Attached files
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, DC 20549
FORM 10-K
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15 (D) OF THE SECURITIES EXCHANGE ACT OF 1934 |
FOR THE FISCAL YEAR ENDED DECEMBER 31, 2009
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(D) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Commission File Number 0-33489
ZYMOGENETICS, INC.
(exact name of registrant as specified in its charter)
| Washington | 91-1144498 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) | |
|
1201 Eastlake Avenue East, Seattle, WA 98102 (Address of principal executive offices) | ||
Registrant’s telephone number, including area code (206) 442-6600
Securities registered pursuant to Section 12(b) of the Act:
| Title of Each Class |
Name of Each Exchange on Which Registered | |
| Common Stock, no par value | The NASDAQ Stock Market |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark whether the Registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes ¨. No x.
Indicate by check mark if the Registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act.
Yes ¨. No x.
Indicate by check mark whether the Registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months, and (2) has been subject to such filing requirements for the past 90 days.
Yes x. No ¨.
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files).
Yes ¨ No ¨.
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of Registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendments to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer ¨ |
Accelerated filer x | Non-accelerated filer ¨ |
Smaller reporting company ¨ | |||||||
| (Do not check if a smaller reporting company) | ||||||||||
Indicate by check mark whether the Registrant is a shell company (as defined in Rule 12b-2 of the Act).
Yes ¨ No x.
The aggregate market value of the common equity held by non-affiliates computed by reference to the price at which the common equity was last sold as of June 30, 2009 was: $173,248,521.
Common stock outstanding at February 19, 2010: 85,477,565 shares.
DOCUMENTS INCORPORATED BY REFERENCE
| (1) | Portions of the Company’s definitive Proxy Statement for the annual meeting of shareholders to be held on June 17, 2010 are incorporated by reference in Part III. |
Table of Contents
ZYMOGENETICS, INC.
ANNUAL REPORT ON FORM 10-K
For the Year Ended December 31, 2009
| Page No. | ||||
| PART I | ||||
| Item 1. |
2 | |||
| Item 1A. |
24 | |||
| Item 1B. |
45 | |||
| Item 2. |
45 | |||
| Item 3. |
46 | |||
| Item 4. |
46 | |||
| PART II | ||||
| Item 5. |
Market for Registrant’s Common Equity, Related Shareholder Matters and Issuer Purchases of Equity Securities | 47 | ||
| Item 6. |
49 | |||
| Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations | 50 | ||
| Item 7A. |
61 | |||
| Item 8. |
62 | |||
| Item 9. |
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure | 91 | ||
| Item 9A. |
91 | |||
| Item 9B. |
92 | |||
| PART III | ||||
| Item 10. |
92 | |||
| Item 11. |
92 | |||
| Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Shareholder Matters | 92 | ||
| Item 13. |
Certain Relationships and Related Transactions, and Director Independence |
93 | ||
| Item 14. |
93 | |||
| PART IV | ||||
| Item 15. |
94 | |||
| 99 | ||||
1
Table of Contents
This Annual Report on Form 10-K contains, in addition to historical information, forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995 that involve risks and uncertainties. This Act provides a “safe harbor” for forward-looking statements to encourage companies to provide prospective information about themselves. All statements other than statements of historical fact, including statements regarding company and industry prospects and future results of operations, financial position and cash flows, made in this Annual Report on Form 10-K are forward-looking. We use words such as “anticipate,” “believe,” “continue,” “could,” “estimate,” “expect,” “future,” “intend,” “may,” “potential,” “seek,” “should,” “target” and similar expressions, including negatives, to identify forward-looking statements. Forward-looking statements reflect management’s current expectations, plans or projections and are inherently uncertain. Our actual results could differ significantly from the results discussed in the forward-looking statements. Readers are cautioned not to place undue reliance on these forward-looking statements, which speak only as of the date hereof. Factors that could cause or contribute to such differences include those discussed in “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” as well as those discussed elsewhere in this Annual Report on Form 10-K. We undertake no obligation to publicly release any revisions to these forward-looking statements that may be made to reflect events or circumstances after the date hereof or to reflect the occurrence of unanticipated events. Readers are urged, however, to review the information provided in reports that we file from time to time with the Securities and Exchange Commission or otherwise make public.
Overview
We are a biopharmaceutical company focused on the development and commercialization of therapeutic proteins for the treatment of human diseases. In 2009, through a series of strategic initiatives and workforce and cost reductions, we restructured our organization and are now focused on developing and commercializing a limited number of products, which we believe have substantial therapeutic and commercial potential and in which we retain a significant ownership position. Our current portfolio includes one commercial product, RECOTHROM® Thrombin, topical (Recombinant), and three immunology product candidates.
Commercial Product
RECOTHROM. We have developed and are marketing RECOTHROM in the United States for use as a topical hemostat to control moderate bleeding during surgical procedures. Our product, which is a recombinant version of the human blood-clotting protein thrombin, provides an effective and safe alternative to other thrombin products marketed in the United States in forms derived from bovine (cattle) plasma or human plasma. We hold worldwide rights to RECOTHROM, except for Canada, where Bayer Schering Pharma AG is responsible for commercializing the product.
Immunology Product Candidates
PEG-IFN-lambda. Pegylated Interferon-lambda (PEG-IFN-lambda) is being studied in collaboration with Bristol-Myers Squibb Company in a Phase 2 clinical trial for treatment of hepatitis C virus infection. In November 2009, we presented final results from an open-label Phase 1b study in patients with hepatitis C. We hold co-development rights to PEG-IFN-lambda in the United States and Europe and the option to co-promote and share profits on product sales in the United States. Bristol-Myers Squibb is responsible for commercializing the product outside the United States, for which we will receive milestones and royalties on sales.
IL-21. Interleukin-21 (IL-21) is currently being tested in an open-label Phase 2 clinical trial as a potential immunotherapy treatment for metastatic melanoma. In May 2009, we presented interim results from this study, with final results expected to be available in 2010. In addition, we presented results from an open-label Phase 2 study in renal cell carcinoma in May 2009. We hold worldwide rights to IL-21.
2
Table of Contents
IL-31 mAb. Interleukin-31 monoclonal antibody (IL-31 mAb) is currently in preclinical development as a potential treatment for atopic dermatitis. We hold worldwide rights to IL-31 mAb and expect to begin clinical testing for this candidate in 2011.
Our goal is to substantially increase the value of our company by advancing our products and candidates to key value inflection points associated with the achievement of development and/or commercial milestones. We intend to continue to build the market for RECOTHROM in the United States, which we expect will provide net cash flows that we may use to fund the further development of our immunology product candidates. Where appropriate, we intend to enter into strategic collaborations for the commercialization of our immunology product candidates, which we believe will enable us to maximize long-term value of these assets, while leveraging our internal resources and accessing complementary technologies, infrastructure and expertise. For example, we established the PEG-IFN-lambda collaboration with Bristol-Myers Squibb in January 2009, which provided substantial near-term cash and long-term commercial value retention.
We have out-licensed several product candidates previously identified through our discovery research efforts to third parties, including atacicept, FGF-18, IL-22RA mAb and IL-17RC soluble receptor to Merck Serono SA and IL-20 mAb and IL-21 mAb to Novo Nordisk A/S. These candidates are either outside our core area of interest or require levels of capital investment that we could not justify considering our available financial resources. We are not actively involved in the development of these product candidates. We are, however, eligible to receive milestone payments and royalties and, thus, we consider these product candidates to be important assets that could result in the generation of substantial value over the long term.
We intend to maintain strong patent protection for our asset portfolio. We file detailed patent applications with respect to our discoveries covering multiple patentable inventions, typically including composition of matter, method of making and method of use claims. We have issued patents or pending applications covering RECOTHROM and all of our internal product candidates. In total, we have more than 340 unexpired issued or allowed U.S. patents and over 180 U.S. patent applications pending. Outside of the United States, we have more than 790 issued or allowed foreign patents.
We were incorporated in the state of Washington in 1981. From 1988 to 2000, we were a wholly owned subsidiary of Novo Nordisk, one of the world’s largest producers of therapeutic proteins. In November 2000, as part of a restructuring by Novo Nordisk, we became an independent company. In February 2002, we completed our initial public offering. In addition to RECOTHROM, we have contributed to the discovery or development of seven recombinant protein products currently on the market. Our principal executive offices are located at 1201 Eastlake Avenue East, Seattle, Washington, 98102. Our telephone number is (206) 442-6600. Our website is www.zymogenetics.com. At the Investor Relations section of this website, we make available free of charge our annual report on Form 10-K, our annual proxy statement, our quarterly reports on Form 10-Q, any current reports on Form 8-K, and any amendments to these reports, as soon as reasonably practicable after we electronically file them with, or furnish them to, the Securities and Exchange Commission (SEC). The information on our website is not a part of, and is not incorporated into, this annual report on Form 10-K. In addition to our website, the SEC maintains an Internet site at www.sec.gov that contains reports, proxy and information statements, and other information regarding us and other issuers that file electronically with the SEC.
Commercial Product and Product Pipeline
Our current focus is the continued commercialization of our first product, RECOTHROM, and the development of three product candidates: PEG-IFN-lambda in partnership with Bristol-Myers Squibb, IL-21, and IL-31 mAb. We have out-licensed several product candidates that are outside of our core areas of interest or for which we could not justify the required capital investment. We are eligible to receive milestone payments and royalties related to these assets. The following table summarizes our commercial product and product candidates that are being internally developed or co-developed, as well as out-licensed product candidates.
3
Table of Contents
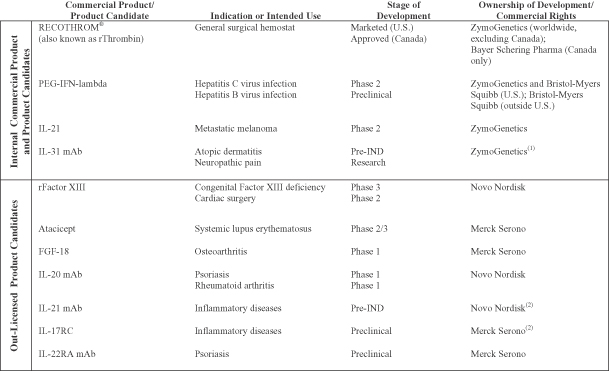
| (1) | Subject to certain opt-in rights granted to Merck Serono. |
| (2) | Subject to certain opt-in rights granted to ZymoGenetics. |
In the preceding table, “Research” refers to the stage in which we analyze the biology and therapeutic potential of proteins using a variety of laboratory methods. “Preclinical” refers to the stage in which safety, pharmacology and proof of efficacy in non-human animal models of specific human disease are evaluated. “Pre-IND” refers to the stage in which investigational new drug enabling preclinical toxicology studies are performed and materials in support of the investigational new drug (IND) and clinical studies are manufactured. “Phase 1” refers to clinical trials designed primarily to determine safety and pharmacokinetics in healthy volunteers or a limited patient population. “Phase 1b” refers to clinical trials designed to demonstrate biomarker or clinical outcome that could be considered for proof of concept in a limited patient population. “Phase 2” refers to clinical trials designed to evaluate preliminary efficacy, further characterize safety and optimize dosing in a limited patient population. “Phase 2/3” refers to large-scale clinical trials designed to establish safety and confirm efficacy in comparison to standard therapies or placebo in a patient population large enough to generate statistically significant results. “Phase 3” refers to clinical trials in a broad patient population with the intention of generating statistical evidence of efficacy and safety to support product approval.
Commercial Product
RECOTHROM®
RECOTHROM Thrombin, topical (Recombinant) was approved by the U.S. Food and Drug Administration (FDA) on January 17, 2008 for use as a general aid to control diffuse (non-arterial) bleeding during surgery. Net RECOTHROM sales in the United States totaled $8.8 million and $28.2 million during the years ended December 31, 2008 and December 31, 2009, respectively. RECOTHROM is available in a 5,000 international unit (IU) vial, a 20,000 IU vial and a 20,000 IU vial co-packaged with a spray applicator kit. Three wholesalers, AmerisourceBergen Corporation, Cardinal Health, Inc. and McKesson Corporation, accounted for approximately 90% of U.S. sales in 2009. If any of these wholesalers ceased distributing RECOTHROM, other wholesalers already distributing RECOTHROM would likely absorb the incremental sales volume with minimal interruption to the business or we would sell directly to hospitals.
4
Table of Contents
Thrombin is a specific blood-clotting enzyme that converts fibrinogen to fibrin, the primary protein contained in newly formed blood clots. Thrombin also promotes clot formation by activating Factor XIII, which cross-links the fibrin molecules and strengthens the newly forming clot. Topical thrombin is widely used to stop diffuse (non-arterial) bleeding occurring during surgical procedures, when control of bleeding by standard surgical techniques, such as direct pressure, ligation, or cautery, is ineffective or impractical. Minimizing bleeding during surgical procedures is important to maintain visibility in the operating field, limit the use of transfused blood products and reduce peri- and post-operative complications. Thrombin is generally sold as a lyophilized powder stored at room temperature, which is dissolved in saline and absorbed onto a surgical sponge, embedded onto a hemostatic pad or sprayed directly for topical application to wounds. Currently, there are three types of topical thrombin available in the United States: bovine (cattle) plasma-derived (Thrombin-JMI® marketed by King Pharmaceuticals), human-plasma derived (Evithrom® marketed by Ethicon, Inc. and the thrombin contained in GELFOAM Plus hemostasis kit marketed by Baxter Healthcare Corporation) and recombinant human thrombin (RECOTHROM marketed by ZymoGenetics). The thrombin market, based on the combined sales to hospitals of RECOTHROM, Thrombin-JMI, Evithrom and GELFOAM Plus, was estimated at $228 million in 2009, with full-year RECOTHROM sales representing approximately 13% market share.
We believe that there are several important advantages to recombinant human thrombin. Some patients may experience allergic reactions to plasma-derived products. Patients could also develop antibodies to bovine plasma-derived thrombin or to bovine Factor V or other protein impurities in the bovine plasma-derived product. In some cases, these antibodies can cross-react with analogous human proteins, creating a bleeding condition that can be difficult to manage and which has been fatal in patients who develop the most severe cases. Use of bovine plasma-derived thrombin in patients with pre-existing antibodies to bovine clotting factors may increase these risks and is, therefore, contraindicated. The package insert for bovine plasma-derived thrombin contains a black box warning, the most serious form of warning the FDA can require for approved products, describing these potential risks. In addition, all human plasma-derived products carry an FDA-mandated warning addressing a potential risk of transmitting infectious and other diseases, including HIV, hepatitis, parvovirus, Creutzfeldt-Jakob disease (CJD) and variant CJD. RECOTHROM, which is human thrombin produced using recombinant DNA technology, is inherently free from these potential risks and its package insert does not have a black box warning or any other warnings associated with the risk of transmitting blood-borne pathogens or infectious diseases.
In August 2009, we submitted a Citizen Petition to the FDA requesting that the FDA remove Thrombin-JMI Thrombin, topical (bovine origin) from the market in the interest of patient safety. The Citizen Petition was prompted by reports of serious or fatal bleeding-related adverse events in surgical patients exposed to bovine thrombin. In January 2010, we received an interim response from the FDA indicating that additional time was needed for the agency to reach a decision and provide a final response to the Citizen Petition. We can provide no assurance that the petition will be granted or that Thrombin-JMI will be removed from the market.
Prior to launch, we established the supply chain for RECOTHROM, from sourcing of critical raw materials and manufacturing to distribution to end customers, and built commercial inventories to satisfy expected market demand and provide what we believe are sufficient levels of safety stock. We have developed a patent-protected two-step process for the manufacture of recombinant thrombin. First, recombinant human prethrombin-1 is produced in mammalian cells. Then, using an enzyme activation step, prethrombin-1 is converted to recombinant human thrombin. The commercial-scale manufacturing process was developed in collaboration with Abbott Laboratories, our commercial manufacturer of the RECOTHROM bulk drug substance.
In December 2009, we announced a restructuring of our U.S. co-promotion agreement with Bayer HealthCare LLC and our license and collaboration agreement with Bayer Schering Pharma AG. Effective December 31, 2009 we ended the co-promotion with Bayer HealthCare such that Bayer HealthCare will no longer participate in the sales and marketing of RECOTHROM in the United States. We are currently in the process of increasing the size of our sales organization and intend to have the additional sales personnel fully trained and in the field by the end of first quarter of 2010. Outside the United States, we regained all rights to RECOTHROM, except for Canada, where the product was approved in December 2009. Bayer Schering Pharma
5
Table of Contents
will market and sell RECOTHROM in Canada and pay us royalties on net sales. Also, in December 2009, Bayer Schering Pharma withdrew the Marketing Authorization Application (MAA) to the European Medicines Agency (EMEA) for approval to market RECOTHROM in Europe. The application was withdrawn in response to indications from the European regulatory authorities that approval would require additional clinical trial data.
Clinical Trials. In September 2006, we completed a pivotal Phase 3 clinical study designed to evaluate the comparative efficacy of RECOTHROM and bovine thrombin, both administered with an absorbable gelatin sponge. The randomized, double-blind study was conducted at 34 sites in the United States and enrolled 411 patients in four surgical settings: spinal surgery, liver resection, peripheral artery bypass and arteriovenous graft construction. Both the primary and secondary endpoints of the study were met. RECOTHROM was shown to have comparable efficacy to bovine thrombin, as measured by the overall percentage of patients achieving hemostasis within 10 minutes. Both treatments were well tolerated and exhibited similar adverse event profiles. RECOTHROM also demonstrated a superior immunogenicity profile to bovine thrombin, based on a significantly lower incidence of post-treatment anti-product antibody development. The study was not powered to detect differences in clinical outcomes based on differences in antibody formation.
In 2007, we completed an open-label, non-comparative Phase 2 clinical study designed to evaluate safety and immunogenicity of RECOTHROM administered using a spray device in patients with burns undergoing autologous skin grafting. The study results demonstrated a safety and immunogenicity profile similar to that observed in the pivotal Phase 3 study.
In 2008, we completed an open-label Phase 3b study designed to evaluate the safety and immunogenicity of RECOTHROM in subjects at increased risk for having anti-bovine thrombin product antibodies as a result of prior surgical history. The study enrolled 205 subjects, 16% of whom had pre-existing antibodies to bovine thrombin. The study results demonstrated that no patients developed antibodies against RECOTHROM. Following a 29-day period after topical RECOTHROM application during a single spinal or vascular surgical procedure, the immunogenicity profile of RECOTHROM did not differ among subjects with or without pre-existing antibodies to bovine thrombin. RECOTHROM was well tolerated and observed adverse events were consistent with those commonly seen in post-surgical settings.
Adverse events observed in clinical trials with RECOTHROM were consistent with those commonly observed in surgical patients. In pooled results from completed clinical trials involving 583 patients exposed to RECOTHROM, the most common adverse events were incision site pain, procedural pain, and nausea. Of the 552 patients for whom complete immunogenicity observations were available, only 5 patients (0.9%) developed specific anti-product antibodies, and none of these antibodies were found to neutralize native human thrombin.
As part of our post-marketing approval commitments, we are conducting an open-label Phase 4 clinical study to evaluate the safety and immunogenicity of re-exposure to RECOTHROM in approximately 30 subjects previously treated with RECOTHROM. We expect to complete patient enrollment in the re-exposure Phase 4 study in 2011. In addition, pursuant to the Pediatric Research Equity Act, we are conducting an open-label Phase 4 clinical study to evaluate the safety of RECOTHROM as an aid to hemostasis in a pediatric population. We completed patient enrollment in the pediatric study in December 2009 and expect to report study results in 2010.
In November 2008, we borrowed $25 million under a $100 million financing arrangement with Deerfield Management. The draw entitles Deerfield to a royalty equal to 2% of RECOTHROM net sales in the United States until repayment, which is due by June 27, 2014. No further draws on the remaining $75 million available for borrowing under the facility were taken prior to expiration of the facility in February 2010.
We own issued U.S. and foreign patents directed to a genetically engineered thrombin precursor termed “prethrombin-1”, methods of producing recombinant human thrombin from prethrombin-1, formulations, and methods of activation and therapeutic use of the protein.
6
Table of Contents
Product Candidates
PEG-IFN-lambda
Interferon-lambda 1 (IFN-lambda 1, also known as IL-29) is a type III interferon that belongs to the 4-helical-bundle cytokine family. Native IFN-lambda 1 is generated in response to a viral infection and exhibits broad cellular anti-viral activity similar to type I interferons, such as interferon-alpha. However, IFN-lambda 1 signals through a receptor that is distinct from the type I interferon receptor and that has a more selective expression pattern compared to the widely expressed receptor for type I interferons. The difference in the receptor tissue distribution suggests that IFN-lambda 1 may serve as an alternative to interferon-alpha based therapy for viral infection by providing comparable antiviral activity with potentially fewer side effects.
In vitro studies have shown that IFN-lambda 1 has antiviral activity against human hepatitis C virus (HCV) in HCV preclinical models. Additionally, we have demonstrated that IFN-lambda 1 induces antiviral gene expression similar to interferon-alpha in primary human hepatocytes. Combined with the significant expression of the receptor for IFN-lambda 1 in liver samples from HCV positive individuals, these data provided the rationale for selecting HCV infection as our first clinical indication. Recent clinical studies have also demonstrated the importance of type III interferons in controlling HCV infection. Correlative data from these studies showed that patients with certain genotypes who had poor response to the current standard of care regimen of interferon-alpha plus ribavirin also produced lesser amounts of endogenous type III interferon, thus illustrating the importance of type III interferons in controlling the viral replication pathway.
Chronic infection with HCV is a leading cause of cirrhosis, liver failure, and hepatocellular carcinoma worldwide. It is estimated that there are over 170 million people worldwide infected with hepatitis C virus. In the United States, an estimated 4.0 million people have been exposed to HCV, and approximately 3.2 million have chronic HCV infection. HCV is associated with an estimated 8,000-10,000 deaths per year and is the main indication for liver transplantation in the United States. The current standard of care for chronic HCV infection involves treatment with the combination of pegylated interferon-alpha and ribavirin. Standard of care therapy has been associated with a number of significant side effects, including flu-like symptoms, anorexia, depression, hemolytic anemia and myelosuppression, which continue to be treatment-limiting factors. With a response rate to the current standard treatment for the most common form of HCV (genotype 1) in the United States of approximately 40%, there remains a need for better tolerated and more effective therapy for HCV infection. Our product candidate, PEG-IFN-lambda, is a pegylated version of the IFN-lambda 1 protein, produced using recombinant DNA technology. Pegylation extends the in vivo half-life of the protein, allowing for convenient dose scheduling, such as once per week.
In January 2009, we entered into an exclusive global collaboration with Bristol-Myers Squibb Company for PEG-IFN-lambda. Under the terms of the collaboration, we will co-develop PEG-IFN-lambda in the United States and Europe with Bristol-Myers Squibb and share development costs. We will have the option to co-promote PEG-IFN-lambda and to share profits on product sales in the United States, while receiving royalties on sales in the rest of the world. We may opt out of the co-development, co-promotion and profit sharing arrangement in the United States, in which case we would no longer be obligated to co-fund development or commercialization activities, and we would receive royalties on worldwide product sales.
In 2007, we completed a randomized, placebo-controlled, dose-escalation Phase 1a clinical trial in healthy volunteers to evaluate the safety, tolerability and pharmacokinetics of a single dose of PEG-IFN-lambda administered subcutaneously. The study enrolled 20 subjects who were randomized to four dose levels of PEG-IFN-lambda, ranging from 0.5 to 7.5 mcg/kg, or placebo. The results from this study demonstrated that administration of a single dose of PEG-IFN-lambda was associated with dose-related pharmacokinetic and pharmacodynamic effects, with evidence of biological activity, including up-regulation of interferon response markers, being observed at dose levels of 1.5 mcg/kg and above. No fever or hematologic effects, which are typically seen with interferon-alpha, were observed at all tested dose levels in this study.
7
Table of Contents
In 2009, we completed a Phase 1b study to evaluate the safety and antiviral effect of repeat dosing of PEG-IFN-lambda administered subcutaneously for four weeks as a single agent or in combination with ribavirin in genotype 1 HCV patients. Final study results were presented in November 2009 at the American Association for the Study of the Liver Diseases (AASLD) annual meeting. A total of 56 patients were enrolled in this three-part study. In Part 1, a total of 24 relapsed HCV patients were enrolled in 4 cohorts, consisting of 6 patients each. PEG-IFN-lambda was administered as a single agent weekly or bi-weekly at dose levels of 1.5 or 3.0 mcg/kg. In Part 2, a total of 25 relapsed HCV patients were treated in 4 cohorts, consisting of 6 or 7 patients each. PEG-IFN-lambda was administered weekly at dose levels of 0.5, 0.75, 1.5, or 2.25 mcg/kg in combination with ribavirin. In Part 3, a total of 7 previously untreated HCV patients were enrolled in a single cohort receiving 1.5 mcg/kg of PEG-IFN-lambda in combination with ribavirin. The results from this study demonstrated anti-viral activity of PEG-IFN-lambda at all dose levels tested in both relapsed and previously untreated HCV patients. A majority of patients across all treatment arms achieved a greater than 2 log reduction in HCV RNA measured by a test that identifies the presence of hepatitis C virus in patient’s blood. Minimal constitutional symptoms or hematologic effects were observed with PEG-IFN-lambda given as a single agent or in combination with ribavirin. The majority of adverse events and laboratory changes were mild or moderate. Dose-limiting elevations in liver enzymes, with or without an increase in bilirubin, were dose-dependent and reversible.
In October 2009, in collaboration with our partner Bristol-Myers Squibb, we initiated a two-part, randomized, controlled Phase 2 study of PEG-IFN-lambda administered subcutaneously for up to 48 weeks in combination with ribavirin in treatment-naïve patients with chronic genotype 1, 2, 3, or 4 HCV infection (the “EMERGE” study). The EMERGE study will evaluate the safety, tolerability and antiviral efficacy of PEG-IFN-lambda and ribavirin compared to PEGASYS® (PEG-IFN alfa-2a) and ribavirin. The primary efficacy endpoint of the study is the proportion of patients who achieve complete early virologic response (cEVR), defined as undetectable levels of HCV RNA after 12 weeks of treatment. A secondary efficacy endpoint is the proportion of patients who achieve sustained virologic response (SVR), defined as undetectable levels of HCV RNA 24 weeks after completed treatment. Part A of the EMERGE study is an open-label study that will explore four fixed doses of PEG-IFN-lambda compared to standard dose of PEGASYS in approximately 55 patients. Up to four doses of PEG-IFN-lambda will be selected from Part A for testing in the second part of the study. Part B of the study will be conducted as a randomized, blinded study and is designed to enroll up to 600 patients. We began enrolling patients in Part A of the study in October 2009 and expect to begin Part B in 2010.
We own issued patents for IFN-lambda 1 polypeptides, polynucleotides, expression vectors, cells, methods of treating a hepatitis infection, and a method of producing IFN-lambda 1, as well as patents on all known related molecules in the type III interferon family. We have filed patent applications for IFN-lambda 1 polypeptides, IFN-lambda 1 fusion proteins, antibodies, methods of expressing and purifying IFN-lambda 1, methods of using IFN-lambda 1 alone and in combination with other therapeutic agents to treat various viral diseases, cancers and autoimmune disorders. We will continue to file patent applications as new inventions are made. As part of our agreement with Bristol-Myers Squibb, we have assigned to Bristol-Myers Squibb a one-half ownership interest in each core patent relating to PEG-IFN-lambda filed outside the United States and a security interest in each core patent relating to PEG-IFN-lambda filed in the United States.
IL-21
IL-21 is a cytokine that activates several types of immune cells thought to be critical in eliminating cancerous or virally infected cells from the body. More specifically, IL-21 enhances the activity of mature natural killer (NK) cells; it has multiple effects on cytotoxic T lymphocyte cells (CTL), including increased activation and proliferation, extended longevity in circulation and improved ability to kill cancerous cells; and it enhances B-cell antibody production.
Preclinical studies have indicated that our recombinant version of IL-21 is an effective therapy in a number of animal models of cancer. In an animal model of metastatic melanoma, IL-21 was associated with significant
8
Table of Contents
anti-tumor activity. Animals in this model develop aggressive metastases to the lung, which can be readily measured. Treatment with IL-21 led to a significant reduction in the number of lung metastases relative to controls. IL-21 also was found to have potent inhibitory activity in other animal models of cancer. These models demonstrated that the in vivo effects of IL-21 were mediated through the activation of CTL and NK cells, which contribute to rejection of the tumors in the animal models. Moreover, this led to establishment of immunological memory, which protected animals from re-challenge with the parent tumor.
We believe that IL-21 could represent a potentially better tolerated and more efficacious immunotherapeutic agent than other cancer immunotherapies, such as interleukin-2 (IL-2) and interferon-alpha. In clinical practice, IL-2 produces durable responses in a very small percentage of patients with metastatic melanoma and metastatic renal cell carcinoma. Accompanying this relatively low level of efficacy are significant toxicities, including vascular leak syndrome and the release of pro-inflammatory cytokines, which profoundly limit the utility of IL-2 in treating disease. These side effects can be so severe that many patients are either hospitalized or stop the therapy before completion of the treatment program. Although somewhat better tolerated, interferon-alpha therapy is associated with significant chronic toxicities limiting its administration and produces a lower overall response rate with fewer complete responses compared to IL-2.
We own worldwide rights to our product candidate, recombinant human IL-21 protein. We had previously out-licensed rights to the IL-21 protein outside North America to Novo Nordisk and entered into a collaborative data sharing and cross-licensing agreement with them. In January 2009, subsequent to a strategic decision by Novo Nordisk to exit all of its oncology development programs, we reacquired rights to the IL-21 protein outside North America. Simultaneously, we and Novo Nordisk terminated a collaborative data sharing and cross-license agreement and a manufacturing agreement, under which Novo Nordisk was supplying clinical materials. As part of this termination, we acquired rights to all patent applications and data generated by Novo Nordisk as well as clinical product manufactured by Novo Nordisk. The reacquisition agreements did not require any upfront payment to Novo Nordisk. However, we will owe milestone payments and royalties to Novo Nordisk upon commercialization of IL-21 outside North America.
Metastatic Melanoma. We are pursuing metastatic melanoma as the lead indication for IL-21. There are an estimated 69,000 new cases of melanoma per year in the United States, with over 8,650 deaths per year attributed to this disease. Metastatic melanoma is essentially an incurable cancer with no established standard of care. Because of poor prognosis of overall survival for patients with advanced stages of metastatic melanoma, with a median of six to nine months, there is a significant unmet need for the development of therapies that can prolong overall survival. Several drugs have previously failed in late-stage clinical trials of metastatic melanoma. Most recently, Nexavar® (a product marketed by Bayer HealthCare AG and Onyx Pharmaceuticals, Inc.) failed to meet its endpoint of improved overall survival in a Phase 3 trial and a Phase 3 trial of elesclomol (Synta Pharmaceuticals Corp.) was suspended due to safety concerns. In October 2005, the FDA granted IL-21 orphan drug status for the treatment of melanoma patients with advanced or aggressive disease.
We are developing IL-21 as a single-agent treatment for metastatic melanoma. In 2007, we initiated an open-label Phase 2 clinical trial of IL-21 in previously untreated patients with metastatic or recurrent melanoma. The study, which is being conducted by the National Cancer Institute of Canada (NCIC), is designed to evaluate two dose levels of IL-21 at 30 and 50 mcg/kg. The interim results from 24 patients, presented at the World Congress on Melanoma annual meeting in May 2009, showed IL-21 to be biologically active, with 7 patients, or 29%, having a partial response and 8 patients, or 33%, having stable disease. Administered at a dose of 30 mcg/kg during three 5- day cycles, IL-21 was well tolerated. The most common adverse events were mild or moderate fatigue and rash. The 50 mcg/kg dose of IL-21 was poorly tolerated, with severe adverse events including neutropenia and skin rash. In August 2009, we completed study enrollment. A total of 30 patients received IL-21 at a dose of 30 mcg/kg and 10 patients received IL-21 at a dose of 50 mcg/kg. Final results from the study are expected to be available in 2010. In collaboration with the NCIC, we expect to initiate a larger randomized study versus DTIC (dacarbazine) in the first half of 2010. The study will evaluate safety and efficacy of IL-21 versus DTIC as first-line therapy in metastatic melanoma.
9
Table of Contents
Metastatic Renal Cell Carcinoma. In 2009, we completed an open-label Phase 2 clinical trial of IL-21 in combination with the tyrosine kinase inhibitor Nexavar in patients with advanced renal cell carcinoma. The study was designed to evaluate the safety, pharmacokinetics and anti-tumor activity of the combination therapy at the IL-21 maximum tolerated dose, previously established at 30 mcg/kg. The final study results were presented at the American Society of Clinical Oncology (ASCO) meeting in May 2009. The results indicated that the combination of IL-21 with Nexavar was well tolerated, with side effects that were manageable in an outpatient setting. The combination therapy was associated with anti-tumor activity both in terms of tumor response and duration of disease control, measured by progression-free survival (PFS). An independent data review completed for 33 patients showed an overall response rate of 21%, with 7 patients having a partial response. Median PFS was 5.7 months. No further investigation of IL-21 in renal cell carcinoma is planned at this time based on an evaluation of the competitive landscape and commercial opportunity.
B-cell Lymphoma. We have explored the use of IL-21 in combination with monoclonal antibodies, such as Rituxan® (a product marketed by Genentech, Inc. and Biogen Idec Inc.), an anti-CD20 antibody, that functions via antibody-dependent cellular cytotoxicity, a process enhanced by IL-21. In 2008, we completed an open-label Phase 1 clinical trial of IL-21 in combination with Rituxan in patients with relapsed low-grade B-cell lymphoma. The final study results, which were presented at the ASCO annual meeting in 2008, demonstrated that the combination of IL-21 at 100 mcg/kg with Rituxan was well tolerated and provided evidence of anti-tumor activity in this heavily pre-treated population, including one confirmed complete response, and three partial responses. No further investigation of IL-21 in B-cell lymphoma is planned.
We own issued patents for IL-21 polypeptides, polynucleotides and methods of using IL-21 to stimulate immune responses, particularly in tumor-bearing subjects as well as to the cell lines and methods of producing the recombinant IL-21 clinical product. We have filed patent applications for pharmaceutical compositions, IL-21 fusion proteins and other methods of using IL-21 for the treatment of disease. We have additional patent applications relating to IL-21 directed to methods for expressing and purifying recombinant IL-21; methods of treating specific cancers and viral diseases; combination therapies for IL-21 and monoclonal antibodies and IL-21 and tyrosine kinase inhibitors; and antagonist IL-21 ligands. We will continue to file patent applications as new inventions are made.
IL-31 mAb
Interleukin-31 (IL-31) is a cytokine derived from T cells. Analysis of IL-31 and IL-31 receptor levels in human and murine disease tissues suggests that IL-31 could play a role in atopic dermatitis (AD) and neuropathic pain. Transgenic animals over-expressing the IL-31 gene develop a severe type of dermatitis that resembles human AD, characterized by a destructive chronic scratching behavior in response to IL-31 mediated itch. Itch is a characteristic of human AD and the scratch response to itch is thought to be a major contributor to the severity of skin damage and disease. Treatment of animals in a murine model of spontaneous AD with a neutralizing antibody against IL-31 results in the reduction of scratching behavior. In addition, data shows that elevated levels of IL-31 mRNA and protein in skin correlate with both mouse and human AD, and that elevated circulating levels of IL-31 in serum correlate with severity of AD in patients. Analysis of peripheral blood T cells from human atopic dermatitis patients provides an association between IL-31 and skin-homing T cells, suggesting that skin diseases, such as AD, may be a promising therapeutic area for inhibition of IL-31.
Atopic dermatitis is a relapsing, chronic, inflammatory skin disorder that affects over 50 million people in the United States, major European countries and Japan. The disease has a high prevalence in children, with as many as 85% of cases developing AD before age 5. Some patients suffer from the disease into adulthood. AD typically resolves over time, but the disease becomes severe for those patients that do not go into remission, representing approximately 10% of the total affected population. Severe AD patients suffer from intense itching resulting in psychological problems, significant sleep loss and skin disfigurement. Current therapies on the market, such as topical corticosteroids, topical calcineurin inhibitors and antihistamines, are not effective for patients with severe disease, have safety concerns with long-term use, and do not target the disease mechanism. We believe that an inhibitor of IL-31 may be an effective therapy for treatment of these severely affected AD patients.
10
Table of Contents
Our product candidate is an IL-31 monoclonal antibody (IL-31 mAb) that has been shown to neutralize the activity of IL-31 in preclinical settings in a highly specific manner. IL-31 mAb is currently in the pre-IND development stage as a potential treatment for AD. We are in the process of manufacturing sufficient quantities of drug to supply toxicology studies, which are expected to begin in 2010, and Phase 1 clinical trials, which we intend to initiate in 2011. We own worldwide rights to IL-31, including protein products that target IL-31 such as monoclonal antibodies, subject to certain opt-in rights held by Merck Serono.
We have issued patents to IL-31 and IL-31 antibodies. We also have filed several patent applications relating to IL-31 and IL-31 antagonists, compositions and uses in disease on a worldwide basis, which cover protein products that target IL-31, including monoclonal antibodies, and therapeutic uses and will continue to file new patent applications as new inventions are made.
Out-licensed Product Candidates
Atacicept (formerly known as TACI-Ig)
Atacicept is a soluble form of the TACI receptor, a member of the tumor necrosis factor receptor family of proteins. Atacicept binds to and inhibits the activity of two ligands, BLyS and APRIL, which are implicated in B-cell survival, maturation and antibody production. We believe that atacicept could represent a more specific immunosuppressive agent for the treatment of autoimmune diseases.
Until August 2008, atacicept was developed jointly by us and Merck Serono SA pursuant to a collaborative development and marketing agreement established in 2001. In 2008, we converted this agreement to a worldwide royalty-bearing license, granting Merck Serono exclusive worldwide development and commercialization rights for atacicept.
Merck Serono is conducting a randomized, double-blind, placebo-controlled Phase 2/3 clinical trial of atacicept in patients with general systemic lupus erythematosus (SLE). In 2008, Merck Serono discontinued a Phase 2/3 study in patients with lupus nephritis. The study was discontinued due to the observation of the increased risk of severe infection, possibly resulting from significant underlying disease activity and the concomitant use of several immunosuppressive agents. In order to obtain regulatory approval in SLE, Merck Serono will need to complete additional studies of atacicept in patients with SLE.
Merck Serono is conducting three Phase 2 studies investigating atacicept in rheumatoid arthritis (RA): one in RA patients with inadequate response to TNF inhibitor therapy, another in RA patients who have not previously received TNF inhibitor therapy, and a third to evaluate the safety and efficacy of atacicept in combination with Rituxan. Preliminary results of the two single-agent atacicept Phase 2 studies confirmed the biological effect of atacicept on immunoglobulin and autoantibody production and no new safety signals were observed. However, the studies did not meet the pre-specified level of disease control activity to support moving directly into Phase 3 clinical testing. Following completion of further exploratory analysis, Merck Serono decided not to initiate further studies of atacicept in RA patients.
In September 2009, Merck Serono voluntarily discontinued two studies investigating atacicept in multiple sclerosis (MS) based upon a recommendation from an Independent Data Monitoring Committee (IDMC). In one of the MS studies, the IDMC observed an increase in MS disease activity in the atacicept treatment arms compared to the placebo arm. No comparable issues have been identified in the Phase 2/3 clinical trial in SLE or in the Phase 2 clinical trials in RA.
IL-21 monoclonal antibody (IL-21 mAb)
IL-21 mAb is a fully human monoclonal antibody we developed as an inhibitor of IL-21. IL-21 is a T-cell derived cytokine that exerts multiple effects on both T-cell and B-cell responses, which can be beneficial in fighting cancer or infections. In some situations, over expression of IL-21 can lead to autoimmune or
11
Table of Contents
inflammatory disease. In particular, IL-21 is a key regulator of two types of T cells: Th17 cells and T follicular helper (TFH) cells. Th17 cells are known to be involved in inflammation. By blocking IL-21 with IL-21 mAb, inflammation may be reduced in a number of diseases that share this pathway, such as psoriasis, Crohn’s disease and rheumatoid arthritis. TFH cells are specialized types of T cells that promote antibody responses from B cells. Blocking IL-21’s effect on B cells may have an impact on human diseases that are driven by antibody responses, such as systemic lupus erythematous (SLE). Murine models of psoriasis, Crohn’s disease (colitis), rheumatoid arthritis and SLE have demonstrated that inhibition of IL-21 leads to significant reductions in disease scores and pathology.
We and Novo Nordisk A/S have been parties to a license agreement for IL-21 since 2001, pursuant to which Novo Nordisk had certain rights to IL-21 outside North America, including monoclonal antibodies targeting IL-21. In December 2009, we and Novo Nordisk amended this license agreement, giving Novo Nordisk worldwide rights to IL-21 mAb and certain other embodiments of IL-21. Under the agreement, we have a right to co-promote the IL-21 mAb product in the United States and receive increased royalties on net sales in the United States if we contribute to Phase 3 clinical development costs. This license does not affect our IL-21 Phase 2 development candidate, which is recombinant human IL-21 protein and as to which we maintain worldwide rights.
IL-21 mAb is currently in the pre-IND stage. We expect to complete the transfer of IL-21 mAb technology and information to Novo Nordisk in 2010, which will enable them to begin clinical testing.
Other Out-licensed Product Candidates
rFactor XIII. rFactor XIII is a recombinant version of a human protein that is involved in blood clotting, and is being developed for the treatment of bleeding disorders. Novo Nordisk acquired rights to this protein from us in October 2004 after we completed several Phase 1 clinical trials in healthy volunteers and in patients with congenital Factor XIII deficiency. Novo Nordisk is conducting a Phase 3 study of rFactor XIII in patients with congenital Factor XIII deficiency and a Phase 2 study in patients undergoing cardiac surgery.
Fibroblast growth factor-18 (FGF-18). FGF-18 is a member of the fibroblast growth factor family of proteins. Our preclinical data suggest that FGF-18 may be useful for healing cartilage damaged by injury or disease. We out-licensed this protein to Merck Serono in October 2004 in conjunction with the strategic alliance. Merck Serono is conducting a Phase 1 clinical trial of FGF-18 for the treatment of osteoarthritis.
IL-17 receptor C (IL-17RC) soluble receptor. IL-17RC is a soluble receptor that binds to both IL-17A and IL-17F, the two most closely related cytokines in the IL-17 family. Both cytokines are highly expressed in a variety of inflammatory and autoimmune diseases, including rheumatoid arthritis, inflammatory bowel disease, multiple sclerosis and transplant rejection. We hypothesize that use of IL-17RC soluble receptor to neutralize the pro-inflammatory properties of IL-17A and IL-17F could have a beneficial therapeutic effect in any or all of these diseases. In August 2008, as part of the restructuring of our relationship, Merck Serono acquired an exclusive development and commercialization license to IL-17RC soluble receptor worldwide, subject to certain opt-in rights that we hold. The product candidate is currently in preclinical development.
IL-20 monoclonal antibody (IL-20 mAb). IL-20 is a member of the IL-10 cytokine family. In September 2001, Novo Nordisk licensed the rights to IL-20 outside North America pursuant to the option and license agreement. In March 2004, they licensed the rights to IL-20 in North America under a separate agreement. Our preclinical data suggest that IL-20 may play an important role in the regulation of skin inflammation and the pathology of psoriasis, as well as other inflammation diseases. Novo Nordisk is currently conducting Phase 1 clinical trials in psoriasis and rheumatoid arthritis patients.
IL-22 receptor subunit alpha monoclonal antibody (IL-22RA mAb). IL-22RA is a cytokine receptor that binds to both IL-20 and IL-22 cytokines and may be a potential target for the treatment of psoriasis. We
12
Table of Contents
out-licensed rights to IL-22RA monoclonal antibodies and to IL-22RA as a target to Merck Serono in October 2004 as part of the strategic alliance. The IL-22RA mAb product candidate is currently in preclinical development by Merck Serono.
Out-licensed Commercial Products
In addition to RECOTHROM, we have contributed to the discovery or development of seven recombinant protein products marketed by other companies.
Augment™ Bone Graft (formerly GEM-OS1TM)/Augment™ Injectable Bone Graft (formerly GEM-OS2TM). Augment Bone Graft/Augment Injectable Bone Graft is a combination of platelet-derived growth factor (PDGF-BB) and a synthetic bone matrix. PDGF-BB is a growth factor that stimulates the growth of a variety of cell types, including bone forming cells. We cloned the gene that codes for platelet-derived growth factor, which we have out-licensed to BioMimetic Therapeutics, Inc. In November 2009, BioMimetic received approval to market Augment Bone Graft as an alternative to the use of autograft in foot and ankle fusion indications in Canada. In addition, BioMimetic completed submission of a modular Product Market Approval (PMA) to the U.S. FDA in February 2010.
Cleactor™ (tPA analog). Cleactor is a modified form of the protein tissue plasminogen activator, marketed in Japan by Eisai for the treatment of myocardial infarction, or heart attacks. In collaboration with Eisai, we developed this modified protein, which has enhanced properties that allow it to be given as a single injection.
GEM 21S® (platelet-derived growth factor). GEM 21S is a combination of a platelet-derived growth factor with a synthetic bone matrix, developed by BioMimetic Therapeutics, Inc. and marketed by Osteohealth Company, a division of Luitpold Pharmaceuticals, Inc. for the treatment of bone loss and gum tissue recession associated with advanced periodontal disease. We cloned the gene that codes for platelet-derived growth factor, the active agent in GEM 21S.
GlucaGen® (glucagon). GlucaGen is a protein therapeutic marketed by Novo Nordisk, Bedford Laboratories and Eisai Co., Ltd. (Eisai) for use as an aid for gastrointestinal motility inhibition and for the treatment of severe hypoglycemia in diabetic patients treated with insulin. In collaboration with Novo Nordisk, we developed a process for the production of this protein that is currently used by Novo Nordisk in the manufacture of GlucaGen.
Insulin and insulin analogs. Insulin and insulin analogs manufactured using recombinant DNA technology are marketed by Novo Nordisk worldwide for the treatment of diabetes. In collaboration with Novo Nordisk, we developed a process for the production of recombinant human insulin in yeast that is used by Novo Nordisk.
NovoSeven® (recombinant Factor VIIa). Factor VIIa is a protein involved in the generation of blood clots, and NovoSeven is marketed worldwide by Novo Nordisk for the treatment of patients with hemophilia and certain other coagulation deficiencies. We cloned the gene that codes for human Factor VII and developed the process for the production of activated recombinant human Factor VII, or recombinant Factor VIIa, which led to the establishment of the manufacturing process that Novo Nordisk currently uses to produce this protein.
Regranex® (platelet-derived growth factor). Regranex, until recently a product of Ethicon, Inc., a Johnson & Johnson Company, is a growth factor approved for the treatment of non-healing diabetic ulcers. In December 2008, One Equity Partners LLC announced the acquisition of Regranex from Ethicon Inc., which will be marketed and distributed by Systagenix Wound Management, a company created by One Equity Partners LLC. We cloned the gene that codes for platelet-derived growth factor and demonstrated the importance of this protein in stimulating wound healing.
13
Table of Contents
We have earned royalties on sales of some of these products. In the aggregate, from sales of these products and other technology licenses, we earned royalties of $1.3 million, $6.3 million, and $6.3 million for the years ended December 31, 2009, 2008, and 2007, respectively.
Commercialization
To commercialize RECOTHROM in the United States, we established our own dedicated commercial operations team with sales and sales operations, marketing, and supply chain and inventory management functions. We believe that the thrombin market, with its concentrated customer base, can be addressed with a relatively small sales force and that our recombinant technology gives us a competitive advantage in the current market.
In June 2007, we entered into a co-promotion agreement with Bayer HealthCare LLC, under which Bayer HealthCare provided sales people and medical liaisons to support RECOTHROM commercialization in the United States. During 2008 and 2009, the combined sales force worked to convert larger bovine thrombin accounts to RECOTHROM, by focusing on key surgeons, clinical pharmacists, operating room nurses and Pharmacy and Therapeutics (P&T) committee members within each account. Three wholesalers, AmerisourceBergen Corporation, Cardinal Health, Inc. and McKesson Corporation, accounted for approximately 90% of U.S. sales in 2009. If any of these wholesalers ceased distributing RECOTHROM, other wholesalers already distributing RECOTHROM would likely absorb the incremental sales volume with minimal interruption to the business or we would sell directly to hospitals.
In December 2009, we amended the U.S. co-promotion whereby Bayer HealthCare will no longer participate in the sales and marketing of RECOTHROM in the United States. We are currently in the process of increasing the size of our sales organization and intend to have the additional sales personnel fully trained and in the field by the end of first quarter 2010.
With our other product candidates, we intend, where appropriate, to enter into strategic collaborations for the commercialization of these candidates. We believe that this approach will enable us to maximize the long-term value of these assets.
Research and Development
In 2009, through a series of strategic initiatives and workforce and cost reductions, we restructured our organization to focus on developing and commercializing a smaller number of product candidates, which we believe have substantial therapeutic and commercial potential and in which we retain a significant ownership position. As part of these changes, we discontinued ongoing immunology and oncology discovery research programs, while retaining the research and development capabilities necessary to support the ongoing development programs for our product candidates.
Our research and development infrastructure draws upon a broad range of skills and technologies, including scientific computing, molecular and cellular biology, animal models of human disease, protein chemistry, antibody generation and engineering, pharmacology and toxicology, clinical development, medical and regulatory affairs, drug formulation, process development and protein manufacturing methods. We believe that this comprehensive approach enables us to effectively and efficiently develop our pipeline of therapeutic proteins.
We have a development organization with the skills and expertise to design and implement clinical trials for multiple product candidates and to file license applications with the FDA and other regulatory agencies. Our in-house development resources include a clinical development group responsible for designing, conducting and analyzing clinical trials. The group includes clinical research, clinical operations, biometrics, medical writing and drug safety. Our preclinical development group provides support in the areas of bioanalytical research and
14
Table of Contents
development, pharmacology (including pharmacogenomics), toxicology, pathology and pharmacokinetics. Our regulatory affairs group develops regulatory strategies and manages communications and submissions to regulatory agencies.
For additional details for research and development activities, refer to the Operating Expenses section under “Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations Overview”.
Manufacturing
RECOTHROM®
We have established a RECOTHROM commercial supply chain, which relies on several single-source vendors. We have entered into a long-term manufacturing agreement with Abbott Laboratories for commercial-scale production of bulk recombinant human thrombin (rThrombin), the active drug substance in RECOTHROM. Under the agreement, Abbott manufactures rThrombin using the two-step process developed by ZymoGenetics, according to specifications developed and agreed upon by both companies. First, recombinant human prethrombin-1 is produced in mammalian cells. Then, using an enzyme activation step, prethrombin-1 is converted to rThrombin. Abbott has committed to annually supply up to a maximum amount, which we believe is sufficient to meet our projected market demand and provide adequate safety stock. We have agreed to purchase annual minimum amounts from Abbott. The agreement terminates in 2018. We have also entered into a manufacturing services agreement with Patheon Italia S.p.A. for fill and finish of rThrombin, which expires in December 2013, and are currently negotiating with an additional fill and finish supplier located in the United States. In addition, we have entered into agreements with two vendors for the final packaging of RECOTHROM and with Cardinal Health SPS, Inc. for third party logistics services. Furthermore, we have entered into agreements with several suppliers of critical raw materials, manufacturing aids, and components for RECOTHROM, some of which are located outside the United States.
Under the terms of the amended license and collaboration agreement, we agreed to supply vials of rThrombin approved for sale in the United States to Bayer Schering Pharma for sale in Canada throughout the term of the agreement.
PEG-IFN-lambda
We manufactured initial clinical supplies of PEG-IFN-lambda in our pilot-scale GMP manufacturing facility, using a high-yield internally developed E. coli process and believe we have adequate supply of product to support clinical development though Phase 2. Under the terms of our co-development and co-promotion agreement with Bristol-Myers Squibb, they will be responsible for all future manufacturing of PEG-IFN-lambda, including product for Phase 3 clinical trials and commercial sale.
IL-21
Our initial clinical supply of IL-21, which is made in E. coli, was manufactured by a third party using a process we developed. Subsequently, Novo Nordisk manufactured clinical materials for Phase 2 and initial Phase 3 development under a manufacturing agreement established in 2007. In January 2009, Novo Nordisk terminated the agreement and we acquired all rights to the manufacturing processes and obtained the existing supply of the product. We will need to identify and enter into an agreement with a third-party contract manufacturer for commercial supply of IL-21.
IL-31 mAb
Our initial supplies of IL-31 mAb product for toxicology studies and Phase 1 clinical trials are currently being manufactured by a third-party contractor.
15
Table of Contents
Manufacturing Changes
In 2009, we discontinued operation of our pilot-scale GMP manufacturing facility that was used to supply materials for toxicology studies and early-stage clinical trials. Going forward, we intend to rely on collaborative partners or third-party contractors for production of all preclinical, clinical and commercial supplies.
Collaborative Relationships
Bristol-Myers Squibb Co-Development and Co-Promotion Agreement for PEG-IFN-lambda
In January 2009, we entered into a co-development/co-promotion and license agreement with Bristol-Myers Squibb Company that covers all members of the type III interferon family, including interferon-lambda 1. Under the terms of the agreement, we are obligated to work exclusively with Bristol-Myers Squibb to develop biopharmaceutical products based on the type III interferon family. Currently, the companies intend to only develop PEG-IFN-lambda, a pegylated version of interferon-lambda 1, which is in development as a treatment for hepatitis C.
As part of the co-development/co-promotion and license agreement, Bristol-Myers Squibb receives an exclusive worldwide license to the core patents relating to the type III interferon family and a co-ownership interest in all core patents relating to the type III interferon family filed outside of the United States. In addition, Bristol-Myers Squibb receives a non-exclusive license to other intellectual property rights relating to the licensed products. We will be responsible for funding the first $100 million of development costs in the United States and Europe, which we expect to incur during Phase 1b and Phase 2 clinical testing, and 20% of all further development costs in the United States and Europe.
In return, during 2009 we have received:
| • | $105 million in license fees; and |
| • | $95 million in milestone payments related to initiation of Phase 2 activities. |
In addition, we may receive:
| • | Additional payments of up to $335 million based on pre-defined development and regulatory milestones for hepatitis C, up to $287 million in development and regulatory milestones for other potential indications, and up to $285 million based on pre-defined annual sales milestones; |
| • | 40% of the profits from the co-commercialization of any type III interferon family product within the United States. We will also be responsible for 40% of any loss from the co-commercialization of any product within the United States, provided that a portion of our share of losses incurred through the initial launch phase will be deferred, and deferred losses will subsequently be deducted from milestones, royalties and our share of profits; and |
| • | Royalties on product sales outside the United States. |
The research and development activities are governed by a steering committee made up of an equal number of representatives from each company. Bristol-Myers Squibb is responsible for all future manufacturing of PEG-IFN-lambda, including product for Phase 3 clinical trials and commercial sale.
We have the right to co-promote or co-fund PEG-IFN-lambda in the United States, and must exercise this right within 30 days after acceptance by the FDA of a Biologics License Application (BLA) filing, in which case we will share any profits or losses in the United States. In certain circumstances, we may opt out of co-promotion while retaining the option to co-fund and share product profits and losses. We have the right to discontinue co-promotion and co-funding in the United States, in which case we would be eligible to receive royalties on product sales in the United States. Under certain restricted circumstances, Bristol-Myers Squibb may terminate our right to co-promote in the United States, provided that, in certain of these circumstances, we will retain the
16
Table of Contents
option to co-fund and share product profits and losses. If Bristol-Myers Squibb terminates our co-promotion right and we do not have the option to co-fund or choose not to exercise that option, we would receive royalties on product sales instead of sharing profits and losses in the United States.
Royalties on sales vary based on annual sales volume and the degree of patent protection provided by the licensed intellectual property. Royalty payments may be reduced if Bristol-Myers Squibb is required to license additional intellectual property from one or more third parties in order to commercialize a product or, in certain circumstances, if product sales suffer from direct competition. Royalty obligations under the agreement continue on a country-by-country basis until the date on which no valid patent claims relating to a product exist or, if the product is not covered by a valid patent claim, eleven years after the date of first sale of the product in that country.
The term of the agreement began on February 26, 2009 and will continue for as long as a type III interferon product is the subject of an active development project or there is an obligation to pay royalties under the agreement.
Bayer License and Collaboration Agreement for rThrombin and U.S. Co-Promotion Agreement for RECOTHROM®
In June 2007, we executed a license and collaboration agreement with Bayer Schering Pharma AG and a U.S. co-promotion agreement with Bayer HealthCare LLC. Under the terms of the license and collaboration agreement, Bayer Schering Pharma was responsible for developing and commercializing rThrombin outside of the United States. Under the co-promotion agreement, Bayer HealthCare agreed to contribute sales people and medical science liaisons for the first three years following the launch of RECOTHROM in the United States. Through December 31, 2009, we received an initial milestone of $30.0 million upon signing of the agreements, an additional $40.0 million upon the U.S. approval of rThrombin, and $6.5 million upon the initial filings for approval in Canada, Europe and Asia.
In December 2009, we executed amendments to both agreements. Pursuant to the amended license and collaboration agreement, Bayer Schering Pharma will develop and commercialize the initial presentations of rThrombin in Canada, where it received marketing approval in December 2009, but will return all other rights to RECOTHROM outside the United States and Canada to us. As part of the agreement, we will also supply vials of rThrombin approved for sales in the United States to Bayer Schering Pharma for sale in Canada for the term of the license. Pursuant to the amended co-promotion agreement, Bayer HealthCare’s active role in promoting RECOTHROM in the United States ceased as of December 31, 2009, but they are entitled to commissions on sales in the United States for up to two years subject to an aggregate maximum amount of $12 million.
Merck Serono Development and Marketing Agreement for Atacicept
In August 2001, we entered into a collaborative development and marketing agreement with Ares Trading S.A., a wholly owned subsidiary of Serono S.A., focused on product candidates derived from two cellular receptors (designated TACI and BCMA) that are involved in the regulation of the human immune system. Following the acquisition of Serono by Merck KGaA in 2007, Serono’s rights under this agreement have been held by Merck Serono S.A. Pursuant to the collaborative development and marketing agreement, the parties had been co-developing atacicept in autoimmune diseases and cancer. In August 2008, we entered into an amended and restated development and marketing agreement, providing Merck Serono with exclusive worldwide rights to develop, market and sell products developed under the agreement, for which we will be entitled to receive milestone fees and royalties on worldwide net sales.
We granted Merck Serono an exclusive license to our intellectual property relating to TACI, BCMA and certain other related technologies to make, use, have made, sell, offer to sell and import products based on TACI and BCMA. Merck Serono is required to pay royalties on sales, which vary based on annual sales volume and the
17
Table of Contents
degree of patent protection provided by the licensed intellectual property. Royalty payments may be reduced if Merck Serono is required to license additional intellectual property from one or more third parties in order to commercialize a product or, in certain circumstances, if product sales suffer from direct competition. Royalty obligations under the agreement continue on a country-by-country basis until the date on which no valid patent claims relating to a product exist or, if the product is not covered by a valid patent claim, 15 years from the date of first sale of the product in that country.
The agreement will continue for as long as a TACI or BCMA product is the subject of an active development project or there is an obligation to pay royalties under the agreement. The agreement provides for an initial fee and milestone payments to be paid by Merck Serono in connection with the development and approval of products, up to an aggregate of $52.5 million of which $15.5 million has been received to date.
Novo Nordisk License Agreements for IL-21
As a result of a series of agreements, we now have worldwide development and commercialization rights for products based on our intellectual property to IL-21 protein and Novo Nordisk has development and commercialization rights for products based on our intellectual property to other embodiments of IL-21, including antibodies to IL-21.
In 2001, Novo Nordisk initially licensed the rights under our intellectual property to various embodiments of IL-21 in territories outside of North America. In 2005, we entered into a collaborative data sharing and cross-license agreement with Novo Nordisk to develop and execute a joint global clinical development plan for the IL-21 protein to achieve regulatory approval of a common product in the companies’ respective territories. In January 2007, the parties also entered into a manufacturing agreement whereby Novo Nordisk agreed to supply us with IL-21 protein for use in clinical trials. In January 2009, the parties restructured their relationship as it relates to IL-21. As part of the restructuring, the parties:
| • | Amended and restated the license agreement to no longer cover the IL-21 protein. However, Novo Nordisk continued to be responsible for developing other embodiments of IL-21, including antibodies to IL-21, outside North America. |
| • | Entered into a license and transfer agreement pursuant to which we received an exclusive license outside North America to the intellectual property rights that Novo Nordisk developed relating to the IL-21 protein, and are obligated to make milestone payments based on approval and sales and pay single-digit royalties on sales of any resulting products outside North America. In addition, we will pay Novo Nordisk a portion of any third-party license fees above a specified threshold. |
| • | Terminated the collaborative data sharing and cross-license agreement. However, our exclusive license in North America to the intellectual property rights that Novo Nordisk developed relating to the IL-21 protein survived termination. |
| • | Terminated the manufacturing agreements and Novo Nordisk transferred to us all manufacturing processes developed and its existing stock of IL-21 protein. |
In December 2009, the parties further amended and restated the license agreement to provide Novo Nordisk worldwide rights to embodiments of IL-21 (other than IL-21 protein), including antibodies to IL-21. Novo Nordisk is obligated to make milestone payments based on the achievement of development milestones and royalties on sales of any resulting products. In addition, we will have a right to co-promote the IL-21 mAb product in the United States and receive double-digit royalties on net sales in the United States if we contribute to Phase 3 clinical development costs directed to achieving regulatory approval in the United States or European Union. We must make an election to contribute to Phase 3 clinical development costs within 90 days following receipt of data from the Phase 2b studies, in which case we will pay a one-time fee of $10 million and 15% of Phase 3 clinical development costs. Royalty obligations under the agreement continue on a country-by-country basis until the date on which no valid patent claims relating to a product exist or, if the product is not covered by
18
Table of Contents
a valid patent claim, 10 years from the date of first sale of the product in that country. Royalty payments may be reduced if Novo Nordisk is required to license additional intellectual property from one or more third parties in order to commercialize a product or, in certain circumstances, if product sales suffer from direct competition.
Merck Serono Strategic Alliance Agreement
In October 2004, we executed a strategic alliance agreement with Serono S.A. to research, develop and commercialize product candidates, including protein and antibody therapeutics, based on a specific portfolio of our proprietary genes. Following the acquisition of Serono in 2007 by Merck KGaA, Serono’s rights under this agreement have been held by Merck Serono SA, an affiliate of Merck KGaA. In August 2008, we amended the strategic alliance agreement, while retaining the original five-year term, which expired in October 2009.
At the time of the original agreement, we executed agreements granting Merck Serono exclusive worldwide licenses to two preclinical candidates, FGF-18 and IL-22RA mAb, and entered into a co-development agreement relating to IL-31 mAb. Subsequently, in June 2007, we entered into a co-development agreement with Merck Serono for the IL-17RC soluble receptor, under the strategic alliance agreement. In connection with the original agreement we received a $20.0 million upfront option fee and $11.25 million in license fees for FGF-18, IL-22RA mAb and IL-31 mAb. In addition, Merck Serono purchased approximately 3.2 million shares of our common stock for a total of $50.0 million, and entered into a related lockup agreement and a standstill agreement.
In connection with the 2008 amendment of the strategic alliance agreement, we amended and restated the co-development agreements for IL-31 mAb and IL-17RC soluble receptor to provide for exclusive licenses to, in the case of IL-31 mAb, ZymoGenetics and, in the case of IL-17RC soluble receptor, Merck Serono. Pursuant to these exclusive licenses we will receive (in the case of IL-17RC soluble receptor) and pay (in the case of IL-31 mAb):
| • | Potential milestone payments related to development progress, regulatory submissions and approvals for product candidates. |
| • | Royalties on worldwide sales of licensed products. The licensee is required to pay royalties on sales, which vary based on annual sales volume and the degree of patent protection provided by the licensed intellectual property. Royalty payments may be reduced if the licensee is required to license additional intellectual property from one or more third parties in order to commercialize a product or, in certain circumstances, if product sales suffer from direct competition. Royalty obligations under the agreements continue on a country-by-country basis until the date on which no valid patent claims relating to a product exist or, if the product is not covered by a valid patent claim, 15 years from the date of first sale of the product in that country. |
In addition, these exclusive licenses provide that if the licensee (whether us or Merck Serono) seeks a partner for the applicable product candidate, the licensor will have the right to opt in to co-develop and co-commercialize the product candidate on pre-negotiated terms, including retroactive and prospective cost sharing, royalties and milestone fees. In addition to having co-development and co-commercialization rights within the United States, Merck Serono would have an exclusive license outside of the United States whether Merck Serono opts in to develop a product candidate of ours or we opt in to develop a product candidate of Merck Serono.
Patents and Proprietary Rights
We seek appropriate patent protection for our proprietary technologies and product candidates by filing patent applications in the United States. We have more than 340 unexpired issued or allowed U.S. patents, and over 180 pending U.S. patent applications. When appropriate, we also seek foreign patent protection and to date have more than 790 issued or allowed foreign patents.
19
Table of Contents
Our patents and patent applications are primarily directed to therapeutic protein-based products. We commonly seek claims directed to compositions of matter for genes and proteins, including antibodies, methods of using and methods of making. When appropriate, we also seek claims to related technologies, such as reagents used in release assays and formulations. We maintain patents and prosecute applications, worldwide, for technologies that we have outlicensed. Similarly, for development projects that are partnered, we work closely with our development partners to coordinate patent efforts, including filings, prosecution, term extension, defense and enforcement. As our development product candidates advance through research and development, we seek to diligently identify and protect new inventions, such as combinations, improvements to methods of manufacturing or purification, and methods of treatment. We also work closely with our scientist personnel to identify and protect new inventions that could eventually add to our development pipeline.
Patents expire, on a country by country basis, at various times depending on various factors, including the filing date of the corresponding patent application(s), the availability of patent term extension and supplemental protection certificates and terminal disclaimers. For our commercial product and each of our product candidates, we have filed or expect to file multiple patent applications and expect to obtain multiple patents. The table below provides expected dates for the first patent expiration in patent portfolios for our commercial product, RECOTHROM, and product candidates in our development pipeline. Each expiration date may be subject to patent term extension, where the length of term extension would not exceed five years under current law and depends on factors such as the amount of time taken by the FDA to review the first marketing approval application of a drug covered by the patent.
| Commercial Product/Product Candidate | First Patent Expiration Date | |
| RECOTHROM | December 2012; expected to extend until July 2015 under patent term extension | |
| PEG-IFN-lambda | September 2021 | |
| IL-21 | March 2020 | |
| IL-31 mAb | January 2023 |
We require our scientific personnel to maintain laboratory notebooks and other research records in accordance with our policies, which are designed to strengthen and support our patent efforts. In addition to our patented intellectual property, we also develop and seek to protect unpatented technology, trade secrets and confidential information, including our genetic sequence database, bioinformatics algorithms, research, preclinical and clinical data, development and manufacturing strategies. Our policy is to require our employees, consultants and advisors to execute a confidentiality and proprietary information agreement before beginning their employment, consulting or advisory relationship with us. These agreements generally provide that the individual must keep confidential and not disclose to other parties any confidential information developed or learned by the individual during the course of their relationship with us except in limited circumstances. These agreements also generally provide that we shall own all inventions conceived by the individual in the course of rendering services to us. These agreements, however, may not provide effective protection of our technology, confidential information or, in the event of unauthorized use or disclosure, may not provide adequate remedies.
As part of our business strategy, we often work with third parties in our research and development activities. Accordingly, disputes may arise about inventorship, ownership and corresponding rights to know-how and inventions resulting from the joint creation or use of intellectual property by us and our corporate partners, licensors, licensees, scientific collaborators and consultants. In addition, other parties may circumvent any proprietary protection we do have. These parties may independently develop equivalent technologies or independently gain access to and disclose substantially equivalent information, and confidentially agreements and material transfer agreements we have entered into with them may not provide us with effective protection.
Refer to “Item 1A. Risk Factors” for additional information relating to our patents and proprietary rights.
20
Table of Contents
Government Regulation
Regulation by government authorities in the United States, Canada, Europe, Japan and other countries is a significant consideration in our ongoing research and product development activities and in the manufacture and marketing of our products and product candidates. Both before and after the approval of our products and product candidates, we, our products, our product candidates, our operations, our facilities, our suppliers, and our contract manufacturers, contract research organizations, and contract testing laboratories are subject to extensive regulation by government authorities in the United States and other countries. The FDA and comparable regulatory bodies in other countries currently regulate therapeutic proteins and related pharmaceutical products as biologics. Biologics cannot be lawfully marketed in the United States without FDA approval. Biologics are subject to extensive pre- and post-market regulation by the FDA, including regulations that govern the collection, testing, manufacture, safety, efficacy, potency, labeling, storage, record keeping, advertising, promotion, sale and distribution of the products. The time required for completing testing and obtaining approvals of our product candidates is uncertain and varies among product candidates for several reasons, but the process takes many years. In addition, we may encounter delays in product development or rejections of product applications due to changes in FDA or foreign regulatory laws or policies during the period of product development and testing. Failure to comply with regulatory requirements may subject us to, among other things, warning letters, civil penalties and criminal prosecution; fines and other monetary penalties; restrictions on product development and production; suspension, delay, rejection, or withdrawal of approvals; injunctions; and the seizure or recall of products. The lengthy process of obtaining regulatory approvals and ensuring compliance with appropriate statutes and regulations requires the expenditure of substantial resources. Any delay or failure, by us or our corporate partners, to obtain regulatory approvals could adversely affect our ability to commercialize product candidates, receive royalty payments and generate sales revenue.
The nature and extent of the governmental pre-market review process for our potential products will vary, depending on the regulatory categorization of particular products. The necessary steps before a new biological product may be marketed in the United States ordinarily include:
| • | nonclinical laboratory and animal tests; |
| • | submission to the FDA of an IND application, which must become effective before clinical trials may commence in the United States; |
| • | completion of adequate and well-controlled human clinical trials to establish the safety and efficacy of the proposed drug for its intended use; |
| • | submission to the FDA of a BLA; |
| • | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance with current Good Manufacturing Practices (cGMP); and |
| • | FDA review and approval of the BLA. |
Nonclinical tests include laboratory evaluation of the product, as well as animal studies to assess the potential safety and efficacy of the product. The results of nonclinical tests, together with extensive manufacturing information, analytical data and proposed clinical trial protocols, are submitted to the FDA as part of an IND application, which must become effective before the initiation of clinical trials. The IND application will automatically become effective 30 days after receipt by the FDA unless before that time the FDA raises concerns or questions and imposes a clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding FDA concerns or questions before clinical trials can proceed. FDA may also impose a clinical hold on an ongoing clinical trial if, for example, safety concerns arise, in which case the trial cannot recommence without FDA authorization.
Clinical trials involve the administration of the product to human subjects under the supervision of qualified personnel. Clinical trials are conducted under protocols that detail the objectives of the trial, inclusion and exclusion criteria, and the parameters to be used to monitor safety and the efficacy criteria to be evaluated.
21
Table of Contents
Protocols for each phase of the clinical trials are submitted to the FDA as part of the original IND application or as an amendment to the IND application. Further, each clinical trial must be reviewed and approved by an independent institutional review board at each participating institution. The institutional review board will consider, among other things, ethical factors and the safety of human subjects, and may approve the protocol as submitted, require changes, or decline to approve it.
Clinical trials generally are conducted in three sequential phases that may overlap. In Phase 1, the potential product is administered to healthy human subjects or patients, or both, to assess safety, metabolism, pharmacokinetics and pharmacological actions, and side effects associated with increasing doses of the drug. Phase 2 usually involves one or more trials to evaluate the efficacy of the potential product for a particular indication or indications in patients with the disease or condition under study, to determine dosage tolerance and optimum dosage, and to further identify possible adverse reactions and safety risks. If a compound appears to be effective and to have an acceptable safety profile in Phase 2 evaluations, Phase 3 trials may be undertaken to evaluate further clinical efficacy, usually in comparison to standard therapies or placebo, generally within a broader patient population with the disease state or condition for which an indication for use will be sought, and at geographically dispersed clinical sites. Phase 1, Phase 2 or Phase 3 testing may not be completed successfully within any specific period of time, if at all, with respect to any of our potential products. Furthermore, we, an institutional review board, the FDA or other regulatory bodies may suspend a clinical trial at any time for any reason, including a finding that the subjects or patients are being exposed to an unacceptable health risk.
The results of pharmaceutical development, nonclinical studies and clinical trials are submitted to the FDA in the form of a BLA for approval of the manufacture, marketing and commercial shipment of the biological product. A BLA contains extensive manufacturing information, and FDA usually inspects each manufacturing facility and quality system to assess compliance with cGMP before approving a BLA. The inspection and approval process require substantial time, effort and resources, and approvals may not be granted on a timely basis, if at all. If FDA determines that the BLA is not acceptable, it may issue a complete response letter outlining the deficiencies and stating what additional information or studies it requires for approval. Notwithstanding the submission of any requested additional testing or information, the FDA ultimately may decide that the application does not satisfy the criteria for approval, and deny the application. If a product is approved, the FDA may limit the indications for which the product may be marketed, require extensive warnings on the product label, impose restrictive distribution or other risk mitigation measures, and /or require post-approval clinical studies. After approval, certain changes to the approved product, such as adding new indications, manufacturing changes, or additional labeling claims, will require submittal of a new BLA or, in some instances, a BLA supplement, for further FDA review and approval.
Some of our product candidates may qualify as orphan drugs under the Orphan Drug provisions of the Food, Drug, and Cosmetic Act. Orphan drug designation must be requested before an application for marketing authorization is submitted. If granted, orphan drug designation applies to a drug for a specific indication and it is possible for more than one drug to receive orphan drug designation for the same indication. If a product which has an orphan drug designation subsequently receives the first FDA approval for the indication for which it has such designation, the product is entitled to orphan exclusivity, which means the FDA may not approve any other application to market the same drug for the same indication for a period of seven years, except in limited circumstances, such as the showing of clinical superiority to the product with orphan exclusivity. The FDA granted IL-21 orphan drug status for the treatment of melanoma patients with advanced or aggressive disease. However, orphan drug designation for IL-21 may not have a positive effect on our revenues even if the product is approved, and it is possible that in the future none of our other product candidates will be designated as an orphan drug by the FDA.
In addition, the FDA regulates the advertising and promotion of biologic products and medical devices, emphasizing areas such as appropriate disclosure of risks, prohibition of off-label promotion, industry-sponsored scientific and educational activities, comparative advertising, and promotional activities involving the Internet.
22
Table of Contents
FDA marketing approval is only applicable in the United States. Marketing approval in foreign countries is subject to the regulations of those countries. The approval procedures vary among countries and can involve additional testing. The time required to obtain approval outside of the United States may differ from that required for FDA approval. There are centralized procedures for filings in the European Union (EU) countries, which allow submission of a single marketing authorization application to obtain approval in the approximately 25 countries of the EU. Outside of the EU, most countries generally have their own procedures and requirements, and compliance with these procedures and requirements may be expensive and time-consuming. Accordingly, there may be delays in obtaining required approvals from foreign regulatory authorities after the relevant applications are filed, if approvals are ultimately received at all.
We are also subject to various federal, state and local laws, regulations, industry guidelines and recommendations relating to employment practices; safe working conditions; laboratory and manufacturing practices; the experimental use of animals; the use and disposal of hazardous or potentially hazardous substances, including radioactive compounds and infectious disease agents; product liability; and unfair competition, including advertising and other promotional efforts. Government regulations that might result from future legislation or administrative action, including additions or changes to environmental laws, may materially affect our business operations and revenues.
Competition
We face competition from a broad range of biotechnology and pharmaceutical companies as well as academic and research institutions. We compete with these entities to develop and obtain proprietary rights to new therapeutic proteins and to commercialize the products we develop from these proteins. Some of our competitors have greater resources and experience than we have in discovering, developing, manufacturing and selling protein-based products. We expect that competition in our field will continue to be intense.
RECOTHROM, which was approved in January 2008 by the FDA for use as a topical hemostat in the United States, faces substantial competition in the topical hemostat market. In addition to RECOTHROM, there are two stand-alone thrombin products currently available in the United States: Thrombin-JMI, a bovine plasma-derived thrombin from King Pharmaceuticals, Inc., and Evithrom, a pooled human plasma-derived thrombin, from Ethicon, Inc., a Johnson & Johnson Company. Also, a number of other topical hemostatic agents are currently available on the market from Johnson & Johnson Wound Management, a division of Ethicon, Inc. and the BioSurgery business unit of Baxter BioScience, including GELFOAM Plus, a gelatin sponge product co-packaged with human-plasma derived thrombin. Furthermore, new products and technologies could be developed in the future to limit or control bleeding during surgeries.
We anticipate that our other product candidates currently in development will face intense competition in their respective therapeutic areas. PEG-IFN-lambda, which we are co-developing with Bristol-Myers Squibb as a potential treatment for hepatitis C virus (HCV), is targeting a market with an established standard of care, i.e., interferon-alpha based therapy plus ribavirin. We believe that PEG-IFN-lambda may be a better alternative to the current standard of care due to the potential for fewer side effects and equivalent or potentially better antiviral activity. However, we would have to compete with two well established interferon-alpha based drugs, Roche’s PEGASYS® and Schering-Plough’s PEG-Intron®. Furthermore, the introduction of a new class of drugs, such as HCV protease inhibitors or polymerase inhibitors or a combination thereof, may result in transition to a different treatment regimen and, if this regimen excludes an interferon component, may represent an additional competitive threat to our product. Two HCV protease inhibitors, Vertex’s telaprevir and Schering-Plough’s boceprevir, are currently in Phase 3 development.
Our other product candidates, such as IL-21 for metastatic melanoma and IL-31 mAb for severe atopic dermatitis, target diseases for which there is a significant unmet medical need; however, this may change with the introduction of new products currently in development by other companies. There are several product candidates currently in Phase 3 development for metastatic melanoma, including ipilimumab, an immunotherapy being developed by Bristol-Myers Squibb, and PLX4032, a targeted therapy being co-developed by Plexxikon Inc. and Roche.
23
Table of Contents
Although we believe that currently we are well positioned to compete effectively with respect to our existing and potential competitors, our ability to compete successfully in the future will depend on many factors, including our ability to:
| • | successfully maintain and expand as appropriate RECOTHROM commercial infrastructure, including the product supply and sales force, and establish commercial infrastructure for other product candidates as necessary; |
| • | develop products that are safer, more efficacious or more convenient to administer than other products in the marketplace; |
| • | leverage our established collaborations and enter into new collaborations to support the development of our products; |
| • | obtain timely regulatory approvals; |
| • | manufacture our products in a cost-effective manner in quantities sufficient to meet market demands; |
| • | obtain adequate reimbursement from government health administration authorities, private health insurers and health maintenance organizations; |
| • | obtain and enforce adequate patent protection for our product candidates and technologies; |
| • | maintain adequate capital levels; and |
| • | attract and retain key personnel. |
Employees
As part of our strategic refocus, we reduced headcount by approximately 33% in April 2009 and by an additional 15% in December 2009. As of December 31, 2009, we had 323 full-time employees, including 176 employees dedicated to research and development and 64 employees dedicated to sales and marketing. Each of our employees had signed confidentiality and intellectual property agreements, and no employees are covered by a collective bargaining agreement. We have never experienced employment-related work stoppages and consider our employee relations to be good.
Website Access to Our SEC Reports
Our Internet address is www.zymogenetics.com. We make our periodic SEC reports (Form 10-Q and Form 10-K), current reports (Form 8-K) and amendments to these reports available free of charge through our website as soon as reasonably practicable after they are filed electronically with the SEC. We may from time to time provide important disclosures to investors by posting them in the investor relations section of our website, as allowed by SEC rules.
You should carefully consider the risks described below together with the other information set forth in this Annual Report on Form 10-K, which could materially affect our business, financial condition or future results. The risks described below are not the only risks facing our company. The risks described below are not the only ones that we face. Additional risks not presently known to us or that we currently deem immaterial may also affect our business, operating results and financial condition.
Risks Related to Our Business
If we fail to increase sales of RECOTHROM® recombinant human thrombin or to successfully develop and commercialize product candidates in our pipeline, we will not meet our financial goals.
24
Table of Contents
Our near-term financial success is highly dependent on our ability to increase revenue from the commercialization of RECOTHROM. The successful commercialization of RECOTHROM will depend on many factors, including the following:
| • | the effectiveness of our product differentiation, marketing, promotion, distribution, sales and pricing strategies and programs, and those of our competitors; |
| • | our ability to maintain effective promotional materials that are acceptable to regulatory officials; |
| • | product demand within the medical community; |
| • | our ability to penetrate the existing thrombin market and develop complementary products; |
| • | the introduction of new alternative topical hemostats that may compete with RECOTHROM and offer desirable features or benefits; |
| • | new data or adverse event information relating to RECOTHROM or any similar products and any resulting regulatory action; |
| • | clinical practice or other guidelines regarding topical hemostats published by professional organizations or specialty groups; |
| • | successfully maintaining a product supply chain to meet demand; |
| • | successfully maintaining a commercial infrastructure, including a sales force; and |
| • | the ability to gain formulary acceptance and favorable formulary positioning in a timely fashion or at all. |
Part of our business strategy involves developing and commercializing new products, such as PEG-IFN-lambda, IL-21 and IL-31 monoclonal antibody (IL-31 mAb), at times through entry into strategic alliances and collaborations. This part of our strategy requires us or our collaborators to conduct clinical trials to support approval of these new products. The success of this component of our strategy will depend on the outcome of these clinical trials, the success of our collaborators, the content and timing of our submissions to regulatory authorities and whether and when those authorities determine to grant approvals. The other element of our business strategy is maximizing the commercial opportunity for RECOTHROM. If we fail to significantly increase sales of RECOTHROM, our ability to successfully complete development and commercialization activities for our other product candidates, and our ability to become profitable in the future, will be adversely affected.
We anticipate incurring additional losses and may not achieve profitability.
As of December 31, 2009, we had an accumulated deficit of $805.2 million. We expect to continue to incur significant losses over the next several years, and we may never become profitable. Although we began generating RECOTHROM sales revenue in 2008, it will be a number of years before we generate revenues from sales of other product candidates, if ever. Our revenues from the sales of RECOTHROM and existing collaborative and licensing arrangements are currently insufficient to fund our operating expenses, and we may never generate revenues sufficient to fund these expenses. In addition, we will continue to incur substantial expenses relating to our product development and commercialization efforts. The development and commercialization of our product candidates will require significant further research, development, testing, regulatory approvals and sales and marketing activities. We will continue to incur substantial operating losses for at least the near term as we continue to support the commercialization of RECOTHROM and as a result of our development activities for the product candidates in our development pipeline. These losses have had and will have an adverse effect on our shareholders’ equity (deficit) and working capital. Even if we become profitable in the future, we may not remain profitable.
25
Table of Contents
If we fail to obtain or generate the capital we need to fund our operations, we will be unable to continue operations.
Our business does not currently generate the cash needed to finance our operations, and we do not expect it to do so in the foreseeable future. We anticipate that we will continue to expend substantial funds on the development of our product candidates, and the amount of these expenditures may increase in the future. We expect to seek additional funding through public or private financings, including equity financings, credit facilities, or through other arrangements, including collaborative and licensing arrangements. Poor financial results, including sales of RECOTHROM being less than expected, unanticipated expenses or unanticipated opportunities that require financial commitments could give rise to additional financing requirements or require us to obtain additional financing sooner than we expect. However, financing may be unavailable when we need it or may be unavailable on acceptable terms, especially in light of the current global economic conditions. If we raise additional funds by issuing equity or convertible debt securities, the percentage ownership of our existing shareholders will be diluted, and these securities may have rights superior to those of our common stock. If we are unable to raise additional funds when we need them, we may be required to delay, scale back or eliminate expenditures for some of our development or commercial programs. We may also be required to grant rights to third parties to develop and commercialize products and product candidates that we would prefer to develop and commercialize ourselves, and such rights may be granted on terms that are not favorable to us. If we were required to grant such rights, the ultimate value of these products or product candidates to us would be reduced. In addition, if our cash and cash equivalents drop below specified levels it may result in the loss of co-promotion rights and U.S. patent rights under our agreement with Bristol-Myers Squibb relating to PEG-IFN-lambda and constitute an event of default under our June 28, 2008 Facility Agreement with Deerfield Private Design Fund, L.P., Deerfield Private Design International, L.P. and Deerfield ZG Corporation, which would result in the principal and accrued and unpaid interest on the loans made under the agreement to become immediately due and payable.
Additionally, a substantial portion of our operating expenses are funded through our collaborative agreements with third parties. For example, as part of the co-development/co-promotion and license agreement with Bristol-Myers Squibb for PEG-IFN-lambda, we received $200.0 million in 2009. To the extent that we lose collaborative partners for a program or a portion of a program that we do not fund internally, or to the extent that we do not receive the funding that we expect from our collaborative agreements, unless we are able to obtain alternative sources of funding, we would be delayed in or unable to continue developing product candidates under the affected program. In addition, to the extent that funding is provided by a collaborator for non-program-specific uses, the loss of significant amounts of collaborative funding could result in the delay, reduction or termination of additional research and development programs, a reduction in capital expenditures or business development and other operating activities, or any combination of these measures, which would harm our business. Subject to the terms of the relevant agreement, each collaborator has the right to terminate its obligation to provide research funding.
We are focused on a limited number of product candidates, and adverse developments with respect to one or more of those candidates could harm our business.
Our business is focused on a limited number of product candidates. We have discontinued discovery research activities in oncology and immunology and have retained only those research capabilities necessary to support the continued development of PEG-IFN-lambda, IL-21, and our IL-31 mAb product candidate. Our focus on a limited number of internal product candidates means that adverse developments with respect to one or more of these candidates could have a more significant adverse impact on our business than if we maintained a broader portfolio of product candidates.
One of our limited number of internal product candidates relates to therapeutic antibodies where we have limited experience and where we may be unsuccessful developing or commercializing a product.
One of our internal product candidates, the IL-31 mAb, involves therapeutic antibody technology, an area where we have limited experience and we may be unsuccessful in our efforts to develop and commercialize this
26
Table of Contents
product candidate. Moreover, we may be unsuccessful in obtaining adequate, if any, patent coverage for our discoveries and therapeutic antibody products. In addition, third parties may own key technology or dominating patents that may prevent us from developing, manufacturing or commercializing therapeutic antibodies.
For example, we are aware of broad patents owned by others relating to the discovery, development, manufacture, use and sale of recombinant humanized antibodies, recombinant humanized single chain antibodies, recombinant human antibodies, and recombinant human single chain antibodies and other technologies. Our IL-31 mAb product candidate may use or include such technologies. While we are investigating and contemplating entering into agreements with certain third parties in order to gain access to their technology, we have no assurance that a license to a particular technology will be available and such third parties may not be willing to grant licenses to the technology. We may be unable to obtain necessary rights to key technologies needed for the discovery, development, production or commercialization of therapeutic antibodies through licensing agreements on terms attractive to us, if at all. If these licenses are not obtained, we might be prevented from developing IL-31 monoclonal antibodies. If we are unsuccessful in our efforts to obtain needed licenses, our ability to develop and commercialize our IL-31 mAb product candidate could be limited. Any patent infringement or other legal claims that might be brought against us may cause us to incur significant expenses, enjoin our development or commercialization of such products, divert the attention of our management and key personnel from other business concerns and, if successfully asserted against us, require us to pay substantial damages.
We are subject to extensive and rigorous governmental regulation including the requirement of approval before our products may be lawfully marketed.
Both before and after the approval or our product and product candidates, we, our product, our product candidates, our operations, our facilities, our suppliers, and our contract manufacturers, contract research organizations, and contract testing laboratories are subject to extensive regulation by governmental authorities in the United States and other countries, with regulations differing from country to country. In the United States, the FDA regulates, among other things, the pre-clinical testing, clinical trials, manufacturing, safety, efficacy, potency, labeling, storage, record keeping, quality systems, advertising, promotion, sale and distribution of therapeutic products. Failure to comply with applicable requirements could result in, among other things, one or more of the following actions: notices of violation, untitled letters, warning letters, fines and other monetary penalties, unanticipated expenditures, delays in approval or refusal to approve a product candidate; product recall or seizure; interruption of manufacturing or clinical trials; operating restrictions; injunctions; and criminal prosecution. We or the FDA, or an institutional review board, may suspend or terminate human clinical trials at any time on various grounds, including a finding that the subjects are being exposed to an unacceptable health risk. Except for RECOTHROM, none of our product candidates has been approved for sale in the United States. Our product candidates, PEG-IFN-lambda, IL-21, IL-31 mAb, or any subsequent product candidates cannot be lawfully marketed in the United States without FDA approval. Any failure to receive the marketing approvals necessary to commercialize our product candidates could harm our business.
The regulatory review and approval process of governmental authorities, which includes the need to conduct nonclinical studies and clinical trials of each product candidate, is lengthy, expensive and uncertain, and regulatory standards may change during the development of a particular product candidate. We are not permitted to market our product candidates in the United States or other countries until we have received requisite regulatory approvals. For example, securing FDA approval requires the submission of a new drug approval (NDA) or biologics license approval (BLA) application to the FDA. The approval application must include extensive nonclinical and clinical data and supporting information to establish the product candidate’s safety and effectiveness for each indication. The approval application must also include significant information regarding the chemistry, manufacturing and controls for the product. The FDA review process typically takes many years to complete and approval is never guaranteed. If a product is approved, FDA may limit the indications for which the product may be marketed, require extensive warnings on the product labeling, impose restricted distribution programs, require expedited reporting of certain adverse events, or require costly ongoing requirements for post-
27
Table of Contents
marketing clinical studies and surveillance or other risk management measures to monitor the safety or efficacy of the product candidate. Markets outside of the United States also have requirements for approval of drug candidates with which we must comply prior to marketing. Obtaining regulatory approval for marketing of a product candidate in one country does not ensure we will be able to obtain regulatory approval in other countries but a failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory process in other countries. Also, any regulatory approval of any of our products or product candidates, once obtained, may be withdrawn.
The FDA may grant orphan drug designation to a drug intended to treat a “rare disease or condition,” which generally is a disease or condition that affects fewer than 200,000 persons in the United States. Orphan drug designation must be requested before submitting an application for marketing authorization. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. If a product which has an orphan drug designation subsequently receives the first FDA approval for the indication for which it has such designation, the product is entitled to orphan exclusivity, which means the FDA may not approve any other application to market the same drug for the same indication for a period of seven years, except in limited circumstances, such as the showing of clinical superiority to the product with orphan exclusivity. Also, competitors may receive approval of other different drugs or biologics for the indications for which the orphan product has exclusivity.
FDA has increased its attention to product safety concerns in light of recent high profile safety issues with certain drug products, in the United States. Moreover, heightened Congressional scrutiny on the adequacy of the FDA’s drug approval process and the agency’s efforts to assure the safety of marketed drugs has resulted in proposed agency initiatives and new legislation addressing drug safety issues. If adopted, any new legislation or agency initiatives could result in delays or increased costs during the period of product development, clinical trials and regulatory review and approval, as well as increased costs to assure compliance with any new post-approval regulatory requirements. Any of these restrictions or requirements could force us to conduct costly studies.
In addition, we, our suppliers, our operations, our facilities, and our contract manufacturers, our contract research organizations, and our contract testing laboratories are required to comply with extensive FDA requirements both before and after approval of our products. For example, we are required to report certain adverse reactions and production problems, if any, to FDA, and to comply with certain requirements concerning advertising and promotion for our products. Also, quality control and manufacturing procedures must continue to conform to current Good Manufacturing Practices (“cGMP”) regulations after approval, and the FDA periodically inspects manufacturing facilities to assess compliance with cGMP. Accordingly, we and others with whom we work must continue to expend time, money, and effort in all areas of regulatory compliance, including manufacturing, production, and quality control. In addition, discovery of safety issues may result in changes in labeling or restrictions on a product manufacturer or BLA holder, including removal of the product from the market.
Our ability to differentiate RECOTHROM in the marketplace may be limited if FDA challenges our promotional and marketing materials.
FDA has authority to regulate advertising and promotional labeling for RECOTHROM under the Federal Food, Drug, and Cosmetic Act and implementing regulations. In general, that authority requires advertising and promotional labeling to be truthful and not misleading, and marketed only for the approved indications. FDA routinely provides its interpretations of that authority in informal communications and also in more formal communications such as untitled letters or warning letters, and although such communications are not final agency decisions, companies may decide not to contest the agency’s interpretations so as to avoid disputes with FDA, even if they believe the claims to be truthful, not misleading and otherwise lawful. FDA has generally given close scrutiny to claims making a comparison of the attributes of one product to another. We have made such claims for RECOTHROM as a means of differentiating RECOTHROM from competitive products containing bovine thrombin, and are aware that King Pharmaceuticals, Inc. has repeatedly complained to FDA
28
Table of Contents
about those claims. In April 2008, we received an untitled letter from FDA identifying concerns with certain comparative statements contained in a press release relating to antibody formation for RECOTHROM and bovine thrombin. We provided our response to the FDA and received a letter from the FDA confirming that the matter was closed in May 2008. Subsequently, in its response filed in October 2009 to a Citizen Petition we filed with FDA in August 2009 requesting that FDA remove King’s bovine thrombin product from the market in the interest of patient safety, King identified comparative claims from our promotional materials that it believes are false and misleading. We believe our claims, including those challenged by King, are truthful, not misleading, and otherwise lawful, but it is possible that the FDA or other regulatory authorities may disagree with our conclusion. If FDA or other regulatory authorities were to challenge our promotional materials or activities, and if we were to elect not to contest such a challenge or were unable to resolve it in a satisfactory manner, our ability to differentiate RECOTHROM in the marketplace could be adversely affected.
Our success with RECOTHROM will depend on our ability to effectively compete with large and well established competitors who have greater resources and broader product lines than we do.
The biotechnology and pharmaceutical field is extremely competitive, and RECOTHROM faces substantial competition from alternative topical hemostats. In the United States, stand-alone plasma-derived thrombin products on the market include Thrombin-JMI, a bovine plasma-derived thrombin sold by King, and Evithrom, a pooled human plasma-derived thrombin sold by Ethicon, Inc., a division of Johnson & Johnson. In addition, Baxter International, Inc. markets the GELFOAM Plus Hemostasis Kit, which is Pfizer Inc.’s GELFOAM sterile sponge co-packaged with human plasma-derived thrombin. Further, a number of companies, including Johnson & Johnson and Baxter International, Inc., currently market other hemostatic agents that may compete with RECOTHROM, including passive agents such as gelatin and collagen pads and flowable hemostats, as well as fibrin sealants and tissue glues. Many of these alternative hemostatic agents are relatively inexpensive and have been widely used for many years. Consequently, physicians and hospital formulary decision-makers may be hesitant to adopt RECOTHROM.
Several companies, some of which have substantial experience and resources, have products or are developing product candidates in the areas we have targeted for our product candidates.
For our product candidates in development, we face competition from other entities involved in the research and development of therapeutic proteins, antibody products and pharmaceuticals, including Human Genome Sciences, Inc., Biolex Therapeutics, Inc., Bristol-Myers Squibb Company, Plexxikon Inc., and Genentech, Inc., among others. A number of our largest competitors are pursuing the development or marketing of pharmaceuticals that address the same diseases that we are pursuing, and it is possible that the number of companies seeking to develop products and therapies for these diseases will increase. We also face competition from entities developing other types of products related to particular diseases or medical conditions, including other biotechnology and pharmaceutical companies, universities, public and private research institutions, government entities and other organizations.
Furthermore, our potential products, if approved and commercialized, may compete against well-established therapeutic protein-based products or well-established antibody products, many of which may be currently reimbursed by government health administration authorities, private health insurers and health maintenance organizations.
Many of our existing and potential competitors have substantially greater research, product development and commercial capabilities and financial, scientific, marketing and human resources than we do. As a result, these competitors may:
| • | succeed in developing therapeutic protein-based products or alternative therapies, earlier than we do; |
| • | obtain approvals for products from the FDA or other regulatory agencies more rapidly than we do; |
29
Table of Contents
| • | obtain patents that block or otherwise inhibit our ability to develop and commercialize our product candidates; |
| • | develop treatments or cures that are safer, more effective, convenient or economical than those we propose to develop; |
| • | devote greater resources to marketing or selling their products; |
| • | introduce products that make the continued development of our potential products uneconomical; |
| • | withstand price competition more successfully than we can; |
| • | negotiate more favorable terms with third-party collaborators, licensees, group purchasing organizations and other large customers; and |
| • | take advantage of acquisitions or other opportunities more readily than we can. |
Because of these and other potential disadvantages, we may be unable to compete effectively with these competitors. All of our product candidates face competition and we expect that competition in our industry will continue to be intense.
Clinical trials may fail to demonstrate the safety and effectiveness of our product candidates, which could delay, limit or prevent their regulatory approval.
Clinical trials involving PEG-IFN-lambda, IL-21, IL-31 mAb or any subsequent product candidates may reveal that those candidates are ineffective, are insufficiently effective given their safety profile, have unacceptable toxicity or safety profiles or have other unacceptable side effects. In addition, data obtained from tests and trials are susceptible to varying interpretations and FDA or other regulatory authorities may interpret the results of our preclinical studies or clinical trials less favorably than we do, which may delay, limit or prevent regulatory approval. Likewise, the results of preliminary studies do not predict clinical success, and larger and later-stage clinical trials may not produce the same results as earlier-stage trials. Frequently, product candidates that have shown promising results in early clinical trials have subsequently suffered significant setbacks in later clinical trials. Similarly, clinical trial results may vary between different arms of a clinical trial for reasons that we cannot adequately explain. In addition, the design of a clinical trial can determine whether its results will support approval of a product and flaws in the design of a clinical trial may not become apparent until the clinical trial is well advanced. We have limited experience in designing clinical trials that have supported the approval of a product and may be unable to do so successfully. Further, clinical trials of potential products often reveal that it is not practical or feasible to continue development efforts. The failure to provide clinical and preclinical data that are adequate to demonstrate to the satisfaction of the regulatory authorities that our product candidates are safe and effective for their proposed use will delay, limit or prevent approval and will prevent us from marketing those products.
If we or others identify previously unknown side effects or safety concerns for RECOTHROM our business could be harmed.
If we or others identify previously unknown side effects, safety concerns, for RECOTHROM or any products perceived to be similar to RECOTHROM, then in any of these circumstances:
| • | sales of RECOTHROM may decrease significantly; |
| • | regulatory approvals for RECOTHROM may be restricted or withdrawn; |
| • | we may decide to, or be required to, initiate a recall or send product warning letters to physicians, pharmacists and hospitals; |
| • | reformulation of the product, additional preclinical or clinical studies, changes in labeling or changes to or re-approvals of manufacturing facilities may be required; |
30
Table of Contents
| • | our reputation in the marketplace may suffer; and |
| • | government investigations and lawsuits, and private party lawsuits, including class action suits, may be brought against us. |
Any of the above occurrences could harm or prevent sales of RECOTHROM, increase our expenses and impair our ability to successfully commercialize RECOTHROM.
Furthermore, now that RECOTHROM is approved in the United States, it is being used in a wider population and in a less rigorously controlled fashion than in clinical studies. It is expected that some patients exposed to RECOTHROM will become sick or die suddenly, that in some or even many of these cases there will not be sufficient information available to rule out RECOTHROM as a contributing factor or cause of sickness or mortality, and that safety reporting from physicians or from us to regulatory authorities may link RECOTHROM to death or other serious adverse effects. As a result, regulatory authorities, healthcare practitioners, third-party payers or patients may perceive or conclude that the use of RECOTHROM is associated with death or other serious adverse effects, any of which could mean that our ability to commercialize RECOTHROM could be adversely affected and our business could be impaired.
We may be required to defend lawsuits and pay damages in connection with alleged or actual harm caused by our products and product candidates.
The design, testing, manufacture and sale of therapeutic products involve an inherent risk of product liability claims and associated adverse publicity, even if the claims arise from use of the product in a manner inconsistent with label or other instructions. In addition, RECOTHROM is and will be used on patients undergoing surgery, where there are significant risks to patients, and adverse outcomes in patients exposed to RECOTHROM could result in lawsuits.
If any of these potential lawsuits against us were to be successful, we may incur significant costs and could be required to make significant modifications to our business. Even if such lawsuits are without merit or otherwise unsuccessful, they could cause adverse publicity, divert management attention and be costly to respond to, and, therefore, could have an adverse effect on our business. Although we maintain product liability and general insurance, our coverage may not be adequate to cover product liability or other claims. We do not know if we will be able to maintain existing or obtain additional product liability insurance on acceptable terms or with adequate coverage against potential liabilities. This type of insurance is expensive and may in the future be unavailable on acceptable terms, if at all. Any product liability claims, whether or not ultimately successful, could have a negative effect on our reputation, stock price, ability to penetrate the market and sell our products and our financial condition and results.
Guidelines, recommendations, codes and other literature published by various organizations, including competitors, may affect our ability to effectively promote and sell RECOTHROM.
Various professional societies, industry trade associations, practice management groups, private health/science foundations, and organizations periodically publish guidelines, codes, recommendations and other literature to the healthcare and patient communities. These organizations have in the past made recommendations about RECOTHROM or products that compete with RECOTHROM, such as the treatment guidelines of the Society of Thoracic Surgeons. Competitors may also conduct and publish the results of clinical trials or support the conduct and publishing of other reviews or analyses aimed at diminishing concerns about their own products or indicating advantages over RECOTHROM. We have no control over the content of many of these publications, even those for which we may provide some form of financial support. For example, from time to time, we make medical education grants to organizations that in some cases result in a publication. The content of these publications or independent medical education programs may not be favorable to RECOTHROM and may negatively impact our ability to penetrate the market.
31
Table of Contents
Pending litigation by our primary competitor for RECOTHROM could have an adverse impact on our business.
King Pharmaceuticals, Inc. is our primary competitor in the stand-alone thrombin market, and they have been aggressive in defending their leading position in that market. In November 2009, King and affiliated entities filed a lawsuit against us in federal district court in Greeneville, Tennessee (near King’s headquarters in Bristol, Tennessee), seeking to prevent us from making certain claims relating to Thrombin-JMI and RECOTHROM and from making certain comparative claims regarding King’s products and our RECOTHROM product. King alleges that we have engaged in unfair competition, false advertising, trademark infringement, and related claims under federal law and Tennessee state law. King also filed motions with the District Court seeking temporary restraining orders and preliminary injunctive relief. On December 10, 2009, the judge denied King’s motions for a preliminary injunction. However, the lawsuit continues and will likely not be resolved for some time. In the lawsuit, King is seeking a permanent injunction, as well as monetary damages, and if they ultimately prevail in the litigation our business could be harmed. Further, King’s litigation efforts, even if they are not successful, will require significant management time and attention, potentially diverting focus from direct efforts to secure additional customers for RECOTHROM.
Our patents and patent applications, including those relating to RECOTHROM, may not result in meaningful protection against competitors, provide us with any competitive advantage, or provide adequate protection or rights for new discoveries, and our competitors may commercialize the discoveries we patent or attempt to patent.
While we hold patents to the manufacture of RECOTHROM, our composition of matter patent protection is limited to a key intermediate in the production of recombinant thrombin. Accordingly, we may be unable to prevent other parties from developing alternate methods of manufacturing recombinant thrombin or from selling recombinant thrombin. If a third party sold recombinant thrombin manufactured using an alternate method of manufacturing, it could impair our business. In addition, after FDA approval of RECOTHROM, we filed an application for patent term extension of our relevant U.S. patents, but thus far we have not received confirmation from the U.S. Patent and Trademark Office that the extension will be granted to the extent we requested, or at all. If we are unable to obtain the requested term extension of our RECOTHROM patents, it could limit our ability to stop competitors and could impair our business. Additionally, we are aware of certain U.S. and European patents and patent applications held by third parties relating to thrombin and to methods of manufacture of thrombin and other recombinant proteins. Based on our analyses of these patents, we believe that we do not and will not infringe these patents and that many of the claims of these patents are invalid or unenforceable; however, the patent holders, courts or other governmental or legal entities may conclude that our products, processes or actions in developing, manufacturing or selling RECOTHROM do infringe one or more valid and enforceable claims of these patents. We may seek licenses to such patents if, in our judgment, such licenses are needed. If any licenses are required, we may be unable to obtain any such licenses on commercially favorable terms, if at all. If these licenses are not obtained, we might be prevented from selling RECOTHROM or from using certain of our technologies for the manufacture of RECOTHROM. Our failure to obtain a license to any technology that we may require may harm our business.
We own or hold exclusive rights to many issued U.S. and foreign patents and pending patent applications related to the development and commercialization of RECOTHROM and our product candidates. These patents and applications cover composition-of-matter for genes, proteins, and antibodies, medical indications, methods of use, methods of making, formulations, technologies and other inventions related to therapeutic proteins and antibodies. Our success will depend in part on our ability to obtain and maintain patent protection for our products and product candidates in the United States and other countries.
Although we diligently seek to identify and protect our important discoveries and inventions, we may fail to file timely patent applications. We cannot guarantee that any patents will issue from any pending or future patent applications owned by or licensed to us. Our pending and future patent applications covering products and product candidates may not meet the statutory requirements for patentability, meaning that our applications may
32
Table of Contents
not result in the issuance of any patents, and, if issued, such patents may not be valid or enforceable. Our rights under any patents may not provide us with sufficient protection against competitive products or otherwise cover commercially valuable products or processes. In addition, because patent applications in the United States are maintained in secrecy for eighteen months after the filing of the applications, and publication of discoveries in the scientific or patent literature often lag behind actual discoveries, we cannot be sure that the inventors of subject matter covered by our patents and patent applications were the first to invent or the first to file patent applications for these inventions.
Our patents may not provide us with any competitive advantage. Although we have a number of issued patents, the discoveries or technologies covered by these patents may not have any value. These issued patents may not provide commercially meaningful protection against competitors, nor may they provide all rights necessary to commercialize our products or product candidates. In addition, we may be unable or not allowed to obtain patent term extension or restoration on patents covering our products in a manner that would provide commercially meaningful protection against competitors.
Other parties may have a dominating or blocking patent position covering a composition of matter, or methods of making or using our products or product candidates. In addition, other parties may be able to design around our issued patents or independently develop products having attributes or uses similar or identical to our patented product candidates. The business model of some companies is to “design around” patented marketed protein-based products by altering the amino acid sequence of the marketed product, thereby avoiding the patent, but maintaining functional equivalence. Similarly, it may be easier to develop equivalent versions of monoclonal antibodies and competitive soluble receptors, or receptor antibodies than to develop equivalent versions of the proteins with which they interact because there is often more than one antibody or receptor that can have the same therapeutic effect. Consequently, any of our existing or future patents that cover monoclonal antibodies or soluble receptors may not provide any meaningful protection against competitors. In addition, other parties may discover uses for genes, proteins or antibodies that are different from the uses described in our patents, and these other uses may be separately patentable. If another party holds a patent on the use of a gene, protein or antibody, then even if we hold the patent covering the composition of matter of the gene, protein or antibody itself, that party might prevent us from promoting and selling any product directed to such use. Also, other parties may have patents covering the composition of matter of genes or proteins for which we have patents covering only methods of use or methods of manufacture. Furthermore, our patents on recombinant proteins or their precursors or methods of manufacturing such proteins, such as our patents covering the precursor to RECOTHROM and its method of manufacture, may not prevent competitors from developing other precursors or methods of manufacturing these proteins.
Third parties may infringe our patents and challenge the validity or enforceability of our patents.
Competitors and other third parties may infringe our patents, or use inventions described in our patent applications. It may be difficult or impossible for us to police third party activities and detect such infringement. For example, we may be unable to discover a competitor’s manufacturing process to determine whether it infringes patent claims to a method of manufacture. Patent litigation is very expensive and time-consuming and is a distraction to management and personnel who are needed to supply evidence and support to litigation efforts. Enforcing our patents against third parties may require significant expenditures regardless of outcome. We may incur substantial expenditures in such patent litigation and the outcome of any lawsuit is uncertain.
Additionally, challenges raised in patent infringement litigation initiated by us or by third parties may result in determinations that our patents have not been infringed or that they are invalid, unenforceable or otherwise subject to limitations. Consequently, third parties, including licensees, may be able to use the discoveries or technologies claimed or described in our patents without paying licensing fees or royalties to us, which could diminish the value of our intellectual property.
33
Table of Contents
Moreover, the issuance of a patent is not conclusive as to its scope, validity or enforceability. Third parties, including our competitors and licensees, may initiate proceedings to limit the scope, validity or enforceability of our patents, including but not limited to inter-partes re-examination proceedings in the U.S. Patent and Trademark Office, opposition proceedings in patent authorities outside of the United States, declaratory judgment proceedings in U.S. courts, or in the event a third party independently makes an invention similar to ours, interference proceedings in the U.S. Patent and Trademark Office to determine priority of invention. Likewise, we may initiate inter-partes proceedings to challenge the scope, validity or enforceability of third-party patents. The outcome of any such proceeding is uncertain and could result in judicial determinations that our patents are invalid, limited in scope, not infringed, or unenforceable, which would impair our business. Participating in such proceedings or other challenges, whether initiated by us or by third parties may require significant expenditures and divert the attention of our management and key personnel from other business concerns, which may also impair our business.
Patent protection for protein-based therapeutic products and other biotechnology inventions is subject to a great deal of uncertainty, and if patent laws or the interpretation of patent laws change, our competitors may be able to develop and commercialize products based on our discoveries.
Patent protection for protein-based therapeutic products is highly uncertain and involves complex legal and factual questions. In recent years, there have been significant changes in patent law, including the legal standards that govern the scope of protein and biotechnology patents. Standards for patentability of full-length and partial genes, and their corresponding proteins, are changing. Court decisions have made it more difficult to obtain patents, by making it more difficult to satisfy the requirements of written description, enablement, utility and non-obviousness, have decreased the availability of injunctions against infringers, have decreased the likelihood of proving willfulness, and have increased the likelihood of challenging the validity of a patent through a declaratory judgment action. Taken together, these decisions make it more difficult and costly for us to obtain, license and enforce our patents. In addition, in recent years, several members of the U.S. Congress have made numerous proposals to change the patent statute. These proposals include measures that, among other things, would expand the ability of third parties to oppose U.S. patents, introduce the “first to file” standard to the U.S. patent system, and limit damages an infringer is required to pay. If the patent statute is changed, the scope, validity and enforceability of our patents may be decreased.
There also have been, and continue to be, policy discussions concerning the scope of patent protection awarded to biotechnology inventions. Social and political opposition to patents on genes and proteins may lead to narrower patent protection, or narrower claim interpretation, for genes, their corresponding proteins and inventions related to their use, formulation and manufacture. Patent protection relating to biotechnology products is also subject to a great deal of uncertainty outside the United States, and patent laws are evolving and undergoing revision in many countries. Changes in, or different interpretations of, patent laws worldwide may result in our inability to obtain or enforce patents, and may allow others to use our discoveries to develop and commercialize competitive products, which would impair our business.
We may be subject to patent infringement claims, which could result in substantial costs and liability and prevent us from commercializing our products and product candidates.
Third parties may claim that our products or product candidates, or processes or related technologies infringe their patents. The risk of infringement claims filed against us is likely to increase as we commercialize products or move product candidates closer to commercialization. Furthermore, we may not have identified or analyzed all U.S. and foreign patents that pose a risk of our infringement.
Any patent infringement or other legal claims that might be brought against us may cause us to incur significant expenses, divert the attention of our management and key personnel from other business concerns and, if successfully asserted against us, require us to pay substantial damages. In addition, as a result of a patent infringement suit, we may be forced to stop or delay developing, manufacturing or selling products or product
34
Table of Contents
candidates that are claimed to infringe a third party’s patent unless that party grants us rights to use its intellectual property. We may be unable to obtain these rights on terms acceptable to us, if at all. Even if we are able to obtain rights to a third party’s patented intellectual property, these rights may be non-exclusive, which would allow our competitors to obtain access to the same intellectual property. Ultimately, we may be unable to commercialize our potential products or may have to cease some of our business operations, which could harm our business.
We may be unable to protect our unpatented proprietary technology and information.
In addition to our patented intellectual property, we also rely on trade secrets and confidential information. We may be unable to effectively protect our rights to such proprietary technology or information. Other parties may independently develop or gain access to equivalent technologies or information and disclose it for others to use. Disputes may arise about inventorship and corresponding rights to know-how and inventions resulting from the joint creation or use of intellectual property by us and our corporate partners, licensees, scientific and academic collaborators and consultants. In addition, confidentiality agreements and material transfer agreements we have entered into with these parties and with employees and advisors may not provide effective protection of our proprietary technology or information or, in the event of unauthorized use or disclosure, may not provide adequate remedies.
We rely to a significant extent, and expect to continue to rely, on obtaining and maintaining third-party relationships to assist us in developing and commercializing our product candidates.
We have historically entered into collaboration arrangements with partners to co-develop and co-commercialize products and expect to continue to pursue similar opportunities. To be successful, we must identify and attract partners whose competencies and priorities complement ours. We must enter into collaboration agreements on terms beneficial to us and integrate and coordinate their processes, resources and capabilities with our own on a continuing basis. We may be unsuccessful in entering into collaboration agreements with acceptable partners or negotiating favorable terms in these agreements or maintaining such relationships so as to benefit from them over time. Also, we may be unsuccessful in integrating the resources, processes, capabilities or priorities of these collaborators on a continuing basis. In addition, our collaborators may prove difficult to work with or less skilled than we expected. If we are unsuccessful in our collaborative efforts, our ability to develop and market our product candidates could be limited.
In January 2009, we entered into a co-development/co-promotion and license agreement with Bristol-Myers Squibb, under which we and Bristol-Myers Squibb will co-develop PEG-IFN-lambda and Bristol-Myers Squibb will be solely responsible for commercializing PEG-IFN-lambda outside of the United States. While we believe that we will be able to continue working effectively with our counterparts at Bristol-Myers Squibb, we have limited experience with them and are unable to accurately predict our ultimate ability to collaborate with them.
Collaboration arrangements require close and frequent communications between several different teams within the respective companies, technology transfer, and in general a collaborative sharing of responsibilities for clinical studies and all other development activities. Difficulties in collaboration arrangements could result in lower than expected revenue, delays in development, loss of market opportunities, and significant deterioration in the value of the related product candidate and our company.
With the termination of the co-promotion arrangement with Bayer HealthCare as of the end of 2009, we are solely responsible for commercialization of RECOTHROM in the United States, and we have limited experience with respect to commercial activities.
Bayer HealthCare’s active participation in the promotion of RECOTHROM in the United States ended as of December 31, 2009, and we are now solely responsible for all sales and marketing activities for RECOTHROM in the United States. Our commercial organization was formed only within the past few years, and we are still
35
Table of Contents
developing capabilities in some key areas. In order to compensate for the loss of the Bayer HealthCare promotional effort, we will need to add additional personnel, and our ability to do so on a timely basis and to rapidly integrate the new personnel into our existing organization is uncertain. Accordingly, while we expect to be able to manage the transition effectively, it is possible that it could take some time before we have a fully effective and optimal sales organization in place, and it is possible that some customer relationships for which Bayer HealthCare was primarily responsible could be negatively impacted.
We rely on contract suppliers to manufacture commercial supplies of RECOTHROM and clinical material for our product candidates and, therefore, we may be unable to effectively control production or obtain adequate supplies, particularly in situations where we rely on a sole source of supply, which could cause delays in product manufacturing, subject us to product shortages or reduce product sales.
We rely and expect to continue to rely on contract manufacturers and suppliers over whom we exercise little control and who may not always be motivated to do what is in our best interests. The manufacture and delivery of sufficient quantities of pharmaceutical products is a time-consuming and complex process. In order to successfully commercialize our products, including RECOTHROM, and continue to develop our product candidates, including PEG-IFN-lambda, we need to contract or otherwise arrange for the necessary manufacturing. For example, we have entered into an agreement with Abbott Laboratories for commercial-scale production of RECOTHROM bulk drug substance and an agreement with Patheon Italia S.p.A., Inc. for fill and finish of the dosage form of RECOTHROM. We have also entered into agreements with several suppliers of critical raw materials, manufacturing aids, and components for RECOTHROM, some of which are located outside the United States. For our PEG-IFN-lambda product candidate, we will rely on our collaborative partner Bristol-Myers Squibb to manufacture supplies for late-stage clinical trials and, if approved, commercial sales.
Reliance on contract manufacturers, other vendors and collaborators limits our control regarding many aspects of the manufacturing and delivery processes and therefore exposes us to a variety of significant risks relating to the following, particularly in situations where we rely on a sole-source manufacturer, vendor or other collaborator as with RECOTHROM:
| • | our ability to schedule production with contract suppliers when needed to supply market demand or clinical trials; |
| • | reliance on contract suppliers for legal and regulatory compliance and quality assurance; |
| • | contract suppliers’ insistence on exclusivity, minimum and/or maximum levels of supply and related restrictions on our ability to increase or decrease supply, including provisions whereby we pay a penalty if we fail to order a minimum amount; |
| • | breach of agreements by contract suppliers; and |
| • | termination, price increases, or non-renewal of agreements by contract suppliers, based on other business priorities, at times that are costly or inconvenient for us. |
Moreover, these contract manufacturers must comply with FDA’s cGMP requirements. These requirements include quality control, quality assurance, and the maintenance of records and documentation. One or more of these suppliers may be unable to comply with these cGMP requirements and with other FDA, state, or foreign regulatory requirements. A failure to comply with these requirements may result in, among other things, notices of violation, untitled letters, warning letters, fines and other monetary penalties, unanticipated expenditures, delays in approval or refusal to approve a product, suspension of production, product seizure or recall, interruption of manufacturing or clinical trials, operating restrictions, withdrawal of product approval injunctions, and criminal prosecution. If the safety of any quantities supplied of RECOTHROM or any other product we commercialize that is manufactured by a contract party is compromised due to that party’s failure to adhere to applicable laws or for other reasons, we may be unable to obtain regulatory approval for or successfully commercialize our products, which would harm our business.
36
Table of Contents
If any of the circumstances described in these risks occur, our product supply could be interrupted resulting in lost or delayed revenues, delayed clinical trials, and significant increase in production costs and cost of goods. While we seek to negotiate effective remedies in our agreements, we may not have an adequate remedy for all performance-related issues. In particular, terminating a manufacturing arrangement entails significant risks associated with identifying an alternative manufacturer, the length of time it takes for an alternative manufacturer to meet the regulatory requirements and the possibility of litigation arising from any alleged breach.
We and the manufacturers of our products rely on suppliers of raw materials used in the production of our products. Some of these materials are available from only one source and others may become available from only one source. In addition, if, for any reason, we are required to engage an additional, second-source or replacement manufacturer or other vendor, the investment of funds and management time could be significant. There are a limited number of manufacturers and other vendors that operate under the FDA’s cGMP regulations capable of manufacturing for us, and we have not established backup manufacturers and suppliers for RECOTHROM or any of our product candidates. Accordingly, if we are unable to maintain third-party manufacturing on commercially reasonable terms, or if we lose a significant supplier used for RECOTHROM or for our other product candidates, we may be unable to market our products, meet certain contractual supply obligations or complete development of our product candidates on a timely basis, if at all. For example, under our agreements with Bayer Schering Pharma, we are required to provide Bayer Schering Pharma with RECOTHROM and may be in breach of the agreement if we cannot make the required deliveries on time.
In addition, some of the inventions and patents licensed to us were initially developed at universities or other not-for-profit institutions with funding support from an agency of the U.S. government. In accordance with federal law, our licensees or we may be required to manufacture in the United States products covered by those patents, unless we can obtain a waiver from the government on the basis that such domestic manufacture is not commercially feasible. We have not attempted to secure any such waivers from the government, and do not know if they will be sought or available if sought. If we are unable to obtain such waivers, if requested, on a timely basis, we might be forced to seek manufacturing arrangements at higher prices, or on otherwise less favorable terms, than might be available to us in the absence of this domestic manufacturing requirement.
We cannot predict whether any of the manufacturers and vendors that we may use will continue to meet our requirements for quality, quantity or timeliness for the manufacture of RECOTHROM, its intermediates or components or for our other product candidates.
We may be unable to generate any revenue from product candidates developed by collaborators or licensees.
We may be unable to derive any value from product candidates developed by or with collaborators or licensees, including Novo Nordisk, Merck Serono and Bristol-Myers Squibb. Our ability to generate revenues from existing or future collaborations and license arrangements is subject to numerous risks, including:
| • | the possibility that our collaborators or licensees lack sufficient financial, technical or other capabilities to develop these product candidates; |
| • | the possibility that our collaborators or licensees choose to scale back or discontinue their development activities due to changes in their strategies, restructuring, mergers or acquisitions or because their view of the commercial market or regulatory landscape in their licensed territory has changed; |
| • | the length of time that it takes for our collaborators or licensees to solve technical problems or achieve various clinical development and regulatory approval milestones; |
| • | differences in opinion about development, clinical and regulatory strategies and timeframes; |
| • | the inability of collaborators or licensees to successfully address any regulatory or technical challenges they may encounter; and |
37
Table of Contents
| • | the possibility that these product candidates may not be effective or may prove to have undesirable side effects, unacceptable toxicities or other characteristics that preclude regulatory approval or prevent or limit commercial use. |
RECOTHROM has not been approved for sale outside of the United States and Canada and we intend to seek new licensing arrangements with one or more third parties, which we will depend on to seek approval for and market and promote RECOTHROM outside the United States.
In the United States, RECOTHROM was approved for marketing on the basis of clinical studies showing non-inferiority to bovine plasma-derived thrombin. The only other country where RECOTHROM has been approved is Canada. In December 2009, our then licensee outside the United States, Bayer Schering Pharma, withdrew the Marketing Authorization Application for RECOTHROM in Europe, due to concerns raised by European regulatory authorities regarding the extent to which the application met the standards described in their guidance applicable to approval of fibrin sealant products. The European Medicines Agency will likely require an additional clinical trial using a comparator other than bovine thrombin in order to support the approval of RECOTHROM, especially because bovine plasma-derived thrombin is not currently on the market in Europe. In addition, other foreign regulatory authorities may not be satisfied with the safety and efficacy data submitted in support of the foreign applications, which could result in either non-approval or a requirement of additional clinical trials or further analysis of existing data. Furthermore, as an element of the foreign approval process, the applicable regulatory authority must be satisfied with the processes and facilities for all stages of the manufacture, packaging and distribution of RECOTHROM, which may include physical inspections of many or all relevant facilities. Any conclusion that there are shortcomings in the processes, facilities, quality control or oversight of contract manufacturers, or other quality assurance procedures related to manufacture, packaging and distribution of the drug could result in a significant delay in or failure to receive foreign approval.
Effective January 1, 2010, our License and Collaboration Agreement with Bayer Schering Pharma was amended to return to ZymoGenetics all rights to develop and commercialize RECOTHROM in territories outside the United States other than Canada, where Bayer Schering Pharma will commercialize RECOTHROM and pay royalties to ZymoGenetics. No form of thrombin is currently sold in Canada and, therefore, Bayer Schering Pharma will have to create a new market for RECOTHROM, an endeavor in which it may not be successful.
We will seek to establish licensing arrangements with one or more third parties to pursue development and approval of RECOTHROM outside the United States and Canada, but our ability to do so on a timely basis on favorable terms is uncertain, if at all. Even if we are able to identify interested third parties and enter into one or more license arrangements in the near future, the ability of any licensee to obtain approval for RECOTHROM in these territories and to successfully commercialize the product, as well as the timing for these activities, is uncertain.
Failure to effectively manage the RECOTHROM supply chain could result in inventory shortages, supply interruptions or inventory obsolescence.
Our supply chain for RECOTHROM, its intermediates and components is particularly complex and involves a number of third parties on several continents. In addition to coordinating the efforts of these third-party contractors, we must navigate the laws and regulations of multiple jurisdictions, and our failure to do so effectively may negatively impact our business. Failure to adequately manage our supply chain could result in inventory shortages or other supply interruptions that could negatively impact RECOTHROM sales and, consequently, negatively impact product revenue.
We have limited expiration dating for RECOTHROM. Consequently, if we are unable to sell at forecasted levels we may have excess RECOTHROM inventory, resulting in inventory obsolescence, increased costs of product sales and ineffective use of our financial resources.
38
Table of Contents
Because we will depend on third parties to conduct certain laboratory tests, clinical trials and other critical services, we have limited control and may encounter delays in our efforts to develop product candidates.
We commonly rely on third parties to conduct laboratory tests, clinical trials and other critical services for us, especially to the extent clinical trials include sites outside the United States. If we are unable to obtain these services on acceptable terms, we may be unable to complete our product development efforts in a timely manner. Also, to the extent we will rely on third parties for laboratory tests and clinical trials, we will have limited control over these activities or may be unable to manage them appropriately, or may become too dependent on these parties. These third parties may not complete the tests or trials on our schedule, and the tests or trials may be methodologically flawed, may not comply with applicable laws or be otherwise defective. We may experience unexpected cost increases that are beyond our control. Problems with the timeliness or quality of the work of a contract research organization may lead us to seek to terminate the relationship and use an alternative service provider. However, making this change may be costly and may delay our trials, and contractual restrictions may make such a change difficult. Additionally, it may be difficult to find a replacement organization that can conduct our trials in an acceptable manner and at an acceptable cost.
We may expand our business through the acquisition of companies or businesses or in-licensing products or product candidates that could disrupt our business and harm our financial condition.
We may in the future seek to expand our products and capabilities by acquiring one or more companies or businesses or in-licensing one or more product candidates. Acquisitions and in-licenses involve numerous risks, which may include:
| • | substantial cash expenditures; |
| • | dilutive issuances of equity securities; |
| • | incurrence of debt and contingent liabilities, some of which may be difficult or impossible to identify at the time of acquisition; |
| • | difficulties in assimilating the operations of the acquired companies; |
| • | diverting our management’s attention away from other business concerns; |
| • | entering markets in which we have limited or no direct experience; and |
| • | potential loss of our key employees or key employees of the acquired companies or businesses. |
Historically, we have not expanded our business through acquisition or in-licensing and, therefore, our experience in making acquisitions and in-licensing is limited. Any acquisition or in-license may not result in short-term or long-term benefits to us. We may incorrectly judge the value or worth of an acquired company or business or in-licensed product candidate. In addition, our future success could depend in part on our ability to manage the rapid growth associated with some of these acquisitions and in-licenses. We may be unable to make the combination of our business with that of acquired businesses or companies or in-licensed product candidates work or be successful. Furthermore, the development or expansion of our business or any acquired business or company or in-licensed product candidate may require a substantial capital investment by us. We may not have the necessary funds or they may be unavailable to us on acceptable terms, if at all. We may also seek to raise funds by selling shares of our stock, which could dilute our current shareholders’ ownership interest, or securities convertible into our stock, which could dilute current shareholders’ ownership interest upon conversion.
The failure to attract or retain key management or other personnel could decrease our ability to discover, develop and commercialize potential products.
We depend on our senior executive officers as well as key scientific, management and other personnel. Only a small number of our key personnel are bound by employment agreements, and those with employment agreements are bound only for a limited period of time. Competition for qualified employees is intense among
39
Table of Contents
pharmaceutical and biotechnology companies. The loss of qualified employees, or an inability to attract, retain and motivate the highly skilled employees required for our activities, could hinder our ability to develop and commercialize our product candidates.
Our restructuring activities in 2009 may place a strain on certain of our remaining personnel, and may have unanticipated effects.
Our restructuring in April 2009 resulted in a workforce reduction of approximately 160 employees, and a further restructuring in December 2009 resulted in an additional workforce reduction of approximately 50 employees. Following these workforce reductions, we had approximately 250 employees at our facility in Seattle, Washington. Our restructuring may yield unanticipated consequences such as attrition beyond our planned reduction in workforce. These workforce reductions may place a significant strain on certain of our remaining personnel. As a result, our ability to respond to unexpected challenges may be impaired and we may be unable to take advantage of new opportunities. In addition, certain of the terminated employees possess specific knowledge or expertise, and that knowledge or expertise may prove to have been important to our operations. In that case, their absence may create significant difficulties. Furthermore, this headcount reduction may subject us to the risk of litigation, which could result in substantial costs to us and could divert management’s time and attention away from business operations.
Environmental and health and safety laws may result in liabilities, expenses and restrictions on our operations.
State and federal laws and regulations and those of foreign jurisdictions regarding environmental protection, hazardous substances and human health and safety may adversely affect our business. The use of hazardous substances in our operations exposes us to the risk of accidental releases. If our operations, including those of our third-party service providers and collaborators, result in contamination of the environment or expose individuals to hazardous substances, we could be liable for damages and fines. Future changes to environmental and health and safety laws could cause us to incur additional expenses or restrict our operations. In addition, the site where our principal headquarters and facilities are located has been listed as a contaminated property by the state of Washington due to its previous use by the city of Seattle as an electricity-generating plant. The city of Seattle has agreed to defend us against and indemnify us for any claims that arise from this pre-existing contamination, except to the extent that we caused the claim through our negligence or intentional fault, or to the extent that we contributed to the contamination that is the subject of the claim, caused an increase in the clean-up costs or failed to comply with our obligations under our agreement with the city of Seattle. This indemnity may be insufficient and we may be subject to environmental liabilities or be prohibited from using or occupying some or all of the property as a result of environmental claims.
If we use biological and hazardous materials in a manner that causes contamination or injury or violates laws, we may be liable for damages.
Our research and development activities and clinical trials involve the use of potentially harmful biological materials, as well as hazardous materials, chemicals, and various radioactive compounds. We cannot completely eliminate the risk of accidental contamination or injury from the distribution, use, storage, handling, or disposal of these materials. In the event of contamination or injury, we could be held liable for damages that result, and any liability could exceed our available financial resources. We do not maintain liability insurance coverage for our handling of biological or hazardous materials. We, our collaborative partners, the third parties that conduct clinical trials on our behalf, and our third-party manufacturers are subject to federal, state, local or foreign laws and regulations governing the use, storage, handling, and disposal of these materials and waste products. The cost of compliance with these laws and regulations could be significant. The failure to comply with any of these laws and regulations could result in significant fines and work stoppages and may harm our business.
40
Table of Contents
We are exposed to financial risks related to foreign currency exchange rates.
Some of our costs and expenses are denominated in foreign currencies and, even if they are not as a matter of contract, vendors may seek concessions in the event that their anticipated economic return is impaired by exchange rate fluctuations. Most of our existing foreign expenses are associated with the manufacture of RECOTHROM and global clinical studies. We are primarily exposed to changes in exchange rates with the Euro. When the U.S. dollar weakens against other currencies, the dollar value of the foreign-currency denominated expense increases, and when the dollar strengthens against other currencies, the dollar value of the foreign-currency denominated expense decreases. Consequently, changes in exchange rates, and in particular a weakening of the U.S. dollar, may adversely affect our operating results. We currently do not hedge against our foreign currency exposure.
Our liquidity, capital resources and results of operations may be adversely affected by declines in the value of our investments in marketable securities.
As of December 31, 2009, we had $174.1 million in cash and cash equivalents, and investments in marketable securities. Until required for use in our business, we invest our cash reserves in bank deposits, money market funds, high-grade corporate notes, asset-backed securities and U.S. government instruments. As of December 31, 2009, we held asset-backed securities with an estimated fair value of $7.5 million, which had an original cost of $10.5 million.
We do not intend to purchase any additional asset-backed securities, but our liquidity, capital resources and results of operations may be adversely affected by further declines in the value of our existing investments in asset-backed securities. These investments may be adversely affected by rating downgrades, deterioration in the underlying collateral or bankruptcies affecting the issuers of such securities, whether caused by instability in the global financial markets, lack of liquidity in the credit and capital markets, or other factors.
Risks Related to Our Industry
If the healthcare system, coverage and reimbursement policies or any other healthcare-related regulations change, the prices of our products and product candidates may fall or our potential sales may decline.
In recent years, U.S. government officials have made numerous proposals to change the healthcare system in the United States. These proposals include measures that would limit or prohibit payments for certain medical procedures and treatments or subject the pricing of pharmaceuticals to government control. Government and other third-party payers increasingly have attempted to control healthcare costs by limiting both coverage and the level of reimbursement of newly approved healthcare products. Increasingly, third-party payors have been challenging the prices charged for products. They may also refuse to provide any coverage of uses of approved products for medical indications other than those for which the FDA has granted marketing approval. The government may adopt future legislative proposals, such as price controls on prescription drugs, and federal, state or private payors for healthcare goods and services may take further action to limit payments for healthcare products and services. Our success depends on the acceptance of our products and product candidates by the medical community. If physicians, hospitals and other providers are unable to obtain adequate coverage and reimbursement for procedures using RECOTHROM, they may be less likely to use it and our business would be adversely impacted. In addition, in certain foreign countries, particularly the countries of the European Union, the pricing of prescription pharmaceuticals is subject to governmental control with many of the same types of challenges as in the United States. Any of these factors could limit our ability to successfully commercialize our potential products.
We may face increased competition from lower-priced products re-imported into the United States from Canada and other countries. The current law, enacted in December 2003, allows the importation of drugs from Canada, but only if the Secretary of Health and Human Services certifies that importation will pose no additional risk to the public’s health and safety. To date, no such certifications have been given. Legislative proposals have
41
Table of Contents
been made to change the law to allow importation without any certification. If this or other new legislation or regulations were passed allowing the reimportation of drugs, it could adversely affect the prices of our potential products.
In addition, there has been much discussion regarding the creation of laws permitting “follow-on” or “generic” versions of biologics. While there is not currently an abbreviated approval pathway for biologics as there is with branded drugs, Congress and the FDA are studying the issue and legislation addressing this issue could be passed as a part of the healthcare reform initiative. An abbreviated pathway for “follow-on” biologics may permit the FDA to rely on clinical data submitted by innovator developers like ourselves when evaluating applications filed by sponsors of follow-on biologics and may not require full or any clinical trials, significantly lowering the risks and financial barriers to entry. The approval of “follow-on” biologics could result in new and increased competition, including competition prior to expiration of our patents covering our products, and related litigation. In addition, if “follow-on” or “generic” versions are permitted, “data exclusivity,” the period during which generic manufacturers may not cite the clinical trial results of the innovator, will become critical. Adoption of a relatively short “data exclusivity” period could result in products that, over the life of such product, are less profitable.
Negative public opinion and increased regulatory scrutiny of genetic and clinical research may limit our ability to conduct our business.
Ethical, social and legal concerns about genetic and clinical research could result in additional regulations restricting or prohibiting some of our activities or the activities of our suppliers and collaborators. In recent years, federal and state agencies, congressional committees and foreign governments have expressed interest in further regulating the biotechnology industry. More restrictive regulations could delay or complicate nonclinical studies or clinical trials, or prevent us from obtaining regulatory approvals or commercializing any products. In addition, animal rights activists may protest our use of animals in research and development and may attempt to disrupt our operations, which could cause us to incur significant expenses and distract management attention from other business concerns.
The marketing and sale of pharmaceutical products and biologics is subject to extensive regulation and aggressive government enforcement, and our corporate compliance program cannot guarantee that we are in compliance with all relevant laws and regulations.
Our activities relating to the sale and marketing of RECOTHROM and any other products we commercialize will be subject to extensive regulation under the U.S. Federal Food, Drug and Cosmetic Act and other federal statutes and associated regulations. These laws and regulations limit the types of marketing claims and other communications we can make regarding marketed products. We are also subject to various U.S. federal and state laws pertaining to healthcare “fraud and abuse,” including anti-kickback and false claims laws. Anti- kickback laws prohibit payments of any kind intended to induce physicians or others either to purchase or arrange for or recommend the purchase of healthcare products or services, including the selection of a particular prescription drug. These laws make certain business practices that are relatively common in other industries illegal in our industry. False claims laws prohibit anyone from knowingly and willingly presenting, or causing to be presented for payment to third-party payors, including Medicare and Medicaid, claims for reimbursed drugs or services that are false or fraudulent. The government has asserted very broad interpretations of these laws against pharmaceutical manufacturers, even though these manufacturers did not directly submit claims for reimbursement to government payors. In addition, regulation is not static and regulatory authorities, including the FDA, evolve in their staff, interpretations and practices and may impose more stringent requirements than currently in effect, which may adversely affect our sales and marketing efforts. In addition, it is possible that action by federal or state regulatory authorities, such as the FDA, or private legal actions related to our sales and marketing efforts could result in additional investigations or legal actions by state attorneys general. Violations of the above laws may be punishable by criminal and/or civil sanctions, including fines and civil monetary penalties, as well as the possibility of exclusion from federal healthcare programs, including Medicare and
42
Table of Contents
Medicaid. Many pharmaceutical and biotechnology companies have in recent years been the target of lawsuits and investigations, by both federal and state governmental authorities, alleging violations of government regulation, including claims asserting violations of the federal False Claims Act, the federal anti-kickback statute, state consumer protection statutes and other violations in connection with off-label promotion of products, pricing, and government price reporting. While we will strive to comply with these complex requirements, the interpretation of these laws as applied to particular sales and marketing practices continues to evolve, and it is possible that our sales and marketing practices might be challenged. Further, although we have taken measures to prevent potential challenges, including through our corporate compliance program, we cannot guarantee that such measures will protect us from future challenges, lawsuits or investigations. Even if such challenges are without merit, they could cause adverse publicity, divert management attention and be costly to respond to, and thus could have a material adverse effect on our business, including impact on our stock price. In addition, our strategic partners and licensees are required to comply with comparably complex requirements in jurisdictions outside the United States.
In order to sell RECOTHROM to federal institutions, such as military hospitals and the Veterans Administration, we must satisfy the requirements of listing on the Federal Supply Schedule and we are required to periodically report product pricing-related information. The calculations used to generate the pricing-related information are complex. If we fail to accurately and timely report product pricing-related information or to comply with any of these or any other laws or regulations, various negative consequences could result, including criminal and/or civil prosecution, substantial criminal and/or civil penalties, exclusion of the approved product from coverage under governmental healthcare programs (including Medicare and Medicaid), costly litigation and restatement of our financial statements. In addition, our efforts to comply with this wide range of laws and regulations are, and will continue to be, time-consuming and expensive.
Risks Related to Ownership of Our Stock
We may issue additional equity securities in the future, which may result in dilution to existing investors.
We believe that our existing cash, cash equivalents and short-term investments will be sufficient to meet our projected operating requirements through at least the next 24 months. However, we may need to raise additional capital in the future. Any additional financing we undertake could impose covenants upon us that restrict our operating flexibility and, to the extent such capital is raised through our issuance of additional equity securities, our then existing shareholders may experience dilution or the new securities may have rights senior to those of our common stock. In addition, to the extent outstanding stock options are exercised, there will be further dilution to investors.
Our operating results are subject to fluctuations that may cause our stock price to decline.
Our operating results have fluctuated in the past and are likely to continue to do so in the future. Our revenues have been unpredictable and could fluctuate due to slow or erratic uptake of RECOTHROM sales or the timing of licensing fees or the achievement of milestones under new or existing licensing and collaborative arrangements. In addition, our expenses may fluctuate from quarter to quarter due to the timing of expenses, particularly with respect to contract manufacturing and clinical and nonclinical testing.
Accordingly, we believe that period-to-period comparisons of our past operating results are not good indicators of our future performance and should not be relied on to predict our future operating results. It is possible that in the future our operating results in a particular quarter or quarters will not meet the expectations of securities analysts or investors, causing the market price of our common stock to decline, perhaps substantially.
Our stock price is volatile and subject to many factors beyond our control.
The market price of our common stock may fluctuate significantly in response to many factors beyond our control, including:
| • | changes in the recommendations of securities analysts or changes in their financial estimates of our operating results; |
43
Table of Contents
| • | recommendations or opinions of journalists, media personalities or market commentators; |
| • | failures in meeting performance expectations of securities analysts or investors; |
| • | acts or omissions of our licensees, collaborators and suppliers; |
| • | changes in the political climate and changes in or uncertainties about federal and state legislation, policies, and programs affecting healthcare and pharmaceuticals; |
| • | fluctuations in the valuations of companies perceived by securities analysts or investors to be comparable to us; and |
| • | share price and volume fluctuations attributable to inconsistent trading volume levels of our shares. |
Furthermore, the stock markets often experience significant price and volume fluctuations that affect the market prices of equity securities of many companies. In particular, there have been high levels of volatility in the market prices of securities of biotechnology companies. These fluctuations often have been unrelated or disproportionate to the operating performance of those companies. These market and industry fluctuations, as well as general economic, political and market conditions such as recessions, interest rate changes or international currency fluctuations, may negatively impact the market price of our common stock.
Certain of our shareholders have significant control of our management and affairs, which they could exercise against other shareholders’ best interests.
Novo Nordisk, together with Warburg Pincus Equity Partners, L.P., beneficially owned an aggregate of approximately 43.7% of our outstanding common stock as of December 31, 2009, with Novo Nordisk beneficially owning approximately 30.1% and Warburg beneficially owning approximately 13.6%. Four of the nine members of our board of directors are representatives or designees of these shareholders pursuant to a shareholders’ agreement. Novo Nordisk, acting independently or together with Warburg, has the ability to significantly influence our management and affairs and matters requiring shareholder approval, including the election of directors and approval of corporate strategy and significant corporate transactions, such as mergers, consolidations or the sale of substantially all of our assets. Consequently, Novo Nordisk, acting independently or together with Warburg, may be able to cause a change in control, as well as delay or prevent a change in control. They may also discourage a potential acquirer from making a tender offer or otherwise attempting to effect a change in control, even if such a change in control would benefit our other shareholders.
Sales of shares of common stock by our significant shareholders could cause the price of our common stock to decline.
Novo Nordisk and Warburg Pincus Equity Partners, L.P. entered into lock-up agreements in connection with the underwritten public offering of our common stock that occurred in January 2010, pursuant to which each has agreed not to sell or transfer any shares of our common stock. These agreements expire on March 8, 2010. Collectively, these shareholders beneficially owned approximately 44% of our outstanding common stock, as of December 31, 2009, and we have previously filed a registration statement covering the resale of the majority of the shares they own. The expiration of the lock-up agreements, or the sale by either shareholder of a substantial number of shares of our common stock or the perception among investors that these sales may occur, could result in the decline of the market price of our common stock.
Provisions in Washington law, our charter documents and executive employment agreements we have entered into may prevent, discourage or delay a change in control.
We are subject to the Washington laws regulating corporate takeovers, which, with limited exceptions, prohibit a “target corporation” from engaging in certain “significant business transactions” for a period of five years after the share acquisition by an acquiring person, unless (i) the prohibited transaction or the acquiring person’s purchase of shares was approved by a majority of the members of the target corporation’s board of directors prior to the acquiring
44
Table of Contents
person’s share acquisition or (ii) the prohibited transaction was both approved by the majority of the members of the target corporation’s board and authorized at a shareholder meeting by at least two-thirds of the outstanding voting shares (excluding the acquiring person’s shares) at or subsequent to the acquiring person’s share acquisition. An “acquiring person” is defined as a person or group of persons that beneficially owns 10% or more of the voting securities of the target corporation. Such prohibited transactions include, among other things:
| • | certain mergers or consolidations with, dispositions of assets to, or issuances of stock to or redemptions of stock from, the acquiring person; |
| • | termination of 5% or more of the employees of the target corporation as a result of the acquiring person’s acquisition of 10% or more of the shares; |
| • | allowing the acquiring person to receive any disproportionate benefit as a shareholder; and |
| • | liquidating or dissolving the target corporation. |
After the five-year period, certain “significant business transactions” are permitted, as long as they comply with certain “fair price” provisions of the Washington statute or are approved by a majority of the outstanding shares other than those of which the acquiring person has beneficial ownership. A corporation may not “opt out” of this statute.
As such, these laws could prohibit or delay mergers or a change in control and may discourage attempts by other companies to acquire us.
In addition, our articles of incorporation and bylaws contain provisions, such as undesignated preferred stock and prohibitions on cumulative voting in the election of directors that could make it more difficult for a third party to acquire us without the consent of our board of directors. Also, our articles of incorporation provide for a staggered board, removal of directors generally only for cause and certain requirements for calling special shareholder meetings. Further, our bylaws require advance notice of shareholder proposals and nominations and impose restrictions on the persons who may call special shareholder meetings. These provisions may have the effect of preventing or hindering any attempts by our shareholders to replace our current board of directors or management.
We may become involved in securities class action litigation that could divert management’s attention and harm our business.
The stock markets have from time to time experienced significant price and volume fluctuations that have affected the market prices for the common stock of biopharmaceutical companies. These broad market fluctuations may cause the market price of our common stock to decline. In the past, securities class action litigation has often been brought against a company following a decline in the market price of its securities. This risk is especially relevant for us because biotechnology and biopharmaceutical companies have experienced significant stock price volatility in recent years. We may become involved in this type of litigation in the future. Litigation often is expensive and diverts management’s attention and resources, which could adversely affect our business.
Item 1B. Unresolved Staff Comments
None.
We are headquartered in Seattle, Washington, where we lease space in several buildings in close proximity to one another. We lease a total of 271,000 square feet in these buildings, as shown in the following table.
| Property |
Square Feet | Use |
Lease Expiration Dates | |||
| Lake Union Steam Plant |
106,000 | Laboratories and offices | May 2019 | |||
| Earl Davie Building |
98,000 | Laboratories, manufacturing and offices | May 2019 | |||
| 1144 Eastlake Building |
67,000 | Offices | April 2019 |
45
Table of Contents
Effective March 2008, we consolidated the existing lease and sublease agreements for the 1144 Eastlake Building into a single lease, under which the lease term was extended to April 2019. Following the reduction of our workforce in 2009, we have ceased using certain space in this building and intend to sublease the unused space. We believe that our existing facilities, excluding the subleased space, will be adequate to fulfill our needs for the foreseeable future.
For additional details on our headquarter lease, refer to “Note 7. Lease Obligation” under Notes to Consolidated Financial Statements.
On November 2, 2009, King Pharmaceuticals, Inc. and affiliated entities (Monarch Pharmaceuticals, Inc., King Pharmaceuticals Research and Development, Inc., and GenTrac, Inc.), or, collectively, King, filed suit against us in the United States District Court for the Eastern District of Tennessee, naming as defendants ZymoGenetics, Inc. and fifty unnamed individuals. King alleges that we have engaged in unfair competition, false advertising, trademark infringement, and related claims under federal law and Tennessee state law. King seeks various forms of relief, including damages and permanent injunctive relief precluding us from making certain representations regarding King’s products and our RECOTHROM product. King also filed motions with the District Court seeking temporary restraining orders and preliminary injunctions with respect to certain comparative advertising claims, use of King trademarks as Google ad words, and certain alleged statements regarding the existence of lawsuits against King. On December 10, 2009 the court denied all three motions for preliminary injunction in their entirety. We dispute the allegations of wrongdoing in King’s complaint and will continue to vigorously defend ourselves in this matter.
Item 4. Submission of Matters to a Vote of Security Holders
On November 10, 2009, we held a Special Meeting of Shareholders to consider a proposal to approve a voluntary stock option exchange program for eligible employees, including executive officers, as set forth in the definitive proxy statement for a Special Meeting of Shareholders filed on October 20, 2009. The stock option exchange program was approved by shareholders at the Special Meeting of Shareholders. Of the votes cast on this matter, 43,884,835 votes were cast for, 10,903,960 votes were cast against; 12,180 votes abstained; and there were 14,460,307 broker non-votes. The offer to exchange expired at 9:00 p.m., Pacific Time on Monday, December 14, 2009. Pursuant to the offer to exchange, eligible stock options to purchase an aggregate of 3,260,763 shares of our stock were tendered, representing 54.7% of the total eligible stock options. All surrendered options were cancelled and in exchange, on December 16, 2009, we granted awards of new stock options to purchase an aggregate of 1,629,632 shares of our common stock under the 2001 Stock Incentive Plan, in accordance with the terms of the offer to exchange. The exercise price of the new stock options is $6.35 (the closing price of our common stock on December 16, 2009 as reported by Nasdaq).
46
Table of Contents
Item 5. Market for Registrant’s Common Equity, Related Shareholder Matters and Issuer Purchases of Equity Securities
Our common stock began trading on the NASDAQ Stock Market under the symbol ZGEN on February 1, 2002. As of February 10, 2010, we had 90 shareholders of record and approximately 7,600 beneficial holders of our stock. We have never paid cash dividends and do not anticipate paying them in the foreseeable future.
The following table sets forth, for the fiscal periods indicated, the range of high and low closing sales prices of our common stock as quoted on the NASDAQ Global Market:
| High | Low | |||||
| Year Ended December 31, 2009 |
||||||
| 1st Quarter |
$ | 5.62 | $ | 2.89 | ||
| 2nd Quarter |
4.85 | 3.50 | ||||
| 3rd Quarter |
6.47 | 4.27 | ||||
| 4th Quarter |
7.28 | 4.65 | ||||
| Year Ended December 31, 2008 |
||||||
| 1st Quarter |
$ | 13.05 | $ | 8.57 | ||
| 2nd Quarter |
10.50 | 7.64 | ||||
| 3rd Quarter |
9.10 | 6.66 | ||||
| 4th Quarter |
6.42 | 2.29 | ||||
The graph on the next page compares the cumulative total shareholder return on our common stock with the cumulative total shareholder return of the CRSP Total Return Index for The NASDAQ Stock Market (U.S. Companies) and the NASDAQ Biotechnology Index, for the period beginning January 1, 2005 and ending on December 31, 2009 (assuming the investment of $100 in our common stock and in each of the other indices on January 1, 2005 and reinvestment of all dividends).
47
Table of Contents
The comparisons in the graph below are based on historical data and are not intended to forecast the possible future performance of our common stock.
| 1/1/05 | 12/31/05 | 12/31/06 | 12/31/07 | 12/31/08 | 12/31/09 | |||||||||||||
| ZymoGenetics, Inc. |
$ | 100.00 | $ | 73.96 | $ | 67.70 | $ | 50.74 | $ | 13.04 | $ | 27.78 | ||||||
| NASDAQ Stock Market (U.S.) |
100.00 | 102.13 | 112.19 | 121.68 | 58.64 | 84.28 | ||||||||||||
| NASDAQ Biotechnology Index |
100.00 | 102.84 | 103.89 | 108.65 | 94.93 | 109.77 | ||||||||||||
48
Table of Contents
Item 6. Selected Financial Data
The following selected financial data should be read in conjunction with the financial statements and notes to the financial statements and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” contained elsewhere in this Form 10-K.
| Years Ended December 31, | ||||||||||||||||||||
| 2009 | 2008 | 2007 | 2006 | 2005 | ||||||||||||||||
| (in thousands, except per share data) | ||||||||||||||||||||
| Statement of Operations Data: |
||||||||||||||||||||
| Revenues |
$ | 136,972 | $ | 73,989 | $ | 38,477 | $ | 25,380 | $ | 42,909 | ||||||||||
| Costs and expenses: |
||||||||||||||||||||
| Costs of product sales |
7,631 | 5,672 | — | — | — | |||||||||||||||
| Research and development |
99,194 | 126,678 | 142,340 | 128,450 | 99,615 | |||||||||||||||
| Selling, general and administrative |
62,202 | 60,238 | 46,890 | 33,224 | 23,321 | |||||||||||||||
| Total costs and expenses |
169,027 | 192,588 | 189,230 | 161,674 | 122,936 | |||||||||||||||
| Loss from operations |
(32,055 | ) | (118,599 | ) | (150,753 | ) | (136,294 | ) | (80,027 | ) | ||||||||||
| Other income (expense) |
(11,289 | ) | 2,358 | 2,609 | 6,292 | 2,000 | ||||||||||||||
| Loss before income tax benefit |
(43,344 | ) | (116,241 | ) | (148,144 | ) | (130,002 | ) | (78,027 | ) | ||||||||||
| Income tax benefit |
363 | — | — | — | — | |||||||||||||||
| Net loss |
$ | (42,981 | ) | $ | (116,241 | ) | $ | (148,144 | ) | $ | (130,002 | ) | $ | (78,027 | ) | |||||
| Basic and diluted net loss per share |
$ | (0.62 | ) | $ | (1.69 | ) | $ | (2.17 | ) | $ | (1.94 | ) | $ | (1.28 | ) | |||||
| Weighted-average shares used in computing basic and diluted net loss per share |
69,069 | 68,696 | 68,156 | 66,917 | 60,928 | |||||||||||||||
| Balance Sheet Data: |
||||||||||||||||||||
| Cash, cash equivalents and short-term investments |
$ | 174,130 | $ | 89,887 | $ | 170,941 | $ | 258,408 | $ | 366,311 | ||||||||||
| Working capital |
110,053 | 75,988 | 118,822 | 239,432 | 343,459 | |||||||||||||||
| Total assets |
319,296 | 210,046 | 263,081 | 347,004 | 453,353 | |||||||||||||||
| Total shareholders’ equity (deficit) |
(3,958 | ) | 23,359 | 114,830 | 235,684 | 333,663 | ||||||||||||||
49
Table of Contents
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
Overview
We are a biopharmaceutical company focused on developing and commercializing therapeutic protein-based products for the treatment of human diseases. The process for taking one of our product candidates to the marketplace is long, complex and very costly. It is difficult to predict the time it will take to reach the market with any given product candidate, but it would not be unusual to span ten years or more and cost hundreds of millions of dollars. It is also a business of attrition; it is expected that, for the industry as a whole, less than 20% of the drug candidates entering human clinical trials will actually make it to the marketplace. For the products that do make it, particularly for those that address previously unmet medical needs, the markets can be significant, with a number of successful products selling in excess of $1.0 billion per year.
In late 2006, we began preparations for the commercial launch of our first product, RECOTHROM®, which was approved by the FDA on January 17, 2008. In 2007, we hired approximately 60 field personnel and additional headquarters-based personnel to support the commercial operations for selling RECOTHROM. We are incurring substantial marketing costs to penetrate the market and to support our selling efforts. We have also built significant levels of inventory in anticipation of expected demand for the product and to minimize the risk of product shortages. These commercialization activities are utilizing substantial cash resources until such time as RECOTHROM sales reach a level, if ever, that will cover our related costs. We recorded net sales revenue of $29.6 million and $8.8 million in 2009 and 2008, respectively, and we anticipate continued increases in RECOTHROM sales over time; however, we cannot be certain of the future rate of market penetration or when, if ever, our revenues will exceed our related costs.
An important element of our business strategy is that we intend to maintain a significant share of the commercial value for our product candidates under development. As a result, we will be required to pay for a portion of the development and commercialization costs for our partnered product candidates, such as PEG-Interferon lambda (PEG-IFN-lambda), and all of the development and commercialization costs for our internal product candidates, such as IL-21 and IL-31 mAb. In some cases, we may out-license our product candidates, as we have done with atacicept, Factor XIII and IL-21 mAb, for example.
Generating the operating capital necessary to fund our business can be challenging. There are a number of potential sources of revenues and cash that we pursue in order to address our funding needs, including the following:
| • | sales of RECOTHROM, which were $29.6 million in 2009, net of all related discounts and allowances; |
| • | research, development and commercialization collaborations, such as the one we have entered into with Bristol-Myers Squibb for PEG-IFN-lambda, which provide revenues while also enabling us to reduce our ongoing research and development expenses; |
| • | licensing of technologies or product candidates, such as atacicept, recombinant Factor XIII and most recently IL-21 mAb, to other companies, which typically provide license fees and potential milestone payments and royalties on sales; |
| • | issuance of equity or equity-based securities such as the offering of common stock we completed in January 2010; |
| • | debt financing, such as the $100.0 million financing arrangement we entered into with Deerfield Management in June 2008; and |
| • | investment income on our cash reserves. |
We expect that it will be at least several years before we can generate enough product-related revenues for our company to reach net income or cash flow breakeven, and we expect to continue to invest significant amounts of cash in developing our business. In the future, we may pursue additional collaborations or license
50
Table of Contents
transactions related to IL-21 and IL-31 mAb in order to realize the full potential of the product candidate, which would likely generate additional cash and reduce our ongoing related expenses. In addition, we are committed to diligently monitoring our operating cost structure and making changes when necessary, as we did in 2009 through corporate restructurings.
It is possible that we will look for opportunities to raise capital by issuing equity or equity-related securities to help fund our company over the next several years. These opportunities may arise at any time, depending on things such as overall market conditions; dynamics in the biotechnology sector of the market; investor appetite for certain types of companies; and fundamental characteristics of our business. At times, it may be difficult to raise capital on terms favorable to our company, if at all. Accordingly, we would expect to raise capital when it is available, as we did in January 2010, not waiting until there is an immediate need. We believe this strategy is important to minimize the financial risks to our company and our shareholders.
Results of Operations
Revenues
Product sales. We received U.S. marketing approval of RECOTHROM on January 17, 2008 for the 5,000 IU vial configuration and on May 27, 2008 for the 20,000 IU vial configurations. Sales of RECOTHROM are recognized as revenue when the product is shipped and title and risk of loss have passed. Product sales are recorded net of provisions for estimated discounts, rebates, chargebacks and returns. Net product sales were $29.6 million for the year ended December 31, 2009 and $8.8 million for the year ended December 31, 2008. The 2009 amount includes $28.2 million in U.S. product sales and $1.4 million of sales to Bayer to support the Canadian launch following the Canadian marketing approval of RECOTHROM in December 2009. The increase in U.S. product sales in 2009 versus the prior year is due to overall market share increases as additional hospitals converted usage from bovine thrombin to RECOTHROM and existing customers increased their purchase volumes of RECOTHROM. For the fourth quarter of 2009, our estimated overall share of the U.S. market for stand-alone thrombin products was 17%, an increase from 7% for the fourth quarter of 2008.
Royalties. We earn royalties on sales of certain products subject to license agreements with other companies. Royalties were $1.3 million for the year ended December 31, 2009 compared to $6.3 million for both years ended December 31, 2008 and 2007. The decrease for 2009 versus 2008 was primarily due to the discontinuation of a minimum royalty obligation payable by BioMimetic Therapeutics, Inc. in December 2008 on its product GEM 21S and the expiration of royalty rights related to BeneFIX, a product of Wyeth Pharmaceuticals, Inc., in December 2008.
Collaborations and licenses. We enter into various collaborative agreements that may generate license, option or other upfront fees with subsequent milestone payments earned upon completion of development milestones. Where we have no continuing performance obligations under an arrangement, we recognize such payments as revenue when contractually due and payment is reasonably assured, as these payments represent the culmination of a separate earnings process. Where we have continuing performance obligations under an arrangement, revenue is recognized using one of two methods. Where we are able to estimate the total amount of services we must provide under the arrangement, revenue is recognized using a proportional performance model. Costs incurred to date compared to total expected costs are used to determine proportional performance, as this is considered to be representative of the delivery of outputs under the arrangement. Revenue recognized at any point in time is limited to cash received and amounts contractually due. Changes in estimates of total expected costs are accounted for prospectively as a change in estimate. Where we cannot estimate the total amount of service that must be provided, a time-based method is used to recognize revenue. Under the time-based method, revenue is recognized over the arrangement’s estimated performance period, starting with the contract’s commencement, but not before the removal of any contingencies for each milestone. From period to period, license fees and milestone payments can fluctuate substantially based on the completion of new licensing or collaborative agreements and the achievement of development-related milestones.
51
Table of Contents
Bristol-Myers Squibb
In January 2009, we entered into a co-development/co-promotion and license agreement with Bristol-Myers Squibb for the type-3 interferon family, with PEG-IFN-lambda being the lead product candidate. We received a total of $200.0 million under the agreement in 2009, consisting of $105.0 million in license fees and two milestone payments related to the initiation of Phase 2 clinical trials. We have profit sharing and co-promotion rights in the United States and will receive royalties on sales outside of the United States. We are also eligible for sales bonuses based on worldwide sales of licensed products. We granted a license to related technology and are obligated to fund the first $100.0 million of costs for development in the United States and Europe, after which we will be responsible for 20% of such costs. We are recognizing revenue using a proportional performance model and are recording revenue related to the license fees and near-term milestone payments through June 2012, which corresponds to the period over which we anticipate satisfying our performance obligations under the agreement.
Novo Nordisk
In December 2009, we amended our license agreement with Novo Nordisk related to IL-21 antagonists, whereby we added certain intellectual property, expanded their commercial rights to include North America and provided exclusive rights to our IL-21 mAb product candidate. In exchange, Novo Nordisk paid us a $24.0 million upfront payment, which was received in December 2009, and will pay future milestone payments and royalties. We are obligated to perform certain transition activities, which are expected to take approximately six months to complete, and to transfer all related patents and technology to Novo Nordisk who will reimburse us for our internal costs and for all our direct external costs for activities. We do not have any substantive obligations beyond the expected six-month transition period. We are recognizing the initial $24.0 million milestone over the expected six-month transition period from December 2009 to May 2010.
Bayer Schering Pharma AG and Bayer HealthCare LLC
In June 2007, we entered into license and collaboration agreement with Bayer Schering Pharma AG and a U.S. co-promotion agreement with Bayer HealthCare LLC (collectively, Bayer) which were significantly amended in December 2009. The original agreements provided Bayer with an exclusive license to develop and sell rThrombin outside the United States and to co-promote rThrombin in the United States for up to four years. Under the original agreement, we recorded all U.S. sales and Bayer was entitled to commissions and sales bonuses on those sales for up to six years, two years longer than the period Bayer co-promoted in the United States. We received a $30.0 million upfront milestone payment in 2007, a $40.0 million milestone in January 2008 upon the U.S. marketing approval of rThrombin, and $6.5 million upon the initial filings for approval in Canada, Europe, and Asia.
In December 2009, we amended the license and collaboration agreement, whereby Bayer returned all license territories to us except Canada, where they received marketing approval in December 2009. As part of the amendment, Bayer will launch RECOTHROM in Canada during 2010 and we will supply them with U.S. approved product. With regards to the U.S. co-promotion agreement, Bayer discontinued co-promotion effective December 31, 2009 and we agreed to pay Bayer up to $12.0 million in commissions based on net sales over the two-year period ending December 31, 2011. Bayer has no further performance obligations to receive the $12.0 million in commission payments. Immediately preceding the effective date of the amendments, we had deferred revenue and guaranteed U.S. bonus payment accruals totaling $46.4 million. Our only substantive obligations subsequent to the amendments are the U.S. commissions of $12.0 million and the supply of U.S. finished drug product for which we have reliable evidence of fair value. Accordingly, we recognized deferred revenue of $34.4 million in the fourth quarter of 2009, leaving a $12.0 million liability recorded for future commission payments.
Collaboration and license revenues were $106.1 million for the year ended December 31, 2009, reflecting an increase of $47.2 million compared to the prior year. The increase resulted primarily from recognition of $38.7 million related to the Bristol-Myers Squibb agreement for PEG-IFN-lambda, $5.0 million related to the Novo
52
Table of Contents
Nordisk IL-21 mAb agreement, $4.2 million related to the accelerated recognition of deferred revenue resulting from the August 2008 restructuring of our agreements with Merck Serono and $34.4 million in December 2009 related to the amended license and U.S. co-promotion agreement with Bayer. These increases were partially offset by a decrease in Bayer collaboration revenue of $11.6 million recognized under the proportional performance model prior to the December 2009 amendment, license revenue of $21.0 million in 2008 related to the Bristol-Myers Squibb agreement for Ig-fusion; and decreases in milestone revenue related to licenses with Novo Nordisk and BioMimetic Therapeutics, Inc.
Collaboration and license revenues were $58.9 million for the year ended December 31, 2008, reflecting an increase of $26.7 million compared to the prior year. The increase resulted primarily from license fees of $21.0 million related to the Bristol-Myers Squibb agreement for Ig-fusion and an increase in the recognition Bayer collaboration revenue. Partially offsetting these increases were milestones earned in 2007 under our IL-21 and IL-31 agreements with Novo Nordisk for which no comparable amounts were earned in 2008.
Costs and expenses
Costs of product sales. Prior to FDA approval of RECOTHROM in January 2008, all third-party manufacturing costs and an allocation of our labor and overhead associated with the manufacturing of RECOTHROM for commercial sale were expensed as research and development costs as incurred. Subsequent to RECOTHROM approval, these costs are recorded as inventory. Costs of product sales include the inventory and distribution costs associated with RECOTHROM product sales revenue and costs incurred subsequent to FDA approval for product that is not expected to be sold. Accordingly, we expect that costs of product sales will be lower as a percentage of product sales revenue during the time we are selling product manufactured prior to approval.
Costs of product sales for 2009 were $7.6 million compared to $5.7 million in 2008. The 2009 costs of product sales includes $1.3 million for product sales to Bayer related to the Canadian launch of RECOTHROM for which we recorded $1.4 million in product sales revenue. The 2008 amount includes a $4.2 million charge for obsolete product not expected to be sold. Excluding these items, costs of product sales as a percentage of net product sales were 22.4% for the year ended December 31, 2009 as compared to 17.3% for the same period in 2008. The percentage was higher in 2009 compared to 2008 primarily because units sold in 2009 included a higher proportion of manufacturing costs incurred subsequent to FDA approval.
Research and development. Research and development expense consists primarily of salaries and benefit expenses, costs of consumables, facility costs, contracted services and stock-based compensation. These amounts are offset by certain cost reimbursements from collaborators. We evaluate cost reimbursements received under our collaboration arrangements based on the underlying nature of the collaboration. Where the collaboration embodies the principles of cost sharing and revenue sharing, cost reimbursements are recorded as reductions to research and development expenses.
A breakdown of research and development expenses is shown in the following table (in thousands):
| 2009 | 2008 | 2007 | ||||||||||
| Salaries and benefits |
$ | 49,814 | $ | 53,457 | $ | 57,731 | ||||||
| Consumables |
6,318 | 10,907 | 11,658 | |||||||||
| Facility costs |
8,704 | 8,886 | 8,934 | |||||||||
| Contracted services |
24,516 | 47,391 | 55,668 | |||||||||
| Depreciation and amortization |
4,621 | 4,902 | 5,421 | |||||||||
| Stock-based compensation |
7,563 | 13,572 | 13,591 | |||||||||
| Subtotal |
101,536 | 139,115 | 153,003 | |||||||||
| Cost reimbursement from collaborators |
(2,342 | ) | (12,437 | ) | (10,663 | ) | ||||||
| Net research and development expense |
$ | 99,194 | $ | 126,678 | $ | 142,340 | ||||||
53
Table of Contents
Salaries and benefits and consumables generally track with changes in our employee base from year to year. The $3.6 million decrease in salaries and benefits in 2009, as compared to 2008, and the corresponding decrease in consumables were related to the April and December 2009 terminations of 184 research and development employees. We recorded severance related charges of $7.0 million in the second quarter of 2009 and $3.6 million in the fourth quarter of 2009. Excluding these severance-related amounts, salaries and benefits declined by 30.0% for the year ended December 31, 2009, as compared to the prior year, reflecting the impact of the headcount reductions. The $4.3 million decrease in salaries and benefits and the corresponding decrease in consumables in 2008, as compared to 2007, was due to the February 2008 termination of 37 research and development employees and costs related to RECOTHROM manufacturing being included in inventory costs subsequent to the January 17, 2008 FDA approval of RECOTHROM instead of being recorded as research and development expense.
Contracted services include the cost of items such as contract research, contract manufacturing, clinical trials, non-clinical studies and payments to collaborators. These costs relate primarily to clinical development programs and can fluctuate substantially from period to period depending on the stage of our various programs. Generally, these external costs increase as a program advances toward commercialization, but there can be periods between major clinical trials or manufacturing campaigns during which costs decline. Our contracted services costs decreased by $22.9 million for the year ended December 31, 2009, as compared to the prior year, primarily due to the discontinuation of our co-development and co-funding obligations under the atacicept collaboration with Merck Serono, which became effective in August 2008 when we converted to a milestone and royalty bearing agreement. These reductions were partially offset by increased costs related to our PEG-IFN-lambda collaboration with Bristol-Myers Squibb.
Contracted services decreased by $8.3 million in 2008 compared to 2007 due to reduced contract manufacturing costs, reflecting the discontinued expensing of pre-approval manufacturing of RECOTHROM bulk drug and finished product inventory after FDA approval in January 2008. This decrease was partially offset by cost increases for other development programs. Our clinical trial costs increased in 2008 as compared to 2007, primarily reflecting the costs incurred for the atacicept lupus nephritis clinical trial that began in late 2007. Payments to collaborators also increased for the same period primarily reflecting our portion of atacicept development costs under our collaboration with Merck Serono prior to the August 2008 conversion to a milestone and royalty bearing agreement.
Cost reimbursements from our collaborators decreased by $10.0 million for the year ended December 31, 2009, as compared to the prior year, primarily due to the discontinuation of our co-development and cost sharing under the atacicept collaboration with Merck Serono, which became effective in August 2008. Additionally, there was a decrease in cost reimbursements from Bayer during this same period. Cost reimbursements increased by $1.8 million in 2008 compared to 2007 primarily related to work performed under the Bayer agreement.
To date, our business needs have not required us to fully allocate all research and development costs among our various programs. However, we track direct labor, contracted services and certain consumable costs by program, which we monitor to assist us in appropriately utilizing our company resources. We also incur indirect costs that are not allocated to specific programs. These costs include indirect labor, certain consumable costs, facility costs, and depreciation and amortization, all of which benefit all of our research and development programs. The following table presents our research and development costs allocated to clinical development, pre-development and discovery research programs, together with the unallocated costs that benefit all programs for the periods presented (in thousands):
| 2009 | 2008 | 2007 | Inception To Date | |||||||||
| Clinical development programs: |
||||||||||||
| Hemostasis |
$ | 11,101 | $ | 18,708 | $ | 39,690 | $ | 209,831 | ||||
| Autoimmunity and oncology |
6,016 | 30,119 | 25,962 | 118,511 | ||||||||
| Antiviral |
20,840 | 5,094 | 5,125 | 39,777 | ||||||||
| Preclinical and research programs |
15,540 | 22,836 | 22,051 | |||||||||
| Unallocated indirect costs |
45,697 | 49,921 | 49,512 | |||||||||
| Total |
$ | 99,194 | $ | 126,678 | $ | 142,340 | ||||||
54
Table of Contents
The following summarizes the reasons for fluctuations in research and development program costs for the three years presented in the table:
| • | Hemostasis clinical development programs (primarily rThrombin) expenses decreased between 2008 and 2009 due to the completion of a Phase 3b rThrombin clinical trial programs. Additionally, all internal and external RECOTHROM commercial product manufacturing costs were recorded as inventory in 2009, while in 2008, certain of these costs were charged to research and development prior to FDA approval on January 17, 2008. The reduction in expense from 2007 to 2008 was primarily due to $19.0 million of RECOTHROM manufacturing costs being charged to expense in 2007 prior to FDA approval. |
| • | Autoimmunity and oncology clinical development program (atacicept and IL-21) expenses decreased significantly between 2008 and 2009 due to the August 2008 amended atacicept agreement with Merck Serono, whereby the collaboration was converted to a license arrangement and Merck Serono became responsible for all subsequent development costs. The increased expense in 2008 compared to 2007 was primarily due to an increase in our share of costs related to the manufacturing of clinical material and clinical trial activity for atacicept. |
| • | Antiviral clinical development program expenses increased significantly in 2009 compared to 2008 primarily due to an increase in both internal and external costs related to PEG-IFN-lambda clinical trials in our collaboration with Bristol-Myers Squibb. |
| • | Preclinical and research program expenses decreased in 2009 compared to 2008 primarily due to the discontinuation of certain preclinical and research programs following our corporate restructuring in April 2009. |
| • | Unallocated indirect expenses decreased in 2009 as compared to 2008 primarily due to the corporate restructurings that occurred in April and December 2009. |
Selling, general and administrative. Selling, general and administrative expense, which consists primarily of salaries and benefit expenses, professional fees and other corporate costs, increased 3% in 2009 as compared to 2008 and 28% in 2008 as compared to 2007. The increase in 2009 as compared to 2008 was primarily due to an increase in commissions payable to Bayer related to RECOTHROM offset by a decrease in legal expenses. The increase in 2008 as compared to 2007 was primarily due to the hiring of our sales force early in the third quarter of 2007 to support the launch and commercialization of RECOTHROM in 2008 and increased sales and marketing activities throughout all of 2008.
Stock-based compensation. Stock-based compensation expense decreased by $8.3 million for the year ended December 31, 2009, as compared to the prior year. The decrease was primarily due to a lower underlying share price for stock options granted in 2008 and 2009, forfeiture of stock options related to the termination of employees in 2009, and the unrestricted stock grants made to all employees in recognition of the FDA approval of RECOTHROM in January 2008. The following table summarizes stock-based compensation expense by expense classification and type of award for the periods presented (in thousands):
| 2009 | 2008 | 2007 | |||||||
| Research and development expense |
|||||||||
| Stock options |
$ | 6,638 | $ | 11,797 | $ | 13,591 | |||
| Restricted stock units |
925 | 1,256 | — | ||||||
| Unrestricted stock grants |
— | 519 | — | ||||||
| 7,563 | 13,572 | 13,591 | |||||||
| Selling, general and administrative expense |
|||||||||
| Stock options |
5,148 | 7,182 | 7,286 | ||||||
| Restricted stock units |
307 | 290 | — | ||||||
| Unrestricted stock grants |
— | 228 | — | ||||||
| 5,455 | 7,700 | 7,286 | |||||||
| Total |
$ | 13,018 | $ | 21,272 | $ | 20,877 | |||
55
Table of Contents
Other Income (Expense)
Investment income. Investment income is generated primarily from investment of our cash in fixed-income securities. The primary factors affecting the amount of investment income that we report are: the amount of cash invested, the effective interest rate, the amount of realized gains or losses on investments sold during the period and the amount of other-than-temporary impairment losses recorded in the period. The decrease in 2009 compared to 2008 was primarily due to a reduced effective interest rate and an other-than-temporary-impairment loss of $1.6 million. The decrease in 2008 as compared to 2007 was primarily due to a lower average cash balance and a lower effective interest rate, as well as realized losses on investments and an other-than-temporary impairment loss of $400,000. The following table shows how each of these factors affected investment income for the three years reported (in thousands, except percentages):
| 2009 | 2008 | 2007 | ||||||||||
| Weighted average amount of cash available to invest |
$ | 122,465 | $ | 119,939 | $ | 207,817 | ||||||
| Effective interest rate |
1.09 | % | 3.72 | % | 4.92 | % | ||||||
| Investment income before gains (losses) |
1,333 | 4,464 | 10,218 | |||||||||
| Net gain (loss) on investments |
3 | (231 | ) | 66 | ||||||||
| Other-than-temporary impairment loss |
(1,638 | ) | (400 | ) | — | |||||||
| Investment income, as reported |
$ | (302 | ) | $ | 3,833 | $ | 10,284 | |||||
Interest expense. We have accounted for a sale-leaseback transaction completed in October 2002 as a financing transaction. Under this method of accounting, an amount equal to the net proceeds of the sale is considered a long-term interest-bearing liability. Rent payments under the leases are considered to be payments toward the liability and are allocated to principal and interest. We recorded related interest expense of $7.6 million for the year ended December 31, 2009 and $7.7 million for both the years ended December 31, 2008 and 2007. In addition, we recorded interest expense of $3.4 million and $933,000 for the years ended December 31, 2009 and 2008, respectively, related to the Deerfield financing arrangement, which represents amortization of the deferred financing costs, including the fair value of the warrants issued; 4.9% interest on the $25.0 million drawn in November 2008; and additional interest expense equal to 2% of RECOTHROM net sales in the United States. beginning upon receipt of the $25.0 million draw.
Gain on sale of fixed assets, net. In August of 2008, we sold undeveloped land near our corporate headquarters for $11.8 million and recognized a gain of $7.0 million.
Liquidity and Capital Resources
As of December 31, 2009, we had cash, cash equivalents and short-term investments of $174.1 million, an increase of $84.2 million from December 31, 2008. The increase was attributable to cash received from Bristol-Myers Squibb totaling $200.0 million in 2009 related to the PEG-IFN-lambda collaboration and license agreement, partially offset by cash used to fund our net loss and to manufacture RECOTHROM. In January 2010, we received $90.9 million in net proceeds from the sale of 16.1 million shares of our common stock. We intend to use these assets to fund our operations and capital expenditures over the next several years. Our cash has been held in a variety of fixed-income securities, including corporate bonds, commercial paper and money market instruments that were investment grade at the time of purchase. Subsequent to our purchase, some asset-backed securities have been downgraded by the major bond rating agencies. Together with the discretionary investment manager responsible for investing our portfolio, we monitor our investments closely and, based on market conditions and our expected working capital requirements, recorded other-than-temporary impairment losses of $1.6 million and $400,000 as of December 31, 2009 and 2008, respectively. We consider all other unrealized losses, totaling $1.1 million as of December 31, 2009, to be temporary.
We expect to fund our future operations using our existing cash; revenues from RECOTHROM sales; cash generated from existing and newly established collaborations and licenses; and public or private financings,
56
Table of Contents
including debt or equity financings. We believe these existing and future cash resources including the proceeds from our January 2010 stock offering will be sufficient to fund our operations for at least the next two years, however, this outlook is dependent upon future events, including the sales performance of RECOTHROM and progress in the development activities for our product candidates.
Cash flows from operating activities
The amount of cash used to fund our operating activities differs from our reported net losses due to the following items:
| • | non-cash items, such as depreciation and amortization of fixed assets, amortization of deferred debt issue costs, gain or loss on sale or disposal of assets, other-than-temporary impairment losses on investments and stock-based compensation, which do not result in uses or sources of cash; |
| • | net realized gains and losses and accretion and amortization of discounts and premiums on short-term investments, which are reflected as sources of cash from investing activities upon maturity or sale of the respective investments; |
| • | changes in receivables, which generally represent temporary timing differences between the recognition of certain revenues and the subsequent receipt of cash payments; |
| • | additions to RECOTHROM inventory subsequent to the January 17, 2008 approval date, which reflect the use of cash but will not be expensed until the related product is sold; |
| • | changes in deferred revenue, which reflect the difference in timing between the receipt of cash from option fees, license fees, other upfront payments and milestone payments, and the subsequent recognition of these amounts as revenue over the period we are contractually required to provide other rights or services that represent continuing obligations; |
| • | the Collaboration Obligation under the Bristol-Myers Squibb agreement to fund the first $100.0 million of certain development costs, which is anticipated to be paid through early 2011; and |
| • | changes in other assets and liabilities, which generally represent temporary timing differences between the recognition of certain expenses and their payment. |
Most of these items do not cause material year-to-year fluctuations in the relationship between our net loss and the amount of net cash used in operating activities. Exceptions are non-cash items, changes in deferred revenue and collaboration obligations and RECOTHROM inventory increases. Substantial license or upfront fees may be received when we enter into new licensing or collaborative agreements and be recorded as deferred revenue, which is then recognized as revenue over a future period. For example, we received $200.0 million in upfront and milestone payments from Bristol-Myers Squibb in 2009, of which $100.0 million was recorded as a collaboration obligation reflecting our responsibility to fund the first $100.0 million of research and development costs incurred for the collaboration. The remaining $100.0 million was recorded as deferred revenue. As of December 31, 2009, the remaining collaboration obligation was $76.0 million, which is expected to be paid through early 2011. The deferred revenue is currently expected to be recognized as revenue through mid-2012. The timing of additional collaboration transactions is expected to be irregular and, accordingly, has the potential to create additional future fluctuations in the relationship between our net loss and the amount of cash used in operating activities.
The supply chain for RECOTHROM involves single source suppliers in various countries. These suppliers often require annual minimum production levels, significant lead times and firm purchase commitments to ensure that manufacturing capacity is available. We have established safety stocks of inventory at each stage in the RECOTHROM manufacturing supply chain in an effort to ensure that sufficient product is available to meet anticipated demand. The purchase of inventory under these arrangements has resulted in a significant use of cash. Our current levels of inventory are higher than necessary to support our current volume of sales; however, work
57
Table of Contents
in process and finished goods inventories have shelf lives of five and three years, respectively. We perform periodic reviews of inventory levels, including reviews of the product expiration dates and forecasted sales, and write-down the value of inventory to reflect any anticipated obsolescence. Such write-downs are reflected as a component of cost of product sales. For example, we recorded an obsolete inventory charge of $4.2 million in the fourth quarter of 2008. At December 31, 2009, we have concluded there is no need to recognize any additional inventory obsolescence.
Cash flows from investing activities
Our most significant use of cash in investing activities is for capital expenditures. We expend a certain amount each year on routine items to maintain the effectiveness of our business, such as to adopt newly developed technologies, expand into new functional areas, adapt our facilities to changing needs or replace obsolete assets. All of the $1.8 million, $4.8 million and $6.4 million expended for purchases of property and equipment for the years ended December 31, 2009, 2008 and 2007, respectively, were of this nature. In August 2008, we sold land for $11.8 million that we had purchased in 2001 and 2002. Cash flows from investing activities also reflect large amounts of cash used to purchase short-term investments and receipts from the sale and maturity of short-term investments. These amounts primarily relate to shifts between cash and cash equivalents and short-term investments. Because we manage our cash usage with respect to our total cash, cash equivalents and short-term investments, we do not consider these cash flows to be important to an understanding of our liquidity and capital resources.
Cash flows from financing activities
In 2008, we borrowed $25.0 million under our debt financing arrangement with Deerfield and incurred related financing costs of $1.2 million. We did not borrow any additional amounts from Deerfield prior to the lapse of the credit facility in February 2010. We received $1.6 million, $454,000 and $5.0 million of proceeds from the exercise of stock options for the years ended December 31, 2009, 2008 and 2007, respectively. The amounts we receive from stock option exercises are dependent upon our stock price and the expiration date of the stock option grants.
We expect to incur substantial additional losses in the coming years as we continue to build the market for RECOTHROM and advance our pipeline candidates, such as PEG-IFN-lambda. It might be some time, if ever, before our RECOTHROM revenues enable us to achieve positive operating cash flow. If at any time our prospects for funding our various initiatives decline, we may decide to look for ways to reduce our ongoing investment. For instance, we might consider discontinuing our funding under existing co-development arrangements, as we did with our atacicept collaboration with Merck Serono in August of 2008. Further, we may establish new co-development arrangements for other product candidates to provide additional funding sources, as we did in early 2009 with our PEG-IFN-lambda collaboration with Bristol-Myers Squibb. Also, we may out-license products, product candidates or certain rights related to products or product candidates that we might otherwise choose to develop and commercialize internally as we did in late 2009 with our IL-21 mAb license agreement with Novo Nordisk. Additionally, we could consider delaying or discontinuing development of product candidates to reduce the level of our related expenditures.
In January 2010, we sold 16.1 million shares of our common stock in an underwritten public offering and received $90.9 million in net proceeds. We believe our existing cash and cash equivalents, short-term investments and the proceeds from our stock offering provide us with sufficient cash resources to fund our operations for at least the next two years; however, this outlook depends on future events, including the sales performance of RECOTHROM and progress in the development activities for PEG-IFN-lambda and our other product candidates. We may seek additional funding through new license and/or collaboration transactions or public or private financings, including debt or equity financings. If we are unable to raise additional funds when we need them, we could be required to delay, scale back or eliminate expenditures for some of our development programs, or grant rights to third parties to develop and market product candidates that we would prefer to develop and market internally, with license terms that are not favorable to us.
58
Table of Contents
Contractual Obligations
At December 31, 2009, we were contractually obligated to make payments as follows (in thousands):
| Payments Due by Period | |||||||||||||||
| Total | Less than 1 Year |
1-3 Years |
4-5 Years |
More than 5 Years | |||||||||||
| Building lease obligations |
$ | 92,091 | $ | 8,437 | $ | 17,771 | $ | 19,037 | $ | 46,846 | |||||
| Operating leases |
28,674 | 2,631 | 5,553 | 5,967 | 14,523 | ||||||||||
| Collaboration obligation |
75,959 | 60,105 | 15,854 | — | — | ||||||||||
| RECOTHROM manufacturing contracts |
57,886 | 10,378 | 18,108 | 9,800 | 19,600 | ||||||||||
| Total |
$ | 254,610 | $ | 81,551 | $ | 57,286 | $ | 34,804 | $ | 80,969 | |||||
The building lease obligations resulted from our 2002 sale-leaseback financing transaction and extends until May 2019. The operating leases relate to office space near our corporate headquarters buildings and expire in April 2019. We have certain renewal provisions at our option, which are not reflected in the above table, for the building leases and the operating leases. The collaboration obligation relates to the collaboration and license agreement we entered into with Bristol-Myers Squibb, which obligates us to fund the first $100.0 million of costs for development in the United States and Europe, after which we will be responsible for 20% of such costs unless we elect to convert to a royalty arrangement. RECOTHROM manufacturing contracts include the manufacture of rThrombin bulk drug and RECOTHROM finished product for commercial sale.
Off-Balance Sheet Arrangements
As of December 31, 2009, we did not have any material off-balance sheet arrangements (as defined by Item 303(a)(4)(ii) of Regulation S-K).
Critical Accounting Estimates
Product sales returns and allowances
We primarily sell RECOTHROM to wholesalers, who in turn sell to hospitals. Sales of RECOTHROM are recognized as revenue when the product is received by the wholesaler and title and risk of loss have passed.
Product sales are recorded net of estimated cash discounts, wholesaler fees for service, chargebacks and GPO fees (collectively gross-to-net adjustments) and returns and are recognized as a reduction in product sales revenue. Gross-to-net adjustments are based on actual amounts allowed plus estimates of the amounts yet to be claimed on previously recorded sales. These estimates take into consideration the terms of our current contracts with group purchasing organizations and wholesalers, levels of wholesaler inventory, known sales trends, historical claims experience and forecasted customer buying patterns. Amounts accrued for gross-to-net adjustments are revised when trends or significant events indicate that an adjustment is appropriate. Accrued amounts are also adjusted to reflect actual results. To date, such adjustments have not been material to our results of operations or financial position.
Accruals for estimated sales returns are recorded in the same period that the related product sales are recorded and are also recognized as reductions in product sales revenue. Returns are estimated by comparing and analyzing inventory information provided by our wholesalers, shipping information and historical return data on a production lot basis. To date, sales returns have been insignificant and the impact of any adjustments have not been material to our results of operations or financial position.
License fees, milestone payments and upfront fees
We enter into various collaborative agreements that generate significant license, option or other upfront fees with subsequent milestone payments earned upon completion of development milestones. Where we have no
59
Table of Contents
continuing performance obligations under an arrangement, we recognize milestone payments as revenue upon receipt, as these payments represent the culmination of a separate earnings process. Where we have continuing performance obligations under an arrangement, revenue is recognized using one of two methods. Where we are able to estimate the total amount of services under the arrangement, revenue is recognized using a proportional performance model. Costs incurred to date compared to total expected costs are used to determine proportional performance, as this is considered to be representative of the delivery of outputs under the arrangement. Revenue recognized at any point in time is limited to cash received and amounts contractually due. Changes in estimates of total expected costs are accounted prospectively as a change in estimate. Where we cannot estimate the total amount of service that is to be provided, a time-based method is used to recognize revenue. Under the time-based method, revenue is recognized over the arrangement’s estimated performance period, starting with the contract’s commencement, but not before the removal of any contingencies for each milestone. Revenue recognition is determined based on the elapsed time compared to the total estimated performance period. Revenue recognized at any point in time is limited to the completed portion of the non-contingent payments received or due.
Inventory obsolescence
We establish provisions for obsolete and excess inventory and include it as a component of costs of product sales. The bulk drug substance form of RECOTHROM has a shelf life of five years and finished (vialed) product has a shelf life of three additional years. The provision for obsolete and excess inventory is evaluated for adequacy at each quarter end based on estimated future product sales, production commitments, and existing inventory levels at all stages of manufacturing.
Investment impairment
We determine the impairment classification of any individual security as either temporary or other-than-temporary. An other-than-temporary impairment for debt securities is recorded when we intend to sell the security, it is more likely than not that we will be required to sell the security before recovery of its amortized cost basis, or we do not expect to recover the security’s entire amortized cost basis. In addition, if we determine that a credit loss exists with respect to an individual debt security, we would record an other-than-temporary impairment if we concluded that we would not be able to recover the entire amortized cost of the security, even if we do not intend to sell the security. The differentiating factors for equity securities, between temporary and other-than-temporary impairments, are primarily the length of the time and the extent to which the fair value has been less than cost, the financial condition and near-term prospects of the issuer and the intent and ability to retain our investment in the issuer for a period of time sufficient to allow for an anticipated recovery in fair value. We record other-than-temporary impairments in gain (loss) on investments, net in our consolidated statements of operations and we record temporary impairments within accumulated other comprehensive (loss) income in our consolidated balance sheets.
Stock-based compensation
In 2009, our assumption for expected stock price volatility was based on the historical volatility of our common stock over a period commensurate with the expected term of the options. Prior to 2009, the expected volatility assumption was based on a blend of historical stock price volatility with the implied volatility of market traded options. In 2008, the calculation of historical volatility was based solely on the trading of our common stock. Prior to 2008, however, historical trading information for our common stock was not available for a long enough period and, thus, was augmented with the historical volatility of similar companies. Furthermore, the implied volatility of market traded options of similar companies was used since the market for options on our common stock was illiquid and could not be relied upon as a source of implied volatility. The risk-free interest rate used in the option valuation model is based on U.S. Treasury zero-coupon issues with remaining terms similar to the expected term of the options. We used historical data to determine an estimate for the expected life of our stock options granted in 2009 and 2008 and used the simplified method for determining the expected term for 2007. We do not anticipate paying any cash dividends in the foreseeable future and therefore an expected
60
Table of Contents
dividend yield of zero is used in the option valuation model. Forfeitures are estimated at the time of grant and revised in subsequent periods if actual forfeitures differ from those estimates. Accordingly, stock-based compensation expense is recorded only for those awards that vest. All stock-based payment awards are amortized on a straight-line basis over the requisite service periods of the awards, which are generally the vesting periods.
Item 7A. Qualitative and Quantitative Disclosures About Market Risk
Until recently, our exposure to market risk has been primarily limited to interest income sensitivity, which is affected by changes in the general level of United States interest rates, particularly because the majority of our investments are in short-term debt securities. The primary objective of our investment activities is to preserve principal while at the same time maximizing the income we receive without significantly increasing risk. To minimize risk, we maintain our portfolio of cash, cash equivalents and short-term investments in a variety of interest-bearing instruments, which have included United States government and agency securities, high-grade United States corporate bonds, asset-backed securities, commercial paper and money market funds.
In 2008 and continuing through 2009, due to deteriorating conditions in the debt markets, our exposure to market risk increased and impacted our investment portfolio. Overall liquidity for many debt issues has declined substantially, meaning that we may realize losses if we are required to liquidate securities upon short notice. Additionally, with respect to asset backed securities, overall economic conditions have generated concerns about the value of underlying assets held as collateral, and highlighted risks associated with insurance policies used to enhance the credit of the related debt issues. To date, we have not experienced defaults on any of our asset backed securities, but we have seen the time for expected repayment increase significantly. In 2009, we revised our investment guidelines to exclude future purchases of asset backed securities; however, we continue to hold several of these securities that were purchased prior to the revision of our guidelines. We continue to monitor these investments closely and, based on market conditions, recorded other-than-temporary impairment losses of $1.6 million and $400,000 in the fourth quarter of 2009 and the third quarter of 2008, respectively.
We have no material foreign currency exposure, nor do we hold derivative financial instruments.
61
Table of Contents
Item 8. Financial Statements and Supplementary Data
| Page in Form 10-K | ||
| 63 | ||
| 64 | ||
| 65 | ||
| Consolidated Statement of Changes in Shareholders’ Equity (Deficit) |
66 | |
| 67 | ||
| 68 –90 | ||
62
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors
and Shareholders of ZymoGenetics, Inc.:
In our opinion, the accompanying consolidated balance sheets and the related consolidated statements of operations, changes in shareholder’s equity (deficit) and cash flows present fairly, in all material respects, the financial position of ZymoGenetics, Inc. and its subsidiaries at December 31, 2009 and 2008, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2009 in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2009, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company’s management is responsible for these financial statements, for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Report on Internal Control over Financial Reporting section under Item 9A. Our responsibility is to express opinions on these financial statements and on the Company’s internal control over financial reporting based on our integrated audits. We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
PricewaterhouseCoopers LLP
Seattle, Washington
February 26, 2010
63
Table of Contents
CONSOLIDATED BALANCE SHEETS
(in thousands)
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| Assets |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 141,634 | $ | 50,088 | ||||
| Short-term investments |
32,496 | 39,799 | ||||||
| Receivables |
7,672 | 11,249 | ||||||
| Inventory |
63,024 | 28,241 | ||||||
| Prepaid expenses |
5,043 | 3,579 | ||||||
| Total current assets |
249,869 | 132,956 | ||||||
| Property and equipment, net |
58,565 | 63,676 | ||||||
| Deferred financing costs |
5,172 | 6,726 | ||||||
| Long-term investment |
2,002 | 1,547 | ||||||
| Other assets |
3,688 | 5,141 | ||||||
| Total assets |
$ | 319,296 | $ | 210,046 | ||||
| Liabilities and Shareholders’ Equity (Deficit) |
||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 14,000 | $ | 8,834 | ||||
| Accrued liabilities |
24,299 | 13,099 | ||||||
| Lease obligations |
928 | 563 | ||||||
| Deferred revenue |
40,484 | 34,472 | ||||||
| Collaboration obligation |
60,105 | — | ||||||
| Total current liabilities |
139,816 | 56,968 | ||||||
| Lease obligations |
67,563 | 67,366 | ||||||
| Debt obligation |
25,000 | 25,000 | ||||||
| Deferred revenue |
63,899 | 33,374 | ||||||
| Collaboration obligation |
15,854 | — | ||||||
| Other long-term liabilities |
11,122 | 3,979 | ||||||
| Commitments and contingencies |
||||||||
| Shareholders’ equity: |
||||||||
| Preferred stock, no par value, 30,000 shares authorized, no shares issued and outstanding |
— | — | ||||||
| Common stock, no par value, 150,000 shares authorized, 69,353 and 68,736 issued and outstanding at December 31, 2009 and 2008, respectively |
801,318 | 786,736 | ||||||
| Non-voting common stock, no par value, 30,000 shares authorized, no shares issued and outstanding |
— | — | ||||||
| Accumulated deficit |
(805,184 | ) | (762,203 | ) | ||||
| Accumulated other comprehensive loss |
(92 | ) | (1,174 | ) | ||||
| Total shareholders’ equity (deficit) |
(3,958 | ) | 23,359 | |||||
| Total liabilities and shareholders’ equity (deficit) |
$ | 319,296 | $ | 210,046 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
64
Table of Contents
CONSOLIDATED STATEMENTS OF OPERATIONS
(in thousands, except per share data)
| Year ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| Revenues: |
||||||||||||
| Product sales, net |
$ | 29,596 | $ | 8,779 | $ | — | ||||||
| Royalties |
1,307 | 6,290 | 6,259 | |||||||||
| Collaborations and licenses |
106,069 | 58,920 | 32,218 | |||||||||
| Total revenues |
136,972 | 73,989 | 38,477 | |||||||||
| Costs and expenses: |
||||||||||||
| Costs of product sales |
7,631 | 5,672 | — | |||||||||
| Research and development |
99,194 | 126,678 | 142,340 | |||||||||
| Selling, general and administrative |
62,202 | 60,238 | 46,890 | |||||||||
| Total costs and expenses |
169,027 | 192,588 | 189,230 | |||||||||
| Loss from operations |
(32,055 | ) | (118,599 | ) | (150,753 | ) | ||||||
| Other income (expense): |
||||||||||||
| Investment income (loss) |
(302 | ) | 3,833 | 10,284 | ||||||||
| Interest expense |
(10,994 | ) | (8,582 | ) | (7,677 | ) | ||||||
| Gain on sale of fixed assets, net |
7 | 7,107 | 2 | |||||||||
| Total other income |
(11,289 | ) | 2,358 | 2,609 | ||||||||
| Loss before income tax benefit |
(43,344 | ) | (116,241 | ) | (148,144 | ) | ||||||
| Income tax benefit |
363 | — | — | |||||||||
| Net loss |
$ | (42,981 | ) | $ | (116,241 | ) | $ | (148,144 | ) | |||
| Basic and diluted net loss per share |
$ | (0.62 | ) | $ | (1.69 | ) | $ | (2.17 | ) | |||
| Weighted-average number of shares used in computing net loss per share |
69,069 | 68,696 | 68,156 | |||||||||
The accompanying notes are an integral part of these consolidated financial statements.
65
Table of Contents
CONSOLIDATED STATEMENT OF CHANGES IN SHAREHOLDERS’ EQUITY (DEFICIT)
(in thousands)
| Common stock | Accumulated deficit |
Accumulated other comprehensive income (loss) |
Total | ||||||||||||||
| Shares | Amount | ||||||||||||||||
| Balance at January 1, 2007 |
67,499 | $ | 732,914 | $ | (497,818 | ) | $ | 588 | $ | 235,684 | |||||||
| Comprehensive loss: |
|||||||||||||||||
| Net loss |
— | — | (148,144 | ) | — | (148,144 | ) | ||||||||||
| Unrealized gain on short-term investment |
— | — | — | 667 | 667 | ||||||||||||
| Unrealized gain on long-term investments |
— | — | — | 701 | 701 | ||||||||||||
| Total comprehensive loss |
(146,776 | ) | |||||||||||||||
| Common stock issued in connection with stock option exercises |
1,029 | 5,045 | — | — | 5,045 | ||||||||||||
| Stock-based compensation expense |
— | 20,877 | — | — | 20,877 | ||||||||||||
| Balance at December 31, 2007 |
68,528 | 758,836 | (645,962 | ) | 1,956 | 114,830 | |||||||||||
| Comprehensive loss: |
|||||||||||||||||
| Net loss |
— | — | (116,241 | ) | — | (116,241 | ) | ||||||||||
| Unrealized loss on short-term investment, net |
— | — | — | (2,163 | ) | (2,163 | ) | ||||||||||
| Unrealized loss reclassified to other-than-temporary impairment |
— | — | — | 400 | 400 | ||||||||||||
| Unrealized loss on long-term investments |
— | — | — | (1,367 | ) | (1,367 | ) | ||||||||||
| Total comprehensive loss |
(119,371 | ) | |||||||||||||||
| Common stock issued in connection with stock option exercises |
151 | 454 | — | — | 454 | ||||||||||||
| Common stock issued in connection with stock awards |
57 | 746 | — | — | 746 | ||||||||||||
| Warrants issued in connection with financing arrangement |
— | 6,174 | — | — | 6,174 | ||||||||||||
| Stock-based compensation expense |
— | 20,526 | — | — | 20,526 | ||||||||||||
| Balance at December 31, 2008 |
68,736 | 786,736 | (762,203 | ) | (1,174 | ) | 23,359 | ||||||||||
| Comprehensive loss: |
|||||||||||||||||
| Net loss |
— | — | (42,981 | ) | — | (42,981 | ) | ||||||||||
| Unrealized loss on short-term investment |
— | — | — | (1,011 | ) | (1,011 | ) | ||||||||||
| Unrealized loss reclassified to other-than-temporary impairment |
— | — | — | 1,638 | 1,638 | ||||||||||||
| Unrealized gain on long-term investments |
— | — | — | 455 | 455 | ||||||||||||
| Total comprehensive loss |
(41,899 | ) | |||||||||||||||
| Common stock issued in connection with stock option exercises |
428 | 1,564 | — | — | 1,564 | ||||||||||||
| Vesting of restricted stock units |
189 | — | — | — | — | ||||||||||||
| Stock-based compensation expense |
— | 13,018 | — | — | 13,018 | ||||||||||||
| Balance at December 31, 2009 |
69,353 | $ | 801,318 | $ | (805,184 | ) | $ | (92 | ) | $ | (3,958 | ) | |||||
The accompanying notes are an integral part of these consolidated financial statements.
66
Table of Contents
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
| Year ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| Operating activities |
||||||||||||
| Net loss |
$ | (42,981 | ) | $ | (116,241 | ) | $ | (148,144 | ) | |||
| Adjustments to reconcile net loss to net cash provided by (used in) operating activities: |
||||||||||||
| Depreciation and amortization |
6,906 | 7,173 | 7,221 | |||||||||
| Amortization of debt issuance costs |
1,544 | 673 | — | |||||||||
| Stock-based compensation |
13,018 | 21,272 | 20,877 | |||||||||
| Inventory impairment |
— | 4,150 | — | |||||||||
| Net gain on disposition of property and equipment |
(7 | ) | (7,107 | ) | (2 | ) | ||||||
| Net realized loss (gain) on sale of short-term investments |
(3 | ) | 231 | (66 | ) | |||||||
| Impairment loss on short-term investments |
1,638 | 400 | — | |||||||||
| Net amortization (accretion) of premium (discount) on short-term investments |
23 | 204 | (769 | ) | ||||||||
| Changes in operating assets and liabilities |
||||||||||||
| Receivables |
3,577 | (4,012 | ) | (1,980 | ) | |||||||
| Inventory |
(34,783 | ) | (32,391 | ) | — | |||||||
| Prepaid expenses |
(1,464 | ) | 1,025 | (879 | ) | |||||||
| Other assets |
1,453 | 1,542 | (750 | ) | ||||||||
| Accounts payable |
5,166 | (3,118 | ) | 4,054 | ||||||||
| Accrued liabilities |
11,200 | (9,856 | ) | 8,833 | ||||||||
| Lease obligations |
562 | 885 | (43 | ) | ||||||||
| Deferred revenue |
36,537 | 26,929 | 23,398 | |||||||||
| Collaboration obligation |
75,959 | — | — | |||||||||
| Other long-term liabilities |
7,143 | (1,404 | ) | 749 | ||||||||
| Net cash provided by (used in) operating activities |
85,488 | (109,645 | ) | (87,501 | ) | |||||||
| Investing activities |
||||||||||||
| Purchases of property and equipment |
(1,801 | ) | (4,802 | ) | (6,442 | ) | ||||||
| Purchases of short-term investments |
(25,072 | ) | (63,397 | ) | (171,094 | ) | ||||||
| Proceeds from sale of property and equipment |
13 | 11,761 | 3 | |||||||||
| Proceeds from sale and maturity of short-term investments |
31,343 | 162,704 | 283,649 | |||||||||
| Net cash provided by investing activities |
4,483 | 106,266 | 106,116 | |||||||||
| Financing activities |
||||||||||||
| Proceeds from debt financing |
— | 25,000 | — | |||||||||
| Proceeds from exercise of stock options |
1,564 | 454 | 5,045 | |||||||||
| Other financing costs |
11 | (1,224 | ) | — | ||||||||
| Net cash provided by financing activities |
1,575 | 24,230 | 5,045 | |||||||||
| Net increase in cash and cash equivalents |
91,546 | 20,851 | 23,660 | |||||||||
| Cash and cash equivalents at beginning of period |
50,088 | 29,237 | 5,577 | |||||||||
| Cash and cash equivalents at end of period |
$ | 141,634 | $ | 50,088 | $ | 29,237 | ||||||
| Supplemental disclosures |
||||||||||||
| Cash paid for interest |
$ | 7,954 | $ | 7,721 | $ | 7,677 | ||||||
| Warrants issued in connection with the Deerfield financing arrangement |
$ | — | $ | 6,174 | $ | — | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
67
Table of Contents
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. Organization and summary of significant accounting policies
Nature of operations
ZymoGenetics, Inc. (the Company) was incorporated in the state of Washington in June 1981 and operated independently until it was acquired in 1988 by Novo Nordisk North America, a wholly owned subsidiary of Novo Nordisk A/S (Novo Nordisk). In November 2000, the Company became independent from Novo Nordisk upon completion of a private placement of Series B mandatorily redeemable convertible preferred stock with an investor consortium. In February 2002, the Company completed an initial public offering of common stock; at which time all Series A and B mandatorily redeemable convertible preferred stock was converted to common stock. Through this and other subsequent stock offerings and stock option exercises, Novo Nordisk’s ownership percentage has been reduced to approximately 30% at December 31, 2009.
ZymoGenetics, Inc. is a biopharmaceutical company focused on the development and commercialization of therapeutic proteins for the treatment of human diseases. In 2009, through a series of strategic initiatives and workforce and cost reductions, the Company restructured its organization and is now focused on developing and commercializing a limited number of products, which it believes has substantial therapeutic and commercial potential and in which it retains a significant ownership position. The Company’s current portfolio includes one commercial product, RECOTHROM® Thrombin, topical (Recombinant) which is approved in the United States and Canada for use as a recombinant topical hemostat to control moderate bleeding during surgical procedures. Additionally, the Company has three immunology product candidates: PEG-Interferon lambda (PEG-IFN-lambda), which is being developed in collaboration with Bristol-Myers Squibb Company in a Phase 2 clinical trial for treatment of hepatitis C virus infection; Interleukin-21 (IL-21) which is being tested in a Phase 2 clinical trial as a potential immunotherapy treatment for metastatic melanoma; and Interleukin-31 monoclonal antibody (IL-31 mAb) which is currently in preclinical development as a potential treatment for atopic dermatitis.
The Company has $174.1 million in cash and cash equivalents and short-term investments as of December 31, 2009 and on January 12, 2010, received an additional $90.9 million in net proceeds from the sale of 16.1 million shares of its common stock (See Note 16). The Company expects to fund its future operations using these cash resources, revenues from RECOTHROM sales, and cash generated from existing and newly established collaborations and licenses and additional public and private financings. The Company believes that it has sufficient cash resources to fund its operations for at least the next two years; however, this outlook is dependent upon future events, including the sales performance of RECOTHROM and progress in the development activities for its product candidates.
Use of estimates
The preparation of financial statements in conformity with generally accepted accounting principles requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities; disclosure of contingent assets and liabilities at the date of the financial statements; and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Cash and cash equivalents
The Company considers all highly liquid investments with a contractual maturity at date of purchase of three months or less to be cash and cash equivalents. The Company invests its cash and cash equivalents with major financial institutions, the amount of which usually exceed federally insured limits. The Company has not experienced any losses on its cash and cash equivalents.
68
Table of Contents
ZYMOGENETICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Available-for-sale securities
Short-term investments
Short-term investments are classified as available-for-sale. Available-for-sale securities are carried at fair value, with the unrealized gains and losses reported as a separate component of shareholders’ equity or deficit. Interest on securities classified as available-for-sale is included in investment income. The amortized cost of debt securities is adjusted for amortization of premiums and accretion of discounts to expected maturity and the amortization is included in interest income. For investments in asset-backed securities, amortization of premiums and accretion of discounts are recognized in interest income using the interest method, adjusted for anticipated prepayments, as applicable. Estimates of expected cash flows are updated periodically and changes are recognized in the calculated effective yield prospectively. Realized gains and losses and declines in value judged to be other-than-temporary on available-for-sale securities are included in investment income. The cost of securities sold is based on the specific identification method.
Long-term investments
Included in other assets is a long-term investment in common shares of BioMimetic Therapeutics, Inc., a company that licensed certain technologies from the Company and made certain payments in shares of common stock. These shares are publicly traded and are adjusted to fair value, with the unrealized gains and losses reported as a separate component of shareholders’ equity or deficit. As of December 31, 2009 and 2008, the unrealized gain on the investment was $1.0 million and $547,000, respectively.
Fair value of financial instruments
The Company records its short-term and long-term investments at fair value. Fair value is the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date (exit price) and establishes a fair value hierarchy based on the inputs used to measure fair value. The three levels of the fair value hierarchy are as follows:
| • | Level 1 – Unadjusted quoted prices in active markets that are accessible at the measurement date for identical, unrestricted assets or liabilities (for example exchange quoted prices); |
| • | Level 2 – Observable inputs, other than Level 1 prices, such as quoted prices for similar assets or liabilities, quoted prices in markets that are not sufficiently active to qualify as Level 1, other observable inputs, or inputs that can be corroborated by observable market data for substantially the full term of the assets or liabilities; and |
| • | Level 3 – Prices or valuation techniques that require inputs that are both significant to the fair value measurement and unobservable (i.e., supported by little or no market activity). |
Assets and liabilities are classified in their entirety based on the lowest level of input that is significant to the fair value measurement. The Company’s assessment of the significance of a particular input to the fair value measurement requires judgment and may affect the valuation of fair value assets and liabilities and their placement within the fair value hierarchy levels.
Inventory
Inventory is stated at the lower of cost or market. Cost includes amounts related to materials, labor and overhead, and is determined on a specific identification basis in a manner which approximates the first-in, first-out (FIFO) method. Inventory balances reflect the cost of post-approval manufacturing activities for
69
Table of Contents
ZYMOGENETICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
RECOTHROM. The manufacturing of RECOTHROM requires multiple steps which are performed by a series of third-party contractors, most of which are single source, based upon the Company’s specifications. As protection against product shortages, the Company maintains safety stocks of inventory at each stage in the manufacturing process and has entered into manufacturing contracts, some of which contain annual minimum purchase commitments. The Company reduces inventory to its estimated net realizable value by reserving for excess and obsolete inventories based on forecasted demand.
Property and equipment
Property and equipment are stated at cost. Additions, betterments and improvements are capitalized and depreciated. When assets are retired or otherwise disposed of, the cost of the assets and related depreciation is eliminated from the accounts and any resulting gain or loss is reflected in the consolidated statements of operations. Depreciation is computed using the straight-line method over the estimated useful lives of the assets, which include five years for furniture and lab equipment, ten years for pilot plant equipment and 40 years for buildings. Expenditures for repairs and maintenance are charged to expense as incurred.
Leasehold improvements are amortized ratably over their estimated useful lives or the remaining term of the lease, whichever is shorter. At December 31, 2009, the Company is amortizing its leasehold improvements over 10 years.
Impairment of long-lived assets
The Company assesses the impairment of long-lived assets whenever events or changes in business circumstances indicate that the carrying amounts of the assets may not be fully recoverable. Measurement of an impairment is required when estimated undiscounted future cash flows expected to result from the use of the asset and its eventual disposition are less than its carrying amount. The amount of a recognized impairment loss is the excess of an asset’s carrying value over its fair value. The Company has not recognized any impairment losses of long-lived assets in 2009, 2008 or 2007.
Revenue recognition
Product sales
Sales of RECOTHROM are recognized as revenue when the product is shipped and title and risk of loss have passed. Product sales are recorded net of provisions for estimated discounts, rebates, chargebacks and returns.
Royalties
The Company earns royalties on certain products marketed by other companies. Royalties on these products are received within 60 days after the end of each calendar quarter. The Company accrues estimated royalties based on historical sales data and the patent life associated with the product. Adjustments are made in the following quarter reflecting the differences between the Company’s estimates and actual reported royalties and, to date, adjustments have not been significant.
Collaborations and licenses
The Company enters into various collaborative agreements that may generate license, option or other upfront fees with subsequent milestone payments earned upon completion of development milestones. Where the Company has no continuing performance obligations under an arrangement, it recognizes such payments as revenue when
70
Table of Contents
ZYMOGENETICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
contractually due and payment is reasonably assured, as these payments represent the culmination of a separate earnings process. Where the Company has continuing performance obligations under an arrangement, revenue is recognized using one of two methods. Where the Company is able to estimate the total amount of services under the arrangement, revenue is recognized using a proportional performance model. Costs incurred to date compared to total expected costs are used to determine proportional performance, as this is considered to be representative of the delivery of outputs under the arrangement. Revenue recognized at any point in time is limited to cash received and amounts contractually due. Changes in estimates of total expected costs are accounted for prospectively as a change in estimate. Where the Company cannot estimate the total amount of service that is to be provided, a time-based method is used to recognize revenue. Under the time-based method, revenue is recognized over the arrangement’s estimated performance period, starting with the contract’s commencement, but not before the removal of any contingencies for each milestone. From period to period, license fees and milestone payments can fluctuate substantially based on the completion of new licensing or collaborative agreements and the achievement of development-related milestones.
Deferred revenue arises from payments received in advance of the culmination of the earnings process. Deferred revenue expected to be recognized within the next twelve months is classified as a current liability. Deferred revenue will be recognized as revenue in future periods when the applicable revenue recognition criteria have been met.
Cost of product sales
Prior to FDA approval, all third party manufacturing costs and an allocation of Company labor and overhead associated with the manufacturing of RECOTHROM for commercial sale were expensed as research and development costs as incurred. Subsequent to RECOTHROM approval, these costs are recorded as inventory. Costs of product sales includes the inventory and distribution costs associated with RECOTHROM product sales and costs incurred subsequent to FDA approval for product that is not expected to be sold before its expiration dating and, thus, considered obsolete. During 2008, a reserve of $4.2 million was included in costs of product sales.
Research and development costs
Research and development costs, consisting of salaries and benefits, costs of consumables, facility costs, contracted services and stock-based compensation, are expensed as incurred. Costs to acquire technologies that are utilized in research and development which have no future use are expensed when incurred. The Company has evaluated its collaboration arrangements and accordingly has recorded research and development cost reimbursements as reductions to research and development expenses or as an increase in collaboration revenue. The reductions to research and development expense were $2.3 million, $12.4 million and $10.7 million in 2009, 2008 and 2007, respectively.
Patent and legal costs
Costs relating to filing, pursuing and defending patent applications are expensed to selling, general and administrative costs as incurred. Other legal costs are expensed as incurred.
Income taxes
The Company records a provision for income taxes using the liability method of accounting for income taxes. Deferred tax assets or liabilities are recorded for all temporary differences between financial and tax reporting. Deferred tax expense or benefit results from the net change during the period of the deferred tax assets and liabilities. A valuation allowance is recorded when it is more likely than not that the deferred tax asset will not be realized.
71
Table of Contents
ZYMOGENETICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Stock-based compensation
The Company recognizes as an expense share-based payment transactions in which the Company receives employee services in exchange for equity instruments of the Company. Fair value is determined using the Black-Scholes valuation method. The Company has elected to use the straight-line method of allocating the fair value of compensation expense over the requisite service period of the related award. Additionally, the Company estimates expected forfeitures and recognizes only the compensation cost for those stock options expected to vest.
The Company recorded the following amounts of stock-based compensation expense for the years ended December 31 (in thousands):
| 2009 | 2008 | 2007 | |||||||
| Research and development expense |
$ | 7,563 | $ | 13,572 | $ | 13,591 | |||
| Selling, general and administrative expense |
5,455 | 7,700 | 7,286 | ||||||
| Total |
$ | 13,018 | $ | 21,272 | $ | 20,877 | |||
Comprehensive income (loss)
Comprehensive income (loss) is the change in shareholders’ equity (deficit) resulting from net income (loss) adjusted for unrealized gains and losses on short-term and long-term investments. Amounts are reclassified from other comprehensive income (loss) into the results of operations when unrealized gains and losses become realized.
Segments
The Company manages and evaluates its operations as one reportable segment.
Guarantees
In the normal course of business, the Company indemnifies other parties, including healthcare providers, wholesalers, collaboration partners, lessors and parties to other transactions with the Company, with respect to certain matters. The Company has agreed to hold the parties harmless against losses arising from a breach of representations and covenants, or out of intellectual property infringement or other claims made against these parties. These agreements may limit the time within which an indemnification claim can be made and the amount of the claim. It is not possible to determine the maximum potential obligation under these indemnification agreements since any claim would be based on the facts and circumstances of the claim and the particular provisions of each agreement.
Concentration of Risks
The Company’s cash and cash equivalents are invested with financial institutions in deposits that usually exceed federally insured limits. The Company has not experienced any losses on its deposits of cash and cash equivalents. Management believes that the institutions are financially sound and, accordingly, that minimal credit risk exists.
The Company is subject to credit risk from its accounts receivable related to product sales, and periodically assesses the financial strength of its customers and establishes allowances for anticipated losses, when necessary. Three wholesalers accounted for approximately 90% and 91% of U.S. sales in 2009 and 2008. The Company
72
Table of Contents
ZYMOGENETICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
believes that if these wholesalers ceased distributing RECOTHROM, other wholesalers already distributing RECOTHROM would absorb the incremental sales volume with minimal interruption to the Company’s business or the Company would sell directly to hospitals.
The Company is dependent on various single source vendors for most steps of its manufacturing process, and some of the key components in the Company’s products come from single or limited sources of supply.
Loss per share
Basic and diluted net loss per share have been computed based on net loss and the weighted-average number of common shares outstanding during the applicable period. The Company has excluded options to purchase common stock, restricted stock units and warrants to purchase common stock, as such potential shares are antidilutive for all periods presented.
The following table presents the calculation of basic and diluted net loss per share for years ended December 31 (in thousands, except per share data):
| 2009 | 2008 | 2007 | ||||||||||
| Net loss |
$ | (42,981 | ) | $ | (116,241 | ) | $ | (148,144 | ) | |||
| Weighted-average shares used in computing basic and diluted net loss per share |
69,069 | 68,696 | 68,156 | |||||||||
| Basic and diluted net loss per share |
$ | (0.62 | ) | $ | (1.69 | ) | $ | (2.17 | ) | |||
| Securities not included in net loss per share calculation: |
||||||||||||
| Options to purchase common stock |
12,977 | 14,385 | 12,702 | |||||||||
| Restricted stock units |
256 | 583 | — | |||||||||
| Warrants to purchase common stock |
1,500 | 1,500 | — | |||||||||
Recent accounting pronouncements
In October 2009, authoritative guidance was provided on revenue arrangements with multiple deliverables. Under this guidance, when vendor specific objective evidence or third party evidence for deliverables in an arrangement cannot be determined, a best estimate of the selling price is required to separate deliverables and allocate arrangement consideration using the relative selling price method. The guidance also includes new disclosure requirements on how the application of the relative selling price method affects the timing and amount of revenue recognition. This guidance is to be applied on a prospective basis for revenue arrangements entered into or materially modified in fiscal years beginning on or after June 15, 2010, with earlier application permitted. The Company is considering whether to adopt this guidance beginning January 1, 2010 and is currently evaluating the impact of the implementation on its results of operations, cash flows and financial condition.
73
Table of Contents
ZYMOGENETICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
2. Short-term investments
Short-term investments consisted of the following (in thousands):
| Amortized Cost |
Gross Unrealized Gains |
Gross Unrealized Losses |
Estimated Fair Value | ||||||||||
| December 31, 2009 |
|||||||||||||
| Type of security: |
|||||||||||||
| Commercial paper and money market |
$ | 11,870 | $ | 8 | $ | (1 | ) | $ | 11,877 | ||||
| Corporate debt securities |
13,198 | — | (73 | ) | 13,125 | ||||||||
| Asset-backed securities |
8,522 | 20 | (1,048 | ) | 7,494 | ||||||||
| $ | 33,590 | $ | 28 | $ | (1,122 | ) | $ | 32,496 | |||||
| Contractual maturity date: |
|||||||||||||
| Less than one year |
$ | 11,870 | $ | 11,877 | |||||||||
| Due in 1-5 years |
14,433 | 14,379 | |||||||||||
| Due in 5-10 years |
— | — | |||||||||||
| Due in 10 years or more |
7,287 | 6,240 | |||||||||||
| $ | 33,590 | $ | 32,496 | ||||||||||
| December 31, 2008 |
|||||||||||||
| Type of security: |
|||||||||||||
| Corporate debt securities |
$ | 7,749 | $ | 4 | $ | (123 | ) | $ | 7,630 | ||||
| Asset-backed securities |
24,804 | — | (1,760 | ) | 23,044 | ||||||||
| U.S. government and agency securities |
8,967 | 158 | — | 9,125 | |||||||||
| $ | 41,520 | $ | 162 | $ | (1,883 | ) | $ | 39,799 | |||||
| Contractual maturity date: |
|||||||||||||
| Less than one year |
$ | 16,765 | $ | 16,805 | |||||||||
| Due in 1-5 years |
13,724 | 13,195 | |||||||||||
| Due in 5-10 years |
— | — | |||||||||||
| Due in 10 years or more |
11,031 | 9,799 | |||||||||||
| $ | 41,520 | $ | 39,799 | ||||||||||
As of December 31, 2009, the weighted average expected maturity dates for all securities did not exceed three years.
In assessing potential impairment of its debt securities, the Company determines if it intends to sell the security, if it is more likely than not that it will be required to sell the security before recovering its amortized cost basis, or if it expects to recover the security’s entire amortized cost basis. In addition, if the Company determines that a credit loss exists with respect to an individual debt security, it would record an other-than-temporary impairment if it concluded that it would not be able to recover the entire amortized cost of the security, even if there was no intent to sell the security. The differentiating factors for equity securities, between temporary and other-than-temporary impairments, are primarily the length of the time and the extent to which the fair value has been less than cost, the financial condition and near-term prospects of the issuer and the intent and ability to retain our investment in the issuer for a period of time sufficient to allow for an anticipated recovery in fair value. In 2009 and 2008, the Company recorded other-than-temporary impairment losses of $1.6 million and $400,000, respectively, on asset-backed security investments.
74
Table of Contents
ZYMOGENETICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
At December 31, 2009, the aggregate estimated fair value of the investments with unrealized losses was as follows (in thousands):
| Fair Value |
Unrealized Loss |
||||||
| Commercial paper and money market |
$ | 2,995 | $ | (1 | ) | ||
| Corporate debt securities |
13,125 | (73 | ) | ||||
| Asset-backed securities |
6,240 | (1,048 | ) | ||||
| $ | 22,360 | $ | (1,122 | ) | |||
The contractual maturities on unrealized loss positions for which other-than-temporary impairments have not been recognized at December 31, 2009, are summarized as follows (in thousands):
| Fair Value |
Unrealized Loss |
||||||
| Less than one year |
$ | 2,995 | $ | (1 | ) | ||
| Greater than one year |
19,365 | (1,121 | ) | ||||
| $ | 22,360 | $ | (1,122 | ) | |||
Realized gains were $4,000, $521,000 and $379,000 for the years ended December 31, 2009, 2008 and 2007, respectively. Realized losses were $0, $752,000 and $313,000 for the years ended December 31, 2009, 2008 and 2007, respectively. Other-than-temporary impairment losses were $1.6 million and $400,000 for the years ended December 31, 2009 and 2008, respectively. Reclassification adjustments reflected in other comprehensive loss for net realized losses were $0, $209,000 and $159,000 for the years ended December 31, 2009, 2008 and 2007, respectively.
Fair value measurements
The Company’s short-term and long-term investments accounted for at fair value as of December 31, 2009 are summarized below (in thousands):
| Level 1 | Level 2 | Level 3 | Total | |||||||||
| Cash equivalents: |
||||||||||||
| Money market funds |
$ | 117,980 | $ | — | $ | — | $ | 117,980 | ||||
| Short-term investments: |
||||||||||||
| Commercial paper and money market |
$ | 11,877 | $ | — | $ | — | $ | 11,877 | ||||
| Corporate debt securities |
— | 13,125 | — | 13,125 | ||||||||
| Asset-backed securities |
— | 7,494 | — | 7,494 | ||||||||
| $ | 11,877 | $ | 20,619 | $ | — | $ | 32,496 | |||||
| Long-term investment: |
||||||||||||
| BMTI common stock |
$ | 2,002 | $ | — | $ | — | $ | 2,002 | ||||
75
Table of Contents
ZYMOGENETICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
3. Inventory
Inventory consisted of the following at December 31 (in thousands):
| 2009 | 2008 | |||||
| Raw materials |
$ | 2,256 | $ | 1,664 | ||
| Work in process |
58,327 | 25,751 | ||||
| Finished goods |
2,441 | 826 | ||||
| Total |
$ | 63,024 | $ | 28,241 | ||
4. Property and equipment
Property and equipment consisted of the following at December 31 (in thousands):
| 2009 | 2008 | |||||||
| Land and buildings |
$ | 71,199 | $ | 71,055 | ||||
| Leasehold improvements |
2,998 | 3,005 | ||||||
| Furniture and equipment |
55,694 | 55,351 | ||||||
| 129,891 | 129,411 | |||||||
| Less: Accumulated depreciation and amortization |
(71,326 | ) | (65,735 | ) | ||||
| $ | 58,565 | $ | 63,676 | |||||
Land and buildings include assets deemed owned in connection with the sale and leaseback financing transaction described in Note 7.
5. Sale of land
In August 2008, the Company sold land located near its corporate headquarters for $11.8 million and recognized a gain of $7.1 million. The gain is included in other income (expense) on the consolidated statement of operations as gain on sale of fixed assets, net.
6. Accrued liabilities
Accrued liabilities consisted of the following at December 31 (in thousands):
| 2009 | 2008 | |||||
| Incentive compensation |
$ | 5,923 | $ | 4,315 | ||
| Severance pay |
5,331 | 55 | ||||
| Vacation pay |
2,936 | 4,043 | ||||
| Contract services |
1,538 | 1,567 | ||||
| Sales discounts and allowances |
2,516 | 1,436 | ||||
| Sales commission to Bayer |
4,675 | — | ||||
| Other |
1,380 | 1,683 | ||||
| $ | 24,299 | $ | 13,099 | |||
76
Table of Contents
ZYMOGENETICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
7. Lease obligation
In October 2002, the Company completed a sale and leaseback transaction involving its headquarter buildings located in Seattle, Washington. The three buildings were sold for a total sale price of $52.3 million. Net proceeds from the transaction amounted to $50.5 million. Simultaneously, the Company agreed to lease the buildings from the purchaser for a period of 15 years, subject to four five-year renewal options. The initial rental payment of $5.1 million per year increases by 3.5% each year during the term. Rent for the renewal terms under these lease agreements will be the greater of fair market value or 90% of the rent for the last year prior to renewal. The Company has provided the lessor a security deposit in the form of pledged securities equal to two months base rent or $1.4 million.
The Company has accounted for the transaction as a financing due to a technical provision within the leases related to condemnation, which could, under remote circumstances, result in continuing ownership involvement by the Company in the three buildings. Under this method of accounting, the net proceeds of the sale are considered to be a long-term interest bearing liability. Rent payments under the leases are considered to be payments toward the liability and are allocated to principal and interest. The Company initially recorded a liability of $50.5 million with an effective annual interest rate of approximately 11%.
In 2003, the Company exercised its option to expand one of the leased buildings and, effective May 2004, the Company assumed occupancy of the new space. The Company incurred total project costs of approximately $21.0 million excluding equipment and received an advance from the landlord of $14.9 million. The advance was included as an addition to the long-term lease obligation with an annual effective interest rate of approximately 12%. At the end of the lease term, the remaining balance of the liability will approximate the net book value of the buildings leased. Upon the completion of the expansion project, the lease terms for all three buildings were reset to 15 years from the date of occupancy of the expansion space.
The Company is required to develop certain space within the expanded facility by June 2011. If this requirement is not satisfied, the Company must post a $1.0 million letter of credit (LOC) made available to the landlord until the lease specifications have been met. If the Company does not develop the space within specification by the end of the 15-year lease term, the landlord will have the right to draw down the full amount of the LOC in satisfaction of this obligation.
The following table presents the Company’s scheduled payments under the lease obligation. In addition, the Company has certain renewal provisions at its option, which are not reflected in the table.
| Year ending December 31, |
|||
| 2010 |
$ | 8,437 | |
| 2011 |
8,733 | ||
| 2012 |
9,038 | ||
| 2013 |
9,355 | ||
| 2014 |
9,682 | ||
| Thereafter |
46,846 | ||
| $ | 92,091 | ||
8. Debt financing
In June 2008, the Company entered into a financing arrangement with Deerfield Management (Deerfield), which was amended on December 31, 2009. Under the amended agreement, the Company could have borrowed up to $100.0 million in four draws of $25.0 million each until February 10, 2010 provided the Company
77
Table of Contents
ZYMOGENETICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
requested the funds by January 19, 2010. Interest accrues on amounts outstanding at a rate of 4.9% per annum, compounded quarterly, and will be due, along with outstanding principal, in June 2014. Accrued interest was $1.5 million as of December 31, 2009 and is included in other long-term liabilities. Each $25.0 million draw entitles Deerfield to a royalty equal to 2% of RECOTHROM net sales in the U.S. subject to certain maximum amounts depending on the amount borrowed. Royalties are payable quarterly. In addition, the Company agreed to issue Deerfield warrants to purchase 1.5 million shares of common stock at $10.34 per share upon the earlier of the first draw or January 2010. The warrants have a six-year term and the Company is obligated to register the common stock issuable under the warrants with the SEC. The Company can repay borrowed amounts in whole or in part at any time, without penalty, and all associated interest and royalty obligations will cease.
In November 2008, the Company borrowed the first $25.0 million under the Deerfield financing arrangement and issued the related 1.5 million warrants. No subsequent borrowing requests were made on or before the January 19, 2010 borrowing request deadline. The Company has calculated the fair value of the initial 1.5 million warrants to be $6.2 million using the Black Scholes option pricing model. The amount was recorded as deferred financing costs with a corresponding increase to common stock.
Deferred financing costs totaling $7.4 million including the $6.2 million fair value of the initial 1.5 million warrants, are being amortized to interest expense through June 2014. During 2009 and 2008, approximately $1.5 million and $673,000 respectively, of amortization was included in interest expense.
Based on the total borrowing of $25.0 million, cumulative royalties are capped at $18.8 million over the term of the loan. During 2009 and 2008, royalty expense was approximately $570,000 and $72,000, respectively.
9. Related party transactions
Novo Nordisk owned approximately 30% of the Company’s outstanding common stock at December 31, 2009, 2008 and 2007, respectively. The following table summarizes revenue earned from Novo Nordisk for the periods presented (in thousands):
| 2009 | 2008 | 2007 | |||||||
| Royalties |
$ | — | $ | 290 | $ | 1,261 | |||
| Collaborations and licenses: |
|||||||||
| Option and license agreements: |
|||||||||
| IL-20 |
3,500 | 2,000 | 1,000 | ||||||
| IL-21 |
5,000 | — | 3,500 | ||||||
| Other |
— | 30 | 2,183 | ||||||
| Factor XIII |
2,500 | 5,000 | 6,820 | ||||||
| Total |
$ | 11,000 | $ | 7,320 | $ | 14,764 | |||
Amounts receivable from Novo Nordisk were approximately $1.2 million and $40,000 at December 31, 2009 and 2008, respectively.
Royalties
The Company historically earned royalties on two products marketed and sold by Novo Nordisk. These royalties ceased in 2008 due to patent expiration.
78
Table of Contents
ZYMOGENETICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Collaborations and licenses
Option and license agreements
In 2000, the Company and Novo Nordisk entered into an option and license agreement, which including subsequent amendments, expired in November 2006. Under the terms of the agreement, Novo Nordisk licensed certain of the Company’s product candidates. Novo Nordisk is responsible for all development activities and obligated to make payments upon the achievement of predefined development milestones and to pay royalties on sales of any resulting products.
IL-20
Milestone payments of $3.5 million, $2.0 million and $1.0 million were earned and recognized as revenue for the years 2009, 2008 and 2007, respectively, as the Company has no continuing performance obligations.
IL-21
In 2001, Novo Nordisk licensed the rights to IL-21 in all territories outside of North America. In 2007, the Company received a milestone payment of $3.5 million and recognized the payment as revenue as the Company had no other significant rights or obligations under this agreement.
In January 2009, Novo Nordisk and the Company restructured their relationship as it relates to IL-21. The Company reacquired Novo Nordisk’s rights to the IL-21 protein, Novo Nordisk retained the rights to develop other embodiments of IL-21, including antibodies to IL-21, outside of North America and will be obligated to pay the Company milestones and royalties on any products developed. As a result, the Company has worldwide development and commercialization rights to the IL-21 protein and will be obligated to pay Novo Nordisk milestone payments based on approval of any IL-21 protein products and pay royalties on any sales outside North America and third party license fees above a certain threshold.
In December 2009, the agreement relating to other embodiments of IL-21 was amended to provide worldwide development and commercialization rights to Novo Nordisk. Under the terms of the revised agreement, Novo Nordisk paid an initial milestone payment of $24.0 million and is obligated to pay the Company milestone payments and royalties on any products developed. In addition, the Company is obligated to perform certain transition activities which are expected to take approximately six months to substantially complete. The Company has no other continuing performance obligations. Because of the substantive transition activities for which the Company is responsible, the Company is recognizing the initial $24.0 million milestone payment over the six-month estimated performance period and $4.0 million was recognized as collaboration and license revenue in December 2009. In addition, the Company earned $1.0 million for transition activities during 2009 which are included in collaboration and license revenue.
Other
During 2008 and 2007, the Company had various additional license agreements in place with Novo Nordisk pursuant to the option and license agreement. Under these agreements, the Company had no continuing performance obligations and recognizes revenue when contractually due.
Factor XIII
In 2004, the Company entered into a license agreement with Novo Nordisk with respect to recombinant Factor XIII whereby Novo Nordisk will develop and commercialize recombinant Factor XIII on a worldwide basis. The Company is entitled to milestones and royalties based on future development and commercial sales of Factor XIII.
79
Table of Contents
ZYMOGENETICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
In 2009, 2008 and 2007, the Company received $2.5 million, $5.0 million and $6.8 million of milestone payments related to Novo Nordisk’s development of Factor XIII. All amounts were recognized as revenue upon receipt since the Company does not have any significant remaining performance obligations.
10. Collaboration and license agreements
Bristol-Myers Squibb
Type III interferon collaboration
In January 2009, the Company entered into a co-development/co-promotion and license agreement with Bristol-Myers Squibb Company (BMS) for the type III interferon family, of which PEG-IFN-lambda is the designated development candidate. Per the agreement, the Company is primarily responsible for the completion of certain Phase 1 and Phase 2 clinical trials while BMS is responsible for certain Phase 2 and Phase 3 clinical trials, clinical and commercial manufacturing, the drug approval process and for commercialization of any approved drugs. The Company is required to fund the first $100.0 million of joint development costs and additional development costs will be funded 80.0% by BMS and 20.0% by the Company. The Company is obligated to exchange enabling technology through the commercialization period and to participate on Executive, Development and Commercialization steering committees. The Company does have the right to cease contributing to all development and commercialization costs at any time, which would convert the agreement into a royalty arrangement. If the Company elects to convert to a royalty arrangement, the Company will still be required to fund the first $100.0 million of development costs and it will no longer participate on any of the steering committees.
The Company’s substantive obligations under this agreement are expected to be satisfied in 2012. As the Company is able to estimate its program costs through the performance period, revenue is being recognized using the proportional performance methodology as a single unit of accounting. The Company received a total of $200.0 million under the agreement in 2009, consisting of $105.0 million in license fees and two milestone payments totaling $95.0 million related to the initiation of Phase 2 clinical trials. The Company recorded $100.0 million as deferred revenue and $100.0 million as a liability (the Collaboration Obligation) due to the Company’s commitment to fund the initial $100.0 million of development costs incurred by both companies. The reimbursement of the Company’s development costs from the Collaboration Obligation, together with the milestones earned and expected to be earned during the performance period, will be recognized as collaboration revenue by the Company based on the percentage of its total allowable program costs incurred to date compared to its total expected allowable program costs over the performance period.
In 2009, the Company recorded collaboration and license revenue of $38.7 million, based on the proportional performance formula described above.
As of December 31, 2009, the remaining balance of the Collaboration Obligation was $76.0 million and deferred revenue was $84.4 million.
Immunoglobulin fusion patents
In October 2008, the Company entered into a binding, nonexclusive, worldwide license with Bristol-Myers Squibb Company (BMS) to the Company’s patents related to immunoglobulin fusion. BMS paid the Company a one-time license payment of $21.0 million. The Company has no future performance obligations under this arrangement and, accordingly, recorded the entire amount as license fee revenue in 2008.
80
Table of Contents
ZYMOGENETICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Bayer Schering Pharma AG and Bayer HealthCare LLC
In June 2007, the Company entered into a License and Collaboration Agreement with Bayer Schering Pharma AG and a U.S. Co-Promotion Agreement with Bayer HealthCare, LLC. (collectively, Bayer). The agreements provided Bayer with an exclusive license to develop and sell rThrombin outside the United States and to co-promote rThrombin in the United States for three or four years. The Company records all U.S. sales and Bayer is entitled to commissions on U.S. sales.
The Company received a $30.0 million upfront milestone payment in 2007 and a $40.0 million milestone payment in February 2008, $20.0 million of which was expected to be repaid to Bayer as U.S. sales bonuses under the co-promotion. In addition, in 2008, the Company received milestone payments of $6.5 million for Bayer’s filing of a marketing authorization application in Europe and a new drug submission in Canada. In 2009, the Company received $500,000 for the filing of a new drug application in Asia. The agreements provided for additional payments based on further regulatory filings, approvals and annual sales thresholds achieved by Bayer outside of the United States.
The Company had a number of substantive obligations under the agreements which were expected to be delivered from the execution date of the agreement through the first quarter of 2014. In addition, the Company agreed to supply bulk drug over the entire term of the License and Collaboration Agreement.
The Company evaluated its substantive obligations under the agreements and determined that there were two separate units of accounting. The first unit of accounting consists of the grant of various licenses, participation on the United States co-promotion committee, research and development support prior to regulatory approval, formation and maintenance of a U.S. sales force and supply of finished drug product until the first quarter of 2014. The second unit of accounting consists of supplying bulk drug product over the term of the agreement. The Company concluded that the combined obligations in the first unit of accounting had value to Bayer on a standalone basis. Regarding the second unit of accounting, the Company has objective and reliable evidence of the fair value of the bulk drug product to be supplied over the term of the License and Collaboration Agreement based on the purchase price paid to the third party supplier of the bulk drug product.
Obligations within the first unit of accounting were to be provided until the first quarter of 2014. Thereafter, the only undelivered element was to provide bulk drug product for the remaining term of the License and Collaboration Agreement. The Company used the residual method to allocate its arrangement consideration between the two units of accounting. The Company has recognized revenue attributable to the first unit of accounting using a proportional performance model as the Company was able to estimate the proportional progress based on the costs expected to be incurred under the agreements. The Company determined based upon the nature and timing of its obligations that cost inputs were the best measure of performance under the arrangements. Revenue attributable to the supply of bulk drug product was to be recognized as the bulk drug is delivered to Bayer provided all other revenue recognition criteria are met.
During 2008 and 2007, the Company recognized $18.0 million and $6.2 million, respectively, of license and collaboration revenue related to the collaboration.
On December 18, 2009, the Company and Bayer substantially amended the License and Collaboration Agreement and the U.S. Co-Promotion Agreement. Under the amended agreements, Bayer discontinued development and approval application activities in all countries other than Canada; returned all rights to rThrombin to the Company except in Canada where Bayer will launch rThrombin during 2010; exited the U.S. co-promotion effective December 31, 2009; and forgave the $20.0 million in guaranteed U.S. sales bonuses which the Company had previously recorded as a liability. As part of the amendments, the Company agreed to
81
Table of Contents
ZYMOGENETICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
pay Bayer a commission on U.S. net sales for the two-year period ending on December 31, 2011 up to a maximum of $12.0 million; to provide finished U.S. approved drug product for Canada at cost plus a fixed profit margin; and to forgive the $3.5 million milestone associated with Canadian approval of rThrombin in December 2009.
Under the amended agreements, as of December 31, 2009, the Company has completed all the deliverables associated with the first unit of accounting, and the Company has a maximum liability to Bayer associated with U.S. commissions of $12.0 million for which Bayer has no performance obligations in order to receive these U.S. commissions.
Immediately preceding the effective date of the amendments, the Company had deferred revenue and liabilities for U.S. bonus payments totaling $46.4 million. Of this amount, $12.0 million will be paid to Bayer until the related commission liability is extinguished. The remaining $34.4 million was recognized as collaboration and license revenue upon the effective date of the amendments.
In 2009, the Company recognized a total of $40.8 million of license and collaboration revenue related to the collaboration. Also during 2009, the Company sold Bayer $1.4 million of finished drug product with a cost of $1.3 million.
Merck Serono
In August 2001, the Company entered into a Collaborative Development and Marketing Agreement with Merck Serono S.A (Merck Serono) for atacicept whereby the companies shared research and development expenses. In September 2004, the Company entered into a Strategic Alliance Agreement with Merck Serono, providing for a strategic research, development and commercialization alliance. Additionally, in a series of related transactions, the Company entered into four other product-related agreements pursuant to which it received upfront fees and potential milestones and royalties.
Effective August 28, 2008, the Company and Merck Serono modified these agreements as follows:
| • | the Collaborative Development and Marketing Agreement for atacicept was amended and converted to an exclusive worldwide license whereby the Company’s responsibility for funding development costs ended and Merck Serono will pay the Company milestone fees and royalties on worldwide net sales. |
| • | the Strategic Alliance Agreement was amended, eliminating the future co-development of product candidates jointly researched and establishing a mechanism by which each company would have alternating options to obtain exclusive rights to such product candidates. |
| • | The two other existing co-development and co-commercialization agreements were amended, providing Merck Serono with an exclusive license and all rights to the IL-17RC soluble receptor and the Company with an exclusive license and all rights to IL-31 mAb in exchange for future milestones and royalties to the other party. |
Under the August 28, 2008 modification, the Company continued to be responsible for certain transitional activities related to atacicept through June 2009 the costs of which were reimbursed by Merck Serono. The Company also had significant remaining performance obligations under the Strategic Alliance Agreement through its expiration in October 2009.
As part of the August 2008 modification discussed above, the Company was relieved of an obligation to reimburse $9.8 million of research and development costs incurred by Merck Serono from June 1, 2008 to August 28, 2008. The forgiveness of these expenses was determined to be additional consideration for the
82
Table of Contents
ZYMOGENETICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
licenses granted to Merck Serono and therefore, the $9.8 million was determined to be incremental revenue which was deferred and recognized as license fee revenue on a straight-line basis from August 28, 2008 through October 12, 2009, the expiration date of the Strategic Alliance Agreement.
The Company recognized collaboration and license revenue of $15.5 million, $11.3 million and $11.0 million in 2009, 2008 and 2007 under these agreements. As of December 31, 2009, all previously deferred revenue under all agreements with Merck Serono has been fully recognized.
11. Retirement plans
Defined contribution
The Company maintains a 401(k) retirement plan covering substantially all of its employees. The plan provides for matching and discretionary contributions by the Company. Contributions were $1.7 million, $2.6 million and $2.3 million for the years ended December 31, 2009, 2008 and 2007, respectively.
Deferred compensation plan
The Company has a Deferred Compensation Plan (DCP) for key employees. Eligible plan participants are designated by the Company’s Board of Directors. The DCP allows participants to defer up to 50% of their annual compensation and up to 100% of any bonus. At December 31, 2009 and 2008, approximately $2.3 million and $3.8 million, respectively, was deferred under the DCP and was recorded both as a long-term asset and a long-term liability.
12. Income taxes
At December 31, 2009, the Company had net operating loss carryforwards of $653.1 million, research and development tax credit carryforwards of $36.3 million and alternative minimum tax credit carryforwards of $1.2 million. The carryforwards are available to offset future tax liabilities. The net operating loss carryforwards will begin to expire from 2021 – 2029, the research and development tax credits expire from 2010 – 2029 and the alternative minimum tax credit will carry forward indefinitely. Utilization of the net operating loss and tax credit carryforwards may be subject to annual limitations under ownership change limitations established by the Internal Revenue Code Section 382. The annual limitations may result in the expiration of net operating loss and tax credit carryforwards before they can be utilized.
Deferred tax assets and liabilities arise from temporary differences between financial and tax reporting. The Company has provided a valuation allowance at December 31, 2009 and 2008 to offset the excess of deferred tax assets over the deferred tax liabilities, due to the uncertainty of realizing the benefits of the net deferred tax asset.
83
Table of Contents
ZYMOGENETICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Deferred tax assets and liabilities were as follows as of December 31 (in thousands):
| 2009 | 2008 | |||||||
| Deferred tax assets: |
||||||||
| Net operating loss carryforwards |
$ | 228,596 | $ | 204,612 | ||||
| Research and development tax credit carryforwards |
36,324 | 34,839 | ||||||
| Alternative minimum tax credit carryforwards |
1,242 | 1,242 | ||||||
| Deferred gain on sale of assets |
9,211 | 8,861 | ||||||
| Deferred revenue |
— | 22,832 | ||||||
| Stock option compensation |
2,276 | 7,332 | ||||||
| Commission payable |
4,228 | — | ||||||
| Other |
8,903 | 9,174 | ||||||
| 290,780 | 288,892 | |||||||
| Deferred tax liabilities: |
||||||||
| Deferred revenue |
— | (4,193 | ) | |||||
| 290,780 | 284,699 | |||||||
| Less: Valuation allowance |
(290,780 | ) | (284,699 | ) | ||||
| Net deferred tax asset |
$ | — | $ | — | ||||
The valuation allowance increased by $6.1 million, $39.5 million and $51.2 million in 2009, 2008 and 2007, respectively, to fully reserve the net deferred tax assets.
In October 2000, the Company entered into a tax sharing agreement with Novo Nordisk. The agreement states that all research and development tax credit carryforwards generated by the Company prior to November 9, 2000 used by the Company to generate a tax benefit in future periods shall be reimbursed to Novo Nordisk. The total amount paid shall not exceed $12.0 million. As of December 31, 2009, the Company has not recognized any of these tax benefits.
The reconciliation between the Company’s effective tax rate and the income tax rate is as follows for the years ended December 31:
| 2009 | 2008 | 2007 | |||||||
| Federal income tax rate |
(35 | )% | (35 | )% | (35 | )% | |||
| Research and development tax credits |
(3 | ) | (3 | ) | (3 | ) | |||
| Valuation allowance |
35 | 38 | 35 | ||||||
| Other |
3 | — | 3 | ||||||
| Effective tax rate |
0 | % | 0 | % | 0 | % | |||
The Company files its income tax return in the U.S. federal jurisdiction. The Company is no longer subject to U.S. federal tax examinations by tax authorities for years before 2006. However, the Internal Revenue Service (IRS) could adjust certain unused tax attributes carried forward from tax years prior to 2006. The Company believes that if subjected to an IRS income tax audit, any assessments would be immaterial to its financial statements. The Company files state tax returns in states where it has tax obligations.
84
Table of Contents
ZYMOGENETICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
A reconciliation of the beginning and ending amount of unrecognized tax benefits is as follows (in thousands):
| Balance at January 1, 2008 |
$ | 1,370 | ||
| Additions based on tax positions related to the current year |
— | |||
| Additions for tax positions of prior years |
— | |||
| Reductions for tax positions of prior years |
(1,370 | ) | ||
| Settlements/Statute of Limitation Lapse |
— | |||
| Balance at January 1, 2009 |
— | |||
| Additions based on tax positions related to the current year |
— | |||
| Additions for tax positions of prior years |
— | |||
| Reductions for tax positions of prior years |
— | |||
| Settlements/Statute of Limitation Lapse |
— | |||
| Balance at December 31, 2009 |
$ | — | ||
When and if applicable, the Company will classify income tax-related interest and penalties as income tax expense in its consolidated statements of operations.
13. Commitments and Contingencies
Operating lease commitments
Historically, the Company had various operating lease agreements for office and parking space in a building near its corporate headquarters in Seattle, Washington. In March 2008, the Company entered into a noncancelable master lease agreement which extended the lease term for all leased space to April 2019. There are certain renewal provisions at the Company’s option, which are not reflected in the following operating lease commitment table. Total annual payments under the lease averages approximately $2.9 million per year over the term.
The following table presents the Company’s commitments for future minimum rental payments under the noncancelable master operating lease (in thousands):
| Year ending December 31, |
|||
| 2010 |
$ | 2,631 | |
| 2011 |
2,727 | ||
| 2012 |
2,826 | ||
| 2013 |
2,930 | ||
| 2014 |
3,037 | ||
| Thereafter |
14,522 | ||
| $ | 28,673 | ||
The master lease agreement provides for scheduled rent increases over its term. The Company is recognizing rent expense on a straight-line basis over the lease term.
The Company reduced its workforce in 2009 and has ceased using certain leased space. Authoritative guidance related to costs associated with exit or disposal activities requires companies to record a liability at the cease use date based on the remaining rental costs for which no economic benefit is derived, reduced by the
85
Table of Contents
ZYMOGENETICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
estimated sublease rentals that could reasonably be obtained. This amount is then discounted using a credit-adjusted risk-free rate. Accordingly, the Company recorded a restructuring liability of $1.0 million as of December 31, 2009. This amount is included in the table above that presents the Company’s commitments for future minimum rental payments under the noncancelable master operating lease.
Gross rental expense for the years ended December 31, 2009, 2008 and 2007, including the restructuring liability established in 2009, was $3.1 million, $1.9 million and $2.0 million, respectively.
Purchase commitments
The Company maintains, with its contract manufacturers, rolling firm orders and annual minimum purchase commitments for RECOTHROM. These orders may be rescheduled or cancelled by the Company under limited conditions and, even then, with certain restrictions and penalties up to the full cost of the production.
The following table presents the Company’s noncancelable annual purchase commitments to its contract manufacturers (in thousands):
| Year ending December 31, |
|||
| 2010 |
$ | 10,378 | |
| 2011 |
13,208 | ||
| 2012 |
4,900 | ||
| 2013 |
4,900 | ||
| 2014 |
4,900 | ||
| Thereafter |
19,600 | ||
| $ | 57,886 | ||
Other commitments
Certain key employees have employment agreements with the Company which provide for salary, health insurance and certain additional severance benefits.
Contingencies
On November 2, 2009, King Pharmaceuticals, Inc. and affiliated entities (Monarch Pharmaceuticals, Inc., King Pharmaceuticals Research and Development, Inc., and GenTrac, Inc.), or, collectively, King, filed suit against the Company in the United States District Court for the Eastern District of Tennessee, naming as defendants the Company and fifty unnamed individuals. King alleges that the Company has engaged in unfair competition, false advertising, trademark infringement, and related claims under federal law and Tennessee state law. King seeks various forms of relief, including damages and permanent injunctive relief precluding the Company from making certain representations regarding King’s products and the Company’s RECOTHROM product. King also filed motions with the District Court seeking temporary restraining orders and preliminary injunctions with respect to certain comparative advertising claims, use of King trademarks as Google ad words, and certain alleged statements regarding the existence of lawsuits against King. On December 10, 2009 the court denied all three motions for preliminary injunction in their entirety. The Company disputes the allegations of wrongdoing in King’s complaint and will continue to vigorously defend itself in this matter. The Company has not recorded a liability related to this suit.
86
Table of Contents
ZYMOGENETICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
14. Stock incentive plans
The Company maintains stock incentive plans (the Plans) that provide for the issuance of incentive stock options, nonqualified stock options, restricted stock and restricted stock units to employees, directors, consultants and other independent contractors who provide services to the Company. The Company’s Board of Directors is responsible for administration of the Plans and determines the term of each option, exercise price and the vesting period.
Stock options
A summary of stock option activity under the Plans is presented below (shares and aggregate intrinsic value in thousands):
| Shares | Weighted- Average Exercise Price |
Weighted- Average Remaining Contractual Term (Years) |
Aggregate Intrinsic Value | ||||||||
| Balance, January 1, 2007 |
11,982 | $ | 12.95 | ||||||||
| Granted |
2,724 | 15.06 | |||||||||
| Exercised |
(1,029 | ) | 4.92 | ||||||||
| Forfeited |
(562 | ) | 17.91 | ||||||||
| Canceled |
(413 | ) | 18.27 | ||||||||
| Balance, December 31, 2007 |
12,702 | $ | 13.66 | 6.6 | $ | 25,295 | |||||
| Granted |
2,631 | 8.18 | |||||||||
| Exercised |
(151 | ) | 3.62 | ||||||||
| Forfeited |
(437 | ) | 15.92 | ||||||||
| Canceled |
(360 | ) | 15.36 | ||||||||
| Balance, December 31, 2008 |
14,385 | $ | 12.65 | 6.3 | $ | 345 | |||||
| Granted |
5,324 | 4.95 | |||||||||
| Exercised |
(428 | ) | 3.66 | ||||||||
| Forfeited |
(2,318 | ) | 10.83 | ||||||||
| Canceled |
(3,986 | ) | 16.74 | ||||||||
| Balance, December 31, 2009 |
12,977 | $ | 8.85 | 6.1 | $ | 13,959 | |||||
| Exercisable, December 31, 2009 |
7,015 | $ | 11.52 | 4.2 | $ | 6,178 | |||||
As of December 31, 2009, there was $13.6 million of total unrecognized compensation costs related to stock options. These costs are expected to be recognized over a weighted average period of 2.3 years.
Estimated fair values of stock options granted have been determined using the Black-Scholes option pricing model with the following assumptions:
| 2009 | 2008 | 2007 | |||||||
| Expected stock price volatility |
55.2 – 61.4 | % | 53.1 – 55.2 | % | 50.2 – 54.8 | % | |||
| Risk-free interest rate |
1.92 – 3.34 | % | 1.63 – 3.72 | % | 3.46 – 4.96 | % | |||
| Expected life of options |
4.2 – 6.4 years | 5.6 – 6.2 years | 6.1 years | ||||||
| Expected dividend yield |
0 | % | 0 | % | 0 | % |
87
Table of Contents
ZYMOGENETICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
In 2009, the Company’s assumption for expected stock price volatility was based on the historical volatility of its common stock over a period commensurate with the expected term of the options. Prior to 2009, the expected volatility assumption was based on a blend of historical stock price volatility with the implied volatility of market traded options. In 2008, the calculation of historical volatility was based solely on the trading of the Company’s common stock. Prior to 2008, however, historical trading information for the Company’s common stock was not available for a long enough period and, thus, was augmented with the historical volatility of similar companies. Furthermore, the implied volatility of market traded options of similar companies was used since the market for options on the Company’s common stock is illiquid and cannot be relied upon as a source of implied volatility. The risk-free interest rate used in the option valuation model is based on U.S. Treasury zero-coupon issues with remaining terms similar to the expected term of the options. The Company used historical data to determine an estimate for the expected life of its stock options granted in 2009 and 2008 and used the simplified method for determining the expected term for options granted prior to January 1, 2008. The Company does not anticipate paying any cash dividends in the foreseeable future and therefore an expected dividend yield of zero is used in the option valuation model. Forfeitures are estimated at the time of grant and revised in subsequent periods if actual forfeitures differ from those estimates. Accordingly, stock-based compensation expense is recorded only for those awards that vest. All stock-based payment awards are amortized on a straight-line basis over the requisite service periods of the awards, which are generally the vesting periods.
A summary of stock option values under the Plans is presented below (in thousands, except per stock option share data):
| 2009 | 2008 | 2007 | |||||||
| Weighted average grant date fair value per stock option share granted |
$ | 2.57 | $ | 4.29 | $ | 8.49 | |||
| Total intrinsic value of stock options exercised |
$ | 860 | $ | 926 | $ | 10,213 | |||
| Total fair value of stock options vested |
$ | 13,132 | $ | 21,161 | $ | 21,681 | |||
Common stock exchange
In December 2009, the Company offered employees the ability to exchange some or all of their outstanding stock options that had an exercise price per share greater than $7.90 for a new stock option to purchase a lesser number of shares based on the following ratios:
| Per Share Exercise Price of Eligible Options |
New Options for Exchanged Options | |
| $7.90 – $9.79 |
1.25 to 1.00 | |
| $9.80 – $16.62 |
1.75 to 1.00 | |
| $16.63 and higher |
2.50 to 1.00 |
A total of 3,260,763 shares were cancelled and in exchange, the Company granted awards of 1,629,632 shares with an exercise price of $6.35 that vest over two and three year periods. In accordance with authoritative guidance, the Company has elected to continue recording expense for the cancelled shares based on the original fair values and vesting terms. The incremental fair value of the newly awarded shares totaled $36,000.
Restricted stock units
In February 2008, the Company granted 620,500 restricted stock units to non-officer employees. These shares vest over a three-year period with one-third vesting on each anniversary of the grant date. The grant date fair value for the restricted stock unit awards was the closing market price of the Company’s common stock on
88
Table of Contents
ZYMOGENETICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
the date of grant, which was $9.26 per share. As of December 31, 2009, total unrecognized compensation costs related to unvested restricted stock units was $1.4 million, which is being expensed on a straight-line basis through February 2011.
A summary of restricted stock unit activity is presented below (number of restricted stock units in thousands):
| Number of Restricted Stock Units |
Weighted Average Grant-Date Fair Value Per Share | |||||
| Outstanding at January 1, 2008 |
— | $ | — | |||
| Granted |
621 | 9.26 | ||||
| Forfeited |
(38 | ) | 9.26 | |||
| Vested |
— | — | ||||
| Outstanding at December 31, 2008 |
583 | 9.26 | ||||
| Granted |
— | — | ||||
| Forfeited |
(138 | ) | 9.26 | |||
| Vested |
(189 | ) | 9.26 | |||
| Outstanding at December 31, 2009 |
256 | $ | 9.26 | |||
Common stock award
In January 2008, the Company issued a total of 57,200 unrestricted shares of common stock to employees of the Company in recognition of FDA approval of RECOTHROM. The share price used to determine compensation was $13.05, based on the closing price on the date the compensation committee of the Company’s board of directors approved the distribution. Additionally, the compensation amount was increased to include payroll taxes paid on behalf of the employees. The entire $1.1 million of costs related to the issuance were expensed in January 2008.
15. Restructuring
In 2008 and 2009, the Company restructured its operations and reduced its workforce by a total of 247 employees. Restructuring charges of $13.1 million and $1.7 million were recorded in 2009 and 2008, respectively. In addition to employee severance, benefits and related costs, the Company ceased using certain rental space as a result of these restructurings. The Company recorded a liability of $1.0 million based on the remaining rental costs for which no economic benefit is derived, reduced by the estimated sublease rentals that could reasonably be obtained. The amount was discounted using a credit-adjusted risk-free rate.
The changes in liabilities related to these restructuring plans for the periods presented are as follows: (in thousands):
| Severance | Lease | Total | |||||||||
| Balance, January 1, 2008 |
$ | — | $ | — | $ | — | |||||
| Severance additions |
1,714 | — | 1,714 | ||||||||
| Severance payments |
(1,659 | ) | — | (1,659 | ) | ||||||
| Balance, December 31, 2008 |
55 | — | 55 | ||||||||
| Severance additions |
13,051 | — | 13,051 | ||||||||
| Severance payments |
(7,775 | ) | — | (7,775 | ) | ||||||
| Lease accrual |
— | 1,000 | 1,000 | ||||||||
| Balance, December 31, 2009 |
$ | 5,331 | $ | 1,000 | $ | 6,331 | |||||
89
Table of Contents
ZYMOGENETICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
16. Subsequent event
On January 12, 2010, the Company sold 16.1 million shares of common stock in an underwritten public offering at $6.00 per share. Net proceeds from the sale, after deducting underwriter discounts and other issuance costs, were $90.9 million.
Novo Nordisk, who owned approximately 30% of the Company’s outstanding stock at December 31, 2009, purchased an additional 1.25 million shares through the offering. After the sale, Novo Nordisk owned approximately 26% of the Company’s outstanding common stock.
17. Quarterly financial results (unaudited)
The following table contains selected statements of operations information, which is unaudited and should be read in conjunction with the Company’s financial statements and related notes included elsewhere in this report. The Company believes that the following unaudited information reflects all normal recurring adjustments necessary for a fair presentation of the information for the periods presented. The operating results for any quarter are not necessarily indicative of results for any future period. Operating results for each quarter of 2009 and 2008 are summarized as follows (in thousands):
| March 31 | June 30 | September 30 | December 31 | |||||||||||||
| 2009 |
||||||||||||||||
| Revenue |
$ | 24,845 | $ | 22,572 | $ | 27,456 | $ | 62,099 | ||||||||
| Net income (loss) |
$ | (18,129 | ) | $ | (26,977 | ) | $ | (11,443 | ) | $ | 13,568 | |||||
| Basic net income (loss) per common share(1) |
$ | (0.26 | ) | $ | (0.39 | ) | $ | (0.17 | ) | $ | 0.20 | |||||
| Diluted net income (loss) per common share(1) |
$ | (0.26 | ) | $ | (0.39 | ) | $ | (0.17 | ) | $ | 0.19 | |||||
| 2008 |
||||||||||||||||
| Revenue |
$ | 13,512 | $ | 12,582 | $ | 11,876 | $ | 36,019 | ||||||||
| Net loss |
$ | (40,896 | ) | $ | (37,380 | ) | $ | (28,790 | ) | $ | (9,176 | ) | ||||
| Basic and diluted net loss per common share(1) |
$ | (0.60 | ) | $ | (0.54 | ) | $ | (0.42 | ) | $ | (0.13 | ) | ||||
| (1) | Basic and diluted net income (loss) per common share for the four quarters of each fiscal year may not sum to the total for the fiscal year because of the different number of shares outstanding during each period. |
90
Table of Contents
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
The Company maintains disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended (the “Exchange Act”)) that are designed to ensure that required information is recorded, processed, summarized and reported within the required timeframe, as specified in the rules set forth by the Securities and Exchange Commission. Our disclosure controls and procedures are also designed to ensure that information required to be disclosed is accumulated and communicated to management, including the Chief Executive Officer and Chief Financial Officer, to allow timely decisions regarding required disclosures.
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures as of December 31, 2009 and, based on this evaluation, our Chief Executive Officer and Chief Financial Officer have concluded that our disclosure controls and procedures were effective as of December 31, 2009.
Management’s Report on Internal Control over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f) and 15d-15(f). Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles.
Internal control over financial reporting cannot provide absolute assurance of achieving financial reporting objectives because of its inherent limitations. Internal control over financial reporting is a process that involves human diligence and compliance and is subject to lapses in judgment and breakdowns resulting from human failures. Internal control over financial reporting also can be circumvented by collusion or improper management override. Because of such limitations, there is a risk that material misstatements may not be prevented or detected on a timely basis by internal control over financial reporting. However, these inherent limitations are known features of the financial reporting process. Therefore, it is possible to design into the process safeguards to reduce, though not eliminate, this risk.
Management conducted an assessment of the effectiveness of our internal control over financial reporting as of December 31, 2009. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control — Integrated Framework. Based on the results of this assessment and on those criteria, management concluded that our internal control over financial reporting was effective as of December 31, 2009.
The effectiveness of the Company’s internal control over financial reporting as of December 31, 2009 has been audited by PricewaterhouseCoopers LLP, an independent registered public accounting firm, as stated in their report which appears herein.
Changes in Internal Control over Financial Reporting
During the fourth fiscal quarter, there were no changes to our internal control over financial reporting that materially affected, or are reasonably likely to materially affect, our internal controls over financial reporting.
91
Table of Contents
None.
Item 10. Directors, Executive Officers and Corporate Governance
(a) The information required by this item with respect to our directors is incorporated by reference to the sections captioned “Proposal I: Election of Directors” and “Report of the Audit Committee” in the proxy statement for our annual meeting of shareholders to be held on June 17, 2010. We expect to file the proxy statement within 120 days of December 31, 2009, our fiscal year end.
(b) The information required by this item with respect to our executive officers is incorporated by reference to the section captioned “Executive Officers” in the proxy statement for our annual meeting of shareholders to be held on June 17, 2010.
(c) The information required by this item with respect to our corporate governance is incorporated by reference to the sections captioned “Corporate Governance,” “Report of the Audit Committee” and “Section 16(a) Beneficial Ownership Reporting Compliance” in the proxy statement for our annual meeting of shareholders to be held on June 17, 2010. We have adopted a Code of Ethics applicable to our chief executive officer, chief financial officer and others responsible for our corporate financial reporting. A copy of the Code of Ethics is available on our website at www.zymogenetics.com.
Item 11. Executive Compensation
The information required by this item with respect to executive compensation is incorporated by reference to the sections captioned “Executive Compensation” and “Corporate Governance” in the proxy statement for our annual meeting of shareholders to be held on June 17, 2010.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Shareholder Matters
The information required by this item with respect to beneficial ownership is incorporated by reference to the section captioned “Security Ownership of Certain Beneficial Owners and Management” in the proxy statement for our annual meeting of shareholders to be held on June 17, 2010.
92
Table of Contents
Equity Compensation Plan Information
The following table provides information regarding our equity compensation plans at December 31, 2009.
| Plan category |
Number of securities to be issued upon exercise of outstanding options, warrants and rights |
Weighted-average exercise price of outstanding options, warrants and rights |
Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)(1) | ||||
| (a) | (b) | (c) | |||||
| Equity compensation plans approved by security holders |
12,977,048 | $ | 8.85 | 4,742,524 | |||
| Equity compensation plans not approved by security holders |
— | — | — | ||||
| Total |
12,977,048 | $ | 8.85 | 4,742,524 | |||
| (1) | Does not include an increase of 2,700,000 shares, effective January 1, 2010, pursuant to a provision of the 2001 Plan that provides for an annual increase effective the first day of each year equal to the least of (i) 2,700,000 shares; (ii) 5% of the outstanding common stock as of the end of the Company’s preceding fiscal year; and (iii) a lesser amount as determined by the Board of Directors. |
Item 13. Certain Relationships and Related Transactions, and Director Independence
(a) The information required by this item with respect to certain relationships and related transactions is incorporated by reference to the section captioned “Certain Transactions” in the proxy statement for our annual meeting of shareholders to be held on June 17, 2010.
(b) The information required by this item with respect to director independence is incorporated by reference to the section captioned “Corporate Governance” in the proxy statement for our annual meeting of shareholders to be held on June 17, 2010.
Item 14. Principal Accountant Fees and Services
The information required by this item is incorporated by reference to the section captioned “Independent Registered Public Accounting Firm” in the proxy statement for our annual meeting of shareholders to be held on June 17, 2010.
93
Table of Contents
Item 15. Exhibits, Financial Statement Schedules
(a) The following documents are filed as part of this Form 10-K:
1. Financial Statements. The following financial statements are contained in Item 8 of this Annual Report on Form 10-K:
| Page in Form 10-K | ||
| 63 | ||
| 64 | ||
| 65 | ||
| 66 | ||
| 67 | ||
| 68 – 90 | ||
2. Financial Statement Schedules
All financial statement schedules have been omitted because the required information is either included in the financial statements or the notes thereto or is not applicable.
3. Exhibits
| Exhibit No. |
Description |
||||
| 3.1 | Amended and Restated Articles of Incorporation of ZymoGenetics, Inc. |
(A | ) | ||
| 3.2 | Articles of Amendment of ZymoGenetics, Inc. |
(C | ) | ||
| 3.3 | Amended and Restated Bylaws. |
(K | ) | ||
| 10.1† | Amended and Restated Employment Agreement, dated February 3, 2005, between ZymoGenetics, Inc. and Bruce L.A. Carter, Ph.D. |
(H | ) | ||
| 10.2† | Amended and Restated 2000 Stock Incentive Plan. |
(A | ) | ||
| 10.3† | 2001 Stock Incentive Plan. |
(A | ) | ||
| 10.4† | Amended and Restated Stock Option Grant Program for Nonemployee Directors under the ZymoGenetics 2001 Stock Incentive Plan. |
(S | ) | ||
| 10.5† | 2001 Stock Incentive Plan, Form of Stock Option Grant Notice. |
(J | ) | ||
| 10.6† | Deferred Compensation Plan for Key Employees. |
(A | ) | ||
| 10.7† | First Amendment to ZymoGenetics Deferred Compensation Plan for Key Employees. |
(M | ) | ||
| 10.8† | Second Amendment to ZymoGenetics Deferred Compensation Plan for Key Employees. |
(M | ) | ||
| 10.9† | Third Amendment to ZymoGenetics Deferred Compensation Plan for Key Employees. |
(M | ) | ||
| 10.10† | Executive Compensation Program (as of January 1,2009) |
(Q | ) | ||
| 10.11† | Amended and Restated Employment Agreement, dated as of June 16, 2009, between ZymoGenetics, Inc. and Stephen Zaruby |
(R | ) | ||
94
Table of Contents
| Exhibit No. |
Description |
||||
| 10.12† | Employment Agreement, dated as of June 8, 2009, between ZymoGenetics, Inc. and Eleanor L. Ramos, M.D. |
(R | ) | ||
| 10.13† | Separation Agreement, effective as of July 23, 2009, between ZymoGenetics, Inc. and Nicole Onetto, M.D. |
(R | ) | ||
| 10.14† | Employment Agreement, dated as of September 28, 2009, between ZymoGenetics, Inc. and Dennis M. Miller, Ph.D. |
(S | ) | ||
| 10.15† | Employment Agreement, dated as of July 3, 2008, between ZymoGenetics, Inc. and Heather L. Franklin |
||||
| 10.16† | Amended and Restated Employment Agreement, dated as of July 3, 2008, between ZymoGenetics, Inc. and James A. Johnson. |
||||
| 10.17† | Amended and Restated Employment Agreement, dated as of July 3, 2008, between ZymoGenetics, Inc. and Darren R. Hamby. |
||||
| 10.18† | Amended and Restated Employment Agreement, dated as of March 12, 2009, between ZymoGenetics, Inc. and Douglas E. Williams, Ph.D. |
||||
| 10.19* | License Agreement, dated December 31, 1998, as amended on February 4, 1999 and October 23, 2000, between ZymoGenetics, Inc. and St. Jude Children’s Research Hospital. |
(A | ) | ||
| 10.20* | Option and License Agreement, effective November 10, 2000, as amended effective as of June 16, 2000 and October 20, 2000, between ZymoGenetics, Inc. and Novo Nordisk A/S. |
(A | ) | ||
| 10.21* | First Amended and Restated Development and Marketing Agreement, dated August 28, 2008, between ZymoGenetics, Inc. and Ares Trading S.A. |
(O | ) | ||
| 10.22* | Exclusive Patent License Agreement, effective December 18, 2002, between ZymoGenetics, Inc. and Aventis Behring GmbH. |
(D | ) | ||
| 10.23 | Shareholders’ Agreement by and among ZymoGenetics, Inc., Novo Nordisk A/S, Novo Nordisk Pharmaceuticals, Inc. and the investors listed on Schedule A thereto, effective as of November 10, 2000. |
(A | ) | ||
| 10.24 | First Amendment to Shareholders’ Agreement by and among ZymoGenetics, Inc., Novo Nordisk A/S, Novo Nordisk Pharmaceuticals, Inc. and the investors listed on Schedule A thereto, dated as of February 4, 2002. |
(B | ) | ||
| 10.25 | Investors’ Rights Agreement by and among ZymoGenetics, Inc., Novo Nordisk Pharmaceuticals, Inc. and the persons listed on Schedule A thereto, effective as of November 10, 2000. |
(A | ) | ||
| 10.26 | Tax Sharing Agreement, effective October 20, 2000, between ZymoGenetics, Inc. and Novo Nordisk of North America, Inc. |
(A | ) | ||
| 10.27 | Lease Agreement, dated October 4, 2002, between ZymoGenetics, Inc. and ARE-1201/1208 Eastlake Avenue, LLC. |
(D | ) | ||
| 10.28 | Amendment No. 2 to Lease Agreement, dated July 19, 2004, between ZymoGenetics, Inc. and ARE-1201/1208 Eastlake Avenue, LLC. |
(F | ) | ||
| 10.29 | Lease Agreement, dated October 4, 2002, between ZymoGenetics, Inc. and ARE-1208 Eastlake Avenue, LLC. |
(D | ) | ||
| 10.30 | Amendment No. 2 to Lease Agreement, dated June 14, 2004, between ZymoGenetics, Inc. and ARE-/1208 Eastlake Avenue, LLC. |
(F | ) | ||
95
Table of Contents
| Exhibit No. |
Description |
||||
| 10.31 | Restated Office Lease Agreement, dated March 1, 2008, between ZymoGenetics, Inc. and 1144 Eastlake LLC. |
(M | ) | ||
| 10.32* | Development and Supply Agreement, dated October 1, 2003, between ZymoGenetics, Inc. and Abbott Laboratories. |
(E | ) | ||
| 10.33* | First Amended and Restated Strategic Alliance Agreement, dated August 28, 2008, between ZymoGenetics, Inc. and Serono Technologies S.A. |
(O | ) | ||
| 10.34* | License Agreement for Recombinant Factor XIII, dated October 4, 2004, among ZymoGenetics, Inc., Novo Nordisk A/S and Novo Nordisk Health Care AG. |
(G | ) | ||
| 10.35* | Amendment No. 1 to License Agreement for Recombinant Factor XIII, dated December 7, 2007, among ZymoGenetics, Inc., Novo Nordisk A/S and Novo Nordisk Health Care AG. |
(L | ) | ||
| 10.36* | License and Transfer Agreement, effective as of January 16, 2009, by and between ZymoGenetics, Inc. and Novo Nordisk A/S |
(Q | ) | ||
| 10.37* | Third Amended and Restated License Agreement for IL-21 Embodiments, dated December 3, 2009, by and between ZymoGenetics, Inc. and Novo Nordisk A/S |
||||
| 10.38* | Manufacturing Service Agreement relating to rhThrombin between Patheon Italia S.p.A. and ZymoGenetics, Inc., executed January 19, 2007. |
(I | ) | ||
| 10.39 | Amendment No. 1 to Manufacturing Service Agreement relating to rhThrombin between Patheon Italia S.p.A. and ZymoGenetics, Inc., executed December 3, 2007 |
(L | ) | ||
| 10.40* | U. S. Co-Promotion Agreement by and between ZymoGenetics, Inc. and Bayer HealthCare, LLC, executed June 18, 2007. |
(J | ) | ||
| 10.41* | Amendment to U. S. Co-Promotion Agreement by and among ZymoGenetics, Inc., ZymoGenetics, LLC and Bayer HealthCare, LLC, executed December 18, 2009. |
||||
| 10.42* | License and Collaboration Agreement by and between ZymoGenetics, Inc. and Bayer Schering Pharma AG, executed June 18, 2007. |
(J | ) | ||
| 10.43* | Amendment to License and Collaboration Agreement by and among ZymoGenetics, Inc., ZymoGenetics, LLC and Bayer Schering Pharma AG, executed December 18, 2009. |
||||
| 10.44* | Facility Agreement dated June 26, 2008 among ZymoGenetics, Inc., Deerfield Private Design Fund, L.P. and Deerfield Private Design International, L.P. |
(N | ) | ||
| 10.45 | Amendment No.1 to Facility Agreement, dated October 22, 2008, among ZymoGenetics, Inc., Deerfield Private Design Fund, L.P., Deerfield Private Design International, L.P. and Deerfield ZG Corporation |
||||
| 10.46 | Amendment No.2 to Facility Agreement, dated December 31, 2009, among ZymoGenetics, Inc., Deerfield Private Design Fund, L.P., Deerfield Private Design International, L.P. and Deerfield ZG Corporation |
||||
| 10.47* | Promissory Note dated June 26, 2008, with issuer ZymoGenetics, Inc. and holder Deerfield Private Design International, L.P. |
(N | ) | ||
| 10.48* | Promissory Note dated June 26, 2008, with issuer ZymoGenetics, Inc. and holder Deerfield Private Design Fund, L.P. |
(N | ) | ||
| 10.49* | Royalty Agreement dated June 26, 2008 among ZymoGenetics, Inc., Deerfield Private Design Fund, L.P. and Deerfield Private Design International, L.P. |
(N | ) | ||
| 10.50 | Registration Rights Agreement dated June 26, 2008 among ZymoGenetics, Inc., Deerfield Private Design Fund, L.P. and Deerfield Private Design International, L.P. |
(N | ) | ||
96
Table of Contents
| Exhibit No. |
Description |
||||
| 10.51* | Form of Warrant to Purchase Common Stock of ZymoGenetics, Inc. relating to the Facility Agreement. |
(N | ) | ||
| 10.52* | Release and License Agreement, dated October 22, 2008, by and between ZymoGenetics and Bristol-Myers Squibb Company |
(P | ) | ||
| 10.53* | Co-Development/Co-Promotion and License Agreement relating to Type-3 Interferon Family, dated January 12, 2009, by and among ZymoGenetics, Inc., ZymoGenetics, LLC and Bristol-Myers Squibb Company |
(Q | ) | ||
| 23.1 | Consent of PricewaterhouseCoopers LLP, independent registered public accounting firm. |
||||
| 31.1 | Certifications of Chief Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
||||
| 31.2 | Certifications of Chief Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
||||
| 32.1 | Certification of Chief Executive Officer and Chief Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
||||
| † | Management contract or compensatory plan or arrangement. |
| * | Portions of these exhibits have been omitted based on a grant of confidential treatment from the Securities and Exchange Commission. The omitted portions of these exhibits have been filed separately with the SEC. |
| (A) | Incorporated by reference to ZymoGenetics, Inc. Registration Statement on Form S-1 (No. 333-69190) filed on September 10, 2001, as amended. |
| (B) | Incorporated by reference to ZymoGenetics, Inc. Quarterly Report on Form 10-Q for the quarter ended March 31, 2002. |
| (C) | Incorporated by reference to ZymoGenetics, Inc. Quarterly Report on Form 10-Q for the quarter ended June 30, 2002. |
| (D) | Incorporated by reference to ZymoGenetics, Inc. Annual Report on Form 10-K for the year ended December 31, 2002. |
| (E) | Incorporated by reference to ZymoGenetics, Inc. Quarterly Report on Form 10-Q for the quarter ended September 30, 2003. |
| (F) | Incorporated by reference to ZymoGenetics, Inc. Quarterly Report on Form 10-Q for the quarter ended June 30, 2004. |
| (G) | Incorporated by reference to ZymoGenetics, Inc. Annual Report on Form 10-K for the year ended December 31, 2004. |
| (H) | Incorporated by reference to ZymoGenetics, Inc. Current Report on Form 8-K dated as of February 3, 2005. |
| (I) | Incorporated by reference to ZymoGenetics, Inc. Quarterly Report on Form 10-Q for the quarter ended March 31, 2007. |
| (J) | Incorporated by reference to ZymoGenetics, Inc. Quarterly Report on Form 10-Q for the quarter ended June 30, 2007. |
| (K) | Incorporated by reference to ZymoGenetics, Inc. Current Report on Form 8-K dated as of November 15, 2007. |
| (L) | Incorporated by reference to ZymoGenetics, Inc. Annual Report on Form 10-K for the year ended December 31, 2007. |
| (M) | Incorporated by reference to ZymoGenetics, Inc. Quarterly Report on Form 10-Q for the quarter ended March 31, 2008. |
| (N) | Incorporated by reference to ZymoGenetics, Inc. Quarterly Report on Form 10-Q for the quarter ended June 30, 2008. |
97
Table of Contents
| (O) | Incorporated by reference to ZymoGenetics, Inc. Quarterly Report on Form 10-Q for the quarter ended September 30, 2008. |
| (P) | Incorporated by reference to ZymoGenetics, Inc. Annual Report on Form 10-K for the year ended December 31, 2008. |
| (Q) | Incorporated by reference to ZymoGenetics, Inc. Quarterly Report on Form 10-Q for the quarter ended March 31, 2009. |
| (R) | Incorporated by reference to ZymoGenetics, Inc. Quarterly Report on Form 10-Q for the quarter ended June 30, 2009. |
| (S) | Incorporated by reference to ZymoGenetics, Inc. Quarterly Report on Form 10-Q for the quarter ended September 30, 2009. |
98
Table of Contents
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Company has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized.
| ZYMOGENETICS, INC. | ||||
| Date: February 26, 2010 |
By: | /s/ DOUGLAS E. WILLIAMS, PH.D. | ||
| Douglas E. Williams, Ph.D. Chief Executive Officer | ||||
Each person whose individual signature appears below hereby authorizes and appoints Douglas E. Williams, Ph.D. and James A. Johnson, and each of them, with full power of substitution and resubstitution and full power to act without the other, as his or her true and lawful attorney-in-fact and agent to act in his or her name, place and stead and to execute in the name and on behalf of each person, individually and in each capacity stated below, and to file, any and all amendments to this Report, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing, ratifying and confirming all that said attorneys-in-fact and agents or any of them or their or his or her substitute or substitutes may lawfully do or cause to be done by virtue thereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this Report has been signed below by the following persons on behalf of the Company and in the capacities and on the dates indicated.
| Signature |
Title |
Date | ||
| /S/ DOUGLAS E. WILLIAMS, PH.D. Douglas E. Williams, Ph.D. |
Chief Executive Officer and Director (Principal Executive Officer) |
February 26, 2010 | ||
| /S/ JAMES A. JOHNSON James A. Johnson |
Executive Vice President, Chief Financial Officer and Treasurer (Principal Accounting and Financial Officer) |
February 26, 2010 | ||
| /S/ BRUCE L.A. CARTER, PH.D. Bruce L.A. Carter, Ph.D. |
Chairman of the Board of Directors |
February 26, 2010 | ||
| /S/ JAMES A. HARPER James A. Harper |
Director |
February 26, 2010 | ||
| /S/ DAVID I. HIRSH, PH.D. David I. Hirsh, Ph.D. |
Director |
February 26, 2010 | ||
| /S/ JONATHAN S. LEFF Jonathan S. Leff |
Director |
February 26, 2010 | ||
| /S/ DAVID H. MACCALLUM David H. MacCallum |
Director |
February 26, 2010 | ||
| /S/ KURT ANKER NIELSEN Kurt Anker Nielsen |
Director |
February 26, 2010 | ||
| /S/ EDWARD E. PENHOET, PH.D. Edward E. Penhoet, Ph.D. |
Director |
February 26, 2010 | ||
| /S/ LARS REBIEN SØRENSEN Lars Rebien Sørensen |
Director |
February 26, 2010 | ||
99