Attached files
| file | filename |
|---|---|
| EX-32 - ACURA PHARMACEUTICALS, INC | v175435_ex32.htm |
| EX-31.1 - ACURA PHARMACEUTICALS, INC | v175435_ex31-1.htm |
| EX-31.2 - ACURA PHARMACEUTICALS, INC | v175435_ex31-2.htm |
| EX-10.30 - ACURA PHARMACEUTICALS, INC | v175435_ex10-30.htm |
UNITED
STATES
SECURITIES
AND EXCHANGE COMMISSION
Washington,
D.C. 20549
FORM
10-K
(Mark
One)
|
x
|
ANNUAL REPORT
PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 FOR
THE FISCAL YEAR ENDED DECEMBER 31,
2009
|
Or
|
¨
|
TRANSITION
REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF
1934
|
For
the transition period from _____ to _____
Commission
file number 1-10113
ACURA
PHARMACEUTICALS, INC.
(Exact
name of registrant as specified in its charter)
|
New
York
|
11-0853640
|
|
(State
or other jurisdiction of Incorporation or organization)
|
(I.R.S.
Employer Identification No.)
|
|
616
N. North Court, Suite 120, Palatine, Illinois
|
60067
|
|
(Address
of principal executive office)
|
(Zip
code)
|
Registrant's
telephone number, including area code: 847 705 7709
Securities
registered pursuant to section 12(b) of the Act:
Common
Stock, par value $0.01 per share
Securities
registered pursuant to section 12(g) of the Act:
(Title of
Class)
None
Indicate
by check mark if the registrant is a well-known seasoned issuer, as defined in
Rule 405 of the Securities Act. Yes o No x
Indicate
by check mark if the registrant is not required to file reports pursuant to
Section 13 or Section 15(d) of the Act. Yes o No x
Indicate
by check mark whether the registrant: (1) has filed all reports required to be
filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the
preceding 12 months (or for such shorter period that the registrant was required
to file such reports), and (2) has been subject to such filing requirements for
the past 90 days. Yes x No o
Indicate
by check mark whether the registrant has submitted electronically and posted on
its corporate Web site, if any, every Interactive Data File required to be
submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this
chapter) during the preceding 12 months (or for such shorter period that the
registrant was required to submit and post such files). Yes o No o
Indicate
by check mark if disclosure of delinquent filers pursuant to Item 405 of
Regulation S-K (§229.405 of this chapter is not contained herein, and will not
be contained, to the best of registrant's knowledge, in definitive proxy or
information statements incorporated by reference in Part III of this Form 10-K
or any amendment to this Form 10-K. x
Indicate
by check mark whether the registrant is a large accelerated filer, an
accelerated filer, a non-accelerated filer, or a smaller reporting
company.
o Large Accelerated
Filer, x
Accelerated Filer, ¨ Non-Accelerated Filer,
¨ Smaller Reporting
Company.
Indicate
by check mark whether the registrant is a shell company (as defined in Rule
12b-2 of the Exchange Act). Yes o No x
Based on
the last sale price on the NASDAQ Capital Market of the Common Stock
on June 30, 2009 ($5.98) (the last
business day of the registrant's most recently completed second fiscal quarter),
the aggregate market value of the voting stock held by non-affiliates of the
registrant was approximately $57,306,507.
As of
February 28, 2010, the registrant had 43,728,626 shares of Common Stock, par
value $0.01, outstanding.
Documents
incorporated by reference: None
Acura
Pharmaceuticals, Inc.
Form
10-K
For
the Fiscal Year Ended December 31, 2009
Tablet
of Contents
|
PAGE
|
||||
|
PART
I
|
||||
|
Item
1.
|
Business
|
3
|
||
|
Item
1A.
|
Risk
Factors
|
21
|
||
|
Item
1B.
|
Unresolved
Staff Comments
|
32
|
||
|
Item
2.
|
Properties
|
32
|
||
|
Item
3.
|
Legal
Proceedings
|
32
|
||
|
Item
4.
|
Reserved
|
33
|
||
|
PART
II
|
33
|
|||
|
Item
5.
|
Market
for Registrant's Common Equity, Related Stockholder Matters and Issuer
Purchases of Equity Securities
|
33
|
||
|
Item
6.
|
Selected
Financial Data
|
34
|
||
|
Item
7.
|
Management's
Discussion and Analysis of Financial Condition and Results of
Operations
|
35
|
||
|
Item
7A.
|
Quantitative
and Qualitative Disclosures About Market Risk
|
43
|
||
|
Item
8.
|
Financial
Statements and Supplementary Data
|
43
|
||
|
Item
9.
|
Changes
in and Disagreement with Accountants on Accounting and Financial
Disclosure
|
43
|
||
|
Item
9A.
|
Controls
and Procedures
|
43
|
||
|
Item
9B.
|
Other
Information
|
46
|
||
|
PART
III
|
||||
|
Item
10.
|
Directors,
Executive Officers and Corporate Governance
|
46
|
||
|
Item
11.
|
Executive
Compensation
|
50
|
||
|
Item
12.
|
Security
Ownership of Certain Beneficial Owners and Management and Related
Stockholder Matters
|
70
|
||
|
Item
13.
|
Certain
Relationships and Related Transactions, and Director
Independence
|
72
|
||
|
Item
14.
|
Principal
Accountant Fees and Services
|
74
|
||
|
PART
IV
|
||||
|
Item
15.
|
Exhibits
and Financial Statement Schedules
|
74
|
||
|
Signatures
|
75
|
|||
|
Index
to Financial Statements
|
F-1
|
|||
2
Forward-Looking
Statements
Certain
statements in this Report constitute "forward-looking statements" within the
meaning of the Private Securities Litigation Reform Act of 1995. Such
forward-looking statements involve known and unknown risks, uncertainties and
other factors which may cause our actual results, performance or achievements to
be materially different from any future results, performance, or achievements
expressed or implied by such forward-looking statements. The most significant of
such factors include, but are not limited to, our ability and the ability of
King Pharmaceuticals Research and Development, Inc. (“King”) (to whom we have
licensed our Aversion®
Technology for certain opioid analgesic products in the United States, Canada
and Mexico) and the ability other pharmaceutical companies, if any, to whom we
may license our Aversion®
Technology, to obtain necessary regulatory approvals and commercialize products
utilizing our Aversion® and
Impede™ Technologies, the ability to avoid infringement of patents, trademarks
and other proprietary rights of third parties, and the ability to fulfill the
U.S. Food and Drug Administration’s (“FDA”) requirements for approving our
product candidates for commercial manufacturing and distribution in the United
States, including, without limitation, the adequacy of the results of the
laboratory and clinical studies completed to date and the results of laboratory
and clinical studies we may complete in the future, to support FDA approval of
our product candidates, the adequacy of the development program for our product
candidates, including whether additional clinical studies will be required to
support FDA approval of our product candidates, changes in regulatory
requirements, adverse safety findings relating to our product candidates, the
risk that the FDA may not agree with our analysis of our clinical studies and
may evaluate the results of these studies by different methods or conclude that
the results of the studies are not statistically significant, clinically
meaningful or that there were human errors in the conduct of the studies or the
risk that further studies of our product candidates are not positive or
otherwise do not support FDA approval, whether or when we are able to obtain FDA
approval of labeling for our product candidates for the proposed indications or
for abuse deterrent features, whether our product candidates will ultimately
deter abuse in commercial settings, and the uncertainties inherent in
scientific research, drug development, laboratory and clinical trials and the
regulatory approval process. Other important factors that may also
affect future results include, but are not limited to: our ability to attract
and retain skilled personnel; our ability to secure and protect our patents,
trademarks and other proprietary rights; litigation or regulatory action that
could require us to pay significant damages or change the way we conduct our
business; our ability to compete successfully against current and future
competitors; our dependence on third-party suppliers of raw materials; our
ability to secure U.S. Drug Enforcement Administration ("DEA") quotas and source
the active ingredients for our products in development; difficulties or delays
in conducting clinical trials for our product candidates or in the commercial
manufacture and supply of our products; and other risks and uncertainties
detailed in this Report. When used in this Report, the words "estimate,"
"project," "anticipate," "expect," "intend," "believe," and similar expressions
identify forward-looking statements.
PART
I
ITEM 1. BUSINESS
Overview
We are a
specialty pharmaceutical company engaged in research, development and
manufacture of product candidates intended to provide abuse deterrent features
and benefits utilizing our proprietary Aversion® and Impede™
Technologies. Our Aversion®
Technology opioid analgesic product candidates are intended to effectively
relieve pain while simultaneously discouraging common methods of opioid product
misuse and abuse, including the:
|
|
·
|
intravenous
injection of dissolved tablets or
capsules;
|
|
|
·
|
nasal
snorting of crushed tablets or capsules;
and
|
|
|
·
|
intentional
swallowing of excess quantities of tablets or
capsules.
|
All of
our opioid product candidates utilize Aversion® Technology and are covered by
two issued U.S. patents, which in combination with our anticipated product
labeling and drug product listing strategies are anticipated to provide our
opioid products with protection from generic competition through the expiration
of our patents in 2025.
In addition to Acurox®, our lead
product candidate, we (and/or our licensee, King) are developing Vycavert®
(hydrocodone bitartrate/niacin/APAP), Acuracet®
(oxycodone HCl/niacin/ APAP) and additional undisclosed opioid product
candidates utilizing our Aversion® Technology. Four opioid product
candidates are licensed to King under our License, Development and
Commercialization Agreement dated October 30, 2007. We are also
developing an undisclosed benzodiazepine tablet product candidate utilizing
Aversion® Technology.
3
In addition to our Aversion®
Technology, as part of our continuing research efforts we are investigating and
developing novel mechanisms to incorporate abuse deterrent characteristics into
abused and misused pharmaceutical products. In this regard we are
engaged in initial laboratory testing of a new product candidate developed with
our novel Impede™ Technology. Impede™ Technology is primarily
intended to inhibit the conversion of pseudoephedrine HCl (a legally
available nasal decongestant) into methamphetamine (an illicit and frequently
abused drug).
Acurox® Tablets,
our lead product candidates, is an orally administered immediate release tablet
containing oxycodone HCl as its sole active analgesic ingredient. On
December 30, 2008, we submitted a 505(b)(2) New Drug Application (“NDA”) for
Acurox®
Tablets to the FDA and on June 30, 2009 we received from the FDA a
Complete Response Letter (“CRL”). The CRL raised issues regarding the
potential abuse deterrent benefits of Acurox®. On
September 2, 2009 we and King met with the FDA and agreed that the data and
evidence supporting the Acurox® Tablets
NDA would be presented to an FDA Advisory Committee. The FDA has not yet
scheduled the Advisory Committee meeting to review the NDA for Acurox® Although
the FDA stated that no new Acurox® clinical
trials are required at this time, we and King are conducting an additional
clinical study (see “Acurox® Tablets
Development Program” below) to further support the abuse deterrent features of
Acurox®.
The misuse and abuse of pharmaceutical
products in general, and opioid analgesics in particular, is a significant
societal problem described as epidemic in nature. It is estimated
that 75 million people in the U.S. suffer from pain, and, according to U.S.
government surveys, 34.9 million people, or more than 10% of the U.S.
population, have used prescription opioid analgesics non-medically at some point
in their lifetime. We expect our Aversion®
Technology opioid product candidates to compete primarily in the market for
immediate release opioid products (“IR Opioid Products”) which are commonly
prescribed for relief of pain for durations generally less than 30
days. In 2009, IMS Health reported 252 million prescriptions
dispensed for opioid analgesic tablets and capsules, of which approximately 236
million were for IR Opioid Products and 16 million were for extended release
opioid tablet and capsule products (“ER Opioid Products”) which are commonly
prescribed for relief of chronic pain for durations ranging from several weeks
to several months or longer. We have contracted, through an independent market
research firm, numerous market research studies including two which surveyed 401
and 435 opioid analgesic prescribing U.S. based physicians,
respectively. These studies revealed that physicians are keenly aware
of opioid analgesic abuse and are personally concerned with the potential impact
of drug abuse on their respective medical practices. Our study of 401
physicians indicated that of the prescriptions likely to be written for our
product candidates that utilize the analgesic oxycodone, 59% will be switched
from immediate release products containing either hydrocodone or oxycodone, with
the remaining 41% being
switched from other currently marketed opioid analgesic products such as
codeine, propoxyphene, morphine, and tramadol. Ninety-four percent
(94%) of 401 physicians surveyed indicated they would either prescribe one of
the Aversion®
Technology products profiled in the market research questionnaire for one
of their last five patients receiving an opioid prescription or they are aware
of a patient in their practice for whom Aversion®
Technology opioid analgesic products would be an appropriate
choice.
We have
established and intend to pursue future strategic alliances and licensing
agreements with pharmaceutical companies to enhance our ability to develop and
commercialize our product candidates. In October 2007, we entered into a
License, Development and Commercialization Agreement with King to develop and
commercialize certain opioid analgesic products utilizing our proprietary
Aversion®
Technology, including Acurox®
Tablets. The King Agreement initially provided King with an exclusive
license in the United States, Canada and Mexico (the “King Territory”) to
Acurox® Tablets
and Acuracet®
(oxycodone HCl/niacin/acetaminophen) Tablets, and an option to license future
opioid analgesic product candidates utilizing our Aversion®
Technology in the King Territory. In May and December 2008, King
exercised its option and licensed an undisclosed opioid analgesic tablet product
and Vycavert® (hydrocodone bitartrate/niacin/acetaminophen) Tablets,
respectively. Under the terms of the King Agreement, King made an
upfront cash payment to us of $30 million. As of February 28, 2010,
we had received an additional $26.2 million from King in the form of milestone
payments, option fees and reimbursement for research and development expenses.
In addition, we are eligible for future regulatory and sales milestone payments,
reimbursement for certain research and development expenses and royalties on
combined annual net sales of all products commercialized under the King
Agreement.
4
We
conduct research, development, laboratory, manufacturing, and warehousing
activities at our operations facility in Culver, Indiana and lease an
administrative office in Palatine, Illinois. In addition to internal
capabilities and activities, we engage numerous clinical research organizations
(“CROs”) with expertise in regulatory affairs, clinical trial design and
monitoring, clinical data management, biostatistics, medical writing, laboratory
testing and related services. Such CROs perform, under our direction,
development and regulatory services relating to our Aversion® and
Impede™ Technology product candidates.
Our
Strategy
Our goal
is to become a leading specialty pharmaceutical company focused on addressing
the growing societal problem of pharmaceutical drug abuse by developing
a broad portfolio of products with abuse deterrent features and
benefits. Specifically, we intend to:
|
|
·
|
Capitalize on our Experience
and Expertise in the Research and Development of Pharmaceutical Products
with Abuse Deterrent Features and Benefits. Our strategy
is to facilitate rapid product development and minimize risk by utilizing
active pharmaceutical ingredients with proven safety and efficacy profiles
with known potential for abuse, and develop new products utilizing our
proprietary Aversion®
and Impede™
Technologies using the FDA’s 505(b)(2) and other regulatory
processes.
|
|
|
·
|
Emerge as a Leader in
Developing and Commercializing Products with Abuse Deterrent Features and
Benefits Able to Uniquely Address the Growing Problem of Abuse of
Prescription Drugs. We believe that Acurox®
and our other opioid product candidates in development have
demonstrated that Aversion®
Technology allows products to provide the analgesic benefit they were
intended to deliver, while simultaneously having features that are
intended to deter misuse and abuse. We believe these benefits
will be attractive to physicians, third party payers, and public advocacy
groups sensitive to the problem of prescription drug
abuse.
|
|
|
·
|
Optimize Shareholder Value and
Temper Risk by Licensing our Product Candidates to Strategically Focused
Pharmaceutical Companies in the U.S. and Other Geographic
Territories. On October 30, 2007, we and King entered
into the King Agreement to develop and commercialize in the United States,
Canada and Mexico opioid analgesic products utilizing Aversion®
Technology, including Acurox ®
Tablets and Acuracet®
Tablets. We believe opportunities exist to enter into similar
agreements with other partners for these same opioid products outside the
King Territory, and in the United States and worldwide for developing
additional Aversion®
Technology and Impede™
Technology product candidates for other abusable drugs such as
tranquilizers, stimulants, sedatives and decongestants. By
licensing our product candidates to strategically focused companies with
expertise and infrastructure in commercialization of pharmaceuticals, we
are able to leverage our expertise, intellectual property rights and
Aversion®
and Impede™
Technologies without the need to build costly sales and manufacturing
infrastructure. We anticipate that our future revenue, if any,
will be derived from milestone and royalty payments related to the
commercialization of products utilizing our Aversion®
and Impede™
Technologies.
|
|
|
·
|
Apply our Aversion® and Impede™
Technologies to
Non-Opioid Drugs that are Subject to Abuse. We intend to
first develop a portfolio of opioid analgesic products, and thereafter we
intend to expand to other pharmaceutical product categories containing
potentially abusable active ingredients such as tranquillizers (brand
products such as Valium®,
Xanax®,
Klonopin® and Ativan®),
stimulants (brand products such as Dexedrine®,
Adderall®,
Ritalin®
and Concerta®),
sedatives (brand products such as Nembutal®,
Butisol®,
and Seconal®)
and decongestants (brand products such as Sudafed®, Zyrtec-D®, Allegra-D®,
and Clarinex-D®). These products, like the opioid analgesics on
which we are currently focused, may also be prone to misuse and
abuse.
|
|
|
·
|
Maintain our Efficient
Internal Cost Structure. We maintain a streamlined and
highly efficient cost structure focused on: (i) selection, formulation
development, laboratory evaluation, manufacture, quality assurance and
stability testing of certain finished dosage form product candidates; (ii)
development and prosecution of our patent applications; and (iii)
negotiation and execution of license and development agreements with
strategically focused pharmaceutical companies. By outsourcing
the high cost elements of our product development and commercialization
process, we believe that we substantially reduce required fixed overhead
and capital investment and thereby reduce our business risk. We
currently do not intend to use a physician focused sales force
to commercialize products on our
own.
|
5
Aversion®
Technology Opioid Product Candidates in Development
Aversion®
Technology opioid analgesic product candidates which have demonstrated Proof of
Concept1 are set
forth in the table below.
|
Our Product Candidates
|
Stage of Development
|
|
|
Acurox® (oxycodone
HCl/niacin) Tablets
|
NDA
submitted to FDA on 12/30/08; Complete Response Letter received 6/30/09;
FDA Advisory Committee meeting pending scheduling.
|
|
|
Acuracet®
(oxycodone HCl/niacin/APAP) Tablets
|
Investigational
New Drug Application ("IND") filed with FDA and active beginning
6-1-08. Testing for NDA submission in
progress
|
|
|
Vycavert®
(hydrocodone bitartrate/niacin/APAP) Tablets
|
Proof
of Concept complete. Testing for IND filing in
progress
|
|
|
4th
(undisclosed opioid analgesic) Tablets
|
Proof
of Concept complete.
|
|
|
5th
(undisclosed opioid analgesic) Tablets
|
Proof
of Concept complete
|
|
|
6th
(undisclosed opioid analgesic) Tablets
|
Proof
of Concept complete
|
|
|
7th
(undisclosed opioid analgesic) Tablets
|
Proof
of Concept complete
|
|
|
8th
(undisclosed opioid analgesic) Tablets
|
Proof
of
Concept complete
|
1 Proof of
concept is attained upon demonstration of certain product stability and
bioavailability parameters defined in the King Agreement. Refer to description
of the King Agreement in this Report. With one exception, King has either
licensed or has an option to license all opioid product candidates listed above
in the U.S., Canada and Mexico.
Aversion®
Technology Overview
Aversion®
Technology is a proprietary platform technology providing abuse deterrent
features and benefits to orally administered pharmaceutical drug products
containing potentially abusable active ingredients. Our focus has
been to utilize our Aversion®
Technology with opioid analgesics administered in tablet form. In
addition, we believe Aversion®
Technology is a versatile technology which may be applicable to non-opioid
active ingredients subject to abuse and administered in tablet or capsule form,
including tranquilizers, sedatives and stimulants (See “Aversion® Technology
Non-Opioid Product Candidates in Development” below).
Aversion®
Technology opioid analgesic product candidates include a unique composition of
active and inactive pharmaceutical ingredients. The opioid active
ingredients are intended to provide effective relief from pain while the
proprietary mixture of inactive ingredients provide non-therapeutic
functionality. When dissolved in water or other solvents, the functional
inactive ingredients quickly form a viscous gel, which increases the difficulty
of extracting the opioid active ingredient in a form and volume suitable for
injection. In addition, the combination of functional inactive
ingredients is intended to induce nasal passage discomfort and disliking effects
if the tablets are pulverized and snorted. Aversion®
Technology opioid product candidates may also include niacin, an active
ingredient in vitamins, cholesterol reducers and nutritional supplements, in
amounts determined by us to be well tolerated when our product candidates are
administered at recommended doses but which are intended to induce temporary
disliking effects as increasing numbers of tablets are swallowed in excess of
the recommended analgesic dose. When Aversion®
Technology is utilized, it is intended that the resulting product provides
the same therapeutic benefits as the non Aversion®
Technology product, while simultaneously discouraging the most common methods of
pharmaceutical product misuse and abuse.
Intended
to Deter I.V. Injection of Opioids Extracted from Dissolved Tablets
Prospective
drug abusers may attempt to dissolve currently marketed opioid-containing
tablets or capsules in water, alcohol, or other common solvents, filter the
dissolved solution, and then inject the resulting fluid intravenously to obtain
euphoric effects. In product candidates utilizing Aversion®
Technology, extracting the active ingredient using generally available solvents,
including water or alcohol, into a volume and form suitable for intravenous
(“I.V.”) injection, converts the tablet into a viscous gel mixture and traps the
active ingredient in the gel. Additionally, it is not possible,
without difficulty, to draw this viscous gel through a needle into a syringe for
I.V. injection. We believe that this gel forming feature will inhibit
prospective I.V. drug abusers from extracting and injecting opioid active
ingredients from product candidates developed utilizing Aversion®
Technology.
6
Intended
to Deter Nasal Snorting
Prospective
drug abusers may crush or grind currently marketed pharmaceutical
opioid-containing tablets or capsules and snort the resulting
powder. The abused active ingredient in the powder is absorbed
through the lining of the nasal passages providing the abuser with a rapid onset
of euphoric effects. Aversion®
Technology products are intended to discourage nasal snorting by burning and
irritating the nasal passages of a prospective drug abuser who crushes and
snorts such products. We believe products which utilize Aversion®
Technology will inhibit prospective nasal drug abusers from snorting crushed
tablets.
Intended
to Deter Swallowing Excess Quantities of Tablets
Niacin,
an active ingredient in vitamins, cholesterol reducers and nutritional
supplements, is included in the majority of our opioid analgesic product
candidates utilizing Aversion®
Technology. We believe that should a person swallow excess quantities
of tablets utilizing Aversion®
Technology with niacin they will experience disliking symptoms,
including intense flushing, itching, sweating and/or chills, headache and a
general feeling of discomfort as a result of the increasing dose of
niacin. It is expected that these niacin-induced disliking symptoms
will begin approximately 10 to 15 minutes after the excess dose is swallowed and
will dissipate approximately 75 to 90 minutes later. In addition, we
believe it is generally recognized by physicians, nurses, and other health care
providers that niacin has a well established safety profile in long term
administration at doses far exceeding the amounts in each product candidate
utilizing Aversion®
Technology with niacin. We believe the undesirable niacin effects at
escalating doses will not prevent, but are expected to deter, swallowing excess
quantities of product candidates utilizing Aversion® Technology with
niacin.
U.S.
Market Opportunity for Opioid Analgesic Products Utilizing Aversion® Technology
The
misuse and abuse of prescription drug products in general, and opioid analgesics
in particular, is a significant societal problem that has been described as
epidemic in nature by Joseph A. Califano, Jr., Chairman and President, National
Center for Addiction and Substance Abuse at Columbia University, July
2005. Results from the 2008 National Survey on Drug Use
and Health, estimated that 34.9 million people, or more than 10% of the
population, have used prescription opioid analgesics non-medically at some point
in their lifetime. In addition, it is estimated that more than 75
million people in the U.S. suffer from pain, which is more than the number of
people with diabetes, heart disease and cancer combined. For many
pain sufferers, opioid analgesics provide their only pain relief. As
a result, opioid analgesics are among the largest prescription drug classes in
the U.S. with over 252 million tablet and capsule prescriptions dispensed in
2009 of which approximately 236 million were for IR Opioid Products and 16
million were for ER Opioid Products. However, physicians and
other health care providers at times are reluctant to prescribe opioid
analgesics for fear of misuse, abuse, and diversion of legitimate prescriptions
for illicit use.
We expect
our Aversion® Technology opioid product candidates to compete primarily in the
IR Opioid Product segment of the US opioid analgesic market, a segment which has
grown at a 4% compounded annual rate over the last five years. On average, an IR
Opioid Product prescription contains approximately 57 tablets or
capsules. According to the 2008 National Survey on Drug Use
and Health, prescription drug abusers have supplanted abusers of all
illicit drugs except marijuana. Of these abused prescription
products, IR Opioid Products, which typically provide rapid onset of analgesia
and require dosing every 4 to 6 hours, comprise the vast majority of this abuse
compared with ER Opioid Products, which release their opioids gradually,
generally over a 12 to 24 hour period. Due to fewer identified
competitors and the significantly larger market for dispensed prescriptions for
IR Opioid Products compared to ER Opioid Products, we have initially focused on
developing IR Opioid Products utilizing Aversion® Technology.
According
to IMS Health, in 2009, sales in the IR Opioid Product segment, comprised of 97%
generic products, were $1.9 billion. Assuming the FDA approves
differentiated label claims of the abuse deterrent features and benefits of our
product candidates, of which no assurance can be given, we anticipate that our
Aversion®
Technology IR Opioid Products will be premium priced compared to generic
products resulting in rapid growth of sales in the IR Opioid Product market
segment.
7
Despite
considerable publicity regarding the abuse of OxyContin® Tablets
and other ER Opioid Products, U.S. government statistics suggest that far more
people have used IR Opioid Products non-medically than ER Opioid
Products. These statistics estimate that nearly 5 times as many
people have misused the IR Opioid Products Vicodin®,
Lortab® and
Lorcet®
(hydrocodone bitartrate/APAP) as have ever abused OxyContin®. We
estimate 60-95% of the 34.9 million lifetime US opioid abusers have
non-medically used the active ingredients in our IR Opioid product
candidates. As indicated in the following chart, the top five abused
opioid products are available only as IR Opioid Products.
Lifetime
Non-Medical Use of Selected Pain Relievers, Age 12 or Older: 2008
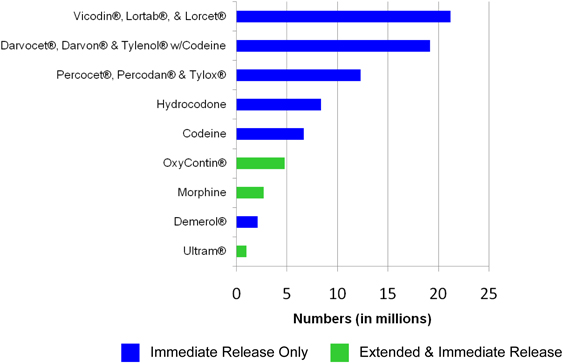
Source: SAMHSA,
Office of Applied Studies, 2008 National Survey on Drug Use and
Health.
We have
commissioned, through an independent market research firm, three physician
market research studies with 282, 401 and 435 opioid prescribing U.S. based
physicians, respectively. A sampling of key findings from these
approximately 1,100 physicians includes:
Physicians
are keenly aware of opioid analgesic abuse
|
|
·
|
The
282 physicians surveyed estimated on average that about one out of six
prescriptions for oxycodone and hydrocodone containing products are
abused.
|
|
|
·
|
94%
of the 435 physicians surveyed experienced at least one suspicious
incident regarding opioid abuse in the past month, while nearly 64%
experienced four or more discretely different incidents regarding opioid
abuse in the past month.
|
Physicians
are personally concerned with opioid abusers impact to their respective
practices
|
|
·
|
Following the survey of 282
physicians, the researchers concluded, “abuse [of opioid analgesics] is a
particular problem for physicians because many are not fully sure who is
abusing these opioids, and they view such abuse as a legal threat to their
practice.” “More than half [of the physicians surveyed] believe
their physician colleagues are more concerned about avoiding state review
[of their opioid prescribing habits] than meeting [professional
association] pain guidelines [for their
patients]”.
|
|
|
·
|
After
the survey of 435 physicians the researchers concluded “the primary motive
for prescribing the Aversion®
Technology product[s] is the concern physicians have about opioid abuse
and the threat it represents to their
practice.”
|
8
Physicians
are favorably inclined toward prescribing opioids with abuse deterrent features
and benefits
|
|
·
|
94%
of the 401 physicians surveyed indicated that they would either prescribe
one of the Aversion®
Technology products profiled in the market research questionnaire for one
of their last five patients receiving an opioid prescription or they are
aware of a patient in their practice for whom Aversion®
Technology products would be an appropriate
choice.
|
|
|
·
|
57%
of the 435 physicians indicated that their opioid analgesic prescribing
would increase if they were more certain they were not aiding
abusers.
|
|
|
·
|
Following
the survey of 401 physicians, the researchers concluded “these
[Aversion®
Technology oxycodone products] would disproportionately replace current
immediate release oxycodone [and oxycodone/acetaminophen] prescriptions,
but would also draw substantial volume from hydrocodone/acetaminophen
products.”
|
Overall,
we believe the availability of opioid analgesics with abuse deterrent features,
including products using our Aversion®
Technology, will greatly impact the selection of products used for relief of
pain. Our market research survey of the 401 physicians indicated that
of the prescriptions likely to be written for our product candidates that
utilize oxycodone, 59% will be switched from immediate release products
containing either oxycodone or hydrocodone, with the remaining 41% being
switched from other currently marketed opioid analgesic products such as
codeine, propoxyphene, morphine and tramadol.
A
majority of pharmaceutical products in the U.S. are paid for by third party
payers such as insurers, pharmacy benefit managers, self-insured companies and
the federal and state governments through Medicare, Medicaid and other
programs. We believe our product candidates will have to demonstrate
a clinical benefit to the patient and/or an economic benefit to third party
payers and/or a benefit to health care providers to receive favored
reimbursement status by the payers.
Independent
estimates have been made assessing the potentially significant cost impact of
prescription opioid abuse to insurers. An analysis of health and
pharmacy insurance claims between 1998 and 2002 for almost 2 million Americans
conducted by Analysis Group, Inc. and others indicated that enrollees with a
diagnosis of opioid abuse had average claims of approximately $14,000 per year
higher than an age-gender matched non-opioid abuse sample. A 2007
report by the Coalition Against Insurance Fraud inflated this excess cost per
patient to more than $16,000 for 2007, and by applying the U.S. government’s
estimated 4.4 million annual opioid abusers, concluded that opioid abuse could
costs health insurers up to $72.5 billion a year.
Acurox®
Tablets Development Program
We
submitted a 505(b)(2) NDA for Acurox®
Tablets to the FDA on December 30, 2008 and received a Complete Response Letter
(“CRL”) on June 30, 2009. On September 2, 2009 we and King met with
the FDA and agreed that the data and evidence supporting the Acurox® Tablets
NDA would be presented to an FDA Advisory Committee. The FDA has not yet
scheduled the Advisory Committee meeting to review the NDA for Acurox®.
The NDA
for Acurox® Tablets
includes results from numerous clinical and laboratory studies assessing the
efficacy and safety of Acurox® Tablets
and to demonstrate abuse deterrent features and benefits, including the data and
results from studies set forth in the table below.
9
|
Studies in the Acurox® Tablets 505(b)(2) NDA Submission
|
Summary of Results
|
|||
|
AP-ADF-101
Phase
I
|
Niacin
dose-response (0-75mg)
|
Identified
appropriate niacin dose in each Acurox® Tablet
|
||
|
AP-ADF-104
Phase
I
|
Bioequivalence
to non Aversion® Technology Reference Listed Drug
|
Acurox®
Tablets are bioequivalent to the Reference Listed Drug
|
||
|
AP-ADF-108
Phase
I
|
Single
dose linearity and food effect
|
Acurox®
Tablets demonstrate single dose linearity. Absorption is
delayed by food.
|
||
|
AP-ADF-109
Phase
I
|
Multi-dose
linearity
|
Acurox®
Tablets demonstrate multi-dose linearity
|
||
|
AP-ADF-106
Phase
I
|
Evaluate
effects of nasal snorting in subjects with a history of snorting and nasal
drug abuse
|
Refer
to summary in this Report
|
||
|
AP-ADF-103
Phase
II
|
Repeat
dose safety and tolerability
|
Confirmed
appropriate niacin dose in each Acurox® Tablet
|
||
|
AP-ADF-107
Phase
II
|
Niacin
dose-response (0-600mg)
|
Confirmed
appropriate niacin dose in each Acurox® Tablet
|
||
|
AP-ADF-102
Phase
II
|
Evaluate
relative dislike of oxycodone HCl/niacin versus oxycodone HCl
alone
|
Refer
to summary in this Report
|
||
|
AP-ADF-111
Phase
II
|
Evaluate
abuse liability of oxycodone HCl/niacin versus oxycodone HCl
alone
|
Refer
to summary in this Report
|
||
|
AP-ADF-105
Phase
III
|
Evaluate
safety and efficacy in relief of moderate to severe pain
|
Refer
to summary in this Report
|
||
|
Extraction
Test
|
Laboratory
test quantifying I.V. abuse deterrent properties
|
Refer
to summary in this Report
|
||
|
Syringe
Test
|
Laboratory
test quantifying I.V. abuse deterrent properties
|
Refer
to summary in this
Report
|
||
Although
the FDA stated that no new Acurox® clinical
trials are required at this time, we and King are performing an additional
clinical study (Refer to Study AP-ADF-114 below) to further support the abuse
deterrent features of Acurox®.
Study AP-ADF-114 or Study
114: Study 114 is a randomized, double-blind, placebo- and
active-controlled study designed to assess the relative abuse potential of
Acurox® Tablets. In Study 114, approximately 47 fasted recreational
drug abuse subjects received single oral doses in five test treatments including
(a) two potentially abused excess doses of Acurox® (oxycodone/niacin) Tablets,
(b) two potentially abused excess oral doses of oxycodone HCl alone, and (c)
placebo. Dosing occurred every 48 hours. Prior to the
first test treatment, each subject first demonstrated, through naloxone
challenge, that they were not dependent on opioids. Also, each
subject demonstrated their ability to distinguish, on a 100-point VAS
like/dislike scale, between placebo and oxycodone doses randomly administered on
a blinded basis. Endpoints assessing abuse potential include
subjective assessments on a 100-point VAS of drug liking/disliking, take drug
again, and global overall drug liking/disliking. Study 114 has
completed enrollment and we are awaiting results of the statistical
analysis.
Study AP-ADF-106 or Study
106: Study 106 was a two part, Phase I, single-center,
single-blind, clinical study in non-dependent subjects with a history of
recreational intranasal opioid use to assess the safety, tolerability and
pharmacodynamic effects of intranasally administered crushed Acurox® Tablets,
crushed generic oxycodone HCl tablets and pure oxycodone HCl
powder. The primary objective of Part 1 (15 subjects enrolled, 13
subjects completed, 14 subjects analyzed) was to determine the maximum tolerated
dose of crushed Acurox® Tablets that subjects could snort in a single
administration. Subjects were administered escalating doses of crushed Acurox®
Tablets (7.5/30 mg) starting at one half tablet and increasing in half tablet
increments on successive days. Part 1 also assessed (over 8 hours
after administration) vital signs, subjective ratings of liking and somatic
(bodily) discomfort, and objective assessments of the nasal cavity determined by
endoscopy and intranasal photography. Part 2 (15 subjects
enrolled, 12 subjects completed, 10 subjects analyzed) assessed the relative
abuse liability of intranasally administered crushed Acurox® Tablets (7.5/30
mg), crushed generic oxycodone HCl immediate release tablets and pure oxycodone
HCl powder, all with a quantity of oxycodone HCl equivalent to the group median
highest tolerated dose of Acurox® Tablets determined in Part 1. Part
2 measurements were made over 12 hours and included vital signs, subjective
measures of liking and somatic (bodily) discomfort, objective assessments of the
nasal cavity and the pharmacokinetics of oxycodone after intranasal
administration.
Part 1 of
the study determined the maximum tolerated intranasal dose of crushed Acurox®
Tablets was 15 mg oxycodone HCl/60 mg niacin (i.e. 2 × 7.5/30 mg Acurox®
Tablets).
10
In Part 2
of the study, nasal administration of 2 crushed Acurox® Tablets
(7.5/30 mg ) resulted in disliking and low positive drug effect on the Overall
Drug Experience and Overall Drug Liking scales. In comparison,
crushed generic oxycodone HCl tablets and oxycodone HCl powder resulted in high
drug liking and positive drug effects with the mean difference being
statistically significant at p ≤ 0.0035. Furthermore, subjects were much less
willing to take crushed Acurox® Tablets
again compared to crushed oxycodone tablets and oxycodone powder (p ≤
0.0007). Intranasal administration of crushed Acurox® Tablets
caused significantly greater negative objective effects for nasal congestion and
discharge (p ≤ 0.0223) and negative subjective effects for nasal burning,
congestion and need to blow nose (p ≤ 0.0038). The results were not
statistically significant for the objective assessment of nasal
irritation.
Examination
of endoscopy and external photography revealed visible differences between the
treatments as only the crushed Acurox®
treatment was associated with white foamy
substance in the interior and middle turbinate of the nose and visible facial
flushing. Oxycodone bioavailability was similar between all three
treatments. In both Parts of the study, no serious adverse events were
reported. All adverse events were classified as mild or moderate with
the most prevalent adverse events being nasal symptoms (congestion, discomfort,
discharge), flushing, tearing and euphoria.
The
principal investigator’s overall conclusion was that, based on Study 106
results, intranasal administration of crushed Acurox® Tablets has a
distinctively lower abuse potential than the same oxycodone HCl dose
administered as crushed generic oxycodone HCl tablets or pure oxycodone HCl
powder.
Study AP-ADF-102 or Study
102: Study 102 was a Phase II, single center, randomized,
double blind crossover design clinical trial in 24 subjects with a history of
opioid abuse with a primary endpoint to assess, whether the subjects disliked
the drug effect they were feeling when varying levels of niacin were
administered in combination with 40 mg of oxycodone HCl compared to 40 mg
oxycodone HCl (alone) and a placebo. Each subject was randomized to a
dosing sequence that included doses of niacin (0, 240, 480 , and 600 mg)
administered in combination with 40 mg oxycodone HCl, while the subjects were
fasted and 600 mg niacin in combination with 40 mg oxycodone HCl administered
following a standardized high-fat meal. Each dosing day, vital sign
measures and subjective and behavioral effects were assessed before dosing
(baseline) and at 0.5, 1, 1.5, 2, 3, 4, 5, 6, and 12 hours after
dosing. After completion of the study, subjects responded to a
Treatment Enjoyment Assessment Questionnaire to select which of the treatments
they would take again. The maximum scale response to the question “Do
you dislike the drug effect you are feeling now?” (i.e., the “Disliking Score”),
was designated as the primary efficacy variable. Study results were
as follows:
|
|
·
|
In
the fasting state, all three doses of niacin in combination with oxycodone
HCl 40mg produced significant (p ≤ .05) Disliking Scores compared to
oxycodone HCl 40mg alone. No other subjective measure was
significantly affected by the niacin addition to
oxycodone.
|
|
|
·
|
The
high fat meal eliminated the niacin effect and also delayed the time to
oxycodone peak blood levels.
|
|
|
·
|
The
addition of niacin to oxycodone HCl alters the subjective response to
oxycodone HCl as indicated by the significant responses on the Disliking
Score. This observation in conjunction with the results from the Treatment
Enjoyment Questionnaire indicates that the addition of niacin reduces the
attractiveness of oxycodone to opiate
abusers.
|
|
|
·
|
There
were no serious adverse events. Niacin produced a dose related attenuation
of pupillary constriction, diastolic blood pressure increase and probably
systolic blood pressure increase produced by oxycodone HCl. The
alterations by niacin on the vital sign responses to oxycodone 40 mg were
minimal, were seen primarily with the 600 mg niacin dose and were not
clinically significant.
|
The principal study investigator’s
overall conclusion was that the results of Study 102 supported the hypothesis
that the addition of niacin to oxycodone HCl in a minimal ratio of 30 mg niacin
to 5 mg oxycodone HCl is aversive compared to oxycodone HCl
alone. The addition of niacin did not alter the safety profile of
oxycodone HCl alone.
11
Study AP-ADF-111 or Study
111: Study 111 is entitled "A Phase II, Single-Center,
Randomized, Double-Blind, Assessment of the Abuse Liability of Acurox®
(oxycodone HCl and niacin) Tablets in Subjects with a History of Opioid
Abuse". In Study 111, 30 fasted subjects with a history of opioid
abuse received a single dose of study drugs every 48 hours for 9 days and were
enrolled in two dosing sequences. The first dosing sequence (Sequence
1) included randomized doses of (i) niacin 240mg alone; (ii) a combination of
oxycodone HCl 40mg with niacin 240mg (4 times the expected recommended dose of
Acurox® Tablets
5/30mg); and (iii) placebo tablets. The objective of Sequence 1 was
to assess the effects of oxycodone HCl on the effects of niacin. The
second dosing sequence (Sequence 2) included randomized doses of (i) a
combination of oxycodone HCl 40mg with niacin 240mg (4 times the expected
recommended dose of Acurox® Tablets
5/30mg) and (ii) oxycodone HCl 40mg alone. Sequence 2 was designed to
assess the abuse liability and abuse deterrence potential of Acurox® Tablets
versus oxycodone HCl alone. On each dosing day, vital sign measures
and subjective and behavioral effects were assessed before dosing (baseline) and
at 0.5, 1, 1.5, 2, 3, 4, 5, 6, and 12 hours after dosing. Vital signs
included measurement of pupil size, blood pressure, heart rate, oral temperature
and respiratory rate. For both Sequence 1 and Sequence 2, subjective
changes were measured with a two item Drug Rating Questionnaire-Subject (DRQS)
and a 40 item short form of the Addiction Research Center Inventory
(ARCI). The ARCI was comprised of three scale scores including the
Morphine Benzedrine Group scale (MBG) measuring euphoria, the LSD/dysphoria
scale measuring somatic/bodily discomfort and dysphoria and the Pentobarbital
Chlorpromazine Alcohol Group scale (PCAG) measuring apathetic
sedation. For Sequence 2 only, in addition to the DRQS and ARCI,
subjects also completed a Street Value Assessment Questionnaire and a Treatment
Enjoyment Assessment Questionnaire.
Sequence 1 results demonstrated that
response to niacin 240 mg alone compared to placebo causes significant dislike
scores (p = .03), and significant LSD/dysphoria scores (p < .001) with these
negative niacin induced effects manifesting rapidly, reaching peak at 0.5-1.5
hours and thereafter diminishing. At 0.5 hours after drug
administration, oxycodone HCl 40 mg has limited effect on niacin-induced
disliking and dysphoric effects. At the one hour observation and
afterward, oxycodone may attenuate niacin-induced disliking and dysphoric
effects.
Sequence 2 demonstrated that the
combination of oxycodone HCl 40mg and niacin 240mg (4 times the expected
recommended dose of Acurox® Tablets
5/30mg) had the potential to be aversive when compared to oxycodone HCl 40mg
alone as shown by statistically significant and clinically meaningful results in
the dislike/like scores (p = .033), the Treatment Enjoyment Assessment scores (p
= .005) and the LSD/dysphoria scores (p<.001). The dislike/like
score at 0.5 hours was designated the primary measure of abuse liability and
abuse deterrence potential for Acurox® Tablets
5/30mg and the Treatment Enjoyment Assessment scores and LSD/dysphoria scores at
0.5 hours were additional measures of the abuse deterrence potential of
Acurox®
Tablets. Subjective measures not achieving statistical significance
included the MBG scores measuring euphoria, the PCAG score measuring apathetic
sedation and the Street Value Assessment Questionnaire score, in which subjects
indicated they would pay more for oxycodone HCl alone compared to Acurox® Tablets
(p=.097).
In this study of 30 subjects with a
history of opioid abuse there were no serious adverse events
reported. Alterations by niacin compared to placebo on vital signs
were minimal and not clinically meaningful. The differences in vital
signs between oxycodone HCl/niacin and niacin alone at 4 times the expected
recommended dose of Acurox® Tablets
were minimal and not clinically meaningful.
Study AP-ADF-105 or Study
105: Study 105 is entitled “A Phase III, Randomized,
Double-blind, Placebo-controlled, Multicenter, Repeat-dose Study of the Safety
and Efficacy of Acurox®
(oxycodone HCl and niacin) Tablets versus Placebo for the Treatment of Acute,
Moderate to Severe Postoperative Pain Following Bunionectomy Surgery in Adult
Patients.” A total of 405 patients were randomized to one of three
treatment arms of approximately 135 patients per arm. One treatment
arm received a dose of two Acurox® Tablets
5/30 mg, a second treatment arm received a dose of two Acurox® Tablets
7.5/30 mg, and the third treatment arm received a dose of two placebo
tablets. Study drugs were administered every 6 hours for 48
hours. The primary endpoint was the sum of the difference in pain
intensity, measured on a 100mm visual analog scale (VAS), compared to baseline
over a 48 hour period (“SPID48”). Prior
to initiating Study 105, the study design, endpoints and statistical analysis
plan were submitted to and agreed by the FDA under a Special Protocol Assessment
and the study was conducted accordingly. Results of Study 105
demonstrate that compared to placebo, Acurox® Tablets
5/30 mg and 7.5/30 mg both met the primary pain relief endpoint with p=.0001 and
p<.0001, respectively. Acurox® Tablets
were generally well tolerated with the most prevalent reported adverse events in
patients receiving Acurox® Tablets
being nausea, vomiting, dizziness, pruritus and flushing; side effects known to
be consistent with opioid and niacin therapies. Most adverse events
were mild or moderate and there were no serious adverse events. Six
patients (2.2%) receiving Acurox® Tablets
withdrew from the study due to treatment–emergent adverse events compared with
no withdrawals due to treatment-emergent adverse events for the placebo
group.
12
Extraction Test - In
consultation with experts in the field, a laboratory protocol for evaluating the
relative IV abuse liability of Acurox® Tablets was designed. To
provide unbiased, scientifically derived and documented study results, we
engaged a laboratory CRO specializing in
pharmaceutical product analysis to execute the protocol. The protocol
was intended to mimic the uncontrolled "real world" environment, except that
professional chemists with access to a wide range of laboratory equipment and
supplies would pose as potential IV drug abusers. As would be the
case with a potential IV drug abuser, these chemists were allowed unrestricted
access to information in developing oxycodone extraction methods and
techniques. The CRO was provided with a list of ingredients (active
and inactive) contained in each test product, allotted up to 80 hours total time
to complete the evaluations and allowed to use any methodology and/or solvents
desired to attempt to extract oxycodone HCl from the tablets in a form suitable
for I.V. injection. The test products were: OxyContin® (oxycodone HCl) Tablets 1 x 40
mg (Purdue Pharma), Oxycodone HCl Tabs 8 x 5 mg (Mallinckrodt), Percocet® (oxycodone HCl/APAP) Tablets 8
x 5/325 mg (Endo) and -Acurox®
(oxycodone HCl/niacin) Tablets 8 x 5/30 mg (Acura). The
results of the Extraction Test suggest that currently marketed oxycodone HCl
containing tablets may be easily dissolved in water in as little as 3 - 10
minutes for potential abuse via IV injection. By contrast, the Extraction Test
simulation suggests that preparing an injectable form of Acurox® Tablets is
difficult and not practical due to the time required (almost 6 hours) and the
inability to obtain a sufficient oxycodone yield that would provide any degree
of euphoric effect to the prospective drug abuser.
Syringe Test - The Syringe
Test was developed to simulate the relative difficulty of abusing dissolved
opioid tablets and capsules via IV injection. The test was designed as a
scientifically reproducible method to quantitatively measure the difficulty of
drawing into a syringe a solution made from tablets or capsules dissolved in
varying types and volumes of solvent. The test utilizes seven (7)
solvents available to potential abusers, the largest syringe barrel available
without a prescription and the largest bore needle in the subcutaneous syringe
set family. The needle and barrel are much larger than typically used
by abusers and represent a worst case scenario (e.g. easier to prepare an IV
injection). The Syringe Test results suggest that preparing an
injectable form of Acurox® Tablets using a variety of available solvents is
impractical due to high solvent volume requirements and the viscous/gelatinous
mixture formed when dissolving Acurox® Tablets in lower volumes of the solvents
tested. Even if a "Theoretically Injectable" solution of crushed
Acurox® Tablets is achieved it would require further processing by the
prospective abuser to separate oxycodone from other tablet ingredients
(including niacin and numerous excipients) and a reduction in total volume to
provide a solution in a volume and form that is practically suitable for IV
injection.
Expectations
for Acurox® Tablets
Product Labeling
The FDA has publicly stated that an
explicit indication or claims of abuse deterrence will not be permitted in
product labeling unless such indication or claims are supported by double blind
controlled clinical studies demonstrating an actual reduction in product abuse
by patients or drug abusers. We believe the cost, time and
practicality of designing and implementing clinical studies adequate to support
explicit labeling claims of abuse deterrence are prohibitive. The FDA
has stated that scientifically derived data and information describing the
physical characteristics of a product candidate and/or the results of laboratory
and clinical studies simulating product abuse may be acceptable to include in
the product label. We intend to include in the labels of our
Aversion®
Technology product candidates both a physical description of the abuse deterrent
characteristics and information from our numerous laboratory and clinical
studies designed to simulate the relative difficulty of abusing our product
candidates. The extent to which such information will be included in
the FDA approved product label will be the subject of our discussions with and
agreement by the FDA as part of the NDA review process for each of our product
candidates. Further, because FDA closely regulates promotional
materials, even if FDA initially approves labeling that includes a description
of the abuse deterrent characteristics of the product, the FDA will continue to
review the acceptability of promotional labeling claims and product advertising
campaigns for our product candidates.
King
Agreement
On October 30, 2007, we and King
Pharmaceuticals Research and Development, Inc. (“King”), a wholly-owned
subsidiary of King Pharmaceuticals, Inc., entered into a License, Development
and Commercialization Agreement (the “King Agreement”) to develop and
commercialize in the United States, Canada and Mexico (the "King Territory")
certain opioid analgesic products utilizing our proprietary Aversion®
Technology. The King Agreement initially provided King with an
exclusive license in the King Territory for Acurox®
(oxycodone HCl/niacin) Tablets and Acuracet®
(oxycodone HCl/niacin/APAP) Tablets, utilizing Aversion®
Technology. In addition, the King Agreement provides King with an
option to license in the King Territory all future opioid analgesic products
developed utilizing Aversion®
Technology. At December 31, 2009, King had exercised its option to license two
additional product candidates including an undisclosed opioid analgesic tablet
product and Vycavert®
(hydrocodone bitartrate/niacin/APAP) Tablets, each of which utilize our
Aversion®
Technology. We are responsible for using commercially reasonable
efforts to develop Acurox® Tablets through regulatory approval by the
FDA. The King Agreement provides that we or King may develop
additional opioid analgesic product candidates utilizing our Aversion®
Technology and, if King exercises its option to license such additional product
candidates, they will be subject to the milestone and royalty payments and other
terms of the King Agreement.
13
Pursuant to the King Agreement, we and
King formed a joint steering committee to oversee development and
commercialization strategies for Aversion® opioid analgesic products licensed to
King. We are responsible for all Acurox® Tablet
development activities, the expenses for which we are reimbursed by King,
through FDA approval of a 505(b)(2) NDA. After NDA approval King will be
responsible for commercializing Acurox® Tablets
in the U.S. With respect to all other products licensed by King
pursuant to the Agreement in all King Territories, King will be responsible, at
its own expense, for development, regulatory, and commercialization
activities. All products developed pursuant to the King Agreement
will be manufactured by King or a third party contract manufacturer under the
direction of King. Subject to the King Agreement, King will have final decision
making authority with respect to all development and commercialization
activities for all licensed products. We have reviewed our participation in the
King-Acura joint steering committee in light of the requirements of Emerging
Issues Task Force, Issue No. 00-21, “Revenue Arrangements with Multiple
Deliverables” (“EITF 00-21”) and concluded that this activity has no standalone
value therefore it does not meet the criteria to be considered a separate unit
of accounting.
At December 31, 2009, we had received
aggregate payments of $56.2 million from King, consisting of a $30.0 million
non-refundable upfront cash payment, $15.2 million in reimbursed research and
development expenses relating to Acurox® Tablets, $6.0 million in fees relating
to King’s exercise of its option to license an undisclosed opioid analgesic
tablet product and Vycavert® Tablets,
and a $5.0 million milestone fee relating to our successful achievement of the
primary endpoints for our pivotal Phase III clinical study for Acurox®
Tablets. The King Agreement also provides for King’s payment to us of
a $3.0 million fee upon King’s exercise of its option for each future opioid
product candidate. In the event that King does not exercise its
option for a future opioid product candidate, King may be required to reimburse
us for certain of our expenses relating to such future opioid product
candidate. Further, we may receive up to $23 million in additional
non-refundable milestone payments for each product candidate licensed to King,
including Acurox® Tablets,
which achieve certain regulatory milestones in specific countries in the King
Territory. We can also receive a one-time $50 million sales milestone
payment upon the first attainment of $750 million in net sales of all of our
licensed products across all King Territories. In addition, for sales
occurring following the one year anniversary of the first commercial sale of the
first licensed product sold, King will pay us a royalty at one of 6 rates
ranging from 5% to 25% based on the level of combined annual net sales for all
products licensed by us to King across all King Territories, with the highest
applicable royalty rate applied to such combined annual sales. King’s
royalty payment obligations expire on a product by product and
country-by-country basis upon the later of (i) the expiration of the last valid
patent claim covering such product in such country, or (ii) fifteen (15) years
from the first commercial sale of such product in such country. No
minimum annual fees are payable by either party under the King
Agreement. Reference is made to Item 9 of Note A of the Notes to
Consolidated Financial Statements included as a part of this Report, entitled
“Revenue Recognition and Deferred Program Fee Revenue” for a description of the
revenue recognition method employed by the Company under the King
Agreement.
The King Agreement expires upon the
expiration of King’s royalty payment and other payment obligations under the
King Agreement. King may terminate the King Agreement in its entirety
or with respect to any product at any time after March 31, 2010, upon the
provision of not less than 12 months’ prior written notice, and in its entirety
if regulatory approval of the NDA for Acurox® Tablets
is not received prior to March 31, 2010 and with respect to a particular product
with respect to a country in which regulatory approval for such product is
withdrawn by a regulatory authority in such country. We do not expect to
receive FDA approval of the NDA for Acurox® Tablets
prior to March 31, 2010. As a result, King may terminate the King
Agreement at any time following such date upon written notice to
us. We may terminate the King Agreement with respect to a product in
the United States in the event such product is not commercially launched by King
within 120 days after receipt of regulatory approval of such product or in its
entirety if King commences any interference or opposition proceeding challenging
the validity or enforceability any of our patent rights licensed to King under
the King Agreement. Either party has the right to terminate the King
Agreement on a product by product and country-by-country basis if the other
party is in material breach of its obligations under the King Agreement relating
to such product and such country, and to terminate the Agreement in its entirety
in the event the other party makes an assignment for the benefit of creditors,
files a petition in bankruptcy or otherwise seeks relief under applicable
bankruptcy laws, in each case subject to applicable cure
periods.
14
In the event of termination, no
payments are due except those royalties and milestones that have accrued prior
to termination under the King Agreement and all licenses under the King
Agreement are terminated. For all Acura terminations and termination
by King where we are not in breach, the King Agreement provides for the
transition of development and marketing of the licensed products from King to
us, including the conveyance by King to us of the trademarks and all regulatory
filings and approvals solely used in connection with the commercialization of
such licensed products and, in certain cases, for King’s supply of such licensed
products for a transitional period at King’s cost plus a mark-up.
The foregoing description of the King
Agreement contains forward-looking statements about Acurox® Tablets,
and other product candidates being developed pursuant to the King
Agreement. As with any pharmaceutical products under development or
proposed to be developed, substantial risks and uncertainties exist in
development, regulatory review and commercialization process. There
can be no assurance that the King Agreement will not be terminated by its terms
prior to receipt of regulatory approval for any product developed pursuant to
the King Agreement. Further, there can be no assurance that any
product developed, in whole or in part, pursuant to the King Agreement will
receive regulatory approval or prove to be commercially
successful. Accordingly, investors in the Company should recognize
that there is no assurance that the Company will receive the milestone payments
or royalty revenues described in the King Agreement or even if such milestones
are achieved, that the related products will be successfully commercialized and
that any royalty revenues payable to us by King will materialize. For
further discussion of other risks and uncertainties associated with the Company,
see Item 1A in this Report under the heading “Risks Factors”.
Patents
and Patent Applications
In April 2007, the United States Patent
and Trademark Office (“USPTO”), issued to us a patent titled “Methods and
Compositions for Deterring Abuse of Opioid Containing Dosage Forms” (the “920
Patent”). The 54 allowed claims in the 920 Patent encompass certain
pharmaceutical compositions intended to deter the most common methods of
prescription opioid analgesic product misuse and abuse. These patented
pharmaceutical compositions include specific opioid analgesics such as oxycodone
HCl and hydrocodone bitartrate among others.
In January 2009, the USPTO issued to us
a patent (the “402 Patent”) with 18 allowed claims. The 402 Patent encompasses
certain combinations of kappa and mu opioid receptor agonists
and other ingredients intended to deter opioid analgesic product misuse and
abuse.
In March 2009, the USPTO issued to us a
patent (the “726 Patent”) with 20 allowed claims. The 726 Patent
encompasses a wider range of abuse deterrent compositions than our 920
Patent.
In addition to our issued U.S. patents,
we have filed multiple U.S. patent applications and international patent
applications relating to compositions containing abusable active pharmaceutical
ingredients. Except for those rights conferred in the King Agreement,
we have retained all intellectual property rights to our Aversion® Technology,
Impede™ Technology, and related product candidates.
Reference is made to Item 1A, “Risk
Factors” for a discussion, among other things, of pending patent applications
owned by third parties including claims that may encompass our Acurox® Tablets
and other product candidates. If such third party patent applications
result in valid and enforceable issued patents containing claims in their
current form, we or our licensees could be required to obtain a license to such
patents, should one be available, or alternatively, to alter our product
candidates to avoid infringing such third-party patents.
Competition
in the Opioid Product Market
We
compete to varying degrees with numerous companies in the pharmaceutical
research, development, manufacturing and commercialization fields. Many of our
competitors have substantially greater financial and other resources and are
able to expend more funds and effort than us in research and development of
their competitive technologies and products. Although a larger company with
greater resources than us will not necessarily have a higher likelihood of
receiving regulatory approval for a particular product or technology as compared
to a smaller competitor, the company with a larger research and development
expenditure will be in a position to support more development projects
simultaneously, thereby potentially improving the likelihood of obtaining
regulatory approval of a commercially viable product or technology than its
smaller rivals.
15
We
believe potential competitors may be developing opioid abuse deterrent
technologies and products. Such potential competitors include, but may not be
limited to, Pain Therapeutics of South San Francisco, CA, (in collaboration with
King Pharmaceuticals Inc.), Purdue Pharma of Stamford, CT, Atlantic
Pharmaceuticals, of Atlanta, GA, Egalet a/s, of Verlose, Denmark and Collegium
Pharmaceuticals, Inc., of Cumberland, RI. These companies appear to
be focusing their development efforts on ER Opioid Products while our lead
product candidate, Acurox® Tablets,
and the majority of our other Aversion®
Technology opioid analgesic product candidates under development, are IR Opioid
Products.
Aversion®
Technology Non-Opioid Product Candidates in
Development
We are developing a benzodiazepine
product candidate utilizing our Aversion® Technology intended for the treatment
of anxiety disorders. According to Drugs of Abuse, published by
the US Drug Enforcement Administration, tranquilizers are abused in manners
similar to opioid analgesics. The 2008 National Survey on Drug Use and
Health estimates that 21.5 million people have abused prescription
tranquilizers (including benzodiazepines) at some point in their lifetime and
5.1 million have abused tranquilizers in the past year.
We have completed a 2-way pilot
crossover pharmacokinetic study in 9 healthy adult subjects of a single oral
dose of the Aversion® Technology benzodiazepine product candidate compared to
the reference listed drug. While the Aversion® Technology product
candidate did not achieve bioequivalence to the reference listed drug in this
pilot study, the Aversion® Technology product candidate demonstrated suitable
bioavailability of the active benzodiazepine ingredient and we believe such
product candidate may demonstrate bioequivalence to the reference listed drug in
a larger pivotal scale study.
The Aversion® Technology benzodiazepine
product candidate is intended to be encompassed by numerous pending patent
applications filed with the USPTO by the Company. However, at this
stage, there can be no assurances that such pending patent applications will
result in issued patent claims encompassing our benzodiazepine product
candidate.
Impede™
Technology Product Candidates in Development
We are engaged in laboratory testing of
a pseudoephedrine HC1 product candidate utilizing our novel Impede™
Technology. Impede™
Technology is primarily intended to inhibit the conversion of pseudoephedrine
HC1 into methamphetamine, an illicit and frequently abused drug.
Most pseudoephedrine containing
products are classified by the FDA for sale Over-The-Counter (without a doctor’s
prescription) and most product formulations do not require the approval of a New
Drug Application by the FDA to initiate commercial distribution and
marketing. In 2006 regulations relating to over-the counter sale of
pseudoephedrine HCl products were amended with the enactment of the federal
Combat Methamphetamine Abuse Act (CMAA). The CMAA was enacted in
response to an alarming increase in and widespread conversion of pseudoephedrine
containing products into methamphetamine. Among other things, the
CMAA requires retail stores to maintain their inventory of pseudoephedrine
containing products in a secured location and restricts the amount of
pseudoephedrine products a store can sell to an individual
customer. Implementation of the CMAA initially reduced the number of
illegal methamphetamine laboratories as, the most commonly used process for
conversion of pseudoephedrine to methamphetamine requires substantial quantities
of pseudoephedrine. However, a newer, more efficient process for
converting pseudoephedrine to methamphetamine requires less
pseudoephedrine. Possibly as a result of the more efficient process,
the DEA reported a 15% increase in clandestine methamphetamine laboratory
seizures in 2008. Impede™ Technology is designed to deter a wide
range of processes of methamphetamine production including both the older and
newer processes.
Government
Regulation
All
pharmaceutical firms, including us, are subject to extensive regulation by the
federal government, principally by the FDA under the Federal Food, Drug and
Cosmetic Act (the “FD&C Act”), and, to a lesser extent, by state and local
governments. Before our products may be marketed in the U.S., they
must be approved by the FDA for commercial
distribution. Additionally, we are subject to extensive regulation by
the DEA under the Controlled Substances Act for research, development and
manufacturing of controlled substances. We are also subject to
regulation under federal, state and local laws, including requirements regarding
occupational safety, laboratory practices, environmental protection and
hazardous substance control, and may be subject to other present and future
local, state, federal and foreign regulations, including possible future
regulations of the pharmaceutical industry. We cannot predict the extent to
which we may be affected by legislative and other regulatory developments
concerning our products and the healthcare industry in general.
16
However,
because of recent developments in the legislative and regulatory framework
within which drug products are reviewed and approved by FDA, approval of drug
products by FDA may be subject to continuing obligations intended to assure safe
use of the products. Specifically, effective March 25, 2008, under
Title IX of Subtitle A of the Food and Drug Administration Amendments Act of
2007 (“FDAAA”), FDA may require a Risk Evaluation and Mitigation Strategy
(“REMS”) to manage known or potential serious risks associated with drugs or
biological products. If FDA finds that a REMS is necessary to ensure that the
benefits of our products outweigh the risks associated with the products, FDA
will require a REMS and, consequently, that we take additional measures to
ensure safe use of the product. Components of a REMS may include, but are not
limited to a Medication Guide, a marketing and sales communication plan,
elements to assure safe product use, a REMS implementation system, and a
timetable for FDA’s assessment of the effectiveness of the REMS.
The
FD&C Act, the Controlled Substances Act and other federal statutes and
regulations govern the testing, manufacture, quality control, export and import,
labeling, storage, record keeping, approval, pricing, advertising, promotion,
sale and distribution of pharmaceutical products. Noncompliance with applicable
requirements both before and after approval, can subject us, our third party
manufacturers and other collaborative partners to administrative and judicial
sanctions, such as, among other things, warning letters, fines and other
monetary payments, recall or seizure of products, criminal proceedings,
suspension or withdrawal of regulatory approvals, interruption or cessation of
clinical trials, total or partial suspension of production or distribution,
injunctions, limitations on or the limitation of claims we can make for our
products, and refusal of the government to enter into supply contracts for
distribution directly by governmental agencies, or delay in approving or refusal
to approve new drug applications. The FDA also has the authority to revoke or
withhold approvals of new drug applications.
The
Federal Controlled Substances Act imposes various registration, record-keeping
and reporting requirements, procurement and manufacturing quotas, labeling and
packaging requirements, security controls and a restriction on prescription
refills on certain pharmaceutical products. Establishments may not handle
controlled drug substances until they have been inspected and registered by the
DEA. Facilities must be equipped to meet DEA security requirements. A
principal factor in determining the particular requirements, if any, applicable
to a product is its actual or potential abuse profile. A pharmaceutical product
may be “scheduled” as a C-I, C-II, C-III, C-IV or C-V controlled substance, with
C-I substances considered to present the highest risk of substance abuse and C-V
substances the lowest. Because of the potential for abuse, opioid analgesic
active pharmaceutical ingredients and finished drug products, including all of
our Aversion® Technology product candidates, are regulated, or scheduled, under
the Controlled Substances Act. Because it contains oxycodone HCl, we
believe that Acurox®
Tablets will be a DEA C-II product.
FDA
approval is required before any "new drug," can be marketed. A "new drug" is one
not generally recognized as safe and effective for its intended use. Our
products are new drugs. Such approval must be based on adequate and well
controlled laboratory and clinical investigations. In addition to providing
required safety and effectiveness data for FDA approval, a drug manufacturer's
practices and procedures must comply with current Good Manufacturing Practices
(“cGMPs”), which apply to the manufacturing, receiving, holding and shipping.
Accordingly, manufacturers must continue to expend time, money and effort in all
applicable areas relating to quality assurance and regulatory compliance,
including production and quality control to comply with cGMPs. Failure to so
comply risks delays in approval of drug products and possible FDA enforcement
actions, such as an injunction against shipment of products, the seizure of
non-complying products, criminal prosecution and/or any of the other possible
consequences described above. We are subject to periodic inspection by the FDA
and DEA.
The
FDA Drug Approval Process
The
process of drug development is complex and lengthy. The activities undertaken
before a new pharmaceutical product may be marketed in the U.S. generally
include, but are not limited to, preclinical studies; submission to the FDA of
an IND, which must become active before human clinical trials commence; adequate
and well-controlled human clinical trials to establish the safety and efficacy
of the product; submission to the FDA of an NDA; acceptance for filing of the
NDA by FDA; satisfactory completion of an FDA pre-approval inspection of the
clinical trial sites and manufacturing facility or facilities at which the both
the active ingredients and finished drug product are produced to assess
compliance with cGMPs; and FDA review and approval of the NDA prior to any
commercial sale and distribution of the product in the U.S.
17
Preclinical
studies include laboratory evaluation of product chemistry and formulation, and
in some cases, animal studies and other studies to preliminarily assess the
potential safety and efficacy of the product candidate. The results of
preclinical studies together with manufacturing information, and analytical data
are then submitted to the FDA as a part of an IND. An IND must become
effective prior to the commencement of human clinical trials. The IND becomes
effective 30 days following its receipt by the FDA unless the FDA objects to, or
otherwise raises concerns or questions and imposes a clinical
hold. In the event that FDA objects to the IND and imposes a clinical
hold, the IND sponsor must address any outstanding FDA concerns or questions to
the satisfaction of the FDA before clinical trials can proceed. There
can be no assurance that submission of an IND will result in FDA authorization
to commence clinical trials. Once an IND is in effect, the protocol
for each clinical trial to be conducted under the IND must be submitted to the
FDA, which may or may not allow the trial to proceed.
Human
clinical trials are typically conducted in three phases that often
overlap:
Phase I: This phase is
typically the first involving human participants, and involves the smallest
number of human participants (typically, 20-50). The investigational
drug is initially introduced into healthy human subjects or patients and tested
for safety, dosage tolerance, absorption, metabolism, distribution and
excretion. In addition, it is sometimes possible to gain a preliminary
indication of efficacy.
Phase II: Once the preliminary
safety and tolerability of the drug in humans is confirmed during phase I, phase
II involves studies in a somewhat larger group of study
subjects. Unlike phase I studies, which typically involve healthy
subjects, participants in phase II studies maybe affected by the disease or
condition for which the product candidate is being developed. Phase
II studies are intended to identify possible adverse effects and safety risks,
to evaluate the efficacy of the product for specific targeted diseases, and to
determine appropriate dosage and tolerance.
Phase III: Phase III trials
typically involve a large numbers of patients affected by the disease or
condition for which the product candidate is being developed.. Phase III
clinical trials are undertaken to evaluate clinical efficacy and safety under
conditions resembling those for which the product will be used in actual
clinical practice after FDA approval of the NDA. Phase III trials are
typically the most costly and time-consuming of the clinical
phases.
Phase IV: Phase IV
trials may be required by FDA after the approval of the NDA for the product, as
a condition of the approval, or may be undertaken voluntarily by the sponsor of
the trial. The purpose of phase IV trials is to continue to evaluate
the safety and efficacy of the drug on a long-term basis and in a much larger
and more diverse patient population than was included in the prior phases of
clinical investigation.
After
clinical trials have been completed, the sponsor must submit an NDA to the FDA
including the results of the preclinical and clinical testing, together with,
among other things, detailed information on the chemistry, manufacturing,
quality controls, and proposed product labeling. There are two types of NDAs; a
505(b)(1) and a 505(b)(2). A 505(b)(1) NDA is also known as a "full NDA" and is
described by section 505(b)(1) of the FD&C Act as an application containing
full reports of investigations of safety and effectiveness, in addition to other
information. The data in a full NDA is either owned by the applicant or are data
for which the applicant has obtained a right of reference. A 505(b)(2)
application is one described under section 505(b)(2) of the FD&C Act as an
application for which information, or one or more of the investigations relied
upon by the applicant for approval "were not conducted by or for the applicant
and for which the applicant has not obtained a right of reference or use from
the person by or for whom the investigations were conducted". This provision
permits the FDA to rely for approval of an NDA on data not developed by the
applicant, such as published literature or the FDA's finding of safety and
effectiveness of a previously approved drug. 505(b)(2) applications are
submitted under section 505(b)(1) of the FD&C Act and are therefore subject
to the same statutory provisions that govern 505(b)(1) applications that require
among other things, "full reports" of safety and effectiveness. The FDA provided
written guidance to us stating that Acurox®
Tablets is a suitable product candidate for submission as a 505(b)(2) NDA and in
February 2009 accepted our Acurox® Tablets NDA for filing.
18
Each NDA
requires a user fee, pursuant to the requirements of the Prescription Drug User
Fee Act (“PDUFA”), as amended. According to FDA’s fee schedule, effective on
October 1, 2009, for the 2010 fiscal year, the user fee for an application fee
requiring clinical data (such as an NDA) is $1,247,200. The FDA
adjusts PDUFA user fees on an annual basis. PDUFA also imposes an annual product
and facility fees. The annual product fee for prescription drugs and
biologics for the 2010 fiscal year is $71,250 and the annual facility fee for
facilities used to manufacture prescription drugs and biologics for the 2010
fiscal year is $425,600. A written request can be submitted for a
waiver of the application fee for the first human drug application that is filed
by a small business, but no waivers for product or establishment fees are
available. Where we are subject to these fees, they are significant expenditures
that may be incurred in the future and must be paid at the time of submission of
each application to FDA. The King Agreement provides that King will
reimburse us for the NDA application fee for Acurox® Tablets and pay user fees
for all products licensed by the Company to King pursuant to the King
Agreement.
After an
NDA is submitted by an applicant, and if it is accepted for filing by the FDA,
the FDA will then review the NDA and, if and when it determines that the data
submitted are adequate to show that the product is safe and effective for its
intended use, the FDA will approve the product for commercial distribution in
the U.S. There can be no assurance that any of our product candidates will
receive FDA approval or that even if approved, they will be approved with
labeling that includes descriptions of its abuse deterrent
features. Moreover, even if our product candidates are approved with
labeling that includes descriptions of the abuse deterrent characteristics of
our products, advertising and promotion for the products will be limited to the
specific claims and descriptions in the FDA approved product
labeling.
The FDA
requires drug manufacturers to establish and maintain quality control procedures
for manufacturing, processing and holding drugs and investigational products,
and products must be manufactured in accordance with defined
specifications. Before approving an NDA, the FDA usually will inspect
the facility(ies) at which the active pharmaceutical ingredients and finished
drug product is manufactured, and will not approve the product unless it finds
that cGMP compliance at those facility(ies) are satisfactory. If the
FDA determines the NDA is not acceptable, the FDA may outline the deficiencies
in the NDA and often will request additional information, thus delaying the
approval of a product. Notwithstanding the submission of any
requested additional testing or information, the FDA ultimately may decide that
the application does not satisfy the criteria for approval. After a
product is approved, changes to the approved product, such as adding new
indications, manufacturing changes, or changes in or additions to the approved
labeling for the product, may require submission of a new NDA or, in some
instances, an NDA supplement, for further FDA review and
approval. Post-approval marketing of products in larger or different
patient populations than those that were studied during development can lead to
new findings about the safety or efficacy of the products. This
information can lead to a product sponsor’s requesting approval for and/or the
FDA requiring changes in the labeling of the product or even the withdrawal of
the product from the market.
The Best
Pharmaceuticals for Children Act, or BPCA, became law in 2002 and was
subsequently reauthorized and amended by FDAAA. The reauthorization
of BPCA provides an additional six months of patent protection to NDA applicants
that conduct acceptable pediatric studies of new and currently-marketed drug
products for which pediatric information would be beneficial, as identified by
FDA in a Pediatric Written Request. The Pediatric Research Equity Act (“PREA”),
became law in 2003, and was also subsequently reauthorized and amended by
FDAAA. The reauthorization of PREA requires that most applications
for drugs and biologics include a pediatric assessment (unless waived or
deferred) to ensure the drugs' and biologics' safety and effectiveness in
children. Such pediatric assessment must contain data, gathered using
appropriate formulations for each age group for which the assessment is
required, that are adequate to assess the safety and effectiveness of the drug
or the biological product for the claimed indications in all relevant pediatric
subpopulations, and to support dosing and administration for each pediatric
subpopulation for which the drug or the biological product is safe and
effective. The pediatric assessments can only be deferred provided
there is a timeline for the completion of such studies. FDA may waive
(partially or fully) the pediatric assessment requirement for several reasons,
including if the applicant can demonstrate that reasonable attempts to produce a
pediatric formulation necessary for that age group have failed. We
have requested a deferral of the pediatric assessment in our Acurox® Tablets NDA
submission.
The terms
of approval of any Acurox® Tablet NDA, including the indication and product
labeling (and, consequently advertising and promotion) may be more restrictive
than what is sought in the NDA or what is desired by
us. Additionally, FDA may condition approval of abuse deterrence
statements and claims for Acurox® Tablets on phase IV clinical studies for
continued assessment of such statements or claims. The Acurox® Tablet
testing and FDA approval process requires substantial time, effort, and
financial resources, and we cannot be sure that any approval will be granted on
a timely basis, if at all.
19
Further,
as a condition of approval of any NDA, FDA may require a REMS to ensure the safe
use and monitoring of any of our products. If required, a REMS may include, but
may not be limited to, use of an FDA-approved Medication Guide and/or Patient
Package Insert, a communication plan for patients or healthcare providers
concerning the drug, a description of elements to assure safe use of the
product, and a timetable for FDA’s assessment of the effectiveness of the
REMS.
In
addition, we, our suppliers and our licensees are required to comply with
extensive FDA requirements both before and after approval. For
example, we or our licensees are required to report certain adverse reactions
and production problems, if any, to the FDA, and to comply with certain
requirements concerning advertising and promotion for our products, which, as
discussed above, may significantly affect the extent to which we can include
statements or claims referencing our abuse deterrent technology in product
labeling and advertising. Also, quality control and manufacturing
procedures must continue to conform to cGMP after approval to avoid the product
being rendered misbranded and/or adulterated under the FD&C Act as a result
of manufacturing problems. In addition, discovery of any material
safety issues may result in changes to product labeling or restrictions on a
product manufacturer, potentially including removal of the product from the
market.
Whether
or not FDA NDA approval in the U.S. has been obtained, approvals from comparable
governmental regulatory authorities in foreign countries must be obtained prior
to the commencement of commercial activities in those countries. The approval
procedure varies in complexity from country to country, and the time required
may be longer or shorter than that required for FDA approval.
Facilities
for Research, Development, and Manufacturing
We
conduct research, development, laboratory, manufacturing and related activities
for product candidates utilizing Aversion® and
Impede™ Technologies at our Culver, Indiana facility. The 28,000
square foot facility is registered with the DEA to perform research, development
and manufacture of certain DEA Scheduled active pharmaceutical ingredients and
finished dosage form products. We obtain quotas for supply of
DEA-scheduled active pharmaceutical ingredients from the DEA and develop
finished drug product candidate dosage forms in our Culver
laboratories. We manufacture clinical trial supplies of drug products
in our Culver facility in volumes sufficient to meet FDA standards for
NDAs. King is responsible for commercial manufacture of the product
candidates licensed under the King Agreement. We expect that all
future product candidates developed by us will be commercially manufactured by
our licensees or other qualified third party contract
manufacturers.
Segment
Reporting
We
operate in one business segment; the research, development and manufacture of
innovative abuse deterrent, orally administered pharmaceutical product
candidates.
Legal
Proceedings
We are
not involved in any material legal proceedings.
Environmental
Compliance
We are subject to regulation under
federal, state and local environmental laws and believe we are in material
compliance with such laws. We incur the usual waste disposal cost associated
with a pharmaceutical research, development and manufacturing
operation.
Raw
Materials
To purchase certain active ingredients
required for our development and manufacture of product candidates utilizing our
Aversion®
Technology, we are required to file for and obtain supply quotas from the DEA.
No assurance can be given that we will be successful in obtaining adequate DEA
quotas in a timely manner. Even assuming adequate and timely DEA quotas, there
can be no assurances that the approved manufacturers of raw materials for our
product candidates will supply us with our requirements for the active or
inactive ingredients required for the development and manufacture of our product
candidates.
20
Subsidiaries
Our Culver, Indiana research,
development, and manufacturing operations are conducted by Acura Pharmaceutical
Technologies, Inc., an Indiana corporation and our wholly-owned
subsidiary.
ITEM
1A. RISK FACTORS
Our
future operating results may vary substantially from anticipated results due to
a number of factors, many of which are beyond our control. The following
discussion highlights some of these factors and the possible impact of these
factors on future results of operations. If any of the following factors
actually occur, our business, financial condition or results of operations could
be materially harmed. In that case, the value of our common stock
could decline substantially.
Risks
Relating to Our Business and Industry
We
have a history of operating losses and may not achieve profitability sufficient
to generate a positive return on shareholders’ investment.
We
had a net loss of $15.8 million for the year ended December 31,
2009, net income of $14.5 million for the year ended December 31,
2008 and a net loss of $4.3 million the year ended December 31,
2007. Our future profitability will depend on several factors,
including:
|
|
·
|
our
receipt of milestone payments and royalties relating to products developed
and commercialized under our license agreement with King (as more fully
described under the caption “Item 1. Business — King Agreement”);
and
|
|
|
·
|
the
receipt of FDA approval and the successful commercialization by King and
other future licensees (if any) of products utilizing our Aversion®
and Impede™ Technologies without infringing the patents and other
intellectual property rights of third
parties.
|
We cannot
assure you that we will ever have a product approved for commercialization by
the FDA or that we or our licensees will bring any product to
market.
We
recognized revenues of $3.8 million and $44.4 million in the years ended
December 31, 2009 and 2008, respectively, from payments received under the King
Agreement. However, we have not yet generated any revenues from
Aversion® Technology product sales. Even if we succeed in
commercializing one or more of our Aversion® Technology product candidates, we
expect to continue using cash reserves for the foreseeable future. Our expenses
may increase in the foreseeable future as a result of continued research and
development of additional product candidates, maintaining and expanding the
scope of our intellectual property, and hiring of additional research and
development staff.
We will
need to generate royalty revenues from product sales to achieve and maintain
profitability. If we cannot successfully develop, obtain regulatory approval for
and commercialize our product candidates licensed to King under the King
Agreement or other product candidates under similar license agreements
anticipated to be negotiated and executed with other pharmaceutical companies,
of which no assurance can be given, we will not be able to generate such royalty
revenues or achieve future profitability. Our failure to achieve or maintain
profitability would have a material adverse impact on the market price of our
common stock.
We
must rely on current cash reserves, technology licensing fees and third party
financing to fund operations.
Pending
the receipt of milestone payments and royalties, if any, under the King
Agreement or under similar license agreements anticipated to be negotiated and
executed with other pharmaceutical companies, of which no assurance can be
given, we must rely on our current cash reserves and third-party financing to
fund operations and product development activities. No assurance can be given
that current cash reserves will be sufficient to fund the continued operations
and development of our product candidates until such time as we generate
additional revenue from the King Agreement or similar license agreements
anticipated to be negotiated and executed with the other pharmaceutical
companies. Moreover, no assurance can be given that we will be
successful in raising additional financing or, if funding is obtained, that such
funding will be sufficient to fund operations until product candidates utilizing
our Aversion® and
Impede™ Technologies may be commercialized.
21
Our
product candidates are unproven and may not be approved by the FDA.
We
are committing a majority of our resources to the development of Acurox® Tablets
and other product candidates utilizing our Aversion®
Technology. In December 2008 we submitted a 505(b)(2) NDA for
Acurox® Tablets
to the FDA and on June 30, 2009 we received the FDA’s CRL. The FDA
intends to have an Advisory Committee review the NDA for Acurox®. There
can be no assurance that the FDA will ultimately approve Acurox® Tablets
or any other product candidate utilizing Aversion®
Technology for commercial distribution. Further there can
be no assurance that other product candidates developed using Aversion® or
Impede™ Technology will achieve the targeted end points in the required clinical
studies or perform as intended in other pre-clinical and clinical studies or
lead to a NDA submission. Our failure to successfully develop and achieve final
FDA approval of a product candidate utilizing Aversion®
Technology will have a material adverse effect on our financial
condition.
Even
if the FDA Approves Acurox® Tablets
for commercial distribution, if Acurox® Tablets
are not approved with labeling describing its abuse deterrent features, we will
be unable to refer to the abuse deterrent characteristics of Acurox® to
promote the product.
Our
strategy for Acurox® Tablets
depends upon our ability to distinguish Acurox® from
other immediate release oxycodone HCl containing products based primarily on
abuse deterrent features. As with all of our product candidates
utilizing Aversion®
Technology, even if Acurox® Tablets
are approved by the FDA, our failure to achieve approval of product labeling
that sufficiently differentiates Acurox® Tablets
from other immediate release oxycodone HCl containing tablets may adversely
affect our business and results of operations. The FDA has publicly stated that
explicit indications or claims of abuse deterrence will not be permitted unless
such indications or claims are supported by double blind controlled clinical
studies demonstrating an actual reduction in product abuse by patients or drug
abusers. Because the cost, time and practicality of designing and implementing
clinical studies adequate to support explicit claims of abuse deterrence are
prohibitive, we are not pursuing and have not conducted the clinical trials
necessary to include an explicit product label claim of abuse
deterrence. Instead, we intend to rely on certain clinical and
laboratory studies to support the inclusion of information about the abuse
deterrent characteristics of Acurox® Tablets
to support promotion by our licensee(s) of the product. We intend to
include in the product labels of our product candidates both a physical
description of the abuse deterrent characteristics and information from our
multiple laboratory and clinical studies designed to simulate the relative
difficulty of abusing our product candidates. However, the extent to which such
information will be included in the FDA approved product label will be the
subject of our discussions with, and agreement by, the FDA as part of the NDA
review process for each of our product candidates. The outcome of
those discussions with the FDA will determine whether we will be able to market
our product candidates with labeling that sufficiently differentiates them from
other products that have comparable therapeutic profiles. If the FDA does not
approve the Acurox® Tablet
labeling with such information, we will not be able to promote Acurox® Tablets
based on its abuse deterrent features, may not be able to differentiate
Acurox® from
other oxycodone HCl containing immediate release products, and may not be able
to charge a premium above the price of such other products which could
materially adversely affect our business and results of operations.
Because
FDA closely regulates promotional materials and other promotional activities,
even if FDA initially approves product labeling that includes a description of
the abuse deterrent characteristics of our product, FDA may object to our
marketing claims and product advertising campaigns.
Relying
on third party contract research organizations ("CROs") may result in delays in
our pre-clinical, clinical or laboratory testing. If pre-clinical, clinical or
laboratory testing for our product candidates are unsuccessful or delayed, we
will be unable to meet our anticipated development and commercialization
timelines.
To obtain
FDA approval to commercially sell and distribute in the U.S. any of our
prescription product candidates, we or our licensees must submit to the FDA an
NDA demonstrating, among other things, that the product candidate is safe and
effective for its intended use. This demonstration requires significant testing.
As we do not possess the resources or employ all the personnel necessary to
conduct such testing, we rely on CROs for the majority of this testing with our
product candidates. As a result, we have less control over our development
program than if we performed the testing entirely on our own. Third parties may
not perform their responsibilities on our anticipated schedule. Delays in our
development programs could significantly increase our product development costs
and delay product commercialization.
22
The
commencement of clinical trials with our product candidates may be delayed for
several reasons, including but not limited to delays in demonstrating sufficient
pre-clinical safety required to obtain regulatory approval to commence a
clinical trial, reaching agreements on acceptable terms with prospective
licensees, manufacturing and quality assurance release of a sufficient supply of
a product candidate for use in our clinical trials and/or obtaining
institutional review board approval to conduct a clinical trial at a prospective
clinical site. Once a clinical trial has begun, it may be delayed, suspended or
terminated by us or regulatory authorities due to several factors, including
ongoing discussions with regulatory authorities regarding the scope or design of
our clinical trials, failure to conduct clinical trials in accordance with
regulatory requirements, lower than anticipated recruitment or retention rate of
patients in clinical trials, inspection of the clinical trial operations or
trial sites by regulatory authorities, the imposition of a clinical hold by FDA,
lack of adequate funding to continue clinical trials, and/or negative or
unanticipated results of clinical trials.
Clinical
trials required by the FDA for commercial approval may not demonstrate safety or
efficacy of our product candidates. Success in pre-clinical testing and early
clinical trials does not assure that later clinical trials will be successful.
Results of later clinical trials may not replicate the results of prior clinical
trials and pre-clinical testing. Even if the results of our pivotal phase III
clinical trials are positive, we and our licensees may have to commit
substantial time and additional resources to conduct further pre-clinical and
clinical studies before we or our licensees can submit NDAs or obtain regulatory
approval for our product candidates.
Clinical
trials are expensive and at times difficult to design and implement, in part
because they are subject to rigorous regulatory requirements. Further, if
participating subjects or patients in clinical studies suffer drug-related
adverse reactions during the course of such trials, or if we, our licensees or
the FDA believes that participating patients are being exposed to unacceptable
health risks, we or our licensees may suspend the clinical trials. Failure can
occur at any stage of the trials, and we or our licensees could encounter
problems causing the abandonment of clinical trials or the need to conduct
additional clinical studies, relating to a product candidate.
Even if
our clinical trials and laboratory testing are completed as planned, their
results may not support commercially viable product label claims. The clinical
trial process may fail to demonstrate that our product candidates are safe and
effective for their intended use. Such failure may cause us or our licensees to
abandon a product candidate and may delay the development of other product
candidates.
We
or our licensees may not obtain required FDA approval; the FDA approval process
is time-consuming and expensive.
The
development, testing, manufacturing, marketing and sale of pharmaceutical
products are subject to extensive federal, state and local regulation in the
United States and other countries. Satisfaction of all regulatory requirements
typically takes years, is dependent upon the type, complexity and novelty of the
product candidate, and requires the expenditure of substantial resources for
research, development and testing. Substantially all of our operations are
subject to compliance with FDA regulations. Failure to adhere to applicable FDA
regulations by us or our licensees would have a material adverse effect on our
operations and financial condition. In addition, in the event we are successful
in developing product candidates for distribution and sale in other countries,
we would become subject to regulation in such countries. Such foreign
regulations and product approval requirements are expected to be time consuming
and expensive.
We or our
licensees may encounter delays or rejections during any stage of the regulatory
review and approval process based upon the failure of clinical or laboratory
data to demonstrate compliance with, or upon the failure of the product
candidates to meet, the FDA’s requirements for safety, efficacy and quality; and
those requirements may become more stringent due to changes in regulatory agency
policy or the adoption of new regulations. After submission of an NDA, or a
505(b)(2) NDA, the FDA may refuse to file the application, deny approval of the
application, require additional testing or data and/or require post-marketing
testing and surveillance to monitor the safety or efficacy of a product. The FDA
commonly takes more than a year to grant final approval for an NDA, or 505(b)(2)
NDA. The Prescription Drug User Fee Act (“PDUFA”) sets time standards for FDA’s
review of NDA’s. The FDA's timelines described in the PDUFA guidance are
flexible and subject to change based on workload and other potential review
issues and may delay the FDA’s review of an NDA. Further, the terms
of approval of any NDA, including the product labeling, may be more restrictive
than we or our licensees desire and could affect the marketability of our
products.
23
Even if
we comply with all the FDA regulatory requirements, we or our licensees may
never obtain regulatory approval for any of our product candidates. If we or our
licensees fail to obtain regulatory approval for any of our product candidates,
we will have fewer commercialized products and correspondingly lower revenues.
Even if regulatory approval of our products is received, such approval may
involve limitations on the indicated uses or promotional claims we or our
licensees may make for our products, or otherwise not permit labeling that
sufficiently differentiates our product candidates from competitive products
with comparable therapeutic profiles but without abuse deterrent
features. Such events would have a material adverse effect on our
operations and financial condition.
The FDA
also has the authority to revoke or suspend approvals of previously approved
products for cause, to debar companies and individuals from participating in the
drug-approval process, to request recalls of allegedly violative products, to
seize allegedly violative products, to obtain injunctions to close manufacturing
plants allegedly not operating in conformity with current Good Manufacturing
Practices (“cGMP”) and to stop shipments of allegedly violative
products. In the event the FDA takes any such action relating to our
products (if any are approved by FDA), such actions would have a material
adverse effect on our operations and financial condition.
We
must maintain FDA approval to manufacture clinical supplies of our product
candidates at our facility; failure to maintain compliance with FDA requirements
may prevent or delay the manufacture of our product candidates and costs of
manufacture may be higher than expected.
We have
installed the equipment necessary to manufacture clinical trial supplies of our
Aversion® and
Impede™ Technology product candidates in tablet formulations at our Culver,
Indiana facility. To be used in clinical trials, all of our product candidates
must be manufactured in conformity with cGMP regulations. All such product
candidates must be manufactured, packaged, and labeled and stored in accordance
with cGMPs. Modifications, enhancements or changes in manufacturing sites of
marketed products are, in many circumstances, subject to FDA approval, which may
be subject to a lengthy application process or which we may be unable to obtain.
Our Culver, Indiana facility, and those of any third-party manufacturers that we
or our licensees may use, are periodically subject to inspection by the FDA and
other governmental agencies, and operations at these facilities could be
interrupted or halted if the FDA deems such inspections are unsatisfactory.
Failure to comply with FDA or other governmental regulations can result in
fines, unanticipated compliance expenditures, recall or seizure of products,
total or partial suspension of production or distribution, suspension of FDA
review of our product candidates, termination of ongoing research,
disqualification of data for submission to regulatory authorities, enforcement
actions, injunctions and criminal prosecution. We do not have the facilities,
equipment or personnel to manufacture commercial quantities of our product
candidates and therefore must rely on our licensees or other qualified third
party companies with appropriate facilities and equipment to contract
manufacture commercial quantities of products utilizing our Aversion® and
Impede™ Technologies. These licensees are also subject to cGMP
regulations.
We
develop our products, and manufacture clinical supplies, at a single
location. Any disruption at this facility could adversely affect our
business and results of operations.
We rely
on our Culver, Indiana facility for developing our product candidates and the
manufacture of clinical supplies of our product candidates. If the
Culver, Indiana facility were damaged or destroyed, or otherwise subject to
disruption, it would require substantial lead-time to repair or
replace. If our Culver facility were affected by a disaster, we would
be forced to rely entirely on CROs while repairs were being
made. Although we believe we possess adequate insurance for
damage to our property and for the disruption of our business from casualties,
such insurance may not be sufficient to cover all of our potential losses and
may not continue to be available to us on acceptable terms, or at
all. Moreover, any disruptions or delays at our Culver, Indiana
facility could impair our ability to develop our product candidates utilizing
the Aversion® or
Impede™ Technologies, which could adversely affect our business and results of
operations.
Our
operations are subject to environmental, health and safety, and other laws and
regulations, with which compliance is costly and which exposes us to penalties
for non-compliance.
Our
business, properties and product candidates are subject to federal, state and
local laws and regulations relating to the protection of the environment,
natural resources and worker health and safety and the use, management, storage
and disposal of hazardous substances, waste and other regulated
materials. Because we own and operate real property, various
environmental laws also may impose liability on us for the costs of cleaning up
and responding to hazardous substances that may have been released on our
property, including releases unknown to us. These environmental laws
and regulations also could require us to pay for environmental remediation and
response costs at third-party locations where we dispose of or recycle hazardous
substances. The costs of complying with these various environmental
requirements, as they now exist or may be altered in the future, could adversely
affect our financial condition and results of operations.
24
If
our licensees do not satisfy their obligations, we will be unable to develop our
licensed product candidates.
On
October 30, 2007, we entered into an Agreement with King (as more fully
described under the caption “Item 1. Business – King Agreement”). At
December 31, 2009 we had received aggregate payments of $56.2 million from King,
consisting of a $30.0 million non-refundable upfront cash payment, $15.2 million
in reimbursed research and development expenses relating to our licensed product
candidates, $6.0 million in option exercise fees relating to King’s exercise of
its option to license an undisclosed opioid analgesic tablet product and
Vycavert® Tablets,
and a $5.0 million milestone fee relating to our successful achievement of the
primary end points for our pivotal Phase III clinical study for Acurox®
Tablets. Our future revenue, if any, will be derived from
milestone payments and royalties under the King Agreement and under similar
license agreements anticipated to be potentially negotiated and executed with
other pharmaceutical companies. No assurance can be given that we
will receive the milestone and royalty payments provided for in the King
Agreement, or that we will be successful in entering into similar agreements
with other pharmaceutical companies to develop and commercialize products
utilizing our Aversion® or
Impede™ Technologies.
As part
of such license agreements, we will not have day-to-day control over the
activities of our licensees with respect to any product candidate. If
a licensee fails to fulfill its obligations under an agreement with us, we may
be unable to assume the development of the product candidate covered by that
agreement or to enter into alternative arrangements with another third-party. In
addition, we may encounter delays in the commercialization of the product
candidate that is the subject of a license agreement. Accordingly, our ability
to receive any revenue from the product candidates covered by such agreements
will be dependent on the efforts of our licensee. We could be involved in
disputes with a licensee, which could lead to delays in or termination of, our
development and commercialization programs and result in time consuming and
expensive litigation or arbitration. In addition, any such dispute could
diminish our licensee’s commitment to us and reduce the resources they devote to
developing and commercializing our products. If any licensee terminates or
breaches its agreement, or otherwise fails to complete its obligations in a
timely manner, our chances of successfully developing or commercializing our
product candidates would be materially adversely effected. Additionally, due to
the nature of the market for our product candidates, it may be necessary for us
to license all or a significant portion of our product candidates to a single
company thereby eliminating our opportunity to commercialize other product
candidates with other licensees.
If
we fail to maintain our license agreement with King, we may have to reduce or
delay our product candidate development.
Our plan
for developing, manufacturing and commercializing Acurox® Tablets and other
opioid analgesic product candidates utilizing our Aversion® Technology currently
requires us to successfully maintain our license agreement with King to advance
our programs and provide funding to support our expenditures on Acurox® Tablets
and other opioid analgesic product candidates. In addition to other customary
termination provisions, the King Agreement provides that King may terminate the
King Agreement in its entirety if FDA approval of the Acurox® Tablets NDA is not
received prior to March 31, 2010. We anticipate FDA approval of the
NDA for Acurox® Tablets will not occur prior to March 31, 2010 as the Advisory
Committee remains unscheduled. As a result, King may terminate the King
Agreement following such date upon written notice to us. If King
elects to terminate the King Agreement, or if we are otherwise unable to
maintain our existing relationship with King, we may have to limit the size or
scope of, or delay or abandon the development of, Acurox® Tablets and other
opioid analgesic product candidates or undertake and fund development of these
product candidates ourselves. If we were required to fund development and
commercialization efforts with respect to Acurox® Tablets and other opioid
analgesic product candidates on our own, we may need to obtain additional
financing, which may not be available on acceptable terms, or at
all.
If
King is not successful in commercializing Acurox® Tablets
and other licensed product candidates incorporating the Aversion®
Technology our revenues and our business will suffer.
Pursuant
to the King Agreement, King is responsible for manufacturing, marketing,
pricing, promoting, selling, and distributing certain of our product candidates
in the US, Canada and Mexico. If such agreement is terminated in
accordance with its terms, including due to a party’s failure to perform its
obligations or responsibilities under the agreement, then we would need to
commercialize the products ourselves for which we currently have no
infrastructure or alternatively enter into a new agreement with another
pharmaceutical company, of which no assurance can be given. In this
event our revenues and/or royalties for these products could be adversely
impacted.
25
King’s
manufacturing facility is currently the sole commercial source of supply for
Acurox® and our other product candidates licensed to King. If King’s
manufacturing facility fails to obtain sufficient DEA quotas for the opioid
active ingredients contained in such product candidates, fails to source
adequate quantities of active and inactive ingredients, fails to comply with
regulatory requirements, or otherwise experiences disruptions in commercial
supply of our product candidates, product revenue and our royalties could be
adversely impacted.
King has
a diversified product line for which Acurox® and our other product candidates
licensed to King will vie for King’s promotional, marketing, and selling
resources. If King fails to commit sufficient promotional, marketing
and selling resources to our products, product revenue and our royalties could
be adversely impacted.
The
market for our opioid product candidates is highly competitive with many
marketed non abuse deterrent brand and generic products and other abuse
deterrent product candidates in development. If King prices our
product candidates inappropriately, fails to position our products properly,
targets inappropriate physician specialties, or otherwise does not provide
sufficient promotional support, product revenue and our royalties could be
adversely impacted.
The market may
not be receptive to products incorporating our Aversion®
Technology.
The
commercial success of our products will depend on acceptance by health care
providers and others that such products are clinically useful, cost-effective
and safe. There can be no assurance given that our products utilizing the
Aversion® or
Impede™ Technologies would be accepted by health care providers and others.
Factors that may materially affect market acceptance of our product candidates
include but are not limited to:
|
|
·
|
the
relative advantages and disadvantages of our products compared to
competitive products;
|
|
|
·
|
the
relative timing to commercial launch of our products compared to
competitive products;
|
|
|
·
|
the
relative safety and efficacy of our products compared to competitive
products;
|
|
|
·
|
the
product labeling approved by the FDA for our
products;
|
· the
willingness of third party payers to reimburse for our prescription products;
and
· the
willingness of consumers to pay for our products.
Our
product candidates, if successfully developed and commercially launched, will
compete with both currently marketed and new products launched in the future by
other companies. Health care providers may not accept or utilize any of our
products. Physicians and other prescribers may not be inclined to
prescribe our products unless our products demonstrate commercially viable
advantages over other products currently marketed for the same
indications. Consumers may not be willing to purchase our
products. If our products do not achieve market acceptance, we may
not be able to generate significant revenues or become profitable.
If
we, our licensees or others identify serious adverse events or deaths relating
to any of our products once on the market, we may be required to withdraw our
products from the market, which would hinder or preclude our ability to generate
revenues.
We or our
licensees are required to report to relevant regulatory authorities all serious
adverse events or deaths involving our product candidates or approved
products. If we, our licensees, or others identify such events,
regulatory authorities may withdraw their approvals of such products; we or our
licensees may be required to reformulate our products; we or our licensees may
have to recall the affected products from the market and may not be able to
reintroduce them onto the market; our reputation in the marketplace may suffer;
and we may become the target of lawsuits, including class actions
suits. Any of these events could harm or prevent sales of the
affected products and could materially adversely affect our business and
financial condition.
26
In
the event that we or our licensees are successful in bringing any products to
market, our revenues may be adversely affected if we fail to obtain insurance
coverage or adequate reimbursement for our products from third-party
payers.
The
ability of our licensees to successfully commercialize our products may depend
in part on the availability of reimbursement for our prescription products from
government health administration authorities, private health insurers, and other
third-party payers and administrators, including Medicaid and Medicare. We
cannot predict the availability of reimbursement for newly-approved products
utilizing our Aversion®
Technology. Third-party payers and administrators, including state Medicaid
programs and Medicare, are challenging the prices charged for pharmaceutical
products. Government and other third-party payers increasingly are limiting both
coverage and the level of reimbursement for new drugs. Third-party
insurance coverage may not be available to patients for any of our products
candidates. The continuing efforts of government and third-party payers to
contain or reduce the costs of health care may limit our commercial opportunity.
If government and other third-party payers do not provide adequate coverage and
reimbursement for any product utilizing our Aversion®
Technology, health care providers may not prescribe them or patients may ask
their health care providers to prescribe competing products with more favorable
reimbursement. In some foreign markets, pricing and profitability of
pharmaceutical products are subject to government control. In the United States,
we expect there may be federal and state proposals for similar controls. In
addition, we expect that increasing emphasis on managed care in the United
States will continue to put pressure on the pricing of pharmaceutical products.
Cost control initiatives could decrease the price that we or our licensees
charge for any of our products in the future. Further, cost control initiatives
could impair our ability or the ability of our licensees to commercialize our
products and our ability to earn revenues from commercialization.
Consolidation
in the healthcare industry could lead to demands for price concessions or to the
exclusion of some suppliers from certain of our markets, which could have an
adverse effect on our business, financial condition or results of
operations.
Because
healthcare costs have risen significantly, numerous initiatives and reforms by
legislatures, regulators and third-party payers to curb these cost increases
have resulted in a trend in the healthcare industry to consolidate product
suppliers and purchasers. As the healthcare industry consolidates,
competition among suppliers to provide products to purchasers has become more
intense. This in turn has resulted and will likely continue to result
in greater pricing pressures and the exclusion of certain suppliers from
important market segments as group purchasing organizations, and large single
accounts continue to use their market power to influence product pricing and
purchasing decisions. We expect that market demand, government
regulation, third-party reimbursement policies and societal pressures will
continue to influence the worldwide healthcare industry, resulting in further
business consolidations, which may exert further downward pressure on the prices
of our anticipated products. This downward pricing pressure may adversely impact
our business, financial condition or results of operations.
Our
success depends on our ability to protect our intellectual
property.
Our
success depends on our ability to obtain and maintain patent protection for
products developed utilizing our technologies, in the United States and in other
countries, and to enforce these patents. The patent positions of pharmaceutical
firms, including us, are generally uncertain and involve complex legal and
factual questions. Notwithstanding our receipt of U.S. Patent No. 7,201,920 and
U.S. Patent No. 7,476,402 from the United States Patent and Trademark Office
(“USPTO”) encompassing our opioid product candidates utilizing our Aversion®
Technology, there is no assurance that any of our patent claims in our other
pending non-provisional and provisional patent applications relating to our
technologies will issue or if issued, that any such patent claims will be valid
and enforceable against third-party infringement or that our products will not
infringe any third-party patent or intellectual property. Moreover, any patent
claims relating to our technologies may not be sufficiently broad to protect the
our products. In addition, issued patent claims may be challenged, potentially
invalidated or potentially circumvented. Our patent claims may not afford us
protection against competitors with similar technology or permit the
commercialization of our products without infringing third-party patents or
other intellectual property rights.
Our
success also depends on our not infringing patents issued to others. We may
become aware of patents belonging to competitors and others that could require
us to obtain licenses to such patents or alter our technologies. Obtaining such
licenses or altering our technology could be time consuming and costly. We may
not be able to obtain a license to any technology owned by or licensed to a
third party that we or our licensees require to manufacture or market one or
more of our products. Even if we can obtain a license, the financial and other
terms may be disadvantageous.
27
Our
success also depends on maintaining the confidentiality of our trade secrets and
know-how. We seek to protect such information by entering into confidentiality
agreements with employees, potential licensees, raw material suppliers, contract
research organizations, potential investors, consultants and other parties.
These agreements may be breached by such parties. We may not be able to obtain
an adequate, or perhaps, any remedy to such a breach. In addition, our trade
secrets may otherwise become known or be independently developed by our
competitors. Our inability to protect our intellectual property or to
commercialize our products without infringing third-party patents or other
intellectual property rights would have a material adverse affect on our
operations and financial condition.
We
may become involved in patent litigation or other intellectual property
proceedings relating to our Aversion® or
Impede™ Technologies or product candidates which could result in liability for
damages or delay or stop our development and commercialization
efforts.
The
pharmaceutical industry has been characterized by significant litigation and
other proceedings regarding patents, patent applications and other intellectual
property rights. The situations in which we may become parties to such
litigation or proceedings may include:
|
|
·
|
litigation
or other proceedings we may initiate against third parties to enforce our
patent rights or other intellectual property
rights;
|
|
|
·
|
litigation
or other proceedings we may initiate against third parties seeking to
invalidate the patents held by such third parties or to obtain a judgment
that our products do not infringe such third parties’
patents;
|
|
|
·
|
litigation
or other proceedings third parties may initiate against us to seek to
invalidate our patents or to obtain a judgment that third party products
do not infringe our patents;
|
|
|
·
|
if
our competitors file patent applications that claim technology also
claimed by us, we may be forced to participate in interference or
opposition proceedings to determine the priority of invention and whether
we are entitled to patent rights on such invention;
and
|
|
|
·
|
if
third parties initiate litigation claiming that our products infringe
their patent or other intellectual property rights, we will need to defend
against such proceedings.
|
The costs
of resolving any patent litigation or other intellectual property proceeding,
even if resolved in our favor, could be substantial. Many of our potential
competitors will be able to sustain the cost of such litigation and proceedings
more effectively than we can because of their substantially greater resources.
Uncertainties resulting from the initiation and continuation of patent
litigation or other intellectual property proceedings could have a material
adverse effect on our ability to compete in the marketplace. Patent litigation
and other intellectual property proceedings may also consume significant
management time.
Our
technologies may be found to infringe claims of patents owned by others. If we
determine or if we are found to be infringing a patent held by another party, we
or our licensees might have to seek a license to make, use, and sell the
patented technologies. In that case, we or our licensees might not be able to
obtain such license on acceptable terms, or at all. The failure to obtain a
license to any technology that may be required would materially harm our
business, financial condition and results of operations. If a legal action is
brought against us, we could incur substantial defense costs, and any such
action might not be resolved in our favor. If such a dispute is resolved against
us, we may have to pay the other party large sums of money and use of our
technology and the testing, manufacturing, marketing or sale of one or more of
our products could be restricted or prohibited. Even prior to resolution of such
a dispute, use of our technology and the testing, manufacturing, marketing or
sale of one or more of our products could be restricted or
prohibited.
We are
aware of a competitor who has suggested to the USPTO that the USPTO should
declare an interference between that competitor’s pending patent application and
one of our U.S. patents for the Aversion®
Technology. We believe that there is no valid basis for declaring
such an interference and that even if such an interference were declared, that
we would prevail. There can be no assurance, however, that such an
interference will not be declared or if declared, that we will ultimately
succeed such that this competitor would not obtain patent claims which could
encompass our lead product candidate and other product candidates in
development.
We are
aware of certain United States and international pending patent applications
owned by third parties with claims potentially encompassing our product
candidates. While we do not expect the claims contained in such pending patent
applications will issue in their present form, there can be no assurance that
such patent applications will not issue as patents with claims encompassing one
or more of our product candidates. If such patent applications result
in valid and enforceable issued patents, containing claims in their current form
or otherwise encompassing our products we or our licensees may be required to
obtain a license to such patents, should one be available, or alternatively,
alter our products so as to avoid infringing such third-party patents. If we or
our licensees are unable to obtain a license on commercially reasonable terms,
or at all, we or our licensees could be restricted or prevented from
commercializing our products. Additionally, any alterations to our products or
our technologies could be time consuming and costly and may not result in
technologies or products that are non-infringing or commercially
viable.
28
We cannot
assure you that our products and/or actions in developing our products will not
infringe third-party patents. Our failure to avoid infringing
third-party patents and intellectual property rights in the development and
commercialization of our products would have a material adverse affect on our
operations and financial condition.
We
may be exposed to product liability claims and may not be able to obtain
adequate product liability insurance.
Our
business exposes us to potential product liability risks, which are inherent in
the testing, manufacturing, marketing and sale of pharmaceutical products.
Product liability claims might be made by patients, or health care providers or
others that sell or consume our products. These claims may be made even with
respect to those products that possess regulatory approval for commercial
sale. We are currently covered by clinical trial product liability
insurance on a claims-made basis. This coverage may not be adequate to cover any
product liability claims. Product liability coverage is expensive. In the
future, we may not be able to maintain or obtain such product liability
insurance at a reasonable cost or in sufficient amounts to protect us against
losses due to product liability claims. Any claims that are not covered by
product liability insurance could have a material adverse effect on our
business, financial condition and results of operations.
The
pharmaceutical industry is characterized by frequent litigation. Those companies
with significant financial resources will be better able to bring and defend any
such litigation. No assurance can be given that we would not become involved in
such litigation. Such litigation may have material adverse consequences to our
financial condition and results of operations.
We
face significant competition which may result in others developing or
commercializing products before or more successfully than we do.
The
pharmaceutical industry is highly competitive and is affected by new
technologies, governmental regulations, health care legislation, availability of
financing, litigation and other factors. If our product candidates receive FDA
approval, they will compete with a number of existing and future drug products
and therapies developed, manufactured and marketed by others. Existing or future
competing products may provide greater therapeutic convenience, clinical or
other benefits for a specific indication than our products, or may offer
comparable performance at lower costs. If our products are unable to capture and
maintain market share, we or our licensees may not achieve significant product
revenues and our financial condition and results of operations will be
materially adversely affected.
We or our
licensees will compete for market share against fully integrated pharmaceutical
companies or other companies that collaborate with larger pharmaceutical
companies, academic institutions, government agencies and other public and
private research organizations. Many of these competitors have products already
approved, marketed or in development. In addition, many of these competitors,
either alone or together with their collaborative partners, operate larger
research and development programs, have substantially greater financial
resources, experience in developing products, obtaining FDA and other regulatory
approvals, formulating and manufacturing drugs, and commercializing products
than we do.
We are
concentrating a substantial majority of our efforts and resources on developing
product candidates utilizing our Aversion®
Technology. The commercial success of products utilizing Aversion®
Technology will depend, in large part, on the intensity of competition, FDA
approved product labeling for our products compared to competitive products, and
the relative timing and sequence for commercial launch of new products by other
companies developing, marketing, selling and distributing products that compete
with the products utilizing Aversion®
Technology. Alternative technologies and non-opioid products are
being developed to improve or replace the use of opioid analgesics. In the event
that such alternatives to opioid analgesics are widely adopted, then the market
for products utilizing our Aversion®
Technology may be substantially decreased thus reducing our ability to generate
future profits.
29
Key
personnel are critical to our business and our success depends on our ability to
retain them.
We are
dependent on our management and scientific team, including Andrew D. Reddick,
our President and Chief Executive Officer, Ron J. Spivey, Ph.D., our Senior
Scientific Advisor, Robert Jones, our Senior Vice President and Chief Operating
Officer, and Albert W. Brzeczko, Ph.D, our Vice President of Technical
Affairs. We may not
be able to attract and retain personnel on acceptable terms given the intense
competition for such personnel among biotechnology, pharmaceutical and
healthcare companies, universities and non-profit research institutions. While
we have employment agreements with certain employees, all of our employees are
at-will employees who may terminate their employment at any time. We do not have
key personnel insurance on any of our officers or employees. The loss of any of
our key personnel, or the inability to attract and retain such personnel, may
significantly delay or prevent the achievement of our product and technology
development and business objectives and could materially adversely affect our
business, financial condition and results of operations.
The
U.S. Drug Enforcement Administration (“DEA”) limits the availability of the
active ingredients used in our product candidates and, as a result, our quota
may not be sufficient to complete clinical trials or may result in development
delays.
The DEA
regulates certain finished drug products and active pharmaceutical ingredients,
including certain opioid active pharmaceutical ingredients and pseudoephedrine
HCl in our current product candidates. Consequently, their manufacture,
research, shipment, storage, sale and use are subject to a high degree of
regulation. Furthermore, the amount of active ingredients we can obtain for our
clinical trials is limited by the DEA and our quota may not be sufficient to
complete clinical trials. There is a risk that DEA regulations may interfere
with the supply of the products used in our clinical trials.
Prior
ownership changes limit our ability to use our tax net operating loss
carryforwards.
Significant
equity restructuring often results in an Internal Revenue Section 382 ownership
change that limits the future use of Net Operating Loss (“NOL”) carryforwards
and other tax attributes. We have determined that an ownership change
(as defined by Section 382 of the Internal Revenue Code) did occur as a result
of restructuring that occurred in 2004. Neither the amount of our NOL
carryforwards nor the amount of limitation of such carryforwards claimed by us
have been audited or otherwise validated by the Internal Revenue Service, which
could challenge the amount we have calculated. The recognition and
measurement of our tax benefit includes estimates and judgment by our
management, which includes subjectivity. Changes in estimates may create
volatility in our tax rate in future periods based on new information about
particular tax positions that may cause management to change its
estimates. If we establish a contingent tax liability reserve, interest
and penalties related to uncertain tax positions would be classified as general
and administrative expenses.
Risks
Relating to Our Common Stock
Our
quarterly results of operations will fluctuate, and these fluctuations could
cause our stock price to decline.
Our
quarterly and annual operating results are likely to fluctuate in the future.
These fluctuations could cause our stock price to decline. The nature of our
business involves variable factors, such as the timing of the research,
development and regulatory submissions of our product candidates that could
cause our operating results to fluctuate. The forecasting of the
timing of sales of our product candidates is difficult due to the uncertainty
inherent in seeking FDA and other necessary approvals for our product
candidates. As a result, in some future quarters or years our
clinical, financial or operating results may not meet the expectations of
securities analysts and investors which could result in a decline in the price
of our stock.
Volatility
in stock prices of other companies may contribute to volatility in our stock
price.
The
market price of our common stock, like the market price for securities of
pharmaceutical and biotechnology companies, has historically been highly
volatile. The stock market from time to time experiences significant price and
volume fluctuations unrelated to the operating performance of particular
companies. Factors, such as fluctuations in our operating results, future sales
of our common stock, announcements of technological innovations or new
therapeutic products by us or our competitors, announcements regarding
collaborative agreements, laboratory or clinical trial results, government
regulation, developments in patent or other proprietary rights, public concern
as to the safety of drugs developed by us or others, changes in reimbursement
policies, comments made by securities analysts and general market conditions may
have a substantial effect on the market price of our common stock. In the past,
following periods of volatility in the market price of a company’s securities,
securities class action litigation has often been instituted. A securities class
action suit against us could result in substantial costs, potential liabilities
and the diversion of management’s attention and resources and result in a
material adverse affect on our financial condition and results of
operations.
30
Our
stock price has been volatile and there may not be an active, liquid trading
market for our common stock.
Our stock
price has experienced significant price and volume fluctuations and may continue
to experience volatility in the future. Factors that have a material
impact on the price of our common stock, in addition to the other issues
described herein, include results of or delays in our pre-clinical and clinical
studies, any delays in, or failure to receive, FDA approval of our product
candidates, the success of our license agreement with King, announcements of
technological innovations or new commercial products by us or others,
developments in patents and other proprietary rights by us or others, future
sales of our common stock by existing stockholders, regulatory developments or
changes in regulatory guidance, the departure of our officers, directors or key
employees, and period-to-period fluctuations in our financial
results. Also, you may not be able to sell your shares at the best
market price if trading in our stock in not active or if the volume is
low. There is no assurance that an active trading market for our
common stock will be maintained on the NASDAQ Capital Market.
The
National Association of Securities Dealers, Inc., or NASD, and the Securities
and Exchange Commission, or SEC, have adopted rules relating to the listing of
publicly traded stock. If we were unable to continue to comply with such rules,
we could be delisted from trading on the NASDAQ Capital Market and thereafter
trading in our common stock, if any, would be conducted through the
Over-the-Counter Bulletin Board of the NASD. As a consequence of such delisting,
an investor would likely find it more difficult to dispose of, or to obtain
quotations as to the price of, our common stock. Delisting of our common stock
from the NASDAQ Capital Market could also result in lower prices per share of
our common stock than would otherwise prevail.
We
do not have a history of paying dividends on our common stock.
Historically
we have not declared and paid any cash dividends on our common
stock. We intend to retain all of our earnings for the
foreseeable future to finance the operation and expansion of our
business. As a result, you may only receive a return on your
investment in our common stock if the market price of our common stock
increases.
GCE
Holdings LLC can control all matters requiring approval by
shareholders.
GCE
Holdings LLC beneficially owns approximately 75.9% of our outstanding common
stock as of December 31, 2009 (calculated in accordance with Rule 13d-3
promulgated under the Securities Exchange Act of 1934, as
amended). As a result, GCE Holdings LLC, in view of its ownership
percentage of our common stock, will be able to control all matters requiring
approval by our shareholders, including the approval or rejection of mergers,
sales or licenses of all or substantially all of our assets, or other business
combination transactions. The interests of GCE Holdings LLC may not always
coincide with the interests of our other shareholders and as such we may take
action in advance of its interests to the detriment of our other
shareholders. Accordingly, you may not be able to influence any
action we take or consider taking, even if it requires a shareholder
vote.
We
are currently a “Controlled Company” within the meaning of the NASDAQ Capital
Market Listing Requirements and, as a result, are exempt from certain corporate
governance requirements.
Because
GCE Holdings LLC controls more than 50% of the voting power of our common stock,
we are currently considered to be a “controlled company” for purposes of a
NASDAQ Capital Market listing requirements. As such, we are
permitted, and have elected, to opt out of the NASDAQ Capital Market listing
requirements that would otherwise require our board of directors to have a
majority of independent directors, our board nominations to be selected, or
recommended for the board’s selection either by a nominating committee comprised
entirely of independent directors or by a majority of independent directors, and
our compensation committee to be comprised entirely of independent
directors. Accordingly, you may not have the same protections
afforded to stockholders of companies that are subject to all of the NASDAQ
Capital Market corporate governance requirements.
31
Any
future sale of a substantial number of shares included in our current
registration statement could depress the trading price of our stock, lower our
value and make it more difficult for us to raise capital.
In
accordance with the terms of the Securities Purchase Agreement dated August 20,
2007 between us and the investors named therein, we filed a registration
statement with the SEC to register the shares included in our Units issued
pursuant to the Securities Purchase Agreement, including shares underlying
warrants included in the Units. In addition, pursuant to the exercise
of previously granted piggyback registration rights, each of GCE Holdings, LLC,
Galen Partners III, L.P., Galen Partners International III, L.P., Galen Employee
Fund III, L.P., Care Capital Investments II, LP, Care Capital Offshore
Investments II, LP and Essex Woodlands Health Ventures V, L.P. have exercised
their piggyback registration rights to include an aggregate of 26,584,016 shares
in such registration statement. As a result, 34,243,273 shares
(representing approximately 64% of our shares outstanding on a fully-diluted
basis – including all derivative securities, whether or not currently
exercisable on a pre-reverse stock basis) were included in the registration
statement for resale by selling stockholders. Such registration
statement was declared effective by the SEC on November 20, 2007. If
some or all of such shares included in such registration statement are sold by
our affiliates and others it may have the effect of depressing the trading price
of our common stock. In addition, such sales could lower our value
and make it more difficult for us to raise capital if needed in the
future.
ITEM
1B. UNRESOLVED STAFF COMMENTS
The
Company has received no written comments regarding periodic or current reports
from the staff of the SEC that were issued 180 days or more preceding the end of
its 2009 fiscal year that remain unresolved.
ITEM
2. PROPERTIES
We lease
from an unaffiliated Lessor, approximately 1,600 square feet of administrative
office space at 616 N. North Court, Suite 120, Palatine, Illinois 60067. The
lease agreement has a term expiring March 31, 2011. The lease
agreement provides for rent, property taxes, common area maintenance, and
janitorial services on an annualized basis of approximately $29,200 per
year. We utilize this lease space for our administrative, marketing
and business development functions.
We
conduct research, development, laboratory, development scale and NDA submission
batch scale manufacturing and other activities relating to developing product
candidates using Aversion® and
Impede™ Technologies at our facility located at 16235 State Road 17, Culver,
Indiana. At this location, our wholly-owned subsidiary Acura Pharmaceutical
Technologies, Inc., owns a 28,000 square foot facility with 7,000 square feet of
warehouse, 10,000 square feet of manufacturing space, 6,000 square feet of
research and development labs and 5,000 square feet of administrative and
storage space. The facility is located on 30 acres of land.
ITEM
3. LEGAL PROCEEDINGS
None.
ITEM
4. (RESERVED)
32
PART
II
ITEM
5. MARKET FOR REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER
PURCHASES OF EQUITY SECURITIES
Market
and Market Prices of Common Stock
Until
February 1, 2008, our common stock was traded on the Over-the-Counter (“OTC”)
Bulletin Board. Commencing February 4, 2008, our common stock was
admitted for trading on the NASDAQ Capital Market under the symbol
“ACUR”.
Set forth
below for the period January 1, 2008 through February 1, 2008 are the high and
low bid prices for our common stock for trading in our common stock on the OTC
Bulletin Board as reported by the OTC Bulletin Board. Such over-the-counter
market quotations reflect inter-dealer prices, without retail mark-up, mark-down
or commission and may not necessarily represent actual
transactions.
|
|
Bid
Prices
|
|||||||
|
Period
|
High
$
|
Low
$
|
||||||
| 2008 Fiscal Year | ||||||||
|
First
Quarter (through February 1, 2008)
|
8.60 | 5.90 | ||||||
Set forth
below for the periods indicated are the high and low sales prices for trading in
our common stock on the NASDAQ Capital Market as reported by the NASDAQ Capital
Market.
|
|
Sale
Prices
|
|||||||
|
Period
|
High
$
|
Low
$
|
||||||
| 2008 Fiscal Year | ||||||||
|
First
Quarter (commencing February 4, 2008)
|
10.50 | 6.75 | ||||||
|
Second
Quarter
|
10.00 | 7.49 | ||||||
|
Third
Quarter
|
8.97 | 5.79 | ||||||
|
Fourth
Quarter
|
7.89 | 3.43 | ||||||
|
2009
Fiscal Year
|
||||||||
|
First
Quarter
|
7.69 | 4.07 | ||||||
|
Second
Quarter
|
9.00 | 4.85 | ||||||
|
Third
Quarter
|
6.51 | 4.85 | ||||||
|
Fourth
Quarter
|
5.79 | 4.00 | ||||||
|
2010
Fiscal Year
|
||||||||
|
First
Quarter (through January 31, 2010)
|
5.54 | 4.85 | ||||||
Holders
There
were approximately 600 holders of record of our common stock on February
28, 2010. This number, however,
does not reflect the ultimate number of beneficial holders of our common
stock.
Dividend
Policy
The
payment of cash dividends is subject to the discretion of our Board of Directors
and is dependent upon many factors, including our earnings, our capital needs
and our general financial condition. Historically we have not paid any cash
dividends.
Securities
Authorized for Issuance under Equity Compensation Plans
Reference
is made to “Item 11 - Executive Compensation - Restricted Stock Unit Award Plan;
and Securities Authorized for Issuance under Equity Compensation
Plans”.
33
ITEM
6. SELECTED FINANCIAL DATA
The
selected consolidated financial data presented below for the years ended
December 31, 2009, 2008, 2007, 2006 and 2005 are derived from our audited
Consolidated Financial Statements. The Consolidated Financial Statements as of
December 31, 2009 and 2008 and for each of the years in the three-year period
ended December 31, 2009, and the reports thereon, are included elsewhere in this
Report. The selected financial information as of December 31, 2007, 2006, and
2005 and for the years ended December 31, 2006 and 2005 are derived from our
audited Consolidated Financial Statements not presented in this
Report.
The
information set forth below is qualified by reference to, and should be read in
conjunction with, the Consolidated Financial Statements and related notes
thereto included elsewhere in this Report and "Item 7. Management's Discussion
and Analysis of Financial Condition and Results of Operations". All share data
gives effect to the 1 for 10 reverse stock split implemented December 5,
2007.
|
OPERATING DATA (in thousands) except per share data
|
2009
|
2008
|
2007
|
2006
|
2005
|
|||||||||||||||
|
Revenues
|
$ | 3,835 | $ | 44,437 | $ | 6,404 | $ | — | $ | — | ||||||||||
|
Operating
expenses:
|
||||||||||||||||||||
|
Research
and development(1)
|
5,673 | 14,322 | 7,169 | 5,172 | 6,265 | |||||||||||||||
|
Marketing,
general and administrative expenses(2)
|
11,662 | 9,133 | 4,141 | 5,654 | 5,296 | |||||||||||||||
|
Interest
expense
|
— | — | (1,207 | ) | (1,140 | ) | (636 | ) | ||||||||||||
|
Interest
income
|
147 | 780 | 268 | 18 | 36 | |||||||||||||||
|
Amortization
of debt discount & deferred private debt offering
costs
|
— | — | (2,700 | ) | (183 | ) | — | |||||||||||||
|
(Loss)
gain on fair value change of conversion features
|
— | — | (3,483 | ) | 4,235 | — | ||||||||||||||
|
(Loss)
gain on fair value change of common stock warrants
|
— | — | (1,905 | ) | 2,164 | — | ||||||||||||||
|
(Loss)
gain on asset disposals
|
(3 | ) | (2 | ) | 22 | (22 | ) | 81 | ||||||||||||
|
Other
(expense) income
|
— | (1 | ) | (3 | ) | (213 | ) | 5 | ||||||||||||
|
(Loss)
income before income tax
|
(13,356 | ) | 21,759 | (13,914 | ) | (5,967 | ) | (12,075 | ) | |||||||||||
|
Income
tax expense (benefit)
|
2,479 | 7,285 | (9,600 | ) | — | — | ||||||||||||||
|
Net
(loss) income
|
$ | (15,835 | ) | $ | 14,474 | $ | (4,314 | ) | $ | (5,967 | ) | $ | (12,075 | ) | ||||||
|
Net
(loss) income per share: Basic
|
$ | (0.35 | ) | $ | 0.32 | $ | (0.11 | ) | $ | (0.75 | ) | $ | (1.81 | ) | ||||||
|
Net
(loss) income per share: Diluted
|
$ | (0.35 | ) | $ | 0.29 | $ | (0.11 | ) | $ | (0.75 | ) | $ | (1.81 | ) | ||||||
|
Weighted
average shares used in computing net income (loss) per
share: Basic
|
45,932 | 45,675 | 39,157 | 34,496 | 6,680 | |||||||||||||||
|
Weighted
average shares used in computing net income (loss) per
share: Diluted
|
45,932 | 49,416 | 39,157 | 34,496 | 6,680 | |||||||||||||||
(1) Includes stock-based
compensation expense of $1,904, $560, $371, $2,067, and $3,325 for years 2009,
2008, 2007, 2006 and 2005, respectively.
(2) Includes stock-based
compensation expense of $7,300, $3,290, $544, $3,517, and $3,133 for years 2009,
2008, 2007, 2006 and 2005, respectively.
|
BALANCE SHEET DATA(3)
(in thousands)
|
2009
|
2008
|
2007
|
2006
|
2005
|
|||||||||||||||
|
Working
capital (deficiency)
|
$ | 28,750 | $ | 35,991 | $ | 22,306 | $ | (28,641 | ) | $ | (2,478 | ) | ||||||||
|
Total
assets
|
31,917 | 42,961 | 45,628 | 1,619 | 1,792 | |||||||||||||||
|
Total
debt, net (4)
|
— | — | — | 28,787 | 7,613 | |||||||||||||||
|
Total
liabilities
|
2,007 | 5,897 | 26,908 | 39,899 | 7,954 | |||||||||||||||
|
Accumulated
deficit
|
(323,221 | ) | (307,386 | ) | (321,860 | ) | (317,543 | ) | (291,616 | ) | ||||||||||
|
Stockholders'
equity (deficit)
|
$ | 29,910 | $ | 37,064 | $ | 18,720 | $ | (38,280 | ) | $ | (6,162 | ) | ||||||||
(3) Reflects impact of $30
million received from King in December, 2007.
(4) Includes estimated fair value of
conversion features of convertible debt outstanding as of December 31,
2006.
34
ITEM
7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF
OPERATIONS
This
discussion and analysis should be read in conjunction with our financial
statements and accompanying notes included elsewhere in this Report. Operating
results are not necessarily indicative of results that may occur in the future
periods. Certain statements in this Report under this Item 7, Item 1,
"Business", Item 1A, “Risk Factors” and elsewhere in this Report constitute
"forward-looking statements" within the meaning of the Private Securities
Litigation Reform Act of 1995. Such forward-looking statements involve known and
unknown risks, uncertainties and other factors which may cause our actual
results, performance or achievements or industry results, to be materially
different from any future results, performance, or achievements expressed or
implied by such forward-looking statements. See page 3 of this Report for a
description of the most significant of such factors.
Company
Overview
We are a
specialty pharmaceutical company engaged in research, development and
manufacture of product candidates intended to provide abuse deterrent features
and benefits utilizing our proprietary Aversion® and
Impede™ Technologies. Our Aversion® Technology opioid analgesic
product candidates are intended to effectively relieve pain while simultaneously
discouraging common methods of opioid product misuse and abuse,
including:
|
|
·
|
intravenous
injection of dissolved tablets or
capsules;
|
|
|
·
|
nasal
snorting of crushed tablets or capsules;
and
|
|
|
·
|
intentional
swallowing of excess quantities of tablets or
capsules.
|
In
addition to Acurox®, our
lead product candidate, we (and/or our licensee, King Pharmaceuticals Research
and Development, Inc.) are developing Vycavert®
(hydrocodone bitartrate/niacin/APAP), Acuracet®
(oxycodone HCl/niacin/APAP) and additional undisclosed opioid product candidates
utilizing Aversion® Technology. Four opioid product candidates are
licensed to King under the King Agreement. We are also developing a
benzodiazepine tablet product candidate utilizing our Aversion®
Technology.
Our research efforts continue to
investigate and develop novel mechanisms to incorporate abuse deterrent
characteristics into abused and misused pharmaceutical products. In
this regard we are engaged in laboratory testing of a new product candidate
developed with our novel Impede™ Technology. Impede™
Technology is primarily intended to inhibit the conversion of pseudoephedrine
HCl (a legally available nasal decongestant) into methamphetamine (an illicit
and frequently abused drug).
Company’s
Present Financial Condition
At
December 31, 2009, we had cash and cash equivalents of $30.2 million compared to
$35.4 million of cash, cash equivalents, and short-term investments at December
31, 2008. We had working capital of $28.8 million at December 31, 2009 compared
to working capital of $36.0 million at December 31, 2008. We had an
accumulated deficit of approximately $323.2 million and $307.4 million at
December 31, 2009 and December 31, 2008, respectively. We had a loss from
operations of approximately $13.5 million and a net loss of $15.8 million for
the year ended December 31, 2009, compared to income from operations of $21.0
million and net income of $14.5 million for the year ended December 21,
2008. As of February 28, 2010 we had cash and cash equivalents of
approximately $28.4 million.
During
the year ended December 31, 2009, we recognized revenues of $3.8 million derived
from the $3.0 million amortized portion of the $30.0 million upfront cash
payment received from King in December 2007 and $0.8 million for reimbursement
of research and development expenses for Acurox®
Tablets licensed to King under the King Agreement. During the year
ended December 31, 2008, we recognized revenues of $44.4 million derived from
the $21.9 million amortized portion of the $30.0 million upfront cash payment
received from King in December 2007, $6.0 million in option exercise fees paid
to us by King for licenses to the third and fourth opioid analgesic product
candidates, $5.0 million in an Acurox®
Tablet development milestone payment received from King, and $11.5 million paid
to us by King for reimbursement of research and development expenses for
Acurox®
Tablets. We have yet to generate any royalty revenues from product
sales. To fund our continued operations, we expect to rely on our
current cash resources and additional payments that may be made under the King
Agreement and under future license agreements with other pharmaceutical company
partners, of which there can be no assurance of obtaining. Our cash
requirements for operating activities may increase in the future as we continue
to conduct pre-clinical studies and clinical trials for our product candidates,
maintain, defend, if necessary and expand the scope of our intellectual
property, hire additional personnel, or invest in other areas.
35
In 2007,
we recognized revenue of $6.4 million derived from $3.4 million which was the
amortized portion of the $30.0 million non-refundable cash payment received from
King in December 2007 and $3.0 million received from King in reimbursement of
our Acurox® Tablet
development expenses incurred under the King Agreement. Our losses
have resulted principally from costs incurred in connection with research and
development activities, salaries and other personnel-related costs and general
corporate expenses. Research and development activities include costs
of pre-clinical studies, clinical trials, and clinical trial product supplies
associated with our product candidates. Salaries and other
personnel-related costs include non-cash, stock-based compensation associated
with stock options and restricted stock units granted to employees and
non-employee directors.
Results
of Operations for the Years Ended December 31, 2009 and 2008
|
December 31
|
||||||||||||||||
|
2009
|
2008
|
Change
|
||||||||||||||
|
|
$000’s
|
$000’s
|
Percent
|
|||||||||||||
| Revenues | ||||||||||||||||
|
Program
fee revenue
|
3,077 | 27,941 | (24,864 | ) | (89 | )% | ||||||||||
|
Collaboration
fee revenue
|
758 | 11,496 | (10,738 | ) | (93 | )% | ||||||||||
|
Milestone
revenue
|
— | 5,000 | (5,000 | ) | (100 | )% | ||||||||||
|
Total
revenue
|
3,835 | 44,437 | (40,702 | ) | (92 | )% | ||||||||||
|
Operating
expenses
|
||||||||||||||||
|
Research
and development expense
|
5,673 | 14,322 | (8,649 | ) | (60 | )% | ||||||||||
|
Marketing,
general and administrative expense
|
11,662 | 9,133 | 2,529 | 28 | % | |||||||||||
|
Total
operating expenses
|
17,335 | 23,455 | (6,120 | ) | (26 | )% | ||||||||||
|
(Loss)
income from operations
|
(13,500 | ) | 20,982 | (34,482 | ) | (164 | )% | |||||||||
|
Other
income (expense)
|
||||||||||||||||
|
Interest
income
|
147 | 780 | (633 | ) | (81 | )% | ||||||||||
|
Loss
on asset disposals
|
(3 | ) | (2 | ) | (1 | ) | 50 | % | ||||||||
|
Other
expense
|
— | (1 | ) | 1 | 100 | % | ||||||||||
|
Total
other income
|
144 | 777 | (633 | ) | (81 | )% | ||||||||||
|
(Loss)
income before income tax
|
(13,356 | ) | 21,759 | (35,115 | ) | (161 | )% | |||||||||
|
Income
tax expense
|
2,479 | 7,285 | (4,806 | ) | (66 | )% | ||||||||||
|
Net
(loss) income
|
(15,835 | ) | 14,474 | (30,309 | ) | (209 | )% | |||||||||
Revenue
In
December 2007, King paid us a $30.0 million upfront fee in connection with the
closing of the King Agreement. Program fee revenue recognized during 2009 from
amortizing this upfront fee was $3.0 million compared to $21.9 million in 2008.
We have assigned an equal portion of the program fee revenue to each of three
product candidates identified under the King Agreement. We have
completed our development activities on 2 of the 3 product candidates and have
fully amortized the portion of the upfront fee for those two product candidates
in 2008. We currently estimate the development period for the third
product candidate to end in December, 2010. Also, included in program fee
revenue in 2008 are two $3.0 million option exercise fees paid by King to us in
May and December 2008, respectively, upon the exercise of its option to license
a third and fourth opioid analgesic product candidate under the King Agreement.
In June 2008, King paid us a $5.0 million milestone payment for successfully
achieving the primary end points in our pivotal Phase III study, AP-ADF-105
for Acurox® Tablets. We had no milestone revenue in 2009.
36
Collaboration
revenue recognized in 2009 was $0.8 million for reimbursement, pursuant to the
King Agreement, of our Acurox® Tablets development and regulatory expenses
incurred during 2009. We invoice King in arrears on a calendar quarter basis for
our reimbursable development and regulatory expenses under the King
Agreement. We expect the amount and timing of collaboration revenue
to fluctuate in relation to the amount and timing of the underlying research and
development expenses. The Company had collaboration revenue of $11.5 million for
2008.
Operating
Expenses
Research
and development expense during 2009 and 2008 were primarily for product
candidates utilizing our Aversion® Technology, including costs of preclinical,
clinical trials, clinical supplies and related formulation and design costs,
salaries and other personnel related expenses, and facility
costs. Included in the 2009 and 2008 results are non-cash stock-based
compensation charges of $1.9 million and $0.6 million, respectively, associated
with the grant of stock options and restricted stock units. Excluding the
stock-based compensation expense, there is a $10.0 million decrease in
development expenses primarily attributable to a reduction of our clinical study
costs. During 2008, we conducted and completed our pivotal Phase III clinical
trial for Acurox®.
Marketing
expenses during 2009 and 2008 consisted of Aversion® Technology customized
market data research studies. Our general and administrative expenses primarily
consisted of legal, audit and other professional fees, corporate insurance, and
payroll.. Included in the 2009 and 2008 results are non-cash
stock-based compensation charges of $7.3 million and $3.3 million, respectively,
associated with the grant of stock options and restricted stock units. Excluding
the stock-based compensation expense, there is a decrease of $1.5 million in
marketing, general and administrative expenses including reductions of $0.9
million in payroll, $0.3 million legal and accounting professional services, and
$0.3 million state franchise taxes.
Other
Income (Expense)
During
2009 and 2008, we had no debt and cash proceeds received pursuant to the King
Agreement were primarily invested in money market funds, U.S. Treasury Bills,
bank commercial paper, and overnight sweep investments, resulting in interest
income of $0.1 million and $0.8 million, respectively.
Income
Tax Expense (Benefit)
Deferred
income taxes have been recognized in prior years for temporary differences
between financial statement and income tax bases of assets and liabilities and
loss carry-forwards for which income tax benefits are expected to be realized in
future years. During 2009 we recorded a valuation allowance to reduce
our net deferred income tax assets to the amount that is more likely than not to
be realized. Net loss for 2009 includes a provision for income tax expense of
$2.5 million for such adjustment to our valuation allowance as we determined
that the utilization of certain deferred tax assets recorded in prior years
cannot be reasonably predicted based upon financial forecast for succeeding
years.
Net
income for 2008 includes a provision for income tax expense of $8.5 million
which has been partially offset by $1.2 million of income tax benefits recorded
from the anticipated utilization of some of our deferred tax assets arising from
net operating loss carryforwards.
37
Results
of Operations for the Years Ended December 31, 2008 and 2007
|
December 31,
|
||||||||||||||||
|
2008
|
2007
|
Change
|
||||||||||||||
|
|
$000’s
|
$000’s
|
Percent
|
|||||||||||||
| Revenues | ||||||||||||||||
|
Program
fee revenue
|
27,941 | 3,427 | 24,514 | 715 | % | |||||||||||
|
Collaboration
fee revenue
|
11,496 | 2,977 | 8,519 | 286 | ||||||||||||
|
Milestone
revenue
|
5,000 | — | 5,000 | 100 | % | |||||||||||
|
Total
revenue
|
44,437 | 6,404 | 38,033 | 594 | % | |||||||||||
|
Operating
expenses
|
||||||||||||||||
|
Research
and development expenses
|
14,322 | 7,169 | 7,153 | 100 | % | |||||||||||
|
Marketing,
general and administrative expenses
|
9,133 | 4,141 | 4,922 | 121 | % | |||||||||||
|
Total
operating expenses
|
23,455 | 11,310 | 12,145 | 107 | % | |||||||||||
|
Income
(loss) from operations
|
20,982 | (4,906 | ) | 25,888 | 528 | % | ||||||||||
|
Other
income (expense)
|
||||||||||||||||
|
Interest
income (expense) net
|
780 | (939 | ) | 1,719 | 183 | % | ||||||||||
|
Amortization
of debt discount
|
— | (2,700 | ) | 2,700 | 100 | % | ||||||||||
|
Loss
on fair value change of conversion features
|
— | (3,483 | ) | 3,483 | 100 | % | ||||||||||
|
Loss
on fair value change of common stock warrants
|
— | (1,905 | ) | 1,905 | 100 | % | ||||||||||
|
(Loss)
gain on asset disposals
|
(2 | ) | 22 | (24 | ) | (109 | )% | |||||||||
|
Other
expense
|
(1 | ) | (3 | ) | 2 | 67 | % | |||||||||
|
Total
other income
|
777 | (9,008 | ) | 9,785 | 109 | % | ||||||||||
|
Income
(loss) before income tax
|
21,759 | (13,914 | ) | 35,673 | 256 | % | ||||||||||
|
Income
tax expense (benefit)
|
7,285 | (9,600 | ) | (16,885 | ) | 232 | % | |||||||||
|
Net
income (loss)
|
14,474 | (4,314 | ) | 18,788 | 436 | % | ||||||||||
Revenue
In
December 2007, King paid us a $30.0 million upfront fee in connection with the
closing of the King Agreement. Program fee revenue recognized during 2008 from
amortizing this upfront fee was $21.9 million compared to $3.4 million in 2007.
We have assigned an equal portion of the program fee revenue to each of three
product candidates identified under the King Agreement. We have
completed our development activities on 2 of the 3 product candidates and have
fully amortized the portion of the upfront fee for those two product candidates
in 2008. We currently estimate the development period for the third
product candidate to end in December, 2010. Also, included in program
fee revenue are two $3.0 million option exercise fees paid by King to us in May
and December 2008, respectively, upon the exercise of its option to license a
third and fourth opioid analgesic product candidate under the King Agreement. In
June 2008, King paid us a $5.0 million milestone payment for successfully
achieving the primary end points in our pivotal Phase III study, AP-ADF-105
for Acurox® Tablets. We had no milestone revenue in 2007.
Collaboration
revenue recognized in 2008 was $11.5 million for reimbursement, pursuant to the
King Agreement, of our Acurox® Tablets development and regulatory expenses
incurred during 2008. We invoice King in arrears on a calendar quarter basis for
our reimbursable development and regulatory expenses under the King
Agreement. We expect the amount and timing of collaboration revenue
to fluctuate in relation to the amount and timing of the underlying research and
development expenses. The Company had collaboration revenue of $3.0 million for
2007 representing such reimbursable expenses from September 19, 2007, the
commencement date for reimbursement purposes set forth in the King Agreement, to
December 31, 2007.
38
Operating
Expenses
Research
and development expense during 2008 and 2007 were primarily for product
candidates utilizing our Aversion® Technology, including costs of preclinical,
clinical trials, clinical trial supplies and related formulation and design
costs, salaries and other personnel related expenses, and facility
costs. Included in the 2008 and 2007 results are non-cash stock-based
compensation charges of $0.6 million and $0.4 million, respectively. Excluding
the stock-based compensation expense, in 2008 there was a $7.0 million increase
in development expenses primarily attributable to increasing clinical study
costs, including our pivotal Phase III clinical trial for Acurox®.
Marketing
expenses during 2008 and 2007 consisted of Aversion® Technology customized
market data research studies. Our general and administrative expenses primarily
consisted of legal, audit and other professional fees, corporate insurance, and
payroll costs. Included in the 2008 and 2007 results are non-cash
stock-based compensation charges of $3.3 million and $0.5 million, respectively,
associated with the grant of stock options and restricted stock units. Excluding
the stock-based compensation expense, there is an increase of $2.2 million in
marketing, general and administrative expenses including $0.8 million of payroll
costs, $0.3 million of legal services, $0.5 million of state franchise taxes,
$0.2 million of tax reserves and $0.1 million of audit and tax
services.
Other
Income (Expense)
During
2008, we had no debt and cash proceeds received pursuant to the King Agreement
were primarily invested in bank commercial paper with maturity dates less than
12 months, money market funds, U.S. Treasury Bills and in overnight sweep
investments, resulting in interest income of $0.8 million.
During
2007 we incurred interest on our $5.0 million Secured Term Note at the variable
rate of prime plus 4.5% through August 19, 2007 and thereafter at the fixed rate
of 10% per annum. Upon the closing of the King Agreement on December
7, 2007, we repaid this Secured Term Note in full. We also incurred
interest on our $10.544 million Senior Secured Convertible Bridge Notes
(collectively, the “Bridge Loans”) at the fixed rate of 10%. On
August 20, 2007, the entire $10.544 million principal amount of the Bridge Loans
was converted into Units consisting of our common stock and warrants in
accordance with our Unit Offering.
Other
expense for 2007 includes a) debt discount amortization expense of $2.7 million
arising from values assigned to conversion features on issuances of bridge
loans, b) $3.5 million loss on fair value changes to amended conversion features
on bridge loans being accounted for as mark-to-market liabilities and c) $1.9
million loss on fair value changes to common stock warrants being accounted for
as mark-to-market liabilities.
Income
Tax Expense (Benefit)
Deferred
income taxes have been recognized in prior years for temporary differences
between financial statement and income tax bases of assets and liabilities and
loss carry-forwards for which income tax benefits are expected to be realized in
future years. At the same time, we recorded a valuation allowance to
reduce net deferred income tax assets to the amount that is more likely than not
to be realized. Net income for 2008 includes a provision for income tax expense
of $8.5 million which has been offset by $1.2 million of income tax benefits
recorded from the anticipated utilization of some of our deferred tax assets
arising from net operating loss carryforwards.
In 2007,
based upon the receipt of the $30 million under the King Agreement we determined
that we will be able to realize deferred income tax assets in the future and
therefore we adjusted the valuation allowance by $9.6 million which is reflected
as a tax benefit in the 2007 statement of operations.
Liquidity
and Capital Resources
At
December 31, 2009, we had cash and cash equivalents of $30.2 million compared to
$35.4 million in cash, cash equivalents, and short-term investments at December
31, 2008. We had working capital of $28.8 million at December 31,
2009 compared to $36.0 million at December 31, 2008. The decrease in our cash
position of $5.2 million and cash flows used in operating activities of $5.0
million for the year ended December 31, 2009, are primarily due to expenses
incurred in developing product candidates not covered by the King
Agreement. The decrease in working capital of $7.2 million is
primarily due to reductions in collaboration revenue receivable from King of
$3.2 million and deferred income taxes of $2.5 million. Cash flows
generated from operating activities were $4.2 million for the year ended
December 31, 2008, primarily representing net income for the period recognizing
certain non-cash items such as deferred program fee revenue, net deferred tax
assets, and charges for stock compensation. Our investing activities
in 2009 included capital expenditures of $0.2 million and net maturities of
short-term investments of $5.0 million.
39
At February 28, 2010, we had cash and
cash equivalents of approximately $28.4 million. We estimate that such cash
reserves will be sufficient to fund the development of Aversion®
Technology product candidates, Impede™ Technology product candidates, and
related operating expenses at least through the next 12 months.
Pending
our receipt of milestone payments and royalties from King related to product
candidates developed under the King Agreement, and other milestone and royalty
payments under similar license agreements anticipated to be negotiated and
executed with other pharmaceutical company partners, of which no assurance can
be given, we must rely on our current cash reserves, including interest income
from the investment of our cash reserves, to fund the development of our
Aversion®
Technology, Impede™ Technology and related ongoing administrative and operating
expenses. Our future sources of revenue, if any, will be derived from milestone
payments and royalties under the King Agreement and under similar license
agreements with other pharmaceutical company partners, for which there can be no
assurance.
The
amount and timing of our future cash requirements will depend on regulatory and
market acceptance of our product candidates and the resources we devote to the
development and commercialization of our product candidates.
The
following table presents our expected cash payments on contractual obligations
outstanding as of December 31, 2009 (in thousands):
|
Payments due by period (in thousands)
|
||||||||||||||||||||
|
Total
|
Less than
1 year
|
1-3
years
|
3-5
years
|
More than
5 years
|
||||||||||||||||
|
Operating
leases
|
$ | 36 | $ | 29 | $ | 7 | $ | — | $ | — | ||||||||||
|
Clinical
studies(1)
|
1,440 | 1,440 | — | — | — | |||||||||||||||
|
Employment
agreements
|
1,009 | 1,009 | — | — | — | |||||||||||||||
|
Total
|
$ | 2,485 | $ | 2,478 | $ | 7 | $ | — | $ | — | ||||||||||
(1)
Approximately $1,197 is expected to be reimbursed to us by King under the
provisions of the King Agreement.
Off-Balance
Sheet Arrangements
We do not engage in
transactions or arrangements with unconsolidated or other special purpose
entities.
Critical
Accounting Policies
The
preparation of our financial statements in accordance with United States
generally accepted accounting principles requires us to make estimates and
assumptions that affect the reported amounts of assets, liabilities, revenues
and expenses in our financial statements and accompanying notes. We evaluate our
estimates on an ongoing basis, including those estimates related to contract
agreements, research collaborations and investments. We base our estimates on
historical experience and various other assumptions that we believe to be
reasonable under the circumstances, the results of which form the basis for
making judgments about the carrying values of assets and liabilities that are
not readily apparent from other sources. Actual results may differ from these
estimates under different assumptions or conditions. The following items in our
financial statements require significant estimates and judgments:
Revenue
Recognition, Deferred Program Fee Revenue and Collaboration Revenue
We
recognize revenue when there is persuasive evidence that an arrangement exists,
delivery has occurred, the price is fixed and determinable, and collection is
reasonably assured. In connection with King Agreement, we recognize program fee
revenue, collaboration revenue and milestone revenue.
Program
fee revenue is derived from amortized upfront payments, such as the $30.0
million upfront payment from King received in December 2007, and license fees,
such as the $3.0 million option exercise fee paid by King to us in each of May
and December 2008 upon the exercise of its option to license a third and fourth
opioid analgesic product candidate under the King Agreement. We have assigned an
equal portion of King’s $30.0 million upfront payment to each of three product
candidates identified in the King Agreement and recognize the upfront payment as
program fee revenue ratably over our estimate of the development period for each
identified product candidate. We recognized $3.1 million, $21.9 million,
and $3.4 million of this program fee revenue in 2009, 2008 and 2007,
respectively.
40
Collaboration
revenue is derived from reimbursement of development expenses, which are
invoiced quarterly in arrears, and are recognized when costs are incurred
pursuant to the King Agreement. The ongoing research and development
services being provided to King under the collaboration are priced at fair value
based upon the reimbursement of expenses incurred pursuant to the collaboration
with King. We recognized $0.8 million, $11.5 million and $3.0 million of
collaboration revenue in 2009, 2008 and 2007, respectively of which $0.4 million
and $3.5 million were current receivables at December 31, 2009 and 2008,
respectively
Milestone
revenue is contingent upon the achievement of certain pre-defined events in the
development of Acurox® Tablets and other product candidates licensed to King
under the King Agreement. Milestone payments from King are recognized as revenue
upon achievement of the “at risk” milestone events, which represent the
culmination of the earnings process related to that milestone. Milestone
payments are triggered either by the results of our research and development
efforts or by events external to us, such as regulatory approval to market a
product. As such, the milestones are substantially at risk at the inception of
the King Agreement, and the amounts of the payments assigned thereto are
commensurate with the milestone achieved. In addition, upon the achievement of a
milestone event, we have no future performance obligations related to that
milestone payment. Each milestone payment is non-refundable and non-creditable
when made. In June 2008, King paid us a $5.0 million milestone payment for
successfully achieving the primary endpoints in our pivotal Phase III
study, AP-ADF-105 for Acurox® Tablets.
Research
and Development
Research
and Development (“R&D”) expenses include internal R&D activities,
external CRO activities, and other activities. Internal R&D activity
expenses include facility overhead, equipment and facility maintenance and
repairs, depreciation, laboratory supplies, pre-clinical laboratory experiments,
depreciation, salaries, benefits, and incentive compensation expenses. CRO
activity expenses include preclinical laboratory experiments and clinical trial
studies. Other activity expenses include clinical trial studies and regulatory
consulting, and regulatory counsel. Internal R&D activities and other
activity expenses are charged to operations as incurred. we make payments
to the CRO's based on agreed upon terms and may include payments in advance of
the study starting date. We review and accrue CRO expenses and clinical trial
study expenses based on work performed and rely upon estimates of those costs
applicable to the stage of completion of a study as provided by the CRO.
Accrued CRO costs are subject to revisions as such trials progress to
completion. Revisions are charged to expense in the period in which the facts
that give rise to the revision become known. Advance payments are amortized to
expense based on work performed. We have entered into several CRO clinical trial
agreements for which the unfunded CRO commitments were $1.4 million and $1.0
million at December 31, 2009 and 2008, respectively, and are expected to be
incurred as subjects are enrolled into the clinical studies.
Income
Taxes
We
account for income taxes under the liability method. Under this method, deferred
income tax assets and liabilities are determined based on differences between
financial reporting and income tax basis of assets and liabilities and are
measured using the enacted income tax rates and laws that will be in effect when
the differences are expected to reverse. Additionally, net operating loss and
tax credit carryforwards are reported as deferred income tax
assets. The realization of deferred income tax assets is dependent
upon future earnings. A valuation allowance against deferred income tax assets
is required if, based on the weight of available evidence, it is more likely
than not that some or all of the deferred income tax assets may not be
realized. We recorded adjustments of $2.5 million increase and $1.2
million decrease to the deferred income tax asset valuation allowance during
2009 and 2008, respectively. These adjustments recognized a $2.5 million tax
expense and a $1.2 million tax benefit from income taxes in our income for 2009
and 2008, respectively. At December 31, 2009, 100% of the deferred income tax
assets are offset by a valuation allowance due to uncertainties with respect to
future utilization of net operating loss carryforwards. If in the future it is
determined that amounts of our deferred income tax assets would likely be
realized, the valuation allowance would be reduced in the period in which such
determination is made and a benefit from income taxes in such period would be
recognized.
41
Stock
Compensation
On
January 1, 2006, we adopted the Financial Accounting Standards Board (“FASB”)
statement on share-based payments. This change in accounting replaces existing
requirements under FASB and eliminates the ability to account for share-based
compensation transaction using the intrinsic value method. The
compensation cost related to share-based payment transactions is now measured
based on fair value of the equity or liability instrument issued. For purposes
of estimating the fair value of each stock option unit on the date of grant, we
utilized the Black-Scholes option-pricing model. The Black-Scholes option
valuation model was developed for use in estimating the fair value of traded
options, which have no vesting restrictions and are fully transferable. In
addition, option valuation models require the input of highly subjective
assumptions including the expected volatility factor of the market price of our
common stock (as determined by reviewing its historical public market closing
prices). Because our employee stock options have characteristics significantly
different from those of trade options and because changes in the subjective
input assumptions can materially affect the fair value estimate, in management’s
opinion, the existing models do not necessarily provide a reliable measure of
the fair value of its employee stock options.
We had
previously accounted for stock-based compensation using the intrinsic value
method when the exercise price of our employee stock options equaled the market
price of the underlying common stock on the date of grant, no compensation
expense was recognized. Accordingly, no compensation expense had been recognized
in the consolidated financial statements in connection with these types of
grants for 2005 and earlier. When the exercise price of our employee
stock options was less than the market price of the underlying common stock on
the date of grant, compensation expense was recognized. Equity instruments
issued to nonemployees in exchange for goods, fees and services are accounted
for under the fair value-based method.
Our
accounting for stock-based compensation for restricted stock units (“RSUs”) has
been based on the fair-value method. The fair value of the RSUs is the market
price of our common stock on the date of grant, less its exercise
cost.
Debt
Discount
For years
2007 and prior, debt discount resulting from the issuance of common stock
warrants in connection with subordinated debt and other notes payable as well as
from beneficial conversion features contained in convertible debt was recorded
as a reduction of the related obligations and was amortized over the remaining
life of the related obligations. Debt discount related to the common stock
warrants issued was determined by a calculation based on the relative fair
values ascribed to such warrants determined by management's use of the
Black-Scholes valuation model.
Conversion
Features and Common Stock Warrants
For year
2007 certain provisions of the amended conversion features contained in our
Bridge Loan Agreements required us to separate the value of the conversion
feature from the debt and record such value as a separate liability which was
marked-to-market at each balance sheet date. We used the Black-Scholes
option-pricing model to compute the estimated fair value of the conversion
features. Marked-to-market adjustments resulted in the recording of further
gains and losses.
As a
result of the amendment to the Bridge Loan Agreements, all outstanding common
stock purchase warrants were fair valued using the Black - Scholes
option-pricing model and recorded as a liability with a corresponding reduction
in additional paid-in capital. This warrant liability was marked-to-market each
balance sheet date which resulted in the recording of further gains and
losses.
Capital
Expenditures
Our
capital expenditures during 2009, 2008, and 2007 were $220,000, $143,000 and
$31,000, respectively. Capital expenditures in each such year were attributable
to the purchase of scientific equipment and improvements to the Culver, Indiana
facility.
42
Impact
of Inflation
We
believe that inflation did not have a material impact on our operations for the
periods reported.
ITEM
7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Some of
the securities that we invest in may be subject to market risk. Our
primary objective in our cash management activities is to preserve principal
while at the same time maximizing income we receive from our
investments. A change in the prevailing interest rates may cause the
principal amount of our investments to fluctuate. We have no holdings
of derivative financial and commodity instruments. As of December 31, 2009, our
investments consisted primarily of investments in U.S. Treasury Bills or in
money market accounts and checking funds with variable, market
rates of interest.
ITEM
8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
The
consolidated financial statements of Acura Pharmaceuticals, Inc. and Subsidiary
and the Report of the Independent Registered Public Accounting Firm thereon, to
be filed pursuant to Item 8 are included in Item 15.
ITEM
9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL
DISCLOSURE
None.
ITEM
9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls
and Procedures. We have conducted an evaluation, under the supervision
and with the participation of our management, including our Chief Executive
Officer and Chief Financial Officer of the effectiveness of the design and
operation of our disclosure controls and procedures pursuant to Exchange Act
Rule 13a-14. Based upon that evaluation, our Chief Executive Officer and Chief
Financial Officer concluded that our disclosure controls and procedures are
effective in timely alerting them to material information relating to the
Company (including our subsidiary) required to be included in our periodic
Securities and Exchange Commission filings. No significant changes were made in
our internal controls or in other factors that could significantly affect these
controls subsequent to the date of their evaluation.
Management’s Report on Internal
Control Over Financial Reporting. Our management is
responsible for establishing and maintaining adequate internal control over
financial reporting. Internal control over financial reporting is
defined in Rule 13a-15(f) and 15d-15(f) promulgated under the Exchange Act as a
process designated by, or under the supervision of, our principal executive and
principal financial officers and effected by our board of directors, management
and other personnel, to provide reasonable assurance regarding the reliability
of financial reporting and the preparation of financial statements for external
purposes in accordance with generally accepted accounting principles and
includes those policies and procedures that:
|
|
·
|
Pertain
to the maintenance of records that in reasonable detail accurately and
fairly reflect the transactions and disposition of our
assets;
|
|
|
·
|
Provide
reasonable assurance that transactions are recorded as necessary to permit
preparation of financial statements in accordance with generally accepted
accounting principles, and that our receipts and expenditures are being
made only in accordance with authorizations of our management and
directors; and
|
|
|
·
|
Provide
reasonable assurance regarding prevention or timely detection of
unauthorized acquisition, use or disposition of our assets that could have
a material effect on the financial
statements.
|
Because
of its inherent limitations, internal control over financial reporting may not
prevent or detect misstatements. Therefore, even those systems
determined to be effective can provide only reasonable assurance with respect to
financial statement preparation and presentation. Also, projections
of any evaluation of effectiveness to future periods are subject to the risk
that controls may become inadequate because of changes in conditions, or that
the degree of compliance with the policies or procedures may
deteriorate.
43
Our
management assessed the effectiveness of our internal control over financial
reporting as of December 31, 2009. In making this assessment,
management used the criteria set forth by the Committee of Sponsoring
Organizations of the Treadway Commission (COSO) in Internal Control – Integrated
Framework. Based on our assessment, management believes that, as of
December 31, 2009, our internal control over financial reporting is effective
based on those criteria.
The
attestation report concerning the effectiveness of our internal control over
financial reporting as of December 31, 2009 issued by BDO Seidman, LLP, an
independent registered public accounting firm, appears at the end of this Item
9A.
Changes in Internal Control Over
Financial Reporting. There was no change in our internal
control over financial reporting that occurred during the Fourth Quarter 2009
that has materially affected, or is reasonably likely to materially affect, our
internal control over financial reporting.
44
REPORT
OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM ON INTERNAL CONTROL OVER
FINANCIAL REPORTING
Board of
Directors and Shareholders
Acura
Pharmaceuticals, Inc.
Palatine,
Illinois
We have
audited Acura Pharmaceuticals, Inc.’s internal control over financial reporting
as of December 31, 2009, based on criteria established in Internal Control—Integrated
Framework issued by the Committee of Sponsoring Organizations of the
Treadway Commission (the COSO criteria). Acura Pharmaceuticals, Inc.’s
management is responsible for maintaining effective internal control over
financial reporting and for its assessment of the effectiveness of internal
control over financial reporting, included in the accompanying Item 9A,
Management’s Report on Internal Control Over Financial Reporting. Our
responsibility is to express an opinion on the company’s internal control over
financial reporting based on our audit.
We
conducted our audit in accordance with the standards of the Public Company
Accounting Oversight Board (United States). Those standards require that we plan
and perform the audit to obtain reasonable assurance about whether effective
internal control over financial reporting was maintained in all material
respects. Our audit included obtaining an understanding of internal control over
financial reporting, assessing the risk that a material weakness exists, and
testing and evaluating the design and operating effectiveness of internal
control based on the assessed risk. Our audit also included performing such
other procedures as we considered necessary in the circumstances. We believe
that our audit provides a reasonable basis for our opinion.
A
company’s internal control over financial reporting is a process designed to
provide reasonable assurance regarding the reliability of financial reporting
and the preparation of financial statements for external purposes in accordance
with generally accepted accounting principles. A company’s internal control over
financial reporting includes those policies and procedures that (1) pertain
to the maintenance of records that, in reasonable detail, accurately and fairly
reflect the transactions and dispositions of the assets of the company;
(2) provide reasonable assurance that transactions are recorded as
necessary to permit preparation of financial statements in accordance with
generally accepted accounting principles, and that receipts and expenditures of
the company are being made only in accordance with authorizations of management
and directors of the company; and (3) provide reasonable assurance
regarding prevention or timely detection of unauthorized acquisition, use, or
disposition of the company’s assets that could have a material effect on the
financial statements.
Because
of its inherent limitations, internal control over financial reporting may not
prevent or detect misstatements. Also, projections of any evaluation of
effectiveness to future periods are subject to the risk that controls may become
inadequate because of changes in conditions, or that the degree of compliance
with the policies or procedures may deteriorate.
In our
opinion, Acura Pharmaceuticals, Inc. maintained, in all material respects,
effective internal control over financial reporting as of December 31, 2009,
based on the COSO criteria.
We also
have audited, in accordance with the standards of the Public Company Accounting
Oversight Board (United States), the consolidated balance sheets of Acura
Pharmaceuticals, Inc. as of December 31, 2009 and December 31, 2008, and the
related consolidated statements of operations, stockholders’ equity (deficit)
and cash flows for each of the three years in the period ended December 31, 2009
and our report dated March 2, 2010 expressed an unqualified opinion on those
consolidated financial statements.
|
/s/
BDO Seidman, LLP
|
|
Chicago,
Illinois
|
|
March
2, 2010
|
45
Item
8B. OTHER INFORMATION
Not
Applicable.
PART
III
ITEM
10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE
GOVERNANCE
Directors
and Executive Officers
|
Name
|
Age
|
Position
|
||
|
Andrew
D. Reddick
|
57
|
President,
Chief Executive Officer and Director
|
||
|
Robert
B. Jones
|
51
|
Senior
Vice President and Chief Operating Officer
|
||
|
Peter
A. Clemens
|
57
|
Senior
Vice President, Chief Financial Officer and Secretary
|
||
|
Albert
W. Brzeczko
|
53
|
Vice
President, Technical Affairs
|
||
|
Robert
A. Seiser
|
46
|
Vice
President, Treasurer and Corporate Controller
|
||
|
James
F. Emigh
|
54
|
Vice
President of Marketing and Administration
|
||
|
Bruce
F. Wesson
|
67
|
Director
|
||
|
William
A. Sumner
|
72
|
Director
|
||
|
Richard
J. Markham
|
59
|
Director
|
||
|
William
G. Skelly
|
59
|
Director
|
||
|
Immanuel
Thangaraj
|
39
|
Director
|
||
|
George
K. Ross
|
|
68
|
|
Director
|
Andrew D.
Reddick has been President and Chief Executive Officer since August, 2003 and a
member of our Board of Directors since August, 2004. From April, 2000 to
September, 2002 Mr. Reddick was Chief Operating Officer and Sr. Vice President
Commercial Operations for Adolor Corporation and from June, 1999 to March, 2000
he served as President of Faulding Laboratories, Inc. Mr. Reddick holds a
Bachelor of Arts degree in Biology from the University of California and a
Masters of Business Administration degree from Duke University.
Robert B.
Jones has been our Senior Vice President and Chief Operating Officer since April
2008. From May, 2003 to March, 2008, Mr. Jones served first as the
Vice President, Finance and then as Vice President, Strategy and Business
Analysis of Adolor Corporation. From November 2000 to May, 2003 he
served as Vice President, Finance and then as Chief Operating Officer of
Opt-E-Script, Inc., a privately held personalized medicine company where Mr.
Jones was responsible for all commercialization activities. Prior to
that, Mr. Jones was Vice President, Sales and Marketing for Purepac
Pharmaceutical Company. Mr. Jones received his M.B.A. from the
University of North Carolina and a B.S. from Cornell University.
Peter A.
Clemens has been Senior Vice President, Chief Financial Officer and Secretary
since April 2004. Mr. Clemens was our Vice President, Chief Financial Officer
and Secretary from February 1998 to March 2004 and a member of our Board of
Directors from June, 1998 to August, 2004. Mr. Clemens is a Certified Public
Accountant and earned a Bachelor of Business Administration degree from the
University of Notre Dame and a Masters of Business Administration from Indiana
University.
Albert W.
Brzeczko, Ph.D., has been Vice President, Technical Affairs since February
2009. From 1999 through 2009, Dr. Brzeczko was Vice President, Global
Pharma New Product Development and Pharma Technologies for International
Specialty Products, Inc., a contract services group specializing in the
development of technologies for the bioenhancement of poorly soluble
drugs. Prior to 1999, Dr. Brzeczko held various positions of
increasing responsibility in pharmaceutical product development with UPM
Pharmaceuticals, Banner Pharmacaps, Mylan Laboratories, and DuPont
Merck. Dr. Brzeczko received a Bachelor of Science degree in
biochemistry and a Ph.D. in pharmaceutical sciences from the University of
Maryland.
Robert A.
Seiser has been a Vice President, Treasurer and Corporate Controller since April
2004. Mr. Seiser joined us in March 1998 as our Treasurer and Corporate
Controller. Mr. Seiser is a Certified Public Accountant and earned a Bachelor of
Business Administration degree from Loyola University of
Chicago.
46
James F.
Emigh has been Vice President of Marketing and Administration since April 2004.
Prior to such time, Mr. Emigh was our Vice President of Sales and Marketing. Mr.
Emigh joined us in May, 1998, serving first as Executive Director of Customer
Relations and then as Vice President of Operations until November, 2002. Mr.
Emigh holds a Bachelor of Pharmacy degree from Washington State University and a
Masters of Business Administration from George Mason University.
Bruce F.
Wesson has been a member of our Board of Directors since March, 1998. Mr. Wesson
has been a Partner of Galen Associates, a health care venture firm, and a
General Partner of Galen Partners III, L.P. since January 1991. Prior to
January, 1991, he was Senior Vice President and Managing Director of Smith
Barney, Harris Upham & Co. Inc., an investment banking firm. He
currently serves on the Boards of Derma Sciences, Inc., Chemtura Corporation,
and as Vice Chairman of the Board of MedAssets, Inc., each a publicly traded
company. Mr. Wesson earned a Bachelor of Arts degree from Colgate University and
a Masters of Business Administration from Columbia University.
William
A. Sumner has been a member of our Board of Directors since August, 1997. From
1974 until his retirement in 1995, Mr. Sumner held various positions within
Hoechst-Roussel Pharmaceuticals, Inc., including Vice President and General
Manager, Dermatology Division from 1991 through 1995, Vice President, Strategic
Business Development, from 1989 to 1991 and Vice President, Marketing from 1985
to 1989. Since his retirement from Hoechst-Roussel Pharmaceuticals, Inc. in
1995, Mr. Sumner has served as a consultant in the pharmaceutical industry. He
currently serves on the Board of 3I, Ingredient Innovations International, a
privately held company. Mr. Sumner earned a Bachelor of Arts degree from
Montclair State University and a Master of Arts degree from the University of
Virginia.
Richard
J. Markham has been a member of our Board of Directors since May, 2006. Since
November, 2004 Mr. Markham has served as a partner at Care Capital, LLC, a
venture capital firm that primarily invests in life sciences companies. From May
2002 until August 2004, Mr. Markham was the Vice Chairman of the Management
Board and Chief Operating Officer of Aventis SA. From December, 1999 until May,
2002 he was the Chief Executive Officer of Aventis Pharma AG. Previously he was
the Chief Executive Officer of Hoechst Marion Roussel, the President and Chief
Operating Officer of Marion Merrell Dow, Inc. and a member of its board of
directors. From 1973 to 1993 Mr. Markham was associated with Merck & Co.
Inc., culminating in his position as President and Chief Operating Officer. Mr.
Markham received a B.S. in Pharmacy and Pharmaceutical Sciences from Purdue
University.
William
G. Skelly has been a member of our Board of Directors since May, 1996 and served
as our Chairman from October, 1996 through June, 2000. Since 1990, Mr. Skelly
has served as Chairman, President and Chief Executive Officer of Central
Biomedia, Inc. and its subsidiary SERA, Inc. From 1985 to 1990, Mr. Skelly
served as President of Martec Pharmaceutical, Inc. Mr. Skelly earned a Bachelor
of Arts degree from Michigan State University and a Masters of Business
Administration from the University of Missouri-Kansas City.
Immanuel
Thangaraj has been a member of our Board of Directors since December, 2002. Mr.
Thangaraj has been a Managing Director of Essex Woodlands Health Ventures, a
venture capital firm specializing in the healthcare industry, since 1997.
Prior to joining Essex Woodlands Health Ventures, he helped establish a
telecommunication services company, for which he served as its CEO. Mr.
Thangaraj holds a Bachelor of Arts and a Masters in Business Administration from
the University of Chicago.
George K.
Ross has been a member of our Board of Directors since January, 2008. Since
April 2002, Mr. Ross has been a consultant to early stage businesses and a
financial investor. Since July 2005 he has served as Executive Director,
Foundations and Partnerships for World Vision U.S. in New York
City. His business career has included senior financial officer
and board member positions with both public and private companies in diverse
industries. Mr. Ross was Executive Vice President and Chief Financial
Officer and a board member of Tier Technologies Inc. from February 1997 to
January 2000, which became a public company during this period. Mr.
Ross is a Certified Public Accountant and earned a Bachelor of Arts degree from
Ohio Wesleyan University and a Masters of Business Administration from Ohio
State University.
Ron J.
Spivey, Ph.D., serves on a part-time (non-executive officer) basis as our Senior
Scientific Advisor since January 1, 2009. From April 2004 through
December 31, 2008 Dr. Spivey was our Senior Vice President and Chief Scientific
Officer. From June, 2003 to March, 2004 Dr. Spivey was President of Gibraltar
Associates, a private consulting services company for the pharmaceutical
industry. From March, 1998 to May, 2002 he served as Vice President, Scientific
Affairs for Alpharma/Purepac Pharmaceuticals. Dr. Spivey holds a Bachelor of
Arts degree from Indiana University and a Ph.D. degree in pharmaceutics from the
University of Iowa.
47
As of the
date of this Report, the Company had 15 full-time employees, nine of whom are
engaged in the research, development and manufacture of product candidates
utilizing the Aversion®
Technology. The remaining employees are engaged in administrative, legal,
accounting, finance, market research, and business development activities. All
of our senior management and most of our other employees have had prior
experience in pharmaceutical or biotechnology companies. None of our employees
is covered by collective bargaining agreements. We believe that our relations
with our employees are good.
Corporate
Governance
Audit
Committee
The Audit
Committee is composed of George K. Ross, Chairman, William A. Sumner and William
G. Skelly. The Audit Committee is responsible for selecting the Company’s
registered independent public accounting firm, approving the audit fee payable
to the auditors, working with independent auditors and other corporate
officials, reviewing the scope and results of the audit by, and the
recommendations of, our independent auditors, approving the services provided by
the auditors, reviewing our financial statements and reporting on the results of
the audits to the Board, reviewing our insurance coverage, financial controls
and filings with the SEC, including, meeting quarterly prior to the filing of
our quarterly and annual reports containing financial statements filed with the
SEC, and submitting to the Board its recommendations relating to our financial
reporting, accounting practices and policies and financial, accounting and
operational controls.
In
assessing the independence of the Audit Committee in 2009, our Board reviewed
and analyzed the standards for independence provided in NASDAQ Marketplace Rule
4200(a)(15) and applicable SEC regulations. Based on this analysis,
our Board has determined that each of Messrs. Ross, Sumner and Skelly satisfies
such standards for independence. Our Board also determined that Mr.
Ross is a “financial expert” as provided in NASDAQ Marketplace Rule
4350(d)(2)(A) and SEC regulations.
The Charter of our Audit Committee is
available on our website, www.acurapharm.com, under the menu item “Audit
Committee Charter” appearing under the “Corporate” tab.
Compensation
Committee
The Compensation Committee is composed
of Richard J. Markham, Chairman, Bruce F. Wesson and Immanuel Thangaraj. This
committee is responsible for consulting with and making recommendations to the
Board of Directors about executive compensation and compensation of
employees. See “Item 11. Executive Compensation –Board
Process” for a summary of the procedures for approving compensation for our
senior management and employees. The Charter of our Compensation
Committee is available on our website, www.acurapharm.com, under the menu item
“Compensation Committee Charter” appearing under the “Corporate”
tab.
Although the listing standards of the
NASDAQ Capital Market specify that the compensation of our executive officers
must be determined, or recommended to the Board, either by a majority of
independent directors or a compensation committee comprised solely of
independent directors, we are relying on the “controlled company” exemption
provided in the listing standards of the NASDAQ Capital Market in having each of
Messrs. Markham, Wesson and Thangaraj as members of the Compensation
Committee.
48
Nominating
Committee
Currently
our entire Board of Directors functions as our nominating committee. As needed,
the Board will perform the functions typical of a nominating committee,
including the identification, recruitment and selection of nominees for election
to our Board. Three of our seven members of the Board (Messrs. Sumner, Skelly
and Ross) are "independent" as that term is defined under the rules of the
NASDAQ Capital Market and SEC regulations and participate with the entire Board
in the consideration of director nominees. We believe that a nominating
committee separate from the Board is not necessary at this time, given our
relative size and the size of our Board and that an additional committee of the
Board would not add to the effectiveness of the evaluation and nomination
process. The Board's process for recruiting and selecting nominees for Board
members, if required, would be to identify individuals who are thought to have
the business background and experience, industry specific knowledge and general
reputation and expertise allowing them to contribute as effective directors to
our governance, and who would be willing to serve as directors of a public
company. To date, we have not engaged any third party to assist in identifying
or evaluating potential nominees. If a possible candidate is identified, the
individual will meet with each member of the Board and be sounded out concerning
his/her possible interest and willingness to serve, and Board members would
discuss amongst themselves the individual's potential to be an effective Board
member. If the discussions and evaluation are positive, the individual would be
invited to serve on the Board. To date, no shareholder has presented any
candidate for Board membership for consideration, and we do not have a specific
policy on shareholder-recommended director candidates. The Board believes its
process for evaluation of nominees proposed by shareholders would be no
different than the process of evaluating any other candidate. The
Board of Directors does not have a policy regarding diversity in identifying
nominees for director.
The
experience, qualifications, attributes or skills that led the Board to conclude
that the current board members, should serve are (i) their pharmaceutical
industry and senior level management experience in the case of Messrs. Reddick,
Skelly, Sumner, Wesson and Markham; (ii) financial and senior level management
expertise in the case of Mr. Ross, and (iii) their experience in overseeing
management as principals of private equity firms in the case of Messrs. Wesson,
Thangaraj and Markham. In addition we are required to elect the three
designees of GCE Holdings LLC– Messrs. Markham, Thangaraj, and Wesson - to our
Board pursuant to the Voting Agreement described in “Item 13. Certain
Relationships and Related Transactions, and Director
Independence”. See also “Item 10. Directors, Executive Officers and
Corporate Governance” for a further description of the experience of our
directors.
Separation
of Roles of Chairman and CEO
Mr.
Markham serves as the Chairman of our Board of Directors and Mr. Reddick serves
as Chief Executive Officer. We believe the separation of offices is
beneficial because a separate chairman (i) can provide the Chief Executive
Officer with guidance and feedback on his performance, (ii) provides a more
effective channel for the Board to express its views on management, (iii) allows
the chairman to focus on shareholder interests and corporate governance while
the Chief Executive Officer manages the Company’s operations. As Mr.
Markham has significant senior level pharmaceutical industry experience, he is
particularly well suited to serve as Chairman.
Board’s
Role in Risk Assessment
The Board
as a whole engages in risk oversight as part of its functions. As an
emerging pharmaceutical development company we face numerous risks identified
under Item 1A-Risk Factors”, many of which are outside of our
control. In addition, the Audit Committee reviews our insurance
coverage and the Board and Audit Committee regularly monitors our liquidity
position and operating expenses and reviews our capital funding
needs. The Company believes the Board leadership structure
effectively enables it to oversee risk management.
Shareholder
Communications to the Board
Shareholders
who wish to send communications to our Board of Directors may do so by sending
them in care of our Secretary at the address on the cover page of this Report.
The envelope containing such communication must contain a clear notation
indicating that the enclosed letter is a "Shareholder-Board Communication" or
"Shareholder-Director Communication" or similar statement that clearly and
unmistakably indicates the communication is intended for the Board. All such
communications must clearly indicate the author as a shareholder and state
whether the intended recipients are all members of the Board or just certain
specified directors. Our Secretary will have the discretion to screen and not
forward to Directors communications which the Secretary determines in his or her
discretion are communications unrelated to our business or our governance,
commercial solicitations, or communications that are offensive, obscene, or
otherwise inappropriate. The Secretary will, however, compile all shareholder
communications which are not forwarded and such communications will be available
to any Director.
49
Code
of Ethics
Our Code
of Ethics applicable to our principal executive officer, principal financial
officer, principal accounting officer and all of our other employees is
available on our website, www.acurapharm.com, under the menu item “Code of
Ethics” appearing under the “Corporate” tab.
Section 16(a) Beneficial Ownership
Reporting Compliance
Section
16(a) of the Securities Exchange Act of 1934, as amended, requires our Directors
and executive officers, and persons who own beneficially more than ten percent
(10%) of our Common Stock, to file reports of ownership and changes of ownership
with the SEC. Copies of all filed reports are required to be furnished to us
pursuant to Section 16(a). Based solely on the reports received by us and on
written representations from reporting persons, we believe that our Directors,
executive officers and greater than ten percent (10%) beneficial owners of our
Common Stock complied with all Section 16(a) filing requirements during the year
ended December 31, 2009.
ITEM
11. EXECUTIVE COMPENSATION
Unless otherwise noted all share and
share price information with respect to our common stock give effect to a 1 for
10 reverse stock split implemented December 5, 2007.
Compensation
Discussion and Analysis
Our
executive compensation program consists of (i) an annual salary and bonus
compensation and (ii) equity incentives represented by the issuance of stock
options and restricted stock units (“RSUs”). The salary, bonuses, and
equity incentives serve to link executive pay to corporate
performance.
Policies
for Allocating Between Various Forms of Compensation
For a
number of years prior to 2007, because we had insufficient cash reserves, our
ability to pay cash bonuses and increase salaries was limited. As a result, we
did not grant cash bonuses or increase salaries to our principal executives in
the three years ended December 31, 2006. Instead we sought to
incentivize our senior management with equity compensation in the form of stock
options and RSUs.
In 2004
and 2005 we issued stock options to our employees with an exercise price at a
discount to the then current trading price for our common
stock. Because our stock price is based on relatively low trading
volume and a small public float, it can fluctuate widely at times. As
a result, we determined that the issuance of RSUs presented a number of
advantages. First, it allows us to reduce the dilutive effect of this
equity-based compensation, as there are fewer shares underlying a restricted
stock award than an equivalent value issued in a stock option
award. Second, the vesting schedule of the RSUs was structured to
minimize the potential excise tax under Section 280G of the Internal Revenue
Code upon a change of control. Third, it is difficult to
set an exercise price for options that accurately reflects the value of the
Company due to the low trading volume and small public float for our common
stock.
As a
result, in 2005 we established a restricted stock unit plan (the “2005 RSU
Plan”) and issued RSUs aggregating 2,750,000 shares to employees. In
addition, RSUs with respect to 100,000 shares were issued to each of our two
independent directors in 2006. In each case, the number of RSUs we
issued was influenced by the closing price of the stock underlying the RSUs on
the date of grant. As a result of an amendment to our RSU Plan
approved by our Board in March 2008, and ratified by our shareholders in April
2008, we increased the number of RSUs available for issuance under the 2005 RSU
Plan from 3 million to 3.5 million. After giving effect to RSU awards
since the adoption of the 2005 RSU Plan, as of the date of this Report, 184,000
shares remain available for RSU grants under such plan.
Following
the completion of our Unit Offering in August 2007 and the consummation of the
King Agreement in December 2007, our cash position improved and we were able to
increase salaries and grant bonuses to our employees as discussed below under
the caption “Salary and
Bonus”. In addition to periodic awards of equity-based
compensation (see “Stock Options” below), our objective is to award merit based
cash bonuses and salary increases on an annual basis going forward. The amounts
and timing of any such awards will be subject to available cash reserves and the
satisfaction of employee performance objectives established by our Chief
Executive Officer and the Compensation Committee. Our equity-based
compensation going forward is targeted to allow senior management as a group to
own between 5% and 10% of our outstanding common stock, so as to align their
interests with shareholders’ interests.
50
In 2007,
no stock options or RSU awards were made to senior management. In
view of our improved cash reserves following the closing of the King Agreement,
and recognizing that no salary increases or bonuses had been awarded to senior
management over the prior four years, the Compensation Committee and the Board
determined that salary increases and bonuses for each executive officer was
appropriate. As part of its analysis the Compensation Committee and
the Board considered the stock option and RSU awards previously made to the
executive officers in 2004 and 2005 and determined that additional equity
incentive compensation was not warranted in 2007.
As
discussed below under the caption “Stock Options”, in 2008 and 2009, we awarded
stock options to employees with such options having an exercise price equal to
the fair market value of our Common Stock on the date of grant. In
2009 we also awarded RSUs to employees. These stock options and RSUs
were awarded in recognition that no equity-based compensation awards had been
granted since 2005 and in an effort to retain valued employees. We
have also issued RSUs and options to executives upon commencement of
employment. For example, in 2009 we awarded 24,000 RSUs and options
to purchase 96,000 shares of Common Stock to Dr. Brzeczko upon commencement of
employment and in 2008 we awarded 50,000 RSUs and 30,000 options to purchase
Common Stock to Mr. Jones upon commencement of employment.
Salary
and Bonus
Each of
Andrew Reddick, Robert Jones and Peter Clemens are parties to employment
agreements, described under the caption “Employment Agreements” below, which
provide the minimum annual base salary to be payable to such officers, subject
to increase at the discretion of the Board. For 2009, the annual
salaries of Messrs. Reddick, Jones and Clemens were $365,000, $290,000 and
$205,000, respectively. In addition, the Reddick, Jones and Clemens
employment agreements provide for annual bonus payments, in the discretion of
the Compensation Committee or the Board, subject to the satisfaction of such
targets, conditions or parameters as may be agreed upon from time to time by the
employee and the Compensation Committee.
The bonus
performance targets for Messrs. Reddick, Spivey, Jones and Clemens for 2008
included advancing Acurox® to
NDA submission, and other product candidates using our Aversion®
Technology to proof of concept, implementing the King Agreement, licensing of
additional products to King through the exercise of King’s options under the
King Agreement and licensing products utilizing our Aversion®
Technology outside of North America. Such
performance targets are both organization and individual goals. In 2008 we advanced several
products using our Aversion®
Technology, licensed two additional products to King, and advanced Acurox® by
submitting a 505(b)(2) NDA to the FDA. Considering these
achievements, we awarded bonuses of $328,500, $315,000, $130,500 and $102,500 to
Messrs. Reddick, Spivey, Jones and Clemens. In addition, to induce
Dr. Spivey to remain as a full time employee through December 31, 2008, we paid
him an additional $315,000 retention bonus on December 31,
2008. Although salaries were increased for 2009 for non-executive
officer employees, no salary increases were granted to Messrs. Reddick, Jones or
Clemens for 2009. Dr. Spivey became a part-time employee in 2009, at
an annual salary of $120,000 per year.
The
material bonus performance targets for 2009 consisted of achieving FDA
approval of the Acurox® Tablet
NDA, advancement of additional product candidates (both opioid and non-opioid)
utilizing Aversion®
Technology, and expansion of our intellectual property portfolio relative to
abuse deterrent technologies. Such performance targets were both
organization and individual goals. We met most but not all
of our 2009 performance targets. We did not receive FDA
approval of the Acurox® Tablet
NDA in 2009, although we did advance additional product candidates through
development and expanded our intellectual property portfolio with the addition
of two issued patents and the filing of new patent
applications. Considering these circumstances we granted bonuses of
$140,000, $112,000 and $74,000, respectively, to Messrs. Reddick, Jones, and
Clemens, which amounts represented substantially less than their 2008 bonus
awards. Recognizing that our executives did not receive a salary
increase for 2009, for 2010 we modestly increased their salaries in the range of
3.2% and 3.6%. As a result, Messrs. Reddick, Jones and Clemens now
receive annual salaries of $377,000, $300,000 and $211,500,
respectively.
51
The
material bonus performance targets for 2010 are similar to the 2009 goals and
include the gaining of FDA agreement to accept resubmission of the Acurox® Tablets
NDA, subsequent FDA approval of such NDA resubmission, advancement of additional
product candidates (both opioid and non-opioid) utilizing our Aversion® and
Impede™ Technologies and enhancement of our intellectual property portfolio for
abuse deterrent technologies. Such performance targets are both
organizational and individual goals for Messrs. Reddick and Jones. Mr. Clemens'
bonus is weighted 50% to the achievement of the foregoing organizational goals
and 50% to the achievement of goals unique to his position as Chief Financial
Officer.
No
compensation will be earned with respect to a performance measure unless a
performance "floor" for that measure is exceeded; the incentive opportunity with
respect to a measure will be earned if the target is achieved; achievement
between the floor and the target results in a lower amount of award with respect
to that performance measure. An amount larger than the incentive
opportunity for each performance measure can be earned, up to and possibly
exceeding a specified limit, for exceeding the target for that
measure. Depending on market conditions and other circumstances,
performance criteria may be modified during the course of the year, and other
performance criteria reweighted. In setting compensation levels, the
Compensation Committee compares our Company to companies of comparable business
focus, market capitalization, technological capabilities and market in which we
compete for executives. As part of this process, the Compensation
Committee and the Board does not use the compensation levels of comparable
companies as benchmarks, rather as a factor in evaluating the compensation
levels of the named, executive officer. To date, compensation
consultants have not been retained by the Compensation Committee or the Board as
part of this process.
In
ascertaining the achieved level of performance against the targets,
the effects of certain extraordinary events, as determined by the Compensation
Committee, such as (i) major acquisitions and divestitures, (ii) significant
one-time charges, and (iii) changes in accounting principles required by the
Financial Accounting Standards Board, are "compensation neutral" for the year in
which they occurred; that is, they are not taken into account in determining the
degree to which the targets are met in that year.
The
Compensation Committee may, after a review of an executive’s performance,
recommend to the
Board that a bonus award be made to such executives based upon other
non-enumerated performance targets (whether or not they are parties to
employment agreements). This could result in the award of salary
increases or bonuses above a targeted range amount.
For our
other executive officers not subject to an employment contract (Messrs.
Brzeczko, Seiser and Emigh), the Compensation Committee will set the annual
salary for such executive officers between December and March and
establish potential bonus compensation that such executives may earn based upon
quantitative and, if applicable, qualitative performance goals established by
the Compensation Committee. For 2008 and 2009 each of Messrs.
Seiser’s and Emigh’s salaries was $160,000. Dr. Brzeczko commenced
employment in February, 2009 at an annual salary of $265,000.
The
salary and bonus performance targets for both Messrs. Emigh and Seiser for 2008
consisted of advancing our Acurox® Tablets
to NDA submission and other product candidates utilizing our Aversion®
Technology through proof of concept, implementing the King Agreement, licensing
of additional products to King through the exercise of King’s options under the
King Agreement, and licensing products utilizing our Aversion®
Technology outside of North America. Such performance targets were both
organization and individual goals. For the reasons stated above in
the case of Messrs. Reddick, Spivey, Jones and Clemens, we paid bonuses of
$56,000 and $40,000 to Messrs. Seiser and Emigh, respectively, for
2008. Although salaries were increased for 2009 for non-executive
officer employees, no salary increases were granted to Messrs. Seiser or Emigh
for 2009.
Messrs.
Brzeczko’s, Seiser’s and Emigh’s performance targets for 2009 were the same as
those for Reddick, Jones and Clemens as stated above. Because only
some of these targets were met we paid lower bonuses to Messrs. Seiser and Emigh
in 2009 than in 2008. In 2009, Messrs. Seiser and Emigh received
bonuses of $21,500 and $20,000 respectively. Similarly Dr. Brzeczko’s
2009 year-end bonus was $33,000, which was less than the maximum bonus target
included in his February 2009 employment offer letter. Messrs.
Brzeczko’s, Seiser’s and Emigh’s organizational bonus performance targets for
2010 are consistent with those for Messrs. Reddick, Jones and Clemens as
outlined above. Such officers' bonuses are weighted 50% to the organizational
performance targets and 50% to the achievement of performance targets
unique to their respective positions. After receiving no salary increases
in 2009, in 2010 we awarded salary increases to Messrs. Brzeczko, Seiser and
Emigh generally consistent with inflation. As a result, for 2010
Messrs. Brzeczko, Seiser and Emigh will receive annual salaries of $273,000,
$165,000 and $165,000, respectively.
52
Stock
Options
A
long-term component of our executive compensation program consists of stock
option grants. The options generally permit the option holder to buy the number
of shares of our Common Stock covered by the option (an "option exercise") at a
price fixed at the time of grant. While we have historically granted
stock options having an exercise price equal to the fair market value of our
Common Stock on the date of grant and continued this practice in 2009, during
2004 and 2005, we issued stock options to our employees at a discount to the
trading price of our common stock. It is our expectation that
discounted stock option grants will occur, if at all, only on an isolated basis
in the future where circumstances warrant. With respect to stock
options grants having an exercise price equal to the market price of our Common
Stock on the date of grant, such options generally gain value only to the extent
our stock price exceeds the option exercise price during the life of the option.
Generally, a portion of the options vest over a period of time if the option
holder remains an employee and expire no later than 10 years after
grant. Executives will generally be subject to limitations in selling
the vested option stock due to securities law considerations, and therefore will
have an incentive to increase shareholder value. In 2007 no option
grants were made to our executive officers as the Compensation Committee and the
Board elected to grant salary increases and bonuses instead based on our
improved cash position and the absence of the award of such cash incentives
during the prior four years.
It is the
Company’s practice to grant stock options to executives upon commencement of
employment. On April 7, 2008, the date Mr. Jones joined us as Senior
Vice President and Chief Operating Officer, he was granted stock options
exercisable for 30,000 shares. See “—Summary Compensation Table and
Discussion of Employment and Incentive Arrangements-Employment
Agreements”. In 2009, we granted stock options to purchase 96,000
shares to Dr. Brzeczko upon his commencement of employment as Vice President of
Technical Affairs. See “—Summary Compensation Table and Discussion of
Employment and Incentive Arrangements. In addition, in May 2008
and April 2009 we granted options for an aggregate of 1,040,000 and 1,030,000
shares, respectively, to our employees, generally, exercisable at fair market
value on the date of grant, which options vest in equal installments over 24
months. The May 2008 option grants included options with
respect to 250,000, 160,000, 160,000, 100,000, 80,000, and 80,000 underlying
shares, to Messrs. Reddick, Spivey, Jones, Clemens, Seiser and Emigh,
respectively, which represented 24%, 15%, 15%, 10%, 8% and 8% of the options
granted to all employees, generally. The April 2009 option grants
included options with respect to 250,000, 160,000, 120,000, 96,000,
and 72,000 underlying shares, to Messrs. Reddick, Jones, Clemens, Seiser and
Emigh, respectively, which represented 24%, 16%, 12%, 9% and 7%, respectively,
of the options granted to all employees, generally. Dr.
Brzeczko was not included in the April 2009 option grants, as he had received an
option grant in February 2009 upon commencement of his employment. It
is likely we will maintain similar but not necessarily identical ratios of
distribution of option awards in the future as we made in 2008 and 2009 to those
persons and/or persons in similar management positions.
Timing
Policies with Respect to Options
We have
no plan or practice to time option grants in coordination with the release of
non-public information and we do not time the release of non-public information
to affect the value of executive compensation. Option grant dates for options
issued to any new executive officers will likely be the starting date of their
employment.
53
Restricted
Stock Units
Another
component of our executive compensation program is the grant of RSUs under our
2005 RSU Plan. A RSU represents a contingent obligation to deliver a
share of our common stock to the holder of the RSU on a distribution
date. Each RSU award made to our executives in 2005 vested one-third
(1/3) upon grant and the balance in equal monthly increments on the first day of
each month beginning January 1, 2006 and ending December 1, 2007. We
will issue the vested shares underlying the RSU awards on the earlier of (i) a
Change of Control (as defined in our 2005 RSU Plan), or (ii) in four annual
installments starting on January 1, 2011. In the event of a Change of
Control, our issuance of the vested shares shall be made in a lump sum
distribution. In the absence of a Change of Control, the issuance of
the vested shares shall be made in four (4) equal installments on each of
January 1, 2011, January 1, 2012, January 1, 2013 and January 1,
2014. Upon our distribution of the vested shares underlying the RSU
awards, the recipients must submit to us the par value of $0.01 per
share. In 2005, we granted Messrs. Reddick, Spivey, Clemens, Seiser
and Emigh RSU awards with respect to 825,000, 660,000, 440,000, 165,000 and
137,500 underlying shares, respectively. Of the RSU awards to
employees noted above in 2005, 30%, 24%, 16%, 6% and 5% were issued to Messrs.
Reddick, Spivey, Clemens, Seiser and Emigh, respectively. In the case
of Messrs. Reddick, Spivey and Clemens, such awards were reflected in their
employment agreements. We may also grant RSUs in connection with
commencement of employment. In April 2008, upon commencement of his
employment, Mr. Jones was granted an RSU award for 50,000 shares, which vested
monthly in 2,500 share installments commencing May 31, 2008. Dr.
Brzeczko and another executive were also awarded RSUs upon commencement of
employment in 2009. No additional RSUs were granted to our executive
officers in 2007 or 2008 as the Compensation Committee and the Board elected to
grant salary increases in 2007, option awards in 2008 and bonuses in 2007 and
2008. In April 2009 we granted an aggregate of 285,000 RSUs to all of
our employees (other than those who had already received awards earlier in the
year upon commencement of employment), including 85,000, 45,000, 30,000, 24,000
and 18,000 to Messrs. Reddick, Jones, Clemens, Seiser and Emigh, respectively,
representing 30%, 16%, 11%, 8% and 6% of RSUs granted to employees
generally. It is likely we will maintain similar but not
necessarily identical ratios of distribution of RSU awards in the future as we
made in 2005 and 2009 to those persons and/or persons in similar management
positions. Equity awards granted to executive upon commencing
employment are considered in and serve to reduce, the annual equity awards that
may be made to such executive in later in such year.
Termination/Severance
Benefits
The
employment agreement of each of Messrs. Reddick, Jones and Clemens provide
severance benefits under certain circumstances. The severance
benefits provided to each such executive differ, but include payments of a pro
rata bonus or non equity incentive compensation, one to two years of salary and
one to two years of benefits. See “Employment Agreements” and
“Quantifying Termination/Change of Control Payments” in this Item
11. We believe severance arrangements for the highest level officers
help them to focus on their respective job functions and give them
comfort that we will not lightly terminate their employment. We
believe these severance benefits were necessary to be able to initially hire and
to retain these executives. In turn Messrs. Reddick, Jones and
Clemens have agreed after their employment with us ends under certain
circumstances not to compete or solicit our employees for hire for a limited
period of time. We believe that such non-compete and non-solicit
provisions are important to protect our business. The severance
benefits are standard in employment contracts and were the results of
negotiations between us and our executives.
The other
executive officers named in the Summary Compensation Table have no contractual
severance benefits if terminated by us other than acceleration of vesting of
their RSUs.
Retirement
Plans
Beginning
in 1998, we have maintained a 401(k) plan that allows us to make both
discretionary and matching contributions, but we have not done so since
inception. We have no pension plans or non-qualified deferred compensation plans
and, as a result, the columns relating to such plans in the Summary Compensation
Table are blank.
Change
in Control
Currently
unexercisable options vest with respect to all underlying shares upon a change
of control (as defined in employment agreements, in the case of Messrs. Reddick,
Jones and Clemens, and in stock option agreements, in the case of Messrs.
Brzeczko, Emigh and Seiser) for all executive officers. In addition,
discounted options that are subject to Section 409A of the Internal Revenue Code
of 1986, as amended (“Section 409A), become exercisable upon a change of control
that qualifies as a change of control under Section 409A. In
addition, RSUs vest with respect to all underlying shares upon a change of
control and are distributed upon a change of control (provided the requirements
of Section 409A are met). In addition, Messrs. Reddick, Jones and
Clemens receive severance and bonuses if they terminate their employment after a
change of control (as defined in their employment agreements), or we terminate
their employment after a change of control. We believe our change of
control provisions incentivize our executives to seek opportunities for us and
realize benefits from a change of control transaction even though such change of
control may lead to the termination of their positions.
54
Tax
Reimbursements
Because
of the excise tax imposed by Internal Revenue Code Section 280G,
our executive officers may be subject to such tax upon the exercise
of options and distributions under RSUs upon a change of control. We
currently have no agreements to reimburse our executive officers for any taxes
imposed as a result of these additional excise taxes. We will pay
taxes incurred by Mr. Reddick on a lump sum distribution of the value of twelve
months of benefits, which he may elect in lieu of continued benefits, in the
event his employment terminates under certain circumstances. We also
allow our employees to elect to have shares withheld upon exercise of options
and upon the exchange of RSUs in satisfaction of the statutory minimum
withholding tax obligations of such employees relating to such option exercises
or RSU exchanges.
Perquisites
and Other Benefits
Our
executive officers receive no perquisites. We have not made either
discretionary or matching contributions to their 401(k) plans, although our plan
provides that we may do so. Our executive officers are not provided
auto allowances and they receive no country club or golf club
memberships. We may, however, consider such perquisites in the
future.
Board
Process
The
Compensation Committee of the Board of Directors approves all compensation and
awards to our executive officers and other employees and thereafter submits its
recommendation to the full Board for approval. All such decisions are
made with the consultation of the Chief Executive Officer, except those relating
to the compensation of the Chief Executive Officer. Except for salary
adjustments and cash bonus and equity awards to the Chief Executive Officer,
these items are generally based upon the recommendation of the Chief Executive
Officer. For example, in 2009, the Chief Executive Officer made
recommendations with respect to bonuses and salary increases for all other
employees (other than himself) and the Compensation Committee and Board adopted
such recommendations. With respect to salary adjustments and cash
bonus and equity items to the Chief Executive Officer, the Compensation
Committee establishes such awards for the Chief Executive Officer subject to
review and approval of the Board.
Summary
Compensation Table and Discussion of Employment and Incentive
Arrangements
The
following table sets forth a summary of the compensation paid by us for services
rendered in all capacities to us during each of the three fiscal years ended
December 31, 2009, to our Chief Executive Officer, Chief Financial Officer and
our next three most highly compensated executive officers (collectively, the
"2009 named executive officers") whose total annual compensation for 2009
exceeded $100,000:
55
Summary
Compensation Table
|
Name and Principal Position
|
Year
|
Base
Salary
($)
|
Bonus
($)
|
Stock
Awards1
($)
|
Option
Awards2
($)
|
Total
($)
|
||||||||||||||||
|
Andrew
D. Reddick
|
2007
|
300,000 | 850,000 | — | — | 1,040,000 | ||||||||||||||||
|
President
& CEO
|
2008
|
365,000 | 328,500 | — | 2,369,600 | 3,063,100 | ||||||||||||||||
|
2009
|
365,000 | 140,000 | 533,800 | 1,504,725 | 2,543,525 | |||||||||||||||||
|
Peter
A. Clemens
|
2007
|
180,000 | 180,000 | — | — | 360,000 | ||||||||||||||||
|
SVP
& CFO
|
2008
|
205,000 | 102,500 | — | 947,840 | 1,255,340 | ||||||||||||||||
|
2009
|
205,000 | 74,000 | 188,400 | 722,268 | 1,189,668 | |||||||||||||||||
|
Robert
B. Jones,
|
2008
|
211,923 | 130,500 | 282,600 | 685,466 | 1,310,489 | ||||||||||||||||
|
SVP
& COO (commenced
|
2009
|
290,000 | 112,000 | 431,500 | 963,024 | 1,796,524 | ||||||||||||||||
|
employment
April 7, 2008)
|
||||||||||||||||||||||
|
Albert
W. Brzeczko
|
||||||||||||||||||||||
|
VP,
Technical Affairs
|
||||||||||||||||||||||
|
(commenced employment
|
2009
|
234,423 | 73,000 | 136,580 | 523,978 | 967,981 | ||||||||||||||||
|
February
9, 2009)
|
||||||||||||||||||||||
|
Robert
A. Seiser
|
2007
|
133,000 | 140,000 | — | — | 273,000 | ||||||||||||||||
|
VP,
Treasurer & Corporate
|
2008
|
160,000 | 56,000 | — | 758,272 | 974,272 | ||||||||||||||||
|
Controller
|
2009
|
160,000 | 21,500 | 150,720 | 577,814 | 910,034 | ||||||||||||||||
(1) The
2008 entries reflect the grant date fair value of RSUs awarded with respect to
50,000 underlying shares in 2008 to Mr. Jones. The 2009 entries
reflect the grant date fair value of RSUs with respect to 85,000, 45,000,
30,000, 24,000 and 24,000 underlying shares issued in 2009 to Messrs. Reddick,
Jones, Clemens, Brzeczko, and Seiser, respectively. Grant date fair
values are computed in accordance with FASB ASC Topic 718, which are the closing
value of the price of our Common Stock on the business day preceding the date of
grant reduced by the $.01 par value payable by a holder upon exchange of an RSU.
In all cases we exclude the possibility of forfeiture. See Note I to
our Financial Statements for a general discussion of assumptions used in
calculating grant date fair-value.
(2) The 2008 entries reflect the
grant date fair value of options with respect to 250,000, 100,000, 190,000, and
80,000 underlying shares issued in 2008 to Messrs. Reddick, Clemens, Jones, and
Seiser, respectively. The 2009 entries reflect the grant date fair value of
options with respect to 250,000, 120,000, 160,000, 96,000, and 96,000 underlying
shares issued in 2009 to Messrs. Reddick, Clemens, Jones, Brzeczko and Seiser,
respectively. Grant date fair values are computed in accordance with
FASB ASC Topic 718. To calculate grant date fair value, we consider an assumed
risk free interest rate and a historical volatility percentage for our Common
Stock. For the option with respect to 30,000 shares issued to Mr.
Jones in April 2008 we used a risk free interest rate of 3.57% and historical
volatility of 142.47%. For other options issued in 2008 we used a
risk free interest rate of 3.85% and historical volatility of
124.84%. For options issued to Dr, Brzeczko in February 2009 we used
a risk free interest rate of 3.07% and historical volatility of
124.19%. For other options issued in 2009 we used a risk free
interest rate of 2.96% and historical volatility of 123.92%. In all cases
we excluded the possibility of forfeiture and calculated values based on 10 year
option terms. See Note I to our Financial Statements for a
general discussion of assumptions used in calculating grant date fair
value.
56
Other
Compensatory Arrangements
Our
executive officers participate in medical, dental, life and disability insurance
plans provided to all of our employees.
Employment
Agreements
Andrew D.
Reddick is employed pursuant to an Employment Agreement effective as of August
26, 2003, as amended, which provides that Mr. Reddick will serve as our Chief
Executive Officer and President for a term currently scheduled to expire
December 31, 2010. The term of the Employment Agreement provides for
automatic one (1) year renewals in the absence of written notice to the contrary
from us or Mr. Reddick at least ninety (90) days prior to the expiration of the
initial term or any subsequent renewal period. Pursuant to an
amendment to the Employment Agreement executed on July 9, 2008, our non-renewal
of the Employment Agreement is considered a termination without Cause for all
purposes under the Employment Agreement. Mr. Reddick’s base salary
under the Employment Agreement is $377,000 (increased by the Board from $365,000
effective January 1, 2010). Pursuant to the Employment Agreement, Mr.
Reddick is entitled to an annual bonus based on the achievement of such targets,
conditions, or parameters as may be set from time to time by the Board of
Directors or the Compensation Committee of the Board of
Directors. For our 2009 fiscal year, Mr. Reddick was awarded a bonus
of $140,000 due to, among other reasons, the achievement of certain of the items
discussed above under the caption “Salary and Bonus”. The Employment
Agreement also provides for our grant in August, 2004 to Mr. Reddick of stock
options exercisable for up to 875,000 shares of Common Stock at an exercise
price of $1.30 per share. Such stock options provide for vesting of
300,000 shares on the date of grant of the option, with the balance vesting in
monthly increments of 25,000 shares at the expiration of each monthly period
thereafter commencing with the month ending August 31, 2004. The
exercise price of $1.30 per share represents a discount to the fair market value
of our Common Stock on the date of grant. On August 12, 2004, the
date of grant of the stock options, the average of the closing bid and asked
prices for our Common Stock was $4.35. Because 450,000 of the
discounted options are subject to Section 409A, in 2007, we established an
exercise schedule to comply with Section 409A for such 450,000 options so that
the options are exercisable (subject to earlier exercisability as set forth in
the table below entitled “Events Affecting Option Vesting and Exercise”) in four
equal installments on January 1 of each of 2011, 2012, 2013 and 2014, provided
that such options may be exercised only in the calendar year in which they first
become exercisable, and in no event later than August 11, 2014. The
Employment Agreement also acknowledges our grant in December, 2005 to Mr.
Reddick of a Restricted Stock Unit Award providing for our issuance of up to
825,000 shares of our Common Stock. The Restricted Stock Unit vested
one-third (1/3) upon grant and the balance in equal monthly increments on the
first day of each month beginning January 1, 2006 and ending December 1,
2007. The vested shares underlying the Restricted Stock Unit Award
will be issued by us on the earlier of (i) a Change in Control (as defined in
our 2005 RSU Plan), or (ii) January 1, 2011. In the event of a Change
in Control, we will issue the vested shares in a lump sum
distribution. In the absence of a Change of Control, the issuance of
the vested shares shall be made in four (4) equal installments on each of
January 1, 2011, January 1, 2012, January 1, 2013 and January 1,
2014. Upon issuance of the shares underlying the Restricted Stock
Unit Award, Mr. Reddick must remit to us the par value of $0.01 per share or
have us withhold shares to satisfy such payment obligation. On December 22,
2005, the date of grant of the Restricted Stock Unit Award, the average of the
closing bid and asked prices of our Common Stock was $3.33, as reported by the
OTCBB. Mr. Reddick has no rights as a stockholder, including no
dividend or voting rights, with respect to the shares underlying the Restricted
Stock Unit Award until we issue the underlying shares. The Employment
Agreement contains standard termination provisions, including upon death,
disability, for Cause, for Good Reason and without Cause. In the
event the Employment Agreement is terminated due to death or disability, we are
required to pay Mr. Reddick, or his designee, a pro rata portion of the annual
bonus that would have been payable to Mr. Reddick during such year assuming full
achievement of the bonus criteria established for such bonus.
57
In the
event that the Employment Agreement is terminated by us without Cause, or by Mr.
Reddick for Good Reason, we are required to pay Mr. Reddick an amount equal to
the bonus for such year, calculated on a pro rata basis assuming full
achievement of the bonus criteria for such year (to the extent it has not
already been paid), as well as Mr. Reddick's base salary for one year (such
salary amount being the "Severance Pay"). In case of termination
without Cause, such severance is payable in equal monthly installments over a
period of twelve (12) months, and in the case of termination by Mr. Reddick for
Good Reason, one-half of such severance is payable six months after termination,
and the remaining half of such severance is payable thereafter in six monthly
installments. In addition, Mr. Reddick is at his option entitled to
continued coverage under our then existing benefit plans, including medical and
life insurance, for twelve (12) months from the date of termination or the value
of such benefits payable in a lump sum thirty days of termination together with
amount needed to pay income tax on such lump sum. The Employment
Agreement permits Mr. Reddick to terminate the Employment Agreement in the event
of a Change in Control (as defined in the Employment Agreement), in which case
such termination is considered to be made without Cause, entitling Mr. Reddick
to the benefits described above, except that (i) the Severance Pay is payable in
a lump sum within six months after the date of termination, and (ii) with
options being treated as set forth in the table below entitled, “Events
Affecting Option Vesting and Exercise.” The Employment Agreement
restricts Mr. Reddick from disclosing, disseminating or using for his personal
benefit or for the benefit of others, confidential or proprietary information
(as defined in the Employment Agreement) and, provided we have not breached the
terms of the Employment Agreement, from competing with us at any time prior to
one year after the termination of his employment with us. In addition
he has agreed not to (and not to cause or direct any person to) hire or solicit
for employment any of our employees or those of our subsidiaries or affiliates
(i) for six (6) months following the termination of his employment by us without
Cause or by him for Good Reason, prior to a Change of Control, (ii) for twelve
(12) months following the termination of his employment for Cause, prior to a
Change of Control, or (iii) twenty-four (24) months following a Change of
Control. On each of May 23, 2008 and April 24, 2009, we granted Mr.
Reddick options to purchase 250,000 shares of our Common Stock exercisable at
the fair market value of our Common Stock at the date of grant and vesting in
equal installments over 24 months. On April 24, 2009 we also granted
Mr. Reddick RSUs with respect to an additional 85,000 underlying shares that
vest in equal installments over 24 months and are exercisable on the same
schedule as the RSUs granted to Mr. Reddick in 2005. The table
entitled “Events Affecting Option Vesting and Exercise,” below summarizes the
vesting and exercisability of Mr. Reddick’s options following a number of
termination scenarios or a Change of Control.
Robert B.
Jones commenced employment with us on April 7, 2008 pursuant to an Employment
Agreement dated March 18, 2008, which provides that Mr. Jones will serve as our
Senior Vice President and Chief Operating Officer for a term currently scheduled
to expire December 31, 2010. The term of the Employment Agreement
provides for automatic one (1) year renewals in the absence of written notice to
the contrary from us (which would give Mr. Jones the right to terminate his
employment for Good Reason) or Mr. Jones at least ninety (90) days prior to the
expiration of the initial term or any subsequent renewal period. Mr.
Jones’ base salary under the Employment Agreement is $300,000 (increased from
$290,000 effective January 1, 2010). Pursuant to the Employment
Agreement Mr. Jones is eligible for annual bonuses of up to thirty percent (30%)
of his base salary on the achievement of such targets, conditions, or parameters
as may be set from time to time by the Board of Directors or the Compensation
Committee of the Board of Directors. In 2009, Mr. Jones was awarded a
bonus of $112,000 due to, among other reasons, the achievement of certain of the
items discussed above under the caption “Salary and Bonus”. The
Employment Agreement provides for our grant in April 2008 to Mr. Jones of stock
options exercisable for up to 30,000 shares of Common Stock at an exercise price
equal to the last sale price of our Common Stock on the last trading day prior
to his April 7, 2008 commencement date. The stock option provides for
vesting of 1,500 shares on the last day of each month commencing May 31, 2008
and as of December 31, 2009 the stock option was fully vested. In
addition, on each of May 23, 2008 and April 24, 2009, we granted Mr. Jones stock
options to purchase 160,000 shares of our Common Stock exercisable at the fair
market value of our Common Stock at the date of grant and vesting in equal
installments over 24 months (subject to earlier exercisability as set forth in
the table below entitled “Events Affecting Option Vesting and
Exercise”). The Employment Agreement also provides for our grant in
April 2008 to Mr. Jones of a Restricted Stock Unit Award providing for our
issuance of up to 50,000 shares of our Common Stock. The Restricted Stock Units
granted to Mr. Jones in 2008 vested 2,500 shares on the last day of each month
commencing May 31, 2008 and as of December 31, 2009 are fully
vested. The vested shares underlying the Restricted Stock Unit Award
will be issued by us on the earlier of (i) a Change in Control (as defined in
our 2005 RSU Plan), or (ii) January 1, 2011. In the event of a Change
in Control, we will issue the vested shares in a lump sum distribution. In the
absence of a Change in Control, the issuance of the vested shares shall be made
in four (4) equal installments on each of January 1, 2011, January 1, 2012,
January 1, 2013 and January 1, 2014. Upon issuance of the shares underlying the
Restricted Stock Unit Award, Mr. Jones must remit to us the par value of $0.01
per share or have us withhold shares to satisfy such payment
obligation. Mr. Jones has no rights as a stockholder, including no
dividend or voting rights, with respect to the shares underlying the Restricted
Stock Unit Award until we issue the shares. The Employment Agreement
contains standard termination provisions, including upon death, disability, for
Cause, for Good Reason and without Cause. In the event that we
terminate the Employment Agreement without Cause or Mr. Jones terminates the
Employment Agreement for Good Reason, we are required to pay Mr. Jones an amount
equal to the bonus for such year, calculated on a pro rata basis assuming full
achievement of the bonus criteria for such year (to the extent it has not
already been paid), as well as Mr. Jones' base salary for one year (such salary
amount being the "Severance Pay"). In case of termination without
Cause and for Good Reason, such Severance Pay is payable in equal monthly
installments over a period of twelve (12) months, with a six month payment delay
for the that portion of the Severance Pay that would cause the payments to fall
outside an exception to the deferred compensation rules requiring certain
severance payments to certain officers of a public company to be made commencing
six months after termination However, if such termination
without Cause or for Good Reason follows within two years of a qualifying Change
of Control then the Severance Pay is payable in a lump sum 31 days after
termination, otherwise if such termination follows a Change of Control by more
than two years then the Severance Pay is payable six months and one day
following termination. In addition, upon a termination without Cause
or for Good Reason any shares remaining unvested under stock options and
restricted stock units granted to Mr. Jones will vest in full and Mr. Jones will
be entitled to continued coverage under our then existing benefit plans,
including medical and life insurance, for twelve (12) months from the date of
termination. The Employment Agreement restricts Mr. Jones from
disclosing, disseminating or using for his personal benefit or for the benefit
of others, confidential or proprietary information (as defined in the Employment
Agreement) and, provided we have not breached the terms of the Employment
Agreement, from competing with us at any time prior to one year after the
termination of his employment with us. In addition, Mr. Jones has
agreed not to (and not to cause or direct any person to) hire or solicit for
employment any of our employees or those of our subsidiaries or
affiliates (i) for six (6) months following the termination of his employment by
us without Cause or by him for Good Reason, prior to a Change of
Control, (ii) for twelve (12) months following the termination of his employment
for Cause, prior to a Change of Control, or (iii) twenty-four (24) months
following a Change of Control. On April 24, 2009 we granted Mr. Jones
RSUs with respect to an additional 40,000 underlying shares that vest in equal
installments over 24 months and are exercisable on the same schedule as the RSUs
granted to Mr. Jones in 2008 The table entitled “Events
Affecting Option Vesting and Exercise,” below, summarizes the vesting and
exercisability of Mr. Jones’ options following a number of termination scenarios
or a Change of Control.
58
Peter A.
Clemens is employed pursuant to an Employment Agreement effective as of March
10, 1998, as amended, which provides that Mr. Clemens will serve as our Senior
Vice President and Chief Financial Officer for a term currently scheduled to
expire December 31, 2010. The term of the Employment Agreement
provides for automatic one (1) year renewals in the absence of written notice to
the contrary from the Company or Mr. Clemens at least ninety (90) days prior to
the expiration of any renewal period. Pursuant to a 2008 amendment to
the Employment Agreement, our non-renewal of the Employment Agreement is
considered as a termination without Cause for all purposes under the Employment
Agreement. Mr. Clemens current base salary under the Employment
Agreement is $211,500 (increased from $205,000 effective January 1,
2010). Under the Employment Agreement, he may also receive an annual
bonus to be determined based on the satisfaction of such targets, conditions or
parameters as may be determined from time to time by the Compensation Committee
of the Board of Directors. In 2009, Mr. Clemens was awarded a bonus of $74,000
due to, among other reasons, the achievement of certain of the items discussed
above under the caption “Salary and Bonus.” The Employment Agreement
also provides for the grant of stock options on March 10, 1998 to purchase
30,000 shares of our Common Stock at an exercise price of $23.75 per share,
which options vest in equal increments of 2,500 option shares at the end of each
quarterly period during the term of the Employment Agreement (as such vesting
schedule may be amended by mutual agreement of Mr. Clemens and the Board of
Directors) In addition, in August 2004, the Company granted stock
options to Mr. Clemens to purchase 37,500 shares of Common Stock at an exercise
price of $1.30 per share, which exercise price represents a discount to the fair
market value of our Common Stock on the date of grant. Such stock
options vest in four equal portions at the end of each annual period commencing
March 9, 2005. Such stock options are exercisable (subject to earlier
exercisability as set forth in the table below entitled “Events Affecting Option
Vesting and Exercise”) in four equal installments on January 1 of each of 2011,
2012, 2013 and 2014, provided that such options may be exercised only in the
calendar year in which they first become exercisable, and in any event no later
than their respective expiration dates. In addition, on May 23, 2008, we granted
Mr. Clemens options to purchase 100,000 shares of our Common Stock and on April
24, 2009 we granted Mr. Clemens options to purchase 120,000 shares of our Common
Stock, in each case at an exercise price equal to the fair market value of our
Common Stock at the date of grant and vesting in equal installments over 24
months (subject to earlier exercisability as set forth in the table below
entitled “Events Affecting Option Vesting and Exercise”). The Employment
Agreement also acknowledges the grant to Mr. Clemens of a Restricted Stock Unit
Award providing for our issuance of up to 440,000 shares of our Common
Stock. The Restricted Stock Unit vests one-third (1/3) upon grant and
the balance in equal monthly increments on the first day of each month beginning
January 1, 2006 and ending December 1, 2007. We will issue the vested
shares underlying the Restricted Stock Unit Award on the earlier of (i) a Change
in Control (as defined in our 2005 RSU Plan), or (ii) January 1,
2011. In the event of a Change in Control, we will issue the vested
shares in a lump sum distribution. In the absence of a Change in
Control, our issuance of the vested shares shall be made in four (4) equal
installments on each of January 1, 2011, January 1, 2012, January 1, 2013 and
January 1, 2014. Upon issuance of the shares underlying the
Restricted Stock Unit Award, Mr. Clemens must remit to us the par value of $0.01
per share or have us withhold shares to satisfy such payment obligation. On
December 22, 2005, the date of grant of the Restricted Stock Unit Award, the
average of the closing bid and asked prices of our Common Stock was $3.33, as
reported by the OTCBB. Mr. Clemens has no rights as a stockholder,
including no dividend or voting rights, with respect to the shares underlying
the Restricted Stock Unit Award until we issue the shares. On April
24, 2009 we granted Mr. Clemens RSUs with respect to an additional 30,000
underlying shares that vest in equal installments over 24 months and are
exercisable on the same schedule as the RSUs granted to Mr. Clemens in
2005. The Employment Agreement contains standard termination
provisions, including upon death, disability, for Cause, for Good Reason and
without Cause. In the event the Employment Agreement is terminated by us without
Cause or by Mr. Clemens for Good Reason, we are required to pay Mr. Clemens an
amount equal to $410,000 or twice his then base salary, whichever is greater,
payable in the case of termination without Cause in a lump sum within 30 days
following termination and in the case of termination for Good
Reason, six months after termination and to continue to provide Mr.
Clemens coverage under our then existing benefit plans, including medical and
life insurance, for a term of 24 months. The Employment Agreement
permits Mr. Clemens to terminate the Employment Agreement in the event of a
Change in Control (as defined in the Employment Agreement), in which case he
would receive the same payments as on a termination for Good
Reason. The Employment Agreement also restricts Mr. Clemens from
disclosing, disseminating or using for his personal benefit or for the benefit
of others confidential or proprietary information (as defined in the Employment
Agreement) and, provided we have not breached the terms of the Employment
Agreement, from competing with us at any time prior to two years after the
earlier to occur of the expiration of the term and the termination of his
employment. In addition, for a period of two (2) years from and after
the effective date of the termination of his employment with us (for any reason
whatsoever), (i) induce or attempt to influence any employee of the Corporation
or any of its subsidiaries or affiliates to leave its employ, or (ii) aid any
person, business, or firm, including a supplier, a competitor, licensor or
customer of or our manufacturer for the Corporation, in any attempt to hire any
person who shall have been employed by us or any of our subsidiaries or
affiliates within the period of one (1) year of the date of any such requested
aid. The table entitled “Events Affecting Option Vesting and
Exercise,” below, summarizes the vesting and exercisability of Mr. Clemens’
options following a number of termination scenarios or a Change of
Control.
59
Events
Affecting Stock Option Vesting and Exercise (For Messrs. Reddick, Jones, and
Clemens)
|
Event
|
Vesting of All
Options (Options
not subject to
Section 409A(1)
are exercisable
upon vesting)
|
Exercisability of Options
not subject to section
409A(1)
|
Exercisability of Options Subject
to Section 409A(1)
|
|||
|
Termination
due to Death
|
No
additional vesting
|
Vested
options immediately exercisable for one year following
termination
|
Vested
options immediately exercisable for the lesser of (a) one year following
termination or (b) the last day of the year in which they become
exercisable
|
|||
|
Termination
by Company Without Cause or by Employee for Good Reason or
following Change of Control (not qualifying under Section
409A)
|
All
options fully vest for Messrs. Reddick and Jones. Mr. Clemens’s
options vest upon termination after Change of Control.
|
Vested
options immediately exercisable for one year following
termination
|
Vested
options exercisable commencing six months after termination for the lesser
of (a) one year following termination or (b) the last day of the year in
which they become exercisable
|
|||
|
Termination
due to Disability
|
No
additional vesting
|
Vested
options immediately exercisable for one year following
termination
|
Vested
options exercisable commencing six months after termination for the lesser
of (a) one year following termination or (b) the last day of the year in
which they become exercisable
|
|||
|
Termination
by the Company for Cause or by executive other than for Good
Reason
|
No
additional vesting
|
Vested
options immediately exercisable for 40 days following
termination
|
Vested
options exercisable commencing six months after termination for the lesser
of (a) 40 days thereafter or (b) the last day of the calendar year in
which they first become exercisable
|
|||
|
Change
of Control
|
|
Options
fully vest
|
|
Vested
options immediately exercisable
|
|
Vested
options exercisable upon Change of Control qualifying under Section 409A
during the year in which the Change of Control
occurs
|
(1) See Footnote 2 to table entitled
“Outstanding Equity Awards at 2009 Year-End” and corresponding text for
identification of options subject to Section 409A. Mr. Jones does not
hold any such options.
Mr.
Seiser and Dr. Brzeczko are not parties to an employment
agreement. Dr. Brzeczko was hired pursuant to an offer
letter. He received a $40,000 signing bonus and is eligible for an
additional annual bonus of up to 35% of his base salary. In 2009 he
received an additional bonus of $33,000. Upon commencement of his
employment on February 9, 2009, he received 24,000 RSUs vesting in equal
installments over 24 months, and stock options exercisable for 96,000 shares of
Common Stock vesting in equal installments over 24 months. Effective
January 1, 2010, Dr. Brzeczko’s annual salary is $273,000 (increased from
$265,000).
Mr.
Seiser is employed at an annual salary of $165,000 (increased from $160,000
effective January 1, 2010). In 2009 he was granted 24,000 RSUs
vesting in equal installments over 24 months, and stock options exercisable for
96,000 shares of Common Stock vesting in equal installments over 24
months.
Stock
Option Plans
We
maintain three stock option plans adopted in 1995, 1998 and 2008,
respectively. In the past we used, and may continue to use, stock
options to attract and retain key employees in the belief that employee stock
ownership and stock-related compensation devices encourage a community of
interest between employees and shareholders.
60
The
1995 Stock Option Plan
The 1995
Stock Option Plan was approved by our shareholders in September,
1995. As of December 31, 2009 incentive stock options (“ISO’s”) to
purchase 19,000 shares and non-qualified options to purchase 6,500 shares were
outstanding under the 1995 Stock Option Plan. In May, 2005 the 1995 Stock Option
Plan expired and the remaining unissued shares allocated to the Plan were
terminated. The average per share exercise price for all outstanding
options under the 1995 Stock Option Plan is $12.27.
The
1998 Stock Option Plan
The 1998
Stock Option Plan was adopted by the Board of Directors in April, 1998 and
approved by our shareholders in June, 1998. The 1998 Stock Option
Plan permits the grant of ISO’s and non-qualified stock options to purchase
shares of our Common Stock. The 1998 Stock Option Plan was amended by
the Board of Directors in April, 1999 to increase the number of shares available
for the grant of options under the Plan from 260,000 to 360,000
shares. Our shareholders ratified the Plan amendment on August 19,
1999. The 1998 Stock Option Plan was further amended by Board of
Directors in April, 2001 to increase the number of shares available for grant of
options under the Plan from 360,000 to 810,000 shares. Our
shareholders ratified the Plan amendment on June 14, 2001. The 1998
Stock Option Plan was further amended by the Board of Directors on May 5, 2004
to increase the number of shares available for grant of options under the Plan
from 810,000 to 2,000,000 shares. Our shareholders ratified the Plan
amendment on August 12, 2004. The 1998 Stock Option Plan was further
amended on February 8, 2006 to make such plan compliant with Section 409A of the
Internal Revenue Code, as amended. Our shareholders ratified the
amendment on December 14, 2006. On June 25, 2009, the 1998 Stock
Option Plan was further amended by our shareholders to allow participants to
require us to withhold Common Stock upon exercise of options for payment of
exercise price and statutory minimum withholding taxes. As of
December 31, 2009, stock options to purchase 1,361,514 shares of Common Stock
had been granted under the 1998 Stock Option Plan. Of such option
grants, 54,750 are ISO’s and 1,306,764 are non-qualified
options. No exercise price of an ISO was set at less than
100% of the fair market value of the underlying Common Stock. The
exercise price of non-qualified options exercisable for 1,174,414 shares of
Common Stock has been set at less than the fair market value on the date of
grant of the underlying Common Stock. Subject to the terms of the
1998 Stock Option Plan, the Board of Directors, or a Committee appointed by the
Board determines the persons to whom grants are made and the vesting, timing,
amounts and other terms of such grant. An employee may not receive ISO’s
exercisable in any one calendar year for shares with a fair market value on the
date of grant in excess of $100,000. No quantity limitations apply to
the grant of non-qualified stock options.
Options
issued to date at a discount under the 1998 Stock Option Plan, which had not
vested as of December 31, 2004, are exercisable (subject to earlier exercise as
described below) in four equal installments on January 1 of each of 2011, 2012,
2013 and 2014. These options are exercisable earlier than stated
above upon a qualifying change of control and upon termination of employment
(generally for a period of 90 days), subject in the case of termination, to a 6
month waiting period prior to exercise for Messrs. Reddick, Clemens, Jones and
Seiser. In no event are these options exercisable outside the
calendar year in which they first become exercisable.
In April,
2008 the 1998 Stock Option Plan expired and the remaining unissued shares
allocated to the Plan were terminated. The average per share exercise
price for all outstanding options under the 1998 Stock Option Plan is
$2.35.
The
2008 Stock Option Plan
The 2008
Stock Option Plan was adopted by the Board of Directors on March 14, 2008 and
approved by our shareholders on April 30, 2008. On June 25, 2009, the
1998 Stock Option Plan was amended to allow participants to require us to
withhold Common Stock upon exercise of options for payment of exercise price and
statutory minimum withholding taxes. The 2008 Stock Option Plan
permits the grant of ISO’s and non-qualified stock options to purchase in the
aggregate up to 6,000,000 shares of our Common Stock. As of December
31, 2009, stock options to purchase 2,284,000 shares of Common Stock had been
granted under the 2008 Stock Option Plan. Of such option grants,
1,005,246 are ISOs and 1,278,754 are non-qualified options. No
exercise price of an ISO or a non-qualified stock option was set at less than
100% of the fair market value of the underlying Common Stock. Subject
to the terms of the 2008 Stock Option Plan, the Board of Directors, or a
Committee appointed by the Board determines the persons to whom grants are made
and the vesting, timing, amounts and other terms of such grant. An employee may
not receive ISO’s exercisable in any one calendar year for shares with a fair
market value on the date of grant in excess of $100,000. No quantity
limitations apply to the grant of non-qualified stock options. The
average per share exercise price for all outstanding options under the 2008
Stock Option Plan is $7.95.
61
Restricted
Stock Unit Award Plan
On
December 22, 2005, the Board of Directors approved our 2005 Restricted Stock
Unit Award Plan (the “2005 RSU Plan”) for our employees and non-employee
directors. The RSU Plan was amended by the Board of Directors on
October 26, 2006 to allow transfer of RSUs under limited
circumstances. We believe that the 2005 RSU Plan did not require
shareholder approval. Nevertheless, on December 14, 2006, our
shareholders ratified the 2005 RSU Plan, as amended, at our 2006 Annual
Shareholders’ Meeting. A RSU represents the contingent obligation of the Company
to deliver a share of our Common Stock to the holder of the RSU on a
distribution date. On March 14, 2008 the Board of Directors adopted and on April
30, 2008 our shareholders ratified an amendment to the 2005 RSU Plan increasing
the number of shares available under the 2005 RSU Plan from 3 million to 3.5
million.
The
purpose of the 2005 RSU Plan is to attract, motivate and retain experienced and
knowledgeable employees by offering additional stock-based compensation and
incentives to defer and potentially enhance their compensation and to encourage
stock ownership in the Company and to attract and retain qualified non-employee
directors. The 2005 RSU Plan is intended to comply with Section 409A
of the Internal Revenue Code of 1986, as amended and is designed to confirm that
compensation deferred under the Plan which is subject to Code Section 409A is
not included in the gross income of 2005 RSU Plan participants until such time
as the shares of Common Stock underlying RSUs are distributed as set forth in
the Plan and Code Section 409A.
The RSU
Plan is administered by our Board of Directors or a Committee appointed by the
Board of Directors. However, with respect to non-employee directors, the Board
administers the Plan, and the Committee has no discretion with respect to any
grants to non-employee directors. RSUs granted under the RSU plan vest on a
schedule determined by the Board of Directors or such Committee as set forth in
a restricted stock unit award agreement. Unless otherwise set forth in such
award agreement, the RSUs fully vest upon a change in control (as defined in the
2005 RSU Plan) of the Company or upon termination of an employee’s employment
without cause or due to death or disability, and in the case of a non-employee
director, such person’s death or disability or if such person is not renominated
as a director (other than for “cause” or refusal to stand for re-election) or is
not elected by our stockholders, if nominated. Vesting of an RSU
entitles the holder thereof to receive a share of Common Stock of the Company on
a distribution date (after payment of the $0.01 par value per
share).
Absent a
change of control, one-fourth of vested shares of Common Stock underlying an RSU
award will be distributed (after payment of $0.01 par value per share) on
January 1 of each of 2011, 2012, 2013 and 2014. If a change in control occurs
(whether prior to or after 2011), the vested shares underlying the RSU award
will be distributed at or about the time of the change in control. No dividends
accrue on the shares underlying the RSUs prior to issuance. The
recipients of RSU awards need not be employees or directors of the Company on a
distribution date.
RSUs may
not be sold, pledged, assigned, hypothecated, transferred, or disposed of in any
manner by the recipients other than by will or by the laws
of descent or distribution and to (i) the spouse, children or grandchildren of
the awardees (the “Immediate Family Members”), (ii) a trust or trusts for the
exclusive benefit of such Immediate Family Members, or (iii) a partnership in
which such Immediate Family Members are the only partners, provided that (x)
there may be no consideration for any such transfer, (y) subsequent transfers of
transferred RSUs shall be prohibited except those made by will or by the laws of
descent or distribution, and (z) such transfer is approved in advance by the
Committee (or Board in absence of a Committee). A married recipient
may generally designate only a spouse as a beneficiary unless spousal consent is
obtained.
Recipients
of RSUs generally will not recognize income when they are awarded RSUs (unless
they elect to recognize income by making a Section 83(b) election). RSU
recipients will recognize ordinary income in an amount equal to the fair market
value of the shares of our Common Stock issued pursuant to a distribution under
the RSU. We will generally be entitled to a tax deduction in the same
amount.
62
As of
December 31, 2009 we had granted RSUs providing for our issuance of up to an
aggregate of 3,333,000 shares of our Common Stock. Of such RSU awards
17,000 were
forfeited in 2009 upon an employee’s termination of
employment. 2,750,000 of such RSU awards vest one-third (1/3) on
grant and the balance vest in equal monthly increments on the first day of each
month beginning January 1, 2006 and ending December 1, 2007. 200,000
of such RSU awards vested 77,778 shares on grant and the balance vested in equal
monthly increments on the first day of March 1, 2006 and ending December 1,
2007. 50,000 of such RSU awards vest at a rate of 2,500 on the last
day of each month commencing May 31, 2008, and as of December 31, 2009, all of
such RSU awards had vested. 24,000 of such RSU awards vest in 24
equal monthly installments on the 9th day of
each month commencing March 9, 2009 and as of December 31, 2009, 10,000 of such
RSU awards had vested. 24,000 of such RSU awards vested in equal
monthly installments on the last day of each month commencing June 11, 2009, and
as of December 31, 2009, 7,000 of such RSU awards vested and 17,000 were
forfeited. 285,000 of such RSU awards vest in equal installments on
the 24th day of
each month commencing May 24, 2009 and as of December 31, 2009, 95,000 of
such RSU awards had vested.
Outstanding
Equity Awards at 2009 Year End
The
following table presents information regarding outstanding stock and stock
option awards at December 31, 2009 for each of the 2009 named executive
officers:
Outstanding
Equity Awards at 2009 Year-End
|
Stock Option Awards
|
Restricted Stock Unit Awards
|
||||||||||||||||||||
|
Name
|
Number of
Securities
Underlying
Unexercised
Options (#)
Exercisable
|
Number of
Securities
Underlying
Unexercised
Options (#)
Unexercisable
|
Option
Exercise
Price
($)
|
Option
Expiration
Date
|
Number of
Shares or
Units
of
Stock That
Have Not
Vested
|
Market Value
of Shares or
Units of
Stock
That
Have Not
Vested ($)(1)
|
|||||||||||||||
|
Andrew
D. Reddick
|
450,000 | (2) | — | $ | 1.30 |
08/12/2014
|
56,667 | $ | 301,468 | ||||||||||||
| 197,917 | 52,083 | $ | 9.87 |
05/23/2018
|
|||||||||||||||||
| 83,333 | 166,667 | $ | 6.29 |
04/23/2019
|
|||||||||||||||||
|
Robert
B. Jones
|
30,000 | — | $ | 8.64 |
04/06/2018
|
30,000 | $ | 159,600 | |||||||||||||
| 126,667 | 33,333 | $ | 9.87 |
05/23/2018
|
|||||||||||||||||
| 53,333 | 106,667 | $ | 6.29 |
04/23/2019
|
|||||||||||||||||
|
Peter
A. Clemens
|
12,500 | — | $ | 18.75 |
02/17/2010
|
20,000 | $ | 106,400 | |||||||||||||
| 10,000 | — | $ | 11.125 |
06/29/2010
|
|||||||||||||||||
| 37,500 | (2) | — | $ | 1.30 |
03/09/2014
|
||||||||||||||||
| 79,167 | 20,833 | $ | 9.87 |
05/23/2018
|
|||||||||||||||||
| 40,000 | 80,000 | $ | 6.29 |
04/23/2019
|
|||||||||||||||||
|
Albert
W. Brzeczko
|
40,000 | 56,000 | $ | 5.70 |
02/08/2019
|
14,000 | $ | 74,480 | |||||||||||||
|
Robert
A. Seiser
|
3,000 | — | $ | 18.75 |
02/17/2010
|
16,000 | $ | 85,120 | |||||||||||||
| 4,000 | — | $ | 11.125 |
06/29/2010
|
|||||||||||||||||
| 2,500 | — | $ | 24.60 |
11/15/2011
|
|||||||||||||||||
| 24,900 | (2) | — | $ | 1.30 |
03/09/2014
|
||||||||||||||||
| 63,334 | 16,666 | $ | 9.87 |
05/23/2018
|
|||||||||||||||||
| 32,000 | 64,000 | $ | 6.29 |
04/23/2019
|
|||||||||||||||||
(1) Based on the closing price of
$5.33 reported on the Nasdaq Capital Market on December
31, 2009 reduced by the $.01 par value per share that must be paid on the
distribution of shares underlying the RSUs.
(2) See “Stock Option Plans –1998 Stock
Option Plan” for information regarding these options which were issued at a
discount to fair market vale and also see table entitled “Events Affecting Stock
Option Vesting And Exercise” (column labeled “Exercisability of Options Subject
to Section 409A”) with respect to Messrs. Reddick and Clemens and “Potential
Payments Potential Payments Upon Termination or Change in Control - Messrs.
Brzeczko and Seiser”, with respect to Mr. Seiser.
63
Grants
of Plan Based Awards in 2009
The
following table presents information regarding the grant of awards made to our
2009 named executive officers in the last completed fiscal year under our 2005
RSU Plan and 2008 Stock Option Plan:
Grants
of Plan-Based Awards
|
Name
|
Grant
Date
|
Stock awards under
the 2005 Restricted
Stock Unit Award
Plan - Number of
shares of stock
(#)
|
Option awards under
the 2008 Stock Option
Plan- Number of
securities underlying
options
(#)
|
Exercise or
base price
of option
awards
($/Sh)
|
Grant date fair
value of stock and
option awards (2)
|
|||||||||||||
|
Andrew
D. Reddick
|
4/24/2009
|
85,000 | — | (1) | $ | 534,000 | ||||||||||||
|
4/24/2009
|
— | 250,000 | $ | 6.29 | $ | 1,505,000 | ||||||||||||
|
Robert
B. Jones
|
4/24/2009
|
45,000 | — | (1) | $ | 188,000 | ||||||||||||
|
4/24/2009
|
— | 160,000 | $ | 6.29 | $ | 963,000 | ||||||||||||
|
Peter
A. Clemens
|
4/24/2009
|
30,000 | — | (1) | $ | 126,000 | ||||||||||||
|
4/24/2009
|
— | 120,000 | $ | 6.29 | $ | 722,000 | ||||||||||||
|
Albert
W. Brzeczko
|
2/9/2009
|
24,000 | — | (1) | 137,000 | |||||||||||||
|
2/9/2009
|
— | 96,000 | $ | 5.70 | $ | 524,000 | ||||||||||||
|
Robert
A. Seiser
|
4/24/2009
|
24,000 | — | (1) | $ | 151,000 | ||||||||||||
|
4/24/2009
|
— | 96,000 | $ | 6.29 | $ | 578,000 | ||||||||||||
(1) RSUs require the payment
of $0.01 par value per share upon the distribution of the shares underlying the
RSUs.
(2) See Notes 1 and 2 to
Summary Compensation Table for methodology used in computing grant date fair
value. See also Note I to our Financial Statements.
64
Option
Exercises and Stock Vested in 2009
The
following table presents information regarding the exercise of options by our
2009 named executive officers and vesting of awards made to a 2009 named
executive officer under our 2005 RSU Plan that occurred in our last completed
fiscal year.
Option
Exercise and Stock Vested In Fiscal Year 2009
|
Option Awards
|
Stock Awards
|
|||||||||||||||
|
Name
|
Number of
Shares acquired
on exercise
|
Value Realized
on exercise
|
Number of Shares
Vested (#)(1)
|
Value Realized on
Vesting ($)(2)
|
||||||||||||
|
Andrew
D. Reddick
|
425,000 |
(3)
|
$ | 1,258,000 |
(4)
|
28,333 | $ | 156,895 | ||||||||
|
Robert
B. Jones
|
— | — | 35,000 | $ | 251,587 | |||||||||||
|
Peter
A. Clemens
|
— | — | 10,000 | $ | 55,375 | |||||||||||
|
Albert
W. Brzeczko
|
— | — | 10,000 | $ | 58,010 | |||||||||||
|
Robert
A. Seiser
|
— | — | 8,000 | $ | 44,300 | |||||||||||
(1) The vested shares
underlying the RSUs will be issued by us on the earlier of (i) a Change of
Control (as defined in our 2005 Restricted Stock Unit Award Plan), or (ii) in
four annual installments starting on January 1, 2011. In the event of
a Change of Control, our issuance of the vested shares shall be made in a lump
sum distribution. In the absence of a Change of Control, the issuance
of the vested shares shall be issued in four (4) equal installments on each of
January 1, 2011, January 1, 2012, January 1, 2013 and January 1,
2014. Upon our distribution of the vested shares underlying the RSUs,
the recipients must submit to us the par value of $0.01 per
share. The recipients of the RSUs have no rights as a stockholder,
including no dividend or voting rights, with respect to the shares underlying
such awards until the shares are issued by us.
(2) Value is determined by subtracting
the $.01 par value required to be paid on exchange of each share for RSUs from
the closing price of our Common Stock on the Nasdaq Capital Market on each
vesting date (or the preceding closing price if the vesting date is not a
trading date) and multiplying the result by the number of shares underlying the
RSUs that vested on such date and then aggregating those results.
(3) Of the 425,000 options
exercised, 249,353 shares were withheld by the Company for payment of the
exercise price and statutory minimum withholding taxes leaving Mr.
Reddick with a net total of 175,647 common shares. As of
the date of this Report, Mr. Reddick continues to own all 175,647 of such common
shares.
(4) Computed by multiplying
the 425,000 shares acquired on exercise by the sum of the $4.26 closing price of
our stock on the Nasdaq Capital Market on November 23, 2009, the trading day
immediately preceding the date of Mr. Reddick’s option exercise less the $1.30
exercise price per share.
65
Securities
Authorized For Issuance under Equity Compensation Plans
The
following table includes information as of December 31, 2009 relating to our
1995, 1998 and 2008 Stock Option Plans and our 2005 Restricted Stock Unit Award
Plan, which comprise all of our equity compensation plans. The table
provides the number of securities to be issued upon the exercise of outstanding
options and distributions under outstanding Restricted Stock Unit Awards under
such plans, the weighted-average exercise price of outstanding options and the
number of securities remaining available for future issuance under such equity
compensation plans:
Equity
Compensation Plan Information
|
Plan Category
|
Number Of Securities
to Be Issued Upon
Exercise of
Outstanding Options,
Warrants and Rights
(Column a)
|
Weighted-Average
Exercise Price of
Outstanding Options,
Warrants and Rights
(Column b)
|
Number of Securities
Remaining Available for
Future Issuance Under
Equity Compensation Plans
(Excluding Securities
Reflected in Column a
(Column c)
|
|||||||||
|
Stock
Option Equity Compensation Plans Approved by Security
Holders
|
3,671,014 | $ | 5.90 | 3,716,000 | ||||||||
|
Stock
Option Equity Compensation Plans Not Approved by Security
Holders
|
— | — | — | |||||||||
|
Restricted
Stock Unit Equity Compensation Plans Approved by Security
Holders
|
3,316,000 | $ | 0.01 | 184,000 | ||||||||
|
Restricted
Stock Unit Equity Compensation Plans Not Approved by Security
Holders
|
— | — | — | |||||||||
|
TOTAL
|
6,987,014 | $ | 3.10 | 3,900,000 | ||||||||
Potential
Payments Upon Termination or Change in Control
Messrs. Brzeczko and
Seiser
Options. If
a change of control occurs (which constitutes a change of control under the
stock option agreements) previously unvested options vest and become exercisable
with respect to all underlying shares (relating to 52,000 and 64,000 shares for
Messrs. Brzeczko and Mr. Seiser, respectively, as of December 31,
2009). Messrs. Brzeczko and Seiser would realize a benefit of
$284,000 and $385,000 from such option vesting if such change of control had
occurred on December 31, 2009. Upon the occurrence of a change of
control that meets the requirements of Section 409A of the Internal Revenue Code
or upon termination of employment, stock options granted to Mr. Seiser to
purchase 24,900 shares of Common Stock become exercisable in full.
RSUs. If
a change of control occurs (which constitutes a change of control under the 2005
RSU Plan) then Messrs. Brzeczko’s and Seiser’s previously unvested RSUs vest
respect to all underlying shares. Messrs. Brzeczko and Seiser would
realize a benefit of $80,000 and $100,000 from such vesting if such change of
control had occurred on December 31, 2009. Upon the occurrence of a
change of control that meets the requirements of Section 409A of the Internal
Revenue Code, the RSUs are fully distributable for shares upon payment of the
$.01 par value per share, instead of under their normal distribution
schedule.
The
dollar benefits described above are the compensation cost for such awards that
would have been recognized in 2009 in our financial statements in accordance
with FASB ASC TOPIC 718, had such accelerated vesting/distribution
occurred.
66
Messrs. Reddick, Jones and
Clemens
Based
upon a hypothetical triggering date of December 31, 2009, the quantifiable
benefits for Messrs. Andrew Reddick, Robert Jones and Peter Clemens upon a
termination/change of control would have been as set forth the table
below:
|
Triggering
Event
|
Executive
|
Severance
|
Bonus
|
Value
of
Options
Vesting (4)
|
Value
of
RSUs
Vesting
(5)
|
Medical,
Dental,
Health,
Disability
and
Life
Insurance
Benefits
|
Total
(7)
|
|||||||||||||||||||
|
Termination
by Company without Cause or by Employee for Good Reason or after a Change
of Control
|
Andrew
D. Reddick
|
$ | 365,000 | (1)(8) | — | (3) | $ | 469,000 | $ | 356,000 | $ | 26,270 | (6) | $ | 1,216,270 | |||||||||||
|
Robert
B. Jones
|
290,000 | (1)(8) | — | (3) | 312,000 | 188,415 | 26,270 | (6) | $ | 816,685 | ||||||||||||||||
|
Peter
A. Clemens
|
410,000 | (2)(9) | — | (3) |
188,000
(with respect to Change of Control, only payable upon termination after
Change of Control)
|
126,000 | 52,540 | (10) | $ | 776,540 | ||||||||||||||||
|
Termination
for Death
|
Andrew
D. Reddick
|
— | — | (3) | — | 356,000 | $ | 50,000 | $ | 406,000 | ||||||||||||||||
|
Robert
B. Jones
|
— | — | — | 188,000 | $ | 50,000 | $ | 238,000 | ||||||||||||||||||
|
Peter
A. Clemens
|
— | — | — | 126,000 | $ | 50,000 | $ | 176,000 | ||||||||||||||||||
|
Termination
for Disability
|
Andrew
D. Reddick
|
— | — | (3) | — | 356,000 | — | $ | 356,000 | |||||||||||||||||
|
Robert
B. Jones
|
— | — | — | 188,000 | $ | 188,000 | ||||||||||||||||||||
|
Peter
A. Clemens
|
— | — | — | 126,000 | — | $ | 126,000 | |||||||||||||||||||
|
Termination
with Cause
|
Andrew
D. Reddick
|
— | — | — | — | — | — | |||||||||||||||||||
|
Robert
B. Jones
|
— | — | — | — | — | — | ||||||||||||||||||||
|
Peter
A. Clemens
|
— | — | — | — | — | — | ||||||||||||||||||||
|
Change
of Control Without Termination
|
Andrew
D. Reddick
|
— | — | 469,000 | 356,000 | — | $ | 825,000 | ||||||||||||||||||
|
Robert
B. Jones
|
— | — | 312,000 | 188,000 | — | $ | 500,000 | |||||||||||||||||||
|
Peter
A. Clemens
|
— | — | 188,000 | 126,000 | — | $ | 314,000 | |||||||||||||||||||
The terms
"Change of Control", "Cause", and "Good Reason" have the meanings in the listed
executive’s employment agreements.
(1) In the case of termination without
Cause, payable in 12 monthly installments. In the case of termination
for Good Reason, one half of amount is payable six months and one day after
termination, and remaining amount is payable thereafter in six monthly
installments. In the case of termination after a Change of Control,
amount is payable in a lump sum six months and one day after
termination.
(2) In the case of termination without
Cause, payable in a lump sum within 30 days after termination. In the
case of termination for Good Reason and termination after Change of Control,
amount is payable in a lump sum six months and one day after
termination.
(3) Payable in a lump sum within 30
days after termination. Because bonuses were paid prior to December
31, 2009, named executives would not have been entitled to any additional
bonuses upon termination at December 31, 2009.
(4) The dollar amount reported is the
compensation cost for such awards that would have been recognized in 2009 in our
financial statements in accordance with FASB ASC TOPIC 718 had the unvested
stock options at December 31, 2009 vested at such date. See
“Employment Agreements” for a description of the exercise periods following
termination.
67
(5) The
dollar amount reported is the compensation cost for such awards that would have
been recognized in 2009 in our financial statements in accordance with FASB ASC
TOPIC 718, had the unvested RSUs at December 31, 2009 vested at such
date
(6) Represents the value of medical,
dental, disability and life insurance for the twelve months following
termination and a tax gross up for such amounts. Payable in lump sum
within 30 days after termination. Assumes executive has selected lump
sum payment option, in lieu of continued benefits. This amount is
estimated.
(7) Excludes accrued
vacation.
(8) Represents one year of salary, at
the rate in effect on December 31, 2009.
(9) Represents two years of base
salary, at the rate in effect on December 31, 2009.
(10) Represents the
estimated value of medical, dental, disability and life insurance for the
twenty-four months following termination. Payable in lump sum
within thirty days after termination.
Director
Compensation
The
following table sets forth a summary of the compensation paid by us to our
Directors (other than Andrew Reddick, whose compensation, is reflected in the
Summary Compensation Table) for services rendered in all capacities to us during
the fiscal year ended December 31, 2009:
2009
DIRECTOR COMPENSATION
|
Director
|
Fees Earned or Paid in
Cash ($)
|
Stock Awards
($)(1)
|
Option Awards ($)(2)
|
Total ($)
|
||||||||||||
|
William
G. Skelly
|
27,500 | — | 107,283 | 134,783 | ||||||||||||
|
William
A. Sumner
|
26,500 | — | 107,283 | 133,783 | ||||||||||||
|
Bruce
F. Wesson
|
24,000 | — | 107,283 | 131,283 | ||||||||||||
|
Richard
J. Markham
|
28,500 | — | 107,283 | 110,133 | ||||||||||||
|
Immanuel
Thangaraj
|
20,000 | (3) | — | 107,283 | 127,283 | |||||||||||
|
George
K. Ross
|
32,000 | — | 107,283 | 139,283 | ||||||||||||
(1) Messrs. Skelly and Sumner each held
fully vested RSUs with respect to 100,000 underlying shares, as of December 31,
2009. Messrs. Wesson, Markham, Thangaraj and Ross held no
RSUs.
(2) Messrs. Skelly, Sumner,
Wesson, Markham, Thangaraj and Ross, held vested options with respect to,
47,500, 34,000, 30,000, 30,000, 30,000 and 30,000 underlying shares,
respectively, as of December 31, 2009. Each was awarded options to
purchase 15,000 shares of our Common Stock on January 2, 2009 at an exercise
price of $7.48. The dollar amounts represent the grant date fair
value of such options awarded in 2009 in accordance with FASB ASC Topic 718 and
exclude the possibility of forfeiture. In computing the grant date
fair value under a Black-Scholes model and used a risk free interest rate of
2.46%, historical volatility of 124.16% and an option term of 10
years. See Note I to our Financial Statements for a general discussion of our
assumptions.
(3) Committee and board
meeting attendance fees waived.
68
Our
Director compensation program provides for a $20,000 annual retainer for each
non-employee Director (and an additional annual retainer of $5,000 for the
chairperson of the Audit Committee and $2,500 for each other Committee
chairperson), a $1,000 fee for each Board meeting attended in person ($500 if
attended telephonically), and a $500 fee for each Committee meeting attended
($250 if attended telephonically). The annual retainer fees
are payable in four equal installments at the end of each calendar quarter
during the year. In
addition, non-employee Directors will receive an annual grant of options to
purchase 15,000 shares of our Common Stock. The stock options have a
term of 10 years and have an exercise price equal to the closing price of our
Common Stock on the first trading day of the year of grant as reported by the
NASDAQ Capital Market. The stock options vest in equal installments at the end
of each calendar quarter during the year of grant. Directors who are
also our employees receive no additional or special remuneration for their
services as Directors. We also reimburse Directors for
travel and lodging expenses, if any, incurred in connection with attendance at
Board meetings.
Compensation
Committee Interlocks and Insider Participation
During
2009 there were no Compensation Committee interlocks or insider participation in
compensation decisions.
Compensation
Committee Report
The
following report of the Compensation Committee is not deemed to be “soliciting
material” or to be “filed” with the Commission or subject to Regulation 14A or
14C [17 CFR 240.14a-1 et seq. or 240.14c-1
et seq.], other than as
specified, or to the liabilities of Section 18 of the Exchange Act [15 U.S.C.
78r].
The
Compensation Committee has reviewed and discussed the Compensation Discussion
and Analysis in this Report with Company management. Based on such
review and discussions, the Compensation Committee recommended to the Board of
Directors that the Compensation Discussion and Analysis be included in this
Report.
69
ITEM
12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED
STOCKHOLDER MATTERS
The
following table sets forth information regarding the beneficial ownership of the
Common Stock, as of February 1, 2010, for individuals or entities in the
following categories: (i) each of the Company's Directors and nominees for
Directors; (ii) the Company’s principal executive officer, the Company’s
principal financial officer and the next three highest paid executive officers
of the Company whose total annual compensation for 2009 exceeded $100,000 (the
"2009 named executive officers"); (iii) all Directors and executive officers as
a group; and (iv) each person known by the Company to be a beneficial owner of
more than 5% of the Common Stock. Unless indicated otherwise, each of the
shareholders has sole voting and investment power with respect to the shares
beneficially owned.
|
Name of Beneficial Owner
|
Amount
Owned
|
Percent of
Class (1)
|
||||||
|
GCE
Holdings LLC,
c/o Galen Partners III,
L.P.
680 Washington Boulevard,
Stamford, CT 06901
|
34,564,956 |
(2)
|
75.9 | % | ||||
|
Vivo
Ventures Fund VI, L.P.
575
High St, Suite 201
Palo
Alto, CA 9430131
|
2,450,000 |
(3)
|
5.5 | % | ||||
|
Andrew
D. Reddick
|
969,397 |
(4)
|
2.2 | % | ||||
|
Robert
B. Jones
|
250,000 |
(5)
|
* | |||||
|
William
G. Skelly
|
52,250 |
(6)
|
* | |||||
|
Bruce
F. Wesson
|
47,750 |
(2)(7)
|
* | |||||
|
William
A. Sumner
|
56,750 |
(8)
|
* | |||||
|
Peter
A. Clemens
|
211,747 |
(9)
|
* | |||||
|
Richard
J. Markham
|
33,750 |
(2)(10)
|
* | |||||
|
Immanuel
Thangaraj
|
43,750 |
(2)(11)
|
* | |||||
|
Robert
A. Seiser
|
151,733 |
(12)
|
* | |||||
|
Albert
W. Brzeczko
|
52,000 |
(13)
|
* | |||||
|
George
K. Ross
|
33,750 |
(14)
|
* | |||||
|
All
Officers and Directors as a Group (12 persons)
|
2,045,610 |
(15)
|
4.5 | % | ||||
*
Represents less than 1% of the outstanding shares of the Company's Common
Stock.
(1) Shows
percentage ownership assuming (i) such party converts all of its currently
convertible securities or securities convertible within 60 days of February 1,
2010 into the Company's common stock, and (ii) no other Company security holder
converts any of its convertible securities. No shares held by any
Director or 2009 named executive officer has been pledged as collateral
security.
(2) GCE
Holdings LLC, a Delaware limited liability company, was the assignee of all of
the our preferred stock (prior to its conversion into common stock) and bridge
loans entered into in 2005, 2006 and 2007 (prior to their conversion into common
stock and warrants) formerly held by each of Galen Partners III, L.P., Galen
Partners International III, L.P., Galen Employee Fund III, L.P. (collectively,
“Galen”), Care Capital Investments II, LP, Care Capital Offshore Investments II,
LP (collectively, “Care Capital”) and Essex Woodlands Health Ventures Fund V,
L.P. (“Essex”). Galen, Care Capital and Essex own approximately 39.8%, 30.6% and
29.6%, respectively, of the membership interests in GCE Holdings LLC. The
following natural persons exercise voting, investment and dispositive rights
over our securities held of record by GCE Holdings LLC: (i) Galen Partners III,
L.P., Galen Partners International III, L.P. and Galen Employee Fund III, L.P.:
Bruce F. Wesson, L. John Wilkerson, David W. Jahns, and Zubeen Shroff; (ii) Care
Capital Investments II, LP and Care Capital Offshore Investments II, LP: Jan
Leschly, Richard Markham, Argeris Karabelas and David Ramsay; and (iii) Essex
Woodlands Health Ventures Fund V, L.P.: Immanuel Thangaraj, James L. Currie and
Martin P. Sutter. Pursuant to a Voting Agreement among us, GCE Holdings LLC and
certain other shareholders, GCE Holdings LLC has the right to designate three of
the seven members of the Company’s Board of Directors. The Board
designees of GCE Holdings LLC are Immanuel Thangaraj, Richard Markham and Bruce
Wesson. Amounts for GCE Holdings, LLC include 1,786,481 shares
underlying warrants, exercisable at $3.40 per share. Excludes 381,381
shares and warrants to purchase 14,999 shares held by Galen, 136,178 shares and
warrants to purchase 34,500 shares held by Essex; and 10,923 shares, options to
purchase 40,000 shares and warrants to purchase 15,000 shares held by Care
Capital.
70
(3)
Includes shares held by an affiliated fund. Includes warrants to
purchase 450,000 shares exercisable at $3.40 per share held by Vivo Ventures
Fund VI, L.P. and an affiliated fund (collectively,
“Vivo”). Number of shares give effect to the transfer of
warrants to purchase 496,364 and 3,636 shares from Vivo Ventures Fund VI, L.P.
and Vivo Ventures VI Affiliates Fund, L.P., respectively, to Warrant Strategies
Fund, LLC on November 30, 2007 but are otherwise current as of November 20,
2007. The information with respect to Vivo is based solely on our
knowledge of our sale of securities to them and our knowledge of the warrant
transfer stated above.
(4)
Includes 793,750 shares subject to stock options exercisable within 60 days of
February 1, 2010, of which 450,000 shares are subject to fully vested options,
exercisable commencing January 1, 2011 or upon termination of employment or a
change of control of the Company. Excludes 910,000 restricted
stock unit awards (“RSUs”) granted to Mr. Reddick. Mr. Reddick has no
rights as a stockholder, including no dividend or voting rights, with respect to
the shares underlying the RSUs until the shares are issued by the Company
pursuant to the terms of Company’s 2005 Restricted Stock Unit Plan.
(5)
Includes 250,000 shares subject to stock options exercisable within 60 days of
February 1, 2010. Excludes 95,000 RSUs granted to Mr.
Jones Mr. Jones has no rights as a stockholder, including no
dividend or voting rights, with respect to the shares underlying the RSUs until
the shares are issued by the Company pursuant to the terms of Company’s 2005
Restricted Stock Unit Plan.
(6)
Includes 51,250 shares subject to stock options exercisable within 60 days of
February 1, 2010. Excludes 100,000 RSUs granted to Mr.
Skelly. Mr. Skelly has no rights as a stockholder, including no
dividend or voting rights, with respect to the shares underlying the RSUs until
the shares are issued by the Company pursuant to the terms of the Company’s 2005
Restricted Stock Unit Plan.
(7)
Includes 47,750 shares subject to stock options exercisable within 60 days of
February 1, 2010. Mr. Wesson’s holdings do not include securities
held by GCE or by Galen.
(8)
Includes 36,750 shares subject to stock options exercisable within 60 days of
February 1, 2010. Excludes 100,000 RSUs granted to Mr.
Sumner. Mr. Sumner has no rights as a stockholder, including no
dividend or voting rights, with respect to the shares underlying the RSUs until
the shares are issued by the Company pursuant to the terms of the Company’s 2005
Restricted Stock Unit Plan.
(9)
Includes 206,667 shares subject to stock options exercisable with 60 days of
February 1, 2010, of which 37,500 shares are subject to fully vested
options, exercisable commencing January 1, 2011 or upon termination of
employment or a change of control of the
Company. Excludes 470,000 RSUs granted to Mr. Clemens (of
which 453,750 will have vested within 60 days of February 1,
2010). Mr. Clemens has no rights as a stockholder, including no
dividend or voting rights, with respect to the shares underlying the RSUs until
the shares are issued by the Company pursuant to the terms of Company’s 2005
Restricted Stock Unit Plan. Includes 5,080 shares held by minor
children.
(10)
Includes 33,750 shares subject to stock options exercisable within 60 days of
February 1, 2010. Mr. Markham’s holdings do not include amounts held
by GCE or Care Capital of which Mr. Markham disclaims beneficial
ownership.
(11)
Includes 43,750 shares subject to stock options exercisable within 60 days of
February 1, 2010. Mr. Thangaraj’s holdings do not include securities
held by GCE or by Essex. Mr. Thangaraj disclaims beneficial ownership in
securities held by GCE and Essex except to the extent of his pecuniary interest
therein.
71
(12)
Includes 151,733 shares subject to stock options exercisable within 60 days of
February 1, 2010, of which 24,900 shares are subject to fully vested options
exercisable commencing January 1, 2011 or upon termination of employment or a
change of control of the Company. Excludes 189,000 RSUs granted to
Mr. Seiser. Mr. Seiser has no rights as a stockholder, including no
dividend or voting rights, with respect to the shares underlying the RSUs until
the shares are issued by the Company pursuant to the terms of Company’s 2005
Restricted Stock Unit Plan.
(13) Includes
52,000 shares subject to stock options exercisable within 60 days of February 1,
2010. Excludes 24,000 RSUs granted to Dr. Brzeczko (13,000 of which
will have vested within 60 days of February 1, 2010). Dr. Brzeczko
has no rights as a stockholder, including no dividend or voting rights, with
respect to the shares underlying the RSUs until the shares are issued by the
Company pursuant to the terms of Company’s 2005 Restricted Stock Unit
Plan.
(14)
Includes 33,750 shares subject to stock options exercisable within 60 days of
February 1, 2010.
(15)
Includes 2,045,610 shares which Directors and executive officers have the right
to acquire within 60 days of February 1, 2010 through exercise of outstanding
stock options, of which 512,300 shares subject to fully vested options, are
exercisable commencing January 1, 2011 or upon termination of employment or a
change of control of the Company. Includes securities (other than
RSUs) held by James Emigh, our Vice President, Marketing and Administration, in
addition to the officers and directors listed above.
ITEM
13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR
INDEPENDENCE
Certain
Relationships and Related Transactions
GCE
Holdings LLC, our 75.9% stockholder (“GCE”) was
the assignee of all shares of the Company’s preferred stock (prior
to conversion of such preferred stock into common stock) formerly
held by each of Galen Partners III, L.P., Galen Partners International III,
L.P., Galen Employee Fund III, L.P. (collectively, “Galen”), Care Capital
Investments II, LP, Care Capital Offshore Investments II, LP (collectively,
“Care Capital”) and Essex Woodlands Health Ventures V, L.P., (“Essex” and
together with Galen and Care, the “VC Investors”). Galen, Care and Essex own
39.8%, 30.6% and 29.6%, respectively, of the membership interest in GCE. Messrs.
Wesson, Markham and Thangaraj, each a Director, exercise investment control over
the membership interests in GCE held by Galen, Care and Essex, respectively, and
correspondingly exercise investment control over our common stock held by
GCE.
As a
condition to the completion of our 2004 debenture offering, we, the investors in
our 2004 debentures and the holders of our outstanding 5% convertible senior
secured debentures due March 31, 2006 issued by us during the period from 1998
through 2003 executed a certain Voting Agreement dated as of February 6, 2004
(the "Voting Agreement"). The Voting Agreement provided that each of Galen, Care
and Essex (collectively, the "Lead 2004 Debenture Investors") had the right to
designate for nomination one member of our Board of Directors, and that the Lead
Debenture 2004 Investors collectively may designate one additional member of the
Board (collectively, the "Designees"). In connection with the conversion of our
preferred shares into common stock completed in November 2005, the Voting
Agreement was amended to reflect the conveyance by each of Galen, Care and Essex
of their holdings in our preferred shares (prior to their conversion into common
stock) to GCE. After giving effect to a further amendment in January 2008, the
Voting Agreement, as amended, provides that our Board of Directors shall be
comprised of not more than seven (7) members, three (3) of whom shall be
designees of GCE, one of whom shall be our CEO and three of whom shall be
independent directors. The designees of GCE are Messrs. Wesson, Markham and
Thangaraj.
72
On August
20, 2007, we entered into a Securities Purchase Agreement with GCE Holdings LLC,
our controlling shareholder, and the other investors named therein
(collectively, the “Unit Investors”), pursuant to which the
Unit Investors purchased units consisting of four shares of common
stock and a warrant to purchase one share of common stock. In
accordance with the requirements of the Securities Purchase Agreement, we filed
a registration statement with the SEC for purposes of registering the resale of
the shares of common stock issued as part of the Units and the shares of common
stock issuable upon exercise of the warrants issued as part of the Units (the
“Registration Statement”). The Registration Statement was declared
effective by the SEC on November 20, 2007. We must exercise best
efforts to keep the Registration Statement effective until the earlier of (i)
the date that all shares of common stock and shares of common stock underlying
Warrants covered by the Registration Statement have been sold, or (ii) the fifth
anniversary of the Registration Statement, provided that the period during which
the Registration Statement must be kept effective can be shortened to not less
than two years by agreement of holders of registrable
securities. Shares of common stock eligible for sale under Rule
144(k) of the Securities Act of 1933, as amended, need not be included in the
Registration Statement. Under certain circumstances, if shares are
excluded from the Registration Statement by the SEC, we may be required to file
one or more additional Registration Statements for the excluded
shares. Subject to certain exceptions, for each day that we fail to
keep the Registration Statement effective, we must pay each Investor 0.05% of
the purchase price of securities covered by the Registration Statement and held
by such Unit Investor at such time, up to a maximum of 9.9% of the amount paid
by a Unit Investor for the Units.
The
requirement in the Securities Purchase Agreement to file the Registration
Statement triggered the piggyback registration rights granted to certain holders
of shares of our common stock and warrants exercisable for common stock pursuant
to an Amended and Restated Registration Rights Agreement dated as of February 6,
2004, as amended. GCE Holdings LLC, Galen Partners III, L.P., Galen
Partners International III, L.P., Galen Employee Fund III, L.P., Care Capital
Investments II, LP, Care Capital Offshore Investments II, LP and Essex Woodlands
Health Ventures V, L.P. exercised their piggyback registration rights under such
Agreement. As a result, an aggregate of 26,584,016 shares of common stock
and shares underlying warrants held by such shareholders (after giving effect to
our 1 for 10 reverse stock split effected December 5, 2007) were included in the
Registration Statement.
Our Board has not adopted formalized
written policies and procedures for the review or approval of related party
transactions. As a matter of practice, however, our Board has required that all
related party transactions, including, without limitation, each of the
transactions described above in this Item 13, be subject to review and approval
by a committee of independent directors established by the Board. The Board’s
practice is to evaluate whether a related party (including a director, officer,
employee, GCE Holdings, Galen, Care, Essex or other significant shareholder)
will have a direct or indirect interest in a transaction in which we may be a
party. Where the Board determined that such proposed transaction involves a
related party, the Board formally establishes a committee comprised solely of
independent directors to review and evaluate such proposed transaction (the
“Independent Committee”). The Independent Committee is authorized to review any
and all information it deems necessary and appropriate to evaluate the fairness
of the transaction to us and our shareholders (other than the interested related
party to such transaction), including meeting with management, retaining third
party experts (including counsel and financial advisors if determined necessary
and appropriate by the Independent Committee) and evaluating alternative
transactions, if any. The Independent Committee is also empowered to negotiate
the terms of such proposed related party transaction on our behalf. The proposed
related party transaction may proceed only following the approval and
recommendation of the Independent Committee. Following the Independent
Committee’s approval, the related party transaction is subject to final review
and approval of the Board as a whole, with any interested director abstaining
from such action.
Each of
the transactions described above in this Item 13 were subject to the review,
evaluation, negotiation and approval of an Independent Committee of the Board.
In each of such case, the Independent Committee was comprised of Messrs. Sumner
and Skelly.
Director
Independence
In
assessing the independence of our Board members, our Board has reviewed and
analyzed the standards for independence required under the NASDAQ Capital
Market, including NASDAQ Marketplace Rule 4200(a)(15), and applicable SEC
regulations. Based on this analysis, our Board has determined that
each of Messrs. William A. Sumner, William Skelly and George Ross meet the
standards for independence provided in the listing requirements of the NASDAQ
Capital Market and SEC regulations. As a result, three of our seven
Board members meet such standards of independence. Although the
listing standards of the NASDAQ Capital Market specify that a majority of a
listed issuer’s board of directors must be comprised of independent directors,
we are relying upon an exemption for “controlled companies” provided in the
listing standards for the NASDAQ Capital Market. A “controlled
company” is a company of which more than 50% of the voting power is held by an
individual, a group or another company. Based on GCE Holdings LLC’s
ownership of approximately 76% of our common stock, we are considered a
controlled company under the rules of the NASDAQ Capital Market and are relying
upon this exemption in having less than a majority of independent directors on
our Board.
73
With
respect to our Board committees, our Board has determined that the members of
our Compensation committee do not meet the standards for independence described
above.
ITEM
14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
Our
registered independent public accounting firm is BDO Seidman, LLP. The fees
billed by this firm in 2009 and 2008 were as follows:
|
2009
|
2008
|
|||||||
|
Audit
Fees
|
$ | 118,100 | $ | 131,764 | ||||
|
Audit-Related
Fees
|
— | — | ||||||
|
Total
Audit and Audit-Related Fees
|
118,100 | 131,764 | ||||||
|
Tax
Fees
|
62,255 | 180,677 | ||||||
|
All
Other Fees
|
— | — | ||||||
|
Total
for BDO Seidman, LLP
|
$ | 180,355 | $ | 312,441 | ||||
Audit
Fees include professional services rendered in connection with the annual audit
of our financial statements and with our audit of internal control over
financial reporting, and the review of the financial statements included in our
Form 10-Qs for the related periods. Additionally, Audit Fees include other
services that only an independent registered public accounting firm can reasonably provide, such as
services associated with our SEC registration statements or other documents
filed with the SEC or used in connection with financing activities. We had no
Audit-Related Fees which would include accounting consultations related to
accounting, financial reporting or disclosure matters not classified as "Audit
Fees."
Tax Fees
include tax compliance, tax advice and tax planning services. These services
related to the preparation of various state income tax returns, and our federal
income tax return, and reviews of IRC Section 382.
Audit
Committee's Pre-Approval Policies and Procedures
Consistent
with policies of the SEC regarding auditor independence and the Audit Committee
Charter, the Audit Committee has the responsibility for appointing, setting
compensation and overseeing the work of the registered independent public
accounting firm (the “Firm”). The Audit Committee's policy is to pre-approve all
audit and permissible non-audit services provided by the Firm. Pre-approval is
detailed as to the particular service or category of services and is generally
subject to a specific budget. The Audit Committee may also pre-approve
particular services on a case-by-case basis. In assessing requests for services
by the Firm, the Audit Committee considers whether such services are consistent
with the Firm’s independence, whether the Firm is likely to provide the most
effective and efficient service based upon their familiarity with the Company,
and whether the service could enhance the Company's ability to manage or control
risk or improve audit quality.
All of
the audit-related, tax and other services provided by BDO Seidman in 2009 and
2008 and related fees (as described in the captions above) were approved in
advance by the Audit Committee.
PART
IV
ITEM
15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
(a) The
following documents are filed as part of this report:
1. All
Financial Statements: See Index to Financial Statements
2. Financial
Statement Schedules: None
3. Exhibits: See
Index to Exhibits
74
SIGNATURES
Pursuant
to the requirements of Section 13 or 15(d) of the Securities Exchange Act of
1934, the registrant has duly caused this report to be signed on its behalf by
the undersigned, thereunto duly authorized.
|
Date: March
2, 2010
|
ACURA
PHARMACEUTICALS, INC.
|
|
|
By:
|
ANDREW D. REDDICK
|
|
|
Andrew
D. Reddick
President
and Chief Executive Officer
(Principal
Executive Officer)
|
||
Pursuant
to the requirements of the Securities Exchange Act of 1934, this report has been
signed below by the following persons on behalf of the registrant and in the
capacities and on the dates indicated.
|
Signature
|
Title(s)
|
Date
|
||
|
/s/Andrew D. Reddick
|
President,
Chief Executive Officer and Director
|
March
2, 2010
|
||
|
Andrew
D. Reddick
|
(Principal
Executive Officer)
|
|||
|
/s/Peter A. Clemens
|
Senior
Vice President and Chief Financial Officer
|
March 2, 2010 | ||
|
Peter
A. Clemens
|
(Principal
Financial and Accounting Officer)
|
|
||
|
/s/William G. Skelly
|
Director
|
March
2, 2010
|
||
|
William
G. Skelly
|
||||
|
/s/Bruce F Wesson
|
Director
|
March
2, 2010
|
||
|
Bruce
F. Wesson
|
||||
|
/s/William A. Sumner
|
Director
|
March
2, 2010
|
||
|
William
A. Sumner
|
||||
|
/s/Richard J. Markham
|
Director
|
March
2, 2010
|
||
|
Richard
J. Markham
|
||||
|
/s/Immanuel Thangaraj
|
Director
|
March
2, 2010
|
||
|
Immanuel
Thangaraj
|
||||
|
/s/George K. Ross
|
|
Director
|
|
March
2, 2010
|
|
George
K. Ross
|
75
|
Page
|
|
|
Report
of Independent Registered Public Accounting Firm
|
F-2
|
|
Consolidated
Balance Sheets
|
F-3
|
|
Consolidated
Statements of Operations
|
F-4
|
|
Consolidated
Statements of Stockholders' Equity (Deficit)
|
F-5
|
|
Consolidated
Statements of Cash Flows
|
F-6
|
|
Notes
to Consolidated Financial Statements
|
F-7
to F-19
|
F-1
REPORT
OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Board of
Directors and Shareholders
Acura
Pharmaceuticals, Inc.
Palatine,
Illinois
We have
audited the accompanying consolidated balance sheets of Acura Pharmaceuticals,
Inc. as of December 31, 2009 and 2008 and the related consolidated
statements of operations, stockholders’ equity (deficit), and cash flows for
each of the three years in the period ended December 31, 2009. These
financial statements are the responsibility of the Company’s
management. Our responsibility is to express an opinion on these
financial statements based on our audits.
We
conducted our audits in accordance with the standards of the Public Company
Accounting Oversight Board (United States). Those standards require
that we plan and perform the audit to obtain reasonable assurance about whether
the financial statements are free of material misstatement. An audit
includes examining, on a test basis, evidence supporting the amounts and
disclosures in the financial statements, assessing the accounting principles
used and the significant estimates made by management, as well as evaluating the
overall presentation of the financial statements. We believe that our audits
provide a reasonable basis for our opinion.
In our
opinion, the consolidated financial statements referred to above present fairly,
in all material respects, the financial position of Acura Pharmaceuticals,
Inc. at December 31, 2009 and 2008, and the results of its operations and
its cash flows for each of the three years in the period ended December 31,
2009, in conformity with accounting principles generally accepted in the United
States of America.
We also
have audited, in accordance with the standards of the Public Company Accounting
Oversight Board (United States), the effectiveness of Acura Pharmaceuticals,
Inc.’s internal control over financial reporting as of December 31, 2009, based
on criteria established in Internal Control – Integrated
Framework issued by the Committee of Sponsoring Organizations of the
Treadway Commission (COSO) and our report dated March 2, 2010 expressed an
unqualified opinion thereon.
/s/ BDO
Seidman, LLP
Chicago,
Illinois
March 2,
2010
F-2
ACURA
PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED
BALANCE SHEETS
DECEMBER
31, 2009 and 2008
(in
thousands except par values)
|
2009
|
2008
|
|||||||
|
Assets
|
||||||||
|
Current
assets
|
||||||||
|
Cash
and cash equivalents
|
$ | 30,174 | $ | 30,398 | ||||
|
Short-term
investments
|
- | 5,039 | ||||||
|
Collaboration
revenue receivable
|
357 | 3,529 | ||||||
|
Prepaid
insurance
|
193 | 283 | ||||||
|
Prepaid
expenses and other current assets
|
33 | 148 | ||||||
|
Deferred
income taxes
|
- | 2,491 | ||||||
|
Total
current assets
|
30,757 | 41,888 | ||||||
|
Property,
plant and equipment, net
|
1,160 | 1,073 | ||||||
|
Total
assets
|
$ | 31,917 | $ | 42,961 | ||||
|
Liabilities
and Stockholders’ Equity
|
||||||||
|
Current
liabilities
|
||||||||
|
Accounts
payable
|
$ | - | $ | 382 | ||||
|
Deferred
program fee revenue
|
1,555 | 4,632 | ||||||
|
Accrued
expenses
|
452 | 883 | ||||||
|
Total
current liabilities
|
2,007 | 5,897 | ||||||
|
Commitments
and contingencies (Note J)
|
||||||||
|
Stockholders’
equity
|
||||||||
|
Common
stock - $.01 par value; 100,000 shares authorized; 43,728 and 42,723
shares issued and outstanding in 2009 and 2008,
respectively
|
437 | 427 | ||||||
|
Additional
paid-in capital
|
352,694 | 344,023 | ||||||
|
Accumulated
deficit
|
(323,221 | ) | (307,386 | ) | ||||
|
Total
stockholders’ equity
|
29,910 | 37,064 | ||||||
|
Total
liabilities and stockholders’ equity
|
$ | 31,917 | $ | 42,961 | ||||
See
accompanying notes to the consolidated financial statements.
F-3
ACURA
PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED
STATEMENTS OF OPERATIONS
YEARS
ENDED DECEMBER 31, 2009, 2008 and 2007
(in
thousands except per share data)
|
2009
|
2008
|
2007
|
||||||||||
|
Revenues
|
||||||||||||
|
Program
fee revenue
|
$ | 3,077 | $ | 27,941 | $ | 3,427 | ||||||
|
Collaboration
revenue
|
758 | 11,496 | 2,977 | |||||||||
|
Milestone
revenue
|
- | 5,000 | - | |||||||||
|
Total
revenue
|
3,835 | 44,437 | 6,404 | |||||||||
|
Operating
expenses
|
||||||||||||
|
Research
and development expense
|
5,673 | 14,322 | 7,169 | |||||||||
|
Marketing,
general and administrative expense
|
11,662 | 9,133 | 4,141 | |||||||||
|
Total
operating expenses
|
17,335 | 23,455 | 11,310 | |||||||||
|
(Loss)
income from operations
|
(13,500 | ) | 20,982 | (4,906 | ) | |||||||
|
Other
income (expense)
|
||||||||||||
|
Interest
income
|
147 | 780 | 268 | |||||||||
|
Interest
expense
|
- | - | (1,207 | ) | ||||||||
|
Amortization
of debt discount
|
- | - | (2,700 | ) | ||||||||
|
Loss
on fair value change of conversion features
|
- | - | (3,483 | ) | ||||||||
|
Loss
on fair value change of common stock warrants
|
- | - | (1,905 | ) | ||||||||
|
Other
(expense) income
|
(3 | ) | (3 | ) | 19 | |||||||
|
Total
other income (expense)
|
144 | 777 | (9,008 | ) | ||||||||
|
(Loss)
income before income tax
|
(13,356 | ) | 21,759 | (13,914 | ) | |||||||
|
Income
tax expense (benefit)
|
2,479 | 7,285 | (9,600 | ) | ||||||||
|
Net
(loss) income
|
(15,835 | ) | 14,474 | (4,314 | ) | |||||||
|
Deemed
dividend from modification of debt
|
- | - | (3 | ) | ||||||||
|
Net
(loss) income applicable to common stockholders
|
$ | (15,835 | ) | $ | 14,474 | $ | (4,317 | ) | ||||
|
Earnings
(loss) per share
|
||||||||||||
|
Basic
|
$ | (0.35 | ) | $ | 0.32 | $ | (0.11 | ) | ||||
|
Diluted
|
$ | (0.35 | ) | $ | 0.29 | $ | (0.11 | ) | ||||
|
Weighted
average shares
|
||||||||||||
|
Basic
|
45,932 | 45,675 | 39,157 | |||||||||
|
Diluted
|
45,932 | 49,416 | 39,157 | |||||||||
See
accompanying notes to the consolidated financial statements.
F-4
ACURA
PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED
STATEMENTS OF STOCKHOLDERS' EQUITY (DEFICIT)
YEARS
ENDED DECEMBER 31, 2009, 2008 and 2007
(in
thousands except par values)
|
Common
Stock
$0.01
Par Value
|
Additional
Paid-in
|
Accumulated
|
||||||||||||||||||
|
Shares
|
$
Amount
|
Capital
|
Deficit
|
Total
|
||||||||||||||||
|
Balance
at Dec. 31, 2006
|
33,099 | $ | 331 | $ | 278,932 | $ | (317,543 | ) | $ | (38,280 | ) | |||||||||
|
Net
loss for the year ended Dec. 31, 2007
|
- | - | - | (4,314 | ) | (4,314 | ) | |||||||||||||
|
Deemed
dividend related to debt modification
|
- | - | - | (3 | ) | (3 | ) | |||||||||||||
|
Reclassification
of conversion feature value
|
- | - | 21,086 | - | 21,086 | |||||||||||||||
|
Reclassification
of common stock warrant value
|
- | - | 12,453 | - | 12,453 | |||||||||||||||
|
Conversion
feature value of issued debt
|
- | - | 1,789 | - | 1,789 | |||||||||||||||
|
Other
stock-based compensation
|
- | - | 915 | - | 915 | |||||||||||||||
|
Net
proceeds from unit offering
|
5,556 | 56 | 14,090 | - | 14,146 | |||||||||||||||
|
Conversion
of bridge loan notes, net
|
3,905 | 39 | 9,961 | - | 10,000 | |||||||||||||||
|
Issuance
of common shares for exercise of options
|
31 | - | 116 | - | 116 | |||||||||||||||
|
Issuance
of common shares for interest
|
84 | 1 | 811 | - | 812 | |||||||||||||||
|
Issuance
of common shares for cashless exercise of warrants
|
32 | - | - | - | - | |||||||||||||||
|
Reverse
stock split
|
(1) | - | - | - | - | |||||||||||||||
|
Balance
at Dec. 31, 2007
|
42,706 | $ | 427 | $ | 340,153 | $ | (321,860 | ) | $ | 18,720 | ||||||||||
|
Net
income for the year ended Dec. 31, 2008
|
- | - | 14,474 | 14,474 | ||||||||||||||||
|
Other
stock-based compensation
|
- | - | 3,850 | - | 3,850 | |||||||||||||||
|
Issuance
of common shares for exercise of warrant
|
17 | - | 20 | - | 20 | |||||||||||||||
|
Balance
at Dec. 31, 2008
|
42,723 | $ | 427 | $ | 344,023 | $ | (307,386 | ) | $ | 37,064 | ||||||||||
|
Net
loss for the year ended Dec. 31, 2009
|
- | - | - | (15,835 | ) | (15,835 | ) | |||||||||||||
|
Other
stock-based compensation
|
- | - | 9,204 | - | 9,204 | |||||||||||||||
|
Issuance
of common shares for cashless exercise of warrants
|
730 | 7 | (7 | ) | - | - | ||||||||||||||
|
Issuance
of common shares for exercise of warrant
|
50 | 1 | 159 | - | 160 | |||||||||||||||
|
Issuance
of common shares for cashless exercise of options and payroll
taxes
|
225 | 2 | (685 | ) | - | (683 | ) | |||||||||||||
|
Balance
at Dec. 31, 2009
|
43,728 | $ | 437 | $ | 352,694 | $ | (323,221 | ) | $ | 29,910 | ||||||||||
See
accompanying notes to the consolidated financial statements.
F-5
ACURA
PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED
STATEMENTS OF CASH FLOWS
YEARS
ENDED DECEMBER 31, 2009, 2008, and 2007
(in
thousands, except supplemental data)
|
2009
|
2008
|
2007
|
||||||||||
|
Cash
flows from operating activities:
|
||||||||||||
|
Net
(loss) income
|
$ | (15,835 | ) | $ | 14,474 | $ | (4,314 | ) | ||||
|
Adjustments
to reconcile net (loss) income to net cash (used in) provided by operating
activities:
|
||||||||||||
|
Depreciation
and amortization
|
130 | 143 | 130 | |||||||||
|
Amortization
of debt discount
|
- | - | 2,700 | |||||||||
|
Loss
on the fair value change of conversion features
|
- | - | 3,483 | |||||||||
|
Loss
on the fair value change of common stock warrants
|
- | - | 1,905 | |||||||||
|
Non-cash
stock compensation expense
|
9,204 | 3,850 | 915 | |||||||||
|
(Gain)
loss on asset disposals
|
(3 | ) | 1 | (22 | ) | |||||||
|
Common
stock issued for interest
|
- | - | 812 | |||||||||
|
Deferred
income taxes
|
2,479 | 7,109 | (9,600 | ) | ||||||||
|
Change
in fixed asset impairment reserve
|
- | (29 | ) | - | ||||||||
|
Changes
in assets and liabilities
|
||||||||||||
|
Collaboration
revenue receivable
|
3,172 | (553 | ) | (2,977 | ) | |||||||
|
Prepaid
expenses and other current assets
|
222 | 207 | (398 | ) | ||||||||
|
Other
assets and deposits
|
- | - | 7 | |||||||||
|
Accounts
payable
|
(382 | ) | 382 | - | ||||||||
|
Accrued
expenses
|
(1,113 | ) | 549 | 5 | ||||||||
|
Deferred
program fee revenue
|
(3,077 | ) | (21,942 | ) | 26,574 | |||||||
|
Net
cash (used in) provided by operating activities
|
(5,203 | ) | 4,191 | 19,220 | ||||||||
|
Cash
flows from investing activities:
|
||||||||||||
|
Purchases
of short-term investments
|
- | (26,039 | ) | - | ||||||||
|
Maturities
of short-term investments
|
5,039 | 21,000 | - | |||||||||
|
Capital
expenditures
|
(220 | ) | (143 | ) | (31 | ) | ||||||
|
Proceeds
from asset disposals
|
- | 1 | 22 | |||||||||
|
Net
cash provided by (used in) investing activities
|
4,819 | (5,181 | ) | (9 | ) | |||||||
|
Cash
flows from financing activities:
|
||||||||||||
|
Common
stock issued for warrant exercise
|
160 | - | - | |||||||||
|
Proceeds
from issuance of senior secured bridge term notes
|
- | - | 2,696 | |||||||||
|
Repayments
on secured term note
|
- | - | (5,000 | ) | ||||||||
|
Net
proceeds from the unit offering
|
- | - | 14,146 | |||||||||
|
Proceeds
from exercise of stock options
|
- | - | 119 | |||||||||
|
Proceeds
from exercise of warrant
|
- | 20 | - | |||||||||
|
Payments
on capital lease obligations
|
- | - | (32 | ) | ||||||||
|
Net
cash provided by financing activities
|
160 | 20 | 11,929 | |||||||||
|
Net
(decrease) increase in cash and cash equivalents
|
(224 | ) | (970 | ) | 31,140 | |||||||
|
Cash
and cash equivalents at beginning of period
|
30,398 | 31,368 | 228 | |||||||||
|
Cash
and cash equivalents at end of period
|
$ | 30,174 | $ | 30,398 | $ | 31,368 | ||||||
|
Cash
paid during the period:
|
||||||||||||
|
Interest
|
$ | - | $ | 2 | $ | 395 | ||||||
|
Income
taxes
|
$ | 108 | $ | 82 | $ | - | ||||||
See
accompanying notes to the consolidated financial statements.
F-6
ACURA
PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED
STATEMENTS OF CASH FLOWS (CONTINUED)
YEAR
ENDED DECEMBER 31, 2009, 2008, and 2007
Supplemental
disclosures of noncash investing and financing activities presented on a reverse
stock split basis:
Year ended December 31,
2009
|
1.
|
Warrants
to purchase 1,479,000 shares of common stock were exercised at exercise
prices of $2.70 and $3.40 per share in a cashless exercise transactions
resulting in the issuance of 730,000 shares of common
stock.
|
|
2.
|
Options
to purchase 525,000 shares of common stock were exercised at exercise
prices of $1.30 per share in cashless exercise transactions and after
withholding shares for statutory minimum payroll taxes calculated at
$685,000, the transactions resulted in the issuance of 225,000 shares of
common stock.
|
Year ended December 31,
2008
|
1.
|
Impaired
fixed assets with a $52,000 net book value were disposed and a $29,000
reduction in the impairment allowance was favorably
recognized.
|
|
2.
|
A
$1,177,000 valuation allowance against deferred income tax assets was
removed which resulted in an equal amount recorded as a benefit against
current income tax expense.
|
Year ended December 31,
2007
|
1.
|
The
Company issued 47,552 shares of common stock valued at $460,000 as payment
of the accrued interest due on Senior Secured Convertible Bridge Term
Notes Payable.
|
|
2.
|
The
Company issued 36,150 shares of common stock valued at $352,000 as payment
of accrued interest due on Secured Term Note
Payable.
|
|
3.
|
Warrants
to purchase an aggregate 58,000 shares of common stock were exercised at
exercise prices between $1.20 and $6.60 per share in a series of cashless
exercise transactions resulting in the issuance of aggregate 31,361 shares
of common stock.
|
|
4.
|
The
issuance of $896,000 Senior Secured Convertible Bridge Term Notes during
the period January 1, 2007 through March 29, 2007 included conversion
features measured at $849,000, which resulted in the recording of an equal
amount of debt discount and conversion feature
liabilities.
|
|
5.
|
The
change in all separated conversion feature’s fair value through March 30,
2007 resulted in a loss of $3,483,000. Due to a debt agreement
modification on March 30, 2007, the then current conversion feature fair
value of $21,086,000 was reclassified from liabilities to
equity.
|
|
6.
|
The
issuance of $1,800,000 of Senior Secured Bridge Term Notes included
conversion features measured at $1,552,000, which resulted in a recording
of an equal amount of debt discount to
equity.
|
|
7.
|
The
change in the common stock warrants’ fair value through the earlier of
their exercise date or March 30, 2007 resulted in a loss of 1,668,000. Due
to a debt agreement modification on March 30, 2007, the then current fair
value of all 1,592,100 outstanding common stock warrants of $12,307,000
was reclassified from liabilities to equity, as was $146,000 of such value
related to warrants exercised during the
period.
|
|
8.
|
Anti-dilution
provisions in certain warrant grants were triggered resulting in a loss of
$236,000 with an equal amount recorded against
equity.
|
|
9.
|
Senior
Secured Convertible Bridge Term Notes Payable of $10,544,000, less
unamortized debt discount of $544,000 was converted into 3,905,184 shares
of common stock.
|
See
accompanying notes to the consolidated financial statements.
F-7
ACURA
PHARMACEUTICALS, INC. AND SUBSIDIARY
NOTES
TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER
31, 2009, 2008 and 2007
NOTE
A - DESCRIPTION OF BUSINESS AND SUMMARY OF ACCOUNTING POLICIES
Acura
Pharmaceuticals, Inc., a New York corporation, and its subsidiary (the “Company”
or “We”) is a specialty pharmaceutical company engaged in research, development
and manufacture of product candidates intended to provide abuse deterrent
features and benefits utilizing our proprietary Aversion® and
Impede™ Technologies.
Amounts
presented are rounded to the nearest thousand, where indicated, except share and
per share data. The equity amounts and all share and per share data of the
Company have been adjusted to reflect a one-for-ten reverse stock split on
December 5, 2007.
Summary
of Significant Accounting Policies
A summary
of the significant accounting policies consistently applied in the preparation
of the accompanying consolidated financial statements follows.
1. Principles
of Consolidation
The
consolidated financial statements include the accounts of the Company and its
wholly-owned subsidiary, Acura Pharmaceutical Technologies, Inc. All
significant intercompany accounts and transactions are eliminated in
consolidation. We have evaluated subsequent events through the time of filing
this Form 10-K with the SEC on March 2, 2010.
2. Cash
and Cash Equivalents
The
Company considers all highly liquid securities with an original maturity of
three months or less to be cash equivalents. Our cash and cash equivalent
balances can consist of U.S. Treasury Bills or money market accounts and
checking funds with variable, market rates of interest. From time to time,
amounts may exceed the Federal Reserve insurance limits however we believe our
credit risk exposure is not material. We believe the financial risks associated
with these instruments are minimal and we have not experienced any losses
from our investments in these securities. Our cash and cash equivalents are
governed by our investment policy as approved by our Board of Directors. The
carrying amount of cash and cash equivalents approximates its fair value due to
its short-term nature.
3. Short-Term
Investments
The
Company’s entire portfolio of short-term investments at December 31, 2008
consisted of depository bank commercial paper and was accounted for as “held to
maturity securities”. The investments matured and were sold in
2009.
4. Concentration of Credit
Risk
The
Company invests its excess cash in accordance with the investment policy
approved by our Board of Directors that seeks both liquidity and safety of
principal. The policy provides for investments in instruments issued by the
United States government and by commercial institutions with strong investment
grade credit ratings and places restrictions on maturity terms and
concentrations by type and issuer.
5. Use
of Estimates in Consolidated Financial Statements
The
preparation of consolidated financial statements in conformity with accounting
principles generally accepted in the United States of America requires
management to make estimates and use assumptions that affect the reported
amounts of assets and liabilities and disclosure of contingent liabilities at
the date of the consolidated financial statements, as well as the reported
amounts of revenues and expenses during the reporting period. Actual results
could differ from those estimates. Management periodically evaluates estimates
used in the preparation of the consolidated financial statements for continued
reasonableness. Appropriate adjustments, if any, to the estimates used are made
prospectively based on such periodic evaluations.
F-8
6. Inventories
The
Company had no inventories at each of December 31, 2009 and
2008. Purchases of active pharmaceutical ingredients and raw
materials required for the Company’s development and clinical trial manufacture
of product candidates utilizing its Aversion® or Impede™ Technologies are
expensed as incurred. To purchase certain active pharmaceutical ingredients
required for our development and manufacture, we are required to file for and
obtain quotas annually from the U.S. Drug Enforcement Agency.
7. Property,
Plant and Equipment
Property,
plant and equipment are recorded at cost. Depreciation is recorded on a
straight-line basis over the estimated useful lives of the related assets.
Leasehold improvements are amortized on a straight-line basis over the shorter
of their useful lives or the terms of their respective leases. Betterments are
capitalized and maintenance and repairs are charged to operations as
incurred. The estimated lives of the major classification of
depreciable assets are:
|
Building
and improvements
|
10
- 40 years
|
|
Land
improvements
|
20
- 40 years
|
|
Machinery
and equipment
|
7
- 10 years
|
|
Scientific
equipment
|
5
- 10 years
|
|
Computer
hardware and software
|
3
- 10 years
|
|
Office
equipment
|
5
- 10 years
|
8.
Debt
Debt
Discount
For years
2007 and prior, debt discount resulting from the issuance of common stock
warrants in connection with subordinated debt and other notes payable as well as
from beneficial conversion features contained in convertible debt was recorded
as a reduction of the related obligations and was amortized over the remaining
life of the related obligations. Debt discount related to the common stock
warrants issued was determined by a calculation based on the relative fair
values ascribed to such warrants determined by management's use of the
Black-Scholes valuation model.
Debt
Conversion Features and Common Stock Warrants
For years
2007 and prior, certain provisions of the amended conversion features contained
in the Company’s Bridge Loan Agreements required the Company to separate the
value of the conversion feature from the debt and record such value as a
separate liability which was marked-to-market at each balance sheet date. The
Company used the Black-Scholes option-pricing model to compute the estimated
fair value of the conversion features. Marked-to-market adjustments resulted in
the recording of further gains and losses.
As a
result of the amendment to the Bridge Loan Agreements, all outstanding common
stock purchase warrants were accounted for at fair value using the Black -
Scholes option-pricing model and recorded as a liability with a corresponding
reduction in additional paid-in capital. This warrant liability was
marked-to-market each balance sheet date which resulted in the recording of
further gains and losses. This practice ceased in 2007, see Note F.
9.
Revenue
Recognition, Deferred Program Fee Revenue and Collaboration
Revenue
We
recognize revenue when there is persuasive evidence that an arrangement exists,
delivery has occurred, the price is fixed and determinable, and collection is
reasonably assured. In connection with our License, Development, and
Commercialization Agreement dated October 30, 2007 (the “King Agreement”) with
King Pharmaceuticals Research and Development, Inc. (“King”), we recognize
program fee revenue, collaboration revenue and milestone revenue.
Program
fee revenue is derived from amortized upfront payments, such as the $30.0
million upfront payment from King received in December 2007, and license fees,
such as the $3.0 million option exercise fee paid by King to us in each of May
and December 2008 upon the exercise of its option to license a third and fourth
opioid analgesic product candidate under the King Agreement. We have assigned an
equal portion of King’s $30.0 million upfront payment to each of three product
candidates identified in the King Agreement and recognize the upfront payment as
program fee revenue ratably over our estimate of the development period for each
identified product candidate. We expect to recognize the remainder of the
program fee revenue for the third product candidate ratably over its remaining
development period which we currently estimate to end in December 2010. We
recognized $3.1 million, $21.9 million, and $3.4 million of this program fee
revenue in 2009, 2008 and 2007, respectively.
F-9
Collaboration
revenue is derived from reimbursement of development expenses, which are
invoiced quarterly in arrears, and are recognized when costs are incurred
pursuant to the King Agreement. The ongoing research and development
services being provided to King under the collaboration are priced at fair value
based upon the reimbursement of expenses incurred pursuant to the collaboration
with King. We recognized $0.8 million, $11.5 million and $3.0 million of
collaboration revenue in 2009, 2008 and 2007, respectively of which $0.4 million
and $3.5 million were current receivables at December 31, 2009 and 2008,
respectively.
Milestone
revenue is contingent upon the achievement of certain pre-defined events in the
development of Acurox® Tablets
and other product candidates licensed to King under the King Agreement.
Milestone payments from King are recognized as revenue upon achievement of the
“at risk” milestone events, which represent the culmination of the earnings
process related to that milestone. Milestone payments are triggered either by
the results of our research and development efforts or by events external to us,
such as regulatory approval to market a product. As such, the milestones are
substantially at risk at the inception of the King Agreement, and the amounts of
the payments assigned thereto are commensurate with the milestone achieved. In
addition, upon the achievement of a milestone event, we have no future
performance obligations related to that milestone payment. Each milestone
payment is non-refundable and non-creditable when made. In June 2008, we
recognized milestone revenue when King paid us a $5.0 million payment for
successfully achieving the primary endpoints in our pivotal Phase III
study, AP-ADF-105 for Acurox®
Tablets.
10. Research
and Development
Research
and Development (“R&D”) expenses include internal R&D activities,
external contract research organization (“CRO”) activities, and other
activities. Internal R&D activity expenses include facility overhead,
equipment and facility maintenance and repairs, depreciation, laboratory
supplies, pre-clinical laboratory experiments, depreciation, salaries, benefits,
and incentive compensation expenses. CRO activity expenses include preclinical
laboratory experiments and clinical trial studies. Other activity expenses
include clinical trial studies and regulatory consulting, and regulatory
counsel. Internal R&D activities and other activity expenses are charged to
operations as incurred. The Company makes payments to the CRO's based on
agreed upon terms and may include payments in advance of the study starting
date. The Company reviews and accrues CRO expenses and clinical trial study
expenses based on work performed and relies upon estimates of those costs
applicable to the stage of completion of a study as provided by the CRO.
Accrued CRO costs are subject to revisions as such trials progress to
completion. Revisions are charged to expense in the period in which the facts
that give rise to the revision become known. Advance payments are amortized to
expense based on work performed. The Company has entered into several CRO
clinical trial agreements pursuant to which the unfunded CRO commitments were
$1.4 million at December 31, 2009 and are expected to be incurred as subjects
are enrolled into the clinical studies.
11. Income
Taxes
The
Company accounts for income taxes under the liability method. Under this method,
deferred income tax assets and liabilities are determined based on differences
between financial reporting and income tax basis of assets and liabilities and
are measured using the enacted income tax rates and laws that will be in effect
when the differences are expected to reverse. Additionally, net operating loss
and tax credit carryforwards are reported as deferred income tax
assets. The realization of deferred income tax assets is dependent
upon future earnings. A valuation allowance is required against deferred income
tax assets if, based on the weight of available evidence, it is more likely than
not that some or all of the deferred income tax assets may not be
realized. During 2009 the Company determined it was more likely than
not that it would not be able to realize its recorded deferred income tax assets
and recorded an adjustment of $2.5 million to the deferred income tax asset
valuation allowance and recognized an expense from income taxes in such
period. During 2008 and 2007, the Company determined it was more
likely than not that it would be able to realize some of its deferred income tax
assets in the near future, and recorded adjustments of $1.2 million and $8.6
million to the deferred income tax asset valuation allowance, respectively.
These adjustments recognized a benefit from income taxes in our income for such
periods. At both December 31, 2009 and 2008, 100% of all remaining net deferred
income tax assets were offset by a valuation allowance due to uncertainties with
respect to future utilization of net operating loss carryforwards. If in the
future it is determined that additional amounts of our deferred income tax
assets would likely be realized, the valuation allowance would be reduced in the
period in which such determination is made and an additional benefit from income
taxes in such period would be recognized.
F-10
12. Earnings
Per Share
The
computation of basic earnings (loss) per share of common stock is based upon the
weighted average number of common shares outstanding during the period,
including shares related to vested restricted stock units (See Note I). The
computation of diluted earnings (loss) per share is based on the same number of
shares used in the basic share calculation adjusted for the effect of other
potentially dilutive securities. No such adjustments were made for 2009 or 2007
as their effects would be antidilutive.
|
Year
ended December 31,
|
||||||||||||
|
(in
thousands except per share data)
|
2009
|
2008
|
2007
|
|||||||||
|
Basic
earnings per share
|
||||||||||||
|
Numerator:
|
||||||||||||
|
Net
(loss) income
|
$ | (15,835 | ) | $ | 14,474 | $ | (4,314 | ) | ||||
|
Deemed
dividend from modification of debt
|
- | - | (3 | ) | ||||||||
|
Net
(loss) income applicable to common stockholders
|
$ | (15,835 | ) | $ | 14,474 | $ | (4,317 | ) | ||||
|
Denominator:
|
||||||||||||
|
Common
shares (weighted)
|
42,912 | 42,719 | 36,656 | |||||||||
|
Vested
restricted stock units (weighted)
|
3,020 | 2,956 | 2,501 | |||||||||
|
Weighted
average number of shares outstanding
|
45,932 | 45,675 | 39,157 | |||||||||
|
Basic
earnings (loss) per common share
|
$ | (0.35 | ) | $ | 0.32 | $ | (0.11 | ) | ||||
|
Diluted
earnings (loss) per share
|
||||||||||||
|
Denominator:
|
||||||||||||
|
Common
shares (weighted)
|
42,912 | 42,719 | 36,656 | |||||||||
|
Vested
restricted stock units (weighted)
|
3,020 | 2,952 | 2,501 | |||||||||
|
Stock
options
|
- | 1,443 | - | |||||||||
|
Common
stock warrants
|
- | 2,302 | - | |||||||||
|
Weighted
average number of shares outstanding
|
45,932 | 49,416 | 39,157 | |||||||||
|
Diluted
earnings (loss) per common share
|
$ | (0.35 | ) | $ | 0.29 | $ | (0.11 | ) | ||||
|
Excluded
potentially dilutive securities:
|
||||||||||||
|
Common
stock issuable (1):
|
||||||||||||
|
Employee
and director stock options
|
3,671 | 1,149 | 1,858 | |||||||||
|
Common
stock warrants
|
2,380 | - | 3,972 | |||||||||
|
Non-vested
restricted stock units
|
204 | 30 | - | |||||||||
|
Total
excluded dilutive shares
|
6,255 | 1,179 | 4,820 | |||||||||
(1)
Number of shares issuable represents those securities which were either i)
nonvested at year end or ii) were vested but antidilutive. The number of shares
is based on maximum number of shares issuable on exercise or conversion of the
related securities as of year end. Such amounts have not been adjusted for the
treasury stock method or weighted average outstanding calculations as required
if the securities were dilutive.
13.
Stock-Based
Compensation
The
Company has four stock-based compensation plans covering stock options and
restricted stock units for its employees and directors, which are described more
fully in Note I.
The
Company measures its compensation cost related to share-based payment
transactions based on fair value of the equity or liability instrument issued.
For purposes of estimating the fair value of each stock option unit on the date
of grant, the Company utilizes the Black-Scholes option-pricing model. The
Black-Scholes option valuation model was developed for use in estimating the
fair value of traded options, which have no vesting restrictions and are fully
transferable. In addition, option valuation models require the input of highly
subjective assumptions including the expected volatility factor of the market
price of the Company’s common stock (as determined by reviewing its historical
public market closing prices). Because the Company’s employee stock options have
characteristics significantly different from those of trade options and because
changes in the subjective input assumptions can materially affect the fair value
estimate, in management’s opinion, the existing models do not necessarily
provide a reliable measure of the fair value of its employee stock
options.
F-11
The
Company’s accounting for stock-based compensation for restricted stock units
(“RSUs”) has been based on the fair-value method. The fair value of the RSUs is
the market price of the Company’s common stock on the date of grant, less its
exercise cost.
14.
Accounting
Developments
In
October 2009, the FASB issued an amendment to its previously released guidance
on revenue arrangements with multiple deliverables. This guidance becomes
effective for the Company at the beginning of its 2011 fiscal year. The
pronouncement addresses how to determine whether an arrangement involving
multiple deliverables contains more than one unit of accounting and how the
arrangement consideration should be allocated among the separate units of
accounting. The pronouncement may be applied retrospectively or prospectively
for new or materially modified arrangements and early adoption is permitted. The
Company is currently assessing the impact of adopting this
guidance.
In June
2009, the FASB Accounting Standards Codification (“Codification”) was
issued. The Codification will become the source of authoritative
accounting principles recognized by the FASB to be applied by nongovernmental
entities in the preparation of financial statements in conformity with generally
accepted accounting principles. The Codification explicitly recognizes rules and
interpretive releases of the Securities and Exchange Commission (“SEC”) under
federal securities laws as authoritative generally accepted accounting
principles for SEC registrants. The Company adopted this statement on September
30, 2009 and its adoption did not have an effect on our consolidated financial
statements.
In May
2009, the FASB established the general standards of accounting for and
disclosure of subsequent events. In addition, this statement requires disclosure
of the date through which an entity has evaluated subsequent events and the
basis for that date. The Company adopted this standard on July 1, 2009 and has
provided the new disclosures as required.
In March
2008, the Financial Accounting Standards Board (“FASB”) issued a statement for
disclosures about derivative instruments and hedging activities intended to
improve financial reporting about derivative instruments and hedging activities
by requiring enhanced disclosures to enable investors to better understand their
effects on an entity’s financial position, financial performance, and cash
flows. The Company adopted this statement on January 1, 2009 with no
effect on the Company’s consolidated financial statements as we had no
derivative or hedging activities.
In
December 2007, the FASB issued guidance on collaborative arrangements, which is
effective for calendar-year companies beginning January 1, 2009. The
pronouncement clarified the manner in which costs, revenues and sharing payments
made to, or received by, a partner in a collaborative arrangement should be
presented in the income statement and set forth certain disclosures that should
be required in the partners’ financial statements. The implementation of this
standard did not have an impact on the Company’s consolidated financial
statements.
In
December 2007, the FASB issued a statement to establish accounting and reporting
standards for the noncontrolling interest in a subsidiary and for the
deconsolidation of a subsidiary. The Company adopted this statement on January
1, 2009 with no effect on the Company’s consolidated financial statements as we
did not have any noncontrolling interests in a subsidiary.
In
December 2007, the FASB issued a statement on business combinations. The
statement defines the acquirer as the entity that obtains control of one or more
businesses in the business combination, establishes the acquisition date as the
date the acquirer achieves control and requires the acquirer to recognize the
assets and liabilities assumed and any non-controlling interest at their fair
values as of the acquisition date. The statement requires, among other things,
that the acquisition related costs be recognized separately from the
acquisition. The Company adopted this guidance on January 1, 2009 for business
combinations for which the acquisition date is on or after January 1,
2009.
F-12
In June
2007, FASB issued guidance on "Accounting for Nonrefundable Advance Payments for
Goods or Services Received for Use in Future Research and Development
Activities" which was effective for fiscal years beginning after December 15,
2007. Nonrefundable advance payments for goods or services that will be
used or rendered for future research and development activities should be
deferred and capitalized. Such amounts should be recognized as an expense when
the related goods are delivered or services are performed, or when goods or
services are no longer expected to be provided. The adoption of this guidance
did not have an impact on the Company's consolidated
financial statements.
NOTE
B – LICENSE, DEVELOPMENT, AND COMMERCIALIZATION AGREEMENT
On
October 30, 2007, we and King Pharmaceuticals Research and Development, Inc.
(“King”), a wholly-owned subsidiary of King Pharmaceuticals, Inc., entered into
a License, Development and Commercialization Agreement (the “King Agreement”) to
develop and commercialize in the United States, Canada and Mexico (the "King
Territory") certain opioid analgesic products utilizing our proprietary
Aversion® Technology. In addition, the King Agreement provides King
with an option to license in the King Territory all future opioid analgesic
products developed utilizing Aversion®
Technology. At December 31, 2009, King had exercised its option to license two
additional product candidates including an undisclosed opioid analgesic tablet
product and Vycavert®
(hydrocodone bitartrate/niacin/APAP) Tablets, each of which utilize our
Aversion®
Technology. We are responsible for using commercially reasonable
efforts to develop Acurox® Tablets through regulatory approval by the
FDA. The King Agreement provides that we or King may develop
additional opioid analgesic product candidates utilizing our Aversion®
Technology and, if King exercises its option to license such additional product
candidates, they will be subject to the milestone and royalty payments and other
terms of the King Agreement.
At
December 31, 2009, we had received aggregate payments of $56.2 million from
King, consisting of a $30.0 million non-refundable upfront cash payment, $15.2
million in reimbursed research and development expenses relating to Acurox®
Tablets, $6.0 million in fees relating to King’s exercise of its option to
license an undisclosed opioid analgesic tablet product and Vycavert® Tablets,
and a $5.0 million milestone fee relating to our successful achievement of the
primary endpoints for our pivotal Phase III clinical study for Acurox®
Tablets. The King Agreement also provides for King’s payment to us of
a $3.0 million fee upon King’s exercise of its option for each future opioid
product candidate. In the event that King does not exercise its
option for a future opioid product candidate, King may be required to reimburse
us for certain of our expenses relating to such future opioid product
candidate. Further, we may receive up to $23 million in additional
non-refundable milestone payments for each product candidate licensed to King,
including Acurox® Tablets,
which achieve certain regulatory milestones in specific countries in the King
Territory. We can also receive a one-time $50 million sales milestone
payment upon the first attainment of $750 million in net sales of all of our
licensed products across all King Territories. In addition, for sales
occurring following the one year anniversary of the first commercial sale of the
first licensed product sold, King will pay us a royalty at one of 6 rates
ranging from 5% to 25% based on the level of combined annual net sales for all
products licensed by us to King across all King Territories, with the highest
applicable royalty rate applied to such combined annual sales. King’s
royalty payment obligations expire on a product by product and
country-by-country basis upon the later of (i) the expiration of the last valid
patent claim covering such product in such country, or (ii) fifteen (15) years
from the first commercial sale of such product in such country.
The King
Agreement expires upon the expiration of King’s royalty payment and other
payment obligations under the King Agreement. King may terminate the
King Agreement in its entirety or with respect to any product at any time after
March 31, 2010, upon the provision of not less than 12 months’ prior written
notice, and in its entirety if regulatory approval of the NDA for Acurox® Tablets
is not received prior to March 31, 2010 and with respect to a particular product
with respect to a country in which regulatory approval for such product is
withdrawn by a regulatory authority in such country. We do not expect to
receive FDA approval of the NDA for Acurox® Tablets
prior to March 31, 2010. As a result, King may terminate the King
Agreement at any time following such date upon written notice to
us. We may terminate the King Agreement with respect to a product in
the United States in the event such product is not commercially launched by King
within 120 days after receipt of regulatory approval of such product or in its
entirety if King commences any interference or opposition proceeding challenging
the validity or enforceability any of our patent rights licensed to King under
the King Agreement.
F-13
NOTE
C – PREFERRED SHARES
As of the
date of this Report, the Company has no issued or authorized preferred
shares. Prior to June 2008, the Company was authorized to issue
various series of convertible preferred stock. In November 2005 all of the
Company’s issued and outstanding preferred shares were automatically and
mandatorily converted into the Company’s common stock in accordance with the
terms of the Company’s Restated Certification of Incorporation and in June 2009,
the Company amended its Restated Certificate of Incorporation to eliminate all
authorized preferred stock.
NOTE
D – PROPERTY, PLANT AND EQUIPMENT
Property,
plant and equipment are summarized as follows:
|
December
31,
|
||||||||
|
(in
thousands)
|
2009
|
2008
|
||||||
|
Building
and improvements
|
$ | 1,465 | $ | 1,385 | ||||
|
Land
and improvements
|
161 | 161 | ||||||
|
Machinery
and equipment
|
26 | 23 | ||||||
|
Scientific
equipment
|
569 | 476 | ||||||
|
Computer
hardware and software
|
251 | 225 | ||||||
|
Office
equipment
|
27 | 52 | ||||||
|
Other
personal property
|
60 | 60 | ||||||
| 2,559 | 2,382 | |||||||
|
Less
accumulated depreciation and amortization
|
(1,399) | (1,309) | ||||||
|
Total
property, plant and equipment, net
|
$ | 1,160 | $ | 1,073 | ||||
Depreciation
and amortization expense for the years ended December 31, 2009, 2008 and 2007
was $130,000, $143,000, and $130,000, respectively.
NOTE
E – ACCRUED EXPENSES
Accrued
expenses are summarized as follows:
|
December
31,
|
||||||||
|
(in
thousands)
|
2009
|
2008
|
||||||
|
Payroll,
payroll taxes and benefits
|
$ | 89 | $ | 77 | ||||
|
Professional
services
|
160 | 168 | ||||||
|
Franchise
taxes
|
21 | 144 | ||||||
|
Property
taxes
|
19 | 39 | ||||||
|
State
income taxes
|
- | 94 | ||||||
|
Clinical
and regulatory services
|
75 | 173 | ||||||
|
Other
fees and services
|
88 | 188 | ||||||
| $ | 452 | $ | 883 | |||||
NOTE
F – NOTES PAYABLE
Convertible
Bridge Term Notes
A
November 2006 amendment of the conversion feature on all of the then outstanding
Bridge Loans, coupled with the requirement under current accounting guidance to
separate the value of the conversion feature from the debt, required the Company
to record the value of the amended conversion feature on that outstanding debt
as a liability. Upon revaluing the aggregate conversion features on all
outstanding Bridge Loans as of March 30, 2007 (the date immediately before
further amendment to the Bridge Loans), the Company recorded the resulting
increase in value as a $3.48 million loss. The increase in the Company’s common
stock trading price from December 31, 2006 to March 30, 2007 resulted in the
increase in the value of the conversion liability. The Bridge Loan amendment on
March 30, 2007 limited the conversion price of the post-October 2006 loans to no
lower than $2.10 per share. With this limit in place, the outstanding
conversion feature no longer had to be reflected as Company liabilities. As
such, the Company recorded a $21.1 million reclassification of that liability to
additional paid-in capital.
To
compute the estimated value of the conversion features just prior to the
reclassification described above, the Company used the Black-Scholes
option-pricing model with the following assumptions on these dates:
F-14
|
Mar 30,
2007
|
||||
|
Company
stock price
|
$ | 8.50 | ||
|
Exercise
price
|
(1 | ) | ||
|
Expected
dividend
|
0.0 | % | ||
|
Risk
–free interest rate
|
5.07 | % | ||
|
Expected
volatility
|
none
|
|||
|
Contracted
term
|
1
day
|
|||
(1) The
conversion price per share used to estimate fair value of the Bridge Loan
conversion rights was equal to the fixed conversion price per share set forth
above for each of the specified Bridge Loan amounts. While the Bridge Loan
Agreements provide for other than fixed conversion prices under certain
circumstances, the Company has judged that the fixed conversion prices will most
likely be the lowest price per share under any of the circumstances and the
lender would therefore select such fixed price for their
conversion.
The
conversion features related to additional $1.8 million Bridge Note issuances in
2007 were not required to be separated and accounted for at fair value. However,
based on the conversion price of those notes, the issuances did include
beneficial conversion features whereby the common stock to be issued upon
conversion would be worth more than the underlying debt if converted upon
issuance. That incremental value, computed as $1.5 million was
recorded as additional paid-in capital and as debt discount, which would be
amortized over the term of the notes.
NOTE
G – COMMON STOCK WARRANTS
As a
result of a November 2006 amendment to the Company’s then outstanding Bridge
Loan Agreements, the Company’s outstanding common stock purchase warrants
commenced being accounted for as marked-to-market liability. Upon revaluing
these warrants just before their exercise or as of March 30, 2007 (the date
immediately before further amendment to the Bridge Loan Agreements), the Company
recorded the resulting increase in value as a $1.7 million loss. The increase in
the Company’s common stock trading price from December 31, 2006 to March 30,
2007 resulted in the increase in the value of the recorded warrant liability.
The Bridge Loan agreement amendment on March 30, 2007 limited the conversion
price of the Bridge Loans, and with this limit in place, the outstanding common
stock purchase warrants were no longer required to be accounted for as a
liability. As such, the Company recorded a combined $12.5 million
reclassification of that liability to additional paid-in capital during the
first quarter of 2007.
The
Company has outstanding common stock purchase warrants at December 31, 2009
exercisable for an aggregate 2,380,000 shares of common stock, all of which
contained cashless exercise features. Warrants for 64,000 and 2,316,000 shares
at an exercise price of $1.29 and $3.40 per share, respectively, will expire in
May 2010 and August 2014, respectively, if unexercised. These warrants have a
weighted average remaining term of 4.5 years and a weighted average exercise
price of $3.34.
NOTE
H – INCOME TAXES
Provision
for Income Taxes
The reconciliation between the
statutory federal income tax rate and the Company's effective income tax rate is
as follows:
|
December
31,
|
||||||||||||
|
(in
thousands)
|
2009
|
2008
|
2007
|
|||||||||
|
Tax
(benefit) at U.S. 34% statutory rate
|
$ | (4,541 | ) | $ | 7,398 | $ | (4,731 | ) | ||||
|
Current
state tax (benefit), net of Federal effect
|
(416 | ) | 1,518 | (413 | ) | |||||||
|
Research
tax credits
|
(100 | ) | (129 | ) | (220 | ) | ||||||
|
Wage
reported stock compensation
|
(587 | ) | - | - | ||||||||
|
Fair
value change of conversion feature fair value
|
- | - | 1,184 | |||||||||
|
Fair
value change of warrant
|
- | - | 648 | |||||||||
|
Debt
discount amortization
|
- | - | 918 | |||||||||
|
Financing
costs
|
- | - | 311 | |||||||||
|
Other
|
(330 | ) | (325 | ) | (64 | ) | ||||||
| (5,974 | ) | 8,462 | (2,367 | ) | ||||||||
|
Increase
(decrease) in valuation allowance
|
8,453 | (1,177 | ) | (7,233 | ) | |||||||
|
Provision
(benefit) for income taxes
|
$ | 2,479 | $ | 7,285 | $ | (9,600 | ) | |||||
F-15
Tax
expense for 2009 is deferred taxes, 2008 is comprised of $0.2 million of current
state taxes and $7.1 million of deferred taxes, and for 2007 the entire $9.6
million tax benefit is deferred taxes.
Deferred Tax Assets and Valuation
Allowance
Deferred
tax assets reflect the tax effects of net operating losses (“NOLs”), tax credit
carryovers, and temporary differences between the carrying amounts of assets and
liabilities for financial reporting purposes and income tax purposes. The most
significant item of our deferred tax assets is derived from our Federal NOLs. We
have approximately $23.1 million federal income tax benefit at December 31, 2009
derived from $68 million Federal NOLs at the 34% federal income tax rate,
available to offset future taxable income, some of which have limitations for
use as prescribed under internal revenue code IRC Section 382. Our
NOLs will expire in varying amounts between 2010 and 2029 if not
used. Income tax benefits of $3.0 million and $1.4 million from NOLs
under valuation allowance expired in 2009 and 2007, respectively. The components
of our deferred tax assets are as follows:
|
December
31,
|
||||||||
|
(in
thousands)
|
2009
|
2008
|
||||||
|
Deferred
tax assets:
|
||||||||
|
Estimated
future value of NOLs
|
||||||||
|
-
Federal
|
$ | 23,114 | $ | 22,920 | ||||
|
-
State
|
3,862 | 2,365 | ||||||
|
Research
tax credits
|
194 | 754 | ||||||
|
Deferred
program fee revenue
|
611 | 1,819 | ||||||
|
Share-based
compensation
|
10,505 | 7,364 | ||||||
|
Other,
net
|
41 | 98 | ||||||
|
Total
deferred taxes
|
38,327 | 35,320 | ||||||
|
Valuation
allowance
|
(38,327) | (32,829) | ||||||
|
Net
deferred tax assets
|
$ | - | $ | 2,491 | ||||
Realization
of deferred tax assets is dependent upon future earnings, if any, the timing and
amount of which may be uncertain. Valuation allowances are placed on
deferred tax assets when uncertainty exist on their near term utilization.
Periodic reviews are made by us on the valuation allowances and fluctuations can
occur. Those fluctuations are reflected as income tax expenses or benefits in
the period they occur. Prior to 2007 we were uncertain of achieving future
earnings and accordingly, we offset 100% of deferred tax assets with a valuation
allowance. In 2007, based upon the economics of the King Agreement, we concluded
that it was likely that we would realize sufficient future earnings to enable us
to utilize at least $9.6 million of these deferred tax assets and we reduced the
valuation allowance by that amount. Similarly, in 2008, we concluded that it was
likely that we would be able to utilize at least another $1.2 million of these
deferred tax assets and we reduced the valuation allowance by that amount and
$11.9 million of deferred tax assets were used to offset our 2008 federal and
state tax liabilities. In 2009, we increased the valuation allowance
by $2.5 million for the then available deferred tax assets and placed a
valuation allowance against the current year’s operating results.
Uncertainty
in Income Taxes
On
January 2007 we adopted FASB’s statement regarding accounting for uncertainty in
income taxes which defined the threshold for recognizing the benefits of
tax-return positions in the financial statements as "more-likely-than-not" to be
sustained by the taxing authorities. Our adoption of the standard did not result
in establishing a contingent tax liability reserve or a corresponding charge to
retained earnings. At December 31, 2009, 2008 or 2007, we had no liability for
income tax associated with uncertain tax positions. The Company’s practice will
be to recognize interest and penalties related to uncertain tax positions in
interest expense and other expense, respectively. The Company files federal and
state tax returns. In the normal course of business the Company is subject to
examination by taxing authorities. With few exceptions, the Company believes it
is no longer subject to U.S. federal and state income tax examinations for years
before 2006.
NOTE
I – EMPLOYEE BENEFIT PLANS
401(k) and Profit-Sharing
Plan
The
Company has a 401(k) and Profit-Sharing Plan (the “Plan”) for all employees.
Employees may elect to make a basic contribution of up to 15% of their annual
earnings. The Plan provides that the Company can make discretionary matching
contributions equal to 25% of the first 6% of employee contributions for an
aggregate employee contribution of 1.5%, along with a discretionary
profit-sharing contribution. The Company did not contribute matching or profit
sharing contributions for the Plan in years 2009, 2008 and 2007.
F-16
Stock Option
Plans
The
Company maintains various stock option plans. A summary of the Company's stock
option plans as of December 31, 2009, 2008, and 2007, and for the years
then ended consisted of the following:
|
Years Ended December 31,
|
||||||||||||||
|
2009
|
2008
|
2007
|
||||||||||||
|
Number
of
Options
(000’s)
|
Weighted
Average
Exercise
Price
|
Number
of
Options
(000’s)
|
Weighted
Average
Exercise
Price
|
Number
of
Options
(000’s)
|
Weighted
Average
Exercise
Price
|
|||||||||
|
Outstanding,
beginning
|
2,968
|
$
|
4.93
|
1,858
|
$
|
2.60
|
1,899
|
$
|
2.60
|
|||||
|
Granted
|
1,312
|
6.38
|
1,160
|
9.58
|
-
|
-
|
||||||||
|
Exercised
|
(525)
|
1.30
|
-
|
-
|
(31)
|
3.80
|
||||||||
|
Forfeited
or expired
|
(84)
|
7.95
|
(50)
|
23.69
|
(10)
|
3.60
|
||||||||
|
Outstanding,
ending
|
3,671
|
$
|
5.90
|
2,968
|
$
|
4.93
|
1,858
|
$
|
2.60
|
|||||
|
Options
exercisable
|
2,712
|
$
|
5.49
|
2,215
|
$
|
3.26
|
1,827
|
$
|
2.56
|
|||||
The
following table summarizes information about nonvested stock options outstanding
at December 31, 2009:
|
Number of
Options Not
Exercisable
(000)’s
|
Weighted
Average
Fair
Value
|
|||||||
|
Outstanding at
December 31, 2008
|
754 | $ | 6.88 | |||||
|
Granted
|
1,312 | 6.06 | ||||||
|
Vested
|
(1,039 | ) | 7.89 | |||||
|
Forfeited
or expired
|
(68 | ) | 6.02 | |||||
|
Outstanding
at December 31, 2009
|
959 | $ | 6.77 | |||||
The
Company estimated the option’s fair value on the date of grant using the Black -
Scholes option-pricing model. Black-Scholes utilizes assumptions related to
volatility, the risk-free interest rate, the dividend yield (which is assumed to
be zero, as the Company has not paid any cash dividends) and employee exercise
behavior. Expected volatilities utilized in the Black-Scholes model are based on
the historical volatility of the Company’s common stock price. The risk-free
interest rate is derived from the U.S. Treasury yield curve in effect at the
time of grant. The expected life of the grants is derived from historical
factors. No options were granted in 2007.
The
assumptions used in the Black Scholes model to determine fair value for the 2009
and 2008 stock option grants were:
|
2009
|
2008
|
||
|
Dividend
yield
|
0.0%
|
0.0%
|
|
|
Risk-free
interest rate
|
2.4%
to 3.1%
|
3.63%
|
|
|
Average
volatility
|
124%
|
141%
|
|
|
Forfeitures
|
0.0%
|
0.0%
|
|
|
Expected
holding period
|
10
years
|
10
years
|
|
|
Weighted
average grant date fair value
|
$
6.06
|
$
6.88
|
F-17
As of
December 31, 2009, 2008, and 2007 the aggregate intrinsic value of the option
awards vested was $4.8 million, $10.5 million, and $8.1 million, respectively.
In addition, the aggregate intrinsic value of option awards exercised during the
years ended December 31, 2009 and 2007 was $1.7 million and $0.5 million,
respectively. The total remaining unrecognized compensation cost related to the
unvested option awards at December 31, 2009 was $6.4 million and is expected to
be recognized over the next seven month weighted average remaining requisite
service period. The total fair value of the option awards that vested during the
years ended December 31, 2009, 2008 and 2007 was $8.2 million, $3.7 million, and
$0.1 million, respectively and was recognized as stock-based compensation.
Stock-based compensation from option awards in the amount of $1.7 million and
$0.6 million is included in research and development expense in the years ended
December 31, 2009 and 2008, respectively. There was no stock-based compensation
from option awards included in research and development expense during 2007.
Stock-based compensation from option awards in the amount of $6.5 million, $3.1
million and $0.1 million is included in general and administrative expenses in
the years ended December 31, 2009, 2008 and 2007, respectively.
Restricted Stock Unit Award
Plan
The
Company has a Restricted Stock Unit Award Plan (“2005 RSU Plan”) for its
employees and non-employee directors. Vesting of an RSU entitles the holder
thereof to receive a share of common stock of the Company on a distribution
date.
At
December 31, 2007, an aggregate of 2.95 million RSUs were outstanding and fully
vested. During 2009 and 2008, 333,000 and 50,000 RSUs were granted,
respectively, and 142,000 and 20,000 RSUs vested, respectively. During 2009,
17,000 RSU awards were canceled. At December 31, 2009 and 2008, 3.32 million and
3.0 million RSUs were outstanding, respectively, and 3.11 million and 2.97
million RSUs were vested, respectively. The RSUs have a weighted average fair
value of $3.75 per share at December 31, 2009.
The
stock-based compensation cost to be incurred on the RSUs is the RSU’s fair
value, which is the market price of the Company’s common stock on the date of
grant, less its exercise cost. The fair value of the RSU grants made in 2009 and
2008 was $2.1 million and $0.4 million, respectively. The total remaining
unrecognized compensation cost related to the unvested RSU awards amounted to
$1.3 million at December 31, 2009. The Company recognized compensation cost from
the RSU awards of $1.0 million, $0.2 million, and $0.8 million during the years
ended December 31, 2009, 2008 and 2007, respectively. Stock-based
compensation from RSU awards in the amount of $0.2 million and $0.4 million is
included in research and development expense in the years ended December 31,
2009 and 2007, respectively. There was no stock-based compensation from RSU
awards included in research and development expense during 2008. Stock-based
compensation from RSU awards in the amount of $0.8 million, $0.2 million, and
$0.4 million is included in general and administrative expense in the years
ended December 31, 2009, 2008 and 2007, respectively. No related tax benefits
were recorded in calendar year 2009, 2008, or 2007.
As of
December 31, 2009, 2008 and 2007, the aggregate intrinsic value of the RSU
awards outstanding and vested was $16.6 million, $21.8 million and $18.0
million, respectively. As defined in the 2005 RSU Plan, including a change in
control of the Company or upon termination of an employee’s employment with the
Company without cause, vesting will accelerate and the RSUs will fully vest.
Absent a change of control, one-fourth of vested shares of common stock
underlying an RSU award will be distributed (after payment of $0.01 par value
per share) on January 1 of each of 2011, 2012, 2013 and 2014. If a change in
control occurs (whether prior to or after 2011), the vested shares underlying
the RSU award will be distributed at or about the time of the change in
control.
NOTE
J – COMMITMENTS AND CONTINGENCIES
Employment
Agreements
Each of
Andrew D. Reddick, Robert B. Jones and Peter A. Clemens are parties to
employment agreements containing similar terms expiring December 31, 2010, which
provide annual salary of $377,000, $300,000 and $212,000, respectively, plus the
payment of annual bonuses, in the discretion of the Company’s Compensation
Committee or Board of Directors, based on the achievement of targets,
conditions, or parameters as set by the Compensation Committee or the Board of
Directors.
Statutory
Minimum Withholding Tax Obligations
Under our
stock option plans and our 2005 RSU plan, our employees may elect to have shares
withheld upon exercise of options and upon the exchange of RSUs in satisfaction
of the statutory minimum withholding tax obligations of such employees relating
to such option exercises or RSU exchanges.
F-18
Financial
Advisor Agreement
In
connection with the Company’s August 2007 Unit Offering, the Company is
obligated to pay a fee to the Company’s then financial advisor upon each
exercise of the warrants issued in the Unit Offering, in proportion to the
number of warrants exercised. The maximum amount of such remaining fee assuming
100% exercise of the remaining 1,339,000 warrants is $273,000. The Company has
not reflected this obligation as a liability in its consolidated financial
statements as the payment is contingent upon the timing and exercise of the
warrants by each of the warrant holders. Such fee, if any, will be
paid to the financial advisor and be offset against the equity proceeds as the
warrants are exercised.
NOTE
K – QUARTERLY FINANCIAL DATA (UNAUDITED)
Selected
quarterly consolidated financial data is shown below.
|
(in
thousands except share data)
|
Three
Month Period Ended
|
|||||||||||||||
|
Mar.
31
|
Jun.
30
|
Sept.
30
|
Dec.
31
|
|||||||||||||
|
Calendar
Year 2009
|
||||||||||||||||
|
Total
revenue (a)
|
$ | 1,380 | $ | 897 | $ | 808 | $ | 750 | ||||||||
|
Loss
from operations
|
(2,197 | ) | (3,256 | ) | (3,970 | ) | (4,077 | ) | ||||||||
|
Net
loss (b)
|
(1,277 | ) | (6,520 | ) | (3,954 | ) | (4,084 | ) | ||||||||
|
Loss
per common share
|
||||||||||||||||
|
Basic
|
$ | (0.03 | ) | $ | (0.14 | ) | $ | (0.09 | ) | $ | (0.09 | ) | ||||
|
Diluted
|
$ | (0.03 | ) | $ | (0.14 | ) | $ | (0.09 | ) | $ | (0.09 | ) | ||||
|
Mar.
31
|
Jun.
30
|
Sept.
30
|
Dec.
31
|
|||||||||||||
|
Calendar
Year 2008
|
||||||||||||||||
|
Total
revenue (a)
|
$ | 17,084 | $ | 15,685 | $ | 3,880 | $ | 7,788 | ||||||||
|
Income
(loss) from operations
|
12,132 | 11,227 | (3,186 | ) | 809 | |||||||||||
|
Net
income (loss) (b)
|
7,449 | 6,870 | 3,148 | (2,993 | ) | |||||||||||
|
Income
(loss) per common share
|
||||||||||||||||
|
Basic
|
$ | 0.16 | $ | 0.15 | $ | 0.07 | $ | (0.07 | ) | |||||||
|
Diluted
|
$ | 0.15 | $ | 0.13 | $ | 0.06 | $ | (0.07 | ) | |||||||
(a) See
Note A(9) for revenue recognition.
(b) The
Company recorded valuation adjustments on its deferred income tax assets and
recorded income tax expense of $2.5 million and $3.6 million in quarters
ended Jun. 30, 2009 and Dec. 31, 2008, respectively, and recorded
income tax benefit of $5.0 million in the quarter ended Sept. 30,
2008.
F-19
EXHIBIT
INDEX
The
following exhibits are included as a part of this Annual Report on Form 10-K or
incorporated herein by reference.
|
Exhibit
Number
|
Exhibit
Description
|
|
|
3.1
|
Restated
Certificate of Incorporation of the Registrant (incorporated by reference
to Exhibit 3.1 to the Registrant’s Form 8-K filed on June 25,
2009)
|
|
|
3.2
|
Certificate
of Amendment Reverse Splitting Common Stock and restating but not changing
text of part of Article III of Restated Certificate of Incorporation
(incorporated by Reference to Exhibit 3.1 to the Form 8-K filed December
4, 2007).
|
|
|
3.3
|
Restated
Bylaws of the Registrant (incorporated by reference to Exhibit 3.1 to the
Form 8-K filed on March 3, 2009).
|
|
|
10.1
|
License,
Development and Commercialization Agreement by and between the Registrant
and King Pharmaceuticals Research and Development, Inc. (incorporated by
reference to Exhibit 10.1 of the Form 8-K filed on November 2, 2007)
(confidential treatment has been requested for portions of this
Exhibit).
|
|
|
10.2
|
Securities
Purchase Agreement dated as of August 20, 2007 (“PIPE SPA”) among the
Registrant, Vivo Ventures Fund VI, L.P., Vivo Ventures VI Affiliates Fund,
L.P. (collectively “Vivo”), GCE Holdings LLC, and certain other
signatories thereto (incorporated by reference to Exhibit 10.1 to the Form
8-K filed on August 21, 2007).
|
|
|
10.3
|
Form
of Warrant dated as of August 20, 2007 issued pursuant to the PIPE SPA
(incorporated by reference to Exhibit 4.1 to the Form 8-K filed on August
21, 2007).
|
|
|
10.4
|
Form
of Warrants dated May 5, 2003 issued to Galen Partners III, L.P., Galen
Partners International, III, L.P., Galen Employee Fund III, L.P., Essex
Woodlands Health Ventures Fund V, L.P. and Care Capital Investments II, LP
and others) (incorporated by reference to Exhibit 10.6 to the October 2007
S-3).
|
|
|
10.5
|
Amended
and Restated Voting Agreement dated as of February 6, 2004 among the
Registrant, Care Capital Investments II, LP, Essex Woodlands Health
Ventures Fund V, L.P., Galen Partners III, L.P., and others
(incorporated by reference to Exhibit 10.5 of the Form 8-K filed on
February 10, 2004 (the “February 2004 Form 8-K”)).
|
|
|
10.6
|
Joinder
and Amendment to Amended and Restated Voting Agreement dated November 9,
2005 between the Registrant, GCE Holdings, Essex Woodlands Health Ventures
Fund V, L.P., Care Capital Investments II, LP, Galen Partners III, L.P.
and others (incorporated by reference to Exhibit 10.1 to the Form 8-K
filed November 10, 2005).
|
|
|
10.7
|
Second
Amendment to Amended and Restated Voting Agreement dated as of January 24,
2008 between the Registrant and GCE Holdings, LLC (incorporated by
reference to Exhibit 10.1 to the Form 8-K filed January 28,
2008).
|
|
|
10.8
|
Registrant’s
1995 Stock Option and Restricted Stock Purchase Plan (incorporated by
reference to Exhibit 4.1 to the Registrant's Registration Statement on
Form S-8, File No. 33-98396).
|
|
|
10.9
|
Registrant’s
1998 Stock Option Plan, as amended (incorporated by reference to Appendix
C to the Registrant’s Proxy Statement filed on May 12,
2009).
|
E-1
|
Exhibit
Number
|
Exhibit
Description
|
|
|
10.10
|
Registrant’s
2005 Restricted Stock Unit Award Plan, as amended (incorporated by
reference to Appendix B to the Registrant’s Proxy Statement filed on April
2, 2008).
|
|
|
10.11
|
Registrant’s
2008 Stock Option Plan, as amended on June 25, 2009 (incorporated by
reference to Appendix B to our Proxy Statement filed on May 12,
2009)
|
|
|
10.12
|
Executive
Employment Agreement dated as of August 26, 2003 between the Registrant
and Andrew D. Reddick (“Reddick”) (incorporated by reference to Exhibit
10.2 to the Form 10-Q for the quarter ended June 30, 2004 (the “June 2004
10-Q”)).
|
|
|
10.13
|
Amendment
to Executive Employment Agreement between the Registrant and Reddick,
dated May 27, 2004 (incorporated by reference to Exhibit 10.4 to the June
2004 10-Q).
|
|
|
10.14
|
Second
Amendment to Executive Employment Agreement between the Registrant and
Reddick, dated May 24, 2005 incorporated by reference to Exhibit 10.116 to
the Form 10-K for the year ending December 31, 2005 filed on February 21,
2006 (the “2005 Form 10-K”)).
|
|
|
10.15
|
Third
Amendment to Executive Employment Agreement between the Registrant and
Reddick, dated December 22, 2005 (incorporated by reference to Exhibit
10.1 to the Form 8-K filed December 23, 2005 (the “December 2005 Form
8-K”)).
|
|
|
10.16
|
Fourth
Amendment to Executive Employment Agreement between the Registrant and
Reddick dated December 16, 2007 (incorporated by reference to Exhibit
10.20 to the Form 10-K for the year ending December 31, 2007, filed on
March 5, 2008).
|
|
|
10.17
|
Fifth
Amendment to Executive Employment Agreement between the Registrant and
Reddick executed July 9, 2008 (incorporated by reference to
Exhibit 10.1 to our Form 8-K filed on July 10,
2008)
|
|
|
10.18
|
Executive
Employment Agreement dated as of April 5, 2004 between the Registrant and
Ron J. Spivey (incorporated by reference to Exhibit 10.3 to the June 2004
10-Q).
|
|
|
10.19
|
Amendment
to Executive Employment Agreement dated December 22, 2005 between
Registrant and Ron J. Spivey (incorporated by reference to Exhibit 10.2 to
the December 2005 Form 8-K).
|
|
|
10.20
|
Second
Amendment to Executive Employment Agreement dated December 19, 2007
between the Registrant and Ron J. Spivey (incorporated by reference to
Exhibit 10.23 to the Form 10-K for the year ending December 31, 2007,
filed on March 5, 2008).
|
|
|
10.21
|
Third
Amendment to Employment Amendment to Executive Employment Agreement
executed July 9, 2008 (incorporated by reference to Exhibit 10.2 to our
Form 8-K filed on July 10, 2008).
|
|
|
10.22
|
Amended
and Restate Employment Agreement effective as of January 1, 2009 between
the registrant and Ron J. Spivey (incorporated by reference to
Exhibit 10.3 to our Form 8-K filed on July 10,
2008)
|
|
|
10.23
|
Employment
Agreement dated as of March 10, 1998 between the Registrant and Peter
Clemens (“Clemens”) (incorporated by reference to Exhibit 10.44 to the
Form 10-K for the period ending December 31, 2007, filed on April 15,
1998).
|
|
|
10.24
|
First
Amendment to Employment Agreement made as of June 28, 2000 between the
Registrant and Clemens (incorporated by reference to Exhibit 10.44A to the
Registrant’s 2005 Form
10-K).
|
E-2
|
Exhibit
Number
|
Exhibit
Description
|
|
|
10.25
|
Second
Amendment to Executive Employment Agreement between Registrant and
Clemens, dated as of January 5, 2005 (incorporated by reference to Exhibit
99.1 to the Registrant's Form 8-K filed January 31,
2005).
|
|
|
10.26
|
Third
Amendment to Executive Employment Agreement dated December 22, 2005
between Registrant and Clemens (incorporated by reference to Exhibit 10.3
to the December 2005 Form 8-K).
|
|
|
10.27
|
Fourth
Amendment to Executive Employment Agreement dated December 16, 2007
between Registrant and Clemens (incorporated by reference to Exhibit 10.28
to the Form 10-K for the year ending December 31, 2007, filed on March 5,
2008).
|
|
|
10.28
|
Fifth
Amendment to Executive Employment Agreement executed July 9, 2008 between
Registrant and Clemens (incorporated by reference to Exhibit
10.4 to our Form 8-K filed on July 10, 2008)
|
|
|
10.29
|
Employment
Agreement dated as of March 18, 2008 between the Registrant and Robert B.
Jones (incorporated by reference to Exhibit 10.1 to our Form
8-K filed on March 24, 2008)
|
|
|
*10.30
|
Consulting
Agreement dated as of December 10, 2009 between Registrant and Garth
Boehm
|
|
|
14.1
|
Code
of Ethics (incorporated by reference to Exhibit 14.1 of the Form 8-K filed
on December 10, 2007).
|
|
|
21
|
Subsidiaries
of the Registrant (incorporated by reference to the Form 10-K for the
fiscal year ended December 31, 2006 filed on March 15,
2007).
|
|
|
*23.1
|
Consent
of BDO Seidman LLP, Independent Registered Public Accounting
Firm.
|
|
|
*31.1
|
Certification
of Periodic Report by Chief Executive Officer pursuant to Rule 13a-14 and
15d-14 of the Securities Exchange Act of 1934.
|
|
|
*31.2
|
Certification
of Periodic Report by Chief Financial Officer pursuant to Rule 13a-14 and
15d-14 of the Securities Exchange Act of 1934.
|
|
|
*32
|
Certification
of Chief Executive Officer and Chief Financial Officer pursuant to 18
U.S.C. Section 1350, as adopted pursuant to Section 906 of the
Sarbanes-Oxley Act of
2002.
|
*Filed or
furnished herewith.
E-3
CONSENT
OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Acura
Pharmaceuticals, Inc.
Palatine,
Illinois
We hereby
consent to the incorporation by reference in the Registration Statements on Form
S-8 (Nos. 333-151653, 333-151620, 333-133172, 333-123615, 333-63288, and
33-98356) and on Form S-3 (No. 333-146416) of Acura Pharmaceuticals, Inc. of our
reports dated March 2, 2010, relating to the consolidated financial statements,
and the effectiveness of Acura Pharmaceuticals, Inc.’s internal control over
financial reporting, which appear in this Form 10-K.
/s/ BDO
Seidman, LLP
Chicago,
Illinois
March 2,
2010
E-4