing, advertising, medical affairs and other commercial support expenditures. We continue to record revenue from the sales of GLUMETZA product and pay Santarus a promotion fee equal to 80% of the gross margin earned from net sales of GLUMETZA product in the United States. The promotion fee will be reduced to 75% of gross margin beginning in the fourth quarter of 2010.
Santarus is responsible for all costs associated with its sales force and for all other marketing expenses associated with its promotion of GLUMETZA. We are responsible for overseeing product manufacturing and supply. A joint commercialization committee has been formed to oversee and guide the strategic direction of the GLUMETZA alliance.
Pursuant to the terms of the promotion agreement, we retain the option to co-promote GLUMETZA product in the future to obstetricians and gynecologists. The promotion agreement will continue in effect until the expiration of the last-to-expire patent or patent application with a valid claim in the territory covering a GLUMETZA product, unless terminated sooner.
Publicis Selling Solutions. In February 2008, we entered into a professional detailing services agreement with Publicis Selling Services pursuant to which approximately 33 part-time Publicis sales representatives detailed GLUMETZA to physicians. The arrangement with Publicis ended in September 2008.
King Pharmaceuticals. In June 2006, we entered into a promotion agreement with King Pharmaceuticals pursuant to which we granted King the co-exclusive right to promote GLUMETZA in the United States. Under the agreement, King was required to promote GLUMETZA to physicians in the United States through its sales force, to deliver a minimum number of annual detail calls to potential GLUMETZA prescribers, and to maintain a sales force of a minimum size. In consideration for its promotion of GLUMETZA, King received a promotion fee equal to fifty percent of gross margin of GLUMETZA product sales. Out-of-pocket marketing expenses were shared with King at an agreed-upon ratio, in which Depomed's share was lower than King's.
In October 2007, we and King terminated the promotion agreement. Pursuant to the termination agreement, King paid us $29.7 million in termination and other fees, and fulfilled its GLUMETZA promotion obligations through December 31, 2007.
Biovail. We licensed US and Canadian rights to GLUMETZA to Biovail in 2002. In 2005, we received a $25.0 million license fee payment from Biovail under our original license agreement following FDA approval of GLUMETZA. In December 2005, we and Biovail entered into an amended and restated license agreement relating to GLUMETZA. The amended and restated license agreement supersedes our April 27, 2004 amended license and development agreement with Biovail.
Pursuant to the amended and restated license agreement, Biovail has an exclusive license in Canada to manufacture and market the 500mg GLUMETZA, and we receive royalties of six percent of Canadian net sales of the 500mg GLUMETZA. We also receive payments from Biovail equal to one percent of Canadian net sales of the 1000mg GLUMETZA. The royalty payable by Biovail on net sales of the 500mg GLUMETZA was increased to ten percent for the period from June 30, 2007 to December 28, 2007, and returned to six percent when we obtained regulatory approval in the United States of the 1000mg formulation of GLUMETZA.
In December 2005, we also entered into a manufacturing transfer agreement and a supply agreement with Biovail related to the 1000mg GLUMETZA. Under those agreements, we received an exclusive license to market the 1000mg GLUMETZA in the United States, and an exclusive license to the "GLUMETZA" trademark in the United States for the purpose of marketing GLUMETZA. We purchase the 1000mg GLUMETZA exclusively from Biovail under the supply agreement, subject to back-up manufacturing rights in our favor. If we exercise our back-up manufacturing rights,
17
compensation to Biovail will change from a supply-based arrangement to royalties of six percent of net sales of the 1000mg GLUMETZA in the United States (or, if less, thirty percent of royalties and other similar payments from our licensees) under the manufacturing transfer agreement.
We also pay Biovail royalties of one percent of net sales of the 500mg GLUMETZA in the United States (or, if less, five percent of royalties and other similar payments from our licensees).
LG Life Sciences. In August 2004, we entered into a license and distribution agreement granting LG Life Sciences an exclusive license to our 500mg extended-release formulation of metformin in the Republic of Korea. LG Life Sciences launched the product, known as Novamet GR, in Korea in 2006.
We received a $0.6 million upfront license fee from LG in connection with entering into the agreement. In November 2006, we amended the agreement to provide for a $0.5 million milestone payment from LG with respect to LG's approval to market Novamet GR in the Republic of Korea, rather than a $0.7 million payment, as reflected in the original agreement. We received the $0.5 million payment in November 2006, net of applicable Korean withholding taxes, and commenced negotiations to further amend the agreement to formally grant LG a license to manufacture Novamet GR in exchange for royalties on net sales of Novamet GR in Korea, and to remove the provisions of the original agreement providing for the supply of Novamet GR tablets by us to LG. In January 2007, we completed our negotiations with LG, and entered into an amended license agreement implementing the provisions.
Proquin® XR
General
Proquin XR is a once-daily formulation of the antibiotic ciprofloxacin for uncomplicated urinary tract infections. We developed Proquin XR, and the FDA approved it for marketing in the United States in May 2005. Esprit Pharma (Esprit) licensed marketing rights to Proquin XR in the United States in July 2005, and launched the product in the United States in November 2005. We terminated our agreement with Esprit in July 2007 related to Proquin XR and relaunched Proquin XR with Watson in September 2007. Our agreement with Watson terminated on December 31, 2009. We are seeking to divest Proquin XR in the United States.
Proquin XR Collaboration and Licensing Arrangements
Watson Pharma. In July 2007, following the termination of our Proquin XR licensing arrangement with Esprit Pharma described below, we entered into a promotion agreement with Watson Pharma, a subsidiary of Watson Pharmaceuticals, granting Watson a co-exclusive right to promote Proquin XR to the urology specialty and long-term care facilities in the United States. In September 2007, we amended the agreement to also grant Watson a co-exclusive right to promote Proquin XR to the ob/gyn specialty. Under the agreement, Watson was required to deliver a minimum number of annual sales detail calls and maintain a sales force of a minimum size, and received a promotion fee equal to an agreed upon portion of gross margin attributable to the urology and ob/gyn specialties and long-term care facilities above an agreed upon baseline level. We began selling Proquin XR in September 2007 and Watson began promotion in October 2007.
In February 2009, we amended the agreement with Watson and were no longer obligated to pay Watson a promotion fee beginning in the first quarter of 2009, and Watson performed a specified number of physician details in the first quarter of 2009.
Under the provisions of the agreement, Watson elected to terminate the agreement early, and the promotion agreement terminated effective December 31, 2009.
18
Esprit Pharma. In July 2005, we entered into an exclusive license and marketing agreement with Esprit Pharma, Inc. pursuant to which we granted Esprit exclusive US marketing and distribution rights to Proquin XR. The agreement obligated Esprit to pay us $50.0 million in license fees; $30.0 million, which was paid in 2005, and two $10.0 million installments in July 2006 and in July 2007. The agreement also provided for royalty payments to us of 15 percent to 25 percent of Proquin XR net sales, based on escalating net sales, subject to minimum royalty obligations of $4.6 million in 2006, $5.0 million in 2007, and in subsequent years, the minimum royalty was to increase in line with annual increases in the consumer price index beginning in 2008. We also had a supply agreement with Esprit under which we provided Esprit with commercial quantities of Proquin XR.
In July 2006, we amended the license agreement with Esprit to, among other matters, extend to December 2006 the due date on the $10.0 million license fee payment to Depomed that had been due in July 2006, to credit royalties paid to us for sales made in the fourth quarter of 2005 of Proquin XR against Esprit's $4.6 million minimum royalty obligation in 2006, and to establish a joint marketing team to periodically review and discuss all aspects of the commercialization of Proquin XR.
In December 2006, we delivered a notice to Esprit regarding alleged breaches by Esprit of the license agreement. The alleged breaches related to Esprit's failure to make a $10.0 million license fee payment due to the Company in December 2006, and to use commercially reasonable efforts to market Proquin XR. In connection with the notice, we filed a demand for binding arbitration. Subsequent to the delivery of the notice and demand for arbitration, Esprit paid us the $10.0 million license fee payment in December 2006 and we withdrew without prejudice the notice and demand for arbitration. We also agreed to commence, in January 2007, discussions with Esprit toward a mutually agreeable, long-term restructuring of the license agreement.
In July 2007, we entered into a termination and assignment agreement with Esprit terminating the exclusive license and marketing agreement, and the related supply and co-promotion agreements. Upon entering into the termination and assignment agreement, the marketing and distribution rights in the United States for Proquin XR were transferred back to us and Esprit paid us $17.5 million, representing (i) a $10.0 million payment in respect of the final license payment that would have been due to us in July 2007 under the exclusive license and marketing agreement; (ii) a $2.5 million payment in respect to a pro-rated portion minimum royalties for 2007; and (iii) a $5.0 million termination fee. The termination and assignment agreement removed Esprit's future minimum royalty obligations to us. Under the termination and assignment agreement, Esprit remains responsible for all returns and rebates associated with Proquin XR product distributed by Esprit.
Rottapharm/Madaus. In November 2005, we entered into a distribution and supply agreement for Proquin XR in Europe with a privately owned specialty pharmaceutical company, Madaus S.r.l., that was acquired by Rottapharm (Rottapharm) in June 2007. Under the terms of the agreement, we granted an exclusive right to Rottapharm for the commercialization of Proquin XR in Europe and agreed to supply Rottapharm with commercial quantities of Proquin XR tablets in bulk form. In March 2006, Rottapharm filed a Marketing Authorization Application for Proquin XR with the Medical Products Agency in Sweden. In July 2008, the Medical Products Agency in Sweden approved the Marketing Authorization.
In April 2009, we and Rottapharm entered into an amended and restated license agreement for Proquin XR in Europe, which amended the parties' distribution and supply agreement originally entered into in November 2005 and subsequently amended in November 2006. Under the amended and restated license agreement, we will no longer be obligated to supply commercial quantities of Proquin XR tablets in bulk form to Rottapharm as contemplated under the distribution and supply agreement, and we will now receive royalties on net sales of Proquin XR in Europe sold by Rottapharm. We are obligated to provide regulatory and manufacturing support and consultation for up to an agreed upon
19
number of hours per month through December 31, 2010 as reasonably requested by Rottapham. The term of the amended and restated license agreement is through July 2023.
OUR DRUG DELIVERY TECHNOLOGIES
The AcuForm technology is based on our proprietary oral drug delivery technologies and is designed to include formulations of drug-containing polymeric tablets that allow multi-hour delivery of an incorporated drug. Although our formulations are proprietary, the polymers utilized in the AcuForm technology are commonly used in the food and drug industries and are included in the list of inert substances approved by the FDA for use in oral pharmaceuticals. By using different formulations of the polymers, we believe that the AcuForm technology is able to provide continuous, controlled delivery of drugs of varying molecular complexity and solubility. With the use of different polymers and polymers of varying molecular weight, our AcuForm tablet technology can deliver drugs by diffusion, tablet erosion, or from a bi-layer matrix. In addition, our technology allows for the delivery of more than one drug from a single tablet. If taken with a meal, these polymeric tablets remain in the stomach for an extended period of time to provide continuous, controlled delivery of an incorporated drug.
The AcuForm technology's design is based in part on principles of human gastric emptying and gastrointestinal transit. Following a meal, liquids and small particles flow continuously from the stomach into the intestine, leaving behind the larger undigested particles until the digestive process is complete. As a result, drugs in liquid or dissolved form or those consisting of small particles tend to empty rapidly from the stomach and continue into the small intestine and on into the large intestine, often before the drug has time to act locally or to be absorbed in the stomach and/or upper small intestine. The drug-containing polymeric tablets of the AcuForm technology are formulated into easily swallowed shapes and are designed to swell upon ingestion. The tablets attain a size after ingestion sufficient to be retained in the stomach for multiple hours during the digestive process while delivering the drug content at a controlled rate. After drug delivery is complete, the polymeric tablet dissolves and becomes a watery gel, which is safely eliminated through the intestine sight unseen.
20
The following graphic demonstrates the operation of the AcuForm technology.
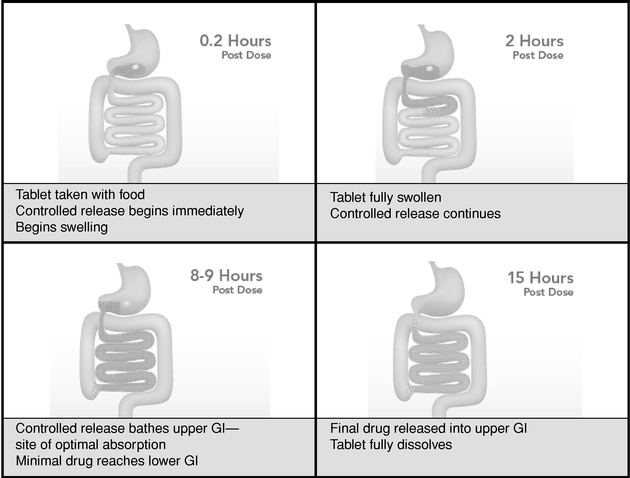
The AcuForm technology is designed to address certain limitations of drug delivery and to provide for orally-administered, conveniently-dosed, cost-effective drug therapy that provides continuous, controlled-delivery of a drug over a multi-hour period. We believe that the AcuForm technology can provide one or more of the following advantages over conventional methods of drug administration:
- •
- Greater Patient and Caregiver Convenience. We believe that
the AcuForm technology may offer once-daily or reduced frequency dosing for certain drugs that are currently required to be administered several times daily. Less frequent dosing promotes
compliance with dosing regimens. Patient noncompliance with dosing regimens has been associated with increased costs of medical therapies by prolonging treatment duration, increasing the likelihood of
secondary or tertiary disease manifestation and contributing to over-utilization of medical personnel and facilities. By improving patient compliance, providers and third-party payors may
reduce unnecessary expenditures and improve therapeutic outcomes.
- •
- Enhanced Safety and Efficacy through Controlled Delivery. We believe that the AcuForm technology may improve the ratio of therapeutic effect to toxicity by decreasing the initial peak concentrations of a drug associated with toxicity, while maintaining levels of the drug at therapeutic, subtoxic concentrations for an extended period of time. Many drugs demonstrate optimal efficacy when concentrations are maintained at therapeutic levels over an extended period of time. When a drug is administered intermittently, the therapeutic concentration is often exceeded for some period after which concentrations fall below therapeutic levels. Excessively high concentrations are a major cause of side effects and subtherapeutic concentrations are ineffective.
21
- •
- Proprietary Reformulation of Generic Products. We believe
that the AcuForm technology may offer the potential to produce improved formulations of off-patent drugs. These proprietary formulations may be differentiated from existing generic
products by virtue of reduced dosing requirements, improved efficacy, decreased toxicity or additional indications.
- •
- More Efficient Gastrointestinal Drug Absorption. We
believe that the AcuForm technology can be used for improved oral administration of drugs that are inadequately absorbed when delivered as conventional tablets or capsules. Many drugs are primarily
absorbed in the stomach, duodenum or upper small intestine regions, through which drugs administered in conventional oral dosage forms transit quickly. In contrast, the AcuForm technology is designed
to be retained in the stomach, allowing for constant multi-hour flow of drugs to these regions of the gastrointestinal tract. Accordingly, for such drugs, we believe that the AcuForm
technology offers a significantly enhanced opportunity for increased absorption. Unlike some insoluble drug delivery systems, the polymer comprising the AcuForm technology dissolves at the end of its
useful life and is passed through the gastrointestinal tract and eliminated.
- •
- Gastric Delivery for Local Therapy and Absorption. We
believe that the AcuForm technology can be used to deliver drugs which can efficiently eradicate gastrointestinal-dwelling microorganisms, such as H.
pylori, the bacterium which is a cause of most peptic ulcers.
- •
- Rational Drug Combinations. We believe that the AcuForm technology may allow for rational combinations of drugs with different biological half-lives. Physicians frequently prescribe multiple drugs for treatment of a single medical condition. Single product combinations have not been considered feasible because the different biological half-lives of these combination drugs would result in an overdosage of one drug and/or an underdosage of the other. By appropriately incorporating different drugs into an AcuForm technology we believe that we can provide for the release of each incorporated drug continuously at a rate and duration (dose) appropriately adjusted for the specific biological half-lives of the drugs. We believe that future rational drug combination products using the AcuForm technology have the potential to simplify drug administration, increase patient compliance, and reduce medical costs.
COMPETITION
GLUMETZA. GLUMETZA competes against immediate release metformin, which is marketed primarily by generic manufacturers. GLUMETZA also competes against both branded and generic extended-release versions of metformin, such as Bristol-Myers Squibb's Glucophage XR and Shinogi/Sciele Pharma's Fortamet. Generic extended-release metformin manufacturers include Barr Pharmaceuticals, Inc., ANDRX Corporation, Mylan Laboratories, Inc., Watson Pharmaceuticals, Inc. and Teva Pharmaceutical Industries, Ltd., among others.
GLUMETZA also competes against oral type 2 diabetes medications other than metformin, such as Takeda's Actos (pioglitazone hydrochloride), GlaxoSmithKline's Avandia (risiglitazon), Pfizer's Glucotrol (sulfonylurea) and Merck's Januvia (sitagliptin), among others.
SeradaTM for Menopausal Hot Flashes. If approved, Serada for hot flashes will compete against HRT, such as Pfizer's Premarin (estrogens) and Prempro (a combination of estrogens and a progestin) products, and anti-depressant medications prescribed off-label. Pfizer's anti-depressant drug candidate, Pristiq, is in pre-registration and Pfizer's Aprela (estrogens) is in Phase 3 clinical trials for the treatment of hot flashes. We are aware that Pfizer has non-exclusively licensed from the University of Rochester rights to develop a hot flash product containing pregabalin under the same patent we have sublicensed exclusive rights to develop a menopausal hot flash product containing gabapentin. Accordingly, Pfizer may develop a competing hot flash product. Bionovo's Menerba completed Phase 2 clinical trials and is expected to commence Phase 3 clinical trials in 2010 for the treatment of hot flashes.
22
DM-1796 for Neuropathic Pain. Gabapentin is currently marketed by Pfizer as Neurontin and by several generic manufacturers for adjunctive therapy for epileptic seizures and for postherpetic pain. In addition, Pfizer's product, Lyrica™ (pregabalin), has been approved for marketing in the United States and the European Union for the treatment of postherpetic neuralgia, diabetic neuropathy, partial seizures and fibromyalgia.
If approved, DM-1796 will compete against other neuropathic pain treatments, such as anti-depressants, other anti-convulsants, local anesthetics used as regional nerve blockers, anti-arrythmics and opiods. We are also aware of at least one company that is developing a product of gabapentin for the neuropathic pain market. To our knowledge, we are the only company currently in clinical trials with a sustained release formulation of gabapentin for the United States market.
DM-3458 for GERD. Any GERD product we develop will compete against the antacids, foaming agents, H2 blockers, proton pump inhibitors and prokinetics described above under "GERD Treatments; Target Market".
DM-1992 for Parkinson's Disease. If approved, DM-1992 will compete against Sinemet, a combination of levodopa and carbidopa product for treatment of Parkinson's disease and syndrome sold by Merck as well as generic Sinemet sold by various generic manufacturers.
Drug Delivery Technologies. Other companies that have oral drug delivery technologies competitive with the AcuForm technology include Elan Corporation, Bristol-Myers Squibb, Teva Pharmaceutical Industries, Ltd , ALZA Corporation (a subsidiary of Johnson & Johnson), SkyePharma plc, Biovail Corporation, Flamel Technologies S.A., Ranbaxy Laboratories, Ltd., and Intec Pharma, all of which develop oral tablet products designed to release the incorporated drugs over time. Each of these companies has patented technologies with attributes different from ours, and in some cases with different sites of delivery to the gastrointestinal tract.
General. We believe that we compete favorably in the markets described above on the basis of the safety and efficacy of our products and product candidates, and in some cases on the basis of the price of our products. However, competition in pharmaceutical products and drug delivery technologies is intense, and we expect competition to increase. There may be other companies developing products competitive with ours of which we are unaware. Competing product or technologies developed in the future may prove superior to our products or technologies, either generally or in particular market segments. These developments could make our products or technologies noncompetitive or obsolete.
Most of our principal competitors have substantially greater financial, sales, marketing, personnel and research and development resources than we do. In addition, many of our potential collaborative partners have devoted, and continue to devote, significant resources to the development of their own products and drug delivery technologies.
23
PATENTS AND PROPRIETARY RIGHTS
Our material issued United States patents and the products and product candidates they cover are as follows:
United States Patent No. |
Expiration Date | Product(s) and Product Candidate(s) Covered |
|||
|---|---|---|---|---|---|
| 6,340,475 | September 19, 2016 | Glumetza 500mg | |||
| Proquin XR | |||||
| DM-1796 | |||||
| Serada | |||||
6,635,280 |
September 19, 2016 |
Glumetza 500mg |
|||
| Proquin XR | |||||
| DM-1796 | |||||
| Serada | |||||
6,723,340 |
October 25, 2021 |
Glumetza 500mg |
|||
| DM-1796 | |||||
| Serada | |||||
6,488,962 |
June 20, 2020 |
Glumetza 500mg |
|||
| Glumetza 1000mg | |||||
| Proquin XR | |||||
| DM-1796 | |||||
| Serada | |||||
5,972,389 |
September 19, 2016 |
Proquin XR |
|||
7,438,927 |
February 26, 2024 |
DM-1796 |
|||
| Serada | |||||
6,310,098 |
(1) |
July 21, 2020 |
Serada |
||
- (1)
- We have an exclusive sublicense from PharmaNova, under United States Patent No. 6,310,098, held by the University of Rochester, to develop and commercialize in the United States a product that contains gabapentin as its active pharmaceutical ingredient, and is indicated for the treatment of menopausal hot flashes.
Our success will depend in part on our ability to obtain and maintain patent protection for our technologies and to preserve our trade secrets. Our policy is to seek to protect our proprietary rights, by among other methods, filing patent applications in the United States and foreign jurisdictions to cover certain aspects of our technology. In addition to those patents noted on the above table, we have nineteen patent applications pending in the United States. We have also prepared and continue to prepare patent applications relating to our expanding technology for filing in the United States and abroad. We have also applied for patents in numerous foreign countries. Some of those countries have granted our applications and other applications are still pending. Our pending patent applications may lack priority over others' applications or may not result in the issuance of patents. Even if issued, our patents may not be sufficiently broad to provide protection against competitors with similar technologies and may be challenged, invalidated or circumvented, which could limit our ability to stop competitors from marketing related products or may not provide us with competitive advantages against competing products. We also rely on trade secrets and proprietary know-how, which are difficult to protect. We seek to protect such information, in part, through entering into confidentiality agreements with employees, consultants, collaborative partners and others before such persons or entities have access to our proprietary trade secrets and know-how. These confidentiality agreements
24
may not be effective in certain cases, due to, among other things, the lack of an adequate remedy for breach of an agreement or a finding that an agreement is unenforceable. In addition, our trade secrets may otherwise become known or be independently developed by competitors.
Our ability to develop our technologies and to make commercial sales of products using our technologies also depends on not infringing others' patents or other intellectual property rights. We are not aware of any intellectual property claims against us. However, the pharmaceutical industry has experienced extensive litigation regarding patents and other intellectual property rights. For example, Pfizer has initiated several suits against companies marketing generic gabapentin products, claiming that these products infringe Pfizer's patents. The results of this litigation could adversely impact the commercialization of pharmaceutical products that contain gabapentin as an active pharmaceutical ingredient. Also, we are aware that patents issued to third parties relating to extended release drug formulations or particular pharmaceutical compounds could in the future be asserted against us, although we believe that we do not infringe any valid claim of any patents. If claims concerning any of our products were to arise and it was determined that these products infringe a third party's proprietary rights, we could be subject to substantial damages for past infringement or be forced to stop or delay our activities with respect to any infringing product, unless we can obtain a license, or we may have to redesign our product so that it does not infringe upon others' patent rights, which may not be possible or could require substantial funds or time. Such a license may not be available on acceptable terms, or at all. Even if we, our collaborators or our licensors were able to obtain a license, the rights may be nonexclusive, which would give our competitors access to the same intellectual property. In addition, any public announcements related to litigation or interference proceedings initiated or threatened against us, even if such claims are without merit, could cause our stock price to decline.
From time to time, we may become aware of activities by third parties that may infringe our patents. Infringement by others of our patents may reduce our market shares (if a related product is approved) and, consequently, our potential future revenues and adversely affect our patent rights if we do not take appropriate enforcement action. We may need to engage in litigation in the future to enforce any patents issued or licensed to us or to determine the scope and validity of third-party proprietary rights. Our issued or licensed patents may not be held valid by a court of competent jurisdiction. Whether or not the outcome of litigation is favorable to us, defending a lawsuit takes significant time, may be expensive and may divert management attention from other business concerns. We may also be required to participate in interference proceedings declared by the United States Patent and Trademark Office for the purpose of determining the priority of inventions in connection with our patent applications or other parties' patent applications. Adverse determinations in litigation or interference proceedings could require us to seek licenses which may not be available on commercially reasonable terms, or at all, or subject us to significant liabilities to third parties. If we need but cannot obtain a license, we may be prevented from marketing the affected product.
MANUFACTURING
We have established internal manufacturing facilities that are in compliance with current good manufacturing practices, to manufacture supplies for our Phase 1 and Phase 2 clinical trials.
We are responsible for the supply and distribution of GLUMETZA, and we have entered into a manufacturing agreement with Patheon Puerto Rico Inc., as our sole supplier for tablets of the 500mg strength of GLUMETZA. We have two qualified suppliers for the active pharmaceutical ingredient in GLUMETZA. However, we obtain the active pharmaceutical ingredient to GLUMETZA on a purchase order basis only. We have a supply agreement with Biovail, as our sole supplier for the 1000mg formulation of GLUMETZA.
25
We are also responsible for supply and distribution of Proquin XR. For the manufacture of Proquin XR tablets, we have entered into a manufacturing agreement with Patheon Puerto Rico Inc., as our sole supplier. We purchase the active ingredient for Proquin XR from Uquifa Mexico, S.A., a sole supplier to us, on a purchase order basis.
Should DM-1796 be approved for marketing by the FDA, we will be responsible for the supply of DM-1796 product to Solvay until November 2012. We have obtained clinical and validation batches of Serada and DM-1796 from Patheon Puerto Rico Inc., our third-party manufacturer and sole supplier of Serada and DM-1796 product. We currently have no long-term supply arrangements for Serada and DM-1796 and obtain Serada and DM-1796 product on a purchase order basis. We have two qualified suppliers for the active pharmaceutical ingredient in Serada and DM-1796. However, we obtain the active pharmaceutical ingredient to Serada and DM-1796 on a purchase order basis only.
We also obtain polyethylene oxide, one of the excipients common to GLUMETZA, Proquin XR, Serada and DM-1796, on a purchase order basis from Dow Chemical, a sole source for polyethylene oxide. We currently have no long-term supply arrangement with respect to polyethylene oxide.
Applicable current Good Manufacturing Practices (cGMP) requirements and other rules and regulations prescribed by foreign regulatory authorities will apply to the manufacture of products using the AcuForm technology. We will depend on the manufacturers of products using the AcuForm technology to comply with cGMP and applicable foreign standards. Any failure by a manufacturer of products using the AcuForm technology to maintain cGMP or comply with applicable foreign standards could delay or prevent the initial or continued commercial sale of our products.
MARKETING AND SALES
In 2004, we announced our determination to evolve from a solely product development focused company to an integrated specialty pharmaceutical company, with sales and marketing of our own products. Preliminary staffing for these activities began in 2005. From 2006 through 2008, we enhanced our internal sales and marketing capabilities through the hiring of additional sales and marketing employees and the engagement of consultants. In 2009, following the results of our Serada Breeze 1 and Breeze 2 Phase 3 clinical trials, we reduced our sales and marketing headcount as the commercial infrastructure needed for Serada has been delayed. We anticipate the build-up of our commercial infrastructure will continue over the next several years.
In 2007 and 2008, our commercial organization transitioned to us the GLUMETZA marketing efforts previously undertaken by King and directed the efforts of a temporary contract sales organization. In July 2008, we entered into a promotion agreement with Santarus for GLUMETZA, and our commercial organization has transitioned the promotion and marketing efforts for GLUMETZA to Santarus, who began promotion in October 2008.
Our sales and marketing personnel are also engaged in preparation for the eventual commercialization of Serada for hot flashes, and commercial and marketing assessments of existing and potential product candidates.
All marketing activities associated with GLUMETZA and Proquin XR, as well as marketing activities related to any other products for which we obtain regulatory approval, will be subject to numerous federal and state laws governing the marketing and promotion of pharmaceutical products. The FDA regulates post-approval promotional labeling and advertising to ensure that they conform with statutory and regulatory requirements. In addition to FDA restrictions, the marketing of prescription drugs is subject to laws and regulations prohibiting fraud and abuse under government healthcare programs. For example, the federal healthcare program antikickback statute prohibits giving things of value to induce the prescribing or purchase of products that are reimbursed by federal healthcare programs, such as Medicare and Medicaid. In addition, federal false claims laws prohibit any
26
person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government. Under this law, the federal government in recent years has brought claims against drug manufacturers alleging that certain marketing activities caused false claims for prescription drugs to be submitted to federal programs. Many states have similar statutes or regulations, which apply to items and services reimbursed under Medicaid and other state programs, or, in some states, regardless of the payer. If we, or our collaborative partners, fail to comply with applicable FDA regulations or other laws or regulations relating to the marketing of our products, we could be subject to criminal prosecution, civil penalties, seizure of products, injunction, exclusion of our products from reimbursement under government programs, as well as other regulatory actions against our product candidates, our collaborative partners or us.
GOVERNMENT REGULATION
Numerous governmental authorities in the United States and other countries regulate our research and development activities and those of our collaborative partners. Governmental approval is required of all potential pharmaceutical products using the AcuForm technology and the manufacture and marketing of products using the AcuForm technology prior to the commercial use of those products. The regulatory process takes several years and requires substantial funds. If new products using the AcuForm technology do not receive the required regulatory approvals or if such approvals are delayed, our business would be materially adversely affected. There can be no assurance that the requisite regulatory approvals will be obtained without lengthy delays, if at all.
In the United States, the FDA rigorously regulates pharmaceutical products, including any drugs using the AcuForm technology. If a company fails to comply with applicable requirements, the FDA or the courts may impose sanctions. These sanctions may include civil penalties, criminal prosecution of the company or its officers and employees, injunctions, product seizure or detention, product recalls and total or partial suspension of production. The FDA may withdraw approved applications or refuse to approve pending new drug applications, premarket approval applications, or supplements to approved applications.
We generally must conduct preclinical testing on laboratory animals of new pharmaceutical products prior to commencement of clinical studies involving human beings. These studies evaluate the potential efficacy and safety of the product. We then submit the results of these studies to the FDA as part of an Investigational New Drug Application, which must become effective before beginning clinical testing in humans.
Typically, human clinical evaluation involves a time-consuming and costly three-phase process:
- •
- In Phase 1, we conduct clinical trials with a small number of subjects to determine a drug's early safety profile
and its pharmacokinetic pattern.
- •
- In Phase 2, we conduct limited clinical trials with groups of patients afflicted with a specific disease in order
to determine preliminary efficacy, optimal dosages and further evidence of safety.
- •
- In Phase 3, we conduct large-scale, multi-center, comparative trials with patients afflicted with a target disease in order to provide enough data to statistically evaluate the efficacy and safety of the product candidate, as required by the FDA.
The FDA closely monitors the progress of each phase of clinical testing. The FDA may, at its discretion, re-evaluate, alter, suspend or terminate testing based upon the data accumulated to that point and the FDA's assessment of the risk/benefit ratio to patients. The FDA may also require additional clinical trials after approval, which are known as Phase 4 trials.
27
The results of the preclinical and clinical testing are submitted to the FDA in the form of a New Drug Application, or NDA, for approval prior to commercialization. An NDA requires that our products are compliant with cGMP. Failure to achieve or maintain cGMP standards for products using the AcuForm technology would adversely impact their marketability. In responding to an NDA, the FDA may grant marketing approval, request additional information or deny the application. Failure to receive approval for any products using the AcuForm technology would have a material adverse effect on the Company.
The FDA regulates not only prescription and over-the-counter drugs approved by NDAs, but also over-the-counter products that comply with monographs issued by the FDA. These regulations include:
- •
- cGMP requirements;
- •
- general and specific over-the-counter labeling requirements (including warning statements);
- •
- advertising restrictions; and
- •
- requirements regarding the safety and suitability of inactive ingredients.
In addition, the FDA may inspect over-the-counter products and manufacturing facilities. A failure to comply with applicable regulatory requirements may lead to administrative or judicially imposed penalties. If an over-the-counter product differs from the terms of a monograph, it will, in most cases, require FDA approval of an NDA for the product to be marketed.
Foreign regulatory approval of a product must also be obtained prior to marketing the product internationally. Foreign approval procedures vary from country to country. The time required for approval may delay or prevent marketing in certain countries. In certain instances we or our collaborative partners may seek approval to market and sell certain products outside of the United States before submitting an application for United States approval to the FDA. The clinical testing requirements and the time required to obtain foreign regulatory approvals may differ from that required for FDA approval. Although there is now a centralized European Union (EU) approval mechanism in place, each EU country may nonetheless impose its own procedures and requirements. Many of these procedures and requirements are time-consuming and expensive. Some EU countries require price approval as part of the regulatory process. These constraints can cause substantial delays in obtaining required approval from both the FDA and foreign regulatory authorities after the relevant applications are filed, and approval in any single country may not meaningfully indicate that another country will approve the product.
EMPLOYEES
As of March 5, 2010, we had 73 full-time employees. At December 31, 2009, we had 75 full-time employees. None of our employees are represented by a collective bargaining agreement, nor have we experienced any work stoppage. We believe that our relations with our employees are good.
28
In addition to other information in this report, the following factors should be considered carefully in evaluating an investment in our securities. If any of the risks or uncertainties described in this Form 10-K actually occurs, our business, results of operations or financial condition could be materially adversely affected. The risks and uncertainties described in this Form 10-K are not the only ones facing us. Additional risks and uncertainties of which we are unaware or currently deem immaterial may also become important factors that may harm our business.
We depend on Solvay Pharmaceuticals for certain aspects of the development and commercialization of DM-1796, which subjects us to risks related to Solvay and its business that are outside our control.
In January 2009, our exclusive license agreement with Solvay Pharmaceuticals Inc. related to the development and commercialization of DM-1796 became effective. The license agreement grants Solvay exclusive rights to develop and commercialize DM-1796 in the United States, Canada and Mexico for pain indications. We depend on Solvay to obtain regulatory approval and complete any further development of DM-1796, and to commercialize the product in the licensed territories, which subjects us to a number of risks, including the following:
- •
- we may not be able to control the amount and timing of resources that Solvay devotes to the development or
commercialization of DM-1796;
- •
- we and Solvay may not be successful in our efforts to obtain regulatory approval of DM-1796 in a timely
manner, or at all;
- •
- Solvay may experience financial difficulties; or
- •
- Solvay may change its business strategy, due to a combination with another company or for another reason unrelated to the DM-1796 commercial opportunity, in a manner that adversely affects the development or commercialization of DM-1796.
Abbott Laboratories completed its acquisition of Solvay's pharmaceutical business in February 2010. Accordingly, Abbott is now responsible for the DM-1796 license arrangement through a subsidiary. We do not yet know whether Abbott will be as committed to the development and commercialization of DM-1796 as Solvay, and we are not likely to know until some time following the completion of the acquisition. However, it is possible that Abbott's acquisition of Solvay's pharmaceutical business may adversely affect the development or commercialization of DM-1796.
Our prior clinical trials evaluating SeradaTM for menopausal hot flashes failed to meet all of their primary endpoints and there can be no assurance this product will be approved for marketing.
In October 2009, our Phase 3 trials evaluating Serada for menopausal hot flashes failed to meet all of their primary endpoints. In December 2009, we met and discussed with the FDA the results of the trials and any additional clinical development that may be required to complete the program and obtain regulatory approval to market Serada in the United States. Based on the guidance from the meeting with the FDA, we expect to initiate an additional Phase 3 trial for Serada by the end of April 2010. The trial will be known as Breeze 3. There can be no assurance the results of the Breeze 3 trial will demonstrate the product candidate is sufficiently safe and effective to obtain approval for marketing.
We will incur significant additional expenses and will not know for at least one to two years whether the drug is safe and effective such that it could be approved for marketing. Clinical development is a long, expensive and uncertain process and is subject to delays. The positive or encouraging results of prior clinical trial are not necessarily indicative of the results we will obtain in later clinical trials. Accordingly, our additional Phase 3 trial may not demonstrate that Serada is
29
effective for menopausal hot flashes. In addition, data obtained from pivotal clinical trials are susceptible to varying interpretations, which could delay, limit or prevent regulatory approval.
Many other factors could delay or result in termination of our clinical trials, including:
- •
- negative or inconclusive results;
- •
- patient noncompliance with the protocol;
- •
- adverse medical events or side effects among patients during the clinical trials;
- •
- FDA inspections of our clinical operations; and
- •
- real or perceived lack of effectiveness or safety of the product candidate.
We depend heavily on Santarus, Inc. for the successful commercialization of GLUMETZA in the United States.
In July 2008, we entered into a promotion agreement with Santarus, Inc. pursuant to which Santarus will promote GLUMETZA in the United States through its sales force beginning in the fourth quarter of 2008. Under the agreement, in exchange for promotion fees, Santarus is required to market and promote GLUMETZA to physicians in the United States, to deliver annual detail calls to potential GLUMETZA prescribers, and to maintain a sales force of a minimum size. Although we have retained rights to promote GLUMETZA to obstetricians/gynecologists, or ob/gyns, and to retain revenues from incremental sales generated by ob/gyns we call upon, ob/gyns generally do not prescribe significant amounts of metformin products. In addition, we do not have any immediate plans to establish a sales force, or contract with a third party to act as our sales force, for the purpose of exercising our GLUMETZA co-promotion rights. Accordingly, the success of the commercialization of GLUMETZA will depend in large part on Santarus' marketing and promotion efforts. Factors that may affect the success of our promotion arrangement with Santarus include the following:
- •
- Santarus may acquire or develop alternative products;
- •
- Santarus may pursue higher-priority programs, or change the focus of its marketing programs;
- •
- Santarus may in the future choose to devote fewer resources to GLUMETZA;
- •
- GLUMETZA may fail to achieve greater market acceptance;
- •
- The outcome of our ongoing litigation against Lupin Limited seeking to prevent Lupin from marketing a generic version of
GLUMETZA in the United States;
- •
- The outcome of Santarus' ongoing litigation against Par Pharmaceutical, Inc. (Par) seeking to prevent Par from
marketing generic versions of Santarus' Zegerid products; and
- •
- Santarus may fail to comply with its obligations under our promotion agreement.
Any of the preceding factors could affect Santarus' commitment to the collaboration, which, in turn, could adversely affect the commercial success of GLUMETZA. Any failure to successfully commercialize GLUMETZA could have a material adverse effect on our business, financial conditions, results of operations and cash flows.
The timing and any potential negative outcome in the ongoing patent litigation with Lupin could adversely affect our financial condition and results of operations as it could result in the introduction of generic products prior to the expiration of the patents for GLUMETZA, as well as in significant legal expenses and diversion of management time.
In November 2009, we filed a lawsuit in the United States District Court for the Northern District of California against Lupin for infringement of U.S. Patent Nos. 6,340,475; 6,488,962; 6,635,280; and
30
6,723,340 listed in the Orange Book for GLUMETZA. The lawsuit is in response to an Abbreviated New Drug Application (ANDA) filed by Lupin with the FDA regarding Lupin's intent to market generic versions of the 500mg and 1000mg strengths of GLUMETZA prior to the expiration date of the asserted patents.
We commenced the lawsuit against Lupin within the applicable 45 day period required to automatically stay, or bar, the FDA from approving Lupin's ANDA for 30 months or until a district court decision that is adverse to the asserted patents, whichever may occur earlier. The 30-month stay expires in May 2012. If the litigation is still ongoing after expiration of the applicable 30-month stay, the termination of the stay could result in the introduction of one or more products generic to GLUMETZA prior to resolution of the litigation.
Although we intend to vigorously defend and enforce our patent rights, we are not able to predict the timing or outcome of the litigation. The court may render its decision at any time after the filing of the post-trial briefs, which may be before or after the expiration of the 30-month stay. An adverse outcome in this litigation could result in one or more generic versions of GLUMETZA being launched before the expiration of the listed patents, which could adversely affect our ability to successfully execute our business strategy to maximize the value of GLUMETZA and would negatively impact our financial condition and results of operations, including causing a significant decrease in our revenues and cash flows. Regardless of how the litigation is ultimately resolved, the litigation may be costly, time-consuming and distracting to management, which could have a material adverse effect on our business.
The development of drug candidates is inherently uncertain and we cannot be certain that any of our product candidates will be approved for marketing or, if approved, will achieve market acceptance.
We have the following programs in clinical development: DM-1796 for neuropathic pain, Serada for menopausal hot flashes, and DM-1992 for Parkinson's. We also have other product candidates in earlier stages of development.
Our own product candidates and those of our collaborative partners are subject to the risk that any or all of them may be found to be ineffective or unsafe, or otherwise may fail to receive necessary regulatory clearances. Additionally, clinical trial results in earlier trials may not be indicative of results that will be obtained in subsequent larger trials, as was the case with the Phase 3 trial for DM-1796 for the treatment of postherpetic neuralgia that we completed in 2007, and with the Phase 3 trials evaluating Serada for menopausal hot flashes we completed in October 2009.
We are unable to predict whether any of these product candidates will receive regulatory clearances or be successfully manufactured or marketed. Further, due to the extended testing and regulatory review process required before marketing clearance can be obtained, the time frames for commercialization of any products are long and uncertain. Even if these other product candidates receive regulatory clearance, our products may not achieve or maintain market acceptance. Also, substantially all of our product candidates use the Acuform technology. If it is discovered that the Acuform technology could have adverse effects or other characteristics that indicate it is unlikely to be effective as a delivery system for drugs or therapeutics, our product development efforts and our business would be significantly harmed.
We are expecting operating losses in the future.
To date, we have recorded revenues from license fees, product sales, royalties, collaborative research and development arrangements and feasibility studies. For the years ended December 31, 2009, 2008 and 2007, we recorded total revenues of $57.7 million, $34.8 million, and $65.6 million, respectively. For the years ended December 31, 2009 and 2008 we incurred net losses of $22.0 million and $15.3 million, respectively. The termination of our license agreement with Esprit in July 2007,
31
including the accelerated recognition of previously deferred revenue under the arrangement, and termination fees received associated with the termination of our promotion agreement with King resulted in our reaching profitability in 2007. However, as we continue our research and development efforts, preclinical testing and clinical trial activities, we anticipate that we will incur operating losses in fiscal year 2010. Therefore, we expect our cumulative losses to increase. These losses, among other things, have had, and we expect that they will continue to have, an adverse impact on our total assets, shareholders' equity and working capital.
Our operating results may fluctuate and affect our stock price.
The following factors will affect our operating results and may result in a material adverse effect on our stock price:
- •
- announcements and results regarding clinical trial results and plans for our drug candidates, including
DM-1796 and Serada;
- •
- filings and other regulatory actions related to DM-1796, Serada and our other product candidates;
- •
- the degree of commercial success of GLUMETZA;
- •
- adverse events related to our products, including recalls;
- •
- interruptions of manufacturing or supply;
- •
- the outcome of our patent infringement litigation against Lupin for GLUMETZA;
- •
- developments concerning proprietary rights, including patents, infringement allegations and litigation matters;
- •
- variations in revenues obtained from collaborative agreements, including milestone payments, royalties, license fees and
other contract revenues;
- •
- decisions by collaborative partners to proceed or not to proceed with subsequent phases of a collaboration or program;
- •
- market acceptance of the Acuform technology;
- •
- adoption of new technologies by us or our competitors;
- •
- the introduction of new products by our competitors;
- •
- manufacturing costs and difficulties;
- •
- third-party reimbursement policies; and
- •
- the status of our compliance with the provisions of the Sarbanes-Oxley Act of 2002.
As a result of these factors, our stock price may continue to be volatile and investors may be unable to sell their shares at a price equal to, or above, the price paid. Additionally, any significant drops in our stock price, such as the one we experienced following the announcement of our Serada Phase 3 trial results in October 2009, could give rise to shareholder lawsuits, which are costly and time consuming to defend against and which may adversely affect our ability to raise capital while the suits are pending, even if the suits are ultimately resolved our favor.
32
Our collaborative arrangements may give rise to disputes over commercial terms, contract interpretation and ownership of our intellectual property and may adversely affect the commercial success of our products.
We currently have collaboration arrangements with Solvay, Santarus and Covidien. In addition, we have in the past and may in the future enter into other collaborative arrangements, some of which have been based on less definitive agreements, such as memoranda of understanding, material transfer agreements, options or feasibility agreements. We may not execute definitive agreements formalizing these arrangements. Collaborative relationships are generally complex and may give rise to disputes regarding the relative rights, obligations and revenues of the parties, including the ownership of intellectual property and associated rights and obligations, especially when the applicable collaborative provisions have not been fully negotiated and documented. Such disputes can delay collaborative research, development or commercialization of potential products, and can lead to lengthy, expensive litigation or arbitration. The terms of collaborative arrangements may also limit or preclude us from developing products or technologies developed pursuant to such collaborations. Additionally, the collaborators under these arrangements might breach the terms of their respective agreements or fail to prevent infringement of the licensed patents by third parties. Moreover, negotiating collaborative arrangements often takes considerably longer to conclude than the parties initially anticipate, which could cause us to enter into less favorable agreement terms that delay or defer recovery of our development costs and reduce the funding available to support key programs.
We may be unable to enter into future collaborative arrangements on acceptable terms, which would harm our ability to develop and commercialize our current and potential future products. Further, even if we do enter into collaboration arrangements, it is possible that our collaborative partners may not choose to develop and commercialize products using the Acuform technology. Other factors relating to collaborations that may adversely affect the commercial success of our products include:
- •
- any parallel development by a collaborative partner of competitive technologies or products;
- •
- arrangements with collaborative partners that limit or preclude us from developing products or technologies;
- •
- premature termination of a collaboration agreement; or
- •
- failure by a collaborative partner to devote sufficient resources to the development and commercial sales of products using the Acuform technology.
Our collaborative arrangements do not necessarily restrict our collaborative partners from competing with us or restrict their ability to market or sell competitive products. Our current and any future collaborative partners may pursue existing or other development-stage products or alternative technologies in preference to those being developed in collaboration with us. Our collaborative partners may also terminate their collaborative relationships with us or otherwise decide not to proceed with development and commercialization of our products.
We have limited in-house sales and marketing resources, which we will require in order to successfully promote products through our own sales force.
If Serada or another product we develop or acquire is approved for marketing in the United States, we may choose to promote the product with our own sale force or through a contract sales organization. We also have rights to promote GLUMETZA through our own sales force, or through third parties, and we have retained co-promotion rights for certain product candidates our collaborative partners may develop. We currently have no sales force and limited marketing and sales staff. The success of our own promotion efforts for Serada, GLUMETZA and any other product candidates that receive regulatory approval that we choose to market or co-market, will require that we substantially
33
enhance our in-house marketing and sales force with technical expertise, or make arrangements with third parties to perform these services for us. The development of the infrastructure associated with these activities involves substantial resources, and considerable attention of our management and key personnel. To the extent that we enter into marketing and sales arrangements with other companies, our revenues will depend on the efforts of others. These efforts may not be successful. If we fail to fully develop marketing and sales capabilities, or enter into arrangements with third parties, our revenues may suffer.
We depend on our marketing partners for the successful commercialization of GLUMETZA in Canada and Korea, and of Proquin XR in Europe.
We have licensed exclusive marketing rights to the 500mg GLUMETZA in Canada to Biovail, and in Korea to LG Life Sciences. Biovail launched the 500mg GLUMETZA in Canada in November 2005, and LG launched a 500mg product in Korea in 2006 under the trade name Novamet GR. We have also entered into a license agreement with Madaus, a company acquired by Rottapharm in June 2007, related to the commercialization of Proquin XR in Europe. If our international commercial partners fail to successfully commercialize products we have licensed to them, our future revenues may be adversely affected.
Our credit facility contains operating covenants that may restrict our business and financing activities.
We entered into a $15.0 million credit facility with Oxford Finance Corporation and General Electric Capital Corporation in June 2008. We have drawn $9.4 million under the facility and will not make any further draws under the facility. As of December 31, 2009, we have $6.1 million of principal outstanding under the facility. The credit facility is secured by a pledge of all of our assets other than intellectual property, and contains a variety of operational covenants, including limitations on our ability to incur liens or additional debt, make dispositions, pay dividends, redeem our stock, make certain investments and engage in certain merger, consolidation or asset sale transactions and transactions with affiliates, among other restrictions. Any future debt financing we enter into may involve similar or more restrictive covenants affecting our operations. Our borrowings under the credit facility or any future debt financing will need to be repaid, which creates additional financial risk for our company, particularly if our business, or prevailing financial market conditions, are not conducive to paying off or refinancing our outstanding debt obligations. Furthermore, our failure to comply with the covenants in the credit facility could result in an event of default that, if not cured or waived, could result in the acceleration of all or a substantial portion of our debt, which could have a material adverse effect on our cash position and significantly harm our business.
Our existing resources may not be sufficient to fund our operations until such time as we may be able to consistently generate sufficient revenues to support our operations.
Our existing capital resources may not be sufficient to fund our operations until such time as we may be able to generate sufficient revenues to consistently support our operations. We have limited credit facilities and, except for our common stock purchase agreement with Azimuth, we have no other committed sources of capital. Any additional development of Serada for menopausal hot flashes or other clinical development programs may require considerable financial resources, should we choose to invest in those programs. To the extent that our capital resources are insufficient to meet our future capital requirements, in order to continue our development programs, we will have to raise additional funds through the sale of our equity securities, through debt financing, or from development and licensing arrangements. We may be unable to raise such additional capital on favorable terms, or at all. If we raise additional capital by selling our equity or convertible debt securities, the issuance of such
34
securities could result in dilution of our shareholders' equity positions. If adequate funds are not available we may have to:
- •
- delay, postpone or terminate clinical trials;
- •
- significantly curtail commercialization of our marketed products or other operations; and/or
- •
- obtain funds through entering into collaboration agreements on unattractive terms.
The global economic downturn may adversely affect our business.
The economic downturn that has affected the global economy over the past several fiscal quarters may have a material adverse effect on our liquidity and financial condition and our ability to raise additional funds, whether pursuant to our existing or future financing arrangements. In addition, if these developments negatively impact the ability of our collaborative partners to develop, manufacture, promote or commercialize our products and product candidates, our revenues may suffer and our business, financial condition and results of operations could be materially and adversely affected. Similarly, any negative impact of an economic downturn or recession on our potential collaborative partners could adversely affect the terms on which collaborative partnerships may be available to us, if at all.
We may be unable to protect our intellectual property and may be liable for infringing the intellectual property of others.
Our success will depend in part on our ability to obtain and maintain patent protection for our products and technologies, and to preserve our trade secrets. Our policy is to seek to protect our proprietary rights, by, among other methods, filing patent applications in the United States and foreign jurisdictions to cover certain aspects of our technology. We currently hold thirteen issued United States patents, and have fifteen patent applications pending in the United States. In addition, we are preparing patent applications relating to our expanding technology for filing in the United States and abroad. We have also applied for patents in numerous foreign countries. Some of those countries have granted our applications and other applications are still pending. Our pending patent applications may lack priority over others' applications or may not result in the issuance of patents. Even if issued, our patents may not be sufficiently broad to provide protection against competitors with similar technologies and may be challenged, invalidated or circumvented, which could limit our ability to stop competitors from marketing related products or may not provide us with competitive advantages against competing products. We also rely on trade secrets and proprietary know-how, which are difficult to protect. We seek to protect such information, in part, through entering into confidentiality agreements with employees, consultants, collaborative partners and others before such persons or entities have access to our proprietary trade secrets and know-how. These confidentiality agreements may not be effective in certain cases, due to, among other things, the lack of an adequate remedy for breach of an agreement or a finding that an agreement is unenforceable. In addition, our trade secrets may otherwise become known or be independently developed by competitors.
Our ability to develop our technologies and to make commercial sales of products using our technologies also depends on not infringing others' patents or other intellectual property rights. We are not aware of any intellectual property claims against us. However, the pharmaceutical industry has experienced extensive litigation regarding patents and other intellectual property rights. For example, Pfizer has initiated several suits against companies marketing generic gabapentin products, claiming that these products infringe Pfizer's patents. The results of this litigation could adversely impact the commercialization of pharmaceutical products that contain gabapentin as an active pharmaceutical ingredient. Also, patents issued to third parties relating to sustained release drug formulations or particular pharmaceutical compounds could in the future be asserted against us, although we believe that we do not infringe any valid claim of any patents. If claims concerning any of our products were to
35
arise and it was determined that these products infringe a third party's proprietary rights, we could be subject to substantial damages for past infringement or be forced to stop or delay our activities with respect to any infringing product, unless we can obtain a license, or we may have to redesign our product so that it does not infringe upon others' patent rights, which may not be possible or could require substantial funds or time. Such a license may not be available on acceptable terms, or at all. Even if we, our collaborators or our licensors were able to obtain a license, the rights may be nonexclusive, which would give our competitors access to the same intellectual property. In addition, any public announcements related to litigation or interference proceedings initiated or threatened against us, even if such claims are without merit, could cause our stock price to decline.
From time to time, we may become aware of activities by third parties that may infringe our patents. Infringement by others of our patents may reduce our market shares (if a related product is approved) and, consequently, our potential future revenues and adversely affect our patent rights if we do not take appropriate enforcement action. We may need to engage in litigation in the future to enforce any patents issued or licensed to us or to determine the scope and validity of third-party proprietary rights. Our issued or licensed patents may not be held valid by a court of competent jurisdiction. Whether or not the outcome of litigation is favorable to us, defending a lawsuit takes significant time, may be expensive and may divert management attention from other business concerns. We may also be required to participate in interference proceedings declared by the United States Patent and Trademark Office for the purpose of determining the priority of inventions in connection with our patent applications or other parties' patent applications. Adverse determinations in litigation or interference proceedings could require us to seek licenses which may not be available on commercially reasonable terms, or at all, or subject us to significant liabilities to third parties. If we need but cannot obtain a license, we may be prevented from marketing the affected product.
Our licensed patent covering the use of gabapentin to treat hot flashes associated with menopause is a method-of-use patent, which increases the risk that prescriptions for gabapentin to treat hot flashes in menopausal women could be written for, or filled with, generic gabapentin.
We have an exclusive sublicense from PharmaNova, Inc. to a patent held by the University of Rochester to develop and commercialize in the United States a gabapentin product for the treatment of hot flashes associated with menopause. Because a method-of-use patent, such as the patent we have sublicensed from PharmaNova, covers only a specified use of a particular compound, not a particular composition of matter, we cannot prevent others from commercializing gabapentin. Accordingly, physicians could prescribe another manufacturer's gabapentin to treat hot flashes in menopausal women rather than Serada, or pharmacists could seek to fill prescriptions for Serada with another manufacturer's gabapentin. Although any such "off-label" use would violate our licensed patent, effectively monitoring compliance with our licensed patent may be difficult and costly.
It is difficult to develop a successful product. If we do not continue to develop successful products, our financial position and liquidity will be adversely affected.
The drug development process is costly, time-consuming and subject to unpredictable delays and failures. Before we or others make commercial sales of products using the Acuform technology, other than GLUMETZA and Proquin XR, we, our current and any future collaborative partners will need to:
- •
- conduct preclinical and clinical tests showing that these products are safe and effective; and
- •
- obtain regulatory approval from the FDA or foreign regulatory authorities.
We will have to curtail, redirect or eliminate our product development programs if we or our collaborative partners find that:
- •
- the Acuform technology has unintended or undesirable side effects; or
- •
- product candidates that appear promising in preclinical or early-stage clinical studies do not demonstrate efficacy in later-stage, larger scale clinical trials.
Even when or if our products obtain regulatory approval, successful commercialization requires:
- •
- market acceptance;
- •
- cost-effective commercial scale production; and
- •
- reimbursement under private or governmental health plans.
36
Any material delay or failure in the governmental approval process and/or the successful commercialization of our potential products would adversely impact our financial position and liquidity.
If we do not achieve our projected development and commercialization goals in the timeframes we announce and expect, the commercialization of our product candidates may be delayed and our business will be harmed and our stock price may decline.
For planning purposes, we sometimes estimate the timing of the accomplishment of various scientific, clinical, regulatory and other product development and commercialization goals. These milestones may include our expectations regarding the commercial launch of our products by us or our licensees, and the commencement or completion of scientific studies and clinical trials and the submission or approval of regulatory filings. From time to time, we may publicly announce the expected timing of some of these milestones, such as the completion of an ongoing clinical trial, the initiation of other clinical programs, or the commercial launch of products. All of these milestones are based on a variety of assumptions. The actual timing of these milestones can vary considerably from our estimates depending on numerous factors, some of which are beyond our control, including:
- •
- the efforts of our marketing partners with respect to the commercialization of our products;
- •
- the rate of progress, costs and results of our clinical trial and research and development activities, including the
extent of scheduling conflicts with participating clinicians and clinical institutions and our ability to identify and enroll patients who meet clinical trial eligibility criteria;
- •
- our receipt of approvals by the FDA and other regulatory agencies and the timing thereof;
- •
- other actions by regulators;
- •
- our ability to access sufficient, reliable and affordable supplies of components used in the manufacture of our product
candidates, including materials for our Acuform technology;
- •
- our available capital resources; and
- •
- the costs of ramping up and maintaining manufacturing operations, as necessary.
If we fail to achieve our announced milestones in the timeframes we announce and expect, our business and results of operations may be harmed and the price of our stock may decline.
We depend on clinical investigators and clinical sites to enroll patients in our clinical trials and other third parties to manage the trials and to perform related data collection and analysis, and, as a result, we may face costs and delays outside of our control.
We rely on clinical investigators and clinical sites to enroll patients and other third parties to manage our trials and to perform related data collection and analysis. However, we may be unable to control the amount and timing of resources that the clinical sites that conduct the clinical testing may devote to our clinical trials. If our clinical investigators and clinical sites fail to enroll a sufficient number of patients in our clinical trials or fail to enroll them on our planned schedule, we will be unable to complete these trials or to complete them as planned, which could delay or prevent us from obtaining regulatory approvals for our product candidates.
Our agreements with clinical investigators and clinical sites for clinical testing and for trial management services place substantial responsibilities on these parties, which could result in delays in, or termination of, our clinical trials if these parties fail to perform as expected. For example, if any of our clinical trial sites fail to comply with FDA-approved good clinical practices, we may be unable to use the data gathered at those sites. If these clinical investigators, clinical sites or other third parties do not carry out their contractual duties or obligations or fail to meet expected deadlines, or if the quality
37
or accuracy of the clinical data they obtain is compromised due to their failure to adhere to our clinical protocols or for other reasons, our clinical trials may be extended, delayed or terminated, and we may be unable to obtain regulatory approval for, or successfully commercialize, our product candidates.
If we are unable to obtain or maintain regulatory approval, we will be limited in our ability to commercialize our products, and our business will be harmed.
The regulatory process is expensive and time consuming. Even after investing significant time and expenditures on clinical trials, we may not obtain regulatory approval of our product candidates. Data obtained from pre-clinical studies such as toxicology studies and from human clinical trials are susceptible to varying interpretations, which could delay, limit or prevent regulatory approval, and the FDA may not agree with our methods of clinical data analysis or our conclusions regarding safety and/or efficacy. Significant clinical trial delays would impair our ability to commercialize our products and could allow our competitors to bring products to market before we do. In addition, changes in regulatory policy for product approval during the period of product development and regulatory agency review of each submitted new application may cause delays or rejections. Even if we receive regulatory approval, this approval may entail limitations on the indicated uses for which we can market a product.
For example, the active ingredients in the products utilizing our Acuform delivery technology that are being developed pursuant to our collaboration with Covidien include acetaminophen in combination with opiates. In connection with concerns that consumers may inadvertently take more than the recommended daily dose of acetaminophen, potentially causing liver damage, an FDA advisory committee has recommended that prescription products containing acetaminophen in combination with prescription analgesics (including opiates) should include black box warnings and/or be removed from the market. The FDA is evaluating the recommendations and has indicated that such an evaluation will take some time. The FDA is not required to accept advisory committee recommendations. Covidien's ability or willingness to develop and market the products subject to our collaboration may be adversely affected by actions of the FDA in response to the advisory committee recommendations.
Further, with respect to our approved products, once regulatory approval is obtained, a marketed product and its manufacturer are subject to continual review. The discovery of previously unknown problems with a product or manufacturer may result in restrictions on the product, manufacturer or manufacturing facility, including withdrawal of the product from the market. Manufacturers of approved products are also subject to ongoing regulation, including compliance with FDA regulations governing current Good Manufacturing Practices (cGMP). Failure to comply with manufacturing regulations can result in, among other things, warning letters, fines, injunctions, civil penalties, recall or seizure of products, total or partial suspension of production, refusal of the government to renew marketing applications and criminal prosecution.
Pharmaceutical marketing is subject to substantial regulation in the United States.
All marketing activities associated with GLUMETZA and Proquin XR, as well as marketing activities related to any other products for which we obtain regulatory approval, will be subject to numerous federal and state laws governing the marketing and promotion of pharmaceutical products. The FDA regulates post-approval promotional labeling and advertising to ensure that they conform to statutory and regulatory requirements. In addition to FDA restrictions, the marketing of prescription drugs is subject to laws and regulations prohibiting fraud and abuse under government healthcare programs. For example, the federal healthcare program antikickback statute prohibits giving things of value to induce the prescribing or purchase of products that are reimbursed by federal healthcare programs, such as Medicare and Medicaid. In addition, federal false claims laws prohibit any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government. Under this law, the federal government in recent years has brought claims against drug manufacturers alleging that certain marketing activities caused false claims for prescription drugs to be
38
submitted to federal programs. Many states have similar statutes or regulations, which apply to items and services reimbursed under Medicaid and other state programs, or, in some states, regardless of the payer. If we, or our collaborative partners, fail to comply with applicable FDA regulations or other laws or regulations relating to the marketing of our products, we could be subject to criminal prosecution, civil penalties, seizure of products, injunction, and exclusion of our products from reimbursement under government programs, as well as other regulatory actions against our product candidates, our collaborative partners or us.
The approval process outside the United States is uncertain and may limit our ability to develop, manufacture and sell our products internationally.
To market any of our products outside of the United States, we and our collaborative partners, including Madaus, are subject to numerous and varying foreign regulatory requirements, implemented by foreign health authorities, governing the design and conduct of human clinical trials and marketing approval for drug products. The approval procedure varies among countries and can involve additional testing, and the time required to obtain approval may differ from that required to obtain FDA approval. The foreign regulatory approval process includes all of the risks associated with obtaining FDA approval set forth above, and approval by the FDA does not ensure approval by the health authorities of any other country, nor does the approval by foreign health authorities ensure approval by the FDA.
If we or our marketing partners are unable to obtain acceptable prices or adequate reimbursement for our products from third-party payers, we will be unable to generate significant revenues.
In both domestic and foreign markets, sales of our product candidates will depend in part on the availability of adequate reimbursement from third-party payers such as:
- •
- government health administration authorities;
- •
- private health insurers;
- •
- health maintenance organizations;
- •
- pharmacy benefit management companies; and
- •
- other healthcare-related organizations.
If reimbursement is not available for our products or product candidates, demand for these products may be limited. Further, any delay in receiving approval for reimbursement from third-party payers would have an adverse effect on our future revenues. Third-party payers are increasingly challenging the price and cost-effectiveness of medical products and services. Significant uncertainty exists as to the reimbursement status of newly approved healthcare products, including pharmaceuticals. Our products may not be considered cost effective, and adequate third-party reimbursement may be unavailable to enable us to maintain price levels sufficient to realize an acceptable return on our investment.
Federal and state governments in the United States and foreign governments continue to propose and pass new legislation designed to contain or reduce the cost of healthcare. Existing regulations affecting pricing may also change before many of our product candidates are approved for marketing. Cost control initiatives could decrease the price that we receive for any product we may develop.
We may be unable to compete successfully in the pharmaceutical product and drug delivery system industries.
Other companies that have oral drug delivery technologies competitive with the Acuform technology include Elan Corporation, Bristol-Myers Squibb, IVAX Corporation (a subsidiary of TEVA
39
Pharmaceutical Industries, Ltd.), Johnson & Johnson, SkyePharma plc, Biovail Corporation, Flamel Technologies S.A., Ranbaxy Laboratories, Ltd., and Intec Pharma, all of which develop oral tablet products designed to release the incorporated drugs over time. Each of these companies has patented technologies with attributes different from ours, and in some cases with different sites of delivery to the gastrointestinal tract.
Bristol-Myers Squibb is currently marketing a sustained release formulation of metformin, Glucophage XR, with which GLUMETZA competes. The limited license that Bristol-Myers Squibb obtained from us under our November 2002 settlement agreement extends to certain current and internally-developed future compounds, which may increase the likelihood that we will face competition from Bristol-Myers Squibb in the future on products in addition to GLUMETZA. Several other companies, including Barr Pharmaceuticals, Inc., Mylan Laboratories, Inc. and Teva Pharmaceutical Industries, Ltd. have received FDA approval for and are selling a controlled-release metformin product.
Bayer Corporation developed a once-daily ciprofloxacin product for the treatment of urinary tract infections, which is currently marketed by Schering-Plough Corporation. There are also generic versions of that product on the market. There may be other companies developing products competitive with GLUMETZA and Proquin XR of which we are unaware.
Gabapentin is currently marketed by Pfizer as Neurontin for adjunctive therapy for epileptic seizures and for postherpetic pain. Pfizer's basic U.S. patents relating to Neurontin have expired, and numerous companies have received approval to market generic versions of the immediate release product. In addition, Pfizer has developed Lyrica® (pregabalin), which has been approved for marketing in the United States and the European Union.
Competition in pharmaceutical products and drug delivery systems is intense. We expect competition to increase. Competing technologies or products developed in the future may prove superior to the Acuform technology or products using the Acuform technology, either generally or in particular market segments. These developments could make the Acuform technology or products using the Acuform technology noncompetitive or obsolete.
Most of our principal competitors have substantially greater financial, sales, marketing, personnel and research and development resources than we do. In addition, many of our potential collaborative partners have devoted, and continue to devote, significant resources to the development of their own drug delivery systems and technologies.
We depend on third parties who are single source suppliers to manufacture GLUMETZA, Proquin XR and our other product candidates. If these suppliers are unable to manufacture GLUMETZA, Proquin XR or our product candidates, our business will be harmed.
We are responsible for the supply and distribution of GLUMETZA, and Patheon, Puerto Rico Inc. is our sole supplier for tablets of the 500mg strength of GLUMETZA pursuant to a supply agreement we entered into with MOVA Pharmaceuticals in December 2006. Biovail is our sole supplier for the 1000mg formulation GLUMETZA. We will be unable to manufacture GLUMETZA in a timely manner if we are unable to obtain GLUMETZA 500mg tablets from our contract manufacturer, active pharmaceutical ingredient from suppliers, or excipient suppliers, or GLUMETZA 1000mg tablets from Biovail.
We are also responsible for supply and distribution of Proquin XR. For the manufacture of Proquin XR tablets, we have entered into an agreement with Patheon, Puerto Rico, Inc., as our sole supplier. We purchase the active ingredient for Proquin XR from Uquifa Mexico, S.A., a sole supplier to us, on a purchase order basis. If we are unable, for whatever reason, to obtain the active pharmaceutical ingredient or Proquin XR tablets from our contract manufacturers, we may be unable to manufacture Proquin XR in a timely manner, if at all.
40
Although we have obtained clinical batches of DM-1796 and Serada from a contract manufacturer, we currently have no long-term supply arrangement with respect to DM-1796 and Serada. Any failure to obtain clinical supplies of DM-1796 and Serada could adversely affect these clinical development programs.
We depend on third parties to manufacture our products, which could adversely affect our ability to deliver our products to market on a timely or competitive basis.
We do not have, and we do not intend to establish in the foreseeable future, internal commercial scale manufacturing capabilities. Rather, we intend to use the facilities of third parties to manufacture products for clinical trials and commercialization. Our dependence on third parties for the manufacture of products using the Acuform technology may adversely affect our ability to deliver such products on a timely or competitive basis. The manufacturing processes of our third party manufacturers may be found to violate the proprietary rights of others. If we are unable to contract for a sufficient supply of required products on acceptable terms, or if we encounter delays and difficulties in our relationships with manufacturers, the market introduction and commercial sales of our products will be delayed, and our future revenue will suffer.
A successful product liability claim against us could materially harm our business.
Our business involves exposure to potential product liability risks that are inherent in the development and production of pharmaceutical products. We have obtained product liability insurance for clinical trials currently underway and forecasted 2009 sales of our products, but:
- •
- we may be unable to obtain product liability insurance for future trials;
- •
- we may be unable to obtain product liability insurance for future products;
- •
- we may be unable to maintain product liability insurance on acceptable terms;
- •
- we may be unable to secure increased coverage as the commercialization of the Acuform technology proceeds; or
- •
- our insurance may not provide adequate protection against potential liabilities.
Our inability to obtain adequate insurance coverage at an acceptable cost could prevent or inhibit the commercialization of our products. Defending a lawsuit would be costly and significantly divert management's attention from conducting our business. If third parties were to bring a successful product liability claim or series of claims against us for uninsured liabilities or in excess of insured liability limits, our business, financial condition and results of operations could be materially harmed.
Our success is dependent in large part upon the continued services of our CEO and senior management with whom we do not have employment agreements.
Our success is dependent in large part upon the continued services of our President and Chief Executive Officer, Carl A. Pelzel, and other members of our executive management team, and on our ability to attract and retain key management and operating personnel. We do not have agreements with Mr. Pelzel or any of our other executive officers that provide for their continued employment with us. We may have difficulty filling open senior scientific, financial and commercial positions. Management, scientific and operating personnel are in high demand in our industry and are often subject to competing offers. The loss of the services of one or more members of management or key employees or the inability to hire additional personnel as needed could result in delays in the research, development and commercialization of our products and potential product candidates.
41
If we choose to acquire new and complementary businesses, products or technologies, we may be unable to complete these acquisitions or to successfully integrate them in a cost effective and non-disruptive manner.
Our success depends on our ability to continually enhance and broaden our product offerings in response to changing customer demands, competitive pressures and technologies. Accordingly, we may in the future pursue the acquisition of complementary businesses, products or technologies instead of developing them ourselves. We have no current commitments with respect to any acquisition or such investment. We do not know if we would be able to successfully complete any acquisitions, or whether we would be able to successfully integrate any acquired business, product or technology or retain any key employees. Integrating any business, product or technology we acquire could be expensive and time consuming, disrupt our ongoing business and distract our management. If we were to be unable to integrate any acquired businesses, products or technologies effectively, our business would suffer. In addition, any amortization or charges resulting from the costs of acquisitions could harm our operating results.
We have implemented certain anti-takeover provisions.
Certain provisions of our articles of incorporation and the California General Corporation Law could discourage a third party from acquiring, or make it more difficult for a third party to acquire, control of our company without approval of our board of directors. These provisions could also limit the price that certain investors might be willing to pay in the future for shares of our common stock. Certain provisions allow the board of directors to authorize the issuance of preferred stock with rights superior to those of the common stock. We are also subject to the provisions of Section 1203 of the California General Corporation Law which requires a fairness opinion to be provided to our shareholders in connection with their consideration of any proposed "interested party" reorganization transaction.
We have adopted a shareholder rights plan, commonly known as a "poison pill". The provisions described above, our poison pill and provisions of the California General Corporation Law may discourage, delay or prevent a third party from acquiring us.
Increased costs associated with corporate governance compliance may significantly impact our results of operations.
Changing laws, regulations and standards relating to corporate governance, public disclosure and compliance practices, including the Sarbanes-Oxley Act of 2002, new SEC regulations and NASDAQ Global Market rules, are creating uncertainty for companies such as ours in understanding and complying with these laws, regulations and standards. As a result of this uncertainty and other factors, devoting the necessary resources to comply with evolving corporate governance and public disclosure standards has resulted in and may in the future result in increased general and administrative expenses and a diversion of management time and attention to compliance activities. We also expect these developments to increase our legal compliance and financial reporting costs. In addition, these developments may make it more difficult and more expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced coverage or incur substantially higher costs to obtain coverage. Moreover, we may be unable to comply with these new rules and regulations on a timely basis.
These developments could make it more difficult for us to attract and retain qualified members of our board of directors, or qualified executive officers. We are presently evaluating and monitoring regulatory developments and cannot estimate the timing or magnitude of additional costs we may incur as a result. To the extent these costs are significant, our selling, general and administrative expenses are likely to increase.
42
If we sell shares of our common stock under our equity line of credit arrangement or in other future financings, existing common shareholders will experience immediate dilution and, as a result, our stock price may go down.
We may from time to time issue additional shares of common stock at a discount from the current trading price of our common stock. As a result, our common shareholders at that time will experience immediate dilution upon the purchase of any shares of our common stock sold at such discount. For example, in December 2006, we entered into a common stock purchase agreement with Azimuth Opportunity Ltd., pursuant to which we may sell shares of common stock at a discount to the prevailing market price ranging from approximately 3.8% to 6.4%, excluding an additional placement agent fee of approximately 1.1% payable by us on the gross offering proceeds. In addition, as other capital raising opportunities present themselves, we may enter into financing or similar arrangements in the future, including the issuance of debt securities, preferred stock or common stock. If we issue common stock or securities convertible into common stock, our common shareholders will experience dilution and this dilution will be greater if we find it necessary to sell securities at a discount to prevailing market prices.
If we are unable to satisfy regulatory requirements relating to internal controls, our stock price could suffer.
Section 404 of the Sarbanes-Oxley Act of 2002 requires companies to conduct a comprehensive evaluation of their internal control over financial reporting. At the end of each fiscal year, we must perform an evaluation of our internal control over financial reporting, include in our annual report the results of the evaluation, and have our external auditors publicly attest to such evaluation. If material weaknesses were found in our internal controls in the future, if we fail to complete future evaluations on time, or if our external auditors cannot attest to our future evaluations, we could fail to meet our regulatory reporting requirements and be subject to regulatory scrutiny and a loss of public confidence in our internal controls, which could have an adverse effect on our stock price.
Business interruptions could limit our ability to operate our business.
Our operations are vulnerable to damage or interruption from computer viruses, human error, natural disasters, telecommunications failures, intentional acts of vandalism and similar events. In particular, our corporate headquarters are located in the San Francisco Bay area, which has a history of seismic activity. We have not established a formal disaster recovery plan, and our back-up operations and our business interruption insurance may not be adequate to compensate us for losses that occur. A significant business interruption could result in losses or damages incurred by us and require us to cease or curtail our operations.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
We currently lease approximately 55,000 square feet of laboratory and office facilities located at 1330, 1360 and 1430 O'Brien Drive, Menlo Park, California. The lease terms for each of these facilities are through January 31, 2012, and include options exercisable by the Company to further extend any of the lease terms for an additional five years. From March 2008 through June 2009, we subleased approximately 9,000 square feet of this space.
We expect that these facilities will accommodate our growth for the next year.
43
Depomed v. Lupin (U.S. Generic GLUMETZA Litigation)
In November 2009, we filed a lawsuit against Lupin for infringement of the patents listed in the Orange Book for the our GLUMETZA product. The lawsuit is in response to an Abbreviated New Drug Application filed by Lupin with the FDA regarding Lupin's intent to market generic versions of 500mg and 1000mg dosage strengths of GLUMETZA prior to the expiration of the four listed patents (U.S. Patent Nos. 6,340,475; 6,488,962; 6,635,280; and 6,723,340). The Company has commenced the lawsuit within the 45 days required to automatically stay, or bar, the FDA from approving Lupin's ANDA for 30 months or until a district court decision that is adverse to the patents, whichever may occur earlier. An adverse outcome in this matter could substantially weaken our U.S. intellectual property.
Biovail and Depomed v. Apotex (Canadian Generic GLUMETZA Litigation)
In December 2007, Apotex, Inc. (Apotex) filed the Canadian equivalent of an Abbreviated New Drug Application in Canada seeking approval to market a generic version of the 500mg formulation of GLUMETZA in Canada.
In February 2010, Apotex received clearance from the Minister of Health in Canada to market the generic version of the 500mg formulation of GLUMETZA. Also in February 2010, Biovail and Depomed filed a complaint in the Federal Court in Canada against Apotex for infringement of the Company's Canadian Patent No. 2,290,624.
An adverse outcome in this matter could substantially weaken our Canadian intellectual property.
General
We may from time to time become party to actions, claims, suits, investigations or proceedings arising from the ordinary course of our business, including actions with respect to intellectual property claims, breach of contract claims, labor and employment claims, and other matters. Although actions, claims, suits, investigations and proceedings are inherently uncertain and their results cannot be predicted with certainty, other than the matters set forth above, we are not currently involved in any matters that we believe may have a material adverse effect on our business, financial position, results of operations or cash flow. However, regardless of the outcome, litigation can have an adverse impact on us because of associated cost and diversion of management time.
44
EXECUTIVE AND OTHER OFFICERS OF THE REGISTRANT
Our executive and other officers of the company and their ages as of March 5, 2010 are as follows:
Name
|
Age | Position | ||
|---|---|---|---|---|
| Executive Officers: | ||||
| Carl A. Pelzel | 59 | President and Chief Executive Officer | ||
| Tammy L. Cameron | 44 | Vice President, Finance | ||
| Matthew M. Gosling | 39 | Vice President and General Counsel | ||
| Michael Sweeney, M.D. | 49 | Vice President, Research & Development | ||
Other Officers: |
||||
| Kera Alexander | 53 | Vice President, Administration and Human Resources | ||
| William Callahan | 51 | Vice President, Operations | ||
| Thadd M. Vargas | 44 | Senior Vice President, Business Development | ||
| Shay Weisbrich | 57 | Vice President, Marketing |
Carl A. Pelzel has served as President and Chief Executive Officer and as a member of the board of directors since August 2007. Mr. Pelzel joined Depomed in June 2005 as Vice President, Marketing and Commercial Development and was promoted to the position of Executive Vice President and Chief Operating Officer in September 2005. Before joining Depomed, Mr. Pelzel was Senior Vice President, Global Commercial Operations at Chiron Corporation from June 2003 to September 2004. Prior to joining Chiron, Mr. Pelzel was President and Chief Executive Officer of Invenux Inc., a privately-held biopharmaceutical company from March 2001 to November 2002. Mr. Pelzel also spent 11 years with GlaxoSmithKline in senior-level sales, marketing and international operational positions, including Country Manager of Hong Kong and China. He spent 13 years with American Home Products, focused primarily on their antibiotics business. During his career, he directed the launch of five major pharmaceutical products, many on a global basis. Mr. Pelzel has a B.A. degree from Hartwick College of Oneonta, New York and a Masters degree in Natural Sciences from the University of Paris.
Kera Alexander has served as Vice President, Administration and Human Resources since October 2007, after having served as Senior Director, Corporate Administration and Human Resources since March 2005. Ms. Alexander joined the Company in February 1997. Prior to joining Depomed, Ms. Alexander held various positions at ALZA Corporation, including Manager, Shareholder Relations. Ms. Alexander received her Professional in Human Resources (PHR) certification in 2005.
Tammy L. Cameron has served Vice President, Finance since December 2008, after having served as Controller since July 2007. In October 2007, Ms. Cameron was named interim principal financial and accounting officer. From January 2005 to June 2007, Ms. Cameron served as the Controller of Adeza Biomedical Corporation, a publicly-traded medical device company. From 2001 to 2005, Ms. Cameron served as the Director of Finance and Administration of Timi3 Systems, a venture-backed medical device company. Prior to Timi3 Systems, Ms. Cameron served as Director of Treasury and External Reporting of KeraVision, Inc., a publicly-traded medical device company and as a member of Ernst & Young's audit practice. She is a Certified Public Accountant and holds a B.A. degree from California State University, East Bay.
William Callahan joined Depomed in 2002 and has served as Vice President, Operations since November 2009. Mr. Callahan previously held the position of Senior Director of Operations. Mr. Callahan has spent most of his more than 20-year career in emerging growth companies, establishing and growing operations groups, and participating in the launch of numerous new products. Prior to Depomed, Mr. Callahan held director of operations positions at Avocet Medical, Cygnus Therapeutics, Applied Biosystems and Johnson & Johnson Lifescan. He received a B.S. degree in Chemistry from San Francisco State University.
45
Matthew M. Gosling has served as Vice President and General Counsel since June 2006. Before joining Depomed, Mr. Gosling was a partner at Heller Ehrman LLP, a national law firm, where he served a nine-year tenure as a corporate transactional attorney. Mr. Gosling received his law degree from the University of Chicago and holds a B.A. degree from Trinity University, San Antonio, Texas.
Michael Sweeney, M.D. joined Depomed as Vice President of Research and Development in December 2007. Before joining Depomed, Dr. Sweeney was Vice President of Medical Affairs at CV Therapeutics from August 2003 to September 2007. Prior to CV Therapeutics, Dr. Sweeney spent 11 years at Pfizer Pharmaceuticals in New York and the U.K. where he held a variety of senior-level medical and marketing positions, including Director of Marketing, Viagra Worldwide Team and Global Urology Medical Group Leader for Pfizer's urological products Viagra, Cardura and Detrol. Prior to Pfizer, he served as a senior clinical pharmacologist and a medical advisor at Zeneca PLC. Dr. Sweeney received his M.D. degree from Manchester University in the U.K. together with post graduate diplomas in Pharmaceutical Medicine and Pharmacoepidemiology, the latter from the University of London. He also is a Fellow of the Royal College of Physicians of Edinburgh.
Thadd M. Vargas has served as Senior Vice President of Business Development since December 2008, after having served as the Company's Vice President of Business Development since December 2002. Before joining the company, Mr. Vargas was Vice President of Finance at Worldres.com, Inc., Director of Finance at Kosan Biosciences, Inc. and Director of Business Development at Anergen, Inc. Prior to Anergen, Mr. Vargas was a member of Ernst & Young's life sciences audit practice. Mr. Vargas holds a B.A. degree in Business Economics from the University of California at Santa Barbara.
Shay Weisbrich joined Depomed as Vice President, Marketing in May 2009. Before joining the Depomed, Ms. Weisbrich was Vice President, Marketing at CV Therapeutics from May 2008 to May 2009. Prior to CV Therapeutics, Ms. Weisbrich was General Manager, Diabetes Marketing at Takeda Pharmaceuticals from April 2005 to April 2008. Ms. Weisbrich spent over 20 years at Pharmacia & Upjohn (now Pfizer) in various marketing and sales management positions, including Global Director, Strategic Marketing, Women's Health Care. She also has assumed a number of vice president positions in marketing, sales and business development at Novo Nordisk, KV Pharmaceuticals/Ther-Rx Corporation, and Gilead Sciences. Ms. Weisbrich received her B.A. and M.A. degrees in Communications Arts & Sciences from Western Michigan University.
46
ITEM 5. MARKET FOR THE REGISTRANT'S COMMON EQUITY, RELATED SHAREHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our common stock trades on the NASDAQ Global Market (NASDAQ) under the symbol "DEPO". The following table sets forth, for the periods indicated, the intraday high and low prices of our common stock as reported by the NASDAQ from January 1, 2008 to December 31, 2009.
| |
High | Low | |||||
|---|---|---|---|---|---|---|---|
2008 |
|||||||
First Quarter |
$ | 4.03 | $ | 2.70 | |||
Second Quarter |
$ | 3.94 | $ | 3.16 | |||
Third Quarter |
$ | 4.42 | $ | 2.95 | |||
Fourth Quarter |
$ | 3.70 | $ | 1.02 | |||
2009 |
|||||||
First Quarter |
$ | 2.68 | $ | 1.70 | |||
Second Quarter |
$ | 3.25 | $ | 1.95 | |||
Third Quarter |
$ | 4.37 | $ | 2.89 | |||
Fourth Quarter |
$ | 6.36 | $ | 3.00 | |||
On March 5, 2010, the closing price of our common stock was $3.16. As of March 5, 2010, there were approximately 40 shareholders of record of our common stock, one of which is Cede & Co., a nominee for Depository Trust Company, or DTC. All of the shares of common stock held by brokerage firms, banks and other financial institutions as nominees for beneficial owners are deposited into participant accounts at DTC, and are therefore considered to be held of record by Cede & Co. as one shareholder.
Dividends
We have never declared or paid any cash dividends on our common stock. We currently intend to retain any future earnings to finance the growth and development of our business. The credit facility we entered into in June 2008 contains covenants which limit our ability to pay dividends. We do not anticipate paying any cash dividends in the foreseeable future.
Issuer Purchases of Securities
We did not repurchase any of our equity securities during the fourth quarter of the year ended December 31, 2009.
47
Stock Price Performance Graph
The following graph compares total shareholder returns of Depomed for the past five years to two indices: the NASDAQ Composite Index and the NASDAQ Biotechnology Index. The total return for Depomed's common stock and for each index assumes the reinvestment of all dividends, although cash dividends have never been declared on Depomed's common stock.
COMPARISON OF 5 YEAR CUMULATIVE TOTAL RETURN*
Among Depomed, Inc., The NASDAQ Composite Index
And The NASDAQ Biotechnology Index
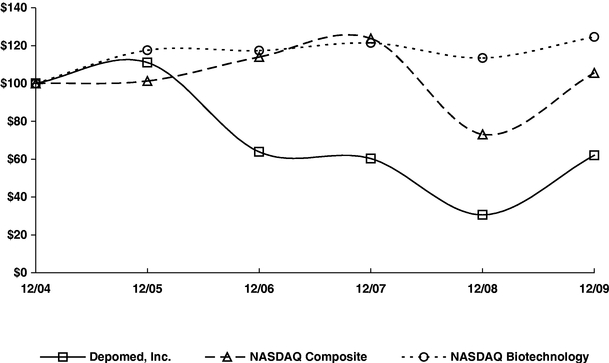
- *
- $100
invested on 12/31/04 in stock or index, including reinvestment of dividends.
Fiscal year ending December 31.
48
ITEM 6. SELECTED FINANCIAL DATA
The data set forth below is not necessarily indicative of the results of future operations and should be read in conjunction with the financial statements and the notes included elsewhere in this annual report on Form 10-K and also with "ITEM 7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS."
| |
Year Ended December 31, | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2009 | 2008(1) | 2007(2) | 2006 | 2005 | ||||||||||||
Statement of Operations Data (in thousands): |
|||||||||||||||||
Total revenues |
$ | 57,728 | $ | 34,842 | $ | 65,582 | $ | 9,551 | $ | 4,405 | |||||||
Total costs and expenses |
79,800 | 51,937 | 18,044 | 51,158 | 30,916 | ||||||||||||
Gain on litigation settlement |
— | 7,500 | — | — | — | ||||||||||||
Gain on termination of Esprit agreements |
— | — | 5,000 | — | — | ||||||||||||
Gain on termination of King agreement |
— | — | 29,584 | — | — | ||||||||||||
Income (loss) from operations |
(22,072 | ) | (17,095 | ) | 47,538 | (41,607 | ) | (26,511 | ) | ||||||||
Gain from extinguishment of debt |
— | — | — | — | 1,059 | ||||||||||||
Net income (loss) before income taxes |
(22,023 | ) | (15,301 | ) | 49,811 | (39,576 | ) | (24,467 | ) | ||||||||
Benefit from (provision for) income taxes |
15 | (1 | ) | (592 | ) | (83 | ) | — | |||||||||
Net income (loss) |
(22,008 | ) | (15,302 | ) | 49,219 | (39,659 | ) | (24,467 | ) | ||||||||
Deemed dividend on preferred stock |
— | (541 | ) | (685 | ) | (665 | ) | (842 | ) | ||||||||
Net income (loss) applicable to common stock shareholders |
(22,008 | ) | (15,843 | ) | 48,534 | (40,324 | ) | (25,309 | ) | ||||||||
Basic net income (loss) per share applicable to common stock shareholders |
$ | (0.43 | ) | $ | (0.32 | ) | $ | 1.06 | $ | (0.97 | ) | $ | (0.64 | ) | |||
Diluted net income (loss) per share applicable to common stock shareholders |
$ | (0.43 | ) | $ | (0.32 | ) | $ | 1.05 | $ | (0.97 | ) | $ | (0.64 | ) | |||
Shares used in computing basic net income (loss) per share |
51,519,912 | 48,778,764 | 45,951,127 | 41,517,661 | 39,821,182 | ||||||||||||
Shares used in computing diluted net income (loss) per share |
51,519,912 | 48,778,764 | 46,353,207 | 41,517,661 | 39,821,182 | ||||||||||||
| |
December 31, | ||||||||||||||||
2009 |
2008 |
2007 |
2006 |
2005 |
|||||||||||||
Balance Sheet Data |
|||||||||||||||||
Cash, cash equivalents and marketable securities |
$ | 81,759 | $ | 82,059 | $ | 69,523 | $ | 33,558 | $ | 59,073 | |||||||
Total assets |
91,581 | 95,084 | 80,645 | 52,617 | 66,414 | ||||||||||||
Deferred revenue, non-current portion |
41,306 | 33,209 | 20,763 | 57,483 | 51,421 | ||||||||||||
Long-term obligations, non-current portion |
2,170 | 5,775 | — | — | — | ||||||||||||
Series A convertible preferred stock |
— | — | 12,015 | 12,015 | 12,015 | ||||||||||||
Accumulated deficit |
(172,202 | ) | (150,194 | ) | (134,892 | ) | (184,111 | ) | (144,452 | ) | |||||||
Total shareholders' equity (deficit) |
15,726 | 33,153 | 45,520 | (27,289 | ) | 6,761 | |||||||||||
- (1)
- Total
costs and expenses, income from operations, net income before income taxes, net income, net income applicable to common stock shareholders and net
income per share in 2008 include a $7.5 million gain on litigation related to our settlement with IVAX.
- (2)
- Total costs and expenses, income from operations, net income before income taxes, net income, net income applicable to common stock shareholders and net income per share in 2007 include (a) a $5.0 million gain on termination of our agreements with Esprit related to Proquin XR and (b) a $29.6 million gain on termination of our promotion agreement with King related to GLUMETZA.
49
ITEM 7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
OVERVIEW
Depomed is a specialty pharmaceutical company focused on the development and commercialization of differentiated products that address large and growing markets and are based on proprietary oral drug delivery technologies. In 2009, we completed Phase 3 clinical trials for two product candidates. In October 2009, we announced that DM-1796, an extended release formulation of gabapentin for the treatment of postherpetic neuralgia that we have licensed to Abbott Laboratories' subsidiary, Solvay Pharmaceuticals, Inc. (Solvay) met its Phase 3 clinical trial primary endpoint with statistical significance. We expect a New Drug Application (NDA) for DM-1796 to be filed with the FDA in March 2010. Also in October 2009, we announced the results of Breeze 1 and Breeze 2, our Phase 3 clinical trials for Serada, our proprietary extended release formulation of gabapentin for the treatment of menopausal hot flashes. The higher dose formulation of Serada evaluated in the studies met five of eight co-primary endpoints across the two studies, while the lower dose formulation evaluated met four of eight co-primary endpoints. In December 2009, we met with the FDA to discuss the results of the Breeze 1 and Breeze 2 studies and any additional clinical development that may be required to obtain approval to market Serada in the United States. Based on guidance from the meeting with the FDA, we expect to initiate one additional Phase 3 trial for Serada by the end of April 2010.
We seek to optimize the use and value of our product candidates and drug delivery technologies in three ways. First, we are seeking to assemble a number of pharmaceutical products that can be highly differentiated from immediate release versions of the compounds upon which they are based and may be promoted together within a specialty pharmaceutical field, such as women's health care providers. Our development of Serada, and our retention of co-promotion rights within the obstetrics/gynecology field in our commercialization arrangements with Covidien, Ltd. (Covidien) and Santarus, Inc. (Santarus), are examples of this aspect of our business strategy. Second, we out-license product candidates after we have increased their value through our formulation and clinical development efforts. Our DM-1796 license and development arrangement with Solvay is an example of this strategy. Third, we enter into collaborative partnerships with other companies where the unique capabilities of our technology can provide superior value to a partner's product candidate, resulting in greater value for Depomed than traditional fee-for-service arrangements. Our license and development arrangement with Covidien and our license agreement with Merck & Co., Inc. (Merck) are examples of this strategy.
We have developed two products which have been approved by the FDA and are currently marketed. GLUMETZA is a once-daily treatment for adults with type 2 diabetes that we commercialize in the United States with Santarus. Proquin® XR (ciprofloxacin hydrochloride) is a once-daily treatment for uncomplicated urinary tract infections.
CRITICAL ACCOUNTING POLICIES AND ESTIMATES
A detailed discussion of our significant accounting policies can be found in Note 1 of the Notes to Financial Statements, and the impact and risks associated with our accounting policies are discussed throughout this Annual Report on Form 10-K and in the footnotes to the financial statements. Critical accounting policies are those that require significant judgment and/or estimates by management at the time that financial statements are prepared such that materially different results might have been reported if other assumptions had been made. We consider certain accounting policies related to revenue recognition, accrued liabilities, stock-based compensation and use of estimates to be critical policies. These estimates form the basis for making judgments about the carrying values of assets and liabilities. We base our estimates and judgments on historical experience and on various other
50
assumptions that we believe to be reasonable under the circumstances. Actual results could differ materially from these estimates.
We believe the following policies to be the most critical to an understanding of our financial condition and results of operations because they require us to make estimates, assumptions and judgments about matters that are inherently uncertain.
Revenue Recognition
We recognize revenue from the sale of GLUMETZA and Proquin XR products, and from license fees, milestones and royalties earned on license agreements and collaborative arrangements. Revenue arrangements with multiple elements are divided into separate units of accounting if certain criteria are met, including whether the delivered element has stand-alone value to the customer and whether there is objective and reliable evidence of the fair value of the undelivered items. The consideration received is allocated among the separate units based on their respective fair values, and the applicable revenue recognition criteria are applied to each of the separate units. Revenue is recognized when there is persuasive evidence that an arrangement exists, delivery has occurred and title has passed, the price is fixed or determinable and we are reasonably assured of collecting the resulting receivable.
Product Sales
We sell GLUMETZA product to wholesalers and retail pharmacies that is subject to rights of return within a period beginning six months prior to, and ending twelve months following product expiration. We began shipping GLUMETZA product to customers in the third quarter of 2006. We currently recognize revenue on shipments of GLUMETZA product at the time title transfers to our customers, which occurs at the time our product is shipped to the customer. Prior to the third quarter of 2008, we were unable to reasonably estimate expected returns of the product at the time of shipment, and therefore, deferred revenue on product shipments of GLUMETZA until the product was dispensed through patient prescriptions. The quantity of prescription units dispensed was estimated based on analysis of third-party information, including third-party market research data and information obtained from wholesalers with respect to inventory levels and inventory movement. Based on the shipment trends, prescription trends and product returns history for GLUMETZA over two years through the third quarter of 2008 and based on an analysis of return rates of companies with products that have similar characteristics and similar return policies within the metformin prescription market, we concluded that we had the information needed to reasonably estimate product returns during the third quarter of 2008. Beginning in the third quarter of 2008, we began recognizing revenue for GLUMETZA sales as revenue at the time of shipment to our customers. Consequently, in 2008, we recognized a one time increase of $6.3 million in net product sales of GLUMETZA, representing product sales previously deferred, net of estimated product returns, managed care and Medicaid rebates, wholesaler and retail pharmacy fees and discounts, chargebacks and prompt payment discounts. Deferred costs related to shipments of GLUMETZA previously deferred were also recognized to cost of sales. This change resulted in a one-time $5.3 million reduction of net loss for 2008. Revenues from GLUMETZA product sales are recorded net of estimated product returns, managed care rebates, wholesaler and retail pharmacy fees and discounts, prompt payment discounts, patient support programs, chargebacks, and Medicaid rebates. These gross-to-net sales adjustments are recognized in the same period the related revenue is recognized and are based on estimates of the amounts earned or to be claimed on the related sales.
We obtained the rights back from Esprit Pharma (Esprit) to market Proquin XR product in July 2007, and began selling Proquin XR to wholesalers and retail pharmacies in October 2007. Given our limited history of selling Proquin XR and declining prescription demand of Proquin XR, we currently cannot reliably estimate expected returns of the product at the time of shipment. Accordingly, we defer recognition of revenue on product shipments of Proquin XR until the right of return no longer exists,
51
which occurs at the earlier of the time Proquin XR units are dispensed through patient prescriptions or expiration of the right of return. We estimate patient prescriptions dispensed using an analysis of third-party information, including third-party market research data and information obtained from wholesalers with respect to inventory levels and inventory movement. We have not had significant history estimating the number of patient prescriptions dispensed for Proquin XR. If we underestimate or overestimate patient prescriptions dispensed for a given period, adjustments to revenue may be necessary in future periods. We have a deferred revenue balance of $1.6 million at December 31, 2009 related to Proquin XR product shipments that have not been recognized as revenue, which is net of wholesaler and retail pharmacy fees and discounts and prompt payment discounts. We will recognize revenue for Proquin XR upon the earlier of prescription units dispensed or expiration of the right of return until we can reliably estimate product returns, at which time we will record a one-time increase in net revenue related to the recognition of revenue previously deferred. In addition, the costs of manufacturing Proquin XR associated with the deferred revenue are recorded as deferred costs, which are included in inventory, until such time the deferred revenue is recognized. Revenues from Proquin XR product sales are recorded net of estimated wholesaler and retail pharmacy fees and discounts, prompt payment discounts, chargebacks and Medicaid rebates. These gross-to-net sales adjustments are recognized in the same period the related revenue is recognized and are based on estimates of the amounts earned or to be claimed on the related sales.
Product Sales Allowances
We recognize product sales allowances as a reduction of product sales in the same period the related revenue is recognized. Product sales allowances are based on amounts owed or to be claimed on the related sales. These estimates take into consideration the terms of our agreements with customers, historical product returns, rebates or discounts taken, estimated levels of inventory in the distribution channel, the shelf life of the product, and specific known market events, such as competitive pricing and new product introductions. If actual future results vary from the our estimates, we may need to adjust these estimates, which could have an effect on product sales and earnings in the period of adjustment. Our product sales allowances include:
- •
- Product Returns—We allow customers to return product for credit on returned product that is within six months
before and up to one year after its product expiration date
- •
- Managed Care Rebates—We offer discounts under contracts with certain managed care providers who do not
purchase directly from us. We generally pay managed care rebates one to two months after the quarter in which prescriptions subject to the rebate are filled.
- •
- Wholesaler and Retail Pharmacy Discounts—We offer contractually determined discounts to certain wholesale
distributors and retail pharmacies that purchase directly from us. These discounts are either taken off-invoice at the time of shipment or paid to the customer on a quarterly basis one to
two months after the quarter in which product was shipped to the customer.
- •
- Prompt Pay Discounts—We offer cash discounts to our customers, generally 2% of the sales price, as an
incentive for prompt payment. Based on our experience, the Company expects its customers to comply with the payment terms to earn the cash discount.
- •
- Medicaid Rebates—We participate in Medicaid rebate programs, which provide assistance to certain low-income patients based on each individual state's guidelines regarding eligibility and services. Under the Medicaid rebate programs, we pay a rebate to each participating state, generally two to three months after the quarter in which prescriptions subject to the rebate are filled.
52
- •
- Chargebacks—We provide discounts to authorized users of the Federal Supply Schedule (FSS) of the General
Services Administration under an FSS contract with the Department of Veterans Affairs. These federal entities purchase products from the wholesale distributors at a discounted price, and the wholesale
distributors then charge back to us the difference between the current retail price and the price the federal entity paid for the product.
- •
- Patient Discount Programs—We offer loyalty card programs for GLUMETZA in which patients receive certain discounts off their prescription at participating retail pharmacies. The discounts are reimbursed by the Company approximately one to two months after the prescription subject to the discount is filled.
We believe our estimates related to gross-to-net sales adjustments for wholesaler and retail pharmacy fees and discounts, prompt payment discounts, patient support programs and chargebacks do not have a high degree of estimation complexity or uncertainty as the related amounts are settled within a relatively short period of time. We believe that our estimated product return allowances and estimated managed care rebates for GLUMETZA require a high degree of judgment and are subject to change based on our experience and certain quantitative and qualitative factors.
Beginning in the third quarter of 2008, we began recognizing GLUMETZA product sales at time title transfers to our customer, and provide for an estimate of future product returns at that time. We monitor actual return history on an individual product lot basis since product launch, which provides us with a basis to reasonably estimate future product returns, taking into consideration the shelf life of product, shipment and prescription trends, estimated distribution channel inventory levels, the return rates of companies with products that have similar characteristics and similar return policies within the metformin prescription market, as well as consideration of the introduction of competitive products. The shelf life of the 500mg GLUMETZA is currently 48 months from the date of tablet manufacture. On product launch in August 2006 and through the second quarter of 2008, the shelf life of 500mg GLUMETZA product shipped was 36 months from the date of tablet manufacture. Since product launch in June 2008 and through December 31, 2009, the 1000mg GLUMETZA shelf life on product shipped to customers was 24 months from the date of tablet manufacture. In 2009, based on our review of actual product returns through the end of 2009, we increased our estimate for GLUMETZA product returns. This resulted in a decrease of product sales in of approximately $0.5 million in 2009 related to sales made in prior periods.
Because of the shelf lives of the GLUMETZA product and our return policy of issuing credits on returned product that is within six months before and up to one year after its product expiration date, there may be a significant period of time between when the product is shipped and when we issue credits on returned product. Accordingly, we may have to adjust these estimates, which could have an effect on product sales and earnings in the period of adjustments. A 1% increase or decrease in our returns reserve estimate for the year ended December 31, 2009 would have a cumulative financial statement impact of $1.1 million.
Our rebate accruals for GLUMETZA are based on definitive contractual agreements with managed care providers related to the dispensing of GLUMETZA under their respective medical benefit plans. Rebates are accrued at the time of sale, and typically are paid out several months after the sale. We estimate rebates based on current and anticipated future patient usage, the applicable contractual rebate rate amounts, and our estimates of the quantity of product in the distribution channel.
Our product sales allowances and related accruals are evaluated each reporting period and adjusted when trends or significant events indicate that a change in estimate is appropriate. Such changes in estimate could materially affect our results of operations of financial position.
53
A rollforward of our product sales allowances for the three years ended December, 31, 2009 is as follows:
(in thousands)
|
Contract Sales Discounts(1) |
Product Returns(2) |
Cash Discounts | Total | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Balance at December 31, 2006 |
$ | 302 | $ | — | $ | 69 | $ | 371 | ||||||
Revenue Allowances: |
||||||||||||||
Provision related to current period sales(2) |
1,448 | — | 279 | 1,727 | ||||||||||
Recorded to balance sheet(2) |
(333 | ) | — | 19 | (314 | ) | ||||||||
Payments and credits related to sales made in current period |
(713 | ) | — | (232 | ) | (945 | ) | |||||||
Payments and credits related to sales made in prior periods |
(302 | ) | — | (69 | ) | (371 | ) | |||||||
Balance at December 31, 2007 |
402 | — | 66 | 468 | ||||||||||
Revenue Allowances: |
||||||||||||||
Provision related to current period sales(2) |
5,010 | 1,583 | 762 | 7,355 | ||||||||||
Recorded to balance sheet(2) |
(422 | ) | — | (103 | ) | (525 | ) | |||||||
Payments and credits related to sales made in current period |
(2,299 | ) | (177 | ) | (612 | ) | (3,088 | ) | ||||||
Payments and credits related to sales made in prior periods |
(402 | ) | — | (66 | ) | (468 | ) | |||||||
Balance at December 31, 2008 |
2,289 | 1,406 | 47 | 3,742 | ||||||||||
Revenue Allowances: |
||||||||||||||
Provision related to current period sales(2) |
7,645 | 2,177 | 936 | 10,758 | ||||||||||
Provision related to sales made in prior years(3) |
— | 526 | — | 526 | ||||||||||
Recorded to balance sheet(2) |
22 | — | (1 | ) | 21 | |||||||||
Payments and credits related to sales made in current period |
(3,485 | ) | (234 | ) | (836 | ) | (4,555 | ) | ||||||
Payments and credits related to sales made in prior periods |
(2,075 | ) | (511 | ) | (47 | ) | (2,633 | ) | ||||||
Balance at December 31, 2009 |
$ | 4,396 | $ | 3,364 | $ | 99 | $ | 7,859 | ||||||
- (1)
- Includes
wholesaler fees and retail discounts, launch discounts, patient support programs, managed care rebates, Medicaid rebates, and chargebacks.
- (2)
- Beginning
in the third quarter of 2008, we began recognizing GLUMETZA product sales at the time title transfers to our customer, and began providing for an
estimate of future product returns at that time. Through December 31, 2009, the Company was unable to reasonably estimate expected returns of product at the time of shipment of Proquin XR
product. Accordingly, the Company currently defers recognition of revenue on product shipments of Proquin XR, and deferred recognition of revenue prior to the third quarter of 2008 on product
shipments of GLUMETZA, until the earlier of when units were dispensed through patient prescriptions or expiration of the right of return. Product sales allowances related to revenue that has been
deferred are recorded on the balance sheet as a reduction of the related deferred revenue, and recognized within the income statement as a reduction of product sales in the same period the related
revenue is recognized.
- (3)
- Adjustment represents an increase in estimate of future product returns.
54
License and Collaborative Arrangements
Revenue from license and collaborative arrangements, including license fees creditable against future royalty obligations (if any), of the licensee, is recognized when an arrangement is entered into if we have substantially completed our obligations under the terms of the arrangement and our remaining involvement is inconsequential and perfunctory. If we have significant continuing involvement under such an arrangement, license fees are deferred and recognized over the estimated performance period. License fee and collaborative payments received in excess of amounts earned are classified as deferred revenue until earned.
We recognize milestone payments upon the achievement of specified milestones if (1) the milestone is substantive in nature, and the achievement of the milestone was not reasonably assured at the inception of the agreement and (2) the fees are nonrefundable. Milestone payments received in excess of amounts earned are classified as deferred revenue until earned.
Research and Development Expense and Accruals
Research and development expenses include related salaries, clinical trial costs, consultant fees and allocations of corporate costs. All such costs are charged to research and development expense as incurred. These expenses result from our independent research and development efforts as well as efforts associated with collaborations. Our expense accruals for clinical trials are based on estimates of the services received from clinical trial centers and clinical research organizations. If possible, we obtain information regarding unbilled services directly from service providers. However, we may be required to estimate these services based on information available to our product development or administrative staff. If we underestimate or overestimate the activity associated with a study or service at a given point in time, adjustments to research and development expenses may be necessary in future periods. Historically, our estimated accrued liabilities have approximated actual expense incurred.
Stock-Based Compensation
We use the Black-Scholes option valuation model to estimate the fair value of stock options and Employee Stock Purchase Plan (ESPP) shares. The Black-Scholes model requires the input of highly subjective assumptions. The most significant assumptions are our estimates of the expected volatility and the expected term of the award. For our volatility assumption, we use the historical volatility of our common stock over the expected term of the options.
As our historical share option exercise experience did not provide a reasonable basis upon which to estimate expected term, we estimated the expected term of options granted by taking the average of the vesting term and the contractual term of the option, as illustrated by the simplified method, and we used the simplified method to estimate the expected term for fiscal years 2006 and 2007. However, we elected to no longer utilize the simplified method after December 31, 2007. At January 1, 2008, we concluded again that our historical share option exercise experience did not provide a reasonable basis upon which to estimate expected term because of limited exercise history. For options granted after January 1, 2008, we have estimated the expected term by using the weighted average of a peer group of companies that grant options with similar vesting provisions. The expected terms used for options granted in 2008 and 2009 are 5.04 years and 5.1 years respectively.
As required, we review our valuation assumptions at each grant date and, as a result, we are likely to change our valuation assumptions used to value employee stock-based awards granted in future periods. Employee and director stock-based compensation costs are to be recognized over the vesting period of the award, and we have elected to use the straight-line attribution method.
Forfeitures are to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. We estimated forfeitures based on historical experience.
55
Fair Value Measurements
Effective January 1, 2008, the Company adopted the provisions of the Fair Value Measurements topic of the Codification, which defines fair value and provides guidance for using fair value to measure certain assets and liabilities. This topic applies whenever other standards require or permit assets or liabilities to be measured at fair value but does not expand the use of fair value in any new circumstances. Accordingly, the carrying amounts of certain of our financial instruments, including cash equivalents and investments, continue to be valued at fair value on a recurring basis. This also requires expanded disclosure of the effect on earnings for items measured using unobservable data, establishes a fair value hierarchy that prioritizes the inputs used to measure fair value and requires separate disclosure by level within the fair value hierarchy.
Fair value is the price that would be received to sell an asset or paid to transfer a liability (an exit price) in an orderly transaction between market participants at the measurement date. We utilize market data or assumptions that we believe market participants would use in pricing assets or liabilities, including assumptions about risk and the risks inherent in the inputs to the valuation technique. These inputs can be readily observable, market corroborated or generally unobservable. We apply the market approach valuation technique for fair value measurements and maximize the use of observable inputs and minimize the use of unobservable inputs. All of our cash equivalents and marketable securities are valued using quoted prices in active markets and are valued at Level 1 or Level 2 within the fair value hierarchy.
RESULTS OF OPERATIONS
Revenues
Total revenues are summarized in the following table (in thousands):
| |
2009 | 2008 | 2007 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
Product sales: |
|||||||||||
GLUMETZA |
$ | 34,570 | $ | 30,526 | $ | 12,489 | |||||
Proquin XR |
524 | 525 | 13 | ||||||||
Total product sales |
35,094 | 31,051 | 12,502 | ||||||||
Royalties: |
|||||||||||
GLUMETZA |
244 | 371 | 207 | ||||||||
Teva |
1,289 | 1,211 | — | ||||||||
Proquin XR |
— | — | 2,500 | ||||||||
Total royalties |
1,533 | 1,582 | 2,707 | ||||||||
License and collaborative revenue: |
|||||||||||
Merck |
10,000 | — | — | ||||||||
DM-1796 |
6,160 | — | — | ||||||||
GLUMETZA |
2,504 | 1,930 | 2,359 | ||||||||
AcuForm technology |
2,332 | 202 | 500 | ||||||||
Proquin XR |
85 | 13 | 47,508 | ||||||||
Other collaborative |
20 | 64 | 6 | ||||||||
Total license and collaborative revenue |
21,101 | 2,209 | 50,373 | ||||||||
Total revenues |
$ | 57,728 | $ | 34,842 | $ | 65,582 | |||||
56
Product sales
GLUMETZA
The increases in GLUMETZA product sales in 2009 from 2008 was primarily driven by increased penetration in the metformin prescription market resulting from the promotion efforts by our promotion partner, Santarus, and to a lesser extent, price increases in 2009. We began selling the 1000mg GLUMETZA in June 2008. Accordingly, 2009 included a full-year of selling the 1000mg GLUMETZA as compared to a partial year in 2008.
As noted above under "CRITICAL ACCOUNTING POLICIES—Revenue Recognition", beginning in the third quarter of 2008, we began to recognize GLUMETZA product sales at the time title transfers to our customer, and provide for an estimate of future product returns at that time. This resulted in a one-time increase for the year ended December 31, 2008, of $6.3 million in net product sales of GLUMETZA, representing product sales previously deferred, net of estimated product returns, managed care and Medicaid rebates, wholesaler and retail pharmacy fees and discounts, chargebacks and prompt payment discounts. The additional increases in 2008 over 2007 included increased penetration in the metformin prescription market and price increases in 2008.
Since October 2008, GLUMETZA has been promoted by Santarus. From February 2008 through September 2008, we promoted GLUMETZA through a contract sales organization. In October 2007, we terminated our promotion agreement related to GLUMETZA with King Pharmaceuticals, who promoted GLUMETZA since its launch in August 2006, and King's promotion obligations ended in December 2007.
Product sales for GLUMETZA relative to its current runrate will depend in part on the success of our promotion partner, Santarus, as well any price adjustments.
Proquin XR
In October 2007, we re-launched Proquin XR with Watson, and began selling to wholesalers and retail pharmacies. We defer recognition of revenue on product shipments of Proquin XR until the right of return no longer exists, which occurs at the earlier of the time Proquin XR units are dispensed through patient prescriptions or expiration of the right of return. At December 31, 2009, we have a deferred revenue balance, which is classified as a liability on the balance sheet, of $1.6 million associated with the deferral of revenue on Proquin XR product shipments, which is net of estimated wholesaler fees, retail pharmacy discounts and prompt payment discounts.
The increase in Proquin XR product sales in 2008 as compared to 2007 is primarily due to a longer period of time of promotion by Watson, which began promotion in October 2007.
In February 2009, we amended our promotion agreement with Watson, pursuant to which Watson performed a specified number of details in the first quarter of 2009. The agreement with Watson terminated effective December 31, 2009, and we currently have no sales force or promotion partner promoting Proquin XR to physicians. Accordingly, we expect product sales for Proquin XR relative to its current runrate to decrease as a result of a decrease in promotional efforts.
We are seeking to divest Proquin XR in the United States.
Royalties
GLUMETZA
GLUMETZA royalties relate to royalties we received from Biovail based on net sales of GLUMETZA in Canada and royalties we received from LG based on net sales of LG's version of
57
GLUMETZA, Novamet GR, in Korea. We began receiving royalties from Biovail in the first quarter of 2006 and from LG in the first quarter of 2007.
Teva
In April 2008, we entered into a settlement and license agreement with Teva related to the patent infringement lawsuit against Teva affiliates IVAX Corporation and IVAX Pharmaceuticals, Inc. we initiated in January 2006 related to Teva's generic Glucophage XR tablets. In connection with the settlement and license agreement we were entitled to receive up to a total of $2.5 million in future royalties on Teva's generic Glucophage XR product in the United States. For the year ended December 31, 2009, we recognized $1.3 million in royalty revenue related to this arrangement. As of December 31, 2009, a cumulative total of $2.5 million in royalties has been recognized to date, with no royalties remaining under the aggregate cap.
Proquin XR
Our agreements with Esprit provided for royalty payments by Esprit to us of 15 percent to 25 percent of Proquin XR net sales in the United States, based on escalating net sales and subject to certain minimum royalty amounts. Esprit's minimum royalty amount for 2006 was $4.6 million and under our amended license agreement entered into in July 2006, amounts paid by Esprit for 2005 royalties were creditable against the 2006 minimum royalty obligation. Net sales of Proquin XR by Esprit for 2005 and 2006 did not reach levels that would obligate Esprit to pay an amount greater than the minimum royalty obligation, and accordingly, Esprit paid us $4.6 million in total royalties for 2005 and 2006. In July 2007, we terminated our license agreement with Esprit, and Esprit paid us $2.5 million in royalties, representing a pro-rated amount of minimum royalties that would have been due to us under the original agreements. Esprit has no further obligations to pay us royalties on Proquin XR sales.
License and Collaborative Revenue
GLUMETZA
GLUMETZA license revenue for the year ended December 31, 2009 consisted primarily of license revenue recognized from the $25.0 million license fee received from Biovail in July 2005 and from the $12.0 million upfront fee received from Santarus in July 2008. We are recognizing the $25.0 million license fee payment from Biovail as revenue ratably until October 2021, which represents the estimated length of time our obligations exist under the arrangement related to royalties we are obligated to pay Biovail on net sales of GLUMETZA in the United States and for our obligation to use Biovail as our sole supplier of the 1000mg GLUMETZA. We are recognizing the $12.0 million upfront payment from Santarus as revenue ratably until October 2021, which represents the estimated length of time our obligations exist under the arrangement related to promotion fees we are obligated to pay Santarus for GLUMETZA in the United States.
We received a $0.6 million upfront license fee from LG in August 2004 and a $0.5 million milestone payment in November 2006 with respect to LG's approval to market LG's extended-release metformin product that incorporates our AcuForm technology, Novamet GR, in the Republic of Korea. These payments were originally deferred and amortized as license revenue over the estimated length of time we were obligated to provide assistance in development and manufacturing. In January 2007, we amended our agreement with LG, granted LG a license to certain of the Company's intellectual property rights to manufacture the 500mg Novamet GR in exchange for royalties on net sales of Novamet GR in Korea, and removed the provisions of the original agreement providing for the supply of 500mg Novamet GR tablets by us to LG. Under the amended agreement, we no longer have continuing performance obligations to LG, and recognized the remaining $0.9 million of previously
58
deferred license revenue in the first quarter of 2007. As a result of this one time recognition in 2007, GLUMETZA license revenue decreased in 2008 as compared to 2007.
Proquin XR
Our license agreement with Esprit for Proquin XR provided for $50.0 million in license fees from Esprit. We received $30.0 million in license fees in July 2005 and an additional $10.0 million in December 2006. The final $10.0 million installment was paid in July 2007. The first $40.0 million in license fees received were recognized as revenue ratably commencing on our receipt of the fees through June 2020, which represented the length of time we were obligated to manufacture Proquin XR under our Proquin XR supply agreement with Esprit. In July 2007, we and Esprit terminated the license and supply agreements, and all deferred revenue related to license fees previously received from Esprit was fully recognized as revenue in July 2007, resulting in total recognition of $46.1 million of license revenue related to our agreements with Esprit in the third quarter of 2007.
AcuForm Technology
In November 2008, we entered into a license agreement with Covidien granting Covidien worldwide rights to utilize our AcuForm technology for the exclusive development of four products containing acetaminophen in combination with opiates. In 2008, Covidien paid us a total of $5.5 million in upfront fees, representing a $4.0 million upfront license fee and a $1.5 million upfront payment for formulation work to be performed by Depomed under the agreement. The entire $5.5 million is being accounted for as a single unit of accounting and being amortized ratably through November 2011, which is the length of time Depomed is obligated to perform formulation work under the agreement. In October 2009, the first formulation was completed by us and delivered to Covidien, which triggered a $0.5 million milestone payment from Covidien to us in October 2009.
In February 2007, we received $0.5 million from Biovail upon entering into a license and development agreement with Biovail granting Biovail an option to license our AcuForm drug delivery technology to develop and commercialize up to two pharmaceutical products. We had no continuing performance obligations under the agreement and recognized the entire upfront license fee as revenue in the first quarter of 2007.
DM-1796
DM-1796 license revenue for the three year ended December 31, 2009 relates to the $25.0 million upfront payment received from Solvay under our license agreement granting Solvay exclusive rights to develop and commercialize DM-1796 in the United States, Canada and Mexico for pain indications. We are recognizing the $25.0 million upfront payment received from Solvay as revenue ratably until January 2013, which represents the expected maximum length of time our development and supply obligations exist under the agreement.
Merck
Merck license revenue for the year ended December 31, 2009 relates to the $10.0 million upfront payment received from Merck in August 2009 under our non-exclusive license agreement granting Merck a license to certain patents related to the Company's metformin extended release technology to be used in developing fixed dose combinations of sitagliptin and extended release metformin. As the Company has no continuing obligations under the agreement, the $10.0 million upfront payment was fully recognized as license revenue on receipt in the third quarter of 2009 as we have no continuing obligations under the agreement.
59
Other Collaborative
Other collaborative revenue in 2009 and 2008 represents a grant received by the Michael J. Fox Foundation in relation to our DM-1992 product candidate for Parkinson's Disease.
Cost of Sales
Cost of sales consists of costs of the active pharmaceutical ingredient, contract manufacturing and packaging costs, inventory write-downs, product quality testing, internal employee costs related to the manufacturing process, distribution costs and shipping costs related to our product sales of GLUMETZA and Proquin XR. Total costs of sales are summarized in the following table (in thousands):
| |
2009 | 2008 | 2007 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Cost of sales |
$ | 5,257 | $ | 5,772 | $ | 2,597 | ||||
Cost of sales decreased in 2009 compared to 2008 primarily as a result of recognition of a $0.6 million provision for slow-moving Proquin XR inventory in 2008 based on Proquin XR inventory levels and expiration dates held by the Company in excess of the Company's expectations on future prescription demand.
Cost of sales increased in 2008 over 2007 primarily as a result of an increase in GLUMETZA product sales. As noted above under "CRITICAL ACCOUNTING POLICIES—Revenue Recognition", beginning in the third quarter of 2008, we began to recognize GLUMETZA product sales at time title transfers to our customer. This resulted in a one-time increase of approximately $1.0 million in cost of sales in 2008.
The costs of manufacturing associated with deferred revenue on Proquin XR product shipments are recorded as deferred costs, which are included in inventory, until such time the deferred revenue is recognized.
Research and Development Expense
Our research and development expenses currently include costs for scientific personnel, supplies, equipment, outsourced clinical and other research activities, consultants, depreciation, facilities and utilities. The scope and magnitude of future research and development expenses cannot be predicted at this time for our product candidates in the early phases of research and development, as it is not possible to determine the nature, timing and extent of clinical trials and studies, the FDA's requirements for a particular drug and the requirements and level of participation, if any, by potential partners. As potential products proceed through the development process, each step is typically more extensive, and therefore more expensive, than the previous step. Success in development therefore, generally results in increasing expenditures until actual product launch. Total research and development expense for the each of the three years ended December 31, 2009 were as follows (in thousands):
| |
2009 | 2008 | 2007 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Research and development expense |
$ | 34,298 | $ | 27,268 | $ | 23,337 | ||||
Dollar change from prior year |
7,030 | 3,931 | ||||||||
Percentage change from prior year |
26 | % | 17 | % | ||||||
From 2007 through 2009, the majority of our research and development expense was related to DM-1796 and Serada programs. In September 2008 we started our two Phase 3 clinical trials for Serada for the treatment of menopausal hot flashes. In February 2009, we completed enrollment of our Breeze 1 Phase 3 clinical trial for Serada. In March 2009, we completed enrollment of our Breeze 2 Phase 3 clinical trial for Serada. The treatment duration of the Breeze 1 study was six months, and the
60
treatment duration of the Breeze 2 study was three months. The trials were completed in October 2009.
In March 2008, we initiated dosing of the first patient in a Phase 3 clinical trial for DM-1796 for post-herpetic neuralgia. The treatment duration was ten weeks. In June 2009, we completed enrollment of our clinical trial for DM-1796 and the trial was completed in October 2009.
The increase in research and development expense in 2009 from 2008 was primarily due to increased clinical research organization expenses related to the completion of our Breeze 1 and Breeze 2 Phase 3 clinical trials for Serada.
The increase in research and development expense in 2008 from 2007 was primarily due to higher clinical research organization expenses with the commencement of Breeze 1 and Breeze 2 Phase 3 clinical trials for Serada and commencement of our Phase 3 clinical trial for DM-1796 for the treatment of postherpetic neuralgia.
We plan to conduct a single additional pivotal Phase 3 trial evaluating Serada for the treatment of menopausal hot flashes. The company expects to initiate the trial, which will be known as Breeze 3, by the end of April 2010 and to complete the trial by the end of the first quarter of 2011. While we expect to continue to incur significant research and development expenses resulting from the progress of Breeze 3, we expect total research and development expenses to decrease in 2010 from 2009 as a result of completion of the DM-1796 Phase 3 clinical trial during 2009.
We categorize our research and development expense by project. The table below shows research and development costs for our major clinical development programs, as well as other expenses associated with all other projects in our product pipeline.
| |
2009 | 2008 | 2007 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
DM-1796 |
$ | 11,768 | $ | 13,636 | $ | 13,928 | ||||
Serada |
15,146 | 7,463 | 4,114 | |||||||
Other projects |
7,384 | 6,169 | 5,295 | |||||||
Total research and development expenses |
$ | 34,298 | $ | 27,268 | $ | 23,337 | ||||
61
The following table summarizes our principal product development initiatives as of March 2010. In addition to the products listed in the table below, from time to time we may enter into feasibility studies with collaborative partners that, if successful, may be followed by definitive agreements to advance development of the product candidate.
| Program | Potential Indications | Development Status | ||
|---|---|---|---|---|
| Serada™ | Menopausal hot flashes | Initial Phase 3 trials (Breeze 1 and Breeze 2) complete. | ||
Second Phase 3 trial (Breeze 3) expected to begin April 2010. |
||||
DM-1796 |
Postherpetic neuralgia |
Second Phase 3 trial Complete. |
||
NDA filing expected in Q1 2010 |
||||
Diabetic peripheral neuropathy |
Phase 2 trial complete. |
|||
DM-3458 |
Gastroesophageal reflux disease (GERD) |
Proof of concept studies completed. |
||
DM-1992 |
Parkinson's disease |
Phase 1 study complete. |
||
Second Phase 1 study expected in 2010. |
We expect that the pharmaceutical products that we develop internally will take, on average, from four to eight years to research, develop and obtain FDA approval in the United States, assuming that we are successful. We generally must conduct preclinical testing on laboratory animals of new pharmaceutical products prior to commencement of clinical studies involving human beings. These studies evaluate the potential efficacy and safety of the product. We then submit the results of these studies to the FDA as part of an Investigational New Drug Application, or IND, which, if successful, allows the opportunity for clinical study of the potential new medicine.
Typically, human clinical evaluation involves a time-consuming and costly three-phase process:
- •
- In Phase 1, we conduct clinical trials with a small number of subjects to determine a drug's early safety profile
and its blood concentration profile over time. A Phase 1 trial for our average potential product may take 6 to 12 months to plan and complete.
- •
- In Phase 2, we conduct limited clinical trials with groups of patients afflicted with a specific disease in order
to determine preliminary efficacy, optimal dosages and further evidence of safety. A Phase 2 trial for our average potential product may take 9 to 18 months to plan and complete.
- •
- In Phase 3, we conduct large-scale, multi-center, comparative trials with patients afflicted with a target disease in order to provide enough data to statistically evaluate the efficacy and safety, as required by the FDA. A Phase 3 trial for our average potential product may take 1 to 3 years to plan and complete.
The most significant expenses associated with clinical development derive from Phase 3 trials as they tend to be the longest and largest studies conducted during the drug development process.
The successful development of pharmaceutical products is highly uncertain. The FDA closely monitors the progress of each phase of clinical testing. The FDA may, at its discretion, re-evaluate, alter, suspend or terminate testing based upon the data accumulated to that point and the FDA's assessment of the risk/benefit ratio to patients. The FDA may also require additional clinical trials after
62
approval, which are known as Phase 4 trials. Various statutes and regulations also govern or influence the manufacturing, safety, labeling, storage and record keeping for each product. The lengthy process of seeking FDA approvals, and the subsequent compliance with applicable statutes and regulation, require the expenditure of substantial resources.
Selling, General and Administrative Expense
Selling, general and administrative expenses primarily consist of personnel expenses to support our administrative and operating activities, marketing and promotion expenses associated with Serada, GLUMETZA and Proquin XR, facility costs and professional expenses, such as legal and accounting fees. Total selling, general and administrative expenses, as compared to the prior year, were as follows:
| |
2009 | 2008 | 2007 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
Selling, general and administrative expense: |
|||||||||||
Promotion fee expense |
$ | 23,589 | $ | 4,841 | $ | 3,012 | |||||
Other selling, general and administrative expense |
16,656 | 21,556 | 23,682 | ||||||||
Total selling, general and administrative expense |
$ | 40,245 | $ | 26,397 | $ | 26,694 | |||||
Dollar change from prior year |
13,848 | (297 | ) | ||||||||
Percentage change from prior year |
52 | % | (1 | %) | |||||||
The increase in selling, general and administrative expense in 2009 as compared to 2008 was primarily driven by a full year of GLUMETZA promotion fees for 2009 as compared to one quarter in 2008, as we began paying Santarus promotion fees beginning with the fourth quarter of 2008. Promotion fee expense related to the Santarus agreement was $23.6 million for the year ended December 31, 2009 compared to $4.7 million for the fourth quarter of 2008. The Company also incurred $0.1 million in promotion fee expense related to the Watson agreement during the year ended December 31, 2008. The increase in promotion fee expense was partially offset by decreases in headcount costs and other sales and marketing expenses for GLUMETZA, as a majority of those efforts have been transferred to Santarus.
The slight decrease in selling, general and administrative expense in 2008 from 2007 was primarily due to a decrease of $2.5 million in legal expenses primarily resulting from the resolution of our patent infringement case against IVAX in April 2008, offset by an increase of $1.7 million in GLUMETZA promotion fees in 2008 to Santarus relative to GLUMETZA promotion fees in 2007 to King.
Our selling, general and administrative expenses in future periods may increase if GLUMETZA gross margin increases. GLUMETZA promotion fees payable to Santarus are calculated as a percentage of GLUMETZA gross margin. The promotion fee currently is 80% of GLUMETZA gross margin and will be reduced to 75% of gross margin beginning in the fourth quarter of 2010.
Interest Income and Expense
| |
2009 | 2008 | 2007 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Interest and other income |
$ | 1,050 | $ | 2,349 | $ | 2,273 | ||||
Interest expense |
(1,001 | ) | (555 | ) | — | |||||
Net interest income (expense) |
$ | 49 | $ | 1,794 | $ | 2,273 | ||||
Interest and other income decreased during in 2009 as compared to 2008 as a result of lower interest rates on our investments. Interest and other income slightly increased in 2008 from 2007 as a result of higher investment balances, which were partially offset by lower interest rates on our investments.
Interest expense relates to interest on the credit facility we entered into in June 2008 with General Electric Capital Corporation and Oxford Finance Corporation.
63
Gain on Settlement with TEVA Pharmaceuticals USA, Inc.
In April 2008, we entered into a settlement and license agreement with Teva related to the patent infringement lawsuit we filed against Teva affiliates IVAX Corporation and IVAX Pharmaceuticals, Inc. The settlement agreement provided for a one-time payment to Depomed of $7.5 million, which has been classified a gain within operating income for the year ended December 31, 2008.
Gain on Termination of King Promotion Agreement
In conjunction with the termination and assignment agreement entered into with King in October 2007, we received a $29.7 million termination payment from King, of which $29.6 million has been classified as a gain within operating income for the year ended December 31, 2007.
Gain on Termination of Esprit Pharma Agreement
In conjunction with the termination and assignment agreement entered into with Esprit in July 2007, we received a $5.0 million termination payment from Esprit, which has been classified as a gain within operating income for the year ended December 31, 2007.
Series A Preferred Stock and Deemed Dividends
In January 2000, the Company issued 12,015 shares of Series A Preferred Stock at a price of $1,000 per share. The Series A Preferred Stock accrued a dividend of 7% per annum, compounded semi-annually and payable in shares of Series A Preferred Stock. The Series A Preferred Stock was convertible at anytime between January 2002 and January 2006 into the Company's common stock. The original conversion price of the Series A Preferred Stock was $12.00; however, as a result of the Company's March 2002 and October 2003 financings, the conversion price had been adjusted to $9.51 per share. In December 2004, the Company entered into an agreement with the Series A Preferred shareholder to resolve a misunderstanding between the Company and the shareholder relating primarily to prior adjustments to the conversion price of the Series A Preferred Stock. Pursuant to the agreement, among other matters, the Company agreed to adjust the conversion price to $7.50 per share. The Company and the shareholder also agreed to binding interpretations of certain other terms related to the Series A Preferred Stock conversion price.
Prior to December 2004, the amounts calculated as Series A Preferred stock dividends were accounted for as an adjustment to the conversion price following EITF Issue No. 98-5, Accounting for Convertible Securities with Beneficial Conversion Features or Contingently Adjustable Conversion Ratios (Issue No. 98-5). As a result of the modifications to the preferred stock agreement in December 2004, the Company determined that a "significant modification" of the agreement had been made, and, therefore, a new "commitment date" for accounting purposes had been established on December 10, 2004. The Company measured the difference between the carrying value of the preferred stock and the fair value of the modified preferred stock pursuant to EITF Topic No. D-42, The Effect on the Calculation of Earnings per Share for the Redemption or Induced Conversion of Preferred Stock and determined that the fair value of the modified security was less than the carrying value of the security prior to the modification. The Company also evaluated the effective conversion rate, after considering the reset rate of $7.50 per share in addition to the common stock issuable upon conversion of the unpaid, accumulated dividends. The fair value of the underlying common stock on December 10, 2004 was $5.06 per share. The Company determined that the conversion rate, after including the effect of the unpaid dividends, did not result in a beneficial conversion feature, which could have had the effect of also providing a deemed dividend to the preferred shareholder. However, an anti-dilution provision of the Series A Preferred Stock was triggered by the Company's January 2005 financing, which adjusted the conversion price of the Series A Preferred Stock to $7.12. As a result of the adjusted conversion price and an increase in the amount of common stock issuable upon conversion of the Series A
64
Preferred Stock due to additional accumulated dividends, the Series A Preferred Stock now contains a "beneficial conversion feature" subject to recognition pursuant to Issue No. 98-5.
In conjunction with the modification of the agreement, the Company issued a warrant to the Series A Preferred shareholder. The value of the warrant was considered in determining the value of the modified security. The warrant was convertible into shares of the Company's common stock during the period between January 2006 and January 2009. The conversion price of the warrant initially was $7.12, which was equal to the Series A Preferred Stock conversion price in effect as of January 20, 2006. The conversion price of the warrant decreased by approximately 4.8% per year during the conversion period, such that the number of shares of the Company's common stock issuable upon conversion of the warrant increased by approximately 5.1% per year. The conversion of the warrant could be satisfied only by surrender of the outstanding shares of Series A Preferred Stock.
The Series A Preferred Stock accrued dividends through January 20, 2006, which is the date the warrant initially became exercisable. As a result of the issuance of the warrant, the preferred stock could be surrendered in exchange for common stock for an additional three years through January 20, 2009. As long as the Series A Preferred Stock remained outstanding, the number of shares into which the warrant could be converted increased as the conversion price of the warrant decreased resulting in additional deemed dividends on the Series A Preferred Stock. For the years ended December 31, 2009, 2008 and 2007 we recognized Series A Preferred Stock deemed dividends of approximately zero, $0.5 million and $0.7 million, respectively, attributable to the beneficial conversion feature from the accrued dividends and decreasing warrant price.
In October 2008, the holder of the Series A Preferred Stock and warrant exercised its warrant to acquire shares of the Company's common stock by surrendering its 18,158 shares of Series A Preferred Stock in exchange for 2,914,526 shares of the Company's common stock. The warrant was exercised in accordance with its terms and without any cash payment to the company, and together with surrender of the Series A Preferred Stock, was convertible into the Company's common stock at a conversion price of $6.23 per share on the date of exercise.
LIQUIDITY AND CAPITAL RESOURCES
| |
As of December 31, | ||||||
|---|---|---|---|---|---|---|---|
| |
2009 | 2008 | |||||
Cash, cash equivalents and marketable securities (in thousands) |
$ | 81,759 | $ | 82,059 | |||
In August 2009, we received a $10.0 million upfront payment from Merck related to our non-exclusive license agreement with Merck. In February 2009, we received a $25.0 million upfront payment from Solvay related to our license agreement for DM-1796 for PHN.
Since inception through December 31, 2009, we have financed our product development efforts and operations primarily from private and public sales of equity securities, upfront license and termination fees from collaborative and license partners, and product sales.
In December 2006, we entered into a common stock purchase agreement with Azimuth Opportunity, Ltd., pursuant to which Azimuth is committed to purchase, from time to time and at our sole discretion, up to the lesser of (a) $30.0 million of our common stock, or (b) 8,399,654 shares of common stock. In August 2008, the agreement was amended and the term of the agreement was extended until December 2010. Sales to Azimuth under the agreement, if any, will be made at a price equal to the average closing price of our common stock over a given pricing period, minus a discount ranging from approximately 3.8% to 6.4%, which varies based on a threshold price set by us. Upon each sale of the our common stock to Azimuth under the agreement, we have also agreed to pay Reedland Capital Partners a placement fee equal to approximately 1.1% of the aggregate dollar amount of common stock purchased by Azimuth. Azimuth is not required to purchase our common
65
stock when the price of our common stock is below $2 per share. As of December 31, 2009, we have not sold any common stock to Azimuth under this common stock purchase agreement.
In June 2008, we entered into a credit facility with GECC and Oxford, to allow us capital flexibility as we funded our clinical trials. The credit facility was available in up to three tranches. The first tranche of $3.8 million was advanced to us upon the closing of the loan agreement. In July 2008, we received the second tranche of $5.6 million. The third tranche of $5.6 million was not drawn and it is no longer available to us, and GECC and Oxford waived the 2% unused line fee related to the third tranche.
We paid interest only on the first tranche for the first six months at an interest rate of 11.59%. Thereafter, we are required to pay the principal on the first tranche, plus interest at such rate, in 30 equal monthly installments. The second tranche was interest-only through December 31, 2008, with principal and interest payable thereafter in 30 equal monthly installments and has an interest rate of 11.59%. As of December 31, 2009, the outstanding balance on the credit facility was approximately $6.1 million at an interest rate of 11.59%.
Our obligations under the loan agreement with GECC and Oxford are secured by interests in all of our personal property, and proceeds from any intellectual property, but not by our intellectual property. The loan agreement contains affirmative and negative covenants with which we must comply. The loan agreement imposes restrictions with regards to additional indebtedness, liens, various fundamental changes (including mergers and acquisitions), payments, investments, transactions with affiliates, and other limitations customary in secured credit facilities. As of December 31, 2009, we were in compliance with such covenants. The loan agreement provides that events of default will exist in certain circumstances, including failure to make payment of principal or interest on the loans when required, failure to perform certain obligations under the loan agreement and related documents, defaults in certain other indebtedness and certain other events. Upon an event of default, the principal amount of the loan may become due immediately.
As of December 31, 2009, we have accumulated net losses of $172.2 million. We expect to continue to incur operating losses in 2010. We anticipate that our existing capital resources will permit us to meet our capital and operational requirements through at least the end of 2011. We base this expectation on our current operating plan, which may change as a result of many factors.
Our cash needs may also vary materially from our current expectations because of numerous factors, including:
- •
- sales of our marketed products;
- •
- expenditures related to our commercialization and development efforts, including arrangements we make for the
commercialization of Serada, if the product is approved for marketing;
- •
- financial terms of definitive license agreements or other commercial agreements we enter into, if any;
- •
- results of research and development efforts;
- •
- changes in the focus and direction of our research and development programs;
- •
- technological advances;
- •
- results of clinical testing, requirements of the FDA and comparable foreign regulatory agencies; and
- •
- acquisitions or investment in complimentary businesses, products or technologies.
66
We will need substantial funds of our own or from third parties to:
- •
- conduct research and development programs;
- •
- commercialize any products we market;
- •
- conduct preclinical and clinical testing; and
- •
- manufacture (or have manufactured) and market (or have marketed) our marketed products and product candidates.
Our existing capital resources may not be sufficient to fund our operations until such time as we may be able to generate sufficient revenues to support our operations. We have limited credit facilities and, except for the common stock purchase agreement with Azimuth, we have no other committed sources of capital. To the extent that our capital resources are insufficient to meet our future capital requirements, we will have to raise additional funds through the sale of our equity securities or from development and licensing arrangements to continue our development programs. We may be unable to raise such additional capital on favorable terms, or at all. If we raise additional capital by selling our equity or convertible debt securities, the issuance of such securities could result in dilution of our shareholders' equity positions. If adequate funds are not available we may have to:
- •
- delay, postpone or terminate clinical trials;
- •
- significantly curtail commercialization of our marketed products or other operations; and/or
- •
- obtain funds through entering into collaboration agreements on unattractive terms.
The inability to raise additional capital required to fund our operations would have a material adverse effect on our company.
The following table summarizes our cash flow activities (in thousands):
| |
2009 | 2008 | 2007 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Cash provided by operating activities |
$ | 1,838 | $ | 3,351 | $ | 14,661 | ||||
Cash provided by (used in) investing activities |
4,126 | (5,123 | ) | (35,986 | ) | |||||
Cash (used in) provided by financing activities |
(1,270 | ) | 9,525 | 21,125 | ||||||
Cash provided by operating activities in 2009 was primarily as a result of the $25.0 million upfront payment received from Solvay in 2009, offset by our net loss for the year. Cash provided by operating activities in 2008 was primarily due to an increase in deferred revenue as a result of the $12.0 million upfront payment received from Santarus and $5.5 million from Covidien during 2008 and an increase in accrued payables, which were offset by our net loss for the year. Cash provided by operations for 2007 primarily consisted of our net income adjusted for stock-based compensation, depreciation expense and movements in working capital, including recognition of previously deferred revenue.
Cash provided by investing activities in 2009 was approximately $4.1 million and consisted primarily of a net decrease in marketable securities to fund our operations. Cash used in investing activities in 2008 was due to a net increase in marketable securities of $5.1 million resulting from investment of proceeds from our credit facility, upfront payments received from Santarus and Covidien, as well the settlement payment we received from Teva. Cash used in investing activities in 2007 was due to a net increase in marketable securities of $35.8 million resulting from investment of termination fees received from King and the final license fee payment and termination fee received from Esprit.
Cash used in financing activities in 2009 primarily consisted of $3.3 million in principal payments on our credit facility offset by $2.0 million of cash proceeds from exercises of stock options and purchases of common stock under our employee stock purchase plan. Cash provided by financing activities in 2008 consisted of proceeds from our credit facility and $0.5 million in cash proceeds from
67
exercises of stock options and purchases of common stock under our employee stock purchase plan. Cash provided from financing activities in 2007 consisted of $20.0 million in proceeds from our registered direct offering of 5,300,000 shares of common stock for $3.78 per share in April 2007 and $1.2 million in cash proceeds from exercises of stock options and purchases of common stock under our employee stock purchase plan.
Contractual Obligations
As of December 31, 2009, our contractual obligations are shown in the following table (in thousands):
| |
Less than 1 year | 1-3 years | 3-5 years | Total | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Operating leases |
$ | 1,614 | $ | 1,773 | $ | $ | 3,387 | ||||||
Long-term debt (principal) |
3,845 | 2,243 | — | 6,088 | |||||||||
Long-term debt (interest portion) |
512 | 82 | — | 594 | |||||||||
Purchase commitments |
1,924 | — | — | 1,924 | |||||||||
|
$ | 7,895 | $ | 4,098 | $ | — | $ | 11,993 | |||||
At December 31, 2009, we had non-cancelable purchase orders and minimum purchase obligations of approximately $1.3 million under our manufacturing agreement with Patheon Puerto Rico, Inc. for the manufacture of 500mg GLUMETZA, and $0.6 million under our supply agreement with Biovail for the supply of 1000mg GLUMETZA. The amounts disclosed only represent minimum purchase requirements. Actual purchases are expected to exceed these amounts.
The contractual obligations reflected in this table exclude $3.0 million of contingent milestone payments we may be obligated to pay in the future under our sublicense agreement with PharmaNova. The payments relate to various milestones for the product candidate under the sublicense agreement, including submission to the FDA of an NDA, and FDA approval of an NDA. The above table also excludes any future royalty payments we may be required to pay on products we have licensed or any promotion fees associated with our promotion agreement with Santarus.
OFF-BALANCE SHEET ARRANGEMENTS
We do not have any off-balance sheet arrangements that are reasonably likely to have a current or future material effect on our financial condition, results of operations, liquidity, capital expenditures or capital resources.
RECENTLY ISSUED ACCOUNTING PRONOUNCEMENTS
In August 2009, the FASB issued Accounting Standards Update No. 2009-05, Measuring Liabilities at Fair Value (ASU 2009-05). ASU 2009-05 amends Accounting Standards Codification Topic 820, Fair Value Measurements. Specifically, ASU 2009-05 provides clarification that in circumstances in which a quoted price in an active market for the identical liability is not available, a reporting entity is required to measure fair value using one or more of the following methods: 1) a valuation technique that uses a) the quoted price of the identical liability when traded as an asset or b) quoted prices for similar liabilities or similar liabilities when traded as assets and/or 2) a valuation technique that is consistent with the principles of Topic 820 of the Accounting Standards Codification. ASU 2009-05 also clarifies that when estimating the fair value of a liability, a reporting entity is not required to adjust to include inputs relating to the existence of transfer restrictions on that liability. The adoption of this standard did not have a material impact on the Company's financial statements.
68
In September 2009, the FASB Emerging Issues Task Force, or EITF reached a consensus on ASC Update 2009-13 (Topic 605), Multiple-Deliverable Revenue Arrangements, or ASC Update 2009-13. ASC Update 2009-13 applies to multiple-deliverable revenue arrangements that are currently within the scope of the Revenue Recognition—Multiple-Element Arrangements topic of the Codification. ASC Update 2009-13 provides principles and application guidance on whether multiple deliverables exist, how the arrangement should be separated and the consideration allocated. ASC Update 2009-13 requires an entity to allocate revenue in an arrangement using estimated selling prices of deliverables if a vendor does not have vendor-specific objective evidence or third-party evidence of selling price. ASC Update 2009-13 eliminates the use of the residual method and requires an entity to allocate revenue using the relative selling price method and also significantly expands the disclosure requirements for multiple-deliverable revenue arrangements. ASC Update 2009-13 should be applied on a prospective basis for revenue arrangements entered into or materially modified in fiscal years beginning on or after June 15, 2010, with earlier application permitted. The Company is currently evaluating the potential impact of the adoption of ASC Update 2009-13 on the Company's financial position or results of operations for future collaborations arrangements.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Interest Rate Risk
We consider all highly liquid investments with an original maturity (at date of purchase) of three months or less to be cash equivalents. At December 31, 2009, our marketable securities available for sale consisted of U.S. Treasury bills, U.S. government agency debt securities, U.S. corporate debt and commercial paper securities with maturity dates of less than two years. Our investments in U.S. corporate debt and commercial paper securities consist primarily of investments in investment grade corporate bonds and notes. Our investments in U.S. Treasury and government debt securities consist of low risk government agency bonds typically with a rating of A or higher. Our operating results have not been sensitive to changes in the general level of interest rates in the United States, particularly because most of our marketable securities are invested in short-term debt instruments.
As of December 31, 2009, the principal amounts, fair values and related weighted-average interest rates of our investments in debt securities classified as marketable securities available-for-sale were as follows:
| |
Duration | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
Less than 1 year | 1 to 2 years | Total | |||||||
Principal amount |
$ | 42,877 | $ | 12,027 | $ | 54,904 | ||||
Fair value |
$ | 42,922 | $ | 12,016 | $ | 54,938 | ||||
Average interest rate |
0.96 | % | 1.83 | % | 1.15 | % | ||||
Foreign Currency Risk
We have not had any significant transactions in foreign currencies, nor did we have any significant balances that were due or payable in foreign currencies at December 31, 2009. Accordingly, significant changes in foreign currency rates would not have a material impact on our financial position and results of operations.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
The financial statements required by this item are set forth beginning on page 79 of this report and are incorporated herein by reference.
69
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
(a) Conclusion Regarding the Effectiveness of Disclosure Controls and Procedures
At the end of the period covered by this report, we carried out an evaluation, under the supervision and with the participation of our management, including our principal executive officer and principal accounting and financial officer, of the effectiveness of the design and operation of our disclosure controls and procedures, as such term is defined under Rule 13a-15(e) promulgated under the Securities Exchange Act of 1934, as amended (the Exchange Act). Based on this evaluation, our principal executive officer and our principal accounting and financial officer concluded that our disclosure controls and procedures were effective as of December 31, 2009 to ensure that information to be disclosed by us in this Annual Report on Form 10-K was recorded, processed summarized and reported within the time periods specified in the Securities and Exchange Commission's rules and Form 10-K.
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in our Exchange Act reports is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission's rules and forms and that such information is accumulated and communicated to our management, including our chief executive officer and principal accounting and financial officer, as appropriate, to allow for timely decisions regarding required disclosure. There were no changes in our internal controls over financial reporting during the year ended December 31, 2009 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
We intend to review and evaluate the design and effectiveness of our disclosure controls and procedures on an ongoing basis and to correct any material deficiencies that we may discover. Our goal is to ensure that our management has timely access to material information that could affect our business. While we believe the present design of our disclosure controls and procedures is effective to achieve our goal, future events affecting our business may cause us to modify our disclosure controls and procedures. In designing and evaluating the disclosure controls and procedures, management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and management is required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
(b) Management's Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f). Under the supervision and with the participation of our management, including our principal executive officer and interim principal accounting and financial officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on our evaluation under the framework in Internal Control—Integrated Framework, our management concluded that our internal control over financial reporting was effective as of December 31, 2009.
70
Report of Independent Registered Public Accounting Firm
The
Board of Directors and Shareholders
Depomed, Inc.
We have audited Depomed, Inc.'s internal control over financial reporting as of December 31, 2009, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). Depomed, Inc.'s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management's Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the company's internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company's internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company's internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company's assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Depomed, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2009, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the balance sheets of Depomed, Inc. as of December 31, 2009 and 2008, and the related statements of operations, shareholders' equity, and cash flows for each of the three years in the period ended December 31, 2009 of Depomed, Inc. and our report dated March 8, 2010 expressed an unqualified opinion thereon.
/s/ ERNST & YOUNG LLP
Palo
Alto, California
March 8, 2010
71
None
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required by this Item with respect to executive officers is set forth in Part I of this report and the information with respect to directors and corporate governance matters is incorporated by reference to the information set forth under the caption "Election of Directors" in the company's Proxy Statement for the 2010 Annual Meeting of Shareholders.
The section entitled "Compliance Under Section 16(a) of the Securities Exchange Act of 1934" appearing in the Proxy Statement for the 2010 Annual Meeting of Shareholders sets forth the information concerning compliance by officers, directors and 10% shareholders of the company with Section 16 of the Exchange Act of 1934 and is incorporated herein by reference.
ITEM 11. EXECUTIVE COMPENSATION
The information required by this Item is incorporated herein by reference to the information set forth under the caption "Executive Compensation" in the Proxy Statement for the 2010 Annual Meeting of Shareholders.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED SHAREHOLDER MATTERS
The information required by this Item is incorporated herein by reference to the information set forth under the caption "Security Ownership of Certain Beneficial Owners and Management and Related Shareholder Matters" in the Proxy Statement for the 2010 Annual Meeting of Shareholders.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required by this Item is incorporated herein by reference to the information set forth under the captions "Directors" and "Certain Relationships and Related Transactions" in the Proxy Statement for the 2010 Annual Meeting of Shareholders.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
The information required by this Item is incorporated herein by reference to the information set forth under the caption "Principal Accountant Fees and Services" in the Proxy Statement for the 2010 Annual Meeting of Shareholders.
72
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
(a)
1. Financial Statements
Report
of Independent Registered Public Accounting Firm
Balance Sheets
Statements of Operations
Statements of Cash Flows
Statement of Shareholders' Equity
Notes to Financial Statements
2. Financial Statement Schedules
Schedule II is included on page 120 of this report. All other schedules are omitted because they are not required or the required information is included in the financial statements or notes thereto.
3. Exhibits:
| Exhibit | Footnote | Description of Document | ||||
|---|---|---|---|---|---|---|
| 3.1 | (1 | ) | Amended and Restated Articles of Incorporation | |||
| 3.2 | (2 | ) | Certificate of Amendment to Amended and Restated Articles of Incorporation | |||
| 3.4 | (3 | ) | Certificate of Determination of Series RP Preferred Stock of the company | |||
| 3.5 | (4 | ) | Bylaws, as amended | |||
| 4.11 | (5 | ) | Rights Agreement, dated as of April 21, 2005, between the company and Continental Stock Transfer and Trust Company as Rights Agent | |||
| 10.1 | (6 | ) | 1995 Stock Option Plan, as amended | |||
| 10.2 | (7 | ) | Form of Incentive Stock Option Agreement under 1995 Stock Option Plan | |||
| 10.3 | (7 | ) | Form of Nonstatutory Stock Option Agreement under 1995 Stock Option Plan | |||
| 10.4 | (7 | ) | Form of Exercise Notice under 1995 Stock Option Plan | |||
| 10.5 | (1 | ) | Agreement re: Settlement of Lawsuit, Conveyance of Assets and Assumption of Liabilities dated August 28, 1995 by and among Depomed Systems, Inc., Dr. John W. Shell and M6 Pharmaceuticals, Inc. | |||
| 10.6 | (8 | ) | Form of Indemnification Agreement between the Company and its directors and executive officers | |||
| 10.7 | (9 | ) | Settlement and Release Agreement, dated as of November 22, 2002, between the Company and Bristol-Myers Squibb Company | |||
| 10.8 | (10 | ) | Lease extension agreement dated April 30, 2003 between the Company and Menlo Business Park LLC | |||
| 10.9 | (10 | ) | Lease agreement dated April 30, 2003 between the Company and Menlo Park Business Park LLC | |||
| 10.10 | (11 | ) | 2004 Equity Incentive Plan, as amended | |||
| 10.11 | (12 | ) | 2004 Employee Stock Purchase Plan, as amended | |||
| 10.12 | (13 | ) | Agreement, dated as of December 10, 2004, between the Company and Kings Road Investments, Ltd. | |||
| 10.13 | (14 | ) | Offer Letter, dated June 14, 2005, between the Company and Carl Pelzel | |||
| 10.14+ | (15 | ) | Technology Transfer and Commercial Manufacturing Agreement dated October 18, 2005 between the Company and MOVA Pharmaceutical Corporation | |||
| 10.15+ | (15 | ) | Amended and Restated License Agreement dated December 13, 2005 between the Company and Biovail Laboratories International SRL | |||
73
| Exhibit | Footnote | Description of Document | ||||
|---|---|---|---|---|---|---|
| 10.16+ | (15 | ) | Supply Agreement dated December 13, 2005 between the Company and Biovail Laboratories International SRL | |||
| 10.17+ | (15 | ) | Manufacturing Transfer Agreement dated December 13, 2005 between the Company and Biovail Laboratories International SRL | |||
| 10.18 | (16 | ) | Description of Non-employee Director Compensation Policy, as amended | |||
| 10.19 | (17 | ) | Bonus Plan of the Company, as amended | |||
| 10.20 | (18 | ) | Form of Management Continuity Agreement between the Company and certain officers of the Company | |||
| 10.21 | (19 | ) | Offer Letter, dated June 14, 2006, between the Company and Matthew Gosling | |||
| 10.22 | (8 | ) | Lease Agreement dated July 28, 2006 between the Company and Menlo Business Park, LLC | |||
| 10.23 | (8 | ) | Lease Extension Agreement dated July 28, 2006 between the Company and Menlo Business Park, LLC | |||
| 10.24 | (8 | ) | Second Lease Extension Agreement dated July 28, 2006 between the Company and Menlo Business Park, LLC | |||
| 10.25+ | (7 | ) | Sublicense Agreement dated October 13, 2006 between the Company and PharmaNova, Inc. | |||
| 10.26 | (20 | ) | Common Stock Purchase Agreement dated December 11, 2006 between the Company and Azimuth Opportunity Ltd. dated December 11, 2006. | |||
| 10.27+ | (7 | ) | Commercial Manufacturing Agreement dated December 19, 2006 between the Company and MOVA Pharmaceutical Corporation | |||
| 10.28 | (11 | ) | Amendment to Supply Agreement dated June 30, 2007 between the Company and Biovail Laboratories International SRL | |||
| 10.29 | (21 | ) | Offer Letter, dated August 24, 2007, between the Company and Carl A. Pelzel | |||
| 10.30 | (23 | ) | Amendment to Offer Letter, dated February 18, 2008, between the Company and Carl A. Pelzel | |||
| 10.31 | (23 | ) | Offer Letter, dated November 19, 2007, between the Company and Michael Sweeney, M.D. | |||
| 10.32+ | (12 | ) | Settlement and License Agreement dated April 4, 2008 between the Company and Teva Pharmaceuticals USA, Inc. | |||
| 10.33 | (12 | ) | Lease Extension Agreement dated March 18, 2008 between the Company and Menlo Business Park, LLC | |||
| 10.34 | (12 | ) | Second Lease Extension Agreement dated March 18, 2008 between the Company and Menlo Business Park, LLC | |||
| 10.35 | (12 | ) | Third Lease Extension Agreement dated March 18, 2008 between the Company and Menlo Business Park, LLC | |||
| 10.36 | (12 | ) | Loan and Security Agreement dated June 27, 2008 between the Company, General Electric Capital Corporation and Oxford Finance Corporation | |||
| 10.37+ | (12 | ) | Promotion Agreement dated July 21, 2008 between the Company and Santarus, Inc. | |||
| 10.38+ | (12 | ) | Amendment No. 1 to Common Stock Purchase Agreement between the Company and Azimuth Opportunity Ltd., dated as of August 8, 2008. |
|||
| 10.39+ | (22 | ) | Exclusive License Agreement between the Company and Solvay Pharmaceuticals, Inc., dated as of November 19, 2008. | |||
| 23.1 | Consent of Independent Registered Public Accounting Firm | |||||
| 24.1 | Power of Attorney (See signature page) | |||||
| 31.1 | Certification pursuant to Rule 13a-14(a) under the Securities Exchange Act of 1934 of Carl A. Pelzel | |||||
| 31.2 | Certification pursuant to Rule 13a-14(a) under the Securities Exchange Act of Tammy L. Cameron | |||||
74
| Exhibit | Footnote | Description of Document | ||||
|---|---|---|---|---|---|---|
| 32.1 | Certification pursuant to 18 U.S.C. Section 1350 of Carl A. Pelzel | |||||
| 32.2 | Certification pursuant to 18 U.S.C. Section 1350 of Tammy L. Cameron | |||||
- (1)
- Incorporated
by reference to the Company's registration statement on Form SB-2 (File No. 333-25445)
- (2)
- Incorporated
by reference to the Company's Form 10-K filed on March 31, 2003
- (3)
- Incorporated
by reference to the Company's Form 10-Q filed on May 10, 2005
- (4)
- Incorporated
by reference to the Company's Form 8-K filed on April 19, 2005
- (5)
- Incorporated
by reference to the Company's Form 8-A filed on April 22, 2005
- (6)
- Incorporated
by reference to the Company's registration statement on Form S-8 (File No. 333-101796) filed on
December 12, 2002
- (7)
- Incorporated
by reference to the Company's Form 10-K filed on March 16, 2007
- (8)
- Incorporated
by reference to the Company's Form 10-Q filed on November 9, 2006
- (9)
- Incorporated
by reference to the Company's Form 8-K/A dated November 22, 2002 and filed on December 23, 2002
- (10)
- Incorporated
by reference to the Company's Form 10-Q filed on August 14, 2003
- (11)
- Incorporated
by reference to the Company's Form 10-Q filed on August 7, 2007
- (12)
- Incorporated
by reference to the Company's Form 10-Q filed on August 8, 2008
- (13)
- Incorporated
by reference to the Company's Form 8-K filed on December 14, 2004
- (14)
- Incorporated
by reference to the Company's Form 8-K filed on June 17, 2005
- (15)
- Incorporated
by reference to the Company's Form 10-K filed on March 16, 2006
- (16)
- Incorporated
by reference to the Company's Form 8-K filed on March 29, 2006 and the Company's Form 8-K filed
on December 12, 2006
- (17)
- Incorporated
by reference to the Company's Form 8-K filed on April 12, 2006
- (18)
- Incorporated
by reference to the Company's Form 10-Q filed on May 5, 2009
- (19)
- Incorporated
by reference to the Company's Form 8-K filed on June 30, 2006
- (20)
- Incorporated
by reference to the Company's Form 8-K filed on December 12, 2006
- (21)
- Incorporated
by reference to the Company's Form 8-K filed on August 27, 2007
- (22)
- Incorporated
by reference to the Company's Form 10-K filed on March 6, 2009
- (23)
- Incorporated
by reference to the Company's Form 10-Q filed on May 7, 2008
- +
- Confidential treatment granted
75
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the issuer, a corporation organized and existing under the laws of the State of California, has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized, in the City of Menlo Park, State of California, on the 8th day of March 2010.
| DEPOMED, INC. | ||||
By |
/s/ CARL A. PELZEL Carl A. Pelzel President and Chief Executive Officer |
|||
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below hereby constitutes and appoints Carl A. Pelzel and Tammy L. Cameron, and each of them acting individually, as his true and lawful attorneys-in-fact and agents, each with full power of substitution, for him in any and all capacities, to sign any and all amendments to this report on Form 10-K and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, with full power of each to act alone, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully for all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, or his or their substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this Annual Report on Form 10-K has been signed by the following persons in the capacities and on the dates indicated.
Signature
|
|
|
||
|---|---|---|---|---|
| /s/ CARL A. PELZEL Carl A. Pelzel |
President and Chief Executive Officer (Principal Executive Officer) |
March 8, 2010 | ||
/s/ TAMMY L. CAMERON Tammy L. Cameron |
Vice President, Finance (Principal Accounting and Financial Officer) |
March 8, 2010 |
||
/s/ PETER D. STAPLE Peter D. Staple |
Chairman of the Board of Directors |
March 8, 2010 |
||
/s/ G. STEVEN BURRILL G. Steven Burrill |
Director |
March 8, 2010 |
||
/s/ KAREN A. DAWES Karen A. Dawes |
Director |
March 8, 2010 |
||
/s/ JAMES A. SCHOENECK James A. Schoeneck |
Director |
March 8, 2010 |
||
/s/ CRAIG R. SMITH, M.D. Craig R. Smith, M.D. |
Director |
March 8, 2010 |
||
/s/ JULIAN N. STERN Julian N. Stern |
Director |
March 8, 2010 |
||
/s/ DAVID B. ZENOFF, D.B.A. David B. Zenoff, D.B.A. |
Director |
March 8, 2010 |
76
DEPOMED, INC.
INDEX TO FINANCIAL STATEMENTS
DEPOMED, INC. FINANCIAL STATEMENTS
77
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The
Board of Directors and Shareholders
Depomed, Inc.
We have audited the accompanying balance sheets of Depomed, Inc. as of December 31, 2009 and 2008, and the related statements of operations, shareholders' equity, and cash flows for each of the three years in the period ended December 31, 2009. Our audits also included the financial statement schedule listed in the Index at Item 15(a). These financial statements and schedule are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements and schedule based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Depomed, Inc. at December 31, 2009 and 2008, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2009, in conformity with U.S. generally accepted accounting principles. Also, in our opinion, the related financial statement schedule, when considered in relation to the basic financial statements taken as a whole, presents fairly in all material respects the information set forth therein.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Depomed, Inc.'s internal control over financial reporting as of December 31, 2009, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated March 8, 2010 expressed an unqualified opinion thereon.
/s/ ERNST & YOUNG LLP
Palo
Alto, California
March 8, 2010
78
DEPOMED, INC.
BALANCE SHEETS
(in thousands, except share amounts)
| |
December 31, | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| |
2009 | 2008 | |||||||
ASSETS |
|||||||||
Current assets: |
|||||||||
Cash and cash equivalents |
$ | 26,821 | $ | 22,127 | |||||
Marketable securities |
42,922 | 59,932 | |||||||
Accounts receivable |
4,840 | 3,099 | |||||||
Unbilled accounts receivable |
93 | 576 | |||||||
Inventories |
2,565 | 2,849 | |||||||
Prepaid and other current assets |
1,185 | 5,404 | |||||||
Total current assets |
78,426 | 93,987 | |||||||
Marketable securities, long-term |
12,016 | — | |||||||
Property and equipment, net |
942 | 900 | |||||||
Other assets |
197 | 197 | |||||||
|
$ | 91,581 | $ | 95,084 | |||||
LIABILITIES AND SHAREHOLDERS' EQUITY |
|||||||||
Current liabilities: |
|||||||||
Accounts payable and accrued liabilities |
15,222 | 12,848 | |||||||
Deferred product sales |
1,635 | 1,702 | |||||||
Deferred license revenue |
11,184 | 4,362 | |||||||
Other current liabilities |
414 | 110 | |||||||
Current portion of long-term debt |
3,747 | 3,356 | |||||||
Total current liabilities |
32,202 | 22,378 | |||||||
Deferred license revenue, non-current portion |
41,306 | 33,209 | |||||||
Long-term debt, net of current portion |
2,170 | 5,775 | |||||||
Other long-term liabilities |
177 | 569 | |||||||
Commitments |
|||||||||
Shareholders' equity: |
|||||||||
Preferred stock, no par value, 5,000,000 shares authorized; Series A convertible preferred stock, 25,000 shares designated, 18,158 shares issued and surrendered, and zero shares outstanding at December 31, 2009 and 2008 |
— | — | |||||||
Common stock, no par value, 100,000,000 shares authorized; 52,200,358 and 51,171,377 shares issued and outstanding at December 31, 2009 and 2008, respectively |
187,895 | 183,196 | |||||||
Accumulated deficit |
(172,202 | ) | (150,194 | ) | |||||
Accumulated other comprehensive gain |
33 | 151 | |||||||
Total shareholders' equity |
15,726 | 33,153 | |||||||
|
$ | 91,581 | $ | 95,084 | |||||
See accompanying Notes to Financial Statements.
79
DEPOMED, INC.
STATEMENTS OF OPERATIONS
(in thousands, except share and per share amounts)
| |
Year Ended December 31, | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2009 | 2008 | 2007 | |||||||||
Revenues: |
||||||||||||
Product sales |
$ | 35,094 | $ | 31,051 | $ | 12,502 | ||||||
Royalties |
1,533 | 1,582 | 2,707 | |||||||||
License and milestone revenue |
21,081 | 2,145 | 50,367 | |||||||||
Collaborative and other revenue |
20 | 64 | 6 | |||||||||
Total revenues |
57,728 | 34,842 | 65,582 | |||||||||
Costs and expenses: |
||||||||||||
Cost of sales |
5,257 | 5,772 | 2,597 | |||||||||
Research and development expense |
34,298 | 27,268 | 23,337 | |||||||||
Selling, general and administrative expense: |
||||||||||||
Promotion fee expense |
23,589 | 4,841 | 3,012 | |||||||||
Other selling, general and administrative |
16,656 | 21,556 | 23,682 | |||||||||
Total selling, general and administrative expense |
40,245 | 26,397 | 26,694 | |||||||||
Gain on termination of King agreement |
— | — | (29,584 | ) | ||||||||
Gain on termination of Esprit Pharma agreement |
— | — | (5,000 | ) | ||||||||
Gain on litigation settlement |
— | (7,500 | ) | — | ||||||||
Total costs and expenses |
79,800 | 51,937 | 18,044 | |||||||||
Income (loss) from operations |
(22,072 | ) | (17,095 | ) | 47,538 | |||||||
Other income (expenses): |
||||||||||||
Interest and other income |
1,050 | 2,349 | 2,273 | |||||||||
Interest expense |
(1,001 | ) | (555 | ) | — | |||||||
Total other income (expenses) |
49 | 1,794 | 2,273 | |||||||||
Net income (loss) before income taxes |
(22,023 | ) | (15,301 | ) | 49,811 | |||||||
Benefit from (provision for) income taxes |
15 | (1 | ) | (592 | ) | |||||||
Net income (loss) |
(22,008 | ) | (15,302 | ) | 49,219 | |||||||
Deemed dividend on preferred stock |
— | (541 | ) | (685 | ) | |||||||
Net income (loss) applicable to common stock shareholders |
$ | (22,008 | ) | $ | (15,843 | ) | $ | 48,534 | ||||
Basic net income (loss) applicable to common stock shareholders per share |
$ | (0.43 | ) | $ | (0.32 | ) | $ | 1.06 | ||||
Diluted net income (loss) applicable to common stock shareholders per share |
$ | (0.43 | ) | $ | (0.32 | ) | $ | 1.05 | ||||
Shares used in computing basic net income (loss) per share |
51,519,912 | 48,778,764 | 45,951,127 | |||||||||
Shares used in computing diluted net income (loss) per share |
51,519,912 | 48,778,764 | 46,353,207 | |||||||||
See accompanying Notes to Financial Statements.
80
DEPOMED, INC.
STATEMENTS OF SHAREHOLDERS' EQUITY
(in thousands, except share amounts)
| |
|
|
|
|
|
Accumulated Other Comprehensive Income (Loss) |
|
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Preferred Stock | Common Stock | |
|
|||||||||||||||||||
| |
Accumulated Deficit |
Shareholders' Equity (Deficit) |
|||||||||||||||||||||
| |
Shares | Amount | Shares | Amount | |||||||||||||||||||
Balances at Dec. 31, 2006 |
18,158 | $ | 12,015 | 42,029,411 | $ | 144,820 | $ | (184,111 | ) | $ | (13 | ) | $ | (27,289 | ) | ||||||||
Issuance of common stock, net of issuance costs |
— | — | 5,300,000 | 19,966 | — | — | 19,966 | ||||||||||||||||
Issuance of common stock upon exercise of options |
— | — | 268,554 | 778 | — | — | 778 | ||||||||||||||||
Issuance of common stock upon exercise of warrants |
— | — | 27,988 | — | — | — | — | ||||||||||||||||
Issuance of common stock under employee stock purchase plan |
— | — | 136,731 | 382 | — | — | 382 | ||||||||||||||||
Issuance of common stock to employees |
— | — | 100,000 | 364 | — | — | 364 | ||||||||||||||||
Issuance of common stock to consultants for services |
— | — | 2,845 | 10 | — | — | 10 | ||||||||||||||||
Stock-based compensation |
— | — | — | 1,967 | — | — | 1,967 | ||||||||||||||||
Comprehensive income (loss): |
|||||||||||||||||||||||
Net income (loss) |
— | — | — | — | 49,219 | — | 49,219 | ||||||||||||||||
Unrealized gain on available-for-sale securities |
— | — | — | — | — | 123 | 123 | ||||||||||||||||
Comprehensive income (loss) |
49,342 | ||||||||||||||||||||||
Balances at Dec. 31, 2007 |
18,158 | $ | 12,015 | 47,865,529 | $ | 168,287 | $ | (134,892 | ) | $ | 110 | $ | 45,520 | ||||||||||
Surrender of preferred stock and exercise of related warrants |
(18,158 | ) | (12,015 | ) | 2,914,526 | 12,015 | — | — | — | ||||||||||||||
Issuance of common stock upon exercise of options |
— | — | 30,614 | 80 | — | — | 80 | ||||||||||||||||
Issuance of common stock upon exercise of warrants |
— | — | 160,476 | — | — | — | — | ||||||||||||||||
Issuance of common stock under employee stock purchase plan |
— | — | 200,232 | 362 | — | — | 362 | ||||||||||||||||
Stock-based compensation |
— | — | — | 2,452 | — | — | 2,452 | ||||||||||||||||
Comprehensive income (loss): |
|||||||||||||||||||||||
Net income (loss) |
— | — | — | — | (15,302 | ) | — | (15,302 | ) | ||||||||||||||
Unrealized gain on available-for-sale securities |
— | — | — | — | — | 41 | 41 | ||||||||||||||||
Comprehensive income (loss) |
(15,261 | ) | |||||||||||||||||||||
Balances at Dec. 31, 2008 |
— | $ | — | 51,171,377 | $ | 183,196 | $ | (150,194 | ) | $ | 151 | $ | 33,153 | ||||||||||
Issuance of common stock upon exercise of options |
— | — | 723,985 | 1,702 | — | — | 1,702 | ||||||||||||||||
Issuance of common stock under employee stock purchase plan |
— | — | 274,996 | 340 | — | — | 340 | ||||||||||||||||
Issuance of common stock to employees |
— | — | 30,000 | 54 | — | — | 54 | ||||||||||||||||
Stock-based compensation |
— | — | — | 2,603 | — | — | 2,603 | ||||||||||||||||
Comprehensive income (loss): |
|||||||||||||||||||||||
Net income (loss) |
— | — | — | — | (22,008 | ) | — | (22,008 | ) | ||||||||||||||
Unrealized loss on available-for-sale securities |
— | — | — | — | — | (118 | ) | (118 | ) | ||||||||||||||
Comprehensive income (loss) |
(22,126 | ) | |||||||||||||||||||||
Balances at Dec. 31, 2009 |
— | $ | — | 52,200,358 | $ | 187,895 | $ | (172,202 | ) | $ | 33 | $ | 15,726 | ||||||||||
See accompanying Notes to Financial Statements.
81
DEPOMED, INC
STATEMENTS OF CASH FLOWS
(in thousands)
| |
Year Ended December 31, | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2009 | 2008 | 2007 | ||||||||||
Operating Activities |
|||||||||||||
Net income (loss) |
$ | (22,008 | ) | $ | (15,302 | ) | $ | 49,219 | |||||
Adjustments to reconcile net income (loss) to net cash provided by operating activities: |
|||||||||||||
Depreciation and amortization |
808 | 1,148 | 865 | ||||||||||
Employee and director stock-based compensation |
2,614 | 2,369 | 2,235 | ||||||||||
Stock-based compensation issued to consultants |
42 | 83 | 106 | ||||||||||
Changes in assets and liabilities: |
|||||||||||||
Accounts receivable |
(1,259 | ) | (51 | ) | 5,459 | ||||||||
Inventories |
283 | 415 | 1,220 | ||||||||||
Other current assets |
4,219 | (2,987 | ) | 338 | |||||||||
Accounts payable and other accrued liabilities |
2,485 | 6,064 | (6,318 | ) | |||||||||
Accrued compensation |
(197 | ) | 1,044 | (260 | ) | ||||||||
Deferred revenue |
14,851 | 10,568 | (38,203 | ) | |||||||||
Net cash provided by operating activities |
1,838 | 3,351 | 14,661 | ||||||||||
Investing Activities |
|||||||||||||
Purchase of property and equipment |
(629 | ) | (257 | ) | (156 | ) | |||||||
Purchases of marketable securities |
(144,567 | ) | (101,607 | ) | (67,476 | ) | |||||||
Maturities of marketable securities |
96,396 | 85,582 | 28,655 | ||||||||||
Sales of marketable securities |
52,926 | 11,159 | 2,991 | ||||||||||
Net cash provided by (used in) investing activities |
4,126 | (5,123 | ) | (35,986 | ) | ||||||||
Financing Activities |
|||||||||||||
Proceeds from long-term debt |
— | 9,400 | — | ||||||||||
Principal payments on long-term debt |
(3,312 | ) | — | — | |||||||||
Debt issuance costs |
— | (316 | ) | — | |||||||||
Proceeds from issuance of common stock |
2,042 | 441 | 21,125 | ||||||||||
Net cash provided by (used in) financing activities |
(1,270 | ) | 9,525 | 21,125 | |||||||||
Net increase (decrease) in cash and cash equivalents |
4,694 | 7,753 | (200 | ) | |||||||||
Cash and cash equivalents at beginning of year |
22,127 | 14,374 | 14,574 | ||||||||||
Cash and cash equivalents at end of year |
$ | 26,821 | $ | 22,127 | $ | 14,374 | |||||||
Supplemental Disclosure of Cash Flow Information |
|||||||||||||
Cash paid during the period for: |
|||||||||||||
Interest |
$ | 935 | $ | 409 | $ | — | |||||||
Taxes |
$ | (6 | ) | $ | 631 | $ | 5 | ||||||
See accompanying Notes to Financial Statements.
82
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS
NOTE 1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Organization
Depomed is a specialty pharmaceutical company focused on the development and commercialization of differentiated products that address large and growing markets and are based on proprietary oral drug delivery technologies. In 2009, the Company completed Phase 3 clinical trial for two product candidates. In October 2009, the Company announced that DM-1796, an extended release formulation of gabapentin for the treatment of postherpetic neuralgia that is licensed to Abbott Laboratories' subsidiary, Solvay Pharmaceuticals, Inc. (Solvay) met its primary endpoint with statistical significance. A New Drug Application (NDA) for DM-1796 is expected to be filed by Solvay with the FDA in March of 2010. Also in October 2009, the Company announced the results of Breeze 1 and Breeze 2, its Phase 3 clinical trials for SeradaTM, an extended release formulation of gabapentin for the treatment of menopausal hot flashes. The Company expects to commence another Phase 3 clinical trial for Serada in April 2010. In addition, the Company has other product candidates in earlier stages of development.
The Company has developed two commercial products. GLUMETZA® (metformin hydrochloride extended release tablets) is a once-daily treatment for adults with type 2 diabetes that we commercialize in the United States with Santarus, Inc. (Santarus). Proquin® XR (ciprofloxacin hydrochloride) is a once-daily treatment for uncomplicated urinary tract infections.
Use of Estimates
The preparation of financial statements in conformity with U.S. generally accepted accounting principles requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Actual results could differ from those estimates.
Revenue Recognition
The Company recognizes revenue from the sale of its products, royalties earned, and on payments received and services performed under contractual arrangements. Revenue arrangements with multiple elements are divided into separate units of accounting if certain criteria are met, including whether the delivered element has stand-alone value to the customer and whether there is objective and reliable evidence of the fair value of the undelivered items. The consideration received is allocated among the separate units based on their respective fair values, and the applicable revenue recognition criteria are applied to each of the separate units.
Revenue is recognized when there is persuasive evidence that an arrangement exists, delivery has occurred and title has passed, the price is fixed or determinable and the Company is reasonably assured of collecting the resulting receivable.
- •
- Product Sales:
- •
- GLUMETZA: The Company sells GLUMETZA product to wholesalers and retail pharmacies that is subject to rights of return up to twelve months after product expiration. The Company began shipping GLUMETZA product to customers in the third quarter of 2006. Prior to the third quarter of 2008, the Company was unable to reasonably estimate expected returns of the product at the time of shipment, and therefore, deferred revenue on product shipments of GLUMETZA until the product was dispensed through patient prescriptions. The quantity of prescription units dispensed was estimated based on analysis
83
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
- •
- Proquin XR: The Company sells Proquin XR product to wholesalers and retail
pharmacies that is subject to rights of return up to twelve months after product expiration. Given the Company's limited history of selling Proquin XR and decreasing prescription demand for Proquin
XR, the Company currently cannot reliably estimate expected returns of the product at the time of shipment. Accordingly, the Company defers recognition of revenue on product shipments of Proquin XR
until the right of return no longer exists, which occurs at the earlier of the time Proquin XR units are dispensed through patient prescriptions or expiration of the right of return. The Company
estimates patient prescriptions dispensed using an analysis of third-party information, including third-party market research data and information obtained from wholesalers with respect to inventory
levels and inventory movement. As a result of this policy, the Company has a deferred revenue balance of $1.6 million at December 31, 2009 related to Proquin XR product shipments that
have not been recognized as revenue, which is net of wholesaler fees, retail pharmacy discounts and prompt payment discounts. The Company will recognize revenue upon the earlier to occur of
prescription units dispensed or expiration of the right of return until it can reliably estimate product returns, at which time the Company will record a one-time increase in net revenue
related to the recognition of revenue previously deferred. In addition, the costs of manufacturing Proquin XR associated with the deferred revenue are recorded as deferred costs, which are included in
inventory, until such time the related deferred revenue is recognized.
- •
- Product Sales Allowances—The Company recognizes products sales allowances as a reduction of product sales in the same period the related revenue is recognized. Product sales allowances are based on amounts owed or to be claimed on the related sales. These estimates take into consideration the terms of the Company's agreements with customers, historical product returns, rebates or discounts taken, estimated levels of inventory in the distribution channel, the shelf life of the product, and specific known market events, such as competitive pricing and new product introductions. If actual future results vary, the Company may need to adjust these estimates,
of third-party information, including third-party market research data and information obtained from wholesalers with respect to inventory levels and inventory movement. Based on the shipment trends, prescription trends and product returns history for GLUMETZA over two years through the third quarter of 2008 and based on an analysis of return rates of companies with products that have similar characteristics and similar return policies within the metformin prescription market, the Company concluded it had the information needed to reasonably estimate product returns during the third quarter of 2008. Beginning in the third quarter of 2008, the Company began recognizing revenue for GLUMETZA sales at the time of shipment to its customers. Consequently, in 2008, the Company recognized a one time increase of $6.3 million in net product sales of GLUMETZA, representing product sales previously deferred, net of estimated product returns, managed care and Medicaid rebates, wholesaler and retail pharmacy discounts, chargebacks and prompt payment discounts. This change resulted in a one-time $5.3 million reduction to net loss and decreased net loss per share by $0.11 for the year ended December 31, 2008.
84
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
- •
- Product Returns—The Company estimates product returns on sales of GLUMETZA. The Company allows customers to
return product that is within six months before and up to one year after its product expiration date. The shelf life of the 500mg GLUMETZA is currently 48 months from the date of tablet
manufacture. On product launch in August 2006 and through the second quarter of 2008, the shelf life of 500mg GLUMETZA product shipped was 36 months from the date of tablet manufacture. Since
product launch in June 2008 and through December 31, 2009, the 1000mg GLUMETZA shelf life on product shipped to customers was 24 months from the date of tablet manufacture. The Company
monitors actual return history on an individual product lot basis since product launch, which provides it with a basis to reasonably estimate future product returns, taking into consideration the
shelf life of product, shipment and prescription trends, estimated distribution channel inventory levels, the return rates of companies with products that have similar characteristics and similar
return policies within the metformin prescription market, as well as consideration of the introduction of competitive products. In 2009, based on its review of actual product returns through the end
of 2009, the Company increased its estimate for GLUMETZA product returns. This resulted in a decrease of product sales revenue of approximately $0.5 million for the year ended
December 31, 2009 related to sales made in prior periods.
- •
- Managed Care Rebates—The Company offers rebates under contracts with certain managed care customers. The
Company establishes an accrual equal to its estimates of future managed care rebates attributable to sales and recognizes the estimated rebates as a reduction of revenue in the same period the related
revenue is recognized. The Company estimates its managed care rebates based on the terms of each agreement, estimated levels of inventory in the distribution channel, and historical and expected
future utilization of product by the managed care organization.
- •
- Wholesaler and Retail Pharmacy Discounts—The Company offers discounts to certain wholesale distributors and
retail pharmacies based on contractually determined rates. The Company accrues the discount on shipment to the respective wholesale distributors and retail pharmacies and recognizes the discount as a
reduction of revenue in the same period the related revenue is recognized.
- •
- Prompt Pay Discounts—The Company offers cash discounts to its customers, generally 2% of the sales price, as
an incentive for prompt payment. Based on the Company's experience, the Company expects its customers to comply with the payment terms to earn the cash discount. The Company accounts for cash
discounts by reducing accounts receivable by the full amount and recognizes the discount as a reduction of revenue in the same period the related revenue is recognized.
- •
- Medicaid Rebates—The Company participates in Medicaid rebate programs, which provide assistance to certain low-income patients based on each individual state's guidelines regarding eligibility and services. Under the Medicaid rebate programs, the Company pays a rebate to each participating state, generally two to three months after the quarter in which prescriptions subject to the rebate are filled. The Company estimates and accrues Medicaid
which could have an effect on earnings in the period of adjustment. The Company's product sales allowances include:
85
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
- •
- Chargebacks—The Company provides discounts to authorized users of the Federal Supply Schedule (FSS) of the
General Services Administration under an FSS contract negotiated by the Department of Veterans Affairs. These federal entities purchase products from the wholesale distributors at a discounted price,
and the wholesale distributors then charge back to the Company the difference between the current retail price and the price the federal entity paid for the product. The Company estimates and accrues
chargebacks based on estimated wholesaler inventory levels, current contract prices and historical chargeback activity.
- •
- Patient Discount Programs—The Company offers loyalty card programs to patients for GLUMETZA in which patients
receive certain discounts at participating retail pharmacies that are reimbursed by the Company. The Company estimates and accrues future redemptions based on historical redemption activity.
- •
- Royalties—Royalties are recognized as earned in accordance with the contract terms when royalties from licensees can be reliably measured and collectibility is reasonably assured.
rebates based on product pricing, current rebates and changes in the level of discounts the Company offers that may affect the level of Medicaid discount, historical and estimated future percentages of product sold to Medicaid recipients and estimated levels of inventory in the distribution channel.
- •
- License and Collaborative Arrangements—Revenue from license and collaborative arrangements is recognized when the Company has substantially completed its obligations under the terms of the arrangement and the Company's remaining involvement is inconsequential and perfunctory. If the Company has significant continuing involvement under such an arrangement, license and collaborative fees are recognized over the estimated performance period. The Company recognizes milestone payments upon the achievement of specified milestones if (1) the milestone is substantive in nature, and the achievement of the milestone was not reasonably assured at the
In April 2008, the Company entered into a settlement and license agreement with Teva Pharmaceuticals USA, Inc. (Teva) in which the Company was entitled to receive royalties from Teva on sales by Teva or its affiliates of generic Glucophage®XR in the United States, subject to a $2.5 million aggregate royalty cap that was met during 2009. The royalties were calculated as a percentage of sales by Teva of generic Glucophage XR in the United States, as reported by a third-party market research company. The Company accrued royalties from Teva each quarter based on Teva's sales of generic Glucophage XR reported by the third-party market research company for that quarter. See Note 2 of the Notes to Financial Statements for further information on the settlement and license agreement with Teva.
Royalties received under the Company's agreements with Biovail Laboratories s.r.l. (Biovail) and LG Life Sciences (LG) are recognized when the royalty payments are received as they cannot reliably be estimated.
The Company recognized royalties under its license agreement with Esprit Pharma, Inc. (Esprit) based on Esprit's sales of Proquin XR, net of any estimated returns, discounts, rebates and chargebacks, subject to minimum annual royalties. The license agreement with Esprit was terminated in July 2007. See Note 2 of the Notes to Financial Statements for additional information with respect to the termination agreement with Esprit.
86
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
inception of the agreement and (2) the fees are nonrefundable. License, milestones and collaborative fee payments received in excess of amounts earned are classified as deferred revenue until earned.
Stock-Based Compensation
The compensation expense for stock-based compensation is based on the single-option approach, includes an estimate for forfeitures and is recognized over the vesting term of the options using the straight-line method. Depomed estimates forfeitures based on historical experience.
As the historical share option exercise experience did not provide a reasonable basis upon which to estimate expected term, the company estimated the expected term of options granted by taking the average of the vesting term and the contractual term of the option, as illustrated by the simplified method through December 31, 2007. At January 1, 2008, the Company concluded again that its historical share option exercise experience did not provide a reasonable basis upon which to estimate expected term because of the Company's limited exercise history and elected to no longer utilize the simplified method. For options granted after January 1, 2008, the Company has estimated the expected term by using the weighted average terms of a peer group of companies that grant options with similar vesting provisions. The expected term used for options granted in 2009 is 5.1 years. See Note 10 of the Notes to Financial Statements for further information regarding Depomed's stock-based compensation expense
Research and Development Expense and Accruals
Research and development expenses include related salaries, contractor fees, clinical trial costs, facilities costs, depreciation costs, administrative expenses and allocations of corporate costs. All such costs are charged to research and development expense as incurred. These expenses result from the Company's independent research and development efforts as well as efforts associated with collaborations. The Company reviews and accrues clinical trial expenses based on work performed, which relies on estimates of total costs incurred based on patient enrollment, completion of patient studies and other events. The Company follows this method since reasonably dependable estimates of the costs applicable to various stages of a research agreement or clinical trial can be made. Accrued clinical costs are subject to revisions as trials progress to completion. Revisions are charged to expense in the period in which the facts that give rise to the revision become known.
Shipping and Handling Costs
Shipping and handling costs incurred for inventory purchases and product shipments are recorded in cost of sales in the Statements of Operations.
Advertising Costs
Costs associated with advertising are expensed on first showing. Advertising expense for the years ended December 31, 2009, 2008 and 2007 were $0.7 million, $1.8 million and $2.2 million, respectively. At December 31, 2009, the Company had approximately $0.1 million in prepaid samples classified within prepaid expenses and other current assets.
87
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Comprehensive Income (Loss)
Comprehensive income (loss) is comprised of net income (loss) and other comprehensive income (loss). Other comprehensive income (loss) includes certain changes in equity of the Company that are excluded from net loss. Unrealized holding gains and losses on the Company's available-for-sale securities are reported separately in shareholders' equity and included in accumulated other comprehensive income (loss). Comprehensive income (loss) for the years ended December 31, 2009, 2008 and 2007 has been reflected in the Statements of Shareholders' Equity.
Cash, Cash Equivalents and Marketable Securities
The Company considers all highly liquid investments with an original maturity (at date of purchase) of three months or less to be cash equivalents. Cash and cash equivalents consist of cash on deposit with banks, money market instruments and commercial paper. The Company places its cash, cash equivalents and marketable securities with high quality, U.S. government and financial institutions and, to date, has not experienced material losses on any of its balances. The Company records cash and cash equivalents at amortized cost, which approximates the fair value. All marketable securities are classified as available-for-sale since these instruments are readily marketable. These securities are carried at fair value, which is based on readily available market information, with unrealized gains and losses included in accumulated other comprehensive income (loss) within shareholders' equity. The Company uses the specific identification method to determine the amount of realized gains or losses on sales of marketable securities. We regularly review all of our investments for other-than-temporary declines in fair value. Our review includes the consideration of the cause of the impairment including the creditworthiness of the security issuers, the number of securities in an unrealized loss position, as well as the severity and duration of the unrealized losses. When we determine that the decline in fair value of an investment is below our accounting basis and this decline is other-than-temporary, we reduce the carrying value of the security we hold and record a loss in the amount of such decline. Realized gains or losses have been insignificant and are included in "interest and other income" in the Statement of Operations.
Accounts Receivable
Trade accounts receivable are recorded net of allowances for cash discounts for prompt payment. To date the Company has not recorded a bad debt allowance due to the fact that the majority of its product revenue comes from sales to a limited number of financially sound companies. The need for bad debt allowance is evaluated each reporting period based on our assessment of the credit worthiness of our customers.
Inventories
Inventories are stated at the lower of cost or market with cost determined by specific manufactured lot. Inventories consist of costs of the active pharmaceutical ingredient, contract manufacturing and packaging costs. The Company writes-off the value of inventory for potentially excess, dated or obsolete inventories based on an analysis of inventory on hand and on firm purchase commitments.
88
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Property and Equipment
Property and equipment are stated at cost, less accumulated depreciation and amortization (See Note 5 of the Notes to Financial Statements). Depreciation is calculated using the straight-line method over the estimated useful lives of the respective assets, as follows:
| Furniture and office equipment | 3-5 years | |
| Laboratory equipment | 3-5 years | |
| Leasehold improvements | Shorter of estimated useful life or lease term |
Impairment of Long-Lived Assets
The Company identifies and records impairment losses, as circumstances dictate, on long-lived assets used in operations when events and circumstances indicate that the assets might be impaired and the discounted cash flows estimated to be generated by those assets are less than the carrying amounts of those assets. If the undiscounted future cash flows are less than the carrying amount of the asset, an impairment loss, measured as the excess of the carrying value of the asset over its estimated fair value, will be recognized. The cash flow estimates used in such calculations are based on management's best estimates, using appropriate and customary assumptions and projections at the time. No such impairments have been identified with respect to the Company's long-lived assets, which consist primarily of property and equipment.
Net Income (Loss) Per Common Share
Basic net income (loss) per share is calculated by dividing the net income (loss) by the weighted-average number of common shares outstanding for the period. Diluted net income (loss) per share is computed by dividing the net income (loss) by the weighted-average number of common shares outstanding for the period, plus dilutive potential common shares for the period determined using the treasury-stock method. For purposes of this calculation, options to purchase stock and warrants are considered to be potential common shares and are only included in the calculation of diluted net
89
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
income (loss) per share when their effect is dilutive. Basic and diluted earnings per share are calculated as follows:
| |
2009 | 2008 | 2007 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| |
(In thousands, except per share amounts) |
||||||||||
Numerator: |
|||||||||||
Net income (loss) |
$ | (22,008 | ) | $ | (15,843 | ) | $ | 48,534 | |||
Denominator: |
|||||||||||
Weighted-average common shares outstanding |
51,520 | 48,779 | 45,951 | ||||||||
Denominator for basic net income (loss) per share |
51,520 | 48,779 | 45,951 | ||||||||
Dilutive effect of: |
|||||||||||
Options to purchase common stock |
— | — | 246 | ||||||||
Warrants |
— | — | 156 | ||||||||
Denominator for diluted net income (loss) per share |
51,520 | 48,779 | 46,353 | ||||||||
Basic net income (loss) per share |
$ | (0.43 | ) | $ | (0.32 | ) | $ | 1.06 | |||
Diluted net income (loss) per share |
$ | (0.43 | ) | $ | (0.32 | ) | $ | 1.05 | |||
For the years ended December 31, 2009, 2008 and 2007, 5.6 million, 5.6 million and 7.0 million common stock equivalents, respectively, were not included in dilutive shares because their effect is anti-dilutive.
Income Taxes
Deferred tax assets and liabilities are measured based on differences between the financial reporting and tax basis of assets and liabilities using enacted rates and laws that are expected to be in effect when the differences are expected to reverse. The Company records a valuation allowance for the full amount of deferred assets, which would otherwise be recorded for tax benefits relating to operating loss and tax credit carryforwards, as realization of such deferred tax assets cannot be determined to be more likely than not. See Note 14 of the Notes to the Financial Statements for further discussion on income taxes.
Segment Information
The Company operates in one operating segment and has operations solely in the United States. To date, all of the Company's revenues from product sales are related to sales of GLUMETZA and Proquin XR in the United States. The Company has recognized license and royalty revenue from license agreements in the territories of the United States, Europe, Canada and Korea.
Concentration of Risk
The Company invests cash that is currently not being used for operational purposes in accordance with its investment policy in low risk debt securities of the U.S. Treasury, U.S. government sponsored agencies and very highly rated banks and corporations. The Company is exposed to credit risk in the event by default by the institutions holding the cash equivalents and available-for sale securities to the extent recorded on the balance sheet.
90
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
The Company is subject to credit risk from its accounts receivable related to product sales. The majority of the Company's trade accounts receivable arises from product sales in the United States. Three wholesale distributors represented 38%, 35% and 21% of GLUMETZA and Proquin XR shipments for the year ended December 31, 2009. These three customers individually comprised 48%, 30% and 15%, respectively, of GLUMETZA and Proquin XR accounts receivable as of December 31, 2009. Three wholesale distributors represented 36%, 35% and 19% of GLUMETZA and Proquin XR shipments for the year ended December 31, 2008. These three customers individually comprised 30%, 42% and 19%, respectively, of GLUMETZA and Proquin XR accounts receivable as of December 31, 2008. To date, the Company has not experienced any losses with respect to the collection of its accounts receivable and believes that all of its past due accounts receivable are collectible. Accounts receivable balances related to product sales were $4.9 million and $2.5 million for the years ended December 31, 2009 and 2008, respectively.
The Company relies on a single third-party manufacturer in Puerto Rico to manufacture 500mg GLUMETZA and Proquin XR. The Company relies on a single third-party manufacturer in Canada to manufacture 1000mg GLUMETZA. The Company also relies on two third-party suppliers for the supply of metformin hydrochloride, the active pharmaceutical ingredient in GLUMETZA and a single third-party supplier for the supply of ciprofloxacin hydrochloride, the active pharmaceutical ingredient in Proquin XR.
Recently Issued Accounting Standards
In August 2009, the FASB issued Accounting Standards Update No. 2009-05, Measuring Liabilities at Fair Value (ASU 2009-05). ASU 2009-05 amends Accounting Standards Codification Topic 820, Fair Value Measurements. Specifically, ASU 2009-05 provides clarification that in circumstances in which a quoted price in an active market for the identical liability is not available, a reporting entity is required to measure fair value using one or more of the following methods: 1) a valuation technique that uses a) the quoted price of the identical liability when traded as an asset or b) quoted prices for similar liabilities or similar liabilities when traded as assets and/or 2) a valuation technique that is consistent with the principles of Topic 820 of the Accounting Standards Codification. ASU 2009-05 also clarifies that when estimating the fair value of a liability, a reporting entity is not required to adjust to include inputs relating to the existence of transfer restrictions on that liability. The adoption of this standard did not have a material impact on the Company's financial statements.
In September 2009, the FASB Emerging Issues Task Force, or EITF reached a consensus on Accounting Standards Codification Update 2009-13 (Topic 605), Multiple-Deliverable Revenue Arrangements (ASU 2009-13). ASU 2009-13 applies to multiple-deliverable revenue arrangements that are currently within the scope of the Revenue Recognition—Multiple-Element Arrangements topic of the Codification. ASU 2009-13 provides principles and application guidance on whether multiple deliverables exist, how the arrangement should be separated and the consideration allocated. ASU 2009-13 requires an entity to allocate revenue in an arrangement using estimated selling prices of deliverables if a vendor does not have vendor-specific objective evidence or third-party evidence of selling price. ASU 2009-13 eliminates the use of the residual method and requires an entity to allocate revenue using the relative selling price method and also significantly expands the disclosure requirements for multiple-deliverable revenue arrangements. ASU 2009-13 should be applied on a prospective basis for revenue arrangements entered into or materially modified in fiscal years beginning on or after June 15, 2010, with earlier application permitted. The Company is currently evaluating the potential impact of the adoption of ASU 2009-13 on the Company's financial position or results of operations for future collaborations arrangements.
91
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 2. LICENSE AND COLLABORATIVE ARRANGEMENTS
Solvay Pharmaceuticals, Inc.
In November 2008, the Company entered into an Exclusive License Agreement with Solvay Pharmaceuticals, Inc. granting Solvay exclusive rights to develop and commercialize DM-1796 for pain indications in the United States, Canada and Mexico for pain indications. The agreement became effective in January 2009, upon clearance of the transaction under Hart-Scott-Rodino (HSR) Antitrust Improvements Act of 1976.
Pursuant to the agreement, Solvay paid the Company a $25.0 million upfront fee in February 2009. The Company is also eligible to receive milestone payments for acceptance and FDA approval of the New Drug Application for DM-1796 for PHN, and sales milestone payments upon reaching certain sales milestones. Solvay will pay Depomed royalties of 14 to 20 percent of net product sales, depending on the level of product sales.
The Company was responsible for completion of the Phase 3 clinical trial for DM-1796 in PHN, which was completed in 2009, and will be responsible for certain other regulatory support activities through NDA approval. Solvay is responsible for the NDA filing and has the option to develop DM-1796 in further pain indications other than PHN. If Solvay elects to develop DM-1796 in fibromyalgia, the Company has a right of first negotiation for co-promote rights in the obstetrics/gynecology field upon fibromyalgia indication regulatory approval.
The license agreement will expire with the last to expire of the Company's patents covering DM-1796, subject to early termination in certain circumstances. The Company will be responsible for the manufacture of DM-1796 for up to four years from the effective date of the license agreement, pursuant to a supply agreement to be entered into by Depomed and Solvay.
The Company is recognizing the $25.0 million upfront payment ratably over the period of the Company's development and supply obligations under the agreement which is estimated to be through January 2013. For the year ended December 31, 2009, the Company recognized $6.2 million in license revenue under the arrangement. The remaining deferred revenue balance is $18.8 million as of December 31, 2009.
Abbott Laboratories (Abbott) acquired the pharmaceutical business of Solvay in a transaction that closed in February 2010. Accordingly, following the closing of the transaction, Abbott is responsible for the Company's DM-1796 license arrangement with Solvay, either directly or through a subsidiary.
Santarus, Inc.
In July 2008, the Company entered into a promotion agreement with Santarus, Inc. granting Santarus exclusive rights to promote GLUMETZA in the United States. Santarus paid the Company a $12.0 million upfront fee, and based on the achievement of specified levels of annual GLUMETZA net product sales, Santarus may be required to pay the Company additional one-time sales milestones totaling up to $16.0 million.
Santarus began promotion of GLUMETZA in October 2008. Santarus is required to meet certain minimum promotion obligations during the term of the agreement, and is required to make certain minimum marketing, advertising, medical affairs and other commercial support expenditures. The Company continues to record revenue from the sales of GLUMETZA product, and starting in October 2008, began paying Santarus a promotion fee equal to 80% of the gross margin earned from net sales
92
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 2. LICENSE AND COLLABORATIVE ARRANGEMENTS (Continued)
of GLUMETZA product in the United States. The promotion fee will be reduced to 75% of gross margin beginning in the fourth quarter of 2010. For the years ended December 31, 2009 and 2008, the Company recognized $23.6 million and $4.7 million, respectively, in promotion fee expense under the agreement, which is classified within selling, general and administrative expense.
Santarus is responsible for all costs associated with its sales force and for all other marketing expenses associated with its promotion of GLUMETZA product. Depomed is responsible for overseeing product manufacturing and supply. A joint commercialization committee has been formed to oversee and guide the strategic direction of the GLUMETZA alliance.
Pursuant to the terms of the promotion agreement, Depomed retains the option to co-promote GLUMETZA product in the future to obstetricians and gynecologists. The promotion agreement will continue in effect until the expiration of the last-to-expire patent or patent application with a valid claim in the territory covering a GLUMETZA product, unless terminated sooner.
The Company is recognizing the $12.0 million upfront payment ratably until October 2021, which represents the estimated length of time the Company's obligations exist under the arrangement for promotion fees it is obligated to pay Santarus. For the years ended December 31, 2009 and 2008, the Company recognized $0.9 million and $0.4 million respectively in license revenue related to the amortization of the upfront payment. The remaining deferred revenue balance is $10.7 million as of December 31, 2009.
Merck & Co., Inc.
In July 2009, the Company entered into a non-exclusive license agreement with Merck & Co., Inc. (Merck) granting Merck a license to certain patents related to the Company's metformin extended release technology to be used in developing fixed dose combinations of sitagliptin and extended release metformin.
Under terms of the agreement, Merck received a non-exclusive license as well as other rights to certain Depomed patents directed to metformin extended release technology. In exchange, the Company received a $10.0 million upfront fee in August 2009. The Company is also eligible to receive a milestone payment upon filing of the New Drug Application for the therapeutic candidate, as well as modest single digit royalties on any net product sales for an agreed-upon period. Merck will also be granted a right of reference to the New Drug Application covering the Company's GLUMETZA product in Merck's regulatory filings covering fixed dose combinations of sitagliptin and extended release metformin. The Company has no development obligations under the agreement. The Company recognized the entire $10.0 million upfront payment as license revenue in the third quarter of 2009.
Covidien
In November 2008, the Company entered into a license agreement with Mallinckrodt, Inc., a subsidiary of Covidien, Ltd. (Covidien) granting Covidien worldwide rights to utilize the Company's AcuForm® technology for the exclusive development of four products containing acetaminophen in combination with opiates. In 2008, Covidien paid the Company a total of $5.5 million in upfront fees, representing a $4.0 million upfront license fee and a $1.5 million upfront payment for formulation work to be performed by Depomed under the agreement. Under the agreement, the Company may also
93
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 2. LICENSE AND COLLABORATIVE ARRANGEMENTS (Continued)
receive certain developmental milestone payments, if achieved, and is also entitled to receive royalties on sales of the products.
In October 2009, the first formulation was completed by the Company and delivered to Covidien, which triggered a $0.5 million milestone payment from Covidien received in October 2009. Because the non-refundable milestone was achieved and substantive in nature, and achievement was not reasonably assured at the inception of the agreement, the Company recognized the entire $0.5 million milestone payment as revenue in the fourth quarter of 2009.
In December 2009, the Company received a second $0.5 million milestone payment from Covidien related to the development of a formulation for the second product candidate under the agreement. Although the milestone payment was received by the Company, the development of the second formulation was not complete as of December 31, 2009, and the Company has additional formulation work to perform in 2010 in order to achieve the milestone. Accordingly, the Company has not recognized the second $0.5 million milestone payment as revenue, and the balance is included in deferred license revenue at December 31, 2009.
The $5.5 million in upfront payments is being accounted for as a single unit of accounting and being amortized ratably through November 2011, which is the length of time Depomed is obligated to perform formulation work under the agreement. For the years ended December 31, 2009 and 2008, the Company recognized $1.8 million and $0.2 million respectively in license revenue under the agreement. The remaining deferred revenue balance is $3.5 million as of December 31, 2009.
Watson Pharma, Inc.
In July 2007, the Company entered into a promotion agreement with Watson Pharma, Inc. (Watson) granting Watson a co-exclusive right to promote Proquin XR to the urology specialty and to long-term care facilities in the United States. In September 2007, the agreement was amended to also grant Watson a co-exclusive right to promote Proquin XR to the obstetrics/gynecology (ob/gyn) specialty. The Company re-launched Proquin XR in September 2007 and Watson commenced promotion in October 2007.
Watson was required to deliver a minimum number of annual sales detail calls and maintain a sales force of a minimum size and received a promotion fee equal to an agreed upon portion of gross margin attributable to the urology and ob/gyn specialties and long-term care facilities above an agreed upon baseline level.
In February 2009, the Company and Watson Pharma, Inc. (Watson) further amended the promotion agreement between the parties, pursuant to which Watson performed a specified number of physician details during the first quarter of 2009, and the Company was no longer obligated to pay Watson promotion fees beginning in 2009, and thereafter.
Under the provisions of the agreement, Watson elected to terminate the agreement early, and the promotion agreement terminated effective December 31, 2009.
The Company recognized promotion fee expense under the agreement of approximately $0.1 million for the year ended December 31, 2008 and zero for the years ended December 31, 2009 and 2007, respectively.
94
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 2. LICENSE AND COLLABORATIVE ARRANGEMENTS (Continued)
Settlement with TEVA Pharmaceuticals USA, Inc.
In April 2008, the Company entered into a settlement and license agreement with Teva related to the patent infringement lawsuit filed by the Company against Teva affiliates IVAX Corporation and IVAX Pharmaceuticals, Inc. The settlement agreement provided for a one-time payment to the Company of $7.5 million, which the Company received in April 2008, and for a non-exclusive license in favor of Teva (including IVAX) to continue to market its generic Glucophage XR product in the United States. The $7.5 million one-time payment received by the Company was recognized as a gain on litigation settlement within operating income during the second quarter of 2008.
The Company also received ongoing royalty payments from Teva on sales by Teva (including IVAX) of generic Glucophage XR in the United States, which was calculated as a percentage of sales, as reported by a third-party market research company. The royalty was subject to a $2.5 million aggregate cap, which was met during the third quarter of 2009. For the years ended December 31, 2009 and 2008, the Company recognized $1.3 and $1.2 million in royalty revenue related to this arrangement, respectively. As of December 31, 2009, a cumulative total of $2.5 million in royalties has been recognized to date under the settlement, with no royalties remaining under the aggregate cap.
King Pharmaceuticals, Inc.
In June 2006, the Company entered into a promotion agreement with King Pharmaceuticals, Inc. (King), pursuant to which King was granted the co-exclusive right to promote GLUMETZA in the United States. Under the agreement, King was required to promote GLUMETZA to physicians in the United States through its sales force, to deliver a minimum number of annual detail calls to potential GLUMETZA prescribers, and to maintain a sales force of a minimum size. In consideration for King's promotion of GLUMETZA, the Company was required to pay King a promotion fee equal to fifty percent of gross margin, which was defined in the agreement as sales of GLUMETZA, net of actual returns, estimated discounts, estimated rebates and estimated chargebacks, minus cost of goods sold.
In October 2007, the Company and King terminated the promotion agreement and King paid the Company $29.7 million in termination fees. As a result of the agreement termination and related termination fee, the Company recognized a gain of $29.6 million within operating income in the fourth quarter of 2007. Beginning in the fourth quarter of 2007, the Company was no longer obligated to pay King promotion fees on sales of GLUMETZA in the United States. The Company recognized $3.0 million in promotion fee expense for the year ended December 31, 2007 under the agreement, which has been classified in selling, general and administrative expense.
Esprit Pharma, Inc.
In July 2005, the Company entered into an exclusive license agreement with Esprit to market and distribute Proquin XR in the United States. The agreement was amended in July 2006. In connection with the license agreement, the Company also entered into a related supply agreement with Esprit, pursuant to which the Company supplied commercial quantities of Proquin XR to Esprit.
The license agreement obligated Esprit to pay the Company $50.0 million in license fees, of which $30.0 million was paid in July 2005 and $10.0 million was paid in December 2006. The remaining $10.0 million was due in July 2007. The license fee payments received were scheduled to be recognized
95
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 2. LICENSE AND COLLABORATIVE ARRANGEMENTS (Continued)
as revenue ratably until June 2020, which represented the length of time that the Company was obligated to manufacture Proquin XR for Esprit or its licensees.
The license agreement also provided for royalty payments by Esprit to the Company of 15 percent to 25 percent of Proquin XR net sales, based on escalating net sales and subject to certain minimum royalty amounts. Esprit's minimum royalty obligation for 2007 was $5.0 million, and in subsequent years was $5.0 million per year, subject to annual increases in the consumer price index beginning in 2008.
In July 2007, the Company entered into a termination and assignment agreement with Esprit terminating the exclusive license agreement and related supply agreement. Upon entering into the termination and assignment agreement, the marketing and distribution rights in the United States for Proquin XR reverted back to the Company and Esprit paid the Company $17.5 million, representing (i) a $10.0 million payment in respect of the final license payment that would have been due to the Company in July 2007 under the license agreement; (ii) a $2.5 million payment in respect of a pro-rated portion of minimum royalties for 2007 under the license agreement; and (iii) a $5.0 million termination fee. Esprit has no future royalty obligations to the Company.
As a result of termination of the license and supply agreements with Esprit, the Company no longer has continuing obligations to Esprit. Accordingly, all deferred revenue related to license fees previously received from Esprit was fully recognized as revenue in July 2007, resulting in recognition of approximately $36.1 million of license revenue. In addition, the final $10.0 million payment received in July 2007 was fully recognized as license revenue on receipt, resulting in total recognition of $46.1 million of license revenue in 2007. The royalty payment of $2.5 million was recognized as royalty revenue and the $5.0 million termination fee has been classified as a gain within operating income 2007.
The Company recognized $47.5 million of license revenue related to these upfront fees for the year ended December 31, 2007. The Company recognized $2.5 million of royalty revenue under the agreements for the year ended December 31, 2007.
Biovail Laboratories International
GLUMETZA
In May 2002, the Company entered into a development and license agreement granting Biovail Laboratories Incorporated (Biovail) an exclusive license in the United States and Canada to manufacture and market GLUMETZA. Under the terms of the agreement, the Company was responsible for completing the clinical development program in support of the 500mg GLUMETZA. In April 2003, Biovail submitted a New Drug Application to the U.S. Food and Drug Administration (FDA) for approval and in July 2005, Biovail received FDA approval to market GLUMETZA in the United States. In accordance with the license agreement, Biovail paid a $25.0 million license fee payment to the Company.
In April 2004, the Company and Biovail amended the GLUMETZA license agreement. Under the amended agreement, the Company would receive royalties on sales of Biovail's 1000mg metformin HCl tablet in the United States and Canada in exchange for allowing Biovail to use the Company's clinical data for its Metformin GR, a 500mg metformin HCl tablet, to support and accelerate regulatory submissions for Biovail's 1000mg tablet and to establish equivalence between the two dosage forms. In
96
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 2. LICENSE AND COLLABORATIVE ARRANGEMENTS (Continued)
May 2005, Biovail received a Notice of Compliance for the 500mg and 1000mg strengths of GLUMETZA from the Therapeutic Products Directorate of Canada to market the products in Canada.
In October 2005, the Company delivered a notice of breach to Biovail and subsequently filed suit in respect of its license agreement with Biovail, related to the failure of Biovail to make the first commercial sale of the 500mg strength GLUMETZA within 120 days of approval in each of Canada and the United States as required in the license agreement. In December 2005, the Company settled its dispute with Biovail and entered into an amended license agreement whereby the Company granted to Biovail an exclusive license in Canada to manufacture and market the 500mg formulation of GLUMETZA and the Company established its right to manufacture and market the 500mg GLUMETZA in the United States and internationally with the exception of Canada. The Company will recognize the $25.0 million license fee payment as revenue ratably until October 2021, which represents the estimated length of time the Company's obligations exist under the arrangement related to royalties it is obligated to pay Biovail on net sales of the 500mg GLUMETZA in the United States and to use Biovail as the Company's sole supplier of the 1000mg GLUMETZA. The Company recognized $1.6 million, $1.5 million and $1.5 million of license revenue related to the amortization of this upfront fee for each of the years ended December 31, 2009, 2008 and 2007, respectively. The remaining deferred revenue balance related to the $25.0 million upfront payment was $18.9 million as of December 31, 2009.
Under the agreement, Biovail is obligated to pay the Company royalties of six percent on Canadian net sales of the 500mg GLUMETZA and one percent on Canadian net sales of the 1000mg GLUMETZA. In July 2007, the royalty percentage increased to ten percent on Canadian net sales of the 500mg GLUMETZA, and returned to six percent in January 2008, on FDA approval of the 1000mg formulation of GLUMETZA in the United States. The Company recognized royalty revenue under the agreement of $0.2 million, $0.3 million, and $0.2 million for the years ended December 31, 2009, 2008 and 2007, respectively.
The Company is obligated to pay Biovail royalties of one percent on net sales of the 500mg GLUMETZA in the United States. The Company recognized royalty expense under the agreement of $0.3 million, $0.3 million and $0.1 million for the years ended December 31, 2009, 2008 and 2007, respectively.
As part of the same settlement, Biovail granted the Company an exclusive license to market the 1000mg GLUMETZA in the United States. The Company is obligated to purchase the 1000mg GLUMETZA exclusively from Biovail, subject to back-up manufacturing rights in the Company's favor. If the Company exercises its back-up rights, compensation to Biovail will change from a supply-based arrangement to royalties of six percent on net sales of the 1000mg GLUMETZA. The Company began selling the 1000mg GLUMETZA in the United States in June 2008.
Technology License
In February 2007, the Company entered into a license and development agreement with Biovail granting Biovail an option to license the AcuForm drug delivery technology to develop and commercialize up to two pharmaceutical products. Biovail did not exercise its options under the agreement, and in August 2008, the option to license the AcuForm drug delivery technology to develop and commercialize up to two pharmaceutical products expired.
97
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 2. LICENSE AND COLLABORATIVE ARRANGEMENTS (Continued)
Pursuant to the agreement, Biovail paid the Company an upfront fee of $0.5 million in February 2007, and the $0.5 million upfront license fee was recognized as license revenue in the first quarter of 2007.
LG Life Sciences, Ltd.
In August 2004, the Company entered into a license and distribution agreement granting LG Life Sciences (LG) an exclusive license to LG's version of the 500mg GLUMETZA in the Republic of Korea. LG launched the product in Korea, known as Novamet GR (extended release metformin tablets) in 2006.
Upon signing of the agreement, LG paid the Company a $0.6 million upfront license fee. In November 2006, both parties amended the agreement and LG paid the Company a $0.5 million milestone payment in respect of LG's approval to market GLUMETZA in the Republic of Korea. Through December 31, 2006, the upfront license fee and milestone payment were initially deferred and were being amortized over a period of eight years, which represented the estimated length of time the Company was obligated to provide assistance in development and manufacturing.
In January 2007, the Company and LG Life Sciences further amended the parties' license and distribution agreement and the Company granted LG a license to certain of the Company's intellectual property rights to manufacture Novamet GR in exchange for royalties on net sales of Novamet GR in Korea, and to remove the provisions of the original agreement providing for the supply of 500mg Novamet GR tablets by the Company to LG. Under the amended agreement, the Company no longer has continuing performance obligations to LG that are other than inconsequential or perfunctory and accordingly, the remaining $0.9 million of previously deferred revenue was recognized as license revenue in the first quarter of 2007.
The Company recognized $0.9 million of license revenue related to the agreements for the year ended December 31, 2007.
Rottapharm/Madaus S.r.l.
In November 2005, the Company entered into a distribution and supply agreement for Proquin XR in Europe with a privately owned specialty pharmaceutical company, Madaus S.r.l, that was acquired by Rottapharm in June 2007. Under the terms of the agreement, the Company granted an exclusive right to Madaus for the commercialization of Proquin XR in Europe. The agreement was amended in April 2009 and removed provisions obligating the Company to supply commercial quantities of tablets to Rottapharm, but obligates the Company to provide regulatory and manufacturing support and consultation for up to an agreed upon number of hours through December 31, 2010. The Company will receive royalties on net sales of Proquin XR by Rottapharm in Europe. The term of the amended agreement is through July 2023.
In January 2006, Madaus paid the Company a $0.2 million license fee, which is being amortized ratably through December 2010. In March 2006, Madaus filed a Marketing Authorization Application (MAA) for Proquin XR with the Medical Products Agency in Sweden, and received approval in July 2008. The Company has recognized approximately $84,000, $13,000 and zero in license revenue under the agreement for the years ended December 31, 2009, 2008 and 2007, respectively. The remaining deferred revenue balance is approximately $0.1 million as of December 31, 2009.
98
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 2. LICENSE AND COLLABORATIVE ARRANGEMENTS (Continued)
In August 2008, Rottapharm/Madaus paid the Company an advance payment of $0.3 million intended for future product supply. The $0.3 million advance payment will now be applied toward future royalties due to the Company under the amended and restated license agreement.
PharmaNova, Inc.
In October 2006, Depomed entered into a sublicense agreement with PharmaNova, Inc. Pursuant to the agreement, PharmaNova has granted the Company an exclusive sublicense, under a United States patent held by the University of Rochester, to develop and commercialize a product in the United States containing the compound gabapentin as its active pharmaceutical ingredient which is indicated for the treatment of hot flashes associated with menopause in women.
The Company paid PharmaNova an upfront license fee of $0.5 million and paid an additional $0.5 million upon dosing of the first patient in the Company's Phase 3 trials for the product. The Company is required to pay PharmaNova $1.0 million upon submission to the FDA of a New Drug Application for the product, and $2.0 million upon FDA approval of an NDA. The agreement also provides for royalty payments to PharmaNova on net sales of the product, and for milestone payments upon achievement of annual net sales in excess of certain thresholds. The Company also paid PharmaNova consultancy fees of $0.3 million over a ten month period beginning in November 2006. The Company has recognized zero, $0.5 million and $0.2 million of research and development expense under the agreement for the years ended December 31, 2009, 2008 and 2007, respectively.
Supernus Pharmaceuticals, Inc.
In September 2006, Depomed entered into a collaboration agreement with Supernus Pharmaceuticals, Inc. to develop through a Phase 1 study a product candidate leveraging the Company's AcuForm drug delivery technology. The cost and ownership of the program will be shared between the parties equally. The collaboration agreement includes provisions pursuant to which the parties may negotiate and enter into a definitive agreement for the further development and for commercialization, by either or both parties, of the product candidate. The feasibility phase of the collaboration was completed in April 2008 and both parties have elected not to continue to develop the product candidate. The Company recognized approximately $4,000, $0.2 million and $0.7 million of research and development expense under the agreement for the years ended December 31, 2009, 2008 and 2007, respectively.
99
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 3. MARKETABLE SECURITIES
Securities classified as available-for-sale as of December 31, 2009 and 2008 are summarized below (in thousands). Estimated fair value is based on quoted market prices for these investments.
December 31, 2009
|
Amortized Cost |
Gross Unrealized Gains |
Gross Unrealized Losses |
Fair Value | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
U.S. debt securities: |
||||||||||||||||
Total included in cash and cash equivalents |
$ | 21,186 | $ | — | $ | — | $ | 21,186 | ||||||||
Total maturing within 1 year and included in marketable securities: |
||||||||||||||||
Commercial paper |
— | — | — | — | ||||||||||||
U.S. corporate debt securities |
7,900 | 1 | — | 7,901 | ||||||||||||
U.S. government agency debt securities |
12,989 | 40 | — | 13,029 | ||||||||||||
U.S. Treasury securities |
21,988 | 9 | (5 | ) | 21,992 | |||||||||||
Total maturing between 1 and 2 years and included in marketable securities: |
||||||||||||||||
Commercial paper |
— | — | — | — | ||||||||||||
U.S. corporate debt securities |
— | — | — | — | ||||||||||||
U.S. government agency debt securities |
12,027 | 1 | (12 | ) | 12,016 | |||||||||||
U.S. Treasury securities |
— | — | — | — | ||||||||||||
Total available-for-sale |
$ | 79,090 | $ | 51 | $ | (17 | ) | $ | 76,124 | |||||||
December 31, 2008
|
Amortized Cost |
Gross Unrealized Gains |
Gross Unrealized Losses |
Fair Value | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
U.S. debt securities: |
||||||||||||||||
Total included in cash and cash equivalents |
$ | 20,155 | $ | — | $ | — | $ | 20,155 | ||||||||
Total maturing within 1 year and included in marketable securities: |
||||||||||||||||
Commercial paper |
2,984 | 7 | — | 2,991 | ||||||||||||
U.S. corporate debt securities |
7,648 | 5 | (6 | ) | 7,647 | |||||||||||
U.S. government agency debt securities |
18,893 | 92 | — | 18,985 | ||||||||||||
U.S. Treasury securities |
30,256 | 53 | — | 30,309 | ||||||||||||
Total maturing between 1 and 2 years and included in marketable securities: |
||||||||||||||||
Commercial paper |
— | — | — | — | ||||||||||||
U.S. corporate debt securities |
— | — | — | — | ||||||||||||
U.S. government agency debt securities |
— | — | — | — | ||||||||||||
U.S. Treasury securities |
— | — | — | — | ||||||||||||
Total available-for-sale |
$ | 79,936 | $ | 157 | $ | (6 | ) | $ | 80,087 | |||||||
At December 31, 2009, the Company had fourteen securities in an unrealized loss position.
The following table shows the gross unrealized losses and fair value of the Company's investments with unrealized losses that are not deemed to be other-than-temporarily impaired, aggregated by
100
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 3. MARKETABLE SECURITIES (Continued)
investment category and length of time that individual securities have been in a continuous unrealized loss position, at December 31, 2009 (in thousands):
| |
Less than 12 months | 12 months or greater | Total | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
U.S. Debt Securities
|
Fair Value | Gross Unrealized Losses |
Fair Value | Gross Unrealized Losses |
Fair Value | Gross Unrealized Losses |
|||||||||||||
Commercial paper |
$ | — | $ | — | $ | — | $ | — | $ | — | $ | — | |||||||
U.S. corporate debt securities |
3,999 | — | — | — | 3,999 | — | |||||||||||||
U.S. government agency debt securities |
8,005 | (12 | ) | — | — | 8,005 | (12 | ) | |||||||||||
U.S. Treasury securities |
13,964 | (5 | ) | — | — | 13,964 | (5 | ) | |||||||||||
Total available-for-sale |
$ | 25,968 | $ | (17 | ) | $ | — | $ | — | $ | 25,968 | $ | (17 | ) | |||||
The gross unrealized losses above were caused by interest rate increases. No significant facts or circumstances have arisen to indicate that there has been any deterioration in the creditworthiness of the issuers of the securities held by the Company. Based on the Company's review of these securities, including the assessment of the duration and severity of the related unrealized losses and the Company's ability and intent to hold the investments until maturity, there were no other-than-temporary impairments for these securities as of December 31, 2009.
Effective January 1, 2008, the Company adopted the provisions of the Fair Value Measurements topic of the Codification. This defines fair value, establishes a framework for measuring fair value under generally accepted accounting principles and enhances disclosures about fair value measurements. Fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value must maximize the use of observable inputs and minimize the use of unobservable inputs. The standard describes a fair value hierarchy based on three levels of inputs, of which the first two are considered observable and the last unobservable, that may be used to measure fair value which are the following:
- •
- Level 1: Quoted prices in active markets for identical assets or liabilities.
- •
- Level 2: Inputs other than Level 1 that are observable, either directly or indirectly, such as quoted prices
for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of
the assets or liabilities.
- •
- Level 3: Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities.
The adoption of this topic did not have a material impact on the Company's financial position, results of operations or cash flows.
101
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 3. MARKETABLE SECURITIES (Continued)
The following table represents the Company's fair value hierarchy for its financial assets measured at fair value on a recurring basis as of December 31, 2009 (in thousands):
| |
Level 1 | Level 2 | Level 3 | Total | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Money market funds |
$ | 21,186 | $ | — | $ | — | $ | 21,186 | |||||
Commercial paper |
— | — | — | — | |||||||||
U.S. corporate debt securities |
— | 7,901 | — | 7,901 | |||||||||
U.S. government agency debt securities |
— | 25,045 | — | 25,045 | |||||||||
U.S. Treasury securities |
— | 21,992 | — | 21,992 | |||||||||
Total |
$ | 21,186 | $ | 54,938 | $ | — | $ | 76,124 | |||||
The following table represents the Company's fair value hierarchy for its financial assets measured at fair value on a recurring basis as of December 31, 2008 (in thousands):
| |
Level 1 | Level 2 | Level 3 | Total | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Money market funds |
$ | 20,155 | $ | — | $ | — | $ | 20,155 | |||||
Commercial paper |
— | 2,991 | — | 2,991 | |||||||||
U.S. corporate debt securities |
— | 7,647 | — | 7,647 | |||||||||
U.S. government agency debt securities |
— | 18,985 | — | 18,985 | |||||||||
U.S. Treasury securities |
— | 30,309 | — | 30,309 | |||||||||
Total |
$ | 20,155 | $ | 59,932 | $ | — | $ | 80,087 | |||||
There are no financial liabilities measured at fair value on a recurring basis as of December 31, 2009 and December 31, 2008
NOTE 4. INVENTORIES
Inventories relate to the manufacture of the Company's GLUMETZA and Proquin XR products. Inventories are stated at the lower of cost or market and consist of the following (in thousands):
| |
December 31, 2009 | December 31, 2008 | |||||
|---|---|---|---|---|---|---|---|
Raw materials |
$ | 49 | $ | 266 | |||
Work-in-process |
— | 127 | |||||
Finished goods |
2,453 | 2,392 | |||||
Deferred costs |
63 | 64 | |||||
Total |
$ | 2,565 | $ | 2,849 | |||
Deferred costs represent the costs of Proquin XR product shipped for which recognition of revenue has been deferred.
102
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 5. PROPERTY AND EQUIPMENT
Property and equipment consists of the following (in thousands):
| |
December 31, 2009 | December 31, 2008 | |||||
|---|---|---|---|---|---|---|---|
Furniture and office equipment |
$ | 1,029 | $ | 991 | |||
Laboratory equipment |
4,396 | 4,301 | |||||
Leasehold improvements |
3,151 | 3,049 | |||||
Construction in Progress |
398 | 41 | |||||
|
8,974 | 8,382 | |||||
Less accumulated depreciation |
(8,032 | ) | (7,482 | ) | |||
Property and equipment, net |
$ | 942 | $ | 900 | |||
There was no property and equipment included under capitalized leases as of December 31, 2009 or December 31, 2008. Depreciation expense was $0.6 million, $1.0 million and $1.1 million for the years ended December 31, 2009, 2008 and 2007, respectively.
NOTE 6. ACCOUNTS PAYABLE AND ACCRUED LIABILITIES
Accounts payable and accrued liabilities consist of the following (in thousands):
| |
December 31, 2009 | December 31, 2008 | |||||
|---|---|---|---|---|---|---|---|
Accounts payable |
$ | 709 | $ | 559 | |||
Accrued compensation |
2,404 | 2,601 | |||||
Accrued clinical trial expense |
221 | 661 | |||||
Accrued rebates and sales discounts |
4,396 | 2,289 | |||||
Allowance for product returns |
3,364 | 1,406 | |||||
Accrued promotion fee |
1,752 | 1,435 | |||||
Other accrued liabilities |
2,376 | 3,897 | |||||
Total accounts payable and accrued liabilities |
$ | 15,222 | $ | 12,848 | |||
103
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 7. DEFERRED REVENUE
Deferred revenue consists of the following (in thousands):
| |
December 31, 2009 | December 31, 2008 | ||||||
|---|---|---|---|---|---|---|---|---|
Deferred revenue, current portion: |
||||||||
Deferred product sales |
$ | 1,635 | $ | 1,702 | ||||
Deferred license revenue, current portion: |
||||||||
Solvay |
6,245 | — | ||||||
Biovail |
1,598 | 1,598 | ||||||
Santarus |
905 | 905 | ||||||
Covidien |
2,333 | 1,833 | ||||||
Madaus |
103 | 26 | ||||||
Deferred license revenue, current portion |
11,184 | 4,362 | ||||||
Deferred revenue, current portion |
12,819 | 6,064 | ||||||
Deferred license revenue, non-current portion: |
||||||||
Solvay |
12,592 | — | ||||||
Biovail |
17,296 | 18,894 | ||||||
Santarus |
9,787 | 10,691 | ||||||
Covidien |
1,631 | 3,464 | ||||||
Madaus |
— | 160 | ||||||
Deferred license revenue, non-current portion |
41,306 | 33,209 | ||||||
Total deferred revenue |
$ | 54,125 | $ | 39,273 | ||||
Deferred product sales as of December 31, 2009 and 2008 relate to Proquin XR product shipments that have not been recognized as revenue in accordance with the Company's revenue recognition policy for Proquin XR.
Deferred license revenue relates to upfront payments received by the Company under license and marketing agreements with its partners. At December 31, 2009, deferred license revenue consisted primarily of upfront license fee payments received from Solvay, Biovail, Santarus and Covidien. At December 31, 2008, deferred license revenue consisted primarily of upfront license fee payments received from Biovail, Santarus and Covidien.
In December 2004, the Company received a $25.0 million license fee payment under its agreements with Biovail. The $25.0 million license fee is being recognized as revenue ratably until October 2021, which represents the estimated length of time our obligations exist under the arrangement related to royalties we are obligated to pay Biovail on net sales of GLUMETZA in the United States and for our obligation to use Biovail as our sole supplier of the 1000mg GLUMETZA.
In July 2008, the Company received a $12.0 million upfront payment under its promotion agreement with Santarus. The Company is recognizing the $12.0 million upfront payment ratably until October 2021, which represents the estimated length of time the Company's obligations exist under the arrangement related to promotion fees it is obligated to pay Santarus.
In November 2008, the Company received a $5.5 million in upfront payment under its license agreement with Covidien. The Company is recognizing the $5.5 million upfront payment as revenue
104
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 7. DEFERRED REVENUE (Continued)
ratably until November 2011, which represents the estimated length of time the Company's formulation development obligations exist under the agreement. In December 2009, the Company received a $0.5 million milestone payment from Covidien related to the development of a formulation for the second product candidate under the agreement. Although the milestone payment was received by the Company, the development of the second formulation is not complete, and the Company has additional formulation work to perform in order to achieve the milestone. Accordingly, the Company has not recognized the second $0.5 million payment as revenue, and the balance is included in deferred license revenue at December 31, 2009.
In February 2009, the Company received a $25.0 million upfront payment under its exclusive license agreement with Solvay. The Company is recognizing the $25.0 million upfront payment ratably over the period of the Company's development and supply obligations under the agreement which is estimated to be through January 2013.
NOTE 8. LONG-TERM DEBT
In June 2008, the Company entered into a loan and security agreement with General Electric Capital Corporation, as agent (GECC), and Oxford Finance Corporation (Oxford) that provided the Company with a $15.0 million credit facility. The credit facility was available in up to three tranches. The first tranche of $3.8 million was advanced to the Company upon the closing of the loan agreement. The second tranche of $5.6 million was advanced to the Company in July 2008. The third tranche of $5.6 million was not drawn and is no longer available to the Company, and GECC and Oxford waived the 2% unused line fee related to the unused portion of the credit facility.
The Company paid interest on the first tranche for the first six months at an interest rate of 11.59%. Beginning in January 2009, the Company began principal payments on the first tranche, plus interest at such rate, which will be paid in 30 equal monthly installments. The second tranche was interest-only through December 31, 2008, with principal and interest payable thereafter in 30 equal monthly installments at an interest rate of 11.59%. Interest expense, which includes amortization of the debt issuance costs, was $1.0 million and $0.6 million for the years ended December 31, 2009 and 2008 respectively.
As of December 31, 2009, the outstanding balance under the facility was $6.1 million, and the unamortized portion of the debt issuance costs was $0.2 million. Future contractual principal and interest payments are as follows (in thousands):
| |
Principal | Interest | |||||
|---|---|---|---|---|---|---|---|
2010 |
$ | 3,845 | $ | 512 | |||
2011 |
2,243 | 82 | |||||
Total |
$ | 6,088 | $ | 594 | |||
105
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 8. LONG-TERM DEBT (Continued)
The Company has the right to voluntarily prepay any tranches received under the facility, in full or in part. Upon any voluntary prepayment of any of the tranches, the Company will be required to pay the lenders, a prepayment premium equal to: (i) 5% on such prepayment amount, if such prepayment is made within 14 months after the closing date, (ii) 4% on such prepayment amount, if such prepayment is made more than 14 months after the closing date but within 29 months after the closing date, and (ii) 3% on such prepayment amount, if such prepayment is made more than 29 months after the closing date, but on or before the maturity date of the respective tranche.
The obligations of the Company under the loan agreement are secured by interests in all of the Company's personal property, and proceeds from any intellectual property, but not by the Company's intellectual property.
The credit facility contains affirmative and negative covenants with which the Company must comply with, and imposes restrictions with regards to additional indebtedness, liens, various fundamental changes (including mergers and acquisitions), payments, investments, transactions with affiliates, and other limitations customary in secured credit facilities. The Company was in compliance with such covenants as of December 31, 2009.
NOTE 9. COMMITMENTS AND CONTINGENCIES
Leases
The Company leases its facilities under non-cancelable operating leases that expire in January 2012 with options to extend the lease terms for an additional five years. The leases are subject to annual increases on the anniversary of the commencement dates. Rent expense was $1.4 million, $1.2 million and $1.2 million years ended December 31, 2009, 2008 and 2007, respectively.
As of December 31, 2009 future minimum payments under operating leases for facilities and equipment were as follows (in thousands):
2010 |
$ | 1,614 | ||
2011 |
1,636 | |||
2012 |
137 | |||
Total |
$ | 3,387 | ||
Manufacturing Agreements
The Company has entered into a manufacturing arrangement with MOVA Pharmaceuticals, a subsidiary of Patheon, Inc. (Patheon) pursuant to which Patheon will manufacture commercial quantities of GLUMETZA and Proquin XR for the Company. The Company also has a supply agreement with Biovail in which Biovail provides the Company with commercial quantities of the 1000mg GLUMETZA. As of December 31, 2009 the Company has non-cancelable purchase orders and minimum purchase obligations for 2010 totaling approximately $1.9 million under these arrangements.
106
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 10. STOCK-BASED COMPENSATION
The Company uses the Black-Scholes option valuation model to determine the fair value of stock options and employee stock purchase plan (ESPP) shares. The determination of the fair value of stock-based payment awards on the date of grant using an option valuation model is affected by the Company's stock price as well as assumptions which include the Company's expected term of the award, the expected stock price volatility, risk-free interest rate and expected dividends over the expected term of the award.
As described in Note 1 of the Notes to Financial Statements, the Company has concluded that its historical share option exercise experience does not provide a reasonable basis upon which to estimate expected term. In 2007, the Company estimated the expected term of options granted by taking the average of the vesting term and the contractual term of the option, as illustrated by the simplified method. In 2008 and 2009, the Company estimated the expected term by using the weighted average terms of a peer group of companies that grant options with similar vesting provisions. The Company estimates the volatility of its common stock price by using the historical volatility over the expected term of the options. The Company bases the risk-free interest rate on U.S. Treasury zero-coupon issues with terms similar to the expected term of the options as of the date of grant. The Company does not anticipate paying any cash dividends in the foreseeable future and therefore uses an expected dividend yield of zero in the option valuation model.
The Company used the following assumptions to calculate the fair value of option grants for the years ended December 31, 2009, 2008 and 2007:
| |
2009 | 2008 | 2007 | |||
|---|---|---|---|---|---|---|
Employee and Director Stock Options |
||||||
Risk-free interest rate |
1.64 – 2.82% | 1.61 – 3.02% | 3.42 – 5.09% | |||
Dividend yield |
None | None | None | |||
Expected option term (in years) |
5.10 | 5.04 | 5.25 – 6.06 | |||
Expected stock price volatility |
65.3 – 71.1% | 58.2 – 64.4% | 53.2 – 63.9% |
The Company used the following assumptions to calculate the fair value of purchase rights granted under the ESPP for the years ended December 31, 2009, 2008 and 2007:
| |
2009 | 2008 | 2007 | |||
|---|---|---|---|---|---|---|
Employee Stock Purchase Plan |
||||||
Risk-free interest rate |
0.15 – 0.97% | 0.44 – 2.51% | 2.90 – 4.98% | |||
Dividend yield |
None | None | None | |||
Expected option term (in years) |
0.5 – 2.0 | 0.5 – 2.0 | 0.5 – 2.0 | |||
Expected stock price volatility |
86.5 – 107.8% | 65.8 – 114.2% | 59.6 – 83.6% |
107
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 10. STOCK-BASED COMPENSATION (Continued)
The following table presents stock-based compensation expense recognized for stock options, stock awards and the ESPP in the Company's Statements of Operations (in thousands):
| |
2009 | 2008 | 2007 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Cost of sales |
$ | 23 | $ | 23 | $ | 24 | ||||
Research and development expense |
885 | 718 | 695 | |||||||
Selling, general and administrative expense |
1,749 | 1,711 | 1,622 | |||||||
Total |
$ | 2,657 | $ | 2,452 | $ | 2,341 | ||||
The weighted-average grant date fair value of options granted during the years ended December 31, 2009, 2008 and 2007 were $1.26, $1.62 and $1.62, respectively. The weighted-average grant date fair value of purchase rights granted under the ESPP during the years ended December 31, 2009, 2008 and 2007 were $1.39, $1.05 and $1.57, respectively. The total intrinsic value of options exercised during the years ended December 31, 2009, 2008 and 2007 were $1.1 million, $0.1 million and $0.2 million, respectively. The total fair value of options that vested during the years ended December 31, 2009, 2008 and 2007 was $2.1 million, $2.4 million and $1.6 million, respectively. At December 31, 2009, Depomed had $2.1 million of total unrecognized compensation expense, net of estimated forfeitures, related to stock option plans that will be recognized over an average vesting period of 2.0 years. Cash received from stock option exercises was $1.7 million, $0.1 million and $0.8 million for the years ended December 31, 2009, 2008 and 2007, respectively.
1995 Stock Option Plan
The Company's 1995 Stock Option Plan (the 1995 Plan) was adopted by the Board of Directors and approved by the shareholders in September 1995, and has been subsequently amended. The 1995 Plan provided for the grant to employees of the Company, including officers, of incentive stock options, and for the grant of nonstatutory stock options to employees, directors and consultants of the Company. The number of shares authorized under the 1995 Plan is 4,700,000 shares, of which zero are available for future issuance at December 31, 2009. In May 2004, the 1995 Plan was terminated with respect to grants of new stock options and all options which expire or are forfeited will be retired from the pool.
Generally, the exercise price of all incentive stock options and nonstatutory stock options granted under the 1995 Plan must be at least 100% and 85%, respectively, of the fair value of the common stock of the Company on the grant date. The term of incentive and nonstatutory stock options may not exceed 10 years from the date of grant. An option shall be exercisable on or after each vesting date in accordance with the terms set forth in the option agreement. The right to exercise an option generally vests over four years at the rate of at least 25% by the end of the first year and then ratably in monthly installments over the remaining vesting period of the option.
108
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 10. STOCK-BASED COMPENSATION (Continued)
The following table summarizes the activity for the three years ended December 31, 2009 under the 1995 Plan:
| |
Shares | Weighted- Average Exercise Price |
||||||
|---|---|---|---|---|---|---|---|---|
Options outstanding at December 31, 2006 |
3,071,402 | $ | 4.31 | |||||
Options exercised |
(266,949 | ) | 2.89 | |||||
Options forfeited |
(990,316 | ) | 5.13 | |||||
Options expired |
(251,767 | ) | 3.82 | |||||
Options outstanding at December 31, 2007 |
1,562,370 | $ | 4.12 | |||||
Options exercised |
(21,905 | ) | 2.51 | |||||
Options forfeited |
— | — | ||||||
Options expired |
(143,578 | ) | 7.42 | |||||
Options outstanding at December 31, 2008 |
1,396,887 | $ | 3.80 | |||||
Options exercised |
(418,397 | ) | 2.33 | |||||
Options forfeited |
— | — | ||||||
Options expired |
(193,790 | ) | 5.07 | |||||
Options outstanding at December 31, 2009 |
784,700 | $ | 4.28 | |||||
Options exercisable and expected to become exercisable at December 31, 2009 |
784,700 |
$ |
4.28 |
|||||
Options exercisable at December 31, 2009 |
784,700 |
$ |
4.28 |
|||||
| |
Weighted- Average Remaining Contractual Term (Years) |
Aggregate Intrinsic Value (in thousands) |
|||||
|---|---|---|---|---|---|---|---|
Options outstanding at December 31, 2009 |
1.48 | $ | 246 | ||||
Options exercisable and expected to become exercisable at December 31, 2009 |
1.48 |
$ |
246 |
||||
Options exercisable at December 31, 2009 |
1.48 |
$ |
246 |
||||
109
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 10. STOCK-BASED COMPENSATION (Continued)
Information regarding the stock options outstanding at December 31, 2009 under the 1995 Plan is summarized below:
Range of Exercise Prices
|
Number Outstanding |
Weighted- Average Remaining Contractual Term (Years) |
Weighted- Average Exercise Price (Outstanding) |
Number Exercisable |
Weighted- Average Exercise Price (Exercisable) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
$1.71 – $2.90 |
172,300 | 2.05 | $ | 1.92 | 172,300 | $ | 1.92 | |||||||||
$3.40 – $3.75 |
197,000 | 0.64 | 3.66 | 197,000 | 3.66 | |||||||||||
$4.25 – $5.25 |
165,250 | 0.44 | 4.43 | 165,250 | 4.43 | |||||||||||
$5.80 – $6.76 |
245,150 | 2.42 | 6.27 | 245,150 | 6.27 | |||||||||||
$7.32 |
5,000 | 4.21 | 7.32 | 5,000 | 7.32 | |||||||||||
|
784,700 | 1.48 | $ | 4.28 | 784,700 | $ | 4.28 | |||||||||
2004 Equity Incentive Plan
The Company's 2004 Equity Incentive Plan (the 2004 Plan) was adopted by the Board of Directors and approved by the shareholders in May 2004. The 2004 Plan provides for the grant to employees of the Company, including officers, of incentive stock options, and for the grant of nonstatutory stock options to employees, directors and consultants of the Company. The number of shares authorized under the 2004 Plan at December 31, 2009 was 6,750,000 shares, of which 1,444,051 were available for future issuance.
Generally, the exercise price of all incentive stock options and nonstatutory stock options granted under the 2004 Plan must be at least 100% and 85%, respectively, of the fair value of the common stock of the Company on the grant date. The term of incentive and nonstatutory stock options may not exceed 10 years from the date of grant. An option shall be exercisable on or after each vesting date in accordance with the terms set forth in the option agreement. The right to exercise an option generally vests over four years at the rate of at least 25% by the end of the first year and then ratably in monthly installments over the remaining vesting period of the option.
110
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 10. STOCK-BASED COMPENSATION (Continued)
The following tables summarize the activity for the three years ended December 31, 2009 under the 2004 Plan:
| |
Shares | Weighted- Average Exercise Price |
||||||
|---|---|---|---|---|---|---|---|---|
Options outstanding at December 31, 2006 |
2,142,943 | $ | 5.49 | |||||
Options granted at fair market value |
2,445,185 | 2.81 | ||||||
Options exercised |
(1,605 | ) | 3.84 | |||||
Options forfeited |
(1,083,317 | ) | 4.82 | |||||
Options outstanding at December 31, 2007 |
3,503,206 | $ | 3.82 | |||||
Options granted at fair market value |
917,071 | 3.08 | ||||||
Options exercised |
(8,709 | ) | 2.87 | |||||
Options forfeited |
(189,413 | ) | 4.16 | |||||
Options outstanding at December 31, 2008 |
4,222,155 | $ | 3.65 | |||||
Options granted at fair market value |
1,267,000 | 2.21 | ||||||
Options exercised |
(305,588 | ) | 2.38 | |||||
Options forfeited |
(341,762 | ) | 3.48 | |||||
Options expired |
(5,194 | ) | 5.30 | |||||
Options outstanding at December 31, 2009 |
4,836,611 | $ | 3.34 | |||||
Options exercisable and expected to become exercisable at December 31, 2009 |
4,506,683 |
$ |
3.40 |
|||||
Options exercisable at December 31, 2009 |
3,108,490 |
$ |
3.76 |
|||||
| |
Weighted- Average Remaining Contractual Term (Years) |
Aggregate Intrinsic Value (in thousands) |
|||||
|---|---|---|---|---|---|---|---|
Options outstanding at December 31, 2009 |
6.88 | $ | 3,156 | ||||
Options exercisable and expected to become exercisable at December 31, 2009 |
6.74 | $ | 2,819 | ||||
Options exercisable at December 31, 2009 |
5.94 | $ | 1,594 | ||||
Information regarding the stock options outstanding at December 31, 2009 under the 2004 Plan is summarized below:
Range of Exercise Prices
|
Number Outstanding |
Weighted- Average Remaining Contractual Term (Years) |
Weighted- Average Exercise Price (Outstanding) |
Number Exercisable |
Weighted- Average Exercise Price (Exercisable) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
$1.49 – $1.98 |
1,642,346 | 8.03 | $ | 1.87 | 951,598 | $ | 1.89 | |||||||||
$2.05 – $3.31 |
1,170,420 | 7.78 | 2.73 | 568,848 | 2.98 | |||||||||||
$3.38 – $4.37 |
975,608 | 6.99 | 3.86 | 598,690 | 3.99 | |||||||||||
$4.40 – $6.29 |
1,029,237 | 3.94 | 5.80 | 970,687 | 5.84 | |||||||||||
$6.36 – $7.78 |
19,000 | 4.80 | 7.48 | 18,667 | 7.50 | |||||||||||
|
4,836,611 | 6.88 | $ | 3.34 | 3,108,490 | $ | 3.76 | |||||||||
111
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 11. SHAREHOLDERS' EQUITY
Series A Preferred Stock
In January 2000, the Company issued 12,015 shares of Series A Preferred Stock at a price of $1,000 per share. The Series A Preferred Stock accrued a dividend of 7% per annum, compounded semi-annually and payable in shares of Series A Preferred Stock. The Series A Preferred Stock was convertible at anytime between January 2002 and January 2006 into the Company's common stock. The original conversion price of the Series A Preferred Stock was $12.00; however, as a result of the Company's March 2002 and October 2003 financings, the conversion price had been adjusted to $9.51 per share. In December 2004, the Company entered into an agreement with the Series A Preferred shareholder to resolve a misunderstanding between the Company and the shareholder relating primarily to prior adjustments to the conversion price of the Series A Preferred Stock. Pursuant to the agreement, among other matters, the Company agreed to adjust the conversion price to $7.50 per share. The Company and the shareholder also agreed to binding interpretations of certain other terms related to the Series A Preferred Stock conversion price.
Prior to December 2004, the amounts calculated as Series A Preferred stock dividends were accounted for as an adjustment to the conversion price following EITF Issue No. 98-5, Accounting for Convertible Securities with Beneficial Conversion Features or Contingently Adjustable Conversion Ratios (Issue No. 98-5). As a result of the modifications to the preferred stock agreement in December 2004, the Company determined that a "significant modification" of the agreement had been made, and, therefore, a new "commitment date" for accounting purposes had been established on December 10, 2004. The Company measured the difference between the carrying value of the preferred stock and the fair value of the modified preferred stock pursuant to EITF Topic No. D-42, The Effect on the Calculation of Earnings per Share for the Redemption or Induced Conversion of Preferred Stock and determined that the fair value of the modified security was less than the carrying value of the security prior to the modification. The Company also evaluated the effective conversion rate, after considering the reset rate of $7.50 per share in addition to the common stock issuable upon conversion of the unpaid, accumulated dividends. The fair value of the underlying common stock on December 10, 2004 was $5.06 per share. The Company determined that the conversion rate, after including the effect of the unpaid dividends, did not result in a beneficial conversion feature, which could have had the effect of also providing a deemed dividend to the preferred shareholder. However, an anti-dilution provision of the Series A Preferred Stock was triggered by the Company's January 2005 financing, which adjusted the conversion price of the Series A Preferred Stock to $7.12. As a result of the adjusted conversion price and an increase in the amount of common stock issuable upon conversion of the Series A Preferred Stock due to additional accumulated dividends, the Series A Preferred Stock now contained a "beneficial conversion feature" subject to recognition pursuant to Issue No. 98-5.
In conjunction with the modification of the agreement, the Company issued a warrant to the Series A Preferred shareholder. The value of the warrant was considered in determining the value of the modified security. The warrant was convertible into shares of the Company's common stock during the period between January 2006 and January 2009. The conversion price of the warrant initially was $7.12, which was equal to the Series A Preferred Stock conversion price in effect as of January 20, 2006. The conversion price of the warrant decreased by approximately 4.8% per year during the conversion period, such that the number of shares of the Company's common stock issuable upon conversion of the warrant increased by approximately 5.1% per year. The conversion of the warrant could be satisfied only by surrender of the outstanding shares of Series A Preferred Stock.
112
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 11. SHAREHOLDERS' EQUITY (Continued)
The Series A Preferred Stock accrued dividends through January 20, 2006, which is the date the warrant initially became exercisable. As a result of the issuance of the warrant, the preferred stock could be surrendered in exchange for common stock for an additional three years through January 20, 2009. As long as the Series A Preferred Stock remained outstanding, the number of shares into which the warrant could be converted increased as the conversion price of the warrant decreased resulting in additional deemed dividends on the Series A Preferred Stock. For the years ended December 31, 2009, 2008 and 2007 the Company recognized Series A Preferred Stock deemed dividends of approximately zero, $0.5 million and $0.7 million, respectively, attributable to the beneficial conversion feature from the accrued dividends and decreasing warrant price.
In October 2008, the holder of the Series A Preferred Stock and warrant exercised its warrant to acquire shares of the Company's common stock by surrendering its 18,158 shares of Series A Preferred Stock in exchange for 2,914,526 shares of the Company's common stock. The warrant was exercised in accordance with its terms and without any cash payment to the company, and together with surrender of the Series A Preferred Stock, was convertible into the Company's common stock at a conversion price of $6.23 per share on the date of exercise.
Employee Stock Purchase Plan
In May 2004, the ESPP was approved by the shareholders. The ESPP is qualified under Section 423 of the Internal Revenue Code. The ESPP is designed to allow eligible employees to purchase shares of the Company's common stock through periodic payroll deductions. The price of the common stock purchased under the ESPP must be equal to at least 85% of the lower of the fair market value of the common stock on the commencement date of each offering period or the specified purchase date. The number of shares authorized for issuance under the ESPP as of December 31, 2009 was 1,000,000, of which 106,727 shares were available for future issuance.
In 2009, the Company sold 274,996 shares of its common stock under the ESPP. The shares were purchased at a weighted average purchase price of $1.24 with proceeds of approximately $0.3 million. In 2008, the Company sold 200,232 shares of its common stock under the ESPP. The shares were purchased at a weighted average purchase price of $1.81 with proceeds of approximately $0.4 million.
Warrant and Option Exercises
During 2009 and 2008, the Company issued zero and 160,476 shares of common stock, respectively, to warrant holders subject to a cashless exercise feature of the exercised warrants. As of December 31, 2009, the Company has no warrants outstanding.
Employees and consultants exercised options to purchase 723,985 shares of the Company's common stock with net proceeds to the Company of approximately $1.7 million during the year ended December 31, 2009. Employees and consultants exercised options to purchase 30,614 shares of the Company's common stock with net proceeds to the Company of approximately $0.1 million during the year ended December 31, 2008.
Shareholder Rights Plan
On April 21, 2005, the Company adopted a shareholder rights plan, (the Rights Plan). Under the Rights Plan, the Company distributed one preferred share purchase right for each share of common
113
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 11. SHAREHOLDERS' EQUITY (Continued)
stock outstanding at the close of business on May 5, 2005. If a person or group acquires 20% or more of the Company's common stock in a transaction not pre-approved by the Company's Board of Directors, each right will entitle its holder, other than the acquirer, to buy additional shares of the Company's common stock at 50% of its market value, as defined in the Rights Plan. In addition, if an unapproved party acquires more than 20% of the Company's common stock, and the Company is later acquired by the unapproved party or in a transaction in which all shareholders are not treated alike, shareholders with unexercised rights, other than the unapproved party, will be entitled to receive upon exercise of the rights, common stock of the merger party or asset buyer with a value of twice the exercise price of the rights. Each right also becomes exercisable for one one-thousandth of a share of the Company's Series RP preferred stock at the right's then current exercise price ten days after an unapproved third party makes, or announces an intention to make, a tender offer or exchange offer that, if completed, would result in the unapproved party acquiring 20% or more of the Company's common stock. The Board of Directors may redeem the rights for a nominal amount before an event that causes the rights to become exercisable. The rights will expire on April 21, 2015.
Equity Line of Credit
In December 2006, the Company entered into a common stock purchase agreement with Azimuth Opportunity, Ltd., pursuant to which Azimuth is committed to purchase up to the lesser of (a) $30,000,000 of the Company's common stock, or (b) 8,399,654 shares of common stock, which was equal to the number of shares that is one less than 20% of the issued and outstanding shares of the Company's common stock as of December 11, 2006. The term of the original agreement was 24 months. In August 2008, the agreement was amended and the agreement was extended an additional 24 months until December 2010. From time to time over the term of the Purchase Agreement, and at the Company's discretion, the Company may present Azimuth with draw down notices requiring Azimuth to purchase a specified dollar amount of shares of its common stock, subject to certain limits and so long as specified conditions are met. The shares of common stock will be sold at a discount ranging from 3.8% to 6.4%, which varies based on a threshold price set by the Company. Upon each sale of the Company's common stock to Azimuth under the agreement, the Company has also agreed to pay Reedland Capital Partners a placement fee equal to 1.1% of the aggregate dollar amount of common stock purchased by Azimuth. Azimuth is not required to purchase the Company's common stock when the price of the Company's common stock is below $2 per share. As of December 31, 2009, the Company has not sold any shares under this common stock purchase agreement.
NOTE 12. RELATED PARTY TRANSACTIONS
Retirement of John W. Fara, Ph.D.
In August 2007, John W. Fara, Ph.D. retired from his positions as President, Chief Executive Officer and Chairman of the Company. Dr. Fara continued to serve as a member of the Company's Board of Directors until May 2008. The Company entered into a consulting agreement with Dr. Fara in August 2007, pursuant to which Dr. Fara provided consulting services to the Company through December 31, 2009. From August 2007 through December 31, 2008, the Company paid Dr. Fara $20,833 per month for his consulting services, reimbursed Dr. Fara for COBRA and life insurance premiums. For the years ended December 31, 2009, 2008 and 2007 the Company incurred expense of approximately zero, $250,000 and $90,000, respectively, associated with this consulting agreement.
114
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 12. RELATED PARTY TRANSACTIONS (Continued)
During the period of his consultancy, Dr. Fara continued to vest in all of his currently unvested stock options, and his vested stock options remained exercisable. For the years ended December 31, 2009, 2008 and 2007, the Company recognized approximately $30,000, $74,000 and $67,000, respectively, in stock compensation expense associated with these awards.
Retirement of John F. Hamilton
In October 2007, John F. Hamilton retired from his position as Vice President, Finance and Chief Financial Officer of the Company. The Company entered into a letter agreement with the Mr. Hamilton, pursuant to which the Company made a $190,000 lump sum payment to Mr. Hamilton. Options to purchase the Company's common stock held by Mr. Hamilton on retirement were cancelled in October 2007 and were exchanged for 100,000 fully vested shares of common stock pursuant to the Company's 2004 Equity Incentive Plan.
The Company treated the issuance of fully vested shares of common stock in exchange for the cancellation of Mr. Hamilton's options as a modification of the terms of the cancelled options for accounting purposes. The Company recognized approximately $364,000 of stock-based compensation expense related to this transaction in 2007, which represented the remaining unrecognized compensation costs associated with Mr. Hamilton's cancelled options on the date of settlement.
The Company also entered into a consulting agreement with Mr. Hamilton, and paid Mr. Hamilton $25,667 per month for his consulting services from October 10, 2007 through October 10, 2008. For the years ended December 31, 2009, 2008 and 2007, the Company incurred expense of approximately zero, $248,000 and $64,000, respectively, associated with this consulting agreement.
NOTE 13. REDUCTION IN FORCE
In October 2009, the Company reduced its workforce by six employees, or approximately 7% of its full-time staff to align its workforce with its anticipated staffing needs. The total cost of the workforce reduction was approximately $0.4 million, which consists of cash payments for severance, medical insurance and outplacement services and was recognized as expense during the year ended December 31, 2009. Severance expense of approximately $0.3 million and $0.1 million was recognized in research and development expense and selling, general and administrative expense, respectively, for the year ended December 31, 2009.
In September 2007, the Company reduced its workforce by 25 employees, or approximately 25% of its full-time staff, to conserve cash and align its workforce with its anticipated staffing needs. The total cost of the workforce reduction was approximately $0.7 million, which consisted of cash payments for severance, medical insurance and outplacement services and was recognized as expense during the year ended December 31, 2007. Severance expense of approximately $0.4 million and $0.3 million was recognized in research and development expense and selling, general and administrative expense, respectively, for the year ended December 31, 2007.
115
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 14. INCOME TAXES
The provision for (benefit from) income taxes consists of the following (in thousands):
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2009 | 2008 | 2007 | |||||||
Current: |
||||||||||
Federal |
$ | (18 | ) | $ | (5 | ) | $ | 470 | ||
State |
(2 | ) | 1 | 118 | ||||||
Foreign |
5 | 5 | 4 | |||||||
|
(15 | ) | 1 | 592 | ||||||
Deferred: |
||||||||||
Federal |
— | — | — | |||||||
State |
— | — | — | |||||||
Foreign |
— | — | — | |||||||
|
— | — | — | |||||||
Total provision for (benefit from) income taxes |
$ | (15 | ) | $ | 1 | $ | 592 | |||
A reconciliation of income taxes at the statutory federal income tax rate to the actual tax rate included in the statements of operations is as follows (in thousands):
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2009 | 2008 | 2007 | |||||||
Tax at federal statutory rate |
$ | (7,488 | ) | $ | (5,202 | ) | $ | 16,936 | ||
State tax, net of federal benefit |
(2 | ) | 1 | 118 | ||||||
Foreign tax |
5 | 5 | 4 | |||||||
Net operating losses not benefited (benefited) |
6,774 | 4,687 | (17,364 | ) | ||||||
Federal—AMT |
— | — | 470 | |||||||
Other |
696 | 510 | 428 | |||||||
|
$ | (15 | ) | $ | 1 | $ | 592 | |||
The Company's tax provisions for the year ended December 31, 2009 and 2008 is due to foreign taxes withheld on license revenue related to the Company's agreement with LG by the Republic of Korea offset by refundable credits. No provisions for federal income taxes have been recorded for the years ended December 31, 2009 and 2008 due to operating losses. For the year ended December 31, 2007, federal and state income tax provisions were based on the Company's alternative minimum tax, as the Company utilized net operating loss carryforwards.
As of December 31, 2009, the Company has net operating loss carryforwards for federal income tax purposes of approximately $94.0 million, which expire in the years 2021 through 2029 and federal research and development tax credits of approximately $5.7 million which expire in the years 2011 through 2029. Net operating loss carryforwards for state income tax purposes are approximately $77.0 million, which expire in the years 2013 through 2029 and state research and development tax credits are approximately $5.0 million which have no expiration date. The Company has federal and state alternative minimum tax credit carryforwards of $0.5 million and $0.1 million, respectively, which
116
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 14. INCOME TAXES (Continued)
have no expiration date. Additionally, the Company has foreign tax credit carryforwards of $0.2 million, which begin to expire in 2014.
Utilization of the Company's net operating loss and credit carryforwards may be subject to a substantial annual limitation due to ownership change limitations provided by the Internal Revenue Code of 1986 and similar state provisions. The annual limitation may result in the expiration of net operating losses and credits before utilization.
Deferred income taxes reflect the net tax effects of net operating loss and tax credit carryovers and temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of the Company's deferred tax assets are as follows (in thousands):
| |
Year Ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2009 | 2008 | |||||
Deferred Tax Assets: |
|||||||
Net operating loss carryforwards |
$ | 35,500 | $ | 33,600 | |||
Tax carryforwards |
7,100 | 6,100 | |||||
In-process research and development |
2,300 | 2,700 | |||||
Capitalized research expenses |
700 | 1,100 | |||||
Deferred revenue |
13,900 | 8,900 | |||||
Other, net |
4,900 | 2,900 | |||||
Total deferred tax assets |
64,400 | 55,300 | |||||
Valuation allowance for deferred tax assets |
(64,400 | ) | (55,300 | ) | |||
Deferred tax assets, net |
$ | — | $ | — | |||
Realization of deferred tax assets is dependent upon future earnings, if any, the timing and amount of which are uncertain. Accordingly, the net deferred tax assets have been fully offset by a valuation allowance. The valuation allowance increased by $9.1 million during the year ended December 31, 2009 and decreased by $1.4 million and $19.3 million during the years ended December 31, 2008 and 2007, respectively.
At December 31, 2009, the portion of the federal and state net operating loss carryforwards related to stock option deductions is approximately $2.2 million, which is not included in the Company's gross or net deferred tax assets, and will be recorded to equity when it has the effect of reducing taxes payable.
On January 1, 2007, the Company adopted the provisions for Accounting for Uncertainty in Income Taxes, which clarifies the accounting for uncertainty in tax positions. The Company did not recognize any adjustment to the liability for uncertain tax positions and therefore did not record any adjustment to the beginning balance of retained earnings on the balance sheet.
The Company files income tax returns in the United States federal jurisdiction and in various states, and the tax returns filed for the years 1995 through 2009 have not been examined and the applicable statutes of limitation have not expired with respect to those returns. Because of net
117
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 14. INCOME TAXES (Continued)
operating loss and research credit carryovers, substantially all of the Company's tax years remain open to examination.
Interest and penalties, if any, related to unrecognized tax benefits, would be recognized as income tax expense by the Company. The Company did not have any accrued interest or penalties associated with any unrecognized tax benefits.
The following table summarizes the activity related to our unrecognized tax benefits for the two years ended December 31, 2009 (in thousands):
Unrecognized tax benefits—January 1, 2008 |
$ | 2,892 | ||
Gross decreases—prior year tax positions |
(384 | ) | ||
Gross increases—current year tax positions |
286 | |||
Settlements with taxing authorities |
— | |||
Expiration of statute of limitations |
— | |||
Unrecognized tax benefits—December 31, 2008 |
$ | 2,794 | ||
Gross decreases—prior year tax positions |
(28 | ) | ||
Gross increases—current year tax positions |
460 | |||
Settlements with taxing authorities |
— | |||
Expiration of statute of limitations |
— | |||
Unrecognized tax benefits—December 31, 2009 |
$ | 3,226 | ||
Though the Company's unrecognized tax benefits may change during the next year for items that arise in the ordinary course of business, the Company does not expect any such change to be significant.
NOTE 15. SUMMARIZED QUARTERLY DATA (UNAUDITED)
The following tables set forth certain statements of operations data for each of the eight quarters beginning with the quarter ended March 31, 2008 through the quarter ended December 31, 2009 (in thousands). This quarterly information is unaudited, but has been prepared on the same basis as the annual financial statements and, in the opinion of management, reflects all adjustments, consisting only of normal recurring adjustments necessary for a fair representation of the information for the periods
118
DEPOMED, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
NOTE 15. SUMMARIZED QUARTERLY DATA (UNAUDITED) (Continued)
presented. Operating results for any quarter are not necessarily indicative of results for any future period.
| |
2009 Quarter Ended | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
March 31 | June 30 | September 30 | December 31 | |||||||||
Product sales |
$ | 6,840 | $ | 8,408 | $ | 9,859 | $ | 9,987 | |||||
Total revenues |
9,872 | 11,608 | 23,014 | 13,234 | |||||||||
Gross margin on product sales |
5,808 | 7,179 | 8,492 | 8,358 | |||||||||
Income (loss) from operations |
(10,183 | ) | (9,590 | ) | 1,416 | (3,715 | ) | ||||||
Net income (loss) |
(10,155 | ) | (9,615 | ) | 1,373 | (3,611 | ) | ||||||
Net income (loss) applicable to common stock shareholders |
(10,155 | ) | (9,615 | ) | 1,373 | (3,611 | ) | ||||||
Basic net income (loss) per share |
$ | (0.20 | ) | $ | (0.19 | ) | $ | 0.03 | $ | (0.07 | ) | ||
Diluted net income (loss) per share |
$ | (0.20 | ) | $ | (0.19 | ) | $ | 0.03 | $ | (0.07 | ) | ||
| |
2008 Quarter Ended | ||||||||||||
March 31 |
June 30 |
September 30 |
December 31 |
||||||||||
Product sales |
$ | 5,226 | $ | 5,519 | $ | 13,011 | $ | 7,295 | |||||
Total revenues |
5,701 | 6,315 | 14,111 | 8,715 | |||||||||
Gross margin on product sales |
4,017 | 4,557 | 10,615 | 6,090 | |||||||||
Gain on litigation settlement |
— | (7,500 | ) | — | — | ||||||||
Income (loss) from operations |
(8,084 | ) | 2,932 | (533 | ) | (11,409 | ) | ||||||
Net income (loss) |
(7,281 | ) | 3,480 | (271 | ) | (11,229 | ) | ||||||
Net income (loss) applicable to common stock shareholders |
(7,456 | ) | 3,300 | (454 | ) | (11,232 | ) | ||||||
Basic net income (loss) per share |
$ | (0.16 | ) | $ | 0.07 | $ | (0.01 | ) | $ | (0.22 | ) | ||
Diluted net income (loss) per share |
$ | (0.16 | ) | $ | 0.07 | $ | (0.01 | ) | $ | (0.22 | ) | ||
NOTE 16. SUBSEQUENT EVENTS
We evaluated our subsequent events through March 8, 2010, when the financial statements were issued.
Acquisition of Solvay by Abbott
Abbott acquired the pharmaceutical business of Solvay in a transaction that closed in February 2010. Accordingly, Abbott is now responsible for the Company's DM-1796 license arrangement with Solvay, either directly or through a subsidiary.
119
SCHEDULE II: VALUATION AND QUALIFYING ACCOUNTS
(in thousands)
| |
|
Additions | |
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Description
|
Balance at Beginning of Year |
Charged as a Reduction to Revenue(1) |
Change in Deferred Revenue(1) |
Deductions(2) | Balance at End of Year |
||||||||||||
Sales & return allowances, discounts, chargebacks and rebates: |
|||||||||||||||||
Year ended December 31, 2009 |
$ | 3,742 | $ | 11,284 | $ | 21 | $ | (7,188 | ) | $ | 7,859 | ||||||
Year ended December 31, 2008 |
468 | 7,355 | (525 | ) | (3,556 | ) | 3,742 | ||||||||||
Year ended December 31, 2007 |
371 | 1,727 | (314 | ) | (1,316 | ) | 468 | ||||||||||
- (1)
- Additions
to sales discounts and allowances are recorded as a reduction of deferred revenue until such time revenue is recognized.
- (2)
- Deductions to sales discounts and allowances relate to discounts or allowances actually taken or paid.
120